-
GEBIET DER ERFINDUNG
-
Die
vorliegende Erfindung betrifft neue substituierte Imidazolverbindungen
mit wertvollen pharmakologischen Eigenschaften, insbesondere gegen
entzündliche
Erkrankungen und allergische Zustände. Erfindungsgemäße Verbindungen
sind Antagonisten der Histaminrezeptoren. Einige sind Antagonisten
der Histamin-H1-Rezeptoren. Einige sind Antagonisten der
Histamin-H3-Rezeptoren. Einige sind Antagonisten
von sowohl den H1- als auch den H3-Rezeptoren, in anderen Worten duale H1- und H3-Rezeptorantagonisten.
Die in dieser Anmeldung offenbarte Erfindung beansprucht die Priorität der vorläufigen Anmeldung
mit dem Aktenzeichen 60/230,053, eingereicht am 20. September 2000,
und ist verwandt mit derjenigen in den anhängigen vorläufigen Anmeldungen mit den
Aktenzeichen Nr. 60/234,039, 60/234 040 und Nr. 60/234 038, alle
eingereicht am 20. September 2000.
-
HINTERGRUND DER ERFINDUNG
-
Die
Histaminrezeptoren H1, H2 und
H3 sind gut identifizierte Formen. Die H1-Rezeptoren sind jene, die die Reaktion
vermitteln, die durch konventionelle Antihistamine antagonisiert
wird. H1-Rezeptoren sind beispielsweise
im Ileum, der Haut und der glatten Bronchialmuskeln von Menschen
und anderen Säugern
vorhanden. Ein wohl bekannter Antagonist von H1-Rezeptoren
ist Loratadin, im Handel erhältlich
unter der Handelsbezeichnung CLARITIN® von
Schering-Plough Corporation, Madison, New Jersey, USA. Durch H2-Rezeptor-vermittelte Reaktionen stimuliert Histamin
die Magensäuresekretion
bei Säugern
und den chronotropen Effekt im isoliertem Herzvorhof von Säugern.
-
H3-Rezeptorstellen finden sich an sympathischen
Nerven, wo sie sympathische Neurotransmission modulieren und eine
Vielfalt von Endorganreaktionen unter Kontrolle des sympathischen
Nervensystems abschwächen.
H3-Rezeptoraktivierung durch Histamin schwächt spezifisch
die Norepinephrinausschüttung
an Widerstands- und Kapazitätsgefäße ab, was
Vasodilatation (Gefäßerweiterung)
herbeiführt.
-
US 4,767,778 (Arrang et
al.) offenbart bestimmte Imidazole, die sich wie Agonisten des H
3-Rezeptors im Rattenhirn verhalten.
EP-A2-0 420 396 (Smith
Kline & French
Laboratories Limited) und Howson et al. offenbaren Imidazolderivate
mit einer Amidingruppe als H
3-Agonisten.
Van der Groot et al. (Eur. J. Med. Chem. (1992) Band 27, Seiten
511–517)
beschreibt Isothioharnstoffanaloga von Histamin als potente Agonisten
oder Antagonisten des Histamin-H
3-Rezeptors,
und diese Isothioharnstoffanaloga von Histamin überschneiden sich teilweise
mit jenen der beiden bereits zitierten Druckschriften. Clapham et
al. ["Ability of
Histamine-H
3 Receptor Antagonists to Improve
Cognition and to Increase Acetylcholine Release in vivo in the Rat", British Assn. for Psychopharmacology,
25–28
Juli (1993), berichtet in J. Psychopharmacol. (Abstr. Book), A17],
beschreiben die Fähigkeit
von Histamin-H
3-Rezeptoren, die Wahrnehmung
zu verbessern und die Freisetzung von Acetylcholin in vivo bei der
Ratte zu erhöhen.
Clapham et al. ["Ability
of the selective Histamine-H
3 Receptor Antagonist
Thioperamide to improve Short-term Memory and Reversal Learning
in the Rat", Brit.
J. Pharm. Suppl., 1993, Abstract 652] stellt Ergebnisse vor, die
zeigen, dass Thioperamid das Kurzzeitgedächtnis und Umkehrlernen bei der
Ratte verbessern kann und bringen dies in Zusammenhang mit der Beteiligung
von H
3-Rezeptoren bei der Mo dulation der
kognitiven Funktion. Yokoyama et al. ["Effect of Thioperamide, a Histamine-H
3 Receptor Antagonist, an Electrically Induced
Convulsions in Mice",
Eur. J. Pharmacol. (1993), Band 234, Seiten 129–133] berichtet, wie Thioperamid
die Dauer jeder Krampfphase vermindert und die Elektrokrampfschwelle
erhöht, und
schlagen weiter vor, dass diese und andere Befunde die Hypothese
stützen,
dass das zentrale histaminerge System an der Inhibierung von Anfällen beteiligt
ist. Die internationale Patentveröffentlichung Nr.
WO 9301812-A1 (SmithKline
Beecham PLC) beschreibt die Verwendung von S-[3-(4(5)-Imidazolyl)propyl]isothioharnstoff
als Histamin-H
3-Antagonist, insbesondere
zur Behandlung kognitiver Störungen,
z. B. Morbus Alzheimer und altersbedingter nachlassender Gedächtnisleistung.
Schlicker et al. ["Novel
Histamine-H
3 Receptor Antagonists: Affinities
in an H
3 Receptor Einding Assay and Potencies
in Two Functional H
3 Receptor Models", British J. Pharmacol.,
(1994), Band 112, 1043–1048
beschreiben eine Reihe von Imidazolylalkylverbindungen, bei denen
die Imidazolylalkylgruppe an eine Guanidingruppe, eine Estergruppe,
eine Amidgruppe, eine Thioamidgruppe und eine Harnstoffgruppe gebunden
ist, und verglichen diese mit Thioperamid. Leurs et al. ["The Histamine-H
3-receptor:
A Target for Developing New Drugs", Progr. Drug Res. (1992), Band 39,
Seiten 127–165]
und Lipp et al. ["Pharmacochemistry
of H
3-receptors" in The Histamine Receptor, Herausgeber: Schwartz
und Haas, Wiley-Liss, New York (1992), Seiten 57–72] besprechen eine Vielfalt
synthetischer H
3-Rezeptorantagonisten, und
Lipp et al. (ibid.) haben die notwendigen strukturellen Anforderungen
an einen H
3-Rezeptorantagonisten vorgeschlagen.
-
WO 95/14007 beansprucht
H
3-Rezeptorantagonisten mit der Formel
wobei
A, m, n, R
1 und R
2 darin
definiert sind. Von diesen Verbindungen wird offenbart, dass sie
brauchbar zur Behandlung verschiedener Störungen sind, insbesondere solcher,
die durch allergieinduzierte Reaktionen hervorgerufen werden.
-
WO 93/12093 offenbart Imidazolylmethylpiperazine
und Diazepine als H
3-Antagonisten. Die US-Patentanmeldung
mit dem Aktenzeichen Nr. 08/965 754, eingereicht am 7. November
1997, offenbart imidazolylalkylsubstituierte heterocyclische Ringverbindungen
als H
3-Rezeptorantagonisten. Die US-Patentanmeldung mit
dem Aktenzeichen Nr. 08/966 344, eingereicht am 7. November 1997,
offenbart Phenylalkylimidazole als H
3-Rezeptorantagonisten.
-
WO 96/29315 (
PCT/FR96/00432 ) offenbart bestimmte
N-Imidazolylalkylverbindungen, die gebundene Phenyleinheiten enthalten.
-
H3-Rezeptorantagonisten werden ebenfalls offenbart
in: H. Stark et al., Eur. J. of Pharmaceutical Sciences (1995) 3,
95–104;
H. Stark et al., J. Med. Chem., (1996) 39, 1157–1163; H. Stark et al., Arch.
Pharm. Pharm. Med. Chem., (1998) 331, 211–218; und A. Sasse et al.,
Bioorganic & Medicinal
Chem., (2000) 8, 1139, 1139–1149.
-
Es
wird auch auf J. R. Bagley et al., Journal of Medicinal Chemistry,
(1991), Band 34, 827–841,
verwiesen, worin unter anderem N-(imidazolylalkyl)substituierte
cyclische Aminverbindungen offenbart sind, die als Analgetika brauchbar
sind, wie die Aminverbindung mit der Formel:
-
Die
anhängige
US-Patentanmeldung mit dem Aktenzeichen Nr. 09/173,642, eingereicht
16. Oktober 1998 (R. Wolin et al.), offenbart N-(imidazolylalkyl)substituierte
cyclische Aminverbindungen mit H3-Antagonistaktivität.
-
A.
Huls et al., Bioorg. & Med.
Chem. Letters, 6 (1996), 2013–2018
offenbaren Imidazolverbindungen, die Diphenylethereinheiten enthalten,
als H
3-Rezeptorantagonisten. Es wird zudem
offenbart, dass die Verbindungen H
1-Rezeptorantagonistaktivität haben.
Eine beispielhafte Verbindung aus jener Veröffentlichung ist:
wobei R
1 und
R
2 wie dort definiert sind.
-
A.
Buschauer, J. Med. Chem., 32 (1989), 1963–1970 offenbart unter anderem
H
2-Rezeptorantagonisten des Typs:
wobei Ar
1 und
Ar
2 Phenyl und/oder Pyridyl sein können.
EP-A1-448 765 (veröffentlicht
am 30. März
1990) offenbart Neuropeptid-Y-Antagonist-Imidazole des Typs:
wobei Ar
1 und
Ar
2 Phenyl und/oder Pyridyl sein können.
-
WO 98-58646 (übertragen
auf Novo Nordisk A/S) offenbart Somatostatin-SSTR4-Rezeptorantagonistverbindungen
des Typs:
worin
m 2-6 ist; n 1-3 ist; p 1-6 ist; R
1 und
R
2 unabhängig
H oder C
1-C
6-Alkyl
sind, das gegebenenfalls mit Halogen, Amino, Hydroxy, Alkoxy oder
Aryl substituiert ist; X S, O, NH, NCOPh oder N(CN) ist; A Aryl
ist, das gegebenenfalls mit Halogen, Amino, Hydroxy, Nitro, C
1-6-Alkyl, C
1-6-Alkoxy
oder Arylsubstituiert ist; und B und D unabhängig Aryl sind, das gegebenenfalls
mit Halogen, Amino, Hydroxy, C
1-6-Alkyl,
C
1-6-Alkoxy oder Aryl substituiert ist.
-
Es
ist in der Literatur über
Verbindungen berichtet worden, die Aktivität gegen sowohl H1-
als auch H2-Rezeptoren zeigen, d. h. duale
Antagonisten gegen H1- und H2-Rezeptoren.
So berichten beispielsweise F. Schulze et al., European J. of Pharmaceutical
Sciences, 6 (1998), 177–186, über kombinierte
H1/H2-Rezeptorantagonisten.
Zu anderen Druckschriften in dieser Kategorie gehören F. Schulze
et al., Arch. Pharm. (Weinheim), 327 (1994), 455–462; C. Wolf et al., Arch.
Pharm. Pharm. Med. Chem., 329 (1996), 87–94; und C. Wolf et al., European
J. of Pharmaceutical Sciences, 6 (1998), 177–186. Über Nicht-Imidazolhistamin-H3-Liganden, insbesondere substituierte Benzothiazolderivate
als H3-Antagonisten und H1-Blockierungsaktivitäten sind
von K. Walczynski et al, Il Farmaco, 54 (1999), 684–694 berichtet
worden.
-
Es
wäre nützlich, über Verbindungen
zu verfügen,
die als Antagonisten von sowohl den H
1-
als auch den H
3-Histaminrezeptoren therapeutisch
wirksam sind. Die einzige derartige berichtete Aktivität war mittels einer
Kombination zweier verschiedener chemischer Strukturen, wobei eine
Aktivität
gegen H
1-Rezeptoren und die andere Aktivität gegen
H
3-Rezeptoren zeigte. So offenbart beispielsweise
US 5,869,479 (ausgegeben am
9. Februar 1999, Schering Corporation) die Kombination eines Histamin-H
1-Rezeptorantagonisten und eines Histamin-H
3-Rezeptorantagonisten zur Behandlung allergieinduzierter
Reaktionen der Luftwege.
-
Die
anhängige
vorläufige
Patentanmeldung mit dem Aktenzeichen Nr. 60/234.039, eingereicht
am 20. September 2000, offenbart neue Imidazolverbindungen mit H3- sowie duale H1-
und H3-Antagonistaktivität. Die dort
offenbarten Verbindungen haben allgemeine Formeln, in denen ein
Imidazol mittels einer oder mehreren Intermediäreinheiten an eine tricyclische
Einheit gebunden ist, wobei mindestens eine Intermediäreinheit
oder -einheiten eine cyclische Einheit ist.
-
Die
anhängige
vorläufige
Patentanmeldung mit dem Aktenzeichen Nr. 60/234.038, eingereicht
am 20. September 2000, offenbart neue Imidazolverbindungen mit H3- sowie dualer H1-
und H3-Antagonistaktivität. Die dort
offenbarten Verbindungen haben allgemeine Formeln, in denen ein
Imidazol mittels einer oder mehreren Intermediäreinheiten an eine tricyclische
Einheit gebunden ist, wobei die Intermediäreinheit oder -einheiten alle
acyclische Einheiten sind.
-
Die
anhängige
vorläufige
Patentanmeldung mit dem Aktenzeichen Nr. 60/234.040, eingereicht
am 20. September 2000, offenbart neue Imidazolverbindungen mit H3- sowie dualer H1-
und H3-Antagonistaktivität. Die dort
offenbarten Verbindungen haben allgemeine Formeln, in denen ein
Imidazol mittels einer oder mehreren Intermediäreinheiten an zwei cyclische
Einheiten gebunden ist, wobei die Intermediäreinheit oder -einheiten acyclisch
ist bzw. sind.
-
Es
wäre ein
willkommener Beitrag zu der Technik, über neue substituierte Imidazolverbindungen
zu verfügen.
-
Es
wäre brauchbar,
wenn dieselbe chemische Struktur duale Aktivität gegen sowohl H1-
als auch H3-Rezeptoren zeigen würde.
-
Es
wäre brauchbar, über neue
substituierte Imidazole zu verfügen,
die Aktivität
gegen sowohl H1- als auch H3-Rezeptoren
zeigen.
-
US 5,801,175 (ausgegeben
am 1. September 1998, Rechtsnachfolger: Schering Corporation) offenbart
Verbindungen mit der folgenden allgemeinen Strukturformel als Inhibitoren
der G-Proteinfunktion
und zur Behandlung proliferierender Erkrankungen:
worin A, B, W, X, Z, R
1 und R
2 dort definiert
sind. Dort sind Imidazole sowie andere Typen von Verbindungen offenbart.
-
US 5,719,148 (ausgegeben
am 17. Februar 1998, Rechtsnachfolger: Schering Corporation),
US 4,826,853 (ausgegeben
am 2. Mai 1989, Rechtsnachfolger: Schering Corporation) und
WO 96/30363 (veröffentlicht
am 3. Oktober 1996, Rechtsnachfolger: Schering Corporation) offenbaren
Verbindungen mit der folgenden allgemeinen Strukturformel als Inhibitoren
der G-Proteinfunktion und zur Behandlung proliferierender Erkrankungen:
wobei die verschiedenen Elemente
dort definiert sind. Dort sind Imidazole sowie andere Typen von
Verbindungen offenbart. Auf die bereits genannten
US 5,801,175 und
US 4,826,853 und
WO 96/30363 wird Bezug genommen.
-
Es
ist nun gefunden worden, dass bestimmte Imidazolverbindungen, die
in den oben genannten
US 5,801,175 und
US 4,826,853 und
WO 96/30363 offenbart sind
oder worauf dort Bezug genommen wurde, überraschenderweise H
3- sowie duale H
1-
und H
3-Antagonistaktivität zeigen. Die vorliegende Anmeldung
offenbart überraschende
Potenz und Verwendung dieser Imidazole, die die allgemeine Formel
haben, in der ein Imidazol über
eine oder mehrere Intermediäreinheit(en)
an eine tricyclische Einheit gebunden ist, wobei mindestens eine
der Intermediäreinheit(en)
eine cyclische Einheit ist. Die H
3-Aktivität sowie
duale H
1/H
3-Aktivität dieser
Verbindungen ist bislang noch nicht offenbart worden.
-
ZUSAMMENFASSUNG DER ERFINDUNG
-
Diese
Erfindung liefert in einer Ausführungsform
substituierte Imidazolverbindungen mit H
3-Antagonistaktivität sowie
dualer H
1- und H
3-Antagonistaktivität. Die erfindungsgemäßen Verbindungen
sind substituierte Imidazole, in denen ein Imidazol mittels einer
oder mehreren Intermediäreinheiten
an eine tricyclische Einheit gebunden ist, wobei mindestens eine
Intermediäreinheit
oder -einheiten eine cyclische Einheit ist. Die Verbindungen haben
die in Formel I gezeigte allgemeine Struktur:
in der f = 0, 1 oder 2 ist;
X
und Y unabhängig
ausgewählt
sind aus der Gruppe bestehend aus N, CH oder N-Oxid;
G eine
Einheit ausgewählt
aus der Gruppe bestehend aus den Einheiten II, III und IV ist, wobei
das obere Ende von II, III und IV mit der tricyclischen Einheit
verbunden ist und das untere Ende von II, III und IV an M gebunden
ist:
wobei
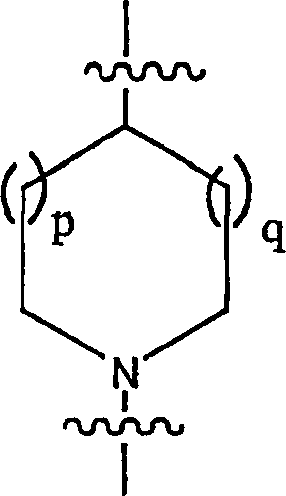
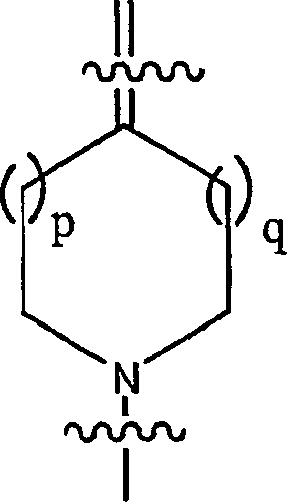
s = t = 1 oder 2; und p = q = 0, 1 oder 2;
M eine Einheit ausgewählt aus
der Gruppe bestehend aus C
1-C
8-Alkyl;
-C(O)-(CH
2)
y; -(CH
2)
x-A-(CH
2)
y-; -C(O)-O-(CH
2)
d- und -C(O)-NR
3(CH
2)
d-
ist; wobei A = O, S(O)
r- und -NR
4-;
n = 0, 1, 2 oder 3 ist;
x eine
ganze Zahl im Bereich von 2 bis 5 ist;
y eine ganze Zahl im
Bereich von 0 bis 5 ist;
d eine Zahl im Bereich von 0 bis 5
ist; r = 0, 1 oder 2 ist;
R
1 und R
2 in irgendeiner Zahl von 1–3 vorhanden
sind und unabhängig
ausgewählt
sind aus der Gruppe bestehend aus Wasserstoff, niederem Alkyl, niederem
Alkoxy, Halogen, OCF
3, OCHF
2,
-OH und -N(R
4)
2;
R
3 ausgewählt
ist aus der Gruppe bestehend aus Wasserstoff, niederem Alkyl und
Poly(halogen)niederem Alkyl;
R
4 ausgewählt ist
aus Wasserstoff, niederem Alkyl, Poly(halogen)-niederem Alkyl, und
R
5 H, (C
1-C
6)-Alkyl oder OH ist.
-
Die
folgenden Begriffe haben hier die angegebenen Bedeutungen:
niederes
Alkyl (einschließlich
der Alkylanteile von niederem Alkoxy) – stehen für eine geradkettige oder verzweigte
gesättigte
Kohlenwasserstoffkette mit 1 bis 6, vorzugsweise 1 bis 4 Kohlenstoffatomen;
Halogen – steht
für Fluor,
Chlor, Brom und Iod;
Der Begriff "substituiert" bezieht sich, wenn nicht anders definiert,
auf chemisch geeignete Substitution mit Einheiten wie beispielsweise
Alkyl, Alkoxy, -CF3, Halogen oder Aryl.
Es sei darauf hingewiesen, dass der Begriff "Alkyl", wenn dies geeignet erscheint, Alkylen
einschließt,
wobei "Alkylen" als eine Alkylgruppe
definiert ist, bei der eines der Wasserstoffatome durch eine Bindung
ersetzt wurde.
-
Zu
dieser Erfindung gehören
auch Tautomere, Enantiomere und andere optische Isomere der Verbindungen
der Formel I sowie pharmazeutisch annehmbare Salze und Solvate davon.
-
Ein
weiteres Merkmal der Erfindung sind pharmazeutische Zusammensetzungen,
die als aktiven Bestandteil eine Verbindung der Formel I (oder deren
Salz, Solvat oder Isomere) zusammen mit einem pharmazeutisch annehmbaren
Träger
oder Hilfsstoff enthalten.
-
Die
Erfindung liefert auch Verfahren zur Herstellung von Verbindungen
der Formel I sowie Verfahren zur Behandlung von Erkrankungen, wie
beispielsweise Entzündung,
Allergie, Erkrankungen des Gastrointestinaltrakts, Herz-Kreislauf-Erkrankungen
oder Störungen
des zentralen Nervensystems sowie allergieinduzierten Reaktionen
der Luftwege (z. B. der oberen Luftwege), Schwellung der Nase sowie
Fettleibigkeit. Die Behandlungsverfahren beinhalten das Verabreichen
einer therapeutisch wirksamen Menge einer Verbindung der Formel
I oder pharmazeutischen Zusammensetzungen, die eine Verbindung der
Formel I enthalten, an einen Säugerpatienten
(einschließlich
Mensch und Tieren), der an der Erkrankung oder den Erkrankungen
leidet.
-
DETAILLIERTE BESCHREIBUNG
DER ERFINDUNG
-
Die
vorliegende Erfindung liefert in einer Ausführungsform neue Imidazolverbindungen
der Formel I als Verbindungen, die H
1-Antagonistaktivität oder H
3-Antagonistaktivität oder duale H
1-
und H
3-Antagonistaktivität zeigen.
wobei die verschiedenen
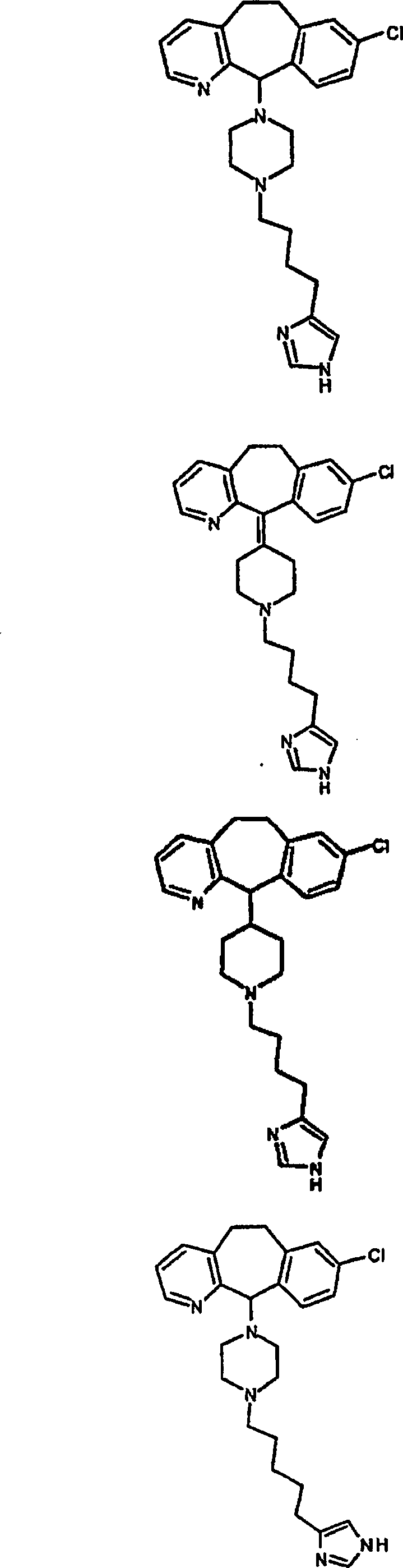
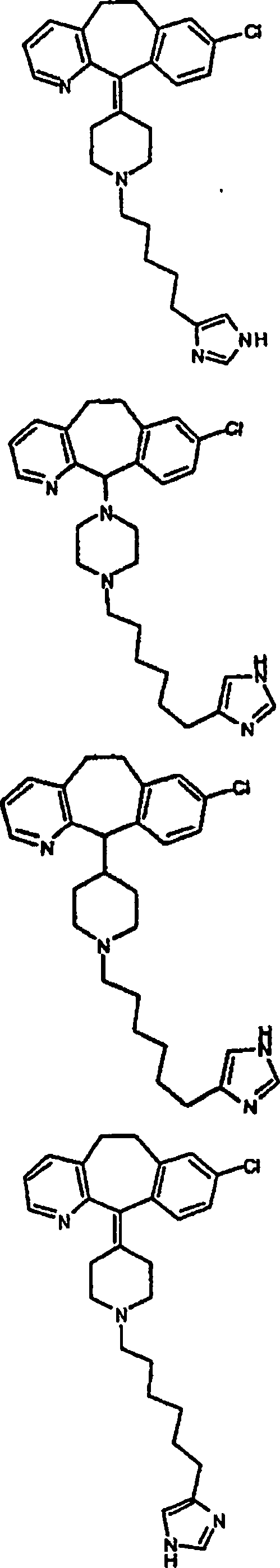
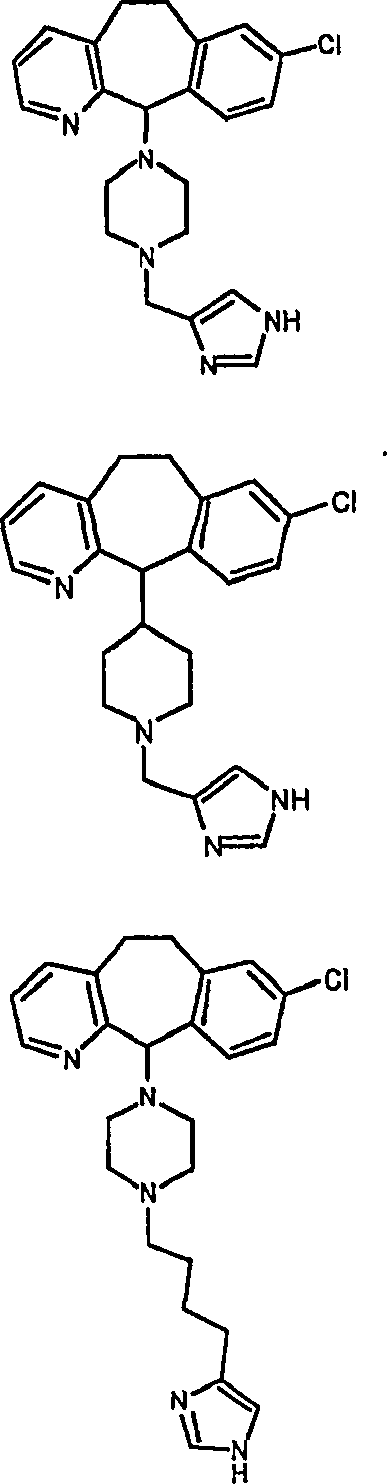
Symbole wie oben definiert sind. Nachfolgend werden repräsentative
Verbindungen der Erfindung gezeigt, die H
3-Antagonistaktivität zeigen:
-
Zu
einigen Beispielen für
Verbindungen, die sowohl H
1- als auch H
3-Aktivität
zeigen, gehören:
-
Zu
repräsentativen
Verbindungen, die H
1-Aktivität zeigen,
gehören:
-
Die
erfindungsgemäßen Verbindungen
sind basisch und bilden pharmazeutisch annehmbare Salze mit organischen
und anorganischen Säuren.
Beispiele für
geeignete Säuren
für die
Salzbil dung sind Salz-, Schwefel-, Phosphor-, Essig-, Citronen-,
Oxal-, Malon-, Salicyl-, Äpfel-,
Fumar-, Bernstein-, Ascorbin-, Malein-, Methansulfonsäure und
andere Mineral- und Carbonsäuren,
die Fachleuten wohl bekannt sind. Die Salze werden hergestellt,
indem die freie Basenform mit einer ausreichenden Menge der gewünschten
Säure kontaktiert wird,
um in konventioneller Weise ein Salz zu produzieren. Die freien
Basenformen können
durch Behandlung des Salzes mit einer geeigneten verdünnten wässrigen
Basenlösung
regeneriert werden, wie mit verdünntem wässrigem
Natriumhydroxid, Kaliumcarbonat, Ammoniak oder Natriumbicarbonat.
Die freien Basenformen unterscheiden sich in bestimmten physikalischen
Eigenschaften etwas von ihren jeweiligen Salzformen, wie Löslichkeit
in polaren Lösungsmitteln,
die Salze sind ansonsten für
erfindungsgemäße Zwecke
jedoch zu ihren jeweiligen freien Basenformen äquivalent.
-
In
Abhängigkeit
von den Substituenten der erfindungsgemäßen Verbindungen ist es vielleicht
möglich, auch
Salze mit Basen zu bilden. Wenn es beispielsweise Carbonsäuresubstituenten
in dem Molekül
gibt, können
Salze mit anorganischen sowie mit organischen Basen gebildet werden,
wie beispielsweise NaOH, KOH, NH4OH, Tetraalkylammoniumhydroxid
und dergleichen.
-
Wie
bereits gesagt schließt
die Erfindung auch Tautomere, Enantiomere und andere Stereoisomere der
Verbindungen ein. Wie der Fachmann weiß, können bestimmte Imidazolverbindungen
in tautomeren Formen vorliegen. Solche Variationen werden als innerhalb
des Umfangs der Erfindung liegend angesehen.
-
Eine
weitere Ausführungsform
der Erfindung offenbart ein Verfahren zur Herstellung der oben offenbarten
substituierten Imidazole. Die Verbindungen können nach mehreren Verfahren
hergestellt werden, die in der Technik wohl bekannt sind. Bei einem
Verfahren können
der Imidazolanteil (hier der Einfachheit halber als "die linke Komponente" bezeichnet) und
der Di arylanteil (hier der Einfachheit halber als "die rechte Komponente" bezeichnet) getrennt
hergestellt werden. Die linke Komponente und die rechte Komponente
können
an sie gebundene reaktive Einheiten enthalten, wobei die Einheiten
unter geeigneten Reaktionsbedingungen miteinander umgesetzt werden
können.
Die linke Komponente kann somit beispielsweise ein Carbethoxyende
enthalten, und die rechte Komponente kann ein Aminende aufweisen.
Unter geeigneten Reaktionsbedingungen können die beiden Komponenten
miteinander umgesetzt werden, wodurch ein Imidazol erhalten wird,
das eine Diarylalkyleinheit enthält,
die über
eine verlängerte
Amidkette gebunden ist. Andere substituierte Imidazole können in ähnlicher
Weise hergestellt werden.
-
Die
Isolierung der Verbindung in verschiedenen Stufen der Reaktion kann
durch Standardtechniken erreicht werden, wie beispielsweise Filtration,
Verdampfen von Lösungsmittel
und dergleichen. Die Reinigung des Produkts, Intermediats und dergleichen
kann auch nach Standardtechniken erfolgen, wie Umkristallisation,
Destillation, Sublimation, Chromatographie, Überführung in ein geeignetes Derivat,
das umkristallisiert und in die Ausgangsverbindung rückverwandelt
werden kann, und dergleichen. Solche Techniken sind Fachleuten wohl
bekannt.
-
Die
so hergestellten Verbindungen können
auf ihre Zusammensetzung und Reinheit analysiert werden sowie durch
Standardanalysetechniken charakterisiert werden, wie beispielsweise
Elementaranalyse, NMR, Massenspektroskopie und IR-Spektrum.
-
Die
erfindungsgemäßen Verbindungen
können
nach bekannten Verfahren, wie beispielsweise E. A. Brown et al.,
British J. Pharm., (1986) Band 80, 569, leicht zur Bestimmung der
Aktivität
an sowohl H
1- als auch H
3-Rezeptoren
bewertet werden. Die H
3-Aktivität kann beispielsweise
durch den Meerschweinchenhirn-Membranassay
und den neuronalen Ileumkontraktionsassay am Meerschweinchen bestimmt
werden, die beide in
US 5,352,707 beschrieben
sind. Ein weiterer brauchbarer Assay auf H
3-Aktivität verwendet
Rattenhirnmembranen und ist von West et al. ("Identification of Two H
3-Histamine
Receptor Subtypes",
Molecular Pharmacology, (1990), Vol. 33, 610–613, beschrieben. Es wurde
gefunden, dass mehrere der vorliegenden Verbindungen hohe H
1- und
H
3-Antagonistaktivität hatten, was in dem folgenden
Abschnitt BEISPIELE ausführlicher
erörtert wird.
-
In
einer anderen Ausführungsform
liefert diese Erfindung pharmazeutische Zusammensetzungen, die die
oben beschriebenen erfindungsgemäßen Imidazole
als Wirkbestandteil enthalten. Die pharmazeutischen Zusammensetzungen
enthalten im Allgemeinen außerdem
pharmazeutisch annehmbares Trägerverdünnungsmittel,
Hilfsmittel oder pharmazeutisch annehmbaren Träger (hier kollektiv als Trägermaterialien
bezeichnet). Solche pharmazeutischen Zusammensetzungen besitzen
wegen ihrer H1- und H3-Antagonistaktivität Nutzen
zur Behandlung von Allergie, Entzündung, Nasenschleimhautschwellung,
Bluthochdruck, Glaukom, Schlafstörungen,
Zuständen
der Hyper- und Hypomotilität
des Gastrointestinaltrakts, Hypo- und Hyperaktivität des zentralen
Nervensystems, Morbus Alzheimer, Schizophrenie, Migräne, Fettleibigkeit
und ähnlichen
Erkrankungen.
-
Die
vorliegende Erfindung offenbart in einer weiteren Ausführungsform
Verfahren zur Herstellung von pharmazeutischen Zusammensetzungen,
die die erfindungsgemäßen Imidazolverbindungen
als aktiven Bestandteil enthalten. In den erfindungsgemäßen pharmazeutischen
Zusammensetzungen und Verwendungen werden die Wirkbestandteile in
der Regel mit geeigneten Trägermaterialien
gemischt verabreicht, die in Bezug auf die vorgesehene Form der
Verabreichung in geeigneter Weise gewählt sind, d. h. orale Tabletten,
Kapseln (entweder fest-gefüllt,
halbfest-gefüllt
oder flüssig-gefüllt), Pulver
zur Auflösung,
ora le Gele, Elixiere, dispergierbare Körnchen, Sirupe, Suspensionen
und dergleichen und gemäß konventioneller
pharmazeutischer Praxis sind. Zur oralen Verabreichung in Form von
Tabletten oder Kapseln kann die aktive Arzneimittelkomponente beispielsweise
mit beliebigem oralen, nicht-toxischen, pharmazeutisch annehmbaren,
inerten Träger
kombiniert werden, wie Lactose, Stärke, Sucrose, Cellulose, Magnesiumstearat,
Dicalciumphosphat, Calciumsulfat, Talkum, Mannit, Ethylalkohol (flüssige Formen)
und dergleichen. Der Mischung können
zudem, falls gewünscht
oder erforderlich, auch geeignete Schmiermittel, Sprengmittel und
Färbungsmittel
zugegeben werden. Pulver und Tabletten können aus etwa 5 bis etwa 95
erfindungsgemäßer Zusammensetzung
zusammengesetzt sein.
-
Zu
geeigneten Bindemittel gehören
Stärke,
Gelatine, natürliche
Zucker, Maissüßungsmittel,
natürliche und
synthetische Gummis, wie Akaziengummi, Natriumalginat, Carboxymethylcellulose,
Polyethylenglykol und Wachse. Unter den Schmiermitteln können zur
Verwendung in diesen Dosierformen Borsäure, Natriumbenzoat, Natriumacetat,
Natriumchlorid und dergleichen erwähnt werden. Sprengmittel schließen Stärke, Methylcellulose,
Guar Gummi und dergleichen ein. Süßungs- und Aromatisierungsmittel
und Konservierungsmittel können
auch eingeschlossen werden, wo dies angebracht ist. Einige der oben
aufgeführten
Begriffe, nämlich Sprengmittel,
Verdünnungsmittel,
Schmiermittel, Bindemittel und dergleichen, werden nachfolgend detaillierter erörtert.
-
Die
erfindungsgemäßen Zusammensetzungen
können
zudem zu Formen mit verzögerter
Freisetzung formuliert werden, um für die kontrollierte Freisetzung
von einer oder mehreren beliebigen der Verbindungen oder Wirkbestandteilen
zu sorgen, um die therapeutischen Effekte zu optimieren, d. h. Antihistaminaktivität und dergleichen.
Geeignete Dosierformen zur verzögerten Freisetzung
schließen
Schichtentabletten, die Schichten mit unterschiedlichen Zerfallgeschwindigkeiten
enthalten, oder Polymermatrizes mit verzögerter Freisetzung, die mit
den Wirkkomponenten imprägniert
und zu Tablettenform geformt sind, oder Kapseln ein, die solche
imprägnierten
oder verkapselten porösen
polymeren Matrizes enthalten.
-
Zubereitungen
in flüssiger
Form schließen
Lösungen,
Suspensionen und Emulsionen ein. Als Beispiel können Wasser oder Wasser-Propylenglykol-Lösungen für die parenterale
Injektion oder Zugabe von Süßungsmitteln
und Opazifizierungsmitteln für
orale Lösungen,
Suspensionen und Emulsionen genannt werden. Zubereitungen in flüssiger Form
können
auch Lösungen
für intranasale
Verabreichung einschließen.
-
Aerosolzubereitungen,
die zur Inhalation geeignet sind, können Lösungen und Feststoffe in Pulverform
einschließen,
die in Kombination mit einem pharmazeutisch annehmbaren Träger wie
inertem komprimiertem Gas, z. B. Stickstoff, vorliegen können.
-
Zur
Herstellung von Zäpfchen
wird ein niedrig schmelzendes Wachs, wie eine Mischung aus Fettsäureglyceriden,
wie Kakaobutter, zuerst geschmolzen und der aktive Bestandteil darin
homogen dispergiert, wie durch Rühren
oder ähnliches
Mischen. Die geschmolzene homogene Mischung wird dann in zweckmäßig bemessene
Formen gegossen, abkühlen
gelassen und dadurch verfestigt.
-
Ebenfalls
eingeschlossen sind Zubereitungen in fester Form, die kurz vor Gebrauch
in Zubereitungen in flüssiger
Form für
orale oder parenterale Verabreichung überführt werden sollen. Solche flüssigen Formen schließen Lösungen,
Suspensionen und Emulsionen ein.
-
Die
erfindungsgemäßen Verbindungen
können
auch transdermal verabreichbar sein. Die transdermalen Zusammensetzungen
können die
Form von Cremes, Lotionen, Aerosolen und/oder Emulsionen annehmen,
und können
einem Transdermalpflaster vom Matrix- oder Reservoirtyp zugefügt werden,
wie in der Technik zu diesem Zweck konventionell ist.
-
Die
Verbindung wird vorzugsweise oral verabreicht.
-
Die
pharmazeutische Zubereitung liegt vorzugsweise in Einheitsdosisform
vor. In einer solchen Form wird die Zubereitung in geeignet bemessene
Einzeldosen unterteilt, die geeignete Mengen der aktiven Komponente
enthalten, z. B. eine wirksame Menge, um den gewünschten Zweck zu erreichen.
-
Die
Menge der erfindungsgemäßen aktiven
Zusammensetzung in einer Einzeldosis der Zubereitung kann im Allgemeinen
gemäß der speziellen
Anwendung von etwa 1,0 mg bis etwa 1000 mg, vorzugsweise etwa 1,0
bis etwa 950 mg, insbesondere etwa 1,0 bis etwa 500 mg und in der
Regel etwa 1 bis etwa 250 mg eingestellt werden. Die tatsächlich verwendete
Dosis kann gemäß dem Alter
des Patienten, seinem Geschlecht, Gewicht und dem Scheseregrad des
behandelten Zustands variiert werden. Solche Techniken sind Fachleuten
wohl bekannt.
-
Die
humane orale Dosierform, die die Wirkbestandteile enthält, kann
im Allgemeinen ein oder zwei Mal täglich verabreicht werden. Die
Menge und Frequenz der Verabreichung wird gemäß dem Urteil des behandelnden
Arztes geregelt. Ein allgemein empfohlenes Tagesdosierschema für die orale
Verabreichung kann im Bereich von etwa 1,0 mg bis etwa 1000 mg pro
Tag in Einzel- oder unterteilten Dosen liegen.
-
Kapsel
bezieht sich auf einen speziellen Behälter oder eine spezielle Umhüllung, der
bzw. die aus Methylcellulose, Polyvinylalkoholen oder denaturierten
Gelatinen oder Stärke
hergestellt ist, um die aktiven Bestandteile aufweisenden Zusammensetzungen
zu halten oder zu enthalten. Hartschalenkapseln sind in der Regel
aus Gemischen aus relativ gelreichen festen Knochengelatinen und
Schweinehautgelatinen hergestellt. Die Kapsel selbst kann kleine
Mengen an Farbstoffen, Opazifizierungsmitteln, Weichmachern und
Konservierungsmitteln enthalten.
-
Tablette
bezieht sich auf eine komprimierte oder geformte feste Dosierform,
die die Wirkbestandteile mit geeigneten Verdünnungsmitteln enthält. Die
Tablette kann durch Kompression von Mischungen oder Granulationen
hergestellt werden, die durch Nassgranulierung, Trockengranulierung
oder durch Verdichtung erhalten werden.
-
Orale
Gele bezieht sich darauf, dass die Wirkbestandteile in einer hydrophilen
halbfesten Matrix dispergiert oder solubilisiert sind.
-
Pulver
zur Auflösung
beziehen sich auf Pulvergemische, die die Wirkbestandteile und geeignete
Verdünnungsmittel
enthalten, die in Wasser oder Säften
suspendiert werden können.
-
Verdünnungsmittel
bezieht sich auf Substanzen, die üblicherweise den größeren Anteil
der Zusammensetzung oder Dosierform ausmachen. Zu geeigneten Verdünnungsmitteln
gehören
Zucker, wie Lactose, Sucrose, Mannit und Sorbit, Stärken, die
von Weizen, Mais, Reis und Kartoffel stammen, und Cellulosen, wie mikrokristalline
Cellulose. Die Menge an Verdünnungsmittel
in der Zusammensetzung kann im Bereich von etwa 10 bis etwa 90 Gew.-%
der Gesamtzusammensetzung, vorzugsweise etwa 25 bis etwa 75 Gew.-%,
insbesondere etwa 30 bis etwa 60 Gew.-%, besonders bevorzugt etwa
12 bis etwa 60% liegen.
-
Sprengmittel
beziehen sich auf Materialien, die der Zusammensetzung zugefügt werden,
um dazu beizutragen, dass sie aus einanderbricht (zerfällt; gesprengt
wird) und die Wirkstoffe freisetzt. Geeignete Sprengmittel schließen Stärken, "kaltwas serlösliche" modifizierte Stärken wie
Natriumcarboxymethylstärke;
natürliche und
synthetische Gummis wie Johannisbrotfrüchte, Karaya, Guar, Tragakanth
und Agar, Cellulosederivate wie Methylcellulose und Natriumcarboxymethylcellulose;
mikrokristalline Cellulosen und vernetzte mikrokristalline Cellulosen
wie Natriumcroscarmellose, Alginate, wie Alginsäure und Natriumalginat, Tone,
wie Bentonite, und Brausemischungen ein. Die Sprengmittelmenge in
der Zusammensetzung kann im Bereich von etwa 2 bis etwa 15 Gew.-%
der Zusammensetzung, insbesondere etwa 4 bis etwa 10 Gew.-% liegen.
-
Bindemittel
bezieht sich auf Substanzen, die Pulver binden oder "verkleben", und macht sie durch
Bildung von Körnchen
kohäsiv,
die so als "Kleber" in der Formulierung
wirken. Bindemittel fügen
Kohäsionsfestigkeit
hinzu, die bereits in dem Verdünnungsmittel
oder Volumenerhöhungsmittel
zur Verfügung
steht. Geeignete Bindemittel schließen Zucker, wie Sucrose, Stärken, die
von Weizen, Mais, Reis und Kartoffel abgeleitet sind, natürliche Gummis
wie Akaziengummis, Gelatine und Tragakanth, Derivate von Tang, wie
Alginsäure, Natriumalginat
und Ammoniumcalciumalginat, Cellulosematerialien, wie Methylcellulose
und Natriumcarboxymethylcellulose und Hydroxypropylmethylcellulose,
Polyvinylpyrrolidon und anorganische Materialien, wie Magnesiumaluminiumsilikat,
ein. Die Menge an Bindemittel in der Zusammensetzung kann im Bereich
von etwa 2 bis etwa 20 Gew.-% der Zusammensetzung, insbesondere
etwa 3 bis etwa 10 Gew.-%, besonders bevorzugt etwa 3 bis etwa 6
Gew.-% liegen.
-
Schmiermittel
bezieht sich auf eine Substanz, die der Dosierform zugegeben wird,
damit die Tablette, Körner,
usw., nachdem sie komprimiert worden sind, aus der Form oder dem
Stempel gelöst
werden können, indem
Reibung oder Verschleiß herabgesetzt
werden. Geeignete Schmiermittel schließen Metall stearate, wie Magnesiumstearat,
Calciumstearat oder Kaliumstearat, Stearinsäure, Wachse mit hohem Schmelzpunkt
und Wasserlösliche
Schmiermittel wie Natriumchlorid, Natriumbenzoat, Natriumacetat,
Natriumoleat, Polyethylenglykole und d,l-Leucin ein. Schmiermittel
werden üblicherweise
in der allerletzten Stufe vor der Kompression zugefügt, da sie
auf den Oberflächen
der Körnchen
und zwischen diesen und den Teilen der Tablettenpresse vorhanden
sein müssen.
Die Menge an Schmiermittel in der Zusammensetzung kann im Bereich
von etwa 0,2 bis etwa 5 Gew.-% der Zusammensetzung, insbesondere
etwa 0,5 bis etwa 2 Gew.-%, besonders bevorzugt etwa 0,3 bis etwa
1,5 Gew.-% liegen.
-
Gleitmittel
sind Materialien, die Zusammenbacken verhindern und die Fließcharakteristika
von Granulationen verbessern, so dass das Fließen glatt und gleichförmig erfolgt.
Geeignete Gleitmittel schließen
Siliciumdioxid und Talkum ein. Die Gleitmittelmenge in der Zusammensetzung
kann im Bereich von etwa 0,1 bis etwa 5 Gew.-% der Gesamtzusammensetzung,
insbesondere etwa 0,5 bis etwa 2 Gew.-% liegen.
-
Färbungsmittel
sind Hilfsstoffe, die der Zusammensetzung oder der Dosierform Färbung verleihen. Solche
Hilfsstoffe können
Lebensmittelfarbstoffe und auf einem geeigneten Adsorbens wie Ton
oder Aluminiumoxid adsorbierte Lebensmittelfarbstoffe einschließen. Die
Menge des Färbungsmittels
kann von etwa 0,1 bis etwa 5 Gew.-% der Zusammensetzung, vorzugsweise
etwa 0,1 bis etwa 1% variieren.
-
Bioverfügbarkeit
bezieht sich auf die Rate und das Maß, bis zu dem der aktive Arzneimittelbestandteil oder
die therapeutische Einheit von einer verabreichten Dosierform in
den systemischen Kreislauf absorbiert wird, verglichen mit einem
Standard oder einer Kontrolle.
-
Konventionelle
Verfahren zur Herstellung von Tabletten sind bekannt. Zu solchen
Verfahren gehören Trockenverfahren,
wie direkte Kompression, und Kompression einer Granulation, die
durch Verdichtung hergestellt ist, oder Nassverfahren oder andere
Spezialverfahren. Konventionelle Verfahren zur Herstellung anderer
Verabreichungsformen, wie beispielsweise Kapseln, Zäpfchen und
dergleichen, sind auch wohl bekannt.
-
Eine
andere Ausführungsform
der Erfindung offenbart die Verwendung der oben offenbarten pharmazeutischen
Zusammensetzungen zur Behandlung von Erkrankungen wie beispielsweise
Allergie, Entzündung, Nasenschleimhautschwellung,
Bluthochdruck, Glaukom, Schlafstörungen,
Zustände
von Hyper- und Hypomotilität
des Gastrointestinaltrakts, Hypo- und Hyperaktivität des zentralen
Nervensystems, Morbus Alzheimer, Schizophrenie, Migräne, Fettleibigkeit
und dergleichen. Bei dem Verfahren wird einem Säugerpatienten mit einer derartigen
Erkrankung oder derartigen Erkrankungen, der dieser Behandlung bedarf,
eine therapeutisch wirksame Menge der erfindungsgemäßen pharmazeutischen
Zusammensetzung verabreicht.
-
Fachleute
werden erkennen, dass der Begriff "obere Luftwege" die oberen Atemwege bedeutet, d. h. Nase,
Rachen und dazugehörige
Strukturen.
-
Fachleuten
werden erkennen, dass sowohl an Materialien als auch an Verfahren
viele Modifikationen, Variationen und Änderungen vorgenommen werden
können.
Diese Modifikationen, Varianten und Veränderungen sollen in dem Geist
und Umfang der vorliegenden Erfindung liegen.
-
Die
folgenden Beispiele werden zur näheren
Erläuterung
der vorliegenden Erfindung gegeben. Sie dienen nur zu veranschaulichenden
Zwecken und werden nicht als den Umfang der Erfindung in irgendeiner Weise
einschränkend
angesehen.
-
BEISPIELE
-
Wenn
nicht anders angegeben, haben die folgenden Abkürzungen in den folgenden Beispielen
die angegebenen Bedeutungen:
- DBU
- = 1,8-Diazabicydo[5.4.0]undec-7-en
- DBN
- = 1,5-Diazabicyclo[4.3.0]non-5-en
- EDCI
- = 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid
- HOBT
- = 1-Hydroxybenzotriazol
- DCC
- = Dicyclohexylcarbodiimid
- Dibal-H
- = Diisobutylaluminumhydrid
- LAH
- = Lithiumaluminumhydrid;
- NaBH(OAc)3
- = Natriumtriacetoxyborhydrid
- NaBH4
- = Natriumborhydrid
- NaBH3CN
- = Natriumcyanborhydrid
- LDA
- = Lithiumdiisopropylamid
- p-TsOH
- = p-Toluolsulfonsäure
- m-CPBA
- = m-Chlorperbenzoesäure
- TMAD
- = N,N,N',N'-Tetramethylazodicarboxamid;
- CSA
- = Camphersulfonsäure
- NaHMDS
- = Natriumhexamethyldisilylazid
- HRMS
- = hochauflösende Massenspektroskopie
- HPLC
- = Hochleistungsflüssigchromatographie
- LRMS
- = niedrigauflösende Massenspektroskopie
- nM
- = nanomolar
- Ki
- = Inhibierungskonstante
für den
Substrat/Rezeptor-Komplex,
pA2 = –logEC50, wie in J. Hey, Eur. J. Pharmacol., (1995),
Vol. 294, 329–335
definiert.
- Ci/mmol
- = Curie/mmol (ein
Maß für die spezifische
Aktivität)
- Tr
- = Triphenylmethyl
- Tris
- = Tris(hydroxymethyl)aminomethan
-
I. ALLGEMEINES SYNTHESEVERFAHREN 'A': REDUKTIVE AMINIERUNG
-
I. ALLGEMEINES VERFAHREN 'A': Reduktive Aminierung
-
-
Beispiel 1. Herstellung von 1-(Triphenylmethyl)-1H-imidazol-4-carboxaldehyd (2):
-
-
Zu
einer gerührten
Suspension von Aldehyd 1 (Aldrich Chemicals, Milwaukee, Wisconsin,
USA) (35,0 g, 0,364 Mol) und Triethylamin (55,8 ml; 0,400 Mol) in
Dichlormethan (2 L) wurde eine Lösung
von Triphenylmethylchlorid in Dichlormethan (600 ml) gegeben, während die
Reaktionstemperatur mit einem Kühlbad
auf ungefähr
15°C gehalten
wurde. Die resultierende Lösung wurde
auf Raumtemperatur erwärmen
gelassen und 19 Stunden gerührt.
Die Lösung
wurde mit einer Lösung
von gesättigter
Salzlösung
und Wasser (1:3,5; 3 × 600 ml),
anschließend
mit Salzlösung
(1 × 800
ml) gewaschen. Sie wurde über
Natriumsulfat getrocknet, filtriert, um das Trockenmittel zu entfernen,
und das Lösungsmittel
wurde im Vakuum entfernt, um das gewünschte tritylierte Produkt
als schmutzigweißen
Feststoff zu erhalten, Schmelzpunkt 186,5–194°C. [Triturierung dieses Produkts
mit Ether ergab ein cremefarbenes Pulver mit einem Schmelzpunkt
von 195–197°C.]
-
Beispiel 2. Herstellung von of 4-[(Z)-4-(Phenylmethoxy)-1-butenyl]-1-(triphenylmethyl)-1H-imidazol
(3):
-
-
Zu
einer mechanisch gerührten
Lösung
des Aldehyds 2 in trockenem Tetrahydrofuran (1 L) wurde (3-Benzyloxypropyl)triphenylphosphoniumbromid
(30,02 g, 0,0611 mmol) gegeben. Die resultierende Suspension wurde
auf 15°C
gekühlt,
und danach wurde eine 1,0 M Lösung
(61,4 ml, 0,0614 mmol) Kalium-t-butoxid in Tetrahydrofuran im Verlauf
von fünf
Minuten zugegeben. Die Reaktionsmischung wurde auf Raumtemperatur
erwärmen
gelassen und 2 Stunden gerührt.
Die Reaktionsmischung wurde durch Celite filtriert; der Filter wurde
mit Tetrahydrofuran (2 × 150
ml) gewaschen; das Filtrat und die Wäschen wurden kombiniert und
mit Ether (800 ml) verdünnt
und erneut durch frische Celite filtriert. Das Filtrat wurde unter
Vakuumkonzentriert, und der Rückstand
wurde an Silikagel chromatographiert, wobei mit einem Gradienten
von Hexanen-Ethylacetat (3:1 → 2:1)
elu iert wurde, um die Titelverbindung als blassgelbes Pulver zu
erhalten, Schmelzpunkt 101–104°C. FABMS
471 (MH+; 6%); 243 (Ph3C+; 100%).
-
Beispiel 3. Herstellung von 1-(Triphenylmethyl)-1H-imidazol-4-butanol (4):
-
-
Eine
Mischung des olefinischen Ethers 3 (18,27 g, 0,0388 mmol) in wasserfreiem
Methanol (350 ml), 1,0 M etherischer Salzsäure (38,8 ml, 0,0388 mmol)
und 10% Palladium-auf-Kohle-Katalysator
wurde mit 48 psi 30 Minuten auf einer Parr-Schüttelapparatur
hydriert. Die Reaktionsmischung wurde dann durch Celite filtriert
und der Filterkuchen mit Methanol gewaschen. Die kombinierten Filtrate
und Wäschen
wurden konzentriert und unter Hochvakuum getrocknet, um den Titelalkohol
als Hydrochlorid als schmutzigweißen Feststoff zu erhalten,
Schmelzpunkt 144–146°C.
-
Beispiel 4. Herstellung von ω-[1-(Triphenylmethyl)-1H-imidazol-4-yl]-butanal
(5):
-
-
In
einem trockenen Kolben, der zur Bereitstellung einer Inertgasatmosphäre ausgerüstet war,
wurde eine Lösung
von Oxalylchlorid (2,18 ml, 0,0250 Mol) in trockenem Dichlormethan
(50 ml) hergestellt und in einem CO2-Aceton-Bad
auf –60°C abge kühlt. Eine
Lösung
von Dimethylsulfoxid (3,60 ml, 0,0507 mmol) in trockenem Dichlormethan
(10 ml) wurde tropfenweise im Verlauf von 5 bis 10 Minuten zugegeben,
während
die Reaktionstemperatur auf –55
bis –60°C gehalten
wurde. Es wurde weitere 5 Minuten bei –60°C gerührt, danach wurde eine Lösung von
Alkohol-Hydrochlorid (4) (8,67 g, 0,0207 mol) in trockenem Dichlormethan
(140 ml) im Verlauf von 15 bis 20 Minuten zugefügt, während die Reaktionstemperatur
im Bereich von –55
bis –60°C gehalten
wurde. Die Mischung wurde eine weitere Stunde bei –60°C gerührt, danach
wurde unverdünntes Triethylamin
(17,6 ml, 0,126 mmol) mit einer solchen Rate zugegeben, dass die
Reaktionstemperatur auf –55 bis –60°C gehalten
wurde. Die Reaktion wurde bei dieser Temperatur 5 Minuten gerührt. Das
Kühlbad
wurde entfernt und weitere 1,5 Stunden bei Raumtemperatur gerührt. Die
Reaktionsmischung wurde mit Wasser (4 × 50 ml), danach Salzlösung (75
ml) gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet und Lösungsmittel unter Vakuum entfernt,
um ein viskoses Öl
zu ergeben. Um jegliches restliche Triethylaminhydrochlorid zu entfernen,
wurde das restliche Öl
in Diethylether (100 ml) gelöst,
mit Wasser (1 × 30
ml; 2 × 10
ml), danach mit Salzlösung
(30 ml) gewaschen und über
wasserfreiem Magnesiumsulfat getrocknet. Das Lösungsmittel wurde unter Vakuum
entfernt, um den Titelaldehyd als viskoses gelbes Öl zu erhalten,
das zur weiteren Verwendung ausreichend rein war. FABMS: 381 (MH+; 10%); 243 (Ph3C+; 100%).
-
Beispiel 5. Herstellung von Ethyl-5-[1-(triphenylmethyl)-1H-imidazol-4-yl]-4-Z-pentenoat:
-
-
- (i) Herstellung von (Ethoxycarbonylprop-1-yl)triphenylphosphoniumbromid
(a): Eine Mischung aus Triphenylphosphin (24,6 g, 0,0936 Mol) und
Ethyl-4-brombutyrat (14,4 ml, 0,101 Mol) wurde über einen Zeitraum von 15 bis
20 Minuten von Raumtemperatur auf 105°C erwärmt, danach wurde das Erwärmen auf
105° 10 Minuten
fortgesetzt. Die Lösung
wurde abkühlen
gelassen, während
sie jedoch noch war, wurde vorsichtig Diethylether (50 ml) über einen
Kühler
zugegeben. Das resultierende Gummi wurde trituriert, um ein weißes Pulver
zu erhalten. Der Überstand
wurde dekantiert, frischer Diethylether (50 ml) wurde zugegeben,
und das Triturieren wurde 10 Minuten fortgesetzt. Das Produkt wurde
filtriert, der Kuchen mit Diethylether gewaschen und dann Lösungsmittel
unter Vakuum aus den kombinierten Filtraten und Wäschen entfernt,
um eine Mischung aus Öl
und Feststoffen zu erhalten. Diese Mischung wurde auf 100°C erwärmt, vorsichtig mit
Diethylether (2 × 55
ml) behandelt und die oben beschriebene Sequenz aus Triturieren,
Filtrieren und Konzentrieren wiederholt. Die beiden aus diesem Verfahren
erhaltenen Chargen der weißen
Feststoffe wurden kombiniert, mit Toluol (150 ml) trituriert und
filtriert. Die aufgefangenen Feststoffe wurden mit Toluol gewaschen
und unter Hochvakuum getrocknet, um das Titelsalz zu erhalten, Schmelzpunkt
177 bis 179°C, FABMS:
377 (M+ für Kation; 84%).
- (ii) Herstellung von Ethyl-5-[1-(triphenylmethyl)-1H-imidazol-4-yl]-4-Z-pentenoat:
Unter einer Stickstoffatmosphäre
wurde das Triphenylphosphoniumsalz (a) (14,0 g, 0,0305 Mol) zu einer
gerührten
Lösung
von Aldehyd 2 (9,81 g, 0,029 Mol) in Tetrahydrofuran (500 ml) gegeben.
Die resultierende Suspension wurde auf 0–5°C abgekühlt, 1 M Kalium-t-butoxid in
Tetrahydrofuran (31 ml, 0,031 Mol) im Verlauf von 3 bis 5 Minuten
zugegeben und die Mischung 20 Minuten bei 0–5°C gerührt. Der Reaktionsmischung
wurde Celite zugefügt,
kurz gerührt
und abfil triert. Der Filterkuchen wurde mit Diethylether und anschließend Dichlormethan
gewaschen. Die kombinierten Filtrate und Wäschen wurden unter Vakuum konzentriert.
Das restliche Öl
wurde an Silikagel chromatographiert und mit einem Gradienten von
Hexanen-Ethylacetat (3:1 → 2:1) eluiert,
um die Titelverbindung als weißen
Feststoff zu ergeben, Schmelzpunkt 90–92,5°C. FABMS: 437 (MH+;
3%); 243 (Ph3C+;
100%).
-
Beispiel 6. Herstellung von 5-[1-(Triphenylmethyl)-1H-imidazol-4-yl]-4-Z-pentenal:
-
-
Zu
einer gerührten
Lösung
des Esters (671 mg, 1,54 mmol) in trockenem Dichlormethan (12 ml),
die sich in einem Kältebad
befand, wurde im Verlauf von ungefähr 4 Minuten eine 1 M Lösung von
DIBAL-H in Toluol (3,08 ml, 3,08 mmol) gegeben, während die
Reaktionstemperatur auf –55
bis –60°C gehalten
wurde. Nachdem 8 bis 10 Minuten bei –58°C gerührt worden war, wurde die Reaktion
durch die Zugabe von Methanol (0,4 ml) und Wasser (6 ml) gequencht.
Die Reaktionsmischung wurde auf Raumtemperatur erwärmen gelassen.
Der gallertartige Niederschlag wurde durch Filtration durch Celite
entfernt. Der Filterkuchen wurde mit Dichlormethan gewaschen, und
die kombinierten Filtrate und Wäschen
wurden über
wasserfreiem Magnesiumsulfat getrocknet. Das Trockenmittel wurde
abfiltriert und Lösungsmittel
unter vermindertem Druck entfernt, um den Titelaldehyd als weißes Pulver
zu erhalten, Schmelzpunkt 117,5–120°C. FABMS:
393 (MH+; 12%); 243 (Ph3C+; 100%)
-
Beispiel 7. Herstellung von [1-(Triphenylmethyl)-1H-imidazol-4-yl]-pentanal (6):
-
-
Eine
Mischung des ungesättigten
Aldehyds (5,42 g, 13,8 mmol) und 5% Palladium-auf-Kohle-Katalysator
(0,50 g) in wasserfreiem Methanol (130 ml) wurde 30 Minuten mit
30–35
psi auf einer Parr-Schüttelapparatur
hydriert. Der Katalysator wurde durch Celite filtriert und das Filtrat
unter vermindertem Druck eingedampft. Der Rückstand wurde unter Hochvakuum
getrocknet, um die Titelverbindung als gelbes viskoses Öl oder Glas
zu erhalten, das ausreichend rein für die weitere Chemie war. FABMS:
395 (MH+; 5%); 243 (Ph3C+; 100%).
-
Beispiel 8. Herstellung von Ethyl-6-[1-(triphenylmethyl)-1H-imidazol-4-yl]-5-Z-hexenoat:
-
-
Aldehyd
2 (12,4 g; 0,0367 Mol) wurde unter einer Stickstoffatmosphäre zu einer
kräftig
gerührten
Teilsuspension von 4-Carboethoxybutyltriphenylphosphoniumbromid
(von Lancaster Chemicals, Windham, New Hampshire, USA) (20,2 g;
0,0408 mol) in Tetrahydrofuran (630 ml) gegeben. Die Suspension
wurde gerührt, bis
der Aldehyd gelöst
war. Die resultierende Mischung wurde auf 0–5°C abgekühlt und im Verlauf von 10 Minuten
1 M Kalium-t-butoxid in Tetrahydrofuran (40,8 ml, 0,0408 Mol) zu gegeben,
und die Mischung wurde 40 Minuten bei 0–5°C, danach 30 Minuten bei 5–10°C kräftig gerührt. Es
wurde trockenes Dichlormethan (100 ml) zugegeben, um jegliche Salzbeschichtung
an den Wänden
des Kolbens zu lösen,
danach wurde die Reaktionsmischung auf 20°C erwärmen gelassen. Celite wurde
zu der Reaktionsmischung gegeben, die kurz gerührt und filtriert wurde, und
der Filterkuchen wurde mit Dichlormethan gewaschen. Die kombinierten
Filtrate und Wäschen
wurden unter Vakuum konzentriert. Das restliche Öl wurde an Silikagel chromatographiert
und mit einem Gradienten von Hexanen-Ethylacetat (5:1 → 2:1) eluiert,
um die Titelverbindung als weißes
Pulver zu ergeben, Schmelzpunkt 78,5–85°C. FABMS: 451 (MH+;
2%); 243 (Ph3C+;
100%).
-
Beispiel 9. Herstellung von 6-[1-(Triphenylmethyl)-1H-imidazol-4-yl]-5-Z-hexenal:
-
-
Zu
einer gerührten
Lösung
des Titel-Esters aus Beispiel 8 (3,98 g, 8,83 mmol) in trockenem
Dichlormethan (50 ml), die sich in einem Kältebad befand, wurde im Verlauf
von ungefähr
15 Minuten eine 1,0 M Lösung
von DIBAL-H in Toluol (17,7 ml, 17,7 mmol) gegeben, während die
Reaktionstemperatur auf –55
bis -60°C
gehalten wurde. Nachdem 45 Minuten bei -58°C bis -60°C gerührt worden war, wurde die Reaktion durch
die Zugabe von Methanol (2,3 ml) und Wasser (34 ml) gequencht. Die
Reaktionsmischung wurde auf Raumtemperatur erwärmen gelassen und dann durch
Celite filtriert. Der Filterkuchen wurde mit Dichlormethan gewaschen,
und die kombinierten Filtrate und Wäschen wurden über wasserfreiem
Magnesiumsulfat getrocknet. Das Trockenmittel wurde filtriert und
Lösungsmittel
unter redu ziertem Druck verdampft, um den Titel-Aldehyd als viskoses Öl zu erhalten,
das ausreichend rein für
die in Beispiel 10 beschriebene Verwendung war. FABMS: 515 (Verunreinigung;
4%); 407 (MH+; 2%); 243 (Ph3C+; 100%).
-
Beispiel 10. Herstellung von ω-[1-(Triphenylmethyl)-1H-imidazol-4-yl]-hexanal
(7):
-
-
Eine
Mischung des ungesättigten
Aldehyds aus Beispiel 9 (3,41 g, 8,39 mmol) und 5% Palladium-auf-Kohle-Katalysator
(0,3 g) in wasserfreiem Methanol (50 ml) wurde 45 Minuten mit 30–35 psi
auf einer Parr-Schüttelapparatur
hydriert. Der Katalysator wurde durch Celite filtriert, das Filtrat
unter reduziertem Druck eingedampft und das restliche Öl an Silikagel
chromatographiert. Die Eluierung mit einem Gradienten aus Hexanen-Ethylacetat
(2:1 → 1:1 → 1:2) ergab
die Titelverbindung, die nach Trocknen im Hochvakuum als weißer, etwas
wachsartiger, hygroskopischer Feststoff, Schmelzpunkt 78–80,5°C, isoliert
wurde. FABMS: 451 (MH+).
-
Beispiel 11. Herstellung von 1-(8-Chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-[(1H-imidazol-4-yl)-methyl]-piperazin:
-
-
- (i) Zu einer gerührten Lösung von 8-Chlor-6,11-dihydro-11-(1-piperazinyl)-5H-benzo[5,6]cyclohepta[1,2-b]pyridin
(8) (6,40 g; 0,020 mol) (offenbart in der veröffentlichten Anmeldung WO 95/10516 , 20. April 1995)
und 1 H-Imidazol-4-carboxaldehyd (2) (1,96 g; 0020 mol) in wasserfreiem
Methanol (100 mL) wurde wasserfreies Magnesiumsulfat (4,91 g) gegeben.
Die resultierende Suspension wurde bei Raumtemperatur 45 Minuten
gerührt.
Während
die Reaktionstemperatur mittels eines Wasserbades auf 25–30°C gehalten wurde,
wurde festes Natriumcyanoborhydrid (4,05 g von 95%; 0,0612 Mol)
zugegeben und die resultierende Mischung 4 Stunden bei Raumtemperatur
rühren
gelassen. Es wurden weitere 0,64 g (0,0067 Mol) Aldehyd 2 zugegeben,
und es wurde weitere 16 Stunden bei Raumtemperatur gerührt. Die
Reaktionsmischung wurde mit Methanol (22 ml) verdünnt und
filtriert. Der Filterkuchen wurde mit Methanol gewaschen, das Filtrat
und die Wäschen
wurden kombiniert und das Lösungsmittel
unter reduziertem Druck entfernt. Der Rückstand wurde unter Hochvakuum
getrocknet, um einen schmutzigweißen glasartigen Feststoff zu erhalten.
Dieses Glas wurde erneut in Dichlormethan (175 ml)-Methanol (30
ml) gelöst
und mit Wasser (1 × 25
ml) gewaschen. Die organische Lösung
wurde über
was serfreiem Magnesiumsulfat getrocknet, filtriert und Lösungsmittel
unter vermindertem Druck entfernt. Der Rückstand wurde an Silikagel
chromatographiert und mit einem Gradienten von Dichlormethan-Methanol-konzentriertem
Ammoniumhydroxid (95:5:0,1 → 90:10:0,1 → 85:15:0,1 → 80:20:0,1)
eluiert, was nach dem Trocknen unter Hochvakuum die Titelverbindung
als weißes
Glas ergab. FABMS: 394, 396 (MH+; 100, 39%);
228 (67%).
- (ii) Zu einer gerührten
Lösung
der freien Basenform der Titelverbindung (5,48 g, 0,0139 Mol) in
Methanol (200 ml) wurde eine etwa 3,4 M Lösung (12,3 ml, 0,0417 Mol)
etherischem Chlorwasserstoff gegeben. Das Lösungsmittel wurde unter reduziertem
Druck entfernt und die restlichen Feststoffe mit Ether (150 ml)
gerührt.
Der Feststoff wurde filtriert, unter einem Kautschukdamm getrocknet
und wieder in Methanol (100 ml) gelöst. Die Lösung wurde mit ~3,4 M Lösung (10
ml, 0,034 Mol) von etherischem Chlorwasserstoff behandelt. Das Lösungsmittel
wurde unter reduziertem Druck entfernt und die restlichen Feststoffe
mit Ether (150 ml) gerührt.
Die Feststoffe wurden abfiltriert, teilweise unter einem Kautschukdamm
getrocknet und dann weiter unter Hochvakuum getrocknet, um das Hydrochloridsalz
der Titelverbindung als weißes
Pulver zu ergeben, danach als 2,8 Hydrochlorid-Hemihydrat analysiert,
Schmelzpunkt 190,5–192°C (Zersetzung; wurde
bei etwa 180°C
dunkel). C22H24ClN5·2,8
HCl·0,5
H2O. FABMS: 394, 396 (MH+;
64/25%).
-
Beispiel 12. Herstellung von 8-Chlor-6,11-dihydro-11-[4-[4-(1H-imidazol-4-yl)butyl]-1-piperazinyl]-5H-benzo[5,6]cyclohepta[1,2-b]pyridin:
-
-
- (i) Zu einer gerührten Lösung von 8-Chlor-6,11-dihydro-11-(1-piperazinyl)-5H-benzo[5,6]cyclohepta[1,2-b]pyridin
(8) (348 mg; 1,11 mol), ω-[1-(Triphenylmethyl)-1H-imidazol-4-yl]-butanal
(5) (459 mg; 1,21 mmol) und Methansulfonsäure (0,07 ml; 1,11 mmol) in
wasserfreiem Methanol (15 ml) wurde wasserfreies Magnesiumsulfat
(267 mg) gegeben. Die resultierende Suspension wurde bei Raumtemperatur
30 Minuten gerührt.
Es wurde eine 1,0 M Lösung
von Natriumcyanoborhydrid in Tetrahydrofuran (0,78 ml; 0,78 mmol) zugegeben
und die resultierende Mischung 4,5 Stunden bei Raumtemperatur rühren gelassen.
Die Mischung wurde durch Celite filtriert und das Filtrat unter
vermindertem Druck eingedampft. Der Rückstand wurde in Dichlormethan
(20 ml) gelöst
und nacheinander mit 1,1 M Natriumbicarbonat (10 ml), Wasser (3 × 10 ml)
und Salzlösung
(15 ml) gewaschen. Die organische Phase wurde über wasserfreiem Magnesiumsulfat
getrocknet, filtriert und Lösungsmittel
unter vermindertem Druck entfernt. Der gummiartige Rückstand wurde
an Silikagel chromatographiert, wobei mit einem Gradienten von Dichlormethan-Methanol-konzentriertem
Ammoniumhydroxid (95:5:0,1 → 90:10:0,1)
eluiert wurde, um den Imidazol-N-tritylierten Vorläufer der
Titelverbindung als glasartigen Feststoff zu erhalten. Das Produkt
wurde nach dem Trocknen im Hochvakuum ohne weitere Behandlung oder
Charakterisierung wie folgt Detritylierung unterzogen:
- (ii) Eine Methanollösung
(75 ml) des N-tritylierten Produkts (512 mg; 0,755 mmol) der vorhergehenden
Stufe und 1,0 M Salzsäure
in Ether (8,7 ml, 8,7 mol) wurden 5 Stunden unter einer Stickstoffatmosphäre unter Rückfluss
gehalten. Lösungsmittel
wurde unter reduziertem Druck entfernt, und der Rückstand
wurde mit Ether trituriert. Die Mischung wurde filtriert und der
aufgefangene Feststoff mit Ether gewaschen und unter Hochvakuum
getrocknet, um das HCl-Salz der Titelverbindung als weißen Feststoff
zu erhalten, Schmelzpunkt 165–17°C (Zersetzung).
C25H30ClN5·2,8
HCl·1,9
H2O. FABMS: 436, 438 (MH+;
49/18%).
-
Indem
jeweils die homologen Aldehyde 6 (aus Beispiel 7) und 7 (aus Beispiel
10) in den vorhergehenden Reaktionsverfahren substituiert wurden,
wurden die folgenden Analoga der Titelverbindung hergestellt:
8-Chlor-6,11-dihydro-11-[4-[5-(1H-imidazol-4-yl)pentyl]-1-piperazinyl]-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-trihydrochlorid·1,15 Hydrat·0,5 Diethylether
C26H32ClN5·3
HCl·1,15
H2O·0,5
C4H10O. FABMS: 450,
452 (MH+; 100/35%).
8-Chlor-6,11-dihydro-11-[4-[6-(1H-imidazol-4-yl)hexyl]-1-piperazinyl]-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-trihydrochlorid·0,5 Hydrat·0,5 Methanolat
C27H34ClN5·3
HCl·0,5
H2O·0,5
CH4O. FABMS: 464, 466 (MH+; 8/3%).
-
Beispiel 13. Herstellung von 8-Chlor-6,11-dihydro-11-[1-[5-(1H-imidazol-4-yl)pentyl]-4-piperidinyl]-5H-benzo[5,6]cyclohepta[1,2-b]pyridin:
-
-
- (i) Zu einer gerührten Lösung von 8-Chlor-6,11-dihydro-11-(4-piperazinyl)-5H-benzo[5,6]cyclohepta[1,2-b]pyridin
(9) (499 mg; 1,59 mol) (offenbart in der veröffentlichten Anmeldung WO 95/10516 , 20. April 1995), ω-[1-(Triphenylmethyl)-1H-imidazol-4-yl]-butanal (6)
(699 mg; ~90% rein, 1,59 mmol) und Methansulfonsäure (0,10 ml; 1,59 mmol) in
wasserfreiem Methanol (100 ml) wurde wasserfreies Magnesiumsulfat (380
mg) gegeben. Die resultierende Suspension wurde bei Raumtemperatur
45 Minuten gerührt.
Es wurde eine 1,0 M Lösung
von Natriumcyanoborhydrid in Tetrahydrofuran (1,12 ml; 1,12 mmol)
zugegeben und die resultierende Mischung 22 Stunden bei Raumtemperatur
gerührt.
Die Reaktionsmischung wurde durch Celite filtriert und das Lösungsmittel
unter vermindertem Druck verdampft. Der Rückstand wurde in Dichlormethan
(25 ml) gelöst
und nacheinander mit 1,1 M Natriumbicarbonat (15 ml) und Wasser
(2 × 10
ml) gewaschen. Die organischen Phasen wurden über wasserfreiem Magnesiumsulfat
getrocknet, filtriert, das Lösungsmittel
unter vermindertem Druck verdampft, und es wurde unter Hochvakuum
getrocknet, um einen rosa glasartigen Feststoff zu erhalten. Der
resultierende Feststoff wurde an Silikagel chromatographiert, wobei
mit einem Gradienten von 3,9 M Ammoniak in Methanol-Di chlormethan
(3:97 → 5:95)
eluiert wurde, um den Imidazol-N-tritylierten
Vorläufer
der Titelverbindung zu erhalten, der nach Trocknen unter Hochvakuum
ein schmutzigweißer
Schaum war, Schmelzpunkt 75–78°C (schmolz
zu einem viskosen Gummi). FABMS: 451 (MH+;
20%); 243 (Ph3C+;
100%).
- (ii) Eine Methanollösung
(35 ml) des N-tritylierten Produkts (426 mg; 0,675 mmol) der vorhergehenden
Stufe und 1,0 M Salzsäure
in Ether (7,8 ml, 7,8 mmol) wurden 12,5 Stunden unter einer Stickstoffatmosphäre unter
Rückfluss
gehalten. Das Lösungsmittel
wurde unter vermindertem Druck entfernt und der Rückstand zwischen
Dichlormethan (25 ml) und 1,1 M Natriumbicarbonatlösung (25
ml) entfernt. Die wässrige
Lösung wurde
mit Dichlormethan (2 × 20
ml) extrahiert. Das Lösungsmittel
wurde aus den kombinierten Extrakten entfernt, um einen Glasrückstand
zu erhalten, der an Silikagel chromatographiert wurde, wobei mit
einem Gradient von 3,9 M Ammoniak in Methanol-Dichlormethan (5:95 → 10:90)
eluiert wurde, um die Titelverbindung zu erhalten, die nach Trocknen
unter Hochvakuum ein weißes
Pulver war, Schmelzpunkt 65–68°C (schmolz
zu einem viskosen Gummi). C27H33ClN4·1,5
H2O. CIMS: 449 (MH+;
100%); 477 ([M + C2H5]+; 24%).
- (iii) Zu einer gerührten
Lösung
der Titelverbindung (128 mg; 0,285 mmol) in Methanol (5 ml) wurde
eine 1,0 M Lösung
(1,0 ml; 1,0 mmol) von etherischem Chlorwasserstoff gegeben. Das
Lösungsmittel
wurde unter vermindertem Druck entfernt, der Rückstand wurde im Hochvakuum über Phosphorpentoxid
getrocknet, um das Trihydrochloridsalz der Titelverbindung als weißes Pulver
zu erhalten, Schmelzpunkt 164–170°C (Zersetzung
zu einem viskosen Gummi). C27H33ClN4·3
HCl·H2O·0,5
CH4O (MeOH). FABMS: 449 (MH+; 55%).
-
Indem
jeweils die homologen Aldehyde 5 (Beispiel 4) und 7 (Beispiel 10)
in den vorhergehenden Reaktionsverfahren substi tuiert wurden, wurden
die folgenden Analoga der Titelverbindung hergestellt:
8-Chlor-6,11-dihydro-11-[1-[4-(1H-imidazol-4-yl)butyl]-4-piperidinyl]-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-trihydrochlorid·1,1 Hydrat,
Schmelzpunkt 175–178° C (Zersetzung).
C26H31ClN4·3
HCl·1,1
H2O. FABMS: 435, 437 (MH+;
7/2%).
8-Chlor-6,11-dihydro-11-[1-[6-(1H-imidazol-4-yl)hexyl]-4-piperidinyl]-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-trihydrochlorid·1,5 Hydrat.
C28H35ClN4·3
HCl·1,5
H2O. FABMS: 463, 465 (MH+;
100/44%).
-
Beispiel 14. Herstellung von 8-Chlor-6,11-dihydro-11-[1-[6-(1
H-imidazol-4-yl)hexyl]-4-piperidinyliden]-5H-benzo[5,6]cyclohepta[1,2-b]pyridin:
-
-
- (i) Zu einer gerührten Lösung von 8-Chlor-6,11-dihydro-11-(4-piperidinyliden)-5H-benzo[5,6]cyclohepta[1,2-b]pyridin
(10) (284 mg; 0,914 mol) (offenbart in der veröffentlichten Anmeldung WO 95/10516 , 20. April
1995), -[1-(Triphenylmethyl)-1H-imidazol-4-yl]-hexanal
(7) (393 mg; 0,962 mmol) und Methansulfonsäure (0,06 ml; 0,914 mmol) in
wasserfreiem Methanol (10 ml) wurde wasserfreies Magnesiumsulfat
(220 mg). gegeben. Die resultierende Suspension wurde bei Raumtemperatur
30 Minuten gerührt.
Es wurde eine 1,0 M Lösung
von Natriumcyanoborhydrid in Tetrahydrofuran (0,64 ml; 0,64 mmol)
zugegeben und die re sultierende Mischung 23 Stunden bei Raumtemperatur
rühren
gelassen. Die Reaktionsmischung wurde durch Celite filtriert, und
das Filtrat wurde unter reduziertem Druck eingedampft, um das Mesylatsalz
des Imidazol-N-tritylierten Vorläufers
der Titelverbindung als rosa Glas zu erhalten, das ohne weitere
Reinigung detrityliert wurde. FABMS: 703 (MH+;
35%); 243 (Ph3C+;
100%).
- (ii) Eine Methanollösung
(10 ml) des N-tritylierten Mesylatsalzprodukts (828 mg; 0,675 mmol)
der vorhergehenden Stufe und 1,0 M Salzsäure in Ether (10 ml, 10 mmol)
wurden 10 Stunden unter einer Stickstoffatmosphäre unter Rückfluss gehalten. Das Lösungsmittel
wurde unter vermindertem Druck entfernt und der Rückstand
zwischen Dichlormethan (25 ml) und 1,1 M Natriumbicarbonatlösung (25
ml) entfernt. Die wässrige
Lösung
wurde mit Dichlormethan (2 × 20
ml) extrahiert. Die kombinierten Extrakte wurden mit Wasser (3 × 20 ml)
gewaschen, über
wasserfreiem Magnesiumsulfat getrocknet, filtriert und das Lösungsmittel
aus dem Filtrat entfernt, um ein gelbes Glas zu erhalten. Dieser
Feststoff wurde an Silikagel chromatographiert, wobei mit einem
Gradienten von 3,9 M Ammoniak in Methanol-Dichlormethan (5:95 → 10:90) eluiert wurde, um die
Titelverbindung als freie Base zu erhalten. FABMS: 461 (MH+; 11%).
- (iii) Zu einer gerührten
Lösung
der Titelverbindung (142 mg; 0,297 mmol) in Methanol (3 ml) wurde
eine 1,0 M Lösung
(1,0 ml; 1,0 mmol) von etherischem Chlorwasserstoff gegeben. Das
Lösungsmittel
wurde unter reduziertem Druck entfernt, und der Rückstand
wurde im Hochvakuum über
Phosphorpentoxid getrocknet, um das Trihydrochloridsalz der Titelverbindung
als hellrosa Pulver zu erhalten. C28H33ClN4·3 HCl·0,6 H2O·0,5
CH4O (MeOH). FABMS: 461 (MH+;
100%).
-
Indem
jeweils die homologen Aldehyde 5 (Beispiel 4) und 6 (Beispiel 7)
in den vorhergehenden Reaktionsverfahren substi tuiert wurden, wurden
die folgenden Analoga der Titelverbindung hergestellt:
8-Chlor-6,11-dihydro-11-[1-[4-(1H-imidazol-4-yl)butyl]-4-piperidinyliden]-5H-benzo[5,6]cyclohepta[1,2-b]pyridin·2,8 Hydrochlorid·Dihydrat,
Schmelzpunkt 162–165°C (Zersetzung).
C26H29ClN4·2,8
HCl·2
H2O. CIMS: 433, 435 (MH+;
100/36%); 461, 463 ([M + C2H5]+; 16/6%).
8-Chlor-6,11-dihydro-11-[1-[5-(1H-imidazol-4-yl)pentyl]-4-piperidinyliden]-5H-benzo[5,6]cyclohepta[1,2-b]pyridin·2,9 Hydrochlorid·0,5 Methanolat.
C27H31ClN4·2,9
HCl·H2O·0,5
CH4O (MeOH). FABMS: 447 (MH+;
100%).
-
Beispiel 15:
-
-
- (i) Verbindungen (11) und (12) wurden hergestellt,
indem im Handel erhältliches
4-Imidazolcarboxaldehyd mit nach bekannten Verfahren mit CH3I umgesetzt wurde.
-
Durch
Substituieren analoger Aldehyde (11) und (12) und Umsetzung des
Verfahrens nach Beispiel 12 wurden die folgenden Verbindungen hergestellt:
1-(8-Chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-[(1-methyl-1H-imidazol-4-yl)-methyl]-piperazin,
4 Hydrochlorid. C23H26ClN5·HCl
und
1-(8-Chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-[(1-methyl-1H-imidazol-5-yl)-methyl]-piperazin·4 Hydrochlorid.
C23H26ClN5·4
HCl.
-
Beispiel 16. Herstellung von 1-(8-Chlor-6,11-dihydro-5H-benzo
[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-[3-(1H-imidazol-4-yl)- 1-oxo-propyl]piperazin
(14):
-
-
(i) Herstellung von 1-(8-Chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-[(E)-3-(1H-imidazol-4-yl)-1-oxo-2-propenyl]piperazin
(13):
-
Zu
einer Suspension von Urocansäure
(1,38 g; 10,0 mmol) (von Aldrich Chemicals) in N,N-Dimethylformamid
(175 ml) wurden unter einer inerten Atmosphäre 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid
(1,92 g; 10,0 mmol) und Hydroxybenzotriazol (1,08 g; 8 mmol) gegeben.
Die resultierende Mischung wurde auf 40°C erwärmt und weiter gerührt, bis
sich alle Feststoffe gelöst
hatten (etwa 10 Minuten). Zu der resultierenden Lösung wurde
8-Chlor-6,11-dihydro-11-(1-piperazinyl)-5H-benzo[5,6]cyclohepta[1,2-b]pyridin (8)
(2,51 g; 8,0 mmol) gegeben, und die resultierende Lösung wurde
bei Raumtemperatur 18,5 Stunden rühren gelassen. Dann wurde Wasser
(250 μl,
13,9 mmol) zugefügt,
und die Lösung
wurde kurz gerührt,
bevor unter reduziertem Druck konzentriert wurde. Das restliche Öl wurde
zwischen Dichlormethan (100 ml) und Wasser (100 ml) partitioniert.
Der organische Extrakt wurde durch Filtration mit wasserfreiem Magnesiumsulfat
getrocknet, wobei das Lösungsmittel
unter reduziertem Druck entfernt wurde. Der Rückstand wurde an Silikagel chromatographiert,
wobei mit Dichlormethan-Methanol-konzentriertem Ammoniumhydroxid
(90:9:0,5) eluiert wurde, um die Titelverbindung als gelbes Pulver
zu erhalten, das sich bei 160°C
zu einem schaumig Gummi zersetzte. SIMS: 434 (MH
+;
50%).
- (ii) Eine Mischung des ungesättigten
Amids 13 (200 mg, 0,463 mmol) und 10% Palladium-auf-Kohle-Katalysator
(40 mg) in wasserfreiem Methanol (40 ml) wurde 2,5 Stunden mit 50
psi auf einer Parr-Schüttelapparatur
hydriert. Es wurde eine zweite Portion (20 mg) Katalysator zugegeben
und weitere 7 Stunden weiter mit 50 psi hydriert. Es wurde eine
dritte Portion (20 mg) Katalysator zugegeben und die letzten 2 Stunden weiter
hydriert. Der Katalysator wurde durch Celite filtriert, das Lösungsmittel
wurde unter vermindertem Druck eingedampft und der Rückstand
unter Hochvakuum getrocknet, um die freie Basenform der Titelverbindung
als gelbes Pulver zu erhalten. FABMS: 436 (MH+;
55%); 402 (8%); 228 (63%).
- (iii) Die freie Basenform der Titelverbindung (155 mg; 0,356
mmol) wurde in einer Mischung von Dichlormethan (0,75 ml) und Diethylether
(0,75 ml) gelöst.
Die trübe
Lösung
wurde durch einen Spritzenfilter (0,45 μm) filtriert, und das Filtrat
wurde mit 1,0 M etherischem Chlorwasserstoff (1,8 ml, 1,8 mmol)
behandelt. Der sich bildende hygroskopische Niederschlag wurde aus
Methanol-Ethylacetat kristallisiert, um das Salz der Titelverbindung
als weißes
Pulver zu erhalten, das sich bei 165,5°C zersetzte und laut Analyse C24H26ClN5O·2,35 HCl·2,2 H2O·0,033
C4H8O2 (EtOAc)
war. FABMS: 436 (MH+; 100%); 434 (15 %);
402 (13%); 228 (70%).
-
Beispiel 17. 4-(8-Chlor-5,6-dihydro-11H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yliden)-1-[3-(1H-imidazol-4-yl)-1-oxopropyl]-piperidin
(16):
-
(i) Herstellung von 4-(8-Chlor-5,6-dihydro-11H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-[(E)-3-(1H-imidazol-4-yl)-1-oxo-2-propenyl]piperidin
(15):
-
Zu
einer Lösung
von Urocansäure
(von Aldrich Chemicals) (1,8 g; 12,9 mmol) in wasserfreiem N,N-Dimethylformamid
(65 ml) wurden 8-Chlor-6,11-dihydro-11-(4-piperidinyliden)-5H-ben zo[5,6]cyclohepta[1,2-b]pyridin
(10) (4 g; 12,9 mmol), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid
(2,15 g; 11,2 mmol) und Hydroxybenzotriazol. (1,74 g; 12,9 mmol)
gegeben.
-
-
Die
Reaktionsmischung wurde bei Raumtemperatur 48 Stunden gerührt. Es
wurde Wasser zugegeben und mehrfach mit Ethylacetat extrahiert.
Die organischen Phasen wurden kombiniert und mit Salzlösung gewaschen.
Die organischen Phasen wurden über
Natriumsulfat getrocknet und konzentriert. Der Rückstand wurde durch Flash-Säulenchromatographie
gereinigt, um Verbindung (15) (24%) zu ergeben.
- (ii)
Eine Mischung des ungesättigten
Amids (15) (1,3 g, 3,0 mmol) und 5 Palladium-auf-Kohle-Katalysator (1,3
g) in Methanol (40 ml) wurde bei Raumtemperatur unter einem Druck
von 60 psi hydriert. Die Reaktion wurde durch Dünnschichtchro matographie überwacht.
Als das Ausgangsmaterial verschwand, wurde die Reaktionsmischung
durch ein Celitebett filtriert und unter vermindertem Druck konzentriert, und durch
präparative
Dünnschichtchromatographie
gereinigt, um die Titelverbindung (16) zu ergeben (C25H25ClN4·2HCl·0,3 C4H10O·2 H2O) (54%).
-
Beispiel 18. Herstellung von 8-Chlor-5,6-dihydro-11-[1-[3-(1H-imidazol-4-yl)propyl]-4-piperidinyliden]-11H-benzo[5,6]cyclohepta[1,2-b]pyridin
(17):
-
Zu
einer Lösung
von Verbindung (16) (0,65 g; 1,5 mmol) in wasserfreiem Tetrahydrofuran
(70 ml) wurde eine 1,0 M Lösung
von Lithiumaluminiumhydrid in Tetrahydrofuran (4,6 ml; 4,6 mmol)
gegeben. Die Mischung wurde bei Raumtemperatur 1,75 Stunde gerührt, danach
mit Ethylether verdünnt
und durch tropfenweise Zugabe von gesättigter Ammoniumchloridlösung gequencht.
-
-
zu
gesättigter
Ammoniumchloridlösung.
Zu Carbonat und danach mit Ethylacetat extrahiert. Die kombinierten
organischen Extrakte wurden dann über Natriumsulfat getrocknet,
filtriert und konzentriert. Es wurde mittels Flash-Chromatographie
gereinigt, um Titelverbindung (17) zu ergeben, (C25H27ClN4·3HCl·1,8 H2O) (79%).
-
Beispiel 19. Herstellung von 8-Chlor-6,11-dihydro-11-[4-[2-(1H-imidazol-4-yl)ethyl]-1-piperazinyl]-5H-benzo[5,6]cyclohepta[1,2-b]pyridin:
-
- (i) Zu einer Lösung von Verbindung (8) (0,1
g; 0,32 mmol) in wasserfreiem Dichlormethan (15 ml), die auf 0°C gekühlt war,
wurden die tritylgeschützte
Urocansäure
(0,14 g, 0,38 mmol), 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid
(0,092 g; 0,48 mmol) und Hydroxybenzotriazol (0,065 g; 0,48 mmol)
gegeben. Die Reaktion wurde 3 Stunden bei Raumtemperatur gerührt, danach
mit gesättigter
Lösung
von Natriumcarbonat gequencht. Die wässrige Phase wurde mit Ethylacetat
ex trahiert. Die kombinierten organischen Phasen wurden dann mit
Salzlösung
gewaschen, über Kaliumcarbonat
getrocknet, filtriert und konzentriert. Das Produkt wurde durch
Flash-Chromatographie gereinigt, wobei mit einem Gradienten von
4 M Ammoniak in Methanol-Dichlormethan
(1:9 bis 5:95) eluiert wurde, um Verbindung (18) (77%) zu ergeben.
- (ii) Zu einer Lösung
von Verbindung (18) (1,09 g; 1,6 mmol) in Tetrahydrofuran (5 ml)
wurde eine 1,0 M Lösung
von Lithiumaluminiumhydrid in Tetrahydrofuran (4,8 ml; 4,8 mmol)
gegeben. Die Reaktion wurde 3 Stunden bei Raumtemperatur gerührt, danach
mit Diethylether verdünnt
und mit gesättigtem
wässrigem Natriumsulfat
gequencht. Der Feststoff wurde mit Ethylacetat extrahiert. Die kombinierten
organischen Phasen wurden über
Kaliumcarbonat getrocknet, filtriert und konzentriert.
-
-
Der
Rückstand
wurde durch Flash-Chromatographie gereinigt, wobei mit einem Gradienten
von 4 M Ammoniak in Methanol-Dichlormethan
(1:9–5:95)
eluiert wurde (85%).
- (iii) Nach einem ähnlichen
Verfahren wie in Beispiel 12 wurde das Produkt aus Stufe 2 ditrityliert,
um das HCl-Salz der Titelverbindung als weißen Feststoff zu ergeben, (C23H26ClN5·4 HCl).
-
Beispiel 20. Herstellung einer Mischung
aus 4-(8-Chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-1-[3-(1H-imidazol-4-yl)-1-oxopropyl]piperidin
(21) und 4-(6,11-Dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-1-[3-(1H-imidazol-4-yl)-1-oxopropyl]piperidin
(22)
-
-
(i) Herstellung von 4-(8-Chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-blpyridin-11-yl)-1-[(E)-3-1H-imidazol-4-yl]-1-oxo-2-propenyl]piperidin
(20):
-
Zu
einer Suspension von Urocansäure
(258 mg; 1,87 mmol) in N,N-Dimethylformamid (32,5 ml) wurden unter
einer inerten Atmosphäre
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimidhydrochlorid (358 mg;
1,87 mmol) und Hydroxybenzotriazol (201 mg; 1,49 mmol) gegeben.
Die resultierende Mischung wurde auf 40°C erwärmt und weiter gerührt, bis
sich alle Feststoffe gelöst
hatten (etwa 10 Minuten). Zu der resultierenden Lösung wurde
8-Chlor-6,11-dihydro-11-(4-piperidinyl)-5H-benzo[5,6]cyclohepta[1,2-b]pyridin
(9) (467 mg; 1,49 mmol) gegeben, und die resultierende Lösung wurde
bei Raumtemperatur 17 Stunden rühren
gelassen. Dann wurde Wasser (67 μl,
3,7 mmol) zugefügt,
die Lösung
wurde kurz gerührt
und dann unter reduziertem Druck konzentriert. Das restliche Öl wurde
zwischen Dichlormethan (100 ml) und Wasser (50 ml) partitioniert.
Es wurde mit kleinen Volumina Methanol behandelt, und die resultierende
Mi schung wurde 2 Stunden oder nach Bedarf stehen gelassen, um Trennung
der emulgierten Phasen zu erhalten. Der organische Extrakt wurde
durch Filtration durch ein Silikagelkissen in einem gesinterten
Glastrichter getrocknet, wobei mit Dichlormethan-Methanol-konzentriertem
Ammoniumhyroxid (90:9:0,5) eluiert wurde. Dann wurde das Lösungsmittel
unter reduziertem Druck von dem Filtrat entfernt, um die Titelverbindung
als flaumigen bräunlichen
Feststoff zu erhalten. CIMS: 433, 435 (MH+;
100/47%); 461 ([M + C2H5]+; 18%).
-
(ii) Herstellung einer Mischung aus 4-(8-Chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-1-[3-(1H-imidazol-4-yl]-1-oxopropyl]piperidin
und 4-(6,11-Dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-1-[3-(1H-imidazol-4-yl)-1-oxopropyl]piperidin
(21, 22):
-
-
Eine
Mischung des ungesättigten
Amids 20 (480 mg; 1,11 mmol), 1,0 M etherischem Chlorwasserstoff (1,1
ml; 1,1 mmol) und 10 Palladium-auf-Kohle-Katalysator (216 mg) in
wasserfreiem Methanol (40 ml) wurde 20 Stunden bei 50 psi auf einer
Parr-Schüttelapparatur
hydriert. Der Katalysator wurde durch Celite filtriert, das Lösungsmittel
unter vermindertem Druck verdampft und der restliche Feststoff zwischen
Dichlormethan (30 ml) und gesättigtem
wässrigem
Natriumbicarbonat (20 ml) partitioniert. Die wässrige Phase wurde mit Dichlormethan
(10 ml) extrahiert. Die organischen Extrakte wurden kombiniert,
mit Wasser (20 ml), anschließend
Salzlösung
(20 ml) gewaschen und durch Filtration durch wasserfreies Natriumsulfat
getrocknet. Das Lösungsmittel
wurde unter reduziertem Druck von dem Filtrat entfernt, und der
Rückstand
wurde im Hochvakuum getrocknet, um einen flaumigen weißen Feststoff
zu erhalten, der in einer Vielfalt von DC-Systemen (variierte Anteile Dichlormethan-Methanol-Ammoniumhydroxid)
einen homogenen Einzelspot mit Spuren von geringfügigen Verunreinigungen
zeigte. Spektroskopische und Elementaranalysedaten zeigten, dass
das Produkt eine ungefähre
2:1-Mischung des erwarteten Doppelbindungs-Reduktionsprodukts (M1)
und seines dechlorierten Derivats (M2) war.
FABMS: 435, 437 (M1H+;
70/25%); 401 (M2H+;
100%). Die spektroskopischen und Elementaranalysen waren im Einklang
mit den folgenden Formeln: C25H27ClN4O·0,5
und C25H28N4O·H2O·0,125
CH2Cl2.
-
Beispiel 21. Herstellung von 1-(8-Chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-[3-(1H-imidazol-4-yl)propylipiperazin
(23):
-
-
- (i) Zu einer Lösung von 1-(8-Chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-[3-(1H-imidazol-4-yl)-1-oxopropyl]piperazin
(14) (310 mg; 0,711 mmol) in Tetrahydrofuran (10 ml), die unter
einer inerten Atmosphäre
gerührt
wurde, wurde mittels Spritze in kleinen Portionen im Verlauf von
ungefähr
3 Minuten eine 1,0 M Lösung
von Lithiumaluminiumhydrid in Tetrahydrofuran (1,60 ml; 1,60 mmol)
gegeben. Die Re aktionslösung
wurde bei Raumtemperatur 3 Stunden gerührt. Die Reaktion wurde unter
kräftigem Rühren durch
vorsichtige tropfenweise Zugabe von Wasser (61 μl), gefolgt von nacheinander
15% wässrigem
Natriumhydroxid (61 μl)
und Wasser (183 μl)
gequencht. Der resultierende Niederschlag wurde filtriert und Lösungsmittel
unter reduziertem Druck aus dem Filtrat entfernt. Der Rückstand
wurde zwischen Dichlormethan (25 ml) und Wasser (20 ml) partitioniert.
Die organische Phase wurde mit Salzlösung (20 ml) gewaschen, durch
wasserfreies Natriumsulfat filtriert und Lösungsmittel aus dem Filtrat
entfernt, um die freie Basenform der Titelverbindung als gelbes
Pulver zu erhalten. Dieses wurde an Silikagel chromatographiert,
wobei mit Dichlormethan-Methanol-konzentriertem Ammoniumhydroxid
(90:9:0,5) eluiert wurde, um die Titelverbindung als schmutzigweißes Pulver
zu erhalten, das sich bei etwa 91–98°C zu einem schaumig Gummi zersetzte.
CIMS: 422,424 (MH+; 100/32%); 450, 452 ([M
+ C2H5]+;
14/6%).
- (ii) Zu einer kräftig
gerührten
Lösung
der obigen freien Base (66 mg; 0,152 mmol) in Dichlormethan (2,75 ml)
wurde Diethylether (8 ml) gegeben, gefolgt von 0,115 M etherischer
Maleinsäure
(2,75 ml; 0,316 mmol). Der resultierende Niederschlag wurde ungefähr 5 Minuten
in dem Reaktionsmedium trituriert. Der Überstand wurde dekantiert und
frischer Ether (15 ml) zugefügt.
Dieses Verfahren wurde zwei weitere Male wiederholt. Der hygroskopische
Feststoff wurde dann rasch filtriert und im Hochvakuum über Phosphorpentoxid
getrocknet, um eine Maleatsalzform der Titelverbindung zu erhalten.
FABMS: 422, 424 (MH+; 86/31%); 228, 230(53/19%).
-
Die
spektroskopischen und Elementaranalysen waren im Ein klang mit den
folgenden Formeln: C24H28ClN5·2
C4H4O4·0,8 H2O·0,6
C4H10O (Et2O).
-
Beispiel 22. Herstellung einer Mischung
aus 8-Chlor-6,11-dihydro-11-[1-[3-(1H-imidazol-4-yl)propyl]-4-piperidinyl]-5H-benzo[5,6]cyclohepta[1,2-b]pyridin
(24) und 6,11-Dihydro-11-[1-[3-(1H-imidazol-4-yl]propyl]-4-piperidinyl]-5H-benzo[5,6]cyclohepta[1,2-b]pyridin
(25):
-
-
- (i) Zu einer Lösung der Mischung von ungesättigten
Amiden (21, 22) (275 mg; 0,414 mmol) in Tetrahydrofuran (8 ml),
die unter einer inerten Atmosphäre
gerührt
worden war, wurde im Verlauf von 3 bis 4 Minuten in kleinen Portionen
mittels Spritze eine 1,0 M Lösung
von Lithiumaluminiumhydrid in Tetrahydrofuran (1,28 ml; 1,28 mmol)
gegeben. Die Reaktion wurde bei Raumtemperatur 4 Stunden gerührt. Die
kräftig
gerührte Reaktion
wurde durch vorsichtige tropfenweise Zugabe von Wasser (49 μl), gefolgt
von nacheinander 15 wässrigem
Natriumhydroxid (49 μl)
und Wasser (147 μl)
gequencht. Der resultierende Niederschlag wurde filtriert und Lösungsmittel
unter reduziertem Druck aus dem Filtrat entfernt. Der Rückstand
wurde zwischen Dichlormethan (20 ml) und Wasser (20 ml) partitioniert.
Die organische Phase wurde mit Salzlösung (20 ml) gewaschen, durch
wasserfreies Natriumsulfat filtriert und das Lösungsmittel unter reduziertem
Druck aus dem Filtrat entfernt. Der Rückstand wurde an Silikagel
chromatographiert, wobei mit Di chlormethan-Methanol-konzentriertem
Ammoniumhydroxid (90:9:0,5) eluiert wurde, um die freie Basenform
der Titelmischung als weißes
Pulver zu erhalten. Die spektroskopischen und Elementaranalysen
waren im Einklang mit der folgenden Formel: C25H29ClN4·0,75 C25H30N4·1,25 H2O.
- (ii) Zu einer kräftig
gerührten
Lösung
der obigen freien Base (85 mg; 0,203 mmol) in Dichlormethan (1 ml) wurde
Diethylether (3 ml) gegeben, gefolgt von 0,115 M etherischer Maleinsäure (5,29
ml; 0,608 mmol). Der resultierende Niederschlag wurde ungefähr 5 Minuten
in dem Reaktionsmedium trituriert. Der Überstand wurde dekantiert und
frischer Ether (3 ml) zugefügt.
Dieses Verfahren wurde weitere zwei Male wiederholt, dann wurde
der hygroskopische Feststoff rasch abfiltriert und unter Hochvakuum
getrocknet, um die Maleatsalzform der Titelmischung zu erhalten.
Spektroskopische und Elementaranalysedaten zeigten, dass das Produkt
eine ungefähre
1,5:1-Mischung des
chlorierten Reduktionsprodukts (M1) und
seines dechlorierten Derivats (M2) war.
FABMS: 421, 423 (M1H+;
65/29%); 387 (M2H+;
100%). Die spektroskopischen und Elementaranalysen waren im Einklang
mit der folgenden Formel: C25H29ClN4·0,68
C25H30N4·3,6 C4H4O4·4 H2O·0,5
C4H10O (Et2O).
-
Beispiel 23. Herstellung von 1-Trityl-4-chlormethylimidazol:
-
-
- (i) Im Handel erhältliches 4-Hydroxymethylimidazolhydrochlorid
(von Aldrich) und Triphenylmethylchlorid wurden gemäß Literaturverfahren
(Kelley, J. Med. Chem., 20 (5), 721 (1977) umgesetzt.
- (ii) Das Produkt von Beispiel 23 (i) wurde in Benzol (464 ml)
suspendiert und bis zur Trockne azeotrop destilliert, indem 50 ml
Benzol abdestilliert wurden. Dann wurde Triethylamin (27,76 ml)
zugefügt.
Die resultierende Mischung wurde auf 0°C abgekühlt und tropfenweise im Verlauf
von 10 Minuten Thionylchlorid zugegeben. Nach 35 Minuten wurden
Ethylacetat und Wasser zugegeben. Die wässrige Phase wurde abgetrennt
und mit Ethylacetat gewaschen. Die kombinierten organischen Extrakte
wurden mit gesättigter
Natriumbicarbonatlösung,
danach Salzlösung
gewaschen, über
Kaliumcarbonat getrocknet, filtriert und konzentriert. Trocknen
unter Hochvakuum und Lichtschutz ergab die Titelverbindung (b).
-
Beispiel 24. Herstellung von 8-Chlor-6,11-dihydro-11-[1H-imidazol-4-ylmethyl)-4-piperidinyl]-5H-benzo[5,6]cyclohepta(1,2-b)pyridin
(26):
-
-
- (i) Zu einer Lösung von Verbindung (9) (0,5
g; 1,9 mmol) in wasserfreiem Dichlormethan (10 ml) wurden das tritylgeschützte Imidazolchlorid
(b) aus Beispiel 23 (0,68 g; 1,9 mmol) und Triethylamin (0,264 ml;
1,9 mmol) gegeben. Die Reaktion wurde bei Raumtemperatur über Nacht
gerührt.
Der Mischung wurden nacheinander Wasser (10 ml) und Ethylacetat
(20 ml) zugegeben. Die wässrige
Phase wurde abgetrennt und danach mit Ethylacetat (2 × 10 ml)
extrahiert. Die kombinierten organischen Phasen wurden über Magnesiumsulfat
getrocknet, filtriert und konzentriert. Der Rückstand wurde durch Flash-Chromatographie über SiO2 gereinigt (36%).
- (ii) Unter Verwendung eines ähnlichen
Verfahrens wie in Beispiel 12 wurde der Titelvorläufer entschützt, um das
HCl-Salz der Titelverbindung
als weißen
Feststoff (26) zu ergeben, C23H25ClN4·3
HCl·1,5
H2O.
-
Die
folgende Verbindung wurde durch Substituieren von Verbindung (10)
(Beispiel 14) in dem vorhergehenden Reaktionsverfahren hergestellt:
8-Chlor-6,11-dihydro-11-(1-(4-imidazolylmethyl)-4-piperidyliden)-5H-benzo[5,6]cyclohepta[1,2-b]pyridin,
C23H23ClN4·3
HCl.
-
Beispiel 25. Herstellung der Verbindung
(27):
-
Dies
ist in der veröffentlichten
Anmeldung
WO 95/10516 offenbart.
-
Beispiel 26. Herstellung der Verbindung
(28):
-
-
Beispiel 27. Herstellung von 11-[4-[(1H-Imidazol-4-yl)methyl]-1-piperidinyl]-8-chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin
(29):
-
-
Verbindungen
(27) und (28) wurden kombiniert und nach dem in Beispiel 24 angegebenen
Verfahren umgesetzt. Das Produkt wurde als HCl-Salz der Titelverbindung
(29) erhalten, C23H25ClN4·3
HCl·2
H2O CIMS: 393 (MH+)
-
Beispiel 28. 4-(1H-Imidazol-4-yl)butyl-4-(8-chlor-6,11-dihy
dro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-1-piperazin carboxylat
(31):
-
-
- (i) Zu einer gerührten Suspension von Natriumhydrid
(200 mg einer 60 Dispersion; 5 mmol) in trockenem Tetrahydrofuran
(25 ml) wurde im Verlauf von 10 Minuten portionsweise Alkohol-Hydrochlorid (4)
(1,05 g; 2,5 mmol) gegeben. Die resultierende Suspension wurde bei
Raumtemperatur 3,5 Minuten gerührt.
Das Piperazinderivat (8) (706 mg; 2,25 mmol) wurde zugegeben, die
Mischung mit trockenem Tetrahydrofuran (25 ml) verdünnt und
die resultierende Suspension über
Nacht bei Raumtemperatur rühren
gelassen. Triethylamin (0,35 ml; 2,5 mmol) wurde zugegeben, gefolgt
von der Zugabe von einer Lösung
von Triphosgen (252 mg; 0,849 mmol) in trockenem Dichlormethan (2,5
ml) (milde Exotherme) im Verlauf von etwa 4 Minuten. Die resultierende Suspension
wurde 6 Stunden bei Raumtemperatur gerührt, danach wurden die Feststoffe filtriert
und mit Ethylacetat gewaschen. Das Filtrat und die Wäschen wurden
kombiniert und Lösungsmittel unter
reduziertem Druck entfernt. Chromatographie des Rückstands
an Silikagel, wobei mit einem Gradienten von Ethylacetat-Methanol
(98:2 → 95:5)
eluiert wurde, ergab das tritylierte Titelprodukt (31) als weißen glasartigen
Schaum. ESIMS: 722 (MH+; 85%); 243 (Ph3C+; 100%). Eine
polarere Fraktion, die dem symmetrischen Harnstoff (30) entspricht,
wurde auch isoliert.]
- (ii) Eine Mischung des obigen tritylierten Carbamat (31) (310
mg; 0,430 mmol) und 15 wässrige
Salzsäure (10
ml) in Methanol (10 ml) wurde 1,5 Stunden unter Rückfluss
gehalten. 6M HCl (4 ml) und Methanol (1 ml) wurde zugefügt und eine
weitere Stunde unter Rückfluss
gehalten. Die Lösungsmittel
wurden unter vermindertem Druck entfernt. Der Rückstand wurde zwischen Dichlormethan
(25 ml) und 1,1 M wässrigem
Natriumbicarbonat (13 ml) partitioniert. Die wässrige Phase wurde mit Dichlormethan
(3 × 25
ml) extrahiert und die kombinierten organischen Extrakte mit Wasser
(2 ×)
und Salzlösung
gewaschen. Die organischen Phasen wurden über wasserfreiem Magnesiumsulfat
getrocknet, das Trockenmittel wurde filtriert, das Lösungsmittel
unter vermindertem Druck entfernt und der Rückstand unter Hochvakuum getrocknet,
um das Titelprodukt (32) als freie Base zu erhalten. FABMS: 460
(MH+; 100%).
- (iii) Zu einer Lösung
der freien Base des Titelprodukts (98,4 mg; 0,205 mmol) in Methanol
(4,5 ml) wurde 1,0 M etherischer Chlorwasserstoff (0,72 ml; 0,72
mmol) gegeben. Das Lösungsmittel
wurde unter reduziertem Druck entfernt, und der Rückstand
wurde im Hochvakuum über
Phosphorpentoxid getrocknet, um das Hydrochloridsalz des Titelprodukts
als blassgelben Schaum zu erhalten. CIMS: 480 (MH+;
97%); 251 (54%); 228 (100%). Die spektroskopischen und Elementaranalysen
waren im Einklang mit der folgenden Formel: C26H30ClN5O2·2,6 HCl·4 H2O·0,7
CH4O (MeOH).
-
Beispiel 29. Herstellung von (+)-(8-Chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-[(1H-imidazol-4-yl)methyl]piperazin
(33) und (–)-(8-Chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-[(1H-imidazol-4-yl)me
thyl]piperazin (34):
-
- (i) Eine heiße (Dampfbad) Mischung der
racemischen Piperazinverbindung (8) (1 g) in 8% wässrigem
Acetonitril (10 ml) wurde filtriert, was etwa 0,33 g Rückstand
hinterließ.
Zu dem Filtrat wurde eine heiße
Lösung von
N-Acetyl-L-Leucin (0,55 g) in 8% wässrigem Acetonitril (10 ml)
gegeben, gefolgt von 8% wässrigem Acetonitril
(8 ml). Die Lösung
wurde stehen gelassen und abkühlen
gelassen. Die Kristalle wurden nach 24,5 Stunden filtriert und trocknen
gelassen.
-
-
Die
Kristalle wurden dann mit Kaliumcarbonat (0,11 g; 0,8 mmol), Wasser
(10 ml) und Dichlormethan (10 ml) gerührt. Die wässrige Phase wurde abgetrennt
und mit Dichlormethan (5 ml) extrahiert. Die kombinierten Extrakte
wurden mit Salzlösung
gewaschen, über
Natriumsulfat getrocknet, filtriert und zu einem weißen Schaum
konzentriert. Das Verfahren wurde zwei weitere Male mit der ursprünglichen
racemischen Mischung wiederholt, um insgesamt 0,59 g mit 82–90% ee
zu ergeben. Dieses Material wurde dann in der gleichen Weise zurückgeführt, um
0,13 g mit 100% ee zu ergeben. Die kombinierten Mutterlaugen wurden
wie oben konzentriert und alkalisch gemacht, danach in der gleichen
Weise zwei Mal mit N-Acetyl-D-leucin behandelt, um 0,39 g mit 100%
ee des entgegengesetzten Enantiomers zu ergeben.
- (ii)
Indem die jeweiligen (+)- und (–)-Enantiomere
der Verbindung (i) mit dem tritylgeschützten Chlorimidazol nach dem
in Beispiel 13 beschriebenen Verfahren umgesetzt wurden, wurden
die folgenden Verbindungen hergestellt:
(+)-(8-Chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-[(1H-imidazol-4-yl)methyl]piperazin,
C22H24ClN5·4
HCl3H2O CIMS: 394
(MH+).
(–)-8-Chlor-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-11-yl)-4-[(1H-imidazol-4-yl)methyl]piperazin,
C22H24ClN5·4
HCl·4
H2O CIMS: 394 (MH+).
-
Beispiel 30. Herstellung von 4-(8-Chlor-6,11-dihydro-5H-benzo
[5,6]cyclohepta[1,2-b]pyridin-11-yl)-N-[4-(1H-imidazol-4-yl) butyl-1-piperazincarboxamid
(36):
-
-
- (i) Eine Lösung
von Verbindung 8 (47,7 mg; 0,152 mmol) und Triethylamin (0,025 ml;
0,177 mmol) in CH2Cl2 (0,5
ml) wurde tropfenweise im Verlauf von 10 Minuten bei 20°C zu einer
gerührten
Lösung
von Triphosgen (16,4 mg; 0,0555 mmol) in CH2Cl2 (0,4 ml) gegeben. Nachdem die Reaktionsmischung
weitere 20 Minuten bei Raumtemperatur gerührt worden war, wurde tropfenweise
im Verlauf von 7 Minuten bei 20°C
eine Lösung
des Amins (Literaturverbindung: R. Wolin et al., Bioorg. Med. Chem.
Lett. 8 (1998) 2157–162)
(57,2 mg; 0,15 mmol) und Triethylamin (0,015 ml; 0,177 mmol) in
Dichlormethan (0,5 ml) zugegeben. Die grüngelbe Reaktion wurde bei Raumtemperatur
20,5 Stunden gerührt.
Die rohe Reaktionsmischung wurde auf Silikagel geladen und chromatographiert,
wobei mit einem Gradienten von Ethylacetat-Methanol (95:5 → 90:10)
eluiert wurde, um das tritylierte Produkt als Feststoff (35) zu
erhalten. FABMS: 721 (MH+).
-
-
- (ii) Durch Verwendung eines ähnlichen
Verfahrens wie in Beispiel 12 wurde der Tritylvorläufer entschützt, um
das HCl-Salz der
Titelverbindung (36) als hellbraunen Feststoff zu ergeben. FABMS:
479 (MH+). HRMS: (C26H32ClN6O) berechnet
479,2326, gemessen 479,2320.
-
Allgemeines Verfahren für den H1-Rezeptorbindungsassay:
-
Das
verwendete Verfahren basierte auf demjenigen, das in V. T. Tran
et al., "Histamine
H1 receptors identified in mammalian brain
membranes with [H-3]mepyramine",
Proc. Natl. Acad. Sci. U.S.A. 75 (1978) 6290–6294, offenbart ist.
-
I. Gewebepräparationsprotokoll für Histamin-H1-Rezeptorbindungsassay:
-
- 1. Die Gewebequelle war das Hirn männlicher
Sprague-Dawley-Ratten. Diese wurden erworben, gestrippt und gefroren
(erhältlich
von Rockland Corporation, Gilbertsville, Pennsylvania, USA). Der
verwendete Puffer war eiskalter 50 mM Tris-HCl, pH 7,5. (Der pH-Wert
wurde bei 25°C
ermittelt).
- 2. Die Hirne wurden auf einer Kunststoffhülle auf dem Arbeitstisch ausgebreitet
und 10 bis 15 Minuten tauen gelassen. Danach wurde alles eiskalt
gehalten.
- 3. Zwei Hirne wurden in jedes 50 ml Zentrifugenröhrchen mit
rundem Boden gegeben, und es wurden 25 ml Puffer zugegeben. Dann
wurden sie mit einem Polytron (von Brinkmann Instruments, Westburg,
New York, USA), der mit einer FT-10-Spitze. ausgestattet war, mit
der Einstellung 6 30 Sekunden lang aufgebrochen.
- 4. Das Volumen in dem Röhrchen
wurde auf 45 ml gebracht und gemischt, und das Teilchenmaterial
wurde mit 1000 g (3000 UpM, SS-34-Rotor) 10 Minuten zentrifugiert,
um Kerne und nicht zerbrochene Zellen zu entfernen.
- 5. Die Pellets wurden verworfen und die Überstände 10 Minuten mit 50.000 g
(20 000 UpM, SS-34-Rotor) zentrifugiert.
- 6. Die Hochgeschwindigkeitspellets wurden in einem Volumen Trispuffer,
das dem ursprünglichen
entsprach (4 ml), erneut suspendiert, die Inhalte aller Röhrchen wurden
zusammengegeben und eine Probe für
den BCA-Proteinassay entnommen. Das Material wurde aufgeteilt, 45
ml pro Röhrchen
mit Rundboden, und die Resuspension wurde erneut zentrifugiert.
Die Ausbeute an Protein betrug ungefähr 20 mg/Hirn, so dass es etwa
40 mg Protein pro Röhrchen
waren.
- 7. Die Pellets wurden bei –80°C eingefroren.
-
II. H1-Histaminrezeptorbindungsassay:
-
Materialien:
96-Mulden-Tiefmuldenpolypropylenplatten, [3H]-Pyrilamin,
20–30
Ci/mmol von DuPont NEN Life Science Products, Boston, Massachusetts,
USA), Chlorpheniraminmaleat (von Schering-Plough Corporation, Kenilworth,
New Jersey, USA) als Standard, gelagert als gefrorene 10–5,
10–6,
10–7,
10–8 M
Lösungen.
- 1. FDCL und die Vergleichsverbindungen für den Assay
wurden unabhängig
durch Vortexieren oder, falls erforderlich, durch Ultraschallbehandlung
mit 1 mg/ml DMSO solubilisiert. Die erste Verdünnung, 100-fach, erfolgte in
50 mM Tris-HCl, pH 7,5, bei Raumtemperatur. Die drei oder vier nachfolgenden
10-fachen seriellen
Verdünnungen
erfolgten in 1% DMSO/50 mM Tris-HCl,
pH 7,5. Arzneimittellösungen
und Assayplatten wurden während
des Verlaufs des Assayaufbaus auf Raumtemperatur gehalten.
- 2. Testverbindungen wurden bei vier oder fünf Konzentrationen untersucht:
1, 0,1, 0,01, 0,001 und 0,0001 μg/ml.
Zwanzig μl
Arzneimittellösung
wurden in jede der drei Mulden pipettiert. Ein Chlorpheniraminmaleatstandard
wurde bei 10–9 bis 10–6 M
untersucht, wobei 20 μl
von jeder der entsprechenden Lösungen
als Dreifachproben in Mulden pipettiert wurden. Die gesamte und
unspezifische Bindung (10–6 M Chlorpheniraminmaleat)
wurde mindestens als Vierfachversuch ermittelt. Für die Gesamtbindung
wurden 20 μl
Puffer pipettiert, und für
die unspezifische Bindung wurden 20 μl 10–5 M
Chlorpheniraminmaleat in jede Mulde pipettiert.
- 3. [3H]Pyrilamin wurde ungefähr 2000-fach
mit eiskalter mM Tris-HCl, pH 7,5 (auf eine Arbeitskonzentration von
20–25
nM) verdünnt
und auf Eis gegeben.
- 4. Ein gefrorenes Gewebepellet wurde in einem Wasserbad mit
25°C aufgetaut,
erneut in 50 mM Tris-HCl, pH 7,5, in 1,7–2 mg/ml durch kurzes Aufbrechen
auf dem Polytron suspendiert und auf Eis gegeben.
- 5. Zwanzig μl
von verdünntem
[3H]Pyrilamin wurden in jede Mulde gegeben.
- 6. 150 μl
Gewebesuspension wurden in jede Mulde gegeben.
- 7. Der obere Bereich der Platte wurde abgedeckt und sie wurde
30 Minuten in ein Schüttelwasserbad
mit 25°C
(etwa 60 Oszillationen/Minute) gegeben.
- 8. Die Proben wurden mit einem Tomtec Mach 2 Ernter (erhältlich von
Tomtec Corporation, Orange, Connecticut, USA) durch eine GF/B-Filtermatte
(von Wallac, Inc., Gaithersburg, Maryland, USA) filtriert, die in 0,3%
Polyethylenimin vorgeweicht war. Jede Probe wurde drei Mal mit eiskaltem
50 mM Tris-HCl, pH 7,5, gewaschen, 20 Sekunden auf der Tomtec getrocknet
und 3–4
Minuten in einem Mikrowellenofen auf einem Papierhandtuch getrocknet.
Der Filter wurde mit Wachs-Szintillationsmittel der Marke MELTILEX
(von Wallac Corporation) imprägniert
und mit einem Betaplate Szintillationszähler (von Wallac Corporation)
gezählt.
- 9. Die spezifische Bindung wurde als Unterschied zwischen gesamter
und unspezifischer Bindung bestimmt. Die prozentuale Inhibierung
in Gegenwart von Inhibitor oder Standard wurde mit der folgenden
Formel bestimmt: [1-(Probebindung – unspezifische
Bindung)/spezifische Bindung] × 100
-
Bei
Verbindungen, die mehr als 50% bei 1 μg/ml inhibieren, wurde der IC50-Wert aus Näherungskonzentrationen interpoliert.
Der Wert wurde unter Verwendung des Formelgewichts der Verbindung
in einen Wert in nM umgerechnet, und der Ki-Wert
wurde unter Verwendung der Gleichung von Cheng und Prusoff berechnet:
(Ki = IC50/(1 +
[L]/KD), [Y -C. Cheng und W. H. Prusoff, "Relationship between
the inhibitory constant (Kj) and the concentration of inhibitor
which causes 50 per cent inhibition (IC50)
of an enzymatic reaction",
Biochem. Pharmacol. 22 (1973) 3099–3108]. Ein niedriger Wert
von Ki zeigt größere Bindungsaffinität.
-
Allgemeines Verfahren für den H3-Rezeptorbindungsassay:
-
Die
Quelle für
die H3-Rezeptoren in diesem Experiment war
Meerschweinchenhirn. Die Tiere wogen 400–600 g. Das Hirngewebe wurde
mit einer Lösung
von 50 mM Tris, pH 7,5, homogenisiert. Die Endkonzentration von
Gewebe in dem Homogenisierungspuffer betrug 10% Gew./Vol. Die Homogenisate
wurden mit 1000 g 10 Minuten zentrifugiert, um Gewebeklumpen und
Trümmer
zu entfernen. Die resultierenden Überstände wurden dann mit 50.000 × g 20 Minuten
zentrifugiert, um die Membranen zu sedimentieren, die danach drei Mal
in Homogenisierungspuffer gewaschen wurden (50.000 × g jeweils
20 Minuten). Die Membranen wurden gefroren und bis zum Gebrauch
bei –70°C gelagert.
-
Alle
zu testenden Verbindungen wurden in DMSO gelöst und danach mit dem Bindungspuffer
(50 mM Tris, pH 7,5) mit 0,1 DMSO verdünnt, so dass die Endkonzentration
2 μg/ml
betrug. Dann wurden den Reaktionsröhrchen Membranen zugefügt (400 μg Protein).
Die Reaktion wurde durch Zugabe von 3 nM [
3H]R-α-Methylhistamin
(8,8 Ci/mmol) oder 3 nM [
3H]N
α-Methylhistamin
(80 Ci/mmol) gestartet und unter Inkubation bei 30°C 30 Minuten
fortgesetzt. Gebundener Ligand wurde durch Filtration von ungebundenem
Ligand getrennt, und die Menge an radioaktivem Ligand, der an die
Membranen gebunden war, wurde durch Flüssigszintillationsspektrometrie
quantifiziert. Alle Inkubationen wurden doppelt durchgeführt, und
die Standardabweichung war immer weniger als 10 Verbindungen, die
mehr als 70 der spezifischen Bindung von radioaktivem Liganden an
den Rezeptor inhibierten, wurden seriell verdünnt, um ein K
i (nM)
zu bestimmen. Die Ergebnisse sind in Tabelle 1 für das HCl-Salz der gezeigten
Verbindungen angegeben. Tabelle
1
-
Es
ist aus diesen Testergebnissen für
den versierten Fachmann offensichtlich, dass die erfindungsgemäßen Verbindungen
zur Behandlung von Entzündung,
Allergie, Erkrankungen des Gastrointestinaltrakts, Herz-Kreislauf-Erkrankung,
Nasenschwellung, Erkrankungen des zentralen Nervensystems und ähnlichen
Erkrankungen nützlich
sind, die bereits angegeben wurden.
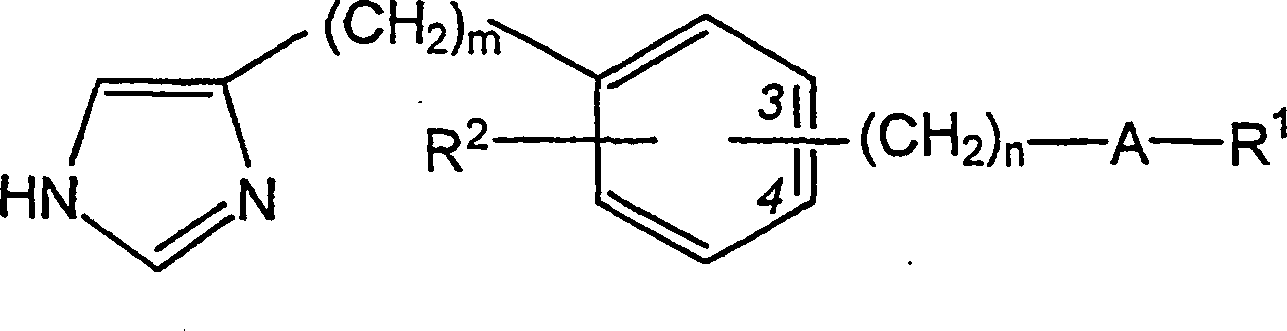
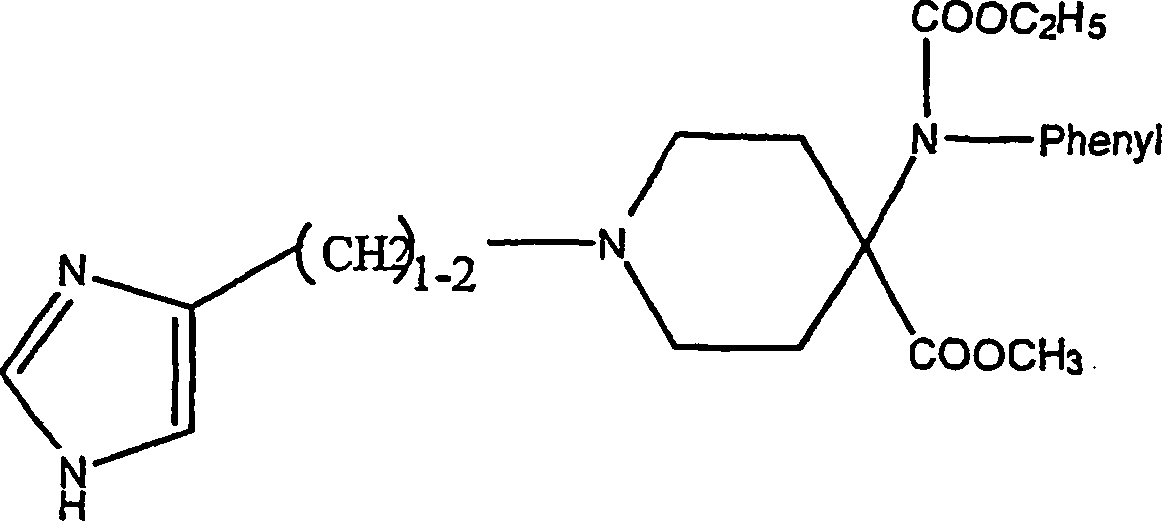
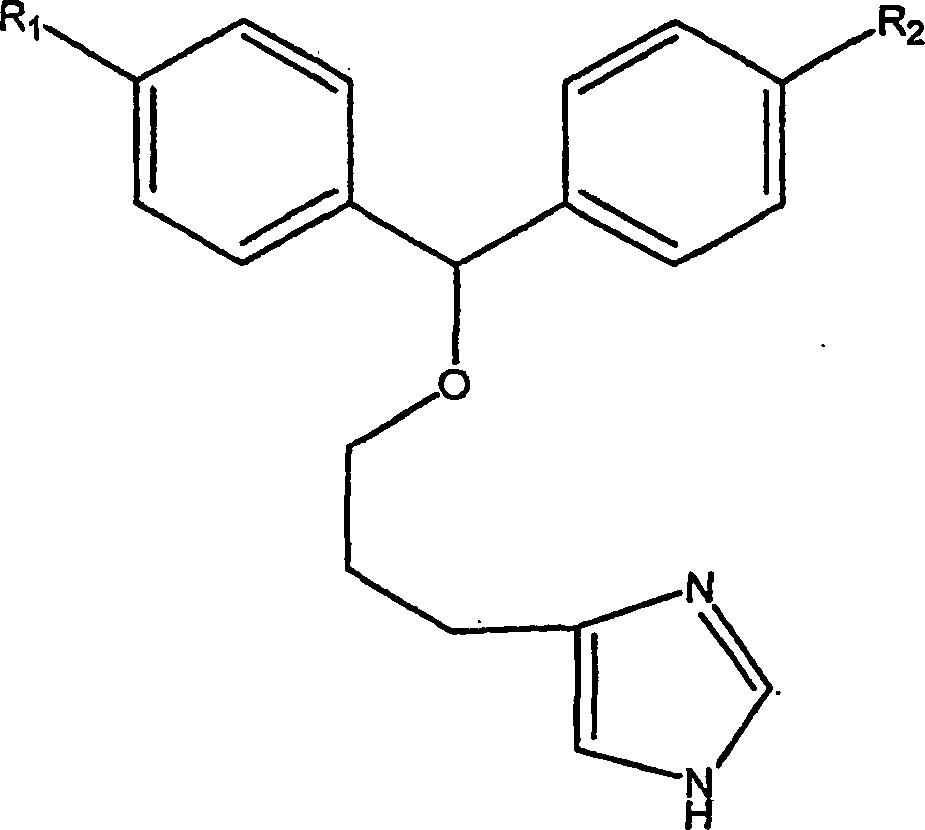
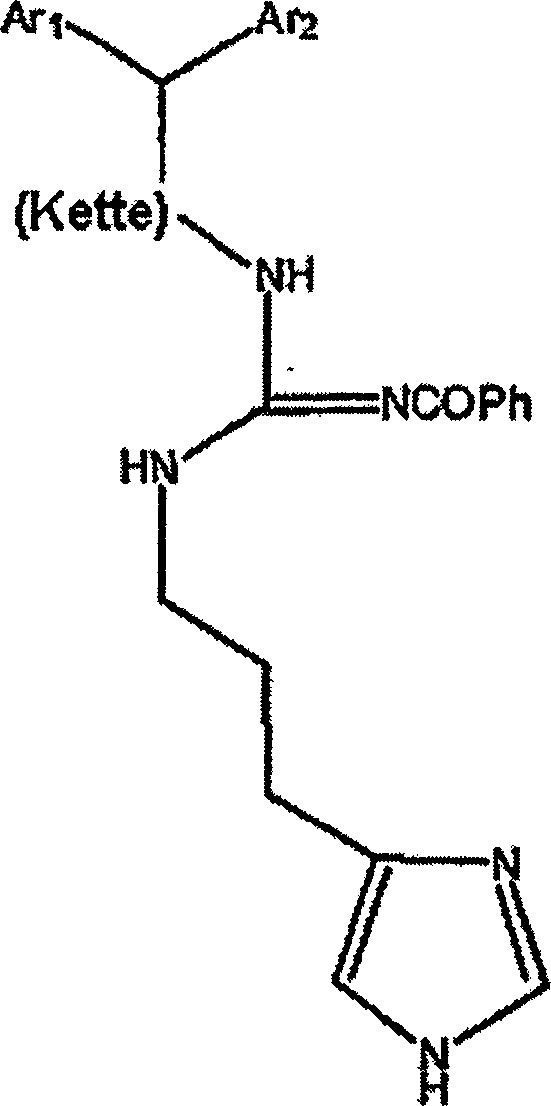

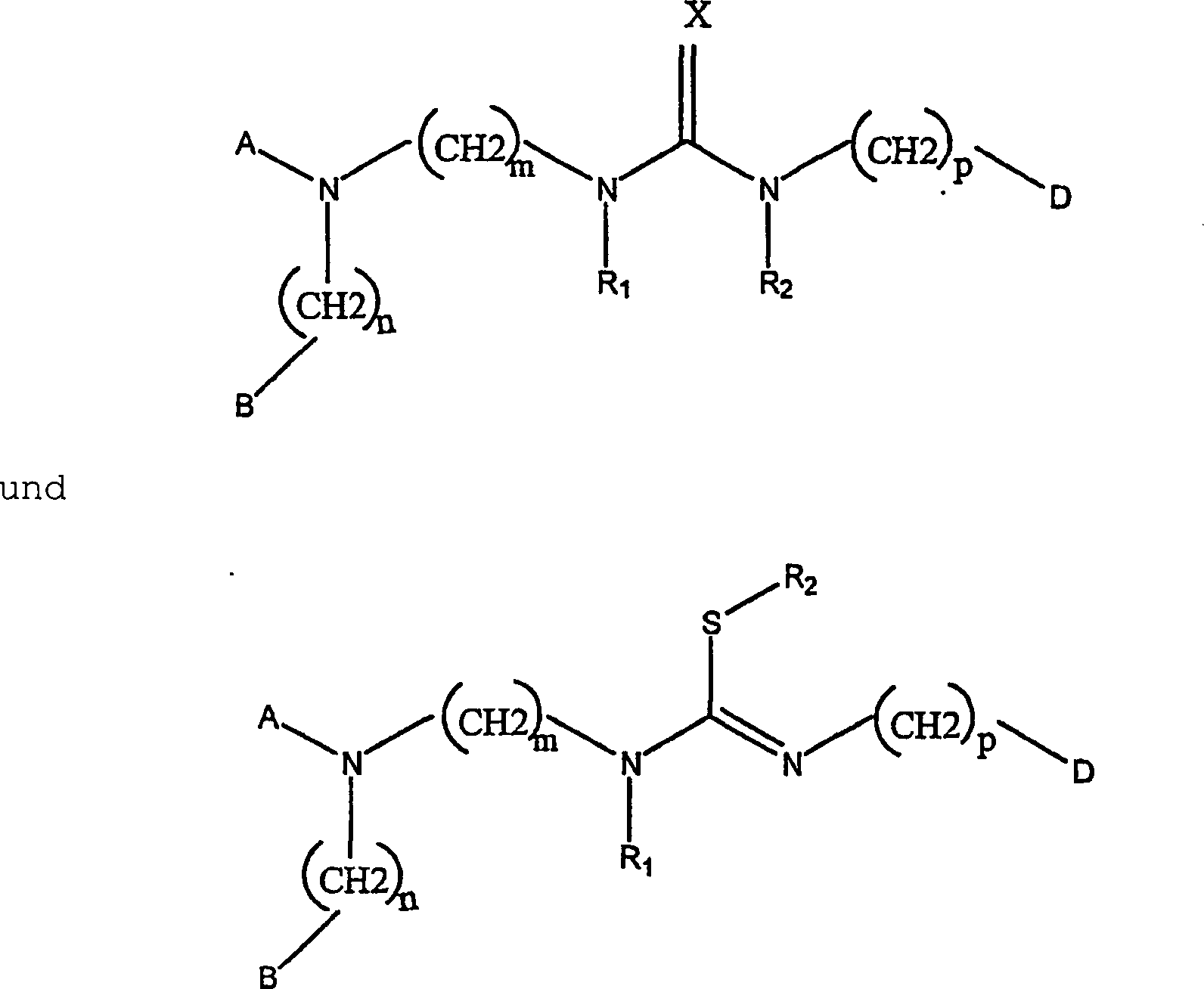



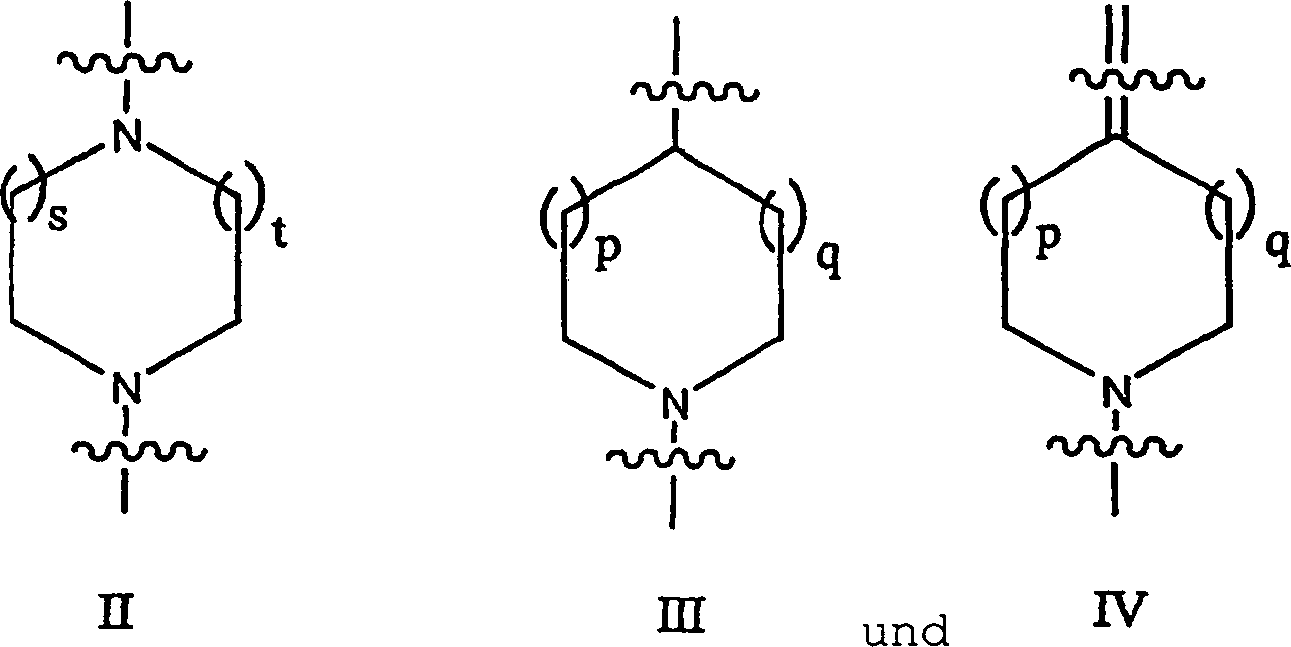



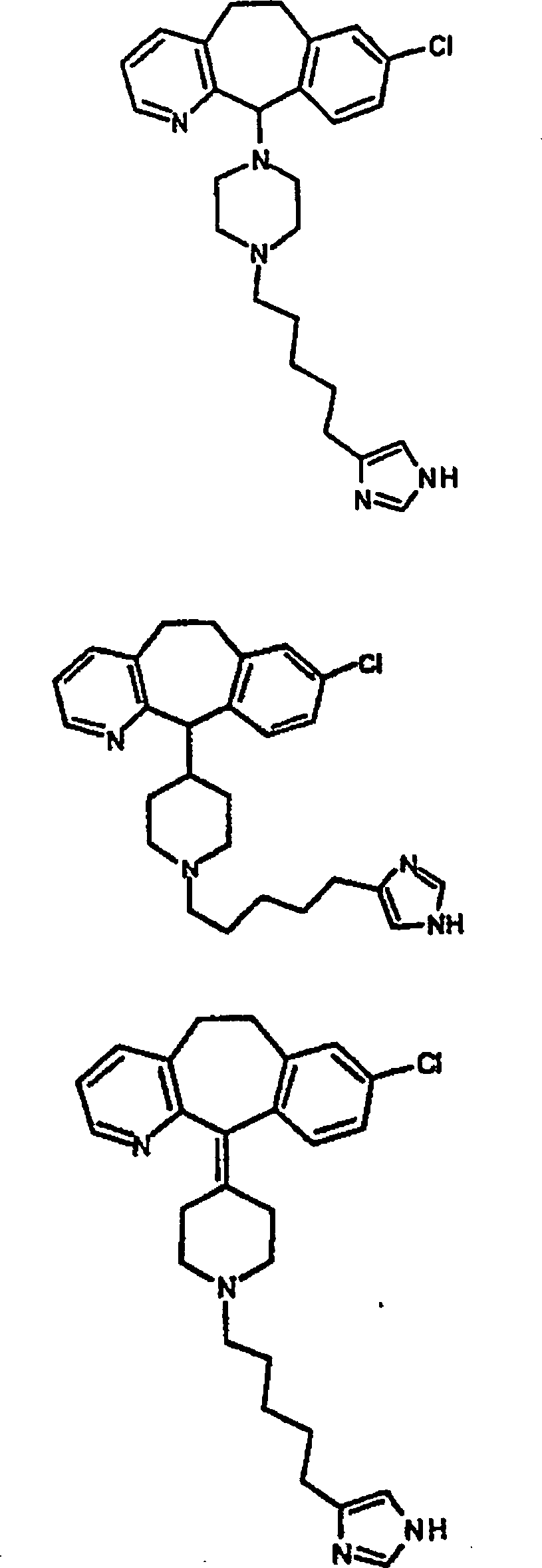

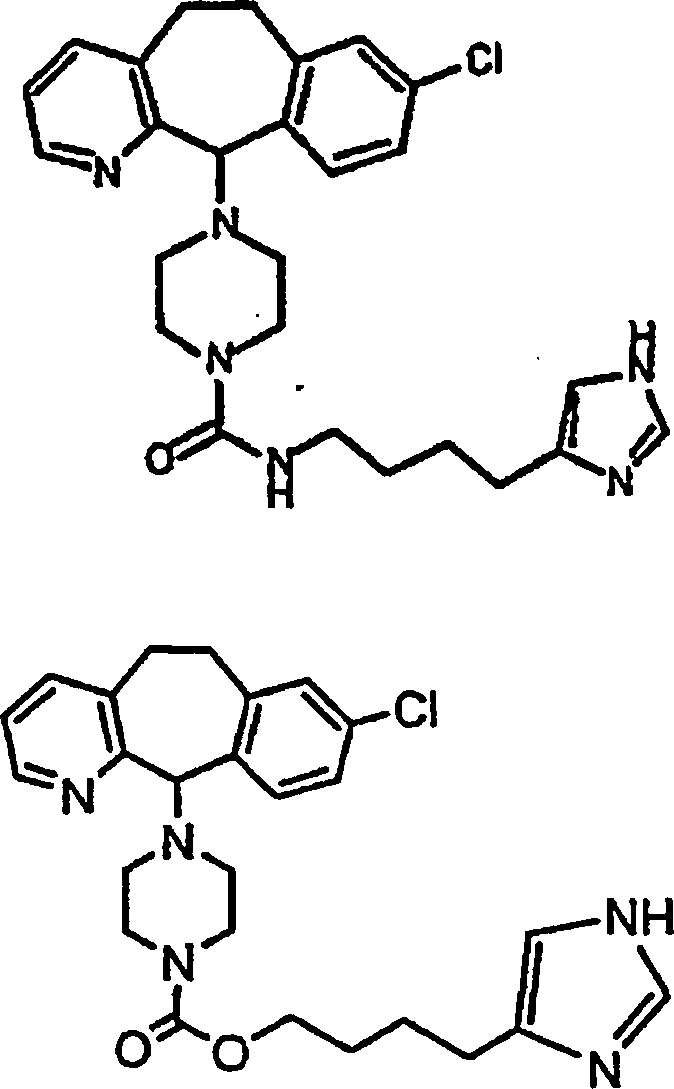
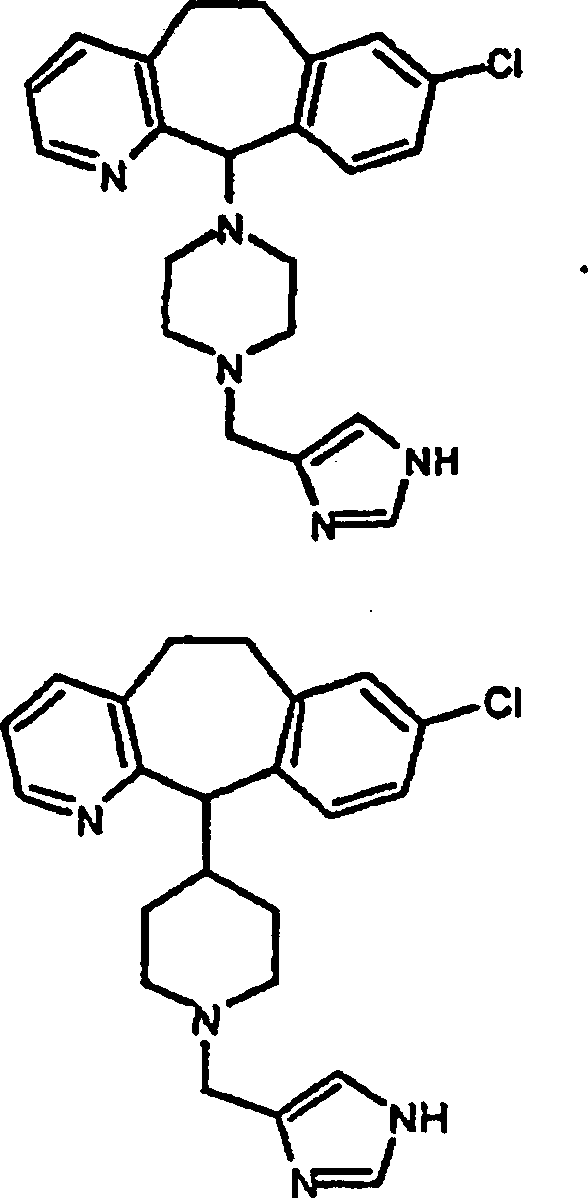
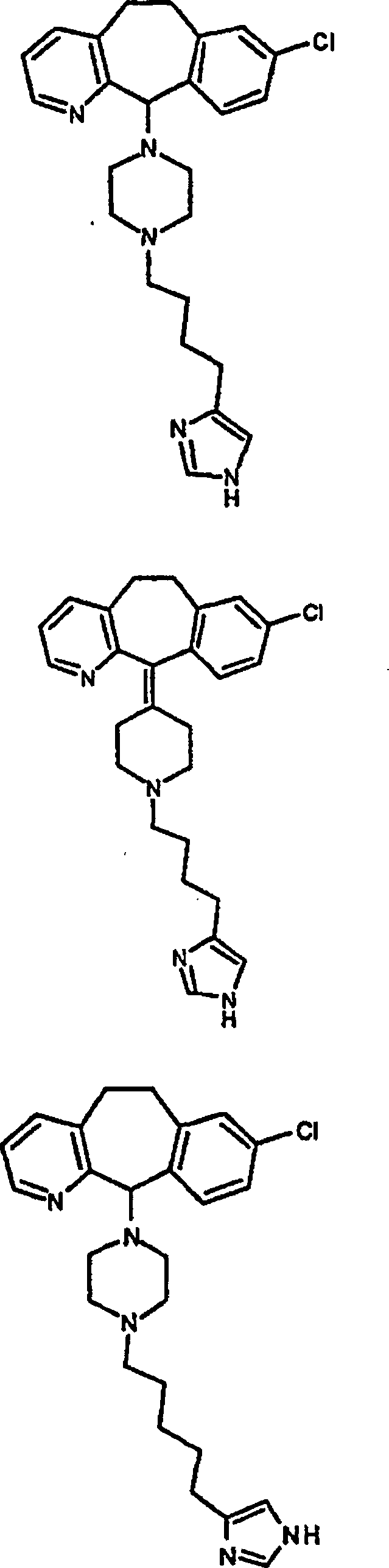
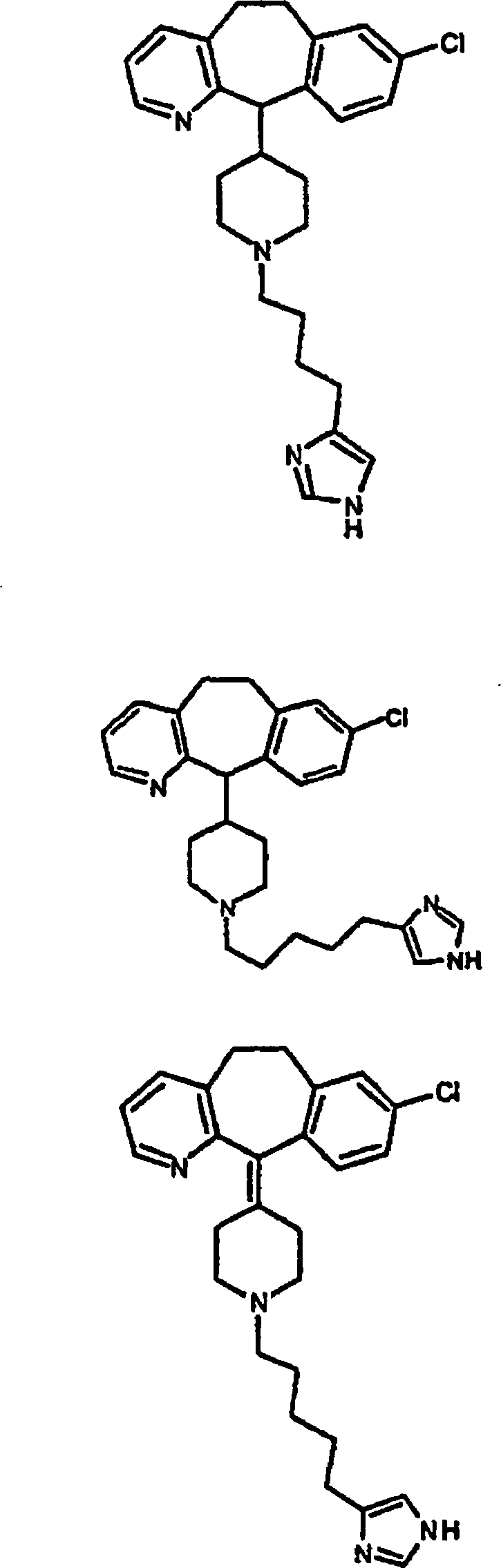
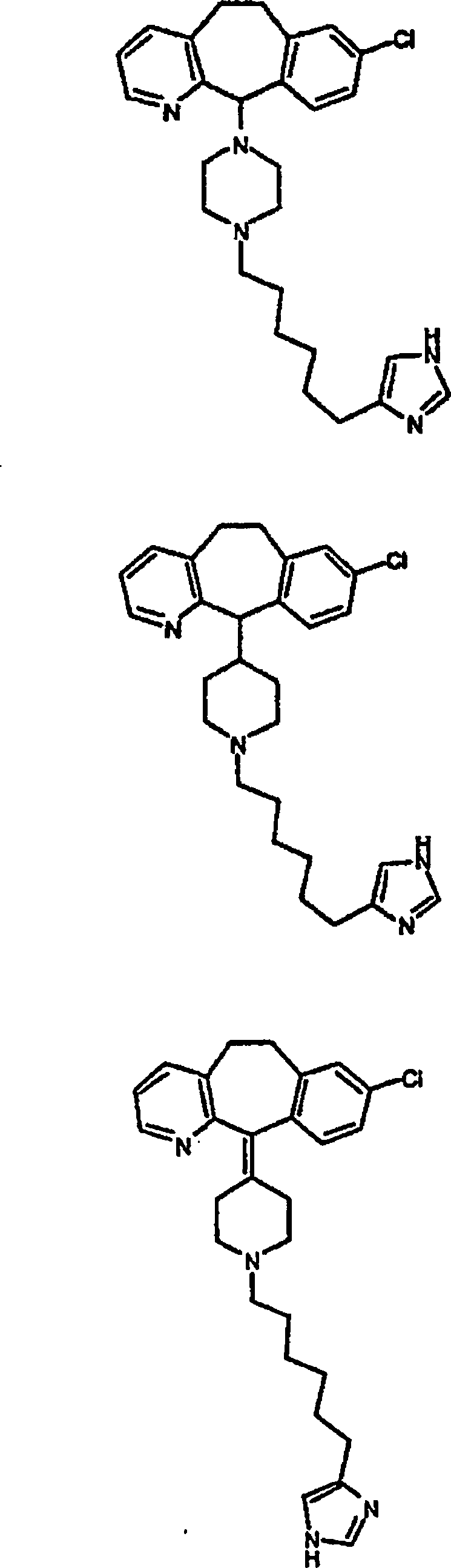
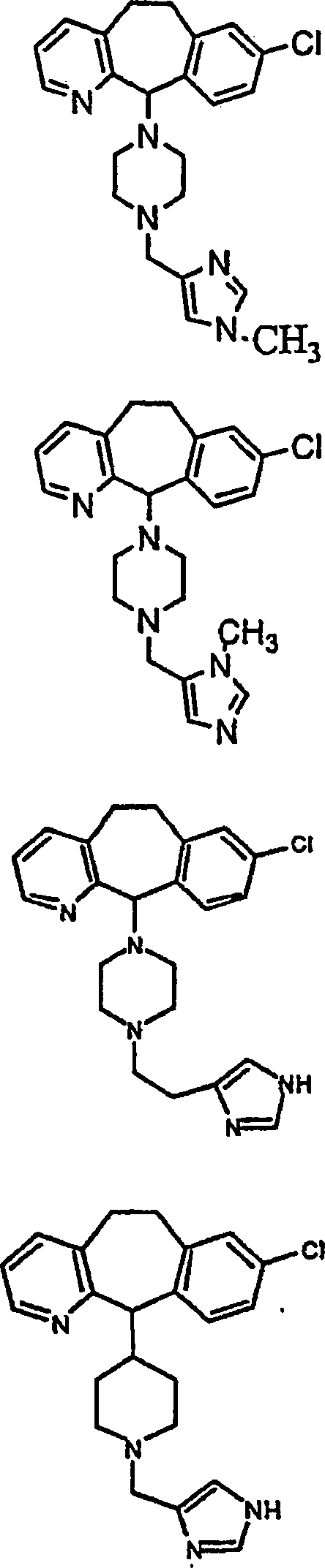
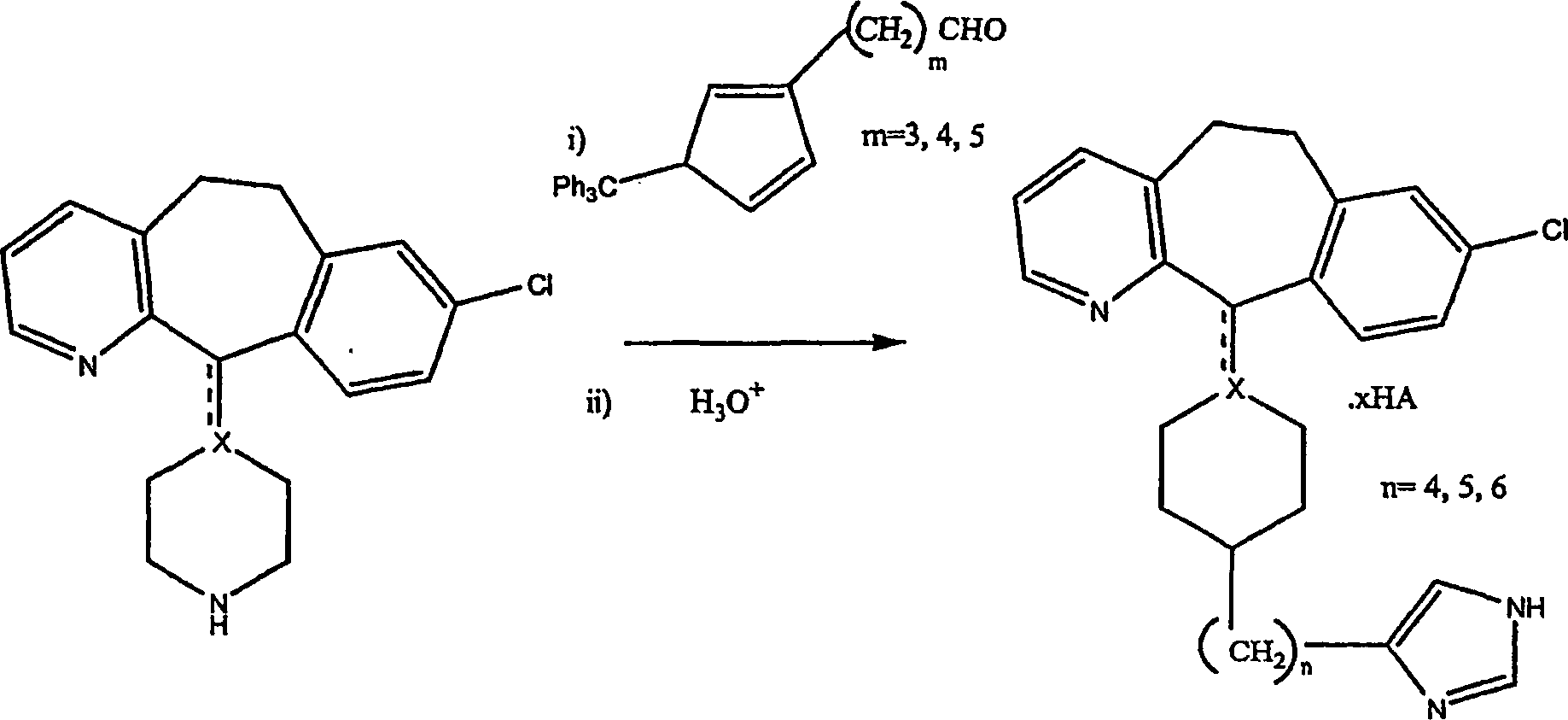

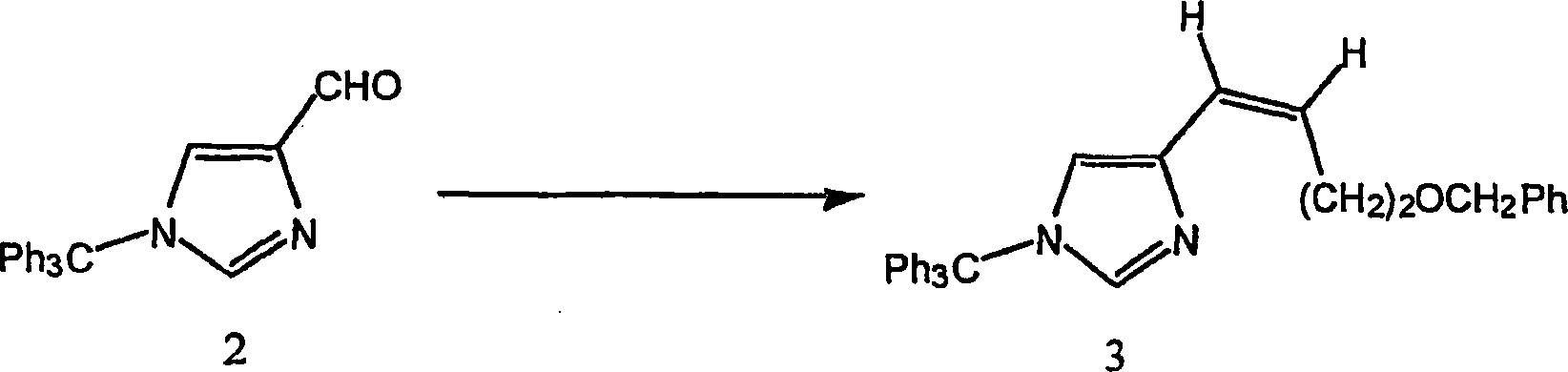

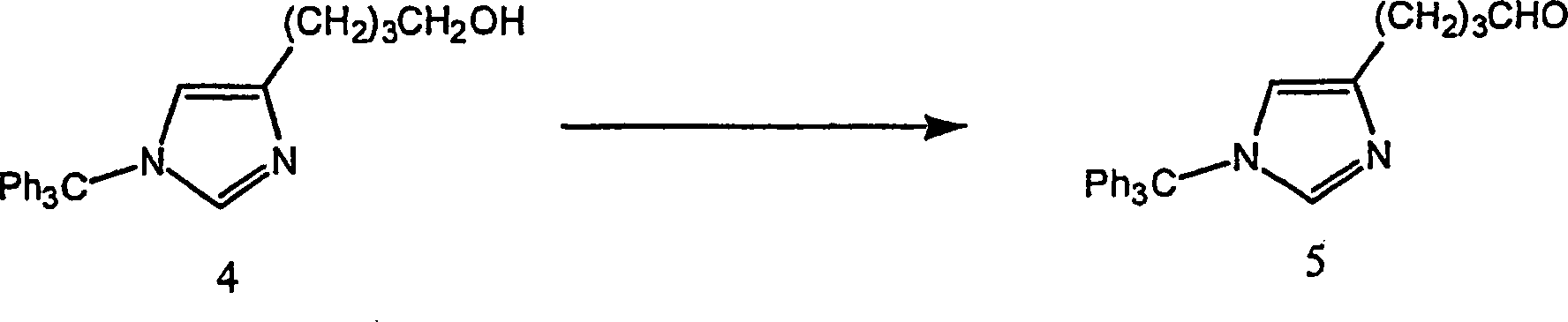


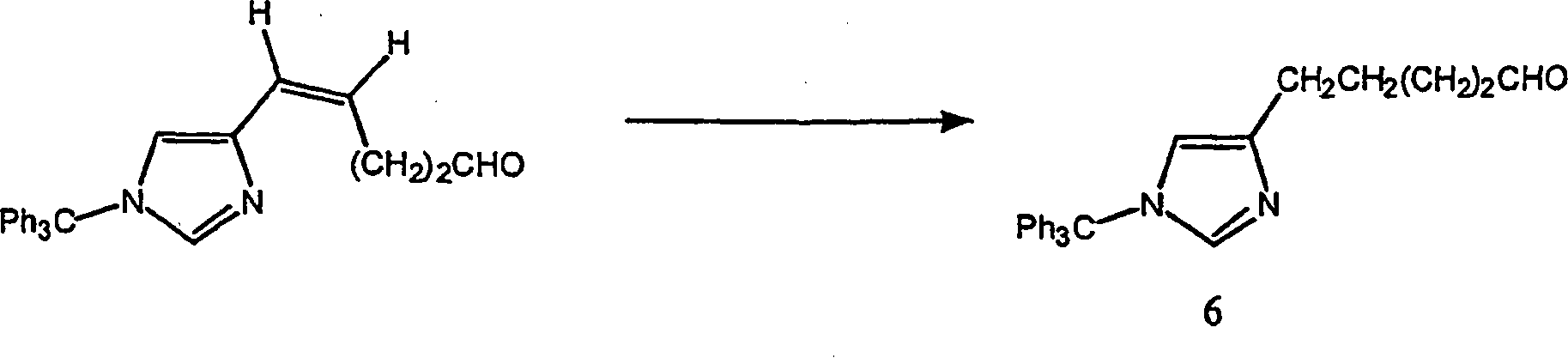
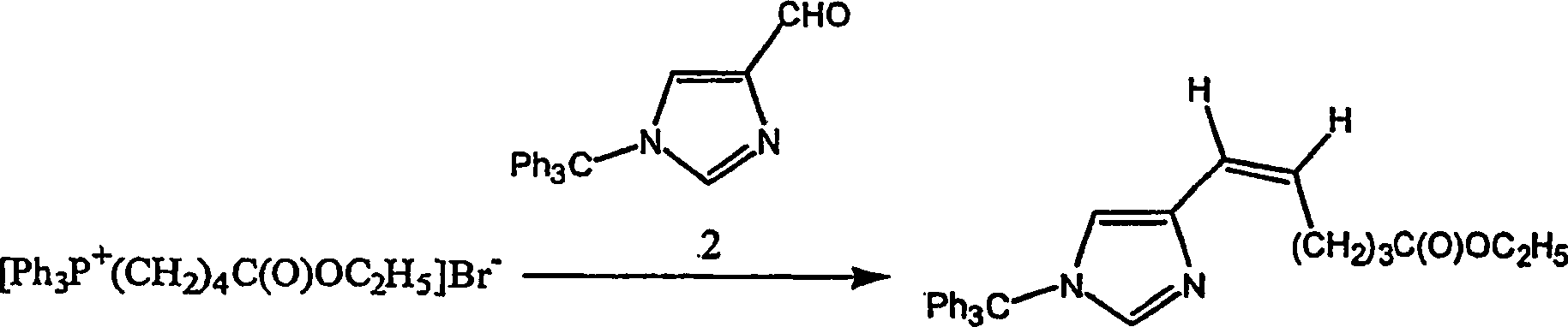


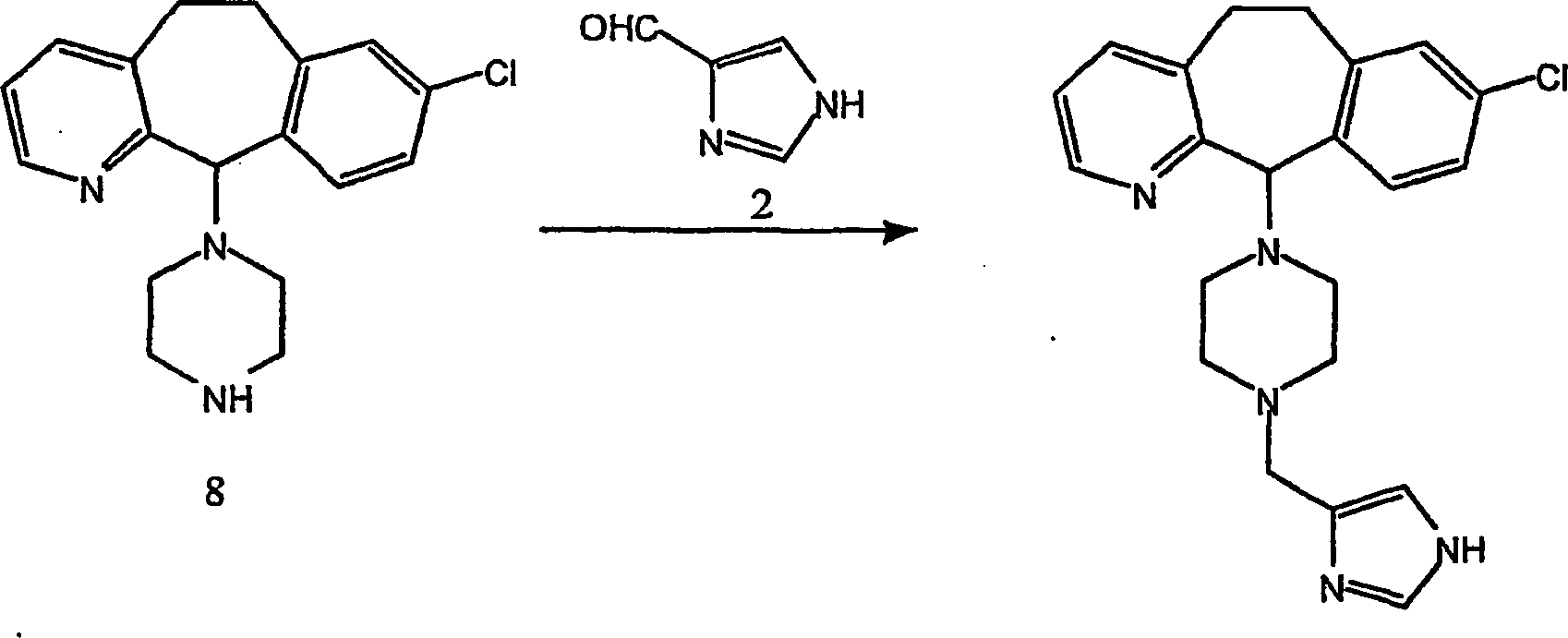

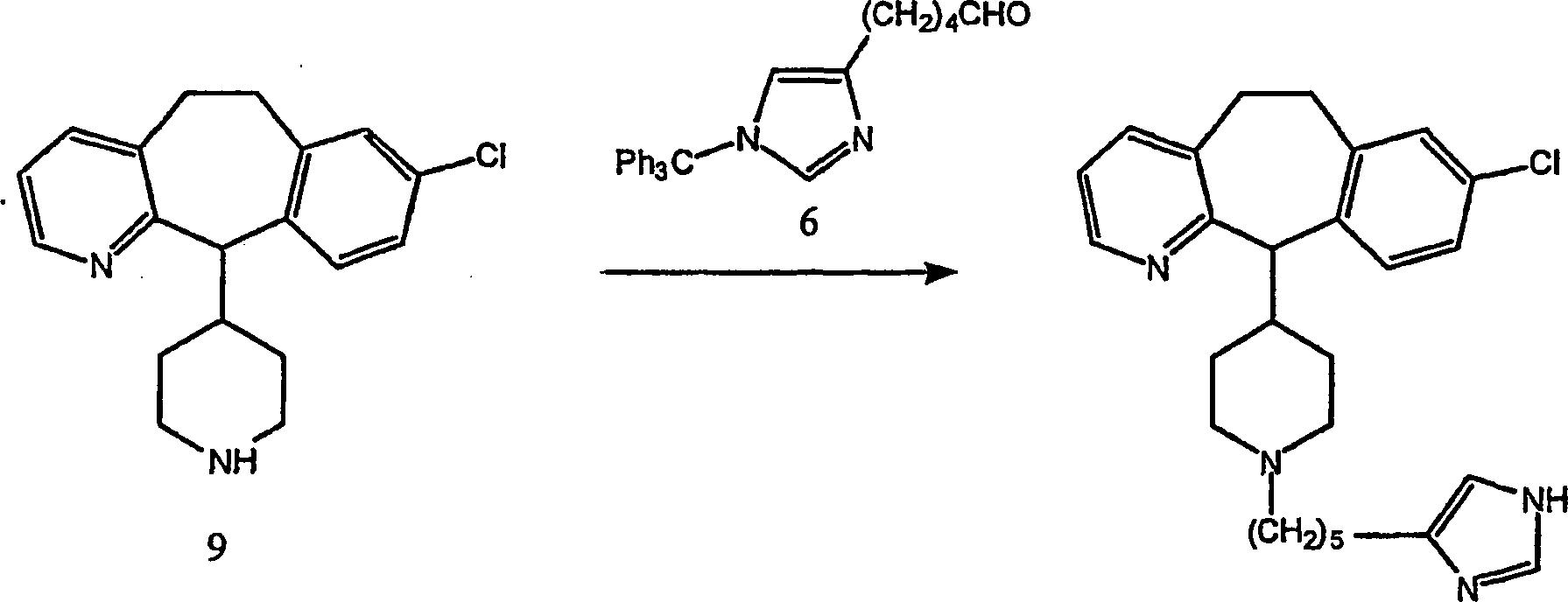
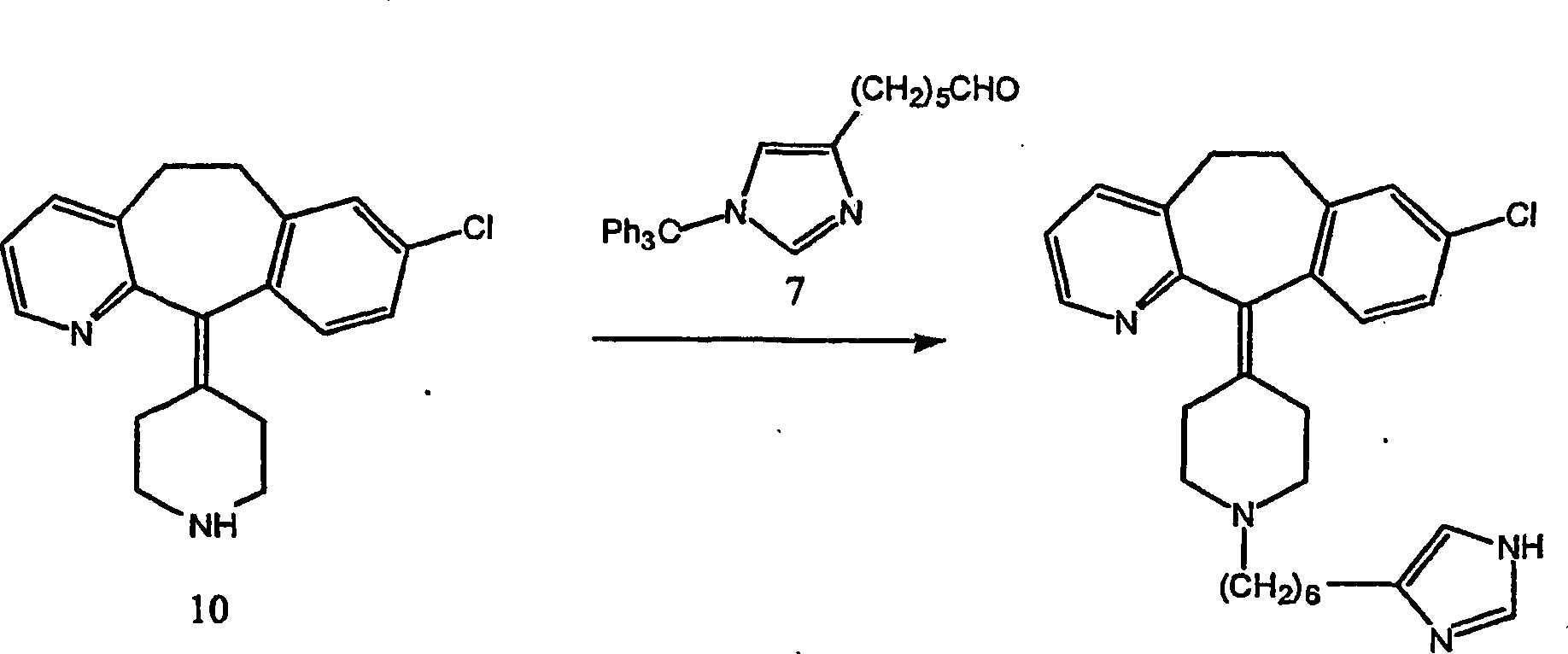



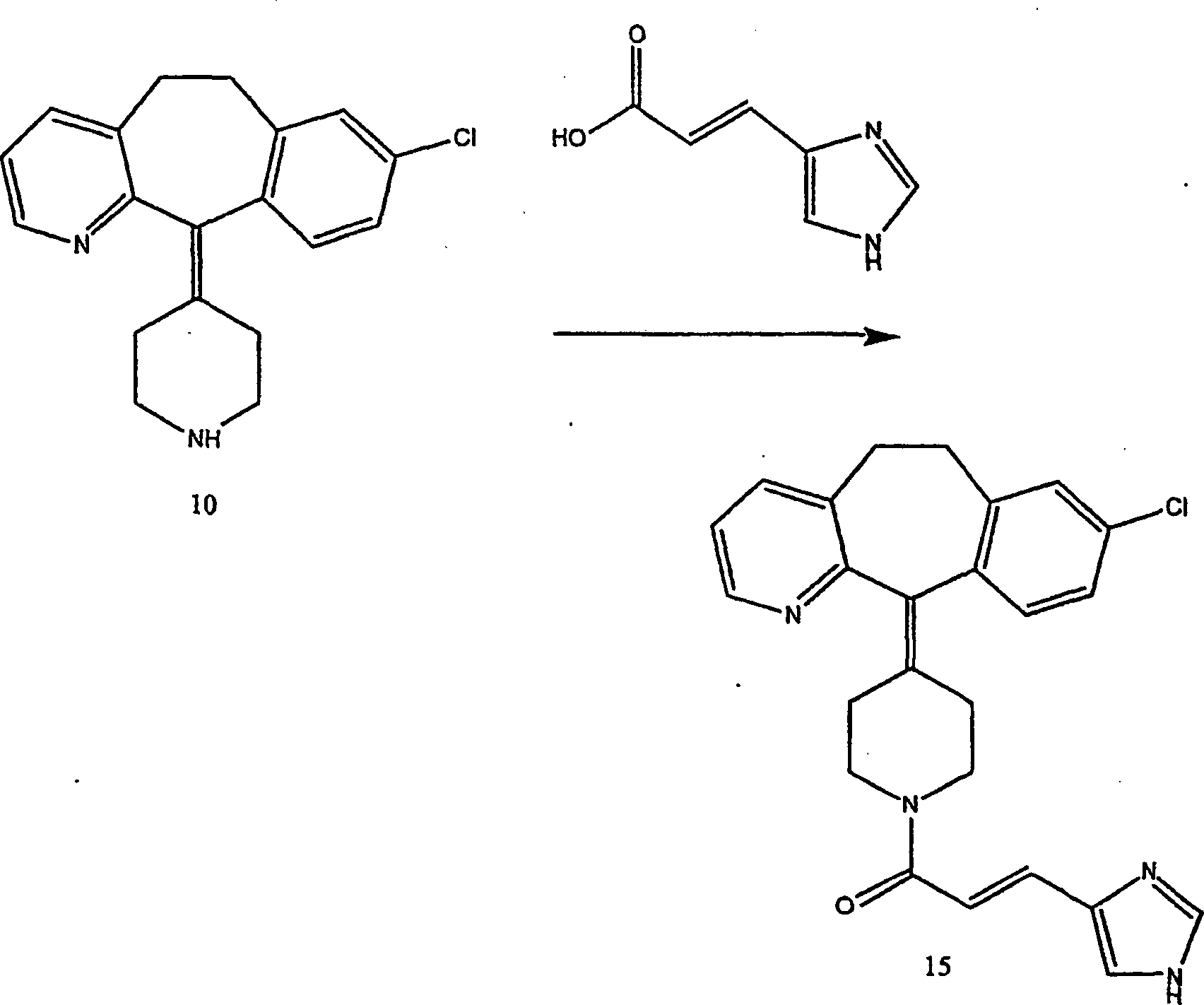
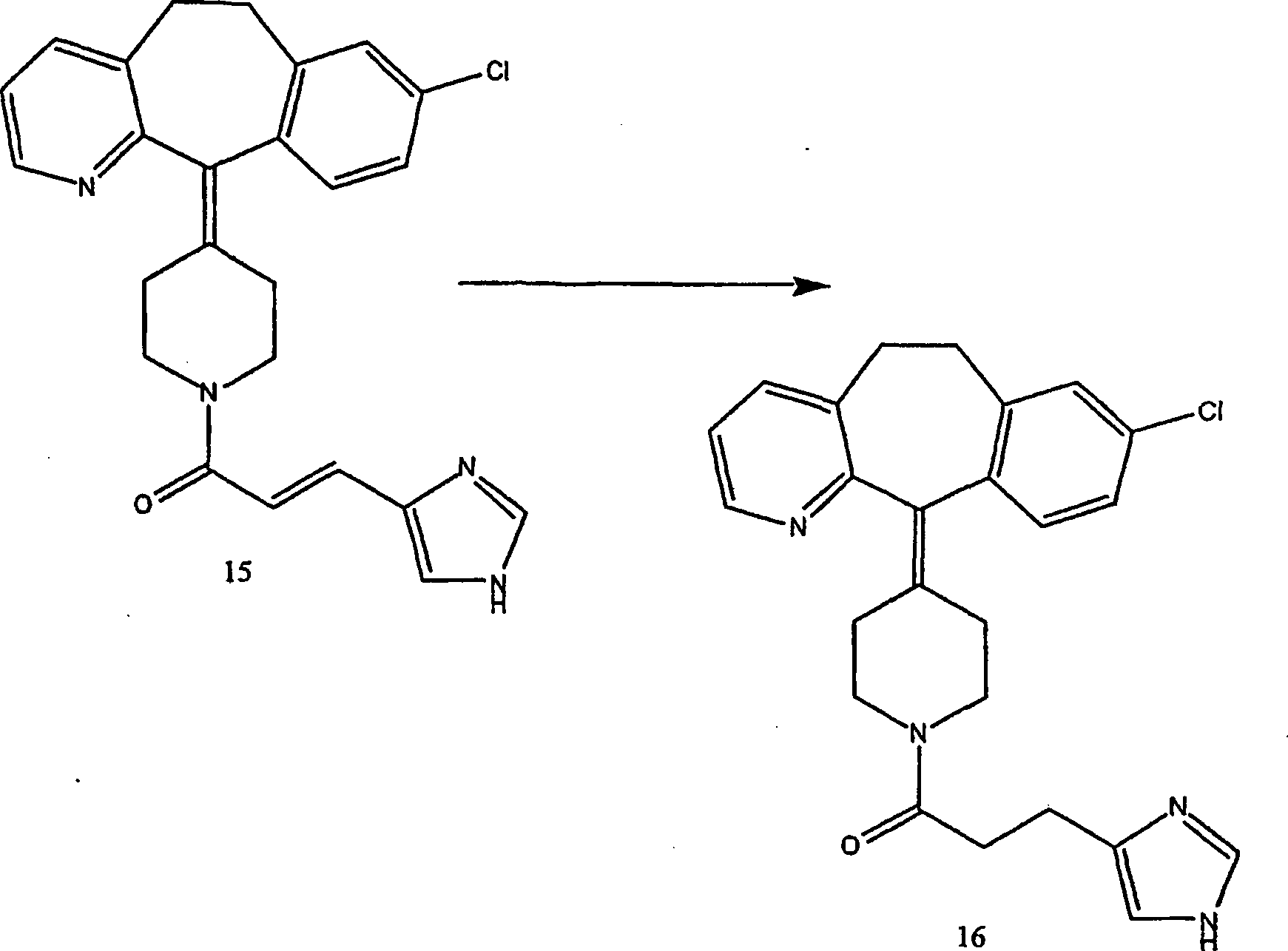
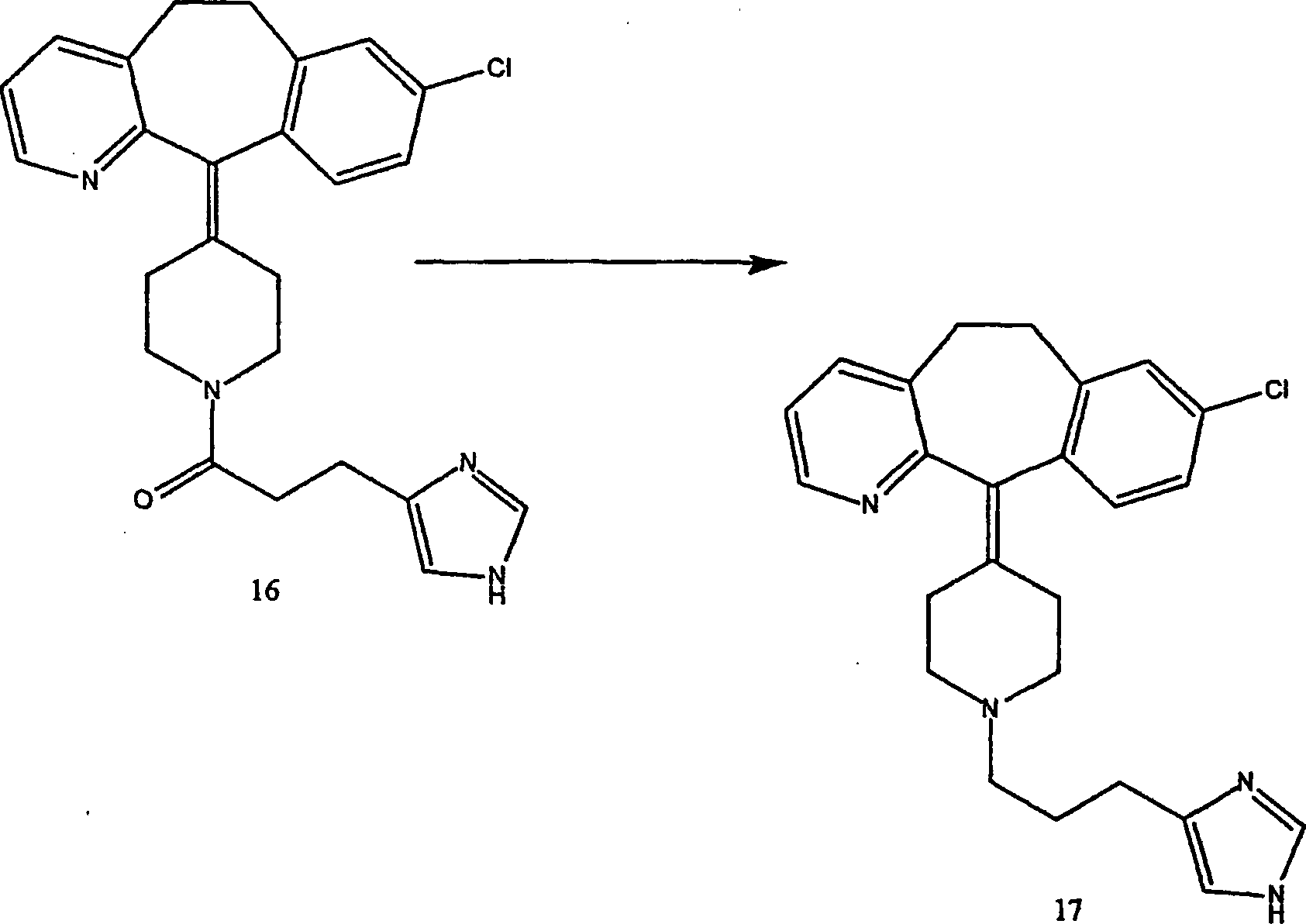
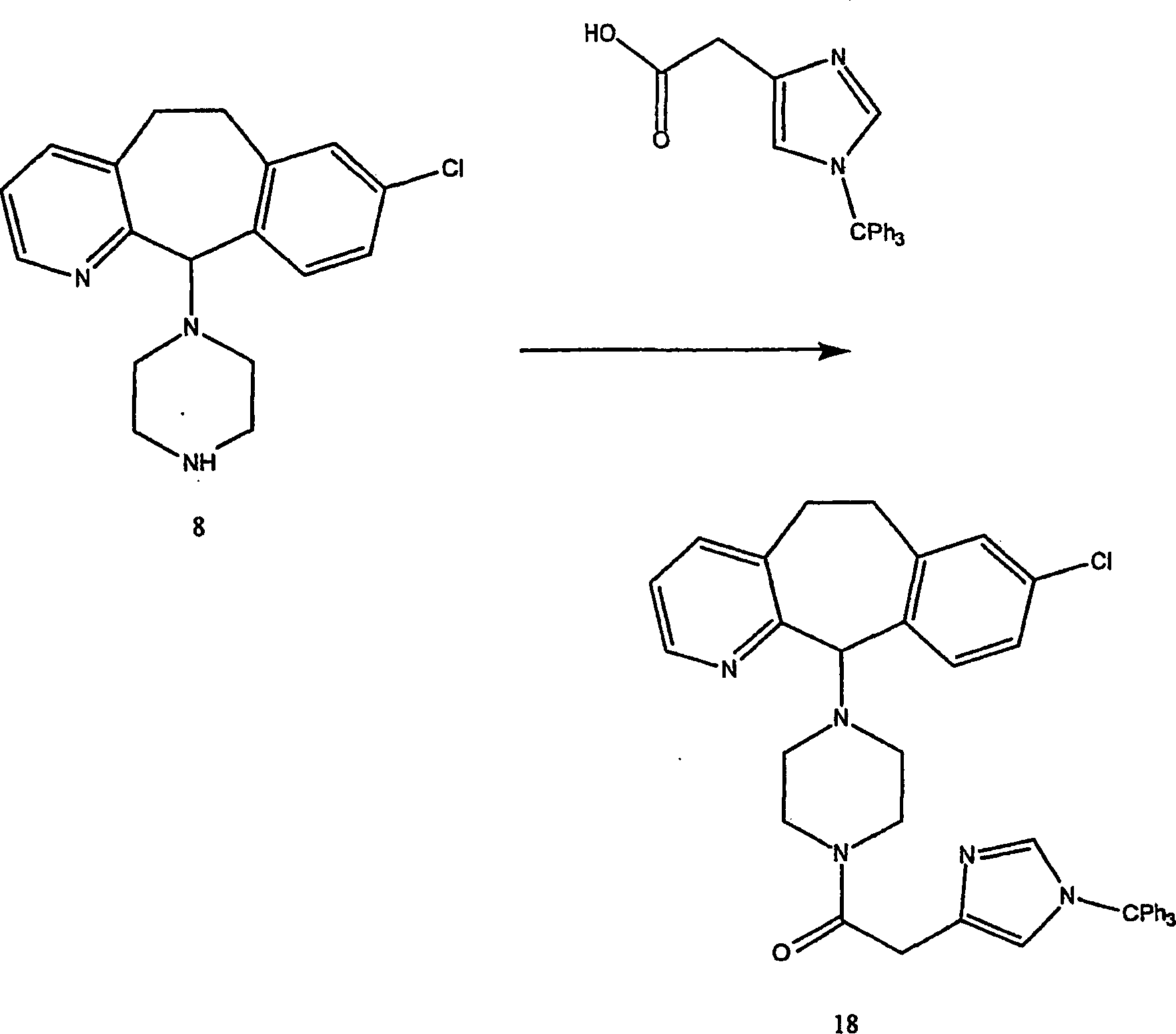
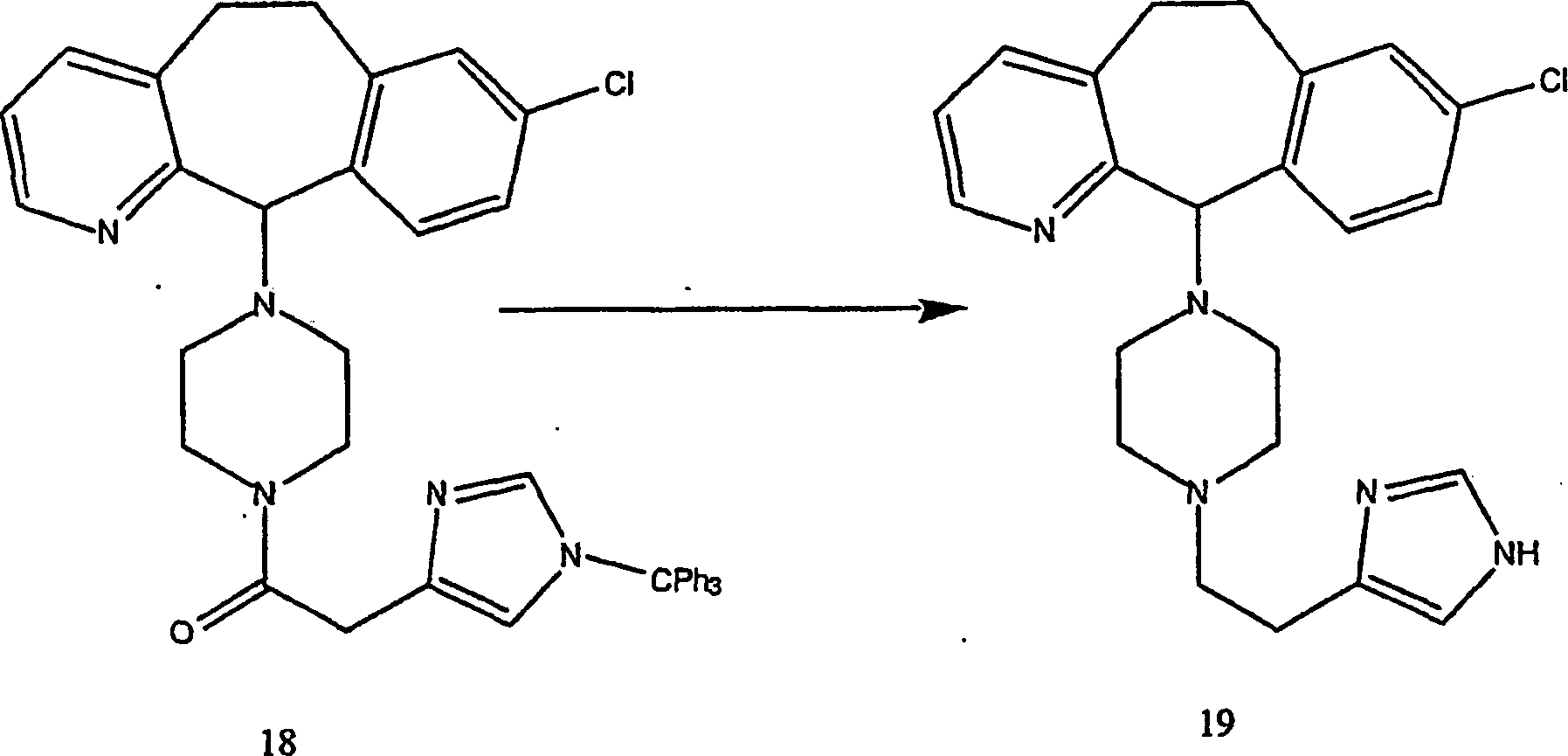
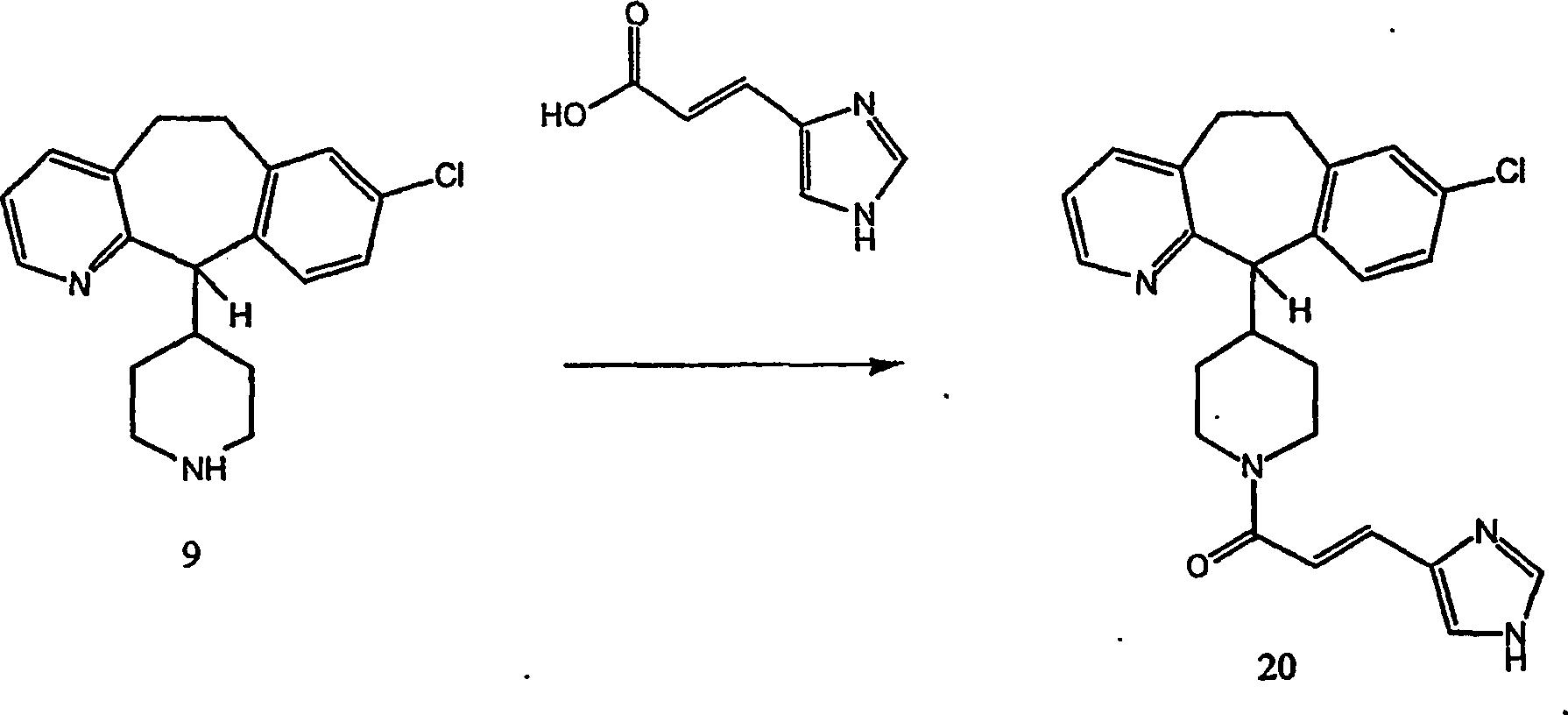

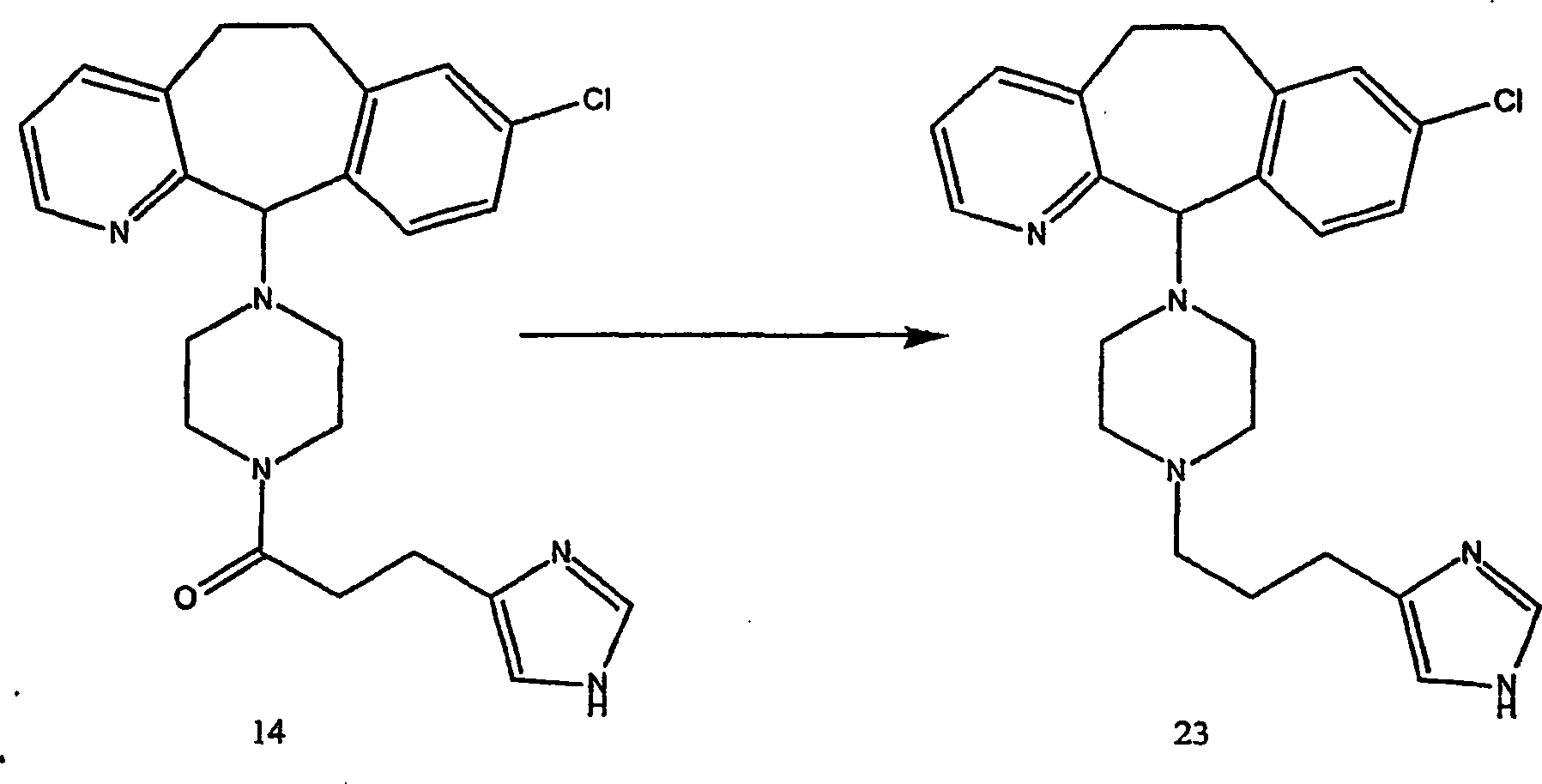
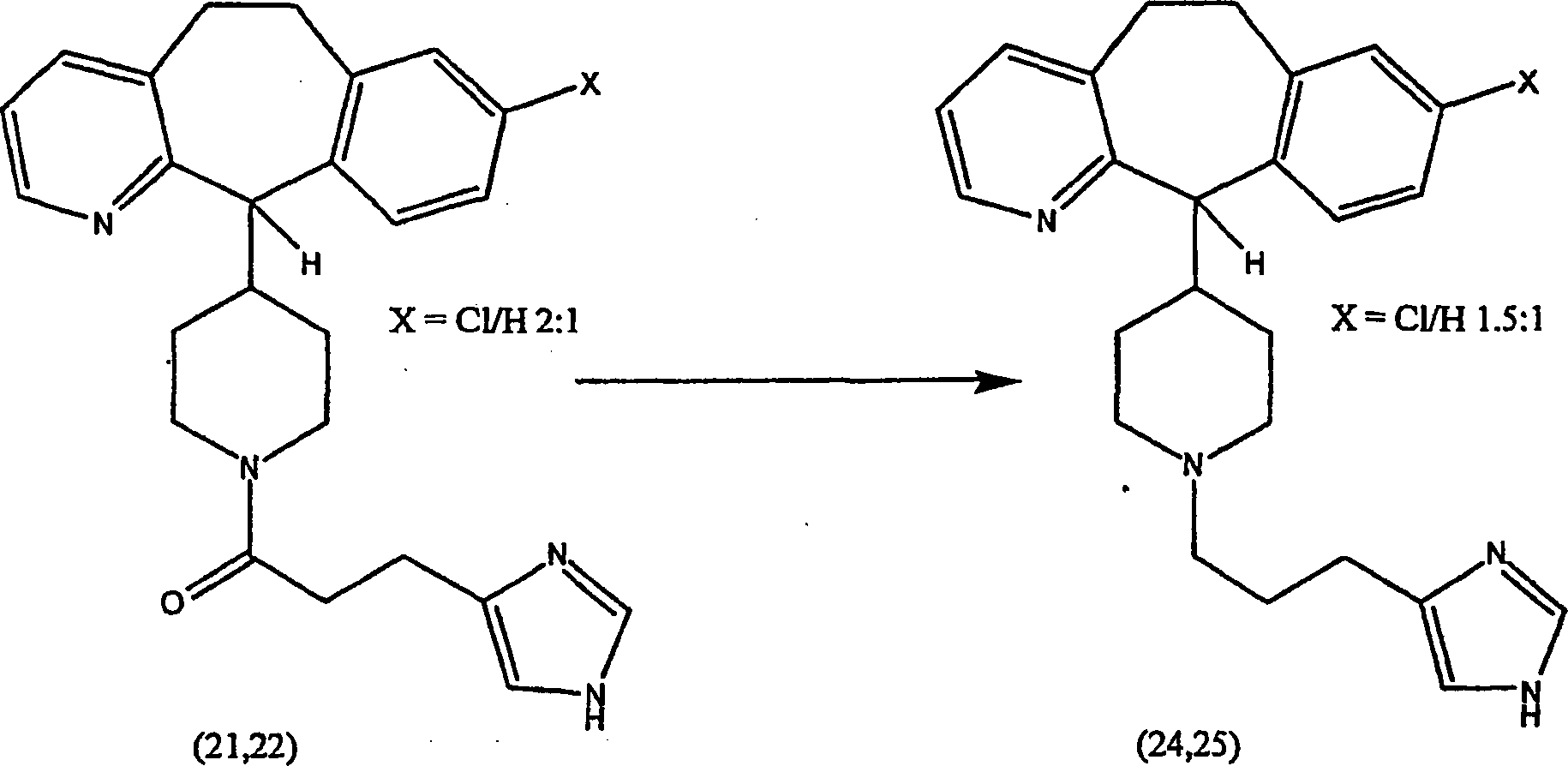


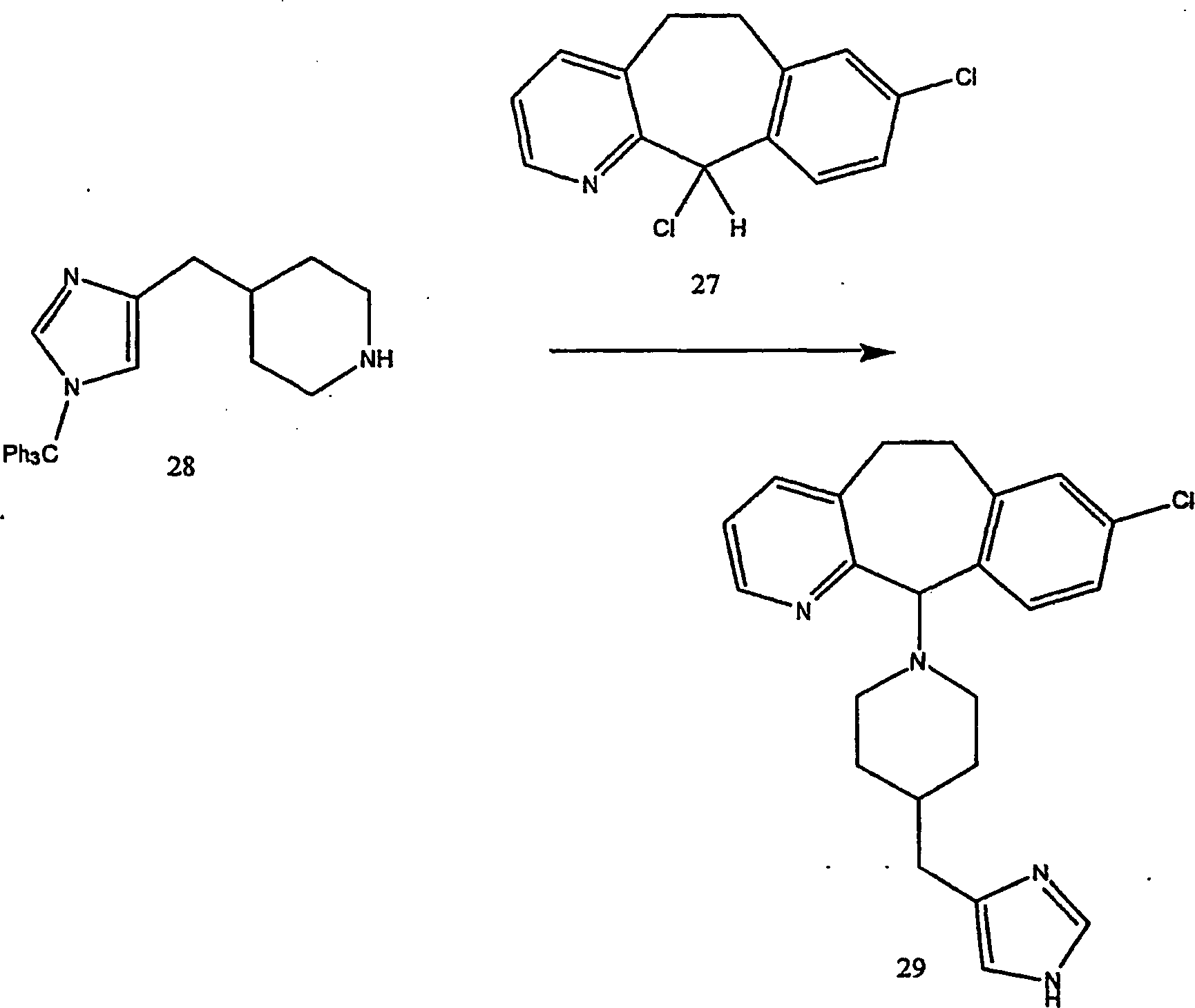

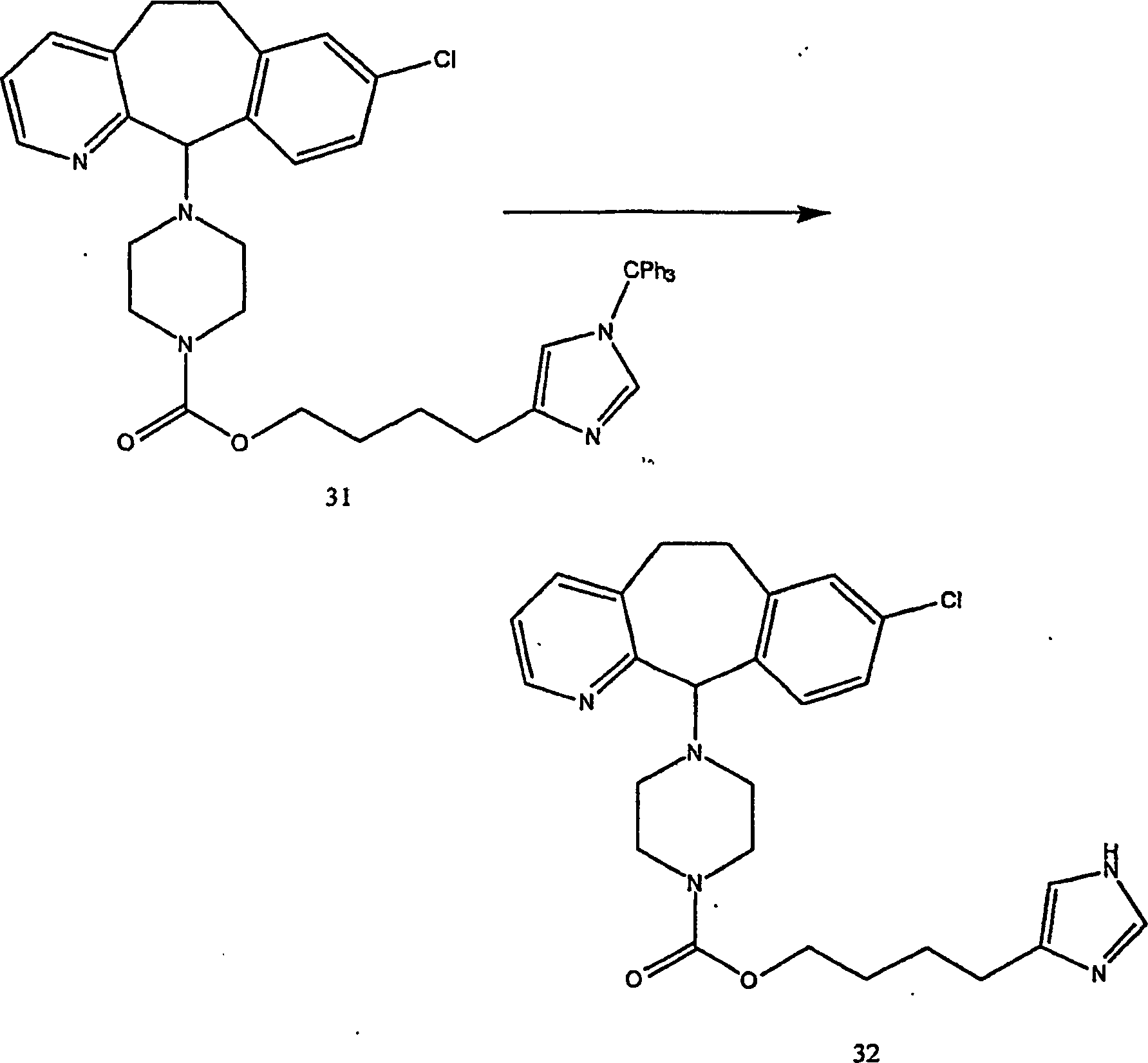

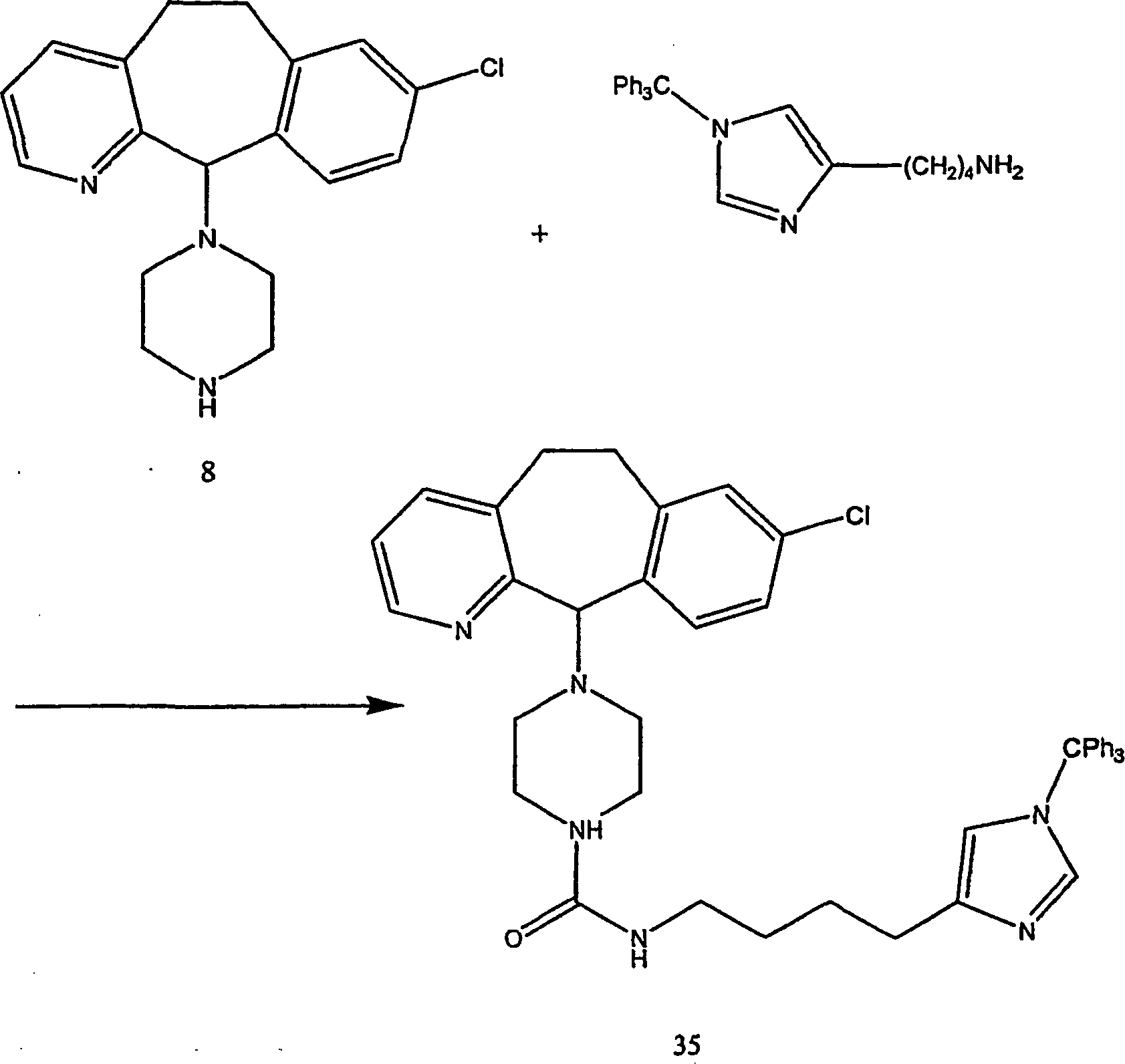
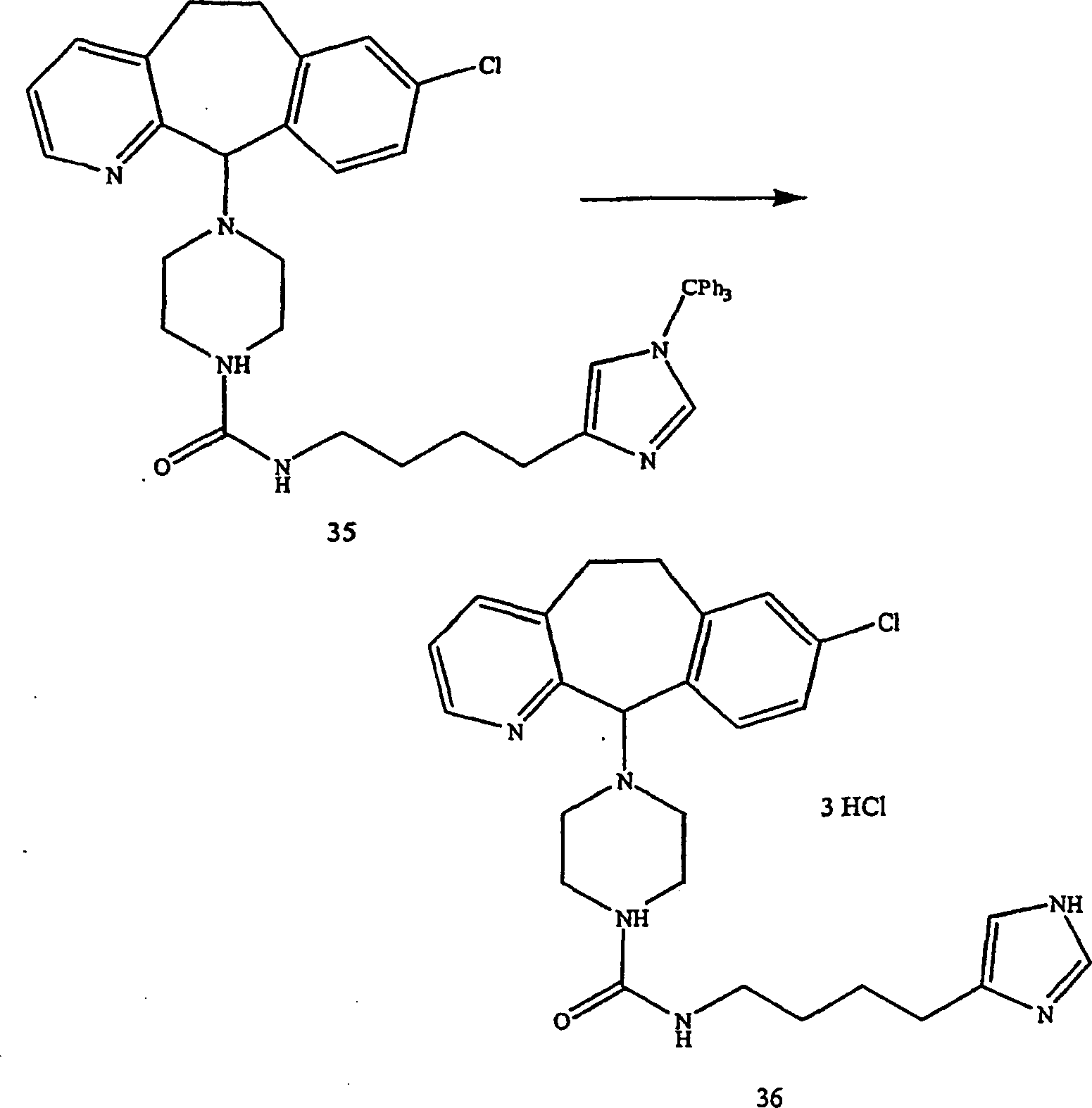

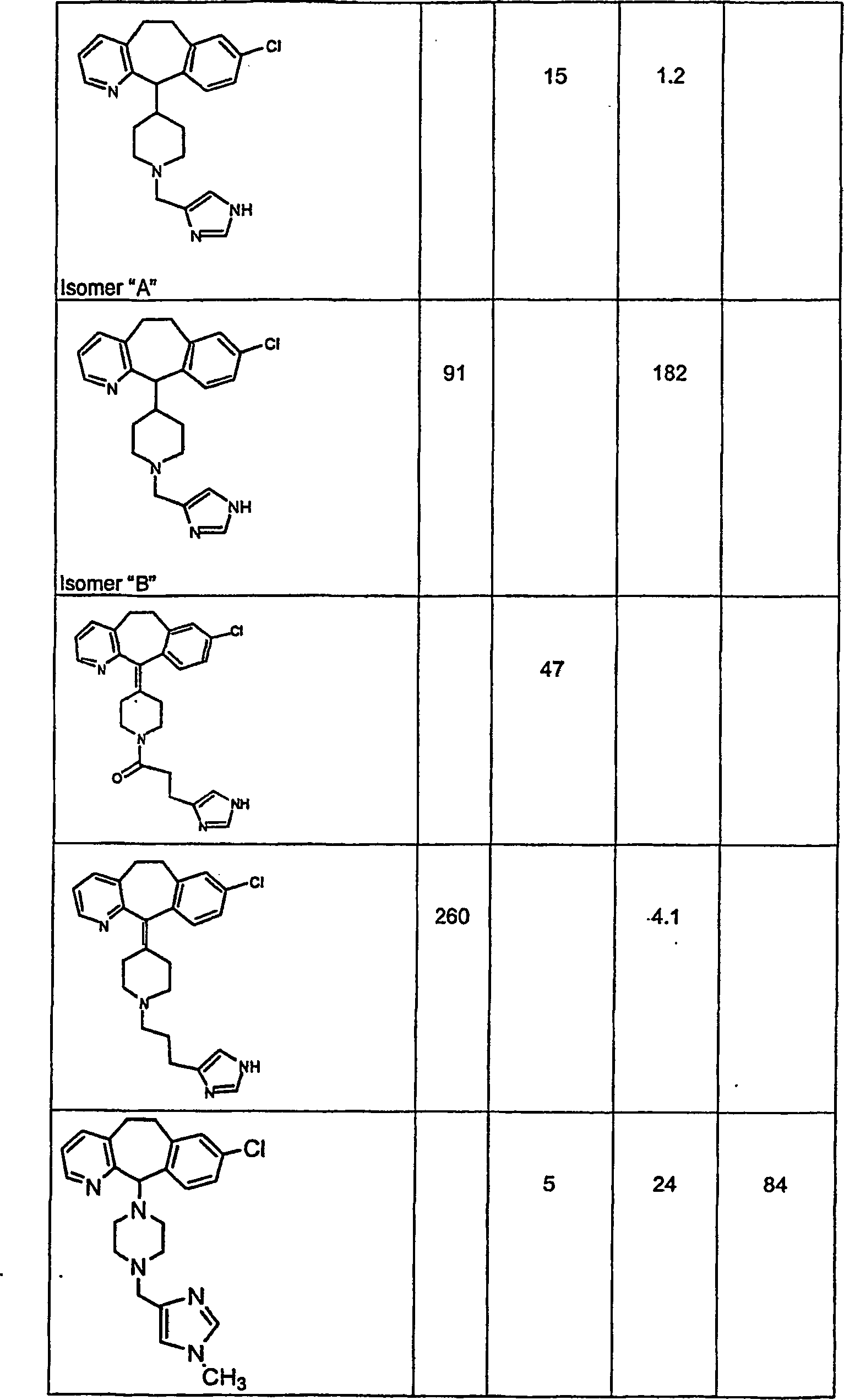

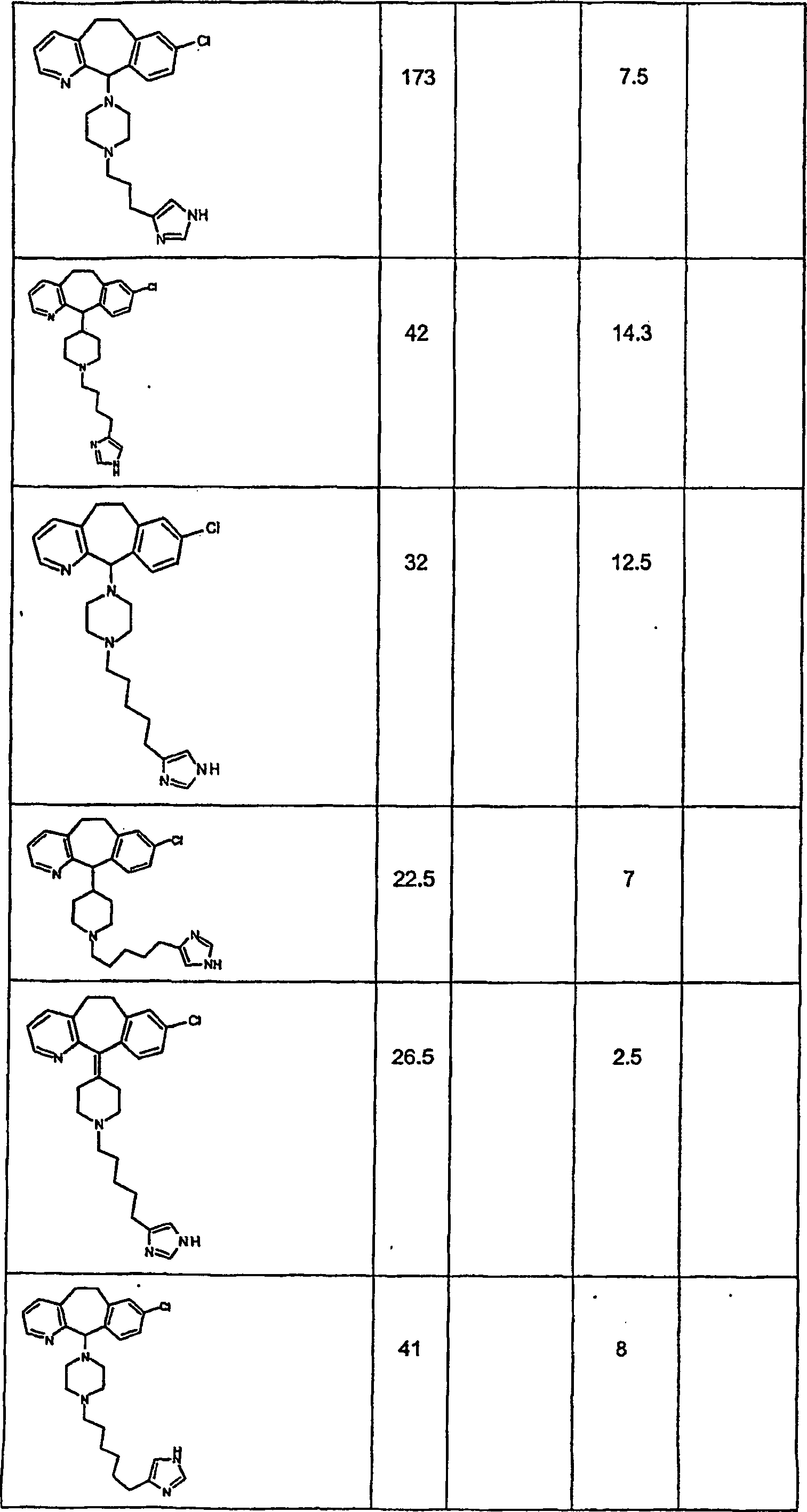
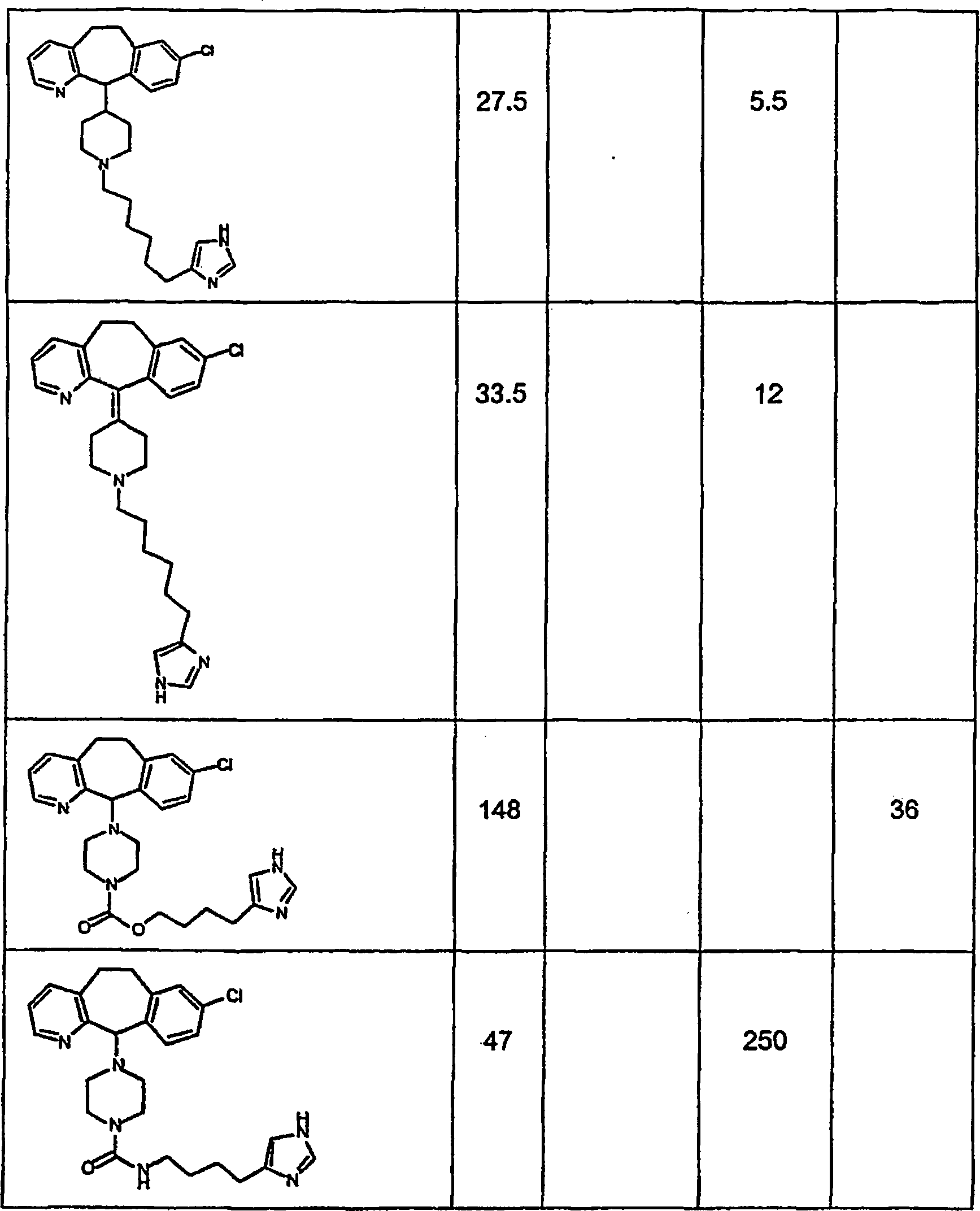
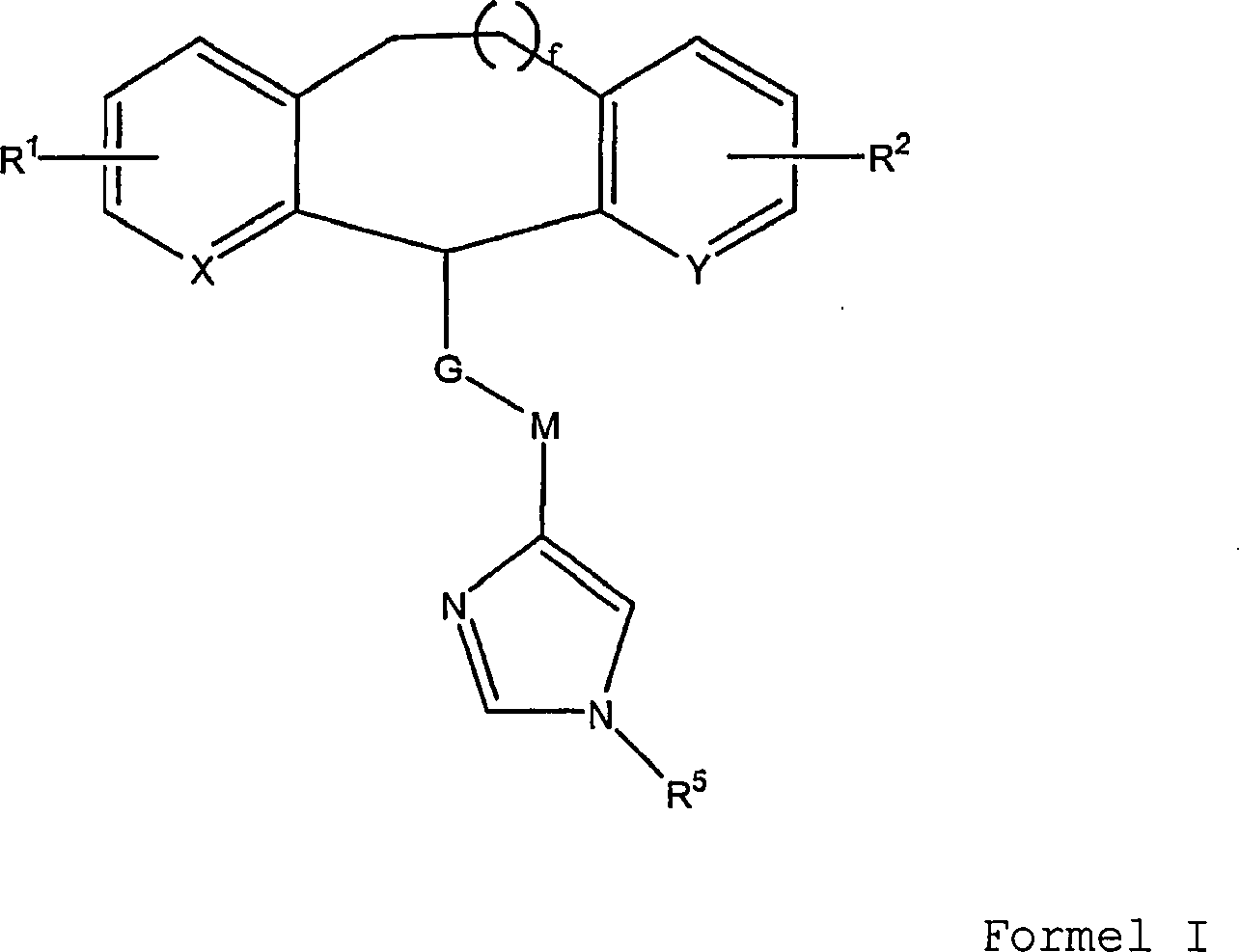 wobei s = t = 1 oder 2; und p = q = 0, 1 oder 2; M eine Einheit ausgewählt aus der Gruppe bestehend aus C1-C3-Alkyl; -C(O)-(CH2)y-; -(CH2)x-A-(CH2)-; -C(O)-O-(CH2)d- und -C(O)-NR3(CH2)d- ist; A = O, S(O)r- und -NR4- ist; n = 0, 1, 2 oder 3 ist; x eine ganze Zahl im Bereich von 2 bis 5 ist; y eine ganze Zahl im Bereich von 0 bis 5 ist; d eine Zahl im Bereich von 0 bis 5 ist; r = 0, 1 oder 2 ist; R1 und R2 jeweils 1–3 Mal vorkommen können und unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, C1-C6-Alkyl, -O-(C1-C6)-Alkyl, Halogen, OCF3, OCHF2, -OH, und -N(R4)2; R3 ausgewählt ist aus der Gruppe bestehend aus Wasserstoff, C1-C6-Alkyl und Polyhalogen-C1-C6-alkyl; R4 ausgewählt ist aus der Gruppe bestehend aus Wasserstoff, C1-C6-Alkyl, Polyhalogen-C1-C6-alkyl und R5 H, -(C1-C6)-Alkyl oder OH ist.
wobei s = t = 1 oder 2; und p = q = 0, 1 oder 2; M eine Einheit ausgewählt aus der Gruppe bestehend aus C1-C3-Alkyl; -C(O)-(CH2)y-; -(CH2)x-A-(CH2)-; -C(O)-O-(CH2)d- und -C(O)-NR3(CH2)d- ist; A = O, S(O)r- und -NR4- ist; n = 0, 1, 2 oder 3 ist; x eine ganze Zahl im Bereich von 2 bis 5 ist; y eine ganze Zahl im Bereich von 0 bis 5 ist; d eine Zahl im Bereich von 0 bis 5 ist; r = 0, 1 oder 2 ist; R1 und R2 jeweils 1–3 Mal vorkommen können und unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, C1-C6-Alkyl, -O-(C1-C6)-Alkyl, Halogen, OCF3, OCHF2, -OH, und -N(R4)2; R3 ausgewählt ist aus der Gruppe bestehend aus Wasserstoff, C1-C6-Alkyl und Polyhalogen-C1-C6-alkyl; R4 ausgewählt ist aus der Gruppe bestehend aus Wasserstoff, C1-C6-Alkyl, Polyhalogen-C1-C6-alkyl und R5 H, -(C1-C6)-Alkyl oder OH ist.