-
Diese
Anmeldung beansprucht die Priorität der Provisorischen Anmeldung
60/156870, eingereicht am 30. September 1999, die hierin durch diesen
Hinweis einbezogen ist.
-
Gebiet der Erfindung
-
Diese
Erfindung betrifft bestimmte Alkylendiaminsubstituierte Heterocyclen,
die selektiv und stark an Säuger-Neuropeptid-Y-(NPY)-Rezeptoren
binden. Diese Erfindung betrifft auch pharmazeutische Zusammensetzungen,
die solche Verbindungen umfassen. Sie betrifft weiterhin die Verwendung
solcher Verbindungen bei der Herstellung eines Arzneimittels für das Behandeln
von physiologischen Störungen,
die mit einem Überschuss
an Neuropeptid Y verbunden sind, insbesondere Essstörungen,
einige psychiatrische Störungen
und bestimmte cardiovaskuläre
Erkrankungen.
-
Hintergrund der Erfindung
-
Neuropeptid
Y (NPY) ist ein Peptid mit 36 Aminosäuren, das zuerst 1982 isoliert
wurde, und sich anschließend
als größtenteils über die
Spezies konserviert erwies. Es gehört zu einer großen Familie
von Peptiden, die unter anderem Peptid YY (PYY) und Pankreaspeptid
(PP) einschließt.
NPY wird als das im Säugerhirn am
reichlichsten vorliegende Peptid angenommen. Es wurde auch in sympathischen
Neuronen gefunden, und NPY-enthaltende
Fasern wurden in peripheren Geweben, wie rund um die Arterien des
Herzens, dem Atmungstrakt, dem Gastrointestinaltrakt und dem genitourinären Trakt,
gefunden. Zentrale Einspritzung von NPY ruft eine Vielzahl von physiologischen
Reaktionen, wie Stimulierung des Essens, Erhöhung der Fetteinlage rung, Entwicklung
von Blutzucker und Insulin, anxiolytisches Verhalten, Verminderung
von lokomotorischer Aktivität,
Hormonfreisetzung, Blutdruckerhöhung,
Verminderung der Körpertemperatur
und Katalepsie, hervor. In dem cardiovaskulären System wird von NPY angenommen,
dass es in die Regulierung des Koronartonus einbezogen ist, während über PYY
mitgeteilt wird, dass es im Gastrointestinaltrakt die Inhibierung
von Magensäuresekretion,
pankreatischer Exokrinsekretion und Gastrointestinalmotilität verursacht.
Diese Wirkungen scheinen durch verschiedene NPY-Rezeptoren selektiv
vermittelt zu werden, die gegenwärtig
die Y1-, Y2-, Y3-, Y4-, Y5- und Y6-Untertypen,
zusätzlich
zu dem hypothetischen Y1-artigen Untertyp,
einschließen.
Selektive Peptidagonisten und -antagonisten wurden für die Meisten
der Untertypen identifiziert, jedoch wurden nur einige selektive
Nicht-Peptid-Antagonisten mitgeteilt. Die Y1- und Y5-Rezeptoruntertypen
scheinen in die Appetitregulierung einbezogen zu sein, jedoch deren
relativer Beitrag zur Modulation von Nahrungsaufnahme und Energieverbrauch
bleibt unklar. Das Auffinden von nicht-peptidischen Antagonisten
der Y1- und Y5-Rezeptoren
stellt neue therapeutische Mittel bereit, die weniger zu Unzulänglichkeiten
der Peptidantagonisten neigen; nämlich beispielsweise
zu schlechter metabolischer Stabilität, geringer oraler Bioverfügbarkeit
und schlechter Hirnpermeabilität,
für die
Behandlung von Fettsucht und cardiovaskulärer Erkrankung.
-
Die
für die
Behandlung von psychiatrischen Störungen zu verwendenden Verbindungen
werden unter anderem in WO98/03510, WO99/38868 und WO99/40091 offenbart.
-
Kurzdarstellung der Erfindung
-
Die
vorliegende Erfindung stellt auch ein allgemeines Verfahren bereit,
wodurch mono-, bi- oder tricyclische Heterocyclen modifiziert werden
können,
um starke Antagonisten an dem NPY1-Rezeptor
zu erhalten.
-
Die
vorliegende Erfindung stellt neue, starke, nicht-peptidische Antagonisten von NPY-Rezeptoren, insbesondere
NPY1-Rezeptoren,
entwickelt aus einer Auswahl von mono-, bi- oder tricyclischen Heterocyclenkernen,
bereit.
-
Die
durch die Verbindungen der Formel II-XV (nachstehend gezeigt) umfassten
heterocyclischen Kerne wurden vorangehend beschrieben. Die Modifizierungen,
die an diesen heterocyclischen Kerne gemacht wurden, um starke und
selektive NPY1-Antagonisten zu erhalten, sind völlig neu.
-
Verbindungen,
die mit dem Y1-Rezeptor in Wechselwirkung
treten und die Aktivität
des Neuropeptids Y an jenen Rezeptoren inhibieren, sind beim Behandeln
von physiologischen Störungen,
die mit einem Überschuss
an Neuropeptid Y verbunden sind, einschließlich Essstörungen, wie beispielsweise
Fettsucht und Bulimie, und bestimmten cardiovaskulären Störungen,
beispielsweise Hypertension, verwendbar.
-
Die
Erfindung betrifft neue Verbindungen, Zusammensetzungen und Verfahren
für die
Herstellung eines Arzneimittels für die Behandlung von physiologischen
Störungen,
die mit einem Überschuss
an Neuropeptid Y verbunden sind. Die neuen Verbindungen, die durch
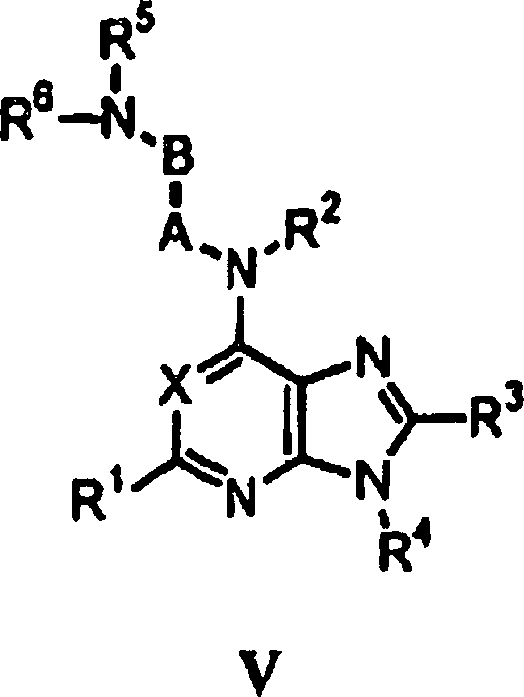
die vorliegende Erfindung umfasst werden, sind jene der Formel I–XV.
worin
X N oder CR
14 darstellt,
R
1 ausgewählt ist
aus H, C
1-C
6-Alkyl,
C
3-C
10-Cycloalkyl,
(C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
2-C
6-Alkenyl, C
2-C
6-Al kinyl, Cyano,
Halogen, C
1-C
6-Halogen-alkyl,
OR
7, C
1-C
6-Alkyl-OR
7; C
1-C
6-Cyanoalkyl,
NR
8R
9, C
1-C
6-Alkyl-NR
8R
9;
R
2 H oder C
1-C
6-Alkyl darstellt;
A (CH
2)
m darstellt, worin m 1, 2 oder 3 ist und
gegebenenfalls mono- oder di-substituiert ist bei jedem Auftreten mit
C
1-C
6-Alkyl, C
3-C
10-Cycloalkyl,
(C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
1-C
6-Alkenyl, C
1-C
6-Alkinyl, Cyano, Halogen, C
1-C
6-Halogenalkyl,
OR
7, C
1-C
6-Alkyl-OR
7; C
1-C
6-Cyanoalkyl,
NR
8R
9, C
1-C
6-Alkyl-NR
8R
9,
B (CH
2)
n darstellt, worin n 1, 2 oder 3 ist und
gegebenenfalls mono- oder di-substituiert ist bei jedem Auftreten mit
C
1-C
6-Alkyl, C
3-C
10-Cycloalkyl,
(C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
2-C
6-Alkenyl, C
2-C
6-Alkinyl, Cyano, Halogen, C
1-C
6-Halogenalkyl,
OR
7, C
1-C
6-Alkyl-OR
7; C
1-C
6-Cyanoalkyl,
NR
8R
9, C
1-C
6-Alkyh-NR
8R
9,
R
3 unabhängig ausgewählt ist
bei jedem Auftreten aus H, C
1-C
6-Alkyl,
C
3-C
10-Cycloalkyl,
(C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
2-C
6-Alkenyl, C
2-C
6-Alkinyl, Cyano, Halogen, C
1-C
6-Halogenalkyl,
OR
7, C
1-C
6-Alkyl-OR
7, C
1-C
6-Cyanoalkyl,
NR
8R
9, C
1-C
6-Alkyl-NR
8R
9;
R
4 ausgewählt ist
aus Aryl oder Heteroaryl, jeder gegebenenfalls substituiert mit
1 bis 5 Substituenten, unabhängig
ausgewählt
bei jedem Auftreten aus C
1-C
6-Alkyl,
C
3-C
10-Cycloalkyl, C
3-C
10-Cycloalkenyl,
(C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
1-C
6-Alkenyl, Halogen,
C
1-C
6-Halogenalkyl,
Trifluormethylsulfonyl, OR
7, C
1-C
6-Alkyl-OR
7, NR
8R
9, C
1-C
6-Alkyl-NR
8R
9, CONR
8R
9, C
1-C
6-Alkyl-CONR
8R
9, COOR
7, C
1-C
6-Alkyl-COOR
7, CN, C
1-C
6-Alkyl-CN, SO
2NR
8R
9, SO
2R
7, Aryl, Heteroaryl, Heterocycloalkyl, 3-,
4- oder 5-(2-Oxo-1,3-oxazolidinyl), mit der Maßgabe, dass mindestens eine
der Positionen ortho oder para zu dem Bindungspunkt des Aryl- oder
Heteroarylrings an dem heterocyclischen Kern substituiert ist;
R
5 ausgewählt
ist aus:
C
1-C
6-Alkyl,
(C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
2-C
6-Alkenyl, C
2-C
6-Alkinyl, wobei
jeder davon mit 1 bis 5 Gruppen sub stituiert ist, unabhängig ausgewählt bei
jedem Auftreten aus Halogen, C
1-C
2-Halogenalkyl, OR
7,
Cyano, NR
8R
9, CONR
8R
9, COOR
7, SO
2NR
8R
9, SO
2R
7,
NR
11COR
12, NR
11SO
2R
7;
C
1-C
6-Arylalkyl, C
1-C
6-Heteroarylalkyl,
C
5-C
8-Arylcycloalkyl,
oder C
5-C
8-Heteroarylcycloalkyl,
worin Aryl Phenyl oder Naphthyl darstellt, und Heteroaryl 2-, 3-
oder 4-Pyridyl, 2-, 4- oder 5-Pyrimidinyl, Triazinyl, 1-, 2- oder 4-Imidazolyl, 2-, 4-
oder 5-Oxazolyl, Isoxazolyl, Indolyl, Pyrazolyl, Chinolyl, Isochinolyl,
2-, 4- oder 5-Thiazolyl, Benzothiadiazolyl, 1-, 3- oder 4-Pyrazolyl,
1-, 3- oder 4-Triazolyl,
2-Triazinyl, 2-Pyrazinyl, 2- oder 3-Furanyl, 2- oder 3-Thienyl, 2- oder 3-Benzothienyl,
oder 1-, 2- oder 5-Tetrazolyl
darstellt, wobei jedes davon gegebenenfalls mit 1 bis 5 Substituenten
substituiert ist, unabhängig
ausgewählt
bei jedem Auftreten aus C
1-C
6-Alkyl, C
3-C
10-Cycloalkyl,
C
3-C
10-Cycloalkenyl, (C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
1-C
6-Alkenyl, Halogen,
C
1-C
6-Halogenalkyl,
Trifluormethylsulfonyl, OR
7, NR
8R
9, C
1-C
6-Alkyl-OR
7, C
1-C
6-Alkyl-NR
8R
9, CONR
8R
9, COOR
7, CN, SO
2NR
8R
9, SO
2R
7, Aryl, Heteroaryl, Heterocycloalkyl, 3-,
4- oder 5-(2-Oxo-1,3-oxazolidinyl),
mit der Maßgabe, dass
2 benachbarte Substituenten gegebenenfalls einen C
3-C
10-Cycloalkylring, einen C
3-C
10-Cycloalkenylring oder einen Heterocycloalkylring
bilden können;
C
3-C
10-Cycloalkyl,
(C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
3-C
10-Cycloalkenyl, C
2-C
6-Alkenyl, C
2-C
6-Alkinyl, wobei jedes davon gegebenenfalls
mit 1 bis 6 Substituenten substituiert ist, unabhängig ausgewählt bei
jedem Auftreten aus C
1-C
6-Alkyl,
C
3-C
10-Cycloalkyl, C
3-C
10-Cycloalkenyl, (C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl,
C
1-C
6-Alkenyl, Halogen,
C
1-C
6-Halogenalkyl,
OR
7, NR
8R
9, mit der Maßgabe, dass, wenn zwei Substituenten
OR
7 oder NR
8R
9 geminal an demselben Kohlenstoff angeordnet
sind, R
7 nicht H darstellt und sie zusammen
einen C
2-C
4-Ketal-, Oxazolin-,
Oxazolidin-, Imidazolin- oder Imidazolidinheterocyclus bilden können, C
1-C
6-Alkyl-OR
7, C
1-C
6-Alkyl-NR
8R
9, CONR
8R
9, COOR
7, CN, Oxo, Hydroximino, C
1-C
6-Alkoximino, SO
2NR
8R
9, SO
2R
7, Heterocyclo alkyl, Aryl, Heteroaryl, worin
Aryl oder Heteroaryl gegebenenfalls mit 1 bis 5 Substituenten substituiert
ist, unabhängig
ausgewählt
bei jedem Auftreten aus C
1-C
6-Alkyl,
C
3-C
10-Cycloalkyl,
C
3-C
10-Cycloalkenyl,
(C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
1-C
6-Alkenyl, Halogen, C
1-C
6-Halogenalkyl, Trifluormethylsulfonyl, OR
7, NR
8R
9,
C
1-C
6-Alkyl-OR
7, C
1-C
6-Alkyl-NR
8R
9, CONR
8R
9, COOR
7, CN, SO
2NR
8R
9, SO
2R
7, Aryl, Heteroaryl, Heterocycloalkyl, 3-, 4-
oder 5-(2-Oxo-1,3-oxazolidinyl, mit der Maßgabe, dass 2 benachbarte Substituenten
gegebenenfalls zusammen einen C
3-C
10-Cycloalkylring,
einen C
3-C
10-Cycloalkenylring
oder einen Heterocycloalkylring bilden können;
Aryl oder Heteroaryl,
gegebenenfalls substituiert mit 1 bis 5 Substituenten, unabhängig ausgewählt bei
jedem Auftreten aus Halogen C
1-C
6-Alkyl, C
3-C
10-Cycloalkyl, C
3-C
10-Cycloalkenyl, (C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
1-C
6-Alkenyl, Halogen, C
1-C
6-Halogenalkyl,
Trifluormethylsulfonyl, OR
7, NR
8R
9, C
1-C
6-Alkyl-OR
7,
C
1-C
6-Alkyl-NR
8R
9, CONR
8R
9, COOR
7, CN, SO
2NR
8R
9, SO
2R
7, Aryl, Heteroaryl, Heterocycloalkyl, 3-,
4- oder 5-(2-Oxo-1,3-oxazolidinyl), mit der Maßgabe, dass 2 benachbarte Substituenten
gegebenenfalls zusammen einen C
3-C
10-Cycloalkylring, einen C
3-C
10-Cycloalkenylring
oder einen Heterocycloalkylring bilden können;
oder
3- oder
4-Piperidinyl, 3-Pyrrolidinyl, 3- oder 4-Tetrahydropyranyl, 3-Tetrahydrofuranyl,
3- oder 4-Tetrahydropyranyl, 3-Tetrahydrofuranyl, 3- oder 4-Tetrahydrothiopyranyl,
3- oder 4-(1,1-Dioxo)-tetra-hydrothiopyranyl,
1-Azabicyclo[4.4.0]decyl, 8-Aza-bicyclo[3.2.1]octanyl, Norbornyl,
Chinuclidinyl, jeweils gegebenenfalls substituiert mit 1 bis 5 Substituenten,
unabhängig
ausgewählt
bei jedem Auftreten aus R
7, C
1-C
6-Alkyl-OR
7, C
1-C
6-Alkyl-NR
8R
9, CONR
8R
9, COOR
7;
R
6 ausgewählt ist
aus H, C
1-C
6-Alkyl,
C
3-C
10-Cycloalkyl,
(C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
2-C
4-Alkenyl, C
1-C
6-Arylalkyl, C
1-C
6-Heteroarylalkyl,
worin Aryl oder Heteroaryl gegebenenfalls mit 1 bis 5 Substituenten substituiert
sind, unabhängig
ausgewählt
bei jedem Auftreten aus Halogen, C
1-C
6-Halogenalkyl,
OR
13, NR
8R
9, C
1-C
6-Alkyl-OR
13, C
1-C
6-Alkyl-NR
8R
9, CONR
8R
9, COOR
7, CN, SO
2NR
8R
9, SO
2R
7;
R
7 H, C
1-C
6-Alkyl, C
3-C
10-Cycloalkyl,
C
3-C
10-Cycloalkenyl,
(C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
1-C
3-Halogenalkyl,
oder
Heterocycloalkyl, C
1-C
8-Alkylsulfonyl,
Arylsulfonyl, Heteroarylsulfonyl, C
1-C
8-Alkanoyl, Aroyl, Heteroaroyl, Aryl, Heteroaryl,
C
1-C
6-Arylalkyl
oder C
1-C
6-Heteroarylalkyl,
jeweils gegebenenfalls substituiert mit 1 bis 5 Substituenten, unabhängig ausgewählt bei
jedem Auftreten aus Halogen, C
1-C
6-Halogenalkyl,
OR
13, NR
8R
9, C
1-C
6-Alkyl-OR
13, C
1-C
6-Alkyl-NR
8R
9, CONR
8R
9, COOR
13, CN, SO
2NR
8R
9, SO
2R
13, mit der Maßgabe, dass, wenn R
7 SO
2R
13 darstellt,
R
13 nicht H sein darf;
R
8 und
R
9 unabhängig
ausgewählt
sind bei jedem Auftreten aus H, C
1-C
6-Alkyl , C
3-C
10-Cycloalkyl , C
2-C
6-Alkenyl , C
3-C
10-Cycloalkenyl,
C
2-C
6-Alkinyl, Heterocycloalkyl,
C
1-C
8-Alkanoyl,
Aroyl, Heteroaroyl, Aryl, Heteroaryl, C
1-C
6-Arylalkyl oder C
1-C
6-Heteroarylalkyl, oder R
8 und
R
9, zusammengenommen, einen C
3-C
6-Aminocarbocyclus oder einen C
2-C
5-Aminoheterocyclus bilden können, jeweils
egebenenfalls substituiert bei jedem Auftreten mit C
1-C
6-Alkgyl, C
3-C
10-Cycloalkyl, C
3-C
10-Cycloalkenyl, (C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
1-C
3-Halogenalkyl oder Heterocycloalkyl, C
1-C
8-Alkylsulfonyl,
Arylsulfonyl, Heteroarylsulfonyl, C
1-C
8-Alkanoyl, Aroyl, Heteroaroyl, Aryl, Heteroaryl,
C
1-C
6-Arylalkyl
oder C
1-C
6-Heteroarylalkyl;
R
11 ausgewählt
ist aus H, C
1-C
6-Alkyl,
C
3-C
10-Cycloalkyl,
(C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl;
R
12 ausgewählt
ist aus H, Aryl, Heteroaryl, C
1-C
6-Alkyl,
C
3-C
10-Cycloalkyl,
(C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, gegebenenfalls
substituiert mit OR
7, NR
8R
9, C
3-C
6-Aminocarbocyclus
oder C
2-C
5-Aminoheterocyclus;
R
13 unabhängig
ausgewählt
ist bei jedem Auftreten aus H, C
1-C
6-Alkyl, C
3-C
10-Cycloalkyl, (C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl,
C
2-C
6-Alkenyl, C
2-C
6-Alkinyl, C
1-C
6-Halogenalkyl,
mit der Maßgabe,
dass für
SO
2NR
8R
9, SO
2R
13, R
13 nicht
H sein darf;
R
14 H, C
1-C
6-Alkyl, C
3-C
10-Cycloalkyl, (C
3-C
10-Cycloalkyl)-C
1-C
6-alkyl, C
2-C
4-Alkenyl, C
2-C
4-Alkinyl, Halogen oder CN darstellt;
mit
der Maßgabe,
dass die Verbindung keine Verbindung ist, worin X=N, A=CH
2, B=CH
2, R
1=CH
3, R
2=Et,
R
3=H, R
4=2-Brom-4-(2-propyl)phenyl,
R
5=CH
3 und R
6=CH
3;
oder
ein pharmazeutisch verträgliches
Salz oder Hydrat davon.
-
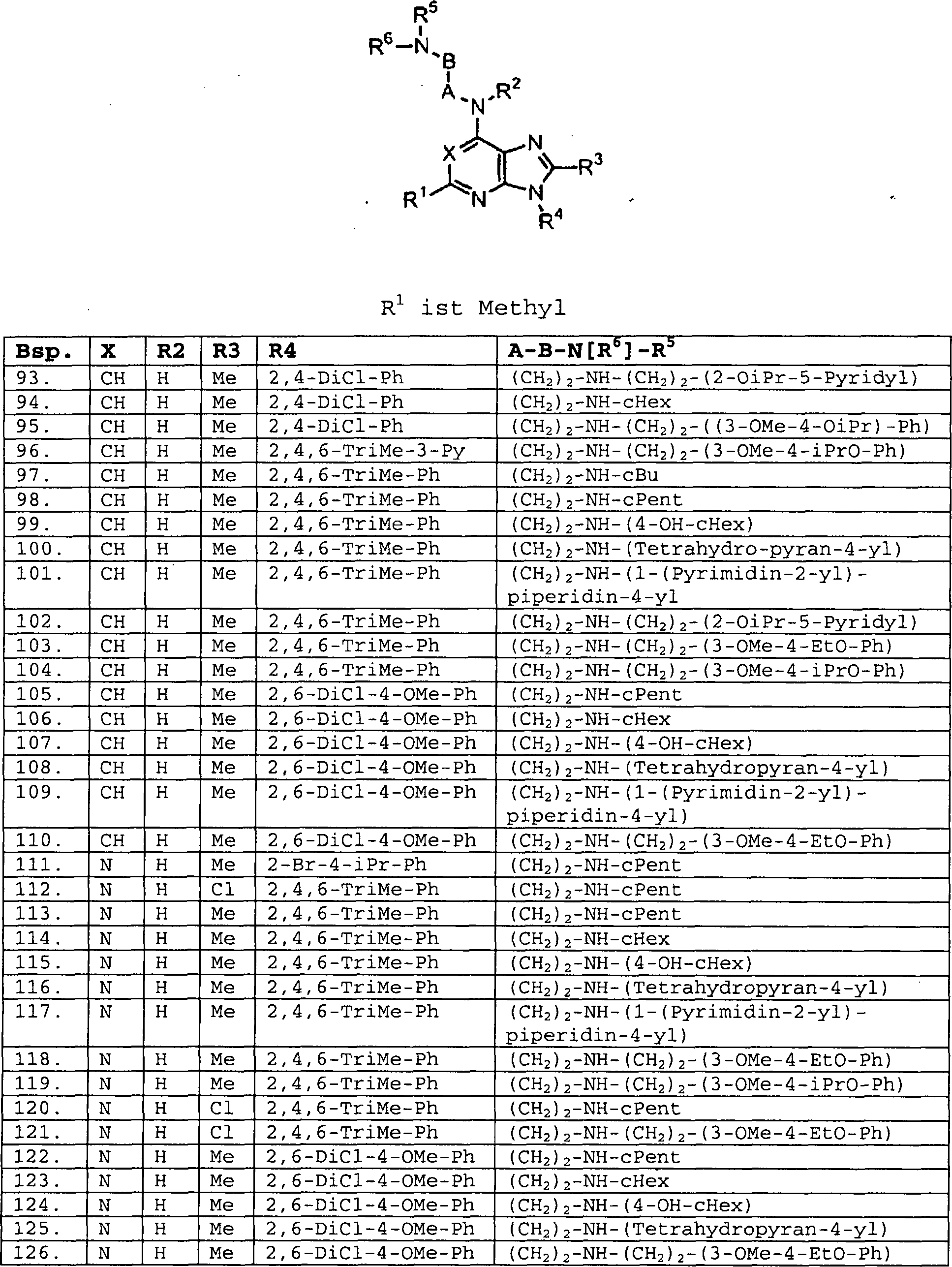
Bevorzugte
Verbindungen der vorliegenden Erfindung sind jene, worin X N oder
CH darstellt, R1 H, C1-C6-Alkyl, C3-C10-Cycloalkyl
oder (C3-C10-Cycloalkyl)-C1-C6-alkyl darstellt;
R6 H, C1-C6-Alkyl, C3-C10-Cycloalkyl oder (C3-C10-Cycloalkyl)-C1-C6-alkyl
darstellt.
-
Diese
Erfindung umfasst auch in zusätzlichen
Ausführungsformen
die neuen Verbindungen der Formel V, und die Salze und Solvate davon,
sowie die pharmazeutischen Formulierungen, die eine Verbindung der
Formel V umfassen, oder ein pharmazeutisch verträgliches Salz oder Solvat davon,
in Kombination mit einem oder mehreren pharmazeutisch verträglichen
Trägern,
Exzipienten oder Verdünnungsmitteln
davon.
-
Diese
Erfindung umfasst auch Verfahren für die Herstellung eines Arzneimittels
zum Behandeln von physiologischen Störungen, die mit einem Überschuss
an Neuropeptid Y verbunden sind, wie Ess- und cardiovaskuläre Störungen,
wobei das Verfahren Verabreichen an einen Säuger bei Behandlungsbedarf
einer wirksamen Menge einer Verbindung der Formel V umfasst.
-
Diese
Erfindung umfasst auch Verfahren zum selektiven Inhibieren des Bindens
von NPY1-Rezeptoren, das In-Kontakt-Bringen einer Verbindung
der Formel V mit neuronalen Zellen umfasst, wobei die Verbindung
in einer wirksamen Menge vorliegt, um eine ausreichende Konzentration
zum Inhibieren des Bindens von NPY1-Rezeptoren
in vitro herzustellen.
-
Als
solche stellt die vorliegende Erfindung auch ein Verfahren zum Umwandeln
von heterocyclischen Kernen der Formel Va bereit, worin X, R1, R3, R4,
R15 und R16 wie
vorstehend defi niert sind, in Verbindungen, die stark und selektiv
mit NPY1-Rezeptoren
in Wechselwirkung treten, durch Substitution der n-Position des Heterocyclus
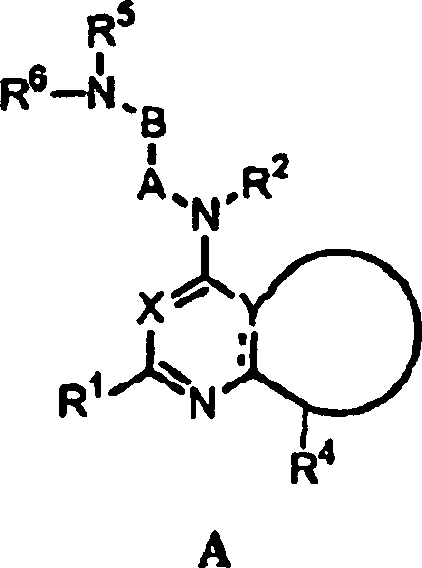
der Formel A mit einer Diamingruppe der Formel N[R2]-A-B-N[R6]-R5, worin R2, A, B, R6 und R5 wie in Anspruch 1 definiert sind.
-
-
Beschreibung der Erfindung
im Einzelnen
-
Diese
Erfindung betrifft bestimmte Alkylendiaminsubstituierte Heterocyclen,
die selektiv und stark Säuger-Neuropeptid-Y-(NPY)-Rezeptoren
binden, jene der Formel V, die neue und nützliche Neuropeptid-Y-Rezeptor-Antagonisten
darstellen.
-
In
der vorliegenden Beschreibung können
die Verbindungen der Formel V generisch unter Formel A beschrieben
werden, worin R1, R2,
R4, R5, R6, X, A und B vorstehend definiert sind.
-
-
In
bestimmten Situationen können
die Verbindungen der Formel V ein oder mehrere asymmetrische Kohlenstoffatome
enthalten, sodass die Verbindungen in verschiedenen stereoisomeren
Formen vorliegen können.
Diese Verbindungen können
beispielsweise Racemate oder optisch aktive Formen sein. In diesen
Situationen können
die einzelnen Enantiomeren; d.h. optisch aktive Formen, durch asymmetrische
Synthese oder durch Auftrennung der Racemate erhalten werden. Die
Auftrennung der Racemate kann beispielsweise durch herkömmliche
Verfahren, wie Kristallisation in Gegenwart eines Auftrennungsmittels,
oder Chromatographie, unter Anwendung von beispielsweise chiraler
HPLC-Säule,
ausgeführt
werden.
-
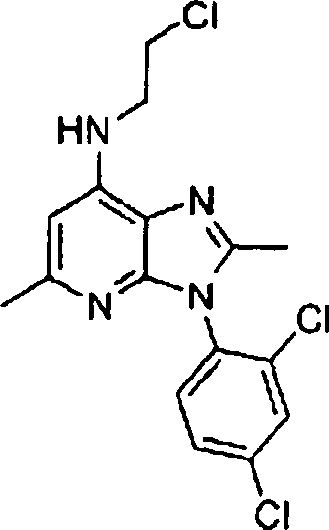
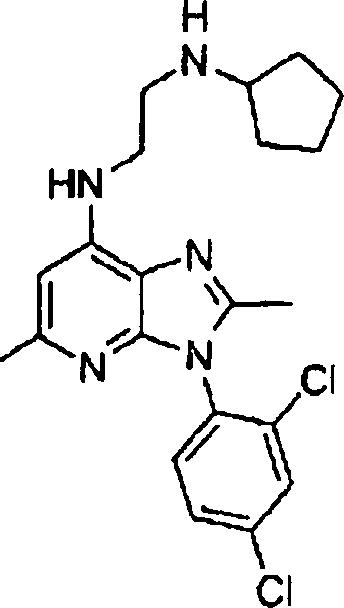
Repräsentative
Verbindungen der vorliegenden Erfindung, die durch die Formel V
umfasst werden, schließen
ein, sind jedoch nicht auf die Verbindungen in Beispielen 5–7 und 93–126 und
deren pharmazeutisch verträgliche
Säureadditionssalze
begrenzt. Wenn die erfindungsgemäße Verbindung
als ein Säureadditionssalz
erhalten wird, kann die freie Base außerdem durch Basifizieren einer
Lösung
des sauren Salzes erhalten werden. Wenn umgekehrt das Produkt eine
freie Base darstellt, kann ein Additionssalz, insbesondere ein pharmazeutisch
verträgliches
Additionssalz, durch Auflösen
der freien Base in einem geeigneten organischen Lösungsmittel
und Behandeln der Lösung
mit einer Säure,
gemäß herkömmlichen
Verfahren zum Herstellen von Säureadditionssalzen
aus Basenverbindungen, hergestellt werden.
-
Nicht-toxische
pharmazeutische Salze schließen
Salze von Säuren,
wie Salz-, Phosphor-, Bromwasserstoff-, Schwefel-, Sulfin-, Ameisensäure-, Toluolsulfon-,
Methansulfon-, Salpeter-, Benzoe-, Zitronen-, Wein-, Äpfel-, Hydrojod-,
Alkan-, wie Essig-, HOOC-(CH2)n-COOH-,
-Säure,
worin n 0–4
ist, und dergleichen ein. Der Fachmann wird eine breite Vielzahl
von nicht-toxischen, pharmazeutisch verträglichen Additionssalzen kennen.
-
Wenn
eine Verbindung in verschiedenen tautomeren Formen vorliegt, ist
die Erfindung nicht auf eines der speziellen Tautomeren begrenzt.
Die Erfindung schließt
alle tautomeren Formen einer Verbindung ein.
-
"Heteroatom" bedeutet in der
vorliegenden Erfindung Sauerstoff oder Schwefel, oder ein Stickstoffatom,
gegebenenfalls substituiert mit C1-C6-Niederalkyl, C1-C6-Arylalkyl, C1-C10-Cycloalkyl,
(C3-C10-Cycloalkyl)-C1-C6-alkyl, C2-C8-Alkanoyl, C1-C6-Sulfonyl.
-
"Alkyl", "Niederalkyl" oder "C1-C6-Alkyl" bedeuten
in der vorliegenden Erfindung gerad- oder verzweigtkettige Alkylgruppen
mit 1–6
Kohlenstoffatomen, wie beispielsweise Methyl, Ethyl, Propyl, Isopropyl,
n-Butyl, sec-Butyl, tert-Butyl,
Pentyl, 2-Pentyl, Isopentyl, Neopentyl, Hexyl, 2-Hexyl, 3-Hexyl
und 3-Methylpentyl.
-
"Cycloalkyl" oder "C3-C10-Cycloalkyl" bedeuten in der vorliegenden Erfindung
Alkylgruppen mit 3–10 Kohlenstoffatomen,
die ein mono-, bi- oder polycyclisches Ringsystem bilden, wie beispielsweise
Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl, Cycloheptyl, Norbornyl
und dergleichen.
-
"(Cycloalkyl)alkyl", "Nieder(cycloalkyl)alkyl" oder (C3-C10-Cycloalkyl)-C1-C6-alkyl bedeutet in der vorliegenden Erfindung
ein gerader oder verzweigter Alkylsubstituent, der aus 1 bis 6 Kohlenstoffatomen
gebildet wird, gebunden an ein mono-, bi- oder polycyclisches Ringsystem
mit 3–10
Kohlenstoffatomen, wie beispielsweise Cyclopropylmethyl, Cyclobutylmethyl,
Cyclopentylmethyl, Cyclohexylmethyl, Cycloheptylmethyl und dergleichen.
-
Der
Begriff "C2-C6-Alkenyl" bedeutet in der
vorliegenden Erfindung Kohlenwasserstoffketten mit 2 bis 6 Kohlenstoffatomen
in gerader oder verzweigter Anordnung und enthaltend eine oder mehrere
ungesättigte Kohlenstoff-Kohlenstoff-Doppelbindungen,
die an einem beliebigen stabilen Punkt entlang der Kette auftreten können, wie
beispielsweise Ethenyl, Allyl, Isopropenyl und dergleichen.
-
"Cycloalkenyl" oder "C3-C10-Cycloalkenyl" bedeuten in der vorliegenden Erfindung
Alkylgruppen mit 3–10
Kohlenstoffatomen, die ein mono-, bi- oder polycyclisches Ringsystem
mit 3–10
Kohlenstoffatomen bilden und eine oder mehrere Kohlenstoff-Kohlenstoff-Doppelbindungen
enthalten, die an jedem stabilen Punkt in dem Ring auftreten können, wie
beispielsweise Cyclopentenyl, Cyclohexenyl oder Cycloheptenyl.
-
Der
Begriff "C2-C6-Alkinyl" bedeutet in der
vorliegenden Erfindung Kohlenwasserstoffketten mit 2 bis 6 Kohlenstoff atomen
in gerader oder verzweigter Anordnung und enthaltend eine oder mehrere
ungesättigte Kohlenstoff-Kohlenstoff-Dreifachbindungen,
die an jedem stabilen Punkt entlang der Kette auftreten können, wie
beispielsweise Ethinyl, Propargyl und dergleichen.
-
Der
Begriff "Aryl" bedeutet in der
vorliegenden Erfindung eine monocyclische oder bicyclische aromatische
Gruppe mit vorzugsweise 6 bis 10 Kohlenstoffatomen, wie beispielsweise
Phenyl oder Naphthyl.
-
Der
Begriff "Heteroaryl" bedeutet in der
vorliegenden Erfindung eine Arylgruppe, worin ein oder mehrere des/der
Ringkohlenstoffatoms/atome gegen ein Heteroatom ersetzt ist/sind.
Solche Gruppen haben vorzugsweise 4 bis 10 Kohlenstoffatome und
1 bis 4 Heteroatome, wie beispielsweise Pyridyl, Pyrimidinyl, Triazinyl,
Imidazolyl, Oxazolyl, Isoxazolyl, Indolyl, Pyrrolyl, Pyrazolyl,
Chinolinyl, Isochinolinyl, Thiazolyl, Benzothiadiazolyl, Triazolyl,
Triazinyl, Pyrazinyl, Furanyl, Thienyl, Benzothienyl, Benzofuranyl,
Tetrazolyl.
-
Der
Begriff "Heterocyclyl", "Heterocyclus" oder "Heterocycloalkyl" bedeutet in der
vorliegenden Erfindung eine gesättigte
oder teilweise gesättigte
Heteroarylgruppe.
-
"C1-C6-Arylalkyl" oder "C1-C6-Heteroarylalkyl" bedeutet in der vorliegenden Erfindung
eine verzweigte oder geradkettige Alkylgruppe mit 1–6 Kohlenstoffatomen
und substituiert an einem der Kohlenstoffatome durch gegebenenfalls
substituierten Aryl- oder Heteroarylring, wie beispielsweise Benzyl,
Phenethyl, Methylpyridyl, Ethylpyridyl und dergleichen.
-
"C5-C8-Arylcycloalkyl" bedeutet in der vorliegenden Erfindung
Cycloalkylgruppen mit 5–8
Kohlenstoffatomen und kondensiert an eine Arylgruppe, wie beispielsweise
1,2,3,4-Tetrahydronaphthalinyl,
2,3-Dihydrobenzothienyl oder 2,3-Dihydrobenzofuranyl.
-
"C5-C8-Heteroarylcycloalkyl" bedeutet in der vorliegenden Erfindung
Cycloalkylgruppen mit 5–8
Kohlenstoffatomen, kondensiert an eine Heteroarylgruppe, wie beispielsweise 1,2,3,4-Tetrahydrochinolyl,
2,3-Dihydrobenzothienyl, 2,3-Dihydrobenzofuranyl oder Indolinyl.
-
"Alkoxy", "C1-C6-Alkoxy" oder "C1-C6-Alkyloxy" bedeutet in der vorliegenden Erfindung
gerade oder verzweigtkettige Alkoxygruppen mit 1–6 Kohlenstoffatomen, wie beispielsweise
Methoxy, Ethoxy, Propoxy, Isopropoxy, n-Butoxy, sec-Butoxy, tert-Butoxy,
Pentoxy, 2-Pentyl, Isopentoxy, Neopentoxy, Hexoxy, 2-Hexoxy, 3-Hexoxy
und 3-Methylpentoxy.
-
"Cycloalkoxy", "C3-C10-Cycloalkoxy" oder "C3-C10-Cycloalkyloxy" bedeutet in der vorliegenden Erfindung
eine Gruppe, die durch ein Sauerstoffatom gebildet wird, das an
ein mono-, bi- oder polycyclisches Ringsystem mit 3–10 Kohlenstoffatomen
gebunden ist, wie beispielsweise Cyclopropoxy, Cyclobutoxy, Cyclopentoxy,
Cyclohexoxy oder Cycloheptoxy.
-
"(Cycloalkyl)alkyloxy", "(C3-C10-Cycloalkyl)-C1-C6-alkoxy" oder "(C3-C10-Cycloalkyl)-C1-C6-alkyloxy" bedeutet in der vorliegenden Erfindung
eine Gruppe, die durch ein Sauerstoffatom gebildet wird, das an
eine 1–6 Kohlenstoffkette
gebunden ist, die an ein mono-, bi- oder polycyclisches Ringsystem
mit 3–10
Kohlenstoffatomen gebunden ist, wie beispielsweise Cyclopropylmethyloxy,
Cyclobutylmethyloxy, Cyclopentylmethyloxy, Cyclohexylmethyloxy,
Cycloheptylmethyloxy und dergleichen.
-
"C3-C6-Aminocarbocyclus" bedeutet eine cyclische Aminogruppe,
die durch einen Stickstoff, der in einem Ring mit 3 bis 6 Kohlenstoffatomen
enthalten ist, wie beispielsweise Azetidino, Pyrrolidino, Piperidino, Perhydroazepino,
gebildet wurde.
-
"C2-C5-Aminoheterocyclus" bedeutet eine cyclische Aminogruppe,
die durch einen Stickstoff, der in einem Ring mit 2 bis 5 Kohlenstoffatomen
enthalten ist, und ein weiteres Heteroatom gebildet wird, wie beispielsweise
Morpholino, Thiomorpholino, Piperazino.
-
Die
Begriffe "Halo" oder "Halogen" bedeuten in der
vorliegenden Erfindung Fluor, Chlor, Brom oder Jod.
-
"Halogenalkyl" ist vorgesehen,
sowohl verzweigtes als auch geradkettiges Alkyl mit der ausgewiesenen
Anzahl an Kohlenstoffatomen, substituiert mit 1 oder mehreren Halogenen,
einzuschließen.
-
Der
Begriff "C2-C8-Alkanoyl" bedeutet eine Acylgruppe
mit 2 bis 8 Kohlenstoffatomen in einer linearen, verzweigten oder
C3-C10-Cycloalkylanordnung,
gegebenenfalls substituiert mit 1 bis 5 Substituenten, unabhängig ausgewählt bei
jedem Auftreten aus Halogen, Trifluormethyl, OR7,
NR8R9, CONR8R9, COOR7 oder CN.
-
Der
Begriff "C1-C6-Alkylsulfonyl" bedeutet eine Alkylsulfonylgruppe,
die 1 bis 6 Kohlenstoffatome in einer linearen, verzweigten oder
C3-C7-Cycloalkylanordnung
enthält.
-
Der
Begriff "substituiert" bedeutet, dass ein
oder mehrere Wasserstoffatom(e) an dem bezeichneten Atom durch die
ausgewiesene Gruppe ersetzt ist/sind, vorausgesetzt, dass die Bindigkeit
des bezeichneten Atoms nicht überschritten
ist und dass sich eine chemisch stabile Verbindung aus der Substitution
ergibt.
-
Eine
stabile Verbindung ist hierin als jene definiert, die isoliert,
charakterisiert und auf biologische Aktivität getestet werden kann.
-
Der
Begriff "Oxo" (d.h. =O) weist
aus, dass zwei geminale Wasserstoffatome durch eine Doppelbindungs-Sauerstoffgruppe
ersetzt sind.
-
"Heterocyclischer
Kern" bedeutet in
der vorliegenden Erfindung die nachstehende Struktur der Formel Va,
worin X, R1, R3,
R4, R15 und R16 vorstehend definiert sind.
-
-
In
der vorliegenden Erfindung werden einige der Gruppen, die speziell
vorstehend erwähnt
wurden, wie nachstehend definiert:
- 2-On-1,3-oxazolidinyl
ist
- 1-Aza-bicyclo[4.4.0]decyl ist
- -8-Azabicyclo[3.2.1]octanyl ist
- (1,1-Dioxo)tetrahydrothiopyranyl ist
- Norbornyl ist
- Chinuclidinyl ist
-
Sofern
nicht anders ausgewiesen, kann der Bindungspunkt an einem beliebigen
stabilen Punkt entlang der vorstehend erwähnten Ringe auftreten.
-
In
der vorliegenden Erfindung bezeichnet der Begriff "stark" im Zusammenhang
mit NPY1-Rezeptor-Antagonisten eine Bindungsaffinität mit einem
Ki-Wert von weniger als 10 Mikromolar, vorzugsweise weniger als
1 Mikromolar und bevorzugter weniger als 100 Nanomolar bei dem Human-NPY1-Bindungsassay.
-
In
der vorliegenden Erfindung bezeichnet der Begriff "selektiv" im Zusammenhang
mit NPY1-Rezeptor-Antagonisten eine Bindungsaffinität mit einem
Ki-Wert bei dem humanen NPY1-Bindungsassay,
das 10-fach, vorzugsweise 100-fach und bevorzugter 1000-fach, weniger
als der Ki von der gleichen Verbindung ist, die in einem weiteren
Rezeptorbindungsassay gemessen wurde, insbesondere dem NPY5- und CRF1-Rezeptor-Bindungsassay.
Bindungsassays für
die NPY5- und CRF1-Rezeptoren
wurden beispielsweise in J. Clin. Invest., 102, 2136 (1998) bzw.
in Endocrinology 116, 1653 (1985) beschrieben.
-
Da
die Verbindungen der Formel V Antagonisten des Y1-Rezeptors darstellen,
sind sie bei der Behandlung einer brei ten Vielzahl von klinischen
Zuständen
von Wert, die durch das Vorliegen eines Überschusses an Neuropeptid
Y charakterisiert sind. Somit stellt die Erfindung Verfahren für die Behandlung
oder Verhinderung einer physiologischen Störung, die mit einem Überschuss
an Neuropeptid Y verbunden ist, bereit, wobei das Verfahren Verabreichen
an einen Säuger
bei Behandlungsbedarf einer wirksamen Menge einer Verbindung der
Formel V oder eines pharmazeutisch verträglichen Salzes, Solvats oder
Prodrugs davon umfasst. Der Begriff "physiologische Störung, die mit einem Überschuss
an Neuropeptid Y" verbunden
ist, umfasst jene Störungen,
die mit einer ungeeigneten Stimulierung von Neuropeptid-Y-Rezeptoren
verbunden sind, ungeachtet der tatsächlichen Menge an örtlich vorliegendem
Neuropeptid Y. Diese physiologischen Störungen können einschließen: Störungen oder
Erkrankungen, die das Herz, die Blutgefäße oder das Nierensystem betreffen, wie
Vasospasmus, Herzversagen, Schock, Herzhypertrophie, erhöhter Blutdruck,
Angina, Herzinfarkt, plötzlicher
Herztod, Arrhythmie, periphere vaskuläre Erkrankung und anormale
Nierenzustände,
wie beeinträchtigter Fluidfluss,
anormaler Massentransport oder Niereninsuffizienz; Zustände bezüglich erhöhter sympathischer Nervenaktivität, beispielsweise
während
oder nach Herzkranzarterienchirurgie, und Operationen und chirurgischer
Eingriff in den Gastrointestinaltrakt; zerebrale Erkrankungen und
Erkrankungen bezüglich
des zentralen Nervensystems, wie Hirnschlag, Neurodegeneration,
Epilepsie, Schlaganfall, und Zustände bezüglich Schlaganfall, zerebraler
Vasospasmus und Hämorrhagie,
Depression, Angstzustand, Schizophrenie und Demenz; Zustände bezüglich Schmerz
oder Nocizeption; Erkrankungen bezüglich anormaler Gastrointestinalmotilität und -sekretion,
wie verschiedene Formen von Ileus, Harninkontinenz und Crohn'scher Krankheit;
anormale Trink- und Essensaufnahmestörungen, wie Fettsucht, Anorexia,
Bulimie und metabolische Störungen;
Erkrankungen bezüglich
sexueller Dysfunktion und reproduktiver Störungen; Zustände oder
Störungen
im Zusammenhang mit Entzündung;
Atemwegserkrankungen, wie Asthma und Zustände bezüglich Asthma und Bronchokonstriktion;
und Erkrankungen bezüglich
anormaler Hormonfreisetzung, wie luteinisierendes Hormon, Wachstumshormon,
Insulin und Prolactin. Siehe US-Patent 5 504 094.
-
Pharmazeutische
Zubereitungen
-
Die
Verbindungen der allgemeinen Formel V können oral, örtlich, parenteral, durch Inhalation
oder Spray oder rektal in Dosierungseinheitsformulierungen, die
herkömmliche,
nichttoxische, pharmazeutisch verträgliche Träger, Hilfsmittel und Vehikel
enthalten, verabreicht werden. Der wie hierin verwendete Begriff
parenteral schließt
subkutane Injektionen, intravenöse,
intramuskuläre,
intrasternale Injektion oder Infusionstechniken ein. Zusätzlich wird
eine pharmazeutische Formulierung bereitgestellt, umfassend eine
Verbindung der allgemeinen Formel V und einen pharmazeutisch verträglichen
Träger.
Eine oder mehrere Verbindungen der allgemeinen Formel V können in
Verbindung mit einem oder mehreren nicht-toxischen pharmazeutisch
verträglichen
Trägern
und/oder Verdünnungsmitteln
und/oder Hilfsmitteln und, falls erwünscht, anderen Wirkstoffen
vorliegen. Die pharmazeutischen Zusammensetzungen, die die Verbindungen
der allgemeinen Formel V enthalten, können in einer Form vorliegen,
die zur oralen Verwendung geeignet ist, beispielsweise als Tabletten,
Pastillen (troches), Pastillen (lozenges), wässrige oder ölige Suspensionen,
dispergierbare Pulver oder Granulate, Emulsion, Hart- oder Weichkapseln,
oder Sirupe oder Elixiere.
-
Die
zur oralen Verwendung vorgesehenen Zusammensetzungen können gemäß beliebigen
Verfahren hergestellt werden, die dem Fachmann für die Herstellung von pharmazeutischen
Zusammensetzungen bekannt sind, und solche Zusammensetzungen können ein
oder mehrere Mittel, ausgewählt
aus Süßungsmitteln, Geschmacksmitteln,
Färbemitteln
und Konservierungsmitteln, enthalten, um pharmazeutisch elegante
und wohlschmeckende Zubereitungen bereitzustellen. Tabletten enthalten
den Wirkstoff in Anmischung bzw. Beimengung mit nicht-toxischen
pharmazeutisch verträglichen
Exzipienten, die für
die Herstellung von Tabletten geeignet sind. Diese Exzipienten können beispielsweise
inerte Verdünnungsmittel,
wie Calciumcarbonat, Natriumcarbonat, Lactose, Calciumphosphat oder
Natriumphosphat; Granulierungs- und Sprengmittel, wie beispielsweise
Maisstärke
oder Alginsäure,
Bindungsmittel, beispielsweise Stärke, Gelatine oder Acacia,
und Gleitmittel, beispielsweise Magnesiumstearat, Stearinsäure oder
Talkum, sein. Die Tabletten können
unbeschichtet sein oder sie können
durch bekannte Techniken, um den Zerfall oder die Absorption im
Gastrointestinaltrakt zu verzögern,
beschichtet sein und dadurch eine verzögerte Wirkung über einen
längeren
Zeitraum bereitstellen. Beispielsweise kann ein Zeitverzögerungsmaterial,
wie Glycerylmonostearat oder Glyceryldistearat, angewendet werden.
-
Formulierungen
zur oralen Verwendung können
als Hartgelatinekapseln dargereicht werden, wobei der Wirkstoff
mit einem inerten, festen Verdünnungsmittel,
beispielsweise Calciumcarbonat, Calciumphosphat oder Kaolin, vermischt
wird, oder als Weichgelatinekapseln, wobei der Wirkstoff mit Wasser
oder einem Ölmedium,
beispielsweise Erdnussöl,
flüssigem
Paraffin oder Olivenöl,
vermischt wird.
-
Wässrige Suspensionen
enthalten die wirksamen Materialien in Anmischung mit Exzipienten,
die für die
Herstellung von wässrigen
Suspensionen geeignet sind. Solche Exzipienten sind suspendierende
Mittel, beispielsweise Natriumcarboxymethylcellulose, Methylcellulose,
Hydropropylmethylcellulose, Natriumalginat, Polyvinylpyrrolidon,
Tragacanthgummi und Akaziengummi; dispergierende oder benetzende
Mittel können
natürlich
vorkommendes Phosphatid, beispielsweise Lezithin, oder Kondensationsprodukte
von einem Alkylenoxid mit Fettsäuren,
beispielsweise Polyoxyethylenstearat, oder Kondensationsprodukte
von Ethylenoxid mit langkettigen aliphatischen Alkoholen, beispielsweise
Heptadecaethylenoxycetanol, oder Kondensationsprodukte von Ethylenoxid
mit Teilestern, abgeleitet von Fettsäuren und einem Hexit, wie Polyoxyethylensorbitmonooleat,
oder Kondensationsprodukten von Ethylenoxid mit Teilestern, abgeleitet
von Fettsäuren
und Hexitanhydriden, beispielsweise Polyethylensorbitanmonostearat,
sein. Die wässrigen
Suspensionen können
auch ein oder mehrere Konservierungsmittel, beispielsweise p-Hydroxybenzoesäureethyl-
oder -n-propylester, ein oder mehrere färbende Mittel, ein oder mehrere
Geschmacksmittel und ein oder mehrere Süßungsmittel, wie Saccharose
oder Saccharin, sein.
-
Ölige Suspensionen
können
durch Suspendieren der Wirkstoffe in einem Pflanzenöl, beispielsweise Erdnussöl, Olivenöl, Sesamöl oder Kokosnussöl, oder
in einem Mineralöl,
wie flüssigem
Paraffin, formuliert werden. Die öligen Suspensionen können ein
Verdickungsmittel, beispielsweise Bienenwachs, hartes Paraffin oder
Cetylalkohol, enthalten. Süßungsmittel,
wie jene, vorstehend angeführten,
und Geschmacksmittel können
zugesetzt werden, um schmackhafte orale Zubereitungen bereitzustellen.
Diese Zusammensetzungen können
durch die Zugabe eines Antioxidationsmittels, wie Ascorbinsäure, konserviert
werden.
-
Dispergierbare
Pulver und Granulate, die zur Herstellung einer wässrigen
Suspension durch Zusatz von Wasser geeignet sind, versehen den Wirkstoff
in Anmischung mit einem dispergierenden oder benetzenden Mittel,
Suspendierungsmittel und einem oder mehreren Konservierungsmitteln.
Geeignete dispergierende oder Netzmittel und Suspendierungsmittel
werden beispielhaft durch jene, die bereits vorstehend erwähnt wurden,
angegeben. Zusätzliche
Exzipienten, beispielsweise Süßungs-,
Geschmacks- und färbende
Mittel können
auch vorliegen.
-
Die
erfindungsgemäßen pharmazeutischen
Zusammensetzungen können
auch in Form von Öl-in-Wasser-Emulsionen
vorliegen. Die ölige
Phase kann ein Pflanzenöl,
beispielsweise Olivenöl
oder Erdnussöl,
oder ein Mineralöl,
beispielsweise flüssiges
Paraffin oder Gemische von diesen, sein. Geeignete Emulgatoren können natürlich vorkommende
Gummis, beispielsweise Akaziengummi oder Tragacanthgummi, natürlich vorkommende
Phosphatide, beispielsweise Sojabohne, Lezithin, und Ester oder
Teilester, die von Fettsäuren
und Hexit abgeleitet sind, Anhydride, beispielsweise Sorbitanmonooleat,
und Kondensationsprodukte der Teilester mit Ethylenoxid, beispielsweise
Polyoxyethylensorbitanmonooleat, sein. Die Emulsionen können auch
Süßungs- und
Geschmacksmittel enthalten.
-
Sirupe
und Elixiere können
mit Süßungsmitteln,
beispielsweise Glycerin, Propylenglycol, Sorbit oder Saccharose,
formuliert werden. Solche Formulierungen können auch ein Milderungsmittel,
ein Konservierungsmittel und Geschmacks- und färbende Mittel enthalten. Die
pharmazeutischen Zusammensetzungen können in Form einer sterilen,
injizierbaren wässrigen
oder ölartigen
Suspension vorliegen. Diese Suspension kann gemäß dem bekannten Stand der Technik,
unter Anwendung von geeigneten dispergierenden oder Benetzungsmitteln
und suspendierenden Mitteln, die vorstehend erwähnt wurden, formuliert werden.
Die sterile, injizierbare Zubereitung kann auch eine sterile, injizierbare
Lösung
oder Suspension in einem nicht-toxischen, parenteral
verträglichen
Verdünnungs-
oder Lösungsmittel,
beispielsweise als eine Lösung
in 1,3-Butandiol, sein. Unter den verträglichen Trägern und Lösungsmitteln, die angewendet
werden können,
sind Wasser, Ringer's
Lösung
und isotonische Natriumchloridlösung.
Zusätzlich
werden sterile, fixierte Öle
herkömmlicherweise als
ein Lösungsmittel
oder suspendierendes Medium angewendet. Für diesen Zweck kann jedes milde,
fixierte Öl
angewendet werden, einschließlich
synthetische Mono- oder Diglyceride. Außerdem finden Fettsäuren, wie Ölsäure, bei
der Zubereitung von injizierbaren Zubereitungen Anwendung.
-
Die
Verbindungen der allgemeinen Formel V können auch in Form von Suppositorien
zur rektalen Verabreichung des Arzneistoffs verabreicht werden.
Diese Zusammensetzungen können
durch Vermischen des Arzneistoffs mit einem geeigneten nicht reizenden
Exzipienten, der bei gewöhnlichen
Temperaturen fest jedoch bei der Rektaltemperatur flüssig ist,
hergestellt wer den und werden deshalb im Rektum unter Freisetzung des
Arzneistoffs schmelzen. Solche Materialien sind Kakaobutter und
Polyethylenglycole.
-
Verbindungen
der allgemeinen Formel V können
parenteral in einem sterilen Medium verabreicht werden. Der Arzneistoff
kann in Abhängigkeit
von dem Träger
und der angewendeten Konzentration entweder suspendiert oder in
dem Träger
gelöst
sein. Vorteilhafterweise können
Hilfsmittel, wie Lokalanästhetika,
Konservierungsmittel und puffernde Mittel, in dem Träger gelöst sein.
-
Dosierungsspiegel
in der Größenordnung
von etwa 0,1 mg bis etwa 50 mg pro Kilogramm Körpergewicht pro Tag sind bei
der Behandlung der vorstehend ausgewiesenen Zustände (etwa 0,5 mg bis etwa 3
g pro Patient pro Tag) verwendbar, obwohl in einigen Umständen höhere Mengen,
beispielsweise bis zu 140 mg/kg/Tag, geeignet sein können. Die
Menge des Wirkstoffs, der mit den Trägermaterialien zur Herstellung
einer Einzeldosierungsform kombiniert werden kann, wird in Abhängigkeit
von dem zu behandelnden Wirt und dem besonderen Verabreichungsmodus
variieren. Dosierungseinheitsformen werden im Allgemeinen zwischen
etwa 1 mg bis etwa 500 mg des Wirkstoffs enthalten.
-
Die
Häufigkeit
der Dosierung kann auch in Abhängigkeit
von der angewendeten Verbindung und der besonderen zu behandelnden
Krankheit variieren. Jedoch ist für die Behandlung der meisten
Essstörungen ein
Dosierungsregime von 4-mal täglich
oder weniger bevorzugt. Für
die Behandlung von Stress und Depression ist ein Dosierungsregime
von 1- oder 2-mal täglich
besonders bevorzugt.
-
Es
ist jedoch selbstverständlich,
dass der spezifische Dosierungsspiegel für jeden einzelnen Patienten von
einer Vielzahl von Faktoren, einschließlich der Wirkung der spezifischen
angewendeten Verbindung, dem Alter, Körpergewicht, der Allgemeingesundheit,
dem Geschlecht, der Nahrung, Verabreichungszeit, Verabreichungsweg
und Ausscheidungsrate, Arznei stoffkombination und der Schwere der
besonderen Krankheit unterliegenden Therapie abhängen wird.
-
Bevorzugte
erfindungsgemäße Verbindungen
werden bestimmte pharmakologische Eigenschaften aufweisen. Solche
Eigenschaften schließen
orale Bioverfügbarkeit,
niedrige Toxizität,
niedriges Serumproteinbinden und erwünschte In-vitro- und In-vivo-Halbwertszeiten
ein, sind jedoch nicht darauf begrenzt. Das Durchdringen der Blut-Hirn-Schranke
der angewendeten Verbindungen, um ZNS-Störungen zu behandeln, ist notwendig,
da niedrige Hirnspiegel der angewendeten Verbindungen zum Behandeln
von peripheren Störungen
häufig
bevorzugt sind.
-
Assays
können
verwendet werden, um diese erwünschten
pharmakologischen Eigenschaften vorherzusagen. Assays, die zum Vorhersagen
von Bioverfügbarkeit
verwendet werden, schließen
Transport über
humane Intestinalzelleneinschichten, einschließlich Caco-2-Zellen-Einschichten,
ein. Die Toxizität
für kultivierte Hepatozyten
kann angewendet werden, um die Verbindungstoxizität vorherzusagen.
Das Durchdringen der Blut-Hirn-Schranke
einer Verbindung beim Menschen kann aus den Hirnspiegeln der Verbindung
bei Labortieren, denen die Verbindung intravenös verabreicht wurde, vorhergesagt
werden.
-
Serumproteinbinden
kann aus Albuminbindungsassays vorhergesagt werden. Solche Assays
werden in einer Untergruppe von Oravcová, et al. (Journal of Chromatography
B 1996, 677, 1–27)
beschrieben.
-
Die
Verbindungshalbwertszeit ist invers proportional der Häufigkeit
der Dosierung einer Verbindung. In-vitro-Halbwertszeiten von Verbindungen können aus
Assays von mikrosomaler Halbwertszeit, wie von Kuhnz und Gieschen
(Drug Metabolism and Disposition 1998, 26, 1120–1127) beschrieben, vorhergesagt
werden.
-
Wie
vorstehend erörtert,
zeigen bevorzugte erfindungsgemäße Verbindungen
gute Wirksamkeit bei Standard-in-vitro-NPY-Rezeptor-Bindungsassays, insbesondere
dem folgenden, wie in Beispiel 261 ausgewiesem Assay. Bezugnahmen
hierin auf "Standard-in-vitro-Rezeptor-Bindungsassay" sollen sich auf
das folgende, wie in Beispiel 261 definierte Protokoll beziehen.
Im Allgemeinen zeigen bevorzugte erfindungsgemäße Verbindungen einen Ki-Wert von etwa 1 Mikromolar oder weniger,
noch bevorzugter eine Ki von etwa 100 Nanomolar
oder weniger, bevorzugter einen Ki-Wert
von etwa 10 Nanomolar oder weniger oder auch 1 Nanomolar oder weniger,
in einem solchen definierten Standard in-vitro-NPY-Rezeptor-Bindungsassay,
wie beispielhaft in folgendem Beispiel 261 ausgewiesen.
-
In
einem geeigneten Fall können
die erfindungsgemäßen Verbindungen
in Kombination mit anderen wirksamen Mitteln angewendet werden.
Die Erfindung stellt deshalb auch pharmazeutische Kombinationszusammensetzungen
bereit, die eine therapeutisch wirksame Menge einer Zusammensetzung
umfassen, umfassend: (a) eine erste Verbindung, wobei die erste
Verbindung eine wie vorstehend beschriebene Verbindung eines Prodrugs
davon ist oder ein pharmazeutisch verträgliches Salz der Verbindung
oder von dem Prodrug; und (b) eine zweite Verbindung, wobei die
zweite Verbindung ein β3-Agonist, ein Thyromimetikum, ein Essverhalten
modifizierendes Mittel oder ein NPY-Antagonist; und ein pharmazeutischer
Träger,
Vehikel oder Verdünnungsmittel
ist. Schließlich
stellt deshalb die Erfindung auch ein Kit bereit, umfassend: (a)
eine erste Verbindung, wobei die erste Verbindung eine wie vorstehend
beschriebene Verbindung darstellt, ein Prodrug davon oder ein pharmazeutisch
verträgliches
Salz der Verbindung oder von dem Prodrug; (b) eine zweite Verbindung, wobei
die zweite Verbindung ein β3-Agonist,
ein Thyromimetikum, ein Essverhalten modifizierendes Mittel oder ein
NPY-Antagonist ist; und einen pharmazeutischen Träger, Vehikel,
Verdünnungsmittel;
und (c) Mittel zum Enthalten der ersten und zweiten Einheitsdosierungsformen,
wobei die Mengen der ersten und zweiten Verbindungen sich aus einem
therapeutischen Effekt ergeben.
-
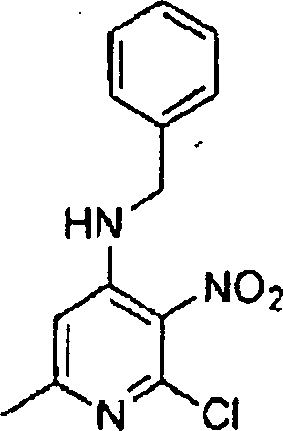
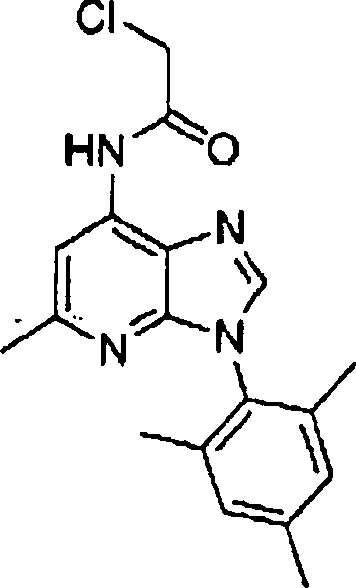
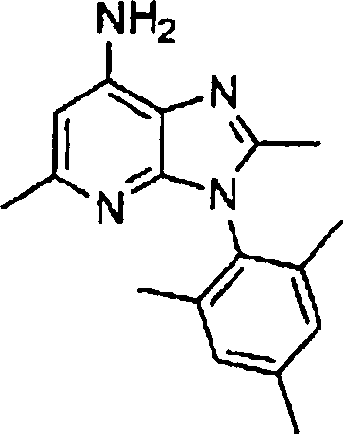
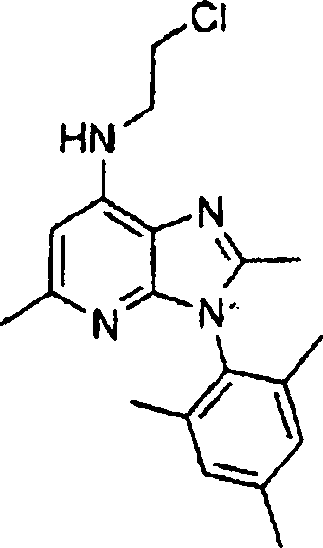
Hestellung
von Alkylendiamin-substituierten Heterocyclen der Formel V
-
Die
Herstellung dieser heterocyclischen Kerne, die in Formel V angegeben
sind, kann gemäß den in nachstehenden
Literaturstellen und jenen, die darin zitiert wurden, beschriebenen
Verfahren ausgeführt
werden:
Formel V: WO 9533750, J. Med. Chem. 42(5), 833–848 (1999);
Bioorg. Med. Chem. Lett., 9(7), 967–972 (1999).
-
Eine
Erläuterung
der Herstellungsverfahren der erfindungsgemäßen Verbindungen wird in den
nachstehenden Schemata angegeben. Insbesondere liefert Austausch
einer Abgangsgruppe Z, wie in Formel 10 (Schema 1), durch das geeignet
substituierte Amin oder geeignete Substitution einer heterocyclischen
Aminogruppe, wie in Formel 21 (Schema 7), allgemeine Verfahren,
um die heterocyclischen Kerne der vorliegenden Erfindung in Verbindungen
umzuwandeln, die stark mit dem NPY1-Rezeptor
in Wechselwirkung treten. Solche Transformationen können verschiedene,
aufeinander folgende chemische Schritte erfordern. In jenen wird
der Fachmann erkennen, dass die Ausgangsmaterialien variiert und
zusätzliche
Schritte angewendet werden können,
um Verbindungen, die durch die vorliegende Erfindung umfasst werden,
herzustellen. Die Offenbarungen von allen Artikeln und Literaturstellen,
die in dieser Anmeldung erwähnt
werden, einschließlich
Patente, sind hierin durch diesen Hinweis einbezogen.
-
-
Wie
in Schema 1 erläutert,
können
Verbindungen der Formel A aus Zwischenproduktverbindungen der Formel
10, worin Z Halogen (vorzugsweise Chlor oder Brom), Alkansulfonyloxy,
Arylsulfonyloxy oder Halogenalkansulfonyloxy darstellt, und worin
R1, R2, R4, R5, R6,
X, Y, A und B wie vorstehend definiert sind, unter Anwendung der
nachstehend ausgewiesenen Verfahren hergestellt werden.
-
Verbindungen
der Formel 10 reagieren mit einem Amin der Formel H2N-A-B-N[R6]-R5, worin A, B,
R5 und R6 wie vorstehend
definiert sind, in Gegenwart oder Abwesenheit einer Base, in Gegenwart
oder Abwesenheit eines inerten Lösungsmittels,
bei Reaktionstemperaturen im Bereich von –78°C bis 250°C, um Verbindungen der Formel
A zu erzeugen. Basen können
Alkalimetallhydride (vorzugsweise Natriumhydrid), Alkalimetallalkoxide
(1–6 Kohlenstoffatome)
(vorzugsweise Natriummethoxid, Natriumethoxid oder Natrium-tert-butoxid),
Erdalkalimetallhydride, Alkalimetalldialkylamide (vorzugsweise Lithiumdiisopropylamid),
Alkalimetallcarbonate, Alkalimetallbicarbonate, Alkalimetall-bis-(trialkylsilyl)amide
(vorzugsweise Lithium- oder Natrium(trimethylsilyl)amid), Trialkylamine
(vorzugsweise N,N-Diisopropyl-N-ethylamin
oder Triethylamin), Arylamine (vorzugsweise 4-Dimethylanilin), oder
heteroaromatische Amine (vorzugsweise Pyridin) einschließen, sind
jedoch nicht darauf begrenzt. Inerte Lösungsmittel können Alkylalkohole
(1–8 Kohlenstoffatome)
(vorzugsweise Methanol, Ethanol oder tert-Butanol), Niederalkannitrile
(1–6 Kohlenstoffe)
(vorzugsweise Acetonitril), Dialkylether (vorzugsweise Diethylether),
cyclische Ether (vorzugsweise Tetrahydrofuran oder 1,4-Dioxan),
N,N-Dialkylformamide (vorzugsweise Dimethylformamid), N,N-Dialkylacetamide
(vorzugsweise Dimethylacetamid), cyclische Amide (vorzugsweise N-Methylpyrrolidin-2-on),
Dialkylsulfoxide (vorzugsweise Dimethylsulfoxid), aromatische Kohlenwasserstoffe
(vorzugsweise Benzol oder Toluol), oder Halogenalkane (1–10 Kohlenstoffatome und
1–10 Halogenatome)
(vorzugsweise CH2Cl2) einschließen, sind
jedoch nicht darauf begrenzt. Bevorzugte Reaktionstemperaturen liegen
im Bereich von 0°C
bis 140°C.
-
-
Alternativ
können,
wie in Schema 2 erläutert,
Verbindungen der Formel A durch zuerst Umsetzen einer Verbindung
der Formel 10 mit einem Aminoalkohol der Formel H2N-A-B-OH,
worin A und B wie vorstehend definiert sind, in Gegenwart oder Abwesenheit
einer Base, in Gegenwart oder Abwesenheit eines inerten Lösungsmittels,
bei Reaktionstemperaturen im Bereich von –8°C bis 250°C, um Zwischenprodukte der Formel
11 zu erzeugen, erhalten werden. Umsetzen einer Verbindung der Formel
11 mit einem Halogenierungsmittel oder Sulfonylierungsmittel, in
Gegenwart oder Abwesenheit einer Base, in Gegenwart oder Abwesenheit
eines inerten Lösungsmittels,
bei Reaktionstemperaturen im Bereich von –78°C bis 250°C, um Produkte der Formel 12a
bereitzustellen (wo Z Halogen, Alkansulfonyloxy, Arylsulfonyloxy
oder Halogenalkansulfonyloxy darstellt) oder 12b, wenn A und B beide
CH2 darstellen und X CR14 darstellt.
Halogenierungsmittel schließen
SOCl2, POCl3, PCl3, PCl5, POBr3, PBr3, PBr5, CCl4/PPh3 ein, sind jedoch nicht darauf begrenzt.
Sulfonylierungsmittel schließen
Alkansufonylhalogenide oder -anhydride (vorzugsweise Methansulfonylchlorid
oder Methansulfonsäureanhydrid),
Arylsulfonylhalogenide oder -anhydride (wie p-Toluolsulfonylchlorid oder -anhydrid),
oder Halogenalkylsulfonylhalogenide oder -anhydride (vorzugsweise
Trifluormethansulfonsäureanhydrid)
ein, sind jedoch nicht darauf begrenzt. Basen können Trialkylamine (vorzugsweise
N,N-Diisopropyl-N-ethylamin
oder Triethylamin), bicyclische Amidine (vorzugsweise DBU), Aniline
(vorzugsweise N-Dimethylanilin), oder heteroaromatische Amine (vorzugsweise
Pyridin) einschließen,
sind jedoch nicht darauf begrenzt. Inerte Lösungsmittel können Niederalkannitrile
(1–6 Kohlenstoffe)
(vorzugsweise Acetonitril), Dialkylether (vorzugsweise Diethylether),
cyclische Ether (vorzugsweise Tetrahydrofuran oder 1,4-Dioxan),
N,N-Dialkylformamide (vorzugsweise Dimethylformamid), N,N-Dialkylacetamide
(vorzugsweise Dimethylacetamid), cyclische Amide (vorzugsweise N-Methylpyrrolidin-2-on),
Dialkylsulfoxide (vorzugsweise Dimethylsulfoxid), aromatische Kohlenwasserstoffe
(vorzugsweise Benzol oder Toluol), oder Halogenalkane mit 1–10 Kohlenstoffatomen
und 1–10 Halogenatomen
(vorzugsweise CH2Cl2)
einschließen,
sind jedoch nicht darauf begrenzt. Bevorzugte Reaktionstemperaturen
liegen im Bereich von –20°C bis 100°C. Verbindungen
der Formel 12a oder 12b können
dann mit einem Amin der Formel HN[R6]-R5, worin R5 und R6 wie vorstehend definiert sind, zur Bildung
einer Verbindung der Formel A umgesetzt werden. Basen können Alkalimetallhydride
(vorzugsweise Natriumhydrid), Alkalimetallalkoxide (1–6 Kohlenstoffatome)
(vorzugsweise Natriummethoxid, Natriumethoxid oder Natriumtert-butoxid),
Erdalkalimetallhydride, Alkalimetalldialkylamide (vorzugsweise Lithiumdiisopropylamid),
Alkalimetallcarbonate, Alkalimetallbicarbonate, Alkalimetall-bis-(trialkylsilyl)amide
(vorzugsweise Lithium- oder Natrium(trimethylsilyl)amid), Trialkylamine
(vorzugsweise N,N-Diisopropyl-N-ethylamin oder Triethylamin), Arylamine
(vorzugsweise 4-Dimethyl anilin), oder heteroaromatische Amine (vorzugsweise
Pyridin) einschließen, sind
jedoch nicht darauf begrenzt. Inerte Lösungsmittel können Alkylalkohole
(1–8 Kohlenstoffatome)
(vorzugsweise Methanol, Ethanol oder tert-Butanol), Niederalkannitrile
(1–6 Kohlenstoffatome)
(vorzugsweise Acetonitril), Dialkylether (vorzugsweise Diethylether),
cyclische Ether (vorzugsweise Tetrahydrofuran oder 1,4-Dioxan),
N,N-Dialkylformamide (vorzugsweise Dimethylformamid), N,N-Dialkylacetamide
(vorzugsweise Dimethylacetamid), cyclische Amide (vorzugsweise N-Methylpyrrolidin-2-on),
Dialkylsulfoxide (vorzugsweise Dimethylsulfoxid), aromatische Kohlenwasserstoffe
(vorzugsweise Benzol oder Toluol), oder Halogenalkane (1–10 Kohlenstoffatome
und 1–10
Halogenatome) (vorzugsweise CH2Cl2) einschließen, sind jedoch nicht darauf
begrenzt. Bevorzugte Reaktionstemperaturen liegen im Bereich von
0°C bis
140°C.
-
-
Eine
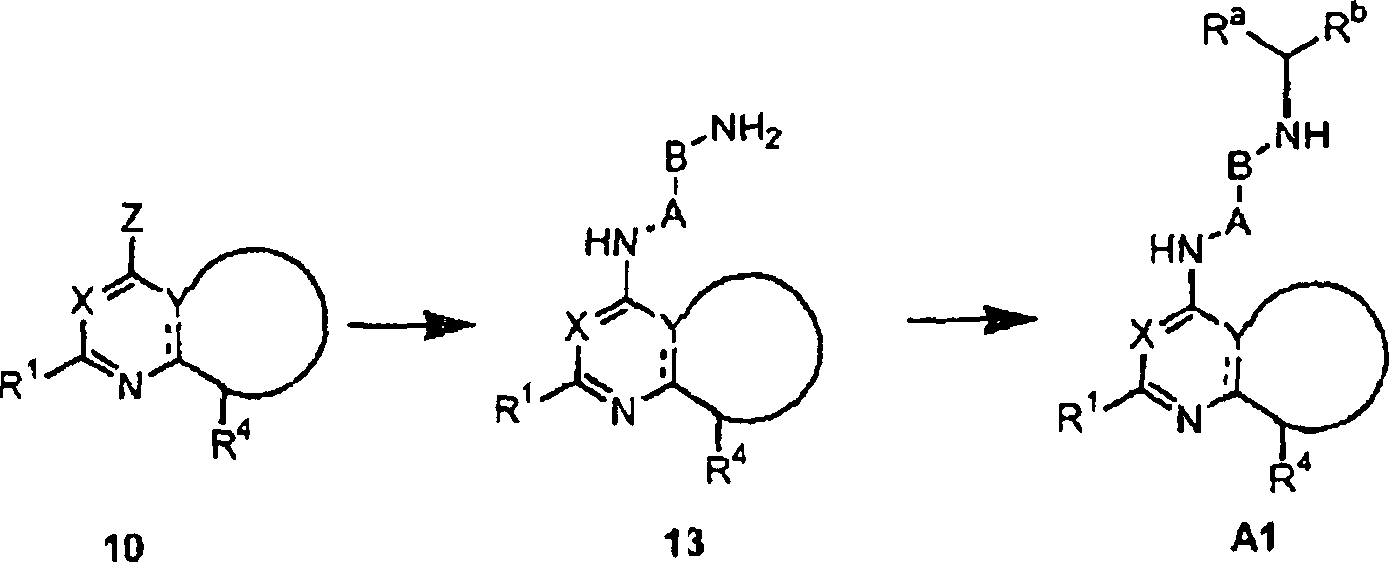
Untergruppe von Verbindungen der Formel A, beschrieben unter Formel
A1 (Schema 3), kann durch zuerst Umsetzen einer Verbindung der Formel
10 mit einem Diamin der Formel H2N-A-B-NH2, worin A und B wie vorstehend definiert
sind, in Gegenwart oder Abwesenheit einer Base, in Gegenwart oder
Abwesenheit eines inerten Lösungsmittels,
bei Reaktionstemperaturen im Bereich von –78°C bis 250°C erhalten werden, um Zwischenprodukte
der Formel 13 zu erzeugen. Die Reaktion einer Verbindung der Formel
13 mit einem Keton der Formel Ra-C=O-Rb in Gegenwart eines Reduktionsmittels liefert
eine Verbindung der Formel A1, worin die Gruppierung Ra-CH-Rb R5 in Formel A,
wie vorstehend definiert, entspricht. Reduktionsmittel schließen Alkalimetall-
oder Erdalkalimetallborhydride (vorzugsweise Lithium- oder Natriumborhydrid),
Boran (vorzugsweise komplexiert mit Dimethylsulfid oder Tetrahydrofuran),
Dialkylborane (wie Diisoamylboran), Alkalimetallaluminiumhydride
(vorzugsweise Lithiumaluminiumhydrid), Alkalimetall(trialkoxy)aluminiumhydride (wie
Triethoxyaluminiumhydrid), Dialkylaluminiumhydride (wie Diisobutylaluminiumhydrid),
Alan (vorzugsweise komplexiert mit Dimethylamin) ein, sind jedoch
nicht darauf begrenzt. Inerte Lösungsmittel
können
Alkylalkohole (1–6
Kohlenstoffatome) (vorzugsweise Methanol, Ethanol oder tert-Butanol), Dialkylether
(vorzugsweise Diethylether), cyclische Ether (vorzugsweise Tetrahydrofuran
oder 1,4-Dioxan), aromatische Kohlenwasserstoffe (vorzugsweise Benzol
oder Toluol) einschließen,
sind jedoch nicht darauf begrenzt. Bevorzugte Reaktionstemperaturen
liegen im Bereich von –78°C bis 100°C.
-
-
Alternativ
kann eine Untergruppe von Verbindungen der Formel A, beschrieben
unter Formel A2 (Schema 4), durch zuerst Umsetzen einer Verbindung
der Formel 13 mit einer aktivierten Säure der Formel Rc-C=O-Z,
worin Z Halogen (vorzugsweise Chlor), O-Acyl (vorzugsweise O-C=O-Rc) darstellt, in Gegenwart oder Abwesenheit
einer Base, in Gegenwart oder Abwesenheit eines inerten Lösungsmittels,
bei Reaktionstemperaturen im Bereich von –78°C bis 250°C erhalten werden, um ein Amidzwi schenprodukt
der Formel 14 zu erzeugen. Die Reaktion einer Verbindung der Formel
14 mit einem Reduktionsmittel liefert eine Verbindung der Formel
A2, worin die Gruppierung Rc-CH2 R5 in Formel A, wie vorstehend definiert,
entspricht. Reduktionsmittel schließen Alkalimetall- oder Erdalkalimetallborhydride
(vorzugsweise Lithium- oder Natriumborhydrid), Boran (vorzugsweise
komplexiert mit Dimethylsulfid oder Tetrahydrofuran), Dialkylborane
(wie Diisoamylboran), Alkalimetallaluminiumhydride (vorzugsweise
Lithiumaluminiumhydrid), Alkalimetall(trialkoxy)aluminiumhydride
(wie Triethoxyaluminiumhydrid), Dialkylaluminiumhydride (wie Diisobutylaluminiumhydrid),
Alan (vorzugsweise komplexiert mit Dimethylamin) ein, sind jedoch
nicht darauf begrenzt. Inerte Lösungsmittel
können Alkylalkohole
(1–6 Kohlenstoffatome)
(vorzugsweise Methanol, Ethanol oder tert-Butanol), Dialkylether
(vorzugsweise Diethylether), cyclische Ether (vorzugsweise Tetrahydrofuran
oder 1,4-Dioxan), aromatische Kohlenwasserstoffe (vorzugsweise Benzol
oder Toluol) einschließen,
sind jedoch nicht darauf begrenzt. Bevorzugte Reaktionstemperaturen
liegen im Bereich von –78°C bis 100°C.
-
-
Alternativ
kann eine Untergruppe von Verbindungen der Formel A, beschrieben
unter Formel A3 (Schema 5), durch zuerst Umsetzen einer Verbindung
der Formel 10 mit einem Amin der Formel H2N-A-CH(ORc)(ORd), worin A
vorstehend definiert ist und Rc und Rd C1-C6-Niederalkyle
darstellen, oder, zusammengenommen, eine Ketalgruppe, wie beispielsweise
eine Dioxan- oder Dioxolangruppe, vervollständigen, in Gegenwart oder Abwesenheit
einer Base, in Gegenwart oder Abwesenheit eines inerten Lösungsmittels,
bei Reaktionstemperaturen im Bereich von –78°C bis 250°C erhalten werden, um Verbindungen
der Formel 15 zu erzeugen. Basen können Alkalimetallhydride (vorzugsweise
Natriumhydrid), Alkalimetallalkoxide (1–6 Kohlenstoffatome) (vorzugsweise
Natriummethoxid, Natriumethoxid oder Natrium-tert-butoxid), Erdalkalimetallhydride,
Alkalimetalldialkylamide (vorzugsweise Lithiumdiisopropylamid),
Alkalimetallcarbonate, Alkalimetallbicarbonate, Alkalimetall-bis-(trialkylsilyl)amide
(vorzugsweise Lithium- oder Natrium(trimethylsilyl)amid), Trialkylamine
(vorzugsweise N,N-Diisopropyl-N-ethylamin oder Triethylamin), Arylamine
(vorzugsweise 4-Dimethylanilin), oder heteroaromatische Amine (vorzugsweise
Pyridin) einschließen,
sind jedoch nicht darauf begrenzt. Inerte Lösungsmittel können Alkylalkohole
(1–8 Kohlenstoffatome)
(vorzugsweise Methanol, Ethanol oder tert-Butanol), Niederalkannitrile
(1–6 Kohlenstoffatome)
(vorzugsweise Acetonitril), Dialkylether (vorzugsweise Diethylether),
cyclische Ether (vorzugsweise Tetrahydrofuran oder 1,4-Dioxan),
N,N-Dialkylformamide (vorzugsweise Dimethylformamid), N,N-Dialkylacetamide
(vorzugsweise Dimethylacetamid), cyclische Amide (vorzugsweise N-Methylpyrrolidin-2-on), Dialkylsulfoxide
(vorzugsweise Dimethylsulfoxid), aromatische Kohlenwasserstoffe
(vorzugsweise Benzol oder Toluol), oder Halogenalkane (1–10 Kohlenstoffatome und
1–10 Halogenatome)
(vorzugsweise CH2Cl2)
einschließen,
sind jedoch nicht darauf begrenzt. Verbindungen der Formel 15 reagieren
mit einer protischen Säure
in Gegenwart oder Abwesenheit eines inerten Lösungsmittels bei Reaktionstemperaturen
im Bereich von –78°C bis 250°C, gefolgt
von wässriger
Aufarbeitung, um Verbindungen der Formel 16 zu erzeugen. Inerte
Lösungsmittel
können
Dial kylether (vorzugsweise Diethylether), cyclische Ether (vorzugsweise
Tetrahydrofuran oder 1,4-Dioxan), N,N-Dialkylformamide (vorzugsweise
Dimethylformamid), N,N-Dialkylacetamide (vorzugsweise Dimethylacetamid),
cyclische Amide (vorzugsweise N-Methylpyrrolidin-2-on), Dialkylsulfoxide
(vorzugsweise Dimethylsulfoxid), aromatische Kohlenwasserstoffe
(vorzugsweise Benzol oder Toluol), oder Halogenalkane (1–10 Kohlenstoffatome
und 1–10
Halogenatome) (vorzugsweise CH2Cl2) einschließen, sind jedoch nicht darauf
begrenzt. Protische Säuren
schließen
Ameisensäure,
Essigsäure,
Trifluoressigsäure,
Salzsäure,
Methansulfonsäure
ein, sind jedoch nicht darauf begrenzt. Alternativ können Verbindungen
der Formel 16 durch Oxidation von Verbindungen der Formel 11, worin
B= CH2, erhalten werden. Oxidationsmittel
schließen Übergangsmetalloxide,
wie CrO3 oder MnO2, Pyridin-Chrom-Komplexe,
wie CrO3.C5H5N, Pyridiniumdichromat oder Pyridiniumchlorochromat,
oder ein Oxalylchlorid-DMSO-Triethylamin-Reagenz (Swern-Oxidation)
ein, sind jedoch nicht darauf begrenzt. Verbindungen der Formel
16 reagieren mit Aminen der Formel H2N-R5, worin R5 vorstehend
definiert ist, in Gegenwart eines Reduktionsmittels, in Gegenwart
oder Abwesenheit eines inerten Lösungsmittels,
in Gegenwart oder Abwesenheit einer protischen Säure, bei Temperaturen im Bereich
von –78°C bis 100°C, unter
Gewinnung von Verbindungen der Formel A3. Reduktionsmittel schließen Alkalimetall-
oder Erdalkalimetallborhydride (vorzugsweise Lithium- oder Natriumborhydrid),
Boran (vorzugsweise komplexiert mit Dimethylsulfid oder Tetrahydrofuran),
Dialkylborane (wie Diisoamylboran), Alkalimetallaluminiumhydride
(vorzugsweise Lithiumaluminiumhydrid), Alkalimetall(trialkoxy)aluminiumhydride
(wie Triethoxyaluminiumhydrid), Dialkylaluminiumhydride (wie Diisobutylaluminiumhydrid),
Alan (vorzugsweise komplexiert mit Dimethylamin) ein, sind jedoch
nicht darauf begrenzt. Inerte Lösungsmittel
können
Alkylalkohole (1–6
Kohlenstoffatome) (vorzugsweise Methanol, Ethanol oder tert-Butanol),
Dialkylether (vorzugsweise Diethylether), cyclische Ether (vorzugsweise
Tetrahydrofuran oder 1,4-Dioxan), aromatische Kohlenwasserstoffe
(vorzugsweise Benzol oder Toluol) einschließen, sind jedoch nicht darauf
begrenzt.
-
Verbindungen
der Formel 10, die heterocyclische Kerne der Formel I–XV umfassen,
werden gemäß den in
den nachstehend angeführten
Literaturstellen beschriebenen Verfahren hergestellt.
-
Verbindungen
der Formel 10a, die als Ausgangsmaterial für die Herstellung von Verbindungen
der Formel I verwendet werden, können
gemäß Schema
6 erhalten werden.
-
-
Verbindungen
der Formel 17, worin E, F und G vorstehend definiert sind und Z
Halogen darstellt, reagieren mit einer Verbindung der Formel R4-CH2-CN, worin R4 vorstehend definiert ist, in Gegenwart
von Base, in Gegenwart oder Abwesenheit eines inerten Lösungsmittels,
bei Reaktionstemperaturen im Bereich von –78°C bis 100°C, um Verbindungen der Formel
18 zu erzeugen. Basen können
Alkalimetallhydride (vorzugsweise Natriumhydrid), Alkalimetallalkoxide
(1–6 Kohlenstoffatome)
(vorzugsweise Natriummethoxid, Natriumethoxid oder Natrium tert-butoxid),
Erdalkalimetallhydride, Alkalimetalldialkylamide (vorzugsweise Lithiumdiisopropylamid),
Alkalimetallcarbonate, Alkalimetallbicarbonate, Alkalimetall-bis-(trialkylsilyl)amide
(vorzugsweise Lithium- oder Natrium(trimethylsilyl)amid), Trialkylamine
(vorzugsweise N,N-Diisopropyl-N-ethylamin oder Triethylamin), Arylamine
(vorzugsweise 4-Dimethylanilin), oder heteroaromatische Amine (vorzugsweise Pyridin)
einschließen,
sind jedoch nicht darauf begrenzt. Inerte Lösungsmittel können Alkylalkohole
(1–8 Kohlenstoffatome)
(vorzugsweise Methanol, Ethanol oder tert-Butanol), Dialkylether
(vorzugsweise Diethylether), cyclische Ether (vorzugsweise Tetrahydrofuran
oder 1,4-Dioxan), oder Dialkylsulfoxide (vorzugsweise Dimethylsulfoxid)
einschließen,
sind jedoch nicht darauf begrenzt. Bevorzugte Reaktionstemperaturen
liegen im Bereich von 0°C
bis 100°C.
Verbindungen der Formel 18 reagieren dann mit einer Verbindung der
Formel Z-CH2-COORc,
worin Z Halogen (vorzugsweise Chlor oder Brom) darstellt, Alkansulfonyloxy,
Arylsulfonyloxy oder Halogenalkansulfonyloxy darstellt und Rc C1-C6-Alkyl
darstellt, in Gegenwart von Base, in Gegenwart oder Abwesenheit
eines inerten Lösungsmittels,
bei Reaktionstemperaturen im Bereich von –78°C bis 100°C, um Verbindungen der Formel
19 zu erzeugen. Basen können
Alkalimetallhydride (vorzugsweise Natriumhydrid), Alkalimetallalkoxide
(1–6 Kohlenstoffatome)
(vorzugsweise Natriummethoxid, Natriumethoxid oder Natrium-tert-butoxid),
Erdalkalimetallhydride, Alkalimetalldialkylamide (vorzugsweise Lithiumdiisopropylamid),
Alkalimetallcarbonate, Alkalimetallbicarbonate, Alkalimetall-bis-(trialkylsilyl)amide
(vorzugsweise Lithium- oder Natrium(trimethylsilyl)amid), Trialkylamine
(vorzugsweise N,N-Diisopropyl-N-ethylamin
oder Triethylamin), Arylamine (vorzugsweise 4-Dimethylanilin), oder
heteroaromatische Amine (vorzugsweise Pyridin) einschließen, sind jedoch nicht darauf begrenzt. Inerte Lösungsmittel
können
Alkylalkohole (1–8
Kohlenstoffatome) (vorzugsweise Methanol, Ethanol oder tert-Butanol), Dialkylether
vorzugsweise Diethylether), cyclische Ether (vorzugsweise Tetrahydrofuran
oder 1,4-Dioxan), oder Dialkylsulfoxide (vorzugsweise Dimethylsulfoxid)
einschließen,
sind jedoch nicht darauf begrenzt. Bevorzugte Reaktionstemperaturen
liegen im Bereich von 0°C bis
100°C. Verbindungen
der Formel 20, worin X CR14 darstellt und
R1 vorstehend definiert ist, können in
zwei aufeinander folgenden Schritten aus Verbindungen der Formel
19 erhalten werden, und können
zuerst mit einer Verbindung der Formel R1C(ORc)-CH2-R14,
worin Rc Methyl oder Ethyl darstellt, in
Gegenwart von Base, in Gegenwart oder Abwesenheit eines inerten
Lösungsmittels,
bei Reaktionstemperaturen im Bereich von –78°C bis 100°C, umgesetzt werden, um ein
nicht isoliertes Iminzwischenprodukt zu erzeugen. Basen können Alkalimetallhydride
(vorzugsweise Natriumhydrid), Alkalimetallalkoxide (1–6 Kohlenstoffatome)
(vorzugsweise Natriummethoxid, Natriumethoxid oder Natrium-tert-butoxid),
Erdalkalimetallhydride, Alkalimetalldialkylamide (vorzugsweise Lithiumdiisopropylamid),
Alkalimetallcarbonate, Alkalimetallbicarbonate, Alkalimetall-bis-(trialkylsilyl)amide
(vorzugsweise Lithium- oder Natrium(trimethylsilyl)amid), Trialkylamine
(vorzugsweise N,N-Diisopropyl-N-ethylamin oder Triethylamin), Arylamine
(vorzugsweise 4-Dimethylanilin), oder heteroaromatische Amine (vorzugsweise
Pyridin) einschließen,
sind jedoch nicht darauf begrenzt. Inerte Lösungsmittel können Alkylalkohole
(1–8 Kohlenstoffatome)
(vorzugsweise Methanol, Ethanol oder tert-Butanol), Dialkylether
(vorzugsweise Diethylether), cyclische Ether (vorzugsweise Tetrahydrofuran
oder 1,4-Dioxan), oder Dialkylsulfoxide (vorzugsweise Dimethylsulfoxid)
einschließen,
sind jedoch nicht darauf begrenzt. Bevorzugte Reaktionstemperaturen
liegen im Bereich von 0°C
bis 100°C.
In einem zweiten Schritt werden die Iminzwischenprodukte zu einer
Verbindung der Formel 20 (X = CR14) in Gegenwart
von Base, in Gegenwart oder Abwesenheit eines inerten Lösungsmittels,
bei Reaktionstemperaturen im Bereich von –78°C bis 100°C cyclisiert. Basen können Alkalimetallhydride
(vorzugsweise Natriumhydrid), Alkalimetallalkoxide (1–6 Kohlenstoffatome)
(vorzugs weise Natriummethoxid, Natriumethoxid oder Natrium-tert-butoxid),
Erdalkalimetallhydride, Alkalimetalldialkylamide (vorzugsweise Lithiumdiisopropylamid),
Alkalimetallcarbonate, Alkalimetallbicarbonate, Alkalimetall-bis-(trialkylsilyl)amide
(vorzugsweise Lithium- oder Natrium(trimethylsilyl)amid), Trialkylamine
(vorzugsweise N,N-Diisopropyl-N-ethylamin oder Triethylamin), Arylamine
(vorzugsweise 4-Dimethylanilin), oder heteroaromatische Amine (vorzugsweise
Pyridin) einschließen,
sind jedoch nicht darauf begrenzt. Inerte Lösungsmittel können Alkylalkohole
(1–8 Kohlenstoffatome)
(vorzugsweise Methanol, Ethanol oder tert-Butanol), Dialkylether
(vorzugsweise Diethylether), cyclische Ether (vorzugsweise Tetrahydrofuran
oder 1,4-Dioxan), oder Dialkylsulfoxide (vorzugsweise Dimethylsulfoxid)
einschließen,
sind jedoch nicht darauf begrenzt. Bevorzugte Reaktionstemperaturen
liegen im Bereich von 0°C
bis 100°C.
-
Verbindungen
der Formel 20, worin X N darstellt und R1 vorstehend
definiert ist, können
durch Umsetzen von Verbindungen der Formel 19 mit einer Verbindung
der Formel R1-C=O-Rc,
worin Rc Halogen, Cyano, Niederalkoxy (1–6 Kohlenstoffatome)
oder Niederalkanoyloxy (1–6
Kohlenstoffatome) darstellt, in Gegenwart einer Base in einem inerten
Lösungsmittel
bei Reaktionstemperaturen im Bereich von –78°C bis 200°C erhalten werden. Basen können Alkalimetallhydride
(vorzugsweise Natriumhydrid), Alkalimetallalkoxide (1–6 Kohlenstoffatome)
(vorzugsweise Natriummethoxid, Natriumethoxid oder Natrium-tert-butoxid),
Erdalkalimetallhydride, Alkalimetalldialkylamide (vorzugsweise Lithiumdiisopropylamid),
Alkalimetallcarbonate, Alkalimetallhydroxide, Alkalimetall-bis-(trialkylsilyl)amide
(vorzugsweise Lithium- oder Natrium(trimethylsilyl)amid), Trialkylamine
(vorzugsweise N,N-Diisopropyl-N-ethylamin oder Triethylamin), bicyclische
Amidine (vorzugsweise DBU), oder heteroaromatische Amine (vorzugsweise
Pyridin) einschließen,
sind jedoch nicht darauf begrenzt. Inerte Lösungsmittel können Alkylalkohole
(1–8 Kohlenstoffatome)
(vorzugsweise Methanol, Ethanol oder tert-Butanol), Niederalkannitrile
(1–6 Kohlenstoffatome)
(vorzugswe,ise Acetonitril), Dialkylether (vorzugsweise Diethylether),
cyclische Ether (vorzugsweise Tetrahydrofuran oder 1,4-Dioxan),
N,N-Dialkylformamide (vorzugsweise Dimethylformamid), N,N-Dialkylacetamide
(vorzugsweise Dimethylacetamid), cyclische Amide (vorzugsweise N-Methylpyrrolidin-2-on), Dialkylsulfoxide
(vorzugsweise Dimethylsulfoxid), aromatische Kohlenwasserstoffe
(vorzugsweise Benzol oder Toluol) einschließen, sind jedoch nicht darauf
begrenzt. Verbindungen der Formel 10a, worin Z Halogen, Alkansulfonyloxy,
Arylsulfonyloxy oder Halogenalkansulfonyloxy darstellt und X N darstellt,
können
durch Umsetzen einer Verbindung der Formel 20 mit einem Halogenierungsmittel
oder Sulfonylierungsmittel, in Gegenwart oder Abwesenheit einer
Base, in Gegenwart oder Abwesenheit eines inerten Lösungsmittels,
bei Reaktionstemperaturen im Bereich von –78°C bis 250°C hergestellt werden. Halogenierungsmittel
schließen
SOCl2, POCl3, PCl3, PCl5, POBr3, PBr3 oder PBr5 ein, sind jedoch nicht darauf begrenzt.
Sulfonylierungsmittel schließen
Alkansulfonylhalogenide oder -anhydride (vorzugsweise Methansulfonylchlorid
oder Methansulfonsäureanhydrid),
Arylsulfonylhalogenide oder -anhydride (wie p-Toluolsulfonylchlorid
oder -anhydrid), oder Halogenalkylsulfonylhalogenide oder -anhydride
(vorzugsweise Trifluormethansulfonsäureanhydrid) ein, sind jedoch
nicht darauf begrenzt. Basen können
Trialkylamine (vorzugsweise N,N-Diisopropyl-N-ethylamin oder Triethylamin),
bicyclische Amidine (vorzugsweise DBU), Aniline (vorzugsweise N-Dimethylanilin),
oder heteroaromatische Amine (vorzugsweise Pyridin) einschließen, sind
jedoch nicht darauf begrenzt. Inerte Lösungsmittel können Niederalkannitrile
(1–6 Kohlenstoffatome)
(vorzugsweise Acetonitril), Dialkylether (vorzugsweise Diethylether),
cyclische Ether (vorzugsweise Tetrahydrofuran oder 1,4-Dioxan),
N,N-Dialkylformamide
(vorzugsweise Dimethylformamid), N,N-Dialkylacetamide (vorzugsweise
Dimethylacetamid), cyclische Amide (vorzugsweise N-Methylpyrrolidin-2-on),
Dialkylsulfoxide (vor zugsweise Dimethylsulfoxid), aromatische Kohlenwasserstoffe
(vorzugsweise Benzol oder Toluol), oder Halogenalkane mit 1–10 Kohlenstoffatomen
und 1–10
Halogenatomen (vorzugsweise CH2Cl2) einschließen, sind jedoch nicht darauf
begrenzt. Bevorzugte Reaktionstemperaturen liegen im Bereich von –20°C bis 100°C.
-
Alternativ
können
einige der Verbindungen der Formel A aus Verbindungen der Formel
21, wie in Schema 7 erläutert,
erhalten werden.
-
-
Verbindungen
der Formel 21, worin X, R1 und R4 vorstehend definiert sind, reagieren mit
einer Verbindung der Formel Z-B-(C=O)-Rc,
worin B vorstehend definiert ist, Z Halogen (vorzugsweise Chlor
oder Brom), Alkansulfonyloxy, Arylsulfonyloxy oder Halogenalkansulfonyloxy
darstellt und Rc Halogen, Cyano, Niederalkoxy
(1–6 Kohlenstoffatome)
oder Niederalkanoyloxy (1–6
Kohlenstoffatome) darstellt, in Gegenwart oder Abwesenheit einer
Base, in Gegenwart oder Abwesenheit eines inerten Lösungsmittels,
bei Reaktionstemperaturen im Bereich von –78°C bis 200°C, zur Herstellung von Verbindungen
der Formel 22. Basen können
Trialkylamine (vorzugsweise N,N-Diisopropyl-N-ethylamin oder Triethylamin), bicyclische
Amidine (vorzugswei se DBU), Aniline (vorzugsweise N-Dimethylanilin),
oder heteroaromatische Amine (vorzugsweise Pyridin) einschließen, sind
jedoch nicht darauf begrenzt. Inerte Lösungsmittel können Niederalkannitrile
(1–6 Kohlenstoffatome)
(vorzugsweise Acetonitril), Dialkylether (vorzugsweise Diethylether),
cyclische Ether (vorzugsweise Tetrahydrofuran oder 1,4-Dioxan),
N,N-Dialkylformamide (vorzugsweise Dimethylformamid), N,N-Dialkylacetamide
(vorzugsweise Dimethylacetamid), cyclische Amide (vorzugsweise N-Methylpyrrolidin-2-on),
Dialkylsulfoxide (vorzugsweise Dimethylsulfoxid), aromatische Kohlenwasserstoffe
(vorzugsweise Benzol oder Toluol), oder Halogenalkane mit 1–10 Kohlenstoffatomen
und 1–10
Halogenatomen (vorzugsweise CH2Cl2) einschließen, sind jedoch nicht darauf
begrenzt. Bevorzugte Reaktionstemperaturen liegen im Bereich von –20°C bis 100°C.
-
Amide
der Formel 22 können
mit einem Amin der Formel NH[R6]R5, worin R6 und R5 vorstehend definiert sind, in Gegenwart
oder Abwesenheit einer Base, in Gegenwart oder Abwesenheit eines
inerten Lösungsmittels,
bei Reaktionstemperaturen im Bereich von –78°C bis 200°C, umgesetzt werden, um Verbindungen
der Formel 23 herzustellen. Basen können Alkalimetallhydride (vorzugsweise
Natriumhydrid), Alkalimetallalkoxide (1–6 Kohlenstoffatome) (vorzugsweise
Natriummethoxid, Natriumethoxid oder Natrium-tert-butoxid), Erdalkalimetallhydride,
Alkalimetalldialkylamide (vorzugsweise Lithiumdiisopropylamid),
Alkalimetallcarbonate, Alkalimetallbicarbonate, Alkalimetall-bis-(trialkylsilyl)amide
(vorzugsweise Lithium- oder Natrium(trimethylsilyl)amid), Trialkylamine
(vorzugsweise N,N-Diisopropyl-N-ethylamin
oder Triethylamin), Arylamine (vorzugsweise 4-Dimethylanilin) oder
heteroaromatische Amine (vorzugsweise Pyridin) einschließen, sind
jedoch nicht darauf begrenzt. Inerte Lösungsmittel können Alkylalkohole
(1–8 Kohlenstoffatome)
(vorzugsweise Methanol, Ethanol oder tert-Butanol), Niederalkannitrile
(1–6 Kohlenstoffatome)
(vorzugsweise Acetonitril), Dialkylether (vorzugsweise Diethylether),
cyclische Ether (vorzugsweise Tetrahydrofuran oder 1,4-Dioxan), N,N-Dialkylformamide
(vorzugsweise Dimethylformamid), N,N-Dialkylacetamide (vorzugsweise
Dimethylacetamid), cyclische Amide (vorzugsweise N-Methylpyrrolidin-2-on),
Dialkylsulfoxide (vorzugsweise Dimethylsulfoxid), aromatische Kohlenwasserstoffe
(vorzugsweise Benzol oder Toluol), oder Halogenalkane (1–10 Kohlenstoffatome und
1–10 Halogenatome)
(vorzugsweise CH2Cl2)
einschließen,
sind jedoch nicht darauf begrenzt. Bevorzugte Reaktionstemperaturen
liegen im Bereich von 0°C
bis 140°C.
-
Verbindungen
der Formel 23 können
mit einem Reduktionsmittel, in Gegenwart oder Abwesenheit eines
inerten Lösungsmittels,
unter Bereitstellung von Verbindungen der Formel A, wie vorstehend
definiert, umgesetzt werden. Reduktionsmittel schließen Alkalimetall-
oder Erdalkalimetallborhydride (vorzugsweise Lithium- oder Natriumborhydrid),
Boran (vorzugsweise komplexiert mit Dimethylsulfid oder Tetrahydrofuran),
Dialkylborane (wie Di-isoamylboran), Alkalimetallaluminiumhydride
(vorzugsweise Lithiumaluminiumhydrid), Alkalimetall(trialkoxy)aluminiumhydride
(wie Triethoxyaluminiumhydrid), Dialkylaluminiumhydride (wie Di-isobutylaluminiumhydrid),
Alan (vorzugsweise komplexiert mit Dimethylamin) ein, sind jedoch
nicht darauf begrenzt. Inerte Lösungsmittel
können
Alkylalkohole (1–6
Kohlenstoffatome) (vorzugsweise Methanol, Ethanol oder tert-Butanol), Dialkylether
(vorzugsweise Diethylether), cyclische Ether (vorzugsweise Tetrahydrofuran
oder 1,4-Dioxan), aromatische Kohlenwasserstoffe (vorzugsweise Benzol
oder Toluol) einschließen,
sind jedoch nicht darauf begrenzt. Bevorzugte Reaktionstemperaturen
liegen im Bereich von –78°C bis 100°C.
-
Alternativ
können
Verbindungen der Formel 22 zuerst unter experimentellen Bedingungen,
die ähnlich zu
jenen für
die Umwandlung von Verbindungen der Formel 23 zu Verbindungen der
Formel A sind, reduziert werden, um Verbindungen der Formel 24 zu
erzeugen. Verbindungen der Formel 24 können dann mit Verbindungen
der Formel NH[R6]R5,
wie vorstehend definiert, unter experimentellen Bedingungen, die ähnlich zu
jenen, die für
die Umwandlung von Verbindungen der Formel 22 zu Verbindungen der Formel
23 sind, umgesetzt werden, um Verbindungen der Formel A zu erzeugen.
-
Andere
Verbindungen der Formel 10 und der Formel 21, die als Ausgangsmaterial
für die
Herstellung von Verbindungen der Formel V verwendet werden, können gemäß Verfahren
erhalten werden, die in der nachstehenden Literaturstelle beschrieben
werden:
Formel V: WO 9533750, J. Med. Chem. 42(5), 833–848 (1999);
Bioorg. Med. Chem. Lett., 9(7), 967–972 (1999).
-
BEISPIELE
-
Die
nachstehenden Beispiele, die zur weiteren Erläuterung der Reaktionsschemata
bereitgestellt werden, sind nicht als die Erfindung begrenzend aufzufassen.
-
Die
Herstellung der erfindungsgemäßen Verbindungen
durch die vorstehend erwähnten
Verfahren wird weiterhin durch die nachstehenden Beispiele erläutert, und
jene, die in den Tabellen geschildert werden, welche nicht als die
Erfindung in Umfang oder Gedanken bezüglich der spezifischen Verfahren
und in ihnen beschriebenen Verbindungen begrenzend aufzufassen sind.
Handelsübliche
Reagenzien werden ohne weitere Reinigung verwendet. THF bezieht
sich auf Tetrahydrofuran. LDA bezieht sich auf Lithiumdiisopropylamid und
DBU bezieht sich auf 1,8-Diazabicyc-lo[5.4.0]undec-7-en.
Raum- oder Umgebungstemperatur bezieht sich auf 20°C bis 25°C. Aufkonzentrierung
impliziert die Anwendung eines Rotationsverdampfers. DC bezieht sich
auf Dünnschicht-Chromatographie.
Massenspektraldaten wurden entweder durch CI- oder APCI-Verfahren
erhalten. Üblicherweise
verwendete Abkürzungen
sind: Ph ist Phenyl, Me ist Methyl, Et ist Ethyl, Pr ist n-Propyl,
iPr ist Isopropyl, Bu ist Butyl, iBu ist Isobutyl (CH2-CHMe2), tBu ist tert-Butyl, cBu ist Cyclobutyl,
Pent ist n-Pentyl, cPent ist Cyclopentyl, cHex ist Cyclohexyl, Py
ist Pyridyl, MeOH bedeutet Methanol, EtOH bedeutet Ethanol, EtOAc
bedeutet Essigsäureethylester,
Et2O bedeutet Diethylether, CH2Cl2 bedeutet Methylenchlorid, DMSO bedeutet Dimethylsulfoxid,
NMP bedeutet N-Methylpyrrolidon, THF bedeutet Tetrahydrofuran, DMF bedeutet
Dimethylformamid, BSP bedeutet Beispiel. Die speziellen heterocyclischen
Kerne, die generisch unter Formel A beschrieben werden, sind in
der Tabelle unter Formel ausgewiesen, was sich auf den in Formel V
definierten heterocyclischen Kern bezieht.
-
BEISPIEL 5
-
A.
4-Chlor-6-methyl-3-nitro-pyridin-2-ol
-
Man
gibt 1-Ethylpropylamin (9,00 g, 76,4 mMol) zu einer Lösung von
4-Hydroxy-6-methyl-3-nitropyridon (10,0 g, 58,8 mMol) in wasserfreiem
MeOH (20 ml). Man rührt
das Gemisch, bis es klar ist, dann konzentriert man im Vakuum auf.
Man gibt den Rückstand
portionsweise zu Phosphoroxychlorid (70 ml). Man rührt bei
Umgebungstemperatur für
10 h und gießt
auf Eiswasser. Nach Rühren
für 20
min filtriert man den Niederschlag und trocknet über Na2SO4, unter Gewinnung der Titelverbindung als
einen gelben Feststoff.
-
B.
4-Benzylamino-6-methyl-3-nitro-pyridin-2-ol
-
Man
rührt eine
Lösung
von 4-Chlor-6-methyl-3-nitropyridin-2-ol (10,0 g, 53,0 mMol) und
Benzylamin (17,3 g, 159 mMol) in EtOH (30 ml) bei Umgebungstemperatur
für 12
h, dann konzentriert man im Vakuum auf. Man gießt den erhaltenen Rückstand
in Wasser, säuert
mit 1N HCl auf pH 3 an und filtriert.
-
Man
wäscht
den Niederschlag einige Male mit Wasser, um die Titelverbindung
als einen gelben Feststoff zu erhalten.
-
C.
4-Benzylamino-6-methyl-3-nitro-2-chlor-pyridin
-
Man
erhitzt eine Lösung
von 4-Benzylamino-6-methyl-3-nitro-pyridin-2-ol
(13,70 g, 52,8 mMol) und Tetramethylammoniumchlorid (6,0 g, 52,8
mMol) unter Rückfluss
in Phosphoroxychlorid (15 ml) für
5 h. Nach Kühlen
entfernt man das überschüssige Phosphoroxychlorid
im Vakuum. Man verreibt den Rückstand
in Eiswasser (400 ml), unter Gewinnung der Titelverbindung als einen
braunen Feststoff.
-
D.
6-Methyl-3-nitro-4-[benzylamino](2-pyridyl)}(2,4,6-trimethylphenyl)amin
-
Man
gibt p-Toluolsulfonsäure
(11,0 g, 56,3 mMol) zu einer Lösung
von 4-Benzylamino-6-methyl-3-nitro-2-chlor-pyridin (13,0 g, 46,8
mMol) und 2,4,6-Trimethylanilin (6,58 g, 46,8 mMol) in 350 ml Toluol.
Man erhitzt das erhaltene Gemisch für 14 h unter Rückfluss,
unter Verwendung einer Dean-Stark-Falle. Nach Kühlen auf Umgebungstemperatur
verdünnt
man die Lösung
mit EtOAc (200 ml) und wäscht
nacheinander mit gesättigter
wässriger
NaHCO3 und gesättigter wässriger NaCl. Man trennt die
organische Schicht ab, trocknet über Na2SO4, fil triert und
konzentriert im Vakuum auf. Man reinigt durch Flash-Säulenchromatographie
(2% Methanol in CH2Cl2),
unter Gewinnung der Titelverbindung als einen dunkelgelben Feststoff.
-
E.
3-Amino-6-methyl-4-[benzylamino](2-pyridyl)(2,4,6-trimethylphenyl)amin
-
Man
gibt Eisenpulver (7,0 g) zu einer Lösung von 6-Methyl-3-nitro-4-[benzylamino](2-pyridyl)(2,4,6-trimethylphenyl)amin
(8,75 g, 23,2 mMol) in Essigsäure
(14 ml) und MeOH (70 ml). Man erhitzt das erhaltene Gemisch für 1 h auf
70°C, kühlt auf
Umgebungstemperatur und filtriert durch eine Lage Celite. Man dampft
das Filtrat unter vermindertem Druck zur Trockne ein. Man behandelt
den Rückstand
mit 1N wässriger
NaOH und extrahiert mit EtOAc. Man wäscht den EtOAc-Extrakt mit
Wasser und gesättigter
wässriger
NaCl. Man trennt die organische Schicht ab, trocknet über Na2SO4, filtriert und
konzentriert auf, unter Gewinnung der Titelverbindung als einen
braunen Feststoff, der ohne weitere Reinigung verwendet wird.
-
F.
[5-Methyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4b]pyridin-7-yl]benzylamin
-
Man
behandelt eine Lösung
von 3-Amino-6-methyl-4-[benzylamino](2-pyridyl)(2,4,6-trimethylphenyl)amin
(0,80 g, 2,31 mMol) und Orthoameisensäuretriethylester (8,0 ml, 48,5
mMol) in Dimethylacetamid (9 ml) mit 12 M HCl (0,4 ml). Man rührt das
Gemisch bei Umgebungstemperatur für 14 h, verdünnt mit
EtOAc (50 ml) und wäscht
mit wässriger
NaHCO3 (50 ml) und gesättigter wässriger NaCl (50 ml). Man trocknet
die organische Schicht über
Na2SO4, filtriert
und konzentriert auf. Man reinigt durch präparative DC (10% Methanol in CH2Cl2 mit 0,1% Ammoniumhydroxid),
unter Gewinnung der Titelverbindung als einen Feststoff.
-
G.
5-Methyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-ylamin
-
Man
gibt Palladiumhydroxid (0,05 g) zu einer Lösung von [2,5-Dimethyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-yl]benzylamin
(0,15 g, 0,42 mMol) in Essigsäure
(10 ml). Man hydriert das Gemisch bei einem Druck von 50 psi für 20 h.
Man filtriert das Reaktionsgemisch durch Celite, dampft unter vermindertem Druck
zur Trockne ein, verdünnt
mit EtOAc und wäscht
nacheinander mit wässriger
NaHCO3 und wässriger NaCl. Man trocknet
die organische Schicht über
Na2SO4, filtriert
und konzentriert auf. Man reinigt durch Flash-Säulenchromatographie (3% MeOH
in CH2Cl2), unter
Gewinnung der Titelverbindung als einen braunen Feststoff.
-
H.
N-[5-Methyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-yl]-2-chloracetamid
-
Man
behandelt eine Lösung
von 5-Methyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-ylamin (0,05
g, 1,9 mMol) und N,N-Diisopropylethylamin (0,04 ml, 0,21 mMol) in
1,2-Dichlorethan
(5 ml) mit Chloracetylchlorid (0,017 ml, 0,21 mMol). Man erhitzt
die Lösung
3 h unter Rückfluss,
kühlt auf
Umgebungstemperatur und gießt
in eine wässrige
Kaliumcarbonatlösung.
Man extrahiert das erhaltene Gemisch mit CH2Cl2 und wäscht
mit gesättigter
wässriger
NaCl. Man trennt die organische Schicht ab, trocknet über Na2SO4, filtriert und konzentriert
auf. Man reinigt durch präparative
DC (5% MeOH in CH2Cl2),
unter Gewinnung der Titelverbindung als einen gelben Feststoff.
-
I.
(2-Chlorethyl)[5-methyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-yl]-amin
-
Man
behandelt eine Lösung
von N-[5-Methyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-yl]-2-chloracetamid
(0,04 g, 0,10 mMol) in THF (5 ml) mit Boran-Methylsulfid-Komplex (0,03 ml,
0,30 mMol). Man erhitzt das Gemisch für 8 h unter Rückfluss
und stoppt bei Umgebungstemperatur mit einem großen Überschuss an MeOH, gefolgt
von 6N HCl (2 ml). Man erhitzt erneut das Gemisch für 1 h unter
Rückfluss, konzentriert
dann unter vermindertem Druck auf, unter Gewinnung der Titelverbindung
als einen gelben Feststoff.
-
J.
(2-{[2-(4-Ethoxy-3-methoxyphenyl)ethyl]amino}ethyl)[5-methyl-3-(2,4,6-Lrimethylphenyl)imidazolo[5,4-b]pyridin-7-yl]amin
-
Man
erhitzt eine Lösung
von (2-Chlorethyl)[5-methyl-3-(2,4,6,-trimethylphenyl)imidazolo[5,4-b]pyridin-7-yl]-amin
(0,030 g, 0,09 mMol) und 4-Ethoxy-3-methoxyphenethylamin (0,10 g,
0,55 mMol) in trockenem NMP (2 ml) für 16 h auf 110 °C. Man gießt das gekühlte Gemisch
auf Wasser (20 ml) und extrahiert zweimal mit EtOAc (20 ml). Man
wäscht
die vereinigten Extrakte mit Salzlösung (30 ml), trocknet und
dampft im Vakuum ein. Man reinigt durch Flash-Säulenchromatographie (10% MeOH
in CH2Cl2 mit 0,1%
Ammoniumhydroxid), unter Gewinnung der Titelverbindung als einen
Feststoff.
-
BEISPIEL 6
-
A.
[2,5-Dimethyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-yl]benzylamin
-
Man
erhitzt eine Lösung
von 3-Amino-6-methyl-4-[benzylamino](2-pyridyl)(2,4,6-trimethylphenyl)amin (2,50
g, 7,21 mMol), Orthoessigsäuretriethylester
(2,6 ml, 14,4 mMol) und Camphersulfonsäure (250 mg) in Toluol (50
ml) 14 h unter Rückfluss.
Nach Kühlen
auf Umgebungstemperatur konzentriert man das Gemisch im Vakuum auf,
verdünnt
mit Wasser (100 ml) und extrahiert mit EtOAc. Man wäscht den
EtOAc-Extrakt nacheinander mit Wasser und gesättigter wässriger NaCl, trocknet über Na2SO4, filtriert und
konzentriert auf. Man reinigt durch Flash-Säulenchromatographie (5% Methanol
in CH2Cl2), unter
Gewinnung der Titelverbindung als einen weißen Feststoff.
-
B.
2,5-Dimethyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-ylamin
-
Man
gibt Palladiumhydroxid (2,0 g) zu einer Lösung von [2,5-Dimethyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-yl]benzylamin
(1,50 g, 4,05 mMol) in Essigsäure
(10 ml). Man hydriert das Gemisch bei einem Druck von 40 psi für 20 h. Man
filtriert das Reaktionsgemisch durch Celite, dampft unter vermindertem Druck
zur Trockne ein, verdünnt
mit EtOAc und wäscht
nacheinander mit wässriger
NaHCO3 und wässriger NaCl. Man trocknet
die organische Schicht über
Na2SO4, filtriert
und konzentriert auf. Man reinigt durch Flash-Säulenchromatographie (3% MeOH
in CH2Cl2), unter
Gewinnung der Titelverbindung als einen weißen Feststoff.
-
C.
N-[2,5-Dimethyl-3-(2,4,6-trimethylphenyl)imidazo-lo[5,4-b]pyridin-7-yl]-2-chloracetamid
-
Man
behandelt eine Lösung
von 2,5-Dimethyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-ylamin
(0,8 g, 2,85 mMol) und N,N-Diisopropylethylamin (0,54 ml, 3,13 mMol)
in 1,2-Dichlorethan (10 ml) mit Chloracetylchlorid (0,25 ml, 3,13
mMol). Man erhitzt die Lösung
5 h unter Rückfluss,
kühlt auf
Umgebungstemperatur und gießt
in eine wässrige
Kaliumcarbonatlösung.
Man extrahiert das erhaltene Gemisch mit CH2Cl2 und wäscht
mit gesättigter
wässriger
NaCl. Man trennt die organische Schicht ab, trocknet über Na2SO4, filtriert und konzentriert
auf. Man reinigt durch präparative
DC (5% MeOH in CH2Cl2),
unter Gewinnung der Titelverbindung als einen braunen Feststoff.
-
D.
[2,5-Dimethyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-yl](2-chlorethyl)amin
-
Man
behandelt eine Lösung
von N-[2,5-Dimethyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-yl]-2-chloracetamid
(0,91 g, 2,55 mMol) in THF (5 ml) mit Boran-Methylsulfid-Komplex (0,8 ml,
7,65 mMol). Man erhitzt das Gemisch 18 h unter Rückfluss und stoppt bei Umgebungstemperatur
mit einem großen Überschuss
an MeOH, gefolgt von 6N HCl (5 ml). Man erhitzt das Gemisch erneut
für 1 h
unter Rückfluss,
dann konzentriert man unter vermindertem Druck auf, unter Gewinnung
der Titelverbindung als einen weißen, kristallinen Feststoff.
-
E.
[2,5-Dimethyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-yl][2-(cyclohexylamino)ethyl]amin
-
Man
erhitzt eine Lösung
von [2,5-Dimethyl-3-(2,4,6-trimethylphenyl)imidazolo[5,4-b]pyridin-7-yl](2-chlorethyl)amin
(0,04 g, 0,12 mMol) und Cyclohexylamin (0,13 ml g, 1,16 mMol) in
trockenem NMP (2 ml) bei 80°C
für 20
h. Man gießt
das gekühlte
Gemisch auf Wasser (20 ml) und extrahiert zweimal mit Essigsäureethylester
(20 ml). Man wäscht
die vereinigten Extrakte mit Salzlösung (20 ml), trocknet und
dampft im Vakuum ein. Man reinigt durch Flash-Säulenchromatographie (10% Methanol
in CH2Cl2 mit 0,1%
Ammoniumhydroxid), unter Gewinnung der Titelverbindung als einen
weißen
Feststoff.
-
BEISPIEL 7
-
A.
3-(2,4-Dichlorphenyl)-2,5-dimethylimidazolo[5,4-b]pyridin-7-ylamin
-
Man
rührt eine
Lösung
von [3-(2,4-Dichlorphenyl)-2,5-dimethylimidazolo[5,4-b]pyridin-7-yl](4-methoxybenzyl)amin
(4,23 g, 10 mMol), Anisol (3,2 ml, 29,6 mMol), Trifluoressigsäure (80
ml) und konzentrierter Schwefelsäure
(2 ml) bei Umgebungstemperatur für
14 h. Man gibt das erhaltene Gemisch tropfenweise zu Eiswasser und
verdünnt
mit NaHCO3. Man filtriert den erhaltenen
Niederschlag und wäscht
sorgfältig
mit Wasser, unter Gewinnung der Titelverbindung als einen braunen
Feststoff.
-
B.
N-[3-(2,4-Dichlorphenyl)-2,5-dimethylimidazolo[5,4-b]pyridin-7-yl]-2-chloracetamid
-
Man
behandelt eine Lösung
von 3-(2,4-Dichlorphenyl)-2,5-dimethylimidazolo[5,4-b]pyridin-7-ylamin (0,2
g, 0,65 mMol) und N,N-Diisopropylethylamin (0,12 ml, 0,72 mMol)
in 1,2-Dichlorethan (10 ml) mit Chloracetylchlorid (0,06 ml, 30,72
mMol). Man erhitzt die Lösung
2 h unter Rückfluss,
kühlt auf
Umgebungstemperatur und gießt
in eine wässrige
Kaliumcarbonatlösung.
Man extrahiert das erhaltene Gemisch mit CH2Cl2 und wäscht
mit gesättigter
wässriger
NaCl. Man trennt die organische Schicht ab, trocknet über Na2SO4, filtriert und konzentriert
auf. Man reinigt durch präparative
DC (5% Methanol in CH2Cl2),
unter Gewinnung der Titelverbindung als einen gelben Feststoff.
-
C.
[3-(2,4-Dichlorphenyl)-2,5-dimethylimidazolo[5,4-b]pyridin-7-yl](2-chlorethyl)amin
-
Man
behandelt eine Lösung
von N-[3-(2,4-Dichlorphenyl)-2,5-dimethylimidazolo[5,4-b]pyridin-7-yl]-2-chloracetamid
(1,0 g, 2,6 mMol) in THF (5 ml) mit Boran-Methylsulfid-Komplex (0,8
ml, 7,8 mMol). Man erhitzt das Gemisch 3 h unter Rückfluss
und stoppt bei Umgebungstemperatur mit einem großen Überschuss an MeOH, gefolgt
von 5 N HCl (2 ml) . Man erhitzt das Gemisch erneut für 1 h unter
Rückfluss,
dann konzentriert man unter vermindertem Druck auf. Man reinigt
durch Flash-Säulenchromatographie
(5% EtOAc-Hexan), unter Gewinnung der Titelverbindung als einen
beigefarbenen Feststoff.
-
D.
[3-(2,4-Dichlorphenyl-2,5-dimethylimidazolo[5,4-b]pyridin-7-yl][2-(cyclopentylamino)ethyl]amin
-
Man
erhitzt eine Lösung
von [3-(2,4-Dichlorphenyl)-2,5-dimethylimidazolo[5,4-b]pyridin-7-yl](2-chlorethyl)amin
(0,08 g, 0,22 mMol) und Cyclopentylamin (0,2 ml g, 0,22 mMol) in
trockenem NMP (3 ml) bei 80°C für 20 h.
Man gießt
das gekühlte
Gemisch auf Wasser (30 ml) und extrahiert zweimal mit EtOAc (50
ml). Man wäscht
die vereinigten Extrakte mit Salzlösung (50 ml), trocknet und
dampft im Vakuum ein. Man reinigt durch präparative DC (10% MeOH in CH2Cl2), unter Gewinnung
der Titelverbindung als ein gelbes Öl.
-
-
Beispiel 261 Charakterisierung
von NPY-Rezeptor-Wechselwirkungen
und In-vivo-Funktion
-
A. Assay für humane
NPY1-Rezeptor-Bindungs-Aktivität:
-
Verbindungen
werden auf die Wirksamkeit, unter Anwendung des nachstehenden Verfahrens,
bewertet: Ein cDNA codierendes Human-NPY1 (SEQ ID NR:1) wird in
der geeigneten Orientierung zur Expression in den kommerziellen
Expressionsvektor pBacPAK9 (Clontech, Palo Alto, Kalif.) zur Expression
in Sf9-Zellen ligiert. Jeder baculovirale Expressionsvektor wird
zusammen mit BACULOGOLD DNA (BD PharMingen, San Diego, Kalif.) in
Sf9-Zellen co-transfiziert.
Der Sf9-Zellen-Kulturüberstand
wird drei Tage nach Transfektion geerntet. Der Rekombinantenvirusenthaltende Überstand
wird in Hink's TNM-FH
Insektenmedium (JRH Biosciences, Kansas City), ergänzt mit
Grace's Salzen und
mit 4,1 mM L-Gln, 3,3 g/l LAH, 3,3 g/l ultrafiltriertem Hefestolat
und 10%igem Wärme-inaktiviertem
fötalem
Rinderserum (nachstehend hierin "Insektenmedium") und Plaque, bewertet
auf rekombinante Plaques, seriell verdünnt. Nach vier Tagen werden
rekombinante Plaques ausgewählt
und in 1 ml Insektenmedium zur Verstärkung geerntet. Jeder ml Volumen
von rekombinantem Baculovirus (bei Passage 0) wird verwendet, um
einen getrennten T25 Kolben, enthaltend 2 × 106 Sf9-Zellen
in 5 ml Insektenmedium, zu infizieren. Nach fünf Tagen Inkubation bei 27°C wird das Überstandsmedium
für jede der
T25 Infektionen zur Verwendung als Passage 1 Inokulum geerntet.
Rekombinante baculovirale Klone werden dann einer zweiten Vermehrungsrunde
unterzogen, unter Verwendung von 1 ml Passage, 1 Stamm zu infizieren
von 1 × 108 Zellen in 100 ml Insektenmedium, geteilt
in 2 T175 Kolben. Achtundvierzig Stunden nach Infektion wird Passage
2 Medium für
jeweils 100 ml Prep und Plaque, bewertet für den Titer, geerntet. Das
Zellsediment aus der zweiten Runde der Vermehrung wird auf Affinitätsbinden
von radiomarkiertem Liganden (siehe nachstehend) zum Verifizieren
von rekombinanter Rezeptorexpression, bewertet. Eine dritte Runde
der Vermehrung wird dann gestartet, unter Anwendung von M.O.I. von
0,1, um einen Liter Sf9-Zellen zu infizieren. Vierzig Stunden nach
Infektion wird das Überstandsmedium
geerntet, um Passage 3-baculoviralen
Stamm zu ergeben und das Zellpellet wird auf Affinitätsbinden
bewertet. Der Titer von Passage 3-baculoviralem Stamm wird durch
Plaqueassay und ein M.O.I. bestimmt und Inkubation-Zeit-Verlaufsversuch
(ITC) wird ausgeführt, um
die Bedingungen für
optimale Rezeptorexpression zu bestimmen.
-
Log-Phase
Sf9-Zellen werden mit Stämmen
von rekombinantem Baculovirus, der die jeweiligen Proteine (beispielsweise
humane NPY1- und drei g-Proteine) codiert, infiziert, gefolgt von
Züchten
in Insektenmedium bei 27°C.
72 h nach Infektion wird eine Probe der Zellsuspension auf Lebensfähigkeit
durch Trypan-Blaufarbstoffausschluss analysiert, und die verbleibenden
Sf9-Zellen werden über
Zentrifugierung (3000 U/min/10 Minuten/4°C) geerntet.
-
B. Herstellung von gereinigten
Membranen:
-
Sf9-Zellpellets werden
in homogenisiertem Puffer (10 mM HEPES, 250 mM Saccharose, 0,5 ☐g/ml Leupeptin,
2 ☐g/ml Aprotinin, 200 ☐M PMSF und 2,5 mM EDTA,
pH 7,4) resuspendiert und unter Anwendung eines POLYTRON Homogenisators
(Einstellung 5 für
30 Sekunden) homogenisiert. Das Homogenisat wird zentrifugiert (536 × G/10 Minuten/4°C), um die
Kerne zu pelletisieren. Der isolierte Membranen enthaltende Überstand
wird zu einem sauberen Zentrifugenröhrchen dekantiert, zentrifugiert
(48 000 × G/30
Minuten, 4°C) und
in 30 ml oder vorzugsweise 20 ml Homogenisierungspuffer resuspendiert.
Dieser Zentrifugierungs- und Resuspensionsschritt wird zweimal wiederholt.
Das Endpellet wird in eiskaltem Dulbecco's PBS, enthaltend 5 mM EDTA, resuspendiert
und in gefrorenen Aliquoten, bis benötigt, bei –80°C gelagert. Die Proteinkonzentration
der erhaltenen Membranzubereitung wird unter Verwendung des Bradford-Protein-Assays
(Bio-Rad Laboratories, Hercules, Kalif.) gemessen. Hierdurch ergibt
sich eine 1-Liter-Kultur von Zellen, die typischerweise 50–100 mg
des gesamten Membranproteins ergibt.
-
Co-Infektion von GTPγ35S
Bindungsassay:
-
Vier
baculovirale Expressionsvektorstämme
werden verwendet, um eine Kultur von Sf9-Zellen mit einem MOI von
1:1:1:1 zu infizieren. Diese Vier bestanden aus einem Vektor, der
den Human-NPY1-Rezeptor codiert und einen anderen, kommerziell erhältlichen
baculoviralen Expressionsvektorstamm, der jeweils an den drei Untereinheiten
eines heterotrimeren G-Proteins codiert, insbesondere werden die
G-Protein codierenden Virusstämme
von BIOSIGNAL Inc., Montreal, erhalten; und sind 1) ein Gα G-Protein
Untereinheit codierender Virusstamm (entweder der Ratten Ga12 G-Protein
codierende Virusstamm BIOSIGNAL #V5J008 oder der Ratten Gα0 G-Protein
codierende Virusstamm BIOSIGNAL #V5H010), 2) ein Rinder-β1-G-Protein-codierender
Virusstamm (BIOSIGNAL #V5H012) und 3) ein Human-γ2-G-Protein codierender Virusstamm
(BIOSIGNAL #V6B003). Agonisten-stimuliertes GTPγ35S-Binden
an gereinigten Membranen wird unter Verwendung von hNPY 1–36 (American
Peptide Co., Sunnyvale, Kalif.) als Agonist bewertet, um funktionelle
Aktivität,
wie gemessen durch GTPγ35S-Binden,
zu ermitteln.
-
GTPγ35S
Bindungsassay:
-
Gereinigte
Sf9-Zellmembranen werden durch Dounce-Homogenisierung (enger Reiber)
in GTPγ35S-Bindungsassaypuffer
(50 mM Tris pH 7,0, 120 mM NaCl, 2 mM MgCl2,
2 mM EGTA, 0,1% BSA, 0,1 mM Bacitracin, 100 KIU/ml Aprotinin, 5 μM GDP) resuspendiert
und in Reaktionsröhrchen
bei einer Konzentration von 30 μg/Reaktionsröhrchen versetzt.
Nach Zugeben von ansteigenden Dosen des Agonisten hNPY 1–36 (American
Peptide Co., Sunnyvale, Kalif.) ergaben sich Reaktionen, durch die
Zugabe von 100 pM GTPγ35S gestartet. Nach einer 30-minütigen Inkubation
bei Umgebungstemperatur werden die Reaktionen durch Vakuumfiltration über GF/C-Filter
(vorher voll gesogen in Waschpuffer, 0,1% BSA) beendet, gefolgt
von Waschen mit eiskaltem Waschpuffer (50 mM Tris pH 7,0, 120 mM
NaCl).
-
Gebundenes
GTPγ35S wird durch Flüssig-Szintillationsspektrometrie
der gewaschenen Filter bestimmt. Nichtspezifisches Binden wird unter
Verwendung von 10 mM GTPγS
bestimmt. Die Daten werden im Allgemeinen als % Maximalreaktion
ausgedrückt
und werden durch Bestimmen des maximal Agonisten stimulierter Prozentsatz
oberhalb Basisstimulierung, abgeleitet. Computeranalyse kann geeigneterweise
verwendet werden, um geschätzte
EC50-, IC50- und
Ki-Werte von GTPγ35S-Bindungsversuchsdaten,
unter Anwendung von beispielsweise SigmaPlot-Software, zu berechnen. Die Bindungsaffinität der bevorzugten,
erfindungsgemäßen Verbindungen,
ausgedrückt
als Ki-Werte, liegt im Bereich von etwa
0,1 Nanomolar bis etwa 5 Mikromolar. Besonders bevorzugte Verbindungen
ergeben einen Ki-Wert von weniger als 100
Nanomolar, besonders bevorzugt weniger als 10 Nanomolar.
-
Assay für das Affinitätsbinden
von radiomarkiertem Liganden:
-
Gereinigte
Membranen werden mit PBS gewaschen und durch mildes Pipettieren
in Bindungspuffer (50 mM Tris(HCl), 5 mM KCl, 120 mM NaCl, 2 mM
CaCl2, 1 mM MgCl2,
0,1% Rinderserumalbumin (BSA), pH 7,4) resuspendiert. Membranen
(5 μg) werden
zu silikonisierten (Sigmacote, Sigma) Polypropylenröhrchen, zusätzlich zu
0,050 nM [125I]NPY (Schwein, New England Nuclear Corp., Boston,
Mass.), zur Vervollständigung
der Analyse, oder 0,010–0,500
nM [125I]NPY (Schwein) zur Sättigungsanalyse,
zugegeben. Zur Bewertung von Guanin-Nucleotid-Effekten auf Rezeptoraffinität wird GTP
bei einer Endkonzentration von 100 μM zugegeben. Kalte Austausche
werden bei Konzentrationen im Bereich von 10–12 M bis 10–6 M zugesetzt,
unter Gewinnung eines Endvolumens von 0,250 ml. Nichtspezifisches
Binden wird in Gegenwart von 1 μM
NPY (human, American Peptide Co., Sunnyvale, CA) und die Zählungen
für weniger
als 10% vom Gesamtbinden bestimmt. Nach 2 Stunden Inkubation bei
Umgebungstemperatur wird die Reaktion durch schnelle Vakuumfiltration
beendet. Die Proben werden über
vorgesogene GF/C-Whatmanfilter (1,0% Polyethy lenimin für 2 Stunden) filtriert
und 2-mal mit 5 ml kaltem Bindungspuffer, dem BSA fehlt, gespült. Die
verbleibende, gebundene Radioaktivität wird durch Gamma-Zählung gemessen.
Um den Bmax, Kd und Ki zu schätzen,
werden die Ergebnisse von Bindungsversuchen unter Verwendung von
SigmaPlot-Software (SPSS Science, Chicago, Ill.) analysiert. Die
Bindungsaffinität
der erfindungsgemäßen Verbindungen,
ausgedrückt
als ein Ki-Wert, liegt im Bereich von etwa 0,1 Nanomolar bis etwa
10 Mikromolar. Die besonders bevorzugten erfindungsgemäßen Verbindungen
haben einen Ki-Wert von weniger als 100 Nanomolar und eine Bindungsselektivität von >100-fach, bezogen auf
die anderen G-Protein-gekuppelten Rezeptoren, einschließlich NPY5- und
CRF1-Rezeptoren.
-
C. In-vivo-Analyse – Nahrungsentzug
-
Personen.
-
Natürbelassene
und erfahrene männliche
Sprague-Dawley-Versuchsratten (Sasco, St. Louis, Mo.) mit einem
Gewicht von 210–300
g am Beginn des Versuchs werden verwendet. Die Tiere werden in Edelstahl-Hängekäfigen in
einer Temperatur- (22°C ± 2) und
Feuchtigkeits-(40–70%
RH) gesteuerten Tierhaltungseinrichtung bei einem 12:12-Stunden
Hell-Dunkel-Zyklus
zu Dritt gehalten. Nahrung (Standard Rat Chow, PMI Feeds Inc., #5012)
und Wasser sind nach Belieben verfügbar.
-
Apparatur.
-
Verbrauchsdaten
werden gesammelt, während
die Tiere in Nalgene Metabolic-Käfigen
(Modell #650-0100) gehalten werden. Jeder Käfig umfasst Teilvorrichtungen,
die aus klarem Polymethylpenten (PMP), Polycarbonat (PC) oder Edelstahl
(SS) hergestellt wurden. Alle Teile sind für schnelle und genaue Datensammlung
und zum Reinigen zerlegbar. Ein ganzer zylinderförmiger Kunststoff- und SS-Käfig ruht
auf einem SS-Stand und beherbergt ein Tier.
-
Das
Tier ist in der runden Oberen Kammer (PC)-Anordnung (12 cm hoch
und 20 cm im Durchmesser) enthalten und ruht auf einem SS-Boden.
Zwei Teilvorrichtungen werden an der oberen Kammer befestigt. Die erste
Vorrichtung besteht aus einer SS-Fütterungskammer
(10 cm lang, 5 cm hoch und 5 cm breit) mit einem PC-Fütterungsfach,
das am Boden angebracht ist. Das Fütterungsfach hat zwei Kammern:
eine Nahrungslagerungskammer mit dem Fassungsvermögen von
ungefähr
50 g von pulverisiertem Rattenfutter und eine Nahrungsüberlaufkammer.
Dem Tier wird Zugang zu dem pulverisierten Futter durch eine Öffnung in
dem SS-Boden der Fütterungskammer
erlaubt. Der Boden der Fütterungskammer
erlaubt nicht den Zugang zu dem Futter, das in die Überflusskammer
abfällt.
-
Die
zweite Anordnung schließt
eine Wasserflaschenhalterung, eine PC-Wasserflasche (100 ml Fassungsvermögen) und
ein graduiertes Wasserüberlaufsammelrohr
ein. Die Wasserflaschenhalterung trichtert jedes übergelaufene
Wasser in das Wasserüberlaufsammelrohr.
-
Die
untere Kammer besteht aus einem PMP-Trennkegel, dem PMP-Sammeltrichter,
PMP-Flüssigkeit (Urin)-Sammelrohr
und einem PMP-Feststoff (Fäkalien)-Sammelrohr.
Der Trennkegel ist an dem Oberen des Trichters befestigt, der wiederum
am Boden der oberen Kammer befestigt ist. Der Urin läuft von
dem Trennkegel auf die Wände
des Sammeltrichters und in das Urinsammelrohr. Der Trennkegel trennt
auch die Fäkalien und
trichtert sie in das Fäkaliensammelrohr.
-
Der
Nahrungsverbrauch, Wasserverbrauch und das Körpergewicht werden mit einer
transportierbaren Advanced-Waage von Ohaus (± 0,1 g Genauigkeit) gemessen.
-
Verfahren.
-
Vor
dem Testtag werden die Tiere auf der Testapparatur durch Anordnen
jedes Tiers in einem metabolischen Käfig für 1 Stunde gewöhnen lassen.
Am Versuchstag wird den Tieren Nahrung entzogen, die vorangehende
Nacht gewogen und Behandlungsgruppen zugeordnet. Die Zuordnungen
erfolgen unter Anwendung eines quasi statistischen Verfahrens, unter
Verwendung der Körpergewichte,
zum Sicherstellen, dass die Behandlungsgruppen ähnliches mittleres Körpergewicht
hatten. Den Tieren werden dann entweder Träger (0,5% Methylcellulose)
oder Arzneistoff (eine erfindungsgemäße Verbindung) verabreicht.
Zu der Zeit wird das Futterfach mit pulverisiertem Futter ge füllt, die
gefüllte
Wasserflasche und die leeren Urin- und Fäkaliensammelröhren werden
gewogen. Zwei Stunden nach Arzneistoffbehandlung wird jedes Tier
gewogen und in einen metabolischen Käfig gesetzt. Nach einer Stunde
Testsitzung werden die Tiere entfernt und das Körpergewicht erhalten. Die Nahrungs- und Wasserbehälter werden
dann gewogen und die Nahrungs- und Wasserverbrauchsdaten aufgezeichnet.
-
Arzneistoffe.
-
Arzneistoff,
suspendiert in Träger,
oder Träger
allein, als eine Kontrolle, wird oral (PO), unter Anwendung eines
Abgaberöhrchens,
verbunden über
eine 3- oder 5 ml-Spritze mit einem Volumen von 10 ml/kg, verabreicht.
Arzneistoff wird zu einer homogenen Suspension durch Rühren und
Ultrabeschallung für
mindestens 1 Stunde vor dem Dosieren hergestellt.
-
Statistische Analysen.
-
Die
mittleren und Standardfehler des Durchschnitts (SEM) für Nahrungsverbrauch,
Wasserverbrauch und Körpergewichtsveränderung
werden erhalten. Ein-Weg-Varianz-Analyse
unter Anwendung von Systat (5.2.1) wird zum Testen der Gruppenunterschiede
verwendet. Ein signifikanter Effekt wird als mit einem p-Wert von <0,05 definiert.
-
Die
nachstehenden Parameter werden definiert: Körpergewichtsveränderung
ist der Unterschied zwischen dem Körpergewicht des Tiers, unmittelbar
vor der Anordnung in den metabolischen Käfig, und seinem Körpergewicht
am Ende einer Ein-Stunden-Test-Sitzung.
Nahrungsverbrauch ist der Unterschied in dem Gewicht des Nahrungsfachs
vor dem Testen und dem Gewicht nach der 1-Stunden-Test-Sitzung.
Der Wasserverbrauch ist der Unterschied in dem Gewicht der Wasserflasche
vor dem Testen und dem Gewicht nach der Ein-Stunden-Sitzung. Bevorzugte
erfindungsgemäße Verbindungen
vermindern die Nahrungsaufnahme und den Körpergewichtszuwachs, vorzugsweise
zu einem statistisch signifikanten Grad, wie durch parametrische Standardanalyse,
wie einen Student T-Test, bestimmt.
-
BEISPIEL 262
-
Herstellung von radiomarkierten,
erfindungsgemäßen Sondenverbindungen
-
Die
erfindungsgemäßen Verbindungen
werden als radiomarkierte Sonden durch Ausführen ihrer Synthese, unter
Anwendung von Vorstufen, die mindestens ein Atom, das ein Radioisotop
darstellt, umfassen, hergestellt. Das Radioisotop wird vorzugsweise
ausgewählt
aus mindestens einem von Kohlenstoff (vorzugsweise 14C),
Wasserstoff (vorzugsweise 3H), Schwefel
(vorzugsweise 35S) oder Jod (vorzugsweise 125I). Solche radiomarkierten Sonden werden
geeigneterweise durch einen Radioisotopenhersteller, der auf übliche Synthesen für radiomarkierte
Sondenverbindungen spezialisiert ist, synthetisiert. Solche Hersteller
schließen
Amersham Corporation, Arlington Heights, Ill.; Cambridge Isotope
Laboratories, Inc. Andover, MA; SRI International, Menlo Park, CA;
Wizard Laboratories, West Sacramento, CA; ChemSyn Laboratories,
Lexena, KS; American Radiolabeled Chemicals, Inc., St. Louis, MO;
und Moravek Biochemicals Inc., Brea, CA, ein.
-
Tritium-markierte
Sondenverbindungen werden auch herkömmlicherweise katalytisch über Platin-katalysierten
Austausch in tritiierter Essigsäure,
säurekatalysierten
Austausch in tritiierter Trifluoressigsäure oder heterogen katalysierten
Austausch mit Tritiumgas hergestellt. Solche Zubereitungen werden
auch herkömmlicherweise
als ein übliches
Radiomarkieren durch jeden der Hersteller, der in dem vorangehenden
Absatz angeführt
ist, unter Verwendung der erfindungsgemäßen Verbindung als Substrat,
ausgeführt.
Zusätzlich
können bestimmte
Vorstufen Tritium-Halogen-Austausch mit Tritiumgas, Tritiumgasreduktion
von ungesättigten
Bindungen, oder Reduktion unter Verwendung von Natriumbortritid,
falls geeignet, unterzogen werden.
-
BEISPIEL 263
-
Rezeptor Autoradiographie
-
Rezeptor-Autoradiographie
(Rezeptormapping) wird in vitro, wie von Kuhar in Abschnitten 8.1.1
bis 8.1.9 von Current Protocols in Pharmacology (1998) John Wiley & Sons, New York,
beschrieben, unter Anwendung von radiomarkierten, erfindungsgemäßen Verbindungen,
hergestellt wie in dem vorangehenden Beispiel beschrieben, ausgeführt.
-
Die
Erfindung und die Art und das Verfahren zur Herstellung und die
Anwendung derselben werden nun in solchen vollständigen, deutlichen, präzisen und
exakten Begriffen beschrieben, dass jeder Fachmann, den es betrifft,
in die Lage versetzt wird, dieselben herzustellen und zu verwenden.