CN1368954A - 水杨酸酰胺衍生物 - Google Patents
水杨酸酰胺衍生物 Download PDFInfo
- Publication number
- CN1368954A CN1368954A CN00811487A CN00811487A CN1368954A CN 1368954 A CN1368954 A CN 1368954A CN 00811487 A CN00811487 A CN 00811487A CN 00811487 A CN00811487 A CN 00811487A CN 1368954 A CN1368954 A CN 1368954A
- Authority
- CN
- China
- Prior art keywords
- formula
- expression
- salicylamide derivatives
- salicylamide
- derivatives
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- SKZKKFZAGNVIMN-UHFFFAOYSA-N Salicilamide Chemical class NC(=O)C1=CC=CC=C1O SKZKKFZAGNVIMN-UHFFFAOYSA-N 0.000 title claims abstract description 55
- 229940121363 anti-inflammatory agent Drugs 0.000 claims abstract description 9
- 239000002260 anti-inflammatory agent Substances 0.000 claims abstract description 9
- 239000003018 immunosuppressive agent Substances 0.000 claims abstract description 9
- 230000014509 gene expression Effects 0.000 claims description 74
- 150000001875 compounds Chemical class 0.000 claims description 43
- 238000006243 chemical reaction Methods 0.000 claims description 32
- 238000002360 preparation method Methods 0.000 claims description 31
- 150000003839 salts Chemical class 0.000 claims description 14
- 230000004913 activation Effects 0.000 claims description 10
- -1 carboxylic acid halides Chemical class 0.000 claims description 9
- 239000000470 constituent Substances 0.000 claims description 9
- 239000000825 pharmaceutical preparation Substances 0.000 claims description 6
- 125000000217 alkyl group Chemical group 0.000 claims description 5
- 240000000203 Salix gracilistyla Species 0.000 claims description 4
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 claims description 4
- 239000003112 inhibitor Substances 0.000 claims description 4
- 238000006722 reduction reaction Methods 0.000 claims description 4
- NAZDVUBIEPVUKE-UHFFFAOYSA-N 2,5-dimethoxyaniline Chemical compound COC1=CC=C(OC)C(N)=C1 NAZDVUBIEPVUKE-UHFFFAOYSA-N 0.000 claims description 3
- 125000005843 halogen group Chemical group 0.000 claims description 3
- 238000006735 epoxidation reaction Methods 0.000 claims description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 2
- 230000000694 effects Effects 0.000 abstract description 12
- 239000003814 drug Substances 0.000 abstract description 6
- 238000000034 method Methods 0.000 abstract description 6
- 229940079593 drug Drugs 0.000 abstract description 4
- 230000002401 inhibitory effect Effects 0.000 abstract description 2
- 239000000543 intermediate Substances 0.000 abstract description 2
- 239000004480 active ingredient Substances 0.000 abstract 1
- 229940125721 immunosuppressive agent Drugs 0.000 abstract 1
- 230000007076 release of cytoplasmic sequestered NF-kappaB Effects 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 93
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 66
- 239000000243 solution Substances 0.000 description 26
- 239000002904 solvent Substances 0.000 description 17
- 238000005406 washing Methods 0.000 description 15
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 15
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 14
- 238000000862 absorption spectrum Methods 0.000 description 14
- 230000002829 reductive effect Effects 0.000 description 14
- 108010057466 NF-kappa B Proteins 0.000 description 13
- 102000003945 NF-kappa B Human genes 0.000 description 13
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 12
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 10
- 239000003795 chemical substances by application Substances 0.000 description 10
- 238000001035 drying Methods 0.000 description 10
- 239000007787 solid Substances 0.000 description 10
- 238000003756 stirring Methods 0.000 description 10
- 238000012360 testing method Methods 0.000 description 9
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 8
- 102000008186 Collagen Human genes 0.000 description 7
- 108010035532 Collagen Proteins 0.000 description 7
- 241000699666 Mus <mouse, genus> Species 0.000 description 7
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 7
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 7
- 239000007864 aqueous solution Substances 0.000 description 7
- 210000004027 cell Anatomy 0.000 description 7
- 238000002143 fast-atom bombardment mass spectrum Methods 0.000 description 7
- 239000000203 mixture Substances 0.000 description 7
- 238000001291 vacuum drying Methods 0.000 description 7
- 108060001084 Luciferase Proteins 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 235000017557 sodium bicarbonate Nutrition 0.000 description 6
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 6
- LIKMAJRDDDTEIG-UHFFFAOYSA-N 1-hexene Chemical compound CCCCC=C LIKMAJRDDDTEIG-UHFFFAOYSA-N 0.000 description 5
- 239000005089 Luciferase Substances 0.000 description 5
- 230000002917 arthritic effect Effects 0.000 description 5
- 108010005774 beta-Galactosidase Proteins 0.000 description 5
- 239000000376 reactant Substances 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 230000008961 swelling Effects 0.000 description 5
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 108700008625 Reporter Genes Proteins 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- 102100040247 Tumor necrosis factor Human genes 0.000 description 4
- 102000005936 beta-Galactosidase Human genes 0.000 description 4
- 210000003194 forelimb Anatomy 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 210000003141 lower extremity Anatomy 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 239000013612 plasmid Substances 0.000 description 4
- 230000000452 restraining effect Effects 0.000 description 4
- 229910000033 sodium borohydride Inorganic materials 0.000 description 4
- 239000012279 sodium borohydride Substances 0.000 description 4
- 239000006188 syrup Substances 0.000 description 4
- 235000020357 syrup Nutrition 0.000 description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 4
- 238000001890 transfection Methods 0.000 description 4
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- 238000013016 damping Methods 0.000 description 3
- 239000012530 fluid Substances 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 238000007670 refining Methods 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 108010002352 Interleukin-1 Proteins 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- SEQKRHFRPICQDD-UHFFFAOYSA-N N-tris(hydroxymethyl)methylglycine Chemical compound OCC(CO)(CO)[NH2+]CC([O-])=O SEQKRHFRPICQDD-UHFFFAOYSA-N 0.000 description 2
- 241001460678 Napo <wasp> Species 0.000 description 2
- GQPLMRYTRLFLPF-UHFFFAOYSA-N Nitrous Oxide Chemical compound [O-][N+]#N GQPLMRYTRLFLPF-UHFFFAOYSA-N 0.000 description 2
- 206010039361 Sacroiliitis Diseases 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 2
- 102000040945 Transcription factor Human genes 0.000 description 2
- 108091023040 Transcription factor Proteins 0.000 description 2
- 150000001263 acyl chlorides Chemical class 0.000 description 2
- 239000012752 auxiliary agent Substances 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- WORJEOGGNQDSOE-UHFFFAOYSA-N chloroform;methanol Chemical group OC.ClC(Cl)Cl WORJEOGGNQDSOE-UHFFFAOYSA-N 0.000 description 2
- 229920001436 collagen Polymers 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 210000003414 extremity Anatomy 0.000 description 2
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 239000003701 inert diluent Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 230000010534 mechanism of action Effects 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000012046 mixed solvent Substances 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 230000034190 positive regulation of NF-kappaB transcription factor activity Effects 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 239000012266 salt solution Substances 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 238000010898 silica gel chromatography Methods 0.000 description 2
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 2
- 235000019345 sodium thiosulphate Nutrition 0.000 description 2
- 235000014347 soups Nutrition 0.000 description 2
- 238000010025 steaming Methods 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- 210000003857 wrist joint Anatomy 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- UFDULEKOJAEIRI-UHFFFAOYSA-N (2-acetyloxy-3-iodophenyl) acetate Chemical compound CC(=O)OC1=CC=CC(I)=C1OC(C)=O UFDULEKOJAEIRI-UHFFFAOYSA-N 0.000 description 1
- KUWPCJHYPSUOFW-YBXAARCKSA-N 2-nitrophenyl beta-D-galactoside Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1OC1=CC=CC=C1[N+]([O-])=O KUWPCJHYPSUOFW-YBXAARCKSA-N 0.000 description 1
- GNFTZDOKVXKIBK-UHFFFAOYSA-N 3-(2-methoxyethoxy)benzohydrazide Chemical compound COCCOC1=CC=CC(C(=O)NN)=C1 GNFTZDOKVXKIBK-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 206010002198 Anaphylactic reaction Diseases 0.000 description 1
- 208000008822 Ankylosis Diseases 0.000 description 1
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 1
- FGUUSXIOTUKUDN-IBGZPJMESA-N C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 Chemical compound C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 FGUUSXIOTUKUDN-IBGZPJMESA-N 0.000 description 1
- RGJOEKWQDUBAIZ-IBOSZNHHSA-N CoASH Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCS)O[C@H]1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-IBOSZNHHSA-N 0.000 description 1
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- YPINZEGNLULHHT-UHFFFAOYSA-N Fujimycin Natural products COC1CC(CCC1O)C=C(/C)C2OC(=O)C3CCCCCN3C(=O)C(=O)C4(O)OC(C(CC4C)OC)C(OC)C(C)CC(=CC(CC=C)C(=O)CC(O)C2C)C YPINZEGNLULHHT-UHFFFAOYSA-N 0.000 description 1
- 102000009331 Homeodomain Proteins Human genes 0.000 description 1
- 108010048671 Homeodomain Proteins Proteins 0.000 description 1
- 101000611183 Homo sapiens Tumor necrosis factor Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 102000012960 Immunoglobulin kappa-Chains Human genes 0.000 description 1
- 108010090227 Immunoglobulin kappa-Chains Proteins 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 description 1
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- 108090001007 Interleukin-8 Proteins 0.000 description 1
- 241000581650 Ivesia Species 0.000 description 1
- 206010023198 Joint ankylosis Diseases 0.000 description 1
- 102000011931 Nucleoproteins Human genes 0.000 description 1
- 108010061100 Nucleoproteins Proteins 0.000 description 1
- 108010001859 Proto-Oncogene Proteins c-rel Proteins 0.000 description 1
- 102000000850 Proto-Oncogene Proteins c-rel Human genes 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-N Salicylic acid Natural products OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 1
- UZMAPBJVXOGOFT-UHFFFAOYSA-N Syringetin Natural products COC1=C(O)C(OC)=CC(C2=C(C(=O)C3=C(O)C=C(O)C=C3O2)O)=C1 UZMAPBJVXOGOFT-UHFFFAOYSA-N 0.000 description 1
- 239000007997 Tricine buffer Substances 0.000 description 1
- 108010000134 Vascular Cell Adhesion Molecule-1 Proteins 0.000 description 1
- 102100023543 Vascular cell adhesion protein 1 Human genes 0.000 description 1
- ZBIKORITPGTTGI-UHFFFAOYSA-N [acetyloxy(phenyl)-$l^{3}-iodanyl] acetate Chemical compound CC(=O)OI(OC(C)=O)C1=CC=CC=C1 ZBIKORITPGTTGI-UHFFFAOYSA-N 0.000 description 1
- 230000003187 abdominal effect Effects 0.000 description 1
- WPDDCDPPSHTJDH-UHFFFAOYSA-N acetic acid [2-[(2,5-dimethoxyanilino)-oxomethyl]phenyl] ester Chemical compound COC1=CC=C(OC)C(NC(=O)C=2C(=CC=CC=2)OC(C)=O)=C1 WPDDCDPPSHTJDH-UHFFFAOYSA-N 0.000 description 1
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 230000036783 anaphylactic response Effects 0.000 description 1
- 208000003455 anaphylaxis Diseases 0.000 description 1
- 230000000947 anti-immunosuppressive effect Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000007503 antigenic stimulation Effects 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 150000001510 aspartic acids Chemical class 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 229910052728 basic metal Inorganic materials 0.000 description 1
- 150000003818 basic metals Chemical class 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 230000021164 cell adhesion Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- RGJOEKWQDUBAIZ-UHFFFAOYSA-N coenzime A Natural products OC1C(OP(O)(O)=O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-UHFFFAOYSA-N 0.000 description 1
- 239000005516 coenzyme A Substances 0.000 description 1
- 229940093530 coenzyme a Drugs 0.000 description 1
- 230000000536 complexating effect Effects 0.000 description 1
- ZTOGYGRJDIWKAQ-UHFFFAOYSA-N cyclohexene-1,2-diamine Chemical compound NC1=C(N)CCCC1 ZTOGYGRJDIWKAQ-UHFFFAOYSA-N 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- KDTSHFARGAKYJN-UHFFFAOYSA-N dephosphocoenzyme A Natural products OC1C(O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 KDTSHFARGAKYJN-UHFFFAOYSA-N 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- KCFYHBSOLOXZIF-UHFFFAOYSA-N dihydrochrysin Natural products COC1=C(O)C(OC)=CC(C2OC3=CC(O)=CC(O)=C3C(=O)C2)=C1 KCFYHBSOLOXZIF-UHFFFAOYSA-N 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 238000004945 emulsification Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000012447 hatching Effects 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical group 0.000 description 1
- 239000003973 paint Substances 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000003180 prostaglandins Chemical class 0.000 description 1
- RYVMUASDIZQXAA-UHFFFAOYSA-N pyranoside Natural products O1C2(OCC(C)C(OC3C(C(O)C(O)C(CO)O3)O)C2)C(C)C(C2(CCC3C4(C)CC5O)C)C1CC2C3CC=C4CC5OC(C(C1O)O)OC(CO)C1OC(C1OC2C(C(OC3C(C(O)C(O)C(CO)O3)O)C(O)C(CO)O2)O)OC(CO)C(O)C1OC1OCC(O)C(O)C1O RYVMUASDIZQXAA-UHFFFAOYSA-N 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229960001967 tacrolimus Drugs 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D303/00—Compounds containing three-membered rings having one oxygen atom as the only ring hetero atom
- C07D303/02—Compounds containing oxirane rings
- C07D303/12—Compounds containing oxirane rings with hydrocarbon radicals, substituted by singly or doubly bound oxygen atoms
- C07D303/14—Compounds containing oxirane rings with hydrocarbon radicals, substituted by singly or doubly bound oxygen atoms by free hydroxyl radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D303/00—Compounds containing three-membered rings having one oxygen atom as the only ring hetero atom
- C07D303/02—Compounds containing oxirane rings
- C07D303/36—Compounds containing oxirane rings with hydrocarbon radicals, substituted by nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/336—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having three-membered rings, e.g. oxirane, fumagillin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/60—Salicylic acid; Derivatives thereof
- A61K31/609—Amides, e.g. salicylamide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/28—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton
- C07C237/38—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton having the nitrogen atom of the carboxamide group bound to a carbon atom of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/28—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton
- C07C237/40—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a non-condensed six-membered aromatic ring of the carbon skeleton having the nitrogen atom of the carboxamide group bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D303/00—Compounds containing three-membered rings having one oxygen atom as the only ring hetero atom
- C07D303/02—Compounds containing oxirane rings
- C07D303/12—Compounds containing oxirane rings with hydrocarbon radicals, substituted by singly or doubly bound oxygen atoms
- C07D303/18—Compounds containing oxirane rings with hydrocarbon radicals, substituted by singly or doubly bound oxygen atoms by etherified hydroxyl radicals
- C07D303/20—Ethers with hydroxy compounds containing no oxirane rings
- C07D303/22—Ethers with hydroxy compounds containing no oxirane rings with monohydroxy compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D303/00—Compounds containing three-membered rings having one oxygen atom as the only ring hetero atom
- C07D303/02—Compounds containing oxirane rings
- C07D303/12—Compounds containing oxirane rings with hydrocarbon radicals, substituted by singly or doubly bound oxygen atoms
- C07D303/32—Compounds containing oxirane rings with hydrocarbon radicals, substituted by singly or doubly bound oxygen atoms by aldehydo- or ketonic radicals
Abstract
Description
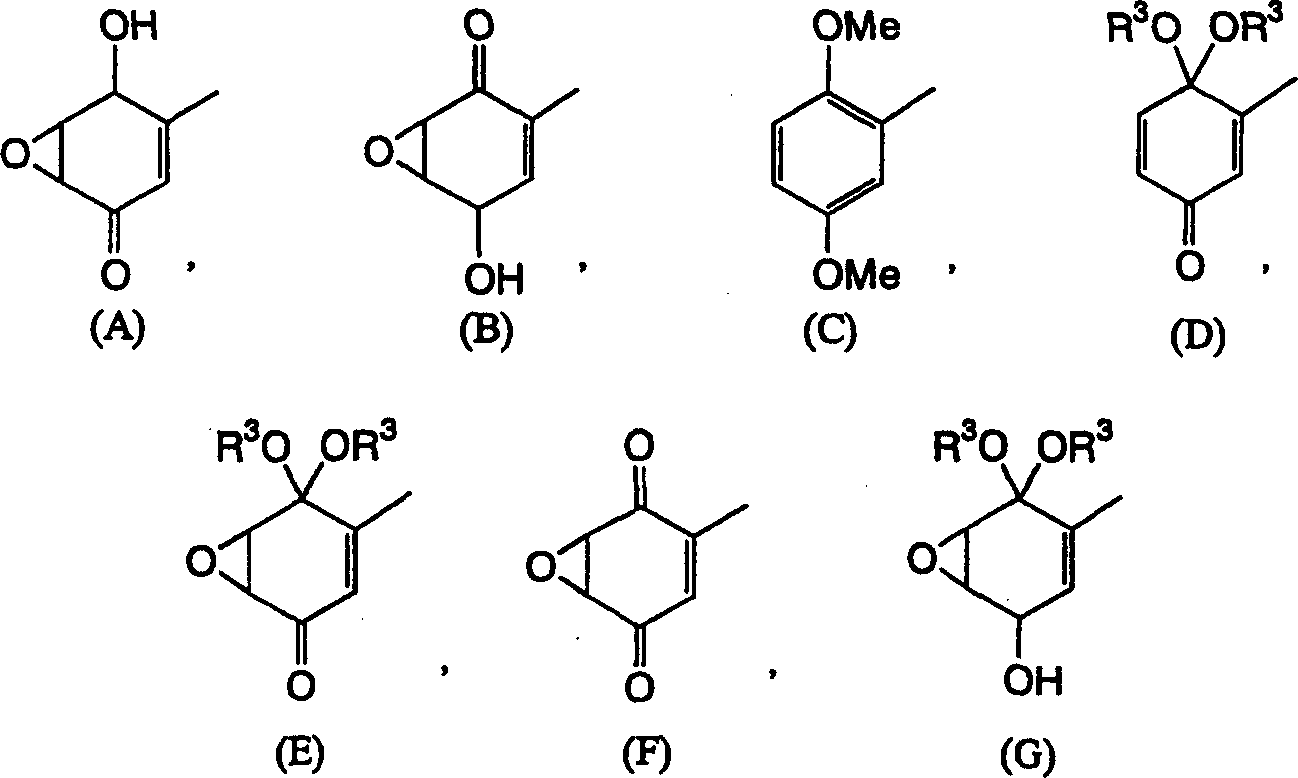
















Claims (17)



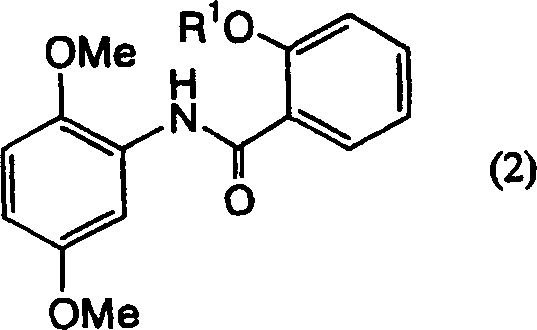





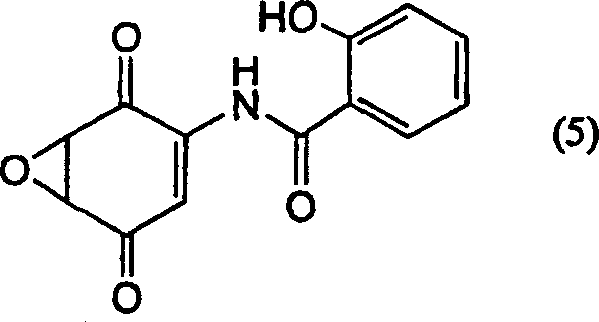


Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP227389/1999 | 1999-08-11 | ||
| JP227389/99 | 1999-08-11 | ||
| JP22738999 | 1999-08-11 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1368954A true CN1368954A (zh) | 2002-09-11 |
| CN1185212C CN1185212C (zh) | 2005-01-19 |
Family
ID=16860062
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB008114870A Expired - Lifetime CN1185212C (zh) | 1999-08-11 | 2000-08-09 | 水杨酸酰胺衍生物 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US6566394B1 (zh) |
| EP (1) | EP1219596B1 (zh) |
| JP (1) | JP4691295B2 (zh) |
| KR (1) | KR100709317B1 (zh) |
| CN (1) | CN1185212C (zh) |
| AT (1) | ATE344234T1 (zh) |
| AU (1) | AU774221B2 (zh) |
| CA (1) | CA2393883C (zh) |
| DE (1) | DE60031705T2 (zh) |
| WO (1) | WO2001012588A1 (zh) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102414190A (zh) * | 2009-03-27 | 2012-04-11 | 普罗菲克图斯生物科学股份有限公司 | NF-kB的抑制剂 |
| CN103588731A (zh) * | 2013-12-02 | 2014-02-19 | 深圳万和制药有限公司 | 水杨酸酰胺衍生物及制备方法 |
| CN103588732A (zh) * | 2013-12-02 | 2014-02-19 | 深圳万和制药有限公司 | 水杨酸酰胺衍生物的结晶 |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE548031T1 (de) * | 2000-12-18 | 2012-03-15 | Inst Med Molecular Design Inc | Inhibitoren gegen die produktion und freisetzung entzündungfördernder zytokine |
| DE10222895A1 (de) * | 2002-05-23 | 2003-12-11 | Bosch Gmbh Robert | Hochdruckspeicher für Kraftstoffeinspritzsysteme mit integriertem Druckregelventil |
| US20060019958A1 (en) * | 2002-06-05 | 2006-01-26 | Susumu Muto | Immunity-related protein kinase inhibitors |
| EP1510207A4 (en) * | 2002-06-05 | 2008-12-31 | Inst Med Molecular Design Inc | THERAPEUTIC MEDICAMENT AGAINST DIABETES |
| CN1658849A (zh) * | 2002-06-05 | 2005-08-24 | 株式会社医药分子设计研究所 | Ap-1和nfat活化抑制剂 |
| US20060122243A1 (en) * | 2002-06-06 | 2006-06-08 | Susumu Muto | Antiallergic |
| EA009051B1 (ru) * | 2002-06-06 | 2007-10-26 | Инститьют Оф Медисинал Молекьюлар Дизайн. Инк. | О-замещенные гидроксиарильные производные |
| JPWO2003103654A1 (ja) * | 2002-06-10 | 2005-10-06 | 株式会社医薬分子設計研究所 | NF−κB活性化阻害剤 |
| CA2488974A1 (en) * | 2002-06-10 | 2003-12-18 | Institute Of Medicinal Molecular Design, Inc. | Medicament for treatment of cancer |
| WO2003103657A1 (ja) * | 2002-06-11 | 2003-12-18 | 株式会社医薬分子設計研究所 | 神経変性疾患治療剤 |
| CN1674881A (zh) * | 2002-06-26 | 2005-09-28 | 学校法人庆应义塾 | 含NF-κB抑制剂的医药组合物 |
| WO2004044463A1 (ja) * | 2002-11-12 | 2004-05-27 | Nok Corporation | ゴム状弾性部品 |
| WO2004072056A1 (ja) * | 2003-02-14 | 2004-08-26 | Keio University | 医薬組成物 |
| AU2004263025A1 (en) * | 2003-08-06 | 2005-02-17 | Signal Creation Inc. | Macrophage activation inhibitor |
| JP2007182397A (ja) * | 2006-01-05 | 2007-07-19 | Keio Gijuku | NF−κB阻害剤 |
| WO2010127058A1 (en) * | 2009-05-01 | 2010-11-04 | Profectus Biosciences, Inc. | Benzamide and naphthamide derivatives inhibiting nuclear factor- kap pa (b) - (nf-kb) |
| WO2010148042A1 (en) | 2009-06-18 | 2010-12-23 | Profectus Biosciences, Inc. | Oxabicyclo [4.1.o]hept-b-en-s-ylcarbamoyl derivatives inhibiting the nuclear factor-kappa(b) - (nf-kb) |
| RU2703735C1 (ru) * | 2018-11-09 | 2019-10-22 | Общество с ограниченной ответственностью "ПеритонТрит" | Фармацевтическая композиция, содержащая DHMEQ или его аналоги |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6445738A (en) * | 1987-08-13 | 1989-02-20 | Fujikura Ltd | Production of optical fiber preform |
| JPH09157266A (ja) * | 1995-12-04 | 1997-06-17 | Microbial Chem Res Found | 新規抗生物質エポキシキノマイシンaおよびbとその製造法 |
| JPH1045738A (ja) * | 1996-07-29 | 1998-02-17 | Microbial Chem Res Found | 抗生物質エポキシキノマイシンcおよびdとその製造法ならびに抗リウマチ剤 |
-
2000
- 2000-08-09 CN CNB008114870A patent/CN1185212C/zh not_active Expired - Lifetime
- 2000-08-09 JP JP2001516889A patent/JP4691295B2/ja not_active Expired - Fee Related
- 2000-08-09 EP EP00951914A patent/EP1219596B1/en not_active Expired - Lifetime
- 2000-08-09 WO PCT/JP2000/005332 patent/WO2001012588A1/ja active IP Right Grant
- 2000-08-09 DE DE60031705T patent/DE60031705T2/de not_active Expired - Lifetime
- 2000-08-09 US US10/049,158 patent/US6566394B1/en not_active Expired - Fee Related
- 2000-08-09 AT AT00951914T patent/ATE344234T1/de not_active IP Right Cessation
- 2000-08-09 KR KR1020027001552A patent/KR100709317B1/ko not_active IP Right Cessation
- 2000-08-09 CA CA2393883A patent/CA2393883C/en not_active Expired - Fee Related
- 2000-08-09 AU AU64727/00A patent/AU774221B2/en not_active Ceased
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102414190A (zh) * | 2009-03-27 | 2012-04-11 | 普罗菲克图斯生物科学股份有限公司 | NF-kB的抑制剂 |
| CN102414190B (zh) * | 2009-03-27 | 2015-06-03 | 普罗菲克图斯生物科学股份有限公司 | NF-κB的抑制剂 |
| CN103588731A (zh) * | 2013-12-02 | 2014-02-19 | 深圳万和制药有限公司 | 水杨酸酰胺衍生物及制备方法 |
| CN103588732A (zh) * | 2013-12-02 | 2014-02-19 | 深圳万和制药有限公司 | 水杨酸酰胺衍生物的结晶 |
| CN103588732B (zh) * | 2013-12-02 | 2015-03-25 | 深圳万和制药有限公司 | 水杨酸酰胺衍生物的结晶 |
| CN103588731B (zh) * | 2013-12-02 | 2015-09-02 | 深圳万和制药有限公司 | 水杨酸酰胺衍生物及制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1219596A1 (en) | 2002-07-03 |
| JP4691295B2 (ja) | 2011-06-01 |
| KR100709317B1 (ko) | 2007-04-20 |
| AU774221B2 (en) | 2004-06-17 |
| CN1185212C (zh) | 2005-01-19 |
| KR20020031402A (ko) | 2002-05-01 |
| AU6472700A (en) | 2001-03-13 |
| DE60031705D1 (de) | 2006-12-14 |
| EP1219596B1 (en) | 2006-11-02 |
| WO2001012588A1 (fr) | 2001-02-22 |
| CA2393883C (en) | 2010-07-06 |
| DE60031705T2 (de) | 2007-10-04 |
| CA2393883A1 (en) | 2001-02-22 |
| US6566394B1 (en) | 2003-05-20 |
| ATE344234T1 (de) | 2006-11-15 |
| EP1219596A4 (en) | 2004-08-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1368954A (zh) | 水杨酸酰胺衍生物 | |
| CN1052005C (zh) | 咪唑并吡啶、其制法和药用 | |
| CN1310904C (zh) | 三环烷基异羟肟酸酯,它们的制备和它们作为细胞增殖抑制剂的应用 | |
| CN1301260A (zh) | 烟曲霉醇衍生物及其制备方法 | |
| CN1568311A (zh) | 作为高选择性的环加氧酶-2抑制剂的1h-吲哚衍生物 | |
| CN1039011C (zh) | 雪花胺衍生物及其制备方法和其作为药剂的用途 | |
| CN1129216A (zh) | 三环取代异羟肟酸衍生物 | |
| CN1036389C (zh) | 4-氨基-2,6-二甲基苯磺酰基硝基甲烷的苯乙酰衍生物或其制备方法 | |
| FR2463774A1 (fr) | Derives du 2-phenylimidazo(2,1-b)benzothiazole | |
| CN1015334B (zh) | 新苯并唑衍生物的制备方法 | |
| CN1027264C (zh) | 粟籽豆精胺苷的制备方法 | |
| US11827643B2 (en) | Pharmaceutical intermediates and methods for preparing the same | |
| CN1136807A (zh) | N-取代的氮杂二环庚烷衍生物、它们的制备及应用 | |
| CN1040976A (zh) | 取代的β-二酮的制备方法 | |
| CN1025857C (zh) | 制备具有药理性质的噻唑并[5,4-b][1,5]苯并二氮杂的方法 | |
| CN1014992B (zh) | 喹唑啉二酮和吡啶并嘧啶二酮的制备方法 | |
| EP1873140A1 (fr) | Nouveaux dérivés naphtaleniques, leur procédé de préparation et les compositions pharmaceutiques qui les contiennent | |
| FR3062851A1 (fr) | Procede de preparation monotope de composes organo-iodes | |
| CN1045582A (zh) | [5(6)(苯并异-,苯并异噻-或吲唑-3基)-1h-苯并咪唑-2-基]氨基甲酸酯 | |
| CN1098258C (zh) | 异香豆素衍生物及其在医药上的应用 | |
| CN1051546C (zh) | 新的1,1-双杂唑基烷烃衍生物及其作为神经保护剂的用途 | |
| CN1636997A (zh) | 取代的三环香豆素类化合物、其制备及抗hiv的应用 | |
| CN1536992A (zh) | 2-杂环-1,2-乙二醇的氨基甲酸酯 | |
| Wang et al. | An Efficient Enantioselective Synthesis of Florfenicol Based on Sharpless Asymmetric Dihydroxylation | |
| CN1736994A (zh) | 一种含氮杂环β-榄香烯酰胺类衍生物其制备及应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| ASS | Succession or assignment of patent right |
Owner name: CREATIVE SIGNAL CO., LTD. Free format text: FORMER OWNER: KEIO UNIVERSITY Effective date: 20060908 |
|
| C41 | Transfer of patent application or patent right or utility model | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20060908 Address after: Kanagawa Patentee after: Univ Keio Address before: Tokyo, Japan Patentee before: Keio University |
|
| ASS | Succession or assignment of patent right |
Owner name: SHENZHEN WANHE PHARMACEUTICAL CO., LTD. Free format text: FORMER OWNER: UNIV KEIO Effective date: 20110921 |
|
| C41 | Transfer of patent application or patent right or utility model | ||
| COR | Change of bibliographic data |
Free format text: CORRECT: ADDRESS; TO: 518057 SHENZHEN, GUANGDONG PROVINCE |
|
| TR01 | Transfer of patent right |
Effective date of registration: 20110921 Address after: 518057, Guangdong, Nanshan District science and Technology Park, Shenzhen hi tech, a No. 8 Vanward pharmaceutical Park, 6 floor Patentee after: Shenzhen Wanhe Pharmaceutical Co., Ltd. Address before: Japan Kanagawa Prefecture Patentee before: Univ Keio |
|
| C56 | Change in the name or address of the patentee | ||
| CP02 | Change in the address of a patent holder |
Address after: 518057 Nanshan District high tech Zone, Guangdong, China, a high-tech Vanward Medical Park Patentee after: Shenzhen Wanhe Pharmaceutical Co., Ltd. Address before: 518057, Guangdong, Nanshan District science and Technology Park, Shenzhen hi tech, a No. 8 Vanward pharmaceutical Park, 6 floor Patentee before: Shenzhen Wanhe Pharmaceutical Co., Ltd. |
|
| CX01 | Expiry of patent term |
Granted publication date: 20050119 |
|
| CX01 | Expiry of patent term |