CN1136855C - 用于从患者体内除去胆汁盐的交联聚合物 - Google Patents
用于从患者体内除去胆汁盐的交联聚合物 Download PDFInfo
- Publication number
- CN1136855C CN1136855C CNB951935224A CN95193522A CN1136855C CN 1136855 C CN1136855 C CN 1136855C CN B951935224 A CNB951935224 A CN B951935224A CN 95193522 A CN95193522 A CN 95193522A CN 1136855 C CN1136855 C CN 1136855C
- Authority
- CN
- China
- Prior art keywords
- alkyl
- formula
- polymer
- polymer composition
- copolymer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/74—Synthetic polymeric materials
- A61K31/785—Polymers containing nitrogen
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/74—Synthetic polymeric materials
- A61K31/785—Polymers containing nitrogen
- A61K31/787—Polymers containing nitrogen containing heterocyclic rings having nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/74—Synthetic polymeric materials
- A61K31/795—Polymers containing sulfur
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/52—Amides or imides
- C08F220/54—Amides, e.g. N,N-dimethylacrylamide or N-isopropylacrylamide
- C08F220/60—Amides, e.g. N,N-dimethylacrylamide or N-isopropylacrylamide containing nitrogen in addition to the carbonamido nitrogen
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S210/00—Liquid purification or separation
- Y10S210/902—Materials removed
- Y10S210/903—Nitrogenous
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Polymers & Plastics (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- External Artificial Organs (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
一种用离子交换从患者体内除去胆汁盐的方法,包括给患者服用治疗有效量的一种或多种特征为具有式(1)重复单元高交联聚合物或其共聚物,其中n为整数,R1是H或C1-C8烷基;M是(a)或-Z-R2;Z是O,NR3,S,或(CH2)m;m=0-10;R3是H或C1-C8烷基;及R2是(b)或(c),其中P=0-10,每个R4,R5,和R6各自为H,C1-C8烷基,或芳基,该聚合物服用后无毒并且稳定。
Description
本发明的背景技术
本发明涉及从患者体内除去胆汁盐。
从患者体内螯合和除去胆汁盐(例如胆酸盐,甘胆酸盐,甘鹅胆酸盐,中磺胆酸盐和脱氧胆酸盐)可以降低患者的胆固醇水平。经过消化系统服用后除去胆汁盐的离子交换树脂己经用于该目的。除去胆汁盐将导致体内产生更多的胆汁盐。因为胆汁盐的生物前体是胆固醇,所以胆固醇代谢为胆汁盐就伴随有病人体内的胆固醇的减少。
本发明的技术方案
第一方面,本发明特征在于通过离子交换从患者体内除去胆汁盐的方法,包括给患者服用无毒和稳定的治疗有效量的一种或多种高交联聚合物。聚合物的特征在于具有下式重复单元或其共聚物。
其中n为整数;R1是H或C1-C8烷基(可以是直链或支链、取代的或未取代的,例如甲基);M是
 或-Z-R2;Z是O,NR3,S,或(CH2)m;m=0-10;R3是H或C1-C8烷基(可以是直链或支链、取代的或未取代的,例如甲基);及R2是或
或-Z-R2;Z是O,NR3,S,或(CH2)m;m=0-10;R3是H或C1-C8烷基(可以是直链或支链、取代的或未取代的,例如甲基);及R2是或
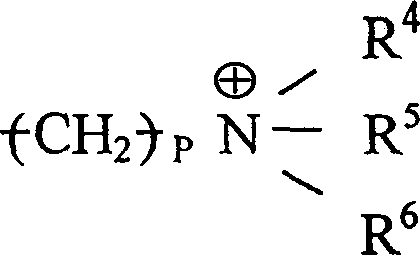
其中P=0-10,且每个R4、R5和R6各自是H,C1-C8烷基(可以是直链或支链,取代的或未取代的,例如甲基),或芳基(例如具有一个或多个环并且可被取代或未被取代,例如苯基,萘基,咪唑基,或吡啶基)。
“无毒”意指当治疗有效量被消化时,聚合物或任何释放到体内的离子都是无害的。优选地,实际上这种释放到体内的离子对患者是有利的。例如,可交换的离子为天然营养物如氨基酸。
“稳定”意指当聚合物以治疗有效量被吸收时不会被溶解,也不分解形成有害的副产物,并且基本上保持完整以便将完成了离子交换的离子排出体外。
在优选实例中,聚合物通过多功能交联共聚单体交联而完成,其中共聚单体的量约为单位总重量的1-25%(更优选2.5-20%)。
聚合物还优选包括一种或多种疏水性共聚单体,例如苯乙烯,乙烯基萘,乙基乙烯基苯,丙烯酰胺和甲基丙烯酰胺的N-烷基和N-芳基衍生物,丙烯酸烷基酯和芳基酯,甲基丙烯酸烷基酯和芳基酯,4-乙烯基联苯基,4-乙烯基茴香醚,4-氨基苯乙烯,和任何这些共聚单体的氟化衍生物(例如对一氟苯乙烯,全氟苯乙烯,六氟异丙基丙烯酸酯,六氟丁基甲基丙烯酸酯,或十七氟癸基甲基丙烯酸酯)。烷基优选为C1-C15烷基,可以是直链,支链或环状的(例如环己基),并且可进一步被取代或未取代。芳基优选具有一外或多个环并且可被取代或未取代,例如苯基,萘基,咪唑基,或吡啶基。聚合物还可以包括一种或多种带正电荷的共聚单体,例如乙烯基吡啶,二甲氨基甲基苯乙烯,或乙烯基咪唑。
第一个优选的聚合物实例的特征在于具有下式重复单元或其共聚物
该聚合物还可以包括以下一种或多种下述成份作为共聚单体:n-丁基甲基丙烯酰胺,六氟丁基甲基丙烯酸酯,十七氟癸基甲基丙烯酸酯,苯乙烯或其氟化衍生物,2-乙烯基萘,4-乙烯基咪唑,乙烯基吡啶,乙基甲基丙烯酸酯三甲铵,乙基丙烯酸酯三甲铵,4-乙烯基联苯基,4-乙烯基茴香醚,或4-氨基苯乙烯。
第二个优选聚合物的实例的特征在于具有下式重复单元或其共聚物
该聚合物可以包括以下一种或多种下述成份作为共聚单体:异丙基丙烯酰胺,苯乙烯或其氟化物,六氟异丙基丙烯酸酯,和乙基甲基丙烯酸酯三甲铵。
第三个优选聚合物的实例的特征在于具有下式重复单元或其共聚物
该聚合物还可以包括苯乙烯或其氟化衍生物作为共聚单体。
第四个优选聚合物的实例的特征在于具有下式重复单元或其共聚物。
第五个优选聚合物的实例的特征在于具有下式重复单元或其共聚物。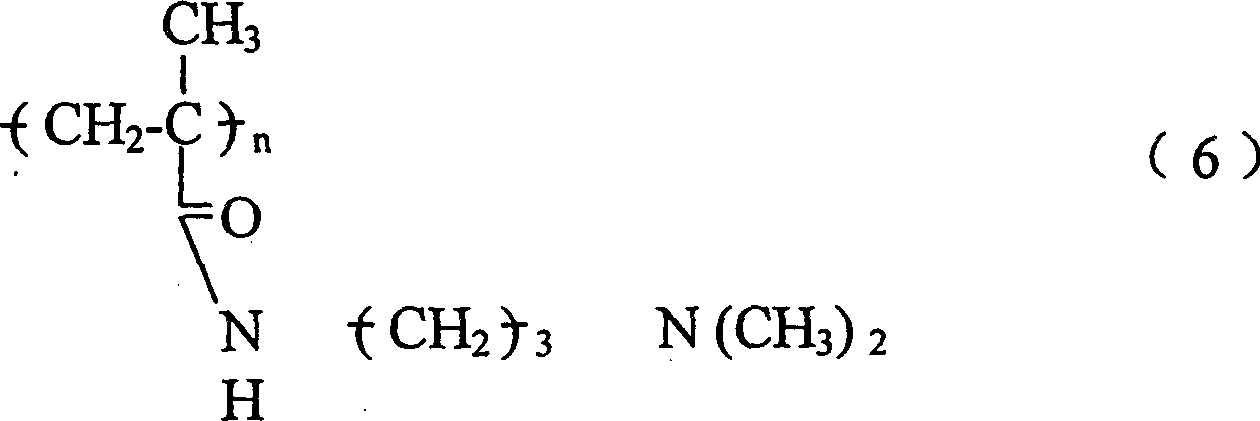
第六个优选聚合物的实例的特征在于具有下式重复单元或其共聚物
该聚合物还可以包括乙基乙烯基苯乙烯作为共聚单体。
第七个优选聚合物实例的特征在于具有下式重复单元或其共聚物。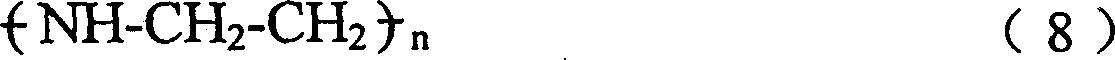
第八个优选聚合物实例的特征在于具有下式重复单元或其共聚物
该聚合物还可以包括苯乙烯或其氟化衍生物作为共聚单体。
第二方面,本发明特征在于通过离子交换从患者体内除去胆汁盐的方法,包括给患者服用的治疗有效量的一种或多种高交联聚合物,该聚合物具有下式重复单元或其共聚物,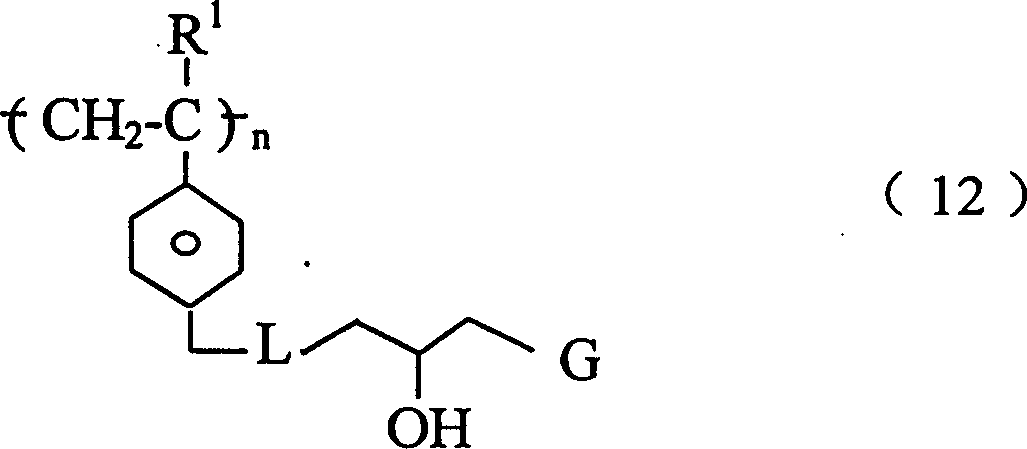
其中n为整数;R1是H或C1-C8烷基;L是-NH-或
G是
或
 并且,每个R2、R3和R4各自为H,C1-C8烷基,或芳基,该聚合物服用后无毒并且稳定。
并且,每个R2、R3和R4各自为H,C1-C8烷基,或芳基,该聚合物服用后无毒并且稳定。
在优选实例中,聚合物通过多功能交联共聚单体交联,其中共聚单体的量约为单体总重量的1-25%(更优选2.5-20%)。该聚合物还优选包括一种或多种上述疏水性共聚单体。
第一种优选聚合物的实例的特征在于具有下式重复单元或其共聚物该聚合物还可以包括苯乙烯或其氟化衍生物作为共聚单体。
第二种优选聚合物的实例的特征在于具有下式重复单元或其共聚物。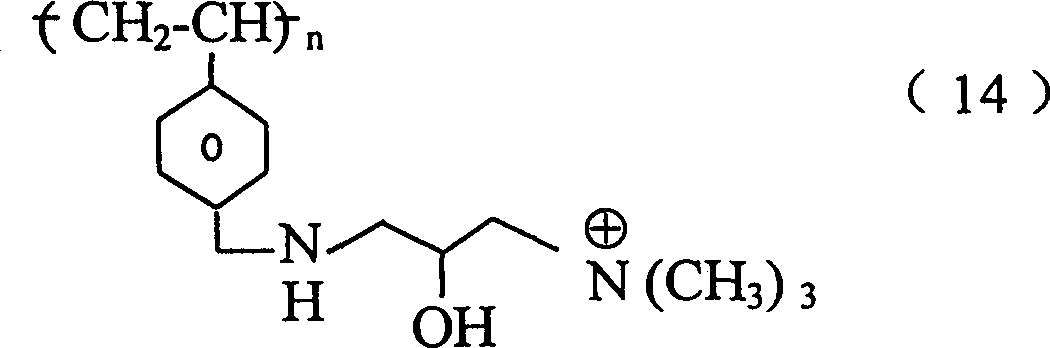
根据本发明第一和第二方面,聚合物可以带有正电荷,或者在服用后的生理pH下能变为带电荷。对于后者,带电荷的离子在服用后结合能与胆汁盐交换的带负电荷的平衡离子。如果聚合物带有正电荷,则该聚合物可被提供一种或多种可交换的平衡离子。适宜平衡离子的实例包括Cl-,Br-,CH3OSO3 -,HSO4 -,HO4 2-,HCO3 -,CO3 2-,乙酸盐,乳酸盐,琥珀酸盐,丙酸盐,丁酸盐,抗坏血酸盐,柠檬酸盐,马来酸盐,叶酸盐,氨基酸衍生物,核苷酸,类脂,或磷脂。平衡离子之间可以相同或不同。例如,聚合物可以含有二种不同类型的平衡离子,这两种离子被交换用于除去胆汁盐。也可以服用一种以上聚合物,这些聚合物各自具有结合不同的其固有电荷平衡离子。
本发明还包括用于除去胆汁盐的治疗组合物,该组合物包括治疗有效量的一种或多种上述聚合物。
另一方面,本发明涉及高交联聚合物组合物,该组合物包括具有下重复单元的聚合物
其中R1是H或甲基,Q是-NH-(CH2)3-或-O-(CH2)2,n为整数,以及至少一种选自乙烯基萘,乙烯基咪唑,苯乙烯氟化衍生物,氟化烷基甲基丙烯酸酯的附加共聚单体。
在这方面的一些优选实例中,R1是甲基,Q是-NH-(CH2)3-。该聚合物还可以包括作为共聚单体的乙基丙烯酸酯三甲铵或乙基甲基丙烯酸酯三甲铵。在另一个优选实例中,Q是-O-(CH2)2。
适宜的氟化苯乙烯衍生物的实例包括对-氟苯乙烯和五氟苯乙烯。适宜的氟化烷基甲基丙烯酸酯的实例包括六氟丁基甲基丙烯酸酯和十七氟癸基甲基丙烯酸酯。
在本发明的另一方面,还涉及高交联聚合物组合物,该组合物包括特征为具有下述重复单元的聚合物,
其中R1是H或甲基,Q是-NH-(CH2)3-或-O-(CH2)2,n为整数,以及作为附加共聚单体的(a)苯乙烯(b)乙基丙烯酸酯三甲铵或乙基甲基丙烯酸酯三甲铵,其中R1是甲基,Q是-NH-(CH2)3-。
再一方面,本发明涉及合成具有疏水和亲水单元高交联聚合物的方法,该方法包括在醇溶剂存在下与亲水性单体或疏水性单体反应。
再又一方面,本发明涉及从患者体内除去胆汁盐的方法,该方法包括给患者服用治疗有效量的以下物质的反应产物:
(a)具有下式重复单元的一种或多种高交联聚合物: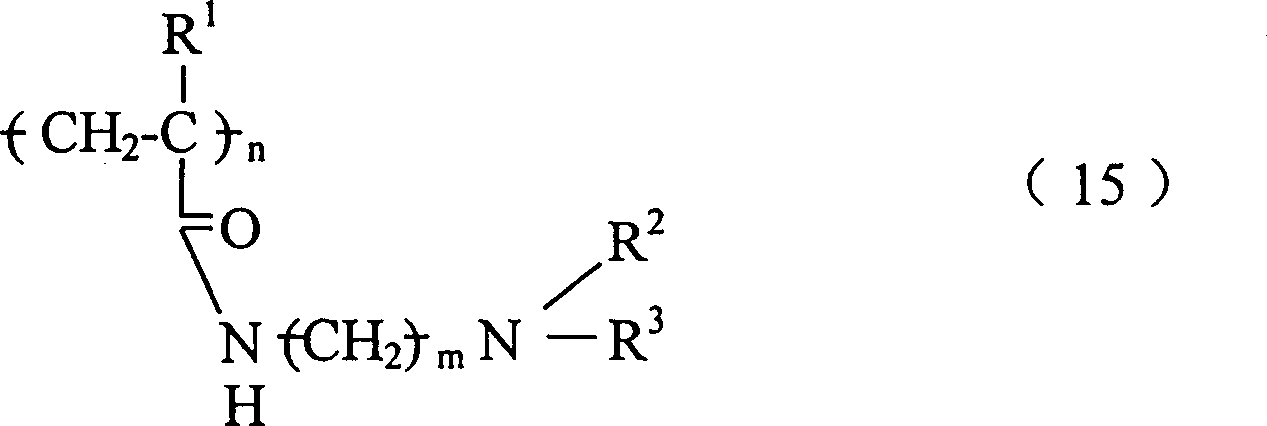
及其盐和共聚物,其中n和m是整数,每个R1、R2和R3各自是H,或C1-C8烷基;及
(b)至少一种烷基化试剂。该反应产物服用后无毒并且稳定。
“盐”意指在重复单元中胺氮被质子化后得到的带正电荷氮原子与它的带负电荷的平衡离子相结合。
“烷基化试剂”意指与交联聚合物反应时,以共价键在一个或多个聚合物氮原子上连接有烷基或其衍生物(例如芳烷基,羟基烷基,烷基铵盐,烷基酰胺,或它们的结合物)。
在优选实例中,这种反应产物通过多功能交联共聚单体交联,其中共聚单体约为单体总重量的1-25%(更优选2.5-20%)。
第一个优选聚合物的特征在于具有下式重复单元: 或其盐或其共聚物。
或其盐或其共聚物。
第二个优选聚合物的特征在于具有下式重复单元:或其盐或其共聚物。
优选烷基化试剂具有式RX,其中R是C1-C20烷基,C1-C20羟基烷基,C1-C20芳烷基,C1-C20烷基铵,或C1-C20烷基酰氨基,X包括一种或多种亲电子离去基团。“亲电子离去基团”意指在烷基化期间,可被交联聚合物中的氮原子置换的基团。优选离去基团的实例包括卤化物,环氧化物,甲苯磺酸盐,甲磺酸盐基团。例如,对于环氧基团,烷基化反应将引起三员环氧环开环。
优选烷基化试剂的实例包括C1-C20烷基卤化物(例如正丁基卤,正-己基卤,正-辛基卤,正-癸基卤,正-十二烷基卤,正-十四烷基卤,正-十八烷基卤,及其结合物);C1-C20卤代烷(例如1,10-二卤代癸烷;C1-C20羟基烷基卤(例如11-卤代-1-十一烷醇);C1-C20芳烷基卤(例如苄基卤);C1-C20烷基卤铵盐(例如(4-卤代丁基)三甲铵盐,(6-卤代己基)三甲铵盐,(8-卤代辛基)三甲铵盐,(10-卤代癸基)三甲铵盐,(12-卤代-十二烷基)三甲铵盐及它们的结合物);C1-C20烷基环氧铵盐(例如(缩水甘油基丙基)三甲铵盐);和C1-C20环氧烷基酰胺(例如N-(2,3-环氧丙烷)丁酰胺,N-(2,3-环氧丙烷)己酰胺,及其结合物)。
聚合物与至少二种烷基化试剂反应是特别优选的。在一个优选实例中,烷基化试剂之一是式RX,其中R是C1-C20烷基,X包括一种或多种亲水性离去基团(例如烷基卤),另一个烷基化试剂是式R’X,其中R’是C1-C20烷基铵基,X包括一种或多种亲水性离去基团(例如烷基卤化铵盐)。
在另一个优选实例中,烷基化试剂之一是RX,其中R是C1-C20烷基,X包括一种或多种亲水性离去基团(例如烷基卤),另一个烷基化试剂是R’X,其中R’是C1-C20羟基烷基,X包括一种或多种亲水性离去基团(例如羟基烷基卤)。
再一个优选实例中,烷基化试剂之一是C1-C20二卤代烷,另一个烷基化试剂是C1-C20烷基铵盐。
本发明提供了一种从患者体内除去胆汁盐的有效治疗方法(从而降低患者胆固醇的水平)。以治疗有效量服用这种组合物是无毒的并用稳定。
本发明还提供了具有亲水性和疏水性单元的聚合物有效合成,即该反应在醇溶剂存在下进行,这种醇溶剂由于其链转移性质通常认为不是好的聚合溶剂。
其它特征和优点将从以下优选实施方案及权利要求中看清楚。
本发明的最佳实施例组合物
优选聚合物具有在上述发明概述部分给出的结构式。该聚合物是高交联的。高交联水平使得聚合物完全不溶并且限制烷基化反应产物仅在胃肠道有活性。因此,该组合物是非系统活性的,这样将减小对患者的副作用。
聚合物优选通过在聚合期间在反应混合物中加入交联共聚单体进行交联。适宜交联共聚单体的实例包括二丙烯酸酯和二甲基丙烯酸酯(例如二丙烯酸乙二醇酯,二丙烯酸丙二醇酯,二丙烯酸丁二醇酯,二甲基丙烯酸乙二醇酯,二甲基丙烯酸丙二醇酯,二甲基丙烯酸丁二醇酯,二甲基丙烯酸聚乙二醇酯,二丙烯酸聚乙二醇酯),亚甲双丙烯酰胺,亚甲双甲基丙烯酰胺,亚乙双丙烯酰胺,亚乙双甲基丙烯酰胺,亚乙(H3CH2)双甲基丙烯酰胺,亚乙基双丙烯酰胺,二乙烯基苯,双酚A二甲基丙烯酸酯,和双酚A二丙烯酸酯。这些交联共聚单体即可以是商品也可用Mandeville等人“Process for Adjusting Ion Concentration in a Patient andCompositions Therefor,”U.S.S.N.08/065,113,申请日1993年5月20(转让给本专利申请相同受让人,这里引作参考)所述方法制备。典型地,交联剂的量为交联剂和单体总重量的1.0-25%,优选为2.5-20%。
优选地,聚合物包括一种或多种增加聚合物总疏水性的共聚单体。由于胆汁盐是疏水的,疏水性单体可以使聚合物与胆汁盐之间相互作用的选择性达到最大。
适宜的疏水性共聚单体的实例包括,例如丙烯酰胺,甲基丙烯酰胺,及其N-烷基(例如甲基,乙基,异丙基,丁基,己基,十二烷基,环己基,二环己基)和N-芳基(例如苯基,二苯基)衍生物;烷基和芳基丙烯酸酯及甲基丙烯酸酯(例如乙基,丙基,丁基,十二烷基),及其氟化衍生物(例如六氟异丙基丙烯酸酯,六氟丁基甲基丙烯酸酯,十七氟癸基丙烯酸酯);苯乙烯及其衍生物(例如二甲氨基甲基苯乙烯,4氨基苯乙烯,及其氟化衍生物,例如对-氟苯乙烯,五氟苯乙烯);乙基乙烯基苯;乙烯基萘;乙烯基吡啶;乙烯基咪唑;4-乙烯基联苯,4,4-乙烯基茴香醚;及其结合物。在制备这些聚合物中所用疏水性共聚单体的量为1-75%重量,优选3-65%。
所需疏水水平也可以通过适当选择交联共聚单体而方便地获得。例如二乙烯基苯是适宜的交联共聚单体并且也是疏水性的。另外,在二乙烯基苯中的主要“杂质”是乙基乙烯基苯,这种疏水性的可聚合的共聚单体也对聚合物总疏水性作出贡献。其它疏水性交联共聚单体包括双酚A二丙烯酸酯和双酚A二甲基丙烯酸酯。
交联聚合物可与一种或多种烷基化试剂反应。优选的烷基化试剂的实例己在发明概述中给出。A.聚合物的制备实施例1.聚(甲基丙烯酰氨基丙基三甲氯化铵)(聚MAPTAC)的制备
向1000mL的3颈园底烧瓶中加入下列物质:甲基丙烯酰氨基丙基三甲基氯化铵(MAPTAC)(40mL,50%水溶液,21g),二甲基丙烯酸乙二醇酯交联共聚单体(5.00g,4.76mL),乙酸乙酯(200mL),2-丙醇(200mL)。所得溶液是澄清的。接着加入聚合引发剂AIBN(0.1g)并将反应混合物加热至65℃。当温度达到65℃时,用氮气将溶液脱气5分钟,这时溶液变为混蚀,说明聚合正在进行。将反应在65℃再保持3小时,然后冷却至室温。
将所得聚合物(坚硬而粘)与500mL水合并以便软化聚合物,然后将其转移至混合器中并与1500mL 2-丙醇共混并离心。滗析混合物并借助100mL水转移至另一个混合机中。加入800mL2-丙醇并将混合物渗合,沉降然后滗析。将混合物与1000mL2-丙醇合并,掺合,过滤并真空干燥,得到12.6g聚合物。
用类似方法制备与0.5%亚甲双甲基丙烯酰胺交联共聚单体交联的聚MAPTAC,与10%亚甲双甲基丙烯酰胺交联共聚单体交联的聚MAPTAC;和与10%二乙烯基苯交联共聚单体交联的聚MAPTAC。实施例2.聚(乙烯胺)的制备
第一步为亚乙基双乙酰胺的制备。将乙酰胺(118g),乙醛(44.06g),乙酸铜(0.2g),和水(300mL)装在1L带有冷凝器、温度计和机械搅拌的三颈烧瓶中。加入浓HCl(34mL)并将混合物加热至45-50℃搅拌24小时。真空除去水得到稠淤渣,将其冷却至5℃时结晶。加入丙酮(200mL)并搅拌几分钟,滤出固体并弃去。将丙醇冷却至0℃并滤出固体。将该固体用500mL丙酮冲洗并空气干燥18小时,得到31.5g亚乙基双乙酰胺。
第二步为从亚乙基双乙酰胺制备乙烯基乙酰胺,将亚乙基双乙酰胺(31.05g),碳酰钙(2g)和硅藻土541(2g)装入带有温度计、机械搅拌和顶部Vigreux柱的蒸馏头的500mL三颈烧瓶中。通过加热烧瓶至180-225℃在35mmHg真空蒸馏混合物。收集除了产物之外还含有大部分乙酰胺(由NMR测定)的单一组分(10.8g)。将该固体产物溶于异丙醇(30mL)以形成聚合用粗乙烯基乙酰胺溶液。
将粗乙烯基乙酰胺溶液(15mL),二乙烯基苯(1g,技术级,纯度55%,混合异构体),和AIBN(0.3g)混合并在氮气氛下加热至回流90分钟,形成固体沉淀。冷却溶液,加入异丙醇(50mL),并用离心收集固体。用异丙醇冲洗该固体两次,用水冲洗一次,并在真空箱中干燥得到0.8g聚(乙烯基乙酰胺),该物质用于制备以下的聚(乙烯胺)。
将聚(乙烯基乙酰胺)(0.79g)装入含有水(25mL)和浓HCl(25mL)的100mL单颈烧瓶中。将混合物回流5天,并滤出固体,用水冲洗一次,用异丙醇冲洗两次,在真空箱中干燥得到0.77g产物。红外光谱分析表明还有大量酰胺(1656cm-1)存在,没有形成很多的胺(1606cm-1)。将反应物(约0.84g)悬浮在NaOH(46g)和水(46g)中并加热至沸点(约140℃)。由于发泡温度被降低,保持在约100℃2小时。加水(100mL)并过滤收集固体。用水冲洗固体一次后将其悬浮在水(500mL)中并用乙酸调节pH至5。再滤出固体,用水、异丙醇冲洗,并在真空箱中干燥得到0.51g产物。红外光谱分析表明已经开成大量胺。实施例3.聚(3-二甲氨基丙基丙烯酰胺)(DMAPA)的制备
在100mL三颈烧瓶中,将二甲氨基丙基丙烯酰胺(10g)和亚甲双丙烯酰胺交联共聚单体(1.1g)溶解在50mL水中。将该溶液在氮气下搅拌10分钟。将过硫酸钾(0.3g)和偏亚硫酸氢钠(0.3g)分别溶于2-3mL水中然后混合。几秒钟之后仍在氮气下将上述溶液加到单体溶液中。这时立即形成胶状物并被置过夜。除去胶状物并与500mL异丙醇混合。滤出固体并用丙酮冲洗三次。滤出白色固体粉末并在真空箱中干燥得到6.1g。实施例4.聚(二甲氨基丙基丙烯酰胺盐酸化物)(DMAPA·HCl)的制备
将二甲氨基丙基丙烯酰胺(20.10g)溶解在水(100mL)中并用浓HCl中和至pH6.95。加入亚甲双丙烯酰胺交联共聚单体(2.2g)和水(100mL)并温热(34°)以便溶解。在搅拌下加入过硫酸钾(0.2g)和偏亚硫酸氢钾(0.2g)。凝胶化后,将溶液放置6小时,并与异丙醇(600mL)混合3次,真空箱中干燥得到14.47g标题聚合物。
用类似方法制备与10%亚甲双甲基丙烯酰胺交联共聚单体交联的聚DMAPA·HCl。实施例5.聚(二甲氨基丙基甲基丙烯酰胺盐酸化物)(DMAPMA.HCl)
的制备
将二甲氨基丙基甲基丙烯酸胺(20.0g)溶解在水(100mL)中并用浓HCl中和至pH6.94。加入亚甲双丙烯酰胺交联共聚单体(2.2g)并将该溶液温热(39℃)以便溶解。在搅拌和氮气氛下加入过硫酸钾(0.3g)和偏亚硫酰氢钾(0.3g)。凝胶化后,将溶液放置过夜,并与异丙醇(500mL)混合两次,真空箱中干燥得到27.65g产物。将一些固体(3.2g;以-80/+200目筛子过筛)在水(100mL)中搅拌50分钟。再加水(100mL)并将溶液搅拌36分钟。离心收集固体,再悬浮在水(400mL)中,搅拌15分钟,再离心收集固体。最后将固体悬浮在水(500mL)中,搅拌90分钟,过滤收集。将固体在真空箱中干燥得出0.28g标题聚合物。实施例6.聚(甲基丙烯酰氨基丙基三甲基氯化铵)共聚(正-丁基甲基丙
烯酰胺)(MAPTAC共聚-BuMA)的制备
共聚单体正-丁基甲基丙烯酰胺(BuMA)制备如下:
在1L烧瓶中,将甲基丙烯酰氯(48.4mL,52.3g,0.500mol)溶解在四氢呋喃(300mL)中并将其放在冰浴中。在保持温度5-15℃时滴加含有丁胺(36.6g)和三乙胺(55.6g)的溶液。将溶液搅拌5分钟后,滤出固体三乙胺盐酸化物并弃去。从母液中真空除去溶剂并将所得黄色油用于下步制备,无需进一步纯化。产生71.58g BuMA共聚单体。
向1000mL三颈园底烧瓶中加入下列物质:甲基丙烯酰氨基丙基三甲基氯化铵(MAPTAC)(108mL,50%水溶液,56.8g),二甲基丙烯酸乙二醇酯交联共聚单体(19.62g),BuMA共聚单体(12.12g)和2-丙醇(850mL)。所得溶液为澄清的。接着将反应混合物加热至40℃,同时用氮气脱气。当溶液达到40℃时,加入催化剂,即过硫酸钾(0.75g)和偏硫酸氢钾(0.75g)的25mL水溶液。这时溶液立即开始变混浊,表明聚合正在进行。将反应在40℃保持24小时,然后冷却至室温。
过滤所得聚合物并在漏斗上用异丙醇洗涤,真空干燥得到64.54g标题聚合物。
将用于试验的聚合物每次用800mL水洗涤共两次,随后每次用500mL甲醇洗涤两次,得到34.5g纯化过的聚合物。
交联的MAPTAC共聚-BuMA共聚物也被用作为交联共聚单体的二甲基丙烯酸丙二醇酯制备,而不是用二甲基丙烯酸乙二醇酯制备,具体制备如下:
向1000mL三颈园底烧瓶中加入以下物质:甲基丙烯酰氨基丙基三甲基氯化铵(MAPTAC)(60mL,50%水溶液,31.5g),二甲基丙烯酸丙二醇酯交联共聚单体(9.81g),BuMA共聚单体(6.06g),和2-丙醇(300mL)。所得溶液是澄清的。接着将反应混合物加热至70℃,同时用氮气脱气。当溶液被加热至70℃时,加入催化剂AIBN(0.50g)。这时溶液立即开始变混浊,说明聚合正在进行。将反应在70℃保持6小时然后冷却至室温。
过滤所得聚合物并在漏斗上用异丙醇洗涤,真空干燥得到23.3g聚合物。
用类似方法并通过调整起始单体的比例制备与24%二甲基丙烯酸乙二醇酯交联共聚单体交联的MAPTAC共聚BuMA(5%),与0.5%亚甲双甲基丙烯酰胺交联共聚单体交联的MAPTAC共聚BuMA(2%)和与22%二甲基丙烯酸丙二醇酯交联共聚单体交联的MAPTAC共聚BuMA(14%)。实施例7.聚(甲基丙烯酰氨基丙基三甲基氯化铵)共聚(苯乙烯)
(MAPTAC共聚苯乙烯)的制备
向1000mL三颈园底烧瓶中加入以下物质,甲基丙烯酰氨基丙基三甲基氯化铵(MAPTAC)(60mL,50%水溶液,31.5g),二乙烯基苯交联共聚单体(2.00g),苯乙烯共聚单体(1.75g),和2-丙醇(300mL)。所得溶液是澄清的。接着将反应混合物加热至60℃,并用氮气脱气。当溶液温度达到60℃时,加入催化剂AIBN(0.50g)。这时该溶液立即开始变混浊,说明聚合正在进行。将反应在60℃保持24小时,然后冷却至室温。7小时后,该混合物变得非常粘稠,为了更好搅拌加入100mL异丙醇。
过滤所得聚合物并在漏斗上用异丙醇洗涤,真空干燥得到30.9g标题聚合物。
将用于试验的聚合物每次用1000mL水洗涤两次,接着每次用800mL甲醇洗涤两次得到纯化过的28.0g聚合物。
用类似方法并通过变化起始单体的比例制备与7.5%二甲基丙烯酸丁二醇酯交联共聚单体交联的MAPTAC共聚-苯乙烯(19%),与7%二乙烯基苯共聚单体交联的MAPTAC共聚-苯乙烯(23%),与6%二乙烯基苯共聚单体交联的MAPTAC共聚-苯乙烯(30%),和与6%二乙烯基苯共聚单体交联的MAPTAC共聚-苯乙烯(38%)。实施例8.聚(甲基丙烯酰氨基丙基三甲基氯化铵)共聚(乙烯基萘)
(MAPTAC共聚-VN)的制备
向1000mL三颈园底烧瓶中加入以下物质:甲基丙烯酰氨基丙基三甲基氯化铵(MAPTAC)(40mL的50%水溶液,21.0g),二乙烯基苯交联共聚单体(2.25g),2-乙烯基萘共聚单体(10.5g),和2-丙醇(320mL)。所得溶液是澄清的。接着将反应混合物加热至65℃,同时用氮气脱气。当溶液达到65℃时,加入催化剂AIBN(0.50g)。这时溶液立即开始变混浊,说明聚合正在进行。将反应在65℃保持20小时,然后冷却至室温。
过滤所得聚合物并用异丙醇在漏斗上洗涤,然后立即在400mL蒸馏水中浆化。将混合物搅拌0.5小时然后过滤。重复水洗涤一次。将滤饼在400mL甲醇中浆化并搅拌0.5小时。过滤混合物并重复甲醇浆化一次。真空干燥得到22.11g,65.5%标题聚合物。
用类似方法通过改变起始单体的比例制备与5%二乙烯基苯交联共聚单体交联的MAPTAC共聚-VN(39%)。实施例9.聚(甲基丙烯酰氨基丙基三甲基氯化铵)共聚(1-乙烯基咪唑)
(MAPTAC共聚-VI)的制备
向1000mL三颈园底烧瓶中加入以下物质:甲基丙烯酰氨基丙基三甲基氯化铵(MAPTAC)(40mL的50%水溶液,21.0g),二乙烯基苯交联共聚单体(2.25g),1-乙烯基咪唑共聚单体(12.54g),和2-丙醇(300mL)。所得溶液是澄清的。接着将反应混合物加热至65℃,同时用氮气脱气。当溶液达到65℃时,加入催化剂AIBN(0.50g)。这时溶液立即开始变混浊,说明聚合正在进行。将反应在65℃保持20小时,然后冷却至室温。
过滤所得聚合物并用异丙醇在漏斗上洗涤,然后立即在400mL蒸馏水中浆化。将混合物搅拌0.5小时然后过滤。重复水洗涤一次。将滤饼在400mL甲醇中浆化并搅拌0.5小时。过滤混合物并重复甲醇浆化一次。真空干燥得到7.34g,20.5%标题聚合物。实施例10.聚(三甲基氯化铵丙烯酸乙酯)共聚(苯乙烯)(TMAEAC共聚-
Sty)的制备
向1000mL三颈园底烧瓶中加入以下物质:三甲基氯化铵丙烯酸乙酯(TMAEAC)(99.4mL的50%水溶液,53.0g),二乙烯基苯交联共聚单体(7.00g),苯乙烯共聚单体(40.0g),和2-丙醇(800mL)。所得溶液是澄清的。接着将反应混合物加热至65℃,同时用氮气脱气。当溶液达到65℃时,加入催化剂AIBN(1.50g)。这时溶液立即开始变混浊,说明聚合正在进行。将反应在65℃保持6小时,冷却至60℃并再搅拌18小时,然后冷却至室温。
过滤所得聚合物并用异丙醇在漏斗上洗涤,然后立即在1000mL蒸馏水中浆化。将混合物搅拌0.5小时然后加入800mL甲醇并再搅拌0.5小时。将混合物沉降并滗析上清液,得到750mL剩余物。将剩余物再用750mL甲醇浆化并搅拌0.5小时。每次用800mL甲醇将甲醇浆化和滗析重复两次。随后加入800mL异丙醇,将混合物搅拌0.5小时并过滤。最后加入600mL异丙醇并搅拌0.5小时。真空干燥滤液得到49.2g,49.2%标题聚合物。
用类似方法通过改变起始单体的比例制备与8%二乙烯基苯交联共聚单体交联的TMAEAC共聚-苯乙烯(31%)和与6%二乙烯基苯交联共聚单体交联的TMAEAC共聚-苯乙烯(46%)。实施例11.聚(三甲基氯化铵甲基丙烯酸乙酯)共聚(苯乙烯)(TMAEMC
共聚-苯乙烯)的制备
向1000mL三颈园底烧瓶中加入以下物质:三甲基氯化铵甲基丙烯酸乙酯(TMAEMAC)(38.8mL的50%水溶液,21.7g),二乙烯基苯交联共聚单体(3.72g),苯乙烯共聚单体(15.66g),和2-丙醇(2500mL)。所得溶液是澄清的。接着将反应混合物加热至65℃,同时用氮气脱气。当溶液达到65℃时,加入催化剂AIBN(0.50g)。这时溶液立即开始变混浊,说明聚合正在进行。2小时后混合物变得非常粘稠,再加入100mL异丙醇。5小时后,混合物再次变得非常粘稠,所以再加入100mL异丙醇。将反应在65℃保持6小时,然后冷却至室温。
过滤所得聚合物并用异丙醇在漏斗上洗涤,然后立即在1000mL蒸馏水中浆化。将混合物搅拌0.5小时然后转移至混合器中并混合5分钟。过滤聚合物浆状物并加入1000mL蒸馏水并再将混合物搅拌0.5小时。过滤混合物,每次用500mL甲醇浆化滤饼两次。真空干燥滤液得到30.2g,75.9%标题聚合物。
用类似方法通过改变起始单体的比例制备与4%二乙烯基苯交联共聚单体交联的TMAEMC共聚-苯乙烯(58%),与4%二乙烯基苯交联共聚单体交联的TMAEMC共聚-苯乙烯(33%)和与4%二乙烯基苯交联共聚单体交联的TMAEMC共聚-苯乙烯(24%)。实施例12.聚(甲基丙烯酰氨基丙基-3-(三甲基氯化铵)),共聚(2,3,4,5,6-
五氟乙烯基苯乙烯)(MAPTAC共聚-StyF5)的制备
向1000mL三颈园底烧瓶中加入以下物质:甲基丙烯酰氨基丙基-3-(三甲基氯化铵)(MAPTAC)(24.5mL的50%水溶液,13.00g),二乙烯基苯交联共聚单体(1.00g),五氟苯乙烯共聚单体(6.00g),和2-丙醇(150mL)和AIBN(0.50g)。所得溶液是澄清的。接着将反应混合物加热至65℃,同时用氮气脱气。很短一段时间后溶液开始变混浊,说明聚合正在进行。5小时后混合物变得非常粘稠,所以再加入100mL异丙醇。将反应在65℃保持24小时,然后冷却至室温。
过滤所得聚合物并用异丙醇在漏斗上洗涤,然后立即在500mL蒸馏水中浆化。将混合物搅拌0.5小时,加入500mL蒸馏水并将混合物搅拌0.5小时。过滤聚合物,并将滤饼每次用300mL甲醇浆化两次。真空干燥滤液得到7.74g标题聚合物。
用类似方法通过改变起始单体的比例制备与5%二乙烯基苯交联共聚单体交联的MAPTAC共聚-StyF5(20%),与5%二乙烯基苯交联共聚单体交联的MAPTAC共聚-StyF5(40%)和与5%二乙烯基苯交联共聚单体交联的MAPTAC共聚-StyF5(45%)。实施例13.聚(甲基丙烯酰氨基丙基-3-(三甲基氯化铵)),共聚2-(三甲基
氯化铵)甲基丙烯酸乙酯共聚-苯乙烯(MAPTAC共聚-
TMAEMC共聚-Sty)的制备
向1000mL三颈园底烧瓶中加入以下物质:甲基丙烯酰氨基丙基-3-(三甲基氯化铵)(MAPTAC)(10.40g的50%水溶液,5.20g),2-(三甲基氯化铵)甲基丙烯酸乙酯(TMAEMC)(4.86g,70%水溶液,3.40g),二乙烯基苯交联共聚单体(1.00g),苯乙烯共聚单体(10.40g),2-丙醇(150mL)和AIBN(0.50g)。所得溶液是澄清的。接着将反应混合物加热至70℃,同时用氮气脱气。很短一段时间后溶液开始变混浊,说明聚合正在进行。将反应在70℃保持24小时,然后冷却至室温。
过滤所得聚合物并用异丙醇在漏斗上洗涤,然后立即在500mL甲醇中浆化。将混合物搅拌0.5小时。过滤聚合物浆状物并加入400mL蒸馏水并再将混合物搅拌0.5小时。过滤混合物并重复水浆化。过滤混合物并将滤饼每次用400mL甲醇浆化两次。真空干燥滤液得到5.39g标题聚合物。
用类似方法通过改变起始单体的比例制备与5%二乙烯基苯交联共聚单体交联的MAPTAC共聚-TMAEMC(34%)共聚-Sty(36%),与5%二乙烯基苯交联共聚单体交联的MAPTAC共聚-TMAEMC(31%)共聚-Sty(41%),与5%二乙烯基苯交联共聚单体交联的MAPTAC共聚-TMAEMC(28%)共聚-Sty(46%),与5%二乙烯基苯交联共聚单体交联的MAPTAC共聚-TMAEMC(23%)共聚-Sty(48%),与4%二乙烯基苯交联共聚单体交联的MAPTAC共聚-TMAEMC(26%)共聚-Sty(52%),与4%二乙烯基苯交联共聚单体交联的MAPTAC共聚-TMAEMC(17%)共聚-Sty(53%),与4%二乙烯基苯交联共聚单体交联的MAPTAC共聚-TMAEMC(15%)共聚-Sty(55%),与4%二乙烯基苯交联共聚单体交联的MAPTAC共聚-TMAEMC(13%)共聚-Sty(61.5%)。实施例14.聚(三甲基氯化铵甲基丙烯酸乙酯共聚-(异丙基丙烯酰胺))
(TMAEMAC共聚-IPA)的制备
首先,共聚单体异丙基丙烯酰胺(IPA)的制备如下:
在1L烧瓶中将丙烯酰氯(63mL,70.2g,0.775mol)溶解于四氢呋喃(200mL)并置于冰浴。滴加含异丙胺(127.7mL,88.67g,1.50mol)溶液,保持温度在5-15℃。将溶液搅拌10分钟后将固体异丙胺盐酸化物滤除。从母液中真空除去溶剂,得到几乎无色的油,经过放置使其固化,无需进一步纯化即可用于下面标题共聚物的制备。
向1000mL三颈园底烧瓶中加入以下物质:三甲基氯化铵甲基丙烯酸乙酯(76.5mL的50%水溶液,41.18g,0.213mol),亚甲基双丙烯酰胺交联共聚单体(2.40g),IPA共聚单体(4.52g,0.070mol),和水(200mL)。所得溶液是澄清的。搅拌反应混合物,同时用氮气脱气。当溶液脱气时加入过硫酸钾(0.3g)和焦亚磷酸钾(0.3g)组成的催化剂。聚合反应2分钟后开始,3分钟后凝结成胶。
第二天早晨,将该胶转到混合机中并加入1000mL水。混合几秒钟后聚合物吸进全部的水。膨胀的聚合物分几批用异丙醇混合几次以将其脱水。过滤所得聚合物并用异丙醇在漏斗上洗涤,真空干燥滤液得到36.8g标题共聚物。实施例15.聚(甲基丙烯酰氨基丙基-3-(三甲基氯化铵))共聚-(乙烯基吡啶)
(MAPTAC共聚-VP)的制备
向1000mL三颈园底烧瓶中加入以下物质:甲基丙烯酰氨基丙基-3-(三甲基氯化铵)(MAPTAC)(40mL的50%水溶液,21.0g),二乙烯基苯交联共聚单体(2.25g),乙烯基吡啶(14.0g,0.133mol),浓盐酸(11mL,0.133mol),2-丙醇(300mL),和AIBN(0.67g)。所得溶液是澄清的。接着,将反应混合物加热至60℃,同时用氮气脱气。短时间后该溶液开始混浊,表明聚合反应正在进行。将反应混合物在60℃保持20小时,然后冷却至室温。
过滤所得聚合物并在漏斗上用异丙醇洗涤,然后立即在1000mL蒸馏水中浆化。将混合物搅拌1小时。过滤聚合物浆液,在漏斗上用甲醇洗涤,然后在600mL甲醇中浆化1小时。过滤并空气干燥,得到20.4g共聚物。实施例16.聚(三甲基氯化铵甲基丙烯酸乙酯)共聚-(对氟苯乙烯)
(TMAEMC共聚-F1Sty)的制备
向500mL烧瓶中加入以下物质:三甲基氯化铵甲基丙烯酸乙酯(TMAEMC)(11.0g的70%水溶液,7.70g),二乙烯基苯交联共聚单体(0.50g),对氟苯乙烯共聚单体(4.00g),2-丙醇(125mL)和AIBN(0.25g)。所得溶液是澄清的。接着,将反应混合物加热至65℃,同时用氮气脱气。溶液马上开始混浊,表明聚合反应正在进行。反应在65℃保持6小时,然后冷却至室温。
通过滗析除去溶剂,聚合物立即在250mL蒸馏水中浆化。搅拌该混合物0.5小时,然后滗析。如此用水浆化3次。最后,用400mL甲醇浆化聚合物。过滤并真空干燥,得到5.42g,44.4%标题共聚物。
用类似方法通过改变起始单体的比例制备与4%二乙烯基苯交联共聚单体交联的TMAEMC共聚-F1Sty(24%)。实施例17.聚(甲基丙烯酰氨基丙基-3-(三甲基氯化铵))共聚-(六氟甲基丙
烯酸丁酯)(MAPTAC共聚-F6BMA)的制备
向1000mL三颈园底烧瓶中加入以下物质:甲基丙烯酰氨基丙基-3-(三甲基氯化铵)(MAPTAC)(28.5mL的50%水溶液,15.0g),二乙烯基苯交联共聚单体(1.00g),六氟甲基丙烯酸丁酯(4.00g),2-丙醇(150mL)和AIBN(0.50g)。所得溶液是澄清的。接着,将反应混合物加热至60℃,同时用氮气脱气。短时间后溶液开始混浊,表明聚合反应正在进行。反应在60℃保持24小时,然后冷却至室温。
将所得聚合物过滤并在漏斗上用异丙醇洗涤,随即用500mL蒸馏水浆化。将混合物搅拌1小时。过滤聚合物浆液,并再用水浆化一次。将聚合物在500mL甲醇中浆化1小时并过滤,并重复一次。最后将该混合物在400mL异丙醇中浆化并搅拌过夜。过滤及空气干燥,得到7.52g标题共聚物。实施例18.聚(三甲基氯化铵丙烯酸乙酯)共聚-(六氟丙烯酸异丙酯)
(TMAEAC共聚-F6IA)的制备
向1000mL三颈园底烧瓶中加入以下物质:三甲基氯化铵丙烯酸乙酯(30.0mL的50%水溶液,15.0g),二乙烯基苯交联共聚单体(1.00g),F6IPA(4.00g),AIBN(0.50g)和2-丙醇(150mL)。所得溶液是澄清的。将反应混合物搅拌,同时用氮气脱气,并加热至60℃。18小时后反应混合物允许被冷却至室温,滗析除去溶剂。剩下的聚合物在400mL水中浆化,搅拌1小时后过滤。再用水浆化一次,然后用甲醇浆化两次。最后,将聚合物在200mL异丙醇中浆化,搅拌2小时并过滤。空气干燥,得到5.59g标题聚合物。实施例19.聚(甲基丙烯酰氨基丙基-3-(三甲基氯化铵))共聚-(十七氟甲基
丙烯酸癸酯)(MAPTAC共聚-F17DecMA)的制备
向1000mL三颈园底烧瓶中加入以下物质:甲基丙烯酰氨基丙基-3-(三甲基氯化铵)(MAPTAC)(28.5mL的50%水溶液,15.0g),二乙烯基苯交联共聚单体(1.00g),十七氟甲基丙烯酸癸酯(4.00g),2-丙醇(1 50mL)和AIBN(0.40g)。所得溶液是澄清的。然后,将反应混合物加热至65℃,同时用氮气脱气。短时间后溶液开始混浊,表明聚合反应正在进行。4小时后反应混合物已经非常稠,再加入100mL异丙醇,将反应在65℃保持18小时,然后冷却至室温。
过滤所得聚合物,并在漏斗上用异丙醇洗涤,然后立即在600mL蒸馏水中浆化。将混合物搅拌1小时。过滤聚合物浆液,再用水浆化一次。然后将混合物在500mL甲醇中浆化1小时并过滤。空气干燥,得到17.73g标题聚合物。实施例20.聚(甲基丙烯酰氨基丙基-3-(三甲基氯化铵))共聚-(2-(三甲基氯
化铵)丙烯酸乙酯),共聚-苯乙烯(MAPTAC共聚-TMAEAC共
聚-Sty)的制备
向1000mL三颈园底烧瓶中加入以下物质:甲基丙烯酰氨基丙基-3-(三甲基氯化铵)(MAPTAC)(10.00g的50%水溶液,5.00g),2-(三甲基氯化铵)丙烯酸乙酯(TMAEAC)(6.00g的50%水溶液,3.00g),二乙烯基苯交联共聚单体(1.00g),苯乙烯(11.00g),2-丙醇(150mL)和AIBN(0.25g)。所得溶液是澄清的。然后,将反应混合物加热至70℃,同时用氮气脱气。短时间后溶液开始混浊,表明聚合反应正在进行。将反应在70℃保持24小时,然后冷却至室温。
过滤所得聚合物,并在漏斗上用异丙醇洗涤,然后立即在500mL甲醇中浆化。将混合物搅拌0.5小时。沉降和滗析聚合物浆液。加入200mL蒸馏水并将混合物搅拌0.5小时。滗析混合物,然后再用400mL水浆化。滗析混合物,聚合物每次用200mL甲醇浆化两次。过滤并空气干燥,得到2.76g标题共聚物。
用类似方法通过改变起始单体的比例制备与5%二乙烯基苯交联共聚单体交联的MAPTAC共聚-TMAEAC(10%)共聚-Sty(60%)。实施例21.聚(2-(三甲基氯化铵)丙烯酸乙酯),共聚-(2,3,4,5,6-五氟苯乙烯)
(TMAEAC共聚-StyF5)的制备
向1000mL三颈园底烧瓶中加入以下物质:2-(三甲基氯化铵)丙烯酸乙酯(TMAEAC)(24.0mL的50%水溶液,13.00g),二乙烯基苯交联共聚单体(1.00g),五氟苯乙烯(6.00g),2-丙醇(150mL)和AIBN(0.50g)。所得溶液是澄清的。然后,将反应混合物加热至65℃,同时用氮气脱气。短时间后溶液开始混浊,表明聚合反应正在进行。2小时后混合物已经非常稠,因此加入100mL异丙醇。将反应在65℃保持22小时,然后冷却至室温。
过滤所得聚合物,并在漏斗上用异丙醇洗涤,然后立即在400mL蒸馏水中浆化。将混合物搅拌0.5小时。将聚合物浆液过滤并加入600mL蒸馏水,再将混合物搅拌0.5小时。过滤混合物,滤饼在400mL甲醇中浆化。过滤并空气干燥,得到7.26g标题共聚物。
用类似方法通过改变起始单体的比例制备与5%二乙烯基苯交联共聚单体交联的TMAEAC共聚-StyF5(20%)。实施例22.聚(2-(三甲基氯化铵)甲基丙烯酸乙酯),共聚-(2,3,4,5,6-五氟苯
乙烯)(TMAEMC共聚-StyF5)的制备
向1000mL三颈园底烧瓶中加入以下物质:2-(三甲基氯化铵)甲基丙烯酸乙酯(TMAEMC)(19.52mL的70%水溶液,13.66g),二乙烯基苯交联共聚单体(1.00g),五氟苯乙烯(9.18g),2-丙醇(150mL)和AIBN(0.40g)。所得溶液是澄清的。然后,将反应混合物加热至70℃,同时用氮气脱气。短时间后溶液开始混浊,表明聚合反应正在进行。1.5小时后混合物已经非常稠,因此加入50mL异丙醇。将反应在70℃保持5小时,然后冷却至室温。
过滤所得聚合物,并在漏斗上用异丙醇洗涤,然后立即在500mL蒸馏水中浆化。将混合物搅拌0.25小时。将聚合物浆液过滤并加入500mL蒸馏水,再将混合物搅拌0.25小时。聚合物再用水浆化一次。过滤混合物,滤饼在每次300mL的甲醇中浆化三次。过滤并空气干燥,得到1.26g标题共聚物。
用类似方法通过改变起始单体的比例制备与4%二乙烯基苯交联共聚单体交联的TMAEMC共聚-StyF5(24%)以及与4%二乙烯基苯交联共聚单体交联的TMAEMC共聚-StyF5(39%)。实施例23.聚(吖丙啶)的制备
将聚吖丙啶(120g的50%水溶液;Scientific Polymer Products)溶解于水(250mL)。滴加表氯醇(22.1mL)。将溶液加热至60℃4小时。之后,形成胶体。移出胶体,用水(1.5L)调合,并滤出固体,用水(3L)漂洗三次和用异丙醇(3L)漂洗两次。所得胶体在真空炉中干燥,得到81.2g标题聚合物。实施例24.聚(甲基丙烯酰氨基丙基-3-(三甲基氯化铵)),共聚-(2-(三甲基
氯化铵)甲基丙烯酸乙酯),共聚-(2,3,4,5,6-五氟苯乙烯)
(MAPTAC共聚-TMAEMC共聚-StyF5)的制备
向1000mL三颈园底烧瓶中加入以下物质:甲基丙烯酰氨基丙基-3-(三甲基氯化铵)(MAPTAC)(10.00g的50%水溶液,5.00g),2-(三甲基氯化铵)甲基丙烯酸乙酯(TMAEMC)(5.71g的70%水溶液,4.00g),二乙烯基苯交联共聚单体(1.00g),五氟苯乙烯(10.00g),2-丙醇(1 50mL)和AIBN(0.50g)。所得溶液是澄清的。然后,将反应混合物加热至70℃,同时用氮气脱气。短时间后溶液开始混浊,表明聚合反应正在进行。将反应在70℃保持24小时,然后冷却至室温。
过滤所得聚合物,并在漏斗上用异丙醇洗涤,然后立即在500mL甲醇中浆化。将混合物搅拌0.25小时。过滤浆状聚合物,然后每次用300mL水浆化,共三次。将最后一次浆状聚合物搅合5分钟。过滤混合物,然后每次用300mL甲醇浆化滤饼,共两次。过滤并真空干燥,得到9.74g共聚物。实施例25.聚(2-(三甲基氯化铵)丙烯酸乙酯),共聚-(2-(三甲基氯化铵)甲
基丙烯酸乙酯),共聚-苯乙烯(TMAEAC共聚-TMAEMC共聚
-Sty)的制备
向1000mL三颈园底烧瓶中加入以下物质:2-(三甲基氯化铵)丙烯酸乙酯(TMAEAC)(6.00g的50%水溶液,3.00g),2-(三甲基氯化铵)甲基丙烯酸乙酯(TMAEMC)(4.29g的70%水溶液,3.00g),二乙烯基苯交联共聚单体(1.00g),苯乙烯(13.00g),2-丙醇(150mL)和AIBN(0.50g)。所得溶液是澄清的。然后,将反应混合物加热至70℃,同时用氮气脱气。短时间后溶液开始混浊,表明聚合反应正在进行。将反应在70℃保持24小时,然后冷却至室温。
滗析所得聚合物,然后立即在500mL甲醇中浆化。将混合物搅拌0.5小时。过滤浆状聚合物并加入500mL蒸馏水。将混合物搅拌0.5小时并混合10分钟。将混合物沉降并滗析水。重复水浆化两次,每次用400mL甲醇浆化滗析剩余物,并每次沉降和滗析。真空干燥得到8.03g标题共聚物。实施例26.聚(甲基丙烯酰氨基丙基-3-(三甲基氯化铵)),共聚-(2-(三甲基
氯化铵)丙烯酸乙酯),共聚-(2,3,4,5,6-五氟苯乙烯)(MAPTAC
共聚-TMAEAC共聚-StyF5)的制备
向1000mL三颈园底烧瓶中加入以下物质:甲基丙烯酸乙酯-3-(三甲基氯化铵)(MAPTA)(8.00g,50%水溶液,4.00g),2-(三甲基氯化铵)丙烯酸乙酯(TMAEMA)(6.00g的50%水溶液,3.00g),二乙烯基苯交联共聚单体(1.00g),五氟苯乙烯(12.00g),2-丙醇(150mL)和AIBN(0.50g)。所得溶液是澄清的。然后,将反应混合物加热至70℃,同时用氮气脱气。短时间后溶液开始混浊,表明聚合反应正在进行。将反应在70℃保持24小时,然后冷却至室温。
过滤所得聚合物,并在漏斗上用异丙醇洗涤,然后立即在400mL甲醇中浆化。将混合物搅拌0.5。将浆状聚合物过滤然后每次用250mL水浆化,共两次。将最后一次浆状聚合物混合5分钟。过滤混合物,然后每次用250mL甲醇浆化,共两次。真空干燥滤液得到7.80g共聚物。实施例27.聚(2-(三甲基氯化铵)丙烯酸乙酯),共聚-(2-(三甲基氯化铵)甲
基丙烯酸乙酯),共聚-(2,3,4,5,6-五氟苯乙烯)(TMAEAC共聚
-TMAEMC共聚-Sty F5)的制备
向1000mL三颈园底烧瓶中加入以下物质:2-(三甲基氯化铵)丙烯酸乙酯(TMAEAC)(6.00g的50%水溶液,3.00g),2-(三甲基氯化铵)甲基丙烯酸乙酯(TMAEMC)(4.29g的,70%水溶液,3.00g),二乙烯基苯交联共聚单体(1.00g),五氟苯乙烯(13.00g),2-丙醇(150mL)和AIBN(0.50g)。所得溶液是澄清的。然后,将反应混合物加热至70℃,同时用氮气脱气。短时间后溶液开始混浊,表明聚合反应正在进行。将反应在70℃保持24小时,然后冷却至室温。
滗析所得聚合物,然后立即在400mL甲醇中浆化。将混合物搅拌0.5小时。过滤浆状聚合物,然后每次用200mL水浆化,共两次。将第二次浆状聚合物混合5分钟。过滤混合物并每次用200mL甲醇浆化,共两次。真空干燥得到6.87g标题共聚物。实施例28.聚(甲基丙烯酰氨基丙基三甲基氯化铵)共聚(4-乙烯基联苯)
(MAPTAC共聚-VBPh)的制备
向1000mL三颈园底烧瓶中加入以下物质:甲基丙烯酰氨基丙基三甲基氯化铵(MAPTAC)(10.49g的50%水溶液,0.0475mol),4-乙烯基联苯(VBPh)(9.01g,0.050mol),二乙烯基苯交联共聚单体(1.47g),2-丙醇(150mL),聚合引发剂AIBN(0.25g)。所得混合物含有不溶的VBPh(加热后溶解)。然后,将反应混合物加热至70℃,同时用氮气脱气。短时间后溶液开始混浊,表明聚合反应正在进行。将反应在70℃保持24小时小时,然后冷却至室温。
过滤所得聚合物,并在漏斗上用异丙醇洗涤,然后立即在200mL甲醇中浆化,接着搅拌1小时。过滤浆状聚合物,然后重复甲醇浆化步骤。接着每次用200mL水浆化聚合物,共两次。过滤所得混合物然后将滤饼然后每次用200mL甲醇浆化,共两次。过滤所得混合物并真空干燥得到9.59g共聚物。实施例29.聚(甲基丙烯酰氨基丙基三甲基氯化铵)共聚(4-乙烯基联苯)
(MAPTAC共聚-VA)的制备
向1000mL三颈园底烧瓶中加入以下物质:甲基丙烯酰氨基丙基三甲基氯化铵(MAPTAC)(10.49g的50%水溶液,0.0475mol),4-乙烯基茴香醚(VA)(6.71g,0.050mol),二乙烯基苯交联共聚单体(1.30g),2-丙醇(200mL),聚合引发剂AIBN(0.40g)。然后,将所得澄清溶液加热至70℃,同时用氮气脱气。几小时后溶液开始混浊,表明聚合反应正在进行。将反应在70℃保持36小时,然后冷却至室温。
过滤所得聚合物,并在漏斗上用异丙醇洗涤,然后立即在200mL甲醇中浆化,接着搅拌1小时。过滤浆状聚合物。接着每次用200mL水浆化聚合物两次。过滤所得混合物,将滤饼用200mL甲醇浆化,然后用200异丙醇浆化。过滤所得混合物并真空干燥得到5.19g共聚物。实施例30.聚[(N-(4-甲基苯乙烯)-N’-(3-三甲基氯化铵-2-羟基丙基)哌嗪]
的制备
第一步是制备4-(哌嗪基甲基)苯乙烯的反应。
向500mL烧瓶中加入乙烯基苄基氯(7.63g,0.050mol),哌嗪(8.61g,0.100mol)和异丙醇(50mL)。将所得混合物在70℃加热45分钟,然后慢慢冷却至室温以便形成结晶物质的浆。将该浆状物放在冰箱中约3小时,然后过滤。将固体哌嗪盐酸盐真空干燥,然后称重(5.55g)并弃去。在旋转蒸发器上将母液浓缩至约25mL并加入乙酸乙酯(50mL)。将所得混合物放在冰箱中约10分钟,然后将第二次得到的哌嗪盐酸盐过滤并弃去。在旋转蒸发器上蒸发母液至干得到10.35g粗4-(哌嗪甲基)苯乙烯,将其用于制备以下N-(4-甲基苯乙烯)-N’-(3-三甲基氯化铵-2-羟基丙基)哌嗪,并且无需进一步纯化。向500mL烧瓶中加入4-(哌嗪甲基)苯乙烯(10.35g,约0.45mol),缩水甘油基三甲基氯化铵(7.58g,0.045mol)和异丙醇(50mL)。将所得混合物加热至60℃并搅拌20小时,然后冷却至室温,并用于以下聚合反应且无需进一步纯化。
将二乙烯基苯交联共聚单体(0.93g)加到0.40mol N-(4-甲基苯乙烯)-N’-(3-三甲基氯化铵-2-羟基丙基)哌嗪中。用氮气将所得溶液脱气,然后加入聚合引发剂AIBN(0.20g)。将温度升至70℃继续用氮气脱气并放置3小时,这时大量的交联聚合物沉淀。然后将混合物冷却至40℃并过滤。
过滤所得聚合物,并在漏斗上用异丙醇洗涤,然后立即在200mL甲醇中浆化,接着搅拌1小时。过滤浆状聚合物,然后重复甲醇浆化步骤。每次用200mL水浆化聚合物,共两次。接着过滤,将滤饼每次用200mL甲醇浆化,共两次,真空干燥得到7.90g共聚物。实施例31.聚(N-(4-甲基苯乙烯)-N’-(3-三甲基氯化铵-2-羟基丙基)哌嗪,
共聚苯乙烯的制备
向0.022mol N-(4-甲基苯乙烯)-N’-(3-三甲基氯化铵-2-羟基丙基)哌嗪(按实施例30所述制备)中加入苯乙烯共聚单体(2.29g,0.022mol)和二乙烯基苯交联共聚单体(0.50g,0.004mol)。用氮气将所得溶液脱气,然后加入聚合引发剂AIBN(0.3g)。将温度升至70℃,继续用氮气脱气并放置4小时,这时大量的交联聚合物沉淀。然后添加25mL异丙醇并继续加热19小时。之后,将混合物冷却至室温并过滤,得到固体聚合物。
过滤所得聚合物,并在漏斗上用异丙醇洗涤,然后立即在200mL甲醇中浆化。接着,将混合物搅拌0.5小时。过滤浆状聚合物,每次用250mL水浆化聚合物,共两次。过滤混合物,滤饼在200mL甲醇中浆化,过滤,然后在200mL异丙醇中浆化。过滤并真空干燥,得到4.98g共聚物。
含有2∶1摩尔比的苯乙烯:季铵单体也被用类似方法制备。实施例32.聚(N-(3-三甲基氯化铵-2-羟基丙基)-4-氨基苯乙烯的制备
第一步是按下面所述制备4-氨基甲基苯乙烯。
向250mL烧瓶中加入乙烯基苄基氯(7.63g,0.050mol),浓氨水(9.8mL)和异丙醇(40mL)。将混合物搅拌1星期,这时大量结晶物质(氯化铵)沉淀下来。滤出固体并用异丙醇洗涤。母液在旋转蒸发器上蒸发直到闻不到氨味。加入异丙醇(50mL),然后将混合物冷冻几小时。冷冻之后,已经沉淀的4-氨基甲基苯乙烯氯化铵被滤出,得到4-氨基甲基苯乙烯,无需进一步纯化即可使用。
在500mL烧瓶中加入4-氨基甲基苯乙烯(0.050mol),缩水甘油基三甲基氯化铵(7.62g,0.050mol)和水(约2mL)形成溶液,并加入二乙烯基交联共聚单体(0.98g)。将所得溶液在70℃加热5小时,之后,加入少量水(约5-10mL)以溶解一些沉淀的盐。加入聚合引发剂AIBN(0.20g),同时用氮气给溶液脱气。反应混合物这时已变得非常稠,需要添加异丙醇使聚合物可以搅拌。
在70℃搅拌约16小时后,将混合物冷却至室温并过滤。所得聚合物在漏斗上用甲醇洗涤,然后立即在200mL甲醇中浆化。接着,将所得混合物搅拌1小时。过滤浆状聚合物,用甲醇再重复一次浆化过程。然后,聚合物用每次200mL甲醇浆化,共两次。过滤混合物,滤饼在200mL甲醇中浆化。过滤并真空干燥,得到8.68g聚合物。实施例33.用1-碘辛烷烷基化剂将与亚甲基双甲基丙烯酰胺交联的聚(二
甲氨基丙基甲基丙烯酰胺)烷基化
用实施例5所述方法制备的与亚甲基双甲基丙烯酰胺交联的聚(二甲氨基丙基甲基丙烯酰胺)(1.0g)悬浮在甲醇(100mL)中,并加入氢氧化钠(0.2g)。搅拌15分钟后,加入1-碘辛烷(1.92mL),并将混合物在60℃搅拌20小时。然后,冷却混合物,滤出固体。通过将固体悬浮在异丙醇(500mL)中来洗涤它。之后,搅拌1小时并通过过滤收集。用氯化钠水溶液(500mL的1M溶液)重复洗涤过程两次,用水(500mL)洗涤,共两次,用异丙醇(500mL)洗涤一次。在50℃真空炉中干燥24小时,得到0.1g烷基化产品。实施例34.用1-碘辛烷烷基化剂将与亚甲基双甲基丙烯酰胺交联的聚(二
甲氨基丙基丙烯酰胺)烷基化
根据实施例33所述方法将用实施例4所述方法制备的与亚甲基双甲基丙烯酰胺交联的聚(二甲氨基丙基丙烯酰胺)(10g)烷基化,得到2.95g烷基化产品。实施例35.聚(2-(甲基丙烯酰氨基)乙基三甲基碘化铵)共聚-苯乙烯的制
备
第一步是按下面所述制备2-(N’,N’-二甲基氨基)-N-乙基甲基丙烯酰胺盐酸盐。
向1000mL烧瓶中加入甲基丙烯酰氯(52.3g,0.5mol)和四氢呋喃(300mL)。将溶液冷却至10℃以下,然后滴加N,N-二甲基氨基乙胺(30.5g,0.35mol)的四氢呋喃(100mL),同时保持温度在8-10℃。添加完成后过滤混合物,用冷四氢呋喃洗涤,真空干燥,得到65.69g 2-(N’,N’-二甲基氨基)-N-乙基甲基丙烯酰胺盐酸盐。
下一步是如下所述制备2-(甲基丙烯酰氨基)乙基三甲基碘化铵。
将氢氧化钾(15.4g,0.24mol)和甲醇(240mL)放入500mL烧瓶,然后将混合物充分搅拌使氢氧化钾完全溶解。在溶液中加入2-(N’,N’-二甲基氨基)-N-乙基甲基丙烯酰胺盐酸盐(46.35g,0.24mol),并将所得混合物搅拌0.5小时。过滤混合物以除去氯化钾。滤液在旋转蒸发器中浓缩。将异丙醇(400mL)和甲基碘(18.7mL,42.6g,0.30mol)加到浓缩的滤液中,然后将混合物在室温搅拌过夜。第二天早晨,滤出的固体产物,用异丙醇洗涤,真空干燥,得到61.08g 2-(甲基丙烯酰氨基)乙基三甲基碘化铵,为白色晶状固体。
接着,将2-(甲基丙烯酰氨基)乙基三甲基碘化铵(12.24g,0.050mol),苯乙烯(5.2g,0.050mol),二乙烯基苯交联共聚单体(0.65g,0.005mol),异丙醇(150mL),水(20mL),和聚合引发剂AIBN(0.4g)加到1000mL烧瓶中。用氮气给所得溶液脱气,同时加热至70℃。然后,将溶液在70℃及氮气下搅拌24小时,之后,将其冷却至室温。这时,滗析溶剂,然后将200mL甲醇加到烧瓶中形成浆液,并搅拌过夜。过滤产物,然后加到搅拌器中与500mL水混合。搅合所得混合物15分钟,然后过滤。依次用水(200mL)和甲醇(200mL)洗涤留下的固体物质。过滤并真空干燥,得到3.22g标题聚合物。聚合物试验A.人造肠液的制备试验方法1
将碳酸钠(1.27g)和氯化钠(1.87g)溶解于400mL蒸馏水中。向此溶液加入纯胆汁酸混合物,该混合物由牛磺胆酸(0.138g,0.24mmol),甘氨胆酸(0.292g,0.60mmol),甘氨脱氧胆酸(0.085g,0.18mmol)和甘氨鹅脱氧胆酸(glycochenodeoxycholic)(0.085g,0.18mmol)组成。用乙酸将溶液的pH调至7.20。该溶液被用于多种聚合物的试验。该溶液中全部胆汁盐的浓度为3毫摩尔,该浓度近似等于十二指肠中正常体液的浓度。
聚合物试验如下。
在40mL离心管中加入0.25g聚合物和上文制备的20mL人造小肠液。将混合物在保持于37℃的水浴中搅拌3小时。将混合物离心,并过滤有点儿混浊的上清液。通过用3a-羟基甾族脱氢酶进行酶测定来分析滤液的总3-羟基甾族化合物含量(如下所述)。试验方法2
将碳酸钠(1.27g)和氯化钠(1.87g)溶解于400mL蒸馏水中。在此溶液中加入甘氨胆酸(1.95g,4.0mmol)或甘氨鹅脱氧胆酸(1.89g,4.0mmol),制成10mM溶液。用乙酸将溶液的pH调至6.8。这些储备溶液被用于多种聚合物的试验。
聚合物试验如下。
在14mL离心管中加入10mg聚合物和从10mM储液(制备如上)制备的,浓度为0.1-10mM的10mL胆汁盐溶液,以及适量无胆汁盐缓冲液。将混合物在保持于37℃的水浴中搅拌3小时。过滤混合物。通过用3α-羟基甾族脱氢酶进行酶测定来分析滤液的总3-羟基甾族化合物含量(如下所述)。
总胆汁盐含量的酶测定
制备四种储液。
溶液1:Tris-HCl缓冲液,含0.133M Tris,0.666mM EDTA,
pH9.5。
溶液2:肼水合物溶液,含1M pH为9.5的肼水合物。
溶液3:NAD+溶液,含7mM NAD+,pH7.0。
溶液4:HSD溶液,含2单位/mL的Tris-HCl缓冲液(0.03M Tris,
1mM EDTA),pH7.2。
在3mL比色杯中加入1.5mL溶液1,1.0mL溶液2,0.3mL溶液3,0.1mL溶液4和0.1mL上述聚合物试验得到的上清液/滤液。将该溶液置于UV-VIS分光光度计并测量其在340nm处的NADH吸收(O.D.)。胆汁盐浓度由上面制备的人造肠液稀释度刻度曲线来测定。
前面所述的全部聚合物都参加了上述一个或两个试验,并且全部对从人造肠液中除去胆汁盐有效。使用
本发明聚合物可以约1mg/kg/天至约10g/kg/天剂量给患者口服给药。具体的剂量取决于每个患者的情况,如患者的重量和所要除去的胆汁盐的范围。聚合物可以水合物或非水合物形式给药,如果需要,还可以添加调味剂以增加患者的接受能力。另外可以添加的成份还有如人造色素。
适于给药的实例包括丸剂,片剂,囊剂和粉剂(如喷洒在食物上)。这些丸剂,片剂,囊剂,或粉剂可以用有保护能力的物质包衣,该物质保护组合物不受患者胃中的胃酸影响,以获得足够长时间不被分解地抵达患者小肠。该聚合物可以独自或与药物上可接受的载体物质如碳酸镁或乳糖一起给药,或聚合物与磷脂形成微胞。
其它具体说明包括在本发明的权利要求中。
Claims (29)
1.一种用于制造通过离子交换从患者体内除去胆汁盐的药物的聚合物组合物,包括治疗有效量的下述物质的反应产物:
(a)一种或多种具有下式重复单元的交联聚合物: 或其盐和其共聚合物,其中n为整数;R1是H或C1-C8烷基;M是
或-Z-R2;Z为O,NR3,S,或(CH2)m;m=0-10;R3是H或C1-C8烷基;及R2是或
或其盐和其共聚合物,其中n为整数;R1是H或C1-C8烷基;M是
或-Z-R2;Z为O,NR3,S,或(CH2)m;m=0-10;R3是H或C1-C8烷基;及R2是或
 其中P=0-10,并且R4和R5,是H;R6是H,C1-C8烷基,或芳基;和
其中P=0-10,并且R4和R5,是H;R6是H,C1-C8烷基,或芳基;和
(b)至少一种烷基化试剂,其中所述聚合物是通过多官能交联试剂交联的,所述试剂的量占单体总重量的1-25%重量比;所述的烷基化试剂具有式RX,其中R包括C1-C20烷基,C1-C20羟基烷基,C1-C20芳烷基,C1-C20烷基铵,或C1-C20烷基酰氨基,及X包括一种或多种亲电子离去基团。
2.根据权利要求1所述的聚合物组合物,其中所述的聚合物是通过多功能交联共聚单体交联的。
3.根据权利要求2所述的聚合物组合物,其中所述的交联共聚单体为单体总重量的2.5-20%。
4.一种用于制造通过离子交换从患者体内除去胆汁盐的药物的聚合物组合物,包括治疗有效量的下述物质的反应产物,(a)一种或多种交联聚合物,包括下式表示的重复单元: 或其盐和其共聚物,其中n是整数,p=0-10,R1是H或C1到C8烷基基团,R4和R5每个是H,C1到C8烷基或芳基;(b)至少两种烷基化试剂,其中(i)一种所述的烷基化试剂具有化学式RX,其中R是C1到C20烷基,X是一个或多个亲电性离去基团,另一种所述的烷基化试剂具有化学式R’X,其中R’是C1到C20烷基铵基团,X是一个或多个亲电子离去基团;或(ii)一种所述的烷基化试剂具有化学式RX,其中R是C1到C20烷基,X是一个或多个亲电性离去基团,另一种所述的烷基化试剂具有化学式R’X,其中R’是C1到C20羟烷基,X是一个或多个亲电子离去基团;(iii)一种所述烷基化试剂是C1到C20二卤代烷烃,另一种所述的烷基化试剂是C1到C20烷基铵盐。
或其盐和其共聚物,其中n是整数,p=0-10,R1是H或C1到C8烷基基团,R4和R5每个是H,C1到C8烷基或芳基;(b)至少两种烷基化试剂,其中(i)一种所述的烷基化试剂具有化学式RX,其中R是C1到C20烷基,X是一个或多个亲电性离去基团,另一种所述的烷基化试剂具有化学式R’X,其中R’是C1到C20烷基铵基团,X是一个或多个亲电子离去基团;或(ii)一种所述的烷基化试剂具有化学式RX,其中R是C1到C20烷基,X是一个或多个亲电性离去基团,另一种所述的烷基化试剂具有化学式R’X,其中R’是C1到C20羟烷基,X是一个或多个亲电子离去基团;(iii)一种所述烷基化试剂是C1到C20二卤代烷烃,另一种所述的烷基化试剂是C1到C20烷基铵盐。
5.根据权利要求4所述的聚合物组合物,其中所述的重复单元(a)具有下式或其盐或其共聚物。
6.根据权利要求4所述的聚合物组合物,其中所述的重复单元(a)具有下式或其盐或其共聚物。
7.一种用于制造通过离子交换从患者体内除去胆汁盐的药物的聚合物组合物,包括治疗有效量的下述物质的反应产物,(a)一种或多种交联聚合物,包括下式表示的重复单元: 或其盐或其共聚物,其中R1是H或C1到C8烷基,n是整数,和p=0-10;以及(b)至少两种烷基化试剂,其中(i)一种所述的烷基化试剂具有化学式RX,其中R是C1到C20烷基,X是一个或多个亲电性离去基团,另一种所述的烷基化试剂具有化学式R’X,其中R’是C1到C20烷基铵基团,X是一个或多个亲电子离去基团;或(ii)一种所述的烷基化试剂具有化学式RX,其中R是C1到C20烷基,X是一个或多个亲电性离去基团,另一种所述的烷基化试剂具有化学式R’X,其中R’是C1到C20羟烷基,X是一个或多个亲电子离去基团;(iii)一种所述烷基化试剂是C1到C20二卤代烷烃,另一种所述的烷基化试剂是C1到C20烷基铵盐。
或其盐或其共聚物,其中R1是H或C1到C8烷基,n是整数,和p=0-10;以及(b)至少两种烷基化试剂,其中(i)一种所述的烷基化试剂具有化学式RX,其中R是C1到C20烷基,X是一个或多个亲电性离去基团,另一种所述的烷基化试剂具有化学式R’X,其中R’是C1到C20烷基铵基团,X是一个或多个亲电子离去基团;或(ii)一种所述的烷基化试剂具有化学式RX,其中R是C1到C20烷基,X是一个或多个亲电性离去基团,另一种所述的烷基化试剂具有化学式R’X,其中R’是C1到C20羟烷基,X是一个或多个亲电子离去基团;(iii)一种所述烷基化试剂是C1到C20二卤代烷烃,另一种所述的烷基化试剂是C1到C20烷基铵盐。
8.一种用于制造通过离子交换从患者体内除去胆汁盐的药物的聚合物组合物,包括治疗有效量的下述物质的反应产物,(a)一种或多种交联聚合物,包括下式表示的重复单元: 或其盐或其共聚物,其中R1是H或C1到C8烷基,n是整数,p=1-10;(b)至少一种烷基化试剂,所述的烷基化试剂具有式RX,其中R包括C1-C20烷基,C1-C20羟基烷基,C1-C20芳烷基,C1-C20烷基铵,或C1-C20烷基酰氨基,及X包括一种或多种亲电子离去基团。
或其盐或其共聚物,其中R1是H或C1到C8烷基,n是整数,p=1-10;(b)至少一种烷基化试剂,所述的烷基化试剂具有式RX,其中R包括C1-C20烷基,C1-C20羟基烷基,C1-C20芳烷基,C1-C20烷基铵,或C1-C20烷基酰氨基,及X包括一种或多种亲电子离去基团。
9.根据权利要求1所述的聚合物组合物,其中所述的重复单元(a)具有下式 或其盐和其共聚合物,其中Z为NR3或(CH2)m,R2是或
并且R1、R3、R4、R5、R6、n、m,和p如权利要求1所定义。
或其盐和其共聚合物,其中Z为NR3或(CH2)m,R2是或
并且R1、R3、R4、R5、R6、n、m,和p如权利要求1所定义。
10.根据权利要求1,7或8所述的聚合物组合物,其中所述的聚合物还含有一种或多种可交换的平衡物。
11.根据权利要求10所述的聚合物组合物,其中至少所述平衡物之一包括Cl-,Br-,CH3OSO3 -,HSO4 -,SO4 2-,HCO3 -,CO3 2-,乙酸盐,乳酸盐,琥珀酸盐,丙酸盐,丁酸盐,抗坏血酸盐,柠檬酸盐,马来酸盐,叶酸盐,氨基酸衍生物,核苷酸,类脂,磷脂。
12.根据权利要求11所述的聚合物组合物,其中所述的X包括卤化物,环氧化物,苯甲磺酸盐,或甲磺酸盐基团。
13.根据权利要求12所述的聚合物组合物,其中所述的烷基化试剂包括C4-C20烷基卤。
14.根据权利要求12所述的聚合物组合物,其中所述的烷基化试剂包括C1-C20烷基卤铵盐。
15.根据权利要求12所述的聚合物组合物,其中所述的烷基化试剂是C1-C20二卤代烷基,C1-C20羟基烷基卤,C1-C20芳烷基卤,C1-C20烷基环氧基铵盐,或C1-C20环氧基烷基酰胺。
16.一种用于制造通过离子交换从患者体内除去胆汁盐的药物的聚合物组合物,包括治疗有效量的下述物质的反应产物:
(a)一种或多种具有下式重复单元的交联聚合物: 或其盐和其共聚合物,其中n为整数;R1是H或C1-C8烷基;M是
或其盐和其共聚合物,其中n为整数;R1是H或C1-C8烷基;M是 或-Z-R2;Z为O,NR3,S,或(CH2)m;m=0-10;R3是H或C1-C8烷基;及R2是
或-Z-R2;Z为O,NR3,S,或(CH2)m;m=0-10;R3是H或C1-C8烷基;及R2是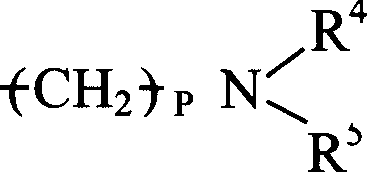 或
或
 其中P=0-10,并且R4,R5或R6每个是H,C1-C8烷基,或芳基;和
其中P=0-10,并且R4,R5或R6每个是H,C1-C8烷基,或芳基;和
(b)至少两种烷基化试剂,所述烷基化试剂之一具有式RX,其中R是C1-C20烷基,X包括一种或多种亲电子离去基团,及另一个烷基化试剂具有式R’X,其中R’是C1-C20烷基铵,X包括一种或多种亲电子电子离去基团,条件是R2包括R4,R5和R3,R4,R5和R3中至少一个是H。
17.一种用于制造通过离子交换从患者体内除去胆汁盐的药物的聚合物组合物,包括治疗有效量的下述物质的反应产物:
(a)一种或多种具有下式重复单元的交联聚合物: 或其盐和其共聚合物,其中n为整数;R1是H或C1-C8烷基;M是
或-Z-R2;Z为O,NR3,S,或(CH2)m;m=0-10;R3是H或C1-C8烷基;及R2是或
其中P=0-10,并且R4,R5或R6每个是H,C1-C8烷基,或芳基;和
或其盐和其共聚合物,其中n为整数;R1是H或C1-C8烷基;M是
或-Z-R2;Z为O,NR3,S,或(CH2)m;m=0-10;R3是H或C1-C8烷基;及R2是或
其中P=0-10,并且R4,R5或R6每个是H,C1-C8烷基,或芳基;和
(b)至少两种烷基化试剂,所述烷基化试剂之一具有式RX,其中R是C1-C20烷基,X包括一种或多种亲电子离去基团,及另一个烷基化试剂具有式R’X,其中R’是C1-C20羟基烷基,X包括一种或多种亲电子离去基团,条件是R2包括R4,R5和R3,R4,R5和R3中至少一个是H。
18.一种用于制造通过离子交换从患者体内除去胆汁盐的药物的聚合物组合物,包括治疗有效量的下述物质的反应产物:
(a)一种或多种具有下式重复单元的交联聚合物:或其盐和其共聚合物,其中n为整数;R1是H或C1-C8烷基;M是
 或-Z-R2;Z为O,NR3,S,或(CH2)m;m=0-10;R3是H或C1-C8烷基;及R2是
或-Z-R2;Z为O,NR3,S,或(CH2)m;m=0-10;R3是H或C1-C8烷基;及R2是 或
或
 其中P=0-10,并且R4,R5或R6每个是H,C1-C8烷基,或芳基;和
其中P=0-10,并且R4,R5或R6每个是H,C1-C8烷基,或芳基;和
(b)至少两种烷基化试剂,所述烷基化试剂之一是C1-C20二卤代烷,而另一个烷基化试剂是C1-C20烷基铵盐,条件是R2包括R4,R5和R3,R4,R5和R3中至少一个是H。
19.一种用于制造通过离子交换从患者体内除去胆汁盐的药物的聚合物组合物,包括给患者服用一种或多种治疗有效量的具有下式重复单元的交联聚合物
或其共聚物,其中n为整数,R1是H或C1-C8烷基;L是
-NH-或
G是
或
并且每个R2,R3和R4各自是H,C1-C8烷基,或芳基。
20.根据权利要求19所述的聚合物组合物,其中所述的聚合物通过多功能交联共聚单体交联,所述共聚单体约为单体总重量的1-25%。
21.根据权利要求20所述的聚合物组合物,其中所述的交联共聚单体为单体总重量的2.5-20%。
22.根据权利要求19所述的聚合物组合物,其中所述的聚合物还包括一种或多种疏水性共聚单体。
23.根据权利要求22所述的聚合物组合物,其中所述的疏水性共聚单体包括苯乙烯及其氟化衍生物;乙基乙烯基苯及其氟化衍生物;丙烯酰胺和甲基丙烯酰胺的N-烷基和N-芳基衍生物及其氟化衍生物;丙烯酸的烷基和芳基酯及其氟化衍生物;甲基丙烯酸的烷基和芳基酯及其氟化衍生物;4-乙烯基联苯及其氟化衍生物;4-乙烯基茴香醚及其氟化衍生物;和4-氨基苯乙烯及其氟化衍生物。
24.根据权利要求19所述的聚合物组合物,其中所述的聚合物的特征在于具有下式重复单元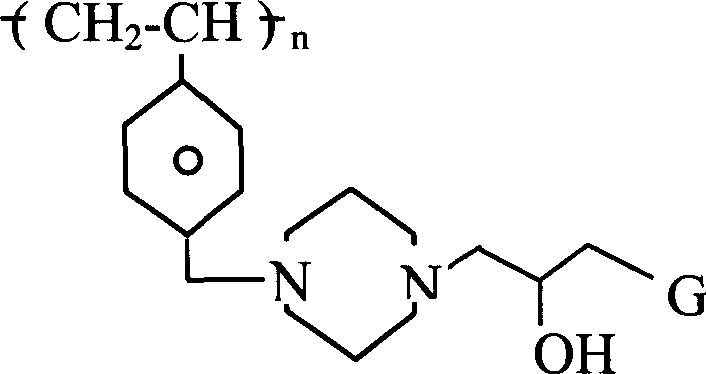
或其共聚物,其中n和G如权利要求19所定义。
25.根据权利要求19所述的聚合物组合物,其中所述的聚合物的特征在于具有下式重复单元
或其共聚物,其中n和G如权利要求19所定义。
26.根据权利要求19所述的聚合物组合物,其中所述的聚合物包括具有下式的重复单元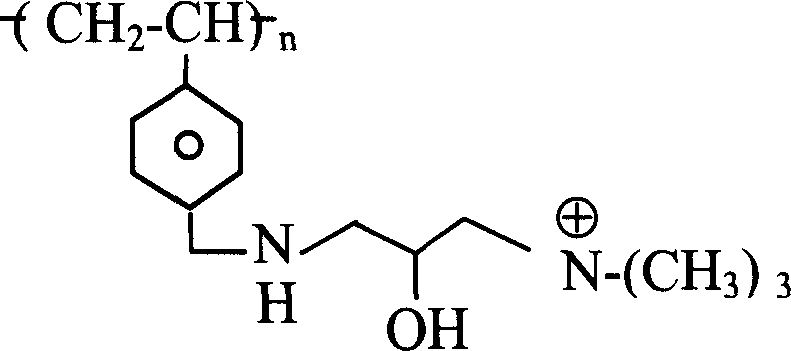
或其共聚物,其中所述的n如权利要求19所定义。
27.根据权利要求19所述的聚合物组合物,还包括作为共聚单体的苯乙烯或其氟化衍生物。
28.根据权利要求19所述的聚合物组合物,其中所述的聚合物包括具有下式的重复单元
或其共聚物,其中所述的n如权利要求19所定义。
29.一种用于制造通过离子交换从患者体内除去胆汁盐的药物的聚合物组合物,包括治疗有效量的下述物质的反应产物:
(a)一种或多种具有下式重复单元的交联聚合物:或其盐和其共聚合物,其中n为整;R1是H或C1-C8烷基;M是
或-Z-R2;Z为O,NR3,S,或(CH2)m;m=0-10;R3是H或C1-C8烷基;及R2是 或
或
 其中P=0-10,并且R4,R5或R6每个是H,C1-C8烷基,或芳基;和
其中P=0-10,并且R4,R5或R6每个是H,C1-C8烷基,或芳基;和
(b)至少一种烷基化试剂,所述烷基化试剂之一具有式RX,其中R包括C4-C20烷基铵,X包括一种或多种亲电子离去基团,条件是R2包括R4,R5和R3,R4,R5和R3中至少一个是H。如权利要求1-29之一的聚合物组合物在制备从患者体内除去胆汁盐的药物中的应用。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/258,477 | 1994-06-10 | ||
| US08/258,477 US5624963A (en) | 1993-06-02 | 1994-06-10 | Process for removing bile salts from a patient and compositions therefor |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1155287A CN1155287A (zh) | 1997-07-23 |
| CN1136855C true CN1136855C (zh) | 2004-02-04 |
Family
ID=22980711
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB951935224A Expired - Lifetime CN1136855C (zh) | 1994-06-10 | 1995-05-24 | 用于从患者体内除去胆汁盐的交联聚合物 |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US5624963A (zh) |
| EP (1) | EP0764177B1 (zh) |
| JP (1) | JPH10501264A (zh) |
| CN (1) | CN1136855C (zh) |
| AT (1) | ATE205508T1 (zh) |
| AU (1) | AU694777B2 (zh) |
| CA (1) | CA2192592C (zh) |
| DE (1) | DE69522687T2 (zh) |
| DK (1) | DK0764177T3 (zh) |
| ES (1) | ES2164152T3 (zh) |
| MX (1) | MX9606172A (zh) |
| NZ (1) | NZ285979A (zh) |
| PT (1) | PT764177E (zh) |
| RU (1) | RU2146266C1 (zh) |
| WO (1) | WO1995034588A1 (zh) |
Families Citing this family (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6129910A (en) * | 1993-06-02 | 2000-10-10 | Geltex Pharmaceuticals, Inc. | Water-insoluble noncrosslinked bile acid sequestrants |
| US5703188A (en) * | 1993-06-02 | 1997-12-30 | Geltex Pharmaceuticals, Inc. | Process for removing bile salts from a patient and compositions therefor |
| US5607669A (en) * | 1994-06-10 | 1997-03-04 | Geltex Pharmaceuticals, Inc. | Amine polymer sequestrant and method of cholesterol depletion |
| US5618530A (en) * | 1994-06-10 | 1997-04-08 | Geltex Pharmaceuticals, Inc. | Hydrophobic amine polymer sequestrant and method of cholesterol depletion |
| US5900475A (en) * | 1994-06-10 | 1999-05-04 | Geltex Pharmaceuticals, Inc. | Hydrophobic sequestrant for cholesterol depletion |
| US5929184A (en) * | 1993-06-02 | 1999-07-27 | Geltex Pharmaceuticals, Inc. | Hydrophilic nonamine-containing and amine-containing copolymers and their use as bile acid sequestrants |
| TW474813B (en) * | 1994-06-10 | 2002-02-01 | Geltex Pharma Inc | Alkylated composition for removing bile salts from a patient |
| FR2757866B1 (fr) * | 1996-12-30 | 2004-12-17 | Catalyse | Polymeres comportant des groupes ammoniums quaternaires, leur utilisation pour la fabrication d'un materiau a propretes antibacteriennes et leurs procedes de preparation |
| US6203785B1 (en) | 1996-12-30 | 2001-03-20 | Geltex Pharmaceuticals, Inc. | Poly(diallylamine)-based bile acid sequestrants |
| DE19705963A1 (de) * | 1997-02-17 | 1998-08-20 | Hoechst Ag | Vernetzte Vinylpolymere mit Gallensäure-Adsorberwirkung |
| US5925379A (en) * | 1997-03-27 | 1999-07-20 | Geltex Pharmaceuticals, Inc. | Interpenetrating polymer networks for sequestration of bile acids |
| US6423754B1 (en) * | 1997-06-18 | 2002-07-23 | Geltex Pharmaceuticals, Inc. | Method for treating hypercholesterolemia with polyallylamine polymers |
| US5900233A (en) * | 1997-10-16 | 1999-05-04 | Day; Charles E. | Epichlorohydrin and 1-(3-aminopropyl) imidazole copolymer and its use in treating irritable bowel syndrome |
| US5985938A (en) * | 1997-11-05 | 1999-11-16 | Geltex Pharmaceuticals, Inc. | Method for reducing oxalate |
| US6726905B1 (en) | 1997-11-05 | 2004-04-27 | Genzyme Corporation | Poly (diallylamines)-based phosphate binders |
| US6566407B2 (en) | 1997-11-05 | 2003-05-20 | Geltex Pharmaceuticals, Inc. | Method for reducing oxalate |
| US6083497A (en) | 1997-11-05 | 2000-07-04 | Geltex Pharmaceuticals, Inc. | Method for treating hypercholesterolemia with unsubstituted polydiallylamine polymers |
| US6299868B1 (en) | 1999-07-14 | 2001-10-09 | Geltex Pharmaceuticals, Inc. | Fat-binding polymers |
| US6264937B1 (en) * | 1998-01-09 | 2001-07-24 | Geltex Pharmaceuticals, Inc. | Fat-binding polymers |
| US7048917B1 (en) | 1998-01-09 | 2006-05-23 | Genzyme Corporation | Fat-binding polymers |
| US6267952B1 (en) | 1998-01-09 | 2001-07-31 | Geltex Pharmaceuticals, Inc. | Lipase inhibiting polymers |
| AU4369499A (en) * | 1998-06-30 | 2000-01-17 | Aventis Research & Technologies Gmbh & Co. Kg | Novel method for producing cross-linked vinyl polymers based on quaternary ammonium groups exhibiting a bile acid adsorbent effect |
| WO2000008063A1 (fr) * | 1998-08-05 | 2000-02-17 | Hisamitsu Pharmaceutical Co., Inc. | Procede de production d'un polymere cationique |
| US6294163B1 (en) | 1998-10-02 | 2001-09-25 | Geltex Pharmaceuticals, Inc. | Polymers containing guanidinium groups as bile acid sequestrants |
| US6271264B1 (en) | 1998-12-01 | 2001-08-07 | Geltex Pharmaceuticals, Inc. | Polymers containing spirobicyclic ammonium moieties as bile acid sequestrants |
| US6190649B1 (en) | 1999-04-23 | 2001-02-20 | Geltex Pharmaceuticals, Inc. | Polyether-based bile acid sequestrants |
| WO2001005408A1 (en) * | 1999-07-14 | 2001-01-25 | Geltex Pharmaceuticals, Inc. | Fat-binding polymers, optionally combined with lipase inhibitors |
| US6733780B1 (en) | 1999-10-19 | 2004-05-11 | Genzyme Corporation | Direct compression polymer tablet core |
| US20020054903A1 (en) * | 1999-10-19 | 2002-05-09 | Joseph Tyler | Direct compression polymer tablet core |
| WO2002081528A1 (en) * | 2001-04-05 | 2002-10-17 | Sekisui Chemical Co., Ltd. | Polymer specifically recognizing bile acid, process for producing the same, bile acid-absorbing polymer and cholesterol-lowering agents |
| EP1923064B1 (en) | 2001-04-18 | 2017-06-28 | Genzyme Corporation | Use of amine polymer for lowering serum glucose |
| CA2444028A1 (en) * | 2001-04-18 | 2002-10-31 | Genzyme Corporation | Methods of treating syndrome x with aliphatic polyamines |
| DE60212819T2 (de) * | 2001-04-18 | 2006-11-23 | Genzyme Corp., Cambridge | Salzformarme polyallylamine |
| WO2002085379A1 (en) * | 2001-04-18 | 2002-10-31 | Geltex Pharmaceuticals, Inc. | Method for improving vascular access in patients with vascular shunts |
| WO2002085380A1 (en) * | 2001-04-18 | 2002-10-31 | Geltex Pharmaceuticals, Inc. | Method for treating gout and reducing serum uric acid |
| AU2002257145B2 (en) * | 2001-04-18 | 2005-06-02 | Genzyme Corporation | Method for lowering serum glucose |
| US7049345B2 (en) * | 2001-06-29 | 2006-05-23 | Genzyme Corporation | Fat-binding polymers |
| US7041280B2 (en) * | 2001-06-29 | 2006-05-09 | Genzyme Corporation | Aryl boronate functionalized polymers for treating obesity |
| AT411463B (de) | 2002-09-03 | 2004-01-26 | Dsm Fine Chem Austria Gmbh | Verfahren zur herstellung von alkylierten n- bzw. amino-, ammonium- oder spirobicyclischen ammoniumgruppen haltigen, vernetzten polymeren |
| WO2004037274A1 (en) * | 2002-10-22 | 2004-05-06 | Genzyme Corporation | Amine polymers for promoting bone formation |
| AT412473B (de) | 2003-01-15 | 2005-03-25 | Dsm Fine Chem Austria Gmbh | Verfahren zur kontinuierlichen trocknung von n- bzw. amino-, ammonium- oder spirobicyclische ammoniumgruppen haltigen polymeren |
| CA2560927C (en) | 2004-03-26 | 2013-05-14 | Mitsubishi Pharma Corporation | Insulin resistance-improving agent |
| US7985418B2 (en) | 2004-11-01 | 2011-07-26 | Genzyme Corporation | Aliphatic amine polymer salts for tableting |
| US20060177415A1 (en) * | 2004-11-01 | 2006-08-10 | Burke Steven K | Once a day formulation for phosphate binders |
| US8986669B2 (en) * | 2005-09-02 | 2015-03-24 | Genzyme Corporation | Method for removing phosphate and polymer used therefore |
| HUE026628T2 (en) | 2005-09-15 | 2016-06-28 | Genzyme Corp | Pouches for amine polymers |
| DE102005063339B4 (de) * | 2005-10-18 | 2012-01-12 | Henkel Ag & Co. Kgaa | Verwendung halogenidarmer Polymerlösungen mit kationischen Aminogruppen |
| EP2016114A2 (en) * | 2006-05-05 | 2009-01-21 | Genzyme Corporation | Amine condensation polymers as phosphate sequestrants |
| WO2007145308A1 (ja) | 2006-06-16 | 2007-12-21 | Mitsubishi Tanabe Pharma Corporation | 糸球体疾患の予防及び/または治療剤 |
| US20100124542A1 (en) * | 2006-07-18 | 2010-05-20 | Genzyme Corporation | Amine dendrimers |
| EP2066293A2 (en) | 2006-09-29 | 2009-06-10 | Genzyme Corporation | Amide dendrimer compositions |
| US8163799B2 (en) * | 2006-12-14 | 2012-04-24 | Genzyme Corporation | Amido-amine polymer compositions |
| JP2010519298A (ja) * | 2007-02-23 | 2010-06-03 | ゲンズイメ コーポレーション | アミンポリマー組成物 |
| JP2010520285A (ja) * | 2007-03-08 | 2010-06-10 | ゲンズイメ コーポレーション | スルホンポリマー組成物 |
| US20100166696A1 (en) * | 2007-04-27 | 2010-07-01 | Dhal Pradeep K | Amido-amine dendrimer compositions |
| JP5043501B2 (ja) * | 2007-05-09 | 2012-10-10 | 日油株式会社 | ホスホリルコリン類似基含有ランダム共重合体 |
| EP2217215A1 (en) * | 2007-12-14 | 2010-08-18 | Genzyme Corporation | Coated pharmaceutical compositions |
| WO2009154747A1 (en) * | 2008-06-20 | 2009-12-23 | Genzyme Corporation | Pharmaceutical compositions |
| US8673272B2 (en) * | 2009-07-27 | 2014-03-18 | Isp Investments Inc. | Ultraviolet-absorbing compounds |
| CN105017481B (zh) * | 2010-02-24 | 2018-10-02 | 瑞立普萨公司 | 用作胆汁酸螯合剂的交联聚乙烯胺、聚烯丙胺和乙烯亚胺 |
| WO2012027331A1 (en) | 2010-08-27 | 2012-03-01 | Ironwood Pharmaceuticals, Inc. | Compositions and methods for treating or preventing metabolic syndrome and related diseases and disorders |
| US20130123433A1 (en) | 2011-11-14 | 2013-05-16 | Formosa Laboratories, Inc. | Method for preparing poly(allylamine) hydrochloride and derivatives therefrom |
| US9475891B2 (en) | 2013-09-19 | 2016-10-25 | Navinta, Llc | Process for the preparation of colesevelam hydrochloride |
| PL229768B1 (pl) | 2015-06-10 | 2018-08-31 | Univ Jagiellonski | Zastosowanie polimeru blokowego zawierającego blok poli- (chlorku 3-metakryloilaminopropylotrimetyloamoniowego) (PMAPTAC) do neutralizacji heparyny |
| GB201515602D0 (en) * | 2015-09-03 | 2015-10-21 | Biocompatibles Uk Ltd | Polymers and microspheres |
| KR102019785B1 (ko) * | 2017-04-11 | 2019-11-04 | 단국대학교 천안캠퍼스 산학협력단 | pH-온도 감응성 공중합체 및 이를 이용한 소장 표적 약물 전달체 |
| AU2020312953B2 (en) | 2019-07-12 | 2024-06-27 | Gc Corporation | Antibacterial polymer particles, composition and article |
| DE102019008024A1 (de) * | 2019-11-18 | 2021-05-20 | Universität Stuttgart | Kationenaustauscher- und Anionenaustauscherpolymere und -(blend)membranen aus hochfluorierte aromatische Gruppen enthaltenden Polymeren mittlels nucleophiler Substitution |
| CN111521705B (zh) * | 2020-05-05 | 2021-04-20 | 大连润生康泰医学检验实验室有限公司 | 一种血清中胆汁酸的富集方法 |
Family Cites Families (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE580490A (fr) * | 1958-07-15 | 1960-01-08 | Merck & Co Inc | Compositions et procédés pour abaisser la teneur en cholestérol du sang |
| US3308020A (en) * | 1961-09-22 | 1967-03-07 | Merck & Co Inc | Compositions and method for binding bile acids in vivo including hypocholesteremics |
| US3383281A (en) * | 1961-09-22 | 1968-05-14 | Merck & Co Inc | Method for binding bile acids in vivo |
| BE756035A (fr) * | 1969-09-12 | 1971-03-11 | Inveresk Res Int | Polymeres pontes |
| US3803237A (en) * | 1969-11-03 | 1974-04-09 | Upjohn Co | Reaction products of polyethylenepolyamines and chlorohydrins or epoxy containing compounds |
| US3980770A (en) * | 1971-06-04 | 1976-09-14 | Pharmacia Aktiebolag | Polymerization products containing amino groups useful in serum cholesterol level control |
| US3923972A (en) * | 1971-10-12 | 1975-12-02 | Monsanto Co | Method of lowering blood cholesterol level |
| US3953406A (en) * | 1973-01-26 | 1976-04-27 | California Institute Of Technology | Water-insoluble, swellable polyurethanes |
| US4027009A (en) * | 1973-06-11 | 1977-05-31 | Merck & Co., Inc. | Compositions and methods for depressing blood serum cholesterol |
| US4217429A (en) * | 1973-06-11 | 1980-08-12 | Merck & Co., Inc. | Poly-[(methylimino)trimethylene] |
| US4205064A (en) * | 1973-06-11 | 1980-05-27 | Merck & Co., Inc. | Bile acid sequestering composition containing poly[{alkyl-(3-ammoniopropyl)imino}-trimethylenedihalides] |
| CS187563B1 (en) * | 1974-02-08 | 1979-02-28 | Petr Strop | Method of preparation of the hydrophilic homogeneous or macroporous annexes |
| CS173201B1 (zh) * | 1974-02-13 | 1977-02-28 | ||
| US4198396A (en) * | 1974-07-03 | 1980-04-15 | Warren-Teed Laboratories, Inc. | Dissolution of gallstones |
| US4016209A (en) * | 1975-04-23 | 1977-04-05 | Merck & Co., Inc. | 3-[N'-(3-Halopropyl)-N-'-methylamino]-N,N,N-trimethyl-1-propanaminium halide and acid addition salts thereof |
| US4071478A (en) * | 1976-06-07 | 1978-01-31 | Merck & Co., Inc. | Controlled partially cross-linked 3,3-ionenes |
| FI67483C (fi) * | 1977-02-17 | 1985-04-10 | Merck & Co Inc | Foerfarande foer framstaellning av ett icke adhesivt friflytande farmakologiskt godtagbart gallsyrakomplexbildande adsorbatpreparat |
| IT1106718B (it) * | 1978-12-21 | 1985-11-18 | Alfa Farmaceutici Spa | Composizioni a base di resine anioniche salificate farmacologicamente attive |
| JPS57142920A (en) * | 1981-03-02 | 1982-09-03 | Mitsubishi Petrochem Co Ltd | Cholesterol depressant |
| JPS5879022A (ja) * | 1981-11-04 | 1983-05-12 | Bitamin Kenkyusho:Kk | 第四級窒素原子を含有する新規な金属架橋高分子化合物、その製法及び該高分子化合物を有効成分とする高脂血症治療剤 |
| US4557930A (en) * | 1982-01-18 | 1985-12-10 | Mitsubishi Petrochemical Co., Ltd. | Anticholesteremic anion exchange resins |
| JPS6090243A (ja) * | 1983-10-25 | 1985-05-21 | Nitto Boseki Co Ltd | 小球状モノアリルアミン橋かけ重合体の製造方法 |
| AU551886B2 (en) * | 1983-11-14 | 1986-05-15 | Nitto Boseki Co. Ltd. | Poly(allylamine) derivatives |
| JPS60106803A (ja) * | 1983-11-14 | 1985-06-12 | Nitto Boseki Co Ltd | アリル尿素重合体の製造方法 |
| CA1220897A (en) * | 1984-01-11 | 1987-04-21 | Kiyoshi Shimizu | Process for producing polymers of monoallylamine |
| JPS60209523A (ja) * | 1984-04-03 | 1985-10-22 | Mitsubishi Petrochem Co Ltd | コレステロ−ル低下剤 |
| OA08092A (fr) * | 1984-05-11 | 1987-03-31 | Bristol Myers Co | Novel bile sequestrant resin and uses. |
| US4837015A (en) * | 1987-03-05 | 1989-06-06 | Carolina Medical Products Company, Inc. | Alkali metal ion-charged, cation exchanger and use thereof to adjust sodium, potassium and calcium body fluid levels |
| US4759923A (en) * | 1987-06-25 | 1988-07-26 | Hercules Incorporated | Process for lowering serum cholesterol using poly(diallylmethylamine) derivatives |
| GB8829088D0 (en) * | 1988-12-13 | 1989-01-25 | Smith Kline French Lab | Compounds |
| US5236701A (en) * | 1989-07-19 | 1993-08-17 | Lowchol Scientific Inc. | Ingestible hydrophilic polymeric amines useful for lowering blood cholesterol |
| GB8928278D0 (en) * | 1989-12-14 | 1990-02-21 | Smith Kline French Lab | Compounds |
| CA2040996A1 (en) * | 1990-05-02 | 1991-11-03 | Robert L. Albright | Composition and method for controlling cholesterol |
| GB9011332D0 (en) * | 1990-05-21 | 1990-07-11 | Smith Kline French Lab | Compounds |
| US5250524A (en) * | 1990-12-06 | 1993-10-05 | Hoechst Aktiengesellschaft | Bile acid derivatives, process for their preparation and use of these compounds as pharmaceuticals |
| IE914179A1 (en) * | 1990-12-07 | 1992-06-17 | Ici Plc | Nitrogen derivatives |
| US5055197A (en) * | 1991-04-05 | 1991-10-08 | Rohm And Haas Company | Process for removing residual monomers and oligemers from amine-containing polymers |
| CA2042870C (en) * | 1991-05-17 | 1996-11-26 | Leon Edward St. Pierre | Metal ion coordinated polyamine resins for the lowering of blood cholesterol |
| CA2116431A1 (en) * | 1991-09-06 | 1993-03-18 | Gurcharn S. Sidhu | Composition and method for reducing cholesterol concentration |
| JP2712056B2 (ja) * | 1992-01-14 | 1998-02-10 | 久光製薬株式会社 | コレステロール低下剤 |
| WO1993025595A1 (en) * | 1992-06-17 | 1993-12-23 | Isp Investments Inc. | Cationic polymer compositions |
| ATE159262T1 (de) * | 1992-07-22 | 1997-11-15 | Hoechst Ag | Hydrophile zentren aufweisende polyvinylamin- derivate, verfahren zu ihrer herstellung sowie die verwendung der verbindungen als arzneimittel, wirkstoffträger und nahrungsmittelhilfsstoff |
| ATE168707T1 (de) * | 1992-08-20 | 1998-08-15 | Du Pont | Vernetzte polymerische ammoniumsalze |
| US5451397A (en) * | 1992-12-21 | 1995-09-19 | Rohm And Haas Company | Bile acid sequestrant |
| AU7047994A (en) * | 1993-06-02 | 1994-12-20 | Geltex Pharmaceuticals, Inc. | Compositions and process for removing bile salts |
-
1994
- 1994-06-10 US US08/258,477 patent/US5624963A/en not_active Expired - Fee Related
-
1995
- 1995-05-24 ES ES95919914T patent/ES2164152T3/es not_active Expired - Lifetime
- 1995-05-24 CN CNB951935224A patent/CN1136855C/zh not_active Expired - Lifetime
- 1995-05-24 EP EP95919914A patent/EP0764177B1/en not_active Expired - Lifetime
- 1995-05-24 DK DK95919914T patent/DK0764177T3/da active
- 1995-05-24 RU RU96124822A patent/RU2146266C1/ru active
- 1995-05-24 DE DE69522687T patent/DE69522687T2/de not_active Expired - Lifetime
- 1995-05-24 AT AT95919914T patent/ATE205508T1/de active
- 1995-05-24 CA CA002192592A patent/CA2192592C/en not_active Expired - Lifetime
- 1995-05-24 NZ NZ285979A patent/NZ285979A/en not_active IP Right Cessation
- 1995-05-24 PT PT95919914T patent/PT764177E/pt unknown
- 1995-05-24 WO PCT/US1995/006542 patent/WO1995034588A1/en not_active Ceased
- 1995-05-24 AU AU25560/95A patent/AU694777B2/en not_active Expired
- 1995-05-24 JP JP8502188A patent/JPH10501264A/ja not_active Ceased
-
1996
- 1996-12-06 MX MX9606172A patent/MX9606172A/es unknown
Also Published As
| Publication number | Publication date |
|---|---|
| WO1995034588A1 (en) | 1995-12-21 |
| ES2164152T3 (es) | 2002-02-16 |
| CA2192592C (en) | 2009-11-24 |
| JPH10501264A (ja) | 1998-02-03 |
| EP0764177A1 (en) | 1997-03-26 |
| DK0764177T3 (da) | 2002-01-14 |
| DE69522687D1 (de) | 2001-10-18 |
| CA2192592A1 (en) | 1995-12-21 |
| HK1001611A1 (zh) | 1998-07-03 |
| DE69522687T2 (de) | 2002-06-20 |
| EP0764177B1 (en) | 2001-09-12 |
| AU694777B2 (en) | 1998-07-30 |
| AU2556095A (en) | 1996-01-05 |
| CN1155287A (zh) | 1997-07-23 |
| MX9606172A (es) | 1998-06-30 |
| NZ285979A (en) | 1998-08-26 |
| RU2146266C1 (ru) | 2000-03-10 |
| US5624963A (en) | 1997-04-29 |
| ATE205508T1 (de) | 2001-09-15 |
| PT764177E (pt) | 2002-03-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1136855C (zh) | 用于从患者体内除去胆汁盐的交联聚合物 | |
| CN1150217C (zh) | 一种交联聚合物及其应用 | |
| US5703188A (en) | Process for removing bile salts from a patient and compositions therefor | |
| CN1511039A (zh) | 降低血浆葡萄糖的方法 | |
| CA2349620C (en) | Use of aliphatic polyamines for reducing oxalate | |
| CN1245425A (zh) | 聚(二烯丙基胺)基胆汁酸多价螯合体 | |
| US6566407B2 (en) | Method for reducing oxalate | |
| CN1044105A (zh) | 化合物 | |
| EP0706399A1 (en) | Compositions and process for removing bile salts | |
| CN1509180A (zh) | 用脂肪族聚胺治疗综合症x的方法 | |
| CN1056056A (zh) | 控制胆固醇的组合物及其控制方法 | |
| CN1103871A (zh) | 胆汁酸螯合剂 | |
| CN101065409A (zh) | 交联胺聚合物 | |
| HK1001611B (zh) | 用於从患者体内除去胆汁盐的交联聚合物 | |
| CN1034550A (zh) | 化合物 | |
| CN1288381A (zh) | 脂肪结合聚合物 | |
| HK1025912A (zh) | 聚(二烯丙基胺)基胆汁酸多价螯合体 | |
| CN1222855A (zh) | 作为抗感染剂的离子性聚合物 | |
| CN1189168A (zh) | 用于清除胆固醇的含疏水杂原子的螯合剂 | |
| HK1110341A (zh) | 交聯胺聚合物 | |
| HK1009820A (zh) | 用於清除胆固醇的含疏水杂原子的螯合剂 | |
| HK1034044B (zh) | 脂肪結合聚合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| ASS | Succession or assignment of patent right |
Owner name: ENZYME CO., LTD. Free format text: FORMER OWNER: GELTEX PHARMACEUTICALS INC. Effective date: 20031010 |
|
| C41 | Transfer of patent application or patent right or utility model | ||
| TR01 | Transfer of patent right |
Effective date of registration: 20031010 Patentee after: Genzyme Corp. Patentee before: Geltex Pharmaceuticals Inc. |
|
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C17 | Cessation of patent right | ||
| CX01 | Expiry of patent term |
Expiration termination date: 20150524 Granted publication date: 20040204 |