CN1103402A - 取代的吡咯类化合物 - Google Patents
取代的吡咯类化合物 Download PDFInfo
- Publication number
- CN1103402A CN1103402A CN94105846A CN94105846A CN1103402A CN 1103402 A CN1103402 A CN 1103402A CN 94105846 A CN94105846 A CN 94105846A CN 94105846 A CN94105846 A CN 94105846A CN 1103402 A CN1103402 A CN 1103402A
- Authority
- CN
- China
- Prior art keywords
- methyl
- indoles
- tetrahydropyridine
- indyl
- pyrroles
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Virology (AREA)
- Endocrinology (AREA)
- Communicable Diseases (AREA)
- Emergency Medicine (AREA)
- Pulmonology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Oncology (AREA)
- Rheumatology (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Tropical Medicine & Parasitology (AREA)
- AIDS & HIV (AREA)
- Pain & Pain Management (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Pyrrole Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Saccharide Compounds (AREA)
- Cephalosporin Compounds (AREA)
Abstract
本发明提供了式I化合物以及式I酸性化合物
与碱形成的可药用盐和式I碱性化合物与酸形成的
可药用盐,它们可用于防治各种疾病,特别是用于防
治炎性疾病、免疫疾病、肿瘤、支气管肺病、皮肤病和
心血管疾病,治疗哮喘、艾滋病或糖尿病并发症或刺
激毛发生长。
其中R1、R2、m和n具有说明书中所给出的意义。
Description
本发明涉及取代的吡咯类化合物。更具体地说,本发明涉及通式Ⅰ的化合物以及式Ⅰ酸性化合物与碱形成的可药用盐和式Ⅰ碱性化合物与酸形成的可药用盐。

其中R1代表低级烷基、低级环烷基、芳基或低级芳烷基;R2代表氢、芳基或低级烷基,所述芳基和烷基可任意被羟基、酰氧基、氨基、单(低级烷基)氨基、二(低级烷基)氨基、羧基、低级烷氧羰基或氨基羰基取代;m和n代表1或2。
本发明的目的是式Ⅰ化合物及其上述盐本身和式Ⅰ化合物及其盐作为治疗活性物质;所述化合物及其盐的制备方法和用于所述方法中的新的中间体;含有所述化合物或其盐的药物和这些药物的制备;所述化合物和其盐在防治疾病方面的用途,特别是防治炎性疾病、免疫疾病、肿瘤、支气管肺病、皮肤病和心血管疾病,治疗哮喘、艾滋病或糖尿病并发症或用于刺激毛发生长,或者用于制备一种药物以防治炎性疾病、免疫疾病、肿瘤、支气管肺病、皮肤病和心血管疾病或防治哮喘、艾滋病或糖尿病并发症或刺激毛发生长。
在此单独或组合使用的术语“低级烷基”是指直链或支链的含有1~6个碳原子的烷基,例如甲基、乙基、丙基、异丙基、丁基、仲丁基、叔丁基、戊基等。单独或组合使用的“低级烷氧基”是指经一个氧原子连接的如前所定义的烷基,例如甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、叔丁氧基等。术语“低级环烷基”是指含有3~6个碳原子的环烷基,即环丙基、环丁基、环戊基和环己基。术语“芳基”是指未取代的苯基或带有一个或多个选自(例如)卤素、低级烷基和低级烷氧基的取代基的苯基,例如对氯苯基、对甲苯基和对甲氧苯基。术语“低级芳烷基”是指一个氢原子已被如前定义的芳基替代的如前定义的低级烷基,例如苄基、2-苯基乙基、对氯苄基、对甲苄基和对甲氧苄基。术语“酰氧基”是指由含有最多至6个碳原子的链烷酸衍生的酰氧基,例如乙酰氧基、丙酰氧基或丁酰氧基,或者是指由可任意被(例如)卤素、低级烷基和/或低级烷氧基取代的芳族羧酸衍生的酰氧基,例如苯甲酰氧基、对氯苯甲酰氧基、对甲苯甲酰氧基和对甲氧苯甲酰氧基。术语“卤素”是指氟、氯、溴或碘。
式Ⅰ化合物含有两个手性碳原子,因此能以外消旋或旋光形式存在。本发明的范围不仅包括外消旋化合物,而且包括旋光异构体。
在式Ⅰ化合物中,R1优选代表低级烷基,特别是含有1~3个碳原子的低级烷基。R2优选代表低级烷基,特别是甲基。m优选代表1,n优选代表2。
特别优选的式Ⅰ化合物是下列化合物:
3-[8(S)-[1(R或S)-氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,和
3-[8(S)-[1(S)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮。
其它优选的式Ⅰ化合物是下列化合物:
3-[8(R或S)-[1(R或S)-氨基乙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[8(R或S)-[1(R或S)-氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[8(R或S)-[1(R或S)-氨基丁基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,和
3-[8(R或S)-[1(R或S)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮。
下列化合物也是优选的式Ⅰ化合物:
3-[8(S)-[1(R)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[8(R或S)-[a(R或S)-氨基苄基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[8(S)-[(R或S)-(氨基)(环戊基)甲基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[2(R或S)-[1(R或S)-氨基-2-甲基丙基]-2,3-二氢-1H-吡啶并[1,2-a]吲哚-9-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[8(RS)-[1(RS)-氨基-2-甲基丙基]-7,8,9,10-四氢-6H-吖庚因并[1,2-a]吲哚-11-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[7(RS)-[1(RS)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-9-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,和
3-[8(S)-[1(S)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-苯基-3-吲哚基)-1H-吡咯-2,5-二酮。
按照本发明所提供的方法,式Ⅰ化合物以及式Ⅰ酸性化合物与碱形成的可药用盐和式Ⅰ碱性化合物与酸形成的可药用盐的制备方法是:脱除通式Ⅱ化合物中由R3表示的保护基,并且根据需要,对获得的式Ⅰ化合物中存在于R2中的反应性取代基在官能上进行修饰,并且必要时,用碱将式Ⅰ酸性化合物转化成可药用盐或者用酸将式Ⅰ碱性化合物转化成可药用盐,

其中R1、R2、m和n具有前述意义,R3代表尿烷保护基。
式Ⅱ中R3表示的尿烷保护基优选是低级烷氧羰基(特别是叔丁氧羰基)或低级芳烷氧羰基(特别是苄氧羰基)。
式Ⅱ化合物中R3表示的尿烷保护基的脱保护反应可按本来公知的方法进行。例如,当R3代表低级烷氧羰基时,可以在一种惰性有机溶剂中采用一种无机酸(如盐酸)或三氟乙酸进行该脱保护反应,所述惰性有机溶剂例如是环醚(如四氢呋喃或二噁烷)、链烷醇(如甲醇或乙醇)、酯(如乙酸乙酯)或者卤代(特别是氯代)烃(如二氯甲烷)。当R3代表芳烷氧羰基时,按本来公知的方法通过氢解进行脱保护反应,例如在催化剂(如钯/炭)存在下利用氢进行氢解。
对获得的式Ⅰ化合物中存在于R2中的反应性取代基在官能上进行修饰,包括:将羧基酯化成低级烷氧羰基,将酰氧基水解成羟基或者将低级烷氧羰基转化成羧基。所有这些修饰反应可按本来公知的方法进行。
可按本来公知的方法,通过用合适的碱处理,将式Ⅰ酸性化合物转化成可药用盐。合适的盐不仅是无机碱衍生的那些盐(例如钠盐、钾盐、钙盐等),而且是有机碱(如乙二胺、单乙醇胺、二乙醇胺等)衍生的那些盐等等。可按本来公知的方法,通过用合适的酸处理,将式Ⅰ碱性化合物转化成可药用盐。合适的盐不仅是无机酸衍生的那些盐(例如盐酸盐、氢溴酸盐、磷酸盐、硫酸盐等),而且是有机酸衍生的那些盐(例如乙酸盐、柠檬酸盐、富马酸盐、酒石酸盐、马来酸盐、甲磺酸盐、对甲苯磺酸盐等)。
上述式Ⅱ的起始原料是新的化合物并且也是本发明的目的。例如,它们可按下法制备:
(a)将通式Ⅲ化合物与草酰氯反应,将所得的通式Ⅳ的活化的乙醛酸与通Ⅳ的亚氨酸酯在强碱存在下缩合并将所得的通式Ⅵ的羟基吡咯啉酮水解和脱水,

其中R1、R3、m和n具有前述意义,

其中R1、R3、m和n具有前述意义,

其中R2具有前述意义,R4代表低级烷基,

其中R1、R2、R3、R4、m和n具有前述意义。
或者
(b)在强碱存在下将前述式Ⅳ的活化的乙醛酸与通式Ⅶ的吲哚乙酸反应并将所得的通式Ⅷ的取代的呋喃二酮转化成相应的式Ⅱ的酰亚胺起始原料,

其中R2具有前述意义,

其中R1、R2、R3、m和n具有前述意义。
式Ⅲ化合物与草酰氯的反应通常在惰性有机溶剂(合适的有卤代脂族烃,例如二氯甲烷)存在下进行。也通常是在约0℃下进行该反应。
式Ⅴ的亚氨酸酯是公知的化合物或者是公知化合物的类似物。式Ⅴ的亚氨酸酯与式Ⅳ的活化的乙醛酸的缩合反应通常在惰性有机溶剂中进行。合适的碱例如是叔胺,例如三乙胺、二异丙基乙胺、4-二甲氨基吡啶、N-乙基吗啉和1,4-二氮杂双环[2,2,2]辛烷以及吡啶。合适的溶剂例如是卤代脂族烃类(如二氯甲烷和氯仿)、任意卤代的芳族烃类(如苯、甲苯和氯苯)、开链和环状醚类(如二甲氧基乙烷、叔丁基·甲基醚和四氢呋喃)、甲酰胺类(如二甲基甲酰胺)、酯类(如乙酸乙酯)以及腈类(如乙腈)。优选在0℃~40℃,尤其在室温进行该缩合反应。另外,优选就地进行该缩合反应。
将式Ⅵ的羟基吡咯啉酮水解并脱水生成式Ⅱ化合物的反应可便利地按下法进行,即用无机酸(如盐酸或硫酸)或者有机酸(如甲磺酸或对甲苯磺酸)处理,或者用酰基化试剂(如三氟乙酸酐)和合适的碱(如吡啶)处理,上述处理通常大约在室温进行。优选将式Ⅵ的羟基吡咯啉酮就地进行水解和脱水。
式Ⅶ的吲哚乙酸是公知化合物或者是公知化合物的类似物,式Ⅶ的吲哚乙酸与式Ⅳ的活化的乙醛酸的反应方法,通常类似于前述有关式Ⅳ的活化的乙醛酸与式Ⅴ的亚氨酸酯的缩合反应的方法。
将式Ⅷ的取代的呋喃二酮转化为期望的式Ⅱ的酰亚胺起始原料的反应通常按下法进行,即在惰性有机溶剂中、在链烷醇(如甲醇)存在下,用六甲基二硅氮烷处理。合适的溶剂例如是卤代脂族烃类(如二氯甲烷和氯仿)、任意卤代的芳族烃类(如苯、甲苯和氯苯)、开链和环状醚类(如二甲氧基乙烷、叔丁基·甲基醚和四氢呋喃)或者甲酰胺类(如二甲基甲酰胺)。该反应优选大约在室温至100℃、尤其在约50℃进行。
可按(例如)下述反应方案Ⅰ中所述方法制备上述式Ⅲ化合物,在反应方案Ⅰ中,R1、m和n具有前述意义。
反应方案Ⅰ

参照反应方案Ⅰ,其中所有的单个步骤均可按常规方法进行。在第一步反应中,例如使用氢氧化钠溶液,将式Ⅸ的乙基酯(一种公知的化合物或公知化合物的类似物)皂化成相应的式Ⅹ的酸。然后将所得的酸酰胺化,例如通过在三乙胺存在下和氯甲酸乙酯反应,随后用氨处理。将所得的式Ⅺ的酰胺用(例如)三氟乙酸酐转化成式Ⅻ的腈。然后将式Ⅻ的腈与式R1-Mg-X的格氏试剂(其中R1具有前述意义,X代表卤素,优选氯)反应,并用复合金属氢化物(如氢化铝锂)将所得的式XⅢ的亚胺还原成式XIV的伯胺。优选就地进行该还原反应。将式XIV的伯胺转化成式Ⅲ化合物,例如通过在碱(如三乙胺)存在下,使式XIV的伯胺与式R3Cl的氯甲酸酯或式R3OR3的酸酐反应,其中R3具有前述意义。
可按(例如)下述反应方案Ⅱ中所述方法制备此后由ⅢA表示的式Ⅲ的同手型化合物,在反应方案Ⅱ中,R1、m和n具有前述意义,并且R5代表低级烷基。
反应方案Ⅱ

在反应方案Ⅱ中所述的各个合成步骤均可按本来公知的方法进行。在第一步反应中,用强酸(如浓硫酸)将式XV的
基酯(一种公知的化合物或公知化合物的类似物)转化成相应的式XVI的羧酸。然后将该酸与N,O-二甲基羟胺缩合,并将所得的式XVⅡ的N-甲氧基-N-甲基羧酰胺与式R1-Mg-X的格式试剂(其中R1具有前述意义,X代表卤素,优选氯)反应,得到式XVⅢ的酮。将该酮与式H2N-OR5的羟胺(其中R5具有前述意义)反应,得到式XIX的肟,用复合金属氢化物(如氢化铝锂)将所述肟还原成式XX的伯胺。后者然后通过酰化反应转化成式ⅢA的同手型起始原料,所述酰化方法类似于反应方案Ⅰ中所述的将式XIV化合物转化成式Ⅲ化合物的方法。
下列反应方案Ⅲ阐明另外一种制备式ⅢA的同手型起始原料的反应路线。在该反应方案中,R1、m和n具有前述意义。并且Ms代表甲磺酰基。
反应方案Ⅲ
参照反应方案Ⅲ,其中所有的单一反应步骤均可按常规方法进行。首先,例如用甲磺酸酐将式XXI的醇磺化,然后用氰化钠将所得的式XXⅡ的甲磺酸酯转化成式XXⅢ的腈。将后者(例如)用氢氧化钠溶液水解成相应的式XXIV的羧酸,在(例如)浓硫酸存在下用甲醇进行甲基化,将式XXIV的羧酸转化成式XXV的甲酯。在强碱(例如二异丙基氨化锂)存在下,将式XXV的甲酯与式R1-X的卤化物(其中R1和X具有前述意义)反应,随后用氢氧化钠溶液处理,得到式ⅩⅩⅤⅠ的羧酸。后者与二苯膦酰基叠氮化物反应,得到式ⅩⅩⅤⅡ的异氰酸酯,通过用盐酸处理,将该异氰酸酯转化成式ⅩⅩ的伯胺。按类似于反应方案Ⅰ中所述的有关方法,通过酰化将式ⅩⅩ的伯胺转化成式ⅢA的同手型化合物。
式Ⅰ化合物和其可药用盐是蛋白激酶抑制剂;它们抑制细胞过程,例如细胞增殖和细胞分泌,并能用于防治疾病,例如防治炎性疾病(如关节炎)、免疫疾病、牛皮癣、接触性皮炎、与器官移植有关的疾病以及肿瘤。式Ⅰ化合物及其盐抑制人免疫缺陷病毒或EB病毒的细胞感染并因此可用于治疗艾滋病和传染性单核细胞增多症。本发明化合物及其盐还抑制平滑肌收缩并因此能用于治疗心血管疾病和支气管肺病。另外,本发明化合物及其盐还可用于哮喘治疗。本发明化合物及其盐还抑制血小板聚集并能用于防治血栓形成。另外,它们还抑制活化的中性白细胞释放介质并因此用于治疗缺血性损伤,例如心脏或大脑中缺血性损伤。再者,它们抑制由升高的葡萄糖水平诱发的神经毒性并因此可用于治疗糖尿病并发症。最后,本发明化合物及其盐刺激毛发生长并因此能用于防治秃发。
通过下述体外试验方法可以证实本发明化合物抑制蛋白激酶C的活性。
使用Takai等人在BBRC 19,1218,(1979)中所描述的试验系统。反应混合物(100μl)含有10μM[γ-32P]ATP、0.2mg/ml(约15μM)富含赖氨酸的组蛋白、0.5mM CaCl2和40μg/ml磷脂酰丝氨酸在25mMTris HCl中的溶液、5mM MgNO3(pH7.5)缓冲液。按照Kikkawa等人在J.Biol.Chem.257,13341(1982)中的方法从大鼠大脑中分离蛋白激酶C(所述酶)。
通过加入所述酶启动反应,将反应在30℃进行10分钟,然后用1ml冰冷却的10%三氯乙酸使反应停止。在玻璃纤维碟上过滤收集酸可沉降的蛋白。然后依次用含有20mM焦磷酸钠的5%三氯乙酸(以除去未反应的ATP)和乙醇洗涤,干燥并计数。有关各个碟的计数被用以测定[γ-32P]ATP中32P与组蛋白的结合。在各个试验化合物浓度下酶阻断的程度可由下式计算: (结合的Cpm+试验化合物+酶)/(结合的Cpm-试验化合物+酶) ×100%
IC50值是在上述试验条件下,能够减少50%的蛋白激酶诱导的32P结合的试验化合物的浓度。
在下表中示出了上述试验中具有代表性的式Ⅰ化合物所得的结果。
表
| 所述实施例的产物 | IC50(nM) |
| 1011 | 3.35.5 |
式Ⅰ化合物和其上述盐可用作药物,例如以药物制剂的形式。可经口服施用药物制剂,药剂形式例如有片剂、包衣片剂、糖锭剂、硬和软明胶胶囊剂、溶液剂、乳剂或悬浮剂。但是,药物制剂也可经直肠(例如以栓剂形式)或非胃肠(例如以溶液注射剂形式)给药。
为了制备药物制剂,可将式Ⅰ化合物和其上述盐与治疗上惰性的无机或有机载体一起配制。例如可用乳糖、玉米淀粉或其衍生物、滑石粉、硬脂酸或其盐等作为片剂、包衣片剂、糖锭剂和硬明胶胶囊剂的载体。软明胶胶囊剂的合适载体(例如)是植物油、蜡、脂肪、半固体和液体多元醇等。但是,依据活性物质的性质,在软明胶胶囊剂的情况下一般可不用载体。制备溶液剂和糖浆剂的合适载体(例如)是水、多元醇、蔗糖、转化糖、葡萄糖等。溶液注射剂的合适载体(例如)是水、醇、多元醇、甘油、植物油等。栓剂的合适载体(例如)是天然油或硬化油、蜡、脂肪、半液体多元醇等。
药物制剂也可含有防腐剂、加溶剂、稳定剂、湿润剂、乳化剂、甜味剂、着色剂、调味剂、改变渗透压的盐、缓冲剂、包衣剂或抗氧化剂。它们还可含有治疗上有活性的其它物质。含有上述式Ⅰ化合物或其盐和治疗上惰性的载体的药物以及制备该药物的方法也是本发明的目的。该方法包括将上述式Ⅰ化合物或其盐与治疗上惰性的载体物质(必要时还可有一种或多种治疗上有活性的其它物质)一起配制成盖仑制剂形式。
如上所述,式Ⅰ化合物及其上述盐可用于防治各种疾病,特别是防治炎性疾病、免疫疾病、支气管肺病、皮肤病和心血管疾病,治疗哮喘、艾滋病或糖尿病并发症或用于刺激毛发生长。剂量可在宽范围内变化,对于每个具体病例当然应根据个体需要作相应调整。一般来说,对成人口服给药时,日剂量约为5mg~500mg应是合适的,虽然当必要时也可超出其上限。日剂量可单次给药或分次给药。
以下实施例阐明本发明:
实施例1
将1.25g(2.32mmol)3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基乙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A)在10ml乙酸乙酯中的溶液用30ml饱和氯化氢的乙酸乙酯溶液处理并在室温下搅拌18小时。将所得固体过滤并干燥,得到1.0g 3-[8(R或S)-[1(R或S)-氨基乙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A),是一种红色固体,熔点:324~325℃。
用作起始原料的3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基乙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A)可制备如下:
(ⅰ)将27g(126mmol)6,7,8,9-四氢吡啶并[1,2-a]吲哚-8-羧酸在30ml水和450ml丙酮中的悬浮液搅拌冷却至0℃,相继用14.7g(145mmol)三乙胺和17.3g(159mmol)氯甲酸乙酯处理。0.5小时后加入6.3ml0.880M氨并持续搅拌1小时。减压除去溶剂将残余物用含水乙醇研制。过滤产物并干燥得到13g 6,7,8,9-四氢吡啶并[1,2-a]吲哚-8-甲酰胺,是一种白色固体,熔点:165~168℃。
(ⅱ)于10℃和搅拌下,将25.7g(126mmol)三氟乙酸酐滴加到26.5g(123mmol)6,7,8,9-四氢吡啶并[1,2-a]吲哚-8-甲酰胺和23.4g(300mmol)吡啶在500ml无水二噁烷中的悬浮液中。加毕,减压除去溶剂并将残余物在甲醇中结晶,得到13g 6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(RS)-甲腈,是一种浅褐色固体,熔点:106~109℃。
(ⅲ)将1.4g(7.2mmol)6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(RS)-甲腈在400ml无水甲苯中的溶液用7ml(21mmol)3M氯化甲基镁的四氢呋喃溶液处理并将所得溶液在氮气氛下加热回流0.5小时。然后将该溶液加到17ml(17mmol)1M氢化铝锂的四氢呋喃溶液中。将所得溶液加热回流15分钟,冷却并滴加约20ml水处理。将沉淀过滤并用100ml乙酸乙酯洗涤,将滤液和洗涤液合并减压蒸发,得到1.55g浅棕色油状物。
将该油状物溶解在70ml无水二氯甲烷中并在0℃和氮气氛下依次用1.5g(15mmol)三乙胺和1.8g(7.4mmol)二碳酸二叔丁酯处理。将该搅拌着的溶液温热至室温并继续搅拌1小时。减压除去溶剂并将产物在硅胶上用乙醚/石油醚(1:2)洗脱进行快速色谱法纯化,得到925mg[1(R或S)-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(R或S)-基)乙基]氨基甲酸叔丁酯,是白色固状非对映体混合物,熔点:114~117℃。
(ⅳ)于0℃用1.28g(10.3mmol)草酰氯滴加处理3.0g(9.57mmol)[1(R或S)-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(R或S)-基)乙基]氨基甲酸叔丁酯在150ml二氯甲烷中的搅拌着的溶液。5分钟后,减压除去溶剂并将残余物溶解在150ml二氯甲烷中并在0℃和氨气氛下用2.94g(11mmol)1-甲基-3-吲哚亚氨代乙酸异丙酯盐酸盐和4.38g(43mmol)三乙胺处理。将溶液温热至室温,搅拌24小时,用水洗涤并用硫酸镁干燥。蒸发除去溶剂并将残余物溶解在30ml吡啶中。将搅拌着的溶液冷却至冰浴温度并滴加1.5ml(10.8mmol)三氟乙酸酐处理。15分钟后减压除去溶剂并将残余物在200ml乙酸乙酯和200ml 0.2M盐酸之间分配。有机层用50ml碳酸氢钠溶液洗涤,用硫酸镁干燥并蒸发至干。将残余物在硅胶上用乙酸乙酯/石油醚(1∶2)洗脱进行快速色谱法纯化,得到1.35g 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基乙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A),为红色固体,熔点:255~7℃。进一步洗脱得到1.38g红色固体状的非对映体B,熔点:230~233℃。
实施例2
按类似于实施例1的第一段所述方法,由按实施例1(ⅳ)所述制备的1.38g 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基乙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体B)制得930mg 3-[8(R或S)-[1(R或S)-氨基乙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体B),是一种红色固体,熔点:254~258℃。
实施例3
按类似于实施例1的第一段所述方法,由180mg 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A),制得115mg 3-[8(R或S)-[1(R或S)-氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体A),是一种红色固体,熔点:321~323℃。
用作起始原料 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A)可制备如下:
(ⅰ)按类似于实施例1(ⅲ)中所述的方法,由500mg(2.5mmol)6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(RS)-甲腈和2.5ml(5mmol)2M溴化乙基镁的四氢呋喃溶液制得480mg[1(R或S)-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(R或S)-基)丙基]氨基甲酸叔丁酯,是白色固体状的非对映体混合物,熔点:153~156℃。
(ⅱ)按类似于实施例1(ⅳ)中所述的方法,由440mg(1.34mmol)[1(R或S)-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(R或S)-基)丙基]氨基甲酸叔丁酯制得180mg 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A),是一种红色胶状物。进一步洗脱得到130mg红色胶状的非对映体B。
实施例4
按类似于实施例1的第一段中所述的方法,由130mg 3-[8(R或S)-1(R或S)-叔丁氧甲酰氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮[非对映体B,按实施例3(ⅱ)所述制备]制得65mg 3-[8(R或S)-1(R或S)-氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体B),是一种红色固体,熔点:245~249℃。
实施例5
按类似于实施例1的第一段中所述的方法,由400mg(0.7mmol) 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基丁基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A)制得310mg 3-[8(R或S)-[1(R或S)-氨基丁基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体A),是一种红色固体,熔点:237~241℃。
用作起始原料的 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基丁基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A)可制备如下:
按类似于实施例1(ⅲ)和(ⅳ)中所述的方法,由1.0g(5mmol)6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(RS)-甲腈和5.0ml(10mmol)2M氯化正丙基镁的乙醚溶液制得470mg 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基丁基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A),是一种红色固体,熔点:227~9℃。进一步洗脱得到285mg红色固体状非对映体B,熔点:169~172℃。
实施例6
按类似于实施例1的第一段中所述的方法,由按实施例5的第二段中所述方法制得的270mg(0.48mmol) 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基丁基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体B)制得210mg 3-[8(R或S)-[1(R或S)-氨基丁基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体B),是一种红色固体,熔点:235~238℃。
实施例7
按类似于实施例1的第一段所述的方法,由400mg(0.7mmol) 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A)制得300mg 3-[8(R或S)-[1(R或S)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体A),是一种红色固体,熔点:254~256℃。
用作起始原料的 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A)可制备如下:
(ⅰ)用10ml(20mmol)2M氯化异丙基镁的四氢呋喃溶液处理2.0g(10mmol)6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(RS)-甲腈在150ml无水甲苯中的溶液并将所得溶液加热回流20分钟。将溶液冷却并滴加24ml(24mmol)1M氢化铝锂的四氢呋喃溶液进行处理。将所得溶液在室温搅拌1.5小时,冷却并滴加约20ml水处理。过滤沉淀,用100ml二氯甲烷洗涤,将合并的滤液减压蒸发,得到7.2g浅棕色油状物。将该油状物溶解在100ml无水二氯甲烷中并在0℃和氮气氛下依次用2.34g(23.4mmol)三乙胺和2.8g(11.6mmol)二碳酸二叔丁酯处理。将搅拌着的溶液温热至室温并继续搅拌70小时。减压除去溶剂并将产物在硅胶上用乙醚/石油醚(1∶2)洗脱进行快速色谱法纯化,得到800mg[1(R或S)-[6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(R或S)-基]-2-甲基丙基]氨基甲酸叔丁酯(非对映体A),是一种白色固体,熔点:118~9℃。进一步洗脱得到420mg白色固体状的非对映体B,熔点:128~129℃。
(ⅱ)用287mg(2.4mmol)草酰氯于0℃滴加处理770mg(2.25mmol)[1(R或S)-[6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(R或S)-基]-2-甲基丙基]氨基甲酸叔丁酯(非对映体A)在30ml二氯甲烷中的搅拌着的溶液。5分钟后减压除去溶剂并将残余物溶解在30ml二氯甲烷中,并在0℃和氮气氛下用686mg(2.57mmol)1-甲基-3-吲哚亚氨代乙酸异丙酯盐酸盐和1.02g(10mmol)三乙胺处理。将溶液温热至室温并搅拌18小时,用50ml水洗涤并用硫酸镁干燥。蒸发除去溶剂并将残余物溶解在10ml吡啶中。将搅拌着的溶液冷却至冰浴温度并滴加664μl(4.8mmol)三氟乙酸酐处理。15分钟后加入50ml乙酸乙酯并将有机层用水和2M盐酸洗涤,用硫酸镁干燥并蒸发至干,将残余物在硅胶上用乙酸乙酯/石油醚(1∶2)洗脱进行快速色谱法纯化,得到460mg 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A),是一种红色固体,熔点:223~224℃。
实施例8
按类似于实施例1第一段所述的方法,由270mg(0.48mmol) 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体B),制得180mg 3-[8(R或S)-[1(R或S)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体B),是一种红色固体,熔点:248~250℃。
用作起始原料的 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体B)可制备如下:
按类似于实施例7(ⅱ)中所述的方法,由按实施例7(ⅰ)中所述制备的[1(R或S)-[6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(R或S)-基]-2-甲基丙基]氨基甲酸叔丁酯(非对映体B)制得270mg 3-[8(R或S)-[1(R或S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体B),是一种红色胶状物。
实施例9
按类似于实施例1的第一段中所述的方法,由1.2g(2.17mmol) 3-[8(S)-[1(R或S)-叔丁氧甲酰氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A),制得850mg 3-[8(S)-[1(R或S)-氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体A),是一种红色固体,熔点:304~308℃。
用作起始原料的3-[8(S)-[1(R或S)-叔丁氧甲酰氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A)可制备如下:
(ⅰ)在20分钟内,用50ml(358mmol)三乙胺和52.0g(298mmol)甲磺酸酐处理50.0g(248mmol)8(S)-羟甲基-6,7,8,9-四氢吡啶并[1,2-a]吲哚在500ml二氯甲烷中的冰冷却的溶液。2小时后加入250ml水并将有机相相继用250ml饱和碳酸氢钠溶液(×2)和200ml水洗涤。然后用硫酸镁干燥有机相并蒸发。用乙醚研制残余固体,过滤并真空干燥,得到65.4g甲磺酸[6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-基]甲基酯,是一种浅粉红色固体,熔点:114~5℃,[α]20 D=-39.7°(c=1%,CH2Cl2)。
(ⅱ)将18.0g(367mmol)氰化钠加到65.0g(233mmol)甲磺酸(6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-基)甲基酯在500ml二甲基甲酰胺中的溶液中,并将混合物在70℃加热24小时。将混合物在1000ml水和600ml乙酸乙酯之间分配。水相用乙酸乙酯(700ml×2)萃取,合并的萃取液用水(500ml×2)洗涤,用硫酸镁干燥并蒸发。将所得的棕色固体溶解在乙酸乙酯中,将溶液经一层硅胶垫过滤。真空除去溶剂并将残余物在甲醇中结晶,得到25.8g 6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-乙腈,是一种浅棕色固体,熔点:100~101℃;[α]20 D=-40.6°(c=0.84%,CH2Cl2)。
(ⅲ)将27.0g(129mmol)6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-乙腈,和120ml 2M氢氧化钠在400ml 1,2-乙二醇中的溶液加热回流4小时。加入400ml乙酸乙酯并将有机相相继用500ml水、150ml 2M盐酸和三份500ml水洗涤,用硫酸镁干燥并蒸发,得到29g[8(S)-(6,7,8,9-四氢吡啶并[1,2-a]吲哚基]乙酸,是一种浅粉红色固体,熔点:118~120℃。
(ⅳ)将29g(127mmol)8(S)-(6,7,8,9-四氢吡啶并[1,2-a]吲哚基)乙酸和5ml浓硫酸在500ml甲醇中的溶液加热回流1小时。将混合物减压浓缩并将产物过滤和干燥,得到28.4g[8(S)-(6,7,8,9-四氢吡啶并[1,2-a]吲哚基)乙酸甲酯,是一种浅粉红色固体,熔点:84~87℃。
(ⅴ)在0℃和氮气氛下,用63ml(100mmol)1.6M正丁基锂的己烷溶液处理60ml无水四氢呋喃中的10g(100mmol)二异丙基胺并搅拌15分钟。将所得溶液冷却至-78℃并滴加14.9g(61.3mmol)[8(S)-6,7,8,9-四氢吡啶并[1,2-a]吲哚基]乙酸甲酯在60ml四氢呋喃中的溶液。30分钟后加入15.6g(100mmol)乙基碘并将混合物搅拌0.5小时。然后再加入8g乙基碘并将混合物温热至室温保持约1小时。将混合物在300ml乙醚和20ml 2M盐酸之间分配。将有机相用水(300ml×3)洗涤,用硫酸镁干燥并蒸发,得到17g浅桔黄色油状物。取样在甲醇中结晶,得到α(R或S)-乙基-6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)乙酸甲酯(非对映体A+B),是一种浅粉红色固体,熔点:87~90℃。
(ⅵ)用130ml 2M氢氧化钠处理17g(63mmol)α(R或S)-乙基-6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)乙酸甲酯(非对映体A+B),在150ml甲醇中的溶液并加热回流18小时。将该冷却后的溶液加到150ml 4M盐酸中,将所得沉淀过滤并在200ml二氯甲烷和100ml水之间分配。分离有机相,用硫酸镁干燥并蒸发,得到15g α(R或S)-6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)乙酸(非对映体A+B),是一种浅粉红色固体。
(ⅶ)在室温和氮气氛下用6.5g(65mmol)三乙胺和18.6g(67.5mmol)二苯膦酰基叠氮化物处理15gα(R或S)-6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)乙酸(非对映体A+B)在450ml甲苯中的搅拌着的溶液。在室温下1小时后,将混合物加热回流0.5小时,冷却并减压除去溶剂。将残余物在硅胶上用乙醚/石油醚(1∶3)洗脱进行快速色谱法纯化,得到14g无色油状异氰酸酯。将该油状物溶解在400ml二噁烷和150ml 2M盐酸中并将所得溶液在室温下搅拌18小时,浓缩溶液并在300ml乙酸乙酯和2M氢氧化钠溶液之间分配。用100ml乙酸乙酯萃取水层,合并的有机溶液用水(400ml×2)洗涤,用硫酸镁干燥并蒸发至干,得到7.7g乳白色泡沫状物。
将该泡沫状物在200ml无水二氯甲烷中用7.6g(75mmol)三乙胺和10.9g(50mmol)二碳酸二叔丁酯处理并将混合物在室温下搅拌3小时。减压除去溶剂并将残余物在硅胶上用乙醚/石油醚(1∶2)洗脱进行快速色谱法纯化,得到4.5g[1(R或S)-[6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-基]丙基]氨基甲酸叔丁酯(非对映体A+B),是一种白色固体,熔点:152~158℃。
(ⅷ)按类似于实施例1(ⅳ)中所述方法,由4.3g(13.1mmol)[1(R或S)-[6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-基]丙基]氨基甲酸叔丁酯(非对映体A+B)制得1.2g 3-[8(S)-[1(R或S)-叔丁氧甲酰氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A)和2.0g非对映体B,两者都是红色固体。
实施例10
按类似于实施例1的第一段中所述的方法,由按实施例9所述制得的2.0g(3.62mmol) 3-[8(S)-[1(R或S)-叔丁氧甲酰氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体B),制得1.28g 3-[8(S)-[1(R或S)-氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体B),是一种红色固体,熔点:247~253℃。
实施例11
按类似于实施例1的第一段中所述的方法,由1.46g 3-[8(S)-[1(S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮制得1.29g 3-[8(S)-[1(S)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐,是一种红色固体,熔点:253~256℃。
用作起始原料的3-[8(S)-[1(S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮可制备如下:
(ⅰ)往40g(114mmol)6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-甲酸1-
 基酯中加入50ml浓硫酸并搅拌所得混合物直至所有的起始原料都已溶解(约20分钟)。将溶液小心倾入1500ml冰-水中并过滤所得沉淀,用石油醚/甲苯(3∶1)洗涤并干燥,得到24.2g 6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-甲酸,是一种白色固体,熔点:251~253℃。
基酯中加入50ml浓硫酸并搅拌所得混合物直至所有的起始原料都已溶解(约20分钟)。将溶液小心倾入1500ml冰-水中并过滤所得沉淀,用石油醚/甲苯(3∶1)洗涤并干燥,得到24.2g 6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-甲酸,是一种白色固体,熔点:251~253℃。
(ⅱ)在0℃下,相继用24ml(138mmol)二异丙基乙胺、13.24g(136mmol)N,O-二甲基盐酸胲、10mg二甲氨基吡啶和23.04g(112mmol)二环己基碳二亚胺处理24.0g(111mmol)6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)甲酸在500ml二氯甲烷中的搅拌着的悬浮液。将所得混合物在室温搅拌18小时并过滤,并将固体用二氯甲烷(100ml×2)洗涤。将合并的滤液蒸发至干并将残余物在硅胶上用乙酸乙酯/石油醚(1:3)洗脱进行快速色谱法纯化,得到22.6g白色固体。取样用乙醚/石油醚研制,得到6,7,8,9-四氢-N-甲氧基-N-甲基-吡啶并[1,2-a]吲哚-8(S)甲酰胺,是一种白色固体,熔点:78~80℃。
(ⅲ)在0℃滴加60ml(120mmol)2M氯化异丙基镁的四氢呋喃溶液处理10.0g(38.7mmol)6,7,8,9-四氢-N-甲氧基-N-甲基-吡啶[1,2-a]吲哚-8(S)-甲酰胺在250ml四氢呋喃中的搅拌着的溶液。将混合物在室温下搅拌18小时并倾入250ml饱和氯化铵溶液中。将水相用乙醚(100ml×4)洗涤并将合并的乙醚萃取液用200ml盐水洗涤,用硫酸镁干燥并蒸发至干。将产物在硅胶上用乙酸乙酯/石油醚(1:3)洗脱进行快速色谱法纯化,得到4.4g异丙基6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-基酮,是一种白色固体,熔点:78~79℃。
(ⅳ)用2.30g(33mmol)盐酸胲和1.0g(25mmol)氢氧化钠在20ml水中的溶液处理4.0g(16.6mmol)异丙基6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-基酮在120ml乙醇中的悬浮液。将所得混合物加热回流3.5小时,冷却并过滤,将所得固体干燥得到3.54g白色固体状的肟。
将该肟溶解在150ml无水四氢呋喃中并用12.5ml(12.5mmol)1M氢化铝锂的四氢呋喃溶液处理。在氮气氛下将所得溶液加热回流3小时,冷却并用150ml水小心处理。依次用200ml乙酸乙酯和两份150ml乙酸乙酯萃取混合物,合并的有机萃取液用硫酸镁干燥并蒸发至干。将残余物溶解在150ml二氯甲烷中并将所得溶液用3ml(21.5mmol)三乙胺和3.4g(15.6mmol)二碳酸二叔丁酯处理并搅拌18小时。用150ml饱和氯化铵溶液洗涤混合物,用硫酸镁干燥并减压蒸发。在硅胶上用乙醚/石油醚(1:3)洗脱进行快速色谱法纯化,得到1.4g[1(R)-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-基)-2-甲基丙基]氨基甲酸叔丁酯,是一种白色固体,熔点:122~124℃。进一步洗脱得到1.1g[1(S)-[6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-基]-2-甲基丙基]氨基甲酸叔丁酯,是一种白色固体,熔点:154~155℃。
(ⅴ)按类似于实施例1(ⅳ)中所述的方法,由1.1g[1(S)-[6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-基]-2-甲基丙基]氨基甲酸叔丁酯制得1.46g3-[8(S)-[1(S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,是一种红色泡沫状物。
实施例12
用类似于实施例1的第一段中所述方法,由0.5g(0.88mmol) 3-[8(S)-[1(S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,得到0.41g 3-[8(S)-[1(R)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐,为一红色固体,熔点:235~242℃。
用作起始原料的3-[8(S)-[1(R)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮制备如下:
用类似于实施例1(ⅳ)中所述方法,由0.94g[1(R)-[6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-基]-2-甲基丙基]氨基甲酸叔丁酯(按实施例11(ⅰ)-(ⅳ)中所述方法制备)制得1.05g 3-[8(S)-[1(R)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,为一红色泡沫状物。
实施例13
用类似于实施例1的第一段中所述方法,由100mg 3-[8(R或S)-[α(R或S)-叔丁氧甲酰氨基苄基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A),得到50mg 3-[8(R或S)-[α(R或S)-氨基苄基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体A),为一红色固体,熔点:234~237℃。
用作起始原料的 3-[8(R或S)-[α(R或S)-叔丁氧甲酰氨基苄基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A)制备如下:
(ⅰ)用类似于实施例1(ⅲ)中所述方法,由1.0g(5.1mmol)6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(RS)-甲腈和3.7ml(11mmol)3M溴化苯基镁的四氢呋喃溶液,制得0.9g[α(R或S)-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(R或S)-基)苄基]氨基甲酸叔丁酯的非对映体混合物,呈白色固体形式,熔点:160~165℃。
(ⅱ)用类似于实施例1(ⅳ)中所述方法,由800mg(2.1mmol)[α(R或S)-(6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(R或S)-基)苄基]氨基甲酸叔丁酯,制得330mg 3-[8(R或S)-[α(R或S)-叔丁氧甲酰氨基苄基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A),为红色胶状物。进一步洗脱得到280mg非对映体B,为红色胶状物。
实施例14
用类似于实施例1的第一段中所述方法,由200mg 3-[8(R或S)-[α(R或S)-叔丁氧甲酰氨基苄基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体B)(按实施例13(ⅱ)中所述制备)制得70mg 3-[8(R或S)-[α(R或S)-氨基苄基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体B),为红色固体,熔点:226~233℃。
实施例15
用类似于实施例1的第一段中所述方法,由200mg 3-[8(S)-[(R或S)-(叔丁氧甲酰氨基)(环戊基)甲基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A),制得150mg 3-[8(S)-[(R或S)-(氨基)(环戊基)甲基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体A),为红色固体,熔点:236~241℃。
用作起始原料的 3-[8(S)-[(R或S)-(叔丁氧甲酰氨基)(环戊基)甲基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮可制备如下:
(ⅰ)用类似于实施例11(ⅲ)中所述方法,由2.0g(7.75mmol)6,7,8,9-四氢-N-甲氧基-N-甲基吡啶并[1,2-a]-吲哚-8(S)甲酰胺和15ml(30mmol)2M氯化环戊基镁在乙醚中的溶液制得1.1g环戊基6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-基酮,为一浅黄色固体,熔点:69℃。
(ⅱ)用类似于实施例11(ⅳ)中所述方法,由1.05g(3.9mmol)环戊基6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-基酮制得330mg 8(S)-[(R或S)-(叔丁氧甲酰氨基)(环戊基)甲基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚(非对映体A),为一白色固体,熔点:140~143℃。进一步洗脱得到430mg非对映体B,为白色固体,熔点:58~63℃。
(ⅲ)用类似于实施例1(ⅳ)中所述方法,由300mg8(S)-[(R或S)-(叔丁氧甲酰氨基)(环戊基)甲基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚(非对映体A),制得200mg 3-[8(S)-[(R或S)-(叔丁氧甲酰氨基)(环戊基)甲基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,为一红色胶状物。
实施例16
用类似于实施例1的第一段中所述方法,由250mg 3-[8(S)-[(R或S)-(叔丁氧甲酰氨基)(环戊基)甲基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体B)制得160mg 3-[8(S)-[(R或S)-(氨基)(环戊基)甲基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体B),为一红色固体,熔点:241~245℃。
用作起始原料的 3-[8(S)-[(R或S)-(叔丁氧甲酰氨基)(环戊基)甲基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮可制备如下:
用类似于实施例1(ⅳ)中所述方法,由400mg 8(S)-[(R或S)-(叔丁氧甲酰氨基)(环戊基)甲基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚(非对映体B)(按实施例15(ⅰ)~(ⅱ)中所述制备)制得250mg 3-[8(S)-[(R或S)-(叔丁氧甲酰氨基)(环戊基)甲基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,为一红色胶状物。
实施例17
用类似于实施例1的第一段中所述方法,由40mg 3-[2(R或S)-[1(R或S)-叔丁氧甲氧氨基-2-甲基丙基]-2,3-二氢-1H-吡咯并[1,2-a]吲哚-9-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体A)制得20mg 3-[2(R或S)-[1(R或S)-氨基-2-甲基丙基]-2,3-二氢-1H-吡咯并[1,2-a]吲哚-9-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体A),为红色固体,熔点:224~230℃。
用作起始原料的 3-[2(R或S)-[1(R或S)-叔丁氧甲酰氨基-2-甲基丙基]-2,3-二氢-1H-吡咯并[1,2-a]吲哚-9-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮可制备如下:
(ⅰ)将8.0g(35mmol)2,3-二氢-1H-吡咯并[1,2-a]吲哚-2(RS)-甲酸乙酯在100ml乙醇和100ml水中的溶液用3.0g(75mmol)氢氧化钠处理。将混合物加热回流15分钟,然后冷却,并用60ml(120mmol)2M盐酸酸化。过滤该悬浮液,将固体用50ml水洗涤,然后干燥,得到5.9g 2,3-二氢-1H-吡咯并[1,2-a]吲哚-2(RS)-甲酸,为白色固体。熔点:171~173℃。
(ⅱ)用类似于实施例11(ⅱ)中所述方法,由4.0g(20mmol)2,3-二氢-1H-吡咯并[1,2-a]吲哚-2(RS)-甲酸制得2.35g 2,3-二氢-N-甲氧基-N-甲基-1H-吡咯并[1,2-a]吲哚-2(RS)-甲酰胺,为一白色固体,熔点:87~88℃。
(ⅲ)将840mg(35mg原子)镁屑在60ml四氢呋喃中的悬浮液中滴加4.4g(37mmol)2-溴丙烯在10ml四氢呋喃中的溶液进行处理。再将该混合物加热回流30分钟,然后冷却到0℃,并在此温度下将其加到2.3g(9.4mmol)2,3-二氢-N-甲氧基-N-甲基-1H-吡咯并[1,2-a]吲哚-2(RS)-甲酰胺在50ml四氢呋喃中的溶液中。在0℃搅拌30分钟后,将混合物倒入200ml饱和氯化铵水溶液中。用200ml乙酸乙酯萃取该溶液,有机相用硫酸镁干燥并过滤。加入石油醚(b.p.40~60℃),得到沉淀,将其过滤并干燥,得到1.6g白色固体。将该固体溶于100ml乙醇中,并在200mg 10%钯-碳上在大气压下氢化1小时。过滤除去催化剂,减压浓缩滤液直到开始结晶。将产物滤出干燥,得到1.55g异丙基2,3-二氢-1H-吡咯并[1,2-a]吲哚-2(RS)-基酮,为白色固体,熔点:104~105℃。
(ⅳ)用类似于实施例11(ⅳ)中所述方法,由1.5g异丙基2,3-二氢-1H-吡咯并[1,2-a]吲哚-2(RS)-基酮,制得到830mg[1(R或S)-[2,3-二氢-1H-吡咯并[1,2-a]吲哚-2(R或S)-基]-2-甲基丙基]-氨基甲酸叔丁酯的非对映体混合物。将该混合物在20ml用氯化氢饱和的乙酸乙酯中搅拌2小时。将得到的固体滤出,并在硅胶上用甲醇/二氯甲烷(1:10)洗脱进行快速色谱法纯化,得到150mg2(R或S)-[1(R或S)-氨基-2-甲基丙基]-2,3-二氢-1H-吡咯并[1,2-a]吲哚盐酸盐(非对映体A),为白色固体形式。进一步洗脱,得到150mg非对映体B,为白色固体。
(ⅴ)将100mg(0.38mmol)2(R或S)-[1(R或S)-氨基-2-甲基丙基]-2,3-二氢-1H-吡咯并[1,2-a]吲哚盐酸盐(非对映体A)在30ml二氯甲烷中的溶液用110mg(0.5mmol)二碳酸二叔丁酯和100mg(1mmol)三乙胺处理并搅拌72小时。将该溶液相继用30ml 1M盐酸和30ml饱和碳酸氢钠水溶液洗涤,然后用硫酸镁干燥。过滤后,减压浓缩滤液,残余物用乙醚/石油醚(b.p.40~60℃)(1:2)洗脱进行快速色谱法纯化,得到100mg[1(R或S)-[2,3-二氢-1H-吡咯并[1,2-a]吲哚-2(R或S)-基]-2-甲基丙基]氨基甲酸叔丁酯(非对映体A),为油状物形式。
(ⅵ)用类似于实施例1的第一段中所述方法,由55mg[1(R或S)-[2,3-二氢-1H-吡咯并[1,2-a]吲哚-2(R或S)-[1(R或S)-叔丁氧甲酰氨基-2-甲基丙基]-2,3-二氢-1H-吡咯并[1,2-a]吲哚-9-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,为红色油状物。
实施例18
用类似于实施例1的第一段中所述方法,由80mg 3-[2(R或S)-[1(R或S)-叔丁氧甲酰氨基-2-甲基丙基]-2,3-二氢-1H-吡咯并[1,2-a]吲哚-9-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮(非对映体B)制得40mg 3-[2(R或S)-氨基-2-甲基丙基]-2,3-二氢-1H-吡咯并[1,2-a]吲哚-9-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐(非对映体B),为红色固体,熔点:220~225℃。
用作起始原料的3-[2(R或S)-[1(R或S)-叔丁氧甲酰氨基-2-甲基丙基]-2,3-二氢-1H-吡咯并[1,2-a]吲哚-9-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮可制备如下:
(ⅰ)用类似于实施例17(ⅴ)中所述方法,由90mg(0.34mmol)2(R或S)-[1(R或S)-氨基-2-甲基丙基]-2,3-二氢-1H-吡咯并[1,2-a]吲哚盐酸盐(非对映体B)(按实施例17(ⅳ)中所述制备)制得100mg[1(R或S)-[2,3-二氢-1H-吡咯并[1,2-a]吲哚-2(R或S)-基]-2-甲基丙基]氨基甲酸叔丁酯(非对映体B),为一油状物。
(ⅱ)用类似于实施例1(ⅳ)中所述方法,由100mg[1(R或S)-[2,3-二氢-1H-吡咯并[1,2-a]吲哚-2(R或S)-基]-2-甲基丙基]氨基甲酸叔丁酯制得80mg3-[2(R或S)-[1(R或S)-叔丁氧甲酰氨基-2-甲基丙基]-2,3-二氢-1H-吡咯并[1,2-a]吲哚-9-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,为一红色油状物。
实施例19
用类似于实施例1的第一段中所述方法,由320mg 3-[8(RS)-[1(RS)-叔丁氧甲酰氨基-2-甲基丙基]-7,8,9,10-四氢-6H-吖庚因并[1,2-a]吲哚-11-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮制得220mg 3-[8(RS)-[1(RS)-氨基-2-甲基丙基]-7,8,9,10-四氢-6H-吖庚因并[1,2-a]吲哚-11-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐,为红色固体,熔点:248~256℃。
用作起始原料的3-[8(RS)-[1(RS)-叔丁氧甲酰氨基-2-甲基丙基]-7,8,9,10-四氢-6H-吖庚因并[1,2-a]吲哚-11-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮可如下制备:
(ⅰ)用类似于实施例11(ⅱ)中所述方法,由1.0g(4.4mmol)7,8,9,10-四氢-6H-吖庚因并[1,2-a]吲哚-8(RS)-甲酸制得0.8g 7,8,9,10-四氢-N-甲氧基-N-甲基-6H-吖庚因并[1,2-a]吲哚-8(RS)-甲酰胺,为白色固体,熔点:134~135℃。
(ⅱ)用类似于实施例17(ⅲ)中所述方法,由0.8g(2.9mmol)7,8,9,10-四氢-N-甲氧基-N-甲基-6H-吖庚因并[1,2-a]吲哚-8(RS)-甲酰胺制得0.56g异丙基7,8,9,10-四氢-6H-吖庚因并[1,2-a]吲哚-8(RS)-基酮,为白色固体,熔点:79~80℃。
(ⅲ)用类似于实施例11(ⅳ)中所述方法,由0.56g(2.2mmol)异丙基7,8,9,10-四氢-6H-吖庚因并[1,2-a]吲哚-8(RS)-基酮制得330mg[1(RS)-[7,8,9,10-四氢-6H-吖庚因并[1,2-a]吲哚-8(RS)-基]-2-甲基丙基]氨基甲酸叔丁酯的非对映体混合物,为白色固体形式,熔点:152~153℃。
(ⅳ)用类似于实施例1(ⅳ)中所述方法,由300mg[1(RS)-[7,8,9,10-四氢-6H-吖庚因并[1,2-a]吲哚-8(RS)-基]-2-甲基丙基]氨基甲酸叔丁酯制得350mg 3-[8(RS)-[1(RS)-叔丁氧甲酰氨基-2-甲基丙基]-7,8,9,10-四氢-6H-吖庚因并[1,2-a]吲哚-11-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,为红色油状物。
实施例20
用类似于实施例1的第一段中所述方法,由800mg 3-[7(RS)-[1(RS)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮制得480mg 3-[7(RS)-[1(RS)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐,为红色固体,熔点:238~244℃。
用作起始原料的3-[7(RS)-[1(RS)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮可制备如下:
(ⅰ)用类似于实施例11(ⅱ)中所述方法,由2.0g(9.3mmol)6,7,8,9-四氢吡啶并[1,2-a]吲哚-7(RS)-甲酸制得1.6g 6,7,8,9-四氢-N-甲氧基-N-甲基吡啶并[1,2-a]吲哚-7(RS)-甲酰胺,为一浅黄色油状物。
(ⅱ)用类似于实施例17(ⅲ)中所述方法,由1.6g6,7,8,9-四氢-N-甲氧基-N-甲基吡啶并[1,2-a]吲哚-7(RS)-甲酰胺制得1.05g异丙基6,7,8,9-四氢吡啶并[1,2-a]吲哚-7(RS)-基酮,为褐色固体,熔点:43~44℃。
(ⅲ)用类似于实施例11(ⅳ)中所述方法,由1.0g异丙基6,7,8,9-四氢吡啶并[1,2-a]吲哚-7(RS)-基酮制得800mg[1(RS)-[6,7,8,9-四氢吡啶并[1,2-a]吲哚-7(RS)-基]-2-甲基丙基]氨基甲酸叔丁酯,为白色固体,熔点:55~57℃(为非对映体的混合物)。
(ⅳ)用类似于实施例1(ⅳ)中所述的方法,由700mg[1(RS)-[6,7,8,9-四氢吡啶并[1,2-a]吲哚-7(RS)-基]-2-甲基丙基]氨基甲酸叔丁酯制得800mg 3-[7(RS)-[1(RS)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,为一红色胶状物。
实施例21
用类似于实施例1的第一段中所述方法,由1.3g 3-[8(S)-[1(S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-苯基-3-吲哚基)-1H-吡咯-2,5-二酮制得1.12g 3-[8(S)-[1(S)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-苯基-3-吲哚基)-1H-吡咯-2,5-二酮盐酸盐,为红色固体,熔点:235~245℃。
用作起始原料的3-[8(S)-[1(S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-苯基-3-吲哚基)-1H-吡咯-2,5-二酮可制备如下:
(ⅰ)将10g(51.8mmol)1-苯基吲哚在100ml无水乙醚中的用冰冷却的溶液在5分钟内通过滴加6ml(68.8mmol)草酰氯在20ml无水乙醚中的溶液进行处理。将混合物搅拌3小时,同时用冰冷却,然后一次性用25ml乙醇处理。搅拌10分钟后,减压除去溶剂,残余固体在60ml乙醇中结晶,得到12.38g 1-苯基吲哚-3-乙醛酸乙酯,为浅黄色固体,熔点:109~110℃。
(ⅱ)将10g(34.1mmol)1-苯基吲哚-3-乙醛酸乙酯和约25g阮内镍在350ml乙醇和150ml水中的混合物加热回流6小时。将悬浮液通过玻璃纤维滤纸过滤,并将固体用四份50ml乙酸乙酯洗涤,同时小心不使固体变干。减压浓缩滤液,残余物在硅胶上用乙酸乙酯/己烷(1:2)洗脱进行快速色谱法纯化,得到6.38g 1-苯基吲哚-3-乙酸乙酯,为黄色油状物。
(ⅲ)将6.3g(22.6mmol)1-苯基吲哚-3-乙酸乙酯在20ml乙醇中的溶液用20ml(40mmol)2M氢氧化钠溶液处理,并放于室温下17小时。减压除去乙醇,水溶液用二份20ml乙醚洗涤。水相用浓盐酸酸化,将得到的悬浮液于0℃贮存2小时,过滤该悬浮液,固体在甲醇/水(2:1)中结晶,得到5.6g 1-苯基吲哚-3-乙酸,为蓝灰色固体,熔点:131~135℃。
(ⅳ)在5分钟内,在氮气氛下滴加0.85ml(9.74mmol)草酰氯在5ml无水乙醚中的溶液,处理3g(8.77mmol)按实施例11(ⅳ)所述制备的[1(S)-[6,7,8,9-四氢吡啶并[1,2-a]吲哚-8(S)-基]-2-甲基丙基]氨基甲酸叔丁酯在50ml无水乙醚中的冰冷却的溶液。再过5分钟后,减压除去溶剂并将残余物溶解在50ml无水二氯甲烷中,在0℃和搅拌下将该溶液滴加到2.2g(8.77mmol)1-苯基吲哚-3-乙酸和3.65ml(26.3mmol)三乙胺在50ml无水二氯甲烷中的混合物中。将该混合物搅拌17小时,然后减压除去溶剂。在硅胶上用乙酸乙酯/己烷(1:2)洗脱进行快速色谱法纯化,然后在乙酸乙酯/己烷中结晶进行纯化,得到1.6g 3-[8(S)-[1(S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-苯基-3-吲哚基)-呋喃-2,5-二酮,是一种橙色固体,熔点:148~150℃。
(ⅴ)用5.35ml(25.4mmol)六甲基二硅氮烷和0.41g(12.8mmol)甲醇处理1.6g(2.54mmol) 3-[8(S)-[1(S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-苯基-3-吲哚基)-呋喃-2,5-二酮在20ml无水N,N-二甲基甲酰胺中的溶液。将该溶液在50℃加热3小时,然后用另外的5.35ml(24.8mmol)六甲基二硅氮烷和0.41g(12.8mmol)甲醇处理。总共6小时后,减压蒸发溶剂并将残余物用20ml甲醇重新蒸发。在硅胶上用乙酸乙酯/己烷(1:2)洗脱进行快速色谱法纯化,得到1.35g 3-[8(S)-[1(S)-叔丁氧甲酰氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-苯基-3-吲哚基)-1H-吡咯-2,5-二酮,是一种红色固体,熔点:165~168℃。
下列实施例阐述含有本发明化合物的一般药物制剂:
实施例A
含有下列成分的片剂可用常规方法生产:
成分 每片
式Ⅰ化合物 5.0mg
乳糖 125.0mg
玉米淀粉 75.0mg
滑石粉 4.0mg
硬脂酸镁 1.0mg
每片重 210.0mg
实施例B
含有下列成分的胶囊剂可用常规方法生产:
成分 每粒
式Ⅰ化合物 10.0mg
乳糖 165.0mg
玉米淀粉 20.0mg
滑石粉 5.0mg
每粒胶囊剂填充物重 200.0mg
Claims (13)
1、通式Ⅰ化合物以及式Ⅰ酸性化合物与碱形成的可药用盐和式Ⅰ碱性化合物与酸形成的可药用盐:
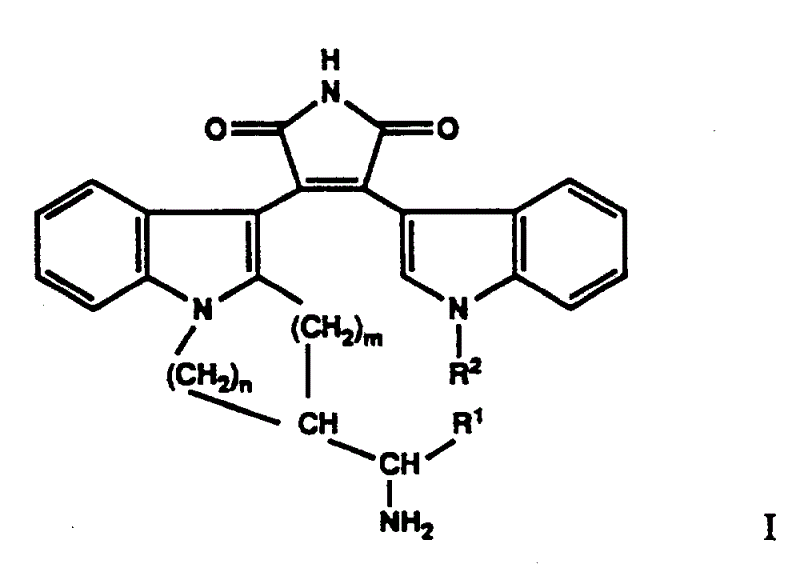
其中R1代表低级烷基、低级环烷基、芳基或低级芳烷基;R2代表氢、芳基或低级烷基,所述芳基和烷基可任意被羟基、酰氧基、氨基、单(低级烷基)氨基、二(低级烷基)氨基、羧基、低级烷氧羰基或氨基羰基取代;m和n代表1或2。
2、权利要求1所述的化合物,其中R1代表低级烷基,特别是含有1~3个碳原子的低级烷基。
3、权利要求1-2中任一权项所述的化合物,其中R2代表低级烷基。
4、权利要求1~3中任一权项所述的化合物,其中m代表1并且n代表2。
5、3-[8(S)-[1(R或S)-氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮。
6、3-[8(S)-[1(S)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮。
7、选自下列的权利要求1所述的化合物:
3-[8(R或S)-[1(R或S)-氨基乙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[8(R或S)-[1(R或S)-氨基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[8(R或S)-[1(R或S)-氨基丁基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,和
3-[8(R或S)-[1(R或S)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮。
8、选自下列的权利要求1所述的化合物:
3-[8(S)-[1(R)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[8(R或S)-[a(R或S)-氨基苄基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[8(S)-[(R或S)-(氨基)(环戊基)甲基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[2(R或S)-[1(R或S)-氨基-2-甲基丙基]-2,3,-二氢-1H-吡啶并[1,2-a]吲哚-9-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[8(RS)-[1(RS)-氨基-2-甲基丙基]-7,8,9,10-四氢-6H-吖庚因并[1,2-a]吲哚-11-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,
3-[7(RS)-[1(RS)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-9-基]-4-(1-甲基-3-吲哚基)-1H-吡咯-2,5-二酮,和
3-[8(S)-[1(S)-氨基-2-甲基丙基]-6,7,8,9-四氢吡啶并[1,2-a]吲哚-10-基]-4-(1-苯基-3-吲哚基)-1H-吡咯-2,5-二酮。
9、通式Ⅱ化合物:

其中R1、R2、m和n具有权利要求1中给出的意义,并且R3代表尿烷保护基。
10、用作治疗活性物质(特别是用作防治炎性疾病、免疫疾病、肿瘤、支气管肺病、皮肤病和心血管疾病的治疗活性物质,作为治疗哮喘、艾滋病或糖尿病并发症的活性物质或者作为刺激毛发生长的活性物质)的权利要求1~8中任一权项所述的化合物。
11、权利要求1~8中任一权项所述化合物的制备方法,该方法包括脱除式Ⅱ化合物中由R3表示的保护基,并且根据需要,对获得的式Ⅰ化合物中存在于R2中的反应性取代基在官能上进行修饰,并且必要时,用碱将式Ⅰ酸性化合物转化成可药用盐或者用酸将式Ⅰ碱性化合物转化成可药用盐,
其中R1、R2、m和n具有权利要求1中给出的意义并且R3代表尿烷保护基。
12、一种药物,特别是一种防治炎性疾病、免疫疾病、肿瘤、支气管肺病、皮肤病或心血管疾病的药物或者是治疗哮喘、艾滋病或糖尿病并发症或刺激毛发生长的药物,所述药物含有权利要求1~8中任一权项所述的化合物和治疗上惰性的载体物质。
13、权利要求1~8中任一权项所述的化合物在制备防治炎性疾病、免疫疾病、肿瘤、支气管肺病、皮肤病或心血管疾病或者防治哮喘、艾滋病或糖尿病并发症或刺激毛发生长的药物方面的用途。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB939309602A GB9309602D0 (en) | 1993-05-10 | 1993-05-10 | Substituted pyrroles |
| GB9309602.2 | 1993-05-10 | ||
| GB9403249.7 | 1994-02-21 | ||
| GB9403249A GB9403249D0 (en) | 1993-05-10 | 1994-02-21 | Substituted pyrroles |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1103402A true CN1103402A (zh) | 1995-06-07 |
| CN1048014C CN1048014C (zh) | 2000-01-05 |
Family
ID=26302877
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN94105846A Expired - Fee Related CN1048014C (zh) | 1993-05-10 | 1994-05-09 | 取代的吡咯类化合物及其制法、药物组合物和用途 |
Country Status (25)
| Country | Link |
|---|---|
| EP (1) | EP0624586B1 (zh) |
| JP (1) | JP2668818B2 (zh) |
| CN (1) | CN1048014C (zh) |
| AT (1) | ATE184007T1 (zh) |
| AU (1) | AU678435B2 (zh) |
| BG (1) | BG98761A (zh) |
| BR (1) | BR9401935A (zh) |
| CA (1) | CA2121567A1 (zh) |
| CZ (1) | CZ112894A3 (zh) |
| DE (1) | DE69420310T2 (zh) |
| DK (1) | DK0624586T3 (zh) |
| ES (1) | ES2137281T3 (zh) |
| FI (1) | FI106961B (zh) |
| GR (1) | GR3031901T3 (zh) |
| HR (1) | HRP940287B1 (zh) |
| HU (1) | HU215954B (zh) |
| IL (1) | IL109547A (zh) |
| IS (1) | IS1690B (zh) |
| LT (1) | LT3288B (zh) |
| LV (1) | LV11173B (zh) |
| NO (1) | NO301230B1 (zh) |
| NZ (1) | NZ260475A (zh) |
| RU (1) | RU2141960C1 (zh) |
| SI (1) | SI0624586T1 (zh) |
| SK (1) | SK53594A3 (zh) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5624949A (en) * | 1993-12-07 | 1997-04-29 | Eli Lilly And Company | Protein kinase C inhibitors |
| US5843935A (en) * | 1993-12-07 | 1998-12-01 | Eli Lilly And Company | Protein kinase C inhibitors |
| US5723456A (en) * | 1993-12-07 | 1998-03-03 | Eli Lilly & Company | Therapeutic treatment for cardiovascular diseases |
| PT657458E (pt) * | 1993-12-07 | 2002-02-28 | Lilly Co Eli | Inibidores de proteina cinase c |
| CA2179650C (en) * | 1993-12-23 | 2007-10-30 | William Francis Heath, Jr. | Bisindolemaleimides and their use as protein kinase c inhibitors |
| DE69505470T2 (de) * | 1994-08-04 | 1999-05-12 | F. Hoffmann-La Roche Ag, Basel | Pyrrolocarbazol |
| BR9710648A (pt) * | 1996-03-20 | 1999-08-17 | Lilly Co Eli | S¡ntese de indolilmaleimidas |
| KR100499190B1 (ko) * | 1996-03-29 | 2006-04-17 | 교와 핫꼬 고교 가부시끼가이샤 | 육모제 |
| US5859261A (en) * | 1997-03-20 | 1999-01-12 | Eli Lilly And Company | Synthesis of indolylmaleimides |
| CZ2003555A3 (en) | 2000-07-27 | 2004-03-17 | F. Hoffmann-La Roche Ag | 3-indolyl-4-phenyl-1h-pyrrole-2,5-dione derivatives as inhibitors of glycogen synthase kinase-3beta |
| CA2462657C (en) * | 2001-10-30 | 2011-04-26 | Novartis Ag | Staurosporine derivatives as inhibitors of flt3 receptor tyrosine kinase activity |
| CA2393720C (en) | 2002-07-12 | 2010-09-14 | Eli Lilly And Company | Crystalline 2,5-dione-3-(1-methyl-1h-indol-3-yl)-4-[1-(pyridin-2-ylmethyl)piperidin-4-yl]-1h-indol-3-yl]-1h-pyrrole mono-hydrochloride |
| WO2005014004A1 (en) | 2003-08-08 | 2005-02-17 | Novartis Ag | Combinations comprising staurosporines |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3803620A1 (de) * | 1988-02-06 | 1989-08-17 | Goedecke Ag | Indolocarbazol-derivate, verfahren zu deren herstellung und diese enthaltende arzneimittel |
| SK278989B6 (sk) * | 1988-02-10 | 1998-05-06 | F. Hoffmann-La Roche Ag | Substituované pyroly, ich použitie na výrobu lieči |
| DE3833008A1 (de) * | 1988-09-29 | 1990-04-05 | Goedecke Ag | Pyrrolocarbozol-derivate, verfahren zu deren herstellung und deren verwendung als arzneimittel |
| MC2096A1 (fr) * | 1989-02-23 | 1991-02-15 | Hoffmann La Roche | Pyrroles substitues |
| CA2046801C (en) * | 1990-08-07 | 2002-02-26 | Peter D. Davis | Substituted pyrroles |
-
1994
- 1994-04-15 AU AU60555/94A patent/AU678435B2/en not_active Ceased
- 1994-04-26 CA CA002121567A patent/CA2121567A1/en not_active Abandoned
- 1994-05-02 AT AT94106812T patent/ATE184007T1/de not_active IP Right Cessation
- 1994-05-02 DK DK94106812T patent/DK0624586T3/da active
- 1994-05-02 DE DE69420310T patent/DE69420310T2/de not_active Expired - Fee Related
- 1994-05-02 SI SI9430273T patent/SI0624586T1/xx unknown
- 1994-05-02 ES ES94106812T patent/ES2137281T3/es not_active Expired - Lifetime
- 1994-05-02 EP EP94106812A patent/EP0624586B1/en not_active Expired - Lifetime
- 1994-05-04 HU HU9401298A patent/HU215954B/hu not_active IP Right Cessation
- 1994-05-04 IL IL10954794A patent/IL109547A/en not_active IP Right Cessation
- 1994-05-05 HR HR9309602.2A patent/HRP940287B1/xx not_active IP Right Cessation
- 1994-05-05 RU RU94015830A patent/RU2141960C1/ru active
- 1994-05-06 NZ NZ260475A patent/NZ260475A/en unknown
- 1994-05-06 NO NO941682A patent/NO301230B1/no not_active IP Right Cessation
- 1994-05-06 CZ CZ941128A patent/CZ112894A3/cs unknown
- 1994-05-09 JP JP6119542A patent/JP2668818B2/ja not_active Expired - Fee Related
- 1994-05-09 CN CN94105846A patent/CN1048014C/zh not_active Expired - Fee Related
- 1994-05-09 IS IS4162A patent/IS1690B/is unknown
- 1994-05-09 LV LVP-94-100A patent/LV11173B/en unknown
- 1994-05-09 LT LTIP1927A patent/LT3288B/lt not_active IP Right Cessation
- 1994-05-09 SK SK535-94A patent/SK53594A3/sk unknown
- 1994-05-09 BR BR9401935A patent/BR9401935A/pt not_active Application Discontinuation
- 1994-05-10 BG BG98761A patent/BG98761A/xx unknown
- 1994-05-10 FI FI942158A patent/FI106961B/fi not_active IP Right Cessation
-
1999
- 1999-11-18 GR GR990402995T patent/GR3031901T3/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1041827C (zh) | 被取代的3-氨基奎宁环化合物的制备方法 | |
| CN1036652C (zh) | 碱式季酰胺,其制备方法及其药物组合物 | |
| CN1028992C (zh) | 1,4,-苯并硫杂吖庚因衍生物的制备方法 | |
| CN1037439C (zh) | 制备具有神经保护作用的吲哚酮及有关衍生物的方法 | |
| CN1065536C (zh) | 2-甲基-噻吩并苯并二氮杂䓬的结晶形式及制备方法 | |
| CN1231478C (zh) | 光学活性的哌啶中间体的制备方法和该中间体 | |
| CN1259951A (zh) | 新化合物 | |
| CN1034176C (zh) | 用于预防和治疗糖尿病并发症的药物组合物的制备方法 | |
| CN1048014C (zh) | 取代的吡咯类化合物及其制法、药物组合物和用途 | |
| CN1040985C (zh) | 苯并氮杂䓬类化合物的制备方法 | |
| CN1093363A (zh) | 取代的吡唑 | |
| CN1187188A (zh) | 旋转异构酶活性的小分子抑制剂 | |
| CN1592623A (zh) | 具有mGluR5拮抗活性的乙炔衍生物 | |
| CN1111245A (zh) | 雪花胺衍生物、其制备方法及其作为药物的用途 | |
| CN1108658A (zh) | 雪花胺衍生物及其制备方法和其作为药剂的用途 | |
| CN1032440A (zh) | 4-氨基吡啶衍生物类 | |
| CN1039416C (zh) | 新的吡啶并[3,2-b][1,5]苯并硫代吖庚因的制备方法 | |
| CN1756757A (zh) | 吡咯并咪唑衍生物、其制备、包含它们的药物组合物及其作为向精神剂的应用 | |
| CN1035663A (zh) | 取代的喹啉衍生物的制备方法 | |
| CN1161120C (zh) | 多环2-氨基噻唑体系在预防或治疗肥胖的药物制备中的用途 | |
| CN1056844C (zh) | 二氧代吡咯并吡咯衍生物 | |
| CN1069969A (zh) | 作为兴奋性氨基酸神经递质拮抗剂的合成芳基多胺 | |
| CN1030753A (zh) | 制备新的取代n-(3-羟基-4-哌啶基)苯甲酰胺的方法 | |
| CN1976694A (zh) | 用于治疗变应性疾病的局部用制剂 | |
| CN1100098A (zh) | 2-氨基-4-喹啉基-二氢吡啶类化合物及其制备和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |