CN1037857C - 利用紫黑链霉菌制备肽衍生物的方法 - Google Patents
利用紫黑链霉菌制备肽衍生物的方法 Download PDFInfo
- Publication number
- CN1037857C CN1037857C CN89102100A CN89102100A CN1037857C CN 1037857 C CN1037857 C CN 1037857C CN 89102100 A CN89102100 A CN 89102100A CN 89102100 A CN89102100 A CN 89102100A CN 1037857 C CN1037857 C CN 1037857C
- Authority
- CN
- China
- Prior art keywords
- acid
- methyl
- ester
- mixture
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images








Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N1/00—Microorganisms, e.g. protozoa; Compositions thereof; Processes of propagating, maintaining or preserving microorganisms or compositions thereof; Processes of preparing or isolating a composition containing a microorganism; Culture media therefor
- C12N1/20—Bacteria; Culture media therefor
- C12N1/205—Bacterial isolates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12R—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES C12C - C12Q, RELATING TO MICROORGANISMS
- C12R2001/00—Microorganisms ; Processes using microorganisms
- C12R2001/01—Bacteria or Actinomycetales ; using bacteria or Actinomycetales
- C12R2001/465—Streptomyces
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Wood Science & Technology (AREA)
- Biophysics (AREA)
- Biotechnology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Zoology (AREA)
- Molecular Biology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Biomedical Technology (AREA)
- Virology (AREA)
- General Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Tropical Medicine & Parasitology (AREA)
- Animal Behavior & Ethology (AREA)
- Microbiology (AREA)
- Veterinary Medicine (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Investigating Or Analyzing Materials By The Use Of Ultrasonic Waves (AREA)
- Ultra Sonic Daignosis Equipment (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
Abstract
公开了制备下式所示的化合物的方法,式中的R是(E)-3-[2-((Z)-1-戊烯基)苯基]丙烯酰,而Me是甲基,该方法包括在营养培养基中培养紫黑链霉菌No.9326CCTCCNo.M89201,并从产生的培养液中回收化合物所说之式为:
Description
本发明涉及利用紫黑链霉菌制备具有药理活性的新的肽衍生物。
更具体地,本发明涉及具有如物质P拮抗作用、神经激肽A(物质K)拮抗作用或类似作用的药理活性的新的肽衍生物及其药物上可接受的盐,涉及它们的制备方法以及含有它们的药物组合物。
本发明的一个目的是提供一种利用紫黑链霉菌制备肽衍生物的方法。
因此,本发明的另一个目的是提供用于治疗和预防气喘等疾病的肽衍生物及其药物上可接受的盐。
本发明进一步的目的是提供含有作为活性成分的肽衍生物或其物上可接受的盐的药物组合物。
本发明更进一步的目的是提供肽衍生物及其药物上可接受的盐用于治疗和预防气喘等疾病的用途。
本发明的目的物肽衍生物能通过下列式(I)来表示。 其中R1是氢或酰基;
其中R1是氢或酰基;
R2是羟基和
R3是羧基或被护羧基,或
R2和R3连接在一起表示一个式:
 的基团;
的基团;
R4是羟基或被护羟基;
R5是羟基或被护羟基;
R6是羟基、被护羟基或低级烷氧基;和
是一个单链或双键。
根据本发明,该新的肽衍生物(I)能通过不同的方法制备。
〔通过合成方法制备〕
方法1
方法2
方法3
方法4
方法5
方法6
方法7
方法8
方法9 其中R1、R2、R3、R4、R5、R6和
各如上述定义,
其中R1、R2、R3、R4、R5、R6和
各如上述定义,
R1a是酰基,
R6a是酰氧基,
R3a是酯化羧基,
R1b是被一个低级链烯基取代的芳(低级)链烯酰基,
R1c是被一个低级烷基取代的芳(低级)链烯酰基,
R6b是低级烷氧基。
该起始化合物(II)和(III)是新的,并能通过下列方法制备。方法A



 其中R1、R4、R5、R6和
各如上述定义,
其中R1、R4、R5、R6和
各如上述定义,
R7是被护羧基,
R8是被护氨基,
R9是被护氨基
R10是被护氨基,
R11是被护羧基,
R12是被护氨基,
R13是被护氨基,
R14是被护氨基。
下文说明制备本发明起始物和目的化合物的方法。
方法1
该化合物(Ia)或其盐能通过将化合物(II)或其盐经过环化反应来制备。
该反应可以按合成环状肽的常规方法如混合酸酐法,活性酯方法,碳化二亚胺法等方法来进行。
通常,该反应在常规溶剂如醇,四氢呋喃,乙酸乙酯,N,N-二甲基甲酰胺,二氯甲烷,氯仿中,或在任何其它对反应无不利影响的溶剂中进行。
该反应温度的要求并不严格,通常反应在冷却至温热条件下进行。
方法2
该混合物(Ia)或其盐能通过将化合物(III)或其盐经过环化反应来制备。
该反应按合成环状肽的常规方法如混合酸酐法,活性酯方法,碳化二亚胺法等方法来进行。
通常,该反应在常规溶剂如醇,四氢呋喃,乙酸乙酯,N,N-二甲基甲酰胺,二氯甲烷,氯仿中,或在任何其它对反应无不利影响的溶剂中进行。
该反应温度的要求并不严格,通常反应在冷却至温热的条件下进行。
方法3
该化合物(Ic)或其盐能通过将化合物(Ib)或其盐经过脱酰反应来制备。该反应的适当方法可以包括如水解,还原等常规方法。
(i)水解:
该水解最好在一种碱或一种酸包括路易氏酸的存在下进行。
合适的碱可以包括无机碱和有机碱如碱金属〔如钠、钾等〕、碱土金属〔如镁、钙等〕或它们的氢氧化物或碳酸盐或碳酸氢盐,三烷基胺〔如三甲胺、三乙胺等〕、甲基吡啶、1,5-二氮杂二环〔4.3.0〕-壬-5-烯、1,4-二氮杂二环〔2.2.2〕辛烷、1,8-二氮杂二环〔5.4.0〕十一-7-烯或类似的碱等。
合适的酸可以包括有机酸〔如甲酸、乙酸、丙酸、三氯乙酸、三氟乙酸等〕和无机酸〔如盐酸、氢溴酸、硫酸、氯化氢、溴化氢等〕。使用路易氏酸如用三卤代乙酸(如三氯乙酸,三氟乙酸等)进行的消除反应最好在阳离子捕集剂(如茴香醚、苯酚等〕存在下进行。
通常该反应在一种溶剂如水、醇〔如甲醇、乙醇等〕二氯甲烷、四氢呋喃或它们的混合物中,或在任何其它对反应无不利影响的溶剂中进行。液态的碱或酸也能用作溶剂。该反应温度的要求并不严格,通常该反应在冷却至温热的条件下进行。
(ii)还原:
还原是按常规方式进行的,包括化学还原和催化还原。
用于化学还原的合适的还原剂是一种金属(如锡、锌、铁等)或金属化合物(如氯化铬、乙酸铬等)与有机酸或无机酸(如甲酸、乙酸、丙酸、三氟乙酸、对甲苯磺酸、盐酸、氢溴酸等)的组合物。
用于催化还原的合适催化剂是一种常规的催化剂如铂催化剂(如铂片、海绵铂、铂黑、胶态铂、氧化铂、铂丝等),钯催化剂(如海绵钯、钯黑、氧化钯、钯-炭、胶态钯、钯-硫酸钡、钯-碳酸钡等),镍催化剂(如还原镍、氧化镍、阮内镍等),钴催化剂(如还原钴、阮内钴等),铁催化剂(如还原铁、阮内铁等),铜催化剂(如还原酮、阮内铜、Ullman铜等)等。通常该还原在一个对反应无不利影响的常规溶剂中进行,这些溶剂包括水、甲醇、乙醇、丙醇、N,N-二甲基甲酰胺、四氢呋喃或它们的混合物。另外,在上述酸是以液态形式用于化学还原的情况下,它们还能用作溶剂。
该还原反应的反应温度要求并不严格,通常该反应在冷却的至温热的条件下进行。
方法4
化合物(Ib)或其盐能通过将化合物(Ic)或其在氨基上的反应衍生物或其盐经过酰化反应来制备。
在化合物(Ic)氨基上的合适反应衍生物可以包括通过化合物(Ic)与羰基化合物如醛、酮或类似物等反应所形成的席夫碱型亚氨基或其互变异构的烯胺型异构体;还包括通过式(Ic)化合物与甲硅烷基化合物如双(三甲基甲硅烷基)乙酰胺、单(三甲基甲硅烷基)乙酰胺、双(三甲基甲硅烷基)脲或类似物反应所形成的甲硅烷基衍生物;还包括化合物(Ic)与三氯化磷或光气等反应所形成的衍生物。
用于该酰化反应的合适酰化剂可以包括常规酰化剂并能用式Ra-OH(XIV)其中R1a如上述定义)来表示,或其反应性衍生物或其盐。
化合物(XIV)的合适的反应衍生物可以包括酰氯、酸酐、活性酰胺和活性酯等。合适的实例可以是酰氯;酰基叠氮;与一个酸混合的酸酐,这个酸如取代的磷酸(如二烷基磷酸、苯基磷酸、二苯基磷酸、二苄基磷酸、卤代磷酸等),二烷基磷酸,亚硫酸,硫代硫酸,硫酸,磺酸(如甲磺酸等),烷基碳酸、脂族羧酸(如新戊酸、戊酸、异戊酸、2-乙基丁酸或三氯乙酸等)或芳族羧酸(如苯甲酸等);对称的酸酐;带有咪唑,4-取代的咪唑,二甲基吡唑,三唑或四唑的活性酰胺;或活性酯(如氰基甲酯、甲氧基甲酯、二甲基亚氨基甲酯
 乙烯酯、炔丙酯、对硝基苯酯、2,4-二硝基苯酯、三氯苯酯、五氯苯酯、甲磺酰基苯酯、苯基偶氮苯基酯、苯基硫酯、对硝基苯基硫酯、对羟甲苯基硫酯、羧甲基硫酯、吡喃酯、吡啶酯、哌啶酯、8-喹啉基硫酯等)、或带一个N-羟基化合物(如N,N-二甲基羟胺、1-羟基-2-(1H)-吡啶酮、N-羟基琥珀酰亚胺、N-羟基苯邻二甲酰亚胺、1-羟基-6-氯-1H-苯并三唑等)的酯。按照所使用的化合物(XIV)的种类,这些反应衍生物能任意从中选择。
乙烯酯、炔丙酯、对硝基苯酯、2,4-二硝基苯酯、三氯苯酯、五氯苯酯、甲磺酰基苯酯、苯基偶氮苯基酯、苯基硫酯、对硝基苯基硫酯、对羟甲苯基硫酯、羧甲基硫酯、吡喃酯、吡啶酯、哌啶酯、8-喹啉基硫酯等)、或带一个N-羟基化合物(如N,N-二甲基羟胺、1-羟基-2-(1H)-吡啶酮、N-羟基琥珀酰亚胺、N-羟基苯邻二甲酰亚胺、1-羟基-6-氯-1H-苯并三唑等)的酯。按照所使用的化合物(XIV)的种类,这些反应衍生物能任意从中选择。
通常该反应在常规的溶剂如醇(如甲醇、乙醇等)、丙酮、二噁烷、乙腈、二氯甲烷、二氯乙烷、四氢呋喃、N,N-二甲基甲酰胺、吡啶中,或在任何其它对反应无不利影响的溶剂中进行。这些常规溶剂也可以以与水的混合物的形式使用。
当在反应中,化合物(XIV)是以游离酸或其盐的形式使用时,该反应最好在常规的缩合剂存在下进行,该缩合剂如:N,N’-二环己基碳二亚胺;N-环己基-N’-吗啉代乙基碳二亚胺;N-环己基-N’(4-二乙氨基环己基)碳二亚胺;N,N-二乙基碳二亚胺;N,N’-二异丙基碳二亚胺;N-乙基-N’-(3-二甲氨基丙基)碳二亚胺;N,N-羰基双-(2-甲基咪唑);1,5-亚戊基乙烯酮-N-环己基亚胺;二苯基乙烯酮-N-环己基亚胺;乙氧基乙炔;1-烷氧基-1-氯乙烯,亚磷酸三烷基酯;多磷酸乙酯;多磷酸异丙酯;磷酰氯;三氯化磷;亚硫酰氯;草酰氯;三苯膦;2-乙基-7-羟苄基异噁唑鎓盐;2-乙基-5-(间磺苯基)异噁唑鎓氢氧化物分子内盐;1-(对氯苯磺酰氧基)-6-氯-1H-苯并三唑;通过N,N-二甲基甲酰胺与亚硫酰氯、光气、氯甲酸三氯甲酯、磷酰氯等反应制备的称为Vilsmeier试剂;或类似物。
该反应还可以在无机碱或有机碱存在下进行,这些碱如碱金属碳酸氢盐、三(低级)烷基胺、吡啶、N-(低级)烷基吗啉、N,N-二(低级)烷基苄胺,或类似物。该反应的温度要求并不严格,通常反应在冷却或室温条件下进行。
方法5
化合物(Ie)或其盐能通过将化合物(Id)或其盐经过酰化反应来制备。
该反应可以参照后面那些实施例2、4、5、7、8、17和18的叙述进行。
方法6
化合物(If)或其盐能通过将化合物(Ia)或其盐经过水解反应来制备。
该水解反应可参照上述方法3进行。
方法7
化合物(Ig)或其盐能通过将化合物(If)或其盐经过酯化反应来制备。用于该反应的酯化试剂可以包括常规酯化剂如醇或其反应等同物(如卤化物、磺酸盐、硫酸盐、重氮化合物等)或类似物。
通常该反应在常规的溶剂如丙酮、二噁烷、醇、二氯甲烷、二氯乙烷、正己烷、四氢呋喃、乙酸乙酯、N,N-二甲基甲酰胺中,或在任何其它对反应无不利影响的溶剂中进行。
方法8
化合物(Ii)或其盐能通过将化合物(Ih)或其盐经过还原来制备。
用于此反应的还原方法可以包括催化还原。
用于催化还原的合适催化剂是常规的一种如铂催化剂〔如铂片、海绵铂、铂黑、胶态铂、氧化铂、铂丝等〕,把催化剂〔如海绵钯、钯黑、氧化铂、钯-炭、钯胶、钯-硫酸钡、钯-碳酸钡等〕,镍催化剂〔如还原镍、氧化镍、阮内镍等〕,钴催化剂〔如还原钴、阮内钴等〕,铁催化剂〔如还原铁、阮内铁等〕,铜催化剂(如还原铜、阮内铜、Ullman铜等〕等。
通常该反应在常规的溶剂如丙酮、二噁烷、醇、四氢呋喃、乙酸乙酯、N,N-二甲基甲酰胺、二甲亚砜中,或在任何其它对反应无不利影响的溶剂中进行。
该反应温度的要求并不严格,通常该反应在冷却的至温热的条件下进行。
方法9
化合物(Ij)或其盐能通过将化合物(Id)或其盐经过烷基化反应来制备。该反应可参照后面实施例19的叙述进行。
方法A
化合物(II)或其盐能通过将化合物(IV)或其盐接方法A所示的合成路线来制备。在所述合成路线中的每步反应能接用于合成肽的常规方法进行。该起始化合物(IV)或其盐能通过在后面叙述的制备中揭示的方法或本文中的类似方法来制备。
方法B
化合物(III)或其盐能通过将化合物(IX)或其盐按方法B所示的合成路线来制备。在所述合成路线中的每步反应能按用于合成肽的常规方法进行。该起始化合物或其盐能通过在后面叙述的制备中揭示的方法或本文中的类似方法来制备。
〔通过发酵来制备〕
本发明的WS-9326A和WS-9326B能通过将属于链霉菌属如紫黑链霉菌No.9326的WS-9326A和/或WS-9326B产生菌株在营养培养基中发酵来制备。
用于制备WS-9326A和WS-9326B的微生物的详细资料将在下面说明。
能用于制备WS-9326A和WS-9326B的微生物是一种属于链霉菌属的WS-9326A和/或WS-9326B产生菌株,其中紫黑链霉菌No.9326是新近从在Suwa City,Nagano Prefecture,Japan收集的土壤样品中分离得到的。
该新近分离的紫黑链霉菌No.9326已保藏于中国典型培养物保藏中心,保藏号为CCTCCNo.M89201,保藏日期为1989年3月28日。
不用说,该新的WS-9326A和WS-9326B的制备并不限于使用本文叙述的特定有机菌,其仅仅是用于说明的目的。本发明还包括使用任何能够产生WS-9326A和WS-9326B的突变体,包括天然突变体以及能从所述有机菌通过如X-射线、紫外线照射的辐射,用N-甲基-N′-硝基-N-亚硝基喹尼啶、2-氨基嘌呤处理等常规方法产生的人工合成突变体。
紫黑链霉菌No.9326具有下列形态、培养、生物和生理特征。
(1)形态特征:
对于这个分类学研究是采用由Shirling和Gottlieb叙述的方法(Shirling,E.B.and D.Gottlieb:Methods for characterization ofStreptomyces species,International Journal of Systematic Bacteriol-ogy,16,313-340,1966)。
用光和电子显微镜对在燕麦粉琼脂、酵母-麦芽萃取物琼脂和无机盐-淀粉琼脂上,于30℃培养14天的培养物进行形态观察。
该无性繁殖的菌丝体发育很好,并且没有断裂菌丝。该气生的菌丝体单轴分支并形成每条链上带有10至30个孢子的孢子螺旋链。该孢子具有光滑的表面并呈0.6-0.8×0.8-1.3μm大小的卵形。没有观察到硬化颗粒、孢子囊和游动孢子。
(2)培养特征:
在由上述的Shirling和Gottlieb以及由Waksman(Waksman,S.A.“The actinomycetes,Vol.2:classification,identification anddescription of genera and species.The Williams and Wilkinsco.,Baltimore,1961)。叙述的十种培养基上观察培养特征。
该培养是在30℃进行21天。用于本研究的色泽名称取自Mehtuen的颜色手册(Kornerup,A.and J.H.Wanscher:Methuen Handbook ofColour,Methuen,London,1978)。结果列于表1。
表1 菌株No.9326的培养特征
培养基 培养特征
酵母-麦芽萃取物 G:好
琼脂 A:多度,带褐色的灰色(6E2)
R:深褐色(7F6)
S:无
燕麦粉琼脂 G:好
A:适度,深褐色(7E3)
R:带褐色的灰色(7F2)
S:无
无机盐-淀粉 G:好
琼脂 A:多度,带褐色的灰色(7E2)
R:浅黄褐色(5E6)
S:无
甘油-天冬酰胺 G:好
琼脂 A:多度,浅灰紫色(19E3)
R:褐色(6E4)
S:无胨-酵母萃取物- G:好铁离子琼脂 A:稀,浅灰白色(1B1)
R:浅黄褐色(5D6)
S:无酪氨酸琼脂 G:好
A:多度,带褐色的灰色(9E2)
R:褐色(6E5)至黑色
S:无葡萄糖-天冬酰胺 G:好琼脂 A:适度,浅兰灰色(19E2)
R:褐色(6F4)
S:天营养琼脂 G:适度
A:适度,带褐色的灰色(9E2)
R:浅灰褐色(5B)至浅黄褐色(5F4)
S:无Bennet琼脂 G:差
A:差,深褐色(6F4)
R:深褐色(6F4)
S:无蔗糖-硝酸盐 G:差琼脂 A:无
R:浅灰褐色(6E3)
S:无注:F=培养,A=气生菌丝体,R=反面颜色,S=可溶色气生的菌丝体由灰色至带褐色的灰色。部分菌落变黑并湿润,并且在大部分琼脂培养基中显示有吸湿的征状。培养的反面呈浅黄褐色、褐色和深褐色。反面菌丝体颜色没有pH敏感性。没有产生类黑素色和其它可溶色。
通过Becker等(Becker,B.,M.P.Lechevalier,R.E.Gordon andH.A.Lechevalier:Rapid Differentiation between Nocardia andStreptomyces by paper chromatography of whole cell hydrolysa-tes:Appl.Microbiol.,12,421-423,1964)和Yamaguchi(Yamag-uchi,T.:Comparison of the cell wall composition of morphol-ogically distinct actinomycetes:J.Bacteriol.,89,444-453,1965)的方法进行细胞壁分析。分析菌株No.9326的全部细胞水解物表明存在二氨基庚二酸,因此,可以认为该菌株的细胞壁是I型。
〔3〕生物和生理特性:
表2和表3分别表示生理特性和碳源的利用情况。
按照Pridham和Gottlieb(Pridham,T.G.and D.Gottlieb:Theutilization of carbon compounds by some Actinomycetales as anaid for species determination:J.Bacteriol.,56,107-114,1948)方法分析碳源的利用情况。
表2 菌株No.9326的生理特性
条件 特性
培养的温度范围 11℃-47℃
培养的最佳温度范围 29℃-31℃
明胶液化试验 阳性
牛奶凝固试验 阴性
牛奶胨化试验 阳性
淀粉水解 阳性
产生类黑素色 阴性
纤维素分解 阴性
表3 菌株No.9326的碳源利用情况
化合物 培育
D-葡萄糖 +
蔗糖 +
D-木糖 +
D-果糖 +
L-鼠李糖 +
棉子糖 +
L-阿拉伯糖 +
肌醇 +
甘露醇 +
注:+表示利用
菌株No.9326的形态和化学特征可以明显地归入链霉菌属。菌株No9326可以同由Bergey′s手册第8版中叙述的链霉菌种相比较(Buchanan,R.E.and N.E.Gibbons:Bergey′s manual of determinative bacte-riology,eight edition.The Williams and Wilkins Co.,Baltimore,1974),也可以同在Shirling′s ISP报告中叙述的链霉菌种相比较〔(Shirling,E.B.and D.Gottlieb:Cooperative deseription oftype culture of Streptomyces.2.由第一次研究描述的菌种Intern.J.Syst.Bacteriol.18:69-189,1968),(Sbirling,E.B.and D.Gottlieb:Cooperative description of type culture of Streptomyces.3.由第二次研究描述的另一菌种。Intern.J.Syst.Bcteriol.18:279-392,1968)和(Shirling,E.B.and Gottlieb:Cooperative descriptionof type culture of Streptomyces.4.由第二次、第三次和第四次研究描述的菌种。Intern.J.Syst.Bacteriol.19:392-512,1969)〕,
并可以同列于“细菌名称审定表”中的菌种比较,(Skerman,V.B.D.V.McGowan&P.H.A.Sneath:Approved list of bacterial namesIntern.J.Syst.Bacteriol.30:225-420,1980),还可以同在其它参考文献中叙述的菌种相比较(Williams,S.T.:M.Goodfellow,G.Alderson,E.M.H.Wellington,P.H.A.Sneathand M.J.Sackin:Numerical classifica-tion of Streptomyces andrelated genera.J.Gen.Microbiol.1291743-1813,1983)和(Dietz,A.:Criteriaforcharacterization of Hygroseopicusstrains In“Actinomycetes;The Boundary Microorganisms”PP183-191 Edited by T.Arai,1976)]。
结果证明菌株No.9326十分类似于紫黑链霉菌。因此菌株No.9326鉴别为紫黑链霉菌并被命名为紫黑链霉菌菌株No.9326。
制备WS-9326A和WS-9326B
本发明的新的WS-9326A和WS-9326B能通过在营养培养基中培养属于链霉菌属(如紫黑链霉菌No.9326,CCTCCNo.89201)的WS-9326A和/或WS-9326B产生菌株来制备。
通常,WS-9326A和WS-9326B能通过在一种含有可吸收的碳源和氮源的含水营养培养基中,并最好在需氧条件下(如摇动培养、浸没培养等),培养WS-9326A和/或WS-9326B产生菌株来制备。
在该营养培养基中优选碳源是烃类如葡萄糖、木糖、半乳糖、甘油、淀粉、糊精等。
可以包括的其它碳源是麦芽糖、鼠李糖、棉子糖、阿拉伯糖、水杨苷、琥珀酸钠等。
优选的氮源是酵母萃取物、胨、谷蛋白粉、棉籽粉、大豆粉、玉米浸液、干酵母、小麦芽、羽毛粉、花生粉等,以及无机的和有机的氮化合物如铵盐(如硝酸铵、硫酸铵、磷酸铵等),尿素、氨基酸等。
该碳源和氮源可以方便地混合使用,并不需要以其纯化的形式使用,因为不纯的物料含有微量生长因子和相当量的矿质营养素,所以也适合于应用。当需要时,在培养基中可以加入矿物盐如碳酸钠或碳酸钙、磷酸钠或磷酸钾、氯化钠或氯化钾、碘化钠或碘化钾、镁盐、铜盐、钴盐等。如有必要,尤其当培养基严重发泡时,可以加入消泡剂如液体石蜡、脂肪油、植物油、矿物油或硅。
就大量制备WS-9326A和WS-9326B的条件而言,优选的是浸没需氧培养条件。小量制备时可以使用在烧瓶或瓶子里进行摇动或表面培养。此外,当在大罐中进行培养时,为避免在制备WS-9326A和WS-9326B的过程中发生滞后培育,最好使用生长形式的微生物用于制备罐接种。因此,首先需要通过接种相应的小量的带有该微生物的孢子或菌丝体的培养基,并培养所述的接种培养基来产生该微生物的生长接种物,然后将该培养的生长接种物无菌地转移至大罐中。其中产生生长接种物的培养基基本上与用于制备WS-9326A和WS-9326B的培养基同,也可不相同。
该培养混合物的搅拌和通气可以用各种方法达到。搅拌可以通过下列方法进行,即使用螺旋桨或类似的机械搅拌设备,旋转或摇动发酵罐,使用各种泵设备或使无菌空气通过培养基。通气可以采用将无菌空气通过该发酵混合物的方法来实现。
通常发酵在约20℃和40℃之间的温度并最好在25-35℃之间进行,发酵时间约50至150小时,这个时间可根据发酵的条件和规模进行变化。
因而能通过一般用于回收其它已知的生物活性物质的常规方法,从培养基中回收产生的WS-9326A和WS-9326B。由于产生的WS-9326A和WS-9326B存在于培养的滤液和菌丝体中,因此,该WS-9326A和WS-9326B能从滤液和菌丝体中分离并纯化,它们是通过下列方法得到的,即将营养液进行过滤或离心,用常规方法如减压浓缩、冻干,用常规溶剂萃取,调节pH,用常规离子处理(如阴离子或阳离子交换树脂,非离子型吸收树脂等),用常规吸收剂(如活性炭、硅酸、硅胶纤维素、氧化铝等),结晶,重结晶等。
按上述方法产生的WS-9326A具有下列物理和化学特性。
(1)形状和色泽:无色粉末
(2)颜色反应:
阳性:硫酸铈反应,
碘蒸气反应,
氯化铁-铁氰化钾反应,
阴性:茚三酮反应,
莫利施氏(Molisch)反应,
氯化铁反应,
埃尔利希反应,
泡利反应
(3)溶解度:
可溶:甲醇、乙醇,
微溶:丙酮、乙酸乙酯,
不溶:水、氯仿。
(4)熔点:187-190℃
(5)旋光率:〔α〕D 23:-84℃(C=1.0,甲醇)
(6)紫外吸收光谱:
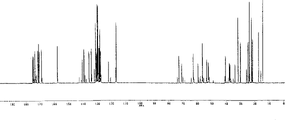
(7)红外吸收光谱:
1610,1560,154O,1530,1510,1440,1380,
1340,1280,124O,1170,1110,1080,1060,
1040,970,920,880,860,830cm-1,其谱图见附图1(8)元素分析:C54H68N8O13·2H2O
实测值:C 60.18,H 6.61,N 10.32
计算值:C 60.43,H 6.76,N 10.44(9)薄层色谱:固定相 展开剂 Rf值硅胶板 氯仿-甲醇 0.38(Merck Art 5715) (5∶1 V/V)RP-18板(Merck) 甲醇-水 0.46
(8∶2,V/V)(10)分子式:C54H68N8O13(11)分子量:FAB-MS:m/z 1037(M+H)+(12)该物质性质:酸性(13)13C核磁共振谱:(100MHz,CD3OD)δ:175.69(s), 174.70(s),173.73(s), 173.38(s),172.89(s), 171.04(s),170.45(s), 167.79(s),167.15(s), 159.20(s),140.05(d), 139.12(s),138.71(s), 135.27(d),134.85(s), 132.11(d),132.03(s), 131.69(d)x2,130.70(d), 129.90(d),129.61(d)x2, 129.22(d)x2,128.55(d), 128.04(d),127.99(d), 127.38(d),126.09(s), 123.70(d),115.63(d)x2, 73.46(d),71.34(d), 62.80(t),59.53(d), 56.91(d),56.76(d), 55.55(d),53.64(d), 52.10(d),39.85(t), 37.18(t),37.09(t), 34.58(q),31.37(t), 24.56(d),23.63(t), 22.71(q),22.52(q), 21.17(q),其谱图见附图2(14)1H核磁共振(400MHz,CD3OD)δ:
7.80 (1H,d,J=8Hz),
7.67 (1H,d,J=16Hz),
7.45-7.14 (9H,m),
7.06 (2H,d,J=8Hz),
6.83 (1H,s),
6.65 (2H,d,J=8Hz),
6.59 (1H,d,J=12Hz),
5.88 (1H,dt,J=12 and 7Hz),
5.55 (1H,m),
5.35 (1H,broad signal),
5.10 (1H,dd,J=3 and 9.5Hz),
4.68 (1H,d,J=10Hz),
4.55 (1H,t,J=6Hz),
4.48 (1H,dd,J=3 and 12Hz),
3.92 (2H,d,J=6Hz),
3.70 (1H,t,J=7.5Hz),
3.62 (1H,m),
3.46 (1H,dd,J=3 and 14Hz),
2.94 (1H,dd,J=3 and 16Hz),
2.89 (3H,s),
2.74 (1H,dd,J=9.5 and 16Hz),
2.69 (1H,dd,J=12 and 14Hz),
2.14 (2H,m),
1.5-1.4 (2H,m),
1.20 (3H,d,J=6Hz),
1.08 (3H,d,J=6Hz),
1.0-0.8 (2H,m)
0.91 (3H,t,J=7Hz),
0.6 (1H,m),
0.53 (3H,d,J=6Hz),
0.51 (3H,d,J=6Hz),
其谱图见附图3
(15)氨基酸分析:
将MS-9326A(5mg)置于封管中,用盐酸(2ml)在110℃水解20小时。混合物蒸发至于,得到水解产物,将其放置在Hitachi 835氨基酸自动分析仪上进行分析。
氨基酸分析结果如下:
苏氨酸(2),亮氨酸(1),苯丙氨酸(1),天冬氨酸(1),丝氨酸(1),甲胺(1)和氨(1)。
关于WS-9326A,应注意到在附图2和3中的13C和1H核磁共振谱表明WS-9326A在CD3OD溶液中至少存在两个稳定的构象,在上述(13)和(14)中叙述的是那些占优势构象的WS-9326A的化学位移。
按上述方法产生的WS-9326B具有下列物理和化学特性:
(1)形状和色泽:无色无定形粉末,
(2)颜色反应:
阳性:硫酸铈反应,
碘蒸气反应,
阴性:茚三酮反应
(3)溶解度:
可溶:甲醇,
微溶:乙醇,
不溶:水、丙酮、乙酸乙酯、氯仿
(4)熔点:165-170℃(分解)
(5)旋光率:〔α〕D 23:-64°(C=1.0,甲醇)
(6)紫外吸收光谱:
(7)分子式:C54H70N8O13
(8)元素分析:C54H70N8O13·2H2O
实测值:C 59.97,H 6.87,N 10.29
计算值:C 60.32,H 6.94,N 10.42
(9)分子量:
FAB-MS:m/z 1061.6(M+Na)+
(10)薄层色谱:
固定相 展开剂 Rf值
硅胶板 氯仿-甲醇 0.38
(Merck Art 5715) (5∶1 V/V)
RP-18 醇-水 0.25
(8∶2,V/V)(11)红外吸收光谱:
1510,1450,1400,1380,1340,1260,
1220,1080,980,920cm-1(12)13C核磁共振谱:
(100MHz,CD3OD)δ174.99(s), 174.54(s),173.60(s) 173.41(s),173.30(s), 171.27(s),170.74(s), 170.19(s),168.69(s), 157.59(s),140.53(d), 139.35(s),139.18(s), 135.76(d),134.17(s), 131.15(d)x2,130.93(d), 130.35(d),129.88(d)x2, 129.39(d)x2,128.70(d), 128.58(s),128.13(d), 127.64(d),127.53(d), 121.99(d),116.45(d)x2, 72.76(d),70.82(d), 62.73(t),62.67(d), 59.35(d),56.33(d)x2, 56.19(d),53.36(d), 52.24(d),40.24(t), 37.55(t),37.08(t), 33.69(t),31.57(t), 29.93(q),24.61(d), 23.70(q),23.59(t), 22.16(q),21.36(q), 17.12(q),14.23(q),其谱图见附图6,(13)1H核磁共振谱:(400MHz,CD3OD)δ:
7.86 (1H,d,J=16Hz),
7.80 (1H,br d,J=8Hz),
7.12-7.42 (11H,m),
6.77 (2H,d,J=8.5Hz),
6.61 (1H,d,J=11.5Hz),
5.88 (1H,dt,J=7.5 and 11.5Hz),
5.08 (1H,dd,J=3.5 and 10Hz),
5.04 (1H,q,J=6.5Hz),
4.66 (1H,dd,J=3.5 and 13Hz),
4.65 (1H,d,J=11.5Hz),
4.56 (1H,dd,J=2.5 and 7Hz),
4.48 (1H,dd,J=4.5 and 11Hz),
4.46 (1H,s),
3.88 (2H,m),
3.64 (2H,m),
3.51 (1H,dd,J=3.5 and 14Hz),
3.17 (1H,dd,J=4.5 and 14Hz),
3.01 (1H,dd,J=11 and 14Hz),
2.94 (1H,dd,J=3.5 and 16Hz),
2.71 (3H,s),
2.71 (1H,dd,J=10 and 16Hz),
2.64 (1H,dd,J=13 and 14Hz),
2.04 (2H,m),
1.43 (2H,m),
1.28 (2H,m),
1.20 (3H,d,J=6Hz),
0.95 (3H,d,J=6.5Hz),
0.87 (3H,t,J=7.5Hz),
0.53 (1H,m),
0.52 (6H,d,J=10.5Hz),其谱图见附图7
关于WS-9326B,应注意到在图6和7中的13C和1H核磁共振谱表明WS-9326B在CD3OD溶液中至少存在两个稳定的构象,在上述(12)和(13)中叙述的是那些占优势构象的WS-9326B的化学位移。
从上述物理和化学特性的分析以及对化学结构鉴定的进一步研究结果已确定WS-9326A和WS-9326B的化学结构,如下所示:
〔(E)-3-〔(=2-((Z)-1-戊烯基)苯基〕丙烯酰基〕
R-:(E)-3-〔2-((Z)-1-戊烯基)苯基〕丙烯酰基〕
目的化合物(I)的合适的药上可接受的盐是常规的无毒性盐,可以包括与碱所成的盐或酸加成盐,例如与无机碱所成的盐如碱金属盐(如锂盐、钠盐、钾盐等),碱土金属盐(如钙盐、镁盐等),铵盐;与有机碱所成的盐,如有机胺盐(如三乙胺盐、吡啶盐、甲基吡啶盐、乙醇胺盐、三乙醇胺盐、二环己基胺盐、N,N′-二苄基乙二胺盐等;无机酸的加成盐(如盐酸盐、氢溴酸盐、硫酸盐、磷酸盐等);有机羧酸或磺酸的加成盐(如甲酸盐、乙酸盐、三氟乙酸盐、马来酸盐、酒石酸盐、甲磺酸盐、苯磺酸盐、对甲苯磺酸盐等);与碱性或酸性氨基酸所成的盐(如精氨酸、天冬氨酸、谷氨酸等)等。
化合物(Ia)-(Ij)、(II)和(III)的合适盐可参照例举的化合物(I)的盐。
在上述和本说明书后来的叙述中,包括在本发明范围内的本发明的合适实施例和各种定义的解释在下文详述。
除另有说明外,术语“低级”是意指1至6个碳原子。
除另有说明外,术语“高级”意指7至20个碳原子。
合适的“酰基”和在术语“酰氨基”中的“酰基”部分可以包括氨基甲酰基,脂族酰基和称为芳酰基的含有芳环的烷基,或称为杂环酰基的含有杂环的酰基。
所述酰基的合适实例可以列举如下:
脂族酰基如低级或高级的烷酰基(如甲酰基、乙酰基、丙酰基、丁酰基、2-甲基苯丙酰基、戊酰基、2,2-二甲基丙酰基、己酰基、庚酰基、辛酰基、壬酰基、癸酰基、十一烷酰基、十二烷酰基、十三烷酰基、十四烷酰基、十五烷酰基、十六烷酰基、十七烷酰基、十八烷酰基、十九烷酰基、二十烷酰基等);
低级或高级烷氧羰基(如甲氧羰基、乙氧羰基、叔丁氧羰基、叔戊氧羰基、庚氧羰基等);
低级或高级链烷磺酰基(如甲磺酰基、乙磺酰基等);
低级或高级烷氧磺酰基(如甲氧磺酰基、乙氧磺酰基等);或类似基团;
芳族酰基如芳酰基(如苯甲酰基,甲苯甲酰基、萘酰基等);
芳(低级)烷酰基〔如苯基(低级)烷酰基(如苯乙酰基、苯丙酰基、苯丁酰基、苯异丁酰基、苯戊酰基、苯己酰基等),萘基(低级)烷酰基(如萘乙酰基、萘丙酰基、萘丁酰基等),等〕;
芳(低级)链烯酰基〔如苯基(低级)链烯酰基(如苯丙烯酰基、苯丁烯酰基、苯基甲基丙烯酰基、苯基戊烯酰基、苯基己烯酰基等),萘基(低级)链烯酰基(如萘基丙烯酰基、萘基了烯酰基、萘基戊烯酰基等),等〕;
芳(低级)烷氧羰基〔如苯基(低级)烷氧羰基(如苄氧羰基等),等〕;
芳氧羰基(如苯氧羰基、萘氧羰基等);
芳氧基(低级)烷酰基(如苯氧乙酰基、苯氧丙酰基等);
芳基乙醛酰基(如苯基乙醛酰基、萘基乙醛酰基、等);
芳基磺酰基(如苯磺酰基、对甲苯磺酰基、等);或类似基团;
杂环酰基如杂环羰基(如噻吩甲酰基、呋喃甲酰基、甲基吡啶甲酰基等,);
杂环(低级)烷酰基(如噻吩基乙酰基、噻吩基丙酰基、噻吩基丁酰基、噻吩基戊酰基、噻吩基己酰基、噻唑基乙酰基、噻二唑基乙酰基、四唑基乙酰基等);
杂环乙醛酰基(如噻唑基乙醛酰基、噻吩基乙醛酰基等,);或类似基团;其中术语“杂环羰基”、“杂环(低级)烷酰基”和“杂环乙醛酰基”中的合适杂环基部分如上述定义,详细讲是指至少含有一个杂原子如氧、硫、氮原子等的饱和或不饱和、单环或多环的杂环基。特别优选的杂环基是下列杂环基:
含有1至4个氮原子的不饱和的3至8元较好的5或6元的单杂环基,如:吡咯基、吡咯啉基、咪唑基、吡唑基、吡啶基及其N-氧化物、二氢吡啶基、嘧啶基、吡嗪基、吡哒嗪基、三唑基(如4H-1,2,4-三唑基、1H-1,2,3-三唑基、2H-1,2,3-三唑基等)、四唑基(如1H-四唑基、2H-四唑基等),等;
含有1至4个氮原子的3至8元(较好为5或6元)的饱和单杂环基如:吡咯烷基、咪唑烷基、哌啶子基、哌嗪基等;
含有1至4个氮原子的未饱和稠合杂环基如吲哚基、异吲哚基、中氮茚基、苄基咪唑基、喹啉基、异喹啉基、吲唑基、苯并三唑基等;
含有1至2个氧原子和1至3个氮原子的未饱和的3至8元(较好为5或6元)单杂环基,如噁唑基、异噁唑基、噁二唑基(如1,2,4-噁二唑基、1,3,4-噁二唑基、1,2,5-噁二唑基等),等;
含有1至2个氧原子和1至3个氧原子的饱和的3至8元,较好为5或6元)单杂环基如吗啉基、斯德酮基等;
含有1至2个氧原子和1至3个氮原子的未饱和的稠合杂环基如:苄基噁唑基、苄基噁二唑基等;
含有1至2个硫原子和1至3个氮原子的未饱和的3至8元(较好为5或6元)单杂环基如:噻唑基、异噻唑基、噻二唑基(如1,2,3-噻二唑基、1,2,4-噻二唑基、1,3,4-噻二唑基、1,2,5-噻二唑基等),二氢噻嗪等;
含有1至2个硫原子和1至3个氮原子的饱和的3至8元(较好为5或6元)单杂环基如噻唑烷基等;
含有1至2个硫原子的未饱和的3至8元(较好为5或6元)单杂环基如噻吩基、二氢-dithiinyl、二氢-dithionyl等);
含有1至2个硫原子和1至3个氮原子的未饱和的稠合杂环基如:苯并噻唑基、苯并噻二唑基等;含有1个氧原子的未饱和的3至8元(较好为5至6元)单杂环基如呋喃基等;
含有1个氧原子和1至2个硫原子的未饱和的3至8元(较好为5或6元)杂单环基如:二氢氧硫杂环己二烯基等;
含有1至2个硫原子的未饱和的稠合杂环基如苯并噻吩基、苯并-dithiinyl等;
含有1个氧原子和1至2个硫原子的未饱和的稠合杂环基如:苄基氧硫杂环己二烯基等。
如上所述的酰基部分可以具有1至10个相同的或不同的合适取代基,如低级烷基(如甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、己基等);低级链烯基(如乙烯基、烯丙基、1-丙烯基、1或2或3-丁烯基、1或2或3或4-戊烯基、1或2或3或4或5-己烯基等);低级烷氧基(如:甲氧基、乙氧基、丙氧基等);低级烷硫基(如:甲硫基、乙硫基等);低级烷氨基(如甲氨基等);环(低级)烷基(如:环戊基、环己基等);环(低级)链烯基(如:环己烯基等);卤原子;氨基;被护氨基;羟基;被护羟基;氰基;硝基;羧基;被护羧基;磺基;氨磺酰基;亚氨基;氧代;氨基(低级)烷基(如氨基甲基、氨基乙基等);氨基甲酰氧基;羟(低级)烷基(如:羟甲基、1或2-羟乙基、1或2或3-羟丙基等);氰基(低级)链烯硫基(如:氰基乙烯硫基等);或类似基团。
在术语“被护羟基”中的羟基保护基可以包括如上述定义的苯基(低级)烷基(如苄基等)、酰基等。
合适的“被护羧基”可以包括酯化的羧基。
酯化羧基的酯部分的合适实例可以是低级烷基酯(如甲酯、乙酯、丙酯、异丙酯、丁酯、异丁酯、叔丁酯、戊酯、己酯、1-环丙基乙基酯等),该酯至少可以具有一个合适的取代基;例如低级烷酰氧基(低级)烷基酯〔如:乙酰氧甲酯、丙酰氧甲酯、丁酰氧甲酯、戊酰氧甲酯、新戊酰氧甲酯、己酰氧甲酯、1-(或2)-乙酰氧乙酯、1(或2或3)-乙酰氧丙酯、1(或2或3或4)-乙酰氧丁酯、1(或2)-丙酰氧乙酯、1(或2或3)-丙酰氧丙酯、1(或2)-丁酰氧乙酯、1(或2)-异丁酰氧乙酯、1(或2)-新戊酰氧乙酯、1(或2)-己酰氧乙酯、异丁酰氧甲酯、2-乙基丁酰氧甲酯、3,3-二甲基丁酰氧甲酯、1(或2)-戊酰氧乙酯等〕,低级烷磺酰基(低级)烷基酯(如2-甲磺酰基乙酯等),单(或2或3)-囟代(低级)烷基酯(如:2-碘代乙酯、2,2,2-三氯乙酯等),低级烷氧羰基氧(低级)烷基酯(如:甲氧羰基氧甲酯、乙氧羰基氧甲酯、2-甲氧羰基氧乙酯、1-乙氧羰基氧乙酯、1-异丙氧羰基氧乙酯等);二苯并呋喃酮亚基(低级)烷基酯,或(5-低级烷基2-氧代-1,3-二噁唑-4-基)(低级)烷基酯〔如:(5-甲基-2-氧代-1,3-二噁唑-4-基)甲酯、(5-乙基-2-氧代-1,3-二噁唑-4-基)甲酯、(5-丙基-2-氧代-1,3-二噁唑-4-基)乙酯等〕;低级链烯基酯(如乙烯酯、烯丙酯等);低级炔基酯(如乙炔酯、丙炔酯等);至少可以具有一个合适取代基的芳(低级)烷基酯如:至少可以具有一个合适取代基的单(或二或三)苯基(低级)烷基酯(如:苄酯、4-甲氧基苄酯、4-硝基苄酯、苯乙酯、三苯甲酯、二苯甲酯、双(甲氧苯基)甲酯、3,4-二甲氧苄酯、4-(羟基-3,5-二-叔丁基苄酯等);至少可以具有一个合适取代基的芳酯(如:苯酯、4-氯苯酯、甲苯酯、叔丁基苯酯、二甲苯酯、2,4,6-三甲苯酯、异丙苯酯等);二苯并呋喃酮酯等。
合适的低级烷氧基可以包括甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、异丁氧基、叔丁氧基、戊氧基、己氧基等。
在术语“被护氨基”中合适的“氨基保护基”可以包括如上所述的酰基等。
在术语“带有低级链烯基取代的芳(低级)链烯酰基”中的合适“芳(低级)链烯酰基”可以包括苯基(低级)链烯酰基(如:苯丙烯酰基、苯丁烯酰基、苯异丁烯酰基、苯戊烯酰基、苯己烯酰基等),萘(低级)链烯酰基(如萘丙烯酰基、萘丁烯酰基、萘戊烯酰基等)等。
在术语“带有低级链烯基取代基的芳(低级)链烯酰基”中的合适“低级链烯基”可以包括乙烯基、烯丙基、1-丙烯基、1或2或3-丁烯基、1或2或3或4-戊烯基、1或2或3或4或5-己烯基等。
在术语“带有低级烷基取代基的芳(低级)烷酰基”中的合适“芳(低级)烷酰基”可以包括苯基(低级)烷酰基(如苯乙酰基、苯丙酰基、苯丁酰基、苯异丁酰基、苯戊酰基、苯己酰基等),萘基(低级)烷酰基(如萘乙酰基、萘丙酰基、萘丁酰基等)等。
在术语“带有低级烷基取代的芳(低级)烷酰基”中的合适“低级烷基”可以包括甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、己基等。
目的化合物(I)的优选实例如下:
R1是氢、芳(低级)烷氧羰基(更优选是苯基(低级)烷氧羰基)、低级烷酰基、高级烷酰基(更优选的是C15-C20烷酰基)、芳酰基(更优选的是苯甲酰基)、杂环(低级)烷酰基(更优选的是噻吩基(低级)烷酰基)、带有低级链烯基取代基的芳(低级)烷酰基(更优选的是带有低级链烯基取代的苯基(低级)链烯基、或带有低级烷基取代的芳(低级)烷酰基(更优选的是带有低级烷基取代的苯基(低级)烷酰基;
R2是羟基和
R3是羧基或酯化羧基(更优选的是低级烷氧基,或R2和R3一起表示一个式
 基团;
基团;
R4是羟基、芳(低级)烷氧基(更优选的是苯基(低级)烷氧基)或酰氧基(更优选的是低级烷酰氧基);
R5是羟基、芳(低级)烷氧基(更优选的是苯基(低级)烷氧基)或酰氧基(更优选的是烷酰氧基);
R6是羟基、低级烷氧基、芳(低级)烷氧基(更优选的是苯基(低级)烷氧基)或酰氧基(更优选的是低级烷酰氧基);和
肽衍生物的生物特性:
肽衍生物(I)及其药物上可接受的盐具有如物质P拮抗作用、神经激肽A(物质K)拮抗作用或类似作用的药理活性,因此能用于治疗和预防气喘等疾病。
作为表明这些药理活性的一个实例,下列说明若干药理试验的资料。
(1)放射配位结合测定
(a)粗膜制备
脑:
使用雌性的Wister鼠(200g),所有试剂均从Sigma化学公司购得。将整个脑子(4g)切成小碎片,用一个超一分散混合器(Yamato MODELKL-21)将该脑碎片置于8倍体积的冰冷却的培养基I(50mM Tris-HCl,pH7.5,5mM MnCl2,0.02%BSA,2μg/ml抑凝乳蛋白酶素,4μg/mlleupeptin和40μg/ml杆菌肽)中均化。该均化物可以在-20℃保存或可以立即用于结合实验。
肺:
将患有白化病的Hartley种雄性豚鼠(600g)用去头法处死。取出气管和肺于-80℃保存直至使用为止。用一个小型混合器(Matsuden MJ-761)将这些组织置于500ml缓冲剂(0.25M蔗糖、50mM Tris-HCl,pH7.5,0.1mM EDTA)中融化并均化。该组织是用一个超-分散混合器(YamatoMODEL LK-21)在固定于10秒钟的最大范围,并在均化中间隔10秒钟冷却一次的情况下进行均化的(总的均化时间是60秒)。该均化物离心除去组织团块(900×g离心10分钟),上清液在14000×g离心20分钟产生称为粗膜部分的片。该片再悬浮干培养基I中,用聚四氟乙烯均化器均化,并在14000×g离心20分钟。该片在-20℃保存。
(b)将3H-物质P结合到制备的膜上
将3H-物质P(1nM,New England Nuclear)与50μl制备膜放于培养基I中,最终体积为250μl,将培养物用细胞采集机(Brandel M-24S)通过一个WhatMann GF/B玻璃纤维滤器(使用前用0.1%聚乙烯亚胺预处理3小时)迅速过滤。然后将滤液在0℃以总量为3ml的洗涤用缓冲剂(50mM Tris-HCl pH7.5)分十次洗涤。以3ml Aquazol-2在Packardsintillation计数器中(Parckard TRI-CARB 4530)计算放射活性。
表4 结合于鼠脑和豚鼠肺膜的特定〔3H〕物质P的WS-9326A
WS-9326B,三乙酰基-WS-9326A或四氢-WS-9326A置换量
IC50(M)
脑 肺
WS-9326A 2.5×10-5 3.8×10-6
三乙酰基-WS-9326A 9.5×10-5 7.7×10-5
WS-9326B 8.8×10-5
四氢-WS-9326A 4.2×10-7
(2)WS-9326A或四氢-WS-9326A对豚鼠气管的作用:
按标准技术用患有白化病的Hartley种成年雄性豚鼠(600g)制备螺旋状的气管带,并显于30ml的有夹套的玻璃纤维槽组织浴中。通过连于多种波动描记器(Bipysiograph 180 system,San-Ei Instrument)的力位移变换器来等距离测定气管带的张力。在静止张力为500mg的条件下,将气管带悬浮于含有下列组合物的温热的(37℃)氧化(95%O2:5%CO2)Tyrode溶液的器官浴中,该组合物为:NaCl 137mM(8g/l),KCl2.7mM(0.2g/l),CaCl2·2H2O 1.8mM(0.264g/l),MgCl2·6H2O 1.02mM(0.208g/l),NaHCO311.9mM(1g/l),NaH2PO4·2H2O 0.42mM(0.66g/1)和葡萄糖5.5mM(1g/l)。将该组织平衡90分钟,然后测试WS-9326A或四氢-WS-9326A抗各种支气管收缩(物质P10-8M和神经激肽A10-9M)的作用。用San-Ei Rectigzaph-85记录仪(San-Ei Instrument)记录该张力。
表5 WS-9326A或四氢-WS-9326A对神经激肽A(NKA)和物质P
(SP)引起的豚鼠气管改缩反应的作用WS-9326A 抑制%(μg/ml) NKA10-9M SP10-8M
3 44% -10%
10 79% 50%
30 100% 63%
WS-9326A IC50(M) 3.5×10-6 9.7×10-6
四氢-WS-9326A IC(M) 1.6×10-6 3.1×10-6
(3)WS-9326A或四氢-WS-9326A对由神经激肽A和辣椒辣素引起的支气管缩小的作用
将体重为300-500g的雄性Hartley种豚鼠用戊巴比妥钠(10mg/动物经腹膜内注射给药)制动。颈静脉进行插管用以施用神经激肽A(或辣椒辣素)和药物。另将一导管插入气管用以人工呼吸。该动物通过微型呼吸泵装置(Harvard B-34,5ml/stroke,60 strokes/minute)进行呼吸。通过由Konzett-Rssler改进的溢流技术测定对于肺膨胀的对抗作用。
下列表明经静脉给予显效药和拮抗药(在0.1%甲基-纤维素-盐水中制备)的情况
15分钟 15分钟 15分钟 15分钟 15分钟 15分钟 15分钟
↑:显效药(神经激肽A或辣椒辣素)
↑:拮抗药(WS-9326A或四氢-WS-9326A)
表6WS-9326A或四氢-WS-9326A对神经激肽A引起的支气管缩小的抑制作用剂量 对神经激肽A(In mol/kg,iv)反应的抑制率(%)10mg/kg,iv 2分钟 17分钟 32分钟 47分钟 62分钟WS-9326A -16.5±5.6 30.2±12.9 45.8±13.8 55.4±8.1 49.9±13.4四氢-WS-9326A 41.0±5.6 100.0±0.0 99.2±0.8 98.4±1.6 100.0±0.0
表7
WS-9326A对辣椒辣素引起的支气管缩小的抑制作用
对辣椒辣素(10n mol/kg,iv)反应的抑制率(%)剂量 2分钟 17分钟 32分钟 47分钟 62分钟10mg/kg,iv 16.6±5.1 40.6±12.2 51.2±10.4 37.2±19.7 47.1±16.7
(4)WS-9326A或四氢-WS-9326A经气管给药对神经激肽A引起的豚鼠支气管缩小的作用。
为测试WS-9326A或四氢-WS-9326A对支气管缩小的吸入作用,将WS-9326A或四氢-WS-9326A溶于DMSO并经气管给药。该方法与上述大致相同。
如表8和表9所示,WS-9326A和四氢-WS-9326A是有高效作用的。
表8
WS-9326A经气管给药对神经激肽A引起的支气管缩小的抑制作用
对神经激肽A**反应的抑制率(%)
剂量* 20分钟 35分钟 50分钟 65分钟 n
0.03mg/kg 32.3 28.4 35.6 35.5 4
0.3 50.6 42.4 44.9 42.0 4
3 73.4 79.4 81.7 77.7 4
ED50mg/kg 0.23 0.29 0.19 0.24
*:WS-9326A溶于DMSO
**:In mol/kg iv
表9四氢-WS-9326A经气管给药对神经激肽A引起的支气管缩小的抑制作用
对神经激肽A**反应的抑制率(%)
剂量* 20分钟 35分钟 50分钟 65分钟 n
0.003mg/kg 5.4 23.5 17.2 12.6 4
0.03 50.0 50.9 51.4 43.0 4
0.3 77.7 75.7 74.6 71.1 4
ED50mg/kg 0.048 0.030 0.037 0.059
*:四氢-WS-9326A溶干DMSO
**:In mol/kg iv
(5)急性毒性
通过将试验化合物以递级的剂量分别对5只ddy鼠经腹膜注射给药来测定WS-9326A在ddy鼠中的急性毒性。该WS-9326A的LD50值高于250mg/kg并低于500mg/kg(500mg/kg>LD50>250mg/kg)。
本发明的药物组合物能以药物制剂的形式例如,以固体、半固体或液体形式使用,它含有肽衍生物(I)或其药物上可接受的盐作为活性成分与适于外用、内用或肠胃外使用的有机的或无机的载体或赋形剂相混合。该活性成分可以是组合物例如:与用于片剂、药丸、胶囊、溶液、乳剂、悬浮剂和任何其它适于使用的形式的常用的无毒性的药物上可接受的载体相结合的组合物。这些能使用的载体是水、葡萄糖、乳糖、阿拉伯树胶、明胶、甘露醇、淀粉糊、硅胶、马铃薯淀粉、尿素和其它适于在制备制剂、固体、半固体或液体形式中使用的载体,并且还可以另外加入辅助剂、稳定剂、增稠剂、和着色剂以及调味剂。该活性目的化合物是根据疾病的治疗方法或病情,以能足以产生所需作用的含量加到该药物组合物中的。
然而,该肽衍生物(I)或其药物上可接受的盐的治疗有效剂量视需治疗的每个病人的年龄和情况而有所不同,通常,该活性成分用于治疗疾病的日剂量为约0.01-1000mg,优选的是0.1-500mg,更优选是0.5-100mg,一般给药的单一剂量是约0.5mg、1mg、5mg、10mg、50mg、100mg、250mg和500mg。
在本说明书中,氨基酸、肽、深护基等是按IUPAC-IUB(Commissionon Biological Nomenclature)用缩写来表示的,这些在本技术领域是通常使用的。
此外,在下列实施例和制备中,除了采用按IUPAC-IUB的缩写之外,还使用其它的缩写。
用于本说明书中的缩写如下:
Thr :L-苏氨酸
Ser :L-丝氨酸
TYr :L-酪氨酸Asn :L-天冬氨酸allo-Thr:L-异苏氨酸D-Phe :D-苯丙氨酸Leu :L-白氨酸Z :苄氧羰基Pac :苯酰基Bzl :苄基Boc :叔丁氧羰基Me :甲基Tce :2,2,2-三氯乙基Mmp :4-甲氧基甲氧苯基Si :叔丁基二甲硅基Ac :乙酰基Et :乙基n-Hex :正己烷
下列实施例和制备例是用于详述本发明。
实施例1
发酵
将一种含有可溶性淀粉(1%)、蔗糖(1%)、葡萄糖(1%)、棉籽粉(1%)、胨(0.5%)、大豆粉(0.5%)和碳酸钙(0.2%)的种子含水培养基(160ml)(用6N氢氧化钠将其pH调至7.0),分别注入20个500ml的三角烧瓶中,并于120℃灭菌30分钟。
将一菌环量斜面培养的紫黑链霉菌(Streptomyces violaceoniger)9326接种上述各瓶培养基,并于30℃,在旋转式摇床(220rpm,位移5.1cm)上培养3天。将所得到的该种子培养基接种到装盛在200升不锈钢发酵罐中由甘油(3%)、大豆粉(0.5%)、磨细大豆粉(1.5%)、碳酸钙(0.2%)和碘化钙(0.001%)组成的160升纯菌发酵培养基内,于30℃,在160升/分的通气量和200转/分搅拌的条件下发酵三天。用HitachiModel 655泵,经高性能液相色谱(HPLC)定量测定发酵液中WS-9326A含量。于1.0ml/分的流量下,采用装有R-ODS-5(YMC-装填的柱)的钢柱(内径4.6mm,长250mm)。
所用的流动相为甲醇和水(8∶2)的混合物,HPLC分析样品的制备如下所述:将等体积的丙酮加入剧烈搅拌着的发酵液中,静置一小时,随后离心,将5μl该上清液注射到Hitachi Model 655样品注射器中。
分离和纯化
将等体积的丙酮加入搅拌着的培养液(150升)中,使该混合物于室温静置一小时,然后过滤。滤液于减压于浓缩至80升,并用1N盐酸将其pH调至7.0,而后用80升乙酸乙酯萃取。萃取液于减压下浓缩至干,并置于硅胶柱(Kieselgel 60,70-230目,Merck,3升)中。用正己烷(10升)、正己烷-乙酸乙酯〔1∶1〕(10升)、乙酸乙酯(20升)洗涤该柱,用丙酮(6升)自该柱洗脱活性物质。减压干燥活性级分,并在硅胶(Kieselgel 60,70-230目,Merck 1.2升)上进行柱层析,用氯仿-甲醇〔20∶1〕(5升)洗涤该柱,用氯仿-甲醇〔10∶1〕的溶液(6升)洗脱目的物质,减压干燥洗脱级分得到一种粉。将该粉溶于少量甲醇并置于NS凝胶柱(Nihon Seimitsu,500ml)中,用甲醇-水〔8∶2〕(2升)洗脱目的物质,减压浓缩至300ml。后用500ml乙酸乙酯萃取,萃取液于减压下浓缩至干,得到一种粉(5g)。将该粉(5g)溶于10ml甲醇(500mg/ml),并用装有D-ODS-5(YMC-装填柱)的钢柱(内径20mm,长250mm)进行HPLC测定。用甲醇和水〔8∶2〕的混合物,以9.9ml/分流量洗脱,减压浓缩如此得到的活性级分,随后用乙酸乙酯萃取,减压浓缩萃取液至干,得到一种WS-9326A的纯白粉。
实施例2
将乙酐(1.5ml)和4-二甲氨基吡啶(1mg)加入WS-9326A-(300mg)的吡啶(4.5ml)的溶液中,使反应混合物于室温静置过夜。将反应混合物蒸发至干,得到一种油,用薄层制备色谱(TLC)(氯仿-甲醇(10∶1)纯化该油状产物。
将所得到的产物用乙醚研制,得到三乙酰-WS-9326A(332mg),为一种白色粉,该三乙酰-WS-9326A的理化性质如下:
(1)形态和颜色:无色粉。
(2)颜色反应:
阳性:硫酸铈反应,硫酸反应,碘蒸汽反应。
阴性:水合茚三酮反应。
(3)溶解性:
可溶:甲醇、二甲亚砜。
微溶:氯仿、乙醚。
不溶:正己烷。
(4)熔点:141-143℃。
(5)比旋:〔α〕D 23:-122°(C=1.0,MeOH)(6)紫外线吸收光谱: (7)分子式:C60H74N8O16(8)元素分析:C60H14N8O16·2H2O
理论值:C 60.09,H 6.56,N 9.34
实测值:C 60.19,H 6.42,N 9.27(9)分子量:FAB-MS:m/z 1163.6(M+H)+(10)薄层色谱法:固定相 展开溶剂 Rf值硅胶板 氯仿-甲醇 0.50(Merck Art 5715) (10∶1,V/V)
乙酸乙酯 0.12(11)红外吸收光谱:
1520,1440,1360,1230,1200,1160,1100,
1060,1040,910cm-1(12)物质的性质:中性物质(13)13C核磁共振谱:
(100MHz,CDCl3-CD3OH(10∶1))δ
174.20(s), 173.23(s),
173.06(s), 171.32(s),
171.02(s), 170.84(s),
169.79(s), 169.59(s),
169.55(s), 168.52(s),
167.03(s), 166.36(s),
151.02(s), 140.74(d),
138.82(s), 138.74(s),
137.12(s), 135.23(d),
133.75(s), 131.31(s),
130.20(d), 129.96(d)x2,
129.34(d), 129.21(d)x2,
128.56(d)x2, 127.24(d),
126.95(d), 126.74(d),
126.63(d), 126.50(d),
122.10(d)x2, 121.29(d),
70.99(d), 69.22(d),
63.73(t), 58.13(d),
56.10(d), 53.22(d),
52.66(d), 52.18(d),
49.93(d), 39.75(t),
39.39(q), 39.06(t),
35.57(t), 30.65(t),
24.26(d), 23.15(q),
22.79(t), 21.42(q),
21.21(q), 20.99(q),
20.83(q), 17.05(q),
16.18(q), 13.82(q),
图4示出了其核磁共振谱图。(14)1H核磁共振谱:
(400MHz,CDCl3-CD3OH(10∶1))δ
8.25(1H,d,J=8Hz),
8.02(1H,d,J=8Hz),
7.88(1H,d,J=16Hz),
7.86 (1H,d,J=8Hz),
7.70 (1H,d,J=6Hz),
7.61 (1H,d,J=8Hz),
7.45 (1H,d,J=7Hz),
7.32-7.15 (6H,m),
7.03 (2H,d,J=8Hz),
7.00-6.94 (3H,m),
6.88-6.79 (4H,m),
6.70 (1H,s),
6.49 (1H,d,J=12Hz),
5.76 (1H,dt,J=12 and 7.5Hz),
5.54 (1H,broad s),
5.50-5.45 (2H,m),
4.93 (1H,m),
4.75 (1H,m),
4.65-4.56 (2H,m),
4.46 (1H,dd,J=6 and 11Hz),
4.31 (1H,t,J=6Hz),
4.22 (1H,m),
4.18 (1H,dd,J=8 and 11Hz),
3.56 (3H,s),
2.90 (1H,dd,J=6 and 16Hz),
2.85-2.80 (2H,m),
2.56 (1H,dd,J=4 and 16Hz),
2.26 (3H,s),
2.00 (3H,s),
1.96-1.89 (2H,m),
1.85 (3H,s),
1.58 (1H,m),
1.35 (3H,d,J=6Hz),
1.32-1.20 (3H,m),
1.07 (3H,d,J=6Hz),
0.84 (1H,m),
0.72 (3H,d,J=6Hz),
0.71 (3H,t,J=7.5Hz),
0.65 (3H,d,J=6Hz),
图5示出了其核磁共振谱图。
实施例3
发酵
将一种含有可溶性淀粉(1%)、蔗糖(1%)、葡萄糖(1%)、棉籽粉(1%)、胨(0.5%)、大豆粉(0.5%)和碳酸钙(0.2%)的种子含水培养基(160ml),分别注入10个500ml三角烧瓶中,并于120℃灭菌30分钟。
将一菌环量斜面培养的紫黑链霉菌9326接种到上述各瓶培养基,并于30℃,在旋转式摇床(220rpm,位移5.1cm)上培养3天。
将所得到的该种子培养基接种到盛在500升不锈钢发酵罐内由可溶性淀粉(1%)、蔗糖(1%)、葡萄糖(1%)、棉籽粉(1%)、胨(0.5%)、大豆粉(0.5%)、碳酸钙(0.2%)、Adekanol LG-109(形变剂,商标:AsahiDenka Co.出售)(0.07%)和硅氧烷KM-70(形变剂,商标:Shin-etsu化学药品公司出售)(0.05%)组成的种子含水培养基(160升)内(该培养基己预先于120℃灭菌30分钟),并于30℃,在160升/分的通气量和200转/分搅拌的条件下发酵一天。
将所得的种子培养液(60升)接种到盛在4000升不锈钢发酵罐内含有甘油(3.0%)、大豆粉(1.0%)、鸡肉骨粉(1.0%)、碳酸钙(0.2%)、碘化钠(0.001%)、Adekand LG-109(0.07%)和硅氧烷KM-70(0.05%)的经灭菌的产物培养基内(该培养基已预先于120℃灭菌30分钟),并于30℃,在3000升/分的通气量和100转/分搅拌的条件下培养四天。
用Hitachi Model 655泵,经高性能液相色谱法(HPLC)控制发酵过程。采用装有反相硅胶“YMC装填的R-ODS-5柱”的钢柱,流量为1.0毫升/分。所用的流动相为45%乙腈水溶液,HPLC分析样品的制备如下:将等体积丙酮加到剧烈搅拌的发酵液中,使混合物静置一小时,随后离心,将5μl该上清液注射到Hitachi Model 655 HPLC的注射器中。
分离和纯化
如此得到的培养液用硅藻土(Perlite Topko#34,商标,Showa化学药品工业有限公司)(15g)纯化,用乙酸乙酯(1600升)萃取菌丝体压块,过滤萃取液。将过滤液(1400升)加到活性炭柱(Sirasagi KL,商标,Takeda药品有限公司)(200升),乙酸乙酯(120升)洗涤该柱,随后用乙酸乙酯-甲醇(5∶1)洗脱,合并活性部分(50升至1030升部分),减压浓缩至45升。将正己烷(120升)加到所得到的搅拌溶液中,使该混合物于室温静置一小时,随后用Silika#600(Chuo Silica有限公司)(3kg)过滤,用正己烷(15升)洗涤如此得到的压块,用甲醇(20升)洗脱目的物质。
减压浓缩洗脱液至干,将残余物(500g)溶于甲醇-乙酸-二氯甲烷〔1∶1∶2〕(4升),并置于硅胶柱(Kieselgel 60,70-230目,70升)中,该柱用甲醇-乙酸-二氯甲烷〔1∶1∶2〕(0.5升)和二氯甲烷(25升)展开。该目的物质用二氯甲烷-甲醇〔10∶1〕和二氯甲烷-甲醇〔8∶1〕洗脱。合并活性级分并减压浓缩,残余物溶于甲醇(1升),将乙腈(9升)加入所得到的搅拌溶液中,使混合物于室温静置一小时,过滤收集所得到的沉淀。将该沉淀步骤重复三次,用乙腈(1升)洗涤如此得到的沉淀并干燥,得到WS-9326A白粉(190g)。合并自这些沉淀步骤中得到的滤液并减压浓缩至干。残余物(11.7g)溶于80%甲醇水溶液,将所得溶液通过活性炭柱(300ml),用80%甲醇水溶液(1升)洗涤该柱并用甲醇(6升)进行洗脱。合并活性级分,并减压浓缩至干,残余物(3.4g)溶于甲醇(12ml),所得溶液置于以50%乙腈平衡的反相硅胶柱(YMC装填的R-354 S-15/30(ODS)柱,50×300mm×2;制造者,Yamamura化学药品工业公司)中。用50%乙腈水溶液展开用Waters HPLC(体系500)的柱。合并含有WS-9326B(3升至3.5升的部分)的洗脱液并浓缩至干,得到一种WS-9326B的白粉(790mg)。
实施例4
向WS-9326A(100mg)的吡啶(1ml)溶液,加入乙酐(0.01ml),将该混合物于室温静置过夜。将该混合物蒸发至干,得到一种油,用薄层制备色谱(TLC)(氯仿-甲醇(9∶1)纯化该油。所得到的产物用乙醚研制,得到单乙酰-WS-9326A(55mg),为无色粉,该单乙酰-WS-9326A的理化性质如下:
(1)形态和颜色:无色粉
(2)分子式:C56H70N8O14
(3)分子量
FAB-MS:m/z 1079.4(M+H)+
(4)薄层色谱法:
固定相
硅胶板 展开溶剂 Rf值
(Merck Art 5715) 氯仿-甲醇 0.17
(10∶1,V/V)
(5)红外吸收光谱:
1360,1190,1170,910cm-1
(6)该物质的性质:中性物质
(7)1H核磁共振谱:
(400MHz,CBCl3-CD3OD(5∶1)):
图8示出了其核磁共振谱图。
实施例5
向WS-9326A(100mg)的吡啶(1ml)溶液加入乙酐(0.03ml),使混合物于室温静置过夜,该混合物蒸发至干得到一种油,该油经制备性TLC(氯仿-甲醇(9∶1))纯化得到二乙酰-WS-9326A(72mg)无色粉,该二乙酰-WS-9326A的理化性质如下:
(1)形态和颜色:无色粉
(2)分子式:C58H72N8O15
(3)分子量:FAB-MS:m/z 1121.4(M+H)
(4)薄层色谱法:
固定相
硅胶板 展开溶剂 Rf值
(Merck Art 5715) 氯仿-甲醇 0.35
(10∶1,V/V)
(5)红外吸收光谱:
1360,1200,1170,1100,1040,980,910
cm-1
(6)该物质的性质:中性物质。
(7)1H核磁共振谱
(400MHz,CDCl3-CD3OD(5∶1)):
图9示出了其核磁共振谱图。
实施例6
将WS-9326A(100mg)溶于甲醇(2ml)中,该溶液在钯黑上(25mg),于室温及一个氢气压下氢化四小时。过滤该混合物,于减压下将滤液能使至干。用乙醚研制所得产物,得到四氢-WS-9326A(92mg),为无色粉,该四氢-WS-9326A的理性化性质如下:
(1)形态和颜色:无色粉
(2)紫外线吸收光谱:
(3)分子式:C54H72N8O13
(4)分子量:FAB-MS:m/z 1041.6(M+H)+
(5)13C核磁共振谱
(100MHz,CD3OD):
图10示出了其核磁共振谱图。
(6)H核磁共振谱
(400MHz),CD3OD):
图11示出了其核磁共振谱图。
实施例7
向四氢-WS-9326A(1100mg)的吡啶(10ml)溶液加入乙酐(3ml)和4-二甲基氨基吡啶(3mg)使反应混合物于室温静置过夜。将该溶液蒸发至干,得到一种油,用硅胶柱层析(氯仿-甲醇(20∶1))纯化该油。所得到的该纯产品用乙醚研制,得到四氢-三乙酰-WS-9326A(998mg),为无色粉。该四氢-三乙酰-WS-9326A的理化性质如下:
(1)形态和颜色:无色粉
(2)紫外线吸收光谱:
(3)分子式:C60H78N8O16
(4)元素分析:C60H78N8O16·2H2O
理论值:C 60.80,H 6.80,N 9.45
实测值:C 61.03,H 6.70,N 9.41(5)分子量:FAB-MS:m/z 1167.6(M+H)+(6)13C核磁共振谱:
(100MHz,CDCl3)δ
173.30(s), 129.23(d),
172.96(s), 128.95(d)x2,
172.90(s), 128.61(d)x2,
172.81(s), 128.52(d),
170.87(s), 126.80(d),
170.56(s), 126.07(d),
170.50(s), 126.01(d),
169.46(s), 125.85(d),
169.16(s), 121.87(d)x2,
168.48(s), 70.46(d),
167.99(s), 69.10(d),
165.52(s), 63.32(t),
150.70(s), 58.28(d),
140.84(s), 56.19(d),
138.93(s), 52.63(d),
138.58(s), 52.07(d),
136.83(s), 51.63(d),
131.04(s), 49.23(d),
129.71(d)x2, 39.30(t),
39.17(q), 22.50(t),
38.31(t), 21.46(q),
36.52(t), 21.00(q),
35.22(t), 20.74(q),
32.60(t), 20.63(q),
31.77(t), 16.77(q),
30.74(t), 16.22(q),
27.66(t), 13.97(q).
24.08(d),
22.82(q),(7)1H核磁共振谱:
(400MHz,CDCl3) δ
8.18 (1H,d,J=8Hz),
7.66 (1H,d,J=8Hz),
7.65 (1H,d,J=8Hz),
7.29 (2H,d,J=8Hz),
7.22 (1H,d,J=8Hz),
7.15-7.02 (12H,m),
6.96 (1H,d,J=7Hz),
6.67 (1H,s),
6.21 (1H,broad s),
5.51 (1H,broad s),
5.43 (1H,m),
5.36 (1H,broad d,J=8Hz),
4.85-4.75 (2H,m),
4.66-4.58 (2H,m),
4.40 (1H,dd,J=11 and 6Hz),
4.34 (1H,m),
4.23 (1H,dd,J=11 and 9Hz),
4.07 (1H,m),
3.53 (3H,s),
3.04-2.84 (5H,m),
2.75-2.50 (4H,m),
2.4 6 (1H,dd,J=16 and 5Hz),
2.28 (3H,s),
1.99 (3H,s),
1.87 (3H,s),
1.66-1.50 (3H,m),
1.37-1.27 (4H,m),
1.27 (3H,d,J=7Hz),
1.19 (1H,m),
1.03 (3H,d,J=7Hz),
0.88 (1H,m),
0.86 (3H,t,J=6Hz),
0.72 (3H,d,J=6Hz),
0.65 (3H,d,J=6Hz)
实施例8
将三乙酰-WS-9326A(100mg)溶于甲醇(3ml)中,该溶液于一个氢气压下,在钯黑(35mg)存在下,于室温氢化3小时。
过滤该混合物,滤液于减压下蒸发至干。
乙醚研制残余物,得到一种化合物(90mg),为无色粉。
该化合物在各个方面性质均与实施例7中所得到的四氢-三乙酰-WS-9326A相同。
根据对上述理化性质的分析和对化学结构鉴定作进一步研究的结果,已鉴定出三乙酰-WS-9326A、单乙酰-HS-9326A、二乙酰-WS-9326A、四氢-WS-9326A和四氢-三乙酰-WS-9326A的化学结构如下:三乙酰-HS-9326A
R-:(E)-3-〔2-((Z)-1-戊烯基)苯基〕丙烯酰单乙酰-WS-9326A
R-:(E)-3-〔2-((Z)-1-戊烯基)苯基〕丙烯酰二乙酰-WS-9326A
R-:(E)-3-〔2-(Z)-1-戊烯基)苯基〕丙烯酰四氢-WS-9326A
〔3-(2-戊苯基)丙酰〕四氢-三乙酰-WS-9326A
R-:3-(2-戊苯基)丙酰
实施例9
目的化合物(1)
于室温下,向起始化合物(a)(3.24g)的二氯甲烷(1000ml)的溶液加入三乙胺(350μl)和1-乙氧羰基-2-乙氧基-1,2-二氢喹啉(6.17g),该混合物于室温搅拌24小时后,蒸发溶剂。将氯仿加入残余物中,混合物用水、1N盐酸、水、饱和碳酸氢钠水溶液和水洗涤。硫酸镁干燥混合物后,过滤,蒸发溶剂。残余物进行“Lober柱”(规格C)色谱层析并用3%甲醇的氯仿洗脱,蒸发含有该目的化合物的部分,得到目的化合物(1)(1.06g)。[α]D 18:-95.3°(C=0.33,CHCl3)IR(CHCl3):1660,1600,1510cm-1NMR(CDCl3,δ):0.88(3H,d,J=6Hz),0.93(3H,d,
J=6Hz),1.09(3H,d,J=6.5Hz),1.37(3H,d,J=6.5),
2.82(3H,s),4.87(2H,s),6.93(2H,d,J=8Hz).
实施例10
目的化合物(1)
向起化合物(a)(42mg)的二氯甲烷(4ml)和N,N-二甲基甲酰胺(0.1ml)溶液加入N-羟基琥珀酰亚胺(20.4mg)和水可溶的碳化二亚胺盐酸化物(8.2mg)。
于室温搅拌15小时后,将水可溶的碳化二亚胺盐酸化物(4mg),于1.5小时内加入该混合物中,直至起始化合物(a)消失为止。
真空除去溶剂,残余物溶于乙酸乙酯(10ml)中,并用稀盐酸和水洗涤。
硫酸镁干燥后,真空除去溶剂,残余物溶于三氟乙酸(1ml)和苯甲醚(0.1ml)。
于室温搅拌30分钟后,真空除去溶剂,残余物溶于N,N-二甲基甲酰胺(2ml),并将该混合物加到吡啶(40ml)中。
于室温搅拌16小时后,真空蒸除溶剂,残余物进行薄层制备色谱(Merck 5744)层析,用氯仿-甲醇(10∶1)展开,得到目的化合物(1)(15.2mg) 。
IR(KBr):1635,1510cm-1
NMR(CD3OD,δ):6.24(1H,s)
[α]D 20:+18.0°(C=0.1,MeOH)
实施例11
目的化合物(1)
起始化合物(a)(240mg),于4psi下,用钯(200m),在甲酸和甲醇(1∶24,10ml)中氢化7小时。
过滤该混合物后,蒸发滤液,得到目的化合物(1)(140mg)。
IR(KBr):1730,1650,1510cm-1
[α]D 21:-21.04°(C=0.1,MeOH)
实施例12
目的化合物(1)
将起始化合物(a)(22mg)溶于氟化氢吡啶(0.8ml)和苯甲醚(0.2ml)在氮气袋中的溶液,室温搅拌一小时后,将一些冰块加到该混合物中,用碳酸氢钠水溶液将pH调至8。将该混合物装入Diaion HP-20柱(10ml),并用水洗涤。用甲醇洗脱该产物,经薄层色谱层析(Merck 5715,氯仿-甲醇-水(3∶1∶0.1,V/V)纯化,得到该目的化合物(1)(13.0mg)。IR(KBr):1635,1510cm-1NMR(CD3OD,δ):7.05(1H,s)[α]D 20:-90.6°(C=0.1,MeOH)TLC∶Rf=0.35[Merck Art 5715,CHCl3-MeOH-H2O
(3∶1∶0.1)]
实施例13
目的化合物(1)
R-:(E)-3-〔2-((Z)-1-戊烯基)苯基〕丙烯酰
向起始化合物(a)(6.0mg)的二氯甲烷(1.5ml)、双(二甲基甲硅烷基)乙酰胺(30μl)和N,N-二甲基甲酰胺(0.3ml)的溶液加入0.02M起始化合物((b)(0.4ml)的溶液,室温搅拌一小时后,将4-二甲基氨基吡啶(0.1mg)加入该混合物中。于30分钟内,将起始化合物(b)加入该混合物直至起始化合物(a)消失为止,将稀盐酸加入该混合物,水洗涤有机层,真空蒸发后,残余物进行薄层制备色谱(Merck 5715)层析,用氯仿-甲醇-水(65∶25∶4V/V)展开,得到该目的化合物(1)(0.2mg)。
该化合物与实施例1所得到的WS-9326A相同。
实施例14 
目的化合物(1)
R-:硬脂酰
(十八烷酰基)
向起始化合物(a)(11mg)的吡啶(1ml)溶液中加入起始化合物(b)的0.02M二氯甲烷(0.6ml)溶液,室温搅拌一小时后,在一小时内,将起始化合物(b)加入该混合物直至起始化合物消失为止。将甲醇(2ml)加入该混合物,真空除溶剂,残余物溶于乙酸乙酯(10ml),并用稀盐酸和水洗涤。硫酸镁干燥后,真空除去溶剂,残余物进行薄层制备色谱(Merck 5715)层析,用氯仿-甲醇-水(3∶1∶0.1,V/V)展开,得到该目的化合物(1)(2.0mg)。
IR(KBr):1640,1510cm-1
实施例15
R-:(E)-3-〔2-((Z)-1-戊烯基)苯基〕丙烯酰
向起始化合物(a)(49.7mg)的吡啶(1ml)溶液,于氮气氛下加入起始化合物(b)的0.1M二氯甲烷(1.2ml)溶液,混合物于室温搅拌3.5小时。向该反应混合物加入乙酸乙酯,水7%乙酸、水和饱和的氧化钠溶液洗涤混合物。硫酸镁干燥后,过滤,蒸发溶剂,残余物进行薄层制备色谱(0.5mm×2)层析,并用20%甲醇的氯仿展开,得到该目的化合物(1)(20.6mg)。
该化合物与实施例3所得到的WS-9326B相同。
实施例16
按照实施例15的相似方法制得下列各化合物。
(1)R-:苯甲酰基[α]D 21:-45.8°(C=0.74,MeOH)mp:176-178℃TLC:Rf=0.48[Merck Art 5715,CHCl3-MeOH(5∶1)]IR(KBr):1720(肩峰),1655,1640cm-1
(2)R-:2-(2-噻吩基)乙酰[α]D 23:-16.8°(C=0.73,MeOH)mp:160-163℃TLC:Rf=0.24[Merck Art 5715,CHCl3-MeOH(5∶1)]IR(KBr):1720(肩峰),1650cm-1
(3)R-:乙酰[α]D 21:-37.4°(C=0.72,MeOH)mp:231-233℃TLC:Rf=0.41[Merck Art 5715,CHCl3-MeOH(5∶1)]IR(KBr):1720(肩峰),1650cm-1
实施例17
目的化合物(1)
向起始化合物(a)(100mg)的吡啶(1ml)溶液加入乙酐(11μl),使混合物于室温静置过夜。
蒸发混合物至干,得到一种油,该油经制备性TLC(CHCl3-MeOH(9∶1)纯化,得到该目的化合物(1)(52mg)。TLC:Rf=0.17[Merck Art 5715,CHCl3-MeOH(10∶1)]IR(Nujol):3300,1760,1730,1650,1530,1510,
1200,1160,1070,910cm-1
实施例18
目的化合物(1)R-:3-(2-戊苯基)丙酰
向起始化合物(a)(100mg)的吡啶(1ml)溶液加入乙酐(25μl),使该混合物于室温静置过夜。
蒸发该混合物至干,得到一种油,该油经制备性TLC(CHCl3-MeOH(9∶1))纯化,得到该目的化合物(1)(78mg)。TLC:Rf=0.36[Merck Art 5715,CHCl3-MeOH(10∶1)]IR(Nujol):3300,1760,1740,1650,1540,1510,
1300,1220,1200,1170,1050,920cm-1
实施例19
R-:3-(2-戊苯基)丙酰
向起始化合物(a)(0.21g)的甲醇(3ml)的溶液中加入重氮甲烷的乙醚溶液(3ml)中,搅拌5分钟后,真空除去溶剂,残余物进行制备性薄层色谱(Merck 5744)层析,并用20%甲醇的氯仿展开,得到该目的化合物(1)(45mg)。
IR(液体石蜡):3300,1730,1645,1530,1510cm-1
实施例20
R-:3-(2-戊苯基)丙酰
向起始化合物(a)(1.0g)的甲醇(15ml)溶液,于0℃下,加入1N氢氧化钠(5ml),搅拌一小时后,将1N盐酸(5ml)加入该溶液,真空除去溶剂,残余物溶解在乙酸乙酯(20ml)和稀盐酸(30ml)的混合物中。用盐水洗涤有机层,硫酸镁干燥,真空蒸发,乙酸乙酯洗涤所得到的固体,得到该目的化合物(1)(0.95g)。
IR(液体石蜡):3300,1710(肩峰),1645,1510cm-1
实施例21
R-:3-(2-戊苯基)丙酰
向起始化合物(a)(1.0g)的甲醇(2ml)和乙酸乙酯(20ml)的溶液加入10%三甲基甲硅烷基二重氮甲烷的己烷溶液中,搅拌5分钟后,真空蒸除溶剂,残余物加到硅胶柱(Merck 7734)(20g)上,氯仿-甲醇(10∶1,V/V)洗脱,得到一种该目的化合物(1)的固体(0.55g)。
IR(液体石蜡):3300,1735,1645,1530,1510cm-1
制备例1
Z-Thr → Z-Thr-OPac
起始化合物(a) 目的化合物(1)
向起始化合物(a)(2.53g)、甲醇(10ml)和水(3ml)的溶液加入碳酸铯(1.63g)。
除去溶剂后,残余物溶于N,N-二甲基甲酰胺,并将该混合物加到苯甲酰甲基溴(1.92g)中,混合物于室温搅拌30分钟,蒸除溶剂,并将残余物溶于乙酸乙酯,水洗涤。该混合物经硫酸镁干燥和过滤后,蒸发溶剂,得到该目的化合物(1)的晶体(3.7g)。
〔α〕D 20:-20.3℃(C=1,CHCl3)
IR(液体石蜡):1745,1690,1545cm-1
制备例2
Z-Thr-OPac +

起始化合物(a) 起始化合物(b)
目的化合物(1)
向起始化合物(a)(1.48g)和起始化合物(b)(2.5g)的二氯甲烷(80ml)的溶液,于0℃加入1-乙基-3-(3-二甲基氨基丙基)碳化二亚胺盐酸化物(0.764g)和4-二甲基氨基吡啶(0.488g),搅拌6小时后,蒸发溶剂。
残余物溶于乙酸乙酯,水、1N盐酸、饱和的碳酸氢钠水溶液和水洗涤混合物。
硫酸镁干燥和过滤后,蒸发溶剂,得到该目的化合物(1)(2.58g)。[α]D 20:+9.76°(C=0.3,CHCl3)IR(CHCl3):1755,1720,1705,1505cm-1NMR(CDCl3,δ):1.37(3H,d,J=6Hz),1.45(9H,s),
3.70(1H,m),3.88(1H,m),4.52(2H,m),4.23
(1H,m),5.17(2H,s),5.37(2H,s)
制备例3
起始化合物(a) 目的化合物(1)
于搅拌下,向起始化合物(a)的90%乙酸水溶液(100ml),加入锌粉(11g),混合物于冰冷却下搅拌2小时,再于室温下搅拌一小时。
混合物过滤后,浓缩滤液,用柠檬酸调pH至2,用乙酸乙酯萃取。
硫酸镁干燥萃取液并过滤后,蒸发溶剂,石油醚洗涤残余物,得到该目的化合物(1)(5.4g)。NMR(CDCl3,δ):1.30(3H,d,J=6Hz),1.40(9H,s),
3.60(1H,m),3.82(1H,m)
制备例4
起始化合物(a) 目的化合物(1)
于0℃,搅拌下,向起始化合物(a)(7.7g)的二氯甲烷(60ml)溶液,加入2,2,2-三氯乙醇(2.105ml)、1-乙基-3-(3-二甲基氨基丙基)碳化二亚胺盐酸化物(4.2g)和4-二甲基氨基吡啶(244mg),混合物于0℃搅拌一小时并蒸发,残余物溶于乙酸乙酯,用水、1N盐酸、水和饱和的碳酸氢钠水溶液洗涤。
硫酸镁干燥混合物并过滤后,蒸发滤液,得到该目的化合物(1)(9.37g)。
[α]D 19:-29.84°(C=0.4,CHCl3)
IR(CHCl3):1755,1690,1610,1510cm-1
制备例5
起始化合物(a) 目的化合物(1)
将起始化合物(a)(9.3g)冷却0℃,加入三氟乙酸(15ml)。
于0℃,搅拌混合物30分钟并蒸发,残余物溶于乙酸乙酯,用水、饱和的碳酸氢钠水溶液和水洗涤,硫酸镁干燥和过滤后,蒸除溶剂,得到该目的化合物(6g)。NMR(CDCl3,δ):2.45(3H,s),3.03(2H,m),3.62
(1H,t,J=7Hz),4.75(2H,m),5.06(2H,s),
6.94(2H,d,J=8Hz),7.15(2H,d,J=8Hz),
7.3-7.5(5H,m)
制备例6
起始化合物(a) 起始化合物(b)
目的化合物(1)
向起始化合物(a)(4.07g)和起始化合物(b)(4.70g)的二氯甲烷(40ml)溶液,于室温下,加入1-乙氧羰基-2-乙氧基-1,2-二氢-喹啉(2.8g),并搅拌混合物24小时,蒸发溶剂、残余物溶于乙酸乙酯,用1N盐酸、水、饱和的碳酸氢钠水溶液和水洗涤。
硫酸镁干燥和过滤后,蒸发溶剂,得到该目的化合物(1)(4.35g)。[α]D 20:-27.88°(C=0.12,CHCl3)IR(CHCl3):1750,1710,1655,1510cm-1NMR(CDCl3,δ):1.18(3H,d,J=6Hz),1.36(9H,s),
2.92(3H,s),4.69(2H,s),4.91(2H,s),
5.01(2H,s),6.80(2H,d,J=8Hz),7.02(2H,d,
J=8Hz)
制备例7 起始化合物(a)
起始化合物(a)
目的化合物(1)
起始化合物(a)(4.35g)冷却至0℃,将其加入三氟乙酸(20ml)中,混合物于0℃搅拌45分钟后,蒸发溶剂,残余物溶于乙酸乙酯,用饱和的碳酸氢钠水溶液和水洗涤,硫酸镁干燥该溶液并过滤后,蒸发溶剂,残余物溶于二氯甲烷(100ml)。
向该溶液加入Boc-Asn(1.2g),1-乙基-3-(3-二甲基氨基丙基碳化二亚胺(990mg)和1-氢化苯并三唑(700mg),混合物于0℃搅拌4小时,并用1N盐酸水、饱和的碳酸氢钠水溶液和水洗涤,蒸发溶剂,得到该目的化合物(1)(4.75g)。[α]D 20:-15.7°(C=0.1,CHCl3)IR(KBr):1740,1690,1645,1510cm-1NMR(CDCl3,δ):1.25(3H,d,J=6Hz),1.43(9H,s),
3.00(3H,s),2.55(1H,m),2.80(1H,m),
3.05(1H,m),3.37(1H,m),3.62(1H,m),
3.82(1H,m),6.88(2H,d,J=8.5Hz),7.09(2H,
d,J=8.5Hz)
制备例8
目的化合物(1)
按照制备例7的相似方法,通过使起始化合物(a)反应,制得该目的化合物。[α]D 22:-13.04°(C=0.11,CHCl3)IR(KBr):1740,1700,1655,1510cm-1NMR(CDCl3,δ):1.18,1.27(each 3H,d,J=6Hz),
1.43(9H,s),2.52(1H,m),2.80(1H,m),3.00
(3H,s),3.36(1H,m),4.99(2H,s),6.87(2H,
d,J=8Hz),7.10(2H,d,J=8Hz)
制备例9
按照制备例7的相似方法,通过使起始化合物(a)反应,制得该目的化合物。[α]D 21:-15.19(C=0.1,CHCl3)IR(KBr):1740,1650,1635,1510cm-1NMR(CDCl3,δ):1.11(3H,d,J=6Hz),1.27(3H,d,
J=6Hz),1.33(9H,s),3.01(3H,s),4.72(2H,
s),4.98(2H,s),6.87(2H,d,J=8Hz),7.10(2H,
d,J=8Hz)
制备例10
目的化合物(1)
按照制备例7的相似方法,通过使起始化合物(a)反应,制得该目的化合物(1)。[α]D 21:-19.07°(C=0.1,CHCl3)IR(KBr):1740,1635,1510cm-1NMR(CDCl3,δ):0.81(6H,m),1.13,1.28(每3H,
d,J=6Hz),1.40(9H,s),3.02(3H,s),4.96(2H,
s),6.87,7.09(每2H,d,J=8Hz)
制备例11
目的化合物(1)
于0℃,向起始化合物(a)(4.2g)的90%乙酸水溶液(80ml)加入锌粉(9g),混合物于0℃搅拌2小时,再于室温搅拌1小时。
过滤混合物后,浓缩滤液。将氯仿加入残余物,混合物用1N盐酸和水洗涤。
硫酸镁干燥混合物并过滤后,蒸发溶剂。残余物在硅胶(150g)上进行柱层析,先用含2%甲醇的氯仿洗脱,再用含8%甲醇的氯仿洗脱,蒸发含有该目的化合物的部分,得到该目的化合物(1)(3.4g)。
[α]D 21:-23.42°(C=0.1,MeOH)
IR(KBr):1635,1510cm-1
制备例12
将起始化合物(a)(3.4g)冷却至0℃,并将其加入三氟乙酸(20ml)中。
于0℃搅拌混合物一小时后,蒸发溶剂,残余物溶于氯化氢的二噁烷溶液中,随后蒸发。将残余物溶于氯仿并用水洗涤,硫酸镁干燥溶液并过滤后,蒸发溶剂,得到目的化合物(1)(3.27g)。
[α]D 21:-18.87°(C=0.12,MeOH)
IR(KBr):1650,1510cm-1
制备例13
于室温下,向氢氧化钾(26.8g)的乙醇(500ml)的溶液,加入甘氨酸(14.6g)和4-甲氧基甲氧基苯甲醛(48.5g),搅拌19小时后,真空蒸发溶剂,残余物溶于水并用盐酸酸化,乙酸乙酯洗涤该溶液,用碳酸氢钠将其pH调至6.0,使其沉淀出白色固体并收集之,得到邻-甲氧甲基-β-羟基酪氨酸(9.2g)。
mp:164-166℃
IR(KBr):1610cm-1
制备例14
向邻-甲氧甲基-β-羟基酪氨酸(21.0g)的1N氢氧化钠(250ml)的溶液加入硫酸二甲酯(16.5g),于90℃搅拌20分钟后,该溶液在冰浴上用稀盐酸酸化,乙酸乙酯洗涤该酸溶液,用1N氢氧化钠调pH至6.0。蒸发后,经过滤收集固体,得到邻-甲氧甲基-N-甲基-β-羟基酪氨酸(5.2g)。
mp:177-178℃
IR(KBr):3100,1600cm-1
制备例15
向邻-甲氧甲基-N-甲基-β-羟基酪氨酸(15.1g)和双(三甲基甲硅烷基)乙酰胺(25ml)的二氯甲烷(150ml)溶液加入2-硝基苯亚磺酰氯(11.2g)的二氯甲烷(50ml)溶液,于0℃搅拌2小时后,将双(三甲基甲硅烷基)乙酰胺(10ml)和2-硝基苯亚磺酰氯(5.6g)加入该溶液。混合物于室温搅拌3小时,加入1N氢氧化钠(200ml),水(300ml)洗涤有机层,合并水溶液。稀盐酸酸化该水溶液后,乙酸乙酯(300ml)萃取该产物,水(100ml×3)洗涤萃取液,硫酸镁干燥该溶液后,真空除溶剂,得到邻-甲氧甲基-N-甲基-N-(2-硝基苯硫代)-β-羟基酪氨酸(20.5g)。
mp:59-60℃
IR(KBr):3400,1700cm-1
制备例16
向邻-甲氧甲基-N-甲基-N-(2-硝基苯硫代)-β-羟基酪氨酸(20.0g)的乙酸乙酯(100ml)溶液,加入重氮甲烷的乙醚(80ml),搅拌10分钟后,真空除去该溶剂,残余物装上硅胶柱(Merck 7734:500g),氯仿洗脱,得到邻-甲氧甲基-N-甲基-N-(2-硝基苯硫代)-β-羟基酪氨酸甲基酯(苏型异构体:8.82g,赤型异构体:6.63g)。苏型异构体
IR(薄膜):3500,2950,1735cm-1TLC∶Rf=0.40[Merck Art 5715,AcOEt-n-Hex(1∶1)]赤型异构体
IR(薄膜):3500,2950,1735cm-1TLC∶Rf=0.31[Merck Art 5715,AcOEt-n-Hex(1∶1)]
制备例17
向邻-甲氧甲基-N-甲基-N-(2-硝基苯硫代)-β-羟基酪氨酸甲基酯(赤型异构体(3.85g)的二氯甲烷(30ml)溶液,加入三乙胺(1.38g),4-二甲基氨基吡啶(0.45g)和苯甲酰氯(1.92g),于室温搅拌16小时后,将3-二甲基氨基丙胺(3.3g)加入该混合物中,真空除去溶剂。残余物溶于乙酸乙酯(30ml),用稀盐酸、碳酸氢钠水溶液和水洗涤,蒸发后,残余物装上硅胶柱(Merck 7734,150g),正己烷-乙酸乙酯(5∶2,V/V)洗脱,得到邻-甲氧甲基-N-甲基-N-(2-硝基苯硫代)-β-苯甲酰氧酪氨酸甲基酯(赤型异构体(4.49克)。
IR(薄膜):2950,1740cm-1
TLC∶Rf=0.23(Merck Art 5715,乙酸乙酯∶正己烷=1∶2)
制备例18
按制备例17的相似方法,制得下列化合物。
邻-甲氧甲基-N-甲基-N-(2-硝基苯硫代)-β-苯甲酰氧酪氨酸甲基酯(苏型异构体)。
熔点:114-115℃
IR(CHCl3):2950,1740cm-1
TLC∶Rf=0.26(Merck Art 5715,乙酸乙酯∶正己烷=1∶2)
制备例19
于0℃,向邻-甲氧甲基-N-甲基-N-(2-硝基苯硫代)-β-苯甲酰氧酪氨酸甲基酯(苏型异构体)(4.94克)的二氯甲烷(50ml)溶液,加入苯硫酚(4.8ml)和三氟乙酸(2.5ml)中,搅拌30分钟后,将碳酸氢钠水溶液加入该混合物中,用碳酸氢钠水溶液和盐水洗涤有机层,蒸发后,残余物装上硅胶柱(Merck 7734,100g),用5%甲醇的氯仿洗脱,得到邻-甲氧甲基-N-甲基-β-苯甲酰氧酪氨酸甲基酯(苏型异构体)(0.32g)。TLC∶Rf=0.31(Merck Art 5715,AcOEt∶n-Hex=1∶1)
制备例20
按照制备例19的相似方法,制得下列化合物。
邻-甲氧甲基-N-甲基-β-苯甲酰氧酪氨酸甲基酯(赤型异构体)。
TLC∶Rf=0.25(Merck Art 5715,AcOEt∶n-Hex=1∶1)
制备例21
向N-苄氧羰基-L-苏氨酸(3.7g)和邻-甲氧甲基-N-甲基-β-苯甲酰氧酪氨酸甲基酯(苏型异构体)(3.11g)的二氯甲烷(50ml)的溶液,加入1,2-二氢-2-乙氧-1-喹啉羧酸乙酯(2.9g),室温搅拌20小时后,真空除去溶剂,残余物溶于乙酸乙酯(50ml),用稀盐酸、碳酸氢钠水溶液和水洗涤。蒸发后,残余物装上硅胶柱(Merck 7734,100g),用正己烷-乙酸乙酯(1∶1,V/V)洗脱,得到N-(N-苄氧羰基-L-苏氨酰)-邻-甲氧甲基-N-甲基-β-苯甲酰氧酪氨酸甲基酯(苏型异构体(2.04g)。
IR(薄膜):3400,2950,1740(肩峰),1720cm-1
TLC∶Rf=0.36(Merck Art 5715,MeOH∶CHCl3=3∶97)
制备例22
按照制备例21的相似方法,制得下列化合物。
N-(N-苄氧羰基-L-苏氨酰)-邻-甲氧甲基-N-甲基-β-苯甲酰氧酪氨酸甲基酯(赤型构异体)。
IR(薄膜):2950,1740,1730(肩峰)cm-1
TLC∶Rf=0.23(Merck Art 5715,AcOEt∶正己烷=1∶2)
制备例23
目的化合物(1)
向β-苯甲酰氧-N-(N-苄氧羰基-L-苏氨酰)-邻-甲氧甲基-N-甲基酪氨酸甲基酯(苏型异构体)(1.20g)的甲苯(20ml)溶液,加入1,8-二氮二环〔5.4.0〕十一碳-7-烯(0.30g),室温搅拌0.5小时后,将7%盐酸(10ml)加入该混合物中,用水和碳酸氢钠水溶液洗涤有机层,硫酸镁干燥有机层后,真空除去溶剂,得到该目的化合物(1)(0.95g)。
IR(薄膜):3400,2950,1720cm-1
〔α〕:-7.7°(C=0.64,MeOH)
也可用β-苯甲酰氧-N-(N-苄氧羰基-L-苏氨酰)-邻-甲氧甲基-N-甲基酪氨酸甲基酯(赤型异构体)代替β-苯甲酰氧-N-(N-苄氧羰基-L-苏氨酰)-邻-甲氧甲基-N-甲基酪氨酸甲基酯(苏型异构体)。
制备例24
自起始化合物(a)(1.0g)的N,N-二甲基甲酰胺(10ml)溶液,加入叔丁基二甲基甲硅烷基氯化物(0.75g)和咪唑(0.34g),室温搅拌16小时后,将乙酸乙酯(30ml)和冰(50g)加入该混合物,用稀盐酸、碳酸氢钠水溶液和水洗涤有机层,真空除溶剂,残余物装上硅胶柱(Merck 7734,30g)氯仿洗脱,得到该目的化合物(1)(1.21g)。
IR(薄膜):2950,1720cm-1
〔α〕D 22:-55.9°(C=0.56,MeOH)
制备例25
向起始化合物(a)(0.95g)的溶液,加入1N氢氧化钠水溶液(4.8ml),于30℃搅拌2天后,真空除去溶剂,残余物溶于乙酸乙酯(20ml),稀盐酸和水洗涤,蒸发得到该目的化合物(1)(0.81g)。
IR(薄膜):3300,2950,1720,1700(肩峰)cm-1
〔α〕D 22:-82.9℃(C=1.06,MeOH)
制备例26
起始化合物(a) 起始化合物(b)
目的化合物(1)
(1)向起始化合物(a)(1.60g)和起始化合物(b)(5.50g)的二氯甲烷(50ml)混合物,加入三乙胺(1.25g)和1,2-二氢-2-乙氧基-1-喹啉羧酸乙酯(3.04g),于室温搅拌15小时后,滤出白色固体,并将起始化合物(b)(2.23g)、三乙胺(0.50g)和1,2-二氢-2-乙氧基-1-喹啉羧酸乙酯(1.24g)加入滤液中。混合物搅拌18小时,真空除去溶剂,残余物溶于乙酸乙酯(50ml),稀盐酸、碳酸氢钠水溶液和水洗涤。真空蒸发后,残余物装上硅胶柱(Merck 7734,100g),正己烷-乙酸乙酯(2∶1,V/V)洗脱,得到该目的化合物(1)(0.87g)。
IR(薄膜):2950,1760,1740(肩峰),1720,1660cm-1
〔α〕D 20:-31.5°(C=1.07,MeOH)
制备例27
起始化合物(a)
目的化合物(1)
将起始化合物(a)(0.85g)的甲苯(100ml)的丙酮(10ml)溶液,于0℃经紫外线灯(100V)处理1.5小时,蒸发后,残余物装上硅胶柱(Merck 7734,50g),正己烷-乙酸乙酯(2∶1,V/V)洗脱,得到该目的化合物(1)(0.18g)。TLC∶Rf=0.22(Merck Art 5715,n-Hex∶AcOEt=2∶1)IR(KBr):3300,1740(肩峰),1640cm-1
制备例28
目的化合物(1)
将起始化合物(a)(0.17g)溶于67%乙酸水溶液(10ml)中,于25℃搅拌28小时后,真空除去溶剂,残余物溶于乙酸乙酯(20ml),碳酸氢钠水溶液和水洗涤。
浓缩后,正己烷洗涤残余物,真空除去溶剂,得到该目的化合物(1)(0.15g)。IR(KBr):3250,1740(肩峰),1635cm-1TLC∶Rf=0.18(Merck Art 5715,n-Hex∶AcOEt=1∶1)
制备例29起始化合物(a) 起始化合物(b)
目的化合物(1)向起始化合物(a)(0.14g)的二氯甲烷(5ml)溶液,加入起始化合物(b)(0.10g)、水可溶的碳化二亚胺盐酸化物(65mg)和4-二甲基氨基吡啶(4mg),于室温搅拌12小时后,将N,N-二甲基氨基丙胺(50mg)加到该混合物中,真空除去溶剂,残余物溶于乙酸乙酯(20ml),稀盐酸和水洗涤。蒸发后,残余物装上硅胶柱(Merck 7734,10g),正己烷-乙酸乙酯(1∶1,V/V)洗脱,得到该目的化合物(0.16g)。IR(KBr):3300,1700,1640,1495cm-1TLC∶Rf=0.38(Merck Art 5715,n-Hex∶AcOEt=1∶1)
制备例30
将起始化合物(a)(145mg)溶于4N-盐酸二噁烷液(3ml)和苯甲醚(0.1ml)的混合物中,室温搅拌30分钟后,真空除去溶剂,残余物溶于二氯甲烷(3ml),向该溶液加入N-叔丁氧羰基-L-天冬酰胺(35mg)、三乙胺(13mg)、1-羟基苯并三唑(18mg)和水可溶的碳化二亚胺盐酸化物(29mg),室温搅拌一小时后,将7%盐酸(5ml)加入该混合物,水洗涤有机层,蒸发后,残余物进行制备性薄层色谱(Merck 5744)层析,用6%甲醇的氯仿展开,得到该目的化合物(1)(110mg)。IR(KBr):3300,1650,1505cm-1TLC∶Rf=0.44(Merck Art 5715,CHCl3∶MeOH=10∶1)
制备例31
起始化合物(a)
目的化合物(1)
将起始化合物(a)(105mg)溶于4N盐酸的二噁烷(3ml)和苯甲醚(0.1ml)的混合物中,室温搅拌30分钟后,真空除去溶剂,残余物溶于二氯甲烷(3ml)中,向该溶液加入N-叔丁氧羰基-L-别苏氨酸(22mg)、三乙胺(9mg)、1-羟基苯并三唑(12mg)和水可溶的碳化二亚胺盐酸化物(19mg),室温搅拌8小时后,将7%盐酸(5ml)加入该混合物中,水洗涤有机层,蒸发后,残余物进行制备性薄层色谱(Merck 5744)层析,用6%甲醇的氯仿展开,得到该目的化合物(1)。TLC∶Rf=0.73(Merck Art 5715,CHCl3∶MeOH=5∶1)IR(KBr):3300,1740(肩峰),1650,1500cm-1
制备例32
目的化合物(1)
向起始化合物(a)(59.5mg)的90%乙酸水溶液(1ml)的溶液加入锌粉(30mg),室温搅拌9小时后,于1小时内,将锌粉(30mg)加入该混合物中,直至起始化合物(a)消失为止。过滤后,真空除去溶剂,残余物溶于乙酸乙酯(10ml),水洗涤,真空蒸发,残余物进行制备性薄层色谱(Merck 5744)层析,乙酸乙酯-丙酮-乙酸-水(6∶3∶1∶1,V/V)展开,得到该目的化合物(1)(43.5mg)。
IR(KBr):3330,1650,1505cm-1
TLC∶Rf=0.16[Merck Art 5715,CHCl3-MeOH-AcOH
(10∶1∶0.1)]
制备例33
向苯二醛(6.7g)的二氯甲熔(30ml)溶液加入乙氧羰基亚甲基三苯基正膦(17.42g),混合物于室温搅拌30分钟,蒸发溶剂,残余物溶于乙醚,混合物过滤后,蒸发滤液。残余物于真空(125℃,0.6mmHg)下蒸馏,得到(E)-3-(2-甲酰苯基)丙烯酸乙酯(6g)。NMR(CDCl3,δ):1.24(3H,t,J=6.5Hz),4.19(2H,q,
J=6.5Hz),6.28(1H,d,J=15Hz),7.5(3H,m),7.77
(1H,m),8.43(1H,d,J=15Hz),10.18(1H,s)
制备例34
于氮气氛下,向丁基三苯基溴化鏻(3.2g)的四氢呋喃(50ml)溶液,加入叔丁醇钾(900mg)并于室温搅拌混合物30分钟。将(E)-3-(2-甲酰苯基丙烯酸乙酯(2.0g)的四氢呋喃(30ml)的溶液加到该混合物中,搅拌混合物一小时,蒸发溶剂后,残余物溶于乙醚,盐水和水洗涤。
硫酸镁干燥该溶液,过滤并蒸发,残余物在硅胶柱(100g)上进行柱层析,正己烷和乙酸乙酯(3∶1)的混合物洗脱,蒸发含有目的化合物的各部分,得到(E)-3-〔2-((Z)-1-戊烯基)苯基〕丙烯酸乙酯(2.00g)。NMR(CDCl3,δ):0.88(3H,t,J=7Hz),1.34(3H,t,
J=6.5Hz),1.42(2H,m),2.05(2H,m),4.27(2H,
q,J=6.5Hz),5.85(1H,dt,J=7,11Hz),6.39(1H,
d,J=16Hz),6.56(1H,d,J=11Hz),7.3(3H,m),
7.61(1H,m),7.92(1H,d,J=16Hz)
制备例35
向(E)-3-〔2-((Z)-1-戊烯基)苯基〕丙烯酸乙酯(2g)的20%甲醇水溶液加入氢氧化钾(2.3g),混合物于60℃搅拌2小时,用盐酸将其pH调至1,乙酸乙酯萃取。硫酸镁干燥萃取液并过滤后,蒸发溶剂,残余物溶于正己烷和乙酸乙酯(4∶1)的混合物中。将该溶液加入二环己胺(1.63ml)中,得到晶体,将该晶体溶于乙酸乙酯并用1N硫酸洗涤,硫酸镁干燥该溶液,过滤和蒸发,得到(E)-3-〔2-((Z)-1-戊烯基)苯基〕丙烯酸(0.92g)。
IR(液体石蜡):1690,1680,1620cm-1
制备例36
将(E)-3-〔2-((Z)-1-戊烯基)苯基〕丙烯酸(1.08g)溶于二氯甲烷(10ml)、草酰氯(0.5ml)和N,N-二甲基甲酰胺(0.05ml)的混合物中,于室温氮氛下搅拌一小时,蒸发溶剂,残余物溶于正己烷,过滤混合物,蒸发滤液,得到(E)-3-〔2-((Z)-1-戊烯基)苯基〕丙烯酰氯化物(1.15g)。IR(纯):1750,1730,1605,1585cm-1NMR(CDCl3,δ):0.88(3H,t,J=6.5Hz),1.45(2H,
m),2.06(2H,m),5.95(1H,dt,J=11,7Hz),6.58
(1H,d,J=11Hz),6.66(1H,d,J=16Hz),7.4(3H,
m),7.69(1H,m),8.12(1H,d,J=16Hz)
实施例22按照实施例14的相似方法,制得下列化合物。
R-:3-(2-戊苯基)丙酰分子量:FAB-MS:m/z 1041.6(M+H)+
Claims (1)
1.制备下式所示的化合物的方法,所说之式为:
其中R是(E)-3-[2-((Z)-1-戊烯基)苯基]丙烯酰,而Me是甲基,
该方法包括在营养培养基中培养紫黑链霉菌No.9326 CCTCC No.M89201,并从产生的培养液中回收化合物。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB8807921.5 | 1988-04-05 | ||
| GB888807921A GB8807921D0 (en) | 1988-04-05 | 1988-04-05 | Ws-9326 & its derivatives |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN 96121791 Division CN1163271A (zh) | 1988-04-05 | 1996-11-30 | 肽衍生物的制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1036795A CN1036795A (zh) | 1989-11-01 |
| CN1037857C true CN1037857C (zh) | 1998-03-25 |
Family
ID=10634574
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN89102100A Expired - Fee Related CN1037857C (zh) | 1988-04-05 | 1989-04-04 | 利用紫黑链霉菌制备肽衍生物的方法 |
Country Status (20)
| Country | Link |
|---|---|
| EP (1) | EP0336230B1 (zh) |
| JP (1) | JP2551142B2 (zh) |
| KR (1) | KR0132567B1 (zh) |
| CN (1) | CN1037857C (zh) |
| AT (1) | ATE148890T1 (zh) |
| AU (1) | AU627453B2 (zh) |
| CA (1) | CA1335662C (zh) |
| DE (1) | DE68927759T2 (zh) |
| DK (1) | DK161789A (zh) |
| ES (1) | ES2097736T3 (zh) |
| FI (1) | FI102547B (zh) |
| GB (1) | GB8807921D0 (zh) |
| GR (1) | GR3023353T3 (zh) |
| HU (1) | HU204853B (zh) |
| IL (1) | IL89734A (zh) |
| NO (1) | NO177714C (zh) |
| PH (1) | PH30960A (zh) |
| RU (2) | RU1826970C (zh) |
| UA (1) | UA26845C2 (zh) |
| ZA (1) | ZA892188B (zh) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0360390A1 (en) * | 1988-07-25 | 1990-03-28 | Glaxo Group Limited | Spirolactam derivatives |
| CA2017156A1 (en) * | 1989-06-02 | 1990-12-02 | Sotoo Asakura | Preparation of fr115224 substance for parenteral administration |
| FR2666335B1 (fr) | 1990-09-05 | 1992-12-11 | Sanofi Sa | Arylalkylamines, procede pour leur preparation et compositions pharmaceutiques les contenant. |
| EP0498069B1 (en) * | 1990-12-21 | 1995-10-25 | Fujisawa Pharmaceutical Co., Ltd. | New use of peptide derivative |
| FR2688219B1 (fr) | 1992-03-03 | 1994-07-08 | Sanofi Elf | Sels d'ammonium quaternaires de composes aromatiques amines, leur preparation et compositions pharmaceutiques les contenant. |
| US5830854A (en) * | 1992-12-14 | 1998-11-03 | Merck Sharp & Dohme, Limited | Method of treating cystic fibrosis using a tachykinin receptor antagonist |
| WO1994020126A1 (en) * | 1993-03-03 | 1994-09-15 | Fujisawa Pharmaceutical Co., Ltd. | Use of peptides for the manufacture of a medicament |
| FR2728169A1 (fr) | 1994-12-19 | 1996-06-21 | Oreal | Utilisation d'un antagoniste de substance p pour le traitement des prurits et des dysesthesies oculaires ou palpebrales |
| FR2728166A1 (fr) | 1994-12-19 | 1996-06-21 | Oreal | Composition topique contenant un antagoniste de substance p |
| FR2728165A1 (fr) | 1994-12-19 | 1996-06-21 | Oreal | Utilisation d'un antagoniste de substance p pour le traitement des rougeurs cutanees d'origine neurogene |
| FR2729954B1 (fr) | 1995-01-30 | 1997-08-01 | Sanofi Sa | Composes heterocycliques substitues, procede pour leur preparation et compositions pharmaceutiques les contenant |
| FR2741262B1 (fr) | 1995-11-20 | 1999-03-05 | Oreal | Utilisation d'un antagoniste de tnf-alpha pour le traitement des rougeurs cutanees d'origine neurogene |
| US20100105688A1 (en) | 2007-01-24 | 2010-04-29 | Glaxo Group Limited | Pharmaceutical compositions comprising 3,5-diamino-6-(2,3-dichlophenyl)-1,2,4-triazine or r(-)-2,4-diamino-5-(2,3-dichlorophenyl)-6-fluoromethyl pyrimidine and an nk1 |
| UA105182C2 (ru) | 2008-07-03 | 2014-04-25 | Ньюрексон, Інк. | Бензоксазины, бензотиазины и родственные соединения, которые имеют ингибирующую nos активность |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4661473A (en) * | 1984-03-27 | 1987-04-28 | Merck & Co., Inc. | Renin inhibitors containing peptide isosteres |
| US4803261A (en) * | 1986-06-27 | 1989-02-07 | The Administrators Of The Tulane Educational Fund | Method for synthesizing a peptide containing a non-peptide |
| JPS6424156A (en) * | 1987-07-17 | 1989-01-26 | Yanmar Diesel Engine Co | Air-fuel ratio control device for gas engine |
| JP2570670B2 (ja) * | 1987-12-28 | 1997-01-08 | 味の素株式会社 | トキソプラズマ増殖抑制剤 |
| GB8802229D0 (en) * | 1988-02-02 | 1988-03-02 | Fujisawa Pharmaceutical Co | Ws-9326 & its derivatives |
-
1988
- 1988-04-05 GB GB888807921A patent/GB8807921D0/en active Pending
-
1989
- 1989-03-22 ZA ZA892188A patent/ZA892188B/xx unknown
- 1989-03-23 ES ES89105225T patent/ES2097736T3/es not_active Expired - Lifetime
- 1989-03-23 DE DE68927759T patent/DE68927759T2/de not_active Expired - Fee Related
- 1989-03-23 AT AT89105225T patent/ATE148890T1/de not_active IP Right Cessation
- 1989-03-23 EP EP89105225A patent/EP0336230B1/en not_active Expired - Lifetime
- 1989-03-24 IL IL8973489A patent/IL89734A/en not_active IP Right Cessation
- 1989-04-03 AU AU32397/89A patent/AU627453B2/en not_active Ceased
- 1989-04-03 FI FI891581A patent/FI102547B/fi not_active IP Right Cessation
- 1989-04-03 NO NO891389A patent/NO177714C/no not_active IP Right Cessation
- 1989-04-04 RU SU894613896A patent/RU1826970C/ru active
- 1989-04-04 CA CA000595640A patent/CA1335662C/en not_active Expired - Fee Related
- 1989-04-04 KR KR1019890004430A patent/KR0132567B1/ko not_active IP Right Cessation
- 1989-04-04 CN CN89102100A patent/CN1037857C/zh not_active Expired - Fee Related
- 1989-04-04 DK DK161789A patent/DK161789A/da not_active Application Discontinuation
- 1989-04-04 UA UA5010972A patent/UA26845C2/uk unknown
- 1989-04-04 PH PH38436A patent/PH30960A/en unknown
- 1989-04-05 HU HU891645A patent/HU204853B/hu not_active IP Right Cessation
- 1989-04-05 JP JP1086484A patent/JP2551142B2/ja not_active Expired - Fee Related
-
1992
- 1992-02-27 RU SU925010972A patent/RU2043365C1/ru active
-
1997
- 1997-05-09 GR GR970401005T patent/GR3023353T3/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1040541C (zh) | 新型多肽化合物的制备方法 | |
| CN1037857C (zh) | 利用紫黑链霉菌制备肽衍生物的方法 | |
| CN85109492A (zh) | 三环化合物,其生产方法以及含该化合物的药物组合物 | |
| CN1057306C (zh) | 新的多肽化合物及其制备方法 | |
| CN1039642C (zh) | 六氢化萘酯衍生物、它们的制备和它们在治疗方面的应用 | |
| CN1161462C (zh) | 环脂肽物的脱酰化方法 | |
| CN1148503A (zh) | 新的肽类化合物在制备药物中的应用 | |
| CN1040054A (zh) | 具有生物活性的新化合物及其制备方法 | |
| CN1041951A (zh) | 新的肽酶抑制剂 | |
| CN1845925A (zh) | 取代的杂环化合物 | |
| CN1051757A (zh) | 新的多肽化合物及其制备方法 | |
| CN1077892C (zh) | 噁唑酮衍生物及其作为抗幽门螺杆菌剂的应用 | |
| CN86101394A (zh) | 氨基酸衍生物的制备方法及其应用 | |
| CN1717493A (zh) | 大环内酯化合物的产生方法 | |
| CN1028530C (zh) | 大环内酯化合物的制备方法 | |
| CN86108977A (zh) | N-酰氨基葡糖苷酸糖苷配基抗菌素a40926族和糖苷配基抗菌素a40926 | |
| CN1886505A (zh) | 参与大环内酯类化合物的羟化作用的dna | |
| CN1013120B (zh) | A-21978c衍生物生产方法改进 | |
| CN1116304C (zh) | 有机化合物 | |
| CN1023758C (zh) | 含抗生素a10255复合物或其因子的饲料组合物 | |
| CN1183248C (zh) | 环状缩肽合成酶及其基因、以及环状缩肽的大规模生产系统 | |
| CN1034498C (zh) | 用于抑制胆固醇生物合成的八氢萘肟衍生物,其制备方法和用途 | |
| CN1043422C (zh) | 多羟基环戊烷衍生物的制备方法 | |
| CN1163271A (zh) | 肽衍生物的制备方法 | |
| CN1753996A (zh) | 参与番红菌素生物合成的基因簇及其用于遗传工程的用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C15 | Extension of patent right duration from 15 to 20 years for appl. with date before 31.12.1992 and still valid on 11.12.2001 (patent law change 1993) | ||
| OR01 | Other related matters | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |