CN101977908A - 用于治疗逆转录病毒性疾病的(吡唑基羰基)咪唑烷酮衍生物 - Google Patents
用于治疗逆转录病毒性疾病的(吡唑基羰基)咪唑烷酮衍生物 Download PDFInfo
- Publication number
- CN101977908A CN101977908A CN2009801095006A CN200980109500A CN101977908A CN 101977908 A CN101977908 A CN 101977908A CN 2009801095006 A CN2009801095006 A CN 2009801095006A CN 200980109500 A CN200980109500 A CN 200980109500A CN 101977908 A CN101977908 A CN 101977908A
- Authority
- CN
- China
- Prior art keywords
- group
- compound
- phenyl
- expression
- halogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 0 CCOC(C(C=C(c(cc1)cc(C(F)(F)F)c1F)O*)=O)=O Chemical compound CCOC(C(C=C(c(cc1)cc(C(F)(F)F)c1F)O*)=O)=O 0.000 description 4
- SMIPYIKAYBMFGJ-GHXNOFRVSA-N CCOC(C(/C=C(/c1cc(Cl)cc(F)c1)\OC)=O)=O Chemical compound CCOC(C(/C=C(/c1cc(Cl)cc(F)c1)\OC)=O)=O SMIPYIKAYBMFGJ-GHXNOFRVSA-N 0.000 description 1
- WYHACGJKOPKIBC-UHFFFAOYSA-N CCOC(c(cc1-c(cc2Br)ccc2F)n[n]1-c1cccc(Cl)c1)=O Chemical compound CCOC(c(cc1-c(cc2Br)ccc2F)n[n]1-c1cccc(Cl)c1)=O WYHACGJKOPKIBC-UHFFFAOYSA-N 0.000 description 1
- TYNSHDYALCZFSU-UHFFFAOYSA-N CCOC(c(cc1-c2cc(F)cc(Cl)c2)n[n]1-c1cccc(Cl)c1)=O Chemical compound CCOC(c(cc1-c2cc(F)cc(Cl)c2)n[n]1-c1cccc(Cl)c1)=O TYNSHDYALCZFSU-UHFFFAOYSA-N 0.000 description 1
- FLHCBDZGRGJMIN-UHFFFAOYSA-N CCOC(c(cc1-c2cc(OCCCCl)ccc2)n[n]1-c1cc(Cl)ccc1)=O Chemical compound CCOC(c(cc1-c2cc(OCCCCl)ccc2)n[n]1-c1cc(Cl)ccc1)=O FLHCBDZGRGJMIN-UHFFFAOYSA-N 0.000 description 1
- ARKNSCZOUWGECH-UHFFFAOYSA-N CCOC(c(cc1-c2cccc(Cl)c2)n[n]1-c(cc1)ccc1F)=O Chemical compound CCOC(c(cc1-c2cccc(Cl)c2)n[n]1-c(cc1)ccc1F)=O ARKNSCZOUWGECH-UHFFFAOYSA-N 0.000 description 1
- AOJASSRSABFEFH-UHFFFAOYSA-N CNCCOc1cccc(-c2cc(C(N(C3)CNC3=O)=O)n[n]2-c2cc(Cl)ccc2)c1 Chemical compound CNCCOc1cccc(-c2cc(C(N(C3)CNC3=O)=O)n[n]2-c2cc(Cl)ccc2)c1 AOJASSRSABFEFH-UHFFFAOYSA-N 0.000 description 1
- GKLNCHMRZYZYQF-UHFFFAOYSA-N COc1cc(-c2cc(C(N(C3)CNC3=O)=O)n[n]2-c2cc(Cl)ccc2)cc(F)c1 Chemical compound COc1cc(-c2cc(C(N(C3)CNC3=O)=O)n[n]2-c2cc(Cl)ccc2)cc(F)c1 GKLNCHMRZYZYQF-UHFFFAOYSA-N 0.000 description 1
- FIZMINOPVWQGJB-UHFFFAOYSA-N COc1cc(-c2cc(C(O)=O)n[n]2-c(cc2Cl)ccc2F)ccc1 Chemical compound COc1cc(-c2cc(C(O)=O)n[n]2-c(cc2Cl)ccc2F)ccc1 FIZMINOPVWQGJB-UHFFFAOYSA-N 0.000 description 1
- BNYGLLYSUWMRDC-UHFFFAOYSA-N N#Cc1cc(-c2cc(C(N(C3)CNC3=O)=O)n[n]2-c(cc2Cl)ccc2F)cc(F)c1 Chemical compound N#Cc1cc(-c2cc(C(N(C3)CNC3=O)=O)n[n]2-c(cc2Cl)ccc2F)cc(F)c1 BNYGLLYSUWMRDC-UHFFFAOYSA-N 0.000 description 1
- FWGZLQUABIOYGD-UHFFFAOYSA-N N#Cc1cc(-c2cc(C(N(C3)CNC3=O)=O)n[n]2-c2cc(Cl)ccc2)cc(F)c1 Chemical compound N#Cc1cc(-c2cc(C(N(C3)CNC3=O)=O)n[n]2-c2cc(Cl)ccc2)cc(F)c1 FWGZLQUABIOYGD-UHFFFAOYSA-N 0.000 description 1
- PHOGHKTZTACIIY-UHFFFAOYSA-N N#Cc1cc(F)cc(-c2cc(C(O)=O)n[n]2-c(cc2)cc(Cl)c2F)c1 Chemical compound N#Cc1cc(F)cc(-c2cc(C(O)=O)n[n]2-c(cc2)cc(Cl)c2F)c1 PHOGHKTZTACIIY-UHFFFAOYSA-N 0.000 description 1
- WHRNEZVRZLZKPX-UHFFFAOYSA-N N#Cc1cccc(-c2cc(C(N(C3)CNC3=O)=O)n[n]2-c(cc2C#N)ccc2F)c1 Chemical compound N#Cc1cccc(-c2cc(C(N(C3)CNC3=O)=O)n[n]2-c(cc2C#N)ccc2F)c1 WHRNEZVRZLZKPX-UHFFFAOYSA-N 0.000 description 1
- WEEVZVDJSICOSX-UHFFFAOYSA-N N#Cc1cccc(-c2cc(C(O)=O)n[n]2-c(cc2C#N)ccc2F)c1 Chemical compound N#Cc1cccc(-c2cc(C(O)=O)n[n]2-c(cc2C#N)ccc2F)c1 WEEVZVDJSICOSX-UHFFFAOYSA-N 0.000 description 1
- OZORSANHRYUQAH-UHFFFAOYSA-N O=C(c(cc1-c(cc2)cc(F)c2F)n[n]1-c1cc(Cl)ccc1)N(C1)CNC1=O Chemical compound O=C(c(cc1-c(cc2)cc(F)c2F)n[n]1-c1cc(Cl)ccc1)N(C1)CNC1=O OZORSANHRYUQAH-UHFFFAOYSA-N 0.000 description 1
- MMERGJUFQJJCSE-UHFFFAOYSA-N O=C(c(cc1-c(cccc2F)c2F)n[n]1-c1cc(Cl)ccc1)N(C1)CNC1=O Chemical compound O=C(c(cc1-c(cccc2F)c2F)n[n]1-c1cc(Cl)ccc1)N(C1)CNC1=O MMERGJUFQJJCSE-UHFFFAOYSA-N 0.000 description 1
- MAUDEIGOWBMZBR-UHFFFAOYSA-N O=C(c(cc1-c2cccc(C(F)(F)F)c2)n[n]1-c(cc1)ccc1Cl)N(C1)CNC1=O Chemical compound O=C(c(cc1-c2cccc(C(F)(F)F)c2)n[n]1-c(cc1)ccc1Cl)N(C1)CNC1=O MAUDEIGOWBMZBR-UHFFFAOYSA-N 0.000 description 1
- HFMWZQLSJYGSHJ-UHFFFAOYSA-N OC(c(cc1-c2cc(OCc3ccccc3)ccc2)n[n]1-c1cc(Cl)ccc1)=O Chemical compound OC(c(cc1-c2cc(OCc3ccccc3)ccc2)n[n]1-c1cc(Cl)ccc1)=O HFMWZQLSJYGSHJ-UHFFFAOYSA-N 0.000 description 1
- ZDKFJNLVOVYFBJ-UHFFFAOYSA-N OCCCOc1cccc(-c2cc(C(N(C3)CNC3=O)=O)n[n]2-c2cc(Cl)ccc2)c1 Chemical compound OCCCOc1cccc(-c2cc(C(N(C3)CNC3=O)=O)n[n]2-c2cc(Cl)ccc2)c1 ZDKFJNLVOVYFBJ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Virology (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Molecular Biology (AREA)
- AIDS & HIV (AREA)
- Tropical Medicine & Parasitology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
本发明涉及新的取代的(吡唑基羰基)咪唑烷酮,它们的制备方法,它们用于治疗和/或预防疾病的应用,以及它们用于制备药物的应用,所述药物用于治疗和/或预防疾病,尤其是在人和/或动物中的逆转录病毒性疾病。
Description
本发明涉及新的取代的(吡唑基羰基)咪唑烷酮,它们的制备方法,它们用于治疗和/或预防疾病的应用,以及它们用于制备用于治疗和/或预防疾病的药物的应用,所述疾病尤其是在人和/或动物中的反转录病毒性疾病。
HIV(人免疫缺陷病毒)导致慢性持续性进展的感染。所述疾病通过从无症状感染到病理性疾病AIDS(获得性免疫缺陷综合征)的各个阶段进展。AIDS是由感染导致的疾病的最后阶段。HIV/AIDS疾病特征在于具有持续性病毒血症的长期临床潜伏阶段,其在最后阶段导致免疫防御的失效。
引入抗-HIV组合疗法在20世纪90年代可以有效减缓疾病的进展并且由此导致显著延长HIV-感染患者的生命预期(Palella等,N.Engl.J.Med.1998,238,853-860)。
目前市售的抗-HIV药物通过抑制关键性的病毒酶逆转录酶(RT)、蛋白酶或整合酶,或抑制HIV进入靶细胞来抑制HI病毒的复制(在Flexner,Nature Reviews Drug Discovery(自然综述药物开发)2007,6,959-966中综述)。存在两类RT抑制剂:核苷和核苷酸RT抑制剂(NRTI)通过DNA聚合中的竞争性抑制或链终止作用起作用。非核苷RT抑制剂(NNRTI)变构性地结合于RT的活性中心邻近的疏水袋并且带来酶中的构象变化。目前获得的蛋白酶抑制剂(PI)封闭病毒蛋白酶的活性中心并由此防止新产生的颗粒成熟为感染性的病毒体。仅有的目前批准的整合酶抑制剂Raltegravir在HIV整合酶的活性中心中结合,并且防止原病毒DNA整合到宿主细胞基因组中。进入抑制剂(融合抑制剂和共同受体拮抗剂)通过与HIV外壳蛋白相互作用或通过阻断细胞共同受体CCR5或CXCR4来防止细胞的HIV感染。
由于抗性病毒的选择,用目前可获得的抗-HIV药物进行的单一疗法在极短的时间内导致治疗的失败,通常使用一些来自不同种类的抗-HIV药物进行组合疗法(highly active antiretroviral therapy(高度活性的抗逆转录病毒疗法)=HAART;Carpenter等,J.Am.Med.Assoc.2000,283,381-390)。
尽管在抗逆转录病毒化学疗法中取得进展,但是最近的研究显示用可获得的药物预期不能根除HIV,并且与其相关,不能治愈HIV感染。潜伏的病毒保持在休眠的淋巴细胞中,并且代表再激活的贮器,并因此用于病毒的重新传播(Finzi et al.,Nature Med.(自然医学)1999,5,512-517;Ramratnam等,Nature Med.(自然医学)2000,6,82-85)。HIV-感染的患者因此一生依赖于有效的抗病毒治疗。尽管有组合疗法,抗性病毒的选择在一些时间后发生。由于对于每种治疗种类的抗性突变特征积累,一种疗法的失败通常意味着整个类别的物质的效果的丧失。这种交叉抗性问题关于NNRTIs种类是最显著的,因为在该情形中,在RT中的单一点突变可能通常足以带来所有NNRTIs效果的丧失(在Kavlick&Mitsuya,Antiretroviral Chemotherapy(抗逆转录病毒化学疗法)(编者De Clercq E.)中综述,2001,ASM出版社,279-312)。
抗性的发展通常由患者的较差的顺应性促进,这是由副作用的不利性质和对于抗-HIV药物的复杂给药方案导致的。
因此存在对于控制HIV感染的新的治疗选择的迫切需要。为此目的,HIV疗法研究的迫切目的是鉴定新的化学先导结构,其涉及HIV复制中的一个新靶标和/或针对逐增数量的临床HIV抗性分离株是有效的。
US 5,624,941和EP 576357描述了作为大麻素受体拮抗剂的吡唑类,EP 418845,EP 554829和WO 04/050632将其特别用于治疗炎性和血栓疾病,WO 03/037274将其作为钠离子通道抑制剂用于治疗疼痛,WO06/015860将其作为腺苷受体配体,用于治疗炎性和阻塞性呼吸系统疾病,EP 1762568和EP 1591443将其作为血小板聚集的抑制剂,WO 07/002559将其作为核受体活性的调节剂,WO 07/020388和WO 05/080343将其作为大麻素受体调节剂,特别用于治疗肥胖症和精神和神经疾病,WO07/009701和EP 1743637将其用于治疗心血管危险因子,WO 2005/002576将其作为各种激酶的抑制剂,DE 102004054666将其用于控制有害植物或用于植物生长调节。
因此,本发明的一个目的是提供具有相同或改善的抗病毒活性的新化合物,用于治疗人和动物中的病毒感染疾病,所述人和动物不具有前述的缺陷。
已经令人惊奇地发现本发明中所述的取代的(吡唑基羰基)咪唑烷酮类具有抗病毒活性。
本发明涉及下式的化合物

其中
R1表示苯基,
其中苯基被1-3个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,羟基,氰基,硝基,三氟甲基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基和(C1-C4)-烷氧基,
其中
(C1-C4)-烷基和(C1-C4)-烷氧基又可以被选自以下系列的原子团相同或不同地取代1-3次:卤素,氰基,羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基,(C3-C7)-环烷基和4-到7-元杂环基,
其中最后提及的环烷基和杂环基原子团每个又可以相同地或不同地被卤素,氰基,(C1-C4)-烷基,三氟甲基,羟基,(C1-C4)-烷氧基,三氟甲氧基,氧代,氨基,单-(C1-C4)-烷基氨基和二-(C1-C4)-烷基氨基取代至多3次,
R2表示苯基,
其中苯基被1-3个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,羟基,氰基,硝基,三氟甲基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基和(C1-C4)-烷氧基,
其中
(C1-C4)-烷基和(C1-C4)-烷氧基又可以被选自以下系列的原子团相同地或不同地取代1-3次:卤素,氰基,羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基,(C3-C7)-环烷基和4-7元杂环基,
其中最后提及的环烷基和杂环基原子团又可以分别相同地或不同地被卤素,氰基,(C1-C4)-烷基,三氟甲基,羟基,(C1-C4)-烷氧基,三氟甲氧基,氧代,氨基,单-(C1-C4)-烷基氨基和二-(C1-C4)-烷基氨基取代至多3次,
或其盐,其溶剂合物或其盐的溶剂合物。
本发明的化合物是式(I)的化合物及其盐、溶剂合物及其盐的溶剂合物;以及作为示例性实施方案的在下文提及的式(I)涵盖的化合物及其盐、溶剂合物及其盐的溶剂合物,如果由式(I)所涵盖并且在下文提及的化合物还不是盐、溶剂合物和所述盐的溶剂合物。
取决于它们的结构,本发明的化合物以立体异构型(对映异构体,非对映异构体)存在。因此,本发明还包括对映异构体或非对映异构体及其各自的混合物。从对映异构体和/或非对映异构体的这些混合物中,可以以已知方式分离立体异构纯的组分。
如果本发明的化合物可以以互变异构形式存在,本发明包括所有的互变异构形式。
优选用于本发明的目的盐是本发明的化合物的生理可接受的盐。然而,本身不适合于药用但是可以用于例如分离或纯化本发明的化合物的盐也被包括在内。
本发明的化合物的生理可接受的盐包括无机酸、羧酸和磺酸的酸加成盐,例如盐酸、氢溴酸、硫酸、磷酸、甲磺酸、乙磺酸、甲苯磺酸、苯磺酸、萘二磺酸、乙酸、三氟乙酸、丙酸、乳酸、酒石酸、苹果酸、柠檬酸、富马酸、马来酸和苯甲酸的盐。
本发明的化合物的生理可接受的盐还包括常用的碱的盐,诸如,作为实例,并且优选地碱金属盐(例如,钠和钾盐),碱土金属盐(例如钙和镁盐)和衍生自氨或具有1-16个碳原子的有机胺的铵盐,所述有机胺诸如作为实例,并且优选地,乙胺、二乙胺、三乙胺、乙基二异丙胺、单乙醇胺、二乙醇胺、三乙醇胺、二环己胺、二甲基氨基乙醇、普鲁卡因、二苄基胺、N-甲基吗啉、精氨酸、赖氨酸、乙二胺和N-甲基哌啶。
对于本发明的目的而言,溶剂合物指通过与溶剂分子的配位作用形成固体或液体状态的复合物的本发明的化合物的那些形式。水合物是溶剂合物的特殊形式,其中配位作用与水发生。
对于本发明的目的而言,除非另外指出,取代基具有下列含义:
烷基以及在烷氧基和烷氧基羰基中的烷基部分表示直链或支链烷基,除非另外指出,包括(C1-C6)-烷基,特别是(C1-C4)-烷基,诸如例如甲基,乙基,丙基,异丙基,丁基,异丁基,叔丁基。
对于本发明的目的,烷氧基优选地表示直链或支链烷氧基原子团,特别是具有1-6个,1-4个或1-3个碳原子的烷氧基。优选具有1-3个碳原子的直链或支链烷氧基原子团。举例和优选地是甲氧基,乙氧基,正丙氧基,异丙氧基,叔丁氧基,正戊氧基和正己氧基。
烷氧基羰基举例并且优选地表示甲氧基羰基,乙氧基羰基,正丙氧基羰基,异丙氧基羰基,叔丁氧基羰基,正戊氧基羰基和正己氧基羰基。
杂环基表示这样的单环杂环原子团,其具有4-7个,优选地5-6个环原子和至多3个,优选地至多2个来自系列N,O,S,SO,SO2的杂原子和/或杂基团,其中氮原子还可以形成N-氧化物。所述杂环可以是饱和的或部分不饱和的。优选具有来自系列O,N和S的多达2个杂原子的5-到7-元、单环饱和的杂环,举例并且优选地是1,4-氧氮杂环庚烷基,吡咯烷-1-基,吡咯烷-2-基,吡咯烷-3-基,四氢呋喃基,四氢噻吩基,吡喃基,1,3-噻唑烷基,哌啶-1-基,哌啶-2-基,哌啶-3-基,哌啶-4-基,噻喃基,吗啉-2-基,吗啉-3-基,吗啉-4-基,硫代吗啉-2-基,硫代吗啉-3-基,硫代吗啉-4-基,全氢化氮杂 基,哌嗪-1-基,哌嗪-2-基。
基,哌嗪-1-基,哌嗪-2-基。
卤素表示氟,氯,溴或碘,除非另外指出,其中优选氟和氯。
单-(C 1 -C 4 )-烷基氨基,用于本发明时,表示具有包括1-4个碳原子的 直链或支链烷基取代基的氨基基团。例如,并且优选地,可以提及甲基氨基,乙基氨基,正丙基氨基,异丙基氨基,正丁基氨基,叔丁基氨基,正戊基氨基和正己基氨基。
二-(C 1 -C 4 )-烷基氨基,用于本发明时,代表具有两个相同或不同分别 包括1-4个碳原子的直链或支链烷基取代基的氨基基团。例如,并且优选 地:可以提及N,N-二甲基氨基,N,N-二乙基氨基,N-乙基-N-甲基氨基,N-甲基-N-正丙基氨基,N-异丙基-N-正丙基氨基,N,N-二异丙基氨基,N-正丁基-N-甲基氨基,N-叔丁基-N-甲基氨基,N-甲基-N-正戊基氨基和N-正己基-N-甲基氨基。
(C 3 -C 7 )-环烷基用于本发明的目的时,代表具有3-7个或3-6个环碳原 子的单环饱和碳环。可以提及,例如并且优选地,环丙基、环丁基、环戊基、环己基和环庚基。
上述给出的原子团定义一般或在优选的范围内应用于式(I)的最终产物并且在每种情形中应用于制备所需的相应的原材料和中间体。
在原子团的各个组合和优选组合中的具体指出的原子团的定义,不管各自指定的原子团组合如何,也被其它组合的原子团的定义所任意替代。
本发明还涉及式(I)的化合物,其中
R1表示苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,羟基,氰基,硝基,三氟甲基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基和(C1-C4)-烷氧基,
R2表示苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,羟基,氰基,硝基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基和(C1-C4)-烷氧基,
其中
(C1-C4)-烷氧基又可以被选自以下系列的原子团相同地或不同地取代1-3次:卤素,氰基,羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基,(C3-C7)-环烷基和4-7元杂环基,
其中最后提及的环烷基和杂环基原子团又可以分别被卤素,氰基,(C1-C4)-烷基,三氟甲基,羟基,(C1-C4)-烷氧基,三氟甲氧基,氧代,氨基,单-(C1-C4)-烷基氨基和二-(C1-C4)-烷基氨基相同地或不同地取代至多3次,
或其盐、其溶剂合物和其盐的溶剂合物。
本发明还涉及式(I)的化合物,其中
R1表示苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,氰基,三氟甲基和甲氧基,
R2表示苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,氰基,三氟甲氧基,甲基和(C1-C3)-烷氧基,其中
(C1-C3)-烷氧基又可以由选自以下系列的原子团相同地或不同地取代1-3次:羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基和4-7元杂环基,
其中最后提及的杂环基基团又可以分别被(C1-C4)-烷基取代,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及式(I)的化合物,其中
R1表示苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,氰基,三氟甲基和甲氧基,
R2表示苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,氰基,三氟甲氧基,甲基和甲氧基,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及式(I)的化合物,其中
R1表示苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素和氰基,
R2表示苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素和氰基,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及下式的化合物:

其中
R3表示氢,卤素,羟基,氰基,硝基,三氟甲基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基或(C1-C4)-烷氧基,
R4表示氢或卤素,
R5表示卤素,羟基,氰基,硝基,三氟甲基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基或(C1-C4)-烷氧基,
其中
(C1-C4)-烷氧基又可以被选自以下系列的原子团相同地或不同地取代1-3次:卤素,氰基,羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基,(C3-C7)-环烷基和4-7元杂环基,
其中最后提及的环烷基和杂环基原子团又可以分别被卤素,氰基,(C1-C4)-烷基,三氟甲基,羟基,(C1-C4)-烷氧基,三氟甲氧基,氧代,氨基,单-(C1-C4)-烷基氨基和二-(C1-C4)-烷基氨基相同地或不同地取代至多3次,
R6表示氢或卤素,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及式(Ia)的化合物,其中
R3表示卤素,羟基,氰基,硝基,三氟甲基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基或(C1-C4)-烷氧基,
R4表示氢或卤素,
R5表示卤素,羟基,氰基,硝基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基或(C1-C4)-烷氧基,
其中
(C1-C4)-烷氧基又可以被选自以下系列的原子团相同地或不同地取代1-3次:卤素,氰基,羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基,(C3-C7)-环烷基和4-7元杂环基,
其中最后提及的环烷基和杂环基原子团又可以分别被卤素,氰基,(C1-C4)-烷基,三氟甲基,羟基,(C1-C4)-烷氧基,三氟甲氧基,氧代,氨基,单-(C1-C4)-烷基氨基和二-(C1-C4)-烷基氨基相同地或不同地取代至多3次,
R6表示氢或卤素,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及式(Ia)的化合物,其中
R3表示卤素,氰基,三氟甲基或甲氧基,
R4表示氢或卤素,
R5表示卤素,氰基,三氟甲氧基,甲基或(C1-C3)-烷氧基,
其中
(C1-C3)-烷氧基又可以被选自以下系列的原子团相同地或不同地取代1-3次:羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基和4-7元杂环基,
其中最后提及的杂环基原子团又可以分别被(C1-C4)-烷基取代,
R6表示氢或卤素,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及式(Ia)的化合物,其中
R3表示卤素,氰基,三氟甲基或甲氧基,
R4表示氢,氯或氟,
R5表示卤素,氰基,三氟甲氧基,甲基或甲氧基,
R6表示氢,氯或氟,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及式(Ia)的化合物,其中
R3表示卤素或氰基,
R4表示氢或氟,
R5表示卤素或氰基,
R6表示氢或氟,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及式(Ia)的化合物,其中
R3表示氯或氰基,
R4表示氟,
R5表示氯或氰基,
R6表示氟,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及式(Ia)的化合物,其中
R3表示氯或氰基,
R4表示氟,
R5表示氯或氰基,
R6表示氢,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及式(Ia)的化合物,其中
R3表示氯或氰基,
R4表示氢,
R5表示氯或氰基,
R6表示氢,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及式(Ia)的化合物,其中
R3表示氯或氰基,
R4表示氢,
R5表示氯或氰基,
R6表示氟,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及式(Ia)的化合物,其中
R3表示卤素,氰基,三氟甲基或甲氧基,
R4表示氢或卤素,
R5表示三氟甲基,
R6表示氟,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及式(Ia)的化合物,其中
R3表示氢,
R4表示氟或氯,
R5表示卤素,氰基,三氟甲氧基,甲基或甲氧基,
R6表示氢或卤素,
或其盐,其溶剂合物和其盐的溶剂合物。
本发明还涉及用于制备式(I)和(Ia)的化合物的方法,其中使下式的化合物

其中
R1和R2如上指定的含义,
与咪唑烷-4-酮或咪唑烷-4-酮的盐反应。
反应通常在惰性溶剂中,在存在脱水剂的情况下发生,当适合时,在存在碱,优选在-30℃至50℃的温度范围内,在大气压下发生。
惰性溶剂的实例是卤代烃,诸如二氯甲烷或三氯甲烷,烃如苯或甲苯,硝基甲烷,四氢呋喃,1,4-二噁烷,二甲基甲酰胺或乙腈。类似地,可以使用溶剂的混合物。特别优选,二氯甲烷,二甲基甲酰胺,四氢呋喃或甲苯。
碱的实例是碱金属碳酸盐,如例如,碳酸钠或碳酸钾,或碳酸氢钠或碳酸氢钾,或有机碱如三烷基胺,例如三乙胺,N-甲基吗啉,N-甲基哌啶,4-二甲基氨基吡啶或二异丙基乙胺。
本文的适合的脱水剂的实例包括碳二亚胺诸如,例如N,N′-二乙基-,N,N,′-二丙基-,N,N′-二异丙基-,N,N′-二环己基碳二亚胺,N-(3-二甲基氨基异丙基)-N′-乙基碳二亚胺盐酸盐(EDC),N-环己基碳二亚胺-N‘-丙氧基甲基-聚苯乙烯(PS-碳二亚胺)或羰基化合物诸如羰二咪唑,或1,2-噁唑鎓(oxazolium)化合物诸如2-乙基-5-苯基-1,2-噁唑鎓3-硫酸盐或2-叔-丁基-5-甲基异噁唑鎓高氯酸盐,或酰基氨基化合物诸如2-乙氧基-1-乙氧基羰基-1,2-二氢喹啉,或丙烷膦酸酐,或氯甲酸异丁酯,或二(2-氧代-3-噁唑烷基)磷酰氯,或O-(苯并三唑-1-基)-N,N,N′,N′-四甲基脲鎓六氟磷酸盐(HBTU),2-(2-氧代-1-(2H)-吡啶基)-1,1,3,3-四甲基脲鎓四氟硼酸盐(TPTU)或O-(7-氮杂苯并三唑-1-基)-N,N,N′,N′-四甲基脲鎓六氟磷酸盐(HATU),或1-羟基苯并三唑(HOBt)或苯并三唑-1-基氧基三-(二甲基氨基)六氟膦酸鏻(BOP),或苯并三唑-1-基氧基三(吡咯烷子基)六氟膦酸鏻(PyBOP),或N-羟基琥珀酰亚胺,或这些与碱的混合物。
缩合优选地用PyBOP,TBTU,或用EDC,在HOBt存在的情况下进行。
在备选方法中,式(II)的化合物最初可以与亚硫酰氯反应,并且在第二个阶段与咪唑烷-4-酮或咪唑烷-4-酮的盐,在存在碱如,例如三乙胺的情况下反应。
通过上述方法制备的式(I)和(Ia)的化合物携带保护基,如果合适,其可以在本领域技术人员已知的条件下去除从而获得式(I)和(Ia)的另外的化合物。
式(II)的化合物是已知的,或可以通过用碱来水解下式的化合物中的酯进行制备:
其中
R1和R2具有上述指定的含义。
用碱来水解酯通常发生在惰性溶剂中,优选地在从室温到溶剂回流的温度范围内,在大气压下进行。
碱的实例是碱金属氢氧化物,如氢氧化钠、氢氧化锂或氢氧化钾,或碱金属碳酸盐如碳酸铯、碳酸钠或碳酸钾,优选氢氧化锂,氢氧化钾或氢氧化钠。
惰性溶剂的实例是卤代烃,如二氯甲烷,三氯甲烷,四氯甲烷,三氯乙烷,四氯乙烷,1,2-二氯乙烷或三氯乙烯,醚如二乙醚,甲基叔丁醚,1,2-二甲氧基乙烷,1,4-二噁烷,四氢呋喃,乙二醇二甲醚或二甘醇二甲醚,醇如甲醇,乙醇,正丙醇,异丙醇,正丁醇或叔丁醇,烃如苯,二甲苯,甲苯,己烷,环己烷或石油馏分,或其它溶剂如二甲基甲酰胺,二甲基乙酰胺,二甲亚砜,乙腈或吡啶,或水,或溶剂的混合物。优选的溶剂是1,4-二噁烷,四氢呋喃和/或甲醇。优选在四氢呋喃中的氢氧化锂或在甲醇中的1,4-二噁烷-水混合物或在甲醇中的氢氧化钾。
已知式(III)的化合物或可以通过在第一阶段使下式的化合物

其中
R2具有上面指定的含义,
与下式的化合物
R1-NH-NH2(V),
或式(V)的化合物的盐反应,
其中
R1具有上面指定的含义,
并且在第二阶段中在乙酸中加热来进行制备。
第一阶段中的反应通常在惰性溶剂中发生,优选在从室温到溶剂的回流的温度范围内,在大气压下发生。
惰性溶剂的实例是醇如甲醇、乙醇、正丙醇、异丙醇、正丁醇、叔丁醇或2-甲氧基乙醇,优选乙醇。
在乙酸中的第二个阶段的反应通常在从室温到乙酸回流的温度范围内,在大气压下发生。反应还可以在甲醇、乙醇或二噁烷中,在从室温到溶剂的回流的温度范围内进行。甲醇、乙醇或二噁烷与乙酸以0.5/99.5-99.5/0.5的体积比率的混合物是适合的。还可以在提及的条件下使用甲醇、乙醇、二噁烷或乙酸与其它酸的混合物,所述酸如,例如盐酸、甲磺酸、对甲苯磺酸、樟脑磺酸或三氟乙酸。反应优选地在乙酸,在回流下进行。
备选地,式(III)的化合物可以通过使下式的化合物

其中R2具有上面指定的含义,
与式(V)的化合物反应而制备。
反应通常在惰性溶剂中,优选地在从室温到溶剂的回流的温度范围内,在大气压下发生,如果合适,在酸的存在下发生。
惰性溶剂的实例是醇如甲醇、乙醇、正丙醇、异丙醇、正丁醇、叔丁醇或2-甲氧基乙醇,或其它溶剂如N,N-二甲基甲酰胺,N,N-二甲基乙酰胺或二甲亚砜。
如果反应在酸的存在下发生,可以将其从开始或在1-4小时的时间后加入反应溶液中,其中在后一种加入酸的情形中,如果合适,将反应溶液加热到达到溶剂的回流的温度。
酸例如是浓的无机酸,或浓羧酸,如例如浓盐酸、浓硝酸、浓硫酸或浓乙酸。
已知式(IV),(V)和(VI)的化合物,或可以通过已知方法从相应的原材料合成。
可以通过举例,通过下列合成方案来举例说明本发明化合物的制备。
合成方案:

本发明的化合物显示原先不能预测的有价值的药理学活性谱。
它们因此适用于用作治疗和/或预防人和动物疾病的药物。
本发明的化合物特征特别在于有利的抗逆转录病毒活性谱。
本发明还涉及本发明的化合物用于治疗和/或预防由逆转录病毒,特别是HI病毒导致的疾病的应用。
本发明还涉及本发明的化合物用于治疗和/或预防疾病,特别是前面提及的疾病的应用。
本发明还涉及本发明的化合物用于制备药物的应用,所述药物用于治疗和/或预防疾病,特别是前面提及的疾病。
本发明还涉及使用治疗有效量的本发明的化合物治疗和/或预防疾病的方法,特别是前面提及的疾病的方法。
可以提及的在人类医学中的适应症范围举例为:
1.)治疗和预防人逆转录病毒感染。
2.)用于治疗和预防HIV-1(人类免疫缺陷病毒;以前称为HTLV III/LAV)和HIV-2诱导的感染和疾病(AIDS)和与之相关的阶段如ARC(AIDS相关综合征)和LAS(淋巴结病综合征)和由该病毒导致的免疫缺陷和脑病。
3.)治疗由单,多或多重耐药HI病毒导致的HIV感染。
表述抗性HI病毒是指例如具有针对核苷抑制剂(NRTI)、非核苷抑制剂(NNRTI)或蛋白酶-抑制剂(PI)的抗性的病毒,或具有针对其它作用原理、例如T20(融合抑制剂)的抗性的病毒。
4.)用于治疗或预防AIDS携带状态。
5.)治疗或预防HTLV-I或HTLV-II感染。
可以提及的在兽医医药中的适应症举例为:
关于下列的感染:
a)羊进行性间质肺炎(在绵羊和山羊中)
b)进行性肺炎病毒(PPV)(在绵羊和山羊中)
c)山羊关节炎脑炎病毒(在绵羊和山羊中)
d)Zwoegerziekte病毒(在绵羊中)
e)感染性贫血病毒(马的)
f)由猫白血病毒导致的感染
g)由猫免疫缺陷病毒(FIV)导致的感染
h)由猿猴免疫缺陷病毒(SIV)导致的感染
上述列出的第2,3和4点在人类医学的适应症范围内是优选的。
所述物质特别适合于控制这样的HI病毒,其显示对于逆转录酶的已知非核苷抑制剂,如例如,依法韦仑或奈韦拉平的抗性。
本发明还涉及包含至少一种本发明的化合物和至少一种或多种另外的活性物质的药物,特别用于治疗和/或预防前面提及的疾病。
本发明的化合物还可以作为联合疗法中的成分与一种或多种在这些治疗领域内具有活性的其它化合物有利地用于,特别是用于上面列出的第2,3和4点。例如,这些化合物可以与有效剂量的抗病毒活性物质组合使用,所述抗病毒活性物质基于下面列出的作用原理:
HIV蛋白酶抑制剂;可以提及的例子有沙奎那韦,茚地那韦,利托那韦,那非那韦,安泼那韦,洛匹那韦,阿扎那韦,呋山那韦,替拉那韦,达芦那韦;
HIV逆转录酶的核苷、核苷酸和非核苷抑制剂;可以提及的例子有齐多夫定,拉米夫定,去羟肌苷,扎西他滨(zalzitabin),司他夫定,拉米夫定,阿巴卡韦,替诺福韦,阿德福韦,恩曲他滨,氨多索韦,apricitabine,racivir,奈韦拉平,地拉韦啶,依法韦仑,依曲韦林(etravirin),利匹韦林(rilpivirin),UK-453,061;
HIV整合酶抑制剂,可以提及的例子有:raltegravir,elvitegravir;
HIV融合抑制剂:可以提及的例子有:恩夫韦肽(enfuvirtide);
CXCR4/CCR5/gp120相互作用的抑制剂;可以提及的例子有:马拉韦罗、维立韦罗、INCB009471、AMD-070;
多蛋白突变的抑制剂;可以提及的实例是bevirimat。
这些选择是为了举例说明可能的组合,而不限于本文列出的实例;原则上,应该将本发明的化合物与抗病毒活性物质的每种组合考虑在本发明的范围内。
本发明的化合物可以具有全身性和/或局部效果。出于该目的,它们可以以适合的方式进行施用,所述方式诸如,例如通过口服的、肠胃外的、肺的、鼻的、舌下的、舌的、颊的、直肠的、皮肤的、透皮的、结膜的、耳的途径,或作为植入物或斯滕特固定模。
对于这些给药途径而言,本发明的化合物可以以适合的给药形式进行施用。
适合于口服给药的是这样的给药形式,其按照现有技术发挥功能并且快速地和/或以改进的方式释放本发明的化合物,并且其包括晶体形式和/或非晶形形式和/或溶解形式的本发明的化合物,例如片剂(无涂层的或包衣片剂,例如具有这样的包衣,所述包衣对胃液具有抗性或延缓溶解或是不溶的并且控制本发明的化合物的释放),快速在口腔中崩解的片剂或薄膜/糯米纸囊剂,薄膜/冻干物(lyophilisates),胶囊(例如硬或软明胶胶囊),糖衣片剂,颗粒剂,微型药片,粉末,乳剂,混悬剂,气溶胶或溶液。
肠胃外给药的进行可以避免吸收步骤(例如,静脉内、动脉内、心脏内、脊髓内或腰椎内),或包含吸收(例如,肌内、皮下、皮内、经皮或腹膜内)。适合于肠胃外给药的给药形式特别是,用于注射和输注的,以溶液、混悬液、乳剂、冻干物或无菌粉末存在的制剂。
适合于其它给药途径的实例是用于吸入的药物形式(特别是粉末吸气器,喷雾器),滴鼻剂,溶液,喷雾剂;通过舌、舌下或颊施用的片剂,薄膜/糯米纸囊剂或胶囊;栓剂,用于耳或眼的制剂,阴道胶囊,水性混悬液(洗剂,振摇混合物),亲脂性混悬液,软膏剂,乳膏剂,透皮治疗系统(例如膏药),乳,糊剂,泡沫,扑粉,植入物或斯滕特固定模。
可以将本发明的化合物转化成为列出的给药形式。其可以以本身已知的方式通过混合以惰性、非毒性的药用的赋形剂来进行。这些赋形剂包括,特别是,载体(例如微晶纤维素,乳糖,甘露醇),溶剂(例如液体聚乙二醇),乳化剂和分散剂或湿润剂(例如十二烷基硫酸钠,聚氧基失水山梨糖醇油酸酯),粘合剂(例如聚乙烯吡咯烷酮),合成的和天然的聚合物(例如白蛋白),稳定剂(例如抗氧化剂诸如例如抗坏血酸),颜料(例如无机颜料诸如例如氧化铁)和味道和/或气味矫正剂。
本发明还涉及这样的药物和将其用于前述目的的应用,所述药物包括至少一种本发明的化合物,以及通常地一种或多种惰性、非毒性药用的赋形剂。
通常,在人和兽医药物中,以每24小时0.1到200mg/kg,优选地1到100mg/kg体重的总量施用本发明的活性成分从而获得需要的效果已经证实是有利的,如果适合时,所述量以数次单剂量的形式施用。单剂量优选包含1到80mg/kg的量的活性成分,特别是1到30mg/kg体重。
然而,可能需要的是,当需要时偏离提及的量,其具体地取决于体重、给药途径、对活性成分的个体反应、制剂的性质和在施用发生过程中的时间或间隔。因此,在某些情形中以少于前述最少量进行的施用可以是充分的,而在其它情形中,必须超过提及的上限。在更大量的施用的情形中,可以推荐将这些分成一天内的多个单独剂量。
在下述测试和实施例中的百分比数据是重量百分比,除非另外指出;份是重量份。液体/液体溶液的溶剂比率、稀释比率和浓缩比率在每种情形中基于体积。陈述“w/v”意为“重量/体积”。因此,例如″10%w/v″意为100ml的溶液或混悬液包含10g物质。
A)实施例
缩写:
aq. 水性,水溶液
conc. 浓缩的
DCI 直接化学电离子化(在MS中)
DMA N,N-二甲基乙酰胺
DMF N,N-二甲基甲酰胺
DMSO 二甲亚砜
EDC N′-(3-二甲基氨基丙基)-N-乙基碳二亚胺x HCl
eq. 当量
ESI 电喷射离子化(在MS中)
h 小时
HATU O-(7-氮杂苯并三唑-1-基)-N,N,N′,N′-四甲基脲鎓六
氟磷酸盐
HPLC 高压高性能液相色谱
LC-MS 偶联的液相色谱-质谱
min 分钟
MS 质谱
NMR 核磁共振分光检定法
PyBOP 苯并三唑-1-基氧基三(吡咯烷子基)磷鎓六氟磷酸
盐
Rt 停留时间(在HPLC中)
RT 室温
TBTU O-(苯并三唑-1-基)-N,N,N′,N′-四甲基脲鎓四氟硼酸
盐
TFA 三氟乙酸
THF 四氢呋喃
TMOF 原甲酸三甲酯
LC-MS/GC-MS方法:
方法1:
MS仪器类型:Micromass ZQ;HPLC仪器类型:HP 1100系列;UV DAD;柱:Phenomenex Gemini 3μ 30mmx3.00mm;洗脱剂A:1l的水+0.5ml的50%甲酸,洗脱剂B:1l乙腈+0.5ml的50%甲酸;梯度:0.0min 90%A→2.5min 30%A→3.0min 5%A→4.5min 5%A;流速:0.0min 1ml/min,2.5min/3.0min/4.5min 2ml/min;烘箱:50℃;UV检测:210nm.
方法2:
仪器:Micromass Quattro LCZ具有HPLC安捷伦系列1100;柱:Phenomenex Synergi 2μHydro-RP Mercury 20mm x 4mm;洗脱剂A:1 l水+0.5ml 50%甲酸,洗脱剂B:1l乙腈+0.5ml 50%甲酸;梯度:0.0min 90%A→2.5min 30%A→3.0min 5%A→4.5min 5%A;流速:0.0min 1ml/min→2.5min/3.0min/4.5min 2ml/min;烘箱:50℃;UV检测:208-400nm.
方法3:
MS仪器类型:Micromass ZQ;HPLC仪器类型:Waters Alliance 2795;柱:Phenomenex Synergi 2μHydro-RP Mercury 20mm x 4mm;洗脱剂A:1 l水+0.5ml 50%甲酸,洗脱剂B:1 l乙腈+0.5ml 50%甲酸;梯度:0.0min 90%A→2.5min 30%A→3.0min 5%A →4.5min 5%A;流速:0.0min 1ml/min→2.5min/3.0min/4.5min 2ml/min;烘箱:50℃;UV检测:210nm.
方法4:
仪器:Micromass Quattro LCZ具有HPLC安捷伦系列1100;柱:Phenomenex Onyx Monolithic C18,100mm x 3mm;洗脱剂A:1l水+0.5ml 50%甲酸,洗脱剂B:1l乙腈+0.5ml 50%甲酸;梯度:0.0min90%A→2min 65%A→4.5min 5%A→6min 5%A;流速:2ml/min;烘箱:40℃;UV检测:208-400nm.
方法5:
仪器:Micromass QuattroPremier具有Waters UPLC Acquity;柱:Thermo Hypersil GOLD 1.9μ50mm x 1mm;洗脱剂A:1 l水+0.5ml 50%甲酸,洗脱剂B:1 l乙腈+0.5ml 50%甲酸;梯度:0.0min 90%A→0.1min90%A→1.5min 10%A→2.2min 10%A;烘箱:50℃;流速:0.33ml/min;UV检测:210nm.
方法6:
MS仪器类型:Waters ZQ;HPLC仪器类型:Waters Alliance 2795;柱:Phenomenex Onyx Monolithic C18,100mm x 3mm;洗脱剂A:1 l水+0.5ml 50%甲酸,洗脱剂B:1 l乙腈+0.5ml 50%甲酸;梯度:0.0min 90%A→2min 65%A→4.5min 5%A→6min 5%A;流速:2ml/min;烘箱:40℃;UV检测:210nm.
方法7:
MS仪器类型:Micromass ZQ;HPLC仪器类型:Waters Alliance 2795;柱:Phenomenex Synergi 2.5μMAX-RP 100A Mercury 20mm x 4mm;洗脱剂A:1l水+0.5ml 50%甲酸,洗脱剂B:1l乙腈+0.5ml 50%甲酸;梯度:0.0min 90%A→0.1min 90%A→3.0min 5%A→4.0min 5%A→4.01min90%A;流速:2ml/min;烘箱:50℃;UV检测:210nm.
方法8:
仪器:Micromass Quattro LCZ具有HPLC安捷伦系列1100;柱:Phenomenex Gemini 3μ30mm x 3.00mm;洗脱剂A:1l水+0.5ml 50%甲酸,洗脱剂B:1l乙腈+0.5ml 50%甲酸;梯度:0.0min 90%A→2.5min 30%A→3.0min 5%A→4.5min 5%A;流速:0.0min 1ml/min,2.5min/3.0min/4.5min 2ml/min;烘箱:50℃;UV检测:208-400nm.
方法9:
仪器:Micromass Quattro LCZ具有HPLC安捷伦系列1100;柱:Phenomenex Synergi 2.5μMAX-RP 100A Mercury 20mm x 4mm;洗脱剂A:1l水+0.5ml 50%甲酸,洗脱剂B:1l乙腈+0.5ml 50%甲酸;梯度:0.0min 90%A→0.1min 90%A→3.0min 5%A→4.0min 5%A→4.1min90%A;流速:2ml/min;烘箱:50℃;UV检测:208-400nm.
方法10:
MS仪器类型:Waters(Micromass)Quattro Micro;HPLC仪器类型:安捷伦1100系列;柱:Thermo Hypersil GOLD 3μ20mm x 4mm;洗脱剂A:1l水+0.5ml 50%甲酸,洗脱剂B:1l乙腈+0.5ml 50%甲酸;梯度:0.0min100%A→3.0min 10%A→4.0min 10%A→4.01min 100%A (流速2.5ml/min)→5.00min 100%A;烘箱:50℃;流速:2ml/min;UV检测:210nm.
方法11:
仪器:Micromass GCT,GC6890;柱:Restek RTX-35,15m x 200μm x0.33μm;恒定氦流速:0.88ml/min;烘箱:70℃;入口:250℃;梯度:70℃,30℃/min→310℃(保持3min).
方法12:
仪器:Waters ACQUITY SQD UPLC系统;柱:Waters Acquity UPLC HSS T31.8μ50x 1mm;洗脱剂A:1l水+0.25ml 99%甲酸,洗脱剂B:1l乙腈+0.25ml 99%甲酸;梯度:0.0min 90%A→1.2min 5%A→2.0min 5%A;烘箱:50℃;流速:0.40ml/min;UV检测:210-400nm.
起始化合物和中间体:
实施例1A
1-(3-氯-5-氟苯基)-4-乙氧基-3,4-二氧代丁-1-烯-1-醇锂

在氩气下,在-78℃提供在60ml二乙醚中的78ml(78mmol)的六甲基二硅叠氮化锂(lithium hexamethyldisilazide)(1N在四氢呋喃中的溶液)溶液,并加入12.5g(72.4mmol)的1-(3-氯-5-氟苯基)乙烷酮在190ml二乙醚中的溶液。在-78℃45分钟后,逐滴加入11.6g(79.7mmol)的草酸二乙酯,并将混合物在室温搅拌过夜。浓缩反应混合物并获得28.6g具有71%纯度的标题化合物(100%理论值),将其进行反应,而无需进一步纯化。
1H-NMR(400MHz,DMSO-d6):δ=7.63(t,1H),7.55-7.50(m,2H),6.33(s,1H),4.14(q,2H),1.24(t,3H).
LC-MS(方法1):Rt=2.65min;MS(ESIpos):m/z=273[M-Li+2H]+.
实施例2A
1-(3,5-二氟苯基)-4-乙氧基-3,4-二氧代丁-1-烯-1-醇锂

以类似于合成实施例1A的化合物的方式,从1-(3,5-二氟苯基)乙烷酮和草酸二乙酯起始制备标题化合物。获得5.43g(65%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.45(d,2H),7.35(t,1H),6.38(s,1H),4.17(q,2H),1.26(t,3H).
LC-MS(方法3):Rt=2.43min;MS(ESIpos):m/z=257[M-Li+2H]+.
实施例3A
1-(3-氯苯基)-4-乙氧基-3,4-二氧代丁-1-烯-1-醇锂

以类似于合成实施例1A的化合物的方式,从1-(3-氯苯基)乙烷酮和草酸二乙酯起始制备标题化合物。获得40.4g(92%理论值)标题化合物。
LC-MS(方法4):Rt=3.91min;MS(ESIpos):m/z=255[M-Li+2H]+.
实施例4A
1-(3-氰基苯基)-4-乙氧基-3,4-二氧代丁-1-烯-1-醇锂

以类似于合成实施例1A的化合物的方式,从3-乙酰基苯腈和草酸二乙酯起始制备标题化合物。获得具有62%纯度(76%理论值)的0.53g标题化合物。
LC-MS(方法5):Rt=1.11min;MS(ESIpos):m/z=246[M-Li+2H]+.
实施例5A
1-(3-氯-4-氟苯基)-4-乙氧基-3,4-二氧代丁-1-烯-1-醇锂
以类似于合成实施例1A的化合物的方式,从1-(3-氯-4-氟苯基)乙烷酮和草酸二乙酯起始制备标题化合物。获得9.7g具有66%纯度(79%理论值)的标题化合物。
LC-MS(方法1):Rt=2.69min;MS(ESIpos):m/z=273[M-Li+2H]+.
实施例6A
4-乙氧基-3,4-二氧代-1-[3-(三氟甲氧基)苯基]丁-1-烯-1-醇锂
以类似于合成实施例1A的化合物的方式,从1-[3-(三氟甲氧基)苯基]乙烷酮和草酸二乙酯起始制备标题化合物。获得7.7g具有65%纯度(80%理论值)的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.84(d,1H),7.70(s,1H),7.56(t,1H),7.45(d,1H),6.41(s,1H),4.15(q,2H),1.25(t,3H).
LC-MS(方法5):Rt=1.39min;MS(ESIpos):m/z=305[M-Li+2H]+.
实施例7A
4-乙氧基-1-[4-氟-3-(三氟甲基)苯基]-3,4-二氧代丁-1-烯-1-醇锂

以类似于合成实施例1A的化合物的方式从1-[4-氟-3-(三氟甲基)苯基]乙烷酮和草酸二乙酯起始制备标题化合物。获得8.7g具有78%纯度(90%理论值)的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.91(s,1H),7.89(d,1H),7.79(d,1H),6.40(s,1H),4.16(q,2H),1.25(t,3H).
LC-MS(方法3):Rt=2.64min;MS(ESIpos):m/z=307[M-Li+2H]+.
实施例8A
4-乙氧基-1-(3-氟苯基)-3,4-二氧代丁-1-烯-1-醇锂

以类似于合成实施例1A的化合物的方式,从1-(3-氟苯基)乙烷酮和草酸二乙酯起始制备标题化合物。获得9.4g(98%理论值)标题化合物。
LC-MS(方法5):Rt=1.22min;MS(ESIpos):m/z=239[M-Li+2H]+.
实施例9A
4-乙氧基-3,4-二氧代-1-[3-(三氟甲基)苯基]丁-1-烯-1-醇锂

以类似于合成实施例1A的化合物的方式,从1-[3-(三氟甲基)苯基]乙烷酮和草酸二乙酯起始制备标题化合物。获得44.2g具有62%纯度(70%理论值)的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.12(d,1H),8.07(s,1H),7.83(d,1H),7.67(t,1H),6.45(s,1H),4.16(q,2H),1.25(t,3H).
LC-MS(方法3):Rt=2.54min;MS(ESIpos):m/z=289[M-Li+2H]+.
实施例10A
4-乙氧基-1-(3-甲氧基苯基)-3,4-二氧代丁-1-烯-1-醇锂

以类似于合成实施例1A化合物的方式,从1-(3-甲氧基苯基)乙烷酮和草酸二乙酯起始制备标题化合物。获得0.62g的具有64%纯度(93%理论值)的标题化合物。
LC-MS(方法5):Rt=1.22min;MS(ESIpos):m/z=251[M-Li+2H]+.
实施例11A
1-(3,4-二氟苯基)-4-乙氧基-3,4-二氧代丁-1-烯-1-醇锂

以类似于合成实施例1A的化合物的方式,从1-(3,4-二氟苯基)乙烷酮和草酸二乙酯起始制备标题化合物。获得8.47g(100%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.79(ddd,1H),7.74-7.68(m,1H),7.47(q,1H),6.40(s,1H),4.16(q,2H),1.26(t,3H).
LC-MS(方法3):Rt=2.40min;MS(ESIpos):m/z=257[M-Li+2H]+.
实施例12A
1-(3-溴-4-氟苯基)-4-乙氧基-3,4-二氧代丁-1-烯-1-醇锂
以类似于合成实施例1A化合物的方式,从1-(3-溴-4-氟苯基)乙烷酮和草酸二乙酯起始制备标题化合物。获得7.8g具有75%纯度(79%理论值)的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.05(dd,1H),7.86(ddd,1H),7.40(t,1H),6.36(s,1H),4.15(q,2H),1.25(t,3H).
LC-MS(方法3):Rt=2.59min;MS(ESIpos):m/z=318[M-Li+2H]+.
实施例13A
4-乙氧基-1-(2-氟苯基)-3,4-二氧代丁-1-烯-1-醇锂

以类似于合成实施例1A化合物的方式,从1-(2-氟苯基)乙烷酮和草酸二乙酯起始制备标题化合物。获得4.5g(82%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.67(dt,1H),7.49-7.42(m,1H),7.27-7.17(m,2H),6.24(s,1H),4.14(q,2H),1.24(t,3H).
LC-MS(方法3):Rt=2.36min;MS(ESIpos):m/z=239[M-Li+2H]+.
实施例14A
1-(2,3-二氟苯基)-4-乙氧基-3,4-二氧代丁-1-烯-1-醇锂
以类似于合成实施例1A的化合物的方式,从1-(2,3-二氟苯基)乙烷酮和草酸二乙酯起始制备标题化合物。获得1.7g具有26%纯度(48%理论值)的标题化合物。
LC-MS(方法3):Rt=2.33min;MS(ESIpos):m/z=257[M-Li+2H]+.
实施例15A
4-乙氧基-1-(3-氟-5-甲氧基苯基)-3,4-二氧代丁-1-烯-1-醇锂

在51ml的甲苯中提供5.00g(29.4mmol)的3-氟-5-甲氧基苯甲酸,加入6.4ml(88.2mmol)的亚硫酰氯,并在回流下加热混合物达2小时。随后浓缩反应混合物,将残余物吸收在二氯甲烷中,加入3.73g(38.2mmol)的N,O-二甲基羟胺盐酸盐,在0℃加入10.7ml(76.4mmol)的三乙胺,并将混合物在室温搅拌过夜。加入水,分离各相,用二氯甲烷进行提取,并将合并的有机相通过硫酸钠干燥,过滤并浓缩。将残余物溶解在105ml的二乙醚中,并在室温在氩气下加入9.6ml(28.6mmol)的甲基镁化溴在二乙醚中的3M溶液,并将所述混合物在回流下加热2小时。随后加入饱和的氯化铵水溶液,用二氯甲烷提取,并将合并的有机相通过硫酸钠干燥、过滤并浓缩。以类似于合成实施例1A的化合物的方式,使粗制产物1-(3-氟-5-甲氧基苯基)乙烷酮与草酸二乙酯反应。获得4.02g具有61%纯度(30%理论值)的标题化合物。
LC-MS(方法5):Rt=1.26min;MS(ESIpos):m/z=269[M-Li+2H]+.
实施例16A
1-[3-(苄氧基)苯基]-4-乙氧基-3,4-二氧代丁-1-烯-1-醇锂

以类似于合成实施例1A的化合物的方式,从1-[3-(苄氧基)苯基]乙烷酮和草酸二乙酯起始制备标题化合物。获得8.1g具有74%纯度(82%理论值)的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.49-7.44(m,2H),7.43-7.36(m,4H),7.36-7.30(m,2H),7.14-7.08(m,1H),6.39(s,1H),5.15(s,2H),4.14(q,2H),1.25(t,3H).
LC-MS(方法2):Rt=2.96min;MS(ESIpos):m/z=327[M-Li+2H]+.
实施例17A
4-乙氧基-1-(3-氟-5-三氟甲基苯基)-3,4-二氧代丁-1-烯-1-醇锂

以类似于合成实施例1A的化合物的方式,从1-[3-氟-5-(三氟甲基)苯基]乙烷酮和草酸二乙酯起始制备标题化合物。获得1.5g具有62%纯度(62%理论值)的标题化合物。
LC-MS(方法5):Rt=1.37min;MS(ESIpos):m/z=307[M-Li+2H]+.
实施例18A
1-(3-溴-5-氟苯基)-4-乙氧基-3,4-二氧代丁-1-烯-1-醇锂

以类似于合成实施例1A的化合物的方式,从1-(3-溴-5-氟苯基)乙烷酮(在US2003/229096A1中描述)和草酸二乙酯起始制备标题化合物。获得4.7g具有62%纯度(74%理论值)的标题化合物。
LC-MS(方法7):Rt=2.29min;MS(ESIpos):m/z=317[M-Li+2H]+.
实施例19A
1-[3,5-双(三氟甲基)苯基]-4-乙氧基-3,4-二氧代丁-1-烯-1-醇锂

以类似于合成实施例1A的化合物方式,从1-[3,5-双(三氟甲基)苯基]乙烷酮和草酸二乙酯起始制备标题化合物。获得6.1g(86%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.36(s,2H),8.23(s,1H),6.48(s,1H),4.18(q,2H),1.27(t,3H).
LC-MS(方法3):Rt=2.88min;MS(ESIneg):m/z=355[M-Li]-.
实施例20A
4-乙氧基-1-(3-氟-5-甲基苯基)-3,4-二氧代丁-1-烯-1-醇锂

以类似于合成实施例1A的化合物的方式,从1-(3-氟-5-甲基苯基)乙烷酮和草酸二乙酯起始制备标题化合物。获得8.88g具有66%纯度(70%理论值)的标题化合物。
LC-MS(方法1):Rt=2.67min;MS(ESIpos):m/z=253[M-Li+2H]+.
实施例21A
1-(3-氯-4-氟苯基)-5-(3-氯-5-氟苯基)-1H-吡唑-3-甲酸乙酯

在350ml的乙醇中提供28.6g的具有71%纯度(72.9mmol)的实施例1A的化合物,加入15.8g(80.2mmol)的3-氯-4-氟苯基肼盐酸盐,将混合物在室温搅拌过夜。浓缩反应混合物,并将残余物吸收在350ml的浓乙酸中,并在回流下加热2小时。将反应混合物加入乙酸乙酯中,并用水和饱和的碳酸氢钠水溶液洗涤。浓缩有机相,并通过急骤色谱法纯化(流动相:环己烷/乙酸乙酯20∶1)。获得22.6g(76%理论值)的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.80(dd,1H),7.58-7.50(m,2H),7.38(ddd,1H),7.31(s,1H),7.27(s,1H),7.19(dt,1H),4.34(q,2H),1.32(t,3H).
LC-MS(方法5):Rt=1.52min;MS(ESIpos):m/z=397[M+H]+.
实施例22A
5-(3-氯-5-氟苯基)-1-(3-氯苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例1A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。获得5.82g标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.61-7.56(m,2H),7.54-7.47(m,2H),7.32(s,1H),7.30(d,1H),7.25(d,1H),7.18(dt,1H),4.35(q,2H),1.32(t,3H).
LC-MS(方法5):Rt=1.51min;MS(ESIpos):m/z=379[M+H]+.
实施例23A
1-(3-氯苯基)-5-(3,5-二氟苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例2A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。在通过急骤色谱法(流动相:环己烷/乙酸乙酯3∶1)纯化后,获得2.58g(68%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.60-7.56(m,2H),7.53-7.47(m,1H),7.37-7.27(m,3H),7.10-7.03(m,2H),4.35(q,2H),1.32(t,3H).
LC-MS(方法3):Rt=2.74min;MS(ESIpos):m/z=363[M+H]+.
实施例24A
1,5-双(3-氯苯基)-1H-吡唑-3-甲酸乙酯

将112g(413mmol)的实施例3A化合物提供在1120ml的乙醇中,加入101g(561mmol)的3-氯苯基肼盐酸盐,并将混合物在室温搅拌过夜。浓缩反应混合物,并将残余物吸收在1120ml的浓乙酸中,并在回流下加热2小时。将混合物留待室温过夜,通过吸滤收集得到的沉淀物并用环己烷搅拌,并将所述沉淀物通过吸滤收集,用环己烷洗涤,并在高真空下干燥。获得126g(80%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.58-7.54(m,2H),7.51-7.43(m,3H),7.40(t,1H),7.30-7.26(m,1H),7.25(s,1H),7.19(d,1H),4.34(q,2H),1.32(t,3H).
LC-MS(方法2):Rt=3.00min;MS(ESIpos):m/z=361[M+H]+.
实施例25A
1,5-双(3-氯-4-氟苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例5A的化合物起始制备标题化合物。获得5.78g标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.78(dd,1H),7.67(dd,1H),7.53(t,1H),7.44(t,1H),7.35(ddd,1H),7.25(s,1H),7.22(ddd,1H),4.34(q,2H),1.32(t,3H).
LC-MS(方法5):Rt=1.51min;MS(ESIpos):m/z=397[M+H]+.
实施例26A
1-(3-氯苯基)-5-[3-(三氟甲氧基)苯基]-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例6A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。获得4.56g(95%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.59-7.46(m,4H),7.46-7.38(m,2H),7.32-7.27(m,2H),7.22-7.19(m,1H),4.35(q,2H),1.32(t,3H).
LC-MS(方法3):Rt=2.92min;MS(ESIpos):m/z=411[M+H]+.
实施例27A
1-(3-氯苯基)-5-[4-氟-3-(三氟甲基)苯基]-1H-吡唑-3-甲酸乙酯
以类似于合成实施例21A的化合物的方式,从实施例7A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。获得3.60g标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.76(d,1H),7.61-7.56(m,3H),7.50(t,1H),7.43(s,1H),7.42(s,1H),7.34-7.30(m,1H),4.35(q,2H),1.33(t,3H).
LC-MS(方法1):Rt=3.05min;MS(ESIpos):m/z=413[M+H]+.
实施例28A
1-(3-氯-4-氟苯基)-5-[3-(三氟甲氧基)苯基]-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例6A的化合物起始制备标题化合物。获得3.50g(66%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.74(dd,1H),7.59-7.51(m,2H),7.44-7.35(m,3H),7.28(s,1H),7.21(s,1H),4.35(q,2H),1.32(t,3H).
LC-MS(方法1):Rt=3.04min;MS(ESIpos):m/z=429[M+H]+.
实施例29A
5-(3-氯-4-氟苯基)-1-(3-氯苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例5A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。获得5.67g标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.64(dd,1H),7.58-7.54(m,2H),7.51-7.42(m,2H),7.29-7.21(m,3H),4.34(q,2H),1.32(t,3H).
LC-MS(方法5):Rt=1.50min;MS(ESIpos):m/z=379[M+H]+.
实施例30A
5-(3-氟苯基)-1-(3-甲氧基苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A化合物的方式,从实施例8A的化合物和3-甲氧基苯基肼盐酸盐起始制备标题化合物。获得30.4g(62%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.44-7.33(m,2H),7.24(dd,1H),7.21(s,1H),7.17(dt,1H),7.08(d,1H),7.04(dd,1H),6.95(t,1H),6.86(dd,1H),4.34(q,2H),3.73(s,3H),1.32(t,3H).
LC-MS(方法1):Rt=2.78min;MS(ESIpos):m/z=341[M+H]+.
实施例31A
1-(3-氯苯基)-5-(3-甲氧基苯基)-1H-吡唑-3-甲酸乙酯
以类似于合成实施例21A化合物的方式,从实施例10A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物,通过制备HPLC(RP18柱;流动相:乙腈/水梯度)进行纯化。获得1.60g标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.57-7.51(m,2H),7.47(t,1H),7.31-7.25(m,2H),7.17(s,1H),6.96(dd,1H),6.89(s,1H),6.80(d,1H),4.34(q,2H),3.69(s,3H),1.32(t,3H).
LC-MS(方法3):Rt=2.68min;MS(ESIpos):m/z=357[M+H]+.
实施例32A
5-(3-氯苯基)-1-[3-(三氟甲基)苯基]-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例3A的化合物和3-(三氟甲基)苯基肼盐酸盐起始制备标题化合物,通过制备HPLC(RP18柱;流动相:乙腈/水梯度)进行纯化。获得2.00g(42%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.85(d,1H),7.75(s,1H),7.71(t,1H),7.64(d,1H),7.50-7.43(m,2H),7.40(t,1H),7.27(s,1H),7.20(d,1H),4.35(q,2H),1.33(t,3H).
LC-MS(方法1):Rt=3.09min;MS(ESIpos):m/z=395[M+H]+.
实施例33A
1-(3-氯苯基)-5-(3,4-二氟苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例11A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。获得2.83g(49%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.58-7.43(m,5H),7.28-7.24(m,1H),7.22(s,1H),7.11-7.06(m,1H),4.34(q,2H),1.32(t,3H).
LC-MS(方法3):Rt=2.72min;MS(ESIpos):m/z=363[M+H]+.
实施例34A
5-(3-溴-4-氟苯基)-1-(3-氯苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例12A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。获得3.68g标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.74(dd,1H),7.58-7.54(m,2H),7.48(t,1H),7.41(t,1H),7.30-7.23(m,3H),4.34(q,2H),1.32(t,3H).
LC-MS(方法1):Rt=3.03min;MS(ESIpos):m/z=423[M+H]+.
实施例35A
5-(3-氯苯基)-1-(4-氟苯基)-1H-吡唑-3-甲酸乙酯

将45.0g(155mmol)的实施例3A化合物提供在400ml的乙醇中,加入34.4g(211mmol)的4-氟苯基肼盐酸盐,并将混合物在室温搅拌过夜。浓缩反应混合物,并将残余物吸收在400ml的浓乙酸中,并在回流下加热2小时。浓缩反应混合物,并将残余物吸收在二氯甲烷中,用饱和的碳酸氢钠水溶液和水洗涤,通过硫酸钠干燥并浓缩。将残余物在二乙醚中,在回流下搅拌,并将沉淀物通过吸滤进行收集。浓缩母液,将所述残余物用环己烷搅拌,并将沉淀物通过吸滤收集。共获得23g(43%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.47-7.39(m,4H),7.39-7.30(m,3H),7.23(s,1H),7.16(dt,1H),4.34(q,2H),1.32(t,3H).
LC-MS(方法1):Rt=2.89min;MS(ESIpos):m/z=345[M+H]+.
实施例36A
1-(3-氯苯基)-5-(2-氟苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A化合物的方式,从实施例13A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。获得2.34g(73%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.56-7.41(m,5H),7.33-7.21(m,3H),7.16(s,1H),4.35(q,2H),1.33(t,3H).
LC-MS(方法1):Rt=2.85min;MS(ESIpos):m/z=345[M+H]+.
实施例37A
1-(4-氟苯基)-5-[3-(三氟甲基)苯基]-1H-吡唑-3-甲酸乙酯
以类似于合成实施例21A化合物的方式,从实施例9A的化合物和4-氟苯基肼盐酸盐起始制备标题化合物。获得4.63g(99%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.74(d,1H),7.64-7.54(m,3H),7.47-7.41(m,2H),7.37-7.30(m,3H),4.34(q,2H),1.32(t,3H).
LC-MS(方法4):Rt=4.10min;MS(ESIpos):m/z=379[M+H]+.
实施例38A
1-(3-氯苯基)-5-(2,3-二氟苯基)-1H-吡唑-3-甲酸乙酯
以类似于合成实施例21A的化合物的方式,从实施例14A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。获得240mg(37%理论值)标题化合物。
1H-NMR(400MHz,CDCl3):δ=7.46(t,1H),7.36-7.31(m,1H),7.28-7.18(m,2H),7.14-7.06(m,3H),7.00-6.94(m,1H),4.47(q,2H),1.43(t,3H).
LC-MS(方法1):Rt=2.91min;MS(ESIpos):m/z=363[M+H]+.
实施例39A
5-(3-氯苯基)-1-(4-氯苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例3A的化合物和4-氯苯基肼盐酸盐起始制备标题化合物。在通过在甲醇中搅拌再次纯化后,通过吸滤收集沉淀物,并在高真空下干燥。获得26.8g(57%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.58-7.53(m,2H),7.49-7.43(m,2H),7.42-7.36(m,3H),7.24(s,1H),7.19-7.14(m,1H),4.34(q,2H),1.32(t,3H).
LC-MS(方法1):Rt=3.07min;MS(ESIpos):m/z=361[M+H]+.
实施例40A
5-(3-氟苯基)-1-(4-氟苯基)-1H-吡唑-3-甲酸乙酯
以类似于合成实施例21A的化合物的方式,从实施例8A的化合物和4-氟苯基肼盐酸盐起始制备标题化合物。获得23.0g(44%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.46-7.37(m,3H),7.37-7.29(m,2H),7.26-7.16(m,3H),7.05(d,1H),4.34(q,2H),1.32(t,3H).
LC-MS(方法1):Rt=2.76min;MS(ESIpos):m/z=329[M+H]+.
实施例41A
1-(3-氯苯基)-5-(3-氟-5-甲氧基苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例15A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。获得2.20g具有68%纯度的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.59-7.55(m,2H),7.50(t,1H),7.31-7.27(m,1H),7.24(s,1H),6.89(dt,1H),6.74-6.69(m,2H),4.34(q,2H),3.70(s,3H),1.32(t,3H).
LC-MS(方法1):Rt=2.80min;MS(ESIpos):m/z=375[M+H]+.
实施例42A
1-(3-氯-4-氟苯基)-5-(3-氟-5-甲氧基苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例15A的化合物和3-氯-4-氟苯基肼盐酸盐起始制备标题化合物。获得2.36g具有82%纯度的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.77(dd,1H),7.54(t,1H),7.36(ddd,1H),7.24(s,1H),6.89(dt,1H),6.75-6.70(m,2H),4.34(q,2H),3.71(s,3H),1.32(t,3H).
LC-MS(方法1):Rt=2.82min;MS(ESIpos):m/z=393[M+H]+.
实施例43A
5-[3-(苄氧基)苯基]-1-(3-氯苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例16A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。获得14.1g标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.56-7.44(m,3H),7.40-7.24(m,7H),7.16(s,1H),7.04(dd,1H),7.01-6.98(m,1H),6.80(d,1H),5.05(s,2H),4.34(q,2H),1.32(t,3H).
LC-MS(方法3):Rt=3.03min;MS(ESIpos):m/z=433[M+H]+.
实施例44A
1-(3-氯苯基)-5-(3-羟基苯基)-1H-吡唑-3-甲酸乙酯

将19.1g(44.1mmol)实施例43A的化合物提供在367ml的浓乙酸中,加入23.5g(11.0mmol)的披钯活性炭(5%),并将所述混合物在室温,在氢气氛下搅拌过夜。随后过滤反应混合物,浓缩滤液,将残余物吸收在二氯甲烷中并用碳酸氢钠水溶液洗涤,将有机相通过硫酸钠干燥,并浓缩。获得12.7g具有63%纯度(53%理论值)的标题化合物。
LC-MS(方法1):Rt=2.41min;MS(ESIpos):m/z=343[M+H]+.
实施例45A
1-(3-氯-4-氟苯基)-5-(3-氰基苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例4A的化合物起始制备标题化合物。获得0.50g(80%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.94-7.91(m,1H),7.87(dt,1H),7.77(dd,1H),7.61-7.49(m,3H),7.35(ddd,1H),7.31(s,1H),4.35(q,2H),1.32(t,3H).
LC-MS(方法1):Rt=2.61min;MS(ESIpos):m/z=370[M+H]+.
实施例46A
1-(3-氯-4-氟苯基)-5-(3-氟苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例8A的化合物起始制备标题化合物。获得0.60g(93%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.75(dd,1H),7.53(t,1H),7.47-7.39(m,1H),7.35(ddd,1H),7.29-7.22(m,3H),7.09-7.05(m,1H),4.34(q,2H),1.32(t,3H).
LC-MS(方法1):Rt=2.77min;MS(ESIpos):m/z=363[M+H]+.
实施例47A
1-(3-氯-4-氟苯基)-5-[3-(三氟甲基)苯基]-1H-吡唑-3-甲酸乙酯
以类似于合成实施例21A的化合物的方式,从实施例9A的化合物起始制备标题化合物。获得5.10g(100%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.80-7.74(m,2H),7.72-7.50(m,4H),7.37(ddd,1H),7.33(s,1H),4.35(q,2H),1.33(t,3H).
LC-MS(方法3):Rt=2.87min;MS(ESIpos):m/z=413[M+H]+.
实施例48A
1-(3-氯-4-氟苯基)-5-[3-氟-5-(三氟甲基)苯基]-1H-吡唑-3-甲酸乙酯
以类似于合成实施例21A的化合物的方式,从实施例17A的化合物起始制备标题化合物。获得2.43g具有36%纯度的标题化合物。
LC-MS(方法1):Rt=2.95min;MS(ESIpos):m/z=431[M+H]+.
实施例49A
1-(3-氯苯基)-5-[3-氟-5-(三氟甲基)苯基]-1H-吡唑-3-甲酸乙酯
以类似于合成实施例21A的化合物的方式,从实施例17A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。获得2.08g标题化合物。
LC-MS(方法5):Rt=1.52min;MS(ESIpos):m/z=413[M+H]+.
实施例50A
1-(3-溴苯基)-5-(3-氯苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例3A的化合物和3-溴苯基肼盐酸盐起始制备标题化合物。获得2.50g具有70%纯度(67%理论值)的标题化合物。
LC-MS(方法1):Rt=2.93min;MS(ESIpos):m/z=405[M+H]+.
实施例51A
1-(3-溴苯基)-5-(3-氯-4-氟苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例1A的化合物和3-溴苯基肼盐酸盐起始制备标题化合物。获得689mg具有76%纯度(77%理论值)的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.73-7.69(m,2H),7.52(dt,1H),7.43(t,1H),7.35-7.30(m,2H),7.25(s,1H),7.18(dt,1H),4.35(q,2H),1.32(t,3H).
LC-MS(方法7):Rt=2.55min;MS(ESIpos):m/z=423[M+H]+.
实施例52A
5-(3-溴-5-氟苯基)-1-(3-氯-4-氟苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例18A的化合物起始制备标题化合物。获得3.87g(89%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.80(dd,1H),7.63(dt,1H),7.55(t,1H),7.41-7.35(m,2H),7.31(s,1H),7.24-7.20(m,1H),4.34(q,2H),1.32(t,3H).
LC-MS(方法1):Rt=2.98min;MS(ESIpos):m/z=441[M+H]+.
实施例53A
1-(3-氯-4-氟苯基)-5-(3-甲氧基苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例10A的化合物起始制备标题化合物。获得0.60g(97%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.74(dd,1H),7.52(t,1H),7.34(ddd,1H),7.29(t,1H),7.17(s,1H),6.96(dd,1H),6.91(t,1H),6.79(d,1H),4.34(q,2H),3.71(s,3H),1.32(t,3H).
LC-MS(方法1):Rt=2.77min;MS(ESIpos):m/z=375[M+H]+.
实施例54A
5-[3,5-双(三氟甲基)苯基]-1-(3-氯苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例19A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。获得2.42g(62%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.15(s,1H),7.96(s,2H),7.63-7.57(m,2H),7.55(s,1H),7.50(t,1H),7.34(d,1H),4.36(q,2H),1.33(t,3H).
LC-MS(方法1):Rt=3.16min;MS(ESIpos):m/z=463[M+H]+.
实施例55A
1-(4-氯苯基)-5-[3-(三氟甲基)苯基]-1H-吡唑-3-甲酸乙酯
以类似于合成实施例21A的化合物的方式,从实施例9A的化合物和4-氯苯基肼盐酸盐起始制备标题化合物。获得4.78g(98%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.76(d,1H),7.67-7.59(m,2H),7.58-7.53(m,3H),7.43-7.38(m,2H),7.33(s,1H),4.35(q,2H),1.33(t,3H).
LC-MS(方法6):Rt=4.11min;MS(ESIpos):m/z=395[M+H]+.
实施例56A
1-(4-氯苯基)-5-(3-氟苯基)-1H-吡唑-3-甲酸乙酯
以类似于合成实施例21A的化合物的方式,从实施例8A的化合物和4-氯苯基肼盐酸盐起始制备标题化合物。获得43.4g(87%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.58-7.53(m,2H),7.46-7.36(m,3H),7.28-7.19(m,3H),7.06(d,1H),4.34(q,2H),1.32(t,3H).
LC-MS(方法6):Rt=3.93min;MS(ESIpos):m/z=345[M+H]+.
实施例57A
1-(3-氯-4-氟苯基)-5-(3-氟-5-甲基苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例20A的化合物起始制备标题化合物。获得2.79g具有78%纯度的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.75(dd,1H),7.53(t,1H),7.34(ddd,1H),7.19(s,1H),7.10(d,1H),7.01(s,1H),6.91(s,1H),4.34(q,2H),2.27(s,3H),1.32(t,3H).
LC-MS(方法1):Rt=2.98min;MS(ESIpos):m/z=377[M+H]+.
实施例58A
1-(3-氯苯基)-5-(3-氟-5-甲基苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A化合物的方式,从实施例20A的化合物和3-氯苯基肼盐酸盐起始制备标题化合物。获得1.75g标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.58-7.53(m,2H),7.48(t,1H),7.29-7.25(m,1H),7.19(s,1H),7.12-7.07(m,1H),7.01(s,1H),6.91-6.86(m,1H),4.34(q,2H),2.27(s,3H),1.32(t,3H).
LC-MS(方法1):Rt=2.97min;MS(ESIpos):m/z=359[M+H]+.
实施例59A
5-(3-氯-4-氟苯基)-1-(3-氰基苯基)-1H-吡唑-3-甲酸乙酯

将100mg(0.236mmol)的实施例51A化合物提供在1ml的1-甲基-2-吡咯烷酮中,加入55.4mg(0.472mmol)的氰化锌和27.3mg(0.024mmol)的四(三苯膦)钯(0),并将混合物在微波照射下,在密闭玻璃容器中在200℃加热30分钟。随后将反应混合物通过硅藻土过滤,并用甲醇洗脱,用1N盐酸水溶液使滤液成为弱酸性,并浓缩,将所述残余物通过制备HPLC(RP18柱;流动相:乙腈/水梯度)进行纯化。获得33mg(34%理论值)标题化合物。
LC-MS(方法1):Rt=2.72min;MS(ESIpos):m/z=370[M+H]+.
实施例60A
5-(3-氯苯基)-1-(3-氰基-4-氟苯基)-1H-吡唑-3-甲酸乙酯
以类似于合成实施例21A的化合物的方式,从实施例3A的化合物和3-氰基-4-氟苯基肼盐酸盐起始制备标题化合物。获得0.76g具有46%纯度(23%理论值)的标题化合物。
LC-MS(方法7):Rt=2.29min;MS(ESIpos):m/z=370[M+H]+.
实施例61A
5-(3-氯苯基)-1-(3-氰基苯基)-1H-吡唑-3-甲酸乙酯
以类似于合成实施例21A化合物的方式,从实施例3A的化合物和3-氰基苯基肼盐酸盐起始制备标题化合物。获得540mg具有69%纯度(25%理论值)的标题化合物。
LC-MS(方法7):Rt=2.24min;MS(ESIpos):m/z=352[M+H]+.
实施例62A
1,5-双(3-氰基苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例4A的化合物和3-氰基苯基肼盐酸盐起始制备标题化合物。获得440mg具有26%纯度的标题化合物。
LC-MS(方法7):Rt=1.95min;MS(ESIpos):m/z=343[M+H]+.
实施例63A
1-(3-氰基-4-氟苯基)-5-(3-氰基苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例4A的化合物和3-氰基-4-氟苯基肼盐酸盐起始制备标题化合物。获得510mg具有27%纯度的标题化合物。
LC-MS(方法7):Rt=2.00min;MS(ESIpos):m/z=361[M+H]+.
实施例64A
5-(3-氯-5-氟苯基)-1-(3-氰基苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A的化合物的方式,从实施例1A的化合物和3-氰基苯基肼盐酸盐起始制备标题化合物。获得1.96g具有55%纯度(86%理论值)的标题化合物。
LC-MS(方法1):Rt=2.71min;MS(ESIpos):m/z=370[M+H]+.
实施例65A
5-(3-氯-5-氟苯基)-1-(3-氰基-4-氟苯基)-1H-吡唑-3-甲酸乙酯

以类似于合成实施例21A化合物的方式,从实施例1A的化合物和3-氰基-4-氟苯基肼盐酸盐起始制备标题化合物。获得2.66g具有49%纯度(100%理论值)的标题化合物。
LC-MS(方法1):Rt=2.75min;MS(ESIpos):m/z=388[M+H]+.
实施例66A
1-(3-氯苯基)-5-[3-(2-羟基乙氧基)苯基]-1H-吡唑-3-甲酸乙酯

将100mg(0.29mmol)的实施例44A化合物提供在2ml的无水丙酮中,加入44.4mg(0.32mmol)的碳酸钾和36.5mg(0.29mmol)的2-溴乙醇,并将所述混合物在回流下加热过夜。随后将反应混合物通过制备HPLC(RP18柱;流动相:乙腈/水梯度,加入0.1%甲酸)进行纯化,并获得13.1mg标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.56-7.44(m,3H),7.30-7.24(m,2H),7.16(s,1H),6.96(dd,1H),6.91-6.89(m,1H),6.78(d,1H),4.85(t,1H),4.34(q,2H),3.91(t,2H),3.66(q,2H),1.32(t,3H).
LC-MS(方法1):Rt=2.35min;MS(ESIpos):m/z=387[M+H]+.
实施例67A
5-{3-[3-(乙酰氧基)丙氧基]苯基}-1-(3-氯苯基)-1H-吡唑-3-甲酸乙酯

将100mg(0.292mmol)的实施例44A的化合物提供在1ml的无水DMF中,加入242mg(1.75mmol)的碳酸钾和158mg(0.875mmol)的1-乙酰氧基-3-溴丙烷,并将所述混合物在90℃搅拌4小时。随后,将反应混合物通过制备HPLC(RP18柱;流动相:乙腈/水梯度,加入0.1%甲酸)进行纯化,获得109mg(84%理论值)标题化合物。
LC-MS(方法1):Rt=2.79min;MS(ESIpos):m/z=443[M+H]+.
实施例68A
1-(3-氯苯基)-5-[3-(3-氯丙氧基)苯基]-1H-吡唑-3-甲酸乙酯

将100mg(0.292mmol)的实施例44A化合物提供在3ml的无水DMF中,加入76.6mg(0.554mmol)的碳酸钾和133mg(0.846mmol)的1-溴-3-氯丙烷,并将混合物在回流下加热4小时。随后将反应混合物通过制备HPLC(RP18柱;流动相:乙腈/水梯度,加入0.1%甲酸)纯化,获得42mg具有49%纯度(17%理论值)的标题化合物。
LC-MS(方法7):Rt=2.53min;MS(ESIpos):m/z=419[M+H]+.
实施例69A
1-(3-氯苯基)-5-[3-(2-甲氧基乙氧基)苯基]-1H-吡唑-3-甲酸乙酯

将100mg(0.292mmol)的实施例44A化合物提供在2ml无水丙酮中,加入44.4mg(0.321mmol)的碳酸钾和40.6mg(0.292mmol)的2-溴乙基甲醚,并将所述混合物在回流下加热过夜。随后将反应混合物通过制备HPLC(RP18柱;流动相:乙腈/水梯度,加入0.1%甲酸)纯化并获得48mg(41%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.56-7.44(m,3H),7.30-7.24(m,2H),7.17(s,1H),6.97(dd,1H),6.93-6.89(m,1H),6.79(d,1H),4.34(q,2H),4.05-4.00(m,2H),3.61-3.57(m,2H),3.28(s,3H),1.32(t,3H).
LC-MS(方法1):Rt=2.68min;MS(ESIpos):m/z=401[M+H]+.
实施例70A
1-(3-氯苯基)-5-[3-(3-吡咯烷-1-基丙氧基)苯基]-1H-吡唑-3-甲酸乙酯

将41.5mg(0.10mmol)的实施例68A化合物和83μl(0.99mmol)的吡咯烷在3ml乙醇中,在80℃搅拌过夜。随后将反应混合物通过制备HPLC(RP18柱;流动相:乙腈/水梯度)纯化。获得18.3mg标题化合物。
LC-MS(方法7):Rt=1.51min;MS(ESIpos):m/z=454[M+H]+.
实施例71A
1-(3-氯-4-氟苯基)-5-(3-氯-5-氟苯基)-1H-吡唑-3-甲酸

将5.11g(12.9mmol)的实施例21A化合物提供在142ml的四氢呋喃中,并且在室温,加入3.08g(129mmol)的氢氧化锂和47ml水。将混合物在室温搅拌过夜,并且随后加入1N盐酸水溶液直到pH为酸性,将所述混合物用乙酸乙酯提取,将有机相用水洗涤,通过硫酸钠干燥,过滤并浓缩。获得4.51g(90%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.2(s,1H),7.78(dd,1H),7.58-7.49(m,2H),7.36(ddd,1H),7.28-7.25(m,1H),7.24(s,1H),7.21-7.16(m,1H).
LC-MS(方法1):Rt=2.52min;MS(ESIpos):m/z=369[M+H]+.
实施例72A
5-(3-氯-5-氟苯基)-1-(3-氯苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例22A的化合物起始制备标题化合物。获得4.18g(78%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.59-7.55(m,2H),7.54-7.45(m,2H),7.30-7.26(m,1H),7.26-7.23(m,2H),7.20-7.15(m,1H).
LC-MS(方法1):Rt=2.49min;MS(ESIpos):m/z=351[M+H]+.
实施例73A
1-(3-氯苯基)-5-(3,5-二氟苯基)-1H-吡唑-3-甲酸

将2.50g(6.89mmol)的实施例23A的化合物提供在62ml的1,4-二噁烷和水的2∶1混合物中,加入35ml(70mmol)的2N氢氧化锂在水中的溶液,并将混合物在室温搅拌过夜。浓缩混合物,并随后将2N盐酸溶液加入残余物中直到pH为酸性,将所述混合物用乙酸乙酯提取,将所述有机相通过硫酸钠干燥,过滤并浓缩。获得2.28g(99%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.2(s,1H),7.59-7.54(m,2H),7.53-7.47(m,1H),7.33(tt,1H),7.30-7.26(m,1H),7.24(s,1H),7.10-7.02(m,2H).
LC-MS(方法3):Rt=2.21min;MS(ESIpos):m/z=335[M+H]+.
实施例74A
1,5-双(3-氯苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例24A的化合物起始制备标题化合物。获得5.97g(94%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.57-7.37(m,6H),7.28-7.24(m,1H),7.21-7.18(m,1H),7.17(s,1H).
LC-MS(方法3):Rt=2.28min;MS(ESIpos):m/z=333[M+H]+.
实施例75A
1,5-双(3-氯-4-氟苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例25A的化合物起始制备标题化合物。获得5.15g(96%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.76(dd,1H),7.65(dd,1H),7.52(t,1H),7.44(t,1H),7.33(ddd,1H),7.23(ddd,1H),7.18(s,1H)
LC-MS(方法1):Rt=2.49min;MS(ESIpos):m/z=369[M+H]+.
实施例76A
1-(3-氯苯基)-5-[3-(三氟甲氧基)苯基]-1H-吡唑-3-甲酸

将10.8g(26.2mmol)的实施例26A的化合物提供在236ml的1,4-二噁烷中,加入236ml(472mmol)的氢氧化锂在水中的2N溶液,并将混合物在70℃搅拌2小时。浓缩所述混合物,并随后将2N盐酸水溶液加入残余物中直到pH为酸性,将所述混合物用二氯甲烷提取,将所述有机相通过硫酸镁干燥,过滤,并浓缩。获得9.90g(99%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.59-7.45(m,4H),7.44-7.38(m,2H),7.31-7.26(m,1H),7.22-7.18(m,2H).
LC-MS(方法1):Rt=2.77min;MS(ESIpos):m/z=383[M+H]+.
实施例77A
1-(3-氯苯基)-5-[4-氟-3-(三氟甲基)苯基]-1H-吡唑-3-甲酸
将3.60g(8.72mmol)的实施例27A的化合物提供在50ml的1,4-二噁烷中,加入50ml(50mmol)的1N氢氧化锂在水中的溶液,并将所述混合物在回流下加热1小时。浓缩所述混合物,用水稀释残余物,随后加入浓盐酸水溶液直到pH是酸性的,将所述混合物用乙酸乙酯提取,并将有机相通过硫酸镁干燥,过滤和浓缩。获得3.30g(98%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.70(d,1H),7.55-7.44(m,3H),7.40(t,1H),7.30(s,1H),7.18(d,1H),6.99(s,1H).
LC-MS(方法3):Rt=2.41min;MS(ESIpos):m/z=385[M+H]+.
实施例78A
1-(3-氯-4-氟苯基)-5-[3-(三氟甲氧基)苯基]-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例28A的化合物起始制备标题化合物。获得3.14g(96%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.72(dd,1H),7.59-7.50(m,2H),7.44-7.34(m,3H),7.23-7.18(m,2H).
LC-MS(方法1):Rt=2.56min;MS(ESIpos):m/z=401[M+H]+.
实施例79A
5-(3-氯-4-氟苯基)-1-(3-氯苯基)-1H-吡唑-3-甲酸
以类似于合成实施例71A的化合物的方式,从实施例29A的化合物起始制备标题化合物。获得5.30g(100%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.63(dd,1H),7.57-7.52(m,2H),7.51-7.42(m,2H),7.28-7.21(m,2H),7.17(s,1H).
LC-MS(方法1):Rt=2.46min;MS(ESIpos):m/z=351[M+H]+.
实施例80A
5-(3-氟苯基)-1-(3-甲氧基苯基)-1H-吡唑-3-甲酸

将34.0g(99.9mmol)的实施例30A的化合物提供在150ml的1,4-二噁烷中,加入150ml(300mmol)的2N氢氧化锂在水中的溶液,并将所述混合物在70℃搅拌1小时。浓缩混合物,随后将浓盐酸水溶液加入残余物中直到pH为酸性,将所述混合物用二氯甲烷提取,并将有机相通过硫酸镁进行干燥,过滤并浓缩。将所述残余物从二乙醚中再结晶。获得26.1g(84%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.0(s,1H),7.45-7.32(m,2H),7.26-7.13(m,3H),7.08(d,1H),7.03(dd,1H),6.95(t,1H),6.85(d,1H),3.72(s,3H).
LC-MS(方法1):Rt=2.33min;MS(ESIpos):m/z=313[M+H]+.
实施例81A
1-(3-氯苯基)-5-(3-甲氧基苯基)-1H-吡唑-3-甲酸
将1.50g(4.20mmol)的实施例31A的化合物提供在20ml的1,4-二噁烷中,加入20ml(20mmol)的1N氢氧化锂在水中的溶液,并将所述混合物在70℃搅拌1小时。浓缩所述混合物,随后将1N盐酸水溶液加入残余物中直到pH为酸性,将所述混合物用乙酸乙酯提取,并将合并的有机相通过硫酸镁干燥,过滤并浓缩。获得1.32g(92%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.56-7.44(m,3H),7.32-7.23(m,2H),7.11(s,1H),6.96(dd,1H),6.88(s,1H),6.80(d,1H),3.69(s,3H).
LC-MS(方法3):Rt=2.14min;MS(ESIpos):m/z=329[M+H]+.
实施例82A
5-(3-氯苯基)-1-[3-(三氟甲基)苯基]-1H-吡唑-3-甲酸

将2.00g(5.07mmol)的实施例32A的化合物提供在37ml的1,4-二噁烷中,加入1.07g(20.0mmol)的氢氧化锂在37ml水的溶液中,并将混合物在室温搅拌过夜。浓缩所述混合物,随后将1N盐酸水溶液加入残余物中直到pH为酸性,将所述混合物用乙酸乙酯提取,并将合并的有机相通过硫酸镁干燥,过滤并浓缩。获得0.74g(40%理论值)标题化合物。
1H-NMR(500MHz,DMSO-d6):δ=7.75(d,1H),7.70-7.62(m,2H),7.54(d,1H),7.45(d,1H),7.42-7.36(m,2H),7.19(d,1H),6.92(s,1H).
LC-MS(方法1):Rt=2.65min;MS(ESIpos):m/z=367[M+H]+.
实施例83A
1-(3-氯苯基)-5-(3,4-二氟苯基)-1H-吡唑-3-甲酸

以类似于合成实施例73A的化合物的方式,从实施例33A的化合物起始制备标题化合物。获得2.49g(96%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.57-7.43(m,5H),7.27-7.22(m,1H),7.16(s,1H),7.12-7.06(m,1H).
LC-MS(方法3):Rt=2.21min;MS(ESIpos):m/z=335[M+H]+.
实施例84A
5-(3-溴-4-氟苯基)-1-(3-氯苯基)-1H-吡唑-3-甲酸

以类似于合成实施例77A的化合物的方式,从实施例34A的化合物起始制备标题化合物。获得3.30g(98%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.72(dd,1H),7.58-7.52(m,2H),7.48(t,1H),7.41(t,1H),7.31-7.22(m,2H),7.17(s,1H).
LC-MS(方法3):Rt=2.34min;MS(ESIpos):m/z=395[M+H]+.
实施例85A
5-(3-氯苯基)-1-(4-氟苯基)-1H-吡唑-3-甲酸

将23.0g(66.7mmol)的实施例35A的化合物提供在100ml的1,4-二噁烷中,加入100ml(200mmol)的2N氢氧化锂在水中的溶液,并将所述混合物在50℃搅拌2小时。浓缩所述混合物,随后加入浓盐酸水溶液直到pH为酸性,将所述混合物用二氯甲烷提取,并将有机相通过硫酸镁进行干燥,过滤并浓缩。将所述残余物在少量二乙醚中搅拌,通过过滤收集,并在高真空下干燥。获得17.8g(84%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.46-7.29(m,7H),7.19-7.14(m,2H).
LC-MS(方法4):Rt=3.46min;MS(ESIpos):m/z=317[M+H]+.
实施例86A
1-(3-氯苯基)-5-(2-氟苯基)-1H-吡唑-3-甲酸

将2.30g(6.67mmol)的实施例36A的化合物提供在32ml的1,4-二噁烷和水的2∶1混合物中,加入17ml(33.4mmol)的2N氢氧化锂在水中的溶液,并将所述混合物在50℃搅拌2小时。浓缩所述混合物,随后将2N盐酸水溶液加入残余物中直到pH为酸性,将所述混合物用乙酸乙酯提取,并将有机相通过硫酸钠干燥,过滤并浓缩。将所述残余物在石油醚和二乙醚的少量混合物中搅拌,通过过滤收集,并在高真空下干燥。获得1.58g(75%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.56-7.40(m,5H),7.34-7.20(m,3H),7.11(s,1H).
LC-MS(方法1):Rt=2.35min;MS(ESIpos):m/z=317[M+H]+.
实施例87A
1-(4-氟苯基)-5-[3-(三氟甲基)苯基]-1H-吡唑-3-甲酸

将4.50g(11.9mmol)的实施例37A的化合物提供在107ml的1,4-二噁烷中,加入107ml(214mmol)的2N氢氧化锂在水中的溶液,并将所述混合物在70℃搅拌2小时。浓缩所述混合物,并随后将2N盐酸水溶液加入残余物中直到pH为酸性,将所述混合物用二氯甲烷提取,并将有机相通过硫酸镁进行干燥,过滤并浓缩。获得4.11g(99%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.73(d,1H),7.64-7.54(m,3H),7.46-7.40(m,2H),7.37-7.29(m,2H),7.25(s,1H).
LC-MS(方法4):Rt=3.53min;MS(ESIpos):m/z=351[M+H]+.
实施例88A
1-(3-氯苯基)-5-(2,3-二氟苯基)-1H-吡唑-3-甲酸

将0.21g(0.526mmol)的实施例38A的化合物提供在3.5ml的甲醇中,并且在室温加入295mg(5.26mmol)的氢氧化钾。将混合物在回流下加热5分钟,随后加入1N盐酸水溶液直到pH是5,并将得到的沉淀物通过吸滤收集,用二乙醚洗涤,并在高真空下干燥。获得145mg(82%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.2(s,1H),7.60-7.43(m,4H),7.33-7.19(m,3H),7.18(s,1H).
LC-MS(方法2):Rt=2.32min;MS(ESIpos):m/z=335[M+H]+.
实施例89A
5-(3-氯苯基)-1-(4-氯苯基)-1H-吡唑-3-甲酸
将26.8g(74.2mmol)的实施例39A的化合物提供在300ml的1,4-二噁烷中,加入56ml(111mmol)的2N氢氧化锂在水中的溶液,并将所述混合物在70℃搅拌2小时。浓缩所述混合物,随后将1N盐酸水溶液加入残余物中直到pH为酸性,将所述混合物用二氯甲烷提取,并将合并的有机相通过硫酸镁干燥,过滤并浓缩。获得24.5g(99%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.58-7.52(m,2H),7.48-7.35(m,5H),7.19-7.14(m,2H).
LC-MS(方法6):Rt=3.51min;MS(ESIpos):m/z=333[M+H]+.
实施例90A
5-(3-氟苯基)-1-(4-氟苯基)-1H-吡唑-3-甲酸
将23.9g(72.8mmol)的实施例40A的化合物提供在100ml的1,4-二噁烷中,加入100ml(200mmol)的2N氢氧化锂在水中的溶液,并将所述混合物在50℃搅拌1小时。浓缩所述混合物,将所述残余物用水稀释,随后加入浓盐酸水溶液直到pH为酸性,将所述混合物用二氯甲烷提取,并将有机相通过硫酸镁进行干燥,过滤并浓缩。将所述残余物在少量二乙醚中搅拌,通过过滤收集,并在高真空下干燥。获得22.0g(100%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.44-7.37(m,3H),7.36-7.28(m,2H),7.22(dt,1H),7.16(dt,1H),7.12(s,1H),7.05(d,1H).
LC-MS(方法1):Rt=2.32min;MS(ESIpos):m/z=301[M+H]+.
实施例91A
1-(3-氯苯基)-5-(3-氟-5-甲氧基苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例41A的化合物起始制备标题化合物。获得1.31g标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.58-7.53(m,2H),7.52-7.46(m,1H),7.30-7.25(m,1H),7.18(s,1H),6.88(dt,1H)6.73-6.69(m,2H),3.70(s,3H).
LC-MS(方法1):Rt=2.37min;MS(ESIpos):m/z=347[M+H]+.
实施例92A
1-(3-氯-4-氟苯基)-5-(3-氟-5-甲氧基苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式(但是其中搅拌6小时),从实施例42A的化合物起始制备标题化合物。获得1.38g标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.76(dd,1H),7.54(t,1H),7.35(ddd,1H),7.17(s,1H),6.89(dt,1H),6.74-6.69(m,2H),3.71(s,3H).
LC-MS(方法7):Rt=1.95min;MS(ESIpos):m/z=365[M+H]+.
实施例93A
1-(3-氯-4-氟苯基)-5-(3-氰基苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例45A的化合物起始制备标题化合物。获得348mg(68%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.91(s,1H),7.87(dt,1H),7.75(dd,1H),7.61-7.49(m,3H),7.33(ddd,1H),7.24(s,1H).
LC-MS(方法1):Rt=2.21min;MS(ESIpos):m/z=342[M+H]+.
实施例94A
1-(3-氯-4-氟苯基)-5-(3-氟苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例46A的化合物起始制备标题化合物。获得550mg(99%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.73(dd,1H),7.52(t,1H),7.46-7.39(m,1H),7.34(ddd,1H),7.28-7.21(m,2H),7.16(s,1H),7.07(dt,1H).
LC-MS(方法1):Rt=2.36min;MS(ESIpos):m/z=335[M+H]+.
实施例95A
1-(3-氯-4-氟苯基)-5-[3-(三氟甲基)苯基]-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例47A的化合物起始制备标题化合物。获得344mg(97%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.82-7.74(m,2H),7.67-7.50(m,4H),7.36(ddd,1H),7.26(s,1H).
LC-MS(方法1):Rt=2.53min;MS(ESIpos):m/z=385[M+H]+.
实施例96A
1-(3-氯-4-氟苯基)-5-[3-氟-5-(三氟甲基)苯基]-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例48A的化合物起始制备标题化合物。获得851mg具有51%纯度的标题化合物。
LC-MS(方法1):Rt=2.58min;MS(ESIpos):m/z=403[M+H]+.
实施例97A
1-(3-氯苯基)-5-[3-氟-5-(三氟甲基)苯基]-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例49A的化合物起始制备标题化合物。获得1.82g(85%理论值)标题化合物。
LC-MS(方法1):Rt=2.55min;MS(ESIpos):m/z=385[M+H]+.
实施例98A
1-(3-溴苯基)-5-(3-氯苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例51A的化合物起始制备标题化合物。获得1.74g标题化合物。
LC-MS(方法1):Rt=2.47min;MS(ESIpos):m/z=377[M+H]+.
实施例99A
1-(3-溴苯基)-5-(3-氯-4-氟苯基)-1H-吡唑-3-甲酸

将688mg的具有76%纯度的实施例51A的化合物(1.23mmol)在29ml的四氢呋喃中提供,并且在室温,加入389mg(16.2mmol)的氢氧化锂和10ml水。将所述混合物在室温搅拌5小时,随后加入1N盐酸水溶液直到pH为酸性,并将所述混合物用乙酸乙酯提取,通过硫酸钠干燥,过滤并浓缩。将残余物在少量环己烷中搅拌,并将沉淀物通过吸滤收集。获得413mg(83%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.72-7.68(m,2H),7.51(dt,1H),7.42(t,1H),7.33-7.29(m,1H),7.26-7.23(m,2H),7.18(dt,1H).
LC-MS(方法1):Rt=2.53min;MS(ESIpos):m/z=395[M+H]+.
实施例100A
5-[3-(苄氧基)苯基]-1-(3-氯苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例43A的化合物起始制备标题化合物。获得60.0g(85%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.55-7.43(m,3H),7.40-7.22(m,7H),7.10(s,1H),7.04(dd,1H),7.00-6.97(m,1H),6.80(d,1H),5.04(s,2H).
LC-MS(方法9):Rt=2.40min;MS(ESIpos):m/z=405[M+H]+.
实施例101A
1-(3-氯-4-氟苯基)-5-(3-氟-5-羟基苯基)-1H-吡唑-3-甲酸

将200mg(0.453mmol)的实施例52A的化合物在氩气下,在1.5ml的1,4-二噁烷和0.9ml的脱气水的混合物中提供,加入152mg(2.72mmol)的氢氧化钾,23.1mg(0.054mmol)的2-二-叔丁基膦基-2’,4’,6’-三异丙基联苯基和16.6mg(0.018mmol)的三(二亚苄基丙酮)二钯(0),并将所述混合物在80℃加热过夜。随后加入1N盐酸水溶液直到pH为酸性,并将反应混合物通过制备HPLC(RP18柱;流动相:乙腈/水梯度)纯化。获得100mg(63%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),10.2(s,1H),7.75(dd,1H),7.54(t,1H),7.33(ddd,1H),7.09(s,1H),6.63(dt,1H),6.59(dt,1H),6.44(t,1H).
LC-MS(方法1):Rt=2.14min;MS(ESIpos):m/z=351[M+H]+.
实施例102A
1-(3-氯-4-氟苯基)-5-(3-甲氧基苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例53A的化合物起始制备标题化合物。获得515mg(93%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.72(dd,1H),7.52(t,1H),7.35-7.26(m,2H),7.11(s,1H),6.96(dd,1H),6.90(t,1H),6.79(d,1H),3.70(s,3H).
LC-MS(方法1):Rt=2.34min;MS(ESIpos):m/z=347[M+H]+.
实施例103A
5-[3,5-双(三氟甲基)苯基]-1-(3-氯苯基)-1H-吡唑-3-甲酸

以类似于合成实施例86A的化合物的方式,从实施例54A的化合物起始制备标题化合物。获得1.78g(79%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),8.15(s,1H),7.96(s,2H),7.62-7.56(m,2H),7.52-7.45(m,2H),7.35-7.30(m,1H).
LC-MS(方法1):Rt=2.80min;MS(ESIpos):m/z=435[M+H]+.
实施例104A
1-(4-氯苯基)-5-[3-(三氟甲基)苯基]-1H-吡唑-3-甲酸

将4.75g(12.0mmol)的实施例55A的化合物在108ml的1,4-二噁烷中提供,加入108ml(216mmol)的2N氢氧化锂在水中的溶液,并将所述混合物在70℃搅拌2小时。浓缩所述混合物,随后将浓盐酸水溶液加入残余物中直到pH为酸性,将所述混合物用二氯甲烷提取,并将有机相通过硫酸镁进行干燥,过滤并浓缩。获得4.40g(100%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.75(d,1H),7.65-7.52(m,5H),7.42-7.36(m,2H),7.25(s,1H).
LC-MS(方法4):Rt=3.71min;MS(ESIpos):m/z=367[M+H]+.
实施例105A
1-(4-氯苯基)-5-(3-氟苯基)-1H-吡唑-3-甲酸

将43.0g(125mmol)的实施例56A的化合物在504ml的1,4-二噁烷中提供,加入94ml(188mmol)的2N氢氧化锂在水中的溶液,并将所述混合物在70℃搅拌2小时。浓缩所述混合物,随后将浓盐酸水溶液加入残余物中直到pH为酸性,将所述混合物用二氯甲烷提取,并将有机相通过硫酸镁进行干燥,过滤并浓缩。获得39.5g(100%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.56-7.51(m,2H),7.46-7.34(m,3H),7.27-7.16(m,2H),7.10(s,1H),7.06(d,1H).
LC-MS(方法1):Rt=2.49min;MS(ESIpos):m/z=317[M+H]+.
实施例106A
1-(3-氯-4-氟苯基)-5-(3-氟-5-甲基苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式(但是伴随搅拌6小时),从实施例57A的化合物起始制备标题化合物。获得2.34g具有79%纯度的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.73(dd,1H),7.52(t,1H),7.32(ddd,1H),7.13(s,1H),7.09(d,1H),7.00(s,1H),6.94-6.89(m,1H),2.27(s,3H).
LC-MS(方法1):Rt=2.52min;MS(ESIpos):m/z=349[M+H]+.
实施例107A
1-(3-氯苯基)-5-(3-氟-5-甲基苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式(伴随搅拌6小时),从实施例58A的化合物起始制备标题化合物。获得1.64g(100%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.1(s,1H),7.57-7.52(m,2H),7.48(t,1H),7.25(ddd,1H),7.13(s,1H),7.09(d,1H),7.01(s,1H),6.89(d,1H),2.27(s,3H).
LC-MS(方法1):Rt=2.49min;MS(ESIpos):m/z=331[M+H]+.
实施例108A
5-(3-氯-4-氟苯基)-1-(3-氰基苯基)-1H-吡唑-3-甲酸
以类似于合成实施例71A的化合物的方式,从实施例59A的化合物起始制备标题化合物。获得21mg具有83%纯度(64%理论值)的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.2(s,1H),7.99-7.95(m,2H),7.70-7.66(m,2H),7.52(dt,1H),7.27(s,1H),7.26-7.23(m,1H),7.20-7.15(m,1H).
LC-MS(方法7):Rt=1.78min;MS(ESIpos):m/z=342[M+H]+.
实施例109A
5-(3-氯苯基)-1-(3-氰基-4-氟苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式(但是伴随搅拌6小时),从实施例60A的化合物起始制备标题化合物。获得672mg具有61%纯度的标题化合物。
LC-MS(方法7):Rt=1.86min;MS(ESIpos):m/z=342[M+H]+.
实施例110A
5-(3-氯苯基)-1-(3-氰基苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式(但是伴随搅拌6小时),从实施例61A的化合物起始制备标题化合物。获得470mg具有77%纯度的标题化合物。
LC-MS(方法7):Rt=1.76min;MS(ESIpos):m/z=324[M+H]+.
实施例111A
1,5-双(3-氰基苯基)-1H-吡唑-3-甲酸
以类似于合成实施例71A的化合物的方式(但是伴随搅拌6小时),从实施例62A的化合物起始制备标题化合物。获得397mg具有36%纯度的标题化合物。
LC-MS(方法7):Rt=1.48min;MS(ESIpos):m/z=315[M+H]+.
实施例112A
1-(3-氰基-4-氟苯基)-5-(3-氰基苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式(但是伴随搅拌6小时),从实施例63A的化合物起始制备标题化合物。获得430mg具有43%纯度的标题化合物。
LC-MS(方法7):Rt=1.58min;MS(ESIpos):m/z=333[M+H]+.
实施例113A
5-(3-氯-5-氟苯基)-1-(3-氰基苯基)-1H-吡唑-3-甲酸
以类似于合成实施例71A的化合物的方式(但是伴随搅拌6小时),从实施例64A的化合物起始制备标题化合物。获得1.21g具有53%纯度的标题化合物。
LC-MS(方法7):Rt=1.83min;MS(ESIpos):m/z=342[M+H]+.
实施例114A
5-(3-氯-5-氟苯基)-1-(3-氰基-4-氟苯基)-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式(但是伴随搅拌6小时),从实施例65A的化合物起始制备标题化合物。获得1.88g具有64%纯度的标题化合物。
LC-MS(方法7):Rt=1.91min;MS(ESIpos):m/z=360[M+H]+.
实施例115A
1-(3-氯苯基)-5-[3-(2-羟基乙氧基)苯基]-1H-吡唑-3-甲酸
以类似于合成实施例71A的化合物的方式,从实施例66A的化合物起始制备标题化合物。获得11mg(82%理论值)标题化合物。
LC-MS(方法1):Rt=1.94min;MS(ESIpos):m/z=359[M+H]+.
实施例116A
1-(3-氯苯基)-5-[3-(3-羟基丙氧基)苯基]-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例67A的化合物起始制备标题化合物。获得99mg具有65%纯度(70%理论值)的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.0(s,1H),7.55-7.43(m,3H),7.30-7.23(m,2H),7.10(s,1H),6.94(dd,1H),6.90-6.86(m,1H),6.77(d,1H),4.52(t,1H),3.97(t,2H),3.50(q,2H),1.82-1.74(m,2H).
LC-MS(方法1):Rt=2.04min;MS(ESIpos):m/z=373[M+H]+.
实施例117A
1-(3-氯苯基)-5-[3-(2-甲氧基乙氧基)苯基]-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式并通过制备HPLC(RP18柱;流动相:乙腈/水梯度,加入0.1%甲酸)进行纯化,从实施例69A的化合物起始制备标题化合物。获得46mg(100%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.0(s,1H),7.55-7.44(m,3H),7.31-7.23(m,2H),7.10(s,1H),6.96(dd,1H),6.91-6.88(m,1H),6.80(d,1H),4.05-4.00(m,2H),3.61-3.54(m,2H),3.28(s,3H).
LC-MS(方法1):Rt=2.23min;MS(ESIpos):m/z=373[M+H]+.
实施例118A
1-(3-氯苯基)-5-[3-(3-吡咯烷-1-基丙氧基)苯基]-1H-吡唑-3-甲酸

以类似于合成实施例71A的化合物的方式,从实施例70A的化合物起始制备标题化合物。获得10mg具有79%纯度(44%理论值)的标题化合物。
LC-MS(方法1):Rt=1.44min;MS(ESIpos):m/z=426[M+H]+.
实施例119A
1-({5-[3-(2-氯乙氧基)-5-氟苯基]-1-(3-氯-4-氟苯基)-1H-吡唑-3-基}羰基)咪唑烷-4-酮

将1.66g(4.73mmol)的实施例101A的化合物,1.04g(5.21mmol)的实施例125A的化合物和3.69g(7.10mmol)的PyBOP提供在80ml的四氢呋喃中,并且在室温,加入1.73ml(9.94mmol)的N,N-二异丙基乙胺。将混合物在室温搅拌过夜,加入水,在真空中去除四氢呋喃,将所述残余物用乙酸乙酯提取,浓缩有机相,并将残余物通过急骤色谱法(流动相:乙酸乙酯/甲醇200/1)进行纯化。获得1.24g(63%理论值)的1-{[1-(3-氯-4-氟苯基)-5-(3-氟-5-羟基苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮。
LC-MS(方法7):Rt=1.59min;MS(ESIpos):m/z=419[M+H]+.
将200mg(0.48mmol)的1-{[1-(3-氯-4-氟苯基)-5-(3-氟-5-羟基苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮提供在2.2ml的四氢呋喃中,并且在室温,加入48μl(0.72mmol)的2-氯乙醇,188mg(0.72mmol)的三苯膦和145mg(0.72mmol)的二异丙基偶氮二羧酸酯。将所述反应混合物在室温搅拌过夜,并随后通过制备HPLC(RP18柱;流动相:乙腈/水梯度)进行纯化。获得24mg(10%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.80(dd,1H),7.54(dt,1H),7.37(ddd,1H),7.20(d,1H),6.95(dt,1H),6.79(s,1H),6.77-6.71(m,1H),5.32(s,0.8H),4.90(s,1.2),4.44(s,1.2H),4.24(t,2H),3.98(s,0.8H),3.90(t,2H).
LC-MS(方法1):Rt=2.40min;MS(ESIpos):m/z=481[M+H]+.
实施例120A
3-(3-{1-(3-氯苯基)-3-[(4-氧代咪唑烷-1-基)羰基]-1H-吡唑-5-基}苯氧基)丙基甲磺酸酯

将3.63g(8.23mmol)的实施例24的化合物提供在24ml的二氯甲烷中,并且在室温,加入2.2ml(19.3mmol)的2,6-二甲基吡啶和0.75ml(9.63mmol)甲磺酰氯。将混合物在室温搅拌过夜,用二氯甲烷稀释,用水和碳酸钠水溶液洗涤,通过硫酸钠干燥并浓缩。获得4.33g具有55%纯度(56%理论值)的标题化合物。
LC-MS(方法1):Rt=2.15min;MS(ESIpos):m/z=519[M+H]+.
实施例121A
2-(3-{1-(3-氯苯基)-3-[(4-氧代咪唑烷-1-基)羰基]-1H-吡唑-5-基}苯氧基)乙基甲磺酸酯

以类似于合成实施例120A的化合物的方式,从实施例23的化合物起始制备标题化合物。获得6.14g具有51%纯度(57%理论值)的标题化合物。
LC-MS(方法1):Rt=2.08min;MS(ESIpos):m/z=505[M+H]+.
实施例122A
N2-苄基甘氨酰胺

将44.2g(0.40mol)的甘氨酰胺盐酸盐在室温,在氩气下,提供在2.21的二氯甲烷中,加入112ml(0.80mol)的三乙胺,并将混合物在室温搅拌过夜。接着加入42.5g(0.40mol)的苯甲醛,用脱水器在回流下加热混合物过夜。浓缩所述混合物,将所述残余物溶解在400ml的四氢呋喃/甲醇(1∶1)中,并且在0℃,分份加入16.7g(0.44mol)的硼氢化钠,并将所述混合物在室温搅拌2天。用抽吸过滤混悬液,浓缩滤液并在高真空下进行干燥。将残余物在乙酸乙酯中搅拌,滤去沉淀物,浓缩滤液,并在甲苯中搅拌残余物过夜。在通过过滤收集固体,并随后在高真空下干燥后,获得56.5g(84%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.36-7.28(m,4H),7.27-7.19(m,1H),3.68-3.64(m,2H),3.03-3.00(m,2H).
LC-MS(方法10):Rt=0.40min;MS(ESIpos):m/z=165[M+H]+.
实施例123A
1-苄基-3-(羟基甲基)咪唑烷-4-酮

将172ml(6.20mol)的37%甲醛溶液加入56.5g(0.34mol)的实施例122A的化合物,并将所述混合物在回流下加热30分钟。将所述混合物用二氯甲烷提取,并将合并的有机相通过硫酸钠干燥,过滤,并浓缩。获得74.5g(100%理论值)标题化合物。
LC-MS(方法5):Rt=0.51min;MS(ESIpos):m/z=207[M+H]+.
实施例124A
1-苄基咪唑烷-4-酮三氟乙酸酯

将74.5g(0.36mol)的实施例123A的化合物在150℃,在高真空下加热6小时,同时蒸馏出挥发性的反应产物。将所述残余物通过HPLC(柱:Sunfire C185μ,250x20mm;洗脱剂:0.2%三氟乙酸/水-乙腈梯度)纯化。获得28.4g(27%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.75(s,1H),7.48-7.39(m,5H),4.41(s,2H),4.22(s,2H),3.54(s,2H).
LC-MS(方法10):Rt=0.94min;MS(ESIpos):m/z=177[M-CF3COOH+H]+.
实施例125A
咪唑烷-4-酮三氟乙酸酯

将28.4g(97.9mmol)的实施例124A的化合物溶解在750ml的乙醇中,并且在氩气下,加入4.5g(42.3mmol)的披钯活性炭(5%)。在室温,在氢气气氛下搅拌所述混合物达24小时。将混悬液通过硅藻土过滤,浓缩并在高真空下干燥。获得19.2g(98%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=10.1(s,2H),8.89(s,1H),4.55(s,2H),3.63(s,2H).
GC-MS(方法11):Rt=3.92min MS(EIpos):m/z=86[M-CF3COOH]+.
实施例126A
3-乙酰基-5-氟苯腈

将824mg(0.900mmol)的三(二亚苄基丙酮)二钯和1.23g(1.98mmol)的外消旋-1,1’-联萘-2,2’-二基双(二苯基磷烷)在氩气气氛下加入在甲苯(300ml)中的9.00g(45.0mmol)的3-溴-5-氟苯腈。在加入19.5g(54.0mmol)的(1-乙氧基乙烯基)三丁基锡烷后,将所述混合物在回流下搅拌过夜。随后浓缩反应混合物并将所述残余物吸收在300ml THF中。在加入100ml的2N盐酸水溶液后,将所述混合物在室温搅拌2小时。随后,将所述反应混合物用饱和的碳酸氢钠水溶液中和,并用乙酸乙酯提取。将合并的有机相通过硫酸镁干燥,过滤,并在真空中浓缩。通过急骤色谱法(流动相:环己烷/乙酸乙酯9∶1)纯化粗制产物。获得7.11g(97%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.28(t,1H),8.18-8.14(m,1H),8.08-8.04(m,1H),2.65(s,3H).
GC-MS(方法11):Rt=3.97min;MS(EIpos):m/z=163[M]+.
实施例127A
4-(3-氰基-5-氟苯基)-2,4-二氧代丁酸乙酯

在-78℃和在氩气气氛下,将5.00g(30.6mmol)的实施例126A的苯乙酮在50ml的二乙醚中的溶液加入42.9ml(42.9mmol,1M,在己烷中)的双(三甲基甲硅烷基)氨基化锂在130ml的二乙醚中。将所述混合物在-78℃搅拌1小时,随后并将5.00ml(36.8mmol)的草酸二乙酯缓慢逐滴加入。将所述混合物缓慢加温到室温并搅拌过夜。将1N盐酸水溶液缓慢加入得到的混悬液中,接着将其用水稀释,并用二氯甲烷提取。通过硫酸镁干燥合并的有机相,过滤并在真空中浓缩。将粗制产物悬浮在二乙醚中,并搅拌30分钟。在过滤和在高真空下干燥后,获得6.50g(81%理论值)作为约9∶1酮/烯醇混合物的标题化合物。
LC-MS(方法12):Rt=0.73,1.04min;MS(ESIneg):m/z=262[M-H]-.
实施例128A
1-(3-氯4-氟苯基)-5-(3-氰基-5-氟苯基)-1H-吡唑-3-甲酸乙酯

将898mg(4.56mmol)的3-氯-4-氟苯基肼盐酸盐加入在15ml的N,N-二甲基乙酰胺中的1.00g(3.80mmol)实施例127A的1,3-二酮化合物,随后将0.190ml的10N盐酸水溶液加入得到的溶液中。将所述混合物在室温搅拌24小时,随后通过制备HPLC(RP18柱;流动相:乙腈/水梯度)直接纯化。获得1.22g(83%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.93(d,1H),7.80(dd,1H),7.72(s,1H),7.58-7.49(m,2H),7.41-7.33(m,2H),4.35(q,2H),1.32(t,3H).
LC-MS(方法5):Rt=1.37min;MS(ESIpos):m/z=388[M+H]+.
实施例129A
1-(3-氯-4-氟苯基)-5-(3-氰基-5-氟苯基)-1H-吡唑-3-甲酸

将12.0ml水和741mg(30.1mmol)氢氧化锂加入在36.0ml的THF中的1.20g(3.10mmol)实施例128A的甲酸酯。将所述混合物在室温搅拌6小时,并随后将1N盐酸水溶液加入反应溶液中。将所述混合物用二氯甲烷提取,并将合并的有机相通过硫酸镁干燥,过滤,并在真空中浓缩。获得1.10g(95%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.2(s,1H),7.97-7.88(m,1H),7.78(dd,1H),7.71(s,1H),7.59-7.46(m,2H),7.35(ddd,1H),7.29(s,1H).
LC-MS(方法5):Rt=1.15min;MS(ESIpos):m/z=360[M+H]+.
实施例130A
1-(3-氯苯基)-5-(3-氰基-5-氟苯基)-1H-吡唑-3-甲酸乙酯

将1.87g(6.28mmol)的具有60%纯度的3-氯苯基肼盐酸盐加入在50ml乙醇中的实施例127A的1.30g(4.83mmol)1,3-二酮化合物,并随后将所述混合物在室温搅拌2小时。随后,将40ml的浓醋酸加入反应溶液中,并将所述混合物在室温搅拌2小时,接着在60℃搅拌1小时。将1N盐酸水溶液加入混合物,将其用二氯甲烷提取。将合并的有机相通过硫酸镁干燥,过滤并在真空中浓缩。将残余物通过急骤色谱法(流动相:环己烷/乙酸乙酯20∶1→1∶1)纯化。获得1.16g(65%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=7.93(d,1H),7.70(s,1H),7.63-7.56(m,2H),7.57-7.41(m,2H),7.37(s,1H),7.28(d,1H),4.35(q,2H),1.33(t,3H).
LC-MS(方法10):Rt=2.51min;MS(ESIpos):m/z=370[M+H]+.
实施例131A
1-(3-氯苯基)-5-(3-氰基-5-氟苯基)-1H-吡唑-3-甲酸

将20.0ml水和453mg(18.9mmol)的氢氧化锂加入在60.0ml THF中的700mg(1.89mmol)的实施例130A的甲酸酯。将所述混合物在室温搅拌过夜,随后将1N盐酸水溶液加入反应溶液中。将所述混合物用二氯甲烷提取,并将合并的有机相通过硫酸镁干燥,过滤并在真空中浓缩。通过制备HPLC(RP18柱;流动相:乙腈/水梯度)纯化粗制产物。获得81%纯度的712mg(89%理论值)标题化合物。
LC-MS(方法10):Rt=2.11min;MS(ESIpos):m/z=342[M+H]+.
实施例132A
1-(3-氰基-4-氟苯基)-5-(3-氰基-5-氟苯基)-1H-吡唑-3-甲酸乙酯

将1.96g(3.65mmol)的具有35%纯度的2-氟-5-肼基苯腈加入在12ml的N,N-二甲基乙酰胺中的800mg(3.04mmol)的实施例127A的1,3-二酮化合物,随后将0.152ml的10N盐酸水溶液加入得到的溶液中。将所述混合物在室温搅拌20小时,随后将其通过制备HPLC(RP18柱;流动相:乙腈/水梯度)直接纯化。获得931mg(61%理论值)具有75%纯度的标题化合物。
LC-MS(方法5):Rt=1.25min;MS(ESIpos):m/z=379[M+H]+.
实施例133A
1-(3-氰基-4-氟苯基)-5-(3-氰基-5-氟苯基)-1H-吡唑-3-甲酸

将10.0ml水和570mg(23.8mmol)的氢氧化锂加入在30.0ml THF中的900mg(1.78mmol)具有75%纯度的实施例132A的甲酸酯。将所述混合物在室温搅拌5小时,随后将1N盐酸水溶液加入反应溶液中。将所述混合物用二氯甲烷进行提取,并将合并的有机相通过硫酸镁干燥,过滤并在真空中浓缩。将粗制产物通过制备HPLC(RP18柱;流动相:乙腈/水梯度)纯化。获得527mg(86%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.09(br.s.,1H),7.93(d,1H),7.81-7.69(m,2H),7.69-7.60(m,1H),7.55(d,1H),7.28(br.s.,1H).
LC-MS(方法5):Rt=1.03min;MS(ESIpos):m/z=351[M+H]+.
实施例134A
5-(3-氰基-5-氟苯基)-1-(3-氰基苯基)-1H-吡唑-3-甲酸乙酯

将985mg(4.56mmol)的具有79%纯度的3-肼基苄腈加入在20ml的N,N-二甲基乙酰胺中的1.00g(3.80mmol)的实施例127A的1,3-二酮化合物,随后将0.190ml的10N盐酸水溶液加入得到的溶液。将所述混合物在室温搅拌20小时,随后通过制备HPLC(RP18柱;流动相:乙腈/水梯度)直接纯化。获得1.22g(80%理论值)具有90%纯度的标题化合物。
LC-MS(方法7):Rt=2.04min;MS(ESIpos):m/z=361[M+H]+.
实施例135A
5-(3-氰基-5-氟苯基)-1-(3-氰基苯基)-1H-吡唑-3-甲酸

将15.0ml水和797mg氢氧化锂(33.3mmol)加入在45.0ml THF中的1.20g(3.00mmol)具有90%纯度的实施例134A的甲酸酯。将所述混合物在室温搅拌5小时,随后将1N盐酸水溶液加入反应溶液中。将所述混合物用二氯甲烷提取,并将合并的有机相通过硫酸镁干燥,过滤并在真空中浓缩。通过制备HPLC(RP18柱;流动相:乙腈/水梯度)纯化粗制产物。获得821mg(82%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=13.23(br.s.,1H),8.01-7.88(m,3H),7.73-7.63(m,3H),7.55(d,1H),7.32(s,1H).
LC-MS(方法7):Rt=1.57min;MS(ESIpos):m/z=332[M+H]+.
示例性实施方案:
实施例1
1-{[1-(3-氯-4-氟苯基)-5-(3-氯-5-氟苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮
将50.0mg(0.14mmol)的实施例71A的化合物,12.8mg(0.15mmol)的咪唑烷-4-酮和106mg(0.20mmol)的PyBOP提供在2.5ml的四氢呋喃中,并且在室温,加入50μl(0.28mmol)的N,N-二异丙基乙胺。将反应混合物在室温搅拌过夜,并随后通过制备HPLC(RP18柱;流动相:乙腈/水梯度纯化。获得49.0mg(83%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.63(s,0.4H),7.85-7.81(m,1H),7.58-7.51(m,2H),7.41-7.34(m,1H),7.29-7.26(m,2H),7.22-7.17(m,1H),5.32(s,0.8H),4.90(s,1.2H),4.43(s,1.2H),3.98(s,0.8H).
LC-MS(方法7):Rt=1.94min;MS(ESIpos):m/z=437[M+H]+.
实施例2
1-{[5-(3-氯-5-氟苯基)-1-(3-氯苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例72A的化合物起始制备标题化合物。获得50mg(84%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.63(s,0.4H),7.63-7.61(m,1H),7.59-7.55(m,1H),7.55-7.47(m,2H),7.33-7.29(m,1H),7.29-7.26(m,1H),7.26-7.24(m,1H),7.21-7.16(m,1H),5.32(s,0.8H),4.90(s,1.2H),4.44(s,1.2H),3.98(s,0.8H).
LC-MS(方法7):Rt=1.90min;MS(ESIpos):m/z=419[M+H]+.
实施例3
1-{[1-(3-氯苯基)-5-(3,5-二氟苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例73A的化合物起始制备标题化合物。获得28mg(73%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.62(s,0.4H),7.61(t,1H),7.59-7.54(m,1H),7.53-7.47(m,1H),7.38-7.27(m,2H),7.27-7.25(m,1H),7.10-7.04(m,2H),5.33(s,0.8H),4.90(s,1.2H),4.44(s,1.2H),3.98(s,0.8H).
LC-MS(方法5):Rt=1.15min;MS(ESIpos):m/z=403[M+H]+.
实施例4
1-{[1,5-双(3-氯苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例74A的化合物起始制备标题化合物。获得110mg(91%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.58(t,1H),7.56-7.52(m,1H),7.51-7.39(m,4H),7.31-7.25(m,1H),7.22-7.18(m,2H),5.34(s,0.8H),4.91(s,1.2H),4.45(s,1.2H),3.98(s,0.8H).
LC-MS(方法9):Rt=2.03min;MS(ESIpos):m/z=401[M+H]+.
实施例5
1-{[1,5-双(3-氯-4-氟苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例75A的化合物起始制备标题化合物。获得23mg(39%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.63(s,0.4H),7.82(dd,1H),7.68(dt,1H),7.53(dt,1H),7.45(t,1H),7.38-7.32(m,1H),7.25-7.19(m,2H),5.33(s,0.8H),4.90(s,1.2H),4.44(s,1.2H),3.98(s,0.8H).
LC-MS(方法7):Rt=1.93min;MS(ESIpos):m/z=437[M+H]+.
实施例6
1-({1-(3-氯苯基)-5-[3-(三氟甲氧基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例76A的化合物起始制备标题化合物。获得24mg(64%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.63(s,0.4H),7.60-7.53(m,3H),7.52-7.46(m,1H),7.45-7.40(m,2H),7.34-7.27(m,1H),7.24-7.22(m,1H),7.21-7.18(m,1H)5.34(s,0.8H),4.91(s,1.2H),4.45(s,1.2H),3.99(s,0.8H).
LC-MS(方法7):Rt=1.97min;MS(ESIpos):m/z=451[M+H]+.
实施例7
1-({1-(3-氯苯基)-5-[4-氟-3-(三氟甲基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例77A的化合物起始制备标题化合物。获得28mg(74%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.63(s,0.4H),7.77(ddd,1H),7.65-7.55(m,3H),7.53-7.46(m,1H),7.44-7.40(m,1H),7.38-7.36(m,1H),7.35-7.29(m,1H),5.33(s,0.8H),4.91(s,1.2H),4.45(s,1.2H),3.99(s,0.8H).
LC-MS(方法5):Rt=1.25min;MS(ESIpos):m/z=453[M+H]+.
实施例8
1-({1-(3-氯-4-氟苯基)-5-[3-(三氟甲氧基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例78A的化合物起始制备标题化合物。获得53mg(91%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.63(s,0.4H),7.78-7.74(m,1H),7.60-7.51(m,2H),7.45-7.35(m,3H),7.24-7.19(m,2H),5.33(s,0.8H),4.91(s,1.2H),4.44(s,1.2H),3.99(s,0.8H).
LC-MS(方法7):Rt=2.02min;MS(ESIpos):m/z=469[M+H]+.
实施例9
1-{[5-(3-氯-4-氟苯基)-1-(3-氯苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例79A的化合物起始制备标题化合物。获得52mg(87%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.62(s,0.4H),7.67-7.63(m,1H),7.61(t,1H),7.57-7.52(m,1H),7.51-7.41(m,2H),7.30-7.21(m,2H),7.21-7.19(m,1H),5.33(s,0.8H),4.91(s,1.2H),4.45(s,1.2H),3.98(s,0.8H).
LC-MS(方法7):Rt=1.89min;MS(ESIpos):m/z=419[M+H]+.
实施例10
1-{[5-(3-氟苯基)-1-(3-甲氧基苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例80A的化合物起始制备标题化合物。获得27mg(70%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.71(s,0.6H),8.62(s,0.4H),7.45-7.34(m,2H),7.27-7.15(m,3H),7.09(d,1H),7.06-7.01(m,1H),6.98-6.95(m,1H),6.92-6.86(m,1H),5.33(s,0.8H),4.90(s,1.2H),4.44(s,1.2H),3.98(s,0.8H),3.72(s,3H).
LC-MS(方法7):Rt=1.63min;MS(ESIpos):m/z=381[M+H]+.
实施例11
1-{[1-(3-氯苯基)-5-(3-甲氧基苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例81A的化合物起始制备标题化合物。获得31mg(84%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.57-7.44(m,3H),7.33-7.25(m,2H),7.14-7.12(m,1H),7.00-6.95(m,1H),6.91-6.87(m,1H),6.83-6.78(m,1H),5.34(s,0.8H),4.90(s,1.2H),4.46(s,1.2H),3.98(s,0.8H),3.70(s,3H).
LC-MS(方法7):Rt=1.73min;MS(ESIpos):m/z=397[M+H]+.
实施例12
1-({5-(3-氯苯基)-1-[3-(三氟甲基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例82A的化合物起始制备标题化合物。获得22mg(58%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.64(s,0.4H),7.86-7.81(m,1H),7.76-7.66(m,3H),7.51-7.47(m,1H),7.46-7.38(m,2H),7.24-7.18(m,2H),5.35(s,0.8H),4.91(s,1.2H),4.46(s,1.2H),3.99(s,0.8H).
LC-MS(方法5):Rt=1.23min;MS(ESIpos):m/z=435[M+H]+.
实施例13
1-{[1-(3-氯苯基)-5-(3,4-二氟苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例83A的化合物起始制备标题化合物。获得16mg(41%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.60(t,1H),7.56-7.44(m,4H),7.30-7.24(m,1H),7.19-7.17(m,1H),7.12-7.06(m,1H),5.34(s,0.8H),4.90(s,1.2H),4.45(s,1.2H),3.98(s,0.8H).
LC-MS(方法5):Rt=1.16min;MS(ESIpos):m/z=403[M+H]+.
实施例14
1-{[5-(3-溴-4-氟苯基)-1-(3-氯苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例84A的化合物起始制备标题化合物。获得26mg(70%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.77-7.72(m,1H),7.60(t,1H),7.57-7.52(m,1H),7.51-7.45(m,1H),7.41(t,1H),7.31-7.24(m,2H),7.21-7.19(m,1H),5.33(s,0.8H),4.90(s,1.2H),4.44(s,1.2H),3.98(s,0.8H).
LC-MS(方法7):Rt=1.92min;MS(ESIpos):m/z=463[M+H]+.
实施例15
1-{[5-(3-氯苯基)-1-(4-氟苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例85A的化合物起始制备标题化合物。获得53mg(42%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.71(s,0.6H),8.63(s,0.4H),7.48-7.31(m,7H),7.20-7.15(m,2H),5.32(s,0.8H),4.90(s,1.2H),4.43(s,1.2H),3.98(s,0.8H).
LC-MS(方法7):Rt=1.77min;MS(ESIpos):m/z=385[M+H]+.
实施例16
1-{[1-(3-氯苯基)-5-(2-氟苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例86A的化合物起始制备标题化合物。获得34mg(88%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.64(s,0.4H),7.57-7.40(m,5H),7.35-7.21(m,3H),7.14-7.11(m,1H),5.38(s,0.8H),4.91(s,1.2H),4.49(s,1.2H),3.99(s,0.8H).
LC-MS(方法5):Rt=1.10min;MS(ESIpos):m/z=385[M+H]+.
实施例17
1-({1-(4-氟苯基)-5-[3-(三氟甲基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例87A的化合物起始制备标题化合物。获得28mg(74%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.63(s,0.4H),7.75(d,1H),7.65-7.56(m,3H),7.49-7.43(m,2H),7.37-7.30(m,2H),7.29-7.26(m,1H),5.33(s,0.8H),4.91(s,1.2H),4.44(s,1.2H),3.99(s,0.8H).
LC-MS(方法5):Rt=1.17min;MS(ESIpos):m/z=419[M+H]+.
实施例18
1-{[1-(3-氯苯基)-5-(2,3-二氟苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例88A的化合物起始制备标题化合物。获得8mg(100%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.64(s,0.4H),7.61-7.43(m,4H),7.35-7.21(m,3H),7.21-7.19(m,1H),5.38(s,0.8H),4.91(s,1.2H),4.49(s,1.2H),3.99(s,0.8H).
LC-MS(方法5):Rt=1.12min;MS(ESIpos):m/z=403[M+H]+.
实施例19
1-{[5-(3-氯苯基)-1-(4-氯苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例89A的化合物起始制备标题化合物。获得31mg(80%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.65(s,0.4H),7.58-7.53(m,2H),7.50-7.38(m,5H),7.20-7.16(m,2H),5.32(s,0.8H),4.90(s,1.2H),4.43(s,1.2H),3.98(s,0.8H).
LC-MS(方法5):Rt=1.22min;MS(ESIpos):m/z=401[M+H]+.
实施例20
1-{[5-(3-氟苯基)-1-(4-氟苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮
以类似于合成实施例1的化合物的方式,从实施例90A的化合物起始制备标题化合物。获得18mg(47%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.63(s,0.4H),7.48-7.39(m,3H),7.37-7.30(m,2H),7.27-7.16(m,3H),7.08-7.04(m,1H),5.32(s,0.8H),4.90(s,1.2H),4.43(s,1.2H),3.98(s,0.8H).
LC-MS(方法7):Rt=1.64min;MS(ESIpos):m/z=369[M+H]+.
实施例21
1-{[1-(3-氯苯基)-5-(3-氟-5-甲氧基苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮
将800mg(2.31mmol)的实施例91A化合物提供在19ml的无水甲苯中,并且在室温,加入0.48ml(6.58mmol)亚硫酰氯,并将所述混合物在回流下加热2.5小时。在冷却后,将所述残余物吸收在6ml二氯甲烷中,并将1/8的溶液在0℃与0.13ml(0.92mmol)三乙胺和92.3mg(0.46mmol)的实施例125A化合物混合,并将其在室温搅拌过夜。浓缩反应混合物,并将所述残余物通过制备HPLC(RP18柱;流动相:乙腈/水梯度)和通过制备薄层色谱法(硅胶;流动相:乙酸乙酯)纯化。获得11mg(9%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.61(s,0.4H),7.59(t,1H),7.57-7.53(m,1H),7.53-7.46(m,1H),7.33-7.26(m,1H),7.21-7.19(m,1H),6.90(dt,1H),6.74-6.69(m,2H),5.33(s,0.8H),4.90(s,1.2H),4.44(s,1.2H),3.98(s,0.8H),3.71(s,3H).
LC-MS(方法1):Rt=2.23min;MS(ESIpos):m/z=415[M+H]+.
实施例22
1-{[1-(3-氯-4-氟苯基)-5-(3-氟-5-甲氧基苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例21的化合物的方式,从实施例92A的化合物起始制备标题化合物。获得29mg(24%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.81(dd,1H),7.54(dt,1H),7.40-7.33(m,1H),7.20-7.18(m,1H),6.90(dt,1H),6.75-6.70(m,2H),5.32(s,0.8H),4.90(s,1.2H),4.44(s,1.2H),3.98(s,0.8H),3.72(s,3H).
LC-MS(方法1):Rt=2.27min;MS(ESIpos):m/z=433[M+H]+.
实施例23
1-({1-(3-氯苯基)-5-[3-(2-羟基乙氧基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

将11.1mg(0.031mmol)的实施例115A化合物,6.8mg(0.034mmol)的实施例125A化合物和24.2mg(0.046mmol)的PyBOP提供在0.6ml的四氢呋喃中,并且在室温,加入11μl(0.065mmol)的N,N-二异丙基乙胺。将所述反应混合物在室温搅拌过夜,并随后通过制备HPLC(RP18柱;流动相:乙腈/水梯度)纯化。获得3.5mg(27%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.57-7.43(m,3H),7.32-7.24(m,2H),7.12-7.10(m,1H),7.00-6.95(m,1H),6.92-6.89(m,1H),6.78(d,1H),5.35(s,0.8H),4.90(s,1.2H),4.85(t,1H),4.46(s,1.2H),3.98(s,0.8H),3.92(t,2H),3.67(q,2H).
LC-MS(方法7):Rt=1.38min;MS(ESIpos):m/z=427[M+H]+.
实施例24
1-({1-(3-氯苯基)-5-[3-(3-羟基丙氧基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例23的化合物的方式,从实施例116A的化合物起始制备标题化合物。获得54mg(69%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.56-7.44(m,3H),7.31-7.24(m,2H),7.12-7.10(m,1H),6.99-6.94(m,1H),6.91-6.88(m,1H),6.80-6.76(m,1H),5.34(s,0.8H),4.90(s,1.2H),4.52(t,1H),4.46(s,1.2H),4.01-3.95(m,2.8H),3.51(q,2H),1.83-1.75(m,2H).
LC-MS(方法1):Rt=1.95min;MS(ESIpos):m/z=441[M+H]+.
实施例25
3-{1-(3-氯-4-氟苯基)-3-[(4-氧代咪唑烷-1-基)羰基]-1H-吡唑-5-基}苯腈

以类似于合成实施例23的化合物的方式,从实施例93A的化合物起始制备标题化合物。将所述产物通过制备薄层色谱法(硅胶;流动相:乙酸乙酯)另外进行纯化,并获得8mg(16%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.63(s,0.4H),7.95-7.92(m,1H),7.89(dt,1H),7.81(dd,1H),7.59(t,1H),7.56-7.49(m,2H),7.38-7.31(m,1H),7.27-7.25(m,1H),5.34(s,0.8H),4.91(s,1.2H),4.45(s,1.2H),3.99(s,0.8H).
LC-MS(方法7):Rt=1.64min;MS(ESIpos):m/z=410[M+H]+.
实施例26
1-{[1-(3-氯-4-氟苯基)-5-(3-氟苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例23的化合物的方式,从实施例94A的化合物起始制备标题化合物。将所述产物通过制备薄层色谱法(硅胶;流动相:乙酸乙酯)另外纯化,并获得10mg(16%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.63(s,0.4H),7.79(dd,1H),7.55(dt,1H),7.48-7.40(m,1H),7.39-7.32(m,1H),7.30-7.23(m,2H),7.19-7.17(m,1H),7.10-7.05(m,1H),5.33(s,0.8H),4.90(s,1.2H),4.45(s,1.2H),3.98(s,0.8H).
LC-MS(方法7):Rt=1.78min;MS(ESIpos):m/z=403[M+H]+.
实施例27
1-({1-(3-氯-4-氟苯基)-5-[3-(三氟甲基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例23的化合物的方式,从实施例95A的化合物起始制备标题化合物。将所述产物通过制备薄层色谱法(硅胶;流动相:乙酸乙酯)另外纯化,并获得17mg(29%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.63(s,0.4H),7.83-7.75(m,2H),7.69-7.50(m,4H),7.41-7.34(m,1H),7.30-7.27(d,1H),5.34(s,0.8H),4.91(s,1.2H),4.45(s,1.2H),3.99(s,0.8H).
LC-MS(方法7):Rt=1.95min;MS(ESIpos):m/z=453[M+H]+.
实施例28
1-({1-(3-氯-4-氟苯基)-5-[3-氟-5-(三氟甲基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例23的化合物的方式,从实施例96A的化合物起始制备标题化合物。获得17mg(27%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.63(s,0.4H),7.84(dd,1H),7.77(d,1H),7.60(d,1H),7.55(dt,1H),7.46-7.37(m,2H),7.37-7.35(m,1H),5.33(s,0.8H),4.91(s,1.2H),4.44(s,1.2H),3.99(s,0.8H).
LC-MS(方法7):Rt=2.01min;MS(ESIpos):m/z=471[M+H]+.
实施例29
1-{[1-(3-氯苯基)-5-{3-[3-(二甲基氨基)丙氧基]苯基}-1H-吡唑-3-基]羰基}咪唑烷-4-酮甲酸酯

将49.6mg(0.096mmol)的实施例120A化合物和0.48ml(0.96mmol)的2M二甲胺在四氢呋喃中的溶液在3ml乙醇中,在80℃搅拌过夜。随后将反应混合物通过制备HPLC纯化(RP18柱;流动相:乙腈/水梯度,加入0.1%甲酸)。获得6.2mg(23%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),8.18(s,1H),7.57-7.43(m,3H),7.31-7.24(m,2H),7.13-7.11(m,1H),6.98-6.93(m,1H),6.88-6.86(m,1H),6.80(d,1H),5.34(s,0.8H),4.90(s,1.2H),4.46(s,1.2H),3.98(s,0.8H),3.92(t,2H),2.33(t,2H),2.15(s,6H),1.82-1.74(m,2H).
LC-MS(方法5):Rt=0.83min;MS(ESIpos):m/z=468[M+H]+.
实施例30
1-({1-(3-氯苯基)-5-[3-氟-5-(三氟甲基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例23的化合物的方式(在制备HPLC中加入0.1%甲酸),从实施例97A的化合物起始制备标题化合物。获得(51%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.63(s,0.4H),7.77(d,1H),7.64-7.55(m,3H),7.53-7.46(m,1H),7.44-7.40(m,1H),7.39-7.29(m,2H),5.33(s,0.8H),4.91(s,1.2H),4.45(s,1.2H),3.99(s,0.8H).
LC-MS(方法1):Rt=2.38min;MS(ESIpos):m/z=453[M+H]+.
实施例31
1-({1-(3-氯苯基)-5-[3-(3-吡咯烷-1-基丙氧基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮甲酸酯
以类似于合成实施例23的化合物的方式,从实施例118A的化合物起始制备标题化合物。获得3.2mg(33%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.71(s,0.6H),8.61(s,0.4H),8.17(s,1H),7.56-7.44(m,3H),7.32-7.24(m,2H),7.13-7.11(m,1H),6.98-6.94(m,1H),6.89-6.86(m,1H),6.82-6.78(m,1H),5.34(s,0.8H),4.90(s,1.2H),4.45(s,1.2H),3.98(s,0.8H),3.94(t,2H),2.49-2.43(m,6H),1.86-1.78(m,2H),1.71-1.66(m,4H).
LC-MS(方法5):Rt=0.86min;MS(ESIpos):m/z=494[M+H]+.
实施例32
1-{[1-(3-溴苯基)-5-(3-氯苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例23的化合物的方式,从实施例98A的化合物起始制备标题化合物。获得122mg(52%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.71-7.65(m,2H),7.51-7.38(m,4H),7.36-7.29(m,1H),7.22-7.17(m,2H),5.34(s,0.8H),4.90(s,1.2H),4.45(s,1.2H),3.98(s,0.8H).
LC-MS(方法5):Rt=1.19min;MS(ESIpos):m/z=445[M+H]+.
实施例33
1-{[1-(3-溴苯基)-5-(3-氯-4-氟苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例23的化合物的方式,从实施例99A的化合物起始制备标题化合物。获得186mg(79%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.73(t,1H),7.69(d,1H),7.52(dt,1H),7.47-7.40(m,1H),7.37-7.31(m,1H),7.28-7.24(m,2H),7.21-7.16(m,1H),5.33(s,0.8H),4.90(s,1.2H),4.44(s,1.2H),3.98(s,0.8H).
LC-MS(方法7):Rt=1.93min;MS(ESIpos):m/z=463[M+H]+.
实施例34
1-{[1-(3-氯苯基)-5-(3-羟基苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

将100mg(0.25mmol)的实施例100A的化合物,25.5mg(0.30mmol)的咪唑烷-4-酮和141mg(0.27mmol)的PyBOP在2ml的四氢呋喃中提供,并且在室温,加入47μl(0.27mmol)的N,N-二异丙基乙胺。将所述反应混合物在室温搅拌过夜,并随后通过制备HPLC(RP18柱;流动相:乙腈/水梯度)纯化。获得83.5mg(72%理论值)的1-({5-[3-(苄氧基)苯基]-1-(3-氯苯基)-1H-吡唑-3-基}羰基)咪唑烷-4-酮。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.56-7.43(m,3H),7.41-7.24(m,7H),7.13-7.11(m,1H),7.07-7.03(m,1H),7.01-6.98(m,1H),6.80(d,1H),5.34(s,0.8H),5.06(s,2H),4.90(s,1.2H),4.46(s,1.2H),3.98(s,0.8H).
LC-MS(方法9):Rt=2.25min;MS(ESIpos):m/z=473[M+H]+.
将317mg(0.671mmol)的1-({5-[3-(苄氧基)苯基]-1-(3-氯苯基)-1H-吡唑-3-基}羰基)咪唑烷-4-酮在5.6ml的浓醋酸中提供,并加入357mg(0.168mmol)的披钯活性炭(5%),并将所述混合物在氢气氛下在室温搅拌过夜。随后,过滤反应混合物,浓缩所述滤液,并将所述残余物通过制备HPLC(RP18柱;流动相:乙腈/水梯度)纯化。获得119mg(46%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=9.67(s,1H),8.71(s,0.6H),8.62(s,0.4H),7.55-7.43(m,3H),7.30-7.16(m,2H),7.03-7.01(m,1H),6.82-6.77(m,1H),6.72-6.65(m,2H),5.34(s,0.8H),4.90(s,1.2H),4.46(s,1.2H),3.98(s,0.8H).
LC-MS(方法1):Rt=1.90min;MS(ESIpos):m/z=383[M+H]+.
实施例35
1-({1-(3-氯苯基)-5-[3-(2-甲氧基乙氧基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例23的化合物的方式,从实施例117A的化合物起始制备标题化合物。为了去除1,1′,1″-磷酰基三吡咯烷,将所述产物吸收在少量四氢呋喃中,将水加入混合物中直到形成沉淀物,在真空中去除四氢呋喃,将所述沉淀物通过吸滤收集,并在高真空下干燥。获得10mg(19%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.56-7.44(m,3H),7.32-7.24(m,2H),7.13-7.11(m,1H),7.00-6.96(m,1H),6.91(s,1H),6.80(d,1H),5.34(s,0.8H),4.90(s,1.2H),4.46(s,1.2H),4.03(t,2H),3.98(s,0.8H),3.60(t,2H),3.28(s,3H).
LC-MS(方法1):Rt=2.13min;MS(ESIpos):m/z=441[M+H]+.
实施例36
1-({1-(3-氯-4-氟苯基)-5-[3-(2-羟基乙氧基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

将1.66g(4.73mmol)的实施例101A的化合物,1.04g(5.21mmol)的实施例125A化合物和3.69g(7.10mmol)的PyBOP提供在80ml的四氢呋喃中,并且在室温,加入1.73ml(9.94mmol)的N,N-二异丙基乙胺。将混合物在室温搅拌过夜,加入水,在真空中去除四氢呋喃,用乙酸乙酯提取残余物,并浓缩,并通过急骤色谱法(流动相:乙酸乙酯/甲醇200/1)纯化残余物。获得1.24g(63%理论值)的1-{[1-(3-氯-4-氟苯基)-5-(3-氟-5-羟基苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮。
LC-MS(方法7):Rt=1.59min;MS(ESIpos):m/z=419[M+H]+.
将100mg(0.239mmol)的1-{[1-(3-氯-4-氟苯基)-5-(3-氟-5-羟基苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮提供在2.5ml的无水丙酮中,并加入36.3mg(0.263mmol)的碳酸钾和149mg(1.19mmol)的2-溴乙醇,并将所述混合物在回流下加热过夜。随后将反应混合物通过制备HPLC(RP18柱;流动相:乙腈/水梯度)纯化。获得30mg(26%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.80(dd,1H),7.53(dt,1H),7.40-7.32(m,1H),7.19-7.17(m,1H),6.90(dt,1H),6.75-6.68(m,2H),5.32(s,0.8H),4.92-4.85(m,2.2H),4.44(s,1.2H),4.01-3.92(m,2.8H),3.65(q,2H).
LC-MS(方法7):Rt=1.53min;MS(ESIpos):m/z=463[M+H]+.
实施例37
1-({1-(3-氯苯基)-5-[3-(3-吗啉-1-基丙氧基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例29的化合物的方式(不加入四氢呋喃),从实施例120A的化合物和吗啉起始制备标题化合物。获得40mg(36%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.56-7.44(m,3H),7.32-7.25(m,2H),7.13-7.11(m,1H),6.96(dd,1H),6.88-6.85(m,1H),6.81(d,1H),5.34(s,0.8H),4.90(s,1.2H),4.46(s,1.2H),3.98(s,0.8H),3.93(t,2H),3.56(t,4H),2.39-2.30(m,6H),1.84-1.75(m,2H).
LC-MS(方法1):Rt=1.62min;MS(ESIpos):m/z=510[M+H]+.
实施例38
1-{[1-(3-氯-4-氟苯基)-5-(3-甲氧基苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例23的化合物的方式,从实施例102A的化合物起始制备标题化合物。获得7mg(12%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.79-7.74(m,1H),7.52(dt,1H),7.38-7.27(m,2H),7.14-7.11(m,1H),7.00-6.95(m,1H),6.93-6.90(m,1H),6.80(d,1H),5.34(s,0.8H),4.90(s,1.2H),4.45(s,1.2H),3.98(s,0.8H),3.71(s,3H).
LC-MS(方法7):Rt=1.77min;MS(ESIpos):m/z=415[M+H]+.
实施例39
1-({1-(3-氯苯基)-5-[3-(3-(甲基氨基)丙氧基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮甲酸酯
以类似于合成实施例29的化合物的方式(不加入四氢呋喃),从实施例120A的化合物和甲胺起始制备标题化合物。获得46mg(44%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.63(s,0.4H),8.28(s,1H),7.56-7.44(m,3H),7.32-7.25(m,2H),7.13-7.11(m,1H),7.00-6.94(m,1H),6.92(t,1H),6.81-6.75(m,1H),5.35(s,0.8H),4.90(s,1.2H),4.46(s,1.2H),4.02-3.96(m,2.8H),2.78(t,2H),2.42(s,3H),1.94-1.85(m,2H).
LC-MS(方法1):Rt=1.62min;MS(ESIpos):m/z=454[M+H]+.
实施例40
1-({1-(3-氯苯基)-5-[3-(3-(乙基氨基)丙氧基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮甲酸酯

以类似于合成实施例29的化合物的方式(不加入四氢呋喃),从实施例120A的化合物和乙胺起始制备标题化合物。获得23mg(21%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.63(s,0.4H),8.29(s,1H),7.56-7.44(m,3H),7.32-7.24(m,2H),7.13-7.11(d,1H),6.99-6.94(m,1H),6.90(t,1H),6.81-6.76(m,1H),5.35(s,0.8H),4.90(s,1.2H),4.46(s,1.2H),4.01-3.96(m,2.8H),2.78-2.64(m,4H),1.90-1.81(m,2H),1.06(t,3H).
LC-MS(方法7):Rt=1.18min;MS(ESIpos):m/z=468[M+H]+.
实施例41
1-({5-[3-(3-氮杂环丁烷-1-基丙氧基)苯基]-1-(3-氯苯基)-1H-吡唑-3-基}羰基)咪唑烷-4-酮甲酸酯

以类似于合成实施例29的化合物的方式(不加入四氢呋喃),从实施例120A的化合物和氮杂环丁烷起始制备标题化合物。获得13mg(11%理论值)标题化合物。
H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),8.18(s,1H),7.56-7.44(m,3H),7.31-7.24(m,2H),7.13-7.11(m,1H),6.97-6.92(m,1H),6.88-6.85(m,1H),6.82-6.78(m,1H),5.34(s,0.8H),4.90(s,1.2H),4.46(s,1.2H),3.98(s,0.8H),3.90(t,2H),3.12(t,4H),2.48-2.43(m,2H),2.01-1.92(m,2H),1.67-1.59(m,2H).
LC-MS(方法1):Rt=1.44min;MS(ESIpos):m/z=480[M+H]+.
实施例42
1-{[1-(3-氯苯基)-5-{3-[2-(甲基氨基)乙氧基]苯基}-1H-吡唑-3-基]羰基)咪唑烷-4-酮甲酸酯

以类似于合成实施例29的化合物的方式(不加入四氢呋喃),从实施例121A的化合物和甲胺起始制备标题化合物。获得26mg(26%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.63(s,0.4H),8.24(s,1H),7.56-7.43(m,3H),7.33-7.25(m,2H),7.13-7.11(m,1H),7.02-6.97(m,1H),6.94-6.91(m,1H),6.86-6.78(m,1H),5.35(s,0.8H),4.91(s,1.2H),4.46(s,1.2H),4.02(t,2H),3.98(s,0.8H),3.19-3.16(m,2H),2.92(t,2H),2.40(s,3H).
LC-MS(方法1):Rt=1.52min;MS(ESIpos):m/z=440[M+H]+.
实施例43
1-{[1-(3-氯苯基)-5-{3-[3-(4-甲基哌嗪-1-基)丙氧基]苯基}-1H-吡唑-3-基]羰基}咪唑烷-4-酮甲酸酯

以类似于合成实施例29的化合物的方式,从实施例120A的化合物和1-甲基哌嗪起始制备标题化合物。获得45mg(38%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),8.16(s,1H),7.56-7.43(m,3H),7.31-7.25(m,2H),7.13-7.11(m,1H),6.98-6.93(m,1H),6.88-6.85(m,1H),6.80(d,1H),5.34(s,0.8H),4.90(s,1.2H),4.45(s,1.2H),3.98(s,0.8H),3.92(t,2H),2.39-2.30(m,10H),2.17(s,3H),1.82-1.74(m,2H).
LC-MS(方法1):Rt=1.40min;MS(ESIpos):m/z=523[M+H]+.
实施例44
1-{[1-(3-氯苯基)-5-{3-[2-(4-甲基哌嗪-1-基)乙氧基]苯基}-1H-吡唑-3-基]羰基}咪唑烷-4-酮二甲酸酯

以类似于合成实施例29的化合物的方式,从实施例121A的化合物和1-甲基哌嗪起始制备标题化合物。获得46mg(38%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),8.15(s,2H),7.56-7.44(m,3H),7.31-7.25(m,2H),7.14-7.12(d,1H),6.99-6.95(m,1H),6.90-6.87(m,1H),6.83-6.79(m,1H),5.34(s,0.8H),4.90(s,1.2H),4.46(s,1.2H),4.02-3.97(m,2.8H),2.63-2.58(m,2H),2.48-2.30(m,8H),2.18(s,3H).
LC-MS(方法1):Rt=1.39min;MS(ESIpos):m/z=509[M+H]+.
实施例45
1-({1-(3-氯苯基)-5-[3-(2-吗啉-4-基乙氧基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮甲酸酯

以类似于合成实施例29的化合物的方式(不加入四氢呋喃),从实施例121A的化合物和吗啉起始制备标题化合物。获得47mg(43%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),8.14(s,1H),7.56-7.44(m,3H),7.32-7.25(m,2H),7.14-7.12(m,1H),6.99-6.95(m,1H),6.90-6.87(m,1H),6.81(d,1H),5.34(s,0.8H),4.90(s,1.2H),4.46(s,1.2H),4.01(t,2H),3.98(s,0.8H),3.56(t,4H),2.61(t,2H),2.42(t,4H).
LC-MS(方法1):Rt=1.58min;MS(ESIpos):m/z=496[M+H]+.
实施例46
1-{[1-(3-氯苯基)-5-(3-{2-[(1-甲基乙基)氨基]乙氧基}苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮甲酸酯

以类似于合成实施例29的化合物的方式(不加入四氢呋喃),从实施例121A的化合物和异丙胺起始制备标题化合物。获得29mg(28%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.63(s,0.4H),8.20(s,1H),7.56-7.44(m,3H),7.33-7.25(m,2H),7.13-7.11(m,1H),7.02-6.97(m,1H),6.94-6.91(m,1H),6.83-6.78(m,1H),5.35(s,0.8H),4.90(s,1.2H),4.46(s,1.2H),4.03-3.97(m,2.8H),2.95-2.85(m,3H),1.03(d,6H).
LC-MS(方法1):Rt=1.42min;MS(ESIpos):m/z=468[M+H]+.
实施例47
1-({5-[3,5-双(三氟甲基)苯基]-1-(3-氯苯基)-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例103A的化合物起始制备标题化合物。获得26mg(72%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.63(s,0.4H),8.16(s,1H),7.96(s,2H),7.66-7.63(m,1H),7.60-7.55(m,1H),7.53-7.46(m,2H),7.38-7.31(m,1H),5.34(s,0.8H),4.92(s,1.2H),4.46(s,1.2H),4.00(s,0.8H).
LC-MS(方法7):Rt=2.14min;MS(ESIpos):m/z=503[M+H]+.
实施例48
1-({1-(4-氯苯基)-5-[3-(三氟甲基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例104A的化合物起始制备标题化合物。获得34mg(92%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.65(s,0.4H),7.77(d,1H),7.67-7.60(m,2H),7.59-7.53(m,3H),7.45-7.39(m,2H),7.29-7.27(m,1H),5.33(s,0.8H),4.91(s,1.2H),4.44(s,1.2H),3.99(s,0.8H).
LC-MS(方法7):Rt=1.95min;MS(ESIpos):m/z=435[M+H]+.
实施例49
1-{[1-(4-氯苯基)-5-(3-氟苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例1的化合物的方式,从实施例105A的化合物起始制备标题化合物。获得15mg(39%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.71(s,0.6H),8.64(s,0.4H),7.58-7.53(m,2H),7.47-7.38(m,3H),7.29-7.16(m,3H),7.07(d,1H),5.33(s,0.8H),4.90(s,1.2H),4.44(s,1.2H),3.98(s,0.8H).
LC-MS(方法5):Rt=1.14min;MS(ESIpos):m/z=385[M+H]+.
实施例50
1-{[1-(3-氯-4-氟苯基)-5-(3-氟-5-甲基苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例23的化合物的方式,使用3.2当量的N,N-二异丙基乙胺,并在制备HPLC中加入0.1%甲酸,从实施例106A的化合物起始制备标题化合物。获得5mg(6%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.81-7.77(m,1H),7.56-7.49(m,1H),7.38-7.31(m,1H),7.16-7.08(m,2H),7.03-7.00(m,1H),6.95-6.90(m,1H),5.33(s,0.8H),4.90(s,1.2H),4.44(s,1.2H),3.98(s,0.8H),2.28(s,3H).
LC-MS(方法1):Rt=2.36min;MS(ESIpos):m/z=417[M+H]+.
实施例51
1-{[1-(3-氯苯基)-5-(3-氟-5-甲基苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例23的化合物的方式,使用3.2当量的N,N-二异丙基乙胺,并在制备HPLC中加入0.1%甲酸,从实施例107A的化合物起始制备标题化合物。获得24mg(19%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.62(s,0.4H),7.59-7.45(m,3H),7.30-7.24(m,1H),7.16-7.08(m,2H),7.02(s,1H),6.93-6.87(m,1H),5.34(s,0.8H),4.90(s,1.2H),4.45(s,1.2H),3.98(s,0.8H),2.28(s,3H).
LC-MS(方法7):Rt=1.87min;MS(ESIpos):m/z=399[M+H]+.
实施例52
1-{[5-(3-氯-4-氟苯基)-1-(3-氰基苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例23的化合物的方式,在制备HPLC中加入0.1%甲酸,从实施例108A的化合物起始制备标题化合物。获得8mg(35%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.64(s,0.4H),8.08-8.05(m,1H),7.98-7.94(m,1H),7.70-7.65(m,2H),7.53(dt,1H),7.30-7.25(m,2H),7.21-7.16(m,1H),5.33(s,0.8H),4.91(s,1.2H),4.46(s,1.2H),3.99(s,0.8H).
LC-MS(方法1):Rt=2.14min;MS(ESIpos):m/z=410[M+H]+.
实施例53
1-{[5-(3-氯苯基)-1-(3-氰基-4-氟苯基)-1H-吡唑-3-基]羰基}咪唑烷-4-酮

以类似于合成实施例23的化合物的方式,使用3.2当量的N,N-二异丙基乙胺,并在制备HPLC中加入0.1%甲酸,从实施例109A的化合物起始制备标题化合物。获得45mg(77%理论值)的标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.65(s,0.4H),8.22-8.17(m,1H),7.77-7.70(m,1H),7.63(t,1H),7.53-7.47(m,2H),7.40(t,1H),7.23-7.21(m,1H),7.16(d,1H),5.34(s,0.8H),4.91(s,1.2H),4.46(s,1.2H),3.99(s,0.8H).
LC-MS(方法1):Rt=2.16min;MS(ESIpos):m/z=410[M+H]+.
实施例54
1-({1-(3-氯-4-氟苯基)-5-[3-氟-5-(2-吡咯烷-1-基乙氧基)苯基]-1H-吡唑-3-基}羰基)咪唑烷-4-酮

以类似于合成实施例29的化合物的方式(在制备HPLC中不加入0.1%甲酸),从实施例119A的化合物和吡咯烷起始制备标题化合物。获得2.5mg(5%理论值)标题化合物。
1H-NMR(500MHz,DMSO-d6):δ=8.74(s,0.6H),8.65(s,0.4H),7.84-7.78(m,1H),7.59-7.51(m,1H),7.41-7.33(m,1H),7.20(s,1H),6.90(d,1H),6.77(d,1H),6.67(s,1H),5.32(s,0.8H),4.90(s,1.2H),4.44(s,1.2H),4.05-3.95(m,2.8H),2.71-2.63(m,2H),2.44(s,4H),1.66(s,4H).
LC-MS(方法1):Rt=1.52min;MS(ESIpos):m/z=516[M+H]+.
实施例55
1-({5-[3-(3-氨基丙氧基)苯基]-1-(3-氯苯基)-1H-吡唑-3-基}羰基)咪唑烷-4-酮甲酸酯

以类似于合成实施例29的化合物的方式,其中在制备HPLC中加入0.1%甲酸,从实施例120A的化合物和氨水在甲醇中的7N溶液起始制备标题化合物。获得10mg(10%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.63(s,0.4H),8.36(s,1H),7.56-7.44(m,3H),7.32-7.24(m,2H),7.12-7.10(m,1H),7.00-6.92(m,2H),6.80-6.75(m,1H),5.35(s,0.8H),4.90(s,1.2H),4.46(s,1.2H),4.04-3.97(m,2.8H),2.81(t,2H),1.92-1.83(m,2H).
LC-MS(方法1):Rt=1.41min;MS(ESIpos):m/z=440[M+H]+.
实施例56
3-{5-(3-氯苯基)-3-[(4-氧代咪唑烷-1-基)羰基]-1H-吡唑-1-基}苯腈

以类似于合成实施例23的化合物的方式,使用3.2当量的N,N-二异丙基乙胺,并且在制备HPLC中加入0.1%甲酸,从实施例110A的化合物起始制备标题化合物。为了去除1,1′,1″-磷酰基三吡咯烷,将所述产物吸收在少量四氢呋喃中,将水加入混合物中直到形成沉淀物,在真空中去除四氢呋喃,并通过吸滤收集沉淀物,在高真空下干燥。获得42mg(65%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.64(s,0.4H),8.05-8.02(m,1H),7.96-7.92(m,1H),7.68-7.63(m,2H),7.52-7.45(m,2H),7.41(t,1H),7.23-7.21(m,1H),7.18(dt,1H),5.35(s,0.8H),4.91(s,1.2H),4.47(s,1.2H),3.99(s,0.8H).
LC-MS(方法1):Rt=2.10min;MS(ESIpos):m/z=392[M+H]+.
实施例57
3,3′-{3-[(4-氧代咪唑烷-1-基)羰基]-1H-吡唑-1,5-二基}二苯腈

以类似于合成实施例23的化合物的方式,使用3.2当量的N,N-二异丙基乙胺并在制备HPLC中加入0.1%甲酸,从实施例111A的化合物起始制备标题化合物。获得16mg(29%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.65(s,0.4H),8.06-8.03(m,1H),7.97-7.87(m,3H),7.67-7.63(m,2H),7.58(t,1H),7.52(dt,1H),7.29-7.27(m,1H),5.36(s,0.8H),4.91(s,1.2H),4.47(s,1.2H),3.99(s,0.8H).
LC-MS(方法5):Rt=0.91min;MS(ESIpos):m/z=383[M+H]+.
实施例58
5-{5-(3-氰基苯基)-3-[(4-氧代咪唑烷-1-基)羰基]-1H-吡唑-1-基}-2-氟苯腈

以类似于合成实施例23的化合物的方式,使用3.2当量的N,N-二异丙基乙胺并在制备HPLC中加入0.1%甲酸,从实施例112A的化合物起始制备标题化合物。为了去除1,1′,1″-磷酰基三吡咯烷,将所述产物吸收在少量四氢呋喃中,将水加入混合物中直到形成沉淀物,在真空中去除四氢呋喃,并将所述沉淀物通过吸滤收集,在高真空下干燥。获得25mg(48%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.66(s,0.4H),8.23-8.18(m,1H),7.97-7.94(m,1H),7.89(dt,1H),7.76-7.70(m,1H),7.66-7.50(m,3H),7.29-7.27(m,1H),5.34(s,0.8H),4.91(s,1.2H),4.46(s,1.2H),3.99(s,0.8H).
LC-MS(方法1):Rt=1.93min;MS(ESIpos):m/z=401[M+H]+.
实施例59
3-{5-(3-氯-5-氟苯基)-3-[(4-氧代咪唑烷-1-基)羰基]-1H-吡唑-1-基}苯腈

以类似于合成实施例23的化合物的方式,使用3.2当量的N,N-二异丙基乙胺,并在制备HPLC中加入0.1%甲酸,从实施例113A的化合物起始制备标题化合物。获得58mg(77%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.72(s,0.6H),8.64(s,0.4H),8.08-8.04(m,1H),7.98-7.94(m,1H),7.70-7.65(m,2H),7.54(dt,1H),7.30-7.28(m,1H),7.28-7.25(m,1H),7.21-7.16(m,1H),5.34(s,0.8H),4.91(s,1.2H),4.46(s,1.2H),3.99(s,0.8H).
LC-MS(方法5):Rt=1.10min;MS(ESIpos):m/z=410[M+H]+.
实施例60
5-{5-(3-氯-5-氟苯基)-3-[(4-氧代咪唑烷-1-基)羰基]-1H-吡唑-1-基}-2-氟苯腈

以类似于合成实施例23的化合物的方式,使用3.2当量的N,N-二异丙基乙胺并在制备HPLC中加入0.1%甲酸,从实施例114A的化合物起始制备标题化合物。获得44mg(48%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.65(s,0.4H),8.24-8.19(m,1H),7.79-7.72(m,1H),7.65(t,1H),7.54(dt,1H),7.32-7.29(m,1H),7.29-7.27(m,1H),7.22-7.17(m,1H),5.33(s,0.8H),4.91(s,1.2H),4.45(s,1.2H),3.99(s,0.8H).
LC-MS(方法5):Rt=1.14min;MS(ESIpos):m/z=428[M+H]+.
实施例61
3-{1-(3-氯苯基)-3-[(4-氧代咪唑烷-1-基)羰基]-1H-吡唑-5-基}-5-氟苯腈

将80.0mg(0.190mmol)的具有81%纯度的实施例131A的化合物,49.5mg(0.247mmol)的实施例125A化合物和175mg(0.337mmol)的PyBOP在3.0ml的四氢呋喃中进行提供,并且在室温,加入125μl(0.179mmol)的N,N-二异丙基乙胺。将混合物在室温搅拌过夜,并将反应溶液用二氯甲烷稀释,随后用1N盐酸水溶液洗涤。在将有机相通过硫酸镁干燥后,过滤,并在真空中浓缩,将粗制产物通过制备HPLC(RP18柱;流动相:乙腈/水梯度)纯化。获得56.0mg(72%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(b r.s.,0.6H),8.62(s,0.4H),7.94(d,1H),7.71(br.s.,1H),7.66-7.44(m,4H),7.37-7.23(m,2H),5.33(s,0.8H),4.91(s,1.2H),4.45(s,1.2H),3.99(s,0.8H).
LC-MS(方法5):Rt=1.09min;MS(ESIpos):m/z=410[M+H]+.
实施例62
3-{1-(3-氯-4-氟苯基)-3-[(4-氧代咪唑烷-1-基)羰基]-1H-吡唑-5-基}-5-氟苯腈

将100mg(0.263mmol)的实施例129A的化合物,57.8mg(0.289mmol)的实施例125A的化合物和205mg(0.394mmol)的PyBOP在5.0ml的四氢呋喃中进行提供,并且在室温,加入146μl(0.841mmol)的N,N-二异丙基乙胺。将混合物在室温搅拌过夜,并将反应溶液用二氯甲烷稀释,随后用1N盐酸水溶液洗涤。在通过硫酸镁干燥有机相后,将其过滤并在真空中浓缩,将粗制产物通过制备HPLC(RP18柱;流动相:乙腈/水梯度)纯化。获得78.0mg(66%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.63(s,0.4H),7.94(d,1H),7.84(d,1H),7.73(d,1H),7.61-7.48(m,2H),7.43-7.25(m,2H),5.33(s,0.8H),4.91(s,1.2H),4.44(s,1.2H),3.99(s,0.8H).
LC-MS(方法1):Rt=2.09min;MS(ESIpos):m/z=428[M+H]+.
实施例63
5-{5-(3-氰基-5-氟苯基)-3-[(4-氧代咪唑烷-1-基)羰基]-1H-吡唑-1-基}-2-氟苯腈

将80.0mg(0.228mmol)的实施例133A的化合物,50.3mg(0.251mmol)的实施例125A的化合物和178mg(0.394mmol)的PyBOP在5.0ml的四氢呋喃中提供,并且在室温,加入127μl(0.731mmol)的N,N-二异丙基乙胺。将混合物在室温搅拌过夜,并将反应溶液用二氯甲烷稀释,随后用1N盐酸水溶液洗涤。在通过硫酸镁干燥有机相后,过滤并在真空中浓缩,通过制备HPLC(RP18柱;流动相:乙腈/水梯度)纯化粗制产物。将获得的固体悬浮在乙腈/二乙醚中,搅拌30分钟并随后过滤。获得20.0mg(21%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.65(s,0.4H),8.27-8.13(m,1H),7.95(d,1H),7.74(d,2H),7.68-7.52(m,2H),7.33(s,1H),5.34(s,0.8H),4.91(s,1.2H),4.46(s,1.2H),3.99(s,0.8H).
LC-MS(方法5):Rt=1.00min;MS(ESIpos):m/z=419[M+H]+.
实施例64
3-{1-(3-氰基苯基)-3-[(4-氧代咪唑烷-1-基)羰基]-1H-吡唑-5-基}-5-氟苯腈

将100mg(0.301mmol)的实施例135A的化合物,66.2mg(0.331mmol)的实施例125A的化合物和235mg(0.451mmol)的PyBOP在5.0ml的四氢呋喃中提供,并且在室温,加入168μl(0.936mmol)的N,N-二异丙基乙胺。将混合物在室温搅拌过夜,并将所述反应溶液用二氯甲烷稀释,随后用1N盐水水溶液洗涤。在通过硫酸镁干燥有机相后,将其过滤并且在真空中浓缩,将粗制产物通过制备HPLC(RP18柱;流动相:乙腈/水梯度)纯化。将获得的固体悬浮在乙腈/二乙醚中,搅拌30分钟并随后过滤。获得18.0mg(15%理论值)标题化合物。
1H-NMR(400MHz,DMSO-d6):δ=8.73(s,0.6H),8.64(s,0.4H),8.07(d,1H),8.00-7.90(m,2H),7.76-7.70(m,1H),7.69-7.62(m,2H),7.56(dd,1H),7.37-7.31(m,1H),5.35(s,0.8H),4.91(s,1.2H),4.46(s,1.2H),3.99(s,0.8H).
LC-MS(方法5):Rt=0.97min;MS(ESIpos):m/z=401[M+H]+.
B)评价生理学活性
缩写:
DMSO 二甲亚砜
FCS 胎牛血清(Biochrom AG,柏林,德国)
PBS 磷酸盐缓冲液
MTP 微量滴定板
ELISA 酶联免疫吸附测定
本发明化合物用于治疗由逆转录病毒导致的疾病的适合性可以在下面的测定系统中显示:
体外测定法
生化逆转录酶测定
根据生产商的信息,使用“Reverse Transcriptase Assay,colorimetric(逆转录酶测定法,比色法)″(Roche Diagnostics GmbH(罗氏诊断有限公司),曼海姆,德国)”。将测试物质溶解在DMSO中,并将其用在以5倍梯级(最终DMSO浓度为1%)稀释的测试中。得到的光度测定评估值(405/492nm)关于阴性对照(没有逆转录酶的混合物)是少于0.1,而关于阳性对照(没有测试物质的混合物)在1.5的区域内。将测试物质的IC50值确定为测量的光密度是阳性对照的50%时的测试物质稀释物的浓度。
发现本发明的化合物抑制逆转录酶活性。将实验数据总结在表A中。
用野生型和抑制剂抗性的HI报道病毒进行的光测定法
将携带lu164基因(萤光素酶164)而不是nef基因的HIV-1NL4-3报道病毒用于该测定法中。通过用相应的原病毒pNL4-3质粒(脂转染胺试剂,Invitrogen,Karlsruhe,德国)转染293T细胞来产生病毒。使用″QuikChangeII XL定点诱变试剂盒”(Stratagene,Cedar Creek,德克萨斯,USA)从原病毒质粒DNA起始,提供在逆转录酶基因中具有限定抗性突变的病毒。特别产生下列突变:A98G,A98G-K103N-V108I,A98S,F227C,F227L,G190A,G190S,K101E,K101Q-K103N,K103N,K103N-F227L,K103N-G190A,K103N-G190S,K103N-M230L,K103N-N348I,K103N-P225H,K103N-V108I,K103N-V108I-P225H,K103N-V179F-Y181C,K103N-Y181C,K103N-Y181C-G190A,L100I,L100I-K103N,L100I-K103N-V179I-Y181C,L100I-K103N-Y181C,L234I,N348I,P225H,P236L,V106A,V106A-E138K,V106A-F227C,V106A-F227L,V106I,V106I-Y188L,V106M,V108I,V179F-Y181C,V179I,V179I-Y181C,Y181C,Y181C-G190A,Y181C-M230L,Y181I,Y188L。用这些报道病毒感染的MT47F2细胞将萤光素酶分泌到培养基中,由此使病毒复制通过发光测定法进行量化。
对于96孔MTP的混合物,使3百万MT4 7F2细胞沉淀,将其悬浮在1ml不含酚红的RPMI 1640培养基(Invitrogen,Karlsruhe,德国)/10%FCS/10% AIM-V(Invitrogen,Karlsruhe,德国)中,并将其与适量的相应HIV-1NL4-3报道病毒在37℃一起温育2小时(沉淀物感染)。随后将未吸附的病毒用PBS洗出,并再次将感染的细胞沉淀,将其悬浮在8ml的不含酚红/2%或10%FCS/10%AIM-V的RPMI 1640培养基中。将其中的80μl吸移到白色96-孔MTP的每个孔中,所述孔具有20μl的适合稀释度的测试物质。为了避免边缘作用,在MTP边缘上的孔没有用于物质稀释。MTP的第二垂直排仅包含感染的细胞(病毒对照),并且第十一垂直排仅包含未感染的细胞(细胞对照),在每个情形中,在不含酚红/2%或10%FCS/10%AIM-V的RPMI 1640培养基中。MTP的其它孔从第三垂直排开始包含各种浓度的本发明的化合物,从所述第三垂直排,测试物质以3倍梯级稀释到第十垂直排37-倍。将测试物质溶解在DMSO中,其中在测试混合物中的最终DMSO浓度最终是1%。将测试混合物在37℃/5%CO2温育5天,并且在加入15μl的Lu164底物(5mg/ml溶解在30μM谷胱甘肽/DMSO,100mM NaCl,1M MES,100mM谷胱甘肽中的腔肠素(coelenterazine))后,通过发光光度法进行评估。得到的值对于病毒对照在1000000 RLUs(相对光单位)的区域内,而对于细胞对照是300-400RLUs。将测试物质的EC50值确定为这样的浓度,在所述浓度下以RLUs测量的病毒复制是未处理的感染细胞的50%。
发现本发明的化合物抑制HIV复制。将实验数据总结在表A中。
用野生型HIV-1进行的PBL和H9测定法
使用Ficoll-Paque Leucosep试管(Greiner Bio-One,Frickenhausen,德国)从血液中分离原代人血淋巴细胞(PBLs),并将其用RPMI 1640培养基(Invitrogen,Karlsruhe,德国)/10%FCS中的植物凝集素(90μg/ml)和白介素-2(40U/ml)刺激3天。
关于用于96-孔MTP的混合物,沉淀3百万PBLs,将其悬浮在1ml的RPMI 1640培养基/10%FCS中,并将其与适量的HIV-1LAI(NIH AIDSResearch&Reference Reagent Program(NIH AIDS研究&参考试剂项目),日尔曼敦,USA)在37℃温育2小时(沉淀物感染)。随后将未吸附的病毒用PBS洗出,并将感染的细胞再次沉淀,将其悬浮在18ml的RPMI 1640培养基/10%FCS/白介素-2(40U/ml)中。将其中的180μl吸移到白色96孔MTP的每个孔中,所述孔具有20μl的适合稀释度的测试物质。备选地,在MTP中制备物质稀释度后,将HIV与细胞一起移入,并不再次洗涤出来(上清液感染)。为了避免边缘效应,未将在MTP边缘的孔用于物质稀释。MTP的第二垂直排仅包含感染的细胞(病毒对照),并且第十一垂直排仅包含未感染的细胞(细胞对照),在每个情形中,在RPMI 1640培养基/10%FCS/白介素-2(40U/ml)中。MTP的其它孔从第三垂直排起始包含各种浓度的本发明的化合物,从所述第三垂直排,测试物质以3倍步骤稀释到第十垂直排37-倍。将测试物质溶解在DMSO中,其中在测试混合物中的最终DMSO浓度最终是1%。将测试混合物在37℃/5%CO2温育。在5和7天后,将50μl的无细胞上清液从每个孔中移出以通过p24ELISA确定存在的p24的量(HIV-1p24CA抗原捕获测定试剂盒,NCI-Frederick Cancer Research and Development Center(NCI-Frederick癌症研究和开发中心),弗雷德里克,USA)。从光度测定评估(450/620nm)得到的值,将测试物质的EC50值确定为这样的浓度,在所述浓度上,p24的量是未处理的感染的细胞的50%。
备选地,使用H9细胞(ATCC,Wesel,德国)替代PBLs用于测试测试物质。根据上述模式,将H9细胞在具有2%或10%FCS的RPMI 1640培养基中在37℃/5%CO2进行温育5天,作为HIV-1LAI上清液感染(20μl的物质稀释液和80μl的细胞/病毒/孔)。随后,将10μl的AlamarBlue(Invitrogen,Karlsruhe,德国)加入每个孔中,并在发生荧光评估(544/590nm)之前,将MTPs在37℃温育3小时。得到的值对于未处理的未感染细胞是约40000,而对于未处理的感染细胞是约7000。在低浓度范围中,将测试物质的EC50值确定为这样的浓度,在所述浓度上,荧光是未处理的未感染细胞的50%(在每个情形中,扣除未处理的感染细胞的值)。此外,在高浓度范围内,将测试物质的CC50值确定为这样的浓度,在所述浓度,荧光是未处理的未感染细胞的50%(在每个情形中,扣除未处理的感染细胞的值)。
发现本发明的化合物抑制HIV复制。将实验数据总结在表A中。
确定测试物质的细胞毒性作用的测定法
为了确定在未感染的细胞中的测试物质的细胞毒性作用,将物质以适当浓度移入透明的96-孔MTPs中,并将其与未感染的细胞一起温育(例如H9,PBLs,THP-1,MT4 7F2,CEM,Jurkat)(与上述测定法类似)。5天后,将每孔1/10体积的AlamarBlue加入测试混合物中,并将MTPs在37℃温育3小时。随后发生荧光评估(544/590nm)。取决于细胞类型,得到的值对于未处理的细胞在20000和40000之间。将测试物质的CC50直到确定为这样的浓度,在所述浓度下荧光是未处理的细胞的50%。没有评估在作用浓度范围内显示细胞毒性发现的测试物质它们的抗病毒活性。
表A:

体内测定法
动物模型:
NOD Scid小鼠,通常5-6周龄,购自商业饲养者(例如,Taconic或Jackson实验室)。将动物在无菌条件(包括草垫和饲料)下保持在隔离器中。
将限定量的细胞(例如5×106T细胞(例如C8166))用适量m.o.i.(例如0.01TCID50)HIV感染。将感染的细胞引入胶原海绵中。将以这种方式预处理的海绵植入小鼠的脊背皮肤下。每天,对小鼠口服、腹膜内、皮下或静脉内处理一次或数次,其中可以在植入前发生第一次处理。处理组通常包括10只小鼠。至少一组用安慰剂处理,至少一组用已知活性的物质(=阳性对照)处理,并且通常将数个组用本发明的物质处理。本发明物质的每日剂量是每kg体重0.01mg和100mg之间。将物质配制在PBS中2%DMSO/0.5%甲基纤维素中,或另一种适合的混合物中,所述混合物促进物质的溶解性。所述处理通常持续4天半。在最后一次施用所述物质后,处死所述动物,并去除海绵。通过胶原酶消化,病毒-感染的细胞获自海绵。
总RNA获自细胞,并且通过关于病毒RNA含量的定量PCR来检查。基于持家基因(例如GAPDH)的量来对病毒RNA的量进行标准化。确定与安慰剂处理的对照组比较的用物质处理后的HIV RNA的量。如果使用携带萤光素酶的HIV,可以另外或作为替代进行萤光素酶测量。在该情形中,从萤光素酶信号的水平确定HIV的量,因为其在该情形中,充当病毒复制的量度。统计学分析通过适合的计算机程序,例如Graph Pad Prism进行。
B)药物代谢动力学性质的评估
体内研究
为了确定体内药物代谢动力学,将测试物质静脉内和口服施用于小鼠、大鼠和狗。用于确定测试物质的药物代谢动力学性质的在静脉内研究中选择的剂量在所有物种中是0.5mg/kg。在口服施用时,将3mg/kg施用于啮齿动物,并将1mg/kg施用于狗。将测试物质配制在99%的血浆,1%DMSO中,以用于向啮齿动物的静脉内施用,并且以不同比例配制在PEG400,乙醇和水中以用于口服施用。将后一种赋形剂用于关于狗的两种施用途径。
在施用测试物质前,将雄性Wistar大鼠进行插管,从而可以在原位在导管的辅助下或通过在不同时间,以2分钟到至多26小时的间隔,对静脉进行穿刺来取血液样品。
将测试物质作为推注静脉内施用于雌性BalbC小鼠,并且在该情形中,通过从2分钟到至多26小时的间隔,穿刺腔静脉来专门获得样品。向雌性小猎犬的施用只通过15分钟的静脉内输注发生。通过在10分钟到26小时的间隔,通过穿刺臂静脉或颈静脉来获得样品。
物质从获得的动物血浆中定量地确定,并将校准样品在血浆中进行调整。通过用乙腈(ACN)沉淀来去除血浆蛋白。随后,使用各种柱,例如Luna C8,LichroCart Purospher Star RP18e,在安捷伦1100LC系统(安捷伦,Santa Clara,加利福尼亚,USA)上将样品通过HPLC进行分级分离。所述HPLC系统通过Turbo离子喷雾(Turbo Ion Spray)接口与API 3000三重四极杆质谱仪(Applied Biosystems(应用生物系统),Darmstadt,德国)偶联。血浆浓度-时间进程的评估通过使用内标和使用校验的动力学分析程序发生。
除了关于确定测试物质在体内的药物代谢动力学参数的研究之外,确定来自在大鼠中混悬液(配制:Tylose混悬液)对比溶液的相对生物利用度以及在小鼠、大鼠和狗中的作用测试之初的高剂量研究和毒性研究。
血浆稳定性
通过将血液置入用Li-肝素包被的monovettes中来新鲜获得来自各种物种(BalbC小鼠,Wistar大鼠,小猎犬和人)的血浆并随后离心。为了确定测试物质的血浆稳定性,将在每种情形中包含血浆中的500ng/ml的2ml在37℃温育。将样品在至多3小时的间隔内,在不同时间取自温育容器。将获得的样品用ACN沉淀从而终止反应并去除血浆蛋白。以等价于体内研究的方式来分析所述样品。
微粒体和肝细胞温育
用各种物种(BalbC小鼠,Wistar大鼠,小猎犬,人)的肝微粒体进行的温育在1.5ml的总体积,在37℃,在修饰的Multiprobe 机器人系统(Canberra Packard)或
机器人系统(Canberra Packard)或 机器人系统(铂尔金爱尔默)中进行。
机器人系统(铂尔金爱尔默)中进行。
温育混合物分别包含0.5μg/ml的测试物质以及0.2-0.5mg/ml微粒体蛋白。此外,加入0.05M磷酸盐缓冲液(pH=7.4),1mM EDTA,5mM 6-磷酸葡萄糖和1.5U/ml来自肠膜样明串珠菌(Leuconostoc Mesenteroides)的6-磷酸葡萄糖脱羟基酶(dehydrogenase)。通过加入NADP+(最终浓度:1mM)来起始微粒体温育。
在每种情形中,将1百万细胞/ml用于确定在新鲜分离的和培养的大鼠、狗和人肝细胞中的测试物质的代谢稳定性。以等价于微粒体测定的方式,在每种情形中,将0.5μg/ml测试物质加入肝细胞中。
在2,5,10,20,30,45和60分钟或在关于更稳定的化合物的2,10,20,30,50,70和90分钟后,从各个温育混合物中去除125μl,加入ACN从而终止酶促反应。离心后,通过LC-MS/MS(API 2000或3000,AppliedBiosystems(应用生物系统))分析样品。从在微粒体温育中的化合物的各个半衰期来计算″CL血液充分-搅拌”和″Fmax充分-搅拌”值。可以通过下式描述底物降解(Houston JB,Utility of in-vitro drug-metabolism data in predicting in-vivo metabolic-clearance(体外药物代谢数据在预测的体内代谢清除中的利用性),Bioch.Pharm.47(9)1469-1479(1994);Obach RS;Baxter JG;Liston TE;Silber BM;Jones BC;MacIntyre F;Rance DJ;Wastall P,The prediction of human pharmacokinetic parameters from preclinical and invitro metabolism data(来自临床前和体外代谢数据的人药物代谢动力学参数的预测),J.Pharmacol.Exp.Ther.283(1)46-58(1997)):
CL′固有[ml/(min·kg)]=(0.693/体外t1/2[min])·(肝重量[g肝/kg体重])·(微粒体蛋白[mg]/肝重量[g])/(微粒体蛋白[mg]/温育体积[ml])。
通过充分-搅拌模型描述血液清除率″CL血液”,忽视蛋白结合(Pang KS;Rowl and M,Hepatic clearance of drugs.I.Theoretical considerations of a″well-stirred″model and a″parallel tube″model.Influence of hepatic blood flow,plasma and blood cell binding,and the hepatocellular enzymatic activity on hepatic drug clearance(“充分搅拌”模型和“并联管”模型的理论考虑。肝血液流动、血浆和血细胞结合的影响,以及肝细胞酶促活性对肝药物清除的影响),J Pharmacokinet Biopharm 5(6):625-53(1977)):
CL血液充分-搅拌[l/(h·kg)]=(QH[l/(h·kg)]·CL′固有[l/(h·kg)])/(QH[l/(h·kg)]+CL′固有[l/(h·kg)]).
对于大鼠,具体的肝重量是32g/kg体重,而肝血流是4.2l/(h·kg)。估计大鼠肝的具体微粒体蛋白含量在40mg/g肝。对于另外的物种的具体外推因子显示在下表中,并且部分基于文献数据和部分基于我们自己的测定。对于肝细胞,将1.1亿/g肝的细胞计数用于关于所有物种的计算。

C.药物组合物的示范性实施方案
可以将本发明的化合物以下列方式转化为药物制剂:
片剂:
组成:
100mg的实施例1的化合物,50mg的乳糖(一水合物),50mg的玉米淀粉(天然的),10mg的聚乙烯吡咯烷酮(PVP 25)(BASF,路德维希港,德国)和2mg的硬脂酸镁。
片剂重量212mg。直径8mm,曲率半径12mm。
制备:
将本发明的化合物、乳糖和淀粉的混合物用PVP在水中的5%浓度的溶液(m/m)成粒。干燥后,将所述颗粒与硬脂酸镁混合达5分钟。使用常规的压片机(见上述关于片剂的形成)压缩该混合物。用于所述压缩的压力的指导是15kN。
可以口服施用的溶液剂:
组成:
500mg的实施例1的化合物,2.5g的聚山梨醇酯和97g的聚乙二醇400。20g的口服溶液对应于本发明化合物的100mg的单一剂量。
制备:
将本发明化合物悬浮在聚乙二醇和聚山梨醇酯的混合物中并伴随搅拌。持续进行搅拌直到完全溶解本发明的化合物。
静脉注射(i.v.)溶液:
将本发明的化合物以低于饱和溶解度的浓度溶解于生理可接受的溶剂(例如等渗盐水,5%葡萄糖溶液,30%PEG 400溶液)中。将该溶液通过过滤除菌并且分散到无菌和无热原的注射容器中。
Claims (26)
1.下式的化合物:
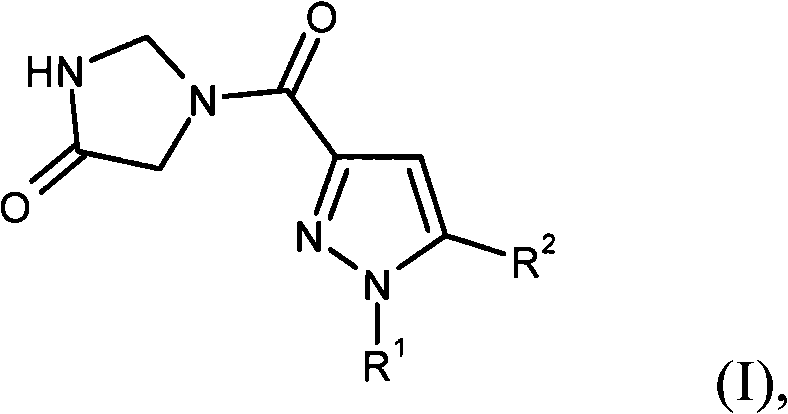
其中
R1表示苯基,
其中苯基被1-3个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,羟基,氰基,硝基,三氟甲基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基和(C1-C4)-烷氧基,
其中
(C1-C4)-烷基和(C1-C4)-烷氧基又可以被选自以下系列的原子团相同或不同地取代1-3次:卤素,氰基,羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基,(C3-C7)-环烷基和4-到7-元杂环基,
其中最后提及的环烷基和杂环基原子团每个又可以相同地或不同地被卤素,氰基,(C1-C4)-烷基,三氟甲基,羟基,(C1-C4)-烷氧基,三氟甲氧基,氧代,氨基,单-(C1-C4)-烷基氨基和二-(C1-C4)-烷基氨基取代至多3次,
R2表示苯基,
其中苯基被1-3个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,羟基,氰基,硝基,三氟甲基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基和(C1-C4)-烷氧基,
其中
(C1-C4)-烷基和(C1-C4)-烷氧基又可以被选自以下系列的原子团相同地或不同地取代1-3次:卤素,氰基,羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基,(C3-C7)-环烷基和4-7元杂环基,
其中最后提及的环烷基和杂环基原子团又可以分别相同地或不同地被卤素,氰基,(C1-C4)-烷基,三氟甲基,羟基,(C1-C4)-烷氧基,三氟甲氧基,氧代,氨基,单-(C1-C4)-烷基氨基和二-(C1-C4)-烷基氨基取代至多3次,
或其盐,其溶剂合物或其盐的溶剂合物之一。
2.根据权利要求1的化合物,其特征在于
R1代表苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,羟基,氰基,硝基,三氟甲基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基和(C1-C4)-烷氧基,
R2表示苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,羟基,氰基,硝基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基和(C1-C4)-烷氧基,
其中
(C1-C4)-烷氧基又可以被选自以下系列的原子团相同地或不同地取代1-3次:卤素,氰基,羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基,(C3-C7)-环烷基和4-7元杂环基,
其中最后提及的环烷基和杂环基原子团又可以分别被卤素,氰基,(C1-C4)-烷基,三氟甲基,羟基,(C1-C4)-烷氧基,三氟甲氧基,氧代,氨基,单-(C1-C4)-烷基氨基和二-(C1-C4)-烷基氨基相同地或不同地取代至多3次,
或其盐、其溶剂合物或其盐的溶剂合物之一。
3.根据权利要求1或2的化合物,其特征在于
R1代表苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,氰基,三氟甲基和甲氧基,
R2表示苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,氰基,三氟甲氧基,甲基和(C1-C3)-烷氧基,其中
(C1-C3)-烷氧基又可以由选自以下系列的原子团相同地或不同地取代1-3次:羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基和4-7元杂环基,
其中最后提及的杂环基基团可以分别又被(C1-C4)-烷基取代,或其盐,其溶剂合物或其盐的溶剂合物之一。
4.根据权利要求1-3中任一项的化合物,其特征在于
R1代表苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,氰基,三氟甲基和甲氧基,
R2表示苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素,氰基,三氟甲氧基,甲基和甲氧基,或其盐,其溶剂合物或其盐的溶剂合物之一。
5.根据权利要求1-4中任一项的化合物,其特征在于
R1代表苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素和氰基,
R2表示苯基,
其中苯基被1-2个取代基取代,其中所述取代基彼此独立地选自由下列组成的组:卤素和氰基,
或其盐,其溶剂合物或其盐的溶剂合物之一。
6.根据权利要求1的化合物,其特征在于它对应于下式

其中
R3表示氢,卤素,羟基,氰基,硝基,三氟甲基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基或(C1-C4)-烷氧基,
R4表示氢或卤素,
R5表示卤素,羟基,氰基,硝基,三氟甲基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基或(C1-C4)-烷氧基,
其中
(C1-C4)-烷氧基又可以被选自以下系列的原子团相同地或不同地取代1-3次:卤素,氰基,羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基,(C3-C7)-环烷基和4-7元杂环基,
其中最后提及的环烷基和杂环基原子团又可以分别被卤素,氰基,(C1-C4)-烷基,三氟甲基,羟基,(C1-C4)-烷氧基,三氟甲氧基,氧代,氨基,单-(C1-C4)-烷基氨基和二-(C1-C4)-烷基氨基相同地或不同地取代至多3次,
R6表示氢或卤素,
或其盐,其溶剂合物或其盐的溶剂合物之一。
7.根据权利要求6的化合物,其特征在于
R3表示卤素,羟基,氰基,硝基,三氟甲基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基或(C1-C4)-烷氧基,
R4表示氢或卤素,
R5表示卤素,羟基,氰基,硝基,三氟甲氧基,三氟甲硫基,(C1-C4)-烷基或(C1-C4)-烷氧基,
其中
(C1-C4)-烷氧基又可以被选自以下系列的原子团相同地或不同地取代1-3次:卤素,氰基,羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基,(C3-C7)-环烷基和4-7元杂环基,
其中最后提及的环烷基和杂环基原子团又可以分别被卤素,氰基,(C1-C4)-烷基,三氟甲基,羟基,(C1-C4)-烷氧基,三氟甲氧基,氧代,氨基,单-(C1-C4)-烷基氨基和二-(C1-C4)-烷基氨基相同地或不同地取代至多3次,
R6表示氢或卤素,
或其盐,其溶剂合物或其盐的溶剂合物之一。
8.根据权利要求6或7的化合物,其特征在于
R3表示卤素,氰基,三氟甲基或甲氧基,
R4表示氢或卤素,
R5表示卤素,氰基,三氟甲氧基,甲基或(C1-C3)-烷氧基,
其中
(C1-C3)-烷氧基又可以被选自以下系列的原子团相同地或不同地取代1-3次:羟基,(C1-C4)-烷氧基,氨基,单-(C1-C4)-烷基氨基,二-(C1-C4)-烷基氨基和4-7元杂环基,
其中最后提及的杂环基原子团又可以分别被(C1-C4)-烷基取代,
R6表示氢或卤素,
或其盐,其溶剂合物或其盐的溶剂合物之一。
9.根据权利要求6-8任一项的化合物,其特征在于
R3表示卤素,氰基,三氟甲基或甲氧基,
R4表示氢,氯或氟,
R5表示卤素,氰基,三氟甲氧基,甲基或甲氧基,
R6表示氢,氯或氟,
或其盐,其溶剂合物或其盐的溶剂合物之一。
10.根据权利要求6-9中任一项的化合物,其特征在于
R3表示卤素或氰基,
R4表示氢或氟,
R5表示卤素或氰基,
R6表示氢或氟,
或其盐,其溶剂合物或其盐的溶剂合物之一。
11.根据权利要求6-10中任一项的化合物,其特征在于
R3表示氯或氰基,
R4表示氟,
R5表示氯或氰基,
R6表示氟,
或其盐,其溶剂合物或其盐的溶剂合物之一。
12.根据权利要求6-10中任一项的化合物,其特征在于
R3表示氯或氰基,
R4表示氟,
R5表示氯或氰基,
R6表示氢,
或其盐,其溶剂合物或其盐的溶剂合物之一。
13.根据权利要求6-10中任一项的化合物,其特征在于
R3表示氯或氰基,
R4表示氢,
R5表示氯或氰基,
R6表示氢,
或其盐,其溶剂合物或其盐的溶剂合物之一。
14.根据权利要求6-10任一项的化合物,其特征在于
R3表示氯或氰基,
R4表示氢,
R5表示氯或氰基,
R6表示氟,
或其盐,其溶剂合物或其盐的溶剂合物之一。
15.根据权利要求6的化合物,其特征在于:
R3表示卤素,氰基,三氟甲基或甲氧基,
R4表示氢或卤素,
R5表示三氟甲基,
R6表示氟,
或其盐,其溶剂合物或其盐的溶剂合物之一。
16.根据权利要求6的化合物,其特征在于
R3表示氢,
R4表示氟或氯,
R5表示卤素,氰基,三氟甲氧基,甲基或甲氧基,
R6表示氢或卤素,
或其盐,其溶剂合物或其盐的溶剂合物之一。
17.用于制备根据权利要求1的式(I)的化合物的方法,其中使下式的化合物

其中
R1和R2具有权利要求1中指定的含义,
与咪唑烷-4-酮或咪唑烷-4-酮的盐反应。
18.根据权利要求1-16中任一项的化合物,其用于治疗和/或预防疾病。
19.根据权利要求1-16中任一项的化合物用于制备药物的应用,所述药物用于治疗和/或预防疾病。
20.根据权利要求1-16中任一项的化合物用于制备药物的应用,所述药物用于治疗和/或预防逆转录病毒性疾病。
21.根据权利要求20的应用,其特征在于所述逆转录病毒性疾病是HI病毒感染。
22.药物,其包含至少一种根据权利要求1-16中任一项的化合物以及至少一种另外的活性成分。
23.药物,其包含至少一种根据权利要求1-16中任一项的化合物以及至少一种惰性、非毒性的药用赋形剂。
24.根据权利要求22或23的药物,其用于治疗和/或预防逆转录病毒性疾病。
25.根据权利要求24的药物,其特征在于所述逆转录病毒性疾病是HI病毒感染。
26.用于控制人和动物中病毒性疾病的方法,所述方法通过施用抗病毒有效量的至少一种根据权利要求1-16中任一项的化合物或根据权利要求22-25中任一项的药物来进行。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102008015033.9 | 2008-03-17 | ||
| DE102008015033A DE102008015033A1 (de) | 2008-03-17 | 2008-03-17 | Substituierte (Pyrazolyl-carbonyl)imidazolidinone und ihre Verwendung |
| PCT/EP2009/001714 WO2009115213A1 (de) | 2008-03-17 | 2009-03-10 | (pyrazolyl-carbonyl) imidazolidinone-derivate zur behandlung retroviraler erkrankungen |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101977908A true CN101977908A (zh) | 2011-02-16 |
| CN101977908B CN101977908B (zh) | 2014-04-23 |
Family
ID=40565083
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN200980109500.6A Expired - Fee Related CN101977908B (zh) | 2008-03-17 | 2009-03-10 | 用于治疗逆转录病毒性疾病的(吡唑基羰基)咪唑烷酮衍生物 |
Country Status (22)
| Country | Link |
|---|---|
| US (1) | US8399682B2 (zh) |
| EP (1) | EP2254885B1 (zh) |
| JP (1) | JP5576853B2 (zh) |
| KR (1) | KR20100124337A (zh) |
| CN (1) | CN101977908B (zh) |
| AP (1) | AP2774A (zh) |
| AR (1) | AR070919A1 (zh) |
| AU (1) | AU2009226656A1 (zh) |
| CA (1) | CA2718685C (zh) |
| CL (1) | CL2009000636A1 (zh) |
| DE (1) | DE102008015033A1 (zh) |
| IL (1) | IL207649A0 (zh) |
| MA (1) | MA32224B1 (zh) |
| MX (1) | MX2010010230A (zh) |
| NZ (1) | NZ588141A (zh) |
| PE (1) | PE20091718A1 (zh) |
| RU (1) | RU2010141578A (zh) |
| TW (1) | TW200951125A (zh) |
| UA (1) | UA102390C2 (zh) |
| UY (1) | UY31717A1 (zh) |
| WO (1) | WO2009115213A1 (zh) |
| ZA (1) | ZA201007361B (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104024242A (zh) * | 2011-11-29 | 2014-09-03 | 爱库利斯有限两合公司 | 甲酰胺取代的杂芳基吡唑及其用途 |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102008015032A1 (de) * | 2008-03-17 | 2009-09-24 | Aicuris Gmbh & Co. Kg | Substituierte Pyrazolamide und ihre Verwendung |
| DE102008015033A1 (de) | 2008-03-17 | 2009-09-24 | Aicuris Gmbh & Co. Kg | Substituierte (Pyrazolyl-carbonyl)imidazolidinone und ihre Verwendung |
| DE102008062863A1 (de) | 2008-12-17 | 2010-06-24 | Aicuris Gmbh & Co. Kg | Substituierte (Thiophenyl-carbonyl)imidazolidinone und ihre Verwendung |
| DE102008062878A1 (de) | 2008-12-17 | 2010-06-24 | Aicuris Gmbh & Co. Kg | Substituierte Furancarboxamide und ihre Verwendung |
| EP2295038B1 (en) * | 2009-09-11 | 2013-05-29 | AiCuris GmbH & Co. KG | Solid dispersion comprising an anti-HIV agent |
| DE102012016908A1 (de) | 2012-08-17 | 2014-02-20 | Aicuris Gmbh & Co. Kg | Tris-(Hetero)Aryl-Pyrazole und ihre Verwendung |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004024147A1 (en) * | 2002-09-16 | 2004-03-25 | Pfizer Limited | 4-(3,5-dicyanophenoxy) pyrazole derivatives for use as transcriptase modulators in the treatment of i.a. hiv |
| WO2004031178A1 (en) * | 2002-10-07 | 2004-04-15 | Pfizer Limited | Pyrazole derivatives |
| CN1527829A (zh) * | 2001-06-04 | 2004-09-08 | - | 作为hiv逆转录酶抑制剂的吡唑衍生物 |
| CN1751028A (zh) * | 2003-02-18 | 2006-03-22 | 弗·哈夫曼-拉罗切有限公司 | 非核苷逆转录酶抑制剂 |
| CN1832928A (zh) * | 2003-06-26 | 2006-09-13 | 诺瓦提斯公司 | 以5元杂环为基础的p38激酶抑制剂 |
Family Cites Families (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB453061A (en) | 1935-03-23 | 1936-09-04 | Charles Howard Twigg | Improvements in and relating to gas heated geysers and water heaters |
| US3806506A (en) | 1971-08-05 | 1974-04-23 | Uniroyal Inc | Furan derivatives,compositions thereof and methods for using same |
| DE3119796A1 (de) | 1981-05-19 | 1982-12-23 | Hoechst Ag, 6000 Frankfurt | Substituierte tryptaminderivate von thienyloxpropanolaminen, verfahren zu ihrer herstellung, pharmazeutische praeparate auf basis dieser verbindungen sowie ihre verwendung |
| FR2538388B1 (fr) | 1982-12-24 | 1985-06-21 | Pharmuka Lab | Nouveaux derives de naphtalene- ou azanaphtalenecarboxamide, leurs procedes de preparation et leur utilisation comme medicaments |
| US5591761A (en) | 1993-05-20 | 1997-01-07 | Texas Biotechnology Corporation | Thiophenyl-, furyl-and pyrrolyl-sulfonamides and derivatives thereof that modulate the activity of endothelin |
| PH27357A (en) | 1989-09-22 | 1993-06-21 | Fujisawa Pharmaceutical Co | Pyrazole derivatives and pharmaceutical compositions comprising the same |
| US5571810A (en) | 1990-06-11 | 1996-11-05 | Fujisawa Pharmaceutical Co., Ltd. | Thiophene derivatives |
| GB9012936D0 (en) | 1990-06-11 | 1990-08-01 | Fujisawa Pharmaceutical Co | Thiophene derivatives,processes for preparation thereof and pharmaceutical composition comprising the same |
| IL104311A (en) | 1992-02-05 | 1997-07-13 | Fujisawa Pharmaceutical Co | Pyrazole derivatives, processes for the preparation thereof and pharmaceutical compositions containing the same |
| FR2692575B1 (fr) | 1992-06-23 | 1995-06-30 | Sanofi Elf | Nouveaux derives du pyrazole, procede pour leur preparation et compositions pharmaceutiques les contenant. |
| JPH084281B2 (ja) | 1992-09-30 | 1996-01-17 | 橋本コーポレイション株式会社 | オンフック後相手の声を拡声する電話装置 |
| FR2728571B1 (fr) | 1994-12-22 | 1997-01-31 | Adir | Nouveaux ethers d'oximes tricycliques, leurs procedes de preparation et les compositions pharmaceutiques qui les contiennent |
| US5696151A (en) | 1995-11-30 | 1997-12-09 | Uniroyal Chemical Company, Inc. | Compounds useful for the inhibition of the replication of HIV-1 and HIV-1 mutants |
| US6143780A (en) | 1999-09-17 | 2000-11-07 | Uniroyal Chemical Company, Inc. | N-arylmethylthioanilide compounds useful for the inhibition of the replication of HIV |
| SK18102002A3 (sk) | 2000-06-28 | 2003-07-01 | Astrazeneca Ab | Substituované chinazolínové deriváty, ich použitie a kompozícia obsahujúca tieto deriváty |
| US6930190B2 (en) | 2001-08-01 | 2005-08-16 | Basell Polyolefine Gmbh | Process for the preparation of heterocyclic pentalene derivatives |
| US7223782B2 (en) | 2001-11-01 | 2007-05-29 | Icagen, Inc. | Pyrazole-amides and -sulfonamides |
| US7005432B2 (en) | 2002-05-16 | 2006-02-28 | Hoffman-La Roche Inc. | Substituted imidazol-pyridazine derivatives |
| US7166619B2 (en) | 2002-08-14 | 2007-01-23 | Ppd Discovery , Inc. | Prenylation inhibitors and methods of their synthesis and use |
| ATE538097T1 (de) | 2002-12-02 | 2012-01-15 | Astellas Pharma Inc | Als cox-i-inhibitoren geeignete pyrazolderivate |
| CA2515119A1 (en) | 2003-02-07 | 2004-08-19 | Daiichi Pharmaceutical Co., Ltd. | Pyrazole derivative |
| US7678788B2 (en) | 2003-02-27 | 2010-03-16 | Neurosearch A/S | Diazabicyclic aryl derivatives |
| FR2856683A1 (fr) | 2003-06-25 | 2004-12-31 | Sanofi Synthelabo | Derives de 4-cyanopyrazole-3-carboxamide, leur preparation et leur application en therapeutique |
| GB0315657D0 (en) * | 2003-07-03 | 2003-08-13 | Astex Technology Ltd | Pharmaceutical compounds |
| WO2005007625A2 (en) | 2003-07-14 | 2005-01-27 | The University Of Tennessee Research Foundation | Heterocyclic amides with anti-tuberculosis activity |
| US7220772B2 (en) * | 2003-09-05 | 2007-05-22 | Pfizer, Inc. | Pyrazole derivatives |
| FR2860792B1 (fr) | 2003-10-10 | 2006-02-24 | Sanofi Synthelabo | Derives de thiophene-2-carboxamide, leur preparation et leur application en therapeutique |
| JP4208925B2 (ja) | 2004-02-20 | 2009-01-14 | アストラゼネカ アクチボラグ | 治療薬 |
| AU2005258397A1 (en) | 2004-07-01 | 2006-01-12 | Daiichi Pharmaceutical Co., Ltd. | Pyrazole derivatives |
| GB0417910D0 (en) | 2004-08-11 | 2004-09-15 | Novartis Ag | Organic compounds |
| JP4942654B2 (ja) | 2004-08-23 | 2012-05-30 | イーライ リリー アンド カンパニー | ヒスタミンh3受容体薬剤、製剤及び治療的使用 |
| DE102004054666A1 (de) | 2004-11-12 | 2006-05-18 | Bayer Cropscience Gmbh | Substituierte Pyrazol-3-carboxamide, Verfahren zur Herstellung und Verwendung als Herbizide und Pflanzenwachstumsregulatoren |
| JP2008523072A (ja) | 2004-12-07 | 2008-07-03 | ルーカス ファーマシューティカルズ, インコーポレイテッド | タンパク質キナーゼの阻害剤 |
| WO2006062982A2 (en) | 2004-12-07 | 2006-06-15 | Locus Pharmaceuticals, Inc. | Urea inhibitors of map kinases |
| AU2005317280A1 (en) | 2004-12-15 | 2006-06-22 | Astrazeneca Ab | Nicotinic acetylcholine receptor ligands |
| AU2006223199A1 (en) | 2005-03-10 | 2006-09-21 | Bayer Pharmaceuticals Corporation | Pyrimidine derivatives for treatment of hyperproliferative disorders |
| AU2006261841B8 (en) | 2005-06-27 | 2012-12-06 | Exelixis Patent Company Llc | Pyrazole based LXR modulators |
| WO2007009701A2 (en) | 2005-07-15 | 2007-01-25 | Laboratorios Del Dr. Esteve, S.A. | Use of substituted pyrazole compounds and combinations thereof for the treatment of the metabolic syndrome |
| EP1743637A1 (en) | 2005-07-15 | 2007-01-17 | Laboratorios Del Dr. Esteve, S.A. | Use of substituted pyrazole compounds and combinations thereof for the treatment of the metabolic syndrome |
| GB0516661D0 (en) | 2005-08-13 | 2005-09-21 | Astrazeneca Ab | Therapeutic agents |
| RU2009117653A (ru) | 2006-10-12 | 2010-11-20 | Янссен Фармацевтика Нв (Be) | Применение производных замещенного пиразинона в качестве лекарственного средства |
| US20100210649A1 (en) | 2006-12-21 | 2010-08-19 | Sloan-Kettering Institute For Cancer Research | Pyridazinones and furan-containing compounds |
| GB0701426D0 (en) | 2007-01-25 | 2007-03-07 | Univ Sheffield | Compounds and their use |
| DE102008015032A1 (de) * | 2008-03-17 | 2009-09-24 | Aicuris Gmbh & Co. Kg | Substituierte Pyrazolamide und ihre Verwendung |
| DE102008015033A1 (de) | 2008-03-17 | 2009-09-24 | Aicuris Gmbh & Co. Kg | Substituierte (Pyrazolyl-carbonyl)imidazolidinone und ihre Verwendung |
-
2008
- 2008-03-17 DE DE102008015033A patent/DE102008015033A1/de not_active Ceased
-
2009
- 2009-03-10 RU RU2010141578/04A patent/RU2010141578A/ru not_active Application Discontinuation
- 2009-03-10 AU AU2009226656A patent/AU2009226656A1/en not_active Abandoned
- 2009-03-10 JP JP2011500075A patent/JP5576853B2/ja not_active Expired - Fee Related
- 2009-03-10 CA CA2718685A patent/CA2718685C/en not_active Expired - Fee Related
- 2009-03-10 EP EP09721416.7A patent/EP2254885B1/de active Active
- 2009-03-10 KR KR1020107023291A patent/KR20100124337A/ko not_active Application Discontinuation
- 2009-03-10 UA UAA201012130A patent/UA102390C2/ru unknown
- 2009-03-10 NZ NZ588141A patent/NZ588141A/en not_active IP Right Cessation
- 2009-03-10 CN CN200980109500.6A patent/CN101977908B/zh not_active Expired - Fee Related
- 2009-03-10 AP AP2010005422A patent/AP2774A/xx active
- 2009-03-10 MX MX2010010230A patent/MX2010010230A/es active IP Right Grant
- 2009-03-10 WO PCT/EP2009/001714 patent/WO2009115213A1/de active Application Filing
- 2009-03-16 TW TW098108355A patent/TW200951125A/zh unknown
- 2009-03-17 PE PE2009000388A patent/PE20091718A1/es not_active Application Discontinuation
- 2009-03-17 AR ARP090100947A patent/AR070919A1/es not_active Application Discontinuation
- 2009-03-17 CL CL2009000636A patent/CL2009000636A1/es unknown
- 2009-03-17 UY UY031717A patent/UY31717A1/es not_active Application Discontinuation
-
2010
- 2010-08-17 IL IL207649A patent/IL207649A0/en unknown
- 2010-09-17 US US12/885,340 patent/US8399682B2/en not_active Expired - Fee Related
- 2010-10-11 MA MA33241A patent/MA32224B1/fr unknown
- 2010-10-14 ZA ZA2010/07361A patent/ZA201007361B/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1527829A (zh) * | 2001-06-04 | 2004-09-08 | - | 作为hiv逆转录酶抑制剂的吡唑衍生物 |
| WO2004024147A1 (en) * | 2002-09-16 | 2004-03-25 | Pfizer Limited | 4-(3,5-dicyanophenoxy) pyrazole derivatives for use as transcriptase modulators in the treatment of i.a. hiv |
| WO2004031178A1 (en) * | 2002-10-07 | 2004-04-15 | Pfizer Limited | Pyrazole derivatives |
| CN1751028A (zh) * | 2003-02-18 | 2006-03-22 | 弗·哈夫曼-拉罗切有限公司 | 非核苷逆转录酶抑制剂 |
| CN1832928A (zh) * | 2003-06-26 | 2006-09-13 | 诺瓦提斯公司 | 以5元杂环为基础的p38激酶抑制剂 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104024242A (zh) * | 2011-11-29 | 2014-09-03 | 爱库利斯有限两合公司 | 甲酰胺取代的杂芳基吡唑及其用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2254885A1 (de) | 2010-12-01 |
| JP2011514366A (ja) | 2011-05-06 |
| WO2009115213A1 (de) | 2009-09-24 |
| CA2718685A1 (en) | 2009-09-24 |
| RU2010141578A (ru) | 2012-04-27 |
| AP2774A (en) | 2013-09-30 |
| AU2009226656A1 (en) | 2009-09-24 |
| MX2010010230A (es) | 2010-11-09 |
| CN101977908B (zh) | 2014-04-23 |
| IL207649A0 (en) | 2010-12-30 |
| JP5576853B2 (ja) | 2014-08-20 |
| AR070919A1 (es) | 2010-05-12 |
| US20110124618A1 (en) | 2011-05-26 |
| UA102390C2 (ru) | 2013-07-10 |
| TW200951125A (en) | 2009-12-16 |
| UY31717A1 (es) | 2009-08-31 |
| DE102008015033A1 (de) | 2009-09-24 |
| KR20100124337A (ko) | 2010-11-26 |
| CA2718685C (en) | 2016-11-29 |
| EP2254885B1 (de) | 2013-12-04 |
| ZA201007361B (en) | 2011-12-28 |
| AP2010005422A0 (en) | 2010-10-31 |
| PE20091718A1 (es) | 2009-12-11 |
| NZ588141A (en) | 2012-05-25 |
| CL2009000636A1 (es) | 2010-05-07 |
| US8399682B2 (en) | 2013-03-19 |
| MA32224B1 (fr) | 2011-04-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101977908B (zh) | 用于治疗逆转录病毒性疾病的(吡唑基羰基)咪唑烷酮衍生物 | |
| CN102083803B (zh) | 氧代杂环取代的羧酸衍生物和它的用途 | |
| CN103998432A (zh) | 双环化合物 | |
| CN110099898A (zh) | 化合物及其用途 | |
| US20030171406A1 (en) | Medicinal compositions containing propenone derivatives | |
| KR101112515B1 (ko) | 이미다졸-5- 카복실산 유도체의 염 및 그 제조방법 및 상기 염으로 구성되는 약학 조성물 | |
| CN105980381A (zh) | 取代的尿嘧啶及其用途 | |
| WO2020221006A1 (zh) | 一种bet蛋白抑制剂、其制备方法及用途 | |
| CN107108638A (zh) | 哌啶基吡唑并嘧啶酮及其用途 | |
| CN104684913B (zh) | 三(杂)芳基吡唑及其应用 | |
| US8324268B2 (en) | Substituted furancarboxamides, and use thereof | |
| TW201900616A (zh) | 經取代之n-芳基乙基-2-胺基喹啉-4-甲醯胺及其用途 | |
| CN105451737B (zh) | 药物组合物及其用途以及所述药物组合物用于按需避孕的施用方法 | |
| US9296720B2 (en) | Carboxamide-substituted heteroaryl-pyrazoles and the use thereof | |
| US8546438B2 (en) | Substituted (thiophenyl-carbonyl)imidazolidinones, and use thereof | |
| US6627643B2 (en) | Methods for preparing sodium-hydrogen exchanger type-1 inhibitors | |
| TWI353245B (en) | Novel aminoidazole derivatives as medicaments and | |
| JP2003504383A (ja) | アルファ−ヒドロキシ−ガンマ−[[(炭素環式−又はヘテロ環式−置換)アミノ]カルボニル]アルカンアミド誘導体及びこれらの使用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20140423 Termination date: 20180310 |