CN100516070C - 制备7-氨基-3-乙烯基头孢烷酸的方法 - Google Patents
制备7-氨基-3-乙烯基头孢烷酸的方法 Download PDFInfo
- Publication number
- CN100516070C CN100516070C CNB2007101147821A CN200710114782A CN100516070C CN 100516070 C CN100516070 C CN 100516070C CN B2007101147821 A CNB2007101147821 A CN B2007101147821A CN 200710114782 A CN200710114782 A CN 200710114782A CN 100516070 C CN100516070 C CN 100516070C
- Authority
- CN
- China
- Prior art keywords
- vinyl
- gvne
- amino
- phenylacetylamino
- preparing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Cephalosporin Compounds (AREA)
Abstract
一种制备7-氨基-3-乙烯基头孢烷酸的方法,是以7-苯乙酰氨基-3-氯甲基头孢烷酸对甲氧苄酯(GCLE)为起始原料,采用Wittig反应,在3位引入乙烯基后得7-苯乙酰氨基-3-乙烯基-4-头孢烷酸对甲氧苄酯,即产物GVNE,再由GVNE通过化学法或酶解法制备7-氨基-3-乙烯基头孢烷酸(7-AVCA),其主要技术特点是GVNE的制备中使用价格较低廉的亚磷酸三乙酯来代替三苯基磷,来实现在GCLE的3位引入乙烯基,制得GVNE的目的。生产方法简单、主要原料成本低、收率高、综合收益效率高,可工业化大规模制备7-AVCA。
Description
技术领域
本发明涉及一种医药中间体的制备方法,尤其涉及一种低成本生产7-氨基-3-乙烯基头孢烷酸的合成方法。
背景技术
7-氨基-3-乙烯基头孢烷酸(7-AVCA)是合成第三代头孢菌素头孢克肟的重要原料。头孢克肟(cefixime)为第三代口服头孢菌素,由日本藤泽药品工业株式会社首次开发上市,商品名Cefspan,1987年通过美国FDA批准,1998年在欧洲发达国家市场中,头孢克肟已超过了头孢呋辛酯成为市场占有率居首位的口服头孢菌素,现今美国、英国、日本及欧洲药典均收载了本品,目前已在80多个国家得到广泛的临床使用。头孢克肟的特点是抗谱广、抗菌作用强、有效浓度持续时间长,具有对β-内酰胺酶稳定、体内分布广,口服生物利用度高的优点,用于泌尿系统、胆道系统、淋病、猩红热、中耳炎、副鼻窦炎的治疗,是国际上销量仅次于阿莫西林的β-内酰胺类抗菌药物,为美国性疾病传播中心和世界卫生组织推荐淋病首选治疗药物,且对治疗泌尿系统及呼吸道系统疾病也有特效,因此受到临床医生和患者的欢迎。并且由于头孢克肟是一个具有多种口服释药技术特征的药物,目前国内已开发并获准上市了6个剂型,分别是胶囊、片剂、分散片、颗粒剂、干混悬剂和咀嚼片剂,不但可用于成年人患者,而且小剂量药物也是专为儿童设计推出的抗生素药物,从而为产品市场拓宽的渠道。到2005年底,在我国原料药生产已经达到19家的规模,从而也拉动了其主要原料7-氨基-3-乙烯基头孢烷酸的发展。国内外文献报道的合成方法有多种,但最常采用的合成方法有:(1)以GCLE为原料,采用Wittig反应,在3位引入乙烯基后得7-苯乙酰氨基-3-7烯基-4-头孢烷酸对甲氧苄酯,即产物GVNE,再经7位裂解和4位脱保护反应制得7-AVCA(化学法);(2)也可以在在3位引入乙烯基制得产物GVNE后,再经酸解脱掉4位对甲氧基苄基,再在青霉素酰化酶作用下裂解掉7位苯乙酰氨基后制得7-AVCA(酶解法),具体合成路线为:

随着市场的不断变化,原有工艺成本已经不能满足市场的需要,一般都存在周期长、三废多、原材料成本高和收率较低等缺点,其中最大的问题就在于目前生产7-AVCA的合成方法中,由于关键中间体GVNE的合成,在GCLE的3位引入乙烯基时,需要使用三苯基磷,一方面,三苯基磷价格昂贵,导致生产成本较高;另一方面,三苯基磷的分子量较大,使得生产中三苯基磷的用量也较大,这也大大提高了生产成本。
发明内容
本发明的目的就是针对现有合成方法所存在的问题,提出一种生产方法简单、主要原料成本低、收率高、综合收益效率高的制备7-氨基-3-乙烯基头孢烷酸(7-AVCA)的方法。
本发明制备7-氨基-3-乙烯基头孢烷酸的方法,是以7-苯乙酰氨基-3-氯甲基头孢烷酸对甲氧苄酯(GCLE)为起始原料,采用Wittig反应,在3位引入乙烯基后得7-苯乙酰氨基-3-乙烯基-4-头孢烷酸对甲氧苄酯,即产物GVNE,再由GVNE制备7-氨基-3-乙烯基头孢烷酸,其特征在于所述GVNE的制备过程为:7-苯乙酰氨基-3-氯甲基头孢烷酸对甲氧苄酯在溶剂体系中,在氮气或氩气保护下,加入催化剂,于0~100℃与亚磷酸三乙酯反应1~24小时,再采用Wittig反应,得产物GVNE粗品,再经水洗,甲醇洗涤,得到白色晶体GVNE,所述的溶剂体系可以是四氢呋喃、二氯甲烷、乙腈、乙酸乙酯、甲醇、乙醇,或是上述溶剂与水组成的混合溶剂体系,所述的催化剂采用NaI、KI或I2。
7-苯乙酰氨基-3-氯甲基头孢烷酸对甲氧苄酯(GCLE)与亚磷酸三乙酯的反应摩尔比为1∶1~1∶1.5,催化剂的用量为7-苯乙酰氨基-3-氯甲基头孢烷酸对甲氧苄酯摩尔用量的0.2~1.5倍,优选0.9~1.2倍。7-苯乙酰氨基-3-氯甲基头孢烷酸对甲氧苄酯与溶剂的重量体积比(g/ml)为1∶5~1∶200,优选1∶5~1∶50。
所述Wittig反应,在3位引入乙烯基,即加入预冷的甲醛溶液,滴加适量碱液,反应1~10小时即得产物GVNE粗品。Witting反应中使用的碱液可以是碳酸钾、碳酸钠、碳酸氢钾、碳酸氢钠、氢氧化钠、氢氧化钾、氨水或三乙胺。甲醛的用量控制在亚磷酸三乙酯摩尔用量的1.0~1.2的范围之内。
本发明主要通过使用价格较低廉的亚磷酸三乙酯来代替三苯基磷,来实现在GCLE的3位引入乙烯基,制得GVNE的目的,这可以大大降低7-氨基-3-乙烯基头孢烷酸(7-AVCA)的生产成本,具体利用亚磷酸三乙酯合成GVNE的改进合成路线为:
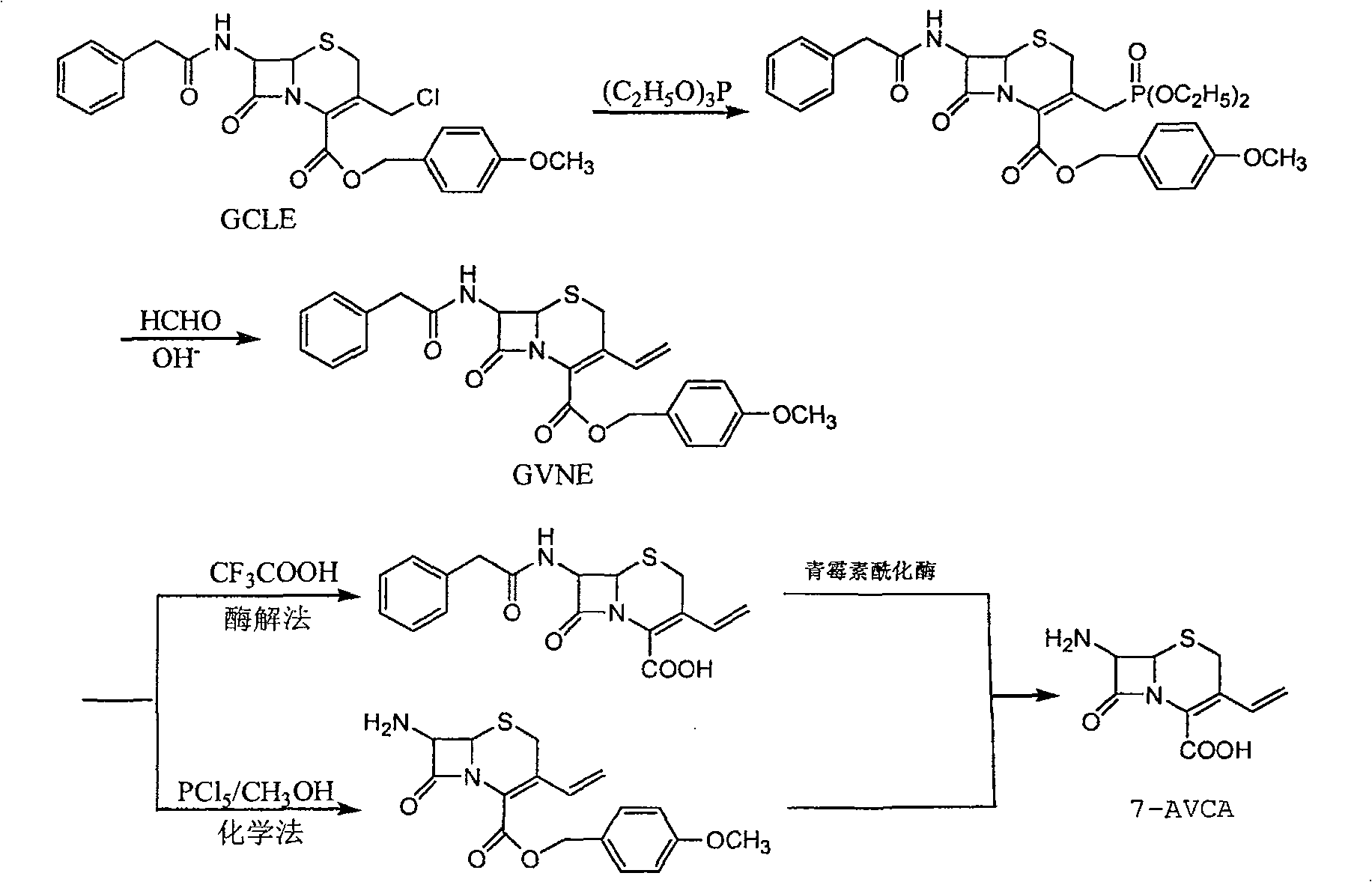
由GVNE制备7-氨基-3-乙烯基头孢烷酸(7-AVCA)仍按照公知技术方法采用化学法或酶解法生产。
化学法:GVNE,再经7位裂解和4位脱保护反应制得7-AVCA。具体工艺是:在二氯甲烷中加入五氯化磷,搅拌加热使物料溶解,加热温度不超过40℃。降温至-40~-20℃,滴加吡啶,控制反应温度在-20~-10℃反应1~4h。然后加入GVNE,控制反应温度在-10~10℃反应1~4h,再滴加预冷甲醇,温度控制在-20~-10℃,滴加完甲醇后再反应1~4h,自然升温至室温,再加入间甲酚于25~50℃反应2~8h。降温水洗,有机相用稀盐酸洗涤,水相用二氯甲烷洗涤,合并水相,用碱液调pH值至3.0,析出产物,再用水、丙酮洗涤产物得白色固体产物7-AVCA,减压干燥。
酶解法:在3位引入乙烯基制得产物GVNE后,再经酸解(三氟乙酸)脱掉4位对甲氧基苄基,再在青霉素酰化酶作用下裂解掉7位苯乙酰氨基后制得7-AVCA。
本发明的优点:
生产方法简单、主要原料成本低、收率高、综合收益效率高,可工业化大规模制备7-AVCA。
具体实施方式
下面列举几种本发明的非限定实施的具体实施例:
实施例1:
在2000L的反应釜中加入250L四氢呋喃,搅拌,通入氮气,降温至10℃,然后依次加入GCLE 48.5Kg、亚磷酸三乙酯16.6Kg、碘化钠15kg,于10~20℃反应4h。加入预冷至10~15℃的37%甲醛水溶液10Kg和水40L,滴加碳酸氢钠溶液,析出结晶。保持pH8.7~8.9,强烈搅拌下,在15~20℃反应3h。加入800L水,继续反应1h、抽滤,滤餠水洗,抽干后移入180L甲醇中,于0~10℃搅拌洗涤1h。抽滤,不超过50℃,真空干燥,得白色固体产品GVNE 38.2Kg,含量96.8%(高效液相色谱面积归一法),熔点180.2~182.4℃。
在2000L反应釜中加入二氯甲烷540L、五氯化磷22.5Kg,搅拌并加热至30℃,使之完全溶解。降温至-25~-20℃,滴加吡啶7.2L,于-20~-10℃反应2h。加入GVNE 30Kg,-5~5℃反应4h。降温至-20℃,缓慢加入甲醇150L,加料完毕后,于-20~-10℃反应4h。自然升温至室温,加入间甲酚150L,30~40℃反应4h。降温至0℃,加入预冷水300L,0~10℃搅拌30min,分层。有机相用盐酸萃取,合并水相,用二氯甲烷洗涤。然后在所得水溶液中加入活性炭2Kg,搅拌30min,过滤,滤液用氢氧化钠溶液调pH至3.0,析出结晶,过滤,滤餠分别用水和丙酮洗涤,真空干燥,得白色固体产品7-AVCA 10.0Kg,含量98.2%(HPLC面积归一法),熔点>200℃。
实施例2:
在2000L的反应釜中加入250L二氯甲烷,搅拌,通入氮气,降温至10℃,然后依次加入GCLE 48.5Kg、亚磷酸三乙酯17.6Kg、碘化钾18kg,于10~20℃反应3h。加入预冷至10~15℃的37%甲醛水溶液12Kg和水60L,滴加碳酸氢钠溶液,析出结晶。保持pH8.7~8.9,强烈搅拌下,在15~20℃反应3h。加入800L水,继续反应1h、抽滤,滤餠水洗,抽干后移入180L甲醇中,于0~10℃搅拌洗涤1h。抽滤,不超过50℃,真空干燥,得白色固体产品GVNE 40.4Kg,含量96.8%(HPLC面积归一法),熔点180.2~182.4℃。
以下同实施例1。
Claims (5)
1、一种制备7-氨基-3-乙烯基头孢烷酸的方法,是以7-苯乙酰氨基-3-氯甲基头孢烷酸对甲氧苄酯为起始原料,采用Wittig反应,在3位引入乙烯基后得7-苯乙酰氨基-3-乙烯基-4-头孢烷酸对甲氧苄酯,即产物GVNE,再由GVNE制备7-氨基-3-乙烯基头孢烷酸,其特征在于所述GVNE的制备过程为:7-苯乙酰氨基-3-氯甲基头孢烷酸对甲氧苄酯在溶剂体系中,在氮气或氩气保护下,加入催化剂,于0~100℃与亚磷酸三乙酯反应1~24小时,再采用Wittig反应,得产物GVNE粗品,再经水洗,甲醇洗涤,得到白色晶体GVNE,所述的溶剂体系是四氢呋喃、二氯甲烷、乙腈、二甲基甲酰胺、乙酸乙酯、甲醇或乙醇,或是上述溶剂与水组成的混合溶剂体系,所述的催化剂采用NaI、KI或I2。
2、根据权利要求1所述的制备7-氨基-3-乙烯基头孢烷酸的方法,其特征在于7-苯乙酰氨基-3-氯甲基头孢烷酸对甲氧苄酯与亚磷酸三乙酯的反应摩尔比为1∶1~1∶1.5,催化剂的用量为7-苯乙酰氨基-3-氯甲基头孢烷酸对甲氧苄酯摩尔用量的0.2~1.5倍。
3、根据权利要求1所述的制备7-氨基-3-乙烯基头孢烷酸的方法,其特征在于所述催化剂的用量为7-苯乙酰氨基-3-氯甲基头孢烷酸对甲氧苄酯摩尔用量的0.9~1.2倍。
4、根据权利要求1所述的制备7-氨基-3-乙烯基头孢烷酸的方法,其特征在于所述Wittig反应中使用的碱液为碳酸钾、碳酸钠、碳酸氢钾、碳酸氢钠、氢氧化钠、氢氧化钾、氨水或三乙胺。
5、根据权利要求1所述的制备7-氨基-3-乙烯基头孢烷酸的方法,其特征在于所述Wittig反应中甲醛的用量控制在亚磷酸三乙酯摩尔用量的1.0~3.0的范围之内。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNB2007101147821A CN100516070C (zh) | 2007-11-30 | 2007-11-30 | 制备7-氨基-3-乙烯基头孢烷酸的方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNB2007101147821A CN100516070C (zh) | 2007-11-30 | 2007-11-30 | 制备7-氨基-3-乙烯基头孢烷酸的方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN101182326A CN101182326A (zh) | 2008-05-21 |
| CN100516070C true CN100516070C (zh) | 2009-07-22 |
Family
ID=39447766
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB2007101147821A Expired - Fee Related CN100516070C (zh) | 2007-11-30 | 2007-11-30 | 制备7-氨基-3-乙烯基头孢烷酸的方法 |
Country Status (1)
| Country | Link |
|---|---|
| CN (1) | CN100516070C (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102212072A (zh) * | 2011-04-21 | 2011-10-12 | 山东天信化工有限公司 | 7-氨基-3-乙烯基头孢烷酸的制备方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102898440A (zh) * | 2012-10-11 | 2013-01-30 | 南通康鑫药业有限公司 | 一种7-苯乙酰氨基-3-乙烯基头孢烷酸对甲氧苄酯的制备工艺 |
| CN103923104B (zh) * | 2014-04-25 | 2016-04-13 | 湖北凌晟药业有限公司 | 7-苯乙酰氨基-3-乙烯基-4-头孢烯酸对甲氧基苄酯的制备方法 |
| CN104073543A (zh) * | 2014-06-06 | 2014-10-01 | 广东立国制药有限公司 | 一种7-氨基-3-乙烯基-3-头孢环-4-羧酸的合成方法 |
| CN107298689B (zh) * | 2017-06-06 | 2019-10-25 | 河北科技大学 | 以三苯胺为双电子供体的D-D-π-A结构的光敏染料及其制备方法和应用 |
| CN111592557A (zh) * | 2020-05-09 | 2020-08-28 | 河北合佳医药科技集团股份有限公司 | 一种7-氨基-3-乙烯基头孢烷酸的一步法环保制备方法 |
-
2007
- 2007-11-30 CN CNB2007101147821A patent/CN100516070C/zh not_active Expired - Fee Related
Non-Patent Citations (3)
| Title |
|---|
| A synthesis of new 3-dialkoxyphosphinylmethyland 3-dihydroxyphosphinylmethyl cephalosporins. Nishide, Kiyoharu et al.Chemical & Pharmaceutical Bulletin,Vol.36 No.(7). 1988 |
| A synthesis of new 3-dialkoxyphosphinylmethyland 3-dihydroxyphosphinylmethyl cephalosporins. Nishide, Kiyoharu et al.Chemical & * |
| Pharmaceutical Bulletin,Vol.36 No.(7). 1988 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102212072A (zh) * | 2011-04-21 | 2011-10-12 | 山东天信化工有限公司 | 7-氨基-3-乙烯基头孢烷酸的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN101182326A (zh) | 2008-05-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN100516070C (zh) | 制备7-氨基-3-乙烯基头孢烷酸的方法 | |
| CN106083691B (zh) | 一种盐酸阿比朵尔一水合物的制备方法 | |
| CN103539803B (zh) | 一种制备头孢曲松钠的方法 | |
| CN103073438B (zh) | 一种盐酸氨溴索化合物的精制方法 | |
| CN102491918B (zh) | 一种丙氨酰谷氨酰胺化合物及其制法 | |
| CN102786431A (zh) | 一种盐酸丙帕他莫的制备方法 | |
| CN102516331A (zh) | 一种加米霉素的制备方法 | |
| CN101830891A (zh) | 一种吡贝地尔的制备方法 | |
| CN102731605B (zh) | 一种醋酸阿比特龙的纯化方法 | |
| CN102633819A (zh) | 一种头孢西丁的制备方法 | |
| CN102351778A (zh) | 一种盐酸阿比朵尔的制备方法 | |
| CN101362732A (zh) | 一种头孢克肟侧链酸的制备方法 | |
| CN102516191B (zh) | 一种利奈唑胺的制备方法 | |
| CN102234313A (zh) | 一种匹多莫德的合成方法 | |
| CN110028418A (zh) | 一种碘佛醇的制备方法 | |
| KR20110110226A (ko) | 세프디니르 산성 복염 화합물 및 제조방법 | |
| JP6086925B2 (ja) | ナルメフェン塩酸塩の回収方法 | |
| CN102267953B (zh) | 合成头孢拉定或Cefroxadine的中间体化合物及其制备方法和应用 | |
| CN104230956B (zh) | 一种头孢西丁的制备方法 | |
| CN102079750B (zh) | 一种头孢唑肟钠化合物及其新方法 | |
| CN105175316B (zh) | 一种制备缓泻剂匹可硫酸钠的方法 | |
| CN103360310A (zh) | 一种西他沙星中间体、西他沙星的制备方法和西他沙星药物组合物 | |
| CN102675385A (zh) | 制备阿霉素13-位腙衍生物的方法 | |
| CN114436880A (zh) | 碘普罗胺中间体的制备方法 | |
| CN102070654A (zh) | 一种头孢硫脒的制备方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20090722 Termination date: 20151130 |
|
| EXPY | Termination of patent right or utility model |