CN100348564C - 二苯乙烯衍生物的制备方法 - Google Patents
二苯乙烯衍生物的制备方法 Download PDFInfo
- Publication number
- CN100348564C CN100348564C CNB2004800254708A CN200480025470A CN100348564C CN 100348564 C CN100348564 C CN 100348564C CN B2004800254708 A CNB2004800254708 A CN B2004800254708A CN 200480025470 A CN200480025470 A CN 200480025470A CN 100348564 C CN100348564 C CN 100348564C
- Authority
- CN
- China
- Prior art keywords
- compound
- formula
- resveratrol
- reaction
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C39/00—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a six-membered aromatic ring
- C07C39/205—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a six-membered aromatic ring polycyclic, containing only six-membered aromatic rings as cyclic parts with unsaturation outside the rings
- C07C39/21—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a six-membered aromatic ring polycyclic, containing only six-membered aromatic rings as cyclic parts with unsaturation outside the rings with at least one hydroxy group on a non-condensed ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C37/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring
- C07C37/01—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring by replacing functional groups bound to a six-membered aromatic ring by hydroxy groups, e.g. by hydrolysis
- C07C37/055—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring by replacing functional groups bound to a six-membered aromatic ring by hydroxy groups, e.g. by hydrolysis the substituted group being bound to oxygen, e.g. ether group
- C07C37/0555—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom of a six-membered aromatic ring by replacing functional groups bound to a six-membered aromatic ring by hydroxy groups, e.g. by hydrolysis the substituted group being bound to oxygen, e.g. ether group being esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C39/00—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a six-membered aromatic ring
- C07C39/205—Compounds having at least one hydroxy or O-metal group bound to a carbon atom of a six-membered aromatic ring polycyclic, containing only six-membered aromatic rings as cyclic parts with unsaturation outside the rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C69/00—Esters of carboxylic acids; Esters of carbonic or haloformic acids
- C07C69/017—Esters of hydroxy compounds having the esterified hydroxy group bound to a carbon atom of a six-membered aromatic ring
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
本发明公开了一种涉及Heck反应新的制备白藜芦醇和Piceatannol和其酯的方法。本发明还公开了此方法中的新的中间体。
Description
本发明涉及一种新的制备苯乙烯基衍生物的方法。更具体的是,本发明涉及一种新的制备白藜芦醇和Piceatannol和其酯的方法。
白藜芦醇(Resveratrol)存在于自然界中,系统名为3,4’,5-三羟基二苯乙烯,它是一种已知化合物,由于它的生物性质(参见例如国际专利申请WO 01/60774),所以备受关注。Picetannol也是一种已知化合物,系统名为3,3’,4,5’-四羟基二苯乙烯。
除其它文献以外,WO 01/60774和Tetrahedron Letter 43(2002)597-598中已经公开了白藜芦醇的制备方法。后一参考文献描述了如下方法,其中,通过多步反应由3,5-二羟基苯甲醛开始,包括3,5-二羟基苯乙烯与4-乙酰氧基-碘化苯的Heck反应制备白藜芦醇,产率为70%。本发明的方法使用更易得到的原料且进行的反应步骤更少,产率更高,因此为制备自藜芦醇和Picetannol和其衍生物(例如酯),提供了一种技术上更吸引人的方法。
一方面,本发明涉及一种制备白藜芦醇和Piceatannol和其酯的方法,该方法包括将式I化合物

与式II的化合物反应
其中,Z和Z1分别为被保护的羟基;Z2是氢或Z1;且A和B其中之一是乙烯基,另一为氯或溴;
以得到式III化合物

其中Z和Z1如上定义,R1是氢或Z1;
由式III的化合物裂解羟基保护基得到白藜芦醇或Piceatannol,如果希望的话,将所得到的白藜芦醇或Piceatannol转化成酯,如果还希望的话,将(E)-白藜芦醇或其酯或者(E)-Piceatannol或其酯异构化以得到相应的(Z)-异构体。
此处使用的术语“白藜芦醇”和“Piceatannol”指(E)和(Z)异构体和其混合物。在优选的方面,本发明涉及(E)异构体的制备方法。更优选的是,本发明涉及(E)白藜芦醇的制备方法。
被保护的羟基Z和Z1上的保护基团可以是任何可解离的保护基团。这种保护基团实例是吸电子基团,例如酰基,比如直链的或分枝的烷酰基,具体的是乙酰基,或芳酰基,例如苯甲酰;碳酸酯基,比如甲氧基羰基、乙氧基羰基和苄氧基羰基;氨基甲酸酯基,例如甲基氨基甲酰基;和磺酸酯,例如甲苯磺酸酯和甲烷磺酸酯。保护基团Z和Z1另外的实例是非吸电子基团,比如缩醛基,例如甲氧基亚甲基、乙氧基亚乙基、甲氧基甲基、苄氧基甲基、四羟基吡喃基、1-乙氧基乙基和1-甲氧基-1-甲基乙基,或甲硅烷醚,比如三甲基甲硅烷基、三异丙基甲硅烷基和叔丁基二甲基甲硅烷基。引入和解离这些保护基团的技术是本领域所已知的,参见例如T.W.Greene和P.G.M.Wuts(des.)Protective Group in Organic Synthesis,第三版,John Wiley 1999 246-287和其中引用的文献。
式I化合物与式II化合物的反应可以在本身已知的Heck反应条件下实施。合适的是,使用约等摩尔量的式I化合物和式II化合物。作为反应溶剂,可以使用任意惰性有机溶剂,这种溶剂的实例是有机溶剂,例如烃,比如甲苯;醚,比如二氧杂环己烷;腈,比如乙腈;酮,比如丙酮和酰胺,比如二甲基甲酰胺或N-甲基吡咯烷酮。而且,该反应适合在碱的存在下实施,该碱可以是无机碱,例如碳酸盐和碳酸氢盐,比如碳酸钠和碳酸氢钠、叔磷酸盐,例如K3PO4,或可以是有机碱,例如胺,比如三乙基胺或二异丙基乙基胺或碱金属的乙酸盐,比如乙酸钠。分别根据反应物I和反应物II以至少等摩尔量使用碱。而且,根据Heck反应的通用条件,可以加入催化剂,该催化剂包括钯源,比如乙酸钯或Pd(dba)2和稳定配合剂,例如膦,比如三邻甲苯基膦,或铵盐,例如四丁基氯化铵。而且,已证明钯催化剂和环状钯(palladacycle),例如肟衍生的钯配合物或反式-二(μ-乙酸)-二[(o-甲苯膦基)苄基]钯II(参见Advanced Synthesis and Catalysis344(2002),172-183和Journal of Organometallic Chemistry 576(1999),23-41)是适宜的钯源。
该反应适于在常压或高压下、在高温下(例如不高于适当压力下的使用溶剂的沸点温度)、且在惰性气氛(例如氩气)中实施。
通过解离保护基团式III所得到的化合物可以转化成白藜芦醇,如果希望的话,通过酯化羟基,可以转化成白藜芦醇酯。被保护羟基的Z和Z1的解离可以通过以上所引用的本身已知的方法来完成。解离被保护羟基(例如乙酰氧基)可以通过例如碱水解,比如在高温下用醇式碱金属氢氧化物处理来完成。在本发明的优选的方面中,被保护羟基是乙酰氧基,其在基本上中性条件下水解。用乙酸铵处理式III化合物,其中Z和Z1或Z、Z1和R1是乙酰氧基,可以实现这种水解,例如通过在大约室温下或不高于沸点高温下将乙酸铵水溶液(不高于60%)加到式III化合物的例如甲醇或乙醇的适当溶剂的溶液中,可参见Tetrahedron 59(2003)1049-1054。解离保护基团以后,通过酸化(碱水解后)和用例如乙酸乙酯的有机溶剂萃取可以从反应混合物中分别离析出白藜芦醇和Piceatannol。如果希望的话,白藜芦醇和Piceatannol通过采用适当的酸酯化可以转化成酯或者通过本身已知的方法转化成其活性衍生物或者它可以进行异构化。
白藜芦醇和Piceatannol的酯分别可以由未取代或取代的直链或分枝的具有1到26个碳原子的烷基衍生,或者由未取代或取代的直链或分枝的具有1到26个碳原子的脂族、芳烷基或芳族羧酸衍生。
尽管通过本发明的方法得到的(E)-白藜芦醇或(E)-Piceatannol是唯一的反应产物或基本上唯一的反应产物,如果希望的话,通过已知的方法(E)-异构体可以异构化成(Z)-异构体,参见例如Agric.Food Chem.43;1995;1820-1823。
在本发明的另一方面中,在以上反应中使用的式I化合物由式IV化合物制备。

其中Z如上定义。
更具体的是,根据本发明的这个方面,式I化合物通过还原式IV化合物得到式V化合物

其中Z如上定义,
并将式V化合物脱水来制备。
可以通过催化氢化,例如采用贵金属催化剂,比如炭负载Pd或Pt,或者活化Ni催化剂,比如Raney Ni在醇(例如甲醇)中的溶液,将式IV化合物还原以形成式V化合物。氢化反应条件不是严格限制的,该氢化反应可以在常压或高压下实施。合适的是,在贵金属催化剂的存在下使用H2在高氢气压力下,例如高达200bar,具体的是,在约10bar到约30bar氢气,和在约20℃到约50℃的温度下实施氢化。式V化合物是新化合物,它同样也是本发明的目的。通过传统的方法,例如通过反应溶液硅胶色谱可以将其离析。式IV化合物是已知的,或者可以由已知的二羟基苯乙酮以本身已知的方式通过保护羟基来制备,见上。通过本身已知的醇转化成烯烃的过程,式V化合物可以转化成式I化合物。合适的是,式V化合物的羟基首先转化成离去基团生成式VI的化合物

其中Y是离去基团,Z如上定义,
接着消去该离去基团。在此反应步骤中离去基团实例是卤素,例如氯或溴;或酯基,具体的是磺酸基,比如对甲苯磺酸基或甲磺酸基,或羧酸酯基,比如乙酰氧基;或碳酸酯;或黄原酸酯。采用传统的方法可以引入和消去离去基团。因此,式V化合物可以与卤化剂反应,例如与三溴化磷,生成式VI化合物,其中Y是溴。采用脱氢卤化剂,比如LiBr/Li2CO3,处理式VI化合物制备式I化合物。式V化合物脱氢生成式I化合物也可以通过以下方法来实现,将羟基与活性磺酸衍生物比如甲磺酰或苯磺酰卤化物或酸酐酯化以生成式VI化合物,其中Y是用于酯化式IV中的羟基的磺酸衍生物的磺酸基部分。而且,式V化合物可以与例如氯甲酸乙酯的氯甲酸烷基酯反应生成式VI化合物,其中Y是(烷基-O-CO-O-)。
式VI化合物(其中Y是酯基),转化成式I化合物适于在适当的惰性、酸稳定有机溶剂中在不高于使用溶剂的沸点的温度下实施,该有机溶剂可以是非极性溶剂,例如烃(比如甲苯),或卤化烃(比如二氯甲烷),或者是极性溶剂,比如酯(例如乙酸乙酯)或胺(例如DMF或NMP)或二甲亚砜。所述转化也可以在气相实施,例如在800℃的温度下,如果希望的话,也可以以连续模式实施。如果Y表示(烷基-OCOO-),那么从化合物VI热解消去离去基团Y特别适合。可以理解,本领域技术人员可以容易确定裂解具体离去基团的合适条件。
式VI化合物是新的化合物,这同样也是本发明的目的。
而且,将式V化合物转化成式I化合物也可以直接通过采用无机酸,比如硫酸或氢化硫酸盐或磷酸;或有机酸,比如甲酸或对-甲苯磺酸;或酸性离子交换树脂处理式V化合物完成,所述酸或树脂可以以催化量或以高达等摩尔量使用。
以下实施例进一步阐明该发明。
实施例1
在500ml的高压釜中装20g(85mmol)3,5-二乙酰氧基苯乙酮、2g炭负载钯(催化剂E101 CA/W 5%,Degussa,Germany)和350ml甲醇。用氢气(3bar)冲洗高压釜三次,搅拌该混合物,在30bar氢压力下实施氢化2小时。用3bar氮气冲洗该溶液两次后,通过Hyflo Super Gel过滤除去催化剂。蒸发溶剂留下19.2g浅棕色油,将其用正乙烷/乙酸乙酯(5∶2)作为洗脱混合物通过硅胶闪蒸色谱进一步纯化得到18.4g 1-(1-羟基乙基)-3,5-二乙酰氧基苯无色油(81mmol,GC 98%)。
实施例2
将10g(42mmol)在80ml DMF中的1-(1-羟基乙基)-3,5-二乙酰氧基苯放到装有磁力搅拌的250ml两口瓶中。用氩气完全冲洗该反应瓶,并将混合物冷却到0℃。将4.06ml(42mmol)三溴化磷在30ml CH2Cl2中的溶液缓缓加入,该反应在0℃下3小时内完成。蒸发CH2Cl2,采用21.38g(0.24mol)溴化锂和20.74g(0.28mol)碳酸锂从中间体1-(1-溴乙基)-3,5-二乙酰氧基苯消去溴化氢。在弱氩气流下,120℃下搅拌该混合物18小时,然后倒入150ml乙酸乙酯中,用300ml冰水萃取成两部分。有机层用盐水和饱和NH4Cl溶液洗涤,MgSO4干燥,得到5.18g(56%)希望的产物,3,5-二乙酰氧基苯乙烯,和3.14g(42%)单乙酰氧基苯乙烯。
将产物混合物溶入10ml吡啶,用2.33ml(21.2mmol)未进一步纯化的乙酸酐在室温下处理15h。将得到的溶液倒入100ml乙酸乙酯中,用100ml 1N HCl萃取三次。有机层用50ml盐水洗涤二次,MgSO4干燥。蒸发溶剂以后,用7∶2(v/v)的正己烷和乙酸乙酯的混合物进行硅胶色谱纯化得到8.04g 3,5-二乙酰氧基苯乙烯无色油(36.5mmol,87%)。
实施例3
a)将0.5g(2.1mmol)1-(1-羟基乙基)-3,5-二乙酰氧基苯放到25ml的反应瓶中,并将其溶于0.31ml(3.78mmol,1.8当量)吡啶中。滴加2.52mmol(1.2当量)乙酸酐,同时搅拌反应混合物。在60℃下,实施转化三小时。该混合物用5ml乙酸乙酯稀释,用5ml 1N HCl萃取一次。有机层用饱和氯化铵溶液洗涤两次,硫酸镁干燥。蒸发溶剂后,得到纯度为99%(由GC测定)的1-(1-乙酰氧乙基)-3,5-二乙酰氧基苯。
b)将20g(71mmol)1-(1-乙酰氧乙基)-3,5-二乙酰氧基苯溶于20ml甲苯中,并将其放入连接到装满Raschig环的热解管上的加料器。将该管加热到500℃,并立即加入纯混合物。醇的转化在两分钟内完成。用甲苯冲洗此柱。用20ml水萃取甲苯中的粗产物。用甲苯再次萃取水溶液,合并有机层,并用硫酸镁干燥,真空除去溶剂得到6.6g 3,5-二乙酰氧基苯乙烯(42%)。
实施例4
a)在100ml平底反应瓶中将12.7g(53mmol)1-(1-羟基)乙基-3,5-二乙酰氧基苯溶入38ml甲苯和5.6g(71mmol)吡啶。在氩气低于50℃的温度下缓慢加入6.7g(62mmol)氯甲酸乙酯。另外搅拌该混合物30分钟。冷却到室温以后,将13ml水加到反应混合物中并将其剧烈搅拌。相分离以后,用13ml 1N NH4Cl洗涤有机层。用水洗涤有机层至中性,真空下蒸发溶剂得到16.27g乙酸3-乙酰氧基-5-(1-乙氧基羧基-乙基)-苯酯(99%)。
b)将20.0g乙酸3-乙酰氧基-5-(1-乙氧基羧基-乙基)-苯酯溶于92.5ml甲苯中,并在475℃下通过装有陶瓷填料的玻璃管连续处理该混合物。将反应混合物冷却到室温以后,真空蒸发溶剂,分离出12.8g(90%)纯3,5-二乙酰氧基苯乙烯。
实施例5
将装有冷凝器和磁力搅拌且连接到氩气管道上的三口瓶放到油浴上,并加入1g(4.5mmol)3,5-二乙酰氧基苯乙烯、0.98g(4.54mmol)4-乙酰氧基溴苯和在20ml DMF中的0.58g(5.5mmol)碳酸氢钠。通过交替用氩气冲洗反应瓶和抽空反应瓶使该混合物脱气。将62.8mg(0.2mmol)三邻甲苯基膦和在脱气的DMF中的15.3mg(0.07mmol)乙酸钯加到反应器中。将得到的溶液加热到100℃,在氩气下回流18小时。通过将反应混合物倒入50ml冰水中用乙酸乙酯萃取来实施纯化。用氯化钠使水层饱和,并用20ml乙酸乙酯再次萃取。合并有机层,用50ml饱和NH4Cl溶液和50ml盐水洗涤,MgSO4干燥,真空蒸发溶剂得到1.76g黄色固体。用在20ml乙酸乙酯中的0.8ml(10mmol)吡啶和1.1g(10mmol)乙酸酐来乙酰化粗产物。80℃下回流该混合物一小时。冷却到室温后,粗产物用30ml乙酸乙酯稀释,用1N HCl萃取四次。用MgSO4干燥有机层,除去溶剂。采用5∶2(v/v)的正己烷和乙酸乙酯混合物作为洗脱剂进行硅胶色谱纯化得到1.2g(E)-3,4’,5-三乙酰氧基二苯乙烯(3.4mmol,75%)。
实施例6
在装有磁力搅拌的反应瓶中加入50mg(0.23mmol,1当量)3,5-二乙酰氧基苯乙烯、49mg(0.23mmol)4-乙酰氧基溴苯、46mg(0.27mmol)NaHCO3和78mg(0.54mmol,2.4当量)四丁基氯化铵。反应物悬在2ml DMF中,用氩气冲洗该混合物然后抽气。实施氩气冲洗抽气三次。加入0.76mg在50μl脱气DMF中的乙酸钯(3μmol,1.5mol%)开始反应,且在100℃下搅拌3小时。对反应混合物接着进行实施例5的步骤得到(E)-3,4’,5-三乙酰氧基二苯乙烯。
实施例7
在装有磁力搅拌的10ml Schlenk管中加入119mg(0.7mmol)4-乙酰氧基氯苯、200mg(0.84mmol,1.2当量)3,5-二乙酰氧苯乙烯、177mg(0.84mmol,1.2当量)三聚磷酸钾、5mg(0.014mmol,2mol%)二金刚烷基-正丁基膦和3.7mg(0.003mmol,0.5mol%)三(二亚苄基丙酮)-二钯氯仿配合物。抽空该管,氩气冲洗三次,在惰性条件下加入2ml脱气DMAc。在120℃下搅拌15.5小时实施该反应。对反应混合物接着进行实施例5的步骤得到(E)-3,4’,5-三乙酰氧基二苯乙烯。
实施例8
在装有磁力搅拌的10ml Schlenk管中加入59mg(0.7mmol)4-乙酰氧基氯苯、100mg(0.84mmol,1.2当量)3,5-二乙酰氧基苯乙烯、125mg(0.38mmol,1.1当量)碳酸铯、5μl三叔丁基膦(0.02mmol,6mol%)和5.57mg(0.01mmol,1.5mol%)三(二亚苄基丙酮)-二钯氯仿配合物。抽空该管,氩气冲洗三次,在惰性条件下加入1ml脱气二氧六环。在120℃下搅拌19小时实施该反应。对反应混合物接着进行实施例5的步骤得到(E)-3,4’,5-三乙酰氧基二苯乙烯。
实施例9
在100ml平底反应瓶中加入10.8g(50mmol)4-乙酰氧基溴苯、11.7g(50mmol)3,5-二乙酰氧基苯乙烯和8.3g(60mmol,1.2当量)碳酸钾。将组分溶于35ml NMP,用氩气冲洗反应瓶。在惰性条件下加入6.9mg(0.05mol%)溶于5ml NMP的乙酰苯酰-肟衍生钯催化剂(CAS No.32679-19-9,又见Adv.Synth.Catal.2002,344,No.2,p173公开的环钯16a)开始Heck反应。在150℃下搅拌反应混合物3小时,接着将其冷却到室温。将50ml乙酸乙酯加到粗产物中,得到的溶液用50ml1N HCl萃取四次。水溶液用50ml乙酸乙酯再萃取两次,合并有机层,用MgSO4干燥,除去溶剂。接着进行实施例5的步骤。3,4’,5-三乙酰氧基二苯乙烯的分离产量为17.7g(94%)。
实施例10
50mg(0.14mmol)(E)-3,4’,5-三乙酰氧基二苯乙烯溶于3ml甲醇中,并用氩气脱气。弱氩气流下滴加8mg(0.14mmol)在0.5ml甲醇中的氢氧化钾,30分钟将混合物加热到65℃。用1N盐酸中和得到的溶液,将其倒入10ml乙酸乙酯中,用5ml盐水萃取3次。用MgSO4干燥有机相,真空除去溶剂得到30mg白藜芦醇(0.13mmol,92%)。
实施例11
将40g(0.168mol)3,5-二乙酰氧-1-(1-羟基)-乙基苯放到250ml平底瓶中,在氩气下缓慢加入19.6ml(0.2mol,1.2当量)乙酸酐和18.9ml(0.2mol,1.2当量)吡啶。1小时将纯混合物加热到80℃。冷却到室温以后,将200ml乙酸乙酯倒入原料中,用100ml 1N HCl萃取该混合物4次。用硫酸镁干燥有机层,真空蒸发溶剂得到42.4g 3,5-二乙酰氧-1-(1-乙酰氧基)-乙基苯(97%)。
实施例12
将6.32g(23mmol)3,5-二乙酰氧-1-(1-羟基)-乙基苯和12.3ml三乙胺(87.73mmol,3.81当量)溶于50ml甲苯并冷却到0℃。用5ml甲苯稀释6.26ml甲烷磺酰氯(79.76mmol,3.46当量),并将该试剂滴加到醇溶液中。0℃下搅拌混合物2.5小时。将40ml饱和氯化铵溶液倒入反应原液。分离有机层,用40ml饱和氯化氨萃取一次,用30ml碳酸氢钠萃取一次,硫酸镁干燥。真空蒸发溶剂得到8.27g纯度为80.7%(GC)的3,5-二乙酰氧基-1-(1-甲基磺酰基)-乙基苯。
实施例13
将5g(13.06mmol)3,5-二乙酰氧基-1-(1-甲基磺酰基)-乙基苯溶于20ml甲苯,并在室温搅拌下缓缓加入1.75ml(12.41mmol,0.95当量)二异丙胺。3.5小时将混合物加热到180℃,冷却到室温以后,用60ml饱和氯化铵溶液萃取两次。此后,用50ml水洗涤有机层,硫酸镁干燥。减压蒸发溶剂得到2.9g纯度为85%(GC测定)的3,5-二乙酰氧基苯乙烯(86%)。
实施例14
将0.57g(3mmol)3,4-二羟基-溴苯(根据Journal of MaterialChemistry 10(7),2000,1519-1526,由商业可得的3,4-二甲氧基-溴苯合成)溶于0.31ml(3.78mmol)吡啶。搅拌的同时将6.6mmol乙酸酐滴加到反应混合物中。在60℃下实施反应3小时。该混合物用5ml乙酸乙酯稀释,用5ml 1N HCl萃取一次。有机层用饱和氯化铵溶液洗涤两次,硫酸镁干燥。蒸发溶剂以后,得到0.79g(2.9mmol,97%)纯度为99%的3,4-二乙酰氧基溴苯。
实施例15
在装有磁力搅拌的10ml Schlenk管中加入218mg(0.8mmol)3,4-二乙酰氧基溴苯、176mg(0.84mmol)3,5-二乙酰氧基苯乙烯、187mg(0.9mmol)碳酸钾和0.22mg(0.1mol%)实施例9使用的乙酰苯酰-肟衍生钯催化剂CAS No.32679-19-9。抽空该管,氩气冲洗三次,在惰性条件下加入1ml脱气DMF。150℃搅拌下19小时实施该反应。接着进行实施例5的程序得到(E)-3,3’,4’,5-四乙酰氧基二苯乙烯。
实施例16
将2.6g(17.5mmol)2-巯基吡啶-1-氧化物溶于30.0ml溴三氯甲烷,并加热至回流。在同一温度下滴加4.04g(15mmol)3,5-二乙酰氧基苯甲酰氯和433mg(2.5mmol)2,2’-偶氮二异丁腈溶于30.0ml溴三氯甲烷的混合物。再回流两小时后,将反应混合物冷却到室温并真空浓缩。用9∶1(v/v)正己烷和乙酸乙酯作为洗脱剂进行硅胶色谱纯化生成2.4g(8.7mmol,58%)3,5-二乙酰氧基溴苯。
实施例17
在装有磁力搅拌的10ml Schlenk管中加入218mg(0.8mmol)3,5-二乙酰氧基溴苯、136mg(0.84mmol)4-乙酰氧基苯乙烯、187mg(0.9mmol)碳酸钾和0.22mg(0.1mol%)实施例9使用的乙酰苯酰-肟衍生钯催化剂CAS No.32679-19-9。抽空该管,氩气冲洗三次,且在惰性条件下加入1ml脱气DMF。150℃下搅拌15小时实施该反应。接着进行实施例4的程序得到(E)-3,4’,5-三乙酰氧基二苯乙烯。
实施例18
将2.10g(6.0mmol)三乙酰氧基二苯乙烯回流溶于19.3ml甲醇,并将39ml乙酸铵溶液(25%)加到该溶液中。在同一温度下搅拌反应混合物3小时,此后蒸馏出甲醇。通过冷却到5℃,产物结晶且可以滤出。得到1.33g(5.82mmol,97%)白藜芦醇纯化合物。用甲基丁基醚萃取纯化母液,并用氨水中和。减压浓缩后,该溶液再用于所描述的反应。
Claims (8)
1.一种制备白藜芦醇和Piceatannol和它们的酯的方法,该方法包括将式I化合物

与式II化合物反应
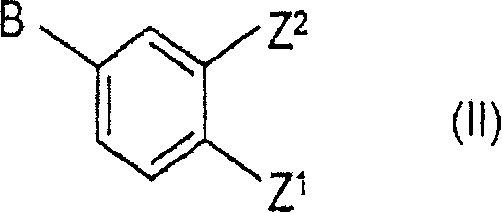
其中,Z和Z1分别为被保护的羟基;Z2是氢或Z1;A和B其中之一是乙烯基,另一为氯或溴;
以得到式III化合物

其中Z和Z1如上定义,R1是氢或Z1;
由所述式III化合物裂解羟基保护基得到白藜芦醇或Piceatannol,如果希望的话,将所得到的白藜芦醇或Piceatannol转化成酯,如果还希望的话,将(E)-白藜芦醇或其酯或者(E)-Piceatannol或其酯异构化以得到相应的(Z)-异构体。
2.如权利要求1所述的方法,其中R1和Z2是氢,得到的是白藜芦醇。
3.如权利要求1或2所述的方法,其中A是乙烯基,B是氯或溴。
4.如权利要求1到3中任意一项所述的方法,其中所述的被保护的羟基是乙酰氧基。
5.如权利要求4所述的方法,其中所述式III化合物中的乙酰氧基在基本上中性条件下水解。
6.如权利要求5所述的方法,其中采用乙酸铵实施所述水解。
7.如权利要求1所述的方法,其中A是乙烯基的所述式I化合物由式IV化合物制备

其中Z是被保护的羟基。
8.如权利要求7所述的方法,其中A是乙烯基的所述式I化合物通过将所述式IV化合物还原得到式V化合物
其中Z是被保护的羟基,
并将所述式V化合物脱水来制备。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP03020123.0 | 2003-09-05 | ||
| EP03020123 | 2003-09-05 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2007101420289A Division CN101117317B (zh) | 2003-09-05 | 2004-08-31 | 二苯乙烯衍生物的制备方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1845889A CN1845889A (zh) | 2006-10-11 |
| CN100348564C true CN100348564C (zh) | 2007-11-14 |
Family
ID=34259163
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNB2004800254708A Expired - Fee Related CN100348564C (zh) | 2003-09-05 | 2004-08-31 | 二苯乙烯衍生物的制备方法 |
| CN2007101420289A Expired - Fee Related CN101117317B (zh) | 2003-09-05 | 2004-08-31 | 二苯乙烯衍生物的制备方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN2007101420289A Expired - Fee Related CN101117317B (zh) | 2003-09-05 | 2004-08-31 | 二苯乙烯衍生物的制备方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7820848B2 (zh) |
| EP (1) | EP1663926A2 (zh) |
| JP (1) | JP4806351B2 (zh) |
| KR (1) | KR101140523B1 (zh) |
| CN (2) | CN100348564C (zh) |
| WO (1) | WO2005023740A2 (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102276426A (zh) * | 2010-11-04 | 2011-12-14 | 上海统益生物科技有限公司 | 白藜芦醇的一种新型合成方法 |
| CN110035992A (zh) * | 2016-12-05 | 2019-07-19 | 拜耳作物科学股份公司 | 用于制备3-取代的2-乙烯基苯基磺酸酯的方法 |
Families Citing this family (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005069998A2 (en) | 2004-01-20 | 2005-08-04 | Brigham Young University Technology Transfer Office | Novel sirtuin activating compounds and methods for making the same |
| FR2904311B1 (fr) * | 2006-07-28 | 2013-01-11 | Clariant Specialty Fine Chem F | Nouveau procede de synthese de derives (e) stilbeniques perm permettant d'obtenir le resveratrol et le piceatannol |
| FR2921921B1 (fr) | 2007-10-03 | 2011-08-19 | Clariant Specialty Fine Chem F | Procede de synthese de composes polyhydroxystilbeniques. |
| KR100878394B1 (ko) * | 2008-01-31 | 2009-01-13 | 한림대학교 산학협력단 | Piceatannol의 합성 |
| DE102008042144A1 (de) | 2008-09-17 | 2010-03-18 | Evonik Degussa Gmbh | Verfahren zur Herstellung von Silbenoiden |
| WO2010079123A2 (en) | 2009-01-06 | 2010-07-15 | Dsm Ip Assets B.V. | Process for resveratrol intermediate |
| CN104725226A (zh) * | 2009-03-04 | 2015-06-24 | 帝斯曼知识产权资产管理有限公司 | 一种制备酰基苯的方法 |
| HUE027734T2 (en) * | 2009-04-03 | 2016-11-28 | Morinaga & Co | Piceatannol-containing compositions and method for producing a piceatannol-containing composition |
| CN101519345B (zh) * | 2009-04-10 | 2012-11-07 | 徐州市心血管病研究所 | 反式-3,5-二羟基-4'-溴丁氧基二苯乙烯及其制法与用途 |
| DE102010023749A1 (de) | 2010-06-14 | 2011-12-15 | Evonik Degussa Gmbh | Zelle und Verfahren zur Herstellung von Resveratrol |
| JP5672963B2 (ja) * | 2010-10-28 | 2015-02-18 | ユーハ味覚糖株式会社 | 新規4−ビニルフェノール重合化合物 |
| JP5703887B2 (ja) * | 2011-03-25 | 2015-04-22 | ユーハ味覚糖株式会社 | 新規レスベラトロール誘導体 |
| JP5673207B2 (ja) * | 2011-02-28 | 2015-02-18 | ユーハ味覚糖株式会社 | 新規レスベラトロール誘導体 |
| JP5742634B2 (ja) * | 2011-09-29 | 2015-07-01 | ユーハ味覚糖株式会社 | 新規ヒドロキシスチルベン誘導体 |
| JP5729134B2 (ja) * | 2011-05-26 | 2015-06-03 | ユーハ味覚糖株式会社 | 新規レスベラトロール誘導体 |
| JP5728972B2 (ja) * | 2011-01-26 | 2015-06-03 | ユーハ味覚糖株式会社 | 新規レスベラトロール誘導体 |
| JP5673025B2 (ja) * | 2010-11-26 | 2015-02-18 | ユーハ味覚糖株式会社 | 新規レスベラトロール誘導体 |
| JP5673091B2 (ja) * | 2010-12-27 | 2015-02-18 | ユーハ味覚糖株式会社 | 新規レスベラトロール誘導体 |
| JP5742589B2 (ja) * | 2011-08-26 | 2015-07-01 | ユーハ味覚糖株式会社 | 新規ヒドロキシスチルベン誘導体 |
| JP5853546B2 (ja) * | 2011-09-29 | 2016-02-09 | ユーハ味覚糖株式会社 | 新規ヒドロキシスチルベン誘導体 |
| WO2012070656A1 (ja) * | 2010-11-26 | 2012-05-31 | ユーハ味覚糖株式会社 | 生理活性を有するヒドロキシスチルベン誘導体の製造方法 |
| EP2468706A1 (en) | 2010-12-15 | 2012-06-27 | DSM IP Assets B.V. | Process for the preparation of resveratrol |
| WO2013003112A1 (en) | 2011-06-27 | 2013-01-03 | The Jackson Laboratory | Methods and compositions for treatment of cancer and autoimmune disease |
| FR2977891A1 (fr) | 2011-07-11 | 2013-01-18 | Centre Nat Rech Scient | Procede de preparation du trans-resveratrol et de ses analogues |
| CN102351658A (zh) * | 2011-08-30 | 2012-02-15 | 昆明制药集团股份有限公司 | 一种制备白皮杉醇的方法 |
| CN103086884A (zh) * | 2012-12-14 | 2013-05-08 | 湖南科源生物制品有限公司 | 乙酰化白藜芦醇的半合成方法 |
| CN103664537B (zh) * | 2013-12-06 | 2015-06-24 | 湖南科源生物制品有限公司 | 一种白藜芦醇的制备方法 |
| TW201702218A (zh) | 2014-12-12 | 2017-01-16 | 美國杰克森實驗室 | 關於治療癌症、自體免疫疾病及神經退化性疾病之組合物及方法 |
| CN105152849A (zh) * | 2015-09-06 | 2015-12-16 | 侯颖 | 一种反式二苯乙烯的合成方法 |
| CN107840792A (zh) * | 2017-10-27 | 2018-03-27 | 常州大学 | 一种白藜芦醇的合成方法 |
| CN108586211B (zh) * | 2018-06-08 | 2021-03-19 | 郑州德瑞医药科技有限公司 | 一种1,1-二芳基乙烯类衍生物的合成方法 |
| JP2019210274A (ja) * | 2018-12-28 | 2019-12-12 | Jfeケミカル株式会社 | 3−アセトキシスチレンの製造方法 |
| CN110172373B (zh) * | 2019-06-14 | 2022-08-05 | 中国农业科学院农产品加工研究所 | 白藜芦醇在花生油中的应用 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020128478A1 (en) * | 2001-03-02 | 2002-09-12 | Krska Shane W. | Process for a carbon-carbon coupling reaction of aryl halides with olefins by heterogeneous catalysts |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2748160A (en) * | 1952-04-15 | 1956-05-29 | Eastman Kodak Co | Process for preparing 2, 5-diacetoxysty-rene and intermediates therefor |
| ES2086165T3 (es) * | 1992-09-09 | 1996-06-16 | Hoechst Ag | Procedimiento para la preparacion de olefinas sustituidas con grupos aromaticos a partir de compuestos cloro-aromaticos. |
| DE4447068A1 (de) * | 1994-12-29 | 1996-07-04 | Hoechst Ag | Verfahren zur Herstellung aromatischer Olefine |
| DE19630581A1 (de) * | 1996-07-30 | 1998-02-05 | Studiengesellschaft Kohle Mbh | Verfahren zur Herstellung von Solvens-stabilisierten Metallkolloiden und trägerfixierten Metallclustern |
| DE19843012A1 (de) * | 1998-09-21 | 2000-03-23 | Studiengesellschaft Kohle Mbh | Verfahren zur Herstellung von Olefinen |
| AU2001241539A1 (en) * | 2000-02-16 | 2001-08-27 | Brigham Young University | Synthesis of resveratrol |
| JP2002179622A (ja) * | 2000-12-08 | 2002-06-26 | Adchemco Corp | 4−アセトキシスチレンの製造方法 |
-
2004
- 2004-08-31 CN CNB2004800254708A patent/CN100348564C/zh not_active Expired - Fee Related
- 2004-08-31 WO PCT/EP2004/009669 patent/WO2005023740A2/en not_active Ceased
- 2004-08-31 CN CN2007101420289A patent/CN101117317B/zh not_active Expired - Fee Related
- 2004-08-31 EP EP04764635A patent/EP1663926A2/en not_active Withdrawn
- 2004-08-31 US US10/570,777 patent/US7820848B2/en not_active Expired - Fee Related
- 2004-08-31 KR KR1020067004514A patent/KR101140523B1/ko not_active Expired - Fee Related
- 2004-08-31 JP JP2006525088A patent/JP4806351B2/ja not_active Expired - Fee Related
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020128478A1 (en) * | 2001-03-02 | 2002-09-12 | Krska Shane W. | Process for a carbon-carbon coupling reaction of aryl halides with olefins by heterogeneous catalysts |
Non-Patent Citations (1)
| Title |
|---|
| A new efficient resveratrol synthesis. Marcella Guiso,Carolina Marra,Angela Farina.tetrahedron letters,No.43. 2002 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102276426A (zh) * | 2010-11-04 | 2011-12-14 | 上海统益生物科技有限公司 | 白藜芦醇的一种新型合成方法 |
| CN110035992A (zh) * | 2016-12-05 | 2019-07-19 | 拜耳作物科学股份公司 | 用于制备3-取代的2-乙烯基苯基磺酸酯的方法 |
| CN110035992B (zh) * | 2016-12-05 | 2022-04-05 | 拜耳作物科学股份公司 | 用于制备3-取代的2-乙烯基苯基磺酸酯的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20070197819A1 (en) | 2007-08-23 |
| WO2005023740A3 (en) | 2005-05-12 |
| EP1663926A2 (en) | 2006-06-07 |
| WO2005023740A2 (en) | 2005-03-17 |
| CN101117317A (zh) | 2008-02-06 |
| KR20060091298A (ko) | 2006-08-18 |
| CN101117317B (zh) | 2011-10-12 |
| KR101140523B1 (ko) | 2012-04-30 |
| CN1845889A (zh) | 2006-10-11 |
| US7820848B2 (en) | 2010-10-26 |
| JP2007504191A (ja) | 2007-03-01 |
| JP4806351B2 (ja) | 2011-11-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN100348564C (zh) | 二苯乙烯衍生物的制备方法 | |
| EP3712130B1 (en) | Method for synthesis of roxadustat and intermediate compounds thereof | |
| Liu et al. | A novel route to olefins from vicinal diols | |
| KR100604699B1 (ko) | 베라프로스트와 이의 염을 제조하는 방법 | |
| CA2200317C (en) | Process for the purification of 2,6-diisopropylphenol | |
| EP1910399A4 (en) | NEW INTERMEDIATES, PROCESSES FOR THEIR PREPARATION AND PROCESS FOR PREPARING COQ10 USING THE SAME NEW INTERMEDIATES | |
| US4689427A (en) | Hydroquinone derivatives useful in the production of vitamin E | |
| KR20010066956A (ko) | 리코펜의 제조 방법 및 그의 중간체 | |
| ZA200602592B (en) | Acid soluble proteins from micellar casein | |
| CN1334803A (zh) | 制备(2s,4r,9s)-八氢-1h-吲哚-2-甲酸的方法及其中间体 | |
| JP2947503B2 (ja) | アリルキノン誘導体の製造方法および中間体 | |
| CN116041358B (zh) | 一种普拉洛芬的制备方法 | |
| JP4584625B2 (ja) | 1,2−シス−2−フルオロシクロプロパン−1−カルボン酸エステル類の製法 | |
| JPS60156666A (ja) | 4−置換−(e)−3−ブテン−2−オ−ル誘導体およびその製造方法 | |
| JP4396025B2 (ja) | 4−メトキシメチル−2,3,5,6−テトラフルオロベンゼンメタノールの製造法 | |
| JPH021823B2 (zh) | ||
| US5087769A (en) | Preparation of 6-substituted-2-vinylnaphthalene | |
| CN119707905A (zh) | 二氢异黄酮衍生物的制备方法 | |
| JP2762643B2 (ja) | 4―ヒドロキシシクロペンテノン誘導体の製造方法 | |
| CN116082386A (zh) | 一种含酯基脂质的合成方法 | |
| JP2002047219A (ja) | ビニルハライド誘導体および製造方法 | |
| JPH01224344A (ja) | 2,6−ジヒドロキシアセトフェノンの製造方法 | |
| JPH078827B2 (ja) | α,β‐不飽和アルデヒドの製造方法 | |
| JP2002212167A (ja) | ジアセチルピリジン誘導体の製造方法 | |
| JPH10265427A (ja) | 6−ヒドロキシ−2−ナフトアルデヒドと関連化合物の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20071114 Termination date: 20140831 |
|
| EXPY | Termination of patent right or utility model |