WO2024195789A1 - 神経オルガノイドの製造方法およびその利用 - Google Patents
神経オルガノイドの製造方法およびその利用 Download PDFInfo
- Publication number
- WO2024195789A1 WO2024195789A1 PCT/JP2024/010698 JP2024010698W WO2024195789A1 WO 2024195789 A1 WO2024195789 A1 WO 2024195789A1 JP 2024010698 W JP2024010698 W JP 2024010698W WO 2024195789 A1 WO2024195789 A1 WO 2024195789A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- organoids
- neural
- culture
- cell
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images












Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0618—Cells of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/30—Nerves; Brain; Eyes; Corneal cells; Cerebrospinal fluid; Neuronal stem cells; Neuronal precursor cells; Glial cells; Oligodendrocytes; Schwann cells; Astroglia; Astrocytes; Choroid plexus; Spinal cord tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61L—METHODS OR APPARATUS FOR STERILISING MATERIALS OR OBJECTS IN GENERAL; DISINFECTION, STERILISATION OR DEODORISATION OF AIR; CHEMICAL ASPECTS OF BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES; MATERIALS FOR BANDAGES, DRESSINGS, ABSORBENT PADS OR SURGICAL ARTICLES
- A61L27/00—Materials for grafts or prostheses or for coating grafts or prostheses
- A61L27/36—Materials for grafts or prostheses or for coating grafts or prostheses containing ingredients of undetermined constitution or reaction products thereof, e.g. transplant tissue, natural bone, extracellular matrix
- A61L27/3641—Materials for grafts or prostheses or for coating grafts or prostheses containing ingredients of undetermined constitution or reaction products thereof, e.g. transplant tissue, natural bone, extracellular matrix characterised by the site of application in the body
- A61L27/3675—Nerve tissue, e.g. brain, spinal cord, nerves, dura mater
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0618—Cells of the nervous system
- C12N5/0619—Neurons
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0618—Cells of the nervous system
- C12N5/0622—Glial cells, e.g. astrocytes, oligodendrocytes; Schwann cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0618—Cells of the nervous system
- C12N5/0623—Stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0697—Artificial constructs associating cells of different lineages, e.g. tissue equivalents
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/02—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving viable microorganisms
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
- G01N33/5058—Neurological cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/115—Basic fibroblast growth factor (bFGF, FGF-2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/155—Bone morphogenic proteins [BMP]; Osteogenins; Osteogenic factor; Bone inducing factor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/70—Enzymes
- C12N2501/72—Transferases [EC 2.]
- C12N2501/727—Kinases (EC 2.7.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/02—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from embryonic cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/45—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from artificially induced pluripotent stem cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2513/00—3D culture
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2533/00—Supports or coatings for cell culture, characterised by material
- C12N2533/50—Proteins
- C12N2533/52—Fibronectin; Laminin
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2533/00—Supports or coatings for cell culture, characterised by material
- C12N2533/90—Substrates of biological origin, e.g. extracellular matrix, decellularised tissue
Definitions
- the present invention relates to a method for producing neural organoids using a culture vessel that has multiple independent cell adhesive regions, and in which cells adhering to each region are present in the same medium.
- the present invention also relates to a transplantation therapy agent containing the neural organoids, and a screening method for a therapeutic or preventive drug for neurodegenerative diseases using the neural organoids.
- iPS cells induced pluripotent stem cells
- ALS amyotrophic lateral sclerosis
- Patent Document 2 a method for inducing differentiation from pluripotent stem cells into neural stem cells or nerve cells using micropatterning technology.
- Patent Document 2 a method for mass-producing neural organoids derived from pluripotent stem cells with stable quality at low cost was not known until now.
- the present invention therefore aims to provide a method for producing neural organoids using a culture vessel that has multiple independent cell adhesive regions and in which cells adhering to each region are present in the same medium, in order to realize the mass and inexpensive production of neural organoids derived from pluripotent stem cells with stable quality. It also aims to provide a transplantation therapy agent containing neural organoids obtained by the production method of the present invention, and a screening method for therapeutic or preventive drugs for neurodegenerative diseases using the neural organoids.
- neural organoids can be produced from uniform cells by simultaneously processing the initial induction stage of neural organoids using this culture vessel.
- the use of this culture vessel can stabilize the quality and improve the production efficiency of neural organoids derived from pluripotent stem cells.
- a method for producing neural organoids comprising: (1) a step of culturing pluripotent stem cells in an adhesion culture in a culture vessel having multiple independent cell adhesive regions, with the cells adhering to each region being present in the same medium, to form a cell population containing neural stem cells; and (2) a step of detaching the cell population containing neural stem cells formed in step (1) and culturing it in suspension.
- the suspension culture in the step (2) includes a step of performing rotation culture, culture under turbulent conditions, culture in a microgravity environment, or culture by the hanging drop method.
- [4] The method according to any one of [1-1] to [3], wherein the neural organoid is a cerebral organoid.
- [5] The method according to any one of [1-1] to [4], wherein the step (1) is carried out for at least 6 days.
- [6] The method according to any one of [1-1] to [5], wherein the cell adhesive region is a region containing an extracellular matrix.
- the cell adhesive region is a region containing a temperature responsive polymer.
- [8] The method according to any one of [1-1] to [7], wherein the number of cell adhesion regions is five or more.
- the agent according to [12-2], wherein the disease or injury caused by a disorder of the telencephalon, diencephalon, midbrain, hindbrain, or spinal cord is selected from the group consisting of Parkinson's disease, Huntington's chorea, Alzheimer's disease, cerebrovascular disease, epilepsy, brain trauma, motor neuron disease, neurodegenerative disease, and injury after brain surgery.
- [13] (1) Culturing the neural organoid according to [11] or cells constituting the neural organoid in the presence or absence of a test substance; (2) evaluating the degree of neurodegeneration of the neural organoid or the cells constituting the neural organoid; and (3) selecting the test substance as a candidate for a therapeutic or preventive drug for neurodegenerative disease when the progression of neurodegeneration of the neural organoid or the cells constituting the neural organoid is suppressed in the presence of the test substance compared to the absence of the test substance in step (2).
- a method for screening a therapeutic or preventive drug for neurodegenerative disease comprising the steps of: [14-1] A method for treating a disease or injury due to a disorder of the telencephalon, diencephalon, midbrain, hindbrain, or spinal cord in a mammal, comprising administering to the mammal an effective amount of the neural organoid described in [11] or an agent described in any one of [12-1] to [12-3].
- the neural organoid described in [11] for use in treating a disease or injury based on a disorder of the telencephalon, diencephalon, midbrain, hindbrain, or spinal cord.
- the present invention provides a method for producing neural organoids using a culture vessel that has multiple independent cell adhesive regions and in which cells adhering to each region are present in the same medium, allowing for the mass production of neural organoids of stable quality at low cost.
- the culture vessel used in the present invention has predetermined cell adhesive regions, which makes it possible to avoid adhesion of adjacent cells due to operations such as medium replacement, and therefore allows for the mass production of neural organoids of stable quality with a single operation.
- the quality of the neural organoids produced by the production method of the present invention is stable (homogeneous), it is expected that transplantation therapy agents containing the neural organoids and screening methods for therapeutic or preventive drugs for neurodegenerative diseases using the neural organoids will lead to the elucidation of the mechanisms of diseases (particularly neurodegenerative diseases), which have been difficult to achieve in the past, as well as the development of drug discovery research and regenerative medicine.
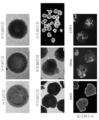
- FIG. 1 shows an overview of the culture method for human cerebral cortex organoids.
- the culture was performed using StemFit (AK02) medium, and the seeding on the TS-02 substrate at a cell density of 15,000 cells/cm 2 was counted as day 0 of culture.
- the cells after seeding reached confluence on the 7th day of culture.
- induction was performed in a medium containing conditions including KSR (knockout serum replacement), SB431542, and IWR-1-endo.
- FIG. 2 shows the induction process and marker expression of cerebral cortical organoids.
- Human cerebral cortical organoids were produced under conditions excluding the differentiation induction signal SB431542 (5 ⁇ M) and the Wnt antagonist IWR-1-endo (3 ⁇ M).
- iPS human induced pluripotent stem cells
- TS-02 spot diameter 1,500 ⁇ m
- the cells on the culture area maintained a confluent state and could be cultured for 20 days. Even after cell detachment, the cell morphology was maintained, and multiple layer structures were formed by the 35th day of culture by performing rotational culture.
- the organoids on the 35th day were evaluated by immunostaining, the expression of neural stem cells (PAX6 positive) and the cortical plate and cerebral cortex layer 5/6 (CTIP2 positive) formed so as to surround them could be confirmed.
- FIG. 3 shows the induction process and marker expression of cerebral cortical organoids.
- Human cerebral cortical organoids were produced under conditions in which the differentiation induction signal SB431542 (5 ⁇ M) and the WNT antagonist IWR-1-endo (3 ⁇ M) were added.
- human iPS cells (201B7) were cultured on TS-02 (spot diameter 1,500 ⁇ m)
- the cells on the culture area maintained a confluent state and could be cultured for 20 days. Even after cell detachment, the cell morphology was maintained, and multiple layer structures were formed by the 35th day of culture by performing rotational culture.
- the organoids on the 35th day were evaluated by immunostaining, the expression of neural stem cells (PAX6 positive) and the cortical plate and cerebral cortex layer 5/6 (CTIP2 positive) formed so as to surround them could be confirmed.
- Figure 4 shows the induction of cerebral cortical organoids using a 96-well plate.
- Human iPS cells Ff-I01s04, Ff-I14s03, and Ff-WJs524 lines
- SB431542 5 ⁇ M
- IWR-1-endo 3 ⁇ M
- FIG. 5 shows the induction of cerebral cortical organoids using TS-02 plates.
- Human iPS cells Ff-I01s04 line, Ff-I14s03 line, Ff-WJs524 line
- No cells exhibiting proliferation failure were observed on days 3, 18, and 35 of differentiation.
- evaluation was performed by immunostaining on day 35, it was confirmed that many of the cells expressed markers for neural stem cells (PAX6 positive) and the cortical plate (CTIP2 positive) that forms surrounding them.
- PAX6 positive neural stem cells
- CTIP2 positive cortical plate
- FIG. 6 shows the evaluation of cerebral cortical organoid induction depending on the difference in the micropatterning area of TS-02.
- a schematic diagram of TS-02 is shown in FIG. 15.
- Human iPS cells (201B7) were induced under the same conditions as in FIG. 3.
- Five types of micropatterning areas on TS-02 were examined, with diameters of 150 ⁇ m, 200 ⁇ m, 500 ⁇ m, 750 ⁇ m, and 1500 ⁇ m.
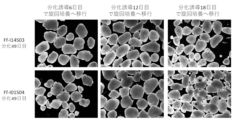
- Figure 7 shows the verification of the difference in the timing of cell detachment from TS-02.
- Human iPS cells (Ff-I01s04 strain, Ff-I14s03 strain) were induced under the same conditions as in Figure 3. Cell detachment was performed on the 6th, 12th, and 18th days after differentiation induction. When the cells after detachment were observed on the 49th day, a layer structure was confirmed in all condition groups. Furthermore, it was found that a layer structure was formed more efficiently when the cells were shifted to rotation culture on the 12th and 18th days after differentiation induction.
- Figure 8 shows how the dish (with unevenness) can homogenize the appearance of cerebral cortical organoids. Human iPS cells (201B7) were cultured for 18 days under the same conditions as in Figure 3.
- the cells were then detached, and at the time of rotational culture, they were cultured on a regular 90 mm dish and on a dish with unevenness (EzSphere). Evaluation on the 35th day of induction showed that the shape of the cerebral cortical organoids in the condition group where rotational culture was performed on EzSphere was more homogenous.
- Figure 9 shows the homogenization of the structural distribution of cerebral cortical organoids using a dish (with unevenness). Human ES cells (Kh-ES1) were cultured under the same conditions as in Figure 8 for comparison. Evaluation by immunostaining was performed on the 35th day of induction, and it was found that the cerebral cortical layer structure was more homogenously distributed throughout the inside of the group that underwent rotational culture on the EzSphere.
- Figure 10 shows the difference in marker expression between human cerebral cortex organoids.
- Human iPS cells (201B7) were cultured under the same conditions as in Figure 3. Cerebral cortical organoids on the 35th day of induction were labeled with antibodies against PAX6 and CTIP2, and the variation in marker expression of each of 18 organoids was examined. It was found that the expression of PAX6 varied by 27.0 ⁇ 12.7%, and the expression of CTIP2 varied by 37.7 ⁇ 10.9%.
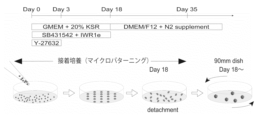
- Figure 11 shows an overview of the culture method for human cerebral cortex organoids. From day 0 to day 18 of induction, a cell suspension (1.2 x 104 cells/ cm2 ) of iPS cells (B7 strain) was seeded and cultured for adhesion.
- FIG. 12 shows the morphology of organoids during the adhesion culture process in the culture method described in Figure 11. After cell seeding, a small number of cells adhered to the adhesion surface, and the cells reached a confluent state over a period of about 7 days. By creating 21 adhesion surfaces in each well of a 24-well plate, it was possible to mass-produce cerebral cortical organoids at once.
- FIG. 13 shows the evaluation results of the structure formed through the rotation culture process in the culture method described in Figure 11. After the 18th day of adhesion culture, the cells were detached and transferred to rotation culture. A structure with a neuroepithelial structure was confirmed at the stage of the 17th day of rotation culture (total culture period 35 days). Immunostaining of these cells revealed that they were cerebral cortical organoids expressing the neural stem cell marker (PAX6) and the cerebral cortex layer 5-6 marker (CTIP2).
- Figure 14 shows the results of a comparison between the culture efficiency by the conventional method and the culture method (micropatterning method) described in Figure 11. Human cerebral cortical organoids were induced by both the conventional method and the micropatterning method. Immunostaining evaluation on the 35th day after differentiation induction showed that the structure formation between organoids was largely variable in the conventional method, whereas a more uniform structure was formed in the micropatterning method.
- FIG. 15 shows a schematic diagram of TS-02.
- present embodiment provides a detailed description of an embodiment of the present invention (hereinafter, simply referred to as the "present embodiment").
- present embodiment is an example for explaining the present invention, and is not intended to limit the present invention to the following content.
- present invention can be implemented with appropriate modifications within the scope of its intent.
- the manufacturing method of the present invention provides a method for producing neural organoids, comprising: (1) a step of adhesively culturing pluripotent stem cells in a culture vessel having a plurality of independent cell adhesive regions, with the cells adhering to each region being present in the same medium, to form a cell population containing neural stem cells; and (2) a step of detaching the cell population containing neural stem cells formed in step (1) and culturing it in suspension.
- Step (1) of the present invention typically comprises the following steps: (1-1) seeding pluripotent stem cells in a culture vessel having a plurality of independent cell adhesive regions such that cells adhering to each region are present in the same medium, and allowing the pluripotent stem cells to adhere to the cell adhesive regions;
- the method includes: (1-2) expanding the pluripotent stem cells adhered in step (1-1); and (1-3) inducing differentiation of the pluripotent stem cells expanded in step (1-2) into a cell population containing neural stem cells.
- cell adhesive region refers to a region in which the adhesiveness of pluripotent stem cells is higher than that of other regions.
- regions in the culture vessel other than the cell adhesive region may be referred to as “cell non-adhesive regions.”
- independent cell adhesive region refers to a state in which each cell adhesive region in the culture vessel is not in contact with any other cell adhesive region.
- cell adhesive region and micropatterning area are used interchangeably.
- cells adhering to each region are present in the same medium
- the paracrine effect causes the cells (or cell populations) present in the cell adhesive regions in the culture vessel to become uniform (homogeneous), resulting in stable (homogeneous) quality of the neural organoids.
- stem cells refer to undifferentiated cells that have the ability to differentiate and proliferate (particularly the ability to self-replicate).
- Stem cells include pluripotent stem cells, multipotent stem cells, unipotent stem cells, etc., depending on their differentiation ability.
- Pluripotent stem cells refer to stem cells that can be cultured in vitro and have the ability to differentiate into all cells that make up a living body (pluripotency). All cells are cells derived from the three germ layers of ectoderm, mesoderm, and endoderm.
- Multipotent stem cells refer to stem cells that have the ability to differentiate into multiple types of tissues and cells, although not all types.
- Unipotent stem cells refer to stem cells that have the ability to differentiate into specific tissues and cells.
- Pluripotent stem cells can be induced from fertilized eggs, cloned embryos, germline stem cells, tissue stem cells, somatic cells, etc.
- pluripotent stem cells include embryonic stem cells (ES cells), EG cells (embryonic germ cells), and induced pluripotent stem cells (iPS cells).
- Muse cells Multi-lineage differentiating Stress Enduring cells obtained from mesenchymal stem cells (MSCs), and GS cells produced from germ cells (e.g. testes) are also included in pluripotent stem cells.
- the pluripotent stem cells are embryonic stem cells or any cells derived from a human embryo
- the cells may be cells produced by destroying an embryo or cells produced without destroying an embryo, but from an ethical point of view, cells produced without destroying an embryo are preferred.
- human embryonic stem cells may be established from a human embryo within 14 days of fertilization.
- Embryonic stem cells were first established in 1981, and have been used to create knockout mice since 1989. Human embryonic stem cells were established in 1998, and are also being used in regenerative medicine. ES cells can be produced by culturing the inner cell population on feeder cells or in a medium containing leukemia inhibitory factor (LIF). Methods for producing ES cells are described in, for example, International Publication No. 96/22362, International Publication No. 02/101057, U.S. Patent No. 5,843,780, U.S. Patent No. 6,200,806, and U.S. Patent No. 6,280,718, etc. Embryonic stem cells are available from designated institutions, and can also be purchased commercially.
- LIF leukemia inhibitory factor
- ntES cells nuclear transfer ES cells
- EG cells can be produced by culturing primordial germ cells in a medium containing mouse stem cell factor (mSCF), LIF, and basic fibroblast growth factor (bFGF) (Cell, 70:841-847, 1992).
- mSCF mouse stem cell factor
- LIF basic fibroblast growth factor
- bFGF basic fibroblast growth factor
- “Induced pluripotent stem cells” are cells in which pluripotency has been induced by reprogramming somatic cells using known methods.
- Specific examples of induced pluripotent stem cells include cells in which pluripotency has been induced by reprogramming somatic cells differentiated into fibroblasts, peripheral blood mononuclear cells, etc., through the expression of multiple genes selected from a group of reprogramming genes including Oct3/4, Sox2, Klf4, Myc (c-Myc, N-Myc, L-Myc), Glis1, Nanog, Sall4, lin28, Esrrb, etc.
- Oct3/4, Sox2, Klf4, Myc (c-Myc, N-Myc, L-Myc), Glis1, Nanog, Sall4, lin28, Esrrb, etc. Yamanaka et al.
- artificial pluripotent stem cells were established from human fibroblasts, and like embryonic stem cells, they have pluripotency and the ability to self-replicate (Cell, 2007, 131(5) pp.861-872; Science, 2007, 318(5858) pp.1917-1920; Nat. Biotechnol., 2008, 26(1) pp.101-106).
- artificial pluripotent stem cells can also be induced from somatic cells by adding compounds (Science, 2013, 341, pp.651-654).
- Somatic cells used in producing induced pluripotent stem cells are not particularly limited, but include tissue-derived fibroblasts, blood cells (e.g., peripheral blood mononuclear cells, T cells, etc.), liver cells, pancreatic cells, intestinal epithelial cells, smooth muscle cells, etc.
- tissue-derived fibroblasts e.g., peripheral blood mononuclear cells, T cells, etc.
- blood cells e.g., peripheral blood mononuclear cells, T cells, etc.
- liver cells e.g., pancreatic cells, intestinal epithelial cells, smooth muscle cells, etc.
- the means for expressing the genes is not particularly limited.
- means for expressing genes include infection methods using viral vectors (e.g., retroviral vectors, lentiviral vectors, Sendai virus vectors, adenoviral vectors, and adeno-associated virus vectors), gene transfer methods using plasmid vectors (e.g., plasmid vectors, episomal vectors) (e.g., calcium phosphate method, lipofection method, retronectin method, and electroporation method), gene transfer methods using RNA vectors (e.g., calcium phosphate method, lipofection method, and electroporation method), and direct protein injection methods.
- viral vectors e.g., retroviral vectors, lentiviral vectors, Sendai virus vectors, adenoviral vectors, and adeno-associated virus vectors
- gene transfer methods using plasmid vectors e.g., plasmid vectors, episomal vectors
- RNA vectors e
- induced pluripotent stem cell lines for example, human induced pluripotent cell lines such as 201B7 cells, 201B7-Ff cells, 253G1 cells, 253G4 cells, 1201C1 cells, 1205D1 cells, 1210B2 cells, and 1231A3 cells established at Kyoto University are available from Kyoto University and iPS Academia Japan Inc.
- established induced pluripotent stem cell lines for example, Ff-I01 cells, Ff-I01s04 cells, Ff-I14 cells, Ff-I14s03 cells, QHJI01s04 cells, and Ff-WJs524 cells established at Kyoto University are available from Kyoto University.
- Pluripotent stem cells may be genetically modified.
- Genetically modified pluripotent stem cells can be produced, for example, by using homologous recombination techniques.
- Genes on chromosomes that can be modified include, for example, cell marker genes, histocompatibility antigen genes, and genes associated with diseases based on damage to nervous system cells (e.g., neurodegenerative diseases, etc.). Modification of target genes on chromosomes can be carried out using methods described in Manipulating the Mouse Embryo, A Laboratory Manual, Second Edition, Cold Spring Harbor Laboratory Press (1994), Gene Targeting, A Practical Approach, IRL Press at Oxford University Press (1993), Biomanual Series 8, Gene Targeting, Creation of Mutant Mice Using ES Cells, Yodosha (1995), etc.
- the genomic gene of the target gene to be modified e.g., a cell marker gene, a gene for a histocompatibility antigen, or a gene associated with a disease (e.g., neurodegenerative disease, etc.)
- a target vector is prepared using the isolated genomic gene for homologous recombination of the target gene.
- the target vector thus prepared is introduced into stem cells, and cells in which homologous recombination has occurred between the target gene and the target vector are selected, thereby producing stem cells in which genes on chromosomes have been modified.
- Methods for isolating the genomic gene of the target gene include known methods described in Molecular Cloning, A Laboratory Manual, Second Edition, Cold Spring Harbor Laboratory Press (1989) and Current Protocols in Molecular Biology, John Wiley & Sons (1987-1997).
- a genomic DNA library screening system manufactured by Genome Systems
- Universal GenomeWalker Kits manufactured by Clontech
- the preparation of a target vector for homologous recombination of a target gene and efficient selection of a homologous recombinant can be performed according to the methods described in Gene Targeting, A Practical Approach, IRL Press at Oxford University Press (1993), Biomanual Series 8, Gene Targeting, Generation of Mutant Mice Using ES Cells, Yodosha (1995), etc. Either a replacement type or an insertion type target vector can be used. Selection methods that can be used include positive selection, promoter selection, negative selection, and poly A selection. Methods for selecting the desired homologous recombinant from the selected cell lines include Southern hybridization and PCR for genomic DNA.
- Pluripotent stem cells that have undergone genome editing can also be used as pluripotent stem cells.
- Gene editing is a technology that intentionally modifies target genes or genomic regions using principles such as site-specific cleavage of genomic DNA strands using nucleases or chemical conversion of bases. Examples of site-specific nucleases include zinc finger nucleases (ZFNs), TALENs, and the CRISPR/Cas9 system. Using genome editing technology, it is possible to create knockout cell lines in which specific genes have been deleted, knock-in cell lines in which a different sequence has been artificially inserted into a specific gene locus, etc.
- Disease-specific pluripotent stem cells may be used as pluripotent stem cells.
- Disease-specific pluripotent stem cells refer to pluripotent stem cells that have a genetic mutation or genetic background involved in the onset of a disease.
- Disease-specific pluripotent stem cells can be produced by establishing induced pluripotent stem cells from patients or close relatives of the target disease using the methods described above, or by modifying the genome of already established pluripotent stem cells using genome editing techniques such as zinc finger nuclease (ZFN), TALEN, and CRISPR/CAS system. Examples of such diseases include neurodegenerative diseases.
- the pluripotent stem cells used in the present invention are, for example, pluripotent stem cells of warm-blooded animals, preferably mammals, and examples thereof include rodents, ungulates, felines, lagomorphs, and primates.
- rodents include mice, rats, hamsters, and guinea pigs.
- ungulates include pigs, cows, goats, horses, and sheep.
- felines include dogs and cats.
- lagomorphs include rabbits.
- "Primates" refers to mammals belonging to the order Primates, and examples of primates include prosimians such as lemurs, lorises, and tree shrews, and anthropoids such as monkeys, apes, and humans.
- Pluripotent stem cells are preferably from rodents (e.g., mice and rats) or primates (e.g., humans and monkeys), and more preferably from humans.
- Adhesion culture refers to culturing cells while maintaining a state in which the cells are attached to a culture vessel or the like.
- the adhesion may be due to physicochemical interactions caused by hydrophobic interactions, electrostatic interactions, van der Waals forces, etc., or may be due to adhesion via cell adhesive proteins such as extracellular matrix.
- the culture vessel used when performing adhesion culture has multiple independent cell adhesive regions, and is configured so that the cells adhering to each region are present in the same medium.
- the culture vessel is not particularly limited as long as it can have the above-mentioned configuration, and examples include flasks, flasks for tissue culture, dishes, dishes for tissue culture, multi-dishes, microplates, microwell plates, micropores, multi-plates, multi-well plates, chamber slides, petri dishes, tubes, trays, culture bags, microcarriers, beads, stack plates, spinner flasks, roller bottles, etc.
- the cell adhesive region of the culture vessel used in the adhesion culture of the present invention is not limited in area or number as long as the desired quality of neural organoids can be obtained in the suspension culture of step (2) of the present invention, but is, for example, 0.005 to 10 mm 2 , preferably 0.01 to 5.0 mm 2 , and more preferably 0.04 to 2.25 mm 2.
- the number of cell adhesive regions in the culture vessel is, for example, 1 or more, preferably 5 or more (10, 50, 100, 200, 300, 400, 500, 1000, or more).
- the shape of the region is not particularly limited, but from the viewpoint of increasing the differentiation induction efficiency, for example, a circle, an ellipse, a rectangle, a regular polygon, etc. can be mentioned.
- the diameter is preferably 150 ⁇ m to 1500 ⁇ m.
- the surface of the cell adhesive region of the culture vessel used in the present invention may be artificially treated in order to improve adhesion to cells (promote cell adhesion).
- the surface of regions other than the cell adhesive region of the culture vessel (non-cell adhesive region) may also be artificially treated in order to reduce adhesion to cells (inhibit cell adhesion).
- artificial treatments include coating treatments with extracellular matrices, polymers (e.g., hydrophilic polymers, hydrophobic polymers, temperature-responsive polymers, etc.), and surface treatments such as gas plasma treatment and positive charge treatment.
- extracellular matrices to which cells adhere include basement membrane preparations, laminin, entactin, collagen, gelatin, etc.
- polymers include polylysine, polyornithine, etc. More specifically, the following treatments may be used.
- the culture vessel used in the present invention contains a hydrophilic polymer layer on its surface
- the cell adhesive region is a region in which part of the hydrophilic polymer layer has been decomposed or modified by either plasma treatment, ultraviolet light treatment, corona discharge treatment or a combination of these
- the cell non-adhesive region has a hydrophilic polymer layer on its surface, thereby making it possible to suppress the adsorption of proteins that contribute to adhesion between the culture vessel and cells in that region.
- the thickness of the layer of the hydrophilic polymer is not particularly limited as long as it is a thickness that allows the above treatment to form cell adhesive regions and cell non-adhesive regions, but is, for example, 10 nm or more, preferably 50 nm or more, more preferably 100 nm or more, and even more preferably 500 nm or more.
- the thickness of the cell adhesive region of the culture vessel is also not particularly limited as long as it allows higher adhesion of pluripotent stem cells compared to the cell non-adhesive region, but is, for example, 1000 nm or less, preferably 500 nm or less, more preferably 100 nm or less, and even more preferably 50 nm or less.
- the method of forming the layer using the hydrophilic polymer can be at least one of a method of forming chemical bonds and a method using physical interactions.
- Methods of forming chemical bonds include methods of forming reactive functional groups such as ultraviolet irradiation, electron beam irradiation, gamma ray irradiation, plasma treatment, and corona treatment. Crosslinking reactions on the substrate surface by organic reactions using ions or radicals as reaction sources are also possible.
- Methods using physical interactions include painting, brush coating, dip coating, spin coating, bar coding, flow coating, spray coating, roll coating, air knife coating, blade coating, gravure coating, microgravure coating, and slot die coating, using a matrix that is highly compatible with the target hydrophilic polymer as a coating material.
- hydrophilic polymer is not particularly limited, but examples include those having polar groups such as hydroxyl groups, amino groups, and polyethylene glycol groups, and those having zwitterionic structures such as betaine structures and phosphorylcholine groups.
- polar groups such as hydroxyl groups, amino groups, and polyethylene glycol groups
- zwitterionic structures such as betaine structures and phosphorylcholine groups.
- a hydroxyl group, a phosphorylcholine group, or a polyethylene glycol group is preferred, a hydroxyl group or a phosphorylcholine group is more preferred, and a phosphorylcholine group is even more preferred.
- the hydrophilic polymer is preferably a random copolymer or block copolymer having both hydrophilic and hydrophobic monomer units, and more preferably a random copolymer having both hydrophilic and hydrophobic monomer units, from the viewpoint of, for example, suppressing the elution of the hydrophilic polymer from the culture vessel and suppressing the impact on quality due to the polymer being mixed into cell aggregates, etc.
- the hydrophilic monomer units are preferably 30 wt% or more, more preferably 40 wt% or more, even more preferably 50 wt% or more, and even more preferably 60 wt% or more.
- the hydrophobic monomer units are preferably 20 wt% or more, more preferably 30 wt% or more, even more preferably 40 wt% or more, and even more preferably 50 wt% or more.
- the hydrophilic monomer units are not particularly limited other than being hydrophilic, but examples thereof include those having an amino group such as 2-dimethylaminoethyl acrylate, 2-dimethylaminoethyl methacrylate, 2-diethylaminoethyl acrylate, 2-diethylaminoethyl methacrylate, and N-[3-(dimethylamino)propyl]acrylamide; those having a betaine such as N-(3-sulfopropyl)-N-methacryloyloxyethyl-N,N-dimethylammonium betaine and N-methacryloyloxyethyl-N,N-dimethylammonium- ⁇ -N-methylcarboxybetaine; hydroxyethyl acrylate, hydroxyethyl methacrylate, and N-(2-hydroxyethyl ) acrylamide, polyethylene glycol monoacrylate, polyethylene glycol monomethacrylate, polyprop
- Examples of phosphorylcholine groups include those having a phosphorylcholine group, such as 3-(meth)acryloyloxypropyl phosphorylcholine, 4-(meth)acryloyloxybutyl phosphorylcholine, 6-(meth)acryloyloxyhexyl phosphorylcholine, 10-(meth)acryloyloxydecyl phosphorylcholine, ⁇ -(meth)acryloyl(poly)oxyethylene phosphorylcholine, 2-acrylamidoethyl phosphorylcholine, 3-acrylamidopropyl phosphorylcholine, 4-acrylamidobutyl phosphorylcholine, 6-acrylamidohexyl phosphorylcholine, 10-acrylamidodecyl phosphorylcholine, and ⁇ -(meth)acrylamido(poly)oxyethylene phosphorylcholine.
- a phosphorylcholine group such as 3-(meth)acryloyloxypropyl phosphorylcholine, 4-(me
- the hydrophobic monomer unit is not particularly limited other than being hydrophobic, but examples include n-butyl acrylate, n-butyl methacrylate, isobutyl acrylate, isobutyl methacrylate, t-butyl acrylate, t-butyl methacrylate, n-hexyl acrylate, n-hexyl methacrylate, n-octyl acrylate, n-octyl methacrylate, n-decyl acrylate, n-decyl methacrylate, n-dodecyl acrylate, n-dodecyl methacrylate, n-tetradecyl acrylate, and n-tetradecyl methacrylate.
- the hydrophilic polymer also preferably contains, for example, a reactive monomer unit from the viewpoint of suppressing elution of the hydrophilic polymer.
- a reactive monomer unit a UV-reactive monomer unit is preferable because it is possible to fix the hydrophilic polymer to the culture vessel with a short treatment, and examples of the reactive monomer unit include 4-azidophenyl acrylate, 4-azidophenyl methacrylate, 2-((4-azidobenzoyl)oxy)ethyl acrylate, 2-((4-azidobenzoyl)oxy)ethyl methacrylate, etc.
- the cell adhesive region may have temperature responsiveness in order to reduce damage caused by detachment of the cultured neural organoid.
- the cells can be cultured at a temperature close to body temperature when cultured on the culture vessel, so the response temperature is preferably 50°C or less, and more preferably 35°C or less.
- the response temperature is particularly preferably 25°C or less.
- the response temperature is preferably 4°C or more, more preferably 10°C or more, and even more preferably 15°C or more.
- the temperature responsiveness may be achieved, for example, by using a temperature responsive polymer described later.
- temperature-responsive polymer refers to a polymer whose degree of hydrophilicity/hydrophobicity changes with temperature.
- a layer containing a temperature responsive polymer with a layer thickness of 1 to 100 nm may be further provided on the surface of the layer containing a hydrophilic polymer.
- a layer containing a temperature responsive polymer with a layer thickness of 1 to 100 nm it is possible to impart temperature responsiveness to the cell adhesive region without impairing the respective characteristics of the cell adhesive region and the cell non-adhesive region formed on the surface of the layer containing a hydrophilic polymer.
- the layer thickness of the layer containing the temperature responsive polymer is more preferably 3 to 50 nm, even more preferably 5 to 40 nm, and most preferably 10 to 35 nm.
- the block copolymer described in the examples below is insoluble in water because it has a water-insoluble block segment, but is a temperature-responsive polymer that has a response temperature at which the degree of hydrophilicity/hydrophobicity changes with temperature change.
- the response temperature of the block copolymer is measured by immersing a substrate coated with the block copolymer in water and measuring the contact angle of air bubbles in water, thereby measuring the degree of hydrophilicity/hydrophobicity of the block copolymer.
- the temperature of the block copolymer is changed by changing the temperature of the water, and after waiting until the temperature stabilizes, the contact angle of the air bubbles is measured again to determine the contact angle at various temperatures.
- the contact angle of the air bubbles is roughly constant at a large value (small contact angle with water), but at the response temperature, the contact angle of the air bubbles becomes small (large contact angle with water), and a curve is obtained in which the contact angle is roughly constant above the response temperature.
- the response temperature can be determined by determining the temperature at which the contact angle at temperatures below the response temperature and the contact angle at temperatures above the response temperature are the average contact angle (midpoint method).
- the temperature range for measurement includes a temperature range of 10°C or more where the contact angle is roughly constant at temperatures below the response temperature, and also includes a temperature range of 10°C or more where the contact angle is roughly constant at temperatures above the response temperature.
- the temperature-responsive polymer is preferably a block copolymer having a water-insoluble block segment and a temperature-responsive block segment.
- a block copolymer As the temperature-responsive polymer, it is possible to increase the mass productivity of the culture vessel and to suppress the contamination of the produced neural organoids with the temperature-responsive polymer.
- the ratio of the constituent units of the temperature-responsive block segment contained in the temperature-responsive polymer is preferably 70 wt% or more, more preferably 80 wt% or more, particularly preferably 90 wt% or more, and most preferably 92 wt% or more, since this is suitable for quickly peeling off the neural organoids from the culture vessel.
- Examples of monomer units constituting the temperature-responsive block segment include (meth)acrylamide compounds such as acrylamide and methacrylamide; N-alkyl-substituted (meth)acrylamide derivatives such as N,N-diethylacrylamide, N-ethylacrylamide, N-n-propylacrylamide, N-n-propylmethacrylamide, N-isopropylacrylamide, N-isopropylmethacrylamide, N-cyclopropylacrylamide, N-cyclopropylmethacrylamide, N-t-butylacrylamide, N-ethoxyethylacrylamide, N-ethoxyethylmethacrylamide, N-tetrahydrofurfurylacrylamide, and N-tetrahydrofurfurylmethacrylamide; N,N-dialkyl-substituted (meth)acrylamide derivatives such as N,N-dimethyl(meth)acrylamide, N,N-ethylmethylacrylamide
- N-n-propylacrylamide and N-proline methyl ester acrylamide are preferred because they are suitable for setting the response temperature of the block copolymer to a temperature lower than room temperature.
- Examples of monomer units constituting the water-insoluble block segment include n-butyl acrylate, n-butyl methacrylate, isobutyl acrylate, isobutyl methacrylate, t-butyl acrylate, t-butyl methacrylate, n-hexyl acrylate, n-hexyl methacrylate, n-octyl acrylate, n-octyl methacrylate, n-decyl acrylate, n-decyl methacrylate, n-dodecyl acrylate, n-dodecyl methacrylate, n-tetradecyl acrylate, and n-tetradecyl methacrylate.
- those having reactive groups are preferred because they are suitable for firmly fixing the block copolymer to the substrate, and examples of such monomer units include 4-azidophenyl acrylate, 4-azidophenyl methacrylate, 2-((4-azidobenzoyl)oxy)ethyl acrylate, and 2-((4-azidobenzoyl)oxy)ethyl methacrylate.
- structures having aromatic rings are preferred because they are suitable for enhancing cell proliferation, and examples include 2-hydroxyphenyl acrylate, 2-hydroxyphenyl methacrylate, 3-hydroxyphenyl acrylate, 3-hydroxyphenyl methacrylate, 4-hydroxyphenyl acrylate, 4-hydroxyphenyl methacrylate, N-(2-hydroxyphenyl)acrylamide, N-(2-hydroxyphenyl)methacrylamide, N-(3-hydroxyphenyl)acrylamide, N-(3-hydroxyphenyl)methacrylamide, N-(4-hydroxyphenyl)acrylamide, N-(4-hydroxyphenyl)methacrylamide, and styrene.
- the water-insoluble block segment may also contain a repeating unit that controls the response temperature of the block copolymer.
- repeating units that control the response temperature of the block copolymer include hydrophilic or hydrophobic components, and are not particularly limited.
- those having an amino group such as 2-dimethylaminoethyl acrylate, 2-dimethylaminoethyl methacrylate, 2-diethylaminoethyl acrylate, 2-diethylaminoethyl methacrylate, and N-[3-(dimethylamino)propyl]acrylamide; those having a betaine such as N-(3-sulfopropyl)-N-methacryloyloxyethyl-N,N-dimethylammonium betaine and N-methacryloyloxyethyl-N,N-dimethylammonium- ⁇ -N-methylcarboxybetaine; hydroxyethyl acrylate, hydroxy
- acrylate, N-(2-hydroxyethyl)acrylamide polyethylene glycol monoacrylate, polyethylene glycol monomethacrylate, polypropylene glycol monoacrylate, polypropylene glycol monomethacrylate, methoxypolyethylene glycol monoacrylate, methoxypolyethylene glycol monomethacrylate, diethylene glycol monomethyl ether acrylate, diethylene glycol monomethyl ether methacrylate, diethylene glycol monoethyl ether acrylate, diethylene glycol monoethyl ether methacrylate, 2-methoxyethyl acrylate, 2-methoxyethyl methacrylate, 2-ethoxyethyl acrylate, 2-ethyl those having a polyethylene glycol group or a methoxyethyl group, such as 2-ethoxyethyl acrylate, 3-butoxyethyl methacrylate, 3-butoxyethyl methacrylate, 3-butoxyethyl methacrylamide, furfury
- Other methods for producing the culture vessel used in the adhesion culture of the present invention include a method in which a substance that promotes or inhibits cell adhesion is coated on a portion of the culture vessel by photolithography or inkjet printing, and a method in which the surface of the culture vessel is treated with plasma, ultraviolet light, or corona discharge, or a combination of these, and then coated with a temperature-responsive polymer.
- the culture vessel used in the adhesion culture of the present invention may be sterilized.
- the sterilization method is not particularly limited, but may be, for example, high-pressure steam sterilization, UV sterilization, gamma ray sterilization, ethylene oxide gas sterilization, etc. From the viewpoint of suppressing denaturation of the block copolymer, for example, high-pressure steam sterilization, UV sterilization, and ethylene oxide gas sterilization are preferred, from the viewpoint of suppressing deformation of the base material, UV sterilization or ethylene oxide gas sterilization is more preferred, and ethylene oxide gas sterilization is even more preferred because of its superior mass productivity of the culture vessel.
- the material used to prepare the culture vessel is not particularly limited, but for example, commonly used glass, polystyrene, polycarbonate, polyethylene terephthalate, polyvinylidene fluoride, polyethylene, polypropylene, polyethylene methacrylate and other polymeric compounds, ceramics, and metals can be used.
- Polystyrene is preferred because of its excellent transparency and ease of molding and surface modification.
- the cell adhesive region of the culture vessel used in the contact culture of the present invention may be coated with an extracellular matrix, for example, a basement membrane preparation, fibronectin, laminin or a fragment thereof, entactin, collagen, gelatin, synthemax, vitronectin, etc.
- the exemplified extracellular matrices may be natural products, may be artificially synthesized by recombinant gene technology, etc., may be fragments cut with restriction enzymes, etc., or may be synthetic proteins or synthetic peptides based on these biological substances.
- the extracellular matrix may be coated in advance on the culture vessel used in step (1), or may be coated by adding it to the medium used in step (1).
- Basis membrane structure refers to a thin membrane-like structure composed of extracellular matrix.
- basement membranes are formed on the basal side of epithelial cells.
- Components of basement membranes include type IV collagen, laminin, heparan sulfate proteoglycan (perlecan), entactin/nidogen, cytokines, growth factors, etc.
- tissue staining such as PAM staining
- immunohistochemistry using antibodies against components of basement membranes (anti-laminin antibodies, anti-type IV collagen antibodies, etc.).
- basement membrane preparation refers to a preparation containing basement membrane components that have the function of controlling epithelial cell-like cell morphology, differentiation, proliferation, movement, functional expression, etc., when desired cells having basement membrane formation ability are seeded and cultured thereon.
- basement membrane components refers to thin membrane-like extracellular matrix molecules that exist between epithelial cell layers and interstitial cell layers, etc., in animal tissues.
- a basement membrane preparation can be prepared, for example, by removing cells having basement membrane formation ability that are adhered to a support via a basement membrane from the support using a solution capable of dissolving the lipids of the cells, an alkaline solution, or the like.
- basement membrane preparations include products commercially available as basement membrane preparations, such as Matrigel (manufactured by Corning) and Geltrex (manufactured by Thermo Fisher Scientific), and those that contain extracellular matrix molecules known as basement membrane components (e.g., laminin, type IV collagen, heparan sulfate proteoglycan, entactin, etc.).
- Laminin is a heterotrimeric molecule consisting of ⁇ , ⁇ , and ⁇ chains, and is an extracellular matrix protein with isoforms that differ in the composition of the subunit chains. Specifically, laminin has about 15 isoforms, which are heterotrimeric combinations of five types of ⁇ chains, four types of ⁇ chains, and three types of ⁇ chains. The names of laminins are determined by combining the numbers of the ⁇ chains ( ⁇ 1- ⁇ 5), ⁇ chains ( ⁇ 1- ⁇ 4), and ⁇ chains ( ⁇ 1- ⁇ 3). For example, laminin composed of a combination of ⁇ 5 chains, ⁇ 1 chains, and ⁇ 1 chains is called laminin 511. Laminins may be natural products, or may be artificially synthesized using recombinant gene technology, or may be synthetic proteins or peptides based on the laminins.
- the laminins or fragments thereof used in the present invention include laminin-111 and fragments thereof containing the E8 region, laminin-211 or fragments thereof containing the E8 region (e.g., iMatrix-211), laminin-121 or fragments thereof containing the E8 region, laminin-221 or fragments thereof containing the E8 region, laminin-332 or fragments thereof containing the E8 region, laminin-3A11 or fragments thereof containing the E8 region, laminin-411 or fragments thereof containing the E8 region (e.g., iMatrix-411), laminin-4 21 or a fragment containing its E8 region, laminin-511 or a fragment containing its E8 region (e.g., iMatrix-511, iMatrix-511 silk), laminin-521 or a fragment containing its E8 region, laminin-213 or a fragment containing its E8 region, laminin-423 or a fragment
- Vitronectin may be a natural product, may be artificially synthesized using recombinant gene technology, or may be a synthetic protein or peptide based on vitronectin.
- Commercially available products that are easy to obtain include, for example, vitronectin, human plasma-derived (manufactured by Wako Pure Chemical Industries, Ltd.), synthemax (manufactured by Corning Incorporated), and Vitronectin (VTN-N) (manufactured by Gibco).
- Fibronectin may be a natural product, may be artificially synthesized using recombinant gene technology, or may be a synthetic protein or peptide based on the fibronectin.
- Commercially available products that are easy to obtain include, for example, fibronectin solution, derived from human plasma (manufactured by Wako Pure Chemical Industries, Ltd.) and Retronectin (manufactured by Takara Bio Inc.).
- the type of collagen is not particularly limited, but for example, type I collagen or type IV collagen can be used.
- the collagen may be a natural product, may be artificially synthesized using recombinant gene technology, or may be a synthetic peptide based on the collagen.
- Commercially available products that are easy to obtain include, for example, collagen I, human (manufactured by Corning Incorporated) and collagen IV, human (manufactured by Corning Incorporated).
- step (1) of the present invention it is preferable to culture cells under feeder-free and xeno-free conditions. It is also preferable to carry out all steps of the production method of the present invention (i.e., steps (1) and (2)) under feeder-free and xeno-free conditions.
- feeder-free means a medium or culture condition that does not contain other cell types (i.e., feeder cells) that play a supporting role and are used to prepare the culture conditions for the cells to be cultured.
- xeno-free means a medium or culture condition that does not contain components derived from organisms different from the organism species of the cells to be cultured.
- the medium used in step (1) is not particularly limited as long as it is a medium (feeder-free medium) that allows the culture of pluripotent stem cells to maintain their undifferentiated state under feeder-free conditions, but preferably contains factors for maintaining their undifferentiated state in order to enable the culture to maintain their undifferentiated state.
- the undifferentiated state maintenance factor is not particularly limited as long as it has the effect of suppressing the differentiation of pluripotent stem cells.
- undifferentiated state maintenance factors commonly used by those skilled in the art include substances acting on the FGF signaling pathway, substances acting on the TGF ⁇ family signaling pathway, insulin, etc.
- substances acting on the FGF signaling pathway include fibroblast growth factors (e.g., bFGF, FGF4, and FGF8).
- substances acting on the TGF ⁇ family signaling pathway include substances acting on the TGF ⁇ signaling pathway and substances acting on the Nodal/Activin signaling pathway.
- substances acting on the TGF ⁇ signaling pathway include TGF ⁇ 1 and TGF ⁇ 2.
- step (1) preferably contains bFGF as an undifferentiated state maintenance factor.
- the undifferentiation maintenance factor used in the present invention is usually a mammalian undifferentiation maintenance factor.
- mammals include those mentioned above. Since the undifferentiation maintenance factor may have cross-reactivity between mammalian species, any mammalian undifferentiation maintenance factor may be used as long as it can maintain the undifferentiated state of the pluripotent stem cells to be cultured, but it is preferable to use an undifferentiation maintenance factor of the same mammalian species as the cells to be cultured.
- human undifferentiation maintenance factors e.g., bFGF, FGF4, FGF8, EGF, Nodal, Activin A, Activin B, TGF ⁇ 1, TGF ⁇ 2, etc.
- bFGF bFGF, FGF4, FGF8, EGF, Nodal, Activin A, Activin B, TGF ⁇ 1, TGF ⁇ 2, etc.
- the undifferentiation maintenance factor used in the present invention is preferably isolated. "Isolated” means that an operation to remove factors other than the target components and cells has been performed, and the undifferentiation maintenance factor has escaped from the naturally occurring state.
- the undifferentiation maintenance factor may be added to the culture medium in advance.
- the concentration of the undifferentiation maintenance factor in the medium used in step (1) of the present invention is a concentration that can maintain the undifferentiated state of the pluripotent stem cells being cultured, and can be appropriately set by a person skilled in the art.
- the concentration is usually 4 ng to 500 ng/ml, preferably 10 ng to 200 ng/ml, and more preferably 30 ng to 150 ng/ml.
- the medium used in step (1) may be, for example, a culture medium obtained by adding culture supplements such as the above-mentioned undifferentiated maintenance factors and non-essential amino acids to a basal medium such as DMEM, Ham's F12, or D-MEM/Ham's F12, Primate ES Cell Medium (manufactured by REPROCELL Co., Ltd.), StemFit AK02N (manufactured by Ajinomoto Co., Inc.), StemFit AK03 (manufactured by Ajinomoto Co., Inc.), mTeSR1 (manufactured by STEMCELL TECHNOLOGIES), TeSR-E8 ( Examples of commercially available undifferentiated state maintenance media include ReproNaive (manufactured by REPROCELL TECHNOLOGIES), ReproXF (manufactured by REPROCELL), ReproFF (manufactured by REPROCELL), ReproFF2 (manufactured by RE
- the seeding of pluripotent stem cells into the culture vessel is not particularly limited as long as the pluripotent stem cells adhere to the cell adhesion region of the culture vessel, but for example, the cells may be seeded in a monodispersed state or cells that have been dispersed to a state close to this.
- Methods for dispersing pluripotent stem cells include, for example, mechanical dispersion treatment, cell dispersion liquid treatment, and cell protective agent addition treatment. These treatments may also be performed in combination.
- Mechanical dispersion methods include pipetting or scraping with a scraper.
- the cell dispersion liquid used in the cell dispersion treatment can be, for example, a solution containing enzymes such as trypsin, collagenase, hyaluronidase, elastase, pronase, DNase, or papain, or a chelating agent such as ethylenediaminetetraacetic acid (EDTA).
- enzymes such as trypsin, collagenase, hyaluronidase, elastase, pronase, DNase, or papain
- EDTA ethylenediaminetetraacetic acid
- Commercially available cell dispersion liquids such as TrypLE Select (Life Technologies) or TrypLE Express (Life Technologies), can also be used.
- cytoprotective agent used in the cytoprotective agent treatment include substances acting on the FGF signaling pathway, heparin, substances acting on the IGF signaling pathway, serum, and serum substitutes.
- a Rho-associated coiled-coil kinase (ROCK) inhibitor also referred to as "ROCK inhibitor” in this specification
- a myosin inhibitor may be added during dispersion.
- ROCK inhibitors examples include (R)-(+)-trans-N-(4-pyridyl)-4-(1-aminoethyl)-cyclohexanecarboxamide.2HCl.H 2 O (Y-27632), Fasudil (HA1077), and H-1152.
- myosin inhibitors include blebbistatin.
- Preferred cell protective agents include ROCK inhibitors from the viewpoint of maintaining undifferentiation.
- the final concentration in the medium as described below is 1 ⁇ M to 50 ⁇ M, preferably 3 ⁇ M to 20 ⁇ M, more preferably 5 ⁇ M to 15 ⁇ M, and even more preferably 8 ⁇ M to 12 ⁇ M.
- the ROCK inhibitor is typically present in the medium during step (1-1), and may be removed, for example, by replacing the medium with one not containing the ROCK inhibitor 24 to 48 hours after the cells are seeded in a culture vessel.
- step (1-1) the extracellular matrix as described above may be added to the medium as described below.
- laminin of the same mammalian species as the cells to be cultured is preferably used.
- human laminin preferably human laminin 511 is used for culturing human pluripotent stem cells.
- the amount of the extracellular matrix to be added to the medium is not particularly limited as long as it contributes to maintaining the undifferentiated state of the pluripotent stem cells, but for example, laminin 511 may be added so that the amount is 0.5 to 2.0 ⁇ g/cm 2 per cell adhesive region of the culture vessel.
- the timing of addition of the extracellular matrix may be prior addition (precoating) to the substrate, or may be simultaneous with the cell seeding.
- the cell seeding density in the culture vessel is not particularly limited as long as the cells can grow, and is typically 1.0 ⁇ 10 1 to 1.0 ⁇ 10 6 cells/cm 2 , preferably 1.0 ⁇ 10 2 cells/cm 2 to 1.0 ⁇ 10 5 cells/cm 2 , more preferably 1.2 ⁇ 10 3 cells/cm 2 to 5.0 ⁇ 10 4 cells/cm 2 , 2.0 ⁇ 10 3 cells/cm 2 to 2.5 ⁇ 10 4 cells/cm 2 , and 3.0 ⁇ 10 3 cells/cm 2 to 5.0 ⁇ 10 3 cells/cm 2 .
- the culture period in step (1-1) is not particularly limited as long as the pluripotent stem cells adhere to the adhesive region of the culture vessel, but is typically 1 to 7 days, preferably 1 to 3 days, and more preferably 1 to 2 days.
- the culture period in step (1-2) is not particularly limited as long as the pluripotent stem cells are expanded in the adhesive region of the culture vessel, but typically, the cells should cover about 50% to 100% of the cell adhesive region of the culture vessel (about 50% confluent to confluent).
- the culture period is 3 to 14 days, preferably 4 to 10 days, and more preferably 5 to 8 days.
- step (1-3) of the present invention is typically performed after the pluripotent stem cells cover about 50% to 100% of the cell adhesive area of the culture vessel (about 50% confluent to confluent).
- Step (1-3) of the present invention is a step of inducing differentiation of the pluripotent stem cells expanded in step (1-2) into a cell population containing neural stem cells (PAX6 positive).
- adhesion culture is typically performed in a medium that does not contain a factor for maintaining undifferentiated state as described above, so that the cells can be spontaneously induced to differentiate into a cell population containing neural stem cells.
- the cell population containing neural stem cells of the present invention may also contain neural progenitor cells.
- step (1-3) of the present invention may be performed in a medium to which a Wnt signal inhibitor and a TGF ⁇ signal inhibitor are not added (medium not containing a Wnt signal inhibitor and a TGF ⁇ signal inhibitor), and in another embodiment, it may be performed in a medium to which a Wnt signal inhibitor and a TGF ⁇ signal inhibitor are added (medium containing a Wnt signal inhibitor and a TGF ⁇ signal inhibitor).
- cells includes “cell populations.”
- a cell population may be composed of one type of cell, or may be composed of two or more types of cells.
- a cell population may be in the form of a cell aggregate such as a spheroid or organoid, as described below.
- the term "cell population” refers to a cell group consisting of two or more cells.
- the cell population may be composed of one type of cell or multiple types of cells.
- the cells constituting the cell population may be suspended in a medium or may be attached to a culture vessel or the like.
- the cells constituting the cell population are attached to a culture vessel or the like.
- the cells constituting the cell population may be single cells, or at least a part of the cell population may be formed by cell adhesion to each other to form a cell population.
- the term "cell population” includes a cell aggregate, which is a mass formed by the gathering of cells and is a mass in which the cells are adhered to each other, and the cell aggregate also includes an embryoid body, a sphere, a spheroid, and an organoid.
- organoid refers to a structure formed by the accumulation of cells, and typically has a structure and function similar to that of an organ in a living body. Whether or not a structure is an organoid can be confirmed, for example, by observing it under a microscope to see if a layer structure is formed or by examining the expression of marker proteins.
- Wnt signal inhibitor there is no particular limitation on the Wnt signal inhibitor, so long as it can suppress signal transduction mediated by Wnt.
- Wnt signal inhibitors include, but are not limited to, IWR-1-endo (4-[(3aR,4S,7R,7aS)-1,3,3a,4,7,7a-hexahydro-1,3-dioxo-4,7-methano-2H-isoindol-2-yl]-N-8-quinolinyl-benzamide), IWP-2, XAV939, Dkk1, Cerberus protein, Wnt receptor inhibitor, soluble Wnt receptor, Wnt antibody, casein kinase inhibitor, and dominant negative Wnt protein.
- TGF ⁇ signal inhibitor is not particularly limited as long as it can suppress signal transduction mediated by TGF ⁇ .
- TGF ⁇ signal inhibitors include, but are not limited to, SB431542 (4-(5-benzol[1,3]dioxol-5-yl-4-pyridin-2-yl-1H-imidazol-2-yl)-benzamide), LY-364947, SB-505, A-83-01, etc.
- the concentrations of the Wnt signal inhibitor and the TGF ⁇ signal inhibitor in the medium are not particularly limited and can be set appropriately as long as they induce differentiation of pluripotent stem cells into a cell population containing neural stem cells.
- the concentration is usually 0.1 to 50 ⁇ M, preferably 0.3 to 5 ⁇ M.
- SB431542 is used as the TGF ⁇ signal inhibitor, the concentration is usually 0.1 to 100 ⁇ M, preferably 1 to 10 ⁇ M.
- An example of a combination of a Wnt signal inhibitor and a TGF ⁇ signal inhibitor is IWR-1-endo and SB431542.
- the medium used in step (1-3) is not particularly limited as long as it does not contain factors for maintaining undifferentiation such as bFGF, and for example, a medium used for culturing animal cells can be prepared as the basal medium.
- the basal medium is not particularly limited as long as it can be used for culturing animal cells, such as BME medium, BGJb medium, CMRL 1066 medium, Glasgow MEM medium, Improved MEM Zinc Option medium, IMDM medium, Medium 199 medium, Eagle MEM medium, ⁇ MEM medium, DMEM medium, Ham's medium, Ham's F-12 medium, RPMI 1640 medium, Fischer's medium, Neurobasal medium, and mixed media thereof.
- Glasgow MEM medium is used.
- the above-mentioned medium can also be used in step (2) of the present invention.
- the medium is preferably a serum-free medium so as not to affect the induction of differentiation from pluripotent stem cells into a cell population containing neural stem cells.
- the medium used in step (1-3) of the present invention may contain a serum substitute.
- the serum substitute may contain, for example, albumin, transferrin, fatty acid, collagen precursor, trace elements, 2-mercaptoethanol or 3'thiolglycerol, or equivalents thereof, as appropriate.
- a serum substitute may be prepared, for example, by the method described in WO98/30679.
- a commercially available serum substitute may be used. Examples of such commercially available serum substitutes include KSR (knockout serum replacement) (manufactured by Invitrogen), Chemically-defined Lipid Concentrated (manufactured by Gibco), and Glutamax (manufactured by Gibco).
- KSR knockout serum replacement
- Cas chemically-defined Lipid Concentrated
- Glutamax manufactured by Gibco.
- the above-mentioned serum substitutes may also be used in step (2) of the present invention.
- the medium may contain other additives to the extent that they do not adversely affect the differentiation induction from pluripotent stem cells to a cell population containing neural stem cells.
- additives include, but are not limited to, insulin, iron sources (e.g., transferrin, etc.), minerals (e.g., sodium selenate, etc.), sugars (e.g., glucose, etc.), organic acids (e.g., pyruvic acid, lactic acid, etc.), serum proteins (e.g., albumin, etc.), amino acids (e.g., L-glutamine, non-essential amino acids, etc.), reducing agents (e.g., 2-mercaptoethanol, etc.), vitamins (e.g., ascorbic acid, d-biotin, etc.), antibiotics (e.g., streptomycin, penicillin, gentamicin, etc.), buffers (e.g., HEPES, etc.), etc.
- the additives described above may also be used in step (2) of the
- the culture period in step (1-3) of the present invention is not particularly limited as long as the pluripotent stem cells are induced to differentiate into a cell population containing neural stem cells (PAX3 positive), but is typically 2 to 30 days, preferably 7 to 25 days, and more preferably 12 to 18 days.
- the cell population containing neural stem cells induced to differentiate is typically detached from the cell adhesive region by a method described below, and is subjected to step (2) of the present invention.
- the culture period in step (1) is typically at least 6 days.
- culture temperature is, for example, about 30 to 40° C., preferably 37° C.
- the CO2 concentration is, for example, 1 to 10%, preferably about 5%.
- the O2 concentration is, for example, about 20%.
- step (2) of the present invention the method for detaching the cell population containing neural stem cells formed in step (1) is not particularly limited as long as neural organoids of the desired quality can be obtained.
- natural detachment by cooling e.g., 4°C
- detachment by treatment with a buffer solution e.g., PBS, etc.
- medium cooled to 4°C or enzymes such as trypsin, collagenase, hyaluronidase, elastase, pronase, DNase, or papain
- a chelating agent such as ethylenediaminetetraacetic acid (EDTA), or detachment by physical treatment with a scraper, etc.
- EDTA ethylenediaminetetraacetic acid
- TrypLE Select manufactured by Life Technologies
- TrypLE Express manufactured by Life Technologies
- Accutase manufactured by Nacalai Tesque
- Accumax manufactured by Nacalai Tesque
- the suspension culture in step (2) of the present invention is a process for inducing differentiation of a cell population containing detached neural stem cells as described above into desired neural organoids (e.g., brain organoids, cerebral organoids, cerebellar organoids, spinal cord organoids, midbrain organoids, choroid plexus organoids, hippocampal organoids, hypothalamic organoids, anterior pituitary organoids, and basal ganglia organoids, etc.).
- desired neural organoids e.g., brain organoids, cerebral organoids, cerebellar organoids, spinal cord organoids, midbrain organoids, choroid plexus organoids, hippocampal organoids, hypothalamic organoids, anterior pituitary organoids, and basal ganglia organoids, etc.
- desired neural organoids e.g., brain organoids, cerebral organoids, cerebellar organoids, spinal cord organoids, midbrain organoids, choroid plexus organoids, hippocampal organoids, hypothalamic organoids, anterior
- tissue culture refers to culture performed under conditions that maintain a state in which cells or cell aggregates are suspended in the culture solution, i.e., culture under conditions that do not allow the formation of strong cell-substratum junctions between the cells or cell aggregates and the culture vessel.
- the differentiation induction into neural organoids as described above can be carried out by a method known per se, by appropriately setting the conditions such as differentiation inducer, culture medium, and culture period.
- the suspension culture in step (2) of the present invention can be appropriately performed using a method known per se, such as static culture, rotation culture, culture under turbulent conditions (environment), culture in a microgravity (including zero gravity) environment, or culture by the hanging drop method, or a combination of these.
- the culture in step (2) of the present invention is a combination of static culture and rotation culture.
- the period of static culture is, for example, 0 to 10 days, preferably 0 to 4 days, after detachment of the cell population containing neural stem cells obtained in step (1).
- the culture in step (2) of the present invention is rotation culture.
- the culture vessel used in the suspension culture of step (2) of the present invention is not particularly limited as long as it is capable of "suspension culture", and a person skilled in the art can appropriately determine the type.
- Examples of such culture vessels include flasks, tissue culture flasks, dishes, Petri dishes, tissue culture dishes, multi-dishes, microplates, microwell plates, micropores, multi-plates, multi-well plates, chamber slides, petri dishes, tubes, trays, culture bags, and roller bottles.
- a bioreactor is exemplified as a container for suspension culture.
- Examples of the shape of the bottom surface of the culture vessel include a U-bottom, a V-bottom, and a ⁇ -bottom.
- the bottom surface of the culture vessel may have, for example, unevenness, depressions, etc. (for example, EZSPHERE (AGC Technoglass Co., Ltd.)). It is preferable that these culture vessels are non-cell-adhesive in order to enable suspension culture.
- a non-cell-adhesive culture vessel one whose surface has not been artificially treated (e.g., coated with an extracellular matrix, etc.) for the purpose of improving adhesion to cells can be used.
- the conditions of the swirling culture are not particularly limited as long as the swirling culture is carried out by a method known per se (e.g., ML Wegscheid. et al., Cell Rep. 2021 Jul 6; 36(1): 109315., etc.) so as to generate a swirling flow.
- the swirling speed is typically 1 rpm to 200 rpm, preferably 50 rpm to 100 rpm.
- the culture under turbulent conditions is not particularly limited as long as it is carried out in an environment that generates turbulence using a method known per se (e.g., ITO Y. et al., Cell 174: 636-648 e18, 2018, etc.).
- the conditions for culturing in a microgravity (including zero gravity) environment are not particularly limited as long as the culture is performed in an environment with lower gravity than that on the ground by a method known per se (e.g., International Publication No. 2019/107546, etc.), and can be performed, for example, using a microgravity environment cell culture device (clinostat) (Yamato Scientific), etc.
- the culture by the hanging drop method is not particularly limited as long as it is performed by a method known per se (e.g., Z. Gania. Et al., Future Pharmacol. 2022, 2(3), 360-376, etc.), in which a medium containing a cell population including neural stem cells is attached to the bottom surface of a plate so that the bottom surface of the attached medium (droplet) becomes hemispherical due to the action of gravity.
- a method known per se e.g., Z. Gania. Et al., Future Pharmacol. 2022, 2(3), 360-376, etc.
- Neural organoids can be produced by culturing the cell population containing neural stem cells obtained in step (1) of the present invention in a differentiation-inducing medium for the desired neural organoids, based on the methods described, for example, in Sasai Y. et al., Cell Stem Cell. 12:520-530 (2013), Sakaguchi H. et al., Nat Commun. 6:8896 (2015), Kadoshima T. et al., Proc Natl Acad Sci U S A. 2014 May 20;111(20):7498, Sakaguchi H. et a., Stem Cell Reports 13 (3), 458-473, 2019, JP 2021-72818, and JP 2020-195349.
- neural organoids can be induced in a self-organizing manner.
- Examples of media for inducing differentiation into neural organoids include, but are not limited to, a medium containing SB431542 and WR-1-endo for inducing brain organoids, N2 medium (prepared by adding N2 supplement to DMEM/F12 medium) for inducing cerebral (cortical) organoids, a medium containing FGF2 and insulin for inducing cerebellar organoids, a medium containing CHIR99021, bFGF, SB431542, and retinoic acid for inducing spinal cord organoids, a medium containing insulin and bFGF for inducing midbrain organoids, a medium containing CHIR and BMP4 for inducing choroid plexus organoids and hippocampal organoids, a medium containing SAG, BMP4, and KSR (and FGF2) for inducing hypothalamic organoids and pituitary (anterior pituitary) organoids, and a medium containing SHH for inducing basal ganglia organoids.
- N2 medium
- induced neural organoid is the desired neural organoid
- the induced neural organoid can be confirmed, for example, by observing the presence or absence of layer structure formation by microscopy or by examining the expression of marker proteins.
- marker proteins for example, in the case of cerebral (cortical) organoids, those that have at least one combination of neural stem cells (PAX6 positive) and the cortical plate (CTIP2 positive) and cortical layer (CTIP2 positive (layer 5/6), SATB2 positive, CUX1 positive, etc.) that are formed surrounding the neural stem cells can be considered to be cerebral cortical organoids.
- the step (2) of the present invention may involve culturing under feeder-free conditions and/or xeno-free conditions for all or part of the period. From the viewpoint of clinical use, it is preferable that the step is carried out under feeder-free and xeno-free conditions for the entire period.
- a neural organoid obtained by the manufacturing method of the present invention is provided.
- the manufacturing method of the present invention may include a step of recovering the obtained target organoid.
- the recovered organoid may be cryopreserved using a cell cryopreservation solution.
- step (2) of the production method of the present invention the culture method, culture conditions, definitions, etc. other than those described above are all incorporated by reference in the contents described above in "(1) Step (1)", "(2) Culture vessel and cells” and "(3) Adherent culture in step (1)".
- the present invention also provides a transplantation therapeutic agent containing a neural organoid produced by "1.
- the production method of the present invention or a cell constituting the neural organoid.
- the transplantation therapeutic agent of the present invention may be administered in a therapeutically effective amount to a subject (e.g., a mammal (e.g., human, mouse, rat, monkey, cow, horse, pig, dog, etc.)) having a disease or injury due to a disorder of the telencephalon, diencephalon, midbrain, hindbrain, spinal cord, etc., for which cell transplantation therapy is desired, and may be appropriately changed depending on factors such as the age, weight, and severity of the disease of the transplantation subject.
- a subject e.g., a mammal (e.g., human, mouse, rat, monkey, cow, horse, pig, dog, etc.)
- a disease or injury due to a disorder of the telencephalon, diencephalon, midbra
- Examples of the disease include Parkinson's disease, Huntington's chorea, Alzheimer's disease, cerebrovascular disorder (symptoms include stroke, more specifically ischemic cerebral disease (cerebral infarction, transient ischemic attack) and hemorrhagic cerebral disease (cerebral hemorrhage, subarachnoid hemorrhage)), epilepsy, brain trauma, motor neuron disease, neurodegenerative disease, etc.
- Examples of damage include damage after brain surgery (e.g., after brain tumor removal).
- HLA genotype matches the transplanted cells to an extent that immune reactions can be suppressed with immunosuppressants, for example, somatic cells with HLA types that match the three loci of HLA-A, HLA-B, and HLA-DR, or four loci including HLA-C, or six loci including HLA-DP and HLA-DQ. If sufficient cells cannot be obtained due to age, constitution, or other reasons, they can be transplanted in a state where they are embedded in a capsule made of polyethylene glycol or silicone, a porous container, etc., to avoid rejection reactions.
- the transplantation therapy of the present invention may contain cells that constitute neural organoids (e.g., brain organoids, cerebral organoids, cerebellar organoids, spinal organoids, midbrain organoids, choroid plexus organoids, hippocampal organoids, hypothalamic organoids, anterior pituitary organoids, and basal ganglia organoids, etc.) obtained by the manufacturing method of the present invention.
- neural organoids e.g., brain organoids, cerebral organoids, cerebellar organoids, spinal organoids, midbrain organoids, choroid plexus organoids, hippocampal organoids, hypothalamic organoids, anterior pituitary organoids, and basal ganglia organoids, etc.
- the "cells that constitute neural organoids” are not particularly limited as long as they are cells that are contained in neural organoids obtained by the manufacturing method of the present invention, and examples of such cells include neural stem cells, neural progenitor cells, nerve cells, glial progenitor cells, and glial cells (e.g., astrocytes, microglia, oligodendrocytes, ependymal cells, Schwann cells, etc.).
- the "cells that constitute neural organoids” may be one cell, a single cell, or a cell population.
- the cells constituting the neural organoids that can be included in the transplantation therapy of the present invention can be obtained as individual cells, single cells, or cell populations by treating the neural organoids obtained by the manufacturing method of the present invention with a cell dispersion solution containing enzymes or the like as described above, by shredding them by physical incision or the like (e.g., using a scalpel, scissors, or a 27-gauge (G) to 21-G injection needle, or the like), or by aspirating them using a pipette or the like.
- a cell dispersion solution containing enzymes or the like as described above
- the cells constituting the neural organoids are similarly stable (homogeneous), and these cells are also suitable for the transplantation therapy of the present invention.
- the transplantation therapy agent of the present invention can be prepared by mixing with a medicamentously acceptable aqueous liquid, for example.
- a method for producing a transplantation therapy agent is also provided, which includes a step of formulating the neural organoid produced by the production method of the present invention.
- Such a production method can also include a step of preparing the neural organoid, a step of storing the neural organoid, etc.
- the medicamentously acceptable aqueous liquid that may be contained in the cell transplantation therapy of the present invention may contain, for example, an appropriate selection of buffering agents, isotonicity agents, pH adjusters, antioxidants, chelating agents, etc., within a range that does not affect the survival rate and physiological activity of the transplanted neural organoid.
- the buffer include phosphate buffer, borate buffer, citrate buffer, tartrate buffer, acetate buffer, amino acids, and epsilon-aminocaproic acid.
- isotonic agent examples include sugars such as D-sorbitol, D-glucose, and D-mannitol; polyhydric alcohols such as glycerin and propylene glycol; salts such as sodium chloride; and boric acid.
- Chelating agents include sodium edetate and citric acid.
- pH adjusters include sodium hydroxide, potassium hydroxide, sodium carbonate, sodium bicarbonate, boric acid or a salt thereof (borax), hydrochloric acid, citric acid or a salt thereof (sodium citrate, sodium dihydrogen citrate, etc.), phosphoric acid or a salt thereof (disodium hydrogen phosphate, potassium dihydrogen phosphate, etc.), acetic acid or a salt thereof (sodium acetate, ammonium acetate, etc.), tartaric acid or a salt thereof (sodium tartrate, etc.), and the like.
- antioxidants include ascorbic acid, glutathione, sodium hydrogen sulfite, dry sodium sulfite, sodium pyrosulfite, and tocopherol.
- medicamentously acceptable aqueous liquids include aqueous liquids such as physiological saline, isotonic solutions containing glucose and other auxiliary drugs (eg, D-sorbitol, D-mannitol, sodium chloride, etc.).
- the transplantation therapy agent of the present invention may be mixed with, for example, a soothing agent (eg, benzalkonium chloride, procaine hydrochloride, etc.), a stabilizer (eg, human serum albumin, polyethylene glycol, etc.), a preservative, an antioxidant, etc.
- the transplantation therapy agent of the present invention is provided in a frozen state under conditions normally used for cryopreservation of cells, and can be thawed when needed.
- it may further contain serum or a substitute thereof, an organic solvent (e.g., DMSO), etc.
- an organic solvent e.g., DMSO
- the concentration of serum or a substitute thereof is not particularly limited, but may be about 1 to about 30% (v/v), preferably about 5 to about 20% (v/v).
- the concentration of the organic solvent is not particularly limited, but may be 0 to about 50% (v/v), preferably about 5 to about 20% (v/v).
- the neural organoids contained in the transplantation therapy of the present invention may be produced using pluripotent stem cells whose genes have been manipulated by genome editing as a raw material. All of the contents described in "1. Production method of the present invention” are incorporated herein by reference for matters necessary for the transplantation therapy of the present invention other than those described above.
- Screening method of the present invention (1) Culturing a neural organoid produced by the production method of the present invention or a cell constituting the neural organoid in the presence or absence of a test substance; (2) evaluating the degree of neurodegeneration of the neural organoid or the cells constituting the neural organoid; and (3) selecting the test substance as a candidate substance for a therapeutic or preventive drug for neurodegenerative disease when the progression of neurodegeneration of the neural organoid or the cells constituting the neural organoid is suppressed in the presence of the test substance compared to the absence of the test substance in step (2).
- the degree of neurodegeneration of neural organoids and the cells that compose them can be evaluated using indicators such as cerebral cortical neuronal markers, layer structure morphology, electrophysiological responsiveness, metabolic products, and expression of apoptosis-related genes.
- the cells constituting the neural organoids used in the screening method of the present invention can be obtained as single cells, single cells, or cell populations by treating the neural organoids obtained by the manufacturing method of the present invention with a cell dispersion solution containing enzymes or the like as described above, by cutting them into pieces by physical incision or the like (e.g., using a scalpel, scissors, or a 27-gauge (G) to 21-G injection needle, or the like), or by aspirating them using a pipette or the like.
- a cell dispersion solution containing enzymes or the like as described above
- the cells constituting the neural organoids are similarly stable (homogeneous), and these cells are also suitable for carrying out the screening method of the present invention.
- test substances used in the screening method of the present invention include, for example, cell extracts, cell culture supernatants, microbial fermentation products, extracts from marine organisms, plant extracts, purified or crude proteins, peptides, non-peptide compounds, synthetic small molecule compounds, and natural compounds.
- the test substances can also be obtained using any of the many approaches in combinatorial library methods known in the art, including (1) biological libraries, (2) synthetic library methods using deconvolution, (3) "one-bead one-compound” library methods, and (4) synthetic library methods using affinity chromatography selection.
- the neural organoids used in the screening method of the present invention may be produced using pluripotent stem cells whose genes have been manipulated by genome editing or disease-specific pluripotent stem cells as raw materials.
- diseases include Parkinson's disease, Huntington's chorea, Alzheimer's disease, cerebrovascular disorders (symptoms include stroke, more specifically ischemic brain disease (cerebral infarction, transient ischemic attack) and hemorrhagic brain disease (cerebral hemorrhage, subarachnoid hemorrhage)), epilepsy, brain trauma, motor neuron disease, neurodegenerative disease, etc.
- a 35 mm diameter dish (manufactured by Sumitomo Bakelite Co., Ltd., product name: PrimeSurface (registered trademark)) coated on the surface with a hydrophilic polymer was covered with a metal mask (manufactured by Mitani Micronics Co., Ltd.) having a plurality (100 pieces) of circular holes with a diameter of 1,500 ⁇ m, and a culture vessel having a cell adhesive region was produced by performing plasma treatment (20 Pa gas pressure, conductive current of 20 mA, irradiation time of 30 seconds) from above the metal mask using a plasma irradiation device (manufactured by Vacuum Device Co., Ltd., product name Plasma Ion Bombarder PIB-20).
- a temperature-responsive block copolymer was synthesized to cover the culture vessel.
- 0.40 g (0.1 mmol) of 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid, 7.11 g (50 mmol) of n-butyl methacrylate, and 33 mg (0.2 mmol) of azobis(isobutyronitrile) were added to a test tube and dissolved in 50 ml of 1,4-dioxane. After degassing for 30 minutes by nitrogen bubbling, the mixture was reacted at 70°C for 24 hours.
- the reaction solvent was distilled off under reduced pressure using a rotary evaporator, and the reaction solution was concentrated.
- the concentrated solution was poured into 250 ml of methanol, and the precipitated yellow oily substance was collected and dried under reduced pressure to obtain an n-butyl methacrylate polymer.
- 0.9 g (0.3 mmol) of the n-butyl methacrylate polymer, 8.14 g (72 mmol) of N-isopropylacrylamide, and 5 mg (0.03 mmol) of azobisisobutyronitrile were added and dissolved in 15 ml of 1,4-dioxane.
- the mixture was reacted at 65° C. for 17 hours. After the reaction was completed, the reaction solvent was diluted with acetone and poured into 500 ml of hexane, and the precipitated solid was collected and dried under reduced pressure. The mixture was also dissolved again in acetone and poured into 500 ml of pure water, and the precipitated solid was collected and dried under reduced pressure to obtain a diblock copolymer of N-isopropylacrylamide and n-butyl methacrylate.
- the block copolymer was dissolved in ethanol to prepare a 0.8 wt% solution. 50 ⁇ l of this solution was dropped onto the culture vessel having the cell adhesive region, and spin-coated at 2000 rpm for 60 seconds. It was then dried at room temperature for 1 hour. It was then immersed in pure water for 24 hours and washed to produce a culture vessel (TS-02) coated with the block copolymer.
- ⁇ Measurement of response temperature of block copolymer> The substrate coated with the block copolymer was immersed in water, and the bubble contact angle ( ⁇ ) (°) was measured at 5°C intervals in the range of 20 to 45°C, the water contact angle (180- ⁇ ) (°) at each temperature was determined, and the response temperature was calculated by the midpoint method. Note that ⁇ was measured by measuring the contact angle of a 3 ⁇ l bubble in water using a contact angle meter DM300 manufactured by Kyowa Interface Science Co., Ltd. The response temperature was 33°C.
- the thickness of the layer of the temperature-responsive polymer coating the culture vessel was determined by calculating the amount of coating per unit area from the surface area of the vessel, the concentration of the polymer in the solution, and the volume of the solution dropped onto the culture vessel, assuming the specific gravity of the polymer to be 1.
- the layer thickness of the temperature-responsive polymer was 29.4 nm.
- the composition of the temperature-responsive polymer was determined by proton nuclear magnetic resonance spectroscopy ( 1 H-NMR) using a nuclear magnetic resonance measurement device (manufactured by JEOL Ltd., product name JNM-GSX400) or carbon nuclear magnetic resonance spectroscopy ( 13 C-NMR) using a nuclear magnetic resonance measurement device (manufactured by Bruker, product name AVANCEIIIHD500).
- 1 H-NMR proton nuclear magnetic resonance spectroscopy
- 13 C-NMR carbon nuclear magnetic resonance spectroscopy
- 13 C-NMR carbon nuclear magnetic resonance spectroscopy
- the composition of the temperature-responsive polymer was 96 wt % N-isopropylacrylamide polymer and 4 wt % n-butyl methacrylate polymer.
- Undifferentiated maintenance culture was performed using StemFit (AK02) medium, and the human pluripotent stem cells that had been cultured and grown were detached by enzyme treatment (Triple Select) and seeded on a TS-02 substrate at a cell density of 14,000 cells/ cm2 or 15,000 cells/ cm2 (culture day 0).
- iMatrix 1.5 ⁇ g/ cm2
- Y-27632 10 ⁇ M
- ROCK inhibitor a ROCK inhibitor
- the differentiation-inducing medium As the differentiation-inducing medium, a conditioned medium based on GMEM medium with NEAA (1%), Sodium Pyruvate (1%), and 2-mercaptoethanol was used. Furthermore, KSR (20%) was added to the differentiation medium to promote neural differentiation, and differentiation induction was performed for 16 to 20 days. After that, the cells (cell population containing neural stem cells) attached to TS-02 were naturally detached using PBS or medium cooled at 4°C.
- a conditioned differentiation medium based on DMEM/F12 medium with the addition of GlutaMAX, CD lipid concentrate (1%), Penicillin/Streptomycin (1%), Amphotericin B (0.1%), and N2-supplement (1%) was used as the differentiation-inducing medium.
- the cells were transferred onto a 90 mm dish (MS-1390R (Sumitomo Bakelite Co., Ltd.)) and cultured statically for 4 days, after which they were switched to rotational culture for more than 13 days ( Figure 1). The medium was changed once every 1 to 3 days.
- Example 1 Induction of cerebral cortical organoids
- human iPS cells (201B7) were cultured on TS-02 (spot diameter 1,500 ⁇ m) for 20 days in adhesion culture, and then rotated for 6 days.
- the tissue structure could be confirmed under a microscope.
- the tissue structure could be confirmed more clearly and in multiple places.
- Example 2 Verification of induction efficiency by examination of differentiation induction signals It was found that the formation of a layered structure in cerebral cortical organoids was also observed by adding the ectoderm induction factor SB431542 (TGF ⁇ signal inhibitor) (5 ⁇ M) and the Wnt antagonist IWR-1-end (Wnt signal inhibitor) (3 ⁇ M) from day 0 to day 18 of differentiation induction under the differentiation induction described in Example 1 above ( Figure 3).
- SB431542 TGF ⁇ signal inhibitor
- Wnt antagonist IWR-1-end Wnt signal inhibitor
- Example 3 Comparison of induction efficiency with 96 well plate Under the differentiation induction of Example 2 above, the induction efficiency of cerebral cortical organoids in both 96 well plate and TS-02 was compared.
- Three types of human iPS cells Ff-I01s04, Ff-I14s03, Ff-WJs524) were used.
- the cerebral cortical layer structure could be confirmed in a part of Ff-I01s04 on the 35th day of differentiation induction.
- the cerebral cortical layer structure could not be confirmed for Ff-I14s03, even though the culture was continued for 35 days.
- Ff-WJs524 the cells died on the 18th day of differentiation induction and the experiment was discontinued ( Figure 4).
- Example 4 Cortical organoid induction efficiency depending on micropatterning area Human iPS cells (201B7) were subjected to differentiation induction as described in Example 2 above, and the micropatterning area of TS-02 was compared to determine whether it had an effect on induction efficiency when the diameter was 150 ⁇ m, 200 ⁇ m, 500 ⁇ m, 750 ⁇ m, and 1500 ⁇ m. As a result, the formation of a layered structure of cortical organoids was observed under all conditions at the time point of the 25th day of differentiation induction, and it was found that the area was unlikely to cause an extreme decrease in differentiation induction efficiency (FIG. 6).
- Example 5 Formation of cerebral cortical organoids by different timing of detaching cells from TS-02 Under the differentiation induction of Example 2 above, the timing of shifting to rotational culture was performed on the 6th, 12th, and 18th days of differentiation induction to investigate how the efficiency of differentiation induction into cerebral cortical organoids changes. Two types of human iPS cells (Ff-I01s04, Ff-I14s03) were used. At the time of the 49th day of differentiation induction, the tissue structure could be confirmed under a microscope in all condition groups.
- Example 6 Homogenization of cerebral cortical organoid size by rotational culture conditions
- Human iPS cells (201B7) were cultured for 18 days under the differentiation induction of Example 2 above, and cerebral cortical organoids were induced under the conditions of rotational culture in a 90 mm dish after cell detachment, and under the conditions of providing an uneven shape on the dish to easily apply physical stimuli during rotation (EZSPHERE (AGC Technoglass)).
- EZSPHERE AAC Technoglass
- Example 7 Variation in marker expression of cerebral cortex organoid Human iPS cells (201B7) were cultured for 35 days under the differentiation induction of Example 2 above, and the variation in the expression of the markers PAX6 and CTIP2 between organoids was examined. After fixing the cerebral cortex organoids on the 35th day of differentiation induction and staining with PAX6 and CTIP2, evaluation was performed on 18 organoids using a cell sorter, and it was found that the expression of PAX6 was 27.0 ⁇ 12.7% and the expression of CTIP2 was 37.7 ⁇ 10.9% (Figure 10).
- Example 8 Induction of cerebral cortex organoids
- cells were cultured in StemFitAK02 until confluent, and then switched to differentiation-inducing medium (GMEM medium) without reseeding.
- GMEM medium differentiation-inducing medium
- FIG. 11 cells were reseeded when switching to differentiation-inducing medium, and then cultured in the same manner as in Example 1.
- the cells were cultured in the presence of Y-27632 for three days.
- cerebral cortical organoids having a neuroepithelial structure were obtained as in Example 1 (FIGS. 12 and 13).
- this cerebral cortical organoid had a highly uniform structure between organoids compared to the conventional method using a 96 well plate (FIG. 15).
- the present invention is useful because it provides a method for producing neural organoids using a culture vessel that has multiple independent cell adhesive regions and in which cells adhering to each region are present in the same medium, allowing for the mass production of neural organoids with stable quality at low cost.
- the culture vessel used in the present invention has predetermined cell adhesive regions, which makes it possible to avoid adhesion of adjacent cells due to operations such as medium replacement, making it possible to produce neural organoids with stable quality in large quantities with a single operation.
- the quality of the neural organoids produced by the production method of the present invention is stable (homogeneous), it is also useful because it is expected that transplantation therapy agents containing the neural organoids and screening methods for therapeutic or preventive drugs for neurodegenerative diseases using the neural organoids will lead to the elucidation of the mechanisms of diseases (especially neurodegenerative diseases), which have been difficult to achieve in the past, and the development of drug discovery research and regenerative medicine.
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biomedical Technology (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- General Health & Medical Sciences (AREA)
- Cell Biology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- Developmental Biology & Embryology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Molecular Biology (AREA)
- Urology & Nephrology (AREA)
- Pharmacology & Pharmacy (AREA)
- Hematology (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Analytical Chemistry (AREA)
- Physics & Mathematics (AREA)
- Virology (AREA)
- Pathology (AREA)
- Toxicology (AREA)
- Tropical Medicine & Parasitology (AREA)
- General Physics & Mathematics (AREA)
- General Chemical & Material Sciences (AREA)
- Ophthalmology & Optometry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020257034462A KR20250162849A (ko) | 2023-03-20 | 2024-03-19 | 신경 오르가노이드의 제조 방법 및 그 이용 |
| CN202480020291.2A CN120958118A (zh) | 2023-03-20 | 2024-03-19 | 神经类器官的制造方法及其利用 |
| EP24774924.5A EP4685230A1 (en) | 2023-03-20 | 2024-03-19 | Method for producing neural organoids and use of same |
| JP2025508575A JPWO2024195789A1 (https=) | 2023-03-20 | 2024-03-19 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023-044570 | 2023-03-20 | ||
| JP2023044570 | 2023-03-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024195789A1 true WO2024195789A1 (ja) | 2024-09-26 |
Family
ID=92841700
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2024/010698 Ceased WO2024195789A1 (ja) | 2023-03-20 | 2024-03-19 | 神経オルガノイドの製造方法およびその利用 |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP4685230A1 (https=) |
| JP (1) | JPWO2024195789A1 (https=) |
| KR (1) | KR20250162849A (https=) |
| CN (1) | CN120958118A (https=) |
| WO (1) | WO2024195789A1 (https=) |
Citations (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| WO1996022362A1 (en) | 1995-01-20 | 1996-07-25 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| WO1998030679A1 (en) | 1997-01-10 | 1998-07-16 | Life Technologies, Inc. | Embryonic stem cell serum replacement |
| US6280718B1 (en) | 1999-11-08 | 2001-08-28 | Wisconsin Alumni Reasearch Foundation | Hematopoietic differentiation of human pluripotent embryonic stem cells |
| US20020103360A1 (en) | 1998-09-01 | 2002-08-01 | Yang Pan | Novel protein related to melanoma-inhibiting protein and uses thereof |
| WO2002101057A1 (fr) | 2001-06-08 | 2002-12-19 | Dnavec Research Inc. | Transfert genique dans des cellules souches embryonnaires de primate a l'aide d'un virus de l'immunodeficience simienne de pseudo type vsv-g utilise comme vecteur |
| JP2013247926A (ja) * | 2012-06-01 | 2013-12-12 | Dainippon Printing Co Ltd | 細胞培養方法及び細胞培養容器 |
| JP2015015943A (ja) * | 2013-06-10 | 2015-01-29 | 大日本印刷株式会社 | 人工多能性幹細胞の分化誘導方法 |
| WO2015076388A1 (ja) | 2013-11-22 | 2015-05-28 | 国立研究開発法人理化学研究所 | 終脳又はその前駆組織の製造方法 |
| JP2018161097A (ja) * | 2017-03-27 | 2018-10-18 | 一般財団法人電力中央研究所 | 培養細胞供試体の作製方法 |
| WO2019035436A1 (ja) * | 2017-08-16 | 2019-02-21 | 東ソー株式会社 | 多能性幹細胞の培養基材及び多能性幹細胞の製造方法 |
| WO2019107546A1 (ja) | 2017-11-30 | 2019-06-06 | 公立大学法人横浜市立大学 | 細胞塊を集合させる方法及び細胞塊を集合させる装置 |
| JP2020195349A (ja) | 2019-06-05 | 2020-12-10 | 公立大学法人奈良県立医科大学 | 脳幹オルガノイドの作製方法 |
| JP2021158960A (ja) | 2020-03-31 | 2021-10-11 | 東ソー株式会社 | 多能性幹細胞から外胚葉系細胞を分化誘導する方法及び製造方法 |
| JP2022002504A (ja) * | 2020-03-23 | 2022-01-11 | 株式会社リコー | 神経細胞が配置された基板の製造方法 |
| JP2023044570A (ja) | 2021-09-17 | 2023-03-30 | ユニソック合同会社 | マッチング支援システム、マッチング支援プログラム及びマッチング支援方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20150076388A (ko) | 2013-12-26 | 2015-07-07 | 두산인프라코어 주식회사 | 건설기계용 투스 어댑터의 용접 방법 |
-
2024
- 2024-03-19 CN CN202480020291.2A patent/CN120958118A/zh active Pending
- 2024-03-19 KR KR1020257034462A patent/KR20250162849A/ko active Pending
- 2024-03-19 JP JP2025508575A patent/JPWO2024195789A1/ja active Pending
- 2024-03-19 WO PCT/JP2024/010698 patent/WO2024195789A1/ja not_active Ceased
- 2024-03-19 EP EP24774924.5A patent/EP4685230A1/en active Pending
Patent Citations (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| US5403484A (en) | 1988-09-02 | 1995-04-04 | Protein Engineering Corporation | Viruses expressing chimeric binding proteins |
| US5571698A (en) | 1988-09-02 | 1996-11-05 | Protein Engineering Corporation | Directed evolution of novel binding proteins |
| WO1996022362A1 (en) | 1995-01-20 | 1996-07-25 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| US6200806B1 (en) | 1995-01-20 | 2001-03-13 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| WO1998030679A1 (en) | 1997-01-10 | 1998-07-16 | Life Technologies, Inc. | Embryonic stem cell serum replacement |
| US20020103360A1 (en) | 1998-09-01 | 2002-08-01 | Yang Pan | Novel protein related to melanoma-inhibiting protein and uses thereof |
| US6280718B1 (en) | 1999-11-08 | 2001-08-28 | Wisconsin Alumni Reasearch Foundation | Hematopoietic differentiation of human pluripotent embryonic stem cells |
| WO2002101057A1 (fr) | 2001-06-08 | 2002-12-19 | Dnavec Research Inc. | Transfert genique dans des cellules souches embryonnaires de primate a l'aide d'un virus de l'immunodeficience simienne de pseudo type vsv-g utilise comme vecteur |
| JP2013247926A (ja) * | 2012-06-01 | 2013-12-12 | Dainippon Printing Co Ltd | 細胞培養方法及び細胞培養容器 |
| JP2015015943A (ja) * | 2013-06-10 | 2015-01-29 | 大日本印刷株式会社 | 人工多能性幹細胞の分化誘導方法 |
| WO2015076388A1 (ja) | 2013-11-22 | 2015-05-28 | 国立研究開発法人理化学研究所 | 終脳又はその前駆組織の製造方法 |
| JP2021072818A (ja) | 2013-11-22 | 2021-05-13 | 国立研究開発法人理化学研究所 | 終脳又はその前駆組織の製造方法 |
| JP2018161097A (ja) * | 2017-03-27 | 2018-10-18 | 一般財団法人電力中央研究所 | 培養細胞供試体の作製方法 |
| WO2019035436A1 (ja) * | 2017-08-16 | 2019-02-21 | 東ソー株式会社 | 多能性幹細胞の培養基材及び多能性幹細胞の製造方法 |
| WO2019107546A1 (ja) | 2017-11-30 | 2019-06-06 | 公立大学法人横浜市立大学 | 細胞塊を集合させる方法及び細胞塊を集合させる装置 |
| JP2020195349A (ja) | 2019-06-05 | 2020-12-10 | 公立大学法人奈良県立医科大学 | 脳幹オルガノイドの作製方法 |
| JP2022002504A (ja) * | 2020-03-23 | 2022-01-11 | 株式会社リコー | 神経細胞が配置された基板の製造方法 |
| JP2021158960A (ja) | 2020-03-31 | 2021-10-11 | 東ソー株式会社 | 多能性幹細胞から外胚葉系細胞を分化誘導する方法及び製造方法 |
| JP2023044570A (ja) | 2021-09-17 | 2023-03-30 | ユニソック合同会社 | マッチング支援システム、マッチング支援プログラム及びマッチング支援方法 |
Non-Patent Citations (30)
| Title |
|---|
| "Bio-Manual Series 8, Gene Targeting, Production of Mutant Mice Using ES Cells", 1995, YODOSHA CO., LTD |
| "Molecular Cloning, A Laboratory Manual", 1989, COLD SPRING HARBOR LABORATORY PRESS |
| CARELL ET AL., ANGEW. CHEM. INT. ED. ENGL., vol. 33, 1994, pages 2061 |
| CELL, vol. 131, no. 5, 2007, pages 861 - 872 |
| CELL, vol. 70, 1992, pages 841 - 847 |
| CHO ET AL., SCIENCE, vol. 261, 1993, pages 1303 - 5 |
| CULL ET AL., PROC. NATL. ACAD. SCI. USA, vol. 89, 1992, pages 1865 - 9 |
| CWIRLA ET AL., PROC. NATL. ACAD. SCI. USA, vol. 87, 1990, pages 6378 - 82 |
| DEWITT ET AL., PROC. NATL. ACAD. SCI. USA, vol. 90, 1993, pages 6909 - 13 |
| ERB ET AL., PROC. NATL. ACAD. SCI. USA, vol. 91, 1994, pages 11422 - 6 |
| FELICI, J. MOL. BIOL., vol. 222, 1991, pages 301 - 10 |
| FODOR, NATURE, vol. 364, 1993, pages 555 - 6 |
| HOUGHTEN, BIO/TECHNIQUES, vol. 13, 1992, pages 412 - 21 |
| ITO Y. ET AL., CELL, vol. 174, pages 636 - 648 |
| KADOSHIMA T ET AL., PROC NATL ACAD SCI U S A., vol. 110, no. 50, 2013, pages 20284 - 20289 |
| KADOSHIMA T. ET AL., PROC NATL ACAD SCI U S A., vol. 111, no. 20, 20 May 2014 (2014-05-20), pages 7498 |
| LAM, ANTICANCER DRUG DES, vol. 12, 1997, pages 145 - 67 |
| LAM, NATURE, vol. 354, 1991, pages 82 - 4 |
| ML WEGSCHEID. ET AL., CELL REP., vol. 36, no. 1, 6 July 2021 (2021-07-06), pages 109315 |
| NAT. BIOTECHNOL., vol. 26, no. 1, 2008, pages 101 - 106 |
| SAKAGUCHI H. ET AL., NAT COMMUN., vol. 6, 2015, pages 8896 |
| SAKAGUCHI H. ET AL., STEM CELL REPORTS, vol. 13, no. 3, 2019, pages 458 - 473 |
| SASAI Y. ET AL., CELL STEM CELL, vol. 12, 2013, pages 520 - 530 |
| SCIENCE, vol. 318, no. 5858, 2007, pages 1917 - 1920 |
| SCIENCE, vol. 341, 2013, pages 651 - 654 |
| SCOTTSMITH, SCIENCE, vol. 249, 1990, pages 404 - 90 |
| See also references of EP4685230A1 |
| YAMANAKA ET AL.: "established induced pluripotent stem cells using mouse cells", CELL, vol. 126, no. 4, 2006, pages 663 - 676 |
| Z GANIA. ET AL., FUTURE PHARMACOL, vol. 2, no. 3, 2022, pages 360 - 376 |
| ZUCKERMANN ET AL., J. MED. CHEM., vol. 37, 1994, pages 1233 - 51 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN120958118A (zh) | 2025-11-14 |
| EP4685230A1 (en) | 2026-01-28 |
| KR20250162849A (ko) | 2025-11-19 |
| JPWO2024195789A1 (https=) | 2024-09-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7788812B2 (ja) | 臨床グレードの網膜色素上皮細胞の再現性のある分化のための方法 | |
| JP7088496B2 (ja) | 網膜組織の製造方法 | |
| CN109312306B (zh) | 产生视网膜组织的方法 | |
| JP6986016B2 (ja) | Macsを用いた幹細胞由来網膜色素上皮の精製 | |
| JP7154546B2 (ja) | 網膜組織の製造方法 | |
| JP7150281B2 (ja) | 連続的な上皮を含む網膜組織の成熟化方法 | |
| JP2022141924A (ja) | 網膜組織及び網膜関連細胞の製造方法 | |
| CN108350421A (zh) | 用于制备视网膜色素上皮细胞的方法 | |
| CN107109367A (zh) | 神经组织的制备方法 | |
| JP2008099662A (ja) | 幹細胞の培養方法 | |
| CN112204134A (zh) | 眼细胞的分化方法及其用途 | |
| WO2019054514A1 (ja) | 網膜組織の製造方法 | |
| CN111094548A (zh) | 基于背侧化信号转导物质或腹侧化信号转导物质的增加视锥细胞或视杆细胞的方法 | |
| WO2024195789A1 (ja) | 神経オルガノイドの製造方法およびその利用 | |
| JP2025511931A (ja) | Ipscの作製のための方法 | |
| HK40129875A (en) | Method for producing retinal tissue | |
| EP4656716A1 (en) | Method for producing retinal tissue | |
| HK40109085A (en) | Method for reproducible differentiation of clinical-grade retinal pigment epithelium cells | |
| EP4259779A1 (en) | Methods for controlled induction of bioengineered neuroepithelial tissues and 3-d neuroepithelial tubes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24774924 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2025508575 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2025508575 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 1020257034462 Country of ref document: KR Free format text: ST27 STATUS EVENT CODE: A-0-1-A10-A15-NAP-PA0105 (AS PROVIDED BY THE NATIONAL OFFICE) |
|
| WWE | Wipo information: entry into national phase |
Ref document number: KR1020257034462 Country of ref document: KR Ref document number: 1020257034462 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024774924 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2024774924 Country of ref document: EP Effective date: 20251020 |
|
| ENP | Entry into the national phase |
Ref document number: 2024774924 Country of ref document: EP Effective date: 20251020 |
|
| ENP | Entry into the national phase |
Ref document number: 2024774924 Country of ref document: EP Effective date: 20251020 |
|
| ENP | Entry into the national phase |
Ref document number: 2024774924 Country of ref document: EP Effective date: 20251020 |
|
| ENP | Entry into the national phase |
Ref document number: 2024774924 Country of ref document: EP Effective date: 20251020 |
|
| ENP | Entry into the national phase |
Ref document number: 2024774924 Country of ref document: EP Effective date: 20251020 |