WO2024029346A1 - 離型フィルム - Google Patents
離型フィルム Download PDFInfo
- Publication number
- WO2024029346A1 WO2024029346A1 PCT/JP2023/026510 JP2023026510W WO2024029346A1 WO 2024029346 A1 WO2024029346 A1 WO 2024029346A1 JP 2023026510 W JP2023026510 W JP 2023026510W WO 2024029346 A1 WO2024029346 A1 WO 2024029346A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- resin layer
- layer
- release
- resin
- release film
- Prior art date
Links
- 229920005989 resin Polymers 0.000 claims abstract description 313
- 239000011347 resin Substances 0.000 claims abstract description 313
- 229920002050 silicone resin Polymers 0.000 claims abstract description 67
- 239000000853 adhesive Substances 0.000 claims abstract description 47
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 43
- 239000010410 layer Substances 0.000 claims description 460
- -1 dimethylsiloxane skeleton Chemical group 0.000 claims description 120
- 150000001875 compounds Chemical class 0.000 claims description 96
- 239000012790 adhesive layer Substances 0.000 claims description 76
- 239000000203 mixture Substances 0.000 claims description 75
- 229920000728 polyester Polymers 0.000 claims description 72
- 239000002245 particle Substances 0.000 claims description 67
- 238000000034 method Methods 0.000 claims description 64
- 229920000642 polymer Polymers 0.000 claims description 50
- 230000001070 adhesive effect Effects 0.000 claims description 47
- 125000000217 alkyl group Chemical group 0.000 claims description 44
- 229920006267 polyester film Polymers 0.000 claims description 44
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims description 36
- 238000011156 evaluation Methods 0.000 claims description 24
- 229920000178 Acrylic resin Polymers 0.000 claims description 19
- 239000004925 Acrylic resin Substances 0.000 claims description 19
- 239000004820 Pressure-sensitive adhesive Substances 0.000 claims description 19
- 230000003287 optical effect Effects 0.000 claims description 18
- 229930192474 thiophene Natural products 0.000 claims description 18
- 229920001225 polyester resin Polymers 0.000 claims description 17
- 239000004645 polyester resin Substances 0.000 claims description 17
- 238000003825 pressing Methods 0.000 claims description 17
- 150000003577 thiophenes Chemical class 0.000 claims description 17
- 239000006082 mold release agent Substances 0.000 claims description 16
- 239000003522 acrylic cement Substances 0.000 claims description 12
- 150000001449 anionic compounds Chemical class 0.000 claims description 6
- 150000002222 fluorine compounds Chemical class 0.000 claims description 5
- 229920005749 polyurethane resin Polymers 0.000 claims description 5
- GOXQRTZXKQZDDN-UHFFFAOYSA-N 2-Ethylhexyl acrylate Chemical compound CCCCC(CC)COC(=O)C=C GOXQRTZXKQZDDN-UHFFFAOYSA-N 0.000 claims description 4
- CQEYYJKEWSMYFG-UHFFFAOYSA-N butyl acrylate Chemical compound CCCCOC(=O)C=C CQEYYJKEWSMYFG-UHFFFAOYSA-N 0.000 claims description 4
- 125000000129 anionic group Chemical group 0.000 claims description 3
- 230000000903 blocking effect Effects 0.000 abstract description 33
- 239000000463 material Substances 0.000 abstract description 16
- 230000008859 change Effects 0.000 abstract description 4
- 239000010408 film Substances 0.000 description 326
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 78
- 238000000576 coating method Methods 0.000 description 71
- 239000011248 coating agent Substances 0.000 description 69
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 67
- 239000000178 monomer Substances 0.000 description 62
- 239000003431 cross linking reagent Substances 0.000 description 43
- 239000001993 wax Substances 0.000 description 34
- 239000002585 base Substances 0.000 description 33
- 229920001296 polysiloxane Polymers 0.000 description 32
- 238000006243 chemical reaction Methods 0.000 description 30
- 238000004519 manufacturing process Methods 0.000 description 28
- 238000005259 measurement Methods 0.000 description 27
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 24
- 239000002253 acid Substances 0.000 description 21
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 20
- 229920002803 thermoplastic polyurethane Polymers 0.000 description 20
- 230000000052 comparative effect Effects 0.000 description 19
- 125000004432 carbon atom Chemical group C* 0.000 description 17
- 239000002904 solvent Substances 0.000 description 17
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 16
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 16
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 16
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 16
- 239000003054 catalyst Substances 0.000 description 16
- 238000001723 curing Methods 0.000 description 15
- 239000007788 liquid Substances 0.000 description 15
- 230000005012 migration Effects 0.000 description 15
- 238000013508 migration Methods 0.000 description 15
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 15
- 125000002947 alkylene group Chemical group 0.000 description 14
- 239000011572 manganese Substances 0.000 description 14
- 125000003342 alkenyl group Chemical group 0.000 description 13
- 238000001035 drying Methods 0.000 description 13
- 229920005862 polyol Polymers 0.000 description 13
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 12
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 12
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 description 12
- 239000002216 antistatic agent Substances 0.000 description 12
- 238000010438 heat treatment Methods 0.000 description 12
- 150000003077 polyols Chemical class 0.000 description 12
- 239000000243 solution Substances 0.000 description 12
- 239000000126 substance Substances 0.000 description 12
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 12
- 230000015572 biosynthetic process Effects 0.000 description 11
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 11
- 229920001577 copolymer Polymers 0.000 description 11
- 229920000058 polyacrylate Polymers 0.000 description 11
- 230000008569 process Effects 0.000 description 11
- 239000002994 raw material Substances 0.000 description 11
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 11
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 10
- QQVIHTHCMHWDBS-UHFFFAOYSA-N isophthalic acid Chemical compound OC(=O)C1=CC=CC(C(O)=O)=C1 QQVIHTHCMHWDBS-UHFFFAOYSA-N 0.000 description 10
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 10
- 239000002344 surface layer Substances 0.000 description 10
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 9
- 125000003277 amino group Chemical group 0.000 description 9
- 239000011230 binding agent Substances 0.000 description 9
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 9
- VZSRBBMJRBPUNF-UHFFFAOYSA-N 2-(2,3-dihydro-1H-inden-2-ylamino)-N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]pyrimidine-5-carboxamide Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)C(=O)NCCC(N1CC2=C(CC1)NN=N2)=O VZSRBBMJRBPUNF-UHFFFAOYSA-N 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 229920000877 Melamine resin Polymers 0.000 description 8
- 239000006096 absorbing agent Substances 0.000 description 8
- 239000002390 adhesive tape Substances 0.000 description 8
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 8
- 238000004458 analytical method Methods 0.000 description 8
- ADCOVFLJGNWWNZ-UHFFFAOYSA-N antimony trioxide Chemical compound O=[Sb]O[Sb]=O ADCOVFLJGNWWNZ-UHFFFAOYSA-N 0.000 description 8
- 239000006185 dispersion Substances 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 8
- 125000000524 functional group Chemical group 0.000 description 8
- 125000005010 perfluoroalkyl group Chemical group 0.000 description 8
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 8
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical compound OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 description 8
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 8
- 150000003609 titanium compounds Chemical class 0.000 description 8
- 239000004372 Polyvinyl alcohol Substances 0.000 description 7
- 238000007792 addition Methods 0.000 description 7
- 125000003118 aryl group Chemical group 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- 230000006866 deterioration Effects 0.000 description 7
- 229920001971 elastomer Polymers 0.000 description 7
- 150000002430 hydrocarbons Chemical group 0.000 description 7
- 239000012948 isocyanate Substances 0.000 description 7
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 7
- 229910052697 platinum Inorganic materials 0.000 description 7
- 238000006116 polymerization reaction Methods 0.000 description 7
- 229920002451 polyvinyl alcohol Polymers 0.000 description 7
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 7
- 239000004970 Chain extender Substances 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- WERYXYBDKMZEQL-UHFFFAOYSA-N butane-1,4-diol Chemical compound OCCCCO WERYXYBDKMZEQL-UHFFFAOYSA-N 0.000 description 6
- 238000004132 cross linking Methods 0.000 description 6
- 238000007865 diluting Methods 0.000 description 6
- WOZVHXUHUFLZGK-UHFFFAOYSA-N dimethyl terephthalate Chemical compound COC(=O)C1=CC=C(C(=O)OC)C=C1 WOZVHXUHUFLZGK-UHFFFAOYSA-N 0.000 description 6
- 150000002009 diols Chemical class 0.000 description 6
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 6
- 238000005227 gel permeation chromatography Methods 0.000 description 6
- 230000009477 glass transition Effects 0.000 description 6
- 229920001519 homopolymer Polymers 0.000 description 6
- 229910052751 metal Inorganic materials 0.000 description 6
- SLCVBVWXLSEKPL-UHFFFAOYSA-N neopentyl glycol Chemical compound OCC(C)(C)CO SLCVBVWXLSEKPL-UHFFFAOYSA-N 0.000 description 6
- 239000013500 performance material Substances 0.000 description 6
- 238000006068 polycondensation reaction Methods 0.000 description 6
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 5
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 5
- 239000002202 Polyethylene glycol Substances 0.000 description 5
- YIMQCDZDWXUDCA-UHFFFAOYSA-N [4-(hydroxymethyl)cyclohexyl]methanol Chemical compound OCC1CCC(CO)CC1 YIMQCDZDWXUDCA-UHFFFAOYSA-N 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 239000000975 dye Substances 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 238000001125 extrusion Methods 0.000 description 5
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 5
- 150000002500 ions Chemical class 0.000 description 5
- 150000002513 isocyanates Chemical class 0.000 description 5
- 239000002184 metal Substances 0.000 description 5
- RXOHFPCZGPKIRD-UHFFFAOYSA-N naphthalene-2,6-dicarboxylic acid Chemical compound C1=C(C(O)=O)C=CC2=CC(C(=O)O)=CC=C21 RXOHFPCZGPKIRD-UHFFFAOYSA-N 0.000 description 5
- 229910052698 phosphorus Inorganic materials 0.000 description 5
- 239000011574 phosphorus Substances 0.000 description 5
- 229920000515 polycarbonate Polymers 0.000 description 5
- 239000004417 polycarbonate Substances 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 230000000379 polymerizing effect Effects 0.000 description 5
- 229920002635 polyurethane Polymers 0.000 description 5
- 239000004814 polyurethane Substances 0.000 description 5
- YPFDHNVEDLHUCE-UHFFFAOYSA-N propane-1,3-diol Chemical compound OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 description 5
- 238000007789 sealing Methods 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- 230000003746 surface roughness Effects 0.000 description 5
- 238000005809 transesterification reaction Methods 0.000 description 5
- 238000012546 transfer Methods 0.000 description 5
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 4
- 238000005160 1H NMR spectroscopy Methods 0.000 description 4
- 229920002799 BoPET Polymers 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 4
- 229910019142 PO4 Inorganic materials 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 239000004793 Polystyrene Substances 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 239000001361 adipic acid Substances 0.000 description 4
- 235000011037 adipic acid Nutrition 0.000 description 4
- 125000001931 aliphatic group Chemical group 0.000 description 4
- 229910021529 ammonia Inorganic materials 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- CDQSJQSWAWPGKG-UHFFFAOYSA-N butane-1,1-diol Chemical compound CCCC(O)O CDQSJQSWAWPGKG-UHFFFAOYSA-N 0.000 description 4
- 238000007334 copolymerization reaction Methods 0.000 description 4
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 description 4
- 238000009998 heat setting Methods 0.000 description 4
- 150000002440 hydroxy compounds Chemical class 0.000 description 4
- 239000004973 liquid crystal related substance Substances 0.000 description 4
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 4
- 150000007974 melamines Chemical class 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 150000003961 organosilicon compounds Chemical class 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 4
- 239000010452 phosphate Substances 0.000 description 4
- 239000000049 pigment Substances 0.000 description 4
- 229920000172 poly(styrenesulfonic acid) Polymers 0.000 description 4
- 229920000447 polyanionic polymer Polymers 0.000 description 4
- 229920005906 polyester polyol Polymers 0.000 description 4
- 229920001451 polypropylene glycol Polymers 0.000 description 4
- 229920002223 polystyrene Polymers 0.000 description 4
- 229940005642 polystyrene sulfonic acid Drugs 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 239000011541 reaction mixture Substances 0.000 description 4
- 230000009257 reactivity Effects 0.000 description 4
- 150000003839 salts Chemical class 0.000 description 4
- 239000002356 single layer Substances 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 239000006097 ultraviolet radiation absorber Substances 0.000 description 4
- 239000008096 xylene Substances 0.000 description 4
- HMUNWXXNJPVALC-UHFFFAOYSA-N 1-[4-[2-(2,3-dihydro-1H-inden-2-ylamino)pyrimidin-5-yl]piperazin-1-yl]-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethanone Chemical compound C1C(CC2=CC=CC=C12)NC1=NC=C(C=N1)N1CCN(CC1)C(CN1CC2=C(CC1)NN=N2)=O HMUNWXXNJPVALC-UHFFFAOYSA-N 0.000 description 3
- QWDQYHPOSSHSAW-UHFFFAOYSA-N 1-isocyanatooctadecane Chemical compound CCCCCCCCCCCCCCCCCCN=C=O QWDQYHPOSSHSAW-UHFFFAOYSA-N 0.000 description 3
- PTBDIHRZYDMNKB-UHFFFAOYSA-N 2,2-Bis(hydroxymethyl)propionic acid Chemical compound OCC(C)(CO)C(O)=O PTBDIHRZYDMNKB-UHFFFAOYSA-N 0.000 description 3
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 3
- OMIGHNLMNHATMP-UHFFFAOYSA-N 2-hydroxyethyl prop-2-enoate Chemical compound OCCOC(=O)C=C OMIGHNLMNHATMP-UHFFFAOYSA-N 0.000 description 3
- RSWGJHLUYNHPMX-UHFFFAOYSA-N Abietic-Saeure Natural products C12CCC(C(C)C)=CC2=CCC2C1(C)CCCC2(C)C(O)=O RSWGJHLUYNHPMX-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 239000004593 Epoxy Chemical class 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 3
- 239000004698 Polyethylene Substances 0.000 description 3
- KHPCPRHQVVSZAH-HUOMCSJISA-N Rosin Natural products O(C/C=C/c1ccccc1)[C@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 KHPCPRHQVVSZAH-HUOMCSJISA-N 0.000 description 3
- BZHJMEDXRYGGRV-UHFFFAOYSA-N Vinyl chloride Chemical compound ClC=C BZHJMEDXRYGGRV-UHFFFAOYSA-N 0.000 description 3
- 150000001735 carboxylic acids Chemical class 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000011247 coating layer Substances 0.000 description 3
- 239000006258 conductive agent Substances 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 238000004945 emulsification Methods 0.000 description 3
- 239000003822 epoxy resin Substances 0.000 description 3
- 230000032050 esterification Effects 0.000 description 3
- 238000005886 esterification reaction Methods 0.000 description 3
- 239000010419 fine particle Substances 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 125000001153 fluoro group Chemical group F* 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 150000002291 germanium compounds Chemical class 0.000 description 3
- 238000007756 gravure coating Methods 0.000 description 3
- XXMIOPMDWAUFGU-UHFFFAOYSA-N hexane-1,6-diol Chemical compound OCCCCCCO XXMIOPMDWAUFGU-UHFFFAOYSA-N 0.000 description 3
- 125000003010 ionic group Chemical group 0.000 description 3
- IQPQWNKOIGAROB-UHFFFAOYSA-N isocyanate group Chemical group [N-]=C=O IQPQWNKOIGAROB-UHFFFAOYSA-N 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 229940097364 magnesium acetate tetrahydrate Drugs 0.000 description 3
- XKPKPGCRSHFTKM-UHFFFAOYSA-L magnesium;diacetate;tetrahydrate Chemical compound O.O.O.O.[Mg+2].CC([O-])=O.CC([O-])=O XKPKPGCRSHFTKM-UHFFFAOYSA-L 0.000 description 3
- 239000011976 maleic acid Substances 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 125000000962 organic group Chemical group 0.000 description 3
- WXZMFSXDPGVJKK-UHFFFAOYSA-N pentaerythritol Chemical compound OCC(CO)(CO)CO WXZMFSXDPGVJKK-UHFFFAOYSA-N 0.000 description 3
- 235000019271 petrolatum Nutrition 0.000 description 3
- 235000011007 phosphoric acid Nutrition 0.000 description 3
- 229920003216 poly(methylphenylsiloxane) Polymers 0.000 description 3
- 229920000647 polyepoxide Polymers 0.000 description 3
- 229920000573 polyethylene Polymers 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 150000005846 sugar alcohols Polymers 0.000 description 3
- KHPCPRHQVVSZAH-UHFFFAOYSA-N trans-cinnamyl beta-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OCC=CC1=CC=CC=C1 KHPCPRHQVVSZAH-UHFFFAOYSA-N 0.000 description 3
- ARCGXLSVLAOJQL-UHFFFAOYSA-N trimellitic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C(C(O)=O)=C1 ARCGXLSVLAOJQL-UHFFFAOYSA-N 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- 229920002554 vinyl polymer Polymers 0.000 description 3
- 238000004804 winding Methods 0.000 description 3
- DSEKYWAQQVUQTP-XEWMWGOFSA-N (2r,4r,4as,6as,6as,6br,8ar,12ar,14as,14bs)-2-hydroxy-4,4a,6a,6b,8a,11,11,14a-octamethyl-2,4,5,6,6a,7,8,9,10,12,12a,13,14,14b-tetradecahydro-1h-picen-3-one Chemical compound C([C@H]1[C@]2(C)CC[C@@]34C)C(C)(C)CC[C@]1(C)CC[C@]2(C)[C@H]4CC[C@@]1(C)[C@H]3C[C@@H](O)C(=O)[C@@H]1C DSEKYWAQQVUQTP-XEWMWGOFSA-N 0.000 description 2
- QPFMBZIOSGYJDE-UHFFFAOYSA-N 1,1,2,2-tetrachloroethane Chemical compound ClC(Cl)C(Cl)Cl QPFMBZIOSGYJDE-UHFFFAOYSA-N 0.000 description 2
- MYRTYDVEIRVNKP-UHFFFAOYSA-N 1,2-Divinylbenzene Chemical compound C=CC1=CC=CC=C1C=C MYRTYDVEIRVNKP-UHFFFAOYSA-N 0.000 description 2
- ALVZNPYWJMLXKV-UHFFFAOYSA-N 1,9-Nonanediol Chemical compound OCCCCCCCCCO ALVZNPYWJMLXKV-UHFFFAOYSA-N 0.000 description 2
- UAJRSHJHFRVGMG-UHFFFAOYSA-N 1-ethenyl-4-methoxybenzene Chemical compound COC1=CC=C(C=C)C=C1 UAJRSHJHFRVGMG-UHFFFAOYSA-N 0.000 description 2
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 2
- IAXFZZHBFXRZMT-UHFFFAOYSA-N 2-[3-(2-hydroxyethoxy)phenoxy]ethanol Chemical compound OCCOC1=CC=CC(OCCO)=C1 IAXFZZHBFXRZMT-UHFFFAOYSA-N 0.000 description 2
- DSKYSDCYIODJPC-UHFFFAOYSA-N 2-butyl-2-ethylpropane-1,3-diol Chemical compound CCCCC(CC)(CO)CO DSKYSDCYIODJPC-UHFFFAOYSA-N 0.000 description 2
- SVTBMSDMJJWYQN-UHFFFAOYSA-N 2-methylpentane-2,4-diol Chemical compound CC(O)CC(C)(C)O SVTBMSDMJJWYQN-UHFFFAOYSA-N 0.000 description 2
- 125000003504 2-oxazolinyl group Chemical group O1C(=NCC1)* 0.000 description 2
- YLZOPXRUQYQQID-UHFFFAOYSA-N 3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]propan-1-one Chemical compound N1N=NC=2CN(CCC=21)CCC(=O)N1CCN(CC1)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F YLZOPXRUQYQQID-UHFFFAOYSA-N 0.000 description 2
- SXFJDZNJHVPHPH-UHFFFAOYSA-N 3-methylpentane-1,5-diol Chemical compound OCCC(C)CCO SXFJDZNJHVPHPH-UHFFFAOYSA-N 0.000 description 2
- XDLMVUHYZWKMMD-UHFFFAOYSA-N 3-trimethoxysilylpropyl 2-methylprop-2-enoate Chemical compound CO[Si](OC)(OC)CCCOC(=O)C(C)=C XDLMVUHYZWKMMD-UHFFFAOYSA-N 0.000 description 2
- YEJRWHAVMIAJKC-UHFFFAOYSA-N 4-Butyrolactone Chemical compound O=C1CCCO1 YEJRWHAVMIAJKC-UHFFFAOYSA-N 0.000 description 2
- SXIFAEWFOJETOA-UHFFFAOYSA-N 4-hydroxy-butyl Chemical group [CH2]CCCO SXIFAEWFOJETOA-UHFFFAOYSA-N 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- 239000004604 Blowing Agent Substances 0.000 description 2
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 229920002284 Cellulose triacetate Polymers 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 229920001609 Poly(3,4-ethylenedioxythiophene) Polymers 0.000 description 2
- 229920002873 Polyethylenimine Polymers 0.000 description 2
- 239000004721 Polyphenylene oxide Substances 0.000 description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 2
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 2
- QYKIQEUNHZKYBP-UHFFFAOYSA-N Vinyl ether Chemical compound C=COC=C QYKIQEUNHZKYBP-UHFFFAOYSA-N 0.000 description 2
- NNLVGZFZQQXQNW-ADJNRHBOSA-N [(2r,3r,4s,5r,6s)-4,5-diacetyloxy-3-[(2s,3r,4s,5r,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6s)-4,5,6-triacetyloxy-2-(acetyloxymethyl)oxan-3-yl]oxyoxan-2-yl]methyl acetate Chemical compound O([C@@H]1O[C@@H]([C@H]([C@H](OC(C)=O)[C@H]1OC(C)=O)O[C@H]1[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O1)OC(C)=O)COC(=O)C)[C@@H]1[C@@H](COC(C)=O)O[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O NNLVGZFZQQXQNW-ADJNRHBOSA-N 0.000 description 2
- 125000002339 acetoacetyl group Chemical group O=C([*])C([H])([H])C(=O)C([H])([H])[H] 0.000 description 2
- 150000008065 acid anhydrides Chemical class 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 125000002723 alicyclic group Chemical group 0.000 description 2
- XXROGKLTLUQVRX-UHFFFAOYSA-N allyl alcohol Chemical compound OCC=C XXROGKLTLUQVRX-UHFFFAOYSA-N 0.000 description 2
- XYLMUPLGERFSHI-UHFFFAOYSA-N alpha-Methylstyrene Chemical compound CC(=C)C1=CC=CC=C1 XYLMUPLGERFSHI-UHFFFAOYSA-N 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 239000012164 animal wax Substances 0.000 description 2
- 239000002518 antifoaming agent Substances 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 238000007611 bar coating method Methods 0.000 description 2
- 125000002511 behenyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 238000005452 bending Methods 0.000 description 2
- QMKYBPDZANOJGF-UHFFFAOYSA-N benzene-1,3,5-tricarboxylic acid Chemical compound OC(=O)C1=CC(C(O)=O)=CC(C(O)=O)=C1 QMKYBPDZANOJGF-UHFFFAOYSA-N 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 229920001400 block copolymer Polymers 0.000 description 2
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 2
- 239000004359 castor oil Substances 0.000 description 2
- 235000019438 castor oil Nutrition 0.000 description 2
- 238000006482 condensation reaction Methods 0.000 description 2
- 238000011109 contamination Methods 0.000 description 2
- 238000003851 corona treatment Methods 0.000 description 2
- 230000001186 cumulative effect Effects 0.000 description 2
- 238000007766 curtain coating Methods 0.000 description 2
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 230000002542 deteriorative effect Effects 0.000 description 2
- AYOHIQLKSOJJQH-UHFFFAOYSA-N dibutyltin Chemical compound CCCC[Sn]CCCC AYOHIQLKSOJJQH-UHFFFAOYSA-N 0.000 description 2
- NZZFYRREKKOMAT-UHFFFAOYSA-N diiodomethane Chemical compound ICI NZZFYRREKKOMAT-UHFFFAOYSA-N 0.000 description 2
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 125000005388 dimethylhydrogensiloxy group Chemical group 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 125000003700 epoxy group Chemical group 0.000 description 2
- UIWXSTHGICQLQT-UHFFFAOYSA-N ethenyl propanoate Chemical compound CCC(=O)OC=C UIWXSTHGICQLQT-UHFFFAOYSA-N 0.000 description 2
- NKSJNEHGWDZZQF-UHFFFAOYSA-N ethenyl(trimethoxy)silane Chemical compound CO[Si](OC)(OC)C=C NKSJNEHGWDZZQF-UHFFFAOYSA-N 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 239000001530 fumaric acid Substances 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- JFCQEDHGNNZCLN-UHFFFAOYSA-N glutaric acid Chemical class OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 2
- 125000006038 hexenyl group Chemical group 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 238000007373 indentation Methods 0.000 description 2
- 230000001678 irradiating effect Effects 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 239000002346 layers by function Substances 0.000 description 2
- 239000002075 main ingredient Substances 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 125000005395 methacrylic acid group Chemical group 0.000 description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 2
- LVHBHZANLOWSRM-UHFFFAOYSA-N methylenebutanedioic acid Natural products OC(=O)CC(=C)C(O)=O LVHBHZANLOWSRM-UHFFFAOYSA-N 0.000 description 2
- 239000004200 microcrystalline wax Substances 0.000 description 2
- 235000019808 microcrystalline wax Nutrition 0.000 description 2
- 230000001617 migratory effect Effects 0.000 description 2
- 239000012184 mineral wax Substances 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 239000012170 montan wax Substances 0.000 description 2
- BDJRBEYXGGNYIS-UHFFFAOYSA-N nonanedioic acid Chemical compound OC(=O)CCCCCCCC(O)=O BDJRBEYXGGNYIS-UHFFFAOYSA-N 0.000 description 2
- OEIJHBUUFURJLI-UHFFFAOYSA-N octane-1,8-diol Chemical compound OCCCCCCCCO OEIJHBUUFURJLI-UHFFFAOYSA-N 0.000 description 2
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 239000011146 organic particle Substances 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 150000002918 oxazolines Chemical class 0.000 description 2
- 235000013873 oxidized polyethylene wax Nutrition 0.000 description 2
- 239000012188 paraffin wax Substances 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- 239000012169 petroleum derived wax Substances 0.000 description 2
- 235000019381 petroleum wax Nutrition 0.000 description 2
- 239000005011 phenolic resin Substances 0.000 description 2
- WLJVNTCWHIRURA-UHFFFAOYSA-N pimelic acid Chemical compound OC(=O)CCCCCC(O)=O WLJVNTCWHIRURA-UHFFFAOYSA-N 0.000 description 2
- 238000009832 plasma treatment Methods 0.000 description 2
- 229920000570 polyether Polymers 0.000 description 2
- 229920000139 polyethylene terephthalate Polymers 0.000 description 2
- 239000005020 polyethylene terephthalate Substances 0.000 description 2
- 239000005056 polyisocyanate Substances 0.000 description 2
- 229920001228 polyisocyanate Polymers 0.000 description 2
- 229920002959 polymer blend Polymers 0.000 description 2
- 229920001843 polymethylhydrosiloxane Polymers 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- HJWLCRVIBGQPNF-UHFFFAOYSA-N prop-2-enylbenzene Chemical compound C=CCC1=CC=CC=C1 HJWLCRVIBGQPNF-UHFFFAOYSA-N 0.000 description 2
- CYIDZMCFTVVTJO-UHFFFAOYSA-N pyromellitic acid Chemical compound OC(=O)C1=CC(C(O)=O)=C(C(O)=O)C=C1C(O)=O CYIDZMCFTVVTJO-UHFFFAOYSA-N 0.000 description 2
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 239000011342 resin composition Substances 0.000 description 2
- 238000007127 saponification reaction Methods 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 239000013464 silicone adhesive Substances 0.000 description 2
- 238000007711 solidification Methods 0.000 description 2
- 230000008023 solidification Effects 0.000 description 2
- 125000004079 stearyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 150000003440 styrenes Chemical class 0.000 description 2
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical compound OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 238000004381 surface treatment Methods 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 239000002562 thickening agent Substances 0.000 description 2
- DVKJHBMWWAPEIU-UHFFFAOYSA-N toluene 2,4-diisocyanate Chemical compound CC1=CC=C(N=C=O)C=C1N=C=O DVKJHBMWWAPEIU-UHFFFAOYSA-N 0.000 description 2
- ZIBGPFATKBEMQZ-UHFFFAOYSA-N triethylene glycol Chemical compound OCCOCCOCCO ZIBGPFATKBEMQZ-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 239000012178 vegetable wax Substances 0.000 description 2
- DTGKSKDOIYIVQL-WEDXCCLWSA-N (+)-borneol Chemical group C1C[C@@]2(C)[C@@H](O)C[C@@H]1C2(C)C DTGKSKDOIYIVQL-WEDXCCLWSA-N 0.000 description 1
- DNIAPMSPPWPWGF-VKHMYHEASA-N (+)-propylene glycol Chemical compound C[C@H](O)CO DNIAPMSPPWPWGF-VKHMYHEASA-N 0.000 description 1
- WYTZZXDRDKSJID-UHFFFAOYSA-N (3-aminopropyl)triethoxysilane Chemical compound CCO[Si](OCC)(OCC)CCCN WYTZZXDRDKSJID-UHFFFAOYSA-N 0.000 description 1
- SZCWBURCISJFEZ-UHFFFAOYSA-N (3-hydroxy-2,2-dimethylpropyl) 3-hydroxy-2,2-dimethylpropanoate Chemical compound OCC(C)(C)COC(=O)C(C)(C)CO SZCWBURCISJFEZ-UHFFFAOYSA-N 0.000 description 1
- GHKBRNPRJNJGIM-SNAWJCMRSA-N (e)-4-butoxy-4-oxobut-2-eneperoxoic acid Chemical compound CCCCOC(=O)\C=C\C(=O)OO GHKBRNPRJNJGIM-SNAWJCMRSA-N 0.000 description 1
- NNOZGCICXAYKLW-UHFFFAOYSA-N 1,2-bis(2-isocyanatopropan-2-yl)benzene Chemical group O=C=NC(C)(C)C1=CC=CC=C1C(C)(C)N=C=O NNOZGCICXAYKLW-UHFFFAOYSA-N 0.000 description 1
- CHRJZRDFSQHIFI-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;styrene Chemical compound C=CC1=CC=CC=C1.C=CC1=CC=CC=C1C=C CHRJZRDFSQHIFI-UHFFFAOYSA-N 0.000 description 1
- FKTHNVSLHLHISI-UHFFFAOYSA-N 1,2-bis(isocyanatomethyl)benzene Chemical compound O=C=NCC1=CC=CC=C1CN=C=O FKTHNVSLHLHISI-UHFFFAOYSA-N 0.000 description 1
- MTZUIIAIAKMWLI-UHFFFAOYSA-N 1,2-diisocyanatobenzene Chemical compound O=C=NC1=CC=CC=C1N=C=O MTZUIIAIAKMWLI-UHFFFAOYSA-N 0.000 description 1
- ZXHZWRZAWJVPIC-UHFFFAOYSA-N 1,2-diisocyanatonaphthalene Chemical compound C1=CC=CC2=C(N=C=O)C(N=C=O)=CC=C21 ZXHZWRZAWJVPIC-UHFFFAOYSA-N 0.000 description 1
- ZGDSDWSIFQBAJS-UHFFFAOYSA-N 1,2-diisocyanatopropane Chemical group O=C=NC(C)CN=C=O ZGDSDWSIFQBAJS-UHFFFAOYSA-N 0.000 description 1
- PXGZQGDTEZPERC-UHFFFAOYSA-N 1,4-cyclohexanedicarboxylic acid Chemical compound OC(=O)C1CCC(C(O)=O)CC1 PXGZQGDTEZPERC-UHFFFAOYSA-N 0.000 description 1
- VZXPHDGHQXLXJC-UHFFFAOYSA-N 1,6-diisocyanato-5,6-dimethylheptane Chemical group O=C=NC(C)(C)C(C)CCCCN=C=O VZXPHDGHQXLXJC-UHFFFAOYSA-N 0.000 description 1
- PWGJDPKCLMLPJW-UHFFFAOYSA-N 1,8-diaminooctane Chemical compound NCCCCCCCCN PWGJDPKCLMLPJW-UHFFFAOYSA-N 0.000 description 1
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 1
- ZTEHOZMYMCEYRM-UHFFFAOYSA-N 1-chlorodecane Chemical compound CCCCCCCCCCCl ZTEHOZMYMCEYRM-UHFFFAOYSA-N 0.000 description 1
- OACXFSZVCDOBKF-UHFFFAOYSA-N 1-chlorodocosane Chemical compound CCCCCCCCCCCCCCCCCCCCCCCl OACXFSZVCDOBKF-UHFFFAOYSA-N 0.000 description 1
- YAYNEUUHHLGGAH-UHFFFAOYSA-N 1-chlorododecane Chemical compound CCCCCCCCCCCCCl YAYNEUUHHLGGAH-UHFFFAOYSA-N 0.000 description 1
- MLRVZFYXUZQSRU-UHFFFAOYSA-N 1-chlorohexane Chemical compound CCCCCCCl MLRVZFYXUZQSRU-UHFFFAOYSA-N 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical compound CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- CNDHHGUSRIZDSL-UHFFFAOYSA-N 1-chlorooctane Chemical compound CCCCCCCCCl CNDHHGUSRIZDSL-UHFFFAOYSA-N 0.000 description 1
- QIVRKSLMUAURGI-UHFFFAOYSA-N 1-isocyanato-1-[2-(1-isocyanatocyclohexyl)propan-2-yl]cyclohexane Chemical compound C1CCCCC1(N=C=O)C(C)(C)C1(N=C=O)CCCCC1 QIVRKSLMUAURGI-UHFFFAOYSA-N 0.000 description 1
- XFEWMFDVBLLXFE-UHFFFAOYSA-N 1-isocyanatodecane Chemical compound CCCCCCCCCCN=C=O XFEWMFDVBLLXFE-UHFFFAOYSA-N 0.000 description 1
- ODDFKRTWSQGENK-UHFFFAOYSA-N 1-isocyanatodocosane Chemical compound CCCCCCCCCCCCCCCCCCCCCCN=C=O ODDFKRTWSQGENK-UHFFFAOYSA-N 0.000 description 1
- YIDSTEJLDQMWBR-UHFFFAOYSA-N 1-isocyanatododecane Chemical compound CCCCCCCCCCCCN=C=O YIDSTEJLDQMWBR-UHFFFAOYSA-N 0.000 description 1
- DYQFCTCUULUMTQ-UHFFFAOYSA-N 1-isocyanatooctane Chemical compound CCCCCCCCN=C=O DYQFCTCUULUMTQ-UHFFFAOYSA-N 0.000 description 1
- RTBFRGCFXZNCOE-UHFFFAOYSA-N 1-methylsulfonylpiperidin-4-one Chemical compound CS(=O)(=O)N1CCC(=O)CC1 RTBFRGCFXZNCOE-UHFFFAOYSA-N 0.000 description 1
- XLPJNCYCZORXHG-UHFFFAOYSA-N 1-morpholin-4-ylprop-2-en-1-one Chemical compound C=CC(=O)N1CCOCC1 XLPJNCYCZORXHG-UHFFFAOYSA-N 0.000 description 1
- JCTXKRPTIMZBJT-UHFFFAOYSA-N 2,2,4-trimethylpentane-1,3-diol Chemical compound CC(C)C(O)C(C)(C)CO JCTXKRPTIMZBJT-UHFFFAOYSA-N 0.000 description 1
- JVYDLYGCSIHCMR-UHFFFAOYSA-N 2,2-bis(hydroxymethyl)butanoic acid Chemical compound CCC(CO)(CO)C(O)=O JVYDLYGCSIHCMR-UHFFFAOYSA-N 0.000 description 1
- DDHUNHGZUHZNKB-UHFFFAOYSA-N 2,2-dimethylpropane-1,3-diamine Chemical compound NCC(C)(C)CN DDHUNHGZUHZNKB-UHFFFAOYSA-N 0.000 description 1
- ZWNMRZQYWRLGMM-UHFFFAOYSA-N 2,5-dimethylhexane-2,5-diol Chemical compound CC(C)(O)CCC(C)(C)O ZWNMRZQYWRLGMM-UHFFFAOYSA-N 0.000 description 1
- LDXJRKWFNNFDSA-UHFFFAOYSA-N 2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)-1-[4-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidin-5-yl]piperazin-1-yl]ethanone Chemical compound C1CN(CC2=NNN=C21)CC(=O)N3CCN(CC3)C4=CN=C(N=C4)NCC5=CC(=CC=C5)OC(F)(F)F LDXJRKWFNNFDSA-UHFFFAOYSA-N 0.000 description 1
- HLIQLHSBZXDKLV-UHFFFAOYSA-N 2-(2-hydroxyethoxy)-1-phenoxyethanol Chemical compound OCCOCC(O)OC1=CC=CC=C1 HLIQLHSBZXDKLV-UHFFFAOYSA-N 0.000 description 1
- JAHNSTQSQJOJLO-UHFFFAOYSA-N 2-(3-fluorophenyl)-1h-imidazole Chemical compound FC1=CC=CC(C=2NC=CN=2)=C1 JAHNSTQSQJOJLO-UHFFFAOYSA-N 0.000 description 1
- OEPOKWHJYJXUGD-UHFFFAOYSA-N 2-(3-phenylmethoxyphenyl)-1,3-thiazole-4-carbaldehyde Chemical compound O=CC1=CSC(C=2C=C(OCC=3C=CC=CC=3)C=CC=2)=N1 OEPOKWHJYJXUGD-UHFFFAOYSA-N 0.000 description 1
- RWLALWYNXFYRGW-UHFFFAOYSA-N 2-Ethyl-1,3-hexanediol Chemical compound CCCC(O)C(CC)CO RWLALWYNXFYRGW-UHFFFAOYSA-N 0.000 description 1
- LCZVSXRMYJUNFX-UHFFFAOYSA-N 2-[2-(2-hydroxypropoxy)propoxy]propan-1-ol Chemical compound CC(O)COC(C)COC(C)CO LCZVSXRMYJUNFX-UHFFFAOYSA-N 0.000 description 1
- TXBCBTDQIULDIA-UHFFFAOYSA-N 2-[[3-hydroxy-2,2-bis(hydroxymethyl)propoxy]methyl]-2-(hydroxymethyl)propane-1,3-diol Chemical compound OCC(CO)(CO)COCC(CO)(CO)CO TXBCBTDQIULDIA-UHFFFAOYSA-N 0.000 description 1
- NDXNCTAJOQSKIO-UHFFFAOYSA-N 2-butyl-2-ethylpentane-1,5-diamine Chemical compound CCCCC(CC)(CN)CCCN NDXNCTAJOQSKIO-UHFFFAOYSA-N 0.000 description 1
- TUQYFIFPADMJFJ-UHFFFAOYSA-N 2-butyl-2-hexylpropane-1,3-diol Chemical compound CCCCCCC(CO)(CO)CCCC TUQYFIFPADMJFJ-UHFFFAOYSA-N 0.000 description 1
- IPJGPNZOIMZKEK-UHFFFAOYSA-N 2-ethyl-4-hydroxy-2-(2-hydroxyethyl)butanoic acid Chemical compound OCCC(CC)(CCO)C(O)=O IPJGPNZOIMZKEK-UHFFFAOYSA-N 0.000 description 1
- GWZMWHWAWHPNHN-UHFFFAOYSA-N 2-hydroxypropyl prop-2-enoate Chemical compound CC(O)COC(=O)C=C GWZMWHWAWHPNHN-UHFFFAOYSA-N 0.000 description 1
- DPNXHTDWGGVXID-UHFFFAOYSA-N 2-isocyanatoethyl prop-2-enoate Chemical compound C=CC(=O)OCCN=C=O DPNXHTDWGGVXID-UHFFFAOYSA-N 0.000 description 1
- JVZZUPJFERSVRN-UHFFFAOYSA-N 2-methyl-2-propylpropane-1,3-diol Chemical compound CCCC(C)(CO)CO JVZZUPJFERSVRN-UHFFFAOYSA-N 0.000 description 1
- CEBKHWWANWSNTI-UHFFFAOYSA-N 2-methylbut-3-yn-2-ol Chemical compound CC(C)(O)C#C CEBKHWWANWSNTI-UHFFFAOYSA-N 0.000 description 1
- SDQROPCSKIYYAV-UHFFFAOYSA-N 2-methyloctane-1,8-diol Chemical compound OCC(C)CCCCCCO SDQROPCSKIYYAV-UHFFFAOYSA-N 0.000 description 1
- JZUHIOJYCPIVLQ-UHFFFAOYSA-N 2-methylpentane-1,5-diamine Chemical compound NCC(C)CCCN JZUHIOJYCPIVLQ-UHFFFAOYSA-N 0.000 description 1
- AAAWJUMVTPNRDT-UHFFFAOYSA-N 2-methylpentane-1,5-diol Chemical compound OCC(C)CCCO AAAWJUMVTPNRDT-UHFFFAOYSA-N 0.000 description 1
- QWGRWMMWNDWRQN-UHFFFAOYSA-N 2-methylpropane-1,3-diol Chemical compound OCC(C)CO QWGRWMMWNDWRQN-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- RAADBCJYJHQQBI-UHFFFAOYSA-N 2-sulfoterephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C(S(O)(=O)=O)=C1 RAADBCJYJHQQBI-UHFFFAOYSA-N 0.000 description 1
- WMRCTEPOPAZMMN-UHFFFAOYSA-N 2-undecylpropanedioic acid Chemical compound CCCCCCCCCCCC(C(O)=O)C(O)=O WMRCTEPOPAZMMN-UHFFFAOYSA-N 0.000 description 1
- KGIGUEBEKRSTEW-UHFFFAOYSA-N 2-vinylpyridine Chemical compound C=CC1=CC=CC=N1 KGIGUEBEKRSTEW-UHFFFAOYSA-N 0.000 description 1
- NECRQCBKTGZNMH-UHFFFAOYSA-N 3,5-dimethylhex-1-yn-3-ol Chemical compound CC(C)CC(C)(O)C#C NECRQCBKTGZNMH-UHFFFAOYSA-N 0.000 description 1
- RNLHGQLZWXBQNY-UHFFFAOYSA-N 3-(aminomethyl)-3,5,5-trimethylcyclohexan-1-amine Chemical compound CC1(C)CC(N)CC(C)(CN)C1 RNLHGQLZWXBQNY-UHFFFAOYSA-N 0.000 description 1
- SDOFNQUTVKJYKH-UHFFFAOYSA-N 3-butoxycarbonylbut-3-eneperoxoic acid Chemical compound C(C(=C)CC(=O)OO)(=O)OCCCC SDOFNQUTVKJYKH-UHFFFAOYSA-N 0.000 description 1
- ZCNAVPLNWUDRJK-UHFFFAOYSA-N 3-hydroxy-2-(hydroxymethyl)-2-methylpropanoic acid;potassium Chemical compound [K].OCC(C)(CO)C(O)=O ZCNAVPLNWUDRJK-UHFFFAOYSA-N 0.000 description 1
- WHNPOQXWAMXPTA-UHFFFAOYSA-N 3-methylbut-2-enamide Chemical compound CC(C)=CC(N)=O WHNPOQXWAMXPTA-UHFFFAOYSA-N 0.000 description 1
- UUEWCQRISZBELL-UHFFFAOYSA-N 3-trimethoxysilylpropane-1-thiol Chemical compound CO[Si](OC)(OC)CCCS UUEWCQRISZBELL-UHFFFAOYSA-N 0.000 description 1
- KBQVDAIIQCXKPI-UHFFFAOYSA-N 3-trimethoxysilylpropyl prop-2-enoate Chemical compound CO[Si](OC)(OC)CCCOC(=O)C=C KBQVDAIIQCXKPI-UHFFFAOYSA-N 0.000 description 1
- UPMLOUAZCHDJJD-UHFFFAOYSA-N 4,4'-Diphenylmethane Diisocyanate Chemical compound C1=CC(N=C=O)=CC=C1CC1=CC=C(N=C=O)C=C1 UPMLOUAZCHDJJD-UHFFFAOYSA-N 0.000 description 1
- NEQFBGHQPUXOFH-UHFFFAOYSA-N 4-(4-carboxyphenyl)benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1C1=CC=C(C(O)=O)C=C1 NEQFBGHQPUXOFH-UHFFFAOYSA-N 0.000 description 1
- DNFUJUFGVPNZMP-UHFFFAOYSA-N 4-hydroxy-2-(2-hydroxyethyl)-2-methylbutanoic acid Chemical compound OCCC(C)(CCO)C(O)=O DNFUJUFGVPNZMP-UHFFFAOYSA-N 0.000 description 1
- SBVKVAIECGDBTC-UHFFFAOYSA-N 4-hydroxy-2-methylidenebutanamide Chemical compound NC(=O)C(=C)CCO SBVKVAIECGDBTC-UHFFFAOYSA-N 0.000 description 1
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 description 1
- NDWUBGAGUCISDV-UHFFFAOYSA-N 4-hydroxybutyl prop-2-enoate Chemical compound OCCCCOC(=O)C=C NDWUBGAGUCISDV-UHFFFAOYSA-N 0.000 description 1
- JYOZFNMFSVAZAW-UHFFFAOYSA-N 4-phenylbut-3-yn-2-ol Chemical compound CC(O)C#CC1=CC=CC=C1 JYOZFNMFSVAZAW-UHFFFAOYSA-N 0.000 description 1
- ZWAPMFBHEQZLGK-UHFFFAOYSA-N 5-(dimethylamino)-2-methylidenepentanamide Chemical compound CN(C)CCCC(=C)C(N)=O ZWAPMFBHEQZLGK-UHFFFAOYSA-N 0.000 description 1
- GLBHAWAMATUOBB-UHFFFAOYSA-N 6,6-dimethylheptane-1,1-diamine Chemical compound CC(C)(C)CCCCC(N)N GLBHAWAMATUOBB-UHFFFAOYSA-N 0.000 description 1
- GZVHEAJQGPRDLQ-UHFFFAOYSA-N 6-phenyl-1,3,5-triazine-2,4-diamine Chemical compound NC1=NC(N)=NC(C=2C=CC=CC=2)=N1 GZVHEAJQGPRDLQ-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 229920006310 Asahi-Kasei Polymers 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 229930185605 Bisphenol Natural products 0.000 description 1
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 241000283153 Cetacea Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 229920001634 Copolyester Polymers 0.000 description 1
- XMSXQFUHVRWGNA-UHFFFAOYSA-N Decamethylcyclopentasiloxane Chemical compound C[Si]1(C)O[Si](C)(C)O[Si](C)(C)O[Si](C)(C)O[Si](C)(C)O1 XMSXQFUHVRWGNA-UHFFFAOYSA-N 0.000 description 1
- OIFBSDVPJOWBCH-UHFFFAOYSA-N Diethyl carbonate Chemical compound CCOC(=O)OCC OIFBSDVPJOWBCH-UHFFFAOYSA-N 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 1
- 239000004716 Ethylene/acrylic acid copolymer Substances 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 1
- 239000005057 Hexamethylene diisocyanate Chemical group 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- NHTMVDHEPJAVLT-UHFFFAOYSA-N Isooctane Chemical compound CC(C)CC(C)(C)C NHTMVDHEPJAVLT-UHFFFAOYSA-N 0.000 description 1
- 239000005058 Isophorone diisocyanate Chemical group 0.000 description 1
- 239000004166 Lanolin Substances 0.000 description 1
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 1
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 1
- GYCMBHHDWRMZGG-UHFFFAOYSA-N Methylacrylonitrile Chemical compound CC(=C)C#N GYCMBHHDWRMZGG-UHFFFAOYSA-N 0.000 description 1
- CNCOEDDPFOAUMB-UHFFFAOYSA-N N-Methylolacrylamide Chemical compound OCNC(=O)C=C CNCOEDDPFOAUMB-UHFFFAOYSA-N 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- MKYBYDHXWVHEJW-UHFFFAOYSA-N N-[1-oxo-1-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propan-2-yl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(C(C)NC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 MKYBYDHXWVHEJW-UHFFFAOYSA-N 0.000 description 1
- NIPNSKYNPDTRPC-UHFFFAOYSA-N N-[2-oxo-2-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)ethyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 NIPNSKYNPDTRPC-UHFFFAOYSA-N 0.000 description 1
- AFCARXCZXQIEQB-UHFFFAOYSA-N N-[3-oxo-3-(2,4,6,7-tetrahydrotriazolo[4,5-c]pyridin-5-yl)propyl]-2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carboxamide Chemical compound O=C(CCNC(=O)C=1C=NC(=NC=1)NCC1=CC(=CC=C1)OC(F)(F)F)N1CC2=C(CC1)NN=N2 AFCARXCZXQIEQB-UHFFFAOYSA-N 0.000 description 1
- OMRDSWJXRLDPBB-UHFFFAOYSA-N N=C=O.N=C=O.C1CCCCC1 Chemical group N=C=O.N=C=O.C1CCCCC1 OMRDSWJXRLDPBB-UHFFFAOYSA-N 0.000 description 1
- HDONYZHVZVCMLR-UHFFFAOYSA-N N=C=O.N=C=O.CC1CCCCC1 Chemical group N=C=O.N=C=O.CC1CCCCC1 HDONYZHVZVCMLR-UHFFFAOYSA-N 0.000 description 1
- 229910017855 NH 4 F Inorganic materials 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 1
- ALQSHHUCVQOPAS-UHFFFAOYSA-N Pentane-1,5-diol Chemical compound OCCCCCO ALQSHHUCVQOPAS-UHFFFAOYSA-N 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- LGRFSURHDFAFJT-UHFFFAOYSA-N Phthalic anhydride Natural products C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 1
- 229920002845 Poly(methacrylic acid) Polymers 0.000 description 1
- 239000004962 Polyamide-imide Substances 0.000 description 1
- 239000004695 Polyether sulfone Substances 0.000 description 1
- 239000004642 Polyimide Substances 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- GOOHAUXETOMSMM-UHFFFAOYSA-N Propylene oxide Chemical compound CC1CO1 GOOHAUXETOMSMM-UHFFFAOYSA-N 0.000 description 1
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 1
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 1
- 229920001807 Urea-formaldehyde Polymers 0.000 description 1
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 1
- 229920002433 Vinyl chloride-vinyl acetate copolymer Polymers 0.000 description 1
- 229920004482 WACKER® Polymers 0.000 description 1
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 1
- GKXVJHDEWHKBFH-UHFFFAOYSA-N [2-(aminomethyl)phenyl]methanamine Chemical compound NCC1=CC=CC=C1CN GKXVJHDEWHKBFH-UHFFFAOYSA-N 0.000 description 1
- XMUZQOKACOLCSS-UHFFFAOYSA-N [2-(hydroxymethyl)phenyl]methanol Chemical compound OCC1=CC=CC=C1CO XMUZQOKACOLCSS-UHFFFAOYSA-N 0.000 description 1
- QLBRROYTTDFLDX-UHFFFAOYSA-N [3-(aminomethyl)cyclohexyl]methanamine Chemical compound NCC1CCCC(CN)C1 QLBRROYTTDFLDX-UHFFFAOYSA-N 0.000 description 1
- ISKQADXMHQSTHK-UHFFFAOYSA-N [4-(aminomethyl)phenyl]methanamine Chemical compound NCC1=CC=C(CN)C=C1 ISKQADXMHQSTHK-UHFFFAOYSA-N 0.000 description 1
- BWVAOONFBYYRHY-UHFFFAOYSA-N [4-(hydroxymethyl)phenyl]methanol Chemical compound OCC1=CC=C(CO)C=C1 BWVAOONFBYYRHY-UHFFFAOYSA-N 0.000 description 1
- MZVQCMJNVPIDEA-UHFFFAOYSA-N [CH2]CN(CC)CC Chemical group [CH2]CN(CC)CC MZVQCMJNVPIDEA-UHFFFAOYSA-N 0.000 description 1
- BJSBGAIKEORPFG-UHFFFAOYSA-N [[6-amino-1,2,3,4-tetramethoxy-4-(methoxyamino)-1,3,5-triazin-2-yl]-methoxyamino]methanol Chemical compound CONC1(N(C(N(C(=N1)N)OC)(N(CO)OC)OC)OC)OC BJSBGAIKEORPFG-UHFFFAOYSA-N 0.000 description 1
- VXPYSZQGFPMWQU-UHFFFAOYSA-L [acetyloxy(diphenyl)stannyl] acetate Chemical compound CC([O-])=O.CC([O-])=O.C=1C=CC=CC=1[Sn+2]C1=CC=CC=C1 VXPYSZQGFPMWQU-UHFFFAOYSA-L 0.000 description 1
- UKLDJPRMSDWDSL-UHFFFAOYSA-L [dibutyl(dodecanoyloxy)stannyl] dodecanoate Chemical compound CCCCCCCCCCCC(=O)O[Sn](CCCC)(CCCC)OC(=O)CCCCCCCCCCC UKLDJPRMSDWDSL-UHFFFAOYSA-L 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 150000003926 acrylamides Chemical class 0.000 description 1
- 150000001253 acrylic acids Chemical class 0.000 description 1
- HFBMWMNUJJDEQZ-UHFFFAOYSA-N acryloyl chloride Chemical compound ClC(=O)C=C HFBMWMNUJJDEQZ-UHFFFAOYSA-N 0.000 description 1
- 125000003647 acryloyl group Chemical group O=C([*])C([H])=C([H])[H] 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 229920000180 alkyd Polymers 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 150000001450 anions Chemical group 0.000 description 1
- 230000003712 anti-aging effect Effects 0.000 description 1
- 229940058905 antimony compound for treatment of leishmaniasis and trypanosomiasis Drugs 0.000 description 1
- 150000001463 antimony compounds Chemical class 0.000 description 1
- 150000004984 aromatic diamines Chemical class 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- AYJRCSIUFZENHW-DEQYMQKBSA-L barium(2+);oxomethanediolate Chemical compound [Ba+2].[O-][14C]([O-])=O AYJRCSIUFZENHW-DEQYMQKBSA-L 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 239000012965 benzophenone Substances 0.000 description 1
- 150000008366 benzophenones Chemical class 0.000 description 1
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 1
- 239000012964 benzotriazole Substances 0.000 description 1
- 150000001565 benzotriazoles Chemical class 0.000 description 1
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- YHWCPXVTRSHPNY-UHFFFAOYSA-N butan-1-olate;titanium(4+) Chemical compound [Ti+4].CCCC[O-].CCCC[O-].CCCC[O-].CCCC[O-] YHWCPXVTRSHPNY-UHFFFAOYSA-N 0.000 description 1
- BMRWNKZVCUKKSR-UHFFFAOYSA-N butane-1,2-diol Chemical compound CCC(O)CO BMRWNKZVCUKKSR-UHFFFAOYSA-N 0.000 description 1
- OWBTYPJTUOEWEK-UHFFFAOYSA-N butane-2,3-diol Chemical compound CC(O)C(C)O OWBTYPJTUOEWEK-UHFFFAOYSA-N 0.000 description 1
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- JHIWVOJDXOSYLW-UHFFFAOYSA-N butyl 2,2-difluorocyclopropane-1-carboxylate Chemical compound CCCCOC(=O)C1CC1(F)F JHIWVOJDXOSYLW-UHFFFAOYSA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 235000001465 calcium Nutrition 0.000 description 1
- 229940043430 calcium compound Drugs 0.000 description 1
- 150000001674 calcium compounds Chemical class 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 239000004204 candelilla wax Substances 0.000 description 1
- 235000013868 candelilla wax Nutrition 0.000 description 1
- 229940073532 candelilla wax Drugs 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 239000004203 carnauba wax Substances 0.000 description 1
- 235000013869 carnauba wax Nutrition 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 238000006757 chemical reactions by type Methods 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- PMMYEEVYMWASQN-IMJSIDKUSA-N cis-4-Hydroxy-L-proline Chemical compound O[C@@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-IMJSIDKUSA-N 0.000 description 1
- HNEGQIOMVPPMNR-IHWYPQMZSA-N citraconic acid Chemical compound OC(=O)C(/C)=C\C(O)=O HNEGQIOMVPPMNR-IHWYPQMZSA-N 0.000 description 1
- 229940018557 citraconic acid Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- LDHQCZJRKDOVOX-NSCUHMNNSA-N crotonic acid Chemical compound C\C=C\C(O)=O LDHQCZJRKDOVOX-NSCUHMNNSA-N 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- PDXRQENMIVHKPI-UHFFFAOYSA-N cyclohexane-1,1-diol Chemical compound OC1(O)CCCCC1 PDXRQENMIVHKPI-UHFFFAOYSA-N 0.000 description 1
- VKIRRGRTJUUZHS-UHFFFAOYSA-N cyclohexane-1,4-diamine Chemical compound NC1CCC(N)CC1 VKIRRGRTJUUZHS-UHFFFAOYSA-N 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- JGFBRKRYDCGYKD-UHFFFAOYSA-N dibutyl(oxo)tin Chemical compound CCCC[Sn](=O)CCCC JGFBRKRYDCGYKD-UHFFFAOYSA-N 0.000 description 1
- 239000012975 dibutyltin dilaurate Substances 0.000 description 1
- ZXDVQYBUEVYUCG-UHFFFAOYSA-N dibutyltin(2+);methanolate Chemical compound CCCC[Sn](OC)(OC)CCCC ZXDVQYBUEVYUCG-UHFFFAOYSA-N 0.000 description 1
- KEIQPMUPONZJJH-UHFFFAOYSA-N dicyclohexylmethanediamine Chemical compound C1CCCCC1C(N)(N)C1CCCCC1 KEIQPMUPONZJJH-UHFFFAOYSA-N 0.000 description 1
- 238000007607 die coating method Methods 0.000 description 1
- 150000001993 dienes Chemical class 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- OTARVPUIYXHRRB-UHFFFAOYSA-N diethoxy-methyl-[3-(oxiran-2-ylmethoxy)propyl]silane Chemical compound CCO[Si](C)(OCC)CCCOCC1CO1 OTARVPUIYXHRRB-UHFFFAOYSA-N 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 125000005442 diisocyanate group Chemical group 0.000 description 1
- KIQKWYUGPPFMBV-UHFFFAOYSA-N diisocyanatomethane Chemical group O=C=NCN=C=O KIQKWYUGPPFMBV-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- IEJIGPNLZYLLBP-UHFFFAOYSA-N dimethyl carbonate Chemical compound COC(=O)OC IEJIGPNLZYLLBP-UHFFFAOYSA-N 0.000 description 1
- 239000004205 dimethyl polysiloxane Substances 0.000 description 1
- JVSWJIKNEAIKJW-UHFFFAOYSA-N dimethyl-hexane Natural products CCCCCC(C)C JVSWJIKNEAIKJW-UHFFFAOYSA-N 0.000 description 1
- 229920005645 diorganopolysiloxane polymer Polymers 0.000 description 1
- ROORDVPLFPIABK-UHFFFAOYSA-N diphenyl carbonate Chemical compound C=1C=CC=CC=1OC(=O)OC1=CC=CC=C1 ROORDVPLFPIABK-UHFFFAOYSA-N 0.000 description 1
- ZZTCPWRAHWXWCH-UHFFFAOYSA-N diphenylmethanediamine Chemical compound C=1C=CC=CC=1C(N)(N)C1=CC=CC=C1 ZZTCPWRAHWXWCH-UHFFFAOYSA-N 0.000 description 1
- SZXQTJUDPRGNJN-UHFFFAOYSA-N dipropylene glycol Chemical compound OCCCOCCCO SZXQTJUDPRGNJN-UHFFFAOYSA-N 0.000 description 1
- 238000002845 discoloration Methods 0.000 description 1
- 238000005401 electroluminescence Methods 0.000 description 1
- 238000001227 electron beam curing Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- AFSIMBWBBOJPJG-UHFFFAOYSA-N ethenyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC=C AFSIMBWBBOJPJG-UHFFFAOYSA-N 0.000 description 1
- 125000001033 ether group Chemical group 0.000 description 1
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 description 1
- 238000007765 extrusion coating Methods 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 238000009501 film coating Methods 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 238000001879 gelation Methods 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 229920000578 graft copolymer Polymers 0.000 description 1
- 238000010559 graft polymerization reaction Methods 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 238000013007 heat curing Methods 0.000 description 1
- 239000012760 heat stabilizer Substances 0.000 description 1
- IUJAMGNYPWYUPM-UHFFFAOYSA-N hentriacontane Chemical compound CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC IUJAMGNYPWYUPM-UHFFFAOYSA-N 0.000 description 1
- YCOZIPAWZNQLMR-UHFFFAOYSA-N heptane - octane Natural products CCCCCCCCCCCCCCC YCOZIPAWZNQLMR-UHFFFAOYSA-N 0.000 description 1
- SXCBDZAEHILGLM-UHFFFAOYSA-N heptane-1,7-diol Chemical compound OCCCCCCCO SXCBDZAEHILGLM-UHFFFAOYSA-N 0.000 description 1
- CGQIJXYITMTOBI-UHFFFAOYSA-N hex-5-enyl(trimethoxy)silane Chemical compound CO[Si](OC)(OC)CCCCC=C CGQIJXYITMTOBI-UHFFFAOYSA-N 0.000 description 1
- HTDJPCNNEPUOOQ-UHFFFAOYSA-N hexamethylcyclotrisiloxane Chemical compound C[Si]1(C)O[Si](C)(C)O[Si](C)(C)O1 HTDJPCNNEPUOOQ-UHFFFAOYSA-N 0.000 description 1
- RRAMGCGOFNQTLD-UHFFFAOYSA-N hexamethylene diisocyanate Chemical group O=C=NCCCCCCN=C=O RRAMGCGOFNQTLD-UHFFFAOYSA-N 0.000 description 1
- SYECJBOWSGTPLU-UHFFFAOYSA-N hexane-1,1-diamine Chemical compound CCCCCC(N)N SYECJBOWSGTPLU-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-M hexanoate Chemical compound CCCCCC([O-])=O FUZZWVXGSFPDMH-UHFFFAOYSA-M 0.000 description 1
- ANJPRQPHZGHVQB-UHFFFAOYSA-N hexyl isocyanate Chemical compound CCCCCCN=C=O ANJPRQPHZGHVQB-UHFFFAOYSA-N 0.000 description 1
- 150000003949 imides Chemical class 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 239000010954 inorganic particle Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 229920000554 ionomer Polymers 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- XJRAOMZCVTUHFI-UHFFFAOYSA-N isocyanic acid;methane Chemical compound C.N=C=O.N=C=O XJRAOMZCVTUHFI-UHFFFAOYSA-N 0.000 description 1
- NIMLQBUJDJZYEJ-UHFFFAOYSA-N isophorone diisocyanate Chemical group CC1(C)CC(N=C=O)CC(C)(CN=C=O)C1 NIMLQBUJDJZYEJ-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229940119170 jojoba wax Drugs 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 229940039717 lanolin Drugs 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 150000002681 magnesium compounds Chemical class 0.000 description 1
- GVALZJMUIHGIMD-UHFFFAOYSA-H magnesium phosphate Chemical compound [Mg+2].[Mg+2].[Mg+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O GVALZJMUIHGIMD-UHFFFAOYSA-H 0.000 description 1
- 239000004137 magnesium phosphate Substances 0.000 description 1
- 229910000157 magnesium phosphate Inorganic materials 0.000 description 1
- 229960002261 magnesium phosphate Drugs 0.000 description 1
- 235000010994 magnesium phosphates Nutrition 0.000 description 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 1
- 150000002697 manganese compounds Chemical class 0.000 description 1
- 238000000691 measurement method Methods 0.000 description 1
- JDSHMPZPIAZGSV-UHFFFAOYSA-N melamine Chemical compound NC1=NC(N)=NC(N)=N1 JDSHMPZPIAZGSV-UHFFFAOYSA-N 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 150000002736 metal compounds Chemical class 0.000 description 1
- 229910001507 metal halide Inorganic materials 0.000 description 1
- 150000005309 metal halides Chemical class 0.000 description 1
- RBQRWNWVPQDTJJ-UHFFFAOYSA-N methacryloyloxyethyl isocyanate Chemical compound CC(=C)C(=O)OCCN=C=O RBQRWNWVPQDTJJ-UHFFFAOYSA-N 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- AYLRODJJLADBOB-QMMMGPOBSA-N methyl (2s)-2,6-diisocyanatohexanoate Chemical group COC(=O)[C@@H](N=C=O)CCCCN=C=O AYLRODJJLADBOB-QMMMGPOBSA-N 0.000 description 1
- ICCDZMWNLNRHGP-UHFFFAOYSA-N methyl-[3-(oxiran-2-ylmethoxy)propyl]-bis(prop-1-en-2-yloxy)silane Chemical compound CC(=C)O[Si](C)(OC(C)=C)CCCOCC1CO1 ICCDZMWNLNRHGP-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- PYLWMHQQBFSUBP-UHFFFAOYSA-N monofluorobenzene Chemical compound FC1=CC=CC=C1 PYLWMHQQBFSUBP-UHFFFAOYSA-N 0.000 description 1
- OMNKZBIFPJNNIO-UHFFFAOYSA-N n-(2-methyl-4-oxopentan-2-yl)prop-2-enamide Chemical compound CC(=O)CC(C)(C)NC(=O)C=C OMNKZBIFPJNNIO-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- DFFZOPXDTCDZDP-UHFFFAOYSA-N naphthalene-1,5-dicarboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1C(O)=O DFFZOPXDTCDZDP-UHFFFAOYSA-N 0.000 description 1
- VAWFFNJAPKXVPH-UHFFFAOYSA-N naphthalene-1,6-dicarboxylic acid Chemical compound OC(=O)C1=CC=CC2=CC(C(=O)O)=CC=C21 VAWFFNJAPKXVPH-UHFFFAOYSA-N 0.000 description 1
- WPUMVKJOWWJPRK-UHFFFAOYSA-N naphthalene-2,7-dicarboxylic acid Chemical compound C1=CC(C(O)=O)=CC2=CC(C(=O)O)=CC=C21 WPUMVKJOWWJPRK-UHFFFAOYSA-N 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- SXJVFQLYZSNZBT-UHFFFAOYSA-N nonane-1,9-diamine Chemical compound NCCCCCCCCCN SXJVFQLYZSNZBT-UHFFFAOYSA-N 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- HMMGMWAXVFQUOA-UHFFFAOYSA-N octamethylcyclotetrasiloxane Chemical compound C[Si]1(C)O[Si](C)(C)O[Si](C)(C)O[Si](C)(C)O1 HMMGMWAXVFQUOA-UHFFFAOYSA-N 0.000 description 1
- 125000004365 octenyl group Chemical group C(=CCCCCCC)* 0.000 description 1
- 238000010943 off-gassing Methods 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000004812 organic fluorine compounds Chemical class 0.000 description 1
- 229920006136 organohydrogenpolysiloxane Polymers 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000004209 oxidized polyethylene wax Substances 0.000 description 1
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 1
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 description 1
- 239000005022 packaging material Substances 0.000 description 1
- 239000003973 paint Substances 0.000 description 1
- 239000000123 paper Substances 0.000 description 1
- 235000019809 paraffin wax Nutrition 0.000 description 1
- UCUUFSAXZMGPGH-UHFFFAOYSA-N penta-1,4-dien-3-one Chemical compound C=CC(=O)C=C UCUUFSAXZMGPGH-UHFFFAOYSA-N 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- 150000003009 phosphonic acids Chemical class 0.000 description 1
- 150000003016 phosphoric acids Chemical class 0.000 description 1
- 238000000016 photochemical curing Methods 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 229920000233 poly(alkylene oxides) Chemical group 0.000 description 1
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 1
- 229920002312 polyamide-imide Polymers 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920006393 polyether sulfone Polymers 0.000 description 1
- 229920001721 polyimide Polymers 0.000 description 1
- 229920001444 polymaleic acid Polymers 0.000 description 1
- 239000002685 polymerization catalyst Substances 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 229920000098 polyolefin Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920000909 polytetrahydrofuran Polymers 0.000 description 1
- 229920000166 polytrimethylene carbonate Polymers 0.000 description 1
- 229920003009 polyurethane dispersion Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- AXLMPTNTPOWPLT-UHFFFAOYSA-N prop-2-enyl 3-oxobutanoate Chemical compound CC(=O)CC(=O)OCC=C AXLMPTNTPOWPLT-UHFFFAOYSA-N 0.000 description 1
- AOHJOMMDDJHIJH-UHFFFAOYSA-N propylenediamine Chemical compound CC(N)CN AOHJOMMDDJHIJH-UHFFFAOYSA-N 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000010526 radical polymerization reaction Methods 0.000 description 1
- 239000002683 reaction inhibitor Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 239000005871 repellent Substances 0.000 description 1
- 230000001846 repelling effect Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000007763 reverse roll coating Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- 229910000077 silane Inorganic materials 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 150000003377 silicon compounds Chemical class 0.000 description 1
- 229920002545 silicone oil Polymers 0.000 description 1
- LMIYOXCFQPAFTN-UHFFFAOYSA-M sodium;3-hydroxy-2-(hydroxymethyl)propane-1-sulfonate Chemical compound [Na+].OCC(CO)CS([O-])(=O)=O LMIYOXCFQPAFTN-UHFFFAOYSA-M 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 230000001360 synchronised effect Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000010345 tape casting Methods 0.000 description 1
- 150000003505 terpenes Chemical class 0.000 description 1
- 235000007586 terpenes Nutrition 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- VOZKAJLKRJDJLL-UHFFFAOYSA-N tolylenediamine group Chemical group CC1=C(C=C(C=C1)N)N VOZKAJLKRJDJLL-UHFFFAOYSA-N 0.000 description 1
- LDHQCZJRKDOVOX-UHFFFAOYSA-N trans-crotonic acid Natural products CC=CC(O)=O LDHQCZJRKDOVOX-UHFFFAOYSA-N 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- JXUKBNICSRJFAP-UHFFFAOYSA-N triethoxy-[3-(oxiran-2-ylmethoxy)propyl]silane Chemical compound CCO[Si](OCC)(OCC)CCCOCC1CO1 JXUKBNICSRJFAP-UHFFFAOYSA-N 0.000 description 1
- SRPWOOOHEPICQU-UHFFFAOYSA-N trimellitic anhydride Chemical compound OC(=O)C1=CC=C2C(=O)OC(=O)C2=C1 SRPWOOOHEPICQU-UHFFFAOYSA-N 0.000 description 1
- JLGNHOJUQFHYEZ-UHFFFAOYSA-N trimethoxy(3,3,3-trifluoropropyl)silane Chemical compound CO[Si](OC)(OC)CCC(F)(F)F JLGNHOJUQFHYEZ-UHFFFAOYSA-N 0.000 description 1
- DQZNLOXENNXVAD-UHFFFAOYSA-N trimethoxy-[2-(7-oxabicyclo[4.1.0]heptan-4-yl)ethyl]silane Chemical compound C1C(CC[Si](OC)(OC)OC)CCC2OC21 DQZNLOXENNXVAD-UHFFFAOYSA-N 0.000 description 1
- BPSIOYPQMFLKFR-UHFFFAOYSA-N trimethoxy-[3-(oxiran-2-ylmethoxy)propyl]silane Chemical compound CO[Si](OC)(OC)CCCOCC1CO1 BPSIOYPQMFLKFR-UHFFFAOYSA-N 0.000 description 1
- TZYULTYGSBAILI-UHFFFAOYSA-M trimethyl(prop-2-enyl)azanium;chloride Chemical compound [Cl-].C[N+](C)(C)CC=C TZYULTYGSBAILI-UHFFFAOYSA-M 0.000 description 1
- ZQYKGADTDCTWSZ-UHFFFAOYSA-N trimethyl-[(prop-2-enoylamino)methyl]azanium;chloride Chemical compound [Cl-].C[N+](C)(C)CNC(=O)C=C ZQYKGADTDCTWSZ-UHFFFAOYSA-N 0.000 description 1
- 229920001567 vinyl ester resin Polymers 0.000 description 1
- FJKIXWOMBXYWOQ-UHFFFAOYSA-N vinyl ethyl ether Natural products CCOC=C FJKIXWOMBXYWOQ-UHFFFAOYSA-N 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000002023 wood Substances 0.000 description 1
- 230000037303 wrinkles Effects 0.000 description 1
- 229910052726 zirconium Inorganic materials 0.000 description 1
- 229910001928 zirconium oxide Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J7/00—Adhesives in the form of films or foils
- C09J7/40—Adhesives in the form of films or foils characterised by release liners
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/36—Layered products comprising a layer of synthetic resin comprising polyesters
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B37/00—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding
- B32B37/14—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding characterised by the properties of the layers
- B32B37/15—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding characterised by the properties of the layers with at least one layer being manufactured and immediately laminated before reaching its stable state, e.g. in which a layer is extruded and laminated while in semi-molten state
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D133/00—Coating compositions based on homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Coating compositions based on derivatives of such polymers
- C09D133/04—Homopolymers or copolymers of esters
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D165/00—Coating compositions based on macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain; Coating compositions based on derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D167/00—Coating compositions based on polyesters obtained by reactions forming a carboxylic ester link in the main chain; Coating compositions based on derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D171/00—Coating compositions based on polyethers obtained by reactions forming an ether link in the main chain; Coating compositions based on derivatives of such polymers
- C09D171/08—Polyethers derived from hydroxy compounds or from their metallic derivatives
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D175/00—Coating compositions based on polyureas or polyurethanes; Coating compositions based on derivatives of such polymers
- C09D175/04—Polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D5/00—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes
- C09D5/24—Electrically-conducting paints
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J133/00—Adhesives based on homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Adhesives based on derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J183/00—Adhesives based on macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon, with or without sulfur, nitrogen, oxygen, or carbon only; Adhesives based on derivatives of such polymers
- C09J183/04—Polysiloxanes
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J191/00—Adhesives based on oils, fats or waxes; Adhesives based on derivatives thereof
- C09J191/06—Waxes
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J7/00—Adhesives in the form of films or foils
- C09J7/20—Adhesives in the form of films or foils characterised by their carriers
- C09J7/22—Plastics; Metallised plastics
- C09J7/25—Plastics; Metallised plastics based on macromolecular compounds obtained otherwise than by reactions involving only carbon-to-carbon unsaturated bonds
- C09J7/255—Polyesters
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J7/00—Adhesives in the form of films or foils
- C09J7/20—Adhesives in the form of films or foils characterised by their carriers
- C09J7/29—Laminated material
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J7/00—Adhesives in the form of films or foils
- C09J7/30—Adhesives in the form of films or foils characterised by the adhesive composition
- C09J7/38—Pressure-sensitive adhesives [PSA]
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B5/00—Optical elements other than lenses
- G02B5/30—Polarising elements
- G02B5/3025—Polarisers, i.e. arrangements capable of producing a definite output polarisation state from an unpolarised input state
-
- G—PHYSICS
- G06—COMPUTING; CALCULATING OR COUNTING
- G06F—ELECTRIC DIGITAL DATA PROCESSING
- G06F3/00—Input arrangements for transferring data to be processed into a form capable of being handled by the computer; Output arrangements for transferring data from processing unit to output unit, e.g. interface arrangements
- G06F3/01—Input arrangements or combined input and output arrangements for interaction between user and computer
- G06F3/03—Arrangements for converting the position or the displacement of a member into a coded form
- G06F3/041—Digitisers, e.g. for touch screens or touch pads, characterised by the transducing means
-
- G—PHYSICS
- G09—EDUCATION; CRYPTOGRAPHY; DISPLAY; ADVERTISING; SEALS
- G09F—DISPLAYING; ADVERTISING; SIGNS; LABELS OR NAME-PLATES; SEALS
- G09F9/00—Indicating arrangements for variable information in which the information is built-up on a support by selection or combination of individual elements
- G09F9/30—Indicating arrangements for variable information in which the information is built-up on a support by selection or combination of individual elements in which the desired character or characters are formed by combining individual elements
- G09F9/301—Indicating arrangements for variable information in which the information is built-up on a support by selection or combination of individual elements in which the desired character or characters are formed by combining individual elements flexible foldable or roll-able electronic displays, e.g. thin LCD, OLED
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B37/00—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding
- B32B37/14—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding characterised by the properties of the layers
- B32B37/24—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding characterised by the properties of the layers with at least one layer not being coherent before laminating, e.g. made up from granular material sprinkled onto a substrate
- B32B2037/243—Coating
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B37/00—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding
- B32B37/14—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding characterised by the properties of the layers
- B32B37/26—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding characterised by the properties of the layers with at least one layer which influences the bonding during the lamination process, e.g. release layers or pressure equalising layers
- B32B2037/268—Release layers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/20—Properties of the layers or laminate having particular electrical or magnetic properties, e.g. piezoelectric
- B32B2307/21—Anti-static
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2457/00—Electrical equipment
- B32B2457/20—Displays, e.g. liquid crystal displays, plasma displays
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J2203/00—Applications of adhesives in processes or use of adhesives in the form of films or foils
- C09J2203/318—Applications of adhesives in processes or use of adhesives in the form of films or foils for the production of liquid crystal displays
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J2301/00—Additional features of adhesives in the form of films or foils
- C09J2301/30—Additional features of adhesives in the form of films or foils characterized by the chemical, physicochemical or physical properties of the adhesive or the carrier
- C09J2301/312—Additional features of adhesives in the form of films or foils characterized by the chemical, physicochemical or physical properties of the adhesive or the carrier parameters being the characterizing feature
Definitions
- the present invention relates to a release film, a pressure-sensitive adhesive sheet, and a method of using the pressure-sensitive adhesive sheet.
- release films based on polyester films have excellent properties such as mechanical strength, dimensional stability, chemical resistance, and optical properties, and are excellent in cost performance, so they are used for various purposes.
- LCD liquid crystal displays
- PDP plasma display panels
- organic EL organic electroluminescence
- One way to make the release film extremely easy to release is to increase the thickness of the release layer to make it easier to release.
- Migration components originating from the layer may transfer to the surface of the adhesive layer, making it difficult to obtain the desired adhesive strength.
- Patent Document 3 a proposal has been made to provide a resin layer containing particles on the surface of the film opposite to the surface on which the release layer is provided.
- the resin layer may be added to the release layer during film roll formation.
- the unevenness of the particles contained in the mold is transferred, changing the release characteristics of the release layer, and it may be difficult to obtain a stable release force.
- the present invention was made in view of the above-mentioned circumstances, and the problem to be solved is to create a release film that has ultra-light releasability for various pressure-sensitive adhesives, yet does not easily change the release characteristics of the release layer due to blocking. , and to provide a film laminate.
- a release film comprising a resin layer (A) on one side of a base film and a resin layer (B) on the other side, and the resin layer (A) is a silicone resin-based release film.
- the release film has a thickness of 0.2 to 2.0 ⁇ m, and the resin layer (B) contains a non-silicone resin release agent.
- a resin layer (A) is provided on at least one side of the base film, and the resin layer (A) is a cured product of a resin layer composition containing a curable silicone resin and an easy release agent;
- the release agent has a dimethylsiloxane skeleton (DM) represented by the following formula (I) and a methylphenylsiloxane skeleton (MP) represented by the following formula (II), and has a thickness of 0.2 to 1.5 ⁇ m.
- DM dimethylsiloxane skeleton
- MP methylphenylsiloxane skeleton
- the surface of the base film opposite to the surface on which the resin layer (A) is provided is provided with a polyester layer containing 0.4 to 1.0% by mass of particles with an average particle size of 1 to 6 ⁇ m. , the release film according to [2] or [3] above.
- the resin layer (A) has an elastic modulus of 500 MPa or less at 25° C. as measured using a nanoindenter device.
- B Polyhydroxy Compound
- C One or more compounds selected from the group consisting of polyurethane resin, polyester resin, and acrylic resin [14] The above [1] and [6] to [13], wherein the surface resistivity of at least one of the resin layer (A) or the resin layer (B) is 1 ⁇ 10 12 ⁇ / ⁇ or less.
- the release film according to any one of the above.
- the peeling force obtained by peeling at 180° at a tensile speed of 0.3 m/min is defined as (F1).
- the resin layer (A) is laminated so as to be in contact with the surface thereof, and a press treatment is performed for 20 hours at a temperature of 40° C., a humidity of 90% RH, and a load of 1 MPa.
- a press treatment is performed for 20 hours at a temperature of 40° C., a humidity of 90% RH, and a load of 1 MPa.
- an acrylic adhesive tape No. 7475 manufactured by Tesa
- the peeling force obtained by peeling at 180° at a tensile speed of 0.3 m/min is defined as (F2).
- the rate of heavy peeling after pressing is determined by the following formula.
- the pressure-sensitive adhesive sheet according to [20] above, wherein the pressure-sensitive adhesive layer is formed from an acrylic pressure-sensitive adhesive composition.
- the release film of the present invention it is possible to provide a release film and a film laminate in which the release properties of the release layer are difficult to change due to blocking while achieving ultra-light release properties with respect to various adhesives. I can do it.
- a release film according to an example of an embodiment of the present invention includes a resin layer (A) on one surface of a base film, and a resin layer (A) on the other surface.
- the release film is preferably wound into a rolled release film (release film roll) and stored in the form of a release film roll. In that case, the release layer (A) may be placed on either the inside or outside of the roll.
- This release film is characterized in that the resin layer (A) on one side contains a silicone resin release agent.
- the thickness of the resin layer (A) is preferably 0.2 to 2.0 ⁇ m, and the elastic modulus of the resin layer (A) at 25° C. measured using a nanoindenter device is preferably 500 MPa or less.
- the resin layer (B) on the other film surface is characterized in that it contains a non-silicone resin mold release agent.
- the resin layer (A) To reduce damage caused by adhesion to the surface, suppress blocking, and prevent heavy peeling of the resin layer (A) and deterioration of the surface appearance at the lower part of the roll where more pressure is applied to the film. I can do it.
- the resin layer (A) achieves ultra-light releasability, it also has good adhesion to the base film because it does not substantially contain particles, and it also does not fall off, so it is suitable for adhesive tapes, etc. Migration can also be reduced.
- the material of the base film in the present release film is not particularly limited as long as it has a film shape.
- it may be made of paper, resin, metal, etc.
- resin is preferred from the viewpoint of mechanical strength and flexibility.
- the resin base film include films made of polymers such as polyethylene, polypropylene, polyester, polystyrene, polycarbonate, polyethersulfone, polyamide, and polyimide.
- polymers such as polyethylene, polypropylene, polyester, polystyrene, polycarbonate, polyethersulfone, polyamide, and polyimide.
- polyester films are particularly preferred because they have excellent physical properties such as heat resistance, flatness, optical properties, and strength.
- the polyester film constituting the laminated film of the present invention may have a single-layer structure or a multi-layer structure. It may be multi-layered and is not particularly limited. In the present invention, a polyester film having at least three layers is preferred. Further, as the polyester film, a biaxially stretched polyester film is preferable from the viewpoint of thinning and dimensional stability.
- the polyester used in the present invention may be a homopolyester or a copolyester.
- a homopolyester one obtained by polycondensing an aromatic dicarboxylic acid and an aliphatic glycol is preferable.
- aromatic dicarboxylic acids include terephthalic acid and 2,6-naphthalene dicarboxylic acid
- aliphatic glycols include ethylene glycol, diethylene glycol, and 1,4-cyclohexanedimethanol.
- Typical polyesters include polyethylene terephthalate and the like.
- the dicarboxylic acid component of the copolymerized polyester includes isophthalic acid, phthalic acid, terephthalic acid, 2,6-naphthalenedicarboxylic acid, adipic acid, sebacic acid, oxycarboxylic acid (for example, p-oxybenzoic acid, etc.), etc.
- the glycol component include one or more of ethylene glycol, diethylene glycol, propylene glycol, butanediol, 4-cyclohexanedimethanol, neopentyl glycol, and the like.
- examples include those obtained by polycondensing a dicarboxylic acid component and a glycol component.
- the dicarboxylic acid component one or more of isophthalic acid, phthalic acid, terephthalic acid, 2,6-naphthalene dicarboxylic acid, adipic acid, sebacic acid, oxycarboxylic acid (for example, p-oxybenzoic acid, etc.) is used.
- the glycol component include one or more of ethylene glycol, diethylene glycol, propylene glycol, butanediol, 4-cyclohexanedimethanol, neopentyl glycol, and the like.
- the copolymerized polyester is preferably a copolymer containing 30 mol % or less of a third component based on the total of the dicarboxylic acid component and the glycol component.
- the main component resin means the resin with the largest mass percentage among the resins constituting the present polyester film, and is 50% by mass or more, or 75% by mass or more, or 90% by mass or more of the resin constituting the present polyester film. This is the case where it accounts for more than 100% by mass or 100% by mass.
- the polymerization catalyst for the polyester film is not particularly limited, and conventionally known compounds can be used, such as antimony compounds, titanium compounds, germanium compounds, manganese compounds, aluminum compounds, magnesium compounds, calcium compounds, etc. It will be done. Among these, titanium compounds and germanium compounds are preferred because they have high catalytic activity, can be polymerized with a small amount, and have a small amount of metal remaining in the film, increasing the brightness of the film. Furthermore, since germanium compounds are expensive, titanium compounds are more preferred.
- the titanium element content is preferably 50 ppm or less, more preferably 1 to 20 ppm, and still more preferably 2 to 10 ppm. If the content of the titanium compound is too large, deterioration of the polyester may be accelerated during the process of melt extruding the polyester, resulting in a film with a strong yellowish tinge. Furthermore, if the content is too low, the polymerization efficiency may be poor, resulting in increased costs or a film having sufficient strength may not be obtained. Further, when using a polyester made of a titanium compound, it is preferable to use a phosphorus compound in order to lower the activity of the titanium compound for the purpose of suppressing deterioration in the melt extrusion process.
- the phosphorus element content is preferably in the range of 1 to 300 mass ppm, more preferably 3 to 200 mass ppm, and still more preferably 5 to 100 mass ppm, based on the amount of polyester to be melt-extruded. If the content of the phosphorus compound is below the above upper limit, it will not cause gelation or foreign matter, and if it is above the above lower limit, the activity of the titanium compound can be sufficiently lowered and coloration will be prevented. It can be suppressed and does not result in a yellowish film.
- the film may be manufactured using polyester with a low content of oligomer components as a raw material.
- polyester with a low content of oligomer components various known methods can be used, such as a method in which solid phase polymerization is performed after producing polyester.
- the amount of precipitation of the oligomer component may be suppressed by forming the polyester film to have a structure of three or more layers, and making the outermost layer of the polyester film a layer using a polyester raw material with a low content of the oligomer component.
- polyester may be obtained by performing esterification or transesterification reaction and then further increasing the reaction temperature and performing melt polycondensation under reduced pressure.
- the ultraviolet absorber is a compound that absorbs ultraviolet rays and is not particularly limited as long as it can withstand the heat added during the polyester film manufacturing process.
- organic ultraviolet absorbers there are organic ultraviolet absorbers and inorganic ultraviolet absorbers, and organic ultraviolet absorbers are preferable from the viewpoint of transparency.
- organic ultraviolet absorbers include, but are not limited to, cyclic iminoesters, benzotriazoles, benzophenones, and the like. From the viewpoint of durability, cyclic iminoester type and benzotriazole type are more preferable. It is also possible to use two or more types of ultraviolet absorbers in combination.
- particles are not particularly limited as long as they can impart slipperiness; specific examples include silica, calcium carbonate, magnesium carbonate, barium carbonate, and sulfuric acid. Examples include inorganic particles such as calcium, calcium phosphate, magnesium phosphate, kaolin, aluminum oxide, zirconium oxide, and titanium oxide, and organic particles such as acrylic resin, styrene resin, urea resin, phenol resin, epoxy resin, and benzoguanamine resin.
- precipitated particles in which a part of a metal compound such as a catalyst is precipitated and finely dispersed during the polyester manufacturing process can also be used.
- silica particles and calcium carbonate particles are particularly preferred because they are effective even in small amounts.
- the average particle size of the particles is preferably in the range of 0.01 to 5 ⁇ m, more preferably in the range of 0.03 to 4 ⁇ m, and still more preferably in the range of 0.05 to 3.0 ⁇ m.
- the average particle size is below the above upper limit, the haze of the film is suppressed to a low level, and the film has slipperiness.
- the particle content in the polyester layer of the polyester film is preferably 5% by mass or less, more preferably 0.001 to 3% by mass, even more preferably 0.01 to 1% by mass, particularly preferably 0. It is .05 to 0.5% by mass. If there are no particles or only a small number of particles, the film will have high transparency and will be a good film, but in order to prevent the slipperiness from decreasing, it is necessary to improve the slipperiness by incorporating particles into the coating layer. It may be necessary to take some measures. Moreover, when the particle content is below the above upper limit, the haze does not become high and sufficient transparency of the film can be ensured.
- the shape of the particles to be used is not particularly limited either, and any of the shapes such as spherical, lumpy, rod-like, flat, etc. can be used, and there are no particular limitations on their hardness, specific gravity, color, etc. Two or more types of these series of particles may be used in combination as necessary.
- the method of adding particles into the polyester layer is not particularly limited, and any conventionally known method may be employed. For example, it can be added at any stage of producing the polyester constituting each layer, but it is preferably added after the esterification or transesterification reaction is completed.
- the polyester film may have a multilayer structure of two or more layers, but a three-layer structure is preferable.
- the polyester layers (surface layers) on both surfaces may contain particles, and the intermediate layer may not contain particles. Therefore, it is possible to improve the transparency of the polyester film while improving slipperiness and anti-blocking properties.
- the polyester constituting each layer is as described above.
- the polyester layer on the surface opposite to the surface on which the mold release layer is provided (anti-mold release surface) (typically, on the side on which the mold release layer is not provided)
- the average particle diameter of the particles used in the polyester layer is preferably 1 to 6 ⁇ m, more preferably 2 to 5 ⁇ m, particularly preferably 3 to 5 ⁇ m.
- the content of particles in the polyester layer is, for example, 0.03 to 1.0% by mass, preferably 0.3 to 1.0% by mass, more preferably 0.4 to 1.0% by mass, and more preferably 0.4 to 1.0% by mass. Among these, 0.4 to 0.8% by mass is particularly preferred.
- the anti-mold release surface has a large maximum cross-sectional height (Rt) and has an appropriate uneven shape, and the release film has anti-blocking properties and release properties. It is possible to achieve both moldability and moldability.
- the single polyester layer may contain particles from the viewpoint of anti-blocking properties and mold release properties, and the average particle diameter and amount of particles added. It is best to set the range as shown above.
- the polyester layer (surface layer) on the side where the release layer is provided may or may not contain particles, but from the viewpoint of slipperiness etc. It is preferable to contain particles from the following.
- the particle content (mass%) in the polyester layer (surface layer) on the side where the release layer is provided is the particle content (% by mass) in the polyester layer on the side opposite to the side (anti-release side) where the release layer is provided. It is preferable that the amount is less than (mass%).
- the particle content (mass%) in the polyester layer on the surface on which the release layer is provided is preferably in the range of 0.001 to 0.8% by mass, more preferably in the range of 0.01 to 0.8% by mass.
- the amount is 5% by weight, more preferably 0.02 to 0.2% by weight.
- the thickness of the polyester film in the present invention is not particularly limited as long as it can be formed into a film, but is preferably 10 to 300 ⁇ m, more preferably 15 to 200 ⁇ m, still more preferably 25 to 125 ⁇ m, and most preferably is 38 to 75 ⁇ m.
- polyester raw material described above is first melted and extruded from a die using an extruder, and the molten sheet is cooled and solidified with a cooling roll to obtain an unstretched sheet.
- a cooling roll in order to improve the flatness of the sheet, it is preferable to increase the adhesion between the sheet and the rotating cooling drum, and an electrostatic application adhesion method or a liquid application adhesion method is preferably employed.
- the obtained unstretched sheet is stretched in one direction using a roll or tenter type stretching machine.
- the stretching temperature is usually 70 to 120°C, preferably 80 to 110°C, and the stretching ratio is usually 2.5 to 7 times, preferably 3.0 to 6 times.
- the film is stretched in a direction perpendicular to the first-stage stretching direction, usually at 70 to 170°C, and at a stretching ratio of usually 2.5 to 7 times, preferably 3.0 to 6 times.
- heat treatment is performed at a temperature of 180 to 270° C. under tension or under 30% relaxation to obtain a biaxially oriented film.
- a method of stretching in one direction in two or more stages can also be adopted. In that case, it is preferable to carry out the stretching so that the final stretching ratios in both directions fall within the above ranges.
- a simultaneous biaxial stretching method can also be employed for producing the polyester film.
- the simultaneous biaxial stretching method is a method in which the above-mentioned unstretched sheet is stretched and oriented simultaneously in the machine direction and the width direction under temperature control, usually at 70 to 120°C, preferably 80 to 110°C, and the stretching ratio is is usually 4 to 50 times, preferably 7 to 35 times, more preferably 10 to 25 times in area magnification. Subsequently, heat treatment is performed at a temperature of 170 to 270° C. under tension or relaxation within 30% to obtain a stretched oriented film.
- conventionally known stretching methods such as a screw method, a pantograph method, and a linear drive method can be employed.
- the resin layer (A) is a layer formed by curing the resin layer (A) composition whose main component is a curable silicone resin, and is disposed on at least one side of the base film described above.
- the resin layer (A) layer can also be said to be a release layer containing a cured product obtained by curing the resin layer (A) composition.
- main component resin means the resin with the largest mass percentage among the resins that make up the resin layer (A) composition, and the resin that makes up the resin layer (A) composition. It is assumed that the content is 50% by mass or more, or 75% by mass or more, or 90% by mass or more, or 100% by mass.
- the resin layer (A) in the present invention preferably contains a curable silicone resin.
- the curable silicone resin may be a resin whose main component is a curable silicone resin, or a modified silicone obtained by graft polymerization with an organic resin such as an acrylic resin, urethane resin, epoxy resin, or alkyd resin. good. Further, when the adhesive layer is made of a silicone adhesive or the like, it is preferable to contain a fluorosilicone resin or the like.
- any existing curing reaction type can be used, such as heat curing types such as addition type and condensation type, and electron beam curing types such as ultraviolet curing type.
- a type silicone resin may also be used in combination.
- the coating form of the curable silicone resin when forming the resin layer (A) there is no particular restriction on the coating form of the curable silicone resin when forming the resin layer (A), and it may be in the form of dissolved in an organic solvent, solvent-free form, or water-based emulsion form. good.
- Solvent-free curable silicone is a silicone resin with a viscosity that allows it to be coated without diluting it with a solvent, and is composed of short polysiloxane chains and has a relatively low molecular weight.
- solvent-based curable silicone is a silicone resin with such high viscosity that it cannot be coated without being diluted with a solvent, and has a relatively high molecular weight compared to solvent-free curable silicone. .
- Solvent-curable silicone is preferred from the viewpoints of good adhesion to the base film, uniform coating appearance with no coating unevenness, and ease of adjusting the thickness of the resin layer (A).
- curable silicone resin containing an alkenyl group examples include those represented by the following general formula (1) as diorganopolysiloxanes.
- R is a monovalent hydrocarbon group having 1 to 10 carbon atoms
- X is an alkenyl group-containing organic group.
- R is a monovalent hydrocarbon group having 1 to 10 carbon atoms, and specifically includes an alkyl group such as a methyl group, an ethyl group, a propyl group, a butyl group, a cycloalkyl group such as a cyclohexyl group, a phenyl group, and a tolyl group.
- alkyl group such as a methyl group, an ethyl group, a propyl group, a butyl group, a cycloalkyl group such as a cyclohexyl group, a phenyl group, and a tolyl group.
- Examples include aryl groups such as, among others, methyl and phenyl groups are particularly preferred.
- X is an alkenyl group-containing organic group preferably having 2 to 10 carbon atoms, specifically vinyl group, allyl group, hexenyl group, octenyl group, acryloylpropyl group, acryloylmethyl group, methacryloylpropyl group, cyclohexenylethyl group. group, vinyloxypropyl group, etc., and vinyl group, hexenyl group, etc. are particularly preferred.
- dimethylsiloxane/methylhexenylsiloxane copolymer with trimethylsiloxy groups endblocked at both molecular chain ends (96 mol% dimethylsiloxane units, 4 mol% methylhexenylsiloxane units), dimethyl endblocked with dimethylvinylsiloxy groups at both molecular chain ends.
- Siloxane/methylhexenylsiloxane copolymer (97 mol% dimethylsiloxane units, 3 mol% methylhexenylsiloxane units), dimethylsiloxane/methylhexenylsiloxane copolymer (95 mol% dimethylsiloxane units) blocked with dimethylhexenylsiloxy groups at both molecular chain ends , methylhexenylsiloxane units (5 mol%).
- the polyorganosiloxane containing SiH groups which is necessary to react with the curable silicone resin containing alkenyl groups and form a stronger silicone release layer, contains hydrogen bonded to silicon atoms in one molecule.
- Organohydrogenpolysiloxanes having at least 2 atoms, preferably 3 or more atoms, which can be linear, branched, cyclic, etc., include compounds represented by the following general formula (2) However, it is not limited to these.
- R 1 is a monovalent hydrocarbon group having 1 to 6 carbon atoms and containing no aliphatic unsaturated bond.
- b is an integer from 0 to 3
- x and y are each integers.
- Specific examples include methylhydrogenpolysiloxane with trimethylsiloxy groups blocked at both molecular chain ends, dimethylsiloxane/methylhydrogensiloxane copolymer with trimethylsiloxy groups blocked at both molecular chain ends, and methyl blocked with dimethylhydrogensiloxy groups at both molecular chain ends.
- Examples include hydrogen polysiloxane and dimethyl siloxane/methyl hydrogen siloxane copolymer with dimethyl hydrogen siloxy groups endblocked at both molecular chain ends.
- DEHESIVE 636 919, 920, 921, 924, and 929, but are not limited to these.
- the resin layer (A) uses a platinum-based catalyst that promotes addition-type reactions.
- This component includes platinum-based compounds such as chloroplatinic acid, alcoholic solution of chloroplatinic acid, complexes of chloroplatinic acid and olefins, complexes of chloroplatinic acid and alkenylsiloxane, platinum black, platinum-supported silica, and platinum-supported activated carbon. is exemplified.
- the content of the curing catalyst in the resin layer (A) is preferably 0.5 to 500 mass ppm as a metal equivalent amount with respect to the curable silicone resin, and more preferably 5 mass ppm or more. It is more preferably 10 mass ppm or more, more preferably 300 mass ppm or less, and even more preferably 200 mass ppm or less.
- the content of the platinum-based catalyst in the release layer is equal to or higher than the above lower limit, sufficient peeling force is obtained, the curing reaction proceeds sufficiently, and problems such as deterioration of the coat surface condition do not occur.
- the platinum-based catalyst content in the resin layer (A) is below the above upper limit, it is not only advantageous in terms of cost, but also increases reactivity and causes problems in the process such as generation of gel foreign matter. do not have.
- acetylene alcohol may be added as a reaction inhibitor in some cases.
- Its components are organic compounds having a carbon-carbon triple bond and a hydroxyl group, preferably 3-methyl-1-butyn-3-ol, 3,5-dimethyl-1-hexyn-3-ol and phenylbutyn-3-ol. It is a compound selected from the group consisting of nols.
- the content of the reaction control agent is preferably 0.001 to 5.0 parts by mass, more preferably 0.01 to 2.0 parts by mass, per 100 parts by mass of the total amount of the resin layer (A) composition (based on nonvolatile components). parts, more preferably 0.05 to 1.5 parts by weight, most preferably 0.1 to 0.5 parts by weight.
- a catalyst can be used in combination with the resin layer (A) constituting the release film for the purpose of promoting hydrolysis and condensation reactions.
- catalysts include organic acids such as acetic acid, butyric acid, maleic acid, and citric acid, inorganic acids such as hydrochloric acid, nitric acid, phosphoric acid, and sulfuric acid, basic compounds such as triethylamine, tetrabutyl titanate, dibutyltin dilaurate, Organic metal salts such as dibutyltin diacetate, dibutyltin dioctate, dibutyltin diolate, diphenyltin diacetate, dibutyltin oxide, dibutyltin dimethoxide, dibutylbis(triethoxysiloxy)tin, dibutyltinbenzyl maleate, KF, NH 4 F
- fluorine element-containing compounds such as.
- the above catalysts may be used alone or in combination of two or more.
- various peeling control agents may be used in combination.
- the content of organopolysiloxane resin, silica particles, silicone species with heavy peeling force, etc. is adjusted appropriately in the resin layer (A) in order to obtain the desired peeling force.
- various low-molecular-weight siloxanes are selected and the content is appropriately adjusted in the resin layer (A) so that the siloxane transfer component exhibits mold release performance.
- low molecular weight siloxane compounds include hexamethylcyclotrisiloxane, octamethylcyclotetrasiloxane, decamethylcyclopentasiloxane, and the like.
- other compounds of these low-molecular cyclic siloxanes include dimethylsiloxane oligomers with trimethylsiloxy groups endblocked at both ends of the molecular chain; dimethylsiloxane oligomers endblocked with dimethylhydroxysiloxy groups at both ends of the molecular chain; May be used in combination.
- the resin layer (A) composition may contain a light release agent if necessary, and has a dimethylsiloxane skeleton (DM) represented by the following formula (3) and a dimethylsiloxane skeleton (DM) represented by the following formula (4).
- Silicone oils having the methylphenylsiloxane skeleton (MP) shown are preferred. Since the light release agent has a dimethylsiloxane skeleton (DM) and a methylphenylsiloxane skeleton (MP), even if it migrates to the adhesive layer to be bonded, it can penetrate into the adhesive layer, reducing adhesive strength. can be reduced.
- the ratio (DM:MP) of the dimethylsiloxane skeleton (DM) represented by the following formula (3) and the methylphenylsiloxane skeleton (MP) represented by the following formula (4) is a molar ratio of 98:2 to 70:30. It is preferably in the range of 95:5 to 80:20, more preferably in the range of 92:8 to 85:15. By setting DM:PM within the above range, the releasability of the release film can be ensured. Further, the weight average molecular weight of the light release agent is preferably less than 10,000. When the weight average molecular weight of the light release agent is less than 10,000, it is advantageous in terms of migration properties and easy release properties.
- low-molecular-weight siloxane compounds are usually contained in the silicone resin in an amount of 0.1 to 15.0% by mass, preferably 0.5 to 10.0% by mass, and more preferably 0.5 to 5.0% by mass as a transitional component. By doing so, the desired light peeling can be achieved.
- the content is 0.1% by mass or more, the migratory component is sufficient and the mold releasability is sufficiently exhibited, and when the content of low molecular weight siloxane is 15.0% by mass or less, the migratory component is excessive. There is no risk of contamination during the process.
- the resin layer (A) composition may contain a diluting solvent, if necessary.
- diluent solvents include aromatic hydrocarbons such as toluene, aliphatic hydrocarbons such as hexane, heptane, and isooctane, esters such as ethyl acetate and butyl acetate, and ketones such as methyl ethyl ketone (MEK) and isobutyl methyl ketone.
- examples include alcohols such as ethanol and 2-propanol, and ethers such as diisopropyl ether and dibutyl ether. These are preferably used singly or in combination, taking into account solubility, coatability, boiling point, etc.
- an organic silicon compound represented by the following general formula (5) in combination with the resin layer (A) in order to improve the adhesion of the coating film to the film.
- X is an organic group having at least one selected from epoxy group, mercapto group , (meth)acryloyl group, alkenyl group, haloalkyl group, and amino group; It has 1 to 10 carbon atoms
- Y is a hydrolyzable group
- d is an integer of 1 or 2
- e is an integer of 2 or 3
- f is an integer of 0 or 1
- d+e+f 4.
- the organosilicon compound represented by the general formula (5) has two (D unit source) or three (T unit source) hydrolyzable groups Y that can form a siloxane bond by hydrolysis/condensation reaction. source) can be used.
- the monovalent hydrocarbon group R 1 has 1 to 10 carbon atoms, with methyl, ethyl, and propyl groups being particularly preferred.
- examples of the hydrolyzable group Y include the following. That is, they include methoxy group, ethoxy group, butoxy group, isopropenoxy group, acetoxy group, butanoxime group, and amino group. These hydrolyzable groups may be used alone or in combination. It is particularly preferable to use a methoxy group or an ethoxy group because it can impart good storage stability to the coating material and has appropriate hydrolyzability.
- organosilicon compound contained in the resin layer (A) examples include vinyltrimethoxysilane, ⁇ -glycidoxypropyltrimethoxysilane, ⁇ -glycidoxypropylmethyldiethoxysilane, ⁇ -(3,4-epoxy cyclohexyl)ethyltrimethoxysilane, ⁇ -mercaptopropyltrimethoxysilane, ⁇ -methacryloxypropyltrimethoxysilane, ⁇ -acryloxypropyltrimethoxysilane, ⁇ -aminopropyltriethoxysilane, 5-hexenyltrimethoxysilane, p Examples include -styryltrimethoxysilane, trifluoropropyltrimethoxysilane, ⁇ -glycidoxypropyltriethoxysilane, and ⁇ -glycidoxypropylmethyldiisopropenoxysilane.
- the resin layer (A) does not substantially contain particles. By not containing substantially any particles in the resin layer (A), it is possible to stabilize the peeling characteristics and to lower the transferability.
- substantially not containing means that the resin layer (A) may contain particles as long as the amount is small enough not to impede the effects of the present invention. For example, if particles are unavoidably mixed, May be included.
- the content of particles in the present resin layer (A) (layer A) is, for example, less than 0.05% by mass, preferably less than 0.01% by mass, more preferably 0.0001% by mass, based on nonvolatile components. less than The range of the content of particles in the present resin layer (A) composition based on nonvolatile components is also the same as the content of particles described above.
- the resin layer (A) composition may optionally include an antifoaming agent, a coating improver, a thickener, an organic lubricant, an antistatic agent, a conductive agent, and an ultraviolet ray.
- an antifoaming agent e.g., sodium sulfate, sodium sulfate, sodium sulfate, sodium sulfate, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium metabisulfite, sodium
- the number average molecular weight (Mn) of the curable silicone resin is preferably 9,000 or more and 350,000 or less.
- the number average molecular weight (Mn) of the curable silicone resin is equal to or higher than the above lower limit value, when the adhesive layer is laminated on the release film, the amount of low molecular weight silicone resin eluted or transferred to the adhesive layer can be reduced. Moreover, by applying the resin layer (A) thickly, it becomes easier to obtain the effect of easy peeling.
- the number average molecular weight (Mn) of the curable silicone resin is below the above upper limit, the viscosity is prevented from increasing and the fluidity of the resin layer (A) composition is prevented from decreasing.
- the number average molecular weight (Mn) of the curable silicone resin is preferably 9,000 or more, more preferably 10,000 or more, even more preferably 20,000 or more, and especially 30,000 or more. is even more preferable. On the other hand, it is preferably 350,000 or less, more preferably 50,000 or less, and even more preferably 40,000 or less.
- the mass average molecular weight (Mw) of the curable silicone resin is preferably 10,000 to 500,000, more preferably 20,000 or more, even more preferably 50,000 or more, and especially 80,000 or more. It is preferable that it is, and it is more preferable that it is 250,000 or less, and even more preferably that it is 100,000 or less.
- the curable silicone resin has a ratio (Mw/Mn) of mass average molecular weight (Mw) to number average molecular weight (Mn), preferably 1.7 to 3.5, more preferably 1.9 to 3.0, and especially More preferably 2.0 to 3.0. By satisfying this range, it can be expected that the crosslinking reaction will proceed efficiently.
- Mn and Mw are values determined by gel permeation chromatography (GPC) measurement using polystyrene standards, and the specific measurement method is using a GPC measuring device.
- the curable silicone resin may be a combination of two or more types of curable silicone resins, and in that case, the number average molecular weight (Mn) and mass average molecular weight (Mw) of the two or more types of curable silicone resins. It is preferable that the average of is within the above range. Note that the average here is a weighted average weighted by the mass of each resin. Further, when using a base resin and a silicone crosslinking agent as the curable silicone resin, it is preferable that the mass average molecular weight (Mn), number average molecular weight (Mw), and Mw/Mn of the base resin are within the above ranges.
- the viscosity of the curable silicone resin at 25° C. when adjusted to 15% by mass by diluting with n-heptane solvent is preferably 1 to 400 mcps, more preferably 5 to 300 mcps, and even more preferably 10 to 200 mcps.
- the viscosity of the curable silicone resin is 1 mcps or more, it is preferable because repelling is suppressed by the appropriate viscosity of the coating liquid and a uniform coat appearance with high visibility can be obtained. If it is 400 mcps or less, the resin layer (A ) The fluidity of the composition can be maintained, and when the resin layer (A) composition is applied, the occurrence of streaky coating unevenness can be suppressed, and the surface of the resin layer (A) can be made smooth. .
- the curable silicone resin was diluted with n-heptane to 15% by mass, and the viscosity of this solution at 25°C was measured using an E-type viscometer (“TVE-22L” manufactured by Toki Sangyo Co., Ltd.). Measured using
- the content of the alkenyl group is preferably 0.4 to 2.5 mol%, more preferably 0.5 to 2 mol% based on the total amount of siloxane components. 0 mol%, more preferably 0.5 to 1.5 mol%.
- the content of the Si--H groups is preferably 0.8 to 2.5 mol%, more preferably 0.8 to 2.5 mol%, based on the total amount of siloxane components. It is 8 to 2.0 mol%, more preferably 1.0 to 2.0 mol%.
- the curable silicone resin may contain an alkenyl group and a Si-H group (also simply referred to as an "H group") in the side chain and/or terminal of the main chain consisting of a siloxane bond in the same structure.
- an alkenyl group and a Si-H group also simply referred to as an "H group”
- the content of alkenyl groups and Si--H groups is preferably within the above range.
- the content of all the siloxane components in the release layer composition can be measured, for example, by 1 H-NMR from the integral ratio of the main chain dimethylsiloxane unit and other units. Further, the content of vinyl groups and Si--H groups is expressed as a percentage of the total amount of functional groups bonded to the siloxane chain, and can be evaluated by measuring 1 H-NMR. However, the method is not limited to this method.
- the thickness of the resin layer (A) is preferably 0.2 to 2.0 ⁇ m, more preferably 0.4 to 1.5 ⁇ m, and even more preferably ranges from 0.4 to 1.2 ⁇ m, most preferably from 0.4 to 1.0 ⁇ m.
- the thickness of the resin layer (A) is less than 0.2 ⁇ m, as will be described later, it is difficult to easily peel the resin layer (A) even if the elastic modulus of the resin layer (A) is within a predetermined range.
- the thickness is more than 2.0 ⁇ m, the transferability of the resin layer (A) component to adhesive tape etc. may increase, or blocking may not be sufficiently prevented.
- the elastic modulus of the resin layer (A) at 25° C. is preferably 500 MPa or less, more preferably 65 to 400 MPa, and even more preferably 80 to 300 MPa. By satisfying these ranges, it is possible to exhibit good releasability to the adhesive layer.
- the elastic modulus of the resin layer (A) can be adjusted by the type of silicone resin used, the thickness of the resin layer (A), etc. For example, as the resin layer (A) becomes thinner, the elastic modulus tends to increase. Moreover, the elastic modulus here is a value measured by a nanoindenter.
- the normal peeling force of the resin layer (A) is preferably 5 g/25 mm or less, more preferably 0.1 to 4 g/25 mm, and even more preferably 0.5 to 3 g/25 mm.
- the normal peeling force is 5 g/25 mm or less, good peelability can be exhibited even for adhesive layers whose elastic modulus has become lower as optical members become larger and thinner.
- the normal peeling force exceeds 5g/25mm, it may become difficult to peel off from the adhesive layer, and the adhesive layer may be transferred to the release film side due to deformation or breakage of the adhesive layer. be.
- the residual adhesion rate of the resin layer (A) is an index of the transferability of the transfer component derived from the resin layer (A) to the adhesive tape or the like to be bonded.
- the resin layer (A) with high migration properties many migration components adhere to the stacked evaluation film, so the peeling force of the adhesive tape bonded to the evaluation film becomes small, and the residual adhesion rate (%) also decreases. Therefore, the residual adhesion rate (%) is preferably high, preferably 80% or more, more preferably 85% or more, and even more preferably 90% or more. If the residual adhesion rate is 80% or more, it can be considered that there is no problem in actual use.
- the rate of heavy peeling of the resin layer (A) after pressing is preferably 100% or less, more preferably 50% or less, even more preferably 20% or less. If the rate of heavy peeling after pressing is 100% or less, when the release film is wound into a roll, the resin layer (A) will not be heavily peeled at the lower part of the roll where pressure is applied by the film. This prevents problems caused by heavy peeling.
- the method for calculating the rate of heavy peeling after pressing is as described in Examples.
- the resin layer (A) may be provided by in-line coating in which the film surface is treated during the film forming process, or it may be provided by off-line coating in which it is applied outside the system on the film once produced. However, it is more preferably formed by off-line coating.
- the film constituting the release film in the present invention may be previously subjected to surface treatment such as corona treatment or plasma treatment.
- the heat treatment is preferably carried out at 120 to 190°C for 3 to 40 seconds, more preferably at 150 to 180°C for 3 to 40 seconds.
- heat treatment and active energy ray irradiation such as ultraviolet irradiation may be used in combination as necessary.
- active energy ray irradiation known devices and energy sources can be used.
- the light source include a fusion (H) lamp, a metal halide lamp, a high-pressure mercury lamp (ozone generation type, ozone-less type), UV-LED, and the like.
- Active energy ray irradiation is not particularly limited, but in the case of ultraviolet irradiation, the integrated light amount is 10 to 3000 mJ/cm 2 , preferably 50 to 2000 mJ/cm 2 , more preferably 100 to 1000 mJ/cm A range of 2 is good.
- the cumulative light amount conversion of ultraviolet irradiation within the above range, curing of the resin layer (A) is promoted, and on the other hand, by not irradiating too much, the resin layer (A) is not destroyed and peels off after irradiation. This prevents the force from becoming too heavy.
- the surface specific resistivity of at least one surface of the resin layer (A) or the resin layer (B) is 1 ⁇ 10 12 ⁇ / ⁇ or less.
- the center line average roughness (Ra(A)) of the surface of the resin layer (A) is preferably 30 nm or less. It is preferable that the center line average roughness (Ra(A)) is 30 nm or less, since this allows for ultra-light peeling. Regarding (Ra(A)), it is more preferably 25 nm or less, especially 20 nm or less. On the other hand, the lower limit is preferably 5 nm or more from the viewpoint of film handling properties.
- the resin layer (B) constituting the release film in the present invention is provided on the surface of the base film opposite to the surface on which the resin layer (A) is provided, and is a resin layer (B) containing a non-silicone release agent. ), more preferably a resin layer (B) containing a non-silicone mold release agent and having antistatic properties.
- the non-silicone mold release agent used in the resin layer (B) is not particularly limited as long as it is a mold release agent other than silicone compounds, and conventionally known mold release agents can be used, such as long-chain mold release agents.
- mold release agents include alkyl group-containing compounds, fluorine compounds, wax, and the like.
- long-chain alkyl compounds and waxes are preferred from the viewpoint of less contamination and excellent blocking reduction, and particularly long-chain alkyl compounds are more preferred from the viewpoint of reducing blocking.
- These mold release agents may be used alone or in combination.
- a silicone compound is a compound having a siloxane bond in its molecule, and includes compounds having various functional groups in the side chain and/or terminal of the main chain consisting of a siloxane bond. Examples include ether groups, hydroxyl groups, amino groups, epoxy groups, carboxylic acid groups, halogen groups such as fluorine, perfluoroalkyl groups, and hydrocarbon groups (alkyl groups, alkenyl groups, aryl groups, various aromatic groups, etc.). It will be done. If the resin layer (B) contains a silicone compound, it will strongly block with the resin layer (A), deforming the surface of the resin layer (A), and deteriorating the peeling characteristics of the resin layer (A). It is difficult to use in release films.
- a long-chain alkyl group-containing compound is a compound having a linear or branched alkyl group having usually 6 or more carbon atoms, preferably 8 or more carbon atoms, and more preferably 12 or more carbon atoms.
- the alkyl group include hexyl group, octyl group, decyl group, lauryl group, octadecyl group, and behenyl group.
- the compound having an alkyl group include various long-chain alkyl group-containing polymer compounds, long-chain alkyl group-containing amine compounds, long-chain alkyl group-containing ether compounds, and long-chain alkyl group-containing quaternary ammonium salts. .
- a polymer compound is preferable. Further, from the viewpoint of effectively obtaining mold release properties, a polymer compound having a long-chain alkyl group in its side chain is more preferable.
- a polymer compound having a long-chain alkyl group in its side chain can be obtained by reacting a polymer having a reactive group with a compound having an alkyl group capable of reacting with the reactive group.
- a polymer having a reactive group examples include a hydroxyl group, an amino group, a carboxyl group, an acid anhydride, and the like.
- compounds having these reactive groups include polyvinyl alcohol, polyethyleneimine, polyethyleneamine, reactive group-containing polyester resin, and reactive group-containing poly(meth)acrylic resin. Among these, polyvinyl alcohol is preferred in consideration of mold releasability and ease of handling.
- the compound having an alkyl group that can react with the above-mentioned reactive group includes, for example, isocyanate containing a long chain alkyl group such as hexyl isocyanate, octyl isocyanate, decyl isocyanate, lauryl isocyanate, octadecyl isocyanate, and behenyl isocyanate, hexyl chloride, and octyl chloride.
- long-chain alkyl group-containing acid chlorides such as decyl chloride, lauryl chloride, octadecyl chloride, and behenyl chloride, long-chain alkyl group-containing amines, and long-chain alkyl group-containing alcohols.
- long-chain alkyl group-containing isocyanates are preferred in consideration of mold releasability and ease of handling, and octadecyl isocyanate is particularly preferred.
- polymer compounds having long-chain alkyl groups in their side chains can also be obtained by polymerizing long-chain alkyl (meth)acrylates or copolymerizing long-chain alkyl (meth)acrylates with other vinyl group-containing monomers.
- long-chain alkyl (meth)acrylates include hexyl (meth)acrylate, octyl (meth)acrylate, decyl (meth)acrylate, lauryl (meth)acrylate, octadecyl (meth)acrylate, and behenyl (meth)acrylate. It will be done.
- Wax is a wax selected from natural waxes, synthetic waxes, and blended waxes.
- Natural waxes include vegetable waxes, animal waxes, mineral waxes, and petroleum waxes.
- vegetable waxes include candelilla wax, carnauba wax, rice wax, wood wax, and jojoba oil.
- animal waxes include beeswax, lanolin, whale wax, and the like.
- mineral waxes include montan wax, ozokerite, ceresin, and the like.
- petroleum waxes include paraffin wax, microcrystalline wax, and petrolatum.
- Examples of synthetic waxes include synthetic hydrocarbons, modified waxes, hydrogenated waxes, fatty acids, acid amides, amines, imides, esters, ketones, and the like.
- Examples of synthetic hydrocarbons include Fischer-Tropsch wax (also known as sozoir wax) and polyethylene wax. Examples include certain of the following polymers: polypropylene, ethylene/acrylic acid copolymer, polyethylene glycol, polypropylene glycol, block or graft combinations of polyethylene glycol and polypropylene glycol, and the like.
- Examples of the modified wax include montan wax derivatives, paraffin wax derivatives, microcrystalline wax derivatives, and the like. The derivative here is a compound obtained by any one of purification, oxidation, esterification, saponification, or a combination thereof.
- Hydrogenated waxes include hydrogenated castor oil and hydrogenated castor oil derivatives.
- the number average molecular weight (Mn) of the synthetic wax is preferably in the range of 500 to 30,000, more preferably 1,000 to 15,000, and even more preferably 2,000 to 8,000, from the viewpoint of stability of properties such as blocking and ease of handling.
- a fluorine compound is a compound containing a fluorine atom in the compound.
- Organic fluorine compounds are preferably used in terms of the appearance of the coating by in-line coating, such as perfluoroalkyl group-containing compounds, polymers of olefin compounds containing fluorine atoms, aromatic fluorine compounds such as fluorobenzene, etc. . From the viewpoint of mold releasability, a compound having a perfluoroalkyl group is preferable. Further, as the fluorine compound, a compound containing a long-chain alkyl compound as described below can also be used.
- Compounds having a perfluoroalkyl group include, for example, perfluoroalkyl (meth)acrylate, perfluoroalkylmethyl (meth)acrylate, 2-perfluoroalkylethyl (meth)acrylate, 3-perfluoroalkylpropyl (meth)acrylate.
- the polymer may be a single compound or a polymer of multiple compounds. Further, from the viewpoint of mold releasability, the perfluoroalkyl group preferably has 3 to 11 carbon atoms. Furthermore, it may be a polymer with a compound containing a long-chain alkyl compound as described later. Furthermore, from the viewpoint of adhesion to the base material, a polymer with vinyl chloride is also preferable.
- an antistatic agent from the viewpoint of preventing the adhesion of foreign substances.
- a compound consisting of thiophene or a thiophene derivative is doped with another anionic compound, or a self-doped polymer having an anion group in a compound consisting of thiophene or a thiophene derivative, or a polymer containing as a component a monomer having an alkyl sulfonate ion as a counter ion. I can do it.
- Compounds consisting of thiophene or thiophene derivatives include polymers doped with other anionic compounds of compounds consisting of thiophene or thiophene derivatives, or self-doped polymers containing anionic groups in compounds consisting of thiophene or thiophene derivatives.
- Examples of the polymer include those obtained by polymerizing a compound of the following formula (6) or the following formula (7) in the presence of a polyanion.
- R 1 and R 2 each independently represent a hydrogen atom, an aliphatic hydrocarbon group having 1 to 20 carbon atoms, an alicyclic hydrocarbon group, an aromatic hydrocarbon group, etc. .
- n represents an integer of 1 to 4.
- polyanions used during polymerization include poly(meth)acrylic acid, polymaleic acid, polystyrenesulfonic acid, and polyvinylsulfonic acid.
- a method for producing such a polymer for example, a method as disclosed in JP-A-7-90060 can be adopted.
- a compound of the above formula (7) in which n is 2 and polystyrene sulfonic acid is used as the polyanion is preferably used.
- polyanions when these polyanions are acidic, some or all of them may be neutralized.
- base used for neutralization ammonia, organic amines, and alkali metal hydroxides are preferable.
- a specific example of a polymer containing as a component a monomer having an alkyl sulfonate ion as a counter ion includes a polymer having a constituent represented by the following formula (8) as a repeating unit. These homopolymers and copolymers, as well as a plurality of other components, may be copolymerized. From the viewpoint of improving antistatic properties, a homopolymer is preferable.
- substituent R 1 is a hydrogen atom or an alkyl group having 1 to 3 carbon atoms
- R 2 is -O- or -NH-
- R 3 is a hydrogen atom or an alkyl group having 1 to 6 carbon atoms.
- Alkylene group or other structure that can establish the structure of formula (8) at least one of R 4 , R 5 , and R 6 is a hydrogen atom, and the other substituents are an alkyl group having 1 to 3 carbon atoms, or an alkyl group having 1 to 3 carbon atoms; Examples thereof include a hydroxyalkyl group having 2 to 3 carbon atoms, and an alkylsulfonic acid ion in which X - has an alkyl group having 1 to 4 carbon atoms.
- polyhydroxy compound in order to improve the coating appearance and antistatic performance.
- polyhydroxy compound it is preferable to use one or more compounds selected from polyglycerin and alkylene oxide adducts to polyglycerin, or derivatives thereof.
- Polyglycerin is a compound represented by the following general formula (9).
- n is 2 or more, and in the present invention, n in the formula is usually in the range of 2 to 20, preferably 3 to 15, and more preferably 3 to 12.
- the alkylene oxide adduct to polyglycerin has a structure in which alkylene oxide is addition-polymerized to the hydroxyl group of polyglycerin represented by the above general formula (9).
- the structure of the alkylene oxide added may be different for each hydroxyl group of the polyglycerin skeleton. Further, it is sufficient that the alkylene oxide or its derivative is added to at least one hydroxyl group in the molecule, and it is not necessary that alkylene oxide or its derivatives be added to all hydroxyl groups.
- Preferred alkylene oxides added to polyglycerin are ethylene oxide or propylene oxide. If the alkylene chain of the alkylene oxide becomes too long, the hydrophobicity becomes strong, the dispersibility in the coating liquid deteriorates, and the antistatic properties and transparency of the resin layer (B) tend to deteriorate. Particularly preferred is ethylene oxide. Further, the number of additions is preferably one in which the number average molecular weight (Mn) of the final compound is in the range of 200 to 2,000, more preferably in the range of 300 to 1,000, and even more preferably in the range of 400 to 900.
- Mn number average molecular weight
- polyglycerin or alkylene oxide adduct to polyglycerin may be used alone or in combination of two or more.
- various conventionally known polymers can be used together as a binder component from the viewpoint of improving coating appearance and transparency.
- polymers include polyester resin, urethane resin, and acrylic resin.
- examples include resin, polyvinyl alcohol, vinyl chloride vinyl acetate copolymer, and the like. From the viewpoint of making the resin layer (B) stronger and reducing blocking, it is preferable to use polyester resin or urethane resin.
- polyester resins include those consisting of the following polyhydric carboxylic acids and polyhydric hydroxy compounds as main components. That is, examples of polyhydric carboxylic acids include terephthalic acid, isophthalic acid, orthophthalic acid, phthalic acid, 4,4'-diphenyldicarboxylic acid, 2,5-naphthalenedicarboxylic acid, 1,5-naphthalenedicarboxylic acid, and 2,6-diphenyldicarboxylic acid.
- polyhydric hydroxy compounds include ethylene glycol, 1,2-propylene glycol, 1,3-propylene glycol, 1,3-propanediol, 1,4-butanediol, 1,6-hexanediol, 2- Methyl-1,5-pentanediol, neopentyl glycol, 1,4-cyclohexanedimethanol, p-xylylene glycol, bisphenol A-ethylene glycol adduct, diethylene glycol, triethylene glycol, Polyethylene glycol, polypropylene glycol, polytetramethylene glycol, polytetramethylene oxide glycol, dimethylolpropionic acid, glycerin, trimethylolpropane, sodium dimethylolethyl sulfonate, dimethylolpropionic acid Potassium etc.
- Polyester resin is a polycondensate of polyhydric carboxylic acid and polyhydric hydroxy compound.
- One or more polyhydric carboxylic acids and polyhydric hydroxy compounds may be appropriately selected from the above-mentioned compounds, and a polyester resin may be synthesized by a conventional polycondensation reaction.
- the polyester resin may be made into an aqueous dispersion, and in that case, a hydrophilic functional group or the like may be appropriately introduced into the polyester resin.
- Urethane resin is a polymer compound that has urethane bonds in its molecules. Urethane resins are usually created by the reaction of polyols and isocyanates. Examples of the polyol include polycarbonate polyols, polyester polyols, polyether polyols, polyolefin polyols, and acrylic polyols, and these compounds may be used alone or in combination.
- the urethane resin may be an aqueous dispersion, in which case a hydrophilic functional group may be appropriately introduced into the polyol, for example.
- Polycarbonate polyols are obtained from polyhydric alcohols and carbonate compounds by dealcoholization reaction.
- polyhydric alcohols include ethylene glycol, 1,2-propylene glycol, 1,3-propylene glycol, 1,2-butanediol, 1,3-butanediol, 1,4-butanediol, and 1,5-pentane.
- Examples include diol, neopentyl glycol, 3-methyl-1,5-pentanediol, 3,3-dimethylolheptane and the like.
- Examples of carbonate compounds include dimethyl carbonate, diethyl carbonate, diphenyl carbonate, and ethylene carbonate.
- polycarbonate polyols obtained from these reactions include poly(1,6-hexylene) carbonate and poly(3- Examples include methyl-1,5-pentylene) carbonate.
- polyester polyols examples include polycarboxylic acids (malonic acid, succinic acid, glutaric acid, adipic acid, pimelic acid, suberic acid, sebacic acid, fumaric acid, maleic acid, terephthalic acid, isophthalic acid, etc.) or their acid anhydrides.
- polyhydric alcohols ethylene glycol, diethylene glycol, triethylene glycol, propylene glycol, dipropylene glycol, tripropylene glycol, butanediol, 1,3-butanediol, 1,4-butanediol, 2,3-butanediol, 2-Methyl-1,3-propanediol, 1,5-pentanediol, neopentyl glycol, 1,6-hexanediol, 3-methyl-1,5-pentanediol, 2-methyl-2,4-pentanediol , 2-methyl-2-propyl-1,3-propanediol, 1,8-octanediol, 2,2,4-trimethyl-1,3-pentanediol, 2-ethyl-1,3-hexanediol, 2 , 5-dimethyl-2,5-hexanediol, 1,9-
- polyether polyols examples include polyethylene glycol, polypropylene glycol, polyethylene propylene glycol, polytetramethylene ether glycol, polyhexamethylene ether glycol, and the like.
- polycarbonate polyols and polyester polyols are more preferably used to improve the adhesion with various functional layers.
- the polyisocyanate compounds used to obtain the urethane resin include aromatic diisocyanates such as tolylene diisocyanate, xylylene diisocyanate, methylene diphenyl diisocyanate, phenylene diisocyanate, naphthalene diisocyanate, tolidine diisocyanate, ⁇ , ⁇ , ⁇ ', ⁇ ' - Aliphatic diisocyanates with aromatic rings such as tetramethylxylylene diisocyanate, methylene diisocyanate, propylene diisocyanate, lysine diisocyanate, trimethylhexamethylene diisocyanate, hexamethylene diisocyanate, cyclohexane diisocyanate, methylcyclohexane diisocyanate, isophorone diisocyanate, dicyclohexyl Examples include alicyclic diisocyanates such as methane diisocyanate and isopropylid
- a chain extender may be used when synthesizing a urethane resin, and there are no particular restrictions on the chain extender as long as it has two or more active groups that react with isocyanate groups. Alternatively, a chain extender having two amino groups can be mainly used.
- chain extender having two hydroxyl groups examples include aliphatic glycols such as ethylene glycol, propylene glycol, butanediol, aromatic glycols such as xylylene glycol and bishydroxyethoxybenzene, and esters such as neopentyl glycol hydroxypivalate.
- Glycols such as glycol can be mentioned.
- chain extenders having two amino groups include aromatic diamines such as tolylene diamine, xylylene diamine, diphenylmethane diamine, ethylene diamine, propylene diamine, hexane diamine, 2,2-dimethyl-1,3- Propanediamine, 2-methyl-1,5-pentanediamine, trimethylhexanediamine, 2-butyl-2-ethyl-1,5-pentanediamine, 1,8-octanediamine, 1,9-nonanediamine, 1,10- Aliphatic diamines such as decanediamine, 1-amino-3-aminomethyl-3,5,5-trimethylcyclohexane, dicyclohexylmethanediamine, isopropylidenecyclohexyl-4,4'-diamine, 1,4-diaminocyclohexane, 1 , 3-bisaminomethylcyclohexane and other alicyclic diamines.
- aromatic diamines such as toly
- the urethane resin in the present invention may use a solvent as a medium, and preferably uses water as a medium.
- a forced emulsification type using an emulsifier there are a forced emulsification type using an emulsifier, a self-emulsification type or water-soluble type in which a hydrophilic group is introduced into the urethane resin, and the like.
- a self-emulsifying type in which an ionic group is introduced into the structure of the urethane resin to form an ionomer is preferable because it has excellent liquid storage stability and the resulting coating layer has excellent water resistance, transparency, and adhesion.
- various ionic groups to be introduced include carboxyl groups, sulfonic acids, phosphoric acids, phosphonic acids, quaternary ammonium salts, etc., but carboxyl groups are preferred.
- carboxyl groups are preferred.
- Various methods can be used to introduce carboxyl groups into the urethane resin at each stage of the polymerization reaction. For example, there is a method in which a resin having a carboxyl group is used as a copolymerization component during prepolymer synthesis, and a method in which a component having a carboxyl group is used as a component such as a polyol, polyisocyanate, or chain extender.
- a carboxyl group-containing diol is used and a desired amount of carboxyl groups is introduced depending on the amount of this component.
- this carboxyl group is preferably in the form of a salt neutralized with ammonia, amine, alkali metals, inorganic alkalis, etc. Particularly preferred are ammonia, trimethylamine, and triethylamine.
- the carboxyl group from which the neutralizing agent is removed during the drying process after coating can be used as a crosslinking reaction site with another crosslinking agent. This not only provides excellent stability in the liquid state before coating, but also makes it possible to further improve the durability, solvent resistance, water resistance, blocking resistance, etc. of the resulting coating layer.
- Acrylic resin is a polymer made of polymerizable monomers including acrylic and methacrylic monomers. These may be homopolymers or copolymers, or copolymers with polymerizable monomers other than acrylic and methacrylic monomers. Also included are copolymers of these polymers and other polymers (eg, polyester, polyurethane, etc.). For example, block copolymers and graft copolymers. That is, the acrylic resin may be an acrylic modified polyester resin or an acrylic modified polyurethane resin. Furthermore, polymers (or mixtures of polymers in some cases) obtained by polymerizing a polymerizable monomer in a polyester solution or a polyester dispersion are also included.
- polymers obtained by polymerizing polymerizable monomers in polyurethane solutions and polyurethane dispersions are also included.
- polymers (in some cases, polymer mixtures) obtained by polymerizing polymerizable monomers in other polymer solutions or dispersions are also included, and these are also referred to herein as acrylic-modified polyester resins, acrylic-modified polyester resins, and acrylic-modified polyester resins.
- acrylic-modified polyester resins acrylic-modified polyester resins
- acrylic-modified polyester resins acrylic-modified polyester resins
- acrylic-modified polyester resins Use polyurethane resin.
- the above-mentioned polyester and polyurethane used in the acrylic resin can be appropriately selected from those exemplified as the above-mentioned polyester and polyurethane as binder components.
- the acrylic resin may contain a hydroxyl group or an amino group in order to further improve the adhesion with the base film.
- the above-mentioned polymerizable monomers are not particularly limited, but typical compounds include various carboxyl group-containing monomers such as acrylic acid, methacrylic acid, crotonic acid, itaconic acid, fumaric acid, maleic acid, and citraconic acid. , and their salts; various such as 2-hydroxyethyl (meth)acrylate, 2-hydroxypropyl (meth)acrylate, 4-hydroxybutyl (meth)acrylate, monobutyl hydroxyl fumarate, monobutyl hydroxy itaconate.
- carboxyl group-containing monomers such as acrylic acid, methacrylic acid, crotonic acid, itaconic acid, fumaric acid, maleic acid, and citraconic acid. , and their salts; various such as 2-hydroxyethyl (meth)acrylate, 2-hydroxypropyl (meth)acrylate, 4-hydroxybutyl (meth)acrylate, monobutyl hydroxyl fumarate, monobutyl hydroxy itaconate
- Hydroxyl group-containing monomers various (meth)acrylic acid esters such as methyl (meth)acrylate, ethyl (meth)acrylate, propyl (meth)acrylate, butyl (meth)acrylate, and lauryl (meth)acrylate; (meth) Various nitrogen-containing compounds such as acrylamide, diacetone acrylamide, N-methylolacrylamide or (meth)acrylonitrile, etc.; various styrene derivatives such as styrene, ⁇ -methylstyrene, divinylbenzene, vinyltoluene, vinyl propionate, etc.
- Various vinyl esters include various silicon-containing polymerizable monomers such as ⁇ -methacryloxypropyltrimethoxysilane and vinyltrimethoxysilane; phosphorus-containing vinyl monomers; various vinyl halides such as vinyl chloride and polypylidene chloride. various conjugated dienes such as butadiene.
- crosslinking agent In order to improve the strength of the resin layer (B), it is also possible to use a crosslinking agent in combination.
- the crosslinking agent conventionally known materials can be used, such as oxazoline compounds, epoxy compounds, melamine compounds, isocyanate compounds, carbodiimide compounds, organosilicon compounds, and the like. From the viewpoint of increasing the strength of the resin layer (B), melamine compounds are more preferred. It is also possible to use two or more types of crosslinking agents together.
- the resin layer (B) in the present release film may contain particles for the purpose of improving the slipperiness of the film, but a more preferred form is that it does not substantially contain particles. By not containing substantially any particles in the resin layer (B), it is possible to stabilize the release characteristics and reduce the migration of the release component.
- substantially not containing means that the resin layer (B) may contain particles as long as the amount is small enough not to impede the effects of the present invention. For example, if particles are unavoidably mixed, May be included.
- the specific content of particles in the resin layer (B) is, for example, less than 0.05% by mass, preferably less than 0.01% by mass, and more preferably less than 0.0001% by mass, based on nonvolatile components. Note that the range of the content of particles in the resin layer (B) based on nonvolatile components is also the same as the above content of particles.
- the resin layer (B) may optionally include an antifoaming agent, a coating property improver, a thickener, an organic lubricant, an antistatic agent, an ultraviolet absorber, an oxidizing agent, etc. It may also contain inhibitors, blowing agents, dyes, pigments, etc.
- the content of the release agent in the nonvolatile components is preferably 10 to 70% by mass.
- the content of the mold release agent is 10% by mass or more, good anti-blocking performance can be obtained.
- the long-chain alkyl group-containing compound is preferably It ranges from 5 to 90% by weight, more preferably from 10 to 70% by weight, even more preferably from 20 to 60% by weight, and most preferably from 20 to 40% by weight.
- the proportion of the long-chain alkyl group-containing compound is within the above range, good anti-blocking performance can be obtained, and by reducing changes in the release properties of the release layer due to blocking, the lower roll of the roll can apply more pressure to the film. Heavy peeling of the mold release layer in some areas can be greatly suppressed.
- the wax When wax is used as a non-silicone mold release agent in the resin layer (B), the wax preferably accounts for 5 to 90% by mass, more preferably 10 to 80% by mass of the total nonvolatile components in the resin layer (B).
- the amount is preferably in the range of 25 to 70% by weight.
- the proportion of wax is within the above range, good anti-blocking performance can be obtained, and by reducing changes in the release characteristics of the release layer due to blocking, the release layer can be used in the lower part of the roll where more pressure is applied to the film. Heavy peeling can be greatly suppressed. Further, when used in combination with a melamine compound, heavy peeling may occur. Therefore, when using wax, care must be taken in combination with crosslinking agents and the like.
- composition combinations include a combination of a long-chain alkyl group-containing compound and a melamine compound, a long-chain alkyl group-containing compound and an oxazoline compound, a long-chain alkyl group-containing compound, a melamine compound, and an oxazoline compound, and a combination of a wax, a melamine compound, and an oxazoline compound. Illustrated.
- wax As a non-silicone mold release agent, the factors (estimated) that cause wax to have a higher rate of heavy release depending on the combination with a crosslinking agent than compounds containing long-chain alkyl groups are considered as follows. In addition to water repellency, wax also has oil repellency. As a result, it is incompatible with the melamine compound to be combined, and the melamine compound becomes more likely to be repelled.
- the heavy peeling rate evaluation of the present invention is performed under high temperature (40°C) and high humidity (90% RH) with moisture present, so the resin layer (combined with wax and melamine compound) B) It is presumed that the heavy release rate increased as a result of easier formation of aggregates on the surface and improved adhesion with the silicone release layer on the opposite surface.
- a release agent was selected with an eye to its oil-repellent effect, which had not been considered with conventional non-silicone release agents, and a release film with a resin layer (B) was constructed.
- the present invention is characterized in that
- the proportion of the compound made of thiophene or a thiophene derivative in the total nonvolatile components of the resin layer (B) is preferably 5 to 50.
- the amount is preferably in the range of 5 to 40% by weight, and even more preferably 5 to 30% by weight. Within the above range, good antistatic properties can be obtained.
- the polyhydroxy compound is preferably 10 to 80% by mass, more preferably 20 to 70% by mass, and even more preferably 30 to 65% by mass, based on the total nonvolatile components of the resin layer (B). % range. Within the above range, good antistatic properties can be obtained.
- the binder component is preferably in the range of 5 to 50% by mass, more preferably 5 to 40% by mass, and even more preferably 5 to 30% by mass.
- the ratio of the binder component is within the above range, the strength of the resin layer (B) is good and blocking can be reduced.
- the proportion of the crosslinking agent in the total nonvolatile components of the resin layer (B) is preferably 5 to 70% by mass, preferably 10 to 60% by mass, and more preferably 15 to 50% by mass. When the proportion of the crosslinking agent is within the above range, the resin layer (B) has good strength and blocking can be reduced.
- the resin layer (B) in the present invention can have the following characteristics.
- the surface roughness (Ra (B)) of the resin layer (B) is preferably 20 nm or more, more preferably 25 to 60 nm, more preferably 30 to 55 nm, and still more preferably 38 to 60 nm.
- the range is 50 nm.
- the thickness is 20 nm or more, blocking is less likely to occur in the roll state after the release layer is provided.
- the surface roughness is 60 nm or less, when the present release film is wound up into a roll, the unevenness of the film is not transferred to the release layer, and the release characteristics of the release layer are maintained.
- the thickness of the resin layer (B) is preferably 0.005 to 0.25 ⁇ m, more preferably 0.008 to 0.15 ⁇ m, and still more preferably 0.01 to 0.10 ⁇ m.
- the non-silicone release agent component in the resin layer (B) migrates to the release layer when the release film is wound up into a roll. If the thickness of the resin layer (B) is 0.005 ⁇ m or more, good anti-blocking properties can be provided. It is assumed that unreacted substances of various compounds, compounds after reaction, or a mixture thereof are present in the resin layer (B).
- the surface specific resistivity of the resin layer (B) surface is preferably 1 ⁇ 10 12 ⁇ / ⁇ or less, more preferably 1 ⁇ 10 10 ⁇ / ⁇ or less, still more preferably 1 ⁇ 10 8 ⁇ / ⁇ or less, and most preferably is less than 1 ⁇ 10 6 ⁇ / ⁇ .
- the specific surface resistivity is preferably 1 ⁇ 10 4 ⁇ / ⁇ or more. The lower the specific surface resistivity of the resin layer (B), the better the antistatic property, and it is possible to suppress the charging of the film during the process and prevent the adhesion of foreign substances.
- the normal peeling force of the resin layer (B) is preferably 400 to 2000 g/25 mm, more preferably 400 to 1500 g/25 mm, and still more preferably 400 to 1000 g/25 mm.
- the normal peeling force of the resin layer (B) is preferably 400 to 2000 g/25 mm, more preferably 400 to 1500 g/25 mm, and still more preferably 400 to 1000 g/25 mm.
- the surface free energy of the resin layer (B) is preferably 50 mN/m or less, more preferably 40 mN/m or less, and even more preferably 30 mN/m or less.
- the surface free energy of the resin layer is preferably 50 mN/m or less, more preferably 40 mN/m or less, and even more preferably 30 mN/m or less.
- the elastic modulus of the resin layer (B) at 25° C. is preferably 500 MPa or more.
- the elastic modulus here is a value measured using a nanoindenter.
- the elastic modulus is 500 MPa or more, when the release film is wound into a roll, heavy peeling of the resin layer (A) will not occur even in the lower part of the roll where pressure is applied by the film.
- the present release film preferably includes an undercoat layer between at least one of the resin layer (A) and the resin layer (B) and the base film.
- the undercoat layer may be provided either between the resin layer (A) and the base film, or between the resin layer (B) and the base film, or may be provided on both.
- the undercoat layer is used not only for the purpose of improving the adhesion between the film and the resin layer (A) or the resin layer (B), but also for the purpose of imparting various functions to the release film.
- the undercoat layer has, for example, an antistatic property to suppress peeling charge in the process of peeling off a functional layer such as an adhesive layer provided on the resin layer (A), and to prevent adhesion of foreign substances, etc., and a high-temperature long film. Examples include oligomer sealing performance for sealing oligomer precipitation from a polyester film when heat treatment is performed for a certain amount of time. In the present release film, it is preferable that the undercoat layer has antistatic properties. Note that the undercoat layer may be a single layer or may have a structure of two or more layers.
- the undercoat layer having antistatic properties may contain, as an antistatic agent, a polymer obtained by doping the above-mentioned thiophene or thiophene derivative with another anionic compound, or an anionic compound in the thiophene or thiophene derivative.
- a self-doped polymer having an ionic group or a polymer containing as a component a monomer having an alkylsulfonic acid ion as a counter ion can be used.
- the undercoat layer contains a compound made of thiophene or a thiophene derivative.
- polyglycerin represented by the general formula (9) one or more types selected from polyglycerin represented by the general formula (9) and alkylene oxide adducts to polyglycerin are used for the purpose of improving antistatic performance. It is preferable to use a compound or a derivative thereof.
- the undercoat layer having antistatic performance may contain a binder component.
- a binder component various conventionally known polymers such as polyester resin, acrylic resin, urethane resin, polyvinyl alcohol resin, etc. can be used. Urethane resin is preferred from the viewpoint of improving the transparency and antistatic properties of the undercoat layer.
- crosslinking agent In order to improve the strength of the undercoat layer, it is also possible to use a crosslinking agent in combination.
- the crosslinking agent conventionally known materials can be used, such as oxazoline compounds, epoxy compounds, melamine compounds, isocyanate compounds, carbodiimide compounds, organosilicon compounds, and the like. From the viewpoint of increasing the strength of the undercoat layer, melamine compounds are more preferred. It is also possible to use two or more types of crosslinking agents in combination.
- the proportion of the compound made of thiophene or a thiophene derivative in the total nonvolatile components of the undercoat layer is preferably 5 to 50% by mass, or more. It is preferably in the range of 5 to 40% by weight, more preferably 5 to 30% by weight. Within the above range, good antistatic properties can be obtained.
- the proportion of one or more compounds selected from polyglycerin and alkylene oxide adducts to polyglycerin or derivatives thereof is preferably 10 to 80% by mass, based on the total nonvolatile components of the undercoat layer.
- the content is more preferably from 20 to 70% by weight, and even more preferably from 30 to 65% by weight. Within the above range, good antistatic properties can be obtained.
- the binder component is preferably in the range of 5 to 50% by mass, more preferably 5 to 40% by mass, and even more preferably 5 to 30% by mass.
- the ratio of the binder component is within the above range, the strength of the undercoat layer will be good, and the antistatic property will be good.
- the crosslinking agent is preferably in the range of 5 to 70% by weight, preferably 10 to 60% by weight, and more preferably 15 to 50% by weight. When the proportion of the crosslinking agent is within the above range, the strength of the undercoat layer will be good and blocking can be reduced.
- the undercoat layer having oligomer sealing property may be provided with an undercoat layer having oligomer sealing property containing an organic compound containing one or more metal elements selected from aluminum, titanium, and zirconium. .
- the undercoat layer having oligomer sealing properties may be provided directly on the base film, or may be provided on the undercoat layer having antistatic properties.
- the thickness of the undercoat layer having antistatic properties is preferably 0.005 ⁇ m or more and 0.25 ⁇ m or less, more preferably 0.008 ⁇ m or more and 0.15 ⁇ m or less, and even more preferably 0.01 ⁇ m or more and 0.10 ⁇ m or less. If the thickness of the undercoat layer is within the above range, good antistatic properties can be imparted.
- the surface specific resistivity of the surface of the undercoat layer having antistatic performance is preferably 1 ⁇ 10 12 ⁇ / ⁇ or less, more preferably 1 ⁇ 10 10 ⁇ / ⁇ or less, and even more preferably 1 ⁇ 10 8 ⁇ / ⁇ or less. , most preferably 1 ⁇ 10 6 ⁇ / ⁇ or less.
- the specific surface resistivity is preferably 1 ⁇ 10 4 ⁇ / ⁇ or more. The lower the specific surface resistivity of the resin layer (B), the better the antistatic property, and it is possible to suppress the charging of the film during the process and prevent the adhesion of foreign substances.
- the resin layer (B) and the undercoat layer are formed by coating the film with a coating solution, and may be formed by in-line coating within the film manufacturing process, or by coating outside the system on the film once manufactured. A so-called off-line coating may be employed. More preferred is in-line coating.
- In-line coating is a method in which coating is performed within the process of producing a polyester film, and specifically, it is a method in which coating is performed at any stage from melt extrusion of polyester to stretching, heat setting, and winding.
- the coating is applied to an unstretched sheet obtained by melting and quenching, a stretched uniaxially oriented film, a biaxially oriented film before heat setting, or a film after heat setting and before winding.
- sequential biaxial stretching a method in which a uniaxially stretched film stretched in the longitudinal direction (longitudinal direction) is coated and then stretched in the transverse direction is particularly excellent.
- film formation and formation of the resin layer (B) can be performed at the same time, which is advantageous in terms of manufacturing costs.
- the stretching is performed after coating, the thickness of the resin layer (B) can be stretched.
- the magnification can also be changed, and thin film coating can be performed more easily than offline coating films.
- the resin layer (B) can be stretched together with the polyester film, thereby making it possible to firmly adhere the resin layer (B) to the polyester film. can.
- the film in the production of biaxially oriented polyester film, by holding the edges of the film with clips and stretching it, the film can be restrained in both the vertical and horizontal directions, and the film can be flattened without wrinkles during the heat setting process.
- the resin layer is prepared by preparing the above-mentioned series of compounds as an aqueous solution or dispersion, and adjusting the solid content concentration (total non-volatile components) to about 0.1 to 50% by mass.
- the base film It is preferable to manufacture the base film by applying the composition onto a polyester film.
- Examples of methods for applying the resin layer (B) composition or undercoat layer composition to the film include air doctor coating, blade coating, rod coating, bar coating, knife coating, squeeze coating, impregnation coating, and reverse roll coating.
- Conventionally known coating methods such as transfer roll coating, gravure coating, kiss roll coating, cast coating, spray coating, curtain coating, calendar coating, and extrusion coating can be used.
- the drying and curing conditions when forming the resin layer (B) and undercoat layer on the film are not particularly limited, and when the resin layer (B) is provided by in-line coating, preferably 70 to 270
- the heat treatment is preferably carried out at a temperature of 3 to 200 degrees Celsius, more preferably 100 to 260 degrees Celsius, even more preferably 110 to 250 degrees Celsius, for 10 to 100 seconds.
- heat treatment is preferably performed at 80 to 200°C for 3 to 40 seconds, more preferably at 100 to 180°C for 3 to 40 seconds. .
- heat treatment and active energy ray irradiation such as ultraviolet irradiation may be used in combination as necessary.
- the film constituting the release film in the present invention may be subjected to surface treatment such as corona treatment or plasma treatment in advance.
- the release film in the present invention may have a laminated structure, and a preferred form includes a form in which an adhesive layer is laminated on the surface of the release film on the resin layer (A) side.
- the present release film is the first release film, an adhesive layer is attached to the resin layer (A) side of the present release film, and an adhesive layer is attached to the resin layer (A) of the present release film.
- An example is one in which a second release film is used as a second release film and has a release force different from that of the main release film, and is bonded to the other surface.
- a more preferable embodiment is one in which the adhesive layer is an optical transparent adhesive sheet (OCA) and the present release film is used as a release film with a lighter peeling force.
- OCA optical transparent adhesive sheet
- the adhesive layer may be a single layer or may have a structure of two or more layers.
- release film having a different release force from the present release film means that the release force of the release film evaluated by the measuring method as described in the examples is the same as that of the resin layer (A) of the present release film. This means that it is different from the side peeling force.
- the release force is greater than that of the resin layer (A) of the present release film, and specifically, the release force is 1.5 to 10. It is preferably about 1.5 to 8 times, more preferably about 1.5 to 8 times, and even more preferably about 1.5 to 6 times.
- the adhesive layer is a layer made of an adhesive composition, and may be an acrylic adhesive composition whose main component is an acrylic resin or a rubber adhesive composition whose main component is rubber.
- the adhesive composition may be a urethane adhesive composition containing a urethane resin as a main component, or a silicone adhesive composition containing a silicone resin as a main component.
- an acrylic adhesive composition containing an acrylic resin as a main component is preferable because the adhesive composition can adjust adhesive strength and peeling force in a well-balanced manner, and is inexpensive.
- main component resin means a resin having the highest mass ratio among the resins constituting the adhesive composition.
- it means a component that accounts for 50% by mass or more, preferably 60% by mass or more, and more preferably 70% by mass or more of the total amount of resin constituting the adhesive composition.
- the upper limit is 100% by mass, but it is usually 99.99% by mass.
- acrylic resin examples of the acrylic resin that is the main component resin of the adhesive composition include (meth)acrylic polymers.
- a (meth)acrylic polymer is a polymer whose main structural unit is an alkyl (meth)acrylic acid ester.
- Examples of the (meth)acrylic acid alkyl ester include methyl (meth)acrylate, ethyl (meth)acrylate, n-propyl (meth)acrylate, isopropyl (meth)acrylate, and n-butyl (meth)acrylate.
- methyl (meth)acrylate is preferred from the viewpoint of compatibility with other (meth)acrylates constituting the (meth)acrylic polymer and heat resistance of the cured resin layer (B).
- the content of the (meth)acrylic acid alkyl ester in the monomers forming the (meth)acrylic polymer is, for example, 50% by mass or more, preferably 60 to 99.99% by mass, more preferably 75% by mass or more. ⁇ 98.9% by weight, more preferably 87 ⁇ 97.8% by weight.
- the (meth)acrylic polymer may have a double bond capable of radical polymerization.
- (Meth)acrylic polymers include (meth)acrylic esters other than (meth)acrylic acid alkyl esters, (meth)acrylic acid, Other compounds having vinyl groups can be copolymerized.
- Examples of (meth)acrylic esters other than the above-mentioned alkyl (meth)acrylates that can be used as copolymerization components include hydroxyalkyl (meth)acrylates, methoxymethyl (meth)acrylates, and (meth)acrylic acids described below.
- (meth)acrylic acid alkoxyalkyl esters such as methoxyethyl, ethoxymethyl (meth)acrylate, ethoxyethyl (meth)acrylate, phenyl (meth)acrylate, benzyl (meth)acrylate, glycidyl (meth)acrylate, Examples include ⁇ -butyrolactone (meth)acrylate.
- Examples of the compound having a vinyl group include acrylamide compounds such as dimethylacrylamide, hydroxyethylacrylamide, and dimethylaminopropylacrylamide, styrene compounds such as styrene, ⁇ -methylstyrene, and p-methoxystyrene, maleic anhydride, and the like. I can do it.
- components (a1) and (a2), which will be described later, other than these can also be used as appropriate.
- the monomers constituting the (meth)acrylic polymer include the acrylic monomer as a copolymerization component at the point where it will react with the crosslinking agent described below. (a1) may also be contained.
- the acrylic monomer (a1) becomes a reaction point for the crosslinked structure when it is copolymerized with other copolymerization components to form an acrylic resin, and is a functional group that can react with the functional group contained in the crosslinking agent described later.
- a monomer containing a group may be used.
- acrylic monomers (a1) include hydroxyl group-containing monomers, amino group-containing monomers, acetoacetyl group-containing monomers, isocyanate group-containing monomers, glycidyl group-containing monomers, and the like. Among these, hydroxyl group-containing monomers are preferably used because they can efficiently undergo a crosslinking reaction with a crosslinking agent.
- the acrylic monomer (a1) may be used alone or in combination of two or more.
- hydroxyl group-containing monomers examples include 2-hydroxyethyl (meth)acrylate, 4-hydroxybutyl (meth)acrylate, 5-hydroxypentyl (meth)acrylate, 6-hydroxyhexyl (meth)acrylate, and 8-hydroxyoctyl (meth)acrylate.
- Acrylic acid hydroxyalkyl esters such as acrylate; Caprolactone-modified monomers such as caprolactone-modified 2-hydroxyethyl (meth)acrylate; Oxyalkylene-modified monomers such as diethylene glycol (meth)acrylate and polyethylene glycol (meth)acrylate; 2-acryloyloxy Monomers containing primary hydroxyl groups such as ethyl 2-hydroxyethylphthalate and N-methylol (meth)acrylamide; 2-hydroxypropyl (meth)acrylate, 2-hydroxybutyl (meth)acrylate, 3-chloro 2-hydroxypropyl (meth)acrylate; ) Secondary hydroxyl group-containing monomers such as acrylate; and tertiary hydroxyl group-containing monomers such as 2,2-dimethyl-2-hydroxyethyl (meth)acrylate.
- hydroxyl group-containing monomers are preferred because they have excellent reactivity with crosslinking agents, and 2-hydroxyethyl acrylate is preferable because it contains fewer impurities such as di(meth)acrylate. It is particularly preferred because it is easy to manufacture.
- hydroxyl group-containing monomer used in the present invention it is also preferable to use one in which the content of di(meth)acrylate, which is an impurity, is 0.5% by mass or less, more preferably 0.2% by mass or less, especially 0.2% by mass or less. It is preferable to use 0.1% by mass or less.
- hydroxyl group-containing monomers include 2-hydroxyethyl acrylate, 2-hydroxypropyl acrylate, and 4-hydroxybutyl acrylate, which are preferred because they have a low molecular weight and are easy to purify.
- amino group-containing monomer examples include t-butylaminoethyl (meth)acrylate, ethylaminoethyl (meth)acrylate, dimethylaminoethyl (meth)acrylate, and diethylaminoethyl (meth)acrylate.
- acetoacetyl group-containing monomer examples include 2-(acetoacetoxy)ethyl (meth)acrylate, allyl acetoacetate, and the like.
- isocyanate group-containing monomer examples include 2-acryloyloxyethyl isocyanate, 2-methacryloyloxyethyl isocyanate, and alkylene oxide adducts thereof.
- glycidyl group-containing monomer examples include glycidyl (meth)acrylate, allylglycidyl (meth)acrylate, and the like.
- the content of the acrylic monomer (a1) in the monomers constituting the (meth)acrylic polymer is preferably 0.01 to 20% by mass, more preferably 0.1 to 10% by mass, and especially 0.01 to 20% by mass, more preferably 0.1 to 10% by mass, and especially .2 to 3% by mass.
- the acrylic monomer (a1) is greater than or equal to the above lower limit, the crosslinking point during crosslinking becomes appropriate, resulting in good cohesive force after crosslinking.
- it is below the above upper limit it is possible to prevent the adhesive strength from decreasing due to the component (a1).
- the monomer constituting the (meth)acrylic polymer may be a copolymer component, if necessary.
- a copolymerizable monomer (a2) other than (a1) may also be contained.
- Examples of the copolymerizable monomer (a2) include phenyl (meth)acrylate, benzyl (meth)acrylate, phenoxyethyl (meth)acrylate, ethoxylated o-phenylphenyl (meth)acrylate, phenoxydiethylene glycol (meth)acrylate, 2 - Hydroxy-3-phenoxypropyl (meth)acrylate, aromatic ring-containing monomers such as styrene, (meth)acryloylmorpholine, amide monomers such as dimethyl (meth)acrylamide, diethyl (meth)acrylamide, (meth)acrylamide, acrylonitrile, Methacrylonitrile, vinyl acetate, vinyl propionate, vinyl stearate, vinyl chloride, vinylidene chloride, alkyl vinyl ether, vinyl toluene, vinyl pyridine, vinyl pyrrolidone, dialkyl itaconate, dialkyl fumarate, allyl alcohol
- the content of the copolymerizable monomer (a2) in the monomers forming the (meth)acrylic polymer is preferably 0 to 20% by mass, more preferably 1 to 15% by mass, especially 2 to 10% by mass. % by mass, and when the content of the copolymerizable monomer (a2) is within the above range, it is possible to prevent the adhesive properties from deteriorating due to the component (a2).
- the adhesive composition may optionally contain a crosslinking agent and other resins constituting the adhesive component (other than the main component resin and the crosslinking agent) (for example, acrylic resin, rubber, silicone resin, etc.). urethane resin).
- a crosslinking agent and other resins constituting the adhesive component for example, acrylic resin, rubber, silicone resin, etc.). urethane resin.
- the adhesive composition can contain a crosslinking agent depending on its curing method.
- the crosslinking agent include isocyanate crosslinking agents, epoxy crosslinking agents, aziridine crosslinking agents, melamine crosslinking agents, aldehyde crosslinking agents, and amine crosslinking agents.
- isocyanate-based crosslinking agents are preferably used from the viewpoint of improving adhesion to the base material or reactivity with acrylic resins.
- the crosslinking agents may be used alone or in combination of two or more.
- the content of the crosslinking agent is preferably 0.01 to 10 parts by weight, more preferably 0.05 to 5 parts by weight, and more preferably 0.1 to 3 parts by weight, based on 100 parts by weight of the main component resin. It is. If the content of the crosslinking agent is within the above range, the cohesive force will not be insufficient, and desired durability can be obtained, while deterioration of flexibility and adhesive strength can be prevented.
- polyfunctional (meth)acrylates include, for example, trimethylolpropane tri(meth)acrylate, ethylene oxide-modified trimethylolpropane tri(meth)acrylate, pentaerythritol tri(meth)acrylate, pentaerythritol tetra(meth)acrylate, Examples include pentaerythritol penta(meth)acrylate, dipentaerythritol hexa(meth)acrylate, and glycerin polyglycidyl ether poly(meth)acrylate.
- the adhesive composition may optionally contain resins constituting the adhesive component other than the main component resin and the crosslinking agent (for example, acrylic resin, rubber, silicone resin, urethane resin). May contain.
- resins constituting the adhesive component other than the main component resin and the crosslinking agent for example, acrylic resin, rubber, silicone resin, urethane resin. May contain.
- tackifiers such as rosin, rosin ester, hydrogenated rosin ester, phenol resin, aromatic modified terpene resin, aliphatic petroleum resin, alicyclic petroleum resin, styrene resin, xylene resin, silane coupling
- Additives such as conventionally known additives such as antistatic agents, colorants, fillers, antiaging agents, ultraviolet absorbers, and functional dyes, as well as compounds that cause coloration or discoloration when exposed to ultraviolet rays or radiation. can be blended.
- the blending amount of these additives is preferably 10% by mass or less, more preferably 5% by mass or less of the entire adhesive composition (based on non-volatile components), and is used as an additive with a molecular weight of less than 10,000. It is preferable to contain as few molecular components as possible from the viewpoint of excellent durability.
- the method of forming the adhesive layer is not particularly limited, and the adhesive layer may be formed by applying the above-mentioned adhesive composition onto the resin layer (A) of the present release film, followed by drying and curing as appropriate.
- the above-mentioned pressure-sensitive adhesive composition is applied onto a base film or a release film having a different release force from the present release film, and after drying and curing as appropriate, a release film having a different release force from the present release film is applied.
- the release layer (A layer) of the present release film may be bonded to the surface opposite to the surface provided with the release film.
- the adhesive composition may be cured depending on the crosslinking agent and the main component resin, and may be cured by heating or by irradiation with light such as ultraviolet rays. Moreover, the above-mentioned pressure-sensitive adhesive composition may be diluted with an organic solvent or the like as appropriate and then applied onto a release layer or the like.
- the thickness of the adhesive layer is not particularly limited.
- the thickness is preferably 10,000 ⁇ m or less, particularly 3,000 ⁇ m or less, and even more preferably 1,000 ⁇ m or less.
- the adhesive layer preferably has an elastic modulus of 6.0 MPa or less at 25°C.
- the elastic modulus here is a value measured using a nanoindenter.
- the elastic modulus of the adhesive layer is more preferably 5.0 MPa or less, still more preferably 4.0 MPa or less, especially 3.0 MPa or less.
- the elastic modulus of the adhesive layer at 25° C. is preferably 0.5 MPa or more, more preferably 1.0 MPa or more, and even more preferably 1.5 MPa or more from the viewpoint of adhesive layer formability.
- Glass transition temperature (Tg) of adhesive layer The glass transition temperature is calculated using the following Fox equation.
- Tg Glass transition temperature (K) of copolymer
- Tga Glass transition temperature (K) of homopolymer of monomer A
- Wa Weight fraction of monomer A
- Tgb Glass transition temperature (K) of homopolymer of monomer B
- Tgn Glass transition temperature (K) of homopolymer of monomer N
- Tg is preferably in the range of -55°C to -70°C, more preferably in the range of -58°C to -68°C.
- the film laminate can be used in a variety of laminate configurations.
- the release film with an adhesive layer may be a film laminate in which an optical member is bonded to the surface of the adhesive layer.
- Such a film laminate can be created by peeling off the release film and bonding the optical member to an adherend using the exposed adhesive layer.
- the optical member include a polarizing plate and a touch sensor.
- it may be an in-vehicle optical member such as a touch panel mounted in an automobile.
- the material and structure of the polarizing plate are arbitrary; for example, a stretched polyvinyl alcohol film using iodine as an alignment dye and a TAC (triacetyl cellulose) film laminated as a protective film has been widely put into practical use as this type of polarizing plate. has been done. Further, the polarizing plate may have a layer structure on its surface having functions such as a hard coat having substantially no retardation, anti-glare, low reflection, and antistatic properties.
- a touch sensor is a component that reacts to the touch when a user touches an image displayed on the screen with a finger or touch pen, and determines the touch point.
- the touch sensor is a component that reacts to the touch and determines the touch point. Examples include methods such as a surface wave method using infrared rays or ultrasonic waves.
- a touch sensor is installed in a display device such as a liquid crystal display panel or an organic EL display.
- Touch sensor films are commonly provided with a patterned transparent conductive layer to perform the function of sensing electrodes.
- the film laminate includes the above-mentioned release film with an adhesive layer and another release film, and has a layered structure in which the release film with an adhesive layer is bonded to the other release film via the adhesive layer.
- a film laminate having such a layered structure is composed of the present release film/adhesive layer/another release film and can be used as a double-sided adhesive sheet.
- Other release films are those in which a release layer is formed on the surface of a base material such as a resin film, and the surface on which the release layer is formed is preferably bonded to an adhesive layer.
- As the release layer of another release film it is preferable to use a release layer other than the above-mentioned main release layer.
- the film laminate having the above structure has a ratio (MB/MA) of the elastic modulus (MA) of the release layer of the present release film to the elastic modulus (MB) of the release layer of the other release film, of 4.
- the range is preferably 30 to 30, more preferably 4 to 25.
- the film laminate to which the other release films mentioned above are bonded is preferably used by peeling off the release film and bonding the exposed adhesive layer surface of the remaining release film with an adhesive layer to an optical member. . After that, another release film may be peeled off from the adhesive layer of the release film with an adhesive layer. Further, after that, the release film can be attached again onto the adhesive layer attached to the optical member.
- the release film of the present invention since the release film of the present invention has ultra-light releasability and low migration properties, it is also possible to use what is called a re-peelable release film.
- the normal peeling force of other release films measured by 180° peeling at a peeling speed of 0.3 m/min is greater than that of the present release film. It is preferably about 2 to 10 times the normal peeling force, more preferably 2 to 6 times.
- the above-mentioned release film and film laminate are preferably used for vehicle mounting.
- X to Y means “more than or equal to X and less than or equal to Y” unless otherwise specified, and also means “preferably greater than It also includes the meaning of "less than”.
- X is any number
- it includes the meaning of "preferably greater than X” unless otherwise specified, and it is written as "less than or equal to Y" (where Y is any number). In this case, unless otherwise specified, it also includes the meaning of "preferably smaller than Y".
- Molecular weight measurement of curable silicone resin Measure the chromatogram using a GPC measuring device, determine the number average molecular weight (Mn) and mass average molecular weight (Mw) based on a calibration curve using standard polystyrene, It is shown in Table 2. Specifically, 4 mg of a sample for measurement was dissolved in 4 mL of THF to obtain a measurement solution, and 100 ⁇ L of the measurement solution was injected into a GPC measuring device for measurement. Tetrahydrofuran (THF) was used as the eluent.
- THF Tetrahydrofuran
- Adhesive peeling force The adhesive composition listed in Table 8 was applied to the release layer surface of the sample film so that the film thickness in a wet state was 2 mil, and heat treated at 150°C for 3 minutes. The adhesive layer was cured to form an adhesive layer (thickness (after drying): 20 ⁇ m).
- the polyester used in the Examples and Comparative Examples was prepared as follows.
- polyester (B) ⁇ Method for producing polyester (B)>
- the method for producing polyester (A) after adding 0.04 parts by mass of ethyl acid phosphate, 0.2 parts by mass of silica particles dispersed in ethylene glycol having an average particle size of 2.3 ⁇ m, and 0.04 parts by mass of antimony trioxide.
- Polyester (B) was obtained using the same method as that for producing polyester (A), except that part by mass was added and the polycondensation reaction was stopped at a time point corresponding to an intrinsic viscosity of 0.65.
- the obtained polyester (B) had an intrinsic viscosity of 0.65.
- a biaxially stretched polyester film having a thickness of 50 ⁇ m was obtained, having a layer (B) thickness (after drying) of 0.05 ⁇ m and an undercoat layer thickness (after drying) of 0.05 ⁇ m.
- coating liquid 1 for resin layer (A) shown in Table 3 was applied to the undercoat layer surface of the obtained polyester film as a resin layer (A) so that the thickness (after drying) was 1.0 ⁇ m. Like No. After coating by a bar coating method using 4 bars, it was dried at 150° C. for 30 seconds to obtain a release film.
- Examples 1-2 to 1-12 A release film was obtained in the same manner as in Example 1-1 except that coating solutions 2 to 12 of the resin layer (B) shown in Table 4 were applied to one side of the longitudinally stretched film.
- the obtained release film has a thin resin layer (A) and an elastic modulus of 500 MPa or less, while the resin layer (B) contains a non-silicone release agent. Although it achieved ultra-light peeling with a peeling force (normal peeling force) of 5 g/25 mm or less, it also achieved blocking resistance with a heavy peeling rate of 100% or less after pressing. Furthermore, the adhesion to the base material was good and the migration property was low.
- coating liquid 1 for resin layer (A) shown in Table 3 was applied to the undercoat layer surface of the obtained polyester film as a resin layer (A) so that the thickness (after drying) was 1.0 ⁇ m. Like No. After coating by a bar coating method using 4 bars, it was dried at 150° C. for 30 seconds to obtain a release film.
- the resulting release film has a thin resin layer (A) and an elastic modulus of 500 MPa or less, while the resin layer (B) contains a non-silicone release agent. Although it achieved ultra-light peeling with a peeling force (normal peeling force) of 5 g/25 mm or less, it also achieved blocking resistance with a heavy peeling rate of 100% or less after pressing. Furthermore, the adhesion to the base material was good and the migration property was low.
- Comparative example 1-1 A release film was obtained in the same manner as in Example 1-1 except that the resin layer (B) and the undercoat layer were not provided on the longitudinally stretched film.
- Comparative example 1-2 A release film was obtained in the same manner as in Example 1-1 except that the resin layer (B) was not provided on the longitudinally stretched film.
- the resulting release films of Comparative Examples 1-1 and 1-2 had a high rate of heavy release after pressing due to the absence of the resin layer (B) containing a non-silicone release agent. There was a concern that blocking would occur.
- Example 1-1 the resin layer (B) composition was manufactured in the same manner as in Example 1 except that the coating liquid 13 containing no non-silicone mold release agent was used to obtain a release film. As shown in Table 5, the obtained release film did not contain a non-silicone release agent in the resin layer (B), so the rate of heavy release after pressing was high and there was concern that blocking would occur. Ta.
- Example 1-1 a release film was obtained in the same manner as in Example 1 except that the coating liquid 2 for the resin layer (A) was used. As shown in Table 5, the resulting release film has a peeling force (normal peeling force) of about 3 of Examples 1-1 to 1-12 because the resin layer (A) has a thin thickness and a high elastic modulus. However, it was not possible to achieve ultra-light peeling, which was twice as expensive. Moreover, the adhesion to the base material was also poor.
- Reference example 1 A release film was obtained in the same manner as in Example 1 except for changing to coating liquid 3 (particle content: 0.19% by mass) for resin layer (A). As shown in Table 5, the resulting release film had particles that made the resin layer (A) brittle, resulting in poor adhesion to the substrate and relatively high migration properties due to particles falling off. there were. In addition, the rate of heavy peeling after pressing was high, and there was a concern that blocking would occur.
- Examples of compounds constituting the resin layer (A) are as follows. a1: Cured silicone resin (silicone resin with vinyl groups introduced into the side chain and/or end of the main chain consisting of siloxane bonds, number average molecular weight: 10600, adjusted to 15% by mass by diluting with n-heptane solvent) Viscosity at 25°C: 1.7 mcps) a2: Curable silicone resin (silicone resin with vinyl groups introduced into the side chains and/or ends of the main chain consisting of siloxane bonds, number average molecular weight: 364,000) and a silicone crosslinking agent (the main chain consisting of siloxane bonds) (Silicone resin with Si-H groups introduced into the side chain and/or terminal) (viscosity at 25°C when adjusted to 15% by mass by diluting with n-heptane solvent: 410 mcps)
- the content ratio (mol%) of each functional group in the curable silicone resin is as follows.
- b1 Silicone crosslinking agent (CL750: manufactured by Momentive Performance Materials)
- c1 Addition type platinum catalyst (CM678: Manufactured by Momentive Performance Materials)
- c2 Addition type platinum catalyst (PL-50T: manufactured by Shin-Etsu Chemical Co., Ltd.)
- d1 Particles (Tospearl 120: Manufactured by Momentive Performance Materials)
- the coating liquid constituting the resin layer (A) is as follows.
- Examples of compounds constituting the resin layer (B) and the undercoat layer are as follows.
- (Release agent) AI Long-chain alkyl group-containing compound 200 parts of xylene and 600 parts of octadecyl isocyanate were added to a four-necked flask and heated while stirring. From the time the xylene began to reflux, 100 parts of polyvinyl alcohol having an average degree of polymerization of 500 and a degree of saponification of 88 mol% was added little by little at 10 minute intervals over about 2 hours. After the addition of polyvinyl alcohol was completed, reflux was further carried out for 2 hours to complete the reaction.
- reaction product When the reaction mixture was cooled to about 80°C and added to methanol, the reaction product was precipitated as a white precipitate, so this precipitate was filtered off, 140 parts of xylene was added, and the mixture was heated to completely dissolve. After repeating the operation several times by adding methanol again to precipitate, the precipitate was washed with methanol, dried and pulverized to obtain the precipitate.
- AII Wax: 300 g of oxidized polyethylene wax with a melting point of 105°C, an acid value of 16 mg KOH/g, a density of 0.93 g/mL, and an average molecular weight of 5000, and ion exchange in a 1.5 L emulsification equipment equipped with a stirrer, a thermometer, and a temperature controller.
- Add 650 g of water, 50 g of decaglycerin monooleate surfactant, and 10 g of 48% potassium hydroxide aqueous solution replace with nitrogen, seal, stir at 150°C for 1 hour, cool to 130°C, and turn the high-pressure homogenizer to 400 atm.
- the wax emulsion was passed under and cooled to 40°C.
- a polyester polyurethane containing 876 parts by mass of (C1a), 244 parts by mass of tolylene diisocyanate, 81 parts by mass of ethylene glycol, and 67 parts by mass of dimethylolpropionic acid was neutralized with ammonia and dispersed in water ( Concentration 20%, viscosity 50mPa ⁇ s at 25°C)
- surfactant A nonionic surfactant having a structure having polyethylene oxide in the side chain, as shown in the following formula.
- m and n are integers indicating the number of moles of ethylene oxide added, and in this case, the average of m+n was 10.
- the coating liquid constituting the resin layer (B) is as follows. Note that Table 3 shows the amounts based on non-volatile components.
- the reaction was stopped at a point corresponding to an intrinsic viscosity of 0.59 dl/g by changing the stirring power of the reaction tank, and the polymer was discharged under nitrogen pressure to form polyester (1) with an intrinsic viscosity of 0.59 dl/g. I got it.
- the temperature was gradually raised from 230°C to 280°C.
- the pressure was gradually reduced from normal pressure to 0.3 mmHg.
- the reaction was stopped at a point corresponding to an intrinsic viscosity of 0.59 dl/g by changing the stirring power of the reaction tank, and the polymer was discharged under nitrogen pressure to form polyester (2) with an intrinsic viscosity of 0.59 dl/g. I got it.
- Polyester (2) was produced in the same manner as polyester (2) except that 10 parts by mass of organic particles (styrene-divinylbenzene: styrene resin) with an average particle size of 4.5 ⁇ m were added instead of silica particles. Polyester (3) having an intrinsic viscosity of 0.60 dl/g was obtained.
- PET-A ⁇ Production method of polyester film>
- a blend of polyester (1) and polyester (2) at a mass ratio of 80/20 is used as the raw material for layer A, and only polyester (1) is used as the raw material for layer B, and polyester (1) and polyester (3) are blended by mass.
- ⁇ Release layer composition> a1 Cured silicone resin (silicone resin with vinyl groups introduced into the side chain and/or end of the main chain consisting of siloxane bonds, number average molecular weight: 10600, adjusted to 15% by mass by diluting with n-heptane solvent) Viscosity at 25°C: 1.7 mcps)
- composition analysis of curable silicone resin Compositional analysis of the curable silicone resin used in Examples and Comparative Examples was performed using 400MHz-NMR (Bruker Avance 400M), and the results are shown in Table 1. 1 H-NMR measurements were carried out at a temperature of 30° C. using CDCl 3 as a solvent and using the peak derived from the methyl group of dimethylsiloxane as a chemical shift standard. Table 1 below shows the content ratio (mol%) of each functional group in the curable silicone resin.
- b1 Crosslinking agent (CL750: manufactured by Momentive Performance Materials)
- c1 Addition type platinum catalyst (CM678: Manufactured by Momentive Performance Materials)
- Example 2-1 The following release layer composition was applied to the flat surface of the A-layer side of PET-A so that the coating amount (after drying) was 1 g/m 2 , and then dried at 150°C for 30 seconds and released. A mold film (sample film) was obtained.
- Example 2-2 to 2-5 The release layer composition was produced in the same manner as in Example 2-1, except that the type and amount of the light release agent were changed as shown in Table 7 below, to obtain a film laminate.
- Example film A release film (sample film) was obtained in the same manner as in Example 1 except that the composition of the release layer was changed to the following release layer composition.
- Comparative Example 1 was at the level of a conventional product (light peel type) and had a large peel force against the adhesive layer.
- Comparative Example 2 contained a light release agent with a large molecular weight in the release layer, so the light release effect was poor.
- Example 3-1 The release layer used in Example 1-1 was evaluated for releasability using the adhesive described below. The results are shown in Table 8.
- Example 3-2 In Example 3-1, evaluation was performed in the same manner as in Example 3-1, except that the mold release layer used in Example 2-1 was used as the mold release layer. The results are shown in Table 8.
- the release film of the present invention it is possible to provide a release film in which the release properties of the release layer are not easily changed due to blocking while achieving ultra-light release properties, and its industrial value is high.
- the release film of the present invention and the film laminate including the release film have features that have both ultra-light peelability and anti-blocking properties, changes in the release properties of the release layer and resin layer ( A) It is suitable for applications where deterioration of the surface appearance is undesirable.For example, it is suitable for various applications such as the production of capacitive touch panels, etc., which are bonded together via an adhesive layer, and optical members (polarizing plates, etc.) used in liquid crystal displays.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Materials Engineering (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Physics & Mathematics (AREA)
- Theoretical Computer Science (AREA)
- General Physics & Mathematics (AREA)
- General Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Optics & Photonics (AREA)
- Human Computer Interaction (AREA)
- Laminated Bodies (AREA)
Abstract
Description
また、近年の粘着剤のトレンドとして、段差吸収性がさらに良好となる点で、低弾性率の粘着層が嗜好される傾向にある。低弾性率の粘着層に対応する場合、離型フィルム剥離時に粘着層表面が変形しやすく、糸引き現象が発生する場合がある。そのため、離型フィルムには更なる軽剥離化、いわゆる、超軽剥離化が必要とされる。
本発明は上記実状に鑑みなされたものであって、その解決課題は、各種の粘着剤に対し超軽剥離性を実現しながらも、ブロッキングにより離型層の剥離特性が変化しにくい離型フィルム、及びフィルム積層体を提供することにある。
[1]基材フィルムの一方の面に樹脂層(A)を備え、他方の面に樹脂層(B)を備えた離型フィルムであり、前記樹脂層(A)は、シリコーン樹脂系離型剤を含み、厚みが0.2~2.0μmであり、前記樹脂層(B)は、非シリコーン樹脂系離型剤を含む、離型フィルム。
[2]基材フィルムの少なくとも片面側に樹脂層(A)を備え、前記樹脂層(A)が、硬化型シリコーン樹脂および軽剥離化剤を含む樹脂層組成物の硬化物であり、前記軽剥離化剤が下記式(I)で示されるジメチルシロキサン骨格(DM)および下記式(II)で示されるメチルフェニルシロキサン骨格(MP)を有し、厚みが0.2~1.5μmである、離型フィルム。


[3]前記樹脂層(A)を設ける面とは反対側のフィルム表面の最大断面高さ(Rt)が、2.0μm以下である、上記[2]に記載の離型フィルム。
[4]前記基材フィルムの前記樹脂層(A)が設けられる面とは反対側の面が、平均粒径1~6μmの粒子を0.4~1.0質量%含有するポリエステル層を備える、上記[2]または[3]に記載の離型フィルム。
[5]前記軽剥離化剤の質量平均分子量が1万以上10万以下である、上記[2]~[4]のいずれかに記載の離型フィルム。
[6]前記樹脂層(A)のナノインデンター装置を用いて測定した、25℃における弾性率が500MPa以下である、上記[1]~[5]のいずれかに記載の離型フィルム。
[7]前記非シリコーン樹脂系離型剤がワックス、長鎖アルキル基含有化合物、フッ素化合物の群から選択される少なくとも1種である、上記[1]または[6]に記載の離型フィルム。
[8]前記樹脂層(B)の不揮発成分中における、離型剤の含有量が10~70質量%である、上記[1]、[6]または[7]に記載の離型フィルム。
[9]前記樹脂層(B)が粒子を実質的に含有しない、上記[1]及び[6]~[8]のいずれかに記載の離型フィルム。
[10]下記評価方法で測定した、樹脂層(A)の常態剥離力が5g/25mm以下である、上記[1]~[9]のいずれかに記載の離型フィルム。
<評価方法>
前記離型フィルムの樹脂層(A)面にアクリル系粘着テープ(Tesa製の「No.7475」)を貼り付けた後、25mm×150mmのサイズにカットし、室温にて1時間放置する。剥離力は引張速度0.3m/minの条件下で180°で剥離を行う。
[11]前記樹脂層(A)あるいは樹脂層(B)の少なくとも一方の層と前記基材フィルムとの間に下引き層を備える、上記[1]及び[6]~[10]のいずれかに記載の離型フィルム。
[12]前記下引き層が帯電防止層である、上記[11]に記載の離型フィルム。
[13]前記下引き層が下記化合物(A)~(C)を含有する、上記[11]又は[12]に記載の離型フィルム。
(A)チオフェンまたはチオフェン誘導体からなる化合物に、他の陰イオン化合物によりドーピングされた重合体、またはチオフェンまたはチオフェン誘導体からなる化合物中に陰イオン基を持ち自己ドープされた重合体
(B)ポリヒドロキシ化合物
(C)ポリウレタン樹脂、ポリエステル樹脂、及びアクリル樹脂からなる群から選ばれる1種以上の化合物
[14]前記樹脂層(A)あるいは樹脂層(B)の少なくとも一方の表面の表面固有抵抗率が1×1012Ω/□以下である、上記[1]及び[6]~[13]のいずれかに記載の離型フィルム。
[15]前記基材フィルムがポリエステルフィルムである、上記[1]~[14]のいずれかに記載の離型フィルム。
[16]前記ポリエステルフィルムが三層構成である、上記[15]に記載の離型フィルム。
[17]以下の方法で評価した前記樹脂層(A)のプレス後重剥離化率が100%以下である、上記[1]~[16]のいずれかに記載の離型フィルム。
<評価方法>
前記樹脂層(A)面にアクリル系粘着テープ(Tesa製の「No.7475」)を貼り付けた後、25mm×150mmのサイズにカットし、室温にて1時間放置する。引張速度0.3m/minの条件下で180°で剥離した剥離力を(F1)とする。
次に、前記樹脂層(A)面に接触するように積層し、温度40℃、湿度90%RH、荷重1MPaで20時間プレス処理を行う。処理後の前記樹脂層(A)面に、アクリル系粘着テープ(Tesa製の「No.7475」)を貼り付けた後、25mm×150mmのサイズにカットし、室温にて1時間放置する。引張速度0.3m/minの条件下で180°で剥離した剥離力を(F2)とする。
次式によってプレス後重剥離化率を求める。
プレス後重剥離化率(%)=(F2-F1)/F1×100
[18]前記樹脂層(A)面の中心線平均粗さ(Ra(A))が30nm以下である、上記[1]~[17]のいずれかに記載の離型フィルム。
[19]前記樹脂層(B)面の中心線平均粗さ(Ra(B))が60nm以下である、上記[1]または[6]~[18]のいずれかに記載の離型フィルム。
[20]上記[1]~[19]の何れかに記載の離型フィルムと、粘着層を介して、他の離型フィルムを備えた、粘着シート。
[21]前記粘着層がアクリル系粘着剤組成物から形成される、上記[20]に記載の粘着シート。
[22]前記粘着層の弾性率(25℃)が6.0MPa以下である、上記[20]又は[21]に記載の粘着シート。
[23]前記粘着層が2-エチルヘキシルアクリレートおよび/またはブチルアクリレートを含む、上記[20]~[22]のいずれかに記載の粘着シート。
[24]光学用透明粘着シート用である、上記[20]~[23]のいずれかに記載の粘着シート。
[25]ディスプレイ用である、上記[1]~[19]のいずれかに記載の離型フィルム。
[26]フォルダブルディスプレイ用である、上記[1]~[19]のいずれかに記載の離型フィルム。
[27]上記[20]~[23]のいずれかに記載の粘着シートにおいて、前記他の離型フィルムを剥がした後、露出する粘着層表面を光学部材に貼り合わせる、粘着シートの使用方法。
[28]前記光学部材が偏光板またはタッチセンサーである、上記[27]に記載の粘着シートの使用方法。
本発明の実施形態の一例に係る離型フィルム(以下、「本離型フィルム」と称することがある。)は、基材フィルムの一方の面に樹脂層(A)を備え、他方の面に樹脂層(B)を備えた離型フィルムである。離型フィルムは、巻き取られたロール状の離型フィルム(離型フィルムロール)とされ、離型フィルムロールの形態で保管などされるのがよい。その場合、離型層(A)はロールの内側および外側のどちら側に配置してもよい。
以上の構成を有する離型フィルムは、ロール状とされると、樹脂層(A)の面が、樹脂層(B)の面と重ね合わされるが、その際、樹脂層(B)により樹脂層(A)表面への密着によるダメージを軽減して、ブロッキングを抑制し、よりフィルムに圧力が加わるロールの下巻き部分での樹脂層(A)の重剥離化および表面の外観の悪化を防ぐことができる。また、樹脂層(A)は、超軽剥離性を実現しながらも、粒子を実質的に含有しないことで基材フィルムに対する密着性も良好で、さらには粒子脱落もないため粘着テープ等への移行性を低減することもできる。
本離型フィルムにおける基材フィルムは、フィルム状を呈するものであれば、その材料を特に限定するものではない。例えば、紙製、樹脂製、金属製などであってもよい。これらの中でも、機械的強度および柔軟性の観点から、樹脂製であることが好ましい。
樹脂製の基材フィルムとしては、例えばポリエチレン、ポリプロピレン、ポリエステル、ポリスチレン、ポリカーボネート、ポリエーテルスルホン、ポリアミド、ポリイミドなどの高分子を膜状に形成したフィルムを挙げることができる。また、フィルム化が可能であれば、これらの材料を混合したもの(ポリマーブレンド)や構成単位を複合化したもの(共重合体)であっても構わない。
上記例示したフィルムの中でも、耐熱性、平面性、光学特性、強度などの物性が優れている点から、ポリエステルフィルムであることが特に好ましい。
本発明の積層フィルムを構成するポリエステルフィルムは単層構成であっても多層構成であってもよく、2層、3層構成以外にも本発明の要旨を越えない限り、4層またはそれ以上の多層であってもよく、特に限定されるものではない。本発明においては、少なくとも3層構成からなるポリエステルフィルムであることが好ましい。また、ポリエステルフィルムとしては二軸延伸ポリエステルフィルムが、薄膜化や寸法安定性の点などから好ましい。
なお、主成分樹脂とは、本ポリエステルフィルムを構成する樹脂の中で最も質量割合の大きい樹脂の意味であり、本ポリエステルフィルムを構成する樹脂の50質量%以上、或いは75質量%以上、或いは90質量%以上、或いは100質量%を占める場合である。
また、チタン化合物によるポリエステルを用いる場合、溶融押出する工程での劣化抑制の目的で、チタン化合物の活性を下げるためにリン化合物を使用することが好ましい。リン化合物としては、ポリエステルの生産性や熱安定性を考慮すると正リン酸が好ましい。リン元素含有量は、溶融押出するポリエステル量に対して、好ましくは1~300質量ppm、より好ましくは3~200質量ppm、さらに好ましくは5~100質量ppmの範囲である。リン化合物の含有量が上記上限値以下であると、ゲル化や異物の原因となることがなく、また、上記下限値以上であると、チタン化合物の活性を十分に下げることができ、着色を抑制できて、黄色味のあるフィルムとなることがない。
また、ポリエステルフィルムが単層で構成される場合でも、その単層のポリエステル層は、耐ブロッキング防止性と離型性の観点から、粒子を含有するよく、その粒子の平均粒径と粒子添加量の範囲を上記のようにするとよい。
次にポリエステルフィルムの製造例について具体的に説明するが、以下の製造例に何ら限定されるものではない。例えば、二軸延伸ポリエステルフィルムを製造する場合、まず先に述べたポリエステル原料を、押出機を用いてダイから溶融押し出しし、溶融シートを冷却ロールで冷却固化して未延伸シートを得る。この場合、シートの平面性を向上させるためシートと回転冷却ドラムとの密着性を高めることが好ましく、静電印加密着法や液体塗布密着法が好ましく採用される。次に得られた未延伸シートを一方向にロールまたはテンター方式の延伸機により延伸する。延伸温度は、通常70~120℃、好ましくは80~110℃であり、延伸倍率は通常2.5~7倍、好ましくは3.0~6倍である。次いで、一段目の延伸方向と直交する方向に、通常70~170℃で、延伸倍率は通常2.5~7倍、好ましくは3.0~6倍で延伸する。引き続き180~270℃の温度で緊張下または30%以内の弛緩下で熱処理を行い、二軸配向フィルムを得る方法が挙げられる。上記の延伸においては、一方向の延伸を2段階以上で行う方法を採用することもできる。その場合、最終的に二方向の延伸倍率がそれぞれ上記範囲となるように行うのが好ましい。
次に本発明における離型フィルムを構成する樹脂層(A)の形成について説明する。
一方、溶剤型の硬化型シリコーンとは、溶剤に希釈しなければ塗工できない程度に粘度の高いシリコーン樹脂であり、無溶剤型の硬化型シリコーンに比べると、比較的高い分子量を有するシリコーンである。
基材フィルムへの密着性が良好となり、且つ、コートムラがない均一なコート外観、且つ、樹脂層(A)の厚みの調整もし易いという観点から、溶剤型硬化型シリコーンであるのが好ましい。
R(3-a)XaSiO-(RXSiO)m-(R2SiO)n-SiXaR(3-a) ・・・(1)
HbR1 (3-b)SiO-(HR1SiO)x-(R1 2SiO)y-SiR1 (3-b)Hb ・・・(2)
また、軽剥離化剤の質量平均分子量は1万未満であることが好ましい。軽剥離化剤の質量平均分子量が1万未満であると移行性および軽剥離性の点で有利である。


Si(X)d(Y)e(R1)f ・・・(5)
[上記式中、Xはエポキシ基、メルカプト基、(メタ)アクリロイル基、アルケニル基、ハロアルキル基およびアミノ基から選ばれる少なくとも1種を有する有機基、R1は一価炭化水素基であり、かつ炭素数1~10のものであり、Yは加水分解性基であり、dは1または2の整数、eは2または3の整数、fは0または1の整数であり、d+e+f=4である]
かかる観点から、硬化型シリコーン樹脂の数平均分子量(Mn)は、9000以上であるのが好ましく、中でも10000以上であるのがより好ましく、中でも20000以上がさらに好ましく、その中でも特に30000以上であるのがさらに好ましい。他方、350000以下であることが好ましく、50000以下であるのがより好ましく、中でも40000以下であることがさらに好ましい。
なお、数平均分子量(Mn)及び質量平均分子量(Mw)は、ポリスチレン基準でゲルパーミエーションクロマトグラフィー(GPC)測定により求めた値であり、具体的な測定方法は、GPC測定装置を使用して、クロマトグラムを測定し、標準ポリスチレンを使用した検量線に基づいて数平均分子量(Mn)及び質量平均分子量(Mw)を求める。具体的には、測定用の試料4mgを、4mLのTHFに溶解して測定溶液とし、測定溶液100μLをGPC測定装置に注入して測定した。溶離液にはテトラヒドロフラン(THF)を使用した。分析には東ソー(株)製「Ecosec8320」を使用し、ガードカラムには東ソー(株)製「TSKgel guardcolumn HXL-L」、カラムには東ソー(株)製「TSKgel GMHXL」を4本連結して使用した。また、オーブンの温度は40℃、THF流量1.0mL/分の条件で分析を行い、検出にはRIを用いた。
硬化型シリコーン樹脂の当該粘度が1mcps以上であれば、塗布液の適度な粘度によりハジキが抑制され、視認性の高い均一なコート外観が得られるため好ましく、400mcps以下であれば、樹脂層(A)組成物の流動性を維持することができ、樹脂層(A)組成物を塗布した際に、筋状のコートムラが生じるのを抑制でき、樹脂層(A)表面を平滑にすることができる。
粘度の測定は、硬化型シリコーン樹脂をn-ヘプタンで15質量%に溶媒希釈し、この溶液の25℃での粘度を、E型粘度計(東機産業(株)製「TVE-22L」)を用いて測定した。
本発明における樹脂層(B)に関して、以下に説明する。
これらの中でも汚染性が少なく、ブロッキング軽減に優れるという点からは長鎖アルキル化合物やワックスが好ましく、特にブロッキング軽減の観点で長鎖アルキル化合物がより好ましい。これらの離型剤は単独で用いてもよいし、複数種使用してもよい。
ポリエステル樹脂は、多価カルボン酸と多価ヒドロキシ化合物との重縮合物である。上記した化合物の中から、多価カルボン酸及び多価ヒドロキシ化合物それぞれを適宜1つ以上選択し、常法の重縮合反応によりポリエステル樹脂を合成すればよい。また、ポリエステル樹脂は、水分散体としてもよく、その場合、ポリエステル樹脂には適宜親水性官能基などを導入してもよい。
また、アクリル樹脂は、基材フィルムとの密着性をより向上させるために、ヒドロキシル基、アミノ基を含有してもよい。
樹脂層(B)において、非シリコーン系離型剤として長鎖アルキル基含有化合物を用いる場合、樹脂層(B)組成物中の全不揮発成分に占める割合として、長鎖アルキル基含有化合物は好ましくは5~90質量%、より好ましくは10~70質量%、更に好ましくは20~60質量%、最も好ましくは20~40質量%の範囲である。長鎖アルキル基含有化合物の割合が上記範囲であると、良好なブロッキング防止性能が得られ、ブロッキングによる離型層の剥離特性の変化を軽減することで、よりフィルムに圧力が加わるロールの下巻き部分での離型層の重剥離化を大きく抑制できる。
好ましい組成物の組み合わせとして、長鎖アルキル基含有化合物とメラミン化合物、長鎖アルキル基含有化合物とオキサゾリン化合物、長鎖アルキル基含有化合物とメラミン化合物とオキサゾリン化合物、ワックスとメラミン化合物とオキサゾリン化合物の組み合わせが例示される。
ワックスは撥水性以外にも、撥油性を有する特徴をもつ。それにより、組み合わせるメラミン化合物との相性が悪く、メラミン化合物がさらにはじきやすい状態になる。
一方、本願発明の重剥離化率評価は、高温(40℃)、高湿度(90%RH)下において、水分が介在した状態でプレス処理されるため、ワックスとメラミン化合物を組み合わせた樹脂層(B)表面には凝集物が生成しやすくなり、反対面のシリコーン離型層との接着性を向上させた結果、重剥離化率が大きくなったものと推察される。
上述の通り、従来の非シリコーン系離型剤では注目していなかった、撥油効果にも着目して、離型剤を選択し、樹脂層(B)を備えた、離型フィルムを構成している点に本願発明の特徴がある。
本発明における樹脂層(B)は、以下の特性を有することができる。
本離型フィルムは、樹脂層(A)あるいは樹脂層(B)の少なくとも一方の層と基材フィルムとの間に下引き層を備えることが好ましい。下引き層は、樹脂層(A)と基材フィルムの間、又は樹脂層(B)と基材フィルムの間のいずれかに備えていてもよく、また両方に備えていてもよい。
なお、本離型フィルムにおいては、良好な帯電防止性能を有する観点から、チオフェンまたはチオフェン誘導体からなる化合物を含有する下引き層であることがより好ましい。
帯電防止性能を有する下引き層の厚みは、好ましくは0.005μm以上0.25μm以下、より好ましくは0.008μm以上0.15μm以下、さらに好ましくは0.01μm以上0.10μm以下である。下引き層の厚みが上記の範囲内であれば、良好な帯電防止性を付与できる。
次に樹脂層(B)および下引き層の形成方法について説明する。
以下に限定するものではないが、例えば逐次二軸延伸においては、特に長手方向(縦方向)に延伸された一軸延伸フィルムにコーティングした後に横方向に延伸する方法が優れている。かかる方法によれば、製膜と樹脂層(B)の形成を同時に行うことができるため製造コスト上のメリットがあり、また、コーティング後に延伸を行うために、樹脂層(B)の厚みを延伸倍率により変化させることもでき、オフラインコーティングフィルムに比べ、薄膜コーティングをより容易に行うことができる。また、延伸前にフィルム上に樹脂層(B)を設けることにより、樹脂層(B)をポリエステルフィルムと共に延伸することができ、それにより樹脂層(B)をポリエステルフィルムに強固に密着させることができる。
さらに、二軸延伸ポリエステルフィルムの製造において、クリップ等によりフィルム端部を把持しつつ延伸することで、フィルムを縦及び横方向に拘束することができ、熱固定工程において、しわ等が入らず平面性を維持したまま高温をかけることができる。それゆえ、塗布後に施される熱処理が他の方法では達成されない高温とすることができるために、オフラインコートと比較して樹脂層(B)および下引き層の硬化がより進み、更に強固な塗膜を作ることができる。
本発明における離型フィルムは、積層構成としてもよく、好ましい形態としては、本離型フィルムの樹脂層(A)側の面に粘着層を積層した形態が挙げられる。より好ましい形態としては、本離型フィルムを第一離型フィルムとし、本離型フィルムの樹脂層(A)側に粘着層、本離型フィルムの樹脂層(A)が貼り合わされた粘着層の他方の面に本離型フィルムとは剥離力の異なる離型フィルムを第二離型フィルムとし、貼り合わされたものが挙げられる。更に好ましい形態としては、粘着層が光学用透明粘着シート(OCA)であり、本離型フィルムを剥離力の軽い側の離型フィルムとして使用・貼り合わされたものが挙げられる。なお、粘着層は、単層であっても、2層以上の構成であってもよい。
また、上記「本離型フィルムとは剥離力の異なる離型フィルム」とは、実施例に記載の通りの測定方法で評価した離型フィルムの剥離力が本離型フィルムの樹脂層(A)側の剥離力とは異なることを意味する。また、本離型フィルムとは剥離力の異なる離型フィルムとしては、本離型フィルムの樹脂層(A)の剥離力より大きいことが好ましく、具体的には、剥離力が1.5~10倍のものが好ましく、1.5~8倍のものがより好ましく、1.5~6倍程度のものが更に好ましい。前記剥離力を満足することで、本離型フィルムの樹脂層(A)が本来剥離する必要のない場面において剥離する不具合を低減することができる。
粘着層に関して、以下に説明する。
粘着層は、粘着剤組成物からなる層であり、アクリル系樹脂を主成分樹脂とするアクリル系粘着剤組成物であっても、ゴムを主成分とするゴム系粘着剤組成物であっても、ウレタン系樹脂を主成分とするウレタン系粘着剤組成物であっても、シリコーン樹脂を主成分とするシリコーン系粘着剤組成物であってもよい。中でも、粘着剤組成物は、粘着力と剥離力をバランス良く調整することができ、且つ、安価である点から、アクリル系樹脂を主成分とするアクリル系粘着剤組成物が好ましい。
なお、上記「主成分樹脂」とは、粘着剤組成物を構成する樹脂の中でも最も質量割合の高い樹脂を意味する。例えば、粘着剤組成物を構成する樹脂全量の50質量%以上、好ましくは60質量%以上、より好ましくは70質量%以上を占める成分を意味する。なお、上限としては100質量%であるが、通常99.99質量%である。
粘着剤組成物の主成分樹脂であるアクリル系樹脂としては、(メタ)アクリル系重合体を挙げることができる。
(メタ)アクリル系重合体は、(メタ)アクリル酸アルキルエステルを主な構成単位とする重合体である。(メタ)アクリル酸アルキルエステルとしては、例えば、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル、(メタ)アクリル酸n-プロピル、(メタ)アクリル酸イソプロピル、(メタ)アクリル酸n-ブチル、(メタ)アクリル酸イソブチル、(メタ)アクリル酸sec-ブチル、(メタ)アクリル酸t-ブチル、(メタ)アクリル酸n-ペンチル、(メタ)アクリル酸n-ヘキシル、(メタ)アクリル酸イソボルニル、(メタ)アクリル酸4-t-ブチルシクロヘキシル、(メタ)アクリル酸トリシクロデカニル、(メタ)アクリル酸ジシクロペンタエニル、(メタ)アクリル酸アダマンチル等を挙げることができる。これらは1種のみで用いても2種以上を組み合わせて用いてもよい。これらの中でも、(メタ)アクリル系重合体を構成する他の(メタ)アクリレートとの相溶性、硬化樹脂層(B)(B)の耐熱性の点から、(メタ)アクリル酸メチルが好ましい。(メタ)アクリル系重合体を形成する単量体中における(メタ)アクリル酸アルキルエステルの含有割合は、例えば50質量%以上であるが、好ましくは60~99.99質量%、より好ましくは75~98.9質量%、さらに好ましくは87~97.8質量%である。なお、(メタ)アクリル系重合体はラジカル重合可能な二重結合を有するものであってもよい。
共重合成分として使用できる前記(メタ)アクリル酸アルキルエステル以外の(メタ)アクリル酸エステルとしては、例えば、後述する(メタ)アクリル酸ヒドロキシアルキル、(メタ)アクリル酸メトキシメチル、(メタ)アクリル酸メトキシエチル、(メタ)アクリル酸エトキシメチル、(メタ)アクリル酸エトキシエチル等の(メタ)アクリル酸アルコキシアルキルエステル、(メタ)アクリル酸フェニル、(メタ)アクリル酸ベンジル、(メタ)アクリル酸グリシジル、(メタ)アクリル酸γ-ブチロラクトン等を挙げることができる。
前記ビニル基を有する化合物としては、ジメチルアクリルアミド、ヒドロキシエチルアクリルアミド、ジメチルアミノプロピルアクリルアミド等のアクリルアミド系化合物、スチレン、α-メチルスチレン、p-メトキシスチレン等のスチレン系化合物、無水マレイン酸等を挙げることができる。
また、これら以外の後述する(a1)成分、(a2)成分も適宜使用することができる。
共重合性モノマー(a2)としては、例えば、フェニル(メタ)アクリレート、ベンジル(メタ)アクリレート、フェノキシエチル(メタ)アクリレート、エトキシ化o-フェニルフェニル(メタ)アクリレート、フェノキシジエチレングリコール(メタ)アクリレート、2-ヒドロキシ-3-フェノキシプロピル(メタ)アクリレート、スチレン等の芳香環含有モノマー、(メタ)アクリロイルモルホリン、ジメチル(メタ)アクリルアミド、ジエチル(メタ)アクリルアミド、(メタ)アクリルアミド等のアミド系モノマー、アクリロニトリル、メタクリロニトリル、酢酸ビニル、プロピオン酸ビニル、ステアリン酸ビニル、塩化ビニル、塩化ビニリデン、アルキルビニルエーテル、ビニルトルエン、ビニルピリジン、ビニルピロリドン、イタコン酸ジアルキルエステル、フマル酸ジアルキルエステル、アリルアルコール、アクリルクロライド、メチルビニルケトン、N-アクリルアミドメチルトリメチルアンモニウムクロライド、アリルトリメチルアンモニウムクロライド、ジメチルアリルビニルケトン等を挙げることができる。
本発明では、フォルダブルディスプレイへの適用を考慮して、粘着剤層への柔軟性を付与する観点から、2ーエチルヘキシルアクリレートおよび/またはブチルアクリレートを含むことが好ましい。
粘着剤組成物は、その硬化方法に応じて架橋剤を含有することができる。
架橋剤としては、例えば、イソシアネート系架橋剤、エポキシ系架橋剤、アジリジン系架橋剤、メラミン系架橋剤、アルデヒド系架橋剤、アミン系架橋剤などを挙げることができる。中でも、基材との密着性向上、あるいはアクリル系樹脂との反応性の点で、イソシアネート系架橋剤が好適に用いられる。架橋剤は、単独で使用してもよいし、2種以上を併用してもよい。
架橋剤の含有量は、主成分樹脂100質量部に対して、0.01~10質量部であることが好ましく、更に好ましくは0.05~5質量部、その中でも0.1~3質量部である。架橋剤の含有量が上記範囲内であれば、凝集力が不足することもなく、所望する耐久性を得ることができる一方、柔軟性および粘着力が低下するのを防ぐことができる。
粘着剤組成物は、上記主成分樹脂、架橋剤以外、必要に応じて、主成分樹脂及び架橋剤以外の粘着剤成分を構成する樹脂(例えば、アクリル系樹脂、ゴム、シリコーン樹脂、ウレタン樹脂)を含有してもよい。
例えば、ロジン、ロジンエステル、水添ロジンエステル、フェノール樹脂、芳香族変性テルペン樹脂、脂肪族系石油樹脂、脂環族系石油樹脂、スチレン系樹脂、キシレン系樹脂等の粘着付与剤、シランカップリング剤、帯電防止剤、着色剤、充填剤、老化防止剤、紫外線吸収剤、機能性色素等の従来公知の添加剤や、紫外線あるいは放射線照射により呈色あるいは変色を起こすような化合物などの添加剤を配合することができる。
これら添加剤の配合量は、粘着剤組成物(不揮発成分基準)全体の10質量%以下であることが好ましく、さらに好ましくは5質量%以下であり、添加剤として分子量が1万よりも低い低分子成分は極力含まないことが耐久性に優れる点で好ましい。
粘着層の形成方法は特に限定されず、上記粘着剤組成物を本剥離フィルムの樹脂層(A)上に塗布した後に、適宜乾燥し、硬化などすることで形成してもよい。また、上記粘着剤組成物を基材フィルムあるいは本剥離フィルムとは剥離力の異なる離型フィルム上に塗布し、適宜乾燥し、硬化などした後に、本剥離フィルムとは剥離力の異なる離型フィルムを備えた面とは反対の面に本剥離フィルムの離型層(A層)を貼り合わせることで形成してもよい。
粘着剤組成物は、架橋剤や、主成分樹脂に応じて硬化させればよく、加熱により硬化してもよいし、紫外線などの光照射により硬化してもよい。また、上記粘着剤組成物は、適宜有機溶剤などに希釈したうえで離型層などの上に塗布してもよい。
(粘着層の厚み)
本粘着層の厚みは、特に限定するものではない。例えば、十分な粘着力を付与したり、粘着剤を貼り合わせる基材の凹凸や段差を埋めたりする点から、0.1μm以上であることが好ましく、中でも0.5μm以上、その中でも1μm以上であるのが更に好ましい。その一方、材料の使用効率や透過度、アウトガスの観点から、10000μm以下であるのが好ましく、中でも3000μm以下、その中でも1000μm以下であるのが更に好ましい。
本発明において粘着層は、25℃における弾性率が、6.0MPa以下であることが好ましい。ここでいう弾性率は、ナノインデンターにより測定された値である。弾性率が6.0MPa以下となることで、粘着層は変形に対する追従性が良好となり、フォルダブルディスプレイ用途に適用可能となる。これらの観点から、粘着層の弾性率は、より好ましくは5.0MPa以下、さらに好ましくは4.0MPa以下、その中でも特に3.0MPa以下である。また、粘着層の25℃における弾性率は、粘着層形成性の観点から、0.5MPa以上が好ましく、1.0MPa以上がより好ましく、1.5MPa以上がさらに好ましい。
ガラス転移温度は下記のFoxの式より算出されるものである。

Tg:共重合体のガラス転移温度(K)
Tga:モノマーAのホモポリマーのガラス転移温度(K)
Wa:モノマーAの重量分率
Tgb:モノマーBのホモポリマーのガラス転移温度(K)
Wb:モノマーBの重量分率
Tgn:モノマーNのホモポリマーのガラス転移温度(K)
Wn:モノマーNの重量分率
(Wa+Wb+・・・+Wn=1)
上記範囲を満足することで、折り曲げのような、変形に対する追従性が良好な粘着層を得ることができる。
本フィルム積層体は、様々な積層構成として使用できる。例えば、上記粘着層付き離型フィルムは、粘着層表面に光学部材を貼り合わせたフィルム積層体としてもよい。このようなフィルム積層体は、本離型フィルムを剥がして、露出された粘着層により光学部材を被着体に貼り合わせることで作成することができる。光学部材としては、例えば、偏光板、タッチセンサーなどを挙げることができる。また、自動車に搭載されるタッチパネルなどの車載用光学部材であってもよい。
偏光板の材料および構成は任意であり、例えば、ヨウ素を配向色素として用いた延伸ポリビニルアルコールフィルムに保護フィルムとしてTAC(トリアセチルセルロース)フィルムを積層したものが、この種の偏光板として広く実用化されている。また、偏光板は、表面に、実質的に位相差を有しないハードコート、防眩、低反射、帯電防止などの機能を持つ層構成を有するものであってもよい。
タッチセンサーは、ユーザが画面に表示される画像を指やタッチペンなどで接触する場合、この接触に反応してタッチ地点を把握する部材であり、センサー技術により、静電容量方式、抵抗膜方式、赤外線または超音波などを利用した表面波方式などの方法が例示される。
一般にタッチセンサーは液晶表示パネル、有機ELなどの表示装置に搭載される。
また、近年、ガラス基板の代替として、フレキシブル性に着目して、基材フィルムを用いる傾向にあり、タッチセンサーフィルムを使用することが好ましい。タッチセンサーフィルムは、感知電極の機能を実行するためのパターン化した透明導電層を設けるのが一般的である。
他の離型フィルムは、樹脂フィルムなどの基材表面に離型層が形成されたものであり、離型層が形成された面が粘着層に貼り合わされるとよい。他の離型フィルムの離型層は、上記した本離型層以外を使用するとよい。
本発明においては、「フィルム」と称する場合でも「シート」を含むものとし、「シート」と称する場合でも「フィルム」を含むものとする。
また、画像表示パネル、保護パネル、タッチパネル等のように「パネル」と表現する場合、板体、シート及びフィルムを包含するものである。
また、「X以上」(Xは任意の数字)と記載した場合、特にことわらない限り「好ましくはXより大きい」の意を包含し、「Y以下」(Yは任意の数字)と記載した場合、特にことわらない限り「好ましくはYより小さい」の意も包含するものである。
ポリエステルに非相溶な他のポリマー成分および顔料を除去したポリエステル1gを精秤し、フェノール/テトラクロロエタン=50/50(質量比)の混合溶媒100mlを加えて溶解させ、30℃で測定した。
遠心沈降式粒度分布測定装置(株式会社島津製作所社製SA-CP3型)を使用して測定した等価球形分布における積算(質量基準)50%の値を平均粒径とした。
裏面からの反射を抑えるために、あらかじめ、試料フィルムの測定裏面に黒テープ(ニチバン(株)製「ビニールテープVT―50」)を貼った。測定には、分光光度計(日本分光(株)製 紫外可視分光光度計「V-670」)を使用して、波長範囲300~800nmでの絶対反射率を、同期モード、入射角5°、N偏光、レスポンス Fast、データ取得間隔1.0nm、バンド幅10nm、走査速度1000m/minの条件で測定した。この測定で得られたデータと、シリコーンの屈折率を1.43として計算したデータとを比較することで、膜厚を求めた。
硬化樹脂層(B)の表面をRuO4で染色し、エポキシ樹脂中に包埋した。その後、超薄切片法により作成した切片をRuO4で染色し、塗布層断面をTEM(株式会社日立ハイテクノロジーズ製 H-7650、加速電圧100kV)を用いて測定した。
低抵抗率計(三菱ケミカル(株)製、「ロレスタGP MCP-T600」)を使用し、温度23℃,相対湿度50%の測定雰囲気でサンプルを30分間調湿後に表面固有抵抗率の測定を行い、1分後の値を表面固有抵抗率とした。抵抗値が測定可能な範囲の上限を超えていた場合は測定不可とした。
JIS B 0601-2001に準拠した表面粗度計((株)小坂研究所製、二次元粗度計「surfcorder SE3500」)を用いて、樹脂層(B)の算術平均粗さ(Ra)を測定した。また、反離型面の最大断面高さ(Rt)を測定した。Rtは、評価長さにおける粗さ曲線の山高さRpの最大値と谷深さRvの最大値との和として求めることができる。
GPC測定装置を使用して、クロマトグラムを測定し、標準ポリスチレンを使用した検量線に基づいて数平均分子量(Mn)及び質量平均分子量(Mw)を求め、表2に示した。具体的には、測定用の試料4mgを、4mLのTHFに溶解して測定溶液とし、測定溶液100μLをGPC測定装置に注入して測定した。溶離液にはテトラヒドロフラン(T HF)を使用した。分析には東ソー(株)製「Ecosec8320」を使用し、ガードカラムには東ソー(株)製「TSKgel guardcolumn HXL-L」、カラムには東ソー(株)製「TSKgel GMHXL」を4本連結して使用した。また、 オーブンの温度は40℃、THF流量1.0mL/分の条件で分析を行い、検出にはRI を用いた。
実施例・比較例で用いた硬化型シリコーン樹脂の組成分析を、400MHz-NMR(Bruker Avance400M)を用いて行いた。1H-NMR測定には、溶媒としてCDCl3を用い、ジメチルシロキサンのメチル基に由来するピークを化学シフト基準として、温度30℃にて行った。
試料フィルムの樹脂層(A)面に、ゴムローラー(2kg)で圧着(2往復)し、粘着テープ(Tesa製「No.7475」)を貼り付けた後、25mm×300mmのサイズにカットし、室温(23℃)にて1時間放置した後の剥離力を測定した。剥離力の測定は、(株)インテスコ製「インテスコモデル2001型」を使用し、引張速度0.3m/minの条件下、試料フィルムを180°で剥離して行った。
試料フィルムを、A4サイズ(210mm×297mm)の大きさに切り取り、その離型層(A層)面に、75μmの2軸延伸PETフィルム(三菱ケミカル(株)製:ダイアホイルT100-75)を重ねて、温度23℃、圧力1MPaの条件で2時間プレスした。そして、「75μmの2軸延伸PETフィルム」単体を評価フィルムとした。
他方、樹脂層(A)を設けていない未処理の基材フィルムに、上記と同じ「75μmの2軸延伸PETフィルム」を重ねて上記と同条件でプレスし、「75μmの2軸延伸PETフィルム」単体を基準フィルムとした。
前記評価フィルムの試料フィルムとの接触面(樹脂層(A)(A層))及び前記基準フィルムの基材フィルムとの接触面に、それぞれゴムローラー(2kg)により圧着(2往復)することで、粘着テープ(日東電工(株)製「No.31B」)を貼り付けた後、50mm×300mmのサイズにカットし、室温にて1時間放置後の剥離力を測定した。
剥離力の測定は、(株)インテスコ製「インテスコモデル2001型」を使用し、引張速度0.3m/minの条件下、評価フィルム及び基準フィルムそれぞれを180°で剥離して行った。
そして、測定した評価フィルムの剥離力及び基準フィルムの剥離力を次の式に代入して残留接着率(%)を求めた。なお、ここでは熱処理等の処理はしていない値である。
残留接着率(%)=(評価フィルムの剥離力/基準フィルムの剥離力)×100
実施例及び比較例の離型フィルムの樹脂層(A)面にアクリル系粘着テープ(Tesa製の「No.7475」)を貼り付けた後、25mm×150mmのサイズにカットし、室温にて1時間放置する。引張速度0.3m/minの条件下で180°で剥離した剥離力を(F1)とする。
次に、実施例及び比較例の離型フィルムの樹脂層(A)面に、もう一方の面(本発明では、樹脂層(B)面)が対向するように配置し、温度40℃、湿度90%RH、荷重1MPaで20時間プレス処理を行う。処理後の前記樹脂層(A)面に、アクリル系粘着テープ(Tesa製の「No.7475」)を貼り付けた後、25mm×150mmのサイズにカットし、室温にて1時間放置する。引張速度0.3m/minの条件下で180°で剥離した剥離力を(F2)とする。
次式によってプレス後重剥離化率を求め、この時の重剥離化率を耐ブロッキング性の指標とした。なお、耐ブロッキング性は、当該重剥離化率が小さいほど良好であると判断される。
プレス後重剥離率(%)=(F2-F1)/F1×100
実施例及び比較例の離型フィルムの離型層面と反離型面とが対向するように配置し、プレス機で圧力10kg/cm2、40℃、80%RHの雰囲気下、20時間加圧をし、耐ブロッキング性評価用の測定用サンプルとした。その後、剥離装置((株)島津製作所製「AGI」)を用いて、引張速度300mm/分、180°剥離の条件で剥離力を測定し、この時の剥離力を耐ブロッキング性の指標とした。なお、耐ブロッキング性は、当該剥離力が小さいほど良好であると判断される。
《評価基準》
A(good):剥離力が20g/25mm以下であり、問題なく剥離可能。
B(poor):剥離力が20g/25mmを超えて、ブロッキングが発生。
恒温恒湿槽内において、試料フィルムを60℃、80%RH雰囲気下、4週間放置した後、試料フィルムを取り出した。その後、試料フィルムの樹脂層(A)面を触手により5回擦り、離型層の脱落程度により、密着性を以下の評価基準によって評価を行った。
《評価基準》
〇:塗膜が白くならず、脱落も見られなかった。
×:塗膜が白くなるあるいは脱落が確認された。
試料フィルムの樹脂層(B)に、ゴムローラー(2kg)により圧着(2往復)することで、アクリル系粘着テープ(Tesa製の「No.7475」)を貼り付けた後、25mm×150mmのサイズにカットし、室温(23℃)にて1時間放置した後の剥離力を測定した。剥離力の測定は、(株)インテスコ製「インテスコモデル2001型」を使用し、引張速度0.3m/minの条件下、試料フィルムを180°で剥離して行った。
23℃、50RH%の測定雰囲気で、接触角計(協和界面科学株式会社製、商品名「DMo-501」)を用いて、試料フィルムの樹脂層に、イオン交換水、ジヨードメタンの液滴を作製し、その接触角を測定した。接触角は、各液を積層ポリエステルフィルムに滴下後60秒後の接触角を測定した。
本方法で得られた、イオン交換水及びジヨードメタンの接触角データについて、各溶媒の表面張力成分値(表1)を用いて、Owens-Wendtの理論式により反射防止層表面の表面自由エネルギーを算出した。

Hysitron社のナノインデンター(TI 950 TriboIndenter)を用いて、室温(25℃)下、押し込み深さ50nmの地点での樹脂層(A)の弾性率を測定した。
Hysitron社のナノインデンター(TI 950 TriboIndenter)を用いて、圧子(conical:球形、曲率半径:10μm)、室温(25℃)下、押し込み深さ500nmの地点での粘着層の弾性率を測定した。
試料フィルムの離型層面に、表8に記載の粘着剤組成物を、湿潤状態での膜厚が2milになるように塗布し、150℃で3分間、加熱処理して硬化させて粘着層(厚み(乾燥後)20μm)を形成した。
テレフタル酸ジメチル100質量部とエチレングリコール60質量部とを出発原料とし 、触媒として酢酸マグネシウム・四水塩0.09質量部を反応器にとり、反応開始温度を150℃とし、メタノールの留去とともに徐々に反応温度を上昇させ、3時間後に230℃とした。4時間後、実質的にエステル交換反応を終了させた。この反応混合物にエチルアシッドフォスフェート0.04質量部を添加した後、三酸化アンチモン0.04質量部を加えて、4時間重縮合反応を行った。すなわち、温度を230℃から徐々に昇温し280℃とした。一方、圧力は常圧より徐々に減じ、最終的には0.3mmHgとした。反応開始後、反応槽の攪拌動力の変化により、固有粘度0.65に相当する時点で反応を停止し、窒素加圧下ポリマーを吐出させ、固有粘度0.65のポリエステル(A)を得た。
ポリエステル(A)の製造方法において、エチルアシッドフォスフェート0.04質量部を添加後、平均粒径2.3μmのエチレングリコールに分散させたシリカ粒子を0.2質量部、三酸化アンチモン0.04質量部を加えて、固有粘度0.65に相当する時点で重縮合反応を停止させた以外は、ポリエステル(A)の製造方法と同様の方法を用いてポリエステル(B)を得た。得られたポリエステル(B)は、固有粘度は0.65であった。
ポリエステル(A)、(B)をそれぞれ90質量%、10質量%の割合で混合した混合原料を最外層(表層)の原料とし、ポリエステル(A)を中間層の原料として、2台の押出機に各々を供給し、各々290℃で溶融した後、25℃に設定した冷却ロール上に、2種3層(表層/中間層/表層=1:18:1の吐出量)の層構成で共押出し冷却固化させて未延伸シートを得た。次いで、ロール周速差を利用してフィルム温度85℃で縦方向に3.4倍延伸した後、縦延伸フィルムの片面に、表4に示す樹脂層(B)の塗布液1を、もう片方の面に、下記下引き層組成物をそれぞれ塗布し、テンターに導き、横方向に120℃で4.5倍延伸し、225℃で熱処理を行ったのち、横方向に2%弛緩し、樹脂層(B)膜厚(乾燥後)が0.05μm、下引き層膜厚(乾燥後)が0.05μmそれぞれ有する厚さ50μmの二軸延伸ポリエステルフィルムを得た。
縦延伸フィルムの片面に表4に示す樹脂層(B)の塗布液2~12を塗布する以外は実施例1-1と同様に製造し、離型フィルムを得た。
ポリエステル(A)、(B)をそれぞれ97質量%、3質量%の割合で混合した混合原料を最外層(表層)の原料とし、ポリエステル(A)を中間層の原料として、2台の押出機に各々を供給し、各々290℃で溶融した後、25℃に設定した冷却ロール上に、2種3層(表層/中間層/表層=1:18:1の吐出量)の層構成で共押出し冷却固化させて未延伸シートを得た。次いで、ロール周速差を利用してフィルム温度85℃で縦方向に3.4倍延伸した後、縦延伸フィルムの片面に、表4に示す樹脂層(B)の塗布液を、もう片方の面に、下記下引き層組成物をそれぞれ塗布し、テンターに導き、横方向に120℃で4.5倍延伸し、225℃で熱処理を行ったのち、横方向に2%弛緩し、樹脂層(B)膜厚(乾燥後)が0.05μm、下引き層膜厚(乾燥後)が0.05μmそれぞれ有する厚さ50μmの二軸延伸ポリエステルフィルムを得た。
縦延伸フィルムに樹脂層(B)および下引き層を設けないこと以外は実施例1―1と同様に製造し、離型フィルムを得た。
縦延伸フィルムに樹脂層(B)を設けないこと以外は実施例1―1と同様に製造し、離型フィルムを得た。
実施例1―1において、樹脂層(B)組成物に関して、非シリコーン系離型剤を含まない塗布液13に変更する以外は実施例1と同様に製造し、離型フィルムを得た。
得られた離型フィルムは表5に示すとおり、樹脂層(B)に非シリコーン系離型剤を含まないために、プレス後重剥離化率が高くなりブロッキングの発生が懸念されるものであった。
実施例1―1において、樹脂層(A)の塗布液2に変更する以外は実施例1と同様に製造し、離型フィルムを得た。
得られた離型フィルムは表5に示すとおり、樹脂層(A)の厚みが薄く、弾性率も高いために、剥離力(常態剥離力)が実施例1―1~1-12の約3倍と高く超軽剥離化を実現できなかった。また、基材密着性も悪いものであった。
実施例1において、樹脂層(A)の塗布液3(粒子含有量:0.19質量%)に変更する以外は実施例1と同様に製造し、離型フィルムを得た。
得られた離型フィルムは表5に示すとおり、粒子が存在することで、樹脂層(A)がもろくなり、そのため基材密着性が悪く、また粒子の脱落により比較的移行性が高いものであった。また、プレス後重剥離化率が高く、ブロッキングの発生が懸念されるものであった。
a1:硬化型シリコーン樹脂(シロキサン結合からなる主鎖の側鎖および/又は末端に、ビニル基が導入されたシリコーン樹脂、数平均分子量:10600、n-ヘプタン溶媒希釈にて15質量%に調整した際の25℃における粘度:1.7mcps)
a2:硬化型シリコーン樹脂(シロキサン結合からなる主鎖の側鎖および/又は末端に、ビニル基が導入されたシリコーン樹脂、数平均分子量:364000)と、シリコーン架橋剤(シロキサン結合からなる主鎖の側鎖および/又は末端に、Si-H基が導入されたシリコーン樹脂)を含有する混合物(n-ヘプタン溶媒希釈にて15質量%に調整した際の25℃における粘度:410mcps)

c1:付加型白金触媒(CM678:モメンティブ・パフォーマンス・マテリアルズ製)
c2:付加型白金触媒(PL―50T:信越化学工業株式会社製)
d1:粒子(トスパール120:モメンティブ・パフォーマンス・マテリアルズ製)

AI: 長鎖アルキル基含有化合物
4つ口フラスコにキシレン200部、オクタデシルイソシアネート600部を加え、攪拌下に加熱した。キシレンが還流し始めた時点から、平均重合度500、ケン化度88モル%のポリビニルアルコール100部を少量ずつ10分間隔で約2時間にわたって加えた。ポリビニルアルコールを加え終わってから、さらに2時間還流を行い、反応を終了した。反応混合物を約80℃まで冷却してから、メタノール中に加えたところ、反応生成物が白色沈殿として析出したので、この沈殿を濾別し、キシレン140部を加え、加熱して完全に溶解させた後、再びメタノールを加えて沈殿させるという操作を数回繰り返した後、沈殿をメタノールで洗浄し、乾燥粉砕して得た。
AII: ワックス
攪拌機、温度計、温度コントローラーを備えた内容量1.5Lの乳化設備に融点105℃、酸価16mgKOH/g、密度0.93g/mL、平均分子量5000の酸化ポリエチレンワックス300g、イオン交換水650gとデカグリセリンモノオレエート界面活性剤を50g、48%水酸化カリウム水溶液10gを加え窒素で置換後、密封し150℃で1時間高速攪拌した後130℃に冷却し、高圧ホモジナイザーを400気圧下で通過させ40℃に冷却したワックスエマルション。
BI:ポリエチレンジオキシチオフェンとポリスチレンスルホン酸からなる導電剤(アグファゲバルト社製 Orgacon ICP1010)を濃アンモニア水で中和してpH=9とした導電剤、不揮発成分;1.2質量%、溶媒;水
CI: 前記式(9)で平均n=4であるポリグリセリン
CII: 前記式(9)で平均n=2であるポリグリセリン骨格にポリエチレンオキサイドが平均4分子付加した化合物。
DI: 下記組成で共重合したポリエステル樹脂の水分散体
モノマー組成:(酸成分)2,6-ナフタレンジカルボン酸/5-ナトリウムスルホイソフタル酸//(ジオール成分)エチレングリコール/ジエチレングリコール=92/8//80/20(モル%)
DII: 下記の組成で重合したウレタン樹脂水分散体
テレフタル酸282質量部、イソフタル酸282質量部、エチレングリコール62質量部、およびネオペンチルグリコール250質量部を成分とするポリエステルポリオールを(C1a)としたとき、(C1a)876質量部、トリレンジイソシアネート244質量部、エチレングリコール81質量部、およびジメチロールプロピオン酸67質量部を構成成分としたポリエステルポリウレタンをアンモニアで中和して水分散させたもの(濃度20%、25℃での粘度50mPa・s)
EI: ヘキサメトキシメチロールメラミン
EII: オキサゾリン基及びポリアルキレンオキシド鎖を有するアクリルポリマー
エポクロス(オキサゾリン基量=4.5mmol/g、株式会社日本触媒製)
FI: 下記式に示す、側鎖にポリエチレンオキサイドを有する構造のノニオン性界面活性剤。

上記式中のm、nはエチレンオキサイドの付加モル数を示す整数であり、ここではm+nの平均が10となるものを用いた。

下引き層を構成する塗布液は以下のとおりである。
(BI)/(CII)/(DII)/(FI)=10質量%/35質量%/50質量%/5質量%

テレフタル酸ジメチル100質量部とエチレングリコール55質量部とを出発原料とし、触媒として酢酸マグネシウム・四水塩0.04質量部を反応器にとり、反応開始温度を150℃とし、メタノールの留去とともに徐々に反応温度を上昇させ、3時間後に230℃とした。4時間後、実質的にエステル交換反応を終了させた。この反応混合物にエチルアシッドフォスフェート0.02質量部を添加した後、三酸化アンチモン0.04質量部を加えて、4時間重縮合反応を行った。すなわち、温度を230℃から徐々に昇温し280℃とした。一方、圧力は常圧より徐々に減じ、最終的には0.3mmHgとした。反応開始後、反応槽の攪拌動力の変化により、固有粘度0.59dl/gに相当する時点で反応を停止し、窒素加圧下ポリマーを吐出させ、固有粘度0.59dl/gのポリエステル(1)を得た。
テレフタル酸ジメチル100質量部とエチレングリコール45質量部とを出発原料とし、触媒として酢酸マグネシウム・四水塩0.06質量部を反応器にとり、反応開始温度を150℃とし、メタノールの留去とともに徐々に反応温度を上昇させ、3時間後に230℃とした。4時間後、実質的にエステル交換反応を終了させた。この反応混合物にエチルアシッドフォスフェート0.03質量部を添加した後、エチレングリコールに分散させた平均粒径2.7μmのシリカ粒子を0.3質量部、三酸化アンチモン0.03質量部を加えて、4時間重縮合反応を行った。すなわち、温度を230℃から徐々に昇温し280℃とした。一方、圧力は常圧より徐々に減じ、最終的には0.3mmHgとした。反応開始後、反応槽の攪拌動力の変化により、固有粘度0.59dl/gに相当する時点で反応を停止し、窒素加圧下ポリマーを吐出させ、固有粘度0.59dl/gのポリエステル(2)を得た。
ポリエステル(2)において、シリカ粒子の代わりに、平均粒径4.5μmの有機粒子(スチレンージビニルベンゼン:スチレン系樹脂)を10質量部添加する以外はポリエステル(2)と同様に製造して、固有粘度0.60dl/gのポリエステル(3)を得た。
[PET-A]
ポリエステル(1)とポリエステル(2)とを質量比で80/20でブレンドしたものをA層、およびポリエステル(1)のみをB層の原料として、ポリエステル(1)とポリエステル(3)とを質量比で95.5/4.5でブレンドしたものをC層押出機にそれぞれを供給し、285℃に加熱溶融し、A層およびC層を最外層(表層)、B層を中間層とする3種三層(A(A層)/B(B層)/C(C層))の層構成で、押出条件で厚み構成比がA/B/C=5/90/5となるよう共押出し、表面温度40~50℃の鏡面冷却ドラムに密着させながら冷却固化させ、未延伸ポリエチレンテレフタレートフィルムを作成した。このフィルムを85℃の加熱ロール群を通過させながら長手方向に3.0倍延伸し、一軸配向フィルムとした。この一軸延伸フィルムに対して、下記に示す硬化樹脂層組成物を両面に塗布し、次いでこのフィルムをテンター延伸機に導き、100℃で幅方向に4.1倍延伸し、さらに235℃で熱処理を施した後、幅方向に2%の弛緩処理を行い、膜厚(乾燥後)が0.05μmの硬化樹脂層を両面に備えた、厚み50μmの二軸配向ポリエステルフィルムを得た。
(A):チオフェン系帯電防止剤
ポリエチレンジオキシチオフェンとポリスチレンスルホン酸からなる導電剤、アグファゲバルト社製「Orgacon ICP1010」を濃アンモニア水で中和してpH=9とした物。
(B)ポリヒドロキシ化合物
平均重合度4のポリグリセリン
(C)下記の組成で共重合した、縮合多環構造を有するポリエステル樹脂の水分散体
モノマー組成:(酸成分)2,6-ナフタレンジカルボン酸/5-ソジウムスルホイソフタル酸//(ジオール成分)エチレングリコール/ジエチレングリコール=92/8//80/20(mol%)
A/B/C=7/10/83(質量%)
a1:硬化型シリコーン樹脂(シロキサン結合からなる主鎖の側鎖および/又は末端に、ビニル基が導入されたシリコーン樹脂、数平均分子量:10600、n-ヘプタン溶媒希釈にて15質量%に調整した際の25℃における粘度:1.7mcps)
実施例・比較例で用いた硬化型シリコーン樹脂の組成分析を、400MHz-NMR(Bruker Avance400M)を用いて行い、結果を表1に示した。1H-NMR測定には、溶媒としてCDCl3を用い、ジメチルシロキサンのメチル基に由来するピークを化学シフト基準として、温度30℃にて行った。下記表1には、硬化型シリコーン樹脂における各官能基の含有割合(mol%)を示した。

c1:付加型白金触媒(CM678:モメンティブ・パフォーマンス・マテリアルズ製)
e1:軽剥離化剤(メチルフェニルシリコーンオイル、Mw:1万未満、フェニル基含有比率;DM:MP=91:9(mol%))
e2:軽剥離化剤(メチルフェニルシリコーンオイル、Mw:21000 フェニル基含有比率;DM:MP=91:9(mol%))
e3:軽剥離化剤(メチルフェニルシリコーンオイル、Mw:71000 フェニル基含有比率;DM:MP=90:10(mol%))
e4:軽剥離化剤 BY24―850(東レ・ダウ製)(ポリジメチルシロキサン、Mw:10万以上、フェニル基含有比率;DM:MP=100:0(mol%))
PET-AのA層側の平坦面に、下記離型層組成物を、塗布量(乾燥後)の厚みが1g/m2になるように塗布した後、150℃で30秒乾燥させ、離型フィルム(試料フィルム)を得た。
主剤:a1;100質量部
架橋剤:b1;3質量部
触媒:c1;3質量部
軽剥離化剤:e1;1質量部
溶媒:トルエン;1000質量部
MEK;9000質量部
離型層組成物に関して、軽剥離化剤の種類及び添加量を下記表7の通りに変更する以外は実施例2-1と同様にして製造し、フィルム積層体を得た。
離型層組成物において、軽剥離化剤を添加しない以外は実施例1と同様に製造し、フィルム積層体を得た。
実施例1において、離型層の組成を下記離型層組成に変更する以外は実施例1と同様に製造し、離型フィルム(試料フィルム)を得た。
主剤:a1;100質量部
架橋剤:b1;3質量部
触媒:c1;3質量部
軽剥離化剤:e4;1質量部
溶媒:トルエン;1000質量部
MEK;9000質量部

*1は熱処理していないことを意味する。
なお、比較例1と各実施例とを粘着テープによる剥離力(常態剥離力)で比較すると、最も軽いタイプでは、比較例1の約6割まで軽剥離化させることができ、超軽剥離化を実現できた。
それに対して、比較例1は、従来品レベル(軽剥離タイプ)であり、粘着剤層に対する剥離力が大きいことがわかった。比較例2は離型層中に分子量の大きい軽剥離化剤を含有しているため、軽剥離効果に乏しい結果であった。
実施例1-1で用いた離型層について、以下に記載する粘着剤を用いて、剥離性について評価した。結果を表8に示す。
粘着剤A:2EHA/BA/HEA=36/59/5質量% (弾性率:2.9MPa、Tg:-59.0℃)
粘着剤B:2EHA/HEA/AAc=92.8/7/0.2質量% (弾性率:2.7MPa、Tg:-66.7℃)
2EHA;2-エチルヘキシルアクリレート
BA;ブチルアクリレート
HEA;ヒドロキシエチルアクリレート
AAc;アクリル酸
実施例3-1において、離型層として実施例2-1で用いた離型層を使用したこと以外は、実施例3-1と同様にして評価した。結果を表8に示す。

Claims (28)
- 基材フィルムの一方の面に樹脂層(A)を備え、他方の面に樹脂層(B)を備えた離型フィルムであり、前記樹脂層(A)は、シリコーン樹脂系離型剤を含み、厚みが0.2~2.0μmであり、前記樹脂層(B)は、非シリコーン樹脂系離型剤を含む、離型フィルム。
- 基材フィルムの少なくとも片面側に樹脂層(A)を備え、
前記樹脂層(A)が、硬化型シリコーン樹脂および軽剥離化剤を含む樹脂層組成物の硬化物であり、前記軽剥離化剤が下記式(I)で示されるジメチルシロキサン骨格(DM)および下記式(II)で示されるメチルフェニルシロキサン骨格(MP)を有し、厚みが0.2~1.5μmである、離型フィルム。
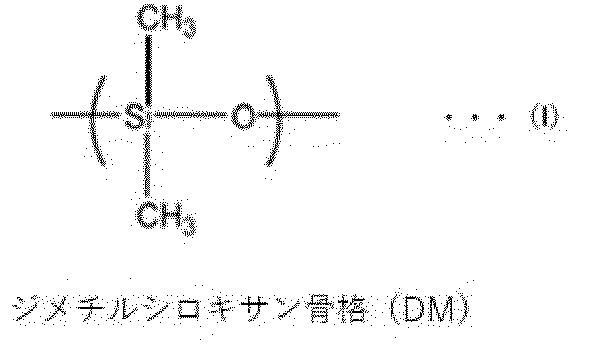

- 前記樹脂層(A)を設ける面とは反対側のフィルム表面の最大断面高さ(Rt)が、2.0μm以下である、請求項2に記載の離型フィルム。
- 前記基材フィルムの前記樹脂層(A)が設けられる面とは反対側の面が、平均粒径1~6μmの粒子を0.4~1.0質量%含有するポリエステル層を備える、請求項2または3に記載の離型フィルム。
- 前記軽剥離化剤の質量平均分子量が1万以上10万以下である、請求項2~4のいずれかに記載の離型フィルム。
- 前記樹脂層(A)のナノインデンター装置を用いて測定した、25℃における弾性率が500MPa以下である、請求項1~5のいずれかに記載の離型フィルム。
- 前記非シリコーン樹脂系離型剤がワックス、長鎖アルキル基含有化合物、フッ素化合物からなる群から選択される少なくとも1種である、請求項1または6に記載の離型フィルム。
- 前記樹脂層(B)の不揮発成分中における、離型剤の含有量が10~70質量%である、請求項1、6または7に記載の離型フィルム。
- 前記樹脂層(B)が粒子を実質的に含有しない、請求項1及び6~8のいずれかに記載の離型フィルム。
- 下記評価方法で測定した、樹脂層(A)の常態剥離力が5g/25mm以下である、請求項1~9のいずれかに記載の離型フィルム。
<評価方法>
前記離型フィルムの樹脂層(A)面にアクリル系粘着テープ(Tesa製の「No.7475」)を貼り付けた後、25mm×150mmのサイズにカットし、室温にて1時間放置する。剥離力は引張速度0.3m/minの条件下で180°で剥離を行う。 - 前記樹脂層(A)あるいは樹脂層(B)の少なくとも一方の層と前記基材フィルムとの間に下引き層を備える、請求項1および6~10のいずれかに記載の離型フィルム。
- 前記下引き層が帯電防止層である、請求項11に記載の離型フィルム。
- 前記下引き層が下記化合物(A)~(C)を含有する、請求項11または12に記載の離型フィルム。
(A)チオフェンまたはチオフェン誘導体からなる化合物に、他の陰イオン化合物によりドーピングされた重合体、またはチオフェンまたはチオフェン誘導体からなる化合物中に陰イオン基を持ち自己ドープされた重合体
(B)ポリヒドロキシ化合物
(C)ポリウレタン樹脂、ポリエステル樹脂、及びアクリル樹脂からなる群から選ばれる1種以上の化合物 - 前記樹脂層(A)あるいは樹脂層(B)の少なくとも一方の表面の表面固有抵抗率が1×1012Ω/□以下である、請求項1および6~13のいずれかに記載の離型フィルム。
- 前記基材フィルムがポリエステルフィルムである、請求項1~14のいずれかに記載の離型フィルム。
- 前記ポリエステルフィルムが三層構成である、請求項15に記載の離型フィルム。
- 以下の方法で評価した前記樹脂層(A)のプレス後重剥離化率が100%以下である、請求項1~16のいずれかに記載の離型フィルム。
<評価方法>
前記樹脂層(A)面にアクリル系粘着テープ(Tesa製の「No.7475」)を貼り付けた後、25mm×150mmのサイズにカットし、室温にて1時間放置する。引張速度0.3m/minの条件下で180°で剥離した剥離力を(F1)とする。
次に、前記樹脂層(A)面に接触するように積層し、温度40℃、湿度90%RH、荷重1MPaで20時間プレス処理を行う。処理後の前記樹脂層(A)面に、アクリル系粘着テープ(Tesa製の「No.7475」)を貼り付けた後、25mm×150mmのサイズにカットし、室温にて1時間放置する。引張速度0.3m/minの条件下で180°で剥離した剥離力を(F2)とする。
次式によってプレス後重剥離化率を求める。
プレス後重剥離化率(%)=(F2-F1)/F1×100 - 前記樹脂層(A)面の中心線平均粗さ(Ra(A))が30nm以下である、請求項1~17のいずれかに記載の離型フィルム。
- 前記樹脂層(B)面の中心線平均粗さ(Ra(B))が60nm以下である、請求項1または6~18のいずれかに記載の離型フィルム。
- 請求項1~19の何れかに記載の離型フィルムと、粘着層を介して、他の離型フィルムを備えた、粘着シート。
- 前記粘着層がアクリル系粘着剤組成物から形成される、請求項20に記載の粘着シート。
- 前記粘着層の弾性率(25℃)が6.0MPa以下である、請求項20または21に記載の粘着シート。
- 前記粘着層が2-エチルヘキシルアクリレートおよび/またはブチルアクリレートを含む、請求項20~22のいずれかに記載の粘着シート。
- 光学用透明粘着シート用である、請求項20~23のいずれかに記載の粘着シート。
- ディスプレイ用である、請求項1~19のいずれかに記載の離型フィルム。
- フォルダブルディスプレイ用である、請求項1~19のいずれかに記載の離型フィルム。
- 請求項20~23のいずれかに記載の粘着シートにおいて、前記他の離型フィルムを剥がした後、露出する粘着層表面を光学部材に貼り合わせる、粘着シートの使用方法。
- 前記光学部材が偏光板またはタッチセンサーである、請求項27に記載の粘着シートの使用方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202380013398.XA CN117916085A (zh) | 2022-08-05 | 2023-07-20 | 脱模薄膜 |
| KR1020247006126A KR102706835B1 (ko) | 2022-08-05 | 2023-07-20 | 이형 필름 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022125548 | 2022-08-05 | ||
| JP2022-125548 | 2022-08-05 | ||
| JP2023015451 | 2023-02-03 | ||
| JP2023-015451 | 2023-02-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024029346A1 true WO2024029346A1 (ja) | 2024-02-08 |
Family
ID=89848830
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2023/026510 WO2024029346A1 (ja) | 2022-08-05 | 2023-07-20 | 離型フィルム |
Country Status (3)
| Country | Link |
|---|---|
| KR (1) | KR102706835B1 (ja) |
| TW (1) | TW202413105A (ja) |
| WO (1) | WO2024029346A1 (ja) |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001003010A (ja) * | 1999-06-16 | 2001-01-09 | Nitto Denko Corp | 感圧性両面接着シート及び感圧性接着部材 |
| JP2003080638A (ja) * | 2001-09-07 | 2003-03-19 | Lintec Corp | 剥離ライナー及びこれを用いた両面テープ |
| JP2011173362A (ja) * | 2010-02-25 | 2011-09-08 | Fujimori Kogyo Co Ltd | 透明性に優れた離型フィルム |
| WO2016158037A1 (ja) * | 2015-03-28 | 2016-10-06 | 三菱樹脂株式会社 | 離型フィルム |
| JP2018130877A (ja) * | 2017-02-15 | 2018-08-23 | 東レフィルム加工株式会社 | 感光性樹脂層転写用離型フィルム、そのロール状物、感光性樹脂層積層フィルム、およびそのロール状物 |
| JP2018161824A (ja) * | 2017-03-27 | 2018-10-18 | 三菱ケミカル株式会社 | 離型フィルム |
| JP2020108937A (ja) * | 2019-01-07 | 2020-07-16 | 三菱ケミカル株式会社 | 離型フィルム |
| JP2022112759A (ja) * | 2021-01-22 | 2022-08-03 | 三菱ケミカル株式会社 | 離型性樹脂シートおよび成形品 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004255704A (ja) | 2003-02-26 | 2004-09-16 | Teijin Dupont Films Japan Ltd | 離形フィルム |
| KR101520762B1 (ko) * | 2013-09-06 | 2015-05-15 | 도레이첨단소재 주식회사 | 실리콘 이형 필름 및 그의 제조방법 |
| JP6147284B2 (ja) | 2015-03-09 | 2017-06-14 | 藤森工業株式会社 | 剥離性に優れた離型フィルム |
| WO2016152369A1 (ja) * | 2015-03-23 | 2016-09-29 | 三菱樹脂株式会社 | 離型フィルム |
| JP6480295B2 (ja) | 2015-09-25 | 2019-03-06 | 藤森工業株式会社 | 剥離性に優れた離型フィルム |
| JP7026588B2 (ja) * | 2018-07-03 | 2022-02-28 | 信越化学工業株式会社 | シリコーン組成物、剥離シート、剥離フィルム、及び、剥離シート及び剥離フィルムの製造方法 |
| JP2022036894A (ja) * | 2020-08-24 | 2022-03-08 | 三菱ケミカル株式会社 | 離型フィルム、粘着層付き離型フィルムおよびフィルム積層体 |
-
2023
- 2023-07-20 KR KR1020247006126A patent/KR102706835B1/ko active IP Right Grant
- 2023-07-20 WO PCT/JP2023/026510 patent/WO2024029346A1/ja active Application Filing
- 2023-07-21 TW TW112127393A patent/TW202413105A/zh unknown
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001003010A (ja) * | 1999-06-16 | 2001-01-09 | Nitto Denko Corp | 感圧性両面接着シート及び感圧性接着部材 |
| JP2003080638A (ja) * | 2001-09-07 | 2003-03-19 | Lintec Corp | 剥離ライナー及びこれを用いた両面テープ |
| JP2011173362A (ja) * | 2010-02-25 | 2011-09-08 | Fujimori Kogyo Co Ltd | 透明性に優れた離型フィルム |
| WO2016158037A1 (ja) * | 2015-03-28 | 2016-10-06 | 三菱樹脂株式会社 | 離型フィルム |
| JP2018130877A (ja) * | 2017-02-15 | 2018-08-23 | 東レフィルム加工株式会社 | 感光性樹脂層転写用離型フィルム、そのロール状物、感光性樹脂層積層フィルム、およびそのロール状物 |
| JP2018161824A (ja) * | 2017-03-27 | 2018-10-18 | 三菱ケミカル株式会社 | 離型フィルム |
| JP2020108937A (ja) * | 2019-01-07 | 2020-07-16 | 三菱ケミカル株式会社 | 離型フィルム |
| JP2022112759A (ja) * | 2021-01-22 | 2022-08-03 | 三菱ケミカル株式会社 | 離型性樹脂シートおよび成形品 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202413105A (zh) | 2024-04-01 |
| KR102706835B1 (ko) | 2024-09-20 |
| KR20240046519A (ko) | 2024-04-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR102535755B1 (ko) | 이형 필름 및 적층체 | |
| TWI752902B (zh) | 離型薄膜 | |
| JP5467752B2 (ja) | 二軸延伸ポリエステルフィルム | |
| JP7031601B2 (ja) | 離型フィルム | |
| CN107429128B (zh) | 粘接膜及其制造方法 | |
| TWI845660B (zh) | 離型膜、膜積層體、彼等之製造方法 | |
| TW201000317A (en) | Readily bondable polyester film | |
| JP6390150B2 (ja) | ポリエステルフィルム | |
| JP7003915B2 (ja) | 離型フィルム | |
| JP7424020B2 (ja) | 離型フィルム | |
| JP2022186741A (ja) | 離型フィルム | |
| JP7020043B2 (ja) | 積層ポリエステルフィルム | |
| KR102706835B1 (ko) | 이형 필름 | |
| JP7383903B2 (ja) | 積層フィルム及びその製造方法 | |
| JP6720600B2 (ja) | 離型フィルム | |
| CN117916085A (zh) | 脱模薄膜 | |
| JP6488819B2 (ja) | 基材レス両面粘着シート | |
| EP4328021A1 (en) | Release film and film laminate | |
| JP7020042B2 (ja) | 積層ポリエステルフィルム | |
| JP2022139376A (ja) | 離型フィルムおよびフィルム積層体 | |
| JP2018069591A (ja) | ハードコートフィルム | |
| JP2023135935A (ja) | 離型フィルム | |
| JP2022155385A (ja) | 積層ポリエステルフィルム | |
| JP6447574B2 (ja) | 積層ポリエステルフィルム |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 20247006126 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202380013398.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 23849901 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2024538917 Country of ref document: JP Kind code of ref document: A |