WO2023054041A1 - 非水電解質二次電池用正極活物質および非水電解質二次電池 - Google Patents
非水電解質二次電池用正極活物質および非水電解質二次電池 Download PDFInfo
- Publication number
- WO2023054041A1 WO2023054041A1 PCT/JP2022/034891 JP2022034891W WO2023054041A1 WO 2023054041 A1 WO2023054041 A1 WO 2023054041A1 JP 2022034891 W JP2022034891 W JP 2022034891W WO 2023054041 A1 WO2023054041 A1 WO 2023054041A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- positive electrode
- active material
- electrode active
- aqueous electrolyte
- composite oxide
- Prior art date
Links
- 239000007774 positive electrode material Substances 0.000 title claims abstract description 69
- 239000011255 nonaqueous electrolyte Substances 0.000 title claims abstract description 30
- 150000001875 compounds Chemical class 0.000 claims abstract description 52
- 239000011163 secondary particle Substances 0.000 claims abstract description 39
- 239000011164 primary particle Substances 0.000 claims abstract description 35
- 229910052791 calcium Inorganic materials 0.000 claims abstract description 24
- 229910052712 strontium Inorganic materials 0.000 claims abstract description 19
- 229910052723 transition metal Inorganic materials 0.000 claims abstract description 18
- 229910052721 tungsten Inorganic materials 0.000 claims abstract description 18
- 229910052750 molybdenum Inorganic materials 0.000 claims abstract description 17
- 229910052719 titanium Inorganic materials 0.000 claims abstract description 17
- 229910052726 zirconium Inorganic materials 0.000 claims abstract description 15
- 239000002905 metal composite material Substances 0.000 claims abstract description 14
- 229910052710 silicon Inorganic materials 0.000 claims abstract description 5
- 229910052742 iron Inorganic materials 0.000 claims abstract description 4
- 229910052758 niobium Inorganic materials 0.000 claims abstract description 4
- 239000000203 mixture Substances 0.000 claims description 40
- 229910052751 metal Inorganic materials 0.000 claims description 14
- 230000002776 aggregation Effects 0.000 claims description 4
- 238000005054 agglomeration Methods 0.000 claims description 3
- 229910052744 lithium Inorganic materials 0.000 abstract description 15
- -1 lithium transition metal Chemical class 0.000 abstract description 11
- 229910005565 NiaMnb Inorganic materials 0.000 abstract 1
- 239000002131 composite material Substances 0.000 description 60
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 28
- 239000010410 layer Substances 0.000 description 26
- 239000011575 calcium Substances 0.000 description 22
- 229910052760 oxygen Inorganic materials 0.000 description 20
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 17
- 238000006243 chemical reaction Methods 0.000 description 17
- 230000015572 biosynthetic process Effects 0.000 description 16
- 238000010304 firing Methods 0.000 description 16
- 238000003786 synthesis reaction Methods 0.000 description 16
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 15
- 239000001301 oxygen Substances 0.000 description 15
- 230000000052 comparative effect Effects 0.000 description 14
- 239000002245 particle Substances 0.000 description 13
- 238000007789 sealing Methods 0.000 description 12
- 239000010936 titanium Substances 0.000 description 12
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 12
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 11
- 239000000920 calcium hydroxide Substances 0.000 description 11
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 11
- 229910052759 nickel Inorganic materials 0.000 description 11
- 230000000694 effects Effects 0.000 description 10
- 229910052748 manganese Inorganic materials 0.000 description 10
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 9
- 238000010438 heat treatment Methods 0.000 description 9
- 239000002184 metal Substances 0.000 description 8
- QGLKJKCYBOYXKC-UHFFFAOYSA-N nonaoxidotritungsten Chemical compound O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1 QGLKJKCYBOYXKC-UHFFFAOYSA-N 0.000 description 8
- 229910001930 tungsten oxide Inorganic materials 0.000 description 8
- 238000001035 drying Methods 0.000 description 7
- 239000003792 electrolyte Substances 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 7
- 239000012535 impurity Substances 0.000 description 7
- 238000000034 method Methods 0.000 description 7
- 150000003839 salts Chemical class 0.000 description 7
- 238000007086 side reaction Methods 0.000 description 7
- UUCCCPNEFXQJEL-UHFFFAOYSA-L strontium dihydroxide Chemical compound [OH-].[OH-].[Sr+2] UUCCCPNEFXQJEL-UHFFFAOYSA-L 0.000 description 7
- 229910001866 strontium hydroxide Inorganic materials 0.000 description 7
- 229910052782 aluminium Inorganic materials 0.000 description 6
- 239000011230 binding agent Substances 0.000 description 6
- 229910000476 molybdenum oxide Inorganic materials 0.000 description 6
- PQQKPALAQIIWST-UHFFFAOYSA-N oxomolybdenum Chemical compound [Mo]=O PQQKPALAQIIWST-UHFFFAOYSA-N 0.000 description 6
- 239000011149 active material Substances 0.000 description 5
- 239000003125 aqueous solvent Substances 0.000 description 5
- 239000006258 conductive agent Substances 0.000 description 5
- 239000010408 film Substances 0.000 description 5
- 229910002804 graphite Inorganic materials 0.000 description 5
- 239000010439 graphite Substances 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 239000007773 negative electrode material Substances 0.000 description 5
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 4
- 239000003575 carbonaceous material Substances 0.000 description 4
- 238000000975 co-precipitation Methods 0.000 description 4
- 230000007423 decrease Effects 0.000 description 4
- JBTWLSYIZRCDFO-UHFFFAOYSA-N ethyl methyl carbonate Chemical compound CCOC(=O)OC JBTWLSYIZRCDFO-UHFFFAOYSA-N 0.000 description 4
- 239000011888 foil Substances 0.000 description 4
- 150000002642 lithium compounds Chemical class 0.000 description 4
- 239000002002 slurry Substances 0.000 description 4
- LLZRNZOLAXHGLL-UHFFFAOYSA-J titanic acid Chemical compound O[Ti](O)(O)O LLZRNZOLAXHGLL-UHFFFAOYSA-J 0.000 description 4
- 238000003466 welding Methods 0.000 description 4
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 3
- 239000002033 PVDF binder Substances 0.000 description 3
- 239000004642 Polyimide Substances 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 239000012046 mixed solvent Substances 0.000 description 3
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 description 3
- 229920002239 polyacrylonitrile Polymers 0.000 description 3
- 229920001721 polyimide Polymers 0.000 description 3
- 229920000098 polyolefin Polymers 0.000 description 3
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 229920005989 resin Polymers 0.000 description 3
- 239000011347 resin Substances 0.000 description 3
- 239000002344 surface layer Substances 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- 229910001928 zirconium oxide Inorganic materials 0.000 description 3
- 229920000178 Acrylic resin Polymers 0.000 description 2
- 239000004925 Acrylic resin Substances 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 229910013870 LiPF 6 Inorganic materials 0.000 description 2
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 2
- 239000004372 Polyvinyl alcohol Substances 0.000 description 2
- 229920002125 Sokalan® Polymers 0.000 description 2
- 229910003514 Sr(OH) Inorganic materials 0.000 description 2
- 229910002367 SrTiO Inorganic materials 0.000 description 2
- 239000006230 acetylene black Substances 0.000 description 2
- 229910021383 artificial graphite Inorganic materials 0.000 description 2
- 239000000292 calcium oxide Substances 0.000 description 2
- ODINCKMPIJJUCX-UHFFFAOYSA-N calcium oxide Inorganic materials [Ca]=O ODINCKMPIJJUCX-UHFFFAOYSA-N 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 238000007600 charging Methods 0.000 description 2
- IEJIGPNLZYLLBP-UHFFFAOYSA-N dimethyl carbonate Chemical compound COC(=O)OC IEJIGPNLZYLLBP-UHFFFAOYSA-N 0.000 description 2
- 238000007599 discharging Methods 0.000 description 2
- 239000002612 dispersion medium Substances 0.000 description 2
- 150000004679 hydroxides Chemical class 0.000 description 2
- IIPYXGDZVMZOAP-UHFFFAOYSA-N lithium nitrate Chemical compound [Li+].[O-][N+]([O-])=O IIPYXGDZVMZOAP-UHFFFAOYSA-N 0.000 description 2
- INHCSSUBVCNVSK-UHFFFAOYSA-L lithium sulfate Chemical compound [Li+].[Li+].[O-]S([O-])(=O)=O INHCSSUBVCNVSK-UHFFFAOYSA-L 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 2
- 239000004810 polytetrafluoroethylene Substances 0.000 description 2
- 229920002451 polyvinyl alcohol Polymers 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 239000002210 silicon-based material Substances 0.000 description 2
- 229920003048 styrene butadiene rubber Polymers 0.000 description 2
- 150000003624 transition metals Chemical class 0.000 description 2
- 229910000838 Al alloy Inorganic materials 0.000 description 1
- 238000004438 BET method Methods 0.000 description 1
- 229910004648 CaMoO3 Inorganic materials 0.000 description 1
- 229910004647 CaMoO4 Inorganic materials 0.000 description 1
- 229910002971 CaTiO3 Inorganic materials 0.000 description 1
- 229910004829 CaWO4 Inorganic materials 0.000 description 1
- 229910002976 CaZrO3 Inorganic materials 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229910000881 Cu alloy Inorganic materials 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- FUJCRWPEOMXPAD-UHFFFAOYSA-N Li2O Inorganic materials [Li+].[Li+].[O-2] FUJCRWPEOMXPAD-UHFFFAOYSA-N 0.000 description 1
- 229910001323 Li2O2 Inorganic materials 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical group [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- HBBGRARXTFLTSG-UHFFFAOYSA-N Lithium ion Chemical compound [Li+] HBBGRARXTFLTSG-UHFFFAOYSA-N 0.000 description 1
- BPQQTUXANYXVAA-UHFFFAOYSA-N Orthosilicate Chemical compound [O-][Si]([O-])([O-])[O-] BPQQTUXANYXVAA-UHFFFAOYSA-N 0.000 description 1
- 239000004962 Polyamide-imide Substances 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 229910002412 SrMoO4 Inorganic materials 0.000 description 1
- 229910002370 SrTiO3 Inorganic materials 0.000 description 1
- 229910004415 SrWO4 Inorganic materials 0.000 description 1
- 239000002174 Styrene-butadiene Substances 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000004931 aggregating effect Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 239000012670 alkaline solution Substances 0.000 description 1
- 229910045601 alloy Inorganic materials 0.000 description 1
- 239000000956 alloy Substances 0.000 description 1
- IZJSTXINDUKPRP-UHFFFAOYSA-N aluminum lead Chemical compound [Al].[Pb] IZJSTXINDUKPRP-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 239000004760 aramid Substances 0.000 description 1
- 229920003235 aromatic polyamide Polymers 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- BRPQOXSCLDDYGP-UHFFFAOYSA-N calcium oxide Chemical compound [O-2].[Ca+2] BRPQOXSCLDDYGP-UHFFFAOYSA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 229910021393 carbon nanotube Inorganic materials 0.000 description 1
- 239000002041 carbon nanotube Substances 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000010280 constant potential charging Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 230000001186 cumulative effect Effects 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- XUCJHNOBJLKZNU-UHFFFAOYSA-M dilithium;hydroxide Chemical compound [Li+].[Li+].[OH-] XUCJHNOBJLKZNU-UHFFFAOYSA-M 0.000 description 1
- 238000002149 energy-dispersive X-ray emission spectroscopy Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 230000020169 heat generation Effects 0.000 description 1
- 229920006015 heat resistant resin Polymers 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 238000002847 impedance measurement Methods 0.000 description 1
- 239000010954 inorganic particle Substances 0.000 description 1
- 238000009413 insulation Methods 0.000 description 1
- 230000010220 ion permeability Effects 0.000 description 1
- 239000003273 ketjen black Substances 0.000 description 1
- HEPLMSKRHVKCAQ-UHFFFAOYSA-N lead nickel Chemical compound [Ni].[Pb] HEPLMSKRHVKCAQ-UHFFFAOYSA-N 0.000 description 1
- 239000011244 liquid electrolyte Substances 0.000 description 1
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 description 1
- 229910052808 lithium carbonate Inorganic materials 0.000 description 1
- PQXKHYXIUOZZFA-UHFFFAOYSA-M lithium fluoride Chemical compound [Li+].[F-] PQXKHYXIUOZZFA-UHFFFAOYSA-M 0.000 description 1
- SIAPCJWMELPYOE-UHFFFAOYSA-N lithium hydride Chemical compound [LiH] SIAPCJWMELPYOE-UHFFFAOYSA-N 0.000 description 1
- 229910001416 lithium ion Inorganic materials 0.000 description 1
- PAZHGORSDKKUPI-UHFFFAOYSA-N lithium metasilicate Chemical compound [Li+].[Li+].[O-][Si]([O-])=O PAZHGORSDKKUPI-UHFFFAOYSA-N 0.000 description 1
- 229910003002 lithium salt Inorganic materials 0.000 description 1
- 159000000002 lithium salts Chemical class 0.000 description 1
- 229910052912 lithium silicate Inorganic materials 0.000 description 1
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium;hydroxide;hydrate Chemical compound [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 239000011572 manganese Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 229910021382 natural graphite Inorganic materials 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000004745 nonwoven fabric Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 229920002312 polyamide-imide Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 229910052814 silicon oxide Inorganic materials 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 239000007784 solid electrolyte Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 238000002216 synchrotron radiation X-ray diffraction Methods 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- 239000002759 woven fabric Substances 0.000 description 1
- 239000004711 α-olefin Substances 0.000 description 1
Images
Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/50—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese
- H01M4/505—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of manganese of mixed oxides or hydroxides containing manganese for inserting or intercalating light metals, e.g. LiMn2O4 or LiMn2OxFy
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/52—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron
- H01M4/525—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides of nickel, cobalt or iron of mixed oxides or hydroxides containing iron, cobalt or nickel for inserting or intercalating light metals, e.g. LiNiO2, LiCoO2 or LiCoOxFy
Definitions
- the present disclosure relates to a positive electrode active material for nonaqueous electrolyte secondary batteries and a nonaqueous electrolyte secondary battery using the active material.
- Patent Document 1 discloses a positive electrode in which an alkaline earth metal and W are present on the surface of secondary particles of a lithium-transition metal composite oxide containing one or more selected from Mn, Ni, and Co as a transition metal element. Active materials are disclosed.
- Patent Document 2 discloses a positive electrode active material containing one or more selected from Mn, Ni, and Co as a transition metal element, and further containing a lithium-transition metal composite oxide in which Ca and W are solid-dissolved. ing.
- Lithium-transition metal composite oxides with a high Ni content are known as high-capacity positive electrode active materials, but when the charge rate is high, a large amount of highly reactive Ni 4+ is present on the surface layer of the composite oxide particles. Therefore, a side reaction with the electrolyte is likely to occur. Decomposition products produced by the side reactions are then deposited on the surfaces of the composite oxide particles, increasing the reaction resistance of the positive electrode.
- An object of the present disclosure is to suppress an increase in reaction resistance in a positive electrode of a non-aqueous electrolyte secondary battery using a positive electrode active material with a high Ni content.
- the lithium-transition metal composite oxide is a secondary particle formed by agglomeration of primary particles, and at least at the interfaces between the primary particles inside the secondary particles, at least one selected from Ca and Sr, A compound containing at least one selected from W, Mo, Ti, and Zr is adhered.
- a non-aqueous electrolyte secondary battery includes a positive electrode containing the positive electrode active material, a negative electrode, and a non-aqueous electrolyte.
- the positive electrode active material in a positive electrode for a non-aqueous electrolyte secondary battery using a positive electrode active material with a high Ni content, side reactions with the electrolyte can be effectively suppressed, and reaction resistance can be kept low. be able to.
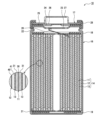
- FIG. 1 is a cross-sectional view of a non-aqueous electrolyte secondary battery that is an example of an embodiment
- the present inventors found that in a high-capacity lithium-transition metal composite oxide having a high Ni content, at least one selected from Ca and Sr is added to the interface between the primary particles inside the secondary particles of the composite oxide. , W, Mo, Ti, and Zr. Lithium-transition metal composite oxides with a high Ni content tend to cause side reactions with the electrolyte, especially at high charging rates, as described above. is protected, and the side reaction with the electrolyte is effectively suppressed.
- the compound may exist on the surface of the secondary particles, but as in Comparative Example 4 described later, the compound exists only on the surface of the secondary particles, and is present on the surface of the primary particles inside the secondary particles. If it does not exist, the effect of suppressing reaction resistance cannot be obtained.
- a cylindrical battery in which the wound electrode body 14 is housed in a bottomed cylindrical outer can 16 will be exemplified. It may be a prismatic battery), a coin-shaped outer can (coin-shaped battery), or an outer body (laminate battery) composed of a laminate sheet including a metal layer and a resin layer. Further, the electrode assembly is not limited to the wound type, and may be a laminated electrode assembly in which a plurality of positive electrodes and a plurality of negative electrodes are alternately laminated with separators interposed therebetween.
- FIG. 1 is a cross-sectional view of a non-aqueous electrolyte secondary battery 10 that is an example of an embodiment.
- the non-aqueous electrolyte secondary battery 10 includes a wound electrode body 14, a non-aqueous electrolyte, and an outer can 16 that accommodates the electrode body 14 and the non-aqueous electrolyte.
- the electrode body 14 has a positive electrode 11, a negative electrode 12, and a separator 13, and has a wound structure in which the positive electrode 11 and the negative electrode 12 are spirally wound with the separator 13 interposed therebetween.
- the outer can 16 is a bottomed cylindrical metal container that is open on one side in the axial direction. In the following description, for convenience of explanation, the side of the sealing member 17 of the battery will be referred to as the upper side, and the bottom side of the outer can 16 will be referred to as the lower side.
- the non-aqueous electrolyte contains a non-aqueous solvent and an electrolyte salt dissolved in the non-aqueous solvent.
- non-aqueous solvents include esters, ethers, nitriles, amides, and mixed solvents of two or more thereof.
- the non-aqueous solvent may contain a halogen-substituted product obtained by substituting at least part of the hydrogen atoms of these solvents with halogen atoms such as fluorine.
- non-aqueous solvents include ethylene carbonate (EC), ethylmethyl carbonate (EMC), dimethyl carbonate (DMC), mixed solvents thereof, and the like.
- a lithium salt such as LiPF 6 is used as the electrolyte salt.
- the non-aqueous electrolyte is not limited to a liquid electrolyte, and may be a solid electrolyte.
- the positive electrode 11, the negative electrode 12, and the separator 13, which constitute the electrode assembly 14, are all strip-shaped elongated bodies, and are alternately laminated in the radial direction of the electrode assembly 14 by being spirally wound.
- the negative electrode 12 is formed with a size one size larger than that of the positive electrode 11 in order to prevent deposition of lithium. That is, the negative electrode 12 is formed longer than the positive electrode 11 in the longitudinal direction and the width direction (transverse direction).
- the separator 13 is at least one size larger than the positive electrode 11, and two separators 13 are arranged so as to sandwich the positive electrode 11, for example.
- the electrode body 14 has a positive electrode lead 20 connected to the positive electrode 11 by welding or the like, and a negative electrode lead 21 connected to the negative electrode 12 by welding or the like.
- Insulating plates 18 and 19 are arranged above and below the electrode body 14, respectively.
- the positive electrode lead 20 extends through the through hole of the insulating plate 18 toward the sealing member 17
- the negative electrode lead 21 extends through the outside of the insulating plate 19 toward the bottom of the outer can 16 .
- the positive electrode lead 20 is connected to the lower surface of the internal terminal plate 23 of the sealing body 17 by welding or the like, and the cap 27, which is the top plate of the sealing body 17 electrically connected to the internal terminal plate 23, serves as the positive electrode terminal.
- the negative electrode lead 21 is connected to the inner surface of the bottom of the outer can 16 by welding or the like, and the outer can 16 serves as a negative electrode terminal.
- a gasket 28 is provided between the outer can 16 and the sealing body 17 to ensure hermeticity inside the battery.
- the outer can 16 is formed with a grooved portion 22 that supports the sealing member 17 and has a portion of the side surface projecting inward.
- the grooved portion 22 is preferably annularly formed along the circumferential direction of the outer can 16 and supports the sealing member 17 on its upper surface.
- the sealing member 17 is fixed to the upper portion of the outer can 16 by the grooved portion 22 and the open end of the outer can 16 that is crimped to the sealing member 17 .
- the sealing body 17 has a structure in which an internal terminal plate 23, a lower valve body 24, an insulating member 25, an upper valve body 26, and a cap 27 are layered in order from the electrode body 14 side.
- Each member constituting the sealing member 17 has, for example, a disk shape or a ring shape, and each member other than the insulating member 25 is electrically connected to each other.
- the lower valve body 24 and the upper valve body 26 are connected at their central portions, and an insulating member 25 is interposed between their peripheral edge portions.
- the positive electrode 11, the negative electrode 12, and the separator 13 that make up the electrode body 14 will be described in detail below, particularly the positive electrode active material that makes up the positive electrode 11.
- the positive electrode 11 includes a positive electrode core 30 and a positive electrode mixture layer 31 provided on the surface of the positive electrode core 30 .
- a foil of a metal such as aluminum or an aluminum alloy that is stable in the potential range of the positive electrode 11, a film having the metal on the surface layer, or the like can be used.
- Positive electrode mixture layer 31 contains a positive electrode active material, a binder, and a conductive agent, and is preferably provided on both surfaces of positive electrode core 30 .
- the positive electrode 11 can be produced, for example, by applying slurry of a positive electrode mixture onto the positive electrode core 30 , drying the coating film, and then compressing it to form the positive electrode mixture layers 31 on both sides of the positive electrode core 30 . .
- binder contained in the positive electrode mixture layer 31 examples include fluororesins such as polytetrafluoroethylene (PTFE) and polyvinylidene fluoride (PVdF), polyacrylonitrile (PAN), polyimide, acrylic resins, and polyolefins. These resins may be used in combination with cellulose derivatives such as carboxymethyl cellulose (CMC) or salts thereof, polyethylene oxide (PEO), and the like.
- the content of the binder is, for example, 0.5 to 2 mass % with respect to the mass of the positive electrode mixture layer 31 .
- Examples of the conductive agent contained in the positive electrode mixture layer 31 include carbon materials such as carbon black, acetylene black, ketjen black, graphite, and carbon nanotubes.
- the content of the conductive agent is, for example, 0.5 to 10 mass % with respect to the mass of the positive electrode mixture layer 31 .
- Specific examples of the layered crystal structure include a layered structure belonging to the space group R-3m and a layered structure belonging to the space group C2/m.
- the above lithium-transition metal composite oxide is secondary particles formed by aggregation of a plurality of primary particles.
- the particle size of the primary particles is, for example, 0.05 ⁇ m to 1 ⁇ m.
- the particle size of primary particles is measured as the diameter of the circumscribed circle in a particle image observed by a scanning electron microscope (SEM).
- SEM scanning electron microscope
- at least one selected from Ca and Sr and at least one selected from W, Mo, Ti, and Zr are present at the interfaces between the primary particles inside the secondary particles of the lithium-transition metal composite oxide. A compound containing seeds is stuck.
- the positive electrode active material contains a composite oxide (Z) as a main component.
- the main component means the component with the highest mass ratio among the constituent components of the positive electrode active material.
- a composite oxide other than the composite oxide (Z) may be used in combination as the positive electrode active material in the positive electrode mixture layer 31, but the content of the composite oxide (Z) is preferably 50% by mass or more. Preferably, it may be substantially 100% by mass.
- the volume-based median diameter (D50) of the composite oxide (Z) is, for example, 3 ⁇ m to 30 ⁇ m, preferably 5 ⁇ m to 25 ⁇ m. Since the composite oxide (Z) is secondary particles formed by agglomeration of primary particles, the D50 of the composite oxide (Z) means the D50 of the secondary particles. D50 means a particle size at which the cumulative frequency is 50% from the smaller particle size in the volume-based particle size distribution, and is also called median diameter.
- the particle size distribution of the composite oxide (Z) can be measured using a laser diffraction particle size distribution analyzer (eg MT3000II manufactured by Microtrack Bell Co., Ltd.) using water as a dispersion medium.
- the BET specific surface area of the composite oxide (Z) is preferably 0.5-3.5 m 2 /g. If the BET specific surface area is within this range, it becomes easy to suppress the reaction resistance without reducing the discharge capacity. If the BET specific surface area is smaller than this range, the reaction area decreases, which may result in a decrease in discharge capacity. On the other hand, when the BET specific surface area is larger than this range, the compound X alone cannot sufficiently cover the surface, so the effect of suppressing the reaction resistance is reduced.
- the BET specific surface area is measured according to the BET method (nitrogen adsorption method) described in JIS R1626.
- the composite oxide (Z) contains 75 mol% Ni with respect to the total number of moles of the elements excluding Li and O.
- Ni content a in the general formula
- the upper limit of the Ni content is preferably 95 mol%. If the Ni content exceeds 95 mol %, it becomes difficult to ensure the stability of the layered structure of the composite oxide (Z), and battery performance such as cycle characteristics deteriorates.
- An example of a suitable range of Ni content is 80-95 mol %, or 85-95 mol %.
- the composite oxide (Z) contains 5 to 25 mol% of Mn relative to the total number of moles of elements excluding Li and O.
- Mn content (b in the general formula) By setting the Mn content (b in the general formula) to 5 mol % or more, the layered structure of the composite oxide (Z) is stabilized while ensuring a high energy density.
- Mn content exceeds 25 mol %, the capacity and high-temperature cycle characteristics deteriorate.
- An example of a suitable range of Mn content is 5 to 20 mol %.
- x which indicates the ratio of Li in the composite oxide (Z), is 0.95 ⁇ x ⁇ 1.05, and more preferably 0.97 ⁇ a ⁇ 1.03.
- x 0.95 or less, the capacity may be lower than when x satisfies the above range.
- x 1.05 or more, a larger amount of Li compound is added than when x satisfies the above range, which may not be economical from the viewpoint of manufacturing cost.
- c which indicates the proportion of Al, shall be 0.02 or less. Since Al does not change its oxidation number during charging and discharging, it is considered that the structure of the transition metal layer is stabilized by being contained in the transition metal layer. On the other hand, if the Al content is too high, it leads to a decrease in capacity.
- the composite oxide (Z) contains Co
- d which indicates the ratio of Co
- Co is an expensive element
- the Co content is 5 mol % or more with respect to the total number of moles of the elements excluding Li and O
- the effect of the compound X to suppress side reactions is reduced.
- the Co content is less than 5 mol %, the effect of compound X becomes significant.
- At least one selected from Ca and Sr and at least one selected from W, Mo, Ti, and Zr are present at the interfaces between the primary particles inside at least the secondary particles of the composite oxide (Z).
- a compound X containing at least one is fixed.
- the compound X exists on the surface of the primary particles inside the secondary particles of the composite oxide (Z), it is thought that the side reaction between the composite oxide (Z) and the electrolyte is effectively suppressed. reaction resistance can be kept low.
- compound X can be confirmed by measuring the cross section of secondary particles using TEM-EDX (transmission microscope-energy dispersive X-ray spectroscopy).
- the compound X may, for example, be scattered on the surface of the primary particles, or may be present in a layer so as to widely cover the surfaces of the primary particles.
- the compound X may further exist on the surface of the secondary particles of the composite oxide (Z). That is, the compound X exists widely on the surface of the primary particles inside and on the surface of the secondary particles.
- the secondary particles of the composite oxide (Z) are formed, for example, by aggregating five or more primary particles, and the surface area of the primary particles is larger inside than on the surface of the secondary particles. Compound X is contained more inside than on the surface of the secondary particles.
- the total amount of Ca and Sr in compound X is preferably 2 mol% or less with respect to the total molar amount of metal elements excluding Li in the composite oxide (Z).
- the total amount of Ca and Sr is 2 mol % or less, the effect of suppressing reaction resistance becomes more pronounced.
- both elements of Ca and Sr may be contained in the compound X, the effect of suppressing the reaction resistance can be obtained if either one of the elements is contained.
- Compound X is preferably an oxide.
- An example of a suitable composition of the oxide is represented by the general formula A ⁇ B ⁇ O ⁇ (wherein 1 ⁇ 2, 1 ⁇ 2, 3 ⁇ 6, A is selected from Ca and Sr). and B is at least one selected from W, Mo, Ti, and Zr).
- a ⁇ B ⁇ O ⁇ wherein 1 ⁇ 2, 1 ⁇ 2, 3 ⁇ 6, A is selected from Ca and Sr.
- B is at least one selected from W, Mo, Ti, and Zr.
- a ⁇ B ⁇ O ⁇ examples include CaWO4 , CaMoO3 , CaMoO4 , CaTiO3 , CaZrO3 , CaZr4O9 , SrWO4 , SrMoO3 , SrMoO4 , SrTiO3 , Sr2TiO4 , SrZrO 3 , SrZr4O9 .
- the content of the element A in A ⁇ B ⁇ O ⁇ is, as described above, 2 mol % with respect to the total molar amount of the metal elements excluding Li in the composite oxide (Z) and A ⁇ B ⁇ O ⁇ .
- the following are preferable.
- the content of element B in A ⁇ B ⁇ O ⁇ is 3 mol% or less with respect to the total molar amount of metals excluding Li in the composite oxide (Z) and A ⁇ B ⁇ O ⁇ . is preferred.
- the content of the elements A and B in A ⁇ B ⁇ O ⁇ is preferably 1 mol % or more. Part of the elements A and B added to form A ⁇ B ⁇ O ⁇ may be dissolved in the composite oxide (Z).
- the manufacturing process of the composite oxide (Z) includes, for example, a first step of obtaining a composite oxide containing Ni or the like, a second step of mixing the composite oxide and a lithium compound to obtain a mixture, A third step of baking the mixture and a fourth step of washing the baked product with water and drying by heating are included.
- the compound X is produced by adding the compound containing the element A and the compound containing the element B during the manufacturing process of the composite oxide (Z), so that the primary particles inside the secondary particles of the composite oxide (Z) It can be stuck to the interface between particles.
- the compound containing element A and the compound containing element B are preferably added in the second step.
- an alkaline solution such as sodium hydroxide is added dropwise to adjust the pH to the alkaline side (eg, 8.5 to 12.5).
- an alkaline solution such as sodium hydroxide
- composite hydroxides containing Ni, Mn, etc. are deposited (coprecipitated).
- firing the composite hydroxide a composite oxide containing Ni, Mn, etc. can be obtained.
- the firing temperature is not particularly limited, it is 300°C to 600°C as an example.
- a lithium compound, a compound containing element A, and a compound containing element B are mixed.
- lithium compounds include Li2CO3 , LiOH , Li2O2 , Li2O , LiNO3 , LiNO2 , Li2SO4 , LiOH.H2O , LiH, and LiF .
- the composite oxide and the lithium compound are preferably mixed at a molar ratio of 1:0.98 to 1:1.12, for example, between the total amount of metal elements excluding Li and Li.
- Examples of compounds containing element A include Ca(OH) 2 , CaO, CaCO 3 , CaSO 4 , Ca(NO 3 ) 2 , Sr(OH) 2 , Sr(OH) 2.8H 2 O, Sr( OH) 2.H 2 O, SrO, SrCO 3 , SrSO 4 , Sr(NO 3 ) 2 and the like. good. Further, these compounds may be pulverized to a particle size of 0.1 to 20 ⁇ m. Compounds containing element B also include hydroxides, oxides, carbonates, sulfates, nitrates, etc. of element B, but in order to reduce the amount of water generated during firing, they are dried and dehydrated. It can be used after Further, these compounds may be pulverized to a particle size of 0.1 to 20 ⁇ m.
- a compound containing a composite oxide and an element A is such that the molar ratio of the total amount of metal elements excluding Li in the composite oxide to the element A is 1:0.0005 to 1:0.03. is preferably mixed with When multiple types of compounds containing element A are used, they are mixed so that the total amount of element A contained in the compounds satisfies the ratio.
- a suitable mixing ratio with the composite oxide is the same for the compound containing the element B.
- the elements A and B are preferably mixed according to the stoichiometric ratio of A ⁇ B ⁇ O ⁇ .
- the firing step of the mixture in the third step includes, for example, a first firing step of firing at 450 ° C. to 680 ° C. under an oxygen stream, and a fired product obtained by the first firing step under an oxygen stream at a temperature exceeding 680 ° C. It is a multi-stage firing process including at least a second firing process of firing at a temperature.
- the temperature is raised to a first set temperature of 680° C. or lower at a first temperature elevation rate of 0.2° C./min to 5.5° C./min.
- the second firing step the temperature is raised to a second set temperature of 900° C. or less at a second temperature increase rate of 0.1° C./min to 3.5° C./min and slower than the first temperature increase rate. .
- a plurality of the first and second heating rates may be set for each predetermined temperature range within the above range.
- the retention time of the first set temperature in the first firing step is preferably 5 hours or less, more preferably 3 hours or less.
- the holding time of the first set temperature is the time for maintaining the first set temperature after reaching the first set temperature, and the holding time may be zero.
- the holding time of the second set temperature in the second firing step is preferably 1 hour to 10 hours, more preferably 1 hour to 5 hours.
- the holding time of the second set temperature is the time for maintaining the second set temperature after reaching the second set temperature. Firing of the mixture is performed, for example, in an oxygen stream with an oxygen concentration of 60% or more, and the flow rate of the oxygen stream is 0.2 mL/min to 4 mL/min per 10 cm 3 of the firing furnace and 0.3 L/min or more per 1 kg of the mixture. and
- the baked product obtained in the third step is washed with water to remove impurities, and the washed baked product is dried by heating. If necessary, the fired product is pulverized, classified, etc., and the D50 of the positive electrode active material is adjusted to the desired range. Drying of the baked product after washing with water may be performed at a temperature of less than 100°C. An example of a suitable drying temperature is 150°C to 250°C. Drying may be performed under vacuum or in the atmosphere. An example of the drying treatment time is 1 hour to 5 hours.
- the negative electrode 12 includes a negative electrode core 40 and a negative electrode mixture layer 41 provided on the surface of the negative electrode core 40 .
- a foil of a metal such as copper or a copper alloy that is stable in the potential range of the negative electrode 12, a film having the metal on the surface layer, or the like can be used.
- the negative electrode mixture layer 41 contains a negative electrode active material and a binder, and is preferably provided on both surfaces of the negative electrode core 40 .
- a negative electrode mixture slurry containing a negative electrode active material, a binder, and the like is applied to the surface of the negative electrode core 40, the coating film is dried, and then the negative electrode mixture layer 41 is compressed to form the negative electrode core. It can be made by forming on both sides of the body 40 .
- the negative electrode mixture layer 41 may contain the same conductive agent as in the case of the positive electrode 11 .
- the negative electrode mixture layer 41 contains, as a negative electrode active material, for example, a carbon material that reversibly absorbs and releases lithium ions.
- a carbon material that reversibly absorbs and releases lithium ions.
- a suitable example of the carbon material is natural graphite such as flake graphite, massive graphite, and earthy graphite, artificial graphite such as massive artificial graphite (MAG), and graphitized mesophase carbon microbeads (MCMB).
- an active material containing at least one of an element that alloys with Li, such as Si and Sn, and a compound containing the element may be used.
- a suitable example of the active material is a silicon material in which Si fine particles are dispersed in a silicon oxide phase or a silicate phase such as lithium silicate.
- a carbon material such as graphite and a silicon material are used together.
- the binder contained in the negative electrode mixture layer 41 fluororesin, PAN, polyimide, acrylic resin, polyolefin, etc. can be used as in the case of the positive electrode 11, but styrene-butadiene rubber (SBR) is used. is preferred.
- the negative electrode mixture layer 41 preferably further contains CMC or a salt thereof, polyacrylic acid (PAA) or a salt thereof, polyvinyl alcohol (PVA), or the like. Among them, it is preferable to use SBR together with CMC or its salt or PAA or its salt.
- the negative electrode mixture layer 41 may contain a conductive agent.
- a porous sheet having ion permeability and insulation is used for the separator 13 .
- porous sheets include microporous thin films, woven fabrics, and non-woven fabrics.
- Suitable materials for the separator 13 include polyolefins such as polyethylene, polypropylene, copolymers of ethylene and ⁇ -olefin, and cellulose.
- the separator 13 may have either a single layer structure or a laminated structure.
- a heat-resistant layer containing inorganic particles, a heat-resistant layer made of a highly heat-resistant resin such as aramid resin, polyimide, polyamideimide, or the like may be formed on the surface of the separator 13 .
- the positive electrode active material, acetylene black, and polyvinylidene fluoride are mixed at a mass ratio of 96:2.5:1.5, and N-methyl-2-pyrrolidone (NMP) is used as a dispersion medium to obtain a positive electrode.
- NMP N-methyl-2-pyrrolidone
- a mixture slurry was prepared.
- the positive electrode material mixture slurry is applied onto a positive electrode core made of aluminum foil, the coating film is dried and compressed, and then the positive electrode core is cut into a predetermined electrode size.
- a positive electrode having an agent layer formed thereon was obtained.
- an exposed portion where the surface of the positive electrode core was exposed was provided on a part of the positive electrode.
- Ethylene carbonate (EC), methyl ethyl carbonate (MEC) and dimethyl carbonate (DMC) were mixed in a volume ratio of 3:3:4 (25° C.).
- a non-aqueous electrolyte was prepared by dissolving LiPF 6 in the mixed solvent so as to have a concentration of 1.2 mol/liter.
- test cell An aluminum lead is attached to the exposed portion of the positive electrode, and a nickel lead is attached to the lithium metal foil as the negative electrode, respectively. A wound electrode body was produced. This electrode assembly was housed in an exterior made of an aluminum laminate sheet, and after the non-aqueous electrolyte was injected thereinto, the opening of the exterior was sealed to obtain a test cell Al.
- Example 2 A test cell A2 was produced in the same manner as in Example 1, except that a predetermined amount of strontium hydroxide was added instead of calcium hydroxide in the synthesis of the positive electrode active material. It was confirmed that the elements shown in Table 1 were contained in the synthesized positive electrode active material. In addition, SrZrO 3 was present in the positive electrode active material, and the presence of Sr and Zr was confirmed at the interfaces between the primary particles inside the secondary particles.
- Example 3 A test cell A3 was produced in the same manner as in Example 1, except that predetermined amounts of strontium hydroxide and titanium hydroxide were added instead of calcium hydroxide and zirconium oxide in the synthesis of the positive electrode active material. It was confirmed that the elements shown in Table 1 were contained in the synthesized positive electrode active material. Moreover, Sr 2 TiO 4 and SrTiO 3 were present in the positive electrode active material, and the presence of Sr and Ti was confirmed at the interfaces between the primary particles inside the secondary particles.
- test cell B1 was prepared in the same manner as in Example 1, except that calcium hydroxide and zirconium oxide were not added in the synthesis of the positive electrode active material.
- Example 4 In the synthesis of the positive electrode active material, a composite hydroxide represented by [Ni 86 Mn 14 ](OH) 2 is used, and a composite oxide obtained by baking the composite hydroxide, lithium hydroxide, and water Calcium oxide and tungsten oxide were mixed such that the total amount of Li, Ni and Mn, Ca and W was in a molar ratio of 1.03:1.00:0.005:0.005.
- the mixture is heated from room temperature to 670° C. at a heating rate of 2.0° C./min under an oxygen stream with an oxygen concentration of 95% (flow rate of 2 mL/min per 10 cm 3 and 5 L/min per 1 kg of the mixture) and fired. After that, the temperature was raised to 780° C.
- a test cell A4 was produced in the same manner as in Example 1, except that the baked product was washed with water to remove impurities, and vacuum-dried at 180° C. for 2 hours to obtain a positive electrode active material.
- the positive electrode active material synthesized in Example 4 contained the elements shown in Table 1.
- CaWO 4 was present in the positive electrode active material, and the presence of Ca and W was confirmed at the interfaces between the primary particles inside the secondary particles.
- Example 5 A test cell A5 was prepared in the same manner as in Example 4, except that predetermined amounts of strontium hydroxide and molybdenum oxide were added instead of calcium hydroxide and tungsten oxide in the synthesis of the positive electrode active material. It was confirmed that the elements shown in Table 1 were contained in the synthesized positive electrode active material. In addition, SrMoO 4 was present in the positive electrode active material, and the presence of Sr and Mo was confirmed at the interfaces between the primary particles inside the secondary particles.
- Example 6 A test cell A6 was prepared in the same manner as in Example 5, except that predetermined amounts of calcium hydroxide and titanium hydroxide were added instead of strontium hydroxide and molybdenum oxide in the synthesis of the positive electrode active material. It was confirmed that the elements shown in Table 1 were contained in the synthesized positive electrode active material. In addition, CaTiO 3 was present in the positive electrode active material, and the presence of Ca and Ti was confirmed at the interfaces between the primary particles inside the secondary particles.
- test cell B2 was prepared in the same manner as in Example 4, except that calcium hydroxide and tungsten oxide were not added in the synthesis of the positive electrode active material.
- test cell B3 was produced in the same manner as in Example 4, except that tungsten oxide was not added in the synthesis of the positive electrode active material.
- Example 4 In the synthesis of the positive electrode active material, the test was performed in the same manner as in Example 4, except that the timing of adding tungsten oxide was changed after the baked product was washed with water, and the drying after washing was performed under atmospheric pressure at 80 ° C. Cell B4 was produced. In the synthesized positive electrode active material, CaWO 4 was present only on the surfaces of the secondary particles, and was not found at the interfaces between the primary particles inside the secondary particles.
- Example 5 A test cell B5 was prepared in the same manner as in Example 4, except that a predetermined amount of magnesium hydroxide was added instead of strontium hydroxide and molybdenum oxide in the synthesis of the positive electrode active material.
- Example 7 A composite hydroxide represented by [Ni 79 Mn 19 Al 1 Co 1 ](OH) 2 obtained by a coprecipitation method was fired at 500° C. for 8 hours to obtain a composite oxide.
- the composite oxide, lithium hydroxide, calcium hydroxide, and tungsten oxide are combined so that the total amount of Li, Ni, Mn, Al, and Co, Ca, and W have a molar ratio of 1.03:1.
- the mixture is heated from room temperature to 650° C. at a heating rate of 2.0° C./min under an oxygen stream with an oxygen concentration of 95% (flow rate of 2 mL/min per 10 cm 3 and 5 L/min per 1 kg of the mixture) and fired.
- a test cell A7 was produced in the same manner as in Example 1, except that the baked product was washed with water to remove impurities, and vacuum-dried at 180° C. for 2 hours to obtain a positive electrode active material.
- the positive electrode active material synthesized in Example 7 contained the elements shown in Table 1.
- CaWO 4 was present in the positive electrode active material, and the presence of Ca and W was confirmed at the interfaces between the primary particles inside the secondary particles.
- Example 8 A test cell A8 was produced in the same manner as in Example 7, except that a predetermined amount of molybdenum oxide was added instead of tungsten oxide in the synthesis of the positive electrode active material. It was confirmed that the elements shown in Table 1 were contained in the synthesized positive electrode active material. In addition, CaMoO 4 was present in the positive electrode active material, and the presence of Ca and Mo was confirmed at the interfaces between the primary particles inside the secondary particles.
- test cell B6 was produced in the same manner as in Example 7, except that calcium hydroxide and tungsten oxide were not added in the synthesis of the positive electrode active material.
- a composite hydroxide represented by [Ni 83 Mn 9 Al 3 Co 5 ](OH) 2 obtained by a coprecipitation method was fired at 500° C. for 8 hours to obtain a composite oxide.
- the composite oxide, lithium hydroxide, calcium hydroxide, and molybdenum oxide are combined so that the total amount of Li, Ni, Mn, Al, and Co, Ca, and Mo have a molar ratio of 1.03:1.
- the mixture is heated from room temperature to 650° C. at a heating rate of 2.0° C./min under an oxygen stream with an oxygen concentration of 95% (flow rate of 2 mL/min per 10 cm 3 and 5 L/min per 1 kg of the mixture) and fired.
- a test cell R1 was produced in the same manner as in Example 1, except that the baked product was washed with water to remove impurities, and vacuum-dried at 180° C. for 2 hours to obtain a positive electrode active material.
- the positive electrode active material synthesized in Reference Example 1 contained the elements shown in Table 1.
- CaMoO 4 was present in the positive electrode active material, and the presence of Ca and Mo was confirmed at the interfaces between the primary particles inside the secondary particles.
- test cell R2 was produced in the same manner as in Reference Example 1, except that calcium hydroxide and molybdenum oxide were not added in the synthesis of the positive electrode active material.
- a composite hydroxide represented by [Ni 80 Mn 10 Co 10 ](OH) 2 obtained by a coprecipitation method was calcined at 500° C. for 8 hours to obtain a composite oxide.
- the composite oxide, lithium hydroxide, strontium hydroxide, and titanium hydroxide were mixed with the total amount of Li, Ni, Mn, and Co, Sr, and Ti in a molar ratio of 1.03:1. It was mixed so that it became 00:0.01:0.01. The mixture is heated from room temperature to 650° C.
- a test cell R3 was produced in the same manner as in Example 1, except that the baked product was washed with water to remove impurities, and vacuum-dried at 180° C. for 2 hours to obtain a positive electrode active material.
- the positive electrode active material synthesized in Reference Example 3 contained the elements shown in Table 1. Moreover, Sr 2 TiO 4 and SrTiO 3 were present in the positive electrode active material, and the presence of Sr and Ti was confirmed at the interfaces between the primary particles inside the secondary particles.
- test cell R4 was prepared in the same manner as in Reference Example 3, except that strontium hydroxide and titanium hydroxide were not added in the synthesis of the positive electrode active material.
- Tables 1 to 4 show the calculated reaction resistance Rct.
- the Rct shown in Table 1 is a relative value when the Rct of the test cell B1 of Comparative Example 1 is 100.
- the Rct shown in Table 2 is a relative value when the Rct of the test cell B2 of Comparative Example 2 is 100.
- Rct shown in Table 3 is a relative value when Rct of test cell B6 of Comparative Example 6 is set to 100.
- Rct of test cell R2 of Reference Example 2 in Table 4 is a relative value when Rct of test cell R1 of Reference Example 1 is 100
- Rct of test cell R4 of Reference Example 4 in Table 4 is a reference example.
- 3 is a relative value when the Rct of the test cell R3 of No. 3 is set to 100.
- the reaction resistance Rct of each of the test cells of the example is kept lower than that of the corresponding test cell of the comparative example.
- element A Ca, Sr
- element B W, Mo, Ti, Zr
- only one of elements A and B is added In the cases (Comparative Examples 3 and 5) and in the case where A ⁇ B ⁇ O ⁇ was present only on the surface of the secondary particles (Comparative Example 4), the effect of suppressing the reaction resistance was not obtained.
- the reaction resistance suppressing effect of A ⁇ B ⁇ O ⁇ decreases. That is, when the Co content is small, particularly when the Co content is less than 5 mol% with respect to the metal elements other than Li in the composite oxide, the reaction resistance is suppressed by A ⁇ B ⁇ O ⁇ . The effect becomes noticeable.
Abstract
実施形態の一例である非水電解質二次電池用正極活物質は、層状構造を有し、一般式LixNiaMnbAlcCodMeO2-y(式中、0.95<x<1.05、0.75≦a≦0.95、0.05≦b≦0.25、0≦c≦0.02、0≦d<0.05、0≦e≦0.03、0≦y≦0.05、a+b+c=1、Mは、Fe、Ti、Si、Nb、Mo、W、およびZrから選択される少なくとも1種の元素)で表されるリチウム遷移金属複合酸化物を含む。リチウム遷移金属複合酸化物の二次粒子内部における一次粒子同士の界面には、CaおよびSrから選択される少なくとも1種と、W、Mo、Ti、およびZrから選択される少なくとも1種とを含有する化合物が固着している。
Description
本開示は、非水電解質二次電池用正極活物質および当該活物質を用いた非水電解質二次電池に関する。
非水電解質二次電池の正極を構成する正極活物質は、容量、サイクル特性等の電池性能に大きく影響することから、正極活物質について多くの検討が行われてきた。例えば、特許文献1には、遷移金属元素としてMn、Ni、およびCoから選択される1種以上を含むリチウム遷移金属複合酸化物の二次粒子表面に、アルカリ土類金属およびWが存在する正極活物質が開示されている。また、特許文献2には、遷移金属元素としてMn、Ni、およびCoから選択される1種以上を含み、さらにCaおよびWが固溶したリチウム遷移金属複合酸化物を含む正極活物質が開示されている。
ところで、Ni含有量が多いリチウム遷移金属複合酸化物は、高容量の正極活物質として知られているが、充電率が高い状態では複合酸化物の粒子表層に反応性の高いNi4+が多く存在するため、電解液との副反応が生じやすい。そして、副反応で生じた分解物が複合酸化物の粒子表面に堆積し、正極の反応抵抗が大きくなる。
本開示の目的は、Ni含有量が多い正極活物質を用いた非水電解質二次電池正極において、反応抵抗の増大を抑制することである。
本開示に係る非水電解質二次電池用正極活物質は、層状構造を有し、一般式LixNiaMnbAlcCodMeO2-y(式中、0.95<x<1.05、0.75≦a≦0.95、0.05≦b≦0.25、0≦c≦0.02、0≦d<0.05、0≦e≦0.03、0≦y≦0.05、a+b+c=1、Mは、Fe、Ti、Si、Nb、Mo、W、およびZrから選択される少なくとも1種の元素)で表されるリチウム遷移金属複合酸化物を含み、リチウム遷移金属複合酸化物は、一次粒子が凝集して形成された二次粒子であり、少なくとも二次粒子の内部における一次粒子同士の界面には、CaおよびSrから選択される少なくとも1種と、W、Mo、Ti、およびZrから選択される少なくとも1種とを含有する化合物が固着していることを特徴とする。
本開示に係る非水電解質二次電池は、上記正極活物質を含む正極と、負極と、非水電解質とを備える。
本開示に係る正極活物質によれば、Ni含有量が多い正極活物質を用いた非水電解質二次電池用正極において、電解液との副反応を効果的に抑制でき、反応抵抗を低く抑えることができる。
本発明者らは、Ni含有量が多い高容量のリチウム遷移金属複合酸化物において、複合酸化物の二次粒子の内部における一次粒子同士の界面に、CaおよびSrから選択される少なくとも1種と、W、Mo、Ti、およびZrから選択される少なくとも1種とを含有する化合物を固着させることで、反応抵抗の増大を特異的に抑制できることを見出した。Ni含有量が多いリチウム遷移金属複合酸化物は、上述の通り、特に充電率が高い状態で電解液との副反応を生じやすいが、上記化合物が一次粒子同士の界面に存在する場合、粒子表面が保護され、電解液との副反応が効果的に抑制されると考えられる。
上記化合物は二次粒子の表面に存在していてもよいが、後述の比較例4のように、上記化合物が二次粒子の表面のみに存在し、二次粒子の内部において一次粒子の表面に存在しない場合は、反応抵抗の抑制効果は得られない。
以下、図面を参照しながら、本開示に係る非水電解質二次電池用正極活物質および当該活物質を用いた非水電解質二次電池の実施形態の一例について詳細に説明する。なお、以下で説明する複数の実施形態、変形例の各構成要素を選択的に組み合わせることは本開示の範囲に含まれている。
以下では、巻回型の電極体14が有底円筒形状の外装缶16に収容された円筒形電池を例示するが、外装体は円筒形の外装缶に限定されず、例えば角形の外装缶(角形電池)や、コイン形の外装缶(コイン形電池)であってもよく、金属層および樹脂層を含むラミネートシートで構成された外装体(ラミネート電池)であってもよい。また、電極体は巻回型に限定されず、複数の正極と複数の負極がセパレータを介して交互に積層された積層型の電極体であってもよい。
図1は、実施形態の一例である非水電解質二次電池10の断面図である。図1に示すように、非水電解質二次電池10は、巻回型の電極体14と、非水電解質と、電極体14および非水電解質を収容する外装缶16とを備える。電極体14は、正極11、負極12、およびセパレータ13を有し、正極11と負極12がセパレータ13を介して渦巻き状に巻回された巻回構造を有する。外装缶16は、軸方向一方側が開口した有底円筒形状の金属製容器であって、外装缶16の開口部は封口体17によって塞がれている。以下では、説明の便宜上、電池の封口体17側を上、外装缶16の底部側を下とする。
非水電解質は、非水溶媒と、非水溶媒に溶解した電解質塩とを含む。非水溶媒には、例えばエステル類、エーテル類、ニトリル類、アミド類、およびこれらの2種以上の混合溶媒等が用いられる。非水溶媒は、これら溶媒の水素の少なくとも一部をフッ素等のハロゲン原子で置換したハロゲン置換体を含有していてもよい。非水溶媒の一例としては、エチレンカーボネート(EC)、エチルメチルカーボネート(EMC)、ジメチルカーボネート(DMC)、およびこれらの混合溶媒等が挙げられる。電解質塩には、例えばLiPF6等のリチウム塩が使用される。なお、非水電解質は液体電解質に限定されず、固体電解質であってもよい。
電極体14を構成する正極11、負極12、およびセパレータ13は、いずれも帯状の長尺体であって、渦巻状に巻回されることで電極体14の径方向に交互に積層される。負極12は、リチウムの析出を防止するために、正極11よりも一回り大きな寸法で形成される。すなわち、負極12は、正極11よりも長手方向および幅方向(短手方向)に長く形成される。セパレータ13は、少なくとも正極11よりも一回り大きな寸法で形成され、例えば正極11を挟むように2枚配置される。電極体14は、溶接等により正極11に接続された正極リード20と、溶接等により負極12に接続された負極リード21とを有する。
電極体14の上下には、絶縁板18,19がそれぞれ配置される。図1に示す例では、正極リード20が絶縁板18の貫通孔を通って封口体17側に延び、負極リード21が絶縁板19の外側を通って外装缶16の底部側に延びている。正極リード20は封口体17の内部端子板23の下面に溶接等で接続され、内部端子板23と電気的に接続された封口体17の天板であるキャップ27が正極端子となる。負極リード21は外装缶16の底部内面に溶接等で接続され、外装缶16が負極端子となる。
外装缶16と封口体17の間にはガスケット28が設けられ、電池内部の密閉性が確保される。外装缶16には、側面部の一部が内側に張り出した、封口体17を支持する溝入部22が形成されている。溝入部22は、外装缶16の周方向に沿って環状に形成されることが好ましく、その上面で封口体17を支持する。封口体17は、溝入部22と、封口体17に対して加締められた外装缶16の開口端部とにより、外装缶16の上部に固定される。
封口体17は、電極体14側から順に、内部端子板23、下弁体24、絶縁部材25、上弁体26、およびキャップ27が積層された構造を有する。封口体17を構成する各部材は、例えば円板形状又はリング形状を有し、絶縁部材25を除く各部材は互いに電気的に接続されている。下弁体24と上弁体26は各々の中央部で接続され、各々の周縁部の間には絶縁部材25が介在している。異常発熱で電池の内圧が上昇すると、下弁体24が上弁体26をキャップ27側に押し上げるように変形して破断することにより、下弁体24と上弁体26の間の電流経路が遮断される。さらに内圧が上昇すると、上弁体26が破断し、キャップ27の開口部からガスが排出される。
以下、電極体14を構成する正極11、負極12、セパレータ13について、特に正極11を構成する正極活物質について詳説する。
[正極]
正極11は、正極芯体30と、正極芯体30の表面に設けられた正極合剤層31とを備える。正極芯体30には、アルミニウム、アルミニウム合金など正極11の電位範囲で安定な金属の箔、当該金属を表層に配置したフィルム等を用いることができる。正極合剤層31は、正極活物質、結着剤、および導電剤を含み、正極芯体30の両面に設けられることが好ましい。正極11は、例えば正極芯体30上に正極合剤のスラリーを塗布し、塗膜を乾燥させた後、圧縮して正極合剤層31を正極芯体30の両面に形成することにより作製できる。
正極11は、正極芯体30と、正極芯体30の表面に設けられた正極合剤層31とを備える。正極芯体30には、アルミニウム、アルミニウム合金など正極11の電位範囲で安定な金属の箔、当該金属を表層に配置したフィルム等を用いることができる。正極合剤層31は、正極活物質、結着剤、および導電剤を含み、正極芯体30の両面に設けられることが好ましい。正極11は、例えば正極芯体30上に正極合剤のスラリーを塗布し、塗膜を乾燥させた後、圧縮して正極合剤層31を正極芯体30の両面に形成することにより作製できる。
正極合剤層31に含まれる結着剤としては、ポリテトラフルオロエチレン(PTFE)、ポリフッ化ビニリデン(PVdF)等のフッ素樹脂、ポリアクリロニトリル(PAN)、ポリイミド、アクリル樹脂、ポリオレフィンなどが例示できる。これらの樹脂と、カルボキシメチルセルロース(CMC)又はその塩等のセルロース誘導体、ポリエチレンオキシド(PEO)などが併用されてもよい。結着剤の含有量は、例えば、正極合剤層31の質量に対して0.5~2質量%である。
正極合剤層31に含まれる導電剤は、カーボンブラック、アセチレンブラック、ケッチェンブラック、黒鉛、カーボンナノチューブ等の炭素材料が例示できる。導電剤の含有量は、例えば、正極合剤層31の質量に対して0.5~10質量%である。
正極活物質は、層状構造を有し、一般式LixNiaMnbAlcCodMeO2-y(式中、0.95<x<1.05、0.75≦a≦0.95、0.05≦b≦0.25、0≦c≦0.02、0≦d<0.05、0≦e≦0.03、0≦y≦0.05、a+b+c=1、Mは、Fe、Ti、Si、Nb、Mo、W、およびZrから選択される少なくとも1種の元素)で表されるリチウム遷移金属複合酸化物を含む。層状の結晶構造の具体例としては、空間群R-3mに属する層状構造、又は空間群C2/mに属する層状構造が挙げられる。
上記リチウム遷移金属複合酸化物は、複数の一次粒子が凝集して形成された二次粒子である。一次粒子の粒径は、例えば、0.05μm~1μmである。一次粒子の粒径は、走査型電子顕微鏡(SEM)により観察される粒子画像において外接円の直径として測定される。また、上記リチウム遷移金属複合酸化物の二次粒子の内部における一次粒子同士の界面には、CaおよびSrから選択される少なくとも1種と、W、Mo、Ti、およびZrから選択される少なくとも1種とを含有する化合物が固着している。
以下、説明の便宜上、上記リチウム遷移金属複合酸化物を「複合酸化物(Z)」とする。また、上記化合物を「化合物X」とする。正極活物質は、複合酸化物(Z)を主成分とする。ここで、主成分とは、正極活物質の構成成分のうち最も質量比率が高い成分を意味する。正極合剤層31には、正極活物質として、複合酸化物(Z)以外の複合酸化物が併用されてもよいが、複合酸化物(Z)の含有量は50質量%以上であることが好ましく、実質的に100質量%であってもよい。
複合酸化物(Z)の体積基準のメジアン径(D50)は、例えば3μm~30μm、好ましくは5μm~25μmである。複合酸化物(Z)は一次粒子が凝集してなる二次粒子であるから、複合酸化物(Z)のD50は二次粒子のD50を意味する。D50は、体積基準の粒度分布において頻度の累積が粒径の小さい方から50%となる粒径を意味し、中位径とも呼ばれる。複合酸化物(Z)の粒度分布は、レーザー回折式の粒度分布測定装置(例えば、マイクロトラック・ベル株式会社製、MT3000II)を用い、水を分散媒として測定できる。
複合酸化物(Z)のBET比表面積は、0.5~3.5m2/gであることが好ましい。BET比表面積が当該範囲内であれば、放電容量を減少させることなく、反応抵抗を抑制することが容易になる。BET比表面積が当該範囲より小さい場合は、反応面積が減少するため、放電容量が低下する場合がある。一方、BET比表面積が当該範囲よりも大きい場合は、化合物Xだけで表面を十分に被覆することができないため、反応抵抗の抑制効果が小さくなる。BET比表面積は、JIS R1626記載のBET法(窒素吸着法)に従って測定される。
複合酸化物(Z)は、上記の通り、Li、Oを除く元素の総モル数に対して75モル%のNiを含有する。Niの含有量(上記一般式中のa)を75モル%以上とすることで、高エネルギー密度の電池が得られる。Ni含有量の上限は、95モル%であることが好ましい。Niの含有量が95モル%を超えると、複合酸化物(Z)の層状構造の安定性を確保することが難しくなり、サイクル特性等の電池性能が低下する。Ni含有量の好適な範囲の一例は、80~95モル%、又は85~95モル%である。
複合酸化物(Z)は、Li、Oを除く元素の総モル数に対して5~25モル%のMnを含有する。Mnの含有量(上記一般式中のb)を5モル%以上とすることで、高エネルギー密度を確保しつつ、複合酸化物(Z)の層状構造が安定化する。他方、Mnの含有量が25モル%を超えると、容量および高温でのサイクル特性が低下する。Mn含有量の好適な範囲の一例は、5~20モル%である。
複合酸化物(Z)中のLiの割合を示すxは、0.95<x<1.05であり、0.97≦a≦1.03であることがより好ましい。xが0.95以下の場合、xが上記範囲を満たす場合と比較して、容量が低下する場合がある。xが1.05以上の場合、xが上記範囲を満たす場合と比較して、Li化合物をより多く添加することになるため、製造コストの観点から経済的ではない場合がある。
複合酸化物(Z)にAlが含有される場合、Alの割合を示すcは0.02以下とされる。Alは、充放電中にも酸化数変化が生じないため、遷移金属層に含有されることで遷移金属層の構造が安定化すると考えられる。一方、Alの含有量を多くし過ぎると、容量低下につながる。
複合酸化物(Z)にCoが含有される場合、Coの割合を示すdは0.05未満とされる。Coは高価な元素であるため、製造コストを考慮すると、Coの含有量を抑えることが好ましい。また、後述の参考例に示すように、Coの含有量がLi、Oを除く元素の総モル数に対して5モル%以上になると、化合物Xによる副反応抑制効果が低下する。言い換えると、Coの含有量が5モル%未満である場合に、化合物Xの効果が顕著になる。
複合酸化物(Z)の少なくとも二次粒子の内部における一次粒子同士の界面には、上記の通り、CaおよびSrから選択される少なくとも1種と、W、Mo、Ti、およびZrから選択される少なくとも1種とを含有する化合物Xが固着している。化合物Xが複合酸化物(Z)の二次粒子の内部において一次粒子の表面に存在する場合、複合酸化物(Z)と電解液の副反応が効果的に抑制されると考えられ、正極11の反応抵抗を低く抑えることができる。
化合物Xの存在は、TEM-EDX(透過型顕微鏡-エネルギー分散型X線分光法)を用いて、二次粒子の断面を測定することにより確認できる。化合物Xは、例えば、一次粒子の表面に点在していてもよく、一次粒子の表面を広く覆うように層状に存在していてもよい。
化合物Xは、さらに、複合酸化物(Z)の二次粒子の表面に存在していてもよい。すなわち、化合物Xは、二次粒子の内部および表面において、一次粒子の表面に広く存在している。複合酸化物(Z)の二次粒子は、例えば、5個以上の一次粒子が凝集して形成されており、一次粒子の表面積は二次粒子の表面よりも内部で大きくなっている。そして、化合物Xは、二次粒子の表面よりも内部に多く含まれている。
化合物X中のCaおよびSrの総量は、複合酸化物(Z)中のLiを除く金属元素の総モル量に対して2モル%以下であることが好ましい。CaおよびSrの総量が2モル%以下である場合、反応抵抗の抑制効果がより顕著になる。化合物X中には、CaおよびSrの両方の元素が含有されていてもよいが、いずれか一方の元素が含有されていれば、反応抵抗の抑制効果が得られる。
化合物Xは、酸化物であることが好ましい。また、酸化物の好適な組成の一例は、一般式AαBβOγ(式中、1≦α≦2、1≦β≦2、3≦γ≦6、AはCaおよびSrから選択される少なくとも1種、BはW、Mo、Ti、およびZrから選択される少なくとも1種)で表される。化合物Xが当該一般式で表される組成の酸化物である場合、反応抵抗の抑制効果がより顕著になる。
AαBβOγの具体例としては、CaWO4、CaMoO3、CaMoO4、CaTiO3、CaZrO3、CaZr4O9、SrWO4、SrMoO3、SrMoO4、SrTiO3、Sr2TiO4、SrZrO3、SrZr4O9が挙げられる。
AαBβOγ中の元素Aの含有量は、上記のように、複合酸化物(Z)およびAαBβOγ中のLiを除く金属元素の総モル量に対して2モル%以下であることが好ましい。AαBβOγ中の元素Bの含有量についても同様に、複合酸化物(Z)およびAαBβOγ中のLiを除く金属の総モル量に対して3モル%以下であることが好ましい。また、AαBβOγ中の元素A,Bの含有量は、1モル%以上であることが好ましい。なお、AαBβOγを形成するために添加された元素A,Bの一部は、複合酸化物(Z)中に固溶していてもよい。
以下、一次粒子同士の界面に化合物Xが固着した複合酸化物(Z)の製造方法の一例について説明する。
複合酸化物(Z)の製造工程には、例えば、Ni等を含有する複合酸化物を得る第1工程と、当該複合酸化物とリチウム化合物とを混合して混合物を得る第2工程と、当該混合物を焼成する第3工程と、焼成物を水洗して加熱乾燥する第4工程とが含まれる。化合物Xは、元素Aを含有する化合物と、元素Bを含有する化合物とを複合酸化物(Z)の製造工程中に添加することにより、複合酸化物(Z)の二次粒子の内部において一次粒子同士の界面に固着させることができる。元素Aを含有する化合物および元素Bを含有する化合物は、上記第2工程で添加されることが好ましい。
第1工程では、例えば、Ni、Mn等を含有する金属塩の溶液を撹拌しながら、水酸化ナトリウム等のアルカリ溶液を滴下し、pHをアルカリ側(例えば、8.5~12.5)に調整することにより、Ni、Mn等を含有する複合水酸化物を析出(共沈)させる。当該複合水酸化物を焼成することにより、Ni、Mn等を含有する複合酸化物が得られる。焼成温度は、特に制限されないが、一例としては300℃~600℃である。
第2工程では、例えば、第1工程で得られた複合酸化物と、リチウム化合物と、元素Aを含有する化合物と、元素Bを含有する化合物とを混合する。リチウム化合物の一例としては、Li2CO3、LiOH、Li2O2、Li2O、LiNO3、LiNO2、Li2SO4、LiOH・H2O、LiH、LiF等が挙げられる。複合酸化物とリチウム化合物は、例えば、Liを除く金属元素の総量と、Liとのモル比が、1:0.98~1:1.12となる比率で混合されることが好ましい。
元素Aを含有する化合物の一例としては、Ca(OH)2、CaO、CaCO3、CaSO4、Ca(NO3)2、Sr(OH)2、Sr(OH)2・8H2O、Sr(OH)2・H2O、SrO、SrCO3、SrSO4、Sr(NO3)2等が挙げられるが、焼成時に発生する水分量を少なくするために、乾燥および脱水してから使用してもよい。また、これらの化合物は粉砕等をして粒子径を0.1~20μmにしてもよい。元素Bを含有する化合物についても同様に、元素Bの水酸化物、酸化物、炭酸塩、硫酸塩、硝酸塩等が挙げられるが、焼成時に発生する水分量を少なくするために、乾燥および脱水してから使用してもよい。また、これらの化合物は粉砕等をして粒子径を0.1~20μmにしてもよい。
複合酸化物と元素Aを含有する化合物は、例えば、複合酸化物中のLiを除く金属元素の総量と、元素Aとのモル比が、1:0.0005~1:0.03となる比率で混合されることが好ましい。元素Aを含有する化合物を複数種用いる場合、化合物中に含まれる元素Aの総量が当該比率を満たすように混合される。複合酸化物との好適な混合比率は、元素Bを含有する化合物についても同様である。また元素AとBは、AαBβOγの化学量論比に合わせて、混合することが好ましい。
第3工程における混合物の焼成工程は、例えば、酸素気流下、450℃~680℃で焼成する第1焼成工程と、第1焼成工程により得られた焼成物を、酸素気流下、680℃を超える温度で焼成する第2焼成工程とを少なくとも含む、多段階焼成工程である。第1焼成工程では、0.2℃/min~5.5℃/minの第1の昇温速度で、680℃以下の第1設定温度まで昇温する。第2焼成工程では、0.1℃/min~3.5℃/minで、且つ第1の昇温速度より遅い第2の昇温速度で、900℃以下の第2設定温度まで昇温する。なお、第1および第2の昇温速度は、上記範囲内において所定の温度領域毎に複数設定してもよい。
第1焼成工程における第1設定温度の保持時間は、5時間以下が好ましく、3時間以下がより好ましい。第1設定温度の保持時間とは、第1設定温度に達した後、第1設定温度を維持する時間であり、保持時間はゼロであってもよい。第2焼成工程における第2設定温度の保持時間は、1時間~10時間が好ましく、1時間~5時間がより好ましい。第2設定温度の保持時間とは、第2設定温度に達した後、第2設定温度を維持する時間である。混合物の焼成は、例えば、酸素濃度60%以上の酸素気流中で行い、酸素気流の流量を、焼成炉10cm3あたり、0.2mL/min~4mL/minおよび混合物1kgあたり0.3L/min以上とする。
第4工程では、第3工程で得られた焼成物を水洗して不純物を除去し、水洗した焼成物を加熱乾燥する。必要により、焼成物の粉砕、分級等を行い、正極活物質のD50を目的とする範囲に調整する。水洗後の焼成物の乾燥は、100℃未満の温度で行われてもよい。好適な乾燥温度の一例は、150℃~250℃である。乾燥処理は、真空下および大気下のいずれで行われてもよい。乾燥処理時間の一例は、1時間~5時間である。
なお、複合酸化物(Z)の二次粒子の内部において一次粒子同士の界面に化合物Xを固着させるためには、元素Aを含有する化合物と、元素Bの含有する化合物とをそれぞれ添加する必要がある。すなわち、元素A,Bの両方を含有する化合物を用いても、二次粒子の内部の一次粒子の表面に化合物Xを存在させることはできない。また、化合物Xは、元素Aが溶融して元素Bを取り込むことにより生成すると考えられ、元素Aを含有する化合物および元素Bの含有する化合物の存在下で、少なくとも600℃以上の温度で熱処理を行わなければ、二次粒子の内部の一次粒子の表面に化合物Xを存在させることはできない。
[負極]
負極12は、負極芯体40と、負極芯体40の表面に設けられた負極合剤層41とを備える。負極芯体40には、銅、銅合金などの負極12の電位範囲で安定な金属の箔、当該金属を表層に配置したフィルム等を用いることができる。負極合剤層41は、負極活物質および結着剤を含み、負極芯体40の両面に設けられることが好ましい。負極12は、例えば、負極芯体40の表面に負極活物質および結着剤等を含む負極合剤スラリーを塗布し、塗膜を乾燥させた後、圧縮して負極合剤層41を負極芯体40の両面に形成することにより作製できる。負極合剤層41には、正極11の場合と同様の導電剤が含まれていてもよい。
負極12は、負極芯体40と、負極芯体40の表面に設けられた負極合剤層41とを備える。負極芯体40には、銅、銅合金などの負極12の電位範囲で安定な金属の箔、当該金属を表層に配置したフィルム等を用いることができる。負極合剤層41は、負極活物質および結着剤を含み、負極芯体40の両面に設けられることが好ましい。負極12は、例えば、負極芯体40の表面に負極活物質および結着剤等を含む負極合剤スラリーを塗布し、塗膜を乾燥させた後、圧縮して負極合剤層41を負極芯体40の両面に形成することにより作製できる。負極合剤層41には、正極11の場合と同様の導電剤が含まれていてもよい。
負極合剤層41には、負極活物質として、例えばリチウムイオンを可逆的に吸蔵、放出する炭素材料が含まれる。炭素材料の好適な一例は、鱗片状黒鉛、塊状黒鉛、土状黒鉛等の天然黒鉛、塊状人造黒鉛(MAG)、黒鉛化メソフェーズカーボンマイクロビーズ(MCMB)等の人造黒鉛などの黒鉛である。また、負極活物質として、Si、Sn等のLiと合金化する元素、および当該元素を含有する化合物の少なくとも一方を含む活物質が用いられてもよい。当該活物質の好適な一例は、酸化ケイ素相又はリチウムシリケート等のシリケート相中にSi微粒子が分散したシリコン材料である。負極活物質には、例えば、黒鉛などの炭素材料とシリコン材料が併用される。
負極合剤層41に含まれる結着剤には、正極11の場合と同様に、フッ素樹脂、PAN、ポリイミド、アクリル樹脂、ポリオレフィン等を用いることもできるが、スチレン-ブタジエンゴム(SBR)を用いることが好ましい。また、負極合剤層41は、さらに、CMC又はその塩、ポリアクリル酸(PAA)又はその塩、ポリビニルアルコール(PVA)などを含むことが好ましい。中でも、SBRと、CMC又はその塩、PAA又はその塩を併用することが好適である。なお、負極合剤層41には、導電剤が含まれていてもよい。
[セパレータ]
セパレータ13には、イオン透過性および絶縁性を有する多孔性シートが用いられる。多孔性シートの具体例としては、微多孔薄膜、織布、不織布等が挙げられる。セパレータ13の材質としては、ポリエチレン、ポリプロピレン、エチレンとαオレフィンの共重合体等のポリオレフィン、セルロースなどが好適である。セパレータ13は、単層構造、積層構造のいずれであってもよい。セパレータ13の表面には、無機粒子を含む耐熱層、アラミド樹脂、ポリイミド、ポリアミドイミド等の耐熱性の高い樹脂で構成される耐熱層などが形成されていてもよい。
セパレータ13には、イオン透過性および絶縁性を有する多孔性シートが用いられる。多孔性シートの具体例としては、微多孔薄膜、織布、不織布等が挙げられる。セパレータ13の材質としては、ポリエチレン、ポリプロピレン、エチレンとαオレフィンの共重合体等のポリオレフィン、セルロースなどが好適である。セパレータ13は、単層構造、積層構造のいずれであってもよい。セパレータ13の表面には、無機粒子を含む耐熱層、アラミド樹脂、ポリイミド、ポリアミドイミド等の耐熱性の高い樹脂で構成される耐熱層などが形成されていてもよい。
以下、実施例により本開示をさらに説明するが、本開示はこれらの実施例に限定されるものではない。
<実施例1>
[正極活物質の合成]
共沈法により得られた[Ni91Mn7Al2(OH)2で表される複合水酸化物を、500℃で8時間焼成して複合酸化物を得た。当該複合酸化物と、水酸化リチウムと、水酸化カルシウムと、酸化ジルコニウムとを、Liと、Ni、Mn、およびAlの総量と、Caと、Zrとのモル比が1.05:1.00:0.005:0.005となるように混合した。当該混合物を、酸素濃度95%の酸素気流下(10cm3あたり2mL/minおよび混合物1kgあたり5L/minの流量)、昇温速度2.0℃/minで室温から650℃まで昇温して焼成した後、昇温速度0.5℃/minで750℃まで昇温して焼成した。この焼成物を水洗して不純物を除去し、180℃で2時間、真空乾燥することにより正極活物質を得た。
[正極活物質の合成]
共沈法により得られた[Ni91Mn7Al2(OH)2で表される複合水酸化物を、500℃で8時間焼成して複合酸化物を得た。当該複合酸化物と、水酸化リチウムと、水酸化カルシウムと、酸化ジルコニウムとを、Liと、Ni、Mn、およびAlの総量と、Caと、Zrとのモル比が1.05:1.00:0.005:0.005となるように混合した。当該混合物を、酸素濃度95%の酸素気流下(10cm3あたり2mL/minおよび混合物1kgあたり5L/minの流量)、昇温速度2.0℃/minで室温から650℃まで昇温して焼成した後、昇温速度0.5℃/minで750℃まで昇温して焼成した。この焼成物を水洗して不純物を除去し、180℃で2時間、真空乾燥することにより正極活物質を得た。
ICP発光分光分析装置(Thermo Fisher Scientific製、iCAP6300)により、得られた正極活物質を測定した結果、Li、O、および不純物元素を除く元素として、後述の表1に示す元素が確認された。また、放射光X線回折測定により、正極活物質中に存在する化合物を同定した結果、CaZrO3の存在が確認された。また、TEM―EDXにより、二次粒子の内部における一次粒子同士の界面に、CaとZrが存在することが確認できた。
[正極の作製]
上記正極活物質と、アセチレンブラックと、ポリフッ化ビニリデンとを、96:2.5:1.5の質量比で混合し、分散媒としてN-メチル-2-ピロリドン(NMP)を用いて、正極合剤スラリーを調製した。次に、アルミニウム箔からなる正極芯体上に正極合剤スラリーを塗布し、塗膜を乾燥、圧縮した後、正極芯体を所定の電極サイズに切断して、正極芯体の両面に正極合剤層が形成された正極を得た。なお、正極の一部に正極芯体の表面が露出した露出部を設けた。
上記正極活物質と、アセチレンブラックと、ポリフッ化ビニリデンとを、96:2.5:1.5の質量比で混合し、分散媒としてN-メチル-2-ピロリドン(NMP)を用いて、正極合剤スラリーを調製した。次に、アルミニウム箔からなる正極芯体上に正極合剤スラリーを塗布し、塗膜を乾燥、圧縮した後、正極芯体を所定の電極サイズに切断して、正極芯体の両面に正極合剤層が形成された正極を得た。なお、正極の一部に正極芯体の表面が露出した露出部を設けた。
[非水電解液の調製]
エチレンカーボネート(EC)と、メチルエチルカーボネート(MEC)と、ジメチルカーボネート(DMC)とを、3:3:4の体積比(25℃)で混合した。当該混合溶媒にLiPF6を1.2モル/リットルの濃度となるように溶解させて、非水電解液を調製した。
エチレンカーボネート(EC)と、メチルエチルカーボネート(MEC)と、ジメチルカーボネート(DMC)とを、3:3:4の体積比(25℃)で混合した。当該混合溶媒にLiPF6を1.2モル/リットルの濃度となるように溶解させて、非水電解液を調製した。
[試験セルの作製]
上記正極の露出部にアルミニウムリードを、負極としてリチウム金属箔にニッケルリードをそれぞれ取り付け、ポリオレフィン製のセパレータを介して正極と負極を渦巻き状に巻回した後、径方向にプレス成形して扁平状の巻回型電極体を作製した。この電極体をアルミラミネートシートで構成される外装体内に収容し、上記非水電解液を注入した後、外装体の開口部を封止して試験セルAlを得た。
上記正極の露出部にアルミニウムリードを、負極としてリチウム金属箔にニッケルリードをそれぞれ取り付け、ポリオレフィン製のセパレータを介して正極と負極を渦巻き状に巻回した後、径方向にプレス成形して扁平状の巻回型電極体を作製した。この電極体をアルミラミネートシートで構成される外装体内に収容し、上記非水電解液を注入した後、外装体の開口部を封止して試験セルAlを得た。
<実施例2>
正極活物質の合成において、水酸化カルシウムの代わりに、所定量の水酸化ストロンチウムを添加したこと以外は、実施例1と同様にして試験セルA2を作製した。合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはSrZrO3が存在し、二次粒子の内部における一次粒子同士の界面においてSrとZrの存在が確認された。
正極活物質の合成において、水酸化カルシウムの代わりに、所定量の水酸化ストロンチウムを添加したこと以外は、実施例1と同様にして試験セルA2を作製した。合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはSrZrO3が存在し、二次粒子の内部における一次粒子同士の界面においてSrとZrの存在が確認された。
<実施例3>
正極活物質の合成において、水酸化カルシウムおよび酸化ジルコニウムの代わりに、所定量の水酸化ストロンチウムおよび水酸化チタンを添加したこと以外は、実施例1と同様にして試験セルA3を作製した。合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはSr2TiO4とSrTiO3が存在し、二次粒子の内部における一次粒子同士の界面においてSrとTiの存在が確認された。
正極活物質の合成において、水酸化カルシウムおよび酸化ジルコニウムの代わりに、所定量の水酸化ストロンチウムおよび水酸化チタンを添加したこと以外は、実施例1と同様にして試験セルA3を作製した。合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはSr2TiO4とSrTiO3が存在し、二次粒子の内部における一次粒子同士の界面においてSrとTiの存在が確認された。
<比較例1>
正極活物質の合成において、水酸化カルシウムおよび酸化ジルコニウムを添加しなかったこと以外は、実施例1と同様にして試験セルB1を作製した。
正極活物質の合成において、水酸化カルシウムおよび酸化ジルコニウムを添加しなかったこと以外は、実施例1と同様にして試験セルB1を作製した。
<実施例4>
正極活物質の合成において、[Ni86Mn14](OH)2で表される複合水酸化物を用い、当該複合水酸化物を焼成して得られる複合酸化物と、水酸化リチウムと、水酸化カルシウムと、酸化タングステンとを、Liと、NiおよびMnの総量と、Caと、Wとのモル比が1.03:1.00:0.005:0.005となるように混合した。当該混合物を、酸素濃度95%の酸素気流下(10cm3あたり2mL/minおよび混合物1kgあたり5L/minの流量)、昇温速度2.0℃/minで室温から670℃まで昇温して焼成した後、昇温速度0.5℃/minで780℃まで昇温して焼成した。この焼成物を水洗して不純物を除去し、180℃で2時間、真空乾燥することにより正極活物質を得たこと以外は、実施例1と同様にして試験セルA4を作製した。
正極活物質の合成において、[Ni86Mn14](OH)2で表される複合水酸化物を用い、当該複合水酸化物を焼成して得られる複合酸化物と、水酸化リチウムと、水酸化カルシウムと、酸化タングステンとを、Liと、NiおよびMnの総量と、Caと、Wとのモル比が1.03:1.00:0.005:0.005となるように混合した。当該混合物を、酸素濃度95%の酸素気流下(10cm3あたり2mL/minおよび混合物1kgあたり5L/minの流量)、昇温速度2.0℃/minで室温から670℃まで昇温して焼成した後、昇温速度0.5℃/minで780℃まで昇温して焼成した。この焼成物を水洗して不純物を除去し、180℃で2時間、真空乾燥することにより正極活物質を得たこと以外は、実施例1と同様にして試験セルA4を作製した。
実施例4で合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはCaWO4が存在し、二次粒子の内部における一次粒子同士の界面においてCaとWの存在が確認された。
<実施例5>
正極活物質の合成において、水酸化カルシウムおよび酸化タングステンの代わりに、所定量の水酸化ストロンチウムおよび酸化モリブデンを添加したこと以外は、実施例4と同様にして試験セルA5を作製した。合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはSrMoO4が存在し、二次粒子の内部における一次粒子同士の界面においてSrとMoの存在が確認された。
正極活物質の合成において、水酸化カルシウムおよび酸化タングステンの代わりに、所定量の水酸化ストロンチウムおよび酸化モリブデンを添加したこと以外は、実施例4と同様にして試験セルA5を作製した。合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはSrMoO4が存在し、二次粒子の内部における一次粒子同士の界面においてSrとMoの存在が確認された。
<実施例6>
正極活物質の合成において、水酸化ストロンチウムおよび酸化モリブデンの代わりに、所定量の水酸化カルシウムおよび水酸化チタンを添加したこと以外は、実施例5と同様にして試験セルA6を作製した。合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはCaTiO3が存在し、二次粒子の内部における一次粒子同士の界面においてCaとTiの存在が確認された。
正極活物質の合成において、水酸化ストロンチウムおよび酸化モリブデンの代わりに、所定量の水酸化カルシウムおよび水酸化チタンを添加したこと以外は、実施例5と同様にして試験セルA6を作製した。合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはCaTiO3が存在し、二次粒子の内部における一次粒子同士の界面においてCaとTiの存在が確認された。
<比較例2>
正極活物質の合成において、水酸化カルシウムおよび酸化タングステンを添加しなかったこと以外は、実施例4と同様にして試験セルB2を作製した。
正極活物質の合成において、水酸化カルシウムおよび酸化タングステンを添加しなかったこと以外は、実施例4と同様にして試験セルB2を作製した。
<比較例3>
正極活物質の合成において、酸化タングステンを添加しなかったこと以外は、実施例4と同様にして試験セルB3を作製した。
正極活物質の合成において、酸化タングステンを添加しなかったこと以外は、実施例4と同様にして試験セルB3を作製した。
<比較例4>
正極活物質の合成において、酸化タングステンの添加のタイミングを焼成物の水洗後に変更し、水洗後の乾燥を大気圧下、80℃の条件で行ったこと以外は、実施例4と同様にして試験セルB4を作製した。合成された正極活物質は、CaWO4が二次粒子の表面のみに存在し、二次粒子の内部における一次粒子同士の界面には確認されなかった。
正極活物質の合成において、酸化タングステンの添加のタイミングを焼成物の水洗後に変更し、水洗後の乾燥を大気圧下、80℃の条件で行ったこと以外は、実施例4と同様にして試験セルB4を作製した。合成された正極活物質は、CaWO4が二次粒子の表面のみに存在し、二次粒子の内部における一次粒子同士の界面には確認されなかった。
<比較例5>
正極活物質の合成において、水酸化ストロンチウムおよび酸化モリブデンの代わりに、所定量の水酸化マグネシウムを添加したこと以外は、実施例4と同様にして試験セルB5を作製した。
正極活物質の合成において、水酸化ストロンチウムおよび酸化モリブデンの代わりに、所定量の水酸化マグネシウムを添加したこと以外は、実施例4と同様にして試験セルB5を作製した。
<実施例7>
共沈法により得られた[Ni79Mn19Al1Co1](OH)2で表される複合水酸化物を、500℃で8時間焼成して複合酸化物を得た。当該複合酸化物と、水酸化リチウムと、水酸化カルシウムと、酸化タングステンとを、Liと、Ni、Mn、Al、およびCoの総量と、Caと、Wとのモル比が1.03:1.00:0.01:0.01となるように混合した。当該混合物を、酸素濃度95%の酸素気流下(10cm3あたり2mL/minおよび混合物1kgあたり5L/minの流量)、昇温速度2.0℃/minで室温から650℃まで昇温して焼成した後、昇温速度0.5℃/minで800℃まで昇温して焼成した。この焼成物を水洗して不純物を除去し、180℃で2時間、真空乾燥することにより正極活物質を得たこと以外は、実施例1と同様にして試験セルA7を作製した。
共沈法により得られた[Ni79Mn19Al1Co1](OH)2で表される複合水酸化物を、500℃で8時間焼成して複合酸化物を得た。当該複合酸化物と、水酸化リチウムと、水酸化カルシウムと、酸化タングステンとを、Liと、Ni、Mn、Al、およびCoの総量と、Caと、Wとのモル比が1.03:1.00:0.01:0.01となるように混合した。当該混合物を、酸素濃度95%の酸素気流下(10cm3あたり2mL/minおよび混合物1kgあたり5L/minの流量)、昇温速度2.0℃/minで室温から650℃まで昇温して焼成した後、昇温速度0.5℃/minで800℃まで昇温して焼成した。この焼成物を水洗して不純物を除去し、180℃で2時間、真空乾燥することにより正極活物質を得たこと以外は、実施例1と同様にして試験セルA7を作製した。
実施例7で合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはCaWO4が存在し、二次粒子の内部における一次粒子同士の界面においてCaとWの存在が確認された。
<実施例8>
正極活物質の合成において、酸化タングステンの代わりに、所定量の酸化モリブデンを添加したこと以外は、実施例7と同様にして試験セルA8を作製した。合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはCaMoO4が存在し、二次粒子の内部における一次粒子同士の界面においてCaとMoの存在が確認された。
正極活物質の合成において、酸化タングステンの代わりに、所定量の酸化モリブデンを添加したこと以外は、実施例7と同様にして試験セルA8を作製した。合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはCaMoO4が存在し、二次粒子の内部における一次粒子同士の界面においてCaとMoの存在が確認された。
<比較例6>
正極活物質の合成において、水酸化カルシウムおよび酸化タングステンを添加しなかったこと以外は、実施例7と同様にして試験セルB6を作製した。
正極活物質の合成において、水酸化カルシウムおよび酸化タングステンを添加しなかったこと以外は、実施例7と同様にして試験セルB6を作製した。
<参考例1>
共沈法により得られた[Ni83Mn9Al3Co5](OH)2で表される複合水酸化物を、500℃で8時間焼成して複合酸化物を得た。当該複合酸化物と、水酸化リチウムと、水酸化カルシウムと、酸化モリブデンとを、Liと、Ni、Mn、Al、およびCoの総量と、Caと、Moとのモル比が1.03:1.00:0.01:0.01となるように混合した。当該混合物を、酸素濃度95%の酸素気流下(10cm3あたり2mL/minおよび混合物1kgあたり5L/minの流量)、昇温速度2.0℃/minで室温から650℃まで昇温して焼成した後、昇温速度0.5℃/minで750℃まで昇温して焼成した。この焼成物を水洗して不純物を除去し、180℃で2時間、真空乾燥することにより正極活物質を得たこと以外は、実施例1と同様にして試験セルR1を作製した。
共沈法により得られた[Ni83Mn9Al3Co5](OH)2で表される複合水酸化物を、500℃で8時間焼成して複合酸化物を得た。当該複合酸化物と、水酸化リチウムと、水酸化カルシウムと、酸化モリブデンとを、Liと、Ni、Mn、Al、およびCoの総量と、Caと、Moとのモル比が1.03:1.00:0.01:0.01となるように混合した。当該混合物を、酸素濃度95%の酸素気流下(10cm3あたり2mL/minおよび混合物1kgあたり5L/minの流量)、昇温速度2.0℃/minで室温から650℃まで昇温して焼成した後、昇温速度0.5℃/minで750℃まで昇温して焼成した。この焼成物を水洗して不純物を除去し、180℃で2時間、真空乾燥することにより正極活物質を得たこと以外は、実施例1と同様にして試験セルR1を作製した。
参考例1で合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはCaMoO4が存在し、二次粒子の内部における一次粒子同士の界面においてCaとMoの存在が確認された。
<参考例2>
正極活物質の合成において、水酸化カルシウムおよび酸化モリブデンを添加しなかったこと以外は、参考例1と同様にして試験セルR2を作製した。
正極活物質の合成において、水酸化カルシウムおよび酸化モリブデンを添加しなかったこと以外は、参考例1と同様にして試験セルR2を作製した。
<参考例3>
共沈法により得られた[Ni80Mn10Co10](OH)2で表される複合水酸化物を、500℃で8時間焼成して複合酸化物を得た。当該複合酸化物と、水酸化リチウムと、水酸化ストロンチウムと、水酸化チタンとを、Liと、Ni、Mn、およびCoの総量と、Srと、Tiとのモル比が1.03:1.00:0.01:0.01となるように混合した。当該混合物を、酸素濃度95%の酸素気流下(10cm3あたり2mL/minおよび混合物1kgあたり5L/minの流量)、昇温速度2.0℃/minで室温から650℃まで昇温して焼成した後、昇温速度0.5℃/minで780℃まで昇温して焼成した。この焼成物を水洗して不純物を除去し、180℃で2時間、真空乾燥することにより正極活物質を得たこと以外は、実施例1と同様にして試験セルR3を作製した。
共沈法により得られた[Ni80Mn10Co10](OH)2で表される複合水酸化物を、500℃で8時間焼成して複合酸化物を得た。当該複合酸化物と、水酸化リチウムと、水酸化ストロンチウムと、水酸化チタンとを、Liと、Ni、Mn、およびCoの総量と、Srと、Tiとのモル比が1.03:1.00:0.01:0.01となるように混合した。当該混合物を、酸素濃度95%の酸素気流下(10cm3あたり2mL/minおよび混合物1kgあたり5L/minの流量)、昇温速度2.0℃/minで室温から650℃まで昇温して焼成した後、昇温速度0.5℃/minで780℃まで昇温して焼成した。この焼成物を水洗して不純物を除去し、180℃で2時間、真空乾燥することにより正極活物質を得たこと以外は、実施例1と同様にして試験セルR3を作製した。
参考例3で合成された正極活物質には、表1に示す元素が含有されることが確認された。また、正極活物質中にはSr2TiO4、SrTiO3が存在し、二次粒子の内部における一次粒子同士の界面においてSrとTiの存在が確認された。
<参考例4>
正極活物質の合成において、水酸化ストロンチウムおよび水酸化チタンを添加しなかったこと以外は、参考例3と同様にして試験セルR4を作製した。
正極活物質の合成において、水酸化ストロンチウムおよび水酸化チタンを添加しなかったこと以外は、参考例3と同様にして試験セルR4を作製した。
[反応抵抗(25℃)の測定]
試験セルを25℃の温度環境下、0.2Itの定電流で電池電圧が4.3V、4.4V、4.5Vになるまで定電流充電を行い、4.3V、4.4V、4.5Vで電流値が1/100Itになるまで定電圧充電させる操作Aと、0.2Itの定電流で電池電圧が2.5Vになるまで定電流放電させる操作Bとを2回繰り返した。その後、再度操作Aを行った各電池に対し、温度25℃、周波数0.01Hz~100000Hz、印加電圧10mVの条件にて、交流インピーダンス測定を行い、Nyquistプロットの等価回路フィッティングにより25℃における反応抵抗Rct(Ω)を求めた。
試験セルを25℃の温度環境下、0.2Itの定電流で電池電圧が4.3V、4.4V、4.5Vになるまで定電流充電を行い、4.3V、4.4V、4.5Vで電流値が1/100Itになるまで定電圧充電させる操作Aと、0.2Itの定電流で電池電圧が2.5Vになるまで定電流放電させる操作Bとを2回繰り返した。その後、再度操作Aを行った各電池に対し、温度25℃、周波数0.01Hz~100000Hz、印加電圧10mVの条件にて、交流インピーダンス測定を行い、Nyquistプロットの等価回路フィッティングにより25℃における反応抵抗Rct(Ω)を求めた。
算出した反応抵抗Rctを表1~4に示す。表1に示すRctは、比較例1の試験セルB1のRctを100としたときの相対値である。表2に示すRctは、比較例2の試験セルB2のRctを100としたときの相対値である。表3に示すRctは、比較例6の試験セルB6のRctを100としたときの相対値である。表4の参考例2の試験セルR2のRctは、参考例1の試験セルR1のRctを100としたときの相対値であり、表4の参考例4の試験セルR4のRctは、参考例3の試験セルR3のRctを100としたときの相対値である。




表1~表3に示すように、実施例の試験セルはいずれも、対応する比較例の試験セルと比べて、反応抵抗Rctが低く抑えられている。正極活物質の合成において、元素A(Ca、Sr)および元素B(W、Mo、Ti、Zr)を添加しない場合(比較例1,2,6)、元素A,Bの一方のみを添加する場合(比較例3,5)、およびAαBβOγが二次粒子の表面のみに存在する場合(比較例4)は、反応抵抗の抑制効果は得られなかった。
表4に示すように、Coの含有量が多くなると、AαBβOγによる反応抵抗の抑制効果が小さくなる。すなわち、Coの含有量が少ない場合、特に複合酸化物中のLiを除く金属元素に対して、Coの含有量が5モル%未満である場合に、AαBβOγによる反応抵抗の抑制効果が顕著になる。
10 非水電解質二次電池、11 正極、12 負極、13 セパレータ、14 電極体、16 外装缶、17 封口体、18,19 絶縁板、20 正極リード、21 負極リード、22 溝入部、23 内部端子板、24 下弁体、25 絶縁部材、26 上弁体、27 キャップ、28 ガスケット、30 正極芯体、31 正極合剤層、40 負極芯体、41 負極合剤層
Claims (5)
- 層状構造を有し、一般式LixNiaMnbAlcCodMeO2-y(式中、0.95<x<1.05、0.75≦a≦0.95、0.05≦b≦0.25、0≦c≦0.02、0≦d<0.05、0≦e≦0.03、0≦y≦0.05、a+b+c=1、Mは、Fe、Ti、Si、Nb、Mo、W、およびZrから選択される少なくとも1種の元素)で表されるリチウム遷移金属複合酸化物を含み、
前記リチウム遷移金属複合酸化物は、一次粒子が凝集して形成された二次粒子であり、
少なくとも前記二次粒子の内部における前記一次粒子同士の界面には、CaおよびSrから選択される少なくとも1種と、W、Mo、Ti、およびZrから選択される少なくとも1種とを含有する化合物が固着している、非水電解質二次電池用正極活物質。 - 前記化合物中のCaおよびSrの総量は、前記リチウム遷移金属複合酸化物中のLiを除く金属元素の総モル量に対して2モル%以下である、請求項1に記載の非水電解質二次電池用正極活物質。
- 前記化合物は、酸化物である、請求項1又は2に記載の非水電解質二次電池用正極活物質。
- 前記酸化物の組成は、一般式AαBβOγ(式中、1≦α≦2、1≦β≦2、3≦γ≦6、AはCaおよびSrから選択される少なくとも1種、BはW、Mo、Ti、およびZrから選択される少なくとも1種)で表される、請求項3に記載の非水電解質二次電池用正極活物質。
- 請求項1~4のいずれか一項に記載の正極活物質を含む正極と、負極と、非水電解質とを備える、非水電解質二次電池。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202280064338.6A CN117999673A (zh) | 2021-09-30 | 2022-09-20 | 非水电解质二次电池用正极活性物质和非水电解质二次电池 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021-160427 | 2021-09-30 | ||
| JP2021160427 | 2021-09-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023054041A1 true WO2023054041A1 (ja) | 2023-04-06 |
Family
ID=85782501
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/034891 WO2023054041A1 (ja) | 2021-09-30 | 2022-09-20 | 非水電解質二次電池用正極活物質および非水電解質二次電池 |
Country Status (2)
| Country | Link |
|---|---|
| CN (1) | CN117999673A (ja) |
| WO (1) | WO2023054041A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024070385A1 (ja) * | 2022-09-30 | 2024-04-04 | パナソニックIpマネジメント株式会社 | 非水電解質二次電池 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012035664A1 (ja) | 2010-09-17 | 2012-03-22 | トヨタ自動車株式会社 | リチウムイオン二次電池 |
| JP2012252807A (ja) * | 2011-05-31 | 2012-12-20 | Toyota Motor Corp | リチウム二次電池 |
| JP2018129221A (ja) | 2017-02-09 | 2018-08-16 | 株式会社Gsユアサ | 非水電解質二次電池用正極活物質、その製造方法、非水電解質二次電池用正極、及び非水電解質二次電池 |
| WO2021106324A1 (ja) * | 2019-11-29 | 2021-06-03 | パナソニックIpマネジメント株式会社 | 非水電解質二次電池用正極活物質、非水電解質二次電池用正極活物質の製造方法、及び非水電解質二次電池 |
| WO2021241075A1 (ja) * | 2020-05-29 | 2021-12-02 | パナソニックIpマネジメント株式会社 | 非水電解質二次電池用正極活物質、及び非水電解質二次電池 |
| WO2022130982A1 (ja) * | 2020-12-18 | 2022-06-23 | パナソニックIpマネジメント株式会社 | 非水電解質二次電池用正極、及び非水電解質二次電池 |
-
2022
- 2022-09-20 WO PCT/JP2022/034891 patent/WO2023054041A1/ja active Application Filing
- 2022-09-20 CN CN202280064338.6A patent/CN117999673A/zh active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012035664A1 (ja) | 2010-09-17 | 2012-03-22 | トヨタ自動車株式会社 | リチウムイオン二次電池 |
| JP2012252807A (ja) * | 2011-05-31 | 2012-12-20 | Toyota Motor Corp | リチウム二次電池 |
| JP2018129221A (ja) | 2017-02-09 | 2018-08-16 | 株式会社Gsユアサ | 非水電解質二次電池用正極活物質、その製造方法、非水電解質二次電池用正極、及び非水電解質二次電池 |
| WO2021106324A1 (ja) * | 2019-11-29 | 2021-06-03 | パナソニックIpマネジメント株式会社 | 非水電解質二次電池用正極活物質、非水電解質二次電池用正極活物質の製造方法、及び非水電解質二次電池 |
| WO2021241075A1 (ja) * | 2020-05-29 | 2021-12-02 | パナソニックIpマネジメント株式会社 | 非水電解質二次電池用正極活物質、及び非水電解質二次電池 |
| WO2022130982A1 (ja) * | 2020-12-18 | 2022-06-23 | パナソニックIpマネジメント株式会社 | 非水電解質二次電池用正極、及び非水電解質二次電池 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024070385A1 (ja) * | 2022-09-30 | 2024-04-04 | パナソニックIpマネジメント株式会社 | 非水電解質二次電池 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN117999673A (zh) | 2024-05-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2022092182A1 (ja) | 非水電解質二次電池 | |
| WO2023054041A1 (ja) | 非水電解質二次電池用正極活物質および非水電解質二次電池 | |
| WO2021039751A1 (ja) | 非水電解質二次電池 | |
| WO2021059728A1 (ja) | 非水電解質二次電池用正極活物質、及び非水電解質二次電池 | |
| JP7324120B2 (ja) | 非水電解質二次電池用正極活物質、及び非水電解質二次電池 | |
| JP7324119B2 (ja) | 非水電解質二次電池用正極活物質、及び非水電解質二次電池 | |
| WO2022209894A1 (ja) | 非水電解質二次電池用正極活物質および非水電解質二次電池 | |
| WO2021241027A1 (ja) | 非水電解質二次電池用正極活物質および非水電解質二次電池 | |
| WO2021024789A1 (ja) | 非水電解質二次電池用正極活物質、及び非水電解質二次電池 | |
| WO2021049198A1 (ja) | 非水電解質二次電池 | |
| WO2021019943A1 (ja) | 非水電解質二次電池 | |
| WO2021153527A1 (ja) | 非水電解質二次電池用正極活物質および非水電解質二次電池 | |
| WO2024062866A1 (ja) | 二次電池用正極活物質および二次電池 | |
| WO2024062848A1 (ja) | 二次電池用正極活物質および二次電池 | |
| WO2023189507A1 (ja) | 非水電解質二次電池用正極活物質および非水電解質二次電池 | |
| WO2023204077A1 (ja) | 非水電解質二次電池用正極活物質および非水電解質二次電池 | |
| WO2024004578A1 (ja) | 非水電解質二次電池 | |
| WO2022070648A1 (ja) | 非水電解質二次電池 | |
| WO2024024364A1 (ja) | 非水電解質二次電池用正極活物質および非水電解質二次電池 | |
| WO2022181264A1 (ja) | 非水電解質二次電池用正極活物質および非水電解質二次電池 | |
| WO2024070385A1 (ja) | 非水電解質二次電池 | |
| WO2024004577A1 (ja) | 非水電解質二次電池用正極活物質および非水電解質二次電池 | |
| WO2023127290A1 (ja) | 非水電解質二次電池用正極活物質 | |
| WO2021039750A1 (ja) | 非水電解質二次電池用正極活物質、及び非水電解質二次電池 | |
| WO2022070898A1 (ja) | 非水電解質二次電池用正極活物質および非水電解質二次電池 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22875912 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023551332 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 2022875912 Country of ref document: EP Effective date: 20240430 |