WO2022009572A1 - 電池用活物質、電池用複合活物質、及び二次電池 - Google Patents
電池用活物質、電池用複合活物質、及び二次電池 Download PDFInfo
- Publication number
- WO2022009572A1 WO2022009572A1 PCT/JP2021/021116 JP2021021116W WO2022009572A1 WO 2022009572 A1 WO2022009572 A1 WO 2022009572A1 JP 2021021116 W JP2021021116 W JP 2021021116W WO 2022009572 A1 WO2022009572 A1 WO 2022009572A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- active material
- battery
- group
- sio
- carbon
- Prior art date
Links
Images
Classifications
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/38—Selection of substances as active materials, active masses, active liquids of elements or alloys
- H01M4/386—Silicon or alloys based on silicon
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/133—Electrodes based on carbonaceous material, e.g. graphite-intercalation compounds or CFx
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/134—Electrodes based on metals, Si or alloys
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/13—Electrodes for accumulators with non-aqueous electrolyte, e.g. for lithium-accumulators; Processes of manufacture thereof
- H01M4/139—Processes of manufacture
- H01M4/1395—Processes of manufacture of electrodes based on metals, Si or alloys
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/362—Composites
- H01M4/364—Composites as mixtures
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/362—Composites
- H01M4/366—Composites as layered products
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/38—Selection of substances as active materials, active masses, active liquids of elements or alloys
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/48—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides
- H01M4/483—Selection of substances as active materials, active masses, active liquids of inorganic oxides or hydroxides for non-aqueous cells
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/36—Selection of substances as active materials, active masses, active liquids
- H01M4/58—Selection of substances as active materials, active masses, active liquids of inorganic compounds other than oxides or hydroxides, e.g. sulfides, selenides, tellurides, halogenides or LiCoFy; of polyanionic structures, e.g. phosphates, silicates or borates
- H01M4/583—Carbonaceous material, e.g. graphite-intercalation compounds or CFx
- H01M4/587—Carbonaceous material, e.g. graphite-intercalation compounds or CFx for inserting or intercalating light metals
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M4/62—Selection of inactive substances as ingredients for active masses, e.g. binders, fillers
- H01M4/624—Electric conductive fillers
- H01M4/625—Carbon or graphite
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M4/00—Electrodes
- H01M4/02—Electrodes composed of, or comprising, active material
- H01M2004/026—Electrodes composed of, or comprising, active material characterised by the polarity
- H01M2004/027—Negative electrodes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Definitions
- the present invention relates to a battery active material which is an amorphous silicon-based material composed of elements containing Si, O, and C, a battery composite active material containing the same, and a secondary battery containing these in a negative electrode.
- secondary batteries such as lithium-ion batteries (sometimes referred to as LIB) are rapidly being developed for electric vehicle (EV) applications and the like.
- LIB lithium-ion batteries
- EV electric vehicle
- the distance that can be traveled on a single charge (cruising distance) is an important item for measuring the performance of an electric vehicle.
- What supports the performance of electric vehicles is the battery performance installed, which is high capacity, high efficiency, and high capacity maintenance rate.
- tin theoretical capacity: 994 mAh / g
- silicon theoretical capacity: 4199 mAh / g
- tin-based materials and silicon materials have a problem that a decrease in charge / discharge capacity cannot be avoided because the active material is pulverized during charging / discharging.
- an active material using particles in which the composition phase represented by the composition formula SiOxCy (0.2 ⁇ x ⁇ 3, 1 ⁇ y ⁇ 5) is dispersed in the carbonaceous material phase (Patent Document 2 below) has a bridging group. It is composed of an active material (Patent Document 3 below) obtained by sintering a crosslinked product obtained by curing reactive silane and siloxane at 700 to 1400 ° C. in an inert atmosphere, silicon, carbon, and oxygen, and has a carbon content of 0.2 to.
- An active substance having an oxygen content of 10 mol%, an oxygen content of 0.5 to 40 mol%, and a Si—C bond of 0.1 to 17.29 mol% or less with respect to a silicon atom (Patent Document 4 below).
- Japanese Unexamined Patent Publication No. 5-144474 Japanese Unexamined Patent Publication No. 2003-197193 Japanese Unexamined Patent Publication No. 2006-062949 Japanese Unexamined Patent Publication No. 2009-283366
- the problem to be solved by the present invention includes an active material for a battery capable of improving the discharge capacity to, for example, 800 mAh / g or more while maintaining good initial Coulomb efficiency and cycle characteristics. It is an object of the present invention to provide a composite active material for a battery and a secondary battery containing these in a negative electrode.
- an amorphous silicon-based material composed of elements containing Si, O, and C includes an active material for a battery having a specific fine structure. We have found that this can solve the above-mentioned problems, and have reached the present invention.
- Item 1 It is an amorphous silicon-based material composed of elements containing Si, O, and C, and is attributed to SiO 2 C 2 and SiO 3 C in the range of chemical shift of 20 ppm to -150 ppm in the solid 29 Si-NMR spectrum.
- Item 2. Item 2. The active material for a battery according to Item 1, wherein the weight reduction start temperature when the active material is thermally decomposed in air is 550 ° C. or higher.
- Item 3. Item 3.
- Item 5. A secondary battery containing the active material according to any one of Items 1 to 4 in the negative electrode.
- Item 6 A composite active material for a battery containing at least one carbon material selected from graphite, low crystalline carbon, and amorphous carbon as a component of the composite active material according to Item 6.
- Item 8. Item 6.
- Item 9. A secondary battery containing the composite active material according to any one of Items 6 to 8 in the negative electrode.
- the secondary battery including the negative electrode containing the active material for a battery of the present invention can maintain good initial Coulomb efficiency and cycle characteristics, and can further improve the discharge capacity to, for example, 800 mAh / g or more.
- the active material for a battery of the present invention is an amorphous silicon-based material composed of elements containing Si, O, and C, and has silicon oxygen carbide (SiOC) as a main component, and each of Si, O, and C. It consists of a three-dimensional network structure consisting of elements. Depending on the type of atom bonded to Si (O or C) and the number of bonds with each atom, it can be mainly divided into three types, which are referred to as SiO 2 C 2 , SiO 3 C, and SiO 4, respectively. I will do it. Si which is bonded to these O and C at various ratios is further randomly bonded to SiOC.
- the active material for a battery of the present invention has a total peak integral value A and SiO 4 attributable to SiO 2 C 2 and SiO 3 C in the range of chemical shift 20 ppm to ⁇ 150 ppm of the solid 29 Si-NMR spectrum, respectively.
- the ratio (A / B) with the attributed peak integral value B is 0.5 or more and 5.0 or less.
- the insertion / desorption reaction with lithium ions can proceed reversibly with SiO 2 C 2 and SiO 3 C, but since SiO 4 produces a silicate compound with a part of lithium ions, insertion of lithium ions. , The elimination reaction may be partially irreversible.
- the electron distribution inside the SiOC fluctuates due to the approach of lithium ions, and electrostatic bonds and coordination bonds are formed between the SiOC and the lithium ions.
- Lithium ions are stored in the skeleton of SiOC. Since these coordination bond energies are relatively low, the desorption reaction of lithium ions is easily carried out. That is, when SiOC is charged and discharged, the lithium ion insertion / desorption reaction can be reversibly caused. Therefore, by grasping this mechanism, the total value of the abundance of SiO 2 C 2 and SiO 3 C with respect to the abundance of SiO 4 strongly contributes to the improvement of the reversible capacity, and the larger the total value, the more. We have found that the reversible capacity is improved and the initial Coulomb efficiency can be improved.
- SiO 4 is an indispensable structure.
- the reversible charge / discharge capacity will be improved, but if it is too large, the improvement of cycle characteristics will be hindered. Will be done.
- the smaller the value the better the cycle characteristics, but if the size is too small, the improvement of the reversible charge / discharge capacity is hindered.
- the ratio of the total value of the abundance of each of specific SiO 2 C 2 and SiO 3 C, which will be described later, and the abundance of SiO 4 is an extremely important factor. Will be.
- the abundance of each of SiO 2 C 2 , SiO 3 C, and SiO 4 can be quantified by solid 29 Si-NMR measurement.
- the structure of the amorphous silicon-based material of the present invention can be identified by measurement of 29 Si-solid-state NMR.
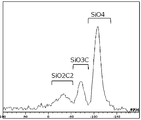
- FIG. 1 shows an example of a solid 29 Si-NMR spectrum in the active material for a battery of the present invention.
- the ratio (A / B) can be obtained by calculating the integrated value of each of the peaks.
- Each integral value in the solid 29 Si-NMR spectrum represents the respective molar ratio in the amorphous silicon-based material. Therefore, the ratio (A / B) means the total molar ratio of SiO 4 , 1 molar ratio of SiO 2 C 2 and SiO 3 C.
- the ratio (A / B), that is, the molar ratio is 0.5 or more and 5.0 or less, good initial Coulomb efficiency and cycle characteristics can be maintained, and the discharge capacity can be improved to, for example, 800 mAh / g or more. Can be done.
- peaks of SiC 4 and SiOC 3 may appear at 40 ppm to -20 ppm. Each peak appears in the vicinity of 10 ppm to -20 for SiC 4 and in the vicinity of 40 ppm to -20 ppm for SiOC 3. There are regions where peaks overlap in this way, and it is difficult to accurately specify quantitative values from the 29 Si-solid-state NMR spectrum. Further, it is known that SiC 4 does not undergo an insertion / desorption reaction with lithium ions. SiOC 3 undergoes an insertion / desorption reaction with lithium ions, but the abundance is generally small.
- the range can be specified by adding the peak integral value of SiOC 3 to the total value A and the peak integral value of SiC 4 to the total value B, which can be used as a condition for improving the charge / discharge characteristics. Can be easily considered.
- the reason than SiC 4 and since it is not realistic to consider the peak integral value of SiOC 3, the sum A of each peak integration value attributed to SiO 2 C 2, SiO 3 C as described above
- the ratio (A / B) of the above to the peak integral value B attributable to SiO 4 is determined to be in the range of 0.5 or more and 5.0 or less.
- the above-mentioned SiOC can be formed by calcining the silane compound which is the above-mentioned SiOC precursor in an atmosphere of an inert gas.
- the silane compound is composed of a Si—O bond and a molecular structure having a Si—C bond as a basic structure.
- the precursor preferably has a molecular structure containing a Si—O bond and a Si—C bond, and more preferably the Si—O bond and the Si—C bond are within the same molecule.
- each independently has a hydrogen atom, a hydroxyl group, a hydrolyzable group, an aliphatic hydrocarbon group, an unsaturated hydrocarbon group, an aromatic hydrocarbon, an aralkyl group, an acyl group, or the following functional group.
- X1, X2, X3, X4, X5 and X6 are all divalent substituents.
- oxygen atoms independently, oxygen atoms, alkylenes, alkens, alkins, divalent aromatics, ethers, ketones, esters, secondary amines, amides, or a combination of these bivalent substitutions.
- L, m, x, y indicate an integer of 0 or more.
- R1 and X1 in the general formulas (S-1), (S-2), (S-3), and (S-4), they are independent and the same. It may be different.
- hydrolyzable group examples include halogen atoms such as chlorine, bromine and iodine, alkoxy groups such as methoxy group, ethoxy group, propoxyl group, butoxy group, pentoxy group and hexylloyl group, asyloxy group, phenoxy group and naphthoxy.
- alkoxy groups such as methoxy group, ethoxy group, propoxyl group, butoxy group, pentoxy group and hexylloyl group, asyloxy group, phenoxy group and naphthoxy.
- alkoxy groups such as methoxy group, ethoxy group, propoxyl group, butoxy group, pentoxy group and hexylloyl group, asyloxy group, phenoxy group and naphthoxy.
- examples thereof include a group, an anthrasenoxy group, a pentasenoxy group, an allyloxy group, an iminooxy group, an alkenyloxy group, an asyl
- hydrolyzable silyl group forms a siloxane bond by a condensation reaction, or the unreacted portion remains as it is or becomes a hydroxyl group.
- a solid polysiloxane compound can be obtained by proceeding with a hydrolysis condensation reaction between hydroxyl groups and unreacted hydrolyzable groups in parallel with the thermal curing reaction.
- Examples of the aliphatic hydrocarbon group include methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group, isopentyl group, neopentyl group and tert-.
- Pentyl group 1-methylbutyl group, 2-methylbutyl group, 1,2-dimethylpropyl group, 1-ethylpropyl group, hexyl group, isohesyl group, 1-methylpentyl group, 2-methylpentyl group, 3-methylpentyl group , 1,1-dimethylbutyl group, 1,2-dimethylbutyl group, 2,2-dimethylbutyl group, 1-ethylbutyl group, 1,1,2-trimethylpropyl group, 1,2,2-trimethylpropyl group, Carbons such as 1-ethyl-2-methylpropyl group, 1-ethyl-1-methylpropyl group, heptyl group, octyl group, decyl group, dodecyl group, myristyl group, palmityl group, stearyl group, isostearyl group, behenyl group, etc.
- Examples thereof include linear or branched alkyl groups having 1 to 30 atoms.
- Examples of the cycloalkyl group include a cycloalkyl group having 3 to 30 carbon atoms such as a cyclopropyl group, a cyclobutyl group, a cyclopentyl group and a cyclohexyl group. Of these, as the cycloalkyl group, a methyl group and an ethyl group are preferable.
- the unsaturated hydrocarbon group includes, for example, an alkenyl group having 2 to 30 carbon atoms such as a vinyl group, an allyl group, and a butenyl group, and a monovalent substituent having a functional group described later, such as a vinyl group and ( Meta) Acryloyl group and the like are mentioned, and among them, a vinyl group and an allyl group are preferable.
- an alkenyl group having 2 to 30 carbon atoms such as a vinyl group, an allyl group, and a butenyl group
- a monovalent substituent having a functional group described later such as a vinyl group and ( Meta) Acryloyl group and the like are mentioned, and among them, a vinyl group and an allyl group are preferable.
- aromatic hydrocarbon group examples include aryl groups having 6 to 30 carbon atoms such as phenyl group, tolyl group, xsilyl group, naphthyl group, styryl group, 4-vinylphenyl group and 3-isopropylphenyl group. Be done. Of these, the phenyl group is preferable as the aromatic hydrocarbon group.
- Examples of the aralkyl group include a benzyl group, a diphenylmethyl group, a naphthylmethyl group and the like.
- a benzyl group is preferred.
- Examples of the acyl group include an aliphatic monocarboxylic acid-based acyl group such as a formyl group, an acetyl group, a propionyl group, a butyryl group, a valeryl group, a pivaloyl group, a lauroyl group, a myritoyl group, a palmitoyl group and a stearoyl group, and Examples thereof include aromatic ring-containing acyl groups such as a benzoyl group and a cinnamoyl group.
- An aliphatic monocarboxylic acid-based acyl group is preferable, and an acetyl group is more preferable.
- Examples of the functional group in the monovalent substituent having the functional group include a vinyl group, a (meth) acrylic group, an amino group, a glycidyl group, and a thiol group.
- Specific examples include vinyl trialkoxysilane, (meth) acrylicoxypropyltrialkoxysilane, aminooxypropyltrialkoxysilane, glycidyloxypropyltrialkoxysilane, glycidyloxypropylmethyldialkoxysilane, epoxycyclohexyltrialkoxysilane, and thiolpropyl.
- Examples thereof include trialkoxysilane and p-styryltrialkoxysilane.
- two or more of the functional groups are present in the polysiloxane segment, more preferably 3 to 200, and even more preferably 3 to 50.
- the cross-linking reaction proceeds, and the solid state allows easy thermal decomposition treatment. ..
- the divalent substituent there are oxygen atoms, alkylenes, alkynes, divalent aromatics, ethers, ketones, esters, secondary amines, and amides.
- a valence substituent can be mentioned. Specific examples include methylene, ethylene, propylene, butylene, pentylene, hexylene, heptene, octylene, nonylene, decylene, cyclohexylene, etc. for alkylenes, and vinylene, isoprene, butene, butadiene, penten, hexene, etc. for alkynes.
- Alkenes such as heptene, octene, nonen, and decene, ethins, propines, butenes, pentins, hexins, etc.
- phenylene, biphenylene, alkylbenzenes, dialkylbenzenes, alkynylbenzenes, dialkynylbenzenes, naphthylenes, etc. are mentioned as divalent aromatics. Can be done. Among them, alkylenes, alkenes and divalent aromatics are preferable, and ethylene and phenylene are particularly preferable.
- the precursor can be produced by a known method, and the following examples (1), (2), and (3) are preferable. However, the manufacturing method is not limited to these.
- a hydrolytic condensation reaction is carried out in an organic solvent with one or more of the above-mentioned substituents and one or more silane compounds having one or more hydrolyzable groups in the presence of a catalyst, if necessary.
- the catalyst is preferably non-catalyst, alkali metal hydroxide, amines and the like.
- an acidic catalyst or an alkaline catalyst can be preferably used, the acidic catalyst is preferably phosphoric acid, hydrochloric acid, sulfuric acid, p-toluenesulfonic acid or the like, and the alkaline catalyst is amines.
- Sodium hydroxide, potassium hydroxide are preferred.
- siloxane compound having a substituent and a hydroxysilyl group and the above-mentioned siloxane compound having a substituent and a vinylsilyl group, an allylsilyl group, etc. are used in an organic solvent using a platinum catalyst such as platinum chloride or an organic platinum complex.
- a platinum catalyst such as platinum chloride or an organic platinum complex.
- the solvent is preferably aromatics, alcohols, ketones, esters, aliphatic amines, aromatic amines, amides, nitriles, sulfides and the like.
- the amorphous silicon-based material has a skeleton portion composed only of carbon together with a SiOC skeleton structure and the like, and some carbon atoms are combined with some Si atoms in the SiOC skeleton. It is bonded and is called free carbon, which is a component that affects charge / discharge characteristics.
- the free carbon is referred to as free carbon F.
- Free carbon F is formed in an aggregate of SiOC composed of SiO 2 C 2 , SiO 3 C, and SiO 4 , and is bonded to a Si atom of a part of the SiO C, so that the SiO C is formed.
- the SiOC active material may expand or contract slightly due to the insertion / desorption reaction of lithium ions, but the presence of free carbon F in the vicinity alleviates the expansion / contraction of the entire active material. , Has the effect of greatly improving the cycle characteristics.
- the free carbon F was formed by thermal decomposition of the precursor silane compound in the atmosphere of the inert gas. Specifically, the free carbon F becomes a carbon component by high-temperature thermal decomposition in an atmosphere in which carbonizable sites in the molecular structure of the silane compound and substituents are inactivated, and some of these carbons are used. It has the characteristic of being bonded to a part of the SiOC skeleton.
- the carbonizable component is not particularly limited, but hydrocarbons are preferable, alkyls, alkylenes, alkenes, alkynes, and aromatics are more preferable, and aromatics are more preferable.
- the amount of free carbon F is an important parameter for the charge / discharge characteristics of the active material. If the carbon content is insufficient, the conductivity may be inferior and the charge / discharge characteristics may be deteriorated. On the other hand, if the amount of carbon in the free carbon F is too large, the theoretical capacity of the free carbon F itself is low, so that the charge / discharge capacity of the entire active material decreases.
- the existence state of the free carbon F can be identified by thermal analysis (TG-DTA). Unlike the C atom in the SiOC skeleton, the free carbon F is easily pyrolyzed in the atmosphere, and the abundance of all free carbon can be determined from the amount of thermogravimetric reduction measured in the presence of air. That is, the total amount of free carbon can be quantified by using a thermogravimetric meter-Differential Thermal Analyzer (TG-DTA). In addition, the thermal decomposition temperature behavior (decomposition reaction start temperature, decomposition reaction end temperature, number of thermal decomposition reaction species, temperature of maximum weight reduction amount in each thermal decomposition reaction species, etc.) obtained from the thermal weight reduction behavior from the measurement.
- TG-DTA thermogravimetric meter-Differential Thermal Analyzer
- the state of free carbon F can be determined using these temperature values.
- the C atom in the SiOC skeleton that is, the carbon atom bonded to the Si atom constituting the SiO 2 C 2 , SiO 3 C, and SiO 4 has a very strong chemical bond and thus has high thermal stability. It is considered that it is high and does not undergo thermal decomposition in the atmosphere within the temperature range measured by the thermal analyzer.
- the free carbon F formed by thermally decomposing the precursor organic silane compound in an inert gas atmosphere has characteristics similar to those of hard carbon, and therefore has a temperature of about 550 ° C to 900 ° C in the atmosphere.
- TG-DTA The maximum temperature under the measurement conditions of TG-DTA is not particularly limited, but in order to completely complete the pyrolysis reaction of all free carbons, TG- under conditions from room temperature (about 25 ° C.) to 1000 ° C. or higher in the air. It is preferable to perform DTA measurement.
- the thermal weight reduction start temperature when the active material is thermally decomposed in air is 550 ° C. or higher.
- the amount of thermogravimetric reduction is preferably 2 wt% or more and 30 wt% or less, and more preferably 3 wt% or more and 25 wt% or less for the above-mentioned reason.
- the state of free carbon can be grasped by Raman spectroscopy in addition to the thermal analysis means.
- the G-band of carbon located near 1590 cm -1 graphite long-period carbon lattice structure
- the D-band of carbon located near 1350 cm -1 graphite short-period carbon lattice with turbulence and defects.
- the peak intensity ratio G / D with respect to the structure is preferably 0.8 or less, and more preferably 0.6 or more and 0.8 or less.
- the composition of the long-period structure and the short-period structure of the free carbon is well-balanced, and the SiOC 3 , SiO 2 C 2 , SiO 3 C, and between SiO 4 and the free carbon It is preferable because electron transfer is promoted.
- the composite active material for a battery of the present invention is not particularly limited as long as the active material for a battery of the present invention is contained as a part of the constituent elements, but graphite, which is generally used as a negative electrode material of a battery as a constituent element, is low. It is preferable to mix at least one carbon material selected from crystalline carbon and amorphous carbon before or after firing. By including these various carbon materials, it is possible to select according to the respective characteristics and exhibit preferable characteristics. As an example, when composited with a highly crystalline carbon material such as graphite, it is preferable because it improves the initial Coulomb efficiency, cycle characteristics (life), and capacity retention rate.
- the ratio of these carbon materials is, for example, 1 to 80% by weight, preferably 5 to 60% by weight, based on the total amount of the composite active material.
- Free carbon G At least one carbon material selected from the graphite, low crystalline carbon, and amorphous carbon has the above-mentioned properties and behaves as a single carbon component, that is, free carbon in the active material. ..
- the free carbon does not have a bond with a Si atom in SiOC, and a part of carbon atoms in the free carbon described in the free carbon F is bonded to a part of Si atom in SiOC, and the SiOC It is different from the carbon component that forms a close presence with. Therefore, hereinafter, the free carbon having no bond with the Si atom in SiOC is referred to as free carbon G.
- the free carbon G improves the initial Coulomb efficiency and the capacity retention rate as described above due to the effect of reducing the resistivity of the entire active material.
- free carbon F there is no choice but to have a limited effect in improving the charge / discharge characteristics of the SiOC active material.
- the composite active material for a battery of the present invention is not particularly limited as long as the active material for a battery of the present invention is contained as a part of the constituent elements, but it is preferable to include the carbon material as described above as the constituent elements, and the carbon material is preferably used.
- a carbon source resin that becomes the carbon material by firing may be mixed and dispersed before firing.
- the carbon source resin is not particularly limited as long as it has good miscibility with the polysiloxane compound and may be carbonized by high-temperature firing in an inert atmosphere, but synthetic resins having aromatic functional groups, natural raw materials, etc. Is preferably used.
- the synthetic resin include phenol resin, polystyrene, polyphenylene ether, polyphenylene sulfide and the like. The use of phenolic resin is more preferable from the viewpoint of inexpensive acquisition and elimination of impurities.
- the composite active material for a battery of the present invention may contain silicon (zero-valent) particles having an average particle size [volume average particle size (D50)] of 150 nm or less as a constituent element.
- the inclusion of such silicon particles has the effect of improving the initial charge / discharge performance (charge / discharge capacity, initial Coulomb efficiency) of the active material as described above.
- the average particle size (D50) of the silicon particles is preferably 10 to 150 nm, more preferably 15 to 120 nm because it correlates with the cycle characteristics of the active material.
- the average particle size of the silicon particles can be measured by a dynamic light scattering method using a laser diffraction / scattering type particle size distribution measuring device or the like.
- the shape of the silicon particles that can be contained in the composite active material for a battery of the present invention is not particularly limited, but it is preferable to have a sheet shape as an example, and the length in the major axis direction is 50 to 300 nm. Moreover, the thickness is preferably 1 to 60 nm. In the present invention, the sheet shape means that the thickness / length (so-called aspect ratio) is 0.5 or less. Large-sized silicon particles having an aspect ratio of more than 0.5 become large lumps and tend to be pulverized during charging / discharging, so that the charging / discharging performance of the active material tends to deteriorate.
- the average particle size can be measured by a dynamic light scattering method, but observation with a transmission electron microscope (TEM) or a field emission scanning electron microscope (FE-SEM), and By using the analysis means, the morphology (size, shape, etc.) of the sample, such as the thickness / length described above, can be observed more easily and accurately.
- the sample can be cut with a focused ion beam (FIB) or the like and the cross section can be observed by FE-SEM, or the sample can be sliced and TEM. By observing, the state of silicon particles can be observed.
- FIB focused ion beam
- the size range of the silicon particles defined in the present invention is a calculation result based on 50 particles of the main part of the sample in the field of view reflected in the TEM image. Since there is a limit to the observation field of view, the silicon particles in the present invention may have a size outside the above range. Further, the silicon particles preferably have a silicon purity of 97% or more, more preferably 99.0% or more.
- the carbon material and the silicon particles are contained as the constituent elements of the composite active material for a battery of the present invention
- the total mass of the precursor is increased.
- the amount of silicon particles added is preferably 3 to 50% by mass
- the solid content of the silane compound is 15 to 85% by mass
- the solid content of the carbon source resin is preferably set to 3 to 70% by mass. It is more preferable to set the solid content addition amount to 8 to 40% by mass, the solid content of the silane compound to 20 to 70% by mass, and the solid content of the carbon source resin to 3 to 60% by mass.
- the negative electrode contains the active material for the battery of the present invention or the composite active material for the battery of the present invention as an essential component, and if necessary, other organic binders (binders), conductive auxiliaries, etc. It is obtained by applying a slurry composed of components onto a collector copper foil to form a thin film.
- the secondary battery of the present invention is not particularly limited as long as the active material for the battery of the present invention or the composite active material for the battery of the present invention is contained in the negative electrode.
- a positive electrode, the negative electrode, and a separator provided as needed are usually wound in a flat spiral shape to form a winding electrode plate group, or these are laminated as a flat plate.
- the structure is such that a laminated electrode plate group or the like is formed, and these electrode plates are enclosed in an exterior body together with an electrolytic solution.
- the secondary battery of the present invention When used in a wet electrolyte secondary battery, for example, the secondary battery of the present invention may be configured by arranging a positive electrode and a negative electrode of the present invention so as to face each other via a separator and injecting an electrolytic solution. can.
- the secondary battery of the present invention can be manufactured by assembling the secondary battery according to this configuration.
- the secondary battery of the present invention having the negative electrode thus obtained contains the active material for the battery of the present invention (or the composite active material for the battery of the present invention), particularly the active material for the battery of the present invention or the battery of the present invention.
- the negative electrode containing the composite active material can maintain good initial Coulomb efficiency and cycle characteristics when used in a lithium ion secondary battery, and has a discharge capacity of, for example, 800 mAh / g or more (half cell measurement voltage range: 0.01-). It can be improved to 1.5V).
- the active material for a battery of the present invention is not particularly limited, but for example, when the silane compound is dissolved in an organic solvent, a step of obtaining a precursor by removing the solvent and drying the organic solvent (precursor). Production), a step of obtaining a calcined product by firing the obtained precursor in an inert atmosphere (firing of the precursor), and a step of obtaining an active substance by crushing the obtained calcined product (active material). It can be manufactured by manufacturing).
- the silane compound organic solvent is desolved and dried.
- the solvent removal and drying work for the purpose of distilling off the organic solvent it has a heating part, a cooling part, etc. with a rotary evaporator, glass that can be distilled under reduced pressure or normal pressure, or metal, etc., and has a heating function and cooling. It is performed by using an apparatus or the like assembled with parts such as a function and a pressure control function, and in the drying operation, a hot air dryer, a vacuum dryer, a spray dryer and the like can be used.
- a step of mixing with the silane compound is required in the production of the carbon material and the precursor containing the silicon particles, and any method is used in that means. Further, there is no problem whether the step is performed before or after the solvent removal, and the process is not particularly limited. Typical examples include a dry method and a wet method, but regardless of the dry method or the wet method, stirring, shaking, vibration, flow stirring by encapsulation of gas, or various other methods may be performed. In the wet method, various dispersion methods can be further used.
- Typical examples include a disper, a homogenizer, a ball mill, a bead mill, a fill mix, an ultrasonic homogenizer, and the like. Further, any dispersant or dispersion aid can be used, and the dispersion method can be used as the dispersion means in that case.
- An alkali metal or an alkaline earth metal compound may be added to the precursor silane compound.
- the alkali metal or alkaline earth metal compound is not particularly limited, and examples thereof include lithium and magnesium metal compounds.
- an alkali metal or an alkaline earth metal compound may be added as a halogen compound, an organic complex compound or the like.
- the step of obtaining the fired product is a step of firing the precursor obtained above at a high temperature in an inert atmosphere. That is, in the firing of the precursor, the thermally decomposable organic component is completely decomposed, and the other main components are precisely controlled under the firing conditions to obtain a fired product suitable for the active material or the composite active material. Specifically, it is a reactive activation site such as a radical terminal of Si, C, O or a reactive coordination bond due to a decomposition reaction of a bond such as Si—O, Si—C present in the precursor silane compound.
- the SiOC skeleton structure is formed by the formation of Si—O, Si—C and the like newly generated by the reaction caused by the reactive activation site. Further, when the carbon source resin is contained, the uniformly dispersed carbon source resin is also carbonized and converted into free carbon G in the three-dimensional structure having a SiOC skeleton.
- the firing conditions are not particularly limited, but the treatment is preferably performed at about 600 ° C. or higher in an inert atmosphere. Generally, firing is performed by heating according to the program setting of the apparatus.
- the maximum temperature reached in firing is the maximum temperature to be set, which strongly affects the structure and performance of the fired product.
- the maximum temperature reached is preferably 1000 ° C to 1300 ° C, more preferably 1050 ° C to 1250 ° C.
- the firing method is not particularly limited, but a reaction device having a heating function in an atmosphere may be used, and treatment by a continuous method or a batch method is possible.
- a fluidized bed reactor, a rotary furnace, a vertical mobile layer reactor, a tunnel furnace, a batch furnace, a rotary kiln, or the like can be appropriately selected according to the purpose.
- the firing is preferably carried out in an atmosphere that does not contain an oxidizing gas, and is preferably carried out in an inert atmosphere such as nitrogen or argon, or in a reducing atmosphere such as a mixed gas of nitrogen / hydrogen, pure hydrogen or carbon monoxide. ..
- a preliminary oxidation treatment may be performed before the firing of the precursor.
- the organic component of the silane compound is more efficiently thermally decomposed, and the specific chemical bond portion can be activated.
- silicon when silicon is contained, a thin oxide film can be imparted on the surface of silicon.
- the chemical stability with respect to the electrolytic solution is increased, exposure of the silicon surface can be prevented, and there is also an effect of suppressing decomposition of the electrolytic solution. Therefore, the cycle characteristics of the active material can be improved by performing the preliminary oxidation treatment.
- the temperature range of 200 ° C. to 450 ° C. is preferable in the atmosphere, and 250 ° C. to 400 ° C. is more preferable.
- the fired product obtained above is crushed and classified as necessary to obtain the active material.
- the pulverization may be performed in one stage up to the target particle size, or may be performed in several stages. For example, when the calcined product is a lump or agglomerated particles of 10 mm or more and a 10 ⁇ m active material is produced, it is roughly crushed with a jaw crusher, a roll crusher, etc. to obtain particles of about 1 mm, and then 100 ⁇ m with a glow mill, a ball mill, etc. Then, pulverize to 10 ⁇ m with a bead mill, jet mill or the like.
- the particles produced by pulverization may contain coarse particles, and in order to remove them, and when removing fine particles to adjust the particle size distribution, classification is performed.
- the classifier to be used is selected according to the purpose such as a wind power classifier or a wet classifier, but when removing coarse particles, a classifying method through a sieve is preferable because the purpose can be surely achieved.
- the negative electrode contains the active material for the battery of the present invention or the composite active material for the battery of the present invention as an essential component, and if necessary, other organic binders (binders), conductive auxiliaries, etc. It is obtained by applying a slurry composed of components onto a collector copper foil to form a thin film.
- the negative electrode in the secondary battery of the present invention is, for example, a stirrer, a ball mill, a super sand mill, a stirrer, a ball mill, a super sand mill, or a binder in which the above-mentioned active material for a battery or the composite active material for a battery of the present invention and a binder which is an organic binder are added together with a solvent. It can be obtained by kneading with a disperser such as a pressure kneader to prepare a negative electrode material slurry, and applying this to a current collector to form a negative electrode layer. It can also be obtained by molding a paste-like negative electrode material slurry into a sheet-like or pellet-like shape and integrating it with a current collector.
- the organic binder is not particularly limited, but is, for example, a styrene-butadiene rubber rubber copolymer (SBR); an ethylenically unsaturated carboxylic acid ester (for example, methyl (meth) acrylate, ethyl (meth) acrylate). , Butyl (meth) acrylate, (meth) acrylonitrile, and hydroxyethyl (meth) acrylate, etc.), and ethylenically unsaturated carboxylic acid (eg, acrylic acid, methacrylic acid, itaconic acid, fumaric acid, maleic acid, etc.).
- SBR styrene-butadiene rubber rubber copolymer
- carboxylic acid ester for example, methyl (meth) acrylate, ethyl (meth) acrylate.
- (Meth) acrylic copolymers such as polyfluorinated vinylidene, polyethylene oxide, polyepichlorohydrin, polyphosphazene, polyacrylonitrile, polyimide, polyamideimide, carboxymethyl cellulose (CMC) and the like.
- organic binder an aqueous binder having high chemical stability can also be adopted.
- organic binders may be dispersed or dissolved in water depending on their physical characteristics, or may be dissolved in an organic solvent such as N-methyl-2-pyrrolidone (NMP).
- NMP N-methyl-2-pyrrolidone
- the content ratio of the organic binder in the negative electrode layer of the negative electrode of the lithium ion secondary battery is preferably 1 to 30% by mass, more preferably 2 to 20% by mass, and 3 to 15% by mass. Is even more preferable.
- the content ratio of the organic binder (binder) is 1% by mass or more, so that the adhesion is good, and the destruction of the negative electrode structure is suppressed by expansion and contraction during charging and discharging. On the other hand, when it is 30% by mass or less, an increase in electrode resistance is suppressed.
- a conductive auxiliary agent may be mixed with the negative electrode material slurry, if necessary.
- the conductive auxiliary agent include carbon black, graphite, acetylene black, oxides and nitrides exhibiting conductivity, and the like.
- the amount of the conductive auxiliary agent used may be about 1 to 15% by mass with respect to the total amount of the active material for batteries (or the composite active material for batteries of the present invention) of the present invention.
- the material and shape of the current collector are not particularly limited, and a strip of copper, nickel, titanium, stainless steel, or the like in the form of a foil, a perforated foil, a mesh, or the like may be used. Further, porous materials such as porous metal (foamed metal) and carbon paper can also be used.
- the method of applying the negative electrode material slurry to the current collector is not particularly limited, but for example, a metal mask printing method, an electrostatic coating method, a dip coating method, a spray coating method, a roll coating method, a doctor blade method, and a gravure coating method.
- Known methods such as a method and a screen printing method can be mentioned.
- After coating it is preferable to perform a rolling process using a flat plate press, a calendar roll, or the like, if necessary.
- the integration of the negative electrode material slurry formed into a sheet-like or pellet-like shape and the current collector can be performed by a known method such as, for example, a roll, a press, or a combination thereof.
- the negative electrode layer formed on the current collector and the negative electrode layer integrated with the current collector are heat-treated according to the organic binder used.
- the organic binder used For example, when a known and commonly used aqueous styrene-butadiene rubber copolymer (SBR) or the like is used, heat treatment may be performed at 100 to 130 ° C., and an organic binder having polyimide or polyamide-imide as a main skeleton may be used. When used, it is preferable to heat-treat at 150 to 450 ° C.
- SBR aqueous styrene-butadiene rubber copolymer
- This heat treatment promotes the removal of the solvent and the hardening of the binder to increase the strength, and the adhesion between the particles and between the particles and the current collector can be improved.
- these heat treatments are preferably performed in an inert atmosphere such as helium, argon or nitrogen, or in a vacuum atmosphere.
- the negative electrode is pressed (pressurized) after the heat treatment.
- the electrode density is preferably 1.0 to 1.8 g / cm 3 , preferably 1.1 to 1.7 g. more preferably / cm 3, more preferably from 1.2 ⁇ 1.6g / cm 3.
- the higher the density the better the adhesion and the volume volume density of the electrode.
- the voids in the electrode will decrease and the effect of suppressing volume expansion such as silicon will be weakened. The characteristics are reduced.
- the positive electrode can be obtained by forming a positive electrode layer on the surface of the current collector in the same manner as the negative electrode.
- a strip-shaped body obtained by forming a metal or alloy such as aluminum, titanium, or stainless steel into a foil shape, a perforated foil shape, a mesh shape, or the like can be used.
- the positive electrode material used for the positive electrode layer is not particularly limited.
- a metal compound, a metal oxide, a metal sulfide, or a conductive polymer material capable of doping or intercalating lithium ions is used. May be used, and is not particularly limited.
- lithium cobalt oxide (LiCoO 2 ), lithium nickel oxide (LiNiO 2 ), lithium manganate (LiMnO 2 ), and composite oxides thereof (LiCoxNyMnzO 2 , x + y + z 1), lithium manganese spinel (LiMn 2 O 4 ).
- Lithium vanadium compounds V 2 O 5 , V 6 O 13 , VO 2 , MnO 2 , TiO 2 , MoV 2 O 8 , TiS 2 , V 2 S 5 , VS 2 , MoS 2 , MoS 3 , Cr 3 O 8 , Cr 2 O 5 , olivine type LiMPO 4 (M: Co, Ni, Mn, Fe), polyacetylene, polyaniline, polypyrrole, polythiophene, polyacetylene and other conductive polymers, porous carbon, etc. are used alone or in combination. be able to.
- M Co, Ni, Mn, Fe
- a non-woven fabric containing a polyolefin as a main component such as polyethylene or polypropylene, a cloth, a micropore film, or a combination thereof can be used. If the structure is such that the positive electrode and the negative electrode of the non-aqueous electrolyte secondary battery to be manufactured do not come into direct contact with each other, it is not necessary to use a separator.
- Electrolytic solution examples include lithium salts such as LiClO 4 , LiPF 6 , LiAsF 6 , LiBF 4 , LiSO 3 CF 3 , ethylene carbonate, propylene carbonate, butylene carbonate, vinylene carbonate, fluoroethylene carbonate, cyclopentanone, and the like.
- lithium salts such as LiClO 4 , LiPF 6 , LiAsF 6 , LiBF 4 , LiSO 3 CF 3 , ethylene carbonate, propylene carbonate, butylene carbonate, vinylene carbonate, fluoroethylene carbonate, cyclopentanone, and the like.
- Non-aqueous solvent of a single substance or a mixture of two or more components such as ethylpropyl carbonate, butylethyl carbonate, dipropyl carbonate, 1,2-dimethoxyethane, tetrahydrofuran, 2-methyltetrachloride, 1,3-dioxolane, methyl acetate, ethyl acetate, etc.
- a so-called organic electrolytic solution dissolved in the above can be used.
- the secondary battery of the present invention is not particularly limited, but is used as a paper type battery, a button type battery, a coin type battery, a laminated type battery, a cylindrical type battery, a square type battery and the like.
- the above-mentioned negative electrode active material of the lithium ion secondary battery of the present invention shall be applied to all electrochemical devices having a charging / discharging mechanism of inserting and removing lithium ions, for example, a hybrid capacitor and a solid lithium secondary battery. Is possible.
- Synthesis Examples 2-7 Synthesis of Polysiloxane Precursor
- trimethoxymethylsilane (MeSi (OMe) 3) and dimethoxydiphenylsilane (Ph2Si (OMe) 2) are used, and trimethoxymethylsilane (MeSi (OMe) 3) and trimethoxyphenylsilane (PhSi (OMe) 3) are used.
- Synthesis Examples 8-9 Precursor Synthesis
- Synthesis Example 1 a mixed solution of trimethoxymethylsilane (MeSi (OMe) 3), dimethoxydiphenylsilane (Ph2Si (OMe) 2), and isopropyl alcohol (IPA) is mixed with polydimethylsiloxane (SiH-containing PDMS) having hydroxysilane. ), 1,3,5,7-Tetramethyl 1,3,5,7-Tetravinylcyclotetrasiloxane, 1,4-bis (dimethylvinylsilyl) benzene, the weight of each is shown in Table 2. The values were all the same as in Synthesis Example 1 except that the mixture was replaced with a mixed solution of 34 g of toluene. The ratio (mol%) of each siloxane in the polysiloxane precursor is as shown in Table 3.
- Synthesis Example 8 polydimethylsiloxane (SiH-containing PDMS) having hydroxysilane, 1,3,5,7-tetramethyl 1,3,5,7-tetravinylcyclotetrasiloxane, 1,4-bis (dimethyl). All were carried out in the same manner as in Synthesis Example 8 except that the weight of vinylsilyl) benzene was set to the value shown in Table 2. The ratio (mol%) of each siloxane in the polysiloxane precursor is as shown in Table 3.
- Examples 1 to 9, Comparative Examples 1 to 4 Preparation of active substance
- the precursors of the silane compounds obtained in Synthesis Examples 1 to 9 and Comparative Synthesis Examples 1 to 4 were fired in a nitrogen stream at 800 ° C. for 2 hours, and then further fired in a nitrogen stream at 1100 ° C. for 6 hours. This was pulverized in a mortar and then further pulverized with a bead mill to a size of about 5 to 10 ⁇ m to obtain each active substance.
- Example 10 Preparation of precursor complex and active material
- the reaction solution after completion of the reaction was subjected to IPA and water distillation at 70 ° C. under reduced pressure, concentrated, and the treatment was completed at NV60%.
- 30 g of artificial graphite powder having an average particle size of 1 micron was added and dispersed by a disper.
- the solvent was completely distilled off at 110 ° C. and then vacuum dried at 110 ° C. to obtain a polysiloxane precursor containing graphite.
- the precursor was calcined at 800 ° C. for 2 hours under a nitrogen stream and then further calcined at 1100 ° C. for 6 hours under a nitrogen stream. This was pulverized in a mortar and then further pulverized with a bead mill to a size of about 5 to 10 ⁇ m to obtain each active substance.
- Example 11 Preparation of precursor complex and active material
- 10 g of a commercially available Si particle having an average particle size of 100 nm was added to a mixed solution in which 30 g of graphite powder having an average particle size of 1 micron was added to 100 g of the concentrated solution after synthesis. gone.
- This slurry was applied onto a copper plate so as to have a film thickness of 60 ⁇ m, and dried on a hot plate heated to 70 ° C. for 5 minutes. Then, it was cut into a circle having a diameter of 14 mm and vacuum dried at 110 ° C. to prepare an electrode as a negative electrode.
Abstract
本発明が解決しようとする課題は、良好な初回クーロン効率とサイクル特性を維持した上、放電容量を例えば800mAh/g以上に向上させることができる電池用活物質、これを含む電池用複合活物質、及びこれらを負極に含む二次電池を提供することである。 本発明の電池用活物質は、Si、O、及びCを含む元素から構成される非晶質珪素系材料であり、固体29Si-NMRスペクトルのケミカルシフト20ppm~-150ppmの範囲において、SiO2C2及びSiO3Cに帰属される各々ピーク積分値の合計値AとSiO4に帰属されるピーク積分値Bとの比率(A/B)が0.5以上5.0以下である。
Description
本発明は、Si、O、及びCを含む元素から構成される非晶質珪素系材料である電池用活物質、これを含む電池用複合活物質、及びこれらを負極に含む二次電池に関する。
近年、スマートフォンなどの携帯電子機器の普及に伴い、小型・高容量二次電池の需要が高まっている。その中でもリチウムイオン電池(LIBと表記する場合がある)などの二次電池は、電気自動車(EV)用途等への展開が急速に進められている。二次電池において、1回の充電で走行可能な距離(航続距離)は、電気自動車の性能をはかる重要項目である。電気自動車の性能を裏付けるものは搭載される電池性能であり、高容量、高効率、高容量維持率のことである。
充放電容量においては従来型の炭素材料(黒鉛の理論容量:372mAh/g)に比べてスズ(理論容量:994mAh/g)、ケイ素(理論容量:4199mAh/g)は、理論容量が高いため多くの検討がなされている。しかしスズ系材料やケイ素材料は、充放電時に活物質の微粉化などが起きるため、充放電容量の低下が回避できない問題がある。問題解決策が数多く提案されてはいるが実用化には至らず、現在なお改善が続けられている。
一方、容量においてはケイ素材料に劣るが、充放電時に生じる副反応挙動等に伴うリチウム吸蔵中の体積膨張等といった材料劣化現象の少ない材料として、ケイ素と炭素の直接結合を有する有機ケイ素化合物の高温焼成物が容量維持率の向上に寄与することが提案されている(下記特許文献1)。
また、ケイ素(Si)と酸素(O)、炭素(C)からなる材料は、一定の充放電容量を保持しながら高い容量維持率の材料開発を行う提案も多くなされている。例えば、炭素質物相中に組成式SiOxCy(0.2≦x≦3、1≦y≦5)で示される組成物相が分散する粒子を用いる活物質(下記特許文献2)、架橋基を有する反応性シラン、シロキサンを硬化させた架橋物を不活性雰囲気下700~1400℃で焼結させた活物質(下記特許文献3)、ケイ素、炭素、酸素からなり、炭素含有量が0.2~10mol%、酸素含有量が0.5~40mol%、Si-C結合がケイ素原子に対して0.1~17.29mol%以下である活物質(下記特許文献4)等である。
しかしながら、前記特許文献1-4に記載の二次電池における活物質では、充放電容量、初回クーロン効率(初回効率)とサイクル特性(容量維持率特性)を同時に改善できた材料がなく、特に放電容量が800mAh/g未満と推定され、ケイ素材料と比べて非常に放電容量が低いことが課題である。
前記実情を鑑み、本発明が解決しようとする課題は、良好な初回クーロン効率とサイクル特性を維持した上、放電容量を例えば800mAh/g以上に向上させることができる電池用活物質、これを含む電池用複合活物質、及びこれらを負極に含む二次電池を提供することである。
本発明者らは前記課題を解決するため、鋭意検討した結果、Si、O、及びCを含む元素から構成される非晶質珪素系材料において、特定の微細構造を有する電池用活物質を含むことで前記課題を解決できることを見出し、本発明に至った。
すなわち本発明は、以下のとおりである。
項1.Si、O、及びCを含む元素から構成される非晶質珪素系材料であり、固体29Si-NMRスペクトルのケミカルシフト20ppm~-150ppmの範囲において、SiO2C2及びSiO3Cに帰属される各々ピーク積分値の合計値AとSiO4に帰属されるピーク積分値Bとの比率(A/B)が0.5以上5.0以下である電池用活物質。
項2. 前記活物質を空気中で熱分解させたときの重量減少開始温度が550℃以上である項1記載の電池用活物質。
項3. 前記活物質中を空気中で熱分解させたときの重量減少量が2wt%以上30wt%以下である項1又は2記載の電池用活物質。
項4. 前記活物質のラマンスペクトルにおいて、1595cm-1付近の位置する炭素のG-バンドと、1320cm-1付近に位置する炭素のD-バンドとのピーク強度比(G/D)が0.8以下である項1~3のいずれか1項に記載の電池用活物質。
項5. 項1~4のいずれか1項に記載の活物質を負極に含む二次電池。
項6. 項1~4のいずれか1項に記載の活物質を構成要素の一部に含む電池用複合活物質。
項7. 項6記載の複合活物質の構成要素に、黒鉛、低結晶性炭素、及び非晶質炭素から選ばれる少なくとも1種の炭素材料を含む電池用複合活物質。
項8. 項6又は7記載の複合活物質の構成要素に、平均粒径150nm以下の珪素粒子を含む 電池用複合活物質。
項9. 項6~8のいずれか1項に記載の複合活物質を負極に含む二次電池。
項1.Si、O、及びCを含む元素から構成される非晶質珪素系材料であり、固体29Si-NMRスペクトルのケミカルシフト20ppm~-150ppmの範囲において、SiO2C2及びSiO3Cに帰属される各々ピーク積分値の合計値AとSiO4に帰属されるピーク積分値Bとの比率(A/B)が0.5以上5.0以下である電池用活物質。
項2. 前記活物質を空気中で熱分解させたときの重量減少開始温度が550℃以上である項1記載の電池用活物質。
項3. 前記活物質中を空気中で熱分解させたときの重量減少量が2wt%以上30wt%以下である項1又は2記載の電池用活物質。
項4. 前記活物質のラマンスペクトルにおいて、1595cm-1付近の位置する炭素のG-バンドと、1320cm-1付近に位置する炭素のD-バンドとのピーク強度比(G/D)が0.8以下である項1~3のいずれか1項に記載の電池用活物質。
項5. 項1~4のいずれか1項に記載の活物質を負極に含む二次電池。
項6. 項1~4のいずれか1項に記載の活物質を構成要素の一部に含む電池用複合活物質。
項7. 項6記載の複合活物質の構成要素に、黒鉛、低結晶性炭素、及び非晶質炭素から選ばれる少なくとも1種の炭素材料を含む電池用複合活物質。
項8. 項6又は7記載の複合活物質の構成要素に、平均粒径150nm以下の珪素粒子を含む 電池用複合活物質。
項9. 項6~8のいずれか1項に記載の複合活物質を負極に含む二次電池。
本発明の電池用活物質を含有する負極を含む二次電池は、良好な初回クーロン効率とサイクル特性を維持でき、さらに放電容量を例えば800mAh/g以上に向上させることができる。
<電池用活物質>
本発明の電池用活物質は、Si、O、及びCを含む元素から構成される非晶質珪素系材料であり、シリコンオキシカーバイド(SiOC)を主成分とし、Si、O、及びCの各元素による三次元ネットワーク構造で成っている。Siと結合する原子の種類(O、又はC)、及びそれぞれの原子との結合の数から、主に3種類に分けることができ、それぞれSiO2C2、SiO3C、及びSiO4と記すこととする。これらO及びCと様々な比率で結合したSiが、さらにランダムに結合したものがSiOCである。本発明の電池用活物質は、固体29Si-NMRスペクトルのケミカルシフト20ppm~-150ppmの範囲において、SiO2C2及びSiO3Cに帰属される各々ピーク積分値の合計値AとSiO4に帰属されるピーク積分値Bとの比率(A/B)が0.5以上5.0以下である。
本発明の電池用活物質は、Si、O、及びCを含む元素から構成される非晶質珪素系材料であり、シリコンオキシカーバイド(SiOC)を主成分とし、Si、O、及びCの各元素による三次元ネットワーク構造で成っている。Siと結合する原子の種類(O、又はC)、及びそれぞれの原子との結合の数から、主に3種類に分けることができ、それぞれSiO2C2、SiO3C、及びSiO4と記すこととする。これらO及びCと様々な比率で結合したSiが、さらにランダムに結合したものがSiOCである。本発明の電池用活物質は、固体29Si-NMRスペクトルのケミカルシフト20ppm~-150ppmの範囲において、SiO2C2及びSiO3Cに帰属される各々ピーク積分値の合計値AとSiO4に帰属されるピーク積分値Bとの比率(A/B)が0.5以上5.0以下である。
リチウムイオン二次電池の充放電過程において、炭素を活物質とする負極の場合を例にした場合、充電時には、炭素とリチウムイオンとが挿入反応により化学結合が生じ炭素がリチウムを補足する。放電時には、炭素に補足されたリチウムが、電子の放出によってリチウムイオンとなり、炭素から離れる脱離反応が行われる。この炭素とリチウムイオンとの挿入、脱離反応の繰り返し、つまり可逆的に反応が進行することによって充放電が行われる。本発明の活物質であるSiOCの場合、SiO2C2、SiO3C、及びSiO4其々がランダムに結合して一つの活物質を形成している。SiO2C2、SiO3Cとは、リチウムイオンとの挿入・脱離反応が可逆的に進行可能であるが、SiO4は、リチウムイオンの一部とシリケート化合物を生じるため、リチウムイオンの挿入、脱離反応が部分的に不可逆になることがある。
さらに具体的に説明を加えると、SiOCは、リチウムイオンの接近によりSiOC内部の電子分布の変動が生じ、SiOCとリチウムイオンの間に静電的な結合や配位結合などが形成されるため、リチウムイオンがSiOCの骨格中に貯蔵される。そしてこれらの配位結合エネルギーは比較的低いため、リチウムイオンの脱離反応が容易に行われる。つまりSiOCが充放電の際にリチウムイオンの挿入・脱離反応を可逆的に起こすことができる。従って、我々はこのメカニズムを捉えることによって、SiO4の存在量に対するSiO2C2、SiO3C、それぞれの存在量の合計値が可逆容量の改善に強く寄与し、その合計値が多いほど、可逆容量が向上し、初回クーロン効率が改善できることを見出すに至った。
一方、SiO4の存在量が多い場合、SiOCとリチウムイオンの間に静電的な結合や配位結合などを形成する際、SiO4とリチウムイオンとの化学反応を容易に起し、化学安定性の高いケイ酸リチウム化合物が生じるため、一部のリチウムイオンが前記のSiO4に固定される。
従って、前記メカニズムからSiO4の存在量を削減することにより、前記ケイ素材料の可逆充放電容量を向上させることができる。しかし、SiO4のように、Siとの結合が全てO原子である場合、Si-Cの結合が1つでも存在する場合に比べて化学結合エネルギーが高く、リチウムイオンがSiO4から脱離しなくとも、ケイ素材料の骨格構造を保持する働きがあると考えられ、SiOCが活物質の容量維持率の向上面で大きく寄与することに繋がる。よって本発明のSiOC活物質において、SiO4は必要不可欠な構造である。
つまりSiO2C2、SiO3C、それぞれの存在量の合計値が、SiO4の存在量に対して、大きい方が可逆充放電容量の向上に繋がるが、大きすぎるとサイクル特性の向上が阻害されることになる。その反面、小さい方がサイクル特性は向上するが、小さすぎると可逆充放電容量の向上が阻害されることになる。このような性質を有している材料がゆえに、後述するような具体的なSiO2C2、SiO3C、それぞれの存在量の合計値と、SiO4の存在量の比率が極めて重要な因子となる。そしてSiO2C2、SiO3C、及びSiO4それぞれの存在量は、固体29Si-NMR測定により定量可能である。
本発明の非晶質珪素系材料の構造は、29Si-固体NMRの測定で同定することができる。該材料の固体29Si-NMRスペクトルにおいて、ケミカルシフト20ppm~-150ppmの範囲にSiO2C2、SiO3Cに帰属される各々ピーク積分値の合計値Aと、SiO4に帰属されるピーク積分値Bとの比率(A/B)が0.5以上5.0以下である範囲を示す活物質である。本発明の電池用活物質における固体29Si-NMRスペクトルの一例を図1に示す。
前記固体29Si-NMRスペクトルでは、図1に例示したように、SiO2C2、SiO3C、及びSiO4に帰属されるピークのケミカルシフトは、おおよそそれぞれ-20ppm~-50ppm、-50ppm~-90ppm、-90ppm~-140ppmに観測されるものである。しかし、それぞれのピークは、SiOCが存在する周囲の環境状態等によりケミカルシフトは変化するため、種々の条件を鑑みたうえで固体29Si-NMRスペクトルを解析した結果、SiO2C2、SiO3C、及びSiO4と帰属されるピークのことを示しており、前述したケミカルシフトの値の範囲内に必ず存在すると限定されるものではなく、種々のサンプルの状態、測定条件から正しく帰属されたピークのそれぞれの積分値を算出することで、前記比率(A/B)を求めることができる。固体29Si-NMRスペクトルにおけるそれぞれの積分値は、非晶質珪素系材料におけるそれぞれのモル比を表す。よって、前記比率(A/B)は、SiO4、1モル比に対するSiO2C2、及びSiO3Cの合計モル比を意味する。前記比率(A/B)が、つまりモル比が0.5以上5.0以下であることで良好な初回クーロン効率とサイクル特性を維持でき、さらに放電容量を例えば800mAh/g以上に向上させることができる。
また、前記29Si-固体NMRスペクトルからは、SiC4、及びSiOC3のピークが40ppm~-20ppmに現れる場合がある。各々のピークは、SiC4が10ppm~-20付近に、SiOC3は40ppm~-20ppm付近に現れる。このようにピークが重複している領域があり、正確に定量値を明示することが29Si-固体NMRスペクトルからは困難である。さらに、SiC4は、リチウムイオンと挿入・脱離反応しないことが知られている。SiOC3は、リチウムイオンと挿入・脱離反応するが、一般的に存在量は少ない。理想的には、SiOC3のピーク積分値を前記合計値Aに加え、SiC4のピーク積分値を前記合計値Bに加える等によって範囲を明示し、充放電特性の向上条件として可能であることが容易に考えられる。しかし、前記理由よりSiC4、及びSiOC3のピーク積分値を考慮することは現実的ではないため、前述のようにSiO2C2、SiO3Cに帰属される各々ピーク積分値の合計値Aと、SiO4に帰属されるピーク積分値Bとの比率(A/B)が0.5以上5.0以下の範囲と定めた。
(前駆体)
前述のSiOCは、前述のSiOC前駆体であるシラン化合物を不活性ガス雰囲気中で焼成することにより形成することができる。該シラン化合物は、Si-O結合、及びSi-C結合を基本構造とする分子構造で構成されている。該前駆体の分子構造の設計・調整を行うことで、不活性ガス雰囲気中での高温焼成後のSiOCの骨格構成をコントロールすることができ、前駆体分子構造と密に相関するSiOC骨格を有する構造体が得られる。
前述のSiOCは、前述のSiOC前駆体であるシラン化合物を不活性ガス雰囲気中で焼成することにより形成することができる。該シラン化合物は、Si-O結合、及びSi-C結合を基本構造とする分子構造で構成されている。該前駆体の分子構造の設計・調整を行うことで、不活性ガス雰囲気中での高温焼成後のSiOCの骨格構成をコントロールすることができ、前駆体分子構造と密に相関するSiOC骨格を有する構造体が得られる。
前記前駆体は、Si-O結合、及びSi-C結合を含む分子構造であることが好ましく、Si-O結合とSi-C結合は同一分子内であることがより好ましい。
一例を示すと、下記一般式(S-1)および/または下記一般式(S-2)、および/または下記一般式(S-3)、および/または下記一般式(S-4)、および/またはこれら下記一般式の組合せを含む構造単位を有するものが好ましい。

(S-1)

(S-2)

(S-3)

(S-4)
(前記一般式(S-1)、(S-2)、(S-3)、(S-4)において、R1、R2、R3、R4、R5、R6は全て一価の置換基であり、例示すると、それぞれ独立して、水素原子、水酸基、加水分解性基、脂肪族炭化水素基、不飽和炭化水素基、芳香族炭化水素類、アラルキル基、アシル基、又は以下の官能基を有する一価の置換基(官能基はビニル基、(メタ)アクリル基、アミノ基、グリシジル基、チオール基)を表す。X1、X2、X3、X4、X5、X6は全て二価の置換基であり、それぞれ独立して、酸素原子、アルキレン類、アルケン類、アルキン類、二価の芳香族類、エーテル類、ケトン類、エステル類、2級アミン類、アミド類、またはこれらを組み合わせた二価の置換基を示す。l、m、x、yは0以上の整数を示す。)
なお、前記一般式(S-1)、(S-2)、(S-3)、(S-4)中のR1やX1等の記号で示す基は、複数有する場合、それぞれ独立し、同一であっても異なっていてもよい。
(前記一般式(S-1)、(S-2)、(S-3)、(S-4)において、R1、R2、R3、R4、R5、R6は全て一価の置換基であり、例示すると、それぞれ独立して、水素原子、水酸基、加水分解性基、脂肪族炭化水素基、不飽和炭化水素基、芳香族炭化水素類、アラルキル基、アシル基、又は以下の官能基を有する一価の置換基(官能基はビニル基、(メタ)アクリル基、アミノ基、グリシジル基、チオール基)を表す。X1、X2、X3、X4、X5、X6は全て二価の置換基であり、それぞれ独立して、酸素原子、アルキレン類、アルケン類、アルキン類、二価の芳香族類、エーテル類、ケトン類、エステル類、2級アミン類、アミド類、またはこれらを組み合わせた二価の置換基を示す。l、m、x、yは0以上の整数を示す。)
なお、前記一般式(S-1)、(S-2)、(S-3)、(S-4)中のR1やX1等の記号で示す基は、複数有する場合、それぞれ独立し、同一であっても異なっていてもよい。
前記加水分解性基としては、例えば、塩素、臭素、ヨウ素等のハロゲン原子、メトキシ基、エトキシ基、プロポキシル基、ブトキシ基、ペントキシ基、ヘキシロイル基等のアルコキシ基、アシロキシ基、フェノキシ基、ナフトキシ基、アントラセノキシ基、ペンタセノキシ基、アリルオキシ基、イミノオキシ基、アルケニルオキシ基、アシロキシ基、アルケニルオキシ基、アミノ基、アミド基、アミノオキシ基、メルカプト基等が挙げられる。中でもメトキシ基、エトキシ基が好ましい。
これらの基が加水分解されることにより加水分解性シリル基は縮合反応によりシロキサン結合の形成、又は未反応部分はそのまま、もしくは水酸基となる。前記熱硬化反応と並行して、水酸基や未反応加水分解性基間で加水分解縮合反応が進行することで、固体状のポリシロキサン化合物を得ることができる。
前記脂肪族炭化水素基としては、例えば、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、ペンチル基、イソペンチル基、ネオペンチル基、tert-ペンチル基、1-メチルブチル基、2-メチルブチル基、1,2-ジメチルプロピル基、1-エチルプロピル基、ヘキシル基、イソヘシル基、1-メチルペンチル基、2-メチルペンチル基、3-メチルペンチル基、1,1-ジメチルブチル基、1,2-ジメチルブチル基、2,2-ジメチルブチル基、1-エチルブチル基、1,1,2-トリメチルプロピル基、1,2,2-トリメチルプロピル基、1-エチル-2-メチルプロピル基、1-エチル-1-メチルプロピル基、ヘプチル基、オクチル基、デシル基、ドデシル基、ミリスチル基、パルミチル基、ステアリル基、イソステアリル基、ベヘニル基等の炭素原子数1~30の直鎖状若しくは分岐鎖状のアルキル基が挙げられる。また、シクロアルキル基としては、例えば、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基等の炭素原子数3~30のシクロアルキル基が挙げられる。中でもシクロアルキル基としては、メチル基、エチル基が好ましい。
前記不飽和炭化水素基としては、例えば、ビニル基、アリル基、ブテニル基等の炭素数2~30のアルケニル基、後述する官能基を有する一価の置換基と重複するが、ビニル基や(メタ)アクリロイル基等が挙げられ、中でもビニル基、アリル基が好ましい。
前記芳香族炭化水素基としては、例えば、フェニル基、トリル基、キシリル基、ナフチル基、スチリル基、4-ビニルフェニル基、3-イソプロピルフェニル基等の炭素原子数6~30のアリール基が挙げられる。中でも芳香族炭化水素基としては、フェニル基が好ましい。
前記アラルキル基としては、例えば、ベンジル基、ジフェニルメチル基、ナフチルメチル基等が挙げられる。ベンジル基が好ましい。前記アシル基としては、例えば、ホルミル基、アセチル基、プロピオニル基、ブチリル基、バレリル基、ピバロイル基、ラウロイル基、ミリストイル基、パルミトイル基、ステアロイル基等の脂肪族モノカルボン酸系アシル基、及び、ベンゾイル基、シンナモイル基等の芳香環含有アシル基が挙げられる。脂肪族モノカルボン酸系アシル基が好ましく、アセチル基がより好ましい。
前記官能基を有する一価の置換基における該官能基としては、例えば、ビニル基、(メタ)アクリル基、アミノ基、グリシジル基、チオール基が挙げられる。具体例としては、ビニルトリアルコキシシラン、(メタ)アクリルオキシプロピルトリアルコキシシラン、アミノオキシプロピルトリアルコキシシラン、グリシジルオキシプロピルトリアルコキシシラン、グリシジルオキシプロピルメチルジアルコキシシラン、エポキシシクロヘキシルトリアルコキシシラン、チオールプロピルトリアルコキシシラン、p-スチリルトリアルコキシシランが挙げられる。該官能基は、ポリシロキサンセグメント中に2個以上存在することが好ましく、3~200個存在することがより好ましく、3~50個存在することが更に好ましい。該官能基を2個以上存在させた状態で、熱分解前にポリシロキサン化合物を加熱処理することにより、架橋反応が進行し、固体状とすることにより、熱分解処理を容易に行うことができる。
前記2価の置換基としては、前述のように酸素原子、アルキレン類、アルキン類、2価の芳香族類、及び、エーテル類、ケトン類、エステル類、2級アミン類、アミド類を有する2価の置換基が挙げられる。其々具体例を挙げると、アルキレン類はメチレン、エチレン、プロピレン、ブチレン、ペンチレン、ヘキシレン、ヘプチレン、オクチレン、ノニレン、デシレン、シクロヘキシレン等、アルキン類はビニレン、イソプレン、ブテン、ブタジエン、ペンテン、ヘキセン、ヘプテン、オクテン、ノネン、デセン等のアルケン、エチン、プロピン、ブチン、ペンチン、ヘキシン等、2価の芳香族類としてフェニレン、ビフェニレン、アルキルベンゼン、ジアルキルベンゼン、アルキニルベンゼン、ジアルキニルベンゼン、ナフチレン等を挙げることができる。中でもアルキレン類、アルケン類、2価の芳香族類が好ましく、エチレン、フェニレンが特に好ましい。
該前駆体は、公知の方法で製造できるが、以下(1)、(2)、(3)として好ましい幾つかの製造方法を例示する。但し、製造方法はこれらに限定されるものではない。
(1)前記した1つ以上の置換基と、1つ以上の加水分解性基を有するシラン化合物を1種類以上と、必要に応じて触媒存在下、有機溶媒中で加水分解縮合反応を行うことによって前駆体としてのシラン化合物を製造する方法。
前記触媒は、加水分解性基がハロゲンの場合は無触媒、アルカリ金属水酸化物、アミン類等が好ましい。他の加水分解性基の場合は、酸性触媒、又はアルカリ性触媒が好適に使用でき、酸性触媒としては、リン酸、塩酸、硫酸、p-トルエンスルホン酸等が好ましく、アルカリ触媒としては、アミン類、水酸化ナトリウム、水酸化カリウム、が好ましい。
前記触媒は、加水分解性基がハロゲンの場合は無触媒、アルカリ金属水酸化物、アミン類等が好ましい。他の加水分解性基の場合は、酸性触媒、又はアルカリ性触媒が好適に使用でき、酸性触媒としては、リン酸、塩酸、硫酸、p-トルエンスルホン酸等が好ましく、アルカリ触媒としては、アミン類、水酸化ナトリウム、水酸化カリウム、が好ましい。
(2)前記した置換基とヒドロキシシリル基を有するシロキサン化合物と、前記した置換基とビニルシリル基、或いはアリルシリル基等を有するシロキサン化合物を塩化白金、有機白金錯体等の白金触媒を用い、有機溶媒中でシリル化反応を行うことによって前駆体としてのシラン化合物を製造する方法。
(3)前記した置換基と加水分解性基、及びヒドロキシシリル化合物と、前記した置換基と加水分解性基、及びビニルシリル基、或いはアリルシリル基等を有するシラン化合物を、塩化白金、有機白金錯体等の白金触媒を用い、有機溶媒中でシリル化反応を行った後、加水分解反応を行う為に、前述した酸性触媒、又はアルカリ触媒を用いてシロキサン結合を形成させることによって前駆体としてのシラン化合物を製造する方法。
前記溶媒は芳香族類、アルコール類、ケトン類、エステル類、脂肪族アミン類、芳香族アミン類、アミド類、ニトリル類、スルフィド類等が好ましい。特にトルエン、キシレン、エチルアルコール、イソプロピルアルコール、ブチルアルコール、メチルエチルケトン、メチルイソブチルケトン、酢酸エチル、酢酸ブチル、トリメチルアミン、トリエチルアミン、トリブチルアミン、ピペリジン、ピリジン、ピロール、イミダゾール、ジメチルホルムアミド、ジメチルアセトアミド、メチルピロリジン、メチルピロリドン、アセトニトリル、ジメチルスルオキシドがより好ましい。
前記溶媒は芳香族類、アルコール類、ケトン類、エステル類、脂肪族アミン類、芳香族アミン類、アミド類、ニトリル類、スルフィド類等が好ましい。特にトルエン、キシレン、エチルアルコール、イソプロピルアルコール、ブチルアルコール、メチルエチルケトン、メチルイソブチルケトン、酢酸エチル、酢酸ブチル、トリメチルアミン、トリエチルアミン、トリブチルアミン、ピペリジン、ピリジン、ピロール、イミダゾール、ジメチルホルムアミド、ジメチルアセトアミド、メチルピロリジン、メチルピロリドン、アセトニトリル、ジメチルスルオキシドがより好ましい。
(フリー炭素F)
本発明の電池用活物質において、非晶質珪素系材料は、SiOC骨格構造等とともに炭素のみで構成される骨格部分を有し、一部の炭素原子がSiOC骨格中の一部のSi原子と結合しており、フリー炭素と呼ばれ、充放電特性に影響を与える成分である。以下、該フリー炭素をフリー炭素Fと記す。フリー炭素Fは、SiO2C2,SiO3C、及びSiO4で構成されるSiOCの集合体中に形成しているものであり、該SiOCの一部のSi原子と結合しているためSiOC内部、及び表面のSi原子とフリー炭素F間の電子伝達がより容易となる。このため充放電時のリチウムイオンの挿入・離脱反応が速やかに進行し、SiOC活物質の充放電特性が向上すると考えることができる。また、リチウムイオンの挿入・脱離反応によって、SiOC活物質は僅かではあるが膨張・収縮することがあるが、フリー炭素Fがその近傍に存在することで活物質全体の膨張・収縮が緩和され、サイクル特性を大きく向上させる効果がある。
本発明の電池用活物質において、非晶質珪素系材料は、SiOC骨格構造等とともに炭素のみで構成される骨格部分を有し、一部の炭素原子がSiOC骨格中の一部のSi原子と結合しており、フリー炭素と呼ばれ、充放電特性に影響を与える成分である。以下、該フリー炭素をフリー炭素Fと記す。フリー炭素Fは、SiO2C2,SiO3C、及びSiO4で構成されるSiOCの集合体中に形成しているものであり、該SiOCの一部のSi原子と結合しているためSiOC内部、及び表面のSi原子とフリー炭素F間の電子伝達がより容易となる。このため充放電時のリチウムイオンの挿入・離脱反応が速やかに進行し、SiOC活物質の充放電特性が向上すると考えることができる。また、リチウムイオンの挿入・脱離反応によって、SiOC活物質は僅かではあるが膨張・収縮することがあるが、フリー炭素Fがその近傍に存在することで活物質全体の膨張・収縮が緩和され、サイクル特性を大きく向上させる効果がある。
前記フリー炭素Fは、前駆体シラン化合物の不活性ガス雰囲気中の熱分解に伴い形成したものである。前記フリー炭素Fは、具体的にはシラン化合物の分子構造中にある炭化可能な部位、及び置換基等が不活性化する雰囲気中で高温熱分解によって炭素成分となり、これらの一部の炭素がSiOC骨格の一部と結合している特徴がある。前記炭化可能な成分は、特に限定されないが、炭化水素が好ましく、アルキル類、アルキレン類、アルケン類、アルキン類、芳香族類がより好ましく、さらに芳香族類であることが好ましい。
前記フリー炭素Fの量は、活物質の充放電特性に対して重要なパラメータである。その炭素量が不十分であれば、導電性に劣り、充放電特性が悪化することがある。一方、フリー炭素Fの炭素量が多すぎると、フリー炭素F自体の理論容量が低いため、活物質全体の充放電容量が低下する。
前記フリー炭素Fの存在状態は、熱分析(TG-DTA)で同定することが可能である。SiOC骨格中のC原子と異なり、フリー炭素Fは、大気中で熱分解されやすく、空気存在下で測定した熱重量減少量により全フリー炭素の存在量を求めることができる。つまり全フリー炭素量は、熱重量示差熱分析装置Thermogravimeter-Differential Thermal Analyzer(TG-DTA)を用いることで定量できる。また、該測定からの熱重量減少挙動より得られる熱分解温度挙動(分解反応開始温度、分解反応終了温度、熱分解反応種の数、各熱分解反応種における最大重量減少量の温度など)の変化も容易に把握でき、これらの温度値を用いてフリー炭素Fの状態を判断することができる。一方、SiOC骨格中のC原子、つまり前記SiO2C2、SiO3C、及びSiO4を構成するSi原子と結合している炭素原子は、非常に強い化学結合を有するために熱安定性が高く、熱分析装置測定温度範囲において大気中で熱分解されることがないと考えられる。また、前記前駆体の有機シラン化合物を不活性ガス雰囲気中の熱分解により形成されるフリー炭素Fは、ハードカーボンと類似する特性を有しているため、大気中において約550℃~900℃の温度範囲に熱分解されることに伴い、急激な重量減少が発生する。TG-DTAの測定条件の最高温度は特に限定されないが、全フリー炭素の熱分解反応を完全に終了させるために、大気中、室温(約25℃)から1000℃以上までの条件下でTG-DTA測定を行うのが好ましい。
本発明の電池用活物質は、一定量以上のフリー炭素Fを含む場合、該活物質を空気中で熱分解させたときの熱重量減少開始温度が550℃以上となることが好ましい。また、フリー炭素Fを含む場合、前述の理由により熱重量減少量が2wt%以上30wt%以下であることが好ましく、3wt%以上25wt%以下であることがより好ましい。
前記活物質がフリー炭素Fを有する場合、前記熱分析手段のほか、ラマン分光測定によってフリー炭素の状態を把握することができる。ラマン分光スペクトルにおいて、1590cm-1付近の位置する炭素のG-バンド(グラファイト長周期炭素格子構造)と、1350cm-1付近に位置する炭素のD-バンド(乱れや欠陥のあるグラファイト短周期炭素格子構造)とのピーク強度比G/Dが0.8以下であることが好ましく、0.6以上0.8以下であることがより好ましい。G/Dがこの範囲であると、フリー炭素の長周期構造と短周期構造の構成バランスがとれた状態となり、前記SiOC3、SiO2C2,SiO3C、及びSiO4とフリー炭素間の電子伝達が促進されるため好ましい。
<電池用複合活物質>
本発明の電池用複合活物質は、本発明の電池用活物質を構成要素の一部に含む限り特に制限されないが、構成要素としてさらに、一般的に電池の負極材に使用される黒鉛、低結晶性炭素、及び非晶質炭素から選ばれる少なくとも1種の炭素材料を、焼成前、又は焼成後に混合させることが好ましい。これら種々の炭素材料を含むことで、それぞれの特性に応じて選択でき好ましい特性を発現できる。一例として、黒鉛等の結晶性の高い炭素材料と複合化させた場合は、初回クーロン効率、サイクル特性(寿命)、及び容量維持率を向上させるため好ましい。これらの炭素材料の割合は、複合活物質全量に対して、例えば1~80重量%、好ましくは5~60重量%である。
本発明の電池用複合活物質は、本発明の電池用活物質を構成要素の一部に含む限り特に制限されないが、構成要素としてさらに、一般的に電池の負極材に使用される黒鉛、低結晶性炭素、及び非晶質炭素から選ばれる少なくとも1種の炭素材料を、焼成前、又は焼成後に混合させることが好ましい。これら種々の炭素材料を含むことで、それぞれの特性に応じて選択でき好ましい特性を発現できる。一例として、黒鉛等の結晶性の高い炭素材料と複合化させた場合は、初回クーロン効率、サイクル特性(寿命)、及び容量維持率を向上させるため好ましい。これらの炭素材料の割合は、複合活物質全量に対して、例えば1~80重量%、好ましくは5~60重量%である。
(フリー炭素G)
前記黒鉛、低結晶性炭素、及び非晶質炭素から選ばれる少なくとも1種の炭素材料は、前述のような特性を有し、該活物質において、単独炭素成分、すなわちフリー炭素としての振る舞いをする。しかし該フリー炭素は、SiOC中のSi原子との結合を有せず、前記フリー炭素Fで述べたフリー炭素中の一部の炭素原子が、SiOC中の一部のSi原子と結合し、SiOCと密接な存在を形成する炭素成分とは異なる。そのため、以下、SiOC中のSi原子との結合を有さないフリー炭素をフリー炭素Gと記す。該フリー炭素Gは、活物質全体の抵抗率低減化効果により、前述のように初回クーロン効率や容量維持率を向上させる。但し前述のフリー炭素Fの特性と比較すると、SiOC活物質の充放電特性改善においては限定的効果と成らざるを得ない。しかし、SiOC中の一部のSi原子と結合したフリー炭素Fの存在を前提として、さらにフリー炭素Gを組合わせることによって、充放電特性をより向上させる炭素成分として重要である。
前記黒鉛、低結晶性炭素、及び非晶質炭素から選ばれる少なくとも1種の炭素材料は、前述のような特性を有し、該活物質において、単独炭素成分、すなわちフリー炭素としての振る舞いをする。しかし該フリー炭素は、SiOC中のSi原子との結合を有せず、前記フリー炭素Fで述べたフリー炭素中の一部の炭素原子が、SiOC中の一部のSi原子と結合し、SiOCと密接な存在を形成する炭素成分とは異なる。そのため、以下、SiOC中のSi原子との結合を有さないフリー炭素をフリー炭素Gと記す。該フリー炭素Gは、活物質全体の抵抗率低減化効果により、前述のように初回クーロン効率や容量維持率を向上させる。但し前述のフリー炭素Fの特性と比較すると、SiOC活物質の充放電特性改善においては限定的効果と成らざるを得ない。しかし、SiOC中の一部のSi原子と結合したフリー炭素Fの存在を前提として、さらにフリー炭素Gを組合わせることによって、充放電特性をより向上させる炭素成分として重要である。
((SiOC/炭素源樹脂/Si)3成分系)
本発明の電池用複合活物質は、本発明の電池用活物質を構成要素の一部に含む限り特に制限されないが、構成要素として前述したような炭素材料を含むことが好ましく、該炭素材料を含有させる方法として前述のように該炭素材料を直接混合させる方法以外に、焼成することによって該炭素材料となるような炭素源樹脂を焼成前に混合・分散させてもよい。該炭素源樹脂は、ポリシロキサン化合物との混和性が良く、また不活性雰囲気中で高温焼成により炭化されることがあれば特に限定されないが、芳香族官能基を有する合成樹脂類や天然原料等を用いることが好ましい。合成樹脂としては、フェノール樹脂、ポリスチレン、ポリフェニレンエーテル、ポリフェニレンスルフィド等が挙げられる。安価入手や不純物排除の観点からフェノール樹脂の使用がより好ましい。
本発明の電池用複合活物質は、本発明の電池用活物質を構成要素の一部に含む限り特に制限されないが、構成要素として前述したような炭素材料を含むことが好ましく、該炭素材料を含有させる方法として前述のように該炭素材料を直接混合させる方法以外に、焼成することによって該炭素材料となるような炭素源樹脂を焼成前に混合・分散させてもよい。該炭素源樹脂は、ポリシロキサン化合物との混和性が良く、また不活性雰囲気中で高温焼成により炭化されることがあれば特に限定されないが、芳香族官能基を有する合成樹脂類や天然原料等を用いることが好ましい。合成樹脂としては、フェノール樹脂、ポリスチレン、ポリフェニレンエーテル、ポリフェニレンスルフィド等が挙げられる。安価入手や不純物排除の観点からフェノール樹脂の使用がより好ましい。
本発明の電池用複合活物質では、構成要素として平均粒径[体積平均粒子径(D50)]150nm以下の珪素(0価)粒子を含んでいてもよい。このような珪素粒子を含むことにより、前述のように活物質の初期充放電性能(充放電容量、初回クーロン効率)を向上させる効果がある。珪素粒子の平均粒径(D50)は、活物質のサイクル特性との相関性があるため、好ましくは10~150nm、さらに好ましくは15~120nmである。珪素粒子の平均粒径は、レーザー回析・散乱式粒子径分布測定装置などを用い動的光散乱法により測定することができる。150nmを超える大きいサイズの珪素粒子は、大きな塊となり、充放電時に微粉化現象が起やすいため、活物質の充放電性能が低下する傾向が想定される。また、10nm未満の小サイズの珪素粒子は細かすぎるため、シリコン粒子同士が凝集しやすくなる傾向がある。そのため、活物質中へ小粒子シリコンを均一に分散させるのが困難となり、また、微小粒子の表面活性エネルギーが高いゆえにシリコン粒子の表面も酸化されやすくなり、活物質の高温焼成で小粒子シリコンの表面上に副生成物などが多くなる傾向もあり、これが充放電性能の大幅な低下に繋がる。
本発明の電池用複合活物質に含むことができる珪素粒子の形状は、特に制限されるものではないが、一例としてシート形状を有することが好ましく、また長軸方向の長さが50~300nm、且つ厚みが1~60nmであることが好ましい。本発明においてシート形状とは、特に厚み/長さ(いわゆるアスペクト比)が0.5以下であることをいう。アスペクト比が0.5を超える大サイズの珪素粒子は、大きな塊となり、充放電時に微粉化現象が起やすいため、活物質の充放電性能が低下する傾向が想定される。
シート形状を有する珪素粒子の形態は、動的光散乱法で平均粒径の測定が可能であるが、透過型電子顕微鏡(TEM)や電界放出型走査電子顕微鏡(FE-SEM)の観察、及び解析手段を用いることで、前述の厚み/長さ等の、サンプルの形態(サイズ、形状など)をより容易かつ精密に観察することができる。なお、シート状シリコンナノ粒子を内包した負極活物質粉末の場合は、サンプルを集束イオンビーム(FIB)等で切断して断面をFE-SEM観察することができ、またはサンプルをスライス加工してTEM観察によりシリコン粒子の状態を観察することができる。本発明に定められる珪素粒子のサイズ範囲は、TEM画像に映る視野内のサンプルの主要部分50粒子をベースにした計算結果である。観察視野の限界もあるゆえ、本発明における珪素粒子が前述の範囲から外れるサイズを有しても構わない。また、前記珪素粒子としては、シリコン純度が97%以上のものが好ましく、99.0%以上がより好ましい。
本発明の電池用複合活物質の構成要素として、前記炭素材料、及び前記珪素粒子を含む場合において、該炭素材料を前駆体中に炭素源樹脂として含有させるときは、前駆体の全重量に対して、珪素粒子の添加量を3~50質量%、シラン化合物の固形分を15~85質量%含有し、炭素源樹脂の固形分を3~70質量%に設定することが好ましく、珪素粒子の固形分添加量を8~40質量%、シラン化合物の固形分を20~70質量%に、炭素源樹脂の固形分を3~60質量%に設定することがより好ましい。
(負極)
本発明の二次電池において負極は、本発明の電池用活物質または本発明の電池用複合活物質を必須成分として、必要に応じてその他の有機結着剤(バインダ)、導電助剤などの成分を含んで構成されるスラリーを集電体銅箔上へ塗布して薄膜とすることにより得られる。
本発明の二次電池において負極は、本発明の電池用活物質または本発明の電池用複合活物質を必須成分として、必要に応じてその他の有機結着剤(バインダ)、導電助剤などの成分を含んで構成されるスラリーを集電体銅箔上へ塗布して薄膜とすることにより得られる。
<二次電池>
本発明の二次電池は、本発明の電池用活物質または本発明の電池用複合活物質を負極に含む限り特に制限はされない。本発明の二次電池は、通常、正極および前記負極と、必要に応じて設けられるセパレータとを、扁平渦巻状に巻回して巻回式極板群としたり、これらを平板状として積層して積層式極板群等としたりし、これら極板群を電解液とともに外装体中に封入した構造である。本発明の二次電池は、例えば、湿式電解質二次電池に用いる場合、正極と、本発明の負極とを、セパレータを介して対向して配置し、電解液を注入することにより構成することができる。この構成に従って二次電池を組み立てることにより本発明の二次電池を製造することができる。
本発明の二次電池は、本発明の電池用活物質または本発明の電池用複合活物質を負極に含む限り特に制限はされない。本発明の二次電池は、通常、正極および前記負極と、必要に応じて設けられるセパレータとを、扁平渦巻状に巻回して巻回式極板群としたり、これらを平板状として積層して積層式極板群等としたりし、これら極板群を電解液とともに外装体中に封入した構造である。本発明の二次電池は、例えば、湿式電解質二次電池に用いる場合、正極と、本発明の負極とを、セパレータを介して対向して配置し、電解液を注入することにより構成することができる。この構成に従って二次電池を組み立てることにより本発明の二次電池を製造することができる。
こうして得られる負極を有する本発明の二次電池は、本発明の電池用活物質(又は本発明の電池用複合活物質)を含むことから、特に本発明の電池用活物質または本発明の電池用複合活物質を含む負極は、リチウムイオン二次電池に使用することにおいて良好な初回クーロン効率とサイクル特性を維持でき、さらに放電容量を例えば800mAh/g以上(ハーフセル測定電圧範囲:0.01-1.5V)に向上させることができる。
<電池用活物質の製造方法>
本発明の電池用活物質は、特に限定されないが、例えば、前記シラン化合物が有機溶剤中に溶解されている場合、該有機溶剤を脱溶媒及び乾燥することで前駆体を得る工程(前駆体の製造)、得られた前駆体を不活性雰囲気中で焼成することにより焼成物を得る工程(前駆体の焼成)、及び得られた焼成物を粉砕することで活物質を得る工程(活物質の製造)により製造することができる。
本発明の電池用活物質は、特に限定されないが、例えば、前記シラン化合物が有機溶剤中に溶解されている場合、該有機溶剤を脱溶媒及び乾燥することで前駆体を得る工程(前駆体の製造)、得られた前駆体を不活性雰囲気中で焼成することにより焼成物を得る工程(前駆体の焼成)、及び得られた焼成物を粉砕することで活物質を得る工程(活物質の製造)により製造することができる。
(前駆体の製造)
前記の前駆体を得る工程では、前記シラン化合物有機溶剤の脱溶剤、及び乾燥を行う。有機溶媒を溜去することを目的とする脱溶剤と乾燥の作業では、ロータリーエバポレータ、減圧又は常圧留去可能なガラス、又は金属等で加熱部、冷却部等を有し、加熱機能、冷却機能、圧力制御機能等の部品を組み立てた装置等を用いて行い、乾燥作業では、熱風乾燥機、減圧乾燥機、噴霧乾燥機等を用いることができる。
前記の前駆体を得る工程では、前記シラン化合物有機溶剤の脱溶剤、及び乾燥を行う。有機溶媒を溜去することを目的とする脱溶剤と乾燥の作業では、ロータリーエバポレータ、減圧又は常圧留去可能なガラス、又は金属等で加熱部、冷却部等を有し、加熱機能、冷却機能、圧力制御機能等の部品を組み立てた装置等を用いて行い、乾燥作業では、熱風乾燥機、減圧乾燥機、噴霧乾燥機等を用いることができる。
本発明の電池用複合活物質の構成要素として、前記炭素材料や、前記珪素粒子を含む前駆体製造において、前記シラン化合物と混合する工程が必要となるが、その手段においてはいかなる方法を用いてもよく、さらに該工程が脱溶剤前、脱溶剤後のどちらの工程で行われても何ら問題はなく、特に限定されるものではない。代表例として、乾式法や湿式法等があるが、乾式法、湿式法問わず、攪拌、振り混ぜ、振動、気体の封入等による流動攪拌、その他種々の方法で行ってよい。湿式法においてはさらに種々の分散方法を用いることができる。代表例としては、ディスパー、ホモジナイザー、ボールミル、ビーズミル、フィルミックス、超音波ホモジナイザー、等を挙げることができる。さらにいかなる分散剤や、分散助剤を用いることも可能であり、その場合の分散手段においても、前記分散方法を用いることができる。
(アルカリ金属化合物の添加)
前記前駆体のシラン化合物に、アルカリ金属やアルカリ土類金属化合物を加えてもよい。アルカリ金属やアルカリ土類金属化合物としては、特に限定されないが、例えばリチウムやマグネシウム金属化合物が挙げられる。また、アルカリ金属やアルカリ土類金属化合物をハロゲン類化合物や有機錯体化合物などとして加えても構わない。前記アルカリ金属やアルカリ土類金属化合物と前記シラン化合物を混合させることで、高温焼成で得られる活物質SiOCの骨格中に存在する電気化学反応の不可逆反応は、アルカリ金属やアルカリ土類金属化合物と反応・化学結合することがあるため、不可逆反応の相対量が低減することに伴い、初回クーロン効率が向上する効果が得られる。
前記前駆体のシラン化合物に、アルカリ金属やアルカリ土類金属化合物を加えてもよい。アルカリ金属やアルカリ土類金属化合物としては、特に限定されないが、例えばリチウムやマグネシウム金属化合物が挙げられる。また、アルカリ金属やアルカリ土類金属化合物をハロゲン類化合物や有機錯体化合物などとして加えても構わない。前記アルカリ金属やアルカリ土類金属化合物と前記シラン化合物を混合させることで、高温焼成で得られる活物質SiOCの骨格中に存在する電気化学反応の不可逆反応は、アルカリ金属やアルカリ土類金属化合物と反応・化学結合することがあるため、不可逆反応の相対量が低減することに伴い、初回クーロン効率が向上する効果が得られる。
(前駆体の焼成)
前記焼成物を得る工程は、前記で得られた前駆体を不活性雰囲気中で、高温焼成する工程である。つまり、前駆体の焼成では、熱分解可能な有機成分を完全分解させ、その他の主成分を焼成条件の精密制御により前記活物質、又は前記複合活物質に適した焼成物を得る。詳細には、前記前駆体シラン化合物中に存在するSi-O、Si-C等といった結合の分解反応による、Si、C、Oのラジカル末端や、反応性配位結合等といった反応性活性化部位の生成、及び該反応性活性化部位が引き起こす反応によって新たに生成するSi-O、Si-C等の結合の構築によって、SiOC骨格構造を形成する。さらに炭素源樹脂を含む場合、均一化分散されていた炭素源樹脂も炭化されることで、SiOC骨格を有する三次元構造体中においてフリー炭素Gに転化される。
前記焼成物を得る工程は、前記で得られた前駆体を不活性雰囲気中で、高温焼成する工程である。つまり、前駆体の焼成では、熱分解可能な有機成分を完全分解させ、その他の主成分を焼成条件の精密制御により前記活物質、又は前記複合活物質に適した焼成物を得る。詳細には、前記前駆体シラン化合物中に存在するSi-O、Si-C等といった結合の分解反応による、Si、C、Oのラジカル末端や、反応性配位結合等といった反応性活性化部位の生成、及び該反応性活性化部位が引き起こす反応によって新たに生成するSi-O、Si-C等の結合の構築によって、SiOC骨格構造を形成する。さらに炭素源樹脂を含む場合、均一化分散されていた炭素源樹脂も炭化されることで、SiOC骨格を有する三次元構造体中においてフリー炭素Gに転化される。
(焼成条件)
焼成条件は、特に制限されないが、不活性雰囲気下、約600℃以上で処理することが好ましい。一般的に焼成は、装置のプログラム設定に沿って加熱することにより行われるものである。焼成における最高到達温度は、設定する最高温度であり、焼成物の構造や性能に強く影響を与えるものである。最高到達温度は1000℃~1300℃であることが好ましく、より好ましくは1050℃~1250℃である。この温度範囲で焼成することにより、非晶質珪素系材料における化学結合状態をより微細化することができ、より優れた充放電特性が得られる。
焼成条件は、特に制限されないが、不活性雰囲気下、約600℃以上で処理することが好ましい。一般的に焼成は、装置のプログラム設定に沿って加熱することにより行われるものである。焼成における最高到達温度は、設定する最高温度であり、焼成物の構造や性能に強く影響を与えるものである。最高到達温度は1000℃~1300℃であることが好ましく、より好ましくは1050℃~1250℃である。この温度範囲で焼成することにより、非晶質珪素系材料における化学結合状態をより微細化することができ、より優れた充放電特性が得られる。
焼成方法は、特に限定されないが、雰囲気中にて加熱機能を有する反応装置を用いればよく、連続法、回分法での処理が可能である。焼成用装置については、流動層反応炉、回転炉、竪型移動層反応炉、トンネル炉、バッチ炉、ロータリーキルン等をその目的に応じ適宜選択することができる。
焼成は、酸化性ガスが含有しない雰囲気下で行うことが好ましく、窒素、アルゴンなどの不活性雰囲気、または窒素/水素の混合ガス、純水素、一酸化炭素などの還元性雰囲気で行うことが好ましい。
(予備酸化処理)
また、前記の前駆体の焼成の前に、予備酸化処理を行っても良い。この予備酸化処理により、シラン化合物の有機成分がより効率よく熱分解されるほか、特定化学結合部分を活性化させることもできる。さらに、珪素を含む場合には、珪素の表面上に薄い酸化膜を付与させることができる。また、電池に用いられるときに電解液に対する化学安定性が高くなり、珪素表面の暴露を防ぐことができ、電解液の分解を抑制する効果もある。このため予備酸化処理を行うことで活物質のサイクル特性を向上させることができる。予備酸化処理条件については、大気中、200℃~450℃の温度範囲が好ましく、250℃~400℃がより好ましい。
また、前記の前駆体の焼成の前に、予備酸化処理を行っても良い。この予備酸化処理により、シラン化合物の有機成分がより効率よく熱分解されるほか、特定化学結合部分を活性化させることもできる。さらに、珪素を含む場合には、珪素の表面上に薄い酸化膜を付与させることができる。また、電池に用いられるときに電解液に対する化学安定性が高くなり、珪素表面の暴露を防ぐことができ、電解液の分解を抑制する効果もある。このため予備酸化処理を行うことで活物質のサイクル特性を向上させることができる。予備酸化処理条件については、大気中、200℃~450℃の温度範囲が好ましく、250℃~400℃がより好ましい。
(活物質の製造)
活物質を得る工程は、前記で得られた焼成物を粉砕し、必要に応じて分級することで活物質を得るものである。粉砕は目的とする粒径まで一段で行っても良いし、数段に分けて行っても良い。例えば焼成物が10mm以上の塊または凝集粒子となっていて、10μmの活物質を作製する場合はジョークラッシャー、ロールクラッシャー等で粗粉砕を行い1mm程度の粒子にした後、グローミル、ボールミル等で100μmとし、ビーズミル、ジェットミル等で10μmまで粉砕する。粉砕で作製した粒子には粗大粒子が含まれる場合がありそれを取り除くため、また、微粉を取り除いて粒度分布を調整する場合は分級を行う。使用する分級機は風力分級機、湿式分級機等目的に応じて使い分けるが、粗大粒子を取り除く場合、篩を通す分級方式が確実に目的を達成できるために好ましい。
活物質を得る工程は、前記で得られた焼成物を粉砕し、必要に応じて分級することで活物質を得るものである。粉砕は目的とする粒径まで一段で行っても良いし、数段に分けて行っても良い。例えば焼成物が10mm以上の塊または凝集粒子となっていて、10μmの活物質を作製する場合はジョークラッシャー、ロールクラッシャー等で粗粉砕を行い1mm程度の粒子にした後、グローミル、ボールミル等で100μmとし、ビーズミル、ジェットミル等で10μmまで粉砕する。粉砕で作製した粒子には粗大粒子が含まれる場合がありそれを取り除くため、また、微粉を取り除いて粒度分布を調整する場合は分級を行う。使用する分級機は風力分級機、湿式分級機等目的に応じて使い分けるが、粗大粒子を取り除く場合、篩を通す分級方式が確実に目的を達成できるために好ましい。
<負極の製造方法>
本発明の二次電池において負極は、本発明の電池用活物質または本発明の電池用複合活物質を必須成分として、必要に応じてその他の有機結着剤(バインダ)、導電助剤などの成分を含んで構成されるスラリーを集電体銅箔上へ塗布して薄膜とすることにより得られる。本発明の二次電池における負極は、例えば、前述の電池用活物質または本発明の電池用複合活物質と、有機結着材であるバインダとを、溶剤とともに撹拌機、ボールミル、スーパーサンドミル、加圧ニーダ等の分散装置により混練して、負極材スラリーを調製し、これを集電体に塗布して負極層を形成することで得ることができる。また、ペースト状の負極材スラリーをシート状、ペレット状等の形状に成形し、これを集電体と一体化することでも得ることができる。
本発明の二次電池において負極は、本発明の電池用活物質または本発明の電池用複合活物質を必須成分として、必要に応じてその他の有機結着剤(バインダ)、導電助剤などの成分を含んで構成されるスラリーを集電体銅箔上へ塗布して薄膜とすることにより得られる。本発明の二次電池における負極は、例えば、前述の電池用活物質または本発明の電池用複合活物質と、有機結着材であるバインダとを、溶剤とともに撹拌機、ボールミル、スーパーサンドミル、加圧ニーダ等の分散装置により混練して、負極材スラリーを調製し、これを集電体に塗布して負極層を形成することで得ることができる。また、ペースト状の負極材スラリーをシート状、ペレット状等の形状に成形し、これを集電体と一体化することでも得ることができる。
(有機粘着剤:バインダ)
前記有機結着剤(バインダ)としては、特に限定されないが、例えば、スチレン-ブタジエンゴム共重合体(SBR);エチレン性不飽和カルボン酸エステル(例えば、メチル(メタ)アクリレート、エチル(メタ)アクリレート、ブチル(メタ)アクリレート、(メタ)アクリロニトリル、およびヒドロキシエチル(メタ)アクリレート等)、およびエチレン性不飽和カルボン酸(例えば、アクリル酸、メタクリル酸、イタコン酸、フマル酸、マレイン酸等)からなる(メタ)アクリル共重合体;ポリ弗化ビニリデン、ポリエチレンオキサイド、ポリエピクロロヒドリン、ポリホスファゼン、ポリアクリロニトリル、ポリイミド、ポリアミドイミド、カルボキシメチルセルロース(CMC)などの高分子化合物が挙げられる。前記有機結着剤としては、化学安定性が高い水性バインダも採用することができる。
前記有機結着剤(バインダ)としては、特に限定されないが、例えば、スチレン-ブタジエンゴム共重合体(SBR);エチレン性不飽和カルボン酸エステル(例えば、メチル(メタ)アクリレート、エチル(メタ)アクリレート、ブチル(メタ)アクリレート、(メタ)アクリロニトリル、およびヒドロキシエチル(メタ)アクリレート等)、およびエチレン性不飽和カルボン酸(例えば、アクリル酸、メタクリル酸、イタコン酸、フマル酸、マレイン酸等)からなる(メタ)アクリル共重合体;ポリ弗化ビニリデン、ポリエチレンオキサイド、ポリエピクロロヒドリン、ポリホスファゼン、ポリアクリロニトリル、ポリイミド、ポリアミドイミド、カルボキシメチルセルロース(CMC)などの高分子化合物が挙げられる。前記有機結着剤としては、化学安定性が高い水性バインダも採用することができる。
これらの有機結着剤(バインダ)は、それぞれの物性によって、水に分散、あるいは溶解したもの、また、N-メチル-2-ピロリドン(NMP)などの有機溶剤に溶解したものがある。リチウムイオン二次電池負極の負極層中の有機結着剤の含有比は、1~30質量%であることが好ましく、2~20質量%であることがより好ましく、3~15質量%であることがさらに好ましい。
有機結着剤(バインダ)の含有比が1質量%以上であることで密着性が良好で、充放電時の膨張・収縮によって負極構造の破壊が抑制される。一方、30質量%以下であることで、電極抵抗の上昇が抑えられる。
(導電助剤)
また、前記負極材スラリーには、必要に応じて、導電助剤を混合してもよい。導電助剤としては、例えば、カーボンブラック、グラファイト、アセチレンブラック、あるいは導電性を示す酸化物や窒化物等が挙げられる。導電助剤の使用量は、本発明の電池用活物質(又は本発明の電池用複合活物質)全量に対して1~15質量%程度とすればよい。
また、前記負極材スラリーには、必要に応じて、導電助剤を混合してもよい。導電助剤としては、例えば、カーボンブラック、グラファイト、アセチレンブラック、あるいは導電性を示す酸化物や窒化物等が挙げられる。導電助剤の使用量は、本発明の電池用活物質(又は本発明の電池用複合活物質)全量に対して1~15質量%程度とすればよい。
(集電体)
前記集電体の材質および形状については、特に限定されず、銅、ニッケル、チタン、ステンレス鋼等を、箔状、穴開け箔状、メッシュ状等にした帯状のものを用いればよい。また、多孔性材料、たとえばポーラスメタル(発泡メタル)やカーボンペーパーなども使用可能である。
前記集電体の材質および形状については、特に限定されず、銅、ニッケル、チタン、ステンレス鋼等を、箔状、穴開け箔状、メッシュ状等にした帯状のものを用いればよい。また、多孔性材料、たとえばポーラスメタル(発泡メタル)やカーボンペーパーなども使用可能である。
前記負極材スラリーを集電体に塗布する方法としては、特に限定されないが、例えば、メタルマスク印刷法、静電塗装法、ディップコート法、スプレーコート法、ロールコート法、ドクターブレード法、グラビアコート法、スクリーン印刷法など公知の方法が挙げられる。塗布後は、必要に応じて平板プレス、カレンダーロール等による圧延処理を行うことが好ましい。また、シート状、ペレット状等の形状に成形された負極材スラリーと集電体との一体化は、例えば、ロール、プレス、もしくはこれらの組み合わせ等、公知の方法により行うことができる。
前記集電体上に形成された負極層および集電体と一体化した負極層は、用いた有機結着剤に応じて熱処理することが好ましい。例えば、公知慣用されている水系のスチレン-ブタジエンゴム共重合体(SBR)などを用いた場合には100~130℃で熱処理すればよく、ポリイミド、ポリアミドイミドを主骨格とした有機結着剤を用いた場合には150~450℃で熱処理することが好ましい。
この熱処理により溶媒の除去、バインダの硬化による高強度化が進み、粒子間及び粒子と集電体間の密着性が向上できる。尚、これらの熱処理は、処理中の集電体の酸化を防ぐため、ヘリウム、アルゴン、窒素等の不活性雰囲気、真空雰囲気で行うことが好ましい。
また、熱処理する後に、負極はプレス(加圧処理)しておくことが好ましい。本発明の電池用活物質(又は本発明の電池用複合活物質)を用いた負極では、電極密度が1.0~1.8g/cm3であることが好ましく、1.1~1.7g/cm3であることがより好ましく、1.2~1.6g/cm3であることがさらに好ましい。電極密度については、高いほど密着性及び電極の体積容量密度が向上する傾向があるが、密度が高すぎると、電極中の空隙が減少することで珪素など体積膨張の抑制効果が弱くなるためサイクル特性が低下する。
<正極>
前記正極は、前記負極と同様にして、集電体表面上に正極層を形成することで得ることができる。この場合の集電体はアルミニウム、チタン、ステンレス鋼等の金属や合金を、箔状、穴開け箔状、メッシュ状等にした帯状のものを用いることができる。
前記正極は、前記負極と同様にして、集電体表面上に正極層を形成することで得ることができる。この場合の集電体はアルミニウム、チタン、ステンレス鋼等の金属や合金を、箔状、穴開け箔状、メッシュ状等にした帯状のものを用いることができる。
前記正極層に用いる正極材料としては、特に制限されない。非水電解質二次電池の中でも、リチウムイオン二次電池を作製する場合には、例えば、リチウムイオンをドーピングまたはインターカレーション可能な金属化合物、金属酸化物、金属硫化物、または導電性高分子材料を用いればよく、特に限定されない。例えば、コバルト酸リチウム(LiCoO2)、ニッケル酸リチウム(LiNiO2)、マンガン酸リチウム(LiMnO2)、およびこれらの複合酸化物(LiCoxNiyMnzO2、x+y+z=1)、リチウムマンガンスピネル(LiMn2O4)、リチウムバナジウム化合物、V2O5、V6O13、VO2、MnO2、TiO2、MoV2O8、TiS2、V2S5、VS2、MoS2、MoS3、Cr3O8、Cr2O5、オリビン型LiMPO4(M:Co、Ni、Mn、Fe)、ポリアセチレン、ポリアニリン、ポリピロール、ポリチオフェン、ポリアセチレン等の導電性ポリマー、多孔質炭素等などを単独或いは混合して使用することができる。
<セパレータ>
前記セパレータとしては、例えば、ポリエチレン、ポリプロピレン等のポリオレフィンを主成分とした不織布、クロス、微孔フィルム又はそれらを組み合わせたものを使用することができる。なお、作製する非水電解質二次電池の正極と負極が直接接触しない構造にした場合は、セパレータを使用する必要はない。
前記セパレータとしては、例えば、ポリエチレン、ポリプロピレン等のポリオレフィンを主成分とした不織布、クロス、微孔フィルム又はそれらを組み合わせたものを使用することができる。なお、作製する非水電解質二次電池の正極と負極が直接接触しない構造にした場合は、セパレータを使用する必要はない。
<電解液>
前記電解液としては、例えば、LiClO4、LiPF6、LiAsF6、LiBF4、LiSO3CF3等のリチウム塩を、エチレンカーボネート、プロピレンカーボネート、ブチレンカーボネート、ビニレンカーボネート、フルオロエチレンカーボネート、シクロペンタノン、スルホラン、3-メチルスルホラン、2,4-ジメチルスルホラン、3-メチル-1,3-オキサゾリジン-2-オン、γ-ブチロラクトン、ジメチルカーボネート、ジエチルカーボネート、エチルメチルカーボネート、メチルプロピルカーボネート、ブチルメチルカーボネート、エチルプロピルカーボネート、ブチルエチルカーボネート、ジプロピルカーボネート、1,2-ジメトキシエタン、テトラヒドロフラン、2-メチルテトラヒドロフラン、1,3-ジオキソラン、酢酸メチル、酢酸エチル等の単体もしくは2成分以上の混合物の非水系溶剤に溶解した、いわゆる有機電解液を使用することができる。
前記電解液としては、例えば、LiClO4、LiPF6、LiAsF6、LiBF4、LiSO3CF3等のリチウム塩を、エチレンカーボネート、プロピレンカーボネート、ブチレンカーボネート、ビニレンカーボネート、フルオロエチレンカーボネート、シクロペンタノン、スルホラン、3-メチルスルホラン、2,4-ジメチルスルホラン、3-メチル-1,3-オキサゾリジン-2-オン、γ-ブチロラクトン、ジメチルカーボネート、ジエチルカーボネート、エチルメチルカーボネート、メチルプロピルカーボネート、ブチルメチルカーボネート、エチルプロピルカーボネート、ブチルエチルカーボネート、ジプロピルカーボネート、1,2-ジメトキシエタン、テトラヒドロフラン、2-メチルテトラヒドロフラン、1,3-ジオキソラン、酢酸メチル、酢酸エチル等の単体もしくは2成分以上の混合物の非水系溶剤に溶解した、いわゆる有機電解液を使用することができる。
<二次電池>
本発明の二次電池は、特に限定されないが、ペーパー型電池、ボタン型電池、コイン型電池、積層型電池、円筒型電池、角型電池などとして使用される。前述した本発明のリチウムイオン二次電池負極活物質は、リチウムイオンを挿入脱離することを充放電機構とする電気化学装置全般、例えば、ハイブリッドキャパシタ、固体リチウム二次電池などにも適用することが可能である。
本発明の二次電池は、特に限定されないが、ペーパー型電池、ボタン型電池、コイン型電池、積層型電池、円筒型電池、角型電池などとして使用される。前述した本発明のリチウムイオン二次電池負極活物質は、リチウムイオンを挿入脱離することを充放電機構とする電気化学装置全般、例えば、ハイブリッドキャパシタ、固体リチウム二次電池などにも適用することが可能である。
以下に、本発明の電池用活物質を実際に作製、評価した一部の代表例を紹介するが、前述のように該作成、評価方法はこれらに限るものではない。
(合成例1:ポリシロキサン前駆体の合成)
トリメトキシメチルシラン(MeSi(OMe)3)21.8g、ジメトキシジフェニルシラン(Ph2Si(OMe)2)9.8gを用い、イソプロピルアルコール(IPA)34gの混合液を窒素雰囲気下で攪拌しながら70℃に加温した。温度が安定したところで純水4.2gと、1%りん酸エステルのIPA溶液0.17gを加え4時間反応させた。その後130℃まで加熱し、IPAと水が完全になくなるまで留去することでポリシロキサン前駆体を得た。ポリシロキサン前駆体における各シロキサンの割合(モル%)は、表3に記載のとおりである。
トリメトキシメチルシラン(MeSi(OMe)3)21.8g、ジメトキシジフェニルシラン(Ph2Si(OMe)2)9.8gを用い、イソプロピルアルコール(IPA)34gの混合液を窒素雰囲気下で攪拌しながら70℃に加温した。温度が安定したところで純水4.2gと、1%りん酸エステルのIPA溶液0.17gを加え4時間反応させた。その後130℃まで加熱し、IPAと水が完全になくなるまで留去することでポリシロキサン前駆体を得た。ポリシロキサン前駆体における各シロキサンの割合(モル%)は、表3に記載のとおりである。
(合成例2~7:ポリシロキサン前駆体の合成)
前記合成例1において、トリメトキシメチルシラン(MeSi(OMe)3)とジメトキシジフェニルシラン(Ph2Si(OMe)2)を、トリメトキシメチルシラン(MeSi(OMe)3)、トリメトキシフェニルシラン(PhSi(OMe)3)、ジメトキシジメチルシラン(Me2Si(OMe)2)、ジメトキシジフェニルシラン(Ph2Si(OMe)2)に代えて、各々の重量を表1に記した値としたこと以外は、全て合成例1と同様に行った。ポリシロキサン前駆体における各シロキサンの割合(モル%)は、表3に記載のとおりである。
前記合成例1において、トリメトキシメチルシラン(MeSi(OMe)3)とジメトキシジフェニルシラン(Ph2Si(OMe)2)を、トリメトキシメチルシラン(MeSi(OMe)3)、トリメトキシフェニルシラン(PhSi(OMe)3)、ジメトキシジメチルシラン(Me2Si(OMe)2)、ジメトキシジフェニルシラン(Ph2Si(OMe)2)に代えて、各々の重量を表1に記した値としたこと以外は、全て合成例1と同様に行った。ポリシロキサン前駆体における各シロキサンの割合(モル%)は、表3に記載のとおりである。
(合成例8~9:前駆体の合成)
前記合成例1において、トリメトキシメチルシラン(MeSi(OMe)3)とジメトキシジフェニルシラン(Ph2Si(OMe)2)、イソプロピルアルコール(IPA)の混合液を、ヒドロキシシランを有するポリジメチルシロキサン(SiH含有PDMS)、1,3,5,7-テトラメチル 1,3,5,7-テトラビニルシクロテトラシロキサン、1,4-ビス(ジメチルビニルシリル)ベンゼンに代えて、各々の重量を表2に示した値とし、トルエン34gとの混合液に代えたこと以外は全て合成例1と同様に行った。ポリシロキサン前駆体における各シロキサンの割合(モル%)は、表3に記載のとおりである。
前記合成例1において、トリメトキシメチルシラン(MeSi(OMe)3)とジメトキシジフェニルシラン(Ph2Si(OMe)2)、イソプロピルアルコール(IPA)の混合液を、ヒドロキシシランを有するポリジメチルシロキサン(SiH含有PDMS)、1,3,5,7-テトラメチル 1,3,5,7-テトラビニルシクロテトラシロキサン、1,4-ビス(ジメチルビニルシリル)ベンゼンに代えて、各々の重量を表2に示した値とし、トルエン34gとの混合液に代えたこと以外は全て合成例1と同様に行った。ポリシロキサン前駆体における各シロキサンの割合(モル%)は、表3に記載のとおりである。
(比較合成例1、2:ポリシロキサン前駆体の合成)
前記合成例1において、トリメトキシメチルシラン(MeSi(OMe)3)とジメトキシジフェニルシラン(Ph2Si(OMe)2)を、トリメトキシメチルシラン(MeSi(OMe)3)、トリメトキシフェニルシラン(PhSi(OMe)3)に変えて、各々の重量を表2に記した値とした以外は全て合成例1と同様に行った。ポリシロキサン前駆体における各シロキサンの割合(モル%)は、表3に記載のとおりである。
前記合成例1において、トリメトキシメチルシラン(MeSi(OMe)3)とジメトキシジフェニルシラン(Ph2Si(OMe)2)を、トリメトキシメチルシラン(MeSi(OMe)3)、トリメトキシフェニルシラン(PhSi(OMe)3)に変えて、各々の重量を表2に記した値とした以外は全て合成例1と同様に行った。ポリシロキサン前駆体における各シロキサンの割合(モル%)は、表3に記載のとおりである。
(比較合成例3、4:前駆体の合成)
前記合成例8において、ヒドロキシシランを有するポリジメチルシロキサン(SiH含有PDMS)、1,3,5,7-テトラメチル 1,3,5,7-テトラビニルシクロテトラシロキサン、1,4-ビス(ジメチルビニルシリル)ベンゼンの重量を表2に示した値としたこと以外は全て合成例8と同様に行った。ポリシロキサン前駆体における各シロキサンの割合(モル%)は、表3に記載のとおりである。
前記合成例8において、ヒドロキシシランを有するポリジメチルシロキサン(SiH含有PDMS)、1,3,5,7-テトラメチル 1,3,5,7-テトラビニルシクロテトラシロキサン、1,4-ビス(ジメチルビニルシリル)ベンゼンの重量を表2に示した値としたこと以外は全て合成例8と同様に行った。ポリシロキサン前駆体における各シロキサンの割合(モル%)は、表3に記載のとおりである。


(実施例1~9、比較例1~4:活物質の作製)
合成例1~9及び比較合成例1~4で得られた各シラン化合物の前駆体を窒素気流下、800℃で2時間焼成後、さらに窒素気流下、1100℃で6時間焼成した。これを乳鉢で粉砕した後、さらにビーズミルで5~10μm程度になるように粉砕して、各活物質を得た。
合成例1~9及び比較合成例1~4で得られた各シラン化合物の前駆体を窒素気流下、800℃で2時間焼成後、さらに窒素気流下、1100℃で6時間焼成した。これを乳鉢で粉砕した後、さらにビーズミルで5~10μm程度になるように粉砕して、各活物質を得た。
(実施例10:前駆体複合物、及び活物質の作製)
合成例2において、反応終了後の反応液を、減圧下70℃でIPA、水を留去、濃縮し、NV60%で処理を終了した。該濃縮液100gに、平均粒径1ミクロンの人造黒鉛粉末を30g加え、ディスパーで分散させた。その後110℃で溶媒を完全に留去した後110℃で真空乾燥し、黒鉛を含むポリシロキサン前駆体を得た。
該前駆体を窒素気流下、800℃で2時間焼成後、さらに窒素気流下、1100℃で6時間焼成した。これを乳鉢で粉砕した後、さらにビーズミルで5~10μm程度になるように粉砕して、各活物質を得た。
合成例2において、反応終了後の反応液を、減圧下70℃でIPA、水を留去、濃縮し、NV60%で処理を終了した。該濃縮液100gに、平均粒径1ミクロンの人造黒鉛粉末を30g加え、ディスパーで分散させた。その後110℃で溶媒を完全に留去した後110℃で真空乾燥し、黒鉛を含むポリシロキサン前駆体を得た。
該前駆体を窒素気流下、800℃で2時間焼成後、さらに窒素気流下、1100℃で6時間焼成した。これを乳鉢で粉砕した後、さらにビーズミルで5~10μm程度になるように粉砕して、各活物質を得た。
(実施例11:前駆体複合物、及び活物質の作製)
実施例10において合成後の濃縮液100gに、平均粒径1ミクロンの黒鉛粉末30gを加えた混合液に、平均粒径100nmのSi粒子市販品10g加えたこと以外は全て実施例10と同様に行った。
実施例10において合成後の濃縮液100gに、平均粒径1ミクロンの黒鉛粉末30gを加えた混合液に、平均粒径100nmのSi粒子市販品10g加えたこと以外は全て実施例10と同様に行った。
(比較例5)
実施例10において用いた合成例2の反応液に変えて、比較合成例3における反応終了後の反応液を用いたこと他は全て実施例10と同様に行った。
実施例10において用いた合成例2の反応液に変えて、比較合成例3における反応終了後の反応液を用いたこと他は全て実施例10と同様に行った。
(比較例6)
実施例11において用いた合成例2の反応液に変えて、比較合成例3における反応終了後の反応液を用いたこと他は全て実施例11と同様に行った。
実施例11において用いた合成例2の反応液に変えて、比較合成例3における反応終了後の反応液を用いたこと他は全て実施例11と同様に行った。
(29Si-NMRの測定及び解析方法)
実施例1~12、及び比較例1~6の各活物質のサンプルを、φ4mm 固体 NMR 試料管に採取し、固体NMR分析装置(JEOL RESONACE製、JNM-ECA600)にて、シングルパルス測定した。得られた固体29Si-NMRスペクトルデータをDelta5にてフーリエ変換したデータを、ACD LabsソフトウェアにてGauss +Lorentz関数を用いて波形分離を行った。波形分離にて得られたピーク面積を元に、ケミカルシフト20ppm~-150ppmの範囲にSiOC3、SiO2C2、SiO3Cに帰属される各々ピーク積分値の合計値Aと、SiO4に帰属されるピーク積分値Bとの比率(A/B)を算出した。算出したA/Bの結果を表4及び5に示す。
実施例1~12、及び比較例1~6の各活物質のサンプルを、φ4mm 固体 NMR 試料管に採取し、固体NMR分析装置(JEOL RESONACE製、JNM-ECA600)にて、シングルパルス測定した。得られた固体29Si-NMRスペクトルデータをDelta5にてフーリエ変換したデータを、ACD LabsソフトウェアにてGauss +Lorentz関数を用いて波形分離を行った。波形分離にて得られたピーク面積を元に、ケミカルシフト20ppm~-150ppmの範囲にSiOC3、SiO2C2、SiO3Cに帰属される各々ピーク積分値の合計値Aと、SiO4に帰属されるピーク積分値Bとの比率(A/B)を算出した。算出したA/Bの結果を表4及び5に示す。
(活物質の特性評価)
実施例1~12、及び比較例1~6の各活物質のサンプルを、熱重量示差熱分析装置TG/TDA(型番「Thermo plus EVO2」、RIGAKU製)で大気中1000℃まで昇温した時、熱分解によって重量減少が開始するときの温度と、該熱分解による重量減少率を遊離炭素(フリー炭素)量とし、該測定結果から熱分解開始温度を求めた。フリー炭素量を表4及び5に示す。
さらに実施例1~9、及び比較例1~4の各活物質のサンプルを、ラマン分光装置(型番「NRS-5500」、日本分光製)より1320cm-1付近(Dバンド)のピーク強度Dと、1595cm-1付近(Gバンド)のピーク強度Gとの比、G/Dを評価した。G/Dを表4及び5に示した。
実施例1~12、及び比較例1~6の各活物質のサンプルを、熱重量示差熱分析装置TG/TDA(型番「Thermo plus EVO2」、RIGAKU製)で大気中1000℃まで昇温した時、熱分解によって重量減少が開始するときの温度と、該熱分解による重量減少率を遊離炭素(フリー炭素)量とし、該測定結果から熱分解開始温度を求めた。フリー炭素量を表4及び5に示す。
さらに実施例1~9、及び比較例1~4の各活物質のサンプルを、ラマン分光装置(型番「NRS-5500」、日本分光製)より1320cm-1付近(Dバンド)のピーク強度Dと、1595cm-1付近(Gバンド)のピーク強度Gとの比、G/Dを評価した。G/Dを表4及び5に示した。
(電極及び電池の作製と評価)
実施例1~11、及び比較例1~6の各活物質800mg、アセチレンブラック(導電助剤)100mg、カーボンオキシメチルセルロース25mg、純水1000mgを混合し、泡取り練太郎を用い2000rpmで、5分攪拌後、0.5分脱泡した。その後スチレンブタジエンゴム(バインダ)75mg、純水 100mgを加えさらに泡取り練太郎を用い2000rpmで、5分攪拌後、0.5分脱泡し、スラリーを得た。
このスラリーを膜厚60μmになるように銅板上に塗布、70℃に加温したホットプレート上で5分間乾燥した。そして直径14mmの円状にカットし、110℃真空乾燥し、負極である電極を作製した。
実施例1~11、及び比較例1~6の各活物質800mg、アセチレンブラック(導電助剤)100mg、カーボンオキシメチルセルロース25mg、純水1000mgを混合し、泡取り練太郎を用い2000rpmで、5分攪拌後、0.5分脱泡した。その後スチレンブタジエンゴム(バインダ)75mg、純水 100mgを加えさらに泡取り練太郎を用い2000rpmで、5分攪拌後、0.5分脱泡し、スラリーを得た。
このスラリーを膜厚60μmになるように銅板上に塗布、70℃に加温したホットプレート上で5分間乾燥した。そして直径14mmの円状にカットし、110℃真空乾燥し、負極である電極を作製した。
前述のように作製した負極とLi金属箔である正極を用い、25μmのポリプロピレン製セパレータを介して前記負極と正極を対向させ、電解液(1mol/LのLiPF6、炭酸ジエチル:炭酸エチレン=1:1(容積比))を吸着させて評価用ハーフ電池を作製した。
下記カットオフ電圧と充放電レートの条件で10回の充放電を行い、充放電特性の評価を行った。初回効率と容量維持率(10回)は以下のようにして求めた。評価結果を表4及び5に示す。
カットオフ電圧範囲:0.005~1.5V
充放電レート:0.1C(初期から3サイクルまで)、0.2C(4サイクル以後)
初回効率(%)=初回放電容量(mAh/g)/初回充電容量(mAh/g)
容量維持率(10回目)=10回目の放電容量(mAh/g)/初回放電容量(mAh/g)
カットオフ電圧範囲:0.005~1.5V
充放電レート:0.1C(初期から3サイクルまで)、0.2C(4サイクル以後)
初回効率(%)=初回放電容量(mAh/g)/初回充電容量(mAh/g)
容量維持率(10回目)=10回目の放電容量(mAh/g)/初回放電容量(mAh/g)



Claims (9)
- Si、O、及びCを含む元素から構成される非晶質珪素系材料であり、
固体29Si-NMRスペクトルのケミカルシフト20ppm~-150ppmの範囲において、SiO2C2及びSiO3Cに帰属される各々ピーク積分値の合計値AとSiO4に帰属されるピーク積分値Bとの比率(A/B)が0.5以上5.0以下である電池用活物質。 - 前記活物質を空気中で熱分解させたときの重量減少開始温度が550℃以上である請求項1記載の電池用活物質。
- 前記活物質中を空気中で熱分解させたときの重量減少量が2wt%以上30wt%以下である請求項1又は2記載の電池用活物質。
- 前記活物質のラマンスペクトルにおいて、1595cm-1付近の位置する炭素のG-バンドと、1320cm-1付近に位置する炭素のD-バンドとのピーク強度比(G/D)が0.8以下である請求項1~3のいずれか1項に記載の電池用活物質。
- 請求項1~4のいずれか1項に記載の活物質を負極に含む二次電池。
- 請求項1~4のいずれか1項に記載の活物質を構成要素の一部に含む電池用複合活物質。
- 請求項6記載の複合活物質の構成要素に、黒鉛、低結晶性炭素、及び非晶質炭素から選ばれる少なくとも1種の炭素材料を含む電池用複合活物質。
- 請求項6又は7記載の複合活物質の構成要素に、平均粒径150nm以下の珪素粒子を含む電池用複合活物質。
- 請求項6~8のいずれか1項に記載の複合活物質を負極に含む二次電池。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP21837320.7A EP4181233A1 (en) | 2020-07-07 | 2021-06-03 | Active material for battery, composite active material for battery, and secondary battery |
| KR1020227041688A KR20230030569A (ko) | 2020-07-07 | 2021-06-03 | 전지용 활물질, 전지용 복합 활물질, 및 이차 전지 |
| CN202180038571.2A CN115668544A (zh) | 2020-07-07 | 2021-06-03 | 电池用活性物质、电池用复合活性物质及二次电池 |
| JP2022534955A JP7201133B2 (ja) | 2020-07-07 | 2021-06-03 | 電池用活物質、電池用複合活物質、及び二次電池 |
| US18/085,741 US20230125241A1 (en) | 2020-07-07 | 2022-12-21 | Active material for battery, composite active material for battery, and secondary battery |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2020-116972 | 2020-07-07 | ||
| JP2020116972 | 2020-07-07 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US18/085,741 Continuation US20230125241A1 (en) | 2020-07-07 | 2022-12-21 | Active material for battery, composite active material for battery, and secondary battery |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022009572A1 true WO2022009572A1 (ja) | 2022-01-13 |
Family
ID=79552377
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2021/021116 WO2022009572A1 (ja) | 2020-07-07 | 2021-06-03 | 電池用活物質、電池用複合活物質、及び二次電池 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20230125241A1 (ja) |
| EP (1) | EP4181233A1 (ja) |
| JP (1) | JP7201133B2 (ja) |
| KR (1) | KR20230030569A (ja) |
| CN (1) | CN115668544A (ja) |
| WO (1) | WO2022009572A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114975997A (zh) * | 2022-05-09 | 2022-08-30 | 东莞理工学院 | 一种锂离子电池负极材料及其制备方法 |
| JP7473098B1 (ja) | 2022-09-01 | 2024-04-23 | Dic株式会社 | 負極活物質前駆体、負極活物質、二次電池および負極活物質の製造方法 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05144474A (ja) | 1991-11-25 | 1993-06-11 | Seiko Electronic Components Ltd | 非水電解質二次電池及びその活物質の製造方法 |
| JP2003197193A (ja) | 2001-12-27 | 2003-07-11 | Toshiba Corp | 非水電解質二次電池用負極活物質、その製造方法、および非水電解質二次電池 |
| JP2006062949A (ja) | 2004-07-30 | 2006-03-09 | Shin Etsu Chem Co Ltd | Si−C−O系コンポジット及びその製造方法並びに非水電解質二次電池用負極材 |
| JP2009283366A (ja) | 2008-05-23 | 2009-12-03 | Sony Corp | 負極およびそれを備えた二次電池 |
| WO2014002602A1 (ja) * | 2012-06-27 | 2014-01-03 | Jnc株式会社 | 二次電池用負極活物質及びその製造方法、それを用いた負極並びにリチウムイオン電池 |
| CN104466142A (zh) * | 2013-09-23 | 2015-03-25 | 北京有色金属研究总院 | 一种锂离子电池用硅/硅氧碳/石墨复合负极材料 |
| JP2018106830A (ja) * | 2016-12-22 | 2018-07-05 | Dic株式会社 | 非水性二次電池負極用活物質の製造方法、非水性二次電池負極用活物質、および非水性二次電池用負極の製造方法 |
| WO2019107336A1 (ja) * | 2017-12-01 | 2019-06-06 | Dic株式会社 | 負極活物質及びその製造方法 |
| WO2020016280A1 (en) * | 2018-07-17 | 2020-01-23 | Commissariat A L'energie Atomique Et Aux Energies Alternatives | Spherical sioc particulate electrode material |
-
2021
- 2021-06-03 KR KR1020227041688A patent/KR20230030569A/ko active Search and Examination
- 2021-06-03 CN CN202180038571.2A patent/CN115668544A/zh active Pending
- 2021-06-03 JP JP2022534955A patent/JP7201133B2/ja active Active
- 2021-06-03 WO PCT/JP2021/021116 patent/WO2022009572A1/ja unknown
- 2021-06-03 EP EP21837320.7A patent/EP4181233A1/en active Pending
-
2022
- 2022-12-21 US US18/085,741 patent/US20230125241A1/en active Pending
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05144474A (ja) | 1991-11-25 | 1993-06-11 | Seiko Electronic Components Ltd | 非水電解質二次電池及びその活物質の製造方法 |
| JP2003197193A (ja) | 2001-12-27 | 2003-07-11 | Toshiba Corp | 非水電解質二次電池用負極活物質、その製造方法、および非水電解質二次電池 |
| JP2006062949A (ja) | 2004-07-30 | 2006-03-09 | Shin Etsu Chem Co Ltd | Si−C−O系コンポジット及びその製造方法並びに非水電解質二次電池用負極材 |
| JP2009283366A (ja) | 2008-05-23 | 2009-12-03 | Sony Corp | 負極およびそれを備えた二次電池 |
| WO2014002602A1 (ja) * | 2012-06-27 | 2014-01-03 | Jnc株式会社 | 二次電池用負極活物質及びその製造方法、それを用いた負極並びにリチウムイオン電池 |
| CN104466142A (zh) * | 2013-09-23 | 2015-03-25 | 北京有色金属研究总院 | 一种锂离子电池用硅/硅氧碳/石墨复合负极材料 |
| JP2018106830A (ja) * | 2016-12-22 | 2018-07-05 | Dic株式会社 | 非水性二次電池負極用活物質の製造方法、非水性二次電池負極用活物質、および非水性二次電池用負極の製造方法 |
| WO2019107336A1 (ja) * | 2017-12-01 | 2019-06-06 | Dic株式会社 | 負極活物質及びその製造方法 |
| WO2020016280A1 (en) * | 2018-07-17 | 2020-01-23 | Commissariat A L'energie Atomique Et Aux Energies Alternatives | Spherical sioc particulate electrode material |
Non-Patent Citations (2)
| Title |
|---|
| DUBEY ROMAIN J.-C., SASIKUMAR PRADEEP VALLACHIRA WARRIAM, CERBONI NOEMI, AEBLI MARCEL, KRUMEICH FRANK, BLUGAN GURDIAL, KRAVCHYK KO: "Silicon oxycarbide-antimony nanocomposites for high-performance Li-ion battery anodes", NANOSCALE, vol. 12, 10 June 2020 (2020-06-10), pages 13540 - 13547, XP055899317, DOI: 10.1039/DONR02930K * |
| WILAMOWSKA, M. ET AL.: "Tailoring of SiOC composition as a way to better performing anodes for Li-ion batteries", SOLID STATE IONICS, vol. 260, 16 April 2014 (2014-04-16), pages 94 - 100, XP028644800, DOI: 10.1016/j.ssi. 2014.03.02 1 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114975997A (zh) * | 2022-05-09 | 2022-08-30 | 东莞理工学院 | 一种锂离子电池负极材料及其制备方法 |
| CN114975997B (zh) * | 2022-05-09 | 2024-04-30 | 东莞理工学院 | 一种锂离子电池负极材料及其制备方法 |
| JP7473098B1 (ja) | 2022-09-01 | 2024-04-23 | Dic株式会社 | 負極活物質前駆体、負極活物質、二次電池および負極活物質の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20230030569A (ko) | 2023-03-06 |
| JPWO2022009572A1 (ja) | 2022-01-13 |
| CN115668544A (zh) | 2023-01-31 |
| EP4181233A1 (en) | 2023-05-17 |
| US20230125241A1 (en) | 2023-04-27 |
| JP7201133B2 (ja) | 2023-01-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7447865B2 (ja) | シリコンナノ粒子及びそれを用いた非水二次電池負極用活物質並びに二次電池 | |
| JP7288198B2 (ja) | 負極活物質及びその製造方法 | |
| US20230125241A1 (en) | Active material for battery, composite active material for battery, and secondary battery | |
| WO2021157459A1 (ja) | 二次電池負極用活物質、負極及び二次電池 | |
| JP2018106830A (ja) | 非水性二次電池負極用活物質の製造方法、非水性二次電池負極用活物質、および非水性二次電池用負極の製造方法 | |
| WO2022172585A1 (ja) | 負極活物質、負極活物質の製造方法及び非水電解質二次電池 | |
| WO2021157460A1 (ja) | 低酸素型ナノ珪素粒子含有スラリー、負極活物質、負極及びリチウムイオン二次電池 | |
| JP7485230B2 (ja) | ナノシリコン、ナノシリコンスラリー、ナノシリコンの製造方法、二次電池用活物質および二次電池 | |
| JP7473098B1 (ja) | 負極活物質前駆体、負極活物質、二次電池および負極活物質の製造方法 | |
| WO2023017587A1 (ja) | 二次電池用材料、負極活物質および二次電池 | |
| JP7088438B1 (ja) | 負極活物質、負極活物質の製造方法及び非水電解質二次電池 | |
| JP7453631B2 (ja) | ケイ素系材料、ケイ素系材料を含む複合材料、二次電池用負極物質および二次電池 | |
| WO2024101263A1 (ja) | 二次電池用複合活物質、及び二次電池 | |
| JP7323089B1 (ja) | 負極活物質、二次電池および負極活物質の製造方法 | |
| WO2023157642A1 (ja) | 二次電池用活物質および二次電池 | |
| WO2023157643A1 (ja) | 二次電池用活物質、二次電池用活物質の製造方法および二次電池 | |
| WO2023162692A1 (ja) | ナノシリコン、ナノシリコンスラリー、ナノシリコンの製造方法、二次電池用活物質および二次電池 | |
| KR20230118085A (ko) | 음극 활물질 및 비수 전해질 이차 전지 | |
| WO2023074099A1 (ja) | 二次電池用複合活物質および二次電池 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21837320 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2022534955 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2021837320 Country of ref document: EP Effective date: 20230207 |