WO2022004701A1 - ガスバリア樹脂組成物、ガスバリア樹脂組成物の製造方法、及び成形体 - Google Patents
ガスバリア樹脂組成物、ガスバリア樹脂組成物の製造方法、及び成形体 Download PDFInfo
- Publication number
- WO2022004701A1 WO2022004701A1 PCT/JP2021/024488 JP2021024488W WO2022004701A1 WO 2022004701 A1 WO2022004701 A1 WO 2022004701A1 JP 2021024488 W JP2021024488 W JP 2021024488W WO 2022004701 A1 WO2022004701 A1 WO 2022004701A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- layer
- resin composition
- gas barrier
- barrier resin
- evoh
- Prior art date
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/26—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers modified by chemical after-treatment
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01G—HORTICULTURE; CULTIVATION OF VEGETABLES, FLOWERS, RICE, FRUIT, VINES, HOPS OR SEAWEED; FORESTRY; WATERING
- A01G27/00—Self-acting watering devices, e.g. for flower-pots
- A01G27/04—Self-acting watering devices, e.g. for flower-pots using wicks or the like
- A01G27/06—Self-acting watering devices, e.g. for flower-pots using wicks or the like having a water reservoir, the main part thereof being located wholly around or directly beside the growth substrate
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01G—HORTICULTURE; CULTIVATION OF VEGETABLES, FLOWERS, RICE, FRUIT, VINES, HOPS OR SEAWEED; FORESTRY; WATERING
- A01G24/00—Growth substrates; Culture media; Apparatus or methods therefor
- A01G24/30—Growth substrates; Culture media; Apparatus or methods therefor based on or containing synthetic organic compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/06—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material
- B32B27/08—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material of synthetic resin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/36—Layered products comprising a layer of synthetic resin comprising polyesters
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B65—CONVEYING; PACKING; STORING; HANDLING THIN OR FILAMENTARY MATERIAL
- B65D—CONTAINERS FOR STORAGE OR TRANSPORT OF ARTICLES OR MATERIALS, e.g. BAGS, BARRELS, BOTTLES, BOXES, CANS, CARTONS, CRATES, DRUMS, JARS, TANKS, HOPPERS, FORWARDING CONTAINERS; ACCESSORIES, CLOSURES, OR FITTINGS THEREFOR; PACKAGING ELEMENTS; PACKAGES
- B65D65/00—Wrappers or flexible covers; Packaging materials of special type or form
- B65D65/38—Packaging materials of special type or form
- B65D65/40—Applications of laminates for particular packaging purposes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/04—Preparation of carboxylic acid esters by reacting carboxylic acids or symmetrical anhydrides onto unsaturated carbon-to-carbon bonds
- C07C67/05—Preparation of carboxylic acid esters by reacting carboxylic acids or symmetrical anhydrides onto unsaturated carbon-to-carbon bonds with oxidation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/02—Ethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F216/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an alcohol, ether, aldehydo, ketonic, acetal or ketal radical
- C08F216/02—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an alcohol, ether, aldehydo, ketonic, acetal or ketal radical by an alcohol radical
- C08F216/04—Acyclic compounds
- C08F216/06—Polyvinyl alcohol ; Vinyl alcohol
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F218/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an acyloxy radical of a saturated carboxylic acid, of carbonic acid or of a haloformic acid
- C08F218/02—Esters of monocarboxylic acids
- C08F218/04—Vinyl esters
- C08F218/08—Vinyl acetate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/12—Hydrolysis
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/005—Processes for mixing polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/20—Compounding polymers with additives, e.g. colouring
- C08J3/203—Solid polymers with solid and/or liquid additives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/18—Manufacture of films or sheets
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/04—Coating
- C08J7/048—Forming gas barrier coatings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/04—Coating
- C08J7/06—Coating with compositions not containing macromolecular substances
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/08—Metals
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/32—Phosphorus-containing compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/34—Silicon-containing compounds
- C08K3/36—Silica
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/38—Boron-containing compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/09—Carboxylic acids; Metal salts thereof; Anhydrides thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/09—Carboxylic acids; Metal salts thereof; Anhydrides thereof
- C08K5/098—Metal salts of carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/13—Phenols; Phenolates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/36—Sulfur-, selenium-, or tellurium-containing compounds
- C08K5/37—Thiols
- C08K5/372—Sulfides, e.g. R-(S)x-R'
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L29/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an alcohol, ether, aldehydo, ketonic, acetal or ketal radical; Compositions of hydrolysed polymers of esters of unsaturated alcohols with saturated carboxylic acids; Compositions of derivatives of such polymers
- C08L29/02—Homopolymers or copolymers of unsaturated alcohols
- C08L29/04—Polyvinyl alcohol; Partially hydrolysed homopolymers or copolymers of esters of unsaturated alcohols with saturated carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L31/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an acyloxy radical of a saturated carboxylic acid, of carbonic acid or of a haloformic acid; Compositions of derivatives of such polymers
- C08L31/02—Homopolymers or copolymers of esters of monocarboxylic acids
- C08L31/04—Homopolymers or copolymers of vinyl acetate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L77/00—Compositions of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Compositions of derivatives of such polymers
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01G—HORTICULTURE; CULTIVATION OF VEGETABLES, FLOWERS, RICE, FRUIT, VINES, HOPS OR SEAWEED; FORESTRY; WATERING
- A01G13/00—Protecting plants
- A01G13/02—Protective coverings for plants; Coverings for the ground; Devices for laying-out or removing coverings
- A01G13/0256—Ground coverings
- A01G13/0268—Mats or sheets, e.g. nets or fabrics
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C51/00—Shaping by thermoforming, i.e. shaping sheets or sheet like preforms after heating, e.g. shaping sheets in matched moulds or by deep-drawing; Apparatus therefor
- B29C51/08—Deep drawing or matched-mould forming, i.e. using mechanical means only
- B29C51/082—Deep drawing or matched-mould forming, i.e. using mechanical means only by shaping between complementary mould parts
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B37/00—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding
- B32B37/14—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding characterised by the properties of the layers
- B32B37/24—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding characterised by the properties of the layers with at least one layer not being coherent before laminating, e.g. made up from granular material sprinkled onto a substrate
- B32B2037/246—Vapour deposition
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/30—Properties of the layers or laminate having particular thermal properties
- B32B2307/31—Heat sealable
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2810/00—Chemical modification of a polymer
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2323/00—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers
- C08J2323/26—Characterised by the use of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Derivatives of such polymers modified by chemical after-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2331/00—Characterised by the use of copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an acyloxy radical of a saturated carboxylic acid, or carbonic acid, or of a haloformic acid
- C08J2331/02—Characterised by the use of omopolymers or copolymers of esters of monocarboxylic acids
- C08J2331/04—Homopolymers or copolymers of vinyl acetate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/18—Oxygen-containing compounds, e.g. metal carbonyls
- C08K3/20—Oxides; Hydroxides
- C08K3/22—Oxides; Hydroxides of metals
- C08K2003/2217—Oxides; Hydroxides of metals of magnesium
- C08K2003/2224—Magnesium hydroxide
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/32—Phosphorus-containing compounds
- C08K2003/321—Phosphates
- C08K2003/324—Alkali metal phosphate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/38—Boron-containing compounds
- C08K2003/387—Borates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2201/00—Properties
- C08L2201/08—Stabilised against heat, light or radiation or oxydation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2201/00—Properties
- C08L2201/14—Gas barrier composition
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2203/00—Applications
- C08L2203/10—Applications used for bottles
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2203/00—Applications
- C08L2203/16—Applications used for films
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2203/00—Applications
- C08L2203/16—Applications used for films
- C08L2203/162—Applications used for films sealable films
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2203/00—Applications
- C08L2203/18—Applications used for pipes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/02—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group
- C08L2205/025—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group containing two or more polymers of the same hierarchy C08L, and differing only in parameters such as density, comonomer content, molecular weight, structure
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/03—Polymer mixtures characterised by other features containing three or more polymers in a blend
- C08L2205/035—Polymer mixtures characterised by other features containing three or more polymers in a blend containing four or more polymers in a blend
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2207/00—Properties characterising the ingredient of the composition
- C08L2207/04—Thermoplastic elastomer
-
- F—MECHANICAL ENGINEERING; LIGHTING; HEATING; WEAPONS; BLASTING
- F16—ENGINEERING ELEMENTS AND UNITS; GENERAL MEASURES FOR PRODUCING AND MAINTAINING EFFECTIVE FUNCTIONING OF MACHINES OR INSTALLATIONS; THERMAL INSULATION IN GENERAL
- F16L—PIPES; JOINTS OR FITTINGS FOR PIPES; SUPPORTS FOR PIPES, CABLES OR PROTECTIVE TUBING; MEANS FOR THERMAL INSULATION IN GENERAL
- F16L9/00—Rigid pipes
- F16L9/12—Rigid pipes of plastics with or without reinforcement
Definitions
- the present invention relates to a gas barrier resin composition, a method for producing a gas barrier resin composition, a molded product using the gas barrier resin composition, and the like.
- Gas barrier materials using resins with excellent ability to block gases such as oxygen are widely used in various applications such as containers, films, sheets, and pipes.
- resin having excellent gas barrier properties polyamide, polyester, polyvinylidene chloride, acrylonitrile copolymer, polyvinylidene fluoride, polychlorotrifluoroethylene, ethylene-vinyl ester copolymer sakenized product and the like are known.
- Patent Document 1 describes an invention of a multilayer plastic container having at least one gas barrier resin layer selected from polyamide, polyester, polyvinyl alcohol, ethylene-vinyl alcohol copolymer, fluorine-containing resin and silicone resin. ..
- Patent Document 2 states that conventional film materials such as biomass-derived polyolefins do not have sufficient qualities such as adhesion, processability, and durability, and a biomass-derived resin for improving such points. Described is an invention of a resin film comprising a biomass-derived resin layer having a specific composition including.
- Patent Document 3 states that a film in which a petroleum-derived resin is replaced with a biomass-derived resin may have reduced impact resistance and the like, and a biomass-derived biomass polyethylene for improving such a point.
- the present invention describes the invention of a laminated film having an intermediate layer containing polyethylene derived from fossil fuel and a propylene-based block copolymer resin.
- gas barrier resins synthesized using biomass-derived raw materials will be commercialized.
- the performance of the biomass-derived synthetic resin may be inferior to that of the fossil fuel-derived synthetic resin. Therefore, when the conventional fossil fuel-derived gas barrier resin is replaced with the biomass-derived gas barrier resin, There is concern that the most important gas barrier property will be reduced. Therefore, it is desired to develop a biomass-derived resin having an excellent gas barrier property comparable to that of a fossil fuel-derived resin.
- the gas barrier resin may be molded into various shapes such as containers and sheets by melt molding. For this reason, it is also important for the gas barrier resin to have a long-running property (long-term operation characteristic) in which defects and the like are unlikely to occur even if melt molding is continuously performed for a long period of time.
- the present invention has been made based on the above circumstances, and an object thereof is to have a high gas barrier property comparable to that derived from fossil fuel and a sufficient long-run property while using a raw material derived from biomass. It is an object of the present invention to provide a gas barrier resin composition having a gas barrier resin composition, a molded product using the gas barrier resin composition, and a method for producing such a gas barrier resin composition.
- the present inventor has synthesized an ethylene-vinyl ester copolymer saken product, which is a kind of gas barrier resin, using a raw material derived from biomass as a monomer and a raw material derived from fossil fuel as a monomer. It was found that it has a high gas barrier property comparable to that of the conventional one having the same structure. On the other hand, regarding the long-run property, the ethylene-vinyl ester copolymer saken product synthesized by using the raw material derived from biomass as a monomer is inferior to the one synthesized by using the raw material derived from fossil fuel as a monomer, which is a gas barrier property. It was also found that it tends to be different from.
- an ethylene-vinyl ester copolymer saken product in which a raw material derived from biomass and a raw material derived from fossil fuel are used in combination is derived from fossil fuel while reducing the environmental load.
- the present invention has been completed by finding that it can exhibit a high gas barrier property similar to that using only the raw materials of the above and also has a sufficient long-running property.
- a part of ethylene and vinyl ester which contains one or more kinds of ethylene-vinyl ester copolymer kendies and is a raw material of the above one or more kinds of ethylene-vinyl ester copolymer kendies, is biomass.
- Gas barrier resin composition of origin the balance of which is derived from fossil fuels;
- the above-mentioned one or more kinds of ethylene-vinyl ester copolymer saken products are the same as the ethylene-vinyl ester copolymer saken product (A) in which at least a part of the raw materials ethylene and vinyl ester is derived from biomass.
- X represents a hydrogen atom, a group represented by a methyl group or R 2 -OH.
- R 1 and R 2 independently represent a single bond, an alkylene group having 1 to 9 carbon atoms or an alkylene oxy group having 1 to 9 carbon atoms, and the alkylene group and the alkylene oxy group are hydroxyl groups, alkoxy groups or It may contain a halogen atom.
- the above-mentioned one or more kinds of ethylene-vinyl ester copolymer kenides are more than the above-mentioned ethylene-vinyl ester copolymer kendies (X) and the above-mentioned ethylene-vinyl ester copolymer kenides (X).
- thermoplastic elastomer (G) Further containing the thermoplastic elastomer (G), the mass ratio of the thermoplastic elastomer (G) to the above-mentioned one or more kinds of ethylene-vinyl ester copolymer saken product is 5/95 or more and 35/65 or less.
- gas barrier resin composition [1]-[19] gas barrier resin composition; [21] Pellets of ethylene-vinyl ester copolymer kenide (A) in which at least a part of ethylene and vinyl ester as raw materials are derived from biomass, and ethylene-vinyl ester copolymer kenide (A) derived from fossil fuel.
- a method for producing a gas barrier resin composition which comprises a step of dry blending with pellets and melt-kneading.
- At least one metal atom (F) selected from the group consisting of pellets of the above-mentioned one or more kinds of ethylene-vinyl ester copolymer saken compounds, pellets of polyamide (E), and magnesium, calcium and zinc.
- a method for producing a gas barrier resin composition comprising a step of dry-blending the pellet of B) and the pellet of the thermoplastic elastomer (G) and melt-kneading the pellet; [24] A molded product comprising a gas barrier layer formed from the gas barrier resin composition according to any one of [1] to [20]; [25] A film or sheet comprising the molded article of [24]; [26] An industrial film or sheet comprising the molded article of [24]; [27] A thermoformed container comprising the molded body of [24]; A blow-molded container comprising the molded article of [28] and [24]; [29] A fuel container comprising the blow-molded container of [28]; [30] A bottle container comprising the blow molded container of [28]; A tube comprising the moldings of [31] and [24]; [32] A pipe comprising the molded body of [24]; A paper container comprising the molded product of [33] and [24]; [34] A single-layer film
- a vapor-deposited film comprising at least one inorganic vapor-deposited layer laminated on the exposed surface of the layer formed from the resin composition; [37] A multilayer structure comprising the thin-film film of [36] and another layer laminated on the inorganic thin-film layer in the thin-film film. [38] A heat-sealing film comprising a single-layer film of [34], a multilayer film of [35], a vapor-deposited film of [36], or a multilayer structure of [37]; [39] A packaging material comprising at least one layer made of the gas barrier resin composition according to any one of [1] to [20]. [40] A medium for plant cultivation containing a molded product formed from the resin composition, wherein the resin composition is the gas barrier resin composition according to any one of [1] to [20], for plant cultivation. Culture medium; Is achieved by providing.
- a gas barrier resin composition having a high gas barrier property comparable to that derived from fossil fuel and a sufficient long-run property while using a raw material derived from biomass, and a molded product using this gas barrier resin composition. Etc., and a method for producing such a gas barrier resin composition can be provided.
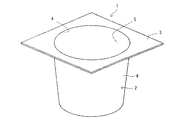
- FIG. 1 is a schematic perspective view showing a cup-shaped container according to an embodiment of the present invention.
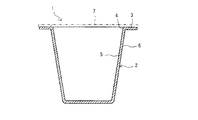
- FIG. 2 is a cross-sectional view of the cup-shaped container of FIG.
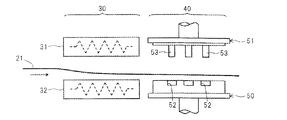
- FIG. 3 is a schematic diagram for explaining a method for manufacturing the cup-shaped container of FIG. 4 (A) to 4 (D) are schematic views for explaining a method for manufacturing the cup-shaped container of FIG. 1.
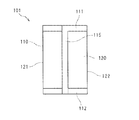
- FIG. 5 is a rear view showing a vertical bag filling seal bag according to an embodiment of the present invention.
- FIG. 6 is a schematic view showing a plant cultivation apparatus according to an embodiment of the present invention.
- FIG. 7 is a graph showing the results of hot tack strength measurement in the examples.
- the gas barrier resin composition of the present invention contains one or more kinds of ethylene-vinyl ester copolymer saken compounds (ethylene-vinyl alcohol copolymer; hereinafter also referred to as "EVOH”), and one or more of the above.
- EVOH ethylene-vinyl alcohol copolymer
- the gas barrier resin composition has a low environmental load because a raw material derived from biomass is partially used.
- EVOH is selected and used as the gas barrier resin, and even when EVOH is synthesized using a raw material derived from biomass, it is the same as that synthesized only from the raw material derived from fossil fuel. It can exhibit the same high gas barrier property as EVOH of the structure.
- EVOH having the same structure means EVOH having the same degree of polymerization, content ratio of each structural unit, presence / absence of denaturation, degree of saponification, and the like.
- EVOH is also used with a raw material derived from fossil fuel, so that the long-run property is sufficient.
- the long-run property in the present specification can be evaluated by the method described in Examples, and can be comprehensively evaluated by the degree of defects, streaks, and coloring when the gas barrier resin composition is continuously formed into a film.
- coloring may not be a particular problem. Therefore, it is evaluated that the long-running property is sufficient if there are few defects and streaks when the film is continuously formed for a predetermined time (for example, 10 hours).
- the biobase degree is an index showing the ratio of biomass-derived raw materials, and in the present specification, it is the biobase carbon content obtained by measuring the concentration of radioactive carbon (14 C) by an accelerator mass spectrometer (AMS).
- the degree of biobase can be specifically measured according to the method described in ASTM D6866-18. That is, when the biobase degree of EVOH is usually more than 0% and less than 100%, it can be said that the ethylene and vinyl esters used as raw materials contain both biomass-derived and fossil fuel-derived.
- Biomass is a resource that is an organic substance derived from animals and plants, excluding fossil fuels (fossil resources). Biomass may be a resource that is an organic matter derived from plants.
- the gas barrier resin composition of the present invention can also be used to track its own products by utilizing the concentration of radiocarbon (14 C).
- Organisms take up and contain a certain amount of radiocarbon (14 C) in the atmosphere during their activity, but when the activity is stopped, the uptake of new 14 C stops and the ratio of 14 C to total carbon decreases.
- isotope sorting occurs when plants fix carbon, and the ratio of 14 C to total carbon differs depending on the plant species. It is known that the ratio of 14 C to total carbon differs depending on the place of origin and age, and a raw material having a ratio of 14 C to total carbon can be obtained depending on the biomass used as a raw material.
- EVOH is used in a wide range of applications, and it is the supplier's responsibility to supply high-quality products to the market. There is also a need for a way to distinguish between our own products and those of other companies for branding.
- EVOH used in the gas barrier layer of a commercially available packaging container is formed into a packaging container by thermoforming, but the ethylene-vinyl ester copolymer saken product is a gel insoluble in a solvent due to the thermal history received during thermoforming. May form. Therefore, even if the packaging container is collected, the EVOH used is extracted with a solvent, and the molecular weight is to be measured, it is often difficult to accurately measure the molecular weight. Therefore, it is not possible to determine whether or not it is EVOH of the company only by analyzing the molded product.
- EVOH is used in films, sheets, containers, etc. as a packaging material for foods, pharmaceuticals, industrial chemicals, pesticides, etc. through many distribution channels. It is also used in fuel tanks for automobiles and other vehicles, tube materials for tires, agricultural films, geomembranes, cushioning materials for shoes, etc., taking advantage of its barrier properties, heat retention properties, stain resistance, and the like. When these materials in which EVOH is used are further discarded, it is difficult to determine from which factory and which production line the such resin and the packaging container after use thereof are manufactured. In addition, it is difficult to conduct a quality survey of the company's products during or after use, and to track the environmental impact after disposal and the degradability into the ground.
- a method of adding a tracer substance to EVOH can be considered.
- the addition of a tracer may cause an increase in cost and a decrease in EVOH performance.
- being able to track in-house products using the concentration of radiocarbon (14 C) can be said to be a very useful effect.
- the gas barrier resin composition of the present invention is a resin composition having a function of suppressing gas permeation.
- the upper limit of the oxygen permeation rate of the gas barrier resin composition of the present invention measured according to the method described in JIS K 7126-2 (isopressure method; 2006) under 20 ° C.-65% RH conditions is 100 mL / 20 ⁇ m /.
- (m 2 ⁇ day ⁇ atm) are preferred, more preferably 50mL ⁇ 20 ⁇ m / (m 2 ⁇ day ⁇ atm), 10mL ⁇ 20 ⁇ m / (m 2 ⁇ day ⁇ atm), 1mL ⁇ 20 ⁇ m / (m 2 ⁇ day ⁇ atm), or 0.5mL ⁇ 20 ⁇ m / (m 2 ⁇ day ⁇ atm) it is more preferred.
- EVOH examples of the form of one or more kinds of EVOH contained in the gas barrier resin composition of the present invention include the following forms (I) and (II).
- (I) One of the forms (II) raw materials of ethylene and vinyl esters containing EVOH (A) in which at least a part of ethylene and vinyl esters as raw materials is derived from biomass and EVOH (B) derived from fossil fuels.
- EVOH may be one in which a part of ethylene unit, vinyl alcohol unit and vinyl ester unit is derived from biomass, and the rest is derived from fossil fuel. That is, EVOH (A) may be EVOH in which at least a part of ethylene unit, vinyl alcohol unit and vinyl ester unit is derived from biomass.
- the EVOH (B) may be an EVOH derived from fossil fuels in all of ethylene units, vinyl alcohol units and vinyl ester units.
- EVOH (A') a part of ethylene unit, vinyl alcohol unit and vinyl ester unit may be derived from biomass, and the balance may be EVOH derived from fossil fuel.
- EVOH (A) is EVOH in which at least a part of ethylene and vinyl esters, which are raw material monomers, is derived from biomass. Since EVOH (A) contains a raw material derived from biomass, the degree of biobase of the gas barrier resin composition of the present invention can be increased and the environmental load can be reduced.
- EVOH (A) is obtained by saponification of a copolymer of ethylene and vinyl ester, which is at least partially derived from biomass.
- the production and saponification of the ethylene-vinyl ester copolymer as a precursor of EVOH (A) shall be carried out by the same known method as the production and saponification of the ethylene-vinyl ester copolymer derived from the conventional fossil fuel.
- the vinyl ester for example, a carboxylic acid vinyl ester such as vinyl acetate, vinyl formate, vinyl propionate, vinyl valerate, vinyl caprate, vinyl laurate, vinyl stearate, vinyl pivalate, vinyl versatic acid can be used. It can be made, and vinyl acetate is preferable.
- Biomass-derived ethylene can be produced by a known method, for example, by purifying bioethanol from a biomass raw material and performing a dehydration reaction.
- biomass raw material waste type, unused type, resource crop type and the like can be used, for example, cellulose type crops (pulp, kenaf, straw, rice straw, used paper, papermaking residue, etc.), wood, charcoal, compost.
- Natural rubber, cotton, sugar cane, okara, fats and oils (rapeseed oil, cottonseed oil, soybean oil, coconut oil, castor oil, etc.), carbohydrate-based crops (corn, potatoes, wheat, rice, rice husks, rice bran, old rice, cassaba, sago palm) , Bagasse, buckwheat, soybean, essential oil (pine root oil, orange oil, eucalyptus oil, etc.), pulp black liquor, vegetable oil residue, etc. can be used.
- the method for producing bioethanol is not particularly limited, and for example, the biomass raw material is pretreated (pressurized hot water treatment, acid treatment, alkali treatment, saccharification treatment using a saccharifying enzyme) as necessary, and then yeast fermentation.
- the bioethanol can be purified through a distillation step and a dehydration step.
- sequential saccharification fermentation in which saccharification and fermentation are carried out in stages may be used, or parallel saccharification fermentation in which saccharification and fermentation are carried out at the same time may be used, but the production efficiency is improved. From the viewpoint, it is preferable to produce bioethanol by parallel saccharification fermentation.
- biomass-derived ethylene may be used, for example, Braskem S. A. Bioethylene derived from sugar cane can be used.
- biomass-derived vinyl ester examples include vinyl esters produced using biomass-derived ethylene.
- methods for producing vinyl ester derived from biomass include a method of reacting ethylene with acetic acid and oxygen molecules using a palladium catalyst, which is a general industrial production method.
- the biomass-derived vinyl ester may be a vinyl ester produced by using a biomass-derived carboxylic acid. If the saponification degree of EVOH (A) is not 100 mol%, the acyl group derived from biomass will remain.
- the lower limit of the ethylene unit content of EVOH (A) is preferably 20 mol%, more preferably 23 mol%, still more preferably 25 mol%. When the ethylene unit content of EVOH (A) is 20 mol% or more, the long-run property tends to increase.
- the upper limit of the ethylene unit content of EVOH (A) is preferably 60 mol%, more preferably 55 mol%, still more preferably 50 mol%. When the ethylene unit content of EVOH (A) is 60 mol% or less, the gas barrier property tends to be better.
- the ethylene unit content of EVOH can be determined by a nuclear magnetic resonance (NMR) method.
- the lower limit of the saponification degree of EVOH (A) is preferably 90 mol%, more preferably 95 mol%, still more preferably 99 mol%.
- the saponification degree of EVOH (A) is 90 mol% or more, the gas barrier property and the long-run property of the gas barrier resin composition of the present invention tend to be better.
- the upper limit of the saponification degree of EVOH (A) may be 100 mol%, and may be 99.97 mol% or 99.94 mol%.
- the degree of saponification of EVOH can be calculated by performing 1 H-NMR measurement and measuring the peak area of hydrogen atoms contained in the vinyl ester structure and the peak area of hydrogen atoms contained in the vinyl alcohol structure.
- At least a part of ethylene and vinyl ester used as a raw material of EVOH (A) is derived from biomass, at least a part of vinyl ester is derived from biomass, and all of vinyl ester is derived from biomass. It is also good. Further, it is preferable that at least a part of ethylene, which is a raw material of EVOH (A), is derived from biomass, and all of ethylene may be derived from biomass. Further, it may be preferable that at least a part of the vinyl ester as a raw material of EVOH (A) and at least a part of ethylene are derived from biomass, and all of the vinyl ester and ethylene may be derived from biomass.
- the lower limit of the ratio of the vinyl alcohol unit derived from biomass in the total vinyl alcohol units (condensate of vinyl ester units) constituting EVOH (A) is preferably 1 mol%, more preferably 5 mol%, and further 10 mol%. Preferably, 25 mol% or 45 mol% is even more preferable, 70 mol%, 90 mol% or 99 mol% may be used, and all vinyl alcohol units constituting EVOH (A) may be derived from biomass. good.
- the upper limit of the proportion of vinyl alcohol units derived from fossil fuels in the total vinyl alcohol units constituting EVOH (A) is preferably 99 mol%, more preferably 95 mol%, still more preferably 90 mol%, or 75 mol% or 55.
- mol% is more preferably 30 mol%, 10 mol% or 1 mol%, and the total vinyl alcohol units constituting EVOH (A) do not contain vinyl alcohol units derived from fossil fuels. May be good.
- the proportion of biomass-derived vinyl alcohol units in the total vinyl alcohol units constituting EVOH (A) is high, the degree of biobase in the gas barrier resin composition of the present invention is increased, and the environmental load tends to be reduced.
- the proportion of vinyl alcohol unit derived from biomass is preferably 5 mol% or more and 95 mol% or less, and 15 mol% or more and 85 mol%. The following is more preferable, 25 mol% or more and 75 mol% or less are further preferable, and 35 mol% or more and 65 mol% or less are particularly preferable.
- the lower limit of the proportion of ethylene units derived from biomass in all ethylene units constituting EVOH (A) is preferably 1 mol%, more preferably 5 mol%, further preferably 10 mol%, and 25 mol% or 45 mol%. Even more preferably, it may be 70 mol%, 90 mol% or 99 mol%, and all ethylene units constituting EVOH (A) may be derived from biomass.
- the upper limit of the proportion of ethylene units derived from fossil fuels in all ethylene units constituting EVOH (A) is preferably 99 mol%, more preferably 95 mol%, further preferably 90 mol%, and 75 mol% or 55 mol%.
- the proportion of biomass-derived ethylene units in all ethylene units constituting EVOH (A) is high, the degree of biobase in the gas barrier resin composition of the present invention is increased, and the environmental load tends to be reduced.
- the proportion of ethylene units derived from biomass is preferably 5 mol% or more and 95 mol% or less, and 15 mol% or more and 85 mol% or less. Is more preferable, 25 mol% or more and 75 mol% or less is further preferable, and 35 mol% or more and 65 mol% or less is particularly preferable.
- the lower limit of the biobase degree of EVOH (A) is preferably 1%, more preferably 5%, further preferably 20%, and particularly preferably 40% from the viewpoint of reducing the environmental load of the gas barrier resin composition of the present invention. Further, the lower limit of the biobase degree of EVOH (A) may be 60%, 80% or 95%.
- the upper limit of the biobase degree of EVOH (A) may be 100%, but 99% is preferable, 95% is more preferable, and 85%, 75% or 65% is preferable from the viewpoint of good long-running property. May be even more preferred.
- EVOH (A) may have a unit derived from a monomer other than ethylene, vinyl ester and a saponified product thereof, as long as the object of the present invention is not impaired.
- the upper limit of the content of the unit derived from the other monomer to all structural units of EVOH (A) is preferably 30 mol%, preferably 20 mol. % Is more preferred, 10 mol% is even more preferred, 5 mol% is even more preferred, and 1 mol% is even more preferred.
- the lower limit of its content may be 0.05 mol% or 0.10 mol%.
- the other monomers include, for example, alkenes such as propylene, butylene, pentene, and hexene; 3-allyloxy-1-propene, 3-acyloxy-1-butene, 4-acyloxy-1-butene, 3,4-diasiloxy.
- -1-Buten 3-Aryloxy-4-methyl-1-butene, 4-Achilloxy-2-methyl-1-butene, 4-Aryloxy-3-methyl-1-butene, 3,4-diasiloxy-2-methyl -1-Buten, 4-Acyloxy-1-Penten, 5-Acyloxy-1-Penten, 4,5-Diacyloxy-1-Penten, 4-Acyloxy-1-hexene, 5-Acyloxy-1-Hexene, 6-Acyloxy Alkenes having ester groups such as -1-hexene, 5,6-diasiloxy-1-hexene, 1,3-diacetoxy-2-methylenepropane, or alkenes thereof; Unsaturated acid or its anhydride, salt, mono or dialkyl ester, etc .; nitriles such as acrylonitrile and methacrylonitrile; amides such as acrylamide and methacrylamide; olefin
- vinylsilane compounds such as vinyltrimethoxysilane, vinyltriethoxysilane, vinyltri ( ⁇ -methoxy-ethoxy) silane, ⁇ -methacryloxypropylmethoxysilane; alkyl vinyl ethers, vinyl ketones, N-vinylpyrrolidone, vinyl chloride, Examples include vinylidene chloride.
- EVOH (A) may be post-denatured by methods such as urethanization, acetalization, cyanoethylation, and oxyalkyleneization.
- EVOH (A) has a modifying group
- EVOH (A) has a structural unit (modifying group) represented by the following formula (I).
- X represents a hydrogen atom, a group represented by a methyl group or R 2 -OH.
- R 1 and R 2 each independently represent a single bond, an alkylene group having 1 to 9 carbon atoms or an alkylene oxy group having 1 to 9 carbon atoms, and the alkylene group and the alkylene oxy group are hydroxyl groups, alkoxy groups or halogen atoms. May include. ]
- X is preferably a group represented by hydrogen or R 2 -OH, more preferably a group represented by R 2 -OH.
- the alkylene group and alkyleneoxy group used as R 1 or R 2 may contain a hydroxyl group, an alkoxy group or a halogen atom.
- R 1 and R 2 are preferably an alkylene group or an alkyleneoxy group having 1 to 5 carbon atoms, and more preferably an alkylene group or an alkyleneoxy group having 1 to 3 carbon atoms.
- structural unit (modifying group) represented by the formula (I) include the following structural units (modifying group) represented by the formulas (II), (III), and (IV). Among them, the structural unit (modifying group) represented by the formula (II) is preferable.
- R 3 and R 4 each independently represent a hydrogen atom or an alkyl group having 1 to 8 carbon atoms, and the alkyl group may contain a hydroxyl group, an alkoxy group or a halogen atom. ]
- R 5 is synonymous with X in formula (I).
- R 6 represents a hydrogen atom or an alkyl group having 1 to 8 carbon atoms, and the alkyl group may contain a hydroxyl group, an alkoxy group or a halogen atom.
- R 7 and R 8 independently represent a hydrogen atom, an alkyl group having 1 to 8 carbon atoms, a cycloalkyl group having 3 to 8 carbon atoms, or a hydroxyl group. Further, a part or all of the alkyl group and the hydrogen atom of the cycloalkyl group may be substituted with a hydroxyl group, an alkoxy group or a halogen atom.
- R 1 in the formula (I) is a single bond and X is a hydroxymethyl group (R 3 and R 4 in the formula (II) are hydrogen atoms).
- EVOH (A) having this structural unit (modifying group) secondary processability such as stretchability and thermoformability tends to be improved without significantly deteriorating the gas barrier property.
- the lower limit of the content is preferably 0.1 mol%, more preferably 0.4 mol%, still more preferably 1.0 mol%.
- the upper limit of the content of the structural unit (modifying group) is preferably 20 mol%, more preferably 10 mol%, further preferably 8 mol%, and particularly preferably 5 mol% from the viewpoint of improving the gas barrier property. ..
- R 1 in the formula (I) is a hydroxymethylene group and X is a hydrogen atom (R 5 and R 6 in the formula (III) are hydrogen atoms).
- EVOH (A) having this structural unit (modifying group) secondary processability such as stretchability and thermoformability tends to be improved without significantly deteriorating the gas barrier property.
- the lower limit of the content is preferably 0.1 mol%, more preferably 0.4 mol%, still more preferably 1.0 mol%.
- the upper limit of the content of the structural unit (modifying group) is preferably 20 mol%, more preferably 10 mol%, further preferably 8 mol%, and particularly preferably 5 mol% from the viewpoint of improving the gas barrier property. ..
- R 1 in the formula (I) is a methylmethyleneoxy group and X is a hydrogen atom.
- EVOH (A) having this structural unit (modifying group) secondary processability such as stretchability and thermoformability tends to be improved without significantly deteriorating the gas barrier property.
- an oxygen atom is bonded to a carbon atom in the main chain. That is, in the formula (IV), it is preferable that one of R 7 and R 8 is a methyl group and the other is a hydrogen atom.
- the lower limit of the content is preferably 0.1 mol%, more preferably 0.5 mol%, still more preferably 1.0 mol%, and 2 9.0 mol% is particularly preferred.
- the upper limit of the content of the structural unit (modifying group) is preferably 20 mol%, more preferably 15 mol%, still more preferably 10 mol% from the viewpoint of improving the gas barrier property.
- EVOH (A) may be used alone or in combination of two or more.
- the lower limit of the melt flow rate (MFR) at 190 ° C. and 2160 g load of EVOH (A) measured in accordance with JIS K7210: 1999 is preferably 0.1 g / 10 minutes, more preferably 0.5 g / 10 minutes. , 1.0 g / 10 minutes is more preferred.
- the upper limit of MFR of EVOH (A) is preferably 30 g / 10 minutes, more preferably 20 g / 10 minutes, and even more preferably 15 g / 10 minutes.
- the lower limit of the melting point of EVOH (A) is preferably 135 ° C, more preferably 150 ° C, and even more preferably 155 ° C. When the melting point of EVOH (A) is 135 ° C. or higher, the gas barrier property tends to be excellent.
- the upper limit of the melting point of EVOH (A) is preferably 200 ° C, more preferably 190 ° C, and even more preferably 185 ° C. When the melting point of EVOH (A) is 200 ° C. or lower, the melt moldability is good, and as a result, the long-run property tends to be enhanced.
- EVOH (B) is an EVOH derived from fossil fuels.
- the EVOH derived from fossil fuel means EVOH synthesized by using a raw material derived from fossil fuel. That is, the EVOH (B) is an EVOH in which all of the raw material monomers, ethylene, vinyl ester, and other monomers used as needed, are derived from fossil fuels. In other words, EVOH (B) is EVOH synthesized without using a biomass-derived raw material.
- the gas barrier resin composition of the present invention contains EVOH (B), sufficient long-running property can be exhibited.
- EVOH (B) is the same as EVOH (A) except that the raw material is derived from fossil fuel, that is, the biobase degree is 0%. That is, the specific and suitable ethylene unit content, saponification degree, type and content derived from other monomers, MFR, melting point, etc. of EVOH (B) are the same as those of EVOH (A).
- EVOH (B) may be used alone or in combination of two or more.
- the lower limit of the mass ratio (A / B) of EVOH (A) and EVOH (B) is preferably 1/99. / 95 is more preferred, 15/85 is even more preferred, and 40/60 is particularly preferred.
- the mass ratio (A / B) may be preferably 60/40 or 75/25.
- the upper limit of the mass ratio (A / B) is preferably 99/1, more preferably 95/5, further preferably 85/15, and even more preferably 60/40 or 40/60.
- the ethylene unit content, saponification degree, MFR, melting point, etc. of EVOH (A) and EVOH (B) may be the same or different. From the viewpoint of enhancing performance such as long-running property, it is preferable that the ethylene unit content, saponification degree, MFR and melting point of EVOH (A) and EVOH (B) are the same or close to each other.
- the difference in ethylene unit content between EVOH (A) and EVOH (B) is preferably 6 mol% or less, more preferably 3 mol% or less, still more preferably 0 mol%.
- the difference in the degree of saponification between EVOH (A) and EVOH (B) is preferably 2 mol% or less, more preferably 1 mol% or less, still more preferably 0 mol%.
- the difference in MFR between EVOH (A) and EVOH (B) is preferably 2 g / 10 minutes or less, more preferably 1 g / 10 minutes or less, and even more preferably 0 g / 10 minutes.
- the difference in melting point between EVOH (A) and EVOH (B) is preferably 7 ° C. or lower, more preferably 3 ° C. or lower, and even more preferably 0 ° C.
- EVOH (A') In EVOH (A'), a part of ethylene and vinyl ester which are raw material monomers is derived from biomass, and the balance of ethylene and vinyl ester which is the raw material monomer is derived from fossil fuel.
- the degree of biobase in the gas barrier resin composition of the present invention tends to be increased and the environmental load can be reduced. Further, since the raw material of EVOH (A') contains a raw material derived from fossil fuel, the long-running property of the gas barrier resin composition of the present invention can be improved.
- EVOH (A') is obtained by saponification of a copolymer of ethylene and vinyl ester, part of which is derived from biomass and the rest of which is derived from fossil fuel.
- the ethylene-vinyl ester copolymer, which is a precursor of EVOH (A') can be produced and saponified by a conventionally known method except that a biomass-derived polymer is used as a part of the raw material.
- biomass-derived ethylene and biomass-derived vinyl ester used for the synthesis of EVOH (A') are the same as those described above as those used for the synthesis of EVOH (A).
- ethylene and vinyl esters used as raw materials for EVOH (A') it is preferable that at least a part of ethylene is derived from biomass, and all of ethylene may be derived from biomass. Further, it is preferable that at least a part of the vinyl ester which is a raw material of EVOH (A') is derived from biomass, and the whole vinyl ester may be derived from biomass.
- the lower limit of the ratio of the vinyl alcohol unit derived from biomass in all the vinyl alcohol units (condensate of vinyl ester unit) constituting EVOH (A') is preferably 1 mol%, more preferably 5 mol%, and 10 mol%. More preferably, 25 mol% or 45 mol% is even more preferred, and it may be 70 mol%, 90 mol% or 99 mol%, and all vinyl alcohol units constituting EVOH (A') are derived from biomass. May be good.
- the upper limit of the ratio of the vinyl alcohol unit derived from fossil fuel to the total vinyl alcohol unit constituting EVOH (A') is preferably 99 mol%, more preferably 95 mol%, further preferably 90 mol%, or 75 mol% or 55 mol% is more preferable, and it may be 30 mol%, 10 mol% or 1 mol%, and the total vinyl alcohol units constituting EVOH (A') do not contain vinyl alcohol units derived from fossil fuels. May be.
- the proportion of biomass-derived vinyl alcohol units in all vinyl alcohol units constituting EVOH (A') is high, the degree of biobase in the gas barrier resin composition of the present invention is increased, and the environmental load tends to be reduced.
- the proportion of vinyl alcohol unit derived from biomass is preferably 5 mol% or more and 95 mol% or less, and 15 mol% or more and 85 mol%.
- the following is more preferable, 25 mol% or more and 75 mol% or less are further preferable, and 35 mol% or more and 65 mol% or less are particularly preferable.
- the lower limit of the proportion of biomass-derived ethylene in all ethylene units constituting EVOH (A') is preferably 1 mol%, more preferably 5 mol%, still more preferably 10 mol%, and 25 mol% or 45 mol%. More preferably, it may be 70 mol%, 90 mol% or 99 mol%, and all ethylene units constituting EVOH (A') may be derived from biomass.
- the upper limit of the proportion of ethylene units derived from fossil fuels in all ethylene units constituting EVOH (A') is preferably 99 mol%, more preferably 95 mol%, further preferably 90 mol%, and 75 mol% or 55 mol.
- the proportion of biomass-derived ethylene units in all ethylene units constituting EVOH (A') is high, the degree of biobase in the gas barrier resin composition of the present invention is increased, and the environmental load tends to be reduced.
- the proportion of ethylene units derived from biomass is preferably 5 mol% or more and 95 mol% or less, and 15 mol% or more and 85 mol% or less. Is more preferable, 25 mol% or more and 75 mol% or less is further preferable, and 35 mol% or more and 65 mol% or less is particularly preferable.
- the lower limit of the biobase degree of EVOH (A') is preferably 1%, more preferably 5%, further preferably 20%, and particularly preferably 40% from the viewpoint of reducing the environmental load of the gas barrier resin composition of the present invention. .. Further, for example, in applications where particularly excellent long-running property is not required, the lower limit of the biobase degree of EVOH (A') may be 60% or 80%.
- the upper limit of the biobase degree of EVOH (A') is preferably 99%, more preferably 95%, and even more preferably 85%, 75% or 65% from the viewpoint of improving long-running property.
- EVOH (A') may have a unit derived from a monomer other than ethylene, vinyl ester and a saponified product thereof, or may be post-denatured, as long as the object of the present invention is not impaired. .. Specific examples of the copolymerization component and post-modification are the same as those of EVOH (A) described above.
- EVOH (A') may be used alone or in combination of two or more.
- melt flow rate (MFR) and melting point of EVOH (A') at 190 ° C. and 2160 g load measured in accordance with JIS K7210: 1999 are the same as those of EVOH (A) described above.
- EVOH (X) and EVOH (Y) One or more types of EVOH preferably contain EVOH (X) and EVOH (Y) having a melting point lower than that of EVOH (X). In other words, it is preferable that one or more kinds of EVOH can be classified into at least two of EVOH (X) and EVOH (Y).
- the gas barrier resin composition of the present invention contains two types of EVOH (EVOH (X) and EVOH (Y)) having different melting points, it tends to be able to maintain good gas barrier properties and moldability.
- Such a gas barrier resin composition is particularly suitable as a material for a multilayer sheet, a packaging material obtained by molding a multilayer sheet by a heat stretching molding method, a container formed by molding a multilayer sheet by a vacuum compressed air molding method, and the like.
- EVOH (X) is composed of any one or more of EVOH (A), EVOH (B) and EVOH (A').
- EVOH (Y) is composed of any one or more of EVOH (A), EVOH (B) and EVOH (A'). The same applies to EVOH (Z) described later.
- EVOH (X) and EVOH (Y) and EVOH (A), EVOH (B) and EVOH (A') is as follows.
- EVOH (X) is an EVOH having a melting point higher than that of EVOH (Y), and is usually an EVOH having the highest melting point among the EVOH contained in the gas barrier resin composition of the present invention.
- the lower limit of the melting point of EVOH (X) is preferably 150 ° C., more preferably 155 ° C., and even more preferably 160 ° C.
- the upper limit of the melting point of EVOH (X) is preferably 200 ° C. When the melting point of EVOH (X) is within the above range, the gas barrier property of the gas barrier resin composition tends to be good.
- the lower limit of the ethylene unit content of EVOH (X) is preferably 20 mol%, more preferably 22 mol%, still more preferably 24 mol%, from the viewpoint of good moldability and long-running property.
- the upper limit of the ethylene unit content of EVOH (X) is preferably 50 mol%, more preferably 48 mol%, still more preferably 46 mol%, from the viewpoint of raising the melting point and improving the gas barrier property.
- the lower limit of the saponification degree of EVOH (X) is preferably 90 mol%, more preferably 95 mol%, still more preferably 99 mol%.
- the saponification degree of EVOH (X) is 90 mol% or more, the gas barrier property and the long-run property of the gas barrier resin composition of the present invention tend to be better.
- the upper limit of the saponification degree of EVOH (X) may be 100 mol%, and may be 99.97 mol% or 99.94 mol%.
- EVOH (X) may have a monomer unit other than ethylene, vinyl ester and a saponified product thereof as long as the object of the present invention is not impaired, but the gas barrier of the gas barrier resin composition of the present invention. From the viewpoint of maintaining high properties, it is preferable not to have other monomer units. Examples of the monomer giving the other monomer unit include the monomers exemplified in the description of EVOH (A). When EVOH (X) has the above other monomer units, the content of EVOH (X) with respect to all structural units is preferably 5 mol% or less, more preferably 3 mol% or less, and further preferably 1 mol% or less. preferable.
- the lower limit of the melt flow rate (MFR) at 190 ° C. and 2160 g load of EVOH (X) measured in accordance with JIS K7210: 1999 is preferably 0.1 g / 10 minutes, more preferably 0.5 g / 10 minutes. , 1.0 g / 10 minutes is more preferred.
- the upper limit of MFR of EVOH (X) is preferably 30 g / 10 minutes, more preferably 20 g / 10 minutes, and even more preferably 15 g / 10 minutes.
- EVOH (Y) is an EVOH having a melting point lower than that of EVOH (X).
- the gas barrier resin composition of the present invention contains EVOH (Y), it tends to exhibit excellent moldability.
- the lower limit of the melting point of EVOH (Y) is preferably 100 ° C, more preferably 105 ° C, and even more preferably 110 ° C.
- the upper limit of the melting point of EVOH (Y) is preferably 180 ° C. When the melting point of EVOH (Y) is within the above range, the gas barrier property of the gas barrier resin composition tends to be good.
- the lower limit of the ethylene unit content of EVOH (Y) is preferably 30 mol%, more preferably 32 mol%, still more preferably 34 mol%, from the viewpoint of lowering the melting point and improving the moldability and long-running property.
- the upper limit of the ethylene unit content of EVOH (Y) is preferably 60 mol%, more preferably 58 mol%, and even more preferably 56 mol% from the viewpoint of improving the gas barrier property.
- the lower limit of the saponification degree of EVOH (Y) is preferably 90 mol%, more preferably 95 mol%, still more preferably 99 mol%.
- the saponification degree of EVOH (Y) is 90 mol% or more, the gas barrier property and the long-run property of the gas barrier resin composition of the present invention tend to be better.
- the upper limit of the saponification degree of EVOH (Y) may be 100 mol%, and may be 99.97 mol% or 99.94 mol%.
- the lower limit of the saponification degree of EVOH (Y) may be 70 mol% or 80 mol% from the viewpoint of improving the moldability, and the upper limit of the saponification degree of EVOH (Y) is the molding. From the viewpoint of enhancing the sex, it may be 98 mol%.
- EVOH (Y) may have a monomer unit other than ethylene, vinyl ester and a saponified product thereof, as in the case of EVOH (X) described above, as long as the object of the present invention is not impaired. From the viewpoint of lowering the melting point of EVOH (Y) and enhancing the moldability of the gas barrier resin composition of the present invention, it may be preferable that EVOH (Y) has another monomer unit (structural unit). be.
- the lower limit of the content of EVOH (Y) with respect to all structural units is preferably 0.1 mol%, more preferably 0.3 mol%. ..
- the upper limit of the content is preferably 15 mol%, more preferably 10 mol%.
- EVOH (Y) may be post-denatured by methods such as urethanization, acetalization, cyanoethylation, and oxyalkyleneization.
- EVOH (Y) has a modifying group
- EVOH (Y) has a structural unit (modifying group) represented by the above formula (I).
- EVOH other than EVOH (Y) may have a structural unit represented by the above formula (I).
- EVOH (Y) has a structural unit (modifying group) represented by the above formula (I), R 1 in the formula (I) is a single bond and X is a hydroxymethyl group (R in the formula (II)). it is preferable 3 and R 4 are hydrogen atoms).
- the lower limit of the content is preferably 0.1 mol%, more preferably 0.4 mol%, still more preferably 1.0 mol%.
- the upper limit of the content of the structural unit (modifying group) is preferably 20 mol%, more preferably 10 mol%, further preferably 8 mol%, and particularly preferably 5 mol% from the viewpoint of improving the gas barrier property. ..
- EVOH (Y) has a structural unit (modifying group) represented by the above formula (I), R 1 in the formula (I) is a hydroxymethylene group and X is a hydrogen atom (R 5 in the formula (III)). And R 6 is a hydrogen atom).
- the lower limit of the content is preferably 0.1 mol%, more preferably 0.4 mol%, still more preferably 1.0 mol%.
- the upper limit of the content of the structural unit (modifying group) is preferably 20 mol%, more preferably 10 mol%, further preferably 8 mol%, and particularly preferably 5 mol% from the viewpoint of improving the gas barrier property. ..
- EVOH (Y) has a structural unit (modifying group) represented by the above formula (I), R 1 is a methyl methylene group in formula (I), it is also preferred that X is a hydrogen atom.
- EVOH (Y) having this structural unit (modifying group) formability such as stretchability and thermoformability tends to be improved without significantly deteriorating the gas barrier property.
- methylmethyleneoxy group an oxygen atom is bonded to a carbon atom in the main chain. That is, in the formula (IV), it is preferable that one of R 7 and R 8 is a methyl group and the other is a hydrogen atom.
- the lower limit of the content is preferably 0.1 mol%, more preferably 0.5 mol%, still more preferably 1.0 mol%, and 2 9.0 mol% is particularly preferred.
- the upper limit of the content of the structural unit (modifying group) is preferably 20 mol%, more preferably 15 mol%, still more preferably 10 mol% from the viewpoint of improving the gas barrier property.
- the preferable range of MFR at 190 ° C. and 2160 g load of EVOH (Y) measured according to JIS K7210: 1999 is the same as that of EVOH (X) described above.
- the ethylene unit content difference (YX) between EVOH (Y) and EVOH (X) is preferably 5 mol% or more, more preferably 7 mol% or more, still more preferably 10 mol% or more. Further, the ethylene unit content difference (YX) may be 25 mol% or less. When the ethylene unit content difference (YX) is in the above range, the moldability tends to be good while showing a good gas barrier property.
- the melting point difference (XY) between EVOH (X) and EVOH (Y) is preferably 15 ° C. or higher, more preferably 18 ° C. or higher.
- the melting point difference (XY) may be 100 ° C. or lower, or 50 ° C. or lower. When the melting point difference (XY) is in the above range, the moldability tends to be good while showing a good gas barrier property.
- the mass ratio (X / Y) of EVOH (X) to EVOH (Y) is preferably 60/40 or more, and more preferably 65/35 or more.
- the mass ratio (X / Y) is preferably 95/5 or less, more preferably 90/10 or less.
- the lower limit of the total content of EVOH (X) and EVOH (Y) with respect to all EVOH contained in the gas barrier resin composition of the present invention is preferably 60% by mass, preferably 70% by mass, 80% by mass, 90% by mass, and 95. In some cases,% by weight or 99% by weight is more preferable. Further, the upper limit of the total content of EVOH (X) and EVOH (Y) with respect to all EVOH contained in the gas barrier resin composition of the present invention may be 100% by mass.
- EVOH may further contain EVOH (Z) having a melting point lower than that of EVOH (Y).
- EVOH EVOH
- one or more kinds of EVOH may be classified into at least three of EVOH (X), EVOH (Y) and EVOH (Z).
- X EVOH
- Y EVOH
- Z EVOH
- a preferred embodiment of EVOH (Z) is similar to EVOH (Y), except that it has a lower melting point than EVOH (Y).
- the content of EVOH (Z) with respect to all EVOH contained in the gas barrier resin composition of the present invention may be, for example, 1% by mass or more and 40% by mass or less.
- the lower limit of the total content of EVOH (X), EVOH (Y) and EVOH (Z) with respect to all EVOH contained in the gas barrier resin composition is preferably 90% by mass, more preferably 95% by mass, and 99% by mass. Is even more preferable.
- the upper limit of the total content of EVOH (X), EVOH (Y) and EVOH (Z) with respect to all EVOH contained in the gas barrier resin composition may be 100% by mass.
- the lower limit of the proportion of one or more EVOHs in all the resins constituting the gas barrier resin composition of the present invention may be, for example, 65% by mass, 70% by mass or 75% by mass, and 80% by mass is used. Preferably, 85% by mass is more preferable, and 90% by mass, 95% by mass, 98% by mass or 99% by mass may be further preferable, and the resin constituting the gas barrier resin composition is substantially one or more. It may be only one kind of EVOH, or it may be only one kind or two or more kinds of EVOH.
- the upper limit of the proportion of one or more EVOHs in all the resins constituting the gas barrier resin composition may be 100% by mass or 95% by mass.
- the lower limit of the ratio occupied by one or more kinds of EVOH in the gas barrier resin composition of the present invention may be, for example, 65% by mass, 70% by mass or 75% by mass, preferably 80% by mass, and 85% by mass. % Is more preferable, and 90% by mass, 95% by mass, 98% by mass or 99% by mass may be further preferable, and the gas barrier resin composition may be substantially composed of only one kind or two or more kinds of EVOH. good.
- the upper limit of the proportion of one or more EVOHs in the gas barrier resin composition may be 100% by mass or 95% by mass.
- the lower limit of the biobase degree of one or more EVOHs contained in the gas barrier resin composition of the present invention is preferably 1%, more preferably 5%, from the viewpoint of reducing the environmental load of the gas barrier resin composition. 20% is more preferable, and 40% is particularly preferable. Further, for example, in applications where particularly excellent long-running property is not required, the lower limit of the biobase degree of the entire EVOH may be 60% or 80%. On the other hand, the upper limit of the biobase degree of the whole EVOH is preferably 99%, more preferably 95%, 85%, 75%, 65%, 55%, 45%, 35% or 25% may be even more preferred.
- the specific and suitable ranges of the ethylene unit content, saponification degree, MFR and melting point of one or more EVOHs contained in the gas barrier resin composition of the present invention are the same as those of EVOH (A) described above. be.
- the gas barrier resin composition of the present invention preferably further contains inorganic particles (C).
- the gas barrier resin composition has inorganic particles (C)
- the fracture resistance of a film or the like containing a layer formed from the gas barrier resin composition tends to be good.
- the inorganic particles (C) when forming a thin-film deposition layer on the surface of a film containing a layer formed from the gas barrier resin composition, it is possible to suppress vapor-film deposition defects and improve the adhesion strength with the vapor-film-deposited layer. May be enhanced.
- the inorganic particles refer to particles containing an inorganic substance as a main component.
- the "main component” refers to a component having the highest content, for example, a component having a content of 50% by mass or more, preferably a component having a content of more than 50% by mass, and a component having a content of 90% by mass or more. Is more preferable.
- the inorganic substance constituting the inorganic particle (C) is preferably an inorganic substance containing at least one element selected from the group consisting of silicon, aluminum, magnesium, zirconium, cerium, tungsten and molybdenum. Among them, an inorganic substance containing at least one element selected from the group consisting of silicon, aluminum and magnesium is more preferable because it is easily available. Examples of the above-mentioned inorganic substances include oxides, nitrides, and oxidative nitrides of the exemplified elements, and oxides are preferable.
- the lower limit of the average particle size of the inorganic particles (C) is preferably 0.5 ⁇ m, more preferably 1.5 ⁇ m, and even more preferably 2.5 ⁇ m.
- the upper limit of the average particle size of the inorganic particles (C) is preferably 10 ⁇ m, more preferably 8 ⁇ m, and even more preferably 5 ⁇ m.
- the lower limit of the content of the inorganic particles (C) is preferably 50 ppm, more preferably 100 ppm, still more preferably 150 ppm.
- the upper limit of the content of the inorganic particles (C) is preferably 5000 ppm, more preferably 4000 ppm, still more preferably 3000 ppm, and even more preferably 2000 ppm or 1000 ppm.
- the gas barrier resin composition is excellent in fracture resistance and ability to suppress vapor deposition defects, and can improve the adhesion strength of the obtained layer with the vapor deposition layer.
- the inorganic particles (C) may contain one kind or two or more kinds of particles. Further, one particle may be formed of one kind or two or more kinds of inorganic substances.
- the lower limit of the proportion of one or more EVOH and the inorganic particles (C) in the gas barrier resin composition of the present invention is preferably 80% by mass, more preferably 90% by mass, still more preferably 95% by mass, and 98% by mass. % Is particularly preferable, and may be 99% by mass, and the gas barrier resin composition may be substantially composed of only one or more kinds of EVOH and inorganic particles (C).
- the gas barrier resin composition of the present invention contains the inorganic particles (C), it tends to be excellent in fracture resistance, blocking resistance, film deposition defect suppressing property, and adhesion to the vapor deposition layer. Therefore, in particular, such a gas barrier resin composition is preferably used for various films such as a single-layer film, a multilayer film, and a vapor-deposited film, and a multilayer structure including a vapor-deposited film.
- the gas barrier resin composition of the present invention preferably further contains the antioxidant (D).
- the gas barrier resin composition contains the antioxidant (D) to improve the oxidation deterioration resistance. Therefore, when the gas barrier resin composition contains the antioxidant (D), the mechanical strength of the molded product such as a pipe formed from the gas barrier resin composition is unlikely to decrease even during long-term use, and the product deteriorates over time. There is a tendency that the occurrence of cracks due to the above can be suppressed.
- the antioxidant (D) is a compound having an antioxidant ability.
- the melting point of the antioxidant (D) is not particularly limited, but is preferably 170 ° C. or lower. When the melting point of the antioxidant (D) is 170 ° C. or lower, it is easy to melt in the extruder when the gas barrier resin composition is produced by melt mixing. Therefore, it is possible to prevent the antioxidant (D) from being localized in the resin composition and coloring the high-concentration portion. Further, the melting point of the antioxidant (D) is preferably 50 ° C. or higher, more preferably 100 ° C. or higher. When the melting point of the antioxidant (D) is 50 ° C. or higher, it is possible to prevent the antioxidant from bleeding out to the surface of the obtained molded product (pipe or the like) and deteriorating the appearance.
- the molecular weight of the antioxidant (D) is preferably 300 or more.
- the molecular weight of the antioxidant (D) is 300 or more, when a molded product is obtained from the gas barrier resin composition of the present invention, the antioxidant bleeds out on the surface and the appearance of the molded product is deteriorated. In addition, the thermal stability of the resin composition is enhanced.
- the molecular weight is more preferably 400 or more, and particularly preferably 500 or more.
- the upper limit of the molecular weight of the antioxidant (D) is not particularly limited, but from the viewpoint of dispersibility, it is preferably 8000 or less, more preferably 6000 or less, further preferably 4000 or less, and particularly preferably 2000 or less.
- antioxidant (D) a compound having a hindered phenol group is preferably used.
- a compound having a hindered phenol group has excellent thermal stability by itself, but also has an ability to capture oxygen radicals that cause oxidative deterioration, and when blended in a gas barrier resin composition as an antioxidant (D). , It is excellent in the effect of preventing oxidative deterioration.
- IRGANOX 1010 manufactured by BASF, melting point 110-125 ° C., molecular weight 1178, pentaerythritol tetrakis [3- (3,5-di-tert-butyl-4-hydroxyphenyl) propionate]
- BASF's "IRGANOX 1076” melting point 50-55 ° C., molecular weight 531 and octadecyl-3- (3,5-di-tert-butyl-4-hydroxyphenyl) propionate
- IRGANOX 1010 manufactured by BASF, melting point 110-125 ° C., molecular weight 1178, pentaeryth
- a compound having a hindered amine group is also preferably used.
- a compound having a hindered amine group is added to a gas barrier resin composition as an antioxidant (D)
- it not only prevents thermal deterioration of EVOH, but also has the effect of capturing aldehydes produced by thermal decomposition of EVOH.
- By reducing the generation of decomposition gas it is possible to suppress the generation of voids or bubbles during molding.
- the point that the odor due to the aldehyde impairs the taste of the contents is also improved.
- a preferred compound having a hindered amine group is a piperidine derivative, and a 2,2,6,6-tetraalkylpiperidine derivative having a substituent at the 4-position is particularly preferable.
- the substituent at the 4-position include a carboxyl group, an alkoxy group and an alkylamino group.
- an alkyl group may be substituted at the N-position of the hindered amine group, it is preferable to use one to which a hydrogen atom is bonded because of its excellent thermal stabilization effect.
- BASF's "TINUVIN 770" melting point 81-85 ° C., molecular weight 481, bis (2,2,6,6-tetramethyl-4-piperidyl) sebacate
- BASF's "TINUVIN 765" liquid Compound, molecular weight 509, bis (1,2,2,6,6-pentamethyl-4-piperidyl) sebacate and 1,2,2,6,6-pentamethyl-4-piperidyl sebacate (mixture)
- BASF "TINUVIN 622LD” melting point 55-70 ° C., molecular weight 3100-4000, dimethyl 1- (2-hydroxyethyl) succinate-4-hydroxy-2,2,6,6-tetramethylpiperidine
- BASF "CHIMASORB 119FL” melting point 130-140 ° C., molecular weight 2000 or more, N, N
- hindered phenol groups or compounds having a hindered amine group may be used alone or in combination of two or more.
- the lower limit of the content of the antioxidant (D) in the gas barrier resin composition of the present invention is preferably 0.01% by mass, more preferably 0.1% by mass, still more preferably 0.3% by mass.
- the upper limit of the content of the antioxidant (D) is preferably 5% by mass, more preferably 3% by mass, still more preferably 1% by mass.
- the lower limit of the proportion of one or more EVOHs and the antioxidant (D) in the gas barrier resin composition of the present invention is preferably 80% by mass, more preferably 90% by mass, still more preferably 95% by mass, 98.
- the mass% is particularly preferable, and it may be 99% by mass, and the gas barrier resin composition may be substantially composed of only one or more kinds of EVOH and the antioxidant (D).
- the gas barrier resin composition of the present invention particularly contains an antioxidant (D)
- the gas barrier resin composition or a molded product using the same has a small environmental load and good gas barrier properties. Moreover, even if it is used for a long period of time, cracks are unlikely to occur due to oxidative deterioration. Therefore, it is suitable for daily necessities, packaging materials, machine parts, durable consumer goods parts, industrial parts, industrial materials and the like used indoors and outdoors. Examples of applications in which the characteristics of the molded body and the like are particularly effectively exhibited include food and drink packaging materials, container packing materials, films, agricultural films, geomembranes, medical infusion bag materials, high-pressure tank materials, and the like.
- Gas tank material fuel container, tube material for tires, cushion material for shoes, inner bag material for bag-in-box, tank material for organic liquid storage, pipe (pipe material for organic liquid transportation (fuel transportation pipe, etc.), for heating)
- Examples include hot water pipe materials (hot water pipe materials for floor heating, etc.), heat-insulating multi-layer pipes, resin wallpaper, plant media, and the like.
- it is suitably used as a film, a pipe, an agricultural film, a plant medium and a geomembrane, which are used outdoors and are easily deteriorated by heat or light.
- the gas barrier resin composition of the present invention preferably further contains polyamide (PA) (E).
- PA polyamide
- PA (E) is a resin containing an amide bond.
- PA (E) can be obtained, for example, by ring-opening polymerization of lactam having a 3-membered ring or more, polycondensation of polymerizable ⁇ -amino acids, polycondensation of dibasic acid and diamine, and the like.
- the polyamide (E) include polycapramid (nylon 6), poly- ⁇ -aminoheptanoic acid (nylon 7), poly- ⁇ -aminononanoic acid (nylon 9), polyundecaneamide (nylon 11), and polylauryl lactam (nylon).
- Polyethylenediamine adipamide (nylon 26), polytetramethylene adipamide (nylon 46), polyhexamethylene adipamide (nylon 66), polyhexamethylene sebacamide (nylon 610), polyhexamethylene dodeca Mido (nylon 612), polyoctamethylene adipamide (nylon 86), polydecamethylene adipamide (nylon 108), caprolactam / lauryllactam copolymer (nylon 6/12), caprolactam / ⁇ -aminononanoic acid co-weight Combined (nylon 6/9), caprolactam / hexamethylene diammonium adipate copolymer (nylon 6/66), lauryl lactam / hexamethylene diammonium adipate copolymer (nylon 12/66), hexamethylene diammonium adipate / hexa Methylene diammonium sebacate copolymer (nylon 26),
- PA (E) may be manufactured using only fossil fuel-derived raw materials or may be manufactured using biomass-derived raw materials.
- Arkema S. A As a polyamide (E) using only biomass-derived raw materials, the company uses polyundecane amide (nylon 11) (“Rilsan PA11”) made from castor oil, as well as biomass-derived raw materials and fossil fuel-derived raw materials.
- Polyamide methylene sebacamide (nylon 610) (“Rilsan S”) is manufactured and sold as the polyamide (C) produced in the above.
- Aromatic dicarboxylic acids such as phthalic acid, xylylene dicarboxylic acid, alkyl-substituted terephthalic acid, alkyl-substituted isophthalic acid, and naphthalenedicarboxylic acid may be used, or modification to polyamide may be performed using these. No.
- polycapramid (nylon 6) is preferable.
- a caprolactam / lauryllactam copolymer (nylon 6/12) is also preferable.
- the content ratio of 6 units of nylon to 12 units of nylon is not particularly limited, but the lower limit of the content of 12 units of nylon is preferably 5% by mass.
- the upper limit of the content of 12 units of nylon is preferably 60% by mass, more preferably 50% by mass.
- the lower limit of the mass ratio (PA (E) / EVOH) of PA (E) to the above-mentioned one or more kinds of ethylene-vinyl ester copolymer saken product in the gas barrier resin composition of the present invention is preferably 5/95. 8/92 is more preferable, and 13/87 is even more preferable.
- the upper limit of the mass ratio (PA (E) / EVOH) is preferably 40/60, more preferably 35/65, even more preferably 30/70, and even more preferably 25/75.
- the mass ratio (PA (E) / EVOH) is 5/95 or more, the retort resistance becomes sufficient, and after heat treatment with hot water or steam on the molded body or the like formed from the gas barrier resin composition. The appearance of is improved. Further, when the mass ratio (PA (E) / EVOH) is 40/60 or less, the gas barrier property is improved and the long-run property is more sufficient.
- the lower limit of the PA (E) content in the gas barrier resin composition of the present invention is preferably 5% by mass, more preferably 7% by mass, from the viewpoint of enhancing the retort resistance.
- the upper limit of the PA (E) content is preferably 40% by mass, more preferably 30% by mass, further preferably 25% by mass, and particularly preferably 15% by mass from the viewpoint of enhancing gas barrier properties and long-running properties.
- the lower limit of the proportion of one or more EVOH and PA (E) in all the resins constituting the gas barrier resin composition of the present invention is preferably 80% by mass, more preferably 90% by mass, and 95% by mass. Further preferably, 98% by mass is particularly preferable, and 99% by mass may be used.
- the resin constituting the gas barrier resin composition may be substantially only one or more kinds of EVOH and PA (E), or may be only one kind or two or more kinds of EVOH and PA (E). good.
- the gas barrier resin composition of the present invention contains PA (E)
- the retort resistance is good, and the retort resistance equivalent to that of EVOH having the same structure synthesized only from fossil fuel-derived raw materials can be exhibited. Therefore, the gas barrier resin composition of the present invention containing PA (E) or the multilayer structure including at least one layer made of the gas barrier resin composition can be suitably used as a packaging material, particularly a packaging material for retort pouches. can.
- the gas barrier resin composition of the present invention preferably contains at least one metal atom (F) selected from the group consisting of magnesium, calcium and zinc.
- F metal atom
- the long-run property at the time of melt molding becomes good.
- a resin composition containing PA with respect to EVOH generally tends to have a reduced long-run property, but the gas barrier resin composition of the present invention contains a metal atom (F) together with PA (E). , Can demonstrate sufficient long-running properties.
- the lower limit of the content of the metal atom (F) in the gas barrier resin composition of the present invention is preferably 1 ppm, more preferably 10 ppm, still more preferably 50 ppm.
- the upper limit of the content of the metal atom (F) is preferably 500 ppm, more preferably 350 ppm, further preferably 300 ppm, and even more preferably 250 ppm.
- the metal atom (F) magnesium is more preferable from the viewpoint of improving the long-running property at the time of melt molding.
- the metal atom (F) may exist as a simple substance of a metal atom or as a constituent atom in a compound, or may exist in the state of a free metal ion.
- the metal atom (F) is a salt of an organic acid such as an aliphatic carboxylic acid, an aromatic carboxylic acid, an aliphatic dicarboxylic acid, an aromatic dicarboxylic acid, a tricarboxylic acid, a tetracarboxylic acid, a hydroxycarboxylic acid, a ketodicarboxylic acid, or an amino acid; Salts of inorganic acids such as sulfuric acid, sulfite, carbonic acid, phosphoric acid; may be contained as hydroxides and the like.
- an aliphatic carboxylic acid metal salt or a hydroxide it is preferably contained as an aliphatic carboxylic acid metal salt or a hydroxide, and more preferably contained as an aliphatic carboxylic acid metal salt having 6 or less carbon atoms or a hydroxide from the viewpoint of compatibility with EVOH.
- the aliphatic carboxylic acid include saturated aliphatic carboxylic acids such as formic acid, acetic acid, propionic acid, butyric acid and caproic acid; and unsaturated aliphatic carboxylic acids such as acrylic acid and methacrylic acid.
- salts or hydroxides of saturated aliphatic carboxylic acids having 1 to 3 carbon atoms are preferable, and acetates or hydroxides are more preferable, from the viewpoint of compatibility with EVOH and the like.
- the metal atom (F) may form a salt with an organic acid or an inorganic acid as described above, and may be a salt as a counter cation of one or more kinds of EVOH alkoxides. May be formed.
- the metal atoms (F) are uniformly dispersed in the entire resin composition. Further, the metal atom (F) may be used alone or in combination of two or more.
- the lower limit of the proportion of one or more EVOH, PA (E) and the metal atom (F) in the gas barrier resin composition is preferably 80% by mass, more preferably 90% by mass, still more preferably 95% by mass. , 98% by mass is particularly preferable, and 99% by mass may be used.
- the gas barrier resin composition may be substantially composed of only one or more EVOH, PA (E) and a metal atom (F), and one or more EVOH, PA (E) and a metal atom. It may be only (F).
- the gas barrier resin composition of the present invention preferably further contains a thermoplastic elastomer (G).
- a thermoplastic elastomer G
- the thermoplastic elastomer (G) may be derived from biomass or fossil fuel as a raw material.
- thermoplastic elastomer (G) is not particularly limited, and polyester-based thermoplastic elastomers, polystyrene-based thermoplastic elastomers, polyolefin-based thermoplastic elastomers, and the like can be used. These may be one kind or a combination of two or more kinds. Above all, from the viewpoint of improving bending resistance, the thermoplastic elastomer (G) is preferably at least one selected from the group consisting of polystyrene-based thermoplastic elastomers and polyolefin-based thermoplastic elastomers.
- polyester-based thermoplastic elastomer examples include a multi-block copolymer comprising polyester as a hard segment in the molecule and a polyether or polyester having a low glass transition temperature (Tg) as a soft segment. Be done.
- the TPEE can be classified into the following types according to the difference in the molecular structure, and among them, the polyester-polyester type TPEE and the polyester-polyester type TPEE are preferable.
- Polyester / polyether type TPEE Generally, it is a thermoplastic elastomer using aromatic crystalline polyester as a hard segment and polyether as a soft segment.
- Polyester / polyester type TPEE It is a thermoplastic elastomer using aromatic crystalline polyester as a hard segment and aliphatic polyester as a soft segment.
- Liquid crystal TPEE It is a thermoplastic elastomer using rigid liquid crystal molecules as hard segments and aliphatic polyester as soft segments.
- polyester segment examples include aromatic dicarboxylic acids such as terephthalic acid, isophthalic acid and naphthalenedicarboxylic acid; alicyclic dicarboxylic acids such as 1,4-cyclohexanedicarboxylic acid; and aliphatic dicarboxylic acids such as succinic acid and adipic acid.
- Polyester segment composed of a dicarboxylic acid component and an aliphatic diol such as ethylene glycol, 1,2-propylene glycol, 1,4-butanediol; and a diol component such as an alicyclic diol such as cyclohexane-1,4-dimethanol.
- polyether segment examples include aliphatic polyether segments such as polyethylene glycol, polypropylene glycol and polybutylene glycol.
- the polystyrene-based thermoplastic elastomer is not particularly limited, but usually includes a styrene monomer polymer block (Hb) as a hard segment and a conjugated diene compound polymer block or a hydrogenated block (Sb) thereof as a soft segment.
- the structure of this styrene-based thermoplastic elastomer is represented by a diblock structure represented by Hb-Sb, a triblock structure represented by Hb-Sb-Hb or Sb-Hb-Sb, and a structure represented by Hb-Sb-Hb-Sb. It may be a tetrablock structure to be formed, or a polyblock structure in which a total of 5 or more Hb and Sb are linearly bonded.
- the styrene-based monomer used in the styrene monomer polymer block (Hb) is not particularly limited, and examples thereof include styrene and its derivatives. Specifically, styrene, ⁇ -methylstyrene, 2-methylstyrene, 4-methylstyrene, 4-propylstyrene, 4-t-butylstyrene, 4-cyclohexylstyrene, 4-dodecylstyrene, 2-ethyl-4- Styrenes such as benzyl styrene, 4- (phenylbutyl) styrene, 2,4,6-trimethylstyrene, monofluorostyrene, difluorostyrene, monochlorostyrene, dichlorostyrene, methoxystyrene, t-butoxystyrene; 1-vinylnaphthalene, 2-Vinyl group-containing
- the conjugated diene compound used for the conjugated diene compound polymer block or the hydrogenated block (Sb) thereof is also not particularly limited, and examples thereof include butadiene, isoprene, 2,3-dimethylbutadiene, pentadiene, and hexadiene. can. Of these, butadiene is preferable.
- the conjugated diene compound may be only one kind or two or more kinds. Further, other co-monomers such as ethylene, propylene, butylene and styrene can be copolymerized. Further, the conjugated diene compound polymer block may be a hydrogenated body which is partially or completely hydrogenated.
- polystyrene-based thermoplastic elastomer examples include styrene-isopregen block copolymer (SI), styrene-butadiene diblock copolymer (SB), styrene-isoprene-styrene triblock copolymer (SIS), and styrene.
- SI styrene-isopregen block copolymer
- SB styrene-butadiene diblock copolymer
- SIS styrene-isoprene-styrene triblock copolymer
- styrene examples include a butadiene / isoprene-styrene triblock copolymer (SB / IS), a styrene-butadiene-styrene triblock copolymer (SBS), and a hydrogenated product thereof.
- a hydrogenated product of a styrene-isopregen block copolymer SEP
- SEB hydrogenated product of a styrene-isoprene-styrene triblock copolymer
- SEPS hydrogenated product of a styrene-isoprene-styrene triblock copolymer
- SEEPS hydrogenated product of a styrene-butadiene / isoprene-styrene triblock copolymer
- SEBS hydrogenated product of a styrene-butadiene-styrene triblock copolymer
- the polyolefin-based thermoplastic elastomer includes a thermoplastic elastomer having a polyolefin block such as polypropylene or polyethylene as a hard segment and a rubber block such as an ethylene-propylene-diene copolymer as a soft segment.
- the thermoplastic elastomer includes a blend type and an implantable type, and the implantable type is preferable in terms of compatibility with EVOH and cost.
- the thermoplastic elastomer (G) contains at least one selected from the group consisting of a non-modified thermoplastic elastomer (g1), a modified thermoplastic elastomer (g2), and a polystyrene-based thermoplastic elastomer containing a halogen atom (g3). Is preferable, and it is more preferable to contain a modified thermoplastic elastomer (g2).
- the inclusion of the modified thermoplastic elastomer (g2) promotes the reaction of one or more EVOHs with the modified thermoplastic elastomer (g2), for example, during melt-kneading, and is a good phase without taking special extrusion conditions.
- the thermoplastic elastomer (G) may contain both the non-modified thermoplastic elastomer (g1) and the modified thermoplastic elastomer (g2), or may consist only of the modified thermoplastic elastomer (g2), and may contain halogen atoms. It may consist only of the contained polystyrene-based thermoplastic elastomer (g3).
- the non-denatured thermoplastic elastomer (g1) the resin mentioned as the above-mentioned thermoplastic elastomer can be used as it is.
- the lower limit of the proportion of the non-modifying thermoplastic elastomer (g1) in the thermoplastic elastomer (G) is preferably 30% by mass, preferably 40% by mass. % Is more preferred, and 85% by mass may be even more preferred.
- the upper limit of the proportion occupied by the non-modified thermoplastic elastomer (g1) may be 100% by mass or 60% by mass.
- the non-modifying thermoplastic elastomer (g1) does not have a site that reacts with one or more kinds of EVOH, when the proportion of the non-modifying thermoplastic elastomer (g1) in the thermoplastic elastomer (G) becomes high, for example.
- the formation of gels and the like tends to be suppressed, and as a result, the long-running property tends to improve.
- the modified thermoplastic elastomer (g2) is preferably an acid-modified thermoplastic elastomer.
- the modified thermoplastic elastomer (g2) can be obtained, for example, by modifying the unmodified thermoplastic elastomer (g1) with an unsaturated carboxylic acid or a derivative thereof.
- the unsaturated carboxylic acid or a derivative thereof include maleic acid, fumaric acid, itaconic acid, maleic anhydride, itaconic anhydride, maleic acid monomethyl ester, maleic acid monoethyl ester, maleic acid diethyl ester, and fumaric acid monomethyl ester. Be done. Of these, it is preferably modified with maleic anhydride.
- thermoplastic elastomer (g2) By using such a modified thermoplastic elastomer (g2), the compatibility between one or more kinds of EVOH and the thermoplastic elastomer (G) is enhanced, and the gas barrier property, bending resistance, etc. tend to be further improved. Become.
- modified thermoplastic elastomer (g2) a modified polyester-based thermoplastic elastomer, a modified polystyrene-based thermoplastic elastomer, a modified polyolefin-based thermoplastic elastomer, or the like can be used. These may be one kind or a combination of two or more kinds.
- the modified thermoplastic elastomer (g2) is preferably a maleic anhydride-modified polyester-based thermoplastic elastomer, a maleic anhydride-modified polystyrene-based thermoplastic elastomer, and a maleic anhydride-modified polyolefin-based thermoplastic elastomer.
- the modified thermoplastic elastomer (g2) is at least one selected from the group consisting of a maleic anhydride-modified polystyrene-based thermoplastic elastomer and an anhydrous maleic acid-modified polyolefin-based thermoplastic elastomer. It is preferable to have.
- the acid value of the acid-modified thermoplastic elastomer is 1 mgKOH / g or more and 50 mgKOH / g or less from the viewpoint of enhancing bending resistance and long-running property.
- the acid value of the thermoplastic elastomer (G) is preferably 0.5 mgKOH / g or more and 25 mgKOH / g or less from the viewpoint of enhancing bending resistance and long-running property.
- the lower limit of the proportion of the modified thermoplastic elastomer (g2) is preferably 5% by mass, more preferably 20% by mass, still more preferably 40% by mass, and the thermoplastic elastomer (G). May be composed only of the modified thermoplastic elastomer (g2).
- the upper limit of the proportion occupied by the modified thermoplastic elastomer (g2) may be 100% by mass or 60% by mass.
- the modified thermoplastic elastomer (g2) enhances the compatibility between one or more types of EVOH and the thermoplastic elastomer (G)
- the proportion of the modified thermoplastic elastomer (g2) in the thermoplastic elastomer (G) increases.
- the compatibility tends to be improved, and as a result, the bending resistance tends to be further improved.
- the mass ratio (g1 / g2) of the unmodified thermoplastic elastomer (g1) to the modified thermoplastic elastomer (g2). ) is preferably more than 0/100, more preferably 20/80 or more, further preferably 40/60 or more, and particularly preferably 80/20 or more.
- the mass ratio (g1 / g2) is preferably 95/5 or less.
- the mass ratio (g1 / g2) is within the above range, the acid value of the thermoplastic elastomer (G) can be easily adjusted, and the gas barrier resin composition of the present invention can be produced with excellent bending resistance and long-running property. It tends to be easier.
- the lower limit of the proportion of the non-modified thermoplastic elastomer (g1) and the modified thermoplastic elastomer (g2) in the thermoplastic elastomer (G) is 90% by mass, whether it is 70% by mass or 80% by mass.
- the thermoplastic elastomer (G) may be substantially composed of only the non-modifying thermoplastic elastomer (g1) and the modified thermoplastic elastomer (g2), and may be composed of the non-modifying thermoplastic elastomer (g1) and the modified heat. It may consist only of a thermoplastic elastomer (g2).
- the thermoplastic elastomer (G) contains a polystyrene-based thermoplastic elastomer (g3) containing a halogen atom.
- the halogen atom may be derived from a polymerization catalyst used in the production of the polystyrene-based thermoplastic elastomer, in which case the halogen atom is contained at the end of the polystyrene-based thermoplastic elastomer.
- halogen atom contained in the polystyrene-based thermoplastic elastomer examples include chlorine, bromine, fluorine, iodine and the like, and among them, chlorine is preferable.
- the content of halogen atoms in the polystyrene-based thermoplastic elastomer is usually 0.005 to 3.000% by mass.
- Halogen atoms in polystyrene-based thermoplastic elastomers can be analyzed using an ion chromatograph.
- the polystyrene-based thermoplastic elastomer (g3) containing a halogen atom is not particularly limited as long as it contains a halogen atom.
- a compound in which a halogen atom is contained in the compound exemplified as the above-mentioned polystyrene-based elastomer is used. be able to.
- a styrene-isobutylene-styrene block copolymer containing a halogen atom is more preferable, and examples thereof include the trade name "SIBSTAR 062T" manufactured by Kaneka Corporation.
- the lower limit of the proportion of the thermoplastic elastomer (g3) containing a halogen atom in the thermoplastic elastomer (G) is preferably 20% by mass, more preferably 50% by mass, still more preferably 90% by mass, and the thermoplastic elastomer (g3).
- G) may be composed of only a polystyrene-based thermoplastic elastomer (g3) containing substantially a halogen atom, or may be composed of only a polystyrene-based thermoplastic elastomer (g3) containing a halogen atom.
- the lower limit of the mass ratio (G / EVOH) of the thermoplastic elastomer (G) to one or more EVOHs in the gas barrier resin composition of the present invention is preferably 5/95, more preferably 8/92, and 12/88. In some cases it is even more preferred, and in other cases 15/85 or 25/75 is even more preferred.
- the upper limit of this mass ratio (G / EVOH) is preferably 35/65, more preferably 30/70, even more preferably 25/75, and even more preferably 15/85.
- the gas barrier resin composition of the present invention contains the thermoplastic elastomer (G), it is preferable that the particles of the thermoplastic elastomer (G) are dispersed in the matrix of one or more kinds of EVOH. That is, the gas barrier resin composition of the present invention has a sea-island structure, the sea phase is mainly composed of one or more kinds of EVOH, and the island phase is mainly composed of a thermoplastic elastomer (G). Is preferable. As described above, since the sea phase is mainly composed of one kind or two or more kinds of EVOH, the flexibility is improved while maintaining the gas barrier property.
- the sea phase is mainly composed of one or more kinds of EVOH
- the island phase is mainly composed of a thermoplastic elastomer (G), from the viewpoint of improving transparency.
- the average particle size of the island phase made of the thermoplastic elastomer (G) is preferably 4.5 ⁇ m or less, more preferably 3.5 ⁇ m or less, further preferably 3.0 ⁇ m or less, particularly preferably 2.5 ⁇ m or less, and 2.0 ⁇ m. The following are the most preferable.
- the average particle size of the thermoplastic elastomer (G) may be 0.1 ⁇ m or more.
- the average particle size of the island phase mainly composed of the thermoplastic elastomer (G) is within the above range, the bending resistance is improved while maintaining the gas barrier property and transparency, and the peeling property in the laminated peeling container described later is further improved. It is preferable because it tends to be.
- the average particle size of the thermoplastic elastomer (G) can be adjusted by adjusting the kneading strength and the composition ratio of one or more kinds of EVOH and the thermoplastic elastomer (G).
- the difference in refractive index between one or more EVOHs and the thermoplastic elastomer (G) is preferably 0.05 or less, more preferably 0.04 or less, and further preferably 0.03 or less. preferable.
- the difference in refractive index may be 0.005 or more. When the difference in refractive index is within the above range, the transparency of the gas barrier resin composition tends to be good.
- thermoplastic elastomer (G) may be used alone or in combination of two or more.
- the lower limit of the proportion of one or more EVOH and the thermoplastic elastomer (G) in all the resins constituting the gas barrier resin composition of the present invention is preferably 80% by mass, more preferably 90% by mass, and 95% by mass. Is more preferable, 98% by mass is particularly preferable, and 99% by mass may be used, and the resin constituting the gas barrier resin composition is substantially composed of only one or more kinds of EVOH and the thermoplastic elastomer (G). It may be composed of only one kind or two or more kinds of EVOH and a thermoplastic elastomer (G).
- the gas barrier resin composition of the present invention contains a thermoplastic elastomer (G), high bending resistance can be exhibited. Therefore, the gas barrier resin composition containing the thermoplastic elastomer (G) or the multilayer structure including at least one layer made of the gas barrier resin composition is for packaging materials, vertical bag-filled seal bags, and bag-in-box contents. It can be suitably used for vessels, laminated peeling containers, tubes, pipes, blow molded containers and the like.
- the gas barrier resin composition of the present invention contains a polystyrene-based thermoplastic elastomer (g3) containing a halogen atom
- the gas barrier resin composition of the present invention preferably contains a halogen scavenger from the viewpoint of improving long-running property.
- the halogen scavenger may be any one having a halogen scavenging ability, for example, a layered inorganic compound having exchangeable ions; an alkaline earth metal compound such as magnesium oxide, calcium hydroxide, magnesium hydroxide and calcium carbonate; zinc oxide. ; Lithium carbonate and the like can be mentioned.
- the halogen scavenger is a layered inorganic compound having commutative ions.
- the halogen ions are incorporated into the layered inorganic compound.
- the layered inorganic compound for example, clay mineral; layered polysilicic acid; layered silicate; layered compound hydroxide; layered phosphate; layered transition metallic oxygen such as titanium niobate, hexaniobate and molybdenate. Salts; layered manganese oxides; layered cobalt oxides and the like can be mentioned, with clay minerals being preferred.
- Examples of the clay minerals include hydrotalcite, zeolite, mica, vermiculite, montmorillonite, biderite, saponite, hectorite and stephensite.
- the clay mineral may be synthetic clay or natural clay.
- hydrotalcite and zeolite are preferable as the clay mineral, and the former is more preferable.
- Examples of hydrotalcite include those represented by the following general formula (i), and examples of zeolites include those represented by the following formula (ii).
- the lower limit of the content is preferably 0.01% by mass, preferably 0.025, from the viewpoint of suppressing rapid crosslinking (gelation) and enhancing long-running properties. % By mass is more preferred.
- the upper limit of the content of the halogen scavenger is preferably 1% by mass, more preferably 0.8% by mass.
- the mass ratio [halogen scavenger / halogen atom] of the content of the halogen scavenger to the content of the halogen atom contained in the polystyrene-based thermoplastic elastomer (g3) containing a halogen atom is. 0.10 or more is preferable, 0.15 or more is more preferable, 0.20 or more is further preferable, and 0.25 or more is particularly preferable.
- the mass ratio [halogen scavenger / halogen atom] is preferably 1.010 or less, more preferably 0.702 or less, further preferably 0.501 or less, and particularly preferably 0.40 or less.
- the gas barrier resin composition of the present invention contains a sulfur compound in an amount of more than 0 ppm and 100 ppm in terms of sulfur atom from the viewpoint of tracking the company's product. Further, the inventors have found that a sulfur compound having a sulfur atom equivalent of 100 ppm or less does not substantially affect the performance of the gas barrier resin composition, and the sulfur compound is suitable as a tracer substance.
- the upper limit of the content of the sulfur compound is more preferably 50 ppm, further preferably 5 ppm, still more preferably 3 ppm, and particularly preferably 0.3 ppm.
- the lower limit of the content of the sulfur compound may be 0.0001 ppm, 0.001 ppm, 0.01 ppm, 0.05 ppm, or 0.1 ppm.
- EVOH containing an organic sulfur compound contained in the raw material of biomass may be obtained.
- the amount of sulfur compound is smaller than that of EVOH derived from biomass. Therefore, when such biomass-derived EVOH is used, it becomes easier to trace the biomass-derived EVOH by comparing the contents of the sulfur compounds.
- the gas barrier resin composition of the present invention contains an organic sulfur compound as a sulfur compound, particularly dimethyl sulfide or dimethyl sulfoxide, tracking becomes easier.
- the content of the sulfur compound is the detection limit value for the biomass-derived ethylene and biomass-derived vinyl ester as raw materials and the obtained EVOH during the production of EVOH. It may be preferable not to carry out excessive purification as described below.
- the gas barrier resin composition of the present invention preferably further contains a carboxylic acid.
- a carboxylic acid melt moldability and coloring resistance at high temperatures can be improved.
- the pH buffering capacity of the gas barrier resin composition is enhanced, and the coloring resistance to acidic substances and basic substances may be improved. Therefore, the pKa of the carboxylic acid is more preferably in the range of 3.5 to 5.5. preferable.
- the lower limit of the content is preferably 30 ppm, more preferably 100 ppm in terms of carboxylic acid root.
- the upper limit of the carboxylic acid content is preferably 1000 ppm, more preferably 600 ppm.
- the content of the carboxylic acid is calculated by titrating an extract obtained by extracting 10 g of the resin composition with 50 ml of pure water at 95 ° C. for 8 hours.
- the content of the carboxylic acid salt present in the extract is not taken into consideration as the content of the carboxylic acid in the resin composition.
- the carboxylic acid may exist as a carboxylic acid ion.
- the carboxylic acid examples include monovalent carboxylic acid and polyvalent carboxylic acid, which may be composed of one kind or a plurality of kinds.
- the melt moldability of the gas barrier resin composition and the coloring resistance at high temperatures may be further improved.
- the multivalent carboxylic acid may have three or more carboxy groups. In this case, the coloring resistance of the gas barrier resin composition of the present invention may be further improved.
- the monovalent carboxylic acid is a compound having one carboxy group in the molecule.
- the pKa of the monovalent carboxylic acid is preferably in the range of 3.5 to 5.5.
- PKa 4.88
- carboxylic acids may have substituents such as hydroxyl groups, amino groups and halogen atoms as long as pKa is in the range of 3.5 to 5.5.
- acetic acid is preferable because it is highly safe and easy to handle.
- a polyvalent carboxylic acid is a compound having two or more carboxy groups in the molecule.
- the pKa of at least one carboxy group is preferably in the range of 3.5 to 5.5.
- the gas barrier resin composition of the present invention preferably further contains a phosphoric acid compound.
- the lower limit of the content thereof is preferably 1 ppm in terms of phosphoric acid root, and more preferably 3 ppm.
- the upper limit of the content is preferably 200 ppm in terms of phosphoric acid root, and more preferably 100 ppm. If a phosphoric acid compound is contained in this range, the thermal stability of the gas barrier resin composition may be improved. In particular, it may be possible to suppress the generation and coloring of gel-like lumps during melt molding for a long period of time.
- the phosphoric acid compound for example, various acids such as phosphoric acid and phosphoric acid and salts thereof can be used.
- the phosphate may be in the form of a first phosphate, a second phosphate, or a third phosphate.
- the cation species of phosphate include alkali metals and alkaline earth metals.
- Specific examples of the phosphoric acid compound include a phosphoric acid compound in the form of sodium dihydrogen phosphate, potassium dihydrogen phosphate, disodium hydrogen phosphate, and dipotassium hydrogen phosphate.
- the gas barrier resin composition of the present invention preferably further contains a boron compound.
- the lower limit of the content thereof is preferably 5 ppm in terms of boron element, more preferably 100 ppm.
- the upper limit of the content is preferably 5000 ppm, more preferably 1000 ppm in terms of boron atom. If a boron compound is contained in this range, the thermal stability of the gas barrier resin composition during melt molding can be improved, and the generation of gel-like lumps may be suppressed. In some cases, the mechanical properties of the obtained molded product can be improved. It is speculated that these effects are due to the chelate interaction between EVOH and the boron compound.
- Examples of the boron compound include boric acid, borate ester, borate, and boron borohydride.
- boric acid include orthoboric acid (H 3 BO 3 ), metaboric acid, and tetraboric acid
- examples of boric acid esters include, for example, trimethyl borate and triethyl borate, boric acid.
- the salt include the above-mentioned alkali metal salts of boric acid, alkaline earth metal salts, and boric acid.
- the gas barrier resin composition of the present invention preferably further contains metal ions.
- the interlayer adhesiveness becomes excellent when a multi-layer molded body, that is, a multi-layer structure is formed.
- the reason for the improvement in interlayer adhesion is not clear, but if the layer adjacent to the layer made of the gas barrier resin composition contains a molecule having a functional group capable of reacting with the hydroxy group of EVOH, both of them are subjected to metal ions. It is considered that the bond formation reaction of is accelerated. Further, by controlling the content ratio of the metal ion and the above-mentioned carboxylic acid, the melt moldability and coloring resistance of the gas barrier resin composition can be improved.
- the lower limit of the content is preferably 1 ppm, more preferably 100 ppm, still more preferably 150 ppm.
- the upper limit of the metal ion content is preferably 1000 ppm, more preferably 400 ppm, still more preferably 350 ppm.
- the content of the metal ion is 1 ppm or more, the interlayer adhesiveness of the obtained multilayer structure tends to be good.
- the content of the metal ion is 1000 ppm or less, the coloring resistance tends to be good.
- the metal ion examples include a monovalent metal ion, a divalent metal ion, and other transition metal ions, which may be composed of one or more kinds. Of these, monovalent metal ions and divalent metal ions are preferable.
- an alkali metal ion is preferable, and examples thereof include lithium, sodium, potassium, rubidium and cesium ions, and sodium or potassium ion is preferable from the viewpoint of industrial availability.
- the alkali metal salt that gives alkali metal ions include aliphatic carboxylates, aromatic carboxylates, carbonates, hydrochlorides, nitrates, sulfates, phosphates and metal complexes. Of these, aliphatic carboxylates and phosphates are preferable because they are easily available, and specifically, sodium acetate, potassium acetate, sodium phosphate and potassium phosphate are preferable.
- divalent metal ions As metal ions, it may be preferable to include divalent metal ions as metal ions.
- the metal ion contains a divalent metal ion, for example, thermal deterioration of EVOH when the trim is recovered and reused is suppressed, and the generation of gel and lumps in the obtained molded product may be suppressed.
- the divalent metal ion include ions of beryllium, magnesium, calcium, strontium, barium and zinc, but magnesium, calcium or zinc ions are preferable from the viewpoint of industrial availability.
- the above-mentioned metal atom (F) exists as an ion, it is included in the divalent metal ion.
- Examples of the divalent metal salt that gives a divalent metal ion include a carboxylate, a carbonate, a hydrochloride, a nitrate, a sulfate, a phosphate and a metal complex, and a carboxylate is preferable.
- the carboxylic acid constituting the carboxylic acid salt is preferably a carboxylic acid having 1 to 30 carbon atoms, and specifically, acetic acid, stearic acid, lauric acid, montanic acid, behenic acid, octyl acid, sebacic acid, ricinolic acid, and the like. Examples thereof include myristic acid and palmitic acid, and acetic acid and stearic acid are preferable.
- the gas barrier resin composition of the present invention is, for example, an antistatic agent, a processing aid, a resin other than the above-mentioned resins, a stabilizer, an ultraviolet absorber, and a plasticizer as long as the effect of the present invention is not impaired.
- the blocking inhibitor examples include oxides, nitrides, and nitride oxides of elements selected from silicon, aluminum, magnesium, zirconium, cerium, tungsten, molybdenum, etc. Among these, silicon oxide is preferable because of its availability. .. When the gas barrier resin composition of the present invention contains a blocking inhibitor, blocking resistance can be enhanced.
- processing aid examples include fluorine-based processing aids such as Arkema's Kynar (trademark) and 3M's Dynamer (trademark).
- fluorine-based processing aids such as Arkema's Kynar (trademark) and 3M's Dynamer (trademark).
- Examples of other resins include various polyolefins (polyethylene, polypropylene, poly1-butene, poly4-methyl-1-pentene, ethylene-propylene copolymers, and polymers of ethylene and ⁇ -olefins having 4 or more carbon atoms.
- Stabilizers for improving melt stability, etc. include hydrotalcite compounds, hindered phenol-based, hindered amine-based heat stabilizers, metal salts of higher aliphatic carboxylic acids (for example, calcium stearate, magnesium stearate, etc.) and the like. Can be mentioned.
- the content thereof is preferably 0.001 to 1% by mass.
- ultraviolet absorber examples include ethylene-2-cyano-3', 3'-diphenylacrylate, 2- (2'-hydroxy-5'-methylphenyl) benzotriazole, and 2- (2'-hydroxy-3'-t. -Butyl-5'-methylphenyl) 5-chlorobenzotriazole, 2-hydroxy-4-methoxybenzophenone, 2,2'-dihydroxy-4-methoxybenzophenone and the like can be mentioned.
- plasticizer examples include dimethyl phthalate, diethyl phthalate, dioctyl phthalate, wax, liquid paraffin, phosphate ester and the like.
- antistatic agent examples include pentaerythritol monostearate, sorbitan monopalmitate, sulfated polyolefins, polyethylene oxide, carbowax and the like.
- lubricant examples include ethylene bisstearoamide and butyl stearate.
- colorant examples include carbon black, phthalocyanine, quinacridone, indoline, azo pigments, red iron oxide and the like.
- filler examples include glass fiber, asbestos, ballastonite, calcium silicate and the like.
- desiccant examples include phosphate (excluding the above phosphate), sodium borate, sodium sulfate, sodium chloride, sodium nitrate, sugar, silica gel, bentonite, molecular sieve, highly water-absorbent resin and the like.
- the water content of the gas barrier resin composition of the present invention is preferably 3.0 parts by mass or less with respect to 100 parts by mass in total of one or more types of EVOH from the viewpoint of preventing the generation of voids during molding. .0 parts by mass or less is more preferable, 0.5 parts by mass or less is further preferable, and 0.3 parts by mass or less is particularly preferable.
- the gas barrier resin composition of the present invention may contain impurities derived from biomass due to EVOH (A) or EVOH (A'). It may contain various impurities, but it tends to contain at least a large amount of metals such as iron and nickel.
- the lower limit of the biobase degree of the gas barrier resin composition of the present invention is preferably 1%, more preferably 5%, further preferably 20%, and particularly preferably 40% from the viewpoint of reducing the environmental load. Further, for example, in applications where particularly excellent long-running property is not required, the lower limit of the biobase degree of the gas barrier resin composition may be 60% or 80%.
- the upper limit of the biobase degree of the gas barrier resin composition is preferably 99%, more preferably 95%, 85%, 75%, 65%, 55%, 45%, 35 from the viewpoint of good long-running property. % Or 25% may be more preferred.
- the biobase degree of this gas barrier resin composition means a value measured in consideration of other resins and the like contained in arbitrary components other than EVOH.
- the method for producing the gas barrier resin composition of the present invention is not particularly limited.
- a gas barrier resin composition containing EVOH (A) and EVOH (B) (1) A method of mixing (dry blending) EVOH (A) pellets, EVOH (B) pellets, and the above-mentioned other components as necessary, and melting and kneading the mixed pellets.
- the pellets of EVOH (A) and the pellets of EVOH (B) After immersing the pellets of EVOH (A) and / or the pellets of EVOH (B) in a solution containing the above-mentioned other components as necessary, the pellets of EVOH (A) and the pellets of EVOH (B) A method of dry blending with pellets and melting and kneading them, (3) The pellets of EVOH (A) and the pellets of EVOH (B) are dry-blended, and when they are melt-kneaded, an aqueous solution containing the above-mentioned other components is added in the middle of the extruder as needed.
- Method (4) A method of blending the molten resin of EVOH (A) and the molten resin of EVOH (B) in a molten state (other components are previously contained in EVOH (A) and / or EVOH (B), if necessary. Even if it is left untouched, it may be liquid-added in the extruder.) And so on.
- the effect of reducing the environmental load and long-running property are achieved by adjusting the mixing ratio of EVOH (A) and EVOH (B) according to the application, performance, etc. It is possible to produce a gas barrier resin composition having a high gas barrier property while considering the balance with the above.
- a ribbon blender, a high-speed mixer conider, a mixing roll, an extruder, an intensive mixer, or the like can be used.
- EVOH (A') is synthesized by a known method, and the obtained EVOH (A') is subjected to other methods as necessary. It can be produced by mixing the components of.
- a method of blending a molten resin of EVOH (A) and a molten resin of EVOH (B) in a molten state after containing inorganic particles (C) other components are EVOH (A) and, if necessary, EVOH (A) and / Or it may be contained in EVOH (B) in advance, or it may be liquid-added in the extruder).
- a method of adding and mixing the inorganic particles (C) alone or as a dispersion liquid using an appropriate solvent can be mentioned.
- the balance between the effect of reducing the environmental load and the long-running property is considered by adjusting the mixing ratio of each component according to the application, performance, etc.
- a method for producing a gas barrier resin composition comprising a step of dry blending an EVOH (A) pellet, an EVOH (B) pellet, and a thermoplastic elastomer (G) pellet and melt-kneading the pellet is suitable for the present invention. It is a form.
- a ribbon blender, a high-speed mixer conider, a mixing roll, an extruder, an intensive mixer, or the like can be used.
- EVOH (A') is synthesized by a known method, and the obtained EVOH (A') is known. It can be produced by mixing inorganic particles (C) or the like by a method and, if necessary, mixing other components by a known method.
- the gas barrier resin composition of the present invention further contains PA (E) and a metal atom (F), as a method for producing such a gas barrier resin composition, one or more kinds of EVOH pellets and PA (E) are used. ) And a compound containing at least one metal atom (F) selected from the group consisting of magnesium, calcium and zinc are mixed and melt-kneaded.
- the form of the compound containing the metal atom (F) at the time of mixing in this production method may be a solid state or an aqueous solution.
- the EVOH pellets, the PA (E) pellets, and the compound containing the metal atom (F) may be mixed in one step or in two or more steps.
- each solid component can be dry-blended.
- EVOH pellets and PA (E) pellets are dry-blended, and when these are melt-kneaded, an aqueous solution containing a metal atom (F) is liquid-added in the middle of the extruder. It can be done by doing something like that.
- the molded product containing the gas barrier resin composition of the present invention is a preferred embodiment of the present invention.
- the gas barrier resin composition may be a single-layer structure molded body, or may be a multi-layered molded body of two or more kinds, that is, a multi-layered structure together with other various base materials and the like.
- Examples of the molding method of the molded product of the present invention include extrusion molding, thermoforming, malformed molding, hollow molding, rotary molding, and injection molding.
- the molded product using the gas barrier resin composition of the present invention has various uses, for example, films, sheets, containers (bags, cups, tubes, trays, bottles, paper cartons, etc.), fuel containers, tanks, pipes, hoses.
- the film, sheet, pipe and hose can be molded by extrusion molding
- the container shape can be molded by injection molding
- the hollow container such as a bottle or tank can be molded by hollow molding or rotary molding.
- the hollow molding include extrusion hollow molding in which a parison is formed by extrusion molding and then blown to form the preform, and injection hollow molding in which a preform is formed by injection molding and then blown to form the preform.
- a method of forming a packaging material such as a multilayer film by extrusion molding and a method of thermoforming a multilayer sheet formed by extrusion molding into a container-shaped packaging material are preferably used.
- the molded product of the present invention may include a gas barrier layer (hereinafter, may be abbreviated as "layer (1)") formed from the gas barrier resin composition of the present invention.
- the molded body may be a single-layer structure, but is preferably a multi-layer structure further including other layers. That is, the multilayer structure A of the present invention includes at least one layer made of the gas barrier resin composition of the present invention.
- a multilayer film, a multilayer sheet, a multilayer pipe, a multilayer structure B and the like, which will be described later, are also included in the form of the multilayer structure A.
- As the lower limit of the number of layers of the multilayer structure A 2 is preferable, and 3 is more preferable.
- the upper limit of the number of layers of the multilayer structure A may be, for example, 1000, 100, 20 or 10.
- a thermoplastic resin layer containing a thermoplastic resin other than the gas barrier resin composition of the present invention as a main component for example, a thermoplastic resin layer containing a thermoplastic resin other than the gas barrier resin composition of the present invention as a main component (hereinafter, “layer (2)””.
- layer (3) an adhesive layer containing an adhesive resin, an anchor coating agent or an adhesive as a main component
- layer (4) an adhesive layer containing an adhesive resin, an anchor coating agent or an adhesive as a main component
- a recovery layer hereinafter, abbreviated.
- layer (4) an inorganic vapor-deposited layer
- layer (6) paper substrate layer
- the multilayer structure A of the present invention has different suitable embodiments depending on its use, but basically includes a layer (1) and a layer (2), and the layer (1), the layer (2) and the layer (3) are preferably included. ) Is more preferable.
- the multilayer structure A is a container, a packaging material, a tube, a pipe, or the like, the layer (2), the layer (3), the layer (1), from the inner surface to the outer surface.
- Layer (3), layer (4), layer (2) in that order (hereinafter referred to as (inside) 2/3/3/4/2 (outside)), (inside) 2/3/1 / 3/2 (outside), (inside) 2/4/3/3/1/3/4/2 (outside), (inside) 4/3/1/3/4 (outside), (inside) 1/3 / 2 (outside), (inside) 2/4/3/3/2 (outside), (inside) 2/1/3/2 (outside), (inside) 2/1/5/2 (outside) ), (Inside) 2/5/1/2 (outside), (inside) 2/5/1/5/2 (outside), (inside) 2/3/1/5/3/2 (outside), (Inside) 2/3/5/1/3/2 (Outside), (Inside) 2/3/1/3/2/6 (Outside), (Inside) 3/1/3/6 (Outside), etc.
- the layer (3) may be omitted, or the layer (4) may be provided instead of the layer (2), and a plurality of layers (1) to (6) are provided.
- the resins constituting each layer may be the same or different.
- the average thickness of the multi-layer structure A or the molded body of the present invention varies depending on the intended use, but is preferably 5 ⁇ m to 15 mm, more preferably 10 ⁇ m to 10 mm.
- the lower limit of the ratio of the average thickness of the layer (1) to the average thickness of the multilayer structure A of the present invention is not particularly limited, and 1% is preferable, and 2% may be more preferable.
- the ratio of the average thickness of the layer (1) is preferably 20%, more preferably 15%. When the ratio of the average thickness of the layer (1) is within the above range, good gas barrier properties and long-run properties are exhibited, and productivity tends to be good.
- the layer (2) is a thermoplastic resin layer containing a thermoplastic resin other than EVOH as a main component.
- the proportion of the thermoplastic resin other than EVOH in the layer (2) is preferably 70% by mass or more, more preferably 80% by mass or more, further preferably 90% by mass or more, particularly preferably 95% by mass or more, and 99% by mass or more.
- the resin constituting the layer (2) may be substantially composed of only a thermoplastic resin other than EVOH.
- thermoplastic resin as the main component of the layer (2) examples include various polyolefins (polyethylene, polypropylene, poly1-butene, poly4-methyl-1-pentene, ethylene-propylene copolymer, ethylene and 4 or more carbon atoms. ⁇ -olefin copolymer, polyolefin and maleic anhydride copolymer, ethylene-vinyl ester copolymer, ethylene-acrylic acid ester copolymer, or grafting these with unsaturated carboxylic acid or a derivative thereof.
- polyolefins polyethylene, polypropylene, poly1-butene, poly4-methyl-1-pentene, ethylene-propylene copolymer, ethylene and 4 or more carbon atoms.
- Modified polyolefins etc.
- various polyamides nylon 6, nylon 6.6, nylon 6/66 copolymer, nylon 11, nylon 12, polymethoxylylen adipamide, etc.
- various polyesters polyethylene terephthalate, polybutylene terephthalate, etc.) , Polyethylene naphthalate, etc.
- Polyvinyl chloride Polyvinylidene chloride, Polystyrene, Polyacrylonitrile, Polyurethane, Polycarbonate, Polyacetal, Polyacrylate and Modified polyvinyl alcohol resin.
- the thermoplastic resin layer may be unstretched, or may be uniaxially or biaxially stretched or rolled.
- polyolefin is preferable in terms of moisture resistance, mechanical properties, economy and heat sealability
- polyamide and polyester are preferable in terms of mechanical properties and heat resistance.
- the lower limit of the MFR of the thermoplastic resin under a load of 190 ° C. and 2,160 g is preferably 0.01 g / 10 minutes, more preferably 0.02 g / 10 minutes.
- the upper limit of the MFR is preferably 0.5 g / 10 minutes, more preferably 0.1 g / 10 minutes, and even more preferably 0.05 g / 10 minutes.
- thermoplastic resin can be used by appropriately selecting it from commercially available products.
- the layer (2) may contain other optional components similar to those of the gas barrier resin composition of the present invention constituting the layer (1) as long as the effects of the present invention are not impaired.
- the lower limit of the average thickness of the layer (2) per layer is preferably 5 ⁇ m, more preferably 20 ⁇ m.
- the upper limit of the average thickness of the layer (2) per layer is preferably 3,000 ⁇ m, more preferably 1,000 ⁇ m.
- the lower limit of the ratio of the average thickness of the layer (2) to the average thickness of the multilayer structure A of the present invention is not particularly limited, and is preferably 10%, more preferably 30%, still more preferably 50%, 70% or 80%. Sometimes.
- the upper limit of the ratio of the average thickness of the layer (2) is not particularly limited, and is preferably 95%, more preferably 90%.
- the layer (3) may be arranged between the layer (1) and the layer (2), and is a layer containing an adhesive resin, an anchor coating agent, or an adhesive as a main component.
- the layer (3) can function as an adhesive layer between the layer (1) and another layer such as the layer (2).
- the adhesive resin is a resin having adhesiveness, and a thermoplastic resin having adhesiveness may be preferable.
- the anchor coating agent and the adhesive may be a resin, may be a non-resin such as a small molecule compound, or may be composed of a plurality of components. Examples of the adhesive resin include carboxylic acid-modified polyolefins and the like.
- the carboxylic acid-modified polyolefin has a carboxy group or an anhydride group thereof obtained by chemically bonding an ethylenically unsaturated carboxylic acid or an anhydride thereof to an olefin polymer by an addition reaction, a graft reaction, or the like.
- An olefinic polymer An olefinic polymer.
- the lower limit of the MFR of the adhesive resin under a load of 190 ° C. and 2,160 g is preferably 0.1 g / 10 minutes, more preferably 0.2 g / 10 minutes, and even more preferably 0.3 g / 10 minutes.
- the upper limit of the MFR is preferably 15 g / 10 minutes, more preferably 10 g / 10 minutes, and even more preferably 5 g / 10 minutes.
- an industrially manufactured commercial product can be used, and examples thereof include trade names "ADMER NF642E", "ADMER AT2235E", and "ADMER NF408E” manufactured by Mitsui Chemicals, Inc. ..
- the layer (3) may contain other optional components similar to the layer (1) in addition to the adhesive resin, the anchor coating agent and the adhesive, as long as the effects of the present invention are not impaired.
- the layer (3) can be formed by applying these to the surface of the layer adjacent to the layer (3) and drying as necessary. Adhesiveness may be improved by performing a surface treatment such as a corona discharge treatment on the coated surface before these coatings.
- the adhesive is not particularly limited, and for example, it is preferable to use a two-component reaction type polyurethane adhesive in which a polyisocyanate component and a polyol component are mixed and reacted.
- the anchor coating agent and the adhesive may be further improved in adhesiveness by adding a small amount of a known silane coupling agent or the like.
- the silane coupling agent include a silane coupling agent having a reactive group such as an isocyanate group, an epoxy group, an amino group, a ureido group and a mercapto group.
- the lower limit of the average thickness of the layer (3) per layer is preferably 1 ⁇ m, more preferably 3 ⁇ m.
- the upper limit of the average thickness of the layer (3) per layer is preferably 300 ⁇ m, more preferably 150 ⁇ m. When the average thickness of the layer (3) per layer is within the above range, it tends to show good adhesiveness at low cost.
- the layer (4) is a layer containing, for example, EVOH, a thermoplastic resin, and an adhesive resin. Further, the layer (4) is preferably formed by using the recovered material of the layer (1), the layer (2) and the layer (3) in the manufacturing process of the multilayer structure A of the present invention. Examples of the recovered product include burrs generated in the manufacturing process of the multilayer structure A, products that have failed the certification, and the like.
- the layer (4) can be used as a substitute for the above-mentioned layer (2), but in general, the mechanical strength of the layer (4) is often lower than that of the layer (2). It is preferable to use the layer (2) and the layer (4) in a laminated manner.
- the layer (4) having a weak strength is arranged on the outer layer side rather than the layer (1).
- layers (4) which are recovery layers can be arranged on both sides of the layer (1).
- the layer (5) is an inorganic thin-film deposition layer.
- the layer (5) is usually a layer having a barrier property against oxygen and water vapor, and is preferably transparent.
- the layer (5) can be formed by depositing an inorganic substance.
- the inorganic substance include a metal (for example, aluminum), a metal oxide (for example, silicon oxide, aluminum oxide), a metal nitride (for example, silicon nitride), a metal nitride oxide (for example, silicon nitride), or a metal carbide.
- Objects (for example, silicon nitride) and the like can be mentioned.
- the layer (5) formed of aluminum oxide, silicon oxide, magnesium oxide, or silicon nitride is preferable from the viewpoint of excellent transparency.
- the method for forming the layer (5) is not particularly limited, and is a vacuum vapor deposition method (for example, resistance heating vapor deposition, electron beam vapor deposition, molecular beam epitaxy method, etc.), a physical vapor deposition method such as a sputtering method or an ion plating method; Thermochemical vapor deposition (eg, catalytic chemical vapor deposition), photochemical vapor deposition, plasma chemical vapor deposition (eg, capacitive coupled plasma, induced coupled plasma, surface wave plasma, electron cyclotron resonance, dual magnetron) , Atomic layer deposition method, etc.), chemical vapor deposition method such as organic metal vapor deposition method, etc.
- a vacuum vapor deposition method for example, resistance heating vapor deposition, electron beam vapor deposition, molecular beam epitaxy method, etc.
- a physical vapor deposition method such as a sputtering method or an ion plating method
- Thermochemical vapor deposition
- the thickness of the layer (5) varies depending on the type of the component constituting the inorganic thin-film deposition layer, but is preferably 0.002 to 0.5 ⁇ m, more preferably 0.005 to 0.2 ⁇ m, and 0.01 to 0.1 ⁇ m. Is more preferable.
- the thickness of the layer (5) is 0.002 ⁇ m or more, the barrier property of the layer (5) to oxygen and water vapor tends to be good. Further, when the thickness of the layer (5) is 0.5 ⁇ m or less, the barrier property after bending of the layer (5) tends to be maintained.
- the inorganic thin-film deposition layer provided in the thin-film deposition film described later is also a suitable form of the layer (5).
- the layer (6) is a paper base material layer.
- the paper base material used for the layer (6) any paper having various types, bending resistance, rigidity, waist, strength, etc. can be used depending on the intended use of the paper container to be applied.
- the main strength material is bleached or unbleached paper, or various types of paper such as pure white roll paper, kraft paper, paperboard, processed paper, and milk base paper can be used.
- the paper base material layer may be a laminate of a plurality of these paper layers.
- the paper substrate layer has a basis weight of 80 to 600 g / m 2 , preferably a basis weight of 100 to 450 g / m 2 , and a thickness of 110 to 860 ⁇ m, preferably 140 to 640 ⁇ m. If the paper substrate layer is thinner than this, the strength as a container is insufficient, and if it is thicker than this, the rigidity becomes too high and processing may be difficult. In addition, for example, characters, figures, symbols, and other desired patterns can be arbitrarily formed on the paper base material layer by a normal printing method.
- the multilayer structure A of the present invention can be produced by a conventionally known molding method such as various melt molding, except that the gas barrier resin composition of the present invention is used.
- a conventionally known molding method such as various melt molding, except that the gas barrier resin composition of the present invention is used.
- the method for melt molding the gas barrier resin composition include extrusion molding, cast molding, inflation extrusion molding, blow molding, injection molding, injection blow molding and the like.
- the film or sheet of the present invention comprises a molded product of the present invention.
- the film means "a film-like soft film having an average thickness of less than 250 ⁇ m"
- the sheet means "a thin plate-like soft film having an average thickness of 250 ⁇ m or more”.
- film or sheet is also referred to as "film or the like”.
- the film or the like of the present invention may be a film or the like made of the molded product of the present invention. That is, one embodiment of the molded product of the present invention may be a film or the like.
- the film or the like of the present invention has a low environmental load and has good gas barrier properties, appearance and productivity.
- the film or the like of the present invention may be a single-layer film composed of only the layer (1), or may be a multilayer film.
- the average thickness of the film or the like of the present invention is preferably, for example, 1 ⁇ m or more and less than 300 ⁇ m, and more preferably 5 ⁇ m or more and less than 100 ⁇ m.
- the film or the like of the present invention can be suitably used as various packaging materials and the like.
- the arithmetic average roughness (Ra) of at least one surface of the film or the like of the present invention measured in accordance with JIS B0601 is preferably 1.0 ⁇ m or less, more preferably 0.8 ⁇ m or less, and further preferably 0.6 ⁇ m or less. It is preferably 0.4 ⁇ m or less, and particularly preferably 0.4 ⁇ m or less.
- the arithmetic mean roughness (Ra) of at least one surface of the film or the like of the present invention is preferably 0.05 ⁇ m or more, more preferably 0.10 ⁇ m or more, further preferably 0.15 ⁇ m or more, and particularly preferably 0.20 ⁇ m or more. .. When the arithmetic average roughness (Ra) of at least one surface of the film or the like of the present invention is within the above range, the fracture resistance is excellent.
- the average length (RSm) of at least one surface contour curve element measured according to JIS B0601 of the film of the present invention is preferably 1000 ⁇ m or less, more preferably 800 ⁇ m or less, further preferably 600 ⁇ m or less, and further preferably 400 ⁇ m. The following are particularly preferred.
- the average length (RSm) of the contour curve element of at least one surface of the film of the present invention is preferably 50 ⁇ m or more, more preferably 100 ⁇ m or more, further preferably 150 ⁇ m or more, and particularly preferably 200 ⁇ m or more.
- the average length (RSm) of the contour curve element on at least one surface of the film or the like of the present invention is within the above range, the fracture resistance is excellent.
- the above-mentioned JIS B0601 represents, for example, JIS B0601: 2001.
- the film or the like of the present invention may be a non-stretched film or the like, but it may be preferable that the film or the like is stretched.
- the strength and the like are improved by being stretched.
- the film or the like of the present invention is a stretched film or the like, the appearance and gas barrier properties are also good because the occurrence of streak-like unevenness that may occur due to stretching is small.
- the film or the like of the present invention can be produced by a known method.
- the method for forming the film or the like is not particularly limited, and examples thereof include a melting method, a solution method, a calendar method, and the like, and the melting method is preferable.
- the melting method include a T-die method (cast method) and an inflation method, and the cast method is preferable.
- the melting temperature in the melting method varies depending on the melting point of the gas barrier resin composition and the like, but is preferably about 150 to 300 ° C. Further, when the film or the like of the present invention has multiple layers, the production method can be produced by a known method, such as a coextrusion method, a dry laminating method, a sand laminating method, an extruded laminating method, a coextruded laminating method, a solution coating method, and the like. Can be adopted.
- the stretching may be uniaxial stretching or biaxial stretching, and biaxial stretching is preferable.
- the biaxial stretching may be either sequential biaxial stretching or simultaneous biaxial stretching.
- the lower limit of the stretch ratio in terms of area is preferably 6 times, more preferably 8 times.
- the upper limit of the draw ratio is preferably 15 times, more preferably 12 times. When the draw ratio is within the above range, the uniformity of the thickness of the film or the like, the gas barrier property, and the mechanical strength can be improved.
- the stretching temperature can be, for example, 60 ° C. or higher and 120 ° C. or lower.
- the method for producing a film or the like of the present invention may include a step of heat-treating the stretched film or the like after the stretching step.
- the heat treatment temperature is usually set to a temperature higher than the stretching temperature, and can be, for example, more than 120 ° C. and 200 ° C. or lower.
- the film or the like of the present invention is suitably used as a material for various packaging containers such as food packaging containers, pharmaceutical packaging containers, industrial chemical packaging containers, and pesticide packaging containers. Further, a heat-shrinkable film or the like and an industrial film or the like, which will be described later, are also included in one embodiment of the film or the like of the present invention.
- the heat-shrinkable film or sheet of the present invention (heat-shrinkable film or heat-shrinkable sheet) comprises the molded body of the present invention.
- the heat-shrinkable film or the like of the present invention may be a heat-shrinkable film or the like made of the molded product of the present invention. That is, one embodiment of the molded product of the present invention may be a heat-shrinkable film or the like.
- the heat-shrinkable film or the like of the present invention has a low environmental load, and has good gas barrier properties, appearance, and productivity.
- heat-shrinkability is imparted by forming a single-layer or multilayer film or the like and then subjecting the film to a stretching step.
- the EVOH used in the layer (1) of the heat-shrinkable film or the like of the present invention preferably has a modifying group (structure) represented by the above formula (I) from the viewpoint of exhibiting good heat-shrinkability.
- the heat-shrinkable film or the like of the present invention is preferably a multilayer film or the like having a layer (1) and a layer (2).
- the thickness of the layer (1) in the multilayer film before stretching is preferably 3 to 250 ⁇ m, more preferably 10 to 100 ⁇ m.
- the thickness of the layer (2) is not particularly limited, and is appropriately selected in consideration of required performance such as moisture permeability, heat resistance, heat sealability, and transparency, and application.
- the total thickness of the multilayer film or the like before stretching is not particularly limited, but is usually 15 to 6000 ⁇ m.
- thermoplastic resin used for the layer (2) in the heat-shrinkable film of the present invention polyolefins such as ethylene-vinyl acetate copolymer, ionomer, and polyethylene are preferably used from the viewpoint of excellent heat-sealing property and heat-shrinkability.
- Polyamide is preferably used from the viewpoint of being used and having excellent mechanical strength such as puncture strength and pinhole resistance.
- thermoplastic resin used for the layer (2) in the heat-shrinkable film of the present invention a configuration in which the polyamide layer and the layer (1) are adjacent to each other is preferably used. With such a configuration, excellent barrier properties and puncture resistance can be obtained. Further, the transparency after shrinkage is excellent as compared with the case where a general-purpose barrier resin is used instead of the layer (1). A configuration in which the adhesive resin layer is not sandwiched between the polyamide layer and the layer (1) is more preferable.
- the polyamide layer / layer (1) / layer (3) / layer (2), layer (2) / polyamide layer / layer (1) / polyamide examples thereof include configurations such as a layer / layer (2), a polyamide layer / layer (1) / polyamide layer / layer (2), and a polyamide layer / polyamide layer / layer (1) / polyamide layer / layer (2).
- the multilayer film or the like can be obtained by various manufacturing methods, and a coextrusion method, a dry laminating method, a sand laminating method, an extrusion laminating method, a coextrusion laminating method, a solution coating method, or the like can be adopted.
- the coextrusion method is a method in which a gas barrier resin composition and another thermoplastic resin are simultaneously extruded from an extruder, laminated under a molten state, and discharged in the form of a multilayer film from a die outlet.
- a method of laminating the layer (1) and the layer (2) with the layer (3) interposed therebetween is preferable.
- the adhesive resin it is preferable to use a polyolefin having a carboxyl group, a carboxylic acid anhydride group or an epoxy group.
- a polyolefin having a carboxyl group, a carboxylic acid anhydride group or an epoxy group is excellent in adhesiveness to a gas barrier resin composition as well as to other thermoplastic resins containing no carboxyl group, carboxylic acid anhydride group or epoxy group.
- the heat-shrinkable film or the like of the present invention is produced by stretching the obtained single-layer or multi-layer film or the like.
- the stretching may be uniaxial stretching or biaxial stretching.
- the biaxial stretching may be simultaneous biaxial stretching or sequential biaxial stretching.
- Examples of the stretching method include a tenter stretching method, a tubular stretching method, and a roll stretching method.
- the heat-shrinkable film or the like of the present invention is preferably stretched at a high magnification. Specifically, a heat-shrinkable film stretched to an area magnification of 7 times or more is particularly suitable.
- the stretching temperature is usually 50 to 130 ° C.
- cross-linking may be performed by irradiation with radiation or the like. From the viewpoint of further enhancing the shrinkage property, it is preferable to quickly cool the film or the like after stretching the film or the like.
- the heat-shrinkable film or the like of the present invention is suitably used as a material for various packaging containers such as food packaging containers, pharmaceutical packaging containers, industrial chemical packaging containers, and pesticide packaging containers.
- the packaging material A of the present invention comprises the film or sheet of the present invention or a heat-shrinkable film or sheet.
- the packaging material A may be a packaging material made of the film or sheet of the present invention or a heat-shrinkable film or sheet. That is, one embodiment of the molded product of the present invention may be a packaging material.
- the packaging material A has a low environmental load, and has good gas barrier properties, appearance, and productivity.
- the packaging material A of the present invention may be a single-layer film or the like, or may be a multilayer film or the like. Further, the multilayer film or the like may further have a layer formed from other than the resin, for example, a paper layer, a metal layer, or the like.
- the packaging material A may be in the form of a film or sheet, or the film or sheet may be secondarily processed. Examples of the packaging material obtained by the secondary processing include (1) a tray cup-shaped container obtained by thermoforming a film or sheet by vacuum forming, vacuum forming, vacuum forming, or the like, (2) a film or.
- Examples thereof include a bottle obtained by performing stretch blow molding on a sheet, a cup-shaped container, (3) a bag-shaped container obtained by heat-sealing a film or a sheet, and the like.
- the secondary processing method is not limited to each of the methods exemplified above, and for example, a known secondary processing method other than the above such as blow molding can be appropriately used.
- the packaging material A of the present invention is used for packaging, for example, foods, beverages, chemicals such as pesticides and pharmaceuticals, medical equipment, machine parts, industrial materials such as precision materials, and clothing.
- the packaging material A is preferably used for applications that require a barrier property against oxygen and applications in which the inside of the packaging material is replaced by various functional gases.
- the packaging material A is formed in various forms depending on the application, for example, a vertical bag filling seal bag, a vacuum packaging bag, a pouch with a spout, a laminated tube container, a lid material for a container, and the like.
- the packaging material A of the present invention may be a vacuum packaging bag.
- An example of a vacuum packaging bag is a bag-shaped container provided with a film or the like of the present invention as a partition wall separating the inside and the outside where the contents are packaged, and the inside is in a depressurized state.
- the two films of the present invention are overlapped with each other, and the peripheral portions of the two films and the like are sealed to each other.
- the partition wall is preferably a multilayer film or the like.
- the vacuum packaging bag can be manufactured using a nozzle type or chamber type vacuum packaging machine.
- the vacuum packaging bag is used for applications where it is desired to wrap in a vacuum state, for example, for storing foods, beverages, etc. Further, the vacuum packaging bag can also be used as an outer packaging material for a vacuum heat insulating body.
- the industrial film or sheet of the present invention comprises a molded body such as a single-layer or multilayer film of the present invention.
- the industrial film or the like may be an industrial film or the like made of the molded product of the present invention. That is, one embodiment of the molded product of the present invention may be an industrial film or the like.
- the industrial film and the like have a low environmental load and have good gas barrier properties, appearance and productivity. Specific examples of industrial films and the like include agricultural films, landfill films and the like, architectural films and the like.
- the industrial film or the like of the present invention is preferably a multilayer film or the like.
- a hydrophobic thermoplastic resin is preferably used for the layer (2) provided in the industrial film or the like for the purpose of preventing deterioration of the gas barrier performance of the layer (1) due to moisture.
- polyolefin-based resins linear low-density polyethylene, low-density polyethylene, ultra-low-density polyethylene, ultra-low-density linear polyethylene, medium-density polyethylene, polyethylene such as high-density polyethylene, and ethylene- ⁇ -.
- Polyolefin resins such as olefin copolymers, polypropylene, ethylene-propylene (block and random) copolymers, polypropylene resins such as propylene- ⁇ -olefin ( ⁇ -olefins with 4 to 20 carbon atoms) copolymers, polybutene , Polypentene, etc .; grafted polyolefins obtained by graft-modifying these polyolefins with unsaturated carboxylic acids or esters thereof, cyclic polyolefin-based resins; ionomers, ethylene-vinyl acetate copolymers, ethylene-acrylic acid copolymers, ethylene-acrylic acid esters.
- Halogenized polyolefins such as copolymers, polyester resins, polyamide resins, polyvinyl chlorides, polyvinylidene chlorides, acrylic resins, polystyrenes, vinyl ester resins, polyester elastomers, polyurethane elastomers, chlorinated polyethylenes, chlorinated polypropylenes, etc.
- Examples thereof include aromatic or aliphatic polyketones.
- polyolefin-based resins are preferable, and polyethylene and polypropylene are particularly preferable in terms of mechanical strength and moldability.
- the lower limit of MFR under a load of 210 ° C. and 2160 g is preferably 1.0 g / 10 minutes, more preferably 2.0 g / 10 minutes.
- the upper limit is preferably 100 g / 10 minutes, more preferably 60 g / 10 minutes.
- the following layer structure can be exemplified as the layer structure of the industrial film or the like of the present invention.
- the layer structure on the left side indicates that the layer is on the outer side (the side exposed to the external environment). 5 layers 1/3/2/3/1, 2/3/3/1/2, 2/3/3/1/3/1 6 layers 2/3/3/2/2 7 layers 2/3/3/3/1/3/2, 2/2/3/3/1/3/2/2
- the layer (1) for the purpose of preventing the deterioration of the oxygen barrier property due to moisture, it is preferable to use the layer (1) as the intermediate layer and the layer (2) as the outer layer.
- a configuration such as 3/1/3/2/2 is more preferable.
- the total thickness of the industrial film or the like of the present invention is usually 5 to 5 mm, preferably 10 to 4.5 mm, more preferably 15 to 4 mm, and particularly preferably 20 to 3.5 mm.
- the thickness of the layer (2) (hydrophobic resin composition layer, etc.) in the industrial film or the like is not particularly limited, but is usually 0.5 to 2.5 mm, preferably 1 to 2 mm, and particularly preferably 1 to 1. It is 1.5 mm.
- the thickness of the layer (1) is not particularly limited, but is preferably in the range of 1 to 20%, preferably 2 to 18%, and more preferably 3 to 15% of the total layer thickness.
- Examples of the above-mentioned architectural film include wallpaper.
- the wallpaper as an embodiment of the industrial film or the like of the present invention has a small environmental load and is excellent in productivity.
- Examples of the landfill film and the like include geomembranes and landfill sheets.
- Geomembrane is a sheet used as a water shield for waste treatment plants.
- the landfill sheet is a sheet that prevents the diffusion of harmful substances generated from industrial waste and the like, and can be used, for example, to prevent the diffusion of radon gas.
- the gas barrier resin composition contains an antioxidant or an ultraviolet resistant agent (ultraviolet absorber, light stabilizer, colorant) or the like from the viewpoint of enabling long-term outdoor use.
- the agricultural film or the like is preferably a multilayer film or the like, and as the layer (2), a hydrophobic thermoplastic resin is preferably used for the purpose of preventing deterioration of the gas barrier performance of the layer (1) due to moisture.
- the layer (2) contains an ultraviolet resistant agent and an adhesive component.
- the ultraviolet resistant agent include an ultraviolet absorber, a light stabilizer, a colorant and the like.
- the blending amount of the above UV resistant agent with respect to the hydrophobic thermoplastic resin is usually 1 to 10% by mass, preferably 2 to 8% by mass, and particularly preferably 3 to 5% by mass with respect to the hydrophobic thermoplastic resin.
- the hydrophobic thermoplastic resin is likely to be deteriorated by ultraviolet rays.
- the mechanical strength of the hydrophobic thermoplastic resin is lowered.
- the adhesive component examples include an aliphatic saturated hydrocarbon resin such as polyisobutene and an alicyclic saturated hydrocarbon resin, and the blending amount with respect to the hydrophobic thermoplastic resin is usually 1 to 30% by mass, preferably 1 to 30% by mass. It is 2 to 20% by mass, particularly preferably 3 to 15% by mass. If the blending amount is appropriate, the films and the like are crimped to each other when wrapping with the agricultural film and the like, and the sealing is easily maintained. If the blending amount is less than the above range, gaps are generated between the films and the like, and air invades the inside, so that the long-term storage property of the contents deteriorates. Further, when the blending amount is larger than the above range, blocking of the multilayer film occurs and it becomes impossible to unwind from the film roll or the like.
- an aliphatic saturated hydrocarbon resin such as polyisobutene and an alicyclic saturated hydrocarbon resin
- the total thickness of the agricultural film or the like is usually 5 to 200 ⁇ m, preferably 10 to 150 ⁇ m, more preferably 15 to 100 ⁇ m, and particularly preferably 20 to 50 ⁇ m.
- the thickness of the layer (2) (hydrophobic resin composition layer, etc.) in the agricultural film or the like is not particularly limited, but is usually 0.5 to 200 ⁇ m, preferably 1 to 100 ⁇ m, and particularly preferably 1 to 10 ⁇ m. be.
- the thickness of the layer (1) is not particularly limited, but is preferably in the range of 1 to 20%, preferably 2 to 18%, and more preferably 3 to 15% of the total layer thickness.
- Agricultural films can be used for various films used in the agricultural field, such as large silo films, soil steaming films, grain storage bags, silage films, vinyl house films, and multi-films.
- a large silo film is a film for wrapping and storing grains. By wrapping the grain in a large silo film and storing it, the grain can be protected from moisture, mold, insects, etc.
- the length of one side of the large silo film is, for example, 1 m or more, and may be 2 m, 3 m, 5 m, or 10 m or more. Further, the large silo film may be processed into a bag shape or the like.
- the soil fumigation film is a film used for preventing the evaporation of a soil fumigant when fumigating soil in a field or the like.
- the grain storage bag is a bag for storing grains in order to protect the grains from moisture, mold, insects, etc., and is also called a hermetic bag or the like.
- the grain storage bag is formed by molding a film having at least one layer made of the gas barrier resin composition of the present invention into a bag shape, a bag shape, or a container shape.
- the grain storage bag is, for example, a square bag, the length of one side thereof is, for example, 0.5 to 2 m.
- the silage film is a film used for manufacturing and packaging silage. Silage obtained by fermenting grass or the like under anaerobic conditions is used as feed for livestock or the like.
- the form of the silo using the silage film is not particularly limited, and various forms such as a bunker silo, an underground (or semi-underground) silo, a bag silo, a tube silo, a stack silo, and a lap silo can be mentioned.
- the grass when making a wrap silo, the grass is first molded into a desired volume (eg 0.1-50 m 3 , preferably 1-30 m 3).
- a desired volume eg 0.1-50 m 3 , preferably 1-30 m 3.
- the size is usually 0.5 to 3 m in diameter, preferably 1 to 2 m, and the height is usually 0.5 to 3 m, preferably 1 to 2 m.
- the silage film is then wrapped around the molded grass using a regular plastic wrap machine to seal the grass.
- the tube of the present invention comprises the molded body of the present invention.
- the tube may be a tube made of the molded product of the present invention. That is, one embodiment of the molded product of the present invention may be a tube.
- the tube has a low environmental load and has good gas barrier properties, appearance and productivity.
- the method for producing the tube of the present invention is not particularly limited, and for example, a method of directly forming a tube by melt molding such as coextrusion molding, co-injection molding, extrusion coating, or heat welding of the film or sheet of the present invention.
- melt molding such as coextrusion molding, co-injection molding, extrusion coating, or heat welding of the film or sheet of the present invention.
- examples thereof include a method of forming into a tube shape, a method of laminating the film or sheet of the present invention with an adhesive, and forming into a tube shape.
- the pipe of the present invention comprises the molded body of the present invention.
- the pipe may be a pipe made of the molded product of the present invention. That is, one embodiment of the molded product of the present invention may be a pipe.
- the pipe has a low environmental load and has good gas barrier properties, appearance and productivity.
- the pipe preferably contains an antioxidant (D) in the gas barrier resin composition.
- the antioxidant is preferably a compound having a hindered amine group and / or a compound having a hindered phenol group from the viewpoint of suppressing oxidative deterioration in use at high temperatures.
- the pipe preferably contains a thermoplastic elastomer (G) in the gas barrier resin composition, and more preferably contains a modified thermoplastic elastomer (g2).
- the use of the pipe is not particularly limited, and it can be used, for example, as a hot water circulation pipe, a heat insulating multi-layer pipe, a fuel pipe, or the like.
- the pipe of the present invention may be a single-layer pipe or a multi-layer pipe, but is preferably a multi-layer pipe.
- the layer structure of the multi-layer pipe the layer structure of the molded body (multi-layer structure A) can be adopted.
- ⁇ Pipe for hot water circulation> When the multi-layer pipe is used as a hot water circulation pipe, a three-layer structure having a layer (2) as the outermost layer (1) / layer (3) / layer (2) is generally adopted.
- a coextrusion coating facility for gas barrier resin composition and adhesive resin to the existing production line for single-layer pipes such as cross-linked polyolefin, it can be easily diverted to the production line for multi-layer pipes, and in fact, many This is because the pipe manufacturer has adopted this configuration.
- Providing a polyolefin layer or the like on both sides of the layer (1) and using the layer (1) as an intermediate layer is effective in preventing scratches on the layer (1).
- a multi-layer pipe is used as a hot water circulation pipe such as a floor heating pipe, it is usually buried under the floor, so the risk of damage to the layer (1) due to physical impact is relatively small, but rather a gas barrier. From the viewpoint of sex, it is desirable to arrange the layer (1) on the outermost layer.
- the gas barrier resin composition shows a large humidity dependence, and the barrier property is lowered under high humidity conditions.
- the layer (1) is mainly located at the farthest place from the inner surface of the pipe in contact with water, which is the most advantageous in terms of the barrier performance of the multi-layer pipe. It has a layered structure.
- the EVOH layer is generally arranged on the outermost layer, it is easily affected by oxidative deterioration because it comes into direct contact with air.
- a gas barrier resin composition containing a compound having a hindered amine group and / or an antioxidant having a hindered phenol group is used, it is arranged on the outermost layer which is less likely to be oxidatively deteriorated even at high temperatures. Therefore, the effect of providing a multi-layer pipe having a good barrier property and reducing the occurrence of cracks due to oxidative deterioration is more effectively exhibited.
- a multi-layer pipe When a multi-layer pipe is used for a heat-insulating multi-layer pipe for district heating and cooling, a three-layer structure (2) / layer (3) / layer (1) in which the layer (1) is arranged inside the layer (2) ( Hereinafter, it may be abbreviated as laminated body 1), or from the viewpoint of preventing scratches on the layer (1), of the layer (2) / layer (3) / layer (1) / layer (3) / layer (2). It is preferable to have a five-layer structure (hereinafter, may be abbreviated as laminated body 2).
- a heat insulating multi-layer pipe for district heating and cooling usually includes a heat insulating foam layer in addition to the layer (1) (a layer formed from a gas barrier resin composition).
- the configuration of the heat insulating multilayer pipe is not particularly limited, but for example, it is preferable to arrange the inner pipe, the heat insulating foam layer surrounding the inner pipe, and the laminated body 1 or 2 as the outer layer in this order.
- the type (material), shape and size of the pipe used for the inner pipe are not particularly limited as long as they can transport a heat medium such as gas or liquid, and the type of heat medium and the use and usage form of the piping material are not particularly limited. It can be appropriately selected according to the above. Specifically, metals such as steel, stainless steel, and aluminum, polyolefins (polyethylene, cross-linked polyethylene (PEX), polypropylene, poly1-butene, poly4-methyl-1-pentene, etc.), and the above-mentioned laminate 1 or 2 and the like. Among these, cross-linked polyethylene (PEX) is preferably used.
- Polyurethane foam polyethylene foam, polystyrene foam, phenol foam, and polyisocyanurate foam can be used as the heat insulating foam, and polyurethane foam is preferably used from the viewpoint of improving heat insulating performance.
- Freon gas various alternative fluorocarbons, water, hydrocarbons chloride, hydrocarbons, carbon dioxide, etc. are used as foaming agents for heat insulating foams, but hydrocarbons, specifically n-, are used from the viewpoint of foaming effect and environmental impact. Pentane and cyclopentane are preferably used.
- a method for manufacturing a heat insulating multi-layer pipe for example, an inner pipe for transporting a heat medium is placed in a pipe-shaped outer layer, the inner pipe is fixed with a spacer to form a double pipe, and then a gap between the inner pipe and the outer layer is formed.
- Examples thereof include a method of injecting various foam stock solutions into the foam to foam and solidify.
- the material of the spacer is not particularly limited, but polyethylene or polyurethane is preferable in order to reduce damage to the inner tube and the outer layer due to the spacer.
- the layer made of the gas barrier resin composition of the present invention further contains the thermoplastic elastomer (G).
- the thermoplastic elastomer (G) By containing the thermoplastic elastomer (G), the crack resistance of the pipe and the like are further enhanced.
- the innermost layer When used for fuel pipes, the innermost layer is formed to be conductive. To that end, the thermoplastic resin in the innermost layer is mixed with a conductive additive, such as carbon black or graphite fiber.
- a conductive additive such as carbon black or graphite fiber.
- the multilayer pipe can be produced, for example, by coextruding a gas barrier resin composition and an adhesive resin on a single-layer pipe such as a crosslinked polyolefin.
- a film in which the gas barrier resin composition and the adhesive resin are melted may be simply coated on the single-layer pipe.
- the adhesive force between the single layer pipe and the coat layer may be insufficient, and the coat layer may peel off during long-term use and lose the gas barrier property.
- it is effective to perform frame treatment and / or corona discharge treatment on the surface of the pipe to be coated before coating.
- multi-layer molding method for manufacturing multi-layer pipes a number of extruders corresponding to the types of resin layers are used, and simultaneous extrusion molding is performed in a layered state in which the flows of the melted resin are overlapped in the extruder. There is a method of carrying out by so-called coextrusion molding. Further, a multi-layer molding method such as dry lamination can also be adopted.
- the method for manufacturing a multi-layer pipe may include a step of cooling with water at 10 to 70 ° C. immediately after molding. That is, it is desirable to solidify the layer (1) by cooling with water at 10 to 70 ° C. after melt molding and before the layer (1) solidifies. If the temperature of the cooling water is too low, cracks due to strain are likely to occur in the layer (1) of the bent portion when the multilayer pipe is bent in the subsequent secondary processing step. The details of the cause of the tendency for cracks to occur due to strain are not clear, but it is presumed that the residual stress in the molded product has an effect. From this viewpoint, the temperature of the cooling water is more preferably 15 ° C. or higher, further preferably 20 ° C. or higher.
- the temperature of the cooling water is more preferably 60 ° C. or lower, further preferably 50 ° C. or lower.
- Various molded bodies can be obtained by secondary processing the multi-layer pipe obtained by the above method.
- the secondary processing method is not particularly limited, and a known secondary processing method can be appropriately used. For example, in a state where the multilayer pipe is heated to 80 to 160 ° C. and then deformed into a desired shape, 1 A method of processing by fixing for 2 minutes to 2 hours can be mentioned.
- the thermoformed container of the present invention comprises the molded body of the present invention.
- the thermoformed container may be a thermoformed container made of the molded product of the present invention. That is, one embodiment of the molded product of the present invention may be a thermoformed container.
- the thermoformed container has a low environmental load and has good gas barrier properties, appearance and productivity.
- the thermoformed container is used in various fields such as foods, cosmetics, medical chemicals, toiletries and the like, where oxygen barrier properties are required.
- the thermoformed container is formed as having an accommodating portion, for example, by thermoforming a single-layer or multi-layer film or sheet.
- the storage part is a part that stores the contents such as food.
- the shape of this accommodating portion is determined according to the shape of the contents.
- the thermoformed container is formed as, for example, a cup-shaped container, a tray-shaped container, a bag-shaped container, a bottle-shaped container, a pouch-shaped container, or the like.
- the form of the accommodating portion can be expressed by the aperture ratio (S) as one index.
- the aperture ratio (S) is a value obtained by dividing the depth of the deepest part of the container by the diameter of the circle having the maximum diameter inscribed in the opening of the container. That is, the aperture ratio (S) means that the larger the value, the deeper the bottom of the container, and the smaller the value, the shallower the bottom of the container.
- the drawing ratio (S) is large, and when it is a tray, the drawing ratio (S) is small.
- the diameter of the inscribed maximum diameter circle is, for example, the diameter of the circle when the opening of the accommodating portion is circular, the minor diameter (minor axis length) when it is elliptical, and short when it is rectangular. The length of the side.
- the suitable value of the aperture ratio (S) differs depending on the film or sheet thickness.
- the drawing ratio (S) is preferably 0.2 or more, more preferably 0.3 or more, still more preferably 0.4 or more.
- the drawing ratio (S) is preferably 0.3 or more, more preferably 0.5 or more, still more preferably 0.8 or more.
- the total thickness I of the other layers laminated on one surface side of the layer (1) and the total thickness O of the other layers laminated on the other surface side of the layer (1) As the lower limit of the thickness ratio (I / O) with, 1/99 is preferable, and 30/70 is more preferable.
- the upper limit of the I / O is preferably 70/30, more preferably 55/45.
- the thickness of all layers or a single layer of the thermoformed container is an average value of the thicknesses measured by observation with an optical microscope for samples cut out from a plurality of locations of the thermoformed container using a microtome.
- the lower limit of the overall average thickness of the thermoformed container of the present invention is preferably 300 ⁇ m, more preferably 500 ⁇ m, and even more preferably 700 ⁇ m.
- the upper limit of the overall average thickness of the thermoformed container is preferably 10,000 ⁇ m, more preferably 8500 ⁇ m, and even more preferably 7,000 ⁇ m.
- the overall average thickness refers to the thickness of all layers in the housing portion of the thermoformed container.
- the multilayer sheet can be formed by using a coextrusion molding apparatus.
- This multilayer sheet can be formed as having a predetermined layer structure by, for example, charging a gas barrier resin composition or another resin forming each layer into separate extruders and co-extruding them with these extruders.
- Extrusion molding of each layer is performed by operating an extruder equipped with a uniaxial screw at a predetermined temperature.
- the temperature of the extruder forming the layer (1) is, for example, 170 ° C. or higher and 260 ° C. or lower.
- the temperature of the extruder forming the layer (2) to the layer (4) is, for example, 150 ° C. or higher and 260 ° C. or lower.
- thermoformed container of the present invention can be formed by heating and softening a multilayer sheet or the like and then molding it into a mold shape.
- thermoforming method for example, vacuum or compressed air is used, and if necessary, a plug is also used to form a mold shape (straight method, drape method, air slip method, snapback method, plug assist method, etc.), press molding. How to do it.
- Various molding conditions such as molding temperature, degree of vacuum, compressed air pressure, and molding speed are appropriately set according to the shape of the plug, the shape of the mold, the properties of the raw material resin, and the like.
- the molding temperature is not particularly limited as long as the resin can be softened sufficiently for molding, and the suitable temperature range differs depending on the configuration of the multilayer sheet or the like.
- This heating temperature is usually lower than the melting point of the resin.
- the lower limit of the heating temperature of a specific multilayer sheet or the like is usually 50 ° C., preferably 60 ° C., more preferably 70 ° C.
- the upper limit of the heating temperature is, for example, 180 ° C., and may be 160 ° C.
- the thermoformed container of the present invention may be provided with at least the layer (1), and may be composed of a single layer or a plurality of layers.
- the layer structure may be appropriately set according to the intended use and the like.
- thermoformed container of the present invention is composed of a plurality of layers, it is preferable to arrange the layer (2) on the outermost layer. That is, from the inner surface to the outer surface of the accommodating portion, layer (2) / layer (3) / layer (1) / layer (3) / layer (2) (hereinafter, "(inner surface) (2) / (3) / (1) / (3) / (2) (outer surface) ”) is preferable from the viewpoint of impact resistance. Further, as a layer structure when the layer (4) which is a recovery layer is included, for example, (inner surface) (2) / (3) / (1) / (3) / (4) / (2) (outer surface).
- a layer configuration may be provided in which the layer (4) is provided instead of the layer (2).
- the resins constituting each layer may be the same or different.
- thermoformed container of the present invention will be specifically described by taking the cup-shaped container shown in FIGS. 1 and 2 as an example.
- the cup-shaped container is only an example of a thermoformed container, and the following description of the cup-shaped container does not limit the scope of the present invention.
- the cup-shaped container 1 of FIGS. 1 and 2 includes a cup body 2 as an accommodating portion and a flange portion 3.
- the cup-shaped container 1 is used by accommodating the contents in the cup body 2 and sealing the lid 7 on the flange portion 3 so as to close the opening 4 of the cup body 2.
- the lid 7 include a resin film, a metal foil, a metal resin composite film, and the like, and among these, a metal resin composite film in which a metal layer is laminated on a resin film is preferable.
- the resin film include a polyethylene film and a polyethylene terephthalate film.
- the metal layer is not particularly limited, and a metal foil and a metal vapor deposition layer are preferable, and an aluminum foil is more preferable from the viewpoint of gas barrier property and productivity.
- the cup-shaped container 1 is usually obtained by thermoforming a multilayer sheet. It is preferable that the multilayer sheet includes at least a layer (1), and another layer is laminated on the layer (1). Examples of other layers include layer (2), layer (3), layer (4) and the like. Specific examples of the layer structure of the multilayer sheet are as described above.
- the cup-shaped container 1 is manufactured by heating a continuous multilayer sheet 21 with a heating device 30 to soften it, and then thermoforming it using a mold device 40.
- the heating device 30 includes a pair of heaters (heater 31 and heater 32), and the continuous multilayer sheet 21 can pass between the heater 31 and the heater 32.
- a device that is heated by a hot press can also be used.
- the mold device 40 is suitable for thermoforming by the plug assist method, and includes a lower mold 50 and an upper mold 51 housed in a chamber (not shown).
- the lower mold 50 and the upper mold 51 can be individually moved in the vertical direction, and the continuous multilayer sheet 21 can pass between the lower mold 50 and the upper mold 51 in a separated state.
- the lower mold 50 has a plurality of recesses 52 for forming the accommodating portion of the cup-shaped container 1.
- the upper die 51 includes a plurality of plugs 53 projecting toward the lower die 50.
- the plurality of plugs 53 are provided at positions corresponding to the plurality of recesses 52 of the lower mold 50. Each plug 53 can be inserted into the corresponding recess 52.
- the continuous multilayer sheet 21 softened by the heating device 30 is brought into close contact with the lower mold 50 by moving the lower mold 50 upward, and the continuous multilayer sheet 21 is brought into close contact with the lower mold 50. Is slightly lifted to give tension to the continuous multilayer sheet 21.
- the plug 53 is inserted into the recess 52 by moving the upper mold 51 downward.
- the upper mold 51 is moved upward to separate the plug 53 from the recess 52, and then the inside of the chamber (not shown) is evacuated to draw the continuous multilayer sheet 21 into the recess 52. Adhere to the inner surface. After that, the shape is fixed by cooling the molded portion by injecting air. Subsequently, as shown in FIG. 4D, the inside of the chamber (not shown) is opened to the atmosphere and the lower mold 50 is moved downward to release the lower mold 50, whereby a primary molded product is obtained. By cutting this primary molded product, the cup-shaped container 1 shown in FIGS. 1 and 2 can be obtained.
- thermoformed container of the present invention is not limited to the above-mentioned form, and the tray-shaped container is also included in the thermoformed container of the present invention.
- the tray-shaped container can also be manufactured by the same method as the cup-shaped container described above.
- the tray-shaped container is suitably used as a food tray or the like.
- the blow-molded container of the present invention includes the molded product of the present invention.
- the blow-molded container may be a blow-molded container made of the molded product of the present invention. That is, one embodiment of the molded product of the present invention may be a blow molded container.
- the blow-molded container has a low environmental load and has good barrier properties, appearance and productivity.
- the blow-molded container can be used for various containers that require gas barrier properties, oil resistance, and the like.
- the blow-molded container of the present invention is, for example, from the inner surface of the container toward the outer surface of the container, the layer (2), the layer (3), the layer (1), the layer (3), the layer (4), and the layer (2).
- (inside) 2/3/3/4/2 (outside)) (inside) 2/3/1/3/2 (outside), (inside) 2/4
- Layered structures such as / 3/4/3/4/2 (outside) and (inside) 4/3/1/3/4 (outside) can be adopted.
- a configuration may include a layer (4) instead of the layer (2), and in the case of an arrangement in which a plurality of layers (1) to (4) are used, the resins constituting each layer are the same but different. You may.
- the blow-molded container of the present invention is preferably manufactured by a manufacturing method including a step of blow-molding using a gas barrier resin composition.
- Blow molding can be performed by a known method such as direct blow molding, injection blow molding, sheet blow molding, and free blow molding.
- a gas barrier resin composition pellet forming the layer (1) and, if necessary, each resin forming each of the other layers are used, and blow molding is performed at a temperature of 100 ° C to 400 ° C by a blow molding machine. Then, the mold is cooled at a temperature of 10 ° C to 30 ° C for 10 seconds to 30 minutes.
- the heating temperature at the time of blow molding may be 150 ° C. or higher, and may be 180 ° C. or 200 ° C. or higher. Further, this heating temperature may be equal to or higher than the melting point of the gas barrier resin composition. On the other hand, the upper limit of this heating temperature may be 350 ° C., and may be 300 ° C. or 250 ° C.
- the blow-molded container of the present invention is used for various purposes such as a fuel container and various bottles.
- the blow molded container of the present invention can be used as a fuel container.
- the fuel container of the present invention may include a filter, a fuel gauge, a baffle plate, and the like.
- the fuel container of the present invention has a low environmental load, good barrier properties, appearance and productivity, and is suitably used as a fuel container.
- the fuel container is a fuel container mounted on an automobile, a motorcycle, a ship, an aircraft, a generator, industrial or agricultural equipment, or a portable fuel container for refueling these fuel containers, and further.
- gasoline particularly oxygen-containing gasoline blended with methanol, ethanol, MTBE, etc.
- methanol, ethanol, MTBE, etc. is mentioned as a fuel, but heavy oil, light oil, kerosene, etc. are also included.
- the fuel container of the present invention is particularly preferably used as a fuel container for oxygen-containing gasoline.
- the blow-molded container of the present invention can be used as a bottle container.
- the bottle container of the present invention may further include a structure other than the blow-molded container of the present invention, such as a cover film and a cap.
- Examples of the method for molding a bottle container of the present invention include direct blow molding and injection blow molding.
- the blow-molded container of the present invention molded into a bottle shape has a low environmental load and has good barrier properties, appearance and productivity, and is therefore preferably used for bottle containers for foods, cosmetics and the like.
- the paper container of the present invention comprises the molded product of the present invention.
- the paper container may be a paper container made of the molded product of the present invention. That is, one embodiment of the molded product of the present invention may be a paper container.
- the paper container is made of a molded body containing a paper base material, and is made by processing it into a shape such as a carton or a cup. Such a paper container can store various beverages and the like for a long period of time.
- the molded product containing the paper substrate can be formed at a high speed by being formed by, for example, extrusion coating by the T-die method.
- the single-layer film of the present invention is a film formed from the gas barrier resin composition of the present invention. That is, the single-layer film is a film consisting only of a layer formed from the gas barrier resin composition of the present invention.
- the single-layer film has a low environmental load and a good gas barrier property. Further, when the single-layer film is formed from the gas barrier resin composition of the present invention containing the inorganic particles (C), the fracture resistance and the like are also good.
- the average thickness of the single-layer film is preferably, for example, 1 ⁇ m or more and less than 300 ⁇ m, and more preferably 5 ⁇ m or more and less than 100 ⁇ m.
- the single-layer film can be suitably used as various packaging materials and the like.
- the preferred range of the arithmetic average roughness (Ra) and the average length (RSm) of the contour curve element of at least one surface of the single-layer film of the present invention measured in accordance with JIS B0601 is the above-mentioned invention. It is the same as the range of film and the like.
- the single-layer film of the present invention may be a non-stretched film or a stretched film, but may be preferably stretched. Stretching tends to improve gas barrier properties, strength, and the like. Further, when the single-layer film of the present invention is a stretched film, the possibility of breaking is low, so that the productivity is good. On the other hand, in the case of non-stretching, the heat fusion property tends to be good. Therefore, when it is used as a heat-sealing film described later, it is preferably unstretched.
- the single-layer film of the present invention can be manufactured by the same method as the above-mentioned manufacturing method of the film or the like of the present invention.
- the single-layer film is suitably used as a material for various packaging containers such as food packaging containers, pharmaceutical packaging containers, industrial chemical packaging containers, and pesticide packaging containers.
- the multilayer film of the present invention is a multilayer film including at least one layer formed from the gas barrier resin composition of the present invention.
- the lower limit of the number of layers of the multilayer film may be 2, but 3 is preferable.
- the upper limit of the number of layers of the multilayer film may be, for example, 1000, 100, 20 or 10.
- the multilayer film is usually obtained by laminating a layer made of the gas barrier resin composition of the present invention and another layer.
- a layer made of a resin other than the gas barrier resin composition of the present invention is an x layer
- a gas barrier resin composition layer of the present invention is a y layer
- an adhesive resin layer is a z layer
- "/" is used as the layer structure of the multilayer film.
- x / y, x / y / x, x / z / y, x / z / y / z / x, x / y / x / y / x, x / Z / y / z / x / z / y / z / x and the like can be mentioned.
- the types may be the same or different.
- a layer using a recovery resin made of scrap such as trim generated during molding may be separately provided, or the recovery resin may be mixed with a layer made of another resin.
- the thickness ratio of the y layer to the total layer thickness is usually 2 to 20% from the viewpoint of moldability, cost and the like.
- the adhesive layer is a layer formed of an adhesive resin or other adhesive.
- thermoplastic resin As the resin used for the x layer, a thermoplastic resin is preferable from the viewpoint of workability and the like.
- thermoplastic resin examples include those exemplified in the description of the multilayer structure A.
- adhesive resin used for the z-layer examples include those exemplified in the description of the multilayer structure A.
- Another layer may be laminated on the multilayer film.
- Examples of the method for obtaining a multilayer film include co-extrusion molding, co-extrusion hollow molding, co-injection molding, extrusion laminating, co-extrusion laminating, dry laminating, solution coating and the like.
- the multilayer film obtained by such a method is further subjected to secondary processing molding after reheating by a method such as vacuum pressure air deep drawing molding, blow molding, press molding, etc. to obtain a desired molded body structure. It's okay.
- the multilayer film is reheated in a range below the melting point of EVOH by a roll stretching method, a pantograph stretching method, an inflation stretching method, or the like, and then uniaxially or biaxially stretched to obtain a stretched multilayer film. You can also.
- Molds using the single-layer film or multilayer film of the present invention include containers (bags, cups, tubes, trays, bottles, etc.), fuel containers, pipes, fibers, food and beverage packaging materials, container packing materials, and medical treatment.
- Cosmetic packaging, dental care packaging, pharmaceutical packaging, packaging child parts (caps, bag-in-box cocks, etc.), pesticide bottles, agricultural films (greenhouse films, soil steaming films) ), Grain storage bag, geomembrane, vacuum insulation board outer bag, wallpaper or decorative board, gas tank for hydrogen, oxygen, etc. can be mentioned.
- the vapor-deposited film of the present invention includes the single-layer film or multilayer film of the present invention and an inorganic vapor-deposited layer.
- the vapor-deposited film of the present invention includes the single-layer film of the present invention or the multilayer film of the present invention including at least one layer formed from the gas barrier resin composition as the outermost layer, and the single-layer film or the multilayer film.
- the film comprises at least one inorganic vapor deposition layer laminated on the exposed surface of the layer formed from the gas barrier resin composition of the present invention.
- the inorganic thin-film deposition layer is directly laminated on the layer formed from the gas barrier resin layer of the present invention.
- the inorganic vapor deposition layer is a layer made of an inorganic substance such as a metal or an inorganic oxide and having a gas barrier property against oxygen and water vapor.
- the gas barrier resin composition of the present invention has a higher affinity for metals and inorganic oxides than ordinary thermoplastic resins, and can form a dense and defect-free inorganic vapor-deposited layer. Further, since the gas barrier resin composition has a gas barrier property, it is possible to suppress a decrease in the gas barrier property even when a defect occurs in the inorganic thin-film deposition layer due to bending or the like.
- the gas barrier resin composition contains the inorganic particles (C)
- the vapor-deposited film of the present invention having a structure in which the inorganic-deposited layer is directly laminated on at least one surface of the layer formed from the gas barrier resin composition is less likely to cause vapor deposition defects and is compatible with the inorganic-deposited layer. Excellent adhesion strength.
- the inorganic vapor deposition layer is preferably either a metal vapor deposition layer containing aluminum as a main component or an inorganic oxide vapor deposition layer containing alumina or silica as a main component.
- a metal-deposited layer is preferable for imparting light-shielding properties, but inorganic oxidation is possible from the viewpoints of visibility of the contents as a packaging material, appropriate range, and suppression of gel and lumps when melt-molding the crushed material.
- a thin-film deposition layer is preferred.
- the metal vapor deposition layer is generally a layer containing aluminum as a main component.
- the content of aluminum atoms in the metal vapor deposition layer is preferably 50 mol% or more, more preferably 70 mol% or more, further preferably 90 mol% or more, and particularly preferably 95 mol% or more.
- the average thickness of the metal vapor deposition layer is preferably 120 nm or less, more preferably 100 nm or less, still more preferably 90 nm or less.
- the average thickness of the metal vapor deposition layer is preferably 25 nm or more, more preferably 35 nm or more, and even more preferably 45 nm or more.
- the average thickness of the metal-deposited layer is an average value of the thicknesses at any 10 points on the cross section of the metal-deposited layer measured by an electron microscope.
- the inorganic oxide vapor deposition layer includes an inorganic oxide such as an oxide such as silicon, aluminum, magnesium, calcium, potassium, tin, sodium, boron, titanium, lead, zirconium, or yttrium, preferably an alumina or silica vapor deposition film. Can be mentioned.
- the average thickness of the inorganic oxide-deposited layer is preferably 60 nm or less, more preferably 50 nm or less, still more preferably 40 nm or less.
- the average thickness of the inorganic oxide-deposited layer is preferably 10 nm or more, more preferably 15 nm or more, and even more preferably 20 nm or more.
- the average thickness of the inorganic oxide-deposited layer is an average value of the thicknesses at any 10 points on the cross section of the inorganic oxide-deposited layer measured by an electron microscope.
- the inorganic vapor deposition layer can be vapor-deposited by a known physical vapor deposition method or chemical vapor deposition method. Specifically, the method illustrated in the description of the multilayer structure A and the like can be mentioned.
- the inorganic thin-film deposition layer can exhibit high gas barrier properties when it is vapor-filmed on a layer with higher dimensional stability, because defects during vapor deposition are less likely to occur.
- the layer formed from the gas barrier resin composition in which the inorganic thin-film deposition layer is laminated is preferably stretched, and it is more preferable to perform heat treatment after stretching.
- the lower limit of the number of layers of the vapor-filmed film of the present invention is 2. That is, the vapor-filmed film is one aspect of the multilayer film of the present invention. Further, the upper limit of the number of layers of the vapor-filmed film may be, for example, 1000, 100, 20, 10, 5 or 4, as in the case of the multilayer film.
- a layer made of a resin other than the gas barrier resin composition of the present invention is an x layer
- a layer formed of the gas barrier resin composition of the present invention is a y layer
- an adhesive resin layer is z.
- the layer and the inorganic vapor-deposited layer are v layers, for example, y / v, v / y / v, x / y / v, x / z / y / v, v / y / z / / / / / / / v, etc. Can be mentioned.
- the vapor-filmed film of the present invention has an inorganic vapor-deposited layer as an outermost layer adjacent to the layer formed from the gas barrier resin composition of the present invention.
- it can also be used as a multilayer structure in which another layer is further laminated on the inorganic thin-film layer of the thin-film film.
- the multilayer structure B of the present invention includes the thin-film film of the present invention and another layer (thermoplastic resin layer or the like) laminated on the inorganic thin-film layer in the thin-film film.
- the other layer is not particularly limited, and examples thereof include layers of various thermoplastic resins exemplified in the description of the multilayer structure A.
- the multilayer structure B may be a multilayer film containing an inorganic thin-film deposition layer.
- the multilayer structure B is included in one form of the multilayer structure A of the present invention described above.
- the lower limit of the number of layers of the multilayer structure B of the present invention is, for example, 3. Further, the upper limit of the number of layers of the multilayer structure B may be, for example, 1000, 100, 20, 10, 5 or 4.
- a layer made of a resin other than the gas barrier resin composition of the present invention is an x layer
- the gas barrier resin composition layer of the present invention is a y layer
- an adhesive resin layer is a z layer.
- the inorganic vapor deposition layer is a v layer, for example, x / v / y, x / v / y / v, x / y / v / x, x / v / y / v / x, x / z / v / y, Examples thereof include x / z / y / v / z / x and x / z / v / y / v / z / x.
- the vapor-filmed film and the multilayer structure B of the present invention are suitably used for packaging materials that require particularly excellent barrier properties.
- packaging materials that require particularly excellent barrier properties.
- containers bags, tubes, lids, etc.
- food and beverage packaging materials medical infusion bag materials, tire tube materials, bag-in-box inner bag materials, cosmetic packaging materials, dental care packaging materials.
- Pharmaceutical packaging materials agricultural films (greenhouse films, soil fumigation films), grain storage bags, geomembranes, vacuum insulation outer bags, and the like.
- the single-layer film, multilayer film, vapor-deposited film or multilayer structure B of the present invention is preferably a film for heat sealing. That is, the heat-sealing film of the present invention includes the single-layer film, the multilayer film, the vapor-deposited film, or the multilayer structure B of the present invention. As a specific layer structure when the heat-sealing film of the present invention has multiple layers, the same layer structure as the above-mentioned layer structure B can be mentioned. In the heat-sealing film, the outermost layer functions as a heat-sealing layer.
- the heat-sealing layer may be a layer made of the single-layer film of the present invention (a layer formed of a gas barrier resin composition), or may be a layer formed of another thermoplastic resin or the like.
- the single-layer film layer of the present invention can be used as a heat-sealing layer.
- the single-layer film of the present invention can effectively suppress the adsorption of scent components and the like. Therefore, the single-layer film of the present invention is a heat-sealing layer, and the packaging material using the heat-sealing film provided with the single-layer film on the outermost layer can efficiently retain the scent component of the contents. ..
- the lower limit of the average ethylene unit content of EVOH constituting the gas barrier resin composition is preferably 25 mol%, more preferably 30 mol%. 40 mol% is more preferred.
- the upper limit of the ethylene unit content is preferably 60 mol%, more preferably 55 mol%.
- the heat-sealing film of the present invention has excellent non-adhesiveness to fragrance components and the like as described above when the single-layer film of the present invention is provided as a heat-sealing layer (outermost layer). It can be suitably used for packaging bags.
- the heat-sealing layers face each other and are pressure-bonded for 1 second under the conditions of a temperature of 185 ° C. and a pressure of 0.1 MPa using a heat plate type heat sealer. Is preferably 10 N / 15 mm or more.
- the heat-sealed layers to be faced with each other may be the same or different.
- the heat-sealing film can obtain excellent heat-sealing strength by heat-sealing, and can be suitably used as a heat-sealing film for packaging bags having high aroma retention.
- the heat seal strength is a value measured in accordance with JIS-Z1707.
- the heat-sealing film of the present invention is formed into a container and a package (bag, cup, tube, tray, bottle, etc.) with the heat-sealing layer inside by heat-sealing the heat-sealing layers. be able to.
- the heat-sealing film can be heat-sealed with high strength, and when the single-layer film of the present invention is provided as a heat-sealing layer, the heat-sealing layer is a fragrance component. Since it is excellent in non-adsorption property, the above-mentioned packaging bag and the like obtained from this can exhibit excellent fragrance-retaining property.
- the heat-sealing method is not particularly limited, and a known method can be used. Examples thereof include a heat-sealing method using a hot plate type heat sealer, an impulse sealer, an ultrasonic sealer, a frictional heat sealer, a dielectric heating sealer, or the like.
- the temperature at the time of heat sealing is not particularly limited, but it is preferable that the temperature is equal to or higher than the melting point of the heat-sealed layer (melting point + 30 ° C.) or lower from the viewpoint of heat sealing strength and prevention of poor appearance.
- the heat-sealing pressure is not particularly limited, but is preferably 0.01 MPa or more and 1.0 MPa or less from the viewpoint of heat-sealing strength and prevention of appearance defects.
- the heat-sealing time is not particularly limited, but is preferably 0.05 seconds or more and 5 seconds or less from the viewpoint of heat-sealing strength and productivity.
- the moisture content of the heat-sealing layer of the heat-sealing film is preferably 0.1% by mass or more and 4% by mass or less. More preferably, it is 0.2% by mass or more and 3% by mass or less.
- this moisture content is not more than the above upper limit, foaming due to vaporization of the moisture in the heat-sealing layer is less likely to occur during heat sealing, and as a result, the appearance of the heat-sealing portion is improved and the heat-sealing property is improved. improves.
- the moisture content is at least the above lower limit, the non-adsorptive property immediately after molding the packaging bag or the like tends to be improved. For example, when the content is filled immediately after molding, the aroma component derived from the content is less likely to be adsorbed by the heat-sealed layer.
- Examples of the contents (packaged object) in the packaging bag include foods, beverages, pharmaceuticals, cosmetics, fragrances, toiletry products and the like containing fragrance components, and in particular, a group consisting of confectionery, tea leaves, coffee, spices and cigarettes. It is preferable that it is at least one selected from the above. Since the packaging bag can be in a form having excellent fragrance-retaining properties as described above, it is particularly suitable as a packaging bag for the above-mentioned foods and the like that require fragrance-retaining properties.
- the packaging material B of the present invention includes the multilayer structure of the present invention (for example, the multilayer structure A).
- the packaging material B may be a packaging material made of the multilayer structure of the present invention.
- the packaging material B has a low environmental load, and has good gas barrier properties, appearance, and productivity.
- the packaging material B of the present invention may have a layer formed from other than resin, for example, a paper layer, a metal layer, or the like.
- the packaging material B may be the multi-layer structure as it is, or the multi-layer structure may be secondary processed.
- Examples of the packaging material obtained by secondary processing include (1) a tray cup-shaped container obtained by thermoforming a multilayer structure by vacuum forming, vacuum forming, vacuum forming, etc., and (2) a multilayer structure. Examples thereof include a bottle obtained by performing stretch blow molding on the body, a cup-shaped container, and (3) a bag-shaped container obtained by heat-sealing the multilayer structure.
- the secondary processing method is not limited to each of the methods exemplified above, and for example, a known secondary processing method other than the above such as blow molding can be appropriately used.
- the packaging material B of the present invention is used for packaging, for example, foods, beverages, chemicals such as pesticides and pharmaceuticals, medical equipment, machine parts, industrial materials such as precision materials, and clothing.
- the packaging material B is preferably used for applications that require a barrier property against oxygen and applications in which the inside of the packaging material is replaced by various functional gases.
- the packaging material B is formed in various forms depending on the application, for example, a vertical bag filling seal bag, a vacuum packaging bag, a pouch with a spout, a laminated tube container, a lid material for a container, and the like.
- the packaging material B when the packaging material B is formed by using a multilayer structure having a layer formed from the gas barrier resin composition containing PA (E), the packaging material B has excellent retort resistance and is therefore boiled and sterilized. It is preferably used as a packaging material for or retort sterilization. Further, when the packaging material B is formed by using a multilayer structure having a layer formed from the gas barrier resin composition containing the thermoplastic elastomer (G), the packaging material B has excellent bending resistance. It is useful as a flexible packaging material. Examples of the flexible packaging material include a vertical bag filling seal bag.
- the vertical bag filling seal bag of the present invention is a vertical bag filling seal bag provided with the multilayer structure of the present invention (for example, the multilayer structure A).
- the vertical bag filling seal bag is often used for packaging, for example, liquids, viscous bodies, powders, solid loose materials, or foods and beverages in which these are combined.
- the vertical bag-filled seal bag has excellent bending resistance, and its gas barrier property is maintained when it receives physical stress such as deformation or impact.
- FIG. 5 shows a form of a vertical bag filling seal bag (Vertical form fill seal pouch).
- the film material 110 is sealed on three sides of the upper end portion 111, the lower end portion 112, and the body portion 115 of the seal bag 101.
- the body portion 115 is arranged at the center portion of the back surface extending from the upper end portion 111 to the lower end portion 112 so as to divide the back surface 120 into two.
- the film material 110 is sealed so that its inner surfaces are in contact with each other. That is, the form of the seal in the body portion 115 is so-called gassho pasting.
- the front surface of the seal bag 101 (a surface having the same shape as the back surface on the opposite side of the back surface), which is not shown in FIG. 5, is not divided by the sealed portion and is usually a content. It is used as a surface for displaying products and products.
- the body portion 115 to be sealed may be arranged at either the side end portions 121 or 122, and in this case, the back surface is not divided by the sealed portion.
- the illustrated seal bag 101 is a vertical type of one film material 110 having a width that is twice the width of the back surface 120 (the total width of the front surface and the width of the back surface) plus the width required for sealing at the body portion 115. It is a bag manufactured by being supplied to a bag making and filling machine.
- the upper end portion 111, the lower end portion 112, and the body portion 115 are all formed as linear sealing portions having no branches.
- One film material is sealed and made into a bag at three parts of the body portion extending vertically to the bag.
- the film material 110 includes the multilayer structure of the present invention.
- the inner container for a bag-in-box of the present invention includes the multilayer structure of the present invention (for example, the multilayer structure A).
- the inner container for the bag-in-box for example, a container in which the liquid injection port is molded from another resin composition and the container body is formed of the above-mentioned multilayer structure can be mentioned.
- the bag-in-box inner container can be created, for example, by heat-sealing the film or sheet of the multilayer structure of the present invention and further heat-sealing the liquid injection port.
- the heat-sealing method normal heat-sealing conditions can be appropriately selected.
- the bag-in-box is, for example, a flexible plastic inner container (inner container for a bag-in-box) provided with a liquid injection port inside a cardboard box.
- the inner container for a bag-in-box is usually exposed to repeated bending during transportation and the like.
- processing such as increasing the thickness of the bent portion or the circumference of the sealing stopper may be performed.
- the inner container for a bag-in-box of the present invention since the multi-layer structure of the present invention is provided, excellent durability can be exhibited.
- the laminated peeling container (delamination container) of the present invention includes the multilayer structure of the present invention (for example, the multilayer structure A).
- the multilayer structure provided in the laminated stripping container further has a layer mainly composed of a polyolefin having no polar functional group, which is directly laminated on one surface of the layer made of the gas barrier resin composition of the present invention. That is, the laminated peeling container has a layer structure in which the layer containing the polyolefin as a main component is directly laminated on one surface of the layer made of the gas barrier resin composition.
- the peelability between the layer made of the gas barrier resin composition of the present invention and the layer containing polyolefin as a main component having no polar functional group becomes good, and it is suitably used as a laminated peeling container. Can be done.
- the proportion of the polyolefin having no polar functional group in the layer containing the polyolefin having no polar functional group as a main component is preferably 70% by mass or more, more preferably 90% by mass or more, still more preferably 95% by mass or more. It is preferable that the layer is composed only of a polyolefin having substantially no polar functional group.
- the layer containing the polyolefin as a main component may be a layer substantially composed of only a polyolefin having no polar functional group.
- the polyolefin having no polar functional group is a homopolymer or a copolymer of an olefin and has no polar functional group.
- Examples of the homopolymer or copolymer of the olefin include linear low-density polyethylene (LLDPE), low-density polyethylene (LDPE), ultra-low-density polyethylene (VLDPE), medium-density polyethylene (MDPE), and high-density polyethylene (high-density polyethylene).
- HDPE high density polyethylene
- LDPE low density polyethylene
- PP polypropylene
- HDPE high density polyethylene
- ethylene-propylene (block or random) copolymer is more preferably used.
- the other layer of the gas barrier resin composition of the present invention is adhered to a layer containing a thermoplastic resin as a main component via an adhesive layer.
- the proportion of the thermoplastic resin in the layer containing the thermoplastic resin as a main component is preferably 70% by mass or more, more preferably 90% by mass or more, further preferably 95% by mass or more, and substantially composed of only the thermoplastic resin. It is preferable that the layer is formed. With such a configuration, the flexibility and strength of the multi-layer structure of the present invention and, by extension, the laminated stripping container can be enhanced.
- the thermoplastic resin constituting the layer containing the thermoplastic resin as a main component is preferably a polyolefin, and this polyolefin may be the same as the polyolefin having no polar functional group, and is an unsaturated carboxylic acid or a derivative thereof. It may be a modified polyolefin modified with. Examples of the unsaturated carboxylic acid or a derivative thereof include maleic acid, fumaric acid, itaconic acid, maleic anhydride, itaconic anhydride, maleic acid monomethyl ester, maleic acid monoethyl ester, maleic acid diethyl ester, and fumaric acid monomethyl ester. Be done. These may be used alone or in combination of two or more.
- Examples of the adhesive resin constituting the adhesive layer include those exemplified as an explanation of the adhesive layer in the multilayer structure A of the present invention.
- the layer made of the gas barrier resin composition of the present invention is y
- the layer mainly composed of another thermoplastic resin is x
- the main component is an adhesive resin.
- Examples of the method for manufacturing the laminated stripping container of the present invention include a coextrusion blow molding method.
- the thickness of the layer made of the gas barrier resin composition of the present invention constituting the laminated stripping container of the present invention is preferably 0.5 ⁇ m or more, more preferably 1 ⁇ m or more, still more preferably 5 ⁇ m or more.
- the thickness of the layer made of the gas barrier resin composition is preferably 1000 ⁇ m or less, more preferably 500 ⁇ m or less, still more preferably 100 ⁇ m or less.
- the thickness of the layer mainly composed of polyolefin having no polar functional group constituting the laminated stripping container of the present invention is preferably 25 ⁇ m or more, more preferably 30 ⁇ m or more, still more preferably 50 ⁇ m or more.
- the strength can be increased by setting the thickness of the layer containing the polyolefin as a main component to 25 ⁇ m or more.
- the thickness of the layer containing the polyolefin as a main component is preferably 5000 ⁇ m or less, more preferably 2000 ⁇ m or less, still more preferably 1000 ⁇ m or less.
- the total thickness of the multilayer structure constituting the laminated stripping container of the present invention is preferably 150 ⁇ m or more, more preferably 200 ⁇ m or more. By setting the total thickness of the multilayer structure to 150 ⁇ m or more, the strength is sufficient and the structure is less likely to be damaged.
- the total thickness of the multilayer structure is preferably 10,000 ⁇ m or less, more preferably 8000 ⁇ m or less, and even more preferably 6000 ⁇ m or less. Flexibility can be enhanced by setting the total thickness of the multilayer structure to 10,000 ⁇ m or less.
- delamination area is preferably 80 cm 2 or more, 150 cm 2 or more preferably between the layer mainly containing polyolefin having no layer with polar functional group composed of the gas barrier resin composition , 200 cm 2 or more is more preferable, and 220 cm 2 or more is particularly preferable.
- delamination area is preferably 300 cm 2 or less, more preferably 280 cm 2 or less, more preferably 260 cm 2 or less.
- the delamination area is determined by cutting the multilayer structure into 300 mm (width) ⁇ 350 mm (length), and in the central portion, a layer made of the gas barrier resin composition and a layer containing polyolefin having no polar functional group as a main component. It can be obtained by forming a peeling port between the two layers and converting the area from the mass of the peeled portion when the tube is inserted into the peeling port by 50 mm and air having a pressure of 0.2 MPa is blown into the peeling port.
- the laminated peeling container of the present invention preferably has a standard peel strength of 1 g / 30 mm or more between a layer made of the gas barrier resin composition and a layer containing a polyolefin having no polar functional group as a main component, preferably 3 g / 30 mm or more. Is more preferable, and 5 g / 30 mm is further preferable.
- the standard peel strength By setting the standard peel strength to 1 g / 30 mm or more, it is possible to suppress the peelability from becoming too high and increase the productivity.
- the standard peel strength is preferably 12 g / 30 mm or less, more preferably 11 g / 30 mm or less, further preferably 9.5 g / 30 mm or less, and particularly preferably 9.0 g / 30 mm or less.
- the laminated stripping container of the present invention has good peelability between the layer made of the gas barrier resin composition and the layer containing a polyolefin having no polar functional group as a main component, and in particular, the gas barrier resin composition.
- the substance contains the thermoplastic elastomer (G), the bending resistance and the like are also good.
- the laminated peeling container of the present invention can be suitably used as a laminated peeling container for foods capable of preventing deterioration of the scent, color, etc. of the contents. From the viewpoint of suppressing the peeling of the oral head, it is preferable that the laminated peeling container of the present invention has a thicker mouth than other portions.
- the thickness of the oral cavity is preferably 0.4 mm or more, more preferably 0.5 mm or more.
- the upper limit of the thickness of the oral cavity may be, for example, 3 mm, and may be 2 mm or 1 mm.
- the multilayer sheet of the present invention includes a barrier layer made of the gas barrier resin composition of the present invention, and a thermoplastic resin layer laminated directly on at least one surface side of the barrier layer or via another layer.
- the specific form, use, etc. of the multilayer sheet may be the same as the specific form, use, etc. of the multilayer film of the present invention described above.
- the multilayer sheet can be suitably used as a material for forming various molded bodies.
- Examples of the method for further molding the molded product using the multilayer sheet of the present invention include a heat stretching molding method, a vacuum forming method, a vacuum forming method, a vacuum forming method, a blow forming method and the like. These moldings are usually carried out in a temperature range below the melting point of EVOH. Among these, the heat stretching molding method and the vacuum compressed air molding method are preferable.
- the heat stretch molding method is a method in which a multilayer sheet is heated and stretched in one direction or a plurality of directions for molding.
- the vacuum compressed air forming method is a method in which a multilayer sheet is heated and formed by using both vacuum and compressed air.
- the packaging material C obtained by molding the multilayer sheet of the present invention by a heat stretching molding method can be easily and surely manufactured, and is excellent in appearance, gas barrier property and the like.
- the packaging material C may be in the form of a sheet, or may be molded into another shape.
- the packaging material C can be used for various purposes similar to the conventionally known packaging materials.
- thermoplastic resin used for the thermoplastic resin layer is preferably a resin that can be stretched within the range of the heat-stretching temperature represented by the following formula (1). X-110 ⁇ Y ⁇ X-10 ... (1)
- X is the melting point (° C.) of EVOH.
- Y is the heating and stretching temperature (° C.).
- the container obtained by molding the multilayer sheet of the present invention by the vacuum compressed air molding method can be easily and surely manufactured, and is excellent in appearance, gas barrier property and the like.
- the container can be used for various purposes similar to the conventionally known container.
- a multilayer sheet is heated and softened, and then molded into a mold shape.
- a molding method vacuum or compressed air is used, and if necessary, a plug is also used to form a mold shape (straight method, drape method, air slip method, snapback method, plug assist method, etc.) or press molding.
- the method and the like can be mentioned.
- Various molding conditions such as molding temperature, degree of vacuum, compressed air pressure or molding speed are appropriately set depending on the shape of the plug, the shape of the mold, the properties of the raw material film and the sheet, and the like.
- the molding temperature is not particularly limited, and may be a temperature at which the resin softens to a sufficient degree for molding.
- the temperature should not be high enough to cause the multilayer sheet to melt due to heating or the unevenness of the metal surface of the heater plate to be transferred to the multilayer sheet, and the shaping is sufficient. It is desirable not to keep the temperature low.
- the temperature of the multilayer sheet is 50 ° C. to 180 ° C., preferably 60 ° C. to 160 ° C.
- the medium for plant cultivation of the present invention is a medium for plant cultivation containing a molded product formed from a resin composition, and the above resin composition is the gas barrier resin composition of the present invention.
- the medium for plant cultivation will also be commercialized as a medium synthesized using raw materials derived from biomass.
- the performance of the synthetic resin derived from biomass may be inferior to that of the synthetic resin derived from fossil fuel, when the conventional medium for plant cultivation derived from fossil fuel is replaced with the medium for plant cultivation derived from biomass, There is concern that the most important plant viability will decline. Therefore, it is desired to develop a biomass-derived resin having excellent plant growth comparable to that of a fossil fuel-derived resin.
- a resin-made culture medium for plant cultivation is often formed into a chip shape by melt molding.
- a chip-shaped plant cultivation medium can be stably and efficiently obtained even if melt molding is continued for a long period of time.
- the long-running property (long-term operation characteristics) of the resin material is important.
- die deposits on the extruder are likely to occur due to melt molding over a long period of time.
- the strand strand-shaped resin material
- the breakage of the strand is also referred to as "thread breakage”.
- the plant cultivation medium of the present invention uses a raw material derived from biomass, but has a sufficient long-running property comparable to that derived from fossil fuel. It is formed from a composition and has high plant growth potential.
- the resin composition (gas barrier resin composition) used for the plant cultivation medium has a low environmental load because a raw material derived from biomass is partially used.
- EVOH is selected and used as a resin having excellent plant growth, and in the plant cultivation medium containing a molded product using EVOH, the EVOH is a raw material derived from biomass. Even when synthesized, it is possible to exhibit the same high plant viability as in the case of EVOH having the same structure synthesized only from fossil fuel-derived raw materials.
- the long-run property of the resin composition used for the medium for plant cultivation can be evaluated by the method described in Examples, and the degree of die deposit and the frequency of yarn breakage when the resin composition is continuously molded. Can be evaluated comprehensively.
- plant growth means having the ability to grow plants, and can be evaluated by the number of enlarged radishes in growing radishes, for example, as in the medium evaluation described in the examples of the present invention.
- the form of the molded product contained in the medium for plant cultivation of the present invention is not particularly limited as long as it is formed from the resin composition (gas barrier resin composition).
- the molded product is usually a melt-molded product of the resin composition.
- the molded product may be a chip (pellet) made of the resin composition, a non-woven fabric made of the resin composition, or the like. From the viewpoint of plant growth efficiency, a chip made of the above resin composition (resin composition chip) is preferable. In this case, for example, a large number of chips can be spread in a container or the like and used as a medium for plant cultivation.
- the chip has, for example, a substantially spherical or substantially disc-shaped shape obtained by hot-cutting a melt of the resin composition, or cutting a strand of the resin composition. It may have a substantially columnar shape obtained, or may have a flake-like or irregular shape.
- the chips are preferably columnar, flat or flake-shaped.
- the size of the molded body such as the chip is not particularly limited, but for example, the maximum length is preferably in the range of 1 to 50 mm, more preferably in the range of 1 to 20 mm. The maximum length can be measured using a caliper.
- the medium for plant cultivation of the present invention may be composed only of a molded product made of the above resin composition, but together with the molded product made of the above resin composition, rock wool, sand, soil, ceramic balls, coconut husk, and bark. , Pete moss, water moss, other components such as other resin media, and the like may be further contained.
- the content of the molded product in the plant cultivation medium is preferably 50% by mass or more, more preferably 80% by mass or more, and further preferably 95% by mass or more.
- the usage form of the medium for plant cultivation of the present invention is not particularly limited, but it is preferable to use it as a medium for nutrient solution cultivation using a culture solution.
- the medium for plant cultivation is used as a medium for nutrient solution cultivation, for example, the medium for plant cultivation is placed in a container such as a pot, the culture medium is added thereto, and then the seeds are sown. It is possible to exemplify a method of transplanting seedlings or seedlings. Further, a method of preparing a cultivation bed in which the plant cultivation medium is spread and transplanting the grown seedlings to the cultivation bed to cultivate various crops can be exemplified.
- the type of plant cultivated using the plant cultivation medium of the present invention is not particularly limited, and examples thereof include flowers, vegetables including root vegetables, fruits, grains, etc., particularly radish, eggplant, burdock, carrot, cucumber, etc. It is preferably used for growing vegetables such as tomatoes, eggplants and peppers. In particular, it has the advantage that it can be suitably used for root vegetables, which were difficult to cultivate with rock wool.
- the present invention also provides a plant cultivation apparatus provided with the above-mentioned medium for plant cultivation of the present invention.
- the plant cultivation apparatus of the present invention is not particularly limited as long as it uses the above-mentioned medium for plant cultivation of the present invention, and configurations other than the medium for plant cultivation are suitable conventionally known plant cultivation apparatus. It may have the configuration of.
- FIG. 6 is a diagram schematically showing a preferred example plant cultivation device 201 of the present invention.
- the plant cultivation device 201 of the example shown in FIG. 6 is a box-shaped object having an opening 203 above, and includes a planter 202 having a drain port 205 at an appropriate height of the side wall 204, and the drain port 205 is provided in the planter 202.
- Water containing nutrients (nutrient solution) 206 is stored up to a height (depth) that does not spill out.
- a shelf 208 is provided on the bottom wall 207 of the planter 202 so that the mounting surface 208a is arranged above the surface of the water 206, and a water absorption sheet 209 is placed on the shelf 208 when viewed from above.
- the water absorbing sheet 209 is a sheet-like material formed of a material such as cellulose fiber, nylon fiber, vinylon fiber, polyester fiber, polyolefin fiber, rayon fiber, aramid fiber, and glass fiber, and the central portion 209a thereof is a shelf 208.
- 20 water 6 is provided on the mounting surface 208a and whose end portion 209b is immersed in water 206 in the planter 202, and 20 water 6 absorbed from the end portion 209b is sent to the central portion 209a. There is.
- the root-proof water-permeable sheet 210 is arranged on the water-absorbing sheet 209 so that the end portion 210a is caught by the upper end 204a of the side wall 204 of the planter 202.
- the root-proof water-permeable sheet 210 is preferably provided when the root vegetables are grown in the plant cultivation device 201, and may not necessarily be provided when the plant 211 to be grown is not a root vegetable.
- the water 206 in the planter 202 is sent to the root-proof water-permeable sheet 210 via the water-absorbing sheet 209.
- the root-proof water-permeable sheet 210 may be a woven fabric composed of a fibrous material, a non-woven fabric, a mat-like material, or, for example, various polyolefins (polyethylene, polypropylene, ethylene-propylene copolymer, ethylene-vinyl alcohol copolymer, ethylene-). Vinyl ester copolymers, ethylene-acrylic acid ester copolymers, or modified polyolefins obtained by modifying these with unsaturated carboxylic acids, graft-modified with derivatives thereof, or modified with maleic anhydride), various nylons (nylon-6).
- the sheet made of the above resin is used as the root-proof water-permeable sheet 210, it is preferable that the sheet has innumerable fine pores and a uniform distribution, and in this case, the maximum diameter of the fine pores is preferably 20 ⁇ m or less.
- the maximum diameter of the micropores By setting the maximum diameter of the micropores to 20 ⁇ m or less, the roots of the plant penetrate the root-proof water-permeable sheet 210 and invade the water-absorbing sheet 209, making it difficult to stick to the roots. Will grow better. Further, when the maximum diameter of the micropores is set to, for example, 5 ⁇ m or more, water is likely to be leached from the water absorption sheet, and the growth of the plant becomes better. Since such a root-proof water-permeable sheet 210 has fine meshes, it has a property that it is difficult for water to pass through and water is easily retained.
- FIG. 6 shows an example in which a plurality of columnar pellets (resin composition chips) 213 formed from the above resin composition are used as the plant cultivation medium 212 of the present invention.
- the present invention also provides a plant cultivation method using the above-mentioned culture medium for plant cultivation of the present invention. That is, the plant cultivation method of the present invention is a plant cultivation method including a step of cultivating using the plant cultivation medium of the present invention.
- the plant cultivation method of the present invention when the above-mentioned medium for plant cultivation of the present invention is used as a medium for hydroponic cultivation, for example, the medium for plant cultivation of the present invention is placed in a container such as a pot, and the culture medium is placed therein.
- a method of sowing or transplanting seedlings can be exemplified.
- a method of preparing a cultivation bed covered with the plant cultivation medium of the present invention and transplanting the grown seedlings to the cultivation bed to cultivate various crops can be exemplified.
- Ethylene unit content and degree of saponification of EVOH For the EVOH pellets obtained by synthesis and the gas barrier resin composition pellets obtained in Examples and Comparative Examples, tetramethylsilane was used as an internal standard and tetra was used as an additive. was dissolved in dimethyl sulfoxide (DMSO) -d 6 containing trifluoroacetic acid (TFA), measured at 80 ° C. using a 500MHz IH-NMR (manufactured by JEOL Ltd. "JMTC-400/54 / SS”), ethylene The unit content and the degree of saponification were measured. Each peak in the spectrum of the above measurement is assigned as follows.
- This treatment liquid was transferred to a 50 mL volumetric flask (manufactured by TPX) and scalpel-up with pure water. Elemental analysis of this solution was performed by an ICP emission spectrophotometer (PerkinElmer's "OPTIMA4300DV"), and the metal atom equivalent amount of metal ions and the phosphorus atom conversion of phosphorus compounds contained in EVOH pellets or gas barrier resin composition pellets. The amount and the boron atom equivalent amount of the boron compound were determined.
- the film-forming defects are "good (A)" when the number of defects is less than 50, “slightly good (B)” when the number of defects is 50 or more and less than 200, and “defective (C)” when the number of defects is 200 or more. Judged as.
- Oxygen permeability A Using the gas barrier resin composition pellets obtained in Examples and Comparative Examples, a single-layer film having a thickness of 20 ⁇ m was formed under the following conditions, and the humidity was adjusted under the conditions of 20 ° C./65% RH, and then the oxygen permeability was adjusted. Oxygen permeability was measured under the condition of 20 ° C./65% RH using a measuring device (“OX-Tran2 / 20” of ModernControl). This measurement was carried out in accordance with JIS K 7126-2 (isopressure method; 2006).
- thermoformed container (formability) Gas barrier resin composition pellets, polypropylene (“Novatec (registered trademark) PP EA7AD” manufactured by Nippon Polypro Co., Ltd.), and adhesive resin (Mitsui) obtained in Examples and Comparative Examples.
- Multilayer sheet (polypropylene / adhesive resin / gas barrier resin composition / adhesive resin / polypropylene) using "Admer (registered trademark) QF551”) manufactured by Kagaku Co., Ltd. and using a 3 type 5 layer coextrusion device under the following conditions. , Thickness ( ⁇ m): 368/16/16/32/16/368).
- Polypropylene resin extruder 32 ⁇ single-screw extruder, GT-32-A type (manufactured by Plastic Engineering Laboratory Co., Ltd.)
- Adhesive resin extruder 25 ⁇ single-screw extruder, P25-18-AC type (manufactured by Osaka Seiki Kogyo Co., Ltd.)
- EVOH resin composition extruder 20 ⁇ extruder, laboratory machine ME type CO-EXT (manufactured by Toyo Seiki Seisakusho Co., Ltd.)
- T-die 300 mm width for 3 types and 5 layers (manufactured by Plastic Engineering Laboratory Co., Ltd.)
- Pick-up speed 1 m / min 30 minutes, 10 hours, and 50 hours after the start of operation, the manufactured
- the molding conditions are shown below.
- the appearance of the cup-shaped thermoformed container obtained visually was evaluated according to the following evaluation criteria. (Appearance evaluation criteria) Good (A): No unevenness or local unevenness was observed. Slightly good (B): Slight unevenness and local unevenness were confirmed. Defective (C): Significant unevenness and local unevenness were confirmed.
- the thin-film deposition defect suppression property of the vapor-filmed films obtained in Examples and Comparative Examples was measured by measuring the number of thin-film deposition defects.
- the deposited film roll toward slitter, unrolled while applying a fluorescent lamp 100W from the film bottom, width 0.5 m, counted visually number evaporation disadvantages in different 10 locations for the region of the length 2m, and the average value per 1 m 2
- the number of vapor deposition defects was determined. Defects in vapor deposition were evaluated according to the following criteria.
- the adhesion strength was determined as follows.
- the gas barrier property of the EVOH layer (layer formed from the gas barrier resin composition) due to the generation of cracks due to oxidative deterioration becomes remarkable, so that the elongation at break becomes remarkable.
- the time when the degree becomes 1/4 can be considered as one of the indexes of the life due to the oxidative deterioration of EVOH at high temperature.
- the time when the elongation at break becomes 1/4 shows the temperature dependence of the Arrhenius type, and when the time (life) at which the elongation at break becomes 1/4 at 80 ° C is to be 100 years or more, the elongation at break at 140 ° C. It is necessary to set the time for the degree to be 1/4 to 210 hours or more.
- Multilayer pipe evaluation High-density polyethylene (“Novatec (registered trademark) HD HE421” manufactured by Nippon Polyethylene Co., Ltd., density 0.956 g / cc, MFR 0.14 g / 10 minutes) was used as the first extruder.
- Each gas barrier resin composition pellet obtained in Examples and Comparative Examples was put into a second extruder, and "Admer (registered trademark) NF408E” manufactured by Mitsui Chemicals Co., Ltd. was put into a third extruder as an adhesive resin.
- a multi-layer pipe having an outer diameter of 20 mm was extruded using a circular die of 3 types and 3 layers, and immediately after that, it was cooled and solidified through a cooling water tank adjusted to 40 ° C.
- the pipes produced 10 hours and 50 hours after the start of operation were cut in the circumferential direction of the pipe, and the thickness of the gas barrier resin composition layer in the circumferential direction was confirmed with a microscope from the cut surface.
- the film-forming defects are "good (A)" when the number of defects is less than 50, “slightly good (B)” when the number of defects is 50 or more and less than 200, and “defective (C)” when the number of defects is 200 or more. Judged as.
- Oxygen permeability B Using the gas barrier resin composition pellets obtained in Examples and Comparative Examples, a single-layer film having a thickness of 20 ⁇ m was formed under the following conditions, and the humidity was adjusted under the conditions of 20 ° C./65% RH, and then the oxygen permeability was adjusted. Oxygen permeability was measured under the condition of 20 ° C./65% RH using a measuring device (“OX-Tran2 / 20” of ModernControl). This measurement was carried out in accordance with JIS K 7126-2 (isopressure method; 2006).
- the film-forming defects are "good (A)" when the number of defects is less than 50, “slightly good (B)” when the number of defects is 50 or more and less than 200, and “defective (C)” when the number of defects is 200 or more. Judged as.
- oxygen permeability measuring device OX-Tran2 / 20” of Mocon Modern Controls.inc
- JIS K 7126-2 isopressure method; 2006
- Polypropylene resin extruder 32 ⁇ single-screw extruder, GT-32-A type (manufactured by Plastic Engineering Laboratory Co., Ltd.)
- Adhesive resin extruder 25 ⁇ single-screw extruder, P25-18-AC type (manufactured by Osaka Seiki Kogyo Co., Ltd.)
- T-die 300 mm width for 3 types and 5 layers (manufactured by Plastic Engineering Laboratory Co., Ltd.)
- Pick-up speed 1 m / min 10 hours after the start of operation, 50 hours after the start of operation, the
- thermoformed container was obtained by thermoforming in 5). The molding conditions are shown below. Heater temperature: 400 ° C Plug: 45 ⁇ x 33mm Mold temperature: 40 ° C The appearance of the obtained cup-shaped thermoformed container was visually confirmed and evaluated according to the following criteria. (Appearance evaluation criteria) A (good): No unevenness or local unevenness was observed B (slightly good): Slight unevenness and local unevenness were confirmed C (poor): Significant unevenness and local unevenness were confirmed
- the mold In the manufacture of blow-molded containers, the mold is cooled at a temperature of 15 ° C. for 20 seconds, and the average thickness of all layers is 1000 ⁇ m ((inside) high-density polyethylene layer / adhesive resin layer / gas barrier resin composition layer / adhesive resin.
- a 3L blow-molded container of layer / high-density polyethylene layer / high-density polyethylene layer (outside) (inside) 340 ⁇ m / 50 ⁇ m / 40 ⁇ m / 50 ⁇ m / 400 ⁇ m / 120 ⁇ m (outside) was molded.
- the bottom surface average diameter of this blow molded container was 100 mm, and the average height was 400 mm.
- Blow-molded containers were collected 3 hours after the start of operation, and streak evaluation was performed by visual inspection of the appearance and observation of a cross-sectional microscope in the circumferential direction. (Streak evaluation criteria) A (good): No streak was observed. B (slightly good): Streak was confirmed. C (defective): Many streaks were confirmed.
- the layer structure of the multilayer film was 90 ⁇ m for the polyethylene resin of the inner and outer layers, 10 ⁇ m each for the adhesive resin, and 20 ⁇ m for the gas barrier resin composition of the intermediate layer.
- the permeability of the model fuel of the obtained multilayer film was measured using a GTR Tech flow-type gas / vapor transmittance measuring device (GTR-30XFKE).
- GTR-30XFKE GTR Tech flow-type gas / vapor transmittance measuring device
- the multilayer film was humidity-controlled at 20 ° C. and 65% RH for 1 month, and the measurement was carried out at 60 ° C.
- (Criteria for determining the amount of die deposits) Good (A): The amount of die deposits is small, and if the die deposits are removed at intervals of 1 hour or more, thread breakage and adhesion of the die deposits to the pellets do not occur. Somewhat good (B): There are die deposits, and if the die deposits are not removed at 30 minute intervals, thread breakage or adhesion of the die deposits to the pellets will occur. Defective (C): There is a significant amount of die deposits, and even if the die deposits are removed at 30 minute intervals, thread breakage or adhesion of degraded material to the pellets occurs.
- Thread breakage frequency The thread breakage frequency was observed according to the following evaluation criteria. (Criteria for determining the frequency of paper breaks) Good (A): Number of thread breaks 0 to 2 times Slightly good (B): Number of thread breaks 3 to 10 times Defective (C): Number of thread breaks 11 times or more
- ethylene ethylene derived from biomass (bioethylene derived from sugar cane manufactured by Braskem SA) is used, and a gas cylinder filled with this ethylene (ethylene purity 96.44%, internal volume 29.502 L, internal pressure 1.8234 MPa). )It was used.
- acetic acid biomass-derived acetic acid (Bioacetate derived from sugarcane produced by Godavari Biorefines Ltd.) was used, vaporized at 220 ° C., and then introduced into the reaction system by steam.
- ethylene derived from biomass bioethylene derived from sugar cane, manufactured by Braskem SA
- V-65 2,2'-azobis (2,4-dimethylvaleronitrile)
- methanol methanol
- the ethylene pressure was maintained at 3.67 MPa and the polymerization temperature was maintained at 65 ° C.
- the mixture was cooled to terminate the polymerization.
- nitrogen gas was bubbled to completely deethylene.
- MeOH was added to the ethylene-vinyl acetate copolymer to prepare a 20% by mass MeOH solution.
- the water-containing pellets of EVOH were put into an acetic acid aqueous solution (bath ratio 20) having a concentration of 1 g / L and washed by stirring for 2 hours. This was deflated, further added to a 1 g / L acetic acid aqueous solution (bath ratio 20), and stirred and washed for 2 hours. After the liquid was removed, the acetic acid aqueous solution was updated and the same operation was performed. After washing with an acetic acid aqueous solution and then deflated, the solution is put into ion-exchanged water (bath ratio 20), stirred and washed for 2 hours, and the operation of deflated is repeated 3 times to purify the catalyst during the saponification reaction.
- Water-containing pellets of EVOH were obtained from which the residue and MeOH used at the time of strand precipitation were removed.
- the water content of the obtained EVOH water-containing pellets was measured with a halogen moisture meter "HR73" manufactured by METTLER CORPORATION and found to be 110% by mass.
- Preparation of pellets> EVOH (A1) is the same method as EVOH (A1-1) pellets, except that the types of ethylene and vinyl acetate as raw materials (raw material monomers) and the contents of phosphoric acid compounds and boron compounds are changed as shown in Table 2.
- EVOH ( A1-12) pellets and EVOH (B1-6) pellets were prepared.
- ethylene derived from fossil fuel ethylene manufactured by Air Liquide Industrial Gas Co., Ltd. was used.
- EVOH (A1-1) pellets to EVOH (A1-9) pellets EVOH (A1-12) pellets, EVOH (A1-13) pellets, EVOH (B1-1) to EVOH (B1-4) pellets and EVOH (B1) -6)
- ethylene unit content and degree of saponification, quantification of carboxylic acid, metal ion, phosphoric acid compound and boron The compounds were quantified and the degree of biobase was measured. The results are shown in Table 2.
- MeOH 2-methylene-1,3-propanediol diacetate
- ethylene was introduced by mixing ethylene derived from biomass and ethylene derived from fossil fuel at 50/50 so that the reaction tank pressure (ethylene pressure) was 5.1 MPa.
- ethylene pressure ethylene pressure
- 50 g of 2,2'-azobis (2,4-dimethylvaleronitrile) (“V-65” manufactured by Wako Pure Chemical Industries, Ltd.) as an initiator is added to a methanol solution.
- V-65 2,2'-azobis (2,4-dimethylvaleronitrile)
- the polymerization was started.
- the ethylene pressure was maintained at 5.1 MPa and the polymerization temperature was maintained at 60 ° C.
- EVOH (B1-5) pellets were prepared and evaluated in the same manner as EVOH (A1-10) pellets except that the types of ethylene and vinyl acetate as raw materials (raw material monomers) were changed as shown in Table 2. The results are shown in Table 2.
- Heater temperature Inlet 900 °C, Outlet 900 °C Gas flow rate: Ar, O 2 300 ml / min each [Analysis system NSX-2100] Measurement mode: TS Parameters: SD-210 Measurement time (timer): 540 seconds (9 minutes) PMT Sensitivity: High-concentration sulfur compounds were identified using gas chromatography (GC) and gas chromatography-mass spectrometry (GC / MS). As a GC detector, an FPD (flame light intensity detector), which is highly sensitive to trace amounts of sulfur compounds and phosphorus compounds, is used to analyze the mass components observed during the retention time when sulfur compounds are detected. By doing so, identification was performed.
- GC gas chromatography
- MS gas chromatography-mass spectrometry
- Example 1-1 After dry-blending EVOH (A1-1) pellets and EVOH (B1-1) pellets at a mass ratio (A1-1 / B1-1) 10/90, a twin-screw extruder (“2D25W” manufactured by Toyo Seiki Seisakusho Co., Ltd.” , 25 mm ⁇ , die temperature 220 ° C., screw rotation speed 100 rpm) was extruded into pellets in a nitrogen atmosphere to obtain gas barrier resin composition pellets of Example 1-1.
- a twin-screw extruder (“2D25W” manufactured by Toyo Seiki Seisakusho Co., Ltd.” , 25 mm ⁇ , die temperature 220 ° C., screw rotation speed 100 rpm) was extruded into pellets in a nitrogen atmosphere to obtain gas barrier resin composition pellets of Example 1-1.
- the ethylene unit content, the degree of saponification, and the quantification of carboxylic acid were determined according to the methods described in the above evaluation methods (1), (3) to (7). Quantification of metal ions, phosphate compounds and boron compounds, measurement or evaluation of biobase degree, long run property and oxygen permeability were carried out. The results are shown in Table 3.
- each gas barrier resin composition of Examples 1-1 to 1-20 uses only a fossil fuel-derived raw material while partially using a biomass-derived raw material (Comparative Example 1-2). It had a high gas barrier property comparable to that of the gas barrier resin composition). Further, the gas barrier resin composition of Example 1-21 has a high gas barrier comparable to that of fossil fuel-derived only (gas barrier resin composition of Comparative Example 1-6) while using a part of the raw material derived from biomass. Maintained sex. The gas barrier resin composition of Example 1-23 has high gas barrier properties comparable to those derived only from fossil fuel (gas barrier resin composition of Comparative Example 1-7) while using a part of the raw material derived from biomass. I was keeping it. Further, each of the gas barrier resin compositions of Examples 1-1 to 1-23 has a film forming defect and a streak evaluation of A or B for the film produced 10 hours after the start of operation, and has sufficient long-running property. It was a thing.
- the gas barrier property does not depend on the biobase degree, whereas the long-run property tends to increase as the biobase degree decreases. It was confirmed that it has specific properties.
- the gas barrier resin compositions of Examples 1-1, 1-2, 1-5 to 1-13, and 1-15 to 1-23 having a biobase degree of 65% or less are 10 hours after the start of operation and 50. The film formation defects, streaks, and roll end coloring of the film produced after hours were evaluated as A or B, and the film had a high long-run property.
- the adsorption amount was measured by the method shown below using a film piece having a size more suitable for the adsorption amount.
- Measurement of adsorption amount To measure the amount of adsorption, use a heat desorption gas chromatograph-mass spectrometer (TCT-GC / MS, TCT: "CP-4020" manufactured by Chromepack, GC / MS: “5793" manufactured by Azilent) as follows. The amount of adsorption was measured by the method of. (Gas sampling method) The film piece was put into a glass chamber heated to 80 ° C.
- a sample was obtained by collecting clean nitrogen from one side at a flow rate of 100 ml / min, attaching a Tenax collecting tube to the other side, and collecting the gas distilled out for 3 minutes.
- the Tenax collection tube was heated to 250 ° C. and desorbed.
- GC introduction method After the desorbed gas was cold trapped at ⁇ 130 ° C., it was heated to 250 ° C. and introduced into the entanglement to measure the adsorption amount.
- a two-component adhesive (Mitsui Chemicals'"A-520" and "A-50") is applied to one side of a biaxially stretched polyester film with an average thickness of 12 ⁇ m (Toray's "Lumilar (registered trademark)"). Then, a multilayer film was produced by laminating a single-layer film having a thickness of 30 ⁇ m used for evaluation of non-adsorption characteristics on the coated surface. The average thickness of the adhesive layer was 4 ⁇ m. Using the obtained multilayer film, a three-way seal pouch was prepared so that the single-layer films were overlapped with each other and heat-sealed, and the heat-sealing strength was measured.
- a three-way seal automatic filling and packaging machine (“KP-109” manufactured by Komak Co., Ltd.) is used to make the pouch, and the seal temperature is set to 180 ° C to continuously supply the multilayer film to a width of 80 mm and a length.
- a 70 mm three-way seal pouch was made at a bag making speed of 100 bags / minute.
- the heat seal strength was measured using a tensile tester (“AGS-H” manufactured by Shimadzu Corporation) under a 50% RH environment at 23 ° C.
- a film piece was prepared by cutting the sealing surface (sealing width 8 mm) in the width direction of the pouch to a width of 15 mm, both ends were fixed with chucks, and the sealing strength when pulled at a tensile speed of 250 m / min was measured. .. (Evaluation of hot tackiness)
- a hot tack tester manufactured by Teller Corporation was used to evaluate the hot tack strength.
- a film piece having a width of 25 mm and a length of 300 mm was prepared from the above-mentioned multilayer film, and the hot tack strength was measured when the sealing temperature was in the range of 100 ° C. to 160 ° C., the sealing pressure was 2.0 MPa, and the sealing time was 1 second.
- Example 1-23 As a result of the heat fusion test, the seal strength of Example 1-23 was 16.0 N / 15 mm. It can be said that this strength is equivalent to the seal strength of 15.8 N / 15 mm in Comparative Example 1-7 using a raw material derived from fossil fuel.
- the hot tack strength measurement results are shown in FIG. 7. As is clear from the measurement results of the hot tack strength, it was found that the single-layer film of the present invention exhibits heat-sealing properties comparable to those of the single-layer film using a raw material derived from fossil fuel.
- EVOH (A2-9) pellets are fed at a rate of 11 kg / hr from the resin feed port of C1, melted, water and oxygen are removed from the vent 1, and epoxy propane is used as the modifier 2 from the hydraulic inlet of C9. Feeding was performed at a rate of 2.4 kg / hr (pressure at the time of feeding: 6 MPa). Then, unreacted epoxy propane was removed from Vent 2 and pelletized to obtain 5 mol% modified EVOH (A2-10) pellets. Regarding the obtained EVOH (A2-10) pellets, the ethylene unit content and saponification degree, melting point, quantification of carboxylic acid, metal ion, and phosphoric acid compound were carried out according to the methods described in the above evaluation methods (1) to (5). And the quantification of boron compounds and the measurement of biobase degree were carried out. The results are shown in Table 4.
- Example 2-1 EVOH (A2-1) pellets and EVOH (B2-1) pellets were used as EVOH (X), and EVOH (B2-2) pellets were used as EVOH (Y), and the mass ratio (A2-1 / B2-1 / B2) was used.
- -2) After dry blending at 10/60/30, extruded pellets in a nitrogen atmosphere using a twin-screw extruder ("2D25W” manufactured by Toyo Seiki Seisakusho Co., Ltd., 25 mm ⁇ , die temperature 220 ° C., screw rotation speed 100 rpm). The gas barrier resin composition pellets of Example 2-1 were obtained.
- each gas barrier resin composition of Examples 2-1 to 2-24 uses only a fossil fuel-derived raw material while partially using a biomass-derived raw material (Comparative Example 2-). It had a high gas barrier property comparable to that of 2 and 2-3 gas barrier resin compositions). Further, each of the gas barrier resin compositions of Examples 2-1 to 2-24 has a film forming defect and a streak evaluation of A or B for the single-layer film produced 10 hours after the start of operation, and has sufficient long-running property. Was to have. Further, the thermoformed container using each of the gas barrier resin compositions of Examples 2-1 to 2-23 has an appearance of A or B after thermoforming even if a multilayer sheet 10 hours after the start of operation is used, which is sufficient. It was shown that it also has good moldability.
- the gas barrier property does not depend on the biobase degree, whereas the long-run property and the moldability tend to increase as the biobase degree decreases. It was confirmed that there is a peculiar property.
- the gas barrier resin compositions of Examples 2-1 and 2-2 and 2-5 to 2-23 having a biobase degree of 65% or less are for films prepared 10 hours and 50 hours after the start of operation.
- the film-forming defects, streak, coloration of the roll end, and the evaluation of the appearance after thermoforming were A or B, and the film had high long-running property and moldability.
- Example 2-25> The result of measuring the sulfur compound content of the gas barrier resin composition pellets obtained in Example 2-6 and identifying them by the same method as the method for measuring EVOH (A) and EVOH (B) pellets.
- the content of the sulfur compound was 0.3 ppm in terms of sulfur atom, and the sulfur compound was dimethyl sulfide.
- the traceability was evaluated using the thermoformed container 30 minutes after the start of operation obtained in the evaluation method (8) thermoformed container evaluation. Specifically, the EVOH layer of the obtained thermoformed container was taken out and used as a traceability sample.
- the biobase degree of the removed EVOH layer was measured according to the method described in the above evaluation method (5) and found to be 10%, which is consistent with the value obtained in the gas barrier resin composition pellets of Example 2-6 and has traceability. It was confirmed that there was. Moreover, when the sulfur compound content of the taken-out EVOH layer was measured and its identification was performed, the sulfur compound was 0.3 ppm in terms of sulfur atom, the sulfur compound was dimethyl sulfide, and the gas barrier of Example 2-6. It was confirmed that there was traceability, which was in agreement with the value obtained with the resin composition pellets.
- EVOH ⁇ Preparation of each pellet of EVOH (A3-1) to EVOH (A3-9) and EVOH (B3-1)> EVOH (A3) is the same method as EVOH (A1-1) pellets, except that the types of ethylene and vinyl acetate as raw materials (raw material monomers) and the contents of phosphoric acid compounds and boron compounds are as shown in Table 7. -2) Pellets-EVOH (A3-9) pellets and EVOH (B3-1) pellets were prepared. As the ethylene derived from fossil fuel, ethylene manufactured by Air Liquide Industrial Gas Co., Ltd. was used.
- Each of the EVOH (A3-1) pellets to the EVOH (A3-9) pellets and the EVOH (B3-1) pellets contains ethylene units according to the methods described in the above evaluation methods (1) and (3) to (5).
- the amount and degree of saponification, the quantification of carboxylic acid, the quantification of metal ions, phosphoric acid compounds and boron compounds, and the degree of biobase were measured. The results are shown in Table 7.
- Example 3-1 EVOH (A3-1) pellets and EVOH (B3-1) pellets in mass ratio (A3-1 / B3-1) 10/90, and synthetic silica as inorganic particles (Fuji Silysia Chemical Ltd. "Silicia 310P"; laser method
- the average particle size (2.7 ⁇ m) measured in 1) was dry-blended using a tumbler so that the content was 300 ppm, and then a twin-screw extruder (“2D25W” manufactured by Toyo Seiki Seisakusho Co., Ltd., 25 mm ⁇ , die temperature 220 ° C.). , Screw rotation speed 100 rpm) was extruded and pelletized in a nitrogen atmosphere to obtain gas barrier resin composition pellets of Example 3-1.
- the obtained gas barrier resin composition pellets are melted at 210 ° C., extruded from a die onto a casting roll, and at the same time, air is blown at a wind speed of 30 m / sec using an air knife to form an unstretched film with a thickness of 170 ⁇ m (single). Layer) was obtained.
- This EVOH unstretched film was brought into contact with warm water at 80 ° C. for 10 seconds and stretched 3.2 times in the vertical direction and 3.0 times in the horizontal direction in a 90 ° C. atmosphere using a tenter type simultaneous biaxial stretching machine.
- the first and 102nd biaxially stretched films were used, and the "batch type vapor deposition equipment EWA-105" manufactured by Nippon Vacuum Technology Co., Ltd. was used at a film running speed of 200 m / min. After vapor-depositing aluminum on one side of the film, the film was wound to obtain a vapor-deposited film roll.
- the obtained vapor-filmed film was evaluated by the method described in the above-mentioned evaluation method (10). The results are shown in Table 9.
- Examples 3-2 to 3-21 Comparative Examples 3-1 to 3-2> Examples 3-2 to 3-21 and the same method as in Example 3-1 except that the type and mass ratio (ratio) of EVOH used and the content of inorganic particles were changed as shown in Table 8.
- Each gas barrier resin composition pellet, biaxially stretched film and vapor-deposited film of Comparative Examples 3-1 to 3-2 were prepared and evaluated. The results are shown in Tables 8 and 9. The vapor-filmed film was not evaluated for Example 3-21.
- each gas barrier resin composition of Examples 3-1 to 3-21 uses only a fossil fuel-derived raw material while partially using a biomass-derived raw material (Comparative Example 3-). It had a high gas barrier property comparable to that of the gas barrier resin composition (2). Further, each of the gas barrier resin compositions of Examples 3-1 to 3-20 was produced with respect to the film breaking resistance of the biaxially stretched film produced 10 hours after the start of operation and 10 hours after the start of operation.
- each gas barrier resin composition of Examples 3-1, 3-2, 3-5 to 3-13, and 3-15 to 3-20 having a biobase degree of 65% or less has passed 10 hours from the start of operation.
- the film rupture resistance of the biaxially stretched film produced later, and the vaporization defect suppression property and adhesion strength of the vaporized film produced using the first biaxially stretched film produced 10 hours after the start of operation were evaluated. It was A or B and had a high long-running property.
- Example 3-21 it was confirmed that the gas barrier resin composition using EVOH had insufficient film breakage resistance when it did not contain inorganic particles.
- ethylene derived from biomass bioethylene derived from sugar cane, manufactured by Braskem SA
- V-65 2,2'-azobis (2,4-dimethylvaleronitrile)
- methanol methanol
- the ethylene pressure was maintained at 4.13 MPa and the polymerization temperature was maintained at 65 ° C.
- the polymerization rate of VAc reached 50%, the mixture was cooled to terminate the polymerization.
- nitrogen gas was bubbled to completely deethylene.
- MeOH was added to the ethylene-vinyl acetate copolymer to prepare a 20% by mass MeOH solution.
- the water-containing pellets of EVOH were put into an acetic acid aqueous solution (bath ratio 20) having a concentration of 1 g / L and washed by stirring for 2 hours. This was deflated, further added to a 1 g / L acetic acid aqueous solution (bath ratio 20), and stirred and washed for 2 hours. After the liquid was removed, the acetic acid aqueous solution was updated and the same operation was performed. After washing with an acetic acid aqueous solution and then deflated, the solution is put into ion-exchanged water (bath ratio 20), stirred and washed for 2 hours, and the operation of deflated is repeated 3 times to purify the catalyst during the saponification reaction.
- Water-containing pellets of EVOH were obtained from which the residue and MeOH used at the time of strand precipitation were removed.
- the water content of the obtained EVOH water-containing pellets was measured with a halogen moisture meter "HR73" manufactured by METTLER CORPORATION and found to be 110% by mass.
- EVOH (A4) is the same method as EVOH (A4-1) pellets, except that the types of ethylene and vinyl acetate as raw materials (raw material monomers) and the contents of phosphoric acid compounds and boron compounds are as shown in Table 10.
- -2) Pellets to EVOH (A4-9) pellets and EVOH (B4-1) to EVOH (B4-4) pellets were prepared.
- ethylene derived from fossil fuel ethylene manufactured by Air Liquide Industrial Gas Co., Ltd. was used.
- Example 4-1 10 parts by mass of EVOH (A4-1) pellets, 90 parts by mass of EVOH (B4-1) pellets, and N, N'-(hexane-1,6-diyl) bis [3- (hexane-1,6-diyl) bis as antioxidant (D). 3,5-Di-tert-butyl-4-hydroxyphenyl) propionamide] (BASF Japan, Inc.
- the obtained gas barrier resin composition pellets of Example 4-1 have an ethylene unit content and ken according to the methods described in the above evaluation methods (1), (3) to (7), (11) and (12).
- the degree of conversion, the quantification of carboxylic acid, the quantification of metal ions, phosphoric acid compounds and boron compounds, the degree of biobase, the long-running property of a single-layer film, the oxygen permeability, the oxidative deterioration resistance, and the multi-layer pipe were measured or evaluated. The results are shown in Tables 11 and 12.
- each gas barrier resin composition of Examples 4-1 to 4-21 uses only a fossil fuel-derived raw material while partially using a biomass-derived raw material (Comparative Example 4-). It had a high gas barrier property comparable to that of the gas barrier resin composition of No. 2). Further, each of the gas barrier resin compositions of Examples 4-1 to 4-21 was evaluated for film-forming defects and streaks of the film produced 10 hours after the start of operation, and for the multilayer pipe produced 10 hours after the start of operation. The streak was evaluated as A or B, and it had sufficient long-running property.
- the gas barrier property does not depend on the biobase degree, whereas the long-run property tends to increase as the biobase degree decreases. It was confirmed that it has specific properties.
- the gas barrier resin compositions of Examples 4-1 and 4-2, 4-5 to 4-13, and 4-15 to 4-21 having a biobase degree of 65% or less are 10 hours after the start of operation and 50. The film formation defects, streaks, and roll end coloring of the film produced after hours were evaluated as A or B, and the film had a high long-run property.
- ethylene derived from biomass bioethylene derived from sugar cane, manufactured by Braskem SA
- methanol 34.7 g of 2,2'-azobis (2,4-dimethylvaleronitrile) ("V-65" from Wako Pure Chemical Industries, Ltd.) as an initiator. It was added as a solution and polymerization was started.
- the ethylene pressure was maintained at 2.87 MPa and the polymerization temperature was maintained at 60 ° C.
- the mixture was cooled to terminate the polymerization.
- nitrogen gas was bubbled to completely deethylene.
- MeOH was added to the ethylene-vinyl acetate copolymer to prepare a 20% by mass MeOH solution.
- the water-containing pellets of EVOH were put into an acetic acid aqueous solution (bath ratio 20) having a concentration of 1 g / L and washed by stirring for 2 hours. This was deflated, further added to a 1 g / L acetic acid aqueous solution (bath ratio 20), and stirred and washed for 2 hours. After the liquid was removed, the acetic acid aqueous solution was updated and the same operation was performed. After washing with an acetic acid aqueous solution and then deflated, the solution is put into ion-exchanged water (bath ratio 20), stirred and washed for 2 hours, and the operation of deflated is repeated 3 times to purify the catalyst during the saponification reaction.
- Water-containing pellets of EVOH were obtained from which the residue and MeOH used at the time of strand precipitation were removed.
- the water content of the obtained EVOH water-containing pellets was measured with a halogen moisture meter "HR73" manufactured by METTLER CORPORATION and found to be 110% by mass.
- EVOH (A5) is the same method as EVOH (A5-1) pellets, except that the types of ethylene and vinyl acetate as raw materials (raw material monomers) and the contents of phosphoric acid compounds and boron compounds are as shown in Table 13. -2) Pellets to EVOH (A5-9) pellets and EVOH (B5-1) to EVOH (B5-4) pellets were prepared.
- ethylene derived from fossil fuel ethylene manufactured by Air Liquide Industrial Gas Co., Ltd. was used.
- Example 5-1 9 parts by mass of EVOH (A5-1) pellets, 81 parts by mass of EVOH (B5-1) pellets, 10 parts by mass of polyamide pellets ("Ny1018A” (nylon 6) manufactured by Ube Kosan Co., Ltd.) (E), and metal.
- the obtained gas barrier resin composition pellets of Example 5-1 have an ethylene unit content and ken according to the methods described in the above evaluation methods (1), (3) to (5), and (13) to (15).
- the degree of conversion, quantification of carboxylic acid, quantification of metal ions, phosphoric acid compounds and boron compounds, biobase degree, long-run property, oxygen permeability and retort resistance were measured or evaluated.
- the quantification of metal ions the amount of metal ions converted into metal atoms including the amount of metal atoms added as metal atoms (F) was determined. The results are shown in Tables 14 and 15.
- Examples 5-2 to 5-27, Comparative Examples 5-1 to 5-8> Except for changing the type and mass ratio (ratio) of EVOH used, the mass ratio of polyamide (E), the type of compound containing the metal atom (F), and the content in terms of metal atom as shown in Table 14. , Each gas barrier resin composition pellet of Examples 5-2 to 5-27 and Comparative Examples 5-1 to 5-8 was prepared and evaluated by the same method as in Example 5-1. The results are shown in Tables 14 and 15.
- each gas barrier resin composition of Examples 5-1 to 5-27 uses only a fossil fuel-derived raw material while partially using a biomass-derived raw material (Comparative Example 5-). It had a high gas barrier property comparable to that of the gas barrier resin composition (2). Further, each of the gas barrier resin compositions of Examples 5-1 to 5-27 has a film forming defect and a streak evaluation of A or B for the film produced 1 hour after the start of operation, and has sufficient long-running property. It was a thing.
- the gas barrier property and the retort resistance do not depend on the degree of biobase, whereas the long-run property tends to increase as the degree of biobase decreases. It was confirmed that there is a peculiar property that it is in.
- the gas barrier resin compositions of Examples 5-1 to 5-3 and 5-5 to 5-27 having a biobase degree of 65% or less are for films prepared 1 hour and 5 hours after the start of operation. The film formation defects and streaks were evaluated as A or B, and they had high long-running properties.
- EVOH ⁇ Preparation of pellets of EVOH (A6-1) to EVOH (A6-9) and EVOH (B6-1) to EVOH (B6-4)> EVOH (A6) is the same method as EVOH (A5-1) pellets, except that the types of ethylene and vinyl acetate as raw materials (raw material monomers) and the contents of phosphoric acid compounds and boron compounds are as shown in Table 16. -1) Pellets to EVOH (A6-9) pellets and EVOH (B6-1) to EVOH (B6-4) pellets were prepared. As the ethylene derived from fossil fuel, ethylene manufactured by Air Liquide Industrial Gas Co., Ltd. was used.
- the modified thermoplastic elastomer (G) used in Examples and Comparative Examples is as follows.
- Example 6-1 After dry-blending 9 parts by mass of EVOH (A6-1) pellets, 81 parts by mass of EVOH (B6-1) pellets and 10 parts by mass of thermoplastic elastomer (G-5) pellets, a twin-screw extruder (Toyo Seiki Co., Ltd.) Extruded pellets were carried out in a nitrogen atmosphere using "2D25W” manufactured by Mfg. Co., Ltd., 25 mm ⁇ , die temperature 220 ° C., screw rotation speed 100 rpm) to obtain gas barrier resin composition pellets of Example 6-1.
- the obtained gas barrier resin composition pellets of Example 1 have an ethylene unit content according to the methods described in the above evaluation methods (1), (3) to (5), (7), (16), and (17). And the degree of saponification, quantification of carboxylic acid, quantification of metal ion, phosphoric acid compound and boron compound, biobase degree, long run property, oxygen permeability and bending resistance were measured or evaluated. The results are shown in Table 18.
- Example 6-1 and Example 6-1 except that the type and content of EVOH used, the type and content of the thermoplastic elastomer (G), and the type, brand and content of the additive were changed as described in Table 17.
- Pellets of each gas barrier resin composition of Examples 6-2 to 6-27 and Comparative Examples 6-1 to 6-5 were prepared and evaluated by the same method. The results are shown in Table 18.
- Irganox 1098 manufactured by BASF Japan Ltd., which is an antioxidant was used as an additive.
- ZHT-4A manufactured by Kyowa Chemical Industry Co., Ltd. which is a halogen scavenger, was used as an additive.
- the various pellets were dry-blended, the above additives were also dry-blended to obtain gas barrier resin composition pellets.
- Example 6-28 Using the gas barrier resin composition pellets obtained in Example 6-13, the oxidative deterioration resistance was evaluated by the method described in the above evaluation method (11). The oxidation resistance deterioration time was 65 hours.
- Example 6-29 Using the gas barrier resin composition pellets obtained in Examples 6-14, the oxidative deterioration resistance was evaluated by the method described in the above evaluation method (11). The oxidation resistance deterioration time was 280 hours. Further, using the gas barrier resin composition pellets obtained in Examples 6-14, a multilayer pipe was produced in the following manner. High-density polyethylene (“Novatec® HD HE421” manufactured by Nippon Polyethylene Co., Ltd., density 0.956 g / cc, MFR 0.14 g / 10 minutes) was used as the first extruder in Example 6-14.
- High-density polyethylene (“Novatec® HD HE421” manufactured by Nippon Polyethylene Co., Ltd., density 0.956 g / cc, MFR 0.14 g / 10 minutes) was used as the first extruder in Example 6-14.
- Each of the obtained gas barrier resin composition pellets was placed in a second extruder, and "Admer (registered trademark) NF408E” manufactured by Mitsui Kagaku Co., Ltd. was placed in a third extruder as an adhesive resin.
- a multi-layer pipe having an outer diameter of 20 mm was extruded using a circular die, and immediately after that, it was cooled and solidified through a cooling water tank adjusted to 40 ° C.
- Example 6-30 Using the gas barrier resin composition pellets obtained in Example 6-1 a film was formed under the following conditions, and the trim was cut to prepare a silage film having a width of 500 mm and a total thickness of 25.5 ⁇ m.
- Equipment 7 types, 7 layers, blown film film forming machine (manufactured by Brampton) -Layer structure and thickness of each layer: Outer layer 1 / Outer layer 2 / Adhesive resin layer 1 / Gas barrier resin composition layer / Adhesive resin layer 2 / Outer layer 3 / Outer layer 4 -Outer layers 1, 4: Linear low-density polyethylene (manufactured by Dow Chemical Co., Ltd., TUFLIN HS-7028 NT7 (MFR 1.0 g / 10 minutes)) 97% by mass, polyisobutene (manufactured by Soltex, PB32) 3% by mass by melt kneading.
- TUFLIN HS-7028 NT7 MFR
- Example 6-31 Using the gas barrier resin composition pellets obtained in Example 6-1 the extruders 1 and 2 used the resin constituting the outer layer A, and the extruders 3 and 4 used the resin constituting the outer layer D to the extruder 5. , 6 is extruded with the resin constituting the adhesive resin layers B and C, and the extruder 7 is extruded with the resin constituting the gas barrier resin composition layer, and the die is 75 mm and the draw ratio is 1.5. The trim was cut to prepare a film for a grain storage bag having a width of 900 mm and a total thickness of 230 ⁇ m in the same manner as in the production conditions of the silage film of 29.
- Example 6-32 [Making a laminated peeling container] Using the gas barrier resin composition pellets obtained in Examples 6-17, a laminated stripping container having a main body and a mouthpiece ((inner surface side) inner surface layer (LLDPE) / adhesive layer (blend of modified polyolefin and LLDPE) / most The outer layer (gas barrier resin composition layer) / outer layer (LLDPE) (outer surface side)) was produced by blow molding under the conditions shown below.
- Outer layer Non-denatured polypropylene (Noblen (registered trademark) FSX16E9, manufactured by Sumitomo Chemical Co., Ltd.)
- Inner layer Three-layer structure of outermost layer / adhesive layer / inner surface layer in order from the outer layer side
- Outer layer Gas barrier resin composition obtained in Example 17
- Adhesive layer Modified polyolefin (Modic (registered trademark) L522, manufactured by Mitsubishi Chemical Corporation) And LLDPE (Harmorex (registered trademark) F325N, manufactured by Japan Polyethylene Corporation) 1: 1 blend
- Inner layer LLDPE (Harmorex (registered trademark) F325N, manufactured by Japan Polyethylene Corporation)
- blow molding conditions By co-extruding each molten resin so as to have the above layer structure, a laminated parison in a molten state was produced. The lip width was adjusted during the production of the laminated parison so that the thickness of the oral cavity became thicker.
- the laminated parison was set in a blow molding die and molded into a desired container shape by a blow molding method. During blow molding, adjustments were made so that the thickness of the mouthpiece was sufficiently thicker than the thickness of the main body.
- the coextrusion conditions were adjusted so that the thickness of both the outer layer and the inner layer excluding the oral cavity was in the range of 70 to 130 ⁇ m and the ratio of the outer layer / inner layer thickness was 0.8 to 1.3. ..
- the conditions for blow molding were blow pressure: 0.4 MPa, mold temperature: 25 ° C., and blow time: 15 seconds.
- a section was prepared by cutting out the oral cavity of the obtained laminated stripping container with a microtome, the section was placed on a slide glass, and the thickness of the oral cavity was measured with an optical microscope. The thickness of the oral cavity was 0.5 mm.
- Preliminary peeling was performed by forming an air introduction hole in the outer layer of the main body of the obtained laminated peeling container and injecting air between the outer layer and the inner layer from the air introduction hole. Air was injected at a pressure of 0.3 MPa for 1.0 second. After pre-peeling, with the air introduction hole closed, the container is crushed with a force of 30 kg to apply pressure to the air between the outer and inner layers, and the interface between the outer and inner layers in the oral cavity. I checked if air leaked from. As a result, no air leakage from the sea surface of the oral cavity was confirmed. Since the gas barrier resin composition layer and the LLDPE layer are directly laminated in the obtained laminated peeling container, the main body portion has an appropriate peeling property at the interface between the gas barrier resin composition layer and the LLDPE layer. It became a container.
- Example 6-33 A laminated parison was produced without adjusting the lip width, and a laminated peeling container was produced by the same method as in Example 6-31 except that the thickness of the oral cavity and the main body was not adjusted during blow molding. , Evaluated. The thickness of the oral cavity was 0.3 mm, and air leakage was confirmed in the peelability test of the oral cavity.
- each gas barrier resin composition of Examples 6-1 to 6-27 uses only a fossil fuel-derived raw material while partially using a biomass-derived raw material (Comparative Example 6-). It had high gas barrier properties and bending resistance comparable to those of (2) gas barrier resin composition, etc.). Further, each of the gas barrier resin compositions of Examples 6-1 to 6-27 has a film forming defect and a streak evaluation of A or B for the film produced 1 hour after the start of operation, and has sufficient long-running property. It was a thing.
- the gas barrier property and the bending resistance do not depend on the degree of biobase, whereas the long-run property tends to increase as the degree of biobase decreases. It was confirmed that there is a peculiar property that it is in.
- the gas barrier resin compositions of Examples 6-1 to 6-3 and 6-5 to 6-27 having a biobase degree of 65% or less are for films prepared 1 hour and 5 hours after the start of operation. The film formation defects, streaks, and roll end coloring were evaluated as A or B, and had high long-running properties.
- Example 7-1 After dry-blending EVOH (A1-1) pellets and EVOH (B1-1) pellets at a mass ratio (A1-1 / B1-1) 10/90, a twin-screw extruder (“2D25W” manufactured by Toyo Seiki Seisakusho Co., Ltd.” , 25 mm ⁇ , die temperature 220 ° C., screw rotation speed 100 rpm) was extruded into pellets in a nitrogen atmosphere to obtain gas barrier resin composition pellets of Example 7-1.
- a twin-screw extruder (“2D25W” manufactured by Toyo Seiki Seisakusho Co., Ltd.” , 25 mm ⁇ , die temperature 220 ° C., screw rotation speed 100 rpm) was extruded into pellets in a nitrogen atmosphere to obtain gas barrier resin composition pellets of Example 7-1.
- the obtained gas barrier resin composition pellets of Example 7-1 were subjected to the ethylene unit content and ken according to the methods described in the above evaluation methods (1), (3) to (5), and (18) to (22). Degree of conversion, quantification of carboxylic acid, quantification of metal ions, phosphate compounds and boron compounds, biobase degree, evaluation of multilayer film, evaluation of shrinkage of heat shrink film, evaluation of multilayer thermoformed container, evaluation of streak of blow molded container and fuel Permeability was measured or evaluated. The results are shown in Tables 19 and 20.
- Examples 7-2 to 7-22 Comparative Examples 7-1 to 7-6> Examples 7-2 to 7-22 and Comparative Examples 7-1 to 7-1 were obtained in the same manner as in Example 7-1 except that the types and mass ratios (ratio) of EVOH used were changed as shown in Table 19.
- Each gas barrier resin composition pellet of 7-6 was prepared and evaluated. For each of the gas barrier resin composition pellets of Examples 7-21 and 7-22, the evaluation of the multilayer thermoformed container, the streak evaluation of the blow molded container, and the measurement or evaluation of the fuel permeability were not performed. The results are shown in Table 20.
- Examples 7-23, Comparative Examples 7-7 and 7-8> Evaluation of co-extruded coated paper
- carton paper thickness 500 ⁇ m, basis weight 400 g / m 2
- the coextrusion structure is low density polyethylene / adhesive layer / gas barrier resin composition layer / adhesive layer / low density polyethylene / carton paper, and the thickness structure is 20/5/5/5/20/500 ⁇ m.
- a feed block and a T-shaped die were used to merge and distribute a low-density polyethylene extruder, an EVOH extruder, an adhesive layer extruder, and resins supplied from the respective extruders.
- the low-density polyethylene is linear low-density polyethylene ("Ultzex (trademark) 2022L” manufactured by Prime Polymer Co., Ltd.)
- the adhesive layer is polypropylene modified with maleic anhydride (“Admer (Admer)” manufactured by Mitsui Chemicals, Inc. Trademark) QF-500 ") was used.
- the temperature conditions of the feed block and the T-shaped die were set to 250 ° C., and the pick-up speed was set to 300 m / min.
- the multilayer films using the gas barrier resin compositions of Examples 7-1 to 7-22 are those derived only from fossil fuels while partially using raw materials derived from biomass. It had a high gas barrier property comparable to that of (multilayer film using the gas barrier resin composition of Comparative Example 7-3). Further, in each of the gas barrier resin compositions of Examples 7-1 to 7-22, the streak evaluation of the multilayer film and the thermoformed container produced 10 hours after the start of operation was A or B, and the start of operation 3 The streak of the blow-molded container prepared after hours was evaluated as A or B, and it had sufficient long-running property. In Examples 7-21 and 7-22 in which heat-shrinkable films using EVOH having a modifying group were prepared, good shrinkage was confirmed.
- the gas barrier property does not depend on the biobase degree, whereas the long-run property tends to increase as the biobase degree decreases. It was confirmed that it has specific properties.
- the gas barrier resin compositions of Examples 7-1, 7-2, 7-5 to 7-13, and 7-15 to 7-22 having a biobase degree of 65% or less are 10 hours after the start of operation and 50.
- the evaluation of streak and roll end coloring of the multilayer film produced after a period of time was A or B, and the multilayer film had a high long-run property.
- Example 8-1 After dry-blending EVOH (A1-1) pellets and EVOH (B1-1) pellets at a mass ratio (A1-1 / B1-1) 10/90, a 30 mm ⁇ twin-screw extruder ("TEX-" manufactured by Japan Steel Works, Ltd. 30SS-30CRW-2V ”) is melt-extruded from a strand die under the following conditions into a strand shape, and the strand-shaped molten resin extruded from the strand die is cooled in a water tank, cut with a pelletizer, and columnar. The resin composition pellets of Example 8-1 having an average diameter of 2.8 mm and an average length of 3.2 mm were obtained. (Conditions for twin-screw extruder) Extrusion temperature: 220 ° C Screw rotation speed: 300 rpm Extruded resin amount: 25 kg / hour Strand die: 3 mm ⁇ , 6 holes
- the ethylene unit content, the degree of saponification, and the carboxylic acid were according to the methods described in the above evaluation methods (1), (3) to (5), and (23). Quantification, quantification of metal ions, phosphate compounds and boron compounds, measurement or evaluation of biobase degree and long-running property. The results are shown in Table 21.
- each of the resin compositions of Examples 8-1 to 8-20 is only derived from fossil fuels while partially using raw materials derived from biomass (). It had plant growth performance comparable to that of Comparative Example 8-2). Further, each of the resin compositions of Examples 8-1 to 8-20 has a long-run property evaluation (die adhesion amount, thread breakage frequency) of A or B when continuously producing resin composition pellets, which is sufficient. It had a long-running property.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Polymers & Plastics (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Environmental Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Manufacturing & Machinery (AREA)
- General Chemical & Material Sciences (AREA)
- Mechanical Engineering (AREA)
- Materials Engineering (AREA)
- Water Supply & Treatment (AREA)
- Laminated Bodies (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
バイオマス由来の原料を用いていながら、化石燃料由来のものと遜色のない高いガスバリア性、及び十分なロングラン性を有するガスバリア樹脂組成物、このガスバリア樹脂組成物を用いた多層構造体、及びこのようなガスバリア樹脂組成物の製造方法の提供。一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物を含み、上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物の原料であるエチレン及びビニルエステルの一部がバイオマス由来であり、残部が化石燃料由来である、ガスバリア樹脂組成物。
Description
本発明は、ガスバリア樹脂組成物、ガスバリア樹脂組成物の製造方法、及びガスバリア樹脂組成物を用いた成形体等に関する。
酸素等のガスを遮断する性能(ガスバリア性)に優れた樹脂を用いたガスバリア材は、容器、フィルム、シート、パイプ等の各種用途に幅広く使用されている。ガスバリア性に優れた樹脂としては、ポリアミド、ポリエステル、ポリ塩化ビニリデン、アクリロニトリル共重合体、ポリフッ化ビニリデン、ポリクロロトリフルオロエチレン、エチレン-ビニルエステル共重合体ケン化物等が知られている。例えば特許文献1には、ポリアミド、ポリエステル、ポリビニルアルコール、エチレン-ビニルアルコール共重合体、フッ素含有樹脂及びシリコーン樹脂から選ばれる少なくとも一種のガスバリア性樹脂層を有する多層プラスチック容器の発明が記載されている。
一方、近年、循環型社会を目指し、カーボンニュートラルなバイオマス由来の原料を用いたバイオプラスチックの需要が高まっている。しかし、バイオマス由来の合成樹脂は、化石燃料由来の合成樹脂と比べて性能が劣る場合があることが知られている。例えば特許文献2には、従来のバイオマス由来のポリオレフィン等のフィルム材は密着性、加工性、耐久性等の品質が十分ではなかったとされ、このような点を改善するための、バイオマス由来の樹脂を含む特定の組成のバイオマス由来樹脂層を備える樹脂フィルムの発明が記載されている。また、特許文献3には、石油由来の樹脂をバイオマス由来の樹脂に置き換えたフィルムは耐衝撃性等が低下する場合があるとされ、このような点を改善するための、バイオマス由来のバイオマスポリエチレンと、化石燃料由来のポリエチレンと、プロピレン系ブロック共重合体樹脂とを含有する中間層を有する積層フィルムの発明が記載されている。
ガスバリア材の用途において、バイオマス由来の原料を用いて合成されたガスバリア樹脂の製品化が期待される。しかし、上述のようにバイオマス由来の合成樹脂は、化石燃料由来の合成樹脂と比べて性能が劣る場合があることから、従来の化石燃料由来のガスバリア樹脂をバイオマス由来のガスバリア樹脂に置き換えた場合、最も重要なガスバリア性が低下することが懸念される。このため、化石燃料由来の樹脂と遜色のない優れたガスバリア性を有するバイオマス由来の樹脂の開発が望まれている。
また、ガスバリア樹脂は、溶融成形によって容器、シート等の各種形状に成形されることがある。このため、ガスバリア樹脂には、長時間に渡る溶融成形を行い続けても、欠陥等が発生し難いといったロングラン性(長時間運転特性)も重要である。
本発明は、以上のような事情に基づいてなされたものであり、その目的は、バイオマス由来の原料を用いていながら、化石燃料由来のものと遜色のない高いガスバリア性、及び十分なロングラン性を有するガスバリア樹脂組成物、このガスバリア樹脂組成物を用いた成形体等、及びこのようなガスバリア樹脂組成物の製造方法を提供することである。
本発明者は、ガスバリア樹脂の一種であるエチレン-ビニルエステル共重合体ケン化物においては、バイオマス由来の原料をモノマーとして用いて合成したものが、化石燃料由来の原料をモノマーとして用いて合成された同一構造の従来のものと遜色ない高いガスバリア性を有することを知見した。一方、ロングラン性に関しては、バイオマス由来の原料をモノマーとして用いて合成したエチレン-ビニルエステル共重合体ケン化物は、化石燃料由来の原料をモノマーとして用いて合成したものと比べて劣るという、ガスバリア性とは異なる傾向にあることもわかった。このようなことから、本発明者らは、バイオマス由来の原料と化石燃料由来の原料とが併用されたエチレン-ビニルエステル共重合体ケン化物であれば、環境負荷を低減させながら、化石燃料由来の原料のみを用いたものと同じような高いガスバリア性を発揮でき、かつ十分なロングラン性も兼ね備えるものとなることを見いだし、本発明の完成に至った。
すなわち本発明は、
[1]一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物を含み、上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物の原料であるエチレン及びビニルエステルの一部がバイオマス由来であり、残部が化石燃料由来である、ガスバリア樹脂組成物;
[2]上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物が、原料であるエチレン及びビニルエステルの少なくとも一部がバイオマス由来であるエチレン-ビニルエステル共重合体ケン化物(A)と、化石燃料由来であるエチレン-ビニルエステル共重合体ケン化物(B)とを含む、[1]のガスバリア樹脂組成物;
[3]上記エチレン-ビニルエステル共重合体ケン化物(A)と上記エチレン-ビニルエステル共重合体ケン化物(B)との質量比(A/B)が1/99~99/1である、[2]のガスバリア樹脂組成物;
[4]上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物が、原料であるエチレン及びビニルエステルの一部がバイオマス由来であり、残部が化石燃料由来であるエチレン-ビニルアルコール共重合体(A’)を含む、[1]のガスバリア樹脂組成物;
[5]上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物のバイオベース度が1%以上99%以下である、[1]~[4]のいずれかのガスバリア樹脂組成物;
[6]バイオベース度が1%以上99%以下である、[1]~[5]のいずれかのガスバリア樹脂組成物;
[7]硫黄化合物を硫黄原子換算で0ppmを超えて100ppm以下含む、[1]~[6]のいずれかのガスバリア樹脂組成物;
[8]上記硫黄化合物が、ジメチルスルフィドまたはジメチルスルホキシドである、[7]のガスバリア樹脂組成物;
[9]上記原料のうちのエチレンの少なくとも一部がバイオマス由来である、[1]~[8]のいずれかのガスバリア樹脂組成物;
[10]上記原料のうちのビニルエステルの少なくとも一部がバイオマス由来である、[1]~[9]のいずれかのガスバリア樹脂組成物;
[11]上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物が、下記一般式(I)で表される変性基を有する、[1]~[10]のいずれかのガスバリア樹脂組成物
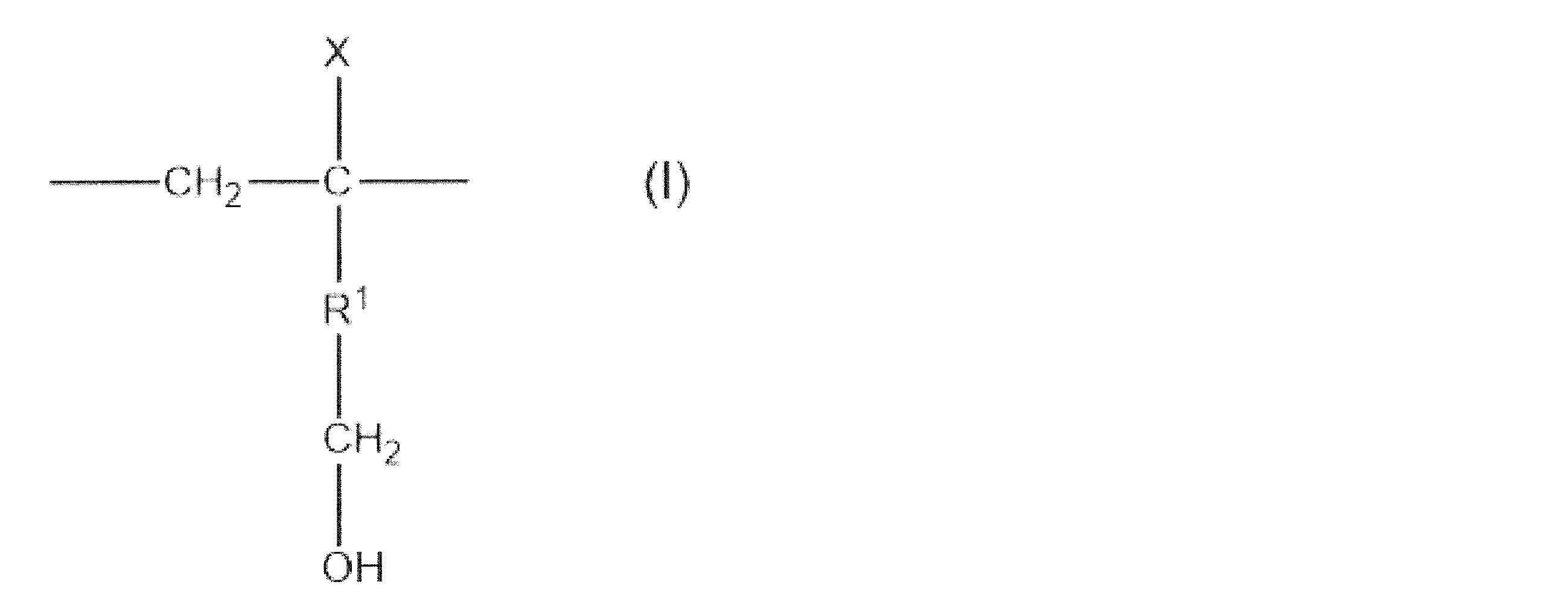
[式(1)中、Xは、水素原子、メチル基又はR2-OHで表される基を表す。R1及びR2は、それぞれ独立して、単結合、炭素数1~9のアルキレン基又は炭素数1~9のアルキレンオキシ基を表し、上記アルキレン基及び上記アルキレンオキシ基は水酸基、アルコキシ基又はハロゲン原子を含んでもよい。];
[12]カルボン酸をカルボン酸根換算で30ppm以上1000ppm以下含む、[1]~[11]のいずれかのガスバリア樹脂組成物;
[13]金属イオンを1ppm以上1000ppm以下含む、[1]~[12]のいずれかのガスバリア樹脂組成物;
[14]リン酸化合物をリン原子換算で1ppm以上200ppm以下含む、[1]~[13]のいずれかのガスバリア樹脂組成物;
[15]ホウ素化合物をホウ素原子換算で5ppm以上5000ppm以下含む、[1]~[14]のいずれかのガスバリア樹脂組成物;
[16]上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物が、エチレン-ビニルエステル共重合体ケン化物(X)、及び上記エチレン-ビニルエステル共重合体ケン化物(X)よりも融点が低いエチレン-ビニルエステル共重合体ケン化物(Y)を含む、[1]~[15]のいずれかのガスバリア樹脂組成物;
[17]無機粒子(C)をさらに含み、無機粒子(C)の含有量が50ppm以上5000ppm以下である、[1]~[16]のいずれかのガスバリア樹脂組成物;
[18]酸化防止剤(D)をさらに含み、酸化防止剤(D)の含有量が0.01質量%以上5質量%以下である、[1]~[17]のいずれかのガスバリア樹脂組成物;
[19]ポリアミド(E)、並びにマグネシウム、カルシウム及び亜鉛からなる群より選ばれる少なくとも1種の金属原子(F)をさらに含み、上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物に対するポリアミド(E)の質量比が5/95以上40/60以下であり、金属原子(F)の含有量が1ppm以上500ppm以下である、[1]~[18]のいずれかのガスバリア樹脂組成物;
[20]熱可塑性エラストマー(G)をさらに含み、上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物に対する熱可塑性エラストマー(G)の質量比が5/95以上35/65以下である、[1]~[19]のいずれかのガスバリア樹脂組成物;
[21]原料であるエチレン及びビニルエステルの少なくとも一部がバイオマス由来であるエチレン-ビニルエステル共重合体ケン化物(A)のペレットと、化石燃料由来であるエチレン-ビニルエステル共重合体ケン化物(B)のペレットとをドライブレンドし、溶融混練する工程を備える、ガスバリア樹脂組成物の製造方法;
[22]上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物のペレットと、ポリアミド(E)のペレットと、マグネシウム、カルシウム及び亜鉛からなる群より選ばれる少なくとも1種の金属原子(F)を含む化合物とを混合し、溶融混練する工程を備える、[19]のガスバリア樹脂組成物の製造方法;
[23]原料であるエチレン及びビニルエステルの少なくとも一部がバイオマス由来であるエチレン-ビニルエステル共重合体ケン化物(A)のペレットと、化石燃料由来であるエチレン-ビニルエステル共重合体ケン化物(B)のペレットと、熱可塑性エラストマー(G)のペレットとをドライブレンドし、溶融混練する工程を備える、ガスバリア樹脂組成物の製造方法;
[24][1]~[20]のいずれかのガスバリア樹脂組成物から形成されるガスバリア層を備える、成形体;
[25][24]の成形体を備える、フィルムまたはシート;
[26][24]の成形体を備える、産業用フィルムまたはシート;
[27][24]の成形体を備える、熱成形容器;
[28][24]の成形体を備える、ブロー成形容器;
[29][28]のブロー成形容器を備える、燃料容器;
[30][28]のブロー成形容器を備える、ボトル容器;
[31][24]の成形体を備える、チューブ;
[32][24]の成形体を備える、パイプ;
[33][24]の成形体を備える、紙容器;
[34][1]~[20]のいずれかのガスバリア樹脂組成物から形成される、単層フィルム;
[35][1]~[20]のいずれかのガスバリア樹脂組成物から形成される層を少なくとも1層備える、多層フィルム;
[36][34]の単層フィルム、または上記ガスバリア樹脂組成物から形成される層の少なくとも1層を最外層として備える[35]の多層フィルムと、上記単層フィルムまたは上記多層フィルムにおける上記ガスバリア樹脂組成物から形成される層の表出面に積層される少なくとも1層の無機蒸着層とを備える、蒸着フィルム;
[37][36]の蒸着フィルムと、上記蒸着フィルムにおける上記無機蒸着層上に積層される他の層とを備える、多層構造体;
[38][34]の単層フィルム、[35]の多層フィルム、[36]の蒸着フィルムまたは[37]の多層構造体を備える、ヒートシール用フィルム;
[39][1]~[20]のいずれかのガスバリア樹脂組成物からなる層を少なくとも1層備える、包装材;
[40]樹脂組成物から形成されている成形体を含む植物栽培用培地であって、上記樹脂組成物が、[1]~[20]のいずれかのガスバリア樹脂組成物である、植物栽培用培地;
を提供することにより達成される。
[1]一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物を含み、上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物の原料であるエチレン及びビニルエステルの一部がバイオマス由来であり、残部が化石燃料由来である、ガスバリア樹脂組成物;
[2]上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物が、原料であるエチレン及びビニルエステルの少なくとも一部がバイオマス由来であるエチレン-ビニルエステル共重合体ケン化物(A)と、化石燃料由来であるエチレン-ビニルエステル共重合体ケン化物(B)とを含む、[1]のガスバリア樹脂組成物;
[3]上記エチレン-ビニルエステル共重合体ケン化物(A)と上記エチレン-ビニルエステル共重合体ケン化物(B)との質量比(A/B)が1/99~99/1である、[2]のガスバリア樹脂組成物;
[4]上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物が、原料であるエチレン及びビニルエステルの一部がバイオマス由来であり、残部が化石燃料由来であるエチレン-ビニルアルコール共重合体(A’)を含む、[1]のガスバリア樹脂組成物;
[5]上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物のバイオベース度が1%以上99%以下である、[1]~[4]のいずれかのガスバリア樹脂組成物;
[6]バイオベース度が1%以上99%以下である、[1]~[5]のいずれかのガスバリア樹脂組成物;
[7]硫黄化合物を硫黄原子換算で0ppmを超えて100ppm以下含む、[1]~[6]のいずれかのガスバリア樹脂組成物;
[8]上記硫黄化合物が、ジメチルスルフィドまたはジメチルスルホキシドである、[7]のガスバリア樹脂組成物;
[9]上記原料のうちのエチレンの少なくとも一部がバイオマス由来である、[1]~[8]のいずれかのガスバリア樹脂組成物;
[10]上記原料のうちのビニルエステルの少なくとも一部がバイオマス由来である、[1]~[9]のいずれかのガスバリア樹脂組成物;
[11]上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物が、下記一般式(I)で表される変性基を有する、[1]~[10]のいずれかのガスバリア樹脂組成物
[12]カルボン酸をカルボン酸根換算で30ppm以上1000ppm以下含む、[1]~[11]のいずれかのガスバリア樹脂組成物;
[13]金属イオンを1ppm以上1000ppm以下含む、[1]~[12]のいずれかのガスバリア樹脂組成物;
[14]リン酸化合物をリン原子換算で1ppm以上200ppm以下含む、[1]~[13]のいずれかのガスバリア樹脂組成物;
[15]ホウ素化合物をホウ素原子換算で5ppm以上5000ppm以下含む、[1]~[14]のいずれかのガスバリア樹脂組成物;
[16]上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物が、エチレン-ビニルエステル共重合体ケン化物(X)、及び上記エチレン-ビニルエステル共重合体ケン化物(X)よりも融点が低いエチレン-ビニルエステル共重合体ケン化物(Y)を含む、[1]~[15]のいずれかのガスバリア樹脂組成物;
[17]無機粒子(C)をさらに含み、無機粒子(C)の含有量が50ppm以上5000ppm以下である、[1]~[16]のいずれかのガスバリア樹脂組成物;
[18]酸化防止剤(D)をさらに含み、酸化防止剤(D)の含有量が0.01質量%以上5質量%以下である、[1]~[17]のいずれかのガスバリア樹脂組成物;
[19]ポリアミド(E)、並びにマグネシウム、カルシウム及び亜鉛からなる群より選ばれる少なくとも1種の金属原子(F)をさらに含み、上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物に対するポリアミド(E)の質量比が5/95以上40/60以下であり、金属原子(F)の含有量が1ppm以上500ppm以下である、[1]~[18]のいずれかのガスバリア樹脂組成物;
[20]熱可塑性エラストマー(G)をさらに含み、上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物に対する熱可塑性エラストマー(G)の質量比が5/95以上35/65以下である、[1]~[19]のいずれかのガスバリア樹脂組成物;
[21]原料であるエチレン及びビニルエステルの少なくとも一部がバイオマス由来であるエチレン-ビニルエステル共重合体ケン化物(A)のペレットと、化石燃料由来であるエチレン-ビニルエステル共重合体ケン化物(B)のペレットとをドライブレンドし、溶融混練する工程を備える、ガスバリア樹脂組成物の製造方法;
[22]上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物のペレットと、ポリアミド(E)のペレットと、マグネシウム、カルシウム及び亜鉛からなる群より選ばれる少なくとも1種の金属原子(F)を含む化合物とを混合し、溶融混練する工程を備える、[19]のガスバリア樹脂組成物の製造方法;
[23]原料であるエチレン及びビニルエステルの少なくとも一部がバイオマス由来であるエチレン-ビニルエステル共重合体ケン化物(A)のペレットと、化石燃料由来であるエチレン-ビニルエステル共重合体ケン化物(B)のペレットと、熱可塑性エラストマー(G)のペレットとをドライブレンドし、溶融混練する工程を備える、ガスバリア樹脂組成物の製造方法;
[24][1]~[20]のいずれかのガスバリア樹脂組成物から形成されるガスバリア層を備える、成形体;
[25][24]の成形体を備える、フィルムまたはシート;
[26][24]の成形体を備える、産業用フィルムまたはシート;
[27][24]の成形体を備える、熱成形容器;
[28][24]の成形体を備える、ブロー成形容器;
[29][28]のブロー成形容器を備える、燃料容器;
[30][28]のブロー成形容器を備える、ボトル容器;
[31][24]の成形体を備える、チューブ;
[32][24]の成形体を備える、パイプ;
[33][24]の成形体を備える、紙容器;
[34][1]~[20]のいずれかのガスバリア樹脂組成物から形成される、単層フィルム;
[35][1]~[20]のいずれかのガスバリア樹脂組成物から形成される層を少なくとも1層備える、多層フィルム;
[36][34]の単層フィルム、または上記ガスバリア樹脂組成物から形成される層の少なくとも1層を最外層として備える[35]の多層フィルムと、上記単層フィルムまたは上記多層フィルムにおける上記ガスバリア樹脂組成物から形成される層の表出面に積層される少なくとも1層の無機蒸着層とを備える、蒸着フィルム;
[37][36]の蒸着フィルムと、上記蒸着フィルムにおける上記無機蒸着層上に積層される他の層とを備える、多層構造体;
[38][34]の単層フィルム、[35]の多層フィルム、[36]の蒸着フィルムまたは[37]の多層構造体を備える、ヒートシール用フィルム;
[39][1]~[20]のいずれかのガスバリア樹脂組成物からなる層を少なくとも1層備える、包装材;
[40]樹脂組成物から形成されている成形体を含む植物栽培用培地であって、上記樹脂組成物が、[1]~[20]のいずれかのガスバリア樹脂組成物である、植物栽培用培地;
を提供することにより達成される。
本発明によれば、バイオマス由来の原料を用いていながら、化石燃料由来のものと遜色のない高いガスバリア性、及び十分なロングラン性を有するガスバリア樹脂組成物、このガスバリア樹脂組成物を用いた成形体等、及びこのようなガスバリア樹脂組成物の製造方法を提供できる。
<ガスバリア樹脂組成物>
本発明のガスバリア樹脂組成物は、一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物(エチレン-ビニルアルコール共重合体;以下、「EVOH」ともいう。)を含み、上記一種又は二種以上のEVOHの原料(原料モノマー)であるエチレン及びビニルエステルの一部がバイオマス由来であり、上記原料(原料モノマー)であるエチレン及びビニルエステルの残部が化石燃料由来である、ガスバリア樹脂組成物である。当該ガスバリア樹脂組成物は、バイオマス由来の原料が一部に用いられていることで、環境負荷が低い。また、当該ガスバリア樹脂組成物は、ガスバリア樹脂としてEVOHを選択して用いており、EVOHはバイオマス由来の原料を用いて合成された場合であっても、化石燃料由来の原料のみから合成された同一構造のEVOHと同等の高いガスバリア性を発揮できる。なお、同一構造のEVOHとは、重合度、各構造単位の含有比率、変性の有無、ケン化度等が同じであるEVOHをいう。さらに、当該ガスバリア樹脂組成物においては、EVOHに化石燃料由来の原料も併用されていることで、ロングラン性も十分なものとなる。なお、本発明がこのような効果を奏する理由は定かではないが、バイオマス由来の原料を用いてEVOHを合成した場合、ガスバリア性には影響を与えないもののロングラン性には影響を与える微量且つ不可避的な不純物が生成又は混入することなどが推測される。
本発明のガスバリア樹脂組成物は、一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物(エチレン-ビニルアルコール共重合体;以下、「EVOH」ともいう。)を含み、上記一種又は二種以上のEVOHの原料(原料モノマー)であるエチレン及びビニルエステルの一部がバイオマス由来であり、上記原料(原料モノマー)であるエチレン及びビニルエステルの残部が化石燃料由来である、ガスバリア樹脂組成物である。当該ガスバリア樹脂組成物は、バイオマス由来の原料が一部に用いられていることで、環境負荷が低い。また、当該ガスバリア樹脂組成物は、ガスバリア樹脂としてEVOHを選択して用いており、EVOHはバイオマス由来の原料を用いて合成された場合であっても、化石燃料由来の原料のみから合成された同一構造のEVOHと同等の高いガスバリア性を発揮できる。なお、同一構造のEVOHとは、重合度、各構造単位の含有比率、変性の有無、ケン化度等が同じであるEVOHをいう。さらに、当該ガスバリア樹脂組成物においては、EVOHに化石燃料由来の原料も併用されていることで、ロングラン性も十分なものとなる。なお、本発明がこのような効果を奏する理由は定かではないが、バイオマス由来の原料を用いてEVOHを合成した場合、ガスバリア性には影響を与えないもののロングラン性には影響を与える微量且つ不可避的な不純物が生成又は混入することなどが推測される。
ここで、本明細書におけるロングラン性とは、実施例に記載の方法で評価することができ、ガスバリア樹脂組成物を連続成膜した際の欠点、ストリーク及び着色の度合いによって総合的に評価できる。但し、用途等によっては、着色は特に問題にならない場合もあることなどから、所定時間(例えば10時間)の連続製膜した際の欠点及びストリークが少なければロングラン性は十分であると評価する。
原料として用いられたエチレン及びビニルエステルがバイオマス由来と化石燃料由来の双方を含むことは、バイオベース度の測定により確認することができる。バイオベース度とは、バイオマス由来原料の割合を表す指標であり本明細書においては、加速器質量分析器(AMS)による放射性炭素(14C)の濃度測定により求められるバイオベース炭素含有率である。バイオベース度は、具体的にはASTM D6866-18に記載の方法に沿って測定することができる。すなわち、通常、EVOHのバイオベース度が0%超100%未満である場合、原料として用いられたエチレン及びビニルエステルがバイオマス由来と化石燃料由来の双方を含むといえる。
「バイオマス」とは、動植物に由来する有機物である資源であって、化石燃料(化石資源)を除いたものをいう。バイオマスは、植物に由来する有機物である資源であってよい。
本発明のガスバリア性樹脂組成物は、放射性炭素(14C)の濃度を利用して自社製品を追跡することも可能である。生物はその活動中に、大気中の放射性炭素(14C)を取り込み一定量含有するが、活動を停止すると新しい14Cの取り込みが止まり、全炭素に対する14Cの比が低下する。また、植物が炭素を固定する際に同位体選別と呼ばれる現象が生じ、植物の種毎に全炭素に対する14Cの比が異なることが知られている。全炭素に対する14Cの比は、産地、年代によっても異なることが知られており、原料とするバイオマスにより、異なる全炭素に対する14Cの比の原料を得ることができる。化石燃料由来の原料には14Cが殆ど残っていないため、例えば、バイオマス由来と化石燃料由来の原料の比を変化させることにより、特定の全炭素に対する14Cの比のEVOHを得ることが可能となり、この全炭素に対する14Cの比を調べることにより、自社製EVOH(ガスバリア性樹脂組成物)の追跡が可能となる。
EVOHは幅広い用途で使用されており、高品質の製品を市場へ供給することはサプライヤーの責務である。また、ブランディングのために自社製品と他社製品を識別する方法が求められている。例えば、市販の包装容器のガスバリア層に用いられているEVOHは、熱成形により包装容器に成形されるが、熱成形時に受ける熱履歴によりエチレン-ビニルエステル共重合体ケン化物は溶媒に不溶なゲルを形成することがある。そのため、包装容器を回収し、使用されているEVOHを溶媒で抽出して、その分子量を測定しようとしても、分子量を正確に測定することが困難な場合が多い。そのため、成形体を分析しただけでは自社のEVOHであるか否かを判別することができない。
EVOHは、多くの流通経路を経て、食品、医薬品、工業薬品、農薬等の包装材料として、フィルム、シート、容器等に利用されている。また、そのバリア性、保温性、耐汚染性等を活かして、自動車等車両の燃料タンク、タイヤ用チューブ材、農業用フィルム、ジオメンブレン、靴用クッション材等の用途にも使用されている。これらのEVOHが使用された材料がさらに廃棄された場合、かかる樹脂やその使用後の包装容器がどの工場、どの製造ラインから製造されたかの判別が困難である。また、使用時あるいは使用後の自社製品の品質調査や廃棄後の環境への影響や地中への分解性などの追跡も困難である。
自社製品の追跡方法の一つとして、例えば、EVOHにトレーサー物質を添加する方法が考えられる。しかしながら、トレーサーの添加はコスト上昇やEVOHの性能低下を起こす場合がある。このような背景において、放射性炭素(14C)の濃度を利用して自社製品を追跡することができることは、非常に有用な効果であると言える。
本発明のガスバリア樹脂組成物は、気体の透過を抑制する機能を有する樹脂組成物である。20℃-65%RH条件下で、JIS K 7126-2(等圧法;2006年)に記載の方法に準じて測定した本発明のガスバリア樹脂組成物の酸素透過速度の上限は、100mL・20μm/(m2・day・atm)が好ましく、50mL・20μm/(m2・day・atm)がより好ましく、10mL・20μm/(m2・day・atm)、1mL・20μm/(m2・day・atm)、又は0.5mL・20μm/(m2・day・atm)がさらに好ましい。
(EVOH)
本発明のガスバリア樹脂組成物に含まれる一種又は二種以上のEVOHの形態としては、以下の(I)及び(II)の形態が挙げられる。
(I)原料であるエチレン及びビニルエステルの少なくとも一部がバイオマス由来であるEVOH(A)と、化石燃料由来であるEVOH(B)とを含む形態
(II)原料であるエチレン及びビニルエステルの一部がバイオマス由来であり、残部が化石燃料由来であるEVOH(A’)を含む形態
本発明のガスバリア樹脂組成物に含まれる一種又は二種以上のEVOHの形態としては、以下の(I)及び(II)の形態が挙げられる。
(I)原料であるエチレン及びビニルエステルの少なくとも一部がバイオマス由来であるEVOH(A)と、化石燃料由来であるEVOH(B)とを含む形態
(II)原料であるエチレン及びビニルエステルの一部がバイオマス由来であり、残部が化石燃料由来であるEVOH(A’)を含む形態
一種又は二種以上のEVOHは、エチレン単位、ビニルアルコール単位及びビニルエステル単位の一部がバイオマス由来であり、残部が化石燃料由来であるものであってよい。すなわち、EVOH(A)は、エチレン単位、ビニルアルコール単位及びビニルエステル単位の少なくとも一部がバイオマス由来のEVOHであってよい。EVOH(B)は、エチレン単位、ビニルアルコール単位及びビニルエステル単位の全てが化石燃料由来のEVOHであってよい。EVOH(A’)は、エチレン単位、ビニルアルコール単位及びビニルエステル単位の一部がバイオマス由来であり、残部が化石燃料由来のEVOHであってよい。
(EVOH(A))
EVOH(A)は、原料モノマーであるエチレン及びビニルエステルの少なくとも一部がバイオマス由来であるEVOHである。EVOH(A)がバイオマス由来の原料を含むことで、本発明のガスバリア樹脂組成物のバイオベース度を高め、環境負荷を低減できる。
EVOH(A)は、原料モノマーであるエチレン及びビニルエステルの少なくとも一部がバイオマス由来であるEVOHである。EVOH(A)がバイオマス由来の原料を含むことで、本発明のガスバリア樹脂組成物のバイオベース度を高め、環境負荷を低減できる。
EVOH(A)は、少なくとも一部がバイオマス由来であるエチレン及びビニルエステルの共重合体のケン化により得られる。EVOH(A)の前駆体となるエチレン-ビニルエステル共重合体の製造及びケン化は、従来の化石燃料由来のエチレン-ビニルエステル共重合体の製造及びケン化と同様の公知の方法により行うことができる。ビニルエステルとしては、例えば酢酸ビニル、ギ酸ビニル、プロピオン酸ビニル、バレリン酸ビニル、カプリン酸ビニル、ラウリン酸ビニル、ステアリン酸ビニル、ピバリン酸ビニル、バーサティック酸ビニル等のカルボン酸ビニルエステルを用いることができ、酢酸ビニルが好ましい。
バイオマス由来のエチレンは、例えばバイオマス原料からバイオエタノールを精製し、脱水反応を行うなど、公知の方法で製造することができる。バイオマス原料としては、廃棄物系、未利用系、資源作物系等を用いることができ、例えば、セルロース系作物(パルプ、ケナフ、麦わら、稲わら、古紙、製紙残渣など)、木材、木炭、堆肥、天然ゴム、綿花、サトウキビ、おから、油脂(菜種油、綿実油、大豆油、ココナッツ油、ヒマシ油など)、炭水化物系作物(トウモロコシ、イモ類、小麦、米、籾殻、米ぬか、古米、キャッサバ、サゴヤシなど)、バガス、そば、大豆、精油(松根油、オレンジ油、ユーカリ油など)、パルプ黒液、植物油カスなどを用いることができる。
バイオエタノールを製造する方法は特に限定されず、例えば、バイオマス原料を必要に応じて前処理(加圧熱水処理、酸処理、アルカリ処理、糖化酵素を用いた糖化処理)した上で、酵母発酵させバイオエタノールを製造した後、蒸留工程及び脱水工程を経てバイオエタノールを精製することができる。バイオエタノール製造の際に、糖化処理を行う場合、糖化と発酵を段階的に行う逐次糖化発酵を用いてもよいし、糖化と発酵を同時に行う並行糖化発酵を用いてもよいが、製造効率の観点から並行糖化発酵にてバイオエタノールを製造することが好ましい。
市販のバイオマス由来のエチレンを使用してもよく、例えばBraskem S.A.製のサトウキビ由来バイオエチレン等を使用できる。
バイオマス由来のビニルエステルとしては、バイオマス由来のエチレンを用いて製造したビニルエステルが挙げられる。バイオマス由来のビニルエステルの製造方法としては、例えば一般的な工業製法であるパラジウム触媒を用いてエチレンと酢酸と酸素分子とを反応させる方法等が挙げられる。また、バイオマス由来のビニルエステルは、バイオマス由来のカルボン酸を用いて製造されたビニルエステルであってもよい。EVOH(A)のケン化度が100モル%では無い場合、バイオマス由来のアシル基が残存することとなる。
EVOH(A)のエチレン単位含有量の下限は20モル%が好ましく、23モル%がより好ましく、25モル%がさらに好ましい。EVOH(A)のエチレン単位含有量が20モル%以上であると、ロングラン性が高まる傾向となる。EVOH(A)のエチレン単位含有量の上限は60モル%が好ましく、55モル%がより好ましく、50モル%がさらに好ましい。EVOH(A)のエチレン単位含有量が60モル%以下であると、ガスバリア性がより良好となる傾向となる。EVOHのエチレン単位含有量は、核磁気共鳴(NMR)法により求めることができる。
EVOH(A)のケン化度の下限は90モル%が好ましく、95モル%がより好ましく、99モル%がさらに好ましい。EVOH(A)のケン化度が90モル%以上であると、本発明のガスバリア樹脂組成物におけるガスバリア性及びロングラン性がより良好となる傾向がある。また、EVOH(A)のケン化度の上限は100モル%であってよく、99.97モル%又は99.94モル%であってもよい。EVOHのケン化度は、1H-NMR測定を行い、ビニルエステル構造に含まれる水素原子のピーク面積と、ビニルアルコール構造に含まれる水素原子のピーク面積とを測定して算出できる。
EVOH(A)の原料として用いられるエチレン及びビニルエステルは、その少なくとも一部がバイオマス由来であり、ビニルエステルの少なくとも一部がバイオマス由来であることが好ましく、ビニルエステルの全部がバイオマス由来であってもよい。また、EVOH(A)の原料であるエチレンの少なくとも一部がバイオマス由来であることも好ましく、エチレンの全部がバイオマス由来であってもよい。また、EVOH(A)の原料であるビニルエステルの少なくとも一部及びエチレンの少なくとも一部がバイオマス由来であることが好ましい場合もあり、ビニルエステル及びエチレンの全てがバイオマス由来であってもよい。
EVOH(A)を構成する全ビニルアルコール単位(ビニルエステル単位のケン化物)におけるバイオマス由来のビニルアルコール単位の割合の下限は、1モル%が好ましく、5モル%がより好ましく、10モル%がさらに好ましく、25モル%又は45モル%がよりさらに好ましく、70モル%、90モル%又は99モル%であってもよく、EVOH(A)を構成する全てのビニルアルコール単位がバイオマス由来であってもよい。EVOH(A)を構成する全ビニルアルコール単位における化石燃料由来のビニルアルコール単位の割合の上限は、99モル%が好ましく、95モル%がより好ましく、90モル%がさらに好ましく、75モル%又は55モル%がよりさらに好ましく、30モル%、10モル%又は1モル%であってもよく、EVOH(A)を構成する全ビニルアルコール単位中に化石燃料由来のビニルアルコール単位が含まれていなくてもよい。EVOH(A)を構成する全ビニルアルコール単位におけるバイオマス由来のビニルアルコール単位の割合が高くなると、本発明のガスバリア樹脂組成物におけるバイオベース度が高まり、環境負荷を低減できる傾向となる。一方、バイオベース度及びロングラン性のバランスに優れるガスバリア樹脂組成物を提供する観点からは、バイオマス由来のビニルアルコール単位の割合は5モル%以上95モル%以下が好ましく、15モル%以上85モル%以下がより好ましく、25モル%以上75モル%以下がさらに好ましく、35モル%以上65モル%以下が特に好ましい。
EVOH(A)を構成する全エチレン単位におけるバイオマス由来のエチレン単位の割合の下限は、1モル%が好ましく、5モル%がより好ましく、10モル%がさらに好ましく、25モル%又は45モル%がよりさらに好ましく、70モル%、90モル%又は99モル%であってもよく、EVOH(A)を構成する全てのエチレン単位がバイオマス由来であってもよい。EVOH(A)を構成する全エチレン単位における化石燃料由来のエチレン単位の割合の上限は、99モル%が好ましく、95モル%がより好ましく、90モル%がさらに好ましく、75モル%又は55モル%がよりさらに好ましく、30モル%、10モル%又は1モル%であってもよく、EVOH(A)を構成する全エチレン単位中に化石燃料由来のエチレン単位が含まれていなくてもよい。EVOH(A)を構成する全エチレン単位におけるバイオマス由来のエチレン単位の割合が高くなると、本発明のガスバリア樹脂組成物におけるバイオベース度が高まり、環境負荷を低減できる傾向となる。一方、バイオベース度及びロングラン性のバランスに優れるガスバリア樹脂組成物を提供する観点からは、バイオマス由来のエチレン単位の割合は5モル%以上95モル%以下が好ましく、15モル%以上85モル%以下がより好ましく、25モル%以上75モル%以下がさらに好ましく、35モル%以上65モル%以下が特に好ましい。
EVOH(A)のバイオベース度の下限は、本発明のガスバリア樹脂組成物の環境負荷を低減する観点から1%が好ましく、5%がより好ましく、20%がさらに好ましく、40%が特に好ましい。さらにEVOH(A)のバイオベース度の下限は、60%であってもよく、80%又は95%であってもよい。また、EVOH(A)のバイオベース度の上限は、100%であってもよいが、ロングラン性を良好とする観点から99%が好ましく、95%がより好ましく、85%、75%又は65%がさらに好ましい場合もある。
EVOH(A)は、本発明の目的が阻害されない範囲で、エチレン、ビニルエステル及びそのケン化物以外の他の単量体由来の単位を有していてもよい。EVOH(A)が上記他の単量体由来の単位を有する場合、上記他の単量体由来の単位のEVOH(A)の全構造単位に対する含有量の上限は30モル%が好ましく、20モル%がより好ましく、10モル%がさらに好ましく、5モル%がよりさらに好ましく、1モル%がよりさらに好ましいこともある。また、EVOH(A)が上記他の単量体由来の単位を有する場合、その含有量の下限は0.05モル%であってもよく、0.10モル%であってもよい。上記他の単量体は、例えば、プロピレン、ブチレン、ペンテン、ヘキセン等のアルケン;3-アシロキシ-1-プロペン、3-アシロキシ-1-ブテン、4-アシロキシ-1-ブテン、3,4-ジアシロキシ-1-ブテン、3-アシロキシ-4-メチル-1-ブテン、4-アシロキシ-2-メチル-1-ブテン、4-アシロキシ-3-メチル-1-ブテン、3,4-ジアシロキシ-2-メチル-1-ブテン、4-アシロキシ-1-ペンテン、5-アシロキシ-1-ペンテン、4,5-ジアシロキシ-1-ペンテン、4-アシロキシ-1-ヘキセン、5-アシロキシ-1-ヘキセン、6-アシロキシ-1-ヘキセン、5,6-ジアシロキシ-1-ヘキセン、1,3-ジアセトキシ-2-メチレンプロパン等のエステル基を有するアルケン又はそのケン化物;アクリル酸、メタクリル酸、クロトン酸、イタコン酸等の不飽和酸又はその無水物、塩、又はモノ若しくはジアルキルエステル等;アクリロニトリル、メタクリロニトリル等のニトリル;アクリルアミド、メタクリルアミド等のアミド;ビニルスルホン酸、アリルスルホン酸、メタアリルスルホン酸等のオレフィンスルホン酸又はその塩;ビニルトリメトキシシラン、ビニルトリエトキシシラン、ビニルトリ(β-メトキシ-エトキシ)シラン、γ-メタクリルオキシプロピルメトキシシラン等ビニルシラン化合物;アルキルビニルエーテル類、ビニルケトン、N-ビニルピロリドン、塩化ビニル、塩化ビニリデン等が挙げられる。
EVOH(A)は、ウレタン化、アセタール化、シアノエチル化、オキシアルキレン化等の手法の後変性がされていてもよい。
EVOH(A)が変性基を有する場合、EVOH(A)が下記式(I)で表される構造単位(変性基)を持つことが好ましい。
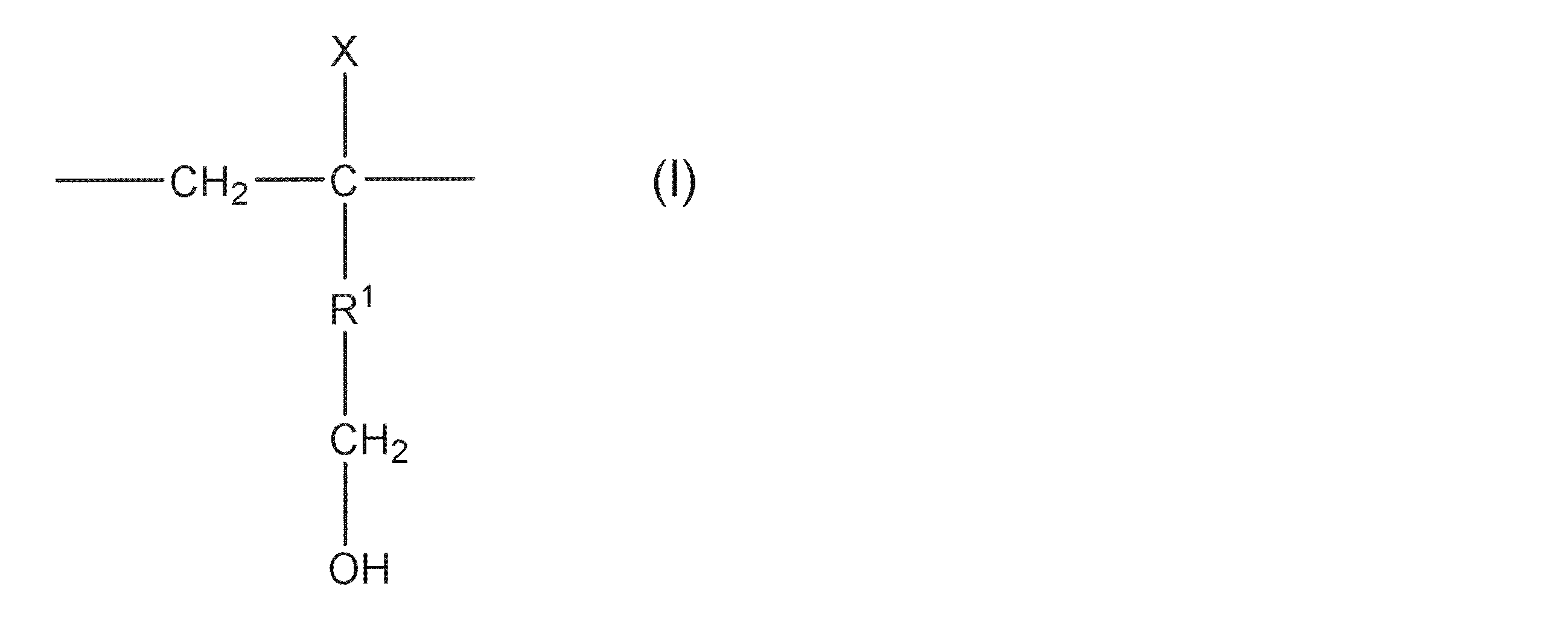
[式(I)中、Xは、水素原子、メチル基又はR2-OHで表される基を表す。R1及びR2は、それぞれ独立に単結合、炭素数1~9のアルキレン基又は炭素数1~9のアルキレンオキシ基を表し、上記アルキレン基及び上記アルキレンオキシ基は水酸基、アルコキシ基又はハロゲン原子を含んでもよい。]
Xは、好ましくは水素原子又はR2-OHで表される基であり、より好ましくはR2-OHで表される基である。
R1又はR2として用いられるアルキレン基及びアルキレンオキシ基は水酸基、アルコキシ基又はハロゲン原子を含んでもよい。R1及びR2は、好ましくは炭素数1~5のアルキレン基又はアルキレンオキシ基であり、より好ましくは炭素数1~3のアルキレン基又はアルキレンオキシ基である。
式(I)で表される構造単位(変性基)の具体例としては、例えば、下記の式(II)、式(III)、及び式(IV)で表される構造単位(変性基)が挙げられ、なか中でも式(II)で表される構造単位(変性基)が好ましい。
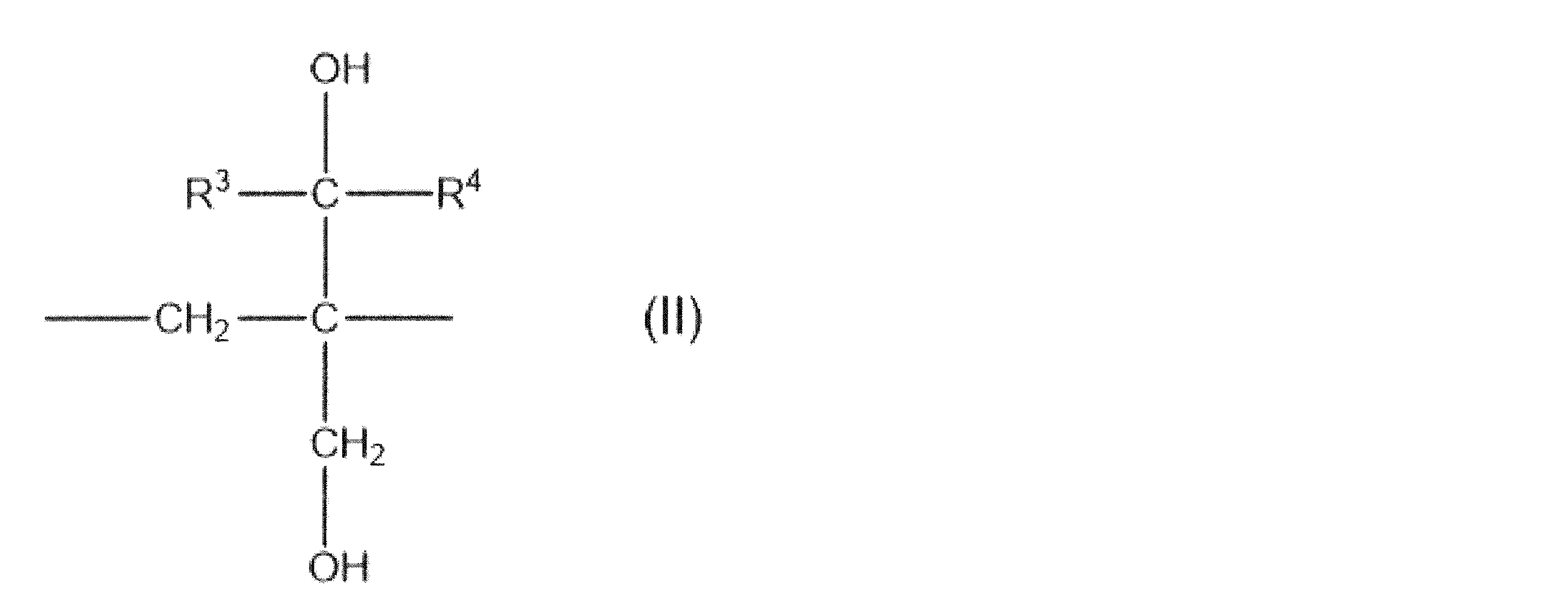
[式(II)中、R3及びR4は、それぞれ独立に水素原子又は炭素数1~8のアルキル基を表し、該アルキル基は水酸基、アルコキシ基又はハロゲン原子を含んでもよい。]
[式(III)中、R5は式(I)中のXと同義である。R6は、水素原子又は炭素数1~8のアルキル基を表し、該アルキル基は水酸基、アルコキシ基又はハロゲン原子を含んでもよい。]

[式(IV)中、R7及びR8は、それぞれ独立して、水素原子、炭素数1~8のアルキル基、炭素数3~8のシクロアルキル基または水酸基を表す。また、上記アルキル基、上記シクロアルキル基が有する水素原子の一部または全部は、水酸基、アルコキシ基またはハロゲン原子で置換されていてもよい。
本発明において、式(I)中のR1が単結合で、Xがヒドロキシメチル基(式(II)中のR3、R4が水素原子)であることが好ましい。この構造単位(変性基)を有するEVOH(A)を用いることで、ガスバリア性を著しく悪化させることなく延伸性、熱成形性等の二次加工性を高められる傾向となる。EVOH(A)が上記構造単位(変性基)を含有する場合、その含有量の下限は0.1モル%が好ましく、0.4モル%がより好ましく、1.0モル%がさらに好ましい。一方、上記構造単位(変性基)の含有量の上限は、ガスバリア性を良好とする観点から20モル%が好ましく、10モル%がより好ましく、8モル%がさらに好ましく、5モル%が特に好ましい。
本発明において、式(I)中のR1がヒドロキシメチレン基、Xが水素原子(式(III)中のR5、R6が水素原子)であることも好ましい。この構造単位(変性基)を有するEVOH(A)を用いることで、ガスバリア性を著しく悪化させることなく延伸性、熱成形性等の二次加工性を高められる傾向となる。EVOH(A)が上記構造単位(変性基)を含有する場合、その含有量の下限は0.1モル%が好ましく、0.4モル%がより好ましく、1.0モル%がさらに好ましい。一方、上記構造単位(変性基)の含有量の上限は、ガスバリア性を良好とする観点から20モル%が好ましく、10モル%がより好ましく、8モル%がさらに好ましく、5モル%が特に好ましい。
本発明において、式(I)中のR1がメチルメチレンオキシ基、Xが水素原子であることも好ましい。この構造単位(変性基)を有するEVOH(A)を用いることで、ガスバリア性を著しく悪化させることなく延伸性、熱成形性等の二次加工性を高められる傾向となる。また、上記メチルメチレンオキシ基は、酸素原子が主鎖の炭素原子に結合している。すなわち、式(IV)中、R7、R8の一方がメチル基であり、他方が水素原子であることが好ましい。EVOH(A)が上記構造単位(変性基)を含有する場合、その含有量の下限は0.1モル%が好ましく、0.5モル%がより好ましく、1.0モル%がさらに好ましく、2.0モル%が特に好ましい。一方、上記構造単位(変性基)の含有量の上限は、ガスバリア性を良好とする観点から20モル%が好ましく、15モル%がより好ましく、10モル%がさらに好ましい。
EVOH(A)は、単独で用いても二種以上併用してもよい。
JIS K7210:1999に準拠して測定した、EVOH(A)の190℃、2160g荷重におけるメルトフローレート(MFR)の下限は、0.1g/10分が好ましく、0.5g/10分がより好ましく、1.0g/10分がさらに好ましい。一方、EVOH(A)のMFRの上限は、30g/10分が好ましく、20g/10分がより好ましく、15g/10分がさらに好ましい。EVOH(A)の190℃、2160g荷重におけるMFRが上記範囲であると、溶融成形性が高まり、結果としてロングラン性が高まる傾向となる。
EVOH(A)の融点の下限は135℃が好ましく、150℃がより好ましく、155℃がさらに好ましい。EVOH(A)の融点が135℃以上であると、ガスバリア性に優れる傾向となる。またEVOH(A)の融点の上限は200℃が好ましく、190℃がより好ましく、185℃がさらに好ましい。EVOH(A)の融点が200℃以下であると、溶融成形性が良好となり、結果としてロングラン性が高まる傾向となる。
(EVOH(B))
EVOH(B)は、化石燃料由来のEVOHである。ここで、化石燃料由来のEVOHとは、化石燃料由来の原料を用いて合成されたEVOHを意味する。すなわち、EVOH(B)は、原料モノマーであるエチレン、ビニルエステル及び必要に応じて用いられるその他の単量体の全てが化石燃料由来であるEVOHである。換言すれば、EVOH(B)は、バイオマス由来の原料を用いずに合成されたEVOHである。本発明のガスバリア樹脂組成物がEVOH(B)を含むことで、十分なロングラン性を発揮することができる。
EVOH(B)は、化石燃料由来のEVOHである。ここで、化石燃料由来のEVOHとは、化石燃料由来の原料を用いて合成されたEVOHを意味する。すなわち、EVOH(B)は、原料モノマーであるエチレン、ビニルエステル及び必要に応じて用いられるその他の単量体の全てが化石燃料由来であるEVOHである。換言すれば、EVOH(B)は、バイオマス由来の原料を用いずに合成されたEVOHである。本発明のガスバリア樹脂組成物がEVOH(B)を含むことで、十分なロングラン性を発揮することができる。
EVOH(B)の具体的及び好適な態様は、原料が化石燃料由来であること、つまりバイオベース度が0%であること以外は、EVOH(A)と同様である。すなわち、EVOH(B)の具体的及び好適なエチレン単位含有量、ケン化度、他の単量体由来の種類及び含有量、MFR、融点等は、EVOH(A)と同様である。
EVOH(B)は、単独で用いても二種以上併用してもよい。
EVOH(A)とEVOH(B)とを併用する上記(I)の形態において、EVOH(A)とEVOH(B)との質量比(A/B)の下限は、1/99が好ましく、5/95がより好ましく、15/85がさらに好ましく、40/60が特に好ましい。質量比(A/B)を1/99以上とすることにより、本発明のガスバリア樹脂組成物のバイオベース度を高め、環境負荷を小さくすることができる。より環境負荷を低減する観点からは、上記質量比(A/B)の下限は、60/40が好ましい場合もあり、75/25が好ましい場合もある。また、質量比(A/B)の上限は99/1が好ましく、95/5がより好ましく、85/15がさらに好ましい場合もあり、60/40又は40/60がよりさらに好ましい場合もある。質量比(A/B)を99/1以下とすることでロングラン性を高めることができる。
EVOH(A)及びEVOH(B)のエチレン単位含有量、ケン化度、MFR及び融点等は、それぞれ同じであっても異なっていてもよい。ロングラン性等の性能を高めることなどからは、EVOH(A)及びEVOH(B)のエチレン単位含有量、ケン化度、MFR及び融点は、それぞれ同一であるか近い値であることが好ましい。EVOH(A)及びEVOH(B)のエチレン単位含有量の差は、6モル%以下が好ましく、3モル%以下がより好ましく、0モル%がさらに好ましい。また、EVOH(A)及びEVOH(B)のケン化度の差は2モル%以下が好ましく、1モル%以下がより好ましく、0モル%がさらに好ましい。また、EVOH(A)及びEVOH(B)のMFRの差は2g/10分以下が好ましく、1g/10分以下がより好ましく、0g/10分がさらに好ましい。また、EVOH(A)及びEVOH(B)の融点の差は7℃以下が好ましく、3℃以下がより好ましく、0℃がさらに好ましい。
(EVOH(A’))
EVOH(A’)は、原料モノマーであるエチレン及びビニルエステルの一部がバイオマス由来であり、上記原料モノマーであるエチレン及びビニルエステルの残部が化石燃料由来であるEVOHである。EVOH(A’)の原料がバイオマス由来の原料を含むことで、本発明のガスバリア樹脂組成物におけるバイオベース度を高め、環境負荷を低減できる傾向となる。また、EVOH(A’)の原料が化石燃料由来の原料を含むことで、本発明のガスバリア樹脂組成物のロングラン性を良好なものとすることができる。
EVOH(A’)は、原料モノマーであるエチレン及びビニルエステルの一部がバイオマス由来であり、上記原料モノマーであるエチレン及びビニルエステルの残部が化石燃料由来であるEVOHである。EVOH(A’)の原料がバイオマス由来の原料を含むことで、本発明のガスバリア樹脂組成物におけるバイオベース度を高め、環境負荷を低減できる傾向となる。また、EVOH(A’)の原料が化石燃料由来の原料を含むことで、本発明のガスバリア樹脂組成物のロングラン性を良好なものとすることができる。
EVOH(A’)は、一部がバイオマス由来であり残部が化石燃料由来であるエチレン及びビニルエステルの共重合体のケン化により得られる。EVOH(A’)の前駆体となるエチレン-ビニルエステル共重合体の製造及びケン化は、原料の一部にバイオマス由来のものが用いられること以外は、従来公知の方法により行うことができる。
EVOH(A’)の合成に用いられるバイオマス由来のエチレン及びバイオマス由来のビニルエステルの例は、EVOH(A)の合成に用いられるものとして上記したものと同様である。
EVOH(A’)のエチレン単位含有量及びケン化度の具体的及び好適な条件は、前述したEVOH(A)と同様である。
EVOH(A’)の原料として用いられるエチレン及びビニルエステルにおいては、エチレンの少なくとも一部がバイオマス由来であることが好ましく、エチレンの全部がバイオマス由来であってもよい。また、EVOH(A’)の原料であるビニルエステルの少なくとも一部がバイオマス由来であることも好ましく、ビニルエステルの全部がバイオマス由来であってもよい。
EVOH(A’)を構成する全ビニルアルコール単位(ビニルエステル単位のケン化物)におけるバイオマス由来のビニルアルコール単位の割合の下限は、1モル%が好ましく、5モル%がより好ましく、10モル%がさらに好ましく、25モル%又は45モル%がさらに好ましく、70モル%、90モル%又は99モル%であってもよく、EVOH(A’)を構成するすべてのビニルアルコール単位がバイオマス由来であってもよい。EVOH(A’)を構成する全ビニルアルコール単位における化石燃料由来のビニルアルコール単位の割合の上限は、99モル%が好ましく、95モル%がより好ましく、90モル%がさらに好ましく、75モル%又は55モル%がさらに好ましく、30モル%、10モル%又は1モル%であってもよく、EVOH(A’)を構成する全ビニルアルコール単位中に化石燃料由来のビニルアルコール単位が含まれていなくてもよい。EVOH(A’)を構成する全ビニルアルコール単位におけるバイオマス由来のビニルアルコール単位の割合が高くなると、本発明のガスバリア樹脂組成物におけるバイオベース度が高まり、環境負荷を低減できる傾向となる。一方、バイオベース度及びロングラン性のバランスに優れるガスバリア樹脂組成物を提供する観点からは、バイオマス由来のビニルアルコール単位の割合は5モル%以上95モル%以下が好ましく、15モル%以上85モル%以下がより好ましく、25モル%以上75モル%以下がさらに好ましく、35モル%以上65モル%以下が特に好ましい。
EVOH(A’)を構成する全エチレン単位におけるバイオマス由来のエチレンの割合の下限は、1モル%が好ましく、5モル%がより好ましく、10モル%がさらに好ましく、25モル%又は45モル%がさらに好ましく、70モル%、90モル%又は99モル%であってもよく、EVOH(A’)を構成する全てのエチレン単位がバイオマス由来であってもよい。EVOH(A’)を構成する全エチレン単位における化石燃料由来のエチレン単位の割合の上限は、99モル%が好ましく、95モル%がより好ましく、90モル%がさらに好ましく、75モル%又は55モル%がよりさらに好ましく、30モル%、10モル%又は1モル%であってもよく、EVOH(A’)を構成する全エチレン単位中に化石燃料由来のエチレン単位が含まれていなくてもよい。EVOH(A’)を構成する全エチレン単位におけるバイオマス由来のエチレン単位の割合が高くなると、本発明のガスバリア樹脂組成物におけるバイオベース度が高まり、環境負荷を低減できる傾向となる。一方、バイオベース度及びロングラン性のバランスに優れるガスバリア樹脂組成物を提供する観点からは、バイオマス由来のエチレン単位の割合は5モル%以上95モル%以下が好ましく、15モル%以上85モル%以下がより好ましく、25モル%以上75モル%以下がさらに好ましく、35モル%以上65モル%以下が特に好ましい。
EVOH(A’)のバイオベース度の下限は、本発明のガスバリア樹脂組成物の環境負荷を低減する観点から1%が好ましく、5%がより好ましく、20%がさらに好ましく、40%が特に好ましい。また、例えば特に優れたロングラン性が要求されない用途などにおいては、EVOH(A’)のバイオベース度の下限は、60%であってもよく、80%であってもよい。一方、EVOH(A’)のバイオベース度の上限は、ロングラン性を良好とする観点から99%が好ましく、95%がより好ましく、85%、75%又は65%がさらに好ましい場合もある。
EVOH(A’)は、本発明の目的が阻害されない範囲で、エチレン、ビニルエステル及びそのケン化物以外の他の単量体由来の単位を有していてもよく、後変性されていてもよい。共重合成分や後変性の具体例としては、前述したEVOH(A)と同様である。
EVOH(A’)は、単独で用いても二種以上併用してもよい。
JIS K7210:1999に準拠して測定した、EVOH(A’)の190℃、2160g荷重におけるメルトフローレート(MFR)及び融点の好適な態様は、前述したEVOH(A)と同様である。
(EVOH(X)及びEVOH(Y))
一種又は二種以上のEVOHは、EVOH(X)、及び上記EVOH(X)よりも融点が低いEVOH(Y)を含むことが好ましい。換言すれば、一種又は二種以上のEVOHは、EVOH(X)及びEVOH(Y)の少なくとも2つに分類できることが好ましい。本発明のガスバリア樹脂組成物が、融点の異なる二種のEVOH(EVOH(X)及びEVOH(Y))を含む場合、良好なガスバリア性及び成形性を維持できる傾向となる。このような当該ガスバリア樹脂組成物は、多層シート、多層シートを加熱延伸成形法により成形してなる包装材、多層シートを真空圧空成形法により成形してなる容器等の材料として特に好適である。
一種又は二種以上のEVOHは、EVOH(X)、及び上記EVOH(X)よりも融点が低いEVOH(Y)を含むことが好ましい。換言すれば、一種又は二種以上のEVOHは、EVOH(X)及びEVOH(Y)の少なくとも2つに分類できることが好ましい。本発明のガスバリア樹脂組成物が、融点の異なる二種のEVOH(EVOH(X)及びEVOH(Y))を含む場合、良好なガスバリア性及び成形性を維持できる傾向となる。このような当該ガスバリア樹脂組成物は、多層シート、多層シートを加熱延伸成形法により成形してなる包装材、多層シートを真空圧空成形法により成形してなる容器等の材料として特に好適である。
EVOH(X)は、EVOH(A)、EVOH(B)及びEVOH(A’)のいずれか1種又は2種以上からなる。同様に、EVOH(Y)は、EVOH(A)、EVOH(B)及びEVOH(A’)のいずれか1種又は2種以上からなる。後述するEVOH(Z)についても同様である。
EVOH(X)及びEVOH(Y)と、EVOH(A)、EVOH(B)及びEVOH(A’)との関係としては、
EVOH(X)及びEVOH(Y)のうちの一方がEVOH(A)であり、他方がEVOH(B)である形態、
EVOH(X)及びEVOH(Y)の少なくとも一方がEVOH(A)又はEVOH(A’)とEVOH(B)との双方を含む形態、
EVOH(X)及びEVOH(Y)がEVOH(A’)である形態
などが挙げられる。
EVOH(X)及びEVOH(Y)のうちの一方がEVOH(A)であり、他方がEVOH(B)である形態、
EVOH(X)及びEVOH(Y)の少なくとも一方がEVOH(A)又はEVOH(A’)とEVOH(B)との双方を含む形態、
EVOH(X)及びEVOH(Y)がEVOH(A’)である形態
などが挙げられる。
(EVOH(X))
EVOH(X)はEVOH(Y)よりも高い融点を有するEVOHであり、通常、本発明のガスバリア樹脂組成物に含まれるEVOHにおいて、最も高い融点を有するEVOHである。当該ガスバリア樹脂組成物がEVOH(X)を含むことで、優れたガスバリア性を有する傾向となる。EVOH(X)の融点の下限は150℃が好ましく、155℃がより好ましく、160℃がさらに好ましい。EVOH(X)の融点の上限は200℃が好ましい。EVOH(X)の融点が上記範囲内であると、当該ガスバリア樹脂組成物のガスバリア性が良好となる傾向となる。
EVOH(X)はEVOH(Y)よりも高い融点を有するEVOHであり、通常、本発明のガスバリア樹脂組成物に含まれるEVOHにおいて、最も高い融点を有するEVOHである。当該ガスバリア樹脂組成物がEVOH(X)を含むことで、優れたガスバリア性を有する傾向となる。EVOH(X)の融点の下限は150℃が好ましく、155℃がより好ましく、160℃がさらに好ましい。EVOH(X)の融点の上限は200℃が好ましい。EVOH(X)の融点が上記範囲内であると、当該ガスバリア樹脂組成物のガスバリア性が良好となる傾向となる。
EVOH(X)のエチレン単位含有量の下限は、成形性及びロングラン性を良好とする観点から、20モル%が好ましく、22モル%がより好ましく、24モル%がさらに好ましい。また、EVOH(X)のエチレン単位含有量の上限は、融点を高くする観点及びガスバリア性を良好とする観点から50モル%が好ましく、48モル%がより好ましく、46モル%がさらに好ましい。
EVOH(X)のケン化度の下限は90モル%が好ましく、95モル%がより好ましく、99モル%がさらに好ましい。EVOH(X)のケン化度が90モル%以上であると、本発明のガスバリア樹脂組成物におけるガスバリア性及びロングラン性がより良好となる傾向がある。また、EVOH(X)のケン化度の上限は100モル%であってよく、99.97モル%又は99.94モル%であってもよい。
EVOH(X)は、本発明の目的が阻害されない範囲で、エチレン、ビニルエステル及びそのケン化物以外の他の単量体単位を有していてもよいが、本発明のガスバリア樹脂組成物のガスバリア性を高く維持する観点から、他の単量体単位を有さないことが好ましい。上記他の単量体単位を与える単量体は、EVOH(A)の説明で例示した単量体等が挙げられる。EVOH(X)が上記他の単量体単位を有する場合、EVOH(X)の全構造単位に対する含有量は、5モル%以下が好ましく、3モル%以下がより好ましく、1モル%以下がさらに好ましい。
JIS K7210:1999に準拠して測定した、EVOH(X)の190℃、2160g荷重におけるメルトフローレート(MFR)の下限は、0.1g/10分が好ましく、0.5g/10分がより好ましく、1.0g/10分がさらに好ましい。一方、EVOH(X)のMFRの上限は、30g/10分が好ましく、20g/10分がより好ましく、15g/10分がさらに好ましい。EVOH(X)の190℃、2160g荷重におけるMFRが上記範囲であると、溶融成形性が高まり、結果としてロングラン性が高まる傾向となる。
(EVOH(Y))
EVOH(Y)は、EVOH(X)よりも低い融点を有するEVOHである。本発明のガスバリア樹脂組成物がEVOH(Y)を含むことで、優れた成形性を示す傾向となる。EVOH(Y)の融点の下限は100℃が好ましく、105℃がより好ましく、110℃がさらに好ましい。EVOH(Y)の融点の上限は180℃が好ましい。EVOH(Y)の融点が上記範囲内であると、当該ガスバリア樹脂組成物のガスバリア性が良好となる傾向となる。
EVOH(Y)は、EVOH(X)よりも低い融点を有するEVOHである。本発明のガスバリア樹脂組成物がEVOH(Y)を含むことで、優れた成形性を示す傾向となる。EVOH(Y)の融点の下限は100℃が好ましく、105℃がより好ましく、110℃がさらに好ましい。EVOH(Y)の融点の上限は180℃が好ましい。EVOH(Y)の融点が上記範囲内であると、当該ガスバリア樹脂組成物のガスバリア性が良好となる傾向となる。
EVOH(Y)のエチレン単位含有量の下限は、融点を低くする観点並びに成形性及びロングラン性を良好とする観点から30モル%が好ましく、32モル%がより好ましく、34モル%がさらに好ましい。また、EVOH(Y)のエチレン単位含有量の上限は、ガスバリア性を良好とする観点から、60モル%が好ましく、58モル%がより好ましく、56モル%がさらに好ましい。
EVOH(Y)のケン化度の下限は90モル%が好ましく、95モル%がより好ましく、99モル%がさらに好ましい。EVOH(Y)のケン化度が90モル%以上であると、本発明のガスバリア樹脂組成物におけるガスバリア性及びロングラン性がより良好となる傾向がある。また、EVOH(Y)のケン化度の上限は100モル%であってよく、99.97モル%又は99.94モル%であってもよい。また、EVOH(Y)のケン化度の下限は、成形性を高める観点から70モル%であっても、80モル%であってもよく、EVOH(Y)のケン化度の上限は、成形性を高める観点から98モル%であってもよい。
EVOH(Y)は、本発明の目的が阻害されない範囲で、上述したEVOH(X)と同様に、エチレン、ビニルエステル及びそのケン化物以外の単量体単位を有していてもよい。EVOH(Y)の融点を下げ、本発明のガスバリア樹脂組成物の成形性を高める観点から、EVOH(Y)が、他の単量体単位(構造単位)を有していることが好ましい場合がある。EVOH(Y)が他の単量体単位(構造単位)を含む場合、EVOH(Y)の全構造単位に対する含有量の下限は、0.1モル%が好ましく、0.3モル%がより好ましい。また、上記含有量の上限は、15モル%が好ましく、10モル%がより好ましい。
EVOH(Y)は、ウレタン化、アセタール化、シアノエチル化、オキシアルキレン化等の手法の後変性がされていてもよい。
EVOH(Y)が変性基を有する場合、EVOH(Y)が上記式(I)で表される構造単位(変性基)を有することが好ましい。なお、EVOH(Y)以外のEVOHが、上記式(I)で表される構造単位を有していてもよい。
EVOH(Y)が上記式(I)で表される構造単位(変性基)を有する場合、式(I)中のR1が単結合で、Xがヒドロキシメチル基(式(II)中のR3及びR4が水素原子)であることが好ましい。この構造単位(変性基)を有するEVOH(Y)を用いることで、ガスバリア性を著しく悪化させることなく延伸性、熱成形性等の成形性を高められる傾向となる。EVOH(Y)が上記構造単位(変性基)を含有する場合、その含有量の下限は0.1モル%が好ましく、0.4モル%がより好ましく、1.0モル%がさらに好ましい。一方、上記構造単位(変性基)の含有量の上限は、ガスバリア性を良好とする観点から20モル%が好ましく、10モル%がより好ましく、8モル%がさらに好ましく、5モル%が特に好ましい。
EVOH(Y)が上記式(I)で表される構造単位(変性基)を有する場合、式(I)中のR1がヒドロキシメチレン基、Xが水素原子(式(III)中のR5及びR6が水素原子)であることも好ましい。この構造単位(変性基)を有するEVOH(Y)を用いることで、ガスバリア性を著しく悪化させることなく延伸性、熱成形性等の成形性を高められる傾向となる。EVOH(Y)が上記構造単位(変性基)を含有する場合、その含有量の下限は0.1モル%が好ましく、0.4モル%がより好ましく、1.0モル%がさらに好ましい。一方、上記構造単位(変性基)の含有量の上限は、ガスバリア性を良好とする観点から20モル%が好ましく、10モル%がより好ましく、8モル%がさらに好ましく、5モル%が特に好ましい。
EVOH(Y)が上記式(I)で表される構造単位(変性基)を有する場合、式(I)中のR1がメチルメチレンオキシ基、Xが水素原子であることも好ましい。この構造単位(変性基)を有するEVOH(Y)を用いることで、ガスバリア性を著しく悪化させることなく延伸性、熱成形性等の成形性を高められる傾向となる。また、上記メチルメチレンオキシ基は、酸素原子が主鎖の炭素原子に結合している。すなわち、式(IV)中、R7及びR8の一方がメチル基であり、他方が水素原子であることが好ましい。EVOH(Y)が上記構造単位(変性基)を含有する場合、その含有量の下限は0.1モル%が好ましく、0.5モル%がより好ましく、1.0モル%がさらに好ましく、2.0モル%が特に好ましい。一方、上記構造単位(変性基)の含有量の上限は、ガスバリア性を良好とする観点から20モル%が好ましく、15モル%がより好ましく、10モル%がさらに好ましい。
JIS K7210:1999に準拠して測定した、EVOH(Y)の190℃、2160g荷重におけるMFRの好適範囲は、前述したEVOH(X)と同様である。
(EVOH(X)とEVOH(Y)との関係)
EVOH(Y)とEVOH(X)とのエチレン単位含有量差(Y-X)は5モル%以上が好ましく、7モル%以上がより好ましく、10モル%以上がさらに好ましい。また、上記エチレン単位含有量差(Y-X)は25モル%以下であってもよい。上記エチレン単位含有量差(Y-X)が上記範囲であると、良好なガスバリア性を示しつつ成形性が良好となる傾向となる。
EVOH(Y)とEVOH(X)とのエチレン単位含有量差(Y-X)は5モル%以上が好ましく、7モル%以上がより好ましく、10モル%以上がさらに好ましい。また、上記エチレン単位含有量差(Y-X)は25モル%以下であってもよい。上記エチレン単位含有量差(Y-X)が上記範囲であると、良好なガスバリア性を示しつつ成形性が良好となる傾向となる。
EVOH(X)とEVOH(Y)との融点差(X-Y)は15℃以上が好ましく、18℃以上がより好ましい。上記融点差(X-Y)は100℃以下であってもよく、50℃以下であってもよい。上記融点差(X-Y)が上記範囲であると、良好なガスバリア性を示しつつ成形性が良好となる傾向となる。
EVOH(X)とEVOH(Y)との質量比(X/Y)は、60/40以上が好ましく、65/35以上がより好ましい。また、上記質量比(X/Y)は95/5以下が好ましく、90/10以下がより好ましい。上記質量比(X/Y)が上記範囲であると良好なガスバリア性を示しつつ成形性が良好となる傾向となる。
本発明のガスバリア樹脂組成物に含まれる全てのEVOHに対するEVOH(X)及びEVOH(Y)の合計含有量の下限は、60質量%が好ましく、70質量%、80質量%、90質量%、95質量%又は99質量%がより好ましい場合がある。また、本発明のガスバリア樹脂組成物に含まれる全てのEVOHに対するEVOH(X)及びEVOH(Y)の合計含有量の上限は100質量%であってよい。
(EVOH(Z))
本発明のガスバリア樹脂組成物において、EVOHは、EVOH(Y)よりも融点が低いEVOH(Z)をさらに含んでいてもよい。換言すれば、一種又は二種以上のEVOHは、EVOH(X)、EVOH(Y)及びEVOH(Z)の少なくとも3つに分類できるものであってよい。当該ガスバリア樹脂組成物がEVOH(Z)を含むと、優れた成形性を示す傾向となる。EVOH(Z)の好適な態様は、EVOH(Y)よりも融点が低いことを除き、EVOH(Y)と同様である。
本発明のガスバリア樹脂組成物において、EVOHは、EVOH(Y)よりも融点が低いEVOH(Z)をさらに含んでいてもよい。換言すれば、一種又は二種以上のEVOHは、EVOH(X)、EVOH(Y)及びEVOH(Z)の少なくとも3つに分類できるものであってよい。当該ガスバリア樹脂組成物がEVOH(Z)を含むと、優れた成形性を示す傾向となる。EVOH(Z)の好適な態様は、EVOH(Y)よりも融点が低いことを除き、EVOH(Y)と同様である。
本発明のガスバリア樹脂組成物に含まれる全てのEVOHに対するEVOH(Z)の含有量としては、例えば1質量%以上40質量%以下であってよい。当該ガスバリア樹脂組成物に含まれる全てのEVOHに対するEVOH(X)、EVOH(Y)及びEVOH(Z)の合計含有量の下限は、90質量%が好ましく、95質量%がより好ましく、99質量%がさらに好ましい。また、当該ガスバリア樹脂組成物に含まれる全てのEVOHに対するEVOH(X)、EVOH(Y)及びEVOH(Z)の合計含有量の上限は100質量%であってよい。
本発明のガスバリア樹脂組成物を構成する全ての樹脂における一種又は二種以上のEVOHが占める割合の下限は、例えば65質量%、70質量%又は75質量%であってもよく、80質量%が好ましく、85質量%がより好ましく、90質量%、95質量%、98質量%又は99質量%がさらに好ましい場合もあり、当該ガスバリア樹脂組成物を構成する樹脂は、実質的に一種又は二種以上のEVOHのみであってもよく、一種又は二種以上のEVOHのみであってもよい。当該ガスバリア樹脂組成物を構成する全ての樹脂における一種又は二種以上のEVOHが占める割合の上限は、100質量%であってもよく、95質量%であってもよい。
また、本発明のガスバリア樹脂組成物における一種又は二種以上のEVOHが占める割合の下限は、例えば65質量%、70質量%又は75質量%であってもよく、80質量%が好ましく、85質量%がより好ましく、90質量%、95質量%、98質量%又は99質量%がさらに好ましい場合もあり、当該ガスバリア樹脂組成物は実質的に一種又は二種以上のEVOHのみから構成されていてもよい。当該ガスバリア樹脂組成物における一種又は二種以上のEVOHが占める割合の上限は、100質量%であってもよく、95質量%であってもよい。
本発明のガスバリア樹脂組成物に含まれる一種又は二種以上のEVOH全体のバイオベース度の下限は、当該ガスバリア樹脂組成物の環境負荷を低減する観点から1%が好ましく、5%がより好ましく、20%がさらに好ましく、40%が特に好ましい。また、例えば特に優れたロングラン性が要求されない用途などにおいては、上記EVOH全体のバイオベース度の下限は、60%であってもよく、80%であってもよい。一方、上記EVOH全体のバイオベース度の上限は、ロングラン性を良好とする観点から99%が好ましく、95%がより好ましく、85%、75%、65%、55%、45%、35%又は25%がさらに好ましい場合もある。
本発明のガスバリア樹脂組成物に含まれる一種又は二種以上のEVOH全体のエチレン単位含有量、ケン化度、MFR及び融点の具体的及び好適範囲は、上述したEVOH(A)の範囲と同様である。
(無機粒子(C))
本発明のガスバリア樹脂組成物は、無機粒子(C)をさらに含むことが好ましい。当該ガスバリア樹脂組成物は、無機粒子(C)を有する場合、当該ガスバリア樹脂組成物から形成される層を含むフィルム等の耐破断性が良好となる傾向となる。また、無機粒子(C)を含むことで、当該ガスバリア樹脂組成物から形成される層を含むフィルムの表面上に蒸着層を形成する際に、蒸着欠点を抑制でき、蒸着層との密着強度を高められる場合がある。さらに、無機粒子(C)を含有することで、形成される層またはフィルムの表面の算術平均粗さ(Ra)を適度なものとし、耐ブロッキング性及び滑り性を向上させる場合もある。ここで、無機粒子とは、無機物を主成分とする粒子をいう。「主成分」とは、最も含有量が多い成分をいい、例えば含有量が50質量%以上の成分をいい、50質量%超の成分であることが好ましく、90質量%以上の成分であることがより好ましい。以下、「主成分」について同様である。
本発明のガスバリア樹脂組成物は、無機粒子(C)をさらに含むことが好ましい。当該ガスバリア樹脂組成物は、無機粒子(C)を有する場合、当該ガスバリア樹脂組成物から形成される層を含むフィルム等の耐破断性が良好となる傾向となる。また、無機粒子(C)を含むことで、当該ガスバリア樹脂組成物から形成される層を含むフィルムの表面上に蒸着層を形成する際に、蒸着欠点を抑制でき、蒸着層との密着強度を高められる場合がある。さらに、無機粒子(C)を含有することで、形成される層またはフィルムの表面の算術平均粗さ(Ra)を適度なものとし、耐ブロッキング性及び滑り性を向上させる場合もある。ここで、無機粒子とは、無機物を主成分とする粒子をいう。「主成分」とは、最も含有量が多い成分をいい、例えば含有量が50質量%以上の成分をいい、50質量%超の成分であることが好ましく、90質量%以上の成分であることがより好ましい。以下、「主成分」について同様である。
無機粒子(C)を構成する無機物は、ケイ素、アルミニウム、マグネシウム、ジルコニウム、セリウム、タングステン及びモリブデンからなる群より選ばれる少なくとも1種の元素を含む無機物が好ましい。中でも、入手が容易であることから、ケイ素、アルミニウム及びマグネシウムからなる群より選ばれる少なくとも1種の元素を含む無機物がより好ましい。上記無機物としては、例示した元素の酸化物、窒化物、酸化窒化物等が挙げられ、酸化物が好ましい。
無機粒子(C)の平均粒子径の下限は、0.5μmが好ましく、1.5μmがより好ましく、2.5μmがさらに好ましい。無機粒子(C)の平均粒子径の上限は、10μmが好ましく、8μmがより好ましく、5μmがさらに好ましい。無機粒子(C)の平均粒子径が上記範囲であると、当該ガスバリア樹脂組成物から形成される層またはフィルムの表面の算術平均粗さ(Ra)が適度なものとなり、耐ブロッキング性及び滑り性が向上する傾向となる。その結果、当該ガスバリア樹脂組成物は、耐破断性、蒸着欠点抑制性及び蒸着層の密着強度を向上できる傾向となる。
無機粒子(C)の含有量の下限は50ppmが好ましく、100ppmがより好ましく、150ppmがさらに好ましい。無機粒子(C)の含有量の上限は5000ppmが好ましく、4000ppmがより好ましく、3000ppmがさらに好ましく、2000ppm又は1000ppmがよりさらに好ましい場合もある。無機粒子(C)の含有量が上記範囲であると、当該ガスバリア樹脂組成物から形成される層またはフィルムの表面の算術平均粗さ(Ra)が適度なものとなり、耐ブロッキング性及び滑り性が向上する。その結果、当該ガスバリア樹脂組成物は、耐破断性及び蒸着欠点抑制性に優れ、また、得られる層の蒸着層との密着強度を向上させることができる。無機粒子(C)としては、1種又は2種以上の粒子を含んでいてもよい。また、1個の粒子が1種又は2種以上の無機物から形成されていてもよい。
本発明のガスバリア樹脂組成物における一種又は二種以上のEVOH及び無機粒子(C)が占める割合の下限は、80質量%が好ましく、90質量%がより好ましく、95質量%がさらに好ましく、98質量%が特に好ましく、99質量%であってもよく、当該ガスバリア樹脂組成物は実質的に一種又は二種以上のEVOH及び無機粒子(C)のみから構成されていてもよい。
本発明のガスバリア樹脂組成物は、無機粒子(C)を含む場合、耐破断性、耐ブロッキング性、蒸着欠点抑制性、及び蒸着層との密着性に優れる傾向にある。従って、特にこのようなガスバリア樹脂組成物は、単層フィルム、多層フィルム、蒸着フィルム等の各種フィルム、及び蒸着フィルムを備える多層構造体に用いることが好適な態様である。
(酸化防止剤(D))
本発明のガスバリア樹脂組成物は、酸化防止剤(D)をさらに含むことが好ましい。当該ガスバリア樹脂組成物は、酸化防止剤(D)を含有することで耐酸化劣化性が向上する。そのため、当該ガスバリア樹脂組成物が酸化防止剤(D)を含むと、当該ガスバリア樹脂組成物から形成されるパイプ等の成形体において、長期使用時においても力学的強度の低下が起こり難く、経年劣化によるクラックの発生を抑制できる傾向となる。
本発明のガスバリア樹脂組成物は、酸化防止剤(D)をさらに含むことが好ましい。当該ガスバリア樹脂組成物は、酸化防止剤(D)を含有することで耐酸化劣化性が向上する。そのため、当該ガスバリア樹脂組成物が酸化防止剤(D)を含むと、当該ガスバリア樹脂組成物から形成されるパイプ等の成形体において、長期使用時においても力学的強度の低下が起こり難く、経年劣化によるクラックの発生を抑制できる傾向となる。
酸化防止剤(D)は、酸化防止能を有する化合物である。酸化防止剤(D)の融点は特に限定されないが、170℃以下であることが好ましい。酸化防止剤(D)の融点が170℃以下であると、溶融混合によりガスバリア樹脂組成物を製造する際に、押出機内で溶融し易くなる。このため、酸化防止剤(D)が樹脂組成物中に局在化して高濃度部分が着色することを抑制できる。また、酸化防止剤(D)の融点は、50℃以上が好ましく、100℃以上がより好ましい場合もある。酸化防止剤(D)の融点が50℃以上であると、得られた成形体(パイプ等)の表面に酸化防止剤がブリードアウトして外観が不良となることを抑制できる。
酸化防止剤(D)の分子量は300以上であることが好ましい。酸化防止剤(D)の分子量が300以上である場合、本発明のガスバリア樹脂組成物から成形体を得た際に、表面に酸化防止剤がブリードアウトして成形体の外観が不良となることを抑制でき、また、樹脂組成物の熱安定性も高まる。上記分子量は400以上がより好ましく、500以上が特に好ましい。一方、酸化防止剤(D)の分子量の上限は特に限定されないが、分散性の観点から、8000以下が好ましく、6000以下がより好ましく、4000以下がさらに好ましく、2000以下が特に好ましい。
酸化防止剤(D)としては、ヒンダードフェノール基を有する化合物が好適に用いられる。ヒンダードフェノール基を有する化合物は、それ自身が熱安定性に優れる一方で、酸化劣化の原因である酸素ラジカルを捕捉する能力があり、酸化防止剤(D)としてガスバリア樹脂組成物に配合した場合、酸化劣化を防止する効果に優れるものである。
ヒンダードフェノール基を有する化合物としては、通常市販されているものを用いることができ、例えば、以下の製品が挙げられる。
(1)BASF社製「IRGANOX 1010」:融点110-125℃、分子量1178、ペンタエリスリトールテトラキス〔3-(3,5-ジ-tert-ブチル-4-ヒドロキシフェニル)プロピオネート〕
(2)BASF社製「IRGANOX 1076」:融点50-55℃、分子量531、オクタデシル-3-(3,5-ジ-tert-ブチル-4-ヒドロキシフェニル)プロピオネート
(3)BASF社製「IRGANOX 1098」:融点156-161℃、分子量637、N,N’-(ヘキサン-1,6-ジイル)ビス〔3-(3,5-ジ-tert-ブチル-4-ヒドロキシフェニル)プロピオンアミド〕
(4)BASF社製「IRGANOX 245」:融点76-79℃、分子量587、トリエチレングリコール-ビス[3-(3-tert-ブチル-5-メチル-4-ヒドロキシフェニル)プロピオネート]
(5)BASF社製「IRGANOX 259」:融点104-108℃、分子量639、1,6-ヘキサンジオール-ビス[3-(3,5-ジ-tert-ブチル-4-ヒドロキシフェニル)プロピオネート]
(6)住友化学工業株式会社製「Sumilizer MDP-s」:融点約128℃、分子量341、2,2’-メチレン-ビス(4-メチル-6-tert-ブチルフェノール)
(7)住友化学工業株式会社製「Sumilizer GM」:融点約128℃、分子量395、2-tert-ブチル-6-(3-tert-ブチル-2-ヒドロキシ-5-メチルベンジル)-4-メチルフェニルアクリレート
(8)住友化学工業株式会社製「Sumilizer GA-80」:融点約110℃、分子量741、3,9-ビス〔2-{3-(3-tert-ブチル-4-ヒドロキシ-5-メチルフェニル)プロピオニルオキシ}-1,1-ジメチルエチル〕-2,4,8,10-テトラオキサスピロ〔5,5〕ウンデカン
(1)BASF社製「IRGANOX 1010」:融点110-125℃、分子量1178、ペンタエリスリトールテトラキス〔3-(3,5-ジ-tert-ブチル-4-ヒドロキシフェニル)プロピオネート〕
(2)BASF社製「IRGANOX 1076」:融点50-55℃、分子量531、オクタデシル-3-(3,5-ジ-tert-ブチル-4-ヒドロキシフェニル)プロピオネート
(3)BASF社製「IRGANOX 1098」:融点156-161℃、分子量637、N,N’-(ヘキサン-1,6-ジイル)ビス〔3-(3,5-ジ-tert-ブチル-4-ヒドロキシフェニル)プロピオンアミド〕
(4)BASF社製「IRGANOX 245」:融点76-79℃、分子量587、トリエチレングリコール-ビス[3-(3-tert-ブチル-5-メチル-4-ヒドロキシフェニル)プロピオネート]
(5)BASF社製「IRGANOX 259」:融点104-108℃、分子量639、1,6-ヘキサンジオール-ビス[3-(3,5-ジ-tert-ブチル-4-ヒドロキシフェニル)プロピオネート]
(6)住友化学工業株式会社製「Sumilizer MDP-s」:融点約128℃、分子量341、2,2’-メチレン-ビス(4-メチル-6-tert-ブチルフェノール)
(7)住友化学工業株式会社製「Sumilizer GM」:融点約128℃、分子量395、2-tert-ブチル-6-(3-tert-ブチル-2-ヒドロキシ-5-メチルベンジル)-4-メチルフェニルアクリレート
(8)住友化学工業株式会社製「Sumilizer GA-80」:融点約110℃、分子量741、3,9-ビス〔2-{3-(3-tert-ブチル-4-ヒドロキシ-5-メチルフェニル)プロピオニルオキシ}-1,1-ジメチルエチル〕-2,4,8,10-テトラオキサスピロ〔5,5〕ウンデカン
酸化防止剤(D)として、ヒンダードアミン基を有する化合物も好適に用いられる。ヒンダードアミン基を有する化合物は、酸化防止剤(D)としてガスバリア樹脂組成物に配合した場合、EVOHの熱劣化を防止するのみにとどまらず、EVOHの熱分解により生成するアルデヒドを捕捉する効果もあり、分解ガスの発生を低減することで成形時のボイドあるいは気泡の発生を抑制することができる。また、アルデヒドを捕捉することにより、本発明のガスバリア樹脂組成物を食品包装容器として用いた際に、アルデヒドによる臭気が内容物の味覚を損ねる点も改善される。
ヒンダードアミン基を有する化合物として好ましいものは、ピペリジン誘導体であり、特に4位に置換基を有する2,2,6,6-テトラアルキルピペリジン誘導体が好ましい。その4位の置換基としては、カルボキシル基、アルコキシ基、アルキルアミノ基が挙げられる。
また、ヒンダードアミン基のN位にはアルキル基が置換していてもよいが、水素原子が結合しているものを用いる方が熱安定効果に優れ好ましい。
ヒンダードアミン基を有する化合物としては、通常市販されているものを用いることができ、例えば、以下の製品を挙げることができる。
(9)BASF社製「TINUVIN 770」:融点81-85℃、分子量481、ビス(2,2,6,6-テトラメチル-4-ピペリジル)セバケート
(10)BASF社製「TINUVIN 765」:液状化合物、分子量509、ビス(1,2,2,6,6-ペンタメチル-4-ピペリジル)セバケート及び1,2,2,6,6-ペンタメチル-4-ピペリジルセバケート(混合物)
(11)BASF社製「TINUVIN 622LD」:融点55-70℃、分子量3100-4000、コハク酸ジメチル・1-(2-ヒドロキシエチル)-4-ヒドロキシ-2,2,6,6-テトラメチルピペリジン重縮合物
(12)BASF社製「CHIMASSORB 119FL」:融点130-140℃、分子量2000以上、N,N’-ビス(3-アミノプロピル)エチレンジアミン・2,4-ビス〔N-ブチル-N-(1,2,2,6,6-ペンタメチル-4-ピペリジル)アミノ〕-6-クロロ-1,3,5-トリアジン縮合物
(13)BASF社製「CHIMASSORB 944LD」:融点100-135℃、分子量2000-3100、ポリ〔〔6-(1,1,3,3-テトラメチルブチル)アミノ-1,3,5-トリアジン-2,4-ジイル〕(2,2,6,6-テトラメチル-4-ピペリジル)イミノ〕ヘキサメチレン(2,2,6,6-テトラメチル-4-ピペジリル)イミノ〕〕
(14)BASF社製「TINUVIN 144」:融点146-150℃、分子量685、ビス(1,2,2,6,6-ペンタメチル-4-ピペリジル)〔〔3,5-ビス(1,1-ジメチルエチル)-4-ヒドリキシフェニル〕メチル〕ブチルマロネート
(15)BASF社製「UVINUL 4050H」:融点157℃、分子量450、N,N’-1,6-ヘキサンジイルビス{N-(2,2,6,6-テトラメチル-4-ピペリジニル)-ホルムアミド}
(16)BASF社製「UVINUL 5050H」:融点104-112℃、分子量約3500、下記構造式を有する化合物
(9)BASF社製「TINUVIN 770」:融点81-85℃、分子量481、ビス(2,2,6,6-テトラメチル-4-ピペリジル)セバケート
(10)BASF社製「TINUVIN 765」:液状化合物、分子量509、ビス(1,2,2,6,6-ペンタメチル-4-ピペリジル)セバケート及び1,2,2,6,6-ペンタメチル-4-ピペリジルセバケート(混合物)
(11)BASF社製「TINUVIN 622LD」:融点55-70℃、分子量3100-4000、コハク酸ジメチル・1-(2-ヒドロキシエチル)-4-ヒドロキシ-2,2,6,6-テトラメチルピペリジン重縮合物
(12)BASF社製「CHIMASSORB 119FL」:融点130-140℃、分子量2000以上、N,N’-ビス(3-アミノプロピル)エチレンジアミン・2,4-ビス〔N-ブチル-N-(1,2,2,6,6-ペンタメチル-4-ピペリジル)アミノ〕-6-クロロ-1,3,5-トリアジン縮合物
(13)BASF社製「CHIMASSORB 944LD」:融点100-135℃、分子量2000-3100、ポリ〔〔6-(1,1,3,3-テトラメチルブチル)アミノ-1,3,5-トリアジン-2,4-ジイル〕(2,2,6,6-テトラメチル-4-ピペリジル)イミノ〕ヘキサメチレン(2,2,6,6-テトラメチル-4-ピペジリル)イミノ〕〕
(14)BASF社製「TINUVIN 144」:融点146-150℃、分子量685、ビス(1,2,2,6,6-ペンタメチル-4-ピペリジル)〔〔3,5-ビス(1,1-ジメチルエチル)-4-ヒドリキシフェニル〕メチル〕ブチルマロネート
(15)BASF社製「UVINUL 4050H」:融点157℃、分子量450、N,N’-1,6-ヘキサンジイルビス{N-(2,2,6,6-テトラメチル-4-ピペリジニル)-ホルムアミド}
(16)BASF社製「UVINUL 5050H」:融点104-112℃、分子量約3500、下記構造式を有する化合物

これらのヒンダードフェノール基又はヒンダードアミン基を有する化合物は単独で使用しても、また、2種以上を併用してもよい。
本発明のガスバリア樹脂組成物における酸化防止剤(D)の含有量の下限は、0.01質量%が好ましく、0.1質量%がより好ましく、0.3質量%がさらに好ましい。酸化防止剤(D)の含有量の上限は、5質量%が好ましく、3質量%がより好ましく、1質量%がさらに好ましい。酸化防止剤(D)の含有量が上記範囲であると、酸化防止剤(D)が良好に分散し、当該ガスバリア樹脂組成物から成形体を得た場合に外観に優れ、かつ良好な耐酸化劣化性、耐熱性等を発揮できる傾向にある。
本発明のガスバリア樹脂組成物における一種又は二種以上のEVOH及び酸化防止剤(D)が占める割合の下限は、80質量%が好ましく、90質量%がより好ましく、95質量%がさらに好ましく、98質量%が特に好ましく、99質量%であってもよく、当該ガスバリア樹脂組成物は実質的に一種又は二種以上のEVOH及び酸化防止剤(D)のみから構成されていてもよい。
本発明のガスバリア樹脂組成物が特に酸化防止剤(D)を含む場合、当該ガスバリア樹脂組成物又はこれを用いた成形体(多層構造体、パイプ等)は、環境負荷が小さく、良好なガスバリア性を示し、また、長期間使用したとしても酸化劣化によりクラックが発生しにくい。このため、屋内外で使用される日用品、包装材、機械部品、耐久消費財部品、産業用部品、産業用資材等に好適である。上記成形体等の特徴が特に効果的に発揮される用途の例としては、飲食品用包装材、容器用パッキング材、フィルム、農業用フィルム、ジオメンブレン、医療用輸液バッグ材、高圧タンク材、ガソリンタンク材、燃料容器、タイヤ用チューブ材、靴用クッション材、バッグインボックス用内袋材、有機液体貯蔵用タンク材、パイプ(有機液体輸送用パイプ材(燃料輸送用パイプ等)、暖房用温水パイプ材(床暖房用温水パイプ材等)、断熱多層パイプ等)、樹脂製壁紙、植物培地等が挙げられる。特に、屋外で使用され、熱や光による劣化が生じ易い、フィルム、パイプ、農業用フィルム、植物培地及びジオメンブレンとして、好適に用いられる。
(ポリアミド(E))
本発明のガスバリア樹脂組成物は、ポリアミド(PA)(E)をさらに含むことが好ましい。当該ガスバリア樹脂組成物がPA(E)を含む場合、耐レトルト性が良好となる。
本発明のガスバリア樹脂組成物は、ポリアミド(PA)(E)をさらに含むことが好ましい。当該ガスバリア樹脂組成物がPA(E)を含む場合、耐レトルト性が良好となる。
PA(E)は、アミド結合を含む樹脂である。PA(E)としては、例えば3員環以上のラクタムの開環重合、重合可能なω-アミノ酸の重縮合、二塩基酸とジアミンとの重縮合等によって得られる。ポリアミド(E)としては、例えばポリカプラミド(ナイロン6)、ポリ-ω-アミノヘプタン酸(ナイロン7)、ポリ-ω-アミノノナン酸(ナイロン9)、ポリウンデカンアミド(ナイロン11)、ポリラウリルラクタム(ナイロン12)、ポリエチレンジアミンアジパミド(ナイロン26)、ポリテトラメチレンアジパミド(ナイロン46)、ポリヘキサメチレンアジパミド(ナイロン66)、ポリヘキサメチレンセバカミド(ナイロン610)、ポリヘキサメチレンドデカミド(ナイロン612)、ポリオクタメチレンアジパミド(ナイロン86)、ポリデカメチレンアジパミド(ナイロン108)、カプロラクタム/ラウリルラクタム共重合体(ナイロン6/12)、カプロラクタム/ω-アミノノナン酸共重合体(ナイロン6/9)、カプロラクタム/ヘキサメチレンジアンモニウムアジペート共重合体(ナイロン6/66)、ラウリルラクタム/ヘキサメチレンジアンモニウムアジペート共重合体(ナイロン12/66)、ヘキサメチレンジアンモニウムアジペート/ヘキサメチレンジアンモニウムセバケート共重合体(ナイロン66/610)、エチレンジアンモニウムアジペート/ヘキサメチレンジアンモニウムアジペート共重合体(ナイロン26/66)、カプロラクタム/ヘキサメチレンジアンモニウムアジペート/ヘキサメチレンジアンモニウムセバケート共重合体(ナイロン6/66/610)、ポリヘキサメチレンイソフタルアミド(ナイロン6I)、ポリヘキサメチレンテレフタルアミド(ナイロン6T)、ヘキサメチレンイソフタルアミド/テレフタルアミド共重合体(ナイロン6I/6T)等が挙げられる。
PA(E)は、化石燃料由来の原料のみを用いて製造されたものであっても良いし、バイオマス由来の原料を用いて製造されたものであっても良い。例えばArkema S.A.社は、バイオマス由来の原料のみを用いたポリアミド(E)として、ヒマシ油を原料としたポリウンデカンアミド(ナイロン11)(「Rilsan PA11」)、並びにバイオマス由来の原料及び化石燃料由来の原料を用いて製造されたポリアミド(C)として、ポリヘキサメチレンセバカミド(ナイロン610)(「Rilsan S」)を製造及び販売している。
また、ジアミンとして2,2,4-トリメチルヘキサメチレンジアミン及び2,4,4-トリメチルヘキサメチレンジアミン等の置換基を導入した脂肪族ジアミン;メチルベンジルアミン、メタキシリレンジアミン等の芳香族アミン等を使用してもよく、またこれらを用いてポリアミドへの変性を行っても構わない。さらに、ジカルボン酸として2,2,4-トリメチルアジピン酸及び2,4,4-トリメチルアジピン酸等の置換基を導入した脂肪族カルボン酸;1,4-シクロヘキサンジカルボン酸等の脂環式ジカルボン酸;フタル酸、キシリレンジカルボン酸、アルキル置換テレフタル酸、アルキル置換イソフタル酸、ナフタレンジカルボン酸等の芳香族ジカルボン酸等を使用してもよく、またこれらを用いてポリアミドへの変性を行っても構わない。
中でも、ポリカプラミド(ナイロン6)が好ましい。その他にも、カプロラクタム/ラウリルラクタム共重合体(ナイロン6/12)も好ましい。この場合、ナイロン6単位とナイロン12単位の含有比は特に限定されないが、ナイロン12単位の含有量の下限としては、5質量%が好ましい。一方、ナイロン12単位の含有量の上限としては、60質量%が好ましく、50質量%がより好ましい。
本発明のガスバリア樹脂組成物における上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物に対するPA(E)の質量比(PA(E)/EVOH)の下限は、5/95が好ましく、8/92がより好ましく、13/87がさらに好ましい。また、質量比(PA(E)/EVOH)の上限は40/60が好ましく、35/65がより好ましく、30/70がさらに好ましく、25/75がよりさらに好ましい。質量比(PA(E)/EVOH)が5/95以上であることにより、耐レトルト性が十分なものとなり、当該ガスバリア樹脂組成物から成形される成形体等に対する熱水又は水蒸気による加熱処理後の外観が向上する。また、質量比(PA(E)/EVOH)が40/60以下であることにより、ガスバリア性が向上し、かつ、ロングラン性がより十分なものとなる。
本発明のガスバリア樹脂組成物におけるPA(E)の含有量の下限は、耐レトルト性を高める観点から、5質量%が好ましく、7質量%がより好ましい。また、PA(E)の含有量の上限は、ガスバリア性及びロングラン性を高める観点から40質量%が好ましく、30質量%がより好ましく、25質量%がさらに好ましく、15質量%特に好ましい。
本発明のガスバリア樹脂組成物を構成する全ての樹脂における一種又は二種以上のEVOH及びPA(E)が占める割合の下限は、80質量%が好ましく、90質量%がより好ましく、95質量%がさらに好ましく、98質量%が特に好ましく、99質量%であってもよい。当該ガスバリア樹脂組成物を構成する樹脂は、実質的に一種又は二種以上のEVOH及びPA(E)のみであってもよく、一種又は二種以上のEVOH及びPA(E)のみであってもよい。
本発明のガスバリア樹脂組成物がPA(E)を含む場合、耐レトルト性が良好となり、化石燃料由来の原料のみから合成された同一構造のEVOHと同等の耐レトルト性を発揮できる。従って、PA(E)を含む本発明のガスバリア樹脂組成物、又は当該ガスバリア樹脂組成物からなる層を少なくとも1層備える多層構造体は、包装材、特にレトルト用の包装材として好適に用いることができる。
(金属原子(F))
本発明のガスバリア樹脂組成物は、マグネシウム、カルシウム及び亜鉛からなる群より選ばれる少なくとも1種の金属原子(F)を含むことが好ましい。当該ガスバリア樹脂組成物が金属原子(F)を含むことで、溶融成形時のロングラン性が良好となる。特に、一般的にEVOHに対してPAを含有させた樹脂組成物はロングラン性が低下する傾向にあるが、本発明のガスバリア樹脂組成物がPA(E)と共に金属原子(F)を含むことで、十分なロングラン性を発揮することができる。
本発明のガスバリア樹脂組成物は、マグネシウム、カルシウム及び亜鉛からなる群より選ばれる少なくとも1種の金属原子(F)を含むことが好ましい。当該ガスバリア樹脂組成物が金属原子(F)を含むことで、溶融成形時のロングラン性が良好となる。特に、一般的にEVOHに対してPAを含有させた樹脂組成物はロングラン性が低下する傾向にあるが、本発明のガスバリア樹脂組成物がPA(E)と共に金属原子(F)を含むことで、十分なロングラン性を発揮することができる。
本発明のガスバリア樹脂組成物における、金属原子(F)の含有量の下限は、1ppmが好ましく、10ppmがより好ましく、50ppmがさらに好ましい。また、金属原子(F)の含有量の上限は、500ppmが好ましく、350ppmがより好ましく、300ppmがさらに好ましく、250ppmがよりさらに好ましい。金属原子(F)の含有量が上記範囲であると、欠点、ストリーク及び着色をバランスよく改善できる傾向となる。金属原子(F)は、溶融成形時のロングラン性がより良好となる観点から、マグネシウムがより好ましい。
金属原子(F)は、金属原子単体又は化合物中の構成原子として存在していてもよく、遊離した金属イオンの状態で存在していてもよい。金属原子(F)は、脂肪族カルボン酸、芳香族カルボン酸、脂肪族ジカルボン酸、芳香族ジカルボン酸、トリカルボン酸、テトラカルボン酸、ヒドロキシカルボン酸、ケトジカルボン酸、アミノ酸等の有機酸の塩;硫酸、亜硫酸、炭酸、リン酸等の無機酸の塩;水酸化物等として含まれていてもよい。中でも、脂肪族カルボン酸金属塩、又は水酸化物として含まれることが好ましく、EVOHとの相溶性の観点から炭素数6以下の脂肪族カルボン酸金属塩、又は水酸化物として含まれることがより好ましい。脂肪族カルボン酸としては、ギ酸、酢酸、プロピオン酸、酪酸、カプロン酸等の飽和脂肪族カルボン酸;アクリル酸、メタクリル酸等の不飽和脂肪族カルボン酸が挙げられる。中でも、EVOH等との相溶性の観点等から、炭素数1~3の飽和脂肪族カルボン酸の塩または水酸化物が好ましく、酢酸塩または水酸化物がより好ましい。
本発明のガスバリア樹脂組成物において、金属原子(F)は、上記のように有機酸又は無機酸と塩を形成していても良いし、一種又は二種以上のEVOHのアルコキシドのカウンターカチオンとして塩を形成していても良い。
本発明のガスバリア樹脂組成物において、金属原子(F)は樹脂組成物全体に均一に分散していることが好ましい。また、金属原子(F)は一種を単独で用いても、二種以上を併用してもよい。
当該ガスバリア樹脂組成物における一種又は二種以上のEVOH、PA(E)及び金属原子(F)が占める割合の下限は、80質量%が好ましく、90質量%がより好ましく、95質量%がさらに好ましく、98質量%が特に好ましく、99質量%であってもよい。当該ガスバリア樹脂組成物は、実質的に一種又は二種以上のEVOH、PA(E)及び金属原子(F)のみであってもよく、一種又は二種以上のEVOH、PA(E)及び金属原子(F)のみであってもよい。
(熱可塑性エラストマー(G))
本発明のガスバリア樹脂組成物は熱可塑性エラストマー(G)をさらに含むことが好ましい。当該ガスバリア樹脂組成物が熱可塑性エラストマー(G)を含む場合、耐屈曲性が良好となる傾向となる。なお、熱可塑性エラストマー(G)は、原料がバイオマス由来であっても、化石燃料由来であってもよい。
本発明のガスバリア樹脂組成物は熱可塑性エラストマー(G)をさらに含むことが好ましい。当該ガスバリア樹脂組成物が熱可塑性エラストマー(G)を含む場合、耐屈曲性が良好となる傾向となる。なお、熱可塑性エラストマー(G)は、原料がバイオマス由来であっても、化石燃料由来であってもよい。
熱可塑性エラストマー(G)としては特に限定されず、ポリエステル系熱可塑性エラストマー、ポリスチレン系熱可塑性エラストマー、ポリオレフィン系熱可塑性エラストマー等を用いることができる。これらは、一種又は二種以上を組み合わせても良い。中でも、耐屈曲性を向上させる観点から、熱可塑性エラストマー(G)は、ポリスチレン系熱可塑性エラストマー及びポリオレフィン系熱可塑性エラストマーからなる群から選択される少なくとも1種であることが好ましい。
上記ポリエステル系熱可塑性エラストマー(以下、TPEEと称することがある)としては、分子中のハードセグメントとしてポリエステルを、ソフトセグメントとしてガラス転移温度(Tg)の低いポリエーテル又はポリエステルを備えるマルチブロックコポリマーが挙げられる。TPEEは、分子構造の違いによって以下のタイプに分けることができ、中でもポリエステル・ポリエーテル型TPEEとポリエステル・ポリエステル型TPEEが好ましい。
(1)ポリエステル・ポリエーテル型TPEE
一般には、ハードセグメントとして芳香族系結晶性ポリエステルを、ソフトセグメントとしてポリエーテルを用いた熱可塑性エラストマーである。
(2)ポリエステル・ポリエステル型TPEE
ハードセグメントとして芳香族系結晶性ポリエステルを、ソフトセグメントとして脂肪族系ポリエステルを用いた熱可塑性エラストマーである。
(3)液晶性TPEE
ハードセグメントとして剛直な液晶分子を、ソフトセグメントとして脂肪族系ポリエステルを用いた熱可塑性エラストマーである。
(1)ポリエステル・ポリエーテル型TPEE
一般には、ハードセグメントとして芳香族系結晶性ポリエステルを、ソフトセグメントとしてポリエーテルを用いた熱可塑性エラストマーである。
(2)ポリエステル・ポリエステル型TPEE
ハードセグメントとして芳香族系結晶性ポリエステルを、ソフトセグメントとして脂肪族系ポリエステルを用いた熱可塑性エラストマーである。
(3)液晶性TPEE
ハードセグメントとして剛直な液晶分子を、ソフトセグメントとして脂肪族系ポリエステルを用いた熱可塑性エラストマーである。
上記ポリエステルセグメントとしては、テレフタル酸、イソフタル酸、ナフタレンジカルボン酸等の芳香族ジカルボン酸;1,4-シクロヘキサンジカルボン酸等の脂環式ジカルボン酸;コハク酸、アジピン酸等の脂肪族ジカルボン酸等のジカルボン酸成分と、エチレングリコール、1,2-プロピレングリコール、1,4-ブタンジオール等の脂肪族ジオール;シクロヘキサン-1,4-ジメタノール等の脂環式ジオール等のジオール成分とからなるポリエステルセグメントが挙げられる。上記ポリエーテルセグメントとしては、ポリエチレングリコール、ポリプロピレングリコール、ポリブチレングリコール等の脂肪族ポリエーテルセグメントが挙げられる。
上記ポリスチレン系熱可塑性エラストマーとしては、特に限定されないが、通常、ハードセグメントとしてスチレンモノマー重合体ブロック(Hb)を、ソフトセグメントとして共役ジエン化合物重合体ブロック又はその水添ブロック(Sb)を備える。このスチレン系熱可塑性エラストマーの構造としては、Hb-Sbで表されるジブロック構造、Hb-Sb-Hb若しくはSb-Hb-Sbで表されるトリブロック構造、Hb-Sb-Hb-Sbで表されるテトラブロック構造、又はHbとSbとが計5個以上直鎖状に結合しているポリブロック構造であってもよい。
上記スチレンモノマー重合体ブロック(Hb)に使用されるスチレン系モノマーとしては、特に限定されず、スチレン及びその誘導体等を挙げることができる。具体的には、スチレン、α―メチルスチレン、2-メチルスチレン、4-メチルスチレン、4-プロピルスチレン、4-t-ブチルスチレン、4-シクロヘキシルスチレン、4-ドデシルスチレン、2-エチル-4-ベンジルスチレン、4-(フェニルブチル)スチレン、2,4,6-トリメチルスチレン、モノフルオロスチレン、ジフルオロスチレン、モノクロロスチレン、ジクロロスチレン、メトキシスチレン、t-ブトキシスチレン等のスチレン類;1-ビニルナフタレン、2-ビニルナフタレン等のビニルナフタレン類等のビニル基含有芳香族化合物;インデン、アセナフチレン等のビニレン基含有芳香族化合物等が挙げられる。中でもスチレンが好ましい。スチレン系モノマーは1種のみでも良く、2種以上であっても良い。
上記共役ジエン化合物重合体ブロック又はその水添ブロック(Sb)に使用される共役ジエン化合物も、特に限定されず、例えば、ブタジエン、イソプレン、2,3-ジメチルブタジエン、ペンタジエン、ヘキサジエン等を挙げることができる。中でも、ブタジエンが好ましい。共役ジエン化合物は1種のみでも良く、2種以上であっても良い。さらに、他の共単量体、例えば、エチレン、プロピレン、ブチレン、スチレンを共重合することもできる。また、共役ジエン化合物重合体ブロックは、部分的又は完全に水素添加されている水素添加体であっても良い。
ポリスチレン系熱可塑性エラストマーの具体例としては、スチレン-イソプレンジブロック共重合体(SI)、スチレン-ブタジエンジブロック共重合体(SB)、スチレン-イソプレン-スチレントリブロック共重合体(SIS)、スチレン-ブタジエン/イソプレン-スチレントリブロック共重合体(SB/IS)、及びスチレン-ブタジエン-スチレントリブロック共重合体(SBS)並びにその水素添加体が挙げられる。中でも、スチレン-イソプレンジブロック共重合体の水素添加体(SEP)、スチレン-ブタジエンジブロック共重合体の水素添加体(SEB)、スチレン-イソプレン-スチレントリブロック共重合体の水素添加体(SEPS)、スチレン-ブタジエン/イソプレン-スチレントリブロック共重合体の水素添加体(SEEPS)、及びスチレン-ブタジエン-スチレントリブロック共重合体の水素添加体(SEBS)からなる群から選択される少なくとも1種が好ましい。
上記ポリオレフィン系熱可塑性エラストマーには、ハードセグメントとしてポリプロピレンやポリエチレン等のポリオレフィンブロックを、ソフトセグメントとしてエチレン-プロピレン-ジエン共重合体等のゴムブロックを備える熱可塑性エラストマー等が含まれる。なお、かかる熱可塑性エラストマーには、ブレンド型とインプラント化型があるが、EVOHとの相溶性及びコストの点でインプラント化型が好ましい。
熱可塑性エラストマー(G)は、無変性熱可塑性エラストマー(g1)、変性熱可塑性エラストマー(g2)及びハロゲン原子を含有するポリスチレン系熱可塑性エラストマー(g3)からなる群より選ばれる少なくとも1種を含むことが好ましく、変性熱可塑性エラストマー(g2)を含有することがより好ましい。変性熱可塑性エラストマー(g2)を含有することにより、例えば溶融混練時に一種又は二種以上のEVOHと変性熱可塑性エラストマー(g2)の反応が促され、特殊な押出条件を取らずとも、良好な相分離構造を有するガスバリア樹脂組成物を得ることができる傾向となる。熱可塑性エラストマー(G)は、無変性熱可塑性エラストマー(g1)及び変性熱可塑性エラストマー(g2)の双方を含有してもよく、変性熱可塑性エラストマー(g2)のみからなってもよく、ハロゲン原子を含有するポリスチレン系熱可塑性エラストマー(g3)のみからなってもよい。無変性熱可塑性エラストマー(g1)は、上記熱可塑性エラストマーとして挙げた樹脂をそのまま使用できる。
本発明のガスバリア樹脂組成物が、無変性熱可塑性エラストマー(g1)を含む場合、熱可塑性エラストマー(G)における無変性熱可塑性エラストマー(g1)が占める割合の下限は30質量%が好ましく、40質量%がより好ましく、85質量%がさらに好ましい場合もある。熱可塑性エラストマー(G)において、無変性熱可塑性エラストマー(g1)が占める割合の上限は100質量%であっても、60質量%であってもよい。無変性熱可塑性エラストマー(g1)は、一種又は二種以上のEVOHと反応する部位を有さないため、熱可塑性エラストマー(G)における無変性熱可塑性エラストマー(g1)の占める割合が高くなると、例えばゲル等の生成を抑制できる傾向となり、結果としてロングラン性が向上する傾向となる。
変性熱可塑性エラストマー(g2)は、酸変性熱可塑性エラストマーであることが好ましい。変性熱可塑性エラストマー(g2)は、例えば、無変性熱可塑性エラストマー(g1)を不飽和カルボン酸又はその誘導体で変性することにより得られる。不飽和カルボン酸又はその誘導体としては、マレイン酸、フマル酸、イタコン酸、無水マレイン酸、無水イタコン酸、マレイン酸モノメチルエステル、マレイン酸モノエチルエステル、マレイン酸ジエチルエステル、フマル酸モノメチルエステルなどが挙げられる。中でも無水マレイン酸により変性されていることが好ましい。このような変性熱可塑性エラストマー(g2)を使用することにより、一種又は二種以上のEVOHと熱可塑性エラストマー(G)との相溶性が高まり、ガスバリア性及び耐屈曲性等がより向上する傾向となる。
変性熱可塑性エラストマー(g2)は、変性ポリエステル系熱可塑性エラストマー、変性ポリスチレン系熱可塑性エラストマー、変性ポリオレフィン系熱可塑性エラストマーなどを用いることができる。これらは、一種又は二種以上を組み合わせても良い。変性熱可塑性エラストマー(g2)は、無水マレイン酸変性ポリエステル系熱可塑性エラストマー、無水マレイン酸変性ポリスチレン系熱可塑性エラストマー、及び無水マレイン酸変性ポリオレフィン系熱可塑性エラストマーであることが好ましい。中でも、耐屈曲性を向上させる観点から、変性熱可塑性エラストマー(g2)が、無水マレイン酸変性ポリスチレン系熱可塑性エラストマー及び無水マレイン酸変性ポリオレフィン系熱可塑性エラストマーからなる群から選択される少なくとも1種であることが好ましい。
変性熱可塑性エラストマー(g2)が酸変性熱可塑性エラストマーである場合、酸変性熱可塑性エラストマーの酸価は、1mgKOH/g以上50mgKOH/g以下であることが、耐屈曲性及びロングラン性を高める観点から好ましい。また、熱可塑性エラストマー(G)の酸価は0.5mgKOH/g以上25mgKOH/g以下であることが、耐屈曲性及びロングラン性を高める観点から好ましい。
熱可塑性エラストマー(G)において、変性熱可塑性エラストマー(g2)が占める割合の下限は5質量%が好ましく、20質量%がより好ましく、40質量%がさらに好ましい場合もあり、熱可塑性エラストマー(G)は変性熱可塑性エラストマー(g2)のみから構成されていてもよい。熱可塑性エラストマー(G)において、変性熱可塑性エラストマー(g2)が占める割合の上限は100質量%であっても、60質量%であってもよい。変性熱可塑性エラストマー(g2)は一種又は二種以上のEVOHと熱可塑性エラストマー(G)との相溶性を高めるため、熱可塑性エラストマー(G)における変性熱可塑性エラストマー(g2)の占める割合が高くなると、相溶性が向上する傾向となり、結果として耐屈曲性がより向上する傾向となる。
熱可塑性エラストマー(G)が無変性熱可塑性エラストマー(g1)及び変性熱可塑性エラストマー(g2)を含む場合、変性熱可塑性エラストマー(g2)に対する無変性熱可塑性エラストマー(g1)の質量比(g1/g2)は0/100超が好ましく、20/80以上がより好ましく、40/60以上がさらに好ましく、80/20以上が特に好ましい場合もある。また、質量比(g1/g2)は95/5以下が好ましい。質量比(g1/g2)が上記範囲内であると、熱可塑性エラストマー(G)の酸価を調整しやすく、本発明のガスバリア樹脂組成物において耐屈曲性及びロングラン性に優れる組成物を製造しやすくなる傾向となる。
熱可塑性エラストマー(G)において無変性熱可塑性エラストマー(g1)及び変性熱可塑性エラストマー(g2)が占める割合の下限は、70質量%であっても、80質量%であっても、90質量%であってもよく、熱可塑性エラストマー(G)は、実質的に無変性熱可塑性エラストマー(g1)及び変性熱可塑性エラストマー(g2)のみからなってもよく、無変性熱可塑性エラストマー(g1)及び変性熱可塑性エラストマー(g2)のみからなってもよい。
熱可塑性エラストマー(G)が、ハロゲン原子を含有するポリスチレン系熱可塑性エラストマー(g3)を含むことも好ましい。当該ハロゲン原子は、ポリスチレン系熱可塑性エラストマーの製造時に使用される重合触媒に由来するものであってもよく、その場合、ハロゲン原子はポリスチレン系熱可塑性エラストマーの末端に含有される。ポリスチレン系熱可塑性エラストマーの末端にハロゲン原子を含有することにより、EVOHと反応し、ポリスチレン系熱可塑性エラストマーのEVOH相中での分散性が向上する傾向となる。ポリスチレン系熱可塑性エラストマーに含有されるハロゲン原子としては、塩素、臭素、フッ素、ヨウ素などが挙げられ、中でも、塩素が好ましい。ポリスチレン系熱可塑性エラストマー中のハロゲン原子の含有量は、通常、0.005~3.000質量%である。ポリスチレン系熱可塑性エラストマー中のハロゲン原子は、イオンクロマトグラフを用いて分析することができる。ハロゲン原子を含有するポリスチレン系熱可塑性エラストマー(g3)としては、ハロゲン原子を含有していれば特に限定されず、例えば、上述したポリスチレン系エラストマーとして例示した化合物にハロゲン原子を含有させた化合物を用いることができる。中でも、ハロゲン原子を含有するスチレン-イソブチレン-スチレンブロック共重合体であることがより好ましく、例えば、株式会社カネカ社製 商品名「SIBSTAR 062T」等が挙げられる。熱可塑性エラストマー(G)におけるハロゲン原子を含有するポリスチレン系熱可塑性エラストマー(g3)が占める割合の下限は20質量%が好ましく、50質量%がより好ましく、90質量%がさらに好ましく、熱可塑性エラストマー(G)は実質的にハロゲン原子を含有するポリスチレン系熱可塑性エラストマー(g3)のみからなってもよく、ハロゲン原子を含有するポリスチレン系熱可塑性エラストマー(g3)のみからなってもよい。
本発明のガスバリア樹脂組成物における一種又は二種以上のEVOHに対する熱可塑性エラストマー(G)の質量比(G/EVOH)の下限は5/95が好ましく、8/92がより好ましく、12/88がさらに好ましい場合もあり、15/85又は25/75がよりさらに好ましい場合もある。質量比(G/EVOH)を上記下限以上とすることで、耐屈曲性等を高めることができる。一方、この質量比(G/EVOH)の上限は35/65が好ましく、30/70がより好ましい場合もあり、25/75がさらに好ましい場合もあり、15/85がよりさらに好ましい場合もある。質量比(G/EVOH)を上記上限以下とすることで、ガスバリア性を高められる傾向となる。
(相分離構造)
本発明のガスバリア樹脂組成物が熱可塑性エラストマー(G)を含む場合、一種又は二種以上のEVOHのマトリックス中に熱可塑性エラストマー(G)の粒子が分散していることが好ましい。すなわち、本発明のガスバリア樹脂組成物は、海島構造を有し、海相が主に一種又は二種以上のEVOHからなり、島相が主に熱可塑性エラストマー(G)からなる海島構造であることが好ましい。このように、海相が主に一種又は二種以上のEVOHからなることで、ガスバリア性を保ちつつ、柔軟性が向上する。
本発明のガスバリア樹脂組成物が熱可塑性エラストマー(G)を含む場合、一種又は二種以上のEVOHのマトリックス中に熱可塑性エラストマー(G)の粒子が分散していることが好ましい。すなわち、本発明のガスバリア樹脂組成物は、海島構造を有し、海相が主に一種又は二種以上のEVOHからなり、島相が主に熱可塑性エラストマー(G)からなる海島構造であることが好ましい。このように、海相が主に一種又は二種以上のEVOHからなることで、ガスバリア性を保ちつつ、柔軟性が向上する。
本発明の樹脂組成物が海島構造を有し、海相が主に一種又は二種以上のEVOHからなり、島相が主に熱可塑性エラストマー(G)からなる場合、透明性を向上させる観点から、熱可塑性エラストマー(G)からなる島相の平均粒子径は4.5μm以下が好ましく、3.5μm以下がより好ましく、3.0μm以下がさらに好ましく、2.5μm以下が特に好ましく、2.0μm以下が最も好ましい。熱可塑性エラストマー(G)の平均粒子径は0.1μm以上であってもよい。主に熱可塑性エラストマー(G)からなる島相の平均粒子径が上記範囲であると、ガスバリア性及び透明性を保ちつつ、耐屈曲性が向上し、さらに後述する積層剥離容器における剥離性が向上する傾向となるため好ましい。熱可塑性エラストマー(G)の平均粒子径は、混錬強度の調整、及び一種又は二種以上のEVOHと熱可塑性エラストマー(G)の組成比により調整できる。
本発明のガスバリア樹脂組成物において、一種又は二種以上のEVOHと熱可塑性エラストマー(G)との屈折率差は0.05以下が好ましく、0.04以下がより好ましく、0.03以下がさらに好ましい。該屈折率差は、0.005以上であってもよい。該屈折率差が上記範囲であると、当該ガスバリア樹脂組成物の透明性が良好になる傾向となる。
熱可塑性エラストマー(G)は1種単独で用いても、2種以上併用してもよい。
本発明のガスバリア樹脂組成物を構成する全ての樹脂における一種又は二種以上のEVOH及び熱可塑性エラストマー(G)が占める割合の下限は80質量%が好ましく、90質量%がより好ましく、95質量%がさらに好ましく、98質量%が特に好ましく、99質量%であってもよく、当該ガスバリア樹脂組成物を構成する樹脂は、実質的に一種又は二種以上のEVOH及び熱可塑性エラストマー(G)のみから構成されていてもよく、一種又は二種以上のEVOH並びに熱可塑性エラストマー(G)のみから構成されていてもよい。
本発明のガスバリア樹脂組成物が熱可塑性エラストマー(G)を含む場合、高い耐屈曲性を発揮することができる。従って、熱可塑性エラストマー(G)を含む当該ガスバリア樹脂組成物、又は当該ガスバリア樹脂組成物からなる層を少なくとも1層備える多層構造体は、包装材、縦製袋充填シール袋、バッグインボックス用内容器、積層剥離容器、チューブ、パイプ、ブロー成形容器等に好適に用いることができる。
(ハロゲン捕捉剤)
本発明のガスバリア樹脂組成物がハロゲン原子を含有するポリスチレン系熱可塑性エラストマー(g3)を含む場合、ロングラン性を向上させる観点から、本発明のガスバリア樹脂組成物はハロゲン捕捉剤を含むことが好ましい。ハロゲン捕捉剤は、ハロゲン捕捉能を有するものであればよく、例えば、交換性イオンを有する層状無機化合物;酸化マグネシウム、水酸化カルシウム、水酸化マグネシウム及び炭酸カルシウムなどのアルカリ土類金属化合物;酸化亜鉛;炭酸リチウムなどが挙げられる。中でも、ハロゲン捕捉剤が交換性イオンを有する層状無機化合物であることが好ましい。層状無機化合物中の層間に存在するイオンがハロゲンイオンと交換されることにより、当該ハロゲンイオンが層状無機化合物に取り込まれる。上記層状無機化合物として、例えば、粘土鉱物;層状ポリ珪酸;層状珪酸塩;層状複水酸化物;層状リン酸塩;チタン・ニオブ酸塩、六ニオブ酸塩及びモリブデン酸塩等の層状遷移金属酸素酸塩;層状マンガン酸化物;層状コバルト酸化物等を挙げることができ、中でも粘土鉱物が好ましい。
本発明のガスバリア樹脂組成物がハロゲン原子を含有するポリスチレン系熱可塑性エラストマー(g3)を含む場合、ロングラン性を向上させる観点から、本発明のガスバリア樹脂組成物はハロゲン捕捉剤を含むことが好ましい。ハロゲン捕捉剤は、ハロゲン捕捉能を有するものであればよく、例えば、交換性イオンを有する層状無機化合物;酸化マグネシウム、水酸化カルシウム、水酸化マグネシウム及び炭酸カルシウムなどのアルカリ土類金属化合物;酸化亜鉛;炭酸リチウムなどが挙げられる。中でも、ハロゲン捕捉剤が交換性イオンを有する層状無機化合物であることが好ましい。層状無機化合物中の層間に存在するイオンがハロゲンイオンと交換されることにより、当該ハロゲンイオンが層状無機化合物に取り込まれる。上記層状無機化合物として、例えば、粘土鉱物;層状ポリ珪酸;層状珪酸塩;層状複水酸化物;層状リン酸塩;チタン・ニオブ酸塩、六ニオブ酸塩及びモリブデン酸塩等の層状遷移金属酸素酸塩;層状マンガン酸化物;層状コバルト酸化物等を挙げることができ、中でも粘土鉱物が好ましい。
上記粘土鉱物として、例えば、ハイドロタルサイト、ゼオライト、雲母、バーミキュライト、モンモリロナイト、バイデライト、サポナイト、ヘクトライト及びスチーブンサイトが挙げられる。粘土鉱物は、合成粘土であっても天然粘土であってもよい。中でも、上記粘土鉱物として、ハイドロタルサイト及びゼオライトが好ましく、前者がより好ましい。ハイドロタルサイトとして、下記の一般式(i)で示されるものなど、ゼオライトとして下記式(ii)で示されるものなどがそれぞれ挙げられる。
Mg1-aAla(OH)2(CO3)a/2・xH2O (i)
Na2O・Al2O3・2SiO2・yH2O (ii)
(式(i)及び(ii)中、xは0~5の数、aは0<a≦0.5を満たす数、yは0~6の数を示す。)
Na2O・Al2O3・2SiO2・yH2O (ii)
(式(i)及び(ii)中、xは0~5の数、aは0<a≦0.5を満たす数、yは0~6の数を示す。)
本発明のガスバリア樹脂組成物がハロゲン捕捉剤を含む場合、急速な架橋(ゲル化)を抑制し、ロングラン性を高める観点から、その含有量の下限は0.01質量%が好ましく、0.025質量%がより好ましい。一方、ハロゲン捕捉剤に由来する凝集物の発生を抑制する観点から、ハロゲン捕捉剤の含有量の上限は1質量%が好ましく、0.8質量%がより好ましい。
本発明のガスバリア樹脂組成物において、ハロゲン原子を含有するポリスチレン系熱可塑性エラストマー(g3)に含有されるハロゲン原子の含有量に対するハロゲン捕捉剤の含有量の質量比[ハロゲン捕捉剤/ハロゲン原子]は0.10以上が好ましく、0.15以上がより好ましく、0.20以上がさらに好ましく、0.25以上が特に好ましい。また、質量比[ハロゲン捕捉剤/ハロゲン原子]は1.010以下が好ましく、0.702以下がより好ましく、0.501以下がさらに好ましく、0.40以下が特に好ましい。質量比[ハロゲン捕捉剤/ハロゲン原子]が上記範囲内であることで、長時間製造時の架橋の進行及びハロゲン捕捉剤の凝集をより抑制でき、余分なハロゲン捕捉剤の使用が抑えられるためコスト低減が可能となる。
本発明のガスバリア樹脂組成物は、硫黄化合物を硫黄原子換算で0ppm超100ppm含んでいることが、自社製品を追跡する観点からより好ましい。また、硫黄原子換算で100ppm以下の硫黄化合物は、ガスバリア樹脂組成物の性能に実質的に影響を与えないことを発明者らは知見しており、硫黄化合物はトレーサー物質として好適である。硫黄化合物の含有量の上限は50ppmがより好ましく、5ppmがさらに好ましく、3ppmがよりさらに好ましく、0.3ppmが特に好ましい。硫黄化合物の含有量の下限は、0.0001ppmであっても、0.001ppmであっても、0.01ppmであっても、0.05ppmであっても、0.1ppmであってもよい。バイオマス由来の原料を用いた場合、バイオマス原料に含まれる有機系硫黄化合物を含むEVOHが得られることがある。一方、化石燃料由来のEVOHは、ナフサのクラッキング時に脱硫していることから、バイオマス由来のEVOHに比して硫黄化合物が少なくなる。そのため、このようなバイオマス由来のEVOHを用いた場合、硫黄化合物の含有量を比較することでバイオマス由来のEVOHの追跡がより容易となる。特に、本発明のガスバリア樹脂組成物が、硫黄化合物として、有機系硫黄化合物、中でもジメチルスルフィドまたはジメチルスルホキシドを含むとき、さらに追跡が容易となる。また、自社製品を追跡する観点などからは、EVOHの製造の際に、原料となるバイオマス由来のエチレン及びバイオマス由来のビニルエステル、並びに得られるEVOHに対して、硫黄化合物の含有量が検出限界値以下となるような過剰な精製を行わないことが好ましい場合がある。
(その他成分)
本発明のガスバリア樹脂組成物はカルボン酸をさらに含有することが好ましい。当該ガスバリア樹脂組成物がカルボン酸を含有すると、溶融成形性や高温下での着色耐性を改善できる。特に、ガスバリア樹脂組成物のpH緩衝能力が高まり、酸性物質や塩基性物質に対する着色耐性を改善できる場合がある点から、カルボン酸のpKaが3.5~5.5の範囲にあることがより好ましい。
本発明のガスバリア樹脂組成物はカルボン酸をさらに含有することが好ましい。当該ガスバリア樹脂組成物がカルボン酸を含有すると、溶融成形性や高温下での着色耐性を改善できる。特に、ガスバリア樹脂組成物のpH緩衝能力が高まり、酸性物質や塩基性物質に対する着色耐性を改善できる場合がある点から、カルボン酸のpKaが3.5~5.5の範囲にあることがより好ましい。
本発明のガスバリア樹脂組成物がカルボン酸を含有する場合、その含有量の下限はカルボン酸根換算で30ppmが好ましく、100ppmがより好ましい。一方、カルボン酸の含有量の上限は1000ppmが好ましく、600ppmがより好ましい。カルボン酸の含有量が30ppm以上であると、高温による着色耐性が良好になる傾向となる。一方、カルボン酸の含有量が1000ppm以下であると、溶融成形性が良好となる傾向となる。カルボン酸の含有量は、樹脂組成物10gを純水50mlで95℃にて8時間抽出して得られる抽出液を滴定することで算出する。ここで、樹脂組成物中のカルボン酸の含有量として、上記抽出液中に存在するカルボン酸塩の含有量は考慮しない。また、カルボン酸はカルボン酸イオンとして存在していてもよい。
カルボン酸としては、1価カルボン酸及び多価カルボン酸が挙げられ、これらは1種又は複数種からなっていてもよい。カルボン酸として1価カルボン酸と多価カルボン酸との両方を含む場合、ガスバリア樹脂組成物の溶融成形性や高温下での着色耐性をより改善できる場合がある。また、多価カルボン酸は、3個以上のカルボキシ基を有してもよい。この場合、本発明のガスバリア樹脂組成物の着色耐性をさらに向上できる場合がある。
1価カルボン酸とは、分子内に1つのカルボキシ基を有する化合物である。1価のカルボン酸のpKaが3.5~5.5の範囲にあることが好ましい。このような1価カルボン酸としては、例えばギ酸(pKa=3.77)、酢酸(pKa=4.76)、プロピオン酸(pKa=4.85)、酪酸(pKa=4.82)、カプロン酸(pKa=4.88)、カプリン酸(pKa=4.90)、乳酸(pKa=3.86)、アクリル酸(pKa=4.25)、メタクリル酸(pKa=4.65)、安息香酸(pKa=4.20)、2-ナフトエ酸(pKa=4.17)が挙げられる。これらのカルボン酸は、pKaが3.5~5.5の範囲にある限り、水酸基、アミノ基、ハロゲン原子といった置換基を有してもよい。中でも、安全性が高く、取扱いが容易であることから酢酸が好ましい。
多価カルボン酸とは、分子内に2つ以上のカルボキシ基を有する化合物である。この場合、少なくとも1つのカルボキシ基のpKaが3.5~5.5の範囲にあることが好ましい。このような多価カルボン酸として、例えばシュウ酸(pKa2=4.27)、コハク酸(pKa1=4.20)、フマル酸(pKa2=4.44)、リンゴ酸(pKa2=5.13)、グルタル酸(pKa1=4.30、pKa2=5.40)、アジピン酸(pKa1=4.43、pKa2=5.41)、ピメリン酸(pKa1=4.71)、フタル酸(pKa2=5.41)、イソフタル酸(pKa2=4.46)、テレフタル酸(pKa1=3.51、pKa2=4.82)、クエン酸(pKa2=4.75)、酒石酸(pKa2=4.40)、グルタミン酸(pKa2=4.07)、アスパラギン酸(pKa=3.90)が挙げられる。
本発明のガスバリア樹脂組成物はリン酸化合物をさらに含有することが好ましい。当該ガスバリア樹脂組成物がリン酸化合物を含有する場合、その含有量の下限はリン酸根換算で1ppmが好ましく、3ppmがより好ましい。一方、上記含有量の上限はリン酸根換算で200ppmが好ましく、100ppmがより好ましい。この範囲でリン酸化合物を含有すると、当該ガスバリア樹脂組成物の熱安定性を改善できる場合がある。特に、長時間に亘って溶融成形を行う際のゲル状ブツの発生や着色を抑制できる場合がある。リン酸化合物としては、例えばリン酸、亜リン酸等の各種の酸やその塩等を用いることができる。リン酸塩は第1リン酸塩、第2リン酸塩、第3リン酸塩のいずれの形であってもよい。リン酸塩のカチオン種として、アルカリ金属、アルカリ土類金属が挙げられる。リン酸化合物として、具体的には、リン酸二水素ナトリウム、リン酸二水素カリウム、リン酸水素二ナトリウム、リン酸水素二カリウムの形でリン酸化合物が挙げられる。
本発明のガスバリア樹脂組成物は、ホウ素化合物をさらに含有することが好ましい。当該ガスバリア樹脂組成物がホウ素化合物を含有する場合、その含有量の下限はホウ素元子換算で5ppmが好ましく、100ppmがより好ましい。一方、上記含有量の上限はホウ素原子換算で5000ppmが好ましく、1000ppmがより好ましい。この範囲でホウ素化合物を含有すると、当該ガスバリア樹脂組成物の溶融成形時の熱安定性を向上でき、ゲル状ブツの発生を抑制できる場合がある。また、得られる成形体の機械物性を向上できる場合もある。これらの効果は、EVOHとホウ素化合物との間にキレート相互作用が発生することに起因すると推測される。ホウ素化合物としては、例えばホウ酸、ホウ酸エステル、ホウ酸塩、水素化ホウ素が挙げられる。具体的には、ホウ酸としては、例えばオルトホウ酸(H3BO3)、メタホウ酸、四ホウ酸が挙げられ、ホウ酸エステルとしては、例えばホウ酸トリメチル、ホウ酸トリエチルが挙げられ、ホウ酸塩としては、例えば上記ホウ酸類のアルカリ金属塩、アルカリ土類金属塩、ホウ砂が挙げられる。
本発明のガスバリア樹脂組成物は、金属イオンをさらに含有することが好ましい。当該ガスバリア樹脂組成物が金属イオンを含有すると、多層の成形体、すなわち多層構造体としたときの層間接着性が優れたものとなる。層間接着性が向上する理由は定かでないが、ガスバリア樹脂組成物からなる層と隣接する層中に、EVOHのヒドロキシ基と反応し得る官能基を有する分子が含まれる場合には、金属イオンによって両者の結合生成反応が加速されると考えられる。また、金属イオンと上記したカルボン酸との含有比率を制御すると、当該ガスバリア樹脂組成物の溶融成形性や着色耐性も改善できる。
本発明のガスバリア樹脂組成物が金属イオンを含有する場合、その含有量の下限は1ppmが好ましく、100ppmがより好ましく、150ppmがさらに好ましい。一方、金属イオンの含有量の上限は1000ppmが好ましく、400ppmがより好ましく、350ppmがさらに好ましい。金属イオンの含有量が1ppm以上であると、得られる多層構造体の層間接着性が良好となる傾向となる。一方、金属イオンの含有量が1000ppm以下であると、着色耐性が良好となる傾向となる。
金属イオンとしては、一価金属イオン、二価金属イオン、その他遷移金属イオンが挙げられ、これらは1種又は複数種からなっていてもよい。中でも一価金属イオン及び二価金属イオンが好ましい。
一価金属イオンとしては、アルカリ金属イオンが好ましく、例えばリチウム、ナトリウム、カリウム、ルビジウム及びセシウムのイオンが挙げられ、工業的な入手容易性の点からはナトリウム又はカリウムのイオンが好ましい。また、アルカリ金属イオンを与えるアルカリ金属塩としては、例えば脂肪族カルボン酸塩、芳香族カルボン酸塩、炭酸塩、塩酸塩、硝酸塩、硫酸塩、リン酸塩及び金属錯体が挙げられる。中でも、脂肪族カルボン酸塩及びリン酸塩が入手容易である点から好ましく、具体的には、酢酸ナトリウム、酢酸カリウム、リン酸ナトリウム及びリン酸カリウムが好ましい。
金属イオンとして二価金属イオンを含むことが好ましい場合もある。金属イオンが二価金属イオンを含むと、例えばトリムを回収して再利用した際のEVOHの熱劣化が抑制され、得られる成形体のゲル及びブツの発生が抑制される場合がある。二価金属イオンとしては、例えばベリリウム、マグネシウム、カルシウム、ストロンチウム、バリウム及び亜鉛のイオンが挙げられるが、工業的な入手容易性の点からはマグネシウム、カルシウム又は亜鉛のイオンが好ましい。なお、上記した金属原子(F)は、イオンとして存在している場合、この二価金属イオンに含まれる。また、二価金属イオンを与える二価金属塩としては、例えばカルボン酸塩、炭酸塩、塩酸塩、硝酸塩、硫酸塩、リン酸塩及び金属錯体が挙げられカルボン酸塩が好ましい。カルボン酸塩を構成するカルボン酸としては、炭素数1~30のカルボン酸が好ましく、具体的には、酢酸、ステアリン酸、ラウリン酸、モンタン酸、ベヘン酸、オクチル酸、セバシン酸、リシノール酸、ミリスチン酸、パルミチン酸等が挙げられ、中でも、酢酸及びステアリン酸が好ましい。
本発明のガスバリア樹脂組成物は、本発明の効果を阻害しない範囲であれば、例えば、ブロッキング防止剤、加工助剤、上述した各樹脂以外の他の樹脂、安定剤、紫外線吸収剤、可塑剤、帯電防止剤、滑剤、着色剤、充填剤、界面活性剤、乾燥剤、酸素吸収剤、架橋剤、各種繊維などの補強剤などのその他成分を含有してもよい。
ブロッキング防止剤としては、ケイ素、アルミニウム、マグネシウム、ジルコニウム、セリウム、タングステン及びモリブデンなどから選ばれる元素の酸化物、窒化物、酸化窒化物等が挙げられ、これらの中でも入手容易性から酸化ケイ素が望ましい。本発明のガスバリア樹脂組成物がブロッキング防止剤を含むことで、耐ブロッキング性を高めることができる。
加工助剤としては、アルケマ社製Kynar(商標)、3M社製ダイナマー(商標)などのフッ素系加工助剤が挙げられる。本発明のガスバリア樹脂組成物が加工助剤を含むことで、ダイリップへの目やに付着を防止できる傾向となる。
他の樹脂としては、例えば各種ポリオレフィン(ポリエチレン、ポリプロピレン、ポリ1-ブテン、ポリ4-メチル-1-ペンテン、エチレン-プロピレン共重合体、エチレンと炭素数4以上のα-オレフィンとの共重合体、ポリオレフィンと無水マレイン酸との共重合体、エチレン-ビニルエステル共重合体、エチレン-アクリル酸エステル共重合体、又はこれらを不飽和カルボン酸もしくはその誘導体でグラフト変性した変性ポリオレフィン等)、各種ポリエステル(ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエチレンナフタレート等)、ポリ塩化ビニル、ポリ塩化ビニリデン、ポリスチレン、ポリアクリロニトリル、ポリウレタン、ポリカーボネート、ポリアセタール、ポリアクリレート及び変性ポリビニルアルコール樹脂が挙げられる。
溶融安定性等を改善するための安定剤としては、ハイドロタルサイト化合物、ヒンダードフェノール系、ヒンダードアミン系熱安定剤、高級脂肪族カルボン酸の金属塩(例えば、ステアリン酸カルシウム、ステアリン酸マグネシウム等)等が挙げられる。本発明のガスバリア樹脂組成物が安定剤を含む場合、その含有量は0.001~1質量%が好ましい。
紫外線吸収剤としては、エチレン-2-シアノ-3’,3’-ジフェニルアクリレート、2-(2’-ヒドロキシ-5’-メチルフェニル)ベンゾトリアゾール、2-(2’-ヒドロキシ-3’-t-ブチル-5’-メチルフェニル)5-クロロベンゾトリアゾール、2-ヒドロキシ-4-メトキシベンゾフェノン、2,2’-ジヒドロキシ-4-メトキシベンゾフェノン等が挙げられる。
可塑剤としては、フタル酸ジメチル、フタル酸ジエチル、フタル酸ジオクチル、ワックス、流動パラフィン、リン酸エステル等が挙げられる。
帯電防止剤としては、ペンタエリスリットモノステアレート、ソルビタンモノパルミテート、硫酸化ポリオレフィン類、ポリエチレンオキシド、カーボワックス等が挙げられる。
滑剤としては、エチレンビスステアロアミド、ブチルステアレート等が挙げられる。
着色剤としては、カーボンブラック、フタロシアニン、キナクリドン、インドリン、アゾ系顔料、ベンガラ等が挙げられる。
充填剤としては、グラスファイバー、アスベスト、バラストナイト、ケイ酸カルシウム等が挙げられる。
乾燥剤としては、リン酸塩(上記リン酸塩を除く)、ホウ酸ナトリウム、硫酸ナトリウム等、塩化ナトリウム、硝酸ナトリウム、砂糖、シリカゲル、ベントナイト、モレキュラーシーブ、高吸水性樹脂等が挙げられる。
本発明のガスバリア樹脂組成物の含水量は、成形加工時のボイドの発生を防ぐ観点から、一種又は二種以上のEVOHの合計100質量部に対して、3.0質量部以下が好ましく、1.0質量部以下がより好ましく、0.5質量部以下がさらに好ましく、0.3質量部以下が特に好ましい。
本発明のガスバリア樹脂組成物は、EVOH(A)又はEVOH(A’)に起因する、バイオマス由来の不純物を含有する場合がある。様々な不純物を含有する可能性があるが、少なくとも鉄及びニッケル等の金属が多く含まれる場合が多い傾向にある。
(ガスバリア樹脂組成物のバイオベース度)
本発明のガスバリア樹脂組成物のバイオベース度の下限は、環境負荷を低減する観点から1%が好ましく、5%がより好ましく、20%がさらに好ましく、40%が特に好ましい。また、例えば特に優れたロングラン性が要求されない用途などにおいては、当該ガスバリア樹脂組成物のバイオベース度の下限は、60%であってもよく、80%であってもよい。一方、当該ガスバリア樹脂組成物のバイオベース度の上限は、ロングラン性を良好とする観点から99%が好ましく、95%がより好ましく、85%、75%、65%、55%、45%、35%又は25%がさらに好ましい場合もある。なお、このガスバリア樹脂組成物のバイオベース度とは、EVOH以外の任意成分に含まれる他の樹脂等も考慮して測定される値をいう。
本発明のガスバリア樹脂組成物のバイオベース度の下限は、環境負荷を低減する観点から1%が好ましく、5%がより好ましく、20%がさらに好ましく、40%が特に好ましい。また、例えば特に優れたロングラン性が要求されない用途などにおいては、当該ガスバリア樹脂組成物のバイオベース度の下限は、60%であってもよく、80%であってもよい。一方、当該ガスバリア樹脂組成物のバイオベース度の上限は、ロングラン性を良好とする観点から99%が好ましく、95%がより好ましく、85%、75%、65%、55%、45%、35%又は25%がさらに好ましい場合もある。なお、このガスバリア樹脂組成物のバイオベース度とは、EVOH以外の任意成分に含まれる他の樹脂等も考慮して測定される値をいう。
<ガスバリア樹脂組成物の製造方法>
本発明のガスバリア樹脂組成物の製造方法としては、特に限定されない。例えばEVOH(A)とEVOH(B)とを含むガスバリア樹脂組成物の場合、
(1)EVOH(A)のペレットと、EVOH(B)のペレットと、必要に応じて上述したその他成分とを混合(ドライブレンド)し、混合されたペレットを溶融混練する方法、
(2)EVOH(A)のペレット及び/又はEVOH(B)のペレットに必要に応じて上述したその他成分等が含まれる溶液に浸漬させた後、EVOH(A)のペレットとEVOH(B)のペレットとをドライブレンドし、これらを溶融混練する方法、
(3)EVOH(A)のペレットとEVOH(B)のペレットとをドライブレンドし、これらを溶融混練する際に、押出機の途中で必要に応じて上述したその他成分を含む水溶液を液添する方法
(4)EVOH(A)の溶融樹脂とEVOH(B)の溶融樹脂とを溶融状態でブレンドする方法(その他成分は、必要に応じてEVOH(A)及び/又はEVOH(B)に予め含ませておいても、押出機内で液添してもよい)
等が挙げられる。
本発明のガスバリア樹脂組成物の製造方法としては、特に限定されない。例えばEVOH(A)とEVOH(B)とを含むガスバリア樹脂組成物の場合、
(1)EVOH(A)のペレットと、EVOH(B)のペレットと、必要に応じて上述したその他成分とを混合(ドライブレンド)し、混合されたペレットを溶融混練する方法、
(2)EVOH(A)のペレット及び/又はEVOH(B)のペレットに必要に応じて上述したその他成分等が含まれる溶液に浸漬させた後、EVOH(A)のペレットとEVOH(B)のペレットとをドライブレンドし、これらを溶融混練する方法、
(3)EVOH(A)のペレットとEVOH(B)のペレットとをドライブレンドし、これらを溶融混練する際に、押出機の途中で必要に応じて上述したその他成分を含む水溶液を液添する方法
(4)EVOH(A)の溶融樹脂とEVOH(B)の溶融樹脂とを溶融状態でブレンドする方法(その他成分は、必要に応じてEVOH(A)及び/又はEVOH(B)に予め含ませておいても、押出機内で液添してもよい)
等が挙げられる。
上記(1)~(4)等の製造方法によれば、用途、性能等に応じて、EVOH(A)とEVOH(B)との混合比率を調整することで環境負荷の低減効果とロングラン性とのバランスを考慮しつつ、高いガスバリア性を有するガスバリア樹脂組成物を製造することができる。これら中でも、(1)EVOH(A)のペレット及びEVOH(B)のペレットをドライブレンドして溶融混練する工程を含むことが好ましい。ペレット同士の混合やペレットと他の成分との混合には、例えばリボンブレンダー、高速ミキサーコニーダー、ミキシングロール、押出機、インテンシブミキサー等を用いることができる。
また、EVOH(A’)を含むガスバリア樹脂組成物の場合、公知の方法でEVOH(A’)を合成し、得られたEVOH(A’)に対して、必要に応じて公知の方法でその他の成分を混合することにより製造することができる。
EVOH(A)とEVOH(B)と無機粒子(C)等(無機粒子(C)、酸化防止剤(D)、PA(E)、金属原子(F)、熱可塑性エラストマー(G)等)とを含むガスバリア樹脂組成物の場合、このようなガスバリア樹脂組成物の製造方法としては、
(1’)EVOH(A)のペレットと、EVOH(B)のペレットと、無機粒子(C)等と必要に応じて上述したその他成分とを混合(ドライブレンド)し、混合されたペレットを溶融混練する方法
(2’)EVOH(A)のペレット及び/又はEVOH(B)のペレットに必要に応じて上述したその他成分等が含まれる溶液に浸漬させた後、EVOH(A)のペレットとEVOH(B)のペレットと無機粒子(C)等とをドライブレンドし、これらを溶融混練する方法
(3’)EVOH(A)のペレットとEVOH(B)のペレットと無機粒子(C)等とをドライブレンドし、これらを溶融混練する際に、押出機の途中で必要に応じて上述したその他成分を含む水溶液を液添する方法
(4’)予めEVOH(A)及び/またはEVOH(B)に無機粒子(C)等を含有させた上で、EVOH(A)の溶融樹脂とEVOH(B)の溶融樹脂とを溶融状態でブレンドする方法(その他成分は、必要に応じてEVOH(A)及び/又はEVOH(B)に予め含ませておいても、押出機内で液添してもよい)
(5’)EVOH(A)及び/またはEVOH(B)を合成する過程で、無機粒子(C)等を単独あるいは適当な溶媒を用いた分散液として添加、混合する方法
等が挙げられる。
(1’)EVOH(A)のペレットと、EVOH(B)のペレットと、無機粒子(C)等と必要に応じて上述したその他成分とを混合(ドライブレンド)し、混合されたペレットを溶融混練する方法
(2’)EVOH(A)のペレット及び/又はEVOH(B)のペレットに必要に応じて上述したその他成分等が含まれる溶液に浸漬させた後、EVOH(A)のペレットとEVOH(B)のペレットと無機粒子(C)等とをドライブレンドし、これらを溶融混練する方法
(3’)EVOH(A)のペレットとEVOH(B)のペレットと無機粒子(C)等とをドライブレンドし、これらを溶融混練する際に、押出機の途中で必要に応じて上述したその他成分を含む水溶液を液添する方法
(4’)予めEVOH(A)及び/またはEVOH(B)に無機粒子(C)等を含有させた上で、EVOH(A)の溶融樹脂とEVOH(B)の溶融樹脂とを溶融状態でブレンドする方法(その他成分は、必要に応じてEVOH(A)及び/又はEVOH(B)に予め含ませておいても、押出機内で液添してもよい)
(5’)EVOH(A)及び/またはEVOH(B)を合成する過程で、無機粒子(C)等を単独あるいは適当な溶媒を用いた分散液として添加、混合する方法
等が挙げられる。
上記(1’)~(5’)等の製造方法によれば、用途、性能等に応じて、各成分の混合比率を調整することで環境負荷の低減効果とロングラン性とのバランスを考慮しつつ、高いガスバリア性を有するガスバリア樹脂組成物を製造することができる。これら中でも、(1)EVOH(A)のペレット、EVOH(B)のペレット及び無機粒子(C)等をドライブレンドして溶融混練する工程を含むことが好ましい。例えば、EVOH(A)のペレットとEVOH(B)のペレットと熱可塑性エラストマー(G)のペレットとをドライブレンドし、溶融混練する工程を備えるガスバリア樹脂組成物の製造方法は、本発明の好適な形態である。ペレット同士の混合やペレットと他の成分との混合には、例えばリボンブレンダー、高速ミキサーコニーダー、ミキシングロール、押出機、インテンシブミキサー等を用いることができる。
また、EVOH(A’)と無機粒子(C)等とを含むガスバリア樹脂組成物の場合、公知の方法でEVOH(A’)を合成し、得られたEVOH(A’)に対して公知の方法で無機粒子(C)等を混合し、必要に応じて公知の方法でその他の成分を混合することにより製造することができる。
本発明のガスバリア樹脂組成物がPA(E)及び金属原子(F)をさらに含む場合、このようなガスバリア樹脂組成物の製造方法としては、一種又は二種以上のEVOHのペレットと、PA(E)のペレットと、マグネシウム、カルシウム及び亜鉛からなる群より選ばれる少なくとも1種の金属原子(F)を含む化合物とを混合し、溶融混練する方法が好ましい。この製造方法における混合の際の金属原子(F)を含む化合物の形態としては、固形状であってもよく、水溶液であってもよい。また、EVOHのペレットと、PA(E)のペレットと、金属原子(F)を含む化合物との混合は、1段階で行ってもよいし、2段階以上で行ってもよい。1段階で混合する場合、例えば固形状の各成分をドライブレンドすることなどにより行うことができる。また、2段階で混合する場合、EVOHのペレットとPA(E)のペレットとをドライブレンドし、これらを溶融混練する際に、押出機の途中で、金属原子(F)を含む水溶液を液添することなどにより行うことができる。
<成形体>
本発明のガスバリア樹脂組成物を含む成形体は、本発明の好適な一実施態様である。当該ガスバリア樹脂組成物は、単層構造の成形体とすることもできるし、他の各種基材等と共に2種以上の多層構造の成形体、すなわち多層構造体とすることもできる。本発明の成形体の成形方法としては、例えば押出成形、熱成形、異形成形、中空成形、回転成形、射出成形が挙げられる。本発明のガスバリア樹脂組成物を用いた成形体の用途は多岐に亘り、例えば、フィルム、シート、容器(袋、カップ、チューブ、トレー、ボトル、紙カートン等)、燃料容器、タンク、パイプ、ホース、繊維、飲食品用包装材、容器用パッキング材、医療用輸液バッグ材、タイヤ用チューブ材、靴用クッション材、バッグインボックス用内袋材、有機液体貯蔵用タンク材、有機液体輸送用パイプ材、暖房用温水パイプ材(床暖房用温水パイプ材等)、化粧品用包装材、デンタルケア用包装材、医薬品用包装材、包材用子部品(キャップ、バッグインボックスのコック部分など)、農薬ボトル、農業用フィルム(温室用フィルム、土壌燻蒸用フィルム)、穀物保管用袋、ジオメンブレン、真空断熱板外袋、壁紙又は化粧板、水素、酸素等のガスタンク等が挙げられる。後述する他の各種用途も、成形体の用途に含まれる。
本発明のガスバリア樹脂組成物を含む成形体は、本発明の好適な一実施態様である。当該ガスバリア樹脂組成物は、単層構造の成形体とすることもできるし、他の各種基材等と共に2種以上の多層構造の成形体、すなわち多層構造体とすることもできる。本発明の成形体の成形方法としては、例えば押出成形、熱成形、異形成形、中空成形、回転成形、射出成形が挙げられる。本発明のガスバリア樹脂組成物を用いた成形体の用途は多岐に亘り、例えば、フィルム、シート、容器(袋、カップ、チューブ、トレー、ボトル、紙カートン等)、燃料容器、タンク、パイプ、ホース、繊維、飲食品用包装材、容器用パッキング材、医療用輸液バッグ材、タイヤ用チューブ材、靴用クッション材、バッグインボックス用内袋材、有機液体貯蔵用タンク材、有機液体輸送用パイプ材、暖房用温水パイプ材(床暖房用温水パイプ材等)、化粧品用包装材、デンタルケア用包装材、医薬品用包装材、包材用子部品(キャップ、バッグインボックスのコック部分など)、農薬ボトル、農業用フィルム(温室用フィルム、土壌燻蒸用フィルム)、穀物保管用袋、ジオメンブレン、真空断熱板外袋、壁紙又は化粧板、水素、酸素等のガスタンク等が挙げられる。後述する他の各種用途も、成形体の用途に含まれる。
成形体の具体的な成形方法として、例えばフィルム、シート、パイプ、ホースは押出成形により、容器形状は射出成形により、ボトルやタンク等の中空容器は中空成形や回転成形により成形できる。中空成形としては、押出成形によりパリソンを成形し、これをブローして成形を行う押出中空成形と、射出成形によりプリフォームを成形し、これをブローして成形を行う射出中空成形が挙げられる。フレキシブル包装材や容器の製造には、押出成形によって多層フィルム等の包装材を成形する方法、押出成形によって成形した多層シートを熱成形して容器状の包装材にする方法が好適に用いられる。
<多層構造体A>
本発明の成形体は、本発明のガスバリア樹脂組成物から形成されるガスバリア層(以下「層(1)」と略記する場合がある。)を備えるものであってよい。当該成形体は、単層の構造体であってもよいが、他の層をさらに備える多層構造体であることが好ましい。すなわち、本発明の多層構造体Aは、本発明のガスバリア樹脂組成物からなる層を少なくとも1層備える。なお、後述する多層フィルム、多層シート、多層パイプ、多層構造体B等も、当該多層構造体Aの形態に含まれる。当該多層構造体Aの層数の下限としては、2が好ましく、3がより好ましい。また、当該多層構造体Aの層数の上限としては、例えば1000であってよく、100であってもよく、20又は10であってもよい。
本発明の成形体は、本発明のガスバリア樹脂組成物から形成されるガスバリア層(以下「層(1)」と略記する場合がある。)を備えるものであってよい。当該成形体は、単層の構造体であってもよいが、他の層をさらに備える多層構造体であることが好ましい。すなわち、本発明の多層構造体Aは、本発明のガスバリア樹脂組成物からなる層を少なくとも1層備える。なお、後述する多層フィルム、多層シート、多層パイプ、多層構造体B等も、当該多層構造体Aの形態に含まれる。当該多層構造体Aの層数の下限としては、2が好ましく、3がより好ましい。また、当該多層構造体Aの層数の上限としては、例えば1000であってよく、100であってもよく、20又は10であってもよい。
本発明の多層構造体Aが有する層(1)以外の層としては、例えば、本発明のガスバリア樹脂組成物以外の熱可塑性樹脂を主成分とする熱可塑性樹脂層(以下「層(2)」と略記する場合がある。)、接着性樹脂、アンカーコーティング剤または接着剤を主成分とする接着層(「以下、「層(3)」と略記する場合がある。)、回収層(以下、「層(4)」と略記する場合がある。)、無機蒸着層(以下「層(5)」と略記する場合がある。)及び紙基材層(以下「層(6)」と略記する場合がある。)等が挙げられる。
本発明の多層構造体Aは、その用途によって好適な態様が異なるが、基本的に層(1)及び層(2)を含むことが好ましく、層(1)、層(2)及び層(3)を含むことがより好ましい。具体的な構成としては、例えば、当該多層構造体Aが容器、包装材、チューブ、パイプ等である場合、内面から外面に向かって、層(2)、層(3)、層(1)、層(3)、層(4)、層(2)の順(以降、(内)2/3/1/3/4/2(外)のように表す)、(内)2/3/1/3/2(外)、(内)2/4/3/1/3/4/2(外)、(内)4/3/1/3/4(外)、(内)1/3/2(外)、(内)2/4/3/1/3/2(外)、(内)2/1/3/2(外)、(内)2/1/5/2(外)、(内)2/5/1/2(外)、(内)2/5/1/5/2(外)、(内)2/3/1/5/3/2(外)、(内)2/3/5/1/3/2(外)、(内)2/3/1/3/2/6(外)、(内)3/1/3/6(外)等の層構造のものを採用することができる。なお、層(2)の種類によっては、層(3)は省略してもよく、層(2)の代わりに層(4)を備える構成でもよく、層(1)~(6)がそれぞれ複数用いられている配置の場合、それぞれの層を構成する樹脂は同一でも異なっていてもよい。
本発明の多層構造体A又は成形体の全体の平均厚みは、目的とする用途によって好適な態様は異なるが、5μm~15mmが好ましく、10μm~10mmがより好ましい。
[層(1)]
本発明の多層構造体Aの平均厚みに対する層(1)の平均厚みの割合の下限は特に限定されず、1%が好ましく、2%がより好ましいこともある。上記層(1)の平均厚みの割合は、20%が好ましく、15%がより好ましい。層(1)の平均厚みの割合が上記範囲内であると、良好なガスバリア性及びロングラン性を示し、生産性が良好となる傾向となる。
本発明の多層構造体Aの平均厚みに対する層(1)の平均厚みの割合の下限は特に限定されず、1%が好ましく、2%がより好ましいこともある。上記層(1)の平均厚みの割合は、20%が好ましく、15%がより好ましい。層(1)の平均厚みの割合が上記範囲内であると、良好なガスバリア性及びロングラン性を示し、生産性が良好となる傾向となる。
[層(2)]
層(2)は、EVOH以外の熱可塑性樹脂を主成分とする熱可塑性樹脂層である。層(2)におけるEVOH以外の熱可塑性樹脂の割合は、70質量%以上が好ましく、80質量%以上がより好ましく、90質量%以上がさらに好ましく、95質量%以上が特に好ましく、99質量%以上であってもよく、層(2)を構成する樹脂としては実質的にEVOH以外の熱可塑性樹脂のみからなってもよい。
層(2)は、EVOH以外の熱可塑性樹脂を主成分とする熱可塑性樹脂層である。層(2)におけるEVOH以外の熱可塑性樹脂の割合は、70質量%以上が好ましく、80質量%以上がより好ましく、90質量%以上がさらに好ましく、95質量%以上が特に好ましく、99質量%以上であってもよく、層(2)を構成する樹脂としては実質的にEVOH以外の熱可塑性樹脂のみからなってもよい。
層(2)の主成分となる熱可塑性樹脂としては、例えば各種ポリオレフィン(ポリエチレン、ポリプロピレン、ポリ1-ブテン、ポリ4-メチル-1-ペンテン、エチレン-プロピレン共重合体、エチレンと炭素数4以上のα-オレフィンとの共重合体、ポリオレフィンと無水マレイン酸との共重合体、エチレン-ビニルエステル共重合体、エチレン-アクリル酸エステル共重合体、又はこれらを不飽和カルボン酸もしくはその誘導体でグラフト変性した変性ポリオレフィン等)、各種ポリアミド(ナイロン6、ナイロン6・6、ナイロン6/66共重合体、ナイロン11、ナイロン12、ポリメタキシリレンアジパミド等)、各種ポリエステル(ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエチレンナフタレート等)、ポリ塩化ビニル、ポリ塩化ビニリデン、ポリスチレン、ポリアクリロニトリル、ポリウレタン、ポリカーボネート、ポリアセタール、ポリアクリレート及び変性ポリビニルアルコール樹脂が挙げられる。かかる熱可塑性樹脂層は無延伸のものであってもよく、一軸又は二軸に延伸又は圧延されていてもよい。中でも、ポリオレフィンは耐湿性、機械的特性、経済性、ヒートシール性の点で、また、ポリアミドやポリエステルは機械的特性、耐熱性の点で好ましい。
上記熱可塑性樹脂の190℃、2,160g荷重下でのMFRの下限は0.01g/10分が好ましく、0.02g/10分がより好ましい。一方、上記MFRの上限は0.5g/10分が好ましく、0.1g/10分がより好ましく、0.05g/10分がさらに好ましい。
上記熱可塑性樹脂は、通常市販品の中から適宜選択して使用できる。また、層(2)は、本発明の効果を損なわない限り、層(1)を構成する本発明のガスバリア樹脂組成物と同様の他の任意成分を含んでいてもよい。
層(2)の1層当たりの平均厚みの下限は、5μmが好ましく、20μmがより好ましい。層(2)の1層当たりの平均厚みの上限は3,000μmが好ましく、1,000μmがより好ましい。層(2)の1層当たりの平均厚みが上記範囲内であると、強度、成形性、外観等が向上する傾向となる。
本発明の多層構造体Aの平均厚みに対する層(2)の平均厚みの割合の下限は特に限定されず、10%が好ましく、30%がより好ましく、50%、70%又は80%がさらに好ましいこともある。上記層(2)の平均厚みの割合の上限は特に限定されず、95%が好ましく、90%がより好ましい。
[層(3)]
層(3)は、層(1)と層(2)との間に配置される場合があり、接着性樹脂、アンカーコーティング剤または接着剤を主成分とする層である。層(3)は、層(1)と層(2)等の他の層との間の接着層として機能させることができる。接着性樹脂は、接着性を有する樹脂であり、接着性を有する熱可塑性樹脂が好ましい場合がある。アンカーコーティング剤及び接着剤は、樹脂であってもよく、低分子化合物等、樹脂以外であってもよく、複数の成分からなるものであってもよい。接着性樹脂としては、カルボン酸変性ポリオレフィン等を挙げることができる。なお、上記カルボン酸変性ポリオレフィンとは、オレフィン系重合体にエチレン性不飽和カルボン酸又はその無水物を付加反応、グラフト反応等により化学的に結合させて得られるカルボキシ基又はその無水物基を有するオレフィン系重合体のことをいう。
層(3)は、層(1)と層(2)との間に配置される場合があり、接着性樹脂、アンカーコーティング剤または接着剤を主成分とする層である。層(3)は、層(1)と層(2)等の他の層との間の接着層として機能させることができる。接着性樹脂は、接着性を有する樹脂であり、接着性を有する熱可塑性樹脂が好ましい場合がある。アンカーコーティング剤及び接着剤は、樹脂であってもよく、低分子化合物等、樹脂以外であってもよく、複数の成分からなるものであってもよい。接着性樹脂としては、カルボン酸変性ポリオレフィン等を挙げることができる。なお、上記カルボン酸変性ポリオレフィンとは、オレフィン系重合体にエチレン性不飽和カルボン酸又はその無水物を付加反応、グラフト反応等により化学的に結合させて得られるカルボキシ基又はその無水物基を有するオレフィン系重合体のことをいう。
上記接着性樹脂の190℃、2,160g荷重下でのMFRの下限は0.1g/10分が好ましく、0.2g/10分がより好ましく、0.3g/10分がさらに好ましい。一方、上記MFRの上限は15g/10分が好ましく、10g/10分がより好ましく、5g/10分がさらに好ましい。なお、このような接着性樹脂は工業的に製造される市販品を用いることができ、例えばいずれも三井化学株式会社製の商品名「ADMER NF642E」「ADMER AT2235E」「ADMER NF408E」などが挙げられる。
なお、層(3)は、本発明の効果を損なわない範囲で、接着性樹脂、アンカーコーティング剤及び接着剤以外に、層(1)と同様の他の任意成分を含んでいてもよい。
アンカーコーティング剤及び接着剤を用いる場合、これらを層(3)に隣接する層の表面に塗布し、必要に応じ乾燥することで層(3)を形成できる。これらの塗布の前に塗布面にコロナ放電処理等の表面処理を行うことによって、接着性を高めることができる場合がある。接着剤としては、特に限定されず、例えば、ポリイソシアネート成分とポリオール成分とを混合し反応させる二液反応型ポリウレタン系接着剤を用いることが好ましい。また、アンカーコーティング剤及び接着剤は、公知のシランカップリング剤などを少量添加することで、さらに接着性を高めることができる場合がある。シランカップリング剤の好適な例としては、イソシアネート基、エポキシ基、アミノ基、ウレイド基、メルカプト基などの反応性基を有するシランカップリング剤を挙げることができる。
層(3)の1層当たりの平均厚みの下限は、1μmが好ましく、3μmがより好ましい。層(3)の1層当たりの平均厚みの上限は300μmが好ましく、150μmがより好ましい。層(3)の1層当たりの平均厚みが上記範囲内であると、低コストで良好な接着性を示す傾向となる。
[層(4)]
層(4)は、例えばEVOH、熱可塑性樹脂、及び接着性樹脂を含有する層である。また、層(4)は、本発明の多層構造体Aの製造工程における層(1)、層(2)及び層(3)の回収物を用いて形成されることが好ましい。回収物としては、当該多層構造体Aの製造工程において発生するバリ、検定の不合格品等が挙げられる。
層(4)は、例えばEVOH、熱可塑性樹脂、及び接着性樹脂を含有する層である。また、層(4)は、本発明の多層構造体Aの製造工程における層(1)、層(2)及び層(3)の回収物を用いて形成されることが好ましい。回収物としては、当該多層構造体Aの製造工程において発生するバリ、検定の不合格品等が挙げられる。
層(4)は、上述の層(2)の代わりとして用いることも可能であるが、一般的には層(2)よりも層(4)の機械的強度が低くなることが多いため、層(2)と層(4)とを積層して用いることが好ましい。本発明の多層構造体Aが容器である場合、外部から衝撃を受けた場合には、容器に応力の集中が生じ、応力集中部において衝撃に対する圧縮応力が容器内層側で働き、破損が起こるおそれがあるため、強度的に弱い層(4)は層(1)よりも外層側に配置することが好ましい。また、バリの発生が多い場合等、多量の樹脂をリサイクルする必要がある場合は、層(1)の両側に回収層である層(4)を配置することもできる。
[層(5)]
層(5)は無機蒸着層である。層(5)は、通常、酸素や水蒸気に対するバリア性を有する層であり、透明性を有することが好ましい。層(5)は無機物を蒸着することで形成できる。無機物としては、金属(例えば、アルミニウム)、金属酸化物(例えば、酸化ケイ素、酸化アルミニウム)、金属窒化物(例えば、窒化ケイ素)、金属窒化酸化物(例えば、酸窒化ケイ素)、または金属炭化窒化物(例えば、炭窒化ケイ素)等が挙げられる。中でも、酸化アルミニウム、酸化ケイ素、酸化マグネシウム、または窒化ケイ素で形成される層(5)が、透明性に優れる観点から好ましい。
層(5)は無機蒸着層である。層(5)は、通常、酸素や水蒸気に対するバリア性を有する層であり、透明性を有することが好ましい。層(5)は無機物を蒸着することで形成できる。無機物としては、金属(例えば、アルミニウム)、金属酸化物(例えば、酸化ケイ素、酸化アルミニウム)、金属窒化物(例えば、窒化ケイ素)、金属窒化酸化物(例えば、酸窒化ケイ素)、または金属炭化窒化物(例えば、炭窒化ケイ素)等が挙げられる。中でも、酸化アルミニウム、酸化ケイ素、酸化マグネシウム、または窒化ケイ素で形成される層(5)が、透明性に優れる観点から好ましい。
層(5)の形成方法は、特に限定されず、真空蒸着法(例えば、抵抗加熱蒸着、電子ビーム蒸着、分子線エピタキシー法等)、スパッタリング法やイオンプレーティング法等の物理気相成長法;熱化学気相成長法(例えば、触媒化学気相成長法)、光化学気相成長法、プラズマ化学気相成長法(例えば、容量結合プラズマ、誘導結合プラズマ、表面波プラズマ、電子サイクロトロン共鳴、デュアルマグネトロン、原子層堆積法等)、有機金属気相成長法等の化学気相成長法が挙げられる。
層(5)の厚さは、無機蒸着層を構成する成分の種類によって異なるが、0.002~0.5μmが好ましく、0.005~0.2μmがより好ましく、0.01~0.1μmがさらに好ましい。層(5)の厚さが0.002μm以上であると、酸素や水蒸気に対する層(5)のバリア性が良好になる傾向となる。また、層(5)の厚さが0.5μm以下であると、層(5)の屈曲後のバリア性が維持される傾向となる。
また、後述する蒸着フィルムに備わる無機蒸着層も、層(5)の好適な形態である。
[層(6)]
層(6)は紙基材層である。層(6)に使用される紙基材としては、適用する紙容器の用途に応じて、種々の賦型性、耐屈曲性、剛性、腰、強度等を有する任意の紙を使用することができ、例えば、主強度材であり、強サイズ性の晒または未晒の紙、あるいは、純白ロール紙、クラフト紙、板紙、加工紙、ミルク原紙等の各種の紙を使用することができる。紙基材層は、これらの紙を複数層重ねてラミネートしたものであってもよい。紙基材層は、坪量80~600g/m2、好ましくは坪量100~450g/m2であり、厚さ110~860μm、好ましくは140~640μmの範囲である。紙基材層がこれより薄いと、容器としての強度が不足し、またこれより厚いと、剛性が高くなりすぎて、加工が困難になり得る。なお、紙基材層には、例えば、文字、図形、記号、その他の所望の絵柄を通常の印刷方式にて任意に形成することができる。
層(6)は紙基材層である。層(6)に使用される紙基材としては、適用する紙容器の用途に応じて、種々の賦型性、耐屈曲性、剛性、腰、強度等を有する任意の紙を使用することができ、例えば、主強度材であり、強サイズ性の晒または未晒の紙、あるいは、純白ロール紙、クラフト紙、板紙、加工紙、ミルク原紙等の各種の紙を使用することができる。紙基材層は、これらの紙を複数層重ねてラミネートしたものであってもよい。紙基材層は、坪量80~600g/m2、好ましくは坪量100~450g/m2であり、厚さ110~860μm、好ましくは140~640μmの範囲である。紙基材層がこれより薄いと、容器としての強度が不足し、またこれより厚いと、剛性が高くなりすぎて、加工が困難になり得る。なお、紙基材層には、例えば、文字、図形、記号、その他の所望の絵柄を通常の印刷方式にて任意に形成することができる。
(多層構造体Aの製造方法)
本発明の多層構造体Aは、本発明のガスバリア樹脂組成物を用いること以外は、各種溶融成形等、従来公知の成形方法により製造することができる。ガスバリア樹脂組成物を溶融成形する方法としては、例えば押出成形、キャスト成形、インフレーション押出成形、ブロー成形、射出成形、射出ブロー成形等が挙げられる。
本発明の多層構造体Aは、本発明のガスバリア樹脂組成物を用いること以外は、各種溶融成形等、従来公知の成形方法により製造することができる。ガスバリア樹脂組成物を溶融成形する方法としては、例えば押出成形、キャスト成形、インフレーション押出成形、ブロー成形、射出成形、射出ブロー成形等が挙げられる。
<フィルムまたはシート>
本発明のフィルムまたはシートは、本発明の成形体を備える。フィルムとは、「平均厚みが250μm未満の膜状の軟質性のもの」をいい、シートとは「平均厚みが250μm以上の薄い板状の軟質性のもの」をいう。熱収縮「フィルムまたはシート」、及び産業用「フィルムまたはシート」におけるフィルムとシートとの区別においても同様である。以下、「フィルムまたはシート」を「フィルム等」とも称する。本発明のフィルム等は、本発明の成形体からなるフィルム等であってよい。すなわち、本発明の成形体の一実施形態は、フィルム等であってよい。本発明のフィルム等は環境負荷が低く、ガスバリア性、外観及び生産性も良好である。本発明のフィルム等は、層(1)のみからなる単層フィルムであってもよく、多層フィルムであってもよい。本発明のフィルム等の平均厚みは、例えば1μm以上300μm未満であることが好ましく、5μm以上100μm未満であることがより好ましい。本発明のフィルム等は、各種包装材などとして好適に用いることができる。
本発明のフィルムまたはシートは、本発明の成形体を備える。フィルムとは、「平均厚みが250μm未満の膜状の軟質性のもの」をいい、シートとは「平均厚みが250μm以上の薄い板状の軟質性のもの」をいう。熱収縮「フィルムまたはシート」、及び産業用「フィルムまたはシート」におけるフィルムとシートとの区別においても同様である。以下、「フィルムまたはシート」を「フィルム等」とも称する。本発明のフィルム等は、本発明の成形体からなるフィルム等であってよい。すなわち、本発明の成形体の一実施形態は、フィルム等であってよい。本発明のフィルム等は環境負荷が低く、ガスバリア性、外観及び生産性も良好である。本発明のフィルム等は、層(1)のみからなる単層フィルムであってもよく、多層フィルムであってもよい。本発明のフィルム等の平均厚みは、例えば1μm以上300μm未満であることが好ましく、5μm以上100μm未満であることがより好ましい。本発明のフィルム等は、各種包装材などとして好適に用いることができる。
本発明のフィルム等の、JIS B0601に準拠して測定される少なくとも一方の表面の算術平均粗さ(Ra)は1.0μm以下が好ましく、0.8μm以下がより好ましく、0.6μm以下がさらに好ましく、0.4μm以下が特に好ましい。本発明のフィルム等の、少なくとも一方の表面の算術平均粗さ(Ra)は0.05μm以上が好ましく、0.10μm以上がより好ましく、0.15μm以上がさらに好ましく、0.20μm以上が特に好ましい。本発明のフィルム等の少なくとも一方の表面の算術平均粗さ(Ra)を上記範囲とすると、耐破断性が優れる。
本発明のフィルム等の、JIS B0601に準拠して測定される少なくとも一方の表面の輪郭曲線要素の平均長さ(RSm)は1000μm以下が好ましく、800μm以下がより好ましく、600μm以下がさらに好ましく、400μm以下が特に好ましい。本発明のフィルム等の、少なくとも一方の表面の輪郭曲線要素の平均長さ(RSm)は50μm以上が好ましく、100μm以上がより好ましく、150μm以上がさらに好ましく、200μm以上が特に好ましい。本発明のフィルム等の少なくとも一方の表面の輪郭曲線要素の平均長さ(RSm)を上記範囲とすると、耐破断性が優れる。なお上記したJIS B0601とは例えばJIS B0601:2001を表す。
本発明のフィルム等は、無延伸フィルム等であってもよいが、延伸されていることが好ましい場合がある。延伸されていることで強度等が向上する。さらに、本発明のフィルム等が延伸フィルム等である場合、延伸に伴って生じうるスジ状のムラの発生が少ないため、外観やガスバリア性等も良好である。
(フィルム等の製造方法)
本発明のフィルム等は公知の方法で製造できる。フィルム等の形成方法としては特に限定されず、例えば溶融法、溶液法、カレンダー法等が挙げられ、溶融法が好ましい。溶融法としては、Tダイ法(キャスト法)、インフレーション法が挙げられ、キャスト法が好ましい。特に、本発明のフィルム等を構成する樹脂組成物をキャスティングロール上に溶融押出するキャスト成形工程、及び上記樹脂組成物から得られる未延伸フィルム等を延伸する工程を備える方法で製造することが好ましい。溶融法の際の溶融温度はガスバリア樹脂組成物の融点等により異なるが、150~300℃程度が好ましい。また、本発明のフィルム等が多層である場合、その製造方法は公知の方法で製造でき、共押出法、ドライラミネート法、サンドラミネート法、押出ラミネート法、共押出ラミネート法、溶液コート法、などを採用できる。
本発明のフィルム等は公知の方法で製造できる。フィルム等の形成方法としては特に限定されず、例えば溶融法、溶液法、カレンダー法等が挙げられ、溶融法が好ましい。溶融法としては、Tダイ法(キャスト法)、インフレーション法が挙げられ、キャスト法が好ましい。特に、本発明のフィルム等を構成する樹脂組成物をキャスティングロール上に溶融押出するキャスト成形工程、及び上記樹脂組成物から得られる未延伸フィルム等を延伸する工程を備える方法で製造することが好ましい。溶融法の際の溶融温度はガスバリア樹脂組成物の融点等により異なるが、150~300℃程度が好ましい。また、本発明のフィルム等が多層である場合、その製造方法は公知の方法で製造でき、共押出法、ドライラミネート法、サンドラミネート法、押出ラミネート法、共押出ラミネート法、溶液コート法、などを採用できる。
延伸は一軸延伸でも二軸延伸でもよく、二軸延伸が好ましい。二軸延伸は、逐次二軸延伸及び同時二軸延伸のいずれでもよい。面積換算の延伸倍率の下限は6倍が好ましく、8倍がより好ましい。延伸倍率の上限は15倍が好ましく、12倍がより好ましい。延伸倍率が上記範囲であると、フィルム等の厚みの均一性、ガスバリア性及び機械的強度の点を向上させることができる。また、延伸温度としては、例えば60℃以上120℃以下とすることができる。
本発明のフィルム等の製造方法は、延伸工程の後に、延伸されたフィルム等を熱処理する工程を備えていてもよい。熱処理温度は、通常、延伸温度よりも高い温度に設定され、例えば120℃超200℃以下とすることができる。
本発明のフィルム等は、食品包装容器、医薬品包装容器、工業薬品包装容器、農薬包装容器等の各種包装容器の材料として好適に用いられる。また、後述する熱収縮フィルム等及び産業用フィルム等も、本発明のフィルム等の一実施形態に含まれる。
<熱収縮フィルムまたはシート>
本発明の熱収縮フィルムまたはシート(熱収縮フィルムまたは熱収縮シート)は、本発明の成形体を備える。本発明の熱収縮フィルム等は、本発明の成形体からなる熱収縮フィルム等であってよい。すなわち、本発明の成形体の一実施形態は、熱収縮フィルム等であってよい。本発明の熱収縮フィルム等は環境負荷が低く、ガスバリア性、外観及び生産性も良好である。本発明の熱収縮フィルム等においては、単層または多層フィルム等を製膜した後、延伸工程に供されることで、熱収縮性が付与される。本発明の熱収縮フィルム等の層(1)に用いられるEVOHは、良好な熱収縮性を示す観点から上記式(I)で表される変性基(構造)を有していることが好ましい。
本発明の熱収縮フィルムまたはシート(熱収縮フィルムまたは熱収縮シート)は、本発明の成形体を備える。本発明の熱収縮フィルム等は、本発明の成形体からなる熱収縮フィルム等であってよい。すなわち、本発明の成形体の一実施形態は、熱収縮フィルム等であってよい。本発明の熱収縮フィルム等は環境負荷が低く、ガスバリア性、外観及び生産性も良好である。本発明の熱収縮フィルム等においては、単層または多層フィルム等を製膜した後、延伸工程に供されることで、熱収縮性が付与される。本発明の熱収縮フィルム等の層(1)に用いられるEVOHは、良好な熱収縮性を示す観点から上記式(I)で表される変性基(構造)を有していることが好ましい。
本発明の熱収縮フィルム等は、層(1)と層(2)とを有する多層フィルム等であることが好適である。このとき、一方の外層に層(1)を、他方の外層に層(2)を配置する構成、又は、層(1)を中間層とし、その両側の外層に層(2)を配置する構成が好ましく、後者がより好適である。層(1)と層(2)とが接着性樹脂層を介して接着されてなることも好ましい。
延伸前の多層フィルム等における、層(1)の厚みは、3~250μmが好適であり、10~100μmがより好適である。一方、層(2)の厚みは特に制限されず、要求される透湿性、耐熱性、ヒートシール性、透明性などの性能や用途を考慮して適宜選択される。延伸前の多層フィルム等の全体の厚みは特に限定されないが、通常15~6000μmである。
本発明の熱収縮フィルム等における層(2)に用いられる熱可塑性樹脂としては、ヒートシール性及び熱収縮性が優れる観点からはエチレン-酢酸ビニル共重合体、アイオノマー、ポリエチレン等のポリオレフィンが好適に用いられ、突刺強度や耐ピンホール性等の機械強度が優れる観点からはポリアミドが好適に用いられる。
本発明の熱収縮フィルム等における層(2)に用いられる熱可塑性樹脂としてポリオレフィンを用いた場合の熱収縮フィルムの構成例として、ポリエチレン層/層(3)/層(1)/層(3)/ポリエチレン層、ポリプロピレン層/層(3)/層(1)/層(2)/ポリプロピレン層、アイオノマー層/層(3)/層(1)/層(3)/アイオノマー層、エチレン-酢酸ビニル共重合体層/層(3)/層(1)/層(3)/エチレン-酢酸ビニル共重合体層等が好適なものとして挙げられる。
本発明の熱収縮フィルム等における層(2)に用いられる熱可塑性樹脂としてポリアミドを用いる場合、ポリアミド層と層(1)とが隣接する構成が好ましく用いられる。このような構成をとることで優れたバリア性と耐突き刺し強度とが得られる。さらに層(1)に代えて汎用のバリア性樹脂を用いた場合に比べ、収縮後における透明性が優れる。ポリアミド層と層(1)との間に接着性樹脂層を挟まない構成がより好ましい。
このようにポリアミド層と層(1)が隣接する場合の構成として、ポリアミド層/層(1)/層(3)/層(2)、層(2)/ポリアミド層/層(1)/ポリアミド層/層(2)、ポリアミド層/層(1)/ポリアミド層/層(2)、ポリアミド層/ポリアミド層/層(1)/ポリアミド層/層(2)などの構成が例示できる。ポリアミド層/層(1)/層(3)/エチレン-酢酸ビニル共重合体層、ポリエチレン層/層(3)/ポリアミド層/層(1)/ポリアミド層/層(3)/ポリエチレン層、ポリアミド層/層(1)/ポリアミド層/層(3)/ポリエチレン層、ポリアミド層/層(3)/ポリアミド層/層(1)/ポリアミド層/層(3)/ポリエチレン層等が好適なものとして挙げられる。
上記多層フィルム等は各種の製造方法によって得ることができ、共押出法、ドライラミネート法、サンドラミネート法、押出ラミネート法、共押出ラミネート法、溶液コート法、などを採用することができる。これらの内、共押出法は、ガスバリア樹脂組成物と、他の熱可塑性樹脂を、押出機より同時に押出して溶融状態下に積層し、ダイス出口から多層フィルム状に吐出する方法である。共押出法で製膜する場合、層(1)と層(2)を層(3)を挟んで積層する方法が好ましい。接着性樹脂としては、カルボキシル基、カルボン酸無水物基又はエポキシ基を有するポリオレフィンを用いることが好ましい。このような接着性樹脂は、ガスバリア樹脂組成物との接着性にも、カルボキシル基、カルボン酸無水物基又はエポキシ基を含有しない他の熱可塑性樹脂との接着性にも優れている。
得られた単層又は多層のフィルム等を延伸することで本発明の熱収縮フィルム等を製造する。延伸は、一軸延伸であってもよいし、二軸延伸であってもよい。二軸延伸は、同時二軸延伸であってもよいし、逐次二軸延伸であってもよい。延伸方法としては、テンター延伸法、チューブラー延伸法、ロール延伸法などが例示される。本発明の熱収縮フィルム等は、高い倍率で延伸されたものであることが好適である。具体的には、面積倍率7倍以上に延伸されてなる熱収縮フィルムが特に好適である。延伸温度は、通常50~130℃である。フィルム等を延伸する前に、放射線照射などによる架橋を施してもよい。収縮性をより高める観点からは、フィルム等を延伸した後に、速やかに冷却することが好適である。
本発明の熱収縮フィルム等は、食品包装容器、医薬品包装容器、工業薬品包装容器、農薬包装容器等の各種包装容器の材料として好適に用いられる。
<包装材A>
本発明の包装材Aは、本発明のフィルムもしくはシートまたは熱収縮フィルムもしくはシートを備える。当該包装材Aは、本発明のフィルムもしくはシートまたは熱収縮フィルムもしくはシートからなる包装材であってよい。すなわち、本発明の成形体の一実施形態は、包装材であってよい。当該包装材Aは、環境負荷が低く、ガスバリア性、外観及び生産性も良好である。
本発明の包装材Aは、本発明のフィルムもしくはシートまたは熱収縮フィルムもしくはシートを備える。当該包装材Aは、本発明のフィルムもしくはシートまたは熱収縮フィルムもしくはシートからなる包装材であってよい。すなわち、本発明の成形体の一実施形態は、包装材であってよい。当該包装材Aは、環境負荷が低く、ガスバリア性、外観及び生産性も良好である。
本発明の包装材Aは、単層フィルム等であってもよく、多層フィルム等であってもよい。また、多層フィルム等は、樹脂以外から形成される層、例えば紙層、金属層等をさらに有していてもよい。当該包装材Aは、フィルムまたはシート形状のままのものであってもよいし、フィルム又はシートが二次加工されたものであってもよい。二次加工することで得られる包装材としては例えば、(1)フィルム又はシートを真空成形、圧空成形、真空圧空成形等、熱成形加工することにより得られるトレーカップ状容器、(2)フィルム又はシートにストレッチブロー成形等を行って得られるボトル、カップ状容器、(3)フィルム又はシートをヒートシールすることにより得られる袋状容器等が挙げられる。なお、二次加工法は、上記に例示した各方法に限定されることなく、例えば、ブロー成形等の上記以外の公知の二次加工法を適宜用いることができる。
本発明の包装材Aは、例えば食品、飲料物、農薬や医薬等の薬品、医療器材、機械部品、精密材料等の産業資材、衣料などを包装するために使用される。特に、当該包装材Aは、酸素に対するバリア性が必要となる用途、包装材の内部が各種の機能性ガスによって置換される用途に好ましく使用される。当該包装材Aは、用途に応じて種々の形態、例えば縦製袋充填シール袋、真空包装袋、スパウト付パウチ、ラミネートチューブ容器、容器用蓋材等に形成される。
(真空包装袋)
本発明の包装材Aは真空包装袋であってもよい。真空包装袋の一例としては、内容物が包装される内部と、外部とを隔てる隔壁として本発明のフィルム等を備え、上記内部が減圧された状態となっている袋状の容器である。真空包装袋においては、例えば本発明の2枚のフィルム等が重なり合わされ、この2枚のフィルム等の周縁部が互いにシールされている。真空包装袋においては、上記隔壁としては、多層フィルム等が好ましい。真空包装袋は、ノズル式又はチャンバー式の真空包装機を用いて製造することができる。
本発明の包装材Aは真空包装袋であってもよい。真空包装袋の一例としては、内容物が包装される内部と、外部とを隔てる隔壁として本発明のフィルム等を備え、上記内部が減圧された状態となっている袋状の容器である。真空包装袋においては、例えば本発明の2枚のフィルム等が重なり合わされ、この2枚のフィルム等の周縁部が互いにシールされている。真空包装袋においては、上記隔壁としては、多層フィルム等が好ましい。真空包装袋は、ノズル式又はチャンバー式の真空包装機を用いて製造することができる。
当該真空包装袋は、真空状態で包装することが望まれる用途、例えば食品、飲料物等の保存に使用される。また、当該真空包装袋は、真空断熱体の外包材として用いることもできる。
<産業用フィルムまたはシート>
本発明の産業用フィルムまたはシート(産業用フィルムまたは産業用シート)は、本発明の単層又は多層フィルム等の成形体を備える。当該産業用フィルム等は、本発明の成形体からなる産業用フィルム等であってよい。すなわち、本発明の成形体の一実施形態は、産業用フィルム等であってよい。当該産業用フィルム等は環境負荷が低く、ガスバリア性、外観及び生産性も良好である。産業用フィルム等の具体例としては、農業用フィルム等、埋立用フィルム等、建築用フィルム等があげられる。
本発明の産業用フィルムまたはシート(産業用フィルムまたは産業用シート)は、本発明の単層又は多層フィルム等の成形体を備える。当該産業用フィルム等は、本発明の成形体からなる産業用フィルム等であってよい。すなわち、本発明の成形体の一実施形態は、産業用フィルム等であってよい。当該産業用フィルム等は環境負荷が低く、ガスバリア性、外観及び生産性も良好である。産業用フィルム等の具体例としては、農業用フィルム等、埋立用フィルム等、建築用フィルム等があげられる。
本発明の産業用フィルム等は、多層フィルム等であることが好ましい。当該産業用フィルム等に備わる層(2)には、水分による層(1)のガスバリア性能の低下を防ぐ目的で、疎水性熱可塑性樹脂が好ましく用いられる。具体的には、ポリオレフィン系樹脂:直鎖状低密度ポリエチレン、低密度ポリエチレン、超低密度ポリエチレン、超低密度直鎖状ポリエチレン、中密度ポリエチレン、高密度ポリエチレンなどのポリエチレン類、およびエチレン-α-オレフィン共重合体などのポリエチレン系樹脂、ポリプロピレン、エチレン-プロピレン(ブロックおよびランダム)共重合体、プロピレン-α-オレフィン(炭素数4~20のα-オレフィン)共重合体などのポリプロピレン系樹脂、ポリブテン、ポリペンテンなど;これらポリオレフィンを不飽和カルボン酸またはそのエステルでグラフト変性したグラフト化ポリオレフィン、環状ポリオレフィン系樹脂;アイオノマー、エチレン-酢酸ビニル共重合体、エチレン-アクリル酸共重合体、エチレン-アクリル酸エステル共重合体、ポリエステル系樹脂、ポリアミド系樹脂、ポリ塩化ビニル、ポリ塩化ビニリデン、アクリル系樹脂、ポリスチレン、ビニルエステル系樹脂、ポリエステルエラストマー、ポリウレタンエラストマー、塩素化ポリエチレン、塩素化ポリプロピレンなどのハロゲン化ポリオレフィン、芳香族または脂肪族ポリケトンなどが挙げられ、中でも機械的強度や成形加工性の点で、ポリオレフィン系樹脂が好ましく、ポリエチレン、ポリプロピレンが特に好ましい。
上記疎水性熱可塑性樹脂の溶融粘度に関し、210℃、2160g荷重下におけるMFRの下限値としては、1.0g/10分であることが好ましく、2.0g/10分であることがより好ましく、上限値としては、100g/10分であることが好ましく、60g/10分であることがより好ましい。このような溶融粘度の疎水性熱可塑性樹脂組成物を用いることで、層乱れのない良好な多層フィルム等を得ることができる。
本発明の産業用フィルム等の層構造としては、以下の層構成が例示できる。層構成は左側のものほど外側(外部の環境にさらされる側)の層となることを表す。
5層 1/3/2/3/1、2/3/1/3/2、2/3/1/3/1
6層 2/3/1/3/2/2
7層 2/3/1/3/1/3/2、2/2/3/1/3/2/2
5層 1/3/2/3/1、2/3/1/3/2、2/3/1/3/1
6層 2/3/1/3/2/2
7層 2/3/1/3/1/3/2、2/2/3/1/3/2/2
特に水分による酸素バリア性の低下を防ぐ目的で、中間層として層(1)を用い、外層として層(2)を用いた構成が好ましく、2/3/1/3/2、2/2/3/1/3/2/2等の構成がより好ましい。
本発明の産業用フィルム等の厚みとしては、その全厚みが通常5~5mm、好ましくは10~4.5mm、より好ましくは15~4mm、特に好ましくは20~3.5mmである。また、産業用フィルム等中の層(2)(疎水性樹脂組成物層等)の厚みは、特に限定しないが、通常0.5~2.5mm、好ましくは1~2mm、特に好ましくは1~1.5mmである。層(1)の厚みは、特に限定しないが、全層厚みの1~20%、好ましくは2~18%、より好ましくは3~15%の範囲であることが好ましい。
上記建築用フィルム等としては、例えば壁紙等があげられる。本発明の産業用フィルム等の一実施形態としての壁紙は、環境負荷が小さく、生産性に優れる。
上記埋立用フィルム等としては、ジオメンブレンやランドフィルシート等があげられる。ジオメンブレンとは、廃棄物処理場などの遮水工として使用されるシートである。また、ランドフィルシートとは、産業廃棄物等から出てくる有害物質の拡散を防止するシートであり、例えば、ラドンガスの拡散を防止するために用いることができる。
上記農業用フィルム等においては、ガスバリア樹脂組成物に酸化防止剤または耐紫外線剤(紫外線吸収剤、光安定剤、着色剤)等を含むことが、屋外での長期使用が可能となる観点から好ましい。上記農業用フィルム等は、多層フィルム等であることが好ましく、層(2)としては、水分による層(1)のガスバリア性能の低下を防ぐ目的で、疎水性熱可塑性樹脂が好ましく用いられる。
層(2)は、耐紫外線剤や粘着性成分を配合することが好ましい。耐紫外線剤としては、例えば紫外線吸収剤、光安定剤、着色剤などが挙げられる。
上記の耐紫外線剤の疎水性熱可塑性樹脂に対する配合量は上記疎水性熱可塑性樹脂に対して通常1~10質量%、好ましくは2~8質量%、特に好ましくは3~5質量%である。配合量が上記範囲より少ない場合、紫外線によって疎水性熱可塑性樹脂が劣化しやすくなる。一方、配合量が上記範囲より多い場合、疎水性熱可塑性樹脂の機械的強度が低下する。
上記の粘着性成分としては、ポリイソブテンなどの脂肪族飽和炭化水素樹脂や、脂環族飽和炭化水素樹脂などが挙げられ、上記疎水性熱可塑性樹脂に対する配合量は通常1~30質量%、好ましくは2~20質量%、特に好ましくは3~15質量%である。配合量が適切であれば上記農業用フィルム等を用いてラッピングする際にフィルム等同士が圧着され、密封が維持されやすくなる。配合量が上記範囲より少ない場合、フィルム等間に隙間が発生し、内部に空気が侵入するため、内容物の長期保管性が悪くなる。また配合量が上記範囲より多い場合、多層フィルムのブロッキングが起こり、フィルムロール等から巻き出すことが出来なくなる。
上記農業用フィルム等の厚みとしては、その全厚みが通常5~200μm、好ましくは10~150μm、より好ましくは15~100μm、特に好ましくは20~50μmである。また、農業用フィルム等中の層(2)(疎水性樹脂組成物層等)の厚みは、特に限定しないが、通常0.5~200μm、好ましくは1~100μm、特に好ましくは1~10μmである。層(1)の厚みは、特に限定しないが、全層厚みの1~20%、好ましくは2~18%、より好ましくは3~15%の範囲であることが好ましい。
農業用フィルムは、例えばラージサイロフィルム、土壌燻蒸用フィルム、穀物保存バッグ、サイレージフィルム、ビニルハウス用フィルム、マルチフィルム等、農業の分野で用いられる各種フィルムに用いることができる。
(ラージサイロフィルム)
ラージサイロフィルムとは、穀物を包んで保存するためのフィルムである。ラージサイロフィルムで穀物を包んで保存することで、穀物を湿気、カビ、虫等から守ることができる。ラージサイロフィルムの一辺の長さは、例えば1m以上であり、2m、3m、5m又は10m以上であってもよい。また、ラージサイロフィルムは袋状等に加工されていてもよい。
ラージサイロフィルムとは、穀物を包んで保存するためのフィルムである。ラージサイロフィルムで穀物を包んで保存することで、穀物を湿気、カビ、虫等から守ることができる。ラージサイロフィルムの一辺の長さは、例えば1m以上であり、2m、3m、5m又は10m以上であってもよい。また、ラージサイロフィルムは袋状等に加工されていてもよい。
(土壌燻蒸用フィルム)
土壌燻蒸用フィルムは、畑等において土壌燻蒸を行うとき、土壌燻蒸剤の蒸散防止のためなどに用いられるフィルムである。
土壌燻蒸用フィルムは、畑等において土壌燻蒸を行うとき、土壌燻蒸剤の蒸散防止のためなどに用いられるフィルムである。
(穀物保存バッグ)
穀物保存バッグは、穀物を湿気、カビ、虫等から守るために、穀物を保存するバッグであり、ヘルメティックバッグ(Hermetic bag)等とも称される。当該穀物保存バッグは、本発明のガスバリア樹脂組成物からなる層を少なくとも一層有するフィルムをバッグ状、袋状、その他容器状に成形したものである。当該穀物保存バッグが例えば方形の袋状である場合、その一辺の長さは例えば0.5~2mである。
穀物保存バッグは、穀物を湿気、カビ、虫等から守るために、穀物を保存するバッグであり、ヘルメティックバッグ(Hermetic bag)等とも称される。当該穀物保存バッグは、本発明のガスバリア樹脂組成物からなる層を少なくとも一層有するフィルムをバッグ状、袋状、その他容器状に成形したものである。当該穀物保存バッグが例えば方形の袋状である場合、その一辺の長さは例えば0.5~2mである。
(サイレージフィルム)
サイレージフィルムとは、サイレージの製造・包装等に用いられるフィルムである。牧草などを嫌気性条件下で発酵させて得られるサイレージは、家畜の飼料等に用いられる。サイレージフィルムを用いるサイロの形態は特に限定されず、バンカーサイロ、地下型(もしくは半地下型)サイロ、バッグサイロ、チューブサイロ、スタックサイロ、ラップサイロなどの種々の形態が挙げられる。
サイレージフィルムとは、サイレージの製造・包装等に用いられるフィルムである。牧草などを嫌気性条件下で発酵させて得られるサイレージは、家畜の飼料等に用いられる。サイレージフィルムを用いるサイロの形態は特に限定されず、バンカーサイロ、地下型(もしくは半地下型)サイロ、バッグサイロ、チューブサイロ、スタックサイロ、ラップサイロなどの種々の形態が挙げられる。
例えばラップサイロを作成する場合には、まず牧草を所望の容量(たとえば0.1~50m3、好ましくは1~30m3)に成形する。例えば円筒状のロールベールサイロの場合、その大きさは通常直径0.5~3m、好ましくは1~2m、高さは通常0.5~3m、好ましくは1~2mである。ついで、通常のラップ巻きつけ機を使用してサイレージフィルムを成形した牧草に巻きつけ、牧草を密封する。
<チューブ>
本発明のチューブは、本発明の成形体を備える。当該チューブは、本発明の成形体からなるチューブであってよい。すなわち、本発明の成形体の一実施形態は、チューブであってよい。当該チューブは、環境負荷が低く、ガスバリア性、外観及び生産性も良好である。
本発明のチューブは、本発明の成形体を備える。当該チューブは、本発明の成形体からなるチューブであってよい。すなわち、本発明の成形体の一実施形態は、チューブであってよい。当該チューブは、環境負荷が低く、ガスバリア性、外観及び生産性も良好である。
本発明のチューブの製造方法は特に限定されず、例えば、共押出成形、共射出成形、押出コーティング等の溶融成形により、直接チューブ状に成形する方法、本発明のフィルムまたはシートを熱溶着してチューブ状に成形する方法、本発明のフィルムまたはシートを、接着剤を用いてラミネートしてチューブ状に成形する方法等が挙げられる。
<パイプ>
本発明のパイプは、本発明の成形体を備える。当該パイプは、本発明の成形体からなるパイプであってよい。すなわち、本発明の成形体の一実施形態は、パイプであってよい。当該パイプは環境負荷が低く、ガスバリア性、外観及び生産性も良好である。当該パイプは、長期使用における酸化劣化を抑制する観点から、ガスバリア樹脂組成物に酸化防止剤(D)を含むことが好ましい。上記酸化防止剤としては、高温での使用における酸化劣化を抑制する観点からヒンダードアミン基を有する化合物及び/またはヒンダードフェノール基を有する化合物であることが好ましい。また、当該パイプは、耐屈曲性の観点から、ガスバリア性樹脂組成物に熱可塑性エラストマー(G)を含むことが好ましく、変性熱可塑性エラストマー(g2)を含むことがより好ましい。
本発明のパイプは、本発明の成形体を備える。当該パイプは、本発明の成形体からなるパイプであってよい。すなわち、本発明の成形体の一実施形態は、パイプであってよい。当該パイプは環境負荷が低く、ガスバリア性、外観及び生産性も良好である。当該パイプは、長期使用における酸化劣化を抑制する観点から、ガスバリア樹脂組成物に酸化防止剤(D)を含むことが好ましい。上記酸化防止剤としては、高温での使用における酸化劣化を抑制する観点からヒンダードアミン基を有する化合物及び/またはヒンダードフェノール基を有する化合物であることが好ましい。また、当該パイプは、耐屈曲性の観点から、ガスバリア性樹脂組成物に熱可塑性エラストマー(G)を含むことが好ましく、変性熱可塑性エラストマー(g2)を含むことがより好ましい。
当該パイプの用途は特に限定されず、例えば温水循環用パイプ、断熱多層パイプ、燃料パイプ等として使用することができる。
本発明のパイプは、単層パイプであってもよく、多層パイプであってもよいが、多層パイプであることが好ましい。多層パイプの層構成としては、上記成形体(多層構造体A)の層構成を採用することができる。
<温水循環用パイプ>
多層パイプが温水循環用パイプとして用いられる場合には、層(2)を最外層とする層(1)/層(3)/層(2)の3層構成が一般的に採用される。これは、既存の架橋ポリオレフィンなど単層パイプの製造ラインに、ガスバリア樹脂組成物と接着性樹脂の共押出コーティング設備を付加することにより、容易に多層パイプの製造ラインに転用でき、実際に多くのパイプメーカーがこの構成を採用しているためである。
多層パイプが温水循環用パイプとして用いられる場合には、層(2)を最外層とする層(1)/層(3)/層(2)の3層構成が一般的に採用される。これは、既存の架橋ポリオレフィンなど単層パイプの製造ラインに、ガスバリア樹脂組成物と接着性樹脂の共押出コーティング設備を付加することにより、容易に多層パイプの製造ラインに転用でき、実際に多くのパイプメーカーがこの構成を採用しているためである。
層(1)の両側にポリオレフィン層などを設けて、層(1)を中間層として使用することは、層(1)の傷付き防止などに有効である。しかしながら、多層パイプを床暖房パイプなどの温水循環用パイプとして用いる場合には、通常床下に埋設されるため、物理的な衝撃による層(1)の傷付きなどのリスクは比較的小さく、むしろガスバリア性の観点から、層(1)を最外層に配することが望ましい。ガスバリア樹脂組成物は大きな湿度依存性を示し、高湿度条件下ではバリア性が低下する。このため、層(1)を最外層に配することにより、主として層(1)が水と接触するパイプ内表面より最も遠い場所に位置することとなり、多層パイプのバリア性能面からは最も有利な層構成となる。一方で、一般的にEVOH層を最外層に配する場合、空気と直接接触するため、酸化劣化の影響を受けやすい。このような環境下において、ヒンダードアミン基を有する化合物及び/またはヒンダードフェノール基を有する酸化防止剤を含むガスバリア樹脂組成物を用いた場合、高温下でも酸化劣化しにくい最外層に配することとなるため、良好なバリア性を有しつつ酸化劣化によるクラックの発生を低減した多層パイプを提供するという効果がより有効に発揮される。
<断熱多層パイプ>
多層パイプが地域冷暖房などの断熱多層パイプに用いられる場合には、層(1)を層(2)より内側に配する層(2)/層(3)/層(1)の3層構成(以下、積層体1と略称することがある)、もしくは層(1)の傷付き防止の観点から層(2)/層(3)/層(1)/層(3)/層(2)の5層構成(以下、積層体2と略称することがある)を有することが好ましい。
多層パイプが地域冷暖房などの断熱多層パイプに用いられる場合には、層(1)を層(2)より内側に配する層(2)/層(3)/層(1)の3層構成(以下、積層体1と略称することがある)、もしくは層(1)の傷付き防止の観点から層(2)/層(3)/層(1)/層(3)/層(2)の5層構成(以下、積層体2と略称することがある)を有することが好ましい。
地域冷暖房などの断熱多層パイプは、層(1)(ガスバリア樹脂組成物から形成される層)に加え、通常、断熱発泡体層をさらに備える。断熱多層パイプの構成は特に限定されないが、例えば、内側から、内管、内管の周りを覆う断熱発泡体層、そして外層として上記積層体1又は2の順で配置されることが好ましい。
内管に使われるパイプの種類(素材)、形状及び大きさは、ガスや液体などの熱媒体を輸送できるものであれば特に限定はなく、熱媒体の種類や、配管材の用途及び使用形態等に応じて適宜選択することができる。具体的には、鋼、ステンレス、アルミニウム等の金属、ポリオレフィン(ポリエチレン、架橋ポリエチレン(PEX)、ポリプロピレン、ポリ1-ブテン、ポリ4-メチル-1-ペンテンなど)、及び上記積層体1又は2などが挙げられ、これらの中でも架橋ポリエチレン(PEX)が好適に用いられる。
断熱発泡体には、ポリウレタンフォーム、ポリエチレンフォーム、ポリスチレンフォーム、フェノールフォーム、ポリイソシアヌレートフォームを用いることができ、断熱性能向上の観点から、ポリウレタンフォームが好適に用いられる。
断熱発泡体の発泡剤としてはフロンガス、各種代替フロン、水、塩化炭化水素、炭化水素、二酸化炭素等が用いられるが、発泡効果、環境への影響の観点から炭化水素、具体的にはn-ペンタン、シクロペンタンが好適に用いられる。
断熱多層パイプの製造方法としては、例えば、熱媒体を輸送する内管を、パイプ状の外層の中に入れて内管をスペーサーで固定し二重管とした後、内管と外層の間隙部に各種発泡体原液を注入し、発泡及び固化させる方法が挙げられる。上記スペーサーの素材は特に限定されないが、スペーサーによる内管及び外層への傷を減らすため、ポリエチレン又はポリウレタンであることが好ましい。
(燃料用パイプ)
本発明のパイプが燃料用パイプに用いられる場合には、本発明のガスバリア樹脂組成物からなる層は、熱可塑性エラストマー(G)をさらに含有することがより好ましい場合がある。熱可塑性エラストマー(G)を含有することにより、パイプの耐クラック性等がより高まる。
本発明のパイプが燃料用パイプに用いられる場合には、本発明のガスバリア樹脂組成物からなる層は、熱可塑性エラストマー(G)をさらに含有することがより好ましい場合がある。熱可塑性エラストマー(G)を含有することにより、パイプの耐クラック性等がより高まる。
燃料用パイプに用いられる場合には、最内側層は導電性であるように形成される。そのために、最内側層の熱可塑性樹脂に、導電性添加物、例えば、カーボンブラックまたはグラファイト繊維が混合される。
(パイプの製造方法)
以下、本発明のパイプ、具体的には多層パイプの製造方法について説明する。多層パイプは、例えば、架橋ポリオレフィンなどの単層パイプの上にガスバリア樹脂組成物と接着性樹脂を共押出コーティングすることにより製造することができる。単層パイプ上にガスバリア樹脂組成物と接着性樹脂の共押出コーティングを実施する際は、単純に単層パイプ上にガスバリア樹脂組成物と接着性樹脂の溶融したフィルムをコートしても良いが、単層パイプとコート層の間の接着力が不十分な場合があり、長期間の使用中にコート層が剥離してガスバリア性を失う可能性がある。その対策としては、コート前にコートするパイプの表面をフレーム処理及び/又はコロナ放電処理することが有効である。
以下、本発明のパイプ、具体的には多層パイプの製造方法について説明する。多層パイプは、例えば、架橋ポリオレフィンなどの単層パイプの上にガスバリア樹脂組成物と接着性樹脂を共押出コーティングすることにより製造することができる。単層パイプ上にガスバリア樹脂組成物と接着性樹脂の共押出コーティングを実施する際は、単純に単層パイプ上にガスバリア樹脂組成物と接着性樹脂の溶融したフィルムをコートしても良いが、単層パイプとコート層の間の接着力が不十分な場合があり、長期間の使用中にコート層が剥離してガスバリア性を失う可能性がある。その対策としては、コート前にコートするパイプの表面をフレーム処理及び/又はコロナ放電処理することが有効である。
多層パイプを製造するためのその他の多層成形方法としては、樹脂層の種類に対応する数の押出機を使用し、この押出機内で溶融された樹脂の流れを重ねあわせた層状態で同時押出成形する、いわゆる共押出成形により実施する方法が挙げられる。また、ドライラミネーションなどの多層成形方法も採用され得る。
多層パイプの製造方法は、成形直後に10~70℃の水で冷却を行う工程を含むとよい。すなわち、溶融成形後、層(1)が固化する前に10~70℃の水で冷却することにより、層(1)を固化させることが望ましい。冷却水の温度が低すぎると、続く二次加工工程において多層パイプを屈曲させる場合に、屈曲部の層(1)に歪みによるクラックが生じやすい。歪みによるクラックが生じやすくなる原因の詳細は明らかでないが、成形物中の残留応力が影響しているものと推測される。この観点から、冷却水の温度は15℃以上がより好ましく、20℃以上がさらに好ましい。一方、冷却水の温度が高すぎても、二次加工の際に屈曲部の層(1)に歪みによるクラックを生じやすい。この原因の詳細も十分に解明されていないが、層(1)の結晶化度が大きくなりすぎるためと推定される。この観点より冷却水の温度は60℃以下がより好ましく、50℃以下がさらに好ましい。
上記の方法で得られた多層パイプを二次加工することにより、各種成形体を得ることができる。二次加工法としては、特に限定されず、公知の二次加工法を適宜用いることができるが、例えば、多層パイプを80~160℃に加熱した後所望の形に変形させた状態で、1分~2時間固定することにより加工する方法が挙げられる。
<熱成形容器>
本発明の熱成形容器は、本発明の成形体を備える。当該熱成形容器は、本発明の成形体からなる熱成形容器であってよい。すなわち、本発明の成形体の一実施形態は、熱成形容器であってよい。当該熱成形容器は、環境負荷が低く、ガスバリア性、外観及び生産性も良好である。当該熱成形容器は、酸素バリア性が要求される用途、例えば食品、化粧品、医化学薬品、トイレタリー等の種々の分野で利用される。この熱成形容器は、例えば単層または多層のフィルムまたはシートを熱成形することで、収容部を有するものとして形成される。
本発明の熱成形容器は、本発明の成形体を備える。当該熱成形容器は、本発明の成形体からなる熱成形容器であってよい。すなわち、本発明の成形体の一実施形態は、熱成形容器であってよい。当該熱成形容器は、環境負荷が低く、ガスバリア性、外観及び生産性も良好である。当該熱成形容器は、酸素バリア性が要求される用途、例えば食品、化粧品、医化学薬品、トイレタリー等の種々の分野で利用される。この熱成形容器は、例えば単層または多層のフィルムまたはシートを熱成形することで、収容部を有するものとして形成される。
(収容部)
収容部は、食品等の内容物を収容する部分である。この収容部の形状は、内容物の形状に対応して決定される。具体的には、当該熱成形容器は、例えばカップ状容器、トレイ状容器、バッグ状容器、ボトル状容器、パウチ状容器等として形成される。
収容部は、食品等の内容物を収容する部分である。この収容部の形状は、内容物の形状に対応して決定される。具体的には、当該熱成形容器は、例えばカップ状容器、トレイ状容器、バッグ状容器、ボトル状容器、パウチ状容器等として形成される。
収容部の形態は、一つの指標として、絞り比(S)で表すことができる。ここで、絞り比(S)とは、容器の最深部の深さを容器の開口に内接する最大径の円の直径で割った値である。すなわち、絞り比(S)は、値が大きいほど底の深い容器であり、値が小さいほど底が浅い容器であることを意味する。例えば、熱成形容器がカップ状である場合には、絞り比(S)が大きく、トレイである場合には絞り比(S)が小さい。なお、内接する最大径の円の直径は、例えば収容部の開口が円形である場合には円の直径、楕円である場合には短径(短軸長さ)、長方形である場合には短辺の長さである。
絞り比(S)は、フィルムまたはシート厚みによって好適値が異なる。本発明の熱成形容器がフィルムを熱成形したものである場合、絞り比(S)としては0.2以上が好ましく、0.3以上がより好ましく、0.4以上がさらに好ましい。一方、当該熱成形容器がシートを成形したものである場合、絞り比(S)としては0.3以上が好ましく、0.5以上がより好ましく、0.8以上がさらに好ましい。
本発明の熱成形容器において、層(1)の一方の面側に積層される他の層の合計厚みIと、層(1)の他方の面側に積層される他の層の合計厚みOとの厚み比(I/O)の下限としては、1/99が好ましく、30/70がより好ましい。また、上記I/Oの上限としては、70/30が好ましく、55/45がより好ましい。なお、熱成形容器の全層又は単層の厚みは、ミクロトームを用いて熱成形容器の複数箇所から切り出したサンプルについて、光学顕微鏡観察により測定した厚みの平均値である。
本発明の熱成形容器の全体平均厚みの下限としては、300μmが好ましく、500μmがより好ましく、700μmがさらに好ましい。また、当該熱成形容器の全体平均厚みの上限としては、10000μmが好ましく、8500μmがより好ましく、7000μmがさらに好ましい。なお、全体平均厚みは、熱成形容器の収容部における全層の厚みをいう。全体平均厚みを上記上限以下とすることで、熱成形容器の製造コストを抑制することができる。一方、全体平均厚みを上記下限以上とすることで、十分な剛性を保ち、熱成形容器が破壊され難くなる。
(熱成形容器に用いる多層シートの製造方法)
本発明の熱成形容器の製造に用いる単層または多層のフィルム等の一つである多層シートの製造方法について説明する。多層シートは、共押出成形装置を用いて形成できる。この多層シートは、例えば各層を形成するガスバリア樹脂組成物や他の樹脂などをそれぞれ別々の押出機に仕込み、これらの押出機で共押出することで所定の層構成を有するものとして形成できる。
本発明の熱成形容器の製造に用いる単層または多層のフィルム等の一つである多層シートの製造方法について説明する。多層シートは、共押出成形装置を用いて形成できる。この多層シートは、例えば各層を形成するガスバリア樹脂組成物や他の樹脂などをそれぞれ別々の押出機に仕込み、これらの押出機で共押出することで所定の層構成を有するものとして形成できる。
各層の押出成形は、一軸スクリューを備えた押出機を所定の温度で運転することにより行われる。層(1)を形成する押出機の温度は、例えば170℃以上260℃以下とされる。また、層(2)~層(4)を形成する押出機の温度は、例えば150℃以上260℃以下とされる。
(熱成形)
本発明の熱成形容器は、多層シート等を加熱して軟化させた後に、金型形状に成形することで形成できる。熱成形方法としては、例えば真空又は圧空を用い、必要によりプラグを併せ用いて金型形状に成形する方法(ストレート法、ドレープ法、エアスリップ法、スナップバック法、プラグアシスト法等)、プレス成形する方法などが挙げられる。成形温度、真空度、圧空の圧力、成形速度等の各種成形条件は、プラグ形状や金型形状、原料樹脂の性質等により適当に設定される。
本発明の熱成形容器は、多層シート等を加熱して軟化させた後に、金型形状に成形することで形成できる。熱成形方法としては、例えば真空又は圧空を用い、必要によりプラグを併せ用いて金型形状に成形する方法(ストレート法、ドレープ法、エアスリップ法、スナップバック法、プラグアシスト法等)、プレス成形する方法などが挙げられる。成形温度、真空度、圧空の圧力、成形速度等の各種成形条件は、プラグ形状や金型形状、原料樹脂の性質等により適当に設定される。
成形温度は、成形するのに十分なだけ樹脂が軟化できる温度であれば特に限定されず、多層シート等の構成によってその好適な温度範囲は異なる。なお、この加熱温度は、通常、樹脂の融点よりも低い。具体的な多層シート等の加熱温度の下限は通常50℃であり、60℃が好ましく、70℃がより好ましい。加熱温度の上限は例えば180℃であり、160℃であってもよい。
(熱成形容器の層構成)
本発明の熱成形容器は、少なくとも層(1)を備えていればよく、単層からなってもよいし複数層からなってもよい。熱成形容器が複数層である場合の層構成は、用途等に応じて適宜設定すればよい。
本発明の熱成形容器は、少なくとも層(1)を備えていればよく、単層からなってもよいし複数層からなってもよい。熱成形容器が複数層である場合の層構成は、用途等に応じて適宜設定すればよい。
本発明の熱成形容器が複数層からなる場合の層構成としては、層(2)を最外層に配置することが好ましい。すなわち、収容部の内表面から外表面に向かって、層(2)/層(3)/層(1)/層(3)/層(2)(以下、「(内表面)(2)/(3)/(1)/(3)/(2)(外表面)」のように表記する)が耐衝撃性の観点から好ましい。また、回収層である層(4)を含む場合の層構成としては、例えば
(内表面)(2)/(3)/(1)/(3)/(4)/(2)(外表面)、
(内表面)(2)/(4)/(3)/(1)/(3)/(4)/(2)(外表面)、
(内表面)(4)/(3)/(1)/(3)/(4)(外表面)
等が挙げられる。なお、これらの層構成において層(2)の代わりに層(4)を備える層構成であってもよい。なお、層(1)~層(4)がそれぞれ複数用いられている場合、それぞれの層を構成する樹脂は同一でも異なっていてもよい。
(内表面)(2)/(3)/(1)/(3)/(4)/(2)(外表面)、
(内表面)(2)/(4)/(3)/(1)/(3)/(4)/(2)(外表面)、
(内表面)(4)/(3)/(1)/(3)/(4)(外表面)
等が挙げられる。なお、これらの層構成において層(2)の代わりに層(4)を備える層構成であってもよい。なお、層(1)~層(4)がそれぞれ複数用いられている場合、それぞれの層を構成する樹脂は同一でも異なっていてもよい。
<カップ状容器>
次に、本発明の熱成形容器について、図1及び図2に示すカップ状容器を例にとって、具体的に説明する。但し、カップ状容器は熱成形容器の一例に過ぎず、以下のカップ状容器の説明は、本発明の範囲を限定するものではない。
次に、本発明の熱成形容器について、図1及び図2に示すカップ状容器を例にとって、具体的に説明する。但し、カップ状容器は熱成形容器の一例に過ぎず、以下のカップ状容器の説明は、本発明の範囲を限定するものではない。
図1及び図2のカップ状容器1は、収容部としてのカップ本体2、及びフランジ部3を備える。このカップ状容器1は、カップ本体2に内容物を収容し、カップ本体2の開口4を塞ぐようにフランジ部3に蓋7をシールすることで使用される。この蓋7としては、例えば樹脂フィルム、金属箔、金属樹脂複合フィルム等が挙げられ、これらの中で、樹脂フィルムに金属層を積層した金属樹脂複合フィルムが好ましい。樹脂フィルムとしては、例えばポリエチレンフィルム、ポリエチレンテレフタレートフィルム等が挙げられる。金属層は特に限定されず、金属箔及び金属蒸着層が好ましく、ガスバリア性及び生産性の観点からアルミニウム箔がより好ましい。
カップ状容器1は、通常、多層シートを熱成形することで得られる。この多層シートは、少なくとも層(1)を備え、この層(1)に他の層が積層されることが好ましい。他の層としては、例えば層(2)、層(3)、層(4)等が挙げられる。多層シートの層構造の具体例は、上述した通りである。
(カップ状容器の製造方法)
カップ状容器1は、図3に示すように連続多層シート21を加熱装置30により加熱して軟化させた後に、金型装置40を用いて熱成形することで製造される。
カップ状容器1は、図3に示すように連続多層シート21を加熱装置30により加熱して軟化させた後に、金型装置40を用いて熱成形することで製造される。
(加熱装置)
加熱装置30は、一対のヒーター(ヒーター31及びヒーター32)を備えるものであり、これらのヒーター31及びヒーター32の間を連続多層シート21が通過可能とされている。なお、加熱装置30としては、熱プレスにより加熱するものを用いることもできる。
加熱装置30は、一対のヒーター(ヒーター31及びヒーター32)を備えるものであり、これらのヒーター31及びヒーター32の間を連続多層シート21が通過可能とされている。なお、加熱装置30としては、熱プレスにより加熱するものを用いることもできる。
(金型装置)
金型装置40は、プラグアシスト法による熱成形に適するものであり、チャンバー(図示略)内に収容される下型50及び上型51を備える。下型50及び上型51は、それぞれ個別に上下方向に移動可能であり、離間状態において、これらの下型50及び上型51の間を連続多層シート21が通過可能とされている。下型50は、カップ状容器1の収容部を形成するための複数の凹部52を有する。上型51は、下型50に向けて突出する複数のプラグ53を備える。複数のプラグ53は、下型50の複数の凹部52に対応した位置に設けられている。各プラグ53は、対応する凹部52に挿入可能である。
金型装置40は、プラグアシスト法による熱成形に適するものであり、チャンバー(図示略)内に収容される下型50及び上型51を備える。下型50及び上型51は、それぞれ個別に上下方向に移動可能であり、離間状態において、これらの下型50及び上型51の間を連続多層シート21が通過可能とされている。下型50は、カップ状容器1の収容部を形成するための複数の凹部52を有する。上型51は、下型50に向けて突出する複数のプラグ53を備える。複数のプラグ53は、下型50の複数の凹部52に対応した位置に設けられている。各プラグ53は、対応する凹部52に挿入可能である。
(熱成形)
まず、図3及び図4(A)に示すように、加熱装置30により軟化させた連続多層シート21に対して、下型50を上動させることで下型50に密着させると共に連続多層シート21を若干持ち上げて連続多層シート21にテンションを付与する。次に、図4(B)に示すように、上型51を下動させることでプラグ53を凹部52に挿入する。
まず、図3及び図4(A)に示すように、加熱装置30により軟化させた連続多層シート21に対して、下型50を上動させることで下型50に密着させると共に連続多層シート21を若干持ち上げて連続多層シート21にテンションを付与する。次に、図4(B)に示すように、上型51を下動させることでプラグ53を凹部52に挿入する。
続いて、図4(C)に示すように、上型51を上動させてプラグ53を凹部52から離間させた後にチャンバー(図示略)内を真空引きし、連続多層シート21を凹部52の内面に密着させる。その後、エアーの噴射により成形部を冷却することで形状を固定する。続いて、図4(D)に示すように、チャンバー(図示略)内を大気開放すると共に下型50を下動させて下型50を離型することで一次成形品が得られる。この一次成形品を切断することで、図1及び図2に示すカップ状容器1が得られる。
<熱成形容器のその他の実施形態>
本発明の熱成形容器は、上述した形態に限定されず、トレイ状容器も本発明の熱成形容器に含まれる。トレイ状容器も、上述したカップ状容器等と同様の方法により製造することができる。当該トレイ状容器は、食品トレイ等として好適に用いられる。
本発明の熱成形容器は、上述した形態に限定されず、トレイ状容器も本発明の熱成形容器に含まれる。トレイ状容器も、上述したカップ状容器等と同様の方法により製造することができる。当該トレイ状容器は、食品トレイ等として好適に用いられる。
<ブロー成形容器>
本発明のブロー成形容器は、本発明の成形体を備える。当該ブロー成形容器は、本発明の成形体からなるブロー成形容器であってよい。すなわち、本発明の成形体の一実施形態は、ブロー成形容器であってよい。当該ブロー成形容器は環境負荷が低く、バリア性、外観及び生産性も良好である。当該ブロー成形容器は、ガスバリア性、耐油性等が要求される各種容器に使用できる。
本発明のブロー成形容器は、本発明の成形体を備える。当該ブロー成形容器は、本発明の成形体からなるブロー成形容器であってよい。すなわち、本発明の成形体の一実施形態は、ブロー成形容器であってよい。当該ブロー成形容器は環境負荷が低く、バリア性、外観及び生産性も良好である。当該ブロー成形容器は、ガスバリア性、耐油性等が要求される各種容器に使用できる。
本発明のブロー成形容器は、例えば、容器内部表面から容器外部表面に向かって、層(2)、層(3)、層(1)、層(3)、層(4)、層(2)の順(以降、(内)2/3/1/3/4/2(外)のように表す)、(内)2/3/1/3/2(外)、(内)2/4/3/1/3/4/2(外)、(内)4/3/1/3/4(外)等の層構造のものを採用することができる。なお、層(2)の代わりに層(4)を備える構成でもよく、層(1)~(4)がそれぞれ複数用いられている配置の場合、それぞれの層を構成する樹脂は同一でも異なっていてもよい。
本発明のブロー成形容器は、ガスバリア樹脂組成物を用いてブロー成形する工程を有する製造方法により製造することが好ましい。ブロー成形は、ダイレクトブロー成形、インジェクションブロー成形、シートブロー成形、フリーブロー成形等の公知の方法により行うことができる。
具体的には、例えば層(1)を形成するガスバリア樹脂組成物ペレット、及び必要に応じて他の各層を形成する各樹脂を用い、ブロー成形機にて100℃~400℃の温度でブロー成形し、金型内温度10℃~30℃で10秒間~30分間冷却する。これにより、ブロー成形された中空容器を成形できる。ブロー成形の際の加熱温度は、150℃以上であってよく、180℃又は200℃以上であってよい。また、この加熱温度は、ガスバリア樹脂組成物の融点以上であってよい。一方、この加熱温度の上限は350℃であってよく、300℃又は250℃であってよい。本発明のブロー成形容器は、燃料容器や各種ボトル等の種々の用途で利用される。
<燃料容器>
本発明のブロー成形容器は、燃料容器として使用できる。本発明の燃料容器はフィルター、残量計、バッフルプレート等を備えていてもよい。本発明の燃料容器は、本発明のブロー成形容器を備えることで、環境負荷が低く、バリア性、外観及び生産性も良好であり、燃料容器として好適に用いられる。ここで、燃料容器とは、自動車、オートバイ、船舶、航空機、発電機、工業用若しくは農業用機器等に搭載された燃料容器、又はこれら燃料容器に燃料を補給するための携帯用燃料容器、さらには、燃料を保管するための容器を意味する。また、燃料としては、ガソリン、特にメタノール、エタノール又はMTBE等をブレンドした含酸素ガソリン等が代表例として挙げられるが、その他、重油、軽油、灯油等も含まれるものとする。これらうち、本発明の燃料容器は、含酸素ガソリン用燃料容器として特に好適に用いられる。
本発明のブロー成形容器は、燃料容器として使用できる。本発明の燃料容器はフィルター、残量計、バッフルプレート等を備えていてもよい。本発明の燃料容器は、本発明のブロー成形容器を備えることで、環境負荷が低く、バリア性、外観及び生産性も良好であり、燃料容器として好適に用いられる。ここで、燃料容器とは、自動車、オートバイ、船舶、航空機、発電機、工業用若しくは農業用機器等に搭載された燃料容器、又はこれら燃料容器に燃料を補給するための携帯用燃料容器、さらには、燃料を保管するための容器を意味する。また、燃料としては、ガソリン、特にメタノール、エタノール又はMTBE等をブレンドした含酸素ガソリン等が代表例として挙げられるが、その他、重油、軽油、灯油等も含まれるものとする。これらうち、本発明の燃料容器は、含酸素ガソリン用燃料容器として特に好適に用いられる。
<ボトル容器>
本発明のブロー成形容器は、ボトル容器として使用できる。本発明のボトル容器は、カバーフィルム、キャップ等、本発明のブロー成形容器以外の構成をさらに備えていてもよい。本発明のボトル容器の成形方法は例えば、ダイレクトブロー成形及びインジェクションブロー成形が挙げられる。ボトル状に成形した本発明のブロー成形容器は、環境負荷が低く、バリア性、外観及び生産性も良好であるため、食品、化粧品などのボトル容器に好適に用いられる。
本発明のブロー成形容器は、ボトル容器として使用できる。本発明のボトル容器は、カバーフィルム、キャップ等、本発明のブロー成形容器以外の構成をさらに備えていてもよい。本発明のボトル容器の成形方法は例えば、ダイレクトブロー成形及びインジェクションブロー成形が挙げられる。ボトル状に成形した本発明のブロー成形容器は、環境負荷が低く、バリア性、外観及び生産性も良好であるため、食品、化粧品などのボトル容器に好適に用いられる。
<紙容器>
本発明の紙容器は、本発明の成形体を備える。当該紙容器は、本発明の成形体からなる紙容器であってよい。すなわち、本発明の成形体の一実施形態は、紙容器であってよい。紙容器は紙基材を含む成形体からなり、カートンあるいはカップ等の形状に加工されることで作成される。かかる紙容器は、各種飲料等を長期に保存することが可能である。
本発明の紙容器は、本発明の成形体を備える。当該紙容器は、本発明の成形体からなる紙容器であってよい。すなわち、本発明の成形体の一実施形態は、紙容器であってよい。紙容器は紙基材を含む成形体からなり、カートンあるいはカップ等の形状に加工されることで作成される。かかる紙容器は、各種飲料等を長期に保存することが可能である。
紙基材を含む成形体は、例えば、Tダイ法による押出コーティングで形成されることにより高速度での製膜が可能である。
<単層フィルム>
本発明の単層フィルムは、本発明のガスバリア樹脂組成物から形成されるフィルムである。すなわち、当該単層フィルムは、本発明のガスバリア樹脂組成物から形成される層のみからなるフィルムである。当該単層フィルムは環境負荷が低く、ガスバリア性も良好である。また、当該単層フィルムは、無機粒子(C)を含む本発明のガスバリア樹脂組成物から形成されている場合、耐破断性等も良好である。当該単層フィルムの平均厚みは、例えば1μm以上300μm未満であることが好ましく、5μm以上100μm未満であることがより好ましい。当該単層フィルムは、各種包装材などとして好適に用いることができる。
本発明の単層フィルムは、本発明のガスバリア樹脂組成物から形成されるフィルムである。すなわち、当該単層フィルムは、本発明のガスバリア樹脂組成物から形成される層のみからなるフィルムである。当該単層フィルムは環境負荷が低く、ガスバリア性も良好である。また、当該単層フィルムは、無機粒子(C)を含む本発明のガスバリア樹脂組成物から形成されている場合、耐破断性等も良好である。当該単層フィルムの平均厚みは、例えば1μm以上300μm未満であることが好ましく、5μm以上100μm未満であることがより好ましい。当該単層フィルムは、各種包装材などとして好適に用いることができる。
本発明の単層フィルムの、JIS B0601に準拠して測定される少なくとも一方の表面の算術平均粗さ(Ra)及び輪郭曲線要素の平均長さ(RSm)の好適範囲は、上記した本発明のフィルム等の範囲と同様である。
本発明の単層フィルムは、無延伸フィルムであってもよく、延伸フィルムであってもよいが、延伸されていることが好ましい場合もある。延伸されていることでガスバリア性や強度等が向上する傾向となる。さらに、本発明の単層フィルムが延伸フィルムである場合、破断する可能性が低いため、生産性が良好である。一方、無延伸である場合は、熱融着性が良好となる傾向がある。従って、後述するヒートシール用フィルムとして用いる場合は、無延伸である方が好ましい。
本発明の単層フィルムは、上記した本発明のフィルム等の製造方法と同様の方法で製造できる。当該単層フィルムは、食品包装容器、医薬品包装容器、工業薬品包装容器、農薬包装容器等の各種包装容器の材料として好適に用いられる。
<多層フィルム>
本発明の多層フィルムは、本発明のガスバリア樹脂組成物から形成される層を少なくとも1層備える多層フィルムである。当該多層フィルムの層数の下限としては、2であってよいが、3が好ましい。また、当該多層フィルムの層数の上限としては、例えば1000であってよく、100であってもよく、20又は10であってもよい。当該多層フィルムは、通常、本発明のガスバリア樹脂組成物からなる層と他の層とを積層して得られる。当該多層フィルムの層構成としては、本発明のガスバリア樹脂組成物以外の樹脂からなる層をx層、本発明のガスバリア樹脂組成物層をy層、接着性樹脂層をz層、「/」を接着層又は直接積層を意味するものとすると、例えばx/y、x/y/x、x/z/y、x/z/y/z/x、x/y/x/y/x、x/z/y/z/x/z/y/z/x等が挙げられる。複数のx層、y層、z層を設ける場合は、その種類は同じでも異なってもよい。また、成形時に発生するトリム等のスクラップからなる回収樹脂を用いた層を別途設けてよいし、回収樹脂を他の樹脂からなる層に混合してもよい。多層フィルムの各層の厚さ構成は、成形性及びコスト等の観点から、全層厚さに対するy層の厚さ比が通常2~20%である。なお、上記接着層は、接着性樹脂又はその他の接着剤から形成される層である。
本発明の多層フィルムは、本発明のガスバリア樹脂組成物から形成される層を少なくとも1層備える多層フィルムである。当該多層フィルムの層数の下限としては、2であってよいが、3が好ましい。また、当該多層フィルムの層数の上限としては、例えば1000であってよく、100であってもよく、20又は10であってもよい。当該多層フィルムは、通常、本発明のガスバリア樹脂組成物からなる層と他の層とを積層して得られる。当該多層フィルムの層構成としては、本発明のガスバリア樹脂組成物以外の樹脂からなる層をx層、本発明のガスバリア樹脂組成物層をy層、接着性樹脂層をz層、「/」を接着層又は直接積層を意味するものとすると、例えばx/y、x/y/x、x/z/y、x/z/y/z/x、x/y/x/y/x、x/z/y/z/x/z/y/z/x等が挙げられる。複数のx層、y層、z層を設ける場合は、その種類は同じでも異なってもよい。また、成形時に発生するトリム等のスクラップからなる回収樹脂を用いた層を別途設けてよいし、回収樹脂を他の樹脂からなる層に混合してもよい。多層フィルムの各層の厚さ構成は、成形性及びコスト等の観点から、全層厚さに対するy層の厚さ比が通常2~20%である。なお、上記接着層は、接着性樹脂又はその他の接着剤から形成される層である。
上記x層に使用される樹脂としては、加工性等の観点から熱可塑性樹脂が好ましい。熱可塑性樹脂としては、多層構造体Aの説明で例示したものなどが挙げられる。上記z層に使用される接着性樹脂としては、多層構造体Aの説明で例示したものなどが挙げられる。多層フィルムには、さらに別の層が積層されていてもよい。
多層フィルムを得る方法としては、例えば共押出成形、共押出中空成形、共射出成形、押出ラミネート、共押出ラミネート、ドライラミネート、溶液コート等が挙げられる。なお、このような方法で得られた多層フィルムに対して、さらに真空圧空深絞成形、ブロー成形、プレス成形等の方法により、再加熱後に二次加工成形を行い、目的とする成形体構造にしてよい。また、多層フィルムに対して、ロール延伸法、パンタグラフ延伸法、インフレーション延伸法等の方法により、EVOHの融点以下の範囲で再加熱後に一軸又は二軸延伸して、延伸された多層フィルムを得ることもできる。
本発明の単層フィルム又は多層フィルムを用いた成形体としては、容器(袋、カップ、チューブ、トレー、ボトル等)、燃料容器、パイプ、繊維、飲食品用包装材、容器用パッキング材、医療用輸液バッグ材、タイヤ用チューブ材、靴用クッション材、バッグインボックス用内袋材、有機液体貯蔵用タンク材、有機液体輸送用パイプ材、暖房用温水パイプ材(床暖房用温水パイプ材等)、化粧品用包装材、デンタルケア用包装材、医薬品用包装材、包材用子部品(キャップ、バッグインボックスのコック部分など)、農薬ボトル、農業用フィルム(温室用フィルム、土壌燻蒸用フィルム)、穀物保管用袋、ジオメンブレン、真空断熱板外袋、壁紙又は化粧板、水素、酸素等のガスタンク等を挙げることができる。
<蒸着フィルム>
本発明の蒸着フィルムは、本発明の単層フィルムまたは多層フィルムと、無機蒸着層とを備える。具体的に本発明の蒸着フィルムは、本発明の単層フィルム、または上記ガスバリア樹脂組成物から形成される層の少なくとも1層を最外層として備える本発明の多層フィルムと、上記単層フィルムまたは多層フィルムにおける本発明のガスバリア樹脂組成物から形成される層の表出面に積層される少なくとも1層の無機蒸着層とを備える。当該蒸着フィルムにおいては、本発明のガスバリア樹脂層から形成される層上に、無機蒸着層が直接積層されている。
本発明の蒸着フィルムは、本発明の単層フィルムまたは多層フィルムと、無機蒸着層とを備える。具体的に本発明の蒸着フィルムは、本発明の単層フィルム、または上記ガスバリア樹脂組成物から形成される層の少なくとも1層を最外層として備える本発明の多層フィルムと、上記単層フィルムまたは多層フィルムにおける本発明のガスバリア樹脂組成物から形成される層の表出面に積層される少なくとも1層の無機蒸着層とを備える。当該蒸着フィルムにおいては、本発明のガスバリア樹脂層から形成される層上に、無機蒸着層が直接積層されている。
無機蒸着層は、金属や無機酸化物などの無機物からなり、酸素や水蒸気に対するガスバリア性を有する層である。本発明のガスバリア樹脂組成物は、通常の熱可塑性樹脂と比べて金属や無機酸化物との親和性が高く、緻密で欠陥のない無機蒸着層を形成することができる。また、該ガスバリア樹脂組成物がガスバリア性を有しているため、屈曲等によって無機蒸着層に欠陥が生じた際にも、ガスバリア性の低下を抑制できる。さらに、該ガスバリア樹脂組成物が無機粒子(C)を含む場合、蒸着欠点が生じ難く、かつ、無機蒸着層との密着強度に優れた蒸着フィルムを製造することができる。したがって、該ガスバリア樹脂組成物から形成される層の少なくとも一方の面上に直接無機蒸着層が積層された構成を有する本発明の蒸着フィルムは、蒸着欠点が生じ難く、かつ、無機蒸着層との密着強度に優れる。
無機蒸着層は、アルミニウムを主成分とする金属蒸着層、及びアルミナ又はシリカを主成分とする無機酸化物蒸着層のいずれかであることが好ましい。遮光性を付与する場合には金属蒸着層が好ましいが、包装材料としての内容物の視認性やレンジ適正、粉砕物を溶融成形する際にゲルやブツの発生を抑制できる観点からは、無機酸化物蒸着層が好ましい。
金属蒸着層は、一般にはアルミニウムを主成分とする層である。金属蒸着層におけるアルミニウム原子の含有量は50モル%以上が好ましく、70モル%以上がより好ましく、90モル%以上がさらに好ましく、95モル%以上が特に好ましい。金属蒸着層の平均厚みは120nm以下が好ましく、100nm以下がより好ましく、90nm以下がさらに好ましい。また、金属蒸着層の平均厚みは25nm以上が好ましく、35nm以上がより好ましく、45nm以上がさらに好ましい。なお、金属蒸着層の平均厚みとは、電子顕微鏡により測定される金属蒸着層断面の任意の10点における厚みの平均値である。
無機酸化物蒸着層は、無機酸化物、例えば、ケイ素、アルミニウム、マグネシウム、カルシウム、カリウム、スズ、ナトリウム、ホウ素、チタン、鉛、ジルコニウム、イットリウム等の酸化物、好ましくはアルミナ又はシリカの蒸着膜が挙げられる。無機酸化物蒸着層の平均厚みは60nm以下が好ましく、50nm以下がより好ましく、40nm以下がさらに好ましい。また、無機酸化物蒸着層の平均厚みは10nm以上が好ましく、15nm以上がより好ましく、20nm以上がさらに好ましい。なお、無機酸化物蒸着層の平均厚みとは、電子顕微鏡により測定される無機酸化物蒸着層断面の任意の10点における厚みの平均値である。
無機蒸着層は、公知の物理的蒸着法や化学的蒸着法により蒸着できる。具体的には、多層構造体Aの説明で例示した方法等が挙げられる。
無機蒸着層は、より寸法安定性の高い層に蒸着された方が蒸着時の欠陥が生じにくく高いガスバリア性を発現することができる。寸法安定性の観点より無機蒸着層を積層するガスバリア樹脂組成物から形成される層は延伸されていることが好ましく、延伸後に熱処理を施すことがさらに好ましい。
本発明の蒸着フィルムの層数の下限は、2である。すなわち、当該蒸着フィルムは、本発明の多層フィルムの一態様である。また、当該蒸着フィルムの層数の上限は、多層フィルムと同様に例えば1000であってよく、100であってもよく、20、10、5又は4であってもよい。
本発明の蒸着フィルムの層構成としては、本発明のガスバリア樹脂組成物以外の樹脂からなる層をx層、本発明のガスバリア樹脂組成物から形成される層をy層、接着性樹脂層をz層、無機蒸着層をv層とすると、例えばy/v、v/y/v、x/y/v、x/z/y/v、v/y/z/x/z/y/v等が挙げられる。
<多層構造体B>
本発明の蒸着フィルムは、本発明のガスバリア樹脂組成物から形成される層に隣接する最外層として無機蒸着層を有する。一方、当該蒸着フィルムの無機蒸着層上に他の層をさらに積層した多層構造体として用いることもできる。すなわち、本発明の多層構造体Bは、本発明の蒸着フィルムと、当該蒸着フィルムにおける無機蒸着層上に積層される他の層(熱可塑性樹脂層等)とを備える。他の層としては特に限定されず、多層構造体Aの説明で例示した各種熱可塑性樹脂の層などが挙げられる。当該多層構造体Bは、無機蒸着層含有多層フィルムであってよい。当該多層構造体Bは、上記した本発明の多層構造体Aの一形態に含まれる。
本発明の蒸着フィルムは、本発明のガスバリア樹脂組成物から形成される層に隣接する最外層として無機蒸着層を有する。一方、当該蒸着フィルムの無機蒸着層上に他の層をさらに積層した多層構造体として用いることもできる。すなわち、本発明の多層構造体Bは、本発明の蒸着フィルムと、当該蒸着フィルムにおける無機蒸着層上に積層される他の層(熱可塑性樹脂層等)とを備える。他の層としては特に限定されず、多層構造体Aの説明で例示した各種熱可塑性樹脂の層などが挙げられる。当該多層構造体Bは、無機蒸着層含有多層フィルムであってよい。当該多層構造体Bは、上記した本発明の多層構造体Aの一形態に含まれる。
本発明の多層構造体Bの層数の下限は、例えば3である。また、当該多層構造体Bの層数の上限は、例えば1000であってよく、100であってもよく、20、10、5又は4であってもよい。
本発明の多層構造体Bの層構成としては、本発明のガスバリア樹脂組成物以外の樹脂からなる層をx層、本発明のガスバリア樹脂組成物層をy層、接着性樹脂層をz層、無機蒸着層をv層とすると、例えばx/v/y、x/v/y/v、x/y/v/x、x/v/y/v/x、x/z/v/y、x/z/y/v/z/x、x/z/v/y/v/z/x等が挙げられる。
本発明の蒸着フィルム及び多層構造体Bは特に優れたバリア性を要求される包装材に好適に用いられる。具体的には容器(袋、チューブ、蓋材等)、飲食品用包装材、医療用輸液バッグ材、タイヤ用チューブ材、バッグインボックス用内袋材、化粧品用包装材、デンタルケア用包装材、医薬品用包装材、農業用フィルム(温室用フィルム、土壌燻蒸用フィルム)、穀物保管用袋、ジオメンブレン、真空断熱板外袋等を挙げることができる。
<ヒートシール用フィルム>
本発明の単層フィルム、多層フィルム、蒸着フィルムまたは多層構造体Bは、ヒートシール用のフィルムであることが好ましい。すなわち、本発明のヒートシール用フィルムは、本発明の単層フィルム、多層フィルム、蒸着フィルムまたは多層構造体Bを備える。本発明のヒートシール用フィルムが多層である場合の具体的層構成としては、上述した多層構造体Bの層構成と同じものが挙げられる。ヒートシール用フィルムにおいては、最表層が熱融着層として機能する。熱融着層は、本発明の単層フィルムからなる層(ガスバリア樹脂組成物から形成される層)であってもよいし、他の熱可塑性樹脂等から形成される層であってもよい。
本発明の単層フィルム、多層フィルム、蒸着フィルムまたは多層構造体Bは、ヒートシール用のフィルムであることが好ましい。すなわち、本発明のヒートシール用フィルムは、本発明の単層フィルム、多層フィルム、蒸着フィルムまたは多層構造体Bを備える。本発明のヒートシール用フィルムが多層である場合の具体的層構成としては、上述した多層構造体Bの層構成と同じものが挙げられる。ヒートシール用フィルムにおいては、最表層が熱融着層として機能する。熱融着層は、本発明の単層フィルムからなる層(ガスバリア樹脂組成物から形成される層)であってもよいし、他の熱可塑性樹脂等から形成される層であってもよい。
本発明のヒートシール用フィルムにおいては、少なくとも1層の本発明の単層フィルムが最表層として設けられていることが好ましい。このような形態のヒートシール用フィルムにおいては、本発明の単層フィルム層が熱融着層として利用できる。本発明の単層フィルムは、上述のように香り成分などの吸着を効果的に抑制できる。したがって、本発明の単層フィルムが熱融着層であり、該単層フィルムを最表層に備えるヒートシール用フィルムを用いた包装材は、内容物の香り成分を効率的に保持することができる。
本発明の単層フィルムが良好な熱融着性を示すためには、ガスバリア樹脂組成物を構成するEVOHの平均のエチレン単位含有量の下限は25モル%が好ましく、30モル%がより好ましく、40モル%がさらに好ましい。上記エチレン単位含有量の上限は60モル%が好ましく、55モル%がより好ましい。
本発明のヒートシール用フィルムは、本発明の単層フィルムが熱融着層(最表層)として設けられている場合、上述のように香り成分等に対する優れた非吸着性を有しており、包装袋用として好適に用いることができる。
本発明のヒートシール用フィルムにおいて、熱融着層(最表層)同士を対面させ、熱板式ヒートシーラーを用いて温度185℃、圧力0.1MPaの条件で1秒間圧着して得られるヒートシール強度が10N/15mm以上であると好ましい。なお、対面させる熱融着層同士は同じであってもよいし異なっていてもよい。当該ヒートシール用フィルムは、ヒートシールにより優れたヒートシール強度を得ることができ、高い保香性を備える包装袋用のヒートシール用フィルムとして好適に用いることができる。なお、このヒートシール強度はJIS-Z1707に準拠して測定した値である。
(包装袋)
本発明のヒートシール用フィルムは、熱融着層同士を重ねてヒートシールすることにより、この熱融着層を内側とした容器及び包装体(袋、カップ、チューブ、トレー、ボトル等)とすることができる。上述のように、当該ヒートシール用フィルムは、高い強度でヒートシールされることができ、また、本発明の単層フィルムが熱融着層として設けられている場合、熱融着層が香り成分の非吸着性に優れているため、これから得られる上記包装袋等は優れた保香性を発揮することができる。
本発明のヒートシール用フィルムは、熱融着層同士を重ねてヒートシールすることにより、この熱融着層を内側とした容器及び包装体(袋、カップ、チューブ、トレー、ボトル等)とすることができる。上述のように、当該ヒートシール用フィルムは、高い強度でヒートシールされることができ、また、本発明の単層フィルムが熱融着層として設けられている場合、熱融着層が香り成分の非吸着性に優れているため、これから得られる上記包装袋等は優れた保香性を発揮することができる。
ヒートシールする方法としては特に限定されず、公知の方法を用いることができ、例えば熱板式ヒートシーラー、インパルスシーラー、超音波シーラー、摩擦熱シーラー、誘電加熱シーラー等によりヒートシールする方法があげられる。ヒートシールする際の温度としては、特に限定されるものではないが、熱融着層の融点以上、(融点+30℃)以下であることがヒートシール強度と外観不良防止の点から好ましい。ヒートシールする圧力としては、特に限定されるものではないが、0.01MPa以上1.0MPa以下であることがヒートシール強度と外観不良防止の点から好ましい。ヒートシールする時間としては、特に限定されるものではないが、0.05秒以上5秒以下であることが、ヒートシール強度と生産性の点から好ましい。
本発明のヒートシール用フィルムをヒートシールして包装袋等を成形する際の、該ヒートシール用フィルムの熱融着層の水分率としては、0.1質量%以上4質量%以下が好ましく、0.2質量%以上3質量%以下がさらに好ましい。ヒートシールの際の熱融着層の水分率を上記範囲とすることで、シール部分の外観の維持や、シール強度の低下を抑えることができる。この水分率が上記上限以下であることで、ヒートシールの際に熱融着層中の水分の気化による発泡が生じにくくなり、その結果、ヒートシール部分の外観が向上したり、ヒートシール性が向上する。また、この水分率が上記下限以上であることで、包装袋等を成形した直後の非吸着性が向上する傾向がある。例えば成形直後に内容物を充填した場合に、内容物由来の香気成分が上記熱融着層に吸着され難くなる。
包装袋における内容物(被包装物)としては、香り成分を含有した例えば食品、飲料、医薬品、化粧品、香料、トイレタリー製品等が挙げられ、特に、菓子、茶葉、コーヒー、香辛料及び煙草からなる群より選択される少なくとも1種であるとよい。当該包装袋は、上述のように優れた保香性を備える形態とすることができるため、特に保香性が必要とされる上記食品等の包装袋として好適である。
<包装材B>
本発明の包装材Bは、本発明の多層構造体(例えば多層構造体A)を備える。当該包装材Bは、本発明の多層構造体からなる包装材であってよい。当該包装材Bは、環境負荷が低く、ガスバリア性、外観及び生産性も良好である。
本発明の包装材Bは、本発明の多層構造体(例えば多層構造体A)を備える。当該包装材Bは、本発明の多層構造体からなる包装材であってよい。当該包装材Bは、環境負荷が低く、ガスバリア性、外観及び生産性も良好である。
本発明の包装材Bは、樹脂以外から形成される層、例えば紙層、金属層等を有していてもよい。当該包装材Bは、多層構造体そのままであってもよく、多層構造体が二次加工されたものであってもよい。二次加工することで得られる包装材としては例えば、(1)多層構造体を真空成形、圧空成形、真空圧空成形等、熱成形加工することにより得られるトレーカップ状容器、(2)多層構造体にストレッチブロー成形等を行って得られるボトル、カップ状容器、(3)多層構造体をヒートシールすることにより得られる袋状容器等が挙げられる。なお、二次加工法は、上記に例示した各方法に限定されることなく、例えば、ブロー成形等の上記以外の公知の二次加工法を適宜用いることができる。
本発明の包装材Bは、例えば食品、飲料物、農薬や医薬等の薬品、医療器材、機械部品、精密材料等の産業資材、衣料などを包装するために使用される。特に、当該包装材Bは、酸素に対するバリア性が必要となる用途、包装材の内部が各種の機能性ガスによって置換される用途に好ましく使用される。当該包装材Bは、用途に応じて種々の形態、例えば縦製袋充填シール袋、真空包装袋、スパウト付パウチ、ラミネートチューブ容器、容器用蓋材等に形成される。
特に、PA(E)を含むガスバリア樹脂組成物から形成された層を有する多層構造体を用いて当該包装材Bが形成されている場合、当該包装材Bは耐レトルト性に優れるため、ボイル殺菌用またはレトルト殺菌用の包装材として好ましく使用される。また、熱可塑性エラストマー(G)を含むガスバリア樹脂組成物から形成された層を有する多層構造体を用いて当該包装材Bが形成されている場合、当該包装材Bは耐屈曲性に優れるため、フレキシブルな包装材として有用である。フレキシブルな包装材としては、例えば縦製袋充填シール袋が挙げられる。
<縦製袋充填シール袋>
本発明の縦製袋充填シール袋は、本発明の多層構造体(例えば多層構造体A)を備える縦製袋充填シール袋である。縦製袋充填シール袋は、例えば、液体、粘稠体、粉体、固形バラ物、又は、これらを組み合わせた食品や飲料物などを包装するために用いられることが多い。当該縦製袋充填シール袋は、耐屈曲性に優れ、変形や衝撃などの物理的ストレスを受けた際にそのガスバリア性が維持される。
本発明の縦製袋充填シール袋は、本発明の多層構造体(例えば多層構造体A)を備える縦製袋充填シール袋である。縦製袋充填シール袋は、例えば、液体、粘稠体、粉体、固形バラ物、又は、これらを組み合わせた食品や飲料物などを包装するために用いられることが多い。当該縦製袋充填シール袋は、耐屈曲性に優れ、変形や衝撃などの物理的ストレスを受けた際にそのガスバリア性が維持される。
縦製袋充填シール袋(Vertical form fill seal pouch)の一形態を図5に示す。図5の縦製袋充填シール袋101は、シール袋101の上端部111、下端部112及び胴体部115の三方でフィルム材110がシールされている。図示したシール袋101では、胴体部115は、背面120を二つに分割するように上端部111から下端部112へと伸びる背面中央部に配置されている。上端部111、下端部112及び胴体部115において、フィルム材110はその内面同士が接触する状態でシールされている。すなわち、胴体部115におけるシールの形態はいわゆる合掌貼りである。図5には示されていないシール袋101の前面(背面の反対側にある背面と同形状の面)は、背面120とは異なり、シールされた部分により分割されておらず、通常は内容物や商品を表示する面として用いられる。なお、シールされる胴体部115は、側端部121、122のいずれかに配置されることもあり、この場合は背面もシールされた部分により分割されることがない。図示したシール袋101は、背面120の幅の2倍(前面の幅と背面の幅の合計)に胴体部115でのシールに要する幅を加えた幅を有する1枚のフィルム材110が縦型製袋充填機に供給されて製造された袋である。上端部111、下端部112及び胴体部115は、いずれも分岐がない直線状のシール部として形成されている。以上のように、縦製袋充填シール袋は、その一形態において、袋の前面及び背面の上辺に相当する上端部、下辺に相当する下端部、上端部から下端部に至るまでこれらの端部に垂直に伸びる胴体部の3つの部位において、1枚のフィルム材がシールされて製袋されている。フィルム材110が、本発明の多層構造体を含む。
<バッグインボックス用内容器>
本発明のバッグインボックス用内容器は、本発明の多層構造体(例えば多層構造体A)を備える。当該バッグインボックス用内容器としては、例えば、液体注入口が他の樹脂組成物から成形され、容器本体が上記多層構造体で形成されたものを挙げることができる。当該バッグインボックス用内容器は、例えば、本発明の多層構造体のフィルムやシートをヒートシールし、さらに液体注入口をヒートシールすることにより作成することができる。ヒートシールする方法は、通常のヒートシール条件を適宜選択することができる。
本発明のバッグインボックス用内容器は、本発明の多層構造体(例えば多層構造体A)を備える。当該バッグインボックス用内容器としては、例えば、液体注入口が他の樹脂組成物から成形され、容器本体が上記多層構造体で形成されたものを挙げることができる。当該バッグインボックス用内容器は、例えば、本発明の多層構造体のフィルムやシートをヒートシールし、さらに液体注入口をヒートシールすることにより作成することができる。ヒートシールする方法は、通常のヒートシール条件を適宜選択することができる。
バッグインボックスは、例えば、ダンボール箱の内部に、液体注入口を設けたフレキシブルなプラスチックの内容器(バッグインボックス用内容器)を収納したものである。バッグインボックス用内容器は、通常、輸送時等に繰り返しの屈曲に晒される。バッグインボックス用内容器成形時には、屈曲箇所や密封栓の周囲の厚みを増すなどの加工を施してもよい。本発明のバッグインボックス用内容器によれば、本発明の多層構造体を備えるため、優れた耐久性を発揮することができる。
<積層剥離容器>
本発明の積層剥離容器(デラミネーション容器)は、本発明の多層構造体(例えば多層構造体A)を備える。当該積層剥離容器に備わる多層構造体は、本発明のガスバリア樹脂組成物からなる層の一方の面に直接積層された、極性官能基を有さないポリオレフィンを主成分とする層をさらに有する。すなわち、当該積層剥離容器は、当該ガスバリア樹脂組成物からなる層の一方の面に上記ポリオレフィンを主成分とする層が直接積層された層構造を有する。このような構成であると、本発明のガスバリア樹脂組成物からなる層と、極性官能基を有さないポリオレフィンを主成分とする層との剥離性が良好となり、積層剥離容器として好適に用いることができる。極性官能基を有さないポリオレフィンを主成分とする層における極性官能基を有さないポリオレフィンが占める割合は、70質量%以上が好ましく、90質量%以上がより好ましく、95質量%以上がさらに好ましく、実質的に極性官能基を有さないポリオレフィンのみから構成される層であることが好ましい。
本発明の積層剥離容器(デラミネーション容器)は、本発明の多層構造体(例えば多層構造体A)を備える。当該積層剥離容器に備わる多層構造体は、本発明のガスバリア樹脂組成物からなる層の一方の面に直接積層された、極性官能基を有さないポリオレフィンを主成分とする層をさらに有する。すなわち、当該積層剥離容器は、当該ガスバリア樹脂組成物からなる層の一方の面に上記ポリオレフィンを主成分とする層が直接積層された層構造を有する。このような構成であると、本発明のガスバリア樹脂組成物からなる層と、極性官能基を有さないポリオレフィンを主成分とする層との剥離性が良好となり、積層剥離容器として好適に用いることができる。極性官能基を有さないポリオレフィンを主成分とする層における極性官能基を有さないポリオレフィンが占める割合は、70質量%以上が好ましく、90質量%以上がより好ましく、95質量%以上がさらに好ましく、実質的に極性官能基を有さないポリオレフィンのみから構成される層であることが好ましい。
上記ポリオレフィンを主成分とする層は、極性官能基を有さないポリオレフィンのみから実質的になる層であってよい。極性官能基を有さないポリオレフィンとは、オレフィンの単独重合体又は共重合体であって、極性官能基を有さないものである。オレフィンの単独重合体又は共重合体としては、例えば、直鎖状低密度ポリエチレン(LLDPE)、低密度ポリエチレン(LDPE)、超低密度ポリエチレン(VLDPE)、中密度ポリエチレン(MDPE)、高密度ポリエチレン(HDPE)、エチレン-プロピレン(ブロック又はランダム)共重合体、ポリプロピレン(PP)、プロピレンと炭素数4~20のα-オレフィンとの共重合体、ポリブテン、ポリペンテン、ポリメチルペンテン等のオレフィンの単独重合体又は共重合体が好適に使用される。中でも、低密度ポリエチレン(LDPE)、ポリプロピレン(PP)、高密度ポリエチレン(HDPE)、エチレン-プロピレン(ブロック又はランダム)共重合体からなる群から選択される少なくとも1種がより好適に使用される。
本発明の積層剥離容器における多層構造体は、本発明のガスバリア樹脂組成物からなる層の他方が、接着層を介して熱可塑性樹脂を主成分とする層と接着されてなることが好適な実施態様である。熱可塑性樹脂を主成分とする層における熱可塑性樹脂が占める割合は、70質量%以上が好ましく、90質量%以上がより好ましく、95質量%以上がさらに好ましく、実質的に熱可塑性樹脂のみから構成される層であることが好ましい。このような構成とすることで、本発明の多層構造体、ひいては積層剥離容器の柔軟性及び強度を高めることができる。熱可塑性樹脂を主成分とする層を構成する熱可塑性樹脂は、ポリオレフィンが好ましく、このポリオレフィンは、上記極性官能基を有さないポリオレフィンと同じものであってもよく、不飽和カルボン酸又はその誘導体で変性された変性ポリオレフィン等であってもよい。不飽和カルボン酸又はその誘導体としては、マレイン酸、フマル酸、イタコン酸、無水マレイン酸、無水イタコン酸、マレイン酸モノメチルエステル、マレイン酸モノエチルエステル、マレイン酸ジエチルエステル、フマル酸モノメチルエステルなどが挙げられる。これらは1種類を単独で用いても、2種以上を組合せて用いてもよい。
接着層を構成する接着性樹脂としては、本発明の多層構造体Aにおける接着層の説明として例示したものなどが挙げられる。
本発明の積層剥離容器(多層構造体)の層構成としては、本発明のガスバリア樹脂組成物からなる層をy、他の熱可塑性樹脂を主成分とする層をx、接着性樹脂を主成分とする層(接着層)をz、極性官能基を有さないポリオレフィンを主成分とする層をχで表す場合、
χ/y
χ/y/z/x
χ/y/z/x/z/y
χ/y/z/x/z/x
χ/y/z/x/z/y/z/x
χ/y/z/x/z/x/z/x
等の構造が挙げられる。
χ/y
χ/y/z/x
χ/y/z/x/z/y
χ/y/z/x/z/x
χ/y/z/x/z/y/z/x
χ/y/z/x/z/x/z/x
等の構造が挙げられる。
本発明の積層剥離容器の製造方法としては、共押出ブロー成形法等が挙げられる。
本発明の積層剥離容器を構成する本発明のガスバリア樹脂組成物からなる層の厚みは0.5μm以上が好ましく、1μm以上がより好ましく、5μm以上がさらに好ましい。当該ガスバリア樹脂組成物からなる層の厚みを0.5μm以上とすることで、ガスバリア性が良好となる傾向となる。また、当該ガスバリア樹脂組成物からなる層の厚みは1000μm以下が好ましく、500μm以下がより好ましく、100μm以下がさらに好ましい。当該ガスバリア樹脂組成物からなる層の厚みを1000μm以下とすることで、柔軟性を高め、剥離性がより良好になる傾向となる。
本発明の積層剥離容器を構成する極性官能基を有さないポリオレフィンを主成分とする層の厚みは25μm以上が好ましく、30μm以上がより好ましく、50μm以上がさらに好ましい。上記ポリオレフィンを主成分とする層の厚みを25μm以上とすることで、強度を高めることができる。また、上記ポリオレフィンを主成分とする層の厚みは5000μm以下が好ましく、2000μm以下がより好ましく、1000μm以下がさらに好ましい。上記ポリオレフィンを主成分とする層の厚みを5000μm以下とすることで、柔軟性を高め、剥離性がより良好になる。
本発明の積層剥離容器を構成する多層構造体の全体の厚みは150μm以上が好ましく、200μm以上がより好ましい。多層構造体の全体の厚みを150μm以上とすることで、強度が十分になり破損し難くなる。また、多層構造体の全体の厚みは10000μm以下が好ましく、8000μm以下がより好ましく、6000μm以下がさらに好ましい。多層構造体の全体の厚みを10000μm以下とすることで、柔軟性を高めることができる。
本発明の積層剥離容器において、当該ガスバリア樹脂組成物からなる層と極性官能基を有さないポリオレフィンを主成分とする層との間におけるデラミ面積は80cm2以上が好ましく、150cm2以上がより好ましく、200cm2以上がさらに好ましく、220cm2以上が特に好ましい。デラミ面積を80cm2以上とすることで、剥離がより十分なものとなる。また、デラミ面積は300cm2以下が好ましく、280cm2以下がより好ましく、260cm2以下がさらに好ましい。デラミ面積を300cm2以下とすることで、剥離性が高くなりすぎるのを抑え、生産性を高めることができる。ここで、デラミ面積は、多層構造体を300mm(幅)×350mm(長さ)に切り出し、中心部における当該ガスバリア樹脂組成物からなる層と極性官能基を有さないポリオレフィンを主成分とする層との層間に剥離口を作製して、上記剥離口にチューブを50mm差し込み圧力0.2MPaのエアーを吹き込んで剥離させた際の剥離部分の質量から面積換算することで求めることができる。
本発明の積層剥離容器は、当該ガスバリア樹脂組成物からなる層と極性官能基を有さないポリオレフィンを主成分とする層との間における標準剥離強度が1g/30mm以上が好ましく、3g/30mm以上がより好ましく、5g/30mmがさらに好ましい。標準剥離強度を1g/30mm以上とすることで、剥離性が高くなりすぎるのを抑え、生産性を高めることができる。また、上記標準剥離強度は12g/30mm以下が好ましく、11g/30mm以下がより好ましく、9.5g/30mm以下がさらに好ましく、9.0g/30mm以下が特に好ましい。標準剥離強度を12g/30mm以下とすることで、十分な剥離性を発揮することができる。
本発明の積層剥離容器は、前述のように当該ガスバリア樹脂組成物からなる層と極性官能基を有さないポリオレフィンを主成分とする層との剥離性が良好であるとともに、特に当該ガスバリア樹脂組成物が熱可塑性エラストマー(G)を含む場合、耐屈曲性等も良好である。本発明の積層剥離容器は、内容物の香り、色などの劣化を防止することのできる食品用の積層剥離容器として好適に用いることができる。なお、口頭部の剥離を抑制する観点から、本発明の積層剥離容器は、口頭部が他の部分と比べて厚みを有していることが好ましい。例えば、口頭部の厚みは、0.4mm以上が好ましく、0.5mm以上がより好ましい。この口頭部の厚みの上限は例えば3mmであってよく、2mm又は1mmであってもよい。
<多層シート>
本発明の多層シートは、本発明のガスバリア樹脂組成物からなるバリア層と、このバリア層の少なくとも一方の面側に直接又は他の層を介して積層される熱可塑性樹脂層とを備える。当該多層シートの具体的形態、用途等は、上述した本発明の多層フィルムの具体的形態、用途等と同様であってよい。当該多層シートは、各種成形体の形成材料として好適に用いることができる。
本発明の多層シートは、本発明のガスバリア樹脂組成物からなるバリア層と、このバリア層の少なくとも一方の面側に直接又は他の層を介して積層される熱可塑性樹脂層とを備える。当該多層シートの具体的形態、用途等は、上述した本発明の多層フィルムの具体的形態、用途等と同様であってよい。当該多層シートは、各種成形体の形成材料として好適に用いることができる。
本発明の多層シートを用いてさらに成形体を成形する方法としては、例えば加熱延伸成形法、真空成形法、圧空成形法、真空圧空成形法、ブロー成形法等が挙げられる。これらの成形は、通常EVOHの融点以下の温度範囲で行われる。これらの中で、加熱延伸成形法及び真空圧空成形法が好ましい。加熱延伸成形法は、多層シートを加熱し、一方向又は複数方向に延伸して成形する方法である。真空圧空成形法は、多層シートを加熱し、真空と圧空を併用して成形する方法である。
<包装材C>
本発明の多層シートを加熱延伸成形法により成形してなる包装材Cは、簡便かつ確実に製造することができ、また外観性、ガスバリア性等に優れる。当該包装材Cはシート状であってもよく、他の形状に成形加工されていてもよい。当該包装材Cは、従来公知の包装材と同様の各種用途に用いることができる。
本発明の多層シートを加熱延伸成形法により成形してなる包装材Cは、簡便かつ確実に製造することができ、また外観性、ガスバリア性等に優れる。当該包装材Cはシート状であってもよく、他の形状に成形加工されていてもよい。当該包装材Cは、従来公知の包装材と同様の各種用途に用いることができる。
上記加熱延伸成形法の場合には、熱可塑性樹脂層に用いる熱可塑性樹脂は、下記式(1)で表される加熱延伸温度の範囲で延伸可能な樹脂であることが好ましい。
X-110≦Y≦X-10 ・・・(1)
X-110≦Y≦X-10 ・・・(1)
上記式(1)中、Xは、EVOHの融点(℃)である。Yは、加熱延伸温度(℃)である。上記包装材Cは、多層シートから加熱延伸成形法を用いて製造する場合、熱可塑性樹脂として上記樹脂を用いることで、外観性をより優れるものにすることができ、またクラック等の欠陥をより抑制することができる。
<容器>
本発明の多層シートを真空圧空成形法により成形してなる容器は、簡便かつ確実に製造することができ、また外観性、ガスバリア性等に優れる。当該容器は、従来公知の容器と同様の各種用途に用いることができる。
本発明の多層シートを真空圧空成形法により成形してなる容器は、簡便かつ確実に製造することができ、また外観性、ガスバリア性等に優れる。当該容器は、従来公知の容器と同様の各種用途に用いることができる。
上記真空圧空成形法では、例えば多層シートを加熱して軟化させた後に、金型形状に成形される。成形方法としては、真空又は圧空を用い、必要によりさらにプラグを併せ用いて金型形状に成形する方法(ストレート法、ドレープ法、エアスリップ法、スナップバック法、プラグアシスト法等)やプレス成形する方法等が挙げられる。成形温度、真空度、圧空の圧力又は成形速度等の各種成形条件は、プラグ形状や金型形状又は原料フィルムやシートの性質等により適切に設定される。
成形温度は特に限定されるものではなく、成形するのに十分な程度に樹脂が軟化する温度であればよい。例えば、多層シートを熱成形する際には、加熱による多層シートの融解が生じたり、ヒーター板の金属面の凹凸が多層シートに転写したりする程度の高温にはせず、かつ賦形が十分でない程度の低温にしないことが望ましい。具体的に、多層シートの温度としては、50℃~180℃、好適には60℃~160℃である。
<植物栽培用培地>
本発明の植物栽培用培地は、樹脂組成物から形成されている成形体を含む植物栽培用培地であって、上記樹脂組成物が、本発明のガスバリア樹脂組成物である。
本発明の植物栽培用培地は、樹脂組成物から形成されている成形体を含む植物栽培用培地であって、上記樹脂組成物が、本発明のガスバリア樹脂組成物である。
植物栽培用培地においても、バイオマス由来の原料を用いて合成された培地の製品化が期待される。しかし、バイオマス由来の合成樹脂は、化石燃料由来の合成樹脂と比べて性能が劣る場合があることから、従来の化石燃料由来の植物栽培用培地をバイオマス由来の植物栽培用培地に置き換えた場合、最も重要な植物生育性が低下することが懸念される。このため、化石燃料由来の樹脂と遜色のない優れた植物生育性を有するバイオマス由来の樹脂の開発が望まれている。また、一般的に樹脂製の植物栽培用培地は、溶融成形によってチップ形状に成形されていることが多い。このため、樹脂製の植物栽培用培地を効率的に製造するためには、長時間に渡る溶融成形を行い続けても、チップ形状の植物栽培用培地が安定して効率的に得られるといった、樹脂材料のロングラン性(長時間運転特性)が重要である。ロングラン性が不十分な樹脂材料を用いた場合、長時間に渡る溶融成形に伴って、押出機のダイ堆積物が生じやすくなる。また、ロングラン性が不十分な樹脂材料を用いた場合、溶融した樹脂材料をストランド状に押し出したときに、ストランド(ストランド状の樹脂材料)が破断し易くなり、生産性が悪い。なお、以下、ストランドが破断することを「断糸」ともいう。
上記のような従来の植物栽培用培地が有する課題に対し、本発明の植物栽培用培地は、バイオマス由来の原料を用いていながら、化石燃料由来のものと遜色のない十分なロングラン性を有する樹脂組成物から形成されており、高い植物生育性を有する。具体的に、当該植物栽培用培地に用いられる樹脂組成物(ガスバリア樹脂組成物)は、バイオマス由来の原料が一部に用いられていることで、環境負荷が低い。また、当該樹脂組成物は、優れた植物生育性を有する樹脂としてEVOHを選択して用いており、EVOHを用いた成形体を含む植物栽培用培地は、そのEVOHがバイオマス由来の原料を用いて合成された場合であっても、化石燃料由来の原料のみから合成された同一構造のEVOHの場合と同等の高い植物生育性を発揮できる。
ここで、植物栽培用培地に用いられる樹脂組成物におけるロングラン性とは、実施例に記載の方法で評価することができ、樹脂組成物を連続成形した際のダイ堆積物及び断糸頻度の度合いによって総合的に評価できる。また、植物生育性とは、植物を育成する性能を有することを意味し、例えば、本発明の実施例記載の培地評価のように、大根の育成における肥大根数等により評価できる。
(成形体、使用方法等)
本発明の植物栽培用培地に含まれる上記成形体は、上記樹脂組成物(ガスバリア樹脂組成物)から形成されているものであれば、その形態に特に制限は無い。上記成形体は、通常、上記樹脂組成物の溶融成形体である。上記成形体は、上記樹脂組成物からなるチップ(ペレット)、上記樹脂組成物からなる不織布等であってもよい。植物の育成効率の観点からは、上記樹脂組成物からなるチップ(樹脂組成物チップ)であることが好ましい。この場合、例えば、多数のチップを容器等に敷き詰めて植物栽培用培地として使用することができる。
本発明の植物栽培用培地に含まれる上記成形体は、上記樹脂組成物(ガスバリア樹脂組成物)から形成されているものであれば、その形態に特に制限は無い。上記成形体は、通常、上記樹脂組成物の溶融成形体である。上記成形体は、上記樹脂組成物からなるチップ(ペレット)、上記樹脂組成物からなる不織布等であってもよい。植物の育成効率の観点からは、上記樹脂組成物からなるチップ(樹脂組成物チップ)であることが好ましい。この場合、例えば、多数のチップを容器等に敷き詰めて植物栽培用培地として使用することができる。
上記チップ(樹脂組成物チップ)は、例えば、上記樹脂組成物の溶融物をホットカットするなどして得られる略球状または略円盤状の形状や、上記樹脂組成物のストランドをカットするなどして得られた略円柱状の形状であってもよいし、フレーク状や不定形の形状であってもよい。上記チップは、柱状、扁平状またはフレーク状であることが好ましい。
また、上記チップ等の成形体のサイズにも特に制限はないが、例えば、最大長さとして1~50mmの範囲内であることが好ましく、1~20mmの範囲内であることがより好ましい。なお当該最大長さはノギスを用いて測定することができる。
上記成形体は、それを23℃の水に24時間浸漬した後の質量をW1とし、当該浸漬後の成形体を乾燥(105℃で24時間乾燥)した後の質量をW0とした際に、以下の式(2)で規定される吸水率が5質量%以上であることが好ましく、10質量%以上であることがより好ましい。吸水率が上記範囲にあることにより、保水性に優れた植物栽培用培地となる。なお、上記の吸水率の上限に特に制限はないが、成形体の強度の観点から300質量%以下であることが好ましい。
吸水率(質量%)=100×(W1-W0)/W0 (2)
吸水率(質量%)=100×(W1-W0)/W0 (2)
本発明の植物栽培用培地は、上記樹脂組成物からなる成形体のみから構成されていてもよいが、上記樹脂組成物からなる成形体とともに、ロックウール、砂、土、セラミックボール、ヤシガラ、バーク、ピートモス、水苔、その他の樹脂培地などの他の部材等をさらに含んでいてもよい。当該植物栽培用培地における上記成形体の含有率は、50質量%以上であることが好ましく、80質量%以上であることがより好ましく、95質量%以上であることがさらに好ましい。
本発明の植物栽培用培地の使用形態に特に制限はないが、培養液を用いた養液栽培用の培地として用いることが好ましい。当該植物栽培用培地を養液栽培用の培地として使用する場合に、その使用方法としては、例えば、当該植物栽培用培地をポット等の容器に入れ、これに培養液を加えた後に、播種したり苗を移植したりする方法などを例示することができる。また、当該植物栽培用培地が敷き詰められた栽培用ベッドを用意し、これに生育した苗を移植して各種作物を栽培する方法なども例示できる。
本発明の植物栽培用培地を用いて栽培する植物の種類に特に制限はなく、たとえば花卉、根菜類を含む野菜、果物類、穀類などが挙げられ、特に大根、さつまいも、ごぼう、にんじん、キュウリ、トマト、ナス、ピーマンなどの野菜の栽培に使用することが好ましい。特に、ロックウールでは栽培が困難であった根菜類にも好適に用いることができるという利点もある。
<植物栽培装置>
本発明はまた、上述した本発明の植物栽培用培地を備える植物栽培装置についても提供する。本発明の植物栽培装置は、上述の本発明の植物栽培用培地を用いたものであるならば特に制限されるものではなく、植物栽培用培地以外の構成は、従来公知の適宜の植物栽培装置の構成を備えていてもよい。
本発明はまた、上述した本発明の植物栽培用培地を備える植物栽培装置についても提供する。本発明の植物栽培装置は、上述の本発明の植物栽培用培地を用いたものであるならば特に制限されるものではなく、植物栽培用培地以外の構成は、従来公知の適宜の植物栽培装置の構成を備えていてもよい。
図6は、本発明の好ましい一例の植物栽培装置201を模式的に示す図である。図6に示す例の植物栽培装置201は、上方に開口203を有する箱状物であり、側壁204の適当な高さに排水口205を有するプランター202を備え、プランター202内に、排水口205から零れ出さない程度の高さ(深さ)にまで、養分を含んだ水(養液)206が収容される。プランター202の底壁207には、水206の面よりも上にその載置面208aが配置されるように棚208が設けられ、棚208の上に、吸水シート209が、上方から見てプランター202の底壁207を殆ど覆うように設けられる。この吸水シート209は、たとえばセルロース繊維、ナイロン繊維、ビニロン繊維、ポリエステル繊維、ポリオレフィン繊維、レーヨン繊維、アラミド繊維、ガラス繊維などの材料で形成されたシート状物であり、その中央部209aは棚208の載置面208a上にあり、かつ、その端部209bがプランター202内の水206に浸かるように設けられ、端部209bから吸収した20水6を、中央部209aに送るように構成されている。
図6に示す例では、吸水シート209上に、その端部210aがプランター202の側壁204の上端204aに引っかかるようにして防根透水シート210が配置される。防根透水シート210は、当該植物栽培装置201で根菜類を生育させる際に設けられることが好ましく、生育させる植物211が根菜類ではない場合には必ずしも設けなくともよい。このような防根透水シート210が設けられる場合、プランター202内の水206は、吸水シート209を介して防根透水シート210へと送られる。
防根透水シート210は、繊維状物で構成される織布、不織布、マット状物、あるいは、たとえば各種ポリオレフィン(ポリエチレン、ポリプロピレン、エチレン-プロピレン共重合体、エチレン-ビニルアルコール共重合体、エチレン-ビニルエステル共重合体、エチレン-アクリル酸エステル共重合体、又はこれらを不飽和カルボン酸で変性、若しくはその誘導体でグラフト変性、若しくは無水マレイン酸で変性した変性ポリオレフィンなど)、各種ナイロン(ナイロン-6、ナイロン-66、ナイロン-6/66共重合体など)、ポリ塩化ビニル、ポリ塩化ビニリデン、ポリエステル、ポリスチレン、ポリアクリロニトリル、ポリウレタン、ポリアセタール及び変性ポリビニルアルコールなどの樹脂からなるシート等であり、親水性、透水性及び柔軟性を有し、根を通さないシートである。上記樹脂からなるシートを防根透水シート210として用いる場合、微細孔を無数にかつ均一な分布で有していることが好ましく、この場合、微細孔の最大径は20μm以下であることが好ましい。微細孔の最大径を20μm以下とすることで、植物の根が防根透水シート210を貫通し、吸水シート209に侵入してからみつきにくくなるため、根が過剰に吸水することが抑制され、植物の成長がより良好になる。また微細孔の最大径をたとえば5μm以上とすることで、吸水シートからの水の浸出が生じ易くなり、植物の生育がより良好になる。このような防根透水シート210は、目が細かいため、水を通しにくく、水が保持されやすくなるという性質を有する。
図6に示す例では、防根透水シート210上に、上述した本発明の植物栽培用培地212が載せられ、その中で植物211が生育される。図6には、本発明の植物栽培用培地212として、上記樹脂組成物から形成されている複数個の円柱状のペレット(樹脂組成物チップ)213が用いられた例が示されている。
<植物栽培方法>
本発明は、上述した本発明の植物栽培用培地を用いた植物栽培方法についても提供する。すなわち本発明の植物栽培方法は、本発明の植物栽培用培地を用いて栽培する工程を備える植物栽培方法である。本発明の植物栽培方法において、上述した本発明の植物栽培用培地を養液栽培用の培地として使用する場合、たとえば本発明の植物栽培用培地をポットなどの容器に入れ、これに培養液を加えた後に、播種したり苗を移植したりする方法などを例示することができる。また本発明の植物栽培用培地が敷き詰められた栽培用ベッドを用意し、これに生育した苗を移植して各種作物を栽培する方法なども例示できる。
本発明は、上述した本発明の植物栽培用培地を用いた植物栽培方法についても提供する。すなわち本発明の植物栽培方法は、本発明の植物栽培用培地を用いて栽培する工程を備える植物栽培方法である。本発明の植物栽培方法において、上述した本発明の植物栽培用培地を養液栽培用の培地として使用する場合、たとえば本発明の植物栽培用培地をポットなどの容器に入れ、これに培養液を加えた後に、播種したり苗を移植したりする方法などを例示することができる。また本発明の植物栽培用培地が敷き詰められた栽培用ベッドを用意し、これに生育した苗を移植して各種作物を栽培する方法なども例示できる。
以下、実施例を示して本発明をさらに具体的に説明するが、本発明はこれらの例によって限定されるものではない。
[評価方法]
(1)EVOHのエチレン単位含有量及びケン化度
合成して得られたEVOHペレット並びに実施例及び比較例で得られたガスバリア樹脂組成物ペレットについて、内部標準物質としてテトラメチルシラン、添加剤としてテトラフルオロ酢酸(TFA)を含むジメチルスルホキシド(DMSO)-d6に溶解し、500MHzの1H-NMR(日本電子株式会社製「JMTC-400/54/SS」)を用いて80℃で測定し、エチレン単位含有量及びケン化度を測定した。
上記測定のスペクトル中の各ピークは、以下のように帰属される。
0.6~1.9ppm:エチレン単位のメチレンプロトン(4H)、ビニルアルコール単位のメチレンプロトン(2H)、酢酸ビニル単位のメチレンプロトン(2H)
1.9~2.0ppm:酢酸ビニル単位のメチルプロトン(3H)
3.1~4.2ppm:ビニルアルコール単位のメチンプロトン(1H)
(1)EVOHのエチレン単位含有量及びケン化度
合成して得られたEVOHペレット並びに実施例及び比較例で得られたガスバリア樹脂組成物ペレットについて、内部標準物質としてテトラメチルシラン、添加剤としてテトラフルオロ酢酸(TFA)を含むジメチルスルホキシド(DMSO)-d6に溶解し、500MHzの1H-NMR(日本電子株式会社製「JMTC-400/54/SS」)を用いて80℃で測定し、エチレン単位含有量及びケン化度を測定した。
上記測定のスペクトル中の各ピークは、以下のように帰属される。
0.6~1.9ppm:エチレン単位のメチレンプロトン(4H)、ビニルアルコール単位のメチレンプロトン(2H)、酢酸ビニル単位のメチレンプロトン(2H)
1.9~2.0ppm:酢酸ビニル単位のメチルプロトン(3H)
3.1~4.2ppm:ビニルアルコール単位のメチンプロトン(1H)
(2)EVOHの融点
合成して得られたEVOHペレットについて、TA Instruments製の示差走査型熱量計「Q2000」を用い、30℃から250℃までを10℃/分の速度で昇温し測定されるピーク温度より融点を求めた。
合成して得られたEVOHペレットについて、TA Instruments製の示差走査型熱量計「Q2000」を用い、30℃から250℃までを10℃/分の速度で昇温し測定されるピーク温度より融点を求めた。
(3)カルボン酸の定量
合成して得られたEVOHペレット又は実施例及び比較例で得られたガスバリア樹脂組成物ペレット20gとイオン交換水100mLとを共栓付き200mL三角フラスコに投入し、冷却コンデンサーを付け、95℃で6時間攪拌抽出した。得られた抽出液にフェノールフタレインを指示薬としてN/50のNaOHで中和滴定し、カルボン酸のカルボン酸根換算の含有量を定量した。なお、リン化合物が含まれる態様においては、後述の評価方法で測定されるリン化合物の含有量を加味して、カルボン酸量を算出した。
合成して得られたEVOHペレット又は実施例及び比較例で得られたガスバリア樹脂組成物ペレット20gとイオン交換水100mLとを共栓付き200mL三角フラスコに投入し、冷却コンデンサーを付け、95℃で6時間攪拌抽出した。得られた抽出液にフェノールフタレインを指示薬としてN/50のNaOHで中和滴定し、カルボン酸のカルボン酸根換算の含有量を定量した。なお、リン化合物が含まれる態様においては、後述の評価方法で測定されるリン化合物の含有量を加味して、カルボン酸量を算出した。
(4)金属イオン、リン酸化合物及びホウ素化合物の定量
合成して得られたEVOHペレット又は実施例及び比較例で得られたガスバリア樹脂組成物ペレット0.5gをテフロン(登録商標)製圧力容器に入れ、ここに濃硝酸5mLを加えて室温で30分間分解させた。30分後蓋をし、湿式分解装置(アクタック社の「MWS-2」)により150℃で10分間、次いで180℃で5分間加熱することで分解を行い、その後室温まで冷却した。この処理液を50mLのメスフラスコ(TPX製)に移し純水でメスアップした。この溶液について、ICP発光分光分析装置(パーキンエルマー社の「OPTIMA4300DV」)により元素分析を行い、EVOHペレット又はガスバリア樹脂組成物ペレットに含まれる、金属イオンの金属原子換算量、リン化合物のリン原子換算量及びホウ素化合物のホウ素原子換算量を求めた。
合成して得られたEVOHペレット又は実施例及び比較例で得られたガスバリア樹脂組成物ペレット0.5gをテフロン(登録商標)製圧力容器に入れ、ここに濃硝酸5mLを加えて室温で30分間分解させた。30分後蓋をし、湿式分解装置(アクタック社の「MWS-2」)により150℃で10分間、次いで180℃で5分間加熱することで分解を行い、その後室温まで冷却した。この処理液を50mLのメスフラスコ(TPX製)に移し純水でメスアップした。この溶液について、ICP発光分光分析装置(パーキンエルマー社の「OPTIMA4300DV」)により元素分析を行い、EVOHペレット又はガスバリア樹脂組成物ペレットに含まれる、金属イオンの金属原子換算量、リン化合物のリン原子換算量及びホウ素化合物のホウ素原子換算量を求めた。
(5)バイオベース度
合成して得られたEVOHペレット並びに実施例及び比較例で得られたガスバリア樹脂組成物ペレットについて、ASTM D6866-18に記載の方法に従い、加速器質量分析器(AMS)により放射性炭素(14C)の濃度を測定し、放射性炭素年代測定の原理に基づいて、バイオベース度を算出した。
合成して得られたEVOHペレット並びに実施例及び比較例で得られたガスバリア樹脂組成物ペレットについて、ASTM D6866-18に記載の方法に従い、加速器質量分析器(AMS)により放射性炭素(14C)の濃度を測定し、放射性炭素年代測定の原理に基づいて、バイオベース度を算出した。
(6)ロングラン性評価A
(6-1)単層フィルム製膜欠点評価
単軸押出装置(株式会社東洋精機製作所の「D2020」;D(mm)=20、L/D=20、圧縮比=3.0、スクリュー:フルフライト)を用い、実施例及び比較例で得られたガスバリア樹脂組成物ペレットから平均厚み20μmの単層フィルムを作製した。このときの各条件は以下に示す通りである。
(単軸押出装置条件)
押出温度:210℃
スクリュー回転数:40rpm
ダイス幅:30cm
引取りロール温度:80℃
引取りロール速度:3.1m/分
上記条件で連続運転して単層フィルムを作製し、運転開始10時間後及び50時間後に作製された各フィルムについて、フィルム長17cm当たりの欠点数をカウントした。上記欠点数のカウントは、フィルム欠点検査装置(フロンティアシステム社の「AI-10」) を用いて行った。なお、このフィルム欠点検査装置における検出カメラは、そのレンズ位置がフィルム面より195mmの距離となるように設置した。製膜欠点は、欠点数が50個未満の場合を「良好(A)」、50個以上200個未満の場合を「やや良好(B)」、200個以上の場合を「不良(C)」として判断した。
(6-1)単層フィルム製膜欠点評価
単軸押出装置(株式会社東洋精機製作所の「D2020」;D(mm)=20、L/D=20、圧縮比=3.0、スクリュー:フルフライト)を用い、実施例及び比較例で得られたガスバリア樹脂組成物ペレットから平均厚み20μmの単層フィルムを作製した。このときの各条件は以下に示す通りである。
(単軸押出装置条件)
押出温度:210℃
スクリュー回転数:40rpm
ダイス幅:30cm
引取りロール温度:80℃
引取りロール速度:3.1m/分
上記条件で連続運転して単層フィルムを作製し、運転開始10時間後及び50時間後に作製された各フィルムについて、フィルム長17cm当たりの欠点数をカウントした。上記欠点数のカウントは、フィルム欠点検査装置(フロンティアシステム社の「AI-10」) を用いて行った。なお、このフィルム欠点検査装置における検出カメラは、そのレンズ位置がフィルム面より195mmの距離となるように設置した。製膜欠点は、欠点数が50個未満の場合を「良好(A)」、50個以上200個未満の場合を「やや良好(B)」、200個以上の場合を「不良(C)」として判断した。
(6-2)単層フィルム外観性の評価
運転開始10時間後及び50時間後に作製されたフィルムについて、目視にて外観性(ストリーク)を下記評価基準により評価した。また、フィルム100mを紙管に巻き取ったロールを作製し、ロールの端部の黄変による外観性(着色)を目視で下記評価基準により評価した。
(ストリークの評価基準)
良好(A):ストリークは認められなかった
やや良好(B):ストリークが確認された
不良(C):多数のストリークが確認された
(ロール端部の着色の評価基準)
良好(A):無色
やや良好(B):黄変
不良(C):著しく黄変
運転開始10時間後及び50時間後に作製されたフィルムについて、目視にて外観性(ストリーク)を下記評価基準により評価した。また、フィルム100mを紙管に巻き取ったロールを作製し、ロールの端部の黄変による外観性(着色)を目視で下記評価基準により評価した。
(ストリークの評価基準)
良好(A):ストリークは認められなかった
やや良好(B):ストリークが確認された
不良(C):多数のストリークが確認された
(ロール端部の着色の評価基準)
良好(A):無色
やや良好(B):黄変
不良(C):著しく黄変
(7)酸素透過度A
実施例及び比較例で得られたガスバリア樹脂組成物ペレットを用いて、下記条件で厚み20μmの単層フィルムを製膜し、20℃/65%RHの条件下で調湿した後、酸素透過度測定装置(ModernControlの「OX-Tran2/20」)を使用し、20℃/65%RHの条件下で酸素透過度を測定した。なお、本測定はJIS K 7126-2(等圧法;2006年)に準拠して実施した。
(単層フィルムの作製)
単軸押出装置(東洋精機製作所の「D2020」、D(mm)=20、L/D=20、圧縮比=3.0、スクリュー:フルフライト)を用い、上記ガスバリア樹脂組成物ペレットから厚み20μmの単層フィルムを作製した。押出条件は以下に示すとおりである。
押出温度:210℃
ダイス幅:30cm
引取りロール温度:80℃
スクリュー回転数:40rpm
引取りロール速度:3.1m/分
実施例及び比較例で得られたガスバリア樹脂組成物ペレットを用いて、下記条件で厚み20μmの単層フィルムを製膜し、20℃/65%RHの条件下で調湿した後、酸素透過度測定装置(ModernControlの「OX-Tran2/20」)を使用し、20℃/65%RHの条件下で酸素透過度を測定した。なお、本測定はJIS K 7126-2(等圧法;2006年)に準拠して実施した。
(単層フィルムの作製)
単軸押出装置(東洋精機製作所の「D2020」、D(mm)=20、L/D=20、圧縮比=3.0、スクリュー:フルフライト)を用い、上記ガスバリア樹脂組成物ペレットから厚み20μmの単層フィルムを作製した。押出条件は以下に示すとおりである。
押出温度:210℃
ダイス幅:30cm
引取りロール温度:80℃
スクリュー回転数:40rpm
引取りロール速度:3.1m/分
(8)熱成形容器(成形性)評価
実施例及び比較例で得られたガスバリア樹脂組成物ペレット、ポリプロピレン(日本ポリプロ株式会社製「ノバテック(登録商標)PP EA7AD」)、及び接着性樹脂(三井化学株式会社製「アドマー(登録商標)QF551」)を用い、3種5層共押出装置を用いて、下記条件にて多層シート(ポリプロピレン/接着性樹脂/ガスバリア樹脂組成物/接着性樹脂/ポリプロピレン、厚み(μm):368/16/32/16/368)を作成した。
(押出機条件)
各樹脂の押出温度:供給部/圧縮部/計量部/ダイ=150℃/150℃/210℃/210℃
ポリプロピレン樹脂の押出機:32φ単軸押出機、GT-32-A型(株式会社プラスチック工学研究所製)
接着性樹脂の押出機:25φ単軸押出機、P25-18-AC型(大阪精機工作株式会社製)
EVOH樹脂組成物の押出機:20φ押出機、ラボ機ME型CO-EXT(株式会社東洋精機製作所製)
Tダイ:300mm幅3種5層用(株式会社プラスチック工学研究所製)
冷却ロールの温度:80℃
引取速度:1m/分
運転開始30分後、10時間後、50時間後に作製された多層シートを採取し、得られた多層シートを熱成形機(株式会社浅野製作所製:真空圧空深絞り成形機「FX-0431-3型」)にて、シート温度を160℃にして、圧縮空気(気圧5kgf/cm2)により、丸カップ形状(金型形状:上部75mmφ、下部60mmφ、深さ75mm、絞り比S=1.0)に熱成形することにより、熱成形容器を得た。成形条件を以下に示す。
ヒーター温度:400℃
プラグ :45φ×65mm
金型温度 :40℃
目視にて得られたカップ形状の熱成形容器の外観を下記評価基準により評価した。
(外観の評価基準)
良好(A):ムラおよび局部的偏肉は認められなかった
やや良好(B):わずかなムラおよび局部的偏肉が確認された
不良(C):著しいムラおよび局部的偏肉が確認された
実施例及び比較例で得られたガスバリア樹脂組成物ペレット、ポリプロピレン(日本ポリプロ株式会社製「ノバテック(登録商標)PP EA7AD」)、及び接着性樹脂(三井化学株式会社製「アドマー(登録商標)QF551」)を用い、3種5層共押出装置を用いて、下記条件にて多層シート(ポリプロピレン/接着性樹脂/ガスバリア樹脂組成物/接着性樹脂/ポリプロピレン、厚み(μm):368/16/32/16/368)を作成した。
(押出機条件)
各樹脂の押出温度:供給部/圧縮部/計量部/ダイ=150℃/150℃/210℃/210℃
ポリプロピレン樹脂の押出機:32φ単軸押出機、GT-32-A型(株式会社プラスチック工学研究所製)
接着性樹脂の押出機:25φ単軸押出機、P25-18-AC型(大阪精機工作株式会社製)
EVOH樹脂組成物の押出機:20φ押出機、ラボ機ME型CO-EXT(株式会社東洋精機製作所製)
Tダイ:300mm幅3種5層用(株式会社プラスチック工学研究所製)
冷却ロールの温度:80℃
引取速度:1m/分
運転開始30分後、10時間後、50時間後に作製された多層シートを採取し、得られた多層シートを熱成形機(株式会社浅野製作所製:真空圧空深絞り成形機「FX-0431-3型」)にて、シート温度を160℃にして、圧縮空気(気圧5kgf/cm2)により、丸カップ形状(金型形状:上部75mmφ、下部60mmφ、深さ75mm、絞り比S=1.0)に熱成形することにより、熱成形容器を得た。成形条件を以下に示す。
ヒーター温度:400℃
プラグ :45φ×65mm
金型温度 :40℃
目視にて得られたカップ形状の熱成形容器の外観を下記評価基準により評価した。
(外観の評価基準)
良好(A):ムラおよび局部的偏肉は認められなかった
やや良好(B):わずかなムラおよび局部的偏肉が確認された
不良(C):著しいムラおよび局部的偏肉が確認された
(9)二軸延伸フィルム評価
(9-1)耐フィルム破断性評価
実施例及び比較例で得られた二軸延伸フィルムについて、2本目から101本目までのフィルムロールをスリッターにかけ、フィルムロールに100N/mの張力をかけて巻きとったときの破断回数を測定し、耐フィルム破断性として評価した。判定基準は以下の通りである。
(判定基準)
A:0~1回/100本
B:2~4回/100本
C:5~7回/100本
D:8~10回/100本
E:11回以上/100本
(9-1)耐フィルム破断性評価
実施例及び比較例で得られた二軸延伸フィルムについて、2本目から101本目までのフィルムロールをスリッターにかけ、フィルムロールに100N/mの張力をかけて巻きとったときの破断回数を測定し、耐フィルム破断性として評価した。判定基準は以下の通りである。
(判定基準)
A:0~1回/100本
B:2~4回/100本
C:5~7回/100本
D:8~10回/100本
E:11回以上/100本
(9-2)酸素透過度
実施例及び比較例で得られた二軸延伸フィルムについて、2本目の二軸延伸フィルムを20℃/65%RHの条件下で調湿した後、酸素透過度測定装置(ModernControlの「OX-Tran2/20」)を使用し、20℃/65%RHの条件下で酸素透過度を測定した。なお、本測定はJIS K 7126-2(等圧法;2006年)に準拠して実施した。
実施例及び比較例で得られた二軸延伸フィルムについて、2本目の二軸延伸フィルムを20℃/65%RHの条件下で調湿した後、酸素透過度測定装置(ModernControlの「OX-Tran2/20」)を使用し、20℃/65%RHの条件下で酸素透過度を測定した。なお、本測定はJIS K 7126-2(等圧法;2006年)に準拠して実施した。
(10)蒸着フィルム評価
(10-1)無機蒸着層の平均厚みの測定
実施例及び比較例で得られた蒸着フィルムをミクロトームでカットし断面を露出させた。この断面を走査型電子顕微鏡(SEM)(エス・アイ・アイナノテクノロジー社の「ZEISS ULTRA 55」)を用いて観察し、反射電子検出器を用いて10点における無機蒸着層の厚みを測定し、平均厚みを求めた。
(10-1)無機蒸着層の平均厚みの測定
実施例及び比較例で得られた蒸着フィルムをミクロトームでカットし断面を露出させた。この断面を走査型電子顕微鏡(SEM)(エス・アイ・アイナノテクノロジー社の「ZEISS ULTRA 55」)を用いて観察し、反射電子検出器を用いて10点における無機蒸着層の厚みを測定し、平均厚みを求めた。
(10-2)蒸着欠点抑制性
実施例及び比較例で得られた蒸着フィルムの蒸着欠点抑制性を、蒸着欠点数の測定により行った。蒸着フィルムロールをスリッターにかけて、フィルム下部から100Wの蛍光灯を当てながら巻きだし、幅0.5m、長さ2mの領域について異なる10箇所で蒸着欠点数を目視で数え、その平均値を1m2あたりの蒸着欠点数とした。蒸着欠点は、以下の基準で評価した。
(判定基準)
A:0~20個/m2
B:21~40個/m2
C:41~60個/m2
D:61~80個/m2
E:81~100個/m2
F:101個以上/m2
実施例及び比較例で得られた蒸着フィルムの蒸着欠点抑制性を、蒸着欠点数の測定により行った。蒸着フィルムロールをスリッターにかけて、フィルム下部から100Wの蛍光灯を当てながら巻きだし、幅0.5m、長さ2mの領域について異なる10箇所で蒸着欠点数を目視で数え、その平均値を1m2あたりの蒸着欠点数とした。蒸着欠点は、以下の基準で評価した。
(判定基準)
A:0~20個/m2
B:21~40個/m2
C:41~60個/m2
D:61~80個/m2
E:81~100個/m2
F:101個以上/m2
(10-3)無機蒸着層と二軸延伸フィルムとの密着強度の測定
得られた蒸着フィルムの無機蒸着層と二軸延伸フィルムとの密着強度を下記方法により測定した。得られた蒸着フィルムの無機蒸着層側の表面に、ドライラミネート用接着剤(三井化学製タケラックA-385/A-50を6/1の質量比で混合し、固形分濃度23質量%の酢酸エチル溶液としたもの)を、バーコーターを用いてコートし、50℃で5分間熱風乾燥させた後、80℃に加熱したニップロールにて、PETフィルム(東洋紡製E5000)とラミネートを行った。このとき、フィルムの半分は、間にアルミホイルを挟むことで貼りあわされない部分を設けた。その後、40℃で72時間養生し、ラミネートフィルムを得た。得られたラミネートフィルムを100mm×15mmの短冊に裁断し、引っ張り試験機により引っ張り速度10mm/分にてT型剥離試験を5回行った。得られた測定値の平均値を密着強度とした。密着強度は、以下のように判定した。なお、剥離界面を目視で確認すると、500g/15mm未満の剥離強度を有する蒸着フィルムにおいては、無機蒸着層と二軸延伸フィルム層との界面で剥離が起こっていた。
(判定基準)
A:500g/15mm以上
B:450以上500g/15mm未満
C:400以上450g/15mm未満
D:350以上400g/15mm未満
E:350g/15mm未満
得られた蒸着フィルムの無機蒸着層と二軸延伸フィルムとの密着強度を下記方法により測定した。得られた蒸着フィルムの無機蒸着層側の表面に、ドライラミネート用接着剤(三井化学製タケラックA-385/A-50を6/1の質量比で混合し、固形分濃度23質量%の酢酸エチル溶液としたもの)を、バーコーターを用いてコートし、50℃で5分間熱風乾燥させた後、80℃に加熱したニップロールにて、PETフィルム(東洋紡製E5000)とラミネートを行った。このとき、フィルムの半分は、間にアルミホイルを挟むことで貼りあわされない部分を設けた。その後、40℃で72時間養生し、ラミネートフィルムを得た。得られたラミネートフィルムを100mm×15mmの短冊に裁断し、引っ張り試験機により引っ張り速度10mm/分にてT型剥離試験を5回行った。得られた測定値の平均値を密着強度とした。密着強度は、以下のように判定した。なお、剥離界面を目視で確認すると、500g/15mm未満の剥離強度を有する蒸着フィルムにおいては、無機蒸着層と二軸延伸フィルム層との界面で剥離が起こっていた。
(判定基準)
A:500g/15mm以上
B:450以上500g/15mm未満
C:400以上450g/15mm未満
D:350以上400g/15mm未満
E:350g/15mm未満
(11)耐酸化劣化性
上記(7)で得られた単層フィルムについて、以下の評価条件にて加熱処理時間を変えた複数のサンプルを測定することで、引張破断伸度の経時変化を評価した。引張破断伸度が加熱処理を行っていないサンプルの1/4になる時間を求め、耐酸化劣化性の指標とした。
評価条件:
所定の時間、140℃に設定した熱風乾燥機内で処理した後、取り出した。その後、20℃の水中に5日間浸漬し、表面水を拭き取って、20℃-65%RHの室内に2週間静置してから、以下の条件で引張強伸度測定を行った。
測定条件
サンプル幅15mm
チャック間隔30mm
引張スピード50mm/分
測定雰囲気20℃-65%RH
上記(7)で得られた単層フィルムについて、以下の評価条件にて加熱処理時間を変えた複数のサンプルを測定することで、引張破断伸度の経時変化を評価した。引張破断伸度が加熱処理を行っていないサンプルの1/4になる時間を求め、耐酸化劣化性の指標とした。
評価条件:
所定の時間、140℃に設定した熱風乾燥機内で処理した後、取り出した。その後、20℃の水中に5日間浸漬し、表面水を拭き取って、20℃-65%RHの室内に2週間静置してから、以下の条件で引張強伸度測定を行った。
測定条件
サンプル幅15mm
チャック間隔30mm
引張スピード50mm/分
測定雰囲気20℃-65%RH
上記評価において、破断伸度が1/4以下になると酸化劣化によるクラックの発生に起因するEVOH層(ガスバリア樹脂組成物から形成される層)のガスバリア性の悪化が顕著になることから、破断伸度が1/4になる時間を、EVOHの高温での酸化劣化による寿命の指標の一つとして考えることができる。破断伸度が1/4になる時間はアレニウス型の温度依存性を示し、80℃において破断伸度が1/4になる時間(寿命)を100年以上にしようとすると、140℃において破断伸度が1/4になる時間を210時間以上にする必要がある。
(12)多層パイプ評価
高密度ポリエチレン(日本ポリエチレン株式会社製「ノバテック(登録商標)HD HE421」、密度0.956g/cc、MFRが0.14g/10分)を1台目の押出機に、実施例及び比較例で得られた各ガスバリア樹脂組成物ペレットを2台目の押出機に、接着性樹脂として三井化学株式会社製「アドマー(登録商標)NF408E」を3台目の押出機に入れ、3種3層の円形ダイを用いて、外径20mmの多層パイプを押出成形し、直後に40℃に調整した冷却水槽を通して冷却して固化させた。多層パイプの層構成は樹脂組成物層が最外層であり、ガスバリア樹脂組成物層/接着性樹脂層/高密度ポリエチレン層=100μm/100μm/2000μmであった。運転開始10時間後及び50時間後に作製されたパイプについて、パイプ円周方向に切断、切断面よりガスバリア樹脂組成物層の円周方向の厚薄を顕微鏡で確認した。厚薄変動が存在する場合はストリークがあると判断し、下記評価基準により評価した。
(ストリークの評価基準)
良好(A):ストリークは確認されなかった
やや良好(B):わずかにストリークが確認された
不良(C):著しいストリークが確認された
高密度ポリエチレン(日本ポリエチレン株式会社製「ノバテック(登録商標)HD HE421」、密度0.956g/cc、MFRが0.14g/10分)を1台目の押出機に、実施例及び比較例で得られた各ガスバリア樹脂組成物ペレットを2台目の押出機に、接着性樹脂として三井化学株式会社製「アドマー(登録商標)NF408E」を3台目の押出機に入れ、3種3層の円形ダイを用いて、外径20mmの多層パイプを押出成形し、直後に40℃に調整した冷却水槽を通して冷却して固化させた。多層パイプの層構成は樹脂組成物層が最外層であり、ガスバリア樹脂組成物層/接着性樹脂層/高密度ポリエチレン層=100μm/100μm/2000μmであった。運転開始10時間後及び50時間後に作製されたパイプについて、パイプ円周方向に切断、切断面よりガスバリア樹脂組成物層の円周方向の厚薄を顕微鏡で確認した。厚薄変動が存在する場合はストリークがあると判断し、下記評価基準により評価した。
(ストリークの評価基準)
良好(A):ストリークは確認されなかった
やや良好(B):わずかにストリークが確認された
不良(C):著しいストリークが確認された
(13)ロングラン性評価B
(13-1)単層フィルム製膜欠点評価
単軸押出装置(株式会社東洋精機製作所の「D2020」;D(mm)=20、L/D=20、圧縮比=3.0、スクリュー:フルフライト)を用い、実施例及び比較例で得られたガスバリア樹脂組成物ペレットから平均厚み20μmの単層フィルムを作製した。このときの各条件は以下に示す通りである。
(単軸押出装置条件)
押出温度:230℃
スクリュー回転数:40rpm
ダイス幅:30cm
引取りロール温度:80℃
引取りロール速度:3.1m/分
上記条件で連続運転して単層フィルムを作製し、運転開始1時間後及び5時間後に作製された各フィルムについて、フィルム長17cm当たりの欠点数をカウントした。上記欠点数のカウントは、フィルム欠点検査装置(フロンティアシステム社の「AI-10」)を用いて行った。なお、このフィルム欠点検査装置における検出カメラは、そのレンズ位置がフィルム面より195mmの距離となるように設置した。製膜欠点は、欠点数が50個未満の場合を「良好(A)」、50個以上200個未満の場合を「やや良好(B)」、200個以上の場合を「不良(C)」として判断した。
(13-1)単層フィルム製膜欠点評価
単軸押出装置(株式会社東洋精機製作所の「D2020」;D(mm)=20、L/D=20、圧縮比=3.0、スクリュー:フルフライト)を用い、実施例及び比較例で得られたガスバリア樹脂組成物ペレットから平均厚み20μmの単層フィルムを作製した。このときの各条件は以下に示す通りである。
(単軸押出装置条件)
押出温度:230℃
スクリュー回転数:40rpm
ダイス幅:30cm
引取りロール温度:80℃
引取りロール速度:3.1m/分
上記条件で連続運転して単層フィルムを作製し、運転開始1時間後及び5時間後に作製された各フィルムについて、フィルム長17cm当たりの欠点数をカウントした。上記欠点数のカウントは、フィルム欠点検査装置(フロンティアシステム社の「AI-10」)を用いて行った。なお、このフィルム欠点検査装置における検出カメラは、そのレンズ位置がフィルム面より195mmの距離となるように設置した。製膜欠点は、欠点数が50個未満の場合を「良好(A)」、50個以上200個未満の場合を「やや良好(B)」、200個以上の場合を「不良(C)」として判断した。
(13-2)単層フィルム外観性の評価
運転開始1時間後及び5時間後に作製されたフィルムについて、目視にて外観性(ストリーク)を下記評価基準により評価した。また、フィルム100mを紙管に巻き取ったロールを作製し、ロールの端部の黄変による外観性(着色)を目視で下記評価基準により評価した。
(ストリークの評価基準)
良好(A):ストリークは認められなかった
やや良好(B):ストリークが確認された
不良(C):多数のストリークが確認された
(ロール端部の着色の評価基準)
良好(A):無色
やや良好(B):黄変
不良(C):著しく黄変
運転開始1時間後及び5時間後に作製されたフィルムについて、目視にて外観性(ストリーク)を下記評価基準により評価した。また、フィルム100mを紙管に巻き取ったロールを作製し、ロールの端部の黄変による外観性(着色)を目視で下記評価基準により評価した。
(ストリークの評価基準)
良好(A):ストリークは認められなかった
やや良好(B):ストリークが確認された
不良(C):多数のストリークが確認された
(ロール端部の着色の評価基準)
良好(A):無色
やや良好(B):黄変
不良(C):著しく黄変
(14)酸素透過度B
実施例及び比較例で得られたガスバリア樹脂組成物ペレットを用いて、下記条件で厚み20μmの単層フィルムを製膜し、20℃/65%RHの条件下で調湿した後、酸素透過度測定装置(ModernControlの「OX-Tran2/20」)を使用し、20℃/65%RHの条件下で酸素透過度を測定した。なお、本測定はJIS K 7126-2(等圧法;2006年)に準拠して実施した。
(単層フィルムの作製)
単軸押出装置(株式会社東洋精機製作所の「D2020」、D(mm)=20、L/D=20、圧縮比=3.0、スクリュー:フルフライト)を用い、上記ガスバリア樹脂組成物ペレットから厚み20μmの単層フィルムを作製した。押出条件は以下に示すとおりである。
押出温度:230℃
ダイス幅:30cm
引取りロール温度:80℃
スクリュー回転数:40rpm
引取りロール速度:3.1m/分
実施例及び比較例で得られたガスバリア樹脂組成物ペレットを用いて、下記条件で厚み20μmの単層フィルムを製膜し、20℃/65%RHの条件下で調湿した後、酸素透過度測定装置(ModernControlの「OX-Tran2/20」)を使用し、20℃/65%RHの条件下で酸素透過度を測定した。なお、本測定はJIS K 7126-2(等圧法;2006年)に準拠して実施した。
(単層フィルムの作製)
単軸押出装置(株式会社東洋精機製作所の「D2020」、D(mm)=20、L/D=20、圧縮比=3.0、スクリュー:フルフライト)を用い、上記ガスバリア樹脂組成物ペレットから厚み20μmの単層フィルムを作製した。押出条件は以下に示すとおりである。
押出温度:230℃
ダイス幅:30cm
引取りロール温度:80℃
スクリュー回転数:40rpm
引取りロール速度:3.1m/分
(15)耐レトルト性評価試験
上記評価方法(14)で得られた厚さ20μmの単層フィルム、二軸延伸ナイロン6フィルム(ユニチカ株式会社製「エンブレム(登録商標)ON」、厚み15μm)及び無延伸ポリプロピレンフィルム(三井化学東セロ株式会社製、ト-セロCP、厚み60μm)をそれぞれA4サイズにカットし、単層フィルムの両面にドライラミネート用接着剤(「タケラック(登録商標)A-385」(三井化学株式会社製)を主剤、「タケネ-ト(登録商標)A-50」(三井化学株式会社製)を硬化剤、希釈液として酢酸エチルを用いたもの)を4.0g/mで塗布し、外層がナイロン6フィルム、内層が無延伸ポリプロピレンフィルムとなるようドライラミネ-トを実施し、得られたラミネートフィルムを80℃で3分間乾燥させて希釈液を蒸発させた。その後、40℃で3日間養生し、3層からなる透明な多層シートを得た。
上記評価方法(14)で得られた厚さ20μmの単層フィルム、二軸延伸ナイロン6フィルム(ユニチカ株式会社製「エンブレム(登録商標)ON」、厚み15μm)及び無延伸ポリプロピレンフィルム(三井化学東セロ株式会社製、ト-セロCP、厚み60μm)をそれぞれA4サイズにカットし、単層フィルムの両面にドライラミネート用接着剤(「タケラック(登録商標)A-385」(三井化学株式会社製)を主剤、「タケネ-ト(登録商標)A-50」(三井化学株式会社製)を硬化剤、希釈液として酢酸エチルを用いたもの)を4.0g/mで塗布し、外層がナイロン6フィルム、内層が無延伸ポリプロピレンフィルムとなるようドライラミネ-トを実施し、得られたラミネートフィルムを80℃で3分間乾燥させて希釈液を蒸発させた。その後、40℃で3日間養生し、3層からなる透明な多層シートを得た。
得られた多層シートを用いて、内寸が12×12cmとなるように四方をシ-ルしたパウチを作製した。内容物は水とした。これをレトルト装置(株式会社日阪製作所製、高温高圧調理殺菌試験機、RCS-40RTGN)を使用して、120℃で20分のレトルト処理を実施した。レトルト処理後、表面水を拭き20℃、65%RHの高温高湿の部屋で1日静置してから、以下の基準で耐レトルト性を評価した。
(耐レトルト性の評価基準)
A(良好):透明性が確保されていた
B(不良):まだらに白化していた
(耐レトルト性の評価基準)
A(良好):透明性が確保されていた
B(不良):まだらに白化していた
(16)ロングラン性評価C
(16-1)単層フィルム製膜欠点評価
単軸押出装置(株式会社東洋精機製作所の「D2020」;D(mm)=20、L/D=20、圧縮比=3.0、スクリュー:フルフライト)を用い、実施例及び比較例で得られたガスバリア樹脂組成物ペレットから平均厚み20μmの単層フィルムを作製した。このときの各条件は以下に示す通りである。
(単軸押出装置条件)
押出温度:210℃
スクリュー回転数:40rpm
ダイス幅:30cm
引取りロール温度:80℃
引取りロール速度:3.1m/分
上記条件で連続運転して単層フィルムを作製し、運転開始1時間後及び5時間後に作製された各フィルムについて、フィルム長17cm当たりの欠点数をカウントした。上記欠点数のカウントは、フィルム欠点検査装置(フロンティアシステム社の「AI-10」)を用いて行った。なお、このフィルム欠点検査装置における検出カメラは、そのレンズ位置がフィルム面より195mmの距離となるように設置した。製膜欠点は、欠点数が50個未満の場合を「良好(A)」、50個以上200個未満の場合を「やや良好(B)」、200個以上の場合を「不良(C)」として判断した。
(16-1)単層フィルム製膜欠点評価
単軸押出装置(株式会社東洋精機製作所の「D2020」;D(mm)=20、L/D=20、圧縮比=3.0、スクリュー:フルフライト)を用い、実施例及び比較例で得られたガスバリア樹脂組成物ペレットから平均厚み20μmの単層フィルムを作製した。このときの各条件は以下に示す通りである。
(単軸押出装置条件)
押出温度:210℃
スクリュー回転数:40rpm
ダイス幅:30cm
引取りロール温度:80℃
引取りロール速度:3.1m/分
上記条件で連続運転して単層フィルムを作製し、運転開始1時間後及び5時間後に作製された各フィルムについて、フィルム長17cm当たりの欠点数をカウントした。上記欠点数のカウントは、フィルム欠点検査装置(フロンティアシステム社の「AI-10」)を用いて行った。なお、このフィルム欠点検査装置における検出カメラは、そのレンズ位置がフィルム面より195mmの距離となるように設置した。製膜欠点は、欠点数が50個未満の場合を「良好(A)」、50個以上200個未満の場合を「やや良好(B)」、200個以上の場合を「不良(C)」として判断した。
(16-2)単層フィルム外観性の評価
運転開始1時間後及び5時間後に作製されたフィルムについて、目視にて外観性(ストリーク)を下記評価基準により評価した。また、フィルム100mを紙管に巻き取ったロールを作製し、ロールの端部の黄変による外観性(着色)を目視で下記評価基準により評価した。
(ストリークの評価基準)
良好(A):ストリークは認められなかった
やや良好(B):ストリークが確認された
不良(C):多数のストリークが確認された
(ロール端部の着色の評価基準)
良好(A):無色
やや良好(B):黄変
不良(C):著しく黄変
運転開始1時間後及び5時間後に作製されたフィルムについて、目視にて外観性(ストリーク)を下記評価基準により評価した。また、フィルム100mを紙管に巻き取ったロールを作製し、ロールの端部の黄変による外観性(着色)を目視で下記評価基準により評価した。
(ストリークの評価基準)
良好(A):ストリークは認められなかった
やや良好(B):ストリークが確認された
不良(C):多数のストリークが確認された
(ロール端部の着色の評価基準)
良好(A):無色
やや良好(B):黄変
不良(C):著しく黄変
(17)耐屈曲性評価
上記(7)で得られた20μmの単層フィルムについて、ASTM F392-74に準じて、テスター産業株式会社製「BE1006恒温槽付ゲルボフレックステスター」を使用し、5℃の環境下、屈曲を100回繰り返した。屈曲後の単層フィルムをろ紙上に置き、フィルム上にインクを塗布した後、ろ紙上に通過したインクの点の数を測定し、かかるインクの点の数を、屈曲後のフィルムのピンホール数とした。測定は各3サンプルについて行い、その平均値を求め下記評価基準により評価した。屈曲後のピンホール数が少ない程、耐屈曲性に優れる。
(耐屈曲性の評価基準)
良好(A):ピンホールの平均値が5未満
やや良好(B):ピンホールの平均値が15未満
不良(C):ピンホールの平均値が15以上
上記(7)で得られた20μmの単層フィルムについて、ASTM F392-74に準じて、テスター産業株式会社製「BE1006恒温槽付ゲルボフレックステスター」を使用し、5℃の環境下、屈曲を100回繰り返した。屈曲後の単層フィルムをろ紙上に置き、フィルム上にインクを塗布した後、ろ紙上に通過したインクの点の数を測定し、かかるインクの点の数を、屈曲後のフィルムのピンホール数とした。測定は各3サンプルについて行い、その平均値を求め下記評価基準により評価した。屈曲後のピンホール数が少ない程、耐屈曲性に優れる。
(耐屈曲性の評価基準)
良好(A):ピンホールの平均値が5未満
やや良好(B):ピンホールの平均値が15未満
不良(C):ピンホールの平均値が15以上
(18)多層フィルムの評価
(18-1)ロングラン性の評価
実施例及び比較例で得られたガスバリア樹脂組成物ペレットを用い、3種5層共押出機を用いて、下記条件にて、多層フィルム(ポリエチレン層/接着性樹脂層/ガスバリア樹脂組成物層/接着性樹脂層/ポリエチレン層、厚み(μm):60/10/10/10/60)を作成した。ポリエチレンとして日本ポリエチレン株式会社製「ノバテック(商標)UF943」を、接着性樹脂として三井化学株式会社製「アドマー(商標)NF528」を用いた。
(押出機条件)
各樹脂の押出温度:供給部/圧縮部/計量部/ダイ=170℃/170℃/210℃/210℃
ポリエチレンの押出機:32φ単軸押出機、GT-32-A型(株式会社プラスチック工学研究所製)
接着性樹脂の押出機:25φ単軸押出機、P25-18-AC型(大阪精機工作株式会社製)
ガスバリア樹脂組成物の押出機:20φ単軸押出機、ラボ機ME型CO-EXT(株式会社東洋精機製作所製)
Tダイ:300mm幅3種5層用(株式会社プラスチック工学研究所製)
冷却ロールの温度:50℃
引取速度:4m/分
運転開始10時間後、50時間後に作製された多層フィルムについて、目視にてストリークの有無を下記評価基準により評価した。また、多層フィルム100mを紙管に巻き取ったロールを作製し、ロールの端部の黄変の有無を目視で下記評価基準により評価した。
(ストリークの評価基準)
A(良好):ストリークは認められなかった
B(やや良好):ストリークが確認された
C(不良):多数のストリークが確認された
(ロール端部の着色の評価基準)
A(良好):無色
B(やや良好):黄変
C(不良):著しく黄変
(18-2)酸素透過度の測定
上記(18-1)で運転開始から30分後に作製された多層フィルムを20℃、65%RHの条件下で調湿した後、酸素透過度測定装置(Mocon Modern Controls.incの「OX-Tran2/20」)を使用し、20℃、65%RHの条件下で、JIS K 7126-2(等圧法;2006年)に記載の方法に準じて酸素透過度を測定した。
(18-1)ロングラン性の評価
実施例及び比較例で得られたガスバリア樹脂組成物ペレットを用い、3種5層共押出機を用いて、下記条件にて、多層フィルム(ポリエチレン層/接着性樹脂層/ガスバリア樹脂組成物層/接着性樹脂層/ポリエチレン層、厚み(μm):60/10/10/10/60)を作成した。ポリエチレンとして日本ポリエチレン株式会社製「ノバテック(商標)UF943」を、接着性樹脂として三井化学株式会社製「アドマー(商標)NF528」を用いた。
(押出機条件)
各樹脂の押出温度:供給部/圧縮部/計量部/ダイ=170℃/170℃/210℃/210℃
ポリエチレンの押出機:32φ単軸押出機、GT-32-A型(株式会社プラスチック工学研究所製)
接着性樹脂の押出機:25φ単軸押出機、P25-18-AC型(大阪精機工作株式会社製)
ガスバリア樹脂組成物の押出機:20φ単軸押出機、ラボ機ME型CO-EXT(株式会社東洋精機製作所製)
Tダイ:300mm幅3種5層用(株式会社プラスチック工学研究所製)
冷却ロールの温度:50℃
引取速度:4m/分
運転開始10時間後、50時間後に作製された多層フィルムについて、目視にてストリークの有無を下記評価基準により評価した。また、多層フィルム100mを紙管に巻き取ったロールを作製し、ロールの端部の黄変の有無を目視で下記評価基準により評価した。
(ストリークの評価基準)
A(良好):ストリークは認められなかった
B(やや良好):ストリークが確認された
C(不良):多数のストリークが確認された
(ロール端部の着色の評価基準)
A(良好):無色
B(やや良好):黄変
C(不良):著しく黄変
(18-2)酸素透過度の測定
上記(18-1)で運転開始から30分後に作製された多層フィルムを20℃、65%RHの条件下で調湿した後、酸素透過度測定装置(Mocon Modern Controls.incの「OX-Tran2/20」)を使用し、20℃、65%RHの条件下で、JIS K 7126-2(等圧法;2006年)に記載の方法に準じて酸素透過度を測定した。
(19)熱収縮フィルムの熱収縮性評価
実施例及び比較例で得られたガスバリア樹脂組成物ペレット、エチレン-酢酸ビニル共重合体(EVA)(三井デュポンポリケミカル株式会社製「エバフレックス(商標)EV340」)、および接着性樹脂(三井化学株式会社性「アドマー(商標)VF500」)を用い、3種5層共押出機を用いて、下記条件にて多層フィルム(層構成:EVA層/接着性樹脂層/ガスバリア樹脂組成物層/接着性樹脂層/EVA層、厚み(μm):300/50/50/50/300)を作製した。
(押出機条件)
各樹脂の押出温度:供給部/圧縮部/計量部/ダイ=170℃/170℃/210℃/210℃
EVAの押出機:32φ単軸押出機、GT-32-A型(株式会社プラスチック工学研究所製)
接着性樹脂の押出機:25φ単軸押出機、P25-18-AC型(大阪精機工作株式会社製)
EVOH樹脂組成物の押出機:20φ単軸押出機、ラボ機ME型CO-EXT(株式会社東洋精機製作所製)
Tダイ:300mm幅3種5層用(株式会社プラスチック工学研究所製)
冷却ロールの温度:50℃
引取速度:1m/分 運転開始から30分後に作製された多層フィルムを用いて、株式会社東洋精機製作所製パンタグラフ式二軸延伸装置にて80℃で30秒間予熱後、延伸倍率3×3倍で同時二軸延伸を行い、熱収縮フィルムを得た。得られた熱収縮フィルムを10cm×10cmにカットし、90℃の熱水に10秒浸漬させて収縮させた後、収縮後のフィルムを目視し、下記の基準により評価した。
(収縮後外観の評価基準)
A:着色も白化も生じることなく、均一に収縮した。
B:着色または白化が生じた。
C:同時二軸延伸時に多層フィルムが破れ、熱収縮フィルムが得られなかった。
実施例及び比較例で得られたガスバリア樹脂組成物ペレット、エチレン-酢酸ビニル共重合体(EVA)(三井デュポンポリケミカル株式会社製「エバフレックス(商標)EV340」)、および接着性樹脂(三井化学株式会社性「アドマー(商標)VF500」)を用い、3種5層共押出機を用いて、下記条件にて多層フィルム(層構成:EVA層/接着性樹脂層/ガスバリア樹脂組成物層/接着性樹脂層/EVA層、厚み(μm):300/50/50/50/300)を作製した。
(押出機条件)
各樹脂の押出温度:供給部/圧縮部/計量部/ダイ=170℃/170℃/210℃/210℃
EVAの押出機:32φ単軸押出機、GT-32-A型(株式会社プラスチック工学研究所製)
接着性樹脂の押出機:25φ単軸押出機、P25-18-AC型(大阪精機工作株式会社製)
EVOH樹脂組成物の押出機:20φ単軸押出機、ラボ機ME型CO-EXT(株式会社東洋精機製作所製)
Tダイ:300mm幅3種5層用(株式会社プラスチック工学研究所製)
冷却ロールの温度:50℃
引取速度:1m/分 運転開始から30分後に作製された多層フィルムを用いて、株式会社東洋精機製作所製パンタグラフ式二軸延伸装置にて80℃で30秒間予熱後、延伸倍率3×3倍で同時二軸延伸を行い、熱収縮フィルムを得た。得られた熱収縮フィルムを10cm×10cmにカットし、90℃の熱水に10秒浸漬させて収縮させた後、収縮後のフィルムを目視し、下記の基準により評価した。
(収縮後外観の評価基準)
A:着色も白化も生じることなく、均一に収縮した。
B:着色または白化が生じた。
C:同時二軸延伸時に多層フィルムが破れ、熱収縮フィルムが得られなかった。
(20)多層熱成形容器評価
実施例及び比較例で得られたガスバリア樹脂組成物ペレット、ポリプロピレン(日本ポリプロ株式会社製「ノバテック(商標)PP EA7AD」)、および接着性樹脂(三井化学株式会社製「アドマー(商標)QF551」)を用い、3種5層共押出装置を用いて、下記条件にて多層フィルム(ポリプロピレン/接着性樹脂/ガスバリア樹脂組成物/接着性樹脂/ポリプロピレン、厚み(μm):368/16/32/16/368)を作成した。
(押出機条件)
各樹脂の押出温度:供給部/圧縮部/計量部/ダイ=150℃/150℃/210℃/210℃
ポリプロピレン樹脂の押出機:32φ単軸押出機、GT-32-A型(株式会社プラスチック工学研究所製)
接着性樹脂の押出機:25φ単軸押出機、P25-18-AC型(大阪精機工作株式会社製)
EVOH樹脂組成物の押出機:20φ押出機、ラボ機ME型CO-EXT(株式会社東洋精機製作所製)
Tダイ:300mm幅3種5層用(株式会社プラスチック工学研究所製)
冷却ロールの温度:80℃
引取速度:1m/分
運転開始10時間後、50時間後に作製された多層シートを採取し、得られた多層シートを熱成形機(株式会社浅野製作所製:真空圧空深絞り成形機「FX-0431-3型」)にて、シート温度を160℃にして、圧縮空気(気圧5kgf/cm2)により、丸カップ形状(金型形状:上部75mmφ、下部60mmφ、深さ38mm、絞り比S=0.5)に熱成形することにより、熱成形容器を得た。成形条件を以下に示す。
ヒーター温度:400℃
プラグ:45φ×33mm
金型温度:40℃
得られたカップ形状の熱成形容器の外観を目視で確認し、下記基準で評価した。
(外観の評価基準)
A(良好):ムラおよび局部的偏肉は認められなかった
B(やや良好):わずかなムラおよび局部的偏肉が確認された
C(不良):著しいムラおよび局部的偏肉が確認された
実施例及び比較例で得られたガスバリア樹脂組成物ペレット、ポリプロピレン(日本ポリプロ株式会社製「ノバテック(商標)PP EA7AD」)、および接着性樹脂(三井化学株式会社製「アドマー(商標)QF551」)を用い、3種5層共押出装置を用いて、下記条件にて多層フィルム(ポリプロピレン/接着性樹脂/ガスバリア樹脂組成物/接着性樹脂/ポリプロピレン、厚み(μm):368/16/32/16/368)を作成した。
(押出機条件)
各樹脂の押出温度:供給部/圧縮部/計量部/ダイ=150℃/150℃/210℃/210℃
ポリプロピレン樹脂の押出機:32φ単軸押出機、GT-32-A型(株式会社プラスチック工学研究所製)
接着性樹脂の押出機:25φ単軸押出機、P25-18-AC型(大阪精機工作株式会社製)
EVOH樹脂組成物の押出機:20φ押出機、ラボ機ME型CO-EXT(株式会社東洋精機製作所製)
Tダイ:300mm幅3種5層用(株式会社プラスチック工学研究所製)
冷却ロールの温度:80℃
引取速度:1m/分
運転開始10時間後、50時間後に作製された多層シートを採取し、得られた多層シートを熱成形機(株式会社浅野製作所製:真空圧空深絞り成形機「FX-0431-3型」)にて、シート温度を160℃にして、圧縮空気(気圧5kgf/cm2)により、丸カップ形状(金型形状:上部75mmφ、下部60mmφ、深さ38mm、絞り比S=0.5)に熱成形することにより、熱成形容器を得た。成形条件を以下に示す。
ヒーター温度:400℃
プラグ:45φ×33mm
金型温度:40℃
得られたカップ形状の熱成形容器の外観を目視で確認し、下記基準で評価した。
(外観の評価基準)
A(良好):ムラおよび局部的偏肉は認められなかった
B(やや良好):わずかなムラおよび局部的偏肉が確認された
C(不良):著しいムラおよび局部的偏肉が確認された
(21)ブロー成形容器のストリーク評価
実施例及び比較例で得られたEVOH樹脂組成物ペレット、高密度ポリエチレン(株式会社プライムポリマー製の「ハイゼックス(商標)8200B」)、及び接着性樹脂(三井化学株式会社の「ADMER(商標)GT-6A」)を用い、鈴木製工所製ブロー成形機TB-ST-6Pにて210℃で、(内側)高密度ポリエチレン層/接着性樹脂層/ガスバリア樹脂組成物層/接着性樹脂層/高密度ポリエチレン層/高密度ポリエチレン層(外側)の3種6層パリソンよりブロー成形容器を作成した。なお、ブロー成形容器の製造においては、金型内温度15℃で20秒間冷却し、全層平均厚み1000μm((内側)高密度ポリエチレン層/接着性樹脂層/ガスバリア樹脂組成物層/接着性樹脂層/高密度ポリエチレン層/高密度ポリエチレン層(外側)=(内側)340μm/50μm/40μm/50μm/400μm/120μm(外側))の3Lブロー成形容器を成形した。このブロー成形容器の底面平均直径は100mm、平均高さは400mmであった。運転開始より3時間経過した後のブロー成形容器を採取し、外観目視及び円周方向の断面顕微鏡観察によるストリーク評価を行った。
(ストリークの評価基準)
A(良好):ストリークは認められなかった。
B(やや良好):ストリークが確認された。
C(不良):多数のストリークが確認された。
実施例及び比較例で得られたEVOH樹脂組成物ペレット、高密度ポリエチレン(株式会社プライムポリマー製の「ハイゼックス(商標)8200B」)、及び接着性樹脂(三井化学株式会社の「ADMER(商標)GT-6A」)を用い、鈴木製工所製ブロー成形機TB-ST-6Pにて210℃で、(内側)高密度ポリエチレン層/接着性樹脂層/ガスバリア樹脂組成物層/接着性樹脂層/高密度ポリエチレン層/高密度ポリエチレン層(外側)の3種6層パリソンよりブロー成形容器を作成した。なお、ブロー成形容器の製造においては、金型内温度15℃で20秒間冷却し、全層平均厚み1000μm((内側)高密度ポリエチレン層/接着性樹脂層/ガスバリア樹脂組成物層/接着性樹脂層/高密度ポリエチレン層/高密度ポリエチレン層(外側)=(内側)340μm/50μm/40μm/50μm/400μm/120μm(外側))の3Lブロー成形容器を成形した。このブロー成形容器の底面平均直径は100mm、平均高さは400mmであった。運転開始より3時間経過した後のブロー成形容器を採取し、外観目視及び円周方向の断面顕微鏡観察によるストリーク評価を行った。
(ストリークの評価基準)
A(良好):ストリークは認められなかった。
B(やや良好):ストリークが確認された。
C(不良):多数のストリークが確認された。
(22)燃料透過度
実施例及び比較例で得られたガスバリア樹脂組成物ペレット、高密度ポリエチレン(株式会社プライムポリマー製の「ハイゼックス(商標)8200B」)及び接着性樹脂(三井化学株式会社の「ADMER(商標)GT-6A」)を用い、上記(5)で用いた3種5層共押出装置及び押出機条件を使用して、多層フィルム(ポリエチレン/接着性樹脂/ガスバリア樹脂組成物/接着性樹脂/ポリエチレン)を作成した。多層フィルムの層構成は、内外層のポリエチレン樹脂が90μm、接着性樹脂が各10μm、中間層のガスバリア樹脂組成物が20μmであった。得られた多層フィルムについて、GTRテック社フロー式ガス・蒸気透過率測定装置(GTR-30XFKE)を用いて、モデル燃料の透過度を測定した。多層フィルムは20℃65%RHで1ヶ月調湿し、測定は60℃で実施した。モデル燃料はCE10ガソリンを用い、その組成はトルエン/イソオクタン/エタノール=45/45/10質量%であった。
実施例及び比較例で得られたガスバリア樹脂組成物ペレット、高密度ポリエチレン(株式会社プライムポリマー製の「ハイゼックス(商標)8200B」)及び接着性樹脂(三井化学株式会社の「ADMER(商標)GT-6A」)を用い、上記(5)で用いた3種5層共押出装置及び押出機条件を使用して、多層フィルム(ポリエチレン/接着性樹脂/ガスバリア樹脂組成物/接着性樹脂/ポリエチレン)を作成した。多層フィルムの層構成は、内外層のポリエチレン樹脂が90μm、接着性樹脂が各10μm、中間層のガスバリア樹脂組成物が20μmであった。得られた多層フィルムについて、GTRテック社フロー式ガス・蒸気透過率測定装置(GTR-30XFKE)を用いて、モデル燃料の透過度を測定した。多層フィルムは20℃65%RHで1ヶ月調湿し、測定は60℃で実施した。モデル燃料はCE10ガソリンを用い、その組成はトルエン/イソオクタン/エタノール=45/45/10質量%であった。
(23)ロングラン性評価D
実施例及び比較例で樹脂組成物ペレットを作製する際に、二軸押出機での溶融押出を長時間連続運転し、長期運転安定性(ロングラン性)の評価を行った。
実施例及び比較例で樹脂組成物ペレットを作製する際に、二軸押出機での溶融押出を長時間連続運転し、長期運転安定性(ロングラン性)の評価を行った。
(23-1)ダイ堆積物付着量
樹脂組成物ペレットの溶融押出を開始してから30分おきにストランドダイ及び得られたペレットを目視により下記評価基準で観察し、ストランドダイに付着する堆積物の量に応じてダイ堆積物の除去を行った。これを24時間連続で実施することでロングラン性を評価した。尚、ペレタイザーで切断する前にストランドが切れてしまった場合(以下「断糸」と表現する場合がある)は速やかにストランドダイ表面を清掃し、運転を再開した。
(ダイ堆積物付着量の判定基準)
良好(A):ダイ堆積物はわずかであり、1時間以上の間隔でダイ堆積物を除去すれば断糸及びダイ堆積物のペレットへの付着が起こらない。
やや良好(B):ダイ堆積物があり、30分の間隔でダイ堆積物を除去しなければ断糸またはダイ堆積物のペレットへの付着が起こる。
不良(C):著しい量のダイ堆積物があり、30分の間隔でダイ堆積物を除去しても、断糸または劣化物のペレットへの付着が起こる。
樹脂組成物ペレットの溶融押出を開始してから30分おきにストランドダイ及び得られたペレットを目視により下記評価基準で観察し、ストランドダイに付着する堆積物の量に応じてダイ堆積物の除去を行った。これを24時間連続で実施することでロングラン性を評価した。尚、ペレタイザーで切断する前にストランドが切れてしまった場合(以下「断糸」と表現する場合がある)は速やかにストランドダイ表面を清掃し、運転を再開した。
(ダイ堆積物付着量の判定基準)
良好(A):ダイ堆積物はわずかであり、1時間以上の間隔でダイ堆積物を除去すれば断糸及びダイ堆積物のペレットへの付着が起こらない。
やや良好(B):ダイ堆積物があり、30分の間隔でダイ堆積物を除去しなければ断糸またはダイ堆積物のペレットへの付着が起こる。
不良(C):著しい量のダイ堆積物があり、30分の間隔でダイ堆積物を除去しても、断糸または劣化物のペレットへの付着が起こる。
(23-2)断糸頻度
断糸頻度を下記評価基準で観察した。
(断紙頻度の判定基準)
良好(A):断糸回数が0~2回
やや良好(B):断糸回数が3~10回
不良(C):断糸回数が11回以上
断糸頻度を下記評価基準で観察した。
(断紙頻度の判定基準)
良好(A):断糸回数が0~2回
やや良好(B):断糸回数が3~10回
不良(C):断糸回数が11回以上
(24)培地評価
2019年4月17日、ハウス内において、得られた植物培地に幅7cm×縦8cm間隔で15穴を空け、大根‘たんしん’の種を1穴あたり3粒直播した。間引きは2019年4月27日に行い、灌水は1日3~6回程度、天候および生育状況に応じて「トンボジョーロ4号」(新輝合成株式会社製)で大塚アグリテクノ株式会社製の養液栽培用肥料「大塚ハウス1号」、「大塚ハウス2号」および「大塚ハウス5号」を混合溶解した養液(N:98.7ppm、P:19.4ppm、K:125.7ppm、Ca:63.0ppm、Mg:13.4ppm、Mn:0.709ppm、B:0.487ppm、Fe:2.025ppm、Cu:0.018ppm、Zn:0.048ppm、Mo:0.019ppm)を与え、収穫調査は2019年6月25日に行った。収穫調査では、生育した15穴に対する肥大根が1g以上の大根の数を数え、肥大根数が12本以上であった場合はA、12本未満であった場合はBとした。
2019年4月17日、ハウス内において、得られた植物培地に幅7cm×縦8cm間隔で15穴を空け、大根‘たんしん’の種を1穴あたり3粒直播した。間引きは2019年4月27日に行い、灌水は1日3~6回程度、天候および生育状況に応じて「トンボジョーロ4号」(新輝合成株式会社製)で大塚アグリテクノ株式会社製の養液栽培用肥料「大塚ハウス1号」、「大塚ハウス2号」および「大塚ハウス5号」を混合溶解した養液(N:98.7ppm、P:19.4ppm、K:125.7ppm、Ca:63.0ppm、Mg:13.4ppm、Mn:0.709ppm、B:0.487ppm、Fe:2.025ppm、Cu:0.018ppm、Zn:0.048ppm、Mo:0.019ppm)を与え、収穫調査は2019年6月25日に行った。収穫調査では、生育した15穴に対する肥大根が1g以上の大根の数を数え、肥大根数が12本以上であった場合はA、12本未満であった場合はBとした。
[酢酸ビニル合成触媒の調製]
上海海源化工科技有限公司製シリカ球体担体HSV-I(球体直径5mm、比表面積160m2/g、吸水率0.75g/g)23g(吸水量19.7g)に、56質量%テトラクロロパラジウム酸ナトリウム水溶液1.5g及び17質量%テトラクロロ金酸四水和物水溶液1.5gを含む担体吸水量相当の水溶液を含浸させた後、メタケイ酸ナトリウム9水和物2.5gを含む水溶液40mLに浸漬し、20時間静置した。続いて、52質量%ヒドラジン水和物水溶液3.3mLを添加し、室温で4時間静置した後、水中に塩化物イオンが無くなるまで水洗し、110℃で4時間乾燥した。得られたパラジウム/金/担体組成物を1.7質量%酢酸水溶液60mLに浸漬し、一晩静置した。次いで、一晩水洗し、110℃で4時間乾燥した。その後、2gの酢酸カリウムの担体吸水量相当水溶液に含浸し、110℃で4時間乾燥することで酢酸ビニル合成触媒を得た。
上海海源化工科技有限公司製シリカ球体担体HSV-I(球体直径5mm、比表面積160m2/g、吸水率0.75g/g)23g(吸水量19.7g)に、56質量%テトラクロロパラジウム酸ナトリウム水溶液1.5g及び17質量%テトラクロロ金酸四水和物水溶液1.5gを含む担体吸水量相当の水溶液を含浸させた後、メタケイ酸ナトリウム9水和物2.5gを含む水溶液40mLに浸漬し、20時間静置した。続いて、52質量%ヒドラジン水和物水溶液3.3mLを添加し、室温で4時間静置した後、水中に塩化物イオンが無くなるまで水洗し、110℃で4時間乾燥した。得られたパラジウム/金/担体組成物を1.7質量%酢酸水溶液60mLに浸漬し、一晩静置した。次いで、一晩水洗し、110℃で4時間乾燥した。その後、2gの酢酸カリウムの担体吸水量相当水溶液に含浸し、110℃で4時間乾燥することで酢酸ビニル合成触媒を得た。
[酢酸ビニルの合成]
<VAM1の合成例>
上記酢酸ビニル合成触媒3mLをガラスビーズ75mLで希釈してSUS316L製反応管(内径22mm、長さ480mm)に充填し、温度150℃、圧力0.6MPaGでエチレン/酸素/水/酢酸/窒素=47.3/6.1/5.6/26.3/14.7(mol%)の割合に混合したガスを流量20NL/時で流通させ、反応を行い、酢酸ビニル(VAM1)を合成した。エチレンには、バイオマス由来のエチレン(Braskem S.A.製、サトウキビ由来のバイオエチレン)を用い、このエチレンが充填されたガスボンベ(エチレン純度96.44%、内容積29.502L、内圧1.8234MPa)を使用した。また、酢酸には、バイオマス由来の酢酸(Godavari Biorefineries Ltd.製、サトウキビ由来のバイオ酢酸)を用い、220℃で気化させてから蒸気で反応系に導入した。
<VAM1の合成例>
上記酢酸ビニル合成触媒3mLをガラスビーズ75mLで希釈してSUS316L製反応管(内径22mm、長さ480mm)に充填し、温度150℃、圧力0.6MPaGでエチレン/酸素/水/酢酸/窒素=47.3/6.1/5.6/26.3/14.7(mol%)の割合に混合したガスを流量20NL/時で流通させ、反応を行い、酢酸ビニル(VAM1)を合成した。エチレンには、バイオマス由来のエチレン(Braskem S.A.製、サトウキビ由来のバイオエチレン)を用い、このエチレンが充填されたガスボンベ(エチレン純度96.44%、内容積29.502L、内圧1.8234MPa)を使用した。また、酢酸には、バイオマス由来の酢酸(Godavari Biorefineries Ltd.製、サトウキビ由来のバイオ酢酸)を用い、220℃で気化させてから蒸気で反応系に導入した。
<VAM2~VAM4の合成>
原料のエチレン及び酢酸を表1に記載の通り、バイオマス由来及び/又は化石燃料由来のものに変更した以外は、VAM1と同様の方法でVAM2~VAM4の各酢酸ビニルを合成した。
原料のエチレン及び酢酸を表1に記載の通り、バイオマス由来及び/又は化石燃料由来のものに変更した以外は、VAM1と同様の方法でVAM2~VAM4の各酢酸ビニルを合成した。
なお、酢酸ビニルの合成に用いた原料としては、下記の原料を使用した。
・バイオマス由来のエチレン :Braskem S.A.製、サトウキビ由来のバイオエチレン
・化石燃料由来のエチレン:エア・リキード工業ガス株式会社製、化石燃料由来のエチレン
・バイオマス由来の酢酸 :Godavari Biorefineries Ltd.製、サトウキビ由来のバイオ酢酸
・化石燃料由来の酢酸 :富士フィルム和光純薬株式会社製、化石燃料由来の酢酸
・バイオマス由来のエチレン :Braskem S.A.製、サトウキビ由来のバイオエチレン
・化石燃料由来のエチレン:エア・リキード工業ガス株式会社製、化石燃料由来のエチレン
・バイオマス由来の酢酸 :Godavari Biorefineries Ltd.製、サトウキビ由来のバイオ酢酸
・化石燃料由来の酢酸 :富士フィルム和光純薬株式会社製、化石燃料由来の酢酸

[EVOHの合成]
<EVOH(A1-1)ペレットの作製>
(エチレン-酢酸ビニル共重合体の重合)
ジャケット、攪拌機、窒素導入口、エチレン導入口及び開始剤添加口を備えた250L加圧反応槽に、VAM1を105kg、及びメタノール(以下、MeOHと称することもある)を32.3kg仕込み、65℃に昇温した後、30分間窒素バブリングして反応槽内を窒素置換した。次いで反応槽圧力(エチレン圧力)が3.67MPaとなるようにエチレンを昇圧して導入した。エチレンには、バイオマス由来のエチレン(Braskem S.A.製、サトウキビ由来のバイオエチレン)を用いた。反応槽内の温度を65℃に調整した後、開始剤として16.8gの2,2’-アゾビス(2,4-ジメチルバレロニトリル)(和光純薬工業社の「V-65」)をメタノール溶液として添加し、重合を開始した。重合中はエチレン圧力を3.67MPaに、重合温度を65℃に維持した。3時間後にVAcの重合率が45%となったところで冷却して重合を停止した。反応槽を開放して脱エチレンした後、窒素ガスをバブリングして脱エチレンを完全に行った。次いで減圧下で未反応のVAcを除去した後、エチレン-酢酸ビニル共重合体にMeOHを添加して20質量%MeOH溶液とした。
<EVOH(A1-1)ペレットの作製>
(エチレン-酢酸ビニル共重合体の重合)
ジャケット、攪拌機、窒素導入口、エチレン導入口及び開始剤添加口を備えた250L加圧反応槽に、VAM1を105kg、及びメタノール(以下、MeOHと称することもある)を32.3kg仕込み、65℃に昇温した後、30分間窒素バブリングして反応槽内を窒素置換した。次いで反応槽圧力(エチレン圧力)が3.67MPaとなるようにエチレンを昇圧して導入した。エチレンには、バイオマス由来のエチレン(Braskem S.A.製、サトウキビ由来のバイオエチレン)を用いた。反応槽内の温度を65℃に調整した後、開始剤として16.8gの2,2’-アゾビス(2,4-ジメチルバレロニトリル)(和光純薬工業社の「V-65」)をメタノール溶液として添加し、重合を開始した。重合中はエチレン圧力を3.67MPaに、重合温度を65℃に維持した。3時間後にVAcの重合率が45%となったところで冷却して重合を停止した。反応槽を開放して脱エチレンした後、窒素ガスをバブリングして脱エチレンを完全に行った。次いで減圧下で未反応のVAcを除去した後、エチレン-酢酸ビニル共重合体にMeOHを添加して20質量%MeOH溶液とした。
(ケン化及び洗浄)
得られたエチレン-酢酸ビニル共重合体ジャケットの20質量%MeOH溶液250kgを、攪拌機、窒素導入口、還流冷却器及び溶液添加口を備えた500L反応槽に入れ、かかる溶液に窒素を吹き込みながら60℃に昇温し、水酸化ナトリウム4kgを濃度2規定のMeOH溶液として添加した。水酸化ナトリウムの添加終了後、系内温度を60℃に保ちながら2時間攪拌してケン化反応を進行させた。2時間経過した後に、再度、同様の方法で水酸化ナトリウムを4kg添加し、2時間加熱攪拌を継続した。その後、酢酸を14kg添加してケン化反応を停止し、イオン交換水50kgを添加した。加熱攪拌しながら反応槽外にMeOHと水を留出させ反応液を濃縮した。3時間経過した後、更にイオン交換水50kgを添加し、EVOHを析出させた。デカンテーションにより析出したEVOHを収集し、ミキサーで粉砕した。得られたEVOH粉末を1g/Lの酢酸水溶液(浴比20:イオン交換水200Lに対し粉末10kgの割合)に投入して2時間攪拌洗浄した。これを脱液し、さらに1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。これを脱液したものを、イオン交換水(浴比20)に投入して攪拌洗浄を2時間行い脱液する操作を3回繰り返して精製を行った。これを60℃で16時間乾燥させることでEVOHの粗乾燥物を25kg得た。
得られたエチレン-酢酸ビニル共重合体ジャケットの20質量%MeOH溶液250kgを、攪拌機、窒素導入口、還流冷却器及び溶液添加口を備えた500L反応槽に入れ、かかる溶液に窒素を吹き込みながら60℃に昇温し、水酸化ナトリウム4kgを濃度2規定のMeOH溶液として添加した。水酸化ナトリウムの添加終了後、系内温度を60℃に保ちながら2時間攪拌してケン化反応を進行させた。2時間経過した後に、再度、同様の方法で水酸化ナトリウムを4kg添加し、2時間加熱攪拌を継続した。その後、酢酸を14kg添加してケン化反応を停止し、イオン交換水50kgを添加した。加熱攪拌しながら反応槽外にMeOHと水を留出させ反応液を濃縮した。3時間経過した後、更にイオン交換水50kgを添加し、EVOHを析出させた。デカンテーションにより析出したEVOHを収集し、ミキサーで粉砕した。得られたEVOH粉末を1g/Lの酢酸水溶液(浴比20:イオン交換水200Lに対し粉末10kgの割合)に投入して2時間攪拌洗浄した。これを脱液し、さらに1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。これを脱液したものを、イオン交換水(浴比20)に投入して攪拌洗浄を2時間行い脱液する操作を3回繰り返して精製を行った。これを60℃で16時間乾燥させることでEVOHの粗乾燥物を25kg得た。
(EVOH含水ペレットの製造)
得られたEVOHの粗乾燥物25kgを、ジャケット、攪拌機及び還流冷却器を備えた100L攪拌槽に入れ、さらに水20kg及びMeOH20gを加え、70℃に昇温して溶解させた。この溶解液を径3mmのガラス管を通して5℃に冷却した重量比で水/MeOH=90/10の混合液中に押し出してストランド状に析出させ、このストランドをストランドカッターでペレット状にカットすることでEVOHの含水ペレットを得た。このEVOHの含水ペレットを濃度1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。これを脱液し、さらに1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。脱液後、酢酸水溶液を更新し同様の操作を行った。酢酸水溶液で洗浄してから脱液したものを、イオン交換水(浴比20)に投入して攪拌洗浄を2時間行い脱液する操作を3回繰り返して精製を行い、ケン化反応時の触媒残渣とストランド析出時に使用したMeOHが除去された、EVOHの含水ペレットを得た。得られたEVOHの含水ペレットの含水率をメトラー社のハロゲン水分計「HR73」で測定したところ、110質量%であった。
得られたEVOHの粗乾燥物25kgを、ジャケット、攪拌機及び還流冷却器を備えた100L攪拌槽に入れ、さらに水20kg及びMeOH20gを加え、70℃に昇温して溶解させた。この溶解液を径3mmのガラス管を通して5℃に冷却した重量比で水/MeOH=90/10の混合液中に押し出してストランド状に析出させ、このストランドをストランドカッターでペレット状にカットすることでEVOHの含水ペレットを得た。このEVOHの含水ペレットを濃度1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。これを脱液し、さらに1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。脱液後、酢酸水溶液を更新し同様の操作を行った。酢酸水溶液で洗浄してから脱液したものを、イオン交換水(浴比20)に投入して攪拌洗浄を2時間行い脱液する操作を3回繰り返して精製を行い、ケン化反応時の触媒残渣とストランド析出時に使用したMeOHが除去された、EVOHの含水ペレットを得た。得られたEVOHの含水ペレットの含水率をメトラー社のハロゲン水分計「HR73」で測定したところ、110質量%であった。
(EVOH(A1-1)ペレットの製造)
得られたEVOHの含水ペレットを酢酸ナトリウム、酢酸、リン酸及びホウ酸が含まれる水溶液(浴比20)に投入し、定期的に攪拌しながら4時間浸漬させた。なお、各成分の濃度は、得られたEVOH(A1-1)ペレットにおける各成分の含有量が表2に記載の通りとなるように調整した。浸漬後に脱液し、空気下で80℃、3時間、及び空気下で130℃、7.5時間乾燥することにより、酢酸ナトリウム、酢酸、リン酸及びホウ酸を含むEVOH(A1-1)ペレットを得た。
得られたEVOHの含水ペレットを酢酸ナトリウム、酢酸、リン酸及びホウ酸が含まれる水溶液(浴比20)に投入し、定期的に攪拌しながら4時間浸漬させた。なお、各成分の濃度は、得られたEVOH(A1-1)ペレットにおける各成分の含有量が表2に記載の通りとなるように調整した。浸漬後に脱液し、空気下で80℃、3時間、及び空気下で130℃、7.5時間乾燥することにより、酢酸ナトリウム、酢酸、リン酸及びホウ酸を含むEVOH(A1-1)ペレットを得た。
<EVOH(A1-2)~EVOH(A1-9)、EVOH(A1-12)、EVOH(A1-13)、EVOH(B1-1)~(B1-4)、EVOH(B1-6)の各ペレットの作製>
原料(原料モノマー)のエチレン及び酢酸ビニルの種類並びにリン酸化合物及びホウ素化合物の含有量を表2に記載の通り変更した以外は、EVOH(A1-1)ペレットと同様の方法で、EVOH(A1-2)ペレット~EVOH(A1-9)ペレット、EVOH(A1-13)ペレット、及びEVOH(B1-1)~EVOH(B1-4)ペレットを作製した。また、原料(原料モノマー)のエチレン及び酢酸ビニルの種類並びにリン酸化合物及びホウ素化合物の含有量を表2に記載の通り変更し、且つエチレン及び酢酸ビニルの使用量を変更することで、EVOH(A1-12)ペレット及びEVOH(B1-6)ペレットを作製した。化石燃料由来のエチレンには、エア・リキード工業ガス株式会社製のエチレンを用いた。
原料(原料モノマー)のエチレン及び酢酸ビニルの種類並びにリン酸化合物及びホウ素化合物の含有量を表2に記載の通り変更した以外は、EVOH(A1-1)ペレットと同様の方法で、EVOH(A1-2)ペレット~EVOH(A1-9)ペレット、EVOH(A1-13)ペレット、及びEVOH(B1-1)~EVOH(B1-4)ペレットを作製した。また、原料(原料モノマー)のエチレン及び酢酸ビニルの種類並びにリン酸化合物及びホウ素化合物の含有量を表2に記載の通り変更し、且つエチレン及び酢酸ビニルの使用量を変更することで、EVOH(A1-12)ペレット及びEVOH(B1-6)ペレットを作製した。化石燃料由来のエチレンには、エア・リキード工業ガス株式会社製のエチレンを用いた。
EVOH(A1-1)ペレット~EVOH(A1-9)ペレット、EVOH(A1-12)ペレット、EVOH(A1-13)ペレット、EVOH(B1-1)~EVOH(B1-4)ペレット及びEVOH(B1-6)ペレットのそれぞれについて、上記評価方法(1)、(3)~(5)に記載の方法に従い、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、並びにバイオベース度の測定を行った。結果を表2に示す。
<EVOH(A1-10)ペレットの作製>
ジャケット、攪拌機、窒素導入口、エチレン導入口及び開始剤添加口を備えた250L加圧反応槽に、酢酸ビニルVAM3及びVAM4を50/50で混合した酢酸ビニルを100kg、メタノール(以下、MeOHと称することがある)を5.7kg、他の単量体として2-メチレン-1,3-プロパンジオールジアセテート(以下、MPDAcと称する)を3kg仕込み、60℃に昇温した後、30分間窒素バブリングして反応槽内を窒素置換した。次いで反応槽圧力(エチレン圧力)が5.1MPaとなるようにバイオマス由来のエチレンと化石燃料由来のエチレンを50/50で混合したエチレンを導入した。反応槽内の温度を60℃に調整した後、開始剤として50gの2,2’-アゾビス(2,4-ジメチルバレロニトリル)(和光純薬工業株式会社製「V-65」)をメタノール溶液として添加し、重合を開始した。重合中はエチレン圧力を5.1MPaに、重合温度を60℃に維持した。また重合中はMPDAcの30wt%MeOH溶液を350mL/15分の添加量で加圧反応槽に連続的に添加した。6.5時間後にVAcの重合率が41%となったところで冷却して重合を停止した。反応槽を開放して脱エチレンした後、窒素ガスをバブリングして脱エチレンを完全に行った。次いで減圧下で未反応のVAcを除去した後、MPDAc由来の構造単位が共重合により導入された変性エチレン-酢酸ビニル共重合体(本明細書中、変性EVAcと称することがある)にMeOHを添加して20質量%MeOH溶液とした。その後のケン化、洗浄、EVOH含水ペレットの製造およびEVOHペレットの製造工程はEVOH(A1-1)ペレットと同様の方法で行い、エチレン単位含有量38モル%、変性基(変性基を含む構造単位)の含有量2.5モル%、ケン化度99モル%以上のEVOH(A1-10)ペレットを作製した。得られたEVOH(A1-10)ペレットについて、上記評価方法(1)、(3)~(5)に記載の方法に従い、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、並びにバイオベース度の測定を行った。結果を表2に示す。
ジャケット、攪拌機、窒素導入口、エチレン導入口及び開始剤添加口を備えた250L加圧反応槽に、酢酸ビニルVAM3及びVAM4を50/50で混合した酢酸ビニルを100kg、メタノール(以下、MeOHと称することがある)を5.7kg、他の単量体として2-メチレン-1,3-プロパンジオールジアセテート(以下、MPDAcと称する)を3kg仕込み、60℃に昇温した後、30分間窒素バブリングして反応槽内を窒素置換した。次いで反応槽圧力(エチレン圧力)が5.1MPaとなるようにバイオマス由来のエチレンと化石燃料由来のエチレンを50/50で混合したエチレンを導入した。反応槽内の温度を60℃に調整した後、開始剤として50gの2,2’-アゾビス(2,4-ジメチルバレロニトリル)(和光純薬工業株式会社製「V-65」)をメタノール溶液として添加し、重合を開始した。重合中はエチレン圧力を5.1MPaに、重合温度を60℃に維持した。また重合中はMPDAcの30wt%MeOH溶液を350mL/15分の添加量で加圧反応槽に連続的に添加した。6.5時間後にVAcの重合率が41%となったところで冷却して重合を停止した。反応槽を開放して脱エチレンした後、窒素ガスをバブリングして脱エチレンを完全に行った。次いで減圧下で未反応のVAcを除去した後、MPDAc由来の構造単位が共重合により導入された変性エチレン-酢酸ビニル共重合体(本明細書中、変性EVAcと称することがある)にMeOHを添加して20質量%MeOH溶液とした。その後のケン化、洗浄、EVOH含水ペレットの製造およびEVOHペレットの製造工程はEVOH(A1-1)ペレットと同様の方法で行い、エチレン単位含有量38モル%、変性基(変性基を含む構造単位)の含有量2.5モル%、ケン化度99モル%以上のEVOH(A1-10)ペレットを作製した。得られたEVOH(A1-10)ペレットについて、上記評価方法(1)、(3)~(5)に記載の方法に従い、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、並びにバイオベース度の測定を行った。結果を表2に示す。
<EVOH(B1-5)ペレットの作製>
原料(原料モノマー)のエチレン及び酢酸ビニルの種類を表2の通り変更した以外は、EVOH(A1-10)ペレットと同様の方法でEVOH(B1-5)ペレットを作製し、評価した。結果を表2に示す。
原料(原料モノマー)のエチレン及び酢酸ビニルの種類を表2の通り変更した以外は、EVOH(A1-10)ペレットと同様の方法でEVOH(B1-5)ペレットを作製し、評価した。結果を表2に示す。
<EVOH(A1-11)ペレットの作製>
冷却コイルを持つ1m3の重合缶にVAM3とVAM4を50/50で混合した酢酸ビニルを500kg、メタノール100kg、アセチルパーオキシド500ppm(対酢酸ビニル)、クエン酸20ppm(対酢酸ビニル)、および他の単量体として3,4-ジアセトキシ-1-ブテンを14kg仕込み、系を窒素ガスで一旦置換した後、バイオマス由来のエチレンと化石燃料由来のエチレンを50/50で混合したエチレンで置換し、エチレン圧が45kg/cm2となるまで圧入して、攪拌しながら67℃まで昇温して、3,4-ジアセトキシ-1-ブテンを15g/分で全量4.5kgを添加しながら重合し、重合率が50%になるまで6時間重合した。反応槽を開放して脱エチレンした後、窒素ガスをバブリングして脱エチレンを完全に行った。次いで減圧下で未反応のVAcを除去した後、3,4-ジアセトキシ-1-ブテン由来の構造単位が共重合により導入された変性エチレン-酢酸ビニル共重合体(本明細書中、変性EVAcと称することがある)にMeOHを添加して20質量%MeOH溶液とした。その後のケン化、洗浄、EVOH含水ペレットの製造およびEVOHペレットの製造工程はEVOH(A1-1)ペレットと同様の方法で行い、エチレン単位含有量38モル%、変性基(変性基を含む構造単位)の含有量2.5モル%、ケン化度99モル%以上のEVOH(A1-11)ペレットを作製した。得られたEVOH(A1-11)ペレットについて、上記評価方法(1)、(3)~(5)に記載の方法に従い、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、並びにバイオベース度の測定を行った。結果を表2に示す。
冷却コイルを持つ1m3の重合缶にVAM3とVAM4を50/50で混合した酢酸ビニルを500kg、メタノール100kg、アセチルパーオキシド500ppm(対酢酸ビニル)、クエン酸20ppm(対酢酸ビニル)、および他の単量体として3,4-ジアセトキシ-1-ブテンを14kg仕込み、系を窒素ガスで一旦置換した後、バイオマス由来のエチレンと化石燃料由来のエチレンを50/50で混合したエチレンで置換し、エチレン圧が45kg/cm2となるまで圧入して、攪拌しながら67℃まで昇温して、3,4-ジアセトキシ-1-ブテンを15g/分で全量4.5kgを添加しながら重合し、重合率が50%になるまで6時間重合した。反応槽を開放して脱エチレンした後、窒素ガスをバブリングして脱エチレンを完全に行った。次いで減圧下で未反応のVAcを除去した後、3,4-ジアセトキシ-1-ブテン由来の構造単位が共重合により導入された変性エチレン-酢酸ビニル共重合体(本明細書中、変性EVAcと称することがある)にMeOHを添加して20質量%MeOH溶液とした。その後のケン化、洗浄、EVOH含水ペレットの製造およびEVOHペレットの製造工程はEVOH(A1-1)ペレットと同様の方法で行い、エチレン単位含有量38モル%、変性基(変性基を含む構造単位)の含有量2.5モル%、ケン化度99モル%以上のEVOH(A1-11)ペレットを作製した。得られたEVOH(A1-11)ペレットについて、上記評価方法(1)、(3)~(5)に記載の方法に従い、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、並びにバイオベース度の測定を行った。結果を表2に示す。
ペレット(A1-1)~ペレット(A1-13)及びペレット(B1-1)~ペレット(B1-6)について、下記記載の方法に従って硫黄化合物の測定を行った。結果(硫黄化合物の硫黄原子換算の含有量及び種類)を表2に示す。
<硫黄化合物含有量の測定>
硫黄化合物の定量は三菱アナリテック製微量窒素硫黄分析装(TS-2100H型)を用いて行い、測定条件は以下の通りとした。
ヒーター温度:Inlet 900℃,Outlet 900℃
ガス流量:Ar,O2各300ml/min
[分析システム NSX-2100]
測定モード:TS
パラメータ:SD-210
測定時間(タイマー):540秒(9分)
PMT感度:高濃度
硫黄化合物の同定は、ガスクロマトグラフィー(GC)と、ガスクロマトグラフィー質量分析法(GC/MS)を用いて行った。GCの検出器としては、微量の硫黄化合物、リン化合物に対して高い感度を示すFPD(炎光光度検出器)を用いて行い、硫黄化合物が検出された保持時間で観測された質量成分を解析することで、同定を行った。
<硫黄化合物含有量の測定>
硫黄化合物の定量は三菱アナリテック製微量窒素硫黄分析装(TS-2100H型)を用いて行い、測定条件は以下の通りとした。
ヒーター温度:Inlet 900℃,Outlet 900℃
ガス流量:Ar,O2各300ml/min
[分析システム NSX-2100]
測定モード:TS
パラメータ:SD-210
測定時間(タイマー):540秒(9分)
PMT感度:高濃度
硫黄化合物の同定は、ガスクロマトグラフィー(GC)と、ガスクロマトグラフィー質量分析法(GC/MS)を用いて行った。GCの検出器としては、微量の硫黄化合物、リン化合物に対して高い感度を示すFPD(炎光光度検出器)を用いて行い、硫黄化合物が検出された保持時間で観測された質量成分を解析することで、同定を行った。

<実施例1-1>
EVOH(A1-1)ペレット及びEVOH(B1-1)ペレットを質量比(A1-1/B1-1)10/90でドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度220℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例1-1のガスバリア樹脂組成物ペレットを得た。
EVOH(A1-1)ペレット及びEVOH(B1-1)ペレットを質量比(A1-1/B1-1)10/90でドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度220℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例1-1のガスバリア樹脂組成物ペレットを得た。
得られた実施例1-1のガスバリア樹脂組成物ペレットについて、上記評価方法(1)、(3)~(7)に記載の方法に従って、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、バイオベース度、ロングラン性並びに酸素透過度の測定又は評価を行った。結果を表3に示す。
<実施例1-2~1-23、比較例1-1~1-7>
用いたEVOHの種類及び質量比(割合)を、表3に記載の通り変更した以外は、実施例1-1と同様の方法で実施例1-2~1-23及び比較例1-1~1-7の各ガスバリア樹脂組成物ペレットを作製し、評価した。結果を表3に示す。
用いたEVOHの種類及び質量比(割合)を、表3に記載の通り変更した以外は、実施例1-1と同様の方法で実施例1-2~1-23及び比較例1-1~1-7の各ガスバリア樹脂組成物ペレットを作製し、評価した。結果を表3に示す。

表3に示されるように、実施例1-1~1-20の各ガスバリア樹脂組成物は、バイオマス由来の原料を一部に用いていながら、化石燃料由来のみのもの(比較例1-2のガスバリア樹脂組成物)と遜色のない高いガスバリア性を有していた。また、実施例1-21のガスバリア樹脂組成物は、バイオマス由来の原料を一部に用いていながら、化石燃料由来のみのもの(比較例1-6のガスバリア樹脂組成物)と遜色のない高いガスバリア性を維持していた。実施例1-23のガスバリア樹脂組成物は、バイオマス由来の原料を一部に用いていながら、化石燃料由来のみのもの(比較例1-7のガスバリア樹脂組成物)と遜色のない高いガスバリア性を維持していた。また、実施例1-1~1-23の各ガスバリア樹脂組成物は、運転開始10時間後に作製されたフィルムについての製膜欠点及びストリークの評価がA又はBであり、十分なロングラン性を有するものであった。
また、実施例及び比較例の結果から、EVOHを用いたガスバリア樹脂組成物において、ガスバリア性はバイオベース度に依存しないのに対し、ロングラン性はバイオベース度が低くなるにつれて高まる傾向にあるという、特異的な性質があることが確認できた。例えば、バイオベース度が65%以下の実施例1-1、1-2、1-5~1-13、1-15~1-23の各ガスバリア樹脂組成物は、運転開始10時間後及び50時間後に作製されたフィルムについての製膜欠点、ストリーク及びロール端部着色の評価がA又はBであり、高いロングラン性を有するものとなった。
<非吸着試験>
実施例1-23及び比較例1-7で得られたガスバリア樹脂組成物ペレットを用いて、下記条件で厚み30μmの単層フィルムを製膜し、ニコチン及びサリチル酸メチルの非吸着特性を評価した。
(単層フィルムの作製)
単軸押出装置(東洋精機製作所の「D2020」、D(mm)=20、L/D=20、圧縮比=3.0、スクリュー:フルフライト)を用い、上記ガスバリア樹脂組成物ペレットから厚み30μmの単層フィルムを作製した。押出条件は以下に示すとおりである。
押出温度:210℃
ダイス幅:30cm
引取りロール温度:80℃
スクリュー回転数:40rpm
引取りロール速度:2.1m/分
(評価サンプルの作製)
ニコチンまたはサリチル酸メチルを40mg秤量後、内容量50cm3の秤量瓶に投入し、そこへ吸着物質に触れない様にU型に加工したSUS網を置いた。この網板の上に上記30μmの単層フィルムから切り出した幅1cm長さ4cm及び幅1cm長さ1cmのフィルム片各1枚を置いて、20℃50%RH環境下で2週間放置した。吸着量により適した大きさのフィルム片を用い、下記に示す方法で吸着量の測定を行った。
(吸着量の測定)
吸着量の測定には熱脱着ガスクロマトグラフ-質量分析計(TCT-GC/MS、TCT:クロムパック社製「CP-4020」、GC/MS:アジレント社製「5973型」)を使用して以下の方法により吸着量を測定した。
(ガス採取方法)
80℃に加熱されたガラス製チャンバーに上記フィルム片を投入した。一方から清浄な窒素を100ml/minの流速で流しながら、もう一方にTenax捕集管を付し、3分間留出してくるガスを採取したものを試料とした。
(ガス脱着条件)
Tenax捕集管を250℃に加熱し脱着した。
(GC導入法)
脱着ガスを-130℃でコールドトラップした後、250℃に加熱して絡むに導入して吸着量を測定した。
実施例1-23及び比較例1-7で得られたガスバリア樹脂組成物ペレットを用いて、下記条件で厚み30μmの単層フィルムを製膜し、ニコチン及びサリチル酸メチルの非吸着特性を評価した。
(単層フィルムの作製)
単軸押出装置(東洋精機製作所の「D2020」、D(mm)=20、L/D=20、圧縮比=3.0、スクリュー:フルフライト)を用い、上記ガスバリア樹脂組成物ペレットから厚み30μmの単層フィルムを作製した。押出条件は以下に示すとおりである。
押出温度:210℃
ダイス幅:30cm
引取りロール温度:80℃
スクリュー回転数:40rpm
引取りロール速度:2.1m/分
(評価サンプルの作製)
ニコチンまたはサリチル酸メチルを40mg秤量後、内容量50cm3の秤量瓶に投入し、そこへ吸着物質に触れない様にU型に加工したSUS網を置いた。この網板の上に上記30μmの単層フィルムから切り出した幅1cm長さ4cm及び幅1cm長さ1cmのフィルム片各1枚を置いて、20℃50%RH環境下で2週間放置した。吸着量により適した大きさのフィルム片を用い、下記に示す方法で吸着量の測定を行った。
(吸着量の測定)
吸着量の測定には熱脱着ガスクロマトグラフ-質量分析計(TCT-GC/MS、TCT:クロムパック社製「CP-4020」、GC/MS:アジレント社製「5973型」)を使用して以下の方法により吸着量を測定した。
(ガス採取方法)
80℃に加熱されたガラス製チャンバーに上記フィルム片を投入した。一方から清浄な窒素を100ml/minの流速で流しながら、もう一方にTenax捕集管を付し、3分間留出してくるガスを採取したものを試料とした。
(ガス脱着条件)
Tenax捕集管を250℃に加熱し脱着した。
(GC導入法)
脱着ガスを-130℃でコールドトラップした後、250℃に加熱して絡むに導入して吸着量を測定した。
試験の結果、実施例1-23の吸着量は、ニコチンが301ng/cm2であり、サリチル酸メチルが220ng/cm2であった。この値は、化石燃料由来の原料を用いた比較例1-7の、ニコチン313ng/cm2、サリチル酸メチル208ng/cm2と同等の値であり、本発明の単層フィルムを用いた場合でも化石燃料由来の原料を用いた場合と比べ遜色ない非吸着性を示すことが分かった。
<熱融着性試験>
平均厚さ12μmの二軸延伸ポリエステルフィルム(東レ社の「ルミラー(登録商標)」)の片面に2液型の接着剤(三井化学社の「A-520」及び「A-50」)を塗布し、塗布面に非吸着特性の評価で使用した厚み30μmの単層フィルムをラミネートすることで多層フィルムを作製した。接着層の平均厚みは4μmであった。
得られた多層フィルムを用い、上記単層フィルム同士が重ね合わされてヒートシールされるように3方シールパウチを作製し、ヒートシール強度を測定した。パウチ作製には3方シール自動充填包装機(株式会社コマック社製「KP-109」)を用い、シール温度を180℃に設定して多層フィルムを連続的に供給する事で幅80mm、長さ70mmの3方シールパウチを製袋速度100袋/分で作製した。23℃50%RH環境下で引張試験機(株式会社島津製作所製「AGS-H」)を使用してヒートシール強度を測定した。サンプルとしてパウチの幅方向のシール面(シール幅8mm)を15mm幅に切断することでフィルム片を作製し、両端をチャックで固定し、引張速度250m/分で引っ張った際のシール強度を測定した。
(ホットタック性の評価)
ホットタック強度の評価にはホットタックテスター(テラーコーポレーション製)を使用した。上記多層フィルムより幅25mm長さ300mmのフィルム片を作製し、シール温度は100℃~160℃の範囲、シール圧力は2.0MPa、シール時間1秒でシールした際のホットタック強度を測定した。
平均厚さ12μmの二軸延伸ポリエステルフィルム(東レ社の「ルミラー(登録商標)」)の片面に2液型の接着剤(三井化学社の「A-520」及び「A-50」)を塗布し、塗布面に非吸着特性の評価で使用した厚み30μmの単層フィルムをラミネートすることで多層フィルムを作製した。接着層の平均厚みは4μmであった。
得られた多層フィルムを用い、上記単層フィルム同士が重ね合わされてヒートシールされるように3方シールパウチを作製し、ヒートシール強度を測定した。パウチ作製には3方シール自動充填包装機(株式会社コマック社製「KP-109」)を用い、シール温度を180℃に設定して多層フィルムを連続的に供給する事で幅80mm、長さ70mmの3方シールパウチを製袋速度100袋/分で作製した。23℃50%RH環境下で引張試験機(株式会社島津製作所製「AGS-H」)を使用してヒートシール強度を測定した。サンプルとしてパウチの幅方向のシール面(シール幅8mm)を15mm幅に切断することでフィルム片を作製し、両端をチャックで固定し、引張速度250m/分で引っ張った際のシール強度を測定した。
(ホットタック性の評価)
ホットタック強度の評価にはホットタックテスター(テラーコーポレーション製)を使用した。上記多層フィルムより幅25mm長さ300mmのフィルム片を作製し、シール温度は100℃~160℃の範囲、シール圧力は2.0MPa、シール時間1秒でシールした際のホットタック強度を測定した。
熱融着性試験の結果、実施例1-23のシール強度は16.0N/15mmであった。この強度は、化石燃料由来の原料を用いた比較例1-7のシール強度15.8N/15mmと同等の強度と言える。また、ホットタック強度測定結果を図7に示す。ホットタック強度の測定結果からも明確である通り、本発明の単層フィルムは、化石燃料由来の原料を用いた単層フィルムと比べ遜色ない熱融着性を示すことが分かった。
[EVOHの合成]
<EVOH(A2-1)~EVOH(A2-9)、EVOH(A2-11)~EVOH(A2-16)、EVOH(B2-1)~(B2-3)の各ペレットの作製>
原料(原料モノマー)のエチレン及び酢酸ビニルの種類、並びにカルボン酸、金属イオン、リン酸化合物及びホウ素化合物の含有量を表4に記載の通りとし、且つエチレン及び酢酸ビニルの使用量を適宜変更した以外は、EVOH(A1-1)ペレットと同様の方法で、EVOH(A2-1)ペレット~EVOH(A2-9)ペレット、EVOH(A2-11)~EVOH(A2-16)、及びEVOH(B2-1)~EVOH(B2-3)ペレットを作製した。化石燃料由来のエチレンには、エア・リキード工業ガス株式会社製のエチレンを用いた。
<EVOH(A2-1)~EVOH(A2-9)、EVOH(A2-11)~EVOH(A2-16)、EVOH(B2-1)~(B2-3)の各ペレットの作製>
原料(原料モノマー)のエチレン及び酢酸ビニルの種類、並びにカルボン酸、金属イオン、リン酸化合物及びホウ素化合物の含有量を表4に記載の通りとし、且つエチレン及び酢酸ビニルの使用量を適宜変更した以外は、EVOH(A1-1)ペレットと同様の方法で、EVOH(A2-1)ペレット~EVOH(A2-9)ペレット、EVOH(A2-11)~EVOH(A2-16)、及びEVOH(B2-1)~EVOH(B2-3)ペレットを作製した。化石燃料由来のエチレンには、エア・リキード工業ガス株式会社製のエチレンを用いた。
EVOH(A2-1)ペレット~EVOH(A2-9)ペレット、EVOH(A2-11)~EVOH(A2-16)、及びEVOH(B2-1)~EVOH(B2-3)ペレットのそれぞれについて、上記評価方法(1)~(5)に記載の方法に従い、エチレン単位含有量及びケン化度、融点、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、並びにバイオベース度の測定を行った。結果を表4に示す。
<EVOH(A2-10)ペレットの合成>
特開2003-231715号公報段落[0158]及び図1に記載の装置を用い、以下の手順でEVOH(A2-10)ペレットを作製した。東芝機械社製TEM-35BS押出機(37mmφ、L/D=52.5)を使用し、バレルC1を水冷し、バレルC2~C3を200℃、バレルC4~C15を240℃に設定し、スクリュー回転数400rpmで運転した。C1の樹脂フィード口からEVOH(A2-9)ペレットを11kg/hrの割合でフィードし、溶融した後、ベント1から水及び酸素を除去し、C9の液圧入口から変性剤2としてエポキシプロパンを2.4kg/hrの割合でフィードした(フィード時の圧力:6MPa)。その後、ベント2から未反応のエポキシプロパンを除去し、ペレタイズすることで、5モル%変性されたEVOH(A2-10)ペレットを得た。得られたEVOH(A2-10)ペレットについて、上記評価方法(1)~(5)に記載の方法に従い、エチレン単位含有量及びケン化度、融点、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、並びにバイオベース度の測定を行った。結果を表4に示す。
特開2003-231715号公報段落[0158]及び図1に記載の装置を用い、以下の手順でEVOH(A2-10)ペレットを作製した。東芝機械社製TEM-35BS押出機(37mmφ、L/D=52.5)を使用し、バレルC1を水冷し、バレルC2~C3を200℃、バレルC4~C15を240℃に設定し、スクリュー回転数400rpmで運転した。C1の樹脂フィード口からEVOH(A2-9)ペレットを11kg/hrの割合でフィードし、溶融した後、ベント1から水及び酸素を除去し、C9の液圧入口から変性剤2としてエポキシプロパンを2.4kg/hrの割合でフィードした(フィード時の圧力:6MPa)。その後、ベント2から未反応のエポキシプロパンを除去し、ペレタイズすることで、5モル%変性されたEVOH(A2-10)ペレットを得た。得られたEVOH(A2-10)ペレットについて、上記評価方法(1)~(5)に記載の方法に従い、エチレン単位含有量及びケン化度、融点、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、並びにバイオベース度の測定を行った。結果を表4に示す。
<EVOH(B2-4)ペレットの合成>
EVOH(A2-10)ペレットの合成における原料EVOHをEVOH(A2-9)ペレットからEVOH(B2-3)ペレットに変更した以外は、EVOH(A2-10)ペレットの合成と同様にしてEVOH(B2-4)ペレットを作製し、測定を行った。結果を表4に示す。
EVOH(A2-10)ペレットの合成における原料EVOHをEVOH(A2-9)ペレットからEVOH(B2-3)ペレットに変更した以外は、EVOH(A2-10)ペレットの合成と同様にしてEVOH(B2-4)ペレットを作製し、測定を行った。結果を表4に示す。
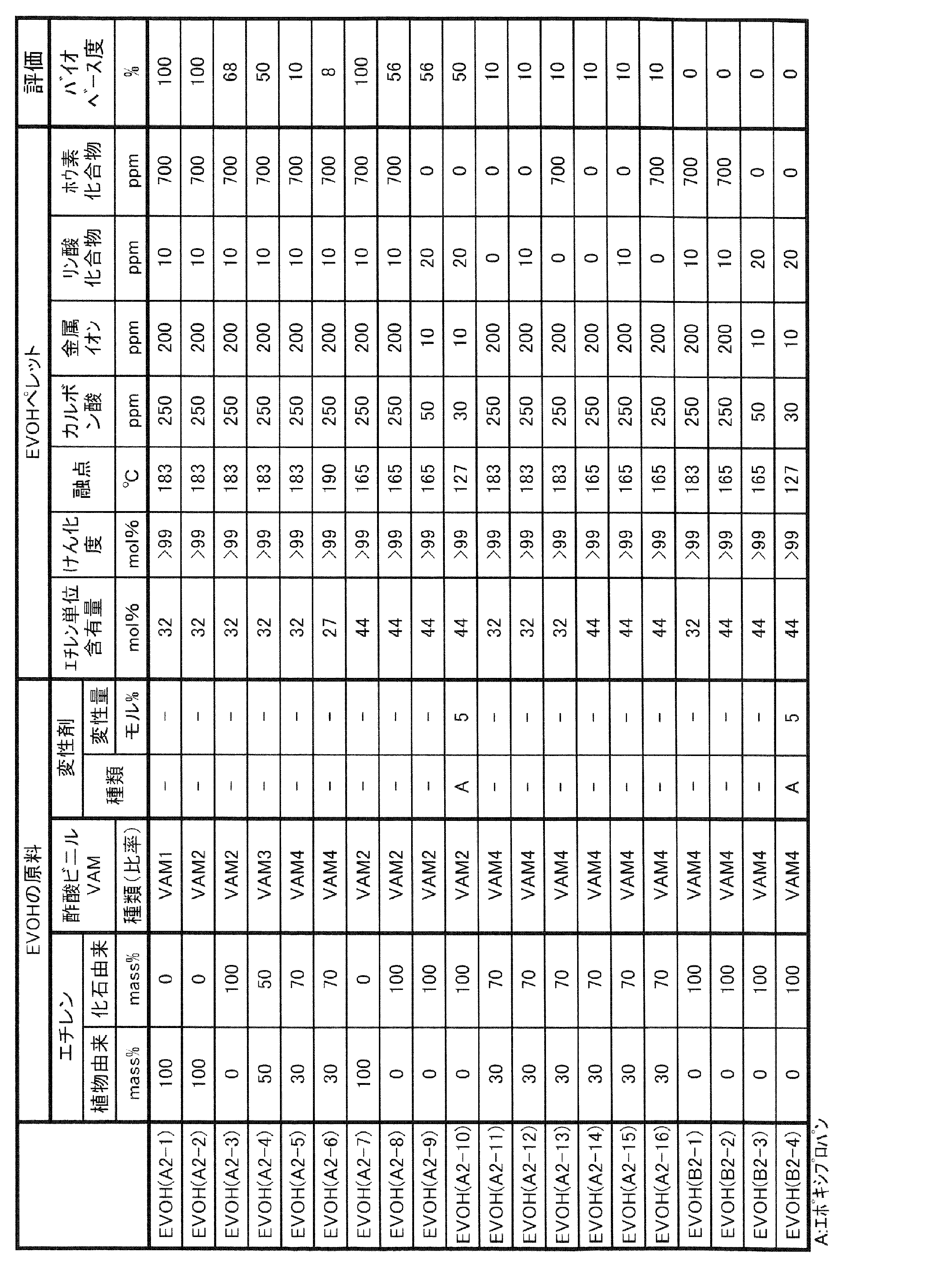
<実施例2-1>
EVOH(X)として、EVOH(A2-1)ペレット及びEVOH(B2-1)ペレットを、EVOH(Y)としてEVOH(B2-2)ペレットを用い、質量比(A2-1/B2-1/B2-2)10/60/30でドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度220℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例2-1のガスバリア樹脂組成物ペレットを得た。
EVOH(X)として、EVOH(A2-1)ペレット及びEVOH(B2-1)ペレットを、EVOH(Y)としてEVOH(B2-2)ペレットを用い、質量比(A2-1/B2-1/B2-2)10/60/30でドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度220℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例2-1のガスバリア樹脂組成物ペレットを得た。
得られた実施例2-1のガスバリア樹脂組成物ペレットについて、上記評価方法(1)、(3)~(8)に記載の方法に従って、ケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、バイオベース度、ロングラン性、酸素透過度並びに熱成形容器評価の測定又は評価を行った。結果を表5、6に示す。
<実施例2-2~2-24、比較例2-1~2-3>
用いたEVOHの種類及び質量比(割合)を、表5に記載の通り変更した以外は、実施例2-1と同様の方法で実施例2-2~2-24及び比較例2-1~2-3の各ガスバリア樹脂組成物ペレットを作製し、評価した。結果を表5、6に示す。
用いたEVOHの種類及び質量比(割合)を、表5に記載の通り変更した以外は、実施例2-1と同様の方法で実施例2-2~2-24及び比較例2-1~2-3の各ガスバリア樹脂組成物ペレットを作製し、評価した。結果を表5、6に示す。


表5、6に示されるように、実施例2-1~2-24の各ガスバリア樹脂組成物は、バイオマス由来の原料を一部に用いていながら、化石燃料由来のみのもの(比較例2-2及び2-3のガスバリア樹脂組成物)と遜色のない高いガスバリア性を有していた。また、実施例2-1~2-24の各ガスバリア樹脂組成物は、運転開始10時間後に作製された単層フィルムについての製膜欠点及びストリークの評価がA又はBであり、十分なロングラン性を有するものであった。また、実施例2-1~2-23の各ガスバリア樹脂組成物を用いた熱成形容器は、運転開始10時間後の多層シートを用いても熱成形後の外観がA又はBであり、十分な成形性も有することが示された。
また、実施例及び比較例の結果から、EVOHを用いたガスバリア樹脂組成物において、ガスバリア性はバイオベース度に依存しないのに対し、ロングラン性及び成形性はバイオベース度が低くなるにつれて高まる傾向にあるという、特異的な性質があることが確認できた。例えば、バイオベース度が65%以下の実施例2-1、2-2、2-5~2-23の各ガスバリア樹脂組成物は、運転開始10時間後及び50時間後に作製されたフィルムについての製膜欠点、ストリーク、ロール端部着色及び熱成形後外観の評価がA又はBであり、高いロングラン性及び成形性を有するものとなった。
また、実施例2-24の結果から、EVOHを用いたガスバリア樹脂組成物において、融点の異なるEVOHを含まない場合には、十分なロングラン性を有していても良好な熱成形性が発現しないことも確認できた。
<実施例2-25>
前記実施例2-6で得られたガスバリア樹脂組成物ペレットの硫黄化合物含有量の測定、及びその同定を、EVOH(A)及びEVOH(B)ペレットを測定した手法と同様の方法で行った結果、硫黄化合物の含有量は硫黄原子換算で0.3ppmであり、硫黄化合物はジメチルスルフィドであった。前記実施例2-6について、上記評価方法(8)熱成形容器評価で得られた運転開始30分後の熱成形容器を用いて、追跡可能性(トレーサビリティ性)の評価を行った。具体的には得られた熱成形容器のEVOH層を取り出し、トレーサビリティ用試料とした。取り出したEVOH層のバイオベース度を上記評価方法(5)に記載の方法に従って測定したところ10%であり、実施例2-6のガスバリア樹脂組成物ペレットで得られた値と一致し、トレーサビリティ性があることが確認された。また、取り出したEVOH層の硫黄化合物含有量の測定、及びその同定を行ったところ、硫黄化合物は硫黄原子換算で0.3ppmであり、硫黄化合物はジメチルスルフィドであり、実施例2-6のガスバリア樹脂組成物ペレットで得られた値と一致し、トレーサビリティ性があることが確認された。
前記実施例2-6で得られたガスバリア樹脂組成物ペレットの硫黄化合物含有量の測定、及びその同定を、EVOH(A)及びEVOH(B)ペレットを測定した手法と同様の方法で行った結果、硫黄化合物の含有量は硫黄原子換算で0.3ppmであり、硫黄化合物はジメチルスルフィドであった。前記実施例2-6について、上記評価方法(8)熱成形容器評価で得られた運転開始30分後の熱成形容器を用いて、追跡可能性(トレーサビリティ性)の評価を行った。具体的には得られた熱成形容器のEVOH層を取り出し、トレーサビリティ用試料とした。取り出したEVOH層のバイオベース度を上記評価方法(5)に記載の方法に従って測定したところ10%であり、実施例2-6のガスバリア樹脂組成物ペレットで得られた値と一致し、トレーサビリティ性があることが確認された。また、取り出したEVOH層の硫黄化合物含有量の測定、及びその同定を行ったところ、硫黄化合物は硫黄原子換算で0.3ppmであり、硫黄化合物はジメチルスルフィドであり、実施例2-6のガスバリア樹脂組成物ペレットで得られた値と一致し、トレーサビリティ性があることが確認された。
[EVOHの合成]
<EVOH(A3-1)~EVOH(A3-9)及びEVOH(B3-1)の各ペレットの作製>
原料(原料モノマー)のエチレン及び酢酸ビニルの種類並びにリン酸化合物及びホウ素化合物の含有量を表7に記載の通りとした以外は、EVOH(A1-1)ペレットと同様の方法で、EVOH(A3-2)ペレット~EVOH(A3-9)ペレット及びEVOH(B3-1)ペレットを作製した。化石燃料由来のエチレンには、エア・リキード工業ガス株式会社製のエチレンを用いた。
<EVOH(A3-1)~EVOH(A3-9)及びEVOH(B3-1)の各ペレットの作製>
原料(原料モノマー)のエチレン及び酢酸ビニルの種類並びにリン酸化合物及びホウ素化合物の含有量を表7に記載の通りとした以外は、EVOH(A1-1)ペレットと同様の方法で、EVOH(A3-2)ペレット~EVOH(A3-9)ペレット及びEVOH(B3-1)ペレットを作製した。化石燃料由来のエチレンには、エア・リキード工業ガス株式会社製のエチレンを用いた。
EVOH(A3-1)ペレット~EVOH(A3-9)ペレット及びEVOH(B3-1)ペレットのそれぞれについて、上記評価方法(1)、(3)~(5)に記載の方法に従い、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、並びにバイオベース度の測定を行った。結果を表7に示す。

<実施例3-1>
EVOH(A3-1)ペレット及びEVOH(B3-1)ペレットを質量比(A3-1/B3-1)10/90、並びに無機粒子として合成シリカ(富士シリシア化学社の「サイリシア310P」;レーザー法で測定された平均粒子径2.7μm)を含有量が300ppmになるようにタンブラーを用いてドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度220℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例3-1のガスバリア樹脂組成物ペレットを得た。
EVOH(A3-1)ペレット及びEVOH(B3-1)ペレットを質量比(A3-1/B3-1)10/90、並びに無機粒子として合成シリカ(富士シリシア化学社の「サイリシア310P」;レーザー法で測定された平均粒子径2.7μm)を含有量が300ppmになるようにタンブラーを用いてドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度220℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例3-1のガスバリア樹脂組成物ペレットを得た。
得られた実施例3-1のガスバリア樹脂組成物ペレットについて、上記評価方法(1)、(3)~(5)に記載の方法に従って、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量及びバイオベース度の測定を行った。結果を表8に示す。
得られたガスバリア樹脂組成物ペレットを210℃にて溶融し、ダイからキャスティングロール上に押出すと同時にエアーナイフを用いて空気を風速30m/秒で吹付け、厚さ170μmの未延伸フィルム(単層)を得た。このEVOH未延伸フィルムを、80℃の温水に10秒接触させ、テンター式同時二軸延伸機を用い、90℃雰囲気下で縦方向に3.2倍、横方向に3.0倍延伸し、さらに170℃に設定したテンター内にて5秒間熱処理を行い、フィルム端部をカットすることにより以下の二軸延伸フィルムロールを得た。
フィルム厚み :12μm
フィルム幅 :50cm
フィルム巻長さ:4,000m
上記二軸延伸フィルム製造開始から10時間経過してから連続して102本の二軸延伸フィルムを採取した。得られた二軸延伸フィルムについて、上記評価方法(9)に記載の方法に従って、二軸延伸フィルム評価を実施した。結果を表9に示す。
フィルム厚み :12μm
フィルム幅 :50cm
フィルム巻長さ:4,000m
上記二軸延伸フィルム製造開始から10時間経過してから連続して102本の二軸延伸フィルムを採取した。得られた二軸延伸フィルムについて、上記評価方法(9)に記載の方法に従って、二軸延伸フィルム評価を実施した。結果を表9に示す。
得られた二軸延伸フィルムロールの内、1本目と102本目の二軸延伸フィルムを用い、日本真空技術社の「バッチ式蒸着設備EWA-105」を使用して、フィルム走行速度200m/分で、フィルム片側にアルミニウムを蒸着させた後に巻き取り、蒸着フィルムロールを得た。得られた蒸着フィルムについて、上記評価方法(10)に記載の方法に従って、蒸着フィルム評価を実施した。結果を表9に示す。
<実施例3-2~3-21、比較例3-1~3-2>
用いたEVOHの種類及び質量比(割合)並びに無機粒子の含有量を、表8に記載の通り変更した以外は、実施例3-1と同様の方法で実施例3-2~3-21及び比較例3-1~3-2の各ガスバリア樹脂組成物ペレット、二軸延伸フィルム及び蒸着フィルムを作製し、評価した。結果を表8、9に示す。なお、実施例3-21に関しては蒸着フィルムの評価を実施しなかった。
用いたEVOHの種類及び質量比(割合)並びに無機粒子の含有量を、表8に記載の通り変更した以外は、実施例3-1と同様の方法で実施例3-2~3-21及び比較例3-1~3-2の各ガスバリア樹脂組成物ペレット、二軸延伸フィルム及び蒸着フィルムを作製し、評価した。結果を表8、9に示す。なお、実施例3-21に関しては蒸着フィルムの評価を実施しなかった。


表8、9に示されるように、実施例3-1~3-21の各ガスバリア樹脂組成物は、バイオマス由来の原料を一部に用いていながら、化石燃料由来のみのもの(比較例3-2のガスバリア樹脂組成物)と遜色のない高いガスバリア性を有していた。また、実施例3-1~3-20の各ガスバリア樹脂組成物は、運転開始より10時間経過した後に作製された二軸延伸フィルムについての耐フィルム破断性、並びに運転開始10時間後に作製された二軸延伸フィルムを用いて作製された蒸着フィルムの無機蒸着層平均厚み、蒸着欠点抑制性及び密着強度がバイオマス由来原料のみのもの(比較例3-1のガスバリア性樹脂組成物)と比較して良好な結果を示しており、十分なロングラン性を有するものであった。
また、実施例及び比較例の結果から、EVOHを用いたガスバリア樹脂組成物において、ガスバリア性はバイオベース度に依存しないのに対し、ロングラン性はバイオベース度が低くなるにつれて高まる傾向にあるという、特異的な性質があることが確認できた。例えば、バイオベース度が65%以下の実施例3-1、3-2、3-5~3-13、3-15~3-20の各ガスバリア樹脂組成物は、運転開始より10時間経過した後に作製された二軸延伸フィルムについての耐フィルム破断性、並びに運転開始10時間後に作製された1本目の二軸延伸フィルムを用いて作製された蒸着フィルムの蒸着欠点抑制性及び密着強度の評価がA又はBであり、高いロングラン性を有するものとなった。
また、実施例3-21の結果から、EVOHを用いたガスバリア樹脂組成物において、無機粒子を含まない場合には、耐フィルム破断性が不十分であることが確認できた。
[EVOHの合成]
<EVOH(A4-1)ペレットの作製>
(エチレン-酢酸ビニル共重合体の重合)
ジャケット、攪拌機、窒素導入口、エチレン導入口及び開始剤添加口を備えた250L加圧反応槽に、VAM1を105kg、及びメタノール(以下、MeOHと称することもある)を10.1kg仕込み、65℃に昇温した後、30分間窒素バブリングして反応槽内を窒素置換した。次いで反応槽圧力(エチレン圧力)が4.13MPaとなるようにエチレンを昇圧して導入した。エチレンには、バイオマス由来のエチレン(Braskem S.A.製、サトウキビ由来のバイオエチレン)を用いた。反応槽内の温度を65℃に調整した後、開始剤として13.2gの2,2’-アゾビス(2,4-ジメチルバレロニトリル)(和光純薬工業社の「V-65」)をメタノール溶液として添加し、重合を開始した。重合中はエチレン圧力を4.13MPaに、重合温度を65℃に維持した。4時間後にVAcの重合率が50%となったところで冷却して重合を停止した。反応槽を開放して脱エチレンした後、窒素ガスをバブリングして脱エチレンを完全に行った。次いで減圧下で未反応のVAcを除去した後、エチレン-酢酸ビニル共重合体にMeOHを添加して20質量%MeOH溶液とした。
<EVOH(A4-1)ペレットの作製>
(エチレン-酢酸ビニル共重合体の重合)
ジャケット、攪拌機、窒素導入口、エチレン導入口及び開始剤添加口を備えた250L加圧反応槽に、VAM1を105kg、及びメタノール(以下、MeOHと称することもある)を10.1kg仕込み、65℃に昇温した後、30分間窒素バブリングして反応槽内を窒素置換した。次いで反応槽圧力(エチレン圧力)が4.13MPaとなるようにエチレンを昇圧して導入した。エチレンには、バイオマス由来のエチレン(Braskem S.A.製、サトウキビ由来のバイオエチレン)を用いた。反応槽内の温度を65℃に調整した後、開始剤として13.2gの2,2’-アゾビス(2,4-ジメチルバレロニトリル)(和光純薬工業社の「V-65」)をメタノール溶液として添加し、重合を開始した。重合中はエチレン圧力を4.13MPaに、重合温度を65℃に維持した。4時間後にVAcの重合率が50%となったところで冷却して重合を停止した。反応槽を開放して脱エチレンした後、窒素ガスをバブリングして脱エチレンを完全に行った。次いで減圧下で未反応のVAcを除去した後、エチレン-酢酸ビニル共重合体にMeOHを添加して20質量%MeOH溶液とした。
(ケン化及び洗浄)
得られたエチレン-酢酸ビニル共重合体ジャケットの20質量%MeOH溶液250kgを、攪拌機、窒素導入口、還流冷却器及び溶液添加口を備えた500L反応槽に入れ、かかる溶液に窒素を吹き込みながら60℃に昇温し、水酸化ナトリウム4kgを濃度2規定のMeOH溶液として添加した。水酸化ナトリウムの添加終了後、系内温度を60℃に保ちながら2時間攪拌してケン化反応を進行させた。2時間経過した後に、再度、同様の方法で水酸化ナトリウムを4kg添加し、2時間加熱攪拌を継続した。その後、酢酸を14kg添加してケン化反応を停止し、イオン交換水50kgを添加した。加熱攪拌しながら反応槽外にMeOHと水を留出させ反応液を濃縮した。3時間経過した後、更にイオン交換水50kgを添加し、EVOHを析出させた。デカンテーションにより析出したEVOHを収集し、ミキサーで粉砕した。得られたEVOH粉末を1g/Lの酢酸水溶液(浴比20:イオン交換水200Lに対し粉末10kgの割合)に投入して2時間攪拌洗浄した。これを脱液し、さらに1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。これを脱液したものを、イオン交換水(浴比20)に投入して攪拌洗浄を2時間行い脱液する操作を3回繰り返して精製を行った。これを60℃で16時間乾燥させることでEVOHの粗乾燥物を25kg得た。
得られたエチレン-酢酸ビニル共重合体ジャケットの20質量%MeOH溶液250kgを、攪拌機、窒素導入口、還流冷却器及び溶液添加口を備えた500L反応槽に入れ、かかる溶液に窒素を吹き込みながら60℃に昇温し、水酸化ナトリウム4kgを濃度2規定のMeOH溶液として添加した。水酸化ナトリウムの添加終了後、系内温度を60℃に保ちながら2時間攪拌してケン化反応を進行させた。2時間経過した後に、再度、同様の方法で水酸化ナトリウムを4kg添加し、2時間加熱攪拌を継続した。その後、酢酸を14kg添加してケン化反応を停止し、イオン交換水50kgを添加した。加熱攪拌しながら反応槽外にMeOHと水を留出させ反応液を濃縮した。3時間経過した後、更にイオン交換水50kgを添加し、EVOHを析出させた。デカンテーションにより析出したEVOHを収集し、ミキサーで粉砕した。得られたEVOH粉末を1g/Lの酢酸水溶液(浴比20:イオン交換水200Lに対し粉末10kgの割合)に投入して2時間攪拌洗浄した。これを脱液し、さらに1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。これを脱液したものを、イオン交換水(浴比20)に投入して攪拌洗浄を2時間行い脱液する操作を3回繰り返して精製を行った。これを60℃で16時間乾燥させることでEVOHの粗乾燥物を25kg得た。
(EVOH含水ペレットの製造)
得られたEVOHの粗乾燥物25kgを、ジャケット、攪拌機及び還流冷却器を備えた100L攪拌槽に入れ、さらに水20kg及びMeOH20gを加え、70℃に昇温して溶解させた。この溶解液を径3mmのガラス管を通して5℃に冷却した重量比で水/MeOH=90/10の混合液中に押し出してストランド状に析出させ、このストランドをストランドカッターでペレット状にカットすることでEVOHの含水ペレットを得た。このEVOHの含水ペレットを濃度1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。これを脱液し、さらに1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。脱液後、酢酸水溶液を更新し同様の操作を行った。酢酸水溶液で洗浄してから脱液したものを、イオン交換水(浴比20)に投入して攪拌洗浄を2時間行い脱液する操作を3回繰り返して精製を行い、ケン化反応時の触媒残渣とストランド析出時に使用したMeOHが除去された、EVOHの含水ペレットを得た。得られたEVOHの含水ペレットの含水率をメトラー社のハロゲン水分計「HR73」で測定したところ、110質量%であった。
得られたEVOHの粗乾燥物25kgを、ジャケット、攪拌機及び還流冷却器を備えた100L攪拌槽に入れ、さらに水20kg及びMeOH20gを加え、70℃に昇温して溶解させた。この溶解液を径3mmのガラス管を通して5℃に冷却した重量比で水/MeOH=90/10の混合液中に押し出してストランド状に析出させ、このストランドをストランドカッターでペレット状にカットすることでEVOHの含水ペレットを得た。このEVOHの含水ペレットを濃度1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。これを脱液し、さらに1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。脱液後、酢酸水溶液を更新し同様の操作を行った。酢酸水溶液で洗浄してから脱液したものを、イオン交換水(浴比20)に投入して攪拌洗浄を2時間行い脱液する操作を3回繰り返して精製を行い、ケン化反応時の触媒残渣とストランド析出時に使用したMeOHが除去された、EVOHの含水ペレットを得た。得られたEVOHの含水ペレットの含水率をメトラー社のハロゲン水分計「HR73」で測定したところ、110質量%であった。
(EVOH(A4-1)ペレットの製造)
得られたEVOHの含水ペレットを酢酸ナトリウム、酢酸及びリン酸が含まれる水溶液(浴比20)に投入し、定期的に攪拌しながら4時間浸漬させた。なお、各成分の濃度は、得られたEVOH(A4-1)ペレットにおける各成分の含有量が表10に記載の通りとなるように調整した。浸漬後に脱液し、空気下で80℃、3時間、及び空気下で130℃、7.5時間乾燥することにより、酢酸ナトリウム、酢酸及びリン酸を含むEVOH(A4-1)ペレットを得た。
得られたEVOHの含水ペレットを酢酸ナトリウム、酢酸及びリン酸が含まれる水溶液(浴比20)に投入し、定期的に攪拌しながら4時間浸漬させた。なお、各成分の濃度は、得られたEVOH(A4-1)ペレットにおける各成分の含有量が表10に記載の通りとなるように調整した。浸漬後に脱液し、空気下で80℃、3時間、及び空気下で130℃、7.5時間乾燥することにより、酢酸ナトリウム、酢酸及びリン酸を含むEVOH(A4-1)ペレットを得た。
<EVOH(A4-2)~EVOH(A4-9)、EVOH(B4-1)~EVOH(B4-4)の各ペレットの作製>
原料(原料モノマー)のエチレン及び酢酸ビニルの種類並びにリン酸化合物及びホウ素化合物の含有量を表10に記載の通りにした以外は、EVOH(A4-1)ペレットと同様の方法で、EVOH(A4-2)ペレット~EVOH(A4-9)ペレット及びEVOH(B4-1)~EVOH(B4-4)ペレットを作製した。化石燃料由来のエチレンには、エア・リキード工業ガス株式会社製のエチレンを用いた。
原料(原料モノマー)のエチレン及び酢酸ビニルの種類並びにリン酸化合物及びホウ素化合物の含有量を表10に記載の通りにした以外は、EVOH(A4-1)ペレットと同様の方法で、EVOH(A4-2)ペレット~EVOH(A4-9)ペレット及びEVOH(B4-1)~EVOH(B4-4)ペレットを作製した。化石燃料由来のエチレンには、エア・リキード工業ガス株式会社製のエチレンを用いた。
EVOH(A4-1)ペレット~EVOH(A4-9)ペレット及びEVOH(B4-1)~EVOH(B4-4)ペレットのそれぞれについて、上記評価方法(1)、(3)~(5)に記載の方法に従い、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、並びにバイオベース度の測定を行った。結果を表10に示す。
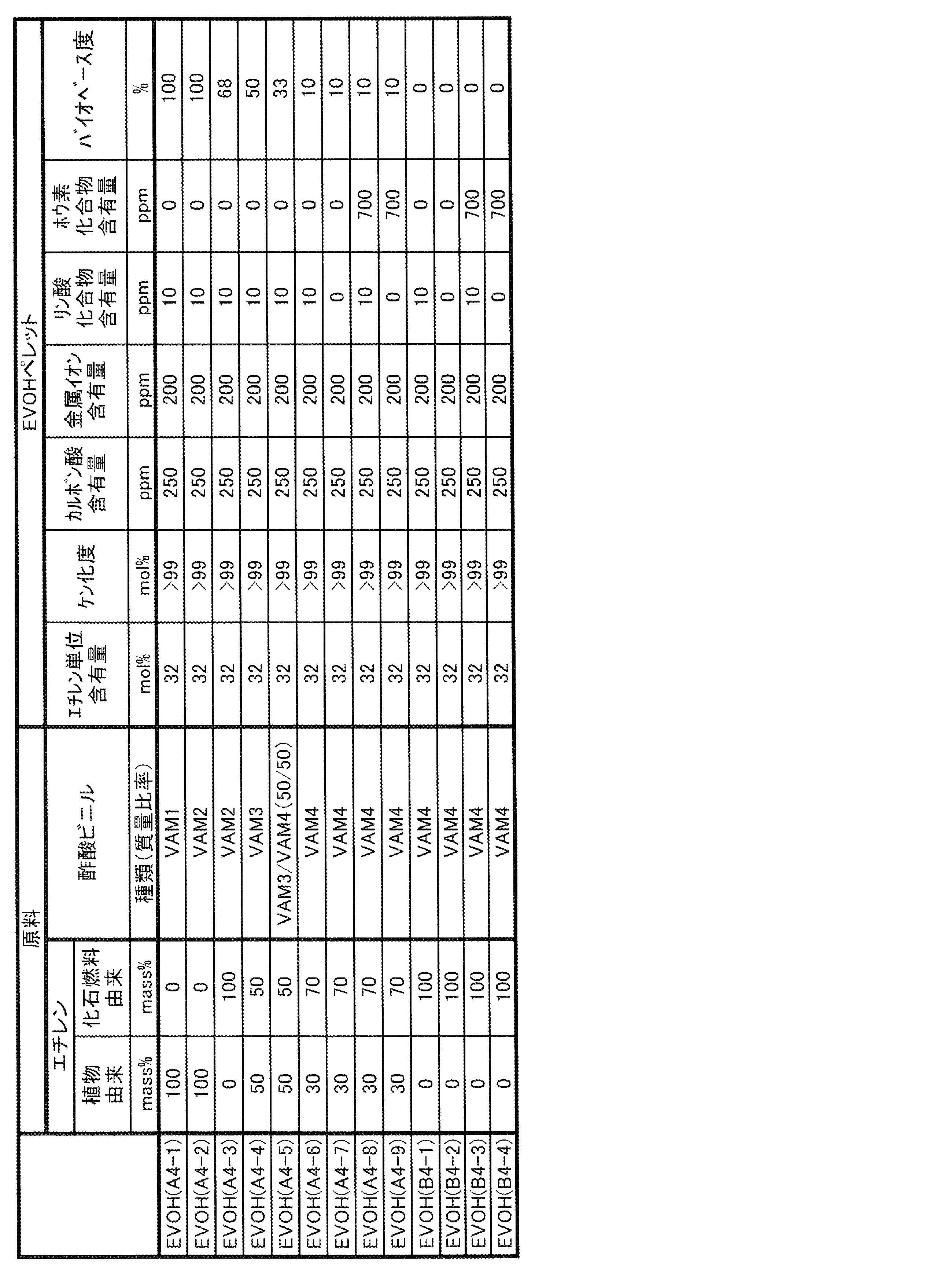
<実施例4-1>
EVOH(A4-1)ペレットを10質量部、EVOH(B4-1)ペレット90質量部、及び酸化防止剤(D)としてN,N’-(ヘキサン-1,6-ジイル)ビス[3-(3,5-ジ-tert-ブチル-4-ヒドロキシフェニル)プロピオンアミド](BASFジャパン社製「Irganox1098」、分子量:637)0.5質量部をドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度220℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例4-1のガスバリア樹脂組成物ペレットを得た。
EVOH(A4-1)ペレットを10質量部、EVOH(B4-1)ペレット90質量部、及び酸化防止剤(D)としてN,N’-(ヘキサン-1,6-ジイル)ビス[3-(3,5-ジ-tert-ブチル-4-ヒドロキシフェニル)プロピオンアミド](BASFジャパン社製「Irganox1098」、分子量:637)0.5質量部をドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度220℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例4-1のガスバリア樹脂組成物ペレットを得た。
得られた実施例4-1のガスバリア樹脂組成物ペレットについて、上記評価方法(1)、(3)~(7)、(11)、(12)に記載の方法に従って、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、バイオベース度、単層フィルムのロングラン性、酸素透過度、耐酸化劣化性並びに多層パイプの測定又は評価を行った。結果を表11、12に示す。
<実施例4-2~4-21、比較例4-1~4-5>
用いたEVOHの種類及び質量比(割合)並びに酸化防止剤(D)の含有量を、表11に記載の通り変更した以外は、実施例4-1と同様の方法で実施例4-2~4-21及び比較例4-1~4-5の各ガスバリア樹脂組成物ペレットを作製し、評価した。結果を表11、12に示す。
用いたEVOHの種類及び質量比(割合)並びに酸化防止剤(D)の含有量を、表11に記載の通り変更した以外は、実施例4-1と同様の方法で実施例4-2~4-21及び比較例4-1~4-5の各ガスバリア樹脂組成物ペレットを作製し、評価した。結果を表11、12に示す。
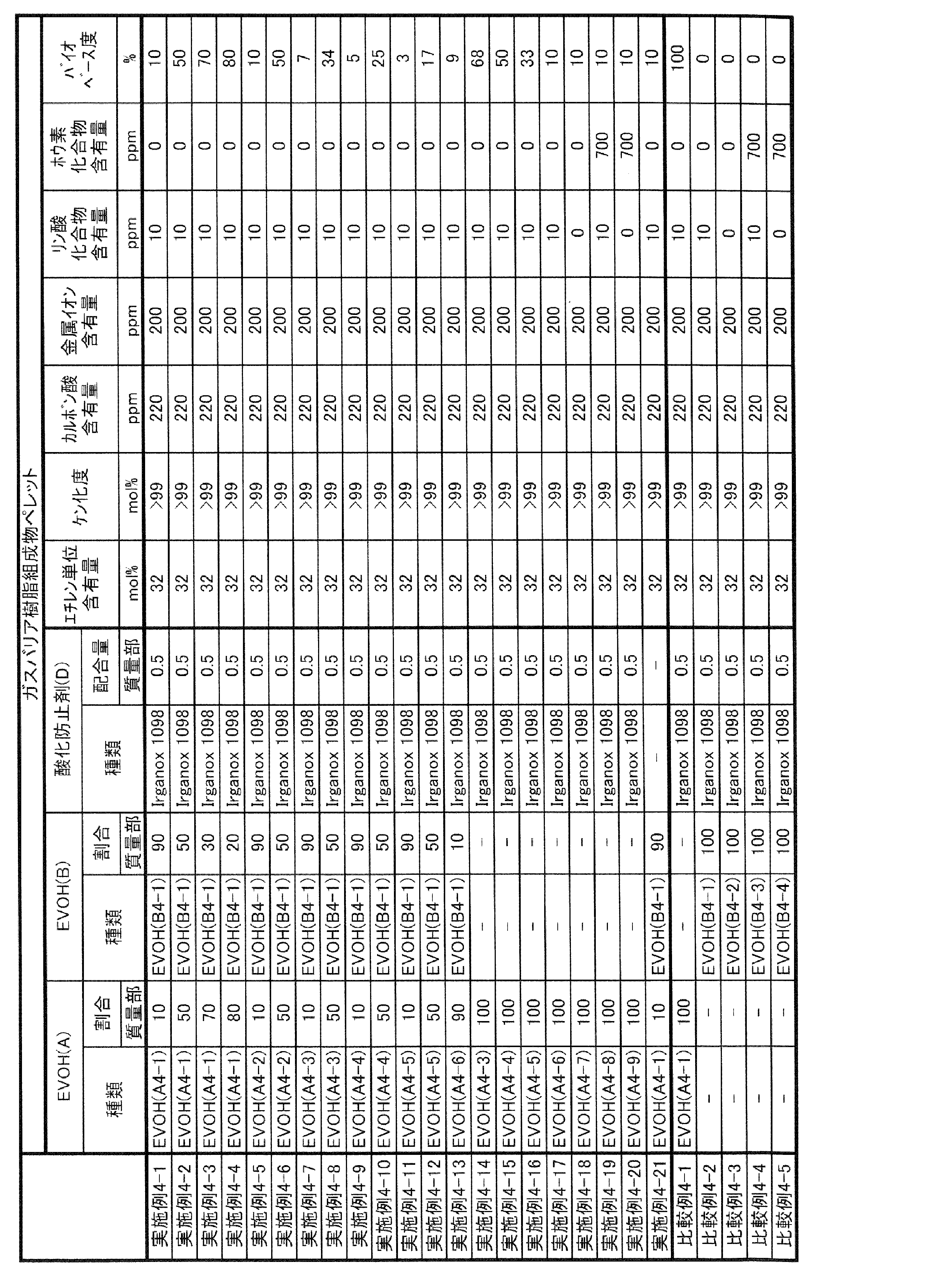
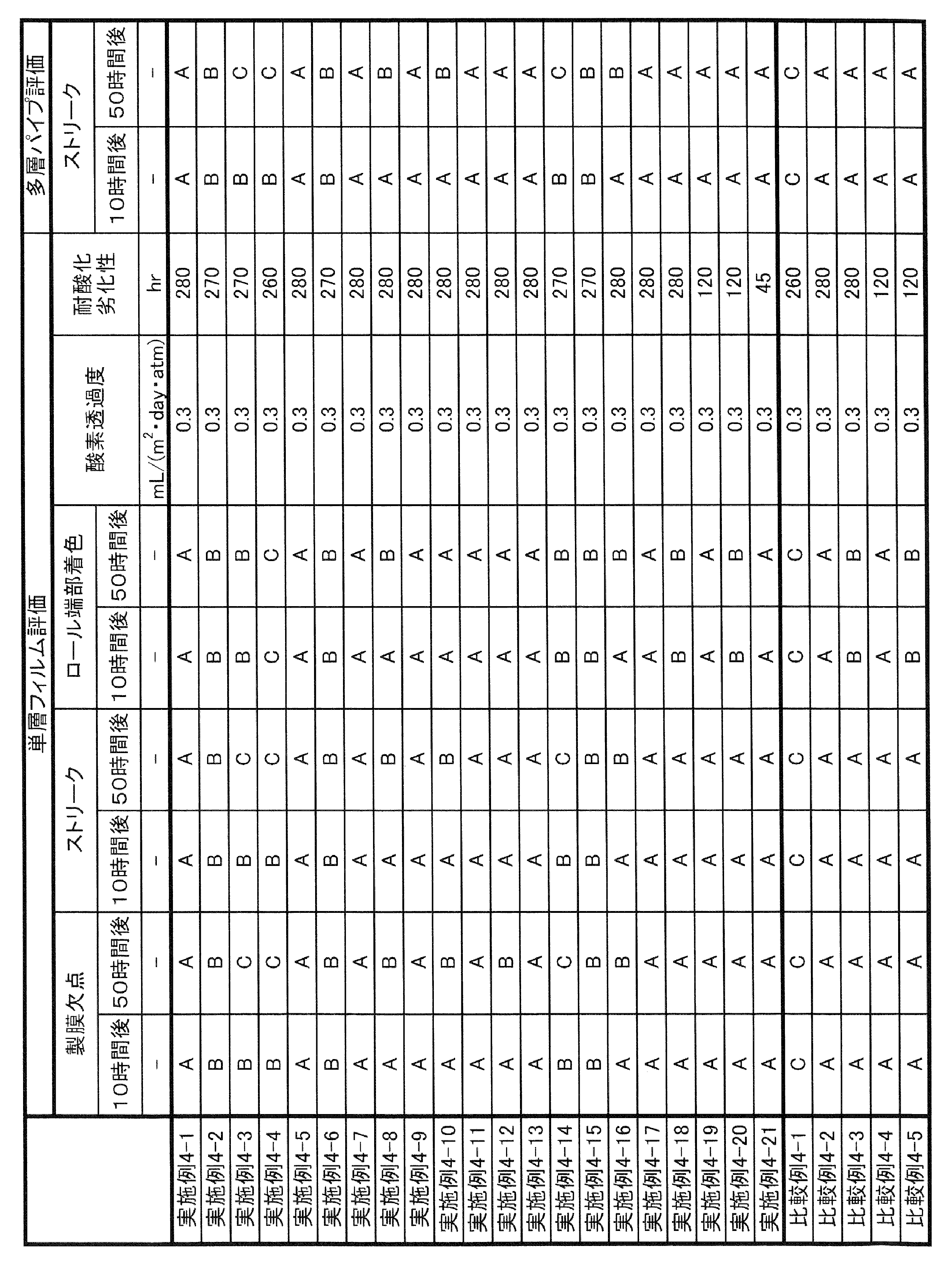
表11、12に示されるように、実施例4-1~4-21の各ガスバリア樹脂組成物は、バイオマス由来の原料を一部に用いていながら、化石燃料由来のみのもの(比較例4-2のガスバリア樹脂組成物等)と遜色のない高いガスバリア性を有していた。また、実施例4-1~4-21の各ガスバリア樹脂組成物は、運転開始10時間後に作製されたフィルムについての製膜欠点及びストリークの評価、並びに運転開始10時間後に作製された多層パイプについてのストリークの評価がA又はBであり、十分なロングラン性を有するものであった。
また、実施例及び比較例の結果から、EVOHを用いたガスバリア樹脂組成物において、ガスバリア性はバイオベース度に依存しないのに対し、ロングラン性はバイオベース度が低くなるにつれて高まる傾向にあるという、特異的な性質があることが確認できた。例えば、バイオベース度が65%以下の実施例4-1、4-2、4-5~4-13、4-15~4-21の各ガスバリア樹脂組成物は、運転開始10時間後及び50時間後に作製されたフィルムについての製膜欠点、ストリーク及びロール端部着色の評価がA又はBであり、高いロングラン性を有するものとなった。
さらに、実施例4-1及び実施例4-21の結果から、ガスバリア性樹脂組成物に含まれる酸化防止剤が耐酸化劣化性の向上に寄与していることが認められた。
[EVOHの合成]
<EVOH(A5-1)ペレットの作製>
(エチレン-酢酸ビニル共重合体の重合)
ジャケット、攪拌機、窒素導入口、エチレン導入口及び開始剤添加口を備えた250L加圧反応槽に、VAM1を105kg、及びメタノール(以下、MeOHと称することもある)を45.7kg仕込み、60℃に昇温した後、30分間窒素バブリングして反応槽内を窒素置換した。次いで反応槽圧力(エチレン圧力)が2.87MPaとなるようにエチレンを昇圧して導入した。エチレンには、バイオマス由来のエチレン(Braskem S.A.製、サトウキビ由来のバイオエチレン)を用いた。反応槽内の温度を60℃に調整した後、開始剤として34.7gの2,2’-アゾビス(2,4-ジメチルバレロニトリル)(和光純薬工業社の「V-65」)をメタノール溶液として添加し、重合を開始した。重合中はエチレン圧力を2.87MPaに、重合温度を60℃に維持した。3時間後にVAcの重合率が45%となったところで冷却して重合を停止した。反応槽を開放して脱エチレンした後、窒素ガスをバブリングして脱エチレンを完全に行った。次いで減圧下で未反応のVAcを除去した後、エチレン-酢酸ビニル共重合体にMeOHを添加して20質量%MeOH溶液とした。
<EVOH(A5-1)ペレットの作製>
(エチレン-酢酸ビニル共重合体の重合)
ジャケット、攪拌機、窒素導入口、エチレン導入口及び開始剤添加口を備えた250L加圧反応槽に、VAM1を105kg、及びメタノール(以下、MeOHと称することもある)を45.7kg仕込み、60℃に昇温した後、30分間窒素バブリングして反応槽内を窒素置換した。次いで反応槽圧力(エチレン圧力)が2.87MPaとなるようにエチレンを昇圧して導入した。エチレンには、バイオマス由来のエチレン(Braskem S.A.製、サトウキビ由来のバイオエチレン)を用いた。反応槽内の温度を60℃に調整した後、開始剤として34.7gの2,2’-アゾビス(2,4-ジメチルバレロニトリル)(和光純薬工業社の「V-65」)をメタノール溶液として添加し、重合を開始した。重合中はエチレン圧力を2.87MPaに、重合温度を60℃に維持した。3時間後にVAcの重合率が45%となったところで冷却して重合を停止した。反応槽を開放して脱エチレンした後、窒素ガスをバブリングして脱エチレンを完全に行った。次いで減圧下で未反応のVAcを除去した後、エチレン-酢酸ビニル共重合体にMeOHを添加して20質量%MeOH溶液とした。
(ケン化及び洗浄)
得られたエチレン-酢酸ビニル共重合体ジャケットの20質量%MeOH溶液250kgを、攪拌機、窒素導入口、還流冷却器及び溶液添加口を備えた500L反応槽に入れ、かかる溶液に窒素を吹き込みながら60℃に昇温し、水酸化ナトリウム4kgを濃度2規定のMeOH溶液として添加した。水酸化ナトリウムの添加終了後、系内温度を60℃に保ちながら2時間攪拌してケン化反応を進行させた。2時間経過した後に、再度、同様の方法で水酸化ナトリウムを4kg添加し、2時間加熱攪拌を継続した。その後、酢酸を14kg添加してケン化反応を停止し、イオン交換水50kgを添加した。加熱攪拌しながら反応槽外にMeOHと水を留出させ反応液を濃縮した。3時間経過した後、更にイオン交換水50kgを添加し、EVOHを析出させた。デカンテーションにより析出したEVOHを収集し、ミキサーで粉砕した。得られたEVOH粉末を1g/Lの酢酸水溶液(浴比20:イオン交換水200Lに対し粉末10kgの割合)に投入して2時間攪拌洗浄した。これを脱液し、さらに1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。これを脱液したものを、イオン交換水(浴比20)に投入して攪拌洗浄を2時間行い脱液する操作を3回繰り返して精製を行った。これを60℃で16時間乾燥させることでEVOHの粗乾燥物を25kg得た。
得られたエチレン-酢酸ビニル共重合体ジャケットの20質量%MeOH溶液250kgを、攪拌機、窒素導入口、還流冷却器及び溶液添加口を備えた500L反応槽に入れ、かかる溶液に窒素を吹き込みながら60℃に昇温し、水酸化ナトリウム4kgを濃度2規定のMeOH溶液として添加した。水酸化ナトリウムの添加終了後、系内温度を60℃に保ちながら2時間攪拌してケン化反応を進行させた。2時間経過した後に、再度、同様の方法で水酸化ナトリウムを4kg添加し、2時間加熱攪拌を継続した。その後、酢酸を14kg添加してケン化反応を停止し、イオン交換水50kgを添加した。加熱攪拌しながら反応槽外にMeOHと水を留出させ反応液を濃縮した。3時間経過した後、更にイオン交換水50kgを添加し、EVOHを析出させた。デカンテーションにより析出したEVOHを収集し、ミキサーで粉砕した。得られたEVOH粉末を1g/Lの酢酸水溶液(浴比20:イオン交換水200Lに対し粉末10kgの割合)に投入して2時間攪拌洗浄した。これを脱液し、さらに1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。これを脱液したものを、イオン交換水(浴比20)に投入して攪拌洗浄を2時間行い脱液する操作を3回繰り返して精製を行った。これを60℃で16時間乾燥させることでEVOHの粗乾燥物を25kg得た。
(EVOH含水ペレットの製造)
得られたEVOHの粗乾燥物25kgを、ジャケット、攪拌機及び還流冷却器を備えた100L攪拌槽に入れ、さらに水20kg及びMeOH20gを加え、70℃に昇温して溶解させた。この溶解液を径3mmのガラス管を通して5℃に冷却した重量比で水/MeOH=90/10の混合液中に押し出してストランド状に析出させ、このストランドをストランドカッターでペレット状にカットすることでEVOHの含水ペレットを得た。このEVOHの含水ペレットを濃度1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。これを脱液し、さらに1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。脱液後、酢酸水溶液を更新し同様の操作を行った。酢酸水溶液で洗浄してから脱液したものを、イオン交換水(浴比20)に投入して攪拌洗浄を2時間行い脱液する操作を3回繰り返して精製を行い、ケン化反応時の触媒残渣とストランド析出時に使用したMeOHが除去された、EVOHの含水ペレットを得た。得られたEVOHの含水ペレットの含水率をメトラー社のハロゲン水分計「HR73」で測定したところ、110質量%であった。
得られたEVOHの粗乾燥物25kgを、ジャケット、攪拌機及び還流冷却器を備えた100L攪拌槽に入れ、さらに水20kg及びMeOH20gを加え、70℃に昇温して溶解させた。この溶解液を径3mmのガラス管を通して5℃に冷却した重量比で水/MeOH=90/10の混合液中に押し出してストランド状に析出させ、このストランドをストランドカッターでペレット状にカットすることでEVOHの含水ペレットを得た。このEVOHの含水ペレットを濃度1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。これを脱液し、さらに1g/Lの酢酸水溶液(浴比20)に投入して2時間攪拌洗浄した。脱液後、酢酸水溶液を更新し同様の操作を行った。酢酸水溶液で洗浄してから脱液したものを、イオン交換水(浴比20)に投入して攪拌洗浄を2時間行い脱液する操作を3回繰り返して精製を行い、ケン化反応時の触媒残渣とストランド析出時に使用したMeOHが除去された、EVOHの含水ペレットを得た。得られたEVOHの含水ペレットの含水率をメトラー社のハロゲン水分計「HR73」で測定したところ、110質量%であった。
(EVOH(A5-1)ペレットの製造)
得られたEVOHの含水ペレットを酢酸ナトリウム、酢酸、リン酸及びホウ酸が含まれる水溶液(浴比20)に投入し、定期的に攪拌しながら4時間浸漬させた。なお、各成分の濃度は、得られたEVOH(A5-1)ペレットにおける各成分の含有量が表13に記載の通りとなるように調整した。浸漬後に脱液し、空気下で80℃、3時間、及び空気下で130℃、7.5時間乾燥することにより、酢酸ナトリウム、酢酸、リン酸及びホウ酸を含むEVOH(A5-1)ペレットを得た。
得られたEVOHの含水ペレットを酢酸ナトリウム、酢酸、リン酸及びホウ酸が含まれる水溶液(浴比20)に投入し、定期的に攪拌しながら4時間浸漬させた。なお、各成分の濃度は、得られたEVOH(A5-1)ペレットにおける各成分の含有量が表13に記載の通りとなるように調整した。浸漬後に脱液し、空気下で80℃、3時間、及び空気下で130℃、7.5時間乾燥することにより、酢酸ナトリウム、酢酸、リン酸及びホウ酸を含むEVOH(A5-1)ペレットを得た。
<EVOH(A5-2)~EVOH(A5-9)、EVOH(B5-1)~EVOH(B5-4)の各ペレットの作製>
原料(原料モノマー)のエチレン及び酢酸ビニルの種類並びにリン酸化合物及びホウ素化合物の含有量を表13に記載の通りとした以外は、EVOH(A5-1)ペレットと同様の方法で、EVOH(A5-2)ペレット~EVOH(A5-9)ペレット及びEVOH(B5-1)~EVOH(B5-4)ペレットを作製した。化石燃料由来のエチレンには、エア・リキード工業ガス株式会社製のエチレンを用いた。
原料(原料モノマー)のエチレン及び酢酸ビニルの種類並びにリン酸化合物及びホウ素化合物の含有量を表13に記載の通りとした以外は、EVOH(A5-1)ペレットと同様の方法で、EVOH(A5-2)ペレット~EVOH(A5-9)ペレット及びEVOH(B5-1)~EVOH(B5-4)ペレットを作製した。化石燃料由来のエチレンには、エア・リキード工業ガス株式会社製のエチレンを用いた。
EVOH(A5-1)ペレット~EVOH(A5-9)ペレット及びEVOH(B5-1)~EVOH(B5-4)ペレットのそれぞれについて、上記評価方法(1)、(3)~(5)に記載の方法に従い、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、並びにバイオベース度の測定を行った。結果を表13に示す。
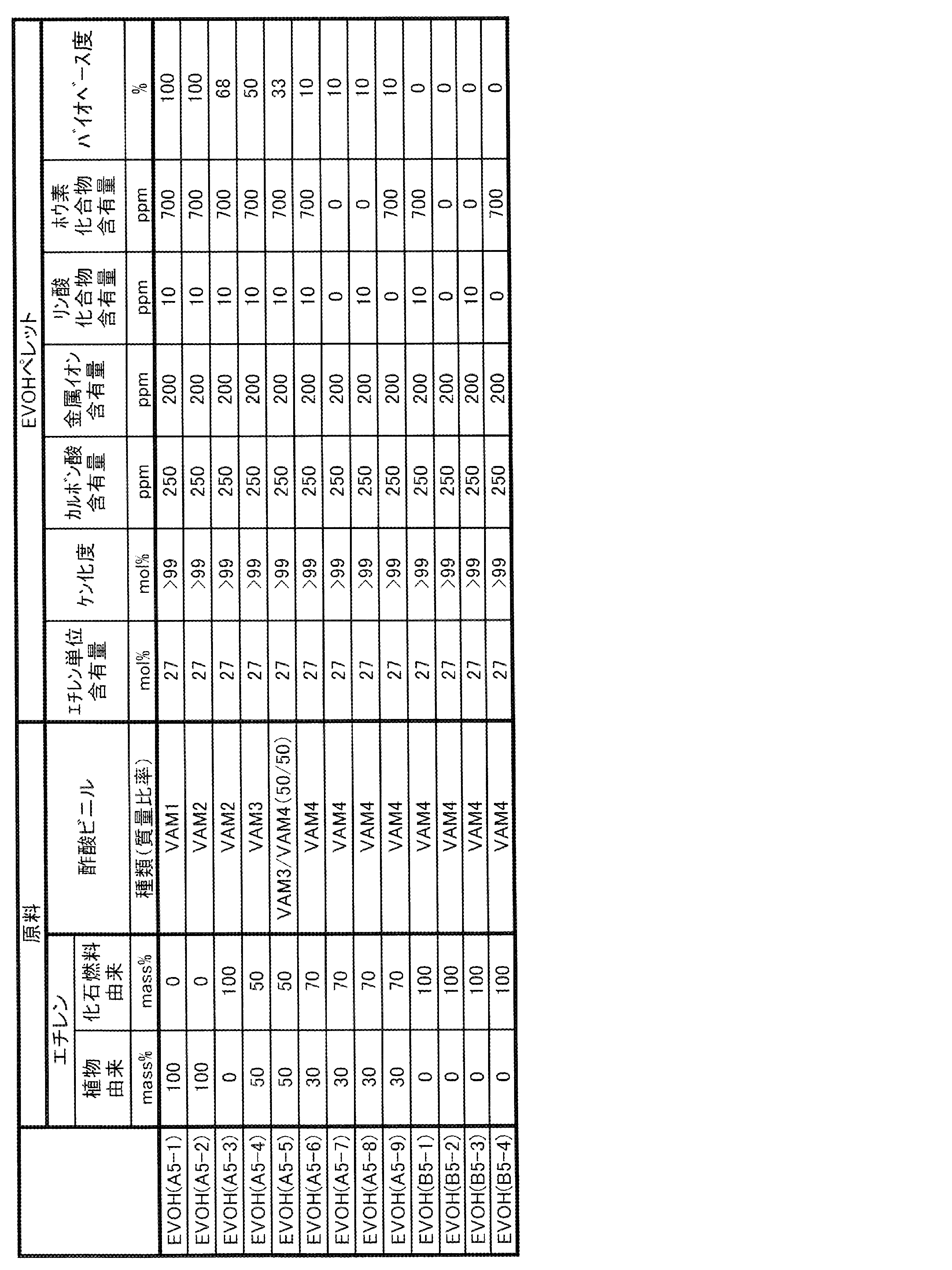
<実施例5-1>
EVOH(A5-1)ペレットを9質量部、EVOH(B5-1)ペレットを81質量部、ポリアミドペレット(宇部興産株式会社製「Ny1018A」(ナイロン6))(E)を10質量部、及び金属原子(F)を含む化合物として酢酸マグネシウムを金属原子換算で100ppm添加してドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度230℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例5-1のガスバリア樹脂組成物ペレットを得た。
EVOH(A5-1)ペレットを9質量部、EVOH(B5-1)ペレットを81質量部、ポリアミドペレット(宇部興産株式会社製「Ny1018A」(ナイロン6))(E)を10質量部、及び金属原子(F)を含む化合物として酢酸マグネシウムを金属原子換算で100ppm添加してドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度230℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例5-1のガスバリア樹脂組成物ペレットを得た。
得られた実施例5-1のガスバリア樹脂組成物ペレットについて、上記評価方法(1)、(3)~(5)、(13)~(15)に記載の方法に従って、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、バイオベース度、ロングラン性、酸素透過度並びに耐レトルト性の測定又は評価を行った。なお、金属イオンの定量に関し、金属原子(F)として添加された金属原子の量も含めて金属イオンの金属原子換算量を求めた。結果を表14、15に示す。
<実施例5-2~5-27、比較例5-1~5-8>
用いたEVOHの種類及び質量比(割合)、ポリアミド(E)の質量比、並びに金属原子(F)を含む化合物の種類及び金属原子換算の含有量を、表14に記載の通り変更した以外は、実施例5-1と同様の方法で実施例5-2~5-27及び比較例5-1~5-8の各ガスバリア樹脂組成物ペレットを作製し、評価した。結果を表14、15に示す。
用いたEVOHの種類及び質量比(割合)、ポリアミド(E)の質量比、並びに金属原子(F)を含む化合物の種類及び金属原子換算の含有量を、表14に記載の通り変更した以外は、実施例5-1と同様の方法で実施例5-2~5-27及び比較例5-1~5-8の各ガスバリア樹脂組成物ペレットを作製し、評価した。結果を表14、15に示す。


表14、15に示されるように、実施例5-1~5-27の各ガスバリア樹脂組成物は、バイオマス由来の原料を一部に用いていながら、化石燃料由来のみのもの(比較例5-2のガスバリア樹脂組成物)と遜色のない高いガスバリア性を有していた。また、実施例5-1~5-27の各ガスバリア樹脂組成物は、運転開始1時間後に作製されたフィルムについての製膜欠点及びストリークの評価がA又はBであり、十分なロングラン性を有するものであった。
また、実施例及び比較例の結果から、EVOHを用いたガスバリア樹脂組成物において、ガスバリア性及び耐レトルト性はバイオベース度に依存しないのに対し、ロングラン性はバイオベース度が低くなるにつれて高まる傾向にあるという、特異的な性質があることが確認できた。例えば、バイオベース度が65%以下の実施例5-1~5-3、5-5~5-27の各ガスバリア樹脂組成物は、運転開始1時間後及び5時間後に作製されたフィルムについての製膜欠点及びストリークの評価がA又はBであり、高いロングラン性を有するものとなった。
また、比較例5-2と比較例5-4の結果から、耐レトルト性の発現にはポリアミド(E)がガスバリア樹脂組成物に含まれる必要があること、比較例5-2と比較例5-3の結果から、金属原子(F)によりロングラン性が高まることが示唆された。
[EVOHの合成]
<EVOH(A6-1)~EVOH(A6-9)、EVOH(B6-1)~EVOH(B6-4)の各ペレットの作製>
原料(原料モノマー)のエチレン及び酢酸ビニルの種類並びにリン酸化合物及びホウ素化合物の含有量を表16に記載の通りとした以外は、EVOH(A5-1)ペレットと同様の方法で、EVOH(A6-1)ペレット~EVOH(A6-9)ペレット及びEVOH(B6-1)~EVOH(B6-4)ペレットを作製した。化石燃料由来のエチレンには、エア・リキード工業ガス株式会社製のエチレンを用いた。
<EVOH(A6-1)~EVOH(A6-9)、EVOH(B6-1)~EVOH(B6-4)の各ペレットの作製>
原料(原料モノマー)のエチレン及び酢酸ビニルの種類並びにリン酸化合物及びホウ素化合物の含有量を表16に記載の通りとした以外は、EVOH(A5-1)ペレットと同様の方法で、EVOH(A6-1)ペレット~EVOH(A6-9)ペレット及びEVOH(B6-1)~EVOH(B6-4)ペレットを作製した。化石燃料由来のエチレンには、エア・リキード工業ガス株式会社製のエチレンを用いた。
EVOH(A6-1)ペレット~EVOH(A6-9)ペレット及びEVOH(B6-1)~EVOH(B6-4)ペレットのそれぞれについて、上記評価方法(1)、(2)~(5)に記載の方法に従い、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、並びにバイオベース度の測定を行った。結果を表16に示す。
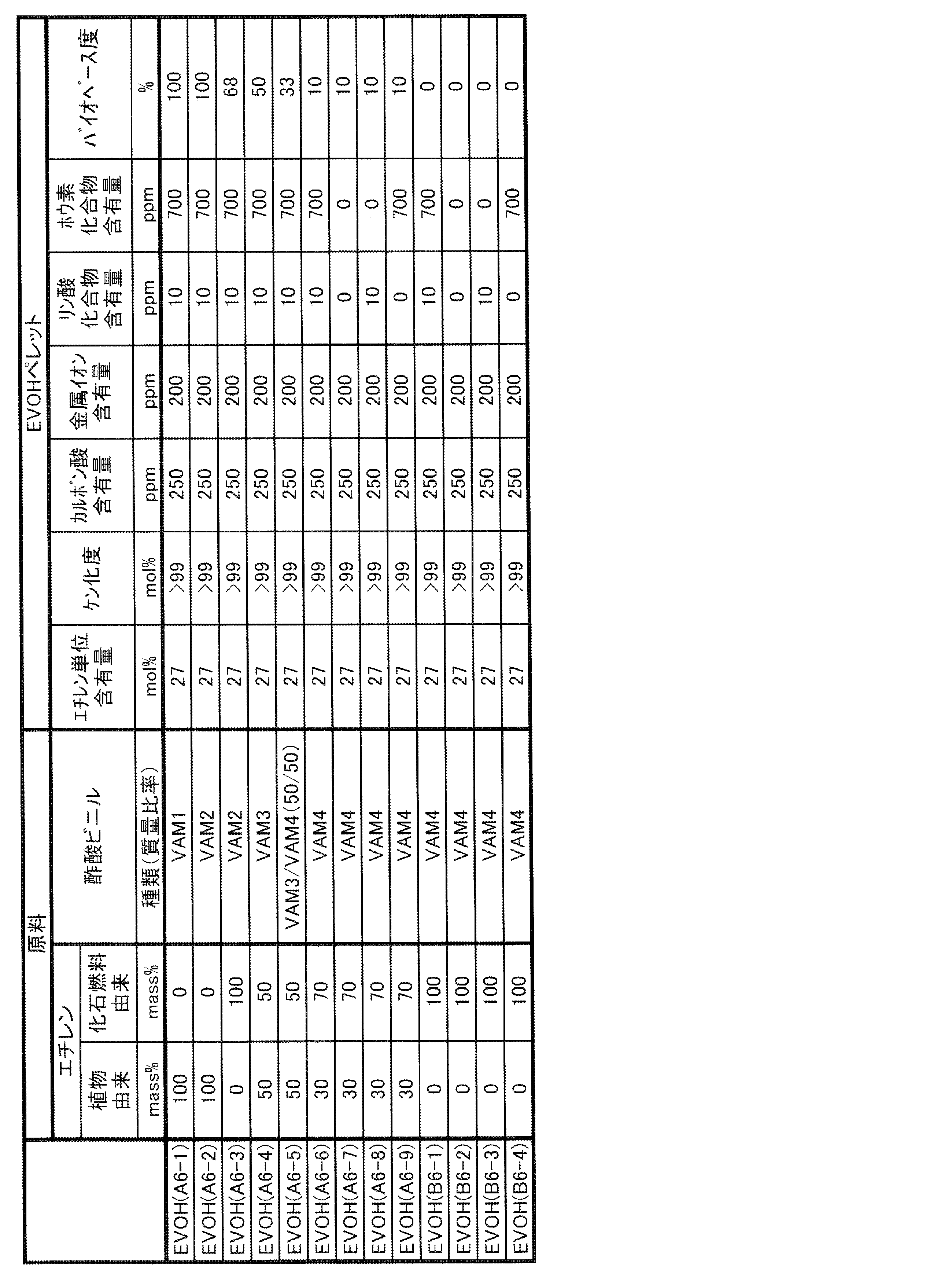
[熱可塑性エラストマー(G)]
実施例及び比較例で用いた変性熱可塑性エラストマー(G)は以下の通りである。
[無変性熱可塑性エラストマー(g1)]
G-3:タフテック(登録商標)H1041(旭化成株式会社製、スチレン系エラストマー樹脂)
G-7:タフマー(登録商標)P0280(三井化学株式会社製、エチレン-プロピレン共重合体)
[変性熱可塑性エラストマー(g2)]
G-1:モディック(登録商標)GQ131(三菱ケミカル株式会社製、不飽和カルボン酸変性ポリエステル系エラストマー樹脂)
G-2:モディック(登録商標)GQ430(三菱ケミカル株式会社製、不飽和カルボン酸変性ポリエステル系エラストマー樹脂)
G-4:タフテック(登録商標)M1911(旭化成株式会社製、不飽和カルボン酸変性スチレン系エラストマー樹脂)
G-5:タフマー(登録商標)MH7020(三井化学株式会社製、無水マレイン酸変性エチレン-ブテン共重合体)
G-6:タフマー(登録商標)MP0610(三井化学株式会社製、無水マレイン酸変性エチレン-プロピレン共重合体)
[ハロゲン原子を含有するスチレン系熱可塑性エラストマー(g3)]
G-8:シブスター(登録商標)062T-FD(株式会社カネカ社製、SIBS;スチレン-イソブチレン-スチレントリブロックコポリマー)
実施例及び比較例で用いた変性熱可塑性エラストマー(G)は以下の通りである。
[無変性熱可塑性エラストマー(g1)]
G-3:タフテック(登録商標)H1041(旭化成株式会社製、スチレン系エラストマー樹脂)
G-7:タフマー(登録商標)P0280(三井化学株式会社製、エチレン-プロピレン共重合体)
[変性熱可塑性エラストマー(g2)]
G-1:モディック(登録商標)GQ131(三菱ケミカル株式会社製、不飽和カルボン酸変性ポリエステル系エラストマー樹脂)
G-2:モディック(登録商標)GQ430(三菱ケミカル株式会社製、不飽和カルボン酸変性ポリエステル系エラストマー樹脂)
G-4:タフテック(登録商標)M1911(旭化成株式会社製、不飽和カルボン酸変性スチレン系エラストマー樹脂)
G-5:タフマー(登録商標)MH7020(三井化学株式会社製、無水マレイン酸変性エチレン-ブテン共重合体)
G-6:タフマー(登録商標)MP0610(三井化学株式会社製、無水マレイン酸変性エチレン-プロピレン共重合体)
[ハロゲン原子を含有するスチレン系熱可塑性エラストマー(g3)]
G-8:シブスター(登録商標)062T-FD(株式会社カネカ社製、SIBS;スチレン-イソブチレン-スチレントリブロックコポリマー)
<実施例6-1>
EVOH(A6-1)ペレット9質量部、EVOH(B6-1)ペレット81質量部及び熱可塑性エラストマー(G-5)のペレット10質量部をドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度220℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例6-1のガスバリア樹脂組成物ペレットを得た。
EVOH(A6-1)ペレット9質量部、EVOH(B6-1)ペレット81質量部及び熱可塑性エラストマー(G-5)のペレット10質量部をドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度220℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例6-1のガスバリア樹脂組成物ペレットを得た。
得られた実施例1のガスバリア樹脂組成物ペレットについて、上記評価方法(1)、(3)~(5)、(7)、(16)、(17)に記載の方法に従って、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、バイオベース度、ロングラン性、酸素透過度並びに耐屈曲性の測定又は評価を行った。結果を表18に示す。
<実施例6-2~6-27、比較例6-1~6-5>
用いたEVOHの種類及び含有量、熱可塑性エラストマー(G)の種類及び含有量、並びに添加剤の種類、銘柄及び含有量を、表17に記載の通り変更した以外は、実施例6-1と同様の方法で実施例6-2~6-27及び比較例6-1~6-5の各ガスバリア樹脂組成物ペレットを作製し、評価した。結果を表18に示す。なお、実施例6-14では、添加剤として酸化防止剤であるBASFジャパン株式会社製のIrganox1098を用いた。また、実施例6-19では、添加剤としてハロゲン捕捉剤である協和化学工業株式会社製のZHT-4Aを用いた。各種ペレットをドライブレンドする際に、上記添加剤もドライブレンドして、ガスバリア樹脂組成物ペレットを得た。
用いたEVOHの種類及び含有量、熱可塑性エラストマー(G)の種類及び含有量、並びに添加剤の種類、銘柄及び含有量を、表17に記載の通り変更した以外は、実施例6-1と同様の方法で実施例6-2~6-27及び比較例6-1~6-5の各ガスバリア樹脂組成物ペレットを作製し、評価した。結果を表18に示す。なお、実施例6-14では、添加剤として酸化防止剤であるBASFジャパン株式会社製のIrganox1098を用いた。また、実施例6-19では、添加剤としてハロゲン捕捉剤である協和化学工業株式会社製のZHT-4Aを用いた。各種ペレットをドライブレンドする際に、上記添加剤もドライブレンドして、ガスバリア樹脂組成物ペレットを得た。
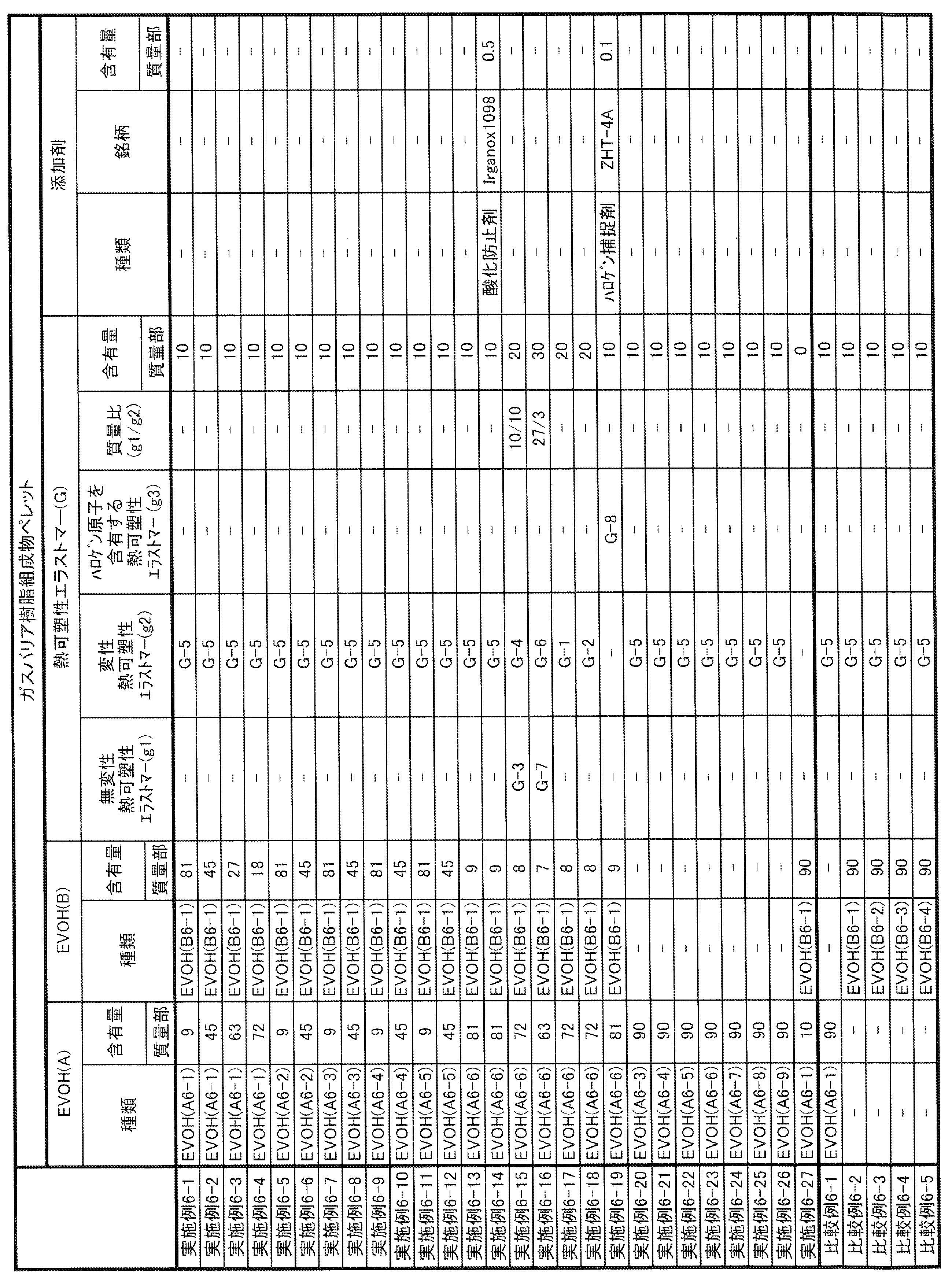
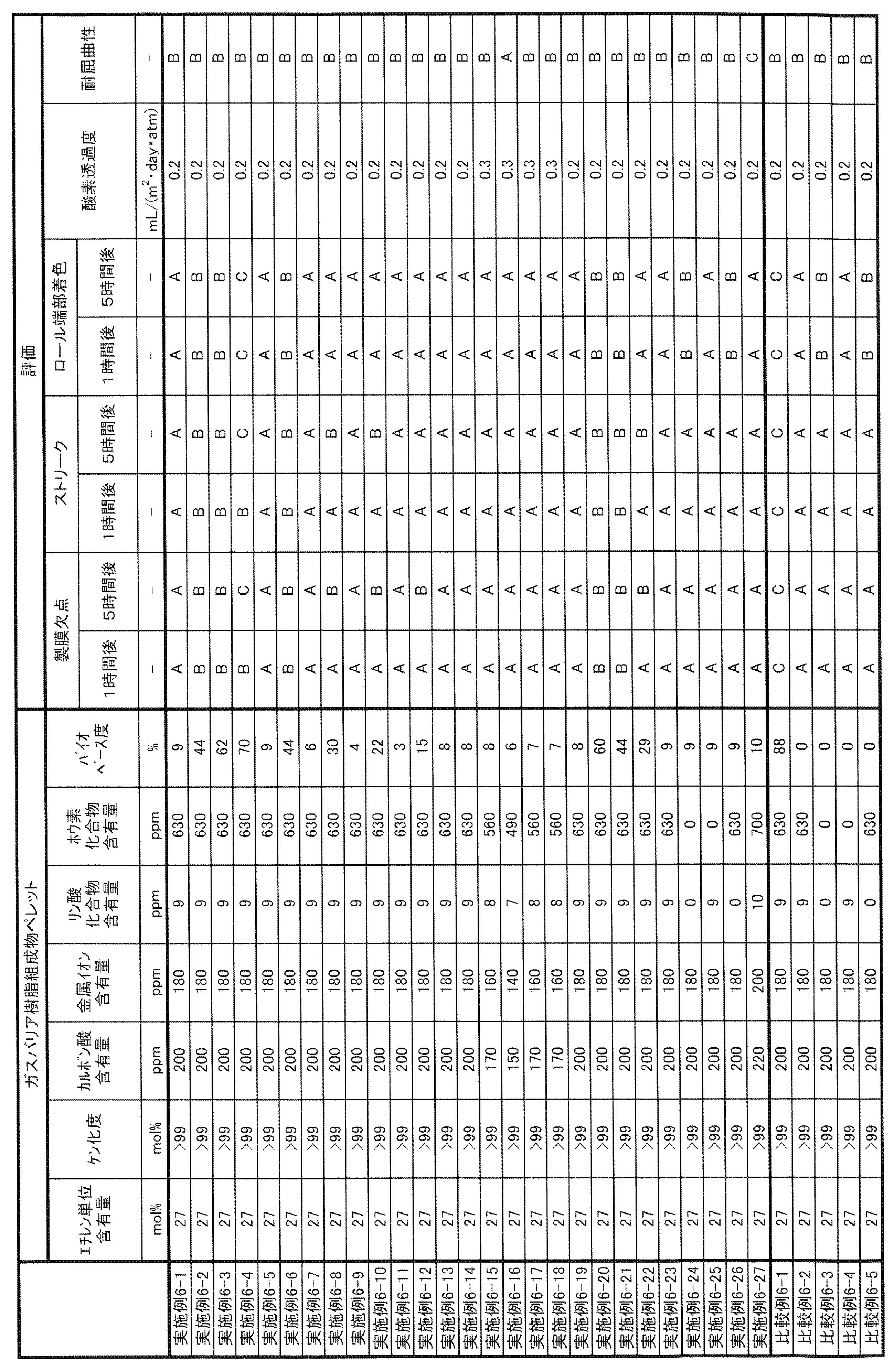
[実施例6-28]
実施例6-13で得られたガスバリア樹脂組成物ペレットを用い、上記評価方法(11)に記載の方法で、耐酸化劣化性を評価した。耐酸化劣化時間は65時間であった。
実施例6-13で得られたガスバリア樹脂組成物ペレットを用い、上記評価方法(11)に記載の方法で、耐酸化劣化性を評価した。耐酸化劣化時間は65時間であった。
[実施例6-29]
実施例6-14で得られたガスバリア樹脂組成物ペレットを用い、上記評価方法(11)に記載の方法で、耐酸化劣化性を評価した。耐酸化劣化時間は280時間であった。
また、実施例6-14で得られたガスバリア樹脂組成物ペレットを用い、以下の要領で多層パイプを作製した。高密度ポリエチレン(日本ポリエチレン株式会社製「ノバテック(登録商標)HD HE421」、密度0.956g/cc、MFRが0.14g/10分)を1台目の押出機に、実施例6-14で得られた各ガスバリア樹脂組成物ペレットを2台目の押出機に、接着性樹脂として三井化学株式会社製「アドマー(登録商標)NF408E」を3台目の押出機に入れ、3種3層の円形ダイを用いて、外径20mmの多層パイプを押出成形し、直後に40℃に調整した冷却水槽を通して冷却して固化させた。多層パイプは問題なく成形でき、その層構成はガスバリア樹脂組成物層が最外層であり、ガスバリア樹脂組成物層/接着性樹脂層/高密度ポリエチレン層=100μm/100μm/2000μmであった。これらの結果から、実施例6-14に記載のガスバリア樹脂組成物は、長期間使用が想定されるパイプ等の用途に好適に使用できることがわかる。
実施例6-14で得られたガスバリア樹脂組成物ペレットを用い、上記評価方法(11)に記載の方法で、耐酸化劣化性を評価した。耐酸化劣化時間は280時間であった。
また、実施例6-14で得られたガスバリア樹脂組成物ペレットを用い、以下の要領で多層パイプを作製した。高密度ポリエチレン(日本ポリエチレン株式会社製「ノバテック(登録商標)HD HE421」、密度0.956g/cc、MFRが0.14g/10分)を1台目の押出機に、実施例6-14で得られた各ガスバリア樹脂組成物ペレットを2台目の押出機に、接着性樹脂として三井化学株式会社製「アドマー(登録商標)NF408E」を3台目の押出機に入れ、3種3層の円形ダイを用いて、外径20mmの多層パイプを押出成形し、直後に40℃に調整した冷却水槽を通して冷却して固化させた。多層パイプは問題なく成形でき、その層構成はガスバリア樹脂組成物層が最外層であり、ガスバリア樹脂組成物層/接着性樹脂層/高密度ポリエチレン層=100μm/100μm/2000μmであった。これらの結果から、実施例6-14に記載のガスバリア樹脂組成物は、長期間使用が想定されるパイプ等の用途に好適に使用できることがわかる。
[実施例6-30]
実施例6-1で得られたガスバリア樹脂組成物ペレットを用い、以下の条件で製膜し、トリムをカットし500mm幅、総厚み25.5μmのサイレージフィルムを作製した。
・装置:7種7層ブローンフィルム製膜機(ブランプトン社製)
・層構成及び各層の厚み:
外層1/外層2/接着性樹脂層1/ガスバリア樹脂組成物層/接着性樹脂層2/外層3/外層4
・外層1、4:直鎖状低密度ポリエチレン(ダウケミカル社製、TUFLIN HS-7028 NT7(MFR1.0g/10分))97質量%、ポリイソブテン(Soltex社製、PB32)3質量%の溶融混練物 各6μm
・外層2、3:直鎖状低密度ポリエチレン(ダウケミカル社製TUFLIN HS-7028 NT7(MFR1.0g/10分))90質量%、ポリイソブテン(Soltex社製、PB32)10質量%の溶融混練物 各4μm
・接着性樹脂層1、2:無水マレイン酸変性直鎖状低密度ポリエチレン(三井化学株式会社製、アドマーNF498) 各2.0μm
・ガスバリア樹脂組成物層:1.5μm
[製膜条件]
押出機
・押出機1:45mmφ単軸押出機(L/D=24)・・・外層1
・押出機2:30mmφ単軸押出機(L/D=24)・・・外層2
・押出機3:30mmφ単軸押出機(L/D=24)・・・外層3
・押出機4:45mmφ単軸押出機(L/D=24)・・・外層4
・押出機5:30mmφ単軸押出機(L/D=24)・・・接着性樹脂層1
・押出機6:30mmφ単軸押出機(L/D=24)・・・接着性樹脂層2
・押出機7:30mmφ単軸押出機(L/D=20)・・・ガスバリア樹脂組成物層
設定温度及び回転数:
・押出機1、4:C1/C2/C3/A=180℃/190℃/205℃/205℃、27rpm
・押出機2、3:C1/C2/C3/A=180℃/190℃/205℃/205℃、69rpm
・押出機5、6:C1/C2/C3/A=190℃/225℃/215℃/220℃、26rpm
・押出機7:C1/C2/C3/A=180℃/210℃/215℃/220℃、19rpm
・ダイ:150mm、設定温度220℃
・延伸比率(Blow up rate):3.09
実施例6-1で得られたガスバリア樹脂組成物ペレットを用い、以下の条件で製膜し、トリムをカットし500mm幅、総厚み25.5μmのサイレージフィルムを作製した。
・装置:7種7層ブローンフィルム製膜機(ブランプトン社製)
・層構成及び各層の厚み:
外層1/外層2/接着性樹脂層1/ガスバリア樹脂組成物層/接着性樹脂層2/外層3/外層4
・外層1、4:直鎖状低密度ポリエチレン(ダウケミカル社製、TUFLIN HS-7028 NT7(MFR1.0g/10分))97質量%、ポリイソブテン(Soltex社製、PB32)3質量%の溶融混練物 各6μm
・外層2、3:直鎖状低密度ポリエチレン(ダウケミカル社製TUFLIN HS-7028 NT7(MFR1.0g/10分))90質量%、ポリイソブテン(Soltex社製、PB32)10質量%の溶融混練物 各4μm
・接着性樹脂層1、2:無水マレイン酸変性直鎖状低密度ポリエチレン(三井化学株式会社製、アドマーNF498) 各2.0μm
・ガスバリア樹脂組成物層:1.5μm
[製膜条件]
押出機
・押出機1:45mmφ単軸押出機(L/D=24)・・・外層1
・押出機2:30mmφ単軸押出機(L/D=24)・・・外層2
・押出機3:30mmφ単軸押出機(L/D=24)・・・外層3
・押出機4:45mmφ単軸押出機(L/D=24)・・・外層4
・押出機5:30mmφ単軸押出機(L/D=24)・・・接着性樹脂層1
・押出機6:30mmφ単軸押出機(L/D=24)・・・接着性樹脂層2
・押出機7:30mmφ単軸押出機(L/D=20)・・・ガスバリア樹脂組成物層
設定温度及び回転数:
・押出機1、4:C1/C2/C3/A=180℃/190℃/205℃/205℃、27rpm
・押出機2、3:C1/C2/C3/A=180℃/190℃/205℃/205℃、69rpm
・押出機5、6:C1/C2/C3/A=190℃/225℃/215℃/220℃、26rpm
・押出機7:C1/C2/C3/A=180℃/210℃/215℃/220℃、19rpm
・ダイ:150mm、設定温度220℃
・延伸比率(Blow up rate):3.09
<ラッピング試験>
得られたサイレージフィルムとリモコンラップマシーンWM1600R(株式会社タカキタ社製)とを用いて、φ120cm×120cmの大きさに成型した牧草のラッピングを行った。得られたサイレージフィルムは牧草を良好にラッピングでき、サイレージフィルムとして適していることを確認した。
得られたサイレージフィルムとリモコンラップマシーンWM1600R(株式会社タカキタ社製)とを用いて、φ120cm×120cmの大きさに成型した牧草のラッピングを行った。得られたサイレージフィルムは牧草を良好にラッピングでき、サイレージフィルムとして適していることを確認した。
[実施例6-31]
実施例6-1で得られたガスバリア樹脂組成物ペレットを用い、押出機1、2にて外層Aを構成する樹脂を、押出機3、4にて外層Dを構成する樹脂を、押出機5、6に接着性樹脂層B、Cを構成する樹脂を、押出機7にガスバリア樹脂組成物層を構成する樹脂をそれぞれ押出し、ダイ:75mm、延伸比率:1.5とした以外は、実施例29のサイレージフィルムの製造条件と同様にして、トリムをカットし900mm幅、総厚み230μmの穀物保存バッグ用フィルムを作製した。得られたフィルムから、400mm×700mmのバッグを作り、穀物保存バッグとした。
・層構成及び各層の厚み:
外層A/接着性樹脂層B/ガスバリア樹脂組成物層/接着性樹脂層C/外層D
(使用樹脂)
・外層A、D:LLDPE;SCLAIR FP120-A(NOVA Chemicals社製) 押出機1、3:各50μm、押出機2、4:各47μm 合計:各97μm
・接着性樹脂層B、C:アドマー(登録商標)NF498A(三井化学株式会社製) 各12μm
・ガスバリア樹脂組成物層:12μm
実施例6-1で得られたガスバリア樹脂組成物ペレットを用い、押出機1、2にて外層Aを構成する樹脂を、押出機3、4にて外層Dを構成する樹脂を、押出機5、6に接着性樹脂層B、Cを構成する樹脂を、押出機7にガスバリア樹脂組成物層を構成する樹脂をそれぞれ押出し、ダイ:75mm、延伸比率:1.5とした以外は、実施例29のサイレージフィルムの製造条件と同様にして、トリムをカットし900mm幅、総厚み230μmの穀物保存バッグ用フィルムを作製した。得られたフィルムから、400mm×700mmのバッグを作り、穀物保存バッグとした。
・層構成及び各層の厚み:
外層A/接着性樹脂層B/ガスバリア樹脂組成物層/接着性樹脂層C/外層D
(使用樹脂)
・外層A、D:LLDPE;SCLAIR FP120-A(NOVA Chemicals社製) 押出機1、3:各50μm、押出機2、4:各47μm 合計:各97μm
・接着性樹脂層B、C:アドマー(登録商標)NF498A(三井化学株式会社製) 各12μm
・ガスバリア樹脂組成物層:12μm
<発芽力試験>
得られた穀物保存バッグに、50kgのコーンを詰めて屋外に180日間保存した。保存後、コーンの発芽力を評価した。保存後のコーンの発芽力は保存前のコーンと同程度であった。
得られた穀物保存バッグに、50kgのコーンを詰めて屋外に180日間保存した。保存後、コーンの発芽力を評価した。保存後のコーンの発芽力は保存前のコーンと同程度であった。
[実施例6-32]
[積層剥離容器の作製]
実施例6-17で得られたガスバリア樹脂組成物ペレットを用い、本体部及び口頭部を有する積層剥離容器((内面側)内面層(LLDPE)/接着層(変性ポリオレフィンとLLDPEのブレンド)/最外層(ガスバリア樹脂組成物層)/外層(LLDPE)(外面側))を以下に示す条件でブロー成形により作製した。
(容器形状)
本体部:φ47mm、高さ110mm
口頭部:φ30mm、高さ16mm
(層構成)
外層:無変性ポリプロピレン(ノーブレン(登録商標)FSX16E9、住友化学株式会社製)
内層:外層側から順に最外層/接着層/内面層の三層構成
最外層:実施例17で得たガスバリア樹脂組成物
接着層:変性ポリオレフィン(モディック(登録商標)L522、三菱ケミカル株式会社製)とLLDPE(ハーモレックス(登録商標)F325N、日本ポリエチレン株式会社製)の1:1ブレンド
内面層:LLDPE(ハーモレックス(登録商標)F325N、日本ポリエチレン株式会社製)
[積層剥離容器の作製]
実施例6-17で得られたガスバリア樹脂組成物ペレットを用い、本体部及び口頭部を有する積層剥離容器((内面側)内面層(LLDPE)/接着層(変性ポリオレフィンとLLDPEのブレンド)/最外層(ガスバリア樹脂組成物層)/外層(LLDPE)(外面側))を以下に示す条件でブロー成形により作製した。
(容器形状)
本体部:φ47mm、高さ110mm
口頭部:φ30mm、高さ16mm
(層構成)
外層:無変性ポリプロピレン(ノーブレン(登録商標)FSX16E9、住友化学株式会社製)
内層:外層側から順に最外層/接着層/内面層の三層構成
最外層:実施例17で得たガスバリア樹脂組成物
接着層:変性ポリオレフィン(モディック(登録商標)L522、三菱ケミカル株式会社製)とLLDPE(ハーモレックス(登録商標)F325N、日本ポリエチレン株式会社製)の1:1ブレンド
内面層:LLDPE(ハーモレックス(登録商標)F325N、日本ポリエチレン株式会社製)
(ブロー成形条件)
上記層構成になるよう各溶融した樹脂を共押出することにより、溶融状態の積層パリソンを作製した。積層パリソン作製時にリップ幅を調整して口頭部の厚みが厚くなるように調整を行った。かかる積層パリソンをブロー成形金型にセットし、ブロー成形法によって所望の容器形状に成形した。ブロー成形の際、口頭部の厚みが本体部の厚みより十分厚くなるように調整を行った。共押出の条件は、口頭部を除く外層と内層の厚さがどちらも70~130μmの範囲内であり且つ外層/内層の厚さの比が0.8~1.3となるように調節した。ブロー成形の条件は、ブロー圧:0.4MPa、金型温度:25℃、ブロー時間:15秒とした。
上記層構成になるよう各溶融した樹脂を共押出することにより、溶融状態の積層パリソンを作製した。積層パリソン作製時にリップ幅を調整して口頭部の厚みが厚くなるように調整を行った。かかる積層パリソンをブロー成形金型にセットし、ブロー成形法によって所望の容器形状に成形した。ブロー成形の際、口頭部の厚みが本体部の厚みより十分厚くなるように調整を行った。共押出の条件は、口頭部を除く外層と内層の厚さがどちらも70~130μmの範囲内であり且つ外層/内層の厚さの比が0.8~1.3となるように調節した。ブロー成形の条件は、ブロー圧:0.4MPa、金型温度:25℃、ブロー時間:15秒とした。
[口頭部の厚み測定]
得られた積層剥離容器の口頭部をミクロトームで切り出すことで切片を作製し、かかる切片をスライドガラスにのせて、光学顕微鏡にて口頭部の厚みを測定した。口頭部の厚みは0.5mmであった。
得られた積層剥離容器の口頭部をミクロトームで切り出すことで切片を作製し、かかる切片をスライドガラスにのせて、光学顕微鏡にて口頭部の厚みを測定した。口頭部の厚みは0.5mmであった。
[口頭部の耐剥離性]
得られた積層剥離容器の本体部の外層に空気導入孔を形成し、この空気導入孔から外層と内層との間に空気を注入することによって予備剥離を行った。空気は、圧力0.3MPaで1.0秒注入した。予備剥離を行った後、空気導入孔を閉鎖した状態で、容器を30kgの力で潰して、外層と内層との間の空気に圧力をかけたときに、口頭部における外層と内層との界面から空気が漏れるかどうか確認した。結果、口頭部海面からの空気漏れが確認されなかった。なお、得られた積層剥離容器は、ガスバリア樹脂組成物層とLLDPE層とが直接積層されているため、ガスバリア樹脂組成物層とLLDPE層との界面において、本体部では、適度な剥離性を有する容器となった。
得られた積層剥離容器の本体部の外層に空気導入孔を形成し、この空気導入孔から外層と内層との間に空気を注入することによって予備剥離を行った。空気は、圧力0.3MPaで1.0秒注入した。予備剥離を行った後、空気導入孔を閉鎖した状態で、容器を30kgの力で潰して、外層と内層との間の空気に圧力をかけたときに、口頭部における外層と内層との界面から空気が漏れるかどうか確認した。結果、口頭部海面からの空気漏れが確認されなかった。なお、得られた積層剥離容器は、ガスバリア樹脂組成物層とLLDPE層とが直接積層されているため、ガスバリア樹脂組成物層とLLDPE層との界面において、本体部では、適度な剥離性を有する容器となった。
[実施例6-33]
リップ幅を調整せずに積層パリソンを作製し、ブロー成形時にも口頭部と本体部の厚みの調整を行わずに成形した以外は実施例6-31と同様の方法で積層剥離容器を作製し、評価を行った。口頭部の厚みは0.3mmであり、口頭部の剥離性試験では空気の漏れが確認された。
リップ幅を調整せずに積層パリソンを作製し、ブロー成形時にも口頭部と本体部の厚みの調整を行わずに成形した以外は実施例6-31と同様の方法で積層剥離容器を作製し、評価を行った。口頭部の厚みは0.3mmであり、口頭部の剥離性試験では空気の漏れが確認された。
実施例6-32のように口頭部に厚みを持たせた容器では、口頭部の剥離が容易に抑制できたが、実施例6-33のように口頭部の厚みが不十分な容器では、口頭部の剥離の抑制が不十分であった。
表17、18に示されるように、実施例6-1~6-27の各ガスバリア樹脂組成物は、バイオマス由来の原料を一部に用いていながら、化石燃料由来のみのもの(比較例6-2のガスバリア樹脂組成物等)と遜色のない高いガスバリア性及び耐屈曲性を有していた。また、実施例6-1~6-27の各ガスバリア樹脂組成物は、運転開始1時間後に作製されたフィルムについての製膜欠点及びストリークの評価がA又はBであり、十分なロングラン性を有するものであった。
また、実施例及び比較例の結果から、EVOHを用いたガスバリア樹脂組成物において、ガスバリア性及び耐屈曲性はバイオベース度に依存しないのに対し、ロングラン性はバイオベース度が低くなるにつれて高まる傾向にあるという、特異的な性質があることが確認できた。例えば、バイオベース度が65%以下の実施例6-1~6-3、6-5~6-27の各ガスバリア樹脂組成物は、運転開始1時間後及び5時間後に作製されたフィルムについての製膜欠点、ストリーク及びロール端部着色の評価がA又はBであり、高いロングラン性を有するものとなった。
さらに、実施例6-28及び実施例6-29により耐酸化劣化性、実施例6-30によりサイレージフィルム、実施例6-31により穀物保存バッグ、実施例6-32及び実施例6-33により積層剥離容器の評価を行ったが、いずれの結果についても優れた実用特性が示唆された。
<実施例7-1>
EVOH(A1-1)ペレット及びEVOH(B1-1)ペレットを質量比(A1-1/B1-1)10/90でドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度220℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例7-1のガスバリア樹脂組成物ペレットを得た。
EVOH(A1-1)ペレット及びEVOH(B1-1)ペレットを質量比(A1-1/B1-1)10/90でドライブレンドした後、二軸押出機(株式会社東洋精機製作所の「2D25W」、25mmφ,ダイ温度220℃,スクリュー回転数100rpm)を用い、窒素雰囲気下で押出しペレット化を行い、実施例7-1のガスバリア樹脂組成物ペレットを得た。
得られた実施例7-1のガスバリア樹脂組成物ペレットについて、上記評価方法(1)、(3)~(5)、(18)~(22)に記載の方法に従って、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、バイオベース度、多層フィルム評価、熱収縮フィルムの収縮性評価、多層熱成形容器評価、ブロー成形容器のストリーク評価並びに燃料透過度の測定又は評価を行った。結果を表19及び表20に示す。
<実施例7-2~7-22、比較例7-1~7-6>
用いたEVOHの種類及び質量比(割合)を、表19に記載の通り変更した以外は、実施例7-1と同様の方法で実施例7-2~7-22及び比較例7-1~7-6の各ガスバリア樹脂組成物ペレットを作製し、評価した。なお、実施例7-21、7-22の各ガスバリア樹脂組成物ペレットについては、多層熱成形容器評価、ブロー成形容器のストリーク評価及び燃料透過度の測定又は評価は行わなかった。結果を表20に示す。
用いたEVOHの種類及び質量比(割合)を、表19に記載の通り変更した以外は、実施例7-1と同様の方法で実施例7-2~7-22及び比較例7-1~7-6の各ガスバリア樹脂組成物ペレットを作製し、評価した。なお、実施例7-21、7-22の各ガスバリア樹脂組成物ペレットについては、多層熱成形容器評価、ブロー成形容器のストリーク評価及び燃料透過度の測定又は評価は行わなかった。結果を表20に示す。
<実施例7-23、比較例7-7および7-8>
(共押出コート紙評価)
基材としてカートン紙(厚み500μm、坪量400g/m2)を用いて、基材上に3種5層の共押出コートを行った。共押出の構成は低密度ポリエチレン/接着層/ガスバリア樹脂組成物層/接着層/低密度ポリエチレン/カートン紙であり、厚み構成は20/5/5/5/20/500μmである。低密度ポリエチレン用押出機、EVOH用押出機、接着層用押出機とそれぞれの押出機から供給される樹脂を合流、分配するフィードブロックとT型ダイスを使用した。低密度ポリエチレンとしては線状低密度ポリエチレン(株式会社プライムポリマー製「ウルトゼックス(商標)2022L」)を、また、接着層としては無水マレイン酸で変性されたポリプロピレン(三井化学株式会社製「アドマー(商標)QF-500」)を使用した。このときのフィードブロック、T型ダイスの温度条件を250℃、引取速度は300m/minとした。運転開始3時間後に作製された共押出コート紙について、目視にて共押出コート面側のストリークの有無を下記評価基準により評価した。
(ストリークの評価基準)
A(良好):ストリークは認められなかった
B(やや良好):ストリークが確認された
C(不良):多数のストリークが確認された
(共押出コート紙評価)
基材としてカートン紙(厚み500μm、坪量400g/m2)を用いて、基材上に3種5層の共押出コートを行った。共押出の構成は低密度ポリエチレン/接着層/ガスバリア樹脂組成物層/接着層/低密度ポリエチレン/カートン紙であり、厚み構成は20/5/5/5/20/500μmである。低密度ポリエチレン用押出機、EVOH用押出機、接着層用押出機とそれぞれの押出機から供給される樹脂を合流、分配するフィードブロックとT型ダイスを使用した。低密度ポリエチレンとしては線状低密度ポリエチレン(株式会社プライムポリマー製「ウルトゼックス(商標)2022L」)を、また、接着層としては無水マレイン酸で変性されたポリプロピレン(三井化学株式会社製「アドマー(商標)QF-500」)を使用した。このときのフィードブロック、T型ダイスの温度条件を250℃、引取速度は300m/minとした。運転開始3時間後に作製された共押出コート紙について、目視にて共押出コート面側のストリークの有無を下記評価基準により評価した。
(ストリークの評価基準)
A(良好):ストリークは認められなかった
B(やや良好):ストリークが確認された
C(不良):多数のストリークが確認された
ガスバリア樹脂組成物として、それぞれ実施例7-19、比較例7-2および比較例7-4の各ガスバリア樹脂組成物ペレットを用いて共押出コート紙評価を行った結果をそれぞれ実施例7-23、比較例7-7および7-8とする。実施例7-23および比較例7-8のストリーク評価はAであったが、比較例7-7のストリーク評価はCであった。

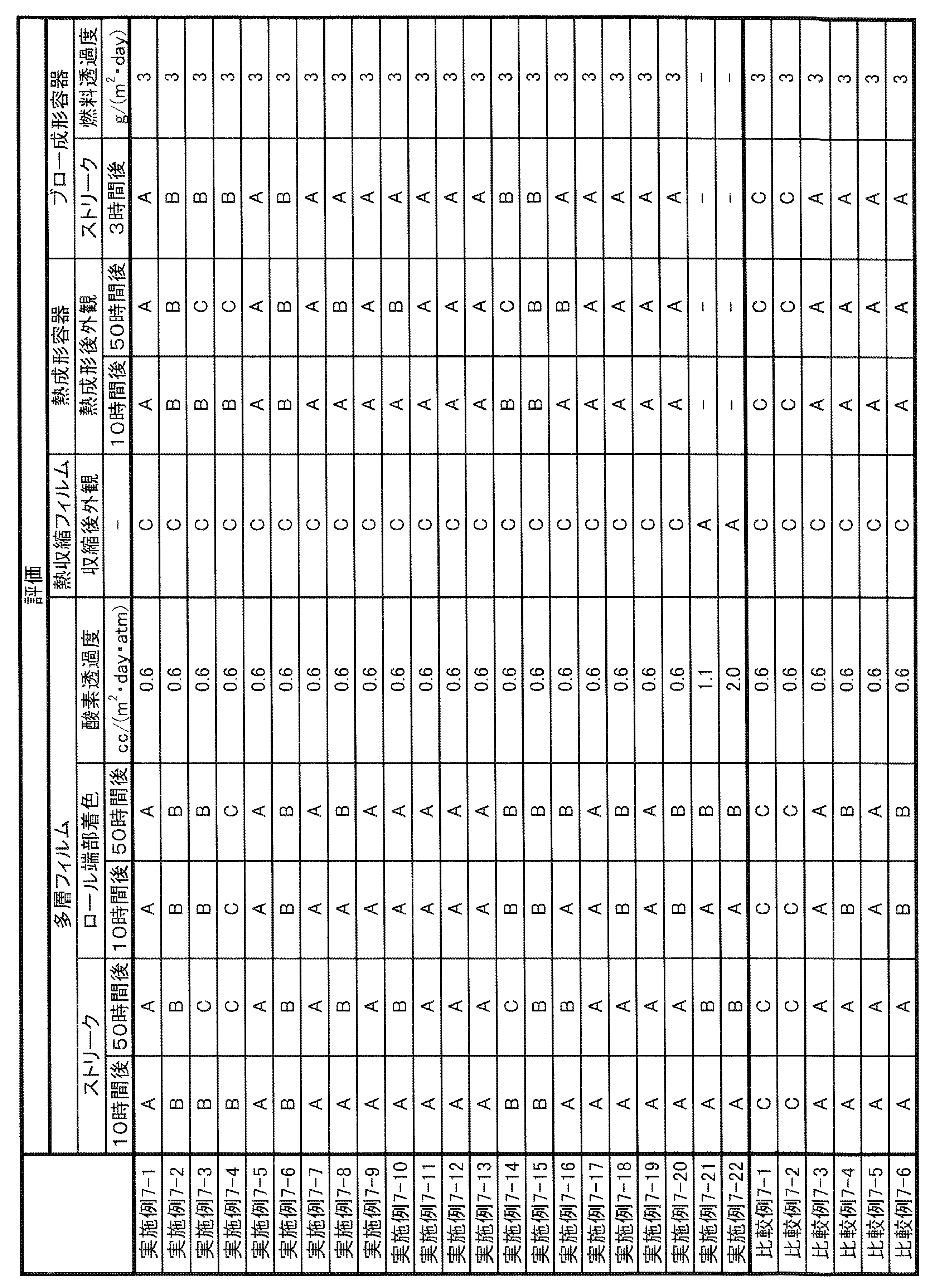
表19、20に示されるように、実施例7-1~7-22の各ガスバリア樹脂組成物を用いた多層フィルムは、バイオマス由来の原料を一部に用いていながら、化石燃料由来のみのもの(比較例7-3のガスバリア樹脂組成物を用いた多層フィルム)と遜色のない高いガスバリア性を有していた。また、実施例7-1~7-22の各ガスバリア樹脂組成物は、運転開始10時間後に作製された多層フィルム及び熱成形容器についてのストリークの評価がA又はBであり、また、運転開始3時間後に作成されたブロー成形容器のストリークの評価がA又はBであり、十分なロングラン性を有するものであった。変性基を有するEVOHを用いた熱収縮フィルムを作成した実施例7-21及び7-22では、良好な収縮性が確認できた。
また、実施例及び比較例の結果から、EVOHを用いたガスバリア樹脂組成物において、ガスバリア性はバイオベース度に依存しないのに対し、ロングラン性はバイオベース度が低くなるにつれて高まる傾向にあるという、特異的な性質があることが確認できた。例えば、バイオベース度が65%以下の実施例7-1、7-2、7-5~7-13、7-15~7-22の各ガスバリア樹脂組成物は、運転開始10時間後及び50時間後に作製された多層フィルムについてのストリーク及びロール端部着色の評価がA又はBであり、高いロングラン性を有するものとなった。
<実施例8-1>
EVOH(A1-1)ペレット及びEVOH(B1-1)ペレットを質量比(A1-1/B1-1)10/90でドライブレンドした後、30mmφ二軸押出機(日本製鋼所社製「TEX-30SS-30CRW-2V」)を用いて下記条件にてストランドダイより、ストランド状に溶融押出し、ストランドダイより押し出されたストランド状の溶融樹脂を水槽にて冷却した後、ペレタイザーで切断し、円柱状の平均直径2.8mm、平均長さ3.2mmの実施例8-1の樹脂組成物ペレットを得た。
(二軸押出装置条件)
押出温度:220℃
スクリュー回転数:300rpm
押出樹脂量:25kg/時間
ストランドダイ:3mmφ、6穴
EVOH(A1-1)ペレット及びEVOH(B1-1)ペレットを質量比(A1-1/B1-1)10/90でドライブレンドした後、30mmφ二軸押出機(日本製鋼所社製「TEX-30SS-30CRW-2V」)を用いて下記条件にてストランドダイより、ストランド状に溶融押出し、ストランドダイより押し出されたストランド状の溶融樹脂を水槽にて冷却した後、ペレタイザーで切断し、円柱状の平均直径2.8mm、平均長さ3.2mmの実施例8-1の樹脂組成物ペレットを得た。
(二軸押出装置条件)
押出温度:220℃
スクリュー回転数:300rpm
押出樹脂量:25kg/時間
ストランドダイ:3mmφ、6穴
得られた実施例8-1の樹脂組成物ペレットについて、上記評価方法(1)、(3)~(5)、(23)に記載の方法に従って、エチレン単位含有量及びケン化度、カルボン酸の定量、金属イオン、リン酸化合物及びホウ素化合物の定量、バイオベース度並びにロングラン性の測定又は評価を行った。結果を表21に示す。
底部より1cmに排水口を設けたプランター(上部幅28.5cm×上部縦46.5cm×深さ26cm、容量28L)を用い、棚の載置面よりも大きく切った吸水シート「ジャームガード」(東洋紡スペシャルティズトレーディング株式会社製)を棚に被せ、余りを底部に折り返してプランターに設置した。さらにその上に防根透水シート(東洋紡スペシャルティズトレーディング株式会社製)をプランター内側に敷いた後、実施例8-1の樹脂組成物ペレットを深さ20cmまで充填し、植物栽培用培地を得た。得られた植物栽培用培地について、上記評価方法(24)に記載の方法に従って、培地評価を行った。結果を表21に示す。
<実施例8-2~8-20、比較例8-1~8-5>
用いたEVOHの種類及び質量比(割合)を、表21に記載の通り変更した以外は、実施例8-1と同様の方法で実施例8-2~8-20及び比較例8-1~8-5の各樹脂組成物ペレットを作製し、評価した。結果を表21に示す。
用いたEVOHの種類及び質量比(割合)を、表21に記載の通り変更した以外は、実施例8-1と同様の方法で実施例8-2~8-20及び比較例8-1~8-5の各樹脂組成物ペレットを作製し、評価した。結果を表21に示す。
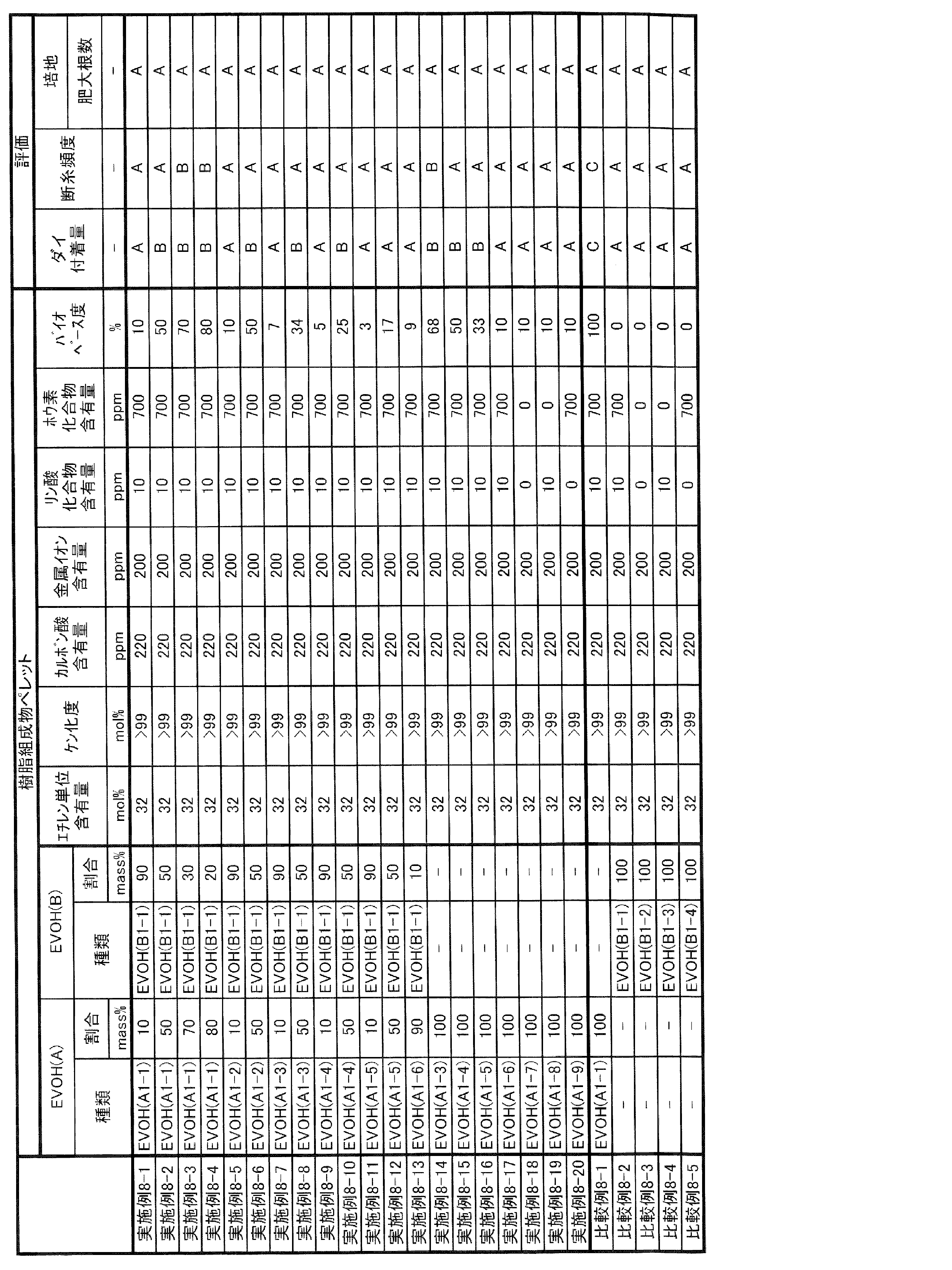
表21に示されるように、実施例8-1~8-20の各樹脂組成物を用いた植物栽培用培地は、バイオマス由来の原料を一部に用いていながら、化石燃料由来のみのもの(比較例8-2)と遜色のない植物生育性能を有していた。また、実施例8-1~8-20の各樹脂組成物は、樹脂組成物ペレットを連続的に製造する際のロングラン性評価(ダイ付着量、断糸頻度)がA又はBであり、十分なロングラン性を有するものであった。
また、実施例及び比較例の結果から、EVOHを用いた樹脂組成物ペレットにおいて、植物の生育性はバイオベース度に依存しないのに対し、ロングラン性はバイオベース度が低くなるにつれて高まる傾向にあるという、特異的な性質があることが確認できた。例えば、バイオベース度が65%以下の実施例8-1、8-2、8-5~8-13、8-15~8-22の各樹脂組成物ペレットは、断糸頻度の評価がAであり、高いロングラン性を有するものとなった。
1 カップ状容器
2 カップ本体
3 フランジ部
4 開口
5 内表面
6 外表面
7 蓋
21 連続多層シート
30 加熱装置
31,32 ヒーター
40 金型装置
50 下型
51 上型
52 凹部
53 プラグ
101 縦製袋充填シール袋
110 フィルム材
111 上端部
112 下端部
115 胴体部
120 背面
121、122 側端部
201 植物栽培装置
202 プランター
203 開口
204 側壁
204a 側壁の上端
205 排水口
206 水
207 底壁
208 棚
208a 棚の載置面
209 吸水シート
209a 吸水シートの中央部
209b 吸水シートの端部
210 防根透水シート
210a 防根透水シートの端部
211 植物
212 植物栽培用培地
213 ペレット(樹脂組成物チップ)
2 カップ本体
3 フランジ部
4 開口
5 内表面
6 外表面
7 蓋
21 連続多層シート
30 加熱装置
31,32 ヒーター
40 金型装置
50 下型
51 上型
52 凹部
53 プラグ
101 縦製袋充填シール袋
110 フィルム材
111 上端部
112 下端部
115 胴体部
120 背面
121、122 側端部
201 植物栽培装置
202 プランター
203 開口
204 側壁
204a 側壁の上端
205 排水口
206 水
207 底壁
208 棚
208a 棚の載置面
209 吸水シート
209a 吸水シートの中央部
209b 吸水シートの端部
210 防根透水シート
210a 防根透水シートの端部
211 植物
212 植物栽培用培地
213 ペレット(樹脂組成物チップ)
Claims (40)
- 一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物を含み、
上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物の原料であるエチレン及びビニルエステルの一部がバイオマス由来であり、残部が化石燃料由来である、ガスバリア樹脂組成物。 - 上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物が、
原料であるエチレン及びビニルエステルの少なくとも一部がバイオマス由来であるエチレン-ビニルエステル共重合体ケン化物(A)と、化石燃料由来であるエチレン-ビニルエステル共重合体ケン化物(B)とを含む、請求項1に記載のガスバリア樹脂組成物。 - 上記エチレン-ビニルエステル共重合体ケン化物(A)と上記エチレン-ビニルエステル共重合体ケン化物(B)との質量比(A/B)が1/99~99/1である、請求項2に記載のガスバリア樹脂組成物。
- 上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物が、
原料であるエチレン及びビニルエステルの一部がバイオマス由来であり、残部が化石燃料由来であるエチレン-ビニルアルコール共重合体(A’)を含む、請求項1に記載のガスバリア樹脂組成物。 - 上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物のバイオベース度が1%以上99%以下である、請求項1~4のいずれか1項に記載のガスバリア樹脂組成物。
- バイオベース度が1%以上99%以下である、請求項1~5のいずれか1項に記載のガスバリア樹脂組成物。
- 硫黄化合物を硫黄原子換算で0ppmを超えて100ppm以下含む、請求項1~6のいずれか1項に記載のガスバリア樹脂組成物。
- 上記硫黄化合物が、ジメチルスルフィドまたはジメチルスルホキシドである、請求項7に記載のガスバリア樹脂組成物。
- 上記原料のうちのエチレンの少なくとも一部がバイオマス由来である、請求項1~8のいずれか1項に記載のガスバリア樹脂組成物。
- 上記原料のうちのビニルエステルの少なくとも一部がバイオマス由来である、請求項1~9のいずれか1項に記載のガスバリア樹脂組成物。
- 上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物が、下記一般式(I)で表される変性基を有する、請求項1~10のいずれか1項に記載のガスバリア樹脂組成物。

- カルボン酸をカルボン酸根換算で30ppm以上1000ppm以下含む、請求項1~11のいずれか1項に記載のガスバリア樹脂組成物。
- 金属イオンを1ppm以上1000ppm以下含む、請求項1~12のいずれか1項に記載のガスバリア樹脂組成物。
- リン酸化合物をリン原子換算で1ppm以上200ppm以下含む、請求項1~13のいずれか1項に記載のガスバリア樹脂組成物。
- ホウ素化合物をホウ素原子換算で5ppm以上5000ppm以下含む、請求項1~14のいずれか1項に記載のガスバリア樹脂組成物。
- 上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物が、エチレン-ビニルエステル共重合体ケン化物(X)、及び上記エチレン-ビニルエステル共重合体ケン化物(X)よりも融点が低いエチレン-ビニルエステル共重合体ケン化物(Y)を含む、請求項1~15のいずれか1項に記載のガスバリア樹脂組成物。
- 無機粒子(C)をさらに含み、無機粒子(C)の含有量が50ppm以上5000ppm以下である、請求項1~16のいずれか1項に記載のガスバリア樹脂組成物。
- 酸化防止剤(D)をさらに含み、酸化防止剤(D)の含有量が0.01質量%以上5質量%以下である、請求項1~17のいずれか1項に記載のガスバリア樹脂組成物。
- ポリアミド(E)、並びにマグネシウム、カルシウム及び亜鉛からなる群より選ばれる少なくとも1種の金属原子(F)をさらに含み、
上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物に対するポリアミド(E)の質量比が5/95以上40/60以下であり、
金属原子(F)の含有量が1ppm以上500ppm以下である、請求項1~18のいずれか1項に記載のガスバリア樹脂組成物。 - 熱可塑性エラストマー(G)をさらに含み、上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物に対する熱可塑性エラストマー(G)の質量比が5/95以上35/65以下である、請求項1~19のいずれか1項に記載のガスバリア樹脂組成物。
- 原料であるエチレン及びビニルエステルの少なくとも一部がバイオマス由来であるエチレン-ビニルエステル共重合体ケン化物(A)のペレットと、化石燃料由来であるエチレン-ビニルエステル共重合体ケン化物(B)のペレットとをドライブレンドし、溶融混練する工程を備える、ガスバリア樹脂組成物の製造方法。
- 上記一種又は二種以上のエチレン-ビニルエステル共重合体ケン化物のペレットと、ポリアミド(E)のペレットと、マグネシウム、カルシウム及び亜鉛からなる群より選ばれる少なくとも1種の金属原子(F)を含む化合物とを混合し、溶融混練する工程を備える、請求項19に記載のガスバリア樹脂組成物の製造方法。
- 原料であるエチレン及びビニルエステルの少なくとも一部がバイオマス由来であるエチレン-ビニルエステル共重合体ケン化物(A)のペレットと、化石燃料由来であるエチレン-ビニルエステル共重合体ケン化物(B)のペレットと、熱可塑性エラストマー(G)のペレットとをドライブレンドし、溶融混練する工程を備える、ガスバリア樹脂組成物の製造方法。
- 請求項1~20のいずれか1項に記載のガスバリア樹脂組成物から形成されるガスバリア層を備える、成形体。
- 請求項24に記載の成形体を備える、フィルムまたはシート。
- 請求項24に記載の成形体を備える、産業用フィルムまたはシート。
- 請求項24に記載の成形体を備える、熱成形容器。
- 請求項24に記載の成形体を備える、ブロー成形容器。
- 請求項28に記載のブロー成形容器を備える、燃料容器。
- 請求項28に記載のブロー成形容器を備える、ボトル容器。
- 請求項24に記載の成形体を備える、チューブ。
- 請求項24に記載の成形体を備える、パイプ。
- 請求項24に記載の成形体を備える、紙容器。
- 請求項1~20のいずれか1項に記載のガスバリア樹脂組成物から形成される、単層フィルム。
- 請求項1~20のいずれか1項に記載のガスバリア樹脂組成物から形成される層を少なくとも1層備える、多層フィルム。
- 請求項34に記載の単層フィルム、または上記ガスバリア樹脂組成物から形成される層の少なくとも1層を最外層として備える請求項35に記載の多層フィルムと、
上記単層フィルムまたは上記多層フィルムにおける上記ガスバリア樹脂組成物から形成される層の表出面に積層される少なくとも1層の無機蒸着層と
を備える、蒸着フィルム。 - 請求項36に記載の蒸着フィルムと、
上記蒸着フィルムにおける上記無機蒸着層上に積層される他の層と
を備える、多層構造体。 - 請求項34に記載の単層フィルム、請求項35に記載の多層フィルム、請求項36に記載の蒸着フィルムまたは請求項37に記載の多層構造体を備える、ヒートシール用フィルム。
- 請求項1~20のいずれか1項に記載のガスバリア樹脂組成物からなる層を少なくとも1層備える、包装材。
- 樹脂組成物から形成されている成形体を含む植物栽培用培地であって、
上記樹脂組成物が、請求項1~20のいずれか1項に記載のガスバリア樹脂組成物である、植物栽培用培地。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US18/012,792 US20230250269A1 (en) | 2020-06-30 | 2021-06-29 | Gas Barrier Resin Composition, Method for Producing Gas Barrier Resin Composition, and Molded Product |
| KR1020237003312A KR20230030654A (ko) | 2020-06-30 | 2021-06-29 | 가스 배리어 수지 조성물, 가스 배리어 수지 조성물의 제조 방법, 및 성형체 |
| EP21834483.6A EP4173823A1 (en) | 2020-06-30 | 2021-06-29 | Gas-barrier resin composition, method for producing gas-barrier resin composition, and molded body |
| CN202180046854.1A CN115715308B (zh) | 2020-06-30 | 2021-06-29 | 阻气树脂组合物、阻气树脂组合物的制造方法和成型体 |
Applications Claiming Priority (20)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2020-113325 | 2020-06-30 | ||
| JP2020113323 | 2020-06-30 | ||
| JP2020-113323 | 2020-06-30 | ||
| JP2020113325 | 2020-06-30 | ||
| JP2020130012 | 2020-07-31 | ||
| JP2020-130011 | 2020-07-31 | ||
| JP2020130000 | 2020-07-31 | ||
| JP2020-130007 | 2020-07-31 | ||
| JP2020-129999 | 2020-07-31 | ||
| JP2020130004 | 2020-07-31 | ||
| JP2020130007 | 2020-07-31 | ||
| JP2020-130000 | 2020-07-31 | ||
| JP2020-130012 | 2020-07-31 | ||
| JP2020129999 | 2020-07-31 | ||
| JP2020130017 | 2020-07-31 | ||
| JP2020-130004 | 2020-07-31 | ||
| JP2020130011 | 2020-07-31 | ||
| JP2020-130017 | 2020-07-31 | ||
| JP2020-205872 | 2020-12-11 | ||
| JP2020205872 | 2020-12-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022004701A1 true WO2022004701A1 (ja) | 2022-01-06 |
Family
ID=79316307
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2021/024488 WO2022004701A1 (ja) | 2020-06-30 | 2021-06-29 | ガスバリア樹脂組成物、ガスバリア樹脂組成物の製造方法、及び成形体 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20230250269A1 (ja) |
| EP (1) | EP4173823A1 (ja) |
| KR (1) | KR20230030654A (ja) |
| CN (1) | CN115715308B (ja) |
| TW (1) | TW202206469A (ja) |
| WO (1) | WO2022004701A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115152580A (zh) * | 2022-06-28 | 2022-10-11 | 重庆交通大学 | 一种废弃工程泥浆软固结体种植基质 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3901606A1 (en) * | 2020-04-20 | 2021-10-27 | Catalytic Instruments GmbH & Co. KG | Thermodenuder and method for removing semi-volatile material and semi-volatile particles from an aerosol |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003231715A (ja) | 2001-05-14 | 2003-08-19 | Kuraray Co Ltd | 変性エチレン−ビニルアルコール共重合体およびその製造方法 |
| JP2007137506A (ja) | 2005-11-22 | 2007-06-07 | Aicello Chemical Co Ltd | 多層プラスチック容器 |
| WO2012108374A1 (ja) * | 2011-02-10 | 2012-08-16 | 株式会社クラレ | 植物栽培用培地 |
| WO2014065380A1 (ja) | 2012-10-26 | 2014-05-01 | 東洋鋼鈑株式会社 | 樹脂フィルム、および樹脂フィルム積層化粧板 |
| JP2018104647A (ja) * | 2016-12-28 | 2018-07-05 | 日本合成化学工業株式会社 | エチレン−ビニルエステル系共重合体ケン化物ペレットおよびその製造方法 |
| WO2018163835A1 (ja) | 2017-03-07 | 2018-09-13 | Dic株式会社 | 積層フィルム及び食品包装袋 |
| JP2019182947A (ja) * | 2018-04-04 | 2019-10-24 | 株式会社クラレ | 樹脂組成物及びその用途 |
| WO2019202405A1 (en) * | 2018-04-16 | 2019-10-24 | Braskem, S.A. | Bio-based eva compositions and articles and methods thereof |
| WO2020071513A1 (ja) * | 2018-10-04 | 2020-04-09 | 株式会社クラレ | 多層構造体及びそれを用いた包装材 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4691513B2 (ja) * | 2002-02-26 | 2011-06-01 | 株式会社クラレ | 樹脂組成物及び多層構造体 |
| JP2014065380A (ja) | 2012-09-25 | 2014-04-17 | Shin Meiwa Ind Co Ltd | 荷受台昇降装置 |
| JP2014200968A (ja) * | 2013-04-03 | 2014-10-27 | 株式会社クレハ | 植物由来の樹脂を含有する層を備える熱収縮性多層フィルム及びその製造方法 |
| JP6574804B2 (ja) | 2017-03-27 | 2019-09-11 | 矢崎総業株式会社 | 防水部品 |
| JP7099019B2 (ja) * | 2018-04-09 | 2022-07-12 | 大日本印刷株式会社 | ポリイミド積層体の製造方法、及びポリイミドフィルムの製造方法 |
-
2021
- 2021-06-29 EP EP21834483.6A patent/EP4173823A1/en active Pending
- 2021-06-29 KR KR1020237003312A patent/KR20230030654A/ko unknown
- 2021-06-29 US US18/012,792 patent/US20230250269A1/en active Pending
- 2021-06-29 WO PCT/JP2021/024488 patent/WO2022004701A1/ja unknown
- 2021-06-29 CN CN202180046854.1A patent/CN115715308B/zh active Active
- 2021-06-30 TW TW110124036A patent/TW202206469A/zh unknown
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003231715A (ja) | 2001-05-14 | 2003-08-19 | Kuraray Co Ltd | 変性エチレン−ビニルアルコール共重合体およびその製造方法 |
| JP2007137506A (ja) | 2005-11-22 | 2007-06-07 | Aicello Chemical Co Ltd | 多層プラスチック容器 |
| WO2012108374A1 (ja) * | 2011-02-10 | 2012-08-16 | 株式会社クラレ | 植物栽培用培地 |
| WO2014065380A1 (ja) | 2012-10-26 | 2014-05-01 | 東洋鋼鈑株式会社 | 樹脂フィルム、および樹脂フィルム積層化粧板 |
| JP2018104647A (ja) * | 2016-12-28 | 2018-07-05 | 日本合成化学工業株式会社 | エチレン−ビニルエステル系共重合体ケン化物ペレットおよびその製造方法 |
| WO2018163835A1 (ja) | 2017-03-07 | 2018-09-13 | Dic株式会社 | 積層フィルム及び食品包装袋 |
| JP2019182947A (ja) * | 2018-04-04 | 2019-10-24 | 株式会社クラレ | 樹脂組成物及びその用途 |
| WO2019202405A1 (en) * | 2018-04-16 | 2019-10-24 | Braskem, S.A. | Bio-based eva compositions and articles and methods thereof |
| WO2020071513A1 (ja) * | 2018-10-04 | 2020-04-09 | 株式会社クラレ | 多層構造体及びそれを用いた包装材 |
Non-Patent Citations (2)
| Title |
|---|
| ANONYMOUS: "Multipurpose use of sugar-Use for intermediate chemical raw materials obtained by fermentation- | Agriculture and Livestock Industries Corporation", AGRICULTURE & LIVESTOCK INDUSTRIES CORPORATION [ ONLINE, 6 March 2010 (2010-03-06), XP055895861, Retrieved from the Internet <URL:https://sugar.alic.go.jp/japan/example_03/example0808a.htm?print=true&css=> [retrieved on 20220228] * |
| SCHMID MARKUS, HAMMANN FELICIA, WINKLER HENNING: "Technofunctional Properties of Films Made From Ethylene Vinyl Acetate/Whey Protein Isolate Compounds : ETHYLENE VINYL ACETATE/WHEY PROTEIN ISOLATE COMPOUNDS", PACKAGING TECHNOLOGY AND SCIENCE, JOHN WILEY & SONS LTD, UK, vol. 27, no. 7, 1 July 2014 (2014-07-01), UK , pages 521 - 533, XP055895902, ISSN: 0894-3214, DOI: 10.1002/pts.2051 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN115152580A (zh) * | 2022-06-28 | 2022-10-11 | 重庆交通大学 | 一种废弃工程泥浆软固结体种植基质 |
| CN115152580B (zh) * | 2022-06-28 | 2023-11-28 | 重庆交通大学 | 一种废弃工程泥浆软固结体种植基质 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN115715308B (zh) | 2024-04-09 |
| KR20230030654A (ko) | 2023-03-06 |
| TW202206469A (zh) | 2022-02-16 |
| US20230250269A1 (en) | 2023-08-10 |
| CN115715308A (zh) | 2023-02-24 |
| EP4173823A1 (en) | 2023-05-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2021166276A1 (ja) | 樹脂組成物、成形体、積層体、熱成形容器、ブロー成形容器、フィルム、農業用フィルム、植物培地及びパイプ | |
| WO2022004701A1 (ja) | ガスバリア樹脂組成物、ガスバリア樹脂組成物の製造方法、及び成形体 | |
| US11766851B2 (en) | Packaging laminate | |
| KR20190040244A (ko) | 산소 장벽 플라스틱 물질 | |
| CN101758959A (zh) | 五层共挤高阻隔液体包装膜材及其制备方法 | |
| CN1318096C (zh) | 包装产品的热处理方法及经热处理的包装产品 | |
| JP7019856B2 (ja) | 成形体、フィルムまたはシート、熱収縮フィルムまたはシート、包装材、産業用フィルムまたはシート、熱成形容器、カップ状容器、トレイ状容器、ブロー成形容器、燃料容器、ボトル容器、チューブ、多層パイプおよび紙容器 | |
| EP3239188B1 (en) | Polymethallyl alcohol copolymer and molding containing same, and method of producing polymethallyl alcohol copolymer | |
| JP6927922B2 (ja) | 架橋性樹脂組成物及び架橋物、並びにそれらの製造方法、並びに多層構造体 | |
| JP7007518B1 (ja) | ガスバリア樹脂組成物、多層構造体、包装材、縦製袋充填シール袋、バックインボックス用内容器、積層剥離容器、多層管、ブロー成形容器及びガスバリア樹脂組成物の製造方法 | |
| JP6680694B2 (ja) | ポリメタアリルアルコール樹脂組成物及びそれを用いた成形体 | |
| WO2022004691A1 (ja) | ガスバリア樹脂組成物、成形体、フィルムまたはシート、包装材、産業用フィルムまたはシート、熱成形容器、カップ状容器、トレイ状容器、ブロー成形容器、燃料容器、ボトル容器、チューブ、多層パイプ及び紙容器 | |
| US10906281B2 (en) | Polymeric film comprising vibration dampening and barrier properties | |
| WO2018180862A1 (ja) | 積層体の製造方法 | |
| JP7007516B1 (ja) | ガスバリア樹脂組成物、単層フィルム、多層フィルム、蒸着フィルム及び多層構造体 | |
| JP7008859B1 (ja) | 単層フィルム、蒸着フィルム、多層フィルム及びヒートシール用フィルム | |
| JP7007517B1 (ja) | ガスバリア樹脂組成物、多層構造体、包装材、及びガスバリア樹脂組成物の製造方法 | |
| WO2023054506A1 (ja) | 樹脂組成物、成形体、多層構造体、熱成形容器、ブロー成形容器及び蒸着フィルム | |
| JP2020055174A (ja) | ポリエチレン積層体およびこれを用いた包装材料 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21834483 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 20237003312 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2021834483 Country of ref document: EP Effective date: 20230130 |