WO2020253882A1 - 作为rock蛋白激酶抑制剂的异喹啉酮的衍生物及其应用 - Google Patents
作为rock蛋白激酶抑制剂的异喹啉酮的衍生物及其应用 Download PDFInfo
- Publication number
- WO2020253882A1 WO2020253882A1 PCT/CN2020/097503 CN2020097503W WO2020253882A1 WO 2020253882 A1 WO2020253882 A1 WO 2020253882A1 CN 2020097503 W CN2020097503 W CN 2020097503W WO 2020253882 A1 WO2020253882 A1 WO 2020253882A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- pharmaceutically acceptable
- isomers
- alkyl
- acceptable salts
- Prior art date
Links
- 0 C1=CC=I*=C1 Chemical compound C1=CC=I*=C1 0.000 description 8
- STPVYJILESOVLO-UHFFFAOYSA-N CC(C)(C)OC(NC(CC1)CN1S(c1cccc2c1C=CNC2=O)(=O)=O)=O Chemical compound CC(C)(C)OC(NC(CC1)CN1S(c1cccc2c1C=CNC2=O)(=O)=O)=O STPVYJILESOVLO-UHFFFAOYSA-N 0.000 description 6
- ANRAZFFVGJPYRI-UHFFFAOYSA-N NC(CC1)CN1S(c1cccc2c1C=CN(COC(Cc1ccccc1)=O)C2=O)(=O)=O Chemical compound NC(CC1)CN1S(c1cccc2c1C=CN(COC(Cc1ccccc1)=O)C2=O)(=O)=O ANRAZFFVGJPYRI-UHFFFAOYSA-N 0.000 description 2
- DHBSHYHRIQGWPS-UHFFFAOYSA-N CC(C)(C)CCC(C)(C)COC(OCN(C=Cc1c2cccc1S(N(CC1)CC1NC(OC(C)(C)C)=O)(=O)=O)C2=O)=O Chemical compound CC(C)(C)CCC(C)(C)COC(OCN(C=Cc1c2cccc1S(N(CC1)CC1NC(OC(C)(C)C)=O)(=O)=O)C2=O)=O DHBSHYHRIQGWPS-UHFFFAOYSA-N 0.000 description 1
- DDVMJVVVNQKNHO-UHFFFAOYSA-N CC(C)(C)OC(NC(CC1)CN1S(c1cccc2c1C=CN(COC(C)=O)C2=O)(=O)=O)=O Chemical compound CC(C)(C)OC(NC(CC1)CN1S(c1cccc2c1C=CN(COC(C)=O)C2=O)(=O)=O)=O DDVMJVVVNQKNHO-UHFFFAOYSA-N 0.000 description 1
- PVKLTPPLROVIRD-UHFFFAOYSA-N CC(C)(C)OC(NC(CC1)CN1S(c1cccc2c1C=CN(COC(Cc1ccccc1)=O)C2=O)(=O)=O)=O Chemical compound CC(C)(C)OC(NC(CC1)CN1S(c1cccc2c1C=CN(COC(Cc1ccccc1)=O)C2=O)(=O)=O)=O PVKLTPPLROVIRD-UHFFFAOYSA-N 0.000 description 1
- BUUSWVZWQHLGHL-UHFFFAOYSA-N CC(C)(C)OC(NC(CC1)CN1S(c1cccc2c1C=CN(COC(c1ccc(C)cc1)=O)C2=O)(=O)=O)=O Chemical compound CC(C)(C)OC(NC(CC1)CN1S(c1cccc2c1C=CN(COC(c1ccc(C)cc1)=O)C2=O)(=O)=O)=O BUUSWVZWQHLGHL-UHFFFAOYSA-N 0.000 description 1
- STUCMBAQCORUTD-UHFFFAOYSA-N CC(C)(C)OC(NC(CC1)CN1S(c1cccc2c1C=CN(COC(c1ccc[s]1)=O)C2=O)(=O)=O)=O Chemical compound CC(C)(C)OC(NC(CC1)CN1S(c1cccc2c1C=CN(COC(c1ccc[s]1)=O)C2=O)(=O)=O)=O STUCMBAQCORUTD-UHFFFAOYSA-N 0.000 description 1
- HQCXVZWQGUVFOB-NRFANRHFSA-N CC(C)(C)OC(N[C@@H](CC1)CN1S(c1cccc2c1C(C)=CN(COC(c1ccc(C)cc1C)=O)C2=O)(=O)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CC1)CN1S(c1cccc2c1C(C)=CN(COC(c1ccc(C)cc1C)=O)C2=O)(=O)=O)=O HQCXVZWQGUVFOB-NRFANRHFSA-N 0.000 description 1
- ZWUGJAONGJADKX-ZDUSSCGKSA-N CC(C)(C)OC(N[C@@H](CC1)CN1S(c1cccc2c1C(C)=CNC2=O)(=O)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](CC1)CN1S(c1cccc2c1C(C)=CNC2=O)(=O)=O)=O ZWUGJAONGJADKX-ZDUSSCGKSA-N 0.000 description 1
- BBJSOJFCLWSIAR-UHFFFAOYSA-N CC(C)C(OCN(C=Cc1c2cccc1S(N(CC1)C3(CC3)C1N)(=O)=O)C2=O)=O Chemical compound CC(C)C(OCN(C=Cc1c2cccc1S(N(CC1)C3(CC3)C1N)(=O)=O)C2=O)=O BBJSOJFCLWSIAR-UHFFFAOYSA-N 0.000 description 1
- DRAFRULNLDTBBA-CYBMUJFWSA-N CC(C)C(OCN(C=Cc1c2cccc1S(N(CC1)C[C@@H]1N)(=O)=O)C2=O)=O Chemical compound CC(C)C(OCN(C=Cc1c2cccc1S(N(CC1)C[C@@H]1N)(=O)=O)C2=O)=O DRAFRULNLDTBBA-CYBMUJFWSA-N 0.000 description 1
- DAEMQFONPFTLAV-UHFFFAOYSA-N CC(OCN(C=Cc1c2cccc1S(N(CC1)CC1N)(=O)=O)C2=O)=O Chemical compound CC(OCN(C=Cc1c2cccc1S(N(CC1)CC1N)(=O)=O)C2=O)=O DAEMQFONPFTLAV-UHFFFAOYSA-N 0.000 description 1
- BDPZFQLKFUONAG-UHFFFAOYSA-N CCCC(OCCl)=O Chemical compound CCCC(OCCl)=O BDPZFQLKFUONAG-UHFFFAOYSA-N 0.000 description 1
- HNUBMUSOXKDQIN-UHFFFAOYSA-N CCCCCC(OCCl)=O Chemical compound CCCCCC(OCCl)=O HNUBMUSOXKDQIN-UHFFFAOYSA-N 0.000 description 1
- PNGLNOHDHZZKMQ-UHFFFAOYSA-N CCCCCC(OCN(C=Cc1c2cccc1S(N(CC1)CC1N)(=O)=O)C2=O)=O Chemical compound CCCCCC(OCN(C=Cc1c2cccc1S(N(CC1)CC1N)(=O)=O)C2=O)=O PNGLNOHDHZZKMQ-UHFFFAOYSA-N 0.000 description 1
- IDKYMTWUBASFEA-UHFFFAOYSA-N CCCCCC(OCN(C=Cc1c2cccc1S(N(CC1)CC1NC(OC(C)(C)C)=O)(=O)=O)C2=O)=O Chemical compound CCCCCC(OCN(C=Cc1c2cccc1S(N(CC1)CC1NC(OC(C)(C)C)=O)(=O)=O)C2=O)=O IDKYMTWUBASFEA-UHFFFAOYSA-N 0.000 description 1
- DULCUDSUACXJJC-UHFFFAOYSA-N CCOC(Cc1ccccc1)=O Chemical compound CCOC(Cc1ccccc1)=O DULCUDSUACXJJC-UHFFFAOYSA-N 0.000 description 1
- HCNXDKGBIDZKNJ-GFCCVEGCSA-N CCOC(OCN(C=Cc1c2cccc1S(N(CC1)C[C@@H]1N)(=O)=O)C2=O)=O Chemical compound CCOC(OCN(C=Cc1c2cccc1S(N(CC1)C[C@@H]1N)(=O)=O)C2=O)=O HCNXDKGBIDZKNJ-GFCCVEGCSA-N 0.000 description 1
- ZKHQSQYLKSSYIP-UHFFFAOYSA-N COC(c1c[o]cc1)=O Chemical compound COC(c1c[o]cc1)=O ZKHQSQYLKSSYIP-UHFFFAOYSA-N 0.000 description 1
- XDRBAVJWSHNLQN-UHFFFAOYSA-N COC(c1ccc2[o]ccc2c1)=O Chemical compound COC(c1ccc2[o]ccc2c1)=O XDRBAVJWSHNLQN-UHFFFAOYSA-N 0.000 description 1
- LPNBBFKOUUSUDB-UHFFFAOYSA-N Cc(cc1)ccc1C(O)=O Chemical compound Cc(cc1)ccc1C(O)=O LPNBBFKOUUSUDB-UHFFFAOYSA-N 0.000 description 1
- VDQALAPYHMXZTJ-UHFFFAOYSA-N Cc(cc1)ccc1C(OCN(C=Cc1c2cccc1S(N(CC1)CC1N)(=O)=O)C2=O)=O Chemical compound Cc(cc1)ccc1C(OCN(C=Cc1c2cccc1S(N(CC1)CC1N)(=O)=O)C2=O)=O VDQALAPYHMXZTJ-UHFFFAOYSA-N 0.000 description 1
- DMAOBLUIJWFUAS-UHFFFAOYSA-N Cc1ccc(C(OCCl)=O)c(C)c1 Chemical compound Cc1ccc(C(OCCl)=O)c(C)c1 DMAOBLUIJWFUAS-UHFFFAOYSA-N 0.000 description 1
- AIUQQDLYQFEDER-SFHVURJKSA-N Cc1ccc(C(OCN(C=C(C)c2c3cccc2S(N(CC2)C[C@H]2N)(=O)=O)C3=O)=O)c(C)c1 Chemical compound Cc1ccc(C(OCN(C=C(C)c2c3cccc2S(N(CC2)C[C@H]2N)(=O)=O)C3=O)=O)c(C)c1 AIUQQDLYQFEDER-SFHVURJKSA-N 0.000 description 1
- OXDOZGCRUOCLFA-UHFFFAOYSA-N NC(CC1)C2(CC2)N1S(c1cccc2c1C=CN(COC(C1CCC1)=O)C2=O)(=O)=O Chemical compound NC(CC1)C2(CC2)N1S(c1cccc2c1C=CN(COC(C1CCC1)=O)C2=O)(=O)=O OXDOZGCRUOCLFA-UHFFFAOYSA-N 0.000 description 1
- QHHVGYYPLSAICC-UHFFFAOYSA-N NC(CC1)CN1S(c1cccc2c1C=CN(COC(c1ccc[s]1)=O)C2=O)(=O)=O Chemical compound NC(CC1)CN1S(c1cccc2c1C=CN(COC(c1ccc[s]1)=O)C2=O)(=O)=O QHHVGYYPLSAICC-UHFFFAOYSA-N 0.000 description 1
- ICXKGAWJFMIZLT-UHFFFAOYSA-N NC(CC1)CN1S(c1cccc2c1C=CN(COC(c1ccccc1)=O)C2=O)(=O)=O Chemical compound NC(CC1)CN1S(c1cccc2c1C=CN(COC(c1ccccc1)=O)C2=O)(=O)=O ICXKGAWJFMIZLT-UHFFFAOYSA-N 0.000 description 1
- GZOJAIJTTXMVEX-UHFFFAOYSA-N NCOC(c1c[o]cc1)=O Chemical compound NCOC(c1c[o]cc1)=O GZOJAIJTTXMVEX-UHFFFAOYSA-N 0.000 description 1
- AFQKQRIDRJMPRB-UHFFFAOYSA-N NCOC(c1ccc2[o]ccc2c1)=O Chemical compound NCOC(c1ccc2[o]ccc2c1)=O AFQKQRIDRJMPRB-UHFFFAOYSA-N 0.000 description 1
- DJRHMVMNVWKQRZ-UHFFFAOYSA-N O=C(c1ccc[s]1)OCc1c[s]c(C(OCCl)=O)c1 Chemical compound O=C(c1ccc[s]1)OCc1c[s]c(C(OCCl)=O)c1 DJRHMVMNVWKQRZ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
- A61K31/4725—Non-condensed isoquinolines, e.g. papaverine containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the invention relates to a series of isoquinolinone derivatives as ROCK protein kinase inhibitors, and their application in preparing drugs for ROCK protein kinase inhibitor-related glaucoma or ocular hypertension diseases. Specifically, it relates to a compound represented by formula (I), its isomers or a pharmaceutically acceptable salt thereof.
- RHO-associated protein kinase which belongs to serine/threonine protein kinase, is the downstream target effector molecule of RHO, and is widely expressed in the human body.
- RHO-related protein kinase (ROCK) is involved in the regulation of myosin light chain (MLC) and is suitable for the treatment of vasodilation.
- MLC myosin light chain
- ROCK kinase can also act on trabecular outflow tract cells, relax trabecular cells, and reduce the resistance of aqueous humor outflow.
- ROCK kinase inhibitors can also promote the repair of corneal endothelial cells and prevent fibrosis, which has huge application prospects.
- Isoquinoline sulfonamide compounds are an important type of ROCK kinase inhibitor. Fasudil and K-115 (patent WO2006057397A1) that have been on the market are both isoquinoline sulfonamide compounds. Fasudil is a new type of drug with extensive pharmacological effects. It is a RHO kinase inhibitor. It expands blood vessels by increasing the activity of myosin light chain phosphatase, reduces the tension of endothelial cells, improves the microcirculation of brain tissue, and does not produce And aggravate the theft of the brain, at the same time can antagonize inflammatory factors, protect nerves against apoptosis, and promote nerve regeneration.
- K-115 The approved and potential applications of K-115 are very wide, including glaucoma, high intraocular pressure, diabetic retinal injury complications, age-related macular degeneration, corneal injury, recovery after cataract and glaucoma surgery, etc., and may be further expanded to Systemic drugs.
- Patent WO2007026664A1 reports a series of compounds with ROCK kinase inhibitory effect, such as control compound 1 and control compound 2.
- the series of compounds have good enzyme activity, but they are in terms of membrane permeability, pharmacokinetics, druggability, etc. Room for improvement.
- the present invention reports a class of similar compounds through structural modification, which significantly improves the properties in this respect.
- the present invention provides a compound represented by formula (I), an isomer thereof or a pharmaceutically acceptable salt thereof,
- T 1 is selected from -(CH 2 ) n -;
- T 2 is selected from -(CH 2 ) m -and -C(R 7 )(R 8 )-;
- R 1 is selected from C 1-16 alkyl, phenyl, C 3-7 cycloalkyl, 3-8 membered heterocycloalkyl and 5-10 membered heteroaryl, the C 1-16 alkyl, phenyl , C 3-7 cycloalkyl, 3-8 membered heterocycloalkyl and 5-10 membered heteroaryl each independently optionally substituted with one, two or three R a;
- R 2 and R 3 are each independently selected from H, F, Cl, Br, I, OH, NH 2 , CN and C 1-3 alkyl;
- R 4 is selected from H, F, Cl, Br, I, OH, NH 2 , CN and a C 1-3 alkyl group optionally substituted with 1, 2 or 3 R b ;
- R 5 is selected from NR 9 R 10 ;
- R 6 is selected from H, F, Cl, Br, I, OH, NH 2 , CN and C 1-3 alkyl;
- R 7 and R 8 are each independently selected from H, F, Cl, Br, I, OH, NH 2 , CN and C 1-3 alkyl optionally substituted with 1, 2 or 3 R c ; or, R 7.
- R 9 and R 10 are each independently selected from H and C 1-3 alkyl optionally substituted with 1, 2 or 3 R e ;
- L is selected from single bond, -O- and -NR 11 -;
- R 11 is from H and C 1-3 alkyl
- n is selected from 0, 1 and 2;
- n 0, 1, 2 and 3;
- R a is selected from F, Cl, Br, I, OH, NH 2 , CN, C 1-3 alkyl and C 1-3 alkoxy, the C 1-3 alkyl and C 1-3 alkoxy Optionally substituted by 1, 2 or 3 R;
- R b , R c , Rd and R e are each independently selected from H, F, Cl, Br, I, OH, NH 2 , CN and C 1-3 alkyl;
- R is selected from F, Cl, Br, I, OH, NH 2 , CN and CH 3 ;
- the 3-8 membered heterocycloalkyl group and the 5-10 membered heteroaryl group each independently comprise 1, 2, 3 or 4 heteroatoms independently selected from -NH-, -O-, -S- and N or Heteroatom group.
- the present invention provides a compound represented by formula (I) or a pharmaceutically acceptable salt thereof,
- T 1 is selected from -(CH 2 ) n -;
- T 2 is selected from -(CH 2 ) m -and -C(R 7 )(R 8 )-;
- R 1 is selected from C 1-16 alkyl, phenyl, C 3-7 cycloalkyl, 3-8 membered heterocycloalkyl and 5-10 membered heteroaryl, the C 1-16 alkyl, phenyl , C 3-7 cycloalkyl, 3-8 membered heterocycloalkyl and 5-10 membered heteroaryl each independently optionally substituted with one, two or three R a;
- R 2 and R 3 are each independently selected from H, F, Cl, Br, I, OH, NH 2 , CN and C 1-3 alkyl;
- R 4 is selected from H, F, Cl, Br, I, OH, NH 2 , CN and a C 1-3 alkyl group optionally substituted with 1, 2 or 3 R b ;
- R 5 is selected from NR 9 R 10 ;
- R 6 is selected from F, Cl, Br, I, OH, NH 2 , CN and C 1-3 alkyl;
- R 7 and R 8 are each independently selected from H, F, Cl, Br, I, OH, NH 2 , CN and C 1-3 alkyl optionally substituted with 1, 2 or 3 R c ; or, R 7.
- R 9 and R 10 are each independently selected from H and C 1-3 alkyl optionally substituted with 1, 2 or 3 R e ;
- L is selected from single bond, -O- and -NR 11 -;
- R 11 is from H and C 1-3 alkyl
- n is selected from 0, 1 and 2;
- n 0, 1, 2 and 3;
- R a is selected from H, F, Cl, Br, I, OH, NH 2 , CN, C 1-3 alkyl and C 1-3 alkoxy, the C 1-3 alkyl and C 1-3 alkane
- the oxy group is optionally substituted with 1, 2 or 3 R;
- R b , R c , Rd and R e are each independently selected from H, F, Cl, Br, I, OH, NH 2 , CN and C 1-3 alkyl;
- R is selected from F, Cl, Br, I, OH, NH 2 , CN and CH 3 ;
- the 3-8 membered heterocycloalkyl group and the 5-10 membered heteroaryl group each independently comprise 1, 2, 3 or 4 heteroatoms independently selected from -NH-, -O-, -S- and N or Heteroatom group.
- R a is selected from F, Cl, Br, I, OH, NH 2, CN, CH 3, CF 3, CH 2 F, CHF 2, CH 2 CH 3 and OCH 3, the other variables As defined in the present invention.
- R a is selected from H, F, Cl, Br, I, OH, NH 2, CN, CH 3, CF 3, CH 2 F, CHF 2, CH 2 CH 3 and OCH 3, Other variables are as defined in the present invention.
- the above-mentioned R 1 is selected from C 1-12 alkyl, phenyl, cyclobutanyl, cyclopentyl, cyclohexane, tetrahydrofuran, tetrahydropyranyl, piperidinyl, Thienyl, furanyl, pyrrolyl and benzofuranyl, the C 1-12 alkyl, phenyl, cyclobutanyl, cyclopentyl, cyclohexyl, tetrahydrofuranyl, tetrahydropyranyl, piperidyl, thienyl, furyl, pyrrolyl, benzofuranyl, and each independently optionally substituted with 1, 2 or 3 R a, the other variables are as defined in the present invention.
- the aforementioned R 1 is selected from CH 3 , CH 2 CH 3 , (CH 2 ) 2 CH 3 , (CH 2 ) 3 CH 3 , (CH 2 ) 4 CH 3 , (CH 2 ) 5 CH 3 , (CH 2 ) 6 CH 3 , (CH 2 ) 10 CH 3 , CH(CH 3 ) 2 , C(CH 3 ) 3 ,
- Other variables are as defined in the present invention.
- the aforementioned R 1 is selected from CH 3 , CH 2 CH 3 , (CH 2 ) 2 CH 3 , (CH 2 ) 3 CH 3 , (CH 2 ) 4 CH 3 , (CH 2 ) 5 CH 3 , (CH 2 ) 6 CH 3 , (CH 2 ) 10 CH 3 , CH(CH 3 ) 2 , C(CH 3 ) 3 ,
- Other variables are as defined in the present invention.
- the aforementioned R 1 is selected from CH 3 , CH 2 CH 3 , (CH 2 ) 2 CH 3 , (CH 2 ) 3 CH 3 , (CH 2 ) 4 CH 3 , (CH 2 ) 5 CH 3 , (CH 2 ) 6 CH 3 , (CH 2 ) 10 CH 3 , CH(CH 3 ) 2 , C(CH 3 ) 3 ,
- Other variables are as defined in the present invention.
- the aforementioned R 1 is selected from CH 3 , CH 2 CH 3 , (CH 2 ) 2 CH 3 , (CH 2 ) 3 CH 3 , (CH 2 ) 4 CH 3 , (CH 2 ) 5 CH 3 , (CH 2 ) 6 CH 3 , (CH 2 ) 10 CH 3 , CH(CH 3 ) 2 , C(CH 3 ) 3 ,
- Other variables are as defined in the present invention.
- the aforementioned R 1 is selected from CH 3 , CH 2 CH 3 , (CH 2 ) 2 CH 3 , (CH 2 ) 3 CH 3 , (CH 2 ) 4 CH 3 , (CH 2 ) 5 CH 3 , (CH 2 ) 6 CH 3 , (CH 2 ) 10 CH 3 , CH(CH 3 ) 2 , C(CH 3 ) 3 ,
- Other variables are as defined in the present invention.
- R 2 and R 3 are independently selected from H, F, Cl, Br, I, OH, and NH 2 , and other variables are as defined in the present invention.
- R 2 and R 3 are independently selected from H, F, Cl, Br, I, OH, and NH 2 , and other variables are as defined in the present invention.
- R 4 is selected from H, F, Cl, Br, I, OH, NH 2 , CN, CH 3 and CH 2 CH 3 , and other variables are as defined in the present invention.
- R 9 and R 10 are independently selected from H, CH 3 and CH 2 CH 3 , and other variables are as defined in the present invention.
- R 5 is selected from NH 2 , NH(CH 3 ) and N(CH 3 ) 2 , and other variables are as defined in the present invention.
- R 6 is selected from H, F, Cl, Br, I, OH, NH 2 , CN and CH 3 , and other variables are as defined in the present invention.
- R 6 is selected from F, Cl, Br, I, OH, NH 2 , CN and CH 3 , and other variables are as defined in the present invention.
- R 7 and R 8 are independently selected from F, Cl, Br, I, OH, NH 2 , CN, and CH 3 , and other variables are as defined in the present invention.
- R 7 and R 8 are independently selected from H, F, Cl, Br, I, OH, NH 2 , CN and CH 3 , and other variables are as defined in the present invention.
- R 7 , R 8 and the atoms connected to them together form a cyclopropyl group optionally substituted with 1, 2, or 3 Rd , and other variables are as defined in the present invention.
- R 7 , R 8 and the atoms connected to them together form a cyclopropyl group, and other variables are as defined in the present invention.
- the above L is selected from single bond, -O-, -NH- and -N(CH 3 )-, and other variables are as defined in the present invention.
- the above-mentioned T 2 is selected from -CH 2 -, -(CH 2 ) 2 -and Other variables are as defined in the present invention.
- Other variables are as defined in the present invention.
- Other variables are as defined in the present invention.
- Other variables are as defined in the present invention.
- the above-mentioned compound, its isomers or pharmaceutically acceptable salts thereof are selected from
- R 1 , R 4 , R 5 and L are as defined in the present invention.
- the above-mentioned compound, its isomers or pharmaceutically acceptable salts thereof are selected from
- R 1 , R 4 and L are as defined in the present invention.
- the present invention also provides a compound represented by the following formula, an isomer thereof or a pharmaceutically acceptable salt thereof, and the compound is selected from
- the above-mentioned compound, its isomer or pharmaceutically acceptable salt thereof is selected from
- the present invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of the above-mentioned compound, its isomers or pharmaceutically acceptable salts thereof as active ingredients and a pharmaceutically acceptable carrier.
- the above-mentioned compound, its isomer or pharmaceutically acceptable salt or the above-mentioned composition is used in the preparation of ROCK protein kinase inhibitor related drugs.
- the aforementioned ROCK protein kinase inhibitor-related drugs are drugs for glaucoma or ocular hypertension.
- pharmaceutically acceptable salt refers to a salt of the compound of the present invention, which is prepared from a compound with specific substituents discovered in the present invention and a relatively non-toxic acid or base.
- the base addition salt can be obtained by contacting the neutral form of the compound with a sufficient amount of base in a pure solution or a suitable inert solvent.
- Pharmaceutically acceptable base addition salts include sodium, potassium, calcium, ammonium, organic ammonia or magnesium salts or similar salts.
- the acid addition salt can be obtained by contacting the neutral form of the compound with a sufficient amount of acid in a pure solution or a suitable inert solvent.
- Examples of pharmaceutically acceptable acid addition salts include inorganic acid salts including, for example, hydrochloric acid, hydrobromic acid, nitric acid, carbonic acid, hydrogen carbonate, phosphoric acid, monohydrogen phosphate, dihydrogen phosphate, sulfuric acid, Hydrogen sulfate, hydroiodic acid, phosphorous acid, etc.; and organic acid salts, the organic acid includes such as acetic acid, propionic acid, isobutyric acid, maleic acid, malonic acid, benzoic acid, succinic acid, suberic acid, Similar acids such as fumaric acid, lactic acid, mandelic acid, phthalic acid, benzenesulfonic acid, p-toluenesulfonic acid, citric acid, tartaric acid and methanesulfonic acid; also include salts of amino acids (such as arginine, etc.) , And salts of organic acids such as glucuronic acid. Certain specific compounds of the present invention contain basic and acidic
- the pharmaceutically acceptable salt of the present invention can be synthesized from the parent compound containing acid or base by conventional chemical methods. Generally, such salts are prepared by reacting these compounds in free acid or base form with a stoichiometric amount of appropriate base or acid in water or an organic solvent or a mixture of both.
- the compounds provided by the present invention also exist in prodrug forms.
- the prodrugs of the compounds described herein easily undergo chemical changes under physiological conditions to transform into the compounds of the invention.
- prodrugs can be converted to the compounds of the present invention by chemical or biochemical methods in the in vivo environment.
- Certain compounds of the present invention may exist in unsolvated or solvated forms, including hydrated forms.
- the solvated form is equivalent to the unsolvated form, and both are included in the scope of the present invention.
- the compounds of the present invention may exist in specific geometric or stereoisomeric forms.
- the present invention contemplates all such compounds, including cis and trans isomers, (-)- and (+)-enantiomers, (R)- and (S)-enantiomers, diastereomers Conformers, (D)-isomers, (L)-isomers, and their racemic mixtures and other mixtures, such as enantiomers or diastereomeric enriched mixtures, all of these mixtures belong to Within the scope of the present invention.
- Additional asymmetric carbon atoms may be present in substituents such as alkyl. All these isomers and their mixtures are included in the scope of the present invention.
- enantiomer or “optical isomer” refers to stereoisomers that are mirror images of each other.
- cis-trans isomer or “geometric isomer” is caused by the inability to rotate freely because of double bonds or single bonds of ring carbon atoms.
- diastereomer refers to a stereoisomer in which a molecule has two or more chiral centers and the relationship between the molecules is not mirror images.
- wedge-shaped solid line keys And wedge-shaped dashed key Represents the absolute configuration of a solid center, with a straight solid line key And straight dashed key Indicates the relative configuration of the three-dimensional center, using wavy lines Represents a wedge-shaped solid line key Or wedge-shaped dotted key Or use wavy lines Represents a straight solid line key And straight dashed key
- the compound of the present invention may be specific.
- tautomer or “tautomeric form” means that at room temperature, the isomers of different functional groups are in dynamic equilibrium and can be transformed into each other quickly. If tautomers are possible (such as in solution), the chemical equilibrium of tautomers can be reached.
- proton tautomers also called prototropic tautomers
- proton migration such as keto-enol isomerization and imine-ene Amine isomerization.
- Valence isomers include some recombination of bonding electrons to carry out mutual transformation.
- keto-enol tautomerization is the tautomerism between two tautomers of pentane-2,4-dione and 4-hydroxypent-3-en-2-one.
- the terms “enriched in one isomer”, “enriched in isomers”, “enriched in one enantiomer” or “enriched in enantiomers” refer to one of the isomers or pairs of
- the content of the enantiomer is less than 100%, and the content of the isomer or enantiomer is greater than or equal to 60%, or greater than or equal to 70%, or greater than or equal to 80%, or greater than or equal to 90%, or greater than or equal to 95%, or 96% or greater, or 97% or greater, or 98% or greater, or 99% or greater, or 99.5% or greater, or 99.6% or greater, or 99.7% or greater, or 99.8% or greater, or greater than or equal 99.9%.
- the term “isomer excess” or “enantiomeric excess” refers to the difference between the relative percentages of two isomers or two enantiomers. For example, if the content of one isomer or enantiomer is 90%, and the content of the other isomer or enantiomer is 10%, the isomer or enantiomer excess (ee value) is 80% .
- optically active (R)- and (S)-isomers and D and L isomers can be prepared by chiral synthesis or chiral reagents or other conventional techniques. If you want to obtain an enantiomer of a compound of the present invention, it can be prepared by asymmetric synthesis or derivatization with chiral auxiliary agents, in which the resulting diastereomeric mixture is separated and the auxiliary group is cleaved to provide pure The desired enantiomer.
- the molecule when the molecule contains a basic functional group (such as an amino group) or an acidic functional group (such as a carboxyl group), it forms a diastereomeric salt with a suitable optically active acid or base, and then passes through a conventional method known in the art The diastereoisomers are resolved, and then the pure enantiomers are recovered.
- the separation of enantiomers and diastereomers is usually accomplished through the use of chromatography, which employs a chiral stationary phase and is optionally combined with chemical derivatization (for example, the formation of amino groups from amines). Formate).
- the compounds of the present invention may contain unnatural proportions of atomic isotopes on one or more of the atoms constituting the compound.
- compounds can be labeled with radioisotopes, such as tritium ( 3 H), iodine-125 ( 125 I), or C-14 ( 14 C).
- deuterated drugs can be formed by replacing hydrogen with heavy hydrogen. The bond formed by deuterium and carbon is stronger than the bond formed by ordinary hydrogen and carbon. Compared with undeuterated drugs, deuterated drugs have reduced toxic side effects and increased drug stability. , Enhance the efficacy, extend the biological half-life of drugs and other advantages. All changes in the isotopic composition of the compounds of the present invention, whether radioactive or not, are included in the scope of the present invention.
- substituted means that any one or more hydrogen atoms on a specific atom are replaced by substituents, and can include deuterium and hydrogen variants, as long as the valence of the specific atom is normal and the substituted compound is stable of.
- oxygen it means that two hydrogen atoms are replaced. Oxygen substitution will not occur on aromatic groups.
- optionally substituted means that it can be substituted or unsubstituted. Unless otherwise specified, the type and number of substituents can be arbitrary on the basis that they can be chemically realized.
- any variable such as R
- its definition in each case is independent.
- the group may optionally be substituted with up to two Rs, and R has independent options in each case.
- combinations of substituents and/or variants thereof are only permitted if such combinations result in stable compounds.
- linking group When the number of a linking group is 0, such as -(CRR) 0 -, it means that the linking group is a single bond.
- substituents When a substituent is vacant, it means that the substituent is absent. For example, when X in AX is vacant, it means that the structure is actually A.
- substituents do not indicate which atom is connected to the substituted group, such substituents can be bonded via any atom.
- a pyridyl group can pass through any one of the pyridine ring as a substituent. The carbon atom is attached to the substituted group.
- the middle linking group L is -MW-, at this time -MW- can be formed by connecting ring A and ring B in the same direction as the reading order from left to right It can also be formed by connecting ring A and ring B in the direction opposite to the reading order from left to right Combinations of the linking groups, substituents, and/or variants thereof are only permitted if such combinations result in stable compounds.
- C 1-16 alkyl is used to mean a linear or branched saturated hydrocarbon group composed of 1 to 16 carbon atoms.
- the C 1-16 alkyl group includes C 1-15 , C 1-14 , C 1-12 , C 1-10 , C 1-9 , C 1-8 , C 1-6 , C 1-5 , C 1-4 , C 1-3 , C 1-2 , C 2-6 , C 2-4 , C 10 , C 8 , C 7 , C 6 and C 5 alkyl, etc.; it can be monovalent (such as methyl Group), divalent (such as methylene) or multivalent (such as methine).
- C 1-16 alkyl groups include, but are not limited to, methyl (Me), ethyl (Et), propyl (including n-propyl and isopropyl), butyl (including n-butyl, isobutyl) , S-butyl and t-butyl), pentyl (including n-pentyl, isopentyl and neopentyl), hexyl, heptyl, octyl, etc.
- C 1-12 alkyl is used to indicate a linear or branched saturated hydrocarbon group composed of 1 to 12 carbon atoms.
- the C 1-12 alkyl group includes C 1-10 , C 1-9 , C 1-8 , C 1-6 , C 1-5 , C 1-4 , C 1-3 , C 1-2 , C 2-6 , C 2-4 , C 10 , C 8 , C 7 , C 6 and C 5 alkyl, etc.; it can be monovalent (such as methyl), divalent (such as methylene) or multivalent ( Such as methine).
- C 1-12 alkyl groups include, but are not limited to, methyl (Me), ethyl (Et), propyl (including n-propyl and isopropyl), butyl (including n-butyl, isobutyl) , S-butyl and t-butyl), pentyl (including n-pentyl, isopentyl and neopentyl), hexyl, heptyl, octyl, etc.
- C 1-3 alkyl is used to indicate a linear or branched saturated hydrocarbon group composed of 1 to 3 carbon atoms.
- the C 1-3 alkyl group includes C 1-2 and C 2-3 alkyl groups, etc.; it can be monovalent (such as methyl), divalent (such as methylene) or multivalent (such as methine) .
- Examples of C 1-3 alkyl include, but are not limited to, methyl (Me), ethyl (Et), propyl (including n-propyl and isopropyl), and the like.
- C 1-3 alkoxy refers to those alkyl groups containing 1 to 3 carbon atoms attached to the rest of the molecule through an oxygen atom.
- the C 1-3 alkoxy group includes C 1-2 , C 2-3 , C 3 and C 2 alkoxy groups and the like.
- Examples of C 1-3 alkoxy include but are not limited to methoxy, ethoxy, propoxy (including n-propoxy and isopropoxy) and the like.
- C 3-7 cycloalkyl means a saturated cyclic hydrocarbon group composed of 3 to 7 carbon atoms, which is a monocyclic ring system, and the C 3-7 cycloalkyl includes C 5 -7 , C 3-4 and C 4-5 cycloalkyl, etc.; it can be monovalent, divalent or multivalent.
- Examples of C 3-7 cycloalkyl include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl and the like.
- C 3-5 cycloalkyl means a saturated cyclic hydrocarbon group composed of 3 to 5 carbon atoms, which is a monocyclic ring system, and the C 3-5 cycloalkyl includes C 3 -4 and C 4-5 cycloalkyl, etc.; it can be monovalent, divalent or multivalent.
- Examples of C 3-5 cycloalkyl include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl and the like.
- the term "3-8 membered heterocycloalkyl" by itself or in combination with other terms means a saturated cyclic group consisting of 3 to 8 ring atoms, with 1, 2, 3 or 4 ring atoms Are heteroatoms independently selected from O, S and N, and the rest are carbon atoms, wherein nitrogen atoms are optionally quaternized, and nitrogen and sulfur heteroatoms can be optionally oxidized (ie, NO and S(O) p , p Is 1 or 2). It includes monocyclic and bicyclic ring systems, where the bicyclic ring system includes spiro, fused, and bridged rings.
- a heteroatom may occupy the connection position of the heterocycloalkyl group with the rest of the molecule.
- the 3-8 membered heterocycloalkyl group includes 3-6 membered, 3-5 membered, 4-6 membered, 5-6 membered, 4-membered, 5-membered and 6-membered heterocycloalkyl group.
- 3-8 membered heterocycloalkyl examples include, but are not limited to, azetidinyl, oxetanyl, thietane, pyrrolidinyl, pyrazolidinyl, imidazolidinyl, tetrahydrothienyl ( Including tetrahydrothiophen-2-yl and tetrahydrothiophen-3-yl, etc.), tetrahydrofuranyl (including tetrahydrofuran-2-yl, etc.), tetrahydropyranyl, piperidinyl (including 1-piperidinyl, 2- Piperidinyl and 3-piperidinyl, etc.), piperazinyl (including 1-piperazinyl and 2-piperazinyl, etc.), morpholinyl (including 3-morpholinyl and 4-morpholinyl, etc.), Dioxanyl, dithiazyl, isoxazolidinyl, isothiazolidiny
- 5-10 membered heteroaryl ring and “5-10 membered heteroaryl group” can be used interchangeably in the present invention.
- the term “5-10 membered heteroaryl group” means a ring consisting of 5 to 10 A cyclic group composed of atoms with a conjugated ⁇ -electron system, wherein 1, 2, 3 or 4 ring atoms are heteroatoms independently selected from O, S and N, and the rest are carbon atoms. It can be a monocyclic, fused bicyclic or fused tricyclic system, where each ring is aromatic.
- the nitrogen and sulfur heteroatoms can be optionally oxidized (ie NO and S(O) p , p is 1 or 2).
- the 5-10 membered heteroaryl group can be attached to the rest of the molecule through a heteroatom or a carbon atom.
- the 5-10 membered heteroaryl groups include 5-8 membered, 5-7 membered, 5-6 membered, 5 membered and 6 membered heteroaryl groups and the like.
- Examples of the 5-10 membered heteroaryl include, but are not limited to, pyrrolyl (including N-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, etc.), pyrazolyl (including 2-pyrazolyl and 3-pyrrolyl, etc.) Azolyl, etc.), imidazolyl (including N-imidazolyl, 2-imidazolyl, 4-imidazolyl and 5-imidazolyl, etc.), oxazolyl (including 2-oxazolyl, 4-oxazolyl and 5- Oxazolyl, etc.), triazolyl (1H-1,2,3-triazolyl, 2H-1,2,3-triazolyl, 1H-1,2,4-triazolyl and 4H-1, 2,4-triazolyl, etc.), tetrazolyl, isoxazolyl (3-isoxazolyl, 4-isoxazolyl and 5-isoxazolyl, etc.), thiazolyl (including 2-thi
- C n-n+m or C n -C n+m includes any specific case of n to n+m carbons, for example, C 1-12 includes C 1 , C 2 , C 3 , C 4, C 5, C 6, C 7, C 8, C 9, C 10, C 11, and C 12, also including any one of n + m to n ranges, for example C 1- 3 comprises a C 1-12 , C 1-6 , C 1-9 , C 3-6 , C 3-9 , C 3-12 , C 6-9 , C 6-12 , and C 9-12, etc.; in the same way, from n to n +m means the number of atoms in the ring is n to n+m, for example, 3-12 membered ring includes 3-membered ring, 4-membered ring, 5-membered ring, 6-membered ring, 7-membered ring, 8-membered ring, 9-membered ring , 10-membered ring, 11-member
- leaving group refers to a functional group or atom that can be replaced by another functional group or atom through a substitution reaction (for example, an affinity substitution reaction).
- representative leaving groups include triflate; chlorine, bromine, iodine; sulfonate groups, such as mesylate, tosylate, p-bromobenzenesulfonate, p-toluenesulfonic acid Esters, etc.; acyloxy groups, such as acetoxy, trifluoroacetoxy and the like.
- protecting group includes but is not limited to "amino protecting group", “hydroxy protecting group” or “thiol protecting group”.
- amino protecting group refers to a protecting group suitable for preventing side reactions at the amino nitrogen position.
- Representative amino protecting groups include, but are not limited to: formyl; acyl, such as alkanoyl (such as acetyl, trichloroacetyl or trifluoroacetyl); alkoxycarbonyl, such as tert-butoxycarbonyl (Boc) ; Arylmethyloxycarbonyl, such as benzyloxycarbonyl (Cbz) and 9-fluorenylmethyloxycarbonyl (Fmoc); arylmethyl, such as benzyl (Bn), trityl (Tr), 1,1-di -(4'-Methoxyphenyl)methyl; silyl groups, such as trimethylsilyl (TMS) and tert-butyldimethyls
- hydroxy protecting group refers to a protecting group suitable for preventing side reactions of the hydroxyl group.
- Representative hydroxy protecting groups include but are not limited to: alkyl groups, such as methyl, ethyl, and tert-butyl; acyl groups, such as alkanoyl groups (such as acetyl); arylmethyl groups, such as benzyl (Bn), p-methyl Oxybenzyl (PMB), 9-fluorenylmethyl (Fm) and diphenylmethyl (diphenylmethyl, DPM); silyl groups such as trimethylsilyl (TMS) and tert-butyl Dimethylsilyl (TBS) and so on.
- alkyl groups such as methyl, ethyl, and tert-butyl
- acyl groups such as alkanoyl groups (such as acetyl)
- arylmethyl groups such as benzyl (Bn), p-methyl Oxybenzyl (P
- the compounds of the present invention can be prepared by a variety of synthetic methods well known to those skilled in the art, including the specific embodiments listed below, the embodiments formed by combining them with other chemical synthesis methods, and those well known to those skilled in the art Equivalent alternatives, preferred implementations include but are not limited to the embodiments of the present invention.
- the solvent used in the present invention is commercially available.
- the present invention uses the following abbreviations: aq stands for water; HATU stands for O-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethylurea hexafluorophosphate ; EDCI stands for N-(3-dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride; m-CPBA stands for 3-chloroperoxybenzoic acid; eq stands for equivalent, equivalent amount; CDI stands for Carbonyl diimidazole; DCM stands for dichloromethane; PE stands for petroleum ether; DIAD stands for diisopropyl azodicarboxylate; DMF stands for N,N-dimethylformamide; DMSO stands for dimethyl sulfoxide; EtOAc stands for ethyl acetate Esters; EtOH stands for ethanol; MeOH stands for methanol; C
- the compound of the present invention significantly increases the exposure of the active drug, and the peak blood concentration and the action time are significantly improved; the compound of the present invention exhibits a greater amplitude of lowering intraocular pressure and a longer duration of action for lowering intraocular pressure; In the high intraocular pressure model, the compound of the present invention showed a good blood pressure lowering effect under different test doses, and at the same time, it has a certain dose correlation.
- the blood pressure lowering amplitude and duration of action are better than K-115; At a high dose of 8 mg/mL, the concentration of metabolites was 0.934 ng/mL after 4 hours of administration; the concentration of metabolites was below the detection limit 8 hours after administration, and the system was highly safe.
- the combined organic phases were washed with saturated brine (100 mL ⁇ 1), dried over anhydrous sodium sulfate, and the filtrate obtained by filtration was concentrated under reduced pressure to obtain a residue.
- Water (50 mL) was added to the residue, the pH was adjusted to 10 with an aqueous Na 2 CO 3 solution, and the mixture was extracted with ethyl acetate (50 mL ⁇ 1), and the aqueous phase was collected.
- the pH of the aqueous phase was adjusted to 4 with 1N aqueous hydrochloric acid solution, and extracted with ethyl acetate (50 mL ⁇ 3).
- the combined organic phase was washed with saturated brine (100 mL ⁇ 1), dried over anhydrous sodium sulfate, filtered, and the obtained filtrate was concentrated under reduced pressure to obtain compound 5g.
- the formate of compound 7 can be adjusted to pH 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 7.
- the formate salt of compound 8 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 8.
- the formate of compound 9 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, then extracted with ethyl acetate and concentrated to dryness to obtain compound 9.
- the formate of compound 10 can be added to the system by adding saturated sodium carbonate aqueous solution to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 10.
- the formate of compound 11 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 11.
- the formate of compound 13 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 13.
- the formate of compound 14 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 14.
- the formate of compound 15 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 15.
- the formate of compound 16 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, then extracted with ethyl acetate and concentrated to dryness to obtain compound 16.
- the formate of compound 17 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 17.
- the formate of compound 18 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 18.
- the formate salt of compound 19 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 19.
- the formate of compound 20 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 20.
- the formate salt of compound 21 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 21.
- the formate of compound 22 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 22.
- the formate of compound 23 can be adjusted to pH 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 23.
- the formate of compound 24 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, then extracted with ethyl acetate and concentrated to dryness to obtain compound 24.
- the formate of compound 25 can be adjusted to pH 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 25.
- the formate of compound 26 can be adjusted to pH 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 26.
- the formate of compound 27 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 27.
- the formate salt of compound 28 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 28.
- the formate salt of compound 29 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 29.
- the formate of compound 30 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 30.
- Trifluoroacetic acid (1.0 mL) was added to the microwave tube, compound 31a (79 mg, 143 ⁇ mol) was added to the microwave tube, and microwaved at 60 degrees Celsius for 1 hour. The solvent was removed by concentration under reduced pressure, and the crude product was purified by high performance liquid chromatography (acidic, formic acid system) to obtain the formate of compound 31.
- the formate of compound 31 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 31.
- the formate of compound 32 can be adjusted to pH 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 32.
- the formate of compound 33 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 33.
- the formate of compound 34 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 34.
- the formate of compound 35 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 35.
- the formate of compound 36 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 36.
- the formate of compound 37 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, then extracted with ethyl acetate and concentrated to dryness to obtain compound 37.
- the formate salt of compound 38 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 38.
- the formate of compound 39 can be adjusted to pH 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 39.
- the formate of compound 40 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 40.
- the formate of compound 41 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 41.
- the formate of compound 42 can be adjusted to pH 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 42.
- the formate of compound 43 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 43.
- the formate of compound 44 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 44.
- the formate of compound 45 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, then extracted with ethyl acetate and concentrated to dryness to obtain compound 45.
- the formate of compound 46 can be adjusted to pH 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 46.
- the formate of compound 47 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 47.
- the formate of compound 48 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 48.
- the formate of compound 49 can be adjusted to pH 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 49.
- the formate of compound 50 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 50.
- the formate of compound 51 can be adjusted to pH 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 51.
- the formate of compound 52 can be added to the system by adding saturated sodium carbonate aqueous solution to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 52.
- the formate salt of compound 53 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 53.
- the formate of compound 54 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 54.
- the hydrochloride salt of compound 56 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 56.
- the formate of compound 57 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, then extracted with ethyl acetate and concentrated to dryness to obtain compound 57.
- the formate of compound 59 can be adjusted to pH 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 59.
- the formate of compound 61 can be adjusted to pH 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain compound 61.
- the formate of compound 62 can be adjusted by adding saturated sodium carbonate aqueous solution to the system to adjust the pH of the reaction solution to 8-9, then extracted with ethyl acetate and concentrated to dryness to obtain compound 62.
- control compound 1 can be adjusted to 8-9 by adding saturated sodium carbonate aqueous solution to the system, and then extracted with ethyl acetate and concentrated to dryness to obtain control compound 1.
- the compound is a prodrug molecule containing an ester functional group, which can be hydrolyzed into an active drug molecule (parent drug) by the action of abundant ester hydrolase in the eye tissue during eye drops administration.
- This experiment detects the speed of the compound producing active pharmaceutical ingredients in the body and the exposure of active pharmaceutical ingredients.
- the solvent used was 1.2% hydroxypropyl methylcellulose E5/20.5% poloxamer P407/1.6% poloxamer P188.
- the dosage of eye drops is 0.5 mg/eye, and both eyes are administered with eye drops.
- Aqueous humor was collected 0.25h, 0.5h, 2h, 4h, 8h, 24h after administration, and aqueous humor samples were prepared. All samples used liquid chromatography coupled with mass spectrometry to quantitatively detect the content of the administered compound in the aqueous humor of experimental animals.
- the measured concentration value used WinNonlin non-compartmental model to calculate the half-life based on the aqueous humor concentration-time data. Peak concentration of water medicine, peak time of aqueous medicine, unit exposure and other parameters.
- test compound prodrug molecule
- active metabolite parent drug molecule
- the compound of the present invention is compared with the control compound 1 and the control compound. 2.
- the exposure of active drugs is significantly increased, and at the same time, the peak blood concentration and action time are significantly increased.
- Rabbits with normal intraocular pressure were used to screen the potential compounds for lowering intraocular pressure through eye drops.
- the compound of the present invention exhibits a greater amplitude of lowering intraocular pressure and a longer duration of lowering intraocular pressure.
- compound 60 at the concentration of 2.5, 5, and 10 mg/mL the intraocular pressure of the animals dropped extremely significantly.
- Example 3 Test of lowering intraocular pressure in New Zealand rabbits with acute ocular hypertension
- the anterior chamber injection of viscoelastics was used to induce acute ocular hypertension in rabbits, and the intraocular pressure-lowering effects of compound 60 and compound 63 at different concentrations were investigated through eye drops.
- the animals in each group were injected with medical sodium hyaluronate gel once in the anterior chamber of the right eye, 100 ⁇ L/eye, to induce the animals to produce high intraocular pressure. 5-15 minutes, 3 and 6 hours after the right eye was modeled, both eyes were given the vehicle, K-115 or the test product (compound 60 at different concentrations), the volume of administration was 50 ⁇ L/eye, before administration, The animal's intraocular pressure was measured 1, 2, 4, 6, 8 and 10 hours after the drug.
- Table 3 The experimental results are shown in Table 3:
- the compound of the present invention showed a good blood pressure lowering effect at different test doses, and at the same time, it has a certain dose correlation.
- the blood pressure reduction amplitude and duration of action are better than K-115.
- a single administration of compound 63 showed better efficacy (highest antihypertensive effect and duration of action) at all tested doses (0.5-8.0 mg/mL), which was significantly better than K-115.
- compound 63 can sustain a significant antihypertensive effect at a dose of 0.5 mg/mL, and is still significantly better than K-115 in the evaluation of the peak (Cmax) antihypertensive effect.
- Evaluation of eye irritation response add the maximum scores of cornea, iris, conjunctiva, edema and secretions to obtain the total score of eye irritation symptoms for each animal eye at each time point.
- scores of eye irritation symptoms calculate the mean value of points for each observation time point and each group of animals, and determine the degree of eye irritation at each time point and each group of animals according to the following table.
- Fluorescein sodium check After each eye irritation test, use a hand-held slit lamp to perform a fluorescein sodium check.
- the scoring standards are as follows:
- the total score of eye irritation response in each group at each time point is less than 3, and the standard classification is non-irritating.
- the fluorescein sodium examination scores of the eyes treated with normal saline, vehicle, K-115 and compound 63 of each group of animals were all lower than 1.
- staining with a corneal fluorescence staining score of 1 appeared at each treatment and individual time point, which was considered as physiological staining.
- K-115 was instilled in eyes at a concentration of 4 mg/mL for 14 consecutive days, 50 ⁇ l/eye/day, without irritation.
- Compound 63 was instilled in the eye at a concentration range of 0.25-4 mg/mL for 14 consecutive days, 50 ⁇ l/eye/day, without irritation.
- BQL means lower than the detection limit.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Ophthalmology & Optometry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description

















































































































































































| 眼刺激反应 | 分值 |
| 角膜 |
| 眼刺激反应 | 分值 |
| 无混浊 | 0 |
| 散在或弥漫性混浊,虹膜清晰可见 | 1 |
| 半透明区易分辨,虹膜模糊不清 | 2 |
| 出现灰白色半透明区,虹膜细节不清,瞳孔大小勉强可见 | 3 |
| 角膜不透明,虹膜无法辨认 | 4 |
| 虹膜 | |
| 正常 | 0 |
| 皱褶明显加深、充血、肿胀,角膜周围轻度充血,瞳孔对光仍有反应 | 1 |
| 出血/肉眼可见坏死/对光无反应(或其中一种) | 2 |
| 结膜 | |
| 充血(指睑结膜和球结膜) | |
| 血管正常 | 0 |
| 血管充血呈鲜红色 | 1 |
| 血管充血呈深红色,血管不易分辨 | 2 |
| 弥漫性充血呈紫红色 | 3 |
| 水肿 | |
| 无水肿 | 0 |
| 轻微水肿(含眼睑) | 1 |
| 明显水肿伴部分眼睑外翻 | 2 |
| 水肿至眼睑近半闭合 | 3 |
| 水肿至眼睑超过半闭合 | 4 |
| 分泌物 | |
| 无分泌物 | 0 |
| 少量分泌物 | 1 |
| 分泌物使眼睑和睫毛潮湿或粘着 | 2 |
| 分泌物使整个眼区潮湿或粘着 | 3 |
| 最大总积分 | 16 |
| 分值 | 评价 |
| 0-3 | 无刺激性 |
| 4-8 | 轻度刺激性 |
| 9-12 | 中度刺激性 |
| 13-16 | 重度刺激性 |


Claims (25)
- 式(I)所示化合物、其异构体或其药学上可接受的盐,
 其中,T 1选自-(CH 2) n-;T 2选自-(CH 2) m-和-C(R 7)(R 8)-;R 1选自C 1-16烷基、苯基、C 3-7环烷基、3-8元杂环烷基和5-10元杂芳基,所述C 1-16烷基、苯基、C 3-7环烷基、3-8元杂环烷基和5-10元杂芳基分别独立地任选被1、2或3个R a取代;R 2和R 3分别独立地选自H、F、Cl、Br、I、OH、NH 2、CN和C 1-3烷基;R 4选自H、F、Cl、Br、I、OH、NH 2、CN和任选被1、2或3个R b取代C 1-3烷基;R 5选自NR 9R 10;R 6选自H、F、Cl、Br、I、OH、NH 2、CN和C 1-3烷基;R 7和R 8分别独立地选自H、F、Cl、Br、I、OH、NH 2、CN和任选被1、2或3个R c取代的C 1-3烷基;或者,R 7、R 8和与它们相连的原子共同构成任选被1、2或3个R d取代的C 3-5环烷基;R 9和R 10分别独立地选自H和任选被1、2或3个R e取代C 1-3烷基;L选自单键、-O-和-NR 11-;R 11自H和C 1-3烷基;n选自0、1和2;m选自0、1、2和3;R a选自F、Cl、Br、I、OH、NH 2、CN、C 1-3烷基和C 1-3烷氧基,所述C 1-3烷基和C 1-3烷氧基任选被1、2或3个R取代;R b、R c、R d和R e分别独立地选自H、F、Cl、Br、I、OH、NH 2、CN和C 1-3烷基;R选自F、Cl、Br、I、OH、NH 2、CN和CH 3;所述3-8元杂环烷基和5-10元杂芳基分别独立地包含1、2、3或4个独立选自-NH-、-O-、-S-和N的杂原子或杂原子团。
其中,T 1选自-(CH 2) n-;T 2选自-(CH 2) m-和-C(R 7)(R 8)-;R 1选自C 1-16烷基、苯基、C 3-7环烷基、3-8元杂环烷基和5-10元杂芳基,所述C 1-16烷基、苯基、C 3-7环烷基、3-8元杂环烷基和5-10元杂芳基分别独立地任选被1、2或3个R a取代;R 2和R 3分别独立地选自H、F、Cl、Br、I、OH、NH 2、CN和C 1-3烷基;R 4选自H、F、Cl、Br、I、OH、NH 2、CN和任选被1、2或3个R b取代C 1-3烷基;R 5选自NR 9R 10;R 6选自H、F、Cl、Br、I、OH、NH 2、CN和C 1-3烷基;R 7和R 8分别独立地选自H、F、Cl、Br、I、OH、NH 2、CN和任选被1、2或3个R c取代的C 1-3烷基;或者,R 7、R 8和与它们相连的原子共同构成任选被1、2或3个R d取代的C 3-5环烷基;R 9和R 10分别独立地选自H和任选被1、2或3个R e取代C 1-3烷基;L选自单键、-O-和-NR 11-;R 11自H和C 1-3烷基;n选自0、1和2;m选自0、1、2和3;R a选自F、Cl、Br、I、OH、NH 2、CN、C 1-3烷基和C 1-3烷氧基,所述C 1-3烷基和C 1-3烷氧基任选被1、2或3个R取代;R b、R c、R d和R e分别独立地选自H、F、Cl、Br、I、OH、NH 2、CN和C 1-3烷基;R选自F、Cl、Br、I、OH、NH 2、CN和CH 3;所述3-8元杂环烷基和5-10元杂芳基分别独立地包含1、2、3或4个独立选自-NH-、-O-、-S-和N的杂原子或杂原子团。 - 根据权利要求1所述化合物、其异构体或其药学上可接受的盐,其中,R a选自F、Cl、Br、I、OH、NH 2、CN、CH 3、CF 3、CH 2F、CHF 2、CH 2CH 3和OCH 3。
- 根据权利要求1所述化合物、其异构体或其药学上可接受的盐,其中,R 1选自C 1-12烷基、苯基、环丁烷基、环戊烷基、环己烷基、四氢呋喃基、四氢吡喃基、哌啶基、噻吩基、呋喃基、吡咯基和苯并呋喃基,所述C 1-12烷基、苯基、环丁烷基、环戊烷基、环己烷基、四氢呋喃基、四氢吡喃基、哌啶基、噻吩基、呋喃基、吡咯基和苯并呋喃基分别独立地任选被1、2或3个R a取代。
- 根据权利要求3所述化合物、其异构体或其药学上可接受的盐,其中,R 1选自CH 3、CH 2CH 3、(CH 2) 2CH 3、(CH 2) 3CH 3、(CH 2) 4CH 3、(CH 2) 5CH 3、(CH 2) 6CH 3、(CH 2) 10CH 3、CH(CH 3) 2、C(CH 3) 3、


- 根据权利要求2或4所述化合物、其异构体或其药学上可接受的盐,其中,R 1选自CH 3、CH 2CH 3、(CH 2) 2CH 3、(CH 2) 3CH 3、(CH 2) 4CH 3、(CH 2) 5CH 3、(CH 2) 6CH 3、(CH 2) 10CH 3、CH(CH 3) 2、C(CH 3) 3、


- 根据权利要求1所述化合物、其异构体或其药学上可接受的盐,其中,R 2和R 3分别独立地选自H、F、Cl、Br、I、OH和NH 2。
- 根据权利要求1所述化合物、其异构体或其药学上可接受的盐,其中,R 4选自H、F、Cl、Br、I、OH、NH 2、CN、CH 3和CH 2CH 3。
- 根据权利要求1所述化合物、其异构体或其药学上可接受的盐,其中,R 9和R 10分别独立地选自H、CH 3和CH 2CH 3。
- 根据权利要求1或8所述化合物、其异构体或其药学上可接受的盐,其中,R 5选自NH 2、NH(CH 3)和N(CH 3) 2。
- 根据权利要求1所述化合物、其异构体或其药学上可接受的盐,其中,R 6选自H、F、Cl、Br、I、OH、NH 2、CN和CH 3。
- 根据权利要求1所述化合物、其异构体或其药学上可接受的盐,其中,R 7和R 8分别独立地选自H、F、Cl、Br、I、OH、NH 2、CN和CH 3。
- 根据权利要求1所述化合物、其异构体或其药学上可接受的盐,其中,R 7、R 8和与它们相连的原子共同构成任选被1、2或3个R d取代的环丙烷基。
- 根据权利要求12所述化合物、其异构体或其药学上可接受的盐,其中,R 7、R 8和与它们相连的原子共同构成环丙烷基。
- 根据权利要求1所述化合物、其异构体或其药学上可接受的盐,其中,L选自单键、-O-、-NH-和-N(CH 3)-。
- 根据权利要求1、12或13任意一项所述化合物、其异构体或其药学上可接受的盐,其中,T 2选自-CH 2-、-(CH 2) 2-和

- 根据权利要求1所述化合物、其异构体或其药学上可接受的盐,其中,结构单元选自


- 根据权利要求1或16所述化合物、其异构体或其药学上可接受的盐,其中,结构单元选自


- 根据权利要求1所述化合物、其异构体或其药学上可接受的盐,其中,结构单元选自CH 3、CH 2CH 3、(CH 2) 2CH 3、(CH 2) 3CH 3、(CH 2) 4CH 3、(CH 2) 5CH 3、(CH 2) 6CH 3、(CH 2) 10CH 3、CH(CH 3) 2、C(CH 3) 3、OCH 3、OCH 2CH 3、O(CH 2) 2CH 3、O(CH 2) 3CH 3、O(CH 2) 4CH 3、O(CH 2) 5CH 3、O(CH 2) 6CH 3、OCH(CH 3) 2、OC(CH 3) 3、N(CH 3) 2、
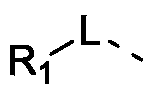


- 根据权利要求1、3~5、7、9或14任意一项所述化合物、其异构体或其药学上可接受的盐,其选自
 其中,R 1如权利要求1、3、4或5所定义;R 4如权利要求1或7所定义;R 5如权利要求1或9所定义;L如权利要求1或14所定义。
其中,R 1如权利要求1、3、4或5所定义;R 4如权利要求1或7所定义;R 5如权利要求1或9所定义;L如权利要求1或14所定义。 - 根据权利要求19所述化合物、其异构体或其药学上可接受的盐,其选自
 其中,R 1、R 4和L如权利要求19所定义。
其中,R 1、R 4和L如权利要求19所定义。 - 下式所示化合物、其异构体或其药学上可接受的盐,



- 根据权利要求21所述化合物、其异构体或其药学上可接受的盐,其选自






- 一种药物组合物,包括作为活性成分的治疗有效量的根据权利要求1~22任意一项所述的化合物、其异构体或其药学上可接受的盐以及药学上可接受的载体。
- 根据权利要求1~22任意一项所述的化合物、其异构体或其药学上可接受的盐或者权利要求23所述的组合物在制备ROCK蛋白激酶抑制剂相关药物上的应用。
- 根据权利要求24所述的应用,其特征在于,所述ROCK蛋白激酶抑制剂相关药物是用于治疗青光眼或高眼压症的药物。
Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US17/621,502 US20230024516A1 (en) | 2019-06-21 | 2020-06-22 | Isoquinolinone derivatives serving as rock protein kinase inhibitors and use thereof |
| CN202080041488.6A CN113906017B (zh) | 2019-06-21 | 2020-06-22 | 作为rock蛋白激酶抑制剂的异喹啉酮的衍生物及其应用 |
| EP20827811.9A EP3988544A4 (en) | 2019-06-21 | 2020-06-22 | ISOQUINOLINONE DERIVATIVES AS ROCK PROTEIN KINASE INHIBITORS AND THEIR USE |
| JP2021576289A JP7398137B2 (ja) | 2019-06-21 | 2020-06-22 | Rockプロテインキナーゼ阻害薬としてのイソキノリンの誘導体及びその使用 |
| AU2020296050A AU2020296050B2 (en) | 2019-06-21 | 2020-06-22 | Isoquinolinone derivatives serving as ROCK protein kinase inhibitors and use thereof |
| MX2021016068A MX2021016068A (es) | 2019-06-21 | 2020-06-22 | Derivados de isoquinolinona como inhibidores de la proteina quinasa rock y usos del mismo. |
| KR1020227002287A KR20220024868A (ko) | 2019-06-21 | 2020-06-22 | Rock단백질 키나아제 억제제인 이소퀴놀리논의 유도체 및 그 응용 |
| CA3144211A CA3144211A1 (en) | 2019-06-21 | 2020-06-22 | Isoquinolinone derivatives serving as rock protein kinase inhibitors and uses thereof |
| ZA2022/00834A ZA202200834B (en) | 2019-06-21 | 2022-01-18 | Isoquinolinone derivatives serving as rock protein kinase inhibitors and use thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201910544202.5 | 2019-06-21 | ||
| CN201910544202 | 2019-06-21 | ||
| CN201911078066 | 2019-11-06 | ||
| CN201911078066.1 | 2019-11-06 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020253882A1 true WO2020253882A1 (zh) | 2020-12-24 |
Family
ID=74040659
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2020/097503 WO2020253882A1 (zh) | 2019-06-21 | 2020-06-22 | 作为rock蛋白激酶抑制剂的异喹啉酮的衍生物及其应用 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20230024516A1 (zh) |
| EP (1) | EP3988544A4 (zh) |
| JP (1) | JP7398137B2 (zh) |
| KR (1) | KR20220024868A (zh) |
| CN (1) | CN113906017B (zh) |
| AU (1) | AU2020296050B2 (zh) |
| CA (1) | CA3144211A1 (zh) |
| MX (1) | MX2021016068A (zh) |
| WO (1) | WO2020253882A1 (zh) |
| ZA (1) | ZA202200834B (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022135421A1 (zh) * | 2020-12-21 | 2022-06-30 | 广州润尔眼科生物科技有限公司 | 一种作为rock蛋白激酶抑制剂的异喹啉酮型化合物的盐型及其制备方法 |
| WO2023241652A1 (zh) * | 2022-06-16 | 2023-12-21 | 广州润尔眼科生物科技有限公司 | 一种药物组合物及其制备方法和应用 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004106325A1 (en) * | 2003-05-29 | 2004-12-09 | Schering Aktiengesellschaft | Prodrugs of 1-(1-hydroxy-5-isoquinolinesulfonyl)homopiperazine |
| WO2006057397A1 (ja) | 2004-11-29 | 2006-06-01 | Kowa Co., Ltd. | (s)-(-)-1-(4-フルオロイソキノリン-5-イル)スルホニル-2-メチル-1,4-ホモピペラジン塩酸塩・二水和物 |
| WO2007026664A1 (ja) | 2005-08-30 | 2007-03-08 | Asahi Kasei Pharma Corporation | スルホンアミド化合物 |
| CN101622243A (zh) * | 2007-02-28 | 2010-01-06 | 旭化成制药株式会社 | 磺酰胺衍生物 |
| CN105085478A (zh) * | 2014-04-28 | 2015-11-25 | 南京明德新药研发股份有限公司 | 异喹啉磺胺衍生物及其药物组合物和制药用途 |
| CN105085525A (zh) * | 2014-04-28 | 2015-11-25 | 南京明德新药研发股份有限公司 | 作为rho激酶抑制剂的异喹啉磺酰衍生物 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0667926B2 (ja) * | 1985-11-12 | 1994-08-31 | 旭化成工業株式会社 | 環状のイソキノリンスルホンアミド誘導体 |
| JP3906284B2 (ja) | 1995-04-14 | 2007-04-18 | 株式会社東芝 | 光ディスクからビデオ及びオーディオを再生する再生システム、その再生方法及び光ディスクの記録方法 |
| JP2006290827A (ja) * | 2005-04-13 | 2006-10-26 | Kowa Co | 緑内障予防又は治療剤 |
| JP4831826B2 (ja) | 2006-10-23 | 2011-12-07 | 日本プラスト株式会社 | ベンチレータ及びベンチレータ装置 |
| GB201107223D0 (en) * | 2011-04-29 | 2011-06-15 | Amakem Nv | Novel rock inhibitors |
| ES2774923T3 (es) * | 2014-04-28 | 2020-07-23 | China Resources Pharmaceutical Holdings Company Ltd | Derivados de isoquinolinsulfonilo como inhibidores de RHO quinasa |
| CN114644618A (zh) * | 2020-12-21 | 2022-06-21 | 广州润尔眼科生物科技有限公司 | 一种作为rock蛋白激酶抑制剂的异喹啉酮型化合物的盐型及其制备方法 |
-
2020
- 2020-06-22 CN CN202080041488.6A patent/CN113906017B/zh active Active
- 2020-06-22 WO PCT/CN2020/097503 patent/WO2020253882A1/zh unknown
- 2020-06-22 KR KR1020227002287A patent/KR20220024868A/ko not_active Application Discontinuation
- 2020-06-22 CA CA3144211A patent/CA3144211A1/en active Pending
- 2020-06-22 EP EP20827811.9A patent/EP3988544A4/en active Pending
- 2020-06-22 US US17/621,502 patent/US20230024516A1/en active Pending
- 2020-06-22 AU AU2020296050A patent/AU2020296050B2/en active Active
- 2020-06-22 MX MX2021016068A patent/MX2021016068A/es unknown
- 2020-06-22 JP JP2021576289A patent/JP7398137B2/ja active Active
-
2022
- 2022-01-18 ZA ZA2022/00834A patent/ZA202200834B/en unknown
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004106325A1 (en) * | 2003-05-29 | 2004-12-09 | Schering Aktiengesellschaft | Prodrugs of 1-(1-hydroxy-5-isoquinolinesulfonyl)homopiperazine |
| WO2006057397A1 (ja) | 2004-11-29 | 2006-06-01 | Kowa Co., Ltd. | (s)-(-)-1-(4-フルオロイソキノリン-5-イル)スルホニル-2-メチル-1,4-ホモピペラジン塩酸塩・二水和物 |
| WO2007026664A1 (ja) | 2005-08-30 | 2007-03-08 | Asahi Kasei Pharma Corporation | スルホンアミド化合物 |
| CN101253166A (zh) * | 2005-08-30 | 2008-08-27 | 旭化成制药株式会社 | 磺酰胺化合物 |
| CN101622243A (zh) * | 2007-02-28 | 2010-01-06 | 旭化成制药株式会社 | 磺酰胺衍生物 |
| CN105085478A (zh) * | 2014-04-28 | 2015-11-25 | 南京明德新药研发股份有限公司 | 异喹啉磺胺衍生物及其药物组合物和制药用途 |
| CN105085525A (zh) * | 2014-04-28 | 2015-11-25 | 南京明德新药研发股份有限公司 | 作为rho激酶抑制剂的异喹啉磺酰衍生物 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022135421A1 (zh) * | 2020-12-21 | 2022-06-30 | 广州润尔眼科生物科技有限公司 | 一种作为rock蛋白激酶抑制剂的异喹啉酮型化合物的盐型及其制备方法 |
| AU2021408195B2 (en) * | 2020-12-21 | 2024-05-02 | Guangzhou Ocusun Ophthalmic Biotechnology Co., Ltd. | Salt form of isoquinolinone type compound as rock protein kinase inhibitor and preparation method therefor |
| WO2023241652A1 (zh) * | 2022-06-16 | 2023-12-21 | 广州润尔眼科生物科技有限公司 | 一种药物组合物及其制备方法和应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2022538086A (ja) | 2022-08-31 |
| CN113906017A (zh) | 2022-01-07 |
| AU2020296050A1 (en) | 2022-02-03 |
| JP7398137B2 (ja) | 2023-12-14 |
| EP3988544A1 (en) | 2022-04-27 |
| CN113906017B (zh) | 2024-07-02 |
| MX2021016068A (es) | 2022-04-20 |
| US20230024516A1 (en) | 2023-01-26 |
| EP3988544A4 (en) | 2023-08-09 |
| ZA202200834B (en) | 2022-10-26 |
| KR20220024868A (ko) | 2022-03-03 |
| CA3144211A1 (en) | 2020-12-24 |
| AU2020296050B2 (en) | 2023-04-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN103562186B (zh) | 作为toll样受体调节剂的取代的苯并氮杂卓 | |
| WO2018137683A1 (zh) | 羊毛甾醇前药化合物及其制备方法和应用 | |
| CN103547577B (zh) | 作为盐皮质激素受体拮抗剂的并环类化合物 | |
| CN109641836A (zh) | 氟化的2-氨基-4-(取代的氨基)苯基氨基甲酸酯衍生物 | |
| TW200911839A (en) | New cyclic peptide compounds | |
| WO2020253882A1 (zh) | 作为rock蛋白激酶抑制剂的异喹啉酮的衍生物及其应用 | |
| JP2022511999A (ja) | Mtorcモジュレーターおよびその使用 | |
| WO2021031999A1 (zh) | 麦考酚酸的前体药物及其制备方法 | |
| EP2488032B1 (en) | Prostaglandin transporter inhibitors and uses thereof | |
| CN108289893A (zh) | 人类血浆激肽释放酶抑制剂 | |
| WO2022135421A1 (zh) | 一种作为rock蛋白激酶抑制剂的异喹啉酮型化合物的盐型及其制备方法 | |
| JP2020536922A (ja) | 眼疾患を治療するためのアザビシクロ及びジアゼピン誘導体 | |
| JP7245397B2 (ja) | 複素環化合物 | |
| TW202321192A (zh) | 化合物、異構體或其藥學上可接受的鹽 | |
| JP2024523552A (ja) | 疾患の処置のためのベータ-ラクタム誘導体 | |
| JP2024517496A (ja) | Rock阻害剤の酸付加塩及び塩の結晶形、組成物並びに薬物用途 | |
| WO2020216240A1 (zh) | 用作血浆激肽释放酶抑制剂的双环烷类化合物 | |
| JP2003146972A (ja) | カルボスチリル誘導体 | |
| AU2021296681A1 (en) | Novel compound, and pharmaceutical composition for preventing or treating resistant cancer, comprising same | |
| WO2021233133A1 (zh) | 用作ret激酶抑制剂的化合物及其应用 | |
| JP7495758B2 (ja) | マクロライド化合物及びその慢性呼吸器疾患の治療用途 | |
| WO2023051753A1 (zh) | 一种rock抑制剂的制备方法、其中间体及中间体的制备方法 | |
| WO2021098817A1 (zh) | 二氧代哌嗪类衍生物、其制备方法及其在医药上的应用 | |
| CN115677830A (zh) | 肽酰胺类化合物及其制备方法与应用 | |
| WO2024051795A1 (zh) | 用作泛素-特异性蛋白酶抑制剂的取代嘌呤酮衍生物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20827811 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3144211 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2021576289 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20227002287 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2020296050 Country of ref document: AU Date of ref document: 20200622 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2020827811 Country of ref document: EP Effective date: 20220121 |