WO2019206120A1 - 甲酰胺类化合物、其制备方法及其应用 - Google Patents
甲酰胺类化合物、其制备方法及其应用 Download PDFInfo
- Publication number
- WO2019206120A1 WO2019206120A1 PCT/CN2019/083829 CN2019083829W WO2019206120A1 WO 2019206120 A1 WO2019206120 A1 WO 2019206120A1 CN 2019083829 W CN2019083829 W CN 2019083829W WO 2019206120 A1 WO2019206120 A1 WO 2019206120A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ring
- alkyl
- group
- cycloalkyl
- compound
- Prior art date
Links
- 0 *C(c1cc(NC(C2CC2)=O)c(CC=C)cn1)=O Chemical compound *C(c1cc(NC(C2CC2)=O)c(CC=C)cn1)=O 0.000 description 7
- QOBZBXGSEOLUOE-UHFFFAOYSA-N Cc(cc(c(C(O)=O)c1)F)c1NC(C1CC1)=O Chemical compound Cc(cc(c(C(O)=O)c1)F)c1NC(C1CC1)=O QOBZBXGSEOLUOE-UHFFFAOYSA-N 0.000 description 1
- ZOOSILUVXHVRJE-UHFFFAOYSA-N O=C(C1CC1)Cl Chemical compound O=C(C1CC1)Cl ZOOSILUVXHVRJE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to the field of pharmaceutical technology, and relates to a carboxamide compound capable of inhibiting ASK1 kinase activity, and to a process for preparing the compound, a pharmaceutical composition comprising the compound as an active ingredient, and a pharmaceutical application thereof.
- the compounds of the present invention are useful as inhibitors of ASK1 kinases for the treatment/prevention of diseases associated with the target, such as inflammatory diseases, metabolic diseases, autoimmune diseases, cardiovascular diseases, neurodegenerative diseases, Cancer and other diseases.
- MAPKs Mitogen-activated protein kinases
- MAP3K MEK kinase
- MAPK2K MAPK kinase
- MAPK MAP kinase
- Apoptosis signal-regulating kinase 1 is a member of the MAP3K family.
- ASK1 is capable of oxidative stress, reactive oxygen species (ROS), lipopolysaccharide (LPS), and tumor necrosis factor (TNF-).
- ROS reactive oxygen species
- LPS lipopolysaccharide
- TNF- tumor necrosis factor
- Various stimuli such as ⁇ ), endoplasmic reticulum (ER) stress, osmotic pressure, inflammation, etc. are first activated, thereby activating MAP2K, and MAP2K is phosphorylated to activate MAPK, such as c-Jun amino terminal protein kinase (c-JunN) -terminal protein kinase, JNK) and p38MAPK.
- c-JunN c-Jun amino terminal protein kinase
- JNK p38MAPK
- ASK1 plays a key role in a variety of cell biology processes, including apoptosis, differentiation, and inflammation (Soga M., Matsuzawa A., Ichijo H., 2012, Int. J. Cell Biol., 2012). :1-5).
- ASK1 activation of ASK1 plays an important role in many diseases, such as inflammatory diseases, metabolic diseases, autoimmune diseases, cardiovascular diseases, neurodegenerative diseases, cancer and other diseases (Soga M., Matsuzawa A. , Ichijo H., 2012, Int. J. Cell Biol., 2012: 1-5; Hayakawa R., Hayakawa T., Takeda K., et al, 2012, Proc. Jpn. Acad. Ser. BPhys. Biol. Sci., 88: 434-453). Therefore, it has been found that a pharmaceutically active molecule having an activity of inhibiting ASK1 will bring significant benefits to patients suffering from the above diseases.
- ASK1 inhibitor patent applications include triazolopyridines WO2009027283, pyrazolo[1,5-A]pyrimidines WO2011041293, aromatic/aromatic heterocyclic amines WO2011008709, heterocyclic amines US20120004267 and thiazolamide Class US20170173031 and so on.
- the activation of ASK1 is associated with a variety of diseases, and its inhibitors have important clinical value and good application prospects as medicines in the field of medicine, but there are currently no approved drugs listed in the world. Therefore, we expect to develop new ASK1 inhibitors to meet unmet clinical needs.
- the present invention provides a novel cycloalkylcarboxamide ASK1 inhibitor for treating/preventing diseases associated with the target, such as inflammatory diseases, metabolic diseases, autoimmune diseases, cardiovascular diseases, Neurodegenerative diseases, cancer and other diseases.
- diseases associated with the target such as inflammatory diseases, metabolic diseases, autoimmune diseases, cardiovascular diseases, Neurodegenerative diseases, cancer and other diseases.
- these compounds or pharmaceutical compositions containing them as active ingredients can maximize the clinical efficacy of these diseases within a safe therapeutic window.
- the invention relates to a cycloalkyl carboxamide compound which inhibits ASK1 kinase activity of Formula I, including pharmaceutically acceptable salts, hydrates, other solvates, stereoisomers thereof and the like Derivatives such as medicines.
- Another aspect of the invention relates to a process for the preparation of the compounds described herein.
- a further aspect of the invention relates to a pharmaceutical composition comprising the compound of the invention as an active ingredient, and to the clinical use of a compound or pharmaceutical composition of the invention for the treatment/prevention of a disease associated with ASK1 kinase, and a compound or combination of the invention Use in the preparation of a medicament for the treatment and/or prevention of a disease associated with ASK1 kinase.
- the present invention also relates to a method of treating and/or preventing a disease associated with ASK1 kinase comprising administering a compound or pharmaceutical composition of the present invention to a subject in need thereof.
- the present invention relates to compounds of formula I, including prodrugs, stereoisomers, pharmaceutically acceptable salts, hydrates or other solvates thereof,
- R 1 is one or more substituents independently selected from the same or different H, halogen, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, NH 2 , COOH, C 1 -C 4 Alkylamino, C 1 -C 4 alkyloxy and Ar 1 ;
- Ar 1 is selected from the group consisting of a benzene ring and a pyridine ring, and the benzene ring and the pyridine ring may be substituted by one or more substituents, which are independently selected from the same or different H, halogen, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, NH 2 , C 1 -C 4 alkylamino and C 1 -C 4 alkyloxy;
- R 2 is one or more substituents independently selected from the same or different H, halogen, CN, C 1 -C 4 alkyl and C 1 -C 4 haloalkyl;
- R 3 is C 1 -C 4 alkyl, C 3 -C 6 cycloalkyl, C 1 -C 4 haloalkyl, halo C 3 -C 6 cycloalkyl, cyano substituted C 1 -C 4 alkyl, a C 3 -C 6 heterocycloalkyl group, a hydroxy-substituted C 1 -C 4 alkyl group or a C 1 -C 4 alkoxy-substituted C 1 -C 4 alkyl group;
- X is selected from C and N;
- A is selected from the group consisting of C 3 -C 7 cycloalkyl and C 3 -C 7 heterocycloalkyl;
- a benzene ring is selected from the group consisting of a benzene ring, a pyridine ring, a thiazole ring, a furan ring, a thiophene ring, a pyrrole ring, a pyrazole ring, an oxazole ring, an isoxazole ring and a quinoline ring, and the aromatic ring may be one or more Substituted by a substituent which is independently selected from the same or different H, halogen, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, NH 2 , C 1 -C 4 alkylamino and C 1 -C 4 alkyl group;
- n is an integer from 1 to 5;
- n is an integer from 1 to 4.
- the invention relates to a compound of formula I, including prodrugs, stereoisomers, pharmaceutically acceptable salts, hydrates or other solvates thereof, wherein
- R 1 is one or more substituents independently selected from the same or different H, halogen, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, NH 2 , COOH, C 1 -C 4 Alkylamino, C 1 -C 4 alkyloxy and Ar 1 ;
- Ar 1 is selected from the group consisting of a benzene ring and a pyridine ring, and the benzene ring and the pyridine ring may be substituted by one or more substituents independently selected from the same or different H, halogen, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, NH 2 , C 1 -C 4 alkylamino and C 1 -C 4 alkyloxy;
- R 2 is one or more substituents independently selected from the same or different H, halogen, CN and C 1 -C 4 alkyl;
- R 3 is C 1 -C 4 alkyl, C 3 -C 6 cycloalkyl, C 1 -C 4 haloalkyl, halo C 3 -C 6 cycloalkyl, cyano substituted C 1 -C 4 alkyl, a C 3 -C 6 heterocycloalkyl group, a hydroxy-substituted C 1 -C 4 alkyl group and a C 1 -C 4 alkoxy-substituted C 1 -C 4 alkyl group;
- X is selected from C and N;
- A is selected from the group consisting of C 3 -C 7 cycloalkyl and C 3 -C 7 heterocycloalkyl;
- B is an aromatic ring, preferably selected from the group consisting of a benzene ring, a pyridine ring, a thiazole ring, a furan ring, a thiophene ring, a pyrrole ring, a pyrazole ring, an oxazole ring, an isoxazole ring, and a quinoline ring, and the aromatic ring can be one Or substituted with a plurality of substituents independently selected from the same or different H, halogen, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, NH 2 , C 1 -C 4 alkane a base amino group and a C 1 -C 4 alkyloxy group;
- n is an integer from 1 to 5;
- n is an integer from 1 to 4.
- the invention relates to a compound of formula I, including prodrugs, stereoisomers, pharmaceutically acceptable salts, hydrates or other solvates thereof, wherein
- R 1 is one or more substituents independently selected from the same or different H, halogen, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, NH 2 , COOH, C 1 -C 4 An alkylamino group and a C 1 -C 4 alkyloxy group;
- R 2 is one or more substituents independently selected from the same or different H, halogen, CN and C 1 -C 4 alkyl;
- R 3 is C 1 -C 4 alkyl, C 3 -C 6 cycloalkyl, C 1 -C 4 haloalkyl, halo C 3 -C 6 cycloalkyl, cyano substituted C 1 -C 4 alkyl, a C 3 -C 6 heterocycloalkyl group, a hydroxy-substituted C 1 -C 4 alkyl group and a C 1 -C 4 alkoxy-substituted C 1 -C 4 alkyl group;
- X is selected from C and N;
- A is selected from the group consisting of C 3 -C 7 cycloalkyl and C 3 -C 7 heterocycloalkyl;
- B is an aromatic ring, preferably selected from the group consisting of a benzene ring, a pyridine ring, a thiazole ring, a furan ring, a thiophene ring, a pyrrole ring, a pyrazole ring, an oxazole ring, an isoxazole ring, and a quinoline ring, and the aromatic ring can be one Or substituted with a plurality of substituents independently selected from the same or different H, halogen, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, NH 2 , C 1 -C 4 alkane a base amino group and a C 1 -C 4 alkyloxy group;
- n is an integer from 1 to 5;
- n is an integer from 1 to 4.
- the invention relates to a compound of formula I, including prodrugs, stereoisomers, pharmaceutically acceptable salts, hydrates or other solvates thereof, wherein
- R 1 is one or more substituents independently selected from the same or different H, halogen, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, NH 2 , COOH, C 1 -C 4 An alkylamino group and a C 1 -C 4 alkyloxy group;
- R 2 is one or more substituents independently selected from the same or different H, halogen, CN and C 1 -C 4 alkyl;
- R 3 is C 1 -C 4 alkyl, C 3 -C 6 cycloalkyl, C 1 -C 4 haloalkyl, halo C 3 -C 6 cycloalkyl and cyano substituted C 1 -C 4 alkyl ;
- X is selected from C and N;
- A is selected from the group consisting of C 3 -C 7 cycloalkyl and C 3 -C 7 heterocycloalkyl;
- B is an aromatic ring, preferably selected from the group consisting of a benzene ring, a pyridine ring, a thiazole ring, a furan ring, a thiophene ring, a pyrrole ring, a pyrazole ring, an oxazole ring, an isoxazole ring, and a quinoline ring, and the aromatic ring can be one Or substituted with a plurality of substituents independently selected from the same or different H, halogen, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, NH 2 , C 1 -C 4 alkane a base amino group and a C 1 -C 4 alkyloxy group;
- n is an integer from 1 to 5;
- n is an integer from 1 to 4.
- the invention relates to a compound of formula I, including prodrugs, stereoisomers, pharmaceutically acceptable salts, hydrates or other solvates thereof, wherein
- R 1 is one or more substituents independently selected from the same or different H, halogen, CN, C 1 -C 4 alkyl, C 1 -C 4 haloalkyl, NH 2 , COOH, C 1 -C 4 An alkylamino group and a C 1 -C 4 alkyloxy group;
- R 2 is one or more substituents independently selected from the same or different H, halogen, CN and C 1 -C 4 alkyl;
- R 3 is C 1 -C 4 alkyl, C 3 -C 6 cycloalkyl, C 1 -C 4 haloalkyl, halo C 3 -C 6 cycloalkyl and cyano substituted C 1 -C 4 alkyl ;
- X is selected from C and N;
- A is selected from C 3 -C 5 cycloalkyl and C 3 -C 5 heterocycloalkyl;
- B is an aromatic ring, preferably selected from the group consisting of a benzene ring, a pyridine ring and a thiazole ring, which may be substituted by one or more substituents independently selected from the same or different H, halogen, CN, C 1- C 4 alkyl, C 1 -C 4 haloalkyl, NH 2 , C 1 -C 4 alkylamino and C 1 -C 4 alkyloxy;
- n is an integer from 1 to 5;
- n is an integer from 1 to 4.
- the invention relates to a compound of formula I, including prodrugs, stereoisomers, pharmaceutically acceptable salts, hydrates or other solvates thereof, wherein
- R 1 is one or more substituents independently selected from the same or different H, halogen, CN and C 1 -C 4 alkyl;
- R 2 is one or more substituents independently selected from the same or different H, halogen, CN, and methyl;
- R 3 is C 1 -C 4 alkyl, C 3 -C 6 cycloalkyl, C 1 -C 4 haloalkyl, halo C 3 -C 6 cycloalkyl or cyano substituted C 1 -C 4 alkyl ;
- X is selected from C and N;
- A is selected from the group consisting of C 3 -C 4 cycloalkyl and C 3 -C 4 heterocycloalkyl;
- B is an aromatic ring, preferably selected from the group consisting of a benzene ring, a pyridine ring and a thiazole ring, which may be substituted by one or more substituents independently selected from the same or different H, halogen, CN, C 1- C 4 alkyl and C 1 -C 4 haloalkyl;
- n is an integer from 1 to 5;
- n is an integer from 1 to 4.
- the invention relates to a compound of formula I, including prodrugs, stereoisomers, pharmaceutically acceptable salts, hydrates or other solvates thereof, wherein
- R 1 is one or more substituents, independently selected from the same or different H, halogen and CN;
- R 2 is one or more substituents independently selected from the same or different H, F, Cl, CN, and methyl;
- R 3 is C 1 -C 4 alkyl, C 3 -C 6 cycloalkyl, C 1 -C 4 haloalkyl, halo C 3 -C 6 cycloalkyl or cyano substituted C 1 -C 4 alkyl ;
- X is selected from C and N;
- A is selected from the group consisting of C 3 -C 4 cycloalkyl and C 3 -C 4 heterocycloalkyl;
- B is an aromatic ring, preferably selected from the group consisting of a benzene ring, a pyridine ring and a thiazole ring, which may be substituted by one or more substituents independently selected from the same or different H, halogen, CN, A Base and CF 3 ;
- n is an integer from 1 to 5;
- n is an integer from 1 to 4.
- the invention relates to a compound of formula I, including prodrugs, stereoisomers, pharmaceutically acceptable salts, hydrates or other solvates thereof, wherein
- R 1 is H
- R 2 is one or more substituents independently selected from the same or different H, F, Cl, CN, and methyl;
- R 3 is C 1 -C 4 alkyl, C 3 -C 6 cycloalkyl, C 1 -C 4 haloalkyl, halo C 3 -C 6 cycloalkyl or cyano substituted C 1 -C 4 alkyl ;
- X is selected from C and N;
- A is selected from C 3 -C 4 cycloalkyl
- B is an aromatic ring, preferably selected from the group consisting of a benzene ring, a pyridine ring and a thiazole ring, which may be substituted by one or more substituents independently selected from the same or different H, halogen, CN, A Base and CF 3 ;
- n 1;
- n is an integer from 1 to 3.
- the invention relates to a compound of formula I, including prodrugs, stereoisomers, pharmaceutically acceptable salts, hydrates or other solvates thereof, wherein
- R 1 is H
- R 2 is one or more substituents independently selected from the same or different H, F, Cl, CN, and methyl;
- R 3 is C 1 -C 4 alkyl, C 3 -C 6 cycloalkyl, C 1 -C 4 haloalkyl, halo C 3 -C 6 cycloalkyl or cyano substituted C 1 -C 4 alkyl ;
- X is selected from C and N;
- A is selected from C 3 -C 4 cycloalkyl
- B is an aromatic ring, preferably selected from the group consisting of a benzene ring, a pyridine ring and a thiazole ring, which may be substituted by one or more substituents independently selected from the same or different H, halogen, CN, A Base and CF 3 ;
- n 1;
- n is an integer from 1 to 2.
- halogen as used in the present invention is fluorine, chlorine, bromine or iodine, preferably fluorine or chlorine.
- Alkyl as used in the present invention includes straight or branched alkyl groups.
- the C 1 -C 4 alkyl group as used in the present invention means an alkyl group having 1 to 4 carbon atoms, preferably methyl, ethyl, propyl or isopropyl, n-butyl or isobutyl or Tert-butyl.
- the alkyl group in the compound of the present invention may be optionally substituted or unsubstituted, and the substituent may include an alkyl group, a halogen, an alkoxy group, a halogenated alkyl group, a cyano group, a hydroxyl group or the like.
- Examples of the alkyl group of the present invention include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, t-butyl and the like.
- the "cycloalkyl group” of the present invention includes a 3-7 membered cycloalkyl group, preferably a cyclopropyl group, a cyclobutyl group, a cyclopentyl group or a cyclohexyl group.
- the cycloalkyl group in the compound of the present invention may be optionally substituted or unsubstituted, and the substituent may include an alkyl group, a halogen, an alkoxy group, a hydrocarbon group, a hydroxyl group and the like.
- heterocycloalkyl group of the present invention includes a 3-7 membered heterocycloalkyl group.
- the heterocycloalkyl group in the compound of the present invention may be optionally substituted or unsubstituted, and the substituent may include an alkyl group, a halogen, an alkoxy group, a halogenated alkyl group, a cyano group, a hydroxyl group or the like.
- alkoxy group refers to a group formed by linking the above alkyl group to an oxygen atom, wherein the oxygen atom has a free bonding ability such as a methoxy group, an ethoxy group, a propoxy group, or a butyl group.
- alkylamino group as used in the present invention means a group formed by linking the above alkyl group to an amino group, such as methylamino group, ethylamino group, 4-dimethylamino group or the like.
- substituted with one or more substituents means substituted by one or more than one substituent, for example, substituted with 1, 2, 3 or 4 substituents; Ground, substituted by 1, 2 or 3 substituents.
- another solvate means a solvate formed with a solvent other than water.
- “Pharmaceutically acceptable” or “pharmaceutically acceptable” as used herein is understood to be suitable for human and animal use within a reasonable medical range, tolerable and without unacceptable side effects including toxicity, allergic reactions. , stimuli and complications.
- the present invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the above formula I, including a prodrug, a stereoisomer, a pharmaceutically acceptable salt, a hydrate or other solvate thereof as an active ingredient.
- the compounds of the present invention may optionally be used in combination with one or more other active ingredients, the respective amounts and ratios of which may be adjusted by one skilled in the art depending on the particular condition and the particular circumstances of the patient, the clinical need, and the like.
- Preparation method The definitions of the following variables are as described above, and the new variables are defined as described in this section.
- the compounds of the formula I and the intermediates involved can be purified by conventional separation methods such as extraction, recrystallization and separation by silica gel column chromatography.
- the 200-300 mesh silica gel and thin layer chromatography silica gel plates used were all produced by Qingdao Ocean Chemical Plant.
- the chemical reagents used are either analytically pure or chemically pure commercial products of the general reagents, and are used without further purification.
- the commercially available II-1 is methylated or ethylated by a usual method such as acid chloride/methanol (CH 3 OH) or ethanol (C 2 H 5 OH), sulfuric acid/CH 3 OH or C 2 H 5 OH.
- II-2 Dissolving II-2 in CH 3 OH or C 2 H 5 OH under the action of common reducing agents including, but not limited to, iron powder/ammonium chloride (Fe/NH 4 Cl) or iron powder/hydrochloric acid, etc.
- II-3 is obtained after reacting at 70-100 ° C for about 2-4 h.
- Dissolve II-3 (home-made or commercially available) in common solvents (including but not limited to dichloromethane (CH 2 Cl 2 ), tetrahydrofuran (THF), N,N'-dimethylformamide (DMF) or pyridine ( In Py), etc.
- the acid chloride II-4 is added dropwise to the above solution under the catalysis of a common base such as triethylamine (TEA) and N,N'-diisopropylethylamine (DIPEA).
- a common base such as triethylamine (TEA) and N,N'-diisopropylethylamine (DIPEA).
- TEA triethylamine
- DIPEA N,N'-diisopropylethylamine
- the carboxylic acid II-4 may be added dropwise to the above solution under the action of a common condensing agent to obtain II-5.
- the II-5 is dissolved in a mixed solvent of CH 3 OH, C 2 H 5 OH or THF and water at room temperature, and is subjected to carboxylic acid by an inorganic base such as lithium hydroxide (LiOH) or sodium hydroxide (NaOH).
- an inorganic base such as lithium hydroxide (LiOH) or sodium hydroxide (NaOH).
- LiOH lithium hydroxide
- NaOH sodium hydroxide
- HATU O-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate
- HBTt 1 -hydroxybenzotriazole
- PyBOP 1H-benzotriazol-1-yloxytripyrrolidinyl hexafluorophosphate
- T 3 P 1-propylphosphoric anhydride
- R ' is OH or Cl; R 4 is alkyl; each of the other variables are as previously defined.
- III-1 Under reflux conditions, commercially available III-1 is reacted with hydrazine hydrate in a suitable protic solvent for about 1-3 hours to obtain III-2; under reflux conditions, III-2 and N,N'-dimethylformate are obtained under reflux conditions.
- the key intermediate III is obtained by reacting for at least 24 h.
- the protic solvent described in the present route may be CH 3 OH, C 2 H 5 OH or the like, but is not limited thereto.
- III'-1 is dissolved in THF or 1,4-dioxane under N 2 protection conditions, and passed through n-butyllithium (n-BuLi) and CO 2 at a low temperature of -70 ° C. The halogen therein is converted to a carboxyl group, and the reaction is carried out for about 1-3 hours to obtain III'-2.
- III'-6 Under the condition of N 2 protection, III'-6 is dissolved in a mixed solvent of 1,4-dioxane and water, benzophenone imine is added, and catalyzed by palladium, common ligand and base The CN coupling reaction was completed, and the reaction was obtained under reflux conditions to give III'-7.
- the key intermediate III is obtained after hydrolysis of III'-7 by dilute hydrochloric acid for about 24 h at room temperature.
- the base is TEA, DIPEA, potassium carbonate (K 2 CO 3 ), cesium carbonate (Cs) 2 CO 3 ) and sodium t-butoxide (t-BuONa), etc., but are not limited thereto;
- the palladium reagent is tribenzylideneacetone dipalladium (Pd 2 (dba) 3 ), Pd 2 (dba) 3 a dichloromethane complex and palladium acetate (Pd(OAc) 2 ), etc., but are not limited thereto; the ligand 4,5-bisdiphenylphosphino-9,9-dimethyloxaxime ( Xantphos), 2-bicyclohexylphosphine-2',6'-dimethoxybiphenyl (Sphos) and 1,1'-binaphthyl-2,2
- the key intermediate II is prepared into an acid chloride by thionyl chloride (SOCl 2 ), oxalyl chloride ((COCl) 2 ), phosphorus trichloride (PCl 3 ) or phosphorus pentachloride (PCl 5 ), followed by intermediate activity
- SOCl 2 thionyl chloride
- (COCl) 2 oxalyl chloride
- PCl 3 phosphorus trichloride
- PCl 5 phosphorus pentachloride
- the compound of the formula I can also be obtained by using a common condensing agent such as HATU, HOBt, PyBOP and T 3 P, etc., but is not limited to the basic catalysts such as TEA, DIPEA and K 2 CO 3 described in this route. But it is not limited to this.
- a common condensing agent such as HATU, HOBt, PyBOP and T 3 P, etc.
- the basic catalysts such as TEA, DIPEA and K 2 CO 3 described in this route. But it is not limited to this.
- the structural formula I of the present invention can also be obtained by starting from the starting materials II and IV-1 by a similar synthetic method as in Scheme 2, as shown in the following Scheme 5; in addition, the formula I can also be obtained from the compound V and the compound of the formula II-4. It is obtained by carrying out a condensation reaction under base catalysis.
- R ' is OH or Cl; R 4 is alkyl; each of the other variables are as previously defined.
- Mass spectrometry conditions instrument Thermo MSQ Plus; ion source ESI (EA + EA-); cone voltage 30V; capillary voltage 3.00KV; source temperature 350 ° C;
- Chromatographic conditions instrument Thermo U3000; detector DAD-3000 (RS) (diode array detector); column Shimadzu Inertsil ODS-HL HP 3 ⁇ m 3.0 ⁇ 100mm; flow rate 0.4mL / min; column temperature 30 ° C; mobile phase CH 3 OH/H 2 O/HCOOH (75/25/0.5).
- Representative compounds I-1 to I-20 were prepared according to the methods described above.
- the present invention is further illustrated by the following specific examples, but the scope of the present invention is not limited to these examples.
- the percentages stated in the present invention are all percentages by weight unless otherwise specified.
- the range of values described in the specification, such as units of measure, reaction conditions, physical state of the compound, or percentage, are intended to provide an unambiguous written reference. It is still possible for a person skilled in the art to use the temperature, concentration, amount, number of carbon atoms, etc. outside of this range or different from a single value in the practice of the present invention, and it is still possible to obtain the desired result.
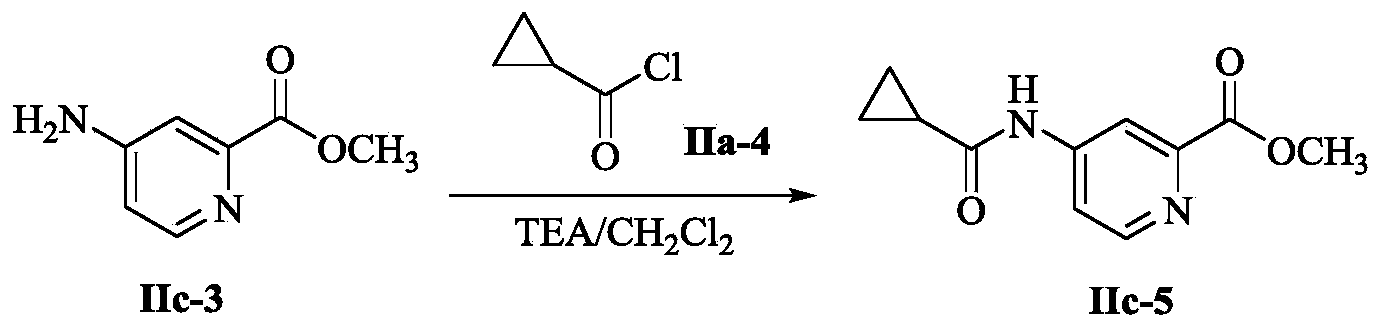
- IIc-3 (1.52g, 10.0mmol, 1.0eq) was dissolved in CH 2 Cl 2 (20mL), was added TEA (4.04g, 40.0mmol, 4.0eq) , an ice / salt bath to 0 °C
- IIa-4 (1.56 g, 15.0 mmol, 1.5 eq) was added dropwise and the obtained mixture was stirred at ambient temperature for 16 h. The reaction was monitored by TLC. The reaction mixture was concentrated. EtOAc mjjjjjj LC-MS MS-ESI (m / z) 221.4 [M + H] +.
- IId-3 (1.84g, 10.0mmol, 1.0eq) was dissolved in CH 2 Cl 2 (20mL), was added TEA (4.04g, 40.0mmol, 4.0eq) , an ice / salt bath to 0 °C, IIa-4 (1.56 g, 15.0 mmol, 1.5 eq) was added and the obtained mixture was stirred at ambient temperature for 16 h. The reaction mixture was concentrated. EtOAc mjjjjjj LC-MS MS-ESI (m / z) 321.2 [M + H] +.
- IIIa-1 (10.00 g, 66.0 mmol, 1.0 eq) was added, followed by methanol (200 mL), hydrazine hydrate (66.07 g, 132.0 mmol, 2.0 eq), and refluxed for 2 h. Most of the solvent of the reaction mixture was concentrated, washed, washed with EtOAc and dried. A white solid, IIIa-2, 10.20 g was obtained.
- MS-ESI m / z
- IIIa-3 (260.0 mg, 1.0 mmol, 1.0 eq) and IIIf-4 (445.5 mg, 5.0 mmol, 5.0 eq) were dissolved in CH 3 CN/AcOH (4/1, 25 mL), and the mixture was heated to reflux for 24 h. solution, extracted with water, 1N NaOH solution was adjusted to pH 10, EtOAc and extracted three times, dried over anhydrous MgSO 4, the organic phase was concentrated to give a yellow solid IIIf 180.0mg (38.0% yield).
- LC-MS MS-ESI (m / z) 233.1 [M + H] +.
- IIIa-3 (3.00 g, 11.5 mmol, 1.0 eq) and commercially available IIIg-4 (3.43 g, 45.8 mmol, 4.0 eq) were dissolved in CH 3 CN/AcOH (4/1, 37.5 mL). After stirring at 92 ° C for 24 h, the reaction was monitored by TLC, cooled to ambient temperature and concentrated. Diluted with water, adjusted to pH 8 with 1N NaOH solution, and concentrated. The resulting solid was washed with CH 2 Cl 2 / CH 3 OH (10 / 1,100mL) slurried, filtered and the filtrate was concentrated to give the crude product as a pale yellow sticky solid IIIg'7.40g.
- Example 22 Compound: 5-(Cyclopropanoylamino)-2-fluoro-4-methyl-N-(6-(4-isopropyl-4H-1,2,4-triazol-3-yl) Preparation of pyridin-2-yl)benzamide I-1
- the IIa (237.0mg, 1.0mmol, 1.0eq) was suspended in SOCl 2 (5mL), heated to 60 °C, for 15min until all starting material was dissolved, and concentrated to give a yellow solid acid chloride was used directly in the next stage of the reaction.
- the acid chloride was dissolved in ultra dry THF (10 mL).
- TEA 0.5 mL
- IIIb 100.0 mg, 0.5 mmol, 1.0 eq
- Fetch III'-2 (8.35g, 40.1mmol, 1.2eq) was dissolved in CH 2 Cl 2 (100mL), at ambient temperature, to which was added HATU (12.7g, 33.4mmol, 1.0eq) , commercially available IIIc -4 (5.00 g, 33.4 mmol, 1.0 eq) and TEA (10.10 g, 100.0 mmol, 3.0 eq).
- the reaction mixture was concentrated and dried (EtOAc mjjjjjjjjjj LC-MS MS-ESI (m / z) 302.0 [MH] -.
- This assay is used to evaluate the in vitro protein level binding inhibitory activity of the compounds of the invention.
- This assay is to comprehensively evaluate the effects of different compounds on ASK1 kinase inhibitory activity.
- This test uses Homogeneous time-resolved fluorescence (HTRF) to evaluate the level of inhibition of the enzymatic activity of recombinant ASK1 in the in vitro reaction system.
- HTRF Homogeneous time-resolved fluorescence
- the basic principle of in vitro enzymatic activity detection is to use a specific substrate labeled with a terminal biotin to phosphorylate under the action of a kinase, a reaction product with an EU 3+ -Cryptate-labeled antibody that recognizes a phosphorylation site, and a XL665-labeled strand. Avidin is mixed. When two fluorescent molecules are simultaneously bound to the substrate, Eu stimulates 620 nM of fluorescence under the excitation of exogenous excitation light (320 nm), while XL665 is excited by energy transfer to produce 665 nm fluorescence.
- Substrate phosphorylation was evaluated by comparing the changes in fluorescence at two wavelengths (620 nm and 665 nm). When different test compounds are added, their inhibition of kinase activity is manifested by a change in the degree of phosphorylation of the substrate, thereby exhibiting a different ratio of fluorescent signals (665/620), and thereby calculating the inhibitory activity of the compound on the kinase.
- the basic detection principle is known in the art (Cisbio, Nature Method 2006, June 23; DOI: 10.1038/NMETH883).
- Human recombinant ASK1 (MAP3K5) kinase, 2X kinase reaction buffer, ATP (10 mM) was purchased from Invitrogen (catalogue number: PR7349B), and HTRF detection kit HTRF KinEASE STK discovery kit was purchased from Cisbio (Cat. No. 62ST0PEB).
- test reagent https://www.cisbio.com/sites/default/files/ressources/cisbio_dd_pi_62ST0PEB.pdf). The details are as follows:
- the kinase reaction buffer (working solution) was prepared as required, and the test compound was diluted to a different concentration gradient (the highest concentration of the compound was 4 ⁇ M) using a kinase reaction buffer.
- a 10 ⁇ L enzymatic reaction system (including 2.5 ⁇ L of the test compound, 5 ⁇ L of the kinase reaction buffer, and 2.5 ⁇ L of the ATP solution (provided by the kit)) was mixed and reacted at room temperature for 1 hour, and the enzymatic reaction was carried out in a 96-well microplate.
- the reaction was simultaneously provided with a control reaction including a positive control to which no test compound was added and a negative control to which no ASK1 kinase was added. Multiple holes were used for all tests.
- the fluorescence signal of each well was detected using a fluorescence detector (TecanSPARK 10M) at an excitation wavelength of 320 nm, and the detected emission wavelengths were 620 nm and 665 nm, respectively.
- Table 2 HTRF method for detection of ASK1 enzymatic inhibition rate of representative compounds of the present invention (single concentration 100 nM)
- Compound Inhibition rate% Compound Inhibition rate% Compound Inhibition rate% Compound Inhibition rate% I-1 54 I-3 51 I-4 32 I-6 13 I-8 twenty four I-9 33 I-10 44 I-11 27 I-13 18 I-14 42 I-16 twenty one I-17 15 I-18 34 I-19 52 I-20 16
Abstract
Description


























































| 化合物 | 抑制率% | 化合物 | 抑制率% | 化合物 | 抑制率% |
| I-1 | 54 | I-3 | 51 | I-4 | 32 |
| I-6 | 13 | I-8 | 24 | I-9 | 33 |
| I-10 | 44 | I-11 | 27 | I-13 | 18 |
| I-14 | 42 | I-16 | 21 | I-17 | 15 |
| I-18 | 34 | I-19 | 52 | I-20 | 16 |
Claims (22)
- 式I化合物,
 包括其前药、立体异构体、药学上可接受的盐、水合物或其它溶剂合物,其中,R 1为一个或多个取代基,独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、COOH、C 1-C 4烷基氨基、C 1-C 4烷基氧基和Ar 1;其中,Ar 1选自苯环和吡啶环,并且所述苯环和吡啶环可被一个或多个取代基取代,所述取代基独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、C 1-C 4烷基氨基和C 1-C 4烷基氧基;R 2为一个或多个取代基,独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基和C 1-C 4卤代烷基;R 3为C 1-C 4烷基、C 3-C 6环烷基、C 1-C 4卤代烷基、卤代C 3-C 6环烷基、氰基取代的C 1-C 4烷基、C 3-C 6杂环烷基、羟基取代的C 1-C 4烷基或C 1-C 4烷氧基取代的C 1-C 4烷基;X选自C和N;A选自C 3-C 7环烷基和C 3-C 7杂环烷基;B为芳香环,优选选自苯环、吡啶环、噻唑环、呋喃环、噻吩环、吡咯环、吡唑环、噁唑环、异噁唑环以及喹啉环,所述芳香环可被一个或多个取代基取代,所述取代基独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、C 1-C 4烷基氨基和C 1-C 4烷基氧基;m为1到5的整数;n为1到4的整数。
包括其前药、立体异构体、药学上可接受的盐、水合物或其它溶剂合物,其中,R 1为一个或多个取代基,独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、COOH、C 1-C 4烷基氨基、C 1-C 4烷基氧基和Ar 1;其中,Ar 1选自苯环和吡啶环,并且所述苯环和吡啶环可被一个或多个取代基取代,所述取代基独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、C 1-C 4烷基氨基和C 1-C 4烷基氧基;R 2为一个或多个取代基,独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基和C 1-C 4卤代烷基;R 3为C 1-C 4烷基、C 3-C 6环烷基、C 1-C 4卤代烷基、卤代C 3-C 6环烷基、氰基取代的C 1-C 4烷基、C 3-C 6杂环烷基、羟基取代的C 1-C 4烷基或C 1-C 4烷氧基取代的C 1-C 4烷基;X选自C和N;A选自C 3-C 7环烷基和C 3-C 7杂环烷基;B为芳香环,优选选自苯环、吡啶环、噻唑环、呋喃环、噻吩环、吡咯环、吡唑环、噁唑环、异噁唑环以及喹啉环,所述芳香环可被一个或多个取代基取代,所述取代基独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、C 1-C 4烷基氨基和C 1-C 4烷基氧基;m为1到5的整数;n为1到4的整数。 - 根据权利要求1所述的式I化合物,其中,R 1为一个或多个取代基,独立地选自相同或不同的H、卤素、CN、C 1-C 4 烷基、C 1-C 4卤代烷基、NH 2、COOH、C 1-C 4烷基氨基、C 1-C 4烷基氧基和Ar 1;其中,Ar 1选自苯环和吡啶环,并且所述苯环和吡啶环可被一个或多个取代基取代,所述取代基独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、C 1-C 4烷基氨基和C 1-C 4烷基氧基;R 2为一个或多个取代基,独立地选自相同或不同的H、卤素、CN和C 1-C 4烷基;R 3为C 1-C 4烷基、C 3-C 6环烷基、C 1-C 4卤代烷基、卤代C 3-C 6环烷基、氰基取代的C 1-C 4烷基、C 3-C 6杂环烷基、羟基取代的C 1-C 4烷基或C 1-C 4烷氧基取代的C 1-C 4烷基;X选自C和N;A选自C 3-C 7环烷基和C 3-C 7杂环烷基;B为芳香环,优选选自苯环、吡啶环、噻唑环、呋喃环、噻吩环、吡咯环、吡唑环、噁唑环、异噁唑环和喹啉,所述芳香环可被一个或多个取代基取代,所述取代基独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、C 1-C 4烷基氨基和C 1-C 4烷基氧基;m为1到5的整数;n为1到4的整数。
- 根据权利要求1所述的式I化合物,其中,R 1为一个或多个取代基,独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、COOH、C 1-C 4烷基氨基和C 1-C 4烷基氧基;R 2为一个或多个取代基,独立地选自相同或不同的H、卤素、CN和C 1-C 4烷基;R 3为C 1-C 4烷基、C 3-C 6环烷基、C 1-C 4卤代烷基、卤代C 3-C 6环烷基、氰基取代C 1-C 4烷基、C 3-C 6杂环烷基、羟基取代的C 1-C 4烷基或C 1-C 4烷氧基取代的C 1-C 4烷基;X选自C和N;A选自C 3-C 7环烷基和C 3-C 7杂环烷基;B为芳香环,优选选自苯环、吡啶环、噻唑环、呋喃环、噻吩环、吡咯环、吡唑环、噁唑环、异噁唑环和喹啉环,所述芳香环可被一个或多个取代基取代,所述取代基独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、C 1-C 4烷基氨基和C 1-C 4烷基氧基;m为1到5的整数;n为1到4的整数。
- 根据权利要求1所述的式I化合物,其中,R 1为一个或多个取代基,独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、COOH、C 1-C 4烷基氨基和C 1-C 4烷基氧基;R 2为一个或多个取代基,独立地选自相同或不同的H、卤素、CN和C 1-C 4烷基;R 3为C 1-C 4烷基、C 3-C 6环烷基、C 1-C 4卤代烷基、卤代C 3-C 6环烷基或氰基取代的C 1-C 4烷基;X选自C和N;A选自C 3-C 7环烷基和C 3-C 7杂环烷基;B为芳香环,优选选自苯环、吡啶环、噻唑环、呋喃环、噻吩环、吡咯环、吡唑环、噁唑环、异噁唑环和喹啉环,所述芳香环可被一个或多个取代基取代,所述取代基独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、C 1-C 4烷基氨基和C 1-C 4烷基氧基;m为1到5的整数;n为1到4的整数。
- 根据权利要求1所述的式I化合物,其中,R 1为一个或多个取代基,独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、COOH、C 1-C 4烷基氨基和C 1-C 4烷基氧基;R 2为一个或多个取代基,独立地选自相同或不同的H、卤素、CN和C 1-C 4烷基;R 3为C 1-C 4烷基、C 3-C 6环烷基、C 1-C 4卤代烷基、卤代C 3-C 6环烷基或氰基取代的C 1-C 4烷基;X选自C和N;A选自C 3-C 5环烷基和C 3-C 5杂环烷基;B为芳香环,优选选自苯环、吡啶环和噻唑环,所述芳香环可被一个或多个取代基取代,所述取代基独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基、C 1-C 4卤代烷基、NH 2、C 1-C 4烷基氨基和C 1-C 4烷基氧基;m为1到5的整数;n为1到4的整数。
- 根据权利要求1所述的式I化合物,其中,R 1为一个或多个取代基,独立地选自相同或不同的H、卤素、CN和C 1-C 4烷基;R 2为一个或多个取代基,独立地选自相同或不同的H、卤素、CN和甲基;R 3为C 1-C 4烷基、C 3-C 6环烷基、C 1-C 4卤代烷基、卤代C 3-C 6环烷基或氰基取代的C 1-C 4烷基;X选自C和N;A选自C 3-C 4环烷基和C 3-C 4杂环烷基;B为芳香环,优选选自苯环、吡啶环和噻唑环,所述芳香环可被一个或多个取代基取代,所述取代基独立地选自相同或不同的H、卤素、CN、C 1-C 4烷基和C 1-C 4卤代烷基;m为1到5的整数;n为1到4的整数。
- 根据权利要求1所述的式I化合物,其中,R 1为一个或多个取代基,独立地选自相同或不同的H、卤素和CN;R 2为一个或多个取代基,独立地选自相同或不同的H、F、Cl、CN和甲基;R 3为C 1-C 4烷基、C 3-C 6环烷基、C 1-C 4卤代烷基、卤代C 3-C 6环烷基或氰基取代的C 1-C 4烷基;X选自C和N;A选自C 3-C 4环烷基和C 3-C 4杂环烷基;B为芳香环,优选选自苯环、吡啶环和噻唑环,所述芳香环可被一个或多个取代基取代,所述取代基独立地选自相同或不同的H、卤素、CN、甲基和CF 3;m为1到5的整数;n为1到4的整数。
- 根据权利要求1所述的式I化合物,其中,R 1为H;R 2为一个或多个取代基,独立地选自相同或不同的H、F、Cl、CN和甲基;R 3为C 1-C 4烷基、C 3-C 6环烷基、C 1-C 4卤代烷基、卤代C 3-C 6环烷基和氰基取代的C 1-C 4烷基;X选自C和N;A选自C 3-C 4环烷基;B为芳香环,优选选自苯环、吡啶环和噻唑环,所述芳香环可被一个或多个取代基取代,所述取代基独立地选自相同或不同的H、卤素、CN、甲基和CF 3;m为1;n为1到3的整数。
- 根据权利要求1所述的式I化合物,其中,R 1为H;R 2为一个或多个取代基,独立地选自相同或不同的H、F、Cl、CN和甲基;R 3为C 1-C 4烷基、C 3-C 6环烷基、C 1-C 4卤代烷基、卤代C 3-C 6环烷基或氰基取代的C 1-C 4烷基;X选自C和N;A选自C 3-C 4环烷基;B为芳香环,优选选自苯环、吡啶环和噻唑环,所述芳香环可被一个或多个取代基取代,所述取代基独立地选自相同或不同的H、卤素、CN、甲基和CF 3;m为1;n为1到2的整数。
- 根据权利要求1所述的式I,其中所述化合物选自:5-(环丙甲酰氨基)-2-氟-4-甲基-N-(6-(4-异丙基-4H-1,2,4-三唑-3-基)吡啶-2-基)苯甲酰胺;5-(环丙甲酰氨基)-2-氟-4-甲基-N-(6-(4-环丙基-4H-1,2,4-三唑-3-基)吡啶-2-基)苯甲酰胺;(S)-5-(环丙甲酰氨基)-2-氟-4-甲基-N-(6-(4-(1,1,1-三氟丙基-2-基)-4H-1,2,4-三唑-3-基)吡啶-2-基)苯甲酰胺;5-(环丙甲酰氨基)-2-氟-4-甲基-N-(6-(4-(2,2,2-三氟乙基)-4H-1,2,4-三唑-3-基)吡啶-2-基)苯甲酰胺;(R)-5-(环丙甲酰氨基)-2-氟-4-甲基-N-(6-(4-(1,1,1-三氟丙基-2-基)-4H-1,2,4-三唑-3-基)吡啶-2-基)苯甲酰胺;(R)-5-(环丙甲酰氨基)-2-氟-N-6-(4-(1-甲氧丙基-2-基)-4H-1,2,4-三唑-3-基)吡啶-2-基)-4-甲基苯甲酰胺;(R)-5-(环丙甲酰氨基)-2-氟-N-6-(4-(1-羟丙基-2-基)-4H-1,2,4-三唑-3-基)吡啶-2-基)-4-甲基苯甲酰胺;(S)-5-(环丙甲酰氨基)-2-氟-4-甲基-N-2-(4-(1,1,1-三氟丙基-2-基)-4H-1,2,4-三唑-3-基)噻唑-4-基)苯甲酰胺;(S)-4-氯-5-(环丙甲酰氨基)-2-氟-N-(6-(4-(1,1,1-三氟丙基-2-基)-4H-1,2,4-三唑-3-基)吡啶-2-基)苯甲酰胺;(S)-4-(环丙甲酰氨基)-N-(6-(4-(1,1,1-三氟丙基-2-基)-4H-1,2,4-三唑-3-基)吡啶-2-基)吡啶-2-甲酰胺;(S)-4-(环丙甲酰氨基)-5-氟-N-(6-(4-(1,1,1-三氟丙基-2-基)-4H-1,2,4-三唑-3-基)吡啶-2-基)吡啶-2-甲酰胺;(S)-5-(环丙甲酰胺基)-2-氟-4-甲基-N-(3-(4-(1,1,1-三氟丙基-2-基)-4H-1,2,4-三唑-3-基)苯基)苯甲酰胺;2-氟-5-(2-(4-氟苯基)环丙基-1-甲酰氨基)-N-(6-(4-异丙基-4H-1,2,4-三唑-3-基)吡啶-2-基)-4-甲基苯甲酰胺;5-(环丁甲酰氨基)-2-氟-N-(6-(4-异丙基-4H-1,2,4-三唑-3-基)吡啶-2-基)-4-甲基苯甲酰胺;2-氟-N-(6-(4-异丙基-4H-1,2,4-三唑-3-基)吡啶-2-基)-4-甲基-5-(1-(三氟甲基)环丙 基-1-甲酰氨基)苯甲酰胺;2-氟-N-(6-(4-异丙基-4H-1,2,4-三唑-3-基)吡啶-2-基)-4-甲基-5-(1-(氟)环丙基-1-甲酰氨基)苯甲酰胺;2-氟-5-((1R,2R)-2-氟环丙基-1-甲酰氨基)-N-(6-(4-异丙基-4H-1,2,4-三唑-3-基)吡啶-2-基)-4-甲基苯甲酰胺;2-氟-5-((1R,2S)-2-氟环丙基-1-甲酰氨基)-N-(6-(4-异丙基-4H-1,2,4-三唑-3-基)吡啶-2-基)-4-甲基苯甲酰胺;(S)-5-(环戊甲酰氨基)-2-氟-4-甲基-N-(6-(4-(1,1,1-三氟丙基-2-基)-4H-1,2,4-三唑-3-基)吡啶-2-基)苯甲酰胺;和(S)-5-(环庚甲酰氨基)-2-氟-4-甲基-N-(6-(4-(1,1,1-三氟丙基-2-基)-4H-1,2,4-三唑-3-基)吡啶-2-基)苯甲酰胺。
- 制备根据权利要求1所述的式I化合物的方法,包括使式II化合物或其酰氯化物与式III化合物在碱催化下进行缩合反应,
 其中,各变量如权利要求1所定义。
其中,各变量如权利要求1所定义。 - 制备根据权利要求1所述的式I化合物的方法,包括以式II化合物和IV-1化合物为起始原料,通过下式所示反应步骤制得,
 其中,各变量如权利要求1所定义。
其中,各变量如权利要求1所定义。 - 制备根据权利要求1所述的式I化合物的方法,包括使式V化合物与式II-4 化合物在碱催化下进行缩合反应,
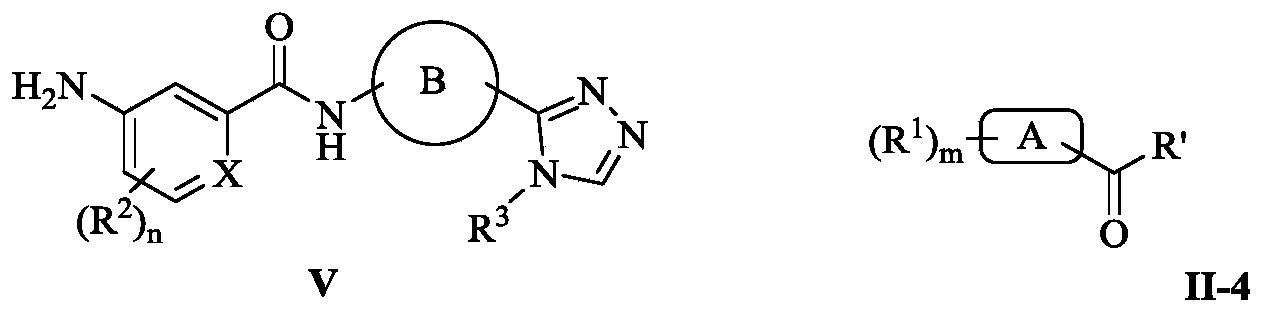 其中,R’为OH或Cl;其它各变量如权利要求1所定义。
其中,R’为OH或Cl;其它各变量如权利要求1所定义。 - 根据权利要求11或13所述的制备方法,其中所述碱催化剂选自TEA、DIPEA和Py。
- 根据权利要求13所述的制备方法,其中所述缩合反应在缩合剂存在下进行,所述缩合剂选自HATU、HOBt、PyBOP和T 3P。
- 根据权利要求11所述的制备方法,其中所述式II化合物由如下合成路线制备,
 其中,R’为OH或Cl;R 4为烷基;其它各变量如权利要求1所定义。
其中,R’为OH或Cl;R 4为烷基;其它各变量如权利要求1所定义。 - 根据权利要求11或12所述的制备方法,其中所述式III化合物由如下合成路线制备,
 其中,各变量如权利要求1所定义。
其中,各变量如权利要求1所定义。 - 一种药物组合物,其包含权利要求1至10中任一项所述的式I化合物以及任选的可药用载体、辅料或稀释剂。
- 权利要求1至10中任一项所述的化合物在制备用于治疗或预防与ASK1激 酶相关的疾病的药物中的应用。
- 根据权利要求19的应用,其中所述与ASK1激酶相关的疾病选自炎性疾病、代谢性疾病、自身免疫性疾病、心血管疾病、神经退行性疾病和癌症。
- 权利要求18所述的药物组合物在制备用于治疗或预防与ASK1靶点相关的疾病的药物中的应用。
- 根据权利要求21所述的应用,其中所述与ASK1靶点相关的疾病选自炎性疾病、代谢性疾病、自身免疫性疾病、心血管疾病、神经退行性疾病、癌症及其它疾病。
Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP19791533.3A EP3789384A4 (en) | 2018-04-28 | 2019-04-23 | FORMAMIDE COMPOUND, PROCESS FOR PREPARATION AND USE |
| BR112020021664-0A BR112020021664A2 (pt) | 2018-04-28 | 2019-04-23 | composto de formamida, método de preparação do mesmo e aplicação do mesmo |
| AU2019260217A AU2019260217B2 (en) | 2018-04-28 | 2019-04-23 | Formamide compound, preparation method therefor and application thereof |
| UAA202007486A UA125056C2 (uk) | 2018-04-28 | 2019-04-23 | Формамідна сполука, спосіб її отримання і її застосування |
| US17/050,443 US20210139454A1 (en) | 2018-04-28 | 2019-04-23 | Formamide compound, preparation method therefor and application thereof |
| MX2020011164A MX2020011164A (es) | 2018-04-28 | 2019-04-23 | Compuesto de formamida, método de preparación para el mismo y aplicación del mismo. |
| CA3098202A CA3098202A1 (en) | 2018-04-28 | 2019-04-23 | Formamide compound, preparation method therefor and application thereof |
| JP2020559445A JP2021519808A (ja) | 2018-04-28 | 2019-04-23 | ホルムアミド類化合物、その調製方法及び応用 |
| KR1020207033670A KR20210005135A (ko) | 2018-04-28 | 2019-04-23 | 포름아미드 화합물, 이의 제조 방법 및 이의 적용 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201810404758.XA CN110407806B (zh) | 2018-04-28 | 2018-04-28 | 甲酰胺类化合物、其制备方法及其应用 |
| CN201810404758.X | 2018-04-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019206120A1 true WO2019206120A1 (zh) | 2019-10-31 |
Family
ID=68294834
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2019/083829 WO2019206120A1 (zh) | 2018-04-28 | 2019-04-23 | 甲酰胺类化合物、其制备方法及其应用 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US20210139454A1 (zh) |
| EP (1) | EP3789384A4 (zh) |
| JP (1) | JP2021519808A (zh) |
| KR (1) | KR20210005135A (zh) |
| CN (1) | CN110407806B (zh) |
| AU (1) | AU2019260217B2 (zh) |
| BR (1) | BR112020021664A2 (zh) |
| CA (1) | CA3098202A1 (zh) |
| MX (1) | MX2020011164A (zh) |
| TW (1) | TWI694824B (zh) |
| UA (1) | UA125056C2 (zh) |
| WO (1) | WO2019206120A1 (zh) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114470217B (zh) * | 2020-11-24 | 2023-06-20 | 深圳微芯生物科技股份有限公司 | 预防和治疗代谢异常或炎症引起的组织损伤的药物组合物 |
| CN114246866B (zh) * | 2021-04-28 | 2023-09-12 | 深圳微芯生物科技股份有限公司 | 用于治疗慢性肾病的药物及其用途 |
| CN113200924B (zh) * | 2021-05-18 | 2022-11-01 | 南开大学 | 一种4-氨基-5-嘧啶甲酰胺类化合物及其制备方法和应用 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009027283A1 (en) | 2007-08-31 | 2009-03-05 | Merck Serono S.A. | Triazolopyridine compounds and their use as ask inhibitors |
| US20110009410A1 (en) | 2009-07-13 | 2011-01-13 | Gilead Sciences, Inc. | Apoptosis signal-regulating kinase inhibitors |
| WO2011041293A1 (en) | 2009-09-30 | 2011-04-07 | Takeda Pharmaceutical Company Limited | Pyrazolo [1, 5-a] pyrimidine derivatives as apoptosis signal-regulating kinase 1 inhibitors |
| US20120004267A1 (en) | 2010-07-02 | 2012-01-05 | Gilead Sciences, Inc. | Apoptosis signal-regulating kinase inhibitors |
| CN104080771A (zh) * | 2012-01-27 | 2014-10-01 | 吉利德科学公司 | 细胞凋亡信号调节激酶抑制剂 |
| CN104918936A (zh) * | 2012-12-21 | 2015-09-16 | 吉联亚科学公司 | 作为细胞凋亡信号调节激酶抑制剂的经取代的吡啶-2-甲酰胺化合物 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2561181T3 (es) * | 2009-02-05 | 2016-02-24 | Takeda Pharmaceutical Company Limited | Compuestos de piridazinona |
| MX2016008254A (es) * | 2013-12-20 | 2016-10-14 | Gilead Sciences Inc | Inhibidores de cinasa reguladora de la señal de apoptosis. |
| US11345676B2 (en) * | 2017-03-27 | 2022-05-31 | Pharmakea, Inc. | Apoptosis signal-regulating kinase 1 (ASK 1) inhibitor compounds |
| JOP20190221A1 (ar) * | 2017-04-05 | 2019-09-23 | Seal Rock Therapeutics Inc | مركبات مثبطات كيناز منظم لإشارات الاستماتة 1 (ask1) واستخداماتها |
-
2018
- 2018-04-28 CN CN201810404758.XA patent/CN110407806B/zh active Active
-
2019
- 2019-04-23 BR BR112020021664-0A patent/BR112020021664A2/pt not_active IP Right Cessation
- 2019-04-23 MX MX2020011164A patent/MX2020011164A/es unknown
- 2019-04-23 UA UAA202007486A patent/UA125056C2/uk unknown
- 2019-04-23 AU AU2019260217A patent/AU2019260217B2/en not_active Ceased
- 2019-04-23 WO PCT/CN2019/083829 patent/WO2019206120A1/zh unknown
- 2019-04-23 US US17/050,443 patent/US20210139454A1/en not_active Abandoned
- 2019-04-23 KR KR1020207033670A patent/KR20210005135A/ko not_active Application Discontinuation
- 2019-04-23 CA CA3098202A patent/CA3098202A1/en not_active Abandoned
- 2019-04-23 JP JP2020559445A patent/JP2021519808A/ja active Pending
- 2019-04-23 EP EP19791533.3A patent/EP3789384A4/en not_active Withdrawn
- 2019-04-25 TW TW108114496A patent/TWI694824B/zh not_active IP Right Cessation
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009027283A1 (en) | 2007-08-31 | 2009-03-05 | Merck Serono S.A. | Triazolopyridine compounds and their use as ask inhibitors |
| CN101878212A (zh) * | 2007-08-31 | 2010-11-03 | 默克雪兰诺有限公司 | 三唑并吡啶化合物及其作为ask抑制剂的用途 |
| US20110009410A1 (en) | 2009-07-13 | 2011-01-13 | Gilead Sciences, Inc. | Apoptosis signal-regulating kinase inhibitors |
| WO2011008709A1 (en) | 2009-07-13 | 2011-01-20 | Gilead Sciences, Inc. | Apoptosis signal-regulating kinase inhibitors |
| CN102482257A (zh) * | 2009-07-13 | 2012-05-30 | 吉利德科学股份有限公司 | 凋亡信号调节激酶抑制剂 |
| WO2011041293A1 (en) | 2009-09-30 | 2011-04-07 | Takeda Pharmaceutical Company Limited | Pyrazolo [1, 5-a] pyrimidine derivatives as apoptosis signal-regulating kinase 1 inhibitors |
| US20120004267A1 (en) | 2010-07-02 | 2012-01-05 | Gilead Sciences, Inc. | Apoptosis signal-regulating kinase inhibitors |
| CN104080771A (zh) * | 2012-01-27 | 2014-10-01 | 吉利德科学公司 | 细胞凋亡信号调节激酶抑制剂 |
| CN104918936A (zh) * | 2012-12-21 | 2015-09-16 | 吉联亚科学公司 | 作为细胞凋亡信号调节激酶抑制剂的经取代的吡啶-2-甲酰胺化合物 |
| US20170173031A1 (en) | 2012-12-21 | 2017-06-22 | Gilead Sciences, Inc. | Apoptosis signal-regulating kinase inhibitors |
Non-Patent Citations (5)
| Title |
|---|
| CARGNELLO M.ROUX P. P., MICROBIOL. MOL. BIOL. REV., vol. 75, 2011, pages 50 - 83 |
| CISBIO, NATURE METHOD, 23 June 2006 (2006-06-23) |
| HAYAKAWA R.HAYAKAWA T.TAKEDA K. ET AL., PROC.JPN.ACAD.SER.BPHYS.BIOL.SCI., vol. 88, 2012, pages 434 - 453 |
| See also references of EP3789384A4 |
| SOGA M.MATSUZAWA A.ICHIJO H., INT. J. CELL BIOL., 2012, pages 1 - 5 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN110407806B (zh) | 2021-08-17 |
| JP2021519808A (ja) | 2021-08-12 |
| UA125056C2 (uk) | 2021-12-29 |
| TW201946623A (zh) | 2019-12-16 |
| TWI694824B (zh) | 2020-06-01 |
| BR112020021664A2 (pt) | 2021-01-26 |
| EP3789384A4 (en) | 2021-12-01 |
| CN110407806A (zh) | 2019-11-05 |
| EP3789384A1 (en) | 2021-03-10 |
| MX2020011164A (es) | 2021-02-09 |
| US20210139454A1 (en) | 2021-05-13 |
| KR20210005135A (ko) | 2021-01-13 |
| AU2019260217A1 (en) | 2020-11-19 |
| AU2019260217B2 (en) | 2021-12-09 |
| CA3098202A1 (en) | 2019-10-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN114605401B (zh) | 一类含氧五元杂环化合物、合成方法、药物组合物及用途 | |
| JP5442717B2 (ja) | 二置換フタラジンヘッジホッグ経路アンタゴニスト | |
| TWI694824B (zh) | 甲醯胺類化合物、其製備方法及其應用 | |
| CN102482283A (zh) | Raf抑制剂化合物及其使用方法 | |
| KR20140072022A (ko) | 키뉴레닌-3-모노옥시게나제 억제제, 약학적 조성물 및 이의 사용 방법 | |
| WO2007036131A1 (fr) | Dérivés de carzole sulfamide et leur procédé de préparation | |
| CA2637573C (en) | 4-(3-benzoylaminophenyl)-6,7-dimethoxy-2-methylaminoquinazoline derivatives | |
| WO2020238785A1 (zh) | 包括甲基和三氟甲基的双取代磺酰胺类选择性bcl-2抑制剂 | |
| CN105705493A (zh) | 喹唑啉衍生物、其制备方法、药物组合物和应用 | |
| KR101908333B1 (ko) | 나프틸아미드계 화합물, 이의 제조 방법과 용도 | |
| WO2016112768A1 (zh) | 一种苯并氧化呋咱组蛋白去乙酰化酶抑制剂及其制备方法和应用 | |
| CN106660970A (zh) | 喹唑啉衍生物 | |
| CN109280032B (zh) | 一种哒嗪酮母核结构的组蛋白去乙酰化酶抑制剂及其制备方法和用途 | |
| WO2012149049A1 (en) | Tyrosine phosphatase inhibitors and uses thereof to modulate the activity of enzymes involved in the pathology of mycobacterium tuberculosis | |
| TW201934547A (zh) | 一種嘧啶類化合物、其製備方法及其醫藥用途 | |
| WO2019085978A1 (zh) | 杂芳基酰胺类化合物、其制备方法、药用组合物及其应用 | |
| TWI404709B (zh) | 4- (3-benzamidophenyl) -6,7-dimethoxy-2-methylamine quinazoline derivatives | |
| WO2014071824A1 (zh) | 4-氨基喹唑啉杂环化合物及其用途 | |
| JP2002322162A (ja) | チアゾリジン誘導体 | |
| CN113061098A (zh) | 酰胺化合物及其衍生物,制备方法、药物组合物和应用 | |
| CN111116565B (zh) | 2-芳基-4-(4-吡唑氧基)吡啶类化合物、其制备方法、药物组合物与应用 | |
| JP2017178811A (ja) | γターン構造を有する化合物及びそれを用いたLSD1阻害剤 | |
| CN116547277A (zh) | P300抑制剂及其在医药上的应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19791533 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3098202 Country of ref document: CA Ref document number: 2020559445 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112020021664 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2019260217 Country of ref document: AU Date of ref document: 20190423 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20207033670 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2019791533 Country of ref document: EP Effective date: 20201130 |
|
| ENP | Entry into the national phase |
Ref document number: 112020021664 Country of ref document: BR Kind code of ref document: A2 Effective date: 20201022 |