WO2019131719A1 - 高脂溶性ホスホラミダイトの製造 - Google Patents
高脂溶性ホスホラミダイトの製造 Download PDFInfo
- Publication number
- WO2019131719A1 WO2019131719A1 PCT/JP2018/047748 JP2018047748W WO2019131719A1 WO 2019131719 A1 WO2019131719 A1 WO 2019131719A1 JP 2018047748 W JP2018047748 W JP 2018047748W WO 2019131719 A1 WO2019131719 A1 WO 2019131719A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- reaction
- compound
- optionally substituted
- capping
- Prior art date
Links
- 0 CC*C(C)NC1(CC(C)(*C)C(C)*C(C)=C)CC1 Chemical compound CC*C(C)NC1(CC(C)(*C)C(C)*C(C)=C)CC1 0.000 description 9
- DFLXKSUJFLXBFO-UHFFFAOYSA-N CCC1C(C)(C2C)C2(C2)C2C2C1C2 Chemical compound CCC1C(C)(C2C)C2(C2)C2C2C1C2 DFLXKSUJFLXBFO-UHFFFAOYSA-N 0.000 description 2
- RKVHNYJPIXOHRW-UHFFFAOYSA-N CC(C)N(C(C)C)P(N(C(C)C)C(C)C)OCCC#N Chemical compound CC(C)N(C(C)C)P(N(C(C)C)C(C)C)OCCC#N RKVHNYJPIXOHRW-UHFFFAOYSA-N 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/22—Amides of acids of phosphorus
- C07F9/24—Esteramides
- C07F9/2454—Esteramides the amide moiety containing a substituent or a structure which is considered as characteristic
- C07F9/2458—Esteramides the amide moiety containing a substituent or a structure which is considered as characteristic of aliphatic amines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/10—Pyrimidine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/20—Purine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/04—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with deoxyribosyl as saccharide radical
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to the preparation of highly lipid soluble phosphoramidites. More particularly, the present invention relates to a method for purifying and producing a highly lipid-soluble phosphoramidite, a solution for capping reaction as a solution containing the lipid-rich phosphoramidite compound, a capping reaction using the solution for capping reaction, and the capping reaction There is also provided a method of producing an oligonucleotide according to a liquid phase method comprising the steps of
- Methods for producing oligonucleotides include the phosphate triester method, the H-phosphonate method, the phosphoramidite method, etc.
- solid phase synthesis solid phase method
- Blockmer synthesis method a method of repeating linking of building blocks of about 2-3 nucleotides
- an oligonucleotide of about 10 bases or so Methods by linking (fragment condensation method) and the like are known.
- Solid-phase synthesis is advantageous in terms of synthesis speed such as using an automatic synthesizer, but synthetic scale-up is limited, and an excessive amount of reaction reagents and reaction raw materials are required. In the middle stage, it is difficult to check the progress of the reaction and to analyze intermediates. On the other hand, the synthesis of oligonucleotides by the liquid phase method is complicated in operation and low in yield, so that it is difficult to synthesize a large amount of oligonucleotides having a high degree of polymerization rapidly.
- oligonucleotide synthesis methods using polyethylene glycol (MPEG) have been reported for the purpose of solving the respective drawbacks of the solid phase method and the liquid phase method (Non-patent documents 1 to 3).
- MPEG polyethylene glycol
- the synthesis example of the oligonucleotide of up to about 20-mer has been reported by this method, the crystallization isolation operation and the recrystallization are required at each step in the elongation cycle, and there is a problem of being complicated.
- N-1 and N + 1 by-products are generated with respect to the target oligonucleotide by, for example, the participation of unreacted products such as unreacted raw materials in the extension step of the oligonucleotide.
- these by-products are also extremely difficult to be separated from the desired product. Therefore, when a synthetic oligonucleotide product is used as a nucleic acid drug, an oligonucleotide is obtained at a purity higher than that of the oligonucleotide obtained by the conventional method by reducing the formation of these N-1 and N + 1 by-products.
- the extension reaction proceeds in competition with the reactive site of the target oligonucleotide chain in the next synthesis cycle.
- Capping reagents have been used in solid phase manufacturing methods to prevent the formation of N-1 oligonucleotides.
- the reaction in the liquid phase system is more reactive than the solid phase method, such as the coupling reaction.
- the solid phase method such as the coupling reaction.
- the reason why the capping reagent is not used in the liquid phase method using a pseudo solid phase protecting group is considered to be due to the following problems, in addition to the above-mentioned reactivity.
- a large excess of acetic anhydride and a base such as a large excess of imidazole derivative are usually used as a capping reagent.
- An additional purification step is required to remove it, and the nature (eg hydrophobicity) of the capping product of the unreacted oligonucleotide chain is intermediate between the oligonucleotide chain and the product of the coupling reaction product.
- the nucleobase is further partially acetylated (mainly 6-amide of adenine, partially 2-amide of guanine, 4-amide of cytidine, 3-position of thymine) to form a diacyl form to form a reaction mixture
- acetylated mainly 6-amide of adenine, partially 2-amide of guanine, 4-amide of cytidine, 3-position of thymine
- each compound in these mixtures gives similar reaction products in the next synthesis cycle, resulting in more complex reaction mixtures, which are analyzed by conventional instrumental analysis methods (eg, TLC, HPLC)
- TLC time difference between the nucleotide
- HPLC HPLC
- Non- Patent Document 6 a quenching agent such as an organic carboxylic acid base salt or alcohol is added immediately after the extension by phosphoramidite as in Non-patent document 1 to improve the purity of the product. Has succeeded (Patent Document 2).
- the capping reagent is also intended to suppress the formation of N-1 by-product generated in oligonucleotide synthesis to improve the purity of the target oligonucleotide product.
- an alkoxyphosphoramidite compound is used in addition to acetic anhydride described above.
- a primary alkoxyphosphoramidite compound having high reactivity is used (patent documents 3, 4 and non-patent documents) 8, 9). Also, within these documents, side reactions of primary alkoxy phosphoramidites and nucleobases (mainly at guanine 6 position carbonyl oxygen) have not been reported.
- Patent Documents 6 and 7 Methods of recovering a phosphorodiamidite compound usable as a raw material of a phosphoramidite compound by adding a poor solvent (for example, alcohols, hydrocarbons, nitriles) to a reaction mixture liquid have been reported (Patent Documents 6 and 7) ). However, it can not be predicted whether the same method can be used for highly reactive primary alkoxy phosphoramidite compounds, and it is considered that appropriate selection of raw material alcohols is required.
- a poor solvent for example, alcohols, hydrocarbons, nitriles
- the present invention relates to a process for the preparation of highly lipid soluble phosphoramidites. More specifically, the present invention provides a simple method for purification and preparation of highly lipid-soluble phosphoramidite which is an alkoxyphosphoramidite compound that can be used as a capping reagent at the time of oligonucleotide synthesis, and highly lipid-soluble phosphoramidite obtained by the method.
- a solution for capping reaction as a solution containing a compound, a capping reaction using the solution for capping reaction, and a method for producing an oligonucleotide by a liquid phase method using a pseudo solid phase protecting group, comprising the step of capping reaction Also provide.
- the present invention provides a highly fat-soluble phosphoramidite which is one of alkoxyphosphoramidite compounds that can be used as a capping reagent at the time of oligonucleotide synthesis, without requiring operations such as column purification, and the types of dissolving solvents and washing solvents, and the same.
- the inventors have found a manufacturing method that can be easily purified by separation operation and the like by sorting the washing order, and can be industrially manufactured on a large scale with high purity.
- the capping reaction is achieved when the solution containing the highly lipid-soluble phosphoramidite obtained by the production method is used as a capping reaction solution in a liquid phase method (in particular, a liquid phase method using a pseudo solid phase protecting group).
- the present invention provides the following aspects, but is not limited thereto.
- R 1 is a C6-C30 alkyloxy group which may be substituted, and the substituent is at least one group selected from a C1-C3 alkyl group or a C3-C6 cycloalkyl group
- Each R 2 is independently an optionally substituted C 1 -C 6 alkyl group, and the substituent is at least one group selected from a C 1 -C 3 alkyl group or a C 3 -C 6 cycloalkyl group
- R 3 is a C6-C30 alkyloxy group which may be substituted, a C1-C8 alkyloxy group which may be substituted, or a C2-C8 alkynyloxy group which may be substituted, wherein
- the substituent in the C6-C30 alkyloxy group which may be substituted is at least one group selected from a C1-C3 alkyl group or
- the aliphatic hydrocarbon solvent used in step (4) is at least one selected from pentane, hexane, heptane or octane, and the nitrile solvent used in step (5) is acetonitrile
- Item [3-1] R 3 is a C1-C8 alkyloxy group which may be substituted, or a C2-C8 alkynyloxy group which may be substituted, and the substituent is a cyano group (CN)
- Item [3-2] The production method according to Item [1] or Item [2], wherein R 3 is a C1-C8 alkyloxy group which may be substituted.
- R 1 is an optionally substituted C 10 -C 30 primary or secondary alkyloxy group, and R 3 is —OCH 2 CH 2 CN, —OCH 3
- the production method according to Item [1] or Item [2], which is a group represented by -OCH 2 CH 3 or Item [4] The process according to any one of Items [1] to [3], wherein each R 2 is an i-propyl group.
- the compound represented by the formula (I) has the following formula: The method according to any one of Items [1] to [4], which is a compound represented by Item [6] Any one of Items [1] to [5], wherein the solution obtained in Step (6) is a solution of a substantially pure phosphoramidite compound containing no H-phosphonate compound or diamidite compound. The manufacturing method as described in a section.
- composition for capping reaction and capping reaction A composition for a capping reaction, containing a solution obtained by the method according to any one of items [1] to [6], and an additive as appropriate.
- Item [8] A reaction solution obtained by coupling and reacting two or more compounds selected from nucleosides, nucleotides, or oligonucleotides with the composition for capping reaction described in item [7], optionally in the presence of an activator.
- a capping method comprising the step of contacting.
- Step (a): In a nonpolar solvent, the pseudo solid phase protecting group is modified via the 3 'hydroxyl group or nucleobase, and the 5' hydroxyl group is blocked with a temporary protecting group removable under acidic conditions Mer (wherein n represents an arbitrary integer of 1 or more) or, when n 0, one hydroxyl group of a linker having two or more hydroxyl groups is modified with a pseudo solid phase protecting group and another A compound blocked with a temporary protecting group whose hydroxyl group can be removed under acidic conditions is reacted with an acid alone or in the presence of a cation scavenger to deblock the temporary protective group of the hydroxyl group and then use a polar solvent.
- a polar solvent wherein n represents an arbitrary integer of 1 or more
- m + 1 polymerizing oligonucleotide where m is any integer of 0 or more
- Step (capping reaction step) Step (d): adding a nucleophile to the capping byproduct of step (c) and reacting, wherein the nucleophile is selected from an alcohol, a phenol, a carboxylic acid or an N-alkylhydroxylamine Selected step (post-treatment step of capping reaction);
- R 5 are each independently an alkyl group optionally substituted by C6-C30
- R 6 is optionally substituted C 1 -C 6 alkyl group, optionally substituted C 3 -C 6 cycloalkyl group, optionally substituted non-aromatic heterocyclic group, optionally substituted An aryl group, an optionally substituted heteroaryl group, an optionally substituted aralkyl group, or an optionally substituted heteroarylalkyl group
- R 7 is an optionally substituted C 1 -C 6 alkylene group or an optionally substituted C 1 -C 6 alkyleneoxy group
- R 8 is an optionally substituted C 1 -C 6 alkylene group, or R 6 and R 7 may be taken together with the nitrogen atom to which R 6 is attached to form an optionally substituted non-aromatic heterocyclic group or an optionally substituted heteroaryl
- R 6 , R 7 and R 8 together with the nitrogen atom to which R 6 is bonded, an optionally substituted non-aromatic heterocyclic group, an optionally substituted non-aromatic heterocyclic ring It may form an alkyl group, an optionally substituted heteroaryl group, or an optionally substituted heteroarylalkyl group, and * Indicates the bonding position with L] Pseudo solid-phase protecting group represented by
- n EWGs is independently an electron withdrawing group
- n R 4 s each independently represent an oxygen atom or a sulfur atom
- n Rs each independently represent a hydrogen atom, a halogen atom, a hydroxy group which may be substituted with a protecting group, a C1-C6 alkoxy group which may be substituted, an organic group which crosslinks to the 4-position carbon atom, Or a cross-linked artificial nucleic acid group
- n is any integer greater than or equal to 0
- X is an acid deprotectable temporary protecting group
- a nucleotide compound represented by The term [14] L has the following formula: [In the formula, * Indicates the bonding position with a hydrophobic group, and ** indicates the bonding position with a phosphorus atom
- Each Base is a nucleobase which may be independently protected
- R each independently represents a hydrogen atom, a halogen atom, a hydroxy group which may be substituted with a protecting group, a
- the high fat-soluble phosphoramidite By the method for producing and purifying the high fat-soluble phosphoramidite of the present invention, the high fat-soluble phosphoramidite can be easily purified and produced in high purity.
- a solution containing the highly lipid-soluble phosphoramidite obtained by the production method of the present invention, or a composition containing the solution and an additive as appropriate can be used as a reagent for capping reaction, and also nucleophilicity after capping reaction. In combination with post-treatment with a reagent, single extension of nucleotide chain length can be efficiently performed.
- the invention relates to compounds of formula (I) [In the formula, R 1 is a C6-C30 alkyloxy group which may be substituted, and the substituent is at least one group selected from a C1-C3 alkyl group or a C3-C6 cycloalkyl group, Each R 2 is independently an optionally substituted C 1 -C 6 alkyl group, and the substituent is at least one group selected from a C 1 -C 3 alkyl group or a C 3 -C 6 cycloalkyl group, And R 3 is a C6-C30 alkyloxy group which may be substituted, a C1-C8 alkyloxy group which may be substituted, or a C1-C8 alkynyloxy group which may be substituted, wherein The substituent in the C6-C30 alkyloxy group which may be substituted is a C1-C3 alky
- a method for producing a compound represented by the formula (hereinafter referred to as "the highly lipid-soluble phosphoramidite compound of the present invention"), which comprises the following steps: (1) reacting an aliphatic alcohol and a trivalent phosphorus compound in an organic solvent in the presence of an activating agent or an organic base; (2) Separately wash the obtained reaction mixture with water; (3) Recover and concentrate the organic layer after step (2) (however, when the organic solvent used in step (1) is a nitrile solvent, steps (2) to (3) are omitted) Also good); (4) Dissolving the residue obtained in step (3) in an aliphatic hydrocarbon solvent (however, if the organic solvent used in step (1) is an aliphatic hydrocarbon solvent, step (2) ) To (4) may be omitted); (5) The aliphatic hydrocarbon solution prepared in step (4) is separated and washed using a nitrile solvent; (6) The aliphatic hydrocarbon-based solution after step (5) is recovered to obtain a phosphoramidite compound as a solution
- the phosphoramidite compound represented by the formula (I) obtained by the production method of the present invention has high affinity to organic solvents (for example, hydrophobic solvents such as aliphatic hydrocarbon solvents), while it has affinity to water. It has low, highly fat-soluble nature.
- organic solvents for example, hydrophobic solvents such as aliphatic hydrocarbon solvents
- the term "optionally substituted C6-C30 alkyloxy group" for R 1 and / or R 3 means a hydrocarbon chain having 6 to 30 carbon atoms bonded to an oxygen atom.
- At least one group selected from a C6 cycloalkyl group, and the alkyloxy group may optionally contain an aromatic ring, a cyano group, a group containing an ester bond, or a group containing an amide bond, Means a group.
- the alkyl group may be linear or branched, but a linear alkyl group is preferable.
- a group that is more hydrophobic than the temporary protecting group X at the 5'-position of the nucleic acid is preferable.
- a C8-C30 primary or secondary alkyloxy group is preferable, a C10-C30 primary or secondary alkyloxy group is more preferable, and a primary or secondary C10-C20 alkyloxy groups are even more preferred.
- One preferable example is a C18 primary or secondary alkyloxy group, particularly a C18 primary alkyloxy group.
- substituent is not limited as long as the reaction used in the present invention (for example, capping reaction, coupling reaction on oligonucleotide synthesis, deprotection reaction, deblocking reaction, oxidation reaction, sulfurization reaction) is not inhibited.
- Non-limiting examples include one or more groups selected from, for example, C1-C6 alkyl groups, or C3-C6 cycloalkyl groups.
- Specific examples of the substituent include methyl, ethyl, isopropyl, cyclopropyl, cyclohexyl and the like.
- C1-C6 alkyl group examples include methyl, ethyl, propyl, isopropyl, butyl, pentyl and hexyl.
- C3-C6 cycloalkyl group cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl is mentioned.
- the “group containing an ester bond” means an alkylcarboxy group, such as methylcarboxy, ethylcarboxy or isopropylcarboxy.
- the group containing an ester bond is intended to include both an acyloxy type group and an alkoxycarbonyl type group.
- the “group containing an amide bond” means an alkylamide group which may be substituted by an alkyl group on a nitrogen atom, and examples thereof include amide, N, N-dimethylamide and N, N-diethylamide.
- the group containing an amide bond is intended to include both an amide type group and a carboxamide type group.
- R 1 is an optionally substituted C 10 -C 30 primary or secondary alkyloxy group
- R 3 is —OCH 2 CH 2 CN -OCH 3 or -OCH 2 CH 3 , with -OCH 2 CH 2 CN being preferred.
- the term "optionally substituted C1-C6 alkyl group" for R 2 means a saturated hydrocarbon chain group having 1 to 6 carbon atoms.
- the alkyl group may be linear or branched.
- a C3 alkyl group is preferable.
- One preferred example is isopropyl.
- the kind of substituent is not limited as long as the reaction used in the present invention (for example, capping reaction, coupling reaction on oligonucleotide synthesis, deprotection reaction, deblocking reaction, oxidation reaction, sulfurization reaction) is not inhibited. Examples include, but are not limited to, one or more groups selected from, for example, a C1-C3 alkyl group and a C3-C6 cycloalkyl group, with a C1-C3 alkyl group being preferred.
- each R 2 is independently an optionally substituted C 1 -C 6 alkyl group, for example an optionally substituted C 3 -C 6 alkyl group is preferred.
- each R 2 is both an i-propyl group.
- the terms "optionally substituted C1-C8 alkyloxy group” and “optionally substituted C2-C8 alkynyloxy group” for R 3 are carbons attached to an oxygen atom It means an alkyl group having 1 to 8 or an alkynyl group having 2 to 8 carbon atoms, which may be optionally substituted with a cyano group.
- Each of the alkyl group and the alkynyl group is preferably a primary or secondary group, and may be either a linear or branched group, but a linear group is preferred.
- a C1-C6 alkyloxy group or a C1-C5 alkynyloxy group is preferable, and a C1-C3 alkyloxy group is more preferable.
- preferable R 3 include, for example, a group represented by -OCH 2 CH 2 CN, -OCH 3 or -OCH 2 CH 3 , and -OCH 2 CH 2 CN is more preferable.
- R 1 is an optionally substituted C 10 -C 30 primary or secondary alkyloxy group
- R 3 is It is a group represented by -OCH 2 CH 2 CN, -OCH 3 or -OCH 2 CH 3 .
- the compounds of formula (I) according to the invention are so-called amidite-type compounds (monoamidite compounds) and can therefore be used as capping reagents in the preparation of the oligonucleotides according to the invention using the phosphoramidite method.
- the compound represented by the formula (I) used in the present invention is a compound having high lipid solubility, that is, high hydrophobicity as compared with a phosphoramidite compound of a nucleotide or oligonucleotide for coupling reaction used in the phosphoramidite method.
- the physical properties eg, affinity with silica gel and ODS modified silica gel
- the physical properties are significantly different from the target oligonucleotide product and the unreacted raw material remaining in the coupling reaction, and thus, the target oligonucleotide product and Unreacted raw material to which the capping reagent is bound can be detected separately.
- the compound represented by the formula (I), which is the capping reagent of the present invention is not reactive because the three-dimensional structure around the phosphoramidite is not crowded compared to the nucleotide phosphoramidite compound for coupling reaction. It has high capping reactivity with the anti-source nucleotides or oligonucleotides, so that the desired coupling reaction can be achieved with a small amount used.
- aliphatic alcohol in step (1) means a hydrocarbon aliphatic alcohol having 6 to 30 carbon atoms, preferably a primary or secondary hydrocarbon aliphatic alcohol, and the is defined as R 1, it may include an aromatic ring, corresponding to aliphatic alcohols to provide the optionally substituted C6-C30 alkyl group.
- Preferred are long-chain primary or secondary aliphatic alcohols having C8 to C30 carbon atoms, more preferably C10 to C30 carbon atoms, and still more preferably C10 to C20 carbon atoms.
- One preferred embodiment is a C18 aliphatic alcohol, in particular a C18 primary aliphatic alcohol.
- trivalent phosphorus compound in step (1) means, for example, alkyl phosphorodiamidite, alkyl chlorophosphoramidite. Specific examples include 2-cyanoethyl N, N, N ', N'-tetraisopropylphosphorodiamidite, and 2-cyanoethyl diisopropyl chlorophosphoroamidite.
- activator in step (1) includes, for example, tetrazole compounds. Specific examples thereof include 1H-tetrazole and 5-ethylthio-1H-tetrazole (abbreviated as ETT), but are not limited thereto.
- organic base in step (1) is used when using alkyl chlorophosphoroamidite as a trivalent phosphorus compound, and examples include triethylamine or diisopropylethylamine.
- organic solvent in step (1) includes, for example, halogen solvents, nitrile solvents and cyclic ether solvents, with halogen solvents being preferred. These organic solvents are preferably anhydrous organic solvents. Specific examples include, but are not limited to, dichloromethane, chloroform, carbon tetrachloride, dioxane, tetrahydrofuran and the like. Dichloromethane and chloroform are preferred.
- the reaction temperature of the reaction of step (1) is usually -20 to 50 ° C.
- the reaction time is usually in the range of 0.1 to 10 hours.
- the molar ratio of the aliphatic alcohol to the trivalent phosphorus compound may be, for example, 0.5 to 1.5 molar equivalents, but preferably the trivalent phosphorus compound is in excess (for example, 1.1 molar equivalents). There is a case.
- step (2) the reaction mixture obtained in step (1) is poured into a separatory funnel, and the organic layer is separated and washed with water (eg, distilled water).
- water eg, distilled water
- the amount of water to be washed is not particularly limited, but can be used at a volume ratio of 0.1 to 5 v / v with respect to the amount of the organic solvent used in step (1).
- Step 3 The organic layer after the washing in step (2) is recovered, and the organic solvent is distilled off using an evaporator or the like under reduced pressure, for example, and concentrated.
- the organic solvent used in the step (1) is a nitrile solvent, carrying out the steps (2) to (3) can be omitted.
- Step 4 An aliphatic hydrocarbon solvent is added to the residue obtained in step (3) to dissolve it.
- aliphatic hydrocarbon solvents include, but are not limited to, pentane, hexane, heptane and octane. Pentane, hexane and heptane are preferred, especially heptane is preferred.
- the amount of the aliphatic hydrocarbon solvent added may be changed according to the solubility of the reaction product high fat-soluble phosphoramidite compound in the aliphatic hydrocarbon solvent used, but it may be an appropriate amount to dissolve, for example, And about 1 to 500 times mL, and typically about 1 to 10 times mL, per 1 mmol of the aliphatic alcohol used in the step (1).
- the organic solvent used in the step (1) is an aliphatic hydrocarbon solvent, carrying out the steps (2) to (4) can be omitted.
- Step 5 The aliphatic hydrocarbon solution prepared in step (4) is poured into a separatory funnel and washed with a nitrile solvent.
- a nitrile solvent is not particularly limited as long as it is a solvent that is phase separated in the organic layer of the solution of the product in the aliphatic hydrocarbon solvent and separated. And acetonitrile, propionitrile and benzonitrile. Acetonitrile is particularly preferred.
- the amount of the nitrile solvent to be washed is not particularly limited, but it can be used at a volume ratio of 0.1 to 10 v / v with respect to the amount of the organic solvent used in the step (1).
- the number of washings is not limited to one, and the presence of H-phosphonate compounds and / or diamidite compounds is confirmed by 31 P NMR measurement of a part of the washed aliphatic hydrocarbon solution. Therefore, the number of times of washing can be easily determined.
- Step 6 The aliphatic hydrocarbon solution obtained after the separation and washing in step 5 is recovered to obtain the desired phosphoramidite compound as a solution.
- the resulting solution can be used for the next reaction step (e.g., capping reaction step) by removing dissolved water.
- the obtained solution can be concentrated, for example, using an evaporator under reduced pressure, if necessary, and used for the next reaction step.
- the production of the highly lipid-soluble phosphoramidite compound represented by the formula (I) of the present invention can be carried out as a post-treatment after the production reaction, and the purification thereof can be carried out only by liquid separation treatment. it can.
- the obtained product contains a small amount (eg, 0.1% or more) of a phosphorous triester compound which is an alkyl trisubstituted compound of the compound of formula (I)
- phosphite triester compounds do not have any reactivity with oligonucleotide raw materials and products, they do not pose a problem when used as capping reagents even if they are contained in compounds of formula (I) .
- the solution of the objective phosphoramidite compound (monoamidite compound) in the aliphatic hydrocarbon solvent obtained in the step (6) uses water in the step (2) and a nitrile solvent in the step (4).
- Oligonucleotide raw materials and H-phosphonate compounds for example, 2-cyanoethyl N, N-diisopropylphosphonamidate
- diamidite compounds, hydrocarbon aliphatic alcohols for raw materials for example, Examples 6
- the present invention adds a solution containing a highly fat-soluble phosphoramidite compound or a suitable additive (for example, a stabilizer), which is produced by the method for producing a highly fat-soluble phosphoramidite of the present invention
- the present invention provides a capping method (hereinafter referred to as “the capping reaction of the present invention") in which the composition is used as a reagent for the capping reaction (hereinafter referred to as "the reagent for the capping reaction of the present invention”).
- the capping reaction is shown below as Reaction Scheme I. Reaction scheme I
- the term "capping” refers to a coupling reaction of two or more compounds selected from nucleosides, nucleotides, or oligonucleotides, typically nucleotides, in the reaction of elongation of nucleotide chains (eg, phosphoramidite In the method, it means protecting the group of the reactive site of the unreacted chain.
- Capping competes with the group of reactive site of the target oligonucleotide chain in the next coupling reaction to prevent the progress of the extension reaction at the reactive site of the oligonucleotide chain remaining before the coupling reaction. It is possible to prevent byproducts of N-1 oligonucleotide.
- the invention is The reaction solution after the coupling reaction of two or more compounds selected from nucleosides, nucleotides or oligonucleotides is appropriately prepared for the capping reaction reagent of the present invention (that is, the highly lipid-soluble phosphoramidite of the present invention) in the presence of an activator.
- the capping reaction reagent of the present invention that is, the highly lipid-soluble phosphoramidite of the present invention
- the capping method of the present invention may further include the step of reacting the capping side reaction product with a nucleophile (same step (d)) after the capping reaction step, if necessary.
- it may further include the step of adding an organic base after the capping reaction step and then reacting with a nucleophile.
- the organic base include, but not limited to, 2,4,6-collidine, 2-picoline, 2,6-lutidine, 1-methylimidazole and pyridine.
- nucleoside refers to a covalent attachment to a sugar moiety (eg, a ribose sugar or the like), a derivative of a sugar moiety, or one having a functional equivalent to a sugar moiety (eg, an analog such as a carbocycle)
- a nucleic acid component comprising a base or a basic group (eg, at least one homocyclic ring, at least one heterocyclic ring, at least one aryl group, etc.).
- a base is attached to the 1 'position of the sugar moiety. Examples of the base include those described later.
- nucleotide refers to an ester of a nucleoside, such as a phosphate ester of a nucleoside.
- oligonucleotide refers to duplex formation by complementary strands of at least two nucleotides, typically three or more (eg, four) nucleotides, and more typically at room temperature.
- a nucleic acid comprising more than 10 possible nucleotides, the total number of nucleotides is designated n in the present invention.
- the oligonucleotide is a chemically modified product thereof (eg, a crosslinked artificial nucleic acid (BNA :), such as a single-stranded nucleotide, or a nucleotide having a 2'-O, 4'-C-methylene bridge in its sugar moiety).
- BNA crosslinked artificial nucleic acid
- BNAs include, for example, Locked Artificial Nucleic Acid (LNA: Locked Nucleic Acid), 2′-O, 4′-C-ethylene-bridged nucleic acid (ENA: 2′-O, 4′-). C-Ethylenebridged (Nucleic Acid), etc.
- LNA Locked Artificial Nucleic Acid
- ENA 2′-O, 4′-C-ethylene-bridged nucleic acid
- C-Ethylenebridged Nucleic Acid
- a specific structure (nucleoside moiety) of BNA including LNA and ENA that can be used in the present invention is described in WO 2016/006697. Indicate citations. Modifications include, but are not limited to, modified backbones of oligonucleotides, and are typically described in Micklefield (2001), "Current Medicinal Chemistry” 8: 1157-1170.
- oligonucleotide consists of any combination of the aforementioned nucleotides and the aforementioned modifications.
- Base is each independently a group selected from an adenine residue, guanine residue, cytosine residue, thymine residue, or uracil residue, and these residues are appropriately selected. It means a group which may be substituted by a protecting group.
- nucleobase residues are: And the groups represented by
- step (c) a reaction solution after the coupling reaction of two or more compounds selected from nucleosides, nucleotides or oligonucleotides is obtained by the method for producing a highly lipid-soluble phosphoramidite of the present invention in the presence of an activator as appropriate. Contacting with a solution containing the highly lipid soluble phosphoramidite compound, or a composition containing the solution and, optionally, additives.
- Two or more compounds selected from nucleosides, nucleotides, or oligonucleotides used in the coupling reaction of step (c) are one after the deblocking reaction after step (a) in reaction scheme I
- the 5'-hydroxyl group is deblocked and the 3'-hydroxyl group of the nucleobase represented by the formula: HO- (N) n -L-Z (where n represents any integer of 1 or more) N-polymerized nucleosides, nucleotides or oligonucleotides wherein at least one group is substituted by a pseudo solid phase protecting group, and the other is a compound of the formula: X- (N) m -Np, wherein m is 0 or more And an oligonucleotide of m + 1 polymerization in which the 5 'hydroxyl group is protected with a temporary protecting group removable under acidic conditions and the 3' hydroxyl group is phosphoramidated. It is.
- the reaction temperature of the capping reaction of step (c) is usually ⁇ 20 to 80 ° C.
- the reaction time is usually in the range of 0 to 30 hours. In addition, it is recommended to carry out in an atmosphere of inert gas.
- the amount of the highly lipid-soluble phosphoramidite compound of the present invention used as the capping reagent is the n -polymerized nucleoside, nucleotide or oligonucleotide represented by the formula: HO- (N) n -L-Z used in the coupling reaction
- the compounding molar ratio is, for example, 0.1 to 1.5 molar equivalents, preferably 0.1 to 0.5 molar equivalents, relative to the amount of
- activators examples include reagents commonly known to be capable of being used in capping reactions using the phosphoramidite method, for example tetrazole (eg 1H- And tetrazole, 5-ethylthio-1H-tetrazole (abbreviated as ETT).
- ETT tetrazole
- the amount of the activator used is, for example, 1.0 to 10.0 molar equivalents, preferably 1.0 to 2., based on the amount of the highly lipid-soluble phosphoramidite compound of the present invention as a capping reagent. Although 0 molar equivalent may be mentioned, an appropriate amount may be added in advance in step (b).
- the capping method of the present invention may further include the step of reacting the capping by-product with a nucleophile after the capping reaction step of step (c), if necessary.
- the nucleophile include compounds selected from alcohol, phenol, carboxylic acid or N-alkylhydroxylamine, and specific examples include ethanol, methanol and N-hydroxysuccinimide, preferably Ethanol is mentioned.
- the amount of the nucleophile used is, for example, 0.1 to 100 molar equivalents, preferably 1 to 20 molar equivalents, based on the amount of the highly lipid-soluble phosphoramidite compound of the present invention as the capping reagent. It can be mentioned.
- the present invention also provides a method of producing an oligonucleotide comprising the steps of the capping method of the present invention.
- reaction scheme II the typical scheme figure of the manufacturing method of the oligonucleotide of this invention is shown to reaction scheme II. Reaction scheme II
- the method for producing the oligonucleotide of the present invention (hereinafter referred to as “the production method of the present invention”) will be described along the description of the above-mentioned reaction scheme II, but it may be a commonly known method for producing oligonucleotides. It is not limited to the reaction scheme of Reaction Scheme II. Specifically, a method for producing an appropriately protected n-mer (that is, n-mer) oligonucleotide to an appropriately protected n + 1 + m-mer (that is, n + 1 + m-mer) oligonucleotide will be described.
- the n-mer oligonucleotide may contain "nucleoside"
- the m + 1-mer oligonucleotide may contain "nucleoside” .
- the method for producing the oligonucleotide of the present invention comprises the following steps (a) to (e).
- Step (a): deblocking step and crystallization step The reaction scheme 1 of step (a) is shown below.
- a compound which is blocked by a removable temporary protecting group for example, a compound represented by the above X- (N) n -L-Z, is reacted in the presence of an acid alone or in the coexistence of a cation scavenger to protect a hydroxyl protecting group.
- Deblocking step deblocking step
- a step of recovering a compound containing the pseudo solid phase protecting group for example, a compound represented by the above H- (N) n -L-Z using a polar solvent (crystallization step), including.
- step (a) The deblocking step reaction in step (a) can be carried out under the reaction conditions described in the literature (for example, WO 2012/157723 A1). The details will be described below.
- the “pseudo-solid phase protecting group” may include the group described in the literature (eg, WO 2012/157723 A1). Examples include, but are not limited to, the quasi-solid phase protecting group described later in the specification.
- the temporary protecting group removable under acidic conditions at the 5'-position hydroxyl group is not particularly limited as long as it is deprotectable under acidic conditions and can be used as a protecting group for hydroxyl group, for example, trityl group, 9- (9-phenyl) xanthenyl group, 9-phenylthioxanthenyl group, 1,1-bis (4-methoxyphenyl) -1-phenylmethyl group (abbreviated as dimethoxytrityl group), 1- (4-methoxyphenyl) And the like), etc. can be mentioned. Dimethoxytrityl and monomethoxytrityl are preferred.
- the reaction solvent used in the deblocking step may be any solvent which does not affect the deblocking reaction, but a solvent having high solubility of the (oligo) nucleotide compound which is a reaction substrate is preferable, for example, a nonpolar solvent is preferable.
- a nonpolar solvent include halogen solvents, aromatic solvents, ester solvents, aliphatic solvents, nonpolar ether solvents, and solvents selected from the group consisting of a combination thereof.
- Examples include dichloromethane, chloroform, 1,2-dichloroethane, benzene, toluene, xylene, mesitylene, hexane, pentane, heptane, nonane, cyclohexane, ethyl acetate, isopropyl acetate, isopropyl acetate, tert-butyl methyl ether, cyclopentyl methyl ether, and The solvent selected from the group consisting of these combinations is mentioned.
- the concentration of the n-mer oligonucleotide in the solvent in the present deblocking step is not particularly limited as long as it is dissolved, but is preferably 1 to 30% by weight.
- the acid that can be used in the present deblocking step include, but are not particularly limited to, for example, trifluoroacetic acid, trichloroacetic acid, trifluoromethanesulfonic acid, trichloroacetic acid, methanesulfonic acid, hydrochloric acid, acetic acid, p-toluenesulfone An acid etc. are mentioned. Trifluoroacetic acid, dichloroacetic acid and trichloroacetic acid are preferred.
- the amount of the acid used can be 1 to 100 mol, preferably 1 to 40 mol, per 1 mol of n-mer (oligo) nucleotide.
- the acid may be used alone or may be used in coexistence with a cation scavenger.
- cation scavengers include, but are not limited to, pyrrole derivatives (eg, pyrrole) and indole derivatives (eg, pyrrole), as long as reprotection with the removed protecting group X and side reactions to the deprotected functional group do not proceed.
- indole furan derivatives can be mentioned.
- the amount of the cation scavenger to be used may be 1 to 50 moles, preferably 5 to 20 moles, per mole of the n-mer oligonucleotide.
- the reaction temperature of the deblocking step is not particularly limited as long as the reaction proceeds, but for example, -10 to 50 ° C. is preferable, and 0 to 40 ° C. is more preferable.
- the reaction time may vary depending on the reaction conditions, for example, the type of n-mer (oligo) nucleotide as a reaction substrate, the type of acid, the type of solvent, the reaction temperature and the like, but may be 5 minutes to 5 hours.
- the step (a) includes the step of crystallizing a compound containing the pseudo solid phase protecting group using a polar solvent as it is or after the deblocking step after the deblocking step to recover the compound.
- polar solvents examples include alcohol solvents and nitrile solvents, and specific examples include methanol or acetonitrile.
- Step (b): Coupling step The reaction scheme 2 of step (b) is shown below. Reaction scheme 2
- step (b) can be carried out under the reaction conditions described in the literature (for example, WO 2012/157723 A1). The details will be described below.
- step (b) After drying the recovered product of step (a), m + 1 unit polymerization in which the 3 'hydroxyl group is phosphoramidated and the 5' hydroxyl group is protected by a temporary protecting group removable under acidic conditions in a nonpolar solvent (oligo N) polymerization of the 5 'hydroxyl group deblocking group obtained in step (a) by addition of nucleotides (here, m represents any integer of 0 or more) and an activator; Step of Condensing Oligonucleotide and Nucleotide Triester Linkage via the 5'-hydroxyl Group (Coupling Step) including.
- oligo N nonpolar solvent
- the upper limit of m is not particularly limited, but is preferably 49 or less, more preferably 29 or less, still more preferably 19 or less, still more preferably 4 or less, and particularly preferably 2 or less (for example, 0). .
- the “m + 1 single-polymerized (oligo) nucleotide protected by a temporary protecting group in which the 3′-hydroxyl group is phosphoramidated and the 5′-hydroxyl group is removable under acidic conditions is the 5 ′ obtained in the step (a).
- (Oligo) nucleotide eg, a compound represented by X- (N) n -L-Z
- a weak acid or an acidic neutralization salt as an activator.
- weak acids or acidic neutralizing salts include pyridine trifluoroacetate, 1H-tetrazole, 5-benzylthio-1H-tetrazole, 4,5-dicyanoimidazole and the like.
- the amount of the weak acid or acidic neutralization salt used is 0 per 1 mole of n-polymerized (oligo) nucleotide in which the protective group at the 5'-hydroxyl group obtained in step (a) is deblocked as a reaction substrate. 1 to 50 molar equivalents can be used, for example, 1 to 5 molar equivalents can be mentioned.
- N-methylimidazole is used for the purpose of suppressing the acidification of the reaction solution. It is preferable to add.
- the amount of the N-methylimidazole used is 0.1 to 10 molar equivalents relative to 1 mole of the n-polymerized (oligo) nucleotide deblocked at the 5'-hydroxyl group obtained in step (a) It can be used, for example, 0.1 to 1 molar equivalent.
- step (a) The compounding ratio with n-polymerized (oligo) nucleotides in which the protective group of the hydroxyl group is deblocked is the n-mer polymerization in which the protective group of the 5 'hydroxyl group obtained in step (a) is deblocked (oligo ) 1 to 10 moles of m + 1 polymerizing (oligo) nucleotide in which the 3 'hydroxyl group is phosphoramidite and the 5' hydroxyl group is protected with a temporary protecting group removable under acidic conditions per 1 mol of the nucleotide) It can be used, preferably 1 to 5 molar equivalents.
- This step is carried out in a solvent that does not affect the coupling reaction.
- the nonpolar solvent similar to the said process (a) is mentioned.
- nitrile solvents for example, acetonitrile
- ketone solvents for example, acetone
- amide solvents for example, N, N-dimethylacetamide
- polar ether solvents for example, 1, 4
- a sulfoxide solvent eg, dimethyl sulfoxide
- the reaction temperature in step (b) is not particularly limited as long as the reaction proceeds, but for example, -20 to 100 ° C. is preferable, and 20 to 50 ° C. is more preferable.
- the reaction time may vary depending on the type of n-mer (oligo) nucleotide to be condensed, the reaction temperature and the like, and may be, for example, 5 minutes to 24 hours.
- the raw material unreacted in step (b) is brought into contact reaction with the highly fat-soluble phosphoramidite compound obtained by the method for producing the highly fat-soluble phosphoramidite compound of the present invention to perform capping reaction
- a post-treatment step may be included, which is a step of reacting the capping by-product with a nucleophile after the capping reaction of step (c).
- step (c) and the step (d) are as described for the above-mentioned capping reaction of the present invention.
- the unreacted starting material in the step (c) for example, an n-mer (oligo) nucleotide from which the 5'-hydroxyl group is deblocked obtained in the step (a) (for example, HO- shown in the reaction scheme 1)
- n -LZ for example, N-LZ
- a capping side reaction product of the step (c) in the step (d) for example, a reaction product in which a capping reagent is added on Base (for example, 6 position oxygen alkoxy phosphite triester of guanine)
- Base for example, 6 position oxygen alkoxy phosphite triester of guanine
- the adduct molecule of the capping reagent is reacted with a nucleophile. It is expected that the removed guanine can be regenerated.
- the reaction with the nucleophile in the present invention is a reaction intended to treat the capping by-product. Thus, it is not intended to quench the reaction solution after the coupling reaction step, as described, for example, in the literature (for example, WO 2017/104836 A1).
- Step (e): oxidation reaction and sulfurization reaction Reaction scheme 3 of step (e) is shown below. Reaction scheme 3
- step (e) can be carried out under the reaction conditions described in the literature (for example, WO 2012/157723 A1). The details will be described below.
- step (e) An oxidant or a sulfurizing agent is added to the reaction solution of step (d) to form a phosphite triester bond of the m + 1 + n-mer oligonucleotide obtained in step (d), a phosphate triester bond or a thiophosphate triester Process of converting into bond respectively (process of oxidation or sulfurization reaction) I will provide a.
- This step is a reaction by adding an oxidizing agent or a sulfurizing agent directly to the reaction solution after the step (d) without isolating the m + 1 + n-mer oligonucleotide obtained in the step (d). You can also do
- the oxidation reaction using an oxidizing agent in step (e) can be carried out according to a method generally known in oligonucleotide synthesis.
- oxidizing agents include, but are not limited to, iodine, (1S)-(+)-(10-camphorsulfonyl) oxazolidine, tert-butyl hydroperoxide (TBHP), 2-butanone peroxide, for example.
- 1,1-dihydroperoxycyclododecane bis (trimethylsilyl) peroxide, m-chloroperbenzoic acid, iodine, (1S)-(+)-(10-camphorsulfonyl) oxazolidine, tert-butyl hydroperoxide (TBHP) ), 2-butanone peroxide and 1,1-dihydroperoxycyclododecane are preferred.
- the sulfurization reaction using a sulfurizing agent in the step (e) can be carried out according to a method generally known in oligonucleotide synthesis.
- sulfiding agents include, but are not limited to, for example, 3-((N, N-dimethylaminomethylidene) amino) -3H-1,2,4-dithiazole-5-thione (DDTT) , 3H-1,2-benzodithiazol-3-one-1,1-dioxide (Beaucage reagent), 3H-1,2-benzothiol-3-one, phenylacetyl disulfide (PADS), tetraethylthiuram disulfide (TETD) ), 3-amino-1,2,4-dithiazol-5-thione (ADTT), sulfur, 3-((N, N-dimethylaminomethylidene) amino) -3H-1,2,4- Dithiazole-5-thione, 3H-1,2-benzodithi
- Sulfurization reaction of the phosphite triester bond of the oligonucleotide converts it to a thiophosphate triester bond. Since thiophosphate has an asymmetric center on the phosphorus atom, it gives an optically active substance. In addition, even if deprotection reaction is carried out in the production process of oligonucleotide thiophosphate, the asymmetric center on the phosphorus atom remains to give an optically active substance.
- the oligonucleotide thiophosphate product becomes a mixture of a plurality of optical isomers, and the purification of the desired product is difficult, so that the synthesis is more pure. Is required.
- the reaction is usually carried out in a solvent.
- the solvent used for the reaction is preferably an anhydrous solvent, and examples thereof include halogen solvents (for example, chloroform), aliphatic solvents (for example, cyclohexane), aromatic solvents (for example, toluene), ester solvents (for example, Ethyl acetate), ether solvents (eg, tert-butyl methyl ether), nitrile solvents (eg, acetonitrile), and mixed solvents of two or more selected from these.
- halogen solvents for example, chloroform
- aliphatic solvents for example, cyclohexane
- aromatic solvents for example, toluene
- ester solvents for example, Ethyl acetate
- ether solvents eg, tert-butyl methyl ether
- nitrile solvents eg, acetonitrile
- the oxidizing agent or sulfiding agent is in a proportion of about 1.0 to about 5.0 moles, preferably about 1.0 to about 1.5 moles, per mole of the phosphoramidite obtained in step (d). .
- the reaction temperature is usually 0 to 50 ° C.
- the reaction time is usually in the range of 0.2 to 1 hour.
- the amount of oxidizing agent or sulfiding agent used may be, for example, 1 to 50 molar equivalents, preferably 1 to 5 molar equivalents, per 1 mol of the (oligo) nucleotide obtained in step (d).
- the reaction temperature is not particularly limited as long as the reaction proceeds, but may be 0 to 100 ° C., preferably 20 to 50 ° C.
- the reaction time may vary depending on the type of oligonucleotide of n + 1 + m-mer, the type of oxidizing agent or sulfiding agent used, the reaction temperature and the like, and may be, for example, 1 minute to 3 hours.
- Step f Precipitation and solid-liquid separation step
- the precipitation and solid-liquid separation operation in step (f) can be performed under the reaction conditions described in the literature (for example, WO 2012/157723 A1). The details will be described below.
- the method of producing the oligonucleotide of the present invention further comprises optionally, a polar solvent may be added to the reaction solution obtained in step (e) to precipitate the m + 1 + n-mer oligonucleotide, which may include steps of solid-liquid separation (precipitation and solid-liquid separation step).
- a polar solvent may be added to the reaction solution obtained in step (e) to precipitate the m + 1 + n-mer oligonucleotide, which may include steps of solid-liquid separation (precipitation and solid-liquid separation step).
- the method for producing the oligonucleotide of the present invention requires an isolation step consisting of precipitation of the product and solid-liquid separation in order to obtain a product m + 1 + n-mer oligonucleotide in order to use a pseudo solid phase protecting group. .
- polar solvents examples include alcohol solvents (eg, methanol, ethanol), nitrile solvents (eg, acetonitrile), ketone solvents (eg, acetone), ether solvents (eg, tetrahydrofuran), amide solvents (eg, , Dimethyl formamide), sulfoxide solvents (eg, dimethyl sulfoxide), water and the like, and mixed solvents of two or more of them.
- Alcohol solvents and nitrile solvents are preferable, and specifically, methanol is preferable.
- the polar solvent may contain water to minimize the loss of the product into the polar solvent.
- the content of water to the polar solvent is, for example, 0 to 10% (v / v), preferably 0 to 8% (v / v).
- step (e) after oxidation or sulfurization is completed in step (e), the excess oxidizing agent or sulfurizing agent is removed by reaction with a trivalent phosphorus reagent (eg, trimethyl phosphite), and then a precipitation solvent is added, or Alternatively, a solution of sodium thiosulfate (hypo) saturated in a precipitation solvent (eg, methanol) may be used during precipitation.
- a trivalent phosphorus reagent eg, trimethyl phosphite
- a precipitation solvent eg, methanol
- the amount of sodium thiosulfate (hypo) to be added depends on the amount of precipitation solvent used and the reaction temperature, and therefore, the reaction with a trivalent phosphorus reagent is more preferable.
- Step g Deprotection step
- the precipitation and solid-liquid separation operation in step (f) can be performed under the reaction conditions described in the literature (for example, WO 2012/157723 A1). The details will be described below.
- the method for producing an oligonucleotide of the present invention further suitably performs deprotection of each protective group after each step according to the type and nature of the protective group to obtain an oligonucleotide It can be isolated and produced.
- the method for producing the oligonucleotide of the present invention further comprises the step of removing all the protecting groups of the m + 1 + n polymerizing oligonucleotide obtained in step (g): step (f), as appropriate. May be included.
- a step of removing all protecting groups of oligonucleotide according to the deprotection method described in Protective Group In Organic Synthesis, 3rd edition, John Willy & Sons publication (1999), etc. It can be performed.
- the method for producing an oligonucleotide of the present invention can be carried out by a liquid phase method by using a nucleotide having a pseudo solid phase protecting group.
- the "pseudo-solid phase protecting group” means that the group is solubilized in a nonpolar solvent by binding to a reaction substrate, allowing reaction in a liquid phase, and the pseudo-solid phase by addition of a polar solvent It is a protecting group that combines the reactivity and the ease of post-treatment, which enables precipitation of the product containing the protecting group to enable solid-liquid separation, and is stable under acidic conditions that can remove the protecting group at the 5 'terminal hydroxyl group.
- pseudo solid phase protecting group examples include the groups disclosed in the literature (for example, WO 2012/157723 A1), and preferred examples of the pseudo solid phase protecting group used in the method of producing the oligonucleotide of the present invention include The residue of the N-alkyl or alkyl alcohol compound represented by the formula (II) can be mentioned.
- the invention is Formula (II): [In the formula, Each R 5 is independently an optionally substituted C 6 -C 30 alkyl group, R 6 is optionally substituted C 1 -C 6 alkyl group, optionally substituted C 3 -C 6 cycloalkyl group, optionally substituted non-aromatic heterocyclic group, optionally substituted An aryl group, an optionally substituted heteroaryl group, an optionally substituted aralkyl group, or an optionally substituted heteroarylalkyl group, R 7 represents an optionally substituted C 1 -C 6 alkylene group or an optionally substituted C 1 -C 6 alkyleneoxy group, R 8 is an optionally substituted C 1 -C 6 alkylene group, or R 6 and R 7 may be taken together with the nitrogen atom to which R 6 is attached to form an optionally substituted non-aromatic heterocyclic group or an optionally substituted heteroaryl group.
- R 6 , R 7 and R 8 together with the nitrogen atom to which R 6 is bonded, an optionally substituted non-aromatic heterocyclic group, an optionally substituted non-aromatic heterocyclic ring It may form an alkyl group, an optionally substituted heteroaryl group, or an optionally substituted heteroarylalkyl group, and * Indicates the bonding position with L] Pseudo solid phase protecting group represented by I will provide a.
- Each R 5 is independently an optionally substituted C 8 -C 24 alkyl group
- R 6 is optionally substituted C 1 -C 3 alkyl group, optionally substituted C 5 -C 6 cycloalkyl group, optionally substituted aryl group, optionally substituted heteroaryl group, or substituted It is selected from an optionally substituted aralkyl group
- R 7 is an optionally substituted C 1 -C 4 alkylene group
- R 8 is a C1-C2 alkylene group which may be substituted
- R 6 and R 7 may be taken together with the nitrogen atom to which R 6 is bonded to form a non-aromatic heterocyclic group which may be substituted, or a heteroaryl group which may be substituted.
- the term “optionally substituted C6-C30 alkyl group” for R 5 is, if appropriate, a primary or C 6-30 primary or optionally substituted substituent. It means a secondary saturated hydrocarbon chain group, preferably a primary saturated hydrocarbon chain group.
- the alkyl group may be linear or branched, but a linear alkyl group is preferable. Specifically, a C8-C30 primary alkyl group is preferable, a C12-C22 primary alkyl group is more preferable, and a C16-C20 primary alkyl group is more preferable.
- One preferable example is a C18 primary alkyl group.
- substituent is not limited as long as the reaction used in the present invention (for example, capping reaction, coupling reaction on oligonucleotide synthesis, deprotection reaction, deblocking reaction, oxidation reaction, sulfurization reaction) is not inhibited.
- examples include, but are not limited to, one or more groups selected from, for example, a C1-C6 alkyl group and a C3-C6 cycloalkyl group, with a C1-C6 alkyl group being preferred.
- substituent include methyl, ethyl, isopropyl, cyclopropyl, cyclohexyl and the like.
- the term "optionally substituted C1-C6 alkyl group" for R 6 means a saturated hydrocarbon chain group having 1 to 6 carbon atoms.
- the alkyl group may be linear or branched.
- a C3 alkyl group is preferable.
- One preferred example is methyl.
- the kind of substituent is not limited as long as the reaction used in the present invention (for example, capping reaction, coupling reaction on oligonucleotide synthesis, deprotection reaction, deblocking reaction, oxidation reaction, sulfurization reaction) is not inhibited.
- Non-limiting examples include one or more groups selected from C1-C3 alkyl groups and C3-C6 cycloalkyl groups.
- the term “optionally substituted C 3 -C 6 cycloalkyl group” for R 6 means an alicyclic saturated hydrocarbon group having 3 to 6 carbon atoms.
- the kind of substituent is not limited as long as the reaction used in the present invention (for example, capping reaction, coupling reaction on oligonucleotide synthesis, deprotection reaction, deblocking reaction, oxidation reaction, sulfurization reaction) is not inhibited.
- the reaction used in the present invention for example, capping reaction, coupling reaction on oligonucleotide synthesis, deprotection reaction, deblocking reaction, oxidation reaction, sulfurization reaction
- C1-C3 alkyl group Specific examples of the cycloalkyl group include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- the term “optionally substituted C1-C6 alkylene group” for R 7 and R 8 means a divalent group of a saturated hydrocarbon chain having 1 to 6 carbon atoms.
- the alkylene group may be linear or branched.
- a C1-C4 alkylene group is preferable, and a C1-C3 alkylene group is more preferable.
- Specific examples include methylene, ethylene, trimethylene (propylene), tetramethylene (n-butylene) and the like.
- substituent the kind of substituent is not limited as long as the reaction used in the present invention (for example, capping reaction, coupling reaction on oligonucleotide synthesis, deprotection reaction, deblocking reaction, oxidation reaction, sulfurization reaction) is not inhibited.
- examples include, but are not limited to, one or more groups selected from, for example, a C1-C3 alkyl group, a C3-C6 cycloalkyl group, and a C6-C10 aryl group.
- substituents include methyl, ethyl, isopropyl, phenyl and the like.
- optionally substituted C1-C6 alkyleneoxy group for R 7 means the above-mentioned "optionally substituted C1-C6 alkylene group" bonded to an oxygen atom. means. Specific examples include methyleneoxy, ethyleneoxy, trimethyleneoxy (propyleneoxy) and the like.
- the term "optionally substituted non-aromatic heterocyclic group" for R 6 , R 7 and R 8 means a sulfur atom, an oxygen atom, and a nitrogen atom (here, oxygen An atom or a nitrogen atom is preferable, and a nitrogen atom is more preferable; and 1 to 4 (here, 1 to 2 are preferable, and 1 is more preferable) hetero atoms selected from the group consisting of 4 To an 8-membered monocyclic or bicyclic non-aromatic heterocyclic group.
- the optionally substituted non-aromatic heterocyclic group may be fused to a cycloalkyl ring, an aromatic hydrocarbon ring or an aromatic heterocyclic ring.
- non-aromatic heterocyclic group which may be substituted include azetidinyl, pyrrolidinyl, tetrahydrofuranyl, imidazolidinyl, thiazolidinyl, isothiazolidinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, tetrahydropyranyl, Homopiperazinyl, homomorpholinyl, 3-azabicyclo [3.1.0] hexyl, 3-azabicyclo [3.2.1] octyl and the like can be mentioned, preferably piperidinyl, piperazinyl, 3-azabicyclo [3.2.1] octyl Etc.
- substituent the kind of substituent is not limited as long as the reaction used in the present invention (for example, capping reaction, coupling reaction on oligonucleotide synthesis, deprotection reaction, deblocking reaction, oxidation reaction, sulfurization reaction) is not inhibited.
- Non-limiting examples include one or more groups selected from C1-C3 alkyl groups, and C3-C6 cycloalkyl groups.
- substituents include methyl, ethyl, isopropyl, cyclopropyl and the like.
- the term “optionally substituted aryl group (or optionally substituted aromatic hydrocarbon cyclic group)” for R 6 , R 7 and R 8 is 6-10 Means a membered monocyclic or bicyclic aromatic hydrocarbon cyclic group.
- the optionally substituted aryl group may be fused to a cycloalkyl ring, a nonaromatic heterocycle, or an aromatic heterocycle.
- Specific examples of the aryl group which may be substituted include phenyl, indenyl, naphthyl, cyclohexanophenyl, pyrrolidine-fused phenyl (indoline), piperidine-fused phenyl and the like, with preference given to phenyl.
- substituent the kind of substituent is not limited as long as the reaction used in the present invention (for example, capping reaction, coupling reaction on oligonucleotide synthesis, deprotection reaction, deblocking reaction, oxidation reaction, sulfurization reaction) is not inhibited.
- Non-limiting examples include one or more groups selected from C1-C3 alkyl groups, and C3-C6 cycloalkyl groups.
- substituents include methyl, ethyl, isopropyl, cyclopropyl and the like.
- the term "optionally substituted heteroaryl group (or optionally substituted aromatic heterocyclic group)" for R 6 , R 7 and R 8 means a sulfur atom , 1 to 4 (here, 1 to 2 are preferably selected) independently selected from the group consisting of an oxygen atom and a nitrogen atom (here, an oxygen atom or a nitrogen atom is preferable, and a nitrogen atom is more preferable) More preferably 5 to 10 membered monocyclic or bicyclic aromatic heterocyclic groups containing one or more hetero atoms).
- the optionally substituted heteroaryl group may be fused to a cycloalkyl ring, a nonaromatic heterocyclic ring, or an aromatic hydrocarbon ring.
- heteroaryl group which may be substituted include pyrrolyl, furanyl, thienyl, imidazolyl, pyrazolyl, oxazolyl, isoxazolyl, thiazolyl, triazolyl, tetrazolyl, oxadiazolyl, pyridinyl, pyrazinyl, pyrimidinyl, pyridazinyl, thiazinyl, triazinyl And indolyl, quinolyl, isoquinolyl, isoindolyl, benzimidazolyl and the like, preferably pyridinyl, pyrazinyl, pyrimidinyl, isoquinolyl, benzimidazolyl and the like.
- substituent the kind of substituent is not limited as long as the reaction used in the present invention (for example, capping reaction, coupling reaction on oligonucleotide synthesis, deprotection reaction, deblocking reaction, oxidation reaction, sulfurization reaction) is not inhibited.
- Non-limiting examples include one or more groups selected from C1-C3 alkyl groups, and C3-C6 cycloalkyl groups.
- substituents include methyl, ethyl, isopropyl, cyclopropyl and the like.
- the term "optionally substituted aralkyl group" for R 6 , R 7 and R 8 means an alkyl group optionally substituted with the above-mentioned optionally substituted aryl group. It means a bonded group.
- Specific examples of the aralkyl group which may be substituted include benzyl, phenethyl and the like, preferably benzyl.
- Examples of the substituent include the substituents described for the above-mentioned optionally substituted alkyl group and the above-mentioned optionally substituted aryl group.
- the term "optionally substituted heteroarylalkyl group" for R 6 , R 7 and R 8 may be optionally substituted for the above-mentioned optionally substituted heteroaryl group. It means a group bound to an alkyl group.
- Specific examples of the heteroarylalkyl group which may be substituted include pyridylmethyl, pyridylethyl and the like, preferably pyridylmethyl.
- substituent include the substituents described for the above-mentioned optionally substituted alkyl group and the above-mentioned optionally substituted heteroaryl group.
- the term "optionally substituted non-aromatic heterocyclic alkyl group" for R 6 , R 7 and R 8 means the above-mentioned optionally substituted non-aromatic heterocyclic ring
- the group means a group bonded to the above-mentioned alkyl group which may be substituted.
- Specific examples of the non-aromatic heterocyclic alkyl group which may be substituted include piperidinylmethyl, piperazinylmethyl, piperidinylethyl, piperazinylethyl, preferably piperidinylmethyl. It can be mentioned.
- the substituent include the substituents described for the above-mentioned optionally substituted non-aromatic heterocyclic group and the above-mentioned optionally substituted alkyl group.
- the soluble part is represented by the following formula (IV): (In the formula, * Indicates a bonding position with L; ** represents a bonding position with a carbonyl group; R 6 , R 7 and R 8 are each as defined in formula (II))
- the part shown by is the following structural formula: The part shown by is mentioned.
- the present invention provides a nucleotide comprising the above-mentioned pseudo solid phase protecting group, such as, for example, the following formula (III): [In the formula, Z is a pseudo solid phase protecting group represented by the above formula (II), L is a linker, Each Base is a nucleobase which may be independently protected.
- n EWGs is independently an electron withdrawing group
- n R 4 s each independently represent an oxygen atom or a sulfur atom
- n Rs each independently represent a hydrogen atom, a halogen atom, a hydroxy group which may be substituted with a protecting group, a C1-C6 alkoxy group which may be substituted, an organic group which crosslinks to the 4-position carbon atom, Or a cross-linked artificial nucleic acid group
- n is any integer greater than or equal to 0
- X is an acid deprotectable temporary protecting group
- halogen atom as R means a fluorine atom, a chlorine atom, a bromine atom and an iodine atom, but a fluorine atom is preferable.
- a substituent in the term “hydroxyl group which may be substituted with a protecting group” as R may be a protecting group known to be usually used in oligonucleotide synthesis, for example, A silyl type protecting group (eg, TOM group (-CH 2 OSi (iPr) 3 )) can be mentioned.
- organic group bridging to the 4-position carbon atom means a group which is 4'-C1-C6 alkoxy-2 ', for example, 4'-CH 2 CH 2 O-2 'is mentioned.
- EWG electron withdrawing group
- acid deprotectable temporary protecting group means a group described herein as "a temporary protecting group removable under acidic conditions of the 5 'hydroxyl group”.
- L in formula (III) is a linker, which means a divalent group linking an (oligo) nucleotide molecule and a pseudo solid phase protecting group.
- the linker does not inhibit each reaction step (for example, capping reaction, coupling reaction on oligonucleotide synthesis, deprotection reaction, deblocking reaction, oxidation reaction, sulfurization reaction) in the method for producing an oligonucleotide of the present invention Is not particularly limited.
- N is, for example, 2 to 100, preferably 10 to 40.
- L is the following formula: [Wherein, * indicates a bonding position to a hydrophobic group, and ** indicates a bonding position to a phosphorus atom,
- Each Base is a nucleobase which may be independently protected, and R each independently represents a hydrogen atom, a halogen atom, a hydroxy group which may be substituted with a protecting group, a C1-C6 alkoxy group which may be substituted, an organic group which crosslinks to the 4-position carbon atom, or a crosslinked structure Type artificial nucleic acid group]
- a linker which is a group selected from any one of the group consisting of
- L has the formula: It is a group selected from any of
- Nucleotides containing the above-described pseudo solid phase protecting group can be used as a starting material for the method of producing an oligonucleotide of the present invention or as a starting material for one cycle of nucleotide extension.
- oligonucleotide production a series of total steps (referred to as "one cycle of nucleotide extension") of extending one nucleotide can be performed including one or two purification steps.
- Second stage vi) The obtained intermediate DMT- (Nps) n + 1 -SR is crystallized using a solvent containing pyrrole and an acid, and pyridine and an alcohol (for example, methanol) to obtain the desired product HO- (Nps) Get n + 1 -SR.
- One cycle of nucleotide elongation in oligonucleotide production 1st stage i) Dissolve the 5 'OH terminal n-mer in a solvent. ii) The 5 'OH terminal n-mer, the m + 1-phosphoramidite compound, and the activating agent are reacted in a solvent to carry out the coupling reaction. The progress of the reaction is confirmed by the disappearance of HO (Nps) n -SR (this is due to the disappearance of HO (Nps) n -SR). ii) After the coupling reaction, the reaction product is reacted with the reagent for capping reaction of the present invention to carry out the capping reaction.
- a nucleophile e.g. an alcohol such as ethanol
- a sulfurization reagent in a solvent containing an organic base (eg, pyridine).
- a phosphite compound e.g, methanol
- kits for example for the preparation and purification of (oligo) nucleotides.
- the kit of the present invention contains at least one reagent for capping reaction of the present invention as a component.
- the kit of the invention comprises (a) a reagent for capping reaction according to the invention; (b) a nucleophile; (c) at least one (oligo) nucleotide compound, for example (oligo) A) a phosphoramidite compound of nucleotides; (d) at least one extendable (oligo) nucleotide compound; (e) at least one activating agent used to extend an oligonucleotide; (f) at least one buffer; And (h) at least one container for filling the kit components) and (d) an instructional document for extending (oligo) nucleotides using the kit components;
- the "buffer” is a commonly used one as long as it does not adversely affect the kit components to be used (eg, degradation of kit components, inhibition of reaction).
- the kit of the present invention is a kit for capping reaction of (oligo) nucleotides, which comprises (a) a reagent for capping reaction of the present invention; (b) (G) a package insert describing the procedure for capping (oligo) nucleotides with the kit components; and (h) at least one container for filling the kit components.
- reagents were appropriately obtained commercially or were prepared according to known methods.
- various measurement instruments were used using instruments that are usually used.
- the reaction solution was transferred to a separatory funnel while washing with 5 mL of dichloromethane, washed with 10 mL of water, and the organic layer was concentrated under reduced pressure.
- 20 mL of hexane was added, and 100 uL of the mixture was collected, and 100 uL of 0.1 M triphenylphosphine dichloromethane solution and 400 uL of CDCl 3 were added to obtain an NMR sample without washing.
- 20 mL of the hexane solution was transferred to a separatory funnel, washing operation was performed with 20 mL of acetonitrile, and the organic layer was transferred to a measuring cylinder.
- Example 2 Preparation of 2-cyanoethyl octadecyl diisopropyl phosphoramidite (50 mmol scale) Under an argon atmosphere, 60 mL of dehydrated dichloromethane and 40 mL of acetonitrile are added to 13.52 g of octadecanol and 1.93 g of tetrazole, stirred and dissolved, and then 3- (bis (diisopropylpropylamino) phosphinoxy) propanenitrile (17.5 mL) ) was added dropwise and stirred for 1 hour. The reaction solution was transferred to a separatory funnel while washing with dichloromethane, washed with water, and the organic layer was concentrated under reduced pressure.
- Example 3 Preparation of 2-cyanoethyl octadecyl diisopropyl phosphoramidite (in the case of using 2-cyanoethyl diisopropyl chlorophosphoramidite as trivalent phosphorus compound) Under argon atmosphere, 6 mL of dehydrated tetrahydrofuran is added to 811 mg of octadecanol and 1.04 mL of diisopropylethylamine, stirred, dissolved and ice cooled, then 2-cyanoethyl diisopropylchlorophosphoramidite (0.74 mL) is added dropwise to Stir for hours.
- reaction solution was transferred to a separatory funnel while washing with dichloromethane, washed with water, and the organic layer was concentrated under reduced pressure.
- the concentrated reaction mixture was dissolved in hexane and washed twice with acetonitrile.
- the hexane layer was concentrated under reduced pressure using an evaporator to obtain 1.08 g of colorless oily 2-cyanoethyl octadecyl diisopropyl phosphoramidite.
- Example 4 Production of 2-cyanoethyl decyl diisopropyl phosphoramidite Under argon atmosphere, dehydrated dichloromethane 2.4 mL and acetonitrile 1.6 mL are added to decanol 381 uL and tetrazole 141 mg, stirred and dissolved, and then 3- (bis (diisopropylpropylamino) phosphinoxy) propanenitrile (0.70 mL, 2.2) The mmol) was added dropwise and stirred for 1 hour. The reaction solution was transferred to a separatory funnel while washing with 5 mL of dichloromethane, washed with 10 mL of water, and the organic layer was concentrated under reduced pressure.
- Example 5 Preparation of 2-cyanoethyl octyl diisopropyl phosphoramidite
- 2.4 mL of dehydrated dichloromethane and 1.6 mL of acetonitrile are added to 314 uL of octanol and 141 mg of tetrazole, stirred and dissolved, and then 3- (bis (diisopropylpropylamino) phosphinoxy) propanenitrile (0.70 mL) is added to the solution. It was added dropwise and stirred for 1 hour.
- the reaction solution was transferred to a separatory funnel while washing with 5 mL of dichloromethane, washed with 10 mL of water, and the organic layer was concentrated under reduced pressure.
- 20 mL of hexane was added, and 100 uL of the mixture was collected, and 100 uL of 0.1 M triphenylphosphine dichloromethane solution and 400 uL of CDCl 3 were added to obtain an NMR sample without washing.
- 20 mL of the hexane solution was transferred to a separatory funnel, washing operation was performed with 20 mL of acetonitrile, and the organic layer was transferred to a measuring cylinder.
- the production and purification method of the present invention extracts and cleans the reaction mixture using different organic solvent systems, and as the lipophilicity of the phosphoramidite compound becomes higher, the by-product is generated during capping. It has been found that substantially high-purity phosphoramidite products (monoamidite compounds) free of H-phosphonate compounds and diamidite compounds which result in the formation of compounds can be recovered in high yield.
- Example 7 Preparation of 2-cyanoethyl 2'-octyldodecanyl diisopropyl phosphoramidite Under an argon atmosphere, 12 mL of dehydrated dichloromethane and 8 mL of acetonitrile are added to 3.56 mL of 2-octyldodecanol and 0.72 g of ETT, stirred and dissolved, and then 3- (bis (diisopropylpropylamino) phosphinoxy) propane nitrile ( 3.5 mL) was added dropwise and stirred for 1 hour. The reaction solution was transferred to a separatory funnel while washing with dichloromethane, washed with water, and the organic layer was concentrated under reduced pressure.
- the concentrated reaction mixture was dissolved in hexane and washed twice with acetonitrile.
- the hexane layer was concentrated under reduced pressure using an evaporator to obtain 3.43 g of colorless oily 2-cyanoethyl octadecyl diisopropyl phosphoramidite.
- Example 8 Production of Phy-CE-phosphoramidite Under argon atmosphere, 12 mL of dehydrated dichloromethane and 8 mL of acetonitrile are added to 2.98 g of compound Phy-OH and 0.39 g of tetrazole, stirred and dissolved, and then 3- (bis (diisopropylpropylamino) phosphinoxy) propane nitrile (3.5) mL) was added dropwise and stirred for 1 hour. The reaction solution was transferred to a separatory funnel while washing with dichloromethane, washed with water, and the organic layer was concentrated under reduced pressure. The concentrated reaction mixture was dissolved in hexane and washed twice with acetonitrile.
- the resulting reaction mixture was diluted with dichloromethane, 150 mL of methanol was added, and the mixture was concentrated under reduced pressure. The solid formed was filtered and washed with methanol to give intermediate DMT-dTpo-SR.
- the obtained intermediate DMT-dTpo-SR was dissolved in 225 mL of dichloromethane in a 500 mL recovery flask, and 5.2 mL of pyrrole and 7.4 mL of dichloroacetic acid were added thereto. The mixture was stirred at room temperature for 15 minutes after which 150 mL of methanol and 8.70 mL of pyridine were added.
- the resulting reaction mixture was diluted with 55 ml of dichloromethane, and then 1.7 mL of pyrrole and 2.3 mL of trifluoroacetic acid were added. The mixture was stirred at room temperature for 15 minutes after which 50 mL of methanol and 4.8 mL of pyridine were added. The mixture was concentrated under reduced pressure, and the resulting solid was filtered, washed with methanol and dried under reduced pressure to obtain 2.42 g of the target compound HO-mAps-mGpo-dTpo-SR.
- reaction formula VI the detrityl reaction formula by acid of guanine 6-position O-highly fat-soluble amidite adduct P (pentavalent) prepared above is shown in the following reaction formula VI.
- Example 12 Solution A: A solution of 0.057 M MMT-mGps-SR in CDCl 3 and a solution B: 0.9 M ETT in CDCl 3 Prepared. 22.1 ul of C 18 H 37 O-phosphoramidite was dispensed into an NMR tube, 437.5 ul of solution A was added and mixed well, and then 125 ul of solution B was added and stirred. Solution B is added after 5 minutes was measured 31 PNMR for specific 31 P signal to the following compounds, respectively.
- FIG. 1 An NMR comparison diagram of a mixture of Compound DMT-diacyl-dA-Su-R and Compound DMT-dA-Su-R (upper stage) and Compound DMT-dA-Su-R (lower stage) is shown in FIG. It can be seen that it is a mixture of about 80% of the diacylated compound DMT-diacyl-dA-Su-R and about 20% of the starting compound DMT-dA-Su-R. In capping with acetic anhydride, the adenine base is It was found to result in a mixture containing the diacylated product.
- a solution containing the highly fat-soluble phosphoramidite compound obtained by the method for producing a highly fat-soluble phosphoramidite compound of the present invention or a composition containing the solution and a suitable additive as a reagent for capping reaction, it is possible to obtain high purity (oligo ) Nucleotides can be produced on a large scale by simple operations. Moreover, according to the method for producing an oligonucleotide of the present invention, a high purity oligonucleotide can be produced on a large scale.
Abstract
本発明は、高脂溶性ホスホラミダイトの精製および製造方法、並びに該高脂溶性ホスホラミダイト化合物を用いるキャッピング反応、並びに該キャッピング反応の工程を含む擬似固相保護基を用いた液相法によるオリゴヌクレオチドの製造方法を提供することを目的とする。本発明はまた、式(I)[式中、R1、R2、およびR3は明細書中に定義する通りである]で示される化合物の製造方法であって、以下の工程:(1)脂肪族アルコールおよび三価リン化合物を、活性化剤または有機塩基の存在下、有機溶媒中で反応する;(2)得られた反応混合物を、水を用いて分液洗浄する;(3)工程(2)後の有機層を回収し、濃縮する(但し、工程(1)で使用する有機溶媒がニトリル系溶媒の場合には、工程(2)~(3)は省略してもよい);(4)工程(3)で得られた残渣を脂肪族炭化水素系溶媒中に溶解する(但し、工程(1)で使用する有機溶媒が脂肪族炭化水素系溶媒の場合には、工程(2)~(4)は省略してもよい);(5)工程(4)で調製した脂肪族炭化水素系溶液を、ニトリル系溶媒を用いて分液洗浄する;(6)工程(5)後の脂肪族炭化水素系溶液を回収して、ホスホラミダイト化合物を溶液として得る、を含む、該製造方法を提供する。また、本発明は、該製造方法によって得られる高脂溶性ホスホラミダイト化合物を含有する溶液または該溶液および適宜添加剤を含む組成物であるキャッピング反応用試薬、該キャッピング反応用試薬を用いるキャッピング反応、並びに該キャッピング反応の工程を含む、擬似固相保護基を用いた(オリゴ)ヌクレオチドの製造方法を提供する。
Description
本発明は、高脂溶性ホスホラミダイトの製造に関する。より詳細には、本発明は、高脂溶性ホスホラミダイトの精製および製造方法、並びに該高脂溶性ホスホラミダイト化合物を含む溶液としてのキャッピング反応用溶液および該キャッピング反応用溶液を用いるキャッピング反応、並びに該キャッピング反応の工程を含む液相法によるオリゴヌクレオチドの製造方法をも提供する。
近年、合成オリゴヌクレオチドを用いた研究が盛んにおこなわれ、その成果としてオリゴ核酸医薬が上市されている。一方で、高純度のオリゴヌクレオチドを大スケールで合成することができる合成法が求められている。
オリゴヌクレオチドの製造方法としては、リン酸トリエステル法、H-ホスホネート法、及びホスホラミダイト法等があり、現在ではホスホラミダイト法を用いた固相合成(固相法)が最も汎用されている。オリゴヌクレオチドの合成には、原料となるヌクレオチドを順次連結する方法、約2~3個のヌクレオチドのビルディングブロック群の連結を繰り返す方法(ブロックマー合成法)、あるいは約10個の塩基程度のオリゴヌクレオチドの連結による方法(フラグメント縮合法)等が知られる。
固相合成(固相法)は、自動合成機を用いる等の合成スピードの面で有利ではあるものの、合成スケールアップが制限され、また過剰量の反応試薬や反応原料が必要となり、特に合成反応の途中段階で反応の進行状況の確認、また中間体の解析なども困難である等の欠点がある。
一方、液相法によるオリゴヌクレオチドの合成は、操作が煩雑であり、また収率が低いことから、重合度の高いオリゴヌクレオチドを大量に且つ迅速に合成することは困難であるという欠点がある。
一方、液相法によるオリゴヌクレオチドの合成は、操作が煩雑であり、また収率が低いことから、重合度の高いオリゴヌクレオチドを大量に且つ迅速に合成することは困難であるという欠点がある。
近年、固相法と液相法とのそれぞれの欠点を解消することを意図して、ポリエチレングリコール(MPEG)を用いるオリゴヌクレオチド合成法が報告されている(非特許文献1~3)。かかる方法により、約20量体までのオリゴヌクレオチドの合成例が報告されているものの、伸張サイクル内各ステップにおいて晶析単離操作や再結晶を必要とし、煩雑であるなどの問題があった。
また、液相製造方法において、ある疎水性基を固相合成法における樹脂を模した可溶性樹脂として採用し、該疎水性基(以下、「擬似固相保護基」と称することもある)を結合したヌクレオシド、つまり、疎水性基結合ヌクレオシドを用いたオリゴヌクレオチドの製造方法が報告されている(特許文献1、および非特許文献4)。かかる方法により、21量体のオリゴヌクレオチドの製造が報告されている。しかし、5’-保護基の脱保護反応、カップリング反応、酸化反応の各工程に晶析単離操作を要し、工程が著しく多いため煩雑であった。
これまでのオリゴヌクレオチドの製造方法では、未反応原料などの未反応物がオリゴヌクレオチドの伸張段階に関与すること等により、目的のオリゴヌクレオチドに対してN-1およびN+1の副生成物が生成する問題点があった。またこれらの副生成物は目的物の生成物と区別して分離することが極めて困難でもあった。よって、合成オリゴヌクレオチド生成物を核酸医薬として用いる場合には、これらN-1およびN+1の副生成物の生成を低減することにより、従来法で得られるオリゴヌクレオチドの純度以上でオリゴヌクレオチドを得ることができる製造方法が求められている。
オリゴヌクレオチドのヌクレオチドとのカップリング反応の後に、未反応オリゴヌクレオチド鎖の反応活性点を保護することにより、次の合成サイクルにおいて目的のオリゴヌクレオチド鎖の反応活性点と競合して伸張反応が進行し、N-1オリゴヌクレオチドが生成することを防止するための、キャッピング試薬が、固相製造方法において使用されている。
しかしながら、擬似固相保護基を用いた液相法によるオリゴヌクレオチド(とりわけ、DNAホスホラミダイト法)製造方法では、液相系での反応であるため、固相法と比べてカップリング反応等の反応性が高く、大過剰量の反応原料や反応試薬を使用する必要がなかった。その為、固相製造方法で行われているような未反応オリゴヌクレオチド鎖のキャッピングのための試薬(キャッピング試薬)は加えられておらず、従ってキャッピング試薬を用いる液相合成反応は報告されていない。
擬似固相保護基を用いた液相法においてキャッピング試薬を用いられていない理由としては、上記反応性に加え、とりわけ以下に挙げる問題点が有る為とも考えられる。固相法によるオリゴヌクレオチド合成において汎用されるホスホラミダイト反応では、通常、キャッピング試薬として、大過剰量の無水酢酸、及び大過剰量のイミダゾール誘導体などの塩基が使用されるが、これらの酸や塩基を除去するために追加の精製工程が必須になること、および未反応オリゴヌクレオチド鎖のキャッピング生成物の性質(例えば、疎水性)がオリゴヌクレオチド鎖とカップリング反応生成目的物の中間の性質をもつこと、及び核酸塩基が更に一部アセチル化(主にアデニンの6-アミド、一部グアニンの2-アミド、シチジンの4-アミド、チミンの3位)されてジアシル体を生成し、反応混合物に成る可能性が有る事が挙げられる(非特許文献5)。その場合、これらの混合物中の各々の化合物から次の合成サイクルで同様の反応生成物を与え、より一層複雑な反応混合物を生じる為、通常の機器分析法(例えば、TLC、HPLC)では解析することが困難になり、オリゴヌクレオチド合成反応における、ヌクレオチドカップリング反応終点(未反応オリゴヌクレオチドの消失)の確認が非常に困難になるという問題点がある。
しかしながら、無水酢酸によるキャッピング工程は、オリゴヌクレオチド(とりわけ、DNA)製造方法においてホスホラミダイトと核酸塩基の副反応物(主にグアニン6位カルボニル酸素における)の収量を抑制する効果も報告されている(非特許文献6)。
また、擬似固相保護基を用いた液相製造方法においても、ホスホラミダイトによる伸張後すぐに、非特許文献1と同様にカルボン酸有機塩基塩やアルコール等のクエンチ剤を加え、生成物の純度向上に成功している(特許文献2)。しかし、カルボン酸有機塩基塩やアルコールは無水酢酸と反応性が有り、追加の精製工程なしに無水酢酸によるキャッピングを行う事は困難であると容易に想像できる。特に、硫化前に無水酢酸、水等の使用により硫化効率が落ちる事も報告されている(非特許文献7)為、アルコール等を添加し、硫化する場合には、硫化効率の確認が不可欠である。
また、擬似固相保護基を用いた液相製造方法においても、ホスホラミダイトによる伸張後すぐに、非特許文献1と同様にカルボン酸有機塩基塩やアルコール等のクエンチ剤を加え、生成物の純度向上に成功している(特許文献2)。しかし、カルボン酸有機塩基塩やアルコールは無水酢酸と反応性が有り、追加の精製工程なしに無水酢酸によるキャッピングを行う事は困難であると容易に想像できる。特に、硫化前に無水酢酸、水等の使用により硫化効率が落ちる事も報告されている(非特許文献7)為、アルコール等を添加し、硫化する場合には、硫化効率の確認が不可欠である。
また、キャッピング試薬は、オリゴヌクレオチド合成において生じるN-1副生成物の生成を抑制して、目的物のオリゴヌクレオチド生成物の純度を向上することを目的としている。キャッピング試薬は、先述の無水酢酸以外にアルコキシホスホラミダイト化合物を使用した例もある。ただし、アルコキシホスホラミダイト化合物は、無水酢酸と比較して極めて高価である為、高い反応性を持つ第1級アルコキシホスホラミダイト化合物が用いられている(特許文献3、4、および非特許文献8、9)。また、これらの文献内では、第1級アルコキシホスホラミダイトと核酸塩基(主にグアニン6位カルボニル酸素における)の副反応は報告されていない。
第1級アルコキシホスホラミダイト化合物をキャッピング試薬として用いた場合、高い反応性により必要等量数が少ない事が期待される。しかし、オリゴ核酸の大量合成においては、かなりの量が必要となり、高純度のアルコキシホスホラミダイト化合物を、より簡便に且つ安全に、大量に製造できる方法の開発が必要である。
反応性が高い第1級アルコキシホスホラミダイト化合物のカラム精製は、通常の安価なアミン共存シリカゲルを用いるのではなく、高価なアミノシリカゲルの使用を要している(特許文献5)。ホスホラミダイト化合物の原料として使用可能なホスホロジアミダイト化合物を、反応混合液に貧溶媒(例えば、アルコール類、炭化水素類、ニトリル類)を加えて回収する方法が報告されている(特許文献6および7)。しかし、同様の手法が、反応性の高い第1級アルコキシホスホラミダイト化合物に用いることができるかどうかは予測しえず、更に原料アルコールの適切な選択が必要となると考えられる。
反応性が高い第1級アルコキシホスホラミダイト化合物のカラム精製は、通常の安価なアミン共存シリカゲルを用いるのではなく、高価なアミノシリカゲルの使用を要している(特許文献5)。ホスホラミダイト化合物の原料として使用可能なホスホロジアミダイト化合物を、反応混合液に貧溶媒(例えば、アルコール類、炭化水素類、ニトリル類)を加えて回収する方法が報告されている(特許文献6および7)。しかし、同様の手法が、反応性の高い第1級アルコキシホスホラミダイト化合物に用いることができるかどうかは予測しえず、更に原料アルコールの適切な選択が必要となると考えられる。
Nucleic Acids Research, 1990, 18, 3155-3159
Tetrahedron Lett. 1991, 32, 27, 3251-3254
Nucleic Acids Research, 1993, 21, (5) 1213-1217
Chem. Eur. J. 2013, 19, 8615-8620
Tetrahedron Letters, 2009, 50, 15, 1751-1753
Nucleic Acids Research, 1986, 14, (16) 6453-6470
Tetrahedron Lett. 2011, 52, 434-437
Tetrahedron Lett. 1994, 35, 46, 8565-8568
Tetrahedron 1997, 53, 28, 9629-9636
本発明は、高脂溶性ホスホラミダイトの製造方法に関する。より詳細には、本発明は、オリゴヌクレオチド合成時にキャッピング試薬として使用可能なアルコキシホスホラミダイト化合物である高脂溶性ホスホラミダイトの簡便な精製および製造方法、並びに該製造方法で得られた高脂溶性ホスホラミダイト化合物を含有する溶液としてのキャッピング反応用溶液、および該キャッピング反応用溶液を用いるキャッピング反応、並びに該キャッピング反応の工程を含む、擬似固相保護基を用いた液相法によるオリゴヌクレオチドの製造方法をも提供する。
本発明は、オリゴヌクレオチド合成時にキャッピング試薬として使用可能なアルコキシホスホラミダイト化合物の1種である高脂溶性ホスホラミダイトを、カラム精製等の操作を要することなく、溶解溶媒および洗浄溶媒の種類、並びにその洗浄順序を選別することで分液操作等によって簡便に精製することができ、工業的に大スケールで高純度に製造することができる製造方法を見出した。また、該製造方法によって得られた該高脂溶性ホスホラミダイトを含有する溶液をキャッピング反応溶液として液相法(特に、擬似固相保護基を用いた液相法)において使用した場合にキャッピング反応を達成することができ、また、副反応が起こるものの、求核剤(例えば、アルコール)を用いて後処理することによって、かかる副生成物の収量を抑制することができることをも見出した。また、オリゴヌクレオチド合成過程にわたって、キャッピング反応後に続く必須工程(オリゴヌクレオチドの酸化反応および/または硫化反応)に対して、かかるキャッピング反応およびその後の後処理方法が悪影響(例えば、反応の阻害)を生じず、目的物の収率が向上することをも見出した。
すなわち、本発明は、以下の態様を提供するが、これに限定されるものではない。
(高脂溶性ホスホラミダイトの製造および精製方法)
項[1] 式(I):
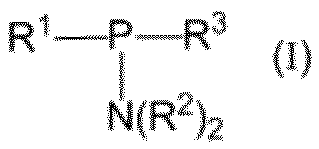
[式中、
R1は、置換されていてもよいC6-C30アルキルオキシ基であり、該置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、
R2は各々独立して、置換されていてもよいC1-C6アルキル基であり、該置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、そして、
R3は、置換されていてもよいC6-C30アルキルオキシ基、置換されていてもよいC1-C8アルキルオキシ基、または置換されていてもよいC2-C8アルキニルオキシ基であり、ここで、置換されていてもよいC6-C30アルキルオキシ基における置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、置換されていてもよいC1-C8アルキルオキシ基、または置換されていてもよいC2-C8アルキニルオキシ基における置換基はシアノ基(CN)である。]
で示される化合物の製造方法であって、以下の工程:
(1)脂肪族アルコールおよび三価リン化合物を、活性化剤または有機塩基の存在下、有機溶媒中で反応する;
(2)得られた反応混合物を、水を用いて分液洗浄する;
(3)工程(2)後の有機層を回収し、濃縮する(但し、工程(1)で使用する有機溶媒がニトリル系溶媒の場合には、工程(2)~(3)は省略してもよい);
(4)工程(3)で得られた残渣を脂肪族炭化水素系溶媒中に溶解する(但し、工程(1)で使用する有機溶媒が脂肪族炭化水素系溶媒の場合には、工程(2)~(4)は省略してもよい);
(5)工程(4)で調製した脂肪族炭化水素系溶液を、ニトリル系溶媒を用いて分液洗浄する;
(6)工程(5)後の脂肪族炭化水素系溶液を回収して、ホスホラミダイト化合物を溶液として得る、
を含む、該製造方法。
項[2] 工程(4)で使用される脂肪族炭化水素系溶媒が、ペンタン、ヘキサン、ヘプタン、またはオクタンから少なくとも1つ選ばれ、そして工程(5)で使用されるニトリル系溶媒が、アセトニトリル、プロピオニトリル、またはベンゾニトリルから少なくとも1つ選ばれる、項[1]に記載の製造方法。
項[3-1] R3が、置換されていてもよいC1-C8アルキルオキシ基、または置換されていてもよいC2-C8アルキニルオキシ基であり、該置換基はシアノ基(CN)である、項[1]または項[2]に記載の製造方法。
項[3-2] R3が、置換されていてもよいC1-C8アルキルオキシ基である、項[1]または項[2]に記載の製造方法。
項[3-3] R1が、置換されていてもよいC10-C30の第1級または第2級のアルキルオキシ基であり、そして、R3が、-OCH2CH2CN、-OCH3または-OCH2CH3で示される基である、項[1]または項[2]記載の製造方法。
項[4] 各R2が共にi-プロピル基である、項[1]~項[3]のいずれか1項に記載の製造方法。
項[5] 式(I)で示される化合物が、下記式:
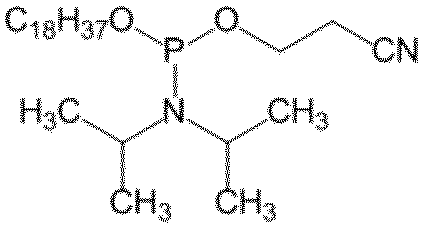
で示される化合物である、項[1]~項[4]のいずれか1項に記載の製造方法。
項[6] 工程(6)で得られる溶液が、H-ホスホネート化合物、ジアミダイト化合物を含まず、実質的に純粋なホスホラミダイト化合物の溶液である、項[1]~項[5]のいずれか1項に記載の製造方法。
項[1] 式(I):
R1は、置換されていてもよいC6-C30アルキルオキシ基であり、該置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、
R2は各々独立して、置換されていてもよいC1-C6アルキル基であり、該置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、そして、
R3は、置換されていてもよいC6-C30アルキルオキシ基、置換されていてもよいC1-C8アルキルオキシ基、または置換されていてもよいC2-C8アルキニルオキシ基であり、ここで、置換されていてもよいC6-C30アルキルオキシ基における置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、置換されていてもよいC1-C8アルキルオキシ基、または置換されていてもよいC2-C8アルキニルオキシ基における置換基はシアノ基(CN)である。]
で示される化合物の製造方法であって、以下の工程:
(1)脂肪族アルコールおよび三価リン化合物を、活性化剤または有機塩基の存在下、有機溶媒中で反応する;
(2)得られた反応混合物を、水を用いて分液洗浄する;
(3)工程(2)後の有機層を回収し、濃縮する(但し、工程(1)で使用する有機溶媒がニトリル系溶媒の場合には、工程(2)~(3)は省略してもよい);
(4)工程(3)で得られた残渣を脂肪族炭化水素系溶媒中に溶解する(但し、工程(1)で使用する有機溶媒が脂肪族炭化水素系溶媒の場合には、工程(2)~(4)は省略してもよい);
(5)工程(4)で調製した脂肪族炭化水素系溶液を、ニトリル系溶媒を用いて分液洗浄する;
(6)工程(5)後の脂肪族炭化水素系溶液を回収して、ホスホラミダイト化合物を溶液として得る、
を含む、該製造方法。
項[2] 工程(4)で使用される脂肪族炭化水素系溶媒が、ペンタン、ヘキサン、ヘプタン、またはオクタンから少なくとも1つ選ばれ、そして工程(5)で使用されるニトリル系溶媒が、アセトニトリル、プロピオニトリル、またはベンゾニトリルから少なくとも1つ選ばれる、項[1]に記載の製造方法。
項[3-1] R3が、置換されていてもよいC1-C8アルキルオキシ基、または置換されていてもよいC2-C8アルキニルオキシ基であり、該置換基はシアノ基(CN)である、項[1]または項[2]に記載の製造方法。
項[3-2] R3が、置換されていてもよいC1-C8アルキルオキシ基である、項[1]または項[2]に記載の製造方法。
項[3-3] R1が、置換されていてもよいC10-C30の第1級または第2級のアルキルオキシ基であり、そして、R3が、-OCH2CH2CN、-OCH3または-OCH2CH3で示される基である、項[1]または項[2]記載の製造方法。
項[4] 各R2が共にi-プロピル基である、項[1]~項[3]のいずれか1項に記載の製造方法。
項[5] 式(I)で示される化合物が、下記式:
項[6] 工程(6)で得られる溶液が、H-ホスホネート化合物、ジアミダイト化合物を含まず、実質的に純粋なホスホラミダイト化合物の溶液である、項[1]~項[5]のいずれか1項に記載の製造方法。
(キャッピング反応用組成物およびキャッピング反応)
項[7] 項[1]~項[6]のいずれか1項に記載の製造方法によって得られる化合物を含有する溶液、および適宜添加剤を含む、キャッピング反応用組成物。
項[8] ヌクレオシド、ヌクレオチド、またはオリゴヌクレオチドから選ばれる2つ以上の化合物をカップリング反応させた後の反応溶液を、適宜アクチベータの存在下、項[7]に記載のキャッピング反応用組成物と接触させる工程を含む、キャッピング方法。
項[9] 更に適宜、キャッピング反応工程後に、キャッピング副反応産物を求核剤と反応させることを含み、ここで、該求核剤は、アルコール、フェノール、カルボン酸、またはN-アルキルヒドロキシルアミンから選ばれる、項[8]に記載のキャッピング方法。
項[10] キャッピング反応工程後に有機塩基を添加し、その後に求核剤と反応させる工程を含む、項[9]に記載のキャッピング方法。
項[7] 項[1]~項[6]のいずれか1項に記載の製造方法によって得られる化合物を含有する溶液、および適宜添加剤を含む、キャッピング反応用組成物。
項[8] ヌクレオシド、ヌクレオチド、またはオリゴヌクレオチドから選ばれる2つ以上の化合物をカップリング反応させた後の反応溶液を、適宜アクチベータの存在下、項[7]に記載のキャッピング反応用組成物と接触させる工程を含む、キャッピング方法。
項[9] 更に適宜、キャッピング反応工程後に、キャッピング副反応産物を求核剤と反応させることを含み、ここで、該求核剤は、アルコール、フェノール、カルボン酸、またはN-アルキルヒドロキシルアミンから選ばれる、項[8]に記載のキャッピング方法。
項[10] キャッピング反応工程後に有機塩基を添加し、その後に求核剤と反応させる工程を含む、項[9]に記載のキャッピング方法。
(オリゴヌクレオチドの製造方法)
項[11]
工程(a):非極性溶媒中において、3’位水酸基もしくは核酸塩基を介して擬似固相保護基が修飾され且つ5’位水酸基が酸性条件下で除去可能な一時保護基でブロックされたn量体(ここで、nは、1以上の任意の整数を示す。)もしくは、n=0の場合、水酸基を2個以上持つリンカーの1つの水酸基が擬似固相保護基で修飾され且つ別の水酸基が酸性条件下で除去可能な一時保護基でブロックされた化合物を、酸の単独もしくはカチオン捕捉剤の共存下で反応させて、水酸基の一時保護基をデブロックし、その後に極性溶媒を用いて該擬似固相保護基を含有する化合物を回収する工程(デブロックおよび晶析工程);
工程(b):工程(a)の回収物を乾燥後、非極性溶媒中において、3’位水酸基がホスホラミダイト化され且つ5’位水酸基が酸性条件下で除去可能な一時保護基で保護されたm+1個重合オリゴヌクレオチド(ここで、mは、0以上の任意の整数を示す。)およびアクチベータを添加して、工程(a)で得られた水酸基の一時保護基がデブロックされた化合物(n個重合オリゴヌクレオチド)と、その5’位水酸基を介してホスファイトトリエステル結合により縮合させる工程(カップリング工程);
工程(c):工程(b)の反応液に、項[1]~[6]に記載のいずれか1項に記載の製造方法によって得られる高脂溶性ホスホラミダイト化合物を含有する溶液、または項[7]に記載のキャッピング反応用組成物を添加し、工程(b)において未反応であった化合物について、その5’位水酸基を介して該高脂溶性化合物のホスファイトトリエステル結合により縮合(キャッピング)させる工程(キャッピング反応工程);
工程(d):工程(c)のキャッピング副反応産物に求核剤を添加して反応させることを含み、ここで、該求核剤は、アルコール、フェノール、カルボン酸またはN-アルキルヒドロキシルアミンから選ばれる、工程(キャッピング反応の後処理工程);
工程(e):工程(d)の反応液に、酸化剤または硫化剤を添加して、工程(b)~(d)で得られたm+1+n量体のオリゴヌクレオチドのホスファイトトリエステル結合を、ホスフェートトリエステル結合またはチオホスフェートトリエステル結合にそれぞれ変換する工程(酸化または硫化反応の工程);
更に適宜、工程(f):工程(e)で得られた反応液に極性溶媒を添加して、m+1+n個重合オリゴヌクレオチドを沈殿させて、固液分離により取得する工程(単離工程);
更に適宜、工程(g):工程(f)で得られたm+1+n個重合オリゴヌクレオチドの保護基を全て除去する工程(脱保護工程);
を含む、オリゴヌクレオチドの製造方法。
項[11]
工程(a):非極性溶媒中において、3’位水酸基もしくは核酸塩基を介して擬似固相保護基が修飾され且つ5’位水酸基が酸性条件下で除去可能な一時保護基でブロックされたn量体(ここで、nは、1以上の任意の整数を示す。)もしくは、n=0の場合、水酸基を2個以上持つリンカーの1つの水酸基が擬似固相保護基で修飾され且つ別の水酸基が酸性条件下で除去可能な一時保護基でブロックされた化合物を、酸の単独もしくはカチオン捕捉剤の共存下で反応させて、水酸基の一時保護基をデブロックし、その後に極性溶媒を用いて該擬似固相保護基を含有する化合物を回収する工程(デブロックおよび晶析工程);
工程(b):工程(a)の回収物を乾燥後、非極性溶媒中において、3’位水酸基がホスホラミダイト化され且つ5’位水酸基が酸性条件下で除去可能な一時保護基で保護されたm+1個重合オリゴヌクレオチド(ここで、mは、0以上の任意の整数を示す。)およびアクチベータを添加して、工程(a)で得られた水酸基の一時保護基がデブロックされた化合物(n個重合オリゴヌクレオチド)と、その5’位水酸基を介してホスファイトトリエステル結合により縮合させる工程(カップリング工程);
工程(c):工程(b)の反応液に、項[1]~[6]に記載のいずれか1項に記載の製造方法によって得られる高脂溶性ホスホラミダイト化合物を含有する溶液、または項[7]に記載のキャッピング反応用組成物を添加し、工程(b)において未反応であった化合物について、その5’位水酸基を介して該高脂溶性化合物のホスファイトトリエステル結合により縮合(キャッピング)させる工程(キャッピング反応工程);
工程(d):工程(c)のキャッピング副反応産物に求核剤を添加して反応させることを含み、ここで、該求核剤は、アルコール、フェノール、カルボン酸またはN-アルキルヒドロキシルアミンから選ばれる、工程(キャッピング反応の後処理工程);
工程(e):工程(d)の反応液に、酸化剤または硫化剤を添加して、工程(b)~(d)で得られたm+1+n量体のオリゴヌクレオチドのホスファイトトリエステル結合を、ホスフェートトリエステル結合またはチオホスフェートトリエステル結合にそれぞれ変換する工程(酸化または硫化反応の工程);
更に適宜、工程(f):工程(e)で得られた反応液に極性溶媒を添加して、m+1+n個重合オリゴヌクレオチドを沈殿させて、固液分離により取得する工程(単離工程);
更に適宜、工程(g):工程(f)で得られたm+1+n個重合オリゴヌクレオチドの保護基を全て除去する工程(脱保護工程);
を含む、オリゴヌクレオチドの製造方法。
(擬似固相保護基およびそれを含有するヌクレオチド化合物)
項[12] 式(II):

[式中、
R5は各々独立して、置換されていてもよいC6-C30のアルキル基であり、
R6は、置換されていてもよいC1-C6アルキル基、置換されていてもよいC3-C6シクロアルキル基、置換されていてもよい非芳香族性複素環式基、置換されていてもよいアリール基、置換されていてもよいヘテロアリール基、置換されていてもよいアラルキル基、または置換されていてもよいヘテロアリールアルキル基から選ばれ、
R7は、置換されていてもよいC1-C6アルキレン基、または置換されていてもよいC1-C6アルキレンオキシ基であり、
R8は、置換されていてもよいC1-C6アルキレン基であり、あるいは、
R6およびR7は、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、または置換されていてもよいヘテロアリール基を形成してもよく、あるいは、
R6、R7およびR8は、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、置換されていてもよい非芳香族性複素環アルキル基、置換されていてもよいヘテロアリール基、または置換されていてもよいヘテロアリールアルキル基を形成してもよく、そして、
*は、Lとの結合位置を示す]
で示される、擬似固相保護基。
項[12] 式(II):
R5は各々独立して、置換されていてもよいC6-C30のアルキル基であり、
R6は、置換されていてもよいC1-C6アルキル基、置換されていてもよいC3-C6シクロアルキル基、置換されていてもよい非芳香族性複素環式基、置換されていてもよいアリール基、置換されていてもよいヘテロアリール基、置換されていてもよいアラルキル基、または置換されていてもよいヘテロアリールアルキル基から選ばれ、
R7は、置換されていてもよいC1-C6アルキレン基、または置換されていてもよいC1-C6アルキレンオキシ基であり、
R8は、置換されていてもよいC1-C6アルキレン基であり、あるいは、
R6およびR7は、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、または置換されていてもよいヘテロアリール基を形成してもよく、あるいは、
R6、R7およびR8は、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、置換されていてもよい非芳香族性複素環アルキル基、置換されていてもよいヘテロアリール基、または置換されていてもよいヘテロアリールアルキル基を形成してもよく、そして、
*は、Lとの結合位置を示す]
で示される、擬似固相保護基。
(ヌクレオチド化合物)
項[13] 式(III):

[式中、
Zは、項[12]に記載の式(II)で示される擬似固相保護基であり、
Lは、リンカーであり、
Baseは、各々独立して保護されていてもよい核酸塩基であり、
n個のEWGは、各々独立して電子吸引性基であり、
n個のR4は、各々独立して酸素原子または硫黄原子であり、
n個のRは、各々独立して水素原子、ハロゲン原子、保護基で置換されていてもよいヒドロキシ基、置換されていてもよいC1-C6アルコキシ基、4位炭素原子に架橋する有機基、または架橋構造型人工核酸基であり、
nは、0以上の任意の整数であり、そして、
Xは、酸により脱保護可能な一時保護基である]
で示される、ヌクレオチド化合物。
項[14] Lが、下記式:

[式中、
*は疎水性基との結合位置、および**はリン原子との結合位置を示し、
Baseは、各々独立して保護されていてもよい核酸塩基であり、そして、
Rは、各々独立して水素原子、ハロゲン原子、保護基で置換されていてもよいヒドロキシ基、置換されていてもよいC1-C6アルコキシ基、4位炭素原子に架橋する有機基、または架橋構造型人工核酸基である]
から選ばれる項[13]に記載のヌクレオチド化合物。
項[13] 式(III):
Zは、項[12]に記載の式(II)で示される擬似固相保護基であり、
Lは、リンカーであり、
Baseは、各々独立して保護されていてもよい核酸塩基であり、
n個のEWGは、各々独立して電子吸引性基であり、
n個のR4は、各々独立して酸素原子または硫黄原子であり、
n個のRは、各々独立して水素原子、ハロゲン原子、保護基で置換されていてもよいヒドロキシ基、置換されていてもよいC1-C6アルコキシ基、4位炭素原子に架橋する有機基、または架橋構造型人工核酸基であり、
nは、0以上の任意の整数であり、そして、
Xは、酸により脱保護可能な一時保護基である]
で示される、ヌクレオチド化合物。
項[14] Lが、下記式:
*は疎水性基との結合位置、および**はリン原子との結合位置を示し、
Baseは、各々独立して保護されていてもよい核酸塩基であり、そして、
Rは、各々独立して水素原子、ハロゲン原子、保護基で置換されていてもよいヒドロキシ基、置換されていてもよいC1-C6アルコキシ基、4位炭素原子に架橋する有機基、または架橋構造型人工核酸基である]
から選ばれる項[13]に記載のヌクレオチド化合物。
本発明の高脂溶性ホスホラミダイトの製造及び精製方法により、高脂溶性ホスホラミダイトを高純度で簡便に精製および製造することができる。また、本発明の製造方法で得られた高脂溶性ホスホラミダイトを含有する溶液、または該溶液および適宜添加剤を含む組成物をキャッピング反応用試薬として使用することができ、またキャッピング反応後の求核試薬を用いた後処理とあわせて、ヌクレオチド鎖長の単一伸張を効率よく行うことができる。
以下に、本発明をさらに詳細に説明する。
本明細書および特許請求の範囲中で使用する用語の定義を示す。特に記述がない限り、本明細書で用いるすべての技術用語及び科学用語は、本発明が属する技術分野の当業者に一般に理解されるのと同じ意味をもつ。本明細書に記載されたものと同様又は同等の任意の方法及び材料は、本発明の実施又は試験において使用することができるが、好ましい方法及び材料を以下に記載する。なお、本明細書において引用された全ての刊行物に記載されている構築物および方法論を、記載および開示する目的で、参照として本明細書に組み込まれる。
本明細書および特許請求の範囲中で使用する用語の定義を示す。特に記述がない限り、本明細書で用いるすべての技術用語及び科学用語は、本発明が属する技術分野の当業者に一般に理解されるのと同じ意味をもつ。本明細書に記載されたものと同様又は同等の任意の方法及び材料は、本発明の実施又は試験において使用することができるが、好ましい方法及び材料を以下に記載する。なお、本明細書において引用された全ての刊行物に記載されている構築物および方法論を、記載および開示する目的で、参照として本明細書に組み込まれる。
(高脂溶性ホスホラミダイトの製造および精製方法)
本発明の1態様によれば、本発明は、式(I):

[式中、
R1は、置換されていてもよいC6-C30アルキルオキシ基であり、該置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、
R2は各々独立して、置換されていてもよいC1-C6アルキル基であり、該置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、そして、
R3は、置換されていてもよいC6-C30アルキルオキシ基、置換されていてもよいC1-C8アルキルオキシ基、または置換されていてもよいC1-C8アルキニルオキシ基であり、ここで、置換されていてもよいC6-C30アルキルオキシ基における置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基であり、置換されていてもよいC1-C8アルキルオキシ基、または置換されていてもよいC2-C8アルキニルオキシ基における置換基はシアノ基(CN)である。]
で示される化合物(以下、「本発明の高脂溶性ホスホラミダイト化合物」と呼称する)の製造方法であって、以下の工程:
(1)脂肪族アルコールおよび三価リン化合物を、活性化剤または有機塩基の存在下、有機溶媒中で反応する;
(2)得られた反応混合物を、水を用いて分液洗浄する;
(3)工程(2)後の有機層を回収し、濃縮する(但し、工程(1)で使用する有機溶媒がニトリル系溶媒の場合には、工程(2)~(3)は省略してもよい);
(4)工程(3)で得られた残渣を脂肪族炭化水素系溶媒中に溶解する(但し、工程(1)で使用する有機溶媒が脂肪族炭化水素系溶媒の場合には、工程(2)~(4)は省略してもよい);
(5)工程(4)で調製した脂肪族炭化水素系溶液を、ニトリル系溶媒を用いて分液洗浄する;
(6)工程(5)後の脂肪族炭化水素系溶液を回収して、ホスホラミダイト化合物を溶液として得る、
を含む、該製造方法(以下、「本発明の高脂溶性ホスホラミダイトの製造方法」と呼称する)を提供する。
本発明の1態様によれば、本発明は、式(I):
R1は、置換されていてもよいC6-C30アルキルオキシ基であり、該置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、
R2は各々独立して、置換されていてもよいC1-C6アルキル基であり、該置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、そして、
R3は、置換されていてもよいC6-C30アルキルオキシ基、置換されていてもよいC1-C8アルキルオキシ基、または置換されていてもよいC1-C8アルキニルオキシ基であり、ここで、置換されていてもよいC6-C30アルキルオキシ基における置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基であり、置換されていてもよいC1-C8アルキルオキシ基、または置換されていてもよいC2-C8アルキニルオキシ基における置換基はシアノ基(CN)である。]
で示される化合物(以下、「本発明の高脂溶性ホスホラミダイト化合物」と呼称する)の製造方法であって、以下の工程:
(1)脂肪族アルコールおよび三価リン化合物を、活性化剤または有機塩基の存在下、有機溶媒中で反応する;
(2)得られた反応混合物を、水を用いて分液洗浄する;
(3)工程(2)後の有機層を回収し、濃縮する(但し、工程(1)で使用する有機溶媒がニトリル系溶媒の場合には、工程(2)~(3)は省略してもよい);
(4)工程(3)で得られた残渣を脂肪族炭化水素系溶媒中に溶解する(但し、工程(1)で使用する有機溶媒が脂肪族炭化水素系溶媒の場合には、工程(2)~(4)は省略してもよい);
(5)工程(4)で調製した脂肪族炭化水素系溶液を、ニトリル系溶媒を用いて分液洗浄する;
(6)工程(5)後の脂肪族炭化水素系溶液を回収して、ホスホラミダイト化合物を溶液として得る、
を含む、該製造方法(以下、「本発明の高脂溶性ホスホラミダイトの製造方法」と呼称する)を提供する。
本発明の製造方法で得られる式(I)で示されるホスホラミダイト化合物は、有機溶媒(例えば、脂肪族炭化水素系溶媒等の疎水性溶媒)との親和性が高く、一方で水との親和性が低い、高脂溶性の性質を有する。
本明細書中、R1および/またはR3についての、用語「置換されていてもよいC6-C30のアルキルオキシ基」とは、酸素原子に結合した、炭素数が6~30の炭化水素鎖基であって、第1級または第2級のアルキルオキシ基が好ましく、これらの基は、適宜、置換基で置換されていてもよく、該置換基は、C1-C6アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、そして、該アルキルオキシ基は、適宜、芳香族環、シアノ基、エステル結合を含む基、またはアミド結合を含む基を含んでいてもよい、基を意味する。該アルキル基は、直鎖または分枝鎖状のいずれでもよいが、直鎖状のアルキル基が好ましい。置換されていてもよいC6~C30アルキルオキシ基としては、核酸5’位の一時保護基Xよりも疎水性である基が好ましい。具体的には、C8-C30の第1級または第2級のアルキルオキシ基が好ましく、C10-C30の第1級または第2級のアルキルオキシ基がより好ましく、第1級または第2級のC10-C20のアルキルオキシ基がより一層好ましい。好適な1具体例としては、C18の第1級または第2級のアルキルオキシ基、特にC18の第1級のアルキルオキシ基が挙げられる。置換基としては、本発明で使用する反応(例えば、キャッピング反応、およびオリゴヌクレオチド合成上のカップリング反応、脱保護反応、デブロック反応、酸化反応、硫化反応)を阻害しない限り置換基の種類は限定されるものではないが、例えばC1-C6アルキル基、またはC3-C6シクロアルキル基から選ばれる1つまたはそれ以上の基を挙げられる。置換基の具体例としては、メチル、エチル、イソプロピル、シクロプロピル、シクロヘキシル等が挙げられる。
「C1-C6アルキル基」としては、例えばメチル、エチル、プロピル、イソプロピル、ブチル、ペンチル及びヘキシルが挙げられる。
「C3-C6シクロアルキル基」としては、シクロプロピル、シクロブチル、シクロペンチル、またはシクロヘキシルが挙げられる。
「エステル結合を含む基」とは、アルキルカルボキシ基を意味し、例えばメチルカルボキシ、エチルカルボキシ、またはイソプロピルカルボキシが挙げられる。ここで、該エステル結合を含む基とは、アシルオキシ型基およびアルコキシカルボニル型基の両方を含むことを意図する。
「アミド結合を含む基」としては、窒素原子上でアルキル基で置換されていてもよいアルキルアミド基を意味し、例えばアミド、N,N-ジメチルアミド、N,N-ジエチルアミドが挙げられる。ここで、該アミド結合を含む基とは、アミド型基およびカルボキサミド型基の両方を含むことを意図する。
本発明の1実施態様によれば、R1が、置換されていてもよいC10-C30の第1級または第2級のアルキルオキシ基であり、そして、R3が、-OCH2CH2CN、-OCH3、または-OCH2CH3で示される基であり、-OCH2CH2CNが好ましい。
本明細書中、R2についての、用語「置換されていてもよいC1-C6アルキル基」とは、炭素数が1~6の飽和炭化水素鎖基を意味する。該アルキル基は、直鎖または分枝鎖状のいずれでもよい。置換されていてもよいC1-C6アルキル基の例としては、C3アルキル基が好ましい。好適な1具体例としては、イソプロピルが挙げられる。置換基としては、本発明で使用する反応(例えば、キャッピング反応、およびオリゴヌクレオチド合成上のカップリング反応、脱保護反応、デブロック反応、酸化反応、硫化反応)を阻害しない限り置換基の種類は限定されるものではないが、例えばC1-C3アルキル基およびC3-C6シクロアルキル基から選ばれる1つまたはそれ以上の基を挙げられ、C1-C3アルキル基が好ましい。
本発明の1実施態様によれば、各R2は各々独立して、置換されていてもよいC1-C6アルキル基であり、例えば置換されていてもよいC3-C6アルキル基が好ましい。
本発明の1実施態様によれば、各R2は共にi-プロピル基である。
本明細書中、R3についての、用語「置換されていてもよいC1-C8アルキルオキシ基」および「置換されていてもよいC2-C8アルキニルオキシ基」とは、酸素原子に結合した、炭素数が1~8のアルキル基または炭素数2~8のアルキニル基であって、適宜、シアノ基で置換されていてもよい基を意味する。該アルキル基および該アルキニル基はいずれも第1級または第2級の基が好ましく、直鎖または分枝鎖状のいずれの基でもよいが、直鎖状の基が好ましい。具体的には、C1~C6のアルキルオキシ基、またはC1~C5のアルキニルオキシ基が好ましく、C1~C3のアルキルオキシ基がより好ましい。
好ましいR3の具体例としては、例えば-OCH2CH2CN、-OCH3または-OCH2CH3で示される基が挙げられ、-OCH2CH2CNがより好ましい。
本発明の1実施態様によれば、式(I)中、R1が、置換されていてもよいC10-C30の第1級または第2級のアルキルオキシ基であり、そして、R3が、-OCH2CH2CN、-OCH3または-OCH2CH3で示される基である。
本発明の1実施態様によれば、式(I)で示される化合物の具体例として、下記式:

で示される化合物が挙げられる。
本発明の式(I)で示される化合物は、いわゆるアミダイト型化合物(モノアミダイト化合物)であり、従って、ホスホラミダイト法を使用する本発明のオリゴヌクレオチドの製造において、キャッピング試薬として使用することができる。ここで、本発明で使用する式(I)で示される化合物は、ホスホラミダイト法で使用するカップリング反応用のヌクレオチドまたはオリゴヌクレオチドのホスホラミダイト化合物と比べて高脂溶性、つまり高疎水性の化合物であることから、目的のオリゴヌクレオチド生成物やカップリング反応で残留する未反応原料とは物理的性質(例えば、シリカゲル及びODS修飾シリカゲルとの親和性)が大きく異なり、よって、目的のオリゴヌクレオチド生成物とキャッピング試薬が結合した未反応原料とを区別して検出することができる。
また、本発明のキャッピング試薬である式(I)で示される化合物は、カップリング反応用のヌクレオチドホスホラミダイト化合物に比べホスホラミダイト周辺の立体が込み合っておらず、反応性が非常に高いため、未反原料のヌクレオチドまたはオリゴヌクレオチドとの高いキャッピング反応性を有し、そのため少量の使用量で目的のカップリング反応を達成することができる。
(工程1)
工程(1)における、用語「脂肪族アルコール」とは、炭素数が6~30個の炭化水素脂肪族アルコール、好ましくは第1級または第2級の炭化水素脂肪族アルコールを意味し、具体的には、R1として定義する、芳香族環を含んでもよく、置換されていてもよいC6-C30アルキルオキシ基を供する脂肪族アルコールに対応する。好ましくは、炭素数がC8~C30個、より好ましくはC10~C30個、より一層好ましくはC10~C20個の長鎖の第1級または第2級の脂肪族アルコールが挙げられる。好適な1具体例としては、C18の脂肪族アルコール、特にC18の第1級脂肪族アルコールが挙げられる。
工程(1)における、用語「脂肪族アルコール」とは、炭素数が6~30個の炭化水素脂肪族アルコール、好ましくは第1級または第2級の炭化水素脂肪族アルコールを意味し、具体的には、R1として定義する、芳香族環を含んでもよく、置換されていてもよいC6-C30アルキルオキシ基を供する脂肪族アルコールに対応する。好ましくは、炭素数がC8~C30個、より好ましくはC10~C30個、より一層好ましくはC10~C20個の長鎖の第1級または第2級の脂肪族アルコールが挙げられる。好適な1具体例としては、C18の脂肪族アルコール、特にC18の第1級脂肪族アルコールが挙げられる。
工程(1)における、用語「三価リン化合物」とは、例えばアルキルホスホロジアミダイト、アルキルクロロホスホロアミダイトを意味する。具体例としては、2-シアノエチルN,N,N',N'-テトライソプロピルホスホロジアミダイト、および2-シアノエチル ジイソプロピルクロロホスホロアミダイトを挙げられる。
工程(1)における、用語「活性化剤」とは、例えばテトラゾール化合物を挙げられる。具体例としては、1H-テトラゾール、5-エチルチオ-1H-テトラゾール(ETTと略す)が挙げられるが、これらに限定されるものではない。
工程(1)における、用語「有機塩基」は、三価リン化合物としてアルキルクロロホスホロアミダイトを使用する場合に使用し、例えば、トリエチルアミン、またはジイソプロピルエチルアミンを挙げられる。
工程(1)における、用語「有機溶媒」とは、例えばハロゲン系溶媒、ニトリル溶媒および環状エーテル系溶媒を挙げられ、ハロゲン系溶媒が好ましい。これらの有機溶媒は、無水の有機溶媒であることが好ましい。具体例としては、ジクロロメタン、クロロホルム、四塩化炭素、ジオキサン、およびテトラヒドロフラン等が挙げられるが、これらに限定されるものではない。ジクロロメタンおよびクロロホルムが好ましい。
工程(1)の反応の反応温度は、通常-20~50℃である。反応時間は、通常0.1~10時間の範囲内である。また、不活性ガスの雰囲気下で行う事が推奨される。
脂肪族アルコールと三価リン化合物との配合モル比は、例えば0.5~1.5モル当量を挙げられるが、好ましくは三価リン化合物が過剰量(例えば、1.1モル当量)である場合が挙げられる。
脂肪族アルコールと三価リン化合物との配合モル比は、例えば0.5~1.5モル当量を挙げられるが、好ましくは三価リン化合物が過剰量(例えば、1.1モル当量)である場合が挙げられる。
(工程2)
工程(2)において、工程(1)において得られた反応混合物を分液漏斗中に注ぎ、有機層を水(例えば、蒸留水)を用いて分液洗浄する。洗浄する水の量は、特に限定されるものではないが、工程(1)で使用した有機溶媒の量に対して容積比が0.1~5 v/vで使用することができる。
工程(2)において、工程(1)において得られた反応混合物を分液漏斗中に注ぎ、有機層を水(例えば、蒸留水)を用いて分液洗浄する。洗浄する水の量は、特に限定されるものではないが、工程(1)で使用した有機溶媒の量に対して容積比が0.1~5 v/vで使用することができる。
(工程3)
工程(2)の洗浄後の有機層を回収し、例えば減圧下でエバポレーター等を用いて有機溶媒を留去して、濃縮する。ここで、前記工程(1)で使用する有機溶媒がニトリル系溶媒の場合には、上記工程(2)~(3)を実施することを省くことができる。
工程(2)の洗浄後の有機層を回収し、例えば減圧下でエバポレーター等を用いて有機溶媒を留去して、濃縮する。ここで、前記工程(1)で使用する有機溶媒がニトリル系溶媒の場合には、上記工程(2)~(3)を実施することを省くことができる。
(工程4)
工程(3)で得られた残渣に、脂肪族炭化水素系溶媒を添加して溶解させる。脂肪族炭化水素系溶媒の具体例としては、ペンタン、ヘキサン、ヘプタン、およびオクタン等が挙げられるが、これらに限定されない。ペンタン、ヘキサン、およびヘプタンが好ましく、特にヘプタンが好ましい。脂肪族炭化水素系溶媒の添加量は、反応生成物の高脂溶性ホスホラミダイト化合物の、使用する脂肪族炭化水素系溶媒に対する溶解度に応じて変わり得るが、溶解するのに適量であればよく、例えば、工程(1)で使用する脂肪族アルコールの1mmol当たり、約1~500倍mLが挙げられ、典型的には約1~10倍mLが挙げられる。ここで、前記工程(1)で使用する有機溶媒が脂肪族炭化水素系溶媒の場合には、上記工程(2)~(4)を実施することを省くことができる。
工程(3)で得られた残渣に、脂肪族炭化水素系溶媒を添加して溶解させる。脂肪族炭化水素系溶媒の具体例としては、ペンタン、ヘキサン、ヘプタン、およびオクタン等が挙げられるが、これらに限定されない。ペンタン、ヘキサン、およびヘプタンが好ましく、特にヘプタンが好ましい。脂肪族炭化水素系溶媒の添加量は、反応生成物の高脂溶性ホスホラミダイト化合物の、使用する脂肪族炭化水素系溶媒に対する溶解度に応じて変わり得るが、溶解するのに適量であればよく、例えば、工程(1)で使用する脂肪族アルコールの1mmol当たり、約1~500倍mLが挙げられ、典型的には約1~10倍mLが挙げられる。ここで、前記工程(1)で使用する有機溶媒が脂肪族炭化水素系溶媒の場合には、上記工程(2)~(4)を実施することを省くことができる。
(工程5)
工程(4)で調製した脂肪族炭化水素系溶液を分液漏斗中に注ぎ、ニトリル系溶媒を用いて洗浄する。ニトリル系溶媒の具体例としては、脂肪族炭化水素系溶媒中の生成物の溶解液の有機層と分液で相分離する溶媒であれば、特に限定されるものではないが、例えば、含水アセトニトリル、アセトニトリル、プロピオニトリル、ベンゾニトリルを挙げられる。アセトニトリルが特に好ましい。洗浄するニトリル系溶媒の量は、特に限定されるものではないが、工程(1)で使用した有機溶媒の量に対して容積比が0.1~10 v/vで使用することができる。あるいは、工程(3)で使用した脂肪族炭化水素系溶媒の量に対して容積比が、0.1~10 v/vで使用することができる。洗浄回数は、1回に限定されるものでは無く、洗浄された脂肪族炭化水素系溶液の一部について31P NMR測定する事で、H-ホスホネート化合物、および/またはジアミダイト化合物の存在を確認することにより容易に洗浄回数を決定できる。
工程(4)で調製した脂肪族炭化水素系溶液を分液漏斗中に注ぎ、ニトリル系溶媒を用いて洗浄する。ニトリル系溶媒の具体例としては、脂肪族炭化水素系溶媒中の生成物の溶解液の有機層と分液で相分離する溶媒であれば、特に限定されるものではないが、例えば、含水アセトニトリル、アセトニトリル、プロピオニトリル、ベンゾニトリルを挙げられる。アセトニトリルが特に好ましい。洗浄するニトリル系溶媒の量は、特に限定されるものではないが、工程(1)で使用した有機溶媒の量に対して容積比が0.1~10 v/vで使用することができる。あるいは、工程(3)で使用した脂肪族炭化水素系溶媒の量に対して容積比が、0.1~10 v/vで使用することができる。洗浄回数は、1回に限定されるものでは無く、洗浄された脂肪族炭化水素系溶液の一部について31P NMR測定する事で、H-ホスホネート化合物、および/またはジアミダイト化合物の存在を確認することにより容易に洗浄回数を決定できる。
(工程6)
工程5の分液洗浄後に得た脂肪族炭化水素溶液を回収して、目的のホスホラミダイト化合物を溶液として得る。
得られた溶液は、溶存水を除くことで次の反応工程(例えば、キャッピング反応工程)に使用することができる。あるいは、得られた溶液を、必要に応じて、例えば減圧下でエバポレーター等を用いて濃縮して、次の反応工程に使用することができる。
工程5の分液洗浄後に得た脂肪族炭化水素溶液を回収して、目的のホスホラミダイト化合物を溶液として得る。
得られた溶液は、溶存水を除くことで次の反応工程(例えば、キャッピング反応工程)に使用することができる。あるいは、得られた溶液を、必要に応じて、例えば減圧下でエバポレーター等を用いて濃縮して、次の反応工程に使用することができる。
上記の通り、本発明の式(I)で示される高脂溶性ホスホラミダイト化合物の製造は、製造反応後の後処理として、その精製は分液処理だけで行うことができ、簡便に製造することができる。分液のみで精製する場合に、得られた生成物中に式(I)の化合物のアルキル3置換体である亜リン酸トリエステル化合物が少量(例えば、0.1%以上)含まれることがあるが、かかる亜リン酸トリエステル化合物はオリゴヌクレオチド原料及び生成物とは反応性が全くないため、式(I)の化合物中に含まれていてもキャッピング試薬として使用する際には問題とならない。また、工程(6)で得られる、脂肪族炭化水素系溶媒中の目的のホスホラミダイト化合物(モノアミダイト化合物)の溶液は、工程(2)での水および工程(4)でのニトリル系溶媒を用いる洗浄操作により、オリゴヌクレオチド原料と、副生成物を生じるH-ホスホネート化合物(例えば、2-シアノエチルN,N-ジイソプロピルホスホナミデート)及び、ジアミダイト化合物、原料の炭化水素脂肪族アルコール(例えば、実施例6参照)を、脂肪族炭化水素系溶液から除去することができ、反応性のある夾雑物を実質的に含まない、目的のモノアミダイト化合物を実質的に純粋な高純度で含有する溶液として得ることができる。
(キャッピング反応用溶液およびキャッピング反応)
本発明の1態様によれば、本発明は、本発明の高脂溶性ホスホラミダイトの製造方法によって製造した、高脂溶性ホスホラミダイト化合物を含有する溶液、または適宜添加剤(例えば、安定化剤)を加えた組成物をキャッピング反応用試薬(以下、「本発明のキャッピング反応用試薬」と呼称する)として用いるキャッピング方法(以下、「本発明のキャッピング反応」と呼称する)を提供する。以下に、キャッピング反応を反応スキームIとして示す。
反応スキームI

本発明の1態様によれば、本発明は、本発明の高脂溶性ホスホラミダイトの製造方法によって製造した、高脂溶性ホスホラミダイト化合物を含有する溶液、または適宜添加剤(例えば、安定化剤)を加えた組成物をキャッピング反応用試薬(以下、「本発明のキャッピング反応用試薬」と呼称する)として用いるキャッピング方法(以下、「本発明のキャッピング反応」と呼称する)を提供する。以下に、キャッピング反応を反応スキームIとして示す。
反応スキームI
本明細書中、用語「キャッピング」とは、ヌクレオチド鎖の伸張の反応において、ヌクレオシド、ヌクレオチド、またはオリゴヌクレオチドから選ばれる2つ以上の化合物、典型的にはヌクレオチド同士のカップリング反応(例えば、ホスホラミダイト法)において未反応鎖の反応活性点の基を保護することを意味する。キャッピングにより、次回のカップリング反応において目的のオリゴヌクレオチド鎖の反応活性点の基と競合して当該カップリング反応以前に残存したオリゴヌクレオチド鎖の反応活性点での伸張反応が進行することを防止することができ、N-1オリゴヌクレオチドの副生を防止することができる。
本発明の1態様によれば、本発明は、
ヌクレオシド、ヌクレオチド、またはオリゴヌクレオチドから選ばれる2つ以上の化合物のカップリング反応後の反応溶液を、適宜アクチベータの存在下、本発明のキャッピング反応用試薬(つまり、本発明の高脂溶性ホスホラミダイトの製造方法で得られた高脂溶性ホスホラミダイト化合物を含有する溶液、または該溶液および適宜添加剤を含有する組成物)と接触させる工程(上記反応スキームI中の工程(c))、
を含むキャッピング方法(以下、「本発明のキャッピング方法」と呼称する)、を提供する。
ヌクレオシド、ヌクレオチド、またはオリゴヌクレオチドから選ばれる2つ以上の化合物のカップリング反応後の反応溶液を、適宜アクチベータの存在下、本発明のキャッピング反応用試薬(つまり、本発明の高脂溶性ホスホラミダイトの製造方法で得られた高脂溶性ホスホラミダイト化合物を含有する溶液、または該溶液および適宜添加剤を含有する組成物)と接触させる工程(上記反応スキームI中の工程(c))、
を含むキャッピング方法(以下、「本発明のキャッピング方法」と呼称する)、を提供する。
本発明の1実施態様によれば、本発明のキャッピング方法は、更に必要に応じて、キャッピング反応工程後に、キャッピング副反応産物を求核剤と反応させる工程(同工程(d))を含み得る。ここで、更に、キャッピング反応工程後に有機塩基を添加し、その後に求核剤と反応させる工程を含み得る。ここで、該有機塩基としては例えば、2,4,6-コリジン、2-ピコリン、2,6-ルチジン、1-メチルイミダゾール、及びピリジンを挙げられるが、これに限定されない。
本明細書中、用語「ヌクレオシド」とは、糖部分(例えば、リボース糖など)、糖部分の誘導体、又は糖部分と官能価が同等なもの(例えば、炭素環等の類似体)と共有結合した塩基または塩基性基(例えば、少なくとも1つの同素環、少なくとも1つの複素環、少なくとも1つのアリール基など)を含む核酸成分を意味する。例えば、ヌクレオシドが糖部分を含む場合には、典型的には塩基が糖部分の1’位に結合する。塩基は、後記のものを挙げられる。
本明細書中、用語「ヌクレオチド」とは、ヌクレオシドのエステル、例えば、ヌクレオシドのリン酸エステルを意味する。
本明細書中、「オリゴヌクレオチド」とは、少なくとも2個のヌクレオチド、典型的には3個以上(例えば、4個)のヌクレオチド、より典型的には室温にて相補鎖による2本鎖形成が可能な10個を超えるヌクレオチドを含む核酸を意味し、ヌクレオチドの総数は、本発明においてnで示す。該オリゴヌクレオチドは、一本鎖のヌクレオチド、又はそれらの糖部分に2’-O,4’-C-メチレン架橋を有するヌクレオチド等の、その化学修飾物(例えば、架橋構造型人工核酸(BNA:Bridged Nucleic Acid)などが挙げられる。BNAとしては、例えば、ロックト人工核酸(LNA:Locked Nucleic Acid)、2’-O,4’-C-エチレン架橋核酸(ENA:2’-O,4’-C-Ethylenebridged Nucleic Acid)などが挙げられる。以下に、本発明に用いることができるLNA及びENAを含むBNAの具体的な構造(ヌクレオシド部分)を、国際公開第2016/006697号公報に記載の図を引用して示す。

修飾物としては、限定されるものではないが、オリゴヌクレオチドの修飾された骨格が挙げられ、典型的には、Micklefield(2001年)の「Current Medicinal Chemistry」8:1157-1170に記載されている、ペプチド核酸(PNA)、ホスホロチオエートDNA、メチルホスホネートDNA等の修飾物が挙げられる。オリゴヌクレオチドは、前記のヌクレオチドおよび前記の修飾物の任意の組み合わせからなる。
本明細書中、「Base」とは、各々独立して、アデニン残基、グアニン残基、シトシン残基、チミン残基、またはウラシル残基から選ばれる基であって、これらの残基は適宜保護基で置換されていてもよい基を意味する。
本発明の好ましい具体的な実施態様において、核酸塩基残基の例としては、下記式:

によって示される基が挙げられる。
(工程(c))
工程(c)においては、ヌクレオシド、ヌクレオチド、またはオリゴヌクレオチドから選ばれる2つ以上の化合物のカップリング反応後の反応溶液を、適宜アクチベータの存在下、本発明の高脂溶性ホスホラミダイトの製造方法で得られる高脂溶性ホスホラミダイト化合物を含有する溶液、または該溶液および適宜添加剤を含む組成物と接触させる工程を含む。
工程(c)においては、ヌクレオシド、ヌクレオチド、またはオリゴヌクレオチドから選ばれる2つ以上の化合物のカップリング反応後の反応溶液を、適宜アクチベータの存在下、本発明の高脂溶性ホスホラミダイトの製造方法で得られる高脂溶性ホスホラミダイト化合物を含有する溶液、または該溶液および適宜添加剤を含む組成物と接触させる工程を含む。
工程(c)のカップリング反応において使用する、ヌクレオシド、ヌクレオチド、またはオリゴヌクレオチドから選ばれる2つ以上の化合物とは、一方が、反応スキームI中の工程(a)後のデブロック反応後の、式:HO-(N)n-L-Z(ここで、nは、1以上の任意の整数を示す。)で示される、5’位水酸基がデブロックされ且つ3’位水酸基、核酸塩基の少なくとも1つの基が擬似固相保護基で置換されたn個重合のヌクレオシド、ヌクレオチド、またはオリゴヌクレオチドであり、他方が、式:X-(N)m-Np(ここで、mは、0以上の任意の整数を示す。)で示される、5’位水酸基が酸性条件下で除去可能な一時保護基で保護され且つ3’位水酸基がホスホラミダイト化されたm+1個重合のオリゴヌクレオチドを挙げられる。
工程(c)のキャッピング反応の反応温度は、通常-20~80℃である。反応時間は、通常0~30時間の範囲内である。また、不活性ガスの雰囲気下で行う事が推奨される。
キャッピング試薬としての本発明の高脂溶性ホスホラミダイト化合物の使用量は、カップリング反応に用いる前記の式:HO-(N)n-L-Zで示されるn個重合のヌクレオシド、ヌクレオチド、またはオリゴヌクレオチドの量に対して、配合モル比が、例えば0.1~1.5モル当量、好ましくは0.1~0.5モル当量が挙げられる。
キャッピング試薬としての本発明の高脂溶性ホスホラミダイト化合物の使用量は、カップリング反応に用いる前記の式:HO-(N)n-L-Zで示されるn個重合のヌクレオシド、ヌクレオチド、またはオリゴヌクレオチドの量に対して、配合モル比が、例えば0.1~1.5モル当量、好ましくは0.1~0.5モル当量が挙げられる。
工程(c)において使用してもよいアクチベータの例としては、ホスホラミダイト方法を用いるキャッピング反応において使用することが可能であることが通常知られている試薬を挙げられるが、例えばテトラゾール(例えば、1H-テトラゾール、5-エチルチオ-1H-テトラゾール(ETTと略す))を挙げられる。該アクチベータの使用量は、キャッピング試薬としての本発明の高脂溶性ホスホラミダイト化合物の使用量に対して、配合モル比が、例えば1.0~10.0モル当量、好ましくは1.0~2.0モル当量が挙げられるが、該当量を予め工程(b)において余分に加えておいてもよい。
(工程(d))
本発明のキャッピング方法は、更に必要に応じて、工程(c)のキャッピング反応工程後に、キャッピング副反応産物を求核剤と反応させる工程を含み得る。ここで、該求核剤の例としては、アルコール、フェノール、カルボン酸またはN-アルキルヒドロキシルアミンから選ばれる化合物が挙げられ、具体的にはエタノール、メタノール、N-ヒドロキシスクシンイミドが挙げられ、好ましくはエタノールが挙げられる。該求核剤の使用量は、キャッピング試薬としての本発明の高脂溶性ホスホラミダイト化合物の使用量に対して、配合モル比が、例えば0.1~100モル当量、好ましくは1~20モル当量が挙げられる。
本発明のキャッピング方法は、更に必要に応じて、工程(c)のキャッピング反応工程後に、キャッピング副反応産物を求核剤と反応させる工程を含み得る。ここで、該求核剤の例としては、アルコール、フェノール、カルボン酸またはN-アルキルヒドロキシルアミンから選ばれる化合物が挙げられ、具体的にはエタノール、メタノール、N-ヒドロキシスクシンイミドが挙げられ、好ましくはエタノールが挙げられる。該求核剤の使用量は、キャッピング試薬としての本発明の高脂溶性ホスホラミダイト化合物の使用量に対して、配合モル比が、例えば0.1~100モル当量、好ましくは1~20モル当量が挙げられる。
(オリゴヌクレオチドの製造方法)
本発明の1態様によれば、本発明は、本発明のキャッピング方法の工程を含むオリゴヌクレオチドの製造方法をも提供する。下記に、本発明のオリゴヌクレオチドの製造方法の代表的な模式図を反応スキームIIに示す。
反応スキームII

本発明の1態様によれば、本発明は、本発明のキャッピング方法の工程を含むオリゴヌクレオチドの製造方法をも提供する。下記に、本発明のオリゴヌクレオチドの製造方法の代表的な模式図を反応スキームIIに示す。
反応スキームII
本発明のオリゴヌクレオチドの製造方法(以下、「本発明の製造方法]と呼称する」を、前記の反応スキームIIの記載に沿って説明するが、一般的に知られるオリゴヌクレオチドの製造方法であればよく、反応スキームIIの反応スキームに限定されるものではない。
具体的には、適宜保護されたn個重合(つまり、n量体)オリゴヌクレオチドから、適宜保護されたn+1+m個重合(つまり、n+1+m量体)オリゴヌクレオチドへの製造方法について説明する。ここで、n=1の場合には、n量体のオリゴヌクレオチドは、「ヌクレオシド」を含み得て、またm=0の場合には、m+1量体のオリゴヌクレオチドは、「ヌクレオシド」を含み得る。
本発明のオリゴヌクレオチドの製造方法は、以下の工程(a)~(e)を含む。
具体的には、適宜保護されたn個重合(つまり、n量体)オリゴヌクレオチドから、適宜保護されたn+1+m個重合(つまり、n+1+m量体)オリゴヌクレオチドへの製造方法について説明する。ここで、n=1の場合には、n量体のオリゴヌクレオチドは、「ヌクレオシド」を含み得て、またm=0の場合には、m+1量体のオリゴヌクレオチドは、「ヌクレオシド」を含み得る。
本発明のオリゴヌクレオチドの製造方法は、以下の工程(a)~(e)を含む。
(工程(a):デブロック工程および晶析工程)
工程(a)の反応スキーム1を下記に示す。
反応スキーム1
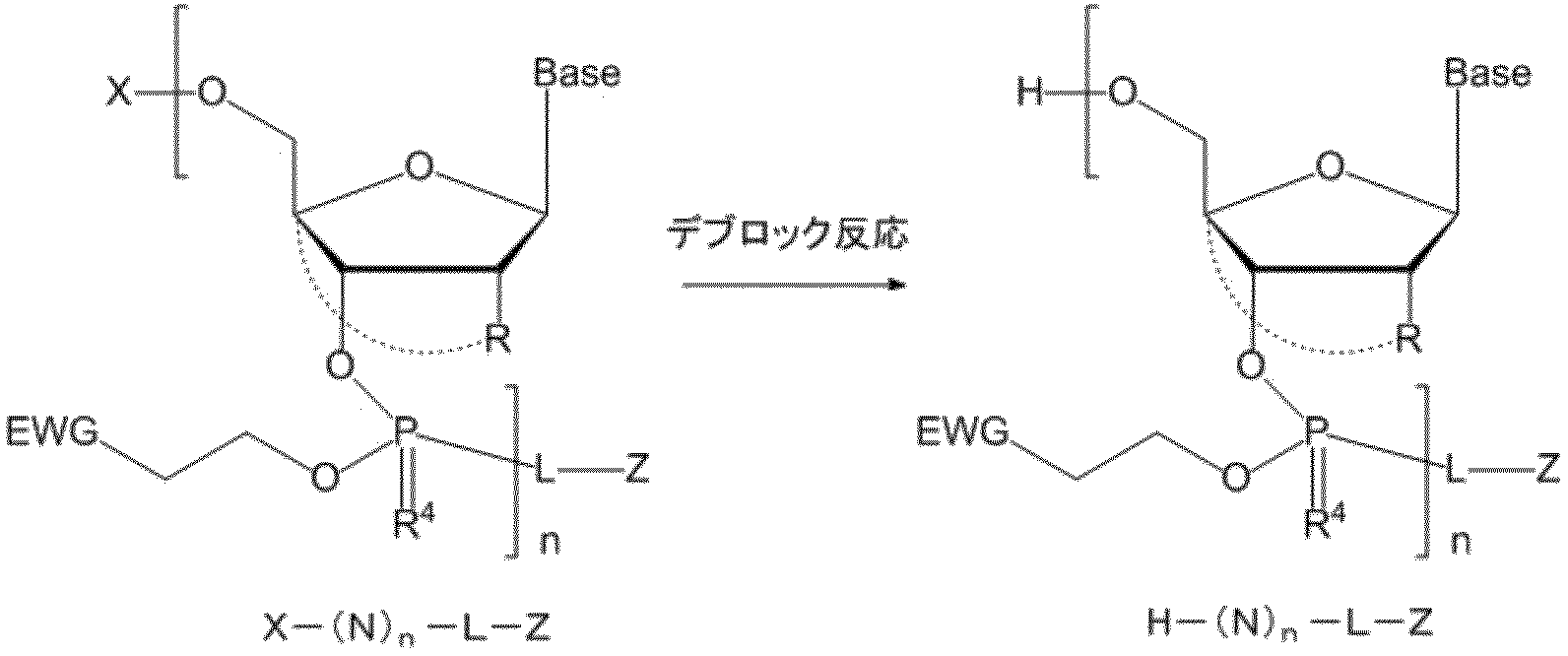
工程(a)において、
非極性溶媒中において、3’位水酸基もしくは核酸塩基が擬似固相保護基で修飾され且つ5’位水酸基が酸性条件下で除去可能な一時保護基でブロックされたn量体(ここで、nは、1以上の任意の整数を示す。)、もしくは、n=0の場合、水酸基を2個以上持つリンカーの1つの水酸基が擬似固相保護基で修飾され且つ別の水酸基が酸性条件下で除去可能な一時保護基でブロックされた化合物、例えば上記X-(N)n-L-Zで示される化合物を、酸の単独もしくはカチオン捕捉剤の共存下で反応させて、水酸基の保護基をデブロックする工程(デブロック工程)、
その後に極性溶媒を用いて該擬似固相保護基を含有する化合物、例えば上記H-(N)n-L-Zで示される化合物を回収する工程(晶析工程)、
を含む。
工程(a)の反応スキーム1を下記に示す。
反応スキーム1
非極性溶媒中において、3’位水酸基もしくは核酸塩基が擬似固相保護基で修飾され且つ5’位水酸基が酸性条件下で除去可能な一時保護基でブロックされたn量体(ここで、nは、1以上の任意の整数を示す。)、もしくは、n=0の場合、水酸基を2個以上持つリンカーの1つの水酸基が擬似固相保護基で修飾され且つ別の水酸基が酸性条件下で除去可能な一時保護基でブロックされた化合物、例えば上記X-(N)n-L-Zで示される化合物を、酸の単独もしくはカチオン捕捉剤の共存下で反応させて、水酸基の保護基をデブロックする工程(デブロック工程)、
その後に極性溶媒を用いて該擬似固相保護基を含有する化合物、例えば上記H-(N)n-L-Zで示される化合物を回収する工程(晶析工程)、
を含む。
工程(a)におけるデブロック工程反応は、文献(例えば、WO2012/157723A1)中に記載の反応条件下で行うことができる。具体的に以下説明する。
本明細書中「擬似固相保護基」とは、文献(例えば、WO2012/157723A1)に記載の基を含み得る。例えば、本明細書中に後述する擬似固相保護基を挙げられるが、これらに限定されるものではない。
5’位水酸基の酸性条件下で除去可能な一時保護基としては、酸性条件下で脱保護可能であり、水酸基の保護基として用いられる基であれば、特に限定はされないが、例えばトリチル基、9-(9-フェニル)キサンテニル基、9-フェニルチオキサンテニル基、1,1-ビス(4-メトキシフェニル)-1-フェニルメチル基(ジメトキシトリチル基と略す)、1-(4-メトキシフェニル)-1,1-ジフェニルメチル基(モノメトキシトリチル基と略す)等を挙げることができる。ジメトキシトリチル基およびモノメトキシトリチル基が好ましい。
デブロック工程に用いられる反応溶媒は、デブロック反応に影響を及ぼさない溶媒であればよいが、反応基質である(オリゴ)ヌクレオチド化合物の溶解度が高い溶媒が好ましく、例えば非極性溶媒が好ましい。非極性溶媒の具体例としては、ハロゲン系溶媒、芳香族系溶媒、エステル系溶媒、脂肪族系溶媒、非極性エーテル系溶媒、及びこれらの組み合わせからなる群より選択される溶媒が挙げられる。具体例としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタン、ベンゼン、トルエン、キシレン、メシチレン、ヘキサン、ペンタン、ヘプタン、ノナン、シクロヘキサン、酢酸エチル、酢酸イソプロピル、tert-ブチルメチルエーテル、シクロペンチルメチルエーテル、及びこれらの組み合わせからなる群より選択される溶媒が挙げられる。
本デブロック工程におけるn量体オリゴヌクレオチドの溶媒中の濃度は、溶解していれば特に限定されるものではないが、好ましくは1~30重量%である。
本デブロック工程に使用することができる酸の具体例としては、特に限定されないが、例えばトリフルオロ酢酸、トリクロロ酢酸、トリフルオロメタンスルホン酸、トリクロロ酢酸、メタンスルホン酸、塩酸、酢酸、p-トルエンスルホン酸等を挙げられる。トリフルオロ酢酸、ジクロロ酢酸、トリクロロ酢酸が好ましい。
酸の使用量は、n量体(オリゴ)ヌクレオチド1モルに対して、1~100モル使用することができ、好ましくは1~40モルが挙げられる。
酸の使用量は、n量体(オリゴ)ヌクレオチド1モルに対して、1~100モル使用することができ、好ましくは1~40モルが挙げられる。
酸は単独で使用することができるが、あるいはカチオン捕捉剤との共存下で使用してもよい。カチオン捕捉剤の例としては、除去された保護基Xによる再保護や脱保護された官能基への副反応が進行しなければ特に限定されないが、ピロール誘導体(例えば、ピロール)、およびインドール誘導体(例えば、インドール)、フラン誘導体を挙げることができる。カチオン捕捉剤の使用量は、n量体オリゴヌクレオチド1モルに対し、1~50モルを使用することができ、好ましくは5~20モルが挙げられる。
デブロック工程の反応温度は、反応が進行すれば特に限定されるものではないが、例えば-10~50℃が好ましく、0~40℃がより好ましい。反応時間は、反応条件、例えば、反応基質としてのn量体(オリゴ)ヌクレオチドの種類、酸の種類、溶媒の種類、反応温度等により変わり得るが、例えば5分~5時間が挙げられる。
工程(a)においては、デブロック工程後に、中和若しくはそのまま極性溶媒を用いて該擬似固相保護基を含有する化合物を晶析して化合物を回収する工程を含む。
極性溶媒の例としては、アルコール系溶媒およびニトリル系溶媒が挙げられ、具体例としてはメタノールまたはアセトニトリルが挙げられる。
(工程(b):カップリング工程)
工程(b)の反応スキーム2を下記に示す。
反応スキーム2
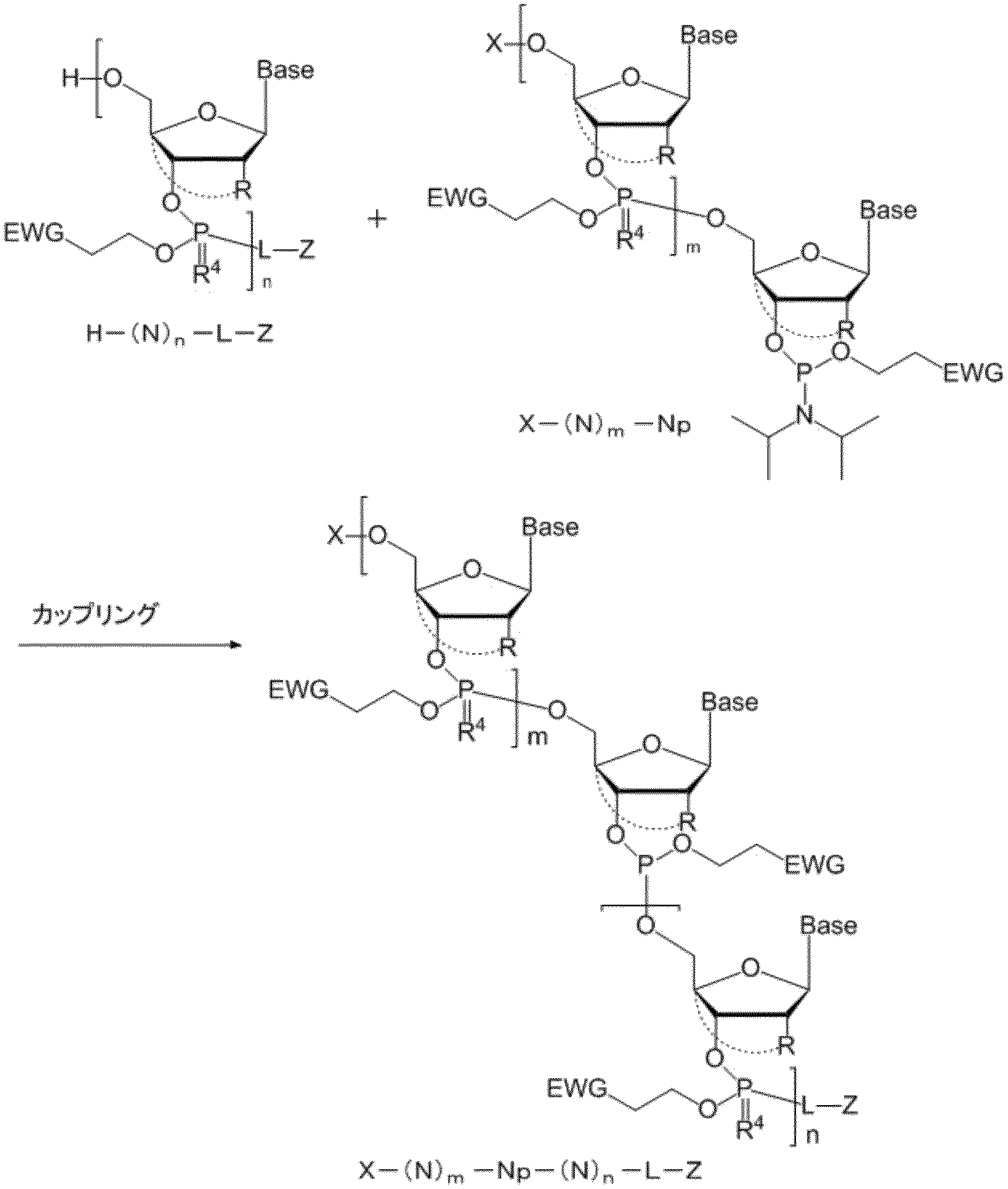
工程(b)の反応スキーム2を下記に示す。
反応スキーム2
工程(b)におけるカップリング工程反応は、文献(例えば、WO2012/157723A1)中に記載の反応条件下で行うことができる。具体的に以下説明する。
工程(b)において、
工程(a)の回収物を乾燥後、非極性溶媒中において、3’位水酸基がホスホラミダイト化され且つ5’位水酸基が酸性条件下で除去可能な一時保護基で保護されたm+1個重合(オリゴ)ヌクレオチド(ここで、mは、0以上の任意の整数を示す。)およびアクチベータを添加して、工程(a)で得られた5’位水酸基の保護基がデブロックされたn個重合(オリゴ)ヌクレオチドと、その5’位水酸基を介して亜リン酸トリエステル結合により縮合させる工程(カップリング工程)
を含む。
工程(a)の回収物を乾燥後、非極性溶媒中において、3’位水酸基がホスホラミダイト化され且つ5’位水酸基が酸性条件下で除去可能な一時保護基で保護されたm+1個重合(オリゴ)ヌクレオチド(ここで、mは、0以上の任意の整数を示す。)およびアクチベータを添加して、工程(a)で得られた5’位水酸基の保護基がデブロックされたn個重合(オリゴ)ヌクレオチドと、その5’位水酸基を介して亜リン酸トリエステル結合により縮合させる工程(カップリング工程)
を含む。
mの上限値は、特に限定されるものではないが、例えば49以下が好ましく、29以下がより好ましく、19以下が更に好ましく、4以下が更に一層好ましく、2以下(例えば、0)が特に好ましい。
「3’位水酸基がホスホラミダイト化され且つ5’位水酸基が酸性条件下で除去可能な一時保護基で保護されたm+1個重合(オリゴ)ヌクレオチド」とは、工程(a)で得られた5’位水酸基の保護基がデブロックされたn個重合(オリゴ)ヌクレオチドとカップリングさせる、(オリゴ)ヌクレオチド(例えば、X-(N)n-L-Zで示される化合物)を意味する。
本カップリング工程においては、アクチベータとして弱酸もしくは酸性中和塩を添加して反応効率を向上させることもできる。弱酸もしくは酸性中和塩の例としては、ピリジン・トリフルオロ酢酸塩、1H-テトラゾール、5-ベンジルチオ-1H-テトラゾール、4,5-ジシアノイミダゾール等が挙げられる。
弱酸もしくは酸性中和塩の使用量は、反応基質としての、工程(a)で得られた5’位水酸基の保護基がデブロックされたn個重合(オリゴ)ヌクレオチドの1モルに対し、0.1~50モル当量使用することができ、例えば1~5モル当量が挙げられる。
弱酸もしくは酸性中和塩の使用量は、反応基質としての、工程(a)で得られた5’位水酸基の保護基がデブロックされたn個重合(オリゴ)ヌクレオチドの1モルに対し、0.1~50モル当量使用することができ、例えば1~5モル当量が挙げられる。
また、本カップリング工程においては、反応液の酸性度が高くなると、一時保護基Xが脱離する副反応が生じるおそれがあるので、反応液の酸性化を抑制する目的でN-メチルイミダゾールを添加することが好ましい。
該N-メチルイミダゾールの使用量は、工程(a)で得られた5’位水酸基の保護基がデブロックされたn個重合(オリゴ)ヌクレオチドの1モルに対し、0.1~10モル当量使用することができ、例えば0.1~1モル当量が挙げられる。
該N-メチルイミダゾールの使用量は、工程(a)で得られた5’位水酸基の保護基がデブロックされたn個重合(オリゴ)ヌクレオチドの1モルに対し、0.1~10モル当量使用することができ、例えば0.1~1モル当量が挙げられる。
「3’位水酸基がホスホラミダイト化され且つ5’位水酸基が酸性条件下で除去可能な一時保護基で保護されたm+1個重合(オリゴ)ヌクレオチド」と、「工程(a)で得られた5’位水酸基の保護基がデブロックされたn個重合(オリゴ)ヌクレオチド」との配合量比は、工程(a)で得られた5’位水酸基の保護基がデブロックされたn個重合(オリゴ)ヌクレオチドの1モルに対して、該3’位水酸基がホスホラミダイト化され且つ5’位水酸基が酸性条件下で除去可能な一時保護基で保護されたm+1個重合(オリゴ)ヌクレオチドを1~10モル使用することができ、好ましくは1~5モル当量である。
本工程は、カップリング反応に影響を及ぼさない溶媒中で行われる。具体的には、前記工程(a)と同様の非極性溶媒が挙げられる。また、上記非極性溶媒に、ニトリル系溶媒(例えば、アセトニトリル)、ケトン系溶媒(例えば、アセトン)、アミド系溶媒(例えば、N,N-ジメチルアセトアミド)、極性エーテル系溶媒(例えば、1,4-ジオキサン)、およびスルホキシド系溶媒(例えば、ジメチルスルホキシド)を、5’位水酸基の一時保護基が除去されたn量体(オリゴ)ヌクレオチドが溶解し得る限り、適宜の割合で混合して用いてもよい。
工程(b)の反応温度は、反応が進行すれば特に限定されないが、例えば-20~100℃が好ましく、20~50℃がより好ましい。反応時間は、縮合させるn量体(オリゴ)ヌクレオチドの種類、反応温度等によって変わり得るが、例えば5分~24時間を挙げられる。
(工程(c)および工程(d))
本発明の1態様によれば、工程(b)において未反応であった原料を、本発明の高脂溶性ホスホラミダイト化合物の製造法によって得られた高脂溶性ホスホラミダイト化合物と接触反応させて、キャッピング反応を行う工程を提供する。
また、本発明の1実施態様によれば、工程(c)のキャッピング反応後に、キャッピング副反応産物を求核剤と反応させる工程である、後処理工程を含んでもよい。
本発明の1態様によれば、工程(b)において未反応であった原料を、本発明の高脂溶性ホスホラミダイト化合物の製造法によって得られた高脂溶性ホスホラミダイト化合物と接触反応させて、キャッピング反応を行う工程を提供する。
また、本発明の1実施態様によれば、工程(c)のキャッピング反応後に、キャッピング副反応産物を求核剤と反応させる工程である、後処理工程を含んでもよい。
工程(c)および工程(d)の詳細については、前記の本発明のキャッピング反応について説明した通りである。
ここで、工程(c)における未反応原料としては例えば、工程(a)で得られた5’位水酸基がデブロックされたn量体(オリゴ)ヌクレオチド(例えば、反応スキーム1に示したHO-(N)n-L-Zで示される化合物)が挙げられる。
ここで、工程(c)における未反応原料としては例えば、工程(a)で得られた5’位水酸基がデブロックされたn量体(オリゴ)ヌクレオチド(例えば、反応スキーム1に示したHO-(N)n-L-Zで示される化合物)が挙げられる。
また、工程(d)における工程(c)のキャッピング副反応産物としては例えば、報告例は無いが、Base上にキャッピング試薬が付加した反応産物(例えば、グアニンの6位酸素アルコキシホスファイトトリエステル体)を挙げることができる。また、かかるBaseの骨格上にキャッピング試薬が付加した反応産物(例えば、前記グアニンの6位酸素アルコキシホスファイトトリエステル体)の場合は、求核剤と反応させることにより、キャッピング試薬の付加分子が除去されたグアニンを再生することができると期待される。
ここで、本発明における求核剤との反応とは、キャッピング副反応産物の処理を意図する反応である。従って、例えば文献(例えば、WO2017/104836A1)に記載されているようなカップリング反応工程後の反応液をクエンチすることを意図するものではない。
ここで、本発明における求核剤との反応とは、キャッピング副反応産物の処理を意図する反応である。従って、例えば文献(例えば、WO2017/104836A1)に記載されているようなカップリング反応工程後の反応液をクエンチすることを意図するものではない。
(工程(e):酸化反応および硫化反応)
工程(e)の反応スキーム3を下記に示す。
反応スキーム3
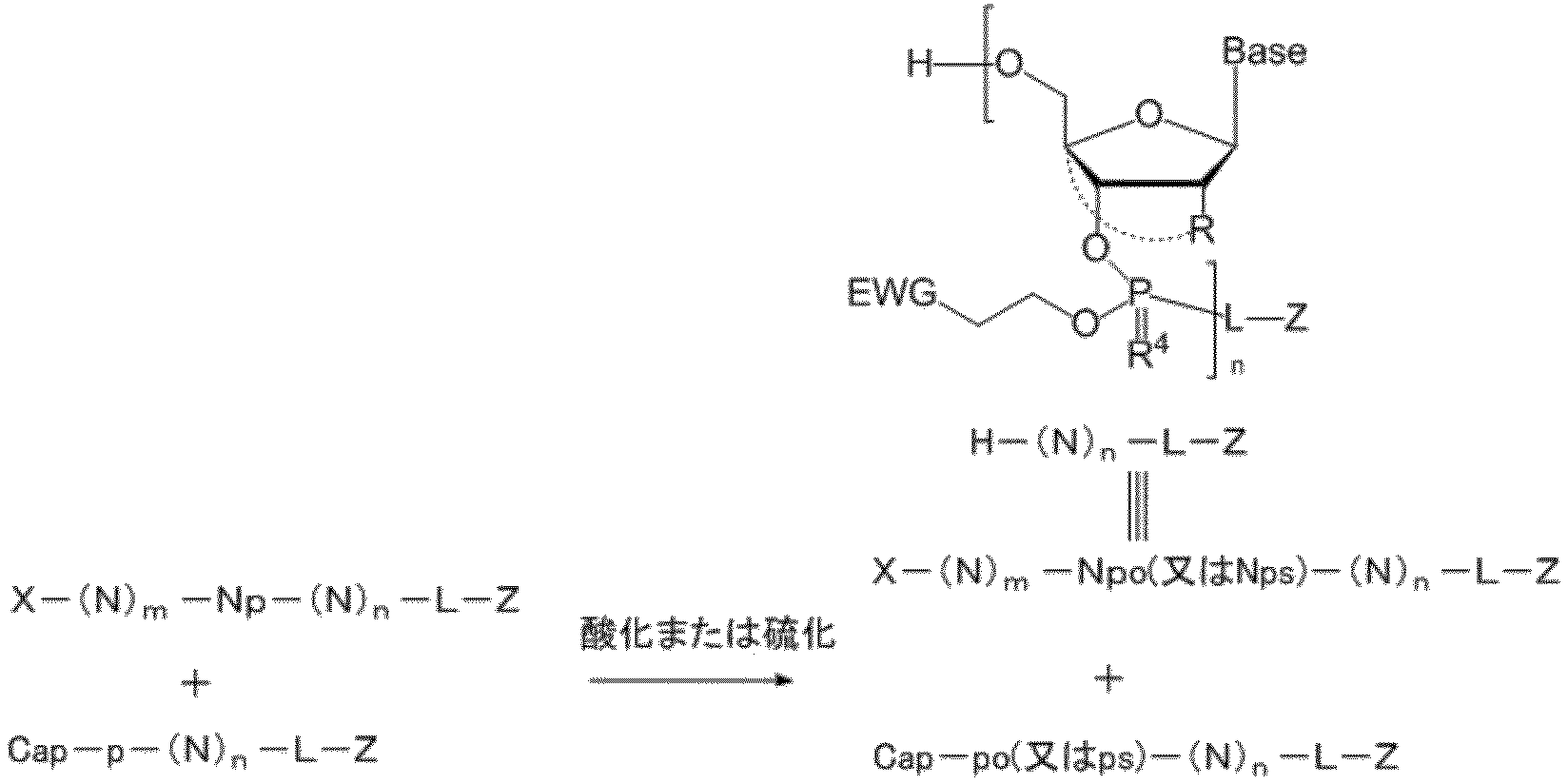
工程(e)の反応スキーム3を下記に示す。
反応スキーム3
工程(e)における酸化反応または硫化反応は、文献(例えば、WO2012/157723A1)中に記載の反応条件下で行うことができる。具体的に以下説明する。
工程(e)において、
工程(d)の反応液に、酸化剤または硫化剤を添加して、工程(d)で得られたm+1+n量体のオリゴヌクレオチドのホスファイトトリエステル結合を、ホスフェートトリエステル結合又はチオホスフェートトリエステル結合にそれぞれ変換する工程(酸化または硫化反応の工程)
を提供する。
工程(d)の反応液に、酸化剤または硫化剤を添加して、工程(d)で得られたm+1+n量体のオリゴヌクレオチドのホスファイトトリエステル結合を、ホスフェートトリエステル結合又はチオホスフェートトリエステル結合にそれぞれ変換する工程(酸化または硫化反応の工程)
を提供する。
本工程は、前記工程(d)で得られたm+1+n量体のオリゴヌクレオチドを単離することなく、該工程(d)後の反応液に、酸化剤または硫化剤を直接に添加するだけで反応を行うこともできる。
工程(e)における酸化剤を用いる酸化反応は、通常オリゴヌクレオチド合成において知られる方法に準じて行うことができる。酸化剤の例としては、特に限定されるものではないが、例えば、ヨウ素、(1S)-(+)-(10-カンファスルホニル)オキサゾリジン、tert-ブチルヒドロペルオキシド(TBHP)、2-ブタノンペルオキシド、1,1-ジヒドロペルオキシシクロドデカン、ビス(トリメチルシリル)ペルオキシド、m-クロロ過安息香酸を挙げられ、ヨウ素、(1S)-(+)-(10-カンファスルホニル)オキサゾリジン、tert-ブチルヒドロペルオキシド(TBHP)、2-ブタノンペルオキシド、1,1-ジヒドロペルオキシシクロドデカンが好ましい。
工程(e)における硫化剤を用いる硫化反応は、通常オリゴヌクレオチド合成において知られる方法に準じて行うことができる。硫化剤の例としては、特に限定されるものではないが、例えば、3-((N,N-ジメチルアミノメチリデン)アミノ)-3H-1,2,4-ジチアゾール-5-チオン(DDTT)、3H-1,2-ベンゾジチアゾール-3-オン-1,1-ジオキシド(Beaucage試薬)、3H-1,2-ベンゾチオール-3-オン、フェニルアセチルジスルフィド(PADS)、テトラエチルチウラムジスルフィド(TETD)、3-アミノ-1,2,4-ジチアゾール-5-チオン(ADTT)、硫黄を挙げられ、3-((N,N-ジメチルアミノメチリデン)アミノ)-3H-1,2,4-ジチアゾール-5-チオン、3H-1,2-ベンゾジチアゾール-3-オン-1,1-ジオキシドが好ましい。
オリゴヌクレオチドのホスファイトトリエステル結合の硫化反応により、チオホスフェートトリエステル結合に変換する。チオホスフェートはリン原子上で不斉中心を有するため、光学活性体を与える。また、オリゴヌクレオチドチオホスフェートの製造工程において脱保護反応を行っても、リン原子上の不斉中心は残存し、光学活性体を与える。そのため、構成するヌクレオチドの各リン原子上に不斉中心が存在するため、オリゴヌクレオチドチオホスフェート生成物は複数の光学異性体の混合物となり、目的物の精製が困難である為、より純度の良い合成が求められる。
反応は、通常溶媒中で行われる。反応に用いられる溶媒としては、無水溶媒が好ましく、例えば、ハロゲン系溶媒(例えば、クロロホルム)、脂肪族系溶媒(例えば、シクロヘキサン)、芳香族系溶媒(例えば、トルエン)、エステル系溶媒(例えば、酢酸エチル)、エーテル系溶媒(例えば、tert-ブチルメチルエーテル)、ニトリル系溶媒(例えば、アセトニトリル)、およびこれらから選ばれる2つ以上の混合溶媒を挙げられる。
酸化剤または硫化剤は、工程(d)で得られたホスホラミダイトの1モルに対して、約1.0~約5.0モル、好ましくは約1.0~約1.5モルの割合である。
反応温度は、通常0~50℃である。反応時間は、通常0.2~1時間の範囲内である。
反応温度は、通常0~50℃である。反応時間は、通常0.2~1時間の範囲内である。
酸化剤または硫化剤の使用量は、例えば工程(d)で得た(オリゴ)ヌクレオチドの1モルに対し、1~50モル当量、好ましくは1~5モル当量を使用することができる。
反応温度は、反応が進行すれば特に限定されるものではないが、0~100℃、好ましくは20~50℃が挙げられる。反応時間は、n+1+m量体のオリゴヌクレオチドの種類、使用する酸化剤または硫化剤の種類、および反応温度等によって変わり得て、例えば1分~3時間が挙げられる。
(工程f:沈殿化および固液分離工程)
工程(f)における沈殿化および固液分離操作は、文献(例えば、WO2012/157723A1)中に記載の反応条件下で行うことができる。具体的に以下説明する。
工程(f)における沈殿化および固液分離操作は、文献(例えば、WO2012/157723A1)中に記載の反応条件下で行うことができる。具体的に以下説明する。
本発明の1実施態様によれば、本発明のオリゴヌクレオチドの製造方法は、更に、
適宜、工程(e)で得られた反応液に極性溶媒を添加して、m+1+n量体オリゴヌクレオチドを沈殿させて、固液分離により取得する工程(沈殿化および固液分離工程)を含み得る。
適宜、工程(e)で得られた反応液に極性溶媒を添加して、m+1+n量体オリゴヌクレオチドを沈殿させて、固液分離により取得する工程(沈殿化および固液分離工程)を含み得る。
本発明のオリゴヌクレオチドの製造方法は、擬似固相保護基を使用するために、生成物m+1+n量体オリゴヌクレオチドを得るために、該生成物の沈殿化および固液分離からなる単離工程を要する。
極性溶媒の例としては、アルコール系溶媒(例えば、メタノール、エタノール)、ニトリル系溶媒(例えば、アセトニトリル)、ケトン系溶媒(例えば、アセトン)、エーテル系溶媒(例えば、テトラヒドロフラン)、アミド系溶媒(例えば、ジメチルホルムアミド)、スルホキシド系溶媒(例えば、ジメチルスルホキシド)、水等、およびこれらの2種以上の混合溶媒が挙げられる。アルコール系溶媒、ニトリル系溶媒が好ましく、具体的にはメタノールが好ましい。
また、該極性溶媒は、生成物の極性溶媒中へのロスを最小限とするために、水を含んでいてもよい。極性溶媒に対する水の含量は、例えば0~10%(v/v)、好ましくは0~8%(v/v)が挙げられる。
また、該極性溶媒は、生成物の極性溶媒中へのロスを最小限とするために、水を含んでいてもよい。極性溶媒に対する水の含量は、例えば0~10%(v/v)、好ましくは0~8%(v/v)が挙げられる。
また、工程(e)において酸化もしくは硫化終了後、三価のリン試薬(例えば、トリメチルホスファイト)と反応させることにより、過剰の酸化剤もしくは硫化剤を取り除いたのちに沈殿化溶媒を加えるか、あるいは、沈殿化の際には沈殿化溶媒(例えば、メタノール)中にチオ硫酸ナトリウム(ハイポ)を飽和させた溶液を使用してもよい。投入されるチオ硫酸ナトリウム(ハイポ)量は、沈殿化溶媒の使用量、反応温度に依存するため、三価のリン試薬との反応がより好ましい。
(工程g:脱保護工程)
工程(f)における沈殿化および固液分離操作は、文献(例えば、WO2012/157723A1)中に記載の反応条件下で行うことができる。具体的に以下説明する。
工程(f)における沈殿化および固液分離操作は、文献(例えば、WO2012/157723A1)中に記載の反応条件下で行うことができる。具体的に以下説明する。
本発明の1実施態様によれば、本発明のオリゴヌクレオチドの製造方法は、更に適宜、各工程の後に、保護基の種類および性質に応じて各保護基の脱保護を行って、オリゴヌクレオチドを単離生成することができる。
本発明の1実施態様によれば、本発明のオリゴヌクレオチドの製造方法は、更に
適宜、工程(g):工程(f)で得られたm+1+n個重合オリゴヌクレオチドの保護基を全て除去する工程を含み得る。
本発明の1実施態様によれば、本発明のオリゴヌクレオチドの製造方法は、更に
適宜、工程(g):工程(f)で得られたm+1+n個重合オリゴヌクレオチドの保護基を全て除去する工程を含み得る。
脱保護の方法としては、例えば、Protective Group In Organic Synthesis、第3版、John Willy & Sons出版(1999年)等の記載されている脱保護方法に従って、オリゴヌクレオチドの全ての保護基を除去する工程を行うことができる。
(反応の進行の確認)
上記オリゴヌクレオチド製造方法の各工程の反応の進行は、一般的な液相有機合成反応の場合と同様の手法により、追跡・確認することができる。例えば、薄層シリカゲルクロマトグラフィー(TLC)および高速液体クロマトグラフィー(HPLC)等によって行うことができる。
上記オリゴヌクレオチド製造方法の各工程の反応の進行は、一般的な液相有機合成反応の場合と同様の手法により、追跡・確認することができる。例えば、薄層シリカゲルクロマトグラフィー(TLC)および高速液体クロマトグラフィー(HPLC)等によって行うことができる。
(擬似固相保護基およびそれを含有するヌクレオチド化合物)
本発明の1態様によれば、本発明のオリゴヌクレオチド製造方法は、擬似固相保護基を有するヌクレオチドを用いることにより、液相方法で行うことができる。
本発明の1態様によれば、本発明のオリゴヌクレオチド製造方法は、擬似固相保護基を有するヌクレオチドを用いることにより、液相方法で行うことができる。
該「擬似固相保護基」とは、該基が反応基質に結合することにより非極性溶媒に可溶化し、液相中での反応が可能となると共に、極性溶媒の添加により該擬似固相保護基を含む生成物が沈殿し、固液分離が可能となる、反応性と後処理の簡便性を兼ね備えた保護基であって、5’末端水酸基の保護基を除去し得る酸性条件で安定な基でありさえすれば、特に限定されない。擬似固相保護基としては、文献(例えば、WO2012/157723A1)に開示された基が挙げられるが、本発明のオリゴヌクレオチドの製造方法において使用される擬似固相保護基の好ましい例としては、下記式(II)で示されるN-アルキル、アルキルアルコール化合物の残基が挙げられる。
本発明の1態様によれば、本発明は、
式(II):
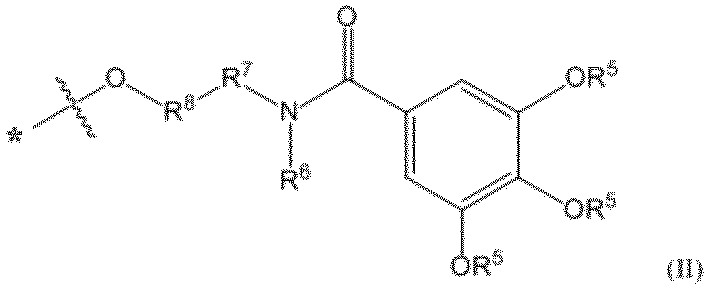
[式中、
R5は各々独立して、置換されていてもよいC6-C30アルキル基であり、
R6は、置換されていてもよいC1-C6アルキル基、置換されていてもよいC3-C6シクロアルキル基、置換されていてもよい非芳香族性複素環式基、置換されていてもよいアリール基、置換されていてもよいヘテロアリール基、置換されていてもよいアラルキル基、または置換されていてもよいヘテロアリールアルキル基から選ばれ、
R7は、置換されていてもよいC1-C6アルキレン基または置換されていてもよいC1-C6アルキレンオキシ基であり、
R8は、置換されていてもよいC1-C6アルキレン基であり、あるいは、
R6およびR7は、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、または置換されていてもよいヘテロアリール基を形成してもよく、あるいは、
R6、R7およびR8は、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、置換されていてもよい非芳香族性複素環アルキル基、置換されていてもよいヘテロアリール基、または置換されていてもよいヘテロアリールアルキル基を形成してもよく、そして、
*は、Lとの結合位置を示す]
で示される擬似固相保護基、
を提供する。
式(II):
R5は各々独立して、置換されていてもよいC6-C30アルキル基であり、
R6は、置換されていてもよいC1-C6アルキル基、置換されていてもよいC3-C6シクロアルキル基、置換されていてもよい非芳香族性複素環式基、置換されていてもよいアリール基、置換されていてもよいヘテロアリール基、置換されていてもよいアラルキル基、または置換されていてもよいヘテロアリールアルキル基から選ばれ、
R7は、置換されていてもよいC1-C6アルキレン基または置換されていてもよいC1-C6アルキレンオキシ基であり、
R8は、置換されていてもよいC1-C6アルキレン基であり、あるいは、
R6およびR7は、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、または置換されていてもよいヘテロアリール基を形成してもよく、あるいは、
R6、R7およびR8は、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、置換されていてもよい非芳香族性複素環アルキル基、置換されていてもよいヘテロアリール基、または置換されていてもよいヘテロアリールアルキル基を形成してもよく、そして、
*は、Lとの結合位置を示す]
で示される擬似固相保護基、
を提供する。
本発明の1実施態様によれば、好ましい擬似固相保護基の例として、
式(II)中、
各R5が独立して、置換されていてもよいC8-C24のアルキル基であり、
R6は、置換されていてもよいC1-C3アルキル基、置換されていてもよいC5-C6シクロアルキル基、置換されていてもよいアリール基、置換されていてもよいヘテロアリール基、または置換されていてもよいアラルキル基から選ばれ、
R7が、置換されていてもよいC1-C4アルキレン基であり、
R8が、置換されていてもよいC1-C2アルキレン基であり、あるいは、
R6およびR7が、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、または置換されていてもよいヘテロアリール基を形成してもよく、あるいは、
R6、R7およびR8が、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、または置換されていてもよい非芳香族性複素環アルキル基を形成してもよい、
擬似固相保護基が挙げられる。
式(II)中、
各R5が独立して、置換されていてもよいC8-C24のアルキル基であり、
R6は、置換されていてもよいC1-C3アルキル基、置換されていてもよいC5-C6シクロアルキル基、置換されていてもよいアリール基、置換されていてもよいヘテロアリール基、または置換されていてもよいアラルキル基から選ばれ、
R7が、置換されていてもよいC1-C4アルキレン基であり、
R8が、置換されていてもよいC1-C2アルキレン基であり、あるいは、
R6およびR7が、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、または置換されていてもよいヘテロアリール基を形成してもよく、あるいは、
R6、R7およびR8が、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、または置換されていてもよい非芳香族性複素環アルキル基を形成してもよい、
擬似固相保護基が挙げられる。
本明細書中、R5についての、用語「置換されていてもよいC6-C30アルキル基」とは、適宜、置換基で置換されていてもよい、炭素数が6~30の第1級または第2級の飽和炭化水素鎖基、好ましくは第1級の飽和炭化水素鎖基を意味する。該アルキル基は、直鎖または分枝鎖状のいずれでもよいが、直鎖状のアルキル基が好ましい。具体的には、C8-C30の第1級アルキル基が好ましく、C12-C22の第1級のアルキル基がより好ましく、C16-C20の第1級のアルキル基がより好ましい。好適な1具体例としては、C18の第1級のアルキル基が挙げられる。置換基としては、本発明で使用する反応(例えば、キャッピング反応、およびオリゴヌクレオチド合成上のカップリング反応、脱保護反応、デブロック反応、酸化反応、硫化反応)を阻害しない限り置換基の種類は限定されるものではないが、例えばC1-C6アルキル基、およびC3-C6シクロアルキル基から選ばれる1つまたはそれ以上の基を挙げられ、C1-C6アルキル基が好ましい。置換基の具体例としては、メチル、エチル、イソプロピル、シクロプロピル、シクロヘキシル等が挙げられる。
本明細書中、R6についての、用語「置換されていてもよいC1-C6アルキル基」とは、炭素数が1~6の飽和炭化水素鎖基を意味する。該アルキル基は、直鎖または分枝鎖状のいずれでもよい。置換されていてもよいC1-C6アルキル基の例としては、C3アルキル基が好ましい。好適な1具体例としては、メチルが挙げられる。置換基としては、本発明で使用する反応(例えば、キャッピング反応、およびオリゴヌクレオチド合成上のカップリング反応、脱保護反応、デブロック反応、酸化反応、硫化反応)を阻害しない限り置換基の種類は限定されるものではないが、例えばC1-C3アルキル基およびC3-C6シクロアルキル基から選ばれる1つまたはそれ以上の基を挙げられる。
本明細書中、R6についての、用語「置換されていてもよいC3-C6シクロアルキル基」とは、炭素数が3~6の脂環式飽和炭化水素基を意味する。置換基としては、本発明で使用する反応(例えば、キャッピング反応、およびオリゴヌクレオチド合成上のカップリング反応、脱保護反応、デブロック反応、酸化反応、硫化反応)を阻害しない限り置換基の種類は限定されるものではないが、例えばC1-C3アルキル基を挙げられる。シクロアルキル基の具体例としては、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシルを挙げることができる。
本明細書中、R7およびR8についての、用語「置換されていてもよいC1-C6アルキレン基」とは、炭素数が1~6の飽和炭化水素鎖の二価基を意味する。該アルキレン基は、直鎖または分枝鎖状のいずれでもよい。置換されていてもよいC1-C6アルキレン基の例としては、C1-C4アルキレン基が好ましく、C1-C3アルキレン基がより好ましい。具体的な例としては、メチレン、エチレン、トリメチレン(プロピレン)、テトラメチレン(n-ブチレン)等が挙げられる。置換基としては、本発明で使用する反応(例えば、キャッピング反応、およびオリゴヌクレオチド合成上のカップリング反応、脱保護反応、デブロック反応、酸化反応、硫化反応)を阻害しない限り置換基の種類は限定されるものではないが、例えばC1-C3アルキル基、C3-C6シクロアルキル基、C6-C10アリール基から選ばれる1つまたはそれ以上の基を挙げられる。置換基の例としては、メチル、エチル、イソプロピル、フェニル等が挙げられる。
本明細書中、R7についての、用語「置換されていてもよいC1-C6アルキレンオキシ基」とは、酸素原子に結合した、上記の「置換されていてもよいC1-C6アルキレン基」を意味する。具体的な例としては、メチレンオキシ、エチレンオキシ、トリメチレンオキシ(プロピレンオキシ)等が挙げられる。
本明細書中、R6、R7およびR8についての、用語「置換されていてもよい非芳香族性複素環式基」とは、硫黄原子、酸素原子、及び窒素原子(ここで、酸素原子または窒素原子が好ましく、窒素原子がより好ましい)からなる群より独立して選ばれる1~4個(ここで、1~2個が好ましく、1個がより好ましい)の異項原子を含む4~8員の単環式または二環式の非芳香族性複素環式基を意味する。該置換されていてもよい非芳香族性複素環基は、シクロアルキル環、芳香族性炭化水素環、または芳香族性複素環と縮合していてもよい。置換されていてもよい非芳香族性複素環式基の具体的な例としては、アゼチジニル、ピロリジニル、テトラヒドロフラニル、イミダゾリジニル、チアゾリジニル、イソチアゾリジニル、ピペリジニル、ピペラジニル、モルホリニル、チオモルホリニル、テトラヒドロピラニル、ホモピペラジニル、ホモモルホリニル、3-アザビシクロ[3.1.0]ヘキシル、3-アザビシクロ[3.2.1]オクチルなどが挙げられ、好ましくは、ピペリジニル、ピペラジニル、3-アザビシクロ[3.2.1]オクチル等が挙げられる。置換基としては、本発明で使用する反応(例えば、キャッピング反応、およびオリゴヌクレオチド合成上のカップリング反応、脱保護反応、デブロック反応、酸化反応、硫化反応)を阻害しない限り置換基の種類は限定されるものではないが、例えばC1-C3アルキル基、およびC3-C6シクロアルキル基から選ばれる1つまたはそれ以上の基を挙げられる。置換基の例としては、メチル、エチル、イソプロピル、シクロプロピル等が挙げられる。
本明細書中、R6、R7およびR8についての、用語「置換されていてもよいアリール基(または、置換されていてもよい芳香族炭化水素環式基)」とは、6-10員の単環または二環式の芳香族炭化水素環式基を意味する。該置換されていてもよいアリール基は、シクロアルキル環、非芳香族性複素環、または芳香族性複素環と縮合していてもよい。置換されていてもよいアリール基の具体的な例としては、フェニル、インデニル、ナフチル、シクロヘキサノフェニル、ピロリジン縮合フェニル(インドリン)、ピペリジン縮合フェニル等が挙げられ、フェニルが好ましい。置換基としては、本発明で使用する反応(例えば、キャッピング反応、およびオリゴヌクレオチド合成上のカップリング反応、脱保護反応、デブロック反応、酸化反応、硫化反応)を阻害しない限り置換基の種類は限定されるものではないが、例えばC1-C3アルキル基、およびC3-C6シクロアルキル基から選ばれる1つまたはそれ以上の基を挙げられる。置換基の例としては、メチル、エチル、イソプロピル、シクロプロピル等が挙げられる。
本明細書中、R6、R7およびR8についての、用語「置換されていてもよいヘテロアリール基(または、置換されていてもよい芳香族性複素環式基)」とは、硫黄原子、酸素原子、及び窒素原子(ここで、酸素原子または窒素原子が好ましく、窒素原子がより好ましい)からなる群より独立して選ばれる1~4個(ここで、1~2個が好ましく、1個がより好ましい)の異項原子を含む5~10員の単環式または二環式の芳香族性複素環式基を意味する。該置換されていてもよいヘテロアリール基は、シクロアルキル環、非芳香族性複素環、または芳香族性炭化水素環と縮合していてもよい。置換されていてもよいヘテロアリール基の具体的な例としては、ピロリル、フラニル、チエニル、イミダゾリル、ピラゾリル、オキサゾリル、イソオキサゾリル、チアゾリル、トリアゾリル、テトラゾリル、オキサジアゾリル、ピリジニル、ピラジニル、ピリミジニル、ピリダジニル、チアジニル、トリアジニル、インドリル、キノリル、イソキノリル、イソインドリル、ベンゾイミダゾリルなどが挙げられ、好ましくはピリジニル、ピラジニル、ピリミジニル、イソキノリル、ベンゾイミダゾリル等が挙げられる。置換基としては、本発明で使用する反応(例えば、キャッピング反応、およびオリゴヌクレオチド合成上のカップリング反応、脱保護反応、デブロック反応、酸化反応、硫化反応)を阻害しない限り置換基の種類は限定されるものではないが、例えばC1-C3アルキル基、およびC3-C6シクロアルキル基から選ばれる1つまたはそれ以上の基を挙げられる。置換基の例としては、メチル、エチル、イソプロピル、シクロプロピル等が挙げられる。
本明細書中、R6、R7およびR8についての、用語「置換されていてもよいアラルキル基」とは、前記置換されていてもよいアリール基が前記置換されていてもよいアルキル基に結合した基を意味する。置換されていてもよいアラルキル基の具体的な例としては、ベンジル、フェネチル等が挙げられ、好ましくはベンジルが挙げられる。置換基としては、前記置換されていてもよいアルキル基および前記置換されていてもよいアリール基について記載する置換基を挙げられる。
本明細書中、R6、R7およびR8についての、用語「置換されていてもよいヘテロアリールアルキル基」とは、前記置換されていてもよいヘテロアリール基が前記置換されていてもよいアルキル基に結合した基を意味する。置換されていてもよいヘテロアリールアルキル基の具体的な例としては、ピリジルメチル、ピリジルエチル等が挙げられ、好ましくはピリジルメチルが挙げられる。置換基としては、前記置換されていてもよいアルキル基および前記置換されていてもよいヘテロアリール基について記載する置換基を挙げられる。
本明細書中、R6、R7およびR8についての、用語「置換されていてもよい非芳香族性複素環アルキル基」とは、前記置換されていてもよい非芳香族性複素環式基が前記置換されていてもよいアルキル基に結合した基を意味する。置換されていてもよい非芳香族性複素環アルキル基の具体的な例としては、ピペリジニルメチル、ピぺラジニルメチル、ピペリジニルエチル、ピぺラジニルエチルが挙げられ、好ましくはピペリジニルメチルが挙げられる。置換基としては、前記置換されていてもよい非芳香族性複素環式基および前記置換されていてもよいアルキル基について記載する置換基を挙げられる。
本発明の1実施態様によれば、上記式(II)中、可溶性部は、以下式(IV):
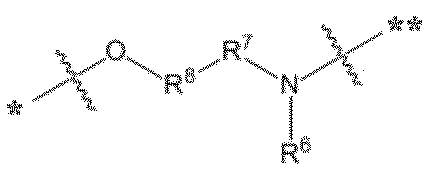
(式中、
*は、Lとの結合位置を示し;
**は、カルボニル基との結合位置を示し;
R6、R7およびR8は各々式(II)で定義する通りである)
で示される部分が、下記の構造式:

によって示される部分が挙げられる。
*は、Lとの結合位置を示し;
**は、カルボニル基との結合位置を示し;
R6、R7およびR8は各々式(II)で定義する通りである)
で示される部分が、下記の構造式:
本発明の1態様によれば、本発明は、上記擬似固相保護基を含むヌクレオチドを提供し、例えば下記式(III):

[式中、
Zは、上記式(II)で示される擬似固相保護基であり、
Lは、リンカーであり、
Baseは、各々独立して保護されていてもよい核酸塩基であり、
n個のEWGは、各々独立して電子吸引性基であり、
n個のR4は、各々独立して酸素原子または硫黄原子であり、
n個のRは、各々独立して水素原子、ハロゲン原子、保護基で置換されていてもよいヒドロキシ基、置換されていてもよいC1-C6アルコキシ基、4位炭素原子に架橋する有機基、または架橋構造型人工核酸基であり、
nは、0以上の任意の整数であり、そして、
Xは、酸により脱保護可能な一時保護基である]
によって示されるヌクレオチドを提供する。
Zは、上記式(II)で示される擬似固相保護基であり、
Lは、リンカーであり、
Baseは、各々独立して保護されていてもよい核酸塩基であり、
n個のEWGは、各々独立して電子吸引性基であり、
n個のR4は、各々独立して酸素原子または硫黄原子であり、
n個のRは、各々独立して水素原子、ハロゲン原子、保護基で置換されていてもよいヒドロキシ基、置換されていてもよいC1-C6アルコキシ基、4位炭素原子に架橋する有機基、または架橋構造型人工核酸基であり、
nは、0以上の任意の整数であり、そして、
Xは、酸により脱保護可能な一時保護基である]
によって示されるヌクレオチドを提供する。
本明細書中、Rとしての、用語「ハロゲン原子」とは、フッ素原子、塩素原子、臭素原子、およびヨウ素原子を意味するが、フッ素原子が好ましい。
本明細書中、Rとしての、用語「保護基で置換されていてもよいヒドロキシル基」における置換基とは、オリゴヌクレオチド合成において通常使用することが知られる保護基であればよいが、例えば、シリル型保護基(例えば、TOM基(-CH2OSi(iPr)3))が挙げられる。
本明細書中、Rとしての、用語「置換されていてもよいC1-C6アルコキシ基」における置換基としては、例えばC1-C6アルコキシ基が挙げられ、具体例としては、2-メトキシエトキシ基が挙げられる。
本明細書中、Rとしての、用語「4位炭素原子に架橋する有機基」とは、4’-C1-C6アルコキシ-2’である基を意味し、例えば、4’-CH2CH2O-2’が挙げられる。
本明細書中、用語「電子吸引性基(EWGと略す)」とは、有機化学上一般的に使用される、電子密度を減弱させる効果を持つ性質を有する置換基を意味する。具体例としては、例えばカルボキシ基、ハロゲン、ニトロ基、エステル基を含むが、これらに限定されるものではない。
本明細書中、用語「酸により脱保護可能な一時保護基」とは、本明細書中に「5’位水酸基の酸性条件下で除去可能な一時保護基」として記載する基を意味する。
本明細書中、式(III)中のLはリンカーであり、これは(オリゴ)ヌクレオチド分子と擬似固相保護基とを連結する二価基を意味する。該リンカーは、本発明のオリゴヌクレオチドの製造方法における各反応工程(例えば、キャッピング反応、およびオリゴヌクレオチド合成上のカップリング反応、脱保護反応、デブロック反応、酸化反応、硫化反応)を阻害しない限りは、特に限定されるものではない。
nは、例えば2~100であり、好ましくは10~40である。
本明細書中、1実施態様によれば、
Lが、下記式:
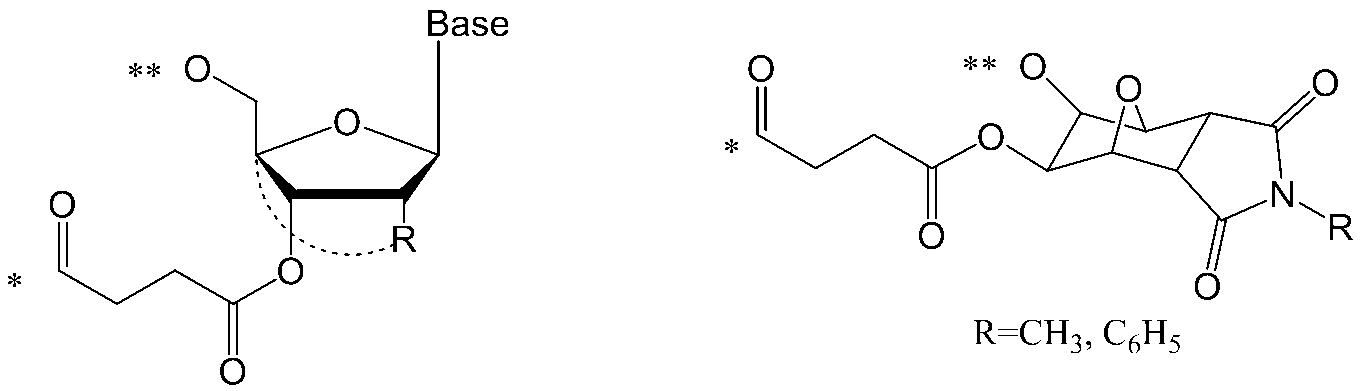
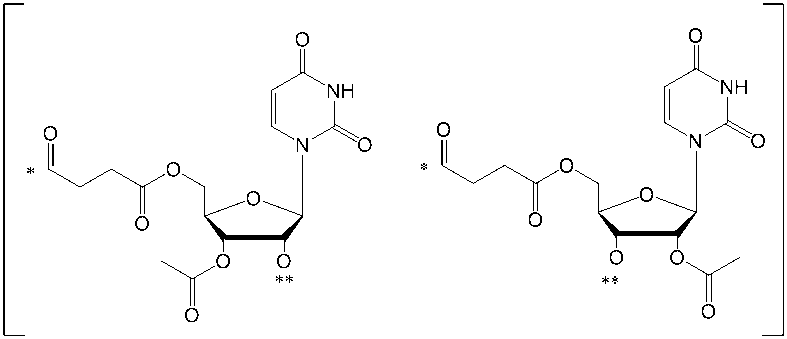
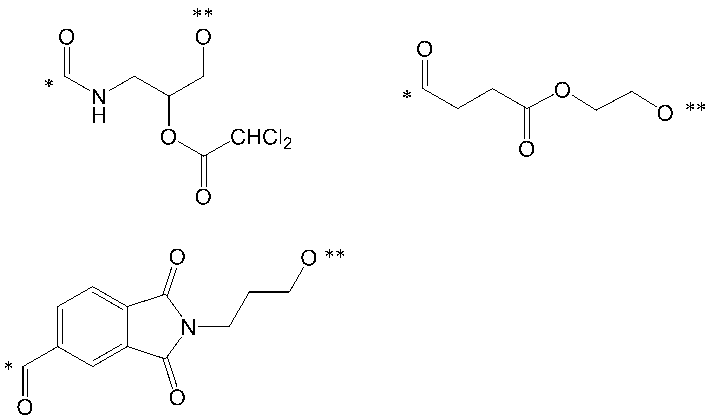
[式中、*は疎水性基との結合位置、および**はリン原子との結合位置を示し、
Baseは、各々独立して保護されていてもよい核酸塩基であり、そして、
Rは、各々独立して水素原子、ハロゲン原子、保護基で置換されていてもよいヒドロキシ基、置換されていてもよいC1-C6アルコキシ基、4位炭素原子に架橋する有機基、または架橋構造型人工核酸基である]
からなる群のいずれか1つから選ばれる基であるリンカーが挙げられる。
Lが、下記式:
Baseは、各々独立して保護されていてもよい核酸塩基であり、そして、
Rは、各々独立して水素原子、ハロゲン原子、保護基で置換されていてもよいヒドロキシ基、置換されていてもよいC1-C6アルコキシ基、4位炭素原子に架橋する有機基、または架橋構造型人工核酸基である]
からなる群のいずれか1つから選ばれる基であるリンカーが挙げられる。
好ましくは、Lは、式:

のいずれかから選ばれる基である。
本明細書中、1実施態様によれば、式(III)によって示されるヌクレオチド化合物の具体例としては、下記式:
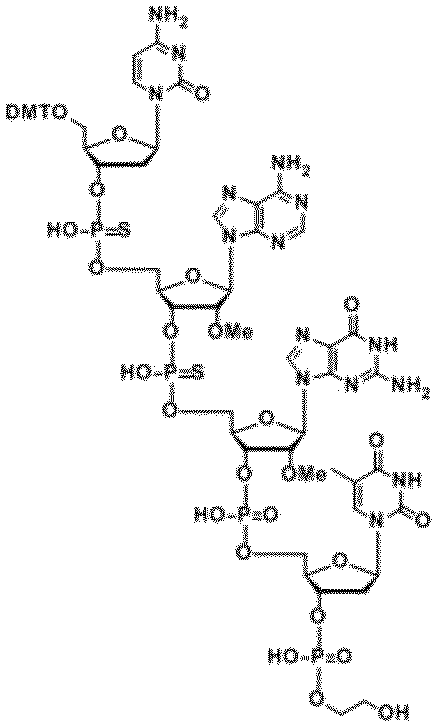
で示されるテトラマー(n=4)を挙げられるが、これらに限定されるものではない。
上記擬似固相保護基を含むヌクレオチドを、本発明のオリゴヌクレオチド製造方法の出発原料として、またはヌクレオチド伸張の1サイクルの出発物質として、使用することができる。
オリゴヌクレオチド製造において、1個分のヌクレオチドを伸張する一連の総工程(「ヌクレオチド伸張の1サイクル」と呼称する)は、1回または2回の精製工程を含んで行うことができる。
オリゴヌクレオチド製造におけるヌクレオチド伸張の1サイクル(2回精製)
1段階目
i)5’OH末端n量体を溶媒に溶解して、
ii)5’OH末端n量体、m+1-ホスホラミダイト化合物、および活性化剤を、溶媒中で反応させて、カップリング反応を行う。
反応の進行は、HO(Nps)n-SR(これは、HO(Nps)n-SRの消失による)の消失により確認する。
ii)カップリング反応後に、反応生成物を本発明のキャッピング反応用試薬と反応させてキャッピング反応を行う。
iv)キャッピング反応後に求核剤(例えば、エタノール等のアルコール)で処理して、カップリング粗生成物を得る。
v)有機塩基(例えばピリジンを)含む溶媒中で、該カップリング反応生成物と硫化試薬とを反応させる。次いで、反応生成物をホスファイト化合物と反応させた後、反応生成物をアルコール(例えば、メタノール)で晶析して、中間体DMT-(Nps)m+1+n-SRを得る。
2段階目
vi)得られた中間体DMT-(Nps) n+1-SRを、ピロールおよび酸と、ピリジンおよびアルコール(例えば、メタノール)を含む溶媒を用いて晶析して、目的物HO-(Nps) n+1-SRを得る。
1段階目
i)5’OH末端n量体を溶媒に溶解して、
ii)5’OH末端n量体、m+1-ホスホラミダイト化合物、および活性化剤を、溶媒中で反応させて、カップリング反応を行う。
反応の進行は、HO(Nps)n-SR(これは、HO(Nps)n-SRの消失による)の消失により確認する。
ii)カップリング反応後に、反応生成物を本発明のキャッピング反応用試薬と反応させてキャッピング反応を行う。
iv)キャッピング反応後に求核剤(例えば、エタノール等のアルコール)で処理して、カップリング粗生成物を得る。
v)有機塩基(例えばピリジンを)含む溶媒中で、該カップリング反応生成物と硫化試薬とを反応させる。次いで、反応生成物をホスファイト化合物と反応させた後、反応生成物をアルコール(例えば、メタノール)で晶析して、中間体DMT-(Nps)m+1+n-SRを得る。
2段階目
vi)得られた中間体DMT-(Nps) n+1-SRを、ピロールおよび酸と、ピリジンおよびアルコール(例えば、メタノール)を含む溶媒を用いて晶析して、目的物HO-(Nps) n+1-SRを得る。
オリゴヌクレオチド製造におけるヌクレオチド伸張の1サイクル(1回精製)
1段階目
i)5’OH末端n量体を溶媒に溶解する。
ii)5’OH末端n量体、m+1-ホスホラミダイト化合物、および活性化剤を、溶媒中で反応させて、カップリング反応を行う。
反応の進行は、HO(Nps)n-SR(これは、HO(Nps)n-SRの消失による)の消失により確認する。
ii)カップリング反応後に、反応生成物を本発明のキャッピング反応用試薬と反応させてキャッピング反応を行う。
iv)キャッピング反応後に求核剤(例えば、エタノール等のアルコール)で処理して、カップリング粗生成物を得る。
v)有機塩基(例えばピリジン)を含む溶媒中で、該カップリング反応生成物と硫化試薬とを反応させる。次いで、反応生成物をホスファイト化合物と反応させた後、ピリジンおよびアルコール(例えば、メタノール)を含む溶媒を用いて晶析して、目的物HO-(Nps) n+1-SRを得る。
1段階目
i)5’OH末端n量体を溶媒に溶解する。
ii)5’OH末端n量体、m+1-ホスホラミダイト化合物、および活性化剤を、溶媒中で反応させて、カップリング反応を行う。
反応の進行は、HO(Nps)n-SR(これは、HO(Nps)n-SRの消失による)の消失により確認する。
ii)カップリング反応後に、反応生成物を本発明のキャッピング反応用試薬と反応させてキャッピング反応を行う。
iv)キャッピング反応後に求核剤(例えば、エタノール等のアルコール)で処理して、カップリング粗生成物を得る。
v)有機塩基(例えばピリジン)を含む溶媒中で、該カップリング反応生成物と硫化試薬とを反応させる。次いで、反応生成物をホスファイト化合物と反応させた後、ピリジンおよびアルコール(例えば、メタノール)を含む溶媒を用いて晶析して、目的物HO-(Nps) n+1-SRを得る。
(キット)
本発明の1態様によれば、本発明は、例えば(オリゴ)ヌクレオチドの製造及び精製のためのキットをも提供する。本発明のキットは、成分として少なくとも1つの本発明のキャッピング反応用試薬を含む。本発明の1実施態様によれば、本発明のキットは、(a)本発明のキャッピング反応用試薬;(b)求核剤;(c)少なくとも1つの(オリゴ)ヌクレオチド化合物、例えば、(オリゴ)ヌクレオチドのホスホラミダイト化合物;(d)少なくとも1つの伸張可能な(オリゴ)ヌクレオチド化合物;(e)オリゴヌクレオチドを伸張するのに用いる少なくとも1つの活性化剤;(f)少なくとも1つの緩衝液;(g)キット成分を用いて(オリゴ)ヌクレオチドを伸張するための操作説明を記載する添付文書;並びに、(h)キット成分を充填するための少なくとも1つの容器、を含む。ここで、「緩衝液」は、使用するキット成分に悪影響(例えば、キット成分の分解、反応の阻害)をもたらさない限り、通常使用されるものが挙げられる。
本発明の1態様によれば、本発明は、例えば(オリゴ)ヌクレオチドの製造及び精製のためのキットをも提供する。本発明のキットは、成分として少なくとも1つの本発明のキャッピング反応用試薬を含む。本発明の1実施態様によれば、本発明のキットは、(a)本発明のキャッピング反応用試薬;(b)求核剤;(c)少なくとも1つの(オリゴ)ヌクレオチド化合物、例えば、(オリゴ)ヌクレオチドのホスホラミダイト化合物;(d)少なくとも1つの伸張可能な(オリゴ)ヌクレオチド化合物;(e)オリゴヌクレオチドを伸張するのに用いる少なくとも1つの活性化剤;(f)少なくとも1つの緩衝液;(g)キット成分を用いて(オリゴ)ヌクレオチドを伸張するための操作説明を記載する添付文書;並びに、(h)キット成分を充填するための少なくとも1つの容器、を含む。ここで、「緩衝液」は、使用するキット成分に悪影響(例えば、キット成分の分解、反応の阻害)をもたらさない限り、通常使用されるものが挙げられる。
また、本発明の1実施態様によれば、本発明のキットは、(オリゴ)ヌクレオチドのキャッピング反応のためのキットであり、これは、(a)本発明のキャッピング反応用試薬;(b)求核剤;(g)キット成分を用いて(オリゴ)ヌクレオチドをキャッピングするための操作説明を記載する添付文書;並びに、(h)キット成分を充填するための少なくとも1つの容器、を含む。
本発明の実施例を、下記に実施例として記載するが、これらに限定されるものではない。
試薬は、適宜、市販のものを入手するか、あるいは公知の方法に従って製造した。また、各種測定機器は、通常使用される機器を用いて使用した。
試薬は、適宜、市販のものを入手するか、あるいは公知の方法に従って製造した。また、各種測定機器は、通常使用される機器を用いて使用した。
高脂溶性ホスホラミダイトの合成
実施例1
2-シアノエチル オクタデシル ジイソプロピルホスホラミダイトの製造

アルゴン雰囲気下、オクタデカノール 541 mg及びテトラゾール141 mgに脱水ジクロロメタン2.4 mL、アセトニトリル1.6 mLを加えて撹拌し溶解させた後、3-(ビス(ジイソプロルピルアミノ)ホスフィノキシ)プロパンニトリル(0.70 mL, 2.2 mmol)を滴下して加えて一時間撹拌した。該反応液をジクロロメタン5 mLで洗い込みながら分液ロートへ移し、水10 mLで洗浄を行い、有機層を減圧濃縮した。濃縮した反応混合物にヘキサン20 mLを加え、混合物の100 uLを採取して、0.1 Mトリフェニルフォスフィンジクロロメタン溶液100 uLとCDCl3400 uLを加えて洗浄なしのNMRサンプルとした。ヘキサン溶液20 mLを分液ロートに移してアセトニトリル20 mLで洗浄操作を行い、有機層をメスシリンダーに移した。得られたヘキサン溶液18 mLにヘキサン2 mLを加え、20 mLのヘキサン溶液から100 uLを採取し、0.1 Mトリフェニルフォスフィンジクロロメタン溶液100 uLを採取してCDCl3を400 uL加えて洗浄一回のNMRサンプルとした。得られたヘキサン溶液20 mLを分液ロートに移してアセトニトリル20 mLで洗浄操作を行い、有機層をメスシリンダーに移した。得られたヘキサン溶液17 mLにヘキサン3 mLを加えた20 mLのヘキサン溶液から100 uL採取し、0.1 Mトリフェニルフォスフィンジクロロメタン溶液100 uLとCDCl3400 uLを加えて洗浄二回のNMRサンプルとした。有機層はエバポレーターで減圧濃縮し、無色油状の2-シアノエチル オクタデシル ジイソプロピルホスホラミダイトを682 mg得た。
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 3H, J = 7.2 Hz), 1.15-1.19 (m, 12H), 1.25-1.34 (m, 90H), 1.58-1.62 (m, 2H), 2.64 (t, 2H, J = 6.4 Hz), 3.55-3.65 (m, 4H), 3.80-3.85 (m, 2H)
31P-NMR (162 MHz CDCl3); δ 146.77
実施例1
2-シアノエチル オクタデシル ジイソプロピルホスホラミダイトの製造
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 3H, J = 7.2 Hz), 1.15-1.19 (m, 12H), 1.25-1.34 (m, 90H), 1.58-1.62 (m, 2H), 2.64 (t, 2H, J = 6.4 Hz), 3.55-3.65 (m, 4H), 3.80-3.85 (m, 2H)
31P-NMR (162 MHz CDCl3); δ 146.77
実施例2
2-シアノエチル オクタデシル ジイソプロピルホスホラミダイトの製造(50 mmol scale)
アルゴン雰囲気下、オクタデカノール 13.52 g及びテトラゾール1.93 gに脱水ジクロロメタン60 mL、アセトニトリル40 mLを加えて撹拌し溶解させた後、3-(ビス(ジイソプロルピルアミノ)ホスフィノキシ)プロパンニトリル(17.5 mL)を滴下して加えて一時間撹拌した。該反応液をジクロロメタンで洗い込みながら分液ロートへ移し、水で洗浄を行い、有機層を減圧濃縮した。濃縮した反応混合物にヘキサンに溶解しアセトニトリルで洗浄操作を2回行なった。ヘキサン層はエバポレーターで減圧濃縮し、無色油状の2-シアノエチル オクタデシル ジイソプロピルホスホラミダイトを20.7g得た
2-シアノエチル オクタデシル ジイソプロピルホスホラミダイトの製造(50 mmol scale)
アルゴン雰囲気下、オクタデカノール 13.52 g及びテトラゾール1.93 gに脱水ジクロロメタン60 mL、アセトニトリル40 mLを加えて撹拌し溶解させた後、3-(ビス(ジイソプロルピルアミノ)ホスフィノキシ)プロパンニトリル(17.5 mL)を滴下して加えて一時間撹拌した。該反応液をジクロロメタンで洗い込みながら分液ロートへ移し、水で洗浄を行い、有機層を減圧濃縮した。濃縮した反応混合物にヘキサンに溶解しアセトニトリルで洗浄操作を2回行なった。ヘキサン層はエバポレーターで減圧濃縮し、無色油状の2-シアノエチル オクタデシル ジイソプロピルホスホラミダイトを20.7g得た
実施例3
2-シアノエチル オクタデシル ジイソプロピルホスホラミダイトの製造(三価リン化合物として2-シアノエチル ジイソプロピルクロロホスホロアミダイトを用いる場合)
アルゴン雰囲気下、オクタデカノール 811 mg及びジイソプロピルエチルアミン1.04 mLに脱水テトラヒドロフラン6 mLを加えて撹拌し溶解、氷冷した後、2-シアノエチル ジイソプロピルクロロホスホロアミダイト(0.74 mL)を滴下して加えて一時間撹拌した。該反応液をジクロロメタンで洗い込みながら分液ロートへ移し、水で洗浄を行い、有機層を減圧濃縮した。濃縮した反応混合物にヘキサンに溶解しアセトニトリルで洗浄操作を2回行なった。ヘキサン層はエバポレーターで減圧濃縮し、無色油状の2-シアノエチル オクタデシル ジイソプロピルホスホラミダイトを1.08 g得た。
2-シアノエチル オクタデシル ジイソプロピルホスホラミダイトの製造(三価リン化合物として2-シアノエチル ジイソプロピルクロロホスホロアミダイトを用いる場合)
アルゴン雰囲気下、オクタデカノール 811 mg及びジイソプロピルエチルアミン1.04 mLに脱水テトラヒドロフラン6 mLを加えて撹拌し溶解、氷冷した後、2-シアノエチル ジイソプロピルクロロホスホロアミダイト(0.74 mL)を滴下して加えて一時間撹拌した。該反応液をジクロロメタンで洗い込みながら分液ロートへ移し、水で洗浄を行い、有機層を減圧濃縮した。濃縮した反応混合物にヘキサンに溶解しアセトニトリルで洗浄操作を2回行なった。ヘキサン層はエバポレーターで減圧濃縮し、無色油状の2-シアノエチル オクタデシル ジイソプロピルホスホラミダイトを1.08 g得た。
実施例4
2-シアノエチル デシル ジイソプロピルホスホラミダイトの製造
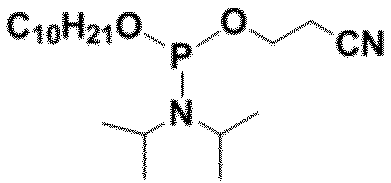
アルゴン雰囲気下、デカノール 381 uL及びテトラゾール141 mgに脱水ジクロロメタン2.4 mL、アセトニトリル1.6 mLを加えて撹拌し溶解させた後、3-(ビス(ジイソプロルピルアミノ)ホスフィノキシ)プロパンニトリル(0.70 mL, 2.2 mmol)を滴下して加えて一時間撹拌した。該反応液をジクロロメタン5 mLで洗い込みながら分液ロートへ移し、水10 mLで洗浄を行い、有機層を減圧濃縮した。濃縮した反応混合物にヘキサン20 mLを加え、混合物の100 uLを採取して、0.1 Mトリフェニルフォスフィンジクロロメタン溶液100 uLとCDCl3400 uLを加えて洗浄なしのNMRサンプルとした。ヘキサン溶液20 mLを分液ロートに移してアセトニトリル20 mLで洗浄操作を行い、有機層をメスシリンダーに移した。得られたヘキサン溶液18 mLにヘキサン2 mLを加え、20 mLのヘキサン溶液から100 uLを採取し、0.1 Mトリフェニルフォスフィンジクロロメタン溶液100 uLを採取してCDCl3を400 uL加えて洗浄一回のNMRサンプルとした。得られたヘキサン溶液20 mLを分液ロートに移してアセトニトリル20 mLで洗浄操作を行い、有機層をメスシリンダーに移した。得られたヘキサン溶液17 mLにヘキサン3 mLを加えた20 mLのヘキサン溶液から100 uL採取し、0.1 Mトリフェニルフォスフィンジクロロメタン溶液100 uLとCDCl3400 uLを加えて洗浄二回のNMRサンプルとした。有機層はエバポレーターで減圧濃縮し、無色油状の2-シアノエチル デシル ジイソプロピルホスホラミダイトを203 mg(収率 28%)得た。
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 3H, J = 7.2 Hz), 1.15-1.19 (m, 12H), 1.25-1.34 (m, 14H), 1.58-1.62 (m, 2H), 2.64 (t, 2H, J = 6.4 Hz), 3.55-3.65 (m, 4H), 3.79-3.84 (m, 2H)
31P-NMR (162 MHz CDCl3); δ 146.75
2-シアノエチル デシル ジイソプロピルホスホラミダイトの製造
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 3H, J = 7.2 Hz), 1.15-1.19 (m, 12H), 1.25-1.34 (m, 14H), 1.58-1.62 (m, 2H), 2.64 (t, 2H, J = 6.4 Hz), 3.55-3.65 (m, 4H), 3.79-3.84 (m, 2H)
31P-NMR (162 MHz CDCl3); δ 146.75
実施例5
2-シアノエチル オクチル ジイソプロピルホスホラミダイトの製造
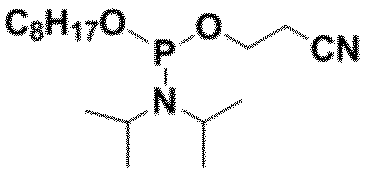
アルゴン雰囲気下、オクタノール 314 uL及びテトラゾール141 mgに脱水ジクロロメタン2.4 mL、アセトニトリル1.6 mLを加えて撹拌し溶解させた後、3-(ビス(ジイソプロルピルアミノ)ホスフィノキシ)プロパンニトリル(0.70 mL)を滴下して加えて一時間撹拌した。該反応液をジクロロメタン5 mLで洗い込みながら分液ロートへ移し、水10 mLで洗浄を行い、有機層を減圧濃縮した。濃縮した反応混合物にヘキサン20 mLを加え、混合物の100 uLを採取して、0.1 Mトリフェニルフォスフィンジクロロメタン溶液100 uLとCDCl3400 uLを加えて洗浄なしのNMRサンプルとした。ヘキサン溶液20 mLを分液ロートに移してアセトニトリル20 mLで洗浄操作を行い、有機層をメスシリンダーに移した。得られたヘキサン溶液18 mLにヘキサン2 mLを加え、20 mLのヘキサン溶液から100 uLを採取し、0.1 Mトリフェニルフォスフィンジクロロメタン溶液100 uLを採取してCDCl3を400 uL加えて洗浄一回のNMRサンプルとした。得られたヘキサン溶液20 mLを分液ロートに移してアセトニトリル20 mLで洗浄操作を行い、有機層をメスシリンダーに移した。得られたヘキサン溶液17 mLにヘキサン3 mLを加えた20 mLのヘキサン溶液から100 uL採取し、0.1 Mトリフェニルフォスフィンジクロロメタン溶液100 uLとCDCl3400 uLを加えて洗浄二回のNMRサンプルとした。有機層はエバポレーターで減圧濃縮し、無色油状の2-シアノエチル オクチル ジイソプロピルホスホラミダイトを54 mg得た。
2-シアノエチル オクチル ジイソプロピルホスホラミダイトの製造
実施例6
2-シアノエチル ヘキシル ジイソプロピルホスホラミダイトの製造
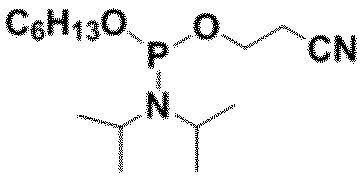
アルゴン雰囲気下、ヘキサノール 249 uL及びテトラゾール141 mgに脱水ジクロロメタン2.4 mL、アセトニトリル1.6 mLを加えて撹拌し溶解させた後、3-(ビス(ジイソプロルピルアミノ)ホスフィノキシ)プロパンニトリル(0.70 mL)を滴下して加えて一時間撹拌した。該反応液をジクロロメタン5 mLで洗い込みながら分液ロートへ移し、水10 mLで洗浄を行い、有機層を減圧濃縮した。濃縮した反応混合物にヘキサン20 mLを加え、混合物の100 uL採取して、0.1 Mトリフェニルフォスフィンジクロロメタン溶液100 uLとCDCl3400 uLを加えて洗浄なしのNMRサンプルとした。ヘキサン溶液20 mLを分液ロートに移してアセトニトリル20 mLで洗浄操作を行い、有機層をメスシリンダーに移した。得られたヘキサン溶液18 mLにヘキサン2 mLを加え、20 mLのヘキサン溶液から100 uLを採取し、0.1 Mトリフェニルフォスフィンジクロロメタン溶液100 uLを採取してCDCl3を400 uL加えて洗浄一回のNMRサンプルとした。得られたヘキサン溶液20 mLを分液ロートに移してアセトニトリル20 mLで洗浄操作を行い、有機層をメスシリンダーに移した。得られたヘキサン溶液17 mLにヘキサン3 mLを加えた20 mLのヘキサン溶液から100 uL採取し、0.1 Mトリフェニルフォスフィンジクロロメタン溶液100 uLとCDCl3400 uLを加えて洗浄二回のNMRサンプルとした。有機層はエバポレーターで減圧濃縮し、無色油状の2-シアノエチル ヘキシル ジイソプロピルホスホラミダイトを24 mg得た。
2-シアノエチル ヘキシル ジイソプロピルホスホラミダイトの製造
上記実施例1~6において製造したホスホラミダイト化合物の精製(抽出及び洗浄を含む)操作による、有機溶液中に含まれるホスホラミダイト化合物(モノアミダイト化合物)の回収率を下記の表1および図1にまとめる。

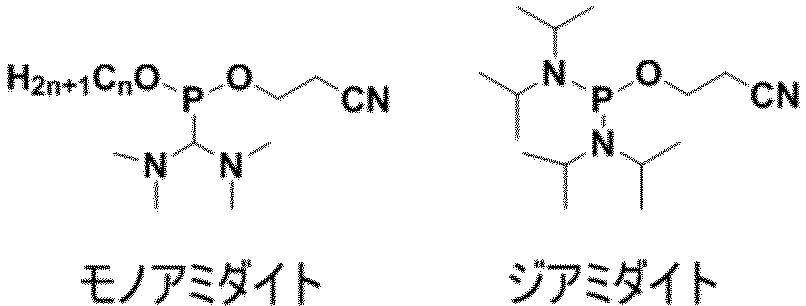
表1及び図1の結果より、本発明の製造及び精製方法は、反応混合物を、異なる有機溶媒系を用いて抽出および洗浄することにより、ホスホラミダイト化合物の脂溶性が高くなるにつれて、キャッピング時に副生成物を生じるH-ホスホネート化合物及びジアミダイト化合物を含まない実質的に高純度のホスホラミダイト生成物(モノアミダイト化合物)を高収率で回収することができることを見出した。
実施例7
2-シアノエチル 2’-オクチルドデカニル ジイソプロピルホスホラミダイトの製造
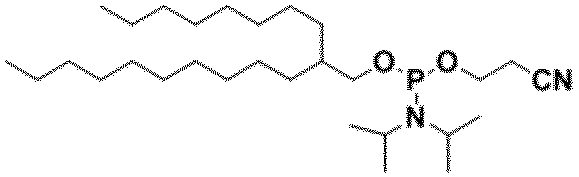
アルゴン雰囲気下、2-オクチルドデカノール 3.56 mL及びETT 0.72 gに脱水ジクロロメタン12 mL、アセトニトリル8 mLを加えて撹拌し溶解させた後、3-(ビス(ジイソプロルピルアミノ)ホスフィノキシ)プロパンニトリル(3.5 mL)を滴下して加えて一時間撹拌した。該反応液をジクロロメタンで洗い込みながら分液ロートへ移し、水で洗浄を行い、有機層を減圧濃縮した。濃縮した反応混合物にヘキサンに溶解しアセトニトリルで洗浄操作を2回行なった。ヘキサン層はエバポレーターで減圧濃縮し、無色油状の2-シアノエチル オクタデシル ジイソプロピルホスホラミダイトを3.43g得た。
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 6H, J =7.2 Hz), 1.16-1.19 (m, 12H), 1.2-1.4 (m, 32H), 1.54 (m, 1H), 2.63 (t, 2H, 6.4 Hz), 3.43-3.49 (m, 1H), 3.52-3.62 (m, 3H), 3.78-3.86 (m, 2H)
31P-NMR (162 MHz CDCl3); δ 146.90
2-シアノエチル 2’-オクチルドデカニル ジイソプロピルホスホラミダイトの製造
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 6H, J =7.2 Hz), 1.16-1.19 (m, 12H), 1.2-1.4 (m, 32H), 1.54 (m, 1H), 2.63 (t, 2H, 6.4 Hz), 3.43-3.49 (m, 1H), 3.52-3.62 (m, 3H), 3.78-3.86 (m, 2H)
31P-NMR (162 MHz CDCl3); δ 146.90
実施例8
Phy-CE-ホスホラミダイトの製造

アルゴン雰囲気下、化合物Phy-OH 2.98g及びテトラゾール 0.39 gに脱水ジクロロメタン12 mL、アセトニトリル8 mLを加えて撹拌し溶解させた後、3-(ビス(ジイソプロルピルアミノ)ホスフィノキシ)プロパンニトリル(3.5 mL)を滴下して加えて一時間撹拌した。該反応液をジクロロメタンで洗い込みながら分液ロートへ移し、水で洗浄を行い、有機層を減圧濃縮した。濃縮した反応混合物にヘキサンに溶解しアセトニトリルで洗浄操作を2回行なった。ヘキサン層はエバポレーターで減圧濃縮し、無色油状の化合物Phy-CE-アミダイトを3.84g得た
1H-NMR (400 MHz CDCl3); δ 0.88-0.99 (m, 15H), 1.01-1.16 (m, 8H), 1.16-1.21 (m, 12H), 1.21-1.32 (m, 9H,), 1.32-1.45 (m, 4H), 1.49-1.55 (m, 2H), 1.55-1.73 (m, 1H), 2.64 (t, 2H, 6.4 Hz), 3.55-3.72 (m, 4H), 3.73-3.79 (m, 2H)
31P-NMR (162 MHz CDCl3); δ 147.13, 147.22
Phy-CE-ホスホラミダイトの製造
1H-NMR (400 MHz CDCl3); δ 0.88-0.99 (m, 15H), 1.01-1.16 (m, 8H), 1.16-1.21 (m, 12H), 1.21-1.32 (m, 9H,), 1.32-1.45 (m, 4H), 1.49-1.55 (m, 2H), 1.55-1.73 (m, 1H), 2.64 (t, 2H, 6.4 Hz), 3.55-3.72 (m, 4H), 3.73-3.79 (m, 2H)
31P-NMR (162 MHz CDCl3); δ 147.13, 147.22
実施例9
エチレングリコラート化合物またはそれらの類似する化合物の合成
エチレングリコラート化合物またはそれらの類似する化合物の合成スキームを以下の反応式IIIに示す。
反応式III
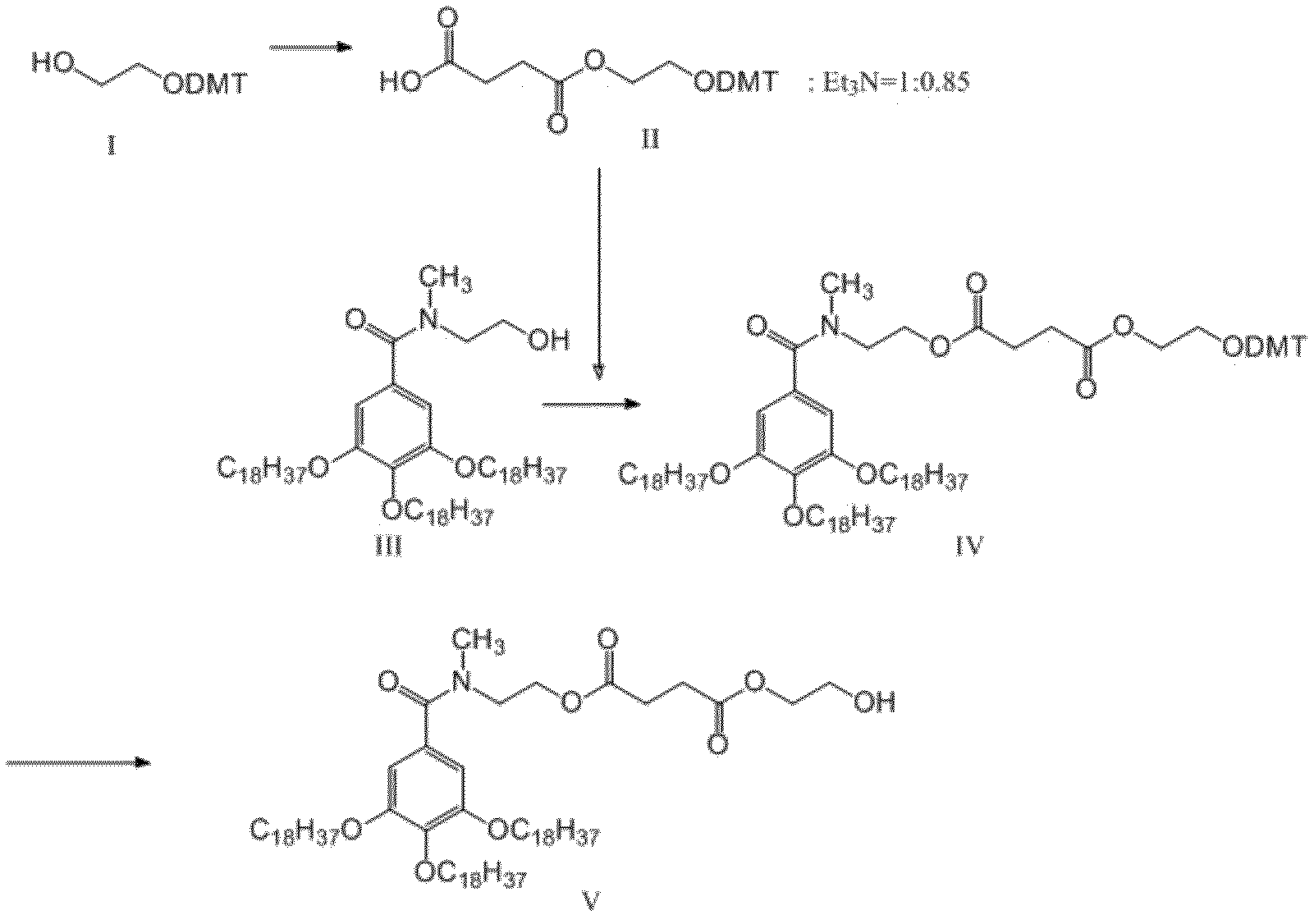
エチレングリコラート化合物またはそれらの類似する化合物の合成
エチレングリコラート化合物またはそれらの類似する化合物の合成スキームを以下の反応式IIIに示す。
反応式III
(化合物IIの製造)
化合物I 8.12 gを脱水ジクロロメタン35 mlに溶解し、該溶液に、トリエチルアミン 14.9 mLおよび無水コハク酸 4.46 gを加えた。該混合物を室温にて終夜攪拌した後にメタノール 2.2 mLを加え、該混合物を更に終夜攪拌した。得られた混合溶液をリン酸トリエチルアミン緩衝液(pH 約6.8)で洗浄した後に、該混合液を減圧濃縮し、続いて減圧下で終夜留去することによって揮発成分を除いて、化合物IIを11.1 g得た。トリエチルアミンの存在比は1H NMR積分値から概算した。
1H-NMR (400 MHz CDCl3); δ 1.20 (t, 7.7H, J = 7.2 Hz), 2.58 (t, 2H, J = 6.8 Hz), 2.68 (t, 2H, J = 6.8 Hz) , 2.94 (q, 5.1H, J = 7.2 Hz), 3.25 (t, 2H, J = 5.0 Hz), 3.79 (s, 6H), 4.23 (t, 2H, J = 5.0 Hz), 6.80-6.85 (m, 4H), 7.15-7.21 (m, 1H), 7.27-7.36 (m. 6H), 7.44-7.46 (m. 2H), 9.65 (brs, 1.9H)
化合物I 8.12 gを脱水ジクロロメタン35 mlに溶解し、該溶液に、トリエチルアミン 14.9 mLおよび無水コハク酸 4.46 gを加えた。該混合物を室温にて終夜攪拌した後にメタノール 2.2 mLを加え、該混合物を更に終夜攪拌した。得られた混合溶液をリン酸トリエチルアミン緩衝液(pH 約6.8)で洗浄した後に、該混合液を減圧濃縮し、続いて減圧下で終夜留去することによって揮発成分を除いて、化合物IIを11.1 g得た。トリエチルアミンの存在比は1H NMR積分値から概算した。
1H-NMR (400 MHz CDCl3); δ 1.20 (t, 7.7H, J = 7.2 Hz), 2.58 (t, 2H, J = 6.8 Hz), 2.68 (t, 2H, J = 6.8 Hz) , 2.94 (q, 5.1H, J = 7.2 Hz), 3.25 (t, 2H, J = 5.0 Hz), 3.79 (s, 6H), 4.23 (t, 2H, J = 5.0 Hz), 6.80-6.85 (m, 4H), 7.15-7.21 (m, 1H), 7.27-7.36 (m. 6H), 7.44-7.46 (m. 2H), 9.65 (brs, 1.9H)
(化合物IIIの製造)
化合物IIIの合成は特開2001-2533986に記載の方法に従い行った。
化合物IIIの合成は特開2001-2533986に記載の方法に従い行った。
(化合物IVの製造)
化合物III 15.7 g、化合物II 10.6 g、および脱水トルエン 100 mLの混合物を、減圧下濃縮乾固した。残渣に脱水クロロホルム100 mlを加え、更にジイソプロピルエチルアミン4.35 mL、HBTU 9.47gおよびDMAP 3.05 gを加えた。該混合物を45℃において一晩攪拌した。得られた反応混合物にメタノール 100 mLを加え、減圧下濃縮した。生じた固形物を濾過し、メタノール洗浄し、減圧下乾燥した。脱水トルエン100 mlとの混合物を、減圧下で濃縮乾固した。残渣に脱水クロロホルム 100 mLを加え、更に2,4,6-トリメチルピリジン 45 mL、1-メチルイミダゾール 30mLおよび無水酢酸 30 mLを加え、該混合物を一晩攪拌した。得られた反応混合物にメタノール 100mLを加え、減圧下濃縮した。生じた固形物を濾過し、メタノール洗浄し、減圧下乾燥して、目的物である化合物IVを22.67 g得た。NMR測定結果及びジメトキシトリチルカチオンによる定量(λ=498 ε=7.0*10-4) 725 umol/g(理論値704 umol/g)よりほぼ完全にエステル化されたと判断した。
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 9 H, J = 7.0 Hz), 1.15-1.39 (m, 84H), 1.40-1.51 (m, 6H), 1.68-1.85 (m, 6H), 2.69 (s, 4H), 3.04(s, 3H) 3.27 (t, 2H, J = 5.0 Hz), 3.5-3.82 (m, 2H) 3.79 (s, 6H), 3.93-3.98 (m, 6H), 4.26 (t, 2H, J = 5.0 Hz), 4.30-4.43 (m, 2H), 6.57 (s, 2H), 6.80-6.85 (m, 4H), 7.17-7.22 (m, 1H), 7.26-7.34 (m. 6H), 7.42-7.46 (m. 2H)
化合物III 15.7 g、化合物II 10.6 g、および脱水トルエン 100 mLの混合物を、減圧下濃縮乾固した。残渣に脱水クロロホルム100 mlを加え、更にジイソプロピルエチルアミン4.35 mL、HBTU 9.47gおよびDMAP 3.05 gを加えた。該混合物を45℃において一晩攪拌した。得られた反応混合物にメタノール 100 mLを加え、減圧下濃縮した。生じた固形物を濾過し、メタノール洗浄し、減圧下乾燥した。脱水トルエン100 mlとの混合物を、減圧下で濃縮乾固した。残渣に脱水クロロホルム 100 mLを加え、更に2,4,6-トリメチルピリジン 45 mL、1-メチルイミダゾール 30mLおよび無水酢酸 30 mLを加え、該混合物を一晩攪拌した。得られた反応混合物にメタノール 100mLを加え、減圧下濃縮した。生じた固形物を濾過し、メタノール洗浄し、減圧下乾燥して、目的物である化合物IVを22.67 g得た。NMR測定結果及びジメトキシトリチルカチオンによる定量(λ=498 ε=7.0*10-4) 725 umol/g(理論値704 umol/g)よりほぼ完全にエステル化されたと判断した。
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 9 H, J = 7.0 Hz), 1.15-1.39 (m, 84H), 1.40-1.51 (m, 6H), 1.68-1.85 (m, 6H), 2.69 (s, 4H), 3.04(s, 3H) 3.27 (t, 2H, J = 5.0 Hz), 3.5-3.82 (m, 2H) 3.79 (s, 6H), 3.93-3.98 (m, 6H), 4.26 (t, 2H, J = 5.0 Hz), 4.30-4.43 (m, 2H), 6.57 (s, 2H), 6.80-6.85 (m, 4H), 7.17-7.22 (m, 1H), 7.26-7.34 (m. 6H), 7.42-7.46 (m. 2H)
(化合物Vの製造)
化合物IV 19.8 gを550mL ジクロロメタンに溶解し、該溶解液にピロール12.8 mLおよびジクロロ酢酸18.3 mLを加えた。該混合物を室温にて15分間攪拌し、反応混合物にメタノール 332 mLおよびピリジン 26.8 mLを加えた。反応混合物を減圧下濃縮し、生じた固形物を濾過し、メタノール洗浄し、減圧下乾燥して、目的物である化合物Vを15.6 g得た。
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 9 H, J = 6.8 Hz), 1.15-1.39 (m, 84H), 1.41-1.51 (m, 6H), 1.68-1.85 (m, 6H), 2.54 (brs, 1H), 2.66 (s, 4H), 3.05(s, 3H) 3.5-3.81 (m, 2H) 3.81 (dd, 2H, J = 9.2, 6.0 Hz), 3.94-3.99 (m, 6H), 4.23 (dd, 2H, J = 9.2, 6.0 Hz), 4.10-4.48 (m, 2H), 6.58 (s, 2H)
化合物IV 19.8 gを550mL ジクロロメタンに溶解し、該溶解液にピロール12.8 mLおよびジクロロ酢酸18.3 mLを加えた。該混合物を室温にて15分間攪拌し、反応混合物にメタノール 332 mLおよびピリジン 26.8 mLを加えた。反応混合物を減圧下濃縮し、生じた固形物を濾過し、メタノール洗浄し、減圧下乾燥して、目的物である化合物Vを15.6 g得た。
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 9 H, J = 6.8 Hz), 1.15-1.39 (m, 84H), 1.41-1.51 (m, 6H), 1.68-1.85 (m, 6H), 2.54 (brs, 1H), 2.66 (s, 4H), 3.05(s, 3H) 3.5-3.81 (m, 2H) 3.81 (dd, 2H, J = 9.2, 6.0 Hz), 3.94-3.99 (m, 6H), 4.23 (dd, 2H, J = 9.2, 6.0 Hz), 4.10-4.48 (m, 2H), 6.58 (s, 2H)
実施例10
オリゴヌクレオチド合成
オリゴヌクレオチドの合成スキームを以下の反応式IVに示す。
反応式IV
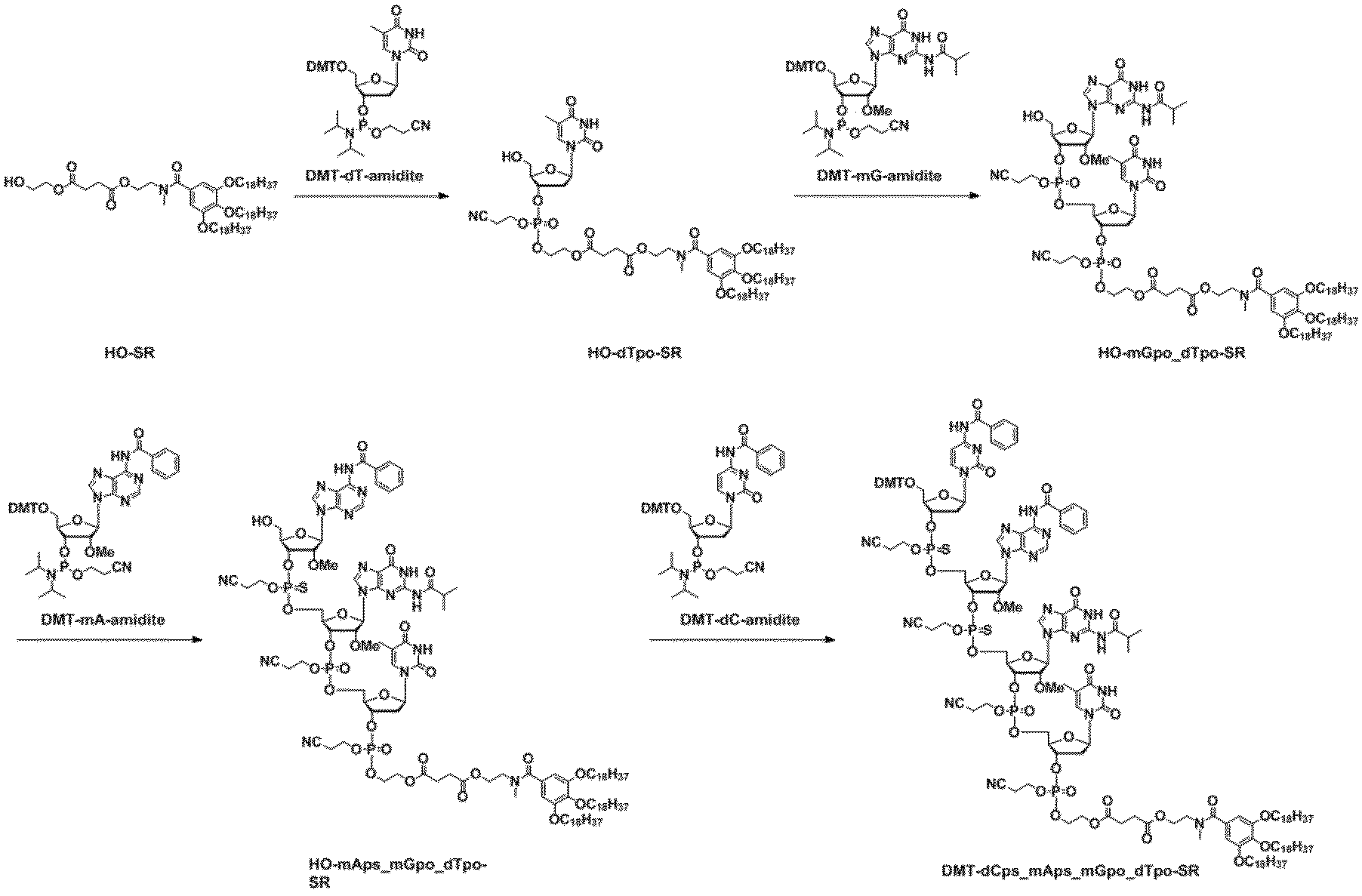
オリゴヌクレオチド合成
オリゴヌクレオチドの合成スキームを以下の反応式IVに示す。
反応式IV
化合物HO-dTpo-SRの合成
200mLナスフラスコ中の2-ヒドロキシエチル 2-(N-メチル-3,4,5-トリス(オクタデシル)ベンゾアミド)エチルスクシネート 3.39 g)に、脱水ジクロロメタン60mL、および化合物DMT-dT-アミダイト 3.35 gを加え溶解後、該溶解液にETT 879 mgを加え、該混合物を室温にて30分間撹拌した。15分後TLCによって基質の消失を確認した。該反応液に、2-シアノエチル オクタデシル ジイソプロピルホスホラミダイト424 uLを加え、反応混合物を15分間撹拌した。該反応混合物にエタノール 876 uLを加え、該混合物を室温にて10分間撹拌した。該混合物に、ピリジン:アセトニトリル=6:4(v/v)の混合溶液 (30 mL)および2-ブタノンペルオキシド(0.1 Mジクロロメタン溶液) 81 mLを加え、該混合物を室温にて15分間撹拌した。該反応混合物に、トリメチルホスファイト 658 uLを加え、該混合物を更に15分間撹拌した。得られた反応混合物をジクロロメタンで希釈後に、メタノール 150 mLを加え、混合物を減圧下濃縮した。生成した固形物を濾過し、メタノール洗浄することによって、中間体DMT-dTpo-SRを得た。
得られた中間体DMT-dTpo-SRを500 mL ナスフラスコ内でジクロロメタン 225 mLに溶解し、そこにピロール 5.2 mL、およびジクロロ酢酸 7.4 mLを加えた。該混合物を室温にて15分間撹拌し、その後にメタノール150 mLおよびピリジン8.70mLを加えた。該混合物を減圧下濃縮し、生成した固形物をろ過し、メタノール洗浄して減圧乾燥することで目的化合物HO-dTpo-SRを4.38 g得た。
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 9H, J = 7.2 Hz), 1.2-1.4 (m, 84H), 1.4-1.5 (m, 6H), 1.60 (s, broad, 1H), 1.71-1.81 (m, 6H), 1.91 (s, 3H), 2.47-2.54 (m, 2H), 2.64-2.71 (m, 4H), 2.80 (t, 2H, J = 6.0 Hz), 3.05 (s, 3H), 3.65-3.81 (m, 2H), 3.84-3.88 (m, 2H), 3.93-3.97 (m, 6H), 4.23-4.40 (m, 9H), 5.12-5.18 (m, 1H), 6.23 (t, 1H, J = 6.8 Hz), 6.58 (s, 2H), 7.52 (s, 1H), 8.14-8.18 (m, 0.5H), 8.52-8.58 (m. 0.5H)
31P-NMR (162 MHz CDCl3); δ -3.15, -3.07
200mLナスフラスコ中の2-ヒドロキシエチル 2-(N-メチル-3,4,5-トリス(オクタデシル)ベンゾアミド)エチルスクシネート 3.39 g)に、脱水ジクロロメタン60mL、および化合物DMT-dT-アミダイト 3.35 gを加え溶解後、該溶解液にETT 879 mgを加え、該混合物を室温にて30分間撹拌した。15分後TLCによって基質の消失を確認した。該反応液に、2-シアノエチル オクタデシル ジイソプロピルホスホラミダイト424 uLを加え、反応混合物を15分間撹拌した。該反応混合物にエタノール 876 uLを加え、該混合物を室温にて10分間撹拌した。該混合物に、ピリジン:アセトニトリル=6:4(v/v)の混合溶液 (30 mL)および2-ブタノンペルオキシド(0.1 Mジクロロメタン溶液) 81 mLを加え、該混合物を室温にて15分間撹拌した。該反応混合物に、トリメチルホスファイト 658 uLを加え、該混合物を更に15分間撹拌した。得られた反応混合物をジクロロメタンで希釈後に、メタノール 150 mLを加え、混合物を減圧下濃縮した。生成した固形物を濾過し、メタノール洗浄することによって、中間体DMT-dTpo-SRを得た。
得られた中間体DMT-dTpo-SRを500 mL ナスフラスコ内でジクロロメタン 225 mLに溶解し、そこにピロール 5.2 mL、およびジクロロ酢酸 7.4 mLを加えた。該混合物を室温にて15分間撹拌し、その後にメタノール150 mLおよびピリジン8.70mLを加えた。該混合物を減圧下濃縮し、生成した固形物をろ過し、メタノール洗浄して減圧乾燥することで目的化合物HO-dTpo-SRを4.38 g得た。
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 9H, J = 7.2 Hz), 1.2-1.4 (m, 84H), 1.4-1.5 (m, 6H), 1.60 (s, broad, 1H), 1.71-1.81 (m, 6H), 1.91 (s, 3H), 2.47-2.54 (m, 2H), 2.64-2.71 (m, 4H), 2.80 (t, 2H, J = 6.0 Hz), 3.05 (s, 3H), 3.65-3.81 (m, 2H), 3.84-3.88 (m, 2H), 3.93-3.97 (m, 6H), 4.23-4.40 (m, 9H), 5.12-5.18 (m, 1H), 6.23 (t, 1H, J = 6.8 Hz), 6.58 (s, 2H), 7.52 (s, 1H), 8.14-8.18 (m, 0.5H), 8.52-8.58 (m. 0.5H)
31P-NMR (162 MHz CDCl3); δ -3.15, -3.07
化合物HO-mGpo-dTpo-SRの合成
化合物HO-dTpo-SR 1.49 gを100mLナスフラスコにおいて脱水ジクロロメタン20mL、および化合物DMT-mG-アミダイト 1.33 gを加え溶解後、該溶解液にETT 293 mgを加え、該混合物を室温にて一晩撹拌した。TLCによって基質の消失を確認した。該反応液に、2-シアノエチル オクタデシル ジイソプロピルホスホラミダイト141 uL加え、反応混合物を30分間撹拌した。該反応混合物にエタノール 292uLを加え、該混合物を室温にて10分間撹拌した。該混合物に、2-ブタノンペルオキシド(6.5 M) 0.55 mLを加え、該混合物を室温にて15分間撹拌した。該反応混合物に、トリメチルホスファイト 328 uLを加え、該混合物を更に15分間撹拌した。得られた反応混合物をジクロロメタン55mlで希釈した後に、ピロール 1.7 mL、およびトリフロロ酢酸 2.3 mLを加えた。該混合物を室温にて15分間撹拌し、その後にメタノール50 mLおよびピリジン4.8mLを加えた。該混合物を減圧下濃縮し、生成した固形物をろ過し、メタノール洗浄して減圧乾燥することで目的化合物HO-mGpo-dTpo-SRを2.00g(収率:定量)得た。
化合物HO-dTpo-SR 1.49 gを100mLナスフラスコにおいて脱水ジクロロメタン20mL、および化合物DMT-mG-アミダイト 1.33 gを加え溶解後、該溶解液にETT 293 mgを加え、該混合物を室温にて一晩撹拌した。TLCによって基質の消失を確認した。該反応液に、2-シアノエチル オクタデシル ジイソプロピルホスホラミダイト141 uL加え、反応混合物を30分間撹拌した。該反応混合物にエタノール 292uLを加え、該混合物を室温にて10分間撹拌した。該混合物に、2-ブタノンペルオキシド(6.5 M) 0.55 mLを加え、該混合物を室温にて15分間撹拌した。該反応混合物に、トリメチルホスファイト 328 uLを加え、該混合物を更に15分間撹拌した。得られた反応混合物をジクロロメタン55mlで希釈した後に、ピロール 1.7 mL、およびトリフロロ酢酸 2.3 mLを加えた。該混合物を室温にて15分間撹拌し、その後にメタノール50 mLおよびピリジン4.8mLを加えた。該混合物を減圧下濃縮し、生成した固形物をろ過し、メタノール洗浄して減圧乾燥することで目的化合物HO-mGpo-dTpo-SRを2.00g(収率:定量)得た。
化合物HO-mAps-mGpo-dTpo-SRの合成
化合物HO-mGpo-dTpo-SR 2.00 g(max.1.00 mmol)を100mLナスフラスコにおいてアルゴン雰囲気下、脱水ジクロロメタン20mL、および化合物DMT-mA-アミダイト 1.35 gを加え溶解後、該溶解液にETT 293 mgを加え、該混合物を室温にて一晩撹拌した。TLCによって基質の消失を確認した。該反応液に、2-シアノエチル オクタデシル ジイソプロピルホスホラミダイト141 uLを加え、反応混合物を30分間撹拌した。該反応混合物にエタノール 292uLを加え、該混合物を室温にて10分間撹拌した。該混合物に、2,4,6-コリジン 714ul、DDTT 582 mgを加え、該混合物を室温にて45分間撹拌した。該反応混合物に、トリメチルホスファイト 328 uLを加え、該混合物を更に15分間撹拌した。得られた反応混合物をジクロロメタン55mlで希釈した後に、ピロール 1.7 mL、およびトリフルオロ酢酸 2.3 mLを加えた。該混合物を室温にて15分間撹拌し、その後にメタノール50 mLおよびピリジン4.8mLを加えた。該混合物を減圧下濃縮し、生成した固形物をろ過し、メタノール洗浄して減圧乾燥することで目的化合物HO-mAps-mGpo-dTpo-SRを2.42g得た。
化合物HO-mGpo-dTpo-SR 2.00 g(max.1.00 mmol)を100mLナスフラスコにおいてアルゴン雰囲気下、脱水ジクロロメタン20mL、および化合物DMT-mA-アミダイト 1.35 gを加え溶解後、該溶解液にETT 293 mgを加え、該混合物を室温にて一晩撹拌した。TLCによって基質の消失を確認した。該反応液に、2-シアノエチル オクタデシル ジイソプロピルホスホラミダイト141 uLを加え、反応混合物を30分間撹拌した。該反応混合物にエタノール 292uLを加え、該混合物を室温にて10分間撹拌した。該混合物に、2,4,6-コリジン 714ul、DDTT 582 mgを加え、該混合物を室温にて45分間撹拌した。該反応混合物に、トリメチルホスファイト 328 uLを加え、該混合物を更に15分間撹拌した。得られた反応混合物をジクロロメタン55mlで希釈した後に、ピロール 1.7 mL、およびトリフルオロ酢酸 2.3 mLを加えた。該混合物を室温にて15分間撹拌し、その後にメタノール50 mLおよびピリジン4.8mLを加えた。該混合物を減圧下濃縮し、生成した固形物をろ過し、メタノール洗浄して減圧乾燥することで目的化合物HO-mAps-mGpo-dTpo-SRを2.42g得た。
化合物DMT-dCps-mAps-mGpo-dTpo-SRの合成
化合物HO-mAps-mGpo-dTpo-SR 2.42 gを、100 mLナスフラスコにおいて、アルゴン雰囲気下、脱水ジクロロメタン20 mL、および化合物DMT-dC-アミダイト 1.27 gを加え、該溶解液にETT 293 mgを加え、該混合物を室温にて撹拌し、HPLCによって基質の消失を確認した後(約2時間後)に該反応液に、2-シアノエチル オクタデシル ジイソプロピルホスホラミダイト141 uLを加え、反応混合物を30分間撹拌した。反応終了後に、該反応混合物にエタノール 292 uLを加え、該混合物を室温にて10分間撹拌した。該混合物に、ピリジン:アセトニトリル=6:4(v/v)の混合溶液 (10 mL)およびDDTT 554 mgを加え、該混合物を室温にて45分間撹拌した。該反応混合物に、トリメチルホスファイト 219 uLを加え、該混合物を更に15分間撹拌した。得られた反応混合物をジクロロメタンで希釈後に、メタノール 55 mLを加え、混合物を減圧下濃縮した。生成した固形物を濾過し、メタノール洗浄することによって、目的化合物DMT-dCps-mAps-mGpo-dTpo-SRを3.15 g得た。
化合物HO-mAps-mGpo-dTpo-SR 2.42 gを、100 mLナスフラスコにおいて、アルゴン雰囲気下、脱水ジクロロメタン20 mL、および化合物DMT-dC-アミダイト 1.27 gを加え、該溶解液にETT 293 mgを加え、該混合物を室温にて撹拌し、HPLCによって基質の消失を確認した後(約2時間後)に該反応液に、2-シアノエチル オクタデシル ジイソプロピルホスホラミダイト141 uLを加え、反応混合物を30分間撹拌した。反応終了後に、該反応混合物にエタノール 292 uLを加え、該混合物を室温にて10分間撹拌した。該混合物に、ピリジン:アセトニトリル=6:4(v/v)の混合溶液 (10 mL)およびDDTT 554 mgを加え、該混合物を室温にて45分間撹拌した。該反応混合物に、トリメチルホスファイト 219 uLを加え、該混合物を更に15分間撹拌した。得られた反応混合物をジクロロメタンで希釈後に、メタノール 55 mLを加え、混合物を減圧下濃縮した。生成した固形物を濾過し、メタノール洗浄することによって、目的化合物DMT-dCps-mAps-mGpo-dTpo-SRを3.15 g得た。
化合物DMT-dCps-mAps-mGpo-dTpo-SRの脱保護及び得られたテトラマーの純度確認
化合物DMT-dCps-mAps-mGpo-dTpo-SR 4 mgをバイアルに加え、28%アンモニア水500 uLを加えて55℃で8時間放置した。下記表2の条件下でのHPLCの分析の結果、純度98.5%であった。
MS (MALDI, TOF) Anal. Calc. for C64H77N15O28P5S2 : 1691.35. Found 1691.52
HPLC分析条件

化合物DMT-dCps-mAps-mGpo-dTpo-SR 4 mgをバイアルに加え、28%アンモニア水500 uLを加えて55℃で8時間放置した。下記表2の条件下でのHPLCの分析の結果、純度98.5%であった。
MS (MALDI, TOF) Anal. Calc. for C64H77N15O28P5S2 : 1691.35. Found 1691.52
HPLC分析条件
DMT-dCps-mAps-mGpo-dTpo-SRの構造式

実施例11
グアニン6位O-高脂溶性アミダイト アダクトのアルコールによる分解
グアニン6位O-高脂溶性アミダイト アダクトの製造を、下記反応式Vに示す。
反応式V
グアニン6位O-高脂溶性アミダイト アダクトのアルコールによる分解
グアニン6位O-高脂溶性アミダイト アダクトの製造を、下記反応式Vに示す。
反応式V
化合物MMT-mGps-SRの合成
2-ヒドロキシエチル 2-(N-メチル-3,4,5-トリス(オクタデシル)ベンゾアミド)エチルスクシネート 1.13 gを、100mLナスフラスコにおいてアルゴン雰囲気下、脱水ジクロロメタン20mL、および化合物MMT-mG-アミダイト 1.26 gを加え溶解後、該溶解液に5-エチルチオ-4H-テトラゾール(略ETT) 293 mgを加え、該混合物を室温にて一晩撹拌した。TLCによって基質の消失を確認した。該反応液に、2-シアノエチル オクタデシル ジイソプロピルホスホラミダイト141 uLを加え、反応混合物を30分間撹拌した。該反応混合物にエタノール 292uLを加え、該混合物を室温にて10分間撹拌した。該混合物に、ピリジン:アセトニトリル=6:4(v/v)の混合溶液 (10 mL)および3-((N,N-ジメチルアミノメチリデン)アミノ)-3H-1,2,4-ジチアゾール-5-チオン(DDTT)582 mgを加え、該混合物を室温にて45分間撹拌した。該反応混合物に、トリメチルホスファイト 218 uL (1.8 mmol)を加え、該混合物を更に15分間撹拌した後にメタノール50 mLを加えた。該混合物を減圧下濃縮し、生成した固形物をろ過し、メタノール洗浄して減圧乾燥することで目的化合物MMT-mGps-SRを1.93g得た。
1H-NMR (400 MHz CDCl3); δ 0.82-0.90 (m, 12H) , 0.95-0.97 (m, 3H)1.20-1.39 (m, 84H), 1.40-1.51 (m, 6H), 1.70-1.82 (m, 6H), 1.89-2.04 (m 1H), 2.57-2.70 (m, 5H), 2.72-2.89 (m, 1H), 3.05 (brs, 3H), 3.24-3.31 (m, 1H), 3.44 (s, 1.5H), 3.45 (s, 1.5H), 3.47-3.53 (m, 1H), 3.75-3.86 (m, 2H), 3.79 (s, 3H), 3.89-3.97 (m, 6H), 4.06-4.45 (m, 9H), 4.70-4.79 (m, 1H), 5.36-5.43 (m, 1H), 5.88 (d, 0.5H, J = 6.8 Hz), 5.90 (d, 0.5H, J = 8.0 Hz), 6.58 (s, 2H), 6.80-6.86 (m, 2H), 7.22-7.38 (m, 8H), 7.46-7.52 (m. 4H), 7.77 (s, 0.5H), 7.78 (s, 0.5H), 8.52 (brs, 0.5H), 8.61 (brs, 0.5H), 12.03 (s, 1H)
31P-NMR (162 MHz CDCl3); δ 67.85, 67.96
2-ヒドロキシエチル 2-(N-メチル-3,4,5-トリス(オクタデシル)ベンゾアミド)エチルスクシネート 1.13 gを、100mLナスフラスコにおいてアルゴン雰囲気下、脱水ジクロロメタン20mL、および化合物MMT-mG-アミダイト 1.26 gを加え溶解後、該溶解液に5-エチルチオ-4H-テトラゾール(略ETT) 293 mgを加え、該混合物を室温にて一晩撹拌した。TLCによって基質の消失を確認した。該反応液に、2-シアノエチル オクタデシル ジイソプロピルホスホラミダイト141 uLを加え、反応混合物を30分間撹拌した。該反応混合物にエタノール 292uLを加え、該混合物を室温にて10分間撹拌した。該混合物に、ピリジン:アセトニトリル=6:4(v/v)の混合溶液 (10 mL)および3-((N,N-ジメチルアミノメチリデン)アミノ)-3H-1,2,4-ジチアゾール-5-チオン(DDTT)582 mgを加え、該混合物を室温にて45分間撹拌した。該反応混合物に、トリメチルホスファイト 218 uL (1.8 mmol)を加え、該混合物を更に15分間撹拌した後にメタノール50 mLを加えた。該混合物を減圧下濃縮し、生成した固形物をろ過し、メタノール洗浄して減圧乾燥することで目的化合物MMT-mGps-SRを1.93g得た。
1H-NMR (400 MHz CDCl3); δ 0.82-0.90 (m, 12H) , 0.95-0.97 (m, 3H)1.20-1.39 (m, 84H), 1.40-1.51 (m, 6H), 1.70-1.82 (m, 6H), 1.89-2.04 (m 1H), 2.57-2.70 (m, 5H), 2.72-2.89 (m, 1H), 3.05 (brs, 3H), 3.24-3.31 (m, 1H), 3.44 (s, 1.5H), 3.45 (s, 1.5H), 3.47-3.53 (m, 1H), 3.75-3.86 (m, 2H), 3.79 (s, 3H), 3.89-3.97 (m, 6H), 4.06-4.45 (m, 9H), 4.70-4.79 (m, 1H), 5.36-5.43 (m, 1H), 5.88 (d, 0.5H, J = 6.8 Hz), 5.90 (d, 0.5H, J = 8.0 Hz), 6.58 (s, 2H), 6.80-6.86 (m, 2H), 7.22-7.38 (m, 8H), 7.46-7.52 (m. 4H), 7.77 (s, 0.5H), 7.78 (s, 0.5H), 8.52 (brs, 0.5H), 8.61 (brs, 0.5H), 12.03 (s, 1H)
31P-NMR (162 MHz CDCl3); δ 67.85, 67.96
化合物MMT-mGps-SR、MMT-mG(psCap)ps-SR混合物の合成
2-ヒドロキシエチル 2-(N-メチル-3,4,5-トリス(オクタデシル)ベンゾアミド)エチルスクシネート 0.56gを、100mLナスフラスコにおいてアルゴン雰囲気下、脱水ジクロロメタン10mL、および化合物MMT-mG-アミダイト 0.63 gを加え溶解後、該溶解液にETT 146 mgを加え、該混合物を室温にて一晩撹拌した。TLCによって基質の消失を確認した。該反応液に、ETT 98 mgを加え、2-シアノエチル オクタデシル ジイソプロピルホスホラミダイト353 uLを加え、反応混合物を30分間撹拌した。該混合物に、ピリジン:アセトニトリル=6:4(v/v)の混合溶液 (5 mL)およびDDTT 485 mgを加え、該混合物を室温にて45分間撹拌した。該反応混合物に、トリメチルホスファイト 137 uLを加え、該混合物を更に15分間撹拌した後にメタノール25 mLを加えた。該混合物を減圧下濃縮し、生成した固形物をろ過し、メタノール洗浄して減圧乾燥することで化合物MMT-mGps-SR及び化合物MMT-mG(psCap)ps-SRの混合物を0.98g得た。
化合物MMT-mGps-SR及び化合物MMT-mG(psCap)ps-SRの混合物
31P-NMR (162 MHz CDCl3); δ 67.85, 67.96, 71.43
2-ヒドロキシエチル 2-(N-メチル-3,4,5-トリス(オクタデシル)ベンゾアミド)エチルスクシネート 0.56gを、100mLナスフラスコにおいてアルゴン雰囲気下、脱水ジクロロメタン10mL、および化合物MMT-mG-アミダイト 0.63 gを加え溶解後、該溶解液にETT 146 mgを加え、該混合物を室温にて一晩撹拌した。TLCによって基質の消失を確認した。該反応液に、ETT 98 mgを加え、2-シアノエチル オクタデシル ジイソプロピルホスホラミダイト353 uLを加え、反応混合物を30分間撹拌した。該混合物に、ピリジン:アセトニトリル=6:4(v/v)の混合溶液 (5 mL)およびDDTT 485 mgを加え、該混合物を室温にて45分間撹拌した。該反応混合物に、トリメチルホスファイト 137 uLを加え、該混合物を更に15分間撹拌した後にメタノール25 mLを加えた。該混合物を減圧下濃縮し、生成した固形物をろ過し、メタノール洗浄して減圧乾燥することで化合物MMT-mGps-SR及び化合物MMT-mG(psCap)ps-SRの混合物を0.98g得た。
化合物MMT-mGps-SR及び化合物MMT-mG(psCap)ps-SRの混合物
31P-NMR (162 MHz CDCl3); δ 67.85, 67.96, 71.43
次に、上記で製造したグアニン6位O-高脂溶性アミダイト アダクトP(5価)の酸による脱トリチル反応式を、下記反応式VIに示す。
反応式VI
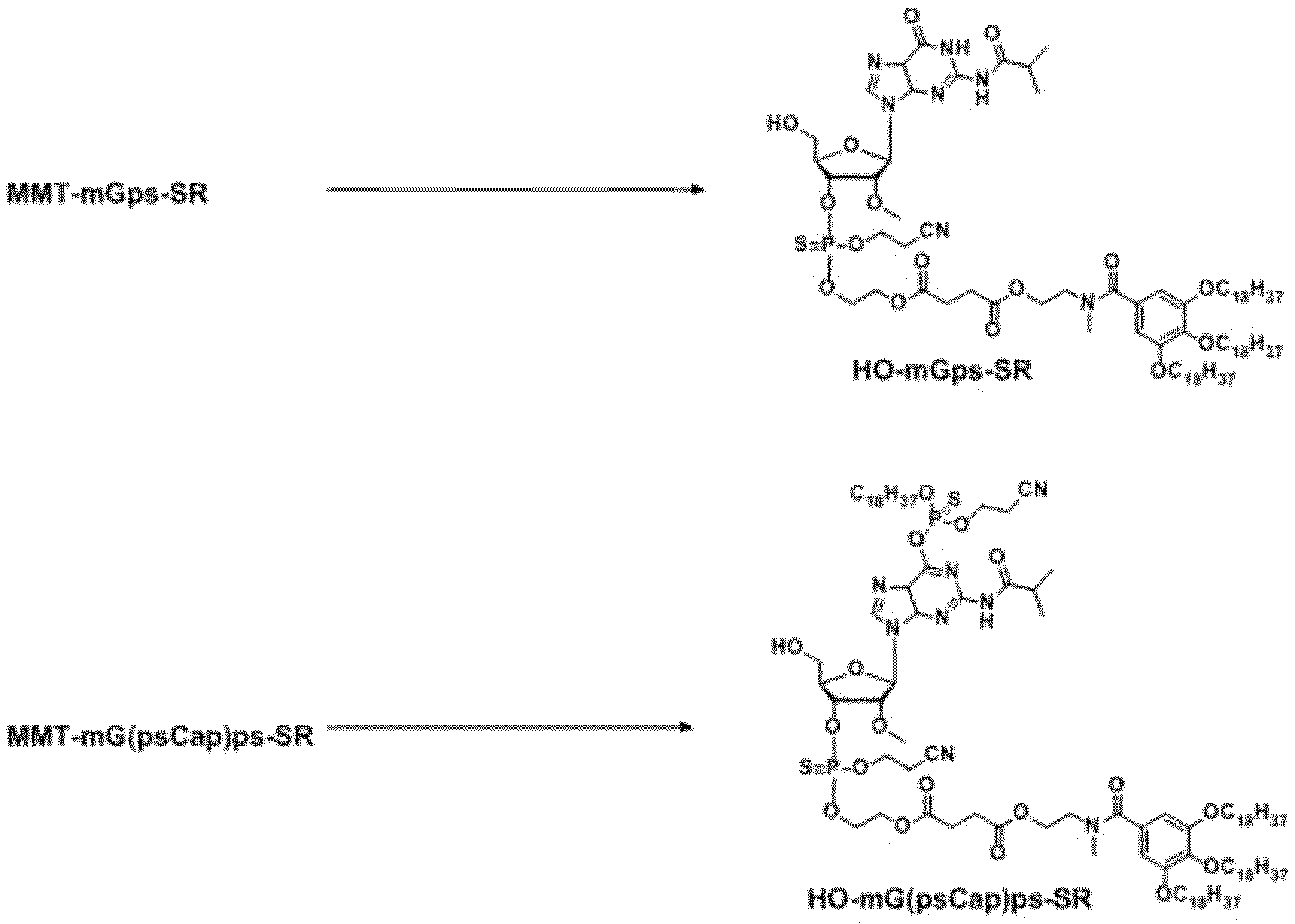
反応式VI
化合物HO-mGps-SRの合成
MMT-mGps-SR 0.380gを50 mL ナスフラスコ内でジクロロメタン 15 mLに溶解し、そこにピロール 0.35 mL、およびトリフロロ酢酸 0.46 mLを加えた。該混合物を室温にて15分間撹拌し、その後にメタノール10 mLおよびピリジン0.97mLを加えた。該混合物を減圧下濃縮し、生成した固形物をろ過し、メタノール洗浄して減圧乾燥することで目的化合物HO-mGps-SRを0.320g得た。
1H-NMR (400 MHz CDCl3); δ 0.88 (t 9H, J = 6.8 Hz) 1.19-1.39 (m, 90H), 1.40-1.51 (m, 6H), 1.70-1.82 (m, 6H), 1.89-2.04 (m 1H), 2.58-2.73 (m, 5H), 2.75-2.85 (m, 2H), 3.07 (brs, 3H), 3.38 (s, 1.5H), 3.39 (s, 1.5H), 3.52-4.06 (m, 10H), 4.12-4.42 (m, 8H), 4.42-4.49 (m, 1H), 4.53-4.58 (m, 1H), 5.32 (dd, 1H, J = 10.8, 4.8 Hz), 5.87 (t, 1H, J = 8.0 Hz), 6.58 (s, 2H), 7.90-7.96 (m, 1H), 8.80-8.96 (m, 1H), 12.13 (s, 1H)
31P-NMR (162 MHz CDCl3); δ 67.55
MMT-mGps-SR 0.380gを50 mL ナスフラスコ内でジクロロメタン 15 mLに溶解し、そこにピロール 0.35 mL、およびトリフロロ酢酸 0.46 mLを加えた。該混合物を室温にて15分間撹拌し、その後にメタノール10 mLおよびピリジン0.97mLを加えた。該混合物を減圧下濃縮し、生成した固形物をろ過し、メタノール洗浄して減圧乾燥することで目的化合物HO-mGps-SRを0.320g得た。
1H-NMR (400 MHz CDCl3); δ 0.88 (t 9H, J = 6.8 Hz) 1.19-1.39 (m, 90H), 1.40-1.51 (m, 6H), 1.70-1.82 (m, 6H), 1.89-2.04 (m 1H), 2.58-2.73 (m, 5H), 2.75-2.85 (m, 2H), 3.07 (brs, 3H), 3.38 (s, 1.5H), 3.39 (s, 1.5H), 3.52-4.06 (m, 10H), 4.12-4.42 (m, 8H), 4.42-4.49 (m, 1H), 4.53-4.58 (m, 1H), 5.32 (dd, 1H, J = 10.8, 4.8 Hz), 5.87 (t, 1H, J = 8.0 Hz), 6.58 (s, 2H), 7.90-7.96 (m, 1H), 8.80-8.96 (m, 1H), 12.13 (s, 1H)
31P-NMR (162 MHz CDCl3); δ 67.55
化合物HO-mGps-SR, HO-mG(psCap)ps-SR混合物の合成
化合物MMT-mGps-SR、およびMMT-mG(psCap)ps-SRの混合物0.380gから上記反応と同様にしてHO-mGps-SR、およびHO-mG(psCap)ps-SRの混合物を0.322g得た。
31P-NMR (162 MHz CDCl3); δ 51.81, 67.55, 67.13
化合物MMT-mGps-SR、およびMMT-mG(psCap)ps-SRの混合物0.380gから上記反応と同様にしてHO-mGps-SR、およびHO-mG(psCap)ps-SRの混合物を0.322g得た。
31P-NMR (162 MHz CDCl3); δ 51.81, 67.55, 67.13
実施例12
NMR tube中でのグアニン6位O-高脂溶性アミダイト アダクトの求核剤による分解の観測
溶液A:0.057M MMT-mGps-SRのCDCl3溶液、及び溶液B:0.9M ETTのCDCl3溶液を調製した。
C18H37O-ホスホラミダイト22.1ulをNMRチューブに分注し、溶液A 437.5ulを加えよく混合後、溶液B 125ulを加え攪拌した。溶液B添加5分後、下記各々の化合物に特有の31Pシグナルについて31PNMRを測定した。
C18H37O-ホスホラミダイト-ETT adduct 由来のシグナル124.9ppmの積分値
MMT-mG(pCap)ps-SR由来のシグナル133.5ppmの積分値
C18H37O-ホスホラミダイト由来のシグナル146.6ppmの積分値
各々の積分値を商の分子にし、MMT-mGps-SR由来のシグナル68.5ppmの積分値を商の分母にした商の値を求め、以下の各種求核剤(ホスホラミダイトに対して2等量)を添加攪拌後5分後、31PNMRを測定し、各々に対応する商の値を比較する事により、それぞれの残存比をもとめた。尚、有機塩基2, 4, 6-コリジン(3等量)を加える場合には、各種求核剤を加える前に添加し、攪拌した。
結果を表3に示す。

酢酸及びN-ヒドロキシスクシンミドは、各々のコリジンの混合溶液を調製後添加した。
この結果、酸性度の低い求核剤全てにおいて、効率よいmG(pCap)由来のシグナルの消失が確認された。また、予め有機塩基2, 4, 6-コリジンを添加し、ETT(酸)による活性化を弱めた状態でも効率よく、mG(pCap)由来のシグナルを消失できることが分かり、本反応が、求核剤がクエンチ剤として作用する反応ではないことを見出した。
NMR tube中でのグアニン6位O-高脂溶性アミダイト アダクトの求核剤による分解の観測
溶液A:0.057M MMT-mGps-SRのCDCl3溶液、及び溶液B:0.9M ETTのCDCl3溶液を調製した。
C18H37O-ホスホラミダイト22.1ulをNMRチューブに分注し、溶液A 437.5ulを加えよく混合後、溶液B 125ulを加え攪拌した。溶液B添加5分後、下記各々の化合物に特有の31Pシグナルについて31PNMRを測定した。
C18H37O-ホスホラミダイト-ETT adduct 由来のシグナル124.9ppmの積分値
MMT-mG(pCap)ps-SR由来のシグナル133.5ppmの積分値
C18H37O-ホスホラミダイト由来のシグナル146.6ppmの積分値
各々の積分値を商の分子にし、MMT-mGps-SR由来のシグナル68.5ppmの積分値を商の分母にした商の値を求め、以下の各種求核剤(ホスホラミダイトに対して2等量)を添加攪拌後5分後、31PNMRを測定し、各々に対応する商の値を比較する事により、それぞれの残存比をもとめた。尚、有機塩基2, 4, 6-コリジン(3等量)を加える場合には、各種求核剤を加える前に添加し、攪拌した。
結果を表3に示す。
この結果、酸性度の低い求核剤全てにおいて、効率よいmG(pCap)由来のシグナルの消失が確認された。また、予め有機塩基2, 4, 6-コリジンを添加し、ETT(酸)による活性化を弱めた状態でも効率よく、mG(pCap)由来のシグナルを消失できることが分かり、本反応が、求核剤がクエンチ剤として作用する反応ではないことを見出した。
実施例13
キャッピングによる、N-1オリゴヌクレオチド副生成物の残存量および求核剤存在下における硫化効率の検証
キャッピング試薬の使用によるN-1オリゴヌクレオチド副生成物の生成の抑制、および求核剤の使用による硫化反応の効率の保持を調べるための反応式を、下記反応式VIIに示す。
反応式VII

キャッピングによる、N-1オリゴヌクレオチド副生成物の残存量および求核剤存在下における硫化効率の検証
キャッピング試薬の使用によるN-1オリゴヌクレオチド副生成物の生成の抑制、および求核剤の使用による硫化反応の効率の保持を調べるための反応式を、下記反応式VIIに示す。
反応式VII
100mLナスフラスコ中の化合物HO-dTpo-SR 0.74 gに、アルゴン雰囲気下、脱水ジクロロメタン10 mL、およびDMT-mUアミダイト 0.42 g を加え、該溶解液にETT 122 mg を加え、該混合物を室温にて2時間撹拌した。該反応液に、2-シアノエチル オクタデシル ジイソプロピルホスホラミダイト 71 uL を加え、反応混合物を45分間撹拌した。反応終了後に、該反応混合物にエタノール 146 uL を加え、該混合物を室温にて10分間撹拌した。該混合物に、ピリジン:アセトニトリル=6:4(v/v)の混合溶液 (5 mL)およびDDTT 335 mg を加え、該混合物を室温にて15分間撹拌した。該反応混合物に、トリメチルホスファイト 133 uL を加え、該混合物を更に15分間撹拌した。得られた反応混合物をジクロロメタンで希釈後に、メタノール 27 mLを加え、混合物を減圧下濃縮した。生成した固形物を濾過し、メタノール洗浄することによって、DMT-mUps-dTpo-SRを1.08 g得た。
化合物DMT-mUps-dTpo-SR 4 mgをバイアルに加え、28%アンモニア水500 uLを加えて、55℃で8時間放置して生じるDMT-mUps-dTpo-EtOHの溶液について、HPLCを用いて、別途核酸合成機で合成したDMT-mUpo-dTpo-EtOH及びDMT-dTps-EtOHを標品として、硫化効率およびN-1生成量を求めた。その結果、DMT-mUpo-dTpo-EtOH、DMT-dTps-EtOH(N-1副生成物)の生成量は目的硫化物DMT-mUps-dTpo-EtOHに対してともに0.1%以下であった。その結果、エタノール添加により硫化効率は落ちないこと及び、キャッピング剤添加により効率よくN-1の生成が抑制されていることが分かった。
化合物DMT-mUps-dTpo-SR 4 mgをバイアルに加え、28%アンモニア水500 uLを加えて、55℃で8時間放置して生じるDMT-mUps-dTpo-EtOHの溶液について、HPLCを用いて、別途核酸合成機で合成したDMT-mUpo-dTpo-EtOH及びDMT-dTps-EtOHを標品として、硫化効率およびN-1生成量を求めた。その結果、DMT-mUpo-dTpo-EtOH、DMT-dTps-EtOH(N-1副生成物)の生成量は目的硫化物DMT-mUps-dTpo-EtOHに対してともに0.1%以下であった。その結果、エタノール添加により硫化効率は落ちないこと及び、キャッピング剤添加により効率よくN-1の生成が抑制されていることが分かった。
実施例14
無水酢酸キャッピングによるアデニン塩基のジアシル化の検証
無水酢酸を用いるキャッピングによる、アデニン塩基のジアシル化の生成を調べるための反応式を、下記反応式VIIIに示す。
反応式VIII

無水酢酸キャッピングによるアデニン塩基のジアシル化の検証
無水酢酸を用いるキャッピングによる、アデニン塩基のジアシル化の生成を調べるための反応式を、下記反応式VIIIに示す。
反応式VIII
化合物 DMT-dA-Sucの合成
化合物DMT-dA 13.2g(carbosynth社製)を脱水ジクロロメタン100mLに溶解し、該溶液に、トリエチルアミン8.4mLおよび無水コハク酸 3.0gを加えた。該混合物を室温にて終夜攪拌し、該反応混合物にメタノール2.0mLを加え、該混合物を更に終夜攪拌した。得られた混合溶液をリン酸トリエチルアミン緩衝液(pH 約6.8)で洗浄し、該混合溶液を減圧濃縮し、続いて減圧下で終夜留去することによって揮発成分を除いて、DMT-dA-Suc17.7g を得た。トリエチルアミンの存在比は1H NMR積分値から概算した。
1H-NMR (400 MHz CDCl3); δ 1.19 (t, 9 H, J = 7.2 Hz), 2.54-2.75 (m, 5H), 2.92(q, 6H, d=7.2), 2.96-3.05 (m, 1H), 3.38-3.49 (m, 1H), 3.74(s, 6H) 4.33-4.38 (m, 1H), 5.52-5.58 (m, 1H), 6.53 (dd, 1H, J=8.4, 5.2 Hz), 6.75-6.83 (m, 4H), 7.16-7.32 (m, 5H), 7.36-7.41 (m. 2H), 7.50-7.56 (m. 2H), 7.57-7.64 (m. 1H), 8.00-8.06 (m. 2H), 8.19 (s. 1H), 8.73 (s. 1H), 9.03 (brs. 1H)
化合物DMT-dA 13.2g(carbosynth社製)を脱水ジクロロメタン100mLに溶解し、該溶液に、トリエチルアミン8.4mLおよび無水コハク酸 3.0gを加えた。該混合物を室温にて終夜攪拌し、該反応混合物にメタノール2.0mLを加え、該混合物を更に終夜攪拌した。得られた混合溶液をリン酸トリエチルアミン緩衝液(pH 約6.8)で洗浄し、該混合溶液を減圧濃縮し、続いて減圧下で終夜留去することによって揮発成分を除いて、DMT-dA-Suc17.7g を得た。トリエチルアミンの存在比は1H NMR積分値から概算した。
1H-NMR (400 MHz CDCl3); δ 1.19 (t, 9 H, J = 7.2 Hz), 2.54-2.75 (m, 5H), 2.92(q, 6H, d=7.2), 2.96-3.05 (m, 1H), 3.38-3.49 (m, 1H), 3.74(s, 6H) 4.33-4.38 (m, 1H), 5.52-5.58 (m, 1H), 6.53 (dd, 1H, J=8.4, 5.2 Hz), 6.75-6.83 (m, 4H), 7.16-7.32 (m, 5H), 7.36-7.41 (m. 2H), 7.50-7.56 (m. 2H), 7.57-7.64 (m. 1H), 8.00-8.06 (m. 2H), 8.19 (s. 1H), 8.73 (s. 1H), 9.03 (brs. 1H)
化合物 DMT-dA-Su-Rの合成
化合物III 15.7g、化合物DMT-dA-Suc 17.7g、に脱水クロロホルム 100mLを加え、更に該混合液にジイソプロピルエチルアミン4.33mL、 HBTU 9.42gおよびDMAP 3.04gを加えた。該混合物を45℃において一晩攪拌した。得られた反応混合物にメタノール 100mLを加え、該混合物を減圧下濃縮した。生じた固形物を濾過し、メタノール洗浄し、減圧下で乾燥して、固形物である化合物DMT-dA-Su-Rを27.9g得た。
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 9 H, J = 6.4 Hz), 1.20-1.38 (m, 84H), 1.41-1.50 (m, 6H), 1.68-1.84 (m, 6H), 2.62-2.74(m, 5H), 2.99-3.12(m, 4H), 3.42-3.52 (m, 3H), 3.68-3.79 (m, 1H) 3.79 (s, 6H), 3.92-3.98 (m, 6H), 4.10-4.51 (m, 3H), 5.53-5.62 (m, 1H), 6.52 (dd, 1H J = 8.4, 5.6 Hz), 6.58 (s, 2H), 6.74-6.84 (m, 4H), 7.16-7.31 (m, 5H), 7.34-7.41 (m. 2H), 7.50-7.58 (m. 2H), 7.59-7.65 (m. 1H), 8.00-8.06 (m. 2H), 8.18 (s. 1H), 8.74 (s. 1H), 8.94(brs. 1H)
化合物III 15.7g、化合物DMT-dA-Suc 17.7g、に脱水クロロホルム 100mLを加え、更に該混合液にジイソプロピルエチルアミン4.33mL、 HBTU 9.42gおよびDMAP 3.04gを加えた。該混合物を45℃において一晩攪拌した。得られた反応混合物にメタノール 100mLを加え、該混合物を減圧下濃縮した。生じた固形物を濾過し、メタノール洗浄し、減圧下で乾燥して、固形物である化合物DMT-dA-Su-Rを27.9g得た。
1H-NMR (400 MHz CDCl3); δ 0.88 (t, 9 H, J = 6.4 Hz), 1.20-1.38 (m, 84H), 1.41-1.50 (m, 6H), 1.68-1.84 (m, 6H), 2.62-2.74(m, 5H), 2.99-3.12(m, 4H), 3.42-3.52 (m, 3H), 3.68-3.79 (m, 1H) 3.79 (s, 6H), 3.92-3.98 (m, 6H), 4.10-4.51 (m, 3H), 5.53-5.62 (m, 1H), 6.52 (dd, 1H J = 8.4, 5.6 Hz), 6.58 (s, 2H), 6.74-6.84 (m, 4H), 7.16-7.31 (m, 5H), 7.34-7.41 (m. 2H), 7.50-7.58 (m. 2H), 7.59-7.65 (m. 1H), 8.00-8.06 (m. 2H), 8.18 (s. 1H), 8.74 (s. 1H), 8.94(brs. 1H)
化合物DMT-diacyl-dA-Su-R、および化合物 DMT-dA-Su-Rの混合物の合成
DMT-dA-Su-Rを27.9gに脱水クロロホルム100mLを加え、更に2,4,6-トリメチルピリジン 45mL、1-メチルイミダゾール 30mLおよび無水酢酸 30mLを加え、該混合物を一晩攪拌した。得られた反応混合物にメタノール 100mLを加え、減圧下濃縮した。生じた固形物を濾過し、メタノール洗浄し、減圧下乾燥して、化合物DMT-diacyl-dA-Su-Rおよび化合物 DMT-dA-Su-Rの混合物を27.8g得た。
DMT-dA-Su-Rを27.9gに脱水クロロホルム100mLを加え、更に2,4,6-トリメチルピリジン 45mL、1-メチルイミダゾール 30mLおよび無水酢酸 30mLを加え、該混合物を一晩攪拌した。得られた反応混合物にメタノール 100mLを加え、減圧下濃縮した。生じた固形物を濾過し、メタノール洗浄し、減圧下乾燥して、化合物DMT-diacyl-dA-Su-Rおよび化合物 DMT-dA-Su-Rの混合物を27.8g得た。
化合物DMT-diacyl-dA-Su-Rおよび化合物 DMT-dA-Su-Rの混合物(上段)、化合物 DMT-dA-Su-R(下段)のNMR比較図を図2に示す。
8割程度のジアシル化された化合物DMT-diacyl-dA-Su-Rと、2割程度の原料化合物 DMT-dA-Su-Rの混合物であることが分かり、無水酢酸によるキャッピングでは、アデニン塩基のジアシル化生成物を含む混合物を生じることが分かった。このことから、オリゴヌクレオチド鎖長が長くなるに従い、オリゴヌクレオチド生成物の組成がより複雑化する事が容易に予想できる。この結果、無水酢酸によるキャッピング工程が、擬似固相保護基を用いた液相法によるオリゴヌクレオチドの製造に導入されると、カップリング工程において反応終点の確認、即ち反応効率の確認が困難に成ることが分かった。
8割程度のジアシル化された化合物DMT-diacyl-dA-Su-Rと、2割程度の原料化合物 DMT-dA-Su-Rの混合物であることが分かり、無水酢酸によるキャッピングでは、アデニン塩基のジアシル化生成物を含む混合物を生じることが分かった。このことから、オリゴヌクレオチド鎖長が長くなるに従い、オリゴヌクレオチド生成物の組成がより複雑化する事が容易に予想できる。この結果、無水酢酸によるキャッピング工程が、擬似固相保護基を用いた液相法によるオリゴヌクレオチドの製造に導入されると、カップリング工程において反応終点の確認、即ち反応効率の確認が困難に成ることが分かった。
本発明の高脂溶性ホスホラミダイト化合物の製造法によって得られる高脂溶性ホスホラミダイト化合物を含有する溶液、または該溶液および適宜添加剤を含む組成物をキャッピング反応用試薬として用いることにより、高純度の(オリゴ)ヌクレオチドを簡便な操作で大スケールで製造することができる。また、本発明のオリゴヌクレオチドの製造方法によれば、高純度のオリゴヌクレオチドを大スケールで製造することができる。
Claims (14)
- 式(I):

R1は、置換されていてもよいC6-C30アルキルオキシ基であり、該置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、
R2は各々独立して、置換されていてもよいC1-C6アルキル基であり、該置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、そして、
R3は、置換されていてもよいC6-C30アルキルオキシ基、置換されていてもよいC1-C8アルキルオキシ基、または置換されていてもよいC2-C8アルキニルオキシ基であり、ここで、置換されていてもよいC6-C30アルキルオキシ基における置換基は、C1-C3アルキル基、またはC3-C6シクロアルキル基から選ばれる少なくとも1つの基であり、置換されていてもよいC1-C8アルキルオキシ基、または置換されていてもよいC2-C8アルキニルオキシ基における置換基はシアノ基(CN)である。]
で示される化合物の製造方法であって、以下の工程:
(1)脂肪族アルコールおよび三価リン化合物を、活性化剤または有機塩基の存在下、有機溶媒中で反応する;
(2)得られた反応混合物を、水を用いて分液洗浄する;
(3)工程(2)後の有機層を回収し、濃縮する(但し、工程(1)で使用する有機溶媒がニトリル系溶媒の場合には、工程(2)~(3)は省略してもよい);
(4)工程(3)で得られた残渣を脂肪族炭化水素系溶媒中に溶解する(但し、工程(1)で使用する有機溶媒が脂肪族炭化水素系溶媒の場合には、工程(2)~(4)は省略してもよい);
(5)工程(4)で調製した脂肪族炭化水素系溶液を、ニトリル系溶媒を用いて分液洗浄する;
(6)工程(5)後の脂肪族炭化水素系溶液を回収して、ホスホラミダイト化合物を溶液として得る、
を含む、該製造方法。 - 工程(4)で使用される脂肪族炭化水素系溶媒が、ペンタン、ヘキサン、ヘプタン、またはオクタンから少なくとも1つ選ばれ、そして工程(5)で使用されるニトリル系溶媒が、アセトニトリル、プロピオニトリル、またはベンゾニトリルから少なくとも1つ選ばれる、請求項1に記載の製造方法。
- R1が、置換されていてもよいC10-C30の第1級または第2級のアルキルオキシ基であり、そして、R3が、-OCH2CH2CN、-OCH3または-OCH2CH3で示される基である、請求項1または2記載の製造方法。
- 各R2が共にi-プロピル基である、請求項1~3のいずれか1項に記載の製造方法。
- 式(I)で示される化合物が、下記式:
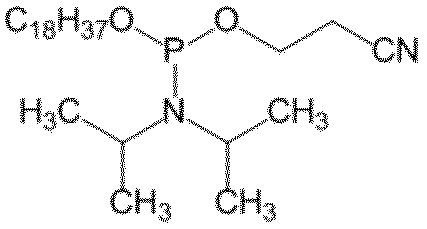
- 工程(6)で得られる溶液が、H-ホスホネート化合物、ジアミダイト化合物を含まず、実質的に純粋なホスホラミダイト化合物の溶液である、請求項1~5のいずれか1項に記載の製造方法。
- 請求項1~6のいずれか1項に記載の製造方法によって得られる化合物を含有する溶液、および適宜添加剤を含む、キャッピング反応用組成物。
- ヌクレオシド、ヌクレオチド、またはオリゴヌクレオチドから選ばれる2つ以上の化合物をカップリング反応させた後の反応溶液を、適宜アクチベータの存在下、請求項7に記載のキャッピング反応用組成物と接触させる工程を含む、キャッピング方法。
- 更に適宜、キャッピング反応工程後に、キャッピング副反応産物を求核剤と反応させることを含み、ここで、該求核剤は、アルコール、フェノール、カルボン酸、またはN-アルキルヒドロキシルアミンから選ばれる、請求項8に記載のキャッピング方法。
- キャッピング反応工程後に有機塩基を添加し、その後に求核剤と反応させる工程を含む、請求項9に記載のキャッピング方法。
- 工程(a):非極性溶媒中において、3’位水酸基もしくは核酸塩基を介して擬似固相保護基で修飾され且つ5’位水酸基が酸性条件下で除去可能な一時保護基でブロックされたn量体(ここで、nは、1以上の任意の整数を示す。)もしくは、n=0の場合、水酸基を2個以上持つリンカーの1つの水酸基が擬似固相保護基で修飾され且つ別の水酸基が酸性条件下で除去可能な一時保護基でブロックされた化合物を、酸の単独もしくはカチオン捕捉剤の共存下で反応させて、水酸基の一時保護基をデブロックし、その後に極性溶媒を用いて該擬似固相保護基を含有する化合物を回収する工程(デブロックおよび晶析工程);
工程(b):工程(a)の回収物を乾燥後、非極性溶媒中において、3’位水酸基がホスホラミダイト化され且つ5’位水酸基が酸性条件下で除去可能な一時保護基で保護されたm+1個重合オリゴヌクレオチド(ここで、mは、0以上の任意の整数を示す。)およびアクチベータを添加して、工程(a)で得られた5’位水酸基の一時保護基がデブロックされたn個重合オリゴヌクレオチドと、その5’位水酸基を介してホスファイトトリエステル結合により縮合させる工程(カップリング工程);
工程(c):工程(b)の反応液に、請求項1~6に記載のいずれか1項に記載の製造方法によって得られる高脂溶性ホスホラミダイト化合物を含有する溶液、または請求項7に記載のキャッピング反応用組成物を添加し、工程(b)において未反応であった化合物について、その5’位水酸基を介して該高脂溶性化合物のホスファイトトリエステル結合により縮合(キャッピング)させる工程(キャッピング反応工程);
工程(d):工程(c)のキャッピング副反応産物に求核剤を添加して反応させることを含み、ここで、該求核剤は、アルコール、フェノール、カルボン酸またはN-アルキルヒドロキシルアミンから選ばれる、工程(キャッピング反応の後処理工程);
工程(e):工程(d)の反応液に、酸化剤または硫化剤を添加して、工程(b)~(d)で得られたm+1+n量体のオリゴヌクレオチドのホスファイトトリエステル結合を、ホスフェートトリエステル結合またはチオホスフェートトリエステル結合にそれぞれ変換する工程(酸化または硫化反応の工程);
更に適宜、工程(f):工程(e)で得られた反応液に極性溶媒を添加して、m+1+n個重合オリゴヌクレオチドを沈殿させて、固液分離により取得する工程(単離工程);
更に適宜、工程(g):工程(f)で得られたm+1+n個重合オリゴヌクレオチドの保護基を全て除去する工程(脱保護工程);
を含む、オリゴヌクレオチドの製造方法。 - 式(II):
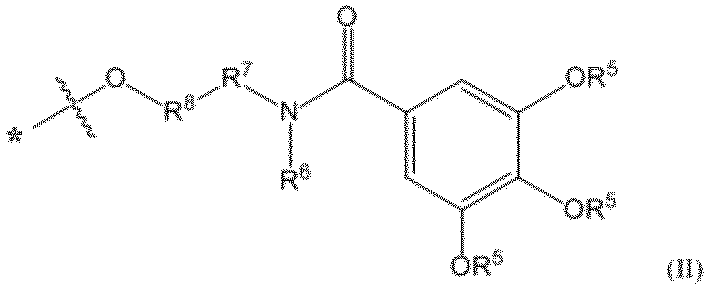
R5は各々独立して、置換されていてもよいC6-C30のアルキル基であり、
R6は、置換されていてもよいC1-C6アルキル基、置換されていてもよいC3-C6シクロアルキル基、置換されていてもよい非芳香族性複素環式基、置換されていてもよいアリール基、置換されていてもよいヘテロアリール基、置換されていてもよいアラルキル基、または置換されていてもよいヘテロアリールアルキル基から選ばれ、
R7は、置換されていてもよいC1-C6アルキレン基、または置換されていてもよいC1-C6アルキレンオキシ基であり、
R8は、置換されていてもよいC1-C6アルキレン基であり、あるいは、
R6およびR7は、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、または置換されていてもよいヘテロアリール基を形成してもよく、あるいは、
R6、R7およびR8は、R6が結合する窒素原子と一緒になって、置換されていてもよい非芳香族性複素環式基、置換されていてもよい非芳香族性複素環アルキル基、置換されていてもよいヘテロアリール基、または置換されていてもよいヘテロアリールアルキル基を形成してもよく、そして、
*は、Lとの結合位置を示す]
で示される、擬似固相保護基。 - 式(III):
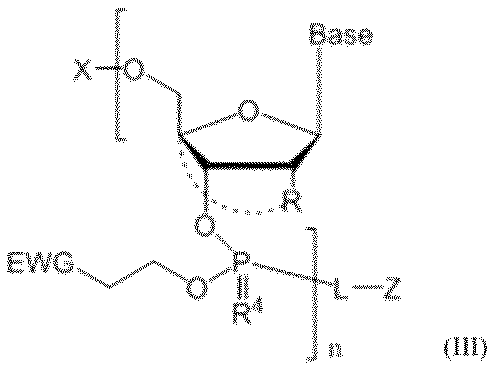
Zは、請求項12に記載の式(II)で示される擬似固相保護基であり、
Lは、リンカーであり、
Baseは、各々独立して保護されていてもよい核酸塩基であり、
n個のEWGは、各々独立して電子吸引性基であり、
n個のR4は、各々独立して酸素原子または硫黄原子であり、
n個のRは、各々独立して水素原子、ハロゲン原子、保護基で置換されていてもよいヒドロキシ基、置換されていてもよいC1-C6アルコキシ基、4位炭素原子に架橋する有機基、または架橋構造型人工核酸基であり、
nは、0以上の任意の整数であり、そして、
Xは、酸により脱保護可能な一時保護基である]
で示される、ヌクレオチド化合物。 - Lが、下記式:

*は疎水性基との結合位置、および**はリン原子との結合位置を示し、
Baseは、各々独立して保護されていてもよい核酸塩基であり、そして、
Rは、各々独立して水素原子、ハロゲン原子、保護基で置換されていてもよいヒドロキシ基、置換されていてもよいC1-C6アルコキシ基、4位炭素原子に架橋する有機基、または架橋構造型人工核酸基である]
から選ばれる、請求項13に記載のヌクレオチド化合物。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019562076A JP7229539B2 (ja) | 2017-12-27 | 2018-12-26 | 高脂溶性ホスホラミダイトの製造 |
| US16/958,100 US11891412B2 (en) | 2017-12-27 | 2018-12-26 | Production of highly fat-soluble phosphoramidite |
| EP18895849.0A EP3733680A4 (en) | 2017-12-27 | 2018-12-26 | Production of highly fat-soluble phosphoramidite |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017252107 | 2017-12-27 | ||
| JP2017-252107 | 2017-12-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019131719A1 true WO2019131719A1 (ja) | 2019-07-04 |
Family
ID=67067449
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/047748 WO2019131719A1 (ja) | 2017-12-27 | 2018-12-26 | 高脂溶性ホスホラミダイトの製造 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US11891412B2 (ja) |
| EP (1) | EP3733680A4 (ja) |
| JP (1) | JP7229539B2 (ja) |
| WO (1) | WO2019131719A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022172994A1 (ja) | 2021-02-12 | 2022-08-18 | 味の素株式会社 | オリゴヌクレオチドの製造方法 |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998020018A1 (en) * | 1996-11-07 | 1998-05-14 | Novartis Ag | Process for the preparation of an oligomeric compound |
| JP2001253986A (ja) | 1998-03-23 | 2001-09-18 | Denki Kagaku Kogyo Kk | 樹脂組成物 |
| WO2004058779A1 (en) | 2002-12-24 | 2004-07-15 | Rhodia Consumer Specialties Limited | Process for preparing phosphorodiamidites |
| WO2005008859A1 (en) | 2003-07-17 | 2005-01-27 | Abb Sp. Z O. O. | A protection system for medium-voltage potential transformers |
| JP2010024228A (ja) | 2008-06-20 | 2010-02-04 | Mitsubishi Chemicals Corp | リン化合物の製造方法及びアリル化合物誘導体の製造方法 |
| JP2010513358A (ja) | 2006-12-22 | 2010-04-30 | エフ.ホフマン−ラ ロシュ アーゲー | オリゴヌクレオチドを合成及び精製するための化合物及び方法 |
| JP2010275254A (ja) | 2009-05-29 | 2010-12-09 | Tokyo Univ Of Agriculture & Technology | 疎水性基結合ヌクレオシド、疎水性基結合ヌクレオシド溶液、及び疎水性基結合オリゴヌクレオチド合成方法 |
| JP2011121881A (ja) | 2009-12-09 | 2011-06-23 | Nippon Shinyaku Co Ltd | ホスホロアミダイト化合物の製造法 |
| WO2012157723A1 (ja) | 2011-05-17 | 2012-11-22 | 味の素株式会社 | オリゴヌクレオチドの製造方法 |
| WO2016006697A1 (ja) | 2014-07-10 | 2016-01-14 | 賢二 中野 | アンチセンス抗悪性腫瘍剤 |
| WO2017057540A1 (ja) * | 2015-09-30 | 2017-04-06 | 塩野義製薬株式会社 | 免疫賦活活性を有する核酸誘導体 |
| WO2017086397A1 (ja) * | 2015-11-17 | 2017-05-26 | 日産化学工業株式会社 | オリゴヌクレオチドの製造方法 |
| WO2017104836A1 (ja) | 2015-12-16 | 2017-06-22 | 味の素株式会社 | オリゴヌクレオチドの製造方法、およびヌクレオシド、ヌクレオチドまたはオリゴヌクレオチド |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10940201B2 (en) | 2015-09-30 | 2021-03-09 | Shionogi & Co., Ltd. | Nucleic acid derivative having immunostimulatory activity |
-
2018
- 2018-12-26 WO PCT/JP2018/047748 patent/WO2019131719A1/ja unknown
- 2018-12-26 JP JP2019562076A patent/JP7229539B2/ja active Active
- 2018-12-26 US US16/958,100 patent/US11891412B2/en active Active
- 2018-12-26 EP EP18895849.0A patent/EP3733680A4/en not_active Withdrawn
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998020018A1 (en) * | 1996-11-07 | 1998-05-14 | Novartis Ag | Process for the preparation of an oligomeric compound |
| JP2001253986A (ja) | 1998-03-23 | 2001-09-18 | Denki Kagaku Kogyo Kk | 樹脂組成物 |
| WO2004058779A1 (en) | 2002-12-24 | 2004-07-15 | Rhodia Consumer Specialties Limited | Process for preparing phosphorodiamidites |
| JP2006512386A (ja) * | 2002-12-24 | 2006-04-13 | ローディア コンシューマー スペシャルティーズ リミテッド | ホスホロジアミダイト生産方法 |
| WO2005008859A1 (en) | 2003-07-17 | 2005-01-27 | Abb Sp. Z O. O. | A protection system for medium-voltage potential transformers |
| JP2010513358A (ja) | 2006-12-22 | 2010-04-30 | エフ.ホフマン−ラ ロシュ アーゲー | オリゴヌクレオチドを合成及び精製するための化合物及び方法 |
| JP2010024228A (ja) | 2008-06-20 | 2010-02-04 | Mitsubishi Chemicals Corp | リン化合物の製造方法及びアリル化合物誘導体の製造方法 |
| JP2010275254A (ja) | 2009-05-29 | 2010-12-09 | Tokyo Univ Of Agriculture & Technology | 疎水性基結合ヌクレオシド、疎水性基結合ヌクレオシド溶液、及び疎水性基結合オリゴヌクレオチド合成方法 |
| JP2011121881A (ja) | 2009-12-09 | 2011-06-23 | Nippon Shinyaku Co Ltd | ホスホロアミダイト化合物の製造法 |
| WO2012157723A1 (ja) | 2011-05-17 | 2012-11-22 | 味の素株式会社 | オリゴヌクレオチドの製造方法 |
| WO2016006697A1 (ja) | 2014-07-10 | 2016-01-14 | 賢二 中野 | アンチセンス抗悪性腫瘍剤 |
| WO2017057540A1 (ja) * | 2015-09-30 | 2017-04-06 | 塩野義製薬株式会社 | 免疫賦活活性を有する核酸誘導体 |
| WO2017086397A1 (ja) * | 2015-11-17 | 2017-05-26 | 日産化学工業株式会社 | オリゴヌクレオチドの製造方法 |
| WO2017104836A1 (ja) | 2015-12-16 | 2017-06-22 | 味の素株式会社 | オリゴヌクレオチドの製造方法、およびヌクレオシド、ヌクレオチドまたはオリゴヌクレオチド |
Non-Patent Citations (14)
| Title |
|---|
| "Protective Group In Organic Synthesis", 1999, JOHN WILLY & SONS |
| CHEM. EUR. J., vol. 19, 2013, pages 8615 - 8620 |
| MICKLEFIELD, CURRENT MEDICINAL CHEMISTRY, vol. 8, 2001, pages 1157 - 1170 |
| NATT FRANÇOIS ET AL.: "Lipocap: a Lipophilic Phosphoramidite-based Capping Reagent", TETRAHEDRON, vol. 53, no. 28, 1997, pages 9629 - 9636, XP004105893 * |
| NUCLEIC ACIDS RESEARCH, vol. 14, no. 16, 1986, pages 6453 - 6470 |
| NUCLEIC ACIDS RESEARCH, vol. 18, 1990, pages 3155 - 3159 |
| NUCLEIC ACIDS RESEARCH, vol. 21, no. 5, 1993, pages 1213 - 1217 |
| PALTE MICHAEL J., ET AL.: "Interaction of Nucleic Acids with the Glycocalyx", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 134, no. 14, 8 March 2012 (2012-03-08), pages 6218 - 6223, XP055623404, ISSN: 0002-7863 * |
| See also references of EP3733680A4 |
| TETRAHEDRON LETT, vol. 32, no. 27, 1991, pages 3251 - 3254 |
| TETRAHEDRON LETT, vol. 35, no. 46, 1994, pages 8565 - 8568 |
| TETRAHEDRON LETT, vol. 52, 2011, pages 434 - 437 |
| TETRAHEDRON LETTERS, vol. 50, no. 15, 2009, pages 1751 - 1753 |
| TETRAHEDRON, vol. 53, no. 28, 1997, pages 9629 - 9636 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022172994A1 (ja) | 2021-02-12 | 2022-08-18 | 味の素株式会社 | オリゴヌクレオチドの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3733680A1 (en) | 2020-11-04 |
| JP7229539B2 (ja) | 2023-02-28 |
| EP3733680A4 (en) | 2021-12-29 |
| JPWO2019131719A1 (ja) | 2021-03-04 |
| US11891412B2 (en) | 2024-02-06 |
| US20210087220A1 (en) | 2021-03-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6281599B2 (ja) | 擬似固相保護基およびヌクレオチド | |
| US8304532B2 (en) | Method for preparing oligonucleotides | |
| CN108473526B (zh) | 寡核苷酸的制备方法以及核苷、核苷酸或寡核苷酸 | |
| CN110831951B (zh) | 多重偶联和氧化方法 | |
| EP2753607A1 (en) | Bicyclo[6.1.0]non-4-yne reagents for chemical modification of oligonucleotides | |
| KR102531388B1 (ko) | 알콕시 페닐 유도체, 뉴클레오시드 보호체 및 뉴클레오티드 보호체, 올리고뉴클레오티드 제조 방법, 및 치환기 제거 방법 | |
| EP2748176B1 (en) | Ionic tags for synthesis of oligoribonucleotides | |
| SG188289A1 (en) | Block synthesis of oligoribonucleotides | |
| WO2019131719A1 (ja) | 高脂溶性ホスホラミダイトの製造 | |
| EP1317466B1 (en) | Synthons for oligonucleotide synthesis | |
| JP2016204316A (ja) | 核酸固相合成用リンカー及び担体 | |
| JP7452549B2 (ja) | ホスホロアミダイト活性化剤 | |
| JP7433684B1 (ja) | 疑似固相保護基、それを用いたヌクレオシド保護体又はオリゴヌクレオチド保護体、オリゴアミダイト前駆体の製造方法 | |
| US20230192751A1 (en) | Method for the preparation of oligonucleotides | |
| WO2024083746A1 (en) | Method and composition for oligonucleotide synthesis | |
| EP4043473A1 (en) | Method for producing nucleic acid oligomers | |
| Yagodkin et al. | Universal linker phosphoramidite | |
| JP2019119739A (ja) | ヌクレオシド誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18895849 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019562076 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2018895849 Country of ref document: EP Effective date: 20200727 |