WO2019000985A1 - 石墨烯复合材料及其制备方法 - Google Patents
石墨烯复合材料及其制备方法 Download PDFInfo
- Publication number
- WO2019000985A1 WO2019000985A1 PCT/CN2018/077169 CN2018077169W WO2019000985A1 WO 2019000985 A1 WO2019000985 A1 WO 2019000985A1 CN 2018077169 W CN2018077169 W CN 2018077169W WO 2019000985 A1 WO2019000985 A1 WO 2019000985A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- graphene
- parts
- graphene oxide
- mass
- pet
- Prior art date
Links
Images







Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/022—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the choice of material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/03—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the shape of the extruded material at extrusion
- B29C48/07—Flat, e.g. panels
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/15—Nano-sized carbon materials
- C01B32/182—Graphene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/12—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from polycarboxylic acids and polyhydroxy compounds
- C08G63/16—Dicarboxylic acids and dihydroxy compounds
- C08G63/18—Dicarboxylic acids and dihydroxy compounds the acids or hydroxy compounds containing carbocyclic rings
- C08G63/181—Acids containing aromatic rings
- C08G63/183—Terephthalic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/78—Preparation processes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/78—Preparation processes
- C08G63/82—Preparation processes characterised by the catalyst used
- C08G63/85—Germanium, tin, lead, arsenic, antimony, bismuth, titanium, zirconium, hafnium, vanadium, niobium, tantalum, or compounds thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G69/00—Macromolecular compounds obtained by reactions forming a carboxylic amide link in the main chain of the macromolecule
- C08G69/02—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids
- C08G69/08—Polyamides derived from amino-carboxylic acids or from polyamines and polycarboxylic acids derived from amino-carboxylic acids
- C08G69/14—Lactams
- C08G69/16—Preparatory processes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G83/00—Macromolecular compounds not provided for in groups C08G2/00 - C08G81/00
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/005—Reinforced macromolecular compounds with nanosized materials, e.g. nanoparticles, nanofibres, nanotubes, nanowires, nanorods or nanolayered materials
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/18—Manufacture of films or sheets
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
- C08K3/042—Graphene or derivatives, e.g. graphene oxides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/16—Solid spheres
- C08K7/18—Solid spheres inorganic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L67/00—Compositions of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Compositions of derivatives of such polymers
- C08L67/02—Polyesters derived from dicarboxylic acids and dihydroxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L77/00—Compositions of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Compositions of derivatives of such polymers
- C08L77/02—Polyamides derived from omega-amino carboxylic acids or from lactams thereof
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01D—MECHANICAL METHODS OR APPARATUS IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS
- D01D1/00—Treatment of filament-forming or like material
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F1/00—General methods for the manufacture of artificial filaments or the like
- D01F1/02—Addition of substances to the spinning solution or to the melt
- D01F1/07—Addition of substances to the spinning solution or to the melt for making fire- or flame-proof filaments
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F1/00—General methods for the manufacture of artificial filaments or the like
- D01F1/02—Addition of substances to the spinning solution or to the melt
- D01F1/09—Addition of substances to the spinning solution or to the melt for making electroconductive or anti-static filaments
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F1/00—General methods for the manufacture of artificial filaments or the like
- D01F1/02—Addition of substances to the spinning solution or to the melt
- D01F1/10—Other agents for modifying properties
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F1/00—General methods for the manufacture of artificial filaments or the like
- D01F1/02—Addition of substances to the spinning solution or to the melt
- D01F1/10—Other agents for modifying properties
- D01F1/106—Radiation shielding agents, e.g. absorbing, reflecting agents
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F6/00—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof
- D01F6/58—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof from homopolycondensation products
- D01F6/62—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof from homopolycondensation products from polyesters
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F6/00—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof
- D01F6/96—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof from other synthetic polymers
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F8/00—Conjugated, i.e. bi- or multicomponent, artificial filaments or the like; Manufacture thereof
- D01F8/04—Conjugated, i.e. bi- or multicomponent, artificial filaments or the like; Manufacture thereof from synthetic polymers
- D01F8/14—Conjugated, i.e. bi- or multicomponent, artificial filaments or the like; Manufacture thereof from synthetic polymers with at least one polyester as constituent
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F8/00—Conjugated, i.e. bi- or multicomponent, artificial filaments or the like; Manufacture thereof
- D01F8/18—Conjugated, i.e. bi- or multicomponent, artificial filaments or the like; Manufacture thereof from other substances
-
- D—TEXTILES; PAPER
- D03—WEAVING
- D03D—WOVEN FABRICS; METHODS OF WEAVING; LOOMS
- D03D15/00—Woven fabrics characterised by the material, structure or properties of the fibres, filaments, yarns, threads or other warp or weft elements used
- D03D15/40—Woven fabrics characterised by the material, structure or properties of the fibres, filaments, yarns, threads or other warp or weft elements used characterised by the structure of the yarns or threads
- D03D15/47—Woven fabrics characterised by the material, structure or properties of the fibres, filaments, yarns, threads or other warp or weft elements used characterised by the structure of the yarns or threads multicomponent, e.g. blended yarns or threads
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2067/00—Use of polyesters or derivatives thereof, as moulding material
- B29K2067/003—PET, i.e. poylethylene terephthalate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2367/00—Characterised by the use of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Derivatives of such polymers
- C08J2367/02—Polyesters derived from dicarboxylic acids and dihydroxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2377/00—Characterised by the use of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Derivatives of such polymers
- C08J2377/02—Polyamides derived from omega-amino carboxylic acids or from lactams thereof
Definitions
- the invention belongs to the field of composite materials, in particular to a graphene composite material and a preparation method thereof, and comprises a graphene/PET nano composite material, a graphene polyester composite fiber for a cord, a graphene/polyester composite fabric, a graphene/PET composite film, Graphene/PET composite sheet, graphene modified polyester blend fabric, graphene modified flame retardant anti-UV polyester fiber.
- PET Polyethylene terephthalate PET is a very important polymer material, which occupies a very large proportion in daily life, such as disposable water bottles, packaging materials, automotive plastics, etc. Polyester is commonly used, so PET is widely present in our lives. If the performance of PET can be further improved or given new performance, it will not only further broaden the application range of PET, but also bring more convenience to human society. In recent years, researchers have upgraded the performance of PET by regulating PET molecular structure, copolymerization, introduction of enhanced phase, synthesis of microscopic morphology such as islands, and control of crystallization behavior, and achieved remarkable results.
- Polyester is an important variety in synthetic fibers. It is made from polyethylene terephthalate (PET), which is made by spinning and post-treatment. Because of its chemical properties, high mechanical strength, light weight, good thermal stability, good hygienic performance, high transparency and easy processing, it is widely used in clothing, bedding, various decorative fabrics, special fabrics for national defense. Such as textiles and other industrial fiber products. Among them, PET industrial yarn is widely used in automobile tires due to its low cost and high strength. In order to further enhance the strength of PET industrial yarn, people use different means to improve.
- Patent 201310043077.2 “Production method of melt direct spinning high modulus low shrinkage polyester industrial filament” adopts the methods of melt liquid phase thickening, melt direct spinning and two-stage drawing to obtain high modulus low shrinkage polyester filament. Used in fields such as cords. In addition to improving the spinning process, it is also possible to increase the strength of the filament by adding a reinforcing material to obtain better performance.
- reinforcing materials are a method that can be produced on a rapid scale and cost-effective.
- Conventional reinforcing materials include metallic materials (nanowires, nanoparticles), inorganic fillers (montmorillonite, titanium dioxide, silicon dioxide, boron nitride, etc.) and carbon. Materials (carbon black, graphite, etc.).
- metallic materials nanowires, nanoparticles
- inorganic fillers montmorillonite, titanium dioxide, silicon dioxide, boron nitride, etc.
- Carbon black, graphite, etc. There are two major defects in conventional reinforcement materials. On the one hand, high addition is required to obtain satisfactory results, but high addition is accompanied by other performance degradation, and it is difficult to achieve overall improvement in performance.
- the enhancement effect is often single. It is not possible to improve multiple performances at the same time. These problems have led to the low cost performance of conventional reinforcement materials and are unsatisfactory.
- the filling reinforcement material must also take into account the influence of dispersion uniformity
- Graphene is a two-dimensional material with atomic thickness, with high specific surface area, excellent mechanical properties, high electrical conductivity, high thermal conductivity and high barrier properties. Moreover, the addition of a small amount of graphene can simultaneously improve the properties of the material, and has an excellent cost performance, which has led to extensive research on composite materials. However, graphene is easily agglomerated and will re-form the graphite stack structure, which reduces its reinforcing effect. Although the dispersibility of graphene and the reduction of graphene stacking can be promoted by adding a dispersing agent and performing surface modification, these methods increase the cost of graphene and introduce new components.
- Patent 201510514154.7 Preparation method of graphene oxide modified PET material
- the addition of water affects the esterification and condensation, and on the other hand, the oxidation in the esterification stage.
- Graphene is reduced, which may result in stacking and reduced performance.
- Patent 201280033203.X polyethylene terephthalate-graphene nanocomposite adds graphene nanosheets to PET polymerization system, multi-layer graphene makes the addition amount higher (2-15%), and because there is no functional group In the presence of graphene, secondary stacking occurs during polymerization to form incompatible defect points.
- Patent 201610111707.9 "PET-based graphene composite material, preparation method thereof and aerostat" firstly oxidize graphene oxide with ethylene glycol, and then esterify or transesterify with PET monomer, and finally polycondensate to obtain composite material, although
- the modification method improves the compatibility of graphene and PET polymerization system, and causes covalent grafting of graphene and PET, but in the esterification process, graphene oxide will still inevitably stack, and the preparation process Complex, the overall production cost is high, not suitable for actual production.
- Patent 201510688803.5 "Preparation method of a military anti-melting antistatic high-strength flame-retardant polyester” modified graphene oxide, dried and then blended with PET to granulate, and spun, although effective for the modification of graphene oxide The agglomeration is reduced, but the agglomeration of graphene in the modified powder after drying cannot be dissociated during the melt extrusion, which may cause clogging of the spinneret and breakage.
- Patent 201610757032.5 "Graphene Polyester Monofilament” treats graphene with a silane coupling agent and then blends it with PET. The coupling agent can improve the interaction between graphene and PET, but it cannot change the state of graphene stacking, and the spinning effect is still not good.
- the preparation of graphene-based polyester fiber at this stage has not been able to fundamentally solve the problem of stacking graphene, thus greatly limiting high-speed, continuous spinning.
- the object of the present invention is to provide a graphene composite material and a preparation method thereof according to the prior art, including a graphene/PET nano composite material, a graphene polyester composite fiber for a cord, a graphene/polyester composite fabric, and a graphene/ PET composite film, graphene/PET composite sheet, graphene modified polyester blend fabric, graphene modified flame retardant anti-UV polyester fiber.
- a graphene/PET nanocomposite consisting of a single layer of graphene sheets and PET, the surface of which is connected to PET molecules by covalent bonds.
- a method for preparing a graphene/PET nanocomposite is prepared by the following steps:
- the atomization drying temperature of the step (1) is 130 to 200 °C.
- the stirring speed in the step (3) is 140 to 200 rpm.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a titanium-based catalyst including an oxide of titanium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the present invention firstly obtains pleated spherical graphene oxide microspheres by using an atomization drying method, and the PET oligomerization of the pleated spherical graphene oxide after the esterification is completed by a reasonably selected carbon-oxygen ratio and graphene oxide size.
- the material can be gradually unfolded and dissociated into flaky graphene oxide.
- the hydroxyl groups and carboxyl groups on the surface of the graphene oxide react with the PET molecules in the system, so that the PET molecules link to the graphene surface, and the two are improved.
- the compatibility of the products also contributes to the improvement of mechanical properties, electrical conductivity and the like.
- Adding graphene oxide after esterification avoids the influence on the esterification process in the first step, is more reasonable in the actual production process, has higher efficiency, lower cost, and avoids the occurrence of graphene oxide in the esterification stage.
- Stacking forms agglomerates.
- For the whole PET polymerization no substance was introduced except for pleated spheroidal graphene oxide.
- the amount of terephthalic acid, ethylene glycol, esterification catalyst and polycondensation catalyst were all in pure PET polymerization process, which minimized the introduction of graphene.
- the influence of process and equipment has broad application prospects.
- the resulting graphene/PET composite has excellent mechanical properties and electrical conductivity and can be used for the preparation of functionalized polyester fibers.
- the invention relates to a graphene polyester composite fiber for a cord, which is obtained by drying, pre-crystallizing, solid phase polycondensation, cooling and high-speed melt spinning of a graphene/PET nano composite material.
- the graphene/PET nanocomposite consists of a single layer of graphene sheet and PET, and the surface of the graphene sheet is connected to the PET molecule by a covalent bond.
- the drying temperature is 170-180 ° C
- the pre-crystallization temperature is 175-185 ° C
- the solid phase polycondensation temperature is 210-220 ° C
- the intrinsic viscosity after solid phase polycondensation is 0.9-1.2
- the cooling temperature is 60-80 ° C
- the spinning The wire temperature was 270 to 290 ° C
- the winding speed was 3,000 to 5,000 m/min
- the draft ratio was 1.5 to 4.
- graphene/PET nanocomposite is prepared by the following steps:
- the atomization drying temperature of the step (1) is 130 to 200 °C.
- the stirring speed in the step (3) is 140 to 200 rpm.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a titanium-based catalyst including an oxide of titanium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the beneficial effects are as follows: (1) The pleated spherical graphene oxide microspheres added after the esterification is completed can be gradually unfolded and dissociated into a single layer of graphene oxide, and the hydroxyl and carboxyl groups on the surface of the graphene oxide are polymerized during PET polymerization.
- the reaction of the PET molecules causes the PET molecules to branch to the graphene surface, which improves the compatibility of the two, and greatly reduces the addition amount of graphene while reducing the stack, so that the method of the invention has high cost performance.
- the viscosity of the melt can be controlled within a suitable range.
- the composite material can be continuously spun at a high speed, and the obtained fiber has high breaking strength and elongation at break, and the heat resistance of the fiber is improved.
- a multifunctional graphene/polyester composite fabric which is obtained by mixing 100 parts by mass of graphene/PET nanocomposites and 0-10 parts of auxiliary agents, spinning, cooling, oiling, drawing , adding, weaving, dyeing, finishing.
- the graphene/PET nanocomposite consists of a single layer of graphene sheet and PET, and the surface of the graphene sheet is connected to the PET molecule by a covalent bond.
- graphene/PET nanocomposite is prepared by the following steps:
- the atomization drying temperature of the step (1) is 130 to 200 °C.
- the stirring speed in the step (3) is 140 to 200 rpm.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a titanium-based catalyst including an oxide of titanium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the auxiliary agent is composed of one or more of an antioxidant, an inorganic filler, a toughening agent, and a gloss improving assistant according to any ratio.
- the spinning temperature is 270 to 290 ° C
- the winding speed is 3,000 to 5,000 m/min
- the draw ratio is 1.5 to 4 times.
- the obtained fiber has a denier of 30 to 600 D.
- the weaving method is knitting using a shuttle loom or a shuttleless loom.
- the beneficial effects are as follows: (1) The pleated spherical graphene oxide microspheres added after the esterification is completed can be gradually unfolded and dissociated into a single layer of graphene oxide, and the hydroxyl and carboxyl groups on the surface of the graphene oxide are polymerized during PET polymerization.
- the reaction of the PET molecules causes the PET molecules to branch to the graphene surface, which improves the compatibility of the two, and greatly reduces the addition amount of graphene while reducing the stack, so that the method of the invention has high cost performance.
- the composite material can be continuously spun at high speed, and the fabric obtained by weaving the fiber has good UV resistance and flame retardancy, and the addition of graphene can significantly improve the electrical conductivity of the fabric. Used as an antistatic cloth.
- the durability of the fabric is good, and the high performance can be maintained after repeated washing, exposure, and smashing.
- the fabric can be reused, and the waste cloth can be recycled and reused, and the anti-UV, flame retardant and other properties can be re-applied.
- a graphene/PET composite film obtained by co-melting and casting a film of 100 parts by mass of a graphene/PET nanocomposite and 0 to 10 parts of an auxiliary agent.
- the graphene/PET nanocomposite consists of a single layer of graphene sheet and PET, and the surface of the graphene sheet is connected to the PET molecule by a covalent bond.
- a method for preparing a graphene/PET composite film which comprises: mixing 100 parts by weight of a graphene/PET nanocomposite and 0 to 10 parts by weight of an auxiliary agent, and then forming a film by melt casting, thereby obtaining The multifunctional graphene/PET composite film of the invention.
- the auxiliary agent is composed of one or more of an antioxidant, an inorganic filler, a toughening agent, and a gloss improving assistant according to any ratio.
- the melt casting film forming temperature is 250 to 280 ° C
- the screw rotation speed is 40 to 300 rpm
- the pulling speed is 1 to 50 m/min.
- graphene/PET nanocomposite is prepared by the following steps:
- the atomization drying temperature of the step (1) is 130 to 200 °C.
- the stirring speed in the step (3) is 140 to 200 rpm.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a titanium-based catalyst including an oxide of titanium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the beneficial effects are as follows: (1) The pleated spherical graphene oxide microspheres added after the esterification is completed can be gradually unfolded and dissociated into a single layer of graphene oxide, and the hydroxyl and carboxyl groups on the surface of the graphene oxide are polymerized during PET polymerization.
- the reaction of the PET molecules causes the PET molecules to branch to the graphene surface, which improves the compatibility of the two, and greatly reduces the addition amount of graphene while reducing the stack, so that the method of the invention has high cost performance.
- the oxygen barrier property of the composite membrane is significantly improved, and it can be used as a protective material and a packaging material.
- the electrical conductivity of the composite film is significantly increased at a high addition amount and can be used as an antistatic material.
- a high-strength, melt-resistant graphene/PET composite sheet obtained by coextruding 100 parts by mass of a graphene/PET nanocomposite and 0 to 10 parts of an auxiliary agent.
- the graphene/PET nanocomposite consists of a single layer of graphene sheet and PET, and the surface of the graphene sheet is connected to the PET molecule by a covalent bond.
- a method for preparing a graphene/PET composite sheet which comprises: mixing 100 parts by weight of a graphene/PET nanocomposite and 0 to 10 parts by weight of an auxiliary agent, and then extruding, thereby obtaining the invention Graphene/PET composite sheet with high temperature and anti-dropping resistance.
- the auxiliary agent is composed of one or more of an antioxidant, an inorganic filler, a toughening agent, and a gloss improving assistant according to any ratio.
- the melt extrusion temperature is 230 to 260 ° C
- the screw rotation speed is 30 to 90 rpm
- the pulling speed is 0.15 to 6 m/min.
- graphene/PET nanocomposite is prepared by the following steps:
- the atomization drying temperature of the step (1) is 130 to 200 °C.
- the stirring speed in the step (3) is 140 to 200 rpm.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a titanium-based catalyst including an oxide of titanium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the lower stack greatly reduces the amount of graphene added, making the process of the invention cost effective.
- the introduction of the graphene oxide polymerization process does not have a significant effect, so the method of the invention is more reasonable, more efficient, and lower in cost in the actual production process.
- (3) The addition of graphene reduces the dripping speed when the sheet is burned, and improves the anti-droplet performance of the material.
- the electrical conductivity of the composite sheet is significantly increased under high addition, and it can be used as an antistatic material.
- a graphene-modified polyester blend fabric obtained by blending 40 to 60 parts by mass of cotton fibers, 30 to 50 parts by mass of graphene/PET composite fibers, and 10 to 20 parts by mass of other fibers.
- the graphene/PET composite fiber is obtained by mixing graphene/PET nanocomposites and 0-10 parts by weight of auxiliary agents, and performing high speed melt spinning, cooling, oiling, drawing, and bombardment.
- the graphene/PET nanocomposite consists of a single layer of graphene sheet and PET, and the surface of the graphene sheet is connected to the PET molecule by a covalent bond.
- graphene/PET nanocomposite is prepared by the following steps:
- the atomization drying temperature of the step (1) is 130 to 200 °C.
- the stirring speed in the step (3) is 140 to 200 rpm.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a titanium-based catalyst including an oxide of titanium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the auxiliary agent is composed of one or more of an antioxidant, an inorganic filler, a toughening agent, and a gloss improving assistant according to any ratio.
- the spinning temperature is 270 to 290 ° C
- the winding speed is 3,000 to 5,000 m/min
- the draw ratio is 1.5 to 4 times.
- the obtained fiber has a denier of 30 to 400D.
- the beneficial effects are as follows: (1) The pleated spherical graphene oxide microspheres added after the esterification is completed can be gradually unfolded and dissociated into a single layer of graphene oxide, and the hydroxyl and carboxyl groups on the surface of the graphene oxide are polymerized during PET polymerization.
- the reaction of the PET molecules causes the PET molecules to branch to the graphene surface, which improves the compatibility of the two, and greatly reduces the addition amount of graphene while reducing the stack, so that the method of the invention has high cost performance.
- the composite material After adding graphene, the composite material can be continuously spun at high speed, blended with traditional natural fabrics (cotton, hemp, wool) and synthetic fabrics (nylon, spandex, aramid), etc., which can retain the original fabric. Comfort, water absorption, gas permeability and other characteristics, and can take advantage of the characteristics of graphene, so that the blended fabric has good UV resistance and flame retardant properties.
- the durability of the fabric is good, and the high performance can be maintained after repeated washing, exposure, and smashing.
- the fabric can be reused, and the waste cloth can be recycled and reused, and the anti-UV, flame retardant and other properties can be re-applied.
- a graphene-modified flame-retardant anti-ultraviolet polyester fiber which is composed of 100 parts by mass of graphene/PET nanocomposite and 0-10 parts of auxiliary agent, after spinning, cooling, oiling, pulling Stretched and wounded.
- the graphene/PET nanocomposite consists of a single layer of graphene sheet and PET, and the surface of the graphene sheet is connected to the PET molecule by a covalent bond.
- a method for preparing graphene modified flame retardant anti-ultraviolet polyester fiber which comprises: mixing 100 parts by weight of graphene/PET nano composite material and 0-10 parts by weight of auxiliary agent, and then spinning, Cooling, oiling, drafting, and winding are obtained.
- the graphene/PET nanocomposite consists of a single layer of graphene sheet and PET, and the surface of the graphene sheet is connected to the PET molecule by a covalent bond.
- graphene/PET nanocomposite is prepared by the following steps:
- the atomization drying temperature of the step (1) is 130 to 200 °C.
- the stirring speed in the step (3) is 140 to 200 rpm.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a titanium-based catalyst including an oxide of titanium, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is a lanthanide catalyst, including an oxide of cerium, an inorganic salt, and an organic compound.
- the auxiliary agent is composed of one or more of an antioxidant, an inorganic filler, a toughening agent, and a gloss improving assistant according to any ratio.
- the spinning temperature is 270 to 290 ° C, and the winding speed is 3000 to 5000 m/min.
- the beneficial effects are as follows: (1) The pleated spherical graphene oxide microspheres added after the esterification is completed can be gradually unfolded and dissociated into a single layer of graphene oxide, and the hydroxyl and carboxyl groups on the surface of the graphene oxide are polymerized during PET polymerization.
- the reaction of the PET molecules causes the PET molecules to branch to the graphene surface, which improves the compatibility of the two, and greatly reduces the addition amount of graphene while reducing the stack, so that the method of the invention has high cost performance.
- a graphene/polyester nanocomposite comprising a polyester and a single-layer graphene sheet uniformly dispersed in a polyester, the surface of the graphene sheet being covalently bonded to the polyester molecule, and the polyester molecule is selected from the group consisting of One or more of propylene terephthalate (PTT), polybutylene terephthalate (PBT), and polybutylene terephthalate (PCT).
- PTT propylene terephthalate
- PBT polybutylene terephthalate
- PCT polybutylene terephthalate
- a method for preparing a graphene/polyester nanocomposite is prepared by the following steps:
- the atomization drying temperature of the step (1) is 130 to 200 °C.
- the diol in the step (2) is one or more of butanediol, propylene glycol, and 1,4-cyclohexanedimethanol.
- the diol in the step (2) is butane diol in an amount of 60 to 76.8 parts by weight.
- the diol in the step (2) is propylene glycol and is added in an amount of 50 to 70 parts by weight.
- the diol in the step (2) is 1,4-cyclohexanedimethanol in an amount of 121.4 to 147.5 parts by weight.
- the catalyst in the step (2) is one or more of an oxide of sodium, titanium, lead, tin, an inorganic salt, and an organic compound.
- the catalyst in the step (3) is one or more of an oxide of cerium, titanium, lead, tin, an inorganic salt, and an organic compound.
- the pleated spherical graphene oxide microspheres are prepared by the atomization drying method, and the pleated spherical graphene oxide can be gradually realized in different polyester oligomers by reasonably selecting the carbon-oxygen ratio and the size of the graphene oxide. Expanded and dissociated into flake graphene oxide.
- the hydroxyl group and carboxyl group on the surface of graphene oxide react with the polyester molecules in the system, so that the polyester molecules link to the surface of graphene, which improves two
- the compatibility of the product also contributes to the improvement of mechanical properties, electrical conductivity, and ultraviolet resistance.
- Adding graphene oxide after esterification avoids the influence on the esterification process in the first step, is more reasonable in the actual production process, has higher efficiency, lower cost, and avoids the occurrence of graphene oxide in the esterification stage.
- Stacking forms agglomerates.
- no substance is introduced except pleated spheroidal graphene oxide, which minimizes the influence of graphene introduction on the process and equipment, and has broad application prospects.
- the resulting graphene/polyester composite has excellent mechanical properties and electrical conductivity and can be used in the preparation of functionalized polyester fibers.
- a method for preparing a graphene/nylon 6 nano composite material is carried out according to the following steps:
- the above dispersion is added to the polycondensation reaction vessel, and the temperature is raised to 250-270 ° C, and the reaction is carried out at 0.5-1 MPa for 2-4 hours; then, the reaction is carried out under vacuum for 4-6 hours to obtain a polymer melt; The polymer melt was subjected to water-cooling granulation to obtain a graphene/nylon 6 nanocomposite.
- the above dispersion was continuously polymerized in a VK tube at a polymerization temperature of 260 ° C and a polymerization time of 20 h.
- the polymer melt was subjected to water-cooling granulation to obtain a graphene/nylon 6 nanometer composite material.
- the temperature of the atomization drying in the step (1) is 130 to 160 °C.
- the beneficial effects are as follows: (1)
- the conventional graphene powders are mostly highly stacked graphene structures, and cannot be dispersed into a single layer of graphene after being added to the polymerization system, and even secondary stacking may occur, thereby reducing the overall performance of the material.
- the invention firstly obtains pleated spherical graphene oxide microspheres by atomization drying method, and the pleat structure greatly reduces the stacking effect between graphene oxide sheets, and pleats by reasonably selecting the carbon-oxygen ratio and the size of graphene oxide.
- the spherical graphene oxide can be gradually unfolded and dissociated in the caprolactam melt, and simultaneously thermally reduced to form a single layer of flake graphene.
- Nylon 6 molecules are gradually grafted onto the surface of graphene during the whole polymerization process, which improves the compatibility of the two, and maintains excellent mechanical properties (such as toughness and spinnability) in the case of high addition. To the extent, it exerts the advantages of graphene enhancement, barrier, and anti-ultraviolet, and has a very low percolation threshold.
- the comprehensive performance is improved, such as mechanical properties, high temperature resistance, anti-UV aging performance, etc.; at the same time, the material toughness is not lost, the molecular weight of the polymer is controllable, and it does not decrease with the increase of the amount of graphene added.
- Graphene is both a nucleating agent and a nano-reinforcing filler in the polymer matrix, and also functions as a UV protection. (3) Graphene has good dispersibility in the polymer matrix, and the transverse dimension of the graphene sheet is large, so the amount of graphene is small (less than 0.5%), and the final product has good processability, and can be industrialized multi-filament bundle high-speed spinning. wire. (4) The whole preparation process is simple and effective, and it is not necessary to modify the existing nylon 6 polymerization equipment, and it is a highly competitive production technology. Since the addition of water is avoided, continuous polymerization can be carried out using a VK tube.
- Figure 1 is a photograph of a graphene/PET nanocomposite prepared by the present invention.
- Figure 3 is a photograph of a graphene polyester conjugate fiber for a cord used in the present invention.
- Figure 4 is a photograph of a multifunctional graphene/polyester composite fabric prepared by the present invention.
- Figure 5 is a photograph of a graphene/PET composite film prepared by the present invention.
- Figure 6 is a photograph of a graphene-modified polyester blend fabric.
- Figure 7 is a photograph of a graphene/polyester nanocomposite prepared by the present invention.
- PET was prepared in accordance with the method of Example 1, except that no pleated spherical graphene oxide was added during the preparation.
- the performance is shown in Table 1.
- Comparative Example 1-1 Comparative Example 1-2, Example 1-1, Example 1-2, Example 1-3, and Comparative Example 1-3, it was found that the carbon oxide ratio and the addition amount of the graphene oxide were maintained. Under constant conditions, the optimum size of the graphene oxide can be selected to obtain the best performance composite.
- the size of the graphene oxide of Comparative Example 1-2 is too small to be an effective reinforcing material by itself, and the graphene oxide of Comparative Examples 1-3 is too large in size and cannot be effectively expanded into flake graphite oxide after being added to the polymerization system.
- the olefin can only be used as a pleated spherical filler to reinforce the composite material, and the tensile strength and modulus increase are small, and the elongation at break is slightly lowered. In the size range of 1 to 50 microns, graphene oxide can be more effectively enhanced as the size increases.
- Comparative Example 1-1 Comparative Example 1-1, Example 1-2, Example 1-4, Comparative Example 1-4, it can be found that the carbon-oxygen ratio is increased, and the performance of the composite material is better, which is due to an increase in the carbon-oxygen ratio, graphene. The defects are less and the performance is better, which makes the composite perform better.
- the carbon-oxygen ratio cannot be too high, otherwise the bonding force between the graphene oxide sheets is too strong, the polymerization is not developed, the reinforcement cannot be effectively enhanced, and the elongation at break is greatly reduced (Comparative Example 4).
- Comparative Example 1-1 Analysis of Comparative Example 1-1, Example 1-2, Example 1-5, Example 1-6, Comparative Example 1-5, it was found that the addition amount of graphene oxide increased, the mechanical properties of the material were improved, and The conductivity is greatly improved. After adding too much graphene oxide, although the electrical conductivity can be further improved, the mechanical properties of the material are degraded, which is due to the excessive stacking of graphene, which reduces the reinforcing effect (Comparative Examples 1-5).
- the composite obtained in the step (3) is dried, pre-crystallized, solid-phase polycondensed, cooled, and melt-spun at a high speed.
- the drying temperature is 175 ° C
- the pre-crystallization temperature is 180 ° C
- the solid phase polycondensation temperature is 215 ° C
- the intrinsic viscosity after solid phase polycondensation is 1.1
- the cooling temperature is 70 ° C
- the spinning temperature is 290 ° C
- the winding speed is 4000 m / Min
- the draft ratio is 3.
- the composite obtained in the step (3) is dried, pre-crystallized, solid-phase polycondensed, cooled, and melt-spun at a high speed.
- the drying temperature is 175 ° C
- the pre-crystallization temperature is 180 ° C
- the solid phase polycondensation temperature is 215 ° C
- the intrinsic viscosity after solid phase polycondensation is 1.1
- the cooling temperature is 70 ° C
- the spinning temperature is 290 ° C
- the winding speed is 4000 m / Min
- the draft ratio is 3.
- the composite obtained in the step (3) is dried, pre-crystallized, solid-phase polycondensed, cooled, and melt-spun at a high speed.
- the drying temperature is 175 ° C
- the pre-crystallization temperature is 180 ° C
- the solid phase polycondensation temperature is 215 ° C
- the intrinsic viscosity after solid phase polycondensation is 1.1
- the cooling temperature is 70 ° C
- the spinning temperature is 290 ° C
- the winding speed is 4000 m / Min
- the draft ratio is 3.
- the composite obtained in the step (3) is dried, pre-crystallized, solid-phase polycondensed, cooled, and melt-spun at a high speed.
- the drying temperature is 175 ° C
- the pre-crystallization temperature is 180 ° C
- the solid phase polycondensation temperature is 215 ° C
- the intrinsic viscosity after solid phase polycondensation is 1.12
- the cooling temperature is 70 ° C
- the spinning temperature is 290 ° C
- the winding speed is 4000 m / Min
- the draft ratio is 3.
- the composite obtained in the step (3) is dried, pre-crystallized, solid-phase polycondensed, cooled, and melt-spun at a high speed.
- the drying temperature is 175 ° C
- the pre-crystallization temperature is 180 ° C
- the solid phase polycondensation temperature is 215 ° C
- the intrinsic viscosity after solid phase polycondensation is 1.14
- the cooling temperature is 70 ° C
- the spinning temperature is 290 ° C
- the winding speed is 4000 m / Min
- the draft ratio is 3.
- PET was prepared in accordance with the method of Example 1, except that no pleated spherical graphene oxide was added during the preparation.
- the performance is shown in Table 2.
- the composite obtained in the step (3) is dried, pre-crystallized, solid-phase polycondensed, cooled, and melt-spun at a high speed.
- the drying temperature is 175 ° C
- the pre-crystallization temperature is 180 ° C
- the solid phase polycondensation temperature is 215 ° C
- the intrinsic viscosity after solid phase polycondensation is 1.1
- the cooling temperature is 70 ° C
- the spinning temperature is 290 ° C
- the winding speed is 4000 m / Min
- the draft ratio is 3.
- the composite obtained in the step (3) is dried, pre-crystallized, solid-phase polycondensed, cooled, and melt-spun at a high speed.
- the drying temperature is 175 ° C
- the pre-crystallization temperature is 180 ° C
- the solid phase polycondensation temperature is 215 ° C
- the intrinsic viscosity after solid phase polycondensation is 1.31
- the cooling temperature is 70 ° C
- the spinning temperature is 290 ° C
- the winding speed is 4000 m / Min
- the draft ratio is 3.
- the composite obtained in the step (3) is dried, pre-crystallized, solid-phase polycondensed, cooled, and melt-spun at a high speed.
- the drying temperature is 175 ° C
- the pre-crystallization temperature is 180 ° C
- the solid phase polycondensation temperature is 215 ° C
- the intrinsic viscosity after solid phase polycondensation is 1.1
- the cooling temperature is 70 ° C
- the spinning temperature is 290 ° C
- the winding speed is 4000 m / Min
- the draft ratio is 3.
- the composite obtained in the step (3) is dried, pre-crystallized, solid-phase polycondensed, cooled, and melt-spun at a high speed.
- the drying temperature is 175 ° C
- the pre-crystallization temperature is 180 ° C
- the solid phase polycondensation temperature is 215 ° C
- the intrinsic viscosity after solid phase polycondensation is 1.37
- the cooling temperature is 70 ° C
- the spinning temperature is 290 ° C
- the winding speed is 4000 m / Min
- the draft ratio is 3.
- Comparative Example 2-4 Analysis of Comparative Example 2-1, Example 2-2, Example 2-3, Comparative Example 2-4, it can be found that the carbon-oxygen ratio increases, and the indexes of the composite fiber increase, which is due to an increase in the carbon-oxygen ratio. Graphene has fewer defects and better performance, which makes the composite perform better. However, the carbon-oxygen ratio cannot be too high, otherwise the bonding force between the graphene oxide sheets is too strong, and the stacking state is maintained during the polymerization, and the spinning holes are blocked, which is difficult to continuously produce (Comparative Examples 2-4).
- the composite obtained in the step (3) is dried, pre-crystallized, solid-phase polycondensed, cooled, and melt-spun at a high speed.
- the drying temperature is 170 ° C
- the pre-crystallization temperature is 175 ° C
- the solid phase polycondensation temperature is 210 ° C
- the intrinsic viscosity after solid phase polycondensation is 0.9
- the cooling temperature is 60 ° C
- the spinning temperature is 290 ° C
- the winding speed is 5000 m / Min
- the draft ratio is 4.
- the graphene polyester composite fiber for cord has good mechanical properties and electrical properties.
- the composite obtained in the step (3) is dried, pre-crystallized, solid-phase polycondensed, cooled, and melt-spun at a high speed.
- the drying temperature is 180 ° C
- the pre-crystallization temperature is 185 ° C
- the solid phase polycondensation temperature is 220 ° C
- the intrinsic viscosity after solid phase polycondensation is 1.2
- the cooling temperature is 80 ° C
- the spinning temperature is 270 ° C
- the winding speed is 3000 m / Min
- the draft ratio is 1.5.
- the graphene polyester composite fiber for cord has good mechanical properties and electrical properties.
- PET was prepared in accordance with the method of Example 1, except that no pleated spherical graphene oxide was added during the preparation.
- the performance is shown in Tables 3 and 4.
- the graphene/polyester composite fabric was obtained, and the specific properties are shown in Tables 3 and 4.
- Comparative Example 3-1 Comparative Example 3-2, Example 3-1, Example 3-2, Example 3-3, and Comparative Example 3-3, it was found that the carbon oxide ratio and the addition amount of the graphene oxide were maintained. In the case of constant, the optimum size of the graphene oxide can be selected to obtain the functional fabric with the best performance.
- the size of the graphene oxide of Comparative Example 3-2 was too small, and the contribution to the improvement of conductivity, UV resistance and flame retardancy was not significant, while the size of graphene oxide of Comparative Example 3 was too large and could not be added to the polymerization system.
- the composite material can only be reinforced as a pleated spherical filler, resulting in a significant decrease in the spinnability and continuity of the material.
- graphene oxide can be more effectively enhanced as the size increases.
- Comparative Example 3-1 Comparative Example 3-1, Example 3-2, Example 3-4, Comparative Example 3-4, it can be found that the carbon-oxygen ratio increases, and the various indexes of the fabric increase, which is due to the increase of the carbon-oxygen ratio, graphite.
- the olefin has fewer defects and its own performance is better, so that the composite material performs better.
- the carbon-oxygen ratio cannot be too high, otherwise the bonding force between the graphene oxide sheets is too strong, and the stacking state is maintained during the polymerization, and the spinning holes are blocked, which is difficult to continuously produce (Comparative Examples 3-4).
- the multifunctional graphene/polyester composite fabric is obtained with good performance.
- the multifunctional graphene/polyester composite fabric is obtained with good performance.
- a graphene/PET composite film was obtained by uniformly mixing 100 parts by mass of a graphene/PET nanocomposite and 0.2 parts by mass of an antioxidant, and forming a film by melt casting.
- the extrusion temperature was 260 ° C
- the screw speed was 100 rpm
- the pulling speed was 8 m/min.
- PET was prepared in accordance with the method of Example 1, except that no pleated spherical graphene oxide was added during the preparation.
- the performance is shown in Tables 5 and 6.
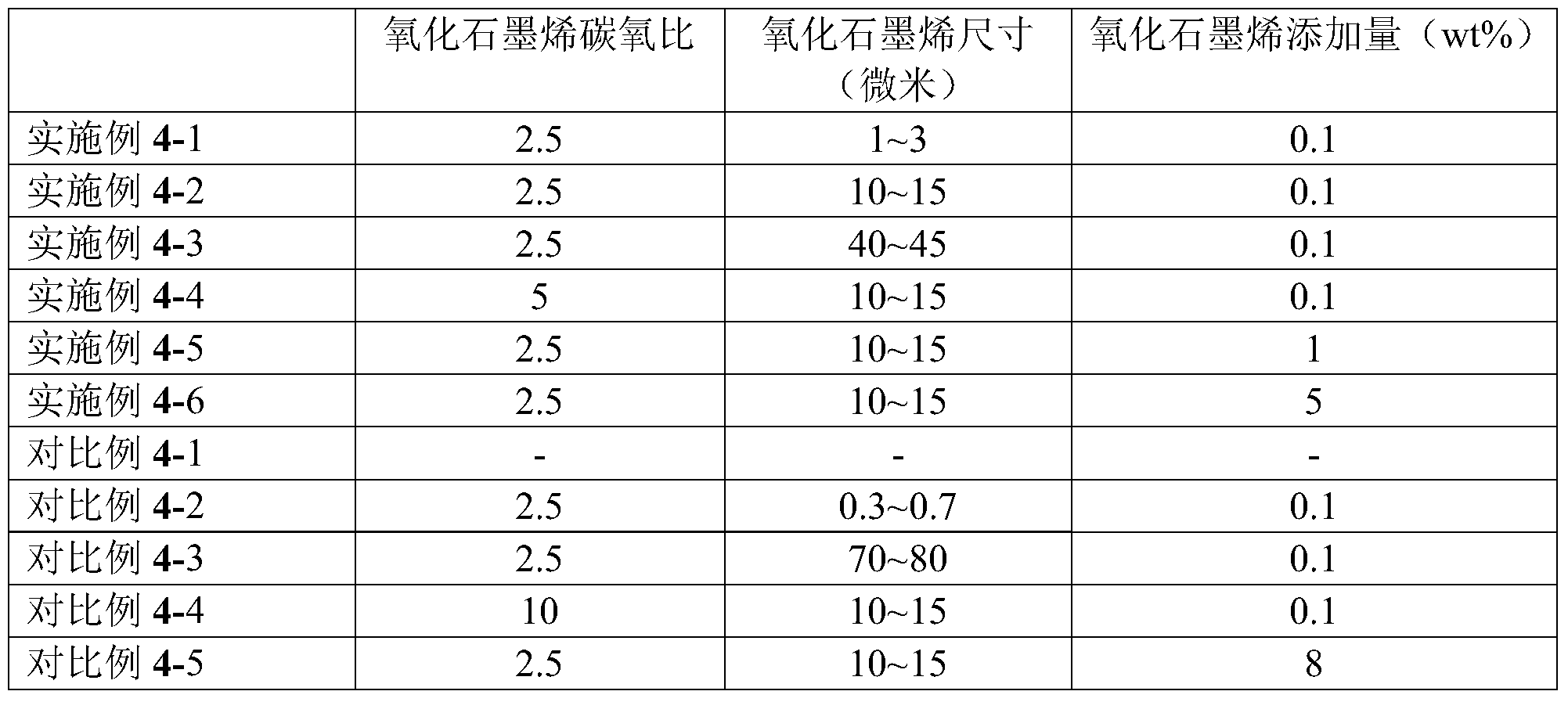
- Comparative Example 4-1 Comparative Example 4-2, Example 4-1, Example 4-2, Example 4-3, and Comparative Example 4-3, it was found that the carbon oxide ratio and the addition amount of the graphene oxide were maintained. Under constant conditions, the optimum size of the graphene oxide can be selected to obtain the best performance composite.
- the size of the graphene oxide in Comparative Example 4-2 was too small, and the reinforcing effect was not remarkable, while the graphene oxide in Comparative Example 4-3 was too large in size and could not be effectively expanded into flake graphene oxide after being added to the polymerization system. It can be used as a pleated spherical filler to enhance the composite material and contribute less to UV protection and barrier properties. In the size range of 1 to 50 microns, graphene oxide can be more effectively enhanced as the size increases.
- Comparative Example 4-1 Example 4-2, Example 4-4, Comparative Example 4-4
- the carbon-oxygen ratio is increased, and the performance of the composite material is better, which is due to an increase in the carbon-oxygen ratio, graphene.
- the defects are less and the performance is better, so that the barrier properties of the composite are better.
- the carbon-oxygen ratio cannot be too high, otherwise the bonding force between the graphene oxide sheets is too strong, and it does not spread during polymerization, and cannot exist in the form of flake graphene in the composite film, and does not have the effect of blocking water oxygen and ultraviolet rays. Even seriously affecting the continuity of film formation (Comparative Example 4-4).
- Comparative Example 4-1 Example 4-2, Example 4-5, Example 4-6, Comparative Example 4-5
- the addition amount of graphene oxide increased, the barrier property of the composite film, and the ultraviolet resistance And the conductivity has increased significantly.
- the conductivity can be further improved, since the graphene is stacked, the film solvent is broken during the casting process, and the uniformity of the film is greatly reduced, and some micropores are generated, which is difficult to achieve a barrier effect.
- Comparative Example 4-5 Comparative Example 4-5.
- a graphene/PET composite film was obtained by melt-extruding 100 parts by mass of graphene/PET nanocomposite.
- the extrusion temperature was 250 ° C
- the screw speed was 40 rpm
- the pulling speed was 1 m/min.
- the graphene/PET composite film was obtained to have good performance.
- a graphene/PET composite film was obtained by melt-extruding 100 parts by mass of graphene/PET nanocomposite.
- the extrusion temperature was 280 ° C
- the screw speed was 300 rpm
- the pulling speed was 50 m/min.
- the graphene/PET composite film was obtained to have good performance.
- PET was prepared in accordance with the method of Example 1, except that no pleated spherical graphene oxide was added during the preparation.
- the performance is shown in Tables 7 and 8.
- Comparative Example 5-1 Comparative Example 5-2, Example 5-1, Example 5-2, Example 5-3, and Comparative Example 5-3, it was found that the carbon oxide ratio and the addition amount of the graphene oxide were maintained. Under constant conditions, the optimum size of the graphene oxide can be selected to obtain the best performance composite.
- the size of the graphene oxide of Comparative Example 5-2 was too small to be an effective reinforcing material by itself, and the graphene oxide of Comparative Example 5-3 was too large in size and could not be effectively expanded into flake graphite oxide after being added to the polymerization system.
- the olefin can only be used as a pleated spherical filler to reinforce the composite material, and the tensile strength and modulus increase are small, and the elongation at break is slightly lowered. In the size range of 1 to 50 microns, graphene oxide can be more effectively enhanced as the size increases.
- Comparative Example 5-1 Example 5-2, Example 5-4, Comparative Example 5-4
- the carbon-oxygen ratio is increased, and the performance of the composite material is better, which is due to an increase in the carbon-oxygen ratio, graphene.
- the defects are less and the performance is better, which makes the composite perform better.
- the carbon-oxygen ratio cannot be too high, otherwise the bonding force between the graphene oxide sheets is too strong, and it does not expand during polymerization, and cannot be effectively enhanced, and the elongation at break is greatly reduced (Comparative Example 5-4).
- Comparative Example 5-5 Analysis of Comparative Example 5-1, Example 5-2, Example 5-5, Example 5-6, Comparative Example 5-5, it was found that the addition amount of graphene oxide increased, the mechanical properties of the material were improved, and the melting The drop rate is greatly reduced and the conductivity is greatly improved. After adding too much graphene oxide, although the flame retardancy and electrical conductivity can be further improved, the mechanical properties of the material are degraded, which is due to the excessive stacking of graphene, which reduces the reinforcing effect and makes the material brittle (Comparative Example 5-5 ).
- the graphene/PET composite sheet has good performance.
- the graphene/PET composite sheet has good performance.
- PET was prepared in accordance with the method of Example 1, except that no pleated spherical graphene oxide was added during the preparation.
- the performance is shown in Table 9.
- the flame retardancy test was carried out by a 45° direction burning rate test.
- the UV protection factor (UPF) value was measured and calculated using an ultraviolet spectrophotometer.
- Comparative Example 6-1 Analysis of Comparative Example 6-1, Comparative Example 6-2, Example 6-1, Example 6-2, Example 6-3, and Comparative Example 3 revealed that the carbon oxide ratio and the amount of addition of graphene oxide were maintained.
- a blend fabric having the best performance can be obtained.
- the size of the graphene oxide of Comparative Example 6-2 was too small, and the contribution to the improvement of conductivity, UV resistance and flame retardancy was not significant, while the graphene oxide of Comparative Example 6-3 was too large in the polymerization system.
- Comparative Example 6-1 Comparative Example 6-1, Example 6-2, Example 6-4, Comparative Example 6-4, it can be found that the carbon-oxygen ratio increases, and the indexes of the fabric increase, which is due to the increase of the carbon-oxygen ratio, graphite.
- the olefin has fewer defects and its own performance is better, so that the composite material performs better.
- the carbon-oxygen ratio cannot be too high, otherwise the bonding force between the graphene oxide sheets is too strong, and the stacking state is maintained during the polymerization, and the spinning holes are blocked, which is difficult to continuously produce (Comparative Example 6-4).
- the addition amount of the pleated spherical graphene oxide, the carbon-oxygen ratio, and the size of the graphene oxide therein are controlled within a reasonable range, and a blended fabric having excellent ultraviolet-proof performance and flame-retardant property can be obtained.
- the graphene-modified polyester blended fabric has good performance.
- a graphene/PET composite fiber is obtained by spinning, cooling, oiling, drawing, and stretching 100 parts by mass of the graphene/PET nanocomposite.
- the spinning temperature was 285 ° C
- the spinning speed was 3600 m/min
- the draw ratio was 4 times
- the denier was 30 D.
- the graphene-modified polyester blended fabric has good performance.
- PET was prepared in accordance with the method of Example 1, except that no pleated spherical graphene oxide was added during the preparation.
- the performance is shown in Tables 10 and 11.
- Comparative Example 7-1 Comparative Example 7-2, Example 7-1, Example 7-2, Example 7-3, and Comparative Example 7-3, it was found that the carbon oxide ratio and the addition amount of the graphene oxide were maintained. Under constant conditions, the optimum size of the graphene oxide can be selected to obtain the best performance composite fiber.
- the size of the graphene oxide of Comparative Example 7-2 was too small to be an effective reinforcing material by itself, and the graphene oxide of Comparative Example 3 was too large in size and could not be effectively expanded into flake graphene oxide after being added to the polymerization system.
- the composite can only be reinforced as a pleated spherical filler, resulting in a significant decrease in the spinnability and continuity of the material. In the size range of 1 to 50 microns, graphene oxide can be more effectively enhanced as the size increases.
- Comparative Example 7-1 Analysis of Comparative Example 7-1, Example 7-2, Example 7-4, and Comparative Example 7-4 revealed that the carbon-oxygen ratio increased and the indexes of the composite fiber increased, which was due to an increase in the carbon-oxygen ratio.
- Graphene has fewer defects and better performance, which makes the composite perform better.
- the carbon-oxygen ratio cannot be too high, otherwise the bonding force between the graphene oxide sheets is too strong, and the stacking state is maintained during the polymerization, and the spinning holes are blocked, which is difficult to continuously produce (Comparative Example 7-4).
- the graphene-modified flame retardant anti-ultraviolet polyester fiber has good performance.
- the graphene-modified flame retardant anti-ultraviolet polyester fiber has good performance.
- the size of graphene oxide (1 ⁇ 50 microns), carbon to oxygen ratio (2.5-5), atomization drying temperature (130 ⁇ 200 ° C) and graphene oxide in the whole system The ratio is the necessary condition for obtaining a graphene/PBT composite with uniform dispersion and superior performance.
- the tensile strength is increased by more than 5% compared with pure PBT, the modulus is increased by more than 10%, and the resistivity is between 10 7 and 10 3 ⁇ m.
- the UV resistance coefficient UPF is higher than 40.
- this embodiment is only a further preferred result.
- the tensile strength and modulus are increased by 25% and 45%, respectively, and the specific resistance is 10 3 ⁇ m.
- the anti-UV coefficient UPF after spinning and weaving into a fabric Above 130.
- the size of graphene oxide (1 ⁇ 50 microns), carbon to oxygen ratio (2.5-5), atomization drying temperature (130 ⁇ 200 ° C) and graphene oxide in the whole system The ratio is the necessary condition for obtaining a graphene/PTT composite with uniform dispersion and superior performance.
- the tensile strength is increased by more than 5% compared with pure PTT, the modulus is increased by more than 8%, and the resistivity is between 10 7 and 10 3 ⁇ m.
- the UV resistance coefficient UPF is higher than 40.
- this embodiment is only a further preferred result.
- the tensile strength and modulus are increased by 20% and 50%, respectively, compared with pure PET, and the electrical resistivity is 10 3 ⁇ m.
- the anti-UV coefficient UPF after spinning and weaving into a fabric Above 140.
- the size of graphene oxide (1 ⁇ 50 microns), carbon to oxygen ratio (2.5-5), atomization drying temperature (130 ⁇ 200 ° C) and graphene oxide in the whole system The ratios are all graphene/PCT composites with uniform dispersion and superior performance.
- the tensile strength is increased by more than 5% compared with pure PCT, the modulus is increased by more than 10%, and the resistivity is between 10 7 and 10 3 ⁇ m.
- the UV resistance coefficient UPF is higher than 40.
- this embodiment is only a further preferred result.
- the tensile strength and modulus are increased by 18% and 39%, respectively, and the specific resistance is 10 3 ⁇ m.
- the anti-UV coefficient UPF after spinning and weaving into a fabric Above 145.
- the size of graphene oxide (1 ⁇ 50 microns), carbon to oxygen ratio (2.5-5), atomization drying temperature (130 ⁇ 200 ° C) and graphene oxide in the whole system The ratios are all graphene/PBT/PTT composites with uniform dispersion and superior performance.
- the tensile strength is increased by more than 8% compared with PBT/PTT without graphene, the modulus is increased by more than 12%, and the resistivity is 10 7 ⁇ 10 3 ⁇ m, after spinning and weaving into a fabric, its UV resistance coefficient UPF is higher than 30.
- this embodiment is only a further preferred result.
- the tensile strength and modulus are 27% and 50% higher than that of the pure PBT, and the specific resistance is 10 3 ⁇ m.
- the anti-UV coefficient UPF after spinning and weaving into a fabric Above 130.
- the graphene/PCT nanocomposite is obtained, and the tensile strength and modulus are increased by 10% and 15%, respectively, compared with the pure PCT, and the electrical resistivity is 10 6 ⁇ m.
- the anti-UV is obtained.
- the coefficient UPF is higher than 50.
- the graphene/PBT nanocomposite is obtained, and the tensile strength and modulus are increased by 15% and 25%, respectively, compared with pure PBT, and the electrical resistivity is 10 7 ⁇ m. After being spun and woven into a fabric, it is UV-resistant. The coefficient UPF is higher than 40.
- the single-layer graphene oxide dispersion is dried by an atomization drying method to obtain pleated spherical graphene oxide having an atomization temperature of 130 ° C, a graphene oxide sheet size of 0.3 to 5 ⁇ m, an average size of 1 ⁇ m, and carbon.
- the oxygen ratio is 5, and the water content is less than 0.1%;
- the filament has a high degree of UV resistance and has a very low percolation threshold.
- the water content is less than 0.1%
- the single-layer graphene oxide dispersion is dried by an atomization drying method to obtain pleated spherical graphene oxide, the atomization temperature is 130 ° C, the average size of the graphene oxide sheet is 5 ⁇ m, and the carbon-oxygen ratio is 3.9.
- the rate is less than 0.1%;
- the resulting graphene/nylon 6 composite has good properties.
- the resulting graphene/nylon 6 composite has good properties.
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Textile Engineering (AREA)
- General Chemical & Material Sciences (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Nanotechnology (AREA)
- Mechanical Engineering (AREA)
- Toxicology (AREA)
- Inorganic Chemistry (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Polyesters Or Polycarbonates (AREA)
- Carbon And Carbon Compounds (AREA)
- Catalysts (AREA)
- Artificial Filaments (AREA)
Abstract
Description













Claims (32)
- 一种石墨烯/PET纳米复合材料,其特征在于,由单层石墨烯片和PET组成,所述石墨烯片表面与PET分子通过共价键相连。
- 一种石墨烯/PET纳米复合材料的制备方法,其特征在于,由以下步骤制备进行:(1)通过雾化干燥法将尺寸为1~50微米的单层氧化石墨烯分散液干燥,得到褶球状氧化石墨烯,其碳氧比为2.5~5;(2)将100重量份对苯二甲酸、48~67重量份乙二醇、0.02重量份醋酸钠充分混合搅拌,在250℃下进行酯化反应;(3)将步骤(1)得到的0.0117~5.85重量份褶球状氧化石墨烯,与0.018重量份催化剂加入步骤(2)得到的酯化产物中,保温搅拌1~3h,之后升温至285℃并抽真空,反应进行至体系不再放热,水冷切粒得到石墨烯/PET纳米复合材料。
- 一种帘子线用石墨烯涤纶复合纤维,其特征在于,所述纤维由石墨烯/PET纳米复合材料经干燥、预结晶、固相缩聚、冷却、高速熔融纺丝得到;所述石墨烯/PET纳米复合材料由单层石墨烯片和PET组成,石墨烯片表面与PET分子通过共价键相连。
- 根据权利要求3所述的复合纤维,其特征在于,所述干燥温度为170~180℃,预结晶温度为175~185℃,固相缩聚温度为210~220℃,固相缩聚后的特性粘度为0.9~1.2,冷却温度为60~80℃,纺丝温度为270~290℃,卷绕速度为3000~5000m/min,牵伸比为1.5~4。
- 根据权利要求3所述的复合纤维,其特征在于,石墨烯/PET纳米复合材料由以下步骤制备进行:(1)通过雾化干燥法将尺寸为1~10微米的单层氧化石墨烯分散液干燥,得到褶球状氧化石墨烯,其碳氧比为2.5~5;(2)将100重量份对苯二甲酸、48~67重量份乙二醇、0.02质量份醋酸钠充分混合搅拌,在250℃下进行酯化反应;(3)将步骤(1)得到的0.117~1.17重量份褶球状氧化石墨烯,与0.018重量份催化剂加入步骤(2)得到的酯化产物中,保温搅拌1~3h,之后升温至285℃并抽真空,反应进行至体系不再放热,水冷切粒得到石墨烯/PET纳米复合材料。
- 一种多功能的石墨烯/涤纶复合织物,其特征在于,所述复合织物由100质量份的石墨烯/PET纳米复合材料和0~10份的助剂混合后,经纺丝、冷却、上油、牵伸、加弹、织布、染色、后整理得到;所述石墨烯/PET纳米复合材料由单层石墨烯片和PET组成,石墨烯片表面与PET分子通过共价键相连。
- 根据权利要求6所述的复合织物,其特征在于,石墨烯/PET纳米复合材料由以下步骤制备进行:(1)通过雾化干燥法将尺寸为1~50微米的单层氧化石墨烯分散液干燥,得到褶球状 氧化石墨烯,其碳氧比为2.5~5;(2)将100重量份对苯二甲酸、48~67重量份乙二醇、0.02质量份醋酸钠充分混合搅拌,在250℃下进行酯化反应;(3)将步骤(1)得到的0.585~5.85重量份褶球状氧化石墨烯,与0.018重量份催化剂加入步骤(2)得到的酯化产物中,保温搅拌1~3h,之后升温至285℃并抽真空,反应进行至体系不再放热,水冷切粒得到石墨烯/PET纳米复合材料。
- 根据权利要求6所述的方法,其特征在于,所述纺丝的温度为270~290℃,纺丝速度为3000~5000m/min,牵伸倍数为1.5~4倍;所得纤维旦数为30~600D;织布方法为采用有梭织机或无梭织机进行编织。
- 一种多功能石墨烯/PET复合膜,其特征在于,所述复合膜由100质量份的石墨烯/PET纳米复合材料和0~10份的助剂共同熔融流延成膜得到;所述石墨烯/PET纳米复合材料由单层石墨烯片和PET组成,石墨烯片表面与PET分子通过共价键相连。
- 一种多功能石墨烯/PET复合膜的制备方法,其特征在于,该方法为:将100重量份的石墨烯/PET纳米复合材料和0~10重量份的助剂混合均匀后,经熔融流延,即得到本发明多功能石墨烯/PET复合膜。
- 根据权利要求10所述的方法,其特征在于,石墨烯/PET纳米复合材料由以下步骤制备进行:(1)通过雾化干燥法将尺寸为1~50微米的单层氧化石墨烯分散液干燥,得到褶球状氧化石墨烯,其碳氧比为2.5~5;(2)将100重量份对苯二甲酸、48~67重量份乙二醇、0.02质量份醋酸钠充分混合搅拌,在250℃下进行酯化反应;(3)将步骤(1)得到的0.0117~5.85重量份褶球状氧化石墨烯,与0.018重量份催化剂加入步骤(2)得到的酯化产物中,保温搅拌1~3h,之后升温至285℃并抽真空,反应进行至体系不再放热,水冷切粒得到石墨烯/PET纳米复合材料。
- 根据权利要求10所述的方法,其特征在于,所述助剂由抗氧化剂、无机填充剂、增韧剂、光泽改善助剂中的一种或多种按照任意配比组成;所述熔融挤出温度为250~280℃,螺杆转速为40~300rpm,牵引速度为1~50m/min。
- 一种高强度耐熔滴的石墨烯/PET复合板材,其特征在于,所述复合板材由100质量份的石墨烯/PET纳米复合材料和0~10份的助剂共同熔融挤出得到;所述石墨烯/PET纳米复合材料由单层石墨烯片和PET组成,石墨烯片表面与PET分子通过共价键相连。
- 一种高强度耐熔滴的石墨烯/PET复合板材的制备方法,其特征在于,该方法为:将100重量份的石墨烯/PET纳米复合材料和0~10重量份的助剂混合均匀后,经熔融挤出,即得到耐高温抗熔滴的石墨烯/PET复合板材。
- 根据权利要求14所述的方法,其特征在于,石墨烯/PET纳米复合材料由以下步骤制备进行:(1)通过雾化干燥法将尺寸为1~50微米的单层氧化石墨烯分散液干燥,得到褶球状氧化石墨烯,其碳氧比为2.5~5;(2)将100重量份对苯二甲酸、48~67重量份乙二醇、0.02质量份醋酸钠充分混合搅拌,在250℃下进行酯化反应;(3)将步骤(1)得到的0.0117~5.85重量份褶球状氧化石墨烯,与0.018重量份催化剂加入步骤(2)得到的酯化产物中,保温搅拌1~3h,之后升温至285℃并抽真空,反应进行至体系不再放热,水冷切粒得到石墨烯/PET纳米复合材料。
- 根据权利要求14所述的方法,其特征在于,所述助剂由抗氧化剂、无机填充剂、增韧剂、光泽改善助剂中的一种或多种按照任意配比组成;所述熔融挤出温度为230~260℃,螺杆转速为30-90rpm,牵引速度为0.15~6m/min。
- 一种石墨烯改性的涤纶混纺织物,其特征在于,所述该混纺织物是由40~60质量份棉纤维、30~50质量份石墨烯/PET复合纤维和10~20质量份其他纤维经混纺得到。
- 根据权利要求17所述的混纺织物,其特征在于,所述石墨烯/PET复合纤维是由石墨烯/PET纳米复合材料和0-10重量份的助剂混合后,经高速熔融纺丝、冷却、上油、牵伸、加弹得到;所述石墨烯/PET纳米复合材料由单层石墨烯片和PET组成,石墨烯片表面与PET分子通过共价键相连。
- 根据权利要求18所述的混纺织物,其特征在于,所述石墨烯/PET纳米复合材料由以下步骤制备进行:(1)通过雾化干燥法将尺寸为1~50微米的单层氧化石墨烯分散液干燥,得到褶球状氧化石墨烯,其碳氧比为2.5~5;(2)将100重量份对苯二甲酸、48~67重量份乙二醇、0.02质量份醋酸钠充分混合搅拌,在250℃下进行酯化反应;(3)将步骤(1)得到的0.117~5.85重量份褶球状氧化石墨烯,与0.018重量份催化剂加入步骤(2)得到的酯化产物中,保温搅拌1~3h,之后升温至285℃并抽真空,反应进行至体系不再放热,水冷切粒得到石墨烯/PET纳米复合材料。
- 一种石墨烯改性的阻燃防紫外涤纶纤维,其特征在于,所述纤维由100质量份的石墨烯/PET纳米复合材料和0~10份的助剂混合后,经纺丝、冷却、上油、牵伸、卷绕得到;所述石墨烯/PET纳米复合材料由单层石墨烯片和PET组成,石墨烯片表面与PET分子通过共价键相连。
- 一种石墨烯改性的阻燃防紫外涤纶纤维的制备方法,其特征在于,该方法为:将100重量份的石墨烯/PET纳米复合材料和0~10重量份的助剂混合均匀后,经熔融挤出,即 得到石墨烯改性的阻燃防紫外涤纶纤维。
- 根据权利要求21所述的方法,其特征在于,石墨烯/PET纳米复合材料由以下步骤制备进行:(1)通过雾化干燥法将尺寸为1~50微米的单层氧化石墨烯分散液干燥,得到褶球状氧化石墨烯,其碳氧比为2.5~5;(2)将100重量份对苯二甲酸、48~67重量份乙二醇、0.02质量份醋酸钠充分混合搅拌,在250℃下进行酯化反应;(3)将步骤(1)得到的0.0117~5.85重量份褶球状氧化石墨烯,与0.018重量份催化剂加入步骤(2)得到的酯化产物中,保温搅拌1~3h,之后升温至285℃并抽真空,反应进行至体系不再放热,水冷切粒得到石墨烯/PET纳米复合材料。
- 根据权利要求2、5、7、11、15、19、21任一项所述的方法,其特征在于,所述步骤(1)的雾化干燥温度为130~200℃。
- 根据权利要求2、5、7、11、15、19、21任一项所述的方法,其特征在于,所述步骤(3)中搅拌速度为140~200转/分。
- 根据权利要求2、5、7、11、15、19、21任一项所述的方法,其特征在于,所述步骤(3)中催化剂为锑系催化剂,包括锑的氧化物、无机盐和有机化合物;或为钛系催化剂,包括钛的氧化物、无机盐和有机化合物;或为锗系催化剂,包括锗的氧化物、无机盐和有机化合物。
- 一种石墨烯/聚酯纳米复合材料,其特征在于,包括聚酯和均匀分散在聚酯中的单层石墨烯片,所述石墨烯片表面与聚酯分子通过共价键相连,聚酯分子选自聚对苯二甲酸丙二醇酯(PTT),聚对苯二甲酸丁二醇酯(PBT),聚对苯二甲酸1,4-环己烷二甲酯(PCT)中的一种或多种。
- 一种石墨烯/聚酯纳米复合材料的制备方法,其特征在于,由以下步骤制备进行:(1)通过雾化干燥法将尺寸为1~50微米的单层氧化石墨烯分散液干燥,得到褶球状氧化石墨烯,其碳氧比为2.5~5;(2)将100重量份对苯二甲酸、50~150重量份二醇、0.01~0.5重量份催化剂充分混合搅拌,在200~260℃下进行酯化反应至无水生成;(3)将步骤(1)得到的0.02~10重量份褶球状氧化石墨烯,与0.01~1重量份催化剂加入步骤(2)得到的酯化产物中,保温搅拌1~3h,之后升温至240~310℃并抽真空,反应进行至体系不再放热,水冷切粒得到石墨烯/聚酯纳米复合材料。
- 根据权利要求27所述的方法,其特征在于,所述步骤(2)中的二醇为丁二醇,添加量优选为60~76.8重量份;或为丙二醇,添加量优选为50~70重量份;或为1,4-环己烷二甲醇,添加量优选为121.4~147.5重量份。
- 根据权利要求27所述的方法,其特征在于,所述步骤(2)中催化剂为钠、钛、铅、锡的氧化物、无机盐和有机化合物中的一种或多种。
- 根据权利要求27所述的方法,其特征在于,所述步骤(3)中催化剂为锑、钛、铅、锡的氧化物、无机盐和有机化合物中的一种或多种。
- 一种石墨烯/尼龙6纳米复合材料的制备方法,其特征在于,按以下步骤进行:(1)通过雾化干燥法将尺寸为1~50微米的单层氧化石墨烯分散液干燥,得到褶球状氧化石墨烯,其碳氧比为2.5~5;(2)将0.01-3.5质量份的褶球状氧化石墨烯和1-3质量份去离子水加入100质量份的己内酰胺熔体中,在80℃下高速(300~500rpm)搅拌混匀形成分散液;(3)在间歇式反应设备或VK管中制备石墨烯/尼龙6纳米复合材料:间歇式反应釜:在氮气保护下,将上述分散液加入缩聚反应釜,并升温至250-270℃,在0.5-1MPa下反应2-4小时;然后在真空下反应4-6小时,得到聚合物熔体;最后将聚合物熔体经水冷造粒得到石墨烯/尼龙6纳米复合材料;VK管:将上述分散液在VK管中连续聚合,聚合温度为260℃,聚合时间为20h,将聚合物熔体经水冷造粒得到石墨烯/尼龙6纳米复合材料。
- 根据权利要求31所述的制备方法,其特征在于,步骤(1)所述雾化干燥的温度为130~160℃。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2020100048A RU2746113C1 (ru) | 2017-06-26 | 2018-02-26 | Графеновый композиционный материал и способ его получения |
| KR1020207002385A KR102284847B1 (ko) | 2017-06-26 | 2018-02-26 | 그래핀 복합재료 및 그 제조방법 |
| JP2019571324A JP6963040B2 (ja) | 2017-06-26 | 2018-02-26 | グラフェン複合材料の製造方法 |
| EP18822649.2A EP3626758B1 (en) | 2017-06-26 | 2018-02-26 | Graphene composite material and preparation method therefor |
| BR112019027930-0A BR112019027930B1 (pt) | 2017-06-26 | 2018-02-26 | Método para preparar um material nanocompósito de grafenopoliéster |
| US16/626,546 US11149129B2 (en) | 2017-06-26 | 2018-02-26 | Graphene composite material and preparation method thereof |
Applications Claiming Priority (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710495004.5 | 2017-06-26 | ||
| CN201710497221.8A CN107163519B (zh) | 2017-06-26 | 2017-06-26 | 一种高强度耐熔滴的石墨烯/pet复合板材及其制备方法 |
| CN201710495022.3A CN107189076B (zh) | 2017-06-26 | 2017-06-26 | 一种多功能的石墨烯/涤纶复合织物及其制备方法 |
| CN201710495017.2A CN107190382B (zh) | 2017-06-26 | 2017-06-26 | 一种石墨烯改性的涤纶混纺织物及其制备方法 |
| CN201710495004.5A CN107326474B (zh) | 2017-06-26 | 2017-06-26 | 一种帘子线用石墨烯涤纶复合纤维及其制备方法 |
| CN201710494271.0A CN107353605B (zh) | 2017-06-26 | 2017-06-26 | 一种多功能石墨烯/pet复合膜及其制备方法 |
| CN201710494462.7A CN107142547B (zh) | 2017-06-26 | 2017-06-26 | 一种石墨烯改性的阻燃防紫外涤纶纤维及其制备方法 |
| CN201710495017.2 | 2017-06-26 | ||
| CN201710494501.3 | 2017-06-26 | ||
| CN201710497221.8 | 2017-06-26 | ||
| CN201710494462.7 | 2017-06-26 | ||
| CN201710494501.3A CN107325268B (zh) | 2017-06-26 | 2017-06-26 | 一种石墨烯/pet纳米复合材料及其制备方法 |
| CN201710495022.3 | 2017-06-26 | ||
| CN201710494271.0 | 2017-06-26 | ||
| CN201710718369.XA CN107513162A (zh) | 2017-08-21 | 2017-08-21 | 一种石墨烯/尼龙6纳米复合材料的制备方法 |
| CN201710718364.7 | 2017-08-21 | ||
| CN201710718364.7A CN107513151B (zh) | 2017-08-21 | 2017-08-21 | 一种石墨烯/聚酯纳米复合材料及其制备方法 |
| CN201710718369.X | 2017-08-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019000985A1 true WO2019000985A1 (zh) | 2019-01-03 |
Family
ID=64740348
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2018/077169 WO2019000985A1 (zh) | 2017-06-26 | 2018-02-26 | 石墨烯复合材料及其制备方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US11149129B2 (zh) |
| EP (1) | EP3626758B1 (zh) |
| JP (1) | JP6963040B2 (zh) |
| KR (1) | KR102284847B1 (zh) |
| BR (1) | BR112019027930B1 (zh) |
| RU (1) | RU2746113C1 (zh) |
| WO (1) | WO2019000985A1 (zh) |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11629420B2 (en) * | 2018-03-26 | 2023-04-18 | Global Graphene Group, Inc. | Production process for metal matrix nanocomposite containing oriented graphene sheets |
| CN110128634B (zh) * | 2019-04-30 | 2021-09-03 | 福建省银河服饰有限公司 | 一种石墨烯改性聚酯切片的制备方法 |
| US20220074078A1 (en) * | 2020-06-25 | 2022-03-10 | Nicholas L. Ciparro | Methods of forming a nanocomposite fiber and related mixture and nanocomposite fibers |
| CN112094421B (zh) * | 2020-08-20 | 2022-12-27 | 湖北中烟工业有限责任公司 | 一种改性还原氧化石墨烯掺杂聚乳酸薄膜的制备方法 |
| CN112876710B (zh) * | 2021-01-18 | 2022-03-11 | 山东阜坤新材料有限公司 | 一种可生物降解的抗菌石墨烯原位聚合共聚酯包装膜及其制备工艺 |
| CN112876660B (zh) * | 2021-01-18 | 2022-04-08 | 山东阜坤新材料有限公司 | 一种石墨烯原位聚合生物降解共聚酯及其制备方法和应用 |
| CN113004678A (zh) * | 2021-02-25 | 2021-06-22 | 深圳市台钜电工有限公司 | 耳机线材料及其制备工艺 |
| KR102518029B1 (ko) * | 2021-03-25 | 2023-04-04 | 김헌상 | 폴리에스터용 마스터 배치 조성물 및 그 조성물이 함유된 폴리에스터 원사 |
| CN113622047B (zh) * | 2021-04-23 | 2023-10-13 | 高碑店市隆泰丰博石墨烯有限公司 | 一种石墨烯基吸湿排汗快速散热的聚酯纤维及其制备方法 |
| CN114182387B (zh) * | 2021-11-08 | 2023-10-03 | 宁夏宁东泰和新材有限公司 | 一种二氧化钛-氧化石墨烯复合改性氨纶的制备方法 |
| WO2023113679A1 (en) | 2021-12-14 | 2023-06-22 | Glenntex Ab | Functionalized graphene structure and nanocomposite comprising such functionalized graphene structure |
| CN114214754A (zh) * | 2021-12-23 | 2022-03-22 | 常州灵达特种纤维有限公司 | 石墨烯改性膨体长丝的制备方法 |
| CN114349511B (zh) * | 2022-01-18 | 2022-11-22 | 南京工业大学 | 一种快速制备高导电石墨烯电磁屏蔽膜的方法 |
| KR102470946B1 (ko) * | 2022-03-18 | 2022-11-29 | 김태식 | 구리-그래핀 합연사 제조방법 및 이로부터 제조된 구리-그래핀 합연사를 포함하는 원단 |
| CN114959942B (zh) * | 2022-07-15 | 2023-02-28 | 浙江桐昆新材料研究院有限公司 | 抗静电防起球透湿粗旦涤纶牵伸丝及其制备方法 |
| KR102534123B1 (ko) * | 2022-08-22 | 2023-05-19 | 주식회사 네오엔프라 | 산화그래핀이 함유된 폴리에스터 원사 |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140080962A1 (en) * | 2011-05-03 | 2014-03-20 | The Board Of Regents For Oklahoma State University | Polyethylene terephthalate-graphene nanocomposites |
| CN105540573A (zh) * | 2016-01-27 | 2016-05-04 | 浙江碳谷上希材料科技有限公司 | 一种高溶解性多褶皱干态氧化石墨烯微球及其制备方法 |
| CN105820519A (zh) * | 2016-02-29 | 2016-08-03 | 洛阳尖端技术研究院 | Pet基石墨烯复合材料、其制备方法及浮空器 |
| CN106884219A (zh) * | 2017-03-07 | 2017-06-23 | 杭州高烯科技有限公司 | 具有永久防紫外线功能的石墨烯/尼龙6织物及其制备方法 |
| CN107142547A (zh) * | 2017-06-26 | 2017-09-08 | 杭州高烯科技有限公司 | 一种石墨烯改性的阻燃防紫外涤纶纤维及其制备方法 |
| CN107163519A (zh) * | 2017-06-26 | 2017-09-15 | 杭州高烯科技有限公司 | 一种高强度耐熔滴的石墨烯/pet复合板材及其制备方法 |
| CN107190382A (zh) * | 2017-06-26 | 2017-09-22 | 杭州高烯科技有限公司 | 一种石墨烯改性的涤纶混纺织物及其制备方法 |
| CN107189076A (zh) * | 2017-06-26 | 2017-09-22 | 杭州高烯科技有限公司 | 一种多功能的石墨烯/涤纶复合织物及其制备方法 |
| CN107325268A (zh) * | 2017-06-26 | 2017-11-07 | 杭州高烯科技有限公司 | 一种石墨烯/pet纳米复合材料及其制备方法 |
| CN107326474A (zh) * | 2017-06-26 | 2017-11-07 | 杭州高烯科技有限公司 | 一种帘子线用石墨烯涤纶复合纤维及其制备方法 |
| CN107353605A (zh) * | 2017-06-26 | 2017-11-17 | 杭州高烯科技有限公司 | 一种多功能石墨烯/pet复合膜及其制备方法 |
| CN107513151A (zh) * | 2017-08-21 | 2017-12-26 | 杭州高烯科技有限公司 | 一种石墨烯/聚酯纳米复合材料及其制备方法 |
| CN107513162A (zh) * | 2017-08-21 | 2017-12-26 | 杭州高烯科技有限公司 | 一种石墨烯/尼龙6纳米复合材料的制备方法 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2007137011A (ru) * | 2005-03-08 | 2009-04-20 | Инвиста Текнолоджиз, С.А.Р.Л. (Ch) | Сложнополиэфирные композиции с высокой стабильностью размеров |
| ITMI20071003A1 (it) * | 2007-05-18 | 2008-11-19 | Polimeri Europa Spa | Compositi a base di polimeri vinilaromatici aventi migliorate proprieta' di isolamento termico e procedimento per la loro preparazione |
| WO2014087992A1 (ja) * | 2012-12-04 | 2014-06-12 | 昭和電工株式会社 | グラフェンシート組成物 |
| KR101482360B1 (ko) * | 2012-12-28 | 2015-01-13 | 주식회사 포스코 | 산화그래핀, 그래핀-고분자 복합체, 그래핀-고분자 복합체 함유 코팅액, 그래핀-고분자 복합체가 코팅된 강판 및 이들의 제조방법 |
| AU2014228255A1 (en) * | 2013-03-15 | 2015-10-01 | Adama Materials, Inc. | Oligomer-grafted nanofillers and advanced composite materials |
| KR102292918B1 (ko) * | 2013-04-18 | 2021-08-24 | 럿거스, 더 스테이트 유니버시티 오브 뉴저지 | 그래핀으로 강화된 고분자 매트릭스 복합체를 제조하기 위한 원 위치 박리방법 |
| AU2015206411B2 (en) * | 2014-01-17 | 2018-04-26 | Qed Labs Inc | Articles with improved flame retardancy and/or melt dripping properties |
| TWI597311B (zh) | 2014-02-05 | 2017-09-01 | Graphene composite fiber and its preparation method | |
| KR102175291B1 (ko) * | 2014-12-30 | 2020-11-09 | 코오롱플라스틱 주식회사 | 폴리에스터 수지 조성물 및 이를 이용한 플라스틱 성형체 제조 |
| CN105017511A (zh) | 2015-08-20 | 2015-11-04 | 浙江万凯新材料有限公司 | 氧化石墨烯改性pet材料的制备方法 |
| US10337124B2 (en) * | 2015-08-26 | 2019-07-02 | Teague Egan | Textile graphene component thermal fiber |
| CN105200547B (zh) * | 2015-10-19 | 2018-06-01 | 南通强生石墨烯科技有限公司 | 一种石墨烯-涤纶纳米复合纤维的制备方法 |
| CN105525381B (zh) | 2015-10-27 | 2018-03-06 | 济南圣泉集团股份有限公司 | 一种含有石墨烯的复合聚酯纤维、其制备方法和用途 |
| CN106832261B (zh) | 2016-12-26 | 2019-07-16 | 伟星集团有限公司 | 一种高性能石墨烯/尼龙6纳米复合材料及其制备方法 |
| CN106868693A (zh) | 2017-04-07 | 2017-06-20 | 江苏工程职业技术学院 | 一种精梳棉与石墨烯涤纶长丝交织多功能床品面料的生产工艺 |
| CN107057058B (zh) | 2017-05-03 | 2019-08-20 | 杭州高烯科技有限公司 | 一种石墨烯/浇铸尼龙复合材料及其制备方法 |
-
2018
- 2018-02-26 BR BR112019027930-0A patent/BR112019027930B1/pt active IP Right Grant
- 2018-02-26 RU RU2020100048A patent/RU2746113C1/ru active
- 2018-02-26 WO PCT/CN2018/077169 patent/WO2019000985A1/zh unknown
- 2018-02-26 JP JP2019571324A patent/JP6963040B2/ja active Active
- 2018-02-26 EP EP18822649.2A patent/EP3626758B1/en active Active
- 2018-02-26 KR KR1020207002385A patent/KR102284847B1/ko active IP Right Grant
- 2018-02-26 US US16/626,546 patent/US11149129B2/en active Active
Patent Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140080962A1 (en) * | 2011-05-03 | 2014-03-20 | The Board Of Regents For Oklahoma State University | Polyethylene terephthalate-graphene nanocomposites |
| CN105540573A (zh) * | 2016-01-27 | 2016-05-04 | 浙江碳谷上希材料科技有限公司 | 一种高溶解性多褶皱干态氧化石墨烯微球及其制备方法 |
| CN105820519A (zh) * | 2016-02-29 | 2016-08-03 | 洛阳尖端技术研究院 | Pet基石墨烯复合材料、其制备方法及浮空器 |
| CN106884219A (zh) * | 2017-03-07 | 2017-06-23 | 杭州高烯科技有限公司 | 具有永久防紫外线功能的石墨烯/尼龙6织物及其制备方法 |
| CN107142547A (zh) * | 2017-06-26 | 2017-09-08 | 杭州高烯科技有限公司 | 一种石墨烯改性的阻燃防紫外涤纶纤维及其制备方法 |
| CN107163519A (zh) * | 2017-06-26 | 2017-09-15 | 杭州高烯科技有限公司 | 一种高强度耐熔滴的石墨烯/pet复合板材及其制备方法 |
| CN107190382A (zh) * | 2017-06-26 | 2017-09-22 | 杭州高烯科技有限公司 | 一种石墨烯改性的涤纶混纺织物及其制备方法 |
| CN107189076A (zh) * | 2017-06-26 | 2017-09-22 | 杭州高烯科技有限公司 | 一种多功能的石墨烯/涤纶复合织物及其制备方法 |
| CN107325268A (zh) * | 2017-06-26 | 2017-11-07 | 杭州高烯科技有限公司 | 一种石墨烯/pet纳米复合材料及其制备方法 |
| CN107326474A (zh) * | 2017-06-26 | 2017-11-07 | 杭州高烯科技有限公司 | 一种帘子线用石墨烯涤纶复合纤维及其制备方法 |
| CN107353605A (zh) * | 2017-06-26 | 2017-11-17 | 杭州高烯科技有限公司 | 一种多功能石墨烯/pet复合膜及其制备方法 |
| CN107513151A (zh) * | 2017-08-21 | 2017-12-26 | 杭州高烯科技有限公司 | 一种石墨烯/聚酯纳米复合材料及其制备方法 |
| CN107513162A (zh) * | 2017-08-21 | 2017-12-26 | 杭州高烯科技有限公司 | 一种石墨烯/尼龙6纳米复合材料的制备方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3626758A4 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20200247974A1 (en) | 2020-08-06 |
| US11149129B2 (en) | 2021-10-19 |
| EP3626758B1 (en) | 2022-04-06 |
| RU2746113C1 (ru) | 2021-04-07 |
| KR20200023420A (ko) | 2020-03-04 |
| EP3626758A4 (en) | 2020-09-16 |
| JP6963040B2 (ja) | 2021-11-05 |
| JP2020525661A (ja) | 2020-08-27 |
| KR102284847B1 (ko) | 2021-08-03 |
| BR112019027930A2 (pt) | 2020-07-14 |
| BR112019027930B1 (pt) | 2023-10-17 |
| EP3626758A1 (en) | 2020-03-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2019000985A1 (zh) | 石墨烯复合材料及其制备方法 | |
| WO2017066937A1 (zh) | 一种石墨烯-涤纶纳米复合纤维的制备方法 | |
| CN107142547B (zh) | 一种石墨烯改性的阻燃防紫外涤纶纤维及其制备方法 | |
| CN105002595B (zh) | 一种含部分还原石墨烯的高分子复合功能纤维及其制备方法 | |
| CA3005917C (en) | Modified fiber and preparation method therefor | |
| CN107190382B (zh) | 一种石墨烯改性的涤纶混纺织物及其制备方法 | |
| CN107189076B (zh) | 一种多功能的石墨烯/涤纶复合织物及其制备方法 | |
| TW200928027A (en) | Fiber and method of forming the same | |
| CN112663167A (zh) | 一种阻燃聚酯纤维及其制备方法 | |
| CN106319680B (zh) | 一种多功能聚酯短纤维的制造方法 | |
| CN107326474B (zh) | 一种帘子线用石墨烯涤纶复合纤维及其制备方法 | |
| CN114921868B (zh) | 一种纳米生物炭改性熔体直纺超细旦聚酯纤维的制备方法 | |
| CN110344160B (zh) | 一种抗菌防静电的运动服面料及其制备方法 | |
| CN111560167A (zh) | 一种抗紫外聚酰胺色母粒及功能纤维的制备方法 | |
| CN107663665B (zh) | 一种高强低收缩特亮绣花线聚酯牵伸丝的制备方法 | |
| KR101235255B1 (ko) | 나노 실리카 입자가 포함된 고강도 폴리에틸렌 멀티필라멘트 연신사의 제조방법 | |
| CN111793849B (zh) | 皮层含羧基功能化的高荧光聚丙烯酸微球的尼龙纤维 | |
| US20240003062A1 (en) | Graphene composite fiber and manufacturing method therefor | |
| KR20190088652A (ko) | 카본블랙 마스터 배치를 포함하는 고강도 폴리에스테르 원사의 제조방법 | |
| CN112680814A (zh) | 一种抗静电涤纶缝纫线专用长丝的制备 | |
| CN110607574A (zh) | 一种高色牢度pps纤维及其制备方法 | |
| JP3688220B2 (ja) | 顔料含有樹脂組成物及びその繊維 | |
| CN108660582B (zh) | 一种石墨烯碳纳米复合布艺材料的制备方法 | |
| CN117402486A (zh) | 一种可纺性高的无机杂化聚酰胺复合物及其制备方法和应用 | |
| KR20120069040A (ko) | 고내열성의 폴리에틸렌테레프탈레이트 나노복합 타이어코드의 제조 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18822649 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019571324 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112019027930 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2018822649 Country of ref document: EP Effective date: 20191220 |
|
| ENP | Entry into the national phase |
Ref document number: 20207002385 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 112019027930 Country of ref document: BR Kind code of ref document: A2 Effective date: 20191226 |