WO2018228475A1 - Syk抑制剂及其使用方法 - Google Patents
Syk抑制剂及其使用方法 Download PDFInfo
- Publication number
- WO2018228475A1 WO2018228475A1 PCT/CN2018/091269 CN2018091269W WO2018228475A1 WO 2018228475 A1 WO2018228475 A1 WO 2018228475A1 CN 2018091269 W CN2018091269 W CN 2018091269W WO 2018228475 A1 WO2018228475 A1 WO 2018228475A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- methyl
- mmol
- amino
- alkyl
- Prior art date
Links
- 0 C**(C)(C)C1=C(*)c2c(*)c(*)c(*)c(*)c2*(*)C1=O Chemical compound C**(C)(C)C1=C(*)c2c(*)c(*)c(*)c(*)c2*(*)C1=O 0.000 description 2
- RECARUFTCUAFPV-UHFFFAOYSA-N C1OCC11CCNCC1 Chemical compound C1OCC11CCNCC1 RECARUFTCUAFPV-UHFFFAOYSA-N 0.000 description 2
- XLSZMDLNRCVEIJ-UHFFFAOYSA-N Cc1c[nH]cn1 Chemical compound Cc1c[nH]cn1 XLSZMDLNRCVEIJ-UHFFFAOYSA-N 0.000 description 2
- LKDJYZBKCVSODK-UHFFFAOYSA-N C(C1)C2NC1CNC2 Chemical compound C(C1)C2NC1CNC2 LKDJYZBKCVSODK-UHFFFAOYSA-N 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N C1CCOCC1 Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- YPWFISCTZQNZAU-UHFFFAOYSA-N C1CCSCC1 Chemical compound C1CCSCC1 YPWFISCTZQNZAU-UHFFFAOYSA-N 0.000 description 1
- GLUUGHFHXGJENI-UHFFFAOYSA-N C1NCCNC1 Chemical compound C1NCCNC1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 1
- YNAVUWVOSKDBBP-UHFFFAOYSA-N C1NCCOC1 Chemical compound C1NCCOC1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 1
- BRNULMACUQOKMR-UHFFFAOYSA-N C1NCCSC1 Chemical compound C1NCCSC1 BRNULMACUQOKMR-UHFFFAOYSA-N 0.000 description 1
- FQPVFKFYCJVHRT-UHFFFAOYSA-N C1OCC1(CCOC1)C1C1C2(COC2)CCSC1 Chemical compound C1OCC1(CCOC1)C1C1C2(COC2)CCSC1 FQPVFKFYCJVHRT-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N C1OCCOC1 Chemical compound C1OCCOC1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- JBYHSSAVUBIJMK-UHFFFAOYSA-N C1OCCSC1 Chemical compound C1OCCSC1 JBYHSSAVUBIJMK-UHFFFAOYSA-N 0.000 description 1
- LOZWAPSEEHRYPG-UHFFFAOYSA-N C1SCCSC1 Chemical compound C1SCCSC1 LOZWAPSEEHRYPG-UHFFFAOYSA-N 0.000 description 1
- OVRKATYHWPCGPZ-UHFFFAOYSA-N CC1CCOCC1 Chemical compound CC1CCOCC1 OVRKATYHWPCGPZ-UHFFFAOYSA-N 0.000 description 1
- AYIXGVABNMIOLK-ZCFIWIBFSA-N CN(CCC1)C[C@@H]1C(O)=O Chemical compound CN(CCC1)C[C@@H]1C(O)=O AYIXGVABNMIOLK-ZCFIWIBFSA-N 0.000 description 1
- KYSKUNOHWWIDDX-UHFFFAOYSA-N CN(c1cc(-c2c[nH]nc2)ccc1N=C1Nc(cc2OC)ccc2N2CCOCC2)C1=O Chemical compound CN(c1cc(-c2c[nH]nc2)ccc1N=C1Nc(cc2OC)ccc2N2CCOCC2)C1=O KYSKUNOHWWIDDX-UHFFFAOYSA-N 0.000 description 1
- OFNWMZRPXFBZPX-UHFFFAOYSA-N CN1C2CNCC1CC2 Chemical compound CN1C2CNCC1CC2 OFNWMZRPXFBZPX-UHFFFAOYSA-N 0.000 description 1
- LHFSJMKERCANGV-JHJMLUEUSA-N CN1CC(C[C@H]2CCNCCC2)SCC1 Chemical compound CN1CC(C[C@H]2CCNCCC2)SCC1 LHFSJMKERCANGV-JHJMLUEUSA-N 0.000 description 1
- PAMIQIKDUOTOBW-UHFFFAOYSA-N CN1CCCCC1 Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 1
- SRUHHGSXPIHREW-GVHYBUMESA-N C[C@H](CO1)C1[IH]N1CCOCC1 Chemical compound C[C@H](CO1)C1[IH]N1CCOCC1 SRUHHGSXPIHREW-GVHYBUMESA-N 0.000 description 1
- RIKMMFOAQPJVMX-UHFFFAOYSA-N Cc1c[nH]nc1 Chemical compound Cc1c[nH]nc1 RIKMMFOAQPJVMX-UHFFFAOYSA-N 0.000 description 1
- QOHGKFZAJXSXMS-UHFFFAOYSA-N Cc1cc(F)c(CCc2ccc(C)cn2)cc1 Chemical compound Cc1cc(F)c(CCc2ccc(C)cn2)cc1 QOHGKFZAJXSXMS-UHFFFAOYSA-N 0.000 description 1
- BKRYFHAXBGUIOI-UHFFFAOYSA-N Cc1cnc(N)[nH]1 Chemical compound Cc1cnc(N)[nH]1 BKRYFHAXBGUIOI-UHFFFAOYSA-N 0.000 description 1
- GUABFMPMKJGSBQ-UHFFFAOYSA-N Cc1cnc(N)[s]1 Chemical compound Cc1cnc(N)[s]1 GUABFMPMKJGSBQ-UHFFFAOYSA-N 0.000 description 1
- LXBGSDVWAMZHDD-UHFFFAOYSA-N Cc1ncc[nH]1 Chemical compound Cc1ncc[nH]1 LXBGSDVWAMZHDD-UHFFFAOYSA-N 0.000 description 1
- VZWOXDYRBDIHMA-UHFFFAOYSA-N Cc1ncc[s]1 Chemical compound Cc1ncc[s]1 VZWOXDYRBDIHMA-UHFFFAOYSA-N 0.000 description 1
- RTLUPHDWSUGAOS-UHFFFAOYSA-N Ic1ccncc1 Chemical compound Ic1ccncc1 RTLUPHDWSUGAOS-UHFFFAOYSA-N 0.000 description 1
- RUKDVLFJSMVBLV-UHFFFAOYSA-N Ic1n[nH]cc1 Chemical compound Ic1n[nH]cc1 RUKDVLFJSMVBLV-UHFFFAOYSA-N 0.000 description 1
- NWAGBIGYWRXBAI-UHFFFAOYSA-N Nc1nc(I)c[s]1 Chemical compound Nc1nc(I)c[s]1 NWAGBIGYWRXBAI-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
Definitions
- the present application belongs to the field of medical technology and relates to a class of Syk inhibitors or pharmaceutically acceptable salts thereof, a process for the preparation thereof and a pharmaceutical composition thereof.
- Syk spleen tyrosine kinase is an intracellular tyrosine protein kinase belonging to the ZAP70 protein kinase family. Syk plays a key role in the early development of B cells, the development of lymphocytes, and the function of mature B cells. In this process it is involved in a variety of signal transduction pathways and does not need to be activated by phosphorylation of Src kinase. In addition to being commonly expressed in hematopoietic stem cells, Syk is expressed in non-hematopoietic cells such as epithelial cells, hepatocytes, fibroblasts, nerve cells, and breast tissues, and has multiple functions.
- Syk PTK dysfunction such as allergic reactions, asthma, inflammation and autoimmune diseases
- Syk is an important mediator of acute or chronic inflammation.
- Activation of Syk is present in several common B-cell malignancies, such as in follicular lymphoma, diffuse large B-cell lymphoma, mantle cell lymphoma, and B-cell chronic lymphocytic leukemia.
- Dependent phosphorylation of Syk The researchers found that inhibition of Syk in follicular lymphoma and diffuse large B-cell lymphoma cells can reduce the phosphorylation level of downstream signaling molecules, thereby inhibiting the proliferation and survival of tumor cells.
- Syk activity can be used to treat a particular type of cancer, including B cell lymphoma and leukemia.
- W is C(R 7 ) or N
- R 1 and R 2 are independently selected from the group consisting of H, halogen, amino, hydroxy, cyano, C 1-6 alkyl, C 3-6 cycloalkyl, 3 to 10 membered heterocycloalkyl, 6 to 12 aryl. Or a 5- to 12-membered heteroaryl group, the amino group, a C 1-6 alkyl group, a C 3-6 cycloalkyl group, a 3 to 10 membered heterocycloalkyl group, a 6 to 12 membered aryl group or a 5 to 12 membered heteroaryl group.
- the base is optionally substituted by R 8 ;
- R 3 , R 4 , R 7 are independently selected from H, halogen, amino, hydroxy, cyano, C 1-6 alkyl, C 3-6 cycloalkyl or 3- to 6-membered heterocycloalkyl, said amino a C 1-6 alkyl group, a C 3-6 cycloalkyl group or a 3-6 membered heterocycloalkyl group optionally substituted by R 9 ;
- R 5 is selected from the group consisting of H, C 1-6 alkyl, C 3-6 cycloalkyl, 3-6 -membered heterocycloalkyl, C 1-6 alkyl C(O), C 3-6 cycloalkyl C ( O), 3- to 6-membered heterocycloalkyl C(O), phenyl C(O), 5- to 6-membered heteroaryl C(O), C 1-6 alkyl SO 2 , C 3-6 naphthenic a base SO 2 , a 3- to 6-membered heterocycloalkyl SO 2 , a phenyl SO 2 or a 5- to 6-membered heteroaryl SO 2 , said C 1-6 alkyl group, C 3-6 cycloalkyl group, 3 to 6 Aminoheterocycloalkyl, C 1-6 alkyl C(O), C 3-6 cycloalkyl C(O), 3- to 6-membered heterocycloalkyl C(O), phenyl C(O), 5 ⁇
- X is selected from a 3 to 12 membered ring in which the H atom is lost at any two positions, and the ring is optionally substituted with R 9 ;
- L is selected from the group consisting of a bond, NH, O, S, SO, SO 2 , C(O), OC(O), C(O)O, C(O)NH, NHSO 2 , SO 2 NH, NHC(O)NH Or NHSO 2 NH;
- R 6 is selected from H, halogen, amino, hydroxy, cyano, C 1-6 alkyl, C 3-10 cycloalkyl or 3 to 10 membered heterocycloalkyl, said amino group, C 1-6 alkyl group, C 3-10 cycloalkyl or 3 to 10 membered heterocycloalkyl is optionally substituted by R 10 ;
- R 8 , R 9 are independently selected from halogen, amino, hydroxy, cyano, C 1-3 alkyl, C 1-3 alkoxy or COOH;
- R 1 and R 2 are selected from a 6- to 12-membered aryl group or a 5- to 12-membered heteroaryl group, and the 6- to 12-membered aryl group or a 5- to 12-membered heteroaryl group is optionally substituted by R 8 .
- R 1 , R 2 are independently selected from H, halo or 5 to 12 membered heteroaryl, and said 5 to 12 membered heteroaryl is optionally substituted by R 8 .
- R 1 , R 2 are independently selected from the group consisting of H, F, Cl, Br, furyl, thienyl, pyrrolyl, pyrazolyl, imidazolyl, pyridyl, pyrimidine Or pyrazinyl, pyrazinyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, tetrazolyl or triazinyl, said furyl, thienyl, pyrrolyl, pyrazolyl, imidazole , pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, tetrazolyl or triazinyl is optionally substituted with R 8.
- R 1 , R 2 are independently selected from H, F, Cl, thiazolyl, pyrazolyl, imidazolyl or pyridyl, said thiazolyl, pyrazolyl, The imidazolyl or pyridyl group is optionally substituted by R 8 .
- R 8 is selected from amino, methyl, ethyl, propyl or isopropyl.
- R 8 is selected from methyl or amino group.
- R 1 is selected from the group consisting of H, F, It may optionally be substituted by R 8 .
- R 1 is selected from the group consisting of H, F,
- R 2 is selected from H, F, Cl or
- R 1 is selected from a 5 to 12 membered heteroaryl; R 2 is selected from H or halo; wherein, R 1 may optionally be substituted with R 8.
- R 1 is selected from the group consisting of furanyl, thienyl, pyrrolyl, pyrazolyl, imidazolyl, pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, thiazolyl, Isothiazolyl, oxazolyl, isoxazolyl, tetrazolyl or triazinyl;
- R 2 is selected from H, F, Cl or Br; wherein R 1 may be optionally substituted by R 8 .
- R 1 is selected from thiazolyl, pyrazolyl, imidazolyl or pyridyl; R 2 is selected from H, F or Cl; wherein R 1 is optionally R 8 replaced.
- R 1 is selected from R 2 is selected from H, F or Cl; wherein R 1 may be optionally substituted by R 8 .
- R 1 is selected from R 2 is selected from H, F or Cl.
- R 1 is selected from H or halo;
- R 2 is selected from 5-12 membered heteroaryl; wherein, R 2 may be optionally substituted with R 8.
- R 1 is selected from H, F, Cl or Br
- R 2 is selected from the group consisting of furyl, thienyl, pyrrolyl, pyrazolyl, imidazolyl, pyridyl, pyrimidinyl And pyridazinyl, pyrazinyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, tetrazolyl or triazinyl; wherein R 2 may be optionally substituted by R 8 .
- R 1 is selected from H or F;
- R 2 is selected from pyrazolyl; wherein, R 2 may be optionally substituted with R 8.
- R 3 , R 4 , R 7 are independently selected from H, halo, C 1-6 alkyl or C 3-6 cycloalkyl, said C 1-6 The alkyl or C 3-6 cycloalkyl group is optionally substituted by R 9 .
- R 3 , R 4 , R 7 are independently selected from H or halogen.
- R 3 , R 4 , R 7 are independently selected from H, F or Cl.
- W is N, and R 3 and R 4 are independently selected from H, F, or Cl.
- R 5 is selected from H, C 1-6 alkyl or C 3-6 cycloalkyl, wherein C 1-6 alkyl or C 3-6 cycloalkyl The choice is replaced by R 9 .
- R 5 is selected from the group consisting of methyl, ethyl, propyl, isopropyl, butyl, isobutyl or tert-butyl, said methyl, ethyl, propyl , isopropyl, butyl, isobutyl or tert-butyl is optionally substituted with R 9.
- R 5 is selected from methyl
- X is selected from benzene rings which lose H atoms in any two positions, Furan ring, thiophene ring, pyrrole ring, pyrazole ring, imidazole ring, pyridine ring, pyrimidine ring, pyridazine ring, pyrazine ring, thiazole ring, isothiazole ring, oxazole ring, isoxazole ring, tetrazole ring or triazine ring, the X may be optionally substituted with R 9.
- One embodiment of the compounds (I) of the present application wherein, X is selected from any two positions lost benzene or pyridine ring H atoms of the X-R 9 may be optionally substituted.
- X is selected from The X may be optionally substituted with R 9 .
- X is selected from
- R 9 is selected from halo, C 1-3 alkyl or a C 1-3 alkoxy group.
- R 9 is selected from F, Cl, methyl, or OCH 3.
- L is selected from the group consisting of a bond, NH, O, S, SO, SO 2 , NHSO 2 , SO 2 NH or NHSO 2 NH.
- L is selected from the group consisting of a bond, NH, O, S, SO or SO 2 .
- L is selected from the group consisting of a bond, NH or SO 2 .
- R 6 is selected from H, amino, C 1-6 alkyl, C 3-6 cycloalkyl or 3 to 10 membered heterocycloalkyl, said amino group, C The 1-6 alkyl group, the C 3-6 cycloalkyl group or the 3- to 10-membered heterocycloalkyl group may be optionally substituted by R 10 .
- R 6 is selected from H, amino, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, cyclopropyl, Butyl, cyclopentyl, cyclohexyl, losing a H atom at any position
- R 6 is selected from the group consisting of H, NH 2 , methyl, isopropyl, cyclobutyl, The NH 2 , methyl, isopropyl, cyclobutyl, It may optionally be substituted by R 10 .
- R 6 is selected from the group consisting of H, NH 2 , methyl,
- the compound of formula (I) has formula (II),
- R 2 is selected from 6 to 12 membered aryl or 5 to 12 membered heteroaryl, and the 6 to 12 membered aryl or 5 to 12 membered heteroaryl is optionally substituted with R 8 ;
- R 1 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , X and L are as defined in the formula (I).
- R 2 is selected from pyrazolyl, which may be optionally substituted by R 8 .
- R 2 is selected from
- the compound of formula (I) has formula (III),
- R 1 , R 3 , R 4 , R 6 , R 7 and X are as defined in formula (I).
- the compound of formula (I) has formula (IV),
- R 1 is selected from 6 to 12 membered aryl or 5 to 12 membered heteroaryl, and R 1 may be optionally substituted by R 8 ;
- R 2 , R 3 , R 4 , R 5 , R 6 , R 8 , X and L are as defined in the formula (I).
- R 1 is selected from a 5 to 12 membered heteroaryl group, the R 1 may be optionally substituted by R 8.
- R 1 is selected from the group consisting of furanyl, thienyl, pyrrolyl, pyrazolyl, imidazolyl, pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, tetrazolyl or triazinyl, of R 1 may optionally be substituted with R 8.
- R 1 is selected from thiazolyl, pyrazolyl, imidazolyl or pyridinyl, of R 1 may optionally be substituted with R 8.
- R 1 is selected from The R 1 may be optionally substituted with R 8 .
- R 1 is selected from
- the compound of formula (I) has a compound of formula (V),
- R 2 , R 3 , R 4 , R 6 , X and L are as defined in the formula (I).
- the compound of formula (I) is selected from the group consisting of:
- the present application is directed to a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I), or a pharmaceutically acceptable salt thereof, of the present application.
- the pharmaceutical compositions of the present application further comprise one or more pharmaceutically acceptable excipients.
- compositions of the present application can be prepared by combining a compound of the present application, a pharmaceutically acceptable salt thereof, with a suitable pharmaceutically acceptable adjuvant, for example, as a solid, semi-solid, liquid or gaseous preparation, such as a tablet.
- a suitable pharmaceutically acceptable adjuvant for example, as a solid, semi-solid, liquid or gaseous preparation, such as a tablet.
- Typical routes of administration of a compound of the present application, a pharmaceutically acceptable salt thereof, or a pharmaceutical composition thereof include, but are not limited to, oral, rectal, transmucosal, enteral administration, or topical, transdermal, inhalation, parenteral, sublingual , intravaginal, intranasal, intraocular, intraperitoneal, intramuscular, subcutaneous, intravenous.
- the pharmaceutical composition of the present application can be produced by a method well known in the art, such as a conventional mixing method, a dissolution method, a granulation method, a sugar-coating method, a grinding method, an emulsification method, a freeze-drying method, and the like.
- the pharmaceutical compositions can be formulated by admixing the active compound with pharmaceutically acceptable excipients known in the art. These excipients enable the compounds of the present application to be formulated into tablets, pills, troches, dragees, capsules, liquids, gels, slurries, suspensions and the like for oral administration to a patient.
- Solid oral compositions can be prepared by conventional methods of mixing, filling or tabletting. For example, it can be obtained by mixing the active compound with a solid adjuvant, optionally milling the resulting mixture, adding other suitable excipients if necessary, and then processing the mixture into granules to give tablets. Or the core of the sugar coating. Suitable excipients include, but are not limited to, binders, diluents, disintegrants, lubricants, glidants, sweeteners or flavoring agents, and the like.
- compositions may also be suitable for parenteral administration, such as sterile solutions, suspensions or lyophilized products in a suitable unit dosage form.
- suitable excipients such as fillers, buffers or surfactants can be used.
- the compounds of formula (I) described herein, or pharmaceutically acceptable salts thereof, can be administered by any suitable route and method, for example by oral or parenteral (e.g., intravenous) administration.
- a therapeutically effective amount of a compound of formula (I) is from about 0.0001 to 20 mg/kg body weight per day, such as from 0.001 to 10 mg/kg body weight per day.
- the dosage frequency of the compound of formula (I) is determined by the needs of the individual patient, for example, once or twice daily, or more times per day. Administration can be intermittent, for example, wherein during a period of several days, the patient receives a daily dose of the compound of formula (I), followed by a period of several days or more, the patient does not receive each of the compounds of formula (I) Daily dose.
- Another object of the present application is to provide a use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described above, for the manufacture of a medicament for the treatment of a Syk receptor related disorder.
- Another aspect of the present application provides a method of treating a Syk receptor related disorder, the method comprising administering a therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition as described above.
- the Syk receptor associated disorder is selected from a cancer or an inflammatory disease.
- the Syk receptor-associated disorder is selected from the group consisting of a B-cell lymphoma, a Hodgkin's lymphoma, a non-Hodgkin's lymphoma, a hairy cell leukemia, multiple myeloma, chronic myeloid leukemia, acute myeloid leukemia, Chronic lymphocytic leukemia, acute lymphocytic leukemia, rheumatoid arthritis, allergic rhinitis, chronic obstructive pulmonary disease (COPD), adult respiratory distress syndrome (ARDs), allergic-induced inflammatory disease, multiple sclerosis, autoimmune Sexual disease, acute inflammatory response, allergic disorder or polycystic kidney disease.
- COPD chronic obstructive pulmonary disease
- ARDs adult respiratory distress syndrome
- pharmaceutically acceptable is for those compounds, materials, compositions and/or dosage forms that are within the scope of sound medical judgment and are suitable for use in contact with human and animal tissues without Many toxic, irritating, allergic reactions or other problems or complications are commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salt refers to a salt of a compound of the present invention prepared from a compound having a particular substituent found herein and a relatively non-toxic acid or base.
- a base addition salt can be obtained by contacting a suitable base with a neutral form of such a compound.
- an acid addition salt can be obtained by contacting a suitable acid with a neutral form of such a compound.
- Certain specific compounds of the present application contain basic and acidic functional groups which can be converted to any base or acid addition salt.
- Certain compounds of the present application may have asymmetric carbon atoms (optical centers) or double bonds. Racemates, diastereomers, geometric isomers, and individual isomers are included within the scope of this application.
- included in the structure of the compound Can be
- included in the structure of the compound Can be or
- the compounds of the present application may exist in specific geometric or stereoisomeric forms. All such compounds are contemplated by the present application, including cis and trans isomers, (-)- and (+)-enantiomers, (R)- and (S)-enantiomers, diastereoisomers , (D)-isomer, (L)-isomer, and racemic mixtures thereof and other mixtures, such as enantiomerically or diastereomeric enriched mixtures, all of which belong to the present Within the scope of the application. Additional asymmetric carbon atoms may be present in the substituents such as alkyl groups. All such isomers, as well as mixtures thereof, are included within the scope of this application.
- optically active (R)- and (S)-isomers as well as the D and L isomers can be prepared by chiral synthesis or chiral reagents or other conventional techniques. If one enantiomer of a compound of the present application is desired, it can be prepared by asymmetric synthesis or by derivatization with a chiral auxiliary wherein the resulting mixture of diastereomers is separated and the auxiliary group cleaved to provide purity. The desired enantiomer.
- a diastereomeric salt is formed with a suitable optically active acid or base, followed by conventional methods well known in the art.
- the diastereomers are resolved and the pure enantiomer is recovered.
- the separation of enantiomers and diastereomers is generally accomplished by the use of chromatography using a chiral stationary phase, optionally in combination with chemical derivatization (eg, formation of an amino group from an amine). Formate).
- the compounds of the present application may contain unnatural proportions of atomic isotopes on one or more of the atoms that make up the compound.
- radiolabeled compounds can be used, such as deuterium (2 H), tritium (3 H), iodine -125 (125 I) or C-14 (14 C). All isotopic compositional changes of the compounds of the present application, whether radioactive or not, are included within the scope of the present application.
- pharmaceutically acceptable excipient refers to those excipients which have no significant irritating effect on the organism and which do not impair the biological activity and properties of the active compound. Suitable excipients are well known to those skilled in the art, such as carbohydrates, waxes, water soluble and/or water swellable polymers, hydrophilic or hydrophobic materials, gelatin, oils, solvents, water, and the like.
- an effective amount or “therapeutically effective amount” refers to a sufficient amount of a drug or agent that achieves the desired effect. The determination of the effective amount will vary from person to person, depending on the age and general condition of the recipient, and also on the particular active substance, and a suitable effective amount in a case can be determined by one skilled in the art based on routine experimentation.
- active ingredient refers to a chemical entity that is effective in treating a target disorder, disease or condition.
- an ethyl group “optionally” substituted with halo refers to an ethyl group may be unsubstituted (CH 2 CH 3), monosubstituted (e.g., CH 2 CH 2 F), polysubstituted (e.g. CHFCH 2 F, CH 2 CHF 2, etc.) or completely substituted (CF 2 CF 3 ). It will be understood by those skilled in the art that for any group containing one or more substituents, no substitution or substitution pattern that is sterically impossible to exist and/or which cannot be synthesized is introduced.
- C mn means having mn carbon atoms in this moiety.
- C 3-10 cycloalkyl means that the cycloalkyl group has 3 to 10 carbon atoms.
- C 0-6 alkylene group means that the alkylene group has 0 to 6 carbon atoms, and when the alkylene group has 0 carbon atoms, the group is a bond.
- C 1-10 means that the group may have 1 carbon atom, 2 carbon atoms, 3 carbon atoms, 4 carbon atoms, 5 carbon atoms, 6 carbon atoms, 7 carbon atoms, 8 One carbon atom, nine carbon atoms or ten carbon atoms.
- substituted means that any one or more hydrogen atoms on a particular atom are replaced by a substituent as long as the valence of the particular atom is normal and the substituted compound is stable.
- any variable eg, R
- its definition in each case is independent.
- the group may optionally be substituted with at most two R, and each case has an independent option.
- combinations of substituents and/or variants thereof are permissible only if such combinations result in stable compounds.
- one of the variables When one of the variables is selected from a bond, it indicates that the two groups to which it is attached are directly linked. For example, when L represents a bond in A-L-Z, the structure is actually A-Z.
- substituent When a substituent is vacant, it means that the substituent is absent. For example, when X is vacant in AX, the structure is actually A. When a bond of a substituent can be cross-linked to two atoms on a ring, the substituent can be bonded to any atom on the ring. When the recited substituents do not indicate which atom is attached to a compound included in the chemical structural formula including but not specifically mentioned, such a substituent may be bonded through any atomic phase thereof. Combinations of substituents and/or variants thereof are permissible only if such combinations result in stable compounds. For example, a structural unit It is indicated that it can be substituted at any position on the cyclohexyl or cyclohexadiene.
- halo or halogen, by itself or as part of another substituent, denotes a fluorine, chlorine, bromine or iodine atom.
- haloalkyl is intended to include monohaloalkyl and polyhaloalkyl; for example, the term “halo C 1-3 alkyl” is intended to include, but is not limited to, trifluoromethyl, 2,2,2-trifluoro. Ethyl and 3-bromopropyl and the like. Examples of haloalkyl groups include, but are not limited to, trifluoromethyl, trichloromethyl, pentafluoroethyl, and pentachloroethyl.
- hydroxy refers to -OH.
- cyano refers to -CN.
- amino refers to -NH 2 .
- alkyl refers to a straight or branched saturated aliphatic hydrocarbon group consisting of a carbon atom and a hydrogen atom, such as methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, ⁇ , ⁇ , etc.
- the specific alkyl group includes all isomeric forms thereof, for example, the propyl group includes -CH 2 CH 2 CH 3 , -CH(CH 3 ) 2 , for example, butyl includes -CH 2 CH 2 CH 2 CH 3 ,- CH(CH 3 )(CH 2 CH 3 ), -C(CH 3 ) 3 , -CH 2 CH(CH 3 ) 2 .
- C 1-8 alkyl refers to an alkyl group having from 1 to 8 carbon atoms.
- C1-6 alkyl refers to an alkyl group having from 1 to 6 carbon atoms.
- C 1-4 alkyl refers to an alkyl group having from 1 to 4 carbon atoms.
- C 1-3 alkyl refers to an alkyl group having from 1 to 3 carbon atoms.
- alkoxy refers to -O-alkyl
- cycloalkyl refers to a monocyclic saturated aliphatic hydrocarbon group consisting solely of carbon atoms and hydrogen atoms, such as a C 3-10 cycloalkyl group, preferably a C 3-6 cycloalkyl group, such as a cyclopropyl group, Cyclobutyl, cyclopentyl, cyclohexyl and the like.
- hetero denotes a hetero atom or a hetero atomic group (ie, a radical containing a hetero atom), including atoms other than carbon (C) and hydrogen (H), and radicals containing such heteroatoms, including, for example, oxygen (O).
- ring means substituted or unsubstituted cycloalkyl, heterocycloalkyl, cycloalkenyl, heterocycloalkenyl, cycloalkynyl, heterocycloalkynyl, aryl or heteroaryl.
- the ring includes a single ring, a ring, a spiral ring, a ring or a bridge ring.
- the number of atoms on the ring is usually defined as the number of elements of the ring.
- “5 to 7-membered ring” means 5 to 7 atoms arranged in a circle.
- the ring optionally contains from 1 to 3 heteroatoms.
- “5- to 7-membered ring” includes, for example, phenyl, pyridine, and piperidinyl.
- heterocycloalkyl refers to a cyclic group that is fully saturated and can exist as a monocyclic, bicyclic or spiro ring. Unless otherwise indicated, the heterocyclic ring is typically a 3 to 10 membered ring containing from 1 to 3 heteroatoms (preferably 1 or 2 heteroatoms) independently selected from sulfur, oxygen and/or nitrogen.
- 3-membered heterocycloalkyl groups include, but are not limited to, oxiranyl, cyclohexylethane, cycloalkylethane, non-limiting examples of 4-membered heterocycloalkyl including, but not limited to, azetidinyl, acetophenan
- Examples of a cyclic group, a thibutyl group, a 5-membered heterocycloalkyl group include, but are not limited to, tetrahydrofuranyl, tetrahydrothiophenyl, pyrrolidinyl, isoxazolidinyl, oxazolidinyl, isothiazolidinyl, thiazolidine
- Examples of the group, imidazolidinyl group, tetrahydropyrazolyl group, 6-membered heterocycloalkyl group include, but are not limited to, piperidinyl, tetrahydropyranyl, tetrahydrothio
- aryl refers to an all-carbon monocyclic or fused polycyclic aromatic ring group having a conjugated ⁇ -electron system.
- an aryl group can have 6-20 carbon atoms, 6-14 carbon atoms or 6-12 carbon atoms.
- Non-limiting examples of aryl groups include, but are not limited to, phenyl, naphthyl, anthracenyl, 1,2,3,4-tetrahydronaphthalene, and the like.
- heteroaryl refers to a monocyclic or fused polycyclic ring system containing at least one ring atom selected from N, O, S, preferably containing 1, 2 or 3 ring atoms selected from N, O or S. The remaining ring atoms are C and have at least one aromatic ring.
- Preferred heteroaryl groups have a single 4 to 8 membered ring, especially a 5 to 8 membered ring, or a plurality of fused rings containing from 6 to 14, especially from 6 to 10 ring atoms.
- heteroaryl groups include, but are not limited to, pyrrolyl, furyl, thienyl, imidazolyl, oxazolyl, pyrazolyl, pyridyl, pyrimidinyl, pyrazinyl, quinolinyl, isoquinolinyl , tetrazolyl, triazolyl, triazinyl, benzofuranyl, benzothienyl, fluorenyl, isodecyl and the like.
- the compounds of the present application can be prepared by a variety of synthetic methods well known to those skilled in the art, including the specific embodiments listed below, combinations thereof with other chemical synthesis methods, and those well known to those skilled in the art. Equivalent alternatives, preferred embodiments include, but are not limited to, embodiments of the present application.
- the temperature is Celsius.
- the solvent used in the present application is commercially available.
- TMSCHN 2 stands for trimethylsilylated diazomethane
- Tf 2 O stands for trifluoromethanesulfonic anhydride
- DMAP 4-dimethylaminopyridine
- Pd 2 (dba) 3 stands for three (two) Benzylidene acetone) dipalladium
- Xantphos represents 4,5-bisdiphenylphosphino-9,9-dimethyloxaxan
- Pd(dppf)Cl 2 represents [1,1'-bis(diphenylphosphine) Base ferrocene] palladium dichloride
- NBS stands for N-bromosuccinimide
- DMF stands for N,N-dimethylformamide
- DMSO stands for dimethyl sulfoxide
- DIEA (DIPEA) stands for N, N-diisopropylethylamine
- Pd(OAc) 2 represents palladium acetate
- Brettphos represents 2-(dic
- Step A 6-(1-Benzyl-1H-pyrazol-3-yl)-3-hydroxy-1-methyl-quinoline-2(1H)-one
- Step B [6-(1-Benzyl-1H-pyrazol-3-yl)-1-methyl-2-oxo-1,2-dihydro-quinolin-3-yl]trifluoromethanesulfonic acid ester
- Step C 6-(1-Benzyl-1H-pyrazol-3-yl)-1-methyl-3-((4-morpholinephenyl)amino)quinoline-2(1H)-one
- reaction solution was diluted with 40 mL of water, extracted twice with 40 mL of dichloromethane.
- Step D 1-Methyl-3-((4-morpholinephenyl)amino)-6-(1H-pyrazol-3-yl)quinoline-2(1H)-one
- Trifluoromethanesulfonic anhydride (39.14 g, 138.73 mmol) was added dropwise to 6-bromo-3-hydroxy-1-methylquinoline-2(1H)-one (23.50 g, 92.49 mmol) at 0 ° C under N2.
- Pyridine (21.95 g, 277.47 mmol) and DMAP (1.13 g, 9.25 mmol) in dichloromethane (400 mL). Stir at 25 ° C for 3 hours. The reaction was quenched with 1N hydrochloric acid and the pH was adjusted to 6. The organic phase was washed with saturated sodium chloride (500 mL) Filtration and evaporation gave the title compound.
- Step D 6-Bromo-1-methyl-3-((5-morpholinepyridin-2-yl)amino)quinolin-2(1H)-one
- 6-Bromo-1-methyl-2-oxo-1,2-dihydroquinolin-3-yl trifluoromethanesulfonate (28.00 g, 81.95 mmol) was added to tetrahydrofuran (300 mL) under nitrogen.
- 5-morpholine pyridin-2-amino (16.15 g, 90.15 mmol)
- Pd 2 (dba) 3 (3.75 g, 4.10 mmol)
- Xantphos (4.74 g, 8.20 mmol)
- cesium carbonate 53.40 g, 163.90 mmol
- Step E 1-Methyl-3-((5-morpholinepyridin-2-yl)amino)-6-(1H-pyrazol-3-yl)quinoline-2(1H)-one
- Step C 1-Methyl-3((5-(tetrahydro-2H-pyran-4-yl)pyridin-2-yl)amino)-6-(1-((2-(trimethylsilyl)) Ethoxy)methyl)-1H-pyrazol-3-yl)quinoline-2(1H)-one
- Step D 1-Methyl-6-(1H-pyrazol-3-yl)-((5-)tetrahydro-2H-pyran-4-yl)pyridin-2-yl)amino)quinoline-2 (1H)-ketone
- Step B 1-(6-Nitropyridin-3-yl)-4-(oxetan-3-yl)piperazine
- Example 4B A mixture of Example 4B (990 mg, 3.75 mmol) and palladium on carbon (100 mg, 10% purity) in methanol (150 mL) was reacted in hydrogen (15 psi) at 50 ° C for 16 hours. After filtration through celite, the filter cake was washed with EtOAc (EtOAc) MS-ESI (m/z): 235 (M+1).
- EtOAc EtOAc
- Step D 6-Chloro-1-methyl-3-[[5-[4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]quinoline-2 (1H)-ketone
- Example 4C To a solution of Example 4C (100 mg, 426.80 ⁇ mol), (6-chloro-1-methyl-2-oxa-1,2-dihydroquinolin-3-yl)trifluoromethanesulfonate (under nitrogen) 145.83 mg, 426.80 ⁇ mol), a solution of cesium carbonate (278.12 mg, 853.60 ⁇ mol) in tetrahydrofuran (5 mL) was added Xantphos (49.39 mg, 85.36 ⁇ mol), Pd 2 (dba) 3 (39.08 mg, 42.68 ⁇ mol). Stir at 80 ° C for 12 hours. The mixture was cooled to room temperature, EtOAc (EtOAc)EtOAc. Purification by column chromatography gave the title compound.
- Step E 1-Methyl-3-[[5-(4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-(1H-pyrazole -3-yl)quinoline-2(1H)-one
- Example 4D (86 mg, 201.92 ⁇ mol), 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyridine Azole (78.36 mg, 403.84 ⁇ mol), cesium carbonate (197.37 mg, 605.76 ⁇ mol) was dissolved in dioxane (8 mL), then Brttphos-Pd (32.26 mg, 40.38 ⁇ mol) was added and stirred at 110 ° C for 15 hours. The mixture was cooled to room temperature, EtOAc (EtOAc)EtOAc. The title compound 4 was obtained by preparative HPLC (trifluoroacetic acid system).
- Step A tert-Butyl 4-hydroxy-4-(trifluoromethyl)piperidine-1-carboxylate
- Trimethyl(trifluoromethyl)silane was added dropwise to a solution of tert-butyl 4-carbonylhexahydropyridine-1-carboxylate (1.2 g, 6.02 mmol) in DMF (10 mL). 3.85g, 27.10mmol), after stirring at 25 ° C for 2 hours, was added with water (100 mL), and the aqueous layer was extracted with ethyl acetate (100 mL ⁇ 3). Washed, dried over sodium sulfate, filtered and evaporated to give title crystal.
- Example 5A (1.6 g, 5.94 mmol) of trifluoroacetic acid (2 mL) and dichloromethane (10 mL) were reacted at 25 ° C for 12 hours under nitrogen. The reaction mixture was evaporated under reduced pressure to give title compound.
- Example 5B (1.67 g, 5.90 mmol), 5-bromo-2-nitropyridine (1.32 g, 6.49 mmol), potassium carbonate (4.08 g, 29.49 mmol), DMF (50 mL) After stirring at ° C for 10 hours, it was diluted with water (50 mL). The aqueous layer was extracted with ethyl acetate (50 mL ⁇ 3). The combined organic layers were washed with EtOAc EtOAc m.
- Step D 1-(6-Aminopyridin-3-yl)-4-(trifluoromethyl)piperidin-4-ol
- Example 5C To a solution of Example 5C (810 mg, 2.78 mmol) in 20 mL of methanol, 10% wet palladium charcoal (81 mg). Then, it was replaced with hydrogen for 3 times, and stirred for 15 hours at 25 ° C under a nitrogen (15 psi) atmosphere.
- Step E 6-Chloro-3-[[5-[4-hydroxy-4-(trifluoromethyl)piperidin-1-yl]pyridin-2-yl]amino]-1-methyl-quinoline- 2(1H)-ketone
- Example 5D (275.24 mg, 1.05 mmol), (6-chloro-1-methyl-2-oxy-3-quinolinyl) trifluoromethanesulfonate (300 mg, 877.99 ⁇ mol), Pd2 (dba) 3 (80.40 mg, 87.80 ⁇ mol), cesium carbonate (572.13 mg, 1.76 mmol), Xantphos (76.20 mg, 131.70 ⁇ mol) in tetrahydrofuran (10.00 mL), and the mixture was stirred at 25 ° C for 4 hours. It was diluted with water (20 mL) and the aqueous layer was extracted with dichloromethane. The combined organic layers were washed with EtOAc EtOAc m.
- Step F 3-((5-(4-Hydroxy-4-(trifluoromethyl)piperidin-1-yl)piperidin-2-yl)amino)-1-methyl-6-(1H-pyridyl Zyrid-3-yl)quinoline-2(1H)-one
- Example 5E (330 mg, 728.70 ⁇ mol), 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyridyl under nitrogen Azole (212.10 mg, 1.09 mmol), [2-(2-aminoethyl)phenyl]-chloro-palladium; biscyclohexyl-[3,6-dimethoxy-2-(2,4,6-three Isopropylbenzene)phenyl]phosphate (58.21 mg, 72.87 ⁇ mol,), cesium carbonate (712.28 mg, 2.19 mmol) in dimethyl sulfoxide (8 mL) and water (2 mL), the mixture was stirred at 120 ° C 10 hour. It was diluted with water (30 mL) and the aqueous layer was extracted with dichloromethane The combined organic layers were washed with EtOAc EtOAc m.
- Step A (E)-N-(4-bromo-3-fluorophenyl)-2-(indolyl)acetamide
- Step E 6-Bromo 7-fluoro-1-methyl-2-oxo-1,2-dihydroquinolin-3-yl-trifluoromethanesulfonate
- Trifluoromethanesulfonic anhydride (1.87 g, 6.62 mmol) was added dropwise to the 6-bromo-7-fluoro-3-hydroxy-1-methylquinoline-2(1H)-one at 1.2 °C under nitrogen. g, 4.41 mmol), pyridine (697.76 mg, 8.82 mmol) and DMAP (538.85 mg, 4.41 mmol) in dichloromethane (20 mL). Stir at 25 ° C for 16 hours. The reaction was quenched with 1N hydrochloric acid and the pH was adjusted to 6. Extract three times with dichloromethane (50 mL). The combined organic layers were washed with saturated sodium s Filtration and evaporation, the ⁇
- Step F 6-Bromo-7-fluoro-1-methyl-3-((5-morpholinepyridin-2-yl)amino)quinoline-2(1H)-one
- Step G 7-Fluoro-1-methyl-3-((5-morpholinepyridin-2-yl)amino)-6-(1H-pyrazol-3-yl)quinoline-2(1H)-one
- NBS (181.54 mg, 1.02 mmol) was added to 4-chloro-1-methyl porphyrin-2,3-dione (200 mg, 1.02 mmol) in acetonitrile (7 mL) and water (7 mL) . After stirring for 12 hours, the reaction mixture was filtered, and the filtered cake was evaporated.
- Step C 6-Bromo-5-chloro-1-methyl-2-oxo-1,2-dihydroquinolin-3-yl-trifluoromethanesulfonate
- Trifluoromethanesulfonic anhydride (1.03 g, 3.65 mmol) was added dropwise to the 6-bromo-5-chloro-3-hydroxy-1-methylquinoline-2(1H)-one (700 mg) at zero temperature under nitrogen. , 2.43 mmol), pyridine (576.64 mg, 7.29 mmol) and DMAP (29.69 mg, 0.243 mmol) in dichloromethane (20 mL). Stir at 25 ° C for 3 hours. The reaction was quenched with 1N hydrochloric acid and the pH was adjusted to 6. Extract three times with dichloromethane (100 mL). The combined organic layers were washed with saturated sodium s Filtration and evaporation, and the ⁇
- Step D 6-Bromo-5-chloro-1-methyl-3-((5-morpholinepyridin-2-yl)amino)quinolin-2(1H)-one
- Step E 5-Chloro-1-methyl-3-((5-morpholinepyridin-2-yl)amino)-6-(1H-pyrazol-3-yl)quinoline-2(1H)-one
- Example 8 can be obtained by referring to the preparation method of Example 6, which is prepared by using 4-bromo-3,5-difluoroaniline.
- Step B 4-Fluoro-1-methyl porphyrin-2,3-dione
- NBS 40.04 g, 224.95 mmol
- 4-fluoro-1-methyl porphyrin-2,3-dione 31.0 g, 173.04 mmol
- acetonitrile 300 mL
- water 600 mL
- Step E 6-Bromo-5-fluoro-1-methyl-2-oxo-1,2-dihydroquinolin-3-yl trifluoromethanesulfonate
- Trifluoromethanesulfonic anhydride (13.48 g, 47.78 mmol) was added dropwise to the 6-bromo-5-fluoro-3-hydroxy-1-methylquinoline-2(1H)-one at 0 ° under nitrogen (10.0) g, 36.76 mmol), pyridine (8.72 g, 110.27 mmol) and DMAP (449.04 mg, 3.68 mmol) in dichloromethane (200 mL). Stir at 15 degrees for 1 hour. The reaction was quenched with water (300 mL) and EtOAc (EtOAc) The organic phase was washed with saturated sodium chloride (250 mL) Filtration and evaporation gave the title compound.
- Step F 6-Bromo-5-fluoro-1-methyl-3-((5-(4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-yl)amino) Quinoline-2(1H)-one
- Step G 5-Fluoro-1-methyl-3-((5-(4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-yl)amino)-6-( 1H-pyrazol-3-yl)quinoline-2(1H)-one
- Step A 6-Bromo-5-fluoro-3-[[5-[4-hydroxy-4-(trifluoromethyl)piperidin-1-yl]pyridin-2-yl]amino]-1-methyl -quinoline-2(1H)-one
- Step B 5-Fluoro-3-[[5-[4-hydroxy-4-(trifluoromethyl)piperidin-1-yl]pyridin-2-yl]amino]-1-methyl-6-( 1H-pyrazol-3-yl)quinoline-2(1H)-one
- Example 10A (220 mg, 426.94 ⁇ mol), 5-(4,4,5,5-tetramethyl 1,3,2-dioxaborolan-2-yl)-1H-pyridine (under nitrogen) 124.27 mg, 640.41 ⁇ mol), Pd(dppf)Cl2 (31.24 mg, 42.69 ⁇ mol), potassium carbonate (177.02 mg, 1.28 mmol) in a mixture of dioxane (8 mL) and water (2 mL) were reacted at 120 ° C for 10 hours. After cooling to room temperature, it was diluted with water (20 mL), EtOAc (EtOAc)EtOAc. Purification by column chromatography gave the title compound 10.
- Example 11 The preparation method of Example 11 can be obtained by referring to the production method of Example 10.
- Step A 7-(6-Nitro-3-pyridine)-2-oxazol-7-azaspiro[3.5]decane
- Step B 5-(2-oxazol-7-azaspiro[3.5]decane-7-yl)pyridin-2-amine
- Example 12A A mixture of Example 12A (1 g, 4.01 mmol) and Raney nickel (34.35 mg, 401.00 ⁇ mol) in methanol (110 mL) was reacted in hydrogen (15 psi) at 30 ° C for 15 hours. After filtration through celite, the filter cake was washed with EtOAc (EtOAc)
- Step C 6-Bromo-5-fluoro-1-methyl-3-[[5-(2-oxazole-7-azaspiro[3.5]decane-7-yl)-2-pyridine]amino] Quinoline-2(1H)-one
- Example 12B To a solution of Example 12B (97.67 mg, 445.40 ⁇ mol), (6-bromo-5-fluoro-1-methyl-2-oxa-3-quinoline) trifluoromethanesulfonate (150.00 mg, 371.17 ⁇ mol), a mixture of cesium carbonate (241.87 mg, 742.34 ⁇ mol) in tetrahydrofuran (5 mL) was added Xantphos (42.95 mg, 74.23 ⁇ mol), Pd 2 (dba) 3 (33.99 mg, 37.12 ⁇ mol). After stirring at 80 ° C for 4 hours, the reaction solution was filtered through a filter paper, and the filter cake was washed three times with a filtrate, and finally, the filter cake was dried to give the title compound.
- Step D 5-Fluoro-1-methyl-3-[[5-(2-oxazole-7-azaspiro[3.5]decane-7-yl)pyridin-2-yl]amino]-6- (1H-pyrazole-3)quinoline-2(1H)-one
- Example 12C 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (131.18 mg, 676.04) under nitrogen. ⁇ mol), potassium carbonate (140.15 mg, 1.01 mmol) dissolved in dioxane (4 mL) and water (1 mL), then Pd(dppf)Cl 2 (24.73 mg, 33.80 ⁇ mol), stirred at 110 ° C 12 hours. The mixture was cooled to room temperature, EtOAc (EtOAc)EtOAc.
- Example 13 The preparation method of Example 13 can be obtained by referring to the production method of Example 12, and preparing with (3R)-3-methylmorpholine.
- the preparation method of Example 14 can be obtained by referring to the preparation method of Example 12, and preparing with (3S)-3-methylmorpholine.
- Example 15 can be prepared by referring to the preparation method of Example 12.
- Example 16 can be prepared by referring to the preparation method of Example 12.
- Example 17 can be prepared by referring to the preparation method of Example 12.
- Step A (3R)-1-(6-nitropyridin-3-yl)piperidine-3-carboxylic acid ethyl ester
- Step B Ethyl (3R)-1-(6-aminopyridin-3-yl)piperidine-3-carboxylate
- Example 18A A mixture of Example 18A (1 g, 3.58 mmol) and EtOAc (30.67 mg) in MeOH (50 mL) After filtration through celite, the filter cake was washed with EtOAc (EtOAc)
- Step C (3R)-1-[6-Bromo-5-fluoro-1-methyl-2-oxa-1,2-dihydroquinolin-3-yl)amino]pyridin-3-yl]piperidin Ethyl pyridine-3-carboxylate
- Example 18B (200 mg, 802.21 ⁇ mol), (6-bromo-5-fluoro-1-methyl-2-oxa-3-quinoline) triflate (356.62 mg, 882.43).
- Xulphos (69.07 mg, 1.2 mmol) in tetrahydrofuran (15 mL) was added Xantphos (69.63 mg, 120.33 ⁇ mol), Pd 2 (dba) 3 (73.46 mg, 80.22 ⁇ mol), and stirred at 15 ° C for 16 hours. . After the addition of water (50 mL), EtOAc EtOAc.
- Step D (3R)-1-[6-[[5-fluoro-1-methyl-2-oxa-6-(1H-pyrazol-3-yl)-1,2-dihydroquinoline- 3-yl]amino]pyridin-3-yl]piperidine-3-carboxylic acid
- Example 1C (350 mg, 695.33 ⁇ mol), 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyridine Azole (148.41 mg, 764.86 ⁇ mol), cesium carbonate (453.10 mg, 1.39 mmol) dissolved in dioxane (8 mL) and water (2 mL), then Pd(dppf)Cl 2 (50.88 mg, 69.53 ⁇ mol) ), stirring at 110 ° C for 12 hours. After cooling to room temperature, the reaction mixture was evaporated to dryness crystals crystals
- Methyl magnesium chloride 70.10 mL, 3 mol was added dropwise to a solution of methyl 6-aminopyridine-3-carboxylic acid methyl ester (3.2 g, 21.03 mmol) in tetrahydrofuran (300 mL) at 0 ° C under nitrogen. After stirring for 15 hours, water (50 mL) was evaporated, evaporated, evaporated, evaporated. Evaporation provided the title compound.
- Step B 6-Bromo-5-fluoro-3-[[5-(2-hydroxypropan-2-yl)pyridin-2-yl]amino]-1-methyl-quinolin-2-one
- Example 20A 400 mg, 2.63 mmol), (6-bromo-5-fluoro-1-methyl-2-oxo-3-quinoline) trifluoromethanesulfonate (1.12 g, 2.76 mmol).
- a mixture of Pd2(dba)3 240.68 mg, 0.263 mmol
- Xantphos 228.12 mg, 0.3945 mmol
- cesium carbonate (1.71 g, 5.26 mmol) in tetrahydrofuran (40 mL) was reacted at 25 ° C for 15 hours. After cooling to room temperature, it was diluted with H.sub.2 (30 mL). EtOAc (EtOAc m. Purification by column chromatography gave the title compound.
- Step C 5-Fluoro-3-[[5-(2-hydroxypropan-2-yl)pyridin-2-yl]amino]-1-methyl-6-(1H-pyridin-3-yl)quinoline -2(1H)-ketone
- Example 20B (100 mg, 0.24615 mmol), 3-(4,4,5,5-tetramethyl 1,3,2-dioxaborolan-2-yl)-1 hydrogen-pyrazole under nitrogen. (52.54 mg, 0.27077 mmol), Pd(dppf)Cl2 (18.01 mg, 0.02462 mmol), a mixture of potassium carbonate (102.06 mg, 0.73845 mmol) in dioxane (2 mL) and water (0.5 mL) stirred at 100 °C After 15 hours, the aqueous layer was diluted with H.sub.2 (20 mL). The combined organic layers were washed with EtOAc EtOAc m.
- Step B 1-(6((diphenylmethylene)amine)pyridin-3-yl)cyclobutanol
- Example 21A To a solution of Example 21A (1 g, 5.45 mmol), benzophenone imine (1.48 g, 8.18 mmol) and cesium carbonate (3.55 g, 10.90 mmol) in dioxane (25 mL) BINAP (339.36 mg, 545 ⁇ mol) and Pd 2 (dba) 3 (249.53 mg, 272.5 ⁇ mol) were stirred at 100 ° C for 12 hours. After cooling to room temperature, it was diluted with water (50 mL), EtOAc (EtOAc m. Purification by column chromatography gave the title compound.
- Example 21B Hydroxylamine hydrochloride (347.45 mg, 5 mmol) was added to a solution of Example 21B (820 mg, 2.5 mmol) and potassium acetate (490.7 mg, 5 mmol) in methanol (10 mL), and the mixture was stirred at 17 ° C for 1 hour, then filtered and filtered. Washing with MeOH (5 mL).
- Step D 6-Bromo-5-fluoro-3-((5-(1-hydroxycyclobutyl)pyridin-2-yl)amine)-1-methylquinolin-2(1H)-one
- Step E 5-Fluoro-3-[[5-(1-hydroxycyclobutyl)pyridin-2-yl]-1-methyl-6-(1H-pyrazol-3-yl)quinoline-2 ( 1H)-ketone
- Example 21D (340 mg, 812.89 ⁇ mol), 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyridyl under N2
- the azole (394.33 mg, 2.03 mmol), potassium carbonate (337.04 mg, 2.44 mmol) was dissolved in dioxane (10 mL) and water (2.5 mL), then Pd(dppf)Cl 2 (59.48 mg, 81.29) ⁇ mol), stirred at 110 ° C for 15 hours; after cooling to room temperature, add 3- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1 Hydrogen-pyrazole (394.33 mg, 2.03 mmol) and Pd(dppf)Cl 2 (59.48 mg, 81.29 ⁇ mol) were further stirred at 110 ° C for 15 hours under nitrogen atmosphere.
- the preparation method of Example 22 can be obtained by referring to the preparation method of Example 21, which is prepared by using the starting material oxetanone.
- Step A 6-Bromo-5-fluoro-3-[[5-(3-fluorooxetan-3-yl)pyridin-2-yl]amino]-1-methyl-quinoline-2 ( 1H)-ketone
- Step B 5-Fluoro-3-[[5-(3-fluorooxetan-3-yl)pyridin-2-yl]amino]-1-methyl-6-(1H-pyrazole-3 -yl)quinoline-2(1H)-one
- Example 23A (130 mg, 307.89 ⁇ mol), 3-(4,4,5,5-tetramethyl 1,3,2-dioxaborolan-2-yl)-1 hydrogen-pyridine under nitrogen. (89.61 mg, 461.84 ⁇ mol), Pd(dppf)Cl2 (22.53 mg, 30.79 ⁇ mol), a mixture of potassium fluoride (53.66 mg, 926.68 ⁇ mol) in dioxane (4 mL) was reacted at 100 ° C for 15 hours. After cooling to rt, EtOAc (EtOAc m. Purification by preparative HPLC gave the title compound 23.
- Step A 2-(6-chloropyridin-3-yl)-1,1,1-trifluoromethyl-propan-2-ol
- Step B 2-[6-(Diphenylmethyleneamino)pyridin-3-yl]-1,1,1-trifluoromethyl-propan-2-ol
- Example 24A (5.5 g, 24.38 mmol), benzophenone imine (6.63 g, 36.57 mmol), Pd2 (dba) 3 (2.23 g, 2.44 mmol), BINAP (2.28 g, 3.66 mmol) And cesium carbonate (15.89 g, 48.76 mmol) in dioxane (100 mL), and the mixture was reacted at 100 ° C for 16 hours. After cooling to room temperature, it was diluted with H.sub.2 (EtOAc (EtOAc) (EtOAc) Purification by column chromatography gave the title compound.
- Step D 6-Bromo-5-fluoro-1-methyl-3-((5-(1,1,1-trifluoro-2-hydroxypropan-2-yl)pyridin-2-yl)amino)quina Porphyrin-2(1H)-one
- Step E 5-Fluoro-1-methyl-6-(1H-pyrazol-3-yl)-3-((5-(1,1,1-trifluoro-2-hydroxypropan-2-yl)) Pyridin-2-yl)amino)quinoline-2(1H)-one
- Step F 5-Fluoro-1-methyl-6-(1H-pyrazol-3-yl)-3-((5-(1,1,1-trifluoro-2-hydroxypropan-2-yl)) Pyridin-2-yl)amino)quinoline-2(1H)-one
- Step B 5-Bromo-N 1 -methylbenzene-1,2-diamine
- Step D 7-(1-Benzyl-1H-pyrazol-4-yl)-1-methylquinoxaline-2,3(1H,4H)-dione
- Step E 7-(1-Benzyl-1H-pyrazol-4-yl)-3-bromo-1-methylquinoxaline-2(1H)-one
- Step F 7-(1-Benzyl-1H-pyrazol-4-yl)-1-methyl-3-((4-morpholinephenyl)amino)quinoxaline-2(1H)-one
- Step G 1-Methyl-3-((4-morpholinephenyl)amino)-7-(1H-pyrazol-4-yl)quinoxaline-2(1H)-one
- Step A 7-(1-Benzyl-1H-pyrazol-3-yl)-1-methylquinoxaline-2,3(1H,4H)-dione
- Step B 7-(1-Benzyl-1H-pyrazol-3-yl)-3-bromo-1-methylquinoxaline-2(1H)-one
- Step C 7-(1-Benzyl-1H-pyrazol-3-yl)-1-methyl-3-((4-morpholinephenyl)amino)quinoxaline-2(1H)-one
- Step D 1-Methyl-3-((4-morpholinephenyl)amino)-7-(1H-pyrazol-3-yl)quinoxaline-2(1H)-one
- Example 28 can be prepared by referring to the preparation method of Example 27.
- Step A 1-methyl-3-((4-morpholinephenyl)amino)-7-(4,4,5,5-tetramethyl-1,3,2-dioxahexrolane- 2-yl)quinoxaline-2(1H)-one
- Step B 1-Methyl-7-(5-methyl-1H-pyrazol-4-yl)-3-((4-morpholinephenyl)amino)quinoxaline-2(1H)-one
- Examples 30-32 can be prepared by referring to the preparation method of Example 29:
- Step C 7-(1-Phenyl-1H-pyrazol-4-yl)-1-methyl-3((4-(4-methylpiperazin-1-yl)phenyl)amino)quinidine Porphyrin-2(1H)-one
- Step D 1-Methyl-3((4-(4-methylpiperazin-1-yl)phenyl)amino)-7-(1H-pyrazol-4-yl)quinoxaline-2 (1H )-ketone
- Example 34 1-Methyl-3-((4-((4-methylpiperazin-1-yl)sulfonyl)phenyl)amino)-7-(1H-pyrazol-4-yl)quina Porphyrin-2(1H)-one
- Step B 4-((4-Methylpiperazin-1-yl)sulfonyl)aniline
- Step C 7-(1-Benzyl-1H-pyrazol-4-yl)-1-methyl-3-((4-((4-methylpiperazin-1-yl)sulfonyl)phenyl) Amino)quinoxaline-2(1H)-one
- the preparation of the step C of Example 34 can be referred to the preparation method of the step C of Example 33.
- Step D 1-Methyl-3-((4-((4-methylpiperazin-1-yl)sulfonyl)phenyl)amino)-7-(1H-pyrazol-4-yl)quinoxaquinone Porphyrin-2(1H)-one
- the preparation of the step D of the example 34 can be referred to the preparation method of the step D of the example 33.
- Example 35 The preparation of Example 35 can be obtained by referring to the production method of Example 33.
- Example 36 3-((4-(4-(ethylsulfonyl)piperazin-1-yl)phenyl)amino)-1-methyl-7-(1H-pyrazol-4-yl)quinoxaline Porphyrin-2(1H)-one
- Example 36 The preparation of Example 36 can be obtained by referring to the production method of Example 33.
- Example 37 1-Methyl-3-((4-(4-methyl-1,4-diazepan-1-yl)phenyl)amino)-7-(1H-pyrazole-4 -yl)quinoxaline-2(1H)-one
- Example 37 The preparation of Example 37 can be obtained by referring to the production method of Example 33.
- Example 38 1-Methyl-3-((4-(piperidin-1-ylsulfonyl)phenyl)amino)-7-(1H-pyrazol-4-yl)quinoxaline-2 (1H )-ketone
- Example 38 The preparation of Example 38 can be obtained by referring to the production method of Example 34.
- Example 39 The preparation of Example 39 can be obtained by referring to the production method of Example 33.
- Example 40 1-Methyl-3-((3-(4-(methylsulfonyl)piperazin-1-yl)phenyl)amino)-7-(1H-pyrazol-4-yl)quinoxaquinone Porphyrin-2(1H)-one
- Example 40 The preparation of Example 40 can be obtained by referring to the production method of Example 33.
- Triethylamine (3.07 mL, 22.15 mmol) and methylamine were added to a solution of 5-bromo-1,3-dichloro-2-nitrobenzene (6 g, 22.15 mmol) in DMF (150 mL)
- the solution (2M, 22.15 mL, 44.3 mmol) was stirred at room temperature for one hour and then heated to 50 ° C for 6 hours. TLC showed the starting material to react completely.
- water (100 mL) and ethyl acetate (150 mL) were added to the mixture. The organic layer was washed with brine, dried over anhydrous sodium sulfate, filtered, and evaporated to give the title compound 5-bromo-3-chloro-N-methyl-2-nitrobenzene.
- Iron powder (3.16 g, 56.5 mmol) was added portionwise to a solution of 5-bromo-3-chloro-N-methyl-2-nitrobenzene (2.5 g, 22.15 mmol) in ethanol (50 mL) and water (50 mL) And acetic acid (0.56 g, 9.42 mmol), stirred at room temperature for one hour and then heated to 60 ° C for 4 hours. TLC showed the starting material to react completely. The mixture was cooled, filtered, and ethyl acetate (150 mL) was evaporated. The organic phase was washed with brine, dried over anhydrous sodium sulfate, filtered, and evaporated to give the title compound 5-bromo-3-chloro-N1-toluene-1,2-diamine.
- Step D 7-Bromo-5-chloro-1-methylquinoxaline-2,3(1H,4H)-dione
- Step F 7-(1-Benzyl-1H-pyrazol-4-yl)-3,5-dichloro-1-methylquinoxaline-2(1H)-one
- Step G 7-(1-Benzyl-1H-pyrazol-4-yl)-5-chloro-1-methyl-3-((4-morpholinephenyl)amino)quinoxaline-2 (1H )-ketone
- Step H 5-Chloro-1-methyl-3-((4-morpholinephenyl)amino)-7-(1H-pyrazol-4-yl)quinoxaline-2(1H)-one
- Example 42A To a solution of Example 42A (20 g, 86.56 mmol), NCS (11.79 g, 88.29 mmol) in DMF (300 mL) at 40 ° C for 18 hours, and diluted with water (500 mL) with ethyl acetate (500 mL ⁇ 2) )extraction. The combined organic layers were washed with EtOAc EtOAc m.
- Step C 5-Bromo-4-chloro-N1-methylbenzene-1,2- hydrazine
- Step D 7-Bromo-6-chloro-1-methylquinoxaline-2,3(1H,4H)-dione
- Step E 7-(1-Benzyl-1H-pyrazol-4-yl)-6-chloro-1-methylquinoxaline-2,3(1H,4H)-dione
- Example 42D (1 g, 3.45 mmol), 1-benzyl-4-(4,4,5,5-tetramethyl 1,3,2-dioxaborolan-2-yl) under N2.
- Pyrazole (1.08 g, 3.8 mmol), Pd(dppf)Cl 2 (282.08 mg, 345.41 ⁇ mol), potassium carbonate (954.79 mg, 6.91 mmol) in DMF (15.00 mL) dioxane (15.00 mL) and water ( In 5.00 mL)
- the mixture was stirred at 100 ° C for 5 hours. Cool to room temperature, dilute with water (100 mL) and filter. The filter cake was purified by column chromatography to give the title compound.
- Step F 7-(1-Benzyl-1H-pyrazol-4-yl)-3,6-dichloro-1-methylquinoxaline-2(1H)-one
- Example 42E 900 mg, 2.45 mmol
- DIEA 265.98 mg, 2.06 mmol
- toluene 9 mL
- EtOAc EtOAc
- Step G 7-(1-Benzyl-1H-pyrazol-4-yl)-6-chloro-1-methyl-3-((4-morpholinephenyl)amino)quinoxaline-2 (1H )-ketone
- Example 42F To a solution of Example 42F (100 mg, 259.57 ⁇ mol), 4-morpholine aniline (92.53 mg, 519.14 ⁇ mol) in acetonitrile (2 mL), the mixture was reacted at 80 ° C for 18 hours, and the reaction mixture was evaporated under reduced pressure. Purification by column chromatography gave the title compound.
- Step H 6-Chloro-1-methyl-3-((4-morpholinephenyl)amino)-7-(1H-pyrazol-4-yl)quinoxaline-2(1H)-one
- Example 42G (80 mg, 151.80 ⁇ mol), t-BuOK (1M, 1.06 mL), DMSO (1.00 mL), and the mixture was stirred at 25 ° C for 4 hours.
- the reaction solution was diluted with ice water (10 mL), stirred for 10 min, and then adjusted to pH 8 with 1M hydrochloric acid. After extraction with methylene chloride (10 mL, EtOAc), EtOAc (EtOAc)
- Example 43 The preparation of Example 43 can be obtained by referring to the preparation method of Example 42.
- Methylamine (21.01 mL, 2.0 mol/L) was added to a solution of 5-bromo-1,3-difluoro-2-nitrobenzene (10.00 g, 42.02 mmol) in DMF (100.00 mL) at 0 °. .
- the reaction solution was stirred at 0 ° C to room temperature for 16 hours.
- the reaction solution was poured into water (500.00 mL)
- the organic phase was washed twice with EtOAc EtOAc EtOAc.
- Oxalyl chloride monoethyl ester (4.36 g, 31.96 mmol) was added dropwise to 5-bromo-3-fluoro-N1-methylbenzene-1,2 diamine (7.00 g, 31.96 mmol) at 0 ° C under N2.
- the mixture was reacted with a solution of triethylamine (8.08 g, 79.89 mmol) in 1,2-dichloroethane (70.00 mL) at room temperature for 2 hours, and a white solid was precipitated, and the mixture was heated to 60 ° C and stirred for 3 hours.
- the reaction mixture was cooled to rt.
- Step D 7-(1-Benzyl-1H-pyrazol-4-yl)-5-fluoro-1-methylquinoxaline-2,3(1H,4H)-dione
- reaction solution was cooled to room temperature, and water (100 mL) was added to the mixture, and the mixture was extracted twice with dichloromethane (100 mL). The organic phase was washed twice with saturated brine (100 mL) and dried over anhydrous sodium sulfate. It was beaten with ethyl acetate to give the title compound.
- Step E 7-(1-Benzyl-1H-pyrazol-4-yl)-3-bromo-5-fluoro-1-methylquinoxaline-2(1H)-one
- Phosphorus tribromide (368.23 mg, 1.28 mmol) was added dropwise to 7-(1-benzyl-1H-pyrazol-4-yl)-5-fluoro-1-methylquinoxaline at 0 °C.
- a solution of 2,3(1H,4H)-dione (300.00 mg, 856.29 ⁇ mol) and DIEA (88.53 mg, 685.03 ⁇ mol) in toluene (8 mL) was stirred at 110 ° C for 1.5 hours under nitrogen.
- the reaction mixture was filtered over EtOAc (EtOAc)EtOAc.
- the organic phase was washed twice with EtOAcqqqqqqqqqm
- Step F 7-(1-Benzyl-1H-pyrazol-4-yl)-5-fluoro-1-methyl-3-((4-morpholinephenyl)amino)quinoxaline-2 (1H )-ketone
- Step G 5-Fluoro-1-methyl-3-((4-morpholinephenyl)amino)-7-(1H-pyrazol-4-yl)quinoxaline-2(1H)-one
- Step A 5-Bromo-4-fluoro-N1-methyl-benzene-1,2- hydrazine
- Zinc powder (7.88 g, 120.45 mmol) was added to a solution of 4-bromo-5-fluoro-N2-methyl-benzene-1,2- hydrazine (6 g, 24.09 mmol) in ethanol (120 mL). And the formic acid amine (7.6 g, 120.45 mmol), after stirring at 50 ° C for 2 hours, the reaction solution was filtered, the filter cake was washed with dichloromethane (500 mL), and the filtrate was washed with water (200 mL) Wash with water (500 mL), dry EtOAc EtOAc
- Step B 7-Bromo-6-fluoro-1-methylquinoxaline-2,3(1H,4H)-dione
- Example 45B (2.4 g, 8.79 mmol), triethylamine (1.33 g, 13.18 mmol), phosphorus bromide (7.56 g, 26.37 mmol), 1,2-dichloroethane (50 mL) After the mixture was stirred at 90 ° C for 6 hours, the reaction mixture was poured into cold sodium hydrogen sulfate (300 mL) and stirred for 10 min.
- Step D 7-Bromo-6-fluoro-1-methyl-3-((4-morpholinephenyl)amino)quinoxaline-2(1H)-one
- Example 45C To a solution of Example 45C (1.95 g, 5.80 mmol), sodium acetate (1.43 g, 17.41 mmol), 4-morpholine aniline (1.24 g, 6.97 mmol) in isopropyl alcohol (30 mL) The reaction was carried out at 100 ° C for 12 hours, the mixture was cooled to room temperature and filtered to give the title compound.
- Step E 6-Fluoro-1-methyl-3-((4-morpholinephenyl)amino)-7-(1H-pyrazol-4-yl)quinoxaline-2(1H)-one
- Example 45D (2.3 g, 5.31 mmol), 4-(4,4,5,5-tetramethyl 1,3,2-dioxaborolan-2-yl)-1H-pyridine a mixture of azole (1.55 g, 7.96 mmol), Pd(dppf)Cl 2 (388.43 mg, 530.85 ⁇ mol), potassium carbonate (2.2 mg, 15.93 mmol) in dioxane (40.00 mL) and water (10.00 mL) Stir at 120 ° C for 10 hours. After cooling to room temperature, the aqueous layer was diluted with water (100 mL). The combined organic layers were washed with EtOAcqqqqqqqqqqqqqqqqqqqqqqqqqqqqqq
- Example 46 can be obtained by referring to the preparation method of Example 44.
- Step C 7-(1-Phenyl-1H-pyrazol-4-yl)-3-((2-fluoro-4-morpholinephenyl)amino)-1-methylquinoxaline-2 (1H )-ketone
- Step D 3-((2-Fluoro-4-morpholinephenyl)amino)-1-methyl-7-(1H-pyrazol-4-yl)quinoxaline-2(1H)-one
- Example 48 The preparation of Example 48 can be obtained by referring to the preparation method of Example 47.
- Example 49 The preparation of Example 49 can be obtained by referring to the preparation method of Example 47.
- Example 50 can be obtained by referring to the preparation method of Example 47.
- Example 51 can be obtained by referring to the preparation method of Example 47.
- Example 52 can be obtained by referring to the preparation method of Example 47.
- Example 53 can be obtained by referring to the preparation method of Example 47 using different amines.
- Example 54 3-[4-(3,8-Diazabicyclo[3.2.1]octane-8-yl)aniline]-1-methyl-7-(1H-pyrazol-4-yl)quina Porphyrin-2(1H)-one
- Step A tert-Butyl-8-(4-nitrophenyl)-3,8-diazabicyclo[3.2.1]octane-3-carboxylate
- Step B tert-Butyl-8-(4-aminophenyl)-3,8-diazabicyclo[3.2.1]octane-3-carboxylate
- Step C tert-Butyl-8-[4-[[6-(1-benzyl-1H-pyrazol-4-yl)-4-methyl-3-oxo-3,4-dihydroquinoxaline ⁇ -2-yl]amino]phenyl]-3,8-diazabicyclo[3.2.1]octane-3-carboxylate
- Step D tert-Butyl-8-[4-[[4-methyl-3-oxo-6-(1H-pyrazol-4-yl)-3,4-dihydroquinoxalin-2-yl Amino]phenyl]-3,8-diazabicyclo[3.2.1]octane-3-carboxylate
- Step E 3-[4-(3,8-Diazabicyclo[3.2.1]octane-8-yl)aniline]-1-methyl-7-(1H-pyrazol-4-yl)quinidine Porphyrin-2(1H)-one
- Step B Ethyl 1-(4-aminophenyl)piperidine-3-carboxylate
- Step C 1-(4-((6-(1-phenyl-1H-pyrazol-4-yl)-4-methyl-3-oxo-3,4-dihydroquinoxaline-2- Ethyl)amino)phenyl)piperidine-3-carboxylic acid ethyl ester
- Step D 1-(4-((4-Hero-3-oxo-6-(1H-pyrazol-4-yl)-3,4-dihydroquinoxalin-2-yl)amino)benzene Piperidine-3-carboxylic acid
- Example 56 1-Methyl-7-(1H-pyrazol-4-yl)-3-((6-((tetrahydro-2H-pyran-4-yl)amino)pyridin-3-yl) Amino)quinoxaline-2(1H)-one
- Step B N 2 -(tetrahydro-2H-pyran-4-yl)pyridine-2,5-diamine
- Step C 7-Bromo-1-methyl-3-((6-((tetrahydro-2H-pyran-4-yl)amino)pyridin-3-yl)amino)quinoxaline-2(1H) -ketone
- Step D 1-Methyl-7-(1H-pyrazol-4-yl)3-((6-((tetrahydro-2H-pyran-4-yl)amino)pyridin-3-yl)amino) Quinoxaline-2(1H)-one
- Step A 7-Imidazo[1,2-a]pyrimidin-3-yl-1-methyl-3-((4-morpholinephenyl)amino)quinoxaline-2(1H)-one
- Step B 7-(2-Amino-1H-imidazol-5-yl)-1-methyl-3-((4-morpholinephenyl)amino)quinoxaline-2(1H)-one
- Step A 1-methyl-3-((4-morpholinephenyl)amino)-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborane-2- Quinoxaline-2(1H)-one
- Step B tert-Butyl (5-(4-methyl-2-((4-morpholinephenyl)amino)-3-oxa-3,4-dihydroquinoxalin-6-yl)thiazole- 2-()((2-(trimethylsilyl)ethoxy)methyl)formate
- Step C 7-(2-Aminothiazol-5-yl)-1-methyl-3-((4-morpholinephenyl)amino)quinoxaline-2(1H)-one
- Example 59 The preparation of Example 59 can be obtained by referring to the preparation method of Example 58.
- HTRF homogeneous time-resolved fluorescence
- SYK tyrosine
- DTT Dithiothreitol
- ATP Adenosine triphosphate
- Magnesium Chloride (MgCl2) (Sigma #63020)
- Manganese chloride (MnCl2) (Sigma#M1787)
- Ethylenediaminetetraacetic acid (Invitrogen #15575-020)
- HEPES Buffer 4-hydroxyethylpiperazine ethanesulfonic acid buffer
- the compound concentration was diluted to 0.74 mM, and the plate was plated using a fully automated microplate pretreatment system at a concentration of 135 nL per well, with a starting concentration of 10 uM, 11 concentration points, and a 3 fold dilution gradient.
- test buffer for dilution, dilute the 5 ⁇ HTRF buffer in the kit to 1 ⁇ , and add the specified amount of dithiothreitol and magnesium chloride solution for use.
- the tyrosinase reaction solution was prepared by using 1 ⁇ HTRF buffer, and the final reaction concentration of tyrosine kinase was 0.0156 ng/ ⁇ L.
- the tyrosinase solution and the tyrosine kinase-substrate-biotin/adenosine triphosphate mixture were added 5 ul each well and incubated at 23 ° C for 1 hour.
- Analytical data Analyze the data using XL-Fit and calculate the IC50 value of the compound.
- lysis buffer trishydroxymethylaminomethane hydrochloride, Invitrogen 15567-1000 ml; sodium chloride, Domestic; sodium deoxycholate, Sigma30970-25G; polyethylene glycol octyl phenyl ether, SigmaT9284-100ml; sodium dodecyl sulfate, SigmaL3771; ethylenediaminetetraacetic acid, Invitrogen 15575-038-100ml; ultrapure water , MilliQ)
- Phosphatase inhibitor mixture 2 (Sigma, P5726-5ML)
- Phosphatase inhibitor mixture 3 (Sigma, P0044-5ML)
- Phosphorylated AKT assay kit Phospho-AKT 1/2/3 (ser473) (TGR Bioscience, EKT002)
- Multi-purpose microplate reader (Envision Reader)
- Ramos cells were diluted with a medium to a density of 5 ⁇ 10 6 /mL, and the diluted cells were added to a 96-well cell culture plate (100 ⁇ L/well) with a lance. The cell plates were placed in a 37 ° C, 5% CO 2 incubator overnight.
- the cells were centrifuged at 1000 rpm for 5 minutes on the next day, the original medium was aspirated by a lance, the serum-free experimental medium was added, and the cell plates were placed in a 37 ° C, 5% CO 2 incubator, and starved overnight. .
- the compound was diluted with dimethyl sulfoxide to have an initial concentration of 5 mM. Three concentration gradients were made using a compound V-well dilution plate with a three-fold gradient.
- the cells were centrifuged at 1000 rpm for 1 minute, and the cell plate was placed in a 37 ° C, 5% CO 2 incubator to allow the compound to act for 1 hour.
- Sheep anti-human immunoglobulin M (F(ab') 2 Goat Anti-Human IgM) (1.2 mg/ml) was diluted to 60 ug/ml with a 1X balanced salt solution preheated at 37 °C.
- IgM stimulated the cells for 10 minutes, centrifuged at 4000 rpm for 5 minutes, allowed the suspended cells to deposit on the bottom of the 96-well plate, gently drained the liquid in the 96-well plate, and blotted the remaining liquid with a paper towel. Note: Try not to get rid of suspended cells.
- ADHP 10-acetyl-3,7-dihydroxyphenazine
- the Envision Reader is a multi-function microplate reader for reading.
- Test Example 1 The results of Test Example 1 and Test Example 2 are shown in Table 1:
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description












































































































Claims (36)
- 式(I)化合物或其药学上可接受的盐,
 其中,W为C(R 7)或N;R 1、R 2独立地选自H、卤素、氨基、羟基、氰基、C 1-6烷基、C 3-6环烷基、3~10元杂环烷基、6~12元芳基或5~12元杂芳基,所述氨基、C 1-6烷基、C 3-6环烷基、3~10元杂环烷基、6~12元芳基或5~12元杂芳基任选地被R 8取代;R 3、R 4、R 7独立地选自H、卤素、氨基、羟基、氰基、C 1-6烷基、C 3-6环烷基或3~6元杂环烷基,所述氨基、C 1-6烷基、C 3-6环烷基或3~6元杂环烷基任选地被R 9取代;R 5选自H、C 1-6烷基、C 3-6环烷基、3~6元杂环烷基、C 1-6烷基C(O)、C 3-6环烷基C(O)、3~6元杂环烷基C(O)、苯基C(O)、5~6元杂芳基C(O)、C 1-6烷基SO 2、C 3-6环烷基SO 2、3~6元杂环烷基SO 2、苯基SO 2或5~6元杂芳基SO 2,所述C 1-6烷基、C 3-6环烷基、3~6元杂环烷基、C 1-6烷基C(O)、C 3-6环烷基C(O)、3~6元杂环烷基C(O)、苯基C(O)、5~6元杂芳基C(O)、C 1-6烷基SO 2、C 3-6环烷基SO 2、3~6元杂环烷基SO 2、苯基SO 2或5~6元杂芳基SO 2任选地被R 9取代;X选自任意两个位置失去H原子的3~12元环,所述环任选地被R 9取代;L选自键、NH、O、S、SO、SO 2、C(O)、OC(O)、C(O)O、C(O)NH、NHSO 2、SO 2NH、NHC(O)NH或NHSO 2NH;R 6选自H、卤素、氨基、羟基、氰基、C 1-6烷基、C 3-10环烷基或3~10元杂环烷基,所述氨基、C 1-6烷基、C 3-10环烷基或3~10元杂环烷基任选地被R 10取代;R 8、R 9独立地选自卤素、氨基、羟基、氰基、C 1-3烷基、C 1-3烷氧基或COOH;R 10选自卤素、氨基、羟基、氰基、卤代C 1-3烷基、COOH、=(O)、C 1-6烷基、C 1-6烷基SO 2、C 3-6环烷基或3~10元杂环烷基;且R 1和R 2中至少有一个选自6~12元芳基或5~12元杂芳基,所述6~12元芳基或5~12元杂芳基任选地被R 8取代。
其中,W为C(R 7)或N;R 1、R 2独立地选自H、卤素、氨基、羟基、氰基、C 1-6烷基、C 3-6环烷基、3~10元杂环烷基、6~12元芳基或5~12元杂芳基,所述氨基、C 1-6烷基、C 3-6环烷基、3~10元杂环烷基、6~12元芳基或5~12元杂芳基任选地被R 8取代;R 3、R 4、R 7独立地选自H、卤素、氨基、羟基、氰基、C 1-6烷基、C 3-6环烷基或3~6元杂环烷基,所述氨基、C 1-6烷基、C 3-6环烷基或3~6元杂环烷基任选地被R 9取代;R 5选自H、C 1-6烷基、C 3-6环烷基、3~6元杂环烷基、C 1-6烷基C(O)、C 3-6环烷基C(O)、3~6元杂环烷基C(O)、苯基C(O)、5~6元杂芳基C(O)、C 1-6烷基SO 2、C 3-6环烷基SO 2、3~6元杂环烷基SO 2、苯基SO 2或5~6元杂芳基SO 2,所述C 1-6烷基、C 3-6环烷基、3~6元杂环烷基、C 1-6烷基C(O)、C 3-6环烷基C(O)、3~6元杂环烷基C(O)、苯基C(O)、5~6元杂芳基C(O)、C 1-6烷基SO 2、C 3-6环烷基SO 2、3~6元杂环烷基SO 2、苯基SO 2或5~6元杂芳基SO 2任选地被R 9取代;X选自任意两个位置失去H原子的3~12元环,所述环任选地被R 9取代;L选自键、NH、O、S、SO、SO 2、C(O)、OC(O)、C(O)O、C(O)NH、NHSO 2、SO 2NH、NHC(O)NH或NHSO 2NH;R 6选自H、卤素、氨基、羟基、氰基、C 1-6烷基、C 3-10环烷基或3~10元杂环烷基,所述氨基、C 1-6烷基、C 3-10环烷基或3~10元杂环烷基任选地被R 10取代;R 8、R 9独立地选自卤素、氨基、羟基、氰基、C 1-3烷基、C 1-3烷氧基或COOH;R 10选自卤素、氨基、羟基、氰基、卤代C 1-3烷基、COOH、=(O)、C 1-6烷基、C 1-6烷基SO 2、C 3-6环烷基或3~10元杂环烷基;且R 1和R 2中至少有一个选自6~12元芳基或5~12元杂芳基,所述6~12元芳基或5~12元杂芳基任选地被R 8取代。 - 权利要求1的化合物,其中R 1、R 2独立地选自H、卤素或5~12元杂芳基,所述5~12元杂芳基任选地被R 8取代。
- 权利要求2的化合物,其中R 1、R 2独立地选自H、F、Cl、Br、呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基或三嗪基,所述呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基或三嗪基任选地被R 8取代。
- 权利要求3的化合物,其中R 1选自H、F、
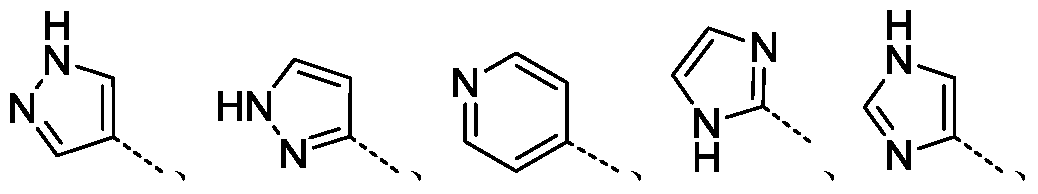 其可任选地被R 8取代。
其可任选地被R 8取代。
- 权利要求4的化合物,其中R 1选自H、F、


- 权利要求3的化合物,其中R 2选自H、F、Cl或

- 权利要求1的化合物,其中R 8选自氨基、甲基、乙基、丙基或异丙基。
- 权利要求1的化合物,其中R 1选自5~12元杂芳基;R 2选自H或卤素;其中,R 1可任选地被R 8取代。
- 权利要求8的化合物,其中R 1选自呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基或三嗪基;R 2选自H、F、Cl或Br;其中,R 1可任选地被R 8取代。
- 权利要求9的化合物,其中R 1选自
 R 2选自H、F或Cl;其中,R 1可任选地被R 8取代。
R 2选自H、F或Cl;其中,R 1可任选地被R 8取代。
- 权利要求10的化合物,其中R 1选自
 R 2选自H、F或Cl。
R 2选自H、F或Cl。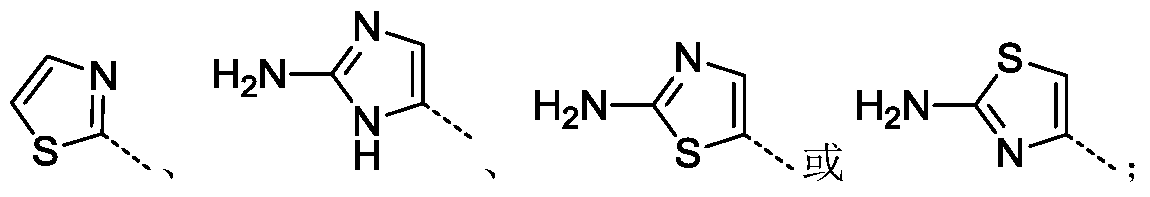
- 权利要求1的化合物,其中R 1选自H或卤素;R 2选自5~12元杂芳基;其中,R 2可任选地被R 8取代。
- 权利要求12的化合物,其中R 1选自H、F、Cl或Br;R 2选自呋喃基、噻吩基、吡咯基、吡唑基、咪唑基、吡啶基、嘧啶基、哒嗪基、吡嗪基、噻唑基、异噻唑基、噁唑基、异噁唑基、四唑基或三嗪基;其中,R 2可任选地被R 8取代。
- 权利要求13的化合物,其中R 1选自H或F;R 2选自

- 权利要求1的化合物,其中R 3、R 4、R 7独立地选自H、卤素、C 1-6烷基或C 3-6环烷基,所述C 1-6烷基或C 3-6环烷基任选地被R 9取代。
- 权利要求15的化合物,其中R 3、R 4、R 7独立地选自H或卤素。
- 权利要求1的化合物,其中R 5选自H、C 1-6烷基或C 3-6环烷基,其中C 1-6烷基或C 3-6环烷基任选被R 9取代。
- 权利要求17的化合物,其中R 5选自甲基、乙基、丙基、异丙基、丁基、异丁基或叔丁基,所述甲 基、乙基、丙基、异丙基、丁基、异丁基或叔丁基任选地被R 9取代。
- 权利要求1的化合物,其中X选自任意两个位置失去H原子的苯环或5~10元杂芳环,所述X可任选地被R 9取代。
- 权利要求19的化合物,其中X选自任意两个位置失去H原子的苯环、
 呋喃环、噻吩环、吡咯环、吡唑环、咪唑环、吡啶环、嘧啶环、哒嗪环、吡嗪环、噻唑环、异噻唑环、噁唑环、异噁唑环、四唑环或三嗪环,所述X可任选地被R 9取代。
呋喃环、噻吩环、吡咯环、吡唑环、咪唑环、吡啶环、嘧啶环、哒嗪环、吡嗪环、噻唑环、异噻唑环、噁唑环、异噁唑环、四唑环或三嗪环,所述X可任选地被R 9取代。
- 权利要求20的化合物,其中X选自所述X可任选地被R 9取代。

- 权利要求21的化合物,其中X选自


- 权利要求1的化合物,其中R 9选自卤素、C 1-3烷基或C 1-3烷氧基。
- 权利要求23的化合物,其中R 9选自F、Cl、甲基或OCH 3。
- 权利要求1的化合物,其中L选自键、NH或SO 2。
- 权利要求1的化合物,其中R 6选自H、氨基、C 1-6烷基、C 3-6环烷基或3~10元杂环烷基,所述氨基、C 1-6烷基、C 3-6环烷基或3~10元杂环烷基可任选地被R 10取代。
- 权利要求26的化合物,其中R 6选自H、氨基、甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、环丙基、环丁基、环戊基、环己基、任意位置失去一个H原子的

 所述氨基、甲基、 乙基、丙基、异丙基、丁基、异丁基、叔丁基、环丙基、环丁基、环戊基、环己基、任意位置失去一个H原子的
所述氨基、甲基、 乙基、丙基、异丙基、丁基、异丁基、叔丁基、环丙基、环丁基、环戊基、环己基、任意位置失去一个H原子的

 任选地被R 10取代。
任选地被R 10取代。
- 权利要求27的化合物,其中R 6选自H、NH 2、甲基、异丙基、环丁基、
 所述NH 2、甲基、异丙基、环丁基、
所述NH 2、甲基、异丙基、环丁基、
 可任选地被R 10取代。
可任选地被R 10取代。
- 权利要求28的化合物,其中R 6选自H、NH 2、甲基、


- 权利要求1的化合物,其中R 10选自卤素、羟基、卤代C 1-3烷基、COOH、=(O)、C 1-6烷基、C 1-6烷基SO 2或3~10元杂环烷基。
- 权利要求30的化合物,其中R 10选自F、Cl、Br、OH、一氟甲基、二氟甲基、三氟甲基、一氟乙基、二氟乙基、三氟乙基、四氟乙基、五氟乙基、一氯甲基、二氯甲基、三氯甲基、COOH、=(O)、甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、SO 2CH 3、SO 2CH 2CH 3、SO 2CH 2CH 2CH 3、SO 2CH(CH 3)CH 3、SO 2CH 2CH 2CH 2CH 3、SO 2CH(CH 3)CH 2CH 3、SO 2CH 2CH(CH 3) 2、SO 2C(CH 3) 3、任意位置失去一个H原子的


- 权利要求31的化合物,其中R 10选自F、OH、三氟甲基、COOH、=(O)、甲基、SO 2CH 3、SO 2CH 2CH 3或

- 权利要求1的化合物,其中式(I)化合物选自以下化合物:



- 一种药物组合物,其包含治疗有效量的权利要求1-33任一项所述的化合物和一种或多种药学上可接受的辅料。
- 权利要求1-33任一项所述的化合物、或权利要求34所述的药物组合物在制备治疗Syk受体相关病症的药物中的应用。
- 一种治疗Syk受体相关病症的方法,包括给予治疗有效量的权利要求1-33任一项所述的化合物、或权利要求34所述的药物组合物。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES18817720T ES2906205T3 (es) | 2017-06-14 | 2018-06-14 | Inhibidor de Syk y método de uso para el mismo |
| EP18817720.8A EP3640247B1 (en) | 2017-06-14 | 2018-06-14 | Syk inhibitor and use method therefor |
| AU2018286247A AU2018286247B2 (en) | 2017-06-14 | 2018-06-14 | Syk inhibitor and use method therefor |
| JP2019568033A JP7299167B2 (ja) | 2017-06-14 | 2018-06-14 | Syk阻害薬及びその使用方法 |
| CA3065114A CA3065114A1 (en) | 2017-06-14 | 2018-06-14 | Syk inhibitor and use method therefor |
| CN201880036803.9A CN110678461B (zh) | 2017-06-14 | 2018-06-14 | Syk抑制剂及其使用方法 |
| US16/619,646 US11091460B2 (en) | 2017-06-14 | 2018-06-14 | Syk inhibitor and use method therefor |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201710448438 | 2017-06-14 | ||
| CN201710448438.X | 2017-06-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018228475A1 true WO2018228475A1 (zh) | 2018-12-20 |
Family
ID=64660192
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2018/091269 WO2018228475A1 (zh) | 2017-06-14 | 2018-06-14 | Syk抑制剂及其使用方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US11091460B2 (zh) |
| EP (1) | EP3640247B1 (zh) |
| JP (1) | JP7299167B2 (zh) |
| CN (1) | CN110678461B (zh) |
| AU (1) | AU2018286247B2 (zh) |
| CA (1) | CA3065114A1 (zh) |
| ES (1) | ES2906205T3 (zh) |
| WO (1) | WO2018228475A1 (zh) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020119785A1 (zh) * | 2018-12-14 | 2020-06-18 | 正大天晴药业集团股份有限公司 | 一种Syk抑制剂的盐及其结晶型 |
| WO2020188015A1 (en) | 2019-03-21 | 2020-09-24 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| WO2023083281A1 (zh) | 2021-11-12 | 2023-05-19 | 正大天晴药业集团股份有限公司 | 一种喹啉酮衍生物治疗免疫性血小板减少症的用途 |
| RU2818103C2 (ru) * | 2018-12-14 | 2024-04-24 | Чиа Тай Тяньцин Фармасьютикал Груп Ко., Лтд. | Соль ингибитора syk и ее кристаллическая форма |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102551354B1 (ko) | 2018-04-20 | 2023-07-04 | 삼성전자 주식회사 | 반도체 발광 소자 및 그 제조 방법 |
| CN112725413B (zh) * | 2021-02-05 | 2022-10-14 | 厦门甄准生物技术有限公司 | 一种宫颈拭子核酸提取的裂解结合液、试剂盒及方法 |
| TW202345806A (zh) | 2022-03-31 | 2023-12-01 | 美商艾伯維有限公司 | 噻唑并〔5,4-b〕吡啶malt-1抑制劑 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1784229A (zh) * | 2003-03-10 | 2006-06-07 | 先灵公司 | 杂环激酶抑制剂:使用方法与合成 |
| WO2015080707A1 (en) * | 2013-11-26 | 2015-06-04 | Gilead Sciences, Inc. | Quinoline derivatives as bromodomain inhibitors |
| WO2017001733A1 (en) * | 2015-07-02 | 2017-01-05 | Orion Corporation | Bicyclic heterocycle derivatives as bromodomain inhibitors |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MXPA05009722A (es) * | 2003-03-10 | 2006-03-09 | Schering Corp | Inhibidores heterociclicos de cinasa: metodos de uso y sintesis. |
| TW200938542A (en) * | 2008-02-01 | 2009-09-16 | Irm Llc | Compounds and compositions as kinase inhibitors |
| WO2010053757A1 (en) * | 2008-10-29 | 2010-05-14 | Gilead Palo Alto, Inc. | 2 -oxoquinoxalin blockers of the late sodium channel |
| PE20130188A1 (es) * | 2009-12-23 | 2013-02-21 | Takeda Pharmaceutical | Pirrolidinonas heteroaromaticas fusionadas como inhibidores de syk |
| EP2545052B1 (en) * | 2010-03-11 | 2014-11-12 | Gilead Connecticut, Inc. | Imidazopyridines syk inhibitors |
| JP5473139B2 (ja) | 2010-06-15 | 2014-04-16 | 富士通テレコムネットワークス株式会社 | 試験装置と試験方法 |
| JP5744021B2 (ja) * | 2010-06-30 | 2015-07-01 | 富士フイルム株式会社 | 新規なニコチンアミド誘導体またはその塩 |
| NZ627113A (en) * | 2012-01-20 | 2016-07-29 | Genosco | Substituted pyrimidine compounds and their use as syk inhibitors |
| ES2450746B2 (es) * | 2012-03-14 | 2015-01-26 | Universidad Complutense De Madrid | Preparación y citotoxicidad de 2-quinolonas |
| US10000482B2 (en) * | 2012-10-19 | 2018-06-19 | Origenis Gmbh | Kinase inhibitors |
| EP2931281B1 (en) * | 2012-12-12 | 2018-01-17 | Merck Sharp & Dohme Corp. | Amino-pyrimidine-containing spleen tyrosine kinase inhibitors |
| CN107709336B (zh) * | 2015-06-12 | 2021-06-22 | 杭州英创医药科技有限公司 | 作为Syk抑制剂和/或Syk-HDAC双重抑制剂的杂环化合物 |
| CN105577525A (zh) | 2015-12-25 | 2016-05-11 | 中兴通讯股份有限公司 | 融合通信交互方法,装置及系统 |
-
2018
- 2018-06-14 CA CA3065114A patent/CA3065114A1/en active Pending
- 2018-06-14 EP EP18817720.8A patent/EP3640247B1/en active Active
- 2018-06-14 ES ES18817720T patent/ES2906205T3/es active Active
- 2018-06-14 WO PCT/CN2018/091269 patent/WO2018228475A1/zh unknown
- 2018-06-14 JP JP2019568033A patent/JP7299167B2/ja active Active
- 2018-06-14 US US16/619,646 patent/US11091460B2/en active Active
- 2018-06-14 CN CN201880036803.9A patent/CN110678461B/zh active Active
- 2018-06-14 AU AU2018286247A patent/AU2018286247B2/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1784229A (zh) * | 2003-03-10 | 2006-06-07 | 先灵公司 | 杂环激酶抑制剂:使用方法与合成 |
| WO2015080707A1 (en) * | 2013-11-26 | 2015-06-04 | Gilead Sciences, Inc. | Quinoline derivatives as bromodomain inhibitors |
| WO2017001733A1 (en) * | 2015-07-02 | 2017-01-05 | Orion Corporation | Bicyclic heterocycle derivatives as bromodomain inhibitors |
Non-Patent Citations (3)
| Title |
|---|
| "Greene's Protective Groups in Organic Synthesis", JOHN WILEY &SONS, INC |
| O'BRIEN, N.J. ET AL.: "Synthesis and Biological Evaluation of Substituted 3-anilino- quinolin-2(1H)-ones as PDK1 Inhibitors", BIOORGANIC & MEDICINAL CHEMISTRY, vol. 22, no. 14, 9 May 2014 (2014-05-09), pages 3781 - 3790, XP029033689 * |
| See also references of EP3640247A4 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020119785A1 (zh) * | 2018-12-14 | 2020-06-18 | 正大天晴药业集团股份有限公司 | 一种Syk抑制剂的盐及其结晶型 |
| RU2818103C2 (ru) * | 2018-12-14 | 2024-04-24 | Чиа Тай Тяньцин Фармасьютикал Груп Ко., Лтд. | Соль ингибитора syk и ее кристаллическая форма |
| WO2020188015A1 (en) | 2019-03-21 | 2020-09-24 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
| WO2023083281A1 (zh) | 2021-11-12 | 2023-05-19 | 正大天晴药业集团股份有限公司 | 一种喹啉酮衍生物治疗免疫性血小板减少症的用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3640247B1 (en) | 2022-01-12 |
| JP7299167B2 (ja) | 2023-06-27 |
| EP3640247A1 (en) | 2020-04-22 |
| AU2018286247B2 (en) | 2021-12-23 |
| CA3065114A1 (en) | 2018-12-20 |
| JP2020523328A (ja) | 2020-08-06 |
| US20200199101A1 (en) | 2020-06-25 |
| CN110678461A (zh) | 2020-01-10 |
| AU2018286247A1 (en) | 2020-01-02 |
| US11091460B2 (en) | 2021-08-17 |
| CN110678461B (zh) | 2021-08-10 |
| ES2906205T3 (es) | 2022-04-13 |
| EP3640247A4 (en) | 2020-10-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN110678461B (zh) | Syk抑制剂及其使用方法 | |
| CN103429585B (zh) | 作为白细胞介素-1受体相关激酶抑制剂的吲唑基三唑衍生物 | |
| JP6663857B2 (ja) | ピラゾロピリジンおよびピラゾロピリミジン | |
| TWI657076B (zh) | 經取代磺醯胺化合物 | |
| CN103702990B (zh) | 2-(2,4,5-取代苯胺)嘧啶衍生物作为egfr调谐子用于治疗癌症 | |
| WO2021143680A1 (zh) | 杂芳基类衍生物及其制备方法和用途 | |
| CN107001377B (zh) | 对呼吸道合胞病毒(rsv)的复制具有抑制活性的哌啶取代的吡唑并[1,5-a]嘧啶衍生物 | |
| CN112638373A (zh) | 细胞周期蛋白依赖性激酶抑制剂 | |
| TW201639829A (zh) | 經取代磺醯胺化合物 | |
| TW202108588A (zh) | α-1 抗胰蛋白酶調節劑 | |
| KR20170131654A (ko) | Usp30 억제제로서의 1-시아노-피롤리딘 화합물 | |
| CN110746424A (zh) | Mk2抑制剂和其用途 | |
| CA2935880A1 (en) | Indazole compounds as irak4 inhibitors | |
| CN115594666B (zh) | 一种磷酸酶降解剂的合成和应用 | |
| JP2019507766A (ja) | 線維症の治療のための新規化合物及びその医薬組成物 | |
| CN113748114A (zh) | 一种喹唑啉化合物及其在医药上的应用 | |
| TW201443023A (zh) | 作爲rock抑制劑之酞□酮及異喹啉酮 | |
| CN101305005A (zh) | 新二氮杂螺烷以及它们用于治疗ccr8介导的疾病的用途 | |
| MX2014008647A (es) | Compuesto de pirazin-carboxamida. | |
| WO2021115457A1 (zh) | 吡唑并[1,5-a]吡啶类化合物及其制备方法和应用 | |
| AU2005213538A1 (en) | Piperidinylcarbonyl-pyrrolidines and their use as melanocortin agonists | |
| CN104066431A (zh) | 吡嗪激酶抑制剂 | |
| TW202039474A (zh) | 吡唑基-氨基-嘧啶基衍生物的苯甲醯胺及其組合物和方法 | |
| WO2022134641A1 (zh) | 芳香杂环类化合物、药物组合物及其应用 | |
| CN105745209A (zh) | 三唑并吡啶化合物、组合物及其使用方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18817720 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3065114 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2019568033 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2018286247 Country of ref document: AU Date of ref document: 20180614 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2018817720 Country of ref document: EP Effective date: 20200114 |