WO2018212228A1 - ポリオルガノシルセスキオキサン、転写用フィルム、インモールド成型品、及びハードコートフィルム - Google Patents
ポリオルガノシルセスキオキサン、転写用フィルム、インモールド成型品、及びハードコートフィルム Download PDFInfo
- Publication number
- WO2018212228A1 WO2018212228A1 PCT/JP2018/018896 JP2018018896W WO2018212228A1 WO 2018212228 A1 WO2018212228 A1 WO 2018212228A1 JP 2018018896 W JP2018018896 W JP 2018018896W WO 2018212228 A1 WO2018212228 A1 WO 2018212228A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- hard coat
- formula
- layer
- curable composition
- Prior art date
Links
Images









Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/14—Polysiloxanes containing silicon bound to oxygen-containing groups
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D183/00—Coating compositions based on macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon, with or without sulfur, nitrogen, oxygen, or carbon only; Coating compositions based on derivatives of such polymers
- C09D183/04—Polysiloxanes
- C09D183/06—Polysiloxanes containing silicon bound to oxygen-containing groups
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C45/00—Injection moulding, i.e. forcing the required volume of moulding material through a nozzle into a closed mould; Apparatus therefor
- B29C45/14—Injection moulding, i.e. forcing the required volume of moulding material through a nozzle into a closed mould; Apparatus therefor incorporating preformed parts or layers, e.g. injection moulding around inserts or for coating articles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/28—Layered products comprising a layer of synthetic resin comprising synthetic resins not wholly covered by any one of the sub-groups B32B27/30 - B32B27/42
- B32B27/283—Layered products comprising a layer of synthetic resin comprising synthetic resins not wholly covered by any one of the sub-groups B32B27/30 - B32B27/42 comprising polysiloxanes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B7/00—Layered products characterised by the relation between layers; Layered products characterised by the relative orientation of features between layers, or by the relative values of a measurable parameter between layers, i.e. products comprising layers having different physical, chemical or physicochemical properties; Layered products characterised by the interconnection of layers
- B32B7/04—Interconnection of layers
- B32B7/12—Interconnection of layers using interposed adhesives or interposed materials with bonding properties
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/22—Di-epoxy compounds
- C08G59/30—Di-epoxy compounds containing atoms other than carbon, hydrogen, oxygen and nitrogen
- C08G59/306—Di-epoxy compounds containing atoms other than carbon, hydrogen, oxygen and nitrogen containing silicon
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/32—Epoxy compounds containing three or more epoxy groups
- C08G59/3254—Epoxy compounds containing three or more epoxy groups containing atoms other than carbon, hydrogen, oxygen or nitrogen
- C08G59/3281—Epoxy compounds containing three or more epoxy groups containing atoms other than carbon, hydrogen, oxygen or nitrogen containing silicon
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/06—Preparatory processes
- C08G77/08—Preparatory processes characterised by the catalysts used
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/20—Polysiloxanes containing silicon bound to unsaturated aliphatic groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/70—Siloxanes defined by use of the MDTQ nomenclature
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/80—Siloxanes having aromatic substituents, e.g. phenyl side groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/04—Coating
- C08J7/043—Improving the adhesiveness of the coatings per se, e.g. forming primers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/04—Coating
- C08J7/046—Forming abrasion-resistant coatings; Forming surface-hardening coatings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/0008—Organic ingredients according to more than one of the "one dot" groups of C08K5/01 - C08K5/59
- C08K5/0025—Crosslinking or vulcanising agents; including accelerators
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/06—Ethers; Acetals; Ketals; Ortho-esters
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D7/00—Features of coating compositions, not provided for in group C09D5/00; Processes for incorporating ingredients in coating compositions
- C09D7/40—Additives
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D7/00—Features of coating compositions, not provided for in group C09D5/00; Processes for incorporating ingredients in coating compositions
- C09D7/40—Additives
- C09D7/60—Additives non-macromolecular
- C09D7/63—Additives non-macromolecular organic
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C45/00—Injection moulding, i.e. forcing the required volume of moulding material through a nozzle into a closed mould; Apparatus therefor
- B29C45/14—Injection moulding, i.e. forcing the required volume of moulding material through a nozzle into a closed mould; Apparatus therefor incorporating preformed parts or layers, e.g. injection moulding around inserts or for coating articles
- B29C45/14827—Injection moulding, i.e. forcing the required volume of moulding material through a nozzle into a closed mould; Apparatus therefor incorporating preformed parts or layers, e.g. injection moulding around inserts or for coating articles using a transfer foil detachable from the insert
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B37/00—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding
- B32B37/14—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding characterised by the properties of the layers
- B32B37/24—Methods or apparatus for laminating, e.g. by curing or by ultrasonic bonding characterised by the properties of the layers with at least one layer not being coherent before laminating, e.g. made up from granular material sprinkled onto a substrate
- B32B2037/243—Coating
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2305/00—Condition, form or state of the layers or laminate
- B32B2305/72—Cured, e.g. vulcanised, cross-linked
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2307/00—Properties of the layers or laminate
- B32B2307/50—Properties of the layers or laminate having particular mechanical properties
- B32B2307/536—Hardness
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B2383/00—Polysiloxanes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31652—Of asbestos
- Y10T428/31663—As siloxane, silicone or silane
Definitions
- the present invention relates to a polyorganosilsesquioxane, a curable composition containing the polyorganosilsesquioxane, and a cured product thereof.
- the present invention also relates to a transfer film (in particular, an in-mold injection molding transfer film) having a hard coat layer formed from a hard coat liquid (hard coat agent) containing the polyorganosilsesquioxane, a hard coat film.
- a transfer film in particular, an in-mold injection molding transfer film having a hard coat layer formed from a hard coat liquid (hard coat agent) containing the polyorganosilsesquioxane, a hard coat film.
- the present invention also relates to an in-mold molded product to which a transfer layer of the transfer film is transferred.
- An in-mold injection molding method is used as a manufacturing method in which the surface of a plastic product is decorated with wood grain and hard coating.
- the in-mold injection molding method forms a release layer on one side of a base film, and laminates a transfer layer (a layer in which a hard coat layer, an anchor coat layer, a colored layer, an adhesive layer, etc. are laminated) on the release layer. Insert the transfer film into the mold, place the base film side in close contact with the inner surface of the mold, close the mold, and inject the molten thermoplastic resin into the mold from the transfer layer side. Then, when the mold is opened and the molded product is taken out, the release layer and the hard coat layer are peeled off to transfer the transfer layer to the outermost surface to obtain a molded product.
- UV acrylic monomer is mainly used (for example, see Patent Document 1).
- nanoparticles are added to the hard coat layer in order to further improve the pencil hardness of the hard coat layer surface.
- the pencil hardness of the transfer film having the hard coat layer using the UV acrylic monomer described above is about 2H, and it cannot be said that the film has sufficient surface hardness yet.
- the curing shrinkage of the hard coat layer may be reduced.
- nanoparticles are added to the hard coat layer, if the compatibility between the nanoparticles and the UV acrylic monomer is poor, there is a problem that the nanoparticles aggregate and the hard coat layer is whitened.
- the surface of the uncured or semi-cured hard coat layer after applying a hard coat solution or the like to the release layer of the base film and drying needs to be tack-free. This is because if the surface has tackiness, the blocking resistance is lowered and it is difficult to wind it on a roll.
- the object of the present invention is to form a hard coat layer having a high surface hardness by an in-mold injection molding method, and to form a tack-free coating film in an uncured or semi-cured stage and to wind it up as a roll
- Another object of the present invention is to provide a polyorganosilsesquioxane suitable as a material for a hard coat layer of a transfer film.
- Another object of the present invention is to form a hard coat layer having a high surface hardness by an in-mold injection molding method, and to form a tack-free coating film at an uncured or semi-cured stage and wind it as a roll. It is to provide a transfer film that can be removed.
- Another object of the present invention is to provide an in-mold molded product having a high surface hardness to which the transfer layer of the transfer film is transferred.
- the use of transfer films having a hard coat layer has been expanding in recent years.
- the hard coat layer of a transfer film has a particularly high surface hardness. It is also required to have excellent heat resistance.
- the hard coat layer in the transfer film using the UV acrylic monomer described above cannot be said to be sufficient from the viewpoint of such heat resistance.
- a hard coat film having a hard coat layer is generally required to have high flexibility and workability in addition to high surface hardness. This is because if the flexibility and workability are poor, the roll-to-roll manufacturing and processing cannot be performed and high production costs are required.
- the present inventors have a silsesquioxane structural unit (unit structure) containing a polymerizable functional group, and a specific structure ratio (a ratio of T3 and T2 forms, a silsesquioxane containing a polymerizable functional group).
- a specific structure ratio a ratio of T3 and T2 forms, a silsesquioxane containing a polymerizable functional group.
- the proportion of structural units) is controlled in a specific range, the number average molecular weight is high, and the molecular weight dispersity is controlled in a specific range.
- R 1 represents a group containing a polymerizable functional group.
- R a is a group containing a polymerizable functional group, a substituted or unsubstituted aryl group, a substituted or unsubstituted aralkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted group.
- An alkyl group, a substituted or unsubstituted alkenyl group, or a hydrogen atom is shown.
- R b represents a group containing a polymerizable functional group, a substituted or unsubstituted aryl group, a substituted or unsubstituted aralkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted group.
- An alkyl group, a substituted or unsubstituted alkenyl group, or a hydrogen atom is shown.
- R c represents a hydrogen atom or an alkyl group having 1 to 4 carbon atoms.
- the molar ratio of the structural unit represented by [the structural unit represented by the formula (I) / the structural unit represented by the formula (II)] is 20 or more and 500 or less, and the total amount of the siloxane structural unit (100 mol%).
- the structural unit represented by the above formula (1) and the following formula (4) [In formula (4), R 1 is the same as that in formula (1). R c is the same as in formula (II). ]
- the number average molecular weight is 2500 to 50000, and the molecular weight dispersity (weight average molecular weight / number average molecular weight) is 1.0 to 4.0.
- a polyorganosilsesquioxane is provided.
- the polyorganosilsesquioxane further comprises the following formula (2) [In the formula (2), R 2 represents a substituted or unsubstituted aryl group, a substituted or unsubstituted aralkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted alkyl group, or a substituted or unsubstituted group.
- R 2 represents a substituted or unsubstituted aryl group, a substituted or unsubstituted aralkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted alkyl group, or a substituted or unsubstituted group.
- An alkenyl group of You may have the structural unit represented by these.
- R 2 may be a substituted or unsubstituted aryl group.
- the polymerizable functional group may be an epoxy group.
- R 1 is represented by the following formula (1a) [In Formula (1a), R 1a represents a linear or branched alkylene group. ]
- a group represented by the following formula (1d) [In formula (1d), R 1d represents a linear or branched alkylene group. ]
- the group represented by these may be sufficient.
- the present invention also provides a curable composition comprising polyorganosilsesquioxane.
- the curable composition may further contain a curing catalyst.
- the curing catalyst may be a photocationic polymerization initiator.
- the curing catalyst may be a thermal cationic polymerization initiator.
- the curing catalyst may be a radical photopolymerization initiator.
- the curing catalyst may be a thermal radical polymerization initiator.
- the curable composition may further contain a vinyl ether compound.
- the curable composition may further contain a vinyl ether compound having a hydroxyl group in the molecule.
- the curable composition may be a curable composition for forming a hard coat layer.
- the present invention also provides a cured product of the curable composition.
- the present invention also provides a transfer film in which a hard coat layer is laminated on a base material and a release layer formed on at least one surface of the base material, the hard coat layer comprising the hard coat layer.
- a transfer film comprising a curable composition for forming a coat layer is provided.
- an anchor coat layer and an adhesive layer may be further laminated in this order on the hard coat layer.
- the transfer film may further include at least one colored layer.
- the hard coat layer may have a thickness of 3 to 150 ⁇ m.
- the transfer film may be a transfer film used for in-mold injection molding.
- the present invention also provides an in-mold molded product in which a layer (transfer layer) excluding the substrate on which the release layer is formed is transferred from the transfer film.
- the present invention is a hard coat film having a base material and a hard coat layer formed on at least one surface of the base material, wherein the hard coat layer is a curable composition for forming the hard coat layer.
- a hard coat film characterized by being a cured product layer of a product.
- the thickness of the hard coat layer may be 1 to 200 ⁇ m.
- the hard coat film may be manufactured in a roll-to-roll manner.
- the hard coat film may have a surface protective film on the surface of the hard coat layer.
- the present invention also includes a step A for feeding a substrate wound in a roll shape, and applying the curable composition for forming a hard coat layer to at least one surface of the fed substrate, and then the curable composition. Including a step B of forming a hard coat layer by curing and a step C of winding the obtained hard coat film again on a roll, and performing steps A to C continuously.
- a method for producing a hard coat film is provided.
- the polyorganosilsesquioxane of the present invention Since the polyorganosilsesquioxane of the present invention has the above-described structure, a high surface is obtained by performing in-mold injection molding using a transfer film having a hard coat layer containing the polyorganosilsesquioxane as an essential component. A molded product coated with a hard coat layer having hardness can be manufactured.
- the uncured or semi-cured hard coat layer containing the polyorganosilsesquioxane of the present invention is tack-free and can be wound and handled in a roll shape, and a transfer film including the hard coat layer can be rolled. Since it can be handled by a toe roll, it can be suitably used for in-mold injection molding. For this reason, the transfer film of the present invention is excellent in both quality and cost.
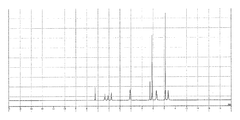
- FIG. 2 is a 1 H-NMR chart of an intermediate epoxy group-containing polyorganosilsesquioxane obtained in Production Example 1.
- FIG. 3 is a 29 Si-NMR chart of the intermediate epoxy group-containing polyorganosilsesquioxane obtained in Production Example 1.
- FIG. 2 is a 1 H-NMR chart of the epoxy group-containing polyorganosilsesquioxane of the present invention obtained in Example 1.
- FIG. 2 is a 29 Si-NMR chart of the epoxy group-containing polyorganosilsesquioxane of the present invention obtained in Example 1.
- FIG. 3 is a 1 H-NMR chart of the epoxy group-containing polyorganosilsesquioxane of the present invention obtained in Example 3.
- FIG. 3 is a 1 H-NMR chart of the epoxy group-containing polyorganosilsesquioxane of the present invention obtained in Example 3.
- FIG. 3 is a 29 Si-NMR chart of the epoxy group-containing polyorganosilsesquioxane of the present invention obtained in Example 3.
- FIG. 2 is a 1 H-NMR chart of an intermediate acrylic group-containing polyorganosilsesquioxane obtained in Production Example 2.
- FIG. 3 is a 29 Si-NMR chart of the intermediate acrylic group-containing polyorganosilsesquioxane obtained in Production Example 2.
- FIG. 4 is a 1 H-NMR chart of the acrylic group-containing polyorganosilsesquioxane of the present invention obtained in Example 4.
- FIG. It is a 29 Si-NMR chart of the acrylic group-containing polyorganosilsesquioxane of the present invention obtained in Example 4.
- the polyorganosilsesquioxane (silsesquioxane) of the present invention has a structural unit represented by the following formula (1); a structural unit represented by the following formula (I) (referred to as “T3 body”).
- the molar ratio of the structural unit represented by the following formula (II) (sometimes referred to as “T2 form”) [the structural unit represented by the formula (I) / the formula (II)
- the structural unit represented by the following formula (1) with respect to the total amount (100 mol%) of the siloxane structural unit and may be described later.
- the ratio (total amount) of the structural unit represented by the formula (4) is 55 to 100 mol%; the number average molecular weight is 2500 to 50000, and the molecular weight dispersity [weight average molecular weight / number average molecular weight] is 1.0 to 4.0.
- the structural unit represented by the above formula (1) is a silsesquioxane structural unit (so-called T unit) generally represented by [RSiO 3/2 ].
- R represents a hydrogen atom or a monovalent organic group, and the same applies to the following.
- the structural unit represented by the above formula (1) is formed by hydrolysis and condensation reaction of a corresponding hydrolyzable trifunctional silane compound (specifically, for example, a compound represented by the following formula (a)). Is done.
- R 1 in the formula (1) represents a group containing a polymerizable functional group (monovalent group). That is, the polyorganosilsesquioxane of the present invention has a cationic curable compound (a compound having a cationic polymerizable functional group) or a radical curable compound (a radical polymerizable functional group having at least a polymerizable functional group in the molecule). Compound).
- the “cationic polymerizable functional group” in the group containing the polymerizable functional group is not particularly limited as long as it has cationic polymerizability, and examples thereof include an epoxy group, an oxetane group, a vinyl ether group, and a vinylphenyl group. Can be mentioned.
- the “radical polymerizable functional group” in the group containing the polymerizable functional group is not particularly limited as long as it has radical polymerizability.
- (meth) acryloxy group, (meth) acrylamide group, vinyl Group, vinylthio group and the like As the polymerizable functional group, an epoxy group, a (meth) acryloxy group, and the like are preferable from the viewpoint of the surface hardness (for example, 4H or more) of the cured product, and an epoxy group is particularly preferable.
- Examples of the group containing the polymerizable functional group include known or commonly used groups having a polymerizable functional group, and are not particularly limited. However, the viewpoint of curability of the curable composition, surface hardness and heat resistance of the cured product, and the like.
- the group represented by the following formula (1a), the group represented by the following formula (1b), the group represented by the following formula (1c), and the group represented by the following formula (1d) are preferable, and more preferable.
- R 1a represents a linear or branched alkylene group.
- the linear or branched alkylene group include a methylene group, a methylmethylene group, a dimethylmethylene group, an ethylene group, a propylene group, a trimethylene group, a tetramethylene group, a pentamethylene group, a hexamethylene group, and a decamethylene group.
- Examples thereof include a linear or branched alkylene group having 1 to 10 carbon atoms.
- R 1a is preferably a linear alkylene group having 1 to 4 carbon atoms or a branched alkylene group having 3 or 4 carbon atoms, more preferably from the viewpoint of the surface hardness or curability of the cured product.
- R 1b represents a linear or branched alkylene group, and examples thereof include the same groups as R 1a .
- R 1b is preferably a linear alkylene group having 1 to 4 carbon atoms or a branched alkylene group having 3 or 4 carbon atoms, more preferably from the viewpoint of the surface hardness or curability of the cured product.
- R ⁇ 1c> shows a linear or branched alkylene group, and the group similar to R ⁇ 1a> is illustrated.
- R 1c is preferably a linear alkylene group having 1 to 4 carbon atoms or a branched alkylene group having 3 or 4 carbon atoms, more preferably from the viewpoint of the surface hardness or curability of the cured product.
- An ethylene group, a trimethylene group, and a propylene group and more preferably an ethylene group and a trimethylene group.
- R 1d represents a linear or branched alkylene group, and examples thereof include the same groups as R 1a .
- R 1d is preferably a linear alkylene group having 1 to 4 carbon atoms or a branched alkylene group having 3 or 4 carbon atoms, more preferably from the viewpoint of the surface hardness or curability of the cured product.
- R 1 in formula (1) is particularly a group represented by the above formula (1a), wherein R 1a is an ethylene group [in particular, 2- (3 ′, 4′-epoxycyclohexyl) Ethyl group] is preferable.
- Examples of the group containing an oxetane group include known or commonly used groups having an oxetane ring, and are not particularly limited. And a group obtained by substituting one or more hydrogen atoms (usually one or more, preferably one hydrogen atom) with an oxetane group.
- Examples of the group containing a vinyl ether group include known or commonly used groups having a vinyl ether group, and are not particularly limited.
- the vinyl ether group itself, an alkyl group (preferably having a carbon number of 1 to 10, more preferably a carbon number).
- a vinyloxymethyl group, a 2- (vinyloxy) ethyl group, a 3- (vinyloxy) propyl group, and the like are preferable.
- Examples of the group containing a vinylphenyl group include known or commonly used groups having a vinylphenyl group, and are not particularly limited.
- the vinylphenyl group itself, an alkyl group (preferably having 1 to 10 carbon atoms, more preferably).
- 4-vinylphenyl group, 3-vinylphenyl group, 2-vinylphenyl group and the like are preferable.
- Examples of the group containing the (meth) acryloxy group include known or conventional groups having a (meth) acryloxy group, and are not particularly limited.
- the (meth) acryloxy group itself, an alkyl group (preferably Is a group formed by substituting a (meth) acryloxy group for a hydrogen atom (usually one or more, preferably one hydrogen atom) of an alkyl group having 1 to 10 carbon atoms, more preferably an alkyl group having 1 to 5 carbon atoms. It is done.
- 2-((meth) acryloxy) ethyl group, 3-((meth) acryloxy) propyl group and the like are preferable.
- Examples of the group containing the (meth) acrylamide group include known or conventional groups having a (meth) acrylamide group, and are not particularly limited.
- the (meth) acrylamide group itself, an alkyl group (preferably having a carbon number) And a group obtained by substituting a (meth) acrylamide group for a hydrogen atom (usually one or more, preferably one hydrogen atom) of 1 to 10, more preferably an alkyl group having 1 to 5 carbon atoms.
- 2-((meth) acrylamide) ethyl group, 3-((meth) acrylamide) propyl group and the like are preferable.
- Examples of the group containing a vinyl group include known or commonly used groups having a vinyl group, and are not particularly limited. And a group obtained by substituting one or more hydrogen atoms (usually one or more, preferably one hydrogen atom) with a vinyl group. From the viewpoint of curability of the curable composition and heat resistance of the cured product, a vinyl group, a vinylmethyl group, a 2-vinylethyl group, a 3-vinylpropyl group, and the like are preferable.
- Examples of the group containing a vinylthio group include known or conventional groups having a vinylthio group, and are not particularly limited.
- the vinylthio group itself, an alkyl group (preferably having 1 to 10 carbon atoms, more preferably having 1 to 10 carbon atoms).
- a vinylthiomethyl group, 2- (vinylthio) ethyl group, 3- (vinylthio) propyl group and the like are preferable.
- R ⁇ 1 > in Formula (1) the group containing an epoxy group and the group containing a (meth) acryloxy group are preferable, Especially, it is group represented by the said Formula (1a), Comprising: R ⁇ 1a> A group which is an ethylene group [among others, 2- (3 ′, 4′-epoxycyclohexyl) ethyl group], 3- (acryloxy) propyl group and 3- (methacryloxy) propyl group are preferable.
- the polyorganosilsesquioxane of the present invention may have only one type of structural unit represented by the above formula (1), or two or more types of structural units represented by the above formula (1). You may have.
- the polyorganosilsesquioxane of the present invention is represented by the following formula (2) in addition to the structural unit represented by the above formula (1) as the silsesquioxane structural unit [RSiO 3/2 ]. You may have a unit.
- the structural unit represented by the above formula (2) is a silsesquioxane structural unit (T unit) generally represented by [RSiO 3/2 ]. That is, the structural unit represented by the above formula (2) is a hydrolysis and condensation reaction of a corresponding hydrolyzable trifunctional silane compound (specifically, for example, a compound represented by the following formula (b)). It is formed by.
- R 2 in the above formula (2) is a substituted or unsubstituted aryl group, a substituted or unsubstituted aralkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted alkyl group, or a substituted or unsubstituted group
- An alkenyl group of As said aryl group, a phenyl group, a tolyl group, a naphthyl group etc. are mentioned, for example.
- Examples of the aralkyl group include a benzyl group and a phenethyl group.
- Examples of the cycloalkyl group include a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, and the like.
- Examples of the alkyl group include linear or branched alkyl groups such as methyl group, ethyl group, propyl group, n-butyl group, isopropyl group, isobutyl group, s-butyl group, t-butyl group, and isopentyl group. Groups.
- alkenyl group linear or branched alkenyl groups, such as a vinyl group, an allyl group, and an isopropenyl group, are mentioned, for example.
- the above-mentioned substituted aryl group, substituted aralkyl group, substituted cycloalkyl group, substituted alkyl group, and substituted alkenyl group are each a hydrogen atom or main chain. Part or all of the case is an ether group, ester group, carbonyl group, siloxane group, halogen atom (fluorine atom, etc.), acrylic group, methacryl group, mercapto group, amino group, and hydroxy group (hydroxyl group). And a group substituted with at least one selected.
- R 2 is preferably a substituted or unsubstituted aryl group, a substituted or unsubstituted alkyl group, a substituted or unsubstituted alkenyl group, more preferably a substituted or unsubstituted aryl group, more preferably a phenyl group. is there.
- the proportion of each of the above silsesquioxane structural units (the structural unit represented by the formula (1), the structural unit represented by the formula (2)) It is possible to adjust appropriately according to the composition of the raw material (hydrolyzable trifunctional silane) for forming the slag.
- the polyorganosilsesquioxane of the present invention is not limited to the structural unit represented by the above formula (1) and the structural unit represented by the formula (2), and is further represented by the above structural formula (1).
- the structural unit etc. which are represented by following formula (3) etc. are mentioned, for example. Can be mentioned.
- the ratio of the structural unit (T3 body) represented by the above formula (I) and the structural unit (T2 body) represented by the above formula (II) [T3 body / T2 body] ] Is 20 or more and 500 or less as described above.
- the lower limit of the ratio [T3 / T2] is preferably 21, more preferably 23, and even more preferably 25.
- the upper limit of the ratio [T3 / T2] is preferably 100, more preferably 50, and still more preferably 40.
- R a in the above formula (I) (formula (I ') in the R a same) and formula (II) in the R b (wherein (II') in the R b versa), respectively, polymerizable A group containing a functional group, a substituted or unsubstituted aryl group, a substituted or unsubstituted aralkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted alkyl group, a substituted or unsubstituted alkenyl group, or hydrogen Indicates an atom.
- R a and R b are the same as R 1 in the above formula (1) and R 2 in the above formula (2).
- R a in the formula (I) and R b in the formula (II) were bonded to silicon atoms in the hydrolyzable trifunctional silane compound used as the raw material of the polyorganosilsesquioxane of the present invention, respectively. It is derived from a group (a group other than an alkoxy group and a halogen atom; for example, R 1 , R 2 and a hydrogen atom in the following formulas (a) to (c)).
- R c in the formula (II) is a hydrogen atom or an alkyl group having 1 to 4 carbon atoms.
- alkyl group having 1 to 4 carbon atoms include linear or branched alkyl groups having 1 to 4 carbon atoms such as a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, and an isobutyl group. .
- the alkyl group represented by R c in the formula (II) is generally an alkoxy group in the hydrolyzable silane compound used as a raw material for the polyorganosilsesquioxane of the present invention (for example, X 1 to X 3 described later). Derived from an alkyl group forming an alkoxy group or the like.
- the ratio [T3 body / T2 body] in the polyorganosilsesquioxane of the present invention can be determined, for example, by 29 Si-NMR spectrum measurement. 29 In the Si-NMR spectrum, the silicon atom in the structural unit (T3 form) represented by the formula (I) is different from the silicon atom in the structural unit (T2 form) represented by the formula (II). In order to show a signal (peak) in (chemical shift), the ratio [T3 body / T2 body] can be obtained by calculating the integration ratio of these respective peaks.
- the polyorganosilsesquioxane of the present invention has a structural unit represented by the above formula (1) and R 1 is a 2- (3 ′, 4′-epoxycyclohexyl) ethyl group.
- R 1 is a 2- (3 ′, 4′-epoxycyclohexyl) ethyl group.
- the signal of the silicon atom in the structure (T3 form) represented by the above formula (I) appears at ⁇ 64 to ⁇ 70 ppm
- the silicon atom in the structure (T2 form) represented by the above formula (II) appears.
- the signal appears at -54 to -60 ppm.
- the ratio [T3 body / T2 body] can be obtained by calculating the integral ratio of the signal (T3 body) of ⁇ 64 to ⁇ 70 ppm and the signal (T2 body) of ⁇ 54 to ⁇ 60 ppm. it can.
- R 1 is a group containing a polymerizable functional group other than 2- (3 ′, 4′-epoxycyclohexyl) ethyl group
- [T3 body / T2 body] can be obtained in the same manner.
- the 29 Si-NMR spectrum of the polyorganosilsesquioxane of the present invention can be measured, for example, with the following apparatus and conditions.
- Measuring apparatus Trade name “JNM-ECA500NMR” (manufactured by JEOL Ltd.)
- Solvent Deuterated chloroform Accumulated times: 1800 times Measurement temperature: 25 ° C
- the ratio [T3 / T2] of the polyorganosilsesquioxane of the present invention is 20 or more and 500 or less, indicating that the abundance of T2 with respect to T3 in the polyorganosilsesquioxane of the present invention. Is relatively small, meaning that the hydrolysis / condensation reaction of silanol is more advanced.
- T2 body examples include a structural unit represented by the following formula (4), a structural unit represented by the following formula (5), a structural unit represented by the following formula (6), and the like.
- R 2 in R 1 and the following formula (5) in the following equation (4) is the same as R 2 in R 1 and the formula in the formula (1) (2).
- R c in the formula (4) to (6) like the R c in Formula (II), a hydrogen atom or an alkyl group having 1 to 4 carbon atoms.
- the polyorganosilsesquioxane of the present invention may have any cage-type, incomplete cage-type, ladder-type, or random-type silsesquioxane structure. You may have in combination.
- the total amount of siloxane structural units in the polyorganosilsesquioxane of the present invention [total siloxane structural units; total amount of M units, D units, T units, and Q units] (100 mol%).
- the proportion (total amount) of the structural unit and the structural unit represented by the above formula (4) is 55 to 100 mol%, preferably 65 to 100 mol%, more preferably 80 to 99, as described above. Mol%.
- hardenability of a curable composition improves and the surface hardness and adhesiveness of hardened
- the ratio of each siloxane structural unit in the polyorganosilsesquioxane of this invention is computable by the composition of a raw material, NMR spectrum measurement, etc., for example.
- the total amount of siloxane structural units in the polyorganosilsesquioxane of the present invention [total siloxane structural units; total amount of M units, D units, T units, and Q units] (100 mol%).
- the proportion (total amount) of the structural units represented by the above formula (5) is not particularly limited, but is preferably 0 to 70 mol%, more preferably 0 to 60 mol%, and still more preferably 0 to It is 40 mol%, particularly preferably 1 to 15 mol%.
- the ratio of the structural unit represented by the formula (1) and the structural unit represented by the formula (4) can be relatively increased by setting the ratio to 70 mol% or less, the curable composition The curability of the cured product is improved, and the surface hardness and adhesiveness of the cured product tend to be higher. On the other hand, when the ratio is 1 mol% or more, the gas barrier property of the cured product tends to be improved.
- the total amount of siloxane structural units in the polyorganosilsesquioxane of the present invention [all siloxane structural units; total amount of M units, D units, T units, and Q units] (100 mol%).
- the ratio (total amount) of the structural unit represented by the structural unit represented by the formula (2), the structural unit represented by the formula (4), and the structural unit represented by the formula (5) is particularly limited. However, it is preferably 60 to 100 mol%, more preferably 70 to 100 mol%, still more preferably 80 to 100 mol%. By setting the above ratio to 60 mol% or more, the surface hardness and adhesiveness of the cured product tend to be higher.
- the number average molecular weight (Mn) in terms of standard polystyrene by gel permeation chromatography of the polyorganosilsesquioxane of the present invention is 2500 to 50000, preferably 2800 to 10000, more preferably 3000 to 8000.
- the number average molecular weight is 2500 or more, the surface of the uncured or semi-cured hard coat layer is likely to be tack-free, the anti-blocking property is improved, and it is easy to wind up on a roll. It can be preferably used as a component of a hard coat layer of a transfer film for injection molding, and the heat resistance, scratch resistance, and adhesion of the cured product are further improved.
- the number average molecular weight to 50000 or less compatibility with other components in the curable composition is improved, and the heat resistance of the cured product is further improved.
- the molecular weight dispersity (Mw / Mn) in terms of standard polystyrene by gel permeation chromatography of the polyorganosilsesquioxane of the present invention is 1.0 to 4.0, preferably 1.1. To 3.0, more preferably 1.2 to 2.5.
- the molecular weight dispersity is 1.0 to 4.0 or less, the surface hardness and adhesiveness of the cured product become higher.
- the molecular weight dispersity is 1.1 or more, it tends to be liquid, and the handleability tends to be improved.
- the number average molecular weight and molecular weight dispersity of the polyorganosilsesquioxane of this invention can be measured with the following apparatus and conditions.
- Measuring device Product name “LC-20AD” (manufactured by Shimadzu Corporation)
- Eluent THF, sample concentration 0.1-0.2% by weight
- Flow rate 1 mL / min
- Detector UV-VIS detector (trade name “SPD-20A”, manufactured by Shimadzu Corporation)
- Molecular weight Standard polystyrene conversion
- the 5% weight loss temperature (T d5 ) in the air atmosphere of the polyorganosilsesquioxane of the present invention is not particularly limited, but is preferably 330 ° C. or higher (for example, 330 to 450 ° C.), more preferably 340 ° C. or higher. More preferably, it is 350 ° C. or higher.
- T d5 The 5% weight loss temperature in the air atmosphere of the polyorganosilsesquioxane of the present invention is not particularly limited, but is preferably 330 ° C. or higher (for example, 330 to 450 ° C.), more preferably 340 ° C. or higher. More preferably, it is 350 ° C. or higher.
- the 5% weight reduction temperature is 330 ° C. or higher, the heat resistance of the cured product tends to be further improved.
- the polyorganosilsesquioxane of the present invention has a ratio [T3 / T2] of 20 or more and 500 or less, a number average molecular weight of 2500 to 50000, and a molecular weight dispersity of 1.0 to 4.0. Therefore, the 5% weight loss temperature is controlled to 330 ° C. or higher.
- the 5% weight reduction temperature is a temperature at the time when 5% of the weight before heating is reduced when heated at a constant rate of temperature increase, and serves as an index of heat resistance.
- the 5% weight loss temperature can be measured by TGA (thermogravimetric analysis) under an air atmosphere at a temperature rising rate of 5 ° C./min.
- the polyorganosilsesquioxane of the present invention can be produced by a known or conventional polysiloxane production method, and is not particularly limited.
- one or two or more hydrolyzable silane compounds are hydrolyzed and It can be produced by a method of condensation.
- a hydrolyzable trifunctional silane compound compound represented by the following formula (a)
- a hydrolyzable trifunctional silane compound for forming the structural unit represented by the above formula (1) is essential. It is necessary to use it as a hydrolyzable silane compound.
- a compound represented by the following formula (a) which is a hydrolyzable silane compound for forming a silsesquioxane structural unit (T unit) in the polyorganosilsesquioxane of the present invention.
- the polyorganosilsesquioxane of the present invention can be produced by hydrolysis and condensation of a compound represented by the following formula (b) and a compound represented by the following formula (c). .
- the compound represented by the above formula (a) is a compound that forms the structural unit represented by the formula (1) in the polyorganosilsesquioxane of the present invention.
- R 1 in the formula (a) represents a group containing a polymerizable functional group, similarly to R 1 in the formula (1). That is, R 1 in the formula (a) is a group represented by the above formula (1a), a group represented by the above formula (1b), a group represented by the above formula (1c), or the above formula (1d).
- X 1 in the above formula (a) represents an alkoxy group or a halogen atom.
- the alkoxy group for X 1 include alkoxy groups having 1 to 4 carbon atoms such as a methoxy group, an ethoxy group, a propoxy group, an isopropyloxy group, a butoxy group, and an isobutyloxy group.
- the halogen atom in X 1 for example, a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom.
- X 1 is preferably an alkoxy group, more preferably a methoxy group or an ethoxy group.
- the three X 1 may be the same or different.
- the compound represented by the above formula (b) is a compound that forms the structural unit represented by the formula (2) in the polyorganosilsesquioxane of the present invention.
- R 2 in formula (b) like the R 2 in the formula (2), a substituted or unsubstituted aryl group, a substituted or unsubstituted aralkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted An alkyl group, or a substituted or unsubstituted alkenyl group.
- R 2 in formula (b) is preferably a substituted or unsubstituted aryl group, a substituted or unsubstituted alkyl group, a substituted or unsubstituted alkenyl group, more preferably a substituted or unsubstituted aryl group, More preferred is a phenyl group.
- X 2 in the above formula (b) represents an alkoxy group or a halogen atom.
- Specific examples of X 2 include those exemplified as X 1 .
- X 2 is preferably an alkoxy group, more preferably a methoxy group or an ethoxy group.
- the three X 2 may be the same or different.
- the compound represented by the above formula (c) is a compound that forms the structural unit represented by the formula (3) in the polyorganosilsesquioxane of the present invention.
- X 3 in the above formula (c) represents an alkoxy group or a halogen atom.
- Specific examples of X 3 include those exemplified as X 1 .
- X 3 is preferably an alkoxy group, more preferably a methoxy group or an ethoxy group.
- the three X 3 may be the same or different.
- hydrolyzable silane compound a hydrolyzable silane compound other than the compounds represented by the above formulas (a) to (c) may be used in combination.
- hydrolyzable trifunctional silane compounds other than the compounds represented by the above formulas (a) to (c) hydrolyzable monofunctional silane compounds that form M units, hydrolyzable bifunctional silanes that form D units
- hydrolyzable tetrafunctional silane compounds that form compounds and Q units.
- the amount and composition of the hydrolyzable silane compound can be appropriately adjusted according to the desired structure of the polyorganosilsesquioxane of the present invention.
- the amount of the compound represented by the above formula (a) is not particularly limited, but is preferably 55 to 100 mol%, more preferably based on the total amount (100 mol%) of the hydrolyzable silane compound to be used. Is from 65 to 100 mol%, more preferably from 80 to 99 mol%.
- the amount of the compound represented by the above formula (b) is not particularly limited, but is preferably 0 to 70 mol%, more preferably based on the total amount (100 mol%) of the hydrolyzable silane compound to be used. Is 0 to 60 mol%, more preferably 0 to 40 mol%, particularly preferably 1 to 15 mol%.
- the ratio of the compound represented by the formula (a) and the compound represented by the formula (b) (the ratio of the total amount) to the total amount (100 mol%) of the hydrolyzable silane compound to be used is not particularly limited.
- the amount is preferably 60 to 100 mol%, more preferably 70 to 100 mol%, still more preferably 80 to 100 mol%.
- hydrolysis and condensation reaction of these hydrolysable silane compounds can also be performed simultaneously, or can also be performed sequentially.
- the order which performs reaction is not specifically limited.
- the hydrolysis and condensation reaction of the hydrolyzable silane compound may be carried out in one step or in two or more steps, but in order to efficiently produce the polyorganosilsesquioxane of the present invention.
- the hydrolysis and condensation reaction is preferably performed in two or more stages (preferably two stages).
- the ratio [T3 body / T2 body] is 5 or more and less than 20 in the first stage hydrolysis and condensation reaction, and the number average molecular weight Polyorganosilsesquioxane having a molecular weight of 1000 to 3000 (hereinafter referred to as “intermediate polyorganosilsesquioxane”).
- the intermediate polyorganosilsesquioxane is further hydrolyzed.
- the first stage hydrolysis and condensation reaction can be performed in the presence or absence of a solvent.
- a solvent examples include aromatic hydrocarbons such as benzene, toluene, xylene and ethylbenzene; ethers such as diethyl ether, dimethoxyethane, tetrahydrofuran and dioxane; ketones such as acetone, methyl ethyl ketone and methyl isobutyl ketone; methyl acetate and ethyl acetate.
- aromatic hydrocarbons such as benzene, toluene, xylene and ethylbenzene
- ethers such as diethyl ether, dimethoxyethane, tetrahydrofuran and dioxane
- ketones such as acetone, methyl ethyl ketone and methyl isobutyl ketone
- methyl acetate and ethyl acetate
- Esters such as isopropyl acetate and butyl acetate; amides such as N, N-dimethylformamide and N, N-dimethylacetamide; nitriles such as acetonitrile, propionitrile and benzonitrile; alcohols such as methanol, ethanol, isopropyl alcohol and butanol Etc. Among them, ketone and ether are preferable.
- a solvent can also be used individually by 1 type and can also be used in combination of 2 or more type.
- the amount of the solvent used in the first stage hydrolysis and condensation reaction is not particularly limited, and the desired reaction time is within the range of 0 to 2000 parts by weight with respect to 100 parts by weight of the total amount of the hydrolyzable silane compound. It can adjust suitably according to etc.
- the first stage hydrolysis and condensation reaction is preferably allowed to proceed in the presence of a catalyst and water.
- the catalyst may be an acid catalyst or an alkali catalyst, but an alkali catalyst is preferable in order to suppress decomposition of a polymerizable functional group such as an epoxy group.
- Examples of the acid catalyst include mineral acids such as hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid and boric acid; phosphoric acid esters; carboxylic acids such as acetic acid, formic acid and trifluoroacetic acid; methanesulfonic acid, trifluoromethanesulfonic acid, p -Sulfonic acids such as toluenesulfonic acid; solid acids such as activated clay; Lewis acids such as iron chloride.
- Examples of the alkali catalyst include alkali metal hydroxides such as lithium hydroxide, sodium hydroxide, potassium hydroxide, and cesium hydroxide; alkaline earth metals such as magnesium hydroxide, calcium hydroxide, and barium hydroxide.
- Hydroxides carbonates of alkali metals such as lithium carbonate, sodium carbonate, potassium carbonate, cesium carbonate; carbonates of alkaline earth metals such as magnesium carbonate; lithium hydrogen carbonate, sodium hydrogen carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate Alkali metal hydrogen carbonates such as cesium hydrogen carbonate; organic acid salts of alkali metals such as lithium acetate, sodium acetate, potassium acetate and cesium acetate (for example, acetate); organic acids of alkaline earth metals such as magnesium acetate Salt (eg acetate); lithium methoxide, sodium methoxy Alkali metal alkoxides such as sodium ethoxide, sodium isopropoxide, potassium ethoxide and potassium t-butoxide; alkali metal phenoxides such as sodium phenoxide; triethylamine, N-methylpiperidine, 1,8-diazabicyclo [5.4 0.0] undec-7-ene, amines such as 1,5-diaza
- the amount of the catalyst used in the first stage hydrolysis and condensation reaction is not particularly limited, and is suitably within the range of 0.002 to 0.200 mol with respect to 1 mol of the total amount of the hydrolyzable silane compound. Can be adjusted.
- the amount of water used in the first stage hydrolysis and condensation reaction is not particularly limited, and is appropriately adjusted within the range of 0.5 to 20 mol with respect to 1 mol of the total amount of the hydrolyzable silane compound. be able to.
- the method for adding water in the hydrolysis and condensation reaction in the first stage is not particularly limited, and the total amount of water to be used (total amount used) may be added all at once or sequentially. Good. When adding sequentially, you may add continuously and may add intermittently.
- reaction conditions for the hydrolysis and condensation reaction in the first stage are selected so that the ratio [T3 / T2] in the intermediate polyorganosilsesquioxane is 5 or more and less than 20. This is very important.
- the reaction temperature of the first stage hydrolysis and condensation reaction is not particularly limited, but is preferably 40 to 100 ° C., more preferably 45 to 80 ° C. By controlling the reaction temperature within the above range, the ratio [T3 / T2] tends to be more efficiently controlled to 5 or more and less than 20.
- the reaction time for the first stage hydrolysis and condensation reaction is not particularly limited, but is preferably 0.1 to 10 hours, more preferably 1.5 to 8 hours.
- the hydrolysis and condensation reaction in the first stage can be performed under normal pressure, or can be performed under pressure or under reduced pressure.
- the atmosphere at the time of performing the first stage hydrolysis and condensation reaction is not particularly limited, for example, under an inert gas atmosphere such as a nitrogen atmosphere or an argon atmosphere, or in the presence of oxygen such as under air. Although it may be present, an inert gas atmosphere is preferred.
- the intermediate polyorganosilsesquioxane is obtained by the hydrolysis and condensation reaction in the first stage. After completion of the first stage hydrolysis and condensation reaction, it is preferable to neutralize the catalyst in order to suppress decomposition of the polymerizable functional group such as ring opening of the epoxy group.
- the intermediate polyorganosilsesquioxane can be separated from, for example, separation means such as water washing, acid washing, alkali washing, filtration, concentration, distillation, extraction, crystallization, recrystallization, column chromatography, or a combination thereof. It may be separated and purified by means or the like.
- the intermediate polyorganosilsesquioxane obtained by the first stage hydrolysis and condensation reaction is subjected to the second stage hydrolysis and condensation reactions to produce the polyorganosilsesquioxane of the present invention. can do.
- the second stage hydrolysis and condensation reaction can be carried out in the presence of a solvent or in the absence.
- the solvents mentioned in the first-stage hydrolysis and condensation reaction can be used.
- the intermediate polyorganosilsesquioxane containing the reaction solvent, extraction solvent, etc. for the first stage hydrolysis and condensation reaction is distilled as it is or partly. You may use what you did.
- a solvent can also be used individually by 1 type and can also be used in combination of 2 or more type.
- the amount used is not particularly limited, and is within the range of 0 to 2000 parts by weight with respect to 100 parts by weight of the intermediate polyorganosilsesquioxane. Thus, it can be appropriately adjusted according to the desired reaction time and the like.
- the second-stage hydrolysis and condensation reaction is preferably allowed to proceed in the presence of a catalyst and water.
- the catalyst the catalyst mentioned in the first stage hydrolysis and condensation reaction can be used.
- an alkali catalyst is preferable.
- alkali metal hydroxides such as sodium hydroxide, potassium hydroxide and cesium hydroxide
- alkali metal carbonates such as lithium carbonate, sodium carbonate, potassium carbonate and cesium carbonate.
- a catalyst can also be used individually by 1 type and can also be used in combination of 2 or more type. Further, the catalyst can be used in a state dissolved or dispersed in water, a solvent or the like.
- the amount of the catalyst used in the second stage hydrolysis and condensation reaction is not particularly limited, and is preferably 0.01 to 10000 ppm, more preferably 0 with respect to the intermediate polyorganosilsesquioxane (1000000 ppm). Within the range of 1 to 1000 ppm, it can be adjusted as appropriate.
- the amount of water used in the second-stage hydrolysis and condensation reaction is not particularly limited, and is preferably 10 to 100,000 ppm, more preferably 100 to 20000 ppm relative to the intermediate polyorganosilsesquioxane (1000000 ppm). Within the range, it can be adjusted as appropriate. If the amount of water used is greater than 100,000 ppm, the polyorganosilsesquioxane ratio [T3 / T2] and the number average molecular weight tend to be difficult to control within a predetermined range.
- the method for adding water in the hydrolysis and condensation reaction in the second stage is not particularly limited, and the total amount of water to be used (total amount used) may be added all at once or sequentially. Good. When adding sequentially, you may add continuously and may add intermittently.
- the ratio [T3 / T2] in the polyorganosilsesquioxane of the present invention is 20 or more and 500 or less, and the number average molecular weight is 2500 to 50000. It is important to select reaction conditions such that The reaction temperature for the hydrolysis and condensation reaction in the second stage varies depending on the catalyst used and is not particularly limited, but is preferably 5 to 200 ° C, more preferably 30 to 100 ° C. By controlling the reaction temperature within the above range, the ratio [T3 body / T2 body] and the number average molecular weight tend to be more efficiently controlled within the desired range.
- the reaction time for the hydrolysis and condensation reaction in the second stage is not particularly limited, but is preferably 0.5 to 1000 hours, more preferably 1 to 500 hours.
- a desired ratio [T3 body / T2 body] the polyorganosilsesquioxane of the present invention having a number average molecular weight can also be obtained.
- the second stage hydrolysis and condensation reaction can be performed under normal pressure, or under pressure or under reduced pressure.
- the atmosphere at the time of performing the hydrolysis and condensation reaction in the second stage is not particularly limited, and for example, under an inert gas atmosphere such as a nitrogen atmosphere or an argon atmosphere, or in the presence of oxygen such as under air. Although it may be present, an inert gas atmosphere is preferred.
- the polyorganosilsesquioxane of the present invention is obtained by the hydrolysis and condensation reaction in the second stage. After completion of the hydrolysis and condensation reaction in the second stage, it is preferable to neutralize the catalyst in order to suppress decomposition of the polymerizable functional group such as ring opening of the epoxy group.
- the polyorganosilsesquioxane of the present invention is combined with, for example, separation means such as water washing, acid washing, alkali washing, filtration, concentration, distillation, extraction, crystallization, recrystallization, column chromatography, and the like. Separation and purification may be performed by separation means or the like.
- the polyorganosilsesquioxane of the present invention has the above-described configuration, an uncured or semi-cured hard coat layer coated with a curable composition containing the polyorganosilsesquioxane as an essential component becomes tack-free. Since the blocking resistance is improved, the film can be wound and handled on a roll. For example, it can be suitably used as a component of a hard coat layer of an in-mold injection transfer film. Further, by curing the curable composition, a cured product having high surface hardness and heat resistance and excellent flexibility and workability can be formed. Moreover, the hardened
- the curable composition of the present invention is a curable composition (curable resin composition) containing the polyorganosilsesquioxane of the present invention as an essential component.
- the curable composition of the present invention further contains other components such as a curing catalyst (particularly a photocationic polymerization initiator and a radical polymerizable initiator), a surface conditioner or a surface modifier. Also good.
- the polyorganosilsesquioxane of the present invention can be used singly or in combination of two or more.
- the content (blending amount) of the polyorganosilsesquioxane of the present invention in the curable composition of the present invention is not particularly limited, but is 70 with respect to the total amount (100% by weight) of the curable composition excluding the solvent.
- the content is preferably not less than 100% by weight and more preferably less than 100% by weight, more preferably 80 to 99.8% by weight, still more preferably 90 to 99.5% by weight.
- the content of the polyorganosilsesquioxane of the present invention is set to less than 100% by weight, it is possible to contain a curing catalyst, thereby allowing the curing of the curable composition to proceed more efficiently. There is a tendency to be able to.
- the ratio of the polyorganosilsesquioxane of the present invention to the total amount (100 wt%) of the cationic curable compound or radical curable compound contained in the curable composition of the present invention is not particularly limited, but is 70 to 100 wt%. It is preferably 75 to 98% by weight, more preferably 80 to 95% by weight. By setting the content of the polyorganosilsesquioxane of the present invention to 70% by weight or more, the surface hardness and adhesiveness of the cured product tend to be further improved.
- the curable composition of the present invention preferably further contains a curing catalyst.
- a curing catalyst it is particularly preferable to include a cationic polymerization initiator or a radical polymerization initiator as a curing catalyst in that the curing time until tack-free can be shortened.
- the cationic polymerization initiator is a compound that can initiate or accelerate the cationic polymerization reaction of a cationically curable compound such as the polyorganosilsesquioxane of the present invention. Although it does not specifically limit as said cationic polymerization initiator, For example, a photocationic polymerization initiator (photoacid generator), a thermal cationic polymerization initiator (thermal acid generator), etc. are mentioned.
- photocationic polymerization initiator known or commonly used photocationic polymerization initiators can be used.
- sulfonium salts salts of sulfonium ions and anions
- iodonium salts salts of iodonium ions and anions
- Selenium salt senium ion and anion salt
- ammonium salt ammonium ion and anion salt
- phosphonium salt phosphonium ion and anion salt
- transition metal complex ion and anion salt etc.
- sulfonium salt examples include [4- (4-biphenylylthio) phenyl] -4-biphenylylphenylsulfonium tris (pentafluoroethyl) trifluorophosphate, triphenylsulfonium salt, tri-p-tolylsulfonium salt, Tri-o-tolylsulfonium salt, tris (4-methoxyphenyl) sulfonium salt, 1-naphthyldiphenylsulfonium salt, 2-naphthyldiphenylsulfonium salt, tris (4-fluorophenyl) sulfonium salt, tri-1-naphthylsulfonium salt, Tri-2-naphthylsulfonium salt, tris (4-hydroxyphenyl) sulfonium salt, diphenyl [4- (phenylthio) phenyl] s
- diphenyl [4- (phenylthio) phenyl] sulfonium salt examples include diphenyl [4- (phenylthio) phenyl] sulfonium hexafluoroantimonate and diphenyl [4- (phenylthio) phenyl] sulfonium hexafluorophosphate. .
- UV9380C trade name “UV9380
- selenium salt examples include triaryl selenium such as triphenyl selenium salt, tri-p-tolyl selenium salt, tri-o-tolyl selenium salt, tris (4-methoxyphenyl) selenium salt, and 1-naphthyldiphenyl selenium salt.
- Salts Diaryl phenacyl selenium salts, diphenyl benzyl selenium salts, diaryl selenium salts such as diphenyl methyl selenium salts; monoaryl selenium salts such as phenyl methyl benzyl selenium salts; trialkyl selenium salts such as dimethyl phenacyl selenium salts, etc. .
- ammonium salt examples include tetramethylammonium salt, ethyltrimethylammonium salt, diethyldimethylammonium salt, triethylmethylammonium salt, tetraethylammonium salt, trimethyl-n-propylammonium salt, and trimethyl-n-butylammonium salt.
- Pyrodium salts such as alkylammonium salts; N, N-dimethylpyrrolidinium salts, N-ethyl-N-methylpyrrolidinium salts; N, N′-dimethylimidazolinium salts, N, N′-diethylimidazolinium salts, etc.
- Imidazolinium salts such as N, N′-dimethyltetrahydropyrimidinium salt, N, N′-diethyltetrahydropyrimidinium salt; N, N-dimethylmorpholinium salt, N, N -Diethylmorpholine Morpholinium salts such as um salt; piperidinium salts such as N, N-dimethylpiperidinium salt and N, N-diethylpiperidinium salt; pyridinium salts such as N-methylpyridinium salt and N-ethylpyridinium salt; N, N 'Imidazolium salts such as dimethylimidazolium salt; Quinolium salts such as N-methylquinolium salt; Isoquinolium salts such as N-methylisoquinolium salt; Thiazonium salts such as benzylbenzothiazonium salt; And the like.
- the phosphonium salts include tetraarylphosphonium salts such as tetraphenylphosphonium salts, tetra-p-tolylphosphonium salts, and tetrakis (2-methoxyphenyl) phosphonium salts; triarylphosphonium salts such as triphenylbenzylphosphonium salts; Examples thereof include tetraalkylphosphonium salts such as benzylphosphonium salt, tributylbenzylphosphonium salt, tetraethylphosphonium salt, tetrabutylphosphonium salt, and triethylphenacylphosphonium salt.
- tetraarylphosphonium salts such as tetraphenylphosphonium salts, tetra-p-tolylphosphonium salts, and tetrakis (2-methoxyphenyl) phosphonium salts
- triarylphosphonium salts such as triphen
- Examples of the salt of the transition metal complex ion include salts of chromium complex cations such as ( ⁇ 5-cyclopentadienyl) ( ⁇ 6-toluene) Cr + and ( ⁇ 5-cyclopentadienyl) ( ⁇ 6-xylene) Cr +. And salts of iron complex cations such as ( ⁇ 5-cyclopentadienyl) ( ⁇ 6-toluene) Fe + and ( ⁇ 5-cyclopentadienyl) ( ⁇ 6-xylene) Fe + .
- anion constituting the above-described salt examples include SbF 6 ⁇ , PF 6 ⁇ , BF 4 ⁇ , (CF 3 CF 2 ) 3 PF 3 ⁇ , (CF 3 CF 2 CF 2 ) 3 PF 3 ⁇ , (C 6 F 5 ) 4 B ⁇ , (C 6 F 5 ) 4 Ga ⁇ , sulfonate anion (trifluoromethanesulfonate anion, pentafluoroethanesulfonate anion, nonafluorobutanesulfonate anion, methanesulfonate anion, benzenesulfonate Anion, p-toluenesulfonate anion, etc.), (CF 3 SO 2 ) 3 C ⁇ , (CF 3 SO 2 ) 2 N ⁇ , perhalogenate ion, halogenated sulfonate ion, sulfate ion, carbonate
- thermal cationic polymerization initiator examples include arylsulfonium salts, aryliodonium salts, allene-ion complexes, quaternary ammonium salts, aluminum chelates, and boron trifluoride amine complexes.
- arylsulfonium salts examples include hexafluoroantimonate salts.
- trade names “SP-66” and “SP-77” manufactured by ADEKA Corporation
- trade names “Sun Aid SI-60L” and “Sun Aid SI-80L” Commercial products such as “Sun-Aid SI-100L” and “Sun-Aid SI-150L” (manufactured by Sanshin Chemical Industry Co., Ltd.) can be used.
- the aluminum chelate examples include ethyl acetoacetate aluminum diisopropylate and aluminum tris (ethyl acetoacetate).
- the boron trifluoride amine complex include boron trifluoride monoethylamine complex, boron trifluoride imidazole complex, and boron trifluoride piperidine complex.
- the radical polymerization initiator is a compound capable of initiating or accelerating the radical polymerization reaction of a radical curable compound such as the polyorganosilsesquioxane of the present invention.
- the radical polymerization initiator is not particularly limited, and examples thereof include a photo radical polymerization initiator and a thermal radical polymerization initiator.
- photo radical polymerization initiator examples include benzophenone, acetophenone benzyl, benzyl dimethyl ketone, benzoin, benzoin methyl ether, benzoin ethyl ether, benzoin isopropyl ether, dimethoxyacetophenone, dimethoxyphenylacetophenone, diethoxyacetophenone, diphenyl disulfite, Orthobenzoyl methyl benzoate, ethyl 4-dimethylaminobenzoate (Nippon Kayaku Co., Ltd., trade name “Kayacure EPA”, etc.), 2,4-diethylthioxanthone (Nippon Kayaku Co., Ltd., trade name) “Kayacure DETX”, etc.), 2-methyl-1- [4- (methyl) phenyl] -2-morpholinopropanone-1 (manufactured by Ciba Gaigi Co., Ltd., trade name “Irgacure 90
- Aminobenzene derivatives such as phenylalkane compounds, tetra (t-butylperoxycarbonyl) benzophenone, benzyl, 2-hydroxy-2-methyl-1-phenyl-propan-1-one, 4,4-bisdiethylaminobenzophenone, 2, 2′-bis (2-chlorophenyl) -4,5,4 ′, 5′-tetraphenyl-1,2′-biimidazole (manufactured by Hodogaya Chemical Co., Ltd., trade name “B-CIM”, etc.), etc.
- phenylalkane compounds tetra (t-butylperoxycarbonyl) benzophenone
- benzyl 2-hydroxy-2-methyl-1-phenyl-propan-1-one
- 4,4-bisdiethylaminobenzophenone 2, 2′-bis (2-chlorophenyl) -4,5,4 ′, 5′-tetraphenyl-1,2′
- 1,6-bis (trichloromethyl) -4- (4-methoxynaphthalen-1-yl) -1,3,5-to Halomethyl triazines compounds such as azine, 2-trichloromethyl-5- (2-benzofuran-2-yl - ethenyl) -1,3,4 oxadiazole, and the like halomethyl oxadiazole compounds, and the like.
- a photosensitizer can be added as needed.
- thermal radical polymerization initiator examples include hydroperoxide, dialkyl peroxide, peroxyester, diacyl peroxide, peroxydicarbonate, peroxyketal, and ketone peroxide (specifically, benzoyl peroxide, t-butylperoxy-2-ethylhexanoate, 2,5-dimethyl-2,5-di (2-ethylhexanoyl) peroxyhexane, t-butylperoxybenzoate, t-butylperoxide, cumene hydro Peroxide, dicumyl peroxide, di-t-butyl peroxide, 2,5-dimethyl-2,5-dibutylperoxyhexane, 2,4-dichlorobenzoyl peroxide, 1,4-di (2-t- Butyl peroxyisopropyl) benzene 1,1-bis (t-butylperoxy) -3,3,5-trimethylcyclohexane
- one type of curing catalyst can be used alone, or two or more types can be used in combination.
- the content (blending amount) of the curing catalyst in the curable composition of the present invention is not particularly limited, but is 0.01 to 3.0 parts by weight with respect to 100 parts by weight of the polyorganosilsesquioxane of the present invention. It is preferably 0.05 to 3.0 parts by weight, more preferably 0.1 to 1.0 parts by weight (for example, 0.3 to 1.0 parts by weight).
- the content of the curing catalyst is 0.01 parts by weight or more, the curing reaction can be efficiently and sufficiently advanced, and the surface hardness and adhesiveness of the cured product tend to be further improved.
- the content of the curing catalyst is 3.0 parts by weight or less, the storability of the curable composition tends to be further improved, and coloring of the cured product tends to be suppressed.
- the curable composition of the present invention further includes a cationic curable compound other than the polyorganosilsesquioxane of the present invention (sometimes referred to as “other cationic curable compounds”) and / or the polyorganosyl of the present invention. It may contain a radical curable compound other than sesquioxane (sometimes referred to as "other radical curable compound”).
- other cationic curable compounds known or conventional cationic curable compounds can be used, and are not particularly limited.
- another cationic curable compound can also be used individually by 1 type, and can also be used in combination of 2 or more type.
- epoxy compound the well-known thru
- numerator can be used, although it does not specifically limit,
- Examples of the alicyclic epoxy compound include known or conventional compounds having one or more alicyclic rings and one or more epoxy groups in the molecule, and are not particularly limited.
- a compound having an epoxy group (referred to as “alicyclic epoxy group”) composed of two adjacent carbon atoms and oxygen atoms constituting the alicyclic ring; (2) the epoxy group is directly bonded to the alicyclic ring by a single bond.
- compounds having an alicyclic ring and a glycidyl ether group in the molecule (glycidyl ether type epoxy compound) and the like.
- Y represents a single bond or a linking group (a divalent group having one or more atoms).
- the linking group include a divalent hydrocarbon group, an alkenylene group in which part or all of a carbon-carbon double bond is epoxidized, a carbonyl group, an ether bond, an ester bond, a carbonate group, an amide group, and the like. And a group in which a plurality of are connected.
- Examples of the divalent hydrocarbon group include a linear or branched alkylene group having 1 to 18 carbon atoms, a divalent alicyclic hydrocarbon group, and the like.
- Examples of the linear or branched alkylene group having 1 to 18 carbon atoms include a methylene group, a methylmethylene group, a dimethylmethylene group, an ethylene group, a propylene group, and a trimethylene group.
- divalent alicyclic hydrocarbon group examples include 1,2-cyclopentylene group, 1,3-cyclopentylene group, cyclopentylidene group, 1,2-cyclohexylene group, 1,3-cyclopentylene group, And divalent cycloalkylene groups (including cycloalkylidene groups) such as cyclohexylene group, 1,4-cyclohexylene group and cyclohexylidene group.
- alkenylene group in the alkenylene group in which part or all of the carbon-carbon double bond is epoxidized include, for example, vinylene group, propenylene group, 1-butenylene group And straight-chain or branched alkenylene groups having 2 to 8 carbon atoms such as 2-butenylene group, butadienylene group, pentenylene group, hexenylene group, heptenylene group, octenylene group and the like.
- the epoxidized alkenylene group is preferably an alkenylene group in which all of the carbon-carbon double bonds are epoxidized, more preferably 2 to 4 carbon atoms in which all of the carbon-carbon double bonds are epoxidized. Alkenylene group.
- alicyclic epoxy compound represented by the above formula (i) include (3,4,3 ′, 4′-diepoxy) bicyclohexyl, and the following formulas (i-1) to (i-10) ) And the like.
- l and m each represents an integer of 1 to 30.
- R ′ in the following formula (i-5) is an alkylene group having 1 to 8 carbon atoms, and among them, a linear or branched chain having 1 to 3 carbon atoms such as a methylene group, an ethylene group, a propylene group, an isopropylene group, etc. -Like alkylene groups are preferred.
- n1 to n6 each represents an integer of 1 to 30.
- Other examples of the alicyclic epoxy compound represented by the above formula (i) include 2,2-bis (3,4-epoxycyclohexyl) propane and 1,2-bis (3,4-epoxycyclohexyl). ) Ethane, 2,3-bis (3,4-epoxycyclohexyl) oxirane, bis (3,4-epoxycyclohexylmethyl) ether and the like.
- Examples of the compound (2) in which the epoxy group is directly bonded to the alicyclic ring with a single bond include compounds represented by the following formula (ii).
- R ′′ is a group obtained by removing p hydroxyl groups (—OH) from the structural formula of p-valent alcohol (p-valent organic group), and p and n each represent a natural number.
- the divalent alcohol [R ′′ (OH) p ] include polyhydric alcohols (such as alcohols having 1 to 15 carbon atoms) such as 2,2-bis (hydroxymethyl) -1-butanol.
- p is preferably 1 to 6
- n is preferably 1 to 30.
- n in each group in () (inside the outer parenthesis) may be the same or different.
- Examples of the compound (3) having an alicyclic ring and a glycidyl ether group in the molecule include glycidyl ethers of alicyclic alcohols (particularly, alicyclic polyhydric alcohols). More specifically, for example, 2,2-bis [4- (2,3-epoxypropoxy) cyclohexyl] propane, 2,2-bis [3,5-dimethyl-4- (2,3-epoxypropoxy) Compound obtained by hydrogenating bisphenol A type epoxy compound such as cyclohexyl] propane (hydrogenated bisphenol A type epoxy compound); bis [o, o- (2,3-epoxypropoxy) cyclohexyl] methane, bis [o , P- (2,3-epoxypropoxy) cyclohexyl] methane, bis [p, p- (2,3-epoxypropoxy) cyclohexyl] methane, bis [3,5-dimethyl-4- (2, 3-Epoxypropoxy)
- aromatic epoxy compound examples include epibis type glycidyl ether type epoxy resins obtained by condensation reaction of bisphenols [for example, bisphenol A, bisphenol F, bisphenol S, fluorene bisphenol and the like] and epihalohydrin; High molecular weight epibis type glycidyl ether type epoxy resin obtained by addition reaction of bis type glycidyl ether type epoxy resin with the above bisphenols; phenols [eg, phenol, cresol, xylenol, resorcin, catechol, bisphenol A, bisphenol F, bisphenol S, etc.] and aldehyde [eg, formaldehyde, acetaldehyde, benzaldehyde, hydroxybenzaldehyde, salicy A novolak alkyl type glycidyl ether type epoxy resin obtained by further condensing polyhydric alcohols obtained by condensation reaction with aldehyde and the like with epihalohydrin; two phenol skeletons are
- Examples of the aliphatic epoxy compound include a glycidyl ether of an alcohol having no q-valent cyclic structure (q is a natural number); a monovalent or polyvalent carboxylic acid [for example, acetic acid, propionic acid, butyric acid, stearic acid, Adipic acid, sebacic acid, maleic acid, itaconic acid, etc.] glycidyl ester; epoxidized oils and fats having double bonds such as epoxidized linseed oil, epoxidized soybean oil, epoxidized castor oil; polyolefins such as epoxidized polybutadiene (poly Epoxidized product of alkadiene).
- a monovalent or polyvalent carboxylic acid for example, acetic acid, propionic acid, butyric acid, stearic acid, Adipic acid, sebacic acid, maleic acid, itaconic acid, etc.
- glycidyl ester e
- Examples of the alcohol having no q-valent cyclic structure include monohydric alcohols such as methanol, ethanol, 1-propyl alcohol, isopropyl alcohol and 1-butanol; ethylene glycol, 1,2-propanediol, 1 Divalent alcohols such as 1,3-propanediol, 1,4-butanediol, neopentyl glycol, 1,6-hexanediol, diethylene glycol, triethylene glycol, tetraethylene glycol, dipropylene glycol, polyethylene glycol, polypropylene glycol; Examples include trihydric or higher polyhydric alcohols such as glycerin, diglycerin, erythritol, trimethylolethane, trimethylolpropane, pentaerythritol, dipentaerythritol, and sorbitol. That.
- the q-valent alcohol may be polyether polyol, polyester polyol, polycarbonate polyo
- oxetane compound examples include known or commonly used compounds having one or more oxetane rings in the molecule, and are not particularly limited.
- the vinyl ether compound may be a known or conventional compound having one or more vinyl ether groups in the molecule, and is not particularly limited.
- 2-hydroxyethyl vinyl ether ethylene glycol monovinyl ether
- 3-hydroxy Propyl vinyl ether 2-hydroxypropyl vinyl ether
- 2-hydroxyisopropyl vinyl ether 4-hydroxybutyl vinyl ether, 3-hydroxybutyl vinyl ether, 2-hydroxybutyl vinyl ether, 3-hydroxyisobutyl vinyl ether, 2-hydroxyisobutyl vinyl ether, 1-methyl-3 -Hydroxypropyl vinyl ether, 1-methyl-2-hydroxypropyl vinyl ether, 1-hydroxymethylpropyl vinyl ether
- 4-hydroxycyclohexyl vinyl ether 1,6-hexanediol monovinyl ether, 1,6-hexanediol divinyl ether, 1,8-octanediol divinyl ether, 1,4-cyclohexanedimethanol monovinyl ether
- the curable composition of the present invention it is preferable to use a vinyl ether compound in combination with the polyorganosilsesquioxane of the present invention as another cationic curable compound.
- a vinyl ether compound in combination with the polyorganosilsesquioxane of the present invention as another cationic curable compound.
- the other cationic curable compound particularly when a vinyl ether compound having one or more hydroxyl groups in the molecule is used, the surface hardness is higher, and further, heat-resistant yellowing (yellowing due to heating hardly occurs).
- a cured product having excellent characteristics can be obtained. For this reason, an in-mold injection molded product and a hard coat film using the cured product of higher quality and higher durability, the transfer film of the present invention can be obtained.
- the number of hydroxyl groups in the molecule of the vinyl ether compound having one or more hydroxyl groups in the molecule is not particularly limited, but is preferably 1 to 4, more preferably 1 or 2.
- examples of vinyl ether compounds having one or more hydroxyl groups in the molecule include 2-hydroxyethyl vinyl ether (ethylene glycol monovinyl ether), 3-hydroxypropyl vinyl ether, 2-hydroxypropyl vinyl ether, 2-hydroxyisopropyl.
- radical curable compound a known or conventional radical curable compound can be used, and is not particularly limited, and examples thereof include (meth) acrylic compounds other than the polyorganosilsesquioxane of the present invention.
- another radical curable compound can also be used individually by 1 type, and can also be used in combination of 2 or more type.
- the (meth) acrylic compound a known or conventional compound having one or more (meth) acrylic groups in the molecule can be used, and is not particularly limited.
- trimethylolpropane tri (meth) acrylate Trimethylolethane tri (meth) acrylate, tetramethylolmethane tri (meth) acrylate, tetramethylolmethanetetra (meth) acrylate, pentaglycerol tri (meth) acrylate, pentaerythritol di (meth) acrylate, pentaerythritol tri (meth) acrylate , Pentaerythritol tetra (meth) acrylate, glycerin tri (meth) acrylate, dipentaerythritol tri (meth) acrylate, dipentaerythritol tetra (meth) acrylate, dipenta Risuri penta (meth) acrylate,
- the content (mixing amount) of other cation curable compounds and / or other radical curable compounds in the curable composition of the present invention is not particularly limited, but the polyorganosilsesquioxane of the present invention and other cations are not limited.
- 50% by weight or less (for example, 0 to 50% by weight) is preferable with respect to the total amount of the curable compound and the other radical curable compound (100% by weight; the total amount of the cationic curable compound and the radical curable compound). It is preferably 30% by weight or less (for example, 0 to 30% by weight), more preferably 10% by weight or less.
- the content (blending amount) of the vinyl ether compound (particularly, the vinyl ether compound having one or more hydroxyl groups in the molecule) in the curable composition of the present invention is not particularly limited, but the polyorganosilsesquioxane of the present invention,
- the amount is preferably 0.01 to 10% by weight, more preferably 0%, based on the total amount of the other cationic curable compound and the other radical curable compound (100% by weight; the total amount of the cationic curable compound and the radical curable compound). 0.05 to 9% by weight, more preferably 1 to 8% by weight.
- the surface hardness of the cured product becomes higher, and the surface hardness is extremely high even when the irradiation amount of active energy rays (for example, ultraviolet rays) is lowered. There is a tendency to get things.
- active energy rays for example, ultraviolet rays
- the content of the vinyl ether compound having one or more hydroxyl groups in the molecule within the above range, in addition to particularly high surface hardness of the cured product, there is a tendency to further improve its heat-resistant yellowing. is there.
- the curable composition of the present invention further includes, as other optional components, precipitated silica, wet silica, fumed silica, calcined silica, titanium oxide, alumina, glass, quartz, aluminosilicate, iron oxide, zinc oxide, calcium carbonate.
- Inorganic fillers such as carbon black, silicon carbide, silicon nitride and boron nitride, inorganic fillers obtained by treating these fillers with organosilicon compounds such as organohalosilanes, organoalkoxysilanes and organosilazanes; silicone resins, epoxy resins , Organic resin fine powders such as fluororesins; fillers such as conductive metal powders such as silver and copper, curing aids, solvents (organic solvents, etc.), stabilizers (antioxidants, ultraviolet absorbers, light stabilizers) , Heat stabilizers, heavy metal deactivators, etc.), flame retardants (phosphorous flame retardants, halogen flame retardants, inorganic flame retardants, etc.), difficult Auxiliaries, reinforcing materials (other fillers, etc.), nucleating agents, coupling agents (silane coupling agents, etc.), lubricants, waxes, plasticizers, mold release agents, impact resistance improvers, hue improvers, clearing
- the curable composition of the present invention is not particularly limited, but can be prepared by stirring and mixing each of the above components at room temperature or while heating as necessary.
- the curable composition of the present invention can be used as a one-component composition in which each component is mixed in advance, for example, two or more components stored separately.
- the curable composition of the present invention is not particularly limited, but is preferably liquid at normal temperature (about 25 ° C.). More specifically, the curable composition of the present invention has a viscosity at 25 ° C. of a liquid diluted to 20% of a solvent [particularly, a curable composition (solution) in which the proportion of methyl isobutyl ketone is 20% by weight]. 300 to 20000 mPa ⁇ s, more preferably 500 to 10000 mPa ⁇ s, and still more preferably 1000 to 8000 mPa ⁇ s. There exists a tendency for the heat resistance of hardened
- the viscosity of the curable composition of the present invention was measured using a viscometer (trade name “MCR301”, manufactured by Anton Paar) with a swing angle of 5%, a frequency of 0.1 to 100 (1 / s), and temperature: It is measured at 25 ° C.
- the curable composition can be cured by advancing the polymerization reaction of the cationic curable compound or radical curable compound (such as the polyorganosilsesquioxane of the present invention) in the curable composition of the present invention, A cured product (sometimes referred to as “cured product of the present invention”) can be obtained.
- the curing method can be appropriately selected from well-known methods and is not particularly limited, and examples thereof include a method of irradiation with active energy rays and / or heating.
- the active energy ray for example, any of infrared rays, visible rays, ultraviolet rays, X-rays, electron beams, ⁇ rays, ⁇ rays, ⁇ rays and the like can be used.
- ultraviolet rays are preferable in terms of excellent handleability.
- Conditions for curing the curable composition of the present invention by irradiation with active energy rays depend on the type and energy of the active energy rays to be irradiated, the shape and size of the cured product, etc. Although it can be appropriately adjusted and is not particularly limited, it is preferably about 1 to 1000 mJ / cm 2 , for example, when irradiated with ultraviolet rays.
- irradiation with active energy rays for example, a high-pressure mercury lamp, an ultrahigh-pressure mercury lamp, a xenon lamp, a carbon arc, a metal halide lamp, sunlight, an LED lamp, a laser, or the like can be used.
- a heating treatment annealing and aging
- conditions for curing the curable composition of the present invention by heating are not particularly limited, but are preferably 30 to 200 ° C., and more preferably 50 to 190 ° C., for example.
- the curing time can be appropriately set.
- the curable composition of the present invention can be cured to form a cured product having high surface hardness and heat resistance and excellent flexibility and workability.
- the curable composition of the present invention particularly includes a “curable composition for forming a hard coat layer” (“hard coat liquid”, “hard coat agent” and the like for forming a hard coat layer in a hard coat film. And may be particularly preferably used.
- the curable composition of the present invention is used as a curable composition for forming a hard coat layer, and a hard coat film having a hard coat layer formed from the composition is flexible while maintaining high hardness and high heat resistance. It can be manufactured and processed by roll-to-roll.
- the curable composition of the present invention is tack-free on the surface of an uncured or semi-cured hard coat layer coated and dried on a release layer provided on a substrate, and has anti-blocking properties. Since it improves, it is possible to handle it by winding it into a roll, and furthermore, a hard coat layer having a high surface hardness can be formed by transferring and curing the hard coat layer on the surface of the molded product. . Accordingly, the curable composition of the present invention can be preferably used as a curable composition for forming a hard coat layer for forming a hard coat layer of a transfer film used for in-mold injection molding. .
- the transfer film of the present invention is a film having a substrate and an uncured or semi-cured hard coat layer on a release layer formed on at least one surface of the substrate, the uncured film
- a semi-cured hard coat layer is formed by the curable composition of the present invention (a curable composition for forming a hard coat layer.
- hard coat agent of the present invention it may be referred to as “hard coat agent of the present invention”.
- uncured means that the polymerizable functional group of the polyorganosilsesquioxane of the present invention contained in the curable composition for forming a hard coat layer (hard coat agent) of the present invention has not undergone a polymerization reaction. means.
- Semi-curing means a state in which a part of the polymerizable functional group undergoes a polymerization reaction and an unreacted polymerizable functional group remains.
- an uncured or semi-cured hard coat layer formed by the curable composition (hard coat agent) of the present invention was simply transferred to a “hard coat layer” and molded product.
- the hard coat layer may be referred to as a “cured hard coat layer”.
- the base material in the transfer film of the present invention is a base material of the transfer film, and refers to a portion constituting other than the transfer layer including the hard coat layer of the present invention.
- the transfer layer is a layer excluding the substrate on which the release layer is formed in the transfer film of the present invention, and refers to a portion transferred to the surface of the molded product.
- a plastic base material such as a plastic base material, a metal base material, a ceramic base material, a semiconductor base material, a glass base material, a paper base material, a wood base material (wood base material), and the base material whose surface is a coating surface Thru
- a plastic substrate (a substrate made of a plastic material) is preferable.
- polyesters such as polyethylene terephthalate (PET) and polyethylene naphthalate (PEN); Polyimide; Polycarbonate; Polyamide; Polyacetal; Polyphenylene oxide; Polyphenylene sulfide; Polyether Sulfone; Polyetheretherketone; Norbornene monomer homopolymer (addition polymer, ring-opening polymer, etc.), Norbornene monomer and olefin monomer copolymer (addition polymer, such as norbornene and ethylene copolymer) And cyclic olefin copolymers such as ring-opening polymers), cyclic polyolefins such as derivatives thereof; vinyl polymers (for example, acrylic resins such as polymethyl methacrylate (PMMA), police Styrene, butadiene resin (ABS resin, etc.); vinylidene polymers (eg, polyvinylidene chloride,
- the plastic substrate it is preferable to use a substrate excellent in heat resistance, moldability, and mechanical strength, more preferably a polyester film (particularly PET, PEN), a cyclic polyolefin film, a polycarbonate film, a TAC film. , PMMA film.
- the plastic substrate is made of an antioxidant, an ultraviolet absorber, a light stabilizer, a heat stabilizer, a crystal nucleating agent, a flame retardant, a flame retardant aid, a filler, a plasticizer, and an impact modifier.
- Other additives such as reinforcing agents, dispersants, antistatic agents, foaming agents, antibacterial agents and the like may be included.
- an additive can also be used individually by 1 type and can also be used in combination of 2 or more type.
- the plastic substrate may have a single-layer configuration or a multilayer (lamination) configuration, and the configuration (structure) is not particularly limited.
- the above-mentioned plastic substrate is a “plastic film / other layer” in which a layer other than the transfer layer of the present invention (sometimes referred to as “other layer”) is formed on at least one surface of the plastic film.
- It may be a plastic substrate having a laminated structure such as “other layer / plastic film / other layer”.
- the other layers include hard coat layers other than the hard coat layer constituting the transfer film of the present invention.
- the above-mentioned plastic material etc. are mentioned, for example.
- Roughening, easy adhesion, antistatic treatment, sandblasting (sandmat treatment), corona discharge treatment, plasma treatment, chemical etching treatment, watermat treatment, flame are applied to part or all of the surface of the plastic substrate.
- Known or conventional surface treatments such as treatment, acid treatment, alkali treatment, oxidation treatment, ultraviolet irradiation treatment, silane coupling agent treatment, etc. may be applied.
- the plastic substrate may be an unstretched film or a stretched film (uniaxially stretched film, biaxially stretched film, etc.).
- the plastic base material is, for example, a method of forming the above plastic material into a film shape to form a plastic base material (plastic film), and if necessary, an appropriate layer (for example, the above-mentioned other layers) with respect to the plastic film.
- a layer or the like, or an appropriate surface treatment for example, a layer or the like, or an appropriate surface treatment.
- a commercial item can also be used as said plastic base material.
- the thickness of the substrate is not particularly limited, and can be appropriately selected from, for example, a range of 0.01 to 10000 ⁇ m. However, from the viewpoint of moldability, shape followability, handleability, etc., 2 to 250 ⁇ m is preferable. 5 to 100 ⁇ m is more preferable, and 20 to 100 ⁇ m is more preferable.
- the release layer in the transfer film of the present invention is a layer that constitutes at least one surface layer of the substrate in the transfer film of the present invention, and is provided to easily peel the transfer layer from the substrate. Is a layer.
- the transfer layer can be reliably and easily transferred from the transfer film to the transfer target (molded product), and the substrate sheet can be reliably peeled off.
- the peel strength between the release layer and the hard coat layer is not particularly limited, but is preferably 30 to 500 mN / 24 mm, more preferably 40 to 300 mN / 24 mm, and still more preferably 50. ⁇ 200 mN / 24 mm.
- the peel strength is within this range, the hard coat layer does not peel during normal handling, and the hard coat tends to be easily peeled simultaneously with the transfer to the molded product.
- the peel strength between the hard coat layer and the release layer of the present invention can be measured according to JIS Z0237.
- the release layer in the transfer film of the present invention may be formed only on one surface (one side) of the substrate, or may be formed on both surfaces (both sides). In addition, the release layer in the transfer film of the present invention may be formed on only a part or on the entire surface of each surface of the substrate.
- a publicly known release agent can be used without particular limitation, and examples thereof include unsaturated ester resins, epoxy resins, epoxy-melamine resins, aminoalkyd resins, At least one selected from acrylic resins, melamine resins, silicon resins, fluorine resins, cellulose resins, urea resin resins, polyolefin resins, paraffin resins, and cycloolefin resins can be used.
- the release layer is preferably a melamine resin or a cycloolefin resin, and in particular, 2-norbornene / ethylene copolymer.
- a cycloolefin copolymer resin (COC resin) such as a coalescence is preferable.
- a publicly known release processing method can be used without any particular limitation.
- the above resin is dispersed or dissolved in a solvent (eg, alcohols such as methanol and butanol, aromatic hydrocarbons such as toluene and xylene, tetrahydrofuran, etc.), bar coat, Mayer bar coat, gravure coat, roll coat, etc.
- a release layer can be formed by coating, drying, and heating at 80 to 200 ° C. by a known coating method.
- the thickness of the release layer is not particularly limited, and can usually be selected from the range of 0.01 to 5 ⁇ m, preferably 0.1 to 3.0 ⁇ m.
- the hard coat layer of the present invention in the transfer film of the present invention is a layer constituting at least one surface layer in the release layer, and is uncured by drying the curable composition (hard coat agent) of the present invention. Or a semi-cured layer partially cured.
- the semi-cured hard coat layer can be formed by partially curing the uncured hard coat layer by the above-mentioned active energy ray irradiation or heating.
- the uncured or semi-cured hard coat layer of the present invention has a low tack property and excellent blocking resistance so that the resin does not adhere when the finger is brought into contact with the surface, and can be wound and handled in a roll shape. It is.
- the hard coat layer of the present invention in the transfer film of the present invention may be formed only on one release layer (one side) of the substrate, or formed on both release layers (both sides). It may be. Further, the hard coat layer of the present invention in the transfer film of the present invention may be formed on only a part of the surface of each of the release layers, or may be formed on the entire surface.
- the method for laminating the hard coat layer of the present invention on the release layer of the transfer film of the present invention is not particularly limited, but the curable composition (hard coat agent) of the present invention is formed on the release layer by a known method. ) Is applied and dried to form an uncured hard coat layer, or further, a semi-hardened hard coat layer is formed by irradiating or heating an activation energy ray to the uncured hard coat layer.
- a coating method of the curable composition (hard coat agent) of the present invention a known coating method can be used without limitation, and examples thereof include bar coater coating, Mayer bar coating, air knife coating, and gravure coating. Examples thereof include offset printing, offset printing, flexographic printing, and screen printing.
- the heating temperature for forming the hard coat layer is not particularly limited, but can be suitably selected from 50 to 200 ° C.
- the heating time is not particularly limited, but can be appropriately selected from 1 to 60 minutes.
- the conditions for irradiating the hard coat layer with the activation energy rays are not particularly limited, and can be appropriately selected from the conditions for forming the above-described cured product, for example.
- the thickness of the hard coat layer in the transfer film of the present invention is not particularly limited, but is preferably 1 to 200 ⁇ m, More preferably, it is 3 to 150 ⁇ m.
- the hard coat layer of the present invention is thin (for example, when the thickness is 5 ⁇ m or less), it is possible to maintain a high surface hardness (for example, a pencil hardness of 5 H or more).
- it is thick for example, when the thickness is 50 ⁇ m or more
- the haze of the hard coat layer in the transfer film of the present invention is not particularly limited, but is preferably 1.5% or less, more preferably 1.0% or less in the case of a thickness of 50 ⁇ m.
- the lower limit of haze is not particularly limited, but is 0.1%, for example.
- the haze of the hard coat layer of the present invention can be measured according to JIS K7136.
- the total light transmittance of the hard coat layer in the transfer film of the present invention is not particularly limited, but is preferably 85% or more, more preferably 90% or more in the case of a thickness of 50 ⁇ m.
- the upper limit of the total light transmittance is not particularly limited, but is 99%, for example.
- an anchor coat layer and an adhesive layer are further laminated in this order on the hard coat layer. Furthermore, when using the transfer film of the present invention as a decorative film, at least one colored layer is laminated. Although the lamination position of the colored layer is not particularly limited, an embodiment in which one layer or two or more layers are preferably laminated between the anchor coat layer and the adhesive layer is preferable.
- the anchor coat layer in the transfer film of the present invention is provided in order to improve the adhesion between the hard coat layer and the adhesive layer or the colored layer.
- the resin for anchor coating of the present invention further includes, as other optional components, wax, silica, plasticizer, leveling agent, surfactant, dispersant, antifoaming agent, ultraviolet absorber, ultraviolet stabilizer, antioxidant, etc.
- wax silica
- plasticizer leveling agent
- surfactant dispersant
- antifoaming agent ultraviolet absorber
- ultraviolet stabilizer antioxidant
- the anchor coat layer is obtained by applying a coating solution obtained by dissolving the above resin in a solvent to the hard coat layer of the present invention by a known coating method such as bar coating, Mayer bar coating, gravure coating, roll coating, and drying. If necessary, it can be formed by heating.
- the temperature for heating when forming the anchor coat layer is not particularly limited, but can be suitably selected from 50 to 200 ° C.
- the heating time is not particularly limited, but can be appropriately selected from 10 seconds to 60 minutes.
- the thickness of the anchor coat layer is usually about 0.1 to 20 ⁇ m, and preferably in the range of 0.5 to 5 ⁇ m.
- the anchor coat layer of the present invention may be formed using a commercially available anchor coat agent.
- the commercially available anchor coating agent include K468HP anchor (epoxy resin anchor coating agent manufactured by Toyo Ink Co., Ltd.) and TM-VMAC (acrylic polyol resin anchor coating agent manufactured by Dainichi Seika Kogyo Co., Ltd.).
- the adhesive layer in the transfer film of the present invention is provided to transfer a transfer layer (including a hard coat layer, an anchor coat layer and a colored layer, which are optionally laminated) to a molded product with good adhesiveness.
- a transfer layer including a hard coat layer, an anchor coat layer and a colored layer, which are optionally laminated
- the adhesive layer include those composed of a heat-sensitive adhesive, a pressure-sensitive adhesive, and the like, but in the present invention, heat sealing that exhibits adhesion to a molded product by heating and pressing as necessary.
- a layer is preferred.
- the resin used for the adhesive layer include acrylic resins, vinyl chloride resins, vinyl acetate resins, vinyl chloride-vinyl acetate copolymer resins, styrene-acrylic copolymer resins, polyester resins, and polyamide resins.
- acrylic resins and vinyl chloride-vinyl acetate copolymer resins are particularly preferable.
- the acrylic resin used in the adhesive layer of the present invention is not particularly limited.
- polymethyl (meth) acrylate, polyethyl (meth) acrylate, polybutyl (meth) acrylate, methyl (meth) acrylate-butyl (meth) examples thereof include acrylic resins such as acrylate copolymers, methyl (meth) acrylate-styrene copolymers, and modified acrylic resins such as fluorine, and these can be used as one kind or a mixture of two or more kinds.
- (meth) acrylic acid alkyl esters such as methyl (meth) acrylate, ethyl (meth) acrylate, butyl (meth) acrylate, 2-ethylhexyl (meth) acrylate, octyl (meth) acrylate, and 2-hydroxyethyl
- Acrylic polyol obtained by copolymerizing (meth) acrylate having a hydroxyl group in the molecule such as (meth) acrylate, 2-hydroxybutyl (meth) acrylate, 2-hydroxy-3-phenoxypropyl (meth) acrylate
- Acrylic polyol obtained by copolymerizing (meth) acrylate having a hydroxyl group in the molecule such as (meth) acrylate, 2-hydroxybutyl (meth) acrylate, 2-hydroxy-3-phenoxypropyl (meth) acrylate
- (meth) acrylic acid alkyl esters such as methyl (meth) acrylate, ethy
- the vinyl chloride-vinyl acetate copolymer resin those having a vinyl acetate content of about 5 to 20% by mass and an average degree of polymerization of about 350 to 900 are usually used. If necessary, the vinyl chloride-vinyl acetate copolymer resin may be further copolymerized with a carboxylic acid such as maleic acid or fumaric acid.
- a subcomponent resin if necessary, other resins, for example, resins such as thermoplastic polyester resins, thermoplastic urethane resins, chlorinated polyethylene, chlorinated polypropylene, and other chlorinated polyolefin resins. May be mixed.
- the adhesive layer one or two or more solutions or emulsions of the above resins that can be applied are applied by a known coating method such as bar coating, Mayer bar coating, gravure coating, roll coating, and dried. And if necessary, it can be formed by heating.
- the temperature at the time of heating at the time of forming the adhesive layer is not particularly limited, but can be suitably selected from 50 to 200 ° C.
- the heating time is not particularly limited, but can be appropriately selected from 10 seconds to 60 minutes.
- the thickness of the adhesive layer is preferably about 0.1 to 10 ⁇ m, more preferably 0.5 to 5 ⁇ m, from the viewpoint that the transfer film can be efficiently transferred to a molded product.
- organic UV absorbers such as benzophenone compounds, benzotriazole compounds, oxalic acid anilide compounds, cyanoacrylate compounds, salicylate compounds, zinc, titanium, cerium, tin, iron, etc.
- An additive of fine particles having an inorganic ultraviolet absorbing ability such as an oxide of the above may be blended.
- a coloring pigment, a white pigment, an extender pigment, a filler, an antistatic agent, an antioxidant, a fluorescent brightening agent, and the like can be used as necessary.
- a commercially available product may be used as the adhesive of the present invention.
- Examples of commercially available adhesives include K588HP adhesive gloss A varnish (vinyl chloride-vinyl acetate copolymer resin adhesive manufactured by Toyo Ink Co., Ltd.) and PSHP780 (acrylic resin adhesive manufactured by Toyo Ink Co., Ltd.). .
- the colored layer in the transfer film of the present invention is provided when a decorative film for transferring a picture layer and / or a concealing layer to a molded product is provided.
- the pattern layer is a layer provided for expressing a pattern such as a pattern or characters
- the concealing layer is generally a solid layer and is provided for concealing coloring such as injection resin. Is a layer.
- a decorative layer may be formed alone in addition to the case of being provided inside the pattern layer in order to enhance the pattern of the pattern layer.
- the pattern layer according to the present invention is a layer provided to express a pattern, a character, and a pattern-like pattern.
- the pattern of the pattern layer is arbitrary, for example, a pattern composed of wood grain, stone grain, cloth grain, sand grain, geometric pattern, character, and the like can be mentioned.
- the colored layer is usually a known printing method such as gravure printing, offset printing, silk screen printing, transfer printing from a transfer sheet, sublimation transfer printing, ink jet printing on the hard coat layer or anchor coat layer. By forming by, it can form between a hard-coat layer and an adhesive bond layer, or between an anchor-coat layer and an adhesive bond layer.
- the thickness of the colored layer is preferably 3 to 40 ⁇ m, more preferably 10 to 30 ⁇ m from the viewpoint of design.
- the binder resin for printing ink used for forming the colored layer include polyester resins, polyurethane resins, acrylic resins, vinyl acetate resins, vinyl chloride-vinyl acetate copolymer resins, and cellulose resins.
- the main component is an acrylic resin alone or a mixture of an acrylic resin and a vinyl chloride-vinyl acetate copolymer resin.
- acrylic resins include polymethyl (meth) acrylate, polyethyl (meth) acrylate, polybutyl (meth) acrylate, methyl (meth) acrylate-butyl (meth) acrylate copolymer, methyl (meth) acrylate-styrene copolymer.
- acrylic resins such as polymers and modified acrylic resins by fluorine, and these can be used as one kind or a mixture of two or more kinds.
- (meth) acrylic acid alkyl esters such as methyl (meth) acrylate, ethyl (meth) acrylate, butyl (meth) acrylate, 2-ethylhexyl (meth) acrylate, octyl (meth) acrylate, and 2-hydroxyethyl
- Acrylic polyol obtained by copolymerizing (meth) acrylate having a hydroxyl group in the molecule such as (meth) acrylate, 2-hydroxybutyl (meth) acrylate, 2-hydroxy-3-phenoxypropyl (meth) acrylate
- Acrylic polyol obtained by copolymerizing (meth) acrylate having a hydroxyl group in the molecule such as (meth) acrylate, 2-hydroxybutyl (meth) acrylate, 2-hydroxy-3-phenoxypropyl (meth) acrylate
- (meth) acrylic acid alkyl esters such as methyl (meth) acrylate, ethy
- the vinyl chloride-vinyl acetate copolymer resin those having a vinyl acetate content of about 5 to 20% by mass and an average degree of polymerization of about 350 to 900 are usually used. If necessary, the vinyl chloride-vinyl acetate copolymer resin may be further copolymerized with a carboxylic acid such as maleic acid or fumaric acid.
- resins such as thermoplastic polyester resins, thermoplastic urethane resins, chlorinated polyethylene, chlorinated polypropylene, and other chlorinated polyolefin resins. May be mixed.
- Examples of the colorant used in the colored layer according to the present invention include aluminum, chromium, nickel, tin, titanium, iron phosphide, copper, gold, silver, brass and other metals, alloys, and scale-like foil powders of metal compounds.
- Pearl made of foil powder such as metallic pigment, mica-like iron oxide, titanium dioxide coated mica, titanium dioxide coated bismuth oxychloride, bismuth oxychloride, titanium dioxide coated talc, fish scale foil, colored titanium dioxide coated mica, basic lead carbonate Glossy (pearl) pigments, fluorescent pigments such as strontium aluminate, calcium aluminate, barium aluminate, zinc sulfide, calcium sulfide, white inorganic pigments such as titanium dioxide, zinc white, antimony trioxide, zinc white, petal, vermilion , Inorganic pigments such as ultramarine, cobalt blue, titanium yellow, yellow lead, carbon black, isoindolinone Chromatography, Hansa Yellow A, quinacridone red, can be mixed Permanent Red 4R, phthalocyanine blue, indanthrene blue RS, (including dyes) organic pigments such as aniline black one or more.
- fluorescent pigments such as strontium aluminate, calcium
- a metal thin film layer or the like may be further formed for the purpose of improving the designability.
- the metal thin film layer can be formed by using a metal such as aluminum, chromium, gold, silver, or copper by a method such as vacuum deposition or sputtering. This metal thin film layer may be provided on the entire surface or may be partially provided in a pattern.
- the printing ink used for forming the colored layer should be appropriately added with an anti-settling agent, a curing catalyst, an ultraviolet absorber, an antioxidant, a leveling agent, a thickener, an antifoaming agent, a lubricant, and the like. Can do.
- the printing ink is provided in a form in which the above components are usually dissolved or dispersed in a solvent.
- Any solvent can be used as long as it dissolves or disperses the binder resin, and an organic solvent and / or water can be used.
- the organic solvent include hydrocarbons such as toluene and xylene, ketones such as acetone and methyl ethyl ketone, esters such as ethyl acetate, cellosolve acetate, and butyl cellosolve acetate, and alcohols.
- the transfer film of the present invention includes a low reflection layer, an antistatic layer, and an ultraviolet absorption layer as required in addition to the above-mentioned base material, release layer, hard coat layer, anchor coat layer, adhesive layer, and colored layer.
- a layer, a near infrared ray blocking layer, an electromagnetic wave absorbing layer, and the like may be laminated in any order.
- the thickness of the transfer film of the present invention is not particularly limited, and can be appropriately selected from the range of 1 to 10000 ⁇ m. Is more preferably from 150 to 150 ⁇ m, further preferably from 25 to 150 ⁇ m.
- the hard coat layer of the transfer film of the present invention is tack-free and excellent in blocking resistance and can be wound and handled in a roll shape, it can be suitably used as a transfer film for in-mold injection molding. it can.
- the transfer film of the present invention was continuously transported by a transport roll or the like into a mold composed of a fixed mold and a movable mold, and the base film side was in contact with the fixed mold surface, and appropriate position adjustment was made. Later, the movable mold moves and clamps. Then, the thermoplastic resin previously melted by heat is injected and filled from the transfer layer side of the transfer film into the mold at high temperature and high pressure, and after quenching, the mold is opened, and the hard coat layer of the present invention is the outermost surface.
- the molded product (in-mold molded product) transferred to the can be taken out.
- the hard coat layer of the present invention When the hard coat layer of the present invention is uncured or semi-cured, the hard coat layer may be cured by irradiation with active energy rays and / or heating.
- the conditions when the hard coat layer is irradiated with active energy rays and / or heated are not particularly limited, and can be appropriately selected from, for example, the conditions when forming the above-described cured product.
- the pencil hardness on the surface of the molded product can be made extremely high. Preferably 5H or more, more preferably 6H or more.
- the pencil hardness can be evaluated according to the method described in JIS K5600-5-4.
- a molded product (in-mold molded product) manufactured by the in-mold injection molding method using the transfer film of the present invention has a very high surface hardness, and the pattern and pattern are clearly transferred. It can be preferably used for any molded product that requires characteristics.
- the transfer film of the present invention can be applied to various exterior molded products that require high surface hardness, scratch resistance, design, and durability, such as car interior and exterior products such as automobile dashboards and housings of home appliances. It can be preferably used.
- the hard coat film of the present invention is a film having a substrate and a hard coat layer formed on at least one surface of the substrate, wherein the hard coat layer is a curable composition (hard It is a hard coat layer (cured product layer of the curable composition of the present invention) formed by a curable composition for forming a coat layer).
- a curable composition hard It is a hard coat layer (cured product layer of the curable composition of the present invention) formed by a curable composition for forming a coat layer).
- the hard coat layer of the present invention in the hard coat film of the present invention may be formed only on one surface (one surface) of the substrate, or may be formed on both surfaces (both surfaces). .
- the hard coat layer of the present invention in the hard coat film of the present invention may be formed on only a part or on the entire surface of each surface of the substrate.
- the base material in the hard coat film of the present invention is a base material of the hard coat film, and refers to a portion other than the hard coat layer of the present invention.
- a base material well-known, such as a plastic base material, a metal base material, a ceramic base material, a semiconductor base material, a glass base material, a paper base material, a wood base material (wood base material), and the base material whose surface is a coating surface Thru
- a plastic substrate (a substrate made of a plastic material) is preferable.
- polyesters such as polyethylene terephthalate (PET) and polyethylene naphthalate (PEN); Polyimide; Polycarbonate; Polyamide; Polyacetal; Polyphenylene oxide; Polyphenylene sulfide; Polyether Sulfone; Polyetheretherketone; Norbornene monomer homopolymer (addition polymer, ring-opening polymer, etc.), Norbornene monomer and olefin monomer copolymer (addition polymer, such as norbornene and ethylene copolymer) And cyclic olefin copolymers such as ring-opening polymers), cyclic polyolefins such as derivatives thereof; vinyl polymers (for example, acrylic resins such as polymethyl methacrylate (PMMA), police Styrene, butadiene resin (ABS resin, etc.); vinylidene polymers (eg, polyvinylidene chloride,
- a substrate excellent in transparency is used as the plastic substrate.
- a polyester film particularly PET, PEN
- a cyclic polyolefin film a polycarbonate film
- TAC film a TAC film
- PMMA film a polymethyl methacrylate film
- the plastic substrate is made of an antioxidant, an ultraviolet absorber, a light stabilizer, a heat stabilizer, a crystal nucleating agent, a flame retardant, a flame retardant aid, a filler, a plasticizer, and an impact modifier.
- Other additives such as reinforcing agents, dispersants, antistatic agents, foaming agents, antibacterial agents and the like may be included.
- an additive can also be used individually by 1 type and can also be used in combination of 2 or more type.
- the plastic substrate may have a single-layer configuration or a multilayer (lamination) configuration, and the configuration (structure) is not particularly limited.
- the above-mentioned plastic substrate is “plastic film / other layer” in which a layer other than the hard coat layer of the present invention (sometimes referred to as “other layer”) is formed on at least one surface of the plastic film.
- it may be a plastic substrate having a laminated structure such as “other layer / plastic film / other layer”.
- the other layers include hard coat layers other than the hard coat layer of the present invention.
- the above-mentioned plastic material etc. are mentioned, for example.
- Roughening, easy adhesion, antistatic treatment, sandblasting (sandmat treatment), corona discharge treatment, plasma treatment, chemical etching treatment, watermat treatment, flame are applied to part or all of the surface of the plastic substrate.
- Known or conventional surface treatments such as treatment, acid treatment, alkali treatment, oxidation treatment, ultraviolet irradiation treatment, silane coupling agent treatment, etc. may be applied.
- the plastic substrate may be an unstretched film or a stretched film (uniaxially stretched film, biaxially stretched film, etc.).
- the plastic base material is, for example, a method of forming the above plastic material into a film shape to form a plastic base material (plastic film), and if necessary, an appropriate layer (for example, the above-mentioned other layers) with respect to the plastic film.
- a layer or the like, or an appropriate surface treatment for example, a layer or the like, or an appropriate surface treatment.
- a commercial item can also be used as said plastic base material.
- the thickness of the substrate is not particularly limited, but can be appropriately selected from a range of 0.01 to 10,000 ⁇ m, for example.
- the hard coat layer of the present invention in the hard coat film of the present invention is a layer constituting at least one surface layer in the hard coat film of the present invention, and the curable composition of the present invention (curable composition for forming a hard coat layer). It is a layer (cured product layer) formed by a cured product (resin cured product) obtained by curing the product.
- the thickness of the hard coat layer of the present invention (the thickness of each hard coat layer when the hard coat layer of the present invention is provided on both surfaces of the substrate) is not particularly limited, but is preferably 1 to 200 ⁇ m, more preferably 3 ⁇ 150 ⁇ m.
- the hard coat layer of the present invention is thin (for example, when the thickness is 5 ⁇ m or less), it is possible to maintain a high surface hardness (for example, the pencil hardness is set to H or more).
- it is thick for example, when the thickness is 50 ⁇ m or more
- the haze of the hard coat layer of the present invention is not particularly limited, but is preferably 1.5% or less, more preferably 1.0% or less in the case of a thickness of 50 ⁇ m.
- the lower limit of haze is not particularly limited, but is 0.1%, for example. By setting the haze to 1.0% or less, for example, the haze tends to be suitable for use in applications that require very high transparency (for example, surface protection sheets for displays such as touch panels).
- the haze of the hard coat layer of the present invention can be measured according to JIS K7136.
- the total light transmittance of the hard coat layer of the present invention is not particularly limited, but is preferably 85% or more, more preferably 90% or more in the case of a thickness of 50 ⁇ m.
- the upper limit of the total light transmittance is not particularly limited, but is 99%, for example. By setting the total light transmittance to 85% or more, for example, it tends to be suitable for use in applications that require extremely high transparency (for example, surface protection sheets for displays such as touch panels).
- the total light transmittance of the hard coat layer of the present invention can be measured according to JIS K7361-1.
- the hard coat film of the present invention may further have a surface protective film on the surface of the hard coat layer of the present invention.
- a surface protective film When the hard coat film of the present invention has a surface protective film, the punchability of the hard coat film tends to be further improved. In the case of having a surface protective film in this way, for example, even if the hardness of the hard coat layer is very high and peeling or cracking from the base material is likely to occur at the time of punching, such a problem occurs. It is possible to perform punching using a Thomson blade without causing it to occur.
- the surface protective film a known or commonly used surface protective film can be used, and is not particularly limited.
- a film having a pressure-sensitive adhesive layer on the surface of a plastic film can be used.
- the plastic film include polyester (polyethylene terephthalate, polyethylene naphthalate, etc.), polyolefin (polyethylene, polypropylene, cyclic polyolefin, etc.), polystyrene, acrylic resin, polycarbonate, epoxy resin, fluorine resin, silicone resin, diacetate resin, Examples thereof include plastic films formed from plastic materials such as triacetate resin, polyarylate, polyvinyl chloride, polysulfone, polyethersulfone, polyetheretherimide, polyimide, and polyamide.
- the adhesive layer examples include acrylic adhesives, natural rubber adhesives, synthetic rubber adhesives, ethylene-vinyl acetate copolymer adhesives, ethylene- (meth) acrylate copolymer adhesives, Examples thereof include a pressure-sensitive adhesive layer formed of one or more known or commonly used pressure-sensitive adhesives such as a styrene-isoprene block copolymer pressure-sensitive adhesive and a styrene-butadiene block copolymer pressure-sensitive adhesive.
- various additives for example, an antistatic agent, a slip agent, etc.
- the plastic film and the pressure-sensitive adhesive layer may each have a single layer configuration, or may have a multilayer (multi-layer) configuration.
- the thickness of a surface protection film is not specifically limited, It can select suitably.
- Examples of the surface protective film include the product name “Sanitek” series (manufactured by Sanei Kaken Co., Ltd.), the product name “E-MASK” series (manufactured by Nitto Denko Corporation), and the product name “Mastak” series (Fujimori Industry (commercially available products such as the product name “Hitarex” series (manufactured by Hitachi Chemical Co., Ltd.) and the product name “Alphan” series (manufactured by Oji F-Tex Co., Ltd.) are available from the market.
- the hard coat film of the present invention can be produced in accordance with a known or commonly used method for producing a hard coat film, and the production method is not particularly limited.
- the hard coat film of the present invention is applied to at least one surface of the substrate. It can be produced by applying a curable composition (a curable composition for forming a hard coat layer), removing the solvent by drying as necessary, and then curing the curable composition (curable composition layer). .
- the conditions for curing the curable composition are not particularly limited, and can be appropriately selected from, for example, the conditions for forming the cured product described above.
- the hard coat layer of the present invention in the hard coat film of the present invention is more than the curable composition of the present invention (a curable composition for forming a hard coat layer) capable of forming a cured product having excellent flexibility and processability. Since it is the formed hard coat layer, the hard coat film of the present invention can be produced by a roll-to-roll method. By producing the hard coat film of the present invention by a roll-to-roll method, the productivity can be remarkably increased. As a method for producing the hard coat film of the present invention by a roll-to-roll method, a known or conventional roll-to-roll method can be adopted, and is not particularly limited.
- the curable composition of the present invention (a curable composition for forming a hard coat layer) to at least one surface of the fed substrate, and then drying the solvent as necessary.
- the step of forming the hard coat layer of the present invention by curing the curable composition (curable composition layer) (step B), and then the obtained hard coat film again in a roll And a step of continuously carrying out these steps (steps A to C).
- the method may include steps other than steps A to C.
- the thickness of the hard coat film of the present invention is not particularly limited, and can be appropriately selected from the range of 1 to 10,000 ⁇ m.
- the pencil hardness of the hard coat layer surface of the present invention of the hard coat film of the present invention is not particularly limited, but is preferably H or more, more preferably 2H or more, and even more preferably 6H or more.
- the pencil hardness can be evaluated according to the method described in JIS K5600-5-4.
- the haze of the hard coat film of the present invention is not particularly limited, but is preferably 1.5% or less, more preferably 1.0% or less.
- the lower limit of haze is not particularly limited, but is 0.1%, for example.
- the haze of the hard coat film of the present invention can be easily controlled within the above range by using, for example, the above-mentioned transparent substrate as a substrate.
- the haze can be measured according to JIS K7136.
- the total light transmittance of the hard coat film of the present invention is not particularly limited, but is preferably 85% or more, more preferably 90% or more.
- the upper limit of the total light transmittance is not particularly limited, but is 99%, for example.
- the total light transmittance of the hard coat film of the present invention can be easily controlled within the above range by using, for example, the above-mentioned transparent substrate as the substrate.
- the total light transmittance can be measured according to JIS K7361-1.
- the hard coat film of the present invention has flexibility while maintaining high hardness and high heat resistance, and can be manufactured and processed in a roll-to-roll system, so it has high quality and high productivity. Also excellent.
- the surface protective film is provided on the surface of the hard coat layer of the present invention, the punching processability is also excellent. For this reason, it can be preferably used for any application that requires such characteristics.
- the hard coat film of the present invention can be used as, for example, a surface protective film for various products, a surface protective film for various product members or parts, and also used as a constituent material for various products, members or parts thereof.
- the products include display devices such as liquid crystal displays and organic EL displays; input devices such as touch panels; solar cells; various home appliances; various electric and electronic products; portable electronic terminals (for example, game machines, personal computers, tablets, Smartphones, mobile phones, etc.) and various electrical and electronic products; various optical devices.
- display devices such as liquid crystal displays and organic EL displays
- input devices such as touch panels; solar cells; various home appliances; various electric and electronic products; portable electronic terminals (for example, game machines, personal computers, tablets, Smartphones, mobile phones, etc.) and various electrical and electronic products; various optical devices.
- portable electronic terminals for example, game machines, personal computers, tablets, Smartphones, mobile phones, etc.
- various electrical and electronic products for example, game machines, personal computers, tablets, Smartphones, mobile phones, etc.
- various optical devices for example, an aspect used in a laminate of a hard coat film and a transparent conductive film in a touch panel, etc. It is done.
- the molecular weight of the product was measured using Alliance HPLC system 2695 (manufactured by Waters), Refractive Index Detector 2414 (manufactured by Waters), column: Tskel GMH HR- M ⁇ 2 (manufactured by Tosoh Corp.), guard column: Tskel guard column H HR L (manufactured by Tosoh Corp.), column oven: COLUMN HEATER U-620 (manufactured by Sugai), solvent: THF, measurement conditions: 40 ° C. Further, the ratio of T2 body to T3 body [T3 body / T2 body] in the product was measured by 29 Si-NMR spectrum measurement using JEOL ECA500 (500 MHz).
- FIG. 1 shows a 1 H-NMR chart and FIG. 2 shows a 29 Si-NMR chart of the obtained intermediate epoxy group-containing polyorganosilsesquioxane.
- Example 1 Production of Epoxy Group-Containing Polyorganosilsesquioxane of the Present Invention (1)
- the intermediate epoxy group-containing polyorganosilsesquioxane obtained in Production Example 1 was placed in a 1000 ml flask (reaction vessel) equipped with a thermometer, a stirrer, a reflux condenser, and a nitrogen inlet tube under a nitrogen stream.
- the molecular weight was measured by sampling at 80 ° C.
- Example 2 Production of Epoxy Group-Containing Polyorganosilsesquioxane of the Present Invention (2) Intermediate epoxy group-containing polyorganosyl obtained in the same manner as in Production Example 1 under a nitrogen stream in a 1000 ml flask (reaction vessel) equipped with a thermometer, a stirrer, a reflux condenser, and a nitrogen inlet tube A mixture (75 g) containing sesquioxane was charged, and 100 ppm (5.6 mg) of potassium carbonate and 2000 ppm (112 mg) of water with respect to the net content (56.2 g) of the intermediate epoxy group-containing polyorganosilsesquioxane. ), And when the molecular weight was measured by sampling when heated at 80 ° C.
- Example 3 Production of Epoxy Group-Containing Polyorganosilsesquioxane of the Present Invention (3) Intermediate epoxy group-containing polyorganosyl obtained in the same manner as in Production Example 1 under a nitrogen stream in a 1000 ml flask (reaction vessel) equipped with a thermometer, a stirrer, a reflux condenser, and a nitrogen inlet tube A mixture (75 g) containing sesquioxane was charged, and 100 ppm (5.6 mg) of potassium carbonate and 2000 ppm (112 mg) of water with respect to the net content (56.2 g) of the intermediate epoxy group-containing polyorganosilsesquioxane. ) When added and heated at 80 ° C.
- Production Example 2 Production of intermediate acrylic group-containing polyorganosilsesquioxane A 1000 ml flask (reaction vessel) equipped with a thermometer, a stirrer, a reflux condenser, and a nitrogen inlet tube was charged with 3- 370 mmol (80 g) of (acryloxy) propyltrimethoxysilane and 320 g of acetone were charged, and the temperature was raised to 50 ° C. After adding 10.144 g of a 5% aqueous potassium carbonate solution (3.67 mmol as potassium carbonate) in 5 minutes, 3670.0 mmol of water (66.08 g) was added over 20 minutes to the mixture thus obtained. Added. There was no significant temperature rise during the addition.
- a 5% aqueous potassium carbonate solution 3.67 mmol as potassium carbonate
- the polycondensation reaction was performed for 2 hours under a nitrogen stream while maintaining the temperature at 50 ° C. Thereafter, simultaneously with cooling the reaction solution, 160 g of methyl isobutyl ketone and 99.056 g of 5% saline were added. This solution was transferred to a 1 L separatory funnel, and 160 g of methyl isobutyl ketone was added again, followed by washing with water.
- aqueous layer is extracted, washed with water until the lower layer solution is neutral, and after the upper layer solution is separated, the solvent is distilled off from the upper layer solution under conditions of 1 mmHg and 50 ° C., and methyl isobutyl ketone 22 71 g of a colorless transparent liquid product (intermediate acrylic group-containing polyorganosilsesquioxane) containing 0.5% by weight was obtained.
- a colorless transparent liquid product intermediate acrylic group-containing polyorganosilsesquioxane
- the ratio [T3 body / T2 body] of the T2 body and T3 body calculated from the 29 Si-NMR spectrum of the product was 13.4.
- FIG. 7 shows a 1 H-NMR chart and FIG. 8 shows a 29 Si-NMR chart of the obtained intermediate acrylic group-containing polyorganosilsesquioxane, respectively.
- Example 4 Production of acrylic group-containing polyorganosilsesquioxane of the present invention (1) Into a 1000 ml flask (reaction vessel) equipped with a thermometer, a stirrer, a reflux condenser, and a nitrogen inlet tube, the intermediate acrylic group-containing polyorganosilsesquioxane obtained in Production Example 2 was placed under a nitrogen stream. Into the intermediate acrylic group-containing polyorgano, 10 ppm (0.55 mg) of potassium hydroxide and 2000 ppm (110 mg) of water are added to the net content of silsesquioxane (55.0 g). When the molecular weight was measured by sampling at the time of heating at 40 ° C.
- Nb / Et (2-norbornene-ethylene copolymer “TOPAS (registered trademark) 6017S-04” manufactured by Topas Advanced Polymers GmbH, glass transition temperature of 178 ° C.) and 1 part by weight of PVDC (polyvinylidene chloride)
- release film A As the base material layer, a biaxially stretched polyethylene terephthalate film ("Embret S50" manufactured by Unitika Ltd., thickness 50 ⁇ m) was used, and the release agent coating solution A was coated on one side of this film by the Mayer bar coating method. The film was dried at a temperature of 100 ° C. for 1 minute to form a release layer having a thickness of 0.3 ⁇ m, and a release film A was obtained.
- Embret S50 manufactured by Unitika Ltd., thickness 50 ⁇ m
- a hard coat coating liquid A is coated on the release layer surface of the release film A by the Mayer bar coating method, dried at a temperature of 80 ° C. for 2 minutes, and further dried at a temperature of 150 ° C. for 8 minutes to give a thickness of 40 ⁇ m.
- a hard coat layer was formed. When the surface of the obtained hard coat layer was touched with a finger, it was confirmed that no resin adhered to the finger and no surface tackiness was exhibited (tack-free).
- K468HP anchor an epoxy resin anchor coat agent manufactured by Toyo Ink Co., Ltd. is coated by the Mayer bar coating method and dried at a temperature of 80 ° C.
- K588HP adhesive gloss A varnish (vinyl chloride-vinyl acetate copolymer resin adhesive manufactured by Toyo Ink) was coated by the Mayer bar coating method and dried at a temperature of 80 ° C. for 30 seconds. An adhesive layer having a thickness of 4 ⁇ m was formed to obtain a transfer film A.
- Transfer film A is placed in the mold of SE130DU-CI (Sumitomo Heavy Industries, Ltd., all-electric two-material injection molding machine), and transparent ABS (Toyolac, grade 920-555, manufactured by Toray Industries, Inc.)
- the molded body 1 having an uncured hard coat layer was obtained by injection molding at 50 ° C. and a resin temperature of 230 ° C.
- the hard coat surface of the obtained hard coat layer uncured molded body 1 is irradiated with ultraviolet rays from a high-pressure mercury lamp (manufactured by Eye Graphics Co., Ltd.) for about 10 seconds (accumulated light amount: about 400 mJ / cm 2 ). After that, an annealing treatment was further performed at 60 ° C. for 1 week to obtain a molded body 1 in which the hard coat layer was cured.
- Comparative Example 1 Production of transfer film B and molded body ( production of transfer film B)
- the hard coat layer was formed by coating Seika Beam HT-S (urethane acrylate hard coat agent manufactured by Dainichi Seika Kogyo Co., Ltd.) by the Mayer bar coating method, drying at 100 ° C. for 1 minute, and then a high-pressure mercury lamp ( Transfer film A except that a semi-hardened hard coat layer having a thickness of 4.5 ⁇ m was formed by UV curing (accumulated light amount: about 30 mJ / cm 2 ) for about 2 seconds with ultraviolet rays from Igraphics.
- a transfer film B was obtained in the same manner.
- R 1 represents a group containing a polymerizable functional group.
- R a is a group containing a polymerizable functional group, a substituted or unsubstituted aryl group, a substituted or unsubstituted aralkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted group.
- An alkyl group, a substituted or unsubstituted alkenyl group, or a hydrogen atom is shown.
- R b represents a group containing a polymerizable functional group, a substituted or unsubstituted aryl group, a substituted or unsubstituted aralkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted group.
- An alkyl group, a substituted or unsubstituted alkenyl group, or a hydrogen atom is shown.
- R c represents a hydrogen atom or an alkyl group having 1 to 4 carbon atoms.
- the molar ratio of the structural unit represented by [the structural unit represented by the formula (I) / the structural unit represented by the formula (II)] is 20 or more and 500 or less, and the total amount of the siloxane structural unit (100 mol%).
- the structural unit represented by the above formula (1) and the following formula (4) [In formula (4), R 1 is the same as that in formula (1). R c is the same as in formula (II). ]
- the number average molecular weight is 2500 to 50000, and the molecular weight dispersity (weight average molecular weight / number average molecular weight) is 1.0 to 4.0. Polyorganosilsesquioxane.
- R 1 is represented by the following formula (1a) [In Formula (1a), R 1a represents a linear or branched alkylene group (preferably an ethylene group, a trimethylene group, more preferably an ethylene group).
- the above R 1 is a group containing a (meth) acryloxy group (preferably a 2-((meth) acryloxy) ethyl group or a 3-((meth) acryloxy) propyl group).
- R 1 is 2- (3 ′, 4′-epoxycyclohexyl) ethyl group], 3- (acryloxy) propyl group or 3- (methacryloxy) propyl group
- R 2 represents a substituted or unsubstituted aryl group, a substituted or unsubstituted aralkyl group, a substituted or unsubstituted cycloalkyl group, a substituted or unsubstituted alkyl group, or a substituted or unsubstituted group.
- the lower limit of the ratio [T3 body / T2 body] of the structural unit (T3 body) represented by the above formula (I) and the structural unit (T2 body) represented by the above formula (II) is 21 ( The polyorganosilsesquioxane according to any one of [1] to [11], which is preferably 23, more preferably 25). [13] The polyorganosilsesquioxy according to any one of the above [1] to [12], wherein the upper limit of the [T3 body / T2 body] is 100 (preferably 50, more preferably 40) Sun.
- the ratio (total amount) of the structural unit represented by the above formula (1) and the structural unit represented by the above formula (4) to the total amount (100 mol%) of the siloxane structural unit is 65 to 100 mol%.
- the ratio (total amount) of the structural unit represented by the above formula (2) and the structural unit represented by the above formula (5) to the total amount (100 mol%) of the siloxane structural unit is 0 to 70 mol%.
- a curable composition comprising the polyorganosilsesquioxane according to any one of [1] to [19].
- the content (blending amount) of the polyorganosilsesquioxane is 70 wt% or more and less than 100 wt% (preferably 80 wt%) with respect to the total amount (100 wt%) of the curable composition excluding the solvent.
- the curable composition according to the above [20] which is ⁇ 99.8 wt%, more preferably 90 to 99.5 wt%.
- the ratio of the polyorganosilsesquioxane to the total amount (100% by weight) of the cationic curable compound or radical curable compound contained in the curable composition is 70 to 100% by weight (preferably 75 to 98).
- the curable composition according to [23], wherein the curing catalyst is a photocationic polymerization initiator.
- the content (blending amount) of the curing catalyst is 0.01 to 3.0 parts by weight (preferably 0.05 to 3.0 parts by weight) with respect to 100 parts by weight of the polyorganosilsesquioxane.
- the content (blending amount) of the vinyl ether compound is the total amount of the cationic curable compound and the radical curable compound in the curable composition ( 100%) is 0.01 to 10% by weight (preferably 0.05 to 9% by weight, more preferably 1 to 8% by weight), and the curability according to [29] or [30] above Composition.
- the curable composition according to any one of [20] to [31] which is a curable composition for forming a hard coat layer.
- the thickness of the base material is 0.01 to 10,000 ⁇ m (preferably 2 to 250 ⁇ m, more preferably 5 to 100 ⁇ m, still more preferably 20 to 100 ⁇ m), according to the above [34] or [35] Transfer film.
- the above [34] to [35], wherein the peel strength between the release layer and the hard coat layer is 30 to 500 mN / 24 mm (preferably 40 to 300 mN / 24 mm, more preferably 50 to 200 mN / 24 mm).
- the transfer film as described in any one of the above.
- the component forming the release layer is an unsaturated ester resin, an epoxy resin, an epoxy-melamine resin, an aminoalkyd resin, an acrylic resin, a melamine resin, a silicon resin, a fluorine resin, At least one selected from a cellulose resin, a urea resin resin, a polyolefin resin, a paraffin resin, and a cycloolefin resin (preferably a cycloolefin resin, particularly preferably a cycloolefin such as a 2-norbornene-ethylene copolymer)
- the transfer film according to any one of [34] to [37], which is a copolymer resin).
- the anchor coat layer is made of phenol resin, alkyd resin, melamine resin, epoxy resin, urea resin, unsaturated polyester resin, urethane resin, thermosetting polyimide, silicone resin, vinyl chloride-vinyl acetate copolymer Any of the above [34] to [46], which is at least one selected from the group consisting of a coalesced resin, acrylic resin, chlorinated rubber, polyamide resin, nitrified cotton resin, and cyclic polyolefin resin (preferably an epoxy resin)
- the transfer film according to one.
- the resin used for the adhesive layer is an acrylic resin, a vinyl chloride resin, a vinyl acetate resin, a vinyl chloride-vinyl acetate copolymer resin, a styrene-acrylic copolymer resin, a polyester resin, and
- Film [50]
- the transfer film has a thickness of 1 to 10000 ⁇ m (preferably 2 to 250 ⁇ m, more preferably 5 to 150 ⁇ m, still more preferably 25 to 150 ⁇ m).
- the transfer film according to one.
- An in-mold molded product obtained by transferring a layer (transfer layer) excluding the substrate on which the release layer is formed from the transfer film according to [52].
- the substrate is a polyester film (particularly, polyethylene terephthalate, polyethylene naphthalate), a cyclic polyolefin film, a polycarbonate film, a triacetyl cellulose film, or a polymethyl methacrylate film. the film.
- a method for producing a film
- the uncured or semi-cured hard coat layer containing the polyorganosilsesquioxane of the present invention is tack-free and can be wound and handled in a roll shape, and a transfer film including the hard coat layer can be rolled. It can be handled with a toe roll. Therefore, the curable composition of the present invention can be preferably used as a curable composition for forming a hard coat layer of a transfer film or a hard coat film used for in-mold injection molding.
Abstract
Description
また、本発明の他の目的は、インモールド射出成型法によって、高い表面硬度を有するハードコート層を形成でき、かつ未硬化又は半硬化の段階ではタックフリーの塗膜を形成してロールとして巻き取り可能な転写用フィルムを提供することにある。
また、本発明の他の目的は、上記転写用フィルムの転写層が転写され、高い表面硬度を有するインモールド成型品を提供することである。
また、ハードコート層を有するハードコートフィルムには、一般に、高い表面硬度に加えて、可とう性や加工性も高いことも求められる。可とう性や加工性に乏しいと、ロールトゥロール方式での製造や加工をすることができず、高い生産コストがかかるからである。

で表される構成単位を有し、下記式(I)
で表される構成単位と、下記式(II)

で表される構成単位のモル比[式(I)で表される構成単位/式(II)で表される構成単位]が20以上500以下であり、シロキサン構成単位の全量(100モル%)に対する上記式(1)で表される構成単位及び下記式(4)

で表される構成単位の割合が55~100モル%であり、数平均分子量が2500~50000、分子量分散度(重量平均分子量/数平均分子量)が1.0~4.0であることを特徴とするポリオルガノシルセスキオキサンを提供する。

で表される構成単位を有していてもよい。
前記ポリオルガノシルセスキオキサンにおいて、前記R1は、下記式(1a)

で表される基、下記式(1b)

で表される基、下記式(1c)

で表される基、又は、下記式(1d)

で表される基であってもよい。
前記硬化性組成物は、さらに、硬化触媒を含んでいてもよい。
前記硬化性組成物において、前記硬化触媒は光カチオン重合開始剤であってもよい。
前記硬化性組成物において、前記硬化触媒は熱カチオン重合開始剤であってもよい。
前記硬化性組成物において、前記硬化触媒は光ラジカル重合開始剤であってもよい。
前記硬化性組成物において、前記硬化触媒は熱ラジカル重合開始剤であってもよい。
前記硬化性組成物は、さらに、ビニルエーテル化合物を含んでいてもよい。
前記硬化性組成物は、さらに、分子内に水酸基を有するビニルエーテル化合物を含んでいてもよい。
前記硬化性組成物は、ハードコート層形成用硬化性組成物であってもよい。
前記転写用フィルムにおいて、前記ハードコート層上に、アンカーコート層及び接着剤層が、この順でさらに積層されていてもよい。
前記転写用フィルムは、さらに、少なくとも1層の着色層を含んでいてもよい。
前記転写用フィルムにおいて、前記ハードコート層の厚さが3~150μmであってもよい。
前記転写用フィルムは、インモールド射出成型に使用される転写用フィルムであってもよい。
また、本発明は、前記の転写用フィルムから前記離型層が形成された基材を除いた層(転写層)が転写されたインモールド成型品を提供する。
前記ハードコートフィルムにおいて、前記ハードコート層の厚さは1~200μmであってもよい。
前記ハードコートフィルムは、ロールトゥロール方式での製造が可能であってもよい。
前記ハードコートフィルムは、前記ハードコート層表面に表面保護フィルムを有していてもよい。
本発明のポリオルガノシルセスキオキサン(シルセスキオキサン)は、下記式(1)で表される構成単位を有し;下記式(I)で表される構成単位(「T3体」と称する場合がある)と、下記式(II)で表される構成単位(「T2体」と称する場合がある)のモル比[式(I)で表される構成単位/式(II)で表される構成単位;「T3体/T2体」と記載する場合がある]が20以上500以下であり;シロキサン構成単位の全量(100モル%)に対する下記式(1)で表される構成単位及び後述の式(4)で表される構成単位の割合(総量)が55~100モル%であり;数平均分子量が2500~50000、分子量分散度[重量平均分子量/数平均分子量]が1.0~4.0であることを特徴とする。


上記重合性官能基を含有する基における「ラジカル重合性官能基」としては、ラジカル重合性を有するものである限り特に限定されず、例えば、(メタ)アクリルオキシ基、(メタ)アクリルアミド基、ビニル基、ビニルチオ基等が挙げられる。
重合性官能基としては、硬化物の表面硬度(例えば、4H以上)の観点から、エポキシ基、(メタ)アクリルオキシ基等が好ましく、エポキシ基が特に好ましい。







測定装置:商品名「JNM-ECA500NMR」(日本電子(株)製)
溶媒:重クロロホルム
積算回数:1800回
測定温度:25℃



測定装置:商品名「LC-20AD」((株)島津製作所製)
カラム:Shodex KF-801×2本、KF-802、及びKF-803(昭和電工(株)製)
測定温度:40℃
溶離液:THF、試料濃度0.1~0.2重量%
流量:1mL/分
検出器:UV-VIS検出器(商品名「SPD-20A」、(株)島津製作所製)
分子量:標準ポリスチレン換算



第2段目の加水分解及び縮合反応は、溶媒の存在下で行うこともできるし、非存在下で行うこともできる。第2段目の加水分解及び縮合反応を溶媒の存在下で行う場合、第1段目の加水分解及び縮合反応で挙げられた溶媒を用いることができる。第2段目の加水分解及び縮合反応の溶媒としては、第1段目の加水分解及び縮合反応の反応溶媒、抽出溶媒等を含む中間体ポリオルガノシルセスキオキサンをそのまま、又は一部留去したものを用いてもよい。なお、溶媒は1種を単独で使用することもできるし、2種以上を組み合わせて使用することもできる。
また、上記反応温度の範囲内にて加水分解及び縮合反応を行いながら適時サンプリングを行って、上記割合[T3体/T2体]、数平均分子量をモニターしながら反応を行うことによって、所望の割合[T3体/T2体]、数平均分子量を有する本発明のポリオルガノシルセスキオキサンを得ることもできる。
本発明の硬化性組成物は、上述の本発明のポリオルガノシルセスキオキサンを必須成分として含む硬化性組成物(硬化性樹脂組成物)である。後述のように、本発明の硬化性組成物は、さらに、硬化触媒(特に光カチオン重合開始剤、ラジカル重合性開始剤)や表面調整剤あるいは表面改質剤等のその他の成分を含んでいてもよい。




本発明の硬化性組成物におけるカチオン硬化性化合物又はラジカル硬化性化合物(本発明のポリオルガノシルセスキオキサン等)の重合反応を進行させることにより、該硬化性組成物を硬化させることができ、硬化物(「本発明の硬化物」と称する場合がある)を得ることができる。硬化の方法は、周知の方法より適宜選択でき、特に限定されないが、例えば、活性エネルギー線の照射、及び/又は、加熱する方法が挙げられる。上記活性エネルギー線としては、例えば、赤外線、可視光線、紫外線、X線、電子線、α線、β線、γ線等のいずれを使用することもできる。中でも、取り扱い性に優れる点で、紫外線が好ましい。
本発明の転写用フィルムは、基材と、該基材の少なくとも一方の表面に形成された離型層上に、未硬化又は半硬化のハードコート層とを有するフィルムであって、上記未硬化又は半硬化のハードコート層が、本発明の硬化性組成物(ハードコート層形成用硬化性組成物。以下、「本発明のハードコート剤」と称する場合がある。)により形成されることを特徴としている。ここで、未硬化とは、本発明のハードコート層形成用硬化性組成物(ハードコート剤)に含まれる本発明のポリオルガノシルセスキオキサンの重合性官能基が重合反応していない状態を意味する。また、半硬化とは、該重合性官能基の一部が重合反応し、未反応の重合性官能基が残存している状態を意味する。なお、本明細書においては、本発明の硬化性組成物(ハードコート剤)により形成された未硬化又は半硬化のハードコート層を、単に「ハードコート層」、成型品に転写・硬化されたハードコート層を、「硬化ハードコート層」と称する場合がある。
また、本発明の転写用フィルムにおける離型層は、上記基材のそれぞれの表面において、一部のみに形成されていてもよいし、全面に形成されていてもよい。
本発明の未硬化又は半硬化のハードコート層は、指を表面に接触させた際に樹脂が付着しない低タック性と優れた耐ブロッキング性を有し、ロール状に巻回して取り扱うことが可能である。
また、本発明の転写用フィルムにおける本発明のハードコート層は、上記離型層のそれぞれの表面において、一部のみに形成されていてもよいし、全面に形成されていてもよい。
ハードコート層を形成する際の加熱温度は、特に限定されないが、好ましくは50~200℃から適宜選択することができる。加熱時間も特に限定されないが、好ましくは1~60分から適宜選択することができる。
ハードコート層に活性化エネルギー線を照射する条件は、特に限定されず、例えば、上述の硬化物を形成する際の条件から適宜選択可能である。
本発明のアンカーコート用樹脂は、さらに、その他任意の成分として、ワックス、シリカ、可塑剤、レベリング剤、界面活性剤、分散剤、消泡剤、紫外線吸収剤、紫外線安定剤、酸化防止剤などの慣用の添加剤を、本発明の効果を損なわない範囲で含んでいてもよい。これらの添加剤は1種を単独で、又は2種以上を組み合わせて使用できる。
アンカーコート層は、上記樹脂を溶媒に溶解した塗工液を、本発明のハードコート層上に、バーコート、メイヤーバーコート、グラビアコート、ロールコート等の公知のコーティング方法で塗布、乾燥して、必要により加熱して形成することができる。
アンカーコート層を形成する際に加熱する場合の温度は、特に限定されないが、好ましくは50~200℃から適宜選択することができる。加熱時間も特に限定されないが、好ましくは10秒~60分から適宜選択することができる。
アンカーコート層の厚みは、通常、0.1~20μm程度であり、好ましくは、0.5~5μmの範囲である。
本発明の接着剤層に用いられるアクリル系樹脂としては、特に限定されないが、例えば、ポリメチル(メタ)アクリレート、ポリエチル(メタ)アクリレート、ポリブチル(メタ)アクリレート、メチル(メタ)アクリレート-ブチル(メタ)アクリレート共重合体、メチル(メタ)アクリレート-スチレン共重合体などのアクリル系樹脂、フッ素などによる変性アクリル樹脂が挙げられ、これらを1種又は2種以上の混合物として用いることができる。この他、メチル(メタ)アクリレート、エチル(メタ)アクリレート、ブチル(メタ)アクリレート、2-エチルヘキシル(メタ)アクリレート、オクチル(メタ)アクリレートなどの(メタ)アクリル酸アルキルエステルと、2-ヒドロキシエチル(メタ)アクリレート、2-ヒドロキシブチル(メタ)アクリレート、2-ヒドロキシ-3-フェノキシプロピル(メタ)アクリレートなどの分子中に水酸基を有する(メタ)アクリル酸エステルと、を共重合させて得られるアクリルポリオールを用いることもできる。また、塩化ビニル-酢酸ビニル系共重合体樹脂としては、通常、酢酸ビニル含有量が5~20質量%程度、平均重合度350~900程度のものが用いられる。必要に応じ、塩化ビニル-酢酸ビニル系共重合体樹脂にさらにマレイン酸、フマル酸などのカルボン酸を共重合させても良い。この他、副成分の樹脂として、必要に応じて、適宜その他の樹脂、例えば、熱可塑性ポリエステル系樹脂、熱可塑性ウレタン系樹脂、塩素化ポリエチレン、塩素化ポリプロピレンなどの塩素化ポリオレフィン系樹脂などの樹脂を混合しても良い。
接着剤層を形成する際に加熱する場合の温度は、特に限定されないが、好ましくは50~200℃から適宜選択することができる。加熱時間も特に限定されないが、好ましくは10秒~60分から適宜選択することができる。
接着剤層の厚みとしては、転写用フィルムを接着性良く、かつ効率的に成型品に転写し得るという点から、0.1~10μm程度が好ましく、0.5~5μmがより好ましい。
着色層は、通常は、上記のハードコート層又はアンカーコート層に印刷インキでグラビア印刷、オフセット印刷、シルクスクリーン印刷、転写シートからの転写印刷、昇華転写印刷、インキジェット印刷などの公知の印刷法により形成することで、ハードコート層と接着剤層との間、又はアンカーコート層と接着剤層との間に形成することができる。着色層の厚みは、意匠性の観点から3~40μmが好ましく、10~30μmがより好ましい。
着色層の形成に用いられる印刷インキは、上記成分の他に、沈降防止剤、硬化触媒、紫外線吸収剤、酸化防止剤、レベリング剤、増粘剤、消泡剤、滑剤などを適宜添加することができる。印刷インキは、上記成分を、通常溶剤に溶解又は分散した態様で提供される。溶剤としては、バインダー樹脂を溶解又は分散させるものであれば良く、有機溶剤及び/又は水を使用することができる。有機溶剤としては、トルエン、キシレンなどの炭化水素類、アセトン、メチルエチルケトンなどのケトン類、酢酸エチル、セロソルブアセテート、ブチルセロソルブアセテートなどのエステル類、アルコール類が挙げられる。
本発明のハードコートフィルムは、基材と、該基材の少なくとも一方の表面に形成されたハードコート層とを有するフィルムであって、上記ハードコート層が、本発明の硬化性組成物(ハードコート層形成用硬化性組成物)により形成されたハードコート層(本発明の硬化性組成物の硬化物層)であることを特徴としている。
温度計、攪拌装置、還流冷却器、及び窒素導入管を取り付けた1000ミリリットルのフラスコ(反応容器)に、窒素気流下で2-(3,4-エポキシシクロヘキシル)エチルトリメトキシシラン277.2ミリモル(68.30g)、フェニルトリメトキシシラン3.0ミリモル(0.56g)、及びアセトン275.4gを仕込み、50℃に昇温した。このようにして得られた混合物に、5%炭酸カリウム水溶液7.74g(炭酸カリウムとして2.8ミリモル)を5分で添加した後、水2800.0ミリモル(50.40g)を20分かけて添加した。なお、添加の間、著しい温度上昇は起こらなかった。その後、50℃のまま、重縮合反応を窒素気流下で5時間行った。
その後、反応溶液を冷却すると同時に、メチルイソブチルケトン137.70gと5%食塩水100.60gとを投入した。この溶液を1Lの分液ロートに移し、再度メチルイソブチルケトン137.70gを投入し、水洗を行った。分液後、水層を抜き取り、下層液が中性になるまで水洗を行い、上層液を分取した後、1mmHg、50℃の条件で上層液から溶媒を留去し、メチルイソブチルケトンを25.04重量%含有する無色透明で液状の生成物(中間体エポキシ基含有ポリオルガノシルセスキオキサン)を75.18g得た。
生成物を分析したところ、数平均分子量は2235であり、分子量分散度は1.54であった。上記生成物の29Si-NMRスペクトルから算出されるT2体とT3体の割合[T3体/T2体]は11.9であった。
得られた中間体エポキシ基含有ポリオルガノシルセスキオキサンの1H-NMRチャートを図1、29Si-NMRチャートを図2にそれぞれ示す。
温度計、攪拌装置、還流冷却器、及び窒素導入管を取り付けた1000ミリリットルのフラスコ(反応容器)に、窒素気流下に製造例1で得られた中間体エポキシ基含有ポリオルガノシルセスキオキサンを含む混合物(75g)を仕込み、中間体エポキシ基含有ポリオルガノシルセスキオキサンの正味含有量(56.2g)に対して水酸化カリウムを100ppm(5.6mg)、水を2000ppm(112mg)添加し、80℃で18時間加熱した時点でサンプリングして分子量を測定したところ、数平均分子量Mnが6000まで上昇しており、その後室温まで冷却し、メチルイソブチルケトンを300mL添加し、水を300mL添加し、水洗を繰り返すことでアルカリ成分を除去して濃縮すると、メチルイソブチルケトンを25重量%含有する無色透明で液状の生成物(本発明のエポキシ基含有ポリオルガノシルセスキオキサン1)を74.5g得た。
生成物を分析したところ、数平均分子量は6176であり、分子量分散度は2.31であった。上記生成物の29Si-NMRスペクトルから算出されるT2体とT3体の割合[T3体/T2体]は50.2であった。
得られたエポキシ基含有ポリオルガノシルセスキオキサン1の1H-NMRチャートを図3、29Si-NMRチャートを図4にそれぞれ示す。
温度計、攪拌装置、還流冷却器、及び窒素導入管を取り付けた1000ミリリットルのフラスコ(反応容器)に、窒素気流下に製造例1と同様の方法で得られた中間体エポキシ基含有ポリオルガノシルセスキオキサンを含む混合物(75g)を仕込み、中間体エポキシ基含有ポリオルガノシルセスキオキサンの正味含有量(56.2g)に対して炭酸カリウムを100ppm(5.6mg)、水を2000ppm(112mg)添加し、80℃で18時間加熱した時点でサンプリングして分子量を測定したところ、数平均分子量Mnが4800まで上昇しており、その後室温まで冷却し、メチルイソブチルケトンを300mL添加し、水を300mL添加し、水洗を繰り返すことでアルカリ成分を除去して濃縮すると、メチルイソブチルケトンを25重量%含有する無色透明で液状の生成物(本発明のエポキシ基含有ポリオルガノシルセスキオキサン2)を74.5g得た。
温度計、攪拌装置、還流冷却器、及び窒素導入管を取り付けた1000ミリリットルのフラスコ(反応容器)に、窒素気流下に製造例1と同様の方法で得られた中間体エポキシ基含有ポリオルガノシルセスキオキサンを含む混合物(75g)を仕込み、中間体エポキシ基含有ポリオルガノシルセスキオキサンの正味含有量(56.2g)に対して炭酸カリウムを100ppm(5.6mg)、水を2000ppm(112mg)添加し、80℃で3時間加熱した時点でサンプリングして分子量を測定したところ、数平均分子量Mnが3500まで上昇しており、その後室温まで冷却し、メチルイソブチルケトンを300mL添加し、水を300mL添加し、水洗を繰り返すことでアルカリ成分を除去して濃縮すると、メチルイソブチルケトンを25重量%含有する無色透明で液状の生成物(本発明のエポキシ基含有ポリオルガノシルセスキオキサン3)を74.5g得た。
生成物を分析したところ、数平均分子量は3500であり、分子量分散度は2.14であった。上記生成物の29Si-NMRスペクトルから算出されるT2体とT3体の割合[T3体/T2体]は21であった。
得られたエポキシ基含有ポリオルガノシルセスキオキサン3の1H-NMRチャートを図5、29Si-NMRチャートを図6にそれぞれ示す。
温度計、攪拌装置、還流冷却器、及び窒素導入管を取り付けた1000ミリリットルのフラスコ(反応容器)に、窒素気流下で3-(アクリルオキシ)プロピルトリメトキシシラン370ミリモル(80g)、及びアセトン320gを仕込み、50℃に昇温した。このようにして得られた混合物に、5%炭酸カリウム水溶液10.144g(炭酸カリウムとして3.67ミリモル)を5分で添加した後、水3670.0ミリモル(66.08g)を20分かけて添加した。なお、添加の間、著しい温度上昇は起こらなかった。その後、50℃のまま、重縮合反応を窒素気流下で2時間行った。
その後、反応溶液を冷却すると同時に、メチルイソブチルケトン160gと5%食塩水99.056gとを投入した。この溶液を1Lの分液ロートに移し、再度メチルイソブチルケトン160gを投入し、水洗を行った。分液後、水層を抜き取り、下層液が中性になるまで水洗を行い、上層液を分取した後、1mmHg、50℃の条件で上層液から溶媒を留去し、メチルイソブチルケトンを22.5重量%含有する無色透明で液状の生成物(中間体アクリル基含有ポリオルガノシルセスキオキサン)を71g得た。
生成物を分析したところ、数平均分子量は2051であり、分子量分散度は1.29であった。上記生成物の29Si-NMRスペクトルから算出されるT2体とT3体の割合[T3体/T2体]は13.4であった。
得られた中間体アクリル基含有ポリオルガノシルセスキオキサンの1H-NMRチャートを図7、29Si-NMRチャートを図8にそれぞれ示す。
温度計、攪拌装置、還流冷却器、及び窒素導入管を取り付けた1000ミリリットルのフラスコ(反応容器)に、窒素気流下に製造例2で得られた中間体アクリル基含有ポリオルガノシルセスキオキサンを含む混合物(71g)を仕込み、中間体アクリル基含有ポリオルガノにシルセスキオキサンの正味含有量(55.0g)に対して水酸化カリウムを10ppm(0.55mg)、水を2000ppm(110mg)添加し、40℃で30時間加熱した時点でサンプリングして分子量を測定したところ、数平均分子量Mnが5693まで上昇しており、その後室温まで冷却し、メチルイソブチルケトンを300mL添加し、水を300mL添加し、水洗を繰り返すことでアルカリ成分を除去して濃縮すると、メチルイソブチルケトンを25重量%含有する無色透明で液状の生成物(本発明のアクリル基含有ポリオルガノシルセスキオキサン)を71g得た。
生成物を分析したところ、数平均分子量は5693であり、分子量分散度は2.58であった。上記生成物の29Si-NMRスペクトルから算出されるT2体とT3体の割合[T3体/T2体]は47.3であった。
得られたアクリル基含有ポリオルガノシルセスキオキサン1の1H-NMRチャートを図9、29Si-NMRチャートを図10にそれぞれ示す。
(離型剤塗工液Aの調製)
Nb/Et(2-ノルボルネン・エチレン共重合体、Topas Advanced Polymers GmbH社製「TOPAS(登録商標)6017S-04」、ガラス転移温度178℃)100重量部及びPVDC(ポリ塩化ビニリデン)1重量部を、固形分濃度が5重量%となるように、トルエン及びテトラヒドロフランの混合溶媒(トルエン/テトラヒドロフラン=70/30(重量比))に添加し、加温して溶解し、離型剤塗工液Aを調製した。
基材層として、二軸延伸ポリエチレンテレフタレートフィルム(ユニチカ(株)製「エンブレットS50」、厚み50μm)を用い、このフィルムの片面に、離型剤塗工液Aをメイヤーバーコーティング法によりコーティングし、100℃の温度で1分間乾燥して、厚み0.3μmの離型層を形成し、離型フィルムAを得た。
実施例3で得られたエポキシ基含有ポリオルガノシルセスキオキサン3(数平均分子量Mn3500)100重量部、CPI-210S(サンアプロ株式会社製光カチオン重合開始剤)1.13重量部を、固形分濃度が70重量%となるように、メチルイソブチルケトンに添加し、ハードコート塗工液Aを調製した。
離型フィルムAの離型層面上に、ハードコート塗工液Aをメイヤーバーコーティング法によりコーティングし、80℃の温度で2分間乾燥し、さらに150℃の温度で8分間乾燥して厚み40μmのハードコート層を形成した。得られたハードコート層の表面を指で触ったところ、指に樹脂が付着せず、表面粘着性を示さない(タックフリーである)ことが確認された。このハードコート層上に、K468HPアンカー(東洋インキ株式会社製エポキシ樹脂系アンカーコート剤)をメイヤーバーコーティング法によりコーティングし、80℃の温度で30秒間乾燥して、厚み1μmのアンカーコート層を形成し、さらにこのアンカーコート層上に、K588HP接着グロスAワニス(東洋インキ製塩化ビニル-酢酸ビニル共重合樹脂系接着剤)をメイヤーバーコーティング法によりコーティングし、80℃の温度で30秒間乾燥して、厚み4μmの接着剤層を形成し、転写フィルムAを得た。
SE130DU-CI(住友重機械工業株式会社製全電動二材射出成型機)の金型内に転写フィルムAを設置し、透明ABS(東レ株式会社製トヨラック、グレード920-555)を、金型温度50℃、樹脂温度230℃で射出成型することにより未硬化のハードコート層を有する成形体1を得た。得られたハードコート層未硬化の成形体1のハードコート面に、高圧水銀ランプ(アイグラフィックス社製)からの紫外線を約10秒間照射(積算光量約400mJ/cm2)して紫外線硬化処理した後、さらに60℃で1週間アニール処理を行うことによりハードコート層が硬化された成形体1を得た。
(転写フィルムBの作製)
ハードコート層の形成を、セイカビームHT-S(大日精化工業株式会社製ウレタンアクリレート系ハードコート剤)をメイヤーバーコーティング法によりコーティングし、100℃の温度で1分間乾燥した後、高圧水銀ランプ(アイグラフィックス社製)からの紫外線で約2秒間UV硬化処理(積算光量約30mJ/cm2)することにより、厚み4.5μmの半硬化のハードコート層を形成したこと以外は転写フィルムAと同様の方法で、転写フィルムBを得た。
(成形体2の作製)
転写フィルムAの代わりに転写フィルムBを用いたこと、および射出成形後の処理を高圧水銀ランプ(アイグラフィックス社製)からの紫外線を約25秒間照射(積算光量約900mJ/cm2)として、半硬化ハードコート層を硬化したこと以外は成形体1と同様の方法でハードコート層が硬化された成形体2を得た。
得られた成形体1及び2の鉛筆硬度を、JIS-K-5600に規定される鉛筆硬度の評価方法に従い評価した。この評価方法による結果を表1に示す。

[1]下記式(1)

で表される構成単位を有し、下記式(I)
で表される構成単位と、下記式(II)

で表される構成単位のモル比[式(I)で表される構成単位/式(II)で表される構成単位]が20以上500以下であり、シロキサン構成単位の全量(100モル%)に対する上記式(1)で表される構成単位及び下記式(4)

で表される構成単位の割合が55~100モル%であり、数平均分子量が2500~50000、分子量分散度(重量平均分子量/数平均分子量)が1.0~4.0であることを特徴とするポリオルガノシルセスキオキサン。
[2]前記重合性官能基が、カチオン重合性官能基、又はラジカル重合性官能基である、前記[1]に記載のポリオルガノシルセスキオキサン。
[3]前記カチオン重合性官能基が、エポキシ基、オキセタン基、ビニルエーテル基、及びビニルフェニル基からなる群から選ばれる少なくとも1種(好ましくはエポキシ基)である、前記[2]に記載のポリオルガノシルセスキオキサン。
[4]前記ラジカル重合性官能基が、(メタ)アクリルオキシ基、(メタ)アクリルアミド基、ビニル基、及びビニルチオ基からなる群から選ばれる少なくとも1種(好ましくは(メタ)アクリルオキシ基)である、前記[2]に記載のポリオルガノシルセスキオキサン。
[5]前記重合性官能基が、エポキシ基、又は(メタ)アクリルオキシ基である、前記[1]~[4]のいずれか1つに記載のポリオルガノシルセスキオキサン。
[6]前記重合性官能基が、エポキシ基である、前記[1]~[5]のいずれか1つに記載のポリオルガノシルセスキオキサン。
[7]前記R1が、下記式(1a)

で表される基、下記式(1b)

で表される基、下記式(1c)

で表される基、又は、下記式(1d)

で表される基である、前記[1]~[6]のいずれか1つに記載のポリオルガノシルセスキオキサン。
[8]前記R1が、(メタ)アクリルオキシ基を含有する基(好ましくは2-((メタ)アクリルオキシ)エチル基、又は3-((メタ)アクリルオキシ)プロピル基)である、前記[1]~[7]のいずれか1つに記載のポリオルガノシルセスキオキサン。
[9]前記R1が、2-(3’,4’-エポキシシクロヘキシル)エチル基]、3-(アクリルオキシ)プロピル基、又は3-(メタクリルオキシ)プロピル基である、前記[1]~[8]のいずれか1つに記載のポリオルガノシルセスキオキサン。
[10]さらに、下記式(2)

で表される構成単位を有する、前記[1]~[9]のいずれか1つに記載のポリオルガノシルセスキオキサン。
[11]前記R2が、置換若しくは無置換のアリール基(好ましくはフェニル基)である、前記[10]に記載のポリオルガノシルセスキオキサン。
[13]前記[T3体/T2体]の上限値が100(好ましくは50、より好ましくは40)である、前記[1]~[12]のいずれか1つに記載のポリオルガノシルセスキオキサン。
[14]シロキサン構成単位の全量(100モル%)に対する、上記式(1)で表される構成単位及び上記式(4)で表される構成単位の割合(総量)が、65~100モル%(好ましくは80~99モル%)である、前記[1]~[13]のいずれか1つに記載のポリオルガノシルセスキオキサン。
[15]シロキサン構成単位の全量(100モル%)に対する、上記式(2)で表される構成単位及び上記式(5)で表される構成単位の割合(総量)が、0~70モル%(好ましくは0~60モル%、より好ましくは0~40モル%、特に好ましくは1~15モル%)である、前記[1]~[14]のいずれか1つに記載のポリオルガノシルセスキオキサン。
[16]シロキサン構成単位の全量(100モル%)に対する、上記式(1)で表される構成単位、上記式(2)で表される構成単位、上記式(4)で表される構成単位、及び上記式(5)で表される構成単位の割合(総量)が、60~100モル%(好ましくは70~100モル%、より好ましくは80~100モル%)である、前記[1]~[15]のいずれか1つに記載のポリオルガノシルセスキオキサン。
[17]数平均分子量(Mn)が、2800~10000(好ましくは3000~8000)である、前記[1]~[16]のいずれか1つに記載のポリオルガノシルセスキオキサン。
[18]分子量分散度(Mw/Mn)が、1.1~3.0(好ましくは1.2~2.5)である、前記[1]~[17]のいずれか1つに記載のポリオルガノシルセスキオキサン。
[19]空気雰囲気下における5%重量減少温度(Td5)が、330℃以上(例えば、330~450℃、好ましくは340℃以上、より好ましくは350℃以上)である、前記[1]~[18]のいずれか1つに記載のポリオルガノシルセスキオキサン。
[21]前記ポリオルガノシルセスキオキサンの含有量(配合量)が、溶媒を除く硬化性組成物の全量(100重量%)に対して、70重量%以上、100重量%未満(好ましくは80~99.8重量%、より好ましくは90~99.5重量%)である、前記[20]に記載の硬化性組成物。
[22]前記硬化性組成物に含まれるカチオン硬化性化合物又はラジカル硬化性化合物の全量(100重量%)に対する前記ポリオルガノシルセスキオキサンの割合が、70~100重量%(好ましくは75~98重量%、より好ましくは80~95重量%)である、前記[20]又は[21]に記載の硬化性組成物。
[23]さらに、硬化触媒を含む、前記[20]~[22]のいずれか1つに記載の硬化性組成物。
[24]前記硬化触媒が光カチオン重合開始剤である、前記[23]に記載の硬化性組成物。
[25]前記硬化触媒が熱カチオン重合開始剤である、前記[23]に記載の硬化性組成物。
[26]前記硬化触媒が光ラジカル重合開始剤である、前記[23]に記載の硬化性組成物。
[27]前記硬化触媒が熱ラジカル重合開始剤である、前記[23]に記載の硬化性組成物。
[28]前記硬化触媒の含有量(配合量)が、前記ポリオルガノシルセスキオキサン100重量部に対して、0.01~3.0重量部(好ましくは0.05~3.0重量部、より好ましくは0.1~1.0重量部、さらに好ましくは0.3~1.0重量部)である、前記[23]~[28]のいずれか1つに記載の硬化性組成物。
[29]さらに、ビニルエーテル化合物を含む、前記[20]~[28]のいずれか1つに記載の硬化性組成物。
[30]さらに、分子内に水酸基を有するビニルエーテル化合物を含む、前記[20]~[29]のいずれか1つに記載の硬化性組成物。
[31]前記ビニルエーテル化合物(特に、分子内に1個以上の水酸基を有するビニルエーテル化合物)の含有量(配合量)が、前記硬化性組成物中のカチオン硬化性化合物とラジカル硬化性化合物の全量(100%)に対して、0.01~10重量%(好ましくは0.05~9重量%、より好ましくは1~8重量%)である、前記[29]又は[30]に記載の硬化性組成物。
[32]ハードコート層形成用硬化性組成物である、前記[20]~[31]のいずれか1つに記載の硬化性組成物。
[33]前記[20]~[32]のいずれか1つに記載の硬化性組成物の硬化物。
[35]前記基材が、ポリエステルフィルム(特に、ポリエチレンテレフタレート、ポリエチレンナフタレート)、環状ポリオレフィンフィルム、ポリカーボネートフィルム、トリアセチルセルロースフィルム、又はポリメチルメタクリレートフィルムである、前記[34]に記載の転写用フィルム。
[36]前記基材の厚みが、0.01~10000μm(好ましくは2~250μm、より好ましくは5~100μm、さらに好ましくは20~100μm)である、前記[34]又は[35]に記載の転写用フィルム。
[37]前記離形層と前記ハードコート層の剥離強度が、30~500mN/24mm(好ましくは40~300mN/24mm、より好ましくは50~200mN/24mm)である、前記[34]~[35]のいずれか1つに記載の転写用フィルム。
[38]前記離型層を形成する成分が、不飽和エステル系樹脂、エポキシ系樹脂、エポキシ-メラミン系樹脂、アミノアルキド系樹脂、アクリル系樹脂、メラミン系樹脂、シリコン系樹脂、フッ素系樹脂、セルロース系樹脂、尿素樹脂系樹脂、ポリオレフィン系樹脂、パラフィン系樹脂、及びシクロオレフィン系樹脂から選ばれる少なくとも一種(好ましくはシクロオレフィン系樹脂、特に好ましくは2-ノルボルネン・エチレン共重合体等のシクロオレフィン共重合体樹脂)である、前記[34]~[37]のいずれか1つに記載の転写用フィルム。
[39]離型層の厚さが、0.01~5μm(好ましくは0.1~3.0μm)である、前記[34]~[38]のいずれか1つに記載の転写用フィルム。
[40]前記ハードコート層の厚さが、1~200μm(好ましくは3~150μm)である、前記[34]~[39]のいずれか1つに記載の転写用フィルム。
[41]前記ハードコート層の50μmの厚みのヘイズが、1.5%以下(好ましくは1.0%以下)である、前記[34]~[40]のいずれか1つに記載の転写用フィルム。
[42]前記ハードコート層の50μmの厚みのヘイズが、0.1%以上である、前記[34]~[41]のいずれか1つに記載の転写用フィルム。
[43]前記ハードコート層の50μmの厚みの全光線透過率が、85%以上(好ましくは90%以上)である、前記[34]~[42]のいずれか1つに記載の転写用フィルム。
[44]前記ハードコート層の50μmの厚みの全光線透過率が、99%以下である、前記[34]~[43]のいずれか1つに記載の転写用フィルム。
[45]前記ハードコート層上に、アンカーコート層及び接着剤層が、この順でさらに積層される、前記[34]~[44]のいずれか1つに記載の転写用フィルム。
[46]さらに、少なくとも1層の着色層を含む、前記[34]~[45]のいずれか1つに記載の転写用フィルム。
[47]前記アンカーコート層が、フェノール樹脂、アルキド樹脂、メラミン系樹脂、エポキシ系樹脂、尿素樹脂、不飽和ポリエステル樹脂、ウレタン系樹脂、熱硬化性ポリイミド、シリコーン樹脂、塩化ビニル-酢酸ビニル共重合体樹脂、アクリル樹脂、塩化ゴム、ポリアミド樹脂、硝化綿樹脂、及び環状ポリオレフィン系樹脂からなる群から選ばれる少なくとも1種(好ましくはエポキシ樹脂)である、前記[34]~[46]のいずれか1つに記載の転写用フィルム。
[48]前記アンカーコート層の厚みが、0.1~20μm(好ましくは、0.5~5μm)である、前記[34]~[47]のいずれか1つに記載の転写用フィルム。
[49]前記接着剤層に用いられる樹脂が、アクリル系樹脂、塩化ビニル系樹脂、酢酸ビニル系樹脂、塩化ビニル-酢酸ビニル系共重合樹脂、スチレン-アクリル系共重合樹脂、ポリエステル系樹脂、及びポリアミド系樹脂からなる群から選ばれる少なくとも1種(好ましくは、アクリル系樹脂、塩化ビニル-酢酸ビニル系共重合樹脂)である、前記[34]~[48]のいずれか1つに記載の転写用フィルム。
[50]前記接着剤層の厚みが、0.1~10μm(好ましくは0.5~5μm)である、前記[34]~[49]のいずれか1つに記載の転写用フィルム。
[51]前記転写用フィルムの厚みが、1~10000μm(好ましくは2~250μm、より好ましくは5~150μmが、さらに好ましくは25~150μm)である、前記[34]~[50]のいずれか1つに記載の転写用フィルム。
[52]インモールド射出成型に使用される転写用フィルムである、前記[34]~[51]のいずれか1つに記載の転写用フィルム。
[53]前記[52]に記載の転写用フィルムから前記離型層が形成された基材を除いた層(転写層)が転写されたインモールド成型品。
[55]前記基材が、ポリエステルフィルム(特に、ポリエチレンテレフタレート、ポリエチレンナフタレート)、環状ポリオレフィンフィルム、ポリカーボネートフィルム、トリアセチルセルロースフィルム、又はポリメチルメタクリレートフィルムである、前記[54]に記載のハードコートフィルム。
[56]前記基材の厚みが、0.01~10000μmである、前記[54]又は[55]に記載のハードコートフィルム。
[57]前記ハードコート層の厚さが1~200μm(好ましくは3~150μm)である、前記[54]~[56]のいずれか1つに記載のハードコートフィルム。
[58]前記ハードコート層の50μmの厚みのヘイズが、1.5%以下(好ましくは1.0%以下)である、前記[54]~[57]のいずれか1つに記載のハードコートフィルム。
[59]前記ハードコート層の50μmの厚みのヘイズが、0.1%以上である、前記[54]~[58]のいずれか1つに記載のハードコートフィルム。
[60]前記ハードコート層の50μmの厚みの全光線透過率が、85%以上(好ましくは90%以上)である、前記[54]~[59]のいずれか1つに記載のハードコートフィルム。
[61]前記ハードコート層の50μmの厚みの全光線透過率が、99%以下である、前記[54]~[60]のいずれか1つに記載のハードコートフィルム。
[62]ロールトゥロール方式での製造が可能な、前記[54]~[61]のいずれか1つに記載のハードコートフィルム。
[63]さらに、前記ハードコート層表面に表面保護フィルムを有する、前記[54]~[62]のいずれか1つに記載のハードコートフィルム。
[64]前記ハードコートフィルムの厚みが、1~10000μmである、前記[54]~[63]のいずれか1つに記載のハードコートフィルム。
[65]前記ハードコートフィルムのヘイズが、1.5%以下(好ましくは1.0%以下)である、前記[54]~[64]のいずれか1つに記載のハードコートフィルム。
[66]前記ハードコートフィルムのヘイズが、0.1%以上である、前記[54]~[65]のいずれか1つに記載のハードコートフィルム。
[67]前記ハードコートフィルムの全光線透過率が、85%以上(好ましくは90%以上)である、前記[54]~[66]のいずれか1つに記載のハードコートフィルム。
[68]前記ハードコートフィルムの全光線透過率が、99%以下である、前記[54]~[67]のいずれか1つに記載のハードコートフィルム。
[65]ロール状に巻いた基材を繰り出す工程Aと、繰り出した基材の少なくとも一方の表面に前記[32]に記載の硬化性組成物を塗布し、次いで、該硬化性組成物を硬化させることによりハードコート層を形成する工程Bと、その後、得られたハードコートフィルムを再びロールに巻き取る工程Cとを含み、工程A~Cを連続的に実施することを特徴とするハードコートフィルムの製造方法。
Claims (26)
- 下記式(1)

で表される構成単位を有し、下記式(I)

で表される構成単位と、下記式(II)

で表される構成単位のモル比[式(I)で表される構成単位/式(II)で表される構成単位]が20以上500以下であり、シロキサン構成単位の全量(100モル%)に対する上記式(1)で表される構成単位及び下記式(4)

で表される構成単位の割合が55~100モル%であり、数平均分子量が2500~50000、分子量分散度(重量平均分子量/数平均分子量)が1.0~4.0であることを特徴とするポリオルガノシルセスキオキサン。 - さらに、下記式(2)

で表される構成単位を有する請求項1に記載のポリオルガノシルセスキオキサン。 - 前記R2が、置換若しくは無置換のアリール基である請求項2に記載のポリオルガノシルセスキオキサン。
- 前記重合性官能基が、エポキシ基である、請求項1~3のいずれか1項に記載のポリオルガノシルセスキオキサン。
- 前記R1が、下記式(1a)

で表される基、下記式(1b)

で表される基、下記式(1c)

で表される基、又は、下記式(1d)

で表される基である請求項1~4のいずれか1項に記載のポリオルガノシルセスキオキサン。 - 請求項1~5のいずれか1項に記載のポリオルガノシルセスキオキサンを含む硬化性組成物。
- さらに、硬化触媒を含む請求項6に記載の硬化性組成物。
- 前記硬化触媒が光カチオン重合開始剤である請求項7に記載の硬化性組成物。
- 前記硬化触媒が熱カチオン重合開始剤である請求項7に記載の硬化性組成物。
- 前記硬化触媒が光ラジカル重合開始剤である請求項7に記載の硬化性組成物。
- 前記硬化触媒が熱ラジカル重合開始剤である請求項7に記載の硬化性組成物。
- さらに、ビニルエーテル化合物を含む請求項6~11のいずれか1項に記載の硬化性組成物。
- さらに、分子内に水酸基を有するビニルエーテル化合物を含む請求項6~12のいずれか1項に記載の硬化性組成物。
- ハードコート層形成用硬化性組成物である請求項6~13のいずれか1項に記載の硬化性組成物。
- 請求項6~14のいずれか1項に記載の硬化性組成物の硬化物。
- 基材と、該基材の少なくとも一方の表面に形成された離型層上に、ハードコート層が積層された転写用フィルムであって、該ハードコート層が、請求項14に記載の硬化性組成物を含むことを特徴とする転写用フィルム。
- 前記ハードコート層上に、アンカーコート層及び接着剤層が、この順でさらに積層される請求項16に記載の転写用フィルム。
- さらに、少なくとも1層の着色層を含む請求項16又は17に記載の転写用フィルム。
- 前記ハードコート層の厚さが3~150μmである請求項16~18のいずれか1項に記載の転写用フィルム。
- インモールド射出成型に使用される転写用フィルムである、請求項16~19のいずれか1項に記載の転写用フィルム。
- 請求項20に記載の転写用フィルムから前記離型層が形成された基材を除いた層(転写層)が転写されたインモールド成型品。
- 基材と、該基材の少なくとも一方の表面に形成されたハードコート層とを有するハードコートフィルムであって、該ハードコート層が、請求項14に記載の硬化性組成物の硬化物層であることを特徴とするハードコートフィルム。
- 前記ハードコート層の厚さが1~200μmである請求項22に記載のハードコートフィルム。
- ロールトゥロール方式での製造が可能な請求項22又は23に記載のハードコートフィルム。
- さらに、前記ハードコート層表面に表面保護フィルムを有する請求項22~24のいずれか1項に記載のハードコートフィルム。
- ロール状に巻いた基材を繰り出す工程Aと、繰り出した基材の少なくとも一方の表面に請求項14に記載の硬化性組成物を塗布し、次いで、該硬化性組成物を硬化させることによりハードコート層を形成する工程Bと、その後、得られたハードコートフィルムを再びロールに巻き取る工程Cとを含み、工程A~Cを連続的に実施することを特徴とするハードコートフィルムの製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019518834A JPWO2018212228A1 (ja) | 2017-05-17 | 2018-05-16 | ポリオルガノシルセスキオキサン、転写用フィルム、インモールド成型品、及びハードコートフィルム |
| CN201880032557.XA CN110621723A (zh) | 2017-05-17 | 2018-05-16 | 聚有机倍半硅氧烷、转印用膜、模内成型品以及硬涂膜 |
| KR1020197036747A KR20200007894A (ko) | 2017-05-17 | 2018-05-16 | 폴리오르가노실세스퀴옥산, 전사용 필름, 인 몰드 성형품 및 하드 코트 필름 |
| US16/614,007 US20200079910A1 (en) | 2017-05-17 | 2018-05-16 | Polyorganosilsesquioxane, transfer film, in-mold molded article, and hard coat film |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017098511 | 2017-05-17 | ||
| JP2017-098511 | 2017-05-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018212228A1 true WO2018212228A1 (ja) | 2018-11-22 |
Family
ID=64273910
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/018896 WO2018212228A1 (ja) | 2017-05-17 | 2018-05-16 | ポリオルガノシルセスキオキサン、転写用フィルム、インモールド成型品、及びハードコートフィルム |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20200079910A1 (ja) |
| JP (1) | JPWO2018212228A1 (ja) |
| KR (1) | KR20200007894A (ja) |
| CN (1) | CN110621723A (ja) |
| TW (1) | TW201902997A (ja) |
| WO (1) | WO2018212228A1 (ja) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2020235274A1 (ja) * | 2019-05-17 | 2020-11-26 | ||
| WO2021060055A1 (ja) * | 2019-09-27 | 2021-04-01 | 富士フイルム株式会社 | ハードコート層形成用組成物、ハードコートフィルム、ハードコートフィルムの製造方法、及びハードコートフィルムを含む物品 |
| WO2022014674A1 (ja) * | 2020-07-16 | 2022-01-20 | 東山フイルム株式会社 | インサート成形用ハードコートフィルムおよびインサート成形品の製造方法 |
| WO2022044968A1 (ja) * | 2020-08-28 | 2022-03-03 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、硬化性組成物、硬化物、ハードコートフィルム、転写用フィルム、及び接着シート |
| WO2022044969A1 (ja) * | 2020-08-28 | 2022-03-03 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、硬化性組成物、硬化物、ハードコートフィルム、転写用フィルム、及び接着シート |
| JP2022537795A (ja) * | 2019-09-25 | 2022-08-30 | エルジー・ケム・リミテッド | 光学積層体およびこれを含むフレキシブルディスプレイ装置 |
| WO2023238836A1 (ja) * | 2022-06-10 | 2023-12-14 | 東亞合成株式会社 | シルセスキオキサン誘導体及びその製造方法、硬化性組成物、ハードコート剤、硬化物、ハードコート、並びに、基材 |
| WO2023238835A1 (ja) * | 2022-06-10 | 2023-12-14 | 東亞合成株式会社 | シルセスキオキサン誘導体及びその製造方法、硬化性組成物、ハードコート剤、硬化物、ハードコート、並びに、基材 |
| TWI837235B (zh) | 2018-12-04 | 2024-04-01 | 日商播磨化成股份有限公司 | 附有硬塗層之模製樹脂及其製造方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11692108B2 (en) * | 2018-08-17 | 2023-07-04 | Sk Innovation Co., Ltd. | Composition for forming hard coating layer, preparation method of hard coating film, and hard coating film prepared using the same |
| US11693154B2 (en) * | 2018-08-23 | 2023-07-04 | Sk Innovation Co., Ltd. | Antireflection hard coating film and preparation method thereof |
| US11693155B2 (en) * | 2018-08-23 | 2023-07-04 | Sk Innovation Co., Ltd. | Antireflection hard coating film and preparation method thereof |
| KR20220128423A (ko) * | 2020-03-27 | 2022-09-20 | 후지필름 가부시키가이샤 | 하드 코트층 형성용 조성물, 하드 코트 필름, 하드 코트 필름의 제조 방법 및 하드 코트 필름을 구비한 물품 |
| CN113665263A (zh) * | 2021-09-10 | 2021-11-19 | 朱宏达 | 一种真空热转印花膜 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008179811A (ja) * | 2006-12-28 | 2008-08-07 | Asahi Kasei Corp | シロキサン誘導体及びその硬化物 |
| WO2015087686A1 (ja) * | 2013-12-13 | 2015-06-18 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、ハードコートフィルム、接着シート、及び積層物 |
| WO2016204014A1 (ja) * | 2015-06-17 | 2016-12-22 | 株式会社ダイセル | 硬化性組成物 |
| JP2017008142A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、硬化性組成物、ハードコートフィルム、及び硬化物 |
| JP2017008144A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、硬化性組成物、ハードコートフィルム、及び硬化物 |
| JP2017008145A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | 硬化性組成物、接着シート、積層物及び装置 |
| JP2017008147A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、硬化性組成物、接着シート、積層物及び装置 |
| JP2017008148A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、硬化性組成物、ハードコートフィルム、及び硬化物 |
| JP2017008143A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | ポリオルガノシルセスキオキサン |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102005002960A1 (de) * | 2005-01-21 | 2006-08-03 | Leibniz-Institut Für Neue Materialien Gemeinnützige Gmbh | Kompositzusammensetzung für mikrogemusterte Schichten mit hohem Relaxationsvermögen, hoher chemischer Beständigkeit und mechanischer Stabilität |
| MY154045A (en) * | 2012-05-25 | 2015-04-27 | Daicel Corp | Curable resin composition and cured product thereof, encapsulating agent, and optical semiconductor device |
| CN104129189B (zh) | 2013-05-02 | 2017-09-29 | 荒川化学工业株式会社 | 转印用装饰膜 |
-
2018
- 2018-05-16 KR KR1020197036747A patent/KR20200007894A/ko not_active Application Discontinuation
- 2018-05-16 US US16/614,007 patent/US20200079910A1/en not_active Abandoned
- 2018-05-16 JP JP2019518834A patent/JPWO2018212228A1/ja not_active Withdrawn
- 2018-05-16 CN CN201880032557.XA patent/CN110621723A/zh active Pending
- 2018-05-16 WO PCT/JP2018/018896 patent/WO2018212228A1/ja active Application Filing
- 2018-05-17 TW TW107116788A patent/TW201902997A/zh unknown
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008179811A (ja) * | 2006-12-28 | 2008-08-07 | Asahi Kasei Corp | シロキサン誘導体及びその硬化物 |
| WO2015087686A1 (ja) * | 2013-12-13 | 2015-06-18 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、ハードコートフィルム、接着シート、及び積層物 |
| WO2016204014A1 (ja) * | 2015-06-17 | 2016-12-22 | 株式会社ダイセル | 硬化性組成物 |
| JP2017008142A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、硬化性組成物、ハードコートフィルム、及び硬化物 |
| JP2017008144A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、硬化性組成物、ハードコートフィルム、及び硬化物 |
| JP2017008145A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | 硬化性組成物、接着シート、積層物及び装置 |
| JP2017008147A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、硬化性組成物、接着シート、積層物及び装置 |
| JP2017008148A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、硬化性組成物、ハードコートフィルム、及び硬化物 |
| JP2017008143A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | ポリオルガノシルセスキオキサン |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI837235B (zh) | 2018-12-04 | 2024-04-01 | 日商播磨化成股份有限公司 | 附有硬塗層之模製樹脂及其製造方法 |
| WO2020235274A1 (ja) * | 2019-05-17 | 2020-11-26 | 富士フイルム株式会社 | ハードコートフィルム、ハードコートフィルムを備えた物品、及び画像表示装置 |
| JPWO2020235274A1 (ja) * | 2019-05-17 | 2020-11-26 | ||
| JP7377261B2 (ja) | 2019-05-17 | 2023-11-09 | 富士フイルム株式会社 | ハードコートフィルム、ハードコートフィルムを備えた物品、及び画像表示装置 |
| JP2022537795A (ja) * | 2019-09-25 | 2022-08-30 | エルジー・ケム・リミテッド | 光学積層体およびこれを含むフレキシブルディスプレイ装置 |
| JP7287599B2 (ja) | 2019-09-25 | 2023-06-06 | エルジー・ケム・リミテッド | 光学積層体およびこれを含むフレキシブルディスプレイ装置 |
| WO2021060055A1 (ja) * | 2019-09-27 | 2021-04-01 | 富士フイルム株式会社 | ハードコート層形成用組成物、ハードコートフィルム、ハードコートフィルムの製造方法、及びハードコートフィルムを含む物品 |
| JPWO2021060055A1 (ja) * | 2019-09-27 | 2021-04-01 | ||
| JP7280963B2 (ja) | 2019-09-27 | 2023-05-24 | 富士フイルム株式会社 | ハードコート層形成用組成物、ハードコートフィルム、ハードコートフィルムの製造方法、及びハードコートフィルムを含む物品 |
| WO2022014674A1 (ja) * | 2020-07-16 | 2022-01-20 | 東山フイルム株式会社 | インサート成形用ハードコートフィルムおよびインサート成形品の製造方法 |
| JP2022031615A (ja) * | 2020-07-16 | 2022-02-22 | 東山フイルム株式会社 | インサート成形用ハードコートフィルムおよびインサート成形品の製造方法 |
| JP6991605B1 (ja) | 2020-07-16 | 2022-02-03 | 東山フイルム株式会社 | インサート成形用ハードコートフィルムおよびインサート成形品の製造方法 |
| WO2022044969A1 (ja) * | 2020-08-28 | 2022-03-03 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、硬化性組成物、硬化物、ハードコートフィルム、転写用フィルム、及び接着シート |
| WO2022044968A1 (ja) * | 2020-08-28 | 2022-03-03 | 株式会社ダイセル | ポリオルガノシルセスキオキサン、硬化性組成物、硬化物、ハードコートフィルム、転写用フィルム、及び接着シート |
| WO2023238836A1 (ja) * | 2022-06-10 | 2023-12-14 | 東亞合成株式会社 | シルセスキオキサン誘導体及びその製造方法、硬化性組成物、ハードコート剤、硬化物、ハードコート、並びに、基材 |
| WO2023238835A1 (ja) * | 2022-06-10 | 2023-12-14 | 東亞合成株式会社 | シルセスキオキサン誘導体及びその製造方法、硬化性組成物、ハードコート剤、硬化物、ハードコート、並びに、基材 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20200079910A1 (en) | 2020-03-12 |
| KR20200007894A (ko) | 2020-01-22 |
| JPWO2018212228A1 (ja) | 2020-03-19 |
| TW201902997A (zh) | 2019-01-16 |
| CN110621723A (zh) | 2019-12-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2018212228A1 (ja) | ポリオルガノシルセスキオキサン、転写用フィルム、インモールド成型品、及びハードコートフィルム | |
| JP6219250B2 (ja) | ポリオルガノシルセスキオキサン、ハードコートフィルム、接着シート、及び積層物 | |
| CN110494466B (zh) | 固化性组合物、固化物及硬涂膜 | |
| JP6795498B2 (ja) | 硬化性組成物、及び成形体 | |
| CN107735253B (zh) | 损伤恢复膜 | |
| WO2018207914A1 (ja) | カールが抑制されたハードコートフィルム及びその製造方法 | |
| CN110546001B (zh) | 层叠体 | |
| CN111093963B (zh) | 成形体 | |
| JP6956517B2 (ja) | 転写用フィルム、及びインモールド成型品 | |
| JP2018177952A (ja) | 硬化性組成物、硬化物及びハードコートフィルム | |
| JP2017008144A (ja) | ポリオルガノシルセスキオキサン、硬化性組成物、ハードコートフィルム、及び硬化物 | |
| JP6737926B2 (ja) | ポリオルガノシルセスキオキサン、ハードコートフィルム、接着シート、及び積層物 | |
| WO2021215106A1 (ja) | ポリオルガノシルセスキオキサン、積層体、及び表面被覆成形体 | |
| WO2022044968A1 (ja) | ポリオルガノシルセスキオキサン、硬化性組成物、硬化物、ハードコートフィルム、転写用フィルム、及び接着シート | |
| JP6595812B2 (ja) | ポリオルガノシルセスキオキサン、硬化性組成物、ハードコートフィルム、及び硬化物 | |
| WO2022044969A1 (ja) | ポリオルガノシルセスキオキサン、硬化性組成物、硬化物、ハードコートフィルム、転写用フィルム、及び接着シート |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18801775 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019518834 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20197036747 Country of ref document: KR Kind code of ref document: A |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 18801775 Country of ref document: EP Kind code of ref document: A1 |