WO2018021518A1 - 光安定性の向上した固形製剤 - Google Patents
光安定性の向上した固形製剤 Download PDFInfo
- Publication number
- WO2018021518A1 WO2018021518A1 PCT/JP2017/027391 JP2017027391W WO2018021518A1 WO 2018021518 A1 WO2018021518 A1 WO 2018021518A1 JP 2017027391 W JP2017027391 W JP 2017027391W WO 2018021518 A1 WO2018021518 A1 WO 2018021518A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- solid preparation
- weight
- active ingredient
- propyl gallate
- light
- Prior art date
Links
- 0 *B*C(CCC1(*)[C@]23CCN(*)[C@@]1C1)[C@]2Oc2c3c1ccc2* Chemical compound *B*C(CCC1(*)[C@]23CCN(*)[C@@]1C1)[C@]2Oc2c3c1ccc2* 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/485—Morphinan derivatives, e.g. morphine, codeine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/02—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/10—Alcohols; Phenols; Salts thereof, e.g. glycerol; Polyethylene glycols [PEG]; Poloxamers; PEG/POE alkyl ethers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/22—Heterocyclic compounds, e.g. ascorbic acid, tocopherol or pyrrolidones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2009—Inorganic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2013—Organic compounds, e.g. phospholipids, fats
- A61K9/2018—Sugars, or sugar alcohols, e.g. lactose, mannitol; Derivatives thereof, e.g. polysorbates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/20—Pills, tablets, discs, rods
- A61K9/2004—Excipients; Inactive ingredients
- A61K9/2022—Organic macromolecular compounds
- A61K9/205—Polysaccharides, e.g. alginate, gums; Cyclodextrin
- A61K9/2054—Cellulose; Cellulose derivatives, e.g. hydroxypropyl methylcellulose
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
Definitions
- the present invention relates to a solid preparation having improved light stability of a solid preparation containing a 4,5-epoxymorphinan derivative or a pharmacologically acceptable acid addition salt thereof.
- Nalfurafine or a pharmacologically acceptable acid addition salt thereof known as a compound having a remarkable antipruritic effect is chemically unstable to heat, light, oxygen, etc.
- a method has been developed to improve mechanical stability.
- Patent Documents 1 and 2 a method of adding a substance selected from specific antioxidants, synergists, sugars or surfactants, and a method of adding sodium thiosulfate, sugars or sugar alcohols and low-substituted hydroxypropylcellulose are reported. (Patent Documents 1 and 2).
- the stability of narfrafin or a pharmacologically acceptable acid addition salt thereof is improved by containing a substance selected from a specific antioxidant, synergist, saccharide or surfactant.
- a substance selected from a specific antioxidant, synergist, saccharide or surfactant As specific dosage forms of solid preparations, tablets and granules are described.
- the solid preparation described in Patent Document 2 contains sodium thiosulfate, sugars or sugar alcohols, and low-substituted hydroxypropylcellulose as stabilizers, so that nalfrafin or a pharmacologically acceptable acid thereof can be obtained. Improves the stability of addition salts.
- the orally disintegrating tablet described in Patent Document 5 is a coating containing a light shielding agent comprising a polyvinyl alcohol-based resin and a specific saccharide on an orally disintegrating tablet containing nalflaphine or a pharmacologically acceptable acid addition salt thereof. To ensure excellent light stability.
- the preparation described in Patent Document 6 is a preparation that improves light stability by blending iron oxide with amlodipine which is unstable to light.
- the light stabilization method described in Patent Document 7 it is described that the light stability is improved by adding a dissolved substance having an absorption behavior for light similar to the active ingredient to be protected.
- Patent Document 1 Although the stability improvement against heat and oxidation of nalfrafin or a pharmacologically acceptable acid addition salt thereof is shown by data, the improvement of the stability against light has not been studied. It was.
- Patent Document 2 Even in Patent Document 2, the stability improvement against heat and oxidation of nalflaphine or a pharmacologically acceptable acid addition salt thereof is shown in the data, but the improvement of the stability against light has not been studied. Only a typical tablet shading coating was applied.
- Patent Documents 3 and 4 the stability against heat and oxidation during storage is shown by data, but the improvement in stability against light is neither described nor suggested.
- the stabilization technology for nalflaphine or a pharmacologically acceptable acid addition salt thereof described in Patent Documents 1 to 4 is a stabilization method during the production of a preparation and / or during long-term storage.
- the stability improvement has not been studied so far.
- the present invention provides a solid preparation containing a 4,5-epoxymorphinan derivative or a pharmacologically acceptable acid addition salt thereof that is stable to light without a light-shielding coating. It is aimed.
- the present invention relates to the following inventions (1) to (5).
- An active ingredient comprising a 4,5-epoxymorphinan derivative represented by the following general formula (I) or a pharmacologically acceptable acid addition salt thereof, n-propyl gallate, sodium bisulfite, dibutylhydroxy
- One or more stabilizers selected from the group consisting of toluene, butylhydroxyanisole, tocopherol and D-isoascorbic acid, and the weight of the active ingredient is 0.00001 to 0 per weight of the solid preparation A solid preparation, wherein the weight of the stabilizer is 0.005 to 5% by weight per weight of the solid preparation.
- R 1 represents cyclopropylmethyl or allyl
- R 2 represents hydrogen, hydroxy, acetoxy or methoxy
- R 3 represents hydrogen, hydroxy, acetoxy or methoxy
- A represents —N ( R 4 ) C ( ⁇ O) — or —N (R 4 ) C ( ⁇ O) O—
- R 4 represents hydrogen or a linear or branched alkyl having 1 to 5 carbon atoms
- B represents a linear alkylene having 1 to 3 carbon atoms, —CH ⁇ CH— or —C ⁇ C—
- R 5 represents hydrogen, phenyl, furyl or thienyl, and the above phenyl, furyl and
- the thienyl hydrogen is alkyl having 1 to 5 carbons, alkoxy having 1 to 5 carbons, alkanoyloxy having 1 to 5 carbons, hydroxy, fluorine, chlorine, bromine, iodine, amino, nitro, cyano, isothiocyanato, tri Flu
- An active ingredient comprising a 4,5-epoxymorphinan derivative represented by the following general formula (I) or a pharmacologically acceptable acid addition salt thereof, a saccharide, n-propyl gallate, hydrogen sulfite
- One or more stabilizers selected from the group consisting of sodium, dibutylhydroxytoluene, butylhydroxyanisole, tocopherol and D-isoascorbic acid, and the weight of the active ingredient is 0 per weight of the solid preparation
- a solid preparation having a weight of 0.0001 to 0.01% by weight and a weight of the stabilizer of 0.005 to 5% by weight of the solid preparation.
- R 1 represents cyclopropylmethyl or allyl
- R 2 represents hydrogen, hydroxy, acetoxy or methoxy
- R 3 represents hydrogen, hydroxy, acetoxy or methoxy
- A represents —N ( R 4 ) C ( ⁇ O) — or —N (R 4 ) C ( ⁇ O) O—
- R 4 represents hydrogen or a linear or branched alkyl having 1 to 5 carbon atoms
- B represents a linear alkylene having 1 to 3 carbon atoms, —CH ⁇ CH— or —C ⁇ C—
- R 5 represents hydrogen, phenyl, furyl or thienyl, and the above phenyl, furyl and
- the thienyl hydrogen is alkyl having 1 to 5 carbons, alkoxy having 1 to 5 carbons, alkanoyloxy having 1 to 5 carbons, hydroxy, fluorine, chlorine, bromine, iodine, amino, nitro, cyano, isothiocyanato, tri Flu
- the light stability of a solid preparation containing a 4,5-epoxymorphinan derivative or a pharmacologically acceptable acid addition salt thereof is improved, and the usefulness as a pharmaceutical can be enhanced.
- the solid preparation in the present invention refers to a medicine containing an active ingredient formulated so as to be solid, for example, tablets (including sublingual tablets, orally disintegrating tablets, and minitablets), hard capsules , Granules, fine granules, powders, dry syrups, pills, troches, or film preparations.
- tablets including sublingual tablets, orally disintegrating tablets, and minitablets
- hard capsules for example, tablets (including sublingual tablets, orally disintegrating tablets, and minitablets), hard capsules , Granules, fine granules, powders, dry syrups, pills, troches, or film preparations.
- the present invention when used for granules, fine granules, powders, dry syrups and film preparations, the complicated process of forming a coating film containing a uniform light-shielding agent on the outer surface of the powdered solid agent is omitted. However, it is preferable because light stability can be secured.
- the active ingredient of the present invention is a 4,5-epoxymorphinan derivative represented by the following general formula (I) or a pharmacologically acceptable acid addition salt thereof.
- R 1 represents cyclopropylmethyl or allyl
- R 2 represents hydrogen, hydroxy, acetoxy or methoxy
- R 3 represents hydrogen, hydroxy, acetoxy or methoxy
- A represents —N ( R 4 ) C ( ⁇ O) — or —N (R 4 ) C ( ⁇ O) O—
- R 4 represents hydrogen or a linear or branched alkyl having 1 to 5 carbon atoms
- B represents a linear alkylene having 1 to 3 carbon atoms, —CH ⁇ CH— or —C ⁇ C—
- R 5 represents hydrogen, phenyl, furyl or thienyl, and the above phenyl, furyl and
- the thienyl hydrogen is alkyl having 1 to 5 carbons, alkoxy having 1 to 5 carbons, alkanoyl
- 4,5-epoxymorphinan derivative represented by the above general formula (I) or a pharmacologically acceptable salt thereof is a compound represented by the following general formula (II): Hydrochloride of (Cyclopropylmethyl) -3,14 ⁇ -dihydroxy-4,5 ⁇ -epoxy-6 ⁇ - [N-methyl-trans-3- (3-furyl) acrylamide] morphinan (hereinafter referred to as “Nalflaphine”) It is.
- Examples of the pharmacologically acceptable acid addition salt include inorganic acid salts such as hydrochloride, sulfate, nitrate, hydrobromide, hydroiodide, phosphate, acetate, lactate, citric acid, and the like. Acid, oxalate, glutarate, malate, tartrate, fumarate, mandelate, maleate, benzoate, phthalate, etc., organic carboxylates, methanesulfonate, ethane Examples thereof include organic sulfonates such as sulfonate, benzenesulfonate, p-toluenesulfonate, camphorsulfonate, and the like. Among them, hydrochloride, hydrobromide, phosphate, tartrate, maleic acid, etc. Salt and methanesulfonate are preferred, and commercially available hydrochloride is most preferred.
- inorganic acid salts such as hydrochloride, sulfate,
- the weight of the 4,5-epoxymorphinan derivative represented by the above general formula (I) or a pharmacologically acceptable acid addition salt thereof as an active ingredient of the present invention is 0.00001 to 0 per weight of the solid preparation. It is preferably 0.011% by weight, more preferably 0.00005 to 0.01% by weight, and even more preferably 0.00025 to 0.01% by weight.
- the active ingredient is more than 0.01% by weight, a solid preparation having sufficient stability to light can be obtained without using a stabilizer.
- the active ingredient is less than 0.00001% by weight, the dosage of the preparation for obtaining a therapeutic effect is increased.
- the range of% by weight of the active ingredient varies depending on the weight of the solid preparation, but typically ranges from 0.01 ⁇ g to 50 ⁇ g of the active ingredient in the daily dose of the solid preparation.
- the stabilizer used in the present invention is one or more stabilizers selected from the group consisting of n-propyl gallate, sodium bisulfite, dibutylhydroxytoluene, butylhydroxyanisole, tocopherol, and D-isoascorbic acid. N-propyl gallate and / or tocopherol are preferred, and n-propyl gallate is more preferred.
- N-propyl gallate is commercially available under the names of propyl gallate, propyl gallate, 3,4,5-trihydroxybenzoic acid propyl ester, and the like.
- Dibutylhydroxytoluene is BHT, 2,6-di-tert-butyl-p-cresol, 3,5-di-tert-butyl-4-hydroxytoluene, 2,6-di-tert-butyl-4-methylphenol It is marketed with names such as.
- Butylhydroxyanisole is commercially available under the names BHA, 3-t-butyl-4-hydroxyanisole, tert-butyl-4-methoxyphenol and the like.
- Tocopherol is commercially available under the names dl- ⁇ -tocopherol, d- ⁇ -tocopherol, d- ⁇ tocopherol, vitamin E and the like.
- the weight of the stabilizer used in the present invention is 0.005 to 5% by weight, preferably 0.005 to 1% by weight, based on the weight of the solid preparation.
- the stabilizer is less than 0.005% by weight per weight of the solid preparation, the light stabilizing effect cannot be sufficiently obtained.
- Exceeding 5% by weight is not preferable because it exceeds the maximum daily use for which safety has been confirmed.
- the solid preparation of the present invention further contains a saccharide.
- the saccharide include saccharides or sugar alcohols, and those that are generally commercially available may be used. Examples thereof include potato starch, sucrose, lactose, mannitol, erythritol, maltose, maltitol, trehalose, sorbitol, xylitol, lactitol and glucose, preferably lactose, erythritol and mannitol.
- a saccharide not only the sweetness of the saccharide is improved from the sweetness but also the 4,5-epoxymorphinan derivative represented by the above general formula (I) or a pharmacologically acceptable acid thereof.
- the storage stability of the addition salt can be improved.
- an antioxidant may be further added in order to suppress decomposition of the active ingredient during production and / or long-term storage.
- an antioxidant includes sodium thiosulfate.
- the solid preparation of the present invention may contain various additives used for production of general preparations in addition to the above-described components.
- additives include an excipient, a disintegrant, a binder, a lubricant, a coating agent, a fluidizing agent, a corrigent, a flavoring agent, a coloring agent, and a sweetening agent.
- disintegrant examples include crospovidone, croscarmellose sodium, carmellose calcium, carboxymethyl starch sodium, and low-substituted hydroxypropylcellulose.
- binder examples include water-soluble polysaccharides such as gelatin, pullulan, carrageenan, xanthan gum, tamarind gum, pectin, sodium alginate and gum arabic, celluloses such as hydroxypropylcellulose, hydroxypropylmethylcellulose and methylcellulose, pregelatinized starch and Examples thereof include starches such as starch paste and synthetic polymers such as polyvinylpyrrolidone, carboxyvinyl polymer and polyvinyl alcohol.
- water-soluble polysaccharides such as gelatin, pullulan, carrageenan, xanthan gum, tamarind gum, pectin, sodium alginate and gum arabic
- celluloses such as hydroxypropylcellulose, hydroxypropylmethylcellulose and methylcellulose
- pregelatinized starch examples include starches such as starch paste and synthetic polymers such as polyvinylpyrrolidone, carboxyvinyl polymer and polyvinyl alcohol.
- Examples of the lubricant include magnesium stearate, calcium stearate, sodium stearyl fumarate, talc, sucrose fatty acid ester, stearic acid, aluminum stearate, potassium sodium tartrate, light anhydrous silicic acid, carnauba wax, carmellose calcium, carme
- Examples include sodium sodium, hydrous silicon dioxide, hydrogenated oil and hydrogenated rapeseed oil.
- the coating agent examples include hydroxypropylmethylcellulose, ethylcellulose, sodium carboxymethylethylcellulose, and polyvinyl alcohol.
- Examples of the fluidizing agent include talc, hydrous silicon dioxide, and light anhydrous silicic acid.
- corrigent examples include glutamic acid, fumaric acid, succinic acid, citric acid, sodium citrate, tartaric acid, malic acid, ascorbic acid, sodium chloride or menthol.
- fragrance examples include orange, vanilla, strawberry or yogurt-flavored fragrance and menthol.
- colorant examples include edible pigments such as titanium oxide, iron sesquioxide, yellow iron sesquioxide, black iron oxide, talc, edible red No. 3, edible yellow No. 5, and edible blue No. 1, and riboflavin.
- sweetening agent examples include aspartame, saccharin, dipotassium glycyrrhizinate and stevia.
- the method for producing a solid preparation of the present invention comprises a step of dissolving or suspending an active ingredient in water or a pharmacologically acceptable solvent, and adding the obtained liquid (solution or suspension) to a carbohydrate. It can be manufactured by a wet granulation method.
- the stabilizer can be added in any step, and can be added in a solid or liquid state.
- the method for adding the stabilizer as a solid is not limited.
- a commercially available stabilizer may be mixed by pulverization or the like as necessary, or water or alcohols such as ethanol and methanol, or a mixed solution thereof.
- a method of adding a stabilizer suspended in the solution is not limited.
- the method of adding the stabilizer as a liquid is not limited.
- the method of adding the stabilizer to water or a pharmacologically acceptable solvent together with the active ingredient and adding the active ingredient to the saccharide And a method of adding a stabilizer after the step of granulating or sizing as appropriate.
- the whole amount may be used in the above step of adding the active ingredient, or the remaining saccharide may be added in the subsequent step using only a part.
- Generally used equipment is used for wet granulation, for example, fluidized bed granulator, rolling fluidized bed granulator, stirring granulator, cylindrical extrusion granulator, wet extrusion granulator, etc. Is mentioned.
- a fluidized bed granulator or a rolling fluidized bed granulator that can be dried while spraying is suitable.
- a volatile solvent such as ethanol is used as a solvent for dissolving or suspending the active ingredient
- a fluidized bed granulator, a rolling fluidized bed granulator, or a stirring granulator is suitable.
- a commonly used mixing apparatus When adding the stabilizer as a solid, a commonly used mixing apparatus is used, and examples thereof include a V-type mixer, a ribbon mixer, and an air blender.
- the light stability in the solid preparation can be further improved by adding yellow ferric oxide, red ferric oxide or black iron oxide as a colorant to the solid preparation.
- the colorant can be added after being suspended in powder or water or a pharmacologically acceptable solvent.
- the solid preparation is a tablet
- compression molding is performed using a commonly used apparatus, such as a single-shot tableting machine or a rotary tableting machine.
- the molding pressure at the time of tableting is not particularly limited as long as it has a tablet hardness that does not cause a problem in handling.
- the solid preparation of the present invention Since the solid preparation of the present invention has improved light stability of the active ingredient, it is easy to handle at the time of dispensing or taking medicine.
- the improvement in stability means that the 4,5-epoxymorphinan derivative represented by the above general formula (I) in the preparation or its pharmacological property as described in International Publication No. 16/052617. This means that the remaining ratio of acid addition salt allowed to be maintained at 90% or more.
- the solid preparation of the present invention is not packaged, the solid preparation is handled in a white fluorescent light (illuminance 2000 lux) environment at a temperature of 25 ° C. and a relative humidity of 51% RH at least 24 hours later (as total illuminance).
- the residual ratio of the active ingredient of 48,000 lux ⁇ hr) is 90% or more, and more preferably the active ingredient after 300 hours (total illuminance of 600,000 lux ⁇ hr) is 90% or more.
- Example 1 Mannitol (Procket, 200SD (registered trademark), 888.8975 parts) manufactured by Rocket Japan Co., Ltd. was charged into a fluidized bed granulator (LAB-1 manufactured by POWREC Co., Ltd.), and an aqueous solution containing 0.0025 parts of nalfrafin hydrochloride was added. To the granulated granules produced by spraying, 0.1 part of ethanol solution of n-propyl gallate (manufactured by Wako Pure Chemical Industries, Ltd., Wako Grade 1) was added and stirred in a mortar. After drying in a hot air dryer at 40 ° C.
- Example 2 100 mg in the same procedure as in Example 1 except that 0.1 part of dl- ⁇ -tocopherol (manufactured by Kanto Chemical Co., Inc., deer grade) was used instead of 0.1 part of n-propyl gallate in Example 1. ⁇ 7 mm tablets were obtained.
- Example 3 (Example 3) Implemented except that 0.1 part of 3-tert-butyl-4-hydroxyanisole (BHA) (manufactured by Nacalai Tesque, EXTRA PURE) was used instead of 0.1 part of n-propyl gallate of Example 1.
- BHA 3-tert-butyl-4-hydroxyanisole
- Example 4 In place of 0.1 part of n-propyl gallate in Example 1, 0.1 part of 2,6-di-t-butyl-p-cresol (BHT) (manufactured by Nacalai Tesque, EXTRA PURE REAGENT) was used. A 100 mg ⁇ 7 mm tablet was obtained in the same procedure except that.
- BHT 2,6-di-t-butyl-p-cresol
- Example 5 Example 1 and Example 1 were used except that an aqueous solution of 0.1 part of D-isoascorbic acid (manufactured by Nacalai Tesque Co., Ltd., GUARANTED REAGENT) was used instead of the ethanol solution of 0.1 part of n-propyl gallate of Example 1. A 100 mg ⁇ 7 mm tablet was obtained in the same procedure.
- D-isoascorbic acid manufactured by Nacalai Tesque Co., Ltd., GUARANTED REAGENT
- Example 6 The same as Example 1 except that 0.1 part of sodium sulfite (made by Wako Pure Chemical Industries, Ltd., special grade) was used in place of the ethanol solution of 0.1 part of n-propyl gallate in Example 1. The procedure yielded 100 mg ⁇ 7 mm tablets.
- Example 2 Example except that 0.1 part of sodium thiosulfate pentahydrate (special grade, manufactured by Kokusan Chemical Co., Ltd.) was used in place of the ethanol solution of 0.1 part of n-propyl gallate in Example 1 In the same manner as in No. 1, 100 mg ⁇ 7 mm tablets were obtained.
- 0.1 part of sodium thiosulfate pentahydrate special grade, manufactured by Kokusan Chemical Co., Ltd.
- Example 3 Similar to Example 1 except that 0.1 part of an aqueous solution of sodium hydrogen sulfite (made by Wako Pure Chemical Industries, Ltd., special grade) was used instead of the ethanol solution of 0.1 part of n-propyl gallate of Example 1. According to the procedure, a 100 mg ⁇ 7 mm tablet was obtained.
- Test Example 1 Tablet light stability test
- the tablets of Examples 1 to 6 and Comparative Examples 1 to 3 were spread so as not to overlap the glass petri dish, and the total illumination was 48,000 lux ⁇ hr and the total illumination was 600,000 in a white fluorescent lamp (illuminance 2000 lux) environment.
- the tablets after lux ⁇ hr were taken out.
- the glass petri dish in which the tablets of Example 1 and Comparative Example 1 were spread was covered with aluminum foil (preserved in light shielding), taken out after irradiation of 48,000 lux ⁇ hr as total illuminance, and the residual ratio of active ingredients by the following HPLC analysis was calculated.
- Injection volume 100 ⁇ L
- Residual rate (%) peak area value of active ingredient in HPLC of sample after light irradiation / peak area value of active ingredient in HPLC of sample before light irradiation ⁇ 100 Formula 1
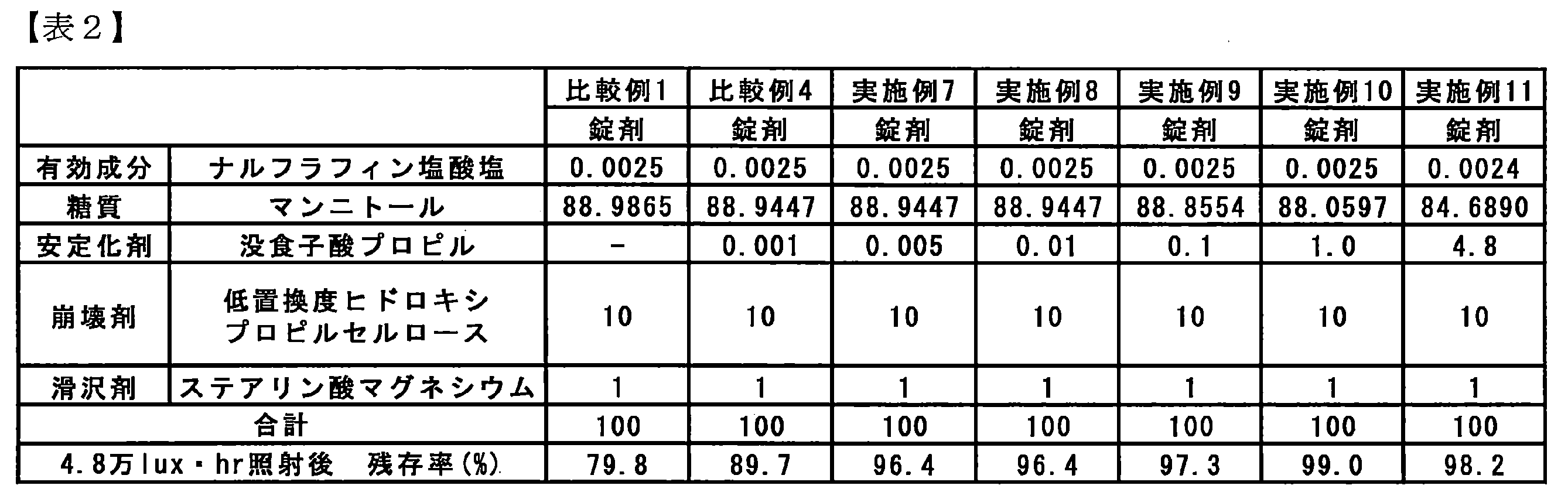
- Table 1 shows the percentages of the components in the tablets of Examples 1 to 6 and Comparative Examples 1 to 3 and the weight percent of each component per weight of the solid preparation and the residual ratio of the active ingredients after light irradiation obtained as a result of HPLC. Shown in
- the tablets of Examples 1 to 6 to which a specific stabilizer was added had a high residual ratio of active ingredients at 48,000 lux ⁇ hr as a total illuminance of 90% or more. It has been shown that stability is improved. Furthermore, regarding Example 1 to which n-propyl gallate was added as a stabilizer, it was shown that a very high residual ratio of active ingredients was maintained even at a total illumination of 600,000 lux ⁇ hr. Moreover, the residual rate of light-shielding preservation
- Example 7 99.5 mg in the same procedure as Comparative Example 4 except that 0.005 part of n-propyl gallate was added to the granulated granule of Comparative Example 4 instead of 0.001 part of n-propyl gallate. ⁇ 7 mm tablets were obtained.
- Example 8 99.5 mg in the same procedure as in Comparative Example 4 except that an ethanol solution of 0.01 part of n-propyl gallate was added to the granulated granule of Comparative Example 4 instead of 0.001 part of n-propyl gallate. ⁇ 7 mm tablets were obtained.
- Example 9 99.6 mg in the same procedure as in Comparative Example 4 except that an ethanol solution of 0.1 part of n-propyl gallate was added to the granulated granule of Comparative Example 4 instead of 0.001 part of n-propyl gallate. ⁇ 7 mm tablets were obtained.
- Example 10 100.5 mg in the same procedure as in Comparative Example 4 except that an ethanol solution of 1.0 part of n-propyl gallate was added to the granulated granule of Comparative Example 4 instead of 0.001 part of n-propyl gallate. ⁇ 7 mm tablets were obtained.
- Example 11 104.5 mg of ⁇ 7 mm in the same procedure as in Comparative Example 4 except that an ethanol solution of 5 parts of n-propyl gallate was added to the granulated granule of Comparative Example 4 instead of 0.001 part of n-propyl gallate. Tablets were obtained.
- Test Example 1 The photostability test shown in Test Example 1 was performed on the tablets of Comparative Example 4 and Examples 7 to 11.
- Table 2 shows the percentages of the components in the tablets of Comparative Examples 1 and 4 and Examples 7 to 11 and the weight percent of each component per weight of the solid preparation and the residual ratio of the active ingredients after light irradiation obtained as a result of HPLC. Shown in
- Example 12 After adding 1.0 part of n-propyl gallate to the granulated granule of Comparative Example 4 and mixing, 10 parts of low-substituted hydroxypropyl cellulose and 1 part of magnesium stearate are mixed to obtain granules for tableting. Obtained. The granules for tableting were made into 100.5 mg 7 mm ⁇ tablets using a tableting machine.
- Example 13 88.5 parts of mannitol is put into a fluidized bed granulator, and a granulated granule is obtained by spraying 30% ethanol aqueous solution in which two kinds of 0.0025 part of nalfurafine hydrochloride and 0.1 part of n-propyl gallate are dissolved. Manufactured.
- Example 14 The granulated granules obtained in Example 13 were mixed with 10 parts of low-substituted hydroxypropylcellulose and 1 part of magnesium stearate to obtain granules for tableting. The granules for tableting were made into 99.5 mg 7 mm ⁇ tablets using a tableting machine.
- Example 15 88.5 parts of mannitol is charged into a fluidized bed granulator and sprayed with an aqueous solution in which 0.0025 part of nalfurafine hydrochloride and 0.1 part of sodium thiosulfate pentahydrate are dissolved to form granulated granules. Manufactured. Next, an ethanol solution of 0.1 part of n-propyl gallate was added to the granulated granules and stirred in a mortar. After drying in a hot air dryer at 40 ° C. for 6 hours, 10 parts of low-substituted hydroxypropylcellulose and 1 part of magnesium stearate were mixed to obtain granules for tableting. The granules for tableting were made into 99.7 mg 7 mm ⁇ tablets using a tableting machine.
- Example 16 100.1 mg in the same procedure as in Example 15 except that an ethanol solution of 0.5 parts of n-propyl gallate was added to the granulated granules of Example 15 instead of 0.1 parts of n-propyl gallate. ⁇ 7 mm tablets were obtained.
- Example 17 100.6 mg of ⁇ 7 mm in the same procedure as in Example 15 except that an ethanol solution of 1 part of n-propyl gallate was added to the granulated granule of Example 15 instead of 0.1 part of n-propyl gallate. Tablets were obtained.
- Example 18 88.5 parts of mannitol are put into a fluidized bed granulator, and 3 kinds of 0.0025 part of nalfurafine hydrochloride, 0.1 part of sodium thiosulfate pentahydrate and 0.1 part of n-propyl gallate are dissolved. The granulated granules were produced by spraying the 30% aqueous ethanol solution.
- Example 19 The granulated granules of Example 18 were mixed with 10 parts of low-substituted hydroxypropyl cellulose and 1 part of magnesium stearate to obtain granules for tableting. The granules for tableting were made into 99.7 mg 7 mm ⁇ tablets using a tableting machine.
- Test Example 2 Production ratio confirmation test of main decomposition product A with respect to active ingredients during production
- the production ratio of the main degradation product A to the active ingredient in the powder obtained by subjecting the granulated granules of Examples 13 and 18 to aeration-dried powder for 30 minutes was calculated by the following HPLC analysis.
- the production ratio of the main decomposition product A was calculated from the following formula 2.
- Production ratio (%) peak area value of main degradation product A in HPLC of sample / peak area value of active ingredient of sample ⁇ 100 Equation 2
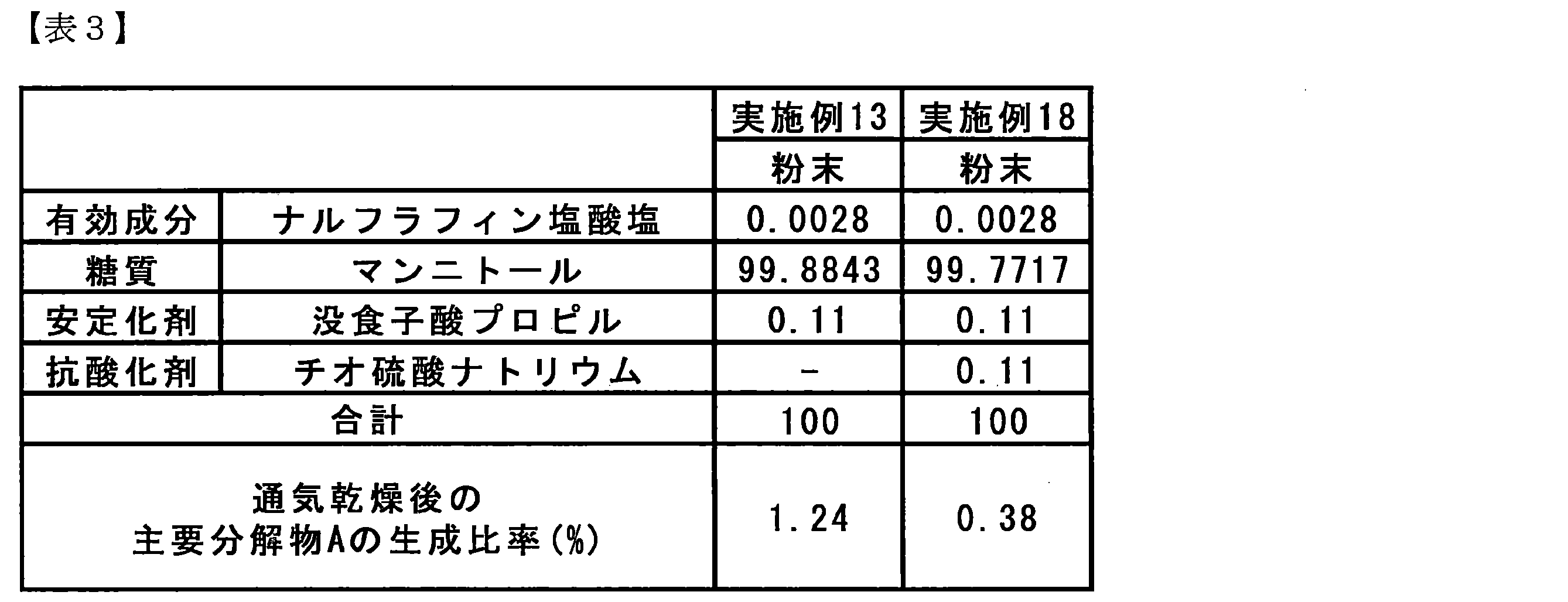
- Table 3 shows the production ratio of the main decomposition product A immediately after production of the granulated granules of Examples 13 and 18.
- Example 18 in which the antioxidant sodium thiosulfate was further added, was shown to significantly suppress the production ratio of the main degradation product during production as compared with Example 13. .
- Test Example 1 The photostability test shown in Test Example 1 was performed on the tablets of Example 12, Examples 14-17, and Example 19. The weight percentage of each component per weight of the components in the tablets and solid preparations of Example 12, Examples 14 to 17 and Example 19, and the residual ratio of the active ingredients after light irradiation obtained as a result of HPLC Table 4 shows.
- Example 12 As shown in Table 4, even when the stabilizer was added as a powder, an excellent light stabilization effect was shown (Example 12). Moreover, it was shown that light stability improves also when sprayed simultaneously with an active ingredient (Example 14). Furthermore, even when used together with the antioxidant sodium thiosulfate, the light stabilizing effect was maintained, and an excellent stabilizing effect was shown regardless of the method of adding the stabilizer and sodium thiosulfate.
- Example 20 99.89 parts of mannitol was placed in a mortar, an aqueous solution of 0.01 part of nalfurafine hydrochloride was added and stirred, and then an ethanol solution of 0.1 part of n-propyl gallate was added and stirred. Then, it dried at 40 degreeC for 12 hours, and was set as the granulated granule.
- Example 21 88.5 parts of mannitol was charged into a fluidized bed granulator, and 30% ethanol aqueous solution in which 0.0025 part of nalfurafine hydrochloride, 0.1 part of sodium thiosulfate and 0.1 part of n-propyl gallate were dissolved. was sprayed to produce granulated granules. Next, 455.65 parts of mannitol was added to 44.35 parts of the granulated granules and mixed using a V-type mixer.
- Test Example 3 Light stability test of powder
- the powders of Comparative Examples 5 to 8 and Examples 20 and 21 were spread thinly on a glass petri dish, and a powder with a total illumination of 48,000 lux ⁇ hr was taken out in a white fluorescent lamp (illuminance 2000 lux) environment.
- the residual ratio of the active ingredient after the light irradiation was calculated by the same calculation formula as Formula 1.
- Pretreatment conditions> Distilled water was added to the powder to suspend or dissolve it, followed by centrifugation, and the supernatant was used as an HPLC sample.
- ⁇ HPLC conditions> The test was performed under the same HPLC conditions as in Test Example 1.
- Test Example 2 The light stability test shown in Test Example 2 was performed on the powders of Comparative Examples 5 to 8 and Examples 20 and 21.
- the percentages of the components in the powders of Comparative Examples 5 to 8 and Examples 20 and 21 and the weight of each component per weight of the solid preparation, and the residual ratio of the active ingredients after light irradiation obtained as a result of HPLC are shown in the table. As shown in FIG.
- Vanillin which is a light absorber described in JP-A-58-57322 was used. Take 99.8975 parts of mannitol in a mortar, add 0.0025 parts of an aqueous solution of nalfraphine hydrochloride and stir, and then add 0.1 part of ethanol solution of vanillin (Wako Pure Chemical Industries, Ltd., Wako Special Grade). Stir. Then, it dried at 40 degreeC for 12 hours, and was set as the granulated granule.
- Example 22 99.7795 parts of mannitol was placed in a mortar, and an aqueous solution of 0.0025 part of nalflaphine hydrochloride was added and stirred. 0.1 part of iron sesquioxide (manufactured by HUNTSMAN, SICOVIT RED30E172) was added, and then an ethanol solution of 0.1 part of n-propyl gallate was added and stirred. Then, it dried at 40 degreeC for 12 hours, and was set as the granulated granule.
- Example 23 100 parts of the granulated granules of Example 21 were placed in a mortar, and 0.01 parts of iron sesquioxide and an aqueous ethanol solution were added and stirred. Then, it dried for 12 hours and was set as the granulated granule.
- Test Example 2 The light stability test shown in Test Example 2 was performed on the powders of Comparative Examples 9 and 10 and Examples 22 and 23.
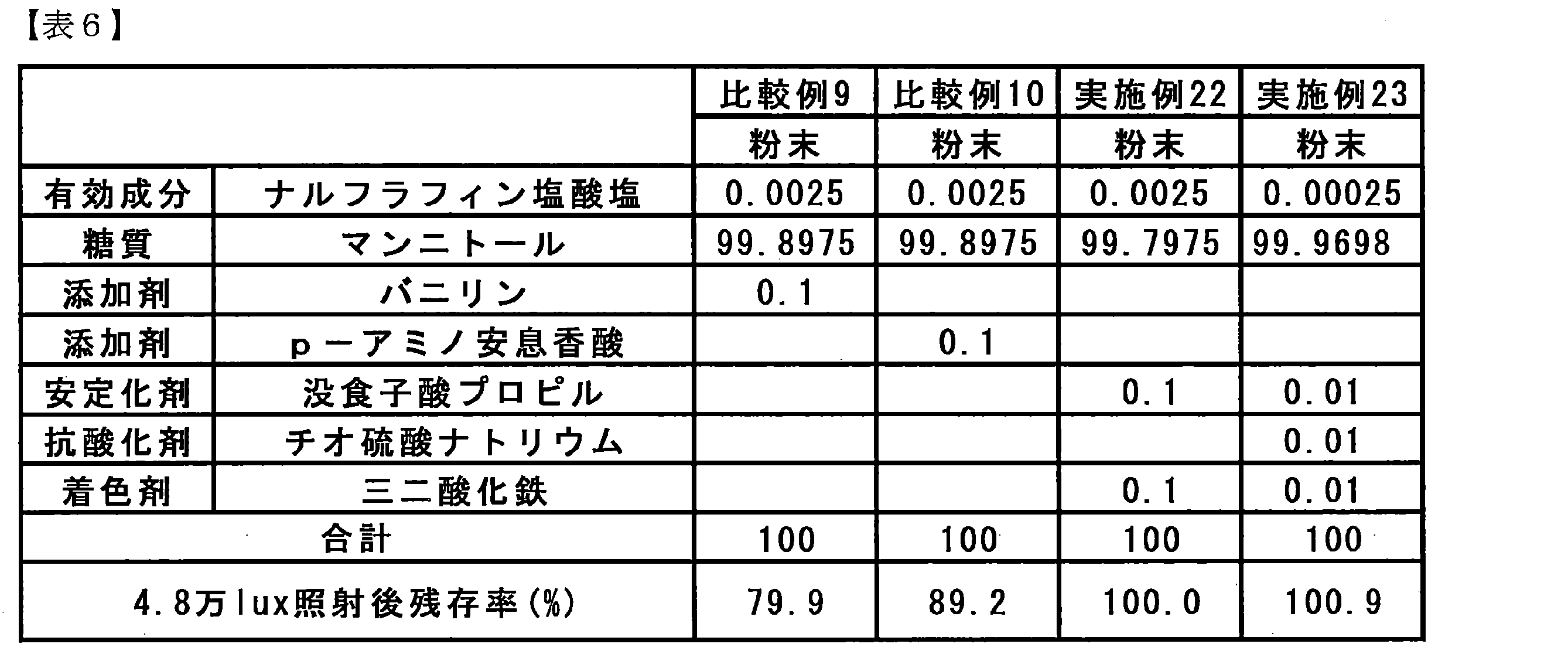
- Table 6 shows the weight percentages per weight of the components and solid preparations in the powders of Comparative Examples 9 and 10 and Examples 22 and 23, and the residual ratio of the active ingredients after light irradiation obtained as a result of HPLC.
- vanillin and p-aminobenzoic acid which are light absorbers described in JP-A-58-57322, are represented by the above-mentioned general formula (I), which is an active ingredient of the present invention.
- the photostabilization effect of 5-epoxymorphinan derivatives or pharmacologically acceptable acid addition salts thereof is insufficient, and it has been shown that effective stabilization methods differ depending on the active ingredient.
- the stabilizer of the present invention as shown in Example 22 it was shown that the light stabilization effect was further improved by containing iron sesquioxide as a colorant.
- Example 23 the amount of iron sesquioxide added is small as compared with 0.1% of the amount of iron sesquioxide added to the composition described in JP-A-2006-306754. It was shown that the light stability of the active ingredient was improved even when the addition rate of iron sesquioxide relative to the solid preparation was 0.01%.
- the light stability of a solid preparation containing a 4,5-epoxymorphinan derivative or a pharmacologically acceptable acid addition salt thereof is improved.
- the dispensing risk can be reduced, patient compliance can be improved, and the therapeutic effect can be improved.
- the manufacturing process can be simplified and the rapid disintegration of the tablet can be ensured.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Biophysics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Inorganic Chemistry (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Emergency Medicine (AREA)
- Dermatology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Biochemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
Description
(1) 下記一般式(I)で示される4,5-エポキシモルヒナン誘導体又はその薬理学的に許容される酸付加塩からなる有効成分と、没食子酸n-プロピル、亜硫酸水素ナトリウム、ジブチルヒドロキシトルエン、ブチルヒドロキシアニソール、トコフェロール及びD-イソアスコルビン酸からなる群から選択される1種以上の安定化剤と、を含有し、上記有効成分の重量は、固形製剤の重量あたり0.00001~0.01重量%であり、上記安定化剤の重量は、固形製剤の重量あたり0.005~5重量%である、固形製剤。

(2)上記安定化剤は、没食子酸n-プロピルである、(1)記載の固形製剤。
(3)チオ硫酸ナトリウムを含有する、(1)又は(2)記載の固形製剤。
(4)黄色三二酸化鉄、赤色三二酸化鉄又は黒酸化鉄を含有する、(1)~(3)のいずれか記載の固形製剤。
(5)糖質を含有する、(1)~(4)のいずれか記載の固形製剤。
(6) 錠剤、顆粒剤、細粒剤、硬カプセル剤、ドライシロップ剤、散剤、丸剤及びトローチ剤からなる群から選択される剤形である、(1)~(5)のいずれか記載の固形製剤。
また、本発明は、下記の(7)~(11)の発明に関するものである。
(7) 下記一般式(I)で示される4,5-エポキシモルヒナン誘導体又はその薬理学的に許容される酸付加塩からなる有効成分と、糖質と、没食子酸n-プロピル、亜硫酸水素ナトリウム、ジブチルヒドロキシトルエン、ブチルヒドロキシアニソール、トコフェロール及びD-イソアスコルビン酸からなる群から選択される1種以上の安定化剤と、を含有し、上記有効成分の重量は、固形製剤の重量あたり0.00001~0.01重量%であり、上記安定化剤の重量は、固形製剤の重量あたり0.005~5重量%である、固形製剤。

(8)上記安定化剤は、没食子酸n-プロピルである、(7)記載の固形製剤。
(9)チオ硫酸ナトリウムを含有する、(7)又は(8)記載の固形製剤。
(10)黄色三二酸化鉄、赤色三二酸化鉄又は黒酸化鉄を含有する、(7)~(9)のいずれか記載の固形製剤。
(11) 錠剤、顆粒剤、細粒剤、硬カプセル剤、ドライシロップ剤、散剤、丸剤及びトローチ剤からなる群から選択される剤形である、(7)~(10)のいずれか記載の固形製剤。


マンニトール(ロケットジャパン株式会社製、ペアリトール200SD(登録商標))88.8975重量部(以下「部」と略記する。以下、特に断らない場合には同様とする。)を流動層造粒機(株式会社パウレック製、LAB-1)に投入し、ナルフラフィン塩酸塩0.0025部の水溶液を噴霧して造粒顆粒を製造した。次に、造粒顆粒に低置換度ヒドロキシプロピルセルロース(信越化学工業株式会社製、LH-11)10部及びステアリン酸マグネシウム(太平化学産業株式会社製)1部を混合して、打錠用顆粒を得た。打錠用顆粒を、打錠機(株式会社菊水製作所製、Correct19)を用いて99.9mgの7mmφ錠剤とした。
マンニトール(ロケットジャパン株式会社製、ペアリトール200SD(登録商標))88.8975部)を流動層造粒機(株式会社パウレック製、LAB-1)に投入し、ナルフラフィン塩酸塩0.0025部の水溶液を噴霧して製造した造粒顆粒に、没食子酸n-プロピル(和光純薬工業株式会社製、和光一級)0.1部のエタノール溶液を添加して乳鉢で攪拌した。熱風乾燥機に40℃、16時間乾燥した後、低置換度ヒドロキシプロピルセルロース10部及びステアリン酸マグネシウム1部を混合して、打錠用顆粒を得た。打錠用顆粒を、打錠機を用いて100mgのφ7mm錠剤とした。
実施例1の没食子酸n-プロピル0.1部の代わりにdl-α-トコフェロール(関東化学株式会社製、鹿特級)0.1部を用いたこと以外は実施例1と同様の手順で100mgのφ7mm錠剤を得た。
実施例1の没食子酸n-プロピル0.1部の代わりに3-t-ブチル-4-ヒドロキシアニソール(BHA)(ナカライテスク株式会社製、EXTRA PURE)0.1部を用いたこと以外は実施例1と同様の手順で100mgのφ7mm錠剤を得た。
実施例1の没食子酸n-プロピル0.1部の代わりに2,6-ジ-t-ブチル-p-クレゾール(BHT)(ナカライテスク株式会社製、EXTRA PURE REAGENT)0.1部を用いたこと以外は同様の手順で100mgのφ7mm錠剤を得た。
実施例1の没食子酸n-プロピル0.1部のエタノール溶液の代わりにD-イソアスコルビン酸(ナカライテスク株式会社製、GUARANTEED REAGENT)0.1部の水溶液を用いたこと以外は実施例1と同様の手順で100mgのφ7mm錠剤を得た。
実施例1の没食子酸n-プロピル0.1部のエタノール溶液の代わりに亜硫酸ナトリウム(和光純薬工業株式会社製、特級)0.1部の水溶液を用いたこと以外は実施例1と同様の手順で100mgのφ7mm錠剤を得た。
実施例1の没食子酸n-プロピル0.1部のエタノール溶液の代わりにチオ硫酸ナトリウム・5水和物(国産化学株式会社製、特級)0.1部の水溶液を用いたこと以外は実施例1と同様の手順で100mgのφ7mm錠剤を得た。
実施例1の没食子酸n-プロピル0.1部のエタノール溶液の代わりに亜硫酸水素ナトリウム(和光純薬工業株式会社製、特級)0.1部の水溶液を用いたこと以外は実施例1と同様の手順で100mgのφ7mm錠剤を得た。
実施例1~6及び比較例1~3の錠剤をガラスシャーレ上に重ならないように広げ、白色蛍光灯(照度2000lux)環境下、総照度として4.8万lux・hr及び総照度として60万lux・hr後の錠剤を取り出した。また、実施例1及び比較例1の錠剤を広げたガラスシャーレをアルミ箔で覆い(遮光保存)、総照度として4.8万lux・hr照射後に取り出し、以下のHPLC分析により有効成分の残存率を算出した。
<前処理条件>
錠剤に25mMリン酸緩衝液/メタノール=40/60(v/v)溶液を加えて崩壊、攪拌した後、遠心分離し上澄みをHPLCサンプルとした。
<HPLC条件>
移動相 : 25mMリン酸緩衝液(pH7.0)/アセトニトリル=60/40(v/v)
カラム : “Capcellpak(登録商標)” MGII(株式会社資生堂製、サイズ:3.0×150mm)
カラム温度 : 40℃
検出波長 : 280nm
注入量 : 100μL
残存率(%)=光照射後のサンプルのHPLCにおける有効成分のピーク面積値/光照射前のサンプルのHPLCにおける有効成分のピーク面積値×100 ・・・式1

マンニトール88.5部を流動層造粒機に投入し、ナルフラフィン塩酸塩0.0025部の水溶液を噴霧して造粒顆粒を製造した。次に、造粒顆粒に没食子酸n-プロピル0.001部のエタノール溶液を添加して乳鉢で攪拌した。熱風乾燥機に40℃、6時間乾燥した後、低置換度ヒドロキシプロピルセルロース10部及びステアリン酸マグネシウム1部を混合して、打錠用顆粒を得た。打錠用顆粒を、打錠機を用いて99.5mgの7mmφ錠剤とした。
比較例4の造粒顆粒に、没食子酸n-プロピル0.001部の代わりに没食子酸n-プロピル0.005部のエタノール溶液を添加したこと以外は比較例4と同様の手順で99.5mgのφ7mm錠剤を得た。
比較例4の造粒顆粒に、没食子酸n-プロピル0.001部の代わりに没食子酸n-プロピル0.01部のエタノール溶液を添加したこと以外は比較例4と同様の手順で99.5mgのφ7mm錠剤を得た。
比較例4の造粒顆粒に、没食子酸n-プロピル0.001部の代わりに没食子酸n-プロピル0.1部のエタノール溶液を添加したこと以外は比較例4と同様の手順で99.6mgのφ7mm錠剤を得た。
比較例4の造粒顆粒に、没食子酸n-プロピル0.001部の代わりに没食子酸n-プロピル1.0部のエタノール溶液を添加したこと以外は比較例4と同様の手順で100.5mgのφ7mm錠剤を得た。
比較例4の造粒顆粒に、没食子酸n-プロピル0.001部の代わりに没食子酸n-プロピル5部のエタノール溶液を添加したこと以外は比較例4と同様の手順で104.5mgのφ7mm錠剤を得た。
比較例4の造粒顆粒に、没食子酸n-プロピル1.0部を添加して混合した後、低置換度ヒドロキシプロピルセルロース10部及びステアリン酸マグネシウム1部を混合して、打錠用顆粒を得た。打錠用顆粒を、打錠機を用いて100.5mgの7mmφ錠剤とした。
マンニトール88.5部を流動層造粒機に投入し、ナルフラフィン塩酸塩0.0025部及び没食子酸n-プロピル0.1部の2種を溶解した30%エタノール水溶液を噴霧して造粒顆粒を製造した。
実施例13で得られた造粒顆粒に、低置換度ヒドロキシプロピルセルロース10部、ステアリン酸マグネシウム1部を混合して、打錠用顆粒を得た。打錠用顆粒を、打錠機を用いて99.5mgの7mmφ錠剤とした。
マンニトール88.5部を流動層造粒機に投入し、ナルフラフィン塩酸塩0.0025部及びチオ硫酸ナトリウム・5水和物0.1部の2種を溶解した水溶液を噴霧して造粒顆粒を製造した。次に、造粒顆粒に没食子酸n-プロピル0.1部のエタノール溶液を添加して乳鉢で攪拌した。熱風乾燥機に40℃、6時間乾燥した後、低置換度ヒドロキシプロピルセルロース10部及びステアリン酸マグネシウム1部を混合して、打錠用顆粒を得た。打錠用顆粒を、打錠機を用いて99.7mgの7mmφ錠剤とした。
実施例15の造粒顆粒に、没食子酸n-プロピル0.1部の代わりに没食子酸n-プロピル0.5部のエタノール溶液を添加したこと以外は実施例15と同様の手順で100.1mgのφ7mm錠剤を得た。
実施例15の造粒顆粒に、没食子酸n-プロピル0.1部の代わりに没食子酸n-プロピル1部のエタノール溶液を添加したこと以外は実施例15と同様の手順で100.6mgのφ7mm錠剤を得た。
マンニトール88.5部を流動層造粒機に投入し、ナルフラフィン塩酸塩0.0025部、チオ硫酸ナトリウム・5水和物0.1部及び没食子酸n-プロピル0.1部の3種を溶解したエタノール30%水溶液を噴霧して造粒顆粒を製造した。
実施例18の造粒顆粒に、低置換度ヒドロキシプロピルセルロース10部及びステアリン酸マグネシウム1部を混合して、打錠用顆粒を得た。打錠用顆粒を、打錠機を用いて99.7mgの7mmφ錠剤とした。
実施例13及び18の造粒顆粒を更に30分間の通気乾燥した粉末中の有効成分に対する主要分解物Aの生成比率を、以下のHPLC分析により算出した。
<前処理条件>
粉末にメタノールを加えて、攪拌した後、遠心分離し上澄みを採取した。採取した溶液をロータリーエバポレーターで濃縮乾固した後、移動層Aで再溶解してHPLCサンプルとした。
<HPLC条件>
移動相A : 50mMリン酸二水素ナトリウム溶液/アセトニトリル=95/5(v/v)
移動相B : 50mMリン酸二水素ナトリウム溶液/アセトニトリル=60/40(v/v)
カラム : YMC-Pack ODS-AM(YMC製、サイズ:4. ×250mm)
カラム温度 : 40℃
検出波長 : 280nm
流速 : 1.0ml/min
生成比率(%)=サンプルのHPLCにおける主要分解物Aのピーク面積値/サンプルの有効成分のピーク面積値×100 ・・・式2

国際公開第99/002158号記載の顆粒剤を以下の通り製造した。乳糖(Pharmatose(登録商標)200M)68.9部及び結晶セルロース(旭化成ケミカルズ株式会社製、セオラス(登録商標)PH-101)31部を乳鉢にとり、ナルフラフィン塩酸塩0.1部の水溶液を添加して攪拌した。その後、40℃、12時間乾燥し、造粒顆粒とした。
国際公開第99/002158号記載の顆粒剤を以下の通り製造した。乳糖(Pharmatose(登録商標)200M)68.8部及び結晶セルロース(旭化成ケミカルズ株式会社製、セオラス(登録商標)PH-101)31部を乳鉢にとり、ナルフラフィン塩酸塩0.1部及びチオ硫酸ナトリウム0.1部の2種を溶解した水溶液を添加して攪拌した。その後、40℃、12時間乾燥し、造粒顆粒とした。
マンニトール99.95部を乳鉢にとり、ナルフラフィン塩酸塩0.05部の水溶液を添加して攪拌した。その後、40℃、12時間乾燥し、造粒顆粒とした。
マンニトール99.99部を乳鉢にとり、ナルフラフィン塩酸塩0.01部の水溶液を添加して攪拌した。その後、40℃、12時間乾燥し、造粒顆粒とした。
マンニトール99.89部を乳鉢にとり、ナルフラフィン塩酸塩0.01部の水溶液を添加して攪拌した後、没食子酸n-プロピル0.1部のエタノール溶液を添加して攪拌した。その後、40℃、12時間乾燥し、造粒顆粒とした。
マンニトール88.5部を流動層造粒機に投入し、ナルフラフィン塩酸塩0.0025部、チオ硫酸ナトリウム0.1部及び没食子酸n-プロピル0.1部の3種を溶解した30%エタノール水溶液を噴霧して造粒顆粒を製造した。次に、造粒顆粒44.35部にマンニトール455.65部を添加して、V型混合機を用いて混合した。
比較例5~8並びに実施例20及び21の粉末をガラスシャーレ上に薄く広げ、白色蛍光灯(照度2000lux)環境下、総照度として4.8万lux・hrの粉末を取り出し、以下のHPLC分析により、光照射後の有効成分の残存率を式1と同様の計算式で算出した。
<前処理条件>
粉末に蒸留水を加えて懸濁又は溶解した後、遠心分離し上澄みをHPLCサンプルとした。
<HPLC条件>
試験例1と同一のHPLC条件で試験した。

特開昭58-57322号記載の光吸収剤であるバニリンを用いた。マンニトール99.8975部を乳鉢にとり、ナルフラフィン塩酸塩0.0025部の水溶液を添加して攪拌した後、バニリン(和光純薬工業株式会社製、和光特級)0.1部のエタノール溶液を添加して攪拌した。その後、40℃、12時間乾燥し、造粒顆粒とした。
特開昭58-57322号記載の光吸収剤であるp-アミノ安息香酸(和光純薬工業株式会社製、和光特級)を用いた。マンニトール99.8975部を乳鉢にとり、ナルフラフィン塩酸塩0.0025部の水溶液を添加して攪拌した後、p-アミノ安息香酸0.1部のエタノール溶液を添加して攪拌した。その後、40℃、12時間乾燥し、造粒顆粒とした。
マンニトール99.7975部を乳鉢にとり、ナルフラフィン塩酸塩0.0025部の水溶液を添加して攪拌した。三二酸化鉄(HUNTSMAN社製、SICOVIT RED30E172)0.1部を添加し、次に没食子酸n-プロピル0.1部のエタノール溶液を添加して攪拌した。その後、40℃、12時間乾燥し、造粒顆粒とした。
実施例21の造粒顆粒100部を乳鉢にとり、三二酸化鉄0.01部及びエタノール水溶液を添加して攪拌した。その後、12時間乾燥し、造粒顆粒とした。
Claims (6)
- 下記一般式(I)で示される4,5-エポキシモルヒナン誘導体又はその薬理学的に許容される酸付加塩からなる有効成分と、没食子酸n-プロピル、亜硫酸水素ナトリウム、ジブチルヒドロキシトルエン、ブチルヒドロキシアニソール、トコフェロール及びD-イソアスコルビン酸からなる群から選択される1種以上の安定化剤と、を含有し、
前記有効成分の重量は、固形製剤の重量あたり0.00001~0.01重量%であり、
前記安定化剤の重量は、固形製剤の重量あたり0.005~5重量%である、固形製剤。

- 前記安定化剤は、没食子酸n-プロピルである、請求項1記載の固形製剤。
- チオ硫酸ナトリウムを含有する、請求項1又は2記載の固形製剤。
- 黄色三二酸化鉄、赤色三二酸化鉄又は黒酸化鉄を含有する、請求項1~3のいずれか一項記載の固形製剤。
- 糖質を含有する、請求項1~4のいずれか一項記載の固形製剤。
- 錠剤、顆粒剤、細粒剤、硬カプセル剤、ドライシロップ剤、散剤、丸剤及びトローチ剤からなる群から選択される剤形である、請求項1~5のいずれか一項記載の固形製剤。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020187031615A KR102427007B1 (ko) | 2016-07-29 | 2017-07-28 | 광안정성이 향상된 고형 제제 |
| CN201780046424.3A CN109562109A (zh) | 2016-07-29 | 2017-07-28 | 光稳定性提高的固体制剂 |
| CA3030392A CA3030392C (en) | 2016-07-29 | 2017-07-28 | Solid preparation having improved light stability |
| JP2017540668A JP7051046B2 (ja) | 2016-07-29 | 2017-07-28 | 光安定性の向上した固形製剤 |
| BR112018075619-9A BR112018075619A2 (pt) | 2016-07-29 | 2017-07-28 | preparação sólida. |
| US16/320,429 US11185509B2 (en) | 2016-07-29 | 2017-07-28 | Solid preparation having improved light stability |
| EP17834520.3A EP3492083A4 (en) | 2016-07-29 | 2017-07-28 | SOLID PREPARATION WITH IMPROVED LIGHT RESISTANCE |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016150238 | 2016-07-29 | ||
| JP2016-150238 | 2016-07-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018021518A1 true WO2018021518A1 (ja) | 2018-02-01 |
Family
ID=61017109
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/027391 WO2018021518A1 (ja) | 2016-07-29 | 2017-07-28 | 光安定性の向上した固形製剤 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US11185509B2 (ja) |
| EP (1) | EP3492083A4 (ja) |
| JP (1) | JP7051046B2 (ja) |
| KR (1) | KR102427007B1 (ja) |
| CN (1) | CN109562109A (ja) |
| BR (1) | BR112018075619A2 (ja) |
| CA (1) | CA3030392C (ja) |
| TW (1) | TWI740994B (ja) |
| WO (1) | WO2018021518A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020143009A (ja) * | 2019-03-06 | 2020-09-10 | 日医工株式会社 | ラメルテオン含有錠剤 |
| EP4062973A4 (en) * | 2019-11-20 | 2023-11-29 | Shionogi & Co., Ltd | SOLID FORMULATION CONTAINING 6,7-UNSATURATED-7-CARBAMOYL MORPHINAND DERIVATIVE |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005002123A (ja) * | 1997-07-11 | 2005-01-06 | Toray Ind Inc | 4,5−エポキシモルヒナン誘導体を含有する安定な医薬品組成物 |
| WO2016195057A1 (ja) * | 2015-06-04 | 2016-12-08 | 東海カプセル株式会社 | ソフトカプセル剤 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3136282A1 (de) | 1981-09-12 | 1983-03-24 | Hoechst Ag | "verfahren zur stabilisierung photoinstabiler arzneistoffe sowie stabilisierte arzneizubereitungen" |
| KR100188318B1 (ko) * | 1993-04-28 | 1999-06-01 | 고오야 마사시 | 안정화된 주사제 및 주사제의 안정화법 |
| JP4214128B2 (ja) | 2005-04-27 | 2009-01-28 | 大日本住友製薬株式会社 | 光安定性の向上した組成物 |
| WO2008133330A1 (ja) | 2007-04-26 | 2008-11-06 | Toray Industries, Inc. | 4,5-エポキシモルヒナン誘導体を含有する安定な固形製剤 |
| US8829019B2 (en) * | 2008-10-24 | 2014-09-09 | Toray Industries, Inc. | Stable tablet containing 4,5-epoxymorphinan derivative |
| TWI455733B (zh) * | 2009-03-30 | 2014-10-11 | Toray Industries | 口腔內崩壞性被覆錠劑 |
| MY172534A (en) | 2010-12-30 | 2019-11-29 | Ardea Biosciences Inc | Polymorphic forms of 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4h-1,2,4-triazol-3-ylthio)acetic acid and uses thereof |
| US10088621B2 (en) * | 2013-12-17 | 2018-10-02 | Samsung Display Co. Ltd. | Light guide panel, backlight unit, and liquid crystal display |
| JP2015172043A (ja) * | 2014-02-20 | 2015-10-01 | 富士カプセル株式会社 | ナルフラフィン塩酸塩を含有するカプセル製剤 |
| JP6247118B2 (ja) * | 2014-03-05 | 2017-12-13 | 東海カプセル株式会社 | カプセル充填組成物 |
| JPWO2016052617A1 (ja) | 2014-09-30 | 2017-09-21 | 国立大学法人 筑波大学 | ナルフラフィン含有局所適用製剤 |
-
2017
- 2017-07-28 KR KR1020187031615A patent/KR102427007B1/ko active IP Right Grant
- 2017-07-28 CN CN201780046424.3A patent/CN109562109A/zh active Pending
- 2017-07-28 EP EP17834520.3A patent/EP3492083A4/en active Pending
- 2017-07-28 JP JP2017540668A patent/JP7051046B2/ja active Active
- 2017-07-28 TW TW106125445A patent/TWI740994B/zh active
- 2017-07-28 CA CA3030392A patent/CA3030392C/en active Active
- 2017-07-28 BR BR112018075619-9A patent/BR112018075619A2/pt not_active Application Discontinuation
- 2017-07-28 US US16/320,429 patent/US11185509B2/en active Active
- 2017-07-28 WO PCT/JP2017/027391 patent/WO2018021518A1/ja unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005002123A (ja) * | 1997-07-11 | 2005-01-06 | Toray Ind Inc | 4,5−エポキシモルヒナン誘導体を含有する安定な医薬品組成物 |
| WO2016195057A1 (ja) * | 2015-06-04 | 2016-12-08 | 東海カプセル株式会社 | ソフトカプセル剤 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3492083A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020143009A (ja) * | 2019-03-06 | 2020-09-10 | 日医工株式会社 | ラメルテオン含有錠剤 |
| EP4062973A4 (en) * | 2019-11-20 | 2023-11-29 | Shionogi & Co., Ltd | SOLID FORMULATION CONTAINING 6,7-UNSATURATED-7-CARBAMOYL MORPHINAND DERIVATIVE |
Also Published As
| Publication number | Publication date |
|---|---|
| US11185509B2 (en) | 2021-11-30 |
| EP3492083A1 (en) | 2019-06-05 |
| BR112018075619A2 (pt) | 2019-04-09 |
| TW201808290A (zh) | 2018-03-16 |
| JP7051046B2 (ja) | 2022-04-11 |
| JPWO2018021518A1 (ja) | 2019-05-23 |
| US20190262270A1 (en) | 2019-08-29 |
| CN109562109A (zh) | 2019-04-02 |
| KR102427007B1 (ko) | 2022-07-29 |
| EP3492083A4 (en) | 2020-03-11 |
| TWI740994B (zh) | 2021-10-01 |
| CA3030392A1 (en) | 2018-02-01 |
| CA3030392C (en) | 2023-12-12 |
| KR20190034140A (ko) | 2019-04-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5578199B2 (ja) | 4,5−エポキシモルヒナン誘導体を含有する安定な固形製剤 | |
| JP5971272B2 (ja) | 4,5−エポキシモルヒナン誘導体を含有する安定な錠剤 | |
| EP2815752B1 (en) | Oral pharmaceutical composition | |
| JP2024033011A (ja) | 医薬組成物 | |
| KR102435064B1 (ko) | 엔테카비르를 유효성분으로 포함하는 약학 제제 및 이의 제조방법 | |
| JP2012149056A (ja) | 新規な安定化固形製剤 | |
| JP2005314413A (ja) | 経口投与用医薬組成物 | |
| JP7051046B2 (ja) | 光安定性の向上した固形製剤 | |
| JP5465867B2 (ja) | 安定なエグアレンナトリウム固形製剤 | |
| KR102206104B1 (ko) | 실로도신을 포함하는 과립물, 및 이를 포함하는 약학적 조성물 및 제형 | |
| JP2015054851A (ja) | 被覆経口固形製剤 | |
| KR20240112968A (ko) | 의약 조성물 | |
| WO2020111089A1 (ja) | 医薬組成物 | |
| JP6131379B1 (ja) | 4,5−エポキシモルヒナン誘導体含有製剤 | |
| WO2018181920A1 (ja) | ナルフラフィンを含有する錠剤化された医薬組成物 | |
| KR20210052495A (ko) | 경구 투여용 의약 조성물 | |
| EP3679926A1 (en) | A method of manufacturing a pharmaceutical composition comprising nefopam and acetaminophen, and the pharmaceutical composition obtained thereby | |
| JP2020045323A (ja) | コハク酸ソリフェナシン含有製剤 | |
| KR100514590B1 (ko) | 프라바스타틴나트륨 및 안정화제로서의 코폴리비돈을함유하는 약제 조성물 | |
| JPWO2014007065A1 (ja) | 固形医薬錠剤およびその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2017540668 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20187031615 Country of ref document: KR Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17834520 Country of ref document: EP Kind code of ref document: A1 |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112018075619 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 3030392 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2017834520 Country of ref document: EP Effective date: 20190228 |
|
| ENP | Entry into the national phase |
Ref document number: 112018075619 Country of ref document: BR Kind code of ref document: A2 Effective date: 20181210 |