WO2017135472A1 - スルホンアミド誘導体及びそれを含有する医薬組成物 - Google Patents
スルホンアミド誘導体及びそれを含有する医薬組成物 Download PDFInfo
- Publication number
- WO2017135472A1 WO2017135472A1 PCT/JP2017/004278 JP2017004278W WO2017135472A1 WO 2017135472 A1 WO2017135472 A1 WO 2017135472A1 JP 2017004278 W JP2017004278 W JP 2017004278W WO 2017135472 A1 WO2017135472 A1 WO 2017135472A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- substituent
- lower alkyl
- alkyl group
- ring
- Prior art date
Links
- 0 CN(C=CC(N1*)=O)C1=O Chemical compound CN(C=CC(N1*)=O)C1=O 0.000 description 2
- AVEQEWZHGGSASH-AWEZNQCLSA-N CC(C(N1c2ccc(C[C@@H](C(OC)=O)N)cc2)=O)=C(C)N(C)C1=O Chemical compound CC(C(N1c2ccc(C[C@@H](C(OC)=O)N)cc2)=O)=C(C)N(C)C1=O AVEQEWZHGGSASH-AWEZNQCLSA-N 0.000 description 1
- YVUPMNFINFXCJV-UHFFFAOYSA-N CC(C)(C)C#CC(Nc(cc1)ccc1S(Nc(cc(c(C(O)=O)c1)F)c1F)(=O)=O)=O Chemical compound CC(C)(C)C#CC(Nc(cc1)ccc1S(Nc(cc(c(C(O)=O)c1)F)c1F)(=O)=O)=O YVUPMNFINFXCJV-UHFFFAOYSA-N 0.000 description 1
- BAJHTRMHUUUDML-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCC1C(Nc(cc1)ccc1S(Nc(c(F)c1)cc(F)c1C(O)=O)(=O)=O)=O)=O Chemical compound CC(C)(C)OC(N(CC1)CCC1C(Nc(cc1)ccc1S(Nc(c(F)c1)cc(F)c1C(O)=O)(=O)=O)=O)=O BAJHTRMHUUUDML-UHFFFAOYSA-N 0.000 description 1
- DUIQJFZTUPHOSG-KRWDZBQOSA-N CC(C)OC([C@H](Cc(cc1)ccc1N(C(C(CCN(C)C1)=C1N1C)=O)C1=O)N)=O Chemical compound CC(C)OC([C@H](Cc(cc1)ccc1N(C(C(CCN(C)C1)=C1N1C)=O)C1=O)N)=O DUIQJFZTUPHOSG-KRWDZBQOSA-N 0.000 description 1
- MLZOBHORMAMIAO-JTQLQIEISA-N CN(C=CC(N1c2ncc(C[C@@H](C(OC)=O)N)cc2)=O)C1=O Chemical compound CN(C=CC(N1c2ncc(C[C@@H](C(OC)=O)N)cc2)=O)C1=O MLZOBHORMAMIAO-JTQLQIEISA-N 0.000 description 1
- ZJHVSXKODLBIEU-YTTGMZPUSA-N CN(c(cncc1)c1C(N1c2cnc(C[C@@H](C(O)=O)NC(c(cc(c(NS(c(cc3)ccc3-c3cnc(C4CCNCC4)nc3)(=O)=O)c3)F)c3F)=O)cc2)=O)C1=O Chemical compound CN(c(cncc1)c1C(N1c2cnc(C[C@@H](C(O)=O)NC(c(cc(c(NS(c(cc3)ccc3-c3cnc(C4CCNCC4)nc3)(=O)=O)c3)F)c3F)=O)cc2)=O)C1=O ZJHVSXKODLBIEU-YTTGMZPUSA-N 0.000 description 1
- VCKWVRVGTRNTLC-UHFFFAOYSA-N COC(c(c(F)c1)cc(F)c1NS(c1cc([nH]cc2)c2cc1)(=O)=O)=O Chemical compound COC(c(c(F)c1)cc(F)c1NS(c1cc([nH]cc2)c2cc1)(=O)=O)=O VCKWVRVGTRNTLC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/63—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/52—Two oxygen atoms
- C07D239/54—Two oxygen atoms as doubly bound oxygen atoms or as unsubstituted hydroxy radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/95—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in positions 2 and 4
- C07D239/96—Two oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/04—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/052—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being six-membered
Definitions
- the present invention relates to a sulfonamide derivative or a pharmaceutically acceptable salt thereof and a pharmaceutical composition containing these compounds as active ingredients.
- the present invention relates to a compound that can be used as a therapeutic or prophylactic agent for inflammatory diseases in which an ⁇ 4 integrin-dependent adhesion process is involved in the pathology.
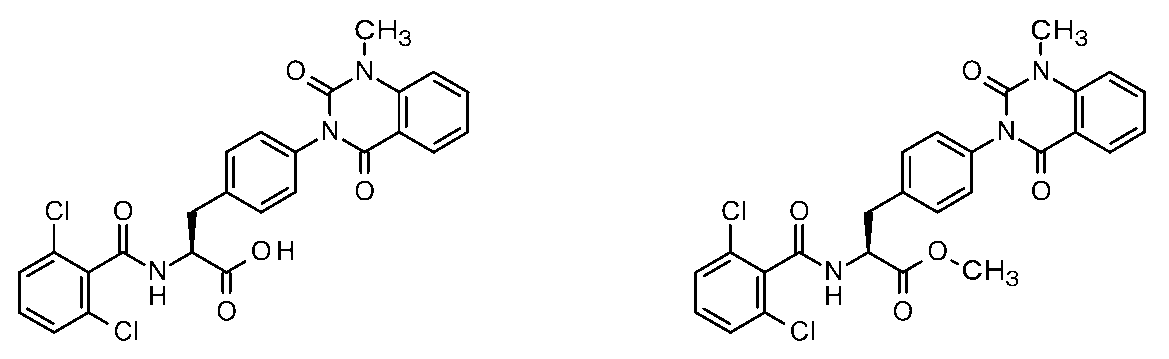
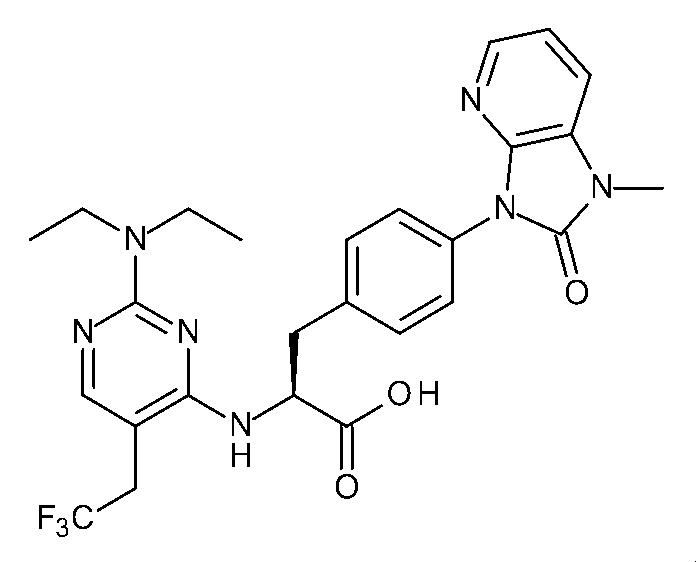
- Patent Document 1 discloses a phenylalanine derivative represented by the following formula or a pharmaceutically acceptable salt thereof, and a typical compound thereof has the following chemical structure.
- Patent Document 1 shows the results of VCAM inhibitory activity (VCAM-1 / ⁇ 4 ⁇ 1 binding assay) and (VCAM-1 / ⁇ 4 ⁇ 7 binding assay).
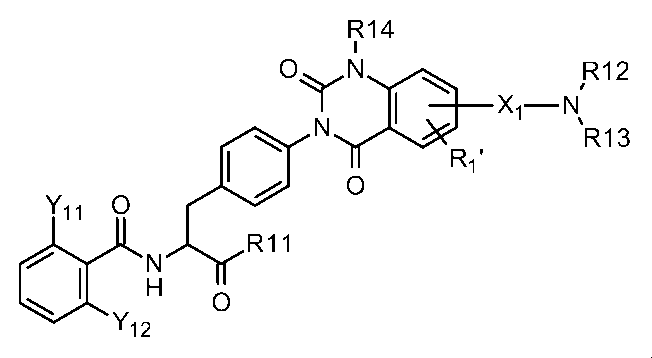
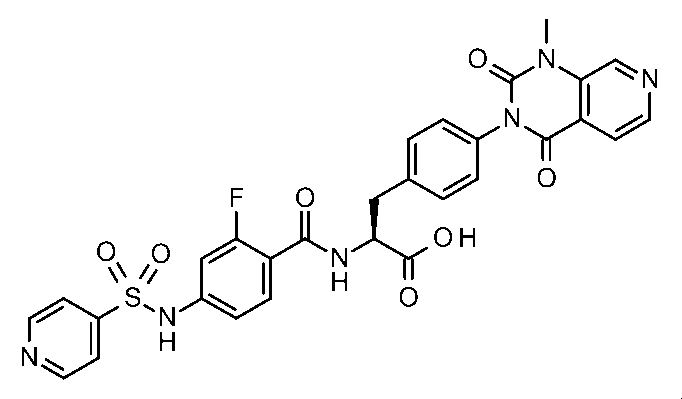
- Patent Document 2 discloses a phenylalanine derivative represented by the following formula having a terminal R12 (R13) N—X1-group or a pharmaceutically acceptable salt thereof. This compound has been shown to have a higher VCAM-1 / ⁇ 4 ⁇ 1 integrin inhibitory activity in the presence of serum than the compound of Example 1 of Patent Document 1.
- Patent Document 3 also discloses a compound having an ⁇ 4 integrin inhibitory action.
- Patent Document 4 (WO2005 / 077915) describes a phenylalanine derivative having an ⁇ 4 integrin inhibitory action as represented by the following formula, but a 2,6-dichlorobenzoyl group or amino acid residue is present at the N-terminus of phenylalanine. A group is bound.
- Patent Document 5 Japanese Patent Application Laid-Open No. 2003-321358 describes a phenylalanine derivative having an ⁇ 4 integrin inhibitory activity represented by the following formula, and the phenylalanine has an N-terminal having a 2,6-dichlorobenzoyl group and the like. Are joined.
- Patent Document 6 (WO01 / 56994) describes a phenylalanine derivative having an ⁇ 4 integrin inhibitory action represented by the following formula, and proline and the like are bonded to the N-terminus of phenylalanine.
- Patent Document 7 (WO 2006/127584) describes a phenylalanine derivative having an ⁇ 4 integrin inhibitory action represented by the following formula, and a pyrimidine ring or the like is directly bonded to the N-terminus of phenylalanine.
- Patent Document 8 (WO01 / 42215) describes a phenylalanine derivative having an ⁇ 4 integrin inhibitory activity represented by the following formula, and the phenylalanine has an N-terminus such as a 2-chloro-6-methylbenzoyl group. Are joined.
- Patent Document 9 (WO2013 / 161904) describes a phenylalanine derivative having an ⁇ 4 ⁇ 7 integrin inhibitory action represented by the following formula. This document shows the results of the VCAM-1 / ⁇ 4 ⁇ 1 integrin binding inhibitory activity evaluation of a specific phenylalanine derivative and the MAdCAM-1 / ⁇ 4 ⁇ 7 integrin binding inhibitory activity evaluation test in the presence of serum. Has a low effect and is described as having a high effect on ⁇ 4 ⁇ 7 integrin.
- Patent Document 10 (WO2015 / 064580) describes a phenylalanine derivative having an ⁇ 4 ⁇ 7 integrin inhibitory action represented by the following formula. This document also shows the results of VCAM-1 / ⁇ 4 ⁇ 1 integrin binding inhibitory activity evaluation of specific phenylalanine derivatives and MAdCAM-1 / ⁇ 4 ⁇ 7 integrin binding inhibitory activity evaluation test in the presence of serum. Has a low effect and is described as having a high effect on ⁇ 4 ⁇ 7 integrin.
- An object of the present invention is to provide a novel compound having an unknown chemical structural formula and having an ⁇ 4 integrin inhibitory action.
- an object of the present invention is to provide a novel compound having a selective ⁇ 4 integrin inhibitory action that has a low effect on ⁇ 4 ⁇ 1 and a high effect on ⁇ 4 ⁇ 7.
- Another object of the present invention is to provide a compound having an ⁇ 4 integrin inhibitory action that may be administered orally.
- Another object of the present invention is to provide a compound having safe ⁇ 4 integrin inhibitory activity.
- Another object of the present invention is to provide a compound having a persistent ⁇ 4 integrin inhibitory activity.
- Another object of the present invention is to provide a novel compound having an ⁇ 4 integrin inhibitory action in human whole blood.
- Another object of the present invention is to provide a pharmaceutical composition containing the above novel compound and a pharmaceutically acceptable carrier.
- Another object of the present invention is to provide a medicament containing the novel compound.
- Another object of the present invention is to provide a therapeutic or prophylactic agent for inflammatory diseases in which an ⁇ 4 ⁇ 7 integrin-dependent adhesion process is involved in the disease state.
- Another object of the present invention is to provide an ⁇ 4 integrin inhibitor.
- the inventors of the present application examined ⁇ 4 integrin inhibitory activity for compounds having various structures.
- a sulfonamide derivative having a specific chemical structure having a heterocyclic group having an acylamino group as a substituent or a sulfonamide group to which a phenyl group is bonded, or a pharmaceutically acceptable salt thereof, is expressed in ⁇ 4 ⁇ 7 integrin in human whole blood. It has been found that the above-mentioned problems can be solved by using these compounds having inhibitory activity.
- R 1 and R 2 are each independently a hydrogen atom, a halogen atom, a lower alkyl group, a lower alkenyl group, a lower alkoxy group, a lower alkoxy lower alkyl group, a halogeno lower alkyl group, a hydroxy group, or a hydroxy lower alkyl group.
- R 1 and R 2 are combined to form a benzene ring which may have a substituent, an alicyclic hydrocarbon having 4 to 7 carbon atoms which may have a substituent, and a substituent.
- a heteroaryl ring or a hetero ring that may have a substituent may be formed,
- R 3 represents a lower alkyl group, e, f, g, and h each independently represent C—H or a nitrogen atom;
- B represents a hydroxy group, an alkoxy group having 1 to 10 carbon atoms, an —O-heterocyclic group, a cilexetiloxy group, or a medoxomiloxy group;
- D represents a benzene ring or a heteroaryl ring which may have a substituent,
- R 4 represents a hydrogen atom or a lower alkyl group,
- R 5 has a lower alkyl group which may have a substituent, a lower alkenyl group which may have
- R 1 and R 2 each independently represent a hydrogen atom, a halogen atom, a lower alkyl group, lower alkenyl group, a lower alkoxy group, a lower alkoxy-lower alkyl group, hydroxy group, or a hydroxy lower alkyl group
- R 1 And R 2 are bonded to each other to form a benzene ring which may have a substituent, an alicyclic hydrocarbon having 4 to 7 carbon atoms which may have a substituent, or a heteroaryl ring which may have a substituent Or may form a heterocyclic ring which may have a substituent
- R 5 has a lower alkyl group which may have a substituent, a lower alkenyl group which may have a substituent, a lower alkylamino group, a phenyl group which may have a substituent, and a substituent.
- R 1 and R 2 are bonded to each other to have a benzene ring which may have a substituent, an alicyclic hydrocarbon having 4 to 7 carbon atoms which may have a substituent, and a substituent.
- a heteroaryl ring or a hetero ring that may have a substituent is a lower alkyl group, a lower alkoxy group, a hydroxy lower alkyl group, an amino group, a lower alkylamino group, and
- R 1 and R 2 each independently represent a hydrogen atom, a lower alkyl group, or a hydroxy lower alkyl group, and R 1 and R 2 may be bonded to each other to have a substituent.
- R 1 and R 2 each independently represent a hydrogen atom, a lower alkyl group, a halogeno lower alkyl group, or a hydroxy lower alkyl group, and R 1 and R 2 may be bonded to each other to have a substituent.
- a good alicyclic hydrocarbon having 4 to 7 carbon atoms, a heteroaryl ring which may have a substituent, or a heterocycle which may have a substituent may be formed.
- R 1 and R 2 may be bonded to each other to have a pyridine which may have a substituent, cyclohexene which may have a substituent, dihydropyran which may have a substituent, A good tetrahydropyridine or an imidazole which may have a substituent is formed, and the substituent is a lower alkyl group, a lower alkoxy group, a hydroxy lower alkyl group, an amino group, a lower alkylamino group, and a lower alkylamino group.
- D represents a benzene ring which may have a substituent, a pyridine ring which may have a substituent, or a thiophene ring which may have a substituent, and the substituent is a halogen atom Or a pharmaceutically acceptable salt thereof.
- D represents a benzene ring which may have a substituent, a pyridine ring which may have a substituent, or a thiophene ring which may have a substituent, and the substituent is a halogen atom.
- R 5 has a substituent
- the substituent is selected from a halogen atom, a cyano group, a hydroxy group, a lower alkyl group, a lower alkoxy group, a trifluoromethyl group, a phenyl group, and a heterocyclic group
- R 4 and R 5 combine to form an optionally substituted heterocyclic ring, and the substituent is selected from a lower alkyl group, a lower alkoxy group, a hydroxy group, and a heterocyclic group
- R 5 represents an optionally substituted lower alkyl group, lower alkylamino group, or optionally substituted heterocyclic group, and the substituent is a halogen atom or a cyano group.
- R 1 and R 2 each independently represent a hydrogen atom, a lower alkyl group, a lower alkoxy group, or a hydroxy lower alkyl group, and R 1 and R 2 are bonded and substituted with a lower alkyl group
- D represents a benzene ring optionally substituted with a halogen atom, or a heteroaryl ring selected from the following formulae, (Wherein, a represents the bonding position with S and b represents the bonding position with N)
- R 4 represents a hydrogen atom
- a novel compound having a chemical structural formula that has not been known so far and having an ⁇ 4 integrin inhibitory action is provided.
- a novel compound having a selective ⁇ 4 integrin inhibitory action that has a low effect on ⁇ 4 ⁇ 1 and a high effect on ⁇ 4 ⁇ 7.
- a compound having an ⁇ 4 integrin inhibitory action that may be orally administered.
- the present invention also provides a compound having safe ⁇ 4 integrin inhibitory activity.
- a compound having a persistent ⁇ 4 integrin inhibitory activity is also provided.
- a novel compound having an ⁇ 4 integrin inhibitory action in human blood is also provided.
- a pharmaceutical composition comprising the novel compound and a pharmaceutically acceptable carrier.
- the pharmaceutical containing the said novel compound is also provided.
- the present invention also provides a therapeutic or prophylactic agent for inflammatory diseases in which an ⁇ 4 ⁇ 7 integrin-dependent adhesion process is involved in the disease state.
- an ⁇ 4 integrin inhibitor is also provided.
- substituent means “substituted or unsubstituted”.
- position and number of substituents are arbitrary and are not particularly limited. When substituted with two or more substituents, these substituents may be the same or different.
- substituents include a halogen atom, a nitro group, a cyano group, a hydroxyl group, a lower alkyl group, a lower alkenyl group, a lower alkynyl group, a lower alkoxy group, a lower alkylthio group, a hydroxy lower alkyl group, a hydroxy lower alkenyl group, and a hydroxy lower group.
- the term “lower” means a group having 1 to 6 carbon atoms
- the “lower alkyl group” means a linear, branched or cyclic alkyl group having 1 to 6 carbon atoms.
- the “lower alkenyl group” refers to a linear or branched alkenyl group having 2 to 6 carbon atoms including each isomer. Examples thereof include a vinyl group, an allyl group, a propenyl group, a butenyl group, a pentenyl group, and a hexenyl group, and a vinyl group, an allyl group, and a propenyl group are preferable.
- the “lower alkynyl group” refers to a linear or branched alkynyl group having 2 to 6 carbon atoms including each isomer.
- an ethynyl group, a propynyl group, a butynyl group, a pentynyl group, a hexynyl group and the like can be mentioned, and an ethynyl group and a propynyl group are preferable.
- halogen atom examples include a fluorine atom, a chlorine atom, a bromine atom and an iodine atom, and a fluorine atom and a chlorine atom are preferable.
- the “lower alkoxy group” refers to an alkoxy group having a linear, branched or cyclic alkyl group having 1 to 6 carbon atoms.
- methoxy group, ethoxy group, n-propoxy group, n-butoxy group, n-pentyloxy group, n-hexyloxy group isopropoxy group, isobutoxy group, sec-butoxy group, tert-butoxy group, cyclopropyloxy Group, a cyclobutoxy group, a cyclopentyloxy group, and a cyclohexyloxy group, and a methoxy group, an ethoxy group, and an n-propoxy group are preferable.
- the “lower alkoxymethyl group” refers to a methyl group that is mono-substituted or further substituted with the above-mentioned “lower alkoxy group”. Examples thereof include a methoxymethyl group, an ethoxymethyl group, an isopropoxymethyl group, a tert-butoxymethyl group, and the like, and a methoxymethyl group and an ethoxymethyl group are preferable.
- the “halogeno lower alkyl group” refers to a lower alkyl group that is mono-substituted or further substituted with the aforementioned “halogen atom”.
- Examples include a trifluoromethyl group, a difluoromethyl group, a monotrifluoromethyl group, a trichloromethyl group, a dichloromethyl group, a monochloromethyl group, a trifluoroethyl group, a pentafluoroethyl group, and the like, preferably a trifluoromethyl group It is.
- hydroxy lower alkyl group refers to a lower alkyl group substituted with a hydroxyl group, and examples thereof include a hydroxymethyl group and a hydroxyethyl group, and a hydroxymethyl group is preferred.
- lower alkylamino group refers to an amino group that is mono-substituted or more than the above-mentioned “lower alkyl group”.
- Examples include methylamino group, ethylamino group, propylamino group, tert-butylamino group, isopropylamino group, dimethylamino group, diethylamino group, dipropylamino group, diisopropylamino group, and methylethylamino group.
- lower alkylamino lower alkyl group refers to a lower alkyl group substituted with the above-mentioned “lower alkyl group” mono- or di-substituted amino group.
- Examples include a methylaminomethyl group, an ethylaminomethyl group, a propylaminomethyl group, an isopropylaminomethyl group, a methylaminoethyl group, an ethylaminoethyl group, a dimethylaminomethyl group, and a methylethylaminomethyl group, preferably methyl An aminomethyl group, an ethylaminomethyl group, and a methylaminoethyl group;
- the “alicyclic hydrocarbon” refers to a cyclic structure composed of carbon atoms and hydrogen atoms, and includes cycloalkanes that are all formed by single bonds and cycloalkenes that may contain double bonds.
- cyclobutane, cyclopentane, cyclohexane, cycloheptane, cyclohexene and the like can be mentioned, and cyclohexane and cyclohexene are preferable.
- heteroaryl ring refers to a 4- to 10-membered aromatic ring containing 1 to 4 heteroatoms selected from oxygen, sulfur and nitrogen atoms as ring atoms.
- Heterocycle refers to a 4- to 10-membered monocyclic to bicyclic heterocycle containing 1 to 4 heteroatoms selected from an oxygen atom, a sulfur atom and a nitrogen atom as ring atoms. Any carbon atom which is a ring atom may be substituted with an oxo group, and a sulfur atom or a nitrogen atom may be oxidized to form an oxide. Further, it may be condensed with a benzene ring.
- heteroaryl group refers to a 4- to 10-membered aromatic ring group containing 1 to 4 heteroatoms selected from an oxygen atom, a sulfur atom and a nitrogen atom as ring atoms.
- Pyridyl group, pyridazinyl group, pyrimidinyl group, pyrazinyl group, furyl group, thienyl group, pyrrolyl group isoxazolyl group, oxazolyl group, isothiazolyl group, thiazolyl group, pyrazolyl group, imidazolyl group, oxadiazolyl group, thiadiazolyl group, triazoyl group, tetrazolyl group , Benzofuranyl group, benzothienyl group, indolyl group, isoindolyl group, benzoxazolyl group, benzisoxazolyl group, benzthiazolyl group, benzisothiazolyl group
- Heterocyclic group refers to a 4- to 10-membered monocyclic to bicyclic heterocyclic group containing 1 to 4 heteroatoms selected from oxygen, sulfur and nitrogen atoms as ring atoms. . Any carbon atom which is a ring atom may be substituted with an oxo group, and a sulfur atom or a nitrogen atom may be oxidized to form an oxide. Further, it may be condensed with a benzene ring.
- the sulfonamide derivative represented by the general formula (I) or a pharmaceutically acceptable salt thereof is preferably the following:
- R 1 and R 2 are each independently preferably a hydrogen atom, a lower alkyl group, a halogeno lower alkyl group or a hydroxy lower alkyl group, more preferably a hydrogen atom or a lower alkyl group.
- a hydrogen atom and a methyl group are particularly preferable.
- R 1 and R 2 are preferably each independently a hydrogen atom, a lower alkyl group, or a hydroxy lower alkyl group.
- the ring formed by combining R 1 and R 2 is preferably pyridine, cyclohexene, dihydropyran, and tetrahydropyridine, more preferably cyclohexene, dihydropyran, and tetrahydropyridine, and dihydropyran. And tetrahydropyridine are particularly preferred.
- R 3 is preferably a lower alkyl group, more preferably an isopropyl group or a methyl group, and particularly preferably a methyl group.
- B is preferably a hydroxy group or a lower alkoxy group, more preferably a hydroxy group, a methoxy group, an ethoxy group, an isopropoxy group, an isobutyloxy group, or a cyclohexyloxy group, and a hydroxy group, a methoxy group, or an ethoxy group.
- B is more preferably a hydroxy group, a methoxy group, an ethoxy group, an isopropoxy group, or a cyclohexyloxy group, and particularly preferably a hydroxy group, a methoxy group, an ethoxy group, or an isopropoxy group.
- D is preferably a benzene ring which may have a substituent or a heteroaryl ring which may have a substituent, more preferably a benzene ring, a pyridine ring or a thiophene ring. A ring is particularly preferred.
- the substituent of D is preferably a halogen atom, a lower alkyl group, a lower alkoxy group or a hydroxy group, more preferably a fluorine atom.
- D when D represents a benzene ring, the substitution position of the aminosulfonyl group and aminocarbonyl group bonded to D is preferably the para position or the meta position, and the para position is particularly preferable.
- R 4 is preferably a hydrogen atom or a lower alkyl group, more preferably a hydrogen atom or a methyl group, and particularly preferably a hydrogen atom.
- R 5 is a lower alkyl group which may have a substituent, a lower alkynyl group which may have a substituent, a lower alkylamino group or a phenyl group which may have a substituent.
- the substituent of R 5 is preferably a halogen atom, a hydroxy group, a lower alkoxy group, a trifluoromethyl group or a phenyl group, particularly preferably a fluorine atom, a hydroxy group, a methoxy group or a trifluoromethyl group.
- the ring formed by combining R 4 and R 5 is preferably a heterocyclic ring which may have a substituent, and pyrimidone and hydantoin are particularly preferable.
- the substituent of the ring formed by combining R 4 and R 5 is preferably a lower alkyl group or a heterocyclic group, and particularly preferably a methyl group or a tetrahydropyranyl group.
- R 1 and R 2 each independently represents a hydrogen atom, a lower alkyl group, a lower alkoxy group, or a hydroxy lower alkyl group, and R 1 and R 2 are bonded and substituted with a lower alkyl group.
- D represents a benzene ring optionally substituted with a halogen atom, or a heteroaryl ring selected from the following formulae, (Wherein, a represents the bonding position with S and b represents the bonding position with N)
- R 4 represents a hydrogen atom
- R 5 may have a substituent selected from the group consisting of a lower alkoxy group, a halogen atom, a hydroxy group and an aryl group, an alkyl group having 2 to 5 carbon atoms, a heteroaryl group, or O as a ring atom
- D is a benzene ring optionally substituted with a halogen atom
- D is bonded to S and N at the para position
- D is E represents a
- the salt may be pharmaceutically acceptable, for example, for an acidic group such as a carboxyl group in the formula
- Organic salts such as ammonium salts, salts with alkali metals such as sodium and potassium, salts with alkaline earth metals such as calcium and magnesium, organic salts such as aluminum salts, zinc salts, triethylamine, ethanolamine, morpholine, piperidine and dicyclohexylamine Examples thereof include salts with amines and salts with basic amino acids such as arginine and lysine.
- salts with inorganic acids such as hydrochloric acid, sulfuric acid, phosphoric acid, acetic acid, citric acid, benzoic acid, maleic acid, fumaric acid, tartaric acid, succinic acid
- organic carboxylic acids such as acids and salts with organic sulfonic acids such as methanesulfonic acid and p-toluenesulfonic acid.
- a method for forming a salt a compound of the general formula (I) and a necessary acid or base are mixed in an appropriate amount ratio in a solvent or a dispersing agent, or cation exchange or anion is performed based on other salt forms. It can also be obtained by ion exchange.
- the compound of the present invention may also contain a solvate of the compound represented by the general formula (I), for example, a hydrate, an alcohol adduct and the like.
- the compound of the present invention includes a prodrug form of the compound represented by formula (I).
- the prodrug of the compound of the present invention is a compound that is converted into a compound represented by the general formula (I) by a reaction with an enzyme, gastric acid, or the like under physiological conditions in vivo, that is, enzymatically causes oxidation, reduction, hydrolysis, etc.
- the prodrug of the compound represented by the general formula (I) are not limited to those exemplified in the compounds of Examples.
- the prodrug may be a compound in which the amino group is acylated, alkylated or phosphorylated (eg, a compound represented by the general formula (I)
- examples thereof include methylated, tert-butylated compounds, etc.
- the prodrug includes acylated, alkylated, phosphorylated, borated.
- the compound represented by the general formula (I) is acetylated, palmitoylated, propano
- a compound having a carboxy group as a prodrug such as a compound having a carboxy group, and a pivaloylated, succinylated, fumarylated, alanylated, dimethylaminomethylcarbonylated compound, etc.
- the prodrug includes a compound in which the carboxy group is esterified with a linear, branched or cyclic alkyl group having 1 to 10 carbon atoms. These compounds are preferably represented by the general formula (I) by a method known per se. ).
- the prodrug of compound (I) changes to compound (I) under physiological conditions as described in Hirokawa Shoten, 1990, “Development of Drugs”, Volume 7, Molecular Design, pages 163 to 198. There may be.
- the present invention includes all isotopes of the compounds represented by formula (I).
- the isotope of the compound of the present invention is one in which at least one atom is substituted with an atom having the same atomic number (number of protons) and a different mass number (sum of the number of protons and neutrons).
- Examples of isotopes contained in the compounds of the present invention include a hydrogen atom, a carbon atom, a nitrogen atom, an oxygen atom, a phosphorus atom, a sulfur atom, a fluorine atom, a chlorine atom, etc., and 2 H, 3 H, 13 C, respectively.
- the isotope of the compound of the present invention can be converted according to a conventional method by replacing the reagent used in the synthesis with a reagent containing the corresponding isotope.
- the compound represented by the general formula (I) or a salt thereof is administered as it is or as various pharmaceutical compositions.
- a dosage form of such a pharmaceutical composition for example, it may be a tablet, powder, pill, granule, capsule, suppository, solution, sugar coating, devoted, or syrup, and a usual formulation aid.
- tablets may contain phenylalanine derivatives, which are the active ingredients of the present invention, known auxiliary substances such as inert diluents such as lactose, calcium carbonate or calcium phosphate, binders such as gum arabic, corn starch or gelatin, alginic acid, corn starch or the like Gelatinized starch and other swelling agents, sweeteners such as sucrose, lactose and saccharin, flavoring agents such as peppermint, red mono oil and cherry, lubricants such as magnesium stearate, talc and carboxymethylcellulose, fats, waxes and semi-solids And liquid gelatin, soft gelatin capsules such as natural or hardened oils and excipients for suppositories, water, alcohol, glycerol, polyols, sucrose, invert sugar, glucose, vegetable oils, etc. Obtained by.
- inert diluents such as lactose, calcium carbonate or calcium phosphate
- binders such as gum arabic
- an inhibitor comprising a compound represented by the general formula (I) or a salt thereof as an active ingredient can be used as a therapeutic or prophylactic agent for an inflammatory disease in which an ⁇ 4 integrin-dependent adhesion process is involved in the disease state.
- inflammatory diseases include, for example, rheumatoid arthritis, inflammatory bowel disease, systemic lupus erythematosus, multiple sclerosis, Sjogren's syndrome, asthma, psoriasis, allergy, diabetes, cardiovascular disease, arteriosclerosis, relapse Examples include stenosis, tumor growth, tumor metastasis, transplant rejection, and / or human immunodeficiency virus infection (see Non-Patent Document 1).
- the dose to be used for the above purpose is determined by the intended therapeutic effect, administration method, treatment period, age, weight, etc., but it is usually given orally as a daily dose for adults by the oral or parenteral route.
- administration 1 ⁇ g to 5 g may be used, and in the case of parenteral administration, 0.01 ⁇ g to 1 g may be used.
- an acylamino group is provided as a substituent of D.
- ⁇ 4 ⁇ 7 integrin inhibitory activity can be obtained in human whole blood.
- the sulfonamide derivative of the present invention moves to the portal vein and the amount of exposure in the circulating blood increases, an effect is obtained.
- the ⁇ 4 ⁇ 7 integrin-dependent adhesion process may be used as a therapeutic or prophylactic agent for inflammatory diseases in which the pathological condition is involved.
- the 2-position and 5-position of phenyl in the phenylalanine moiety are substituted with fluorine atoms.
- the compound represented by the general formula (I) of the present invention includes, for example, an intermediate having a carboxyl group at the terminal represented by the general formula (MI) and an amino group at the terminal represented by the general formula (M-II). And an intermediate having an amidation reaction.
- the amidation reaction is known, and examples thereof include (1) a method using a condensing agent and (2) a method using an acid halide.
- a method using a condensing agent is, for example, a method in which a carboxylic acid and an amine or a salt thereof are mixed with, for example, dichloromethane, tetrahydrofuran (THF), 1,4-dioxane, N, N-dimethylformamide (DMF) or acetonitrile.
- a solvent that does not adversely influence the reaction for example, in the presence or absence of a base such as pyridine, triethylamine or N, N-diisopropylethylamine, for example, 1-hydroxybenzotriazole (HOBt), 1-hydroxy-7
- condensation aids such as azabenzotriazole (HOAt) or N-hydroxysuccinimide (HOSu), for example 1-ethyl-3- (3′-dimethylaminopropyl) carbodiimide (WSC) ), 1,3-dicyclohexylcarbodiimide (DCC)
- DCC 1,3-dicyclohexylcarbodiimide
- 7-azabenzotriazole-1-yl) -N, N, N ' is carried out by reacting with a condensing agent such as N'- tetramethyluronium hexafluorophosphate (HATU).
- a condensing agent such as N'- t
- the carboxylic acid is used in a solvent that does not adversely influence the reaction, such as dichloromethane, or in the absence or presence of a catalyst such as DMF in the absence of a solvent.
- a solvent that does not adversely influence the reaction such as dichloromethane, or in the absence or presence of a catalyst such as DMF in the absence of a solvent.
- an acid halide obtained by reacting with thionyl chloride, oxalyl chloride, thionyl bromide or the like, for example, dichloromethane, THF, or the like in a solvent that does not adversely affect the reaction, for example, pyridine, triethylamine, or The reaction is carried out by reacting with an amine or a salt thereof in the presence of a base such as N, N-diisopropylethylamine.
- the intermediate having a carboxyl group at the terminal represented by the general formula (MI) can be produced, for example, by the following method.
- the production method of a representative compound among the intermediates having a carboxyl group at the terminal represented by the general formula (MI) which is the compound of the present invention is shown below.
- symbols in the formula are the same as defined in the formula (I).
- D represents a phenyl group which may have a substituent selected from the group consisting of a lower alkyl group, a lower alkoxy group and a halogen atom, or a heteroaryl group
- R The intermediate (S7) having a carboxyl group at the terminal where 4 is a hydrogen atom can be synthesized by using, for example, the method described below (Production Method A).
- D 1 represents the above-described substituent represented by D, or a substituent that can be easily converted to D by an operation such as deprotection
- R 21 represents, for example, a lower alkyl group or the like.
- the sulfonyl chloride derivative (S1) and the aniline derivative (S2) are reacted in a solvent that does not adversely affect the reaction such as dichloromethane, acetonitrile, THF, or DMF, for example, in the presence of a base such as pyridine or triethylamine.
- a base such as pyridine or triethylamine
- sulfonamide derivative (S3) for example, a metal catalyst such as palladium carbon, palladium hydroxide, or Raney nickel was used in a solvent that does not adversely affect the reaction such as methanol, ethanol, or isopropyl alcohol.
- the amine derivative (S4) can be synthesized by reacting a metal such as zinc under catalytic reduction reaction or under acidic conditions (for example, hydrochloric acid, acetic acid, or ammonium chloride).
- the obtained amine derivative (S4) and carboxylic acid derivative (S5) are mixed with a solvent that does not adversely affect the reaction, such as dichloromethane, THF, 1,4-dioxane, DMF, or acetonitrile, for example, pyridine, triethylamine, etc. Or in the presence or absence of a base such as N, N-diisopropylethylamine, for example, in the presence or absence of a condensation aid such as HOBt, HOAt or HOSu, for example WSC, DCC or By reacting with a condensing agent such as HATU, it can be derived into the corresponding amide derivative (S6).
- a solvent that does not adversely affect the reaction such as dichloromethane, THF, 1,4-dioxane, DMF, or acetonitrile, for example, pyridine, triethylamine, etc.
- a base such as N, N-diisopropy
- the amide derivative (S6) is used in a solvent that does not adversely influence the reaction such as THF, 1,4-dioxane, methanol or ethanol, for example, using a base such as sodium hydroxide or lithium hydroxide.
- the target carboxylic acid derivative (S7) can be produced by carrying out alkaline hydrolysis, acid hydrolysis using hydrochloric acid or trifluoroacetic acid, for example.
- D is a phenyl group which may have a substituent selected from the group consisting of a lower alkyl group, a lower alkoxy group and a halogen atom, or a terminal which is a heteroaryl group.
- the intermediate body (S7) which has a carboxyl group is compoundable by using the method (manufacturing method B or C) etc. which are described below, for example.
- D 1 represents the above-described substituent represented by D, or a substituent that can be easily converted to D by an operation such as deprotection
- R 21 represents, for example, a lower alkyl group or the like
- X 1 represents a halogen atom such as chlorine, bromine or iodine, or a leaving group such as trifluoromethanesulfonyloxy group.
- the sulfonyl chloride derivative (S8) and the aniline derivative (S2) are reacted in a solvent that does not adversely influence the reaction such as dichloromethane, acetonitrile, THF, or DMF, for example, in the presence of a base such as pyridine or triethylamine.
- a solvent that does not adversely influence the reaction such as dichloromethane, acetonitrile, THF, or DMF, for example, in the presence of a base such as pyridine or triethylamine.
- a metal catalyst such as 1,1′-bis (diphenylphosphinoferrocene) dichloropalladium (II)
- the corresponding boronic acid ester derivative was subsequently obtained.
- the boronic acid ester derivative is removed by treating the boronic acid ester derivative with, for example, sodium periodate or ammonium acetate and water in a solvent that does not adversely affect the reaction, such as acetone.
- the corresponding boronic acid derivative (S10) can be synthesized.
- the obtained boronic acid derivative (S10) and amide derivative (S11) are mixed with a solvent that does not adversely affect the reaction such as dichloromethane, dimethyl sulfoxide (DMSO), or DMF, for example, pyridine, triethylamine, etc.
- a compound (S6) can be synthesized by performing a coupling reaction using a metal catalyst such as copper (II) acetate or copper (II) trifluoromethanesulfonate.
- the target carboxylic acid derivative (S7) can be produced by carrying out alkali hydrolysis or acid hydrolysis using, for example, hydrochloric acid or trifluoroacetic acid.
- D 1 represents the above-described substituent represented by D, or a substituent that can be easily converted to D by an operation such as deprotection
- R 21 represents, for example, a lower alkyl group or the like
- X 1 represents a halogen atom such as chlorine, bromine or iodine, or a leaving group such as trifluoromethanesulfonyloxy group.
- the halogenated aryl derivative (S9) and the amide derivative (S11) are combined with a solvent that does not adversely influence the reaction, such as DMSO, NMP, or DMF, for example, triethylamine, N, N-diisopropylethylamine, or diaza
- a solvent that does not adversely influence the reaction such as DMSO, NMP, or DMF
- a base such as bicycloundecene (DBU)
- DBU bicycloundecene
- a metal catalyst such as copper (I) iodide, copper (I) bromide, or copper (I) chloride, (S6) can be synthesized.
- the target carboxylic acid derivative (S7) can be produced by carrying out alkali hydrolysis or acid hydrolysis using, for example, hydrochloric acid or trifluoroacetic acid.
- the intermediate (S16) having an amino group at the terminal represented by the general formula (M-II), which is the compound of the present invention, can be produced by, for example, the following production methods (Production methods D, E, and F), etc. Can be synthesized.
- Production methods D, E, and F Production methods D, E, and F
- symbols in the formula are the same as defined in the formula (I).
- R 31 is, for example, tert- butoxycarbonyl group, such as benzyloxycarbonyl group, for example, a substituent of the general amines which can removed by manipulation of deprotection such as, wherein X 3 is, for example, chlorine, bromine, represents a leaving group such as a halogen atom or for example, trifluoromethanesulfonyloxy group iodine, wherein B 1 represents, by operation of deprotection such, represents a readily converted can substituents B.
- the halogenated aryl derivative (S12) and a borane derivative such as bis (pinacolato) diborane for example, in a solvent that does not adversely affect the reaction such as DMF, for example, in the presence of a base such as potassium acetate, for example,
- a metal catalyst such as 1,1′-bis (diphenylphosphinoferrocene) dichloropalladium (II)
- the corresponding boronic acid ester derivative is derived, and then the boronic acid obtained
- the boronic ester is deprotected by treating the ester derivative with, for example, sodium periodate or ammonium acetate and water in a solvent that does not adversely affect the reaction, such as acetone.
- the corresponding boronic acid derivative (S13) can be synthesized.
- the obtained boronic acid derivative (S13) and uracil derivative (S14) are mixed with a solvent that does not adversely influence the reaction, such as dichloromethane, dimethyl sulfoxide (DMSO), or DMF, for example, pyridine, triethylamine, etc.
- a solvent that does not adversely influence the reaction such as dichloromethane, dimethyl sulfoxide (DMSO), or DMF, for example, pyridine, triethylamine, etc.
- An amino acid derivative (S15) can be synthesized by performing a coupling reaction in the presence of a base using a metal catalyst such as copper (II) acetate or copper (II) trifluoromethanesulfonate.
- R 31 is, for example, tert- butoxycarbonyl group, such as benzyloxycarbonyl group, for example, a substituent of the general amines which can removed by manipulation of deprotection such as, wherein R 32 and R 33 Each independently represents, for example, a lower alkyl group or a substituent of a general ester such as a benzene ring which may have a substituent, wherein B 1 is converted to B by an operation such as deprotection. Represents a substituent that can be easily converted.
- the catalytic reduction reaction using a metal catalyst such as palladium carbon, palladium hydroxide, or Raney nickel in a solvent that does not adversely affect the reaction such as methanol, ethanol, or isopropyl alcohol.
- a metal catalyst such as palladium carbon, palladium hydroxide, or Raney nickel
- the aniline derivative (S18) can be synthesized by reacting a metal such as zinc under acidic conditions (for example, hydrochloric acid, acetic acid, or ammonium chloride).
- the obtained aniline derivative (S18) and carbamate derivative (S19) are mixed with, for example, triethylamine, pyridine, DBU or the like in a solvent that does not adversely affect the reaction, such as dichloromethane, 1,4-dioxane, THF, or DMF.
- the amino acid derivative (S15) can be synthesized by reacting with the above base. Subsequently, the target carboxylic acid derivative (S16) is produced by deprotecting the amino acid derivative (S15) by, for example, acid hydrolysis using hydrochloric acid or trifluoroacetic acid or hydrogenolysis. Can do.
- R 31 represents a general amine substituent which can be removed by an operation such as deprotection, for example, a tert-butoxycarbonyl group, a benzyloxycarbonyl group, etc., wherein X 3 , X 4 and X 5 each independently represents, for example, a halogen atom such as chlorine, bromine, iodine or the like, or a leaving group such as trifluoromethanesulfonyloxy group, wherein B 1 represents B by the operation such as deprotection.
- halogenated aryl derivative (S20) and the uracil derivative (S14) are mixed with a solvent that does not adversely influence the reaction such as DMSO, NMP, or DMF, for example, triethylamine, N, N-diisopropylethylamine, DBU, or the like.
- a solvent that does not adversely influence the reaction such as DMSO, NMP, or DMF, for example, triethylamine, N, N-diisopropylethylamine, DBU, or the like.
- a compound (S21) is synthesized by performing a coupling reaction using a metal catalyst such as copper (I) iodide, copper (I) bromide, or copper (I) chloride. Can do.
- the obtained compound (S21) and halide (S22) are treated with, for example, tris (di-acid) in the presence of zinc powder activated with, for example, iodine in a solvent that does not adversely affect the reaction, such as DMF.
- a metal catalyst such as benzylideneacetone
- dipalladium (0) dipalladium (0)
- a ligand commonly used in organic synthesis such as 2-dicyclohexyl-2 ′, 6′-dimethoxybiphenyl (SPhos).
- the target carboxylic acid derivative (S16) is produced. be able to.
- tert-butylamine (124 ⁇ l, 1.17 mmol) was added and stirred at room temperature for 3 hours. After the reaction solution was concentrated under reduced pressure, the residue was subjected to reverse phase HPLC using ODS as a filler, and eluted with a mixed solution of water and acetonitrile containing 0.1% (v / v) trifluoroacetic acid. Was lyophilized to give the trifluoroacetate salt of the title compound (15 mg, 10%).
- the mixture was stirred at room temperature for 1 hour and 30 minutes.
- the reaction solution was concentrated under reduced pressure, and water (20 ml) and ethyl acetate (30 ml) were added. Extraction was performed three times with ethyl acetate, and the organic layer was dried over sodium sulfate and concentrated under reduced pressure to obtain the title compound (0.64 g, 75%).
- the residue was subjected to reverse phase HPLC using ODS as a filler, and eluted with a mixed solution of water and acetonitrile containing 0.1% (v / v) trifluoroacetic acid.
- the desired fraction was freeze-dried to obtain the trifluoroacetate salt of the title compound.
- reaction solution was cooled to room temperature, filtered through celite, and concentrated under reduced pressure.
- residue was subjected to reverse phase HPLC using ODS as a filler, and water and acetonitrile containing 0.1% (v / v) trifluoroacetic acid.
- the title compound was obtained by elution with a mixed solution and lyophilization of the desired fraction (0.26 g, 26%).
- reaction solution was concentrated under reduced pressure, the residue was subjected to reverse phase HPLC using ODS as a filler, and eluted with a mixed solution of water and acetonitrile containing 0.1% (v / v) trifluoroacetic acid. Lyophilization gave the title compound (0.20 g, 74%).
- This mixed solution was added to the previously prepared mixed solution, degassed and purged with argon three times, and then stirred at 60 ° C. for 18 hours. After cooling the reaction solution to room temperature and concentrating under reduced pressure, the residue was subjected to reverse phase HPLC using ODS as a filler, and eluted with a mixed solution of water and acetonitrile containing 0.1% (v / v) trifluoroacetic acid. The desired fraction was lyophilized to give the title compound (0.12 g, 45%).
- Step 6 Methyl (2S) -2-amino-3- [6- (1-methyl-2,4-dioxo-7,8-dihydro-5H-pyrano [4,3-d] pyrimidin-3-yl) -3- pyridyl] propanoate
- the compound obtained in (Step 5) (0.12 g, 0.26 mmol) was dissolved in 1,4-dioxane (1.0 ml) and methanol (1.0 ml).
- the compound of [Synthesis Example 30] can be synthesized in the same manner as the compound of [Synthesis Example 25] by using ethyl 3-oxotetrahydran-4-carboxylate in (Step 1) of [Synthesis Example 25]. Can do.
- the compound of [Synthesis Example 34] was prepared in the same manner as the compound of [Synthesis Example 25] by using 6-methyluracil or 5,6-dimethyluracil in (Step 3) of [Synthesis Example 25]. Can be synthesized.
- This mixed solution was added to the previously prepared mixed solution, degassed and purged with argon three times, and then stirred at 60 ° C. for 15 hours.
- the reaction solution was cooled to room temperature, and water (25 ml) and dichloromethane (25 ml) were added. After filtration through celite and extraction twice with dichloromethane, the organic layer was washed with a saturated aqueous sodium chloride solution. The extract was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give the title compound (223 mg, 57.3%).
- the compound of [Synthesis Example 38] can be synthesized in the same manner as the compound of [Synthesis Example 23] by reacting 2-iodopropane in (Step 2) of [Synthesis Example 23].
- [Synthesis Example 38] Methyl (2S) -2-amino-3- [4- (3-isopropyl-4,5-dimethyl-2,6-dioxo-pyrimidin-1-yl) phenyl] propanoate MS (ESI) m / z 360 [M + H] +
- the reaction solution was concentrated under reduced pressure, and the residue was slurry washed with ethyl acetate.
- the obtained solid was dissolved in dichloromethane (50 ml), and N, N-diisopropylethylamine (7.9 ml, 45 mmol) was added. Under ice-cooling, benzoyl chloride (5.3 g, 37 mmol) was added, and the mixture was stirred at room temperature for 12 hours. Water was added to the reaction solution, followed by extraction twice with dichloromethane. The organic layer was washed successively with 0.5M hydrochloric acid and a saturated aqueous sodium chloride solution and dried over anhydrous magnesium sulfate.
- the reaction solution was concentrated under reduced pressure, and the residue was dissolved in acetonitrile (50 ml). Potassium carbonate (4.5 g, 33 mmol) and methyl iodide (2.8 ml, 45 mmol) were sequentially added, and the mixture was stirred at room temperature for 12 hours. Water (30 ml) was added to the reaction solution, and the mixture was stirred at room temperature for 30 minutes. The title compound was obtained by filtering the solid (7.4 g, quant.).
- the reaction solution was cooled to room temperature, and water (25 ml) and dichloromethane (25 ml) were added. After filtration through celite and extraction twice with dichloromethane, the organic layer was washed with a saturated aqueous sodium chloride solution. After drying over anhydrous magnesium sulfate, the mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography to obtain the title compound mixture (1: 1) (0.29 g).
- This mixed solution was added to the previously prepared mixed solution, degassed and purged with argon three times, and then stirred at 60 ° C. for 18 hours.
- the reaction solution was cooled to room temperature, and water (25 ml) and dichloromethane (25 ml) were added. After filtration through celite and extraction twice with dichloromethane, the organic layer was washed with a saturated aqueous sodium chloride solution. After drying over anhydrous magnesium sulfate, the mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography to obtain the title compound (0.21 g).
- Step 6 Synthesis of Methyl (2S) -2-amino-3- [6- (3-methyl-2,6-dioxo-pyrimidin-1-yl) -3-pyridine] propanoate
- the compound (0.21 g, 0.52 mmol) obtained in (Step 5) was dissolved in 1,4-dioxane (2.0 ml) and methanol (1.0 ml).
- 4M Hydrochloric acid / 1,4-dioxane solution (2.0 ml) was added, and the mixture was stirred at room temperature for 5 hr, and concentrated under reduced pressure to give the hydrochloride of the title compound (0.18 g, quant.).
- MS (ESI) m / z 305 [M + H] + .
- This mixed solution was added to the previously prepared mixed solution, degassed and purged with argon three times, and then stirred at 60 ° C. for 18 hours.
- the reaction solution was cooled to room temperature, and water (25 ml) and dichloromethane (25 ml) were added. After filtration through celite and extraction twice with dichloromethane, the organic layer was washed with a saturated aqueous sodium chloride solution. The extract was dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give the title compound (404 mg, 77%). MS (ESI) m / z 447 [M + H] + .
- Step 6 Synthesis of Isopropyl (2S) -2-amino-3- [6- (3,5-dimethyl-2,6-dioxo-pyrimidin-1-yl) -3-pyrylyl] propanoate TFA salt
- the compound (93 mg, 0.21 mmol) obtained in (Step 5) was dissolved in trifluoroacetic acid (3 ml) and stirred at room temperature for 30 minutes. The solvent was concentrated under reduced pressure, water was added and lyophilized to give the title compound (96 mg, 81%).
- Step 6 Synthesis of Methyl 4-((3-amino-1H-indole) -6-sulfonamido) -2,5-difluorobenzoate
- Palladium carbon (30 mg) and triethylamine (3 drops) were added to a methanol (5 ml) solution of the compound (120 mg, 0.29 mmol) obtained in (Step 5), and the mixture was stirred for 1 hour in a hydrogen atmosphere.
- the reaction mixture was filtered and the filtrate was concentrated to give the title compound (108 mg, 98%).
- Step 7 Synthesis of 2,5-difluoro-4-(((3-pivalamido-1H-indole) -6-sulfonamido) benzoic acid
- the title compound was obtained in the same manner as in (Step 4) and (Step 5) of [Synthesis Example 44] using the compound obtained in (Step 6).
- 1 H NMR 400 MHz, CD 3 OD: ⁇ 7.94 (s, 1H), 7.66-7.63 (m, 2H), 7.53-7.45 (m, 2H), 7.40-7.36 (m, 1H), 1.37 (m , 9H); MS (ESI) m / z 452 [M + H] + .
- N, N-diisopropylamine (62 ⁇ l) were sequentially added and stirred at room temperature for 5 hours. Concentrate under reduced pressure, subject the residue to reverse phase HPLC using ODS as a filler, elute with a mixed solution of water and acetonitrile containing 0.1% (v / v) trifluoroacetic acid, and freeze-dry the desired fraction. This gave the title compound (64 mg, 71%).
- N, N-diisopropylamine 33 ⁇ l were sequentially added and stirred at room temperature for 5 hours. Concentrate under reduced pressure, subject the residue to reverse phase HPLC using ODS as a filler, elute with a mixed solution of water and acetonitrile containing 0.1% (v / v) trifluoroacetic acid, and freeze-dry the desired fraction. As a result, the title compound was obtained.
- the solvent was distilled off under reduced pressure, the residue was subjected to reverse phase HPLC using ODS as a filler, and eluted with a mixed solution of water and acetonitrile containing 0.1% (v / v) trifluoroacetic acid.
- the title compound was obtained by lyophilization.
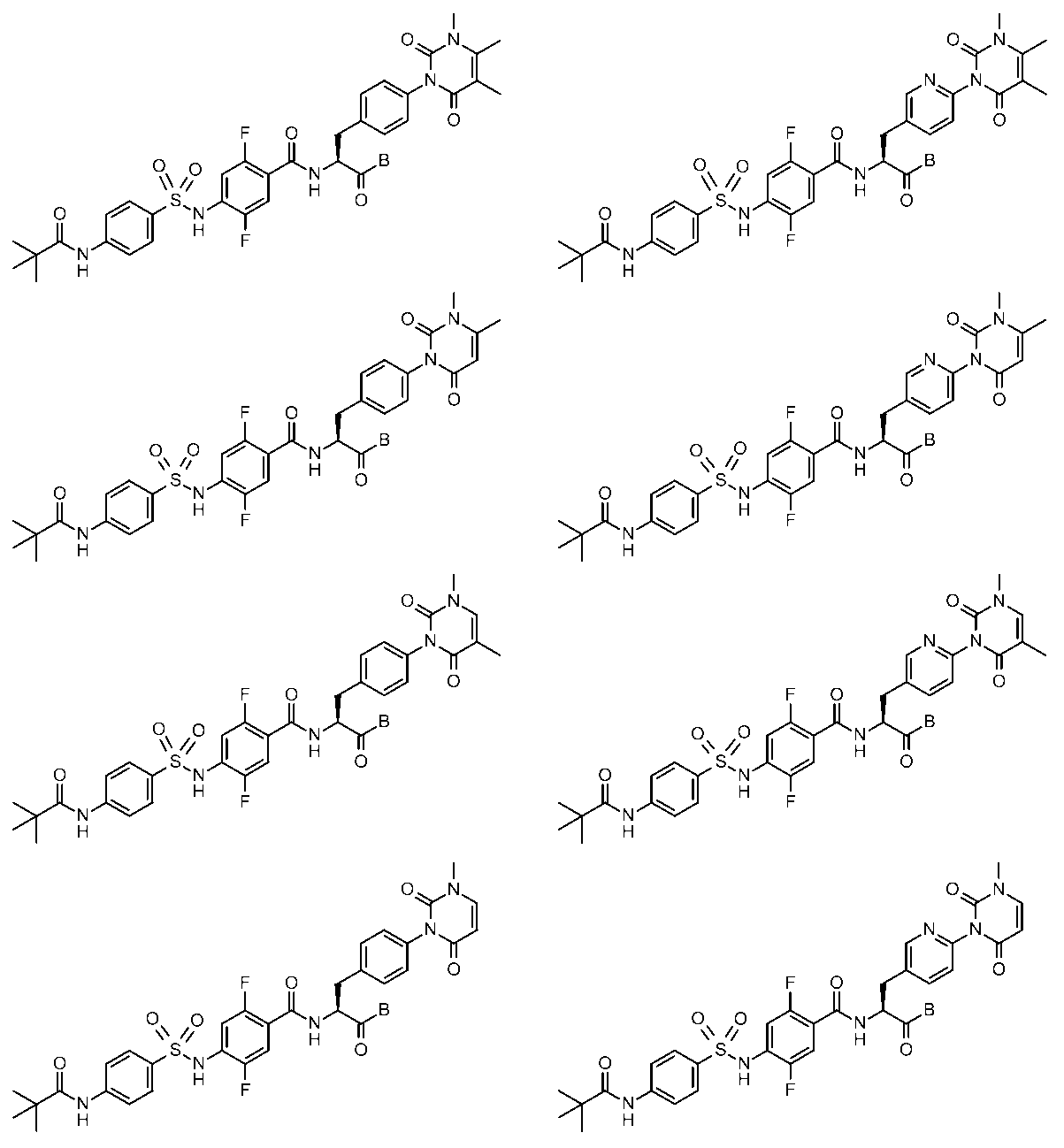
- the compounds shown in Table 1 include any carboxylic acid intermediate selected from [Synthesis Example 1] to [Synthesis Example 21] and [Synthesis Example 43] to [Synthesis Example 46], and [Synthesis Example 22] to By using any amine intermediate selected from [Synthesis Example 42] or a salt thereof, the compound can be synthesized in the same manner as the compound of [Example 1].
- the compounds (P2 to P120) shown in Table 2 are any sulfonamide derivatives selected from A1 to A77 and the corresponding alcohols (methanol, ethanol, n-propyl alcohol, isopropyl alcohol, n-butyl alcohol, isobutyl) Alcohol, cyclopropylmethyl alcohol, tetrahydro-4-pyranol, n-pentyl alcohol, isopentyl alcohol, 3-pentyl alcohol, or cyclohexyl alcohol) in the same manner as the compound of [Example 4]. can do.
- VCAM-1 / ⁇ 4 ⁇ 1 integrin binding inhibitory activity evaluation test Measures the ability of a test substance to inhibit binding of human T cell line Jurkat, which is known to express ⁇ 4 ⁇ 1 integrin, to VCAM-1 did.
- buffer A carbonate buffer, pH 9.6
- Block Ace Snow Brand Milk Products
- Various concentrations of test substances and Jurkat cells (2 ⁇ 10 6 cells / mL) diluted in binding buffer (DMEM containing 40 mM HEPES, 0.2% BSA and 4 mM MnCl 2 ) were added in 100 ⁇ L VCAM-1 / Fc was added to the coated plate (5 ⁇ 10 5 cells / well) and incubated at 30 ° C. for 15-60 minutes. After binding the cells to the wells, unbound cells were removed by washing with PBS. Buffer C (PBS containing 1.5% Triton X-100) was added to the plate at 50 ⁇ L / well to lyse the bound Jurkat cells.
- binding buffer DMEM containing 40 mM HEPES, 0.2% BSA and 4 mM MnCl 2
- Test example 2 (2) MAdCAM-1 / ⁇ 4 ⁇ 7 integrin binding inhibitory activity evaluation test Ability of test substance to inhibit binding of human B cell line cell line RPMI-8866 known to express ⁇ 4 ⁇ 7 integrin to MAdCAM-1 was measured.
- a recombinant mouse MAdCAM-1 / Fc (R & D systems) solution (0.75 ⁇ g / mL) diluted with buffer A (carbonate buffer, pH 9.6) was added. Incubate overnight at 4 ° C. After washing once with PBS, Block Ace (Snow Brand Milk Products) was added at 150 ⁇ L / well and incubated at room temperature for 2 hours.
- Test example 3 (3) MAdCAM-1 / ⁇ 4 ⁇ 7 integrin binding inhibitory activity evaluation test in the presence of serum (1) The ability of test substances to inhibit binding of human B cell line cell line RPMI-8866, known to express ⁇ 4 ⁇ 7 integrin, to MAdCAM-1 was determined. 50 ⁇ L / well of a recombinant mouse MAdCAM-1 / Fc (R & D systems) solution (1 ⁇ g / mL) diluted with buffer A (carbonate buffer, pH 9.6) was added to a 96-well microtiter plate at 4 ° C. Incubated overnight. After washing once with PBS, Block Ace (Snow Brand Milk Products) was added at 150 ⁇ L / well and incubated at room temperature for 2 hours.
- buffer A carbonate buffer, pH 9.6
- the compound of the present invention has selectivity for ⁇ 4 ⁇ 1 having a low effect and ⁇ 4 ⁇ 7 having a high effect. .
- the selectivity of ⁇ 4 ⁇ 1 is low and the effect is high on ⁇ 4 ⁇ 7, so that the action on ⁇ 4 ⁇ 1 that suppresses infiltration of lymphocytes around the whole body is reduced, and it is expressed specifically in the intestinal tract. Since the action on ⁇ 4 ⁇ 7 can be greatly suppressed, there is an advantage that the adaptive disease can be more efficiently treated.
- Test example 4 Mouse MAdCAM-1 / ⁇ 4 ⁇ 7 integrin binding inhibitory activity evaluation test in human whole blood The binding inhibitory activity of T cell ⁇ 4 ⁇ 7 integrin and MAdCAM-1 in human whole blood by the test substance was measured. Blood samples were obtained by donating blood from healthy volunteers. 4 mM MnCl2 solution and various test substance dilutions were added to human whole blood and incubated for 10 minutes. 10 ⁇ g / mL recombinant mouse MAdCAM-1 / Fc (R & D Systems) was added and incubated for a total volume of 50 ⁇ L for 30 minutes. 950 ⁇ L of Lyse / Fix (BD Biosciences) was added, and hemolysis and fixation were performed at 37 ° C.
- Lyse / Fix BD Biosciences
- Test Example 5 The mouse portal vein concentration of the test substance was measured and the oral absorbability was evaluated.
- the test substance was dissolved or uniformly suspended in a 0.5% (w / v) aqueous solution of methylcellulose, and 3 compounds (3 mg / 10 mL / kg) were added to female mice (BALB / cAnNCrlCrlj, 7-9 weeks old) using a gastric tube. ) was orally administered in a cassette. 30 minutes after administration, the abdomen was opened under isoflurane anesthesia, and about 0.2 mL of blood was collected from the portal vein using a syringe treated with DDVP (esterase inhibitor) and heparin sodium, and stored on ice.
- DDVP esterase inhibitor
- the collected blood was centrifuged at 18,000 g ⁇ 3 minutes using a refrigerated centrifuge to obtain a plasma sample. After extracting the test substance with acetonitrile, the plasma concentration was quantified by LC / MS / MS. The plasma concentration was the sum of the test substance and its active metabolite. The calculated plasma concentrations are shown in Table 5.
- Test Example 6 Human MAdCAM-1 / ⁇ 4 ⁇ 7 integrin binding inhibitory activity evaluation test in human whole blood The binding inhibitory activity of T cell ⁇ 4 ⁇ 7 integrin and MAdCAM-1 in human whole blood by the test substance was measured. Blood samples were obtained by donating blood from healthy volunteers. 4 mM MnCl2 solution and various test substance dilutions were added to human whole blood and incubated for 10 minutes. 10 ⁇ g / mL recombinant human MAdCAM-1 / Fc (R & D Systems) was added and incubated for a total of 50 ⁇ L for 30 minutes. 950 ⁇ L of Lyse / Fix (BD Biosciences) was added, and hemolysis and fixation were performed at 37 ° C.
- Lyse / Fix BD Biosciences
- PE Mouse Anti-Human CD4 (BD Pharmigen) was added and incubated for 30 minutes or more. After washing with the medium, the ratio of the MAdCAM-1-positive cell ratio in the CD4-positive cells was measured using flow cytometry. Tests are based on the results of independent tests using 2 to 3 different blood samples. From wells containing no test substance, the absence of ligand is 100% inhibition and the presence of ligand is 0% inhibition. The inhibition rate of MAdCAM-1 binding was determined and the concentration IC 50 resulting in 50% binding inhibition was calculated. Table 6 shows the obtained results.
- Test Example 7 The method described in J Crohns Colitis. 2013 Dec; 7 (11): e533-42. was used as a method for creating a mouse IL-10 ⁇ / ⁇ cell transfer enteritis model. Suspend the test substance in 0.5% aqueous methylcellulose solution at 30, 10, 3, or 1 mg / mL, and use a Terumo syringe (1 mL) and a metal sonde for mice in a volume of 10 ml per kg of animal weight And administered orally. Administration was performed 3 times a day for 14 days. The intestinal weight, which is a medicinal effect index, was measured by the following method.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description



特に、本発明は、α4β1に対しては効果が低く、α4β7に対しては効果が高いという選択性のあるα4インテグリン阻害作用を有する新規化合物を提供することを目的とする。
本発明は、又、経口投与の可能性があるα4インテグリン阻害作用を有する化合物を提供することを目的とする。
本発明は、又、安全性のあるα4インテグリン阻害活性を有する化合物を提供することを目的とする。
本発明は、又、持続性のあるα4インテグリン阻害活性を有する化合物を提供することを目的とする。
本発明は、又、ヒト全血中でα4インテグリン阻害作用を有する新規化合物を提供することを目的とする。
本発明は、又、上記新規化合物と医薬的に許容しうる担体を含有する医薬組成物を提供することを目的とする。
本発明は、又、上記新規化合物を含有する医薬を提供することを目的とする。
本発明は、又、α4β7インテグリン依存性の接着過程が病態に関与する炎症性疾患の治療剤または予防剤を提供することを目的とする。
本発明は、又、α4インテグリン阻害剤を提供することを目的とする。
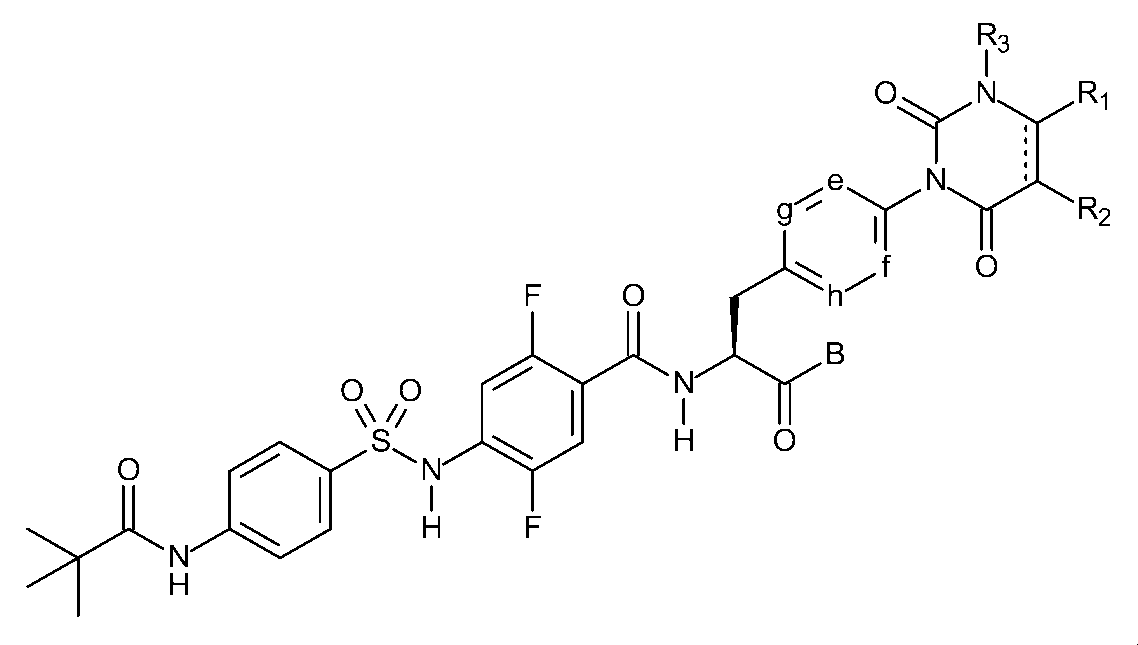
〔1〕下記一般式(I)で示されるスルホンアミド誘導体、又はその医薬的に許容しうる塩。

R1及びR2は、それぞれ独立して、水素原子、ハロゲン原子、低級アルキル基、低級アルケニル基、低級アルコキシ基、低級アルコキシ低級アルキル基、ハロゲノ低級アルキル基、ヒドロキシ基、又は、ヒドロキシ低級アルキル基を表し、R1とR2は結合して、置換基を有しても良いベンゼン環、置換基を有しても良い炭素数4~7の脂環式炭化水素、置換基を有しても良いヘテロアリール環、又は、置換基を有しても良いヘテロ環を形成しても良く、
R3は、低級アルキル基を表し、
e、f、g、及び、hは、それぞれ独立して、C-H、又は、窒素原子を表し、
Bは、ヒドロキシ基、炭素数が1~10のアルコキシ基、-O-ヘテロ環基、シレキセチルオキシ基、又はメドキソミルオキシ基を表し、
Dは、置換基を有しても良い、ベンゼン環又はヘテロアリール環を表し、
R4は、水素原子、又は、低級アルキル基を表し、
R5は、置換基を有しても良い低級アルキル基、置換基を有しても良い低級アルケニル基、置換基を有しても良い低級アルキニル基、低級アルキルアミノ基、置換基を有しても良いフェニル基、置換基を有しても良いヘテロアリール基、又は、置換基を有しても良いヘテロ環基を表し、
R4とR5は結合して、置換基を有しても良いヘテロ環を形成しても良い。)
〔2〕
R1及びR2が、それぞれ独立して、水素原子、ハロゲン原子、低級アルキル基、低級アルケニル基、低級アルコキシ基、低級アルコキシ低級アルキル基、ヒドロキシ基、又は、ヒドロキシ低級アルキル基を表し、R1とR2は結合して、置換基を有しても良いベンゼン環、置換基を有しても良い炭素数4~7の脂環式炭化水素、置換基を有しても良いヘテロアリール環、又は、置換基を有しても良いヘテロ環を形成しても良く、
R5は、置換基を有しても良い低級アルキル基、置換基を有しても良い低級アルケニル基、低級アルキルアミノ基、置換基を有しても良いフェニル基、置換基を有しても良いヘテロアリール基、又は、置換基を有しても良いヘテロ環基を表す、
前記〔1〕に記載されたスルホンアミド誘導体、又はその医薬的に許容し得る塩。
〔3〕
〔4〕R1及びR2は結合して、置換基を有しても良いベンゼン環、置換基を有しても良い炭素数4~7の脂環式炭化水素、置換基を有しても良いヘテロアリール環、又は、置換基を有しても良いヘテロ環を形成し、該置換基が、低級アルキル基、低級アルコキシ基、ヒドロキシ低級アルキル基、アミノ基、低級アルキルアミノ基、及び、低級アルキルアミノ低級アルキル基から選ばれる、前記〔1〕~〔3〕のいずれか1項に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔5〕R1及びR2が、それぞれ独立して、水素原子、低級アルキル基、又は、ヒドロキシ低級アルキル基を表し、R1とR2は結合して、置換基を有しても良い炭素数4~7の脂環式炭化水素、置換基を有しても良いヘテロアリール環、又は、置換基を有しても良いヘテロ環を形成しても良く、該置換基は、低級アルキル基及び低級アルコキシ基から選ばれる、前記〔1〕~〔3〕のいずれか1項に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔6〕
R1及びR2が、それぞれ独立して、水素原子、低級アルキル基、ハロゲノ低級アルキル基、又は、ヒドロキシ低級アルキル基を表し、R1とR2は結合して、置換基を有しても良い炭素数4~7の脂環式炭化水素、置換基を有しても良いヘテロアリール環、又は、置換基を有しても良いヘテロ環を形成しても良く、該置換基は、低級アルキル基及び低級アルコキシ基から選ばれる、前記〔1〕に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔7〕R1及びR2が、それぞれ独立して、水素原子、低級アルキル基、又は、ヒドロキシ低級アルキル基を表す、前記〔1〕~〔3〕のいずれか1項に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔8〕R1とR2が結合して置換基を有しても良いピリジン、置換基を有しても良いシクロヘキセン、置換基を有しても良いジヒドロピラン、置換基を有しても良いテトラヒドロピリジン、又は、置換基を有しても良いイミダゾールを形成し、該置換基が、低級アルキル基、低級アルコキシ基、ヒドロキシ低級アルキル基、アミノ基、低級アルキルアミノ基、及び、低級アルキルアミノ低級アルキル基から選ばれる、前記〔1〕~〔3〕のいずれか1項に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔9〕eが窒素原子を表し、f、g、及び、hが、C-Hを表す、前記〔1〕~〔8〕のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔10〕Bが、ヒドロキシ基、又は、炭素数が1~6のアルコキシ基を表す、前記〔1〕~〔9〕のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔11〕Dの置換基が、ハロゲン原子、低級アルキル基、低級アルコキシ基、及び、ヒドロキシ基から選ばれる、前記〔1〕~〔10〕のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔12〕Dが、置換基を有しても良いベンゼン環、置換基を有しても良いピリジン環、又は、置換基を有しても良いチオフェン環を表し、該置換基が、ハロゲン原子、低級アルキル基、低級アルコキシ基、及び、ヒドロキシ基から選ばれる、前記〔1〕~〔11〕のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔13〕Dが、置換基を有しても良いベンゼン環、置換基を有しても良いピリジン環、又は、置換基を有しても良いチオフェン環を表し、該置換基が、ハロゲン原子を表す、前記〔1〕~〔11〕のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔14〕R4が、水素原子を表す、前記〔1〕~〔13〕のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔15〕R5が、置換基を有する場合における置換基が、ハロゲン原子、シアノ基、ヒドロキシ基、低級アルキル基、低級アルコキシ基、トリフルオロメチル基、フェニル基、及び、ヘテロ環基から選ばれる、前記〔1〕~〔14〕のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔16〕R4とR5が結合して、置換基を有しても良いヘテロ環を形成し、該置換基が、低級アルキル基、低級アルコキシ基、ヒドロキシ基、及び、ヘテロ環基から選ばれる、前記〔1〕~〔14〕のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔17〕R5が、置換基を有しても良い低級アルキル基、低級アルキルアミノ基、又は、置換基を有しても良いヘテロ環基を表し、該置換基が、ハロゲン原子、シアノ基、ヒドロキシ基、低級アルコキシ基、トリフルオロメチル基及びフェニル基から選ばれる、前記〔1〕~〔14〕のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
〔18〕R1及びR2が、それぞれ独立して、水素原子、低級アルキル基、低級アルコキシ基、又は、ヒドロキシ低級アルキル基を表し、R1とR2は結合して、低級アルキル基で置換されていてもよい炭素数4~7の脂環式炭化水素、低級アルキル基で置換されていてもよいヘテロアリール環、又は、低級アルキル基で置換されていてもよいヘテロ環を形成しても良く、
Dは、ハロゲン原子で置換されていてもよいベンゼン環、又は、下記式から選ばれるヘテロアリール環を表し、
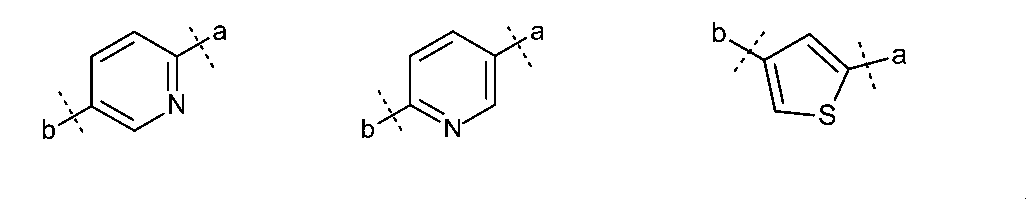
R4が、水素原子を表し、
R5が、低級アルコキシ基、ハロゲン原子、ヒドロキシ基及びアリール基からなる群から選ばれる置換基を有しても良い炭素数2~5のアルキル基、ヘテロアリール基、又は、環原子としてOを含有するヘテロ環基を表し、
但し、
Dがハロゲン原子で置換されていてもよいベンゼン環である場合、Dは、パラ位でSとNとに結合しており、
Dが、

R5が環原子としてOを含有するヘテロ環基である場合、R1とR2は結合してヘテロアリール環を形成し、
R5がヒドロキシ基で置換された炭素数2~5のアルキル基である場合、R5は、下記式により表される、
〔19〕下記式で表される、前記〔1〕又は〔2〕に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。

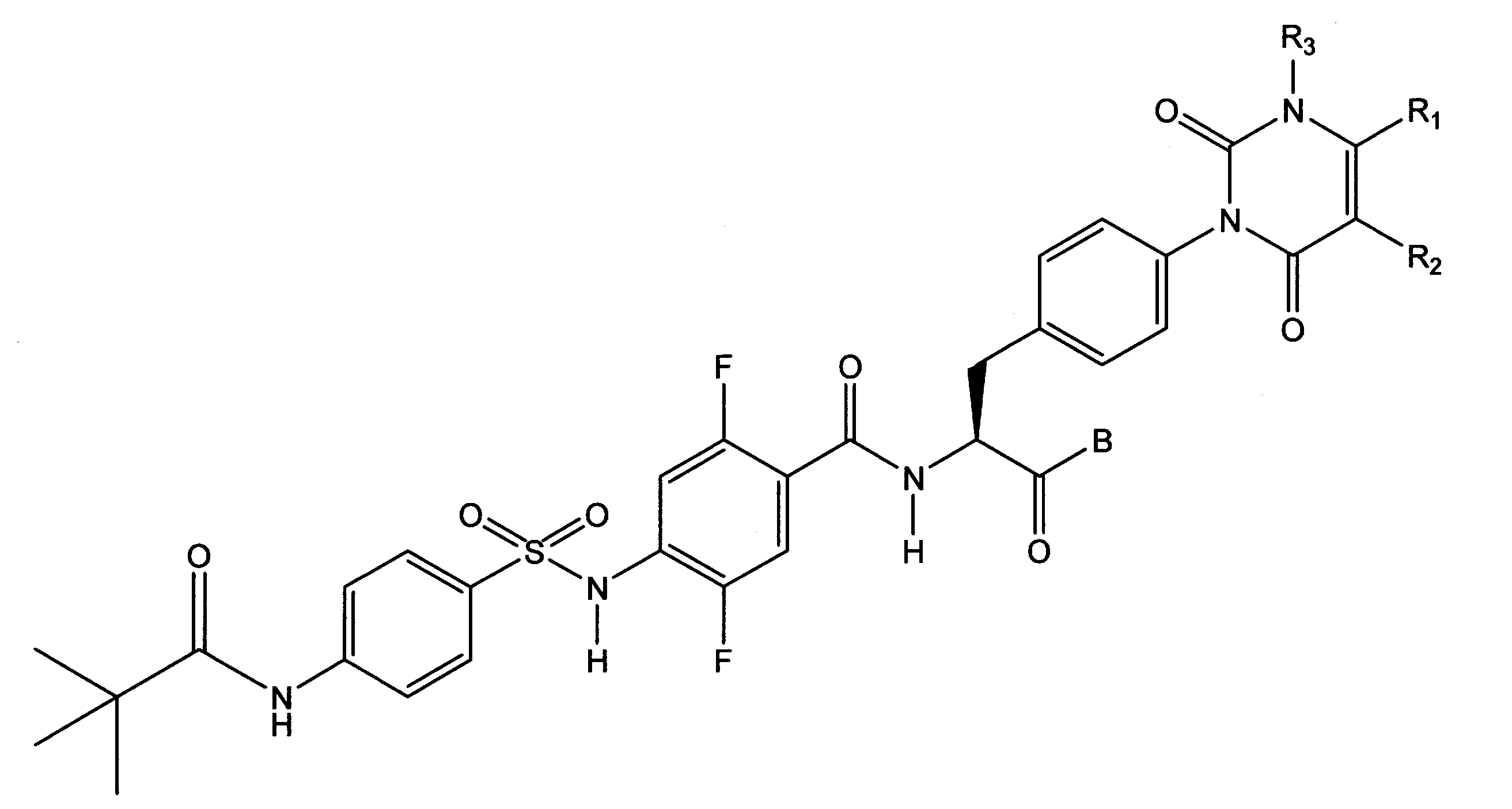
下記式のいずれかで表される、前記〔22〕に記載のスルホンアミド誘導体、又はその医薬的に許容しうる塩。



〔25〕前記〔1〕~〔24〕のいずれかに記載のスルホンアミド誘導体、又はその医薬的に許容し得る塩を含有する医薬組成物。
〔26〕前記〔1〕~〔24〕のいずれかに記載のスルホンアミド誘導体、又はその医薬的に許容し得る塩を含有するα4β7インテグリン依存性の接着過程が病態に関与する炎症性疾患の治療剤又は予防剤。
〔27〕前記〔1〕~〔24〕のいずれかに記載のスルホンアミド誘導体、又はその医薬的に許容し得る塩を含有するα4β7インテグリン阻害剤。
特に、本発明によれば、α4β1に対しては効果が低く、α4β7に対しては効果が高いという選択性のあるα4インテグリン阻害作用を有する新規化合物が提供される。
本発明によれば、又、経口投与の可能性があるα4インテグリン阻害作用を有する化合物が提供される。
本発明によれば、又、安全性のあるα4インテグリン阻害活性を有する化合物が提供される。
本発明によれば、又、持続性のあるα4インテグリン阻害活性を有する化合物が提供される。
本発明によれば、又、ヒトの血液中でα4インテグリン阻害作用を有する新規化合物が提供される。
本発明によれば、又、上記新規化合物と医薬的に許容しうる担体を含有する医薬組成物が提供される。
本発明によれば、又、上記新規化合物を含有する医薬が提供される。
本発明によれば、又、α4β7インテグリン依存性の接着過程が病態に関与する炎症性疾患の治療剤または予防剤が提供される。
本発明によれば、又、α4インテグリン阻害剤が提供される。
「低級アルキニル基」とは、各異性体を含む炭素数2~6の直鎖もしくは分岐鎖状のアルキニル基を示す。例えば、エチニル基、プロピニル基、ブチニル基、ペンチニル基及びヘキシニル基等が挙げられ、好ましくは、エチニル基、プロピニル基である。
「ハロゲノ低級アルキル基」とは、前述の「ハロゲン原子」で一置換、もしくは、それ以上置換された低級アルキル基を示す。例えば、トリフルオロメチル基、ジフルオロメチル基、モノトリフルオロメチル基、トリクロロメチル基、ジクロロメチル基、モノクロロメチル基、トリフルオロエチル基、ペンタフルオロエチル基等が挙げられ、好ましくは、トリフルオロメチル基である。
一般式(I)において、R1及びR2は、それぞれ独立して、水素原子、低級アルキル基、ハロゲノ低級アルキル基、又は、ヒドロキシ低級アルキル基が好ましく、水素原子、又は、低級アルキル基がより好ましく、水素原子、及び、メチル基が特に好ましい。あるいは、R1及びR2は、それぞれ独立して、水素原子、低級アルキル基、又は、ヒドロキシ低級アルキル基が好ましい。
一般式(I)において、R1とR2が結合して形成する環は、ピリジン、シクロヘキセン、ジヒドロピラン、及び、テトラヒドロピリジンが好ましく、シクロヘキセン、ジヒドロピラン、及び、テトラヒドロピリジンがより好ましく、ジヒドロピラン、及び、テトラヒドロピリジンが特に好ましい。
一般式(I)において、R3は、低級アルキル基が好ましく、イソプロピル基、メチル基がより好ましく、メチル基が特に好ましい。
一般式(I)において、e、f、g、及びhは、C-H、又は、窒素原子が好ましく、いずれかひとつが窒素原子であることがより好ましく、e又はfが窒素原子であることが特に好ましい。
一般式(I)において、Bは、ヒドロキシ基、低級アルコキシ基が好ましく、ヒドロキシ基、メトキシ基、エトキシ基、イソプロポキシ基、イソブチルオキシ基、シクロヘキシルオキシ基がより好ましく、ヒドロキシ基、メトキシ基、エトキシ基、イソプロポキシ基、イソブチルオキシ基が特に好ましく、イソブチルオキシ基が最も好ましい。或いは、Bは、ヒドロキシ基、メトキシ基、エトキシ基、イソプロポキシ基、シクロヘキシルオキシ基がより好ましく、ヒドロキシ基、メトキシ基、エトキシ基、イソプロポキシ基、が特に好ましい。
一般式(I)において、Dは、置換基を有しても良いベンゼン環、又は、置換基を有しても良いヘテロアリール環が好ましく、ベンゼン環、ピリジン環、チオフェン環がより好ましく、ベンゼン環が特に好ましい。
一般式(I)において、Dの置換基は、ハロゲン原子、低級アルキル基、低級アルコキシ基、ヒドロキシ基が好ましく、フッ素原子がより好ましい。
一般式(I)において、Dがベンゼン環を表す場合、Dに結合したアミノスルホニル基とアミノカルボニル基の置換位置はパラ位又はメタ位が好ましく、パラ位が特に好ましい。
一般式(I)において、R4は、水素原子、低級アルキル基が好ましく、水素原子、メチル基がより好ましく、水素原子が特に好ましい。
一般式(I)において、R5は、置換基を有しても良い低級アルキル基、置換基を有しても良い低級アルキニル基、低級アルキルアミノ基、置換基を有しても良いフェニル基、置換基を有しても良いヘテロアリール基、置換基を有しても良いヘテロ環基が好ましく、置換基を有しても良い低級アルキル基、低級アルキルアミノ基、置換基を有しても良いフェニル基、置換基を有しても良いヘテロアリール基、置換基を有しても良いヘテロ環基がより好ましく、メチル基、エチル基、tert-ブチル基、3-ペンチル基、シクロプロピル基、tert-ブチルアミド基、ピリジル基、ピペリジル基、テトラヒドロピラニル基が更に好ましく、tert-ブチル基、シクロプロピル基が特に好ましい。
一般式(I)において、R5の置換基は、ハロゲン原子、ヒドロキシ基、低級アルコキシ基、トリフルオロメチル基、フェニル基が好ましく、フッ素原子、ヒドロキシ基、メトキシ基、トリフルオロメチル基が特に好ましい。
一般式(I)において、R4及びR5が結合して形成する環は、置換基を有しても良いヘテロ環が好ましく、ピリミドン、ヒダントインが特に好ましい。
一般式(I)において、R4及びR5が結合して形成する環の置換基は、低級アルキル基、ヘテロ環基が好ましく、メチル基、テトラヒドロピラニル基が特に好ましい。
R1及びR2が、それぞれ独立して、水素原子、低級アルキル基、低級アルコキシ基、又は、ヒドロキシ低級アルキル基を表し、R1とR2は結合して、低級アルキル基で置換されていてもよい炭素数4~7の脂環式炭化水素、低級アルキル基で置換されていてもよいヘテロアリール環、又は、低級アルキル基で置換されていてもよいヘテロ環を形成しても良く、
Dは、ハロゲン原子で置換されていてもよいベンゼン環、又は、下記式から選ばれるヘテロアリール環を表し、

R4が、水素原子を表し、
R5が、低級アルコキシ基、ハロゲン原子、ヒドロキシ基及びアリール基からなる群から選ばれる置換基を有しても良い炭素数2~5のアルキル基、ヘテロアリール基、又は、環原子としてOを含有するヘテロ環基を表し、
但し、
Dがハロゲン原子で置換されていてもよいベンゼン環である場合、Dは、パラ位でSとNとに結合しており、
Dが、
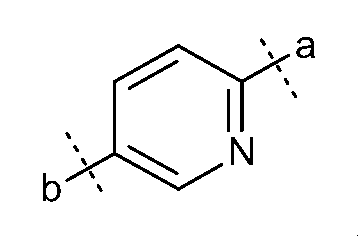
R5が環原子としてOを含有するヘテロ環基である場合、R1とR2は結合してヘテロアリール環を形成し、
R5がヒドロキシ基で置換された炭素数2~5のアルキル基である場合、R5は、下記式により表される、
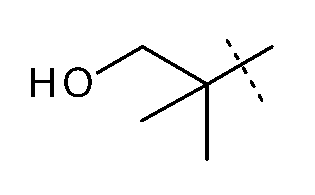
本発明化合物は、一般式(I)で示される化合物のプロドラッグの形態を包含する。本発明化合物のプロドラッグとは、生体内における生理条件下で酵素や胃酸等による反応により一般式(I)で示される化合物に変換する化合物、即ち酵素的に酸化、還元、加水分解等を起こして一般式(I)で示される化合物に変化する化合物、胃酸等により加水分解等を起こして一般式(I)で示される化合物に変化する化合物をいう。一般式(I)で示される化合物のプロドラッグとしては、実施例の化合物に例示されるがこれらに限られない。例えば一般式(I)で示される化合物がアミノ基を有する場合、そのプロドラッグとしては、該アミノ基がアシル化、アルキル化、リン酸化された化合物(例、一般式(I)で示される化合物のアミノ基がエイコサノイル化、アラニル化、ペンチルアミノカルボニル化、(5-メチル-2一オキソ-1,3-ジオキソレン-4-イル)メトキシカルボニル化、テトラヒドロフラニル化、ピロリジルメチル化ピバロイルオキシメチル化、tert-ブチル化された化合物等が挙げられる。一般式(I)で示される化合物がヒドロキシを有する場合、そのプロドラッグとしては、該ヒドロキシがアシル化、アルキル化、リン酸化、ホウ酸化された化合物(例、一般式(I)で示される化合物のヒドロキシがアセチル化、パルミトイル化、プロパノイル化、ピバロイル化、スクシニル化、フマリル化、アラニル化、ジメチルアミノメチルカルボニル化された化合物等が挙げられる。一般式(I)で示される化合物がカルボキシ基を有する場合、そのプロドラッグとしては、該カルボキシ基がエステル化、アミド化された化合物(例、一般式(I)で示される化合物のカルボキシルが、メチルエステル化、エチルエステル化、ノルマルプロピルエステル化、フェニルエステル化、イソプルピルエステル化、イソブチルエステル化、シクロブチルエステル化、シクロペンチルエステル化、シクロヘキシルエステル化、シクロヘプチルエステル化、シクロブチルメチルエステル化、シクロヘキシルメチルエステル化、ノルマルヘキシルエステル化、sec-ブチルエステル化、tert-ブチルエステル化、(4-テトラヒドロピラニル)メチルエステル化、(4-テトラヒドロピラニル)エステル化、カルボキシメチルエステル化、ジメチルアミノメチルエステル化、ピバロイルオキシメチルエステル化、エトキシカルボニルオキシエチルエステル化、フタリジルエステル化、(5-メチル-2一オキソ-1,3-ジオキソレン-4一イル)メチルエステル化、シクロへキシルオキシカルボニルエチルエステル化、メチルアミド化された化合物等が挙げられる。特に、一般式(I)で示される化合物がカルボキシ基を有する場合、そのプロドラッグとしては、該カルボキシ基が、炭素数1~10の直鎖、分岐鎖、又は環状のアルキル基によってエステル化された化合物が好ましい。これらの化合物は自体公知の方法によって一般式(I)で示される化合物から製造することができる。
また、化合物(I)のプロドラッグは、広川書店1990年刊「医薬品の開発」第7巻分子設計163頁から198頁に記載されているような生理的条件で化合物(I)に変化するものであってもよい。
例えば錠剤は、本発明の有効成分であるフェニルアラニン誘導体を既知の補助物質、例えば乳糖、炭酸カルシウムまたは燐酸カルシウム等の不活性希釈剤、アラビアゴム、コーンスターチまたはゼラチン等の結合剤、アルギン酸、コーンスターチまたは前ゼラチン化デンプン等の膨化剤、ショ糖、乳糖またはサッカリン等の甘味剤、ペパーミント、アカモノ油またはチェリー等の香味剤、ステアリン酸マグネシウム、タルクまたはカルボキシメチルセルロース等の滑湿剤、脂肪、ワックス、半固形及び液体のポリオール、天然油または硬化油等のソフトゼラチンカプセル及び坐薬用の賦形剤、水、アルコール、グリセロール、ポリオール、スクロース、転化糖、グルコース、植物油等の溶液用賦形剤と混合することによって得られる。
上記目的のために用いる投与量は、目的とする治療効果、投与方法、治療期間、年齢、体重などにより決定されるが、経口もしくは非経口のルートにより、通常成人一日あたりの投与量として経口投与の場合で1μg~5g、非経口投与の場合で0.01μg~1gを用いるのがよい。
更に、一般式(I)のスルホンアミド誘導体では、フェニルアラニン部分のフェニルの2位と5位が、フッ素原子により置換されている。これにより、α4β1インテグリンに対しては効果が低く、α4β7インテグリンに対して高い阻害活性を得ることができる。
アミド化反応は公知であり、例えば、(1)縮合剤を用いる方法、(2)酸ハロゲン化物を用いる方法等が挙げられる。

一般式(M-I)において、Dが、低級アルキル基、低級アルコキシ基、及び、ハロゲン原子からなる群から選ばれる置換基を有しても良いフェニル基、又は、ヘテロアリール基であり、R4が、水素原子である末端にカルボキシル基を有する中間体(S7)は、例えば、以下に記載する方法(製造方法A)等を用いることで合成することができる。

スルホニルクロリド誘導体(S1)とアニリン誘導体(S2)とを、例えば、ジクロロメタン、アセトニトリル、THF、またはDMF等の本反応に悪影響を及ぼさない溶媒中、例えば、ピリジン、またはトリエチルアミン等の塩基存在下で反応させることでスルホンアミド誘導体(S3)を合成できる。得られたスルホンアミド誘導体(S3)は、例えば、メタノール、エタノール、又はイソプロピルアルコール等の本反応に悪影響を及ぼさない溶媒中、例えば、パラジウムカーボン、水酸化パラジウム、又はラネーニッケル等の金属触媒を用いた接触還元反応、又は酸性条件下(例えば、塩酸、酢酸、又は塩化アンモニウム等)、例えば、亜鉛等の金属を作用させることで、アミン誘導体(S4)を合成することができる。得られたアミン誘導体(S4)とカルボン酸誘導体(S5)とを、例えば、ジクロロメタン、THF、1,4-ジオキサン、DMF又はアセトニトリル等の本反応に悪影響を及ぼさない溶媒中、例えば、ピリジン、トリエチルアミン、又はN,N-ジイソプロピルエチルアミン等の塩基の存在下、又は、非存在下で、例えば、HOBt、HOAt又はHOSu等の縮合補助剤の存在下、又は、非存在下で、例えばWSC、DCC又はHATU等の縮合剤を用いて反応させることにより、対応するアミド誘導体(S6)へと誘導することができる。続いて、アミド誘導体(S6)を、例えば、THF、1,4-ジオキサン、メタノール又はエタノール等の本反応に悪影響を及ぼさない溶媒中、例えば、水酸化ナトリウム、又は水酸化リチウム等の塩基を用いたアルカリ加水分解や、例えば、塩酸、又はトリフルオロ酢酸を用いた酸加水分解等を行うことで、目的とするカルボン酸誘導体(S7)を製造することができる。
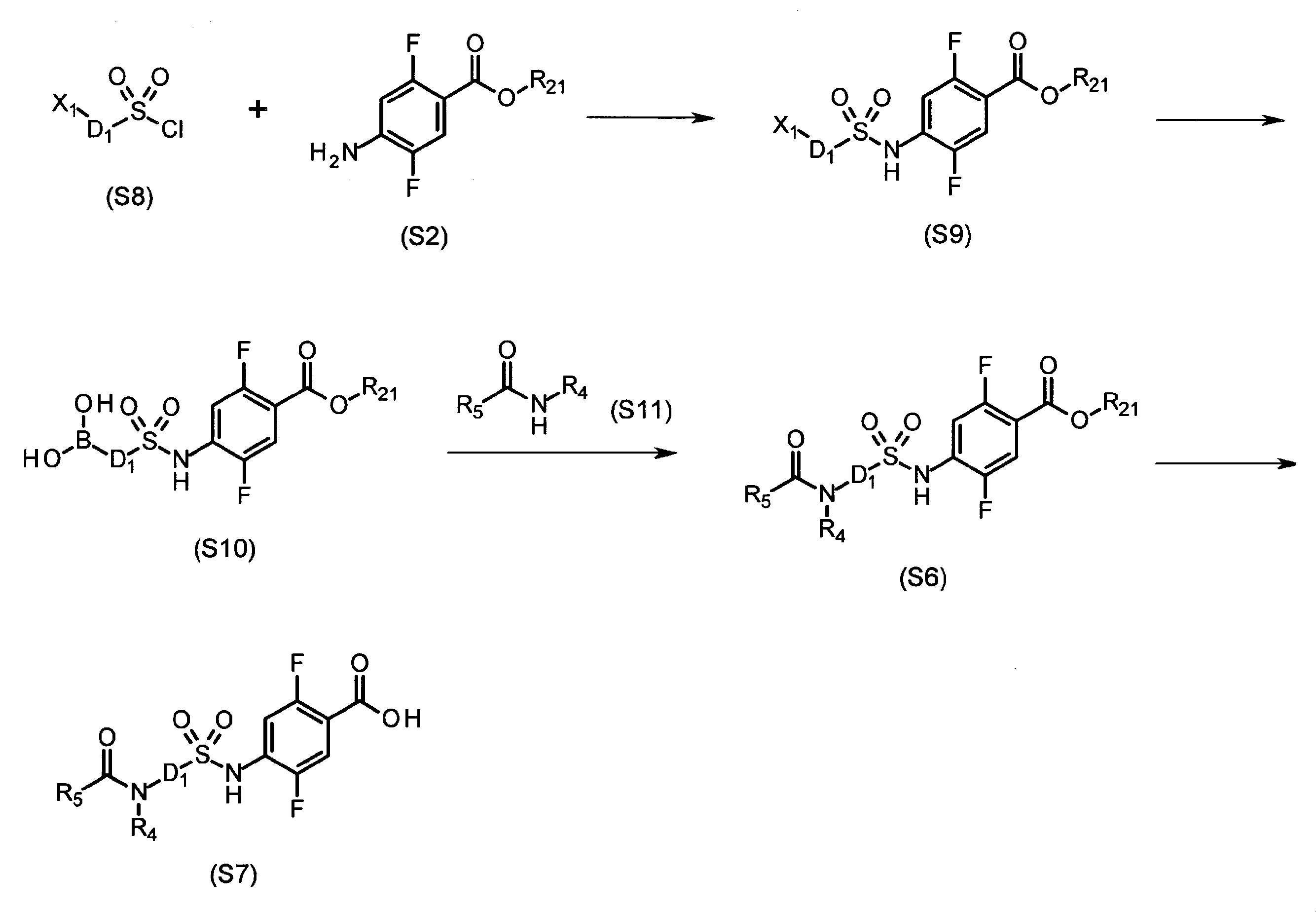
スルホニルクロリド誘導体(S8)とアニリン誘導体(S2)とを、例えば、ジクロロメタン、アセトニトリル、THF、またはDMF等の本反応に悪影響を及ぼさない溶媒中、例えば、ピリジン、またはトリエチルアミン等の塩基存在下で反応させることでスルホンアミド誘導体(S9)を合成できる。得られたスルホンアミド誘導体(S9)と例えば、ビス(ピナコラート)ジボラン等のボラン誘導体とを、例えば、DMF等の本反応に悪影響を及ぼさない溶媒中、例えば、酢酸カリウム等の塩基の存在下、例えば、1,1’-ビス(ジフェニルホスフィノフェロセン)ジクロロパラジウム(II)等の金属触媒を用いてカップリング反応を行うことで、対応するボロン酸エステル誘導体へと誘導し、続いて得られたボロン酸エステル誘導体に対して、例えば、アセトン等の本反応に悪影響を及ぼさない溶媒中、例えば、過ヨウ素酸ナトリウム、又は酢酸アンモニウム、及び、水を加えて処理をすることでボロン酸エステルを脱保護し、対応するボロン酸誘導体(S10)を合成することができる。得られたボロン酸誘導体(S10)とアミド誘導体(S11)とを、例えば、ジクロロメタン、ジメチルスルホキシド(DMSO)、又はDMF等の本反応に悪影響を及ぼさない溶媒中、例えば、ピリジン、又はトリエチルアミン等の塩基存在下、例えば、酢酸銅(II)、又はトリフルオロメタンスルホン酸銅(II)等の金属触媒を用いてカップリング反応を行うことで、化合物(S6)を合成することができる。続いて、化合物(S6)を、例えば、THF、1,4-ジオキサン、メタノール又はエタノール等の本反応に悪影響を及ぼさない溶媒中、例えば、水酸化ナトリウム、又は水酸化リチウム等の塩基を用いたアルカリ加水分解や、例えば、塩酸、又はトリフルオロ酢酸を用いた酸加水分解等を行うことで、目的とするカルボン酸誘導体(S7)を製造することができる。

ハロゲン化アリール誘導体(S9)とアミド誘導体(S11)とを、例えば、DMSO、NMP、又はDMF等の本反応に悪影響を及ぼさない溶媒中、例えば、トリエチルアミン、N,N-ジイソプロピルエチルアミン、又はジアザビシクロウンデセン(DBU)等の塩基存在下、例えば、ヨウ化銅(I)、臭化銅(I)、又は塩化銅(I)等の金属触媒を用いてカップリング反応を行うことにより、化合物(S6)を合成することができる。続いて、化合物(S6)を、例えば、THF、1,4-ジオキサン、メタノール又はエタノール等の本反応に悪影響を及ぼさない溶媒中、例えば、水酸化ナトリウム、又は水酸化リチウム等の塩基を用いたアルカリ加水分解や、例えば、塩酸、又はトリフルオロ酢酸を用いた酸加水分解等を行うことで、目的とするカルボン酸誘導体(S7)を製造することができる。

ハロゲン化アリール誘導体(S12)と例えば、ビス(ピナコラート)ジボラン等のボラン誘導体とを、例えば、DMF等の本反応に悪影響を及ぼさない溶媒中、例えば、酢酸カリウム等の塩基の存在下、例えば、1,1’-ビス(ジフェニルホスフィノフェロセン)ジクロロパラジウム(II)等の金属触媒を用いてカップリング反応を行うことで、対応するボロン酸エステル誘導体へと誘導し、続いて得られたボロン酸エステル誘導体に対して、例えば、アセトン等の本反応に悪影響を及ぼさない溶媒中、例えば、過ヨウ素酸ナトリウム、又は酢酸アンモニウム、及び、水を加えて処理をすることでボロン酸エステルを脱保護し、対応するボロン酸誘導体(S13)を合成することができる。得られたボロン酸誘導体(S13)とウラシル誘導体(S14)とを、例えば、ジクロロメタン、ジメチルスルホキシド(DMSO)、又はDMF等の本反応に悪影響を及ぼさない溶媒中、例えば、ピリジン、又はトリエチルアミン等の塩基存在下、例えば、酢酸銅(II)、又はトリフルオロメタンスルホン酸銅(II)等の金属触媒を用いてカップリング反応を行うことで、アミノ酸誘導体(S15)を合成することができる。続いて、アミノ酸誘導体(S14)を、例えば、塩酸、又はトリフルオロ酢酸を用いた酸加水分解、又は加水素分解等の脱保護を行うことで、目的とするカルボン酸誘導体(S16)を製造することができる。

ニトロ誘導体(S17)を、例えば、メタノール、エタノール、又はイソプロピルアルコール等の本反応に悪影響を及ぼさない溶媒中、例えば、パラジウムカーボン、水酸化パラジウム、又はラネーニッケル等の金属触媒を用いた接触還元反応、又は酸性条件下(例えば、塩酸、酢酸、又は塩化アンモニウム等)、例えば、亜鉛等の金属を作用させることで、アニリン誘導体(S18)を合成することができる。得られたアニリン誘導体(S18)とカルバメート誘導体(S19)とを、例えば、ジクロロメタン、1,4-ジオキサン、THF、又はDMF等の本反応に悪影響を及ぼさない溶媒中、トリエチルアミン、ピリジン、又はDBU等の塩基を用いて反応させることにより、アミノ酸誘導体(S15)を合成することができる。続いて、アミノ酸誘導体(S15)を、例えば、塩酸やトリフルオロ酢酸を用いた酸加水分解、又は加水素分解等の脱保護を行うことで、目的とするカルボン酸誘導体(S16)を製造することができる。

ハロゲン化アリール誘導体(S20)とウラシル誘導体(S14)とを、例えば、DMSO、NMP、又はDMF等の本反応に悪影響を及ぼさない溶媒中、例えば、トリエチルアミン、N,N-ジイソプロピルエチルアミン、又はDBU等の塩基存在下、例えば、ヨウ化銅(I)、臭化銅(I)、又は塩化銅(I)等の金属触媒を用いてカップリング反応を行うことにより、化合物(S21)を合成することができる。得られた化合物(S21)とハロゲン化物(S22)とを、例えば、DMF等の本反応に悪影響を及ぼさない溶媒中、例えば、ヨウ素等によって活性化させた亜鉛粉末存在下、例えば、トリス(ジベンジリデンアセトン)ジパラジウム(0)等の金属触媒と例えば、2-ジシクロヘキシル-2’,6’-ジメトキシビフェニル(SPhos)等の有機合成で一般的に用いられる配位子を用いて、根岸カップリング反応を行うことにより、アミノ酸誘導体(S15)を合成することができる。続いて、アミノ酸誘導体(S15)を、例えば、塩酸、又はトリフルオロ酢酸を用いた酸加水分解、又は加水素分解等の脱保護を行うことで、目的とするカルボン酸誘導体(S16)を製造することができる。

[合成例1]
4-[[4-(2,2-dimethylpropanoylamino)phenyl]sulfonylamino]-2,5-difluoro-benzoic acid

Methyl 2,5-difluoro-4-[(4-nitrophenyl)sulfonylamino]benzoateの合成
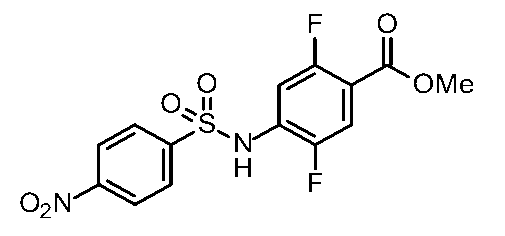
1H NMR (400 MHz, Chloroform-d) δ 8.39 - 8.32 (m, 2H), 8.09 - 8.02 (m, 2H), 7.62 (dd, J = 10.5, 6.2 Hz, 1H), 7.46 (dd, J = 11.0, 6.3 Hz, 1H), 7.08 (s, 1H), 3.90 (s, 3H) ; MS (ESI) m/z 373 [M+H]+
(工程2)
Methyl 4-[(4-aminophenyl)sulfonylamino]-2,5-difluoro-benzoateの合成

1H NMR (400 MHz, Chloroform-d) δ 7.69 - 7.61 (m, 2H), 7.58 (dd, J = 10.7, 6.3 Hz, 1H), 7.38 (dd, J = 11.6, 6.5 Hz, 1H), 6.98 (d, J = 12.5 Hz, 1H), 6.63 (d, J = 8.8 Hz, 2H), 3.89 (s, 3H) ; MS (ESI) m/z 343 [M+H]+
(工程3)
Methyl 4-[[4-(2,2-dimethylpropanoylamino)phenyl]sulfonylamino]-2,5-difluoro-benzoateの合成

MS (ESI) m/z 427 [M+H]+
(工程4)
4-[[4-(2,2-dimethylpropanoylamino)phenyl]sulfonylamino]-2,5-difluoro-benzoic acidの合成

1H NMR (400 MHz, DMSO-d6) δ 10.86 (s, 1H), 9.59 (s, 1H), 7.91 - 7.83 (m, 2H), 7.81 - 7.71 (m, 2H), 7.56 (dd, J = 10.7, 6.5 Hz, 1H), 7.18 (dd, J = 12.0, 6.3 Hz, 1H), 1.22 (s, 8H) ; MS (ESI) m/z 413 [M+H]+
4-[(4-acetamidophenyl)sulfonylamino]-2,5-difluoro-benzoic acid

[合成例3]
4-[[4-(2-ethylbutanoylamino)phenyl]sulfonylamino]-2,5-difluoro-benzoic acid

4-[[4-(tert-butylcarbamoylamino)phenyl]sulfonylamino]-2,5-difluoro-benzoic acid

Methyl 4-[[4-(tert-butylcarbamoylamino)phenyl]sulfonylamino]-2,5-difluoro-benzoateの合成

(工程2)
4-[[4-(tert-butylcarbamoylamino)phenyl]sulfonylamino]-2,5-difluoro-benzoic acidの合成
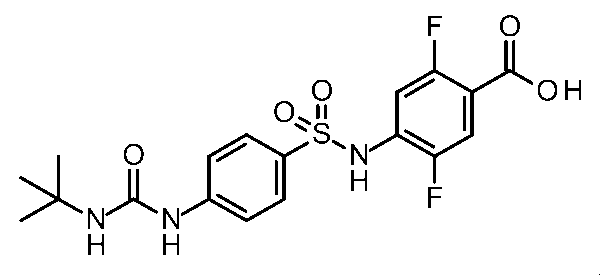
MS (ESI) m/z 428 [M+H]+
4-[[4-[(1-tert-butoxycarbonylpiperidine-4-carbonyl)amino]phenyl]sulfonylamino]-2,5-difluoro-benzoic acid
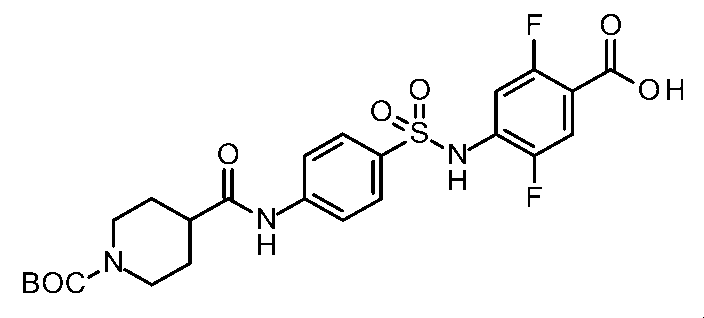
[合成例6]
2,5-difluoro-4-[[4-(pyridine-4-carbonylamino)phenyl]sulfonylamino]benzoic acid

4-[[4-(2,2-dimethylpropanoylamino)-3-fluoro-phenyl]sulfonylamino]-2,5-difluoro-benzoic acid

[合成例8]
2,5-difluoro-4-[[4-(tetrahydropyran-4-carbonylamino)phenyl]sulfonylamino]benzoic acid

2,5-difluoro-4-[[4-[(1-methoxycyclopropanecarbonyl)amino]phenyl]sulfonylamino]benzoic acid
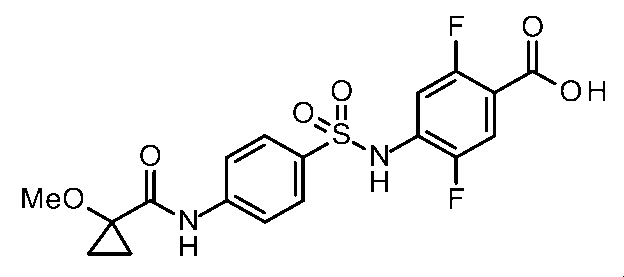
[合成例10]
2,5-difluoro-4-[[4-[(3-hydroxy-2,2-dimethyl-propanoyl)amino]phenyl]sulfonylamino]benzoic acid

4-[[5-(2,2-dimethylpropanoylamino)-2-pyridyl]sulfonylamino]-2,5-difluoro-benzoic acid

[合成例12]
2,5-difluoro-4-[[4-[[1-(trifluoromethyl)cyclopropanecarbonyl]amino]phenyl]sulfonylamino]benzoic acid
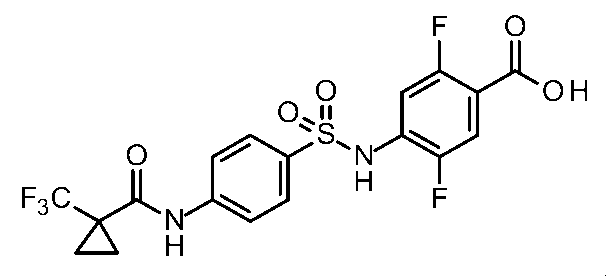
2,5-difluoro-4-[[4-[(1-hydroxycyclopropanecarbonyl)amino]phenyl]sulfonylamino]benzoic acid

[合成例14]
4-[[6-(2,2-dimethylpropanoylamino)-3-pyridyl]sulfonylamino]-2,5-difluoro-benzoic acid

[合成例15]
2,5-difluoro-4-[[4-[(1-phenylcyclopropanecarbonyl)amino]phenyl]sulfonylamino]benzoic acid

2,5-difluoro-4-[[4-(2-oxo-4-tetrahydropyran-4-yl-1-pyridyl)phenyl]sulfonylamino]benzoic acid

Methyl 4-[[4-(4-bromo-2-oxo-1-pyridyl)-4-methylene-cyclohexa-1,5-dien-1-yl]sulfonylamino]-2,5-difluoro-benzoateの合成

MS (ESI) m/z 499 [M+H]+
(工程2)
Methyl 4-[[4-[4-(3,6-dihydro-2H-pyran-4-yl)-2-oxo-1-pyridyl]-4-methylene-cyclohexa-1,5-dien-1-yl]sulfonylamino]-2,5-difluoro-benzoateの合成

MS (ESI) m/z 503 [M+H]+
(工程3)
2,5-difluoro-4-[[4-(2-oxo-4-tetrahydropyran-4-yl-1-pyridyl)phenyl]sulfonylamino]benzoic acidの合成
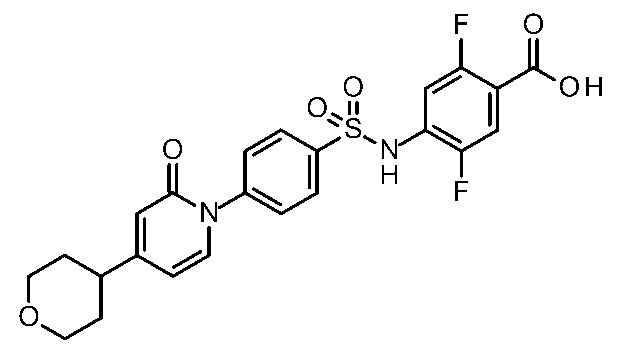
MS (ESI) m/z 491 [M+H]+
4-[[4-[2,2-dimethylpropanoyl(methyl)amino]phenyl]sulfonylamino]-2,5-difluoro-benzoic acid

[合成例18]
2,5-difluoro-4-[[4-(3,4,4-trimethyl-2,5-dioxo-imidazolidin-1-yl)phenyl]sulfonylamino]benzoic acid

[合成例19]
4-[[3-(2,2-dimethylpropanoylamino)phenyl]sulfonylamino]-2,5-difluoro-benzoic acid

[合成例20]
4-[[5-(2,2-dimethylpropanoylamino)-2-thienyl]sulfonylamino]-2,5-difluoro-benzoic acid
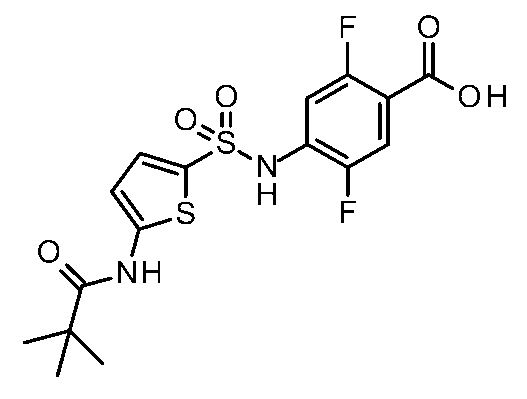
[合成例21]
4-[[4-(2,2-dimethylpropanoylamino)-2-thienyl]sulfonylamino]-2,5-difluoro-benzoic acid

Methyl (2S)-2-amino-3-[4-(1-methyl-2,4-dioxo-pyrido[3,4-d]pyrimidin-3-yl)phenyl]propanoate
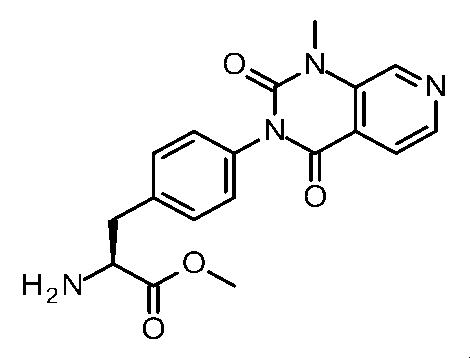
Methyl (2S)-2-amino-3-[4-(3,4,5-trimethyl-2,6-dioxo-pyrimidin-1-yl)phenyl]propanoate

Methyl (2S)-2-(tert-butoxycarbonylamino)-3-[4-(5,6-dimethyl-2,4-dioxo-1H-pyrimidin-3-yl)phenyl]propanoateの合成
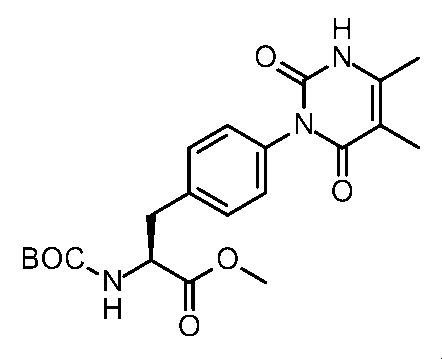
1H NMR (CDCl3, 400 MHz) δ 8.90 (s, 1H), 7.26 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 7.8 Hz, 2H), 5.06 (d, J = 8.7 Hz , 1H), 4.64-4.59 (m, 1H), 3.72 (s, 3H), 3.15 (d, J = 6.3 Hz, 2H), 2.16 (s, 3H), 1.95 (s, 3H), 1.44 (s, 9H).; MS (ESI) m/z 418[M+H]+
(工程2)
Methyl (2S)-2-(tert-butoxycarbonylamino)-3-[4-(3,4,5-trimethyl-2,6-dioxo-pyrimidin-1-yl)phenyl]propanoateの合成

1H NMR (CDCl3, 400 MHz) δ 8.02 (s, 1H), 7.24 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 1H), 5.05 (d, J = 8.0 Hz , 1H), 4.63-4.58 (m, 1H), 3.71 (s, 3H), 3.47 (s, 3H), 3.13 (d, J = 6.0 Hz, 2H), 2.33 (s, 3H), 2.02 (s, 3H), 1.43 (s, 9H).; MS (ESI) m/z 432[M+H]+
(工程3)
Methyl (2S)-2-amino-3-[4-(3,4,5-trimethyl-2,6-dioxo-pyrimidin-1-yl)phenyl]propanoateの合成

1H NMR (CD3OD, 400 MHz) δ 7.31 (d, J = 8.0 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H), 4.31-4.27 (m, 1H), 3.78 (s, 3H), 3.38 (s, 3H), 3.37-3.32 (m, 1H), 3.09-3.03 (m, 1H), 2.30 (s, 3H), 1.91 (s, 3H).; MS (ESI) m/z 332[M+H]+
Isopropyl (2S)-2-amino-3-[4-(1,7-dimethyl-2,4-dioxo-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-3-yl)phenyl]propanoate

Ethyl 1-benzyl-5-[(4-nitrophenoxy)carbonylamino]-3,6-dihydro-2H-pyridine-4-carboxylateの合成

(工程2)
Isopropyl (2S)-3-(4-aminophenyl)-2-(tert-butoxycarbonylamino)propanoateの合成

(工程3)
isopropyl (2S)-3-[4-(7-benzyl-2,4-dioxo-1,5,6,8-tetrahydropyrido[3,4-d]pyrimidin-3-yl)phenyl]-2-(tert-butoxycarbonylamino)propanoateの合成

(工程4)
Isopropyl (2S)-3-[4-(7-benzyl-1,7-dimethyl-2,4-dioxo-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-7-ium-3-yl)phenyl]-2-(tert-butoxycarbonylamino)propanoateの合成

(工程5)
Isopropyl (2S)-2-amino-3-[4-(1,7-dimethyl-2,4-dioxo-6,8-dihydro-5H-pyrido[3,4-d]pyrimidin-3-yl)phenyl]propanoateの合成
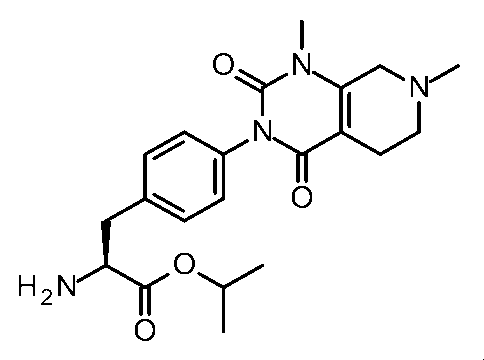
MS (ESI) m/z 401 [M+H]+
Methyl (2S)-2-amino-3-[6-(1-methyl-2,4-dioxo-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-3-yl)-3-pyridyl]propanoate
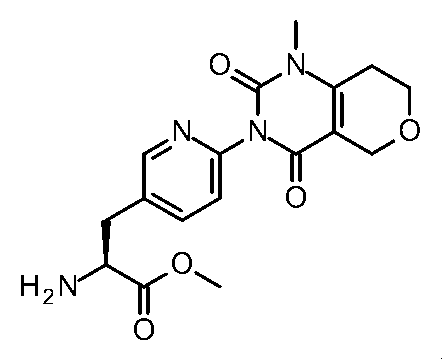
Ethyl 4-amino-3,6-dihydro-2H-pyran-5-carboxylateの合成

(工程2)
1,5,7,8-tetrahydropyrano[4,3-d]pyrimidine-2,4-dioneの合成

(工程3)
3-(5-bromo-2-pyridyl)-1,5,7,8-tetrahydropyrano[4,3-d]pyrimidine-2,4-dioneの合成

(工程4)
3-(5-bromo-2-pyridyl)-1-methyl-7,8-dihydro-5H-pyrano[4,3-d]pyrimidine-2,4-dioneの合成

(工程5)
Methyl (2S)-2-(tert-butoxycarbonylamino)-3-[6-(1-methyl-2,4-dioxo-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-3-yl)-3-pyridyl]propanoateの合成

別の容器に、(工程4)で得られた化合物(0.20g,0.59mmol)を入れ、DMF(1.0ml)に溶解させた。トリス(ジベンジリデンアセトン)ジパラジウム(0)(14mg,0.015mmol)、SPhos(24mg,0.058mmol)を順次加え、10分間撹拌した。この混合溶液を先に調製した混合溶液に加え、脱気とアルゴン置換操作を3回行った後、60℃にて18時間撹拌した。反応溶液を室温まで冷却し、減圧濃縮した後、残渣をODSを充填剤とする逆相HPLCに付し、トリフルオロ酢酸0.1%(v/v)含有する水とアセトニトリルの混合溶液で溶出し、目的のフラクションを凍結乾燥することにより、表題化合物を得た(0.12g,45%)。
(工程6)
Methyl (2S)-2-amino-3-[6-(1-methyl-2,4-dioxo-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-3-yl)-3-pyridyl]propanoateの合成

Methyl (2S)-2-amino-3-[6-(1-methyl-2,4-dioxo-pyrido[3,4-d]pyrimidin-3-yl)-3-pyridyl]propanoate

Isopropyl (2S)-2-amino-3-[4-(1-methyl-2,4-dioxo-7,8-dihydro-5H-pyrano[4,3-d]pyrimidin-3-yl)phenyl]propanoate

[合成例28]
Methyl (2S)-2-amino-3-[4-(6-methoxy-1-methyl-2,4-dioxo-pyrido[3,4-d]pyrimidin-3-yl)phenyl]propanoate

Methyl (2S)-2-amino-3-[4-(3,5-dimethyl-2,6-dioxo-pyrimidin-1-yl)phenyl]propanoate

Methyl (2S)-2-amino-3-[6-(1-methyl-2,4-dioxo-6,8-dihydro-5H-pyrano[3,4-d]pyrimidin-3-yl)-3-pyridyl]propanoate

Isopropyl (2S)-2-amino-3-[4-(1-methyl-2,4-dioxo-6,8-dihydro-5H-pyrano[3,4-d]pyrimidin-3-yl)phenyl]propanoate

[合成例32]
Methyl (2S)-2-amino-3-[4-(3-methyl-2,6-dioxo-pyrimidin-1-yl)phenyl]propanoate
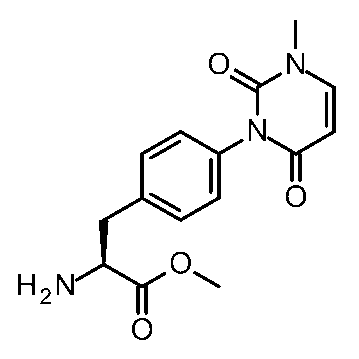
Methyl (2S)-2-amino-3-[4-(3,7-dimethyl-2,6-dioxo-purin-1-yl)phenyl]propanoate

Methyl (2S)-2-amino-3-[6-(3,4,5-trimethyl-2,6-dioxo-pyrimidin-1-yl)-3-pyridyl]propanoate

Methyl (2S)-2-amino-3-[6-(3,4-dimethyl-2,6-dioxo-pyrimidin-1-yl)-3-pyridyl]propanoate

3-(5-bromopyridin-2-yl)-6-methylpyrimidine-2,4(1H,3H)-dioneの合成
MS (ESI) m/z 282 [M+H]+
(工程2)
3-(5-bromopyridin-2-yl)-1,6-dimethylpyrimidine-2,4(1H,3H)-dioneの合成

1H NMR (400 MHz, DMSO-d6) δ 8.72 (d, J = 2.5 Hz, 1H), 8.23 (dd, J = 8.4, 2.6 Hz, 1H), 7.40 (d, J = 8.4 Hz, 1H), 5.76 (s, 1H), 3.32 (s, 3H), 2.32 (s, 3H);MS (ESI) m/z 296 [M+H]+
(工程3)
Methyl (2S)-2-((tert-butoxycarbonyl)amino)-3-(6-(3,4-dimethyl-2,6-dioxo-3,6-dihydropyrimidin-1(2H)-yl)pyridin-3-yl)propanoateの合成

別の容器に、(工程2)で得られた化合物(0.18g, 0.60mmol)を入れ、DMF(1.0ml)に溶解させた。トリス(ジベンジリデンアセトン)ジパラジウム(14mg, 0.015mmol)、SPhos(24mg, 0.058mmol)を順次加え、10分間撹拌した。この混合溶液を先に調製した混合溶液に加え、脱気とアルゴン置換操作を3回行った後、60℃にて18時間撹拌した。反応溶液を室温まで冷却し、減圧濃縮した後、残渣を(工程1)と同様の方法(逆相HPLC分取)にて精製することにより、表題化合物を得た(0.11g, 43%)。
MS (ESI) m/z 419 [M+H]+
(工程4)
Methyl (2S)-2-amino-3-[6-(3,4-dimethyl-2,6-dioxo-pyrimidin-1-yl)-3-pyridyl]propanoateの合成

MS (ESI) m/z 319 [M+H]+
Methyl (2S)-2-amino-3-[4-(3,4-dimethyl-2,6-dioxo-pyrimidin-1-yl)phenyl]propanoate
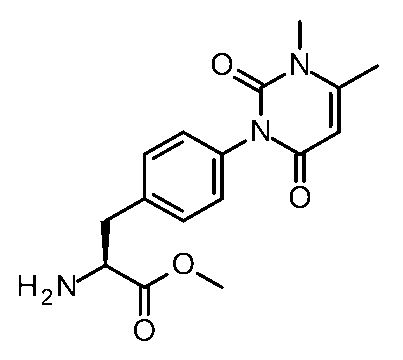
Isopropyl (2S)-2-amino-3-[5-(1-methyl-2,4-dioxo-5,6,7,8-tetrahydroquinazolin-3-yl)-2-pyridyl]propanoate

Ethyl 2-[(4-nitrophenoxy)carbonylamino]cyclohexene-1-carboxylateの合成

MS (ESI) m/z 335[M+H]+
(工程2)
3-(6-bromo-3-pyridyl)-5,6,7,8-tetrahydro-1H-quinazoline-2,4-dioneの合成
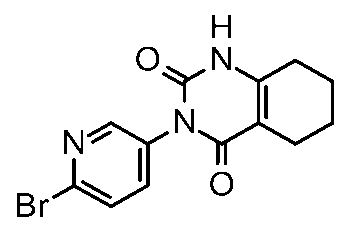
MS (ESI) m/z 322[M+H]+
(工程3)
3-(6-bromo-3-pyridyl)-1-methyl-5,6,7,8-tetrahydroquinazoline-2,4-dioneの合成
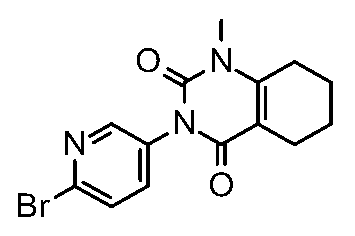
MS (ESI) m/z 336[M+H]+
(工程4)
Isopropyl (2S)-2-(tert-butoxycarbonylamino)-3-[5-(1-methyl-2,4-dioxo-5,6,7,8-tetrahydroquinazolin-3-yl)-2-pyridyl]propanoateの合成
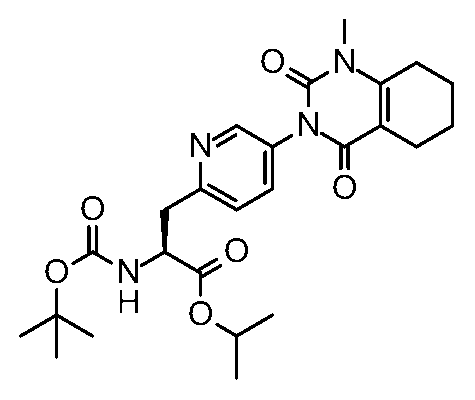
別の容器に、(工程3)で得られた化合物(256mg、0.801mmol)を入れ、DMF(2.0ml)に溶解させた。トリス(ジベンジリデンアセトン)ジパラジウム(0)(18.3mg,0.020mmol)、SPhos(32.9mg,0.0801mmol)を順次加え、10分間撹拌した。この混合溶液を先に調製した混合溶液に加え、脱気とアルゴン置換操作を3回行った後、60℃にて15時間撹拌した。反応溶液を室温まで冷却し、水(25ml)、及びジクロロメタン(25ml)を加えた。セライトろ過し、ジクロロメタンにて2回抽出した後、有機層を飽和塩化ナトリウム水溶液で洗浄した。無水硫酸ナトリウムで乾燥した後、減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィーで精製することにより、表題化合物を得た(223mg、57.3%)。
MS (ESI) m/z 487[M+H]+
(工程5)
Isopropyl (2S)-2-amino-3-[5-(1-methyl-2,4-dioxo-5,6,7,8-tetrahydroquinazolin-3-yl)-2-pyridyl]propanoate塩酸塩の合成
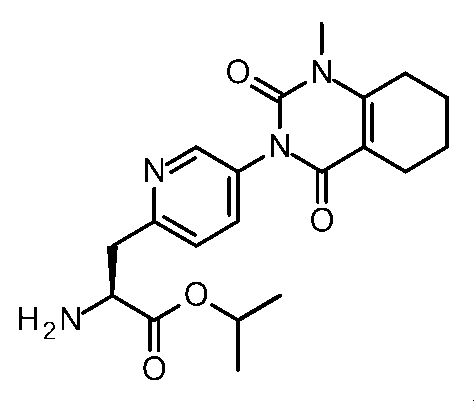
MS (ESI) m/z 387[M+H]+
[合成例38]
Methyl (2S)-2-amino-3-[4-(3-isopropyl-4,5-dimethyl-2,6-dioxo-pyrimidin-1-yl)phenyl]propanoate
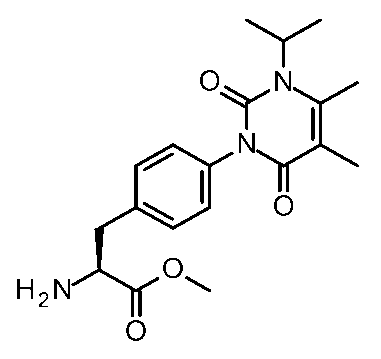
Methyl (2S)-2-amino-3-[4-[5-(hydroxymethyl)-3,4-dimethyl-2,6-dioxo-pyrimidin-1-yl]phenyl]propanoate

Methyl (2S)-2-(tert-butoxycarbonylamino)-3-[4-(5-(ヒドロキシメチル)-6-methyl-2,4-dioxo-1H-pyrimidin-3-yl)phenyl]propanoateの合成
1H NMR (400 MHz, DMSO-d6) δ 11.17 (s, 1H), 7.39 - 7.26 (m, 3H), 7.13 - 7.03 (m, 2H), 4.26 - 4.10 (m, 3H), 3.63 (s, 3H), 3.04 (dd, J = 14.0, 4.8 Hz, 1H), 2.91 (dd, J = 13.8, 10.2 Hz, 1H), 2.20 (s, 3H), 1.35 (s, 9H) ; MS (ESI) m/z 434[M+H]+
(工程2)
Methyl (2S)-2-(tert-butoxycarbonylamino)-3-[4-{5-(hydroxymethyl)-3,4-dimethyl-2,6-dioxo-pyrimidin-1-yl}phenyl]propanoateの合成

MS (ESI) m/z 448[M+H]+
(工程3)
Methyl (2S)-2-amino-3-[4-[5-(hydroxymethyl)-3,4-dimethyl-2,6-dioxo-pyrimidin-1-yl]phenyl]propanoateの合成

MS (ESI) m/z 348[M+H]+
Methyl (2S)-2-amino-3-[6-(1-methyl-2,4-dioxo-5,6,7,8-tetrahydroquinazolin-3-yl)-3-pyridyl]propanoate
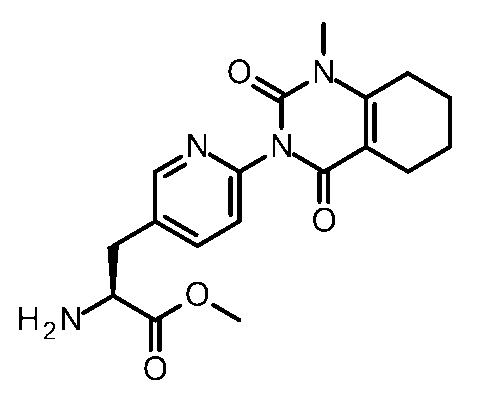
Methyl (2S)-2-amino-3-[6-(3-methyl-2,6-dioxo-pyrimidin-1-yl)-3-pyridyl]propanoate

tert-butyl 3-benzoyl-2,4-dioxo-pyrimidine-1-carboxylateの合成

1H NMR (400 MHz, DMSO-d6) δ 8.15 (dd, J = 8.5, 1.6 Hz, 1H), 8.11 - 8.04 (m, 2H), 7.87 - 7.76 (m, 1H), 7.68 - 7.56 (m, 2H), 5.98 (dd, J = 8.4, 1.7 Hz, 1H), 1.54 (d, J = 1.6 Hz, 9H) ; MS (ESI) m/z 317[M+H]+
(工程2)
3-benzoyl-1-methyl-pyrimidine-2,4-dioneの合成
1H NMR (400 MHz, DMSO-d6) δ 7.96 (dd, J = 8.0, 1.3 Hz, 2H), 7.87 (d, J = 7.9 Hz, 1H), 7.83 - 7.72 (m, 1H), 7.60 (dd, J = 8.4, 7.2 Hz, 2H), 5.81 (dd, J = 7.9, 0.6 Hz, 1H), 3.32 (s, 3H) ; MS (ESI) m/z 231[M+H]+.
(工程3)
1-methyluracilの合成
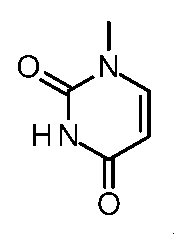
1H NMR (400 MHz, DMSO-d6) δ 11.21 (s, 1H), 7.61 (d, J = 7.8 Hz, 1H), 5.51 (d, J = 7.8 Hz, 1H), 3.22 (s, 3H) ; MS (ESI) m/z 127[M+H]+.
(工程4)
3-(5-bromo-2-pyridyl)-1-methyl-pyrimidine-2,4-dione、及び3-(5-iodo-2-pyridyl)-1-methyl-pyrimidine-2,4-dioneの混合物の合成

(工程5)
Methyl (2S)-2-(tert-butoxycarbonylamino)-3-[6-(3-methyl-2,6-dioxo-pyrimidin-1-yl)-3-pyridyl]propanoateの合成
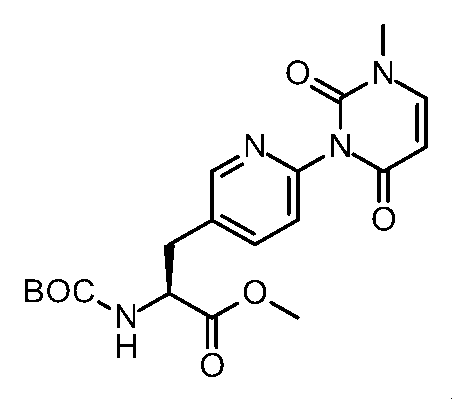
別の容器に、(工程4)で得られた混合物(0.29g)を入れ、DMF(1.0ml)に溶解させた。トリス(ジベンジリデンアセトン)ジパラジウム(0)(22mg,0.024mmol)、SPhos(20mg,0.049mmol)を順次加え、10分間撹拌した。この混合溶液を先に調製した混合溶液に加え、脱気とアルゴン置換操作を3回行った後、60℃にて18時間撹拌した。反応溶液を室温まで冷却し、水(25ml)、及びジクロロメタン(25ml)を加えた。セライトろ過し、ジクロロメタンにて2回抽出した後、有機層を飽和塩化ナトリウム水溶液で洗浄した。無水硫酸マグネシウムで乾燥した後、減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィーで精製することにより、表題化合物を得た(0.21g)。
1H NMR (400 MHz, DMSO-d6) δ 8.42 (d, J = 2.3 Hz, 1H), 7.92 - 7.72 (m, 2H), 7.44 (d, J = 8.1 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 5.77 (d, J = 7.9 Hz, 1H), 4.35 - 4.20 (m, 1H), 3.64 (s, 3H), 3.31 (s, 3H), 3.11 (dd, J = 13.9, 4.8 Hz, 1H), 2.94 (dd, J = 14.1, 10.6 Hz, 1H), 1.34 (s, 9H) ; MS (ESI) m/z 405[M+H]+.
(工程6)
Methyl (2S)-2-amino-3-[6-(3-methyl-2,6-dioxo-pyrimidin-1-yl)-3-pyridyl]propanoateの合成
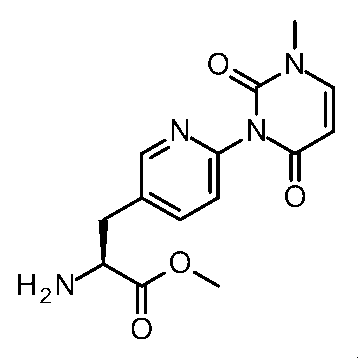
MS (ESI) m/z 305[M+H]+.
Isopropyl (2S)-2-amino-3-[6-(3,5-dimethyl-2,6-dioxo-pyrimidin-1-yl)-3-pyridyl]propanoate TFA salt

tert-butyl 3-benzoyl-5-methyl-2,4-dioxo-3,4-dihydropyrimidine-1(2H)-carboxylateの合成
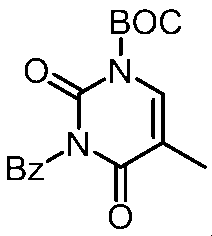
MS (ESI) m/z 331[M+H]+
(工程2)
3-benzoyl-1,5-dimethylpyrimidine-2,4(1H,3H)-dioneの合成

MS (ESI) m/z 245[M+H]+.
(工程3)
1-methyl-thymineの合成
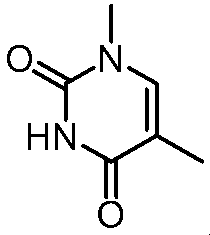
MS (ESI) m/z 141[M+H]+.
(工程4)
3-(5-bromopyridine-2-yl)-1,5-dimethylpyrimidine-2,4(1H,3H)-dioneの合成

1H NMR (400 MHz, DMSO-d6) δ 8.72 (d, J = 2.5 Hz, 1H), 8.24 (dd, J = 8.4, 2.6 Hz, 1H), 7.72 (d, J = 1.4 Hz, 1H), 7.42 (d, J = 8.4 Hz, 1H), 3.29 (s, 3H), 1.83 (s, 3H).
MS (ESI) m/z 296[M+H]+.
(工程5)
Methyl (2S)-2-(tert-butoxycarbonylamino)-3-[6-(3,5-dimethyl-2,6-dioxo-pyrimidin-1-yl)-3-pyridyl]propanoateの合成

別の容器に、(工程4)で得られた化合物(350mg,1.18mmol)を入れ、DMF(5ml)に溶解させた。トリス(ジベンジリデンアセトン)ジパラジウム(0)(27mg,0.03mmol)、SPhos(49mg,0.12mmol)を順次加え、10分間撹拌した。この混合溶液を先に調製した混合溶液に加え、脱気とアルゴン置換操作を3回行った後、60℃にて18時間撹拌した。反応溶液を室温まで冷却し、水(25ml)、及びジクロロメタン(25ml)を加えた。セライトろ過し、ジクロロメタンにて2回抽出した後、有機層を飽和塩化ナトリウム水溶液で洗浄した。無水硫酸マグネシウムで乾燥した後、減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィーで精製することにより、表題化合物を得た(404mg,77%)。
MS (ESI) m/z 447[M+H]+.
(工程6)Isopropyl (2S)-2-amino-3-[6-(3,5-dimethyl-2,6-dioxo-pyrimidin-1-yl)-3-pyridyl]propanoate TFA saltの合成

1H NMR (400 MHz, DMSO-d6) δ 8.58 (s, 3H), 8.45 (d, J = 2.4 Hz, 1H), 7.87 (dd, J = 8.1, 2.4 Hz, 1H), 7.71 (d, J = 1.3 Hz, 1H), 7.36 (d, J = 8.1 Hz, 1H), 4.91 (p, J = 6.2 Hz, 1H), 4.40 (s, 1H), 3.29 (s, 3H), 3.24 (dd, J = 14.3, 5.9 Hz, 1H), 3.11 (dd, J = 14.3, 8.6 Hz, 1H), 1.83 (d, J = 1.0 Hz, 3H), 1.16 (d, J = 6.2 Hz, 3H), 1.06 (d, J = 6.2 Hz, 3H) ; MS (ESI) m/z 347[M+H]+.
4-((4-(4,4-dimethylpent-2-ynamido)phenyl)sulfonamido)-2,5-difluorobenzoic acid

4,4-dimethylpent-2-ynoic acidの合成

1H NMR (300 MHz, CD3OD) δ 7.70 (br, s, 1H), 1.26 (s, 9H).
(工程2)
Methyl 4-((4-(4,4-dimethylpent-2-ynamido)phenyl)sulfonamido)-2,5-difluorobenzoateの合成

(工程3)
4-((4-(4,4-dimethylpent-2-ynamido)phenyl)sulfonamido)-2,5-difluorobenzoic acidの合成

1H NMR (400 MHz, DMSO-d6) δ 13.25 (br, s, 1H), 10.96-10.86 (m, 2H), 7.85-7.74 (m, 4H), 7.61-7.57 (m, 1H), 7.24-7.19 (m, 1H), 1.25 (m, 9H); MS (ESI) m/z 437[M+H]+.
2,5-difluoro-4-((6-pivalamido-1H-indole)-3-sulfonamido)benzoic acid

6-nitro-1H-indole-3-sulfonyl chlorideの合成

1H NMR (400 MHz, CDCl3) δ 9.60 (br, s 1H), 8.51 (d, J = 8.8 Hz, 1H), 8.32 (q, J = 10.8, 6.4 Hz, 1H), 8.24 (d, J = 3.2 Hz, 1H), 8.18 (d, J = 8.8 Hz, 1H).
(工程2)
N-(4-bromo-2,5-difluorophenyl)-6-nitro-1H-indole-3-sulfonamideの合成
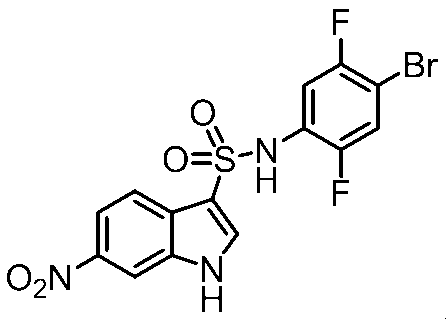
1H NMR (300 MHz, DMSO-d6) δ 12.57 (s, 1H), 10.61 (s, 1H), 8.37 (q, J = 6.4, 5.2 Hz, 1H), 8.10 (d, J = 12.0 Hz, 1H), 7.97 (d, J = 8.0 Hz, 1H), 7.59-7.65 (m, 1H), 7.31-7.37 (m, 1 H) ; MS (ESI) m/z 430[M-H]-.
(工程3)
Methyl 4-((6-amino-1H-indole)-3-sulfonamido)-2,5-difluorobenzoateの合成

1H NMR (400 MHz, DMSO-d6) δ 7.72 (s, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.48 (q, J = 11.2, 6.8 Hz, 1H), 7.37 (q, J = 6.4, 5.2 Hz, 1H), 6.70-6.76 (m, 2H), 3.82 (s, 3H); MS (ESI) m/z 380[M-H]-.
(工程4)
Methyl 2,5-difluoro-4-((6-pivalamido-1H-indole)-3-sulfonamido)benzoateの合成

MS (ESI) m/z 466[M+H]+.
(工程5)
2,5-difluoro-4-((6-pivalamido-1H-indole)-3-sulfonamido)benzoic acidの合成

1H NMR (400 MHz, CD3OD) δ 7.91 (s, 1H), 7.85 (s, 1H), 7.82 (d, J = 3.2 Hz, 1H), 7.47 (q, J = 10.8, 6.8 Hz, 1H), 7.38 (q, J = 12.0, 11.6 Hz, 1H), 7.21 (q, J = 8.8, 8.8 Hz, 1H), 1.30 (s, 9H) ; MS (ESI) m/z 452[M+H]+.
2,5-difluoro-4-((5-pivalamido-1H-indole)-3-sulfonamido)benzoic acid

2,5-difluoro-4-((3-pivalamido-1H-indole)-6-sulfonamido)benzoic acidの合成
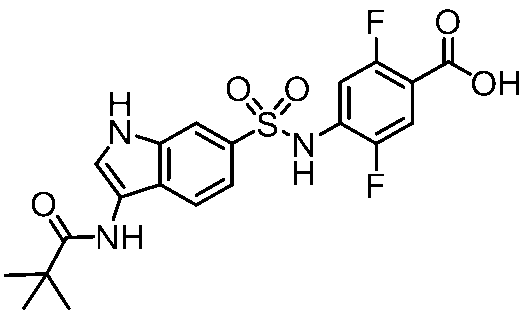
6-Bromo-1-(triisopropylsilyl)-1H-indoleの合成

(工程2)
1-(triisopropylsilyl)-1H-indole-6-sulfonyl chlorideの合成
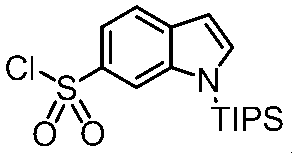
1H NMR (400 MHz, CDCl3) δ 8.21 (s, 1H), 7.78-7.77 (m, 2H), 7.56 (s, 1H), 6.77 (s, 1H), 1.76-1.69 (m, 3H), 1.17 (s, 9H), 1.15 (s, 9H).
(工程3)
N-(4-bromo-2,5-difluorophenyl)-1-(triisopropylsilyl)-1H-indole-6-sulfonamideの合成

1H NMR (300 MHz, DMSO-d6): δ 10.51 (s, 1H), 7.85-7.77 (m, 2H), 7.65-7.25 (m, 4H), 6.77-6.59 (m, 1H), 1.73-1.57 (m, 3H), 1.09 (d, J = 7.5 Hz, 6H), 1.00 (d, J = 10.5 Hz, 12H).
(工程4)
Methyl 2,5-difluoro-4-(1H-indole-6-sulfonamido)benzoateの合成

1H NMR (300 MHz, DMSO-d6): δ 11.65 (s, 1H), 10.91 (s, 1H), 7.97 (s, 1H), 7.74-7.56 (m, 3H), 7.48-7.45 (m, 1H), 7.33-7.28 (m, 1H), 6.56 (s, 1H), 3.78 (s, 3H).
(工程5)
Methyl 2,5-difluoro-4-((3-nitro-1H-indole)-6-sulfonamido)benzoateの合成

1H NMR (300 MHz, DMSO-d6): δ 13.03 (br s, 1H), 11.10 (br s, 1H), 8.89-8.87 (m, 1H), 8.26 (d, J = 8.7 Hz, 1H), 8.07 (s, 1H), 8.82 (dd, J = 8.7 Hz, 1.5 Hz, 1H), 7.65-7.59 (m, 1H), 7.34-7.28 (m, 1H), 3.79 (s, 3H).
(工程6)
Methyl 4-((3-amino-1H-indole)-6-sulfonamido)-2,5-difluorobenzoateの合成

(工程7)
2,5-difluoro-4-((3-pivalamido-1H-indole)-6-sulfonamido)benzoic acidの合成
1H NMR (400 MHz, CD3OD): δ 7.94 (s, 1H), 7.66-7.63 (m, 2H), 7.53-7.45 (m, 2H), 7.40-7.36 (m, 1H), 1.37 (m, 9H) ; MS (ESI) m/z 452[M+H]+.
(2S)-2-[[4-[[4-(2,2-dimethylpropanoylamino)phenyl]sulfonylamino]-2,5-difluoro-benzoyl]amino]-3-[4-(1-methyl-2,4-dioxo-pyrido[3,4-d]pyrimidin-3-yl)phenyl]propanoic acid(A1)

Methyl (2S)-2-[[4-[[4-(2,2-dimethylpropanoylamino)phenyl]sulfonylamino]-2,5-difluoro-benzoyl]amino]-3-[4-(1-methyl-2,4-dioxo-pyrido[3,4-d]pyrimidin-3-yl)phenyl]propanoateの合成
(工程2)
(2S)-2-[[4-[[4-(2,2-dimethylpropanoylamino)phenyl]sulfonylamino]-2,5-difluoro-benzoyl]amino]-3-[4-(1-methyl-2,4-dioxo-pyrido[3,4-d]pyrimidin-3-yl)phenyl]propanoic acidの合成
(2S)-2-[[2,5-difluoro-4-[[4-(piperidine-4-carbonylamino)phenyl]sulfonylamino]benzoyl]amino]-3-[4-(1-methyl-2,4-dioxo-pyrido[3,4-d]pyrimidin-3-yl)phenyl]propanoic acid(A6)

tert-butyl 4-[[4-[[2,5-difluoro-4-[[(1S)-2-methoxy-1-[[4-(1-methyl-2,4-dioxo-pyrido[3,4-d]pyrimidin-3-yl)cyclohexa-2,4-dien-1-yl]methyl]-2-oxo-ethyl]carbamoyl]phenyl]sulfamoyl]phenyl]carbamoyl]piperidine-1-carboxylateの合成

(工程2)
(2S)-2-[[2,5-difluoro-4-[[4-(piperidine-4-carbonylamino)phenyl]sulfonylamino]benzoyl]amino]-3-[4-(1-methyl-2,4-dioxo-pyrido[3,4-d]pyrimidin-3-yl)phenyl]propanoic acidの合成

[実施例3]
(2S)-2-[2,5-difluoro-4-[(4-pivalamidophenyl)sulfonamide]benzamido]-3-[6-(3-methyl-2,6-dioxotetrahydropyrimidin-1(2H)-yl)pyridin-3-yl]propanoic acid(A75)



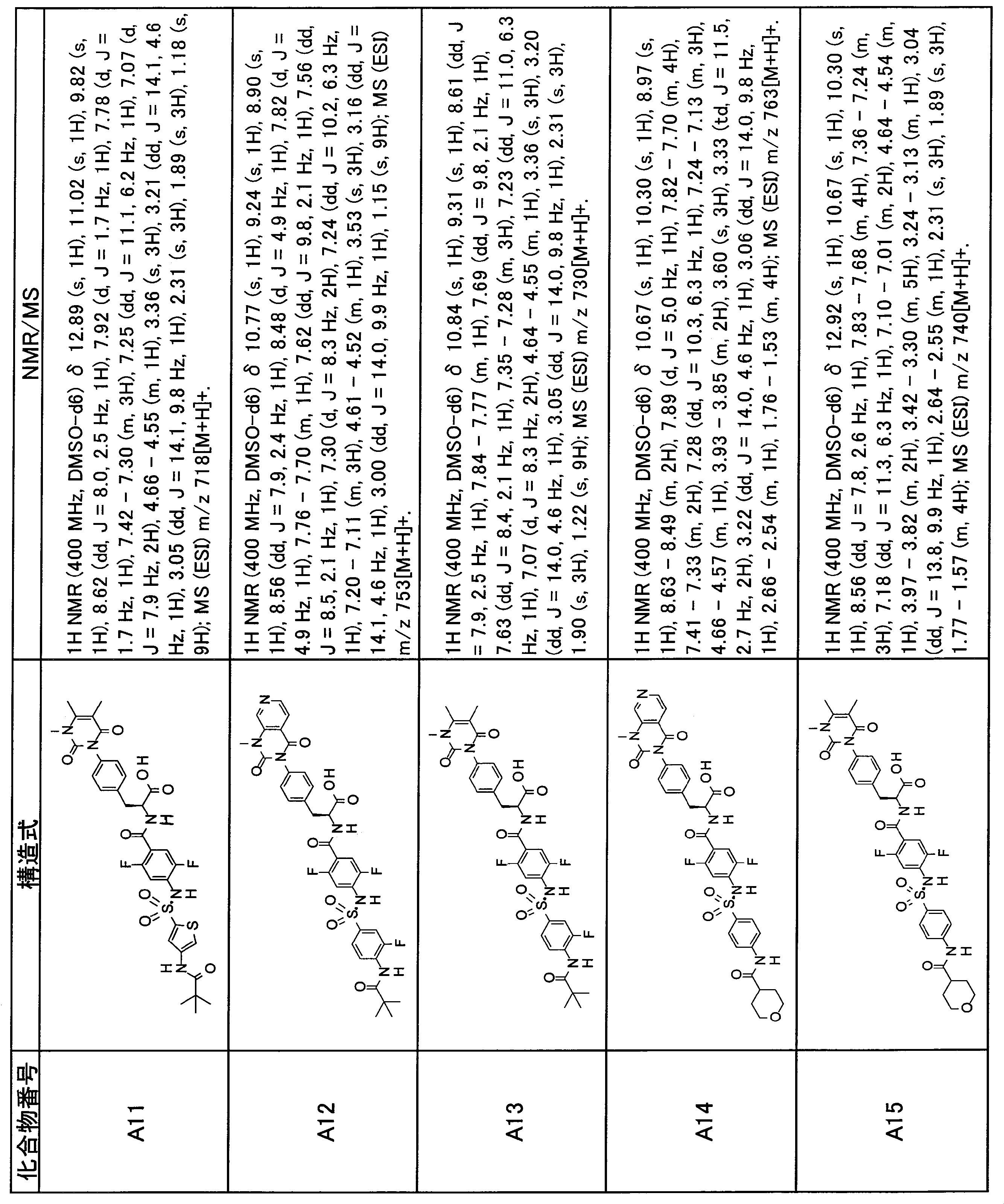








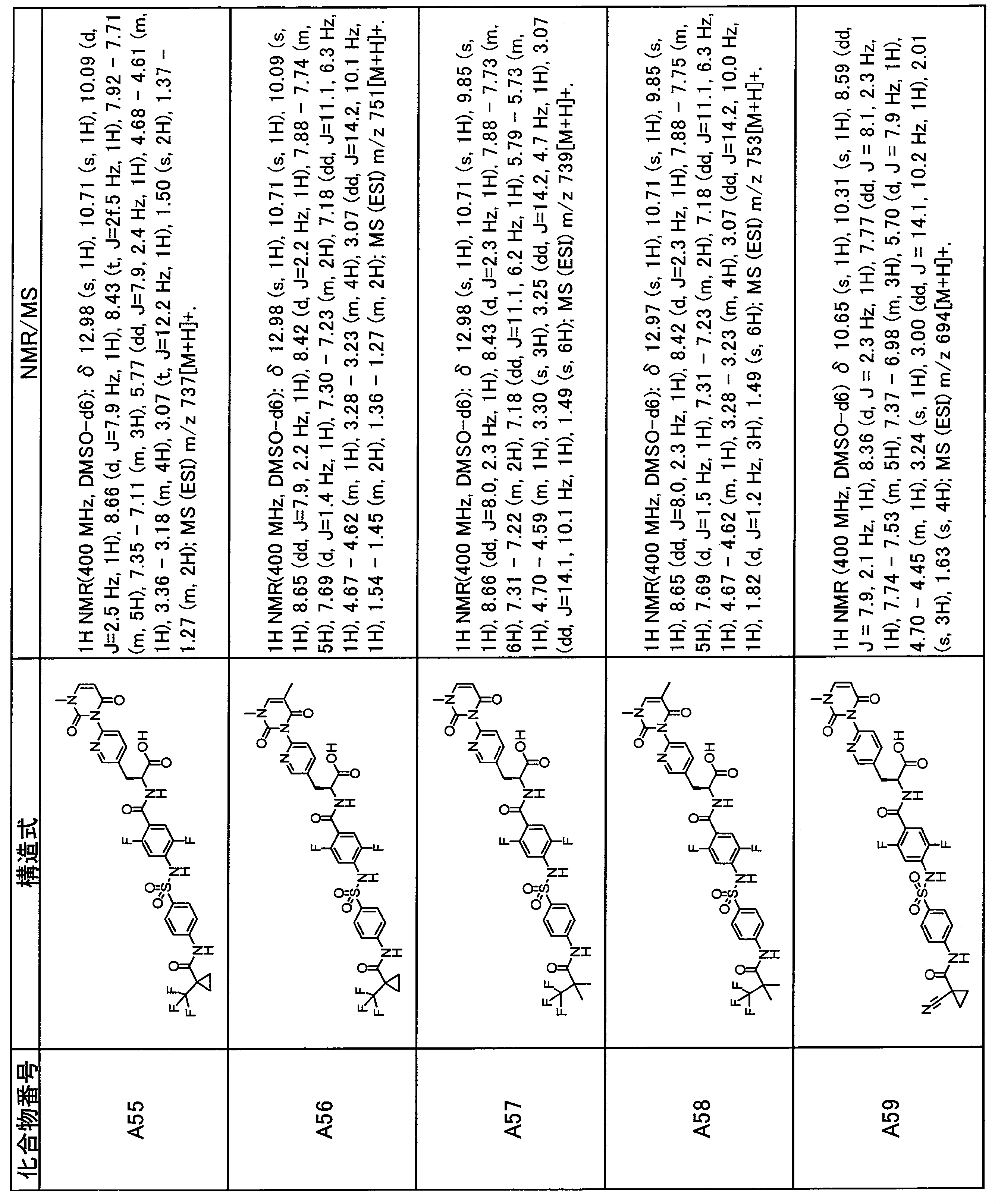
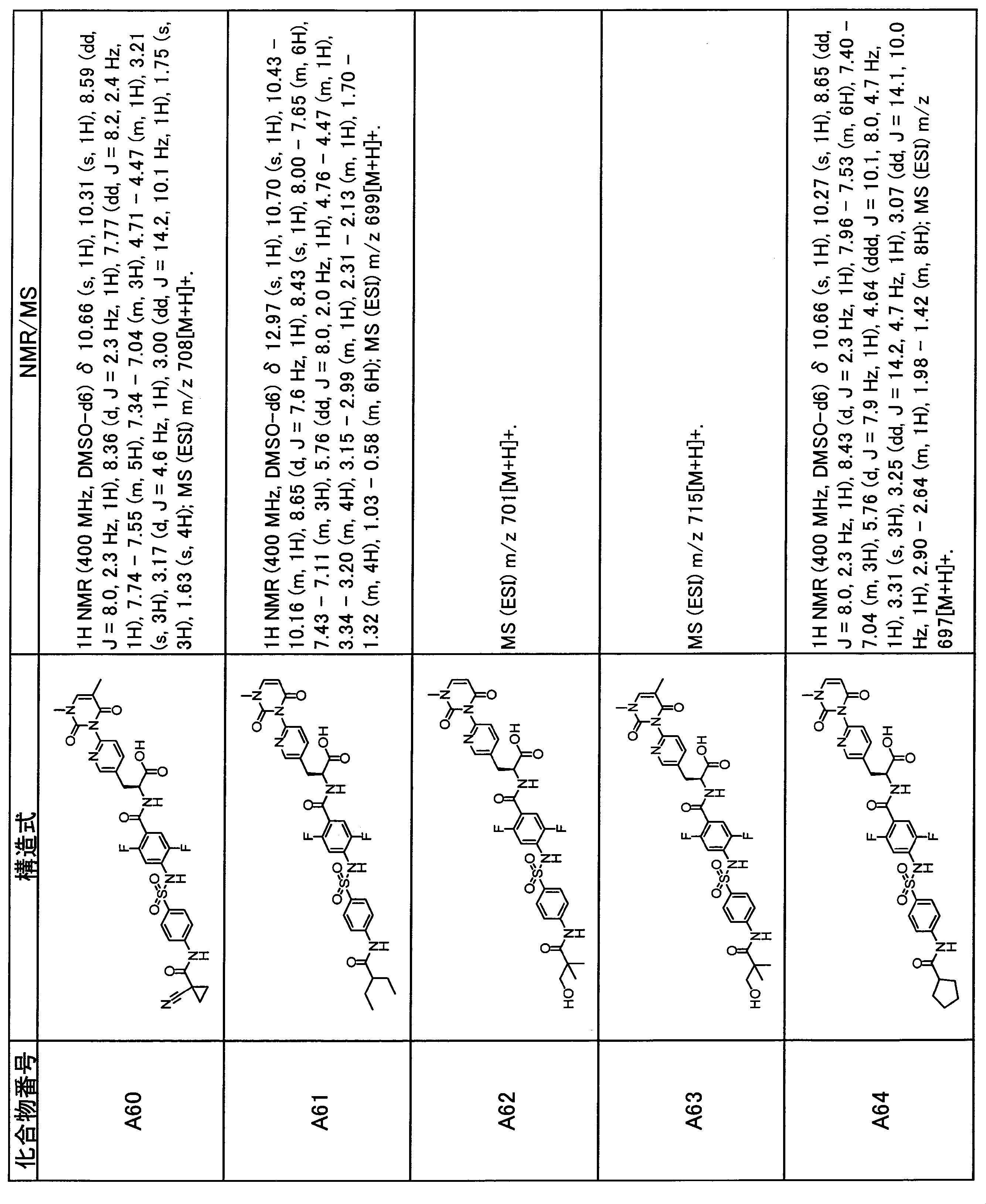


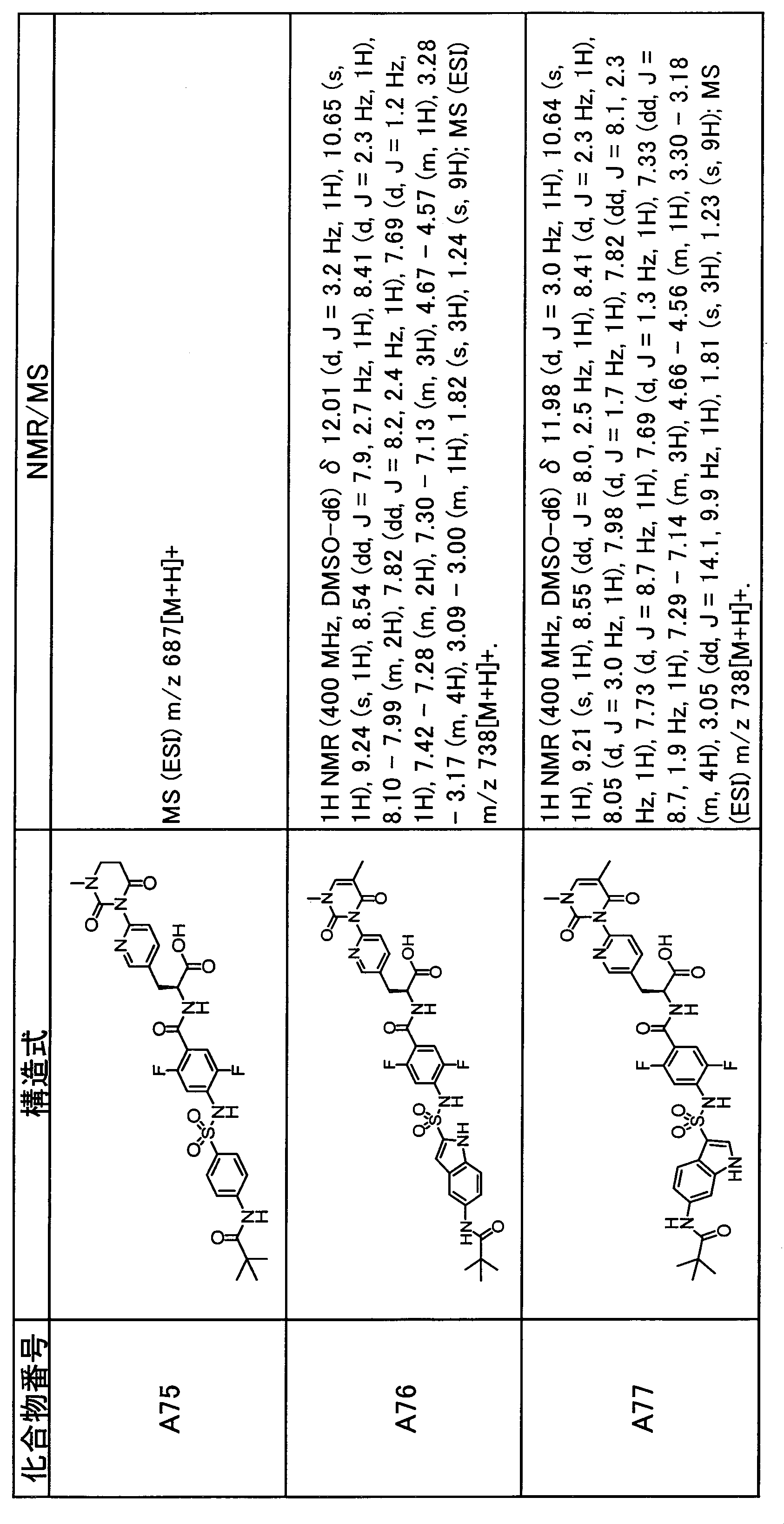
Isopropyl (2S)-2-[[4-(2,2-dimethylpropanoylamino)phenyl]sulfonylamino]-2,5-difluoro-benzoyl]amino]-3-[4-(1-methyl-2,4-dioxo-pyrido[3,4-d]pyrimidin-3-yl)phenyl]propanoate(P1)

Isobutyl (2S)-2-[2,5-difluoro-4-[(4-pivalamidophenyl)sulfonamido]benzamido]-3-[6-(3-methyl-2,6-dioxotetrahydropyrimidin-1(2H)-yl)pyridin-3-yl]propanoate(P121)




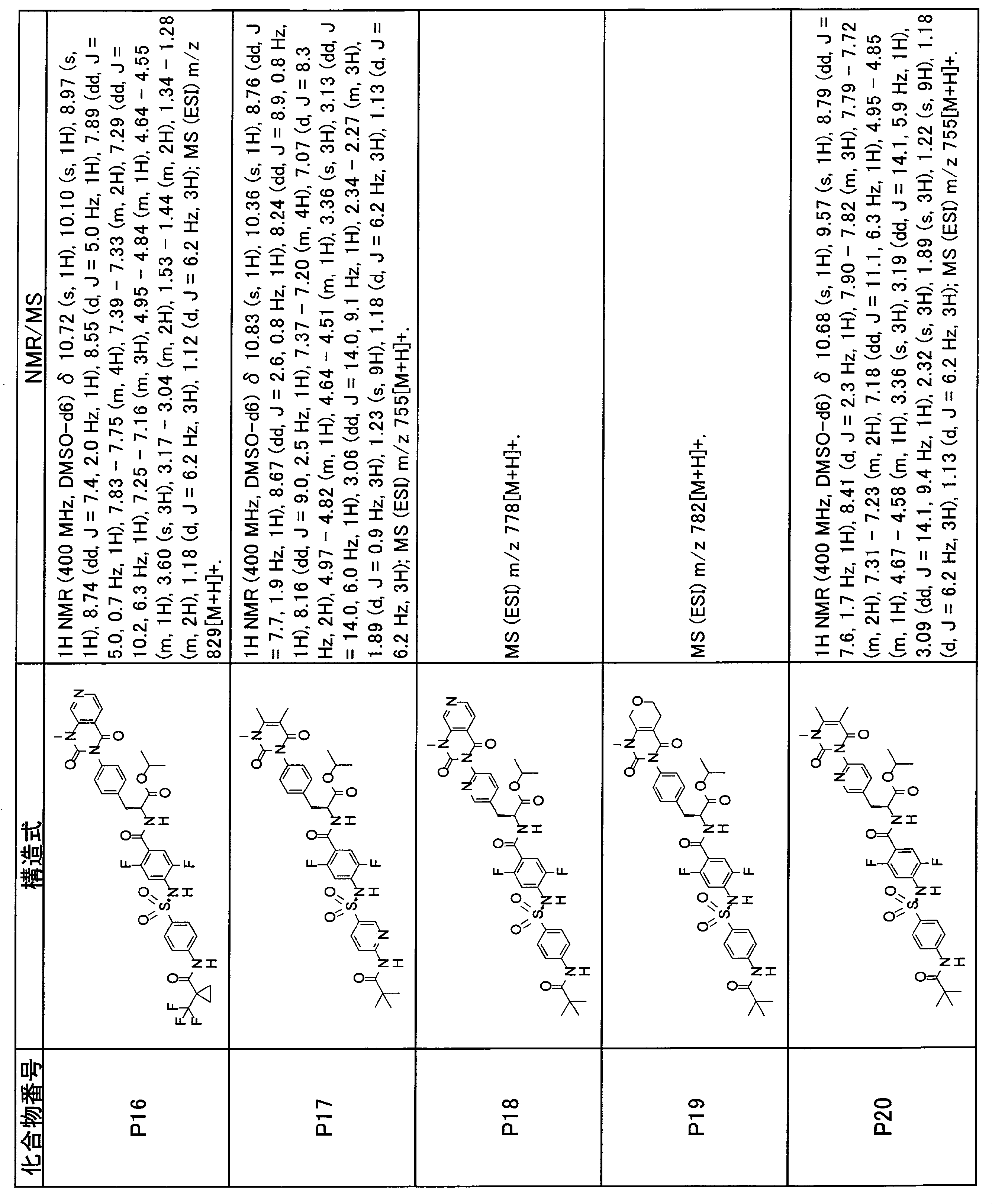

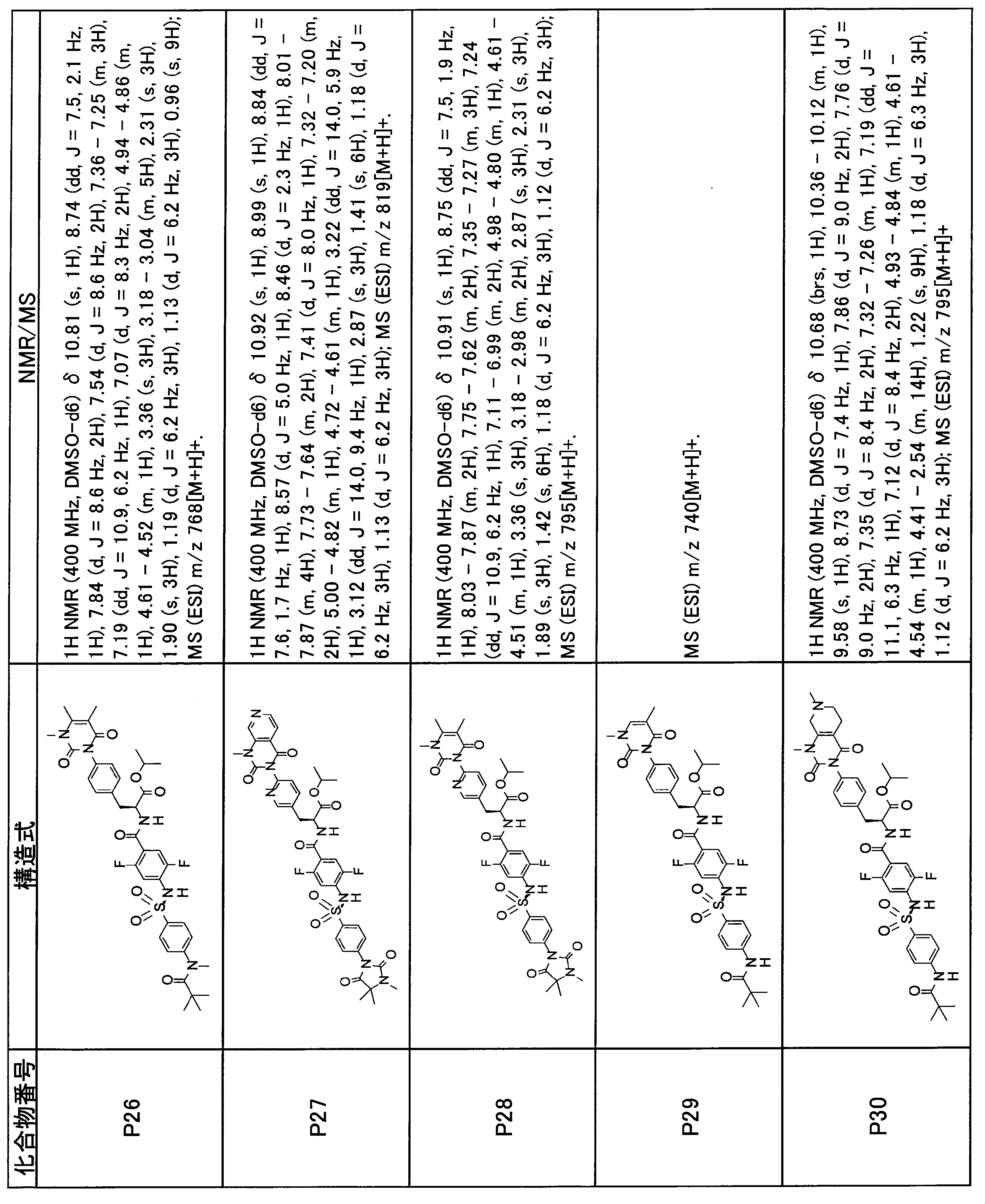

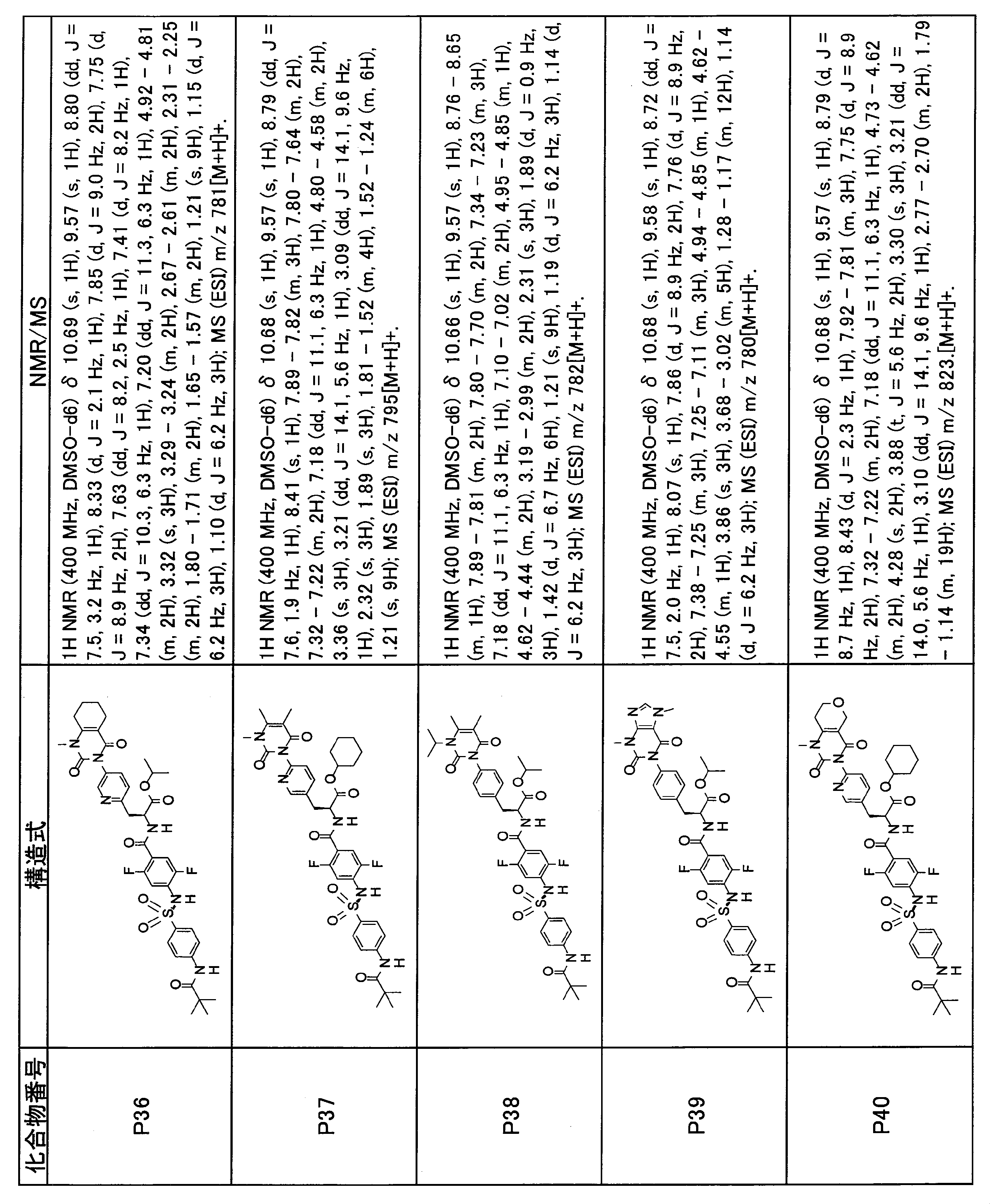


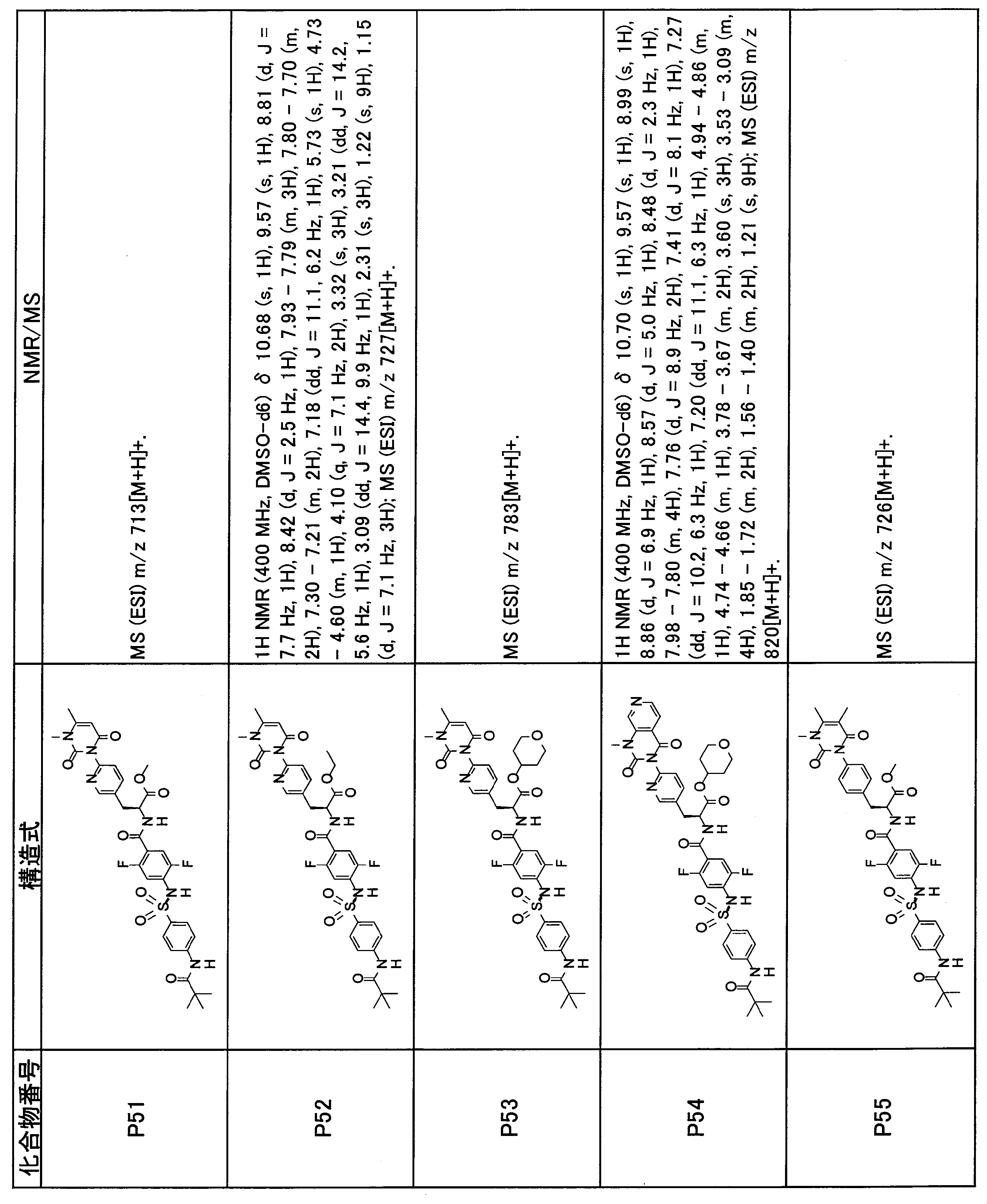












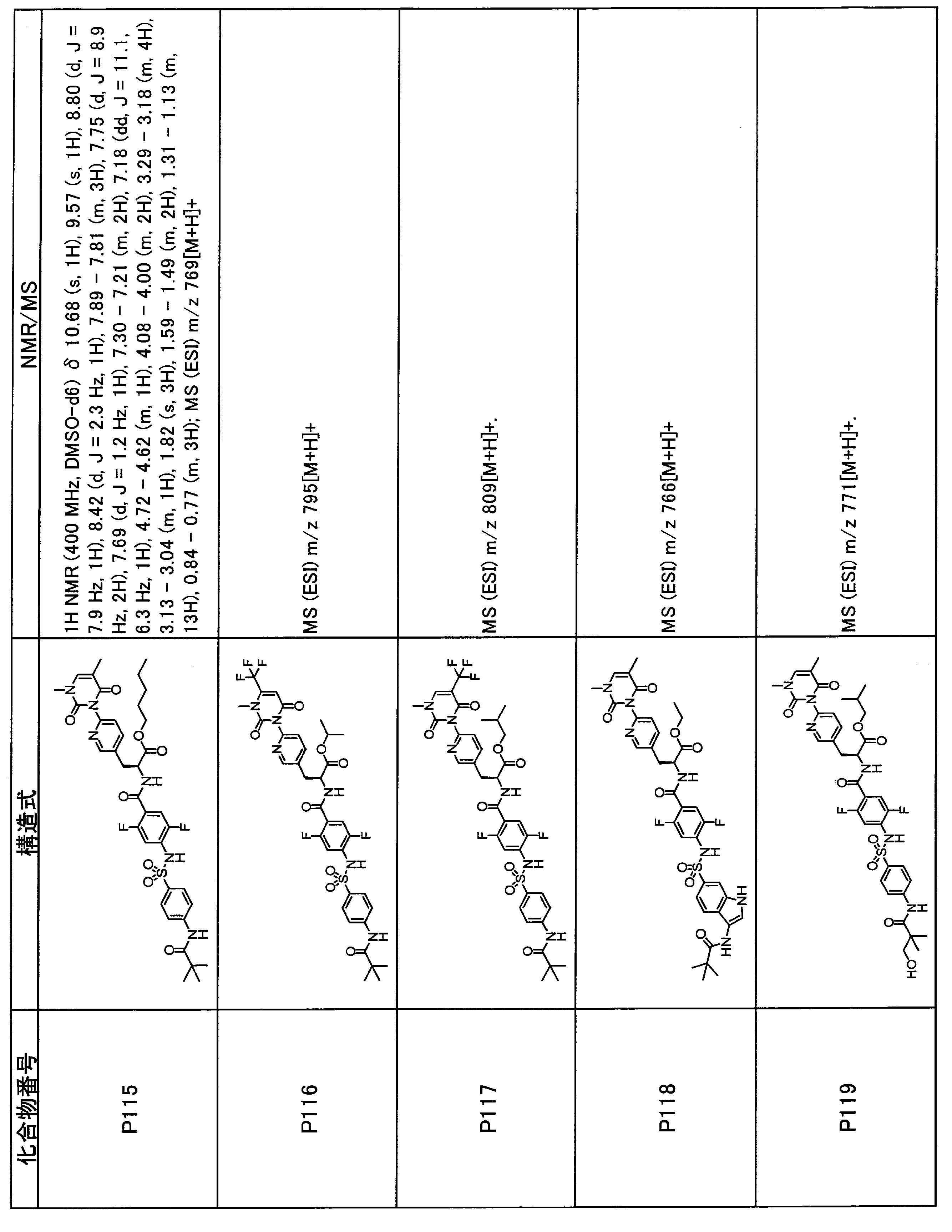
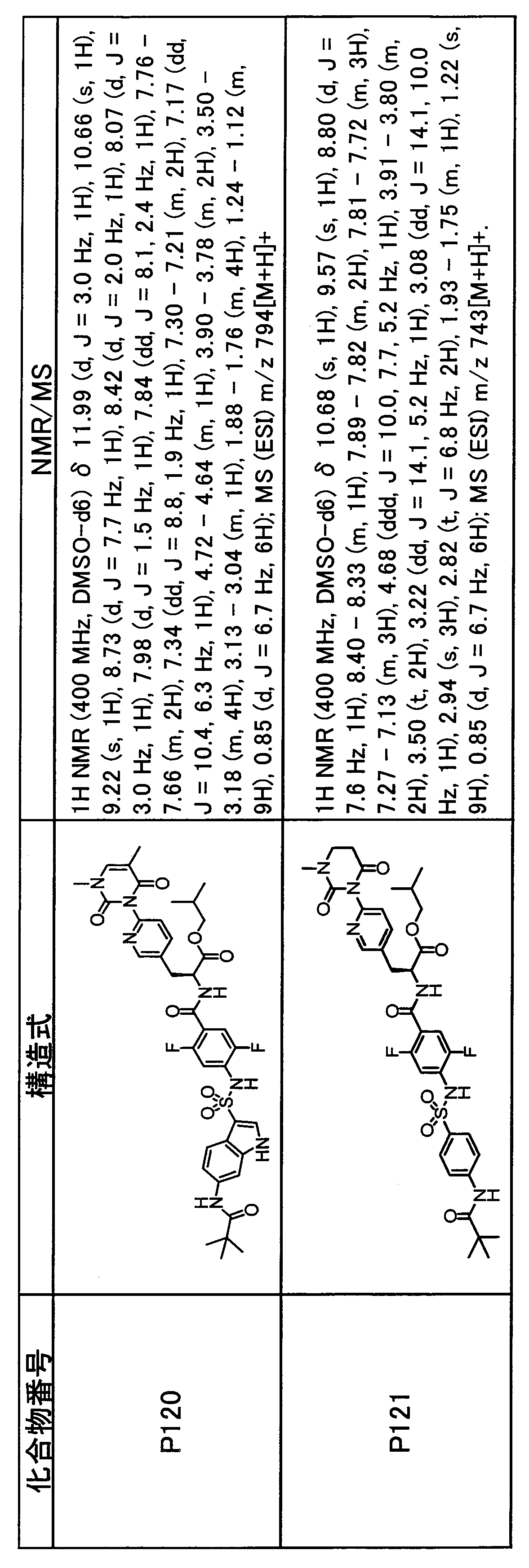
(1)VCAM-1/α4β1インテグリン結合阻害活性評価試験
α4β1インテグリンを発現していることが知られているヒトT細胞系細胞株JurkatのVCAM-1への結合を阻害する試験物質の能力を測定した。
96ウェルのマイクロタイタープレートに、緩衝液A(炭酸緩衝液、pH9.6)で希釈した組み換えヒトVCAM-1/Fc(R&D systems)溶液(1μg/mL)を50μL/ウェル加え、4℃で一晩インキュベートした。PBSで1回洗浄後、ブロックエース(雪印乳業)を150μL/ウェル加え、室温で2時間インキュベートした。除去後に、PBSで1回洗浄を実施した。
結合緩衝液(40 mM HEPES、0.2% BSAおよび4 mM MnCl2を含むDMEM)で希釈した種々の濃度の試験物質及びJurkat細胞(2x106細胞/mL)を、100 μLずつVCAM-1/Fcがコーティングされたプレートに添加し(5x105細胞/ウェル)、30℃で15分~60分間インキュベートした。細胞をウェルに結合させた後、PBSで洗浄することにより、結合していない細胞を除いた。プレートに緩衝液C(1.5% Triton X-100を含むPBS)を50 μL/ウェルで加え、結合したJurkat細胞を溶解した。細胞溶解液30 μLに、30 μLのSubstrate Buffer(Promega、CytoTox 96 Non-Radioactive Cytotoxicity Assay)を加え、室温、暗所で30分反応させた。各々30 μLのStop Solution(Promega、CytoTox 96 Non-Radioactive Cytotoxicity Assay)を加え、プレートリーダーを用いて490 nmの吸光度を測定した。ここで得られた吸光度は、各ウェルの上清に溶出したlactate dehydrogenase(LDH)活性を検出しているものであり、すなわちVCAM-1に結合してプレート上に残ったJurkat細胞の数に比例する。試験はduplicateで行い、試験物質を含まないウェルの吸光度を100%とした時の種々の濃度における細胞の結合率を求め、50%結合阻害をもたらす濃度IC50を計算した。得られた結果を、表3にまとめて示す。
(2)MAdCAM-1/α4β7インテグリン結合阻害活性評価試験
α4β7インテグリンを発現していることが知られているヒトB細胞系細胞株RPMI-8866のMAdCAM-1への結合を阻害する試験物質の能力を測定した。
96ウェルのマイクロタイタープレートに、緩衝液A(炭酸緩衝液、pH9.6)で希釈した組み換えマウスMAdCAM-1/Fc(R&D systems)溶液(0.75 μg/mL)を50 μL/ウェル加え、4℃で一晩インキュベートした。PBSで1回洗浄後、ブロックエース(雪印乳業)を150 μL/ウェル加え、室温で2時間インキュベートした。除去後に、PBSで1回洗浄を実施した。
結合緩衝液(40 mM HEPES、0.2% BSAおよび4 mM MnCl2を含むDMEM)で希釈した種々の濃度の試験物質及びRPMI-8866細胞(2x106細胞/mL)を100 μLずつMAdCAM-1/Fcがコーティングされたプレートに添加し(5x105細胞/ウェル)、30℃で15分~60分間インキュベートした。細胞をウェルに結合させた後、PBSで洗浄することにより、結合していない細胞を除いた。プレートに緩衝液C(1.5% Triton X-100を含むPBS)を50 μL/ウェルで加え、結合したRPMI-8866細胞を溶解した。細胞溶解液30 μLに、30 μLのSubstrate Buffer(Promega、CytoTox 96 Non-Radioactive Cytotoxicity Assay)を加え、室温、暗所で30分反応させた。各々30 μLのStop Solution(Promega、CytoTox 96 Non-Radioactive Cytotoxicity Assay)を加え、プレートリーダーを用いて490 nmの吸光度を測定した。ここで得られた吸光度は、各ウェルの上清に溶出したlactate dehydrogenase(LDH)活性を検出しているものであり、すなわちMAdCAM-1に結合してプレート上に残ったRPMI-8866細胞の数に比例する。試験はduplicateで行い、試験物質を含まないウェルの吸光度を100%とした時の種々の濃度における細胞の結合率を求め、50%結合阻害をもたらす濃度IC50を計算した。得られた結果を、表3にまとめて示す。
(3)血清存在下におけるMAdCAM-1/α4β7インテグリン結合阻害活性評価試験(1)
α4β7インテグリンを発現していることが知られているヒトB細胞系細胞株RPMI-8866のMAdCAM-1への結合を阻害する試験物質の能力を測定した。
96ウェルのマイクロタイタープレートに、緩衝液A(炭酸緩衝液、pH9.6)で希釈した組み換えマウスMAdCAM-1/Fc(R&D systems)溶液(1 μg/mL)を50 μL/ウェル加え、4℃で一晩インキュベートした。PBSで1回洗浄後、ブロックエース(雪印乳業)を150 μL/ウェル加え、室温で2時間インキュベートした。除去後に、PBSで1回洗浄を実施した。
結合緩衝液(40 mM HEPES、0.2% BSAおよび4 mM MnCl2を含むDMEM)で希釈した種々の濃度の試験物質及びRPMI-8866細胞(2x106細胞/mL)を、最終濃度で50%ヒト血清を含むように、100 μLずつMAdCAM-1/Fcがコーティングされたプレートに添加し(5x105細胞/ウェル)、30℃で15分~60分間インキュベートした。細胞をウェルに結合させた後、PBSで洗浄することにより、結合していない細胞を除いた。プレートに緩衝液C(1.5% Triton X-100を含むPBS)を50 μL/ウェルで加え、結合したRPMI-8866細胞を溶解した。細胞溶解液30 μLに、30 μLのSubstrate Buffer(Promega、CytoTox 96 Non-Radioactive Cytotoxicity Assay)を加え、室温、暗所で30分反応させた。各々30 μLのStop Solution(Promega、CytoTox 96 Non-Radioactive Cytotoxicity Assay)を加え、プレートリーダーを用いて490 nmの吸光度を測定した。ここで得られた吸光度は、各ウェルの上清に溶出したlactate dehydrogenase(LDH)活性を検出しているものであり、すなわちMAdCAM-1に結合してプレート上に残ったRPMI-8866細胞の数に比例する。試験はduplicateで行い、試験物質を含まないウェルの吸光度を100%とした時の種々の濃度における細胞の結合率を求め、50%結合阻害をもたらす濃度IC50を計算した。得られた結果を、表3にまとめて示す。
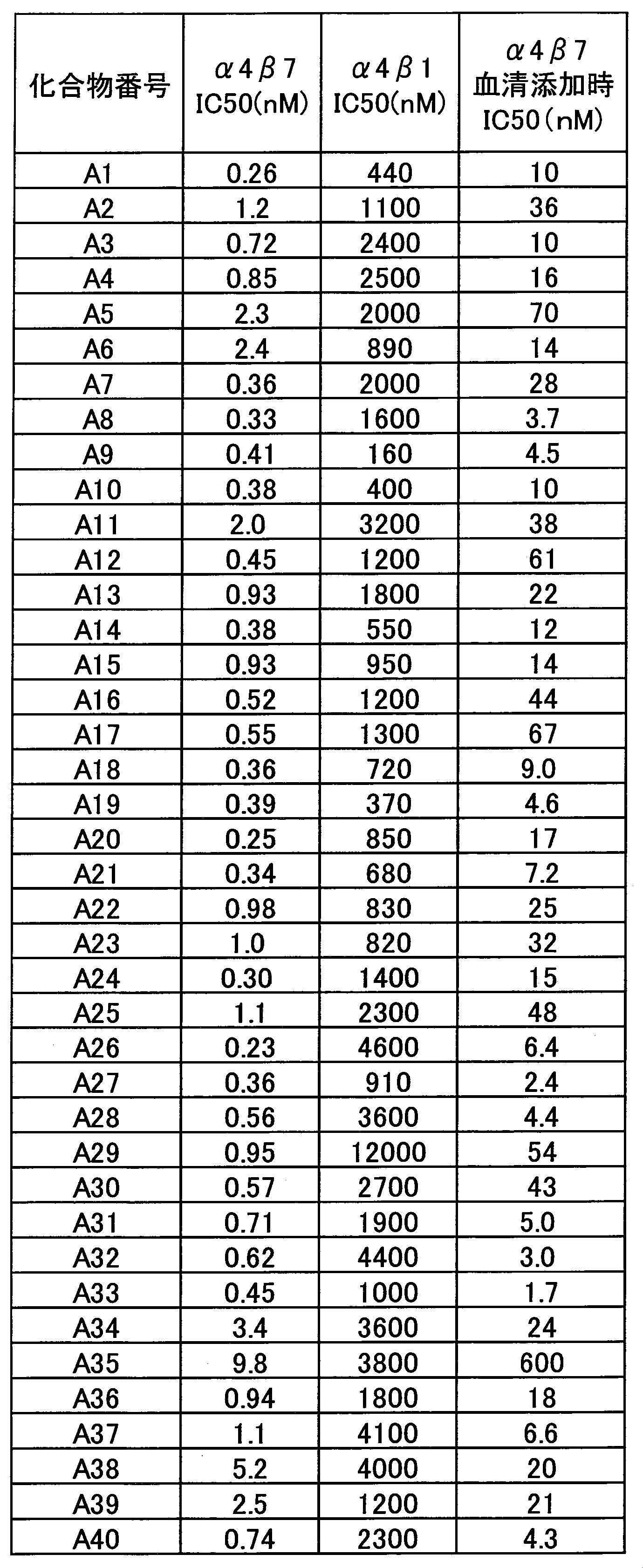
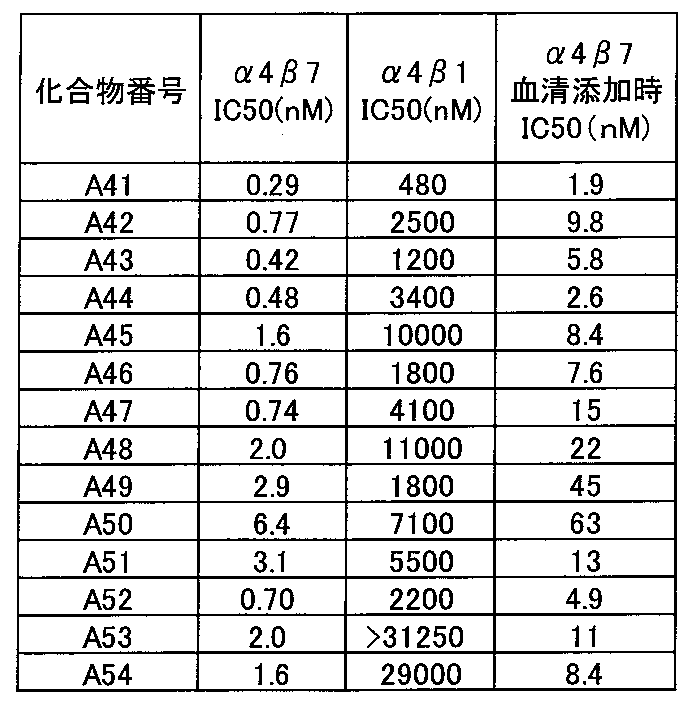
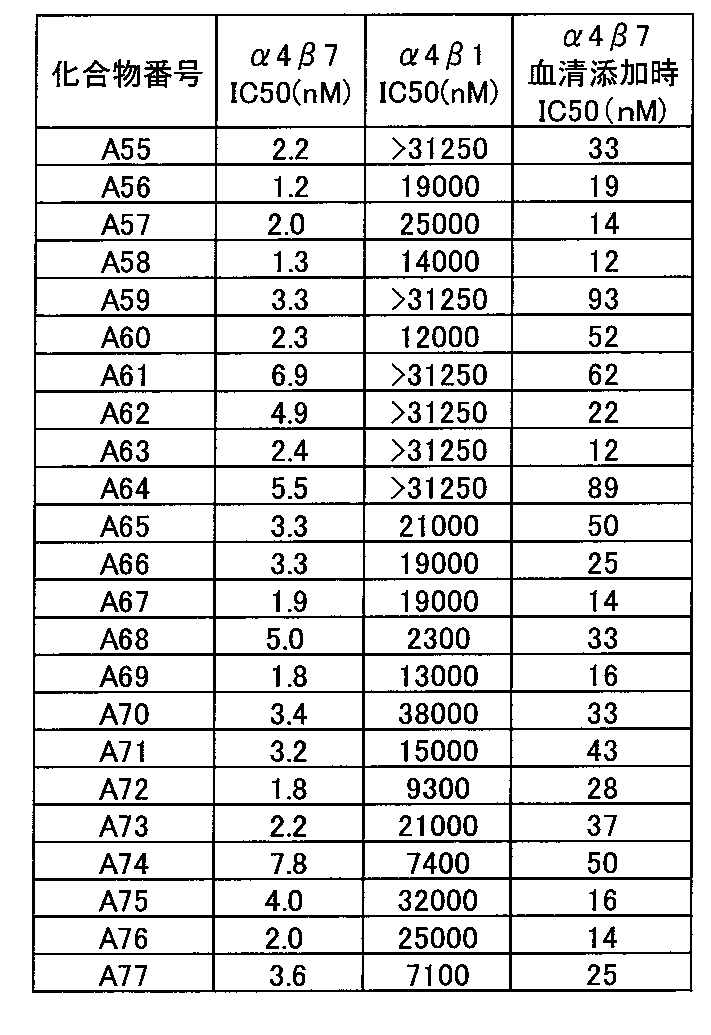
(4)ヒト全血におけるマウスMAdCAM-1/α4β7インテグリン結合阻害活性評価試験
試験物質によるヒト全血中におけるT細胞α4β7インテグリンとMAdCAM-1の結合阻害活性を測定した。血液サンプルは健康人ボランティアの血液提供により入手した。
ヒト全血に、4mM MnCl2溶液と各種試験物質希釈液を添加し10分間インキュベートした。10μg/mLの組み換えマウスMAdCAM-1/Fc(R&D Systems)を添加し、全量50μLとして30分間インキュベートした。Lyse/Fix(BD Biosciences)を950μL添加し、37℃で10分間溶血及び固定した。5分間遠心分離後、上清を除き、10% 非働化ウシ胎仔血清添加RPMI-1640培地(以下、培地とする)を600μL添加し、5分間遠心分離後、上清を除き洗浄した。再び培地で洗浄後、0.625μg/mLのRat Anti-Mouse MAdCAM-1抗体(SouthernBiotech)を添加し、30分以上インキュベートした。培地で洗浄後、50μg/mLのGoat Anti-Rat IgG(H+L)Antibody, FITC(Life Technologies)を添加し、30分以上インキュベートした。培地で洗浄後、10μg/mLのPE Rat Anti-Mouse CD4(BD Pharmigen)を添加し、30分以上インキュベートした。培地で洗浄後、フローサイトメトリーを用いてCD4陽性細胞中に占めるMAdCAM-1陽性細胞率の割合を測定した。
試験は異なる2~3人の血液を用いた独立した試験結果から、試験物質を含まないウェルのうちリガンド無しを阻害100%、リガンド有りを阻害0%としたときの種々の濃度における試験物質のMAdCAM-1結合阻害率を求め、50%結合阻害をもたらす濃度IC50を計算した。得られた結果を、表4に示す。

試験物質のマウス門脈移行濃度を測定し、経口吸収性を評価した。
試験物質を0.5%(w/v)メチルセルロース水溶液に溶解又は均一に懸濁させ、胃ゾンデを用いて雌性マウス(BALB/cAnNCrlCrlj、 7~9週齢)に3化合物(3mg/10mL/kg)をカセット経口投与した。投与30分後にイソフルラン麻酔下にて開腹し、門脈からDDVP(エステラーゼ阻害剤)及びヘパリンナトリウムで処理したシリンジを用いて、約0.2mLを採血し、氷上で保管した。
採取した血液は冷却遠心機を用いて18,000 g x 3分間遠心分離することで血漿サンプルを取得し、アセトニトリルで試験物質を抽出後、LC/MS/MSにて血漿中濃度を定量した。
なお,血漿中濃度は試験物質とその活性代謝物を合算した濃度とした。算出した血漿中濃度を表5に示す。
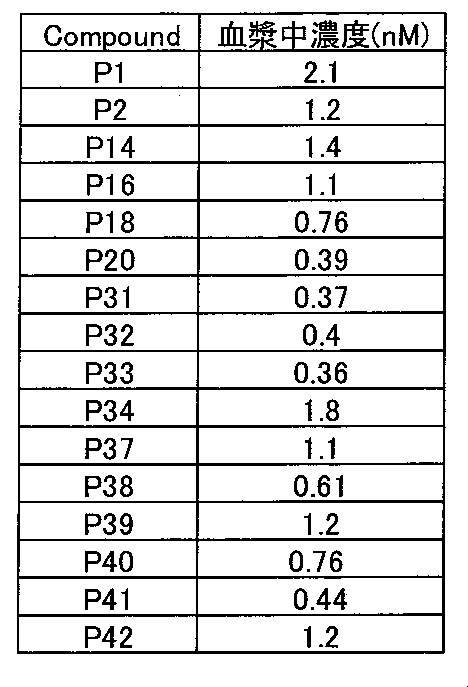
ヒト全血におけるヒトMAdCAM-1/α4β7インテグリン結合阻害活性評価試験
試験物質によるヒト全血中におけるT細胞α4β7インテグリンとMAdCAM-1の結合阻害活性を測定した。血液サンプルは健康人ボランティアの血液提供により入手した。
ヒト全血に、4mM MnCl2溶液と各種試験物質希釈液を添加し10分間インキュベートした。10μg/mLの組み換えヒトMAdCAM-1/Fc(R&D Systems)を添加し、全量50μLとして30分間インキュベートした。Lyse/Fix(BD Biosciences)を950μL添加し、37℃で10分間溶血及び固定した。5分間遠心分離後、上清を除き、10% 非働化ウシ胎仔血清添加RPMI-1640培地(以下、培地とする)を600μL添加し、5分間遠心分離後、上清を除き洗浄した。再び培地で洗浄後、2.5μg/mLのMouse Anti-MAdCAM-1抗体(invitrogen)を添加し、30分以上インキュベートした。培地で洗浄後、3.4μg/mLのGoat Anti-Mouse IgG H&L, FITC(abcam)を添加し、30分以上インキュベートした。培地で洗浄後、0.15μg/mLのPE Mouse Anti-Human CD4(BD Pharmigen)を添加し、30分以上インキュベートした。培地で洗浄後、フローサイトメトリーを用いてCD4陽性細胞中に占めるMAdCAM-1陽性細胞率の割合を測定した。
試験は異なる2~3人の血液を用いた独立した試験結果から、試験物質を含まないウェルのうちリガンド無しを阻害100%、リガンド有りを阻害0%としたときの種々の濃度における試験物質のMAdCAM-1結合阻害率を求め、50%結合阻害をもたらす濃度IC50を計算した。得られた結果を、表6に示す。

マウスIL-10-/-細胞移入腸炎モデルの作成方法は、J Crohns Colitis. 2013 Dec;7(11):e533-42.に記載されている方法を用いた。
被験物質を0.5%メチルセルロース水溶液に30、10、3、または1mg/mLとなるように懸濁し、動物体重1 kgあたり10mlの容量で、テルモシリンジ(1 mL)及びマウス用金属ゾンデを用いて経口投与した。投与は1日3回、14日間行った。
薬効指標である腸管重量の測定は、次の方法で行った。最終剖検日に肛門から盲腸直前までの大腸を摘出し、腸管内容物を生理食塩水で洗浄し、軽く水分を除去した後に重量測定を行った。群毎の腸管重量の平均値について、腸炎発症群を0%、正常群を100%としたときの、それぞれの化合物投与群における抑制率(%)を示した。本評価系を用いて、炎症抑制作用を評価した結果を表7に示した。いずれの化合物も、用量依存的な抑制作用を認めた。

Claims (27)
- 下記一般式(I)で示されるスルホンアミド誘導体、又はその医薬的に許容しうる塩。
(式中、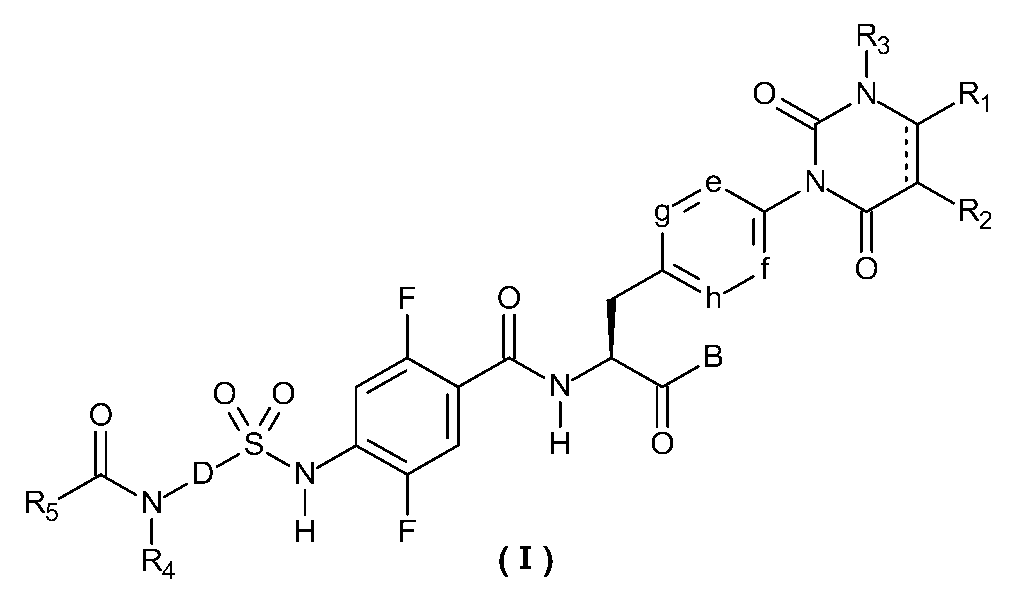
は単結合、又は、二重結合を表し、
R1及びR2は、それぞれ独立して、水素原子、ハロゲン原子、低級アルキル基、低級アルケニル基、低級アルコキシ基、低級アルコキシ低級アルキル基、ハロゲノ低級アルキル基、ヒドロキシ基、又は、ヒドロキシ低級アルキル基を表し、R1とR2は結合して、置換基を有しても良いベンゼン環、置換基を有しても良い炭素数4~7の脂環式炭化水素、置換基を有しても良いヘテロアリール環、又は、置換基を有しても良いヘテロ環を形成しても良く、
R3は、低級アルキル基を表し、
e、f、g、及び、hは、それぞれ独立して、C-H、又は、窒素原子を表し、
Bは、ヒドロキシ基、炭素数が1~10のアルコキシ基、-O-ヘテロ環基、シレキセチルオキシ基、又はメドキソミルオキシ基を表し、
Dは、置換基を有しても良い、ベンゼン環又はヘテロアリール環を表し、
R4は、水素原子、又は、低級アルキル基を表し、
R5は、置換基を有しても良い低級アルキル基、置換基を有しても良い低級アルケニル基、置換基を有しても良い低級アルキニル基、低級アルキルアミノ基、置換基を有しても良いフェニル基、置換基を有しても良いヘテロアリール基、又は、置換基を有しても良いヘテロ環基を表し、
R4とR5は結合して、置換基を有しても良いヘテロ環を形成しても良い。) - R1及びR2が、それぞれ独立して、水素原子、ハロゲン原子、低級アルキル基、低級アルケニル基、低級アルコキシ基、低級アルコキシ低級アルキル基、ヒドロキシ基、又は、ヒドロキシ低級アルキル基を表し、R1とR2は結合して、置換基を有しても良いベンゼン環、置換基を有しても良い炭素数4~7の脂環式炭化水素、置換基を有しても良いヘテロアリール環、又は、置換基を有しても良いヘテロ環を形成しても良く、
R5は、置換基を有しても良い低級アルキル基、置換基を有しても良い低級アルケニル基、低級アルキルアミノ基、置換基を有しても良いフェニル基、置換基を有しても良いヘテロアリール基、又は、置換基を有しても良いヘテロ環基を表す、請求項1に記載されたスルホンアミド誘導体、又はその医薬的に許容し得る塩。 - が二重結合を表す、請求項1又は2に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- R1及びR2は結合して、置換基を有しても良いベンゼン環、置換基を有しても良い炭素数4~7の脂環式炭化水素、置換基を有しても良いヘテロアリール環、又は、置換基を有しても良いヘテロ環を形成し、該置換基が、低級アルキル基、低級アルコキシ基、ヒドロキシ低級アルキル基、アミノ基、低級アルキルアミノ基、及び、低級アルキルアミノ低級アルキル基から選ばれる、請求項1~3のいずれか1項に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- R1及びR2が、それぞれ独立して、水素原子、低級アルキル基、又は、ヒドロキシ低級アルキル基を表し、R1とR2は結合して、置換基を有しても良い炭素数4~7の脂環式炭化水素、置換基を有しても良いヘテロアリール環、又は、置換基を有しても良いヘテロ環を形成しても良く、該置換基は、低級アルキル基及び低級アルコキシ基から選ばれる、請求項1~3のいずれか1項に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- R1及びR2が、それぞれ独立して、水素原子、低級アルキル基、ハロゲノ低級アルキル基、又は、ヒドロキシ低級アルキル基を表し、R1とR2は結合して、置換基を有しても良い炭素数4~7の脂環式炭化水素、置換基を有しても良いヘテロアリール環、又は、置換基を有しても良いヘテロ環を形成しても良く、該置換基は、低級アルキル基及び低級アルコキシ基から選ばれる、請求項1に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- R1及びR2が、それぞれ独立して、水素原子、低級アルキル基、又は、ヒドロキシ低級アルキル基を表す、請求項1~3のいずれか1項に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- R1とR2が結合して置換基を有しても良いピリジン、置換基を有しても良いシクロヘキセン、置換基を有しても良いジヒドロピラン、置換基を有しても良いテトラヒドロピリジン、又は、置換基を有しても良いイミダゾールを形成し、該置換基が、低級アルキル基、低級アルコキシ基、ヒドロキシ低級アルキル基、アミノ基、低級アルキルアミノ基、及び、低級アルキルアミノ低級アルキル基から選ばれる、請求項1~3のいずれか1項に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- eが窒素原子を表し、f、g、及び、hが、C-Hを表す、請求項1~8のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- Bが、ヒドロキシ基、又は、炭素数が1~6のアルコキシ基を表す、請求項1~9のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- Dの置換基が、ハロゲン原子、低級アルキル基、低級アルコキシ基、及び、ヒドロキシ基から選ばれる、請求項1~10のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- Dが、置換基を有しても良いベンゼン環、置換基を有しても良いピリジン環、又は、置換基を有しても良いチオフェン環を表し、該置換基が、ハロゲン原子、低級アルキル基、低級アルコキシ基、及び、ヒドロキシ基から選ばれる、請求項1~11のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- Dが、置換基を有しても良いベンゼン環、置換基を有しても良いピリジン環、又は、置換基を有しても良いチオフェン環を表し、該置換基が、ハロゲン原子を表す、請求項1~11のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- R4が、水素原子を表す、請求項1~13のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- R5が、置換基を有する場合における置換基が、ハロゲン原子、シアノ基、ヒドロキシ基、低級アルキル基、低級アルコキシ基、トリフルオロメチル基、フェニル基、及び、ヘテロ環基から選ばれる、請求項1~14のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- R4とR5が結合して、置換基を有しても良いヘテロ環を形成し、該置換基が、低級アルキル基、低級アルコキシ基、ヒドロキシ基、及び、ヘテロ環基から選ばれる、請求項1~14のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- R5が、置換基を有しても良い低級アルキル基、低級アルキルアミノ基、又は、置換基を有しても良いヘテロ環基を表し、該置換基が、ハロゲン原子、シアノ基、ヒドロキシ基、低級アルコキシ基、トリフルオロメチル基及びフェニル基から選ばれる、請求項1~14のいずれかに記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- R1及びR2が、それぞれ独立して、水素原子、低級アルキル基、低級アルコキシ基、又は、ヒドロキシ低級アルキル基を表し、R1とR2は結合して、低級アルキル基で置換されていてもよい炭素数4~7の脂環式炭化水素、低級アルキル基で置換されていてもよいヘテロアリール環、又は、低級アルキル基で置換されていてもよいヘテロ環を形成しても良く、
Dは、ハロゲン原子で置換されていてもよいベンゼン環、又は、下記式から選ばれるヘテロアリール環を表し、
(式中、aはSとの結合位置を表し、bはNとの結合位置を表す)
R4が、水素原子を表し、
R5が、低級アルコキシ基、ハロゲン原子、ヒドロキシ基及びアリール基からなる群から選ばれる置換基を有しても良い炭素数2~5のアルキル基、ヘテロアリール基、又は、環原子としてOを含有するヘテロ環基を表し、
但し、
Dがハロゲン原子で置換されていてもよいベンゼン環である場合、Dは、パラ位でSとNとに結合しており、
Dが、
で表されるヘテロアリール環である場合、eが窒素原子を表し、
R5が環原子としてOを含有するヘテロ環基である場合、R1とR2は結合してヘテロアリール環を形成し、
R5がヒドロキシ基で置換された炭素数2~5のアルキル基である場合、R5は、下記式により表される、
請求項1又は2に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- 下記式で表される、請求項1又は2に記載されたスルホンアミド誘導体、又はその医薬的に許容しうる塩。
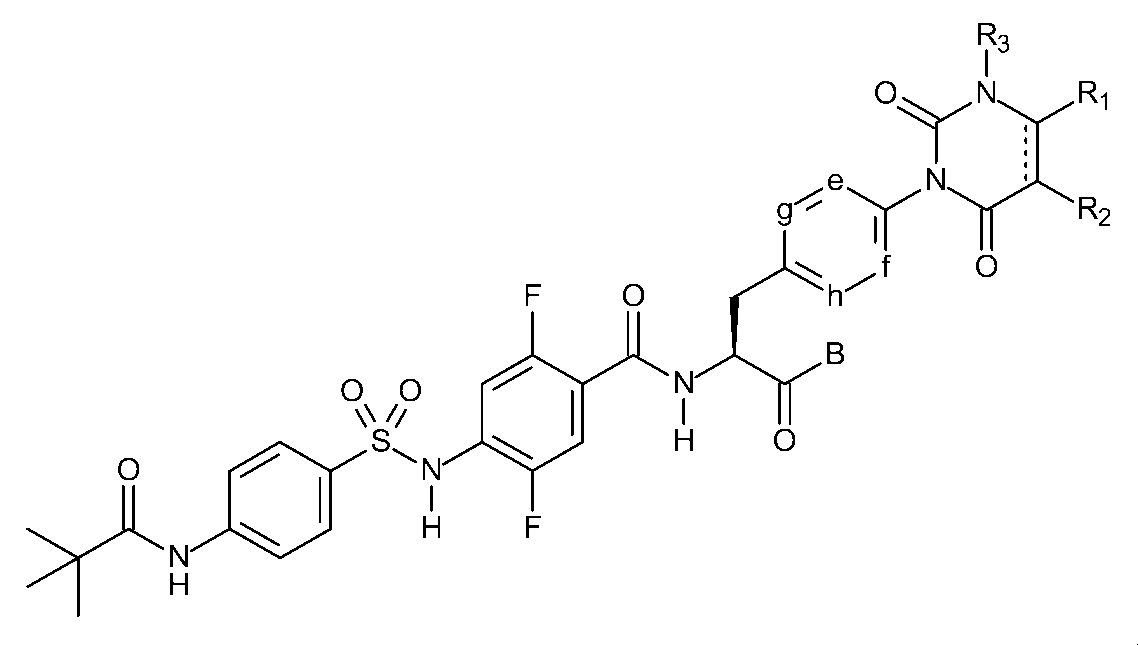
- 下記式で表される、請求項19に記載されたスルホンアミド誘導体、又はその医薬的に許容し得る塩。
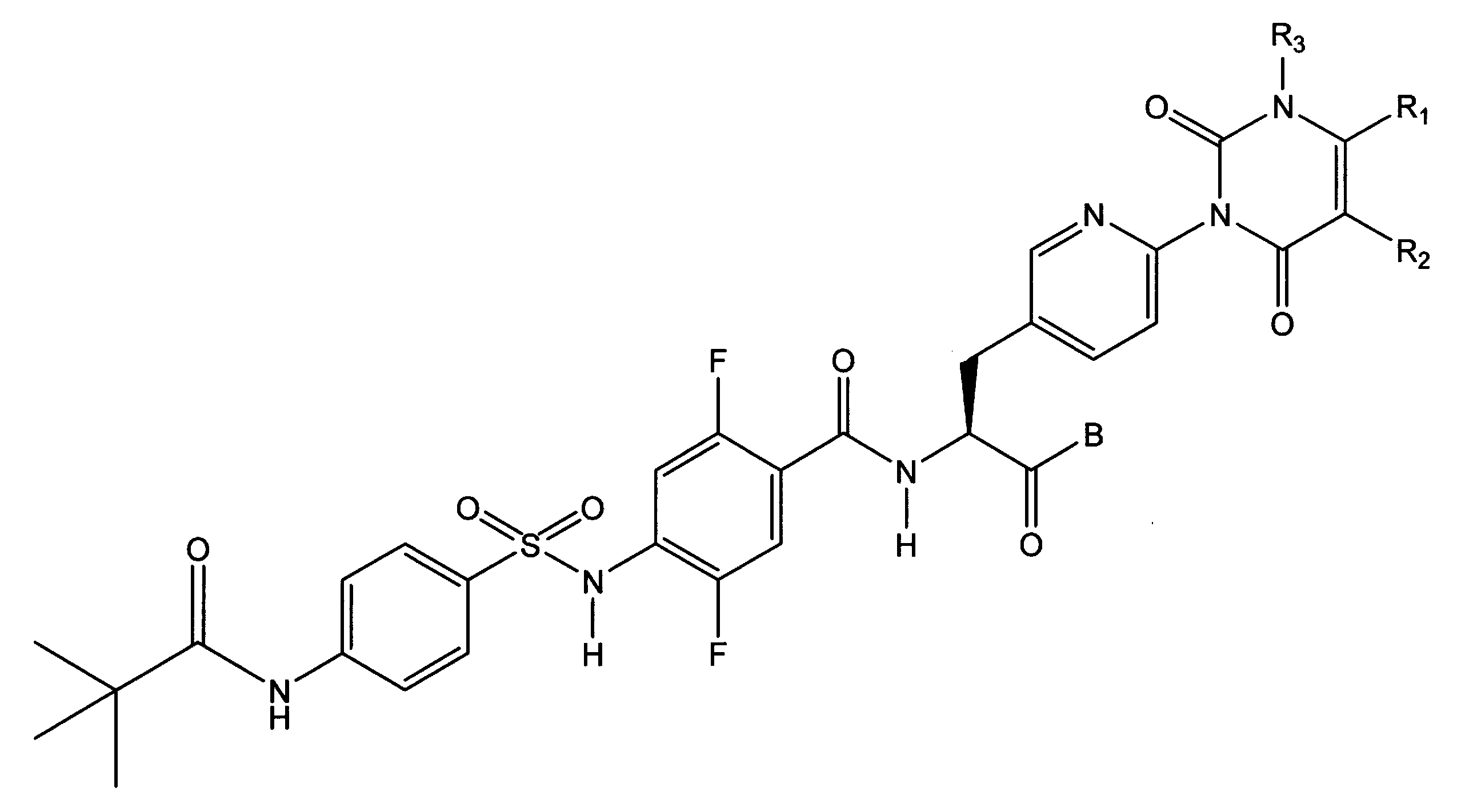
- 下記式で表される、請求項19に記載されたスルホンアミド誘導体、又はその医薬的に許容し得る塩。

- 下記式のいずれかで表される、請求項19に記載のスルホンアミド誘導体、又はその医薬的に許容しうる塩。

- 下記式のいずれかで表される、請求項22に記載のスルホンアミド誘導体、又はその医薬的に許容しうる塩。



- Bが、ヒドロキシ基、メトキシ基、エトキシ基、イソプロポキシ基、又はイソブチルオキシ基である、請求項1~23のいずれか1項に記載のスルホンアミド誘導体、又はその医薬的に許容しうる塩。
- 請求項1~24のいずれかに記載のスルホンアミド誘導体、又はその医薬的に許容し得る塩を含有する医薬組成物。
- 請求項1~24のいずれかに記載のスルホンアミド誘導体、又はその医薬的に許容し得る塩を含有するα4β7インテグリン依存性の接着過程が病態に関与する炎症性疾患の治療剤又は予防剤。
- 請求項1~24のいずれかに記載のスルホンアミド誘導体、又はその医薬的に許容し得る塩を含有するα4β7インテグリン阻害剤。
Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR112018015855-0A BR112018015855B1 (pt) | 2016-02-05 | 2017-02-06 | Derivado de sulfonamida |
| CA3013433A CA3013433A1 (en) | 2016-02-05 | 2017-02-06 | Sulfonamide derivative and pharmaceutical composition containing same |
| EP17747622.3A EP3412660B9 (en) | 2016-02-05 | 2017-02-06 | Sulfonamide derivative and pharmaceutical composition containing same |
| KR1020187025805A KR20180104758A (ko) | 2016-02-05 | 2017-02-06 | 설폰아미드 유도체 및 이를 함유하는 의약 조성물 |
| JP2017565026A JP6820282B2 (ja) | 2016-02-05 | 2017-02-06 | スルホンアミド誘導体及びそれを含有する医薬組成物 |
| US16/075,415 US10562898B2 (en) | 2016-02-05 | 2017-02-06 | Substituted benzenesulfonamides as inhibitors of alpha-4 beta-7 integrin activity |
| DK17747622.3T DK3412660T3 (da) | 2016-02-05 | 2017-02-06 | Sulfonamidderivat og farmaceutisk sammensætning indeholdende samme |
| PL17747622T PL3412660T3 (pl) | 2016-02-05 | 2017-02-06 | Pochodna sulfonamidowa i zawierająca ją kompozycja farmaceutyczna |
| ES17747622T ES2856125T3 (es) | 2016-02-05 | 2017-02-06 | Derivado de sulfonamida y composición farmacéutica que contiene la misma |
| EA201891780A EA036432B1 (ru) | 2016-02-05 | 2017-02-06 | Производные сульфонамида и содержащие их фармацевтические композиции |
| CN201780009898.0A CN108699008B (zh) | 2016-02-05 | 2017-02-06 | 磺酰胺衍生物和含有其的药物组合物 |
| MX2018009459A MX2018009459A (es) | 2016-02-05 | 2017-02-06 | Derivado de sulfonamida y composicion farmaceutica que contiene el mismo. |
| AU2017214589A AU2017214589B2 (en) | 2016-02-05 | 2017-02-06 | Sulfonamide derivative and pharmaceutical composition containing same |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016-021053 | 2016-02-05 | ||
| JP2016021053 | 2016-02-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017135472A1 true WO2017135472A1 (ja) | 2017-08-10 |
Family
ID=59500204
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/004278 WO2017135472A1 (ja) | 2016-02-05 | 2017-02-06 | スルホンアミド誘導体及びそれを含有する医薬組成物 |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US10562898B2 (ja) |
| EP (1) | EP3412660B9 (ja) |
| JP (1) | JP6820282B2 (ja) |
| KR (1) | KR20180104758A (ja) |
| CN (1) | CN108699008B (ja) |
| AU (1) | AU2017214589B2 (ja) |
| CA (1) | CA3013433A1 (ja) |
| DK (1) | DK3412660T3 (ja) |
| EA (1) | EA036432B1 (ja) |
| ES (1) | ES2856125T3 (ja) |
| HU (1) | HUE053354T2 (ja) |
| MX (1) | MX2018009459A (ja) |
| PL (1) | PL3412660T3 (ja) |
| PT (1) | PT3412660T (ja) |
| TW (1) | TWI730042B (ja) |
| WO (1) | WO2017135472A1 (ja) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019147824A1 (en) | 2018-01-26 | 2019-08-01 | Progenity, Inc. | Treatment of a disease of the gastrointestinal tract with a pde4 inhibitor |
| WO2019246455A1 (en) | 2018-06-20 | 2019-12-26 | Progenity, Inc. | Treatment of a disease of the gastrointestinal tract with an integrin inhibitor |
| WO2021174024A1 (en) | 2020-02-28 | 2021-09-02 | First Wave Bio, Inc. | Methods of treating iatrogenic autoimmune colitis |
| US11116760B2 (en) | 2018-10-30 | 2021-09-14 | Gilead Sciences, Inc. | Quinoline derivatives |
| US11174256B2 (en) | 2018-10-30 | 2021-11-16 | Gilead Sciences, Inc. | Imidazopyridine derivatives |
| US11179383B2 (en) | 2018-10-30 | 2021-11-23 | Gilead Sciences, Inc. | Compounds for inhibition of α4β7 integrin |
| US11224600B2 (en) | 2018-10-30 | 2022-01-18 | Gilead Sciences, Inc. | Compounds for inhibition of alpha 4 beta 7 integrin |
| US11345662B2 (en) * | 2016-12-28 | 2022-05-31 | Ucb Pharma Gmbh | Aza (indole)-, benzothiophene-, and benzofuran-3-sulfonamides |
| US11578069B2 (en) | 2019-08-14 | 2023-02-14 | Gilead Sciences, Inc. | Compounds for inhibition of α4 β7 integrin |
| WO2023058645A1 (ja) | 2021-10-05 | 2023-04-13 | Eaファーマ株式会社 | 化合物又はその薬学的に許容される塩の製造方法 |
| WO2024210196A1 (ja) * | 2023-04-06 | 2024-10-10 | Eaファーマ株式会社 | スルホンアミド誘導体を含有する自己乳化組成物および自己乳化製剤 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69920497T2 (de) | 1998-10-26 | 2006-02-09 | The Research Foundation Of State University Of New York | Liponsaürederivate und deren verwendung bei der behandlung von krankheiten |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001042215A1 (en) | 1999-12-06 | 2001-06-14 | F. Hoffmann-La Roche Ag | 4-pyridinyl-n-acyl-l-phenylalanines |
| WO2001056994A1 (en) | 2000-02-04 | 2001-08-09 | Biogen, Inc. | Integrin antagonists |
| WO2002016329A1 (fr) | 2000-08-18 | 2002-02-28 | Ajinomoto Co., Inc. | Nouveaux derives de phenylalanine |
| WO2003070709A1 (fr) | 2002-02-20 | 2003-08-28 | Ajinomoto Co.,Inc. | Nouveau derive de phenylalanine |
| JP2003321358A (ja) | 2002-02-27 | 2003-11-11 | Tanabe Seiyaku Co Ltd | 医薬組成物 |
| WO2005061466A1 (ja) | 2003-12-22 | 2005-07-07 | Ajinomoto Co., Inc. | 新規フェニルアラニン誘導体 |
| WO2005077915A1 (en) | 2004-02-10 | 2005-08-25 | Janssen Pharmaceutica, N.V. | Pyridazinones as antagonists of a4 integrins |
| WO2005113003A2 (en) * | 2004-04-16 | 2005-12-01 | Genentech, Inc. | Method for augmenting b cell depletion |
| WO2006127584A1 (en) | 2005-05-20 | 2006-11-30 | Elan Pharmaceuticals, Inc. | Imidazolone phenylalanine derivatives as vla-4 antagonists |
| JP2007524626A (ja) * | 2003-06-25 | 2007-08-30 | エラン ファーマシューティカルズ,インコーポレイテッド | 関節リウマチを治療する方法および組成物 |
| WO2013161904A1 (ja) | 2012-04-24 | 2013-10-31 | 味の素株式会社 | スルホンアミド誘導体及びその医薬用途 |
| WO2015064580A1 (ja) | 2013-10-29 | 2015-05-07 | 味の素株式会社 | スルホンアミド誘導体及びその医薬用途 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6388084B1 (en) | 1999-12-06 | 2002-05-14 | Hoffmann-La Roche Inc. | 4-pyridinyl-n-acyl-l-phenylalanines |
| DE102013206014A1 (de) * | 2013-04-05 | 2014-10-09 | Dürr Systems GmbH | Energiewandler-System und Baugruppen hierfür |
-
2017
- 2017-02-06 EA EA201891780A patent/EA036432B1/ru not_active IP Right Cessation
- 2017-02-06 MX MX2018009459A patent/MX2018009459A/es unknown
- 2017-02-06 JP JP2017565026A patent/JP6820282B2/ja active Active
- 2017-02-06 AU AU2017214589A patent/AU2017214589B2/en active Active
- 2017-02-06 KR KR1020187025805A patent/KR20180104758A/ko not_active Application Discontinuation
- 2017-02-06 US US16/075,415 patent/US10562898B2/en active Active
- 2017-02-06 EP EP17747622.3A patent/EP3412660B9/en active Active
- 2017-02-06 HU HUE17747622A patent/HUE053354T2/hu unknown
- 2017-02-06 TW TW106103847A patent/TWI730042B/zh active
- 2017-02-06 ES ES17747622T patent/ES2856125T3/es active Active
- 2017-02-06 WO PCT/JP2017/004278 patent/WO2017135472A1/ja active Application Filing
- 2017-02-06 PT PT177476223T patent/PT3412660T/pt unknown
- 2017-02-06 DK DK17747622.3T patent/DK3412660T3/da active
- 2017-02-06 CA CA3013433A patent/CA3013433A1/en active Pending
- 2017-02-06 CN CN201780009898.0A patent/CN108699008B/zh active Active
- 2017-02-06 PL PL17747622T patent/PL3412660T3/pl unknown
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001042215A1 (en) | 1999-12-06 | 2001-06-14 | F. Hoffmann-La Roche Ag | 4-pyridinyl-n-acyl-l-phenylalanines |
| WO2001056994A1 (en) | 2000-02-04 | 2001-08-09 | Biogen, Inc. | Integrin antagonists |
| WO2002016329A1 (fr) | 2000-08-18 | 2002-02-28 | Ajinomoto Co., Inc. | Nouveaux derives de phenylalanine |
| WO2003070709A1 (fr) | 2002-02-20 | 2003-08-28 | Ajinomoto Co.,Inc. | Nouveau derive de phenylalanine |
| JP2003321358A (ja) | 2002-02-27 | 2003-11-11 | Tanabe Seiyaku Co Ltd | 医薬組成物 |
| JP2007524626A (ja) * | 2003-06-25 | 2007-08-30 | エラン ファーマシューティカルズ,インコーポレイテッド | 関節リウマチを治療する方法および組成物 |
| WO2005061466A1 (ja) | 2003-12-22 | 2005-07-07 | Ajinomoto Co., Inc. | 新規フェニルアラニン誘導体 |
| WO2005077915A1 (en) | 2004-02-10 | 2005-08-25 | Janssen Pharmaceutica, N.V. | Pyridazinones as antagonists of a4 integrins |
| WO2005113003A2 (en) * | 2004-04-16 | 2005-12-01 | Genentech, Inc. | Method for augmenting b cell depletion |
| WO2006127584A1 (en) | 2005-05-20 | 2006-11-30 | Elan Pharmaceuticals, Inc. | Imidazolone phenylalanine derivatives as vla-4 antagonists |
| WO2013161904A1 (ja) | 2012-04-24 | 2013-10-31 | 味の素株式会社 | スルホンアミド誘導体及びその医薬用途 |
| WO2015064580A1 (ja) | 2013-10-29 | 2015-05-07 | 味の素株式会社 | スルホンアミド誘導体及びその医薬用途 |
Non-Patent Citations (4)
| Title |
|---|
| "Bunshi Sekkei (Molecular Design)", vol. 7, 1990, HIROKAWA-SHOTEN, article "Iyakuhin no Kaihatsu (Development of Drugs)", pages: 163 - 198 |
| J CROHNS COLITIS, vol. 7, no. 11, December 2013 (2013-12-01), pages e533 - 42 |
| NAT MED, vol. 20, no. 12, December 2014 (2014-12-01), pages 1397 - 1400 |
| See also references of EP3412660A4 |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12071404B2 (en) | 2016-12-28 | 2024-08-27 | Ucb Pharma Gmbh | Aza (indole)-, benzothiophene-, and benzofuran-3-sulfonamides |
| US11820746B2 (en) | 2016-12-28 | 2023-11-21 | Ucb Pharma Gmbh | Aza (indole)-, benzothiophene-, and benzofuran-3-sulfonamides |
| US11345662B2 (en) * | 2016-12-28 | 2022-05-31 | Ucb Pharma Gmbh | Aza (indole)-, benzothiophene-, and benzofuran-3-sulfonamides |
| WO2019147824A1 (en) | 2018-01-26 | 2019-08-01 | Progenity, Inc. | Treatment of a disease of the gastrointestinal tract with a pde4 inhibitor |
| WO2019246455A1 (en) | 2018-06-20 | 2019-12-26 | Progenity, Inc. | Treatment of a disease of the gastrointestinal tract with an integrin inhibitor |
| US11224600B2 (en) | 2018-10-30 | 2022-01-18 | Gilead Sciences, Inc. | Compounds for inhibition of alpha 4 beta 7 integrin |
| US11179383B2 (en) | 2018-10-30 | 2021-11-23 | Gilead Sciences, Inc. | Compounds for inhibition of α4β7 integrin |
| US11174256B2 (en) | 2018-10-30 | 2021-11-16 | Gilead Sciences, Inc. | Imidazopyridine derivatives |
| US11116760B2 (en) | 2018-10-30 | 2021-09-14 | Gilead Sciences, Inc. | Quinoline derivatives |
| US12053462B2 (en) | 2018-10-30 | 2024-08-06 | Gilead Sciences, Inc. | Quinoline derivatives |
| US11578069B2 (en) | 2019-08-14 | 2023-02-14 | Gilead Sciences, Inc. | Compounds for inhibition of α4 β7 integrin |
| WO2021174024A1 (en) | 2020-02-28 | 2021-09-02 | First Wave Bio, Inc. | Methods of treating iatrogenic autoimmune colitis |
| WO2023058645A1 (ja) | 2021-10-05 | 2023-04-13 | Eaファーマ株式会社 | 化合物又はその薬学的に許容される塩の製造方法 |
| WO2024210196A1 (ja) * | 2023-04-06 | 2024-10-10 | Eaファーマ株式会社 | スルホンアミド誘導体を含有する自己乳化組成物および自己乳化製剤 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3412660A1 (en) | 2018-12-12 |
| EA036432B1 (ru) | 2020-11-10 |
| EP3412660A4 (en) | 2019-07-10 |
| US10562898B2 (en) | 2020-02-18 |
| DK3412660T3 (da) | 2020-11-30 |
| EA201891780A1 (ru) | 2019-02-28 |
| CA3013433A1 (en) | 2017-08-10 |
| KR20180104758A (ko) | 2018-09-21 |
| ES2856125T3 (es) | 2021-09-27 |
| CN108699008B (zh) | 2021-10-08 |
| JP6820282B2 (ja) | 2021-01-27 |
| MX2018009459A (es) | 2018-09-21 |
| US20190040059A1 (en) | 2019-02-07 |
| PL3412660T3 (pl) | 2021-04-19 |
| TW201733991A (zh) | 2017-10-01 |
| AU2017214589B2 (en) | 2020-09-03 |
| TWI730042B (zh) | 2021-06-11 |
| AU2017214589A1 (en) | 2018-08-16 |
| EP3412660B1 (en) | 2020-11-18 |
| PT3412660T (pt) | 2021-01-13 |
| BR112018015855A2 (ja) | 2018-12-26 |
| CN108699008A (zh) | 2018-10-23 |
| JPWO2017135472A1 (ja) | 2018-11-29 |
| HUE053354T2 (hu) | 2021-06-28 |
| EP3412660B9 (en) | 2021-04-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6820282B2 (ja) | スルホンアミド誘導体及びそれを含有する医薬組成物 | |
| WO2017135471A1 (ja) | α4β7インテグリン阻害剤 | |
| EP3490989B1 (en) | Covalent inhibitors of pad4 | |
| CN102985417B (zh) | 作为jak1抑制剂的哌啶-4-基氮杂环丁烷衍生物 | |
| EP3510025B1 (en) | Heteroaryl inhibitors of pad4 | |
| JP7253866B1 (ja) | トリアジン誘導体を含有する経口投与する製剤 | |
| US10208055B2 (en) | Pyrazole compounds as modulators of FSHR and uses thereof | |
| US20080021024A1 (en) | Metalloprotease inhibitors | |
| KR101424013B1 (ko) | 1-(3-시아노-1-아이소프로필-인돌-5-일)피라졸-4-카르복실산의 결정형과 그의 제조방법 | |
| US11981680B2 (en) | Substituted thienopyrroles as PAD4 inhibitors | |
| WO2009039321A1 (en) | Prolyl hydroxylase inhibitors | |
| WO2009039323A1 (en) | Prolyl hydroxylase inhibitors | |
| JP2010524962A (ja) | オーロラキナーゼ阻害剤のための創薬法 | |
| CN101821279A (zh) | 某些化学个体、组合物和方法 | |
| JP2016037467A (ja) | スルホンアミド誘導体及びその医薬用途 | |
| WO2009039322A1 (en) | Prolyl hydroxylase inhibitors | |
| US20220348562A1 (en) | Benzimidazole inhibitors of pad enzymes | |
| EP4153175A1 (en) | Piperidine-2,6-diones as small molecule degraders of helios and methods of use | |
| JP2016037468A (ja) | スルホンアミド誘導体及びその医薬用途 | |
| US20230118751A1 (en) | Fused ring compound and application thereof | |
| BR112018015855B1 (pt) | Derivado de sulfonamida | |
| US20240336568A1 (en) | Selective phd1 inhibitor compounds, compositions, and methods of use | |
| WO2010059555A1 (en) | Prolyl hydroxylase inhibitors | |
| CN111559982A (zh) | 2-(2-取代-4-羟基嘧啶-5-甲酰胺基)乙酸类化合物及其制备方法与用途 | |
| CN101296924A (zh) | 作为p38激酶抑制剂的吡唑异喹啉脲衍生物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17747622 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017565026 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3013433 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2018/009459 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112018015855 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2017214589 Country of ref document: AU Date of ref document: 20170206 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201891780 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 20187025805 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020187025805 Country of ref document: KR Ref document number: 2017747622 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2017747622 Country of ref document: EP Effective date: 20180905 |
|
| ENP | Entry into the national phase |
Ref document number: 112018015855 Country of ref document: BR Kind code of ref document: A2 Effective date: 20180802 |