WO2016199906A1 - スルホンアミド誘導体またはその薬学的に許容される酸付加塩 - Google Patents
スルホンアミド誘導体またはその薬学的に許容される酸付加塩 Download PDFInfo
- Publication number
- WO2016199906A1 WO2016199906A1 PCT/JP2016/067405 JP2016067405W WO2016199906A1 WO 2016199906 A1 WO2016199906 A1 WO 2016199906A1 JP 2016067405 W JP2016067405 W JP 2016067405W WO 2016199906 A1 WO2016199906 A1 WO 2016199906A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- hydrogen atom
- pharmaceutically acceptable
- acid addition
- acceptable acid
- Prior art date
Links
- 0 C*C(C(C1=O)=O)=C1NC Chemical compound C*C(C(C1=O)=O)=C1NC 0.000 description 6
- FKPHBNXIUGMZKL-UHFFFAOYSA-N C=CC(NCCN(C(C=C)=O)c1cccc(N)c1)=O Chemical compound C=CC(NCCN(C(C=C)=O)c1cccc(N)c1)=O FKPHBNXIUGMZKL-UHFFFAOYSA-N 0.000 description 1
- XARAICJTVBAUKW-UHFFFAOYSA-N CN(C)C(c1cc(-c(cc2S(=O)=O)ccc2OC)ccc1)=O Chemical compound CN(C)C(c1cc(-c(cc2S(=O)=O)ccc2OC)ccc1)=O XARAICJTVBAUKW-UHFFFAOYSA-N 0.000 description 1
- HYJOHITURDTGFR-UHFFFAOYSA-N CN(C)c1cc(NC(Nc2cc(N)ccc2)=O)ccc1 Chemical compound CN(C)c1cc(NC(Nc2cc(N)ccc2)=O)ccc1 HYJOHITURDTGFR-UHFFFAOYSA-N 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/22—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms
- C07C311/29—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound oxygen atoms having the sulfur atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/18—Sulfonamides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/34—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide
- A61K31/341—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide not condensed with another ring, e.g. ranitidine, furosemide, bufetolol, muscarine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/357—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having two or more oxygen atoms in the same ring, e.g. crown ethers, guanadrel
- A61K31/36—Compounds containing methylenedioxyphenyl groups, e.g. sesamin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
- A61K31/381—Heterocyclic compounds having sulfur as a ring hetero atom having five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4402—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 2, e.g. pheniramine, bisacodyl
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4406—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 3, e.g. zimeldine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4409—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 4, e.g. isoniazid, iproniazid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/21—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/44—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C335/00—Thioureas, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C335/04—Derivatives of thiourea
- C07C335/16—Derivatives of thiourea having nitrogen atoms of thiourea groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C335/20—Derivatives of thiourea having nitrogen atoms of thiourea groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton being further substituted by nitrogen atoms, not being part of nitro or nitroso groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/36—Radicals substituted by singly-bound nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/04—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D233/28—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/30—Oxygen or sulfur atoms
- C07D233/32—One oxygen atom
- C07D233/36—One oxygen atom with hydrocarbon radicals, substituted by nitrogen atoms, attached to ring nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/06—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D239/08—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms directly attached in position 2
- C07D239/10—Oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D243/00—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms
- C07D243/04—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/10—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by doubly bound oxygen or sulphur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/34—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D307/38—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D307/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/44—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D317/46—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D317/48—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring
- C07D317/62—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to atoms of the carbocyclic ring
- C07D317/68—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/26—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D333/30—Hetero atoms other than halogen
- C07D333/36—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/04—Systems containing only non-condensed rings with a four-membered ring
Definitions
- An object of the present invention is to provide a novel compound useful as an excellent orexin receptor agonist.
- Narcolepsy is a sleep disorder caused by the brain's inability to control the sleep / wake cycle. Symptoms of narcolepsy include unbearable sleepiness during the day, weakness-induced seizures (cataplexy) induced by emotions (especially strong joy and surprise), hallucinations during sleep, and paralysis during sleep. In general, it is seriously affected. The prevalence of narcolepsy is estimated to be 0.05-0.2% (0.16-0.18% in Japan), which is not a rare disease.
- Narcolepsy treatment is mainly pharmacotherapy and lifestyle guidance.
- methylphenidate, modafinil and pemoline are used to control daytime sleepiness, and tricyclic antidepressants, selective serotonin reuptake inhibitors (SSRI) and serotonin to control cataplexy.
- SSRI selective serotonin reuptake inhibitor
- SNRI -Noradrenaline reuptake inhibitor
- Orexin is a neuropeptide present in the lateral area of the hypothalamus, and there are two types of peptides: orexin-A and orexin-B (hypolectin 1 and hypolectin 2 (Non-patent Document 1)). These bind to orexin 1 receptor (hereinafter also referred to as OX1R) and orexin 2 receptor (hereinafter also referred to as OX2R), which are G protein-coupled receptors (Non-patent Document 2).
- OX1R orexin 1 receptor
- OX2R orexin 2 receptor
- Non-patent Document 3 mouse model experiments suggested that the function of OX2R is important for maintaining arousal (Non-Patent Documents 4 and 5).
- Orexin receptors are widely present in the brain. Orexin is a peptide and is not useful for pharmaceutical use because of its extremely low permeability through the blood brain barrier. Therefore, it is desired to reduce the molecular weight of orexin receptor agonists.
- a compound having a cyclic guanidine skeleton has been disclosed as a low molecular weight OX2R agonist (Patent Document 1).
- the orexin system is considered not only to regulate sleep / wakefulness as described above, but also to appropriately control feeding behavior according to emotion and energy balance. Fasted mice increase the amount of behavior to search for food by increasing wake time and decreasing sleep time. On the other hand, in orexin receptor-deficient mice, it has been clarified that the awakening time and the amount of behavior do not increase (Non-patent Document 7). Furthermore, OX2R has been shown to be involved in body weight homeostasis by improving leptin sensitivity (Non-patent Document 8). From these facts, orexin receptor (especially OX2R) agonists may be therapeutic agents for diabetes, obesity and metabolic syndrome as well as narcolepsy.
- Non-patent Document 9 spontaneous activity was reduced in septic rats, and it was reported that orexin-containing nerve activity in the hypothalamic periarch area was reduced. There is a report that body temperature increased and recovery of cardiac function was observed after administration (Non-patent Document 10). From these facts, orexin receptor agonists may be therapeutic agents for sepsis.
- An object of the present invention is to provide a novel low molecular weight compound exhibiting orexin receptor agonist activity, which is expected to be useful as an excellent preventive or therapeutic agent such as narcolepsy.
- R 1 is a hydrogen atom, C 1-6 alkoxy, —OH, C 1-6 alkyl or a halogen atom
- X is —C (R 5 ) ⁇ or —N ⁇
- R 5 is a hydrogen atom or C 1-6 alkoxy
- Any one of R 2 , R 3 and R 4 is R 6 , and the other two are each a hydrogen atom
- R 6 is (1) -NR 17 -Y 1 -R 7 (Where Y 1 is —C ( ⁇ O) NR 18 —, —C ( ⁇ S) NH—, —C ( ⁇ NH) NH—, —C ( ⁇ O) O—, —C ( ⁇ O) -, - SO 2 - or -SO 2 -NR 8 - a and, R 8 is a hydrogen atom or C 1-6 alkyl, R 17 is a hydrogen atom or C 1-6 alkyl, R 18 is a hydrogen atom or C 1-6 alkyl, R 17 and R 18
- R 11 is C 1-6 alkoxy or C 6-10 arylamino (where the C 6-10 aryl moiety of C 6-10 arylamino is selected from C 1-6 alkyl and C 1-6 alkoxy) And optionally substituted with 1 to 3 substituents)
- -N NR 12 (Where R 12 is (a) C 1-6 alkyl optionally substituted with —OH, (b) —OH, (c) di (C 1-6 alkyl) amino and (d) C 1— 6 alkoxy - to 1 is selected from carbonylamino is three good C 6-10 aryl which is substituted with a substituent), or (5) -NR 13 R 14 Wherein R 13 is a hydrogen atom, C 1-6 alkyl, C 6-10 aryl-C 1-6 alkyl or C 2-6 alkenyl-carbonyl, R 14 is a hydrogen atom or C 1-6 alkyl (where C 1-6 alkyl is (a) C 1-6 alkoxy-carbonyla
- R 15 is C 1-6 alkyl and R 16 is C 1-6 alkyl, or (2) —C ( ⁇ O) NR Wa R Wb (Wherein R Wa is C 1-6 alkyl (where C 1-6 alkyl may be substituted with phenyl, pyridyl, C 1-6 alkoxy-carbonylamino or di (C 1-6 alkyl) amino) Or phenyl (which may be substituted with di (C 1-6 alkyl) amino) and R Wb is a hydrogen atom or C 1-6 alkyl)] Or a pharmaceutically acceptable acid addition salt thereof (hereinafter also referred to as compound (I)).
- R Wa is C 1-6 alkyl (where C 1-6 alkyl may be substituted with phenyl, pyridyl, C 1-6 alkoxy-carbonylamino or di (C 1-6 alkyl) amino) Or phenyl (which may be substituted with di (C 1-6 alkyl) amino) and R Wb is a hydrogen
- R 15 is C 1-6 alkyl and R 16 is C 1-6 alkyl.
- R 2 is —NH—Y 1 —R 7 (where Y 1 and R 7 are as defined in [1] above), R 3 and R 4 are each a hydrogen atom, The compound of the above-mentioned [1] or a pharmaceutically acceptable acid addition salt thereof.
- Y 1 is —C ( ⁇ O) NH—, —C ( ⁇ S) NH—, —C ( ⁇ NH) NH— or —C ( ⁇ O) O—.
- R 7a is phenyl substituted with —NR 9a R 9b, where R 9a is C 1-6 alkyl and R 9b is C 1-6 alkyl, The symbol is as defined in the above [1]) or a pharmaceutically acceptable acid addition salt thereof according to the above [1].
- R 1 is a hydrogen atom, C 1-6 alkoxy, —OH, C 1-6 alkyl or a halogen atom
- X is —C (R 5 ) ⁇ or —N ⁇
- R 5 is a hydrogen atom or C 1-6 alkoxy
- R 6a is an optionally substituted hydrocarbon group, an optionally substituted heterocyclic group, a group bonded through an oxygen atom, or a group bonded through a sulfur atom
- R 15 is C 1-6 alkyl
- R 16 is C 1-6 alkyl
- a medicament comprising the compound according to any one of [1] to [15] or a pharmaceutically acceptable acid addition salt thereof.
- a sleepiness improving agent comprising the compound according to any one of [1] to [15] or a pharmaceutically acceptable acid addition salt thereof.
- a prophylactic or therapeutic agent for sepsis, severe sepsis or septic shock comprising the compound according to any one of [1] to [15] or a pharmaceutically acceptable acid addition salt thereof.
- a method for treating or preventing narcolepsy comprising administering an effective amount of the compound according to any one of [1] to [15] above or a pharmaceutically acceptable acid addition salt thereof.
- a method for improving drowsiness comprising administering an effective amount of the compound according to any one of [1] to [15] or a pharmaceutically acceptable acid addition salt thereof.
- the compound represented by the formula (I) or (I ′) of the present invention or a pharmaceutically acceptable acid addition salt thereof has excellent OX2R agonist activity.
- FIG. 1 shows a hypnogram for 3 hours after administration of a control substance (5% chromophore-containing physiological saline (Vehicle)) or a test compound (compound of Example 40) to a wild-type mouse (WT mouse).
- a control substance 5% chromophore-containing physiological saline (Vehicle)
- a test compound compound of Example 40
- C 1-6 alkyl means a monovalent linear or branched saturated hydrocarbon group having 1 to 6 carbon atoms, consisting of a carbon atom and a hydrogen atom. Examples include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, neopentyl, hexyl and the like.
- C 1-6 alkoxy means an oxy group to which C 1-6 alkyl is bonded.
- halogen atom in the present specification means a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- C 2-6 alkenyl refers to an unsaturated hydrocarbon having at least one monovalent linear or branched double bond having 2 to 6 carbon atoms, comprising a carbon atom and a hydrogen atom. Means group.
- C 2-6 alkynyl is an unsaturated hydrocarbon group having at least one monovalent straight-chain or branched triple bond having 2 to 6 carbon atoms, consisting of a carbon atom and a hydrogen atom. Means.
- C 3-10 cycloalkyl means a monocyclic or polycyclic aliphatic carbocyclic group having 3 to 10 carbon atoms. Examples include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, adamantyl and the like.
- C 6-10 aryl means a monocyclic or condensed aromatic carbocyclic group having 6 to 10 carbon atoms.
- phenyl, 1-naphthyl, 2-naphthyl and the like can be mentioned.
- it is phenyl.
- the “5- to 10-membered heteroaryl” in the present specification includes, as a ring-constituting atom, one or two kinds selected from an oxygen atom, a sulfur atom and a nitrogen atom in addition to a carbon atom, and 1 to 4 heteroatoms.
- C 1-6 haloalkyl means C 1-6 alkyl substituted with 1 to 5 (preferably 1 to 3) halogen atoms. Examples include trifluoromethyl, 2,2,2-trifluoroethyl, 3,3,3-trifluoropropyl, and the like. Preferably, it is trifluoromethyl.
- C 1-6 alkoxy-carbonyl means a carbonyl group to which C 1-6 alkoxy is bonded. Examples include methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, isopropoxycarbonyl, butoxycarbonyl, isobutoxycarbonyl, tert-butoxycarbonyl and the like.
- C 6-10 arylamino means amino substituted with C 6-10 aryl.
- phenylamino, 1-naphthylamino, 2-naphthylamino and the like can be mentioned.
- it is phenylamino.
- C 1-6 alkyl optionally substituted with -OH herein is meant one C 1-6 alkyl optionally substituted with -OH. Examples thereof include hydroxymethyl, 2-hydroxyethyl, 3-hydroxypropyl and the like.
- di (C 1-6 alkyl) amino means amino substituted with two C 1-6 alkyl.
- C 1-6 alkoxy-carbonylamino means amino substituted with C 1-6 alkoxy-carbonyl. Examples thereof include methoxycarbonylamino, ethoxycarbonylamino, propoxycarbonylamino, isopropoxycarbonylamino, butoxycarbonylamino, isobutoxycarbonylamino, tert-butoxycarbonylamino and the like.
- C 6-10 aryl-C 1-6 alkyl means C 1-6 alkyl substituted with C 6-10 aryl.
- benzyl, phenethyl and the like can be mentioned. Preferably, it is benzyl.
- C 2-6 alkenyl-carbonyl means a carbonyl group to which C 2-6 alkenyl is bonded. Examples thereof include acryloyl and methacryloyl.
- C 2-6 alkenyl-carbonylamino means amino substituted with C 2-6 alkenyl-carbonyl. Examples thereof include acryloylamino and methacryloylamino.
- C 6-10 aryl-C 1-6 alkylaminocarbonyl means an aminocarbonyl substituted with C 6-10 aryl-C 1-6 alkyl.
- benzylaminocarbonyl, phenethylaminocarbonyl and the like can be mentioned. Benzylaminocarbonyl is preferred.
- C 6-10 aryl-carbonyl means a carbonyl group to which C 6-10 aryl is bonded. Examples include benzoyl, 1-naphthoyl, 2-naphthoyl and the like. Benzoyl is preferable.
- a 5- to 7-membered heterocyclic ring containing one nitrogen atom, which is formed by combining R 13 and R 14 together with the nitrogen atom to which they are bonded includes imidazolidine, Examples include piperazine or 1,4-diazepane.
- a 5- to 7-membered heterocyclic ring formed by combining R 17 and R 18 with each other and the nitrogen atom to which they are bonded and adjacent C ( ⁇ O) is imidazolidine-2. -One, hexahydropyrimidin-2-one or 1,3-diazepan-2-one.
- hydrocarbon group of the “optionally substituted hydrocarbon group” in the present specification, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 3-10 cycloalkyl, And C 6-10 aryl.
- heterocyclic group of the “optionally substituted heterocyclic group” in the present specification include 5- to 10-membered heteroaryl, 4- to 10-membered non-aromatic heterocyclic group and the like.
- the “4- to 10-membered non-aromatic heterocyclic group” is, as a ring-constituting atom, one or two kinds selected from oxygen atoms, sulfur atoms and nitrogen atoms in addition to carbon atoms, and 1 to 4 hetero atoms
- a 4- to 10-membered monocyclic or bicyclic saturated or unsaturated non-aromatic heterocyclic group containing an atom is meant.
- groups of the “group bonded through an oxygen atom” in the present specification include an optionally substituted hydrocarbon group or an oxy group (—O—) to which an optionally substituted heterocyclic group is bonded.
- the “group bonded through a sulfur atom” includes an optionally substituted hydrocarbon group, an optionally substituted heterocyclic group, or a group bonded through a nitrogen atom.
- a thio group (—S—), a sulfinyl group (—SO—), a sulfonyl group (—SO 2 —) and the like can be mentioned.
- Examples of the “group bonded through a nitrogen atom” in the present specification include an amino group to which an optionally substituted hydrocarbon group is bonded, an amino group to which an optionally substituted heterocyclic group is bonded, and the like.
- “optionally substituted” means that it may be substituted with, for example, 1 to 5, preferably 1 to 3 substituents selected from the following substituent group.
- R 21k is —OH, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 3-10 cycloalkyl, C 6-10 aryl, C 6-10 aryl-C 1- 6 alkyl, 5 to 10 membered heteroaryl or 4 to 10 membered non-aromatic heterocyclic group, n is 0, 1 or 2), —SO 2 —NR 21m R 21n (Wherein R 21m and R 21n each independently represents a hydrogen atom, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 3-10 cycloalkyl, C 6-10 aryl, C 6-10 aryl-C 1-6 alkyl, 5-10 membered heteroaryl or 4-10 membered non-aromatic heterocyclic group), and C 1-3 alkylenedioxy (eg, methylenedioxy).
- anti-narcolepsy agent means a therapeutic or prophylactic agent for narcolepsy.
- the “drowsiness improving agent” in the present specification means an agent that improves daytime sleepiness due to shift work, jet lag, insomnia or sleep apnea syndrome.
- R 1 is a hydrogen atom, C 1-6 alkoxy, —OH, C 1-6 alkyl or a halogen atom.
- R 1 include a hydrogen atom, methoxy, ethoxy, propoxy, —OH, methyl, chlorine atom and the like.
- R 1 is preferably C 1-6 alkoxy (eg, methoxy, ethoxy, propoxy), and particularly preferably methoxy.
- W is (1) formula (iii):
- W is preferably the formula (iii):
- R 15 is C 1-6 alkyl (eg, methyl), and R 16 is C 1-6 alkyl (eg, methyl)).
- W is preferably —C ( ⁇ O) NR Wa R Wb (Where R Wa is C 1-6 alkyl (eg, methyl, ethyl, propyl, 2,2-dimethylpropyl) (where C 1-6 alkyl is phenyl, pyridyl, C 1-6 alkoxy-carbonylamino) (Eg, optionally substituted with tert-butoxycarbonylamino) or di (C 1-6 alkyl) amino (eg, dimethylamino)) or phenyl (wherein phenyl is di (C 1-6 alkyl) amino R Wb is a hydrogen atom or C 1-6 alkyl (eg, ethyl)) (which may be substituted with (eg, dimethylamino)).
- R Wa is C 1-6 alkyl (eg, methyl, ethyl, propyl, 2,2-dimethylpropyl) (where C 1-6 alkyl is phenyl, pyrid
- X is —C (R 5 ) ⁇ or —N ⁇ , and R 5 is a hydrogen atom or C 1-6 alkoxy. Examples of R 5 include a hydrogen atom or methoxy. R 5 is preferably a hydrogen atom. X is preferably —C (R 5 ) ⁇ , and particularly preferably —CH ⁇ .
- R 2 , R 3 and R 4 are R 6 , and the remaining two are each a hydrogen atom.
- Examples of the combination of R 2 , R 3 and R 4 include the following combinations. (1) R 2 is R 6 , and R 3 and R 4 are each a hydrogen atom. (2) R 3 is R 6 , and R 2 and R 4 are each a hydrogen atom. (3) R 4 is R 6 , and R 2 and R 3 are each a hydrogen atom. Among these, it is preferable that R 2 is R 6 and R 3 and R 4 are each a hydrogen atom.
- R 6 is (1) —NR 17 —Y 1 —R 7 , (2) —NH—Y 2 —R 10 , (3) Formula (ii):
- R 8 is a hydrogen atom or C 1-6 alkyl (eg, methyl).
- R 17 is a hydrogen atom or C 1-6 alkyl (eg, methyl), R 18 is a hydrogen atom or C 1-6 alkyl (eg, methyl); R 17 and R 18 are bonded to each other, and together with the nitrogen atom to which they are bonded and the adjacent C ( ⁇ O), a 5- to 7-membered heterocyclic ring (eg, imidazolidin-2-one, hexahydropyrimidine- 2-one, 1,3-diazepan-2-one) may be formed.
- R 17 and R 18 are preferably each a hydrogen atom.
- R 7 is (A) C 6-10 aryl (eg, phenyl, 1-naphthyl, 2-naphthyl), (B) 5 to 10 membered heteroaryl (eg, pyridyl, thienyl, thiazolyl), (C) C 1-6 alkyl (eg, methyl, tert-butyl), or (d) C 2-6 alkenyl (eg, vinyl) Wherein C 1-6 alkyl and C 2-6 alkenyl may be substituted with one substituent selected from phenyl, furyl and diphenylmethylsulfinyl, C 6-10 aryl and 5- to 10-membered heteroaryl may be optionally substituted with 1 to 4 (preferably 1 to 3, more preferably 1 or 2) R 9 , Each R 9 is independently Halogen atoms (eg, bromine atoms), -NO 2 , -OH, C 1-6 alkyl (eg methyl), C 1-6 haloalkyl (
- R 7 is preferably (A) C 6-10 aryl (eg, phenyl, 1-naphthyl, 2-naphthyl), or (b) 5-10 membered heteroaryl (eg, pyridyl, thienyl, thiazolyl) (Wherein C 6-10 aryl and 5- to 10-membered heteroaryl are optionally substituted by 1 to 4 (preferably 1 to 3, more preferably 1 or 2) R 9 R 9 is as defined above.
- C 6-10 aryl eg, phenyl, 1-naphthyl, 2-naphthyl
- 5-10 membered heteroaryl eg, pyridyl, thienyl, thiazolyl
- R 9 R 9 is as defined above.
- R 7 is particularly preferably —NR 9a R 9b (wherein R 9a is C 1-6 alkyl (eg, methyl), and R 9b is a hydrogen atom or C 1-6 alkyl (eg, methyl)). Phenyl) substituted with
- R 6 is —NH—Y 2 —R 10
- Y 2 is —CH 2 — or a single bond.
- R 10 is (a) C 6-10 aryl (eg, phenyl), or (b) 5 to 10 membered heteroaryl (eg, pyridyl, thienyl, thiazolyl) Wherein C 6-10 aryl and 5- to 10-membered heteroaryl may be optionally substituted with 1 to 4 R 9 , where R 9 is as defined above. .
- R 10 is preferably C 6-10 aryl (eg phenyl) Wherein C 6-10 aryl may be optionally substituted with 1 to 4 (preferably 1 to 3, more preferably 1 or 2) R 9 ,
- Each R 9 is independently Halogen atoms (eg, bromine atoms), -OH, C 1-6 alkyl (eg methyl), C 1-6 haloalkyl (eg, trifluoromethyl), C 1-6 alkoxy (eg, methoxy), or —C ( ⁇ O) NR 9d R 9e (where R 9d is a hydrogen atom or C 1-6 alkyl (eg, methyl), and R 9e is a hydrogen atom Or C 1-6 alkyl (eg, methyl)), or two R 9 together form methylenedioxy).
- R 9d is a hydrogen atom or C 1-6 alkyl (eg, methyl)
- R 9e is a hydrogen atom Or C 1-6 alkyl (eg, methyl)
- R 11 is C 1-6 alkoxy (eg, methoxy) or C 6-10 arylamino (eg, phenylamino) (wherein the C 6-10 aryl moiety of C 6-10 arylamino is C 1-6 alkyl ( Eg, methyl) and C 1-6 alkoxy (eg, methoxy) optionally substituted with 1 to 3 substituents).
- R 12 is (a) C 1-6 alkyl optionally substituted with —OH (eg, methyl), (b) —OH, (c) di (C 1-6 alkyl) amino (eg, dimethylamino). And (d) C 6-10 aryl (eg, phenyl) optionally substituted with 1 to 3 substituents selected from C 1-6 alkoxy-carbonylamino (eg, tert-butoxycarbonylamino) is there.
- R 13 is a hydrogen atom, C 1-6 alkyl (eg, methyl), C 6-10 aryl-C 1-6 alkyl (eg, benzyl) or C 2-6 alkenyl-carbonyl (eg, acryloyl);
- R 14 is a hydrogen atom or C 1-6 alkyl (eg, methyl, ethyl) (where C 1-6 alkyl is (a) C 1-6 alkoxy-carbonylamino (eg, tert-butoxycarbonylamino), ( b) C 2-10 alkenyl-carbonylamino (eg acryloylamino) and (c) C 6-10 aryl-C 1-6 alkylaminocarbonyl optionally substituted by C 1-6 alkyl (eg methyl)
- R 13 and R 14 may be bonded to each other and together with the nitrogen atom to which they are bonded (optionally substituted with one substituent selected from benzylamino
- location further 5 to 7-membered heterocyclic ring containing one nitrogen atom (e.g., imidazolidine) (where, in 5 to 7-membered heterocycle C 1-6 alkyl (e.g., methyl) which may C 6-10 aryl optionally - carbonyl (e.g., benzoyl) to form a well may) be substituted with.
- R 14 is ethyl substituted with C 2-6 alkenyl-carbonylamino
- R 13 is not a hydrogen atom.
- R 14 is preferably a hydrogen atom or C 1-6 alkyl (eg, methyl, ethyl) (where C 1-6 alkyl is (a) C 1-6 alkoxy-carbonylamino (eg, tert-butoxycarbonyl) Amino) and (b) one selected from C 6-10 aryl-C 1-6 alkylaminocarbonyl (eg, benzylaminocarbonyl) optionally substituted with C 1-6 alkyl (eg, methyl) Optionally substituted with a substituent).
- C 1-6 alkyl eg, methyl, ethyl
- C 1-6 alkyl is (a) C 1-6 alkoxy-carbonylamino (eg, tert-butoxycarbonyl) Amino) and (b) one selected from C 6-10 aryl-C 1-6 alkylaminocarbonyl (eg, benzylaminocarbonyl) optionally substituted with C 1-6 alkyl (eg
- R 7a is phenyl substituted with —NR 9a R 9b, where R 9a is C 1-6 alkyl and R 9b is C 1-6 alkyl, The symbol is as defined in the above [1]) or a pharmaceutically acceptable acid addition salt thereof.
- Y 1 is preferably —C ( ⁇ O) NH—, —C ( ⁇ S) NH—, —C ( ⁇ NH) NH— or —C ( ⁇ O) O—, particularly preferably — C ( ⁇ O) NH—.
- R 7 is preferably (A) C 6-10 aryl (eg, phenyl, 1-naphthyl, 2-naphthyl), or (b) 5-10 membered heteroaryl (eg, pyridyl, thienyl, thiazolyl) Wherein C 6-10 aryl and 5- to 10-membered heteroaryl may be optionally substituted with 1 to 4 R 9 , where R 9 is as defined above. .
- R 7 is particularly preferably phenyl substituted with —NR 9a R 9b, where R 9a is C 1-6 alkyl and R 9b is C 1-6 alkyl.
- R 1 is a hydrogen atom, C 1-6 alkoxy, —OH, C 1-6 alkyl or a halogen atom
- X is —C (R 5 ) ⁇ or —N ⁇
- R 5 is a hydrogen atom or C 1-6 alkoxy
- Any one of R 2 , R 3 and R 4 is R 6 , and the other two are each a hydrogen atom
- R 6 is (1) -NR 17 -Y 1 -R 7 (Where Y 1 is —C ( ⁇ O) NR 18 —, —C ( ⁇ S) NH—, —C ( ⁇ NH) NH—, —C ( ⁇ O) O—, —C ( ⁇ O) -, - SO 2 - or -SO 2 -NR 8 - a and, R 8 is a hydrogen atom or C 1-6 alkyl, R 17 is a hydrogen atom or C 1-6 alkyl, R 18 is a hydrogen atom or C atom or C atom or C
- R 11 is C 1-6 alkoxy or C 6-10 arylamino (eg, phenylamino) (where the C 6-10 aryl moiety of C 6-10 arylamino is C 1-6 alkyl and C A group represented by 1) which may be substituted with 1 to 3 substituents selected from 1-6 alkoxy;
- -N NR 12 (Where R 12 is (a) C 1-6 alkyl optionally substituted with —OH, (b) —OH, (c) di (C 1-6 alkyl) amino and (d) C 1— 6 alkoxy - 1 to 3 substituents optionally substituted by C 6-10 aryl is selected from carbonylamino (e.g., phenyl) is), or (5) -NR 13 R 14 Wherein R 13 is a hydrogen atom, C 1-6 alkyl, C 6-10 aryl-C 1-6 alkyl (eg, benzyl) or C 2-6 alkenyl-carbonyl, R 14 is a
- R 15 is C 1-6 alkyl and R 16 is C 1-6 alkyl.
- R 17 and R 18 are each a hydrogen atom.
- R 1 is C 1-6 alkoxy
- R 7a is phenyl substituted with —NR 9a R 9b (where R 9a is C 1-6 alkyl and R 9b is C 1-6 alkyl)
- R Wa is C 1-6 alkyl (where C 1-6 alkyl may be substituted with phenyl, pyridyl, C 1-6 alkoxy-carbonylamino or di (C 1-6 alkyl) amino) or phenyl Where phenyl may be substituted with di (C 1-6 alkyl) amino
- R Wb is a hydrogen atom or C 1-6 alkyl, A compound or a pharmaceutically acceptable acid addition salt thereof.
- R 1 is a hydrogen atom, C 1-6 alkoxy, —OH, C 1-6 alkyl or a halogen atom
- R 5 is a hydrogen atom or C 1-6 alkoxy
- R 6 is (1) -NR 17 -Y 1 -R 7 (Where Y 1 is —C ( ⁇ O) NR 18 —, —C ( ⁇ S) NH—, —C ( ⁇ NH) NH—, —C ( ⁇ O) O—, —C ( ⁇ O) -, - SO 2 - or -SO 2 -NR 8 - a and, R 8 is a hydrogen atom or C 1-6 alkyl, R 17 is a hydrogen atom or C 1-6 alkyl, R 18 is a hydrogen atom or C 1-6 alkyl, R 17 and R 18 are bonded to each other, and together with the nitrogen atom to which they are bonded and the adjacent C ( ⁇ O), a 5- to 7-membered heterocycl
- R 11 is C 1-6 alkoxy or C 6-10 arylamino (eg, phenylamino) (where the C 6-10 aryl moiety of C 6-10 arylamino is C 1-6 alkyl and C A group represented by 1) which may be substituted with 1 to 3 substituents selected from 1-6 alkoxy;
- -N NR 12 (Where R 12 is (a) C 1-6 alkyl optionally substituted with —OH, (b) —OH, (c) di (C 1-6 alkyl) amino and (d) C 1— 6 alkoxy - 1 to 3 substituents optionally substituted by C 6-10 aryl is selected from carbonylamino (e.g., phenyl) is), or (5) -NR 13 R 14 Wherein R 13 is a hydrogen atom, C 1-6 alkyl, C 6-10 aryl-C 1-6 alkyl (eg, benzyl) or C 2-6 alkenyl-carbonyl, R 14 is a
- R 14 is ethyl substituted with C 2-6 alkenyl-carbonylamino, R 13 is not a hydrogen atom
- R 15 is C 1-6 alkyl
- R 16 is C 1-6 alkyl.
- R 1 is a hydrogen atom, C 1-6 alkoxy, —OH, C 1-6 alkyl or a halogen atom
- R 5 is a hydrogen atom or C 1-6 alkoxy
- Y 1 represents —C ( ⁇ O) NR 18 —, —C ( ⁇ S) NH—, —C ( ⁇ NH) NH—, —C ( ⁇ O) O—, —C ( ⁇ O) —, —SO 2 — or —SO 2 —NR 8 —
- R 8 is a hydrogen atom or C 1-6 alkyl
- R 18 is a hydrogen atom
- R 7 is (a) C 6-10 aryl (eg, phenyl, 1-naphthyl, 2-naphthyl), (B) 5 to 10 membered heteroaryl (eg, pyridyl, thienyl, thiazolyl), (C) C 1-6 alkyl, or (d)
- R 1 is a hydrogen atom, C 1-6 alkoxy, —OH, C 1-6 alkyl or a halogen atom
- R 5 is a hydrogen atom or C 1-6 alkoxy
- Y 1 is —C ( ⁇ O) NR 18 —, —C ( ⁇ S) NH— or —C ( ⁇ NH) NH—
- R 8 is a hydrogen atom or C 1-6 alkyl
- R 18 is a hydrogen atom
- R 7 is (a) C 6-10 aryl (eg, phenyl), or (b) 5 to 10 membered heteroaryl (eg, pyridyl, thienyl).
- R 1 is C 1-6 alkoxy
- R 5 is a hydrogen atom
- Y 2 is —CH 2 — or a single bond
- R 10 is C 6-10 aryl (eg, phenyl) Wherein C 6-10 aryl may be optionally substituted with 1 to 4 (preferably 1 to 3, more preferably 1 or 2)
- R 9 is independently Halogen atoms, -OH, C 1-6 alkyl, C 1-6 haloalkyl, C 1-6 alkoxy, or —C ( ⁇ O) NR 9d R 9e (where R 9d is C 1-6 alkyl and R 9e is C 1-6 alkyl), or two R 9 Together form methylenedioxy),
- R 15 is C 1-6 alkyl and R 16 is C 1-6 alkyl.
- R 1 is C 1-6 alkoxy
- R 5 is a hydrogen atom
- R 11 is C 1-6 alkoxy or C 6-10 arylamino (eg, phenylamino) (wherein the C 6-10 aryl moiety of C 6-10 arylamino is C 1-6 alkyl and C 1-6 alkoxy)
- R 15 is C 1-6 alkyl
- R 16 is C 1-6 alkyl.
- R 1 is C 1-6 alkoxy
- R 5 is a hydrogen atom
- R 12 is (a) optionally substituted with -OH C 1-6 alkyl, (b) -OH, (c ) di (C 1-6 alkyl) amino and (d) C 1-6 alkoxy - carbonyl C 6-10 aryl (eg, phenyl) optionally substituted with 1 or 2 substituents selected from amino
- R 15 is C 1-6 alkyl and R 16 is C 1-6 alkyl.
- R 1 is C 1-6 alkoxy;
- R 5 is a hydrogen atom,
- R 13 is a hydrogen atom, C 1-6 alkyl, C 6-10 aryl-C 1-6 alkyl (eg, benzyl) or C 2-6 alkenyl-carbonyl,
- R 14 is a hydrogen atom or C 1-6 alkyl (where C 1-6 alkyl is (a) C 1-6 alkoxy-carbonylamino, (b) C 2-6 alkenyl-carbonylamino and (c) C 1 Whether it is substituted with one substituent selected from C 6-10 aryl-C 1-6 alkylaminocarbonyl (eg, benzylaminocarbonyl) optionally substituted with -6 alkyl) Or R 13 and R 14 are bonded to each other, and together with the nitrogen atom to which they are bonded, a 5- to 7-membered heterocycle containing one nitrogen atom (eg, imidazolidine
- R 14 is ethyl substituted with C 2-6 alkenyl-carbonylamino, R 13 is not a hydrogen atom
- R 15 is C 1-6 alkyl
- R 16 is C 1-6 alkyl.
- R 1 is C 1-6 alkoxy
- R 7 is phenyl substituted with —NR 9a R 9b (where R 9a is C 1-6 alkyl and R 9b is C 1-6 alkyl);
- R 15 is C 1-6 alkyl and
- R 16 is C 1-6 alkyl.
- R 1 is C 1-6 alkoxy
- R 7 is phenyl substituted with —NR 9a R 9b (where R 9a is C 1-6 alkyl and R 9b is C 1-6 alkyl); R 15 is C 1-6 alkyl and R 16 is C 1-6 alkyl] Or a pharmaceutically acceptable acid addition salt thereof.
- R 1 is C 1-6 alkoxy
- R 7 is phenyl substituted with —NR 9a R 9b (where R 9a is C 1-6 alkyl and R 9b is C 1-6 alkyl);
- R 15 is C 1-6 alkyl and
- R 16 is C 1-6 alkyl.
- the pharmaceutically acceptable acid addition salts of the compounds of formula (I) or (I ′) of the present invention include hydrochloride, sulfate, nitrate, hydrobromide, hydroiodide, phosphate Inorganic acid salts such as acetate, lactate, citrate, oxalate, glutarate, malate, tartrate, fumarate, mandelate, maleate, benzoate, phthalate And organic sulfonates such as methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, camphorsulfonate, etc., but these are limited is not.
- hydrochloride hydrobromide, phosphate, tartrate, methanesulfonate, camphorsulfonate are preferable, hydrochloride, tartrate or methanesulfonate is more preferable, and hydrochloride is particularly preferably used. These are also not limiting.
- the compound of the present invention represented by the above formula (I) or (I ′) can be produced by an appropriate method based on the characteristics derived from the basic skeleton and substituent.
- the starting materials and reagents used in the production of these compounds are generally available or described in references such as Organic Reactions (Wiley & Sons), Fieser and Fieser's Reagent for Organic Synthesis (Wiley & Sons), etc. And can be synthesized by methods known to those skilled in the art.
- Specific examples of the method for producing the compound of the present invention represented by the above formula (I) or (I ′) include the methods shown in Schemes 1 to 18.
- Process 1 Compound (IV) can be obtained, for example, by amidating sulfonyl chloride compound (II) with amine compound (III).
- Solvents include halogenated hydrocarbon solvents such as dichloromethane, chloroform, 1,2-dichloroethane, ether solvents such as diethyl ether, tetrahydrofuran (THF), 1,2-dimethoxyethane (DME), dioxane, N, N-dimethyl.
- Aprotic polar solvents such as formamide (DMF) and dimethyl sulfoxide (DMSO) or a mixed solvent thereof can be used.
- dichloromethane or THF is preferably used.
- the sulfonyl chloride compound (II) is used in an amount of 0.5 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to the amine compound (III).
- Examples of the base include sodium carbonate, potassium carbonate, cesium carbonate, potassium phosphate, sodium hydroxide, potassium hydroxide, barium hydroxide, triethylamine, diisopropylethylamine, pyridine, 4-dimethylaminopyridine, and diisopropylethylamine. Triethylamine or pyridine is preferably used.
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 10 minutes to 48 hours.
- the concentration of the substrate (II) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- the amine compound (V) can be obtained, for example, by deprotecting the tert-butoxycarbonyl (Boc) group of the compound (IV) under acidic conditions.
- the solvent halogenated hydrocarbon solvents such as dichloromethane, chloroform and 1,2-dichloroethane, ether solvents such as THF, DME and dioxane, alcohol solvents such as methanol, ethanol and propanol, or a mixed solvent thereof can be used. .
- dichloromethane or dioxane is preferably used.
- the acid examples include organic acids such as trifluoroacetic acid, methanesulfonic acid, p-toluenesulfonic acid, and trifluoromethanesulfonic acid, or inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, and phosphoric acid. Can be used. Usually, trifluoroacetic acid or hydrochloric acid is preferably used. Alternatively, a hydrogen chloride-methanol solution, a hydrogen chloride-THF solution, a hydrogen chloride-ethyl acetate solution, and a hydrogen chloride-dioxane solution obtained by dissolving hydrogen chloride in an organic solvent can be used alone.
- organic acids such as trifluoroacetic acid, methanesulfonic acid, p-toluenesulfonic acid, and trifluoromethanesulfonic acid
- inorganic acids such as hydrochloric acid, hydrobromic acid,
- a hydrogen chloride-methanol solution or a hydrogen chloride-THF solution is used in an amount of 1.0 to 100 equivalents, preferably 3.0 to 10 equivalents, relative to compound (IV).
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours.
- the concentration of the substrate (IV) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- Compound (I-1) can be obtained, for example, by amidating amine compound (V) with acyl halide compound (VI) (wherein X 1 is a halogen atom).
- the solvent include halogenated hydrocarbon solvents such as dichloromethane, chloroform, 1,2-dichloroethane, ether solvents such as diethyl ether, THF, DME, and dioxane, aprotic polar solvents such as DMF and DMSO, or a mixed solvent thereof.
- halogenated hydrocarbon solvents such as dichloromethane, chloroform, 1,2-dichloroethane
- ether solvents such as diethyl ether, THF, DME, and dioxane
- aprotic polar solvents such as DMF and DMSO
- a mixed solvent thereof can be used.
- dichloromethane or THF is preferably used.
- the acyl halide compound (VI) is used in an amount of 0.5 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to the amine compound (V).
- the base include sodium carbonate, potassium carbonate, cesium carbonate, potassium phosphate, sodium hydroxide, potassium hydroxide, barium hydroxide, triethylamine, diisopropylethylamine, pyridine, 4-dimethylaminopyridine, and diisopropylethylamine. Triethylamine or pyridine is preferably used.
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 10 minutes to 48 hours.
- the concentration of the substrate (V) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- Compound (I-1) can be obtained, for example, by amidating amine compound (V) with carboxylic acid (VI) (wherein X 1 is a hydroxy group).
- Solvents include halogenated hydrocarbon solvents such as dichloromethane, chloroform, 1,2-dichloroethane, ether solvents such as diethyl ether, THF, DME, dioxane, aprotic polar solvents such as DMF, DMSO, and ethyl acetate, methanol, Alcohol solvents such as ethanol and propanol, or a mixed solvent thereof can be used.
- dichloromethane or THF is preferably used.
- the carboxylic acid (VI) is used in an amount of 0.5 to 20 equivalents, preferably 0.5 to 10 equivalents, relative to the amine compound (V).
- condensing agent examples include dicyclohexylcarbodiimide, 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (EDCI), benzotriazol-1-yloxytris (dimethylamino) phosphonium hexafluorophosphate (BOP), N, N′-carbonyldiimidazole (CDI), 4- (4,6-dimethoxy-1,3,5-triazin-2-yl) -4-methylmorpholinium chloride (DMT-MM), ⁇ [ (1-Cyano-2-ethoxy-2-oxoethylidene) amino] oxy ⁇ -4-morpholinomethylene ⁇ dimethylammonium hexafluorophosphate (COMU), O- (7-azabenzotriazol-1-yl) -N , N, N ′, N′-tetramethyluronium hexafluorophosphate (HATU)
- the condensing agent is used in an amount of 1.0 to 100 equivalents, preferably 1.0 to 10 equivalents, relative to the amine compound (XXI).
- a base triethylamine, diisopropylethylamine, pyridine, N-methylmorpholine and the like can be used, and triethylamine or diisopropylethylamine is preferably used.
- the base is used in an amount of 3.0 to 100 equivalents, preferably 3.0 to 10 equivalents, relative to the amine compound (V).
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 60 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours. Further, the concentration of the substrate (V) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- Compound (I-2) can be obtained by, for example, reductive alkylation of amine compound (V) with aldehyde compound (VII).
- Solvents include halogenated hydrocarbon solvents such as dichloromethane, chloroform and 1,2-dichloroethane, ether solvents such as diethyl ether, THF, DME and dioxane, aprotic polar solvents such as DMF and DMSO, methanol, ethanol and propanol.
- An alcohol solvent such as acetic acid, water, or a mixed solvent thereof can be used.
- the aldehyde compound (VII) is used in an amount of 0.5 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to the amine compound (V).
- the reducing agent include sodium borohydride, lithium borohydride, sodium cyanoborohydride, sodium triacetoxyborohydride, 2-picoline-borane complex, and the like.
- triethylamine, diisopropylethylamine, pyridine, N-methylmorpholine and the like can be used, and triethylamine or diisopropylethylamine is preferably used.
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 10 minutes to 48 hours. Further, the concentration of the substrate (V) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- Compound (I-3) can be obtained, for example, by subjecting amine compound (V) to a coupling reaction with boronic acid compound (VIII) in the presence of a copper catalyst.
- Solvents include halogenated hydrocarbon solvents such as dichloromethane, chloroform and 1,2-dichloroethane, ether solvents such as THF, DME and dioxane, aprotic polar solvents such as DMF and DMSO, alcohols such as methanol, ethanol and propanol.
- a solvent, an aromatic hydrocarbon solvent such as benzene, toluene, xylene, or a mixed solvent thereof can be used.
- the boronic acid compound (VIII) is used in an amount of 1.0 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to the amine compound (V).
- the copper catalyst include copper (II) acetate.
- the copper catalyst is used in an amount of 0.001 to 1 equivalent, preferably 0.005 to 0.5 equivalent, relative to the amine compound (V).
- a base triethylamine, diisopropylethylamine, pyridine, N-methylmorpholine and the like can be used, and triethylamine or diisopropylethylamine is preferably used.
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours. Further, the concentration of the substrate (V) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- Compound (I-4) can be obtained, for example, by reacting amine compound (V) with isocyanate (IX) or isocyanate (IX) generated from carboxylic acid R 7 —CO 2 H (IX-1). Can do.
- Solvents include halogenated hydrocarbon solvents such as dichloromethane, chloroform and 1,2-dichloroethane, ether solvents such as THF, DME and dioxane, aprotic polar solvents such as DMF and DMSO, and aromatics such as benzene, toluene and xylene.
- a group hydrocarbon solvent or a mixed solvent thereof can be used.
- Isocyanate (IX) is used in an amount of 1.0 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to amine compound (V).
- amine compound (V) When a base is used, triethylamine, diisopropylethylamine, pyridine, N-methylmorpholine and the like can be used, and triethylamine or diisopropylethylamine is preferably used.
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours.
- the concentration of the substrate (V) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- the isocyanate (IX) is generated from the carboxylic acid (IX-1), for example, the carboxylic acid (IX-1) is converted to an acid azide with diphenylphosphoric acid azide (DPPA) and subjected to a Curtius rearrangement reaction under heating conditions. Obtainable.
- Solvents include halogenated hydrocarbon solvents such as dichloromethane, chloroform and 1,2-dichloroethane, ether solvents such as THF, DME and dioxane, aprotic polar solvents such as DMF and DMSO, and aromatics such as benzene, toluene and xylene.
- a group hydrocarbon solvent or a mixed solvent thereof can be used.
- Carboxylic acid (IX-1) is used in an amount of 1.0 to 30 equivalents, preferably 1.0 to 10 equivalents, relative to amine compound (V).
- DPPA is used in an amount of 1.0 to 30 equivalents, preferably 1.0 to 10 equivalents, relative to the carboxylic acid (IX-1).
- triethylamine, diisopropylethylamine, pyridine, N-methylmorpholine and the like can be used, and triethylamine or diisopropylethylamine is preferably used.
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours.
- concentration of the substrate (V) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- Compound (I-5) can be obtained, for example, by reacting amine compound (V) with isothiocyanate (X).
- Solvents include halogenated hydrocarbon solvents such as dichloromethane, chloroform and 1,2-dichloroethane, ether solvents such as THF, DME and dioxane, aprotic polar solvents such as DMF and DMSO, and aromatics such as benzene, toluene and xylene.
- a group hydrocarbon solvent or a mixed solvent thereof can be used.
- the isothiocyanate (X) is used in an amount of 1.0 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to the amine compound (V).
- a base triethylamine, diisopropylethylamine, pyridine, N-methylmorpholine and the like can be used, and triethylamine or diisopropylethylamine is preferably used.
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours.
- the concentration of the substrate (V) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- Compound (I-6) can be obtained, for example, by reacting compound (I-5) with ammonia and 2-iodoxybenzoic acid (IBX).
- a nitrile solvent such as acetonitrile can be used as the solvent.
- As the ammonia an aqueous ammonia solution can be used.
- Ammonia and IBX are used in an amount of 1.0 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to compound (I-6).
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours.
- the concentration of the substrate (I-6) in the reaction system is not particularly limited, but it is usually preferably 0.001 mmol / L to 1 mol / L.
- Compound (I-7) can be obtained, for example, by reacting amine compound (V) with chloroformate (XI).
- Solvents include halogenated hydrocarbon solvents such as dichloromethane, chloroform and 1,2-dichloroethane, ether solvents such as THF, DME and dioxane, aprotic polar solvents such as DMF and DMSO, and aromatics such as benzene, toluene and xylene.
- a group hydrocarbon solvent or a mixed solvent thereof can be used.
- the chloroformate (XI) is used in an amount of 1.0 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to the amine compound (V).
- a base triethylamine, diisopropylethylamine, pyridine, N-methylmorpholine and the like can be used, and triethylamine or diisopropylethylamine is preferably used.
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours.
- the concentration of the substrate (V) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- R 11a is C 1-6 alkoxy
- R 11b is 1 to 3 selected from C 1-6 alkyl and C 1-6 alkoxy
- C 6-10 aryl optionally substituted with 3 substituents.
- Process 1 Compound (I-8) can be obtained, for example, by reacting amine compound (V) with cyclobutene compound (XII).
- Solvents include halogenated hydrocarbon solvents such as dichloromethane, chloroform and 1,2-dichloroethane, ether solvents such as THF, DME and dioxane, aprotic polar solvents such as DMF and DMSO, alcohols such as methanol, ethanol and propanol.
- a solvent, an aromatic hydrocarbon solvent such as benzene, toluene, xylene, or a mixed solvent thereof can be used.
- the cyclobutene compound (XII) is used in an amount of 1.0 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to the amine compound (V).
- a base triethylamine, diisopropylethylamine, pyridine, N-methylmorpholine and the like can be used, and triethylamine or diisopropylethylamine is preferably used.
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours.
- the concentration of the substrate (V) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- Process 2 Compound (I-9) can be obtained, for example, by reacting compound (I-8) in which R 11a is methoxy with amine compound (XIII).
- Solvents include halogenated hydrocarbon solvents such as dichloromethane, chloroform and 1,2-dichloroethane, ether solvents such as THF, DME and dioxane, aprotic polar solvents such as DMF and DMSO, alcohols such as methanol, ethanol and propanol.
- a solvent, an aromatic hydrocarbon solvent such as benzene, toluene, xylene, or a mixed solvent thereof can be used.
- Amine compound (XIII) is used in an amount of 1.0 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to compound (I-8).
- a base triethylamine, diisopropylethylamine, pyridine, N-methylmorpholine and the like can be used, and triethylamine or diisopropylethylamine is preferably used.
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours.
- the concentration of the substrate (I-8) in the reaction system is not particularly limited, but is usually preferably 0.001 mmol / L to 1 mol / L.
- Compound (XX) can be obtained, for example, by subjecting compound (XVIII) to Suzuki coupling with boronic acid compound (XIX).
- the Suzuki coupling reaction is performed in a suitable solvent in the presence of a palladium catalyst and a base, and in the presence or absence of a phosphine ligand.
- Solvents include ether solvents such as THF, DME and dioxane, aprotic polar solvents such as DMF and DMSO, alcohol solvents such as methanol, ethanol and propanol, aromatic hydrocarbon solvents such as benzene, toluene and xylene, water or A mixed solvent thereof can be used.
- a mixed solvent of dioxane, DME, dioxane and water or a mixed solvent of DME and water is preferably used.
- the boronic acid compound (XIX) not only boronic acid but also boronic acid esters such as boronic acid pinacol ester, boronic acid N-methyliminodiacetic acid (MIDA) ester, or potassium trifluoroborate can be used.
- boronic acid or boronic acid pinacol ester is preferably used.
- the boronic acid compound (XIX) is used in an amount of 1.0 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to compound (XVIII).
- the palladium catalyst examples include tetrakis (triphenylphosphine) palladium, palladium acetate, bis (triphenylphosphine) palladium dichloride, bis (dibenzylideneacetone) palladium, bis (diphenylphosphino) ferrocenepalladium dichloride, and the like.
- (Triphenylphosphine) palladium or bis (diphenylphosphino) ferrocenepalladium dichloride is preferably used.
- the palladium catalyst is used in an amount of 0.001 to 1 equivalent, preferably 0.005 to 0.5 equivalent, relative to compound (XVIII).
- Examples of the base include sodium carbonate, potassium carbonate, cesium carbonate, potassium phosphate, sodium hydroxide, potassium hydroxide, barium hydroxide, triethylamine, diisopropylethylamine, and sodium carbonate or potassium carbonate is preferably used.
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 20 to 110 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours.
- the concentration of the substrate (XIX) in the reaction system is not particularly limited, but it is usually preferably 0.001 mmol / L to 1 mol / L.
- the benzyl group of the compound (XX) is deprotected by hydrogenolysis, and the tert-butoxycarbonyl (Boc) group of the obtained compound is deprotected under acidic conditions.
- the hydrogenolysis reaction is carried out in a suitable solvent in the presence of a palladium catalyst and in an atmosphere of hydrogen.
- Solvents include ether solvents such as THF, DME and dioxane, aprotic polar solvents such as DMF, DMSO and ethyl acetate, alcohol solvents such as methanol, ethanol and propanol, and aromatic hydrocarbon solvents such as benzene, toluene and xylene.
- Acetic acid, water or a mixed solvent thereof can be used.
- toluene, THF, and methanol are preferably used.
- the palladium catalyst include palladium (Pd), palladium carbon (Pd / C), palladium hydroxide (Pd (OH) 2 ), palladium hydroxide carbon (Pd (OH) 2 / C), and the like.
- Palladium carbon (Pd / C) is preferably used.
- the palladium catalyst is used in an amount of 0.001 to 1 equivalent, preferably 0.005 to 0.5 equivalent, relative to compound (XX).
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 20 to 110 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours. Further, the concentration of the substrate (XX) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- Deprotection of the tert-butoxycarbonyl (Boc) group can be carried out in the same manner as in Step 2 of Scheme 1.
- Compound (XXIII) is, for example, amine compound (XXI), acyl halide (XXII) (wherein X 1 is a halogen atom) or carboxylic acid (XXII) (wherein X 1 is a hydroxy group) Can be obtained by amidation. Amidation can be carried out in the same manner as in Step 3 in Scheme 1.
- Compound (I-12) can be obtained, for example, by reacting compound (XXIII) with paraformaldehyde.
- a carboxylic acid solvent such as acetic acid can be used.
- Paraformaldehyde is used in an amount of 1.0 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to compound (XXIII).
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours.
- the concentration of the substrate (XXIII) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- Compound (I-13) can be obtained, for example, by diazotizing amine compound (V) and subjecting the obtained diazonium salt to a diazo coupling reaction with compound (XXV).
- Diazotization can be performed by reacting the amine compound (V) with a nitrite such as sodium nitrite in an acidic aqueous solution.
- the nitrite is used in an amount of 1.0 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to the amine compound (V).
- the diazo coupling reaction can be performed by reacting the obtained diazonium salt with compound (XXV).
- Compound (XXV) is used in an amount of 1.0 to 20 equivalents, preferably 1.0 to 10 equivalents, relative to amine compound (V).
- the reaction temperature is usually ⁇ 40 to 150 ° C., preferably 0 to 80 ° C.
- the reaction time is appropriately selected depending on the reaction temperature and other conditions, but is usually about 20 minutes to 48 hours.
- the concentration of the substrate (V) in the reaction system is not particularly limited, but usually 0.001 mmol / L to 1 mol / L is preferable.
- R 1 , X and W are as defined above; R 13a is C 1-6 alkyl, C 6-10 aryl-C 1-6 alkyl or C 2-6 alkenyl-carbonyl; R 14a is (a) C 1-6 alkoxy-carbonylamino, (b) C 2-6 alkenyl-carbonylamino and (c) C 6-10 aryl-C 1 optionally substituted with C 1-6 alkyl. C 1-6 alkyl which may be substituted with one substituent selected from -6alkylaminocarbonyl .
- Compound (I-14) can be obtained, for example, by acylating and / or alkylating amine compound (V). Acylation can be carried out in the same manner as in Step 3 of Scheme 1. Alkylation can be performed in a manner similar to the reductive alkylation of Scheme 2.
- Process 1 Compound (XXVII) can be obtained, for example, by reacting compound (XXVI) with isocyanate (IX) and deprotecting the tert-butoxycarbonyl (Boc) group of the obtained compound.
- the reaction of compound (XXVI) with isocyanate (IX) can be carried out in the same manner as in Scheme 4.
- Deprotection of the tert-butoxycarbonyl (Boc) group can be carried out in the same manner as in Step 2 of Scheme 1.
- Process 2 Compound (XXIX) can be obtained, for example, by amidating compound (XXVII) with sulfonyl chloride compound (XXX). Amidation can be carried out in the same manner as in Step 1 of Scheme 1.
- Process 3 Compound (I-15) can be obtained, for example, by amidating compound (XXIX) with amine compound (XXX). Amidation can be carried out in the same manner as in Step 4 in Scheme 11.
- Process 1 Compound (XXXI) can be obtained, for example, by amidating sulfonyl chloride compound (XVI) with compound (XXIV). Amidation can be carried out in the same manner as in Step 1 of Scheme 1.
- Process 2 Compound (I) can be obtained, for example, by subjecting compound (XXXI) to Suzuki coupling with boronic acid compound (XIX). Suzuki coupling can be performed in the same manner as in Step 2 of Scheme 11.
- Process 1 Compound (I ′) can be obtained, for example, by amidating sulfonyl chloride compound (XXXIII) with compound (XXXIV). Amidation can be carried out in the same manner as in Step 1 of Scheme 1.
- Process 2 Compound (XXXV) can be obtained, for example, by amidating sulfonyl chloride compound (XVI) with compound (XXXIV). Amidation can be carried out in the same manner as in Step 1 of Scheme 1.
- Process 3 Compound (I ′) can be obtained, for example, by subjecting compound (XXXV) to Suzuki coupling with boronic acid compound (XXXVI). Suzuki coupling can be performed in the same manner as in Step 2 of Scheme 11.
- the orexin receptor agonist containing the compound of the present invention is not only effective for humans but also non-human mammals such as mice, rats, hamsters, rabbits, cats, dogs, cows, sheep, monkeys, etc. Also effective.
- the compound of the present invention is not only used as a preventive or therapeutic agent for narcolepsy as described above, but can also be used for the prevention or treatment method for narcolepsy or the manufacture of a medicament for preventing or treating narcolepsy. it can.
- the compound of the present invention can also be used as a sleepiness ameliorating agent or a prophylactic or therapeutic agent for obesity, diabetes, depression, sepsis, severe sepsis or septic shock.
- a sleepiness ameliorating agent or a prophylactic or therapeutic agent for obesity, diabetes, depression, sepsis, severe sepsis or septic shock.
- a preventive or therapeutic agent for narcolepsy a sleepiness improving agent
- a prophylactic or therapeutic agent for obesity, diabetes, depression, sepsis, severe sepsis or septic shock etc.
- May be a free form of the compound of the present invention or its acid addition salt itself, and additives such as excipients, stabilizers, preservatives, buffers, solubilizers, emulsifiers, diluents, isotonic agents and the like.
- Administration forms include oral preparations such as tablets, capsules, granules, powders, syrups, parenteral preparations such as injections, suppositories, and liquids, or topical administration such as ointments, creams, patches, etc. Can be mentioned.
- the preventive or therapeutic agent for narcolepsy of the present invention, the sleepiness-improving agent, or the prophylactic or therapeutic agent for obesity, diabetes, depression, sepsis, severe sepsis or septic shock contains 0.001 to 90 wt. %, Preferably 0.01 to 70% by weight. The amount to be used is appropriately selected according to symptoms, age, body weight, and administration method.
- the narcolepsy preventive or therapeutic agent or sleepiness improving agent of the present invention includes a prophylactic or therapeutic agent for strong daytime sleepiness and dozing, a prophylactic or therapeutic agent for deep sleep disorder, a prophylactic or therapeutic agent for cataplexy, and It can also be used in combination.
- the preventive or therapeutic agent for strong daytime sleepiness and doze include central nervous system stimulants such as methylphenidate, pemoline, and modafinil.
- prophylactic or therapeutic agent for deep sleep disorder examples include sleeping drugs such as triazolam and begetamine B, and anxiolytic drugs.
- prophylactic or therapeutic agents for cataplexy include tricyclic antidepressants such as clomipramine hydrochloride, brotizolam, imipramine hydrochloride, selective serotonin reuptake inhibitors (SSRI) such as fluvoxamine maleate and paroxetine hydrochloride, mil hydrochloride
- SSRI selective serotonin reuptake inhibitors
- SNRI noradrenaline reuptake inhibitors
- Boc tert-butoxycarbonyl
- DME 1,2-dimethoxyethane
- DMF N, N-dimethylformamide
- DPPA diphenyl phosphate azide
- HATU O- (7-azabenzotriazole- 1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate
- IBX 2-iodoxybenzoic acid
- BOP benzotriazol-1-yloxytris (dimethylamino) phosphonium hexafluorophosphoric acid Salt
- the compound name is ChemBioDraw Ultra ver. Named using 12.0.3
- tert-butyl (3-aminophenyl) carbamate (1.22 g) in dichloromethane (4.0 mL) was added to DIPEA (2.80 mL) and 3 ′-(dimethylcarbamoyl) -4-methoxy- [1. , 1′-biphenyl] -3-sulfonyl chloride (1.88 g) in dichloromethane (4.0 mL) was added, and the mixture was stirred at room temperature for 4 hours. The resulting white solid was collected by filtration, and the residue was washed with a hexane solution containing 25% dichloromethane.
- the reaction mixture was diluted with ethyl acetate, and extracted with 1M aqueous hydrochloric acid.
- the organic layer was washed with saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried by adding sodium sulfate, and filtered, and the filtrate was concentrated under reduced pressure.
- N-dimethyl- [1,1′-biphenyl] -3-carboxamide hydrochloride (16.5 mg) under argon atmosphere
- TEA 30.0 ⁇ L
- phenyl isothiocyanate 30.0 mg
- the reaction solution was diluted with chloroform containing 20% methanol, and then extracted with a saturated aqueous ammonium chloride solution.
- tert-butyl (2-((3-aminophenyl) (benzyl) amino) ethyl) carbamate (1.59 g) in dichloromethane (20.0 mL) was added to pyridine (413 ⁇ L) and 5-bromo-2- Methoxybenzenesulfonyl chloride (1.33 g) was added and stirred overnight at room temperature.
- a saturated aqueous sodium hydrogen carbonate solution was added to the reaction solution, and the mixture was extracted with chloroform. The organic layer was dried over sodium sulfate and then filtered, and the filtrate was concentrated.
- the obtained residue was purified by silica gel column chromatography (eluent: ethyl acetate), and tert-butyl (2-((3- (3 ′-(dimethylaminocarbamoyl) -4-methoxy- [1,1′- Biphenyl] -3-ylsulfonamido) phenyl) amino) ethyl) carbamate (950.0 mg) was obtained.
- reaction mixture was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (eluent: ethyl acetate), and 4′-methoxy-N, N-dimethyl-3 ′-(N- (3- (3- ( 3-Methylbenzoyl) imidazolidin-1-yl) phenyl) sulfamoyl)-[1,1′-biphenyl] -3-carboxamide (14.5 mg) was obtained.
- Example 98 The compound of Example 98 described in Table 13 below was also synthesized from the corresponding benzene derivative in the same manner.
- the reaction solution was filtered through Celite, and the filtrate was diluted with chloroform and extracted with a saturated aqueous sodium hydrogen carbonate solution.
- the organic layer was washed with saturated brine, dried by adding sodium sulfate, and after filtration, the filtrate was concentrated under reduced pressure.
- -N, N-dimethyl- [1,1'-biphenyl] -3-carboxamide (14.0 mg) was obtained.
- the reaction solution was concentrated under reduced pressure, and the residue was diluted with chloroform containing 25% methanol, and then extracted with a saturated aqueous sodium hydrogen carbonate solution.
- the organic layer was washed with saturated brine, dried by adding sodium sulfate, and after filtration, the filtrate was concentrated under reduced pressure.
- the reaction solution was concentrated under reduced pressure, and the residue was diluted with chloroform, and then extracted with a saturated aqueous sodium hydrogen carbonate solution.
- the organic layer was washed with saturated brine, dried by adding sodium sulfate, and after filtration, the filtrate was concentrated under reduced pressure.
- Example 120 For Example 120 described in Table 17 below, synthesis was similarly performed from an alkyl halide having a corresponding R group.
- Example 121, Example 122, and Example 124 were prepared in the same manner as in Production Example (4), with the corresponding tert-butyl (2-aminophenyl) carbamate, tert-butyl (4-aminophenyl) carbamate, and tert, respectively. Synthesis was performed using -butyl (6-aminopyridin-2-yl) carbamate.
- Test example 1 Evaluation of agonist activity against OX1R and OX2R A cell line (CHOOX1R), NAFT-luciferase gene, and human OX2R gene, which are expressed constitutively in Chinese hamster ovary-derived cell line CHO cells, NAFT-luciferase gene and human OX1R gene A cell line (CHOOX2R) expressed in E. coli was established. Each cell was seeded in a 96-well multiplate at 10,000 cells / well, and cultured in DMEM medium (Sigma Aldrich) supplemented with 5% FBS (Thermo Scientific) for 48 hours.
- DMEM medium Sigma Aldrich
- FBS Thermo Scientific
- assay buffer containing 20 ⁇ M Fura-2AM (Cayman Chemical) (20 mM HEPES (Sigma Aldrich), Hanks' balanced salt solution (Gibco), 0.1% BSA (Sigma Aldrich), 2.5 mM probenecid 100 ⁇ L of acid (Wako Pure Chemical Industries) was added and incubated for 60 minutes. After removing the buffer containing Fura-2AM, 75 ⁇ L of assay buffer was added. Thereto was added 25 ⁇ L of assay buffer containing the test compound, and the reaction was started.
- the change in intracellular calcium ion concentration due to the reaction was measured by measuring the fluorescence intensity ratio by excitation at two wavelengths of 340 and 380 nm using FDSS7000 (Hamamatsu Photonics).
- the test compound was dissolved in DMSO so that the concentration was 10 mM, and diluted with an assay buffer so that the final concentration was 10 ⁇ 7 M to 10 ⁇ 5 M (the final concentration of DMSO was 1%).
- Tables 21 to 27 show the agonist activity values of the respective compounds.
- Test example 2 Arousal effect of the compound of the present invention on the intraventricular administration to the wild type mouse
- the C57BL / 6 strain wild type mouse (male) was used as the experimental animal.
- Administration and electroencephalogram measurement were started 2 weeks later.
- test compound compound of Example 40
- 5% chromophore-containing physiological saline a compound of Example 40
- mice 5 ⁇ l of test compound (compound of Example 40) (10 nmol; dissolved in 5% chromophore-containing physiological saline) was added at 0.5 ⁇ l / min to ZT6 under isoflurane anesthesia using a microsyringe pump.
- the cannula was injected at a flow rate into the lateral ventricle, and the subsequent electroencephalogram was measured.
- Administration began with control 5% chromophore-containing saline (Vehicle), followed by administration of the test compound.
- the recovery period was defined as one day after administration.
- the results are shown in FIG.
- ZT6 which is a sleep period for mice
- the awakening time was increased in wild-type mice as compared to vehicle administration.
- the compound of the present invention exhibits orexin receptor agonist activity and is useful as a prophylactic or therapeutic agent such as narcolepsy.
- This application is based on a patent application No. 2015-119785 filed on Jun. 12, 2015 in Japan, the contents of which are incorporated in full herein.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Heterocyclic Compounds Containing Sulfur Atoms (AREA)
- Furan Compounds (AREA)
- Pyridine Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
すなわち、本発明は、以下を提供する。
[1] 式(I):

R1は水素原子、C1-6アルコキシ、-OH、C1-6アルキルまたはハロゲン原子であり、
Xは-C(R5)=または-N=であり、
R5は水素原子またはC1-6アルコキシであり、
R2、R3およびR4のいずれか1つはR6であり、残りの2つは、それぞれ水素原子であり、
R6は
(1)-NR17-Y1-R7
(ここで、Y1は-C(=O)NR18-、-C(=S)NH-、-C(=NH)NH-、-C(=O)O-、-C(=O)-、-SO2-または-SO2-NR8-であり、
R8は水素原子またはC1-6アルキルであり、
R17は水素原子またはC1-6アルキルであり、
R18は水素原子またはC1-6アルキルであり、
R17とR18は互いに結合し、それらが結合する窒素原子および隣接するC(=O)と一緒になって、5ないし7員複素環を形成してもよく、
R7は
(a)C6-10アリール、
(b)5ないし10員ヘテロアリール、
(c)C1-6アルキル、または
(d)C2-6アルケニル
(ここで、C1-6アルキルおよびC2-6アルケニルはフェニル、フリルおよびジフェニルメチルスルフィニルから選択される1個の置換基で置換されていてもよく、
C6-10アリールおよび5ないし10員ヘテロアリールは任意に選択される1ないし4個のR9で置換されていてもよく、
R9は、それぞれ独立して、
ハロゲン原子、
-NO2、
-OH、
C1-6アルキル、
C1-6ハロアルキル、
C1-6アルコキシ、
5ないし10員ヘテロアリール、
-NR9aR9b(ここで、R9aは水素原子、C1-6アルキルまたはC1-6アルコキシ-カルボニルであり、R9bは水素原子またはC1-6アルキルである)、
-C(=O)OR9c(ここで、R9cは水素原子またはC1-6アルキルである)、
-C(=O)NR9dR9e(ここで、R9dは水素原子またはC1-6アルキルであり、R9eは水素原子またはC1-6アルキルである)、または
-NH-C(=NR9f)-NHR9g(ここで、R9fは水素原子またはC1-6アルコキシ-カルボニルであり、R9gは水素原子またはC1-6アルコキシ-カルボニルである)であり、または
2個のR9は一緒になってメチレンジオキシを形成する)である)、
(2)-NH-Y2-R10
(ここで、Y2は-CH2-または単結合であり、
R10は
(a)C6-10アリール、または
(b)5ないし10員ヘテロアリール
(ここで、C6-10アリールおよび5ないし10員ヘテロアリールは任意に選択される1ないし4個のR9で置換されていてもよく、R9は前記で定義したとおりである)である)、
(3)式(ii):
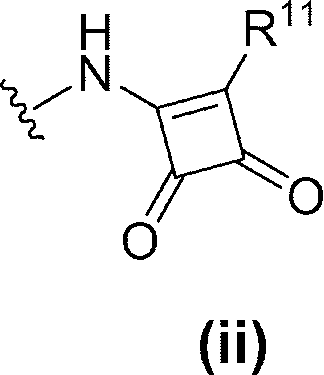
(4)-N=N-R12
(ここで、R12は(a)-OHで置換されていてもよいC1-6アルキル、(b)-OH、(c)ジ(C1-6アルキル)アミノおよび(d)C1-6アルコキシ-カルボニルアミノから選択される1ないし3個の置換基で置換されていてもよいC6-10アリールである)、または
(5)-NR13R14
(ここで、R13は水素原子、C1-6アルキル、C6-10アリール-C1-6アルキルまたはC2-6アルケニル-カルボニルであり、
R14は水素原子またはC1-6アルキル(ここで、C1-6アルキルは(a)C1-6アルコキシ-カルボニルアミノ、(b)C2-6アルケニル-カルボニルアミノおよび(c)C1-6アルキルで置換されていてもよいC6-10アリール-C1-6アルキルアミノカルボニルから選択される1個の置換基で置換されていてもよい)であるか、またはR13とR14は互いに結合し、それらが結合する窒素原子と一緒になって、更に1個の窒素原子を含有する5ないし7員複素環(ここで、5ないし7員複素環はC1-6アルキルで置換されていてもよいC6-10アリール-カルボニルで置換されていてもよい)を形成する。
ただし、R14がC2-6アルケニル-カルボニルアミノで置換されたエチルであるとき、R13は水素原子ではない)であり、
Wは
(1)式(iii):

(2)-C(=O)NRWaRWb
(ここで、RWaはC1-6アルキル(ここで、C1-6アルキルはフェニル、ピリジル、C1-6アルコキシ-カルボニルアミノまたはジ(C1-6アルキル)アミノで置換されていてもよい)またはフェニル(ここで、フェニルはジ(C1-6アルキル)アミノで置換されていてもよい)であり、RWbは水素原子またはC1-6アルキルである)である]
で示される化合物またはその薬学的に許容される酸付加塩(以下、化合物(I)ともいう)。
[2] R17およびR18がそれぞれ水素原子である前記[1]記載の化合物またはその薬学的に許容される酸付加塩。
[3] 式(I-A):

[4] R17およびR18がそれぞれ水素原子である前記[3]記載の化合物またはその薬学的に許容される酸付加塩。
[5] Xが-C(R5)=(ここで、R5は水素原子またはC1-6アルコキシである)である、前記[1]から[4]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩。
[6] R2がR6(ここで、R6は前記[1]で定義したとおりである)であり、R3およびR4がそれぞれ水素原子である、前記[1]から[5]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩。
[7] R6が-NR17-Y1-R7(ここで、R17、Y1およびR7は前記[1]で定義したとおりである)である、前記[1]から[6]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩。
[8] R6が-NH-Y2-R10(ここで、Y2およびR10は前記[1]で定義したとおりである)である、前記[1]から[6]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩。
[9] R6が式(ii):

[10] R6が-N=N-R12(ここで、R12は前記[1]で定義したとおりである)である、前記[1]から[6]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩。
[11] R6が-NR13R14(ここで、R13およびR14は前記[1]で定義したとおりである)である、前記[1]から[6]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩。
[12] Wが式(iii):

Xが-C(R5)=(ここで、R5は水素原子またはC1-6アルコキシである)であり、
R2が-NH-Y1-R7(ここで、Y1およびR7は前記[1]で定義したとおりである)であり、
R3およびR4がそれぞれ水素原子である、
前記[1]記載の化合物またはその薬学的に許容される酸付加塩。
[13] Y1が-C(=O)NH-、-C(=S)NH-、-C(=NH)NH-または-C(=O)O-である、前記[12]記載の化合物またはその薬学的に許容される酸付加塩。
[14] 式(I-B):

[15] 式(I’):

R1は水素原子、C1-6アルコキシ、-OH、C1-6アルキルまたはハロゲン原子であり、
Xは-C(R5)=または-N=であり、
R5は水素原子またはC1-6アルコキシであり、
R6aは置換されていてもよい炭化水素基、置換されていてもよい複素環基、酸素原子を介して結合する基、または硫黄原子を介して結合する基であり、
R15はC1-6アルキルであり、
R16はC1-6アルキルである]
で示される化合物またはその薬学的に許容される酸付加塩。
[16] 前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩を含有する医薬。
[17] 前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩を含有するオレキシン受容体作動薬。
[18] 前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩を含有する抗ナルコレプシー剤。
[19] 前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩を含有する眠気改善剤。
[20] 前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩を含有する、肥満症、糖尿病またはうつ病の予防剤または治療剤。
[21] 前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩を含有する、敗血症、重症敗血症または敗血症性ショックの予防剤または治療剤。
[22] 前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩の有効量を投与することを含むナルコレプシーの治療または予防方法。
[23] 前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩の有効量を投与することを含む眠気の改善方法。
[24] 前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩の有効量を投与することを含む、肥満症、糖尿病またはうつ病の治療または予防方法。
[25] 前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩の有効量を投与することを含む、敗血症、重症敗血症または敗血症性ショックの治療または予防方法。
[26] ナルコレプシーの治療または予防に使用するための、前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩。
[27] 眠気の改善に使用するための、前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩。
[28] 肥満症、糖尿病またはうつ病の治療または予防に使用するための、前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩。
[29] 敗血症、重症敗血症または敗血症性ショックの治療または予防に使用するための、前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩。
[30] オレキシン受容体作動薬;抗ナルコレプシー剤;眠気改善剤;肥満症、糖尿病またはうつ病の予防剤または治療剤;または敗血症、重症敗血症または敗血症性ショックの予防剤または治療剤を製造するための、前記[1]から[15]のいずれか一つに記載の化合物またはその薬学的に許容される酸付加塩の使用。
本明細書中の「C1-6アルキル」は、炭素原子および水素原子からなる、炭素数1から6の一価の直鎖または分岐状の飽和炭化水素基を意味する。例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、ネオペンチル、ヘキシル等が挙げられる。
本明細書中の「C1-6アルコキシ」は、C1-6アルキルが結合したオキシ基を意味する。例えば、メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ、イソブトキシ、tert-ブトキシ等が挙げられる。
本明細書中の「ハロゲン原子」は、フッ素原子、塩素原子、臭素原子またはヨウ素原子を意味する。
本明細書中の「C2-6アルケニル」は、炭素原子および水素原子からなる、炭素数2から6の一価の直鎖または分岐状の少なくとも1個の二重結合を有する不飽和炭化水素基を意味する。例えば、エテニル、1-プロペニル、2-プロペニル、2-メチル-1-プロペニル、1-ブテニル、2-ブテニル、3-ブテニル、3-メチル-2-ブテニル、1-ペンテニル、2-ペンテニル、3-ペンテニル、4-ペンテニル、4-メチル-3-ペンテニル、1-ヘキセニル、3-ヘキセニル、5-ヘキセニル等が挙げられる。
本明細書中の「C2-6アルキニル」は、炭素原子および水素原子からなる、炭素数2から6の一価の直鎖または分岐状の少なくとも1個の三重結合を有する不飽和炭化水素基を意味する。例えば、エチニル、プロピニル、ブチニル、ペンチニル、ヘキシニル等が挙げられる。
本明細書中の「C3-10シクロアルキル」は、炭素数3から10個の単環または多環の脂肪族炭素環式基を意味する。例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、アダマンチル等が挙げられる。
本明細書中の「C6-10アリール」は、炭素数6から10の単環または縮合の芳香族炭素環式基を意味する。例えば、フェニル、1-ナフチル、2-ナフチル等が挙げられる。好ましくは、フェニルである。
本明細書中の「5ないし10員ヘテロアリール」は、環構成原子として、炭素原子以外に酸素原子、硫黄原子および窒素原子から選ばれる1または2種、1ないし4個のヘテロ原子を含む5から10員の単環または二環の芳香族ヘテロ環式基を意味する。例えば、チエニル、フリル、ピロリル、イミダゾリル、ピラゾリル、トリアゾリル、テトラゾリル、オキサゾリル、イソキサゾリル、チアゾリル、イソチアゾリル、ピリジル、ピリダジニル、ピリミジニル、フラザニル、ピラジニル、チアジアゾリル、オキサジアゾリル、ベンゾフリル、ベンゾチエニル、ベンズイミダゾリル、ベンゾチアゾリル、キノリル、イソキノリル、シンノリニル、キナゾリニル、キノキサリニル、フタラジニル、1H-インダゾリル等が挙げられる。
本明細書中の「C1-6ハロアルキル」は、1ないし5個(好ましくは1ないし3個)のハロゲン原子で置換されたC1-6アルキルを意味する。例えば、トリフルオロメチル、2,2,2-トリフルオロエチル、3,3,3-トリフルオロプロピル等が挙げられる。好ましくは、トリフルオロメチルである。
本明細書中の「C1-6アルコキシ-カルボニル」は、C1-6アルコキシが結合したカルボニル基を意味する。例えば、メトキシカルボニル、エトキシカルボニル、プロポキシカルボニル、イソプロポキシカルボニル、ブトキシカルボニル、イソブトキシカルボニル、tert-ブトキシカルボニル等が挙げられる。
本明細書中の「C6-10アリールアミノ」は、C6-10アリールで置換されたアミノを意味する。例えば、フェニルアミノ、1-ナフチルアミノ、2-ナフチルアミノ等が挙げられる。好ましくはフェニルアミノである。
本明細書中の「-OHで置換されていてもよいC1-6アルキル」は、1個の-OHで置換されていてもよいC1-6アルキルを意味する。例えば、ヒドロキシメチル、2-ヒドロキシエチル、3-ヒドロキシプロピル等が挙げられる。
本明細書中の「ジ(C1-6アルキル)アミノ」は、2個のC1-6アルキルで置換されたアミノを意味する。例えば、ジメチルアミノ、ジエチルアミノ、ジプロピルアミノ、ジイソプロピルアミノ、N-エチル-N-メチルアミノ等が挙げられる。
本明細書中の「C1-6アルコキシ-カルボニルアミノ」は、C1-6アルコキシ-カルボニルで置換されたアミノを意味する。例えば、メトキシカルボニルアミノ、エトキシカルボニルアミノ、プロポキシカルボニルアミノ、イソプロポキシカルボニルアミノ、ブトキシカルボニルアミノ、イソブトキシカルボニルアミノ、tert-ブトキシカルボニルアミノ等が挙げられる。
本明細書中の「C6-10アリール-C1-6アルキル」は、C6-10アリールで置換されたC1-6アルキルを意味する。例えば、ベンジル、フェネチル等が挙げられる。好ましくは、ベンジルである。
本明細書中の「C2-6アルケニル-カルボニル」は、C2-6アルケニルが結合したカルボニル基を意味する。例えば、アクリロイル、メタクリロイル等が挙げられる。
本明細書中の「C2-6アルケニル-カルボニルアミノ」は、C2-6アルケニル-カルボニルで置換されたアミノを意味する。例えば、アクリロイルアミノ、メタクリロイルアミノ等が挙げられる。
本明細書中の「C6-10アリール-C1-6アルキルアミノカルボニル」は、C6-10アリール-C1-6アルキルで置換されたアミノカルボニルを意味する。例えば、ベンジルアミノカルボニル、フェネチルアミノカルボニル等が挙げられる。好ましくはベンジルアミノカルボニルである。
本明細書中の「C6-10アリール-カルボニル」は、C6-10アリールが結合したカルボニル基を意味する。例えば、ベンゾイル、1-ナフトイル、2-ナフトイル等が挙げられる。好ましくは、ベンゾイルである。
本明細書において、R13とR14が互いに結合し、それらが結合する窒素原子と一緒になって形成する、更に1個の窒素原子を含有する5ないし7員複素環としては、イミダゾリジン、ピペラジンまたは1,4-ジアゼパンが挙げられる。
本明細書において、R17とR18が互いに結合し、それらが結合する窒素原子および隣接するC(=O)と一緒になって形成する、5ないし7員複素環としては、イミダゾリジン-2-オン、ヘキサヒドロピリミジン-2-オンまたは1,3-ジアゼパン-2-オンが挙げられる。
本明細書中の「置換されていてもよい複素環基」の「複素環基」としては、5ないし10員ヘテロアリール、4ないし10員非芳香族複素環基等が挙げられる。
本明細書中の「4ないし10員非芳香族複素環基」は、環構成原子として、炭素原子以外に酸素原子、硫黄原子および窒素原子から選ばれる1または2種、1ないし4個のヘテロ原子を含む4ないし10員の単環または二環の飽和または不飽和の非芳香族複素環基を意味する。例えば、アゼチジニル、オキセタニル、ピロリジニル、テトラヒドロフリル、イミダゾリジニル、ピラゾリジニル、ピペリジル、テトラヒドロピラニル、テトラヒドロチオピラニル、ピペラジニル、モルホリニル、チオモルホリニル、アゼパニル、オキサゼパニル、ジアゼパニル、ピロリニル、イミダゾリニル、ピラゾリニル等が挙げられる。
本明細書中の「酸素原子を介して結合する基」としては、置換されていてもよい炭化水素基または置換されていてもよい複素環基が結合したオキシ基(-O-)等が挙げられる。
本明細書中の「硫黄原子を介して結合する基」としては、置換されていてもよい炭化水素基、置換されていてもよい複素環基、または窒素原子を介して結合する基が結合したチオ基(-S-)、スルフィニル基(-SO-)またはスルホニル基(-SO2-)等が挙げられる。
本明細書中の「窒素原子を介して結合する基」としては、置換されていてもよい炭化水素基が結合したアミノ、置換されていてもよい複素環基が結合したアミノ等が挙げられる。
本明細書中の「置換されていてもよい」とは、例えば、下記の置換基群から選ばれる1ないし5個、好ましくは1ないし3個の置換基で置換されていてもよいことを意味する。
置換基群
ハロゲン原子、
-NO2、
-CN、
-OH、
オキソ(=O)、
C1-6アルキル、
C1-6ハロアルキル、
C1-6アルコキシ、
C2-6アルケニル、
C2-6アルキニル、
C3-10シクロアルキル、
C6-10アリール、
C6-10アリール-C1-6アルキル、
5ないし10員ヘテロアリール、
4ないし10員非芳香族複素環基、
-NR21aR21b
(ここで、R21aおよびR21bは、それぞれ独立して、水素原子、C1-6アルキル、C1-6アルコキシ-カルボニル、C2-6アルケニル、C2-6アルキニル、C3-10シクロアルキル、C6-10アリール、C6-10アリール-C1-6アルキル、5ないし10員ヘテロアリールまたは4ないし10員非芳香族複素環基である)、
-C(=O)OR21c
(ここで、R21cは水素原子、C1-6アルキル、C2-6アルケニル、C2-6アルキニル、C3-10シクロアルキル、C6-10アリールまたはC6-10アリール-C1-6アルキルである)、
-C(=O)NR21dR21e
(ここで、R21dおよびR21eは、それぞれ独立して、水素原子、C1-6アルキル、C2-6アルケニル、C2-6アルキニル、C3-10シクロアルキル、C6-10アリール、C6-10アリール-C1-6アルキル、5ないし10員ヘテロアリールまたは4ないし10員非芳香族複素環基である)、
-NH-C(=NR21f)-NHR21g
(ここで、R21fは水素原子またはC1-6アルコキシ-カルボニルであり、R21gは水素原子またはC1-6アルコキシ-カルボニルである)、
-OR21h
(ここで、R21hはC1-6ハロアルキル、C2-6アルケニル、C2-6アルキニル、C3-10シクロアルキル、C6-10アリール、C6-10アリール-C1-6アルキル、5ないし10員ヘテロアリールまたは4ないし10員非芳香族複素環基である)、
-C(=O)R21i
(ここで、R21iは水素原子、C1-6アルキル、C2-6アルケニル、C2-6アルキニル、C3-10シクロアルキル、C6-10アリール、C6-10アリール-C1-6アルキル、5ないし10員ヘテロアリールまたは4ないし10員非芳香族複素環基である)、
=N-R21j
(ここで、R21jは水素原子、-OHまたはC1-6アルコキシである)、
-S(O)n-R21k
(ここで、R21kは-OH、C1-6アルキル、C2-6アルケニル、C2-6アルキニル、C3-10シクロアルキル、C6-10アリール、C6-10アリール-C1-6アルキル、5ないし10員ヘテロアリールまたは4ないし10員非芳香族複素環基であり、nは0、1または2である)、
-SO2-NR21mR21n
(ここで、R21mおよびR21nは、それぞれ独立して、水素原子、C1-6アルキル、C2-6アルケニル、C2-6アルキニル、C3-10シクロアルキル、C6-10アリール、C6-10アリール-C1-6アルキル、5ないし10員ヘテロアリールまたは4ないし10員非芳香族複素環基である)、および
C1-3アルキレンジオキシ(例、メチレンジオキシ)。
本明細書中の「眠気改善剤」は、シフトワーク、ジェットラグ、不眠症または睡眠時無呼吸症候群等による日中の眠気を改善する薬剤を意味する。
R1の例としては、水素原子、メトキシ、エトキシ、プロポキシ、-OH、メチル、塩素原子等が挙げられる。
R1は、好ましくは、C1-6アルコキシ(例、メトキシ、エトキシ、プロポキシ)であり、特に好ましくは、メトキシである。
(1)式(iii):

(2)-C(=O)NRWaRWbである。

(ここで、RWaはC1-6アルキル(例、メチル、エチル、プロピル、2,2-ジメチルプロピル)(ここで、C1-6アルキルはフェニル、ピリジル、C1-6アルコキシ-カルボニルアミノ(例、tert-ブトキシカルボニルアミノ)またはジ(C1-6アルキル)アミノ(例、ジメチルアミノ)で置換されていてもよい)またはフェニル(ここで、フェニルはジ(C1-6アルキル)アミノ(例、ジメチルアミノ)で置換されていてもよい)であり、RWbは水素原子またはC1-6アルキル(例、エチル)である)である。
R5の例としては、水素原子またはメトキシが挙げられる。
R5は、好ましくは水素原子である。
Xは、好ましくは、-C(R5)=であり、特に好ましくは、-CH=である。
R2、R3およびR4の組み合わせとしては、以下の組み合わせが挙げられる。
(1)R2がR6であり、R3およびR4がそれぞれ水素原子である。
(2)R3がR6であり、R2およびR4がそれぞれ水素原子である。
(3)R4がR6であり、R2およびR3がそれぞれ水素原子である。
これらのうち、R2がR6であり、R3およびR4がそれぞれ水素原子である場合が好ましい。
(1)-NR17-Y1-R7、
(2)-NH-Y2-R10、
(3)式(ii):

(4)-N=N-R12、または
(5)-NR13R14である。
R6は、好ましくは、
(1)-NH-Y1-R7、
(2)-NH-Y2-R10、
(3)式(ii)で表される基、または
(4)-N=N-R12であり、
特に好ましくは、-NH-Y1-R7である。
Y1は-C(=O)NR18-、-C(=S)NH-、-C(=NH)NH-、-C(=O)O-、-C(=O)-、-SO2-または-SO2-NR8-であり、好ましくは、-C(=O)NH-、-C(=S)NH-、-C(=NH)NH-または-C(=O)O-であり、特に好ましくは、-C(=O)NH-である。
R8は水素原子またはC1-6アルキル(例、メチル)である。
R17は水素原子またはC1-6アルキル(例、メチル)であり、
R18は水素原子またはC1-6アルキル(例、メチル)であり、
R17とR18は互いに結合し、それらが結合する窒素原子および隣接するC(=O)と一緒になって、5ないし7員複素環(例、イミダゾリジン-2-オン、ヘキサヒドロピリミジン-2-オン、1,3-ジアゼパン-2-オン)を形成してもよい。
R17およびR18は、好ましくは、それぞれ水素原子である。
(a)C6-10アリール(例、フェニル、1-ナフチル、2-ナフチル)、
(b)5ないし10員ヘテロアリール(例、ピリジル、チエニル、チアゾリル)、
(c)C1-6アルキル(例、メチル、tert-ブチル)、または
(d)C2-6アルケニル(例、ビニル)
(ここで、C1-6アルキルおよびC2-6アルケニルはフェニル、フリルおよびジフェニルメチルスルフィニルから選択される1個の置換基で置換されていてもよく、
C6-10アリールおよび5ないし10員ヘテロアリールは任意に選択される1ないし4個(好ましくは1ないし3個、より好ましくは1または2個)のR9で置換されていてもよく、
R9は、それぞれ独立して、
ハロゲン原子(例、臭素原子)、
-NO2、
-OH、
C1-6アルキル(例、メチル)、
C1-6ハロアルキル(例、トリフルオロメチル)、
C1-6アルコキシ(例、メトキシ)、
5ないし10員ヘテロアリール(例、ピリジル)、
-NR9aR9b(ここで、R9aは水素原子、C1-6アルキル(例、メチル)またはC1-6アルコキシ-カルボニル(例、tert-ブトキシカルボニル)であり、R9bは水素原子またはC1-6アルキル(例、メチル)である)、
-C(=O)OR9c(ここで、R9cは水素原子またはC1-6アルキル(例、メチル)である)、
-C(=O)NR9dR9e(ここで、R9dは水素原子またはC1-6アルキル(例、メチル)であり、R9eは水素原子またはC1-6アルキル(例、メチル)である)、または
-NH-C(=NR9f)-NHR9g(ここで、R9fは水素原子またはC1-6アルコキシ-カルボニル(例、tert-ブトキシカルボニル)であり、R9gは水素原子またはC1-6アルコキシ-カルボニル(例、tert-ブトキシカルボニル)である)であり、または
2個のR9は一緒になってメチレンジオキシを形成する)である。
(a)C6-10アリール(例、フェニル、1-ナフチル、2-ナフチル)、または
(b)5ないし10員ヘテロアリール(例、ピリジル、チエニル、チアゾリル)
(ここで、C6-10アリールおよび5ないし10員ヘテロアリールは任意に選択される1ないし4個(好ましくは1ないし3個、より好ましくは1または2個)のR9で置換されていてもよく、R9は前記で定義したとおりである)である。
R10は
(a)C6-10アリール(例、フェニル)、または
(b)5ないし10員ヘテロアリール(例、ピリジル、チエニル、チアゾリル)
(ここで、C6-10アリールおよび5ないし10員ヘテロアリールは任意に選択される1ないし4個のR9で置換されていてもよく、R9は前記で定義したとおりである)である。
(ここで、C6-10アリールは任意に選択される1ないし4個(好ましくは1ないし3個、より好ましくは1または2個)のR9で置換されていてもよく、
R9は、それぞれ独立して、
ハロゲン原子(例、臭素原子)、
-OH、
C1-6アルキル(例、メチル)、
C1-6ハロアルキル(例、トリフルオロメチル)、
C1-6アルコキシ(例、メトキシ)、または
-C(=O)NR9dR9e(ここで、R9dは水素原子またはC1-6アルキル(例、メチル)であり、R9eは水素原子またはC1-6アルキル(例、メチル)である)であり、または
2個のR9は一緒になってメチレンジオキシを形成する)である。
R11はC1-6アルコキシ(例、メトキシ)またはC6-10アリールアミノ(例、フェニルアミノ)(ここで、C6-10アリールアミノのC6-10アリール部分はC1-6アルキル(例、メチル)およびC1-6アルコキシ(例、メトキシ)から選択される1ないし3個の置換基で置換されていてもよい)である。
R12は(a)-OHで置換されていてもよいC1-6アルキル(例、メチル)、(b)-OH、(c)ジ(C1-6アルキル)アミノ(例、ジメチルアミノ)および(d)C1-6アルコキシ-カルボニルアミノ(例、tert-ブトキシカルボニルアミノ)から選択される1ないし3個の置換基で置換されていてもよいC6-10アリール(例、フェニル)である。
R13は水素原子、C1-6アルキル(例、メチル)、C6-10アリール-C1-6アルキル(例、ベンジル)またはC2-6アルケニル-カルボニル(例、アクリロイル)であり、
R14は水素原子またはC1-6アルキル(例、メチル、エチル)(ここで、C1-6アルキルは(a)C1-6アルコキシ-カルボニルアミノ(例、tert-ブトキシカルボニルアミノ)、(b)C2-6アルケニル-カルボニルアミノ(例、アクリロイルアミノ)および(c)C1-6アルキル(例、メチル)で置換されていてもよいC6-10アリール-C1-6アルキルアミノカルボニル(例、ベンジルアミノカルボニル)から選択される1個の置換基で置換されていてもよい)であるか、または
R13とR14は互いに結合し、それらが結合する窒素原子と一緒になって、更に1個の窒素原子を含有する5ないし7員複素環(例、イミダゾリジン)(ここで、5ないし7員複素環はC1-6アルキル(例、メチル)で置換されていてもよいC6-10アリール-カルボニル(例、ベンゾイル)で置換されていてもよい)を形成する。ただし、R14がC2-6アルケニル-カルボニルアミノで置換されたエチルであるとき、R13は水素原子ではない。
式(I-A):

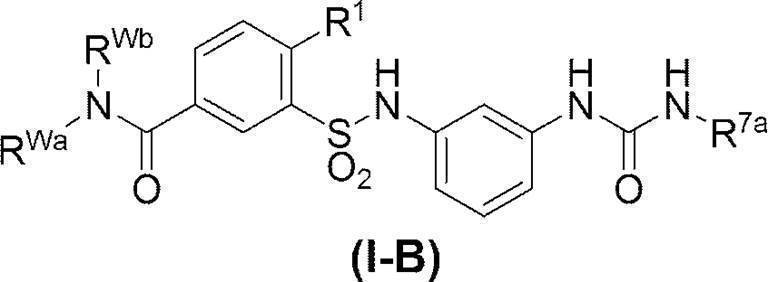
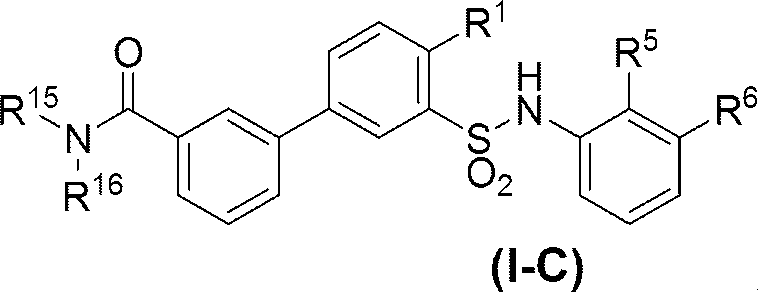





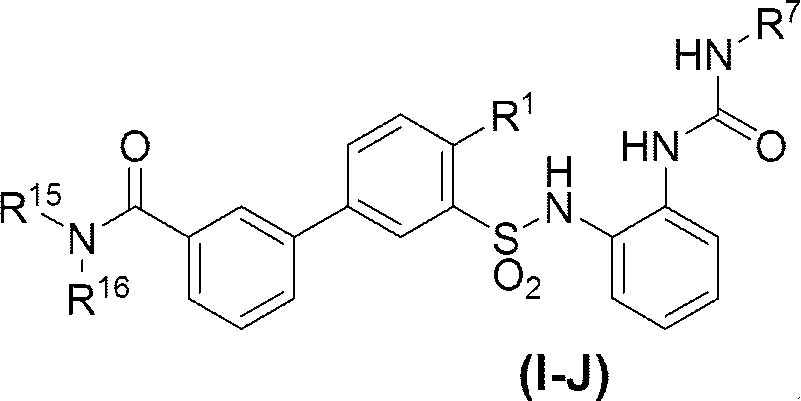
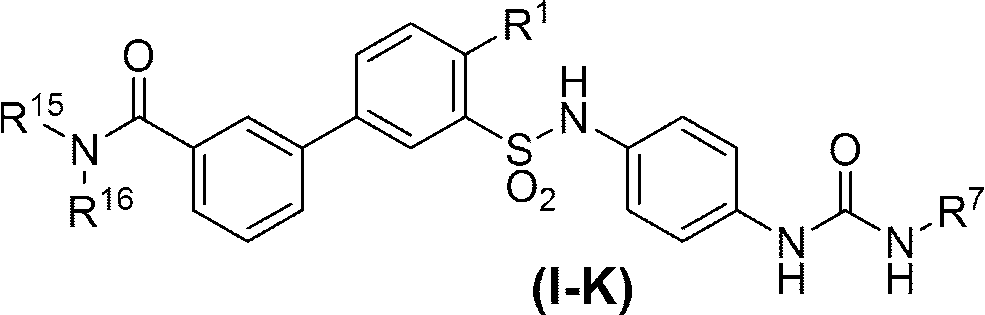

式(I-D)において、
Y1は、好ましくは、-C(=O)NH-、-C(=S)NH-、-C(=NH)NH-または-C(=O)O-であり、特に好ましくは、-C(=O)NH-である。
式(I-D)、(I-J)、(I-K)および(I-L)において、
R7は、好ましくは、
(a)C6-10アリール(例、フェニル、1-ナフチル、2-ナフチル)、または
(b)5ないし10員ヘテロアリール(例、ピリジル、チエニル、チアゾリル)
(ここで、C6-10アリールおよび5ないし10員ヘテロアリールは任意に選択される1ないし4個のR9で置換されていてもよく、R9は前記で定義したとおりである)である。
R7は、特に好ましくは、-NR9aR9b(ここで、R9aはC1-6アルキルであり、R9bはC1-6アルキルである)で置換されたフェニルである。
式(I-A)において、
R1は水素原子、C1-6アルコキシ、-OH、C1-6アルキルまたはハロゲン原子であり、
Xは-C(R5)=または-N=であり、
R5は水素原子またはC1-6アルコキシであり、
R2、R3およびR4のいずれか1つはR6であり、残りの2つは、それぞれ水素原子であり、
R6は
(1)-NR17-Y1-R7
(ここで、Y1は-C(=O)NR18-、-C(=S)NH-、-C(=NH)NH-、-C(=O)O-、-C(=O)-、-SO2-または-SO2-NR8-であり、
R8は水素原子またはC1-6アルキルであり、
R17は水素原子またはC1-6アルキルであり、
R18は水素原子またはC1-6アルキルであり、
R17とR18は互いに結合し、それらが結合する窒素原子および隣接するC(=O)と一緒になって、5ないし7員複素環(例、イミダゾリジン-2-オン、ヘキサヒドロピリミジン-2-オンまたは1,3-ジアゼパン-2-オン)を形成してもよい、
R7は
(a)C6-10アリール(例、フェニル、1-ナフチル、2-ナフチル)、
(b)5ないし10員ヘテロアリール(例、ピリジル、チエニル、チアゾリル)、
(c)C1-6アルキル、または
(d)C2-6アルケニル
(ここで、C1-6アルキルおよびC2-6アルケニルはフェニル、フリルおよびジフェニルメチルスルフィニルから選択される1個の置換基で置換されていてもよく、
C6-10アリールおよび5ないし10員ヘテロアリールは任意に選択される1ないし4個(好ましくは1ないし3個、より好ましくは1または2個)のR9で置換されていてもよく、
R9は、それぞれ独立して、
ハロゲン原子、
-NO2、
-OH、
C1-6アルキル、
C1-6ハロアルキル、
C1-6アルコキシ、
5ないし10員ヘテロアリール(例、ピリジル)、
-NR9aR9b(ここで、R9aは水素原子、C1-6アルキルまたはC1-6アルコキシ-カルボニルであり、R9bは水素原子またはC1-6アルキルである)、
-C(=O)OR9c(ここで、R9cは水素原子またはC1-6アルキルである)、
-C(=O)NR9dR9e(ここで、R9dは水素原子またはC1-6アルキルであり、R9eは水素原子またはC1-6アルキルである)、または
-NH-C(=NR9f)-NHR9g(ここで、R9fは水素原子またはC1-6アルコキシ-カルボニルであり、R9gは水素原子またはC1-6アルコキシ-カルボニルである)であり、または
2個のR9は一緒になってメチレンジオキシを形成する)である)、
(2)-NH-Y2-R10
(ここで、Y2は-CH2-または単結合であり、
R10はC6-10アリール(例、フェニル)
(ここで、C6-10アリールは任意に選択される1ないし4個(好ましくは1ないし3個、より好ましくは1または2個)のR9で置換されていてもよく、
R9は、それぞれ独立して、
ハロゲン原子、
-OH、
C1-6アルキル、
C1-6ハロアルキル、
C1-6アルコキシ、または
-C(=O)NR9dR9e(ここで、R9dはC1-6アルキルであり、R9eはC1-6アルキルである)、または
2個のR9は一緒になってメチレンジオキシを形成する)である)、
(3)式(ii):

(4)-N=N-R12
(ここで、R12は(a)-OHで置換されていてもよいC1-6アルキル、(b)-OH、(c)ジ(C1-6アルキル)アミノおよび(d)C1-6アルコキシ-カルボニルアミノから選択される1ないし3個の置換基で置換されていてもよいC6-10アリール(例、フェニル)である)、または
(5)-NR13R14
(ここで、R13は水素原子、C1-6アルキル、C6-10アリール-C1-6アルキル(例、ベンジル)またはC2-6アルケニル-カルボニルであり、
R14は水素原子またはC1-6アルキル(ここで、C1-6アルキルは(a)C1-6アルコキシ-カルボニルアミノ、(b)C2-6アルケニル-カルボニルアミノおよび(c)C1-6アルキルで置換されていてもよいC6-10アリール-C1-6アルキルアミノカルボニル(例、ベンジルアミノカルボニル)から選択される1個の置換基で置換されていてもよい)であるか、またはR13とR14は互いに結合し、それらが結合する窒素原子と一緒になって、更に1個の窒素原子を含有する5ないし7員複素環(例、イミダゾリジン)(ここで、5ないし7員複素環はC1-6アルキルで置換されていてもよいC6-10アリール-カルボニル(例、ベンゾイル)で置換されていてもよい)を形成する。
ただし、R14がC2-6アルケニル-カルボニルアミノで置換されたエチルであるとき、R13は水素原子ではない)であり、
R15はC1-6アルキルであり、R16はC1-6アルキルである、
化合物またはその薬学的に許容される酸付加塩。
好ましくは、R17およびR18はそれぞれ水素原子である。
式(I-B)において、
R1はC1-6アルコキシであり、
R7aは-NR9aR9b(ここで、R9aはC1-6アルキルであり、R9bはC1-6アルキルである)で置換されたフェニルであり、
RWaはC1-6アルキル(ここで、C1-6アルキルはフェニル、ピリジル、C1-6アルコキシ-カルボニルアミノまたはジ(C1-6アルキル)アミノで置換されていてもよい)またはフェニル(ここで、フェニルはジ(C1-6アルキル)アミノで置換されていてもよい)であり、
RWbは水素原子またはC1-6アルキルである、
化合物またはその薬学的に許容される酸付加塩。
式(I-C)において、
R1は水素原子、C1-6アルコキシ、-OH、C1-6アルキルまたはハロゲン原子であり、
R5は水素原子またはC1-6アルコキシであり、
R6は
(1)-NR17-Y1-R7
(ここで、Y1は-C(=O)NR18-、-C(=S)NH-、-C(=NH)NH-、-C(=O)O-、-C(=O)-、-SO2-または-SO2-NR8-であり、
R8は水素原子またはC1-6アルキルであり、
R17は水素原子またはC1-6アルキルであり、
R18は水素原子またはC1-6アルキルであり、
R17とR18は互いに結合し、それらが結合する窒素原子および隣接するC(=O)と一緒になって、5ないし7員複素環(例、イミダゾリジン-2-オン、ヘキサヒドロピリミジン-2-オンまたは1,3-ジアゼパン-2-オン)を形成してもよい、
R7は
(a)C6-10アリール(例、フェニル、1-ナフチル、2-ナフチル)、
(b)5ないし10員ヘテロアリール(例、ピリジル、チエニル、チアゾリル)、
(c)C1-6アルキル、または
(d)C2-6アルケニル
(ここで、C1-6アルキルおよびC2-6アルケニルはフェニル、フリルおよびジフェニルメチルスルフィニルから選択される1個の置換基で置換されていてもよく、
C6-10アリールおよび5ないし10員ヘテロアリールは任意に選択される1ないし4個(好ましくは1ないし3個、より好ましくは1または2個)のR9で置換されていてもよく、
R9は、それぞれ独立して、
ハロゲン原子、
-NO2、
-OH、
C1-6アルキル、
C1-6ハロアルキル、
C1-6アルコキシ、
5ないし10員ヘテロアリール(例、ピリジル)、
-NR9aR9b(ここで、R9aは水素原子、C1-6アルキルまたはC1-6アルコキシ-カルボニルであり、R9bは水素原子またはC1-6アルキルである)、
-C(=O)OR9c(ここで、R9cは水素原子またはC1-6アルキルである)、
-C(=O)NR9dR9e(ここで、R9dは水素原子またはC1-6アルキルであり、R9eは水素原子またはC1-6アルキルである)、または
-NH-C(=NR9f)-NHR9g(ここで、R9fは水素原子またはC1-6アルコキシ-カルボニルであり、R9gは水素原子またはC1-6アルコキシ-カルボニルである)であり、または
2個のR9は一緒になってメチレンジオキシを形成する)である)、
(2)-NH-Y2-R10
(ここで、Y2は-CH2-または単結合であり、
R10はC6-10アリール(例、フェニル)
(ここで、C6-10アリールは任意に選択される1ないし4個(好ましくは1ないし3個、より好ましくは1または2個)のR9で置換されていてもよく、
R9は、それぞれ独立して、
ハロゲン原子、
-OH、
C1-6アルキル、
C1-6ハロアルキル、
C1-6アルコキシ、または
-C(=O)NR9dR9e(ここで、R9dはC1-6アルキルであり、R9eはC1-6アルキルである)、または
2個のR9は一緒になってメチレンジオキシを形成する)である)、
(3)式(ii):

(4)-N=N-R12
(ここで、R12は(a)-OHで置換されていてもよいC1-6アルキル、(b)-OH、(c)ジ(C1-6アルキル)アミノおよび(d)C1-6アルコキシ-カルボニルアミノから選択される1ないし3個の置換基で置換されていてもよいC6-10アリール(例、フェニル)である)、または
(5)-NR13R14
(ここで、R13は水素原子、C1-6アルキル、C6-10アリール-C1-6アルキル(例、ベンジル)またはC2-6アルケニル-カルボニルであり、
R14は水素原子またはC1-6アルキル(ここで、C1-6アルキルは(a)C1-6アルコキシ-カルボニルアミノ、(b)C2-6アルケニル-カルボニルアミノおよび(c)C1-6アルキルで置換されていてもよいC6-10アリール-C1-6アルキルアミノカルボニル(例、ベンジルアミノカルボニル)から選択される1個の置換基で置換されていてもよい)であるか、またはR13とR14は互いに結合し、それらが結合する窒素原子と一緒になって、更に1個の窒素原子を含有する5ないし7員複素環(例、イミダゾリジン)(ここで、5ないし7員複素環はC1-6アルキルで置換されていてもよいC6-10アリール-カルボニル(例、ベンゾイル)で置換されていてもよい)を形成する。
ただし、R14がC2-6アルケニル-カルボニルアミノで置換されたエチルであるとき、R13は水素原子ではない)であり、
R15はC1-6アルキルであり、R16はC1-6アルキルである、
化合物またはその薬学的に許容される酸付加塩。
式(I-D)において、
R1は水素原子、C1-6アルコキシ、-OH、C1-6アルキルまたはハロゲン原子であり、
R5は水素原子またはC1-6アルコキシであり、
Y1は-C(=O)NR18-、-C(=S)NH-、-C(=NH)NH-、-C(=O)O-、-C(=O)-、-SO2-または-SO2-NR8-であり、
R8は水素原子またはC1-6アルキルであり、
R18は水素原子であり、
R7は
(a)C6-10アリール(例、フェニル、1-ナフチル、2-ナフチル)、
(b)5ないし10員ヘテロアリール(例、ピリジル、チエニル、チアゾリル)、
(c)C1-6アルキル、または
(d)C2-6アルケニル
(ここで、C1-6アルキルおよびC2-6アルケニルはフェニル、フリルおよびジフェニルメチルスルフィニルから選択される1個の置換基で置換されていてもよく、
C6-10アリールおよび5ないし10員ヘテロアリールは任意に選択される1ないし4個(好ましくは1ないし3個、より好ましくは1または2個)のR9で置換されていてもよく、
R9は、それぞれ独立して、
ハロゲン原子、
-NO2、
-OH、
C1-6アルキル、
C1-6ハロアルキル、
C1-6アルコキシ、
5ないし10員ヘテロアリール(例、ピリジル)、
-NR9aR9b(ここで、R9aは水素原子、C1-6アルキルまたはC1-6アルコキシ-カルボニルであり、R9bは水素原子またはC1-6アルキルである)、
-C(=O)OR9c(ここで、R9cは水素原子またはC1-6アルキルである)、
-C(=O)NR9dR9e(ここで、R9dは水素原子またはC1-6アルキルであり、R9eは水素原子またはC1-6アルキルである)、または
-NH-C(=NR9f)-NHR9g(ここで、R9fは水素原子またはC1-6アルコキシ-カルボニルであり、R9gは水素原子またはC1-6アルコキシ-カルボニルである)であり、または
2個のR9は一緒になってメチレンジオキシを形成する)であり、
R15はC1-6アルキルであり、R16はC1-6アルキルである]
で示される化合物またはその薬学的に許容される酸付加塩。
式(I-D)において、
R1は水素原子、C1-6アルコキシ、-OH、C1-6アルキルまたはハロゲン原子であり、
R5は水素原子またはC1-6アルコキシであり、
Y1は-C(=O)NR18-、-C(=S)NH-または-C(=NH)NH-であり、
R8は水素原子またはC1-6アルキルであり、
R18は水素原子であり、
R7は
(a)C6-10アリール(例、フェニル)、または
(b)5ないし10員ヘテロアリール(例、ピリジル、チエニル)
(ここで、C6-10アリールおよび5ないし10員ヘテロアリールは任意に選択される1ないし4個(好ましくは1ないし3個、より好ましくは1または2個)のR9で置換されていてもよく、
R9は、それぞれ独立して、
ハロゲン原子、
-NO2、
-OH、
C1-6アルキル、
C1-6ハロアルキル、
C1-6アルコキシ、
5ないし10員ヘテロアリール(例、ピリジル)、
-NR9aR9b(ここで、R9aは水素原子、C1-6アルキルまたはC1-6アルコキシ-カルボニルであり、R9bは水素原子またはC1-6アルキルである)、
-C(=O)OR9c(ここで、R9cは水素原子またはC1-6アルキルである)、
-C(=O)NR9dR9e(ここで、R9dは水素原子またはC1-6アルキルであり、R9eは水素原子またはC1-6アルキルである)、または
-NH-C(=NR9f)-NHR9g(ここで、R9fは水素原子またはC1-6アルコキシ-カルボニルであり、R9gは水素原子またはC1-6アルコキシ-カルボニルである)であり、または
2個のR9は一緒になってメチレンジオキシを形成する)であり、
R15はC1-6アルキルであり、R16はC1-6アルキルである、
化合物またはその薬学的に許容される酸付加塩。
式(I-E)において、
R1はC1-6アルコキシであり、
R5は水素原子であり、
Y2は-CH2-または単結合であり、
R10はC6-10アリール(例、フェニル)
(ここで、C6-10アリールは任意に選択される1ないし4個(好ましくは1ないし3個、より好ましくは1または2個)のR9で置換されていてもよく、
R9は、それぞれ独立して、
ハロゲン原子、
-OH、
C1-6アルキル、
C1-6ハロアルキル、
C1-6アルコキシ、または
-C(=O)NR9dR9e(ここで、R9dはC1-6アルキルであり、R9eはC1-6アルキルである)、または
2個のR9は一緒になってメチレンジオキシを形成する)であり、
R15はC1-6アルキルであり、R16はC1-6アルキルである、
化合物またはその薬学的に許容される酸付加塩。
式(I-F)において、
R1はC1-6アルコキシであり、
R5は水素原子であり、
R11はC1-6アルコキシまたはC6-10アリールアミノ(例、フェニルアミノ)(ここで、C6-10アリールアミノのC6-10アリール部分はC1-6アルキルおよびC1-6アルコキシから選択される1個の置換基で置換されていてもよい)であり、
R15はC1-6アルキルであり、R16はC1-6アルキルである、
化合物またはその薬学的に許容される酸付加塩。
式(I-G)において、
R1はC1-6アルコキシであり、
R5は水素原子であり、
R12は(a)-OHで置換されていてもよいC1-6アルキル、(b)-OH、(c)ジ(C1-6アルキル)アミノおよび(d)C1-6アルコキシ-カルボニルアミノから選択される1または2個の置換基で置換されていてもよいC6-10アリール(例、フェニル)であり、
R15はC1-6アルキルであり、R16はC1-6アルキルである、
化合物またはその薬学的に許容される酸付加塩。
式(I-H)において、
R1はC1-6アルコキシであり、
R5は水素原子であり、
R13は水素原子、C1-6アルキル、C6-10アリール-C1-6アルキル(例、ベンジル)またはC2-6アルケニル-カルボニルであり、
R14は水素原子またはC1-6アルキル(ここで、C1-6アルキルは(a)C1-6アルコキシ-カルボニルアミノ、(b)C2-6アルケニル-カルボニルアミノおよび(c)C1-6アルキルで置換されていてもよいC6-10アリール-C1-6アルキルアミノカルボニル(例、ベンジルアミノカルボニル)から選択される1個の置換基で置換されていてもよい)であるか、またはR13とR14は互いに結合し、それらが結合する窒素原子と一緒になって、更に1個の窒素原子を含有する5ないし7員複素環(例、イミダゾリジン)(ここで、5ないし7員複素環はC1-6アルキルで置換されていてもよいC6-10アリール-カルボニル(例、ベンゾイル)で置換されていてもよい)を形成する。
ただし、R14がC2-6アルケニル-カルボニルアミノで置換されたエチルであるとき、R13は水素原子ではない)であり、
R15はC1-6アルキルであり、R16はC1-6アルキルである、
化合物またはその薬学的に許容される酸付加塩。
式(I-J)において、
R1はC1-6アルコキシであり、
R7は-NR9aR9b(ここで、R9aはC1-6アルキルであり、R9bはC1-6アルキルである)で置換されたフェニルであり、
R15はC1-6アルキルであり、R16はC1-6アルキルである、
化合物またはその薬学的に許容される酸付加塩。
式(I-K)において、
R1はC1-6アルコキシであり、
R7は-NR9aR9b(ここで、R9aはC1-6アルキルであり、R9bはC1-6アルキルである)で置換されたフェニルであり、
R15はC1-6アルキルであり、R16はC1-6アルキルである]
で示される化合物またはその薬学的に許容される酸付加塩。
式(I-L)において、
R1はC1-6アルコキシであり、
R7は-NR9aR9b(ここで、R9aはC1-6アルキルであり、R9bはC1-6アルキルである)で置換されたフェニルであり、
R15はC1-6アルキルであり、R16はC1-6アルキルである、
化合物またはその薬学的に許容される酸付加塩。
上記式(I)または(I’)で示される本発明の化合物の具体的な製造方法として、例えばスキーム1から18に示す方法を挙げることができる。
スキーム1

工程1
化合物(IV)は、例えば、スルホニルクロリド化合物(II)を、アミン化合物(III)でアミド化することにより得ることができる。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン化炭化水素溶媒、ジエチルエーテル、テトラヒドロフラン(THF)、1,2-ジメトキシエタン(DME)、ジオキサンなどのエーテル溶媒、N,N-ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMSO)などの非プロトン性極性溶媒あるいはそれらの混合溶媒を用いることができる。通常は、ジクロロメタンまたはTHFが好ましく用いられる。スルホニルクロリド化合物(II)はアミン化合物(III)に対し0.5~20当量、好ましくは1.0~10当量用いられる。
塩基としては、例えば、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、リン酸カリウム、水酸化ナトリウム、水酸化カリウム、水酸化バリウム、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、4-ジメチルアミノピリジンなどが挙げられ、ジイソプロピルエチルアミン、トリエチルアミンまたはピリジンが好ましく用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、10分~48時間程度である。また、反応系中の基質(II)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。
アミン化合物(V)は、例えば、化合物(IV)のtert-ブトキシカルボニル(Boc)基を、酸性条件下で脱保護することにより得ることができる。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン化炭化水素溶媒、THF、DME、ジオキサンなどのエーテル溶媒、メタノール、エタノール、プロパノールなどのアルコール溶媒あるいはそれらの混合溶媒を用いることができる。通常は、ジクロロメタンまたはジオキサンが好ましく用いられる。
酸としては、トリフルオロ酢酸、メタンスルホン酸、p-トルエンスルホン酸、トリフルオロメタンスルホン酸などの有機酸、または塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、リン酸などの無機酸を用いることができる。通常は、トリフルオロ酢酸または塩酸が好ましく用いられる。また別法として、塩化水素を有機溶媒に溶解させた塩化水素-メタノール溶液、塩化水素-THF溶液、塩化水素-酢酸エチル溶液、塩化水素-ジオキサン溶液をそれぞれ単独で用いることもできる。その場合、特に塩化水素-メタノール溶液または塩化水素-THF溶液を用いると好ましい結果が得られる。酸は化合物(IV)に対し1.0~100当量、好ましくは3.0~10当量用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(IV)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。
化合物(I-1)は、例えば、アミン化合物(V)を、アシルハライド化合物(VI)(式中、X1はハロゲン原子である)でアミド化することにより得ることができる。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン化炭化水素溶媒、ジエチルエーテル、THF、DME、ジオキサンなどのエーテル溶媒、DMF、DMSOなどの非プロトン性極性溶媒あるいはそれらの混合溶媒を用いることができる。通常は、ジクロロメタンまたはTHFが好ましく用いられる。アシルハライド化合物(VI)はアミン化合物(V)に対し0.5~20当量、好ましくは1.0~10当量用いられる。
塩基としては、例えば、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、リン酸カリウム、水酸化ナトリウム、水酸化カリウム、水酸化バリウム、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、4-ジメチルアミノピリジンなどが挙げられ、ジイソプロピルエチルアミン、トリエチルアミンまたはピリジンが好ましく用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、10分~48時間程度である。また、反応系中の基質(V)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。
また、化合物(I-1)は、例えば、アミン化合物(V)を、カルボン酸(VI)(式中、X1はヒドロキシ基である)でアミド化することにより得ることができる。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン化炭化水素溶媒、ジエチルエーテル、THF、DME、ジオキサンなどのエーテル溶媒、DMF、DMSO、酢酸エチルなどの非プロトン性極性溶媒、メタノール、エタノール、プロパノールなどのアルコール溶媒あるいはそれらの混合溶媒を用いることができる。通常は、ジクロロメタンまたはTHFが好ましく用いられる。カルボン酸(VI)は、アミン化合物(V)に対し0.5~20当量、好ましくは0.5~10当量用いられる。
縮合剤としては、ジシクロヘキシルカルボジイミド、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド 塩酸塩(EDCI)、ベンゾトリアゾール-1-イルオキシトリス(ジメチルアミノ)ホスホニウム ヘキサフルオロリン酸塩(BOP)、N,N’-カルボニルジイミダゾール(CDI)、4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリニウムクロリド(DMT-MM)、{{[(1-シアノ-2-エトキシ-2-オキソエチリデン)アミノ]オキシ}-4-モルホリノメチレン}ジメチルアンモニウム ヘキサフルオロリン酸塩(COMU)、O-(7-アザベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロリン酸塩(HATU)などを用いることができ、特にBOPまたはHATUが好ましく用いられる。縮合剤は、アミン化合物(XXI)に対し1.0~100当量、好ましくは1.0~10当量用いられる。
塩基を用いる場合は、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、N-メチルモルホリン等を用いることができ、トリエチルアミンまたはジイソプロピルエチルアミンが好ましく用いられる。塩基は、アミン化合物(V)に対し3.0~100当量、好ましくは3.0~10当量用いられる。
反応温度は、通常-40~150℃、好ましくは0~60℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(V)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。

化合物(I-2)は、例えば、アミン化合物(V)を、アルデヒド化合物(VII)で還元的アルキル化することにより得ることができる。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン化炭化水素溶媒、ジエチルエーテル、THF、DME、ジオキサンなどのエーテル溶媒、DMF、DMSOなどの非プロトン性極性溶媒、メタノール、エタノール、プロパノールなどのアルコール溶媒、酢酸、水あるいはそれらの混合溶媒を用いることができる。アルデヒド化合物(VII)はアミン化合物(V)に対し0.5~20当量、好ましくは1.0~10当量用いられる。
還元剤としては、水素化ホウ素ナトリウム、水素化ホウ素リチウム、シアノ水素化ホウ素ナトリウム、トリアセトキシ水素化ホウ素ナトリウム、2-ピコリン-ボランコンプレックスなどが挙げられる。
塩基を用いる場合は、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、N-メチルモルホリン等を用いることができ、トリエチルアミンまたはジイソプロピルエチルアミンが好ましく用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、10分~48時間程度である。また、反応系中の基質(V)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。
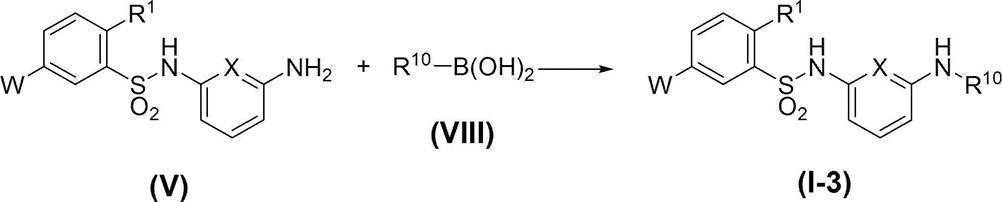
化合物(I-3)は、例えば、アミン化合物(V)を、銅触媒の存在下、ボロン酸化合物(VIII)とのカップリング反応に付すことにより得ることができる。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン化炭化水素溶媒、THF、DME、ジオキサンなどのエーテル溶媒、DMF、DMSOなどの非プロトン性極性溶媒、メタノール、エタノール、プロパノールなどのアルコール溶媒、ベンゼン、トルエン、キシレンなどの芳香族炭化水素溶媒あるいはそれらの混合溶媒を用いることができる。ボロン酸化合物(VIII)は、アミン化合物(V)に対し1.0~20当量、好ましくは1.0~10当量用いられる。
銅触媒としては、酢酸銅(II)などが挙げられる。銅触媒は、アミン化合物(V)に対し0.001~1当量、好ましくは0.005~0.5当量用いられる。
塩基を用いる場合は、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、N-メチルモルホリン等を用いることができ、トリエチルアミンまたはジイソプロピルエチルアミンが好ましく用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(V)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。

化合物(I-4)は、例えば、アミン化合物(V)を、イソシアネート(IX)もしくはカルボン酸R7-CO2H(IX-1)から発生させたイソシアネート(IX)と反応させることにより得ることができる。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン化炭化水素溶媒、THF、DME、ジオキサンなどのエーテル溶媒、DMF、DMSOなどの非プロトン性極性溶媒、ベンゼン、トルエン、キシレンなどの芳香族炭化水素溶媒あるいはそれらの混合溶媒を用いることができる。イソシアネート(IX)は、アミン化合物(V)に対し1.0~20当量、好ましくは1.0~10当量用いられる。
塩基を用いる場合は、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、N-メチルモルホリン等を用いることができ、トリエチルアミンまたはジイソプロピルエチルアミンが好ましく用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(V)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。
カルボン酸(IX-1)からイソシアネート(IX)を発生させる場合、たとえば、カルボン酸(IX-1)をジフェニルリン酸アジド(DPPA)で酸アジド化し、加熱条件下でクルチウス転位反応をすることにより得ることができる。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン化炭化水素溶媒、THF、DME、ジオキサンなどのエーテル溶媒、DMF、DMSOなどの非プロトン性極性溶媒、ベンゼン、トルエン、キシレンなどの芳香族炭化水素溶媒あるいはそれらの混合溶媒を用いることができる。カルボン酸(IX-1)は、アミン化合物(V)に対し1.0~30当量、好ましくは1.0~10当量用いられる。DPPAは、カルボン酸(IX-1)に対し1.0~30当量、好ましくは1.0~10当量用いられる。
塩基を用いる場合は、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、N-メチルモルホリン等を用いることができ、トリエチルアミンまたはジイソプロピルエチルアミンが好ましく用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(V)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。

化合物(I-5)は、例えば、アミン化合物(V)を、イソチオシアネート(X)と反応させることにより得ることができる。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン化炭化水素溶媒、THF、DME、ジオキサンなどのエーテル溶媒、DMF、DMSOなどの非プロトン性極性溶媒、ベンゼン、トルエン、キシレンなどの芳香族炭化水素溶媒あるいはそれらの混合溶媒を用いることができる。イソチオシアネート(X)は、アミン化合物(V)に対し1.0~20当量、好ましくは1.0~10当量用いられる。
塩基を用いる場合は、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、N-メチルモルホリン等を用いることができ、トリエチルアミンまたはジイソプロピルエチルアミンが好ましく用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(V)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。

化合物(I-6)は、例えば、化合物(I-5)を、アンモニアおよび2-ヨードキシ安息香酸(IBX)と反応させることにより得ることができる。
溶媒としては、アセトニトリルなどのニトリル溶媒を用いることができる。アンモニアとしては、アンモニア水溶液を用いることができる。アンモニアおよびIBXは、化合物(I-6)に対し1.0~20当量、好ましくは1.0~10当量用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(I-6)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。

化合物(I-7)は、例えば、アミン化合物(V)を、クロロギ酸エステル(XI)と反応させることにより得ることができる。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン化炭化水素溶媒、THF、DME、ジオキサンなどのエーテル溶媒、DMF、DMSOなどの非プロトン性極性溶媒、ベンゼン、トルエン、キシレンなどの芳香族炭化水素溶媒あるいはそれらの混合溶媒を用いることができる。クロロギ酸エステル(XI)は、アミン化合物(V)に対し1.0~20当量、好ましくは1.0~10当量用いられる。
塩基を用いる場合は、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、N-メチルモルホリン等を用いることができ、トリエチルアミンまたはジイソプロピルエチルアミンが好ましく用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(V)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。
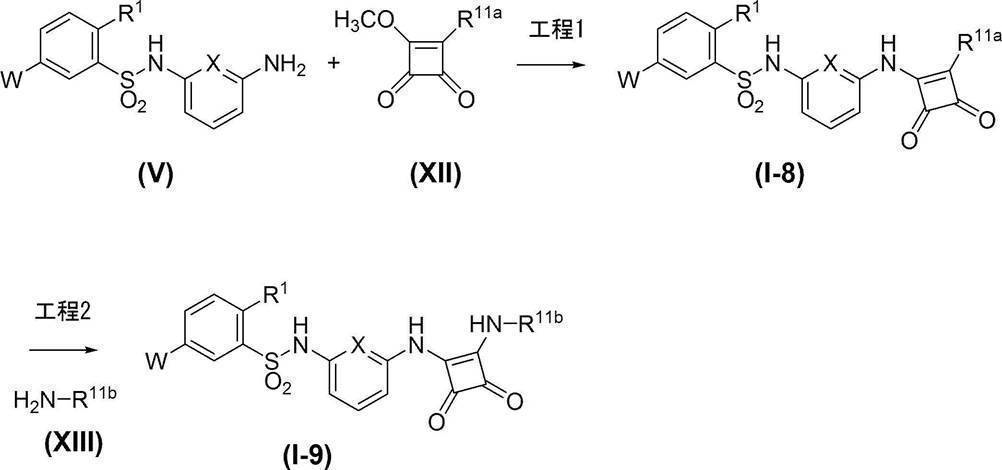
化合物(I-8)は、例えば、アミン化合物(V)を、シクロブテン化合物(XII)と反応させることにより得ることができる。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン化炭化水素溶媒、THF、DME、ジオキサンなどのエーテル溶媒、DMF、DMSOなどの非プロトン性極性溶媒、メタノール、エタノール、プロパノールなどのアルコール溶媒、ベンゼン、トルエン、キシレンなどの芳香族炭化水素溶媒あるいはそれらの混合溶媒を用いることができる。シクロブテン化合物(XII)は、アミン化合物(V)に対し1.0~20当量、好ましくは1.0~10当量用いられる。
塩基を用いる場合は、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、N-メチルモルホリン等を用いることができ、トリエチルアミンまたはジイソプロピルエチルアミンが好ましく用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(V)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。
化合物(I-9)は、例えば、R11aがメトキシである化合物(I-8)を、アミン化合物(XIII)と反応させることにより得ることができる。
溶媒としては、ジクロロメタン、クロロホルム、1,2-ジクロロエタンなどのハロゲン化炭化水素溶媒、THF、DME、ジオキサンなどのエーテル溶媒、DMF、DMSOなどの非プロトン性極性溶媒、メタノール、エタノール、プロパノールなどのアルコール溶媒、ベンゼン、トルエン、キシレンなどの芳香族炭化水素溶媒あるいはそれらの混合溶媒を用いることができる。アミン化合物(XIII)は、化合物(I-8)に対し1.0~20当量、好ましくは1.0~10当量用いられる。
塩基を用いる場合は、トリエチルアミン、ジイソプロピルエチルアミン、ピリジン、N-メチルモルホリン等を用いることができ、トリエチルアミンまたはジイソプロピルエチルアミンが好ましく用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(I-8)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。
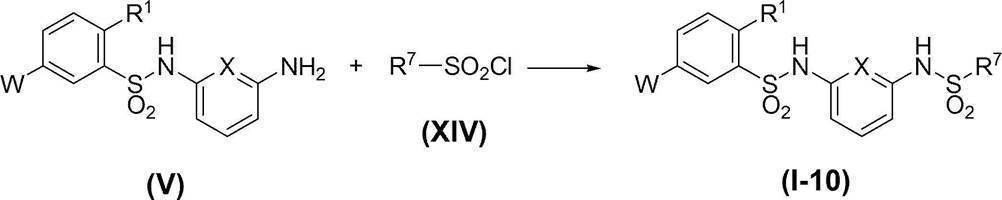
化合物(I-10)は、例えば、アミン化合物(V)を、スルホニルクロリド化合物(XIV)でアミド化することにより得ることができる。
アミド化は、スキーム1の工程1と同様の方法で行うことができる。

化合物(I-11)は、例えば、アミン化合物(V)を、スルホニルクロリド化合物(XV)でアミド化することにより得ることができる。
アミド化は、スキーム1の工程1と同様の方法で行うことができる。

工程1
化合物(XVIII)は、例えば、スルホニルクロリド化合物(XVI)を、アミン化合物(XVII)でアミド化することにより得ることができる。
アミド化は、スキーム1の工程1と同様の方法で行うことができる。
化合物(XX)は、例えば、化合物(XVIII)を、ボロン酸化合物(XIX)で鈴木カップリングすることにより得ることができる。
鈴木カップリング反応は、パラジウム触媒および塩基の存在下、ホスフィンリガンドの存在下または非存在下に、適当な溶媒中で行われる。
溶媒としては、THF、DME、ジオキサンなどのエーテル溶媒、DMF、DMSOなどの非プロトン性極性溶媒、メタノール、エタノール、プロパノールなどのアルコール溶媒、ベンゼン、トルエン、キシレンなどの芳香族炭化水素溶媒、水あるいはそれらの混合溶媒を用いることができる。通常は、ジオキサン、DME、ジオキサンおよび水の混合溶媒またはDMEおよび水の混合溶媒が好ましく用いられる。
ボロン酸化合物(XIX)としては、ボロン酸だけではなく、ボロン酸ピナコールエステル、ボロン酸N-メチルイミノ二酢酸(MIDA)エステルなどのボロン酸エステル、またはトリフルオロボレートカリウム塩も用いることができる。特に、ボロン酸またはボロン酸ピナコールエステルが好ましく用いられる。ボロン酸化合物(XIX)は、化合物(XVIII)に対し1.0~20当量、好ましくは1.0~10当量用いられる。
パラジウム触媒としては、例えば、テトラキス(トリフェニルホスフィン)パラジウム、パラジウムアセテート、ビス(トリフェニルホスフィン)パラジウムジクロリド、ビス(ジベンジリデンアセトン)パラジウム、ビス(ジフェニルホスフィノ)フェロセンパラジウムジクロリドなどが挙げられ、テトラキス(トリフェニルホスフィン)パラジウムまたはビス(ジフェニルホスフィノ)フェロセンパラジウムジクロリドが好ましく用いられる。パラジウム触媒は、化合物(XVIII)に対し0.001~1当量、好ましくは0.005~0.5当量用いられる。
塩基としては、例えば、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、リン酸カリウム、水酸化ナトリウム、水酸化カリウム、水酸化バリウム、トリエチルアミン、ジイソプロピルエチルアミンなどが挙げられ、炭酸ナトリウムまたは炭酸カリウムが好ましく用いられる。
反応温度は、通常-40~150℃、好ましくは20~110℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(XIX)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。
アミン化合物(XXI)は、例えば、化合物(XX)のベンジル基を、加水素分解反応などにより脱保護し、得られた化合物のtert-ブトキシカルボニル(Boc)基を、酸性条件下で脱保護することにより得ることができる。
加水素分解反応は、パラジウム触媒の存在下および水素の雰囲気下に、適当な溶媒中で行われる。
溶媒としては、THF、DME、ジオキサンなどのエーテル溶媒、DMF、DMSO、酢酸エチルなどの非プロトン性極性溶媒、メタノール、エタノール、プロパノールなどのアルコール溶媒、ベンゼン、トルエン、キシレンなどの芳香族炭化水素溶媒、酢酸、水あるいはそれらの混合溶媒を用いることができる。通常は、トルエンやTHF、メタノールが好ましく用いられる。
パラジウム触媒としては、例えば、パラジウム(Pd)、パラジウム炭素(Pd/C)、水酸化パラジウム(Pd(OH)2)、水酸化パラジウム炭素(Pd(OH)2/C)などが挙げられるが、パラジウム炭素(Pd/C)が好ましく用いられる。パラジウム触媒は、化合物(XX)に対し0.001~1当量、好ましくは0.005~0.5当量用いられる。
反応温度は、通常-40~150℃、好ましくは20~110℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(XX)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。
tert-ブトキシカルボニル(Boc)基の脱保護は、スキーム1の工程2と同様の方法で行うことができる。
化合物(XXIII)は、例えば、アミン化合物(XXI)を、アシルハライド(XXII)(式中、X1はハロゲン原子である)またはカルボン酸(XXII)(式中、X1はヒドロキシ基である)でアミド化することにより得ることができる。
アミド化は、スキーム1の工程3と同様の方法で行うことができる。
化合物(I-12)は、例えば、化合物(XXIII)を、パラホルムアルデヒドと反応させることにより得ることができる。
溶媒としては、酢酸などのカルボン酸溶媒を用いることができる。パラホルムアルデヒドは、化合物(XXIII)に対し1.0~20当量、好ましくは1.0~10当量用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(XXIII)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。

化合物(I)は、例えば、スルホニルクロリド化合物(II)を、アミン化合物(XXIV)でアミド化することにより得ることができる。
アミド化は、スキーム1の工程1と同様の方法で行うことができる。

化合物(I-13)は、例えば、アミン化合物(V)をジアゾ化し、得られたジアゾニウム塩を化合物(XXV)とのジアゾカップリング反応に付すことにより得ることができる。
ジアゾ化は、アミン化合物(V)を酸性水溶液中で亜硝酸ナトリウムなどの亜硝酸塩と反応させることにより行うことができる。亜硝酸塩は、アミン化合物(V)に対し1.0~20当量、好ましくは1.0~10当量用いられる。
ジアゾカップリング反応は、得られたジアゾニウム塩を化合物(XXV)と反応させることにより行うことができる。化合物(XXV)は、アミン化合物(V)に対し1.0~20当量、好ましくは1.0~10当量用いられる。
反応温度は、通常-40~150℃、好ましくは0~80℃である。反応時間は、反応温度等の条件に応じて適宜選択されるが、通常、20分~48時間程度である。また、反応系中の基質(V)の濃度は、特に限定されるものではないが、通常、0.001mmol/L~1mol/Lが好ましい。

化合物(I-14)は、例えば、アミン化合物(V)を、アシル化および/またはアルキル化することにより得ることができる。
アシル化は、スキーム1の工程3と同様の方法で行うことができる。アルキル化は、スキーム2の還元的アルキル化と同様の方法で行うことができる。

工程1
化合物(XXVII)は、例えば、化合物(XXVI)を、イソシアネート(IX)と反応させ、得られた化合物のtert-ブトキシカルボニル(Boc)基を脱保護することにより得ることができる。
化合物(XXVI)とイソシアネート(IX)との反応は、スキーム4と同様の方法で行うことができる。tert-ブトキシカルボニル(Boc)基の脱保護は、スキーム1の工程2と同様の方法で行うことができる。
工程2
化合物(XXIX)は、例えば、化合物(XXVII)を、スルホニルクロリド化合物(XXX)でアミド化することにより得ることができる。
アミド化は、スキーム1の工程1と同様の方法で行うことができる。
工程3
化合物(I-15)は、例えば、化合物(XXIX)を、アミン化合物(XXX)でアミド化することにより得ることができる。
アミド化は、スキーム11の工程4と同様の方法で行うことができる。
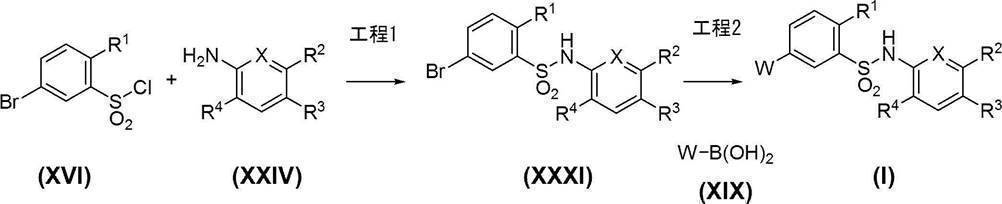
工程1
化合物(XXXI)は、例えば、スルホニルクロリド化合物(XVI)を、化合物(XXIV)でアミド化することにより得ることができる。
アミド化は、スキーム1の工程1と同様の方法で行うことができる。
工程2
化合物(I)は、例えば、化合物(XXXI)を、ボロン酸化合物(XIX)で鈴木カップリングすることにより得ることができる。
鈴木カップリングは、スキーム11の工程2と同様の方法で行うことができる。

化合物(I-16)は、例えば、化合物(XXXII)を、アミン化合物(XXX)でアミド化することにより得ることができる。
アミド化は、スキーム11の工程4と同様の方法で行うことができる。

工程1
化合物(I’)は、例えば、スルホニルクロリド化合物(XXXIII)を、化合物(XXXIV)でアミド化することにより得ることができる。
アミド化は、スキーム1の工程1と同様の方法で行うことができる。
工程2
化合物(XXXV)は、例えば、スルホニルクロリド化合物(XVI)を、化合物(XXXIV)でアミド化することにより得ることができる。
アミド化は、スキーム1の工程1と同様の方法で行うことができる。
工程3
化合物(I’)は、例えば、化合物(XXXV)を、ボロン酸化合物(XXXVI)で鈴木カップリングすることにより得ることができる。
鈴木カップリングは、スキーム11の工程2と同様の方法で行うことができる。
R4がR6であり、R2およびR3がそれぞれ水素原子である化合物(I)は、アミン化合物(III)の代わりに下記のアミン化合物(III-2)を使用して、前記と同様の方法により得ることができる。
本発明の化合物を含有するオレキシン受容体作動薬は、ヒトに対して有効であるだけではなく、ヒト以外の哺乳類、例えばマウス、ラット、ハムスター、ウサギ、ネコ、イヌ、ウシ、ヒツジ、サルなどに対しても有効である。
また本発明の化合物は、上記の通りナルコレプシーの予防剤または治療剤として用いられるだけではなく、ナルコレプシーのための予防または治療方法、あるいはナルコレプシーを予防または治療するための医薬の製造に使用することができる。
さらに本発明の化合物は、眠気改善剤、あるいは肥満症、糖尿病、うつ病、敗血症、重症敗血症または敗血症性ショックなどの予防剤または治療剤としても使用することができる。
本発明の化合物をナルコレプシーの予防剤または治療剤、眠気改善剤あるいは肥満症、糖尿病、うつ病、敗血症、重症敗血症または敗血症性ショックなどの予防剤または治療剤として臨床で使用する際には、薬剤は本発明化合物のフリー体またはその酸付加塩自体でもよく、また、賦形剤、安定化剤、保存剤、緩衝剤、溶解補助剤、乳化剤、希釈剤、等張化剤などの添加剤が適宜混合されてもよい。投与形態としては、錠剤、カプセル剤、顆粒剤、散剤、シロップ剤などによる経口剤、注射剤、坐剤、液剤などによる非経口剤、あるいは軟膏剤、クリーム剤、貼付剤などによる局所投与等を挙げることができる。
本発明のナルコレプシーの予防剤または治療剤、眠気改善剤あるいは肥満症、糖尿病、うつ病、敗血症、重症敗血症または敗血症性ショックなどの予防剤または治療剤は、上記有効成分を0.001~90重量%、好ましくは0.01~70重量%含有することが望ましい。その使用量は、症状、年齢、体重、投与方法に応じて適宜選択されるが、成人に対して、注射剤の場合、有効成分量として1日0.1μg~1g、経口剤の場合1μg~1g、貼付剤の場合1μg~10gであり、それぞれ1回または数回に分けて投与することができる。
また、本発明のナルコレプシーの予防剤または治療剤、あるいは眠気改善剤は、日中の強い眠気と居眠りの予防剤または治療剤、熟眠障害の予防剤または治療剤、カタプレキシーの予防剤または治療剤と組み合わせて用いることもできる。
日中の強い眠気と居眠りの予防剤または治療剤としては、メチルフェニデート、ペモリン、モダフィニルなどの中枢神経賦活薬などを挙げることができる。
熟眠障害の予防剤または治療剤としては、トリアゾラム、ベゲタミンBなどの睡眠薬、および抗不安薬などを挙げることができる。
カタプレキシーの予防剤または治療剤としては、塩酸クロミプラミン、ブロチゾラム、イミプラミン 塩酸塩などの三環性抗うつ薬、フルボキサミン マレイン酸塩、パロキセチン 塩酸塩などの選択的セロトニン再取り込み阻害薬(SSRI)、塩酸ミルナシプラン、デュロキセチン 塩酸塩などのセロトニン・ノルアドレナリン再取り込み阻害薬(SNRI)などを挙げることができる。
Boc:tert-ブトキシカルボニル
Bn:ベンジル
DIPEA:N,N-ジイソプロピルエチルアミン
DME:1,2-ジメトキシエタン
DMF:N,N-ジメチルホルムアミド
DPPA:ジフェニルリン酸アジド
HATU:O-(7-アザベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウム ヘキサフルオロリン酸塩
IBX:2-ヨードキシ安息香酸
BOP:ベンゾトリアゾール-1-イルオキシトリス(ジメチルアミノ)ホスホニウム ヘキサフルオロリン酸塩
Me:メチル
Ph:フェニル
TEA:トリエチルアミン
TFA:2,2,2-トリフルオロ酢酸
化合物名はCambridge社のChemBioDraw Ultra ver.12.0.3を用い命名した。
以下の実施例及び製造例中の「室温」は通常約10℃から約35℃を示す。%は特記しない限り重量パ-セントを示す。


(ii)アルゴン雰囲気下、塩化スルホン酸(870μL)に4’-メトキシ-N,N-ジメチル-[1,1’-ビフェニル]-3-カルボキサミド(1.10g)のジクロロメタン(4.0mL)溶液をゆっくりと加え、氷冷下で10分間撹拌した。反応液を室温まで昇温し、2時間撹拌した後、塩化チオニル(950μL)およびDMF(1.70mL)を加え、室温で終夜撹拌した。反応液を氷に流し込み、1時間撹拌した後、クロロホルムで抽出した。有機層を硫酸ナトリウムで乾燥した後、濾過し、濾液を減圧下にて濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液:酢酸エチル/ヘキサン=1/1→1/0)で精製し、3’-(ジメチルカルバモイル)-4-メトキシ-[1,1’-ビフェニル]-3-スルホニルクロリド(1.40g)を得た。
















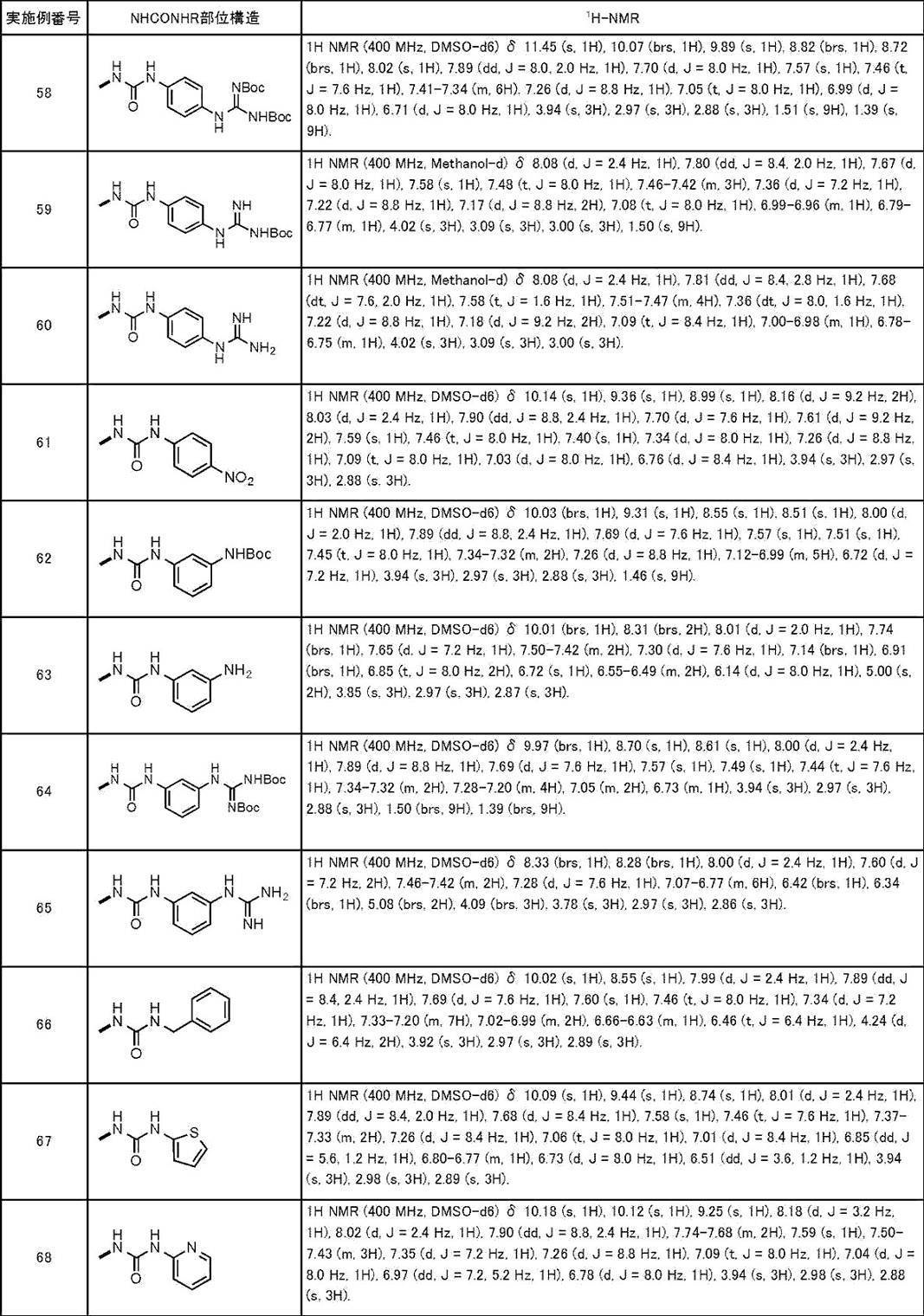








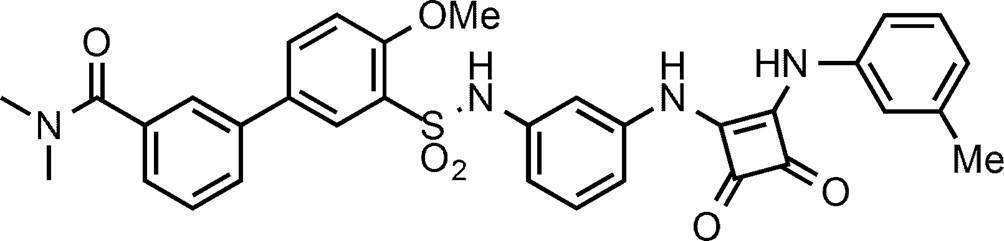
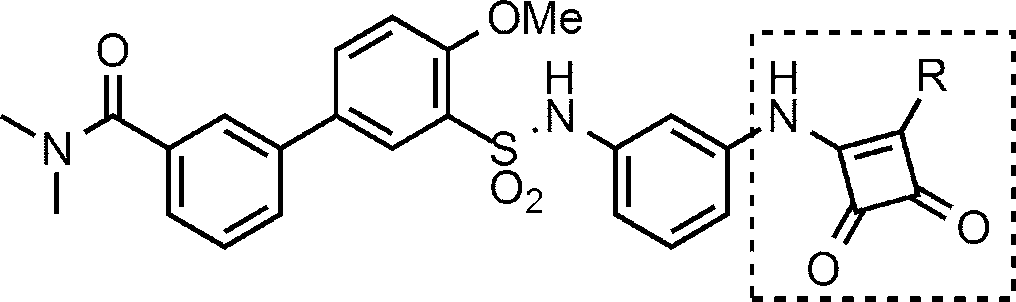







(ii)アルゴン雰囲気下、tert-ブチル (2-((ベンジル)(3-ニトロフェニル)アミノ)エチル)カルバメートのエタノール(50.0mL)および水(20.0mL)混合懸濁液に塩化アンモニウム(6.69g)および鉄粉(4.86g)を加え、2時間加熱還流した。この反応液をセライト濾過し、濾液を濃縮した後、飽和炭酸水素ナトリウム水溶液を加え、クロロホルムで抽出した。有機層を硫酸ナトリウムで乾燥した後、濾過し、濾液を濃縮した。得られた残渣をアミンシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/酢酸エチル=2/1)で精製し、tert-ブチル (2-((3-アミノフェニル)(ベンジル)アミノ)エチル)カルバメート(4.58g)を得た。



(ii)tert-ブチル (2-((3-(3’-(ジメチルアミノカルバモイル)-4-メトキシ-[1,1’-ビフェニル]-3-イルスルホンアミド)フェニル)アミノ)エチル)カルバメート(203.0mg)に10%塩化水素-メタノール溶液(4.0mL)を加え、50℃で2時間撹拌した。この反応液を濃縮することにより、3’-(N-(3-((2-アミノエチル)アミノ)フェニル)スルファモイル)-4’-メトキシ-N,N-ジメチル-[1,1’-ビフェニル]-3-カルボキサミド 二塩酸塩(187.0mg)を得た。






(ii)アルゴン雰囲気下、2-((3-アミノフェニル)アミノ)-N-(3-メチルベンジル)アセトアミド TFA塩(6.8mg)のピリジン(1.0mL)溶液に、3’-(ジメチルカルバモイル)-4-メトキシ-[1,1’-ビフェニル]-3-スルホニルクロリド(6.0mg)を加え、室温で16時間撹拌した。反応液を減圧下で濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=1/9→0/1)で精製し、4'-メトキシ-N,N-ジメチル-3'-(N-(3-((2-((3-メチルベンジル)アミノ)-2-オキソエチル)アミノ)フェニル)スルファモイル)-[1,1'-ビフェニル]-3-カルボキサミド(7.0mg)を得た。

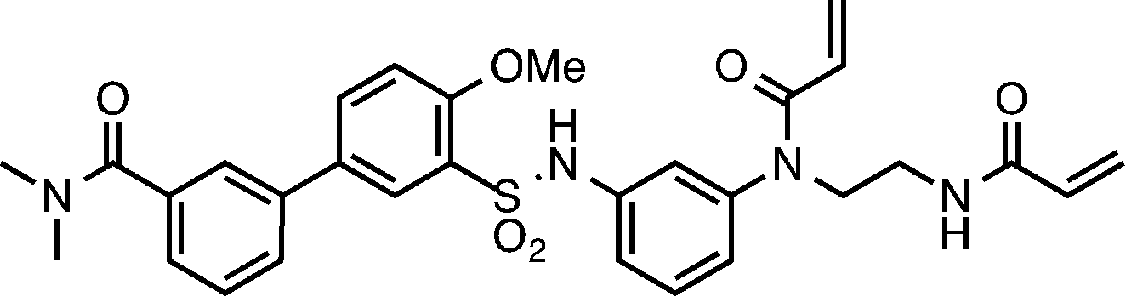


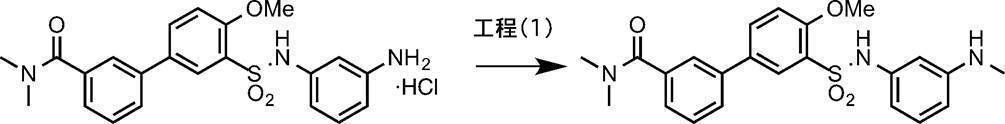

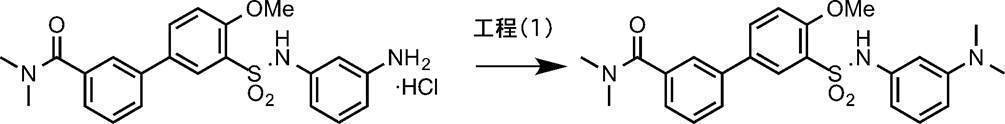






(ii)アルゴン雰囲気下、tert-ブチル (3-(3-(3-(ジメチルアミノ)フェニル)ウレイド)フェニル)カルバメート(1.0g)のジクロロメタン(10.0mL)懸濁液に、TFA(600μL)を加え、終夜撹拌した。反応液を減圧下で濃縮し、残渣を25%メタノール含有クロロホルムで希釈した後、飽和炭酸水素ナトリウム水溶液を加え抽出した。有機層を飽和食塩水で洗浄した後、硫酸ナトリウムを加え乾燥し、濾過後に濾液を減圧下にて濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液:クロロホルム/酢酸エチル=2/1)で精製し、1-(3-アミノフェニル)-3-(3-(ジメチルアミノ)フェニル)ウレア(715.3mg)を得た。





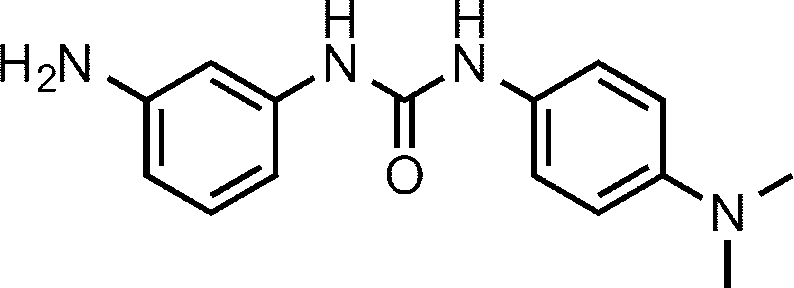
(ii)アルゴン雰囲気下、tert-ブチル (3-(3-(4-(ジメチルアミノ)フェニル)ウレイド)フェニル)カルバメート(1.90g)のジクロロメタン(10.0mL)懸濁液に、TFA(2.00mL)を加え、終夜撹拌した。反応液を減圧下で濃縮し、残渣を15%メタノール含有クロロホルムで希釈した後、飽和炭酸水素ナトリウム水溶液を加え抽出した。有機層を飽和食塩水で洗浄した後、硫酸ナトリウムを加え乾燥し、濾過後に濾液を減圧下にて濃縮し、1-(3-アミノフェニル)-3-(4-(ジメチルアミノ)フェニル)ウレア(1.02g)を得た。本化合物は更なる精製をせずに次の反応に用いた。



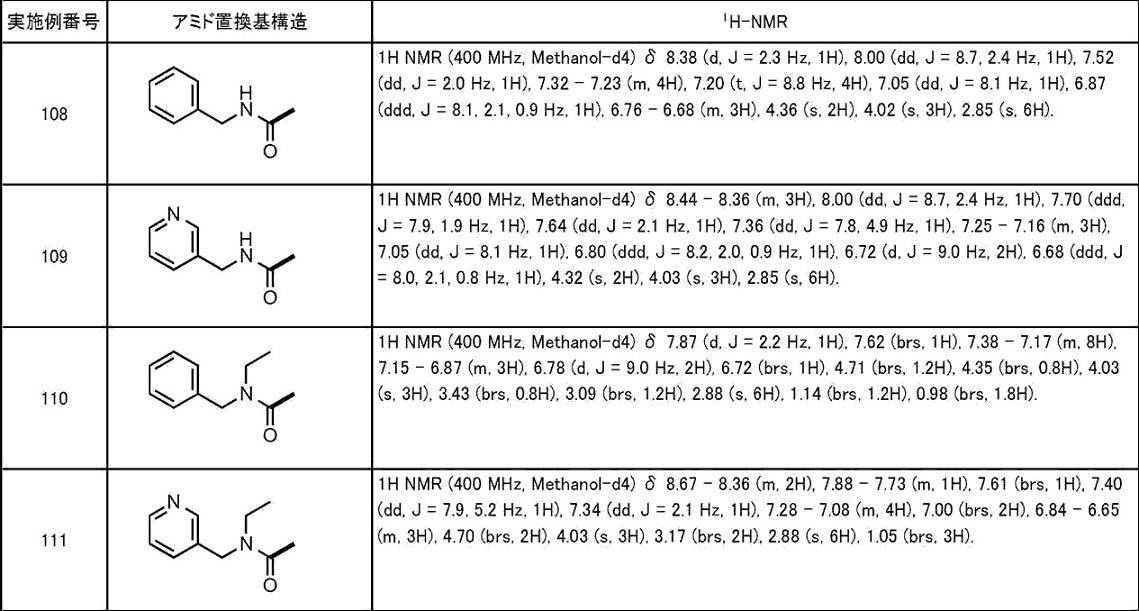


(ii)アルゴン雰囲気下、tert-ブチル (3-(3-(2-(ジメチルアミノ)フェニル)ウレイド)フェニル)カルバメート(1.00g)のジクロロメタン(10.0mL)懸濁液に、TFA(2.00mL)を加え、終夜撹拌した。反応液を減圧下で濃縮し、残渣を15%メタノール含有クロロホルムで希釈した後、飽和炭酸水素ナトリウム水溶液を加え抽出した。有機層を飽和食塩水で洗浄した後、硫酸ナトリウムを加え乾燥し、濾過後に濾液を減圧下にて濃縮し、1-(3-アミノフェニル)-3-(2-(ジメチルアミノ)フェニル)ウレア(725.8mg)を得た。本化合物は更なる精製をせずに次の反応に用いた。











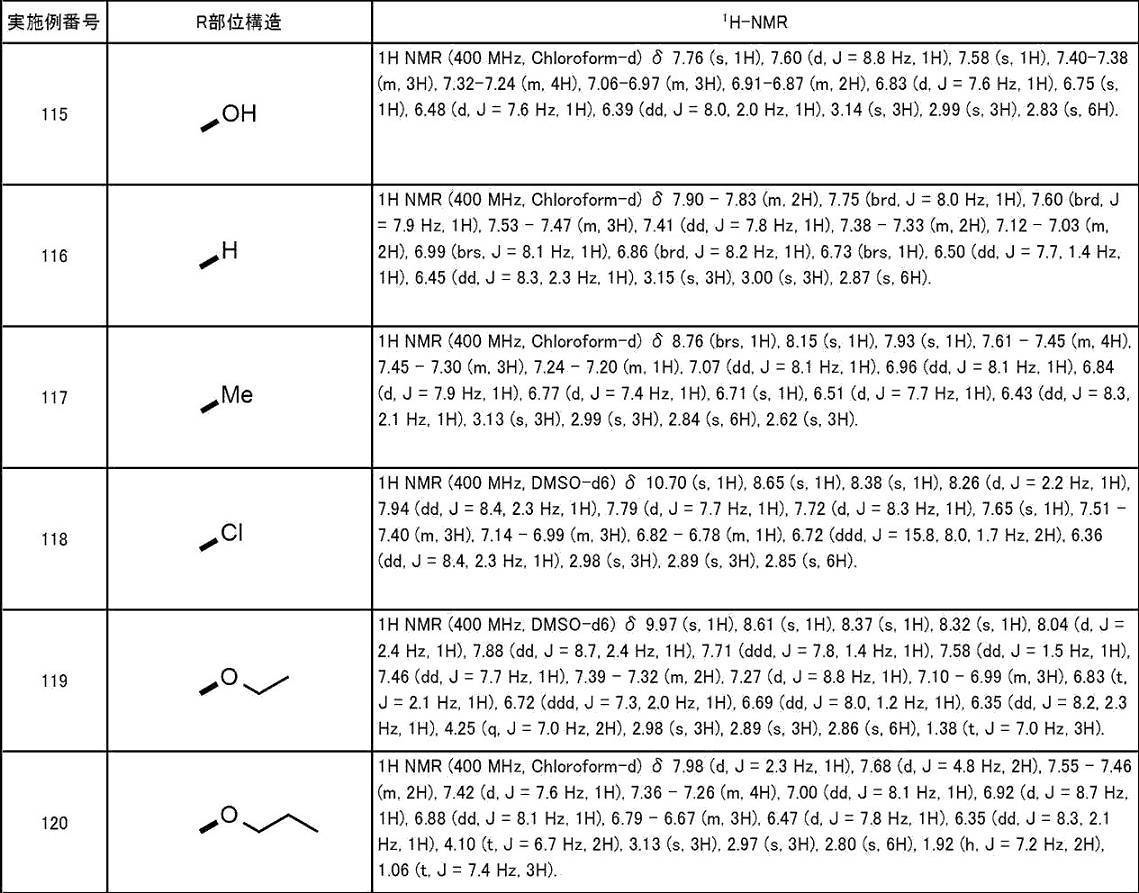
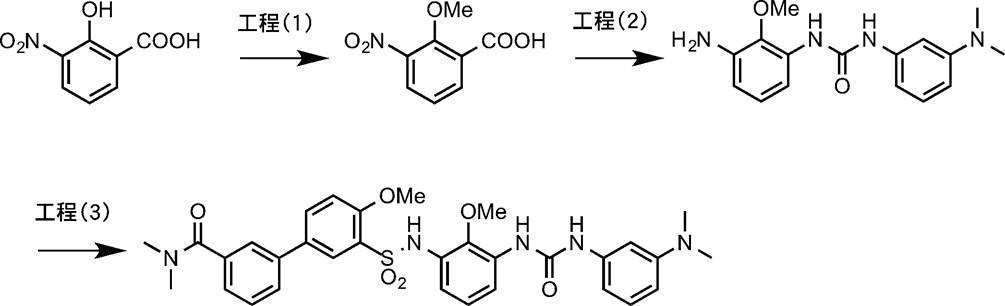

(ii)2-メトキシ-3-ニトロ安息香酸メチル(1.10g)のエタノール(20.0mL)溶液に、1N水酸化ナトリウム水溶液(20.0mL)を加え、室温で終夜撹拌した。反応液に1N塩酸水溶液を加え、酢酸エチルで抽出した。有機層を飽和食塩水で洗浄した後、硫酸マグネシウムを加え乾燥し、濾過後に濾液を減圧下にて濃縮することで、2-メトキシ-3-ニトロ安息香酸(980mg)を得た。本化合物は更なる精製をせずに次の反応に用いた。

(ii)1-(3-ニトロ-2-メトキシフェニル)-3-(3-(ジメチルアミノ)フェニル)ウレア(98.5mg)のメタノール(10.0mL)溶液に、5%パラジウム-活性炭素(10.30mg)を加え、水素雰囲気下、室温で終夜撹拌した。反応液をセライト濾過し、濾液を濃縮した。得られた残渣をシリカゲルカラムクロマトグラフィー(溶出液:ヘキサン/酢酸エチル=9/1→1/4)で精製し、1-(3-アミノ-2-メトキシフェニル)-3-(3-(ジメチルアミノ)フェニル)ウレア(80.5mg)を得た。

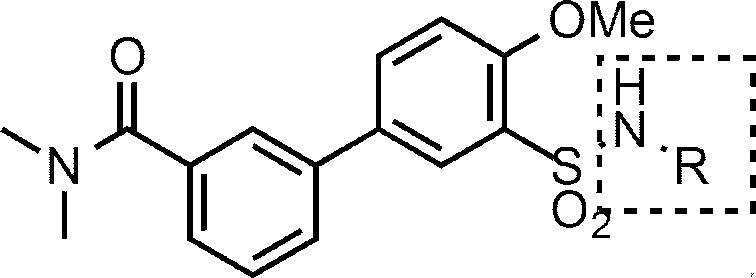



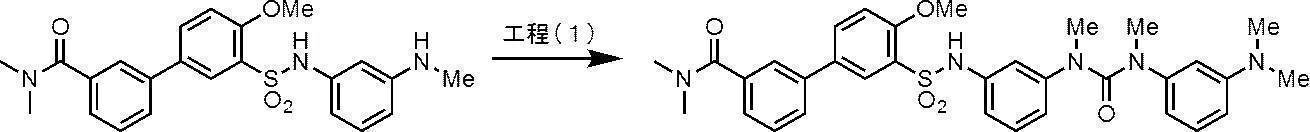











OX1RおよびOX2Rに対する作動活性評価
チャイニーズハムスター卵巣由来細胞株であるCHO細胞にNAFT-ルシフェラーゼ遺伝子およびヒトOX1R遺伝子を恒常的に発現させた細胞株(CHOOX1R)およびNAFT-ルシフェラーゼ遺伝子およびヒトOX2R遺伝子を恒常的に発現させた細胞株(CHOOX2R)を樹立した。それぞれの細胞を96ウェルマルチプレート中に10,000個/ウェルで播種し、5% FBS(サーモサイエンティフィック)添加DMEM培地(シグマアルドリッチ)で48時間培養した。培地を除去後、5μM Fura-2AM(ケイマンケミカル)を含むアッセイ用緩衝液(20mM HEPES(シグマアルドリッチ)、Hanks’ balanced salt solution(ギブコ)、0.1% BSA(シグマアルドリッチ)、2.5mM プロベネシド酸(和光純薬工業))を100μL添加し、60分間インキュベートした。Fura-2AMを含む緩衝液を除去した後、アッセイ用緩衝液75μLを添加した。そこに被験化合物を含むアッセイ用緩衝液25μLを添加し、反応を開始した。反応による細胞内カルシウムイオン濃度の変化は、FDSS7000(浜松ホトニクス)を用いて、340、380nmの二波長励起による蛍光強度比を測定することにより測定した。なお、被験化合物は10mMとなるようにDMSOに溶解し、最終濃度が10-7Mから10-5Mとなるようにアッセイ用緩衝液で希釈した(DMSOの最終濃度は1%)。表21~表27に各化合物の作動活性値を示した。




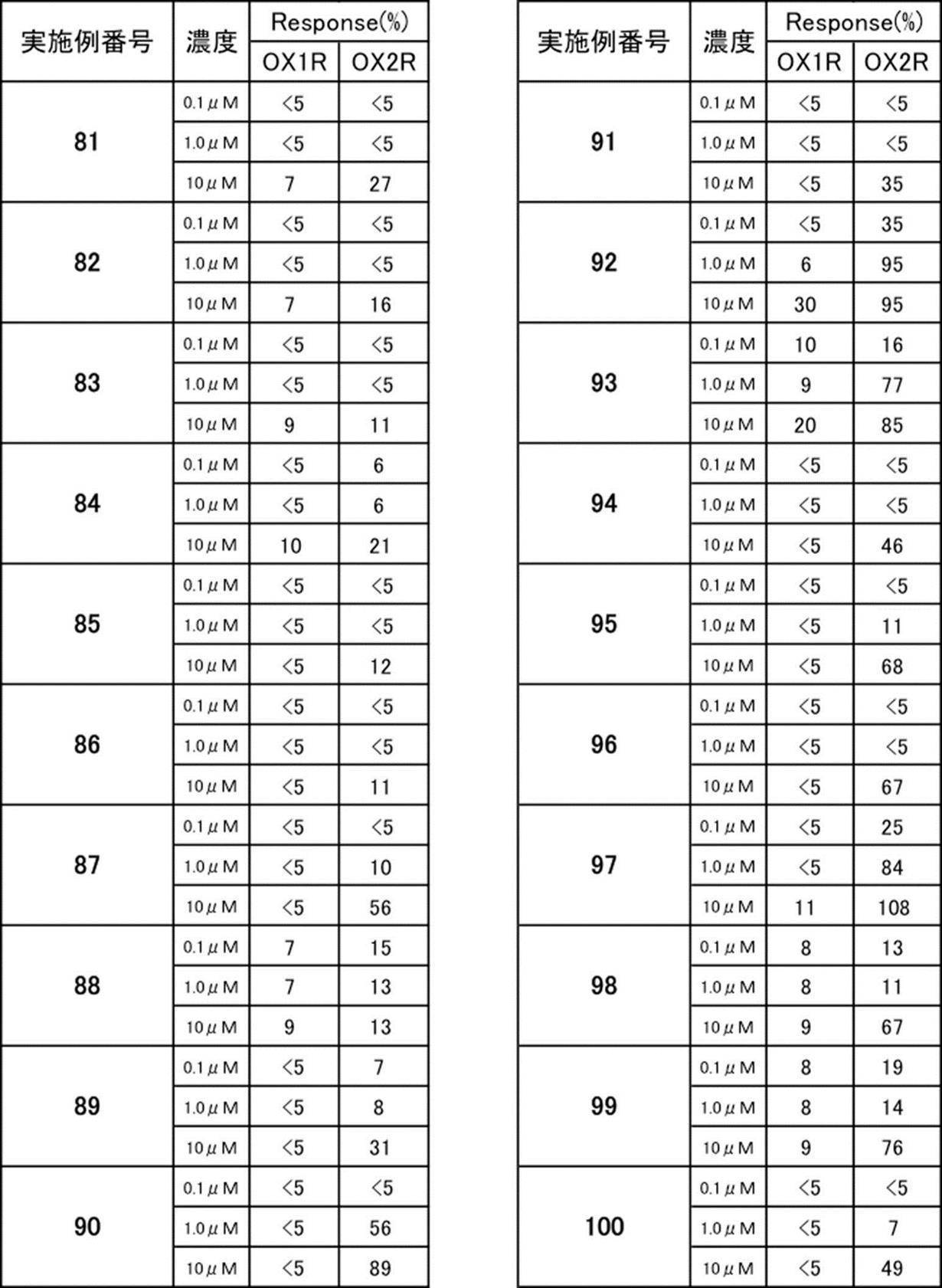

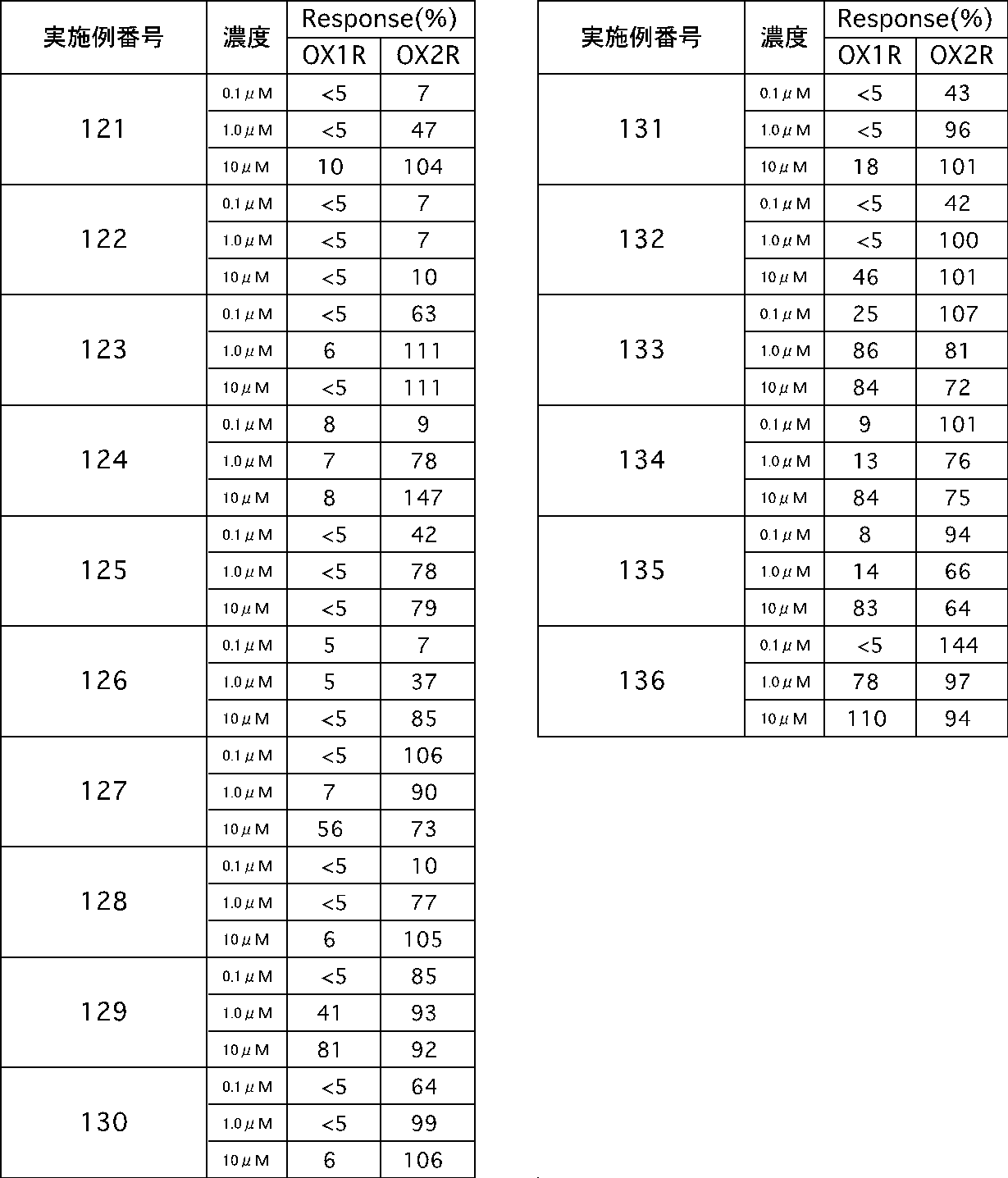
本発明化合物の野生型マウスへの明期脳室内投与による覚醒効果
実験動物は、C57BL/6系統の野生型マウス(雄)を用いた。9週齢前後(±1週間)で、イソフルラン麻酔下で、脳波用電極の頭蓋骨への埋め込み(Bregma:X=1.5;Y=0.6,Lambda:X=1.5;Y=0)、筋電図用電極の僧帽筋への挿入手術を行った。脳室内投与を行うマウスには側脳室へのカニューラ埋め込みも行った(Bregma:X=-0.9;Y=-0.3)。その2週間後から投与、脳波測定を開始した。実験は、明期の始まりを9時(ZT0)、暗期の始まりを21時(ZT12)とする明暗サイクル環境下で行った。
野生型マウスに対して、ZT6に、イソフルラン麻酔下で、マイクロシリンジポンプを用い被験化合物(実施例40の化合物)(10nmol;5%クロモフォア含有生理食塩水に溶解)5μlを0.5μl/minの流速でカニューラを通して側脳室に注入し、その後の脳波筋電図を測定した。投与はコントロールの5%クロモフォア含有生理食塩水(Vehicle)から始め、次に被験化合物の投与を行った。それぞれ、投与後一日を回復期間とした。結果を図1に示す。
マウスにとって睡眠期である明期(ZT6)に被験化合物の脳室内投与を行ったところ、野生型マウスで、Vehicle投与と比べて覚醒時間が増加した。
本出願は、日本国で2015年6月12日に出願された特願2015-119785を基礎としており、その内容は本明細書にすべて包含されるものである。
Claims (29)
- 式(I):

R1は水素原子、C1-6アルコキシ、-OH、C1-6アルキルまたはハロゲン原子であり、
Xは-C(R5)=または-N=であり、
R5は水素原子またはC1-6アルコキシであり、
R2、R3およびR4のいずれか1つはR6であり、残りの2つは、それぞれ水素原子であり、
R6は
(1)-NR17-Y1-R7
(ここで、Y1は-C(=O)NR18-、-C(=S)NH-、-C(=NH)NH-、-C(=O)O-、-C(=O)-、-SO2-または-SO2-NR8-であり、
R8は水素原子またはC1-6アルキルであり、
R17は水素原子またはC1-6アルキルであり、
R18は水素原子またはC1-6アルキルであり、
R17とR18は互いに結合し、それらが結合する窒素原子および隣接するC(=O)と一緒になって、5ないし7員複素環を形成してもよく、
R7は
(a)C6-10アリール、
(b)5ないし10員ヘテロアリール、
(c)C1-6アルキル、または
(d)C2-6アルケニル
(ここで、C1-6アルキルおよびC2-6アルケニルはフェニル、フリルおよびジフェニルメチルスルフィニルから選択される1個の置換基で置換されていてもよく、
C6-10アリールおよび5ないし10員ヘテロアリールは任意に選択される1ないし4個のR9で置換されていてもよく、
R9は、それぞれ独立して、
ハロゲン原子、
-NO2、
-OH、
C1-6アルキル、
C1-6ハロアルキル、
C1-6アルコキシ、
5ないし10員ヘテロアリール、
-NR9aR9b(ここで、R9aは水素原子、C1-6アルキルまたはC1-6アルコキシ-カルボニルであり、R9bは水素原子またはC1-6アルキルである)、
-C(=O)OR9c(ここで、R9cは水素原子またはC1-6アルキルである)、
-C(=O)NR9dR9e(ここで、R9dは水素原子またはC1-6アルキルであり、R9eは水素原子またはC1-6アルキルである)、または
-NH-C(=NR9f)-NHR9g(ここで、R9fは水素原子またはC1-6アルコキシ-カルボニルであり、R9gは水素原子またはC1-6アルコキシ-カルボニルである)であり、または
2個のR9は一緒になってメチレンジオキシを形成する)である)、
(2)-NH-Y2-R10
(ここで、Y2は-CH2-または単結合であり、
R10は
(a)C6-10アリール、または
(b)5ないし10員ヘテロアリール
(ここで、C6-10アリールおよび5ないし10員ヘテロアリールは任意に選択される1ないし4個のR9で置換されていてもよく、R9は前記で定義したとおりである)である)、
(3)式(ii):

(4)-N=N-R12
(ここで、R12は(a)-OHで置換されていてもよいC1-6アルキル、(b)-OH、(c)ジ(C1-6アルキル)アミノおよび(d)C1-6アルコキシ-カルボニルアミノから選択される1ないし3個の置換基で置換されていてもよいC6-10アリールである)、または
(5)-NR13R14
(ここで、R13は水素原子、C1-6アルキル、C6-10アリール-C1-6アルキルまたはC2-6アルケニル-カルボニルであり、
R14は水素原子またはC1-6アルキル(ここで、C1-6アルキルは(a)C1-6アルコキシ-カルボニルアミノ、(b)C2-6アルケニル-カルボニルアミノおよび(c)C1-6アルキルで置換されていてもよいC6-10アリール-C1-6アルキルアミノカルボニルから選択される1個の置換基で置換されていてもよい)であるか、またはR13とR14は互いに結合し、それらが結合する窒素原子と一緒になって、更に1個の窒素原子を含有する5ないし7員複素環(ここで、5ないし7員複素環はC1-6アルキルで置換されていてもよいC6-10アリール-カルボニルで置換されていてもよい)を形成する。
ただし、R14がC2-6アルケニル-カルボニルアミノで置換されたエチルであるとき、R13は水素原子ではない)であり、
Wは
(1)式(iii):

(2)-C(=O)NRWaRWb
(ここで、RWaはC1-6アルキル(ここで、C1-6アルキルはフェニル、ピリジル、C1-6アルコキシ-カルボニルアミノまたはジ(C1-6アルキル)アミノで置換されていてもよい)またはフェニル(ここで、フェニルはジ(C1-6アルキル)アミノで置換されていてもよい)であり、RWbは水素原子またはC1-6アルキルである)である]
で示される化合物またはその薬学的に許容される酸付加塩。 - R17およびR18がそれぞれ水素原子である請求項1記載の化合物またはその薬学的に許容される酸付加塩。
- 式(I-A):

- R17およびR18がそれぞれ水素原子である請求項3記載の化合物またはその薬学的に許容される酸付加塩。
- Xが-C(R5)=(ここで、R5は水素原子またはC1-6アルコキシである)である、請求項1~4のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩。
- R2がR6(ここで、R6は請求項1で定義したとおりである)であり、R3およびR4がそれぞれ水素原子である、請求項1~5のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩。
- R6が-NR17-Y1-R7(ここで、R17、Y1およびR7は請求項1で定義したとおりである)である、請求項1~6のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩。
- R6が-NH-Y2-R10(ここで、Y2およびR10は請求項1で定義したとおりである)である、請求項1~6のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩。
- R6が式(ii):
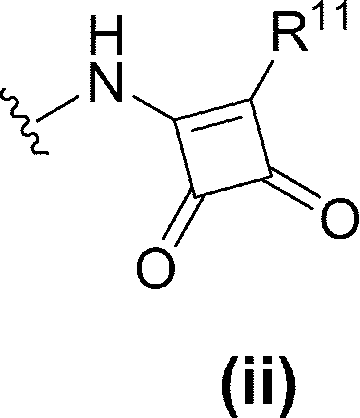
- R6が-N=N-R12(ここで、R12は請求項1で定義したとおりである)である、請求項1~6のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩。
- R6が-NR13R14(ここで、R13およびR14は請求項1で定義したとおりである)である、請求項1~6のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩。
- Wが式(iii):

Xが-C(R5)=(ここで、R5は水素原子またはC1-6アルコキシである)であり、
R2が-NH-Y1-R7(ここで、Y1およびR7は請求項1で定義したとおりである)であり、
R3およびR4がそれぞれ水素原子である、
請求項1記載の化合物またはその薬学的に許容される酸付加塩。 - Y1が-C(=O)NH-、-C(=S)NH-、-C(=NH)NH-または-C(=O)O-である、請求項12記載の化合物またはその薬学的に許容される酸付加塩。
- 式(I-B):

- 式(I’):

R1は水素原子、C1-6アルコキシ、-OH、C1-6アルキルまたはハロゲン原子であり、
Xは-C(R5)=または-N=であり、
R5は水素原子またはC1-6アルコキシであり、
R6aは置換されていてもよい炭化水素基、置換されていてもよい複素環基、酸素原子を介して結合する基、または硫黄原子を介して結合する基であり、
R15はC1-6アルキルであり、
R16はC1-6アルキルである]
で示される化合物またはその薬学的に許容される酸付加塩。 - 請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩を含有する医薬。
- 請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩を含有するオレキシン受容体作動薬。
- 請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩を含有する抗ナルコレプシー剤。
- 請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩を含有する眠気改善剤。
- 請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩を含有する、肥満症、糖尿病またはうつ病の予防剤または治療剤。
- 請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩を含有する、敗血症、重症敗血症または敗血症性ショックの予防剤または治療剤。
- 請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩の有効量を投与することを含むナルコレプシーの治療または予防方法。
- 請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩の有効量を投与することを含む眠気の改善方法。
- 請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩の有効量を投与することを含む、肥満症、糖尿病またはうつ病の治療または予防方法。
- 請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩の有効量を投与することを含む、敗血症、重症敗血症または敗血症性ショックの治療または予防方法。
- ナルコレプシーの治療または予防に使用するための、請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩。
- 眠気の改善に使用するための、請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩。
- 肥満症、糖尿病またはうつ病の治療または予防に使用するための、請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩。
- 敗血症、重症敗血症または敗血症性ショックの治療または予防に使用するための、請求項1~15のいずれか一項に記載の化合物またはその薬学的に許容される酸付加塩。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2989146A CA2989146A1 (en) | 2015-06-12 | 2016-06-10 | Sulfonamide derivative and pharmaceutically acceptable acid addition salt thereof |
| EP16807611.5A EP3309146A4 (en) | 2015-06-12 | 2016-06-10 | SULPHONAMID DERIVATIVE AND PHARMACEUTICALLY ACCEPTABLE ACID ADDITIVE SALT THEREOF |
| JP2017523722A JP6775803B2 (ja) | 2015-06-12 | 2016-06-10 | スルホンアミド誘導体またはその薬学的に許容される酸付加塩 |
| US15/735,835 US10351522B2 (en) | 2015-06-12 | 2016-06-10 | Sulfonamide derivative and pharmaceutically acceptable acid addition salt thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015-119785 | 2015-06-12 | ||
| JP2015119785 | 2015-06-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016199906A1 true WO2016199906A1 (ja) | 2016-12-15 |
Family
ID=57504026
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/067405 WO2016199906A1 (ja) | 2015-06-12 | 2016-06-10 | スルホンアミド誘導体またはその薬学的に許容される酸付加塩 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10351522B2 (ja) |
| EP (1) | EP3309146A4 (ja) |
| JP (1) | JP6775803B2 (ja) |
| CA (1) | CA2989146A1 (ja) |
| WO (1) | WO2016199906A1 (ja) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018164191A1 (ja) | 2017-03-08 | 2018-09-13 | 武田薬品工業株式会社 | 置換ピロリジン化合物およびその用途 |
| WO2018164192A1 (ja) | 2017-03-08 | 2018-09-13 | 武田薬品工業株式会社 | 置換ピロリジン化合物およびその用途 |
| WO2019027003A1 (ja) | 2017-08-03 | 2019-02-07 | 武田薬品工業株式会社 | 複素環化合物およびその用途 |
| WO2019027058A1 (en) | 2017-08-03 | 2019-02-07 | Takeda Pharmaceutical Company Limited | HETEROCYCLIC COMPOUND AND USE THEREOF |
| WO2019112007A1 (ja) * | 2017-12-07 | 2019-06-13 | 国立大学法人 筑波大学 | 鎮痛薬による眠気の予防または治療薬 |
| WO2019117148A1 (ja) * | 2017-12-12 | 2019-06-20 | 国立大学法人 筑波大学 | スルホンアミド誘導体またはその薬学的に許容される酸付加塩 |
| WO2020004537A1 (ja) | 2018-06-29 | 2020-01-02 | 武田薬品工業株式会社 | 複素環化合物およびその用途 |
| WO2020004536A1 (ja) | 2018-06-29 | 2020-01-02 | 武田薬品工業株式会社 | 複素環化合物およびその用途 |
| WO2021106975A1 (ja) * | 2019-11-27 | 2021-06-03 | 武田薬品工業株式会社 | 複素環化合物 |
| US11028048B2 (en) | 2019-01-31 | 2021-06-08 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| US12071434B2 (en) | 2018-12-12 | 2024-08-27 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
| US12091410B2 (en) | 2018-12-12 | 2024-09-17 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR112022009938A2 (pt) | 2019-11-25 | 2022-09-13 | Alkermes Inc | Compostos macrocíclicos substituídos e métodos de tratamento relacionados |
| US12006330B2 (en) | 2020-12-21 | 2024-06-11 | Alkermes, Inc. | Substituted macrocyclic compounds and related methods of treatment |
| US11760747B2 (en) | 2020-12-21 | 2023-09-19 | Alkermes, Inc. | Substituted piperidino compounds and related methods of treatment |
| US20240174605A1 (en) * | 2021-03-03 | 2024-05-30 | Research Triangle Institute | Arylsulfonamides as orexin receptor agonists |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000319249A (ja) * | 1999-03-20 | 2000-11-21 | Bayer Ag | 新規スルホンアミド類 |
| US6153585A (en) * | 1998-07-20 | 2000-11-28 | Tularik Inc. | Arylsulfonanilide derivatives |
| JP2005534684A (ja) * | 2002-07-30 | 2005-11-17 | シェーリング コーポレイション | Cxcケモカインレセプタ配位子としての3,4−二置換シクロブテン−1,2−ジオン |
| JP2006519785A (ja) * | 2003-02-20 | 2006-08-31 | エンサイシブ・ファーマシューティカルズ・インコーポレイテッド | フェニレンジアミンウロテンシン−ii受容体拮抗薬およびccr−9拮抗薬 |
| JP2007536348A (ja) * | 2004-05-05 | 2007-12-13 | ノボ ノルディスク アクティーゼルスカブ | スルホンアミド誘導体 |
| JP2009503106A (ja) * | 2005-08-04 | 2009-01-29 | メルク エンド カムパニー インコーポレーテッド | アミノエタンスルホンアミドオレキシン受容体アンタゴニスト |
| JP2009533419A (ja) * | 2006-04-11 | 2009-09-17 | アクテリオン ファーマシューティカルズ リミテッド | 新規スルホンアミド化合物 |
| JP2011510037A (ja) * | 2008-01-21 | 2011-03-31 | エフ.ホフマン−ラ ロシュ アーゲー | オレキシンアンタゴニストとしてのスルホンアミド |
| WO2014006402A1 (en) * | 2012-07-03 | 2014-01-09 | Heptares Therapeutics Limited | Orexin receptor antagonists |
| WO2015088000A1 (ja) * | 2013-12-12 | 2015-06-18 | 国立大学法人筑波大学 | スルホンアミド誘導体またはその薬学的に許容される酸付加塩 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040097547A1 (en) | 2001-04-16 | 2004-05-20 | Taveras Arthur G. | 3,4-Di-substituted cyclobutene-1,2-diones as CXC-chemokine receptor ligands |
| CA2580690A1 (en) * | 2004-10-21 | 2006-05-04 | Transtech Pharma, Inc. | Bissulfonamide compounds as agonists of galr1, compositions, and methods of use |
| US8258163B2 (en) | 2008-06-04 | 2012-09-04 | Board Of Regents, The University Of Texas System | Small-molecule agonists for type-2 orexin receptor |
| WO2016133160A1 (ja) * | 2015-02-19 | 2016-08-25 | 国立大学法人筑波大学 | スルホンアミド誘導体またはその薬学的に許容される酸付加塩 |
-
2016
- 2016-06-10 CA CA2989146A patent/CA2989146A1/en not_active Abandoned
- 2016-06-10 WO PCT/JP2016/067405 patent/WO2016199906A1/ja active Application Filing
- 2016-06-10 JP JP2017523722A patent/JP6775803B2/ja not_active Expired - Fee Related
- 2016-06-10 EP EP16807611.5A patent/EP3309146A4/en not_active Withdrawn
- 2016-06-10 US US15/735,835 patent/US10351522B2/en not_active Expired - Fee Related
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6153585A (en) * | 1998-07-20 | 2000-11-28 | Tularik Inc. | Arylsulfonanilide derivatives |
| JP2000319249A (ja) * | 1999-03-20 | 2000-11-21 | Bayer Ag | 新規スルホンアミド類 |
| JP2005534684A (ja) * | 2002-07-30 | 2005-11-17 | シェーリング コーポレイション | Cxcケモカインレセプタ配位子としての3,4−二置換シクロブテン−1,2−ジオン |
| JP2006519785A (ja) * | 2003-02-20 | 2006-08-31 | エンサイシブ・ファーマシューティカルズ・インコーポレイテッド | フェニレンジアミンウロテンシン−ii受容体拮抗薬およびccr−9拮抗薬 |
| JP2007536348A (ja) * | 2004-05-05 | 2007-12-13 | ノボ ノルディスク アクティーゼルスカブ | スルホンアミド誘導体 |
| JP2009503106A (ja) * | 2005-08-04 | 2009-01-29 | メルク エンド カムパニー インコーポレーテッド | アミノエタンスルホンアミドオレキシン受容体アンタゴニスト |
| JP2009533419A (ja) * | 2006-04-11 | 2009-09-17 | アクテリオン ファーマシューティカルズ リミテッド | 新規スルホンアミド化合物 |
| JP2011510037A (ja) * | 2008-01-21 | 2011-03-31 | エフ.ホフマン−ラ ロシュ アーゲー | オレキシンアンタゴニストとしてのスルホンアミド |
| WO2014006402A1 (en) * | 2012-07-03 | 2014-01-09 | Heptares Therapeutics Limited | Orexin receptor antagonists |
| WO2015088000A1 (ja) * | 2013-12-12 | 2015-06-18 | 国立大学法人筑波大学 | スルホンアミド誘導体またはその薬学的に許容される酸付加塩 |
Non-Patent Citations (2)
| Title |
|---|
| NAGAHARA T. ET AL.: "Design and synthesis of non-peptide, selective orexin receptor 2 agonists", J. MED. CHEM., vol. 58, no. 20, 2015, pages 7931 - 7937, XP055334462, [retrieved on 20150812] * |
| See also references of EP3309146A4 * |
Cited By (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7012703B2 (ja) | 2017-03-08 | 2022-02-14 | 武田薬品工業株式会社 | 置換ピロリジン化合物およびその用途 |
| WO2018164192A1 (ja) | 2017-03-08 | 2018-09-13 | 武田薬品工業株式会社 | 置換ピロリジン化合物およびその用途 |
| US11034700B2 (en) | 2017-03-08 | 2021-06-15 | Takeda Pharmaceutical Company Limited | Substituted pyrrolidine compound and use thereof |
| US11059780B2 (en) | 2017-03-08 | 2021-07-13 | Takeda Pharmaceutical Company Limited | Substituted pyrrolidine compound and use thereof |
| JPWO2018164191A1 (ja) * | 2017-03-08 | 2020-01-09 | 武田薬品工業株式会社 | 置換ピロリジン化合物およびその用途 |
| JPWO2018164192A1 (ja) * | 2017-03-08 | 2020-01-09 | 武田薬品工業株式会社 | 置換ピロリジン化合物およびその用途 |
| WO2018164191A1 (ja) | 2017-03-08 | 2018-09-13 | 武田薬品工業株式会社 | 置換ピロリジン化合物およびその用途 |
| KR20200035984A (ko) | 2017-08-03 | 2020-04-06 | 다케다 야쿠힌 고교 가부시키가이샤 | 헤테로시클릭 화합물 및 이의 용도 |
| US10428023B2 (en) | 2017-08-03 | 2019-10-01 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| US11319286B2 (en) | 2017-08-03 | 2022-05-03 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and application thereof |
| US10584097B2 (en) | 2017-08-03 | 2020-03-10 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| US11440883B2 (en) | 2017-08-03 | 2022-09-13 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| CN111051295A (zh) * | 2017-08-03 | 2020-04-21 | 武田药品工业株式会社 | 杂环化合物及其用途 |
| WO2019027058A1 (en) | 2017-08-03 | 2019-02-07 | Takeda Pharmaceutical Company Limited | HETEROCYCLIC COMPOUND AND USE THEREOF |
| CN111051295B (zh) * | 2017-08-03 | 2022-09-02 | 武田药品工业株式会社 | 杂环化合物及其用途 |
| WO2019027003A1 (ja) | 2017-08-03 | 2019-02-07 | 武田薬品工業株式会社 | 複素環化合物およびその用途 |
| WO2019112007A1 (ja) * | 2017-12-07 | 2019-06-13 | 国立大学法人 筑波大学 | 鎮痛薬による眠気の予防または治療薬 |
| WO2019117148A1 (ja) * | 2017-12-12 | 2019-06-20 | 国立大学法人 筑波大学 | スルホンアミド誘導体またはその薬学的に許容される酸付加塩 |
| JP7170996B2 (ja) | 2017-12-12 | 2022-11-15 | 国立大学法人 筑波大学 | スルホンアミド誘導体またはその薬学的に許容される酸付加塩 |
| EP3725780A4 (en) * | 2017-12-12 | 2021-06-02 | University of Tsukuba | SULFONAMIDE DERIVATIVE OR PHARMACEUTICAL ADDITIONAL ACID SALT |
| JPWO2019117148A1 (ja) * | 2017-12-12 | 2020-12-03 | 国立大学法人 筑波大学 | スルホンアミド誘導体またはその薬学的に許容される酸付加塩 |
| US11673867B2 (en) | 2017-12-12 | 2023-06-13 | University Of Tsukuba | Sulfonamide derivative or pharmaceutically acceptable acid-addition salt |
| WO2020004536A1 (ja) | 2018-06-29 | 2020-01-02 | 武田薬品工業株式会社 | 複素環化合物およびその用途 |
| JPWO2020004536A1 (ja) * | 2018-06-29 | 2021-07-08 | 武田薬品工業株式会社 | 複素環化合物およびその用途 |
| US11655241B2 (en) | 2018-06-29 | 2023-05-23 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| WO2020004537A1 (ja) | 2018-06-29 | 2020-01-02 | 武田薬品工業株式会社 | 複素環化合物およびその用途 |
| JP7339250B2 (ja) | 2018-06-29 | 2023-09-05 | 武田薬品工業株式会社 | 複素環化合物およびその用途 |
| US12077522B2 (en) | 2018-06-29 | 2024-09-03 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and application thereof |
| US12071434B2 (en) | 2018-12-12 | 2024-08-27 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
| US12091410B2 (en) | 2018-12-12 | 2024-09-17 | Takeda Pharmaceutical Company Limited | Heterocyclic compound |
| US11028048B2 (en) | 2019-01-31 | 2021-06-08 | Takeda Pharmaceutical Company Limited | Heterocyclic compound and use thereof |
| WO2021106975A1 (ja) * | 2019-11-27 | 2021-06-03 | 武田薬品工業株式会社 | 複素環化合物 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6775803B2 (ja) | 2020-10-28 |
| JPWO2016199906A1 (ja) | 2018-06-28 |
| EP3309146A1 (en) | 2018-04-18 |
| CA2989146A1 (en) | 2016-12-15 |
| US10351522B2 (en) | 2019-07-16 |
| EP3309146A4 (en) | 2019-02-27 |
| US20180179151A1 (en) | 2018-06-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2016199906A1 (ja) | スルホンアミド誘導体またはその薬学的に許容される酸付加塩 | |
| ES2337723T3 (es) | Compuesto pentafluorosulfanil sustituido y su utilizacion para la produccion de medicamentos. | |
| JP6440321B2 (ja) | スルホンアミド誘導体またはその薬学的に許容される酸付加塩 | |
| WO2016133160A1 (ja) | スルホンアミド誘導体またはその薬学的に許容される酸付加塩 | |
| KR100468520B1 (ko) | 신규 아마이드계 화합물 및 이를 함유하는 약제학적 조성물 | |
| US7550481B2 (en) | Phenylamide and pyridylamide beta-secretase inhibitors for the treatment of Alzheimer's disease | |
| TWI491595B (zh) | 作為香草類化合物受體之配體之經取代苯基尿素及苯基醯胺 | |
| AU2004293416B2 (en) | Benzylether and benzylamino beta-secretase inhibitors for the treatment of Alzheimer's disease | |
| EP1080069B1 (en) | Anthranilic acid derivatives as inhibitors of the cgmp-phosphodiesterase | |
| BR112017018832B1 (pt) | Profármacos de riluzol e seu uso e composição | |
| EP1322628A2 (en) | Substituted urea neuropeptide y y5 receptor antagonists | |
| CA2681118A1 (en) | Amide derivatives as calcium channel blockers | |
| BRPI0307351B1 (pt) | composto, composição farmacêutica, uso de um composto, e, processo para preparar um composto | |
| EP2054383A2 (en) | Novel compounds as antagonists or inverse agonists at opioid receptors | |
| Gilbert et al. | Design and SAR of novel potassium channel openers targeted for urge urinary incontinence. 2. Selective and potent benzylamino cyclobutenediones | |
| JP2007511612A (ja) | 減少された生体蓄積を有するキナゾリノン化合物 | |
| KR100759744B1 (ko) | 약제로서 아릴렌-카르복시산(2-아미노-페닐)-아미드 유도체 | |
| US20040063643A1 (en) | Bombesin antagonists | |
| JP2007536320A (ja) | シクロヘキシル−1,4−ジアミンの酸誘導体 | |
| EP1560809A1 (de) | Cyclohexyl-harnstoff-derivate | |
| US20090215758A1 (en) | Use of 2,5-Disubstituted Thiazol-4-One Derivatives in Drugs | |
| WO2007080109A1 (en) | Substituded benzyloxy-phenylmethylurea derivatives | |
| Leclerc et al. | Synthesis and structure–activity relationships of novel naphthalenic and bioisosteric related amidic derivatives as melatonin receptor ligands | |
| JP2007536322A (ja) | 鎖長が延ばされ、置換されたシクロヘキシル−1,4−ジアミンの酸誘導体 | |
| JP2005519859A (ja) | N−[(1−ジメチルアミノシクロアルキル)メチル]ベンズアミド誘導体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16807611 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017523722 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2989146 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15735835 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2016807611 Country of ref document: EP |