WO2016159223A1 - チキソトロピー性に優れる硬化性エポキシ樹脂組成物 - Google Patents
チキソトロピー性に優れる硬化性エポキシ樹脂組成物 Download PDFInfo
- Publication number
- WO2016159223A1 WO2016159223A1 PCT/JP2016/060637 JP2016060637W WO2016159223A1 WO 2016159223 A1 WO2016159223 A1 WO 2016159223A1 JP 2016060637 W JP2016060637 W JP 2016060637W WO 2016159223 A1 WO2016159223 A1 WO 2016159223A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mass
- epoxy resin
- resin composition
- core
- monomer
- Prior art date
Links
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L51/00—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers
- C08L51/04—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers grafted on to rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L71/00—Compositions of polyethers obtained by reactions forming an ether link in the main chain; Compositions of derivatives of such polymers
- C08L71/02—Polyalkylene oxides
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J151/00—Adhesives based on graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Adhesives based on derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J163/00—Adhesives based on epoxy resins; Adhesives based on derivatives of epoxy resins
- C09J163/10—Epoxy resins modified by unsaturated compounds
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J175/00—Adhesives based on polyureas or polyurethanes; Adhesives based on derivatives of such polymers
- C09J175/04—Polyurethanes
Definitions
- the present invention relates to a curable epoxy resin composition containing a core-shell polymer, its adhesive, particularly a structural adhesive for vehicles.
- Epoxy resins are used in a wide range of applications, one of which is adhesive. Epoxy resin cured products exhibit excellent mechanical properties, electrical properties, and durability, while curable epoxy resin compositions are used in structural adhesives such as vehicles to improve brittle properties and impact resistance. In some cases, the product contains a core-shell polymer and a blocked isocyanate (Patent Document 1).
- the structural adhesive is also important for handling in the production line and not easily washed off in the shower process existing between the coating process and the curing process (Patent Document 2).
- the composition may partially dissolve, scatter, or deform due to water pressure, leading to deterioration of the application part.
- there is an increase in the viscosity of the curable epoxy resin composition but there is a problem that the coating workability is lowered.
- the curable epoxy resin composition has a so-called thixotropic property that the viscosity at low shear is higher than the viscosity at high shear.
- Patent Documents 1 and 2 do not teach any means for improving thixotropy.
- the present inventors have found that the thixotropic property of the curable epoxy resin composition varies depending on the shell layer component of the core-shell polymer and the amount thereof, and intensively investigate the shell layer of the core-shell polymer. I made it.
- the present invention aims to provide a curable epoxy resin composition that is excellent in thixotropy and further excellent in adhesiveness of the resulting cured product.
- the inventors of the present invention when the shell layer of the core-shell polymer is composed of a specific monomer and a specific amount, the thixotropy of the composition and adhesion of the cured product As a result, the present invention was completed.
- the gist of the present invention is as follows.
- the core layer of (B) is made of any of diene rubber, acrylic rubber, and polysiloxane rubber
- the shell layer of the core-shell polymer (B) is 100% by mass of the total amount of monomers constituting the shell layer, vinyl 10 to 95% by weight of a monomer selected from the group consisting of a cyan monomer and an alkoxyalkyl (meth) acrylate monomer, 0 to 50% by weight of a monomer having an epoxy group, and 0 to 90% by weight of a monomer copolymerizable therewith.
- the shell layer of the core-shell polymer (B) is such that the total amount of monomers constituting the shell layer is 100% by mass, the vinylcyan monomer is 20 to 40% by mass, the monomer having an epoxy group is 3 to 15% by mass, and copolymerized therewith
- the curable epoxy resin composition according to 1) or 60) comprising 60 to 80% by mass of a possible monomer.
- the total amount of monomers constituting the shell layer is 100% by mass
- the alkoxyalkyl (meth) acrylate monomer is 50 to 95% by mass
- the epoxy group-containing monomer is 5 to 50% by mass.
- the curable epoxy resin composition as described in 1).
- the total amount of monomers constituting the shell layer is 100% by mass, the alkoxyalkyl (meth) acrylate monomer is 90 to 95% by mass, and the epoxy group-containing monomer is 5 to 10% by mass.
- a structural adhesive for vehicles comprising the curable epoxy resin composition according to any one of 1) to 14).
- the curable epoxy resin composition of the present invention is excellent in thixotropy and adhesion to a cured product.
- FIG. 1 shows the relationship between the amount of a monomer having an epoxy group of a shell layer of a core-shell polymer, storage stability, and thixotropy in a curable epoxy resin composition of the present invention using a blocked isocyanate having a polypropylene glycol structure.
- FIG. FIG. 2 shows a T-peel bond strength (indicated by T-Peel in the figure) and a shear bond strength (FIG. 2) in the curable epoxy resin composition of the present invention using a blocked isocyanate having a polypropylene glycol structure. It is a figure which shows the relationship between the monomer amount which has an epoxy group of the shell layer of a core-shell polymer.
- FIG. 2 shows a T-peel bond strength (indicated by T-Peel in the figure) and a shear bond strength (FIG. 2) in the curable epoxy resin composition of the present invention using a blocked isocyanate having a polypropylene glycol structure. It is
- FIG. 3 shows the dynamic splitting resistance (indicated as Impact Peel in the figure) and the epoxy group of the shell layer of the core-shell polymer in the curable epoxy resin composition of the present invention using a blocked isocyanate having a polypropylene glycol structure. It is a figure which shows the relationship with the monomer amount which has.
- FIG. 4 shows the relationship between the amount of monomers having an epoxy group in the shell layer of the core-shell polymer, storage stability, and thixotropy in the curable epoxy resin composition of the present invention using a blocked isocyanate having a polytetramethylene glycol structure.
- FIG. 5 shows T-peel adhesive strength (indicated as T-Peel in the figure) and shear adhesive strength in the curable epoxy resin composition of the present invention using a blocked isocyanate having a polytetramethylene glycol structure. It is a figure which shows the relationship between (it shows with Lap Shear in a figure) and the monomer amount which has the epoxy group of the shell layer of a core-shell polymer.
- FIG. 6 shows the dynamic splitting resistance (indicated by “Impact Peel” in the figure) and the shell layer of the core-shell polymer in the curable epoxy resin composition of the present invention using a blocked isocyanate having a polytetramethylene glycol structure. It is a figure which shows the relationship with the monomer amount which has an epoxy group.
- the curable epoxy resin composition of the present invention contains an epoxy resin (A), a core-shell polymer (B), and a blocked isocyanate (C) as essential components.
- Epoxy resin (A)> (A-1) Polyglycidyl ether, polyvalent glycidylamine compound, alicyclic epoxy resin, etc.
- the epoxy resin (A) of the present invention is not particularly limited as long as it is a compound having an epoxy group, but the epoxy used in the present invention
- the resin is preferably an epoxy resin also called polyepoxide.
- the epoxy resin include polyglycidyl ethers (bisphenol A type epoxy resin, bisphenol F type epoxy resin, biphenol type) such as addition reaction products of polychlorophenol and epichlorohydrin such as bisphenol A, bisphenol F, biphenol, and phenol novolak.
- polyepoxides synthesized from polyhydric phenols are disclosed, for example, in US Pat. No. 4,431,782.
- polyepoxides further include those disclosed in US Pat. Nos. 3,804,735, 3,892,819, 3,948,698, 4,147,771, and an epoxy resin handbook (Nikkan Kogyo Shimbun, 1987). Can be mentioned.
- the epoxy resin (A-2) Polyalkylene glycol diglycidyl ether, glycol diglycidyl ether, etc.
- the epoxy resin (A) of the present invention includes polyalkylene glycol diglycidyl ether, glycol diglycidyl ether, diglycidyl of aliphatic polybasic acid. Esters, glycidyl ethers of dihydric or higher polyhydric aliphatic alcohols, and divinylbenzene dioxide can also be used. These are epoxy resins having a relatively low viscosity, and when used in combination with other epoxy resins such as bisphenol A type epoxy resins and bisphenol F type epoxy resins, they function as reactive diluents. The balance of physical properties can be improved.
- the content of these epoxy resins is preferably 0.5 to 20% by mass, more preferably 1 to 10% by mass, and further preferably 2 to 5% by mass in 100% by mass of the epoxy resin (A) component.
- More specific examples of the polyalkylene glycol diglycidyl ether include polyethylene glycol diglycidyl ether and polypropylene glycol diglycidyl ether. More specific examples of the glycol diglycidyl ether include neopentyl glycol diglycidyl ether, 1,4-butanediol diglycidyl ether, 1,6-hexanediol diglycidyl ether, cyclohexanedimethanol diglycidyl ether, and the like. It is done.
- the diglycidyl ester of the aliphatic polybasic acid include dimer acid diglycidyl ester, adipic acid diglycidyl ester, sebacic acid diglycidyl ester, and maleic acid diglycidyl ester.
- the glycidyl ether of the dihydric or higher polyhydric aliphatic alcohol includes trimethylolpropane triglycidyl ether, trimethylolethane triglycidyl ether, castor oil-modified polyglycidyl ether, propoxylated glycerin triglycidyl ether, sorbitol. And polyglycidyl ether.
- epoxy resin (A-3) Epoxy compound obtained by addition reaction of polybasic acids and the like to epoxy resin
- an epoxy resin as described in WO2010 / 098950 is used as the epoxy resin (A) of the present invention.
- Epoxy compounds obtained by addition reaction of polybasic acids and the like can also be used.
- an addition reaction product of a dimer of tall oil fatty acid (dimer acid) and a bisphenol A type epoxy resin can be mentioned.
- Chelate-modified epoxy resins, rubber-modified epoxy resins, and urethane-modified epoxy resins can also be used as the epoxy resin (A) component.
- the epoxy resin that can be used in the present invention is as described above, but generally, an epoxy resin having an epoxy equivalent of 80 to 2000 is exemplified.
- These polyepoxides can be obtained by a well-known method. As a commonly used method, for example, an excess amount of epihalohydrin is reacted with a polyhydric alcohol or polyhydric phenol in the presence of a base.
- a chelate-modified epoxy resin can be used as the epoxy resin (A) of the present invention.
- the chelate-modified epoxy resin is a reaction product of an epoxy resin and a compound containing a chelate functional group (chelate ligand).
- chelate ligand When added to the resin composition of the present invention and used as an adhesive for vehicles, an oily substance The adhesion to the metal substrate surface contaminated with can be improved.
- the chelate functional group is a functional group of a compound having in its molecule a plurality of coordination sites capable of coordinating to metal ions.
- a phosphorus-containing acid group for example, —PO (OH) 2
- a carboxylic acid group for example, — CO 2 H
- sulfur-containing acid groups for example, —SO 3 H
- amino groups and hydroxyl groups particularly, hydroxyl groups adjacent to each other in the aromatic ring.
- chelate ligands include ethylenediamine, bipyridine, ethylenediaminetetraacetic acid, phenanthroline, porphyrin, and crown ether.
- Examples of commercially available chelate-modified epoxy resins include Adeka Resin EP-49-10N manufactured by ADEKA.
- the amount of the chelate-modified epoxy resin used in 100% by mass of the epoxy resin (A) component is preferably 0.1 to 10% by mass, more preferably 0.5 to 3% by mass.
- the rubber-modified epoxy resin is a reaction product obtained by reacting rubber and an epoxy group-containing compound and having an average of 1.1 or more epoxy groups per molecule.
- rubber acrylonitrile butadiene rubber (NBR), styrene butadiene rubber (SBR), hydrogenated nitrile rubber (HNBR), ethylene propylene rubber (EPDM), acrylic rubber (ACM), butyl rubber (IIR), butadiene rubber
- rubber polymers such as polyoxyalkylenes such as polypropylene oxide, polyethylene oxide and polytetramethylene oxide.
- the rubber polymer is preferably one having a reactive group such as an amino group, a hydroxy group, or a carboxyl group at the terminal.
- the rubber-modified epoxy resin used in the present invention may be a product obtained by reacting these rubber-based polymer and epoxy resin at an appropriate blending ratio by a known method.
- acrylonitrile-butadiene rubber-modified epoxy resin and polyoxyalkylene-modified epoxy resin are preferable from the viewpoint of adhesion of the resulting resin composition and impact peel resistance, and acrylonitrile-butadiene rubber-modified epoxy resin is more preferable.
- the acrylonitrile-butadiene rubber-modified epoxy resin may be obtained, for example, by a reaction between a carboxyl group-terminated NBR (CTBN) and a bisphenol A type epoxy resin.
- CBN carboxyl group-terminated NBR
- the polyoxyalkylene-modified epoxy resin may be obtained, for example, by a reaction between an amino group-terminated polyoxyalkylene and a bisphenol A type epoxy resin.
- the content of the acrylonitrile monomer component in 100% by mass of the acrylonitrile-butadiene rubber is preferably 5 to 40% by mass from the viewpoint of the adhesiveness and impact peel resistance of the resulting resin composition. % Is more preferable, and 15 to 30% by mass is even more preferable. From the viewpoint of thixotropy of the obtained resin composition, 20 to 30% by mass is particularly preferable.
- the average number of epoxide reactive end groups per molecule in the rubber is preferably 1.5 to 2.5, more preferably 1.8 to 2.2.
- the number average molecular weight of the rubber is preferably 2,000 to 10,000, more preferably 3,000 to 8,000, and particularly preferably 4,000 to 6,000 in terms of polystyrene measured by GPC.
- the production method of the rubber-modified epoxy resin is not particularly limited, and can be produced, for example, by reacting rubber and an epoxy group-containing compound in a large amount of an epoxy group-containing compound. Specifically, it is preferable to produce by reacting 2 equivalents or more of an epoxy group-containing compound with respect to 1 equivalent of an epoxy-reactive terminal group in the rubber. It is more preferable to react an amount of the epoxy group-containing compound sufficient for the resulting product to be a mixture of an adduct of rubber and an epoxy group-containing compound and a free epoxy group-containing compound.
- the rubber-modified epoxy resin may be produced by heating to a temperature of 100 to 250 ° C.
- the epoxy group-containing compound used in producing the rubber-modified epoxy resin is not particularly limited, but bisphenol A type epoxy resin and bisphenol F type epoxy resin are preferable, and bisphenol A type epoxy resin is more preferable.
- the unreacted epoxy group-containing compound remaining after the reaction is included in the rubber-modified epoxy resin of the present application, It shall not be included.
- the rubber-modified epoxy resin can be modified by pre-reaction with a bisphenol component.
- the bisphenol component used for the modification is preferably 3 to 35 parts by mass, and more preferably 5 to 25 parts by mass with respect to 100 parts by mass of the rubber component in the rubber-modified epoxy resin.
- a cured product obtained by curing a resin composition containing a modified rubber-modified epoxy resin is excellent in adhesion durability after high-temperature exposure and excellent in impact resistance at low temperatures.
- the glass transition temperature (Tg) of the rubber-modified epoxy resin is not particularly limited, but is preferably ⁇ 25 ° C. or lower, more preferably ⁇ 35 ° C. or lower, still more preferably ⁇ 40 ° C. or lower, and particularly preferably ⁇ 50 ° C. or lower.
- the content of the rubber-modified epoxy resin in 100% by mass of the epoxy resin (A) component is preferably 1 to 40% by mass, more preferably 3 to 30% by mass, further preferably 5 to 25% by mass, and 10 to 20% by mass. % Is particularly preferred. If it is less than 1% by mass, the resulting cured product may be brittle and impact-resistant peel adhesion may be low, and if it exceeds 40% by mass, the resulting cured product may have low heat resistance and / or elastic modulus (rigidity).
- the rubber-modified epoxy resins can be used alone or in combination of two or more.
- the urethane-modified epoxy resin is obtained by reacting a compound having reactivity with an isocyanate group and an epoxy group with a urethane prepolymer containing an isocyanate group. Further, it is a reaction product having an average of 1.1 or more epoxy groups per molecule.
- a urethane-modified epoxy resin can be obtained by reacting a hydroxy group-containing epoxy compound with a urethane prepolymer.
- the content of the urethane-modified epoxy resin in 100% by mass of the epoxy resin (A) component is preferably 1 to 40% by mass, more preferably 3 to 30% by mass, further preferably 5 to 25% by mass, and 10 to 20% by mass. % Is particularly preferred. If it is less than 1% by mass, the resulting cured product may be brittle and impact-resistant peel adhesion may be low, and if it exceeds 40% by mass, the resulting cured product may have low heat resistance and / or elastic modulus (rigidity).
- Urethane-modified epoxy resins can be used alone or in combination of two or more.
- epoxy resins (A) bisphenol A type epoxy resins and bisphenol F type epoxy resins are preferable because the obtained cured products have high elastic modulus, excellent heat resistance and adhesiveness, and are relatively inexpensive.
- a type epoxy resin is particularly preferred.
- the epoxy equivalent of the epoxy resin (A) of the present invention is preferably less than 220, more preferably 90 or more and 210 or less, and further preferably 150 or more and 200 or less, from the viewpoint that the obtained cured product has high elastic modulus and heat resistance.
- a bisphenol A type epoxy resin or a bisphenol F type epoxy resin having an epoxy equivalent of less than 220 is preferable because it is liquid at room temperature and the handleability of the resulting resin composition is good.
- a bisphenol A type epoxy resin or a bisphenol F type epoxy resin having an epoxy equivalent of 220 or more and less than 2000 is preferably 40% by mass or less, more preferably 20% by mass or less in 100% by mass of the epoxy resin (A) component.
- the resulting cured product is preferable because of its excellent impact resistance.
- the core-shell polymer (B) of the present invention comprises at least two layers of a core layer and a shell layer. Moreover, the core-shell polymer (B) of the present invention may be composed of three layers of a core layer, an intermediate layer, and a shell layer.
- the volume average particle size of the core-shell polymer (B) of the present invention is preferably 0.03 to 2.10 ⁇ m, more preferably 0.05 to 1.10 ⁇ m, still more preferably 0.05 to 0.30 ⁇ m, and 0.08. Particularly preferred is .about.0.30 .mu.m. In many cases, it is difficult to stably obtain a volume average particle size of less than 0.03 ⁇ m, and when it exceeds 2.10 ⁇ m, the heat resistance and impact resistance of the final molded product may be deteriorated.
- the core layer uses any of diene rubber, acrylic rubber, and polysiloxane rubber.
- the core layer of the core-shell polymer (B) of the present invention contains 50 to 100 mass of at least one monomer (first monomer) selected from natural rubber, diene monomer (conjugated diene monomer) and (meth) acrylate monomer. %, And a rubber elastic body containing 0 to 50% by mass of another copolymerizable vinyl monomer (second monomer), a polysiloxane rubber elastic body, or a combination thereof.
- the core layer of the core-shell polymer (B) has a high toughness-improving effect and an impact-removing adhesiveness-improving effect of the resulting cured product, and due to the low affinity with the matrix resin, A diene rubber using a diene monomer is preferred from the viewpoint that viscosity increase is unlikely to occur. Since a wide variety of polymer designs are possible by combining various monomers, (meth) acrylate rubber is preferred. Moreover, when it is going to improve the impact resistance in low temperature, without reducing the heat resistance of hardened
- diene monomer (conjugated diene monomer) of the first monomer examples include 1,3-butadiene, isoprene, 2-chloro-1,3-butadiene, 2-methyl-1,3-butadiene, and the like. . These diene monomers may be used alone or in combination of two or more.
- the core layer of the core-shell polymer (B) has a high toughness-improving effect and impact-removing adhesiveness-improving effect, and the low affinity with the matrix resin makes it difficult for the viscosity to increase with time due to swelling of the core layer.
- butadiene rubber using 1,3-butadiene and / or butadiene-styrene rubber which is a copolymer of 1,3-butadiene and styrene is preferable, and butadiene rubber is more preferable.
- butadiene-styrene rubber is more preferable because it can increase the transparency of the cured product obtained by adjusting the refractive index.
- Examples of the (meth) acrylate rubber monomer as the first monomer include methyl (meth) acrylate, ethyl (meth) acrylate, butyl (meth) acrylate, 2-ethylhexyl (meth) acrylate, and octyl (meth) acrylate.
- Alkyl (meth) acrylates such as dodecyl (meth) acrylate, stearyl (meth) acrylate, and behenyl (meth) acrylate; aromatic ring-containing (meth) acrylates such as phenoxyethyl (meth) acrylate and benzyl (meth) acrylate; 2 -Hydroxyalkyl (meth) acrylates such as hydroxyethyl (meth) acrylate and 4-hydroxybutyl (meth) acrylate; glycidyl (meth) acrylate, glycidylalkyl (meth) acrylate, etc.
- Glycidyl (meth) acrylates alkoxyalkyl (meth) acrylates; allylalkyl (meth) acrylates such as allyl (meth) acrylate and allylalkyl (meth) acrylate; monoethylene glycol di (meth) acrylate, triethylene glycol di
- Polyfunctional (meth) acrylates such as (meth) acrylate and tetraethylene glycol di (meth) acrylate are exemplified.
- These (meth) acrylate monomers may be used alone or in combination of two or more. Particularly preferred are ethyl (meth) acrylate, butyl (meth) acrylate, and 2-ethylhexyl (meth) acrylate.
- Examples of the other copolymerizable vinyl monomer (second monomer) include, for example, vinyl arenes such as styrene, ⁇ -methyl styrene, monochlorostyrene, dichlorostyrene; vinyl carboxylic acids such as acrylic acid and methacrylic acid; acrylonitrile Vinyl cyanides such as methacrylonitrile; vinyl halides such as vinyl chloride, vinyl bromide, chloroprene; vinyl acetate; alkenes such as ethylene, propylene, butylene, isobutylene; diallyl phthalate, triallyl cyanurate, triallyl And polyfunctional monomers such as isocyanurate and divinylbenzene. These vinyl monomers may be used alone or in combination of two or more. Particularly preferred is styrene.
- the polysiloxane rubber is composed of alkyl or aryl disubstituted silyloxy units such as dimethylsilyloxy, diethylsilyloxy, methylphenylsilyloxy, diphenylsilyloxy, dimethylsilyloxy-diphenylsilyloxy, and the like.
- Examples thereof include polysiloxane polymers and polysiloxane polymers composed of alkyl or aryl 1-substituted silyloxy units such as organohydrogensilyloxy in which a part of the alkyl in the side chain is substituted with a hydrogen atom. These polysiloxane polymers may be used alone or in combination of two or more.
- the polysiloxane polymer portion is 80% by mass or more (more preferably 90%) based on 100% by mass of the entire rubber elastic body in order not to impair the heat resistance of the cured product. (Mass% or more) is preferably contained.
- the core layer of the core-shell polymer (B) preferably has a cross-linked structure introduced from the viewpoint of maintaining dispersion stability in the curable epoxy resin composition.
- a method for introducing a crosslinked structure include a method in which a crosslinkable monomer such as a polyfunctional monomer or a mercapto group-containing compound is added to a polymer component and then polymerized.
- a method for introducing a crosslinked structure into the polysiloxane polymer a method in which a polyfunctional alkoxysilane compound is partially used at the time of polymerization, or a reactive group such as a vinyl reactive group or a mercapto group is added to the polysiloxane polymer. And then adding a vinyl polymerizable monomer or an organic peroxide to cause a radical reaction, or adding a crosslinkable monomer such as a polyfunctional monomer or a mercapto group-containing compound to the polysiloxane polymer, Next, a polymerization method and the like can be mentioned.
- Multifunctional monomer examples include allyl alkyl (meth) acrylates such as allyl (meth) acrylate and allylalkyl (meth) acrylate; allyloxyalkyl (meth) acrylates; (poly) ethylene glycol di (meth) acrylate, Polyfunctional (meth) having two or more (meth) acrylic groups such as butanediol di (meth) acrylate, ethylene glycol di (meth) acrylate, triethylene glycol di (meth) acrylate, tetraethylene glycol di (meth) acrylate Acrylates such as diallyl phthalate, triallyl cyanurate, triallyl isocyanurate, divinylbenzene and the like. Particularly preferred are allyl methacrylate, triallyl isocyanurate, butanediol di (meth) acrylate, and divinylbenzene.
- the core layer of the core-shell polymer (B) of the present invention preferably has rubber properties in order to increase the toughness of the cured product of the curable epoxy resin composition.
- the gel content of the core layer of the core-shell polymer (B) of the present invention is preferably 60% by mass or more, more preferably 80% by mass or more, and 90% by mass or more. More preferably, it is particularly preferably 95% by mass or more.
- Gel content refers to insoluble and soluble when 0.5 g of core-shell polymer obtained by coagulation and drying is immersed in 100 g of toluene and allowed to stand at 23 ° C. for 24 hours, and then insoluble and soluble are separated. It means the ratio of insoluble matter to the total amount of minutes.
- the glass transition temperature of the core layer of the core-shell polymer (B) is preferably 0 ° C. or lower in order to increase the toughness of the resulting cured product.
- -20 ° C or lower is more preferable, -40 ° C or lower is further preferable, and -60 ° C or lower is particularly preferable.
- the Tg of the core layer is preferably greater than 0 ° C, more preferably 20 ° C or more, and even more preferably 50 ° C or more. 80 ° C. or higher is particularly preferable, and 120 ° C. or higher is most preferable.
- At least one monomer having a Tg of a homopolymer of greater than 0 ° C. is 50 to 100 masses. % (Preferably 65 to 99% by mass) and a polymer comprising 0 to 50% by mass (preferably 1 to 35% by mass) of at least one monomer having a homopolymer Tg of 0 ° C. or less. Is mentioned.
- Monomers having a homopolymer Tg greater than 0 ° C. include, for example, unsubstituted vinyl aromatic compounds such as styrene and 2-vinylnaphthalene; vinyl-substituted aromatic compounds such as ⁇ -methylstyrene; 3-methylstyrene, Ring alkylated vinyl aromatic compounds such as 4-methylstyrene, 2,4-dimethylstyrene, 2,5-dimethylstyrene, 3,5-dimethylstyrene, 2,4,6-trimethylstyrene; 4-methoxystyrene, Ring alkoxylated vinyl aromatic compounds such as 4-ethoxystyrene; ring halogenated vinyl aromatic compounds such as 2-chlorostyrene and 3-chlorostyrene; ring ester-substituted vinyl aromatic compounds such as 4-acetoxystyrene; Ring hydroxylated vinyl aromatic compounds such as 4-hydroxystyrene; vinyl benzoate, vinyl Vinyl
- Tg of acrylamide, isopropylacrylamide, N-vinylpyrrolidone, isobornyl methacrylate, dicyclopentanyl methacrylate, 2-methyl-2-adamantyl methacrylate, 1-adamantyl acrylate, 1-adamantyl methacrylate, etc. is 120 ° C. or higher.
- the volume average particle size of the core layer of the core-shell polymer (B) of the present invention is preferably 0.03 to 2 ⁇ m, more preferably 0.05 to 1 ⁇ m. In many cases, it is difficult to stably obtain a volume average particle size of less than 0.03 ⁇ m, and when it exceeds 2 ⁇ m, the heat resistance and impact resistance of the final molded product may be deteriorated.
- the core layer of the core-shell polymer (B) of the present invention is preferably 40 to 97% by mass, more preferably 60 to 95% by mass, still more preferably 70 to 93% by mass, based on 100% by mass of the entire core-shell polymer. Mass% is particularly preferred. If the core layer is less than 40% by mass, the effect of improving the toughness of the cured product may be reduced. When the core layer is larger than 97% by mass, the polymer fine particles tend to aggregate, and the curable epoxy resin composition has a high viscosity and may be difficult to handle.
- the core layer may have a multilayer structure.
- the polymer composition of each layer may be different.
- the core-shell polymer (B) of the present invention may have an intermediate layer between the core layer and the shell layer.
- the following rubber surface cross-linked layer may be formed as the intermediate layer. From the viewpoint of the effect of improving the toughness and the impact resistance improvement effect of the resulting cured product, it is preferable not to contain the following rubber surface cross-linked layer as an intermediate layer.
- the rubber surface cross-linked layer is obtained by polymerizing a rubber surface cross-linked layer component composed of 30 to 100% by mass of a polyfunctional monomer having two or more radical double bonds in the same molecule and 0 to 70% by mass of other vinyl monomers. And an effect of reducing the viscosity of the curable epoxy resin composition of the present invention and an effect of improving the dispersibility of the core-shell polymer (B) component to the epoxy resin (A) component.
- the rubber surface cross-linked layer also has an effect of increasing the cross-linking density of the core layer and increasing the graft efficiency of the shell layer.
- Specific examples of the polyfunctional monomer include the same monomers as the above-mentioned polyfunctional monomer, but preferably allyl methacrylate and triallyl isocyanurate.
- the shell layer of the core-shell polymer (B) is obtained by polymerizing a shell-forming monomer, and improves the compatibility between the core-shell polymer (B) component and the epoxy resin (A) component according to the present invention.
- the shell layer of the core-shell polymer (B) of the present invention is selected from the group consisting of a vinyl cyan monomer and an alkoxyalkyl (meth) acrylate monomer with the total amount of monomers constituting the shell layer being 100% by mass from the viewpoint of high thixotropy. It is preferable that the monomer comprises 10 to 95% by mass of the monomer, 0 to 50% by mass of the monomer having an epoxy group, and 0 to 90% by mass (preferably 5 to 90% by mass) of the monomer copolymerizable therewith.
- the total amount of monomers constituting the shell layer is 100% by mass from the viewpoint of high dynamic splitting resistance while maintaining storage stability.
- a monomer selected from the group consisting of vinyl cyan monomer and alkoxyalkyl (meth) acrylate monomer From 15 to 60% by weight of a monomer selected from the group consisting of vinyl cyan monomer and alkoxyalkyl (meth) acrylate monomer, 1 to 35% by weight of monomer having an epoxy group, and 40 to 85% by weight of monomer copolymerizable therewith Become.
- the shell layer of the core-shell polymer (B) of the present invention has a total amount of monomers composing the shell layer from the viewpoint of maintaining T and having high T-peel adhesive strength and dynamic splitting resistance.
- a total amount of monomers composing the shell layer from the viewpoint of maintaining T and having high T-peel adhesive strength and dynamic splitting resistance.
- vinyl cyan monomer is 20 to 40 mass%
- monomer having an epoxy group is 3 to 15 mass%
- monomer copolymerizable therewith is 60 to 80 mass%.
- the amount of the monomer having an epoxy group in the shell layer is within the above range, the T-shaped peel adhesive strength and the dynamic splitting resistance can be significantly improved as compared with the case where 20% by mass or more is used.
- the total amount of monomers constituting the shell layer is 100% by mass
- the alkoxyalkyl (meth) acrylate monomer is 50 to 95% by mass
- the monomer having an epoxy group is 5 to 50% by mass. %.
- the total amount of monomers constituting the shell layer is 100% by mass
- the alkoxyalkyl (meth) acrylate monomer is 90 to 95% by mass
- the monomer having an epoxy group is 5 to 10%. It consists of mass%.
- Monomers selected from the group consisting of vinylcyan monomers and alkoxyalkyl (meth) acrylate monomers include (meth) acrylonitrile, 2-methoxyethyl (meth) acrylate, 2-ethoxyethyl (meth) acrylate, 2- (2- Ethoxyethoxy) is exemplified.
- Acrylonitrile is particularly preferable from the viewpoint of thixotropy, shear bond strength, T-peel bond strength or dynamic splitting resistance. From the viewpoint of thixotropy, 2-methoxyethyl (meth) acrylate is particularly preferable.
- Examples of the monomer having an epoxy group include allyl glycidyl ether, glycidyl (meth) acrylate, and glycidyl alkyl (meth) acrylate.
- Glycidyl methacrylate is particularly preferable from the viewpoint of thixotropy, shear bond strength, T-peel bond strength or dynamic splitting resistance.
- Monomers that can be copolymerized therewith include styrene, ⁇ -methylstyrene, 1- or 2-vinylnaphthalene, monochlorostyrene, dichlorostyrene, bromostyrene, etc .; aromatic vinyl monomers, methyl (meth) acrylate, ethyl (meth) Examples include alkyl (meth) acrylates such as acrylate, butyl (meth) acrylate, 2-ethylhexyl (meth) acrylate, octyl (meth) acrylate, dodecyl (meth) acrylate, stearyl (meth) acrylate, and behenyl (meth) acrylate. .
- shear bond strength shear bond strength, T-peel bond strength or dynamic splitting resistance
- styrene or methyl methacrylate is more preferable, and styrene and methyl methacrylate are particularly preferable.
- the graft ratio of the shell layer is preferably 70% or more (preferably 80% or more, more preferably 90% or more). When the graft ratio is less than 70%, the viscosity of the liquid resin composition may increase.
- the graft ratio is calculated by first coagulating and dewatering an aqueous latex containing a core-shell polymer, and finally drying to obtain a core-shell polymer powder. Next, after 2 g of core-shell polymer powder is immersed in 100 g of methyl ethyl ketone (MEK) at 23 ° C. for 24 hours, the MEK solubles are separated from the MEK insolubles, and the methanol insolubles are further separated from the MEK solubles. And it calculates by calculating
- MEK methyl ethyl ketone
- the polymer that forms the core layer constituting the core-shell polymer used in the present invention includes at least one monomer (first monomer) selected from diene monomers (conjugated diene monomers) and (meth) acrylate monomers.
- first monomer selected from diene monomers (conjugated diene monomers) and (meth) acrylate monomers.
- the core layer can be formed, for example, by emulsion polymerization, suspension polymerization, microsuspension polymerization, or the like, and for example, the method described in WO2005 / 028546 can be used.
- the formation of the core layer can be produced by, for example, emulsion polymerization, suspension polymerization, microsuspension polymerization, etc.
- the method described in WO2006 / 070664 can be used.
- the intermediate layer can be formed by polymerizing the monomer for forming the intermediate layer by a known radical polymerization.
- the polymerization of the monomer having two or more double bonds is preferably carried out by an emulsion polymerization method.
- the shell layer can be formed by polymerizing a shell layer forming monomer by known radical polymerization.
- the polymerization of the monomer for forming the shell layer is preferably carried out by an emulsion polymerization method, for example, WO2005 Can be produced according to the method described in Japanese Patent No.
- alkyl or aryl sulfonic acid represented by dioctylsulfosuccinic acid and dodecylbenzenesulfonic acid
- alkyl or arylether sulfonic acid alkyl or aryl represented by dodecylsulfuric acid, and the like.
- the dispersion stability of the aqueous latex of the core-shell polymer is not hindered, it is preferable to reduce the amount of emulsifier (dispersant) used. Moreover, an emulsifier (dispersant) is so preferable that the water solubility is high. If the water solubility is high, the emulsifier (dispersant) can be easily removed by washing with water, and adverse effects on the finally obtained cured product can be easily prevented.
- a known initiator that is, 2,2′-azobisisobutyronitrile, hydrogen peroxide, potassium persulfate, ammonium persulfate, or the like can be used as the thermal decomposition type initiator. .
- organic peroxides such as t-butylperoxyisopropyl carbonate, paramentane hydroperoxide, cumene hydroperoxide, dicumyl peroxide, t-butyl hydroperoxide, di-t-butyl peroxide, t-hexyl peroxide, etc.
- Oxides such as inorganic peroxides such as hydrogen peroxide, potassium persulfate, and ammonium persulfate; reducing agents such as sodium formaldehyde sulfoxylate and glucose as necessary; and iron sulfate (II as necessary) ),
- a chelating agent such as disodium ethylenediaminetetraacetate if necessary, and a redox type initiator using a phosphorus-containing compound such as sodium pyrophosphate if necessary.
- the polymerization can be performed even at a low temperature at which the peroxide is not substantially thermally decomposed, and the polymerization temperature can be set in a wide range, which is preferable.
- organic peroxides such as cumene hydroperoxide, dicumyl peroxide, and t-butyl hydroperoxide are preferably used as the redox initiator.
- the amount of the initiator used, or the redox type initiator is used, the amount of the reducing agent / transition metal salt / chelating agent used may be within a known range.
- a known chain transfer agent can be used within a known range.
- a surfactant can be used, but this is also within a known range.
- the conditions such as polymerization temperature, pressure and deoxygenation during the polymerization can be within the known ranges.
- the polymerization of the intermediate layer forming monomer may be performed in one stage or in two or more stages.
- a method of continuously adding it an elastic core layer is added to a reactor in which an intermediate layer forming monomer is charged in advance.
- a method of carrying out polymerization after adding an emulsion of a rubber elastic body to be constituted can be employed.
- the amount of the core-shell polymer (B) is 1 to 50 parts by mass with respect to 100 parts by mass of the epoxy resin (A) from the balance between easy handling of the resulting curable resin composition and toughness improvement effect of the obtained cured product. Is preferable, 10 to 40 parts by mass is more preferable, and 10 to 35 parts by mass is particularly preferable.
- ⁇ Blocked isocyanate (C)> the blocked isocyanate (C) is added for the purpose of further improving performances such as toughness, impact resistance, shearing adhesiveness, and T-peeling adhesiveness.
- a blocked isocyanate (C) component contributes to the improvement of the said performance by using together with a core-shell polymer (B) component.
- the blocked isocyanate (C) of the present invention is an elastomer type, and contains all or part of the terminal isocyanate group of the compound containing a urethane group and / or a urea group and having an isocyanate group at the terminal.
- Compounds capped with various blocking agents having groups In particular, a compound in which all of the terminal isocyanate groups are capped with a blocking agent is preferable.
- Such a compound can be prepared, for example, by reacting an organic polymer having an active hydrogen-containing group at the terminal with an excess of a polyisocyanate compound to have a urethane group and / or a urea group in the main chain and an isocyanate group at the terminal. It is obtained by capping all or part of the isocyanate group with a blocking agent having an active hydrogen group after or simultaneously with the polymer (urethane prepolymer).
- the blocked isocyanate is, for example, the following general formula (1): A- (NR 2 —C ( ⁇ O) —X) a (1)
- a R 2 s are each independently a hydrocarbon group having 1 to 20 carbon atoms.
- A represents the average number of capped isocyanate groups per molecule, 1.1 or more. It is preferably 1.5 to 8, more preferably 1.7 to 6, and particularly preferably 2 to 4.
- X is a residue obtained by removing an active hydrogen atom from a blocking agent described later.
- A is a residue obtained by removing a terminal isocyanate group from an isocyanate-terminated prepolymer.
- the hydrocarbon group may be an aliphatic hydrocarbon group, an alicyclic hydrocarbon group, or an aromatic hydrocarbon group.
- the aliphatic hydrocarbon group include methyl group, ethyl group, propyl group, butyl group, pentyl group, hexyl group, heptyl group, octyl group, nonyl group, decyl group, dodecyl group and the like alkyl group; ethenyl group, propenyl group Alkenyl groups such as butenyl group, pentenyl group, hexenyl group, heptenyl group, octenyl group, nonenyl group, decenyl group, undecenyl group, dodecenyl group, and the like.
- the aliphatic hydrocarbon group may be linear or branched.
- the alicyclic hydrocarbon group include a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, a cycloheptyl group, a cyclooctyl group, a cyclononyl group, a cyclodecyl group, and the like; Group, cyclohexenyl group, cycloheptenyl group, cyclooctenyl group, cyclopentadienyl and the like.
- the aromatic hydrocarbon group include a phenyl group, a naphthyl group, an anthryl group, a phenanthryl group, a biphenyl group, and a terphenyl group.
- the number average molecular weight of the blocked isocyanate is preferably from 2000 to 40000, more preferably from 3000 to 30000, particularly preferably from 4000 to 20000 in terms of polystyrene equivalent molecular weight measured by GPC.
- the molecular weight distribution (ratio of weight average molecular weight to number average molecular weight) is preferably 1 to 4, more preferably 1.2 to 3, and particularly preferably 1.5 to 2.5.
- C-1 Organic polymer having an active hydrogen-containing group at the terminal As the main chain skeleton constituting the organic polymer having an active hydrogen-containing group at the terminal, a polyether polymer, a polyacrylic polymer, a polyester polymer, a polydiene polymer, a saturated hydrocarbon polymer (polyolefin) ) And polythioether polymers.
- Examples of the active hydrogen-containing group constituting the organic polymer having an active hydrogen-containing group at the terminal include a hydroxyl group, an amino group, an imino group, and a thiol group. Among these, a hydroxyl group, an amino group, and an imino group are preferable from the viewpoint of availability, and a hydroxyl group is more preferable from the viewpoint of easy handling (viscosity) of the obtained blocked isocyanate.
- organic polymer having an active hydrogen-containing group at the terminal examples include a polyether polymer having a hydroxyl group at the terminal (polyether polyol), and a polyether polymer having an amino group and / or an imino group at the terminal (polyether amine). ), Polyacryl polyol, polyester polyol, diene polymer having a hydroxyl group at the terminal (polydiene polyol), saturated hydrocarbon polymer having a hydroxyl group at the terminal (polyolefin polyol), polythiol compound, polyamine compound, and the like.
- polyether polyol, polyether amine, and polyacryl polyol are excellent in compatibility with the epoxy resin (A) component, the glass transition temperature of the organic polymer is relatively low, and the resulting cured product has a low temperature. It is preferable because of its excellent impact resistance.
- polyether polyols and polyether amines are more preferred because the resulting organic polymer has a low viscosity and good workability, and polyether polyols are particularly preferred.
- the organic polymer having an active hydrogen-containing group at the terminal used when preparing the urethane prepolymer that is a precursor of blocked isocyanate may be used alone or in combination of two or more.
- the number average molecular weight of the organic polymer having an active hydrogen-containing group at the terminal is preferably from 800 to 7000, more preferably from 1500 to 5000, and particularly preferably from 2000 to 4000 in terms of polystyrene equivalent molecular weight measured by GPC.
- the polyether polymer essentially has the general formula (2): -R 1 -O- (2) (Wherein R 1 is a linear or branched alkylene group having 1 to 14 carbon atoms), and R 1 in the general formula (2) is a carbon atom.
- Specific examples of the repeating unit represented by the general formula (2) include —CH 2 O—, —CH 2 CH 2 O—, —CH 2 CH (CH 3 ) O—, —CH 2 CH (C 2 H 5 ) O—, —CH 2 C (CH 3 ) 2 O—, —CH 2 CH 2 CH 2 CH 2 O— and the like.
- the main chain skeleton of the polyether polymer may be composed of only one type of repeating unit, or may be composed of two or more types of repeating units.
- a polymer composed mainly of a polypropylene glycol having a propylene oxide repeating unit of 50% by mass or more is preferable from the viewpoint of T-shaped peel adhesion strength.
- Polytetramethylene glycol (PTMG) obtained by ring-opening polymerization of tetrahydrofuran is preferred from the viewpoint of dynamic splitting resistance.
- the ratio of the core-shell polymer (B) component to the blocked isocyanate (C) component is 0.1 from the viewpoint of thixotropic properties of the resulting curable epoxy resin composition. Is preferably 10 to 10, more preferably 0.2 to 5, and particularly preferably 0.3 to 1.
- the polyether polyol is a polyether polymer having a hydroxyl group at the terminal, and the polyether amine is a polyether polymer having an amino group or an imino group at the terminal.
- polyacryl polyol examples include a polyol having a (meth) acrylic acid alkyl ester (co) polymer as a skeleton and a hydroxyl group in the molecule.
- a polyacryl polyol obtained by copolymerizing a hydroxyl group-containing (meth) acrylic acid alkyl ester monomer such as 2-hydroxyethyl methacrylate is preferred.
- polyester polyol examples include polybasic acids such as maleic acid, fumaric acid, adipic acid, and phthalic acid, and acid anhydrides thereof, ethylene glycol, propylene glycol, 1,4-butanediol, 1,6-hexanediol, Examples thereof include polymers obtained by polycondensation with polyhydric alcohols such as diethylene glycol, dipropylene glycol and neopentyl glycol in the presence of an esterification catalyst in a temperature range of 150 to 270 ° C. Also included are ring-opening polymers such as ⁇ -caprolactone and valerolactone, and active hydrogen compounds having two or more active hydrogens such as polycarbonate diol and castor oil.
- polybasic acids such as maleic acid, fumaric acid, adipic acid, and phthalic acid
- acid anhydrides thereof ethylene glycol, propylene glycol, 1,4-butanediol, 1,6-hexanedi
- Polydiene polyol examples include polybutadiene polyol, polyisoprene polyol, polychloroprene polyol, and the like, and polybutadiene polyol is particularly preferable.
- Polyolefin polyol examples include polyisobutylene polyol and hydrogenated polybutadiene polyol.
- polyisocyanate compound examples include aromatic polyisocyanates such as toluene (tolylene) diisocyanate, diphenylmethane diisocyanate, and xylylene diisocyanate; fats such as isophorone diisocyanate, hexamethylene diisocyanate, hydrogenated toluene diisocyanate, and hydrogenated diphenylmethane diisocyanate.
- aromatic polyisocyanates such as toluene (tolylene) diisocyanate, diphenylmethane diisocyanate, and xylylene diisocyanate
- fats such as isophorone diisocyanate, hexamethylene diisocyanate, hydrogenated toluene diisocyanate, and hydrogenated diphenylmethane diisocyanate.
- Group polyisocyanates can be mentioned. Among these, aliphatic polyisocyanates are preferable from the viewpoint of heat resistance, and is
- blocking agent examples include a primary amine blocking agent, a secondary amine blocking agent, an oxime blocking agent, a lactam blocking agent, an active methylene blocking agent, an alcohol blocking agent, a mercaptan blocking agent, and an amide.
- an oxime block agent, a lactam block agent, a hydroxy functional (meth) acrylate block agent, and a phenol block agent are preferable, and a hydroxy functional (meth) acrylate block agent and a phenol block agent are more preferable.
- a phenolic blocking agent is more preferable.
- Examples of the primary amine blocking agent include butylamine, isopropylamine, dodecylamine, cyclohexylamine, aniline, benzylamine and the like.
- Examples of the secondary amine blocking agent include dibutylamine, diisopropylamine, dicyclohexylamine, diphenylamine, dibenzylamine, morpholine, piperidine and the like.
- Examples of the oxime blocking agent include formaldoxime, acetoaldoxime, acetoxime, methyl ethyl ketoxime, diacetyl monooxime, cyclohexane oxime and the like.
- lactam blocking agent examples include ⁇ -caprolactam, ⁇ -valerolactam, ⁇ -butyrolactam, ⁇ -butyrolactam and the like.
- active methylene-based blocking agent examples include ethyl acetoacetate and acetylacetone.
- Examples of the alcohol blocking agent include methanol, ethanol, propanol, isopropanol, butanol, amyl alcohol, cyclohexanol, 1-methoxy-2-propanol, ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, propylene glycol monomethyl ether, benzyl alcohol Methyl glycolate, butyl glycolate, diacetone alcohol, methyl lactate, ethyl lactate and the like.
- Examples of the mercaptan-based blocking agent include butyl mercaptan, hexyl mercaptan, decyl mercaptan, t-butyl mercaptan, thiophenol, methylthiophenol, and ethylthiophenol.
- Examples of the amide blocking agent include acetic acid amide and benzamide.
- Examples of the imide-based blocking agent include succinimide and maleic imide.
- Examples of the heterocyclic aromatic compound blocking agent include imidazoles such as imidazole and 2-ethylimidazole, pyrroles such as pyrrole, 2-methylpyrrole, and 3-methylpyrrole, pyridine, 2-methylpyridine, and 4-methyl. Examples thereof include pyridines such as pyridine, and diazabicycloalkenes such as diazabicycloundecene and diazabicyclononene.
- the hydroxy functional (meth) acrylate blocking agent is a (meth) acrylate having one or more hydroxyl groups.
- Specific examples of hydroxy functional (meth) acrylate blocking agents include 2-hydroxyethyl (meth) acrylate, 2-hydroxypropyl (meth) acrylate, 4-hydroxybutyl (meth) acrylate, 2-hydroxybutyl (meth) Acrylate, and the like.
- the phenolic blocking agent contains at least one phenolic hydroxyl group, that is, a hydroxyl group directly bonded to a carbon atom of an aromatic ring.
- the phenolic compound may have two or more phenolic hydroxyl groups, but preferably contains only one phenolic hydroxyl group.
- the phenolic compound may contain other substituents, but these substituents are preferably those that do not react with isocyanate groups under the conditions of the capping reaction, and are preferably alkenyl groups and allyl groups.
- substituents include linear, branched, or cycloalkyl alkyl groups; aromatic groups (eg, phenyl, alkyl-substituted phenyl, alkenyl-substituted phenyl, etc.); aryl-substituted alkyl groups; phenol-substituted alkyl groups Can be mentioned.
- the phenolic blocking agent include phenol, cresol, xylenol, chlorophenol, ethylphenol, allylphenol (especially o-allylphenol), resorcinol, catechol, hydroquinone, bisphenol, bisphenol A, bisphenol AP (1,1- Bis (4-hydroxylphenyl) -1-phenylethane), bisphenol F, bisphenol K, bisphenol M, tetramethylbiphenol and 2,2′-diallyl-bisphenol A, and the like.
- the blocking agent is preferably bonded to the end of the polymer chain of the urethane prepolymer in such a manner that the terminal to which it is bonded no longer has a reactive group.
- the blocking agent may be used alone or in combination of two or more.
- the blocked isocyanate may contain a residue of a crosslinking agent, a residue of a chain extender, or both.
- the blocked isocyanate used in the present invention is preferably a compound obtained by capping a urethane prepolymer containing a polyalkylene glycol structure with a blocking agent, and the urethane prepolymer containing a polypropylene glycol structure is preferably a blocking agent (preferably a phenolic block). It is more preferable that the compound is a compound capped with an agent) or a urethane prepolymer containing a polytetramethylene glycol structure capped with a blocking agent (preferably a phenolic blocking agent).
- the blocked isocyanate can be suitably used in terms of improving thixotropy and adhesion.
- a compound in which a urethane prepolymer containing a polypropylene glycol structure is capped with a blocking agent may be used from the viewpoint of improving T-peel adhesion strength, and a urethane containing a polytetramethylene glycol structure.
- a compound in which a prepolymer is capped with a blocking agent may be used from the viewpoint of improving dynamic splitting resistance.
- the blocked NCO equivalent of blocked isocyanate is, for example, 10 to 500, preferably 50 to 300.
- the epoxide equivalent weight of the blocked isocyanate is, for example, 300 to 900 g / eq, preferably 400 to 800 g / eq.
- a blocked isocyanate having at least one of these characteristics can be suitably used in the present invention.
- Crosslinking agent The molecular weight of the crosslinking agent is preferably 750 or less, more preferably 50 to 500, and a polyol or polyamine compound having at least three hydroxyl groups, amino groups and / or imino groups per molecule.
- Crosslinkers are useful for imparting branching to the blocked isocyanate and increasing the functionality of the blocked isocyanate (ie, the number of capped isocyanate groups per molecule).
- Chain extender The molecular weight of the chain extender is preferably 750 or less, more preferably 50 to 500, and a polyol or polyamine compound having two hydroxyl groups, amino groups and / or imino groups per molecule. Chain extenders are useful for increasing the molecular weight of the blocked isocyanate without increasing functionality.
- crosslinking agent and chain extender include trimethylolpropane, glycerin, trimethylolethane, ethylene glycol, diethylene glycol, propylene glycol, dipropylene glycol, sucrose, sorbitol, pentaerythritol, ethylenediamine, triethanolamine, monoethanol.
- examples include amine, diethanolamine, piperazine, and aminoethylpiperazine.
- Resorcinol catechol, hydroquinone, bisphenol, bisphenol A, bisphenol AP (1,1-bis (4-hydroxylphenyl) -1-phenylethane), bisphenol F, bisphenol K, bisphenol M, tetramethylbiphenol, 2,2 Also included are compounds having two or more phenolic hydroxyl groups, such as' -diallyl-bisphenol A.
- the amount of blocked isocyanate is preferably 1 to 50 parts by mass, more preferably 10 to 40 parts by mass, and particularly preferably 10 to 35 parts by mass with respect to 100 parts by mass of the epoxy resin (A). If it is less than 1 part by mass, the modifying effects such as toughness, impact resistance and adhesiveness may not be sufficient. If it exceeds 50 parts by mass, the resulting cured product may have a low elastic modulus.
- the blocked isocyanate may be used alone or in combination of two or more.
- an epoxy curing agent (D) can be used as necessary.
- the epoxy curing agent (D) so that the adhesive rapidly cures when heated to a temperature of 80 ° C. or higher, preferably 140 ° C. or higher. It is preferred to select the components. On the contrary, it is preferable to select the epoxy curing agent (D) component and the later-described (E) component so that the curing becomes very slow even at room temperature (about 22 ° C.) or at least up to 50 ° C.
- epoxy curing agent (D) component examples include boron trichloride / amine complex, boron trifluoride / amine complex, dicyandiamide, melamine, diallylmelamine, guanamine (for example, acetoguanamine and benzoguanamine), aminotriazole (for example, 3 -Amino-1,2,4-triazole), hydrazides (for example, adipic acid dihydrazide, stearic acid dihydrazide, isophthalic acid dihydrazide, semicarbazide), cyanoacetamide, and aromatic polyamines (for example, diaminodiphenylsulfone).
- boron trichloride / amine complex examples include boron trifluoride / amine complex, dicyandiamide, melamine, diallylmelamine, guanamine (for example, acetoguanamine and benzoguanamine), aminotriazole (for example, 3 -
- dicyandiamide isophthalic acid dihydrazide, adipic acid dihydrazide, or 4,4'-diaminodiphenylsulfone, and dicyandiamide is particularly preferable.
- latent epoxy curing agents such as boron trichloride / amine complex, boron trifluoride / amine complex, dicyandiamide, hydrazide and the like are preferable because the curable epoxy resin composition of the present invention can be made into one component.
- curing agent (D) component may be used independently and may be used together 2 or more types.
- the epoxy curing agent (D) component is used in an amount sufficient to cure the composition. Typically, sufficient curing agent is provided to consume at least 80% of the epoxide groups present in the composition. A large excess over that required for consumption of epoxide groups is usually not necessary.
- the amount of the epoxy curing agent (D) component used is preferably 1 to 40 parts by weight, more preferably 2 to 30 parts by weight, still more preferably 3 to 25 parts by weight with respect to 100 parts by weight of the component (A). ⁇ 20 parts by weight is particularly preferred. If it is less than 1 mass part, the sclerosis
- a hardening accelerator (E) can be used for the curable epoxy resin composition of this invention as needed.
- a hardening accelerator (E) component is a catalyst for accelerating
- the curing accelerator (E) component include ureas such as p-chlorophenyl-N, N-dimethylurea (Monuron), 3-phenyl-1,1-dimethylurea (Phenuron), and 3,4-dichlorophenyl-N.
- N-dimethylurea (Diuron), N- (3-chloro-4-methylphenyl) -N ′, N′-dimethylurea (Chlortoluron), tert-acryl- or alkyleneamines such as benzyldimethylamine, 2, 4,6-tris (dimethylaminomethyl) phenol, 2,4,6-tris (dimethylaminomethyl) phenol incorporated in a poly (p-vinylphenol) matrix, piperidine or derivatives thereof, imidazole derivatives, generally C1- C12 alkylene imidazole or N-aryl Imidazole, such as 2-ethyl-2-methylimidazole or N- butyl imidazole, 6-caprolactam, and the like.
- the catalyst may be encapsulated or may be a potential one that becomes active only when the temperature is raised.
- a hardening accelerator (E) component may be used independently and may be used together 2 or more types.
- the amount of the curing accelerator (E) component used is preferably 0.1 to 10 parts by mass, more preferably 0.2 to 5 parts by mass, and 0.5 to 3 parts by mass with respect to 100 parts by mass of the component (A). Part is more preferable, and 0.8 to 2 parts by mass is particularly preferable. If it is less than 0.1 mass part, the sclerosis
- a filler can be used in the curable epoxy resin composition of the present invention as necessary.
- the filler include reinforcing silica fillers such as dry silica such as hydrophobic fumed silica surface-treated with polydimethylsiloxane, wet silica, aluminum silicate, magnesium silicate, calcium silicate, dolomite and carbon black. Plate-like fillers such as talc and wollastonite; colloidal calcium carbonate, heavy calcium carbonate, magnesium carbonate, titanium oxide, ferric oxide, aluminum fine powder, zinc oxide, activated zinc white and the like.
- a microballoon having an average particle size of up to 200 ⁇ m and a density of up to 0.2 g / cc can also be used.
- the particle size is preferably about 25 to 150 ⁇ m and the density is preferably about 0.05 to about 0.15 g / cc.
- Examples of commercially available microballoons include Dualite manufactured by Dualite Corporation, Expandel manufactured by Akzo Nobel, and Microsphere manufactured by Matsumoto Yushi Seiyaku.
- the curable epoxy resin composition of the present invention includes, as necessary, a monoepoxide such as aliphatic glycidyl ether such as butyl glycidyl ether, phenyl glycidyl ether, cresyl glycidyl ether or 2 as a reactive diluent.
- a monoepoxide such as aliphatic glycidyl ether such as butyl glycidyl ether, phenyl glycidyl ether, cresyl glycidyl ether or 2 as a reactive diluent.
- -It may contain ethylhexyl glycidyl ether, C8-C10 alkyl glycidyl ether, C12-C14 alkyl glycidyl ether, p-tertbutylphenyl glycidyl ether, neodecanoic acid monoglycidyl ether.
- Another compounding component can be used as needed.
- Other ingredients include dehydrating agents such as calcium oxide, colorants such as pigments and dyes, extender pigments, UV absorbers, antioxidants, stabilizers (anti-gelling agents), plasticizers, leveling agents, anti-oxidants.
- dehydrating agents such as calcium oxide
- colorants such as pigments and dyes, extender pigments, UV absorbers, antioxidants, stabilizers (anti-gelling agents), plasticizers, leveling agents, anti-oxidants.
- foaming agents silane coupling agents, antistatic agents, flame retardants, lubricants, viscosity reducers, low shrinkage agents, organic fillers, thermoplastic resins, drying agents, and dispersing agents.
- the thixotropic property of the curable epoxy resin composition of the present invention is preferably 1.00 or more, more preferably 1.1 or more, further preferably 1.2 or more, and 1.25 or more. Even more preferably.
- the upper limit of thixotropy is, for example, about 1.5.
- the thixotropic property can be evaluated by a thixotropic index, a hysteresis loop, or the like.
- the present invention includes a cured product obtained by curing the curable epoxy resin composition.
- the curable epoxy resin composition of the present invention is excellent in thixotropy and can be used as a one-component adhesive.
- the curable epoxy resin composition of the present invention includes adhesives such as structural adhesives for vehicles and aircraft, paints, materials for laminating with glass fibers, materials for laminating with carbon fibers, printed wiring boards, and electrical insulating materials. It is preferably used for such applications. In particular, it is useful as a structural adhesive for vehicles.
- volume average particle diameter of the polybutadiene rubber particles in the polybutadiene rubber latex and the core-shell polymer particles in the core-shell polymer latex was measured using Microtrac UPA150 (manufactured by Nikkiso Co., Ltd.).
- Diluted with deionized water was used as a measurement sample.
- Shear bond strength The shear bond strength was evaluated according to JIS K 6850. Apply a curable epoxy resin composition to two SPCC steel sheets of width 25 mm x length 100 mm x thickness 1.6 mm, and paste them so that the adhesive layer is 25 mm wide x 12.5 mm long x 0.13 mm thick In addition, the specimen was cured for 1 hour under the conditions of 170 ° C. The shear bond strength with the unit of MPa was measured under the measurement conditions of a measurement temperature of 23 ° C. and a test speed of 1.3 mm / min.
- T-shaped peel adhesion strength T-peel adhesion strength was evaluated according to JIS K 6854. Applying a curable epoxy resin composition to two SPCC steel sheets of width 25 mm ⁇ length 200 mm ⁇ thickness 0.5 mm, and bonding them so that the adhesive layer becomes width 25 mm ⁇ length 150 mm ⁇ thickness 0.26 mm, The specimen was cured at 170 ° C. for 1 hour to prepare a test specimen. Under the measurement conditions where the measurement temperature was 23 ° C. and the test speed was 254 mm / min, the T-shaped peel adhesion strength with the unit N / 25 mm was measured.
- the dynamic splitting resistance was evaluated according to ISO 11343. Applying a curable epoxy resin composition to two SPCC steel sheets 20 mm wide x 90 mm long x 0.8 mm thick, and bonding them so that the adhesive layer is 20 mm wide x 30 mm long x 0.26 mm thick, The specimen was cured for 1 hour under the condition of 170 ° C.
- the dynamic splitting resistance with the unit of kN / m was measured under the measurement conditions of a measurement temperature of 23 ° C., an impact energy of 50 J, and an impact speed of 2 m / s.
- Production Example 1-1 Preparation of polybutadiene rubber latex (R-1) In a pressure-resistant polymerization machine, 200 parts by mass of deionized water, 0.03 parts by mass of tripotassium phosphate, disodium ethylenediaminetetraacetate (EDTA) 0.002 Add parts by weight, ferrous sulfate, heptahydrate 0.001 parts by weight, and sodium dodecylbenzenesulfonate (SDBS) 1.55 parts by weight, and thoroughly replace nitrogen with stirring to remove oxygen. Thereafter, 100 parts by mass of butadiene (Bd) was charged into the system, and the temperature was raised to 45 ° C.
- EDTA disodium ethylenediaminetetraacetate
- PPP paramentane hydroperoxide
- SSS sodium formaldehyde sulfoxylate
- 0.0006 parts by mass of EDTA and 0.003 parts by mass of ferrous sulfate / pentahydrate were added at 4, 6 and 8 hours after the start of polymerization.
- the residual monomer was removed by devolatilization under reduced pressure to complete the polymerization, and a polybutadiene rubber latex (R-1) containing polybutadiene rubber as a main component was obtained.
- the volume average particle diameter of the polybutadiene rubber particles contained in the obtained latex was 0.08 ⁇ m.
- Production Example 1-2 Preparation of Polybutadiene Rubber Latex (R-2) 21 parts by mass of polybutadiene rubber latex (R-1) obtained in Production Example 1-1 (including 7 parts by mass of polybutadiene rubber) in a pressure-resistant polymerization machine. ), 185 parts by weight of deionized water, 0.03 part by weight of tripotassium phosphate, 0.002 part by weight of EDTA, and 0.001 part by weight of ferrous sulfate / pentahydrate, and nitrogen is sufficiently stirred. After substituting and removing oxygen, 93 parts by mass of Bd was charged into the system, and the temperature was raised to 45 ° C.
- Polymerization was started by adding 0.02 parts by weight of PHP and subsequently 0.10 parts by weight of SFS. 0.03 part by mass of PHP, 0.0006 part by mass of EDTA, and 0.003 part by mass of ferrous sulfate and heptahydrate were added every 3 hours from the start of polymerization to 24 hours. At 30 hours after polymerization, the residual monomer was removed by devolatilization under reduced pressure to complete the polymerization, and a polybutadiene rubber latex (R-2) containing polybutadiene rubber as a main component was obtained. The volume average particle diameter of the polybutadiene rubber particles contained in the obtained latex was 0.20 ⁇ m.
- Production Example 2-1 Preparation of Core Shell Polymer Latex (L-1) Polybutadiene rubber prepared in Production Example 1 was added to a glass reactor having a thermometer, a stirrer, a reflux condenser, a nitrogen inlet, and a monomer addition device. 250 parts by mass of polybutadiene rubber latex (R-1) containing 83 parts by mass of particles and 65 parts by mass of deionized water were charged and stirred at 60 ° C. while purging with nitrogen.
- Production Example 2-2 to Production Example 2-10 A core-shell polymer latex was prepared in the same manner with the polybutadiene rubber latex and graft monomer in Production Example 2-1 at the levels shown in Table 1.
- Table 1 shows the volume average particle diameters of the core-shell polymers (L-1) to (L-10).
- Production Example 3-2 to Production Example 3-10 The core-shell polymer aqueous latex (L-1) of Production Example 3-1 was replaced with the core-shell polymer aqueous latex (L-2) to (L-10) of Production Example 3-2 to Production Example 3-10.
- Epoxy resins (N-2) to (N-10) in which the core-shell polymer is dispersed were obtained in the same manner as in Production Example 3-1.
- Examples 1 to 16 and Comparative Example 1 In Tables 2 and 3, (A) epoxy resin, (N) epoxy resin in which core-shell polymer is dispersed, (C) blocked isocyanate, (D) epoxy curing agent, (E) curing accelerator, and other compounding raw materials It shows about the compounding composition of the curable epoxy resin composition which mix
- C Blocked isocyanate
- C-1 Adeka Resin QR-9466 (manufactured by ADEKA, blocked isocyanate containing polypropylene glycol structure, block NCO equivalent 220, viscosity 30000 mPa ⁇ s / 25 ° C.)
- C-2 Flexibilizer DY 965 (manufactured by Huntsman, blocked isocyanate containing polytetramethylene glycol structure, Epoxide Eq.
- FIGS. 1 and 4 show the relationship between glycidyl methacrylate (GMA) of the shell layer of the core-shell polymer and storage stability and thixotropy.
- GMA glycidyl methacrylate
- FIG. 1 blocked isocyanate containing a polypropylene glycol structure was used in FIG. 1 (GMA ratio% in the figure corresponds to that used in Examples 1, 5, 6, 7, and 8).
- Block 4 a blocked isocyanate containing a polytetramethylene glycol structure was used (the GMA ratio% in the figure corresponds to that used in Examples 10, 12, 13, 14, and 15).
- glycidyl methacrylate (GMA), T-peel adhesive strength (indicated by T-Peel in the figure) and shear adhesive strength (indicated by Lap Shear in the figure) of the shell layer of the core-shell polymer.
- T-peel adhesive strength (indicated by T-Peel in the figure)
- shear adhesive strength (indicated by Lap Shear in the figure) of the shell layer of the core-shell polymer.
- the shell layer contains a predetermined amount of glycidyl methacrylate (greater than 0% by mass and less than 20% by mass)
- FIG. 2 a blocked isocyanate containing a polypropylene glycol structure is used.
- the GMA ratio% in the figure corresponds to that used in Examples 1, 5, 6, 7, and 8.
- Fig. 5 a blocked isocyanate containing a polytetramethylene glycol structure was used (see figure).
- GMA ratio% in the middle corresponds to those used in Examples 10, 12, 13, 14, 15)).
- 3 and 6 show the relationship between the glycidyl methacrylate (GMA) in the shell layer of the core-shell polymer and the dynamic splitting resistance (indicated as Impact Peel in the figure), and a predetermined amount (0 mass) of glycidyl methacrylate in the shell layer.
- the dynamic splitting resistance can be improved (in FIG. 3, a blocked isocyanate containing a polypropylene glycol structure was used (the GMA ratio% in the figure is Example 1, (This corresponds to the one used in 5, 6, 7, and 8.)
- Fig. 6 a blocked isocyanate containing a polytetramethylene glycol structure was used (the GMA ratio% in the figure is that of Examples 10, 12, 13). , 14, and 15)))).
- a compound in which a urethane prepolymer containing a polypropylene glycol structure is capped with a blocking agent is a compound in which a urethane prepolymer containing a polytetramethylene glycol structure is capped with a blocking agent (preferably a phenolic blocking agent).
- a blocking agent preferably a phenolic blocking agent
- a compound in which a urethane prepolymer containing a polytetramethylene glycol structure is capped with a blocking agent is a compound in which a urethane prepolymer containing a polypropylene glycol structure is capped with a blocking agent (preferably a phenolic blocking agent).
- a blocking agent preferably a phenolic blocking agent
- the storage stability and the dynamic splitting resistance can be improved (see Examples 1 to 16, FIGS. 1, 3, 4, and 6).
Abstract
本発明は、(A)エポキシ樹脂100質量部に対し、(B)コアシェルポリマー1~50質量部および(C)ブロックドイソシアネート1~50質量部を含有する硬化性エポキシ樹脂組成物であって、(B)のコア層が、ジエン系ゴム、アクリル系ゴム、ポリシロキサン系ゴムのいずれかからなり、(B)コアシェルポリマーのシェル層が、シェル層を構成するモノマー全量を100質量%とし、ビニルシアンモノマーおよびアルコキシアルキル(メタ)アクリレートモノマーからなる群より選択されるモノマー10~95質量%、エポキシ基を有するモノマー0~50質量%、およびそれらと共重合可能なモノマー0~90質量%からなる硬化性エポキシ樹脂組成物である。本発明の硬化性エポキシ樹脂組成物は、構造接着剤、特に車両用構造接着剤に好適に使用される。
Description
本発明は、コアシェルポリマーを含む硬化性エポキシ樹脂組成物、その接着剤、特に車両用の構造接着剤に関するものである。
エポキシ樹脂は広範囲の用途で使用されており、その一つに接着剤が挙げられる。エポキシ樹脂硬化物は優れた機械的特性、電気的特性、耐久性を示す一方で、脆性的性質や耐衝撃性などを向上させるために、車両などの構造接着剤においては、硬化性エポキシ樹脂組成物中にコアシェルポリマー及びブロックドイソシアネートを含む場合がある(特許文献1)。
この構造接着剤は、製造ラインでのハンドリング性、塗布工程と硬化工程の間に存在するシャワー工程での洗い落とされにくさ(Wash-off resistance)も重要である(特許文献2)。特に、シャワー工程では、水圧により、組成物が一部溶解したり、飛散したり、変形したりする場合があり、塗布部の劣化に繋がる。この解決策として硬化性エポキシ樹脂組成物の高粘度化が挙げられるが、塗布作業性が低下してしまうという問題があった。
Wash-off resistanceを付与するためには、硬化性エポキシ樹脂組成物が、高せん断時の粘度に対する低せん断時の粘度が高いという、いわゆるチキソトロピー性を有することが効果的であると考えられる。しかしながら、上記特許文献1及び2には、チキソトロピー性を改善する手段が何ら教示されていない。
これに対し、本発明者らは、硬化性エポキシ樹脂組成物において、チキソトロピー性が、コアシェルポリマーのシェル層成分やその量に応じて変化することを見出し、コアシェルポリマーのシェル層について鋭意検討することにした。
本発明は、上述の状況に鑑み、チキソトロピー性に優れ、さらに、得られる硬化物の接着性に優れる硬化性エポキシ樹脂組成物の提供を目的とする。
これに対し、本発明者らは、硬化性エポキシ樹脂組成物において、チキソトロピー性が、コアシェルポリマーのシェル層成分やその量に応じて変化することを見出し、コアシェルポリマーのシェル層について鋭意検討することにした。
本発明は、上述の状況に鑑み、チキソトロピー性に優れ、さらに、得られる硬化物の接着性に優れる硬化性エポキシ樹脂組成物の提供を目的とする。
本発明者らは、エポキシ樹脂、コアシェルポリマー及びブロックドイソシアネートを含有する組成物において、コアシェルポリマーのシェル層を特定のモノマー及び特定の量から構成すると、組成物のチキソトロピー性とその硬化物の接着性が改善されることを見出し、本発明を完成させた。本発明の要旨は以下の通りである。
1)エポキシ樹脂(A)100質量部に対し、コアシェルポリマー(B)1~50質量部およびブロックドイソシアネート(C)1~50質量部を含有する硬化性エポキシ樹脂組成物であって、コアシェルポリマー(B)のコア層が、ジエン系ゴム、アクリル系ゴム、ポリシロキサン系ゴムのいずれかからなり、コアシェルポリマー(B)のシェル層が、シェル層を構成するモノマー全量を100質量%とし、ビニルシアンモノマーおよびアルコキシアルキル(メタ)アクリレートモノマーからなる群より選択されるモノマー10~95質量%、エポキシ基を有するモノマー0~50質量%、およびそれらと共重合可能なモノマー0~90質量%からなることを特徴とする硬化性エポキシ樹脂組成物。
2)それらと共重合可能なモノマーが5~90質量%である1)に記載の硬化性エポキシ樹脂組成物。
3)コアシェルポリマー(B)のシェル層が、シェル層を構成するモノマー全量を100質量%とし、ビニルシアンモノマー20~40質量%、エポキシ基を有するモノマー3~15質量%、およびそれらと共重合可能なモノマー60~80質量%からなる1)または2)に記載の硬化性エポキシ樹脂組成物。
4)コアシェルポリマー(B)のシェル層が、シェル層を構成するモノマー全量を100質量%とし、アルコキシアルキル(メタ)アクリレートモノマー50~95質量%、およびエポキシ基を有するモノマー5~50質量%からなる1)に記載の硬化性エポキシ樹脂組成物。
5)コアシェルポリマー(B)のシェル層が、シェル層を構成するモノマー全量を100質量%とし、アルコキシアルキル(メタ)アクリレートモノマー90~95質量%、およびエポキシ基を有するモノマー5~10質量%からなる1)または4)に記載の硬化性エポキシ樹脂組成物。
6)ビニルシアンモノマーが、アクリロニトリルである1)~3)のいずれかに記載の硬化性エポキシ樹脂組成物。
7)エポキシ基を有するモノマーが、グリシジルメタクリレートである1)~6)のいずれかに記載の硬化性エポキシ樹脂組成物。
8)アルコキシアルキル(メタ)アクリレートモノマーが2-メトキシエチルアクリレートである1)、2)、4)~7)のいずれかに記載の硬化性エポキシ樹脂組成物。
9)コアシェルポリマー(B)のコア層が、ブタジエンゴム、および/または、ブタジエン-スチレンゴムからなる1)~8)のいずれかに記載の硬化性エポキシ樹脂組成物。
10)コアシェルポリマー(B)の体積平均粒子径が0.05~0.30μmである1)~9)のいずれかに記載の硬化性エポキシ樹脂組成物。
11)ブロックドイソシアネート(C)が、ポリプロピレングリコール構造を含むウレタンプレポリマーをブロック剤でキャップした化合物である1)~10)のいずれかに記載の硬化性エポキシ樹脂組成物。
12)ブロックドイソシアネート(C)が、ポリテトラメチレングリコール構造を含むウレタンプレポリマーをブロック剤でキャップした化合物である1)~10)のいずれかに記載の硬化性エポキシ樹脂組成物。
13)更に、エポキシ硬化剤(D)1~40質量部を含有する1)~12)のいずれかに記載の硬化性エポキシ樹脂組成物。
14)更に、硬化促進剤(E)0.1~10質量部を含有する1)~13)のいずれかに記載の硬化性エポキシ樹脂組成物。
15)1)~14)のいずれかに記載の硬化性エポキシ樹脂組成物を硬化した硬化物。
16)1)~14)のいずれかに記載の硬化性エポキシ樹脂組成物を用いてなる構造接着剤。
17)1)~14)のいずれかに記載の硬化性エポキシ樹脂組成物を用いてなる車両用構造接着剤。
本発明の硬化性エポキシ樹脂組成物は、チキソトロピー性や硬化物に対する接着性に優れる。
本発明の硬化性エポキシ樹脂組成物は、エポキシ樹脂(A)、コアシェルポリマー(B)、ブロックドイソシアネート(C)を必須成分として含有する。
<エポキシ樹脂(A)>
(A-1)ポリグリシジルエーテル、多価グリシジルアミン化合物、脂環式エポキシ樹脂等
本発明のエポキシ樹脂(A)は、エポキシ基を有する化合物であれば特に制限されないが、本発明に用いられるエポキシ樹脂はポリエポキシドとも言われるエポキシ樹脂であることが好ましい。前記のエポキシ樹脂としては、ビスフェノールA 、ビスフェノールF 、ビフェノール、フェノール類ノボラック等の多価フェノールとエピクロルヒドリンの付加反応生成物などのポリグリシジルエーテル(ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ビフェノール型エポキシ樹脂、フェノールノボラック型エポキシ樹脂等)、アニリン、ジアミノベンゼン、アミノフェノール、フェニレンジアミン、ジアミノフェニルエーテル等のモノアミンおよび多価アミンより誘導される多価グリシジルアミン化合物、シクロヘキシルエポキシ等の脂環式エポキシ構造を有する脂環式エポキシ樹脂、多価アルコール類とエピクロルヒドリンとの付加反応生成物、これらの一部の水素を臭素等のハロゲン元素で置換したハロゲン化エポキシ樹脂、アリルグリシジルエーテル等の不飽和モノエポキシドを含む単量体を重合して得られるホモポリマーもしくはコポリマーなどが例示される。多価フェノールより合成される多くのポリエポキシドは、例えば米国特許第4431782号に開示されている。ポリエポキシドの例としては更に、米国特許第3804735号、同第3892819号、同第3948698号、同第4014771号、及び、エポキシ樹脂ハンドブック(日刊工業新聞社、昭和62年) に開示されているものが挙げられる。
(A-1)ポリグリシジルエーテル、多価グリシジルアミン化合物、脂環式エポキシ樹脂等
本発明のエポキシ樹脂(A)は、エポキシ基を有する化合物であれば特に制限されないが、本発明に用いられるエポキシ樹脂はポリエポキシドとも言われるエポキシ樹脂であることが好ましい。前記のエポキシ樹脂としては、ビスフェノールA 、ビスフェノールF 、ビフェノール、フェノール類ノボラック等の多価フェノールとエピクロルヒドリンの付加反応生成物などのポリグリシジルエーテル(ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ビフェノール型エポキシ樹脂、フェノールノボラック型エポキシ樹脂等)、アニリン、ジアミノベンゼン、アミノフェノール、フェニレンジアミン、ジアミノフェニルエーテル等のモノアミンおよび多価アミンより誘導される多価グリシジルアミン化合物、シクロヘキシルエポキシ等の脂環式エポキシ構造を有する脂環式エポキシ樹脂、多価アルコール類とエピクロルヒドリンとの付加反応生成物、これらの一部の水素を臭素等のハロゲン元素で置換したハロゲン化エポキシ樹脂、アリルグリシジルエーテル等の不飽和モノエポキシドを含む単量体を重合して得られるホモポリマーもしくはコポリマーなどが例示される。多価フェノールより合成される多くのポリエポキシドは、例えば米国特許第4431782号に開示されている。ポリエポキシドの例としては更に、米国特許第3804735号、同第3892819号、同第3948698号、同第4014771号、及び、エポキシ樹脂ハンドブック(日刊工業新聞社、昭和62年) に開示されているものが挙げられる。
(A-2)ポリアルキレングリコールジグリシジルエーテル、グリコールジグリシジルエーテル等
また、本発明のエポキシ樹脂(A)としては、ポリアルキレングリコールジグリシジルエーテル、グリコールジグリシジルエーテル、脂肪族多塩基酸のジグリシジルエステル、二価以上の多価脂肪族アルコールのグリシジルエーテル、ジビニルベンゼンジオキシドも使用できる。これらは比較的低い粘度を有するエポキシ樹脂であり、ビスフェノールA型エポキシ樹脂やビスフェノールF型エポキシ樹脂等の他のエポキシ樹脂と併用すると、反応性希釈剤として機能し、組成物の粘度と硬化物の物性のバランスを改良する事ができる。
また、本発明のエポキシ樹脂(A)としては、ポリアルキレングリコールジグリシジルエーテル、グリコールジグリシジルエーテル、脂肪族多塩基酸のジグリシジルエステル、二価以上の多価脂肪族アルコールのグリシジルエーテル、ジビニルベンゼンジオキシドも使用できる。これらは比較的低い粘度を有するエポキシ樹脂であり、ビスフェノールA型エポキシ樹脂やビスフェノールF型エポキシ樹脂等の他のエポキシ樹脂と併用すると、反応性希釈剤として機能し、組成物の粘度と硬化物の物性のバランスを改良する事ができる。
これらのエポキシ樹脂の含有量は、エポキシ樹脂(A)成分100質量%中0.5~20質量%が好ましく、1~10質量%がより好ましく、2~5質量%が更に好ましい。前記ポリアルキレングリコールジグリシジルエーテルとしては、より具体的には、ポリエチレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、などが挙げられる。前記グリコールジグリシジルエーテルとしては、より具体的には、ネオペンチルグリコールジグリシジルエーテル、1,4-ブタンジオールジグリシジルエーテル、1,6-ヘキサンジオールジグリシジルエーテル、シクロヘキサンジメタノールジグリシジルエーテルなどが挙げられる。前記脂肪族多塩基酸のジグリシジルエステルとしては、より具体的には、ダイマー酸ジグリシジルエステル、アジピン酸ジグリシジルエステル、セバシン酸ジグリシジルエステル、マレイン酸ジグリシジルエステルなどが挙げられる。前記二価以上の多価脂肪族アルコールのグリシジルエーテルとしては、より具体的には、トリメチロールプロパントリグリシジルエーテル、トリメチロールエタントリグリシジルエーテル、ひまし油変性ポリグリシジルエーテル、プロポキシ化グリセリントリグリシジルエーテル、ソルビトールポリグリシジルエーテルなどが挙げられる。
(A-3)エポキシ樹脂に多塩基酸類等を付加反応させて得られるエポキシ化合物
さらに、本発明のエポキシ樹脂(A)としては、WO2010/098950号公報に記載されているような、エポキシ樹脂に多塩基酸類等を付加反応させて得られるエポキシ化合物も使用できる。例えば、トール油脂肪酸の二量体(ダイマー酸)とビスフェノールA型エポキシ樹脂との付加反応物が挙げられる。また、キレート変性エポキシ樹脂、ゴム変性エポキシ樹脂、及び、ウレタン変性エポキシ樹脂も、エポキシ樹脂(A)成分として使用できる。
さらに、本発明のエポキシ樹脂(A)としては、WO2010/098950号公報に記載されているような、エポキシ樹脂に多塩基酸類等を付加反応させて得られるエポキシ化合物も使用できる。例えば、トール油脂肪酸の二量体(ダイマー酸)とビスフェノールA型エポキシ樹脂との付加反応物が挙げられる。また、キレート変性エポキシ樹脂、ゴム変性エポキシ樹脂、及び、ウレタン変性エポキシ樹脂も、エポキシ樹脂(A)成分として使用できる。
本発明で用いられ得るエポキシ樹脂は前述のようなものであるが、一般的にはエポキシ当量として、80~2000を有するものが挙げられる。これらのポリエポキシドは周知の方法で得ることができるが、通常よく用いられる方法として、例えば、多価アルコールもしくは多価フェノールなどに対して過剰量のエピハロヒドリンを塩基存在下で反応させることが挙げられる。
(A-3-1)キレート変性エポキシ樹脂
本発明のエポキシ樹脂(A)としては、キレート変性エポキシ樹脂が使用できる。キレート変性エポキシ樹脂はエポキシ樹脂とキレート官能基を含有する化合物(キレート配位子)との反応生成物であり、本発明の樹脂組成物に添加して車両用接着剤として用いた場合、油状物質で汚染された金属基材表面への接着性を改善できる。キレート官能基は、金属イオンへ配位可能な配位座を分子内に複数有する化合物の官能基であり、例えば、リン含有酸基(例えば、-PO(OH)2)、カルボン酸基(-CO2H)、硫黄含有酸基(例えば、-SO3H)、アミノ基及び水酸基(特に、芳香環において互いに隣接した水酸基)などが挙げられる。キレート配位子としては、エチレンジアミン、ビピリジン、エチレンジアミン四酢酸、フェナントロリン、ポルフィリン、クラウンエーテル、などが挙げられる。市販されているキレート変性エポキシ樹脂としては、ADEKA製アデカレジンEP-49-10N、などが挙げられる。エポキシ樹脂(A)成分100質量%中のキレート変性エポキシ樹脂の使用量は、好ましくは0.1~10質量%、より好ましくは0.5~3質量%である。
本発明のエポキシ樹脂(A)としては、キレート変性エポキシ樹脂が使用できる。キレート変性エポキシ樹脂はエポキシ樹脂とキレート官能基を含有する化合物(キレート配位子)との反応生成物であり、本発明の樹脂組成物に添加して車両用接着剤として用いた場合、油状物質で汚染された金属基材表面への接着性を改善できる。キレート官能基は、金属イオンへ配位可能な配位座を分子内に複数有する化合物の官能基であり、例えば、リン含有酸基(例えば、-PO(OH)2)、カルボン酸基(-CO2H)、硫黄含有酸基(例えば、-SO3H)、アミノ基及び水酸基(特に、芳香環において互いに隣接した水酸基)などが挙げられる。キレート配位子としては、エチレンジアミン、ビピリジン、エチレンジアミン四酢酸、フェナントロリン、ポルフィリン、クラウンエーテル、などが挙げられる。市販されているキレート変性エポキシ樹脂としては、ADEKA製アデカレジンEP-49-10N、などが挙げられる。エポキシ樹脂(A)成分100質量%中のキレート変性エポキシ樹脂の使用量は、好ましくは0.1~10質量%、より好ましくは0.5~3質量%である。
(A-3-2)ゴム変性エポキシ樹脂
前記ゴム変性エポキシ樹脂は、ゴムとエポキシ基含有化合物とを反応させて得た、1分子当り平均して、エポキシ基を1.1個以上有する反応生成物であり、ゴムとしては、アクリロニトリルブタジエンゴム(NBR)、スチレンブタジエンゴム(SBR)、水素添加ニトリルゴム(HNBR)、エチレンプロピレンゴム(EPDM)、アクリルゴム(ACM)、ブチルゴム(IIR)、ブタジエンゴム、ポリプロピレンオキシドやポリエチレンオキシドやポリテトラメチレンオキシド等のポリオキシアルキレンなどのゴム系重合体を挙げることができる。該ゴム系重合体は、アミノ基、ヒドロキシ基、またはカルボキシル基等の反応性基を末端に有するものが好ましい。これらのゴム系重合体とエポキシ樹脂とを公知の方法により適宜の配合比にて反応させた生成物を本発明に使用されるゴム変性エポキシ樹脂としてもよい。これらの中でも、アクリロニトリル-ブタジエンゴム変性エポキシ樹脂や、ポリオキシアルキレン変性エポキシ樹脂が、得られる樹脂組成物の接着性や耐衝撃剥離接着性の観点から好ましく、アクリロニトリル-ブタジエンゴム変性エポキシ樹脂がより好ましい。なお、アクリロニトリル-ブタジエンゴム変性エポキシ樹脂は、例えば、カルボキシル基末端NBR(CTBN)とビスフェノールA型エポキシ樹脂との反応により得られてもよい。また、ポリオキシアルキレン変性エポキシ樹脂は、例えば、アミノ基末端ポリオキシアルキレンとビスフェノールA型エポキシ樹脂との反応により得られてもよい。
前記アクリロニトリル-ブタジエンゴム100質量%中のアクリロニトリル単量体成分の含有量は、得られる樹脂組成物の接着性や耐衝撃剥離接着性の観点から、5~40質量%が好ましく、10~35質量%がより好ましく、15~30質量%が更に好ましい。得られる樹脂組成物のチキソトロピー性の観点から、20~30質量%が特に好ましい。
前記ゴム変性エポキシ樹脂は、ゴムとエポキシ基含有化合物とを反応させて得た、1分子当り平均して、エポキシ基を1.1個以上有する反応生成物であり、ゴムとしては、アクリロニトリルブタジエンゴム(NBR)、スチレンブタジエンゴム(SBR)、水素添加ニトリルゴム(HNBR)、エチレンプロピレンゴム(EPDM)、アクリルゴム(ACM)、ブチルゴム(IIR)、ブタジエンゴム、ポリプロピレンオキシドやポリエチレンオキシドやポリテトラメチレンオキシド等のポリオキシアルキレンなどのゴム系重合体を挙げることができる。該ゴム系重合体は、アミノ基、ヒドロキシ基、またはカルボキシル基等の反応性基を末端に有するものが好ましい。これらのゴム系重合体とエポキシ樹脂とを公知の方法により適宜の配合比にて反応させた生成物を本発明に使用されるゴム変性エポキシ樹脂としてもよい。これらの中でも、アクリロニトリル-ブタジエンゴム変性エポキシ樹脂や、ポリオキシアルキレン変性エポキシ樹脂が、得られる樹脂組成物の接着性や耐衝撃剥離接着性の観点から好ましく、アクリロニトリル-ブタジエンゴム変性エポキシ樹脂がより好ましい。なお、アクリロニトリル-ブタジエンゴム変性エポキシ樹脂は、例えば、カルボキシル基末端NBR(CTBN)とビスフェノールA型エポキシ樹脂との反応により得られてもよい。また、ポリオキシアルキレン変性エポキシ樹脂は、例えば、アミノ基末端ポリオキシアルキレンとビスフェノールA型エポキシ樹脂との反応により得られてもよい。
前記アクリロニトリル-ブタジエンゴム100質量%中のアクリロニトリル単量体成分の含有量は、得られる樹脂組成物の接着性や耐衝撃剥離接着性の観点から、5~40質量%が好ましく、10~35質量%がより好ましく、15~30質量%が更に好ましい。得られる樹脂組成物のチキソトロピー性の観点から、20~30質量%が特に好ましい。
前記ゴム中の1分子当たりの平均のエポキシド反応性末端基の数は、1.5~2.5個が好ましく、1.8~2.2個がより好ましい。ゴムの数平均分子量は、GPCで測定したポリスチレン換算分子量にて、2000~10000が好ましく、3000~8000がより好ましく、4000~6000が特に好ましい。
ゴム変性エポキシ樹脂の製法は、特に制限は無く、例えば、多量のエポキシ基含有化合物中でゴムとエポキシ基含有化合物とを反応させて製造することができる。具体的には、ゴム中の1当量のエポキシ反応性末端基当たり、2当量以上のエポキシ基含有化合物を反応させて製造することが好ましい。得られる生成物が、ゴムとエポキシ基含有化合物との付加体と、遊離のエポキシ基含有化合物との混合物となるのに十分な量のエポキシ基含有化合物を反応させることがより好ましい。フェニルジメチル尿素やトリフェニルホスフィンなどの触媒の存在下で、100~250℃の温度に加熱することにより、ゴム変性エポキシ樹脂は製造されてもよい。ゴム変性エポキシ樹脂を製造する際に使用されるエポキシ基含有化合物は特に制限は無いが、ビスフェノールA型エポキシ樹脂やビスフェノールF型エポキシ樹脂が好ましく、ビスフェノールA型エポキシ樹脂がより好ましい。なお、本発明において、ゴム変性エポキシ樹脂の製造時に過剰量のエポキシ基含有化合物が使用された場合には、反応後に残存する未反応のエポキシ基含有化合物は、本願のゴム変性エポキシ樹脂には、含まれないものとする。
ゴム変性エポキシ樹脂は、ビスフェノール成分と予備反応させることで改質することができる。改質に使用するビスフェノール成分は、ゴム変性エポキシ樹脂中のゴム成分100質量部に対し、3~35質量部が好ましく、5~25質量部がより好ましい。改質されたゴム変性エポキシ樹脂を含有する樹脂組成物を硬化してなる硬化物は、高温曝露後の接着耐久性に優れ、また、低温時の耐衝撃性にも優れる。
ゴム変性エポキシ樹脂のガラス転移温度(Tg)は、特に制限は無いが、-25℃以下が好ましく、-35℃以下がより好ましく、-40℃以下が更に好ましく、-50℃以下が特に好ましい。
エポキシ樹脂(A)成分100質量%中のゴム変性エポキシ樹脂の含有量は、1~40質量%が好ましく、3~30質量%がより好ましく、5~25質量%が更に好ましく、10~20質量%が特に好ましい。1質量%未満では、得られる硬化物が脆く、耐衝撃剥離接着性が低い場合があり、40質量%より多いと、得られる硬化物の耐熱性や弾性率(剛性)が低い場合がある。
ゴム変性エポキシ樹脂は、単独でまたは2種以上を組み合わせて使用することができる。
ゴム変性エポキシ樹脂は、単独でまたは2種以上を組み合わせて使用することができる。
(A-3-3)ウレタン変性エポキシ樹脂
前記ウレタン変性エポキシ樹脂は、イソシアネート基との反応性を有する基とエポキシ基とを含有する化合物と、イソシアネート基を含有するウレタンプレポリマーを反応させて得た、1分子当り平均して、エポキシ基を1.1個以上有する反応生成物である。例えば、ヒドロキシ基含有エポキシ化合物とウレタンプレポリマーを反応させることにより、ウレタン変性エポキシ樹脂が得られる。
前記ウレタン変性エポキシ樹脂は、イソシアネート基との反応性を有する基とエポキシ基とを含有する化合物と、イソシアネート基を含有するウレタンプレポリマーを反応させて得た、1分子当り平均して、エポキシ基を1.1個以上有する反応生成物である。例えば、ヒドロキシ基含有エポキシ化合物とウレタンプレポリマーを反応させることにより、ウレタン変性エポキシ樹脂が得られる。
エポキシ樹脂(A)成分100質量%中のウレタン変性エポキシ樹脂の含有量は、1~40質量%が好ましく、3~30質量%がより好ましく、5~25質量%が更に好ましく、10~20質量%が特に好ましい。1質量%未満では、得られる硬化物が脆く、耐衝撃剥離接着性が低い場合があり、40質量%より多いと、得られる硬化物の耐熱性や弾性率(剛性)が低い場合がある。
ウレタン変性エポキシ樹脂は、単独でまたは2種以上を組み合わせて使用することができる。
ウレタン変性エポキシ樹脂は、単独でまたは2種以上を組み合わせて使用することができる。
前記のエポキシ樹脂(A)の中でも、ビスフェノールA型エポキシ樹脂やビスフェノールF型エポキシ樹脂は、得られる硬化物の弾性率が高く、耐熱性および接着性に優れ、比較的安価である為好ましく、ビスフェノールA型エポキシ樹脂が特に好ましい。
本発明のエポキシ樹脂(A)のエポキシ当量は、得られる硬化物の弾性率および耐熱性が高いという観点から、220未満が好ましく、90以上210以下がより好ましく、150以上200以下が更に好ましい。特に、エポキシ当量が220未満のビスフェノールA型エポキシ樹脂やビスフェノールF型エポキシ樹脂は、常温で液体であり、得られる樹脂組成物の取扱い性が良い為に好ましい。
エポキシ当量が220以上2000未満のビスフェノールA型エポキシ樹脂やビスフェノールF型エポキシ樹脂を、エポキシ樹脂(A)成分100質量%中に、好ましくは40質量%以下、より好ましくは20質量%以下の範囲で添加すると、得られる硬化物が耐衝撃性に優れる為に好ましい。
<コアシェルポリマー(B)>
本発明のコアシェルポリマー(B)は、コア層とシェル層の少なくとも2層からなる。また、本発明のコアシェルポリマー(B)は、コア層と中間層とシェル層の3層からなるものでもよい。本発明のコアシェルポリマー(B)の体積平均粒子径は、0.03~2.10μmが好ましく、0.05~1.10μmがより好ましく、0.05~0.30μmがさらに好ましく、0.08~0.30μmが特に好ましい。体積平均粒子径が0.03μm未満のものを安定的に得ることは難しい場合が多く、2.10μmを超えると最終成形体の耐熱性や耐衝撃性が悪くなる恐れがある。
本発明のコアシェルポリマー(B)は、コア層とシェル層の少なくとも2層からなる。また、本発明のコアシェルポリマー(B)は、コア層と中間層とシェル層の3層からなるものでもよい。本発明のコアシェルポリマー(B)の体積平均粒子径は、0.03~2.10μmが好ましく、0.05~1.10μmがより好ましく、0.05~0.30μmがさらに好ましく、0.08~0.30μmが特に好ましい。体積平均粒子径が0.03μm未満のものを安定的に得ることは難しい場合が多く、2.10μmを超えると最終成形体の耐熱性や耐衝撃性が悪くなる恐れがある。
以下、各層に関して説明する。
(コア層)
コア層は、ジエン系ゴム、アクリル系ゴム、ポリシロキサン系ゴムのいずれかを使用する。
本発明のコアシェルポリマー(B)のコア層は、天然ゴムやジエン系モノマー(共役ジエン系モノマー)および(メタ)アクリレート系モノマーから選ばれる少なくとも1種のモノマー(第1モノマー)を50~100質量%、および他の共重合可能なビニル系モノマー(第2モノマー)を0~50質量%含んで構成されるゴム弾性体や、ポリシロキサンゴム系弾性体、あるいはこれらを併用したものが挙げられる。コアシェルポリマー(B)のコア層は、得られる硬化物の靱性改良効果および耐衝撃剥離接着性改良効果が高い点、および、マトリックス樹脂との親和性が低い為にコア層の膨潤による経時での粘度上昇が起こり難い点から、ジエン系モノマーを用いたジエン系ゴムが好ましい。多種のモノマーの組合せにより、幅広いポリマー設計が可能なことから、(メタ)アクリレート系ゴムが好ましい。また、硬化物の耐熱性を低下させることなく、低温での耐衝撃性を向上しようとする場合には、弾性コア層はポリシロキサン系ゴムであることが好ましい。なお、本発明において(メタ)アクリレートとは、アクリレートおよび/またはメタクリレートを意味する。
(コア層)
コア層は、ジエン系ゴム、アクリル系ゴム、ポリシロキサン系ゴムのいずれかを使用する。
本発明のコアシェルポリマー(B)のコア層は、天然ゴムやジエン系モノマー(共役ジエン系モノマー)および(メタ)アクリレート系モノマーから選ばれる少なくとも1種のモノマー(第1モノマー)を50~100質量%、および他の共重合可能なビニル系モノマー(第2モノマー)を0~50質量%含んで構成されるゴム弾性体や、ポリシロキサンゴム系弾性体、あるいはこれらを併用したものが挙げられる。コアシェルポリマー(B)のコア層は、得られる硬化物の靱性改良効果および耐衝撃剥離接着性改良効果が高い点、および、マトリックス樹脂との親和性が低い為にコア層の膨潤による経時での粘度上昇が起こり難い点から、ジエン系モノマーを用いたジエン系ゴムが好ましい。多種のモノマーの組合せにより、幅広いポリマー設計が可能なことから、(メタ)アクリレート系ゴムが好ましい。また、硬化物の耐熱性を低下させることなく、低温での耐衝撃性を向上しようとする場合には、弾性コア層はポリシロキサン系ゴムであることが好ましい。なお、本発明において(メタ)アクリレートとは、アクリレートおよび/またはメタクリレートを意味する。
(コア層(ジエン系ゴム))
第1モノマーの前記ジエン系モノマー(共役ジエン系モノマー)としては、例えば、1,3-ブタジエン、イソプレン、2-クロロ-1,3-ブタジエン、2-メチル-1,3-ブタジエンなどが挙げられる。これらのジエン系モノマーは、単独で用いても、2種以上を組み合わせて用いてもよい。
第1モノマーの前記ジエン系モノマー(共役ジエン系モノマー)としては、例えば、1,3-ブタジエン、イソプレン、2-クロロ-1,3-ブタジエン、2-メチル-1,3-ブタジエンなどが挙げられる。これらのジエン系モノマーは、単独で用いても、2種以上を組み合わせて用いてもよい。
コアシェルポリマー(B)のコア層は、靱性改良効果および耐衝撃剥離接着性改良効果が高い点、および、マトリックス樹脂との親和性が低い為にコア層の膨潤による経時での粘度上昇が起こり難い点から、1,3-ブタジエンを用いるブタジエンゴム、および/または、1,3-ブタジエンとスチレンの共重合体であるブタジエン-スチレンゴムが好ましく、ブタジエンゴムがより好ましい。また、ブタジエン-スチレンゴムは、屈折率の調整により得られる硬化物の透明性を高めることができ、より好ましい。
(コア層(アクリル系ゴム))
また、第1モノマーの前記(メタ)アクリレート系ゴムモノマーとしては、例えば、メチル(メタ)アクリレート、エチル(メタ)アクリレート、ブチル(メタ)アクリレート、2-エチルヘキシル(メタ)アクリレート、オクチル(メタ)アクリレート、ドデシル(メタ)アクリレート、ステアリル(メタ)アクリレート、ベヘニル(メタ)アクリレートなどのアルキル(メタ)アクリレート類;フェノキシエチル(メタ)アクリレート、ベンジル(メタ)アクリレートなどの芳香環含有(メタ)アクリレート;2-ヒドロキシエチル(メタ)アクリレート、4-ヒドロキシブチル(メタ)アクリレートなどのヒドロキシアルキル(メタ)アクリレート類;グリシジル(メタ)アクリレート、グリシジルアルキル(メタ)アクリレートなどのグリシジル(メタ)アクリレート類;アルコキシアルキル(メタ)アクリレート類;アリル(メタ)アクリレート、アリルアルキル(メタ)アクリレートなどのアリルアルキル(メタ)アクリレート類;モノエチレングリコールジ(メタ)アクリレート、トリエチレングリコールジ(メタ)アクリレート、テトラエチレングリコールジ(メタ)アクリレートなどの多官能性(メタ)アクリレート類などが挙げられる。これらの(メタ)アクリレート系モノマーは、単独で用いても、2種以上を組み合わせて用いてもよい。特に好ましくはエチル(メタ)アクリレート、ブチル(メタ)アクリレート、2-エチルヘキシル(メタ)アクリレートである。
また、第1モノマーの前記(メタ)アクリレート系ゴムモノマーとしては、例えば、メチル(メタ)アクリレート、エチル(メタ)アクリレート、ブチル(メタ)アクリレート、2-エチルヘキシル(メタ)アクリレート、オクチル(メタ)アクリレート、ドデシル(メタ)アクリレート、ステアリル(メタ)アクリレート、ベヘニル(メタ)アクリレートなどのアルキル(メタ)アクリレート類;フェノキシエチル(メタ)アクリレート、ベンジル(メタ)アクリレートなどの芳香環含有(メタ)アクリレート;2-ヒドロキシエチル(メタ)アクリレート、4-ヒドロキシブチル(メタ)アクリレートなどのヒドロキシアルキル(メタ)アクリレート類;グリシジル(メタ)アクリレート、グリシジルアルキル(メタ)アクリレートなどのグリシジル(メタ)アクリレート類;アルコキシアルキル(メタ)アクリレート類;アリル(メタ)アクリレート、アリルアルキル(メタ)アクリレートなどのアリルアルキル(メタ)アクリレート類;モノエチレングリコールジ(メタ)アクリレート、トリエチレングリコールジ(メタ)アクリレート、テトラエチレングリコールジ(メタ)アクリレートなどの多官能性(メタ)アクリレート類などが挙げられる。これらの(メタ)アクリレート系モノマーは、単独で用いても、2種以上を組み合わせて用いてもよい。特に好ましくはエチル(メタ)アクリレート、ブチル(メタ)アクリレート、2-エチルヘキシル(メタ)アクリレートである。
(第2モノマー)
前記他の共重合可能なビニル系モノマー(第2モノマー)としては、例えば、スチレン、α-メチルスチレン、モノクロロスチレン、ジクロロスチレンなどのビニルアレーン類;アクリル酸、メタクリル酸などのビニルカルボン酸類;アクリロニトリル、メタクリロニトリルなどのビニルシアン類;塩化ビニル、臭化ビニル、クロロプレンなどのハロゲン化ビニル類;酢酸ビニル;エチレン、プロピレン、ブチレン、イソブチレンなどのアルケン類;ジアリルフタレート、トリアリルシアヌレート、トリアリルイソシアヌレート、ジビニルベンゼンなどの多官能性モノマーなどが挙げられる。これらのビニル系モノマーは、単独で用いても、2種以上を組み合わせて用いてもよい。特に好ましくはスチレンである。
前記他の共重合可能なビニル系モノマー(第2モノマー)としては、例えば、スチレン、α-メチルスチレン、モノクロロスチレン、ジクロロスチレンなどのビニルアレーン類;アクリル酸、メタクリル酸などのビニルカルボン酸類;アクリロニトリル、メタクリロニトリルなどのビニルシアン類;塩化ビニル、臭化ビニル、クロロプレンなどのハロゲン化ビニル類;酢酸ビニル;エチレン、プロピレン、ブチレン、イソブチレンなどのアルケン類;ジアリルフタレート、トリアリルシアヌレート、トリアリルイソシアヌレート、ジビニルベンゼンなどの多官能性モノマーなどが挙げられる。これらのビニル系モノマーは、単独で用いても、2種以上を組み合わせて用いてもよい。特に好ましくはスチレンである。
(ポリシロキサン系ゴム)
また、前記ポリシロキサン系ゴムとしては、例えば、ジメチルシリルオキシ、ジエチルシリルオキシ、メチルフェニルシリルオキシ、ジフェニルシリルオキシ、ジメチルシリルオキシ-ジフェニルシリルオキシなどの、アルキル或いはアリール2置換シリルオキシ単位から構成されるポリシロキサン系ポリマーや、側鎖のアルキルの一部が水素原子に置換されたオルガノハイドロジェンシリルオキシなどの、アルキル或いはアリール1置換シリルオキシ単位から構成されるポリシロキサン系ポリマーが挙げられる。これらのポリシロキサン系ポリマーは、単独で用いても、2種以上を組み合わせて用いてもよい。中でも、ジメチルシリルオキシ、メチルフェニルシリルオキシ、ジメチルシリルオキシ-ジフェニルシリルオキシが硬化物に耐熱性を付与する上で好ましく、ジメチルシリルオキシが容易に入手できて経済的でもあることから最も好ましい。コア層がポリシロキサン系ゴムから形成される態様において、ポリシロキサン系ポリマー部位は、硬化物の耐熱性を損なわないために、ゴム弾性体全体を100質量%として80質量%以上(より好ましくは90質量%以上)含有していることが好ましい。
また、前記ポリシロキサン系ゴムとしては、例えば、ジメチルシリルオキシ、ジエチルシリルオキシ、メチルフェニルシリルオキシ、ジフェニルシリルオキシ、ジメチルシリルオキシ-ジフェニルシリルオキシなどの、アルキル或いはアリール2置換シリルオキシ単位から構成されるポリシロキサン系ポリマーや、側鎖のアルキルの一部が水素原子に置換されたオルガノハイドロジェンシリルオキシなどの、アルキル或いはアリール1置換シリルオキシ単位から構成されるポリシロキサン系ポリマーが挙げられる。これらのポリシロキサン系ポリマーは、単独で用いても、2種以上を組み合わせて用いてもよい。中でも、ジメチルシリルオキシ、メチルフェニルシリルオキシ、ジメチルシリルオキシ-ジフェニルシリルオキシが硬化物に耐熱性を付与する上で好ましく、ジメチルシリルオキシが容易に入手できて経済的でもあることから最も好ましい。コア層がポリシロキサン系ゴムから形成される態様において、ポリシロキサン系ポリマー部位は、硬化物の耐熱性を損なわないために、ゴム弾性体全体を100質量%として80質量%以上(より好ましくは90質量%以上)含有していることが好ましい。
(コア層の架橋)
コアシェルポリマー(B)のコア層には、硬化性エポキシ樹脂組成物中での分散安定性を保持する観点から、架橋構造が導入されていることが好ましい。架橋構造の導入方法としては、例えば、ポリマー成分に多官能性モノマーやメルカプト基含有化合物等の架橋性モノマーを添加し、次いで重合する方法などが挙げられる。また、ポリシロキサン系ポリマーに架橋構造を導入する方法としては、重合時に多官能性のアルコキシシラン化合物を一部併用する方法や、ビニル反応性基、メルカプト基などの反応性基をポリシロキサン系ポリマーに導入し、その後ビニル重合性のモノマーあるいは有機過酸化物などを添加してラジカル反応させる方法、あるいは、ポリシロキサン系ポリマーに多官能性モノマーやメルカプト基含有化合物などの架橋性モノマーを添加し、次いで重合する方法などが挙げられる。
コアシェルポリマー(B)のコア層には、硬化性エポキシ樹脂組成物中での分散安定性を保持する観点から、架橋構造が導入されていることが好ましい。架橋構造の導入方法としては、例えば、ポリマー成分に多官能性モノマーやメルカプト基含有化合物等の架橋性モノマーを添加し、次いで重合する方法などが挙げられる。また、ポリシロキサン系ポリマーに架橋構造を導入する方法としては、重合時に多官能性のアルコキシシラン化合物を一部併用する方法や、ビニル反応性基、メルカプト基などの反応性基をポリシロキサン系ポリマーに導入し、その後ビニル重合性のモノマーあるいは有機過酸化物などを添加してラジカル反応させる方法、あるいは、ポリシロキサン系ポリマーに多官能性モノマーやメルカプト基含有化合物などの架橋性モノマーを添加し、次いで重合する方法などが挙げられる。
(多官能性モノマー)
前記多官能性モノマーとしては、アリル(メタ)アクリレート、アリルアルキル(メタ)アクリレート等のアリルアルキル(メタ)アクリレート類;アリルオキシアルキル(メタ)アクリレート類;(ポリ)エチレングリコールジ(メタ)アクリレート、ブタンジオールジ(メタ)アクリレート、エチレングリコールジ(メタ)アクリレート、トリエチレングリコールジ(メタ)アクリレート、テトラエチレングリコールジ(メタ)アクリレート等の(メタ)アクリル基を2個以上有する多官能(メタ)アクリレート類;ジアリルフタレート、トリアリルシアヌレート、トリアリルイソシアヌレート、ジビニルベンゼン等が挙げられる。特に好ましくはアリルメタアクリレート、トリアリルイソシアヌレート、ブタンジオールジ(メタ)アクリレート、及びジビニルベンゼンである。
前記多官能性モノマーとしては、アリル(メタ)アクリレート、アリルアルキル(メタ)アクリレート等のアリルアルキル(メタ)アクリレート類;アリルオキシアルキル(メタ)アクリレート類;(ポリ)エチレングリコールジ(メタ)アクリレート、ブタンジオールジ(メタ)アクリレート、エチレングリコールジ(メタ)アクリレート、トリエチレングリコールジ(メタ)アクリレート、テトラエチレングリコールジ(メタ)アクリレート等の(メタ)アクリル基を2個以上有する多官能(メタ)アクリレート類;ジアリルフタレート、トリアリルシアヌレート、トリアリルイソシアヌレート、ジビニルベンゼン等が挙げられる。特に好ましくはアリルメタアクリレート、トリアリルイソシアヌレート、ブタンジオールジ(メタ)アクリレート、及びジビニルベンゼンである。
(コア層のゲル含量)
本発明のコアシェルポリマー(B)のコア層は、硬化性エポキシ樹脂組成物の硬化物の靱性を高める為に、ゴムとしての性質を有することが好ましい。本発明のコアシェルポリマー(B)のコア層のゲル含量は、硬化物の靭性の観点から、60質量%以上であることが好ましく、80質量%以上であることがより好ましく、90質量%以上であることがさらに好ましく、95質量%以上であることが特に好ましい。ゲル含量とは、凝固、乾燥により得られたコアシェルポリマー0.5gをトルエン100gに浸漬し、23℃で24時間静置した後に不溶分と可溶分を分別したときの、不溶分と可溶分の合計量に対する不溶分の比率を意味する。
本発明のコアシェルポリマー(B)のコア層は、硬化性エポキシ樹脂組成物の硬化物の靱性を高める為に、ゴムとしての性質を有することが好ましい。本発明のコアシェルポリマー(B)のコア層のゲル含量は、硬化物の靭性の観点から、60質量%以上であることが好ましく、80質量%以上であることがより好ましく、90質量%以上であることがさらに好ましく、95質量%以上であることが特に好ましい。ゲル含量とは、凝固、乾燥により得られたコアシェルポリマー0.5gをトルエン100gに浸漬し、23℃で24時間静置した後に不溶分と可溶分を分別したときの、不溶分と可溶分の合計量に対する不溶分の比率を意味する。
(コア層のTg)
本発明において、コアシェルポリマー(B)のコア層のガラス転移温度(以下、単に「Tg」と称する場合がある)は、得られる硬化物の靱性を高める為に、0℃以下であることが好ましく、-20℃以下がより好ましく、-40℃以下が更に好ましく、-60℃以下であることが特に好ましい。
本発明において、コアシェルポリマー(B)のコア層のガラス転移温度(以下、単に「Tg」と称する場合がある)は、得られる硬化物の靱性を高める為に、0℃以下であることが好ましく、-20℃以下がより好ましく、-40℃以下が更に好ましく、-60℃以下であることが特に好ましい。
一方、得られる硬化物の剛性低下を抑制したい場合には、コア層のTgは、0℃よりも大きいことが好ましく、20℃以上であることがより好ましく、50℃以上であることが更に好ましく、80℃以上であることが特に好ましく、120℃以上であることが最も好ましい。
Tgが0℃よりも大きく、得られる硬化物の剛性低下を抑制し得るコア層を形成し得るポリマーとしては、単独重合体のTgが0℃よりも大きい少なくとも1種のモノマーを50~100質量%(好ましくは、65~99質量%)、および単独重合体のTgが0℃以下の少なくとも1種のモノマーを0~50質量%(好ましくは、1~35質量%)含んで構成されるポリマーが挙げられる。
単独重合体のTgが0℃よりも大きいモノマーは、例えば、スチレン、2-ビニルナフタレン等の無置換ビニル芳香族化合物類;α-メチルスチレン等のビニル置換芳香族化合物類;3-メチルスチレン、4-メチルスチレン、2,4-ジメチルスチレン、2,5-ジメチルスチレン、3,5-ジメチルスチレン、2,4,6-トリメチルスチレン等の環アルキル化ビニル芳香族化合物類;4-メトキシスチレン、4-エトキシスチレン等の環アルコキシル化ビニル芳香族化合物類;2-クロロスチレン、3-クロロスチレン等の環ハロゲン化ビニル芳香族化合物類;4-アセトキシスチレン等の環エステル置換ビニル芳香族化合物類;4-ヒドロキシスチレン等の環ヒドロキシル化ビニル芳香族化合物類;ビニルベンゾエート、ビニルシクロヘキサノエート等のビニルエステル類;塩化ビニル等のビニルハロゲン化物類;アセナフタレン、インデン等の芳香族モノマー類;メチルメタクリレート、エチルメタクリレート、イソプロピルメタクリレート等のアルキルメタクリレート類;フェニルメタクリレート等の芳香族メタクリレート;イソボルニルメタクリレート、トリメチルシリルメタクリレート等のメタクリレート類;メタクリロニトリル等のメタクリル酸誘導体を含むメタクリルモノマー;イソボルニルアクリレート、tert-ブチルアクリレート等のある種のアクリル酸エステル;アクリロニトリル等のアクリル酸誘導体を含むアクリルモノマーを挙げることができる。更に、アクリルアミド、イソプロピルアクリルアミド、N-ビニルピロリドン、イソボルニルメタクリレート、ジシクロペンタニルメタクリレート、2-メチル-2-アダマンチルメタクリレート、1-アダマンチルアクリレート及び1-アダマンチルメタクリレート等のTgが120℃以上となるモノマーが挙げられる。
(コア層の平均粒子径)
本発明のコアシェルポリマー(B)のコア層の体積平均粒子径は、0.03~2μmが好ましく、0.05~1μmがより好ましい。体積平均粒子径が0.03μm未満のものを安定的に得ることは難しい場合が多く、2μmを超えると最終成形体の耐熱性や耐衝撃性が悪くなる恐れがある。
本発明のコアシェルポリマー(B)のコア層の体積平均粒子径は、0.03~2μmが好ましく、0.05~1μmがより好ましい。体積平均粒子径が0.03μm未満のものを安定的に得ることは難しい場合が多く、2μmを超えると最終成形体の耐熱性や耐衝撃性が悪くなる恐れがある。
(コア層の比率)
本発明のコアシェルポリマー(B)のコア層は、コアシェルポリマー全体を100質量%として40~97質量%が好ましく、60~95質量%がより好ましく、70~93質量%が更に好ましく、80~90質量%が特に好ましい。コア層が40質量%未満では硬化物の靱性改良効果が低下する場合がある。コア層が97質量%よりも大きいとポリマー微粒子が凝集し易くなり、硬化性エポキシ樹脂組成物が高粘度となり取り扱い難い場合がある。
本発明のコアシェルポリマー(B)のコア層は、コアシェルポリマー全体を100質量%として40~97質量%が好ましく、60~95質量%がより好ましく、70~93質量%が更に好ましく、80~90質量%が特に好ましい。コア層が40質量%未満では硬化物の靱性改良効果が低下する場合がある。コア層が97質量%よりも大きいとポリマー微粒子が凝集し易くなり、硬化性エポキシ樹脂組成物が高粘度となり取り扱い難い場合がある。
(コア層の多層構造)
本発明において、コア層は多層構造であってもよい。また、コア層が多層構造の場合は、各層のポリマー組成が各々相違していてもよい。
本発明において、コア層は多層構造であってもよい。また、コア層が多層構造の場合は、各層のポリマー組成が各々相違していてもよい。
(中間層)
本発明のコアシェルポリマー(B)は、コア層とシェル層の間に中間層を有してもよい。特に、中間層として、以下のゴム表面架橋層を形成させてもよい。得られる硬化物の靱性改良効果および耐衝撃剥離接着性改良効果の点からは、中間層として、以下のゴム表面架橋層を含有しないことが好ましい。
本発明のコアシェルポリマー(B)は、コア層とシェル層の間に中間層を有してもよい。特に、中間層として、以下のゴム表面架橋層を形成させてもよい。得られる硬化物の靱性改良効果および耐衝撃剥離接着性改良効果の点からは、中間層として、以下のゴム表面架橋層を含有しないことが好ましい。
前記ゴム表面架橋層は、同一分子内にラジカル性二重結合を2以上有する多官能性モノマー30~100質量%、及びその他のビニルモノマー0~70質量%からなるゴム表面架橋層成分を重合してなる中間層ポリマーからなり、本発明の硬化性エポキシ樹脂組成物の粘度を低下させる効果、コアシェルポリマー(B)成分のエポキシ樹脂(A)成分への分散性を向上させる効果を有する。また、前記ゴム表面架橋層は、コア層の架橋密度を上げたりシェル層のグラフト効率を高めたりする効果も有する。
前記多官能性モノマーの具体例としては、上述の多官能性モノマーと同じモノマーが例示されるが、好ましくはアリルメタクリレート、トリアリルイソシアヌレートである。
前記多官能性モノマーの具体例としては、上述の多官能性モノマーと同じモノマーが例示されるが、好ましくはアリルメタクリレート、トリアリルイソシアヌレートである。
(シェル層)
コアシェルポリマー(B)のシェル層は、シェル形成用モノマーを重合したものであるが、本発明に係る、コアシェルポリマー(B)成分とエポキシ樹脂(A)成分との相溶性を向上させ、本発明の硬化性エポキシ樹脂組成物、又はその硬化物中においてコアシェルポリマーが一次粒子の状態で分散することを可能にする役割を担うシェルポリマーからなる。
コアシェルポリマー(B)のシェル層は、シェル形成用モノマーを重合したものであるが、本発明に係る、コアシェルポリマー(B)成分とエポキシ樹脂(A)成分との相溶性を向上させ、本発明の硬化性エポキシ樹脂組成物、又はその硬化物中においてコアシェルポリマーが一次粒子の状態で分散することを可能にする役割を担うシェルポリマーからなる。
本発明のコアシェルポリマー(B)のシェル層は、チキソトロピー性が高くなる観点から、シェル層を構成するモノマー全量を100質量%とし、ビニルシアンモノマーおよびアルコキシアルキル(メタ)アクリレートモノマーからなる群より選択されるモノマー10~95質量%、エポキシ基を有するモノマー0~50質量%、それらと共重合可能なモノマー0~90質量%(好ましくは5~90質量%)からなることが好ましい。
より好ましくは、本発明のコアシェルポリマー(B)のシェル層は、貯蔵安定性を維持しつつ、動的割裂抵抗力が高いとの観点から、シェル層を構成するモノマー全量を100質量%とし、ビニルシアンモノマーおよびアルコキシアルキル(メタ)アクリレートモノマーからなる群より選択されるモノマー15~60質量%、エポキシ基を有するモノマー1~35質量%、およびそれらと共重合可能なモノマー40~85質量%からなる。
さらに好ましくは、本発明のコアシェルポリマー(B)のシェル層は、貯蔵安定性を維持しつつ、T字剥離接着強さと動的割裂抵抗力が高いとの観点から、シェル層を構成するモノマー全量を100質量%とし、ビニルシアンモノマー20~40質量%、エポキシ基を有するモノマー3~15質量%、およびそれらと共重合可能なモノマー60~80質量%からなる。特にシェル層中のエポキシ基を有するモノマー量を上記範囲とすると、20質量%以上使用する場合に比べて、T字剥離接着強さと動的割裂抵抗力を有意に改善することができる。
さらに好ましくは、コアシェルポリマー(B)のシェル層は、シェル層を構成するモノマー全量を100質量%とし、アルコキシアルキル(メタ)アクリレートモノマー50~95質量%、およびエポキシ基を有するモノマー5~50質量%からなる。
さらにより好ましくは、コアシェルポリマー(B)のシェル層は、シェル層を構成するモノマー全量を100質量%とし、アルコキシアルキル(メタ)アクリレートモノマー90~95質量%、およびエポキシ基を有するモノマー5~10質量%からなる。
ビニルシアンモノマーおよびアルコキシアルキル(メタ)アクリレートモノマーからなる群より選択されるモノマーとしては、(メタ)アクリロニトリル、2-メトキシエチル(メタ)アクリレート、2-エトキシエチル(メタ)アクリレート、2-(2-エトキシエトキシ)が例示される。チキソトロピー性、せん断接着強さ、T字剥離接着強さあるいは動的割裂抵抗力の観点から、アクリロニトリルが特に好ましい。チキソトロピー性の観点から、2-メトキシエチル(メタ)アクリレートが特に好ましい。
エポキシ基を有するモノマーとしては、アリルグリシジルエーテル、グリシジル(メタ)アクリレート、グリシジルアルキル(メタ)アクリレートが例示される。チキソトロピー性、せん断接着強さ、T字剥離接着強さあるいは動的割裂抵抗力の観点から、グリシジルメタクリレートが特に好ましい。
それらと共重合可能なモノマーとしては、スチレン、α-メチルスチレン、1-又は2-ビニルナフタレン、モノクロロスチレン、ジクロロスチレン、ブロモスチレン等;芳香族ビニルモノマー、メチル(メタ)アクリレート、エチル(メタ)アクリレート、ブチル(メタ)アクリレート、2-エチルヘキシル(メタ)アクリレート、オクチル(メタ)アクリレート、ドデシル(メタ)アクリレート、ステアリル(メタ)アクリレート、ベヘニル(メタ)アクリレート等のアルキル(メタ)アクリレートが例示される。チキソトロピー性、せん断接着強さ、T字剥離接着強さあるいは動的割裂抵抗力の観点から、スチレンあるいはメチルメタクリレートがより好ましく、スチレンおよびメチルメタクリレートが特に好ましい。
(シェル層のグラフト率)
シェル層のグラフト率は、70%以上(好ましくは80%以上、より好ましくは90%以上)であることが好ましい。グラフト率が70%未満の場合には、液状樹脂組成物の粘度が上昇する場合がある。グラフト率の算出方法は、先ず、コアシェルポリマーを含有する水性ラテックスを凝固・脱水し、最後に乾燥してコアシェルポリマーのパウダーを得る。次いで、コアシェルポリマーのパウダー2gをメチルエチルケトン(MEK)100gに23℃で24時間浸漬した後にMEK可溶分をMEK不溶分と分離し、さらにMEK可溶分からメタノール不溶分を分離する。そして、MEK不溶分とメタノール不溶分との合計量に対するMEK不溶分の比率を求めることによって算出する。
シェル層のグラフト率は、70%以上(好ましくは80%以上、より好ましくは90%以上)であることが好ましい。グラフト率が70%未満の場合には、液状樹脂組成物の粘度が上昇する場合がある。グラフト率の算出方法は、先ず、コアシェルポリマーを含有する水性ラテックスを凝固・脱水し、最後に乾燥してコアシェルポリマーのパウダーを得る。次いで、コアシェルポリマーのパウダー2gをメチルエチルケトン(MEK)100gに23℃で24時間浸漬した後にMEK可溶分をMEK不溶分と分離し、さらにMEK可溶分からメタノール不溶分を分離する。そして、MEK不溶分とメタノール不溶分との合計量に対するMEK不溶分の比率を求めることによって算出する。
(コアシェルポリマーの製造方法)
本発明で用いるコアシェルポリマーを構成するコア層を形成するポリマーが、ジエン系モノマー(共役ジエン系モノマー)および(メタ)アクリレート系モノマーから選ばれる少なくとも1種のモノマー(第1モノマー)を含んで構成される場合には、コア層の形成は、例えば、乳化重合、懸濁重合、マイクロサスペンジョン重合などによって製造することができ、例えばWO2005/028546号公報に記載の方法を用いることができる。
本発明で用いるコアシェルポリマーを構成するコア層を形成するポリマーが、ジエン系モノマー(共役ジエン系モノマー)および(メタ)アクリレート系モノマーから選ばれる少なくとも1種のモノマー(第1モノマー)を含んで構成される場合には、コア層の形成は、例えば、乳化重合、懸濁重合、マイクロサスペンジョン重合などによって製造することができ、例えばWO2005/028546号公報に記載の方法を用いることができる。
また、コア層を形成するポリマーがポリシロキサン系ポリマーを含んで構成される場合には、コア層の形成は、例えば、乳化重合、懸濁重合、マイクロサスペンジョン重合などによって製造することができ、例えばWO2006/070664号公報に記載の方法を用いることができる。
中間層は、中間層形成用モノマーを公知のラジカル重合により重合することによって形成することができる。コア層を構成するゴム弾性体をエマルジョンとして得た場合には、二重結合を2以上有するモノマーの重合は乳化重合法により行うことが好ましい。
シェル層は、シェル層形成用モノマーを、公知のラジカル重合により重合することによって形成することができる。コア層、または、コア層を中間層で被覆して構成されるコアシェルポリマー前駆体をエマルジョンとして得た場合には、シェル層形成用モノマーの重合は乳化重合法により行うことが好ましく、例えば、WO2005/028546号公報に記載の方法に従って製造することができる。
乳化重合において用いることができる乳化剤(分散剤)としては、ジオクチルスルホコハク酸やドデシルベンゼンスルホン酸などに代表されるアルキルまたはアリールスルホン酸、アルキルまたはアリールエーテルスルホン酸、ドデシル硫酸に代表されるアルキルまたはアリール硫酸、アルキルまたはアリールエーテル硫酸、アルキルまたはアリール置換燐酸、アルキルまたはアリールエーテル置換燐酸、ドデシルザルコシン酸に代表されるN-アルキルまたはアリールザルコシン酸、オレイン酸やステアリン酸などに代表されるアルキルまたはアリールカルボン酸、アルキルまたはアリールエーテルカルボン酸などの各種の酸類、これら酸類のアルカリ金属塩またはアンモニウム塩などのアニオン性乳化剤(分散剤);アルキルまたはアリール置換ポリエチレングリコールなどの非イオン性乳化剤(分散剤);ポリビニルアルコール、アルキル置換セルロース、ポリビニルピロリドン、ポリアクリル酸誘導体などの分散剤が挙げられる。これらの乳化剤(分散剤)は、単独で用いても、2種以上を組み合わせて用いてもよい。
コアシェルポリマーの水性ラテックスの分散安定性に支障を来さない限り、乳化剤(分散剤)の使用量は少なくすることが好ましい。また、乳化剤(分散剤)は、その水溶性が高いほど好ましい。水溶性が高いと、乳化剤(分散剤)の水洗除去が容易になり、最終的に得られる硬化物への悪影響を容易に防止できる。
乳化重合法を採用する場合には、公知の開始剤、すなわち2,2’-アゾビスイソブチロニトリル、過酸化水素、過硫酸カリウム、過硫酸アンモニウムなどを熱分解型開始剤として用いることができる。
乳化重合法を採用する場合には、公知の開始剤、すなわち2,2’-アゾビスイソブチロニトリル、過酸化水素、過硫酸カリウム、過硫酸アンモニウムなどを熱分解型開始剤として用いることができる。
また、t-ブチルパーオキシイソプロピルカーボネート、パラメンタンハイドロパーオキサイド、クメンハイドロパーオキサイド、ジクミルパーオキサイド、t-ブチルハイドロパーオキサイド、ジ-t-ブチルパーオキサイド、t-ヘキシルパーオキサイドなどの有機過酸化物;過酸化水素、過硫酸カリウム、過硫酸アンモニウムなどの無機過酸化物といった過酸化物と、必要に応じてナトリウムホルムアルデヒドスルホキシレート、グルコースなどの還元剤、および必要に応じて硫酸鉄(II)などの遷移金属塩、さらに必要に応じてエチレンジアミン四酢酸二ナトリウムなどのキレート剤、さらに必要に応じてピロリン酸ナトリウムなどのリン含有化合物などを併用したレドックス型開始剤を使用することもできる。
レドックス型開始剤を用いた場合には、前記過酸化物が実質的に熱分解しない低い温度でも重合を行うことができ、重合温度を広い範囲で設定できるようになり好ましい。中でもクメンハイドロパーオキサイド、ジクミルパーオキサイド、t-ブチルハイドロパーオキサイドなどの有機過酸化物をレドックス型開始剤として用いることが好ましい。前記開始剤の使用量、レドックス型開始剤を用いる場合には前記還元剤・遷移金属塩・キレート剤などの使用量は公知の範囲で用いることができる。また二重結合を2以上有するモノマーを重合するに際しては公知の連鎖移動剤を公知の範囲で用いることができる。追加的に界面活性剤を用いることができるが、これも公知の範囲である。
重合に際しての重合温度、圧力、脱酸素などの条件は、公知の範囲のものが適用できる。また、中間層形成用モノマーの重合は1段で行なっても2段以上で行なってもよい。例えば、弾性コア層を構成するゴム弾性体のエマルジョンに中間層形成用モノマーを一度に添加する方法、連続追加する方法の他、あらかじめ中間層形成用モノマーが仕込まれた反応器に弾性コア層を構成するゴム弾性体のエマルジョンを加えてから重合を実施する方法などを採用することができる。
(コアシェルポリマー(B)の量)
得られる硬化性樹脂組成物の取扱いやすさと、得られる硬化物の靱性改良効果のバランスから、コアシェルポリマー(B)の量は、エポキシ樹脂(A)100質量部に対して、1~50質量部が好ましく、10~40質量部がより好ましく、10~35質量部が特に好ましい。
得られる硬化性樹脂組成物の取扱いやすさと、得られる硬化物の靱性改良効果のバランスから、コアシェルポリマー(B)の量は、エポキシ樹脂(A)100質量部に対して、1~50質量部が好ましく、10~40質量部がより好ましく、10~35質量部が特に好ましい。
<ブロックドイソシアネート(C)>
本発明では、ブロックドイソシアネート(C)は、靭性、耐衝撃性、せん断接着性、及び、T字剥離接着性などの性能を更に向上させる目的で添加する。ブロックドイソシアネート(C)成分は、コアシェルポリマー(B)成分と併用することで上記性能の向上に寄与する。
本発明では、ブロックドイソシアネート(C)は、靭性、耐衝撃性、せん断接着性、及び、T字剥離接着性などの性能を更に向上させる目的で添加する。ブロックドイソシアネート(C)成分は、コアシェルポリマー(B)成分と併用することで上記性能の向上に寄与する。
本発明のブロックドイソシアネート(C)は、エラストマー型であって、ウレタン基および/または尿素基を含有し、かつ、末端にイソシアネート基を有する化合物の当該末端イソシアネート基の全部または一部が活性水素基を有する種々のブロック剤でキャップされた化合物である。特に、当該末端イソシアネート基の全部がブロック剤でキャップされた化合物が好ましい。この様な化合物は、例えば、末端に活性水素含有基を有する有機重合体に、過剰のポリイソシアネート化合物を反応させて、主鎖中にウレタン基および/または尿素基を有し末端にイソシアネート基を有する重合体(ウレタンプレポリマー)とした後、あるいは同時に、該イソシアネート基の全部または一部に、活性水素基を有するブロック剤でキャップすることにより得られる。
前記ブロックドイソシアネートは、例えば、下記一般式(1):
A-(NR2-C(=O)-X)a (1)
(式中、a個のR2は、それぞれ独立に、炭素原子数1~20の炭化水素基である。aはキャップされたイソシアネート基の1分子当たりの平均数を表し、1.1個以上が好ましく、1.5~8個がより好ましく、1.7~6個が更に好ましく、2~4個が特に好ましい。Xは、後述するブロック剤から活性水素原子を除いた残基である。Aは、イソシアネート末端化プレポリマーから末端イソシアネート基を除いた残基である。)で表される。
A-(NR2-C(=O)-X)a (1)
(式中、a個のR2は、それぞれ独立に、炭素原子数1~20の炭化水素基である。aはキャップされたイソシアネート基の1分子当たりの平均数を表し、1.1個以上が好ましく、1.5~8個がより好ましく、1.7~6個が更に好ましく、2~4個が特に好ましい。Xは、後述するブロック剤から活性水素原子を除いた残基である。Aは、イソシアネート末端化プレポリマーから末端イソシアネート基を除いた残基である。)で表される。
上記炭化水素基は、脂肪族炭化水素基、脂環式炭化水素基、芳香族炭化水素基であってもよい。脂肪族炭化水素基としては、メチル基、エチル基、プロピル基、ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ドデシル基等のアルキル基;エテニル基、プロペニル基、ブテニル基、ペンテニル基、ヘキセニル基、ヘプテニル基、オクテニル基、ノネニル基、デセニル基、ウンデセニル基、ドデセニル基等のアルケニル基;等が挙げられる。脂肪族炭化水素基は、直鎖状、分岐鎖状のいずれであってもよい。脂環式炭化水素基としては、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロヘプチル基、シクロオクチル基、シクロノニル基、シクロデシル基等のシクロアルキル基;シクロプロペニル基、シクロブテニル基、シクロペンテニル基、シクロヘキセニル基、シクロヘプテニル基、シクロオクテニル基、シクロペンタジエニル等が挙げられる。芳香族炭化水素基としては、フェニル基、ナフチル基、アントリル基、フェナントリル基、ビフェニル基、ターフェニル基等が挙げられる。
ブロックドイソシアネートの数平均分子量は、GPCで測定したポリスチレン換算分子量にて、2000~40000が好ましく、3000~30000がより好ましく、4000~20000が特に好ましい。分子量分布(重量平均分子量と数平均分子量との比)は、1~4が好ましく、1.2~3がより好ましく、1.5~2.5が特に好ましい。
(C-1 末端に活性水素含有基を有する有機重合体)
末端に活性水素含有基を有する有機重合体を構成する主鎖骨格としては、ポリエーテル系重合体、ポリアクリル系重合体、ポリエステル系重合体、ポリジエン系重合体、飽和炭化水素系重合体(ポリオレフィン)、ポリチオエーテル系重合体などが挙げられる。
末端に活性水素含有基を有する有機重合体を構成する主鎖骨格としては、ポリエーテル系重合体、ポリアクリル系重合体、ポリエステル系重合体、ポリジエン系重合体、飽和炭化水素系重合体(ポリオレフィン)、ポリチオエーテル系重合体などが挙げられる。
(活性水素基)
末端に活性水素含有基を有する有機重合体を構成する活性水素含有基としては、水酸基、アミノ基、イミノ基、チオール基が挙げられる。これらの中でも、入手性の点から、水酸基、アミノ基、イミノ基が好ましく、更に得られるブロックドイソシアネートの取扱い易さ(粘度)の点から、水酸基がより好ましい。
末端に活性水素含有基を有する有機重合体を構成する活性水素含有基としては、水酸基、アミノ基、イミノ基、チオール基が挙げられる。これらの中でも、入手性の点から、水酸基、アミノ基、イミノ基が好ましく、更に得られるブロックドイソシアネートの取扱い易さ(粘度)の点から、水酸基がより好ましい。
末端に活性水素含有基を有する有機重合体としては、末端に水酸基を有するポリエーテル系重合体(ポリエーテルポリオール)、末端にアミノ基および/またはイミノ基を有するポリエーテル系重合体(ポリエーテルアミン)、ポリアクリルポリオール、ポリエステルポリオール、末端に水酸基を有するジエン系重合体(ポリジエンポリオール)、末端に水酸基を有する飽和炭化水素系重合体(ポリオレフィンポリオール)、ポリチオール化合物、ポリアミン化合物などが挙げられる。これらの中でも、ポリエーテルポリオール、ポリエーテルアミン、および、ポリアクリルポリオールは、エポキシ樹脂(A)成分との相溶性に優れ、有機重合体のガラス転移温度が比較的低く、得られる硬化物が低温での耐衝撃性に優れることから好ましい。特に、ポリエーテルポリオールおよびポリエーテルアミンは、得られる有機重合体の粘度が低く作業性が良好である為により好ましく、ポリエーテルポリオールは特に好ましい。
ブロックドイソシアネートの前駆体である前記ウレタンプレポリマーを調製する際に使用する、末端に活性水素含有基を有する有機重合体は、単独で用いてもよく2種以上併用してもよい。
末端に活性水素含有基を有する有機重合体の数平均分子量は、GPCで測定したポリスチレン換算分子量にて、800~7000が好ましく、1500~5000がより好ましく、2000~4000が特に好ましい。
(ポリエーテル系重合体)
前記ポリエーテル系重合体は、本質的に一般式(2):
-R1-O- (2)
(式中、R1は、炭素原子数1から14の直鎖状もしくは分岐アルキレン基である。)で示される繰り返し単位を有する重合体であり、一般式(2)におけるR1は、炭素原子数1から14の、好ましくは2から4の、直鎖状もしくは分岐状アルキレン基が好ましい。一般式(2)で示される繰り返し単位の具体例としては、-CH2O-、-CH2CH2O-、-CH2CH(CH3)O-、-CH2CH(C2H5)O-、-CH2C(CH3)2O-、-CH2CH2CH2CH2O-等が挙げられる。ポリエーテル系重合体の主鎖骨格は、1種類だけの繰り返し単位からなってもよいし、2種類以上の繰り返し単位からなってもよい。特に、プロピレンオキシドの繰り返し単位を50質量%以上有するポリプロピレングリコールを主成分とする重合体から成るものは、T字剥離接着強さの観点で好ましい。また、テトラヒドロフランを開環重合して得られるポリテトラメチレングリコール(PTMG)は、動的割裂抵抗力の観点で、好ましい。
また、コアシェルポリマー(B)成分と併用する際には、得られる硬化性エポキシ樹脂組成物のチキソトロピー性の観点から、コアシェルポリマー(B)成分とブロックドイソシアネート(C)成分の比率は0.1~10であることが好ましく、0.2~5であることがさらに好ましく、0.3~1であることが特に好ましい。
前記ポリエーテル系重合体は、本質的に一般式(2):
-R1-O- (2)
(式中、R1は、炭素原子数1から14の直鎖状もしくは分岐アルキレン基である。)で示される繰り返し単位を有する重合体であり、一般式(2)におけるR1は、炭素原子数1から14の、好ましくは2から4の、直鎖状もしくは分岐状アルキレン基が好ましい。一般式(2)で示される繰り返し単位の具体例としては、-CH2O-、-CH2CH2O-、-CH2CH(CH3)O-、-CH2CH(C2H5)O-、-CH2C(CH3)2O-、-CH2CH2CH2CH2O-等が挙げられる。ポリエーテル系重合体の主鎖骨格は、1種類だけの繰り返し単位からなってもよいし、2種類以上の繰り返し単位からなってもよい。特に、プロピレンオキシドの繰り返し単位を50質量%以上有するポリプロピレングリコールを主成分とする重合体から成るものは、T字剥離接着強さの観点で好ましい。また、テトラヒドロフランを開環重合して得られるポリテトラメチレングリコール(PTMG)は、動的割裂抵抗力の観点で、好ましい。
また、コアシェルポリマー(B)成分と併用する際には、得られる硬化性エポキシ樹脂組成物のチキソトロピー性の観点から、コアシェルポリマー(B)成分とブロックドイソシアネート(C)成分の比率は0.1~10であることが好ましく、0.2~5であることがさらに好ましく、0.3~1であることが特に好ましい。
(ポリエーテルポリオール)
前記ポリエーテルポリオールは、末端に水酸基を有するポリエーテル系重合体であり、前記ポリエーテルアミンは、末端にアミノ基またはイミノ基を有するポリエーテル系重合体である。
前記ポリエーテルポリオールは、末端に水酸基を有するポリエーテル系重合体であり、前記ポリエーテルアミンは、末端にアミノ基またはイミノ基を有するポリエーテル系重合体である。
(ポリアクリルポリオール)
前記ポリアクリルポリオールとしては、(メタ)アクリル酸アルキルエステル(共)重合体を骨格とし、かつ、分子内に水酸基を有するポリオールを挙げることができる。特に、2-ヒドロキシエチルメタクリレート等の水酸基含有(メタ)アクリル酸アルキルエステルモノマーを共重合して得られるポリアクリルポリオールが好ましい。
前記ポリアクリルポリオールとしては、(メタ)アクリル酸アルキルエステル(共)重合体を骨格とし、かつ、分子内に水酸基を有するポリオールを挙げることができる。特に、2-ヒドロキシエチルメタクリレート等の水酸基含有(メタ)アクリル酸アルキルエステルモノマーを共重合して得られるポリアクリルポリオールが好ましい。
(ポリエステルポリオール)
前記ポリエステルポリオールとしては、マレイン酸、フマル酸、アジピン酸、フタル酸等の多塩基酸およびその酸無水物と、エチレングリコール、プロピレングリコール、1,4-ブタンジオール、1,6-へキサンジオール、ジエチレングリコール、ジプロピレングリコール、ネオペンチルグリコール等の多価アルコールとを、エステル化触媒の存在下、150~270℃の温度範囲で重縮合させて得られる重合体が挙げられる。また、ε-カプロラクトン、バレロラクトン等の開環重合物やポリカーボネートジオールやヒマシ油等の活性水素を2個以上有する活性水素化合物等も挙げられる。
前記ポリエステルポリオールとしては、マレイン酸、フマル酸、アジピン酸、フタル酸等の多塩基酸およびその酸無水物と、エチレングリコール、プロピレングリコール、1,4-ブタンジオール、1,6-へキサンジオール、ジエチレングリコール、ジプロピレングリコール、ネオペンチルグリコール等の多価アルコールとを、エステル化触媒の存在下、150~270℃の温度範囲で重縮合させて得られる重合体が挙げられる。また、ε-カプロラクトン、バレロラクトン等の開環重合物やポリカーボネートジオールやヒマシ油等の活性水素を2個以上有する活性水素化合物等も挙げられる。
(ポリジエンポリオール)
前記ポリジエンポリオールとしては、ポリブタジエンポリオール、ポリイソプレンポリオール、ポリクロロプレンポリオールなどを挙げることができ、特に、ポリブタジエンポリオールが好ましい。
前記ポリジエンポリオールとしては、ポリブタジエンポリオール、ポリイソプレンポリオール、ポリクロロプレンポリオールなどを挙げることができ、特に、ポリブタジエンポリオールが好ましい。
(ポリオレフィンポリオール)
前記ポリオレフィンポリオールとしては、ポリイソブチレンポリオール、水添ポリブタジエンポリオールなどを挙げることができる。
前記ポリオレフィンポリオールとしては、ポリイソブチレンポリオール、水添ポリブタジエンポリオールなどを挙げることができる。
(C-2 ポリイソシアネート)
前記ポリイソシアネート化合物の具体例としては、トルエン(トリレン)ジイソシアネート、ジフェニルメタンジイソシアネート、キシリレンジイソシアネート等の芳香族系ポリイソシアネート;イソフォロンジイソシアネート、ヘキサメチレンジイソシアネート、水素化トルエンジイソシアネート、水素化ジフェニルメタンジイソシアネート等の脂肪族系ポリイソシアネートなどを挙げることができる。これらの中でも、耐熱性の点から、脂肪族系ポリイソシアネートが好ましく、更に入手性の点から、イソフォロンジイソシアネートやヘキサメチレンジイソシアネートがより好ましい。
前記ポリイソシアネート化合物の具体例としては、トルエン(トリレン)ジイソシアネート、ジフェニルメタンジイソシアネート、キシリレンジイソシアネート等の芳香族系ポリイソシアネート;イソフォロンジイソシアネート、ヘキサメチレンジイソシアネート、水素化トルエンジイソシアネート、水素化ジフェニルメタンジイソシアネート等の脂肪族系ポリイソシアネートなどを挙げることができる。これらの中でも、耐熱性の点から、脂肪族系ポリイソシアネートが好ましく、更に入手性の点から、イソフォロンジイソシアネートやヘキサメチレンジイソシアネートがより好ましい。
(C-3 ブロック剤)
前記ブロック剤は、例えば、第一級アミン系ブロック剤、第二級アミン系ブロック剤、オキシム系ブロック剤、ラクタム系ブロック剤、活性メチレン系ブロック剤、アルコール系ブロック剤、メルカプタン系ブロック剤、アミド系ブロック剤、イミド系ブロック剤、複素環式芳香族化合物系ブロック剤、ヒドロキシ官能性(メタ)アクリレート系ブロック剤、フェノール系ブロック剤が挙げられる。これらの中でも、オキシム系ブロック剤、ラクタム系ブロック剤、ヒドロキシ官能性(メタ)アクリレート系ブロック剤、フェノール系ブロック剤が好ましく、ヒドロキシ官能性(メタ)アクリレート系ブロック剤、フェノール系ブロック剤がより好ましく、フェノール系ブロック剤が更に好ましい。
前記ブロック剤は、例えば、第一級アミン系ブロック剤、第二級アミン系ブロック剤、オキシム系ブロック剤、ラクタム系ブロック剤、活性メチレン系ブロック剤、アルコール系ブロック剤、メルカプタン系ブロック剤、アミド系ブロック剤、イミド系ブロック剤、複素環式芳香族化合物系ブロック剤、ヒドロキシ官能性(メタ)アクリレート系ブロック剤、フェノール系ブロック剤が挙げられる。これらの中でも、オキシム系ブロック剤、ラクタム系ブロック剤、ヒドロキシ官能性(メタ)アクリレート系ブロック剤、フェノール系ブロック剤が好ましく、ヒドロキシ官能性(メタ)アクリレート系ブロック剤、フェノール系ブロック剤がより好ましく、フェノール系ブロック剤が更に好ましい。
前記第一級アミン系ブロック剤としては、ブチルアミン、イソプロピルアミン、ドデシルアミン、シクロヘキシルアミン、アニリン、ベンジルアミン等が挙げられる。前記第二級アミン系ブロック剤としては、ジブチルアミン、ジイソプロピルアミン、ジシクロヘキシルアミン、ジフェニルアミン、ジベンジルアミン、モルホリン、ピペリジン等が挙げられる。前記オキシム系ブロック剤としては、ホルムアルドキシム、アセトアルドキシム、アセトキシム、メチルエチルケトオキシム、ジアセチルモノオキシム、シクロヘキサンオキシム等が挙げられる。前記ラクタム系ブロック剤としては、ε-カプロラクタム、δ-バレロラクタム、γ-ブチロラクタム、β-ブチロラクタム等が挙げられる。前記活性メチレン系ブロック剤としては、アセト酢酸エチル、アセチルアセトン等が挙げられる。前記アルコール系ブロック剤としては、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、アミルアルコール、シクロヘキサノール、1-メトキシ-2-プロパノール、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、プロピレングリコールモノメチルエーテル、ベンジルアルコール、グリコール酸メチル、グリコール酸ブチル、ジアセトンアルコール、乳酸メチル、乳酸エチル等が挙げられる。前記メルカプタン系ブロック剤としては、ブチルメルカプタン、ヘキシルメルカプタン、デシルメルカプタン、t-ブチルメルカプタン、チオフェノール、メチルチオフェノール、エチルチオフェノール等が挙げられる。前記アミド系ブロック剤としては、酢酸アミド、ベンズアミド等が挙げられる。前記イミド系ブロック剤としては、コハク酸イミド、マレイン酸イミド等が挙げられる。前記複素環式芳香族化合物系ブロック剤としては、イミダゾール、2-エチルイミダゾール等のイミダゾール類、ピロール、2-メチルピロール、3-メチルピロール等のピロール類、ピリジン、2-メチルピリジン、4-メチルピリジン等のピリジン類、ジアザビシクロウンデセン、ジアザビシクロノネン等のジアザビシクロアルケン類が挙げられる。
前記ヒドロキシ官能性(メタ)アクリレート系ブロック剤は、1個以上の水酸基を有する(メタ)アクリレートである。ヒドロキシ官能性(メタ)アクリレート系ブロック剤の具体例としては、2-ヒドロキシエチル(メタ)アクリレート、2-ヒドロキシプロピル(メタ)アクリレート、4-ヒドロキシブチル(メタ)アクリレート、2-ヒドロキシブチル(メタ)アクリレート、等が挙げられる。
前記フェノール系ブロック剤は、少なくとも1個のフェノール性ヒドロキシル基、即ち、芳香環の炭素原子に直接結合したヒドロキシル基を含有する。フェノール性化合物は2個以上のフェノール性ヒドロキシル基を有していてもよいが、好ましくはフェノール性ヒドロキシル基を一つだけ含有する。フェノール性化合物は、他の置換基を含有していてもよいが、これら置換基は好ましくはキャッピング反応の条件下でイソシアネート基と反応しないものであり、アルケニル基、アリル基が好ましい。他の置換基としては、直鎖状、分岐鎖状またはシクロアルキル等のアルキル基;芳香族基(例えば、フェニル、アルキル置換フェニル、アルケニル置換フェニル等);アリール置換アルキル基;フェノール置換アルキル基が挙げられる。フェノール系ブロック剤の具体例としては、フェノール、クレゾール、キシレノール、クロロフェノール、エチルフェノール、アリルフェノール(特にo-アリルフェノール)、レゾルシノール、カテコール、ヒドロキノン、ビスフェノール、ビスフェノールA、ビスフェノールAP(1,1-ビス(4-ヒドロキシルフェニル)-1-フェニルエタン)、ビスフェノールF、ビスフェノールK、ビスフェノールM、テトラメチルビフェノールおよび2,2’-ジアリル-ビスフェノールA、等が挙げられる。
前記ブロック剤は、それが結合する末端がもはや反応性基を有しないような態様で、ウレタンプレポリマーのポリマー鎖の末端に結合していることが好ましい。
前記ブロック剤は、単独で用いてもよく2種以上併用してもよい。
前記ブロックドイソシアネートは、架橋剤の残基、鎖延長剤の残基、または、その両方を含有していてもよい。
前記ブロック剤は、それが結合する末端がもはや反応性基を有しないような態様で、ウレタンプレポリマーのポリマー鎖の末端に結合していることが好ましい。
前記ブロック剤は、単独で用いてもよく2種以上併用してもよい。
前記ブロックドイソシアネートは、架橋剤の残基、鎖延長剤の残基、または、その両方を含有していてもよい。
本発明に使用されるブロックドイソシアネートは、ポリアルキレングリコール構造を含むウレタンプレポリマーをブロック剤でキャップした化合物であることが好ましく、ポリプロピレングリコール構造を含むウレタンプレポリマーをブロック剤(好ましくはフェノール系ブロック剤)でキャップした化合物またはポリテトラメチレングリコール構造を含むウレタンプレポリマーをブロック剤(好ましくはフェノール系ブロック剤)でキャップした化合物であることがより好ましい。上記ブロックドイソシアネートは、チキソトロピー性や接着性を改善する点で好適に使用することができる。
ポリプロピレングリコール構造を含むウレタンプレポリマーをブロック剤(好ましくはフェノール系ブロック剤)でキャップした化合物は、T字剥離接着強さを改善する観点から使用されてもよく、ポリテトラメチレングリコール構造を含むウレタンプレポリマーをブロック剤(好ましくはフェノール系ブロック剤)でキャップした化合物は、動的割裂抵抗力を改善する観点から使用されてもよい。
ポリプロピレングリコール構造を含むウレタンプレポリマーをブロック剤(好ましくはフェノール系ブロック剤)でキャップした化合物は、T字剥離接着強さを改善する観点から使用されてもよく、ポリテトラメチレングリコール構造を含むウレタンプレポリマーをブロック剤(好ましくはフェノール系ブロック剤)でキャップした化合物は、動的割裂抵抗力を改善する観点から使用されてもよい。
ブロックドイソシアネートのブロックNCO当量は、例えば10~500、好ましくは50~300である。ブロックドイソシアネートのエポキシド当量は、例えば300~900g/eq、好ましくは400~800g/eqである。これらの特性の一つを少なくとも備えるブロックドイソシアネートが、本発明で好適に使用できる。
(架橋剤)
前記架橋剤の分子量は750以下が好ましく、より好ましくは50~500であり、かつ、1分子当たり少なくとも3個のヒドロキシル基、アミノ基および/またはイミノ基を有するポリオールまたはポリアミン化合物である。架橋剤はブロックドイソシアネートに分岐を付与し、ブロックドイソシアネートの官能価(即ち、キャップされたイソシアネート基の1分子当たりの数)を増加させるのに有用である。
前記架橋剤の分子量は750以下が好ましく、より好ましくは50~500であり、かつ、1分子当たり少なくとも3個のヒドロキシル基、アミノ基および/またはイミノ基を有するポリオールまたはポリアミン化合物である。架橋剤はブロックドイソシアネートに分岐を付与し、ブロックドイソシアネートの官能価(即ち、キャップされたイソシアネート基の1分子当たりの数)を増加させるのに有用である。
(鎖延長剤)
前記鎖延長剤の分子量は750以下が好ましく、より好ましくは50~500であり、かつ、1分子当たり2個のヒドロキシル基、アミノ基および/またはイミノ基を有するポリオールまたはポリアミン化合物である。鎖延長剤は、官能価を増加させずにブロックドイソシアネートの分子量を上げるのに有用である。
前記鎖延長剤の分子量は750以下が好ましく、より好ましくは50~500であり、かつ、1分子当たり2個のヒドロキシル基、アミノ基および/またはイミノ基を有するポリオールまたはポリアミン化合物である。鎖延長剤は、官能価を増加させずにブロックドイソシアネートの分子量を上げるのに有用である。
前記架橋剤や鎖延長剤の具体例としては、トリメチロールプロパン、グリセリン、トリメチロールエタン、エチレングリコール、ジエチレングリコール、プロピレングリコール、ジプロピレングリコール、スクロース、ソルビトール、ペンタエリスリトール、エチレンジアミン、トリエタノールアミン、モノエタノールアミン、ジエタノールアミン、ピペラジン、アミノエチルピペラジンが挙げられる。また、レゾルシノール、カテコール、ヒドロキノン、ビスフェノール、ビスフェノールA、ビスフェノールAP(1,1-ビス(4-ヒドロキシルフェニル)-1-フェニルエタン)、ビスフェノールF、ビスフェノールK、ビスフェノールM、テトラメチルビフェノール、2,2’-ジアリル-ビスフェノールA等の、2個以上のフェノール性ヒドロキシル基を有する化合物も挙げられる。
(ブロックドイソシアネート(C)の量)
ブロックドイソシアネートの量は、エポキシ樹脂(A)100質量部に対して、1~50質量部が好ましく、10~40質量部がより好ましく、10~35質量部が特に好ましい。1質量部未満では、靱性、耐衝撃性、接着性などの改質効果が十分ではない場合があり、50質量部より多いと、得られる硬化物の弾性率が低くなる場合がある。ブロックドイソシアネートは単独で用いてもよく2種以上併用してもよい。
ブロックドイソシアネートの量は、エポキシ樹脂(A)100質量部に対して、1~50質量部が好ましく、10~40質量部がより好ましく、10~35質量部が特に好ましい。1質量部未満では、靱性、耐衝撃性、接着性などの改質効果が十分ではない場合があり、50質量部より多いと、得られる硬化物の弾性率が低くなる場合がある。ブロックドイソシアネートは単独で用いてもよく2種以上併用してもよい。
<エポキシ硬化剤(D)>
本発明の硬化性エポキシ樹脂組成物には、必要に応じてエポキシ硬化剤(D)を使用することができる。
本発明の硬化性エポキシ樹脂組成物を仮に一成分型接着剤として使用する場合、80℃以上、好ましくは140℃以上の温度まで加熱すると接着剤が急速に硬化するようにエポキシ硬化剤(D)成分を選択するのが好ましい。逆に、室温(約22℃)や少なくとも50℃までの温度では硬化するとしても非常にゆっくりとなるよう、エポキシ硬化剤(D)成分および後述の(E)成分を選択するのが好ましい。
本発明の硬化性エポキシ樹脂組成物には、必要に応じてエポキシ硬化剤(D)を使用することができる。
本発明の硬化性エポキシ樹脂組成物を仮に一成分型接着剤として使用する場合、80℃以上、好ましくは140℃以上の温度まで加熱すると接着剤が急速に硬化するようにエポキシ硬化剤(D)成分を選択するのが好ましい。逆に、室温(約22℃)や少なくとも50℃までの温度では硬化するとしても非常にゆっくりとなるよう、エポキシ硬化剤(D)成分および後述の(E)成分を選択するのが好ましい。
エポキシ硬化剤(D)成分としては、例えば、三塩化ホウ素/アミン錯体、三フッ化ホウ素/アミン錯体、ジシアンジアミド、メラミン、ジアリルメラミン、グアナミン(例えば、アセトグアナミンおよびベンゾグアナミン)、アミノトリアゾール(例えば、3-アミノ-1,2,4-トリアゾール)、ヒドラジド(例えば、アジピン酸ジヒドラジド、ステアリン酸ジヒドラジド、イソフタル酸ジヒドラジド、セミカルバジド)、シアノアセトアミド、並びに、芳香族ポリアミン(例えば、ジアミノジフェニルスルホン)が挙げられる。ジシアンジアミド、イソフタル酸ジヒドラジド、アジピン酸ジヒドラジド、4,4’-ジアミノジフェニルスルホンを用いるのがより好ましく、ジシアンジアミドが特に好ましい。
上記硬化剤の中でも、三塩化ホウ素/アミン錯体、三フッ化ホウ素/アミン錯体、ジシアンジアミド、ヒドラジド等の潜在性エポキシ硬化剤は、本発明の硬化性エポキシ樹脂組成物を一液化できるため好ましい。エポキシ硬化剤(D)成分は、単独で用いてもよく2種以上併用してもよい。
エポキシ硬化剤(D)成分は、組成物を硬化させるのに十分な量で使用する。典型的には、組成物中に存在するエポキシド基の少なくとも80%を消費するのに十分な硬化剤を供給する。エポキシド基の消費に必要な量を超える大過剰量は、通常必要ない。エポキシ硬化剤(D)成分の使用量は、(A)成分100質量部に対して、1~40質量部が好ましく、2~30質量部がより好ましく、3~25質量部が更に好ましく、5~20質量部が特に好ましい。1質量部未満では、本発明の硬化性エポキシ樹脂組成物の硬化性が悪くなる場合がある。40質量部より多いと、本発明の硬化性エポキシ樹脂組成物の貯蔵安定性が悪く、取り扱い難くなる場合がある。
<硬化促進剤(E)>
本発明の硬化性エポキシ樹脂組成物には、必要に応じて、硬化促進剤(E)を使用することができる。
硬化促進剤(E)成分は、エポキシ基と、硬化剤や接着剤の他の成分上のエポキシド反応性基との反応)を促進するための触媒である。
硬化促進剤(E)成分としては、尿素類、例えば、p-クロロフェニル-N,N-ジメチル尿素(Monuron)、3-フェニル-1,1-ジメチル尿素(Phenuron)、3,4-ジクロロフェニル-N,N-ジメチル尿素(Diuron)、N-(3-クロロ-4-メチルフェニル)-N’,N’-ジメチル尿素(Chlortoluron)、tert-アクリル-またはアルキレンアミン、例えば、ベンジルジメチルアミン、2,4,6-トリス(ジメチルアミノメチル)フェノール、ポリ(p-ビニルフェノール)マトリックスに組み込まれた2,4,6-トリス(ジメチルアミノメチル)フェノール、ピペリジンまたはこれらの誘導体、イミダゾール誘導体、一般にC1-C12アルキレンイミダゾールまたはN-アリールイミダゾール、例えば、2-エチル-2-メチルイミダゾールまたはN-ブチルイミダゾール、6-カプロラクタム、等が挙げられる。触媒は封入されていてもよく、あるいは、温度を上げた場合にのみ活性となる潜在的なものでもよい。硬化促進剤(E)成分は、単独で用いてもよく2種以上併用してもよい。
本発明の硬化性エポキシ樹脂組成物には、必要に応じて、硬化促進剤(E)を使用することができる。
硬化促進剤(E)成分は、エポキシ基と、硬化剤や接着剤の他の成分上のエポキシド反応性基との反応)を促進するための触媒である。
硬化促進剤(E)成分としては、尿素類、例えば、p-クロロフェニル-N,N-ジメチル尿素(Monuron)、3-フェニル-1,1-ジメチル尿素(Phenuron)、3,4-ジクロロフェニル-N,N-ジメチル尿素(Diuron)、N-(3-クロロ-4-メチルフェニル)-N’,N’-ジメチル尿素(Chlortoluron)、tert-アクリル-またはアルキレンアミン、例えば、ベンジルジメチルアミン、2,4,6-トリス(ジメチルアミノメチル)フェノール、ポリ(p-ビニルフェノール)マトリックスに組み込まれた2,4,6-トリス(ジメチルアミノメチル)フェノール、ピペリジンまたはこれらの誘導体、イミダゾール誘導体、一般にC1-C12アルキレンイミダゾールまたはN-アリールイミダゾール、例えば、2-エチル-2-メチルイミダゾールまたはN-ブチルイミダゾール、6-カプロラクタム、等が挙げられる。触媒は封入されていてもよく、あるいは、温度を上げた場合にのみ活性となる潜在的なものでもよい。硬化促進剤(E)成分は、単独で用いてもよく2種以上併用してもよい。
硬化促進剤(E)成分の使用量は、(A)成分100質量部に対して、0.1~10質量部が好ましく、0.2~5質量部がより好ましく、0.5~3質量部が更に好ましく、0.8~2質量部が特に好ましい。0.1質量部未満では、本発明の硬化性エポキシ樹脂組成物の硬化性が悪くなる場合がある。10質量部より多いと、本発明の硬化性エポキシ樹脂組成物の貯蔵安定性が悪く、取り扱い難くなる場合がある。
<充填剤>
本発明の硬化性エポキシ樹脂組成物には、必要に応じて、充填剤を使用することができる。充填剤の具体例としては、ポリジメチルシロキサンで表面処理した疎水性ヒュームドシリカなどの乾式シリカ、湿式シリカ、ケイ酸アルミニウム、ケイ酸マグネシウム、ケイ酸カルシウム、ドロマイトおよびカーボンブラックの如き補強性充填材;タルクやウォラストナイト等の板状フィラー;膠質炭酸カルシウム、重質炭酸カルシウム、炭酸マグネシウム、酸化チタン、酸化第二鉄、アルミニウム微粉末、酸化亜鉛、活性亜鉛華等が挙げられる。また、200μmまでの平均粒子径と0.2g/ccまでの密度を有するマイクロバルーンも使用できる。粒子径は好ましくは約25~150μmであり、密度は好ましくは約0.05~約0.15g/ccである。市販されているマイクロバルーンとしては、Dualite Corporation製Dualite、アクゾノーベル社製Expancel、松本油脂製薬製マイクロスフェアーなどが挙げられる。
本発明の硬化性エポキシ樹脂組成物には、必要に応じて、充填剤を使用することができる。充填剤の具体例としては、ポリジメチルシロキサンで表面処理した疎水性ヒュームドシリカなどの乾式シリカ、湿式シリカ、ケイ酸アルミニウム、ケイ酸マグネシウム、ケイ酸カルシウム、ドロマイトおよびカーボンブラックの如き補強性充填材;タルクやウォラストナイト等の板状フィラー;膠質炭酸カルシウム、重質炭酸カルシウム、炭酸マグネシウム、酸化チタン、酸化第二鉄、アルミニウム微粉末、酸化亜鉛、活性亜鉛華等が挙げられる。また、200μmまでの平均粒子径と0.2g/ccまでの密度を有するマイクロバルーンも使用できる。粒子径は好ましくは約25~150μmであり、密度は好ましくは約0.05~約0.15g/ccである。市販されているマイクロバルーンとしては、Dualite Corporation製Dualite、アクゾノーベル社製Expancel、松本油脂製薬製マイクロスフェアーなどが挙げられる。
<反応性希釈剤>
また、本発明の硬化性エポキシ樹脂組成物には、必要に応じ、反応性希釈剤として、モノエポキシド、例えば、脂肪族グリシジルエーテル、例えばブチルグリシジルエーテル、あるいはフェニルグリシジルエーテル、クレジルグリシジルエーテル、2-エチルヘキシルグリシジルエーテル、C8-C10アルキルグリシジルエーテル、C12-C14アルキルグリシジルエーテル、p-tertブチルフェニルグリシジルエーテル、ネオデカン酸モノグリシジルエーテルを含んでいてもよい。
また、本発明の硬化性エポキシ樹脂組成物には、必要に応じ、反応性希釈剤として、モノエポキシド、例えば、脂肪族グリシジルエーテル、例えばブチルグリシジルエーテル、あるいはフェニルグリシジルエーテル、クレジルグリシジルエーテル、2-エチルヘキシルグリシジルエーテル、C8-C10アルキルグリシジルエーテル、C12-C14アルキルグリシジルエーテル、p-tertブチルフェニルグリシジルエーテル、ネオデカン酸モノグリシジルエーテルを含んでいてもよい。
(その他の配合成分)
本発明では、必要に応じて、その他の配合成分を使用することができる。その他の配合成分としては、酸化カルシウムなどの脱水剤、顔料や染料等の着色剤、体質顔料、紫外線吸収剤、酸化防止剤、安定化剤(ゲル化防止剤)、可塑剤、レベリング剤、消泡剤、シランカップリング剤、帯電防止剤、難燃剤、滑剤、減粘剤、低収縮剤、有機質充填剤、熱可塑性樹脂、乾燥剤、分散剤等が挙げられる。
本発明では、必要に応じて、その他の配合成分を使用することができる。その他の配合成分としては、酸化カルシウムなどの脱水剤、顔料や染料等の着色剤、体質顔料、紫外線吸収剤、酸化防止剤、安定化剤(ゲル化防止剤)、可塑剤、レベリング剤、消泡剤、シランカップリング剤、帯電防止剤、難燃剤、滑剤、減粘剤、低収縮剤、有機質充填剤、熱可塑性樹脂、乾燥剤、分散剤等が挙げられる。
本発明の硬化性エポキシ樹脂組成物のチキソトロピー性は、1.00以上であることが好ましく、1.1以上であることがより好ましく、1.2以上であることがさらに好ましく、1.25以上であることがさらにより好ましい。チキソトロピー性の上限は、例えば1.5程度である。チキソトロピー性は、チキソトロピーインデックス、ヒステレシスループ等により評価することができる。例えば、チキソトロピーインデックス(TI)は、回転速度aとbにおける粘度ηaとηbの比(TI=ηb/ηa)として表すことができる。
(硬化物)
本発明には、上記硬化性エポキシ樹脂組成物を硬化した硬化物が含まれる。
本発明には、上記硬化性エポキシ樹脂組成物を硬化した硬化物が含まれる。
(用途)
本発明の硬化性エポキシ樹脂組成物は、チキソトロピー性に優れ、一液型接着剤として使用できる。
本発明の硬化性エポキシ樹脂組成物は、車両や航空機向けの構造用接着剤などの接着剤、塗料、ガラス繊維との積層用材料、炭素繊維との積層用材料、プリント配線基板、電気絶縁材料等の用途に好ましく用いられる。特に、車両用の構造接着剤として有用である。
本発明の硬化性エポキシ樹脂組成物は、チキソトロピー性に優れ、一液型接着剤として使用できる。
本発明の硬化性エポキシ樹脂組成物は、車両や航空機向けの構造用接着剤などの接着剤、塗料、ガラス繊維との積層用材料、炭素繊維との積層用材料、プリント配線基板、電気絶縁材料等の用途に好ましく用いられる。特に、車両用の構造接着剤として有用である。
本願は、2015年3月31日に出願された日本国特許出願第2015-074446号に基づく優先権の利益を主張するものである。2015年3月31日に出願された日本国特許出願第2015-074446号の明細書の全内容が、本願に参考のため援用される。
以下、製造例、実施例および比較例によって、本発明をより詳細に説明するが、本発明はこれらに限定されるものではなく、前後記の趣旨に適合し得る範囲で適宜変更して実施することが可能であり、それらはいずれも本発明の技術的範囲に包含される。
<評価方法>
製造例に記載されたポリブタジエンゴムラテックス中のポリブタジエンゴム粒子、コアシェルポリマーラテックス中のコアシェルポリマー粒子について、それぞれの平均粒子径の測定方法を以下に記載する。
製造例に記載されたポリブタジエンゴムラテックス中のポリブタジエンゴム粒子、コアシェルポリマーラテックス中のコアシェルポリマー粒子について、それぞれの平均粒子径の測定方法を以下に記載する。
(ラテックス中のブタジエンゴム粒子とコアシェルポリマー粒子の体積平均粒子径の測定)
ポリブタジエンゴムラテックス中のポリブタジエンゴム粒子、コアシェルポリマーラテックス中のコアシェルポリマー粒子について、マイクロトラックUPA150(日機装株式会社製)を用いて、体積平均粒子径を測定した。
ポリブタジエンゴムラテックス中のポリブタジエンゴム粒子、コアシェルポリマーラテックス中のコアシェルポリマー粒子について、マイクロトラックUPA150(日機装株式会社製)を用いて、体積平均粒子径を測定した。
脱イオン水で希釈したものを測定試料として用いた。測定は、水、またはメチルエチルケトンの屈折率、およびそれぞれのコアシェルポリマー(B)の屈折率を入力し、計測時間600秒、Signal Levelが0.6~0.8の範囲内になるように試料濃度を調整して行った。
(チキソトロピー性)
E型粘度計を用いて、ずり速度を1(s-1)とした条件で測定した硬化性エポキシ樹脂組成物の粘度をηaとし、ずり速度を2(s-1)とした条件で測定した硬化性エポキシ樹脂組成物の粘度をηbとし、ηbを1としたηaの倍率で評価した。ηbを1としたηaの倍率が高いほど、チキソトロピー性が良好である。
E型粘度計は、BROOKFIELD社製デジタル粘度計DV-II+Pro型を用いて測定した。スピンドルCPE-52を使用し、23℃で測定した。
E型粘度計を用いて、ずり速度を1(s-1)とした条件で測定した硬化性エポキシ樹脂組成物の粘度をηaとし、ずり速度を2(s-1)とした条件で測定した硬化性エポキシ樹脂組成物の粘度をηbとし、ηbを1としたηaの倍率で評価した。ηbを1としたηaの倍率が高いほど、チキソトロピー性が良好である。
E型粘度計は、BROOKFIELD社製デジタル粘度計DV-II+Pro型を用いて測定した。スピンドルCPE-52を使用し、23℃で測定した。
(貯蔵安定性)
E型粘度計を用いて、ずり速度を2(s-1)とした条件で測定した硬化性エポキシ樹脂組成物の貯蔵前粘度をη0とし、40℃で7日間貯蔵した貯蔵後粘度をη1とし、η0を1としたη1の倍率で評価した。η0を1としたη1の倍率が低いほど、貯蔵安定性が良好である。E型粘度計は、BROOKFIELD社製デジタル粘度計DV-II+Pro型を用いて測定した。スピンドルCPE-52を使用し、23℃で測定した。
E型粘度計を用いて、ずり速度を2(s-1)とした条件で測定した硬化性エポキシ樹脂組成物の貯蔵前粘度をη0とし、40℃で7日間貯蔵した貯蔵後粘度をη1とし、η0を1としたη1の倍率で評価した。η0を1としたη1の倍率が低いほど、貯蔵安定性が良好である。E型粘度計は、BROOKFIELD社製デジタル粘度計DV-II+Pro型を用いて測定した。スピンドルCPE-52を使用し、23℃で測定した。
(せん断接着強さ)
JIS K 6850に準じてせん断接着強さを評価した。幅25mm×長さ100mm×厚み1.6mmの2枚のSPCC鋼板に、硬化性エポキシ樹脂組成物を塗布し、接着層が幅25mm×長さ12.5mm×厚み0.13mmとなるように貼りあわせ、170℃の条件下で1時間硬化させ試験体を作製した。
測定温度を23℃、テストスピードを1.3mm/minとした測定条件で、単位をMPaとしたせん断接着強さを測定した。
JIS K 6850に準じてせん断接着強さを評価した。幅25mm×長さ100mm×厚み1.6mmの2枚のSPCC鋼板に、硬化性エポキシ樹脂組成物を塗布し、接着層が幅25mm×長さ12.5mm×厚み0.13mmとなるように貼りあわせ、170℃の条件下で1時間硬化させ試験体を作製した。
測定温度を23℃、テストスピードを1.3mm/minとした測定条件で、単位をMPaとしたせん断接着強さを測定した。
(T字剥離接着強さ)
JIS K 6854に準じてT字剥離接着強さを評価した。幅25mm×長さ200mm×厚み0.5mmの2枚のSPCC鋼板に、硬化性エポキシ樹脂組成物を塗布し、接着層が幅25mm×長さ150mm×厚み0.26mmとなるように貼り合わせ、170℃の条件下1時間硬化させ、試験体を作製した。測定温度を23℃、テストスピードを254mm/minとした測定条件で、単位をN/25mmとしたT字剥離接着強さを測定した。
JIS K 6854に準じてT字剥離接着強さを評価した。幅25mm×長さ200mm×厚み0.5mmの2枚のSPCC鋼板に、硬化性エポキシ樹脂組成物を塗布し、接着層が幅25mm×長さ150mm×厚み0.26mmとなるように貼り合わせ、170℃の条件下1時間硬化させ、試験体を作製した。測定温度を23℃、テストスピードを254mm/minとした測定条件で、単位をN/25mmとしたT字剥離接着強さを測定した。
(動的割裂抵抗力)
ISO 11343に準じて動的割裂抵抗力を評価した。幅20mm×長さ90mm×厚み0.8mmの2枚のSPCC鋼板に、硬化性エポキシ樹脂組成物を塗布し、接着層が幅20mm×長さ30mm×厚み0.26mmとなるように貼りあわせ、170℃の条件下で1時間硬化させ試験体を作製した。
測定温度を23℃、衝撃エネルギーを50J、衝撃スピードを2m/sとした測定条件で、単位をkN/mとした動的割裂抵抗力を測定した。
ISO 11343に準じて動的割裂抵抗力を評価した。幅20mm×長さ90mm×厚み0.8mmの2枚のSPCC鋼板に、硬化性エポキシ樹脂組成物を塗布し、接着層が幅20mm×長さ30mm×厚み0.26mmとなるように貼りあわせ、170℃の条件下で1時間硬化させ試験体を作製した。
測定温度を23℃、衝撃エネルギーを50J、衝撃スピードを2m/sとした測定条件で、単位をkN/mとした動的割裂抵抗力を測定した。
<コアシェルポリマー(B)の製造例、コアシェルポリマーが分散しているエポキシ樹脂(N)の製造例>
製造例1-1および製造例1-2に、コアシェルポリマーのコア層であるポリブタジエンゴムを含むポリブタジエンゴムラテックス(R-1)および(R-2)の調製方法を記載した。製造例2-1から製造例2-10に、コアシェルポリマーラテックス(L-1)~(L-10)の調製方法を記載した。製造例3-1から製造例3-10に、コアシェルポリマーが分散しているエポキシ樹脂(N)の調製方法を記載した。
製造例1-1および製造例1-2に、コアシェルポリマーのコア層であるポリブタジエンゴムを含むポリブタジエンゴムラテックス(R-1)および(R-2)の調製方法を記載した。製造例2-1から製造例2-10に、コアシェルポリマーラテックス(L-1)~(L-10)の調製方法を記載した。製造例3-1から製造例3-10に、コアシェルポリマーが分散しているエポキシ樹脂(N)の調製方法を記載した。
製造例1-1;ポリブタジエンゴムラテックス(R-1)の調製
耐圧重合機中に、脱イオン水200質量部、リン酸三カリウム0.03質量部、エチレンジアミン四酢酸二ナトリウム(EDTA)0.002質量部、硫酸第一鉄・7水和塩0.001質量部、及び、ドデシルベンゼンスルホン酸ナトリウム(SDBS)1.55質量部を投入し、撹拌しつつ十分に窒素置換を行なって酸素を除いた後、ブタジエン(Bd)100質量部を系中に投入し、45℃に昇温した。パラメンタンハイドロパーオキサイド(PHP)0.03質量部、続いてナトリウムホルムアルデヒドスルホキシレート(SFS)0.10質量部を投入し重合を開始した。重合開始から3、5、7時間目それぞれに、パラメンタンハイドロパーオキサイド(PHP)0.025質量部を投入した。また、重合開始4、6、8時間目それぞれに、EDTA0.0006質量部、及び硫酸第一鉄・7水和塩0.003質量部を投入した。重合15時間目に減圧下残存モノマーを脱揮除去して重合を終了し、ポリブタジエンゴムを主成分とするポリブタジエンゴムラテックス(R-1)を得た。得られたラテックスに含まれるポリブタジエンゴム粒子の体積平均粒子径は0.08μmであった。
耐圧重合機中に、脱イオン水200質量部、リン酸三カリウム0.03質量部、エチレンジアミン四酢酸二ナトリウム(EDTA)0.002質量部、硫酸第一鉄・7水和塩0.001質量部、及び、ドデシルベンゼンスルホン酸ナトリウム(SDBS)1.55質量部を投入し、撹拌しつつ十分に窒素置換を行なって酸素を除いた後、ブタジエン(Bd)100質量部を系中に投入し、45℃に昇温した。パラメンタンハイドロパーオキサイド(PHP)0.03質量部、続いてナトリウムホルムアルデヒドスルホキシレート(SFS)0.10質量部を投入し重合を開始した。重合開始から3、5、7時間目それぞれに、パラメンタンハイドロパーオキサイド(PHP)0.025質量部を投入した。また、重合開始4、6、8時間目それぞれに、EDTA0.0006質量部、及び硫酸第一鉄・7水和塩0.003質量部を投入した。重合15時間目に減圧下残存モノマーを脱揮除去して重合を終了し、ポリブタジエンゴムを主成分とするポリブタジエンゴムラテックス(R-1)を得た。得られたラテックスに含まれるポリブタジエンゴム粒子の体積平均粒子径は0.08μmであった。
製造例1-2;ポリブタジエンゴムラテックス(R-2)の調製
耐圧重合機中に、製造例1-1で得たポリブタジエンゴムラテックス(R-1)を21質量部(ポリブタジエンゴム7質量部を含む)、脱イオン水185質量部、リン酸三カリウム0.03質量部、EDTA0.002質量部、及び硫酸第一鉄・7水和塩0.001質量部を投入し、撹拌しつつ十分に窒素置換を行なって酸素を除いた後、Bd93質量部を系中に投入し、45℃に昇温した。PHP0.02質量部、続いてSFS0.10質量部を投入し重合を開始した。重合開始から24時間目まで3時間おきに、それぞれ、PHP0.025質量部、及びEDTA0.0006質量部、及び硫酸第一鉄・7水和塩0.003質量部を投入した。重合30時間目に減圧下残存モノマーを脱揮除去して重合を終了し、ポリブタジエンゴムを主成分とするポリブタジエンゴムラテックス(R-2)を得た。得られたラテックスに含まれるポリブタジエンゴム粒子の体積平均粒子径は0.20μmであった。
耐圧重合機中に、製造例1-1で得たポリブタジエンゴムラテックス(R-1)を21質量部(ポリブタジエンゴム7質量部を含む)、脱イオン水185質量部、リン酸三カリウム0.03質量部、EDTA0.002質量部、及び硫酸第一鉄・7水和塩0.001質量部を投入し、撹拌しつつ十分に窒素置換を行なって酸素を除いた後、Bd93質量部を系中に投入し、45℃に昇温した。PHP0.02質量部、続いてSFS0.10質量部を投入し重合を開始した。重合開始から24時間目まで3時間おきに、それぞれ、PHP0.025質量部、及びEDTA0.0006質量部、及び硫酸第一鉄・7水和塩0.003質量部を投入した。重合30時間目に減圧下残存モノマーを脱揮除去して重合を終了し、ポリブタジエンゴムを主成分とするポリブタジエンゴムラテックス(R-2)を得た。得られたラテックスに含まれるポリブタジエンゴム粒子の体積平均粒子径は0.20μmであった。
製造例2;コアシェルポリマーラテックス(L)の調製
表1に、コアシェルポリマー(L-1)~(L-10)の製造例を製造例2-1~製造例2-10として記した。
表1に、コアシェルポリマー(L-1)~(L-10)の製造例を製造例2-1~製造例2-10として記した。
製造例2-1:コアシェルポリマーラテックス(L-1)の調製
温度計、撹拌機、還流冷却器、窒素流入口、及びモノマーの添加装置を有するガラス反応器に、製造例1で調製したポリブタジエンゴム粒子を83質量部含むポリブタジエンゴムラテックス(R-1)250質量部、及び、脱イオン水65質量部を仕込み、窒素置換を行いながら60℃で撹拌した。EDTA0.004質量部、硫酸第一鉄・7水和塩0.001質量部、及びSFS0.2質量部を加えた後、シェル層を形成するグラフトモノマー17質量部(アクリロニトリル3.9質量部、スチレン5.4質量部、メチルメタクリレート7.7質量部)、およびクメンヒドロパーオキサイド0.04質量部の混合物を2時間かけて連続的に添加しグラフト重合した。添加終了後、更に2時間撹拌して反応を終了させ、コアシェルポリマーのラテックス(L-1)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.10μmであった。
温度計、撹拌機、還流冷却器、窒素流入口、及びモノマーの添加装置を有するガラス反応器に、製造例1で調製したポリブタジエンゴム粒子を83質量部含むポリブタジエンゴムラテックス(R-1)250質量部、及び、脱イオン水65質量部を仕込み、窒素置換を行いながら60℃で撹拌した。EDTA0.004質量部、硫酸第一鉄・7水和塩0.001質量部、及びSFS0.2質量部を加えた後、シェル層を形成するグラフトモノマー17質量部(アクリロニトリル3.9質量部、スチレン5.4質量部、メチルメタクリレート7.7質量部)、およびクメンヒドロパーオキサイド0.04質量部の混合物を2時間かけて連続的に添加しグラフト重合した。添加終了後、更に2時間撹拌して反応を終了させ、コアシェルポリマーのラテックス(L-1)を得た。得られたラテックスに含まれるコアシェルポリマーの体積平均粒子径は0.10μmであった。
製造例2-2~製造例2-10
製造例2-1におけるポリブタジエンゴムラテックス、グラフトモノマーを、表1のような水準として、同様にコアシェルポリマーラテックスを調製した。表1に、コアシェルポリマー(L-1)~(L-10)のそれぞれの体積平均粒子径を示す。
製造例2-1におけるポリブタジエンゴムラテックス、グラフトモノマーを、表1のような水準として、同様にコアシェルポリマーラテックスを調製した。表1に、コアシェルポリマー(L-1)~(L-10)のそれぞれの体積平均粒子径を示す。

製造例3;コアシェルポリマーが分散しているエポキシ樹脂(N)の調製
製造例3-1;コアシェルポリマーが分散しているエポキシ樹脂(N-1)の調製
25℃の1L混合槽に、メチルエチルケトン132gを導入し、撹拌しながら、製造例2-1で得られたコアシェルポリマー40gを含む水性ラテックス(L-1)を132g投入した。均一に混合後、水200gを80g/分の供給速度で投入した。水を供給した後、速やかに撹拌を停止したところ、浮上性の凝集体および有機溶媒を一部含む水相からなるスラリー液を得た。次に、一部の水相を含む凝集体を残し、水相360gを槽下部の払い出し口より排出させた。得られた凝集体にメチルエチルケトン90gを追加して均一に混合し、コアシェルポリマーが分散した溶液を得た。この溶液に、(A)成分であるエポキシ樹脂(A-1:三菱化学社製、JER828EL)80gを混合し、回転式の蒸発装置で揮発分を除去し、コアシェルポリマーが分散しているエポキシ樹脂(N-1)を得た。
製造例3-1;コアシェルポリマーが分散しているエポキシ樹脂(N-1)の調製
25℃の1L混合槽に、メチルエチルケトン132gを導入し、撹拌しながら、製造例2-1で得られたコアシェルポリマー40gを含む水性ラテックス(L-1)を132g投入した。均一に混合後、水200gを80g/分の供給速度で投入した。水を供給した後、速やかに撹拌を停止したところ、浮上性の凝集体および有機溶媒を一部含む水相からなるスラリー液を得た。次に、一部の水相を含む凝集体を残し、水相360gを槽下部の払い出し口より排出させた。得られた凝集体にメチルエチルケトン90gを追加して均一に混合し、コアシェルポリマーが分散した溶液を得た。この溶液に、(A)成分であるエポキシ樹脂(A-1:三菱化学社製、JER828EL)80gを混合し、回転式の蒸発装置で揮発分を除去し、コアシェルポリマーが分散しているエポキシ樹脂(N-1)を得た。
製造例3-2~製造例3-10
製造例3-1のコアシェルポリマーの水性ラテックス(L-1)を、製造例3-2~製造例3-10のコアシェルポリマーの水性ラテックス(L-2)~(L-10)に置き換えて、製造例3-1と同様の方法にて、コアシェルポリマーが分散しているエポキシ樹脂(N-2)~(N-10)を得た。
製造例3-1のコアシェルポリマーの水性ラテックス(L-1)を、製造例3-2~製造例3-10のコアシェルポリマーの水性ラテックス(L-2)~(L-10)に置き換えて、製造例3-1と同様の方法にて、コアシェルポリマーが分散しているエポキシ樹脂(N-2)~(N-10)を得た。
実施例1~16および比較例1
表2および表3に、(A)エポキシ樹脂、(N)コアシェルポリマーが分散しているエポキシ樹脂、(C)ブロックドイソシアネート、(D)エポキシ硬化剤、(E)硬化促進剤、その他配合原料を配合した硬化性エポキシ樹脂組成物の配合組成と、硬化性エポキシ樹脂組成物の硬化物の評価結果について示す。
表2および表3に、(A)エポキシ樹脂、(N)コアシェルポリマーが分散しているエポキシ樹脂、(C)ブロックドイソシアネート、(D)エポキシ硬化剤、(E)硬化促進剤、その他配合原料を配合した硬化性エポキシ樹脂組成物の配合組成と、硬化性エポキシ樹脂組成物の硬化物の評価結果について示す。
また、表2および表3に、(A)エポキシ樹脂100質量部を基準として、(B)コアシェルポリマー、(C)ブロックドイソシアネート、(D)エポキシ硬化剤、(E)硬化促進剤、その他配合原料の質量部を併せて示した。
表2には、(C)ブロックドイソシアネートとして、ポリプロピレングリコール構造を有するブロックドイソシアネートである(C-1)アデカレジンQR-9466を用いた。
表2には、(C)ブロックドイソシアネートとして、ポリプロピレングリコール構造を有するブロックドイソシアネートである(C-1)アデカレジンQR-9466を用いた。
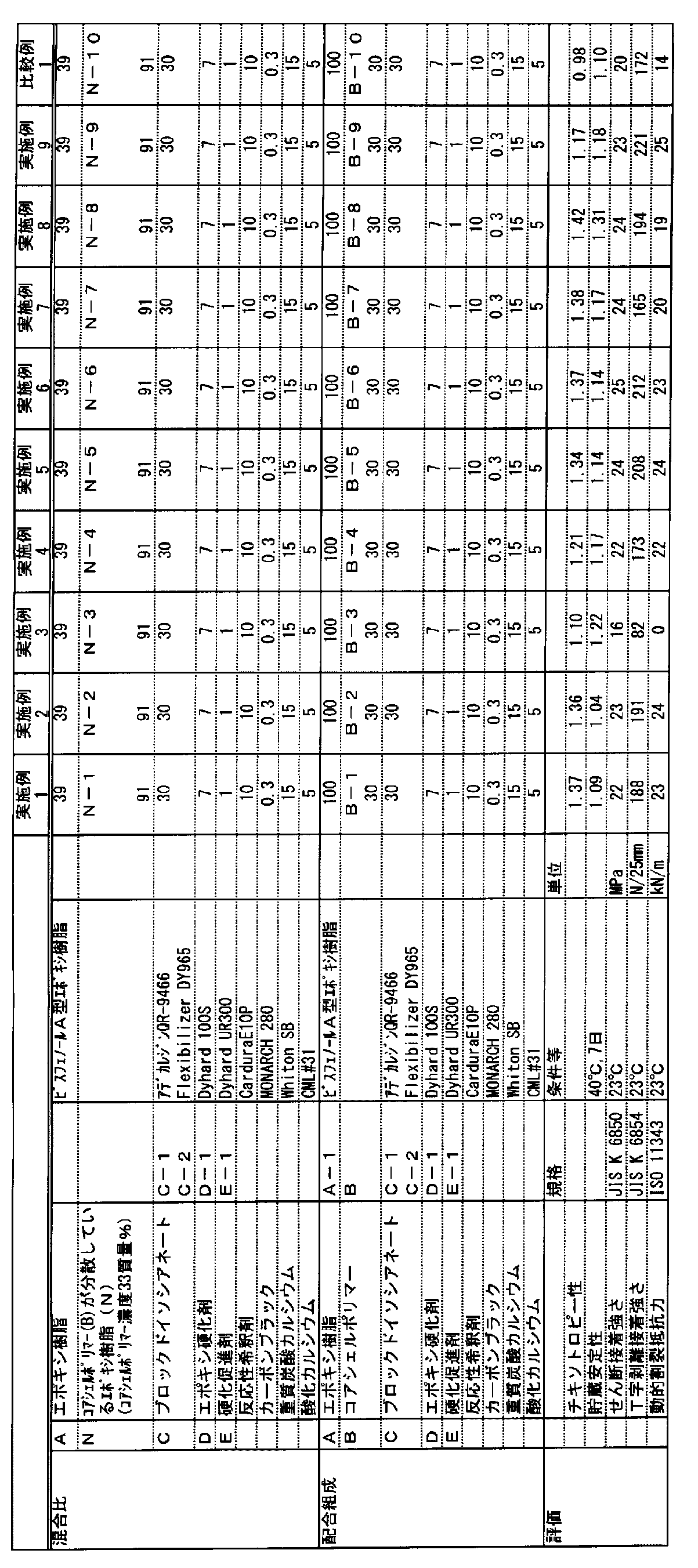
表3には、(C)ブロックドイソシアネートとして、ポリテトラメチレングリコール構造を有するブロックドイソシアネートである(C-2)Flexibilizer DY965を用いた。
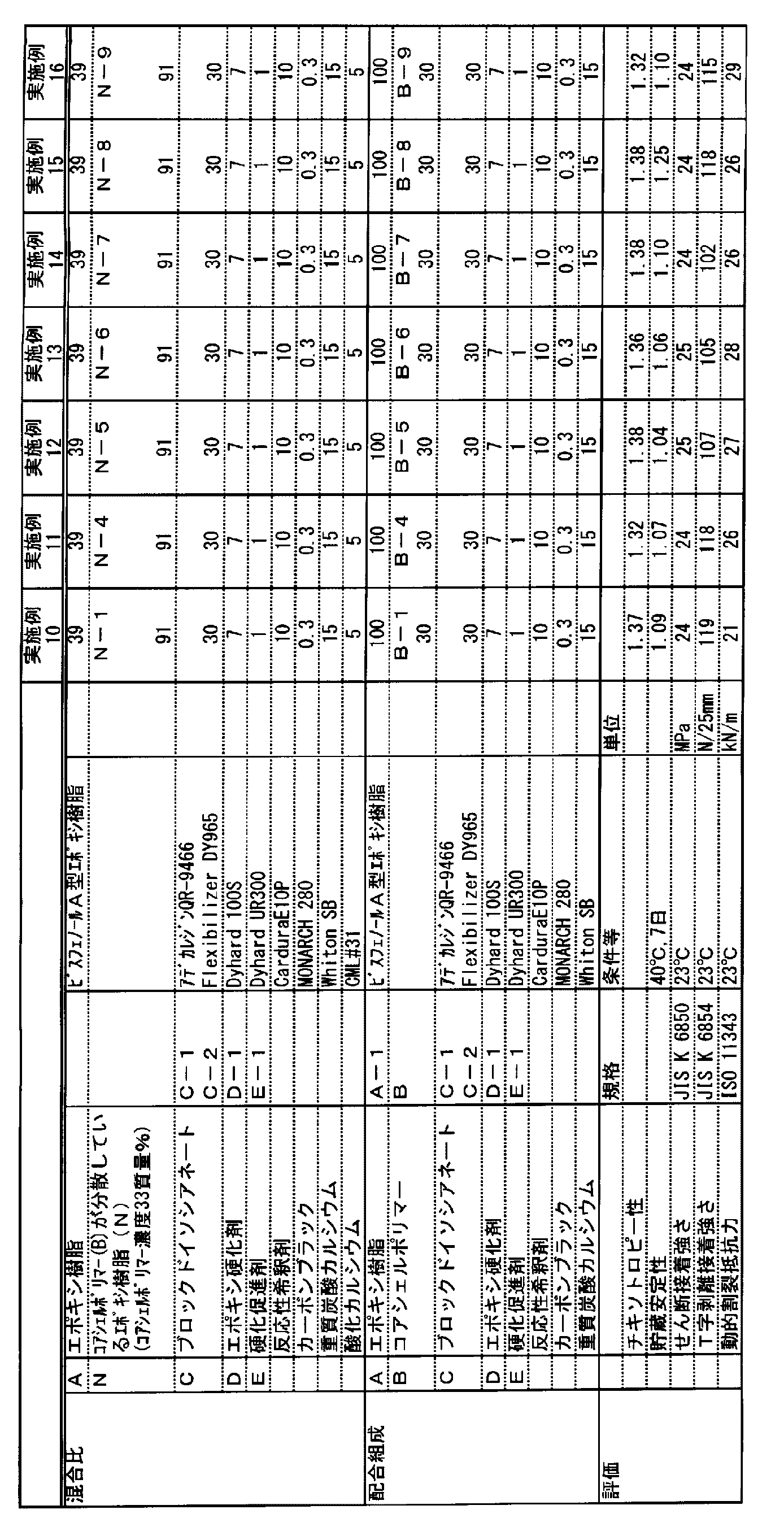
<配合剤>
なお、表2および表3において使用した配合原料を以下に示す。
(A)エポキシ樹脂
(A-1)JER828EL(三菱化学製、ビスフェノールA型エポキシ樹脂)
(N)コアシェルポリマーが分散しているエポキシ樹脂組成物
(N-1)~(N-10);製造例3-1~3-10に記載した。
(B)コアシェルポリマー
(B-1)~(B-10);製造例2-1から製造例2-10に記載したコアシェルポリマーラテックス(L-1)から(L-10)に含まれるコアシェルポリマーである。組成については、表1を参照のこと。
(C)ブロックドイソシアネート
(C-1)アデカレジンQR-9466(ADEKA製、ポリプロピレングリコール構造を含むブロックドイソシアネート、ブロックNCO当量220、粘度30000mPa・s/25℃)
(C-2)Flexibilizer DY 965(Huntsman製、ポリテトラメチレングリコール構造を含むブロックドイソシアネート、Epoxide Eq.Weight558-667[EEW,g/eq]、粘度 4400-12800cP/25℃)
(D)エポキシ硬化剤
(D-1)Dyhard 100S(AlzChem製、ジシアンジアミド)
(E)硬化促進剤
(E-1)Dyhard UR300(AlzChem製、1,1-ジメチル-3-フェニルウレア)
その他配合原料
(重質炭酸カルシウム)ホワイトンSB赤(白石カルシウム製、無処理重質炭酸カルシウム、平均粒子径:1.8μm)
(酸化カルシウム)CML#31(近江化学工業製)
(カーボンブラック)MONARCH 280(Cabot製)
(反応性希釈剤)Cardula E10P(Momentive製、バーサチック酸グリシジルエステル)
なお、表2および表3において使用した配合原料を以下に示す。
(A)エポキシ樹脂
(A-1)JER828EL(三菱化学製、ビスフェノールA型エポキシ樹脂)
(N)コアシェルポリマーが分散しているエポキシ樹脂組成物
(N-1)~(N-10);製造例3-1~3-10に記載した。
(B)コアシェルポリマー
(B-1)~(B-10);製造例2-1から製造例2-10に記載したコアシェルポリマーラテックス(L-1)から(L-10)に含まれるコアシェルポリマーである。組成については、表1を参照のこと。
(C)ブロックドイソシアネート
(C-1)アデカレジンQR-9466(ADEKA製、ポリプロピレングリコール構造を含むブロックドイソシアネート、ブロックNCO当量220、粘度30000mPa・s/25℃)
(C-2)Flexibilizer DY 965(Huntsman製、ポリテトラメチレングリコール構造を含むブロックドイソシアネート、Epoxide Eq.Weight558-667[EEW,g/eq]、粘度 4400-12800cP/25℃)
(D)エポキシ硬化剤
(D-1)Dyhard 100S(AlzChem製、ジシアンジアミド)
(E)硬化促進剤
(E-1)Dyhard UR300(AlzChem製、1,1-ジメチル-3-フェニルウレア)
その他配合原料
(重質炭酸カルシウム)ホワイトンSB赤(白石カルシウム製、無処理重質炭酸カルシウム、平均粒子径:1.8μm)
(酸化カルシウム)CML#31(近江化学工業製)
(カーボンブラック)MONARCH 280(Cabot製)
(反応性希釈剤)Cardula E10P(Momentive製、バーサチック酸グリシジルエステル)
図1、4は、コアシェルポリマーのシェル層のグリシジルメタクリレート(GMA)と貯蔵安定性及びチキソトロピー性との関係を示し、シェル層にグリシジルメタクリレートが少量で含まれると、良好な貯蔵安定性とチキソトロピー性とすることができる(図1ではポリプロピレングリコール構造を含むブロックドイソシアネートを用いた(図中のGMA比率%は、実施例1、5、6、7、8で使用したものに対応する)。図4ではポリテトラメチレングリコ-ル構造を含むブロックドイソシアネートを用いた(図中のGMA比率%は、実施例10、12、13、14、15で使用したものに対応する)。)。
図2、5は、コアシェルポリマーのシェル層のグリシジルメタクリレート(GMA)とT字剥離接着強さ(図中、T-Peelで示す。)及びせん断接着強さ(図中、Lap Shearで示す。)との関係を示し、シェル層にグリシジルメタクリレートが所定量(0質量%超20質量%未満)含まれると、これら特性を改善することができる(図2ではポリプロピレングリコール構造を含むブロックドイソシアネートを用いた(図中のGMA比率%は、実施例1、5、6、7、8で使用したものに対応する)。図5ではポリテトラメチレングリコ-ル構造を含むブロックドイソシアネートを用いた(図中のGMA比率%は、実施例10、12、13、14、15で使用したものに対応する)。)。
図3、6は、コアシェルポリマーのシェル層のグリシジルメタクリレート(GMA)と動的割裂抵抗力(図中、Impact Peelで示す。)との関係を示し、シェル層にグリシジルメタクリレートが所定量(0質量%超20質量%未満)含まれると、動的割裂抵抗力を改善することができる(図3ではポリプロピレングリコール構造を含むブロックドイソシアネートを用いた(図中のGMA比率%は、実施例1、5、6、7、8で使用したものに対応する)。図6ではポリテトラメチレングリコ-ル構造を含むブロックドイソシアネートを用いた(図中のGMA比率%は、実施例10、12、13、14、15で使用したものに対応する)。)。
図2、5は、コアシェルポリマーのシェル層のグリシジルメタクリレート(GMA)とT字剥離接着強さ(図中、T-Peelで示す。)及びせん断接着強さ(図中、Lap Shearで示す。)との関係を示し、シェル層にグリシジルメタクリレートが所定量(0質量%超20質量%未満)含まれると、これら特性を改善することができる(図2ではポリプロピレングリコール構造を含むブロックドイソシアネートを用いた(図中のGMA比率%は、実施例1、5、6、7、8で使用したものに対応する)。図5ではポリテトラメチレングリコ-ル構造を含むブロックドイソシアネートを用いた(図中のGMA比率%は、実施例10、12、13、14、15で使用したものに対応する)。)。
図3、6は、コアシェルポリマーのシェル層のグリシジルメタクリレート(GMA)と動的割裂抵抗力(図中、Impact Peelで示す。)との関係を示し、シェル層にグリシジルメタクリレートが所定量(0質量%超20質量%未満)含まれると、動的割裂抵抗力を改善することができる(図3ではポリプロピレングリコール構造を含むブロックドイソシアネートを用いた(図中のGMA比率%は、実施例1、5、6、7、8で使用したものに対応する)。図6ではポリテトラメチレングリコ-ル構造を含むブロックドイソシアネートを用いた(図中のGMA比率%は、実施例10、12、13、14、15で使用したものに対応する)。)。
ポリプロピレングリコール構造を含むウレタンプレポリマーをブロック剤(好ましくはフェノール系ブロック剤)でキャップした化合物は、ポリテトラメチレングリコール構造を含むウレタンプレポリマーをブロック剤(好ましくはフェノール系ブロック剤)でキャップした化合物よりも、T字剥離接着強さを改善することができる(実施例1~16、図2、4)。
ポリテトラメチレングリコール構造を含むウレタンプレポリマーをブロック剤(好ましくはフェノール系ブロック剤)でキャップした化合物は、ポリプロピレングリコール構造を含むウレタンプレポリマーをブロック剤(好ましくはフェノール系ブロック剤)でキャップした化合物よりも、貯蔵安定性、動的割裂抵抗力を改善することができる(実施例1~16、図1、3、4、6参照)
ポリテトラメチレングリコール構造を含むウレタンプレポリマーをブロック剤(好ましくはフェノール系ブロック剤)でキャップした化合物は、ポリプロピレングリコール構造を含むウレタンプレポリマーをブロック剤(好ましくはフェノール系ブロック剤)でキャップした化合物よりも、貯蔵安定性、動的割裂抵抗力を改善することができる(実施例1~16、図1、3、4、6参照)
Claims (17)
- エポキシ樹脂(A)100質量部に対し、
コアシェルポリマー(B)1~50質量部および
ブロックドイソシアネート(C)1~50質量部
を含有する硬化性エポキシ樹脂組成物であって、
コアシェルポリマー(B)のコア層が、ジエン系ゴム、アクリル系ゴム、ポリシロキサン系ゴムのいずれかからなり、
コアシェルポリマー(B)のシェル層が、シェル層を構成するモノマー全量を100質量%とし、ビニルシアンモノマーおよびアルコキシアルキル(メタ)アクリレートモノマーからなる群より選択されるモノマー10~95質量%、エポキシ基を有するモノマーを0~50質量%、およびそれらと共重合可能なモノマー0~90質量%からなることを特徴とする硬化性エポキシ樹脂組成物。 - それらと共重合可能なモノマーが5~90質量%である請求項1に記載の硬化性エポキシ樹脂組成物。
- コアシェルポリマー(B)のシェル層が、シェル層を構成するモノマー全量を100質量%とし、ビニルシアンモノマー20~40質量%、エポキシ基を有するモノマー3~15質量%、およびそれらと共重合可能なモノマー60~80質量%からなる請求項1または2に記載の硬化性エポキシ樹脂組成物。
- コアシェルポリマー(B)のシェル層が、シェル層を構成するモノマー全量を100質量%とし、アルコキシアルキル(メタ)アクリレートモノマー50~95質量%、およびエポキシ基を有するモノマー5~50質量%からなる請求項1に記載の硬化性エポキシ樹脂組成物。
- コアシェルポリマー(B)のシェル層が、シェル層を構成するモノマー全量を100質量%とし、アルコキシアルキル(メタ)アクリレートモノマー90~95質量%、およびエポキシ基を有するモノマー5~10質量%からなる請求項1または4に記載の硬化性エポキシ樹脂組成物。
- ビニルシアンモノマーが、アクリロニトリルである請求項1~3のいずれかに記載の硬化性エポキシ樹脂組成物。
- エポキシ基を有するモノマーがグリシジルメタクリレートである請求項1~6のいずれかに記載の硬化性エポキシ樹脂組成物。
- アルコキシアルキル(メタ)アクリレートモノマーが2-メトキシエチルアクリレートである請求項1、2、4~7のいずれかに記載の硬化性エポキシ樹脂組成物。
- コアシェルポリマー(B)のコア層が、ブタジエンゴム、および/または、ブタジエン-スチレンゴムからなる請求項1~8のいずれかに記載の硬化性エポキシ樹脂組成物。
- コアシェルポリマー(B)の体積平均粒子径が0.05~0.30μmである請求項1~9のいずれかに記載の硬化性エポキシ樹脂組成物。
- ブロックドイソシアネート(C)が、ポリプロピレングリコール構造を含むウレタンプレポリマーをブロック剤でキャップした化合物である請求項1~10のいずれかに記載の硬化性エポキシ樹脂組成物。
- ブロックドイソシアネート(C)が、ポリテトラメチレングリコール構造を含むウレタンプレポリマーをブロック剤でキャップした化合物である請求項1~10のいずれかに記載の硬化性エポキシ樹脂組成物。
- 更に、エポキシ硬化剤(D)1~40質量部を含有する請求項1~12のいずれかに記載の硬化性エポキシ樹脂組成物。
- 更に、硬化促進剤(E)0.1~10質量部を含有する請求項1~13のいずれかに記載の硬化性エポキシ樹脂組成物。
- 請求項1~14のいずれかに記載の硬化性エポキシ樹脂組成物を硬化した硬化物。
- 請求項1~14のいずれかに記載の硬化性エポキシ樹脂組成物を用いてなる構造接着剤。
- 請求項1~14のいずれかに記載の硬化性エポキシ樹脂組成物を用いてなる車両用構造接着剤。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017510177A JP6694425B2 (ja) | 2015-03-31 | 2016-03-31 | チキソトロピー性に優れる硬化性エポキシ樹脂組成物 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015-074446 | 2015-03-31 | ||
| JP2015074446 | 2015-03-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016159223A1 true WO2016159223A1 (ja) | 2016-10-06 |
Family
ID=57007280
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/060637 WO2016159223A1 (ja) | 2015-03-31 | 2016-03-31 | チキソトロピー性に優れる硬化性エポキシ樹脂組成物 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP6694425B2 (ja) |
| WO (1) | WO2016159223A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20190004562A (ko) * | 2017-07-04 | 2019-01-14 | 삼성에스디아이 주식회사 | 편광판용 점착제 조성물, 이로부터 제조된 편광판용 점착필름, 이를 포함하는 편광판 및 이를 포함하는 광학표시장치 |
| WO2019208569A1 (ja) | 2018-04-27 | 2019-10-31 | 株式会社カネカ | 作業性に優れるポリマー微粒子含有硬化性樹脂組成物を用いる接着方法、及び、該接着方法を用いて得られる積層体 |
| JP2020524728A (ja) * | 2017-06-23 | 2020-08-20 | ディディピー スペシャリティ エレクトロニック マテリアルズ ユーエス インコーポレーテッド | 高温エポキシ接着剤配合物 |
| JP2020132833A (ja) * | 2019-02-26 | 2020-08-31 | 株式会社カネカ | 硬化性エポキシ樹脂組成物、及びそれを用いた積層体 |
| WO2022102467A1 (ja) * | 2020-11-16 | 2022-05-19 | 東レ株式会社 | 熱硬化性エポキシ樹脂組成物とその成形品、繊維強化複合材料、繊維強化複合材料用成形材料、および繊維強化複合材料の製造方法 |
| WO2023195462A1 (ja) * | 2022-04-08 | 2023-10-12 | 株式会社Adeka | 硬化性樹脂組成物 |
| WO2024024247A1 (ja) * | 2022-07-29 | 2024-02-01 | 株式会社ブリヂストン | 接着剤組成物、有機繊維材料、ゴム物品、有機繊維-ゴム複合体及びタイヤ |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3819351B1 (en) * | 2018-07-25 | 2022-09-14 | Lg Chem, Ltd. | Adhesive composition |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005255822A (ja) * | 2004-03-11 | 2005-09-22 | Kaneka Corp | ゴム強化エポキシ樹脂製品 |
| JP2009545656A (ja) * | 2006-07-31 | 2009-12-24 | ヘンケル・アクチェンゲゼルシャフト・ウント・コムパニー・コマンディットゲゼルシャフト・アウフ・アクチェン | 硬化性エポキシ樹脂系粘着組成物 |
| JP2010505020A (ja) * | 2006-09-26 | 2010-02-18 | ロックタイト (アール アンド ディー) リミテッド | 新規付加物およびこれを使う硬化性組成物 |
| WO2010143366A1 (ja) * | 2009-06-09 | 2010-12-16 | 株式会社カネカ | ポリマー微粒子含有ビニルエステル系樹脂組成物、その製造方法、及びその硬化物 |
| JP2014141604A (ja) * | 2013-01-25 | 2014-08-07 | Kaneka Corp | 貯蔵安定性の改善されたポリマー微粒子含有硬化性樹脂組成物 |
| JP2014532800A (ja) * | 2011-11-10 | 2014-12-08 | ダウ グローバル テクノロジーズ エルエルシー | エポキシ硬化用途の潜在性触媒としてのカルボン酸アンモニウム基を有するポリマー |
| WO2015053289A1 (ja) * | 2013-10-11 | 2015-04-16 | 株式会社カネカ | コアシェルポリマー含有エポキシ樹脂組成物、その硬化物、及びその製造方法 |
| WO2015064561A1 (ja) * | 2013-10-29 | 2015-05-07 | 株式会社カネカ | 貯蔵安定性の改善されたポリマー微粒子含有硬化性樹脂組成物 |
-
2016
- 2016-03-31 WO PCT/JP2016/060637 patent/WO2016159223A1/ja active Application Filing
- 2016-03-31 JP JP2017510177A patent/JP6694425B2/ja active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005255822A (ja) * | 2004-03-11 | 2005-09-22 | Kaneka Corp | ゴム強化エポキシ樹脂製品 |
| JP2009545656A (ja) * | 2006-07-31 | 2009-12-24 | ヘンケル・アクチェンゲゼルシャフト・ウント・コムパニー・コマンディットゲゼルシャフト・アウフ・アクチェン | 硬化性エポキシ樹脂系粘着組成物 |
| JP2010505020A (ja) * | 2006-09-26 | 2010-02-18 | ロックタイト (アール アンド ディー) リミテッド | 新規付加物およびこれを使う硬化性組成物 |
| WO2010143366A1 (ja) * | 2009-06-09 | 2010-12-16 | 株式会社カネカ | ポリマー微粒子含有ビニルエステル系樹脂組成物、その製造方法、及びその硬化物 |
| JP2014532800A (ja) * | 2011-11-10 | 2014-12-08 | ダウ グローバル テクノロジーズ エルエルシー | エポキシ硬化用途の潜在性触媒としてのカルボン酸アンモニウム基を有するポリマー |
| JP2014141604A (ja) * | 2013-01-25 | 2014-08-07 | Kaneka Corp | 貯蔵安定性の改善されたポリマー微粒子含有硬化性樹脂組成物 |
| WO2015053289A1 (ja) * | 2013-10-11 | 2015-04-16 | 株式会社カネカ | コアシェルポリマー含有エポキシ樹脂組成物、その硬化物、及びその製造方法 |
| WO2015064561A1 (ja) * | 2013-10-29 | 2015-05-07 | 株式会社カネカ | 貯蔵安定性の改善されたポリマー微粒子含有硬化性樹脂組成物 |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2020524728A (ja) * | 2017-06-23 | 2020-08-20 | ディディピー スペシャリティ エレクトロニック マテリアルズ ユーエス インコーポレーテッド | 高温エポキシ接着剤配合物 |
| JP7018079B2 (ja) | 2017-06-23 | 2022-02-09 | ディディピー スペシャルティ エレクトロニック マテリアルズ ユーエス,エルエルシー | 高温エポキシ接着剤配合物 |
| KR20190004562A (ko) * | 2017-07-04 | 2019-01-14 | 삼성에스디아이 주식회사 | 편광판용 점착제 조성물, 이로부터 제조된 편광판용 점착필름, 이를 포함하는 편광판 및 이를 포함하는 광학표시장치 |
| KR102158871B1 (ko) * | 2017-07-04 | 2020-09-22 | 삼성에스디아이 주식회사 | 편광판용 점착제 조성물, 이로부터 제조된 편광판용 점착필름, 이를 포함하는 편광판 및 이를 포함하는 광학표시장치 |
| WO2019208569A1 (ja) | 2018-04-27 | 2019-10-31 | 株式会社カネカ | 作業性に優れるポリマー微粒子含有硬化性樹脂組成物を用いる接着方法、及び、該接着方法を用いて得られる積層体 |
| US11384267B2 (en) | 2018-04-27 | 2022-07-12 | Kaneka Corporation | Adhesion method employing polymer microparticle-containing curable resin composition having excellent workability, and laminate obtained using said adhesion method |
| US11781047B2 (en) | 2018-04-27 | 2023-10-10 | Kaneka Corporation | Adhesion method employing polymer microparticle-containing curable resin composition having excellent workability, and laminate obtained using said adhesion method |
| JP2020132833A (ja) * | 2019-02-26 | 2020-08-31 | 株式会社カネカ | 硬化性エポキシ樹脂組成物、及びそれを用いた積層体 |
| JP7187347B2 (ja) | 2019-02-26 | 2022-12-12 | 株式会社カネカ | 硬化性エポキシ樹脂組成物、及びそれを用いた積層体 |
| WO2022102467A1 (ja) * | 2020-11-16 | 2022-05-19 | 東レ株式会社 | 熱硬化性エポキシ樹脂組成物とその成形品、繊維強化複合材料、繊維強化複合材料用成形材料、および繊維強化複合材料の製造方法 |
| WO2023195462A1 (ja) * | 2022-04-08 | 2023-10-12 | 株式会社Adeka | 硬化性樹脂組成物 |
| WO2024024247A1 (ja) * | 2022-07-29 | 2024-02-01 | 株式会社ブリヂストン | 接着剤組成物、有機繊維材料、ゴム物品、有機繊維-ゴム複合体及びタイヤ |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6694425B2 (ja) | 2020-05-13 |
| JPWO2016159223A1 (ja) | 2018-02-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2016159224A1 (ja) | 貯蔵安定性に優れる硬化性エポキシ樹脂組成物 | |
| JP6924135B2 (ja) | 耐衝撃剥離接着性の改善されたポリマー微粒子含有硬化性樹脂組成物 | |
| JP6560617B2 (ja) | 貯蔵安定性の改善されたポリマー微粒子含有硬化性樹脂組成物 | |
| JP6476045B2 (ja) | 接着性の改善されたポリマー微粒子含有硬化性樹脂組成物 | |
| JP6463271B2 (ja) | コアシェルポリマー含有エポキシ樹脂組成物、その硬化物、及びその製造方法 | |
| JP6694425B2 (ja) | チキソトロピー性に優れる硬化性エポキシ樹脂組成物 | |
| JP6767758B2 (ja) | 貯蔵安定性および接着性の改善されたポリマー微粒子含有硬化性樹脂組成物 | |
| JP7187347B2 (ja) | 硬化性エポキシ樹脂組成物、及びそれを用いた積層体 | |
| WO2017179536A1 (ja) | 硬化性組成物及び接着剤 | |
| US20210214549A1 (en) | Curable epoxy resin composition and laminate using the same | |
| WO2022138807A1 (ja) | 硬化性樹脂組成物及び接着剤 | |
| JP6722477B2 (ja) | 剥離接着性および耐衝撃剥離接着性の改善されたポリマー微粒子含有硬化性樹脂組成物 | |
| JP2020152775A (ja) | エポキシ樹脂組成物及び接着剤 | |
| WO2019123934A1 (ja) | エポキシ樹脂組成物 | |
| JP2022050820A (ja) | エポキシ樹脂組成物及び接着剤 | |
| JP7277241B2 (ja) | 作業性に優れるポリマー微粒子含有硬化性樹脂組成物を用いる接着方法、及び、該接着方法を用いて得られる積層体 | |
| WO2023249099A1 (ja) | 硬化性樹脂組成物、硬化物、接着剤および積層体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16773103 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017510177 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16773103 Country of ref document: EP Kind code of ref document: A1 |