WO2015108170A1 - シクロブタンテトラカルボン酸及びその無水物の製造方法 - Google Patents
シクロブタンテトラカルボン酸及びその無水物の製造方法 Download PDFInfo
- Publication number
- WO2015108170A1 WO2015108170A1 PCT/JP2015/051149 JP2015051149W WO2015108170A1 WO 2015108170 A1 WO2015108170 A1 WO 2015108170A1 JP 2015051149 W JP2015051149 W JP 2015051149W WO 2015108170 A1 WO2015108170 A1 WO 2015108170A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- acid
- carbon atoms
- alkyl group
- reaction
- Prior art date
Links
- 0 CCC(C(C)(C)*(N=O)=O)=C Chemical compound CCC(C(C)(C)*(N=O)=O)=C 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/09—Preparation of carboxylic acids or their salts, halides or anhydrides from carboxylic acid esters or lactones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C61/00—Compounds having carboxyl groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C61/04—Saturated compounds having a carboxyl group bound to a three or four-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/04—Systems containing only non-condensed rings with a four-membered ring
Definitions
- the present invention relates to a novel process for producing cyclobutanetetracarboxylic acid and its anhydride.
- Tetracarboxylic acid derivatives such as tetracarboxylic acid dialkyl esters are important substances that serve as raw materials for polyamides, polyesters, and polyimides.
- Tetracarboxylic acid derivatives such as tetracarboxylic acid dialkyl esters are important substances that serve as raw materials for polyamides, polyesters, and polyimides.
- a polyimide having a cyclobutane skeleton in the main chain bis (chlorocarbonyl) cyclobutanedicarboxylic acid dimethyl ester and diamine are reacted to obtain polyamic acid methyl ester, and then heated to obtain a polyimide. Examples have been reported (see Non-Patent Document 1).
- the present invention uses, as a raw material, a residue after separation of a target product produced in the production process of a cyclobutanetetracarboxylic acid derivative or the like, and usefully increases cyclobutanetetracarboxylic acid and / or its dianhydride from the residue.
- the object is to provide a new method for obtaining efficiently.
- R 1 is an alkyl group having 1 to 5 carbon atoms
- R 2 is an alkyl group having 1 to 5 carbon atoms
- n is 2 or 4
- R 1 and R 2 are the same Or it may be different
- R 1 is an alkyl group having 1 to 5 carbon atoms
- R 2 is an alkyl group having 1 to 5 carbon atoms
- R 1 and R 2 may be the same or different.
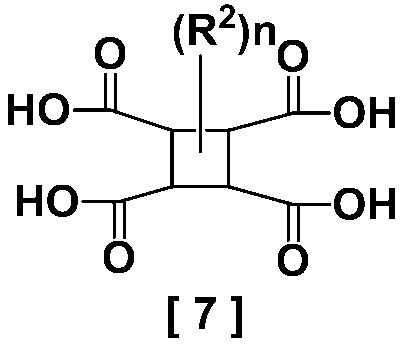
- the formula [7] is represented by the following formula [7-a]
- R 2 represents an alkyl group having 1 to 5 carbon atoms
- the formula [5] is represented by the formula [5-a]
- R 2 is an alkyl group having 1 to 5 carbon atoms.
- a residue obtained after isolation of a target product, which is produced in the process of producing a cyclobutanetetracarboxylic acid derivative or the like, is used as a raw material, and from that, cyclobutanetetracarboxylic acid and / or its dianhydride is a useful product. Can be manufactured easily and with high efficiency.
- the raw material in the production method of the present invention is a compound represented by the following formula [1] or formula [2] contained in a residue produced in the production process such as cyclobutanetetracarboxylic acid dialkyl ester.
- R 1 is an alkyl group having 1 to 5 carbon atoms
- R 2 is an alkyl group having 1 to 5 carbon atoms
- n is 2 or 4.
- R 2 Are preferably the same substituent, and when n is 4, all R 2 are preferably the same, but may be different.
- R 1 and R 2 may be the same or different.
- R 1 is an alkyl group having 1 to 5 carbon atoms, preferably 1 to 3 carbon atoms. Specific examples of the alkyl group include a methyl group, an ethyl group, a normal propyl group, an isopropyl group, a normal butyl group, and a secondary butyl group.
- R 1 preferably has a small number of carbon atoms and is easily detached, and more preferably a methyl group.
- R 2 is an alkyl group having 1 to 5 carbon atoms, preferably 1 to 3 carbon atoms.
- alkyl group examples include a methyl group, an ethyl group, a normal propyl group, an isopropyl group, a normal butyl group, a secondary butyl group, An isobutyl group, a tertiary butyl group, a normal pentyl group, etc. are mentioned.
- n represents an integer of 1 to 4, and is preferably 2.
- Me methyl group, Et: ethyl group, Pr-n: normal propyl group, Pr-iso: isopropyl group, Bu-n: normal butyl group, Bu-sec: secondary butyl group, Bu-iso: isobutyl group, Bu- t: tertiary butyl group, Pen-n: normal pentyl group, OMe: methoxy group, OEt: ethoxy group, OPr-n: normal propyl ether group, OPr-iso: isopropyl ether group, OBu-n: normal butoxy group, OBu-sec: secondary butoxy group, OBu-iso: isobutoxy group, OBu-t: tertiary butoxy group, OPen-n: normal pentyl ether group
- n is 2, and R 2 is an ethyl group, a normal propyl group, an isopropyl group, a normal butyl group, a secondary butyl group, an isobutyl group, a tertiary butyl group, Or in the case of a compound that is a normal pentyl group, Me in b1 to b4 in Table 1 and Table 2 above is Et, Pr-n, Pr-iso, Bu-n, Bu-sec, Bu-iso, Bu-t, Alternatively, compounds substituted with Pen-n can be exemplified.
- Cyclobutanetetracarboxylic acid dialkyl ester is produced by reacting cyclobutanetetracarboxylic dianhydride [5] with an alcohol having 1 to 5 carbon atoms represented by R 1 OH, as shown in the following reaction formula.
- R 1 is an alkyl group having 1 to 5 carbon atoms
- R 2 is an alkyl group having 1 to 5 carbon atoms
- n is 2 or 4.
- R 2 Are preferably the same substituent, and when n is 4, all R 2 are preferably the same, but may be different.
- R 1 and R 2 may be the same or different.
- the above reaction can be performed using alcohol (R 1 OH), which is also a reaction raw material, as a solvent, and other solvents can be used as necessary.
- a solvent is not particularly limited as long as it is inert to the reaction.
- hydrocarbon such as hexane, heptane or toluene, halogenated hydrocarbon such as chloroform, 1,2-dichloroethane or chlorobenzene, diethyl ether or 1 , 4-dioxane, ethers such as ethyl acetate, ketones such as acetone or methyl ethyl ketone, nitriles such as acetonitrile or propionitrile, or mixtures thereof.
- ethyl acetate or acetonitrile is mentioned, More preferably, it is acetonitrile.
- the alcohol (R 1 OH) is generally used in an amount of 2 to 100 times mol, preferably 2 to 40 times mol, more preferably 2 to 20 times mol, relative to the tetracarboxylic dianhydride [5].
- the above reaction proceeds under neutral conditions, but a base or acid may be added.
- the base include inorganic bases such as sodium hydroxide, potassium hydroxide, potassium carbonate or sodium hydrogen carbonate; triethylamine, pyridine, quinoline, 8-quinolinol, 1,10-phenanthroline, bathophenanthroline, bathocuproine, 2, 2'-bipyridyl, 2-phenylpyridine, 2,6-diphenylaminopyridine, 2-dimethylaminopyridine, 4-dimethylaminopyridine, 2- (2-hydroxylethyl) pyridine, N, N-dimethylaniline, 1,8 Organic bases such as diazabicyclo [5,4,0] -7-undene (DBU); metal alkoxides such as sodium methoxide, potassium methoxide or potassium t-butoxide. Preferred is sodium methoxide, potassium methoxide, or pyridine, and more preferred is pyridine.
- DBU diaza
- the acid examples include heteropolyacids such as phosphomolybdic acid and phosphotungstic acid, organic acids such as trimethylborate and triphenylphosphine; inorganic acids such as hydrochloric acid, sulfuric acid, and phosphoric acid; carbonization such as formic acid, acetic acid, and p-toluenesulfonic acid.
- Hydrogen acid; Halogen-based hydrocarbon acids such as trifluoroacetic acid are listed. P-toluenesulfonic acid, phosphoric acid, or acetic acid is preferable, and p-toluenesulfonic acid is more preferable.
- the above base or acid is usually used in an amount of 0 to 100-fold mol, preferably 0.01 to 10-fold mol based on tetracarboxylic dianhydride [5].
- the reaction temperature is not particularly limited but is, for example, ⁇ 90 to 200 ° C., preferably ⁇ 30 to 100 ° C.
- the reaction time is usually 0.05 to 200 hours, preferably 0.5 to 100 hours.
- the selectivity and reaction rate of the formula [1-a] can be improved, and more preferably a basic compound is present.
- Examples of the base or acid used at this time include those exemplified above, and preferred bases or acids and preferred addition amounts are also as described above.
- the target product produced by the reaction can be easily separated.
- the alcohol used is distilled off after completion of the reaction, the precipitated crystals are heated to reflux in an organic solvent, and then cooled to cool the precipitated crystals.
- the organic solvent for example, toluene, acetonitrile, ethyl acetate, ethyl acetate / n-heptane mixed solution, ethyl acetate / alcohol mixed solution, acetonitrile / alcohol mixed solution and the like can be used.
- Acetonitrile, ethyl acetate, ethyl acetate / alcohol mixture, or acetonitrile / alcohol mixture is preferable.
- the alcohol include lower alcohols having 1 to 5 carbon atoms such as methanol, ethanol, propanol, butanol, and isopropanol.
- Primary crystals can be further purified by washing and recrystallization.
- the recrystallization method include a method of adding an organic solvent to the primary crystal and heating, followed by ice cooling, filtration, and drying.
- the organic solvent for example, toluene, acetonitrile, ethyl acetate, ethyl acetate / n-heptane mixed solution, ethyl acetate / various alcohol mixed solution, acetonitrile / various alcohol mixed solution and the like can be used.
- Acetonitrile, ethyl acetate, ethyl acetate / various alcohol mixtures, or acetonitrile / various alcohol mixtures are preferable.
- various alcohols include methanol, ethanol, propanol, butanol, isopropanol and the like.
- the amount of the organic solvent used for obtaining these primary crystals is usually based on the weight when the desired product is obtained in a yield of 100% from the raw material, and preferably 2 to 20 times its amount. . Further, when it is desired to improve the yield, it is preferable to reduce the amount of organic solvent used, and when it is desired to obtain a high purity product, it is preferable to increase the amount of organic solvent used. Considering these yields and purity, 2.5 to 5 times is more preferable.
- a high purity product of the formula [2-a] can be obtained by washing and recrystallizing the filtrate when the primary crystals are obtained. That is, the solvent of the obtained filtrate was distilled off, and the precipitated crystals were heated to reflux in an organic solvent and then cooled, and the precipitated crystals were collected by filtration, washed and dried to obtain the target formula [2-a]. Secondary crystals of high purity are obtained.
- the organic solvent for example, toluene, acetonitrile, ethyl acetate, ethyl acetate / n-heptane mixed solution, ethyl acetate / various alcohol mixed solution, acetonitrile / various alcohol mixed solution and the like can be used.
- Acetonitrile, ethyl acetate, ethyl acetate / various alcohol mixtures, or acetonitrile / various alcohol mixtures are preferable.
- various alcohols include methanol, ethanol, propanol, butanol, isopropanol and the like.
- the purity of the secondary crystal can be further increased by washing and recrystallization.
- the recrystallization method include a method in which an organic solvent is added to the secondary crystal and heated, followed by ice cooling, filtration, and drying.
- the organic solvent for example, toluene, acetonitrile, ethyl acetate, ethyl acetate / n-heptane mixed solution, ethyl acetate / various alcohol mixed solution, acetonitrile / various alcohol mixed solution and the like can be used.
- Acetonitrile, ethyl acetate, ethyl acetate / various alcohol mixtures, or acetonitrile / various alcohol mixtures are preferable.
- various alcohols include methanol, ethanol, propanol, butanol, isopropanol and the like.
- the amount of the organic solvent used for obtaining these secondary crystals is usually the weight obtained by subtracting the weight of the primary crystals taken out above from the weight when the target product was obtained in 100% yield from the raw material.
- the amount is preferably 2 to 20 times the amount. Further, when it is desired to improve the yield, it is preferable to reduce the amount of organic solvent used, and when it is desired to obtain a high purity product, it is preferable to increase the amount of organic solvent used. When considering the yield and purity, the amount of 2.5 to 5 times is more preferable.
- a cyclobutanetetracarboxylic dianhydride represented by the following formula [5-b] is used, and the compound is reacted with an alcohol having 1 to 5 carbon atoms (the above R 1 OH). To produce a compound represented by the formula [2-b].
- R 2 represents an alkyl group having 1 to 5 carbon atoms
- the reaction in the presence of a base or acid can improve the selectivity and reaction rate of the formula [2-b], and more preferably a basic compound is present.
- Examples of the base or acid used at this time include those exemplified above, and preferred bases or acids and preferred addition amounts are also as described above.
- R 1 is an alkyl group having 1 to 5 carbon atoms
- R 2 is an alkyl group having 1 to 5 carbon atoms
- n is 2 or 4.
- R 2 Are preferably the same substituent, and when n is 4, all R 2 are preferably the same, but may be different.
- R 1 and R 2 may be the same or different.
- R 1 is an alkyl group having 1 to 5 carbon atoms
- R 2 is an alkyl group having 1 to 5 carbon atoms
- n is 2 or 4, and R 1 and R 2 are the same Or it may be different
- the solvent in the filtrate produced as a by-product in the separation of the target product produced by the esterification reaction is distilled off, and then the carboxylic acid represented by the formula: RCO 2 H;
- An acidic aqueous solution or a basic aqueous solution is added, and the mixture is preferably heated to reflux, and then cooled, and the precipitated crystals are collected by filtration, washed and dried to obtain high-purity crystals of cyclobutanetetracarboxylic acid [7].
- the carboxylic acid (RCO 2 H) is not particularly limited as long as it is in a liquid state at the reaction temperature.
- an aliphatic carboxylic acid having 1 to 5 carbon atoms such as formic acid, acetic acid, propionic acid and the like is preferable. More preferred is formic acid.
- the acid used in the acidic aqueous solution is not particularly limited as long as it is soluble in water at the reaction temperature.
- heteropolyacids such as phosphomolybdic acid and phosphotungstic acid
- inorganic acids such as hydrochloric acid, sulfuric acid and phosphoric acid
- formic acid And hydrocarbon acids such as acetic acid and p-toluenesulfonic acid
- halogenated hydrocarbon acids such as trifluoroacetic acid.
- hydrochloric acid, sulfuric acid, phosphoric acid, p-toluenesulfonic acid, formic acid or acetic acid and more preferred is hydrochloric acid, sulfuric acid or p-toluenesulfonic acid.
- the amount of the acid used in the acidic aqueous solution is usually 0.01 to 100 times mol, preferably 0.01 to 10 times mol for the tetracarboxylic acid diesters [1] and [2].
- the amount of water used in the acidic aqueous solution is not particularly limited as long as the acid used dissolves at the reaction temperature, but it is usually used in an amount of 1 to 100 times, preferably 1 to 10 times, the acid used.
- the base used in the basic aqueous solution is not particularly limited, but alkali metals such as sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, lithium carbonate; magnesium hydroxide, calcium hydroxide, etc. Examples include alkaline earth metals.
- the amount of the base used in the basic aqueous solution is usually 0.01 to 100 times mol, preferably 0.01 to 10 times mol for the tetracarboxylic acid diesters [1] and [2].
- the amount of water used in the basic aqueous solution is not particularly limited as long as the base used is soluble at the reaction temperature, but is usually 1 to 100 times by mass, preferably 1 to 10 times by mass with respect to the acid to be used. It is.
- the carboxylic acid (RCO 2 H), the acidic aqueous solution, or the basic aqueous solution is usually 2 to 100 times by weight, preferably 2 to 40 times by weight, with respect to the tetracarboxylic acid diesters [1] and [2] in the filtrate. More preferably, it is used 2 to 6 times by weight.
- the reaction may be performed in the presence of an acid which is a catalyst.
- the acid should just be an acid which has comparatively strong acidity, and is not specifically limited.
- the acid include heteropolyacids such as phosphomolybdic acid and phosphotungstic acid; organic acids such as trimethylborate and triphenylphosphine; inorganic acids such as hydrochloric acid, sulfuric acid and phosphoric acid; carbonization such as formic acid, acetic acid and p-toluenesulfonic acid.
- Hydrogen acid; Halogen-based hydrocarbon acids such as trifluoroacetic acid are listed.
- the acid is usually used in an amount of 0 to 100-fold mol, preferably 0.01 to 10-fold mol based on tetracarboxylic acid diester [1] and [2].
- the reaction temperature is not particularly limited but is, for example, ⁇ 90 to 200 ° C., preferably 80 to 130 ° C. Also, in the case of the reaction using carboxylic acid (RCO 2 H), particularly when formic acid is used, the reaction is fast when the alkyl formate generated in the reaction system is excluded from the reaction system, and thus it is generated. It is preferable to carry out at a boiling point higher than that of alkyl formate.
- the reaction time is usually 0.05 to 200 hours, preferably 0.5 to 100 hours.
- cyclobutanetetracarboxylic dianhydride [5] can be obtained by dehydrating the cyclobutanetetracarboxylic acid [7] obtained in [Reaction Formula 1] with a dehydrating agent. .
- the dehydrating agent is not particularly limited as long as the dehydrating agent is in contact with cyclobutanetetracarboxylic acid [7].
- tetracarboxylic acid [7] and the dehydrating agent may be mixed in a solvent.
- the dehydrating agent is preferably a carboxylic acid anhydride such as acetic anhydride, propionic anhydride, trifluoroacetic anhydride, preferably a lower carboxylic acid anhydride having 1 to 3 carbon atoms, more preferably 1 to 2 carbon atoms.
- acetic anhydride is particularly preferable because it is easy to remove after anhydrous and is economically advantageous.
- the amount of the dehydrating agent to be used is not particularly limited, but is preferably 2 to 50 equivalents, particularly preferably 4 to 20 equivalents, with respect to cyclobutanetetracarboxylic acid [7]. If the amount is 2 to 50 equivalents, the anhydride is sufficiently formed and the amount of tetracarboxylic dianhydride [5] obtained does not increase excessively, and the tetracarboxylic dianhydride [5] is obtained in a high yield. ] Can be deposited.
- the tetracarboxylic acid [7] need not be completely dissolved and subjected to an anhydrous reaction in a homogeneous system, and the anhydrous reaction may be performed in a heterogeneous system.
- the heating temperature in the reaction is preferably 30 to 200 ° C., more preferably 40 to 180 ° C., and the higher the reaction temperature, the higher the reaction rate. For this reason, it is preferable to carry out at the reflux temperature of the solvent used.
- the reaction time may be appropriately set according to conditions such as the type of dehydrating agent to be used, temperature, etc., but is preferably 0.5 to 20 hours.
- the anhydride can be sufficiently reacted in this time.
- the cyclobutanetetracarboxylic acid represented by the above formula [7] obtained in the present invention is a novel compound that has not been described in the literature.
- the cyclobutanetetracarboxylic dianhydride [5] can be easily obtained from the above. It can be used for various purposes such as manufacturing.
- the production method of the present invention is used in a wide range of fields because cyclobutanetetracarboxylic acid and / or its dianhydride can be obtained from the residue after separation of the target product produced in the production process such as cyclobutanetetracarboxylic acid derivative. it can.
- the entire contents of the specification, claims, drawings, and abstract of Japanese Patent Application No. 2014-007188 filed on January 17, 2014 are cited herein as disclosure of the specification of the present invention. Incorporated.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description
例えば、主鎖にシクロブタン骨格を有するポリイミドの合成例としては、ビス(クロロカルボニル)シクロブタンジカルボン酸ジメチルエステルとジアミンとを反応させてポリアミド酸メチルエステルを得た後、これを加熱してポリイミドとした例が報告されている(非特許文献1参照)。
特許文献1では、目的のシクロブタンテトラカルボン酸ジアルキルエステル誘導体が高い収率で得られているが、目的物である、シクロブタンテトラカルボン酸ジアルキルエステルを取得する過程で生じる残渣を使用し、かかる残渣から出発物質であるシクロブタンテトラカルボン酸二無水物などの有用物を回収することについては記載されていない。
1.式[1]で表されるテトラカルボン酸ジアルキルエステルと式[2]で表されるテトラカルボン酸ジアルキルエステルとの混合物

を溶媒中で加水分解することによる、式[7]

2.式[7]

3.式[1]及び式[2]がそれぞれ式[1-a]及び式[2-a]

であり、式[7]が下記式[7-a]

4.式[7]が下記式[7-a]


である2記載の製造方法。
5.下記式[7-a]

本発明の製造方法における原料は、シクロブタンテトラカルボン酸ジアルキルエステルなどの製造過程で生じる残渣に含有されている、下記式[1]又は式[2]で表される化合物である。

R1は、炭素数が1~5、好ましくは1~3のアルキル基であり、アルキル基の具体例としては、メチル基、エチル基、ノルマルプロピル基、イソプロピル基、ノルマルブチル基、セカンダリーブチル基、イソブチル基、ターシャリーブチル基、ノルマルペンチル基などが挙げられる。なお、ポリアミド酸エステルを合成した後、イミド化することでポリイミドとして使用する場合は、R1は炭素数が少なく脱離しやすいものが好ましく、より好ましくはメチル基である。
R2は、炭素数1~5、好ましくは1~3のアルキル基であり、アルキル基の具体例としては、メチル基、エチル基、ノルマルプロピル基、イソプロピル基、ノルマルブチル基、セカンダリーブチル基、イソブチル基、ターシャリーブチル基、ノルマルペンチル基などが挙げられる。
nは1~4の整数を表し、好ましくは2である。
なお、以下の表1及び表2において、a1~a4及びb1~b4は、下記式[6]に示したそれぞれの位置を表し、表1及び表2中の記号はそれぞれ以下の意味を示す。
Me:メチル基、Et:エチル基、Pr-n:ノルマルプロピル基、Pr-iso:イソプロピル基、Bu-n:ノルマルブチル基、Bu-sec:セカンダリーブチル基、Bu-iso:イソブチル基、Bu-t:ターシャリーブチル基、Pen-n:ノルマルペンチル基、OMe:メトキシ基、OEt:エトキシ基、OPr-n:ノルマルプロピルエーテル基、OPr-iso:イソプロピルエーテル基、OBu-n:ノルマルブトキシ基、OBu-sec:セカンダリーブトキシ基、OBu-iso:イソブトキシ基、OBu-t:ターシャリーブトキシ基、OPen-n:ノルマルペンチルエーテル基




塩基としては、水酸化ナトリウム、水酸化カリウム、炭酸カリウム又は炭酸水素ナトリウムなどの無機塩基;トリエチルアミン、ピリジン、キノリン、8-キノリノール、1,10-フェナンスロリン、バソフェナンスロリン、バソクプロイン、2,2’-ビピリジル、2-フェニルピリジン、2,6-ジフェニルアミノピリジン、2-ジメチルアミノピリジン、4-ジメチルアミノピリジン、2-(2-ヒドロキシルエチル)ピリジン、N、N-ジメチルアニリン、1,8-ジアザビシクロ[5、4、0]-7-ウンデン(DBU)などの有機塩基;ナトリウムメトキシド、カリウムメトキシド又はカリウムt-ブトキシドなどの金属アルコキシドが挙げられる。好ましくは、ナトリウムメトキシド、カリウムメトキシド、又はピリジンであり、より好ましくは、ピリジンである。
式[1-a]又は式[2-a]で表される化合物は、前記反応式のテトラカルボン酸二無水物[5]として、下記式[5-a]で表されるテトラカルボン酸二無水物を使用することにより製造できる。

このとき、反応温度が低い程、式[1-a]の選択率が向上する。このため、式[1-a]の反応収率を向上させたい場合、より好ましい反応温度は10~30℃である。一方、式[2-a]の反応収率を向上させたい場合、より好ましい反応温度は50~100℃である。

有機溶媒としては、例えば、トルエン、アセトニトリル、酢酸エチル、酢酸エチル・n-ヘプタン混合液、酢酸エチル・各種アルコール混合液、アセトニトリル・各種アルコール混合液等が使用できる。好ましくはアセトニトリル、酢酸エチル、酢酸エチル・各種アルコール混合液、又はアセトニトリル・各種アルコール混合液である。各種アルコールとしてはメタノール、エタノール、プロパノール、ブタノール、イソプロパノール等が挙げられる。

このとき、塩基又は酸の存在下に反応させることで、式[2-b]の選択率及び反応速度を向上させることができ、より好ましくは塩基性化合物を存在させることである。このとき使用する塩基又は酸は前記で例示したものが挙げられ、好ましい塩基又は酸、及び好ましい添加量も前記したとおりである。

本発明では下記の[反応式1]及び[反応式2]に示すように、シクロブタンテトラカルボン酸ジアルキルエステルなどの製造過程で生じる残渣に含有されている、上記[式[1]及び式[2]の化合物を加水分解することでシクロブタンテトラカルボン酸[7]を製造し、さらにこのシクロブタンテトラカルボン酸[7]を無水物化することによりシクロブタンテトラカルボン酸及び/又はその二無水物の製造することができる。

上記[反応式1]の反応は、例えばエステル化反応で生成する目的物の分離の際に副生するろ液中の溶媒を留去した後、式:RCO2Hで表されるカルボン酸、酸性水溶液、又は塩基性水溶液を添加し、好ましくは加熱還流した後、冷却することで析出した結晶を濾取・洗浄し乾燥するとシクロブタンテトラカルボン酸[7]の高純度品の結晶が得られる。
酸性水溶液に用いる酸の使用量はテトラカルボン酸ジエステル[1]及び[2]に対して通常0.01~100倍モル、好ましくは0.01~10倍モル使用される。
酸性水溶液に用いる水の使用量は使用する酸が反応温度で溶解する量であれば特に限定はないが、使用する酸に対して通常1~100質量倍、好ましくは1~10質量倍使用される。
塩基性水溶液に用いる塩基は特に限定されるものではないが、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、炭酸ナトリウム、炭酸カリウム、炭酸リチウムなどのアルカリ金属;水酸化マグネシウム、水酸化カルシウム等のアルカリ土類金属が挙げられる。なかでも好ましくは、水酸化ナトリウム、水酸化カリウム又は水酸化リチウムが挙げられる。
塩基性水溶液に用いる塩基の使用量はテトラカルボン酸ジエステル[1]及び[2]に対して通常0.01~100倍モル、好ましくは0.01~10倍モル使用される。
塩基性水溶液に用いる水の使用量は、使用する塩基が反応温度で溶解する量であれば特に限定されないが、使用する酸に対して通常1~100質量倍、好ましくは1~10倍質量倍である。
カルボン酸(RCO2H)、酸性水溶液、又は塩基性水溶液は、ろ液中のテトラカルボン酸ジエステル[1]及び[2]に対して、通常2~100重量倍、好ましくは2~40重量倍、より好ましくは2~6重量倍使用される。
酸はテトラカルボン酸ジエステル[1]及び[2]に対して通常0~100倍モル、好ましくは0.01~10倍モル使用される。
反応時間は、通常、0.05~200時間、好ましくは0.5~100時間である。
反応における加熱の温度は、好ましくは30~200℃、より好ましくは40~180℃の範囲で行うとよく、反応温度が高いほど反応速度が向上する。このため使用溶媒の還流温度で実施するのが好ましい。
<1H NMR分析条件>
装置 :フーリエ変感型超伝導核磁気共鳴装置(FT-NMR)INOVA-400(Varian社) 400 MHz
溶媒 :DMSO-d6
内標準物質 :テトラメチルシラン(TMS)


1H NMR ( DMSO-d6, δppm ) : 12.49 ( s, 2H ), 3.29( s, 2H ), 1.42 ( s, 6H ).
本測定に用いたHPLC(高速液体クロマトグラフィー)分析は、下記条件にて実施した。
HPLC分析条件;
カラム:Atlantis cd18 (Waters), 5um, 4.6×250mm、オーブン:40℃
溶離液:アセトニトリル/0.5%リン酸水溶液=16/84 検出波長:211nm
流速: 1.0mL/分 サンプル注入量: 10μL サンプル濃度: 1wt%

この結晶は、1H NMR分析結果により、(5-1)であることを確認した。
1H NMR ( DMSO-d6, δppm ) : 3.89 ( s, 2H ), 1.38 ( s, 6H ).
なお、2014年1月17日に出願された日本特許出願2014-007188号の明細書、特許請求の範囲、図面及び要約書の全内容をここに引用し、本発明の明細書の開示として、取り入れるものである。
Claims (10)
- 式[1]で表されるテトラカルボン酸ジアルキルエステルと式[2]で表されるテトラカルボン酸ジアルキルエステルとの混合物を溶媒中で加水分解することによる、式[7]で表されるテトラカルボン酸の製造方法。


- 式:RCOOH(Rは、水素又は、炭素数1~10のアルキルである)を有する有機カルボン酸と反応させて加水分解する、請求項1に記載の製造方法。
- 酸触媒の存在下で加水分解する、請求項1又は2に記載の製造方法。
- 前記溶媒が、炭化水素、ハロゲン化炭化水素、エステル、ケトン、及びニトリルからなる群から選ばれる有機溶媒である、請求項1~3のいずれかに記載の製造方法。
- 式[7]


- 脱水剤の存在下に、40~160℃に加熱して脱水を行う、請求項5に記載の製造方法。
- 脱水剤の炭素数が1~3の低級カルボン無水物である請求項5に記載の製造方法。
- 式[1]及び式[2]が、それぞれ、式[1-a]及び式[2-a]であり、式[7]が下記式[7-a]である請求項1に記載の製造方法。


- 式[7]が下記式[7-a]であり、式[5]が式[5-a]である請求項5に記載の製造方法。


- 下記式[7-a]で表されるテトラカルボン酸化合物。

Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020217002662A KR102272227B1 (ko) | 2014-01-17 | 2015-01-16 | 시클로부탄테트라카르복실산 및 그 무수물의 제조 방법 |
| KR1020167018418A KR102247402B1 (ko) | 2014-01-17 | 2015-01-16 | 시클로부탄테트라카르복실산 및 그 무수물의 제조 방법 |
| JP2015557904A JP6697178B2 (ja) | 2014-01-17 | 2015-01-16 | シクロブタンテトラカルボン酸及びその無水物の製造方法 |
| CN201580004814.5A CN105916833A (zh) | 2014-01-17 | 2015-01-16 | 环丁烷四羧酸及其酸酐的制造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014007188 | 2014-01-17 | ||
| JP2014-007188 | 2014-01-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015108170A1 true WO2015108170A1 (ja) | 2015-07-23 |
Family
ID=53543053
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/051149 WO2015108170A1 (ja) | 2014-01-17 | 2015-01-16 | シクロブタンテトラカルボン酸及びその無水物の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| JP (2) | JP6697178B2 (ja) |
| KR (2) | KR102272227B1 (ja) |
| CN (1) | CN105916833A (ja) |
| TW (1) | TWI653225B (ja) |
| WO (1) | WO2015108170A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6697178B2 (ja) * | 2014-01-17 | 2020-05-20 | 日産化学株式会社 | シクロブタンテトラカルボン酸及びその無水物の製造方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006043519A1 (ja) * | 2004-10-20 | 2006-04-27 | Nissan Chemical Industries, Ltd. | ケージ状シクロブタン酸二無水物及びその製造法 |
| JP2006117673A (ja) * | 2004-10-20 | 2006-05-11 | Eternal Chemical Co Ltd | シクロブタンテトラカルボキシレート化合物及びその製造方法 |
| WO2010092989A1 (ja) * | 2009-02-12 | 2010-08-19 | 日産化学工業株式会社 | テトラカルボン酸誘導体、その製造方法、及び液晶配向剤 |
| JP2013163710A (ja) * | 2012-02-09 | 2013-08-22 | Nissan Chem Ind Ltd | ポリイミド前駆体、ポリイミド、電荷輸送性組成物、及びポリイミド前駆体の製造方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60112736A (ja) * | 1983-11-22 | 1985-06-19 | Mitsubishi Chem Ind Ltd | 桂皮酸類の製造方法 |
| JP6697178B2 (ja) * | 2014-01-17 | 2020-05-20 | 日産化学株式会社 | シクロブタンテトラカルボン酸及びその無水物の製造方法 |
| CN106458825B (zh) * | 2014-05-09 | 2019-07-12 | 日产化学工业株式会社 | 1,3-二取代环丁烷-1,2,3,4-四羧酸及其酸二酐的新型制造方法 |
-
2015
- 2015-01-16 JP JP2015557904A patent/JP6697178B2/ja active Active
- 2015-01-16 CN CN201580004814.5A patent/CN105916833A/zh active Pending
- 2015-01-16 KR KR1020217002662A patent/KR102272227B1/ko active IP Right Grant
- 2015-01-16 KR KR1020167018418A patent/KR102247402B1/ko active IP Right Grant
- 2015-01-16 WO PCT/JP2015/051149 patent/WO2015108170A1/ja active Application Filing
- 2015-01-19 TW TW104101673A patent/TWI653225B/zh active
-
2020
- 2020-01-23 JP JP2020009324A patent/JP2020079247A/ja active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006043519A1 (ja) * | 2004-10-20 | 2006-04-27 | Nissan Chemical Industries, Ltd. | ケージ状シクロブタン酸二無水物及びその製造法 |
| JP2006117673A (ja) * | 2004-10-20 | 2006-05-11 | Eternal Chemical Co Ltd | シクロブタンテトラカルボキシレート化合物及びその製造方法 |
| WO2010092989A1 (ja) * | 2009-02-12 | 2010-08-19 | 日産化学工業株式会社 | テトラカルボン酸誘導体、その製造方法、及び液晶配向剤 |
| JP2013163710A (ja) * | 2012-02-09 | 2013-08-22 | Nissan Chem Ind Ltd | ポリイミド前駆体、ポリイミド、電荷輸送性組成物、及びポリイミド前駆体の製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| G.W.GRIFFIN ET AL.: "The Chemistry of Photodimers of Maleic and Fumaric Acid derivatives. I. Dimethyl Fumarate Dimer", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 83, no. 12, 1961, pages 2725 - 2728, XP002245338 * |
Also Published As
| Publication number | Publication date |
|---|---|
| KR102247402B1 (ko) | 2021-04-30 |
| TWI653225B (zh) | 2019-03-11 |
| JPWO2015108170A1 (ja) | 2017-03-23 |
| CN105916833A (zh) | 2016-08-31 |
| TW201542514A (zh) | 2015-11-16 |
| JP6697178B2 (ja) | 2020-05-20 |
| KR20160108334A (ko) | 2016-09-19 |
| KR102272227B1 (ko) | 2021-07-01 |
| KR20210013332A (ko) | 2021-02-03 |
| JP2020079247A (ja) | 2020-05-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2014201545A (ja) | 2−ヒドロキシメチル−2,3−ジヒドロ−チエノ[3,4−b][1,4]ジオキシン−5,7−ジカルボン酸ジアルキルエステルの製造方法 | |
| WO2006043519A1 (ja) | ケージ状シクロブタン酸二無水物及びその製造法 | |
| CA2889650C (en) | Process and intermediates for preparing lacosamide | |
| US11891351B2 (en) | Synthesis of capsaicin derivatives | |
| JP2020079247A (ja) | シクロブタンテトラカルボン酸及びその無水物の製造方法 | |
| WO2018205299A1 (zh) | 4,5-二取代-1-氢-吡咯(2,3-f)喹啉-2,7,9-三羧酸酯化合物及应用 | |
| JP6260385B2 (ja) | 2−ヒドロキシメチル−2,3−ジヒドロ−チエノ[3,4−b][1,4]ジオキシン−5,7−ジカルボン酸ジアルキルエステルの製造方法 | |
| WO2021242807A1 (en) | Methods for preparing methyl (s)-2-amino-3-(4-(2,3-dimethylpyridin-4-yl)phenyl)propionate and hydrochloric acid salts thereof | |
| JP2005504019A (ja) | イソクマリンを調製するための方法 | |
| EP2880008B1 (en) | Process for preparing spiro[2.5]octane-5,7-dione | |
| WO2018116836A1 (ja) | 重合性化合物の製造方法 | |
| RU2459803C2 (ru) | СПОСОБ ПОЛУЧЕНИЯ 4,4'-(м-ФЕНИЛЕНДИОКСИ)ДИФТАЛОНИТРИЛА | |
| Хузам | SYNTHESIS AND CHARACTERIZATION OF NICOTINE ESTER FROM DINICOTINIC ACID AND 3-AMINO-1-PROPANOL | |
| JP6267221B2 (ja) | 2−トリフルオロメチルイソニコチン酸及びエステルの調製方法 | |
| JP2000327629A (ja) | フェニル酢酸誘導体、ベンゾニトリル誘導体、およびその製造方法 | |
| EP4121416A1 (en) | Improved synthesis of 6-aryl-4-aminopicolinates | |
| JP2011219416A (ja) | 芳香族多価カルボン酸無水物化合物の製造方法 | |
| Pan et al. | A novel method for the synthesis of 2-(3-hydroxy-1-adamantyl)-2-oxoacetic acid | |
| PL232738B1 (pl) | N-[(1R,6S)-4,7,7-trimetylobicyklo[4.1.0]hept-4-en-3-ylo]acetamid i sposób jego otrzymywania | |
| JP2008513371A (ja) | 8−ハロ−1,7−ナフタピリジン誘導体と有機ボロン酸誘導体の反応による6,8−置換−1,7−ナフタピリジン誘導体の製造方法および該方法の中間体 | |
| JP2008528545A (ja) | イソクロマンおよびその誘導体の製造方法 | |
| JP2000327622A (ja) | フェニル酢酸誘導体の製造方法 | |
| US20170369442A1 (en) | Method for preparing 4-cyanopiperidine hydrochloride | |
| JPH115756A (ja) | 2−アルキルアズレン類の製造方法 | |
| PL220142B1 (pl) | (-)-O-n-heksyloksyiminofenchan i sposób jego wytwarzania |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15737423 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015557904 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20167018418 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15737423 Country of ref document: EP Kind code of ref document: A1 |