WO2014115801A1 - α,α-ジフルオロアセトアルデヒドの製造方法 - Google Patents
α,α-ジフルオロアセトアルデヒドの製造方法 Download PDFInfo
- Publication number
- WO2014115801A1 WO2014115801A1 PCT/JP2014/051365 JP2014051365W WO2014115801A1 WO 2014115801 A1 WO2014115801 A1 WO 2014115801A1 JP 2014051365 W JP2014051365 W JP 2014051365W WO 2014115801 A1 WO2014115801 A1 WO 2014115801A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- general formula
- represented
- reaction
- group
- difluoroacetaldehyde
- Prior art date
Links
- 0 *OC(C(F)F)O Chemical compound *OC(C(F)F)O 0.000 description 2
- QUMXAROOVMMPHD-UHFFFAOYSA-N CCOC(C(F)F)OC(C(F)F)O Chemical compound CCOC(C(F)F)OC(C(F)F)O QUMXAROOVMMPHD-UHFFFAOYSA-N 0.000 description 2
- DKNMRIXYSHIIGC-UHFFFAOYSA-N O=CC(F)F Chemical compound O=CC(F)F DKNMRIXYSHIIGC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/41—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by hydrogenolysis or reduction of carboxylic groups or functional derivatives thereof
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Definitions
- the present invention relates to an industrial production method of ⁇ , ⁇ -difluoroacetaldehyde.
- Non-Patent Document 1 Non-Patent Document 1
- Patent Documents 1 and 2 the present applicant has filed patent applications having technical contents similar to those of the present invention.
- the method of using a hydride reducing agent stoichiometrically described in the background art is that the reducing agent is expensive and requires careful handling, and further, the post-treatment is complicated and waste is large. It was unsuitable for production. In addition, extremely low temperature conditions ( ⁇ 78 ° C.) are required to suppress by-production of ⁇ , ⁇ -difluoroethanol due to excessive reduction, which is an industrially difficult method.
- the problem to be solved by the present invention is to provide an industrial production method of ⁇ , ⁇ -difluoroacetaldehyde.
- an object is to find a highly practical production method that replaces hydride reduction.
- hydrogen gas H 2
- ruthenium catalyst particularly the homogeneous catalyst. It has not been.
- the term “homogeneous catalyst” in the present specification is as described in the chemistry dictionary (edited by Michinori Ohki, Toshiaki Osawa, Motoharu Tanaka, Hideaki Chihara, Tokyo Chemical Dojin) and the like.
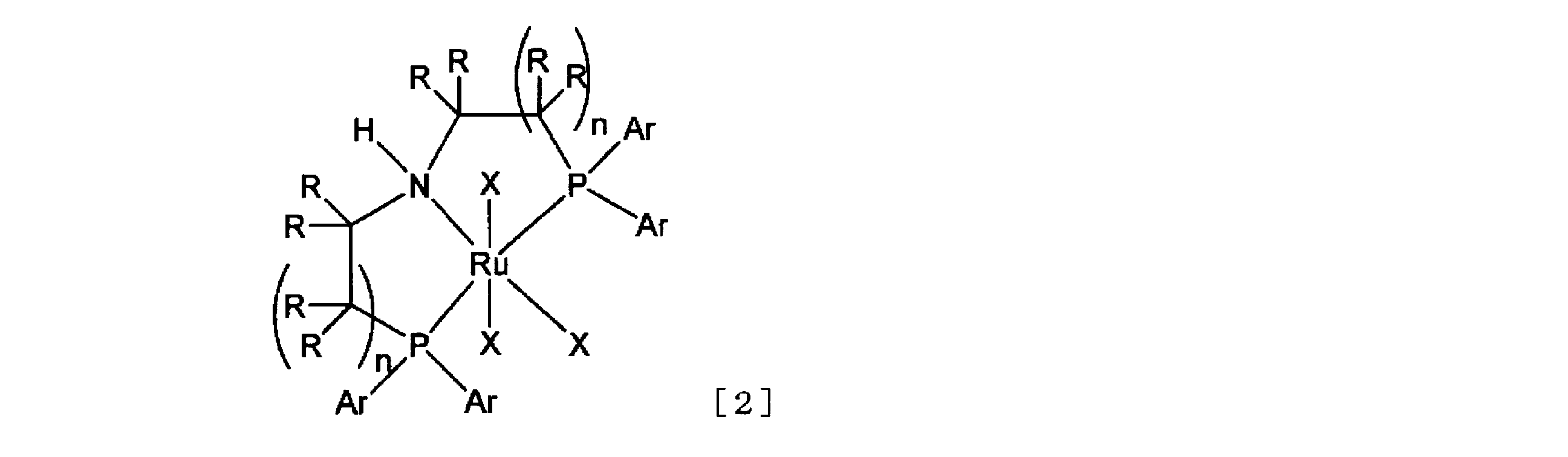
- a ruthenium catalyst preferably a homogeneous ruthenium catalyst, particularly preferably a ruthenium complex represented by the following general formula [2], very preferably the following general formula [ 4] has been found to be an effective hydrogenation catalyst or precursor for partial reduction of ⁇ , ⁇ -difluoroacetic acid esters.
- each R independently represents a hydrogen atom, an alkyl group, a substituted alkyl group, an aromatic ring group or a substituted aromatic ring group
- Ar represents each independently an aromatic ring group or a substituted aromatic ring group
- each X represents an independent group.
- Ph represents a phenyl group.
- the ruthenium complexes of general formula [2] and general formula [4] act as homogeneous ruthenium catalysts.
- Patent Document 1 Prior to this application, the applicant has filed two patent applications with similar technical contents (Patent Document 1 and Patent Document 2).
- the clear difference between the present application and Patent Document 1 is the target product obtained.
- the raw material substrate of the present application is included in ⁇ -fluoroesters which are raw material substrates of Patent Document 1, but the target product of the present application is ⁇ , ⁇ -difluoroacetaldehyde, and the target product of Patent Document 1 is ⁇ -fluoro Alcohols.
- ⁇ , ⁇ -difluoroacetaldehyde which is a partially reduced form of hydrogenation, can be selectively obtained even when the same ruthenium complex is used. I found out.
- ⁇ , ⁇ -difluoroethanol ( ⁇ -fluoroalcohols) is also obtained as a by-product, and the present application does not limit Patent Document 1.
- ⁇ , ⁇ -difluoroethanol which is a by-product of the present application, is significantly different from the target product, ⁇ , ⁇ -difluoroacetaldehyde (including equivalents described later), and can be easily purified and removed. it can. Therefore, the present application is not limited by Patent Document 1 as a method for producing ⁇ , ⁇ -difluoroacetaldehyde.
- R 1 of ⁇ -fluoroesters which are raw material substrates of Patent Document 2 is a hydrogen atom (vs. halogen atom or haloalkyl group).
- the present invention provides the method for producing ⁇ , ⁇ -difluoroacetaldehyde described in [Invention 1] to [Invention 12].
- invention 2 The method according to invention 1, further comprising carrying out the reaction in the presence of a base.
- invention 4 Any of the inventions 1 to 3, wherein the ⁇ , ⁇ -difluoroacetic acid ester represented by the general formula [1] is an ⁇ , ⁇ -difluoroacetic acid ester represented by the general formula [5].
- the method of crab. [Wherein R 2 represents an alkyl group. ]
- invention 7 The invention according to any one of inventions 1 to 4, wherein the reaction is carried out at a reaction temperature of 50 ° C. or lower using an aliphatic hydrocarbon-based, aromatic hydrocarbon-based, halogen-based or ether-based reaction solvent. the method of.
- invention 8 The invention according to any one of inventions 1 to 4, wherein the reaction is carried out at a reaction temperature of 40 ° C. or lower using an aliphatic hydrocarbon-based, aromatic hydrocarbon-based, halogen-based or ether-based reaction solvent. the method of.
- invention 10 Process according to invention 9, characterized in that the ruthenium catalyst is a homogeneous catalyst.
- the production method of the present invention can be an alternative to hydride reduction that is difficult to implement industrially.
- the hydrogenation of the present invention does not require high-pressure equipment, has a high substrate / catalyst ratio, can easily obtain the desired product only by distillation operation after the post-treatment, and has a highly practical production method. It is. Therefore, it is possible to provide an industrial production method of ⁇ , ⁇ -difluoroacetaldehyde that solves the problems of the background art at once.
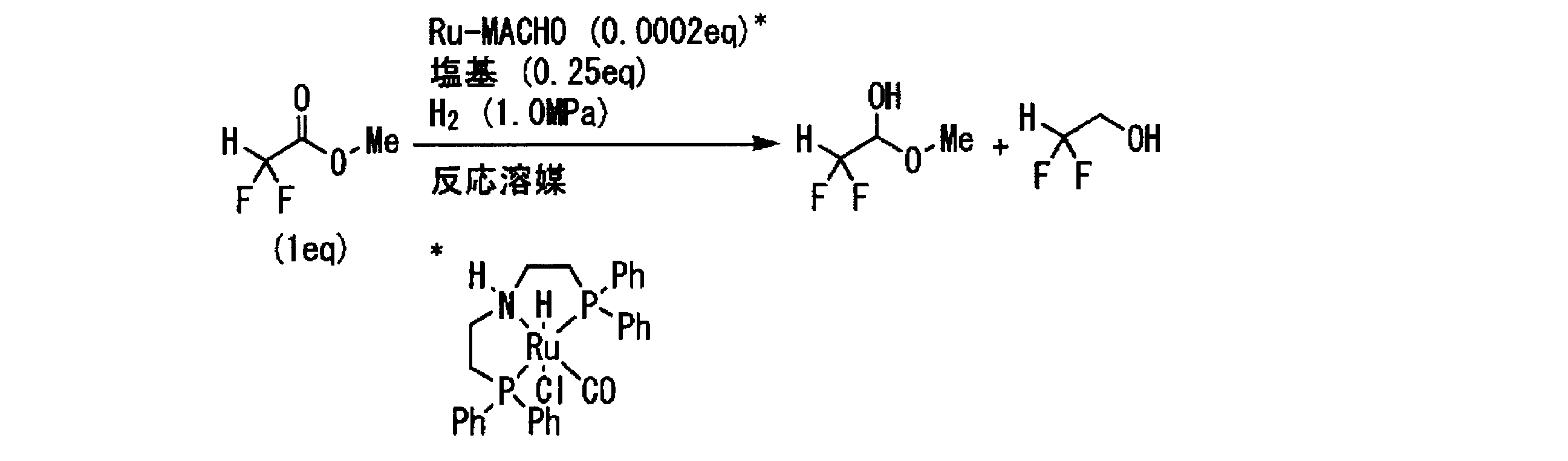
- the ⁇ , ⁇ -difluoroacetic acid ester represented by the general formula [1] is reacted with hydrogen gas in the presence of the ruthenium complex represented by the general formula [2], whereby the general formula [3]
- the indicated ⁇ , ⁇ -difluoroacetaldehyde can be prepared.
- R 1 of the ⁇ , ⁇ -difluoroacetic acid ester represented by the general formula [1] represents an alkyl group or a substituted alkyl group.
- the alkyl group is a linear or branched chain or cyclic group (having 3 or more carbon atoms) having 1 to 18 carbon atoms.
- the substituted alkyl group has a substituent in any number and in any combination on any carbon atom of the alkyl group.
- substituents include halogen atoms such as fluorine, chlorine and bromine, lower alkyl groups such as methyl, ethyl and propyl, lower haloalkyl groups such as fluoromethyl, chloromethyl and bromomethyl, methoxy and ethoxy Lower alkoxy groups such as propoxy group, lower haloalkoxy groups such as fluoromethoxy group, chloromethoxy group and bromomethoxy group, lower alkoxycarbonyl groups such as cyano group, methoxycarbonyl group, ethoxycarbonyl group and propoxycarbonyl group, phenyl group , Aromatic groups such as naphthyl, anthryl, pyrrolyl (including nitrogen protected), pyridyl, furyl, thienyl, indolyl (including nitrogen protected), quinolyl, benzofuryl and benzothienyl , Carboxyl group, carbox Protection of group, amino group, protected amino group, a protected form,
- any carbon-carbon single bond of the above-described alkyl group can be replaced with a carbon-carbon double bond or a carbon-carbon triple bond in any number and in any combination.
- an alkyl group substituted with these unsaturated bonds may have the above-mentioned substituents in the same manner).
- the substituent itself may be involved in the side reaction, but it can be minimized by employing suitable reaction conditions.
- lower means a linear or branched chain or cyclic group (in the case of 3 or more carbon atoms) having 1 to 6 carbon atoms.
- the “aromatic ring group” in the “substituent” is a halogen atom, lower alkyl group, lower haloalkyl group, lower alkoxy group, lower haloalkoxy group, cyano group, lower alkoxycarbonyl group, carboxyl group, A protected group of a carboxyl group, an amino group, a protected group of an amino group, a hydroxyl group, a protected group of a hydroxyl group, and the like can be substituted.
- pyrrolyl, indolyl, carboxyl, amino, and hydroxyl protecting groups are those described in Protective Group in Organic Synthesis, Third Edition, 1999, John Wiley & Sons, Inc. and the like.
- the compound at the stage where it is actually subjected to the reaction falls within the range of ⁇ , ⁇ -difluoroacetic acid ester represented by the general formula [1], it is included in the claims of the present application. It is defined as For example, even when ⁇ , ⁇ -difluoroacetic acid phenyl ester is used as a raw material substrate, it can be considered that the methyl ester is hydrogenated after transesterification with methanol as a reaction solvent. Treated as included in the claims of this application.
- the ⁇ , ⁇ -difluoroacetates represented by the general formula [5] are preferable and are easily available on a large scale.
- the alkyl group represented by R 2 of the ⁇ , ⁇ -difluoroacetic acid ester represented by the general formula [5] is the alkyl group described in R 1 of the ⁇ , ⁇ -difluoroacetic acid ester represented by the general formula [1]. Is the same.
- R in the ruthenium complex represented by the general formula [2] independently represents a hydrogen atom, an alkyl group, a substituted alkyl group, an aromatic ring group or a substituted aromatic ring group.
- the alkyl group and substituted alkyl group are the same as the alkyl group and substituted alkyl group described in R 1 of the ⁇ , ⁇ -difluoroacetic acid ester represented by the general formula [1].
- the aromatic ring group is an aromatic hydrocarbon group having 1 to 18 carbon atoms such as a phenyl group, a naphthyl group and an anthryl group, or a pyrrolyl group (including a nitrogen protector), a pyridyl group, a furyl group, a thienyl group, an indolyl group.
- An aromatic heterocyclic group containing a hetero atom such as a nitrogen atom, oxygen atom or sulfur atom such as a group (including a nitrogen-protected form), a quinolyl group, a benzofuryl group and a benzothienyl group.
- the substituted aromatic ring group has a substituent in any number and in any combination on any carbon atom or nitrogen atom of the aromatic ring group.
- a substituent is the same as the above-mentioned “related substituent”.
- Two Rs at the vicinal position (excluding hydrogen atoms) can adopt a cyclic structure by a covalent bond between carbon atoms. Such covalent bonds include those via a nitrogen atom, oxygen atom or sulfur atom. Among them, all eight hydrogen atoms are preferable (when two n are both 1).
- Ar in the ruthenium complex represented by the general formula [2] independently represents an aromatic ring group or a substituted aromatic ring group.
- the aromatic ring group and the substituted aromatic ring group are the same as the aromatic ring group and the substituted aromatic ring group described in R of the ruthenium complex represented by the general formula [2]. Of these, all four are preferred.
- X of the ruthenium complex represented by the general formula [2] is independently a ligand having a formal charge of ⁇ 1 or 0 [however, the sum of the formal charges of three X is ⁇ 2 (the formal charge of Ru is +2) ].
- the “ligand having a formal charge of ⁇ 1 or 0” is an organic synthesis by Hegedas transition metal (LS Hegedus, original 2nd edition, translated by Shinji Murai, p.4-9, Tokyo Kagaku Dojin, 2001) and postgraduate lectures on organic chemistry.
- BH 4 - and R 5 CO 2 - R 5 is hydrogen Represents an atom, an alkyl group, or a substituted alkyl group, and the alkyl group and the substituted alkyl group are the same as the alkyl group and the substituted alkyl group described in R 1 of the ⁇ , ⁇ -difluoroacetic acid ester represented by the general formula [1]. And so on. Among these, one among three, hydrogen atom, chlorine atom and carbon monoxide are preferable.
- the reaction can be performed in the absence of a base (naturally in the presence of a base). Reaction can also be carried out). Among them, it is preferable that the Cl ligand of the ruthenium complex represented by the general formula [4] is replaced with BH 4 (H—BH 3 ) (see International Publication No. 2011/048727).
- N in the ruthenium complex represented by the general formula [2] independently represents an integer of 1 or 2.
- n 1, it means that the nitrogen atom and the phosphorus atom are bonded through two carbon atoms.
- n is 2, the nitrogen atom and the phosphorus atom are bonded through three carbon atoms.
- two n's are preferably 1.
- Ph of the ruthenium complex represented by the general formula [4] represents a phenyl group.
- the ruthenium complex represented by the general formula [4] is preferable.
- the ruthenium complex represented by the general formula [4] commercially available Ru-MACHO TM (manufactured by Takasago International Corporation) can be used.
- the ruthenium complex represented by the general formula [2] can be produced in the same manner with reference to the production method of Ru-MACHO TM described above.
- what contains organic solvents, such as water and toluene can be used equally, and purity should just be 70% or more, 80% or more is preferable and 90% or more is especially preferable.
- the amount of the ruthenium complex represented by the general formula [2] may be 0.000001 mol or more per 1 mol of the ⁇ , ⁇ -difluoroacetic acid ester represented by the general formula [1]. 005 mol is preferable, and 0.00002 to 0.002 mol is particularly preferable.
- Bases are alkali metal hydrogen carbonates such as lithium hydrogen carbonate, sodium hydrogen carbonate and potassium hydrogen carbonate, alkali metal carbonates such as lithium carbonate, sodium carbonate and potassium carbonate, lithium hydroxide, sodium hydroxide and potassium hydroxide.
- Alkali metal hydroxides such as tetramethylammonium hydroxide, tetraethylammonium hydroxide, tetra-n-propylammonium hydroxide and tetra-n-butylammonium hydroxide, tetraalkylammonium hydroxide, lithium methoxide, sodium methoxide , Potassium methoxide, lithium ethoxide, sodium ethoxide, potassium ethoxide, lithium isopropoxide, sodium isopropoxide, potassium isopropoxide, lithium tert-butoxide, nato Alkali metal alkoxides such as um tert-butoxide and potassium tert-butoxide, organic
- the amount used may be 0.001 mol or more, preferably 0.005 to 5 mol, based on 1 mol of ⁇ , ⁇ -difluoroacetic acid ester represented by the general formula [1], 0.01 ⁇ 3 mol is particularly preferred.
- catalytically active species is derived from the ruthenium complex represented by the general formula [2] in the presence of a base as necessary. Therefore, even when a catalytically active species is prepared in advance (including an isolated species) and subjected to hydrogenation, it is treated as being included in the scope of claims.
- the amount of hydrogen gas used may be 1 mol or more with respect to 1 mol of ⁇ , ⁇ -difluoroacetic acid ester represented by the general formula [1], preferably a large excess, and particularly preferably a large excess under the following pressure. .
- the hydrogen pressure is not particularly limited, but is preferably 2 to 0.001 MPa, particularly preferably 1 to 0.01 MPa.
- Reaction solvents include aliphatic hydrocarbons such as n-hexane, cyclohexane and n-heptane, aromatic hydrocarbons such as toluene, xylene and mesitylene, methylene chloride, 1,2-dichloroethane and ⁇ , ⁇ , ⁇ -tri Halogens such as fluorotoluene, diethyl ether, 1,2-dimethoxyethane, 1,4-dioxane, tetrahydrofuran, 2-methyltetrahydrofuran, tert-butyl methyl ether, diisopropyl ether, diethylene glycol dimethyl ether, anisole and other ethers, methanol, Alcohols such as ethanol, n-propanol, isopropanol, n-butanol, tert-butanol, n-pentanol, n-hexanol and cyclohexanol,
- reaction solvents can be used alone or in combination.
- reaction solvents (alcohol-based, aliphatic hydrocarbon-based, aromatic hydrocarbon-based, halogen-based and ether-based) specified in [Claim 5] to [Claim 8]
- two or more kinds of reaction solvents are combined. Even if it is used, it is defined that it is sufficient if the volume ratio of the specified reaction solvent is the largest.
- Example 11 a mixed solvent of methanol and tert-butyl methyl ether is used.
- Example 11 since the volume ratio of tert-butyl methyl ether is large, an ether-based solvent is used even if the mixed solvent is used. Since it is specified as a reaction solvent and Example 12 has a large volume ratio of methanol, even a mixed solvent is specified as an alcohol-based reaction solvent.
- the alcohol-based reaction solvent (hereinafter referred to as A) has an effect of accelerating the reaction rate, and the aliphatic hydrocarbon-based, aromatic hydrocarbon-based, halogen-based and ether-based reaction solvents (hereinafter referred to as B) are ⁇ , Has the effect of suppressing excessive reduction to ⁇ -difluoroethanol.
- A aliphatic hydrocarbon-based, aromatic hydrocarbon-based, halogen-based and ether-based reaction solvents
- B ⁇
- an embodiment using a mixed solvent of A and B is preferred (see Embodiment 1, Examples 11 to 13), and the volume ratio (A: B, A and B
- the sum may be 100 or more: 40 or less, preferably 70 or more and 30 or less, and particularly preferably 80 or more and 20 or less.
- B can be used alone or in combination with A as a minor component.
- the amount of the reaction solvent used may be 0.03 L (liter) or more per 1 mol of ⁇ , ⁇ -difluoroacetic acid ester represented by the general formula [1], preferably 0.05 to 10 L, preferably 0.07 ⁇ 7 L is particularly preferred.
- the reaction temperature may be + 30 ° C. or less, preferably +25 to ⁇ 50 ° C., particularly preferably +20 to ⁇ 40 ° C., extremely preferably +15 to ⁇ 30 ° C., aliphatic carbonization
- the reaction may be performed at + 50 ° C. or less, preferably +45 to ⁇ 30 ° C., particularly preferably +40 to ⁇ 20 ° C., and +35 to ⁇ 10. C. is very preferred.
- the reaction time may be within 72 hours, and varies depending on the raw material substrate and reaction conditions. Therefore, the progress of the reaction is traced by analytical means such as gas chromatography, liquid chromatography, nuclear magnetic resonance, etc.
- analytical means such as gas chromatography, liquid chromatography, nuclear magnetic resonance, etc.
- the end point should be the point at which almost no recognition is possible.
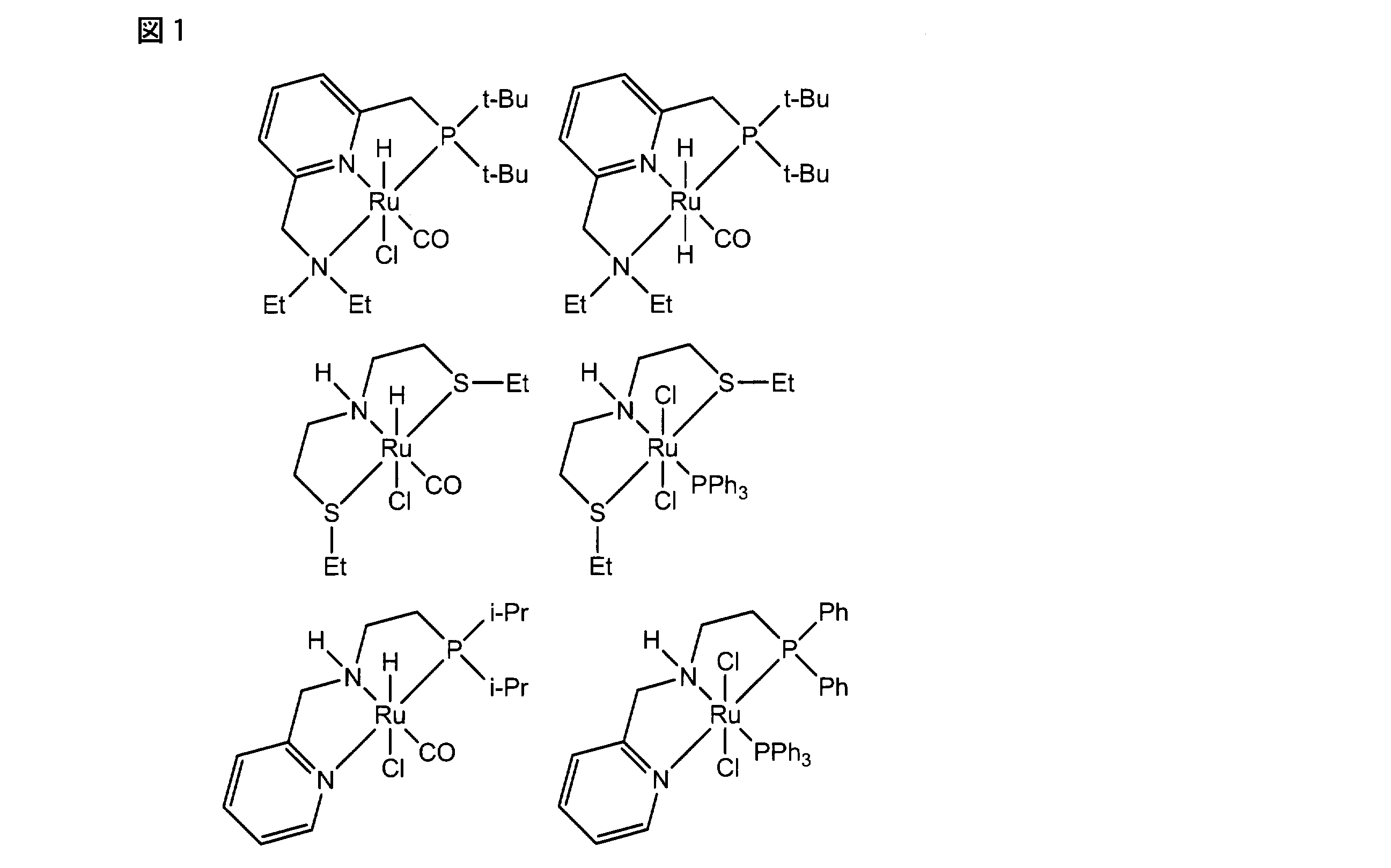
- the hydrogenation catalyst or the precursor used in the present invention is not limited to the ruthenium complex represented by the general formula [2] or the general formula [4], and an existing ruthenium catalyst can be used similarly.
- an existing ruthenium catalyst can be used similarly.
- Angew.Chem.Int.Ed.2013,52,2538-2542, Organanometallics 2012,31,5239-5242, Angew.Chem.Int.Ed.2012,51,2772-2775 and Angew.Chem.Int.Ed Ruthenium catalysts described in .2006, 45, 1113-1115, etc., and typical ones (homogeneous ruthenium catalysts) are shown in FIG.
- ruthenium catalysts are prepared under the same reaction conditions as the ruthenium complex represented by the general formula [2] or the general formula [4] (the amount of the ruthenium catalyst used, if necessary, the presence of a base, the type of base, the use of the base) Volume, amount of hydrogen gas used, hydrogen pressure, reaction solvent, amount of reaction solvent used, reaction temperature, reaction time, post-treatment, and preferred embodiments).
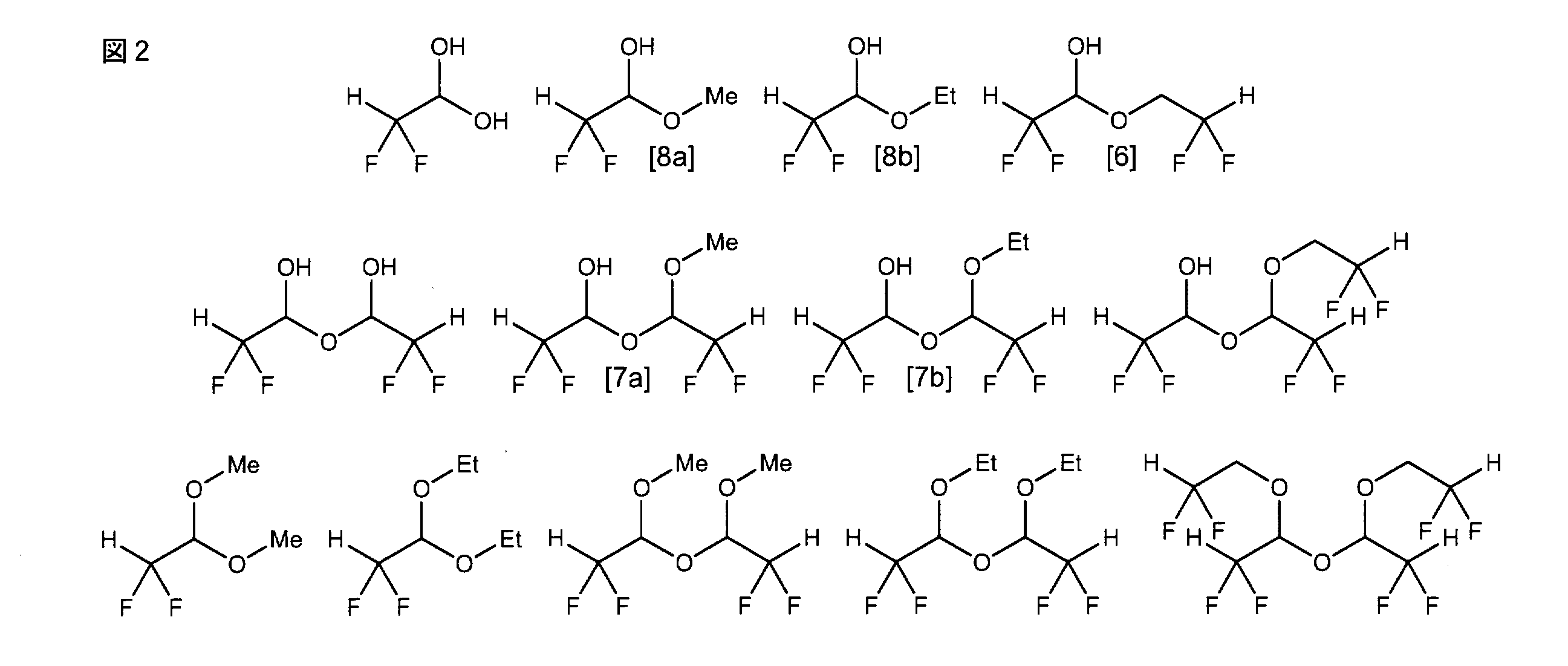
- ⁇ , ⁇ -difluoroacetaldehyde represented by the general formula [3], which is the target product can be obtained by employing a general operation in organic synthesis. Since ⁇ , ⁇ -difluoroacetaldehyde represented by the general formula [3] is an aldehyde in which a difluoromethyl group, which is a strong electron-attracting group, is directly linked, self-polymer, hydrate, hemiacetal, acetal and In many cases, it is obtained as a stable equivalent such as a compound in which these structural features are combined (of course, in some cases, it can also be obtained in the form of an aldehyde).
- the ⁇ , ⁇ -difluoroacetaldehyde represented by the general formula [3] in the claims is defined to include these stable equivalents.
- the alcohol constituting the hemiacetal body and the acetal body is an alkali metal alkoxide used as a base, an alcohol used as a reaction solvent, an ester moiety of a raw material substrate (an ⁇ , ⁇ -difluoroacetate ester represented by the general formula [1]) Derived from OR 1 ) and ⁇ , ⁇ -difluoroethanol by-produced by excessive reduction.
- FIG. 2 Representative examples of stable equivalents of ⁇ , ⁇ -difluoroacetaldehyde represented by the general formula [3] are shown in FIG. 2 (abbreviations / Me; methyl group, Et: ethyl group), but naturally limited to these. It is not a thing.
- hemiacetals of the general formula [8] show good reactivity with respect to various reactions as intermediates in organic synthesis, and in industrial production Is an easy-to-handle compound.
- the stable equivalent of the general formula [6] is a hemiacetal derived from ⁇ , ⁇ -difluoroethanol by-produced by excessive reduction
- the stable equivalent of the general formula [7] ([7a], [7b])
- the equivalent is a dimer obtained by adding the hemiacetal compound of the general formula [8] to ⁇ , ⁇ -difluoroacetaldehyde released by the reverse reaction.
- the stable equivalent of the general formula [6] or the general formula [7] is used when the relative amount of methanol or ethanol relative to the hemiacetal form of the general formula [8] decreases (for example, the main fraction of fractional distillation). This is a by-product compound during recovery. Therefore, in order to obtain a hemiacetal compound represented by the general formula [8] which is a useful compound (intermediate) with high purity (particularly 90 mol% or more), the general formula [6] or the general formula coexisting as a minor component A purification operation for converging (inducing) the stable equivalent of [7] into the hemiacetal of general formula [8] becomes important. Furthermore, from the viewpoint of an industrial production method, a simple purification operation is required.
- R 3 in the stable equivalent of ⁇ , ⁇ -difluoroacetaldehyde represented by the general formula [7] represents a methyl group or an ethyl group.
- R 4 in the hemiacetal form of ⁇ , ⁇ -difluoroacetaldehyde represented by the general formula [8] represents a methyl group or an ethyl group.
- the contact with methanol or ethanol is not particularly limited, and a general method in organic synthesis can be adopted.
- the amount of methanol or ethanol used may be 0.7 mol or more with respect to 1 mol in total of stable equivalents of ⁇ , ⁇ -difluoroacetaldehyde represented by general formula [6] or general formula [7].
- the contact temperature may be ⁇ 20 ° C. or higher, preferably 0 to + 150 ° C., particularly preferably +20 to + 125 ° C.
- the contact time may be within 48 hours, and varies depending on the stable equivalent (general formula [6] or general formula [7]) and the contact conditions. Therefore, analytical means such as gas chromatography, liquid chromatography, nuclear magnetic resonance, etc. By tracking the progress of contact, the end point may be the point when almost no decrease in stable equivalent is observed. This contact may be carried out in the presence of an acid as necessary to smoothly converge to a hemiacetal body (general formula [8]).
- the acid can be the same as the “acid used for neutralization of the reaction completion liquid” described later.
- this contact can be carried out well even in the absence of acid.
- a hydrate can be obtained as a stable equivalent in good yield.
- the crude product ( ⁇ , ⁇ -difluoroacetaldehyde or the stable equivalent) can be purified to high purity by activated carbon treatment, fractional distillation, recrystallization, column chromatography, etc., if necessary.
- the boiling point of the target product is low, the operation of recovering and distilling directly from the reaction-terminated liquid is simple.
- the target product self-polymer, hydrate or hemiacetal, etc.
- the target product may cause a Cannizzaro reaction in the presence of a base.
- the reaction end solution is preliminarily prepared with an organic acid such as formic acid, acetic acid, citric acid, oxalic acid, benzoic acid, methanesulfonic acid and paratoluenesulfonic acid, or an inorganic acid such as hydrogen chloride, hydrogen bromide, nitric acid and sulfuric acid. And neutralizing with (3), and performing recovery distillation (including recovery and washing of the residue with an organic solvent such as tert-butyl methyl ether, diisopropyl ether, methanol and ethanol, and filtration of the generated salt as necessary).
- the target product can be obtained with good yield.
- a crude product obtained by a reaction that gives priority to the selectivity over the conversion rate may be more easily purified, which is a preferred embodiment.
- the selectivity decreases as the conversion rate increases. Therefore, when the reaction is stopped at a conversion rate of 65% or less, the target product can be obtained with a high selectivity (90 to 10 described later: 10 or less).
- the selectivity target product: ⁇ , ⁇ -difluoroethanol, the sum of both compounds being 100
- Particularly preferred is 90 or more and 10 or less.
- the selectivity is 90 or more and 10 or less
- fractional distillation with a high number of theoretical plates is not required to purify the target product (for example, commercialization is possible from the recovered residue in the kettle.
- Example 20 And 21 it may be easier to achieve high-purity fractionation after induction into a hemiacetal compound having a large boiling point difference from ⁇ , ⁇ -difluoroethanol, which is a byproduct, and this is a preferred embodiment (Aspect 3).
- ethanol, n-propanol, isopropanol and n-butanol may be used, ethanol, n-propanol and n-butanol are preferred, and ethanol and n-propanol are particularly preferred.
- a hemiacetal derived from methanol can be highly purified by employing fractional distillation with a high number of theoretical plates.
- the unreacted raw material substrate ⁇ , ⁇ -difluoroacetic acid esters
- the fractional distillation may be performed under a slightly reduced pressure at which the distillation temperature is 70 ° C. or lower, or under atmospheric pressure, and preferably under a slightly reduced pressure at which the distillation temperature is 70 ° C. or lower.
- the recovery and reuse can be performed as a solution of the reaction solvent used or as an azeotropic composition with the reaction solvent.
- the combination with the reaction (aspect 2) giving priority to the selectivity over the conversion ratio is a more preferable aspect (aspect 5). (For embodiments 2-5, see especially Examples 20 and 21.)
- the stable equivalent of ⁇ , ⁇ -difluoroacetaldehyde represented by the general formula [3] is operated in the same manner as in the case of similar stable equivalents of fluoral (such as hydrates and hemiacetals). 1999, Vol. 57, No. 10, p. 102-104) can be used to convert to ⁇ , ⁇ -difluoroacetaldehyde (see Examples 22 and 23).
- the yield of the desired product was about 60% as determined by 19 F-NMR analysis of the methanol solution by the internal standard method (internal standard substance ⁇ , ⁇ , ⁇ -trifluorotoluene). Furthermore, by carrying out fractional distillation, it could be purified to a high purity product (gas chromatography purity 90% or more).
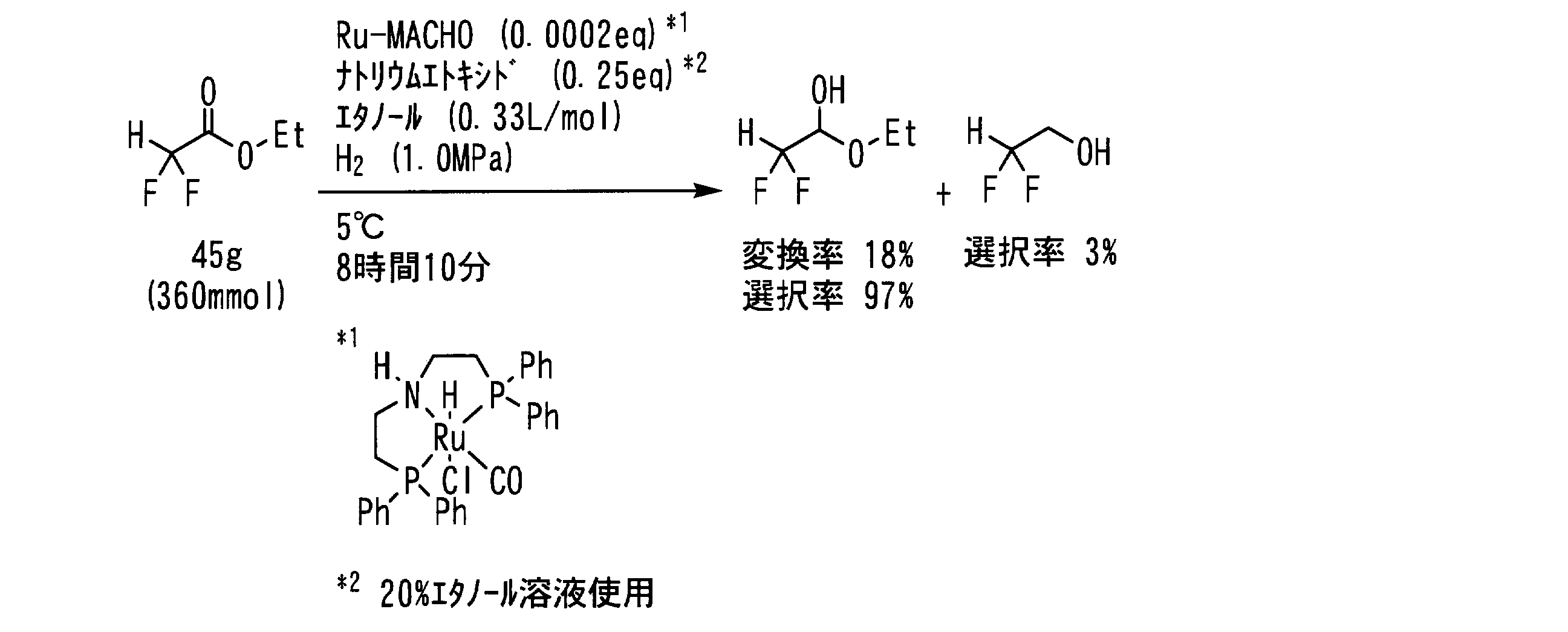
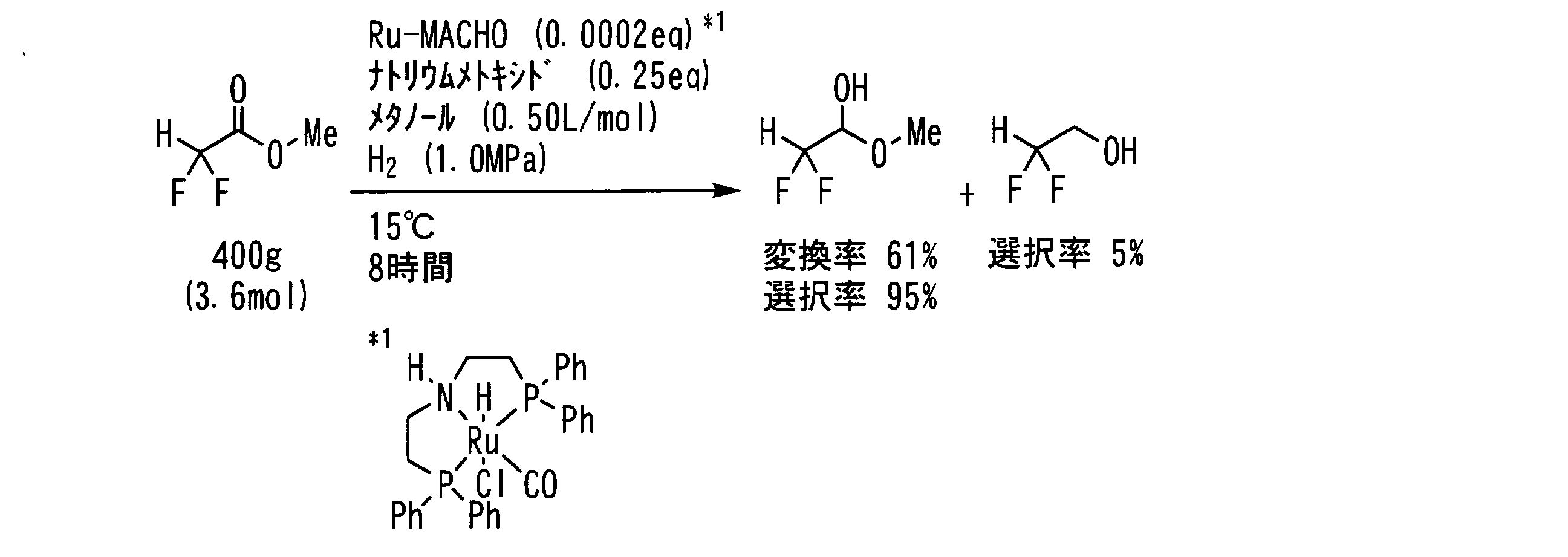
- Example 14 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula: 45 g (360 mmol, 1 eq) of ⁇ , ⁇ -difluoroacetic acid ester represented by the following formula: 47 mg (purity 94.2%, 73 ⁇ mol, 0.0002 eq), 31 g of a 20% ethanol solution of sodium ethoxide (91 mmol, 0.25 eq as sodium ethoxide) and 120 mL of ethanol (0.33 L / mol) Then, the inside of the reaction vessel was replaced with hydrogen gas five times, the hydrogen pressure was set to 1.0 MPa, and the mixture was stirred at 5 ° C. for 8 hours and 10 minutes.
- SUS stainless steel
- Example 15 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula: 450 g (3.6 mol, 1 eq) of ⁇ , ⁇ -difluoroacetic acid ester represented by the following formula: 470 mg (purity 94.2%, 730 ⁇ mol, 0.0002 eq), 170 g of a 28% methanol solution of sodium methoxide (910 mmol, 0.25 eq as sodium methoxide) and 1.2 L of methanol (0.33 L / mol) was added, the inside of the reaction vessel was replaced with hydrogen gas five times, the hydrogen pressure was set to 1.0 MPa, and the mixture was stirred at 15 ° C. for 8 hours and 10 minutes.
- SUS stainless steel
- Example 16 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula: 45 g (360 mmol, 1 eq) of ⁇ , ⁇ -difluoroacetic acid ester represented by the following formula: 47 mg (purity 94.2%, 73 ⁇ mol, 0.0002 eq), 28% methanol solution of sodium methoxide 7.0 g (36 mmol, 0.1 eq as sodium methoxide) and 120 mL of methanol (0.33 L / mol) was added, the inside of the reaction vessel was replaced with hydrogen gas five times, the hydrogen pressure was set to 2.5 MPa, and the mixture was stirred at 15 ° C. for 7 hours.
- SUS stainless steel
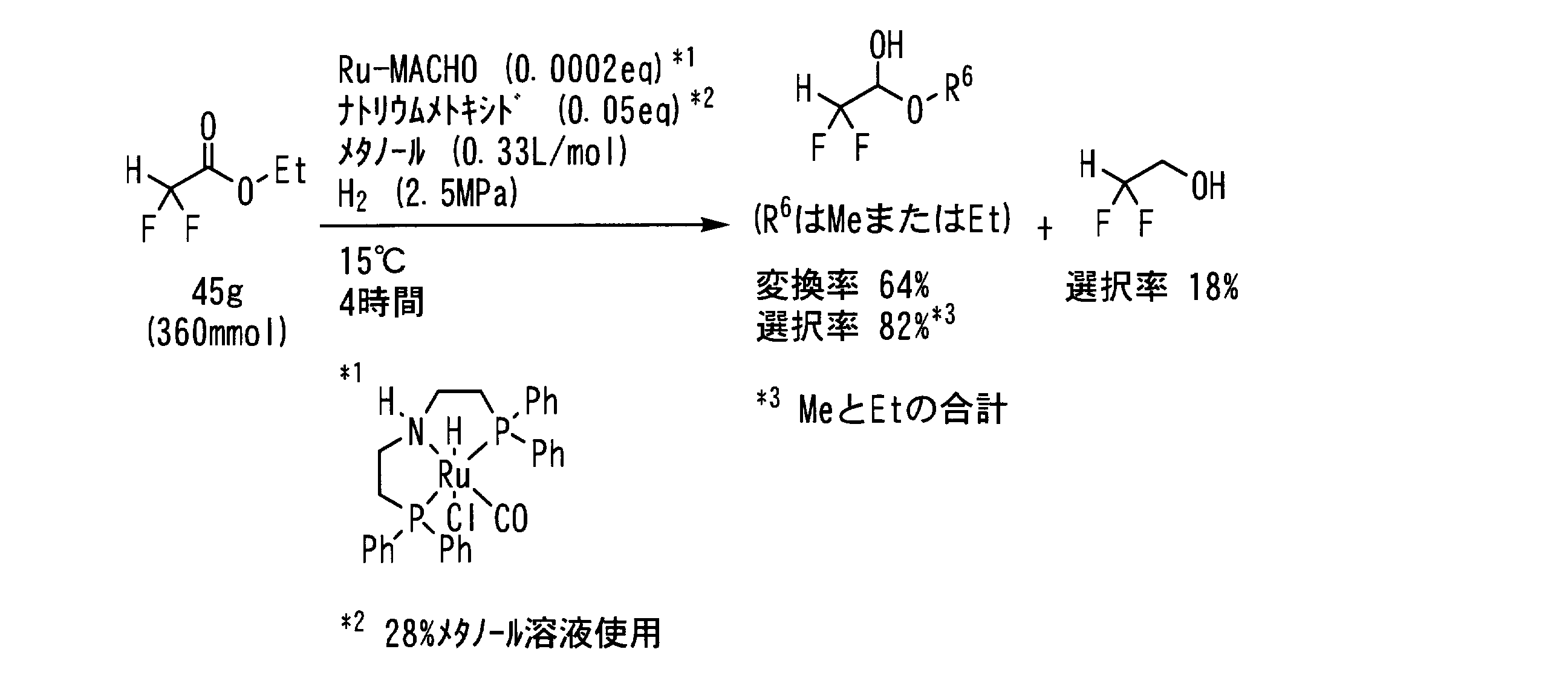
- Example 17 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula: 45 g (360 mmol, 1 eq) of ⁇ , ⁇ -difluoroacetic acid ester represented by the following formula: 47 mg (purity 94.2%, 73 ⁇ mol, 0.0002 eq), 3.5 g of 28% methanol solution of sodium methoxide (18 mmol, 0.05 eq as sodium methoxide) and 120 mL of methanol (0.33 L / mol) was added, the inside of the reaction vessel was replaced with hydrogen gas five times, the hydrogen pressure was set to 2.5 MPa, and the mixture was stirred at 15 ° C. for 4 hours.
- SUS stainless steel
- Example 18 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula: 2.2 g (20 mmol, 1 eq) of ⁇ , ⁇ -difluoroacetic acid ester represented by the following formula: 24 mg (50 ⁇ mol, 0.0025 eq), 350 mg (5.0 mmol, 0.25 eq) potassium methoxide and 10 mL (0.50 L / mol) tert-butyl methyl ether were added, and hydrogen gas was added to the reaction vessel. The hydrogen pressure was set to 1.0 MPa, and the mixture was stirred at 40 ° C. for 15 minutes.
- SUS stainless steel
- Example 19 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula: 400 g (3.6 mol, 1 eq) of ⁇ , ⁇ -difluoroacetic acid ester represented by the following formula: 470 mg (purity 94.2%, 730 ⁇ mol, 0.0002 eq), sodium methoxide 49 g (910 mmol, 0.25 eq) and 1.8 L (0.50 L / mol) of methanol were added to the reaction vessel. The gas was replaced with hydrogen gas five times, the hydrogen pressure was set to 1.0 MPa, and the mixture was stirred at 15 ° C. for 8 hours.
- SUS stainless steel
- Acetic acid 150 g (2.5 mol, 0.23 eq) was added to the reaction end solution, and the product was directly recovered and distilled (oil bath temperature to 66 ° C., reduced pressure to 2.1 kPa) to obtain the desired product (methyl hemiacetal form).
- a methanol solution containing was obtained.
- purified products include methanol, ethanol, ⁇ , ⁇ -difluoroethanol, methyl hemiacetal, ethyl hemiacetal, and the following formula:
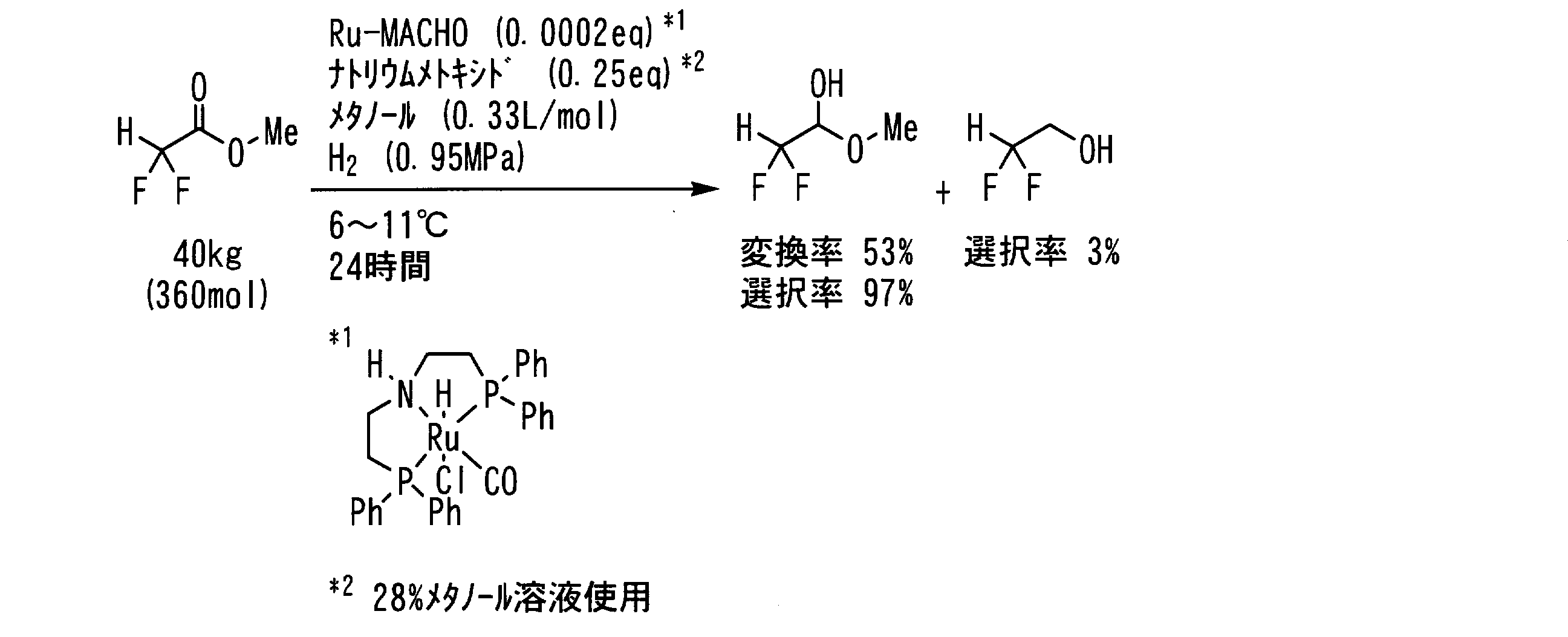
- Example 20 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula: ⁇ , ⁇ -difluoroacetate represented by the formula: 40 kg (360 mol, 1 eq), 47 g of ruthenium complex (purity 94.2%, 73 mmol, 0.0002 eq), 18 kg of a 28% methanol solution of sodium methoxide (91 mol, 0.25 eq as sodium methoxide) and 120 L of methanol (0.33 L / mol) Then, the inside of the reaction vessel was replaced with hydrogen gas five times, the hydrogen pressure was set to 0.95 MPa, and the mixture was stirred at 6 to 11 ° C. for 24 hours.
- SUS stainless steel
- the methanol solution was quantified by an internal standard method of 19 F-NMR (internal standard substance; C 6 F 6 ).
- 19 F-NMR internal standard substance
- C 6 F 6 internal standard substance
- 17 kg (150 mol) of a raw material substrate was contained [overreduced ⁇ , ⁇ -difluoroethanol. Contained 0.30 kg (3.7 mol).
- the high boiling fraction with a high content of this by-product was discarded].
- the recovery rate of the unreacted raw material substrate was as good as about 90%.
- the pressure was reduced to 0.8 kPa) to obtain 170 kg of an ethanol solution (1) containing the target product. Again, neutralize the kettle residue with a small amount of acetic acid (pH 6-7), add 25 kg of ethanol, and perform the same stirring washing and recovery distillation as above to obtain 22 kg of the ethanol solution (2) containing the target product. Obtained.
- the ethanol solutions (1) and (2) were combined and quantified by an internal standard method of 19 F-NMR (internal standard substance; C 6 F 6 ). ) was included in an amount of 500 mol.
- fractional distillation By removing fractions of low boiling points (such as methanol, ethanol and ⁇ , ⁇ -difluoroethanol) from these solutions by fractional distillation (theoretical plate number: 15 plates, distillation temperature: 47 ° C., reduced pressure: up to 24 kPa) [ethyl hemi Acetal body: Distillation is continued by appropriately adding ethanol until the selectivity of methyl hemiacetal body (the sum of both compounds is 100) is 95 or more: 5 or less], the following formula: 70 kg of the residue (purified product-1) mainly comprising a stable equivalent of ⁇ , ⁇ -difluoroacetaldehyde (ethyl hemiacetal) represented by the formula (1) was recovered.
- the content of the dimer derived from the ethyl hemiacetal body is remarkably increased. May increase.
- the purity of ⁇ , ⁇ -difluoroethanol, methyl hemiacetal, ethyl hemiacetal, dimer derived from ⁇ , ⁇ -difluoroethyl hemiacetal and ethyl hemiacetal is 2.7%, respectively. 6%, 78.8%, 3.3%, and 13.6% (ethanol content was 2.3%). Even in such a case, by adopting the above purification operation, it was possible to converge to 1.9%, 1.2%, 91.7%, 0.9%, and 4.3%, respectively. The content is 6.4%).
- Example 21 In a pressure resistant reaction vessel made of stainless steel (SUS), the following formula: ⁇ , ⁇ -difluoroacetate represented by the formula: 45 kg (360 mol, 1 eq), 47 g of ruthenium complex (purity 94.2%, 73 mmol, 0.0002 eq), 18 kg of a 28% methanol solution of sodium methoxide (91 mol, 0.25 eq as sodium methoxide) and 120 L of methanol (0.33 L / mol) Then, the inside of the reaction vessel was replaced with hydrogen gas five times, the hydrogen pressure was set to 1.0 MPa, and the mixture was stirred at 30 ° C. for 15 minutes and further at 20 ° C. for 6 hours.
- SUS stainless steel
- Acetic acid (5.3 kg, 88 mol, 0.24 eq) was added to the reaction-finished solution, and directly recovered and distilled (distillation temperature to 44 ° C., reduced pressure to 31 kPa), whereby unreacted raw material substrate ( ⁇ , ⁇ -difluoro) was obtained.
- 100 kg of a methanol solution containing a total of methyl acetate and ethyl ⁇ , ⁇ -difluoroacetate, the former being major) was recovered.
- the methanol solution was quantified by 19 F-NMR internal standard method (internal standard substance; C 6 F 6 ).
- Example 22 Following formula: The following formula is obtained by thermally decomposing (oil bath temperature to 133 ° C.) a diisopropyl ether solution containing a stable equivalent (methyl hemiacetal) of ⁇ , ⁇ -difluoroacetaldehyde represented by: ⁇ , ⁇ -difluoroacetaldehyde represented by the above was generated as a gas, and a fraction containing diisopropyl ether was recovered.
- the assignment of 1 H and 19 F-NMR analysis of ⁇ , ⁇ -difluoroacetaldehyde is shown below. Furthermore, mass structure analysis of the generated gas suggested the structure of the above formula.
- Example 23 Following formula: By adding diphosphorus pentoxide to a stable equivalent (ethyl hemiacetal form) of ⁇ , ⁇ -difluoroacetaldehyde represented by formula (II) and heating (oil bath temperature to 110 ° C.), the following formula: ⁇ , ⁇ -difluoroacetaldehyde represented by the following formula is generated as a gas and further bubbled into cold methanol to obtain the following formula: A methanol solution of a stable equivalent (methyl hemiacetal form) of ⁇ , ⁇ -difluoroacetaldehyde represented by the following was recovered. The recovery rate was about 80%.
- the ⁇ , ⁇ -difluoroacetaldehyde obtained by the production method of the present invention can be used as an intermediate for medical and agricultural chemicals.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description
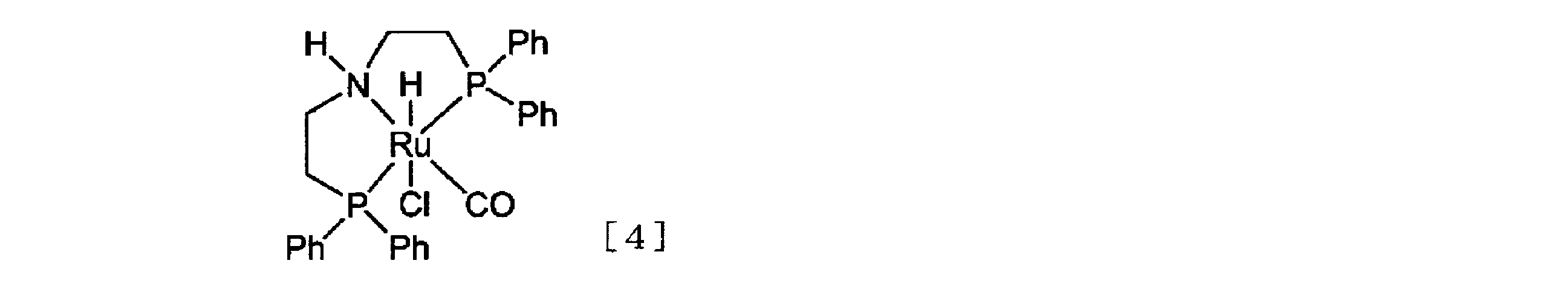
一般式[1]で示されるα,α-ジフルオロ酢酸エステル類を、一般式[2]で示されるルテニウム錯体の存在下に水素ガス(H2)と反応させることにより、一般式[3]で示されるα,α-ジフルオロアセトアルデヒドを製造する方法。



さらに塩基の存在下に反応を行うことを特徴とする、発明1に記載の方法。
一般式[1]で示されるα,α-ジフルオロ酢酸エステル類を、一般式[4]で示されるルテニウム錯体および塩基の存在下に水素ガス(H2)と反応させることにより、一般式[3]で示されるα,α-ジフルオロアセトアルデヒドを製造する方法。



前記一般式[1]で示されるα,α-ジフルオロ酢酸エステル類が、一般式[5]で示されるα,α-ジフルオロ酢酸エステル類であることを特徴とする、発明1乃至発明3の何れかに記載の方法。

アルコール系の反応溶媒を用いて、30℃以下の反応温度で反応を行うことを特徴とする、発明1乃至発明4の何れかに記載の方法。
アルコール系の反応溶媒を用いて、20℃以下の反応温度で反応を行うことを特徴とする、発明1乃至発明4の何れかに記載の方法。
脂肪族炭化水素系、芳香族炭化水素系、ハロゲン系またはエーテル系の反応溶媒を用いて、50℃以下の反応温度で反応を行うことを特徴とする、発明1乃至発明4の何れかに記載の方法。
脂肪族炭化水素系、芳香族炭化水素系、ハロゲン系またはエーテル系の反応溶媒を用いて、40℃以下の反応温度で反応を行うことを特徴とする、発明1乃至発明4の何れかに記載の方法。
一般式[1]で示されるα,α-ジフルオロ酢酸エステル類を、ルテニウム触媒の存在下に水素ガス(H2)と反応させることにより、一般式[3]で示されるα,α-ジフルオロアセトアルデヒドを製造する方法。


ルテニウム触媒が均一系触媒であることを特徴とする、発明9に記載の方法。
一般式[3]で示されるα,α-ジフルオロアセトアルデヒドとβ,β-ジフルオロエタノールとの選択率が、該アルデヒド:該エタノール=90以上:10以下の状態で反応を止め、未反応のα,α-ジフルオロ酢酸エステル類を回収し再利用することを特徴とする、発明1乃至発明10の何れかに記載の方法。
後処理過程にて、一般式[6]もしくは一般式[7]で示されるα,α-ジフルオロアセトアルデヒドの安定等価体をメタノールまたはエタノールと接触させることにより、一般式[8]で示されるα,α-ジフルオロアセトアルデヒドのヘミアセタール体に収束させる精製操作を行うことを特徴とする、発明1乃至発明11の何れかに記載の方法。



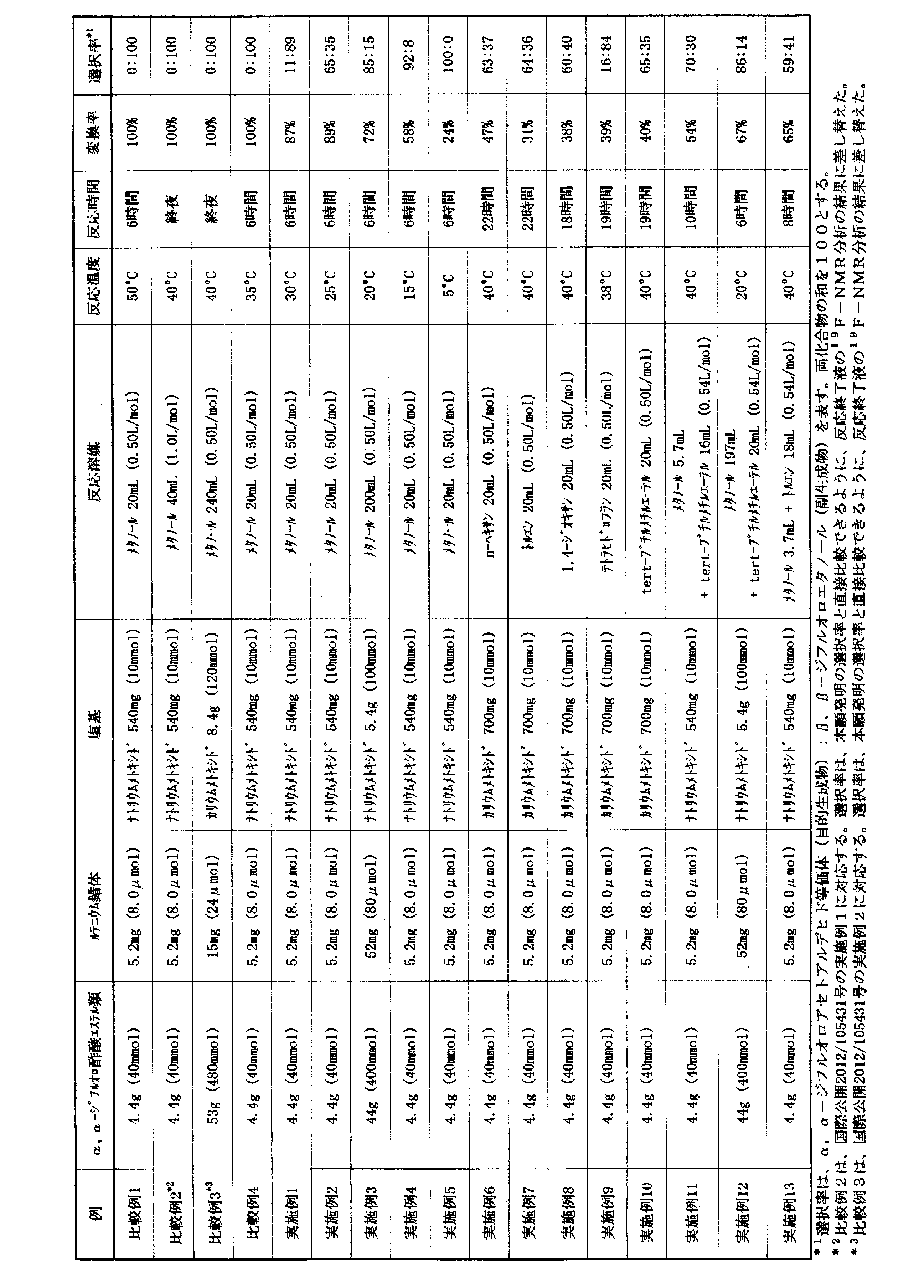
比較例1~4および実施例1~13の一般製造方法を以下に示し、これらの結果を表1に纏めた。
ステンレス鋼(SUS)製耐圧反応容器に、下記式:




実施例3については、反応終了液から直接、回収蒸留することにより、目的生成物を含むメタノール溶液を得た。該メタノール溶液の19F-NMR分析より内部標準法(内部標準物質α,α,α-トリフルオロトルエン)で定量したところ、目的生成物の収率は約60%であった。さらに、分別蒸留を行うことにより、高純度品に精製することができた(ガスクロマトグラフィー純度90%以上)。
ステンレス鋼(SUS)製耐圧反応容器に、下記式:




ステンレス鋼(SUS)製耐圧反応容器に、下記式:



で示されるα,α-ジフルオロアセトアルデヒドの安定等価体(メチルヘミアセタール体とエチルヘミアセタール体の合計)と、過剰に還元された下記式:


ステンレス鋼(SUS)製耐圧反応容器に、下記式:



で示されるα,α-ジフルオロアセトアルデヒドの安定等価体(メチルヘミアセタール体とエチルヘミアセタール体の合計)と、過剰に還元された下記式:


ステンレス鋼(SUS)製耐圧反応容器に、下記式:



で示されるα,α-ジフルオロアセトアルデヒドの安定等価体(メチルヘミアセタール体とエチルヘミアセタール体の合計)と、過剰に還元された下記式:

ステンレス鋼(SUS)製耐圧反応容器に、下記式:





ステンレス鋼(SUS)製耐圧反応容器に、下記式:







メチルヘミアセタール体、エチルヘミアセタール体、β,β-ジフルオロエチルヘミアセタール体およびエチルヘミアセタール体由来の二量体の、1Hと19F-NMR分析の帰属を以下に示す。
[メチルヘミアセタール体]
1H-NMR(基準物質;Me4Si、重溶媒;CDCl3)、δ ppm;3.53(s、3H)、4.62(m、1H)、5.60(dt、1H)、ヒドロキシル基のプロトンは帰属できず。
19F-NMR(基準物質;C6F6、重溶媒;CDCl3)、δ ppm;27.9(ddd、2F)。
[エチルヘミアセタール体]
1H-NMR(基準物質;Me4Si、重溶媒;CDCl3)、δ ppm;1.26(t、3H)、3.63(m、1H)、3.92(m、1H)、4.70(m、1H)、5.60(dt、1H)、ヒドロキシル基のプロトンは帰属できず。
19F-NMR(基準物質;C6F6、重溶媒;CDCl3)、δ ppm;27.9(ddd、2F)。
[β,β-ジフルオロエチルヘミアセタール体]
1H-NMR(基準物質;Me4Si、重溶媒;CDCl3)、δ ppm;3.92(m、2H)、4.79(m、1H)、5.68(dt、1H)、5.91(ddt、1H)、ヒドロキシル基のプロトンは帰属できず。
19F-NMR(基準物質;C6F6、重溶媒;CDCl3)、δ ppm;27.0(ddd、2F)、36.1(dt、2F)。
[エチルヘミアセタール体由来の二量体]
1H-NMR(基準物質;Me4Si、重溶媒;CDCl3)、δ ppm;1.28(t、3H)、3.67(m、1H)、3.88(m、1H)、4.78(m、0.5H)、4.90(m、0.5H)、5.04(m、1H)、5.64(m、2H)、ヒドロキシル基のプロトンは帰属できず。
19F-NMR(基準物質;C6F6、重溶媒;CDCl3)、δ ppm;27.9(m、4F)。
ステンレス鋼(SUS)製耐圧反応容器に、下記式:




上記の反応を計3回繰り返して行うことにより(2ラン目と3ラン目は、それぞれ1ラン目、2ラン目の未反応の原料基質を回収して再利用した)、α,α-ジフルオロ酢酸エステル類1.1kmol(1,090mol)を反応に供したことに相当する、未反応の原料基質を回収した後の釜残(酢酸ナトリウムと目的生成物が含まれる固形物)を得た。釜残にエタノール100kg(2.2kmol、2eq)を加え、10℃以下で攪拌洗浄し、濾過し、固形物を少量の冷エタノールで洗浄し、濾洗液から回収蒸留(油浴温度~50℃、減圧度~0.8kPa)することにより、目的生成物を含むエタノール溶液(1)を170kg得た。再度、釜残を少量の酢酸で中和し(pH6~7)、エタノール25kgを加えて、上記と同様の攪拌洗浄と回収蒸留を行うことにより、目的生成物を含むエタノール溶液(2)を22kg得た。該エタノール溶液(1)と(2)を合わせて19F-NMRの内部標準法(内部標準物質;C6F6)で定量したところ、目的生成物(α,α-ジフルオロアセトアルデヒドの安定等価体の合計)が500mol含まれていた。これらの溶液から分別蒸留(理論段数15段、留出温度~47℃、減圧度~24kPa)にて低沸分(メタノール、エタノールおよびβ,β-ジフルオロエタノール等)を除去することにより[エチルヘミアセタール体:メチルヘミアセタール体の選択率(両化合物の和を100とする)が95以上:5以下になるまで、エタノールを適宜加えて蒸留を継続する]、下記式:



精製品-1に含まれるβ,β-ジフルオロエチルヘミアセタール体とエチルヘミアセタール体由来の二量体の合計に対して、エタノールを1.2eq加え、80℃で2時間攪拌し、単蒸留(留出温度~44℃、減圧度~0.5kPa)することにより(エタノールの含量が多い低沸側の留分と、エチルヘミアセタール体由来の二量体の含量が多い釜残は回収再利用した)、精製品-2を回収率83%で得た(回収率は、蒸留前後のα,α-ジフルオロアセトアルデヒドの安定等価体の合計で計算した)。精製品-2の1Hと19F-NMR分析より、β,β-ジフルオロエタノール、メチルヘミアセタール体、エチルヘミアセタール体、β,β-ジフルオロエチルヘミアセタール体とエチルヘミアセタール体由来の二量体の純度は、それぞれ1.9%、2.1%、92.2%、0.8%、3.0%であった。また、エタノールの含量は6.5%であった。この操作により、メチルヘミアセタール体、β,β-ジフルオロエチルヘミアセタール体とエチルヘミアセタール体由来の二量体の含量を低減でき、所望のエチルヘミアセタール体の含量を向上することができた。
また、上記とは別に、低沸分の除去をエタノールの相対的な共存量が少なく(特に3.0重量%未満)なるまで行うと、エチルヘミアセタール体由来の二量体の含量が格段に増加する場合がある。例えば、β,β-ジフルオロエタノール、メチルヘミアセタール体、エチルヘミアセタール体、β,β-ジフルオロエチルヘミアセタール体とエチルヘミアセタール体由来の二量体の純度が、それぞれ2.7%、1.6%、78.8%、3.3%、13.6%となった(エタノールの含量は2.3%であった)。この様な場合でも上記の精製操作を採用することにより、それぞれ1.9%、1.2%、91.7%、0.9%、4.3%に収束させることができた(エタノールの含量は6.4%)。
ステンレス鋼(SUS)製耐圧反応容器に、下記式:



で示されるα,α-ジフルオロアセトアルデヒドの安定等価体(メチルヘミアセタール体とエチルヘミアセタール体の合計)と、過剰に還元された下記式:


上記の反応を計3回繰り返して行うことにより(2ラン目と3ラン目は、それぞれ1ラン目、2ラン目の未反応の原料基質を回収して再利用した)、α,α-ジフルオロ酢酸エステル類1.1kmol(1,088mol)を反応に供したことに相当する、未反応の原料基質を回収した後の釜残(酢酸ナトリウムと目的生成物が含まれる固形物)を得た。以降は実施例20と同様の操作を行うことにより、下記式:



下記式:


1H-NMR(基準物質;Me4Si、重溶媒;CDCl3)、δ ppm;5.73(t、1H)、9.54(s、1H)。
19F-NMR(基準物質;C6F6、重溶媒;CDCl3)、δ ppm;30.9(d、2F)。
下記式:



Claims (12)
- 一般式[1]で示されるα,α-ジフルオロ酢酸エステル類を、一般式[2]で示されるルテニウム錯体の存在下に水素ガス(H2)と反応させることにより、一般式[3]で示されるα,α-ジフルオロアセトアルデヒドを製造する方法。



- さらに塩基の存在下に反応を行うことを特徴とする、請求項1に記載の方法。
- 一般式[1]で示されるα,α-ジフルオロ酢酸エステル類を、一般式[4]で示されるルテニウム錯体および塩基の存在下に水素ガス(H2)と反応させることにより、一般式[3]で示されるα,α-ジフルオロアセトアルデヒドを製造する方法。
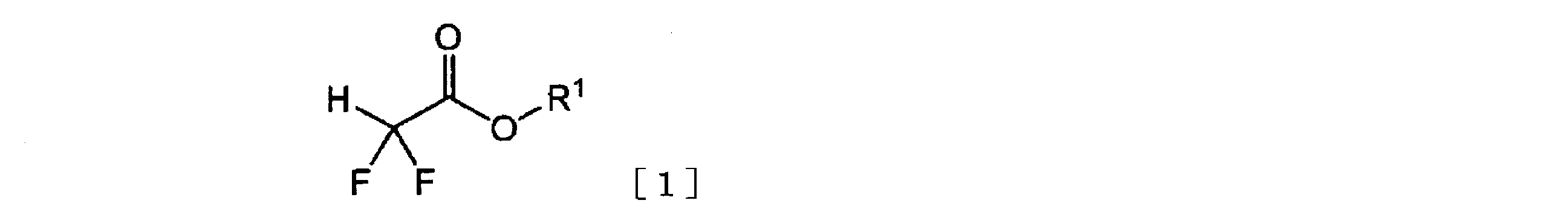
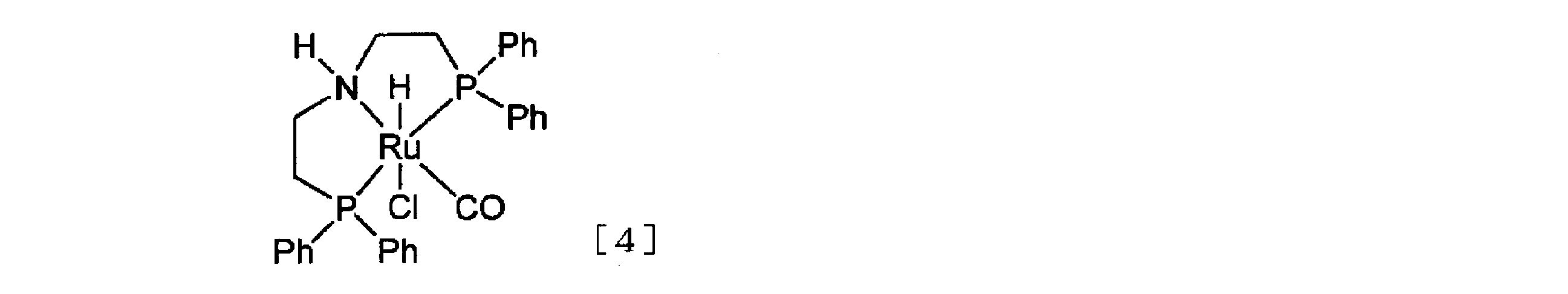

- 前記一般式[1]で示されるα,α-ジフルオロ酢酸エステル類が、一般式[5]で示されるα,α-ジフルオロ酢酸エステル類であることを特徴とする、請求項1乃至請求項3の何れかに記載の方法。

- アルコール系の反応溶媒を用いて、30℃以下の反応温度で反応を行うことを特徴とする、請求項1乃至請求項4の何れかに記載の方法。
- アルコール系の反応溶媒を用いて、20℃以下の反応温度で反応を行うことを特徴とする、請求項1乃至請求項4の何れかに記載の方法。
- 脂肪族炭化水素系、芳香族炭化水素系、ハロゲン系またはエーテル系の反応溶媒を用いて、50℃以下の反応温度で反応を行うことを特徴とする、請求項1乃至請求項4の何れかに記載の方法。
- 脂肪族炭化水素系、芳香族炭化水素系、ハロゲン系またはエーテル系の反応溶媒を用いて、40℃以下の反応温度で反応を行うことを特徴とする、請求項1乃至請求項4の何れかに記載の方法。
- 一般式[1]で示されるα,α-ジフルオロ酢酸エステル類を、ルテニウム触媒の存在下に水素ガス(H2)と反応させることにより、一般式[3]で示されるα,α-ジフルオロアセトアルデヒドを製造する方法。


- ルテニウム触媒が均一系触媒であることを特徴とする、請求項9に記載の方法。
- 一般式[3]で示されるα,α-ジフルオロアセトアルデヒドとβ,β-ジフルオロエタノールとの選択率が、該アルデヒド:該エタノール=90以上:10以下の状態で反応を止め、未反応のα,α-ジフルオロ酢酸エステル類を回収し再利用することを特徴とする、請求項1乃至請求項10の何れかに記載の方法。
- 後処理過程にて、一般式[6]もしくは一般式[7]で示されるα,α-ジフルオロアセトアルデヒドの安定等価体をメタノールまたはエタノールと接触させることにより、一般式[8]で示されるα,α-ジフルオロアセトアルデヒドのヘミアセタール体に収束させる精製操作を行うことを特徴とする、請求項1乃至請求項11の何れかに記載の方法。



Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014558610A JP6217653B2 (ja) | 2013-01-25 | 2014-01-23 | α,α−ジフルオロアセトアルデヒドの製造方法 |
| US14/761,774 US9440900B2 (en) | 2013-01-25 | 2014-01-23 | α,α-difluoroacetaldehyde production method |
| EP14743082.1A EP2949639B1 (en) | 2013-01-25 | 2014-01-23 | Alpha, alpha-difluoroacetaldehyde production method |
| CN201480006088.6A CN104955794B (zh) | 2013-01-25 | 2014-01-23 | α,α‑二氟乙醛的制造方法 |
| US15/227,628 US9765004B2 (en) | 2013-01-25 | 2016-08-03 | α,α-Difluoroacetaldehyde production method |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013012532 | 2013-01-25 | ||
| JP2013-012532 | 2013-01-25 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/761,774 A-371-Of-International US9440900B2 (en) | 2013-01-25 | 2014-01-23 | α,α-difluoroacetaldehyde production method |
| US15/227,628 Continuation US9765004B2 (en) | 2013-01-25 | 2016-08-03 | α,α-Difluoroacetaldehyde production method |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014115801A1 true WO2014115801A1 (ja) | 2014-07-31 |
Family
ID=51227590
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/051365 WO2014115801A1 (ja) | 2013-01-25 | 2014-01-23 | α,α-ジフルオロアセトアルデヒドの製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US9440900B2 (ja) |
| EP (1) | EP2949639B1 (ja) |
| JP (1) | JP6217653B2 (ja) |
| CN (2) | CN107400044B (ja) |
| WO (1) | WO2014115801A1 (ja) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016017318A1 (ja) * | 2014-07-30 | 2016-02-04 | セントラル硝子株式会社 | 2,2-ジフルオロアセトアルデヒドの保存安定性の向上方法 |
| WO2017030074A1 (ja) * | 2015-08-19 | 2017-02-23 | セントラル硝子株式会社 | α-フルオロアルデヒド類の製造方法 |
| WO2017126233A1 (ja) * | 2016-01-21 | 2017-07-27 | セントラル硝子株式会社 | α,α-ジフルオロアセトアルデヒドの製造方法 |
| JP6195028B1 (ja) * | 2017-02-02 | 2017-09-13 | セントラル硝子株式会社 | α,α−ジフルオロアセトアルデヒドアルキルヘミアセタールの保存方法 |
| CN107922301A (zh) * | 2015-08-19 | 2018-04-17 | 中央硝子株式会社 | α‑氟代醛类的制造方法 |
| WO2018180610A1 (ja) * | 2017-03-31 | 2018-10-04 | セントラル硝子株式会社 | α,α-ジフルオロアセトアルデヒドヘミアセタールの製造方法 |
| US11370736B2 (en) | 2019-04-01 | 2022-06-28 | Triad National Security, Llc | Synthesis of fluoro hemiacetals via transition metal-catalyzed fluoro ester and carboxamide hydrogenation |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9944579B2 (en) | 2015-01-15 | 2018-04-17 | Council Of Scientific & Industrial Research | Catalytic hydrogenation process for the synthesis of terminal diols from terminal dialkyl aliphatic esters |
| CN107438592B (zh) * | 2017-02-02 | 2018-09-11 | 中央硝子株式会社 | α,α-二氟乙醛烷基半缩醛的保存方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11501575A (ja) * | 1995-11-08 | 1999-02-09 | ロデイア・シミ | ルテニウム/スズバイメタル触媒の製造法 |
| WO2011048727A1 (ja) | 2009-10-23 | 2011-04-28 | 高砂香料工業株式会社 | 3座配位子を有する新規ルテニウムカルボニル錯体、並びにその製造法及び用途 |
| WO2012105431A1 (ja) | 2011-02-03 | 2012-08-09 | セントラル硝子株式会社 | β-フルオロアルコール類の製造方法 |
| WO2013018573A1 (ja) | 2011-08-03 | 2013-02-07 | セントラル硝子株式会社 | α-フルオロアルデヒド類の製造方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1990458B (zh) * | 2005-12-27 | 2010-08-11 | 上海药明康德新药开发有限公司 | 光学活性的4,4-二氟-2-氨基-丁酸及相应酯的制备方法 |
| CA2671765A1 (en) * | 2006-12-14 | 2008-06-26 | Eli Lilly And Company | 5- [4- (azetidin-3-yloxy) -phenyl] -2-phenyl-5h-thiazolo [5,4-c] pyridin-4-one derivatives and their use as mch receptor |
| CA2832310A1 (en) * | 2011-04-06 | 2012-10-11 | Bayer Intellectual Property Gmbh | Substituted imidazopyridines and intermediates thereof |
-
2014
- 2014-01-23 US US14/761,774 patent/US9440900B2/en not_active Expired - Fee Related
- 2014-01-23 CN CN201710452208.0A patent/CN107400044B/zh active Active
- 2014-01-23 CN CN201480006088.6A patent/CN104955794B/zh active Active
- 2014-01-23 EP EP14743082.1A patent/EP2949639B1/en active Active
- 2014-01-23 JP JP2014558610A patent/JP6217653B2/ja active Active
- 2014-01-23 WO PCT/JP2014/051365 patent/WO2014115801A1/ja active Application Filing
-
2016
- 2016-08-03 US US15/227,628 patent/US9765004B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11501575A (ja) * | 1995-11-08 | 1999-02-09 | ロデイア・シミ | ルテニウム/スズバイメタル触媒の製造法 |
| WO2011048727A1 (ja) | 2009-10-23 | 2011-04-28 | 高砂香料工業株式会社 | 3座配位子を有する新規ルテニウムカルボニル錯体、並びにその製造法及び用途 |
| WO2012105431A1 (ja) | 2011-02-03 | 2012-08-09 | セントラル硝子株式会社 | β-フルオロアルコール類の製造方法 |
| WO2013018573A1 (ja) | 2011-08-03 | 2013-02-07 | セントラル硝子株式会社 | α-フルオロアルデヒド類の製造方法 |
Non-Patent Citations (11)
| Title |
|---|
| "Protective Groups in Organic Synthesis, Third Edition,", 1999, JOHN WILEY & SONS, INC. |
| ANGEW. CHEM. INT. ED., vol. 45, 2006, pages 1113 - 1115 |
| ANGEW. CHEM. INT. ED., vol. 51, 2012, pages 2772 - 2775 |
| ANGEW. CHEM. INT. ED., vol. 52, 2013, pages 2538 - 2542 |
| J. ORG. CHEM. (U.S., vol. 58, 1993, pages 2302 - 2312 |
| JOURNAL OF SYNTHETIC ORGANIC CHEMISTRY, JAPAN, vol. 57, no. 10, 1999, pages 102 - 104 |
| L. S. HEGEDUS: "Transition Metals in the Synthesis of Complex Organic Molecules", 2001, TOKYO KAGAKU DOJIN, pages: 4 - 9 |
| ORGANOMETALLICS, vol. 31, 2012, pages 5239 - 5242 |
| RYOJI NOYORI ET AL.: "Organic Chemistry for Graduate Students", vol. II, 1999, TOKYO KAGAKU DOJIN, article "Molecular Structure & Reaction/Organic Metal Chemistry", pages: 389 - 390 |
| TAKAO IKARIYA ET AL.: "Catalytic Reductive Transformations of Carboxylic and Carbonic Acid Derivatives Using Molecular Hydrogen", ACS CATALYSIS, vol. 2, pages 1718 - 1741, XP055266853 * |
| TAKAO IKARIYA ET AL.: "Practical Selective Hydrogenation of ?-Fluorinated Esters with Bifunctional Pincer-Type Ruthenium(II) Catalysts Leading to Fluorinated Alcohols or Fluoral Hemiacetals", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 135, 13 June 2013 (2013-06-13), pages 9600 - 9603, XP055103702 * |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016017318A1 (ja) * | 2014-07-30 | 2016-02-04 | セントラル硝子株式会社 | 2,2-ジフルオロアセトアルデヒドの保存安定性の向上方法 |
| US9845278B2 (en) | 2014-07-30 | 2017-12-19 | Central Glass Company, Limited | Method for improving preservation stability of 2,2-difluoroacetaldehyde |
| WO2017030074A1 (ja) * | 2015-08-19 | 2017-02-23 | セントラル硝子株式会社 | α-フルオロアルデヒド類の製造方法 |
| CN107922301B (zh) * | 2015-08-19 | 2021-05-28 | 中央硝子株式会社 | α-氟代醛类的制造方法 |
| CN107922301A (zh) * | 2015-08-19 | 2018-04-17 | 中央硝子株式会社 | α‑氟代醛类的制造方法 |
| US10351502B2 (en) | 2016-01-21 | 2019-07-16 | Central Glass Company, Limited | Method for producing α,α-difluoroacetaldehyde |
| WO2017126233A1 (ja) * | 2016-01-21 | 2017-07-27 | セントラル硝子株式会社 | α,α-ジフルオロアセトアルデヒドの製造方法 |
| JP6195028B1 (ja) * | 2017-02-02 | 2017-09-13 | セントラル硝子株式会社 | α,α−ジフルオロアセトアルデヒドアルキルヘミアセタールの保存方法 |
| WO2018016126A1 (ja) | 2017-02-02 | 2018-01-25 | セントラル硝子株式会社 | α,α-ジフルオロアセトアルデヒドアルキルヘミアセタールの保存方法 |
| WO2018180610A1 (ja) * | 2017-03-31 | 2018-10-04 | セントラル硝子株式会社 | α,α-ジフルオロアセトアルデヒドヘミアセタールの製造方法 |
| JPWO2018180610A1 (ja) * | 2017-03-31 | 2020-02-06 | セントラル硝子株式会社 | α,α−ジフルオロアセトアルデヒドヘミアセタールの製造方法 |
| JP7032666B2 (ja) | 2017-03-31 | 2022-03-09 | セントラル硝子株式会社 | α,α-ジフルオロアセトアルデヒドヘミアセタールの製造方法 |
| US11370736B2 (en) | 2019-04-01 | 2022-06-28 | Triad National Security, Llc | Synthesis of fluoro hemiacetals via transition metal-catalyzed fluoro ester and carboxamide hydrogenation |
Also Published As
| Publication number | Publication date |
|---|---|
| CN107400044A (zh) | 2017-11-28 |
| EP2949639B1 (en) | 2018-01-10 |
| EP2949639A4 (en) | 2016-10-19 |
| EP2949639A1 (en) | 2015-12-02 |
| US20160340284A1 (en) | 2016-11-24 |
| CN107400044B (zh) | 2020-08-07 |
| US9765004B2 (en) | 2017-09-19 |
| JP6217653B2 (ja) | 2017-10-25 |
| JPWO2014115801A1 (ja) | 2017-01-26 |
| US20150329455A1 (en) | 2015-11-19 |
| CN104955794A (zh) | 2015-09-30 |
| US9440900B2 (en) | 2016-09-13 |
| CN104955794B (zh) | 2017-06-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6217653B2 (ja) | α,α−ジフルオロアセトアルデヒドの製造方法 | |
| JP5849710B2 (ja) | β−フルオロアルコール類の製造方法 | |
| JP6035918B2 (ja) | α−フルオロアルデヒド類の製造方法 | |
| CA2659153A1 (en) | Processes for preparing intermediate compounds useful for the preparation of cinacalcet | |
| JP5211876B2 (ja) | 高純度2’−トリフルオロメチルプロピオフェノンの製造方法 | |
| JP6669962B2 (ja) | α,α−ジフルオロアセトアルデヒドの製造方法 | |
| JP7032666B2 (ja) | α,α-ジフルオロアセトアルデヒドヘミアセタールの製造方法 | |
| JP2013028590A (ja) | アミノアルコール化合物の製造方法 | |
| JP6488638B2 (ja) | α−フルオロアルデヒド類等価体の製造方法 | |
| JP5854345B2 (ja) | 二量体の製造方法 | |
| JP2011116661A (ja) | フルオロアルキルエーテルの製造方法 | |
| JP2017039707A (ja) | α−フルオロアルデヒド類の製造方法 | |
| JP2011162494A (ja) | 4−ホルミルピペリジンアセタール誘導体の製造方法 | |
| JP5003072B2 (ja) | 3,3,3−トリフルオロプロピオンアルデヒドの製造方法 | |
| WO2017030074A1 (ja) | α-フルオロアルデヒド類の製造方法 | |
| JP2007077031A (ja) | 不飽和ビシナルジオール化合物の製造法 | |
| JP2016023159A (ja) | 長鎖ビシナルジオールの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14743082 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014558610 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14761774 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014743082 Country of ref document: EP |