WO2011102505A1 - 経口用徐放性固形製剤 - Google Patents
経口用徐放性固形製剤 Download PDFInfo
- Publication number
- WO2011102505A1 WO2011102505A1 PCT/JP2011/053643 JP2011053643W WO2011102505A1 WO 2011102505 A1 WO2011102505 A1 WO 2011102505A1 JP 2011053643 W JP2011053643 W JP 2011053643W WO 2011102505 A1 WO2011102505 A1 WO 2011102505A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- preparation
- preparation according
- component
- sustained
- drug
- Prior art date
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 146
- 238000013268 sustained release Methods 0.000 title abstract description 45
- 239000012730 sustained-release form Substances 0.000 title abstract description 45
- 239000007787 solid Substances 0.000 title description 6
- 239000003814 drug Substances 0.000 claims abstract description 95
- 229940079593 drug Drugs 0.000 claims abstract description 84
- 239000002202 Polyethylene glycol Substances 0.000 claims abstract description 26
- 229920001223 polyethylene glycol Polymers 0.000 claims abstract description 26
- 239000004014 plasticizer Substances 0.000 claims abstract description 20
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 claims abstract description 13
- 239000003405 delayed action preparation Substances 0.000 claims abstract description 10
- 238000002156 mixing Methods 0.000 claims abstract description 10
- IYKJEILNJZQJPU-UHFFFAOYSA-N acetic acid;butanedioic acid Chemical compound CC(O)=O.OC(=O)CCC(O)=O IYKJEILNJZQJPU-UHFFFAOYSA-N 0.000 claims abstract description 7
- 150000001875 compounds Chemical class 0.000 claims description 29
- -1 5-chloropyridin-2-yl Chemical group 0.000 claims description 28
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 claims description 25
- 239000011230 binding agent Substances 0.000 claims description 18
- 150000003839 salts Chemical class 0.000 claims description 18
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 claims description 18
- 150000007524 organic acids Chemical class 0.000 claims description 16
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 claims description 14
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 claims description 14
- 239000001863 hydroxypropyl cellulose Substances 0.000 claims description 14
- 239000001530 fumaric acid Substances 0.000 claims description 12
- HGVDHZBSSITLCT-JLJPHGGASA-N Edoxaban Chemical compound N([C@H]1CC[C@@H](C[C@H]1NC(=O)C=1SC=2CN(C)CCC=2N=1)C(=O)N(C)C)C(=O)C(=O)NC1=CC=C(Cl)C=N1 HGVDHZBSSITLCT-JLJPHGGASA-N 0.000 claims description 8
- DOOTYTYQINUNNV-UHFFFAOYSA-N Triethyl citrate Chemical compound CCOC(=O)CC(O)(C(=O)OCC)CC(=O)OCC DOOTYTYQINUNNV-UHFFFAOYSA-N 0.000 claims description 8
- 239000001069 triethyl citrate Substances 0.000 claims description 8
- 235000013769 triethyl citrate Nutrition 0.000 claims description 8
- VMYFZRTXGLUXMZ-UHFFFAOYSA-N triethyl citrate Natural products CCOC(=O)C(O)(C(=O)OCC)C(=O)OCC VMYFZRTXGLUXMZ-UHFFFAOYSA-N 0.000 claims description 8
- OGHNVEJMJSYVRP-UHFFFAOYSA-N carvedilol Chemical group COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=C1C1=CC=CC=C1N2 OGHNVEJMJSYVRP-UHFFFAOYSA-N 0.000 claims description 7
- 239000000783 alginic acid Substances 0.000 claims description 6
- 235000010443 alginic acid Nutrition 0.000 claims description 6
- 229920000615 alginic acid Polymers 0.000 claims description 6
- 229960001126 alginic acid Drugs 0.000 claims description 6
- 229940093429 polyethylene glycol 6000 Drugs 0.000 claims description 6
- 150000004781 alginic acids Chemical class 0.000 claims description 5
- KFDCQAWKWQKJRS-JBLDHEPKSA-N N'-[(1S,2R,4S)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]-4-(1,3,4-oxadiazol-2-yl)cyclohexyl]oxamide Chemical compound CN1CCc2nc(sc2C1)C(=O)N[C@@H]1C[C@H](CC[C@@H]1NC(=O)C(N)=O)c1nnco1 KFDCQAWKWQKJRS-JBLDHEPKSA-N 0.000 claims description 4
- 239000000203 mixture Substances 0.000 abstract description 40
- 230000000144 pharmacologic effect Effects 0.000 abstract description 6
- 210000003750 lower gastrointestinal tract Anatomy 0.000 abstract description 4
- 239000011159 matrix material Substances 0.000 description 42
- 238000007922 dissolution test Methods 0.000 description 37
- 230000007935 neutral effect Effects 0.000 description 37
- 238000009472 formulation Methods 0.000 description 33
- 238000000034 method Methods 0.000 description 30
- 239000002585 base Substances 0.000 description 27
- 238000004090 dissolution Methods 0.000 description 25
- 230000002378 acidificating effect Effects 0.000 description 22
- 239000000243 solution Substances 0.000 description 22
- 229920000642 polymer Polymers 0.000 description 21
- 230000001419 dependent effect Effects 0.000 description 20
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical group CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 18
- 239000012085 test solution Substances 0.000 description 17
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 16
- 239000008187 granular material Substances 0.000 description 16
- 238000010828 elution Methods 0.000 description 14
- 238000012360 testing method Methods 0.000 description 14
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 14
- 229920000639 hydroxypropylmethylcellulose acetate succinate Polymers 0.000 description 13
- 239000003921 oil Substances 0.000 description 13
- 239000007788 liquid Substances 0.000 description 12
- 239000002245 particle Substances 0.000 description 12
- 239000002253 acid Substances 0.000 description 10
- 239000003929 acidic solution Substances 0.000 description 10
- 239000003925 fat Substances 0.000 description 10
- 235000019197 fats Nutrition 0.000 description 10
- 238000005469 granulation Methods 0.000 description 10
- 230000003179 granulation Effects 0.000 description 10
- 238000002844 melting Methods 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- 229940075507 glyceryl monostearate Drugs 0.000 description 9
- 230000008018 melting Effects 0.000 description 9
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 9
- 238000001125 extrusion Methods 0.000 description 8
- 238000004519 manufacturing process Methods 0.000 description 8
- 229920003145 methacrylic acid copolymer Polymers 0.000 description 7
- 239000008118 PEG 6000 Substances 0.000 description 6
- 229920002584 Polyethylene Glycol 6000 Polymers 0.000 description 6
- 239000002775 capsule Substances 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid group Chemical group C(CC(O)(C(=O)O)CC(=O)O)(=O)O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 210000001035 gastrointestinal tract Anatomy 0.000 description 6
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 6
- 210000002784 stomach Anatomy 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 5
- ZUAAPNNKRHMPKG-UHFFFAOYSA-N acetic acid;butanedioic acid;methanol;propane-1,2-diol Chemical compound OC.CC(O)=O.CC(O)CO.OC(=O)CCC(O)=O ZUAAPNNKRHMPKG-UHFFFAOYSA-N 0.000 description 5
- 238000000137 annealing Methods 0.000 description 5
- 239000007864 aqueous solution Substances 0.000 description 5
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 5
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 5
- 229940117841 methacrylic acid copolymer Drugs 0.000 description 5
- 239000008194 pharmaceutical composition Substances 0.000 description 5
- 239000008363 phosphate buffer Substances 0.000 description 5
- 238000011084 recovery Methods 0.000 description 5
- 210000000813 small intestine Anatomy 0.000 description 5
- 238000003860 storage Methods 0.000 description 5
- KMZHZAAOEWVPSE-UHFFFAOYSA-N 2,3-dihydroxypropyl acetate Chemical compound CC(=O)OCC(O)CO KMZHZAAOEWVPSE-UHFFFAOYSA-N 0.000 description 4
- QNZCBYKSOIHPEH-UHFFFAOYSA-N Apixaban Chemical compound C1=CC(OC)=CC=C1N1C(C(=O)N(CC2)C=3C=CC(=CC=3)N3C(CCCC3)=O)=C2C(C(N)=O)=N1 QNZCBYKSOIHPEH-UHFFFAOYSA-N 0.000 description 4
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 4
- 229920000881 Modified starch Polymers 0.000 description 4
- 239000003086 colorant Substances 0.000 description 4
- 229920001577 copolymer Polymers 0.000 description 4
- 230000007423 decrease Effects 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 238000009826 distribution Methods 0.000 description 4
- PSMMNJNZVZZNOI-SJILXJHISA-N edoxaban tosylate hydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.N([C@H]1CC[C@@H](C[C@H]1NC(=O)C=1SC=2CN(C)CCC=2N=1)C(=O)N(C)C)C(=O)C(=O)NC1=CC=C(Cl)C=N1 PSMMNJNZVZZNOI-SJILXJHISA-N 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 239000011521 glass Substances 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 239000003112 inhibitor Substances 0.000 description 4
- 210000002429 large intestine Anatomy 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- 229960003511 macrogol Drugs 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 210000002438 upper gastrointestinal tract Anatomy 0.000 description 4
- CXKLIELNANLEIH-UHFFFAOYSA-N CC(=C)C(O)=O.CC=C(C)C(O)=O Chemical compound CC(=C)C(O)=O.CC=C(C)C(O)=O CXKLIELNANLEIH-UHFFFAOYSA-N 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- 235000013871 bee wax Nutrition 0.000 description 3
- 239000012166 beeswax Substances 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000004203 carnauba wax Substances 0.000 description 3
- 235000013869 carnauba wax Nutrition 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 239000012153 distilled water Substances 0.000 description 3
- 235000013305 food Nutrition 0.000 description 3
- 239000012458 free base Substances 0.000 description 3
- 230000005176 gastrointestinal motility Effects 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 3
- 239000011976 maleic acid Substances 0.000 description 3
- 239000000178 monomer Substances 0.000 description 3
- KGFYHTZWPPHNLQ-AWEZNQCLSA-N rivaroxaban Chemical compound S1C(Cl)=CC=C1C(=O)NC[C@@H]1OC(=O)N(C=2C=CC(=CC=2)N2C(COCC2)=O)C1 KGFYHTZWPPHNLQ-AWEZNQCLSA-N 0.000 description 3
- 230000002459 sustained effect Effects 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- 239000001856 Ethyl cellulose Substances 0.000 description 2
- 229920003134 Eudragit® polymer Polymers 0.000 description 2
- 108010074860 Factor Xa Proteins 0.000 description 2
- 229920003114 HPC-L Polymers 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 229920002701 Polyoxyl 40 Stearate Polymers 0.000 description 2
- 229920001800 Shellac Polymers 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 239000003146 anticoagulant agent Substances 0.000 description 2
- 229940127219 anticoagulant drug Drugs 0.000 description 2
- 229960003886 apixaban Drugs 0.000 description 2
- 229950011103 betrixaban Drugs 0.000 description 2
- XHOLNRLADUSQLD-UHFFFAOYSA-N betrixaban Chemical compound C=1C=C(Cl)C=NC=1NC(=O)C1=CC(OC)=CC=C1NC(=O)C1=CC=C(C(=N)N(C)C)C=C1 XHOLNRLADUSQLD-UHFFFAOYSA-N 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 2
- 239000008116 calcium stearate Substances 0.000 description 2
- 235000013539 calcium stearate Nutrition 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 229960000541 cetyl alcohol Drugs 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 230000001186 cumulative effect Effects 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 210000001198 duodenum Anatomy 0.000 description 2
- 229940088598 enzyme Drugs 0.000 description 2
- 235000019325 ethyl cellulose Nutrition 0.000 description 2
- 229920001249 ethyl cellulose Polymers 0.000 description 2
- GDCRSXZBSIRSFR-UHFFFAOYSA-N ethyl prop-2-enoate;2-methylprop-2-enoic acid Chemical compound CC(=C)C(O)=O.CCOC(=O)C=C GDCRSXZBSIRSFR-UHFFFAOYSA-N 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- 229960003943 hypromellose Drugs 0.000 description 2
- JEIPFZHSYJVQDO-UHFFFAOYSA-N iron(III) oxide Inorganic materials O=[Fe]O[Fe]=O JEIPFZHSYJVQDO-UHFFFAOYSA-N 0.000 description 2
- YOBAEOGBNPPUQV-UHFFFAOYSA-N iron;trihydrate Chemical compound O.O.O.[Fe].[Fe] YOBAEOGBNPPUQV-UHFFFAOYSA-N 0.000 description 2
- 238000004898 kneading Methods 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 239000011812 mixed powder Substances 0.000 description 2
- VYNKVNDKAOGAAQ-RUZDIDTESA-N n-[(1r)-2-[4-(1-methylpiperidin-4-yl)piperazin-1-yl]-2-oxo-1-phenylethyl]-1h-indole-6-carboxamide Chemical compound C1CN(C)CCC1N1CCN(C(=O)[C@H](NC(=O)C=2C=C3NC=CC3=CC=2)C=2C=CC=CC=2)CC1 VYNKVNDKAOGAAQ-RUZDIDTESA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 235000014593 oils and fats Nutrition 0.000 description 2
- 229940056211 paraffin Drugs 0.000 description 2
- 239000012188 paraffin wax Substances 0.000 description 2
- 239000008055 phosphate buffer solution Substances 0.000 description 2
- 229940099429 polyoxyl 40 stearate Drugs 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 229960001148 rivaroxaban Drugs 0.000 description 2
- 239000004208 shellac Substances 0.000 description 2
- 229940113147 shellac Drugs 0.000 description 2
- 235000013874 shellac Nutrition 0.000 description 2
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 2
- 235000012239 silicon dioxide Nutrition 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 230000036962 time dependent Effects 0.000 description 2
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical class OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- TYPXEIGMGZWOGB-UHFFFAOYSA-N 2-[2-(aminomethyl)phenyl]-n-[2-fluoro-4-(2-sulfamoylphenyl)phenyl]-5-(trifluoromethyl)pyrazole-3-carboxamide Chemical compound NCC1=CC=CC=C1N1C(C(=O)NC=2C(=CC(=CC=2)C=2C(=CC=CC=2)S(N)(=O)=O)F)=CC(C(F)(F)F)=N1 TYPXEIGMGZWOGB-UHFFFAOYSA-N 0.000 description 1
- MAOALPSHCIBFJZ-RUZDIDTESA-N 2-[[(2r)-2-[2-[[4-[amino(azaniumylidene)methyl]anilino]methyl]-1-methylbenzimidazol-5-yl]-1-oxo-1-pyrrolidin-1-ylpropan-2-yl]amino]acetate Chemical compound N=1C2=CC([C@@](C)(NCC(O)=O)C(=O)N3CCCC3)=CC=C2N(C)C=1CNC1=CC=C(C(N)=N)C=C1 MAOALPSHCIBFJZ-RUZDIDTESA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- VKPLPDIMEREJJF-UHFFFAOYSA-N 3-methoxybenzamide Chemical compound COC1=CC=CC(C(N)=O)=C1 VKPLPDIMEREJJF-UHFFFAOYSA-N 0.000 description 1
- RSWGJHLUYNHPMX-UHFFFAOYSA-N Abietic-Saeure Natural products C12CCC(C(C)C)=CC2=CCC2C1(C)CCCC2(C)C(O)=O RSWGJHLUYNHPMX-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- WLDHEUZGFKACJH-ZRUFZDNISA-K Amaranth Chemical compound [Na+].[Na+].[Na+].C12=CC=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(O)=C1\N=N\C1=CC=C(S([O-])(=O)=O)C2=CC=CC=C12 WLDHEUZGFKACJH-ZRUFZDNISA-K 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- SGHZXLIDFTYFHQ-UHFFFAOYSA-L Brilliant Blue Chemical compound [Na+].[Na+].C=1C=C(C(=C2C=CC(C=C2)=[N+](CC)CC=2C=C(C=CC=2)S([O-])(=O)=O)C=2C(=CC=CC=2)S([O-])(=O)=O)C=CC=1N(CC)CC1=CC=CC(S([O-])(=O)=O)=C1 SGHZXLIDFTYFHQ-UHFFFAOYSA-L 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- DMQVTIMACMUVQO-UHFFFAOYSA-N CCOC(C=C)=O.CCOC(C(C)=C)=O.CC(C(OC)=O)=C.CN(C)C Chemical compound CCOC(C=C)=O.CCOC(C(C)=C)=O.CC(C(OC)=O)=C.CN(C)C DMQVTIMACMUVQO-UHFFFAOYSA-N 0.000 description 1
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- CKLJMWTZIZZHCS-UHFFFAOYSA-N D-OH-Asp Natural products OC(=O)C(N)CC(O)=O CKLJMWTZIZZHCS-UHFFFAOYSA-N 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 229920003139 Eudragit® L 100 Polymers 0.000 description 1
- 229920003135 Eudragit® L 100-55 Polymers 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- CKLJMWTZIZZHCS-UWTATZPHSA-N L-Aspartic acid Natural products OC(=O)[C@H](N)CC(O)=O CKLJMWTZIZZHCS-UWTATZPHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- KWSAYQLNHUMQLP-ROSVLZJBSA-N N'-[(1S,2R,4S)-4-(dimethylcarbamoyl)-2-[(5-methyl-6,7-dihydro-4H-[1,3]thiazolo[5,4-c]pyridine-2-carbonyl)amino]cyclohexyl]oxamide 4-methylbenzenesulfonic acid hydrate Chemical compound O.Cc1ccc(cc1)S(O)(=O)=O.CN(C)C(=O)[C@H]1CC[C@H](NC(=O)C(N)=O)[C@@H](C1)NC(=O)c1nc2CCN(C)Cc2s1 KWSAYQLNHUMQLP-ROSVLZJBSA-N 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- YIKSCQDJHCMVMK-UHFFFAOYSA-N Oxamide Chemical compound NC(=O)C(N)=O YIKSCQDJHCMVMK-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical group OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- KHPCPRHQVVSZAH-HUOMCSJISA-N Rosin Natural products O(C/C=C/c1ccccc1)[C@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 KHPCPRHQVVSZAH-HUOMCSJISA-N 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 1
- YKTSYUJCYHOUJP-UHFFFAOYSA-N [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] Chemical compound [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] YKTSYUJCYHOUJP-UHFFFAOYSA-N 0.000 description 1
- 125000005396 acrylic acid ester group Chemical group 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- OENHQHLEOONYIE-UKMVMLAPSA-N all-trans beta-carotene Natural products CC=1CCCC(C)(C)C=1/C=C/C(/C)=C/C=C/C(/C)=C/C=C/C=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C OENHQHLEOONYIE-UKMVMLAPSA-N 0.000 description 1
- 229940024546 aluminum hydroxide gel Drugs 0.000 description 1
- WMGSQTMJHBYJMQ-UHFFFAOYSA-N aluminum;magnesium;silicate Chemical compound [Mg+2].[Al+3].[O-][Si]([O-])([O-])[O-] WMGSQTMJHBYJMQ-UHFFFAOYSA-N 0.000 description 1
- SMYKVLBUSSNXMV-UHFFFAOYSA-K aluminum;trihydroxide;hydrate Chemical compound O.[OH-].[OH-].[OH-].[Al+3] SMYKVLBUSSNXMV-UHFFFAOYSA-K 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- 150000007514 bases Chemical class 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 239000011648 beta-carotene Substances 0.000 description 1
- TUPZEYHYWIEDIH-WAIFQNFQSA-N beta-carotene Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CCCC1(C)C)C=CC=C(/C)C=CC2=CCCCC2(C)C TUPZEYHYWIEDIH-WAIFQNFQSA-N 0.000 description 1
- 235000013734 beta-carotene Nutrition 0.000 description 1
- 229960002747 betacarotene Drugs 0.000 description 1
- 238000005282 brightening Methods 0.000 description 1
- 229940067573 brown iron oxide Drugs 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000007765 cera alba Substances 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000000748 compression moulding Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- IJNIQYINMSGIPS-UHFFFAOYSA-N darexaban Chemical compound C1=CC(OC)=CC=C1C(=O)NC1=CC=CC(O)=C1NC(=O)C1=CC=C(N2CCN(C)CCC2)C=C1 IJNIQYINMSGIPS-UHFFFAOYSA-N 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- FSBVERYRVPGNGG-UHFFFAOYSA-N dimagnesium dioxido-bis[[oxido(oxo)silyl]oxy]silane hydrate Chemical compound O.[Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O FSBVERYRVPGNGG-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 210000003722 extracellular fluid Anatomy 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-N glutaric acid Chemical compound OC(=O)CCCC(O)=O JFCQEDHGNNZCLN-UHFFFAOYSA-N 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 230000023597 hemostasis Effects 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 235000012907 honey Nutrition 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- WTFXARWRTYJXII-UHFFFAOYSA-N iron(2+);iron(3+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[O-2].[Fe+2].[Fe+3].[Fe+3] WTFXARWRTYJXII-UHFFFAOYSA-N 0.000 description 1
- SZVJSHCCFOBDDC-UHFFFAOYSA-N iron(II,III) oxide Inorganic materials O=[Fe]O[Fe]O[Fe]=O SZVJSHCCFOBDDC-UHFFFAOYSA-N 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- 235000012245 magnesium oxide Nutrition 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 229910052919 magnesium silicate Inorganic materials 0.000 description 1
- 235000019792 magnesium silicate Nutrition 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical class [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 125000005397 methacrylic acid ester group Chemical group 0.000 description 1
- IWVKTOUOPHGZRX-UHFFFAOYSA-N methyl 2-methylprop-2-enoate;2-methylprop-2-enoic acid Chemical compound CC(=C)C(O)=O.COC(=O)C(C)=C IWVKTOUOPHGZRX-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- TZBAVQKIEKDGFH-UHFFFAOYSA-N n-[2-(diethylamino)ethyl]-1-benzothiophene-2-carboxamide;hydrochloride Chemical compound [Cl-].C1=CC=C2SC(C(=O)NCC[NH+](CC)CC)=CC2=C1 TZBAVQKIEKDGFH-UHFFFAOYSA-N 0.000 description 1
- GOQYKNQRPGWPLP-UHFFFAOYSA-N n-heptadecyl alcohol Natural products CCCCCCCCCCCCCCCCCO GOQYKNQRPGWPLP-UHFFFAOYSA-N 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- LJCNRYVRMXRIQR-UHFFFAOYSA-L potassium sodium tartrate Chemical compound [Na+].[K+].[O-]C(=O)C(O)C(O)C([O-])=O LJCNRYVRMXRIQR-UHFFFAOYSA-L 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 229940088417 precipitated calcium carbonate Drugs 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000012827 research and development Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- BTURAGWYSMTVOW-UHFFFAOYSA-M sodium dodecanoate Chemical compound [Na+].CCCCCCCCCCCC([O-])=O BTURAGWYSMTVOW-UHFFFAOYSA-M 0.000 description 1
- 229940082004 sodium laurate Drugs 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- KHPCPRHQVVSZAH-UHFFFAOYSA-N trans-cinnamyl beta-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OCC=CC1=CC=CC=C1 KHPCPRHQVVSZAH-UHFFFAOYSA-N 0.000 description 1
- UJMBCXLDXJUMFB-UHFFFAOYSA-K trisodium;5-oxo-1-(4-sulfonatophenyl)-4-[(4-sulfonatophenyl)diazenyl]-4h-pyrazole-3-carboxylate Chemical compound [Na+].[Na+].[Na+].[O-]C(=O)C1=NN(C=2C=CC(=CC=2)S([O-])(=O)=O)C(=O)C1N=NC1=CC=C(S([O-])(=O)=O)C=C1 UJMBCXLDXJUMFB-UHFFFAOYSA-K 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- OENHQHLEOONYIE-JLTXGRSLSA-N β-Carotene Chemical compound CC=1CCCC(C)(C)C=1\C=C\C(\C)=C\C=C\C(\C)=C\C=C\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C OENHQHLEOONYIE-JLTXGRSLSA-N 0.000 description 1
Images






Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
- A61K47/38—Cellulose; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/14—Esters of carboxylic acids, e.g. fatty acid monoglycerides, medium-chain triglycerides, parabens or PEG fatty acid esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/34—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyesters, polyamino acids, polysiloxanes, polyphosphazines, copolymers of polyalkylene glycol or poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
Definitions
- the present invention relates to a matrix granular preparation that reliably exerts the main pharmacological effect by oral administration once or twice a day.
- Sustained-release preparations for adjusting the blood concentration of drugs are useful in terms of separation of main pharmacological effects and side effects, improvement in medication compliance such as reduction in the number of administrations due to improved drug efficacy, and medical economics. high. For this reason, several techniques relating to sustained-release preparations have been reported. On the other hand, since the chemical properties of compounds that exhibit the main pharmacological action are diverse, several sustained-release techniques for adapting to the various chemical properties of these compounds have been reported, but still insufficient (For example, refer to Patent Documents 1 and 2).
- An acidic drug refers to an acidic compound in which a free form (a free form that does not form a salt with an acidic group of the drug, such as an addition salt of an alkali or an amine) shows acidity.
- a free form a free form that does not form a salt with an acidic group of the drug, such as an addition salt of an alkali or an amine
- the solubility in the upper part of the gastrointestinal tract such as the stomach is low, and in the case of a salt of an acidic compound (such as an alkali or amine addition salt), the problem is that it becomes a free acid with low solubility in an acidic solution. It is as.
- a basic drug refers to a basic compound in which a free form (a free form such as an acid addition salt that does not form a salt with the basic group of the drug) shows basicity, and dissolves well in a strongly acidic aqueous solution.
- a neutral aqueous solution such as a neutral buffer decreases the solubility. That is, when a basic drug is administered orally, it shows good solubility in an acidic stomach, but the solubility in the lower part of the digestive tract, such as the large intestine, which is neutral and has little water, is greatly reduced and the absorption rate is reduced. Concerned.
- sustained-release preparations for oral administration of basic drugs
- the preparations collapse due to coexistence with food in the acidic environment of the upper gastrointestinal tract, which exhibits high water solubility, and mechanical stress due to gastrointestinal motility, etc.
- the overdose of the drug that occurs at the time is a problem.
- the formulation strength is increased by increasing the sustained release base to avoid overdose of the drug
- To maintain the absorption remains as a problem of sustained-release preparations.
- we are satisfied with the sustained release of basic drugs that can avoid excessive drug release in acidic environments such as the upper gastrointestinal tract and can simultaneously achieve sustained elution in the lower gastrointestinal tract, which is a neutral environment. There is nothing you can do.
- the present inventors have found that a pharmacologically active drug, a pH-dependent polymer base, a plasticizer, and low melting point oils and fats are contained in an acidic environment.
- the present invention was completed by finding a sustained-release matrix granular preparation that avoids dose dumping of the drug underneath and improves neutral dissolution.
- the present invention relates to the following (1) to (44).
- the content of the (B) pH-dependent polymer base in the granular granular preparation in the granular preparation is in the range of 5 to 90% by weight, and any one of (1) to (3) The sustained-release solid preparation described.
- Formulation. (8) The sustained-release matrix granular preparation according to any one of (1) to (7), wherein (D) the low melting point oil or fat is glyceryl monostearate or polyethylene glycol.
- (21) (A) The sustained-release matrix granular preparation according to any one of (1) to (17), wherein the pharmacologically active drug exhibits the following solubility: The lowest solubility in a neutral state (range 7.5>pH> 5) is 0.5 mg / ml or less.
- the pharmacologically active drug is the following ( ⁇ ) -1- (carbazol-4-yloxy) -3-[[2- (o-methoxyphenoxy) ethyl] amino] -2-propanol ; N 1- (5-chloropyridin-2-yl) -N 2 -((1S, 2R, 4S) -4-[(dimethylamino) carbonyl] -2- ⁇ [(5-methyl-4,5,6 , 7-tetrahydrothiazolo [5,4-c] pyridin-2-yl) carbonyl] amino ⁇ cyclohexyl) ethanediamide; and N 1- (5-chloropyridin-2-yl) -N 2 -[(1S, 2R , 4S) -2- ⁇ [(5-Methyl-4,5,6,7-tetrahydrothiazolo [5,4-c] pyridin-2-yl) carbonyl]
- the content of (A) the pharmacologically active drug in the matrix granular preparation in the granular preparation is in the range of 0.1 to 50% by weight, according to any one of (1) to (23) Sustained release solid preparation.
- the sustained-release matrix granular preparation according to any one of (1) to (25), which is 60% or less after 4 hours.
- (29) The preparation according to (28), wherein the content of component (B) in the preparation is 50 to 90% by weight.
- (31) The preparation according to any one of (28) to (30), wherein the component (C) is triethyl citrate.
- the component (A) is ( ⁇ ) -1- (carbazol-4-yloxy) -3-[[2- (o-methoxyphenoxy) ethyl] amino] -2-propanol, N 1- (5-chloropyridin-2-yl) -N 2 -((1S, 2R, 4S) -4-[(dimethylamino) carbonyl] -2- ⁇ [(5-methyl-4,5,6 , 7-tetrahydrothiazolo [5,4-c] pyridin-2-yl) carbonyl] amino ⁇ cyclohexyl) ethanediamide, and N 1- (5-chloropyridin-2-yl) -N 2 -[(1S, 2R, 4S) -2- ⁇ [(5-methyl-4,5,6,7-tetrahydrothiazolo [5,4 -C] pyridin-2-yl) carbonyl] amino ⁇ -4-([1,3,4]
- a sustained-release matrix granular preparation for oral administration containing a pharmacologically active drug represented by Compound (1).
- the sustained-release matrix granular preparation of the present invention has good strength to prevent excessive release in an acidic solution and good dissolution in a neutral solution, so that it extends from the duodenum and small intestine to the lower gastrointestinal tract. , The effect of maintaining the continuous elution of the contained pharmacologically active drug can be obtained.
- the term “acidic solution” means an acidic dissolution test solution used for evaluating dissolution at the upper digestive tract such as the stomach.
- the dissolution test first solution described in the Japanese Pharmacopoeia, US USP 0.1 N hydrochloric acid, 0.01 N hydrochloric acid, Simulated Gastric Fluid without Enzyme and the like described in the pharmacopoeia can be mentioned, but the acidic dissolution test solution is not limited to these.
- the “neutral solution” in the present specification includes, for example, the second dissolution test solution described in the Japanese Pharmacopoeia and phosphorus as the neutral dissolution test solution used for evaluating the dissolution property of the drug in the small intestine, the large intestine and the like.
- the dissolution test solution is not limited to these.
- the above dissolution test solution is prepared by the method described in the pharmacopoeia of each country.
- the pH of these dissolution test solutions is preferably within ⁇ 0.05 of the pH specified for each dissolution test solution when the dissolution test solution is a buffer solution.
- the paddle method using an acidic eluate for evaluating the dissolution property of the sustained-release matrix granular preparation of the present invention in the upper digestive tract is, for example, at 37 ⁇ 0.5 ° C. in 0.01 N hydrochloric acid (900 mL).
- the paddle method includes a method of performing a dissolution test for 2 hours at 50 and 200 revolutions per minute.
- the pharmacologically active drug in the preparation is a basic drug
- the preparation is caused by mechanical stress due to coexistence with food in the acidic environment of the upper gastrointestinal tract showing high water solubility, gastrointestinal motility, etc.
- the average dissolution rate of the pharmacologically active drug in the dissolution test solution can maintain the strength of the preparation and keep the dissolution rate within a certain range even at 200 rotations per minute of the paddle method and / or 50 rotations per minute of the paddle method. Is preferred.
- the difference in the average dissolution rate of pharmacologically active drugs in the dissolution test solution (200 rotations per minute per paddle method-50 rotations per paddle method) when the dissolution test method is performed for 2 hours is preferably 15% or less. 10% or less is more preferable; 5% or less is particularly preferable.
- the ratio of the average dissolution rate of the pharmacologically active drug in the dissolution test solution after 2 hours is preferably 2.0 or less; 1.5 or less Is more preferable; 1.3 or less is particularly preferable.
- USP Apparatus 3 which is a dissolution test method under conditions close to the environment in the human digestive tract, may be used.
- the drug concentration in the solution can be measured using the conditions (test solution, shaking speed, and measurement time) shown in Examples described later.
- the average dissolution rate and dissolution time of a pharmacologically active drug in the dissolution test solution can be calculated using a UV method or the like.
- the “average dissolution rate” may be determined by measuring the dissolution rates of at least 2, preferably 6, and more preferably 12 for one kind of solid preparation, and calculating the average value thereof.
- a drug that exerts a main pharmacological effect in the formulation of a preparation and is relatively low in water solubility is preferable.
- a pharmacologically active drug, a neutral compound means a compound that does not have a group that ionizes and dissociates even in an acidic state or basic state in the molecule, and an acidic compound includes a carboxy group, phenol It means a drug having an acidic group typified by a functional hydroxyl group, phosphoric acid group, sulfonic acid, tetrazolyl group and the like.
- the basic drug means a drug having a basic nitrogen atom represented by amino group, piperidinyl group, piperazinyl group and the like in the molecule.
- a basic drug is particularly suitable.
- Basic drugs have a physicochemical property in which the solubility in the neutral state (7.5>pH> 5) in the small and large intestines is lower than the solubility in the acidic state (pH ⁇ 2) such as the stomach.
- the basic drug refers to a drug whose solubility in the neutral state is lower than the solubility in the acidic state, and the ratio of the decrease in solubility in the neutral state includes the following ranges: Can do.
- (Neutral state solubility) / (Acid state solubility) preferably ranges from 0.00001 to 0.6; (Neutral state solubility) / (Acid state solubility) is more preferably in the range of 0.001 to 0.5; The range of (solubility in neutral state) / (solubility in acidic state) is more preferably in the range of 0.01 to 0.1, but is not limited to these ranges.
- the solubility is more preferably in the range of 10 to 500 ⁇ g / ml.
- the absolute value of the solubility is preferably a drug whose minimum solubility in a neutral state (in the range of 7.5>pH> 5) is reduced to 3 mg / ml or less; more preferably a drug which decreases to 1 mg / ml or less; More preferred is a drug that lowers to 0.5 mg / ml or less.
- pharmacologically active drugs include anticoagulants.
- an activated blood coagulation factor X (FXa) inhibitor is preferable, and specific examples of the FXa inhibitor include the following (a) to (l).
- FXa inhibitor include the following (a) to (l).
- R 1 represents an N, N-dimethylcarbamoyl group or a [1,3,4] oxadiazol-2-yl group.
- the compound represented by the formula [hereinafter sometimes abbreviated as compound (1)] is more preferred.
- Compound (1) may be a free form (free base), a pharmacologically acceptable salt thereof, or a hydrate thereof.
- the salt of the compound represented by the formula (1) include hydrochloride, sulfate, hydrobromide, hydroiodide, phosphate, nitrate, benzoate, methanesulfonate, 2-hydroxyethane.
- maleic acid, hydrochloride, methanesulfonate, and p-toluenesulfonate are preferable, and maleic acid and p-toluenesulfonate are particularly preferable.
- N 1- 5-chloropyridin-2-yl) -N 2 -[(1S, 2R, 4S) -2- ⁇ [(5-methyl -4,5,6,7-tetrahydrothiazolo [5,4-c] pyridin-2-yl) carbonyl] amino ⁇ -4-([1,3,4] oxadiazol-2-yl) cyclohexyl] Ethanediamide monomaleate;
- N 1- (5-chloropyridin-2-yl) -N 2 -((1S, 2R, 4S) -4-[(dimethylamino) carbonyl] -2- ⁇ [(5-methyl-4,5,6 , 7-tetrahydrothiazolo [5,4-c] pyridin-2-yl) carbonyl] amino ⁇ cyclohexyl) ethanediamide;
- N 1- (5-chloropyridin-2-yl) -N 2 -[(1S,
- These compounds (1) can be produced by a method described in the literature or a method analogous thereto (WO2003-000657 pamphlet; WO2003-000680 pamphlet; WO2003-016302 pamphlet; WO2004-058715 pamphlet).
- the free base (free form) of the compound (1) is, for example, the above-mentioned salt (acid addition salt) and / or acid addition salt “acid” and water forming a hydrate. This means a compound excluding “water” in a Japanese product.
- the free base (free form) of compound (1a) and compound (1b) means the following formula (1a-1) and formula (1b-1)
- Examples of “(B) pH-dependent polymer base” in the present invention include polymer bases exhibiting pH-dependent dissolution characteristics used in the pharmaceutical field.
- the “pH-dependent polymer base” can be further divided into an enteric base and a gastric base, and an enteric base is preferable.
- As the enteric base those which are difficult to dissolve in a pH environment such as in the stomach and gradually dissolve in a neutral pH environment such as the small intestine and large intestine which are main absorption sites are preferable.
- the pH-dependent polymer base include the following (1) to (3).
- Methacrylic acid copolymer Here, the methacrylic acid copolymer means a copolymer of two or more monomers selected from the group consisting of methacrylic acid, methacrylic acid ester, acrylic acid and acrylic acid ester, It is not limited by the combination of the said monomers, the number of used monomers, etc.
- Hydroxypropyl methylcellulose acetate succinate (3) Carboxymethyl ethyl cellulose.
- the “(B) pH-dependent polymer base” of the present invention is preferably (1) a methacrylic acid copolymer; and (2) hydroxypropylmethylcellulose acetate succinate; and hydroxypropylmethylcellulose acetate succinate. More preferred.
- Methacrylic acid copolymers include methacrylic acid-methyl methacrylic acid copolymer, (ethyl acrylate-methyl methacrylate-trimethylammonium ethyl methacrylate) copolymer, (methacrylic acid-ethyl acrylate) copolymer, (methacrylic acid) -Methyl methacrylate) copolymers are preferred; methacrylic acid-methyl methacrylic acid copolymers are more preferred.
- methacrylic acid-methyl methacrylic acid copolymer examples include Eudragit (EUDRAGIT), and commercially available products include Eudragit L100-55 and Eudragit L100.
- (2) Hydroxypropyl methylcellulose acetate succinate (HPMCAS) can be purchased from Shin-Etsu Chemical Co., Ltd. as AQOAT (trade name), for example. HPMCAS grades are available in LF, MF, HF, LG, MG and HG, with MF and HF grades being preferred.
- the content of (B) the pH-dependent polymer base in the matrix granular preparation of the present invention is preferably in the range of 5 to 90%; more preferably in the range of 50 to 80%.
- plasticizer (C) in the present invention those used as pharmaceutical additives can be used. Tri (C1 to C6 alkyl) citrate or glycerin monoorganic acid ester is preferred; triethyl citrate And glycerin monoacetate is more preferred; triethyl citrate is particularly preferred.
- the content of the plasticizer (C) in the granular preparation of the present invention is preferably in the range of 1 to 30%; more preferably in the range of 3 to 15%.
- the matrix sustained-release granular preparation of the present invention in addition to the above (A) pharmacologically active drug, (B) pH-dependent polymer base, and (C) plasticizer, (D) low melting point It is characterized by containing fats and oils.
- examples of the (D) low melting point fats and oils in the present invention include glyceryl monostearate and polyethylene glycol; Preferred are glyceryl monostearate and polyethylene glycol; Polyethylene glycol is more preferred.
- the polyethylene glycol is preferably polyethylene glycol having an average molecular weight in the range of 4000 to 12000; more preferably having an average molecular weight of 7000 to 10500. More specifically, polyethylene glycol 6000 (CAS number: 25322-68-3) is preferable as the polyethylene glycol.
- the content of the (D) low melting point fats and oils in the granular preparation of the present invention is preferably in the range of 0.1 to 20%; more preferably in the range of 1 to 6%.
- the matrix sustained-release granular preparation of the present invention may further contain one or more selected from a binder and an organic acid.
- a cellulose derivative is preferable.
- the cellulose derivative include hypromellose (HPMC: hydroxypropylmethylcellulose), hydroxypropylcellulose (HPC), ethylcellulose, methylcellulose and the like; hypromellose and hydroxypropylcellulose are preferable; hydroxypropylcellulose is more preferable.
- HPMC hydroxypropylmethylcellulose
- HPC hydroxypropylmethylcellulose
- HPC hydroxypropylcellulose
- HPC hydroxypropylcellulose
- HPC hydroxypropylcellulose
- HPC hydroxypropylcellulose
- HPC hydroxypropylcellulose
- ethylcellulose methylcellulose and the like
- hypromellose and hydroxypropylcellulose are preferable
- hydroxypropylcellulose is more preferable.
- hydroxypropyl cellulose grades such as L, SL and SSL can be used, and those of L (6.0 to 10 mPa ⁇ s) grade are preferred.
- L 6.0 to 10 mPa ⁇ s
- hydroxypropylcellulose is used as a binding solution, it is usually dissolved in an organic solvent such as water or alcohol and used as a solution.
- the content of the binder in the granular preparation of the present invention is preferably in the range of 0 to 10%; more preferably in the range of 0 to 2%.
- organic acid in the present specification fumaric acid, succinic acid, alginic acid, adipic acid, citric acid, L-aspartic acid, malonic acid, maleic acid, DL-malic acid and tartaric acid are preferable; fumaric acid and alginic acid are more preferable. Fumaric acid is particularly preferred.
- the content of the organic acid in the granular preparation of the present invention is preferably in the range of 0 to 40%; more preferably in the range of 0 to 20%.
- the matrix sustained-release granular preparation includes (A) a pharmacologically active drug, (B) a pH-dependent polymer base, (C) a plasticizer, and (D low-melting point fats and oils, You may mix
- Lubricants include cocoa butter, carnauba wax, hydrous silicon dioxide, dry aluminum hydroxide gel, glycerin fatty acid ester, magnesium silicate, light anhydrous silicic acid, hydrogenated oil, synthetic aluminum silicate, white beeswax, magnesium oxide, sodium tartrate Potassium, sucrose fatty acid ester, stearic acid, calcium stearate, magnesium stearate, stearyl alcohol, polyoxyl 40 stearate, cetanol, soybean hardened oil, gelatin, talc, magnesium carbonate, precipitated calcium carbonate, corn starch, potato starch, fumaric acid
- Examples include sodium stearyl, beeswax, magnesium aluminate metasilicate, sodium laurate, and magnesium sulfate.
- Coloring agents include yellow iron sesquioxide, iron sesquioxide, titanium oxide, orange essence, brown iron oxide, ⁇ -carotene, black iron oxide, edible blue No. 1, edible blue No. 2, edible red No. 2, edible red 3 No., Edible Red No. 102, Edible Yellow No. 4, Edible Yellow No. 5 and the like.
- Examples thereof include medium gold foil, white shellac, paraffin, povidone, macrogol 1500, macrogol 4000, macrogol 6000, beeswax, glyceryl monostearate, and rosin.
- the matrix sustained-release granular preparation of the present invention is not particularly limited as long as it is a granular preparation that can be administered orally, but is preferably a matrix granule. These matrix sustained-release granular preparations are preferably used as capsules filled in capsules.
- the pharmaceutical composition of the present invention comprises (A) a pharmacologically active drug, (B) a pH-dependent polymer base, (C) a plasticizer, (D) a low melting point oil and fat, and if necessary. , One or more selected from binders and organic acids, disintegrating agents, binders, fluidizers, lubricants, colorants, brighteners, etc., and generally used granular preparations It can manufacture by the manufacturing method of.
- the present invention also relates to a method for producing the above sustained release granular pharmaceutical composition, which will be described in detail below.
- the sustained-release granular pharmaceutical composition for oral administration of the present invention comprises the following (E): a step of preparing a [mixture] by mixing a pharmacologically active drug, a pH-dependent polymer base, and fats and oils ; (F): a step of adding a plasticizer, an aqueous solution of a binder and distilled water and stirring to prepare a [dispersion]; (G): a step of producing [kneaded product] from [mixed powder] and [dispersion]; and (H): a step of extruding and granulating [kneaded product] to produce [granule]; It can be produced by the steps (E) to (H).
- the pH-dependent polymer base is preferably hydroxypropyl methylcellulose acetate succinate; the plasticizer is preferably triethyl citrate; the plasticizer is highly pH-dependent. It is preferably used in the range of 5 to 30% by weight with respect to the molecular base.
- Said process (H) is a process of manufacturing [granule] by extruding and granulating [kneaded material]. Here, after extruding [kneaded product], granulation may be performed using a granulator or the like. To produce [granule] from the granulation, annealing is performed using a normal dryer. (Heat treatment) may be performed.
- examples of the dryer include a fluidized bed granulation dryer and a shelf dryer.
- the annealing temperature (heat treatment) and the treatment time may be appropriately changed depending on the drug used.
- the specific annealing (heat treatment) temperature and treatment time it is preferable that the temperature inside the annealing (heat treatment) apparatus is preferably in the range of 40 to 90 ° C .; The range of 15 minutes to 2 hours is preferable, but the range is not limited to this range.
- the obtained [granule] should just arrange a particle size suitably using a sieve.
- the content ratio of the pharmacologically active drug in the pharmaceutical composition of the present invention in the present invention is usually preferably 0.1 to 50% by weight in terms of free form of the pharmacologically active drug, preferably 10 to 25% by weight. % Is more preferable.
- the dosage form of the pharmaceutical composition of the present invention is preferably a capsule filled with granules.
- the content of the pharmacologically active drug contained per capsule is, for example, a compound ( In the case of 1), in terms of free form, it is usually 0.5 to 500 mg; preferably 1 to 100 mg; more preferably 5 to 75 mg; and particularly preferably 15 to 60 mg.
- the sustained-release preparation of the present invention comprises (A) a pharmacologically active drug, (B) hydroxypropylmethylcellulose acetate succinate, (C) a plasticizer, and (D) polyethylene glycol, followed by extrusion granulation. Is a sustained-release preparation.
- the preparation of the present invention is characterized by being obtained by extrusion granulation after mixing the component (A), the component (B), the component (C) and the component (D), for example, the component (A), A preparation obtained by mixing the component (C) and the component (D) and then extruding and granulating the preparation and coating the composition containing the component (B) is not included in the present invention.
- a preparation obtained by mixing the component (A), the component (B), the component (C) and the component (D) and then extruding and granulating the formulation containing the component (B) are encompassed by the present invention.
- Examples of the “pharmacologically active drug” as the component (A) used in the preparation of the present invention include the above-mentioned compounds, and are prodrugs that are converted into pharmacologically active drugs in the body. Also good.
- the content of the component (A) in the preparation of the present invention is preferably 0.1 to 60% by weight, more preferably 1 to 50% by weight, and even more preferably 5 to 35% by weight.
- HPMCAS Hydropropyl methylcellulose acetate succinate
- the average particle diameter (D 50 ) of HPMCAS which is the component (B) used in the preparation of the present invention, is preferably 50 ⁇ m or less, more preferably 20 ⁇ m or less, and even more preferably 10 ⁇ m or less. Particularly preferably, it is 5 ⁇ m or less.
- D 50 means the particle diameter corresponding to the median of the cumulative distribution curve when measured using HELOS (Japan Laser Co., Ltd.), which is a laser diffraction measurement device, The median diameter.
- D 90 refers to a particle diameter corresponding to 90% of the cumulative distribution curve when measured using the HELOS.
- D 90 of 20 ⁇ m means that 90% of the measured powder has a particle size of 20 ⁇ m or less, and the remaining 10% has a particle size of more than 20 ⁇ m.
- the content of the component (B) in the preparation of the present invention is preferably 40 to 90% by weight, more preferably 50 to 80% by weight, and even more preferably 60 to 75% by weight.
- Examples of the “plasticizer” as the component (C) used in the preparation of the present invention include the above-mentioned substances, and preferably triethyl citrate.
- the content of the component (C) in the preparation of the present invention is preferably 5 to 30% by weight.
- Examples of the “polyethylene glycol (PEG)” that is the component (D) used in the preparation of the present invention include the above-mentioned substances, preferably PEG having an average molecular weight of 4000 to 12000, and PEG having an average molecular weight of 7000 to 10500. Is more preferable.
- An example of such a PEG having a molecular weight is polyethylene glycol 6000 (CAS number: 25322-68-3).
- the content of the component (D) in the preparation of the present invention is preferably 1 to 10% by weight, more preferably 2 to 8% by weight, and even more preferably 3 to 6% by weight.
- the preparation of the present invention may further contain a binder and / or an organic acid in addition to the components (A) to (D).
- a binder the above-mentioned substances can be mentioned, and hydroxypropylcellulose or hydroxypropylmethylcellulose is preferable, and hydroxypropylcellulose is more preferable.
- the organic acid include the above-mentioned substances, fumaric acid or alginic acid is preferable, and fumaric acid is more preferable.
- the preparation of the present invention contains a binder and / or an organic acid
- the content of the binder in the preparation is preferably from 0.1 to 10% by weight, more preferably from 0.5 to 2% by weight.
- the acid content is preferably 0.1 to 40% by weight, and more preferably 1 to 20% by weight.
- additives such as the above-mentioned lubricants, colorants, and brighteners may be further blended within a range that does not affect the effects of the present invention.
- the preparation of the present invention is manufactured by extruding and granulating after mixing (A) to (D). More specifically, the components (A), (B), (C) and (D) are mixed, a solution containing a binder or distilled water is added to the mixture, and then kneaded and extruded. Can be granulated and manufactured.
- the obtained preparation may be compression molded as necessary, or may be coated. Mixing, kneading, extrusion granulation, compression molding, coating, and the like may be performed using methods well known in the art. When other additives are blended in the preparation of the present invention, they may be blended in any of the mixing step, kneading step, tableting step or coating step.
- each component is preferably in a matrix form.
- the preparation of the present invention thus obtained has good tablet strength to prevent excessive release in an acidic solution and good dissolution in a neutral solution, as described in the above-mentioned effects of the invention. From the duodenum and the small intestine to the lower gastrointestinal tract, the effect of maintaining the continuous elution of the contained pharmacologically active drug can be obtained.
- PEG 6000 polyethylene glycol 6000 -HPC-L: Hydroxypropylcellulose L grade (manufactured by Nippon Soda
- the recovery rate of the formulation is: (Weight of granule having specific particle size distribution among granulated products obtained by extrusion granulation) / (Weight of kneaded product) ⁇ 100 When it was 80% or more, it was evaluated as ⁇ , and when it was less than 80%, it was evaluated as ⁇ .
- the dissolution test in an acidic solution or a neutral solution was performed as follows. (Dissolution test in acidic solution) The dissolution test was performed at 37 ⁇ 0.5 ° C. in 0.01 N hydrochloric acid (900 mL) at a paddle method at 50 rpm, and the average drug dissolution rate in the eluate was calculated over time.
- Example 1 For Formulations 1 to 4 shown in Table 1, compound (1a) pulverized product and HPMCAS-HF1 were mixed in a vinyl bag for 5 minutes, and 40 mL of 5% HPC-L aqueous solution in which triethyl citrate was dispersed was obtained. And 90 mL of purified water was added and kneaded for 5 minutes with a high speed mixer. The obtained kneaded product was extruded and granulated with an extrusion granulator (dome gran, screen diameter: 1.0 mm ⁇ ), and annealed (80 ° C., 2 hr) with a shelf dryer. The granulated product after annealing was sieved with No. 14 and No. 18 sieves, and granules under No. 14 No. 14 sieve were collected to prepare a preparation. The recovery rate of the preparation is shown in Table 2, and the results of the dissolution test with an acidic solution are shown in FIG.
- Formulations with good recovery were Formulations 2 to 4 to which partially pregelatinized starch, glyceryl monostearate or PEG 6000 was added. However, as shown in FIG. 1, Formula 2 to which partially pregelatinized starch was added had a higher drug dissolution rate in the acidic solution than Formula 1, Formula 3 and Formula 4.
- Example 2 For the formulations 5 to 8 shown in Table 3, a granulated product was prepared in the same manner as in Example 1. The obtained granulated product was sieved with No. 14 and No. 30 sieves, and No. 14 on the No. 30 sieve. The granules were collected to prepare a preparation. Table 4 shows the recovery rate of the preparation. The results of the dissolution test with an acidic solution are shown in FIG.
- Formulations with good recovery were Formulations 6-8 with partially pregelatinized starch, glyceryl monostearate or PEG 6000 added. However, as shown in FIG. 2, in Formulation 6 to which partially pregelatinized starch was added, the drug dissolution rate in the acidic solution was faster than Formulations 5, Formulations 7 and 8.
- Example 3 For the formulations 9 and 10 shown in Table 5, a granulated product was prepared in the same manner as in Example 1. The obtained granulated product was sieved with No. 14 and No. 18 sieves, and No. 14 on the No. 18 sieve. The granules were collected to prepare a preparation. The obtained formulation was sealed in a glass bottle and stored at 40 ° C., 50 ° C. or 60 ° C. for 1 day or 1 week. The preparation immediately after preparation and the preparation after storage were subjected to a dissolution test with a neutral solution, and the results are shown in FIGS.
- Example 4 About the prescription 7 and the prescription 8 shown in Table 3, the obtained preparation was put into the glass bottle, was sealed, and was stored at 50 degreeC for 3 days. The preparation immediately after preparation and the preparation after storage were subjected to a dissolution test with a neutral solution. As a result, formulation 7 added with glyceryl monostearate delayed the dissolution of the drug by storage at 50 ° C. for 3 days, and formulation 8 added with PEG 6000 changed the dissolution behavior even after storage at 50 ° C. for 3 days. Was hardly recognized.
- Example 5 For the formulation 11 shown in Table 6, a granulated product was prepared in the same manner as in Example 1. The obtained granulated product was sieved with No. 14 and No. 30 sieves, and the granules under No. 14 No. 14 sieve were obtained. Was collected to prepare a preparation. The obtained preparation was subjected to a dissolution test in a neutral solution, and the results are shown in FIG.
- Example 6 For the formulation 12 shown in Table 7, a granulated product was prepared in the same manner as in Example 1 (extruded granulated screen diameter was 1.2 mm ⁇ ), and the resulting granulated product was sieved with Nos. 12 and 18 sieves. The granules under No. 12 on No. 18 sieve were collected to prepare a preparation. The obtained preparation was subjected to a dissolution test in a neutral solution, and the results are shown in FIG.
- Example 7 For the formulations 13 and 14 shown in Table 8, a granulated product was prepared in the same manner as in Example 1. The obtained granulated product was sieved with No. 14 and No. 30 sieves, and No. 14 on the No. 30 sieve. The granules were collected to prepare a preparation. The resulting formulation was filled into hydroxypropyl methylcellulose capsules.
- the present invention can be used for the production of a sustained-release matrix granular preparation for oral administration containing a pharmacologically active drug such as compound (1).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Inorganic Chemistry (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
薬物の過量放出を回避し、消化管下部における薬剤の溶出性を改善することにより、1日1回又は2回の経口投与で確実に主薬理効果を発揮する経口投与用の徐放性粒状製剤を提供する。 (A)薬理上活性な薬物、 (B)ヒドロキシプロピルメチルセルロースアセテートサクシネート、 (C)可塑剤、及び、 (D)ポリエチレングリコール、 を混合した後、押し出し造粒して得られる徐放性製剤。
Description
本発明は、1日1回又は2回の経口投与で確実に主薬理効果を発揮する、マトリックス粒状製剤に関する。
薬物の血中濃度を調節するための徐放製剤は、主薬理作用と副作用の分離、薬効の持続性向上による投与回数の削減などの服用コンプライアンス向上、さらに医療経済性などの点から有用性が高い。このことから、徐放製剤に関する技術がいくつかの報告されている。一方、主薬理作用を発現する化合物の化学的性質が多様であることから、それらの化合物の多様な化学的性質に適応するための徐放技術がいくつか報告されているが、未だ十分なものではない(例えば、特許文献1、2参照)。
薬物それ自体の性質としては、大きく中性、酸性、塩基性に大別することができ、それらの中でも水に対する溶解性(溶解度)は化合物ごとに大きく異なり、水溶性の低い化合物では、溶出特性を改善するための製剤設計における問題点が多い。酸性薬物とは、遊離体(アルカリ又はアミンの付加塩など、薬物の酸性基との塩を形成していないフリー体)が酸性を示す酸性化合物を指し、酸性の薬物の問題点としては、酸性溶液、例えば胃などの消化管上部における溶解性が低い問題点があり、酸性化合物の塩(アルカリ又はアミン付加塩など)では、酸性溶液中では、溶解性の低い遊離酸になる点が問題点としてある。また、塩基性薬物とは、遊離体(酸付加塩など、薬物の塩基性基との塩を形成していないフリー体)が塩基性を示す塩基性化合物を指し、強酸性水溶液では良好な溶解性を示すが、中性の緩衝液などの中性水溶液では溶解性が低下することが知られている。すなわち、塩基性薬物を経口投与した場合、酸性である胃では良好な溶解性を示すものの、中性でかつ水分の少ない大腸などの消化管下部での溶解性が大きく低下し吸収率の低下が懸念される。
例えば、塩基性薬物の経口投与用の徐放製剤の設計においては、高い水溶性を示す消化管上部の酸性環境における食物との共存、消化管運動などによる機械的ストレスにより、製剤が崩壊するために生じる薬物の過量放出(dose dumping)が問題となる。さらに、薬物の過量放出を回避するため徐放基剤を増量するなどして製剤強度を高めても、中性領域で水溶性が低下する塩基性薬物を含む製剤の消化管下部での溶出性を改善し、吸収を維持させることが徐放性製剤の問題点として残る。現在までに、塩基性薬物の徐放剤で、消化管上部などの酸性環境での薬物の過量放出を回避し、かつ中性環境である消化管下部での持続溶出を同時に達成できる技術として満足できるものはない。
本発明の課題は、消化管上部、特に胃の酸性環境と食物との共存下での消化管運動による機械的ストレスによる薬物の過量放出(dose dumping)を回避し、かつ中性領域である消化管下部における薬剤の溶出性を改善することにより、1日1回又は2回の経口投与で確実に主薬理効果を発揮する薬物を主薬効成分として含有する経口投与用の徐放性製剤を提供することにある。
本発明者らは、経口投与用の徐放製剤化の検討を重ねた結果、薬理上活性な薬物、pH依存性の高分子基剤、可塑剤、及び低融点油脂類を含有し、酸性環境下での薬物の過量放出(dose dumping)を回避し、かつ中性における溶出性を改善した徐放性マトリックス粒状製剤を見出し、本発明を完成した。
すなわち、本発明は、以下の(1)~(44)に関するものである。
(1):
(A)薬理上活性な薬物;
(B)pH依存性の高分子基剤;
(C)可塑剤;及び
(D)低融点油脂類;
上記(A)~(D)を含有することを特徴とし、押し出し造粒して得られる徐放性マトリックス粒状製剤。
(2):(B)pH依存性の高分子基剤が、腸溶性基剤である(1)に記載の徐放性マトリックス粒状製剤。
(3):腸溶性基剤が、ヒドロキシプロピルメチルセルロースアセテートサクシネートである(2)に記載の徐放性マトリックス粒状製剤。
(4):マトリックス粒状製剤中の(B)pH依存性の高分子基剤の粒状製剤中の含有率が、5~90重量%の範囲である(1)~(3)のいずれか1に記載の徐放性固形製剤。
(5):(C)可塑剤が、クエン酸トリ(C1~C6アルキル)エステル又はグリセリンモノ有機酸エステルである(1)~(4)のいずれか1に記載の徐放性マトリックス粒状製剤。
(6):(C)可塑剤が、クエン酸トリエチル又はグリセリンモノ酢酸エステルである(1)~(4)のいずれか1に記載の徐放性マトリックス粒状製剤。
(7):マトリックス粒状製剤中の(C)可塑剤の粒状製剤中の含有率が、1~30重量%の範囲である(1)~(6)のいずれか1に記載の徐放性固形製剤。
(8):(D)低融点油脂類が、モノステアリン酸グリセリン又はポリエチレングリコールである(1)~(7)のいずれか1に記載の徐放性マトリックス粒状製剤。
(9):(D)低融点油脂類が、ポリエチレングリコールである(1)~(7)のいずれか1に記載の徐放性マトリックス粒状製剤。
(10):ポリエチレングリコールが、平均分子量4000~12000の範囲のポリエチレングリコールである(8)又は(9)に記載のマトリックス粒状製剤。
(11):ポリエチレングリコールが、平均分子量7000~9000の範囲のポリエチレングリコールである(8)又は(9)に記載の徐放性マトリックス粒状製剤。
(12):マトリックス粒状製剤中の(D)低融点油脂類の粒状製剤中の含有率が、0.1~20重量%の範囲である(1)~(11)のいずれか1に記載の徐放性固形製剤。
(13):さらに、結合剤及び有機酸から選ばれる1種又は2種以上を含有する(1)~(12)のいずれか1に記載の徐放性マトリックス粒状製剤。
(14):結合剤が、親水性ゲル形成性高分子物質である(13)に記載の徐放性マトリックス粒状製剤。
(15):親水性ゲル形成性高分子物質が、ヒドロキシプロピルセルロースである(14)に記載の徐放性マトリックス粒状製剤。
(16):有機酸が、フマル酸又はアルギン酸である(13)~(15)のいずれか1に記載の徐放性マトリックス粒状製剤。
(17):有機酸が、フマル酸である(13)~(15)のいずれか1に記載の徐放性マトリックス粒状製剤。
(18):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(17)のいずれか1に記載の徐放性マトリックス粒状製剤:
(中性状態の溶解度)/(酸性状態の溶解度)が、0.00001~0.6の範囲である。
(19):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(17)のいずれか1に記載の徐放性マトリックス粒状製剤:
(中性状態の溶解度)/(酸性状態の溶解度)が、0.001~0.5の範囲である。
(20):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(17)のいずれか1に記載の徐放性マトリックス粒状製剤:
中性状態(7.5>pH>5の範囲)における最低溶解度が、3mg/ml以下である。
(21):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(17)のいずれか1に記載の徐放性マトリックス粒状製剤:
中性状態(7.5>pH>5の範囲)における最低溶解度が、0.5mg/ml以下である。
(22):(A)薬理上活性な薬物が、塩基性薬物である(1)~(21)のいずれか1に記載の徐放性マトリックス粒状製剤。
(23):(A)薬理上活性な薬物が、下記の
(±)-1-(カルバゾール-4-イルオキシ)-3-[[2-(o-メトキシフェノキシ)エチル]アミノ]-2-プロパノール;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド;及び
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド;
からなる群より選ばれる化合物、その薬理上許容される塩、又はそれらの水和物である(1)~(17)のいずれか1に記載の徐放性マトリックス粒状製剤。
(24):マトリックス粒状製剤中の(A)薬理上活性な薬物の粒状製剤中の含有率が、0.1~50重量%の範囲である(1)~(23)のいずれか1に記載の徐放性固形製剤。
(25):マトリックス粒状製剤が、マトリックス顆粒剤である(1)~(24)のいずれか1に記載の徐放性マトリックス粒状製剤。
(26):0.01規定塩酸水溶液(900mL)中、37±0.5℃において、パドル法毎分50回転で溶出試験を行なうとき、薬理上活性な薬剤の平均溶出率が、溶出試験開始後4時間で60%以下である(1)~(25)のいずれか1に記載の徐放性マトリックス粒状製剤。
(27):(E):薬理上活性な薬物、pH依存性の高分子基剤、及び油脂類を混合して[混合物]を調製する工程;
(F):可塑剤、結合剤の水溶液及び蒸留水を添加、撹拌して[分散液]を調製する工程;
(G):[混合末]及び[分散液]から[練合物]を製造する工程;及び
(H):[練合物]を押出し造粒し、[顆粒]を製造する工程;
上記(E)~(H)の工程からなる、(1)~(26)のいずれか1に記載の経口投与用の徐放性マトリックス粒状製剤の製造方法。
(1):
(A)薬理上活性な薬物;
(B)pH依存性の高分子基剤;
(C)可塑剤;及び
(D)低融点油脂類;
上記(A)~(D)を含有することを特徴とし、押し出し造粒して得られる徐放性マトリックス粒状製剤。
(2):(B)pH依存性の高分子基剤が、腸溶性基剤である(1)に記載の徐放性マトリックス粒状製剤。
(3):腸溶性基剤が、ヒドロキシプロピルメチルセルロースアセテートサクシネートである(2)に記載の徐放性マトリックス粒状製剤。
(4):マトリックス粒状製剤中の(B)pH依存性の高分子基剤の粒状製剤中の含有率が、5~90重量%の範囲である(1)~(3)のいずれか1に記載の徐放性固形製剤。
(5):(C)可塑剤が、クエン酸トリ(C1~C6アルキル)エステル又はグリセリンモノ有機酸エステルである(1)~(4)のいずれか1に記載の徐放性マトリックス粒状製剤。
(6):(C)可塑剤が、クエン酸トリエチル又はグリセリンモノ酢酸エステルである(1)~(4)のいずれか1に記載の徐放性マトリックス粒状製剤。
(7):マトリックス粒状製剤中の(C)可塑剤の粒状製剤中の含有率が、1~30重量%の範囲である(1)~(6)のいずれか1に記載の徐放性固形製剤。
(8):(D)低融点油脂類が、モノステアリン酸グリセリン又はポリエチレングリコールである(1)~(7)のいずれか1に記載の徐放性マトリックス粒状製剤。
(9):(D)低融点油脂類が、ポリエチレングリコールである(1)~(7)のいずれか1に記載の徐放性マトリックス粒状製剤。
(10):ポリエチレングリコールが、平均分子量4000~12000の範囲のポリエチレングリコールである(8)又は(9)に記載のマトリックス粒状製剤。
(11):ポリエチレングリコールが、平均分子量7000~9000の範囲のポリエチレングリコールである(8)又は(9)に記載の徐放性マトリックス粒状製剤。
(12):マトリックス粒状製剤中の(D)低融点油脂類の粒状製剤中の含有率が、0.1~20重量%の範囲である(1)~(11)のいずれか1に記載の徐放性固形製剤。
(13):さらに、結合剤及び有機酸から選ばれる1種又は2種以上を含有する(1)~(12)のいずれか1に記載の徐放性マトリックス粒状製剤。
(14):結合剤が、親水性ゲル形成性高分子物質である(13)に記載の徐放性マトリックス粒状製剤。
(15):親水性ゲル形成性高分子物質が、ヒドロキシプロピルセルロースである(14)に記載の徐放性マトリックス粒状製剤。
(16):有機酸が、フマル酸又はアルギン酸である(13)~(15)のいずれか1に記載の徐放性マトリックス粒状製剤。
(17):有機酸が、フマル酸である(13)~(15)のいずれか1に記載の徐放性マトリックス粒状製剤。
(18):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(17)のいずれか1に記載の徐放性マトリックス粒状製剤:
(中性状態の溶解度)/(酸性状態の溶解度)が、0.00001~0.6の範囲である。
(19):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(17)のいずれか1に記載の徐放性マトリックス粒状製剤:
(中性状態の溶解度)/(酸性状態の溶解度)が、0.001~0.5の範囲である。
(20):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(17)のいずれか1に記載の徐放性マトリックス粒状製剤:
中性状態(7.5>pH>5の範囲)における最低溶解度が、3mg/ml以下である。
(21):(A)薬理上活性な薬物が、以下の溶解度を示すものである、(1)~(17)のいずれか1に記載の徐放性マトリックス粒状製剤:
中性状態(7.5>pH>5の範囲)における最低溶解度が、0.5mg/ml以下である。
(22):(A)薬理上活性な薬物が、塩基性薬物である(1)~(21)のいずれか1に記載の徐放性マトリックス粒状製剤。
(23):(A)薬理上活性な薬物が、下記の
(±)-1-(カルバゾール-4-イルオキシ)-3-[[2-(o-メトキシフェノキシ)エチル]アミノ]-2-プロパノール;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド;及び
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド;
からなる群より選ばれる化合物、その薬理上許容される塩、又はそれらの水和物である(1)~(17)のいずれか1に記載の徐放性マトリックス粒状製剤。
(24):マトリックス粒状製剤中の(A)薬理上活性な薬物の粒状製剤中の含有率が、0.1~50重量%の範囲である(1)~(23)のいずれか1に記載の徐放性固形製剤。
(25):マトリックス粒状製剤が、マトリックス顆粒剤である(1)~(24)のいずれか1に記載の徐放性マトリックス粒状製剤。
(26):0.01規定塩酸水溶液(900mL)中、37±0.5℃において、パドル法毎分50回転で溶出試験を行なうとき、薬理上活性な薬剤の平均溶出率が、溶出試験開始後4時間で60%以下である(1)~(25)のいずれか1に記載の徐放性マトリックス粒状製剤。
(27):(E):薬理上活性な薬物、pH依存性の高分子基剤、及び油脂類を混合して[混合物]を調製する工程;
(F):可塑剤、結合剤の水溶液及び蒸留水を添加、撹拌して[分散液]を調製する工程;
(G):[混合末]及び[分散液]から[練合物]を製造する工程;及び
(H):[練合物]を押出し造粒し、[顆粒]を製造する工程;
上記(E)~(H)の工程からなる、(1)~(26)のいずれか1に記載の経口投与用の徐放性マトリックス粒状製剤の製造方法。
(28):(A)薬理上活性な薬物、(B)ヒドロキシプロピルメチルセルロースアセテートサクシネート、(C)可塑剤、及び、(D)ポリエチレングリコール、を混合した後、押し出し造粒して得られる徐放性製剤。
(29):製剤中の(B)成分の含有率が50~90重量%である、(28)に記載の製剤。
(30):製剤中の(B)成分の含有率が55~80重量%である、(28)に記載の製剤。
(31):(C)成分がクエン酸トリエチルである、(28)~(30)いずれか1に記載の製剤。
(32):製剤中の(C)成分の含有率が5~30重量%である、(28)~(31)のいずれか1に記載の製剤。
(33):(D)成分が平均分子量4000~12000の範囲のポリエチレングリコールである、(28)~(32)のいずれか1に記載の製剤。
(34):(D)成分が平均分子量7000~10500の範囲のポリエチレングリコールである、(28)~(32)のいずれか1に記載の製剤。
(35):(D)成分が、ポリエチレングリコール6000である、(28)~(32)のいずれか1に記載の製剤。
(36):製剤中の(D)成分の含有率が1~10重量%である、(28)~(35)のいずれか1に記載の製剤。
(37):製剤中の(D)成分の含有率が2~8重量%である、(28)~(35)のいずれか1に記載の製剤。
(38):さらに、結合剤及び/又は有機酸を含有する、(28)~(37)のいずれか1に記載の製剤。
(39):結合剤がヒドロキシプロピルセルロースである、(38)に記載の製剤。
(40):有機酸が、フマル酸又はアルギン酸である、(38)又は(39)に記載の製剤。
(41):有機酸がフマル酸である、(38)又は(39)に記載の製剤。
(42):(A)成分が塩基性薬物である、(28)~(41)のいずれか1に記載の製剤。
(43):(A)成分が、
(±)-1-(カルバゾール-4-イルオキシ)-3-[[2-(o-メトキシフェノキシ)エチル]アミノ]-2-プロパノール、
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド、及び、
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド、
からなる群より選ばれる化合物、その薬理上許容される塩、又はそれらの水和物である、(28)~(41)のいずれか1に記載の製剤。
(44):製剤中の(A)成分の含有率が、0.1~50重量%である(28)~(43)のいずれか1に記載の製剤。
(29):製剤中の(B)成分の含有率が50~90重量%である、(28)に記載の製剤。
(30):製剤中の(B)成分の含有率が55~80重量%である、(28)に記載の製剤。
(31):(C)成分がクエン酸トリエチルである、(28)~(30)いずれか1に記載の製剤。
(32):製剤中の(C)成分の含有率が5~30重量%である、(28)~(31)のいずれか1に記載の製剤。
(33):(D)成分が平均分子量4000~12000の範囲のポリエチレングリコールである、(28)~(32)のいずれか1に記載の製剤。
(34):(D)成分が平均分子量7000~10500の範囲のポリエチレングリコールである、(28)~(32)のいずれか1に記載の製剤。
(35):(D)成分が、ポリエチレングリコール6000である、(28)~(32)のいずれか1に記載の製剤。
(36):製剤中の(D)成分の含有率が1~10重量%である、(28)~(35)のいずれか1に記載の製剤。
(37):製剤中の(D)成分の含有率が2~8重量%である、(28)~(35)のいずれか1に記載の製剤。
(38):さらに、結合剤及び/又は有機酸を含有する、(28)~(37)のいずれか1に記載の製剤。
(39):結合剤がヒドロキシプロピルセルロースである、(38)に記載の製剤。
(40):有機酸が、フマル酸又はアルギン酸である、(38)又は(39)に記載の製剤。
(41):有機酸がフマル酸である、(38)又は(39)に記載の製剤。
(42):(A)成分が塩基性薬物である、(28)~(41)のいずれか1に記載の製剤。
(43):(A)成分が、
(±)-1-(カルバゾール-4-イルオキシ)-3-[[2-(o-メトキシフェノキシ)エチル]アミノ]-2-プロパノール、
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド、及び、
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド、
からなる群より選ばれる化合物、その薬理上許容される塩、又はそれらの水和物である、(28)~(41)のいずれか1に記載の製剤。
(44):製剤中の(A)成分の含有率が、0.1~50重量%である(28)~(43)のいずれか1に記載の製剤。
本願発明によれば、化合物(1)に代表される薬理上活性な薬物を含有する、経口投与用の徐放性マトリックス粒状製剤が提供される。本発明の徐放性マトリックス粒状製剤は、酸性溶液中における過剰放出を防ぐ良好な強度を持ち、かつ中性溶液中で良好な溶出性を持つことから、十二指腸、小腸から下部消化管に至るまで、含有する薬理上活性な薬物の持続的な溶出を維持する効果が得られる。
以下に本発明を詳細に説明する。
本明細書における「酸性溶液」としては、胃などの消化管上部での溶出性を評価するために用いる酸性の溶出試験液を意味し、例えば、日本薬局方記載の溶出試験第1液、米国薬局方に記載のUSP 0.1規定塩酸、0.01規定塩酸、Simulated Gastric Fluid without Enzyme等を挙げることができが、酸性溶出試験液としては、これらに限られるものではない。
本明細書における「中性溶液」としては、小腸、大腸などにおける薬剤の溶出性を評価するために用いる中性の溶出試験液としては、例えば、日本薬局方記載の溶出試験第2液やリン酸塩緩衝液pH6.8、米国薬局方記載のUSP Phosphate Buffer(pH6.8)、Simulated Interstinal Fluid without Enzyme、欧州薬局方記載のPhosphate Buffer Solution pH6.8等を挙げることができるが、中性溶液の溶出試験液としては、これらに限られるものではない。
上記の溶出試験液は、各国の薬局方等に記載された方法で調製される。これらの溶出試験液のpHは、溶出試験液が緩衝液の場合、各溶出試験液に規定されたpHの±0.05以内となることが好ましい。
本願発明の徐放性マトリックス粒状製剤の、消化管上部における溶出性評価のための酸性溶出液を用いたパドル法としては、例えば0.01規定塩酸(900mL)中、37±0.5℃において、パドル法毎分50回転及び200回転で2時間溶出試験を行う方法を挙げることができる。上記のように、製剤中の薬理上活性な薬物が塩基性薬物の場合には、高い水溶性を示す消化管上部の酸性環境における食物との共存、消化管運動などによる機械的ストレスにより、製剤が崩壊するために生じる薬物の過量放出(dose dumping)の問題がある。したがって、溶出試験液中における薬理上活性な薬物の平均溶出率は、パドル法毎分200回転及び/又はパドル法毎分50回転においても、製剤強度を保ち、一定の範囲に溶出率を抑えるのが好ましい。また、2時間溶出試験方法を行ったときの、溶出試験液中における薬理上活性な薬物の平均溶出率の差(パドル法毎分200回転-パドル法毎分50回転)が15%以下が好ましく;10%以下がより好ましく;5%以下が特に好ましい。さらに、2時間後の溶出試験液中における薬理上活性な薬物の平均溶出率の比(パドル法毎分200回転/パドル法毎分50回転)が、2.0以下が好ましく;1.5以下がより好ましく;1.3以下が特に好ましい。
溶出試験法としては、ヒト消化管内環境に近い条件での溶出試験法であるUSP Apparatus3(Bio-Dis法)を用いてもよい。
溶液中の薬物濃度は、後述の実施例に示す条件(試験液、振とう速度及び測定時間)を用いて測定することができる。溶出試験では、UV法などを用いて、溶出試験液中における薬理上活性な薬物の平均溶出率、溶出時間を算出することができる。
溶液中の薬物濃度は、後述の実施例に示す条件(試験液、振とう速度及び測定時間)を用いて測定することができる。溶出試験では、UV法などを用いて、溶出試験液中における薬理上活性な薬物の平均溶出率、溶出時間を算出することができる。
なお、上記「平均溶出率」とは、1種の固形製剤について、少なくとも2個、好ましくは6個、さらに好ましくは12個の溶出率を測定し、それらの平均値を求めればよい。
本明細書における「薬理上活性な薬物」としては、製剤の処方中で主薬理効果を発揮する薬物であって、比較的水溶性の低い薬物が好ましい。薬理上活性な薬物で、中性化合物としては、分子中に酸性状態又は塩基性の状態においてもイオン化して解離する基を持たない化合物を意味し、また、酸性化合物としては、カルボキシ基、フェノール性水酸基、リン酸基、スルホン酸、テトラゾリル基などに代表される酸性基を有する薬物を意味する。さらに、塩基性薬物としては、分子内にアミノ基、ピペリジニル基、ピペラジニル基などに代表される塩基性窒素原子を有する薬物を意味する。本願発明においては、特に塩基性薬物が好適である。塩基性薬物は、小腸、大腸での中性状態(7.5>pH>5)での溶解度が、胃などの酸性状態(pH≦2)での溶解度と比較して低下する物理化学的性質を有する。
上記のように、塩基性薬物とは、上記の酸性状態における溶解度に比べて中性状態における溶解度が低下する薬物を指し、中性状態における溶解度の低下の割合としては、下記の範囲を挙げることができる。
(中性状態の溶解度)/(酸性状態の溶解度)が、0.00001~0.6の範囲のものが好ましく;
(中性状態の溶解度)/(酸性状態の溶解度)が、0.001~0.5の範囲のものがより好ましく;
(中性状態の溶解度)/(酸性状態の溶解度)が、0.01~0.1の範囲のものがさらに好ましいが、これら範囲になんら限定されるものではない。
上記のように、塩基性薬物とは、上記の酸性状態における溶解度に比べて中性状態における溶解度が低下する薬物を指し、中性状態における溶解度の低下の割合としては、下記の範囲を挙げることができる。
(中性状態の溶解度)/(酸性状態の溶解度)が、0.00001~0.6の範囲のものが好ましく;
(中性状態の溶解度)/(酸性状態の溶解度)が、0.001~0.5の範囲のものがより好ましく;
(中性状態の溶解度)/(酸性状態の溶解度)が、0.01~0.1の範囲のものがさらに好ましいが、これら範囲になんら限定されるものではない。
本明細書における「塩基性薬物」の酸性領域(日本薬局方溶出試験第1液:pH=1.2,20±5℃)での溶解度としては、1~500mg/mlの範囲であり;かつ中性領域(日本薬局方溶出試験第2液:pH=6.8,20±5℃)での溶解度が、0.01~3000μg/mlの範囲が好ましく;
酸性領域(日本薬局方溶出試験第1液:pH=1.2,20±5℃)での溶解度が、1~500mg/mlの範囲であり;かつ中性領域(日本薬局方溶出試験第2液:pH=6.8,20±5℃)での溶解度が、10~500μg/mlの範囲であるものがより好ましい。また、溶解度の絶対値としては、中性状態(7.5>pH>5の範囲)における最低溶解度が3mg/ml以下に低下する薬物が好ましく;1mg/ml以下に低下する薬物がより好ましく;0.5mg/ml以下に低下する薬物がさらに好ましい。
酸性領域(日本薬局方溶出試験第1液:pH=1.2,20±5℃)での溶解度が、1~500mg/mlの範囲であり;かつ中性領域(日本薬局方溶出試験第2液:pH=6.8,20±5℃)での溶解度が、10~500μg/mlの範囲であるものがより好ましい。また、溶解度の絶対値としては、中性状態(7.5>pH>5の範囲)における最低溶解度が3mg/ml以下に低下する薬物が好ましく;1mg/ml以下に低下する薬物がより好ましく;0.5mg/ml以下に低下する薬物がさらに好ましい。
「薬理上活性な薬物」の具体的な例としては、抗凝固剤を挙げることができる。
抗凝固剤としては、活性化血液凝固第X因子(FXa)阻害剤が好ましく、FXa阻害剤の具体例としては、下記の(a)~(l)を挙げることができる。
(a)Darexaban Maleate(tanexaban),(N-[2-ヒドロキシ-6-メトキシベンズアミド]フェニル]-4-(4-メチル-1,4-ジアゼパン-1-イル)ベンズアミド)[薬食審査発1111第1号(平成22年11月11日);Pre-publication copy,Proposed INN: List 101;Research and development pipeline.Yamanouchi Pharmaceutical Co Ltd.Company World Wide Web site,11 Feb 2004,参照];
(b)rivaroxaban,(5-クロロ-N-({(5S)-2-オキソ-3-[4-(3-オキソ-4-モルホリニル)フェニル]-1,3-オキサゾリジン-5-イル}メチル)-2-チオフェンカルボキサミド)[WFO Drug Information,Vol.18,No.3,2004,page 260;Susanne R,et al,J.Med.Chem.,2005,48,5900-5908;D.Kubitza et al,Multiple dose escalation study investigating the pharmacodynamics,safety,and pharmacokinetic of Bay59-7939,an oral,direct Factor Xa inhibitor,in healthy male subjects.Blood,2003,102;Abstract 3004.参照];
(c)apixaban,(1-(4-メトキシフェニル)-7-オキソ-6-[4-(2-オキソピペリジン-1-イル)フェニル]-4,5,6,7-テトラヒドロ-1H-ピラゾロ[3,4-c]ピリジン-3-カルボキサミド)[WFO Drug Information,Vol.20, No.1,2006,page 38;Pinto DJP, Orwat MJ, Lam PYS,et al,Discovery of 1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (Apixaban,BMS-562247),a highly potent,selective,efficacious,and orally bioavailable inhibitor of blood coagulation factor Xa,J.Med.Chem.,50(22),5339-56,2007,参照];
(d)Betrixaban,(N-(5-クロロピリジン-2-イル)-2-[4-(N,N-ジメチルカルバムイミドイル)ベンズアミド]-5-メトキシベンズアミド)[WFO Drug Information,Vol.22,No.3,2008,page 226-227;Zhang P, Huang W,Zhu BY,et al.,Discovery of betrixaban (PRT054021),N-(5-chloropyridin-2-yl)-2-(4-(N,N-dimethylcarbamimidoyl)benzamido)-5-methoxybenzamide,a highly potent,selective, and orally efficacious factor Xa inhibitor,Bioorg Med.Chem.Lett.19(8),2179-85,2009,参照];
(e)AX-1826,[S.Takehana et al.Japanese Journal of Pharmacology 2000,82(Suppl.1),213P;T.Kayahara et al.Japanese Journal of Pharmacology 2000,82(Suppl.1),213P];
(f)HMR-2906,[XVIIth Congress of the International Society for Thrombosis and Haemostasis,Washington D.C.,USA,14-21 Aug 1999;Generating greater value from our products and pipeline.Aventis SA Company Presentation,05 Feb 2004];
(g)Otamixaban,((2R,3R)-2-(3-カルバムイミドイルベンジル)-3-[[4-(1-オキシドピリジン-4-イル)ベンゾイル]アミモ]ブタン酸メチル)[WFO Drug Information,Vol.16,No.3,2002,page 257,参照];
(h)BIBT-986,(プロドラッグ:BIBT-1011)[American Chemical Society-226th National Meeting,New York City,NY,USA,2003];
(i)DPC-602,[J.R.Pruitt et al.J.Med.Chem.2003,46,5298-5313];
(j)LY517717,(N-[(1R)-2-[4-(1-メチル-4-ピペリジニル)-1-ピペラジニル]-2-オキソ-1-フェニルエチル]-1H-インドール-6-カルボキサミド)[S.Young,Medicinal Chemistry-12th RSC-SCI Symposium,7-10 September 2003,Cambridge,UK;M.Wiley et al.228th ACS National Meeting,Philadelphia,August 22-26,2004,MEDI-252&254,参照];
(k)N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド、その薬理上許容される塩、又はそれらの水和物[国際公開2004/058715号パンフレット,参照];及び
(l)N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド、その薬理上許容される塩、又はそれらの水和物[国際公開03/000657号パンフレット;国際公開03/000680号パンフレット;国際公開03/016302号パンフレット,参照]。
(a)Darexaban Maleate(tanexaban),(N-[2-ヒドロキシ-6-メトキシベンズアミド]フェニル]-4-(4-メチル-1,4-ジアゼパン-1-イル)ベンズアミド)[薬食審査発1111第1号(平成22年11月11日);Pre-publication copy,Proposed INN: List 101;Research and development pipeline.Yamanouchi Pharmaceutical Co Ltd.Company World Wide Web site,11 Feb 2004,参照];
(b)rivaroxaban,(5-クロロ-N-({(5S)-2-オキソ-3-[4-(3-オキソ-4-モルホリニル)フェニル]-1,3-オキサゾリジン-5-イル}メチル)-2-チオフェンカルボキサミド)[WFO Drug Information,Vol.18,No.3,2004,page 260;Susanne R,et al,J.Med.Chem.,2005,48,5900-5908;D.Kubitza et al,Multiple dose escalation study investigating the pharmacodynamics,safety,and pharmacokinetic of Bay59-7939,an oral,direct Factor Xa inhibitor,in healthy male subjects.Blood,2003,102;Abstract 3004.参照];
(c)apixaban,(1-(4-メトキシフェニル)-7-オキソ-6-[4-(2-オキソピペリジン-1-イル)フェニル]-4,5,6,7-テトラヒドロ-1H-ピラゾロ[3,4-c]ピリジン-3-カルボキサミド)[WFO Drug Information,Vol.20, No.1,2006,page 38;Pinto DJP, Orwat MJ, Lam PYS,et al,Discovery of 1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (Apixaban,BMS-562247),a highly potent,selective,efficacious,and orally bioavailable inhibitor of blood coagulation factor Xa,J.Med.Chem.,50(22),5339-56,2007,参照];
(d)Betrixaban,(N-(5-クロロピリジン-2-イル)-2-[4-(N,N-ジメチルカルバムイミドイル)ベンズアミド]-5-メトキシベンズアミド)[WFO Drug Information,Vol.22,No.3,2008,page 226-227;Zhang P, Huang W,Zhu BY,et al.,Discovery of betrixaban (PRT054021),N-(5-chloropyridin-2-yl)-2-(4-(N,N-dimethylcarbamimidoyl)benzamido)-5-methoxybenzamide,a highly potent,selective, and orally efficacious factor Xa inhibitor,Bioorg Med.Chem.Lett.19(8),2179-85,2009,参照];
(e)AX-1826,[S.Takehana et al.Japanese Journal of Pharmacology 2000,82(Suppl.1),213P;T.Kayahara et al.Japanese Journal of Pharmacology 2000,82(Suppl.1),213P];
(f)HMR-2906,[XVIIth Congress of the International Society for Thrombosis and Haemostasis,Washington D.C.,USA,14-21 Aug 1999;Generating greater value from our products and pipeline.Aventis SA Company Presentation,05 Feb 2004];
(g)Otamixaban,((2R,3R)-2-(3-カルバムイミドイルベンジル)-3-[[4-(1-オキシドピリジン-4-イル)ベンゾイル]アミモ]ブタン酸メチル)[WFO Drug Information,Vol.16,No.3,2002,page 257,参照];
(h)BIBT-986,(プロドラッグ:BIBT-1011)[American Chemical Society-226th National Meeting,New York City,NY,USA,2003];
(i)DPC-602,[J.R.Pruitt et al.J.Med.Chem.2003,46,5298-5313];
(j)LY517717,(N-[(1R)-2-[4-(1-メチル-4-ピペリジニル)-1-ピペラジニル]-2-オキソ-1-フェニルエチル]-1H-インドール-6-カルボキサミド)[S.Young,Medicinal Chemistry-12th RSC-SCI Symposium,7-10 September 2003,Cambridge,UK;M.Wiley et al.228th ACS National Meeting,Philadelphia,August 22-26,2004,MEDI-252&254,参照];
(k)N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド、その薬理上許容される塩、又はそれらの水和物[国際公開2004/058715号パンフレット,参照];及び
(l)N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド、その薬理上許容される塩、又はそれらの水和物[国際公開03/000657号パンフレット;国際公開03/000680号パンフレット;国際公開03/016302号パンフレット,参照]。
上記の活性化血液凝固第X因子(FXa)阻害剤としては、下記の式(1)

(式中、R1は、N,N-ジメチルカルバモイル基又は[1,3,4]オキサジアゾール-2-イル基を示す。)
で表される化合物[以下、化合物(1)と略する場合がある]が、より好ましい。化合物(1)はフリー体(遊離塩基)、又はその薬理学上許容される塩、それらの水和物であってもよい。
式(1)で表される化合物の塩としては、塩酸塩、硫酸塩、臭化水素酸塩、ヨウ化水素酸塩、燐酸塩、硝酸塩、安息香酸塩、メタンスルホン酸塩、2-ヒドロキシエタンスルホン酸塩、p-トルエンスルホン酸塩、酢酸塩、プロパン酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、グルタル酸塩、アジピン酸塩、酒石酸塩、マレイン酸塩、フマル酸塩、リンゴ酸塩、マンデル酸塩等が挙げられる。
で表される化合物[以下、化合物(1)と略する場合がある]が、より好ましい。化合物(1)はフリー体(遊離塩基)、又はその薬理学上許容される塩、それらの水和物であってもよい。
式(1)で表される化合物の塩としては、塩酸塩、硫酸塩、臭化水素酸塩、ヨウ化水素酸塩、燐酸塩、硝酸塩、安息香酸塩、メタンスルホン酸塩、2-ヒドロキシエタンスルホン酸塩、p-トルエンスルホン酸塩、酢酸塩、プロパン酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、グルタル酸塩、アジピン酸塩、酒石酸塩、マレイン酸塩、フマル酸塩、リンゴ酸塩、マンデル酸塩等が挙げられる。
式(1)で表される化合物の塩としては、マレイン酸、塩酸塩、メタンスルホン酸塩、p-トルエンスルホン酸塩が好ましく、マレイン酸及びp-トルエンスルホン酸塩が特に好ましい。
式(1)で表される化合物として好ましいものとして、以下の
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド モノマレイン酸塩;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド 塩酸塩;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド モノp-トルエンスルホン酸塩;及び
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド モノp-トルエンスルホン酸塩・1水和物;
を挙げることができる。
式(1)で表される化合物として好ましいものとして、以下の
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド モノマレイン酸塩;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド 塩酸塩;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド モノp-トルエンスルホン酸塩;及び
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド モノp-トルエンスルホン酸塩・1水和物;
を挙げることができる。
上記の好ましい化合物の中で、下記の式(1a)[以下、化合物(1a)と略する場合がある]及び式(1b)[以下、化合物(1b)と略する場合がある]

で表される、N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド モノマレイン酸塩(1a);及び
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド モノp-トルエンスルホン酸塩・1水和物(1b)が特に好ましい。
これらの化合物(1)は、文献に記載の方法又はそれに準じる方法によって製造することができる(WO2003-000657号パンフレット;WO2003-000680号パンフレット;WO2003-016302号パンフレット;WO2004-058715号パンフレット)。
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド モノp-トルエンスルホン酸塩・1水和物(1b)が特に好ましい。
これらの化合物(1)は、文献に記載の方法又はそれに準じる方法によって製造することができる(WO2003-000657号パンフレット;WO2003-000680号パンフレット;WO2003-016302号パンフレット;WO2004-058715号パンフレット)。
化合物(1)の遊離塩基(フリー体)とは、化合物(1)と、例えば上記の塩(酸付加塩)、及び/又は水和物を形成している酸付加塩の「酸」及び水和物の「水」を除いた化合物を意味し、例えば化合物(1a)及び化合物(1b)の遊離塩基(フリー体)とは、下記の式(1a-1)及び式(1b-1)
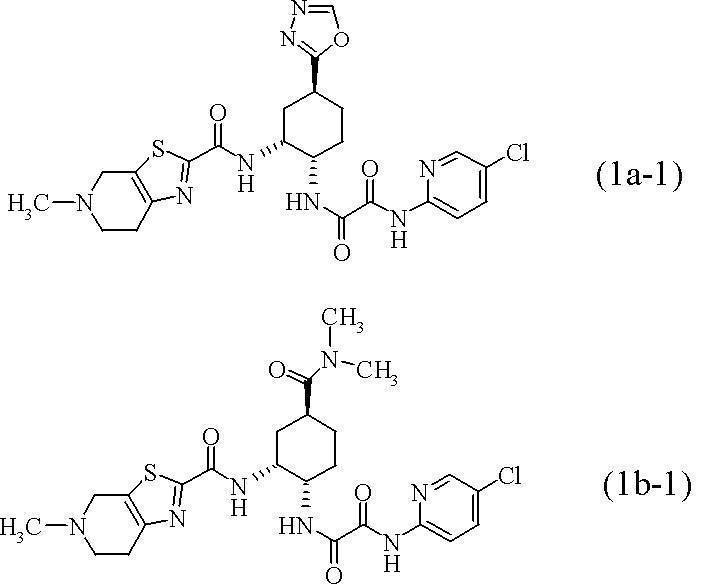
で表されるN1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド(1a-1)、及びN1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド(1b-1)を意味する。
また、本願発明の(A)薬理上活性な薬物の好ましい例として、下記の式(2)[以下、化合物(2)と略する場合がある]

で表される(±)-1-(カルバゾール-4-イルオキシ)-3-[[2-(o-メトキシフェノキシ)エチル]アミノ]-2-プロパノール(3)(CAS番号:72956-09-3)、その薬理上許容される塩、又はそれらの水和物を挙げることができる。
本願発明における「(B)pH依存性の高分子基剤」としては、製剤分野で用いられるpH依存的な溶解特性を示す高分子基剤を挙げることができる。「pH依存性の高分子基剤」は、さらに、腸溶性基剤と胃溶解性基剤に分けることができ、腸溶性基剤が好ましい。腸溶性基剤としては、胃内等のpH環境下で溶解しにくく、主な吸収部位である小腸、大腸など中性のpH環境下で徐々に溶解するものが好ましい。
pH依存性の高分子基剤の具体例としては、以下の(1)~(3)を挙げることができる。
(1)メタアクリル酸系コポリマー:ここで、メタアクリル酸系コポリマーとは、メタクリル酸、メタクリル酸エステル、アクリル酸及びアクリル酸エステルからなる群より選ばれる2種以上のモノマーのコポリマーを意味し、上記モノマーの組み合わせ、使用モノマーの数等によって限定されない。
(2)ヒドロキシプロピルメチルセルロースアセテートサクシネート:
(3)カルボキシメチルエチルセルロース。
本願発明の「(B)pH依存性の高分子基剤」としては、上記の(1)メタアクリル酸系コポリマー;及び(2)ヒドロキシプロピルメチルセルロースアセテートサクシネートが好ましく;ヒドロキシプロピルメチルセルロースアセテートサクシネートがより好ましい。
(1)メタアクリル酸系コポリマーとしては、メタクリル酸-メチルメタクリル酸コポリマー、(アクリル酸エチル-メタクリル酸メチル-メタクリル酸塩化トリメチルアンモニウムエチル)コポリマー、(メタクリル酸-アクリル酸エチル)コポリマー、(メタクリル酸-メタクリル酸メチル)コポリマーが好ましく;メタクリル酸-メチルメタクリル酸コポリマーがより好ましい。メタクリル酸-メチルメタクリル酸コポリマーの好ましい具体例としては、オイドラギット(EUDRAGIT)を挙げることができ、市販品として、オイドラギットL100-55、オイドラギットL100を挙げることができる。
(2)ヒドロキシプロピルメチルセルロースアセテートサクシネート(HPMCAS)は、例えばAQOAT(商品名)として,信越化学株式会社から購入することができる。HPMCASのグレードは、LF、MF、HF、LG、MG及びHGが入手可能であるが、MF及びHFグレードが好ましい。
また、本願発明のpH依存性の高分子基剤の粒径の好ましいものとしては、D50=100以下のものが好ましく;D50=50以下のものがより好ましく;D50=10以下のものがさらに好ましい。また、本願発明のpH依存性の高分子基剤としてヒドロキシプロピルメチルセルロースアセテートサクシネートの粒径の好ましいものとしては、D50=40以下のものが好ましく;D50=20以下のものがより好ましく;D50=10以下のものがさらに好ましく;D50=5以下のものが特に好ましい。
本願発明のマトリックス粒状製剤中における(B)pH依存性の高分子基剤の含有率としては、5~90%の範囲が好ましく;50~80%の範囲がより好ましい。
(1)メタアクリル酸系コポリマー:ここで、メタアクリル酸系コポリマーとは、メタクリル酸、メタクリル酸エステル、アクリル酸及びアクリル酸エステルからなる群より選ばれる2種以上のモノマーのコポリマーを意味し、上記モノマーの組み合わせ、使用モノマーの数等によって限定されない。
(2)ヒドロキシプロピルメチルセルロースアセテートサクシネート:
(3)カルボキシメチルエチルセルロース。
本願発明の「(B)pH依存性の高分子基剤」としては、上記の(1)メタアクリル酸系コポリマー;及び(2)ヒドロキシプロピルメチルセルロースアセテートサクシネートが好ましく;ヒドロキシプロピルメチルセルロースアセテートサクシネートがより好ましい。
(1)メタアクリル酸系コポリマーとしては、メタクリル酸-メチルメタクリル酸コポリマー、(アクリル酸エチル-メタクリル酸メチル-メタクリル酸塩化トリメチルアンモニウムエチル)コポリマー、(メタクリル酸-アクリル酸エチル)コポリマー、(メタクリル酸-メタクリル酸メチル)コポリマーが好ましく;メタクリル酸-メチルメタクリル酸コポリマーがより好ましい。メタクリル酸-メチルメタクリル酸コポリマーの好ましい具体例としては、オイドラギット(EUDRAGIT)を挙げることができ、市販品として、オイドラギットL100-55、オイドラギットL100を挙げることができる。
(2)ヒドロキシプロピルメチルセルロースアセテートサクシネート(HPMCAS)は、例えばAQOAT(商品名)として,信越化学株式会社から購入することができる。HPMCASのグレードは、LF、MF、HF、LG、MG及びHGが入手可能であるが、MF及びHFグレードが好ましい。
また、本願発明のpH依存性の高分子基剤の粒径の好ましいものとしては、D50=100以下のものが好ましく;D50=50以下のものがより好ましく;D50=10以下のものがさらに好ましい。また、本願発明のpH依存性の高分子基剤としてヒドロキシプロピルメチルセルロースアセテートサクシネートの粒径の好ましいものとしては、D50=40以下のものが好ましく;D50=20以下のものがより好ましく;D50=10以下のものがさらに好ましく;D50=5以下のものが特に好ましい。
本願発明のマトリックス粒状製剤中における(B)pH依存性の高分子基剤の含有率としては、5~90%の範囲が好ましく;50~80%の範囲がより好ましい。
本願発明においては、上記の(A)薬理上活性な薬物及び(B)pH依存性の高分子基剤のほかに、さらに(C)可塑剤が含まれることが特徴である。
本願発明における(C)可塑剤としては、医薬品の添加剤として用いられているものが使用可能であるが、クエン酸トリ(C1~C6アルキル)エステル又はグリセリンモノ有機酸エステルが好ましく;クエン酸トリエチル及びグリセリンモノ酢酸エステルがより好ましく;クエン酸トリエチルが特に好ましい。本願発明の粒状製剤中における(C)可塑剤の含有率としては、1~30%の範囲が好ましく;3~15%の範囲がより好ましい。
本願発明のマトリックス徐放粒状製剤においては、上記の(A)薬理上活性な薬物、(B)pH依存性の高分子基剤、及び(C)可塑剤の他に、さらに(D)低融点油脂類が含まれることが特徴である。
本願発明における(D)低融点油脂類としては、モノステアリン酸グリセリン及びポリエチレングリコールなどを挙げることができ;
モノステアリン酸グリセリン及びポリエチレングリコールが好ましく;
ポリエチレングリコールがより好ましい。
本願発明における(D)低融点油脂類としては、モノステアリン酸グリセリン及びポリエチレングリコールなどを挙げることができ;
モノステアリン酸グリセリン及びポリエチレングリコールが好ましく;
ポリエチレングリコールがより好ましい。
ポリエチレングリコールとしては、平均分子量4000~12000の範囲のポリエチレングリコールが好ましく;平均分子量7000~10500のものがより好ましい。また、より具体的にはポリエチレングリコールとしては、ポリエチレングリコール6000(CAS番号:25322-68-3)が好ましい。ポリエチレングリコール6000の一般名はマクロゴール6000(HOCH2(CH2OCH2)nCH2OH:n=165~210)であり、商業的にはマクロゴール6000(旭電化工業旭電化工業、等)、PEG6000(第一工業製薬株式会社)として入手することができる。本願発明の粒状製剤中における(D)低融点油脂類の含有率としては、0.1~20%の範囲が好ましく;1~6%の範囲がより好ましい。
本願発明のマトリックス徐放粒状製剤は、さらに結合剤及び有機酸から選ばれる1種又は2種以上を含んでもよい。
本願発明における結合剤としては、セルロース誘導体が好ましい。セルロース誘導体としては、ヒプロメロース(HPMC:ヒドロキシプロピルメチルセルロース)、ヒドロキシプロピルセルロース(HPC)、エチルセルロース、メチルセルロース等を挙げることができ;ヒプロメロース及びヒドロキシプロピルセルロースが好ましく;ヒドロキシプロピルセルロースがより好ましい。
ヒドロキシプロピルセルロースを用いる場合、用いる粒度に特に制限はなく、例えば、市販の微粉品(100メッシュの篩の通過率が99%:平均粒度=150ミクロン)が使用できる。ヒドロキシプロピルセルロース(HPC)の粘度としては、L,SL、SSL等のグレードのものが使用可能であり、L(6.0~10mPa・s)グレードのものが好ましい。ヒドロキシプロピルセルロースを結合液として用いる場合、通常、水又はアルコールなどの有機溶媒に溶解し、溶液として使用すればよい。本願発明の粒状製剤中における結合剤の含有率としては、0~10%の範囲が好ましく;0~2%の範囲がより好ましい。
ヒドロキシプロピルセルロースを用いる場合、用いる粒度に特に制限はなく、例えば、市販の微粉品(100メッシュの篩の通過率が99%:平均粒度=150ミクロン)が使用できる。ヒドロキシプロピルセルロース(HPC)の粘度としては、L,SL、SSL等のグレードのものが使用可能であり、L(6.0~10mPa・s)グレードのものが好ましい。ヒドロキシプロピルセルロースを結合液として用いる場合、通常、水又はアルコールなどの有機溶媒に溶解し、溶液として使用すればよい。本願発明の粒状製剤中における結合剤の含有率としては、0~10%の範囲が好ましく;0~2%の範囲がより好ましい。
本明細書における有機酸としては、フマル酸、コハク酸、アルギン酸、アジピン酸、クエン酸、L-アスパラギン酸、マロン酸、マレイン酸、DL-リンゴ酸、酒石酸が好ましく;フマル酸及びアルギン酸がより好ましく;フマル酸が特に好ましい。本願発明の粒状製剤中における有機酸の含有率としては、0~40%の範囲が好ましく;0~20%の範囲がより好ましい。
上記のマトリックス徐放粒状製剤には、上記(A)薬理上活性な薬物、(B)pH依存性の高分子基剤、(C)可塑剤、及び(D低融点油脂類及を含み、さらに本発明の効果に影響を与えない範囲内で、滑沢剤、着色剤、光沢化剤等を配合してもよい。
滑沢剤としては、カカオ脂、カルナウバロウ、含水二酸化ケイ素、乾燥水酸化アルミニウムゲル、グリセリン脂肪酸エステル、ケイ酸マグネシウム、軽質無水ケイ酸、硬化油、合成ケイ酸アルミニウム、サラシミツロウ、酸化マグネシウム、酒石酸ナトリウムカリウム、ショ糖脂肪酸エステル、ステアリン酸、ステアリン酸カルシウム、ステアリン酸マグネシウム、ステアリルアルコール、ステアリン酸ポリオキシル40、セタノール、ダイズ硬化油、ゼラチン、タルク、炭酸マグネシウム、沈降炭酸カルシウム、トウモロコシデンプン、バレイショデンプン、フマル酸ステアリルナトリウム、ミツロウ、メタケイ酸アルミン酸マグネシウム、ラウリル酸ナトリウム、硫酸マグネシウム等が挙げられる。
着色剤としては、黄色三二酸化鉄、三二酸化鉄、酸化チタン、オレンジエッセンス、褐色酸化鉄、β-カロチン、黒酸化鉄、食用青色1号、食用青色2号、食用赤色2号、食用赤色3号、食用赤色102号、食用黄色4号、食用黄色5号等を挙げることができる。
光沢化剤としては、カルナウバロウ、硬化油、酢酸ビニル樹脂、サラシミツロウ、酸化チタン、ステアリン酸、ステアリン酸カルシウム、ステアリン酸ポリオキシル40、ステアリン酸マグネシウム、精製セラック、精製パラフィン・カルナウバロウ混合ワックス、セタノール、タルク、中金箔、白色セラック、パラフィン、ポビドン、マクロゴール1500、マクロゴール4000、マクロゴール6000、ミツロウ、モノステアリン酸グリセリン、ロジン等が挙げられる。
本願発明のマトリックス徐放粒状製剤としては、経口投与可能な粒状製剤であればその粒度は特に制限されないが、マトリックス顆粒剤であるのがより好ましい。また、これらのマトリックス徐放粒状製剤は、カプセルに充填したカプセル剤として用いるのが好ましい。
本願発明のマトリックス徐放粒状製剤の製造方法としては、粒状製剤の周知の製造方法を採用することができる。例えば、本願発明の医薬組成物は、(A)薬理上活性な薬物、(B)pH依存性の高分子基剤、(C)可塑剤、(D)低融点油脂類、さらに必要に応じて、結合剤及び有機酸から選ばれる1種又は2種以上、崩壊剤、結合剤、流動化剤、滑沢剤、着色剤及び光沢化剤等を配合し、一般的に用いられている粒状製剤の製造法により製造することができる。
本願発明は上記の徐放性粒状医薬組成物の製造法にも関するものであり、以下に詳細に説明する。
本願発明の経口投与用の徐放性粒状医薬組成物は以下の
(E):薬理上活性な薬物、pH依存性の高分子基剤、及び油脂類を混合して[混合物]を調製する工程;
(F):可塑剤、結合剤の水溶液及び蒸留水を添加、撹拌して[分散液]を調製する工程;
(G):[混合末]及び[分散液]から[練合物]を製造する工程;及び
(H):[練合物]を押出し造粒し、[顆粒]を製造する工程;
上記(E)~(H)の工程によって製造することができる。
ここで、上記工程(G)において、pH依存性の高分子基剤としては、ヒドロキシプロピルメチルセルロースアセテートサクシネートが好ましく;可塑剤としては、クエン酸トリエチルが好ましく;可塑剤を、pH依存性の高分子基剤に対して5~30重量%の範囲で使用することが好ましい。
上記の工程(H)は、[練合物]を押出し造粒し、[顆粒]を製造する工程である。ここで、[練合物]を押し出し造粒後、整粒機など用い、整粒の操作を行ってもよく、整粒から[顆粒]を製造するには、通常の乾燥機を用いてアニーリング(加熱処理)を行えばよい。ここで、乾燥機としては、流動層造粒乾燥機、棚乾燥機等を挙げることができる。また、アニーリング(加熱処理)の温度、処理時間は使用する薬物等によって適宜設定を変更すればよい。具体的なアニーリング(加熱処理)の温度、処理時間としては、アニーリング(加熱処理)の装置内部の温度が40~90℃の範囲で実施するのが好ましく;アニーリング(加熱処理)の処理時間としては、15分~2時間の範囲が好ましいが、なんらこの範囲に限定されるものではない。
また、得られた[顆粒]は、篩を用いて適宜粒度を揃えればよい。
本願発明の経口投与用の徐放性粒状医薬組成物は以下の
(E):薬理上活性な薬物、pH依存性の高分子基剤、及び油脂類を混合して[混合物]を調製する工程;
(F):可塑剤、結合剤の水溶液及び蒸留水を添加、撹拌して[分散液]を調製する工程;
(G):[混合末]及び[分散液]から[練合物]を製造する工程;及び
(H):[練合物]を押出し造粒し、[顆粒]を製造する工程;
上記(E)~(H)の工程によって製造することができる。
ここで、上記工程(G)において、pH依存性の高分子基剤としては、ヒドロキシプロピルメチルセルロースアセテートサクシネートが好ましく;可塑剤としては、クエン酸トリエチルが好ましく;可塑剤を、pH依存性の高分子基剤に対して5~30重量%の範囲で使用することが好ましい。
上記の工程(H)は、[練合物]を押出し造粒し、[顆粒]を製造する工程である。ここで、[練合物]を押し出し造粒後、整粒機など用い、整粒の操作を行ってもよく、整粒から[顆粒]を製造するには、通常の乾燥機を用いてアニーリング(加熱処理)を行えばよい。ここで、乾燥機としては、流動層造粒乾燥機、棚乾燥機等を挙げることができる。また、アニーリング(加熱処理)の温度、処理時間は使用する薬物等によって適宜設定を変更すればよい。具体的なアニーリング(加熱処理)の温度、処理時間としては、アニーリング(加熱処理)の装置内部の温度が40~90℃の範囲で実施するのが好ましく;アニーリング(加熱処理)の処理時間としては、15分~2時間の範囲が好ましいが、なんらこの範囲に限定されるものではない。
また、得られた[顆粒]は、篩を用いて適宜粒度を揃えればよい。
本願発明における(A)薬理上活性な薬物の本発明医薬組成物中の含有比としては、薬理上活性な薬物のフリー体換算で、通常0.1~50重量%が好ましく、10~25重量%がより好ましい。特に、本発明医薬組成物の剤形は、顆粒をカプセルに充填したカプセル剤が好ましいが、その場合、1カプセルあたりに含有される薬理上活性な薬物の含有量は、用いる薬物がたとえば化合物(1)である場合、フリー体換算で、通常0.5~500mgであり;1~100mgが好ましく;5~75mgがより好ましく;15~60mgが特により好ましい。
本願発明の別の態様について以下に説明する。
本願発明の徐放性製剤は、(A)薬理上活性な薬物、(B)ヒドロキシプロピルメチルセルロースアセテートサクシネート、(C)可塑剤、及び(D)ポリエチレングリコール、を混合した後、押し出し造粒して得られる徐放性製剤である。
本発明の製剤は、(A)成分、(B)成分、(C)成分及び(D)成分を混合した後、押し出し造粒して得られることに特徴があり、例えば、(A)成分、(C)成分及び(D)成分を混合した後、押し出し造粒して得られた製剤に(B)成分を含有するコーティングを施した製剤は本発明には包含されない。ただし、例えば、(A)成分、(B)成分、(C)成分及び(D)成分を混合した後、押し出し造粒して得られた製剤に、(B)成分含有するコーティングを施した製剤は本発明に包含される。
本願発明の製剤に使用される(A)成分である「薬理上活性な薬物」としては、上述の化合物を挙げることができ、また体内で薬理上活性な薬物に変換されるプロドラッグであってもよい。本願発明の製剤における(A)成分の含有率は、0.1~60重量%が好ましく、1~50重量%がより好ましく、5~35重量%がさらにより好ましい。
本願発明の製剤に使用される(B)成分である「ヒドロキシプロピルメチルセルロースアセテートサクシネート(HPMCAS)」としては、上述の物質を挙げることができる。HPMCASのグレードは、LF、MF又はHFが好ましく、MF又はHFがより好ましい。
また、本願発明の製剤に使用される(B)成分であるHPMCASの平均粒子径(D50)は、好ましくは50μm以下であり、より好ましくは20μm以下であり、さらにより好ましくは10μm以下であり、特に好ましくは5μm以下である。
なお、本明細書において「D50」とは、レーザー回折式の測定装置であるHELOS(株式会社日本レーザー)を使用して測定した場合に、積算分布曲線の中央値に相当する粒子径、すなわちメジアン径をいう。また、本明細書において「D90」とは、上記HELOSを使用して測定した場合に、積算分布曲線の90%に相当する粒子径をいう。例えば、D90が20μmであるとは、測定した粉体のうち90%が20μm以下の粒子径であり、残りの10%が20μmより大きい粒子径を有していることをいう。
本願発明の製剤における(B)成分の含有率は、40~90重量%が好ましく、50~80重量%がより好ましく、60~75重量%がさらにより好ましい。
本願発明の製剤に使用される(C)成分である「可塑剤」としては、上述の物質を挙げることができ、好ましくはクエン酸トリエチルである。本願発明の製剤における(C)成分の含有率は、5~30重量%が好ましい。
本願発明の製剤に使用される(D)成分である「ポリエチレングリコール(PEG)」としては、上述の物質を挙げることができ、平均分子量4000~12000のPEGが好ましく、平均分子量7000~10500のPEGがより好ましい。かかる分子量のPEGは、例えば、ポリエチレングリコール6000(CAS番号:25322-68-3)を挙げることができる。本願発明の製剤における(D)成分の含有率は、1~10重量%が好ましく、2~8重量%がより好ましく、3~6重量%がさらにより好ましい。
本願発明の製剤は、(A)成分~(D)成分に加えて、さらに結合剤及び/又は有機酸を含有してもよい。結合剤としては、上述の物質を挙げることができ、ヒドロキシプロピルセルロース又はヒドロキシプロピルメチルセルロースが好ましく、ヒドロキシプロピルセルロースがより好ましい。有機酸としては、上述の物質を挙げることができ、フマル酸又はアルギン酸が好ましく、フマル酸がより好ましい。本願発明の製剤が結合剤及び/又は有機酸を含有する場合、製剤における結合剤の含有率は、0.1~10重量%が好ましく、0.5~2重量%がより好ましく、製剤における有機酸の含有率は0.1~40重量%が好ましく、1~20重量%がより好ましい。
本願発明の製剤には、本発明の効果に影響を与えない範囲内で、さらに上述の、滑沢剤、着色剤、及び光沢化剤等の添加剤を配合してもよい。
本願発明の製剤は、(A)~(D)を混合した後に、押し出し造粒することで製造される。より詳細には、(A)成分、(B)成分、(C)成分及び(D)成分を混合し、結合剤を含有する溶液あるいは蒸留水を混合物に添加した後、練合し、押出し造粒して製造することができる。得られた製剤を必要に応じて圧縮成型してもよいし、コーティングを施しても良い。混合、練合、押し出し造粒、圧縮成型、コーティングなどは、当該分野で周知の方法を用いて行えばよい。本願発明の製剤にその他の添加剤を配合する場合、混合工程、練合工程、打錠工程又はコーティング工程のいずれの工程で配合しても良い。
このように得られた製剤は、各成分がマトリックス状になっていることが好ましい。
かくして得られた本願発明の製剤は、上述の発明の効果に記載したように、酸性溶液中における過剰放出を防ぐ良好な錠剤強度を持ち、かつ中性溶液中で良好な溶出性を持つことから、十二指腸、小腸から下部消化管に至るまで、含有する薬理上活性な薬物の持続的な溶出を維持する効果が得られる。
次に実施例を挙げて本願発明を詳細に説明するが、本願発明は何らこれら実施例に限定されるものではない。
実施例において使用する略号は以下の通りである。
・HPMCAS-HF1:ヒドロキシプロピルメチルセルロースアセテートサクシネートHFグレード(D50=5μm、D90=11μm、スクシノイル基の含有率が7.3~8.0%)(信越化学株式会社製)
・HPMCAS-HF2:ヒドロキシプロピルメチルセルロースアセテートサクシネートHFグレード(D50=5μm、D90=11μm、スクシノイル基の含有率が4.0~7.2%)(信越化学株式会社製)
・HPMCAS-MF:ヒドロキシプロピルメチルセルロースアセテートサクシネートMFグレード(D50=5μm、D90=11μm)(信越化学株式会社製)
・PEG6000:ポリエチレングリコール6000
・HPC-L:ヒドロキシプロピルセルロースLグレード(日本曹達株式会社製)
・HPMCAS-HF1:ヒドロキシプロピルメチルセルロースアセテートサクシネートHFグレード(D50=5μm、D90=11μm、スクシノイル基の含有率が7.3~8.0%)(信越化学株式会社製)
・HPMCAS-HF2:ヒドロキシプロピルメチルセルロースアセテートサクシネートHFグレード(D50=5μm、D90=11μm、スクシノイル基の含有率が4.0~7.2%)(信越化学株式会社製)
・HPMCAS-MF:ヒドロキシプロピルメチルセルロースアセテートサクシネートMFグレード(D50=5μm、D90=11μm)(信越化学株式会社製)
・PEG6000:ポリエチレングリコール6000
・HPC-L:ヒドロキシプロピルセルロースLグレード(日本曹達株式会社製)
D50及びD90は、レーザー回折式粒子径分布測定装置HELOS&RODOS(株式会社日本レーザー)を用いて、分散圧3bar、測定レンジR4で測定した。
製剤の回収率は、次の式:
(押し出し造粒で得られた造粒物のうち特定の粒度分布を有する顆粒の重量)/(練合物の重量)×100
で算出し、80%以上の場合は○、80%未満の場合は×と評価した。
(押し出し造粒で得られた造粒物のうち特定の粒度分布を有する顆粒の重量)/(練合物の重量)×100
で算出し、80%以上の場合は○、80%未満の場合は×と評価した。
酸性溶液又は中性溶液中での溶出性試験は次のように行った。
(酸性溶液中での溶出試験)
0.01規定塩酸(900mL)中、37±0.5℃において、パドル法毎分50回転で溶出試験を行い、経時的に溶出液中の薬物平均溶出率を算出した。
(酸性溶液中での溶出試験)
0.01規定塩酸(900mL)中、37±0.5℃において、パドル法毎分50回転で溶出試験を行い、経時的に溶出液中の薬物平均溶出率を算出した。
(中性溶液中での溶出試験)
pH6.8のリン酸緩衝液(900mL)中、37±0.5℃において、パドル法毎分50回転で溶出試験を行い経時的に溶出液中の薬物平均溶出率を算出した。
pH6.8のリン酸緩衝液(900mL)中、37±0.5℃において、パドル法毎分50回転で溶出試験を行い経時的に溶出液中の薬物平均溶出率を算出した。
(実施例1)
表1に示す処方1~4につき、化合物(1a)粉砕品及びHPMCAS-HF1をビニル袋内で5分間混合後、得られた混合物に、クエン酸トリエチルを分散させた5%HPC-L水溶液40mL及び精製水90mLを添加して、ハイスピードミキサーにて5分間練合した。得られた練合物を押出し造粒機(ドームグラン、スクリーン径:1.0mmφ)にて押し出し造粒し、棚乾燥機にてアニーリング(80°C,2hr)した。アニーリング後の造粒物を14号及び18号の篩で篩過し、18号篩上14号篩下の顆粒を回収して製剤を調製した。製剤の回収率を表2に、酸性溶液での溶出試験の結果を図1に示した。
表1に示す処方1~4につき、化合物(1a)粉砕品及びHPMCAS-HF1をビニル袋内で5分間混合後、得られた混合物に、クエン酸トリエチルを分散させた5%HPC-L水溶液40mL及び精製水90mLを添加して、ハイスピードミキサーにて5分間練合した。得られた練合物を押出し造粒機(ドームグラン、スクリーン径:1.0mmφ)にて押し出し造粒し、棚乾燥機にてアニーリング(80°C,2hr)した。アニーリング後の造粒物を14号及び18号の篩で篩過し、18号篩上14号篩下の顆粒を回収して製剤を調製した。製剤の回収率を表2に、酸性溶液での溶出試験の結果を図1に示した。


<試験結果>
回収率が良かった処方は、部分アルファ化デンプン、モノステアリン酸グリセリン又はPEG6000を添加した処方2~4であった。しかし、図1に示す通り、部分アルファ化デンプンを添加した処方2は、処方1、処方3及び処方4と比較して酸性液中での薬物溶出速度が速かった。
回収率が良かった処方は、部分アルファ化デンプン、モノステアリン酸グリセリン又はPEG6000を添加した処方2~4であった。しかし、図1に示す通り、部分アルファ化デンプンを添加した処方2は、処方1、処方3及び処方4と比較して酸性液中での薬物溶出速度が速かった。
(実施例2)
表3に示す処方5~8につき、実施例1と同様に造粒物を調製し、得られた造粒物を14号及び30号の篩で篩過し、30号篩上14号篩下の顆粒を回収して製剤を調製した。製剤の回収率を表4に、
酸性溶液での溶出試験の結果を図2に示した。
表3に示す処方5~8につき、実施例1と同様に造粒物を調製し、得られた造粒物を14号及び30号の篩で篩過し、30号篩上14号篩下の顆粒を回収して製剤を調製した。製剤の回収率を表4に、
酸性溶液での溶出試験の結果を図2に示した。


<試験結果>
回収率が良かった処方は、部分アルファ化デンプン、モノステアリン酸グリセリン又はPEG6000を添加した処方6~8であった。しかし、図2に示す通り、部分アルファ化デンプンを添加した処方6では、処方5、処方7及び8と比較して酸性液中での薬物溶出速度が速かった。
回収率が良かった処方は、部分アルファ化デンプン、モノステアリン酸グリセリン又はPEG6000を添加した処方6~8であった。しかし、図2に示す通り、部分アルファ化デンプンを添加した処方6では、処方5、処方7及び8と比較して酸性液中での薬物溶出速度が速かった。
(実施例3)
表5に示す処方9及び10につき、実施例1と同様に造粒物を調製し、得られた造粒物を14号及び18号の篩で篩過し、18号篩上14号篩下の顆粒を回収して製剤を調製した。得られた製剤をガラス瓶に入れて密閉し、40℃、50℃又は60℃で1日間又は1週間保存した。調製直後の製剤と保存後の製剤について中性溶液での溶出試験を行い、その結果を図3及び4に示した。
表5に示す処方9及び10につき、実施例1と同様に造粒物を調製し、得られた造粒物を14号及び18号の篩で篩過し、18号篩上14号篩下の顆粒を回収して製剤を調製した。得られた製剤をガラス瓶に入れて密閉し、40℃、50℃又は60℃で1日間又は1週間保存した。調製直後の製剤と保存後の製剤について中性溶液での溶出試験を行い、その結果を図3及び4に示した。

<試験結果>
図3に示す通り、モノステアリン酸グリセリンを添加した処方9は、40℃又は50℃で1日保存することにより薬物溶出が遅延した。また、60℃で1日間保存することにより溶出挙動が速くなった。一方、図4に示す通り、PEG6000を添加した処方10は、40℃、50℃又は60℃で1週間保存後も溶出挙動の変化はほとんど認められなかった。
図3に示す通り、モノステアリン酸グリセリンを添加した処方9は、40℃又は50℃で1日保存することにより薬物溶出が遅延した。また、60℃で1日間保存することにより溶出挙動が速くなった。一方、図4に示す通り、PEG6000を添加した処方10は、40℃、50℃又は60℃で1週間保存後も溶出挙動の変化はほとんど認められなかった。
(実施例4)
表3に示した処方7及び処方8について、得られた製剤をガラス瓶に入れて密閉し、50℃で3日間保存した。調製直後の製剤と保存後の製剤について中性溶液での溶出試験を行った。その結果、モノステアリン酸グリセリンを添加した処方7は、50℃で3日間保存することにより薬物溶出が遅延し、PEG6000を添加した処方8は、50℃で3日間保存後も、溶出挙動の変化はほとんど認められなかった。
表3に示した処方7及び処方8について、得られた製剤をガラス瓶に入れて密閉し、50℃で3日間保存した。調製直後の製剤と保存後の製剤について中性溶液での溶出試験を行った。その結果、モノステアリン酸グリセリンを添加した処方7は、50℃で3日間保存することにより薬物溶出が遅延し、PEG6000を添加した処方8は、50℃で3日間保存後も、溶出挙動の変化はほとんど認められなかった。
(実施例5)
表6に示す処方11につき、実施例1と同様に造粒物を調製し、得られた造粒物を14号及び30号の篩で篩下し、30号篩上14号篩下の顆粒を回収して製剤を調製した。得られた製剤について、中性溶液中での溶出試験を行い、その結果を図5に示した。
表6に示す処方11につき、実施例1と同様に造粒物を調製し、得られた造粒物を14号及び30号の篩で篩下し、30号篩上14号篩下の顆粒を回収して製剤を調製した。得られた製剤について、中性溶液中での溶出試験を行い、その結果を図5に示した。

<試験結果>
図5に示すように中性溶液において持続的な薬物溶出挙動が得られた。
図5に示すように中性溶液において持続的な薬物溶出挙動が得られた。
(実施例6)
表7に示す処方12につき、実施例1と同様に造粒物を調製し(ただし押出し造粒のスクリーン径は1.2mmφ)、得られた造粒物を12号及び18号の篩で篩下し、18号篩上12号下の顆粒を回収して製剤を調製した。得られた製剤について、中性溶液中での溶出試験を行い、その結果を図6に示した。
表7に示す処方12につき、実施例1と同様に造粒物を調製し(ただし押出し造粒のスクリーン径は1.2mmφ)、得られた造粒物を12号及び18号の篩で篩下し、18号篩上12号下の顆粒を回収して製剤を調製した。得られた製剤について、中性溶液中での溶出試験を行い、その結果を図6に示した。

<試験結果>
図6に示すように中性溶液において持続的な薬物溶出挙動が得られた。
図6に示すように中性溶液において持続的な薬物溶出挙動が得られた。
(実施例7)
表8に示す処方13及び14につき、実施例1と同様に造粒物を調製し、得られた造粒物を14号及び30号の篩で篩過し、30号篩上14号篩下の顆粒を回収して製剤を調製した。得られた製剤をヒドロキシプロピルメチルセルロースカプセルに充填した。
表8に示す処方13及び14につき、実施例1と同様に造粒物を調製し、得られた造粒物を14号及び30号の篩で篩過し、30号篩上14号篩下の顆粒を回収して製剤を調製した。得られた製剤をヒドロキシプロピルメチルセルロースカプセルに充填した。

<試験結果>
これらのカプセルをヒトに投与した結果、同一薬物量の化合物(1a)を水に溶解させた製剤と比較し、Cmax低減及び24時間後の血中濃度の増大を示し、徐放製剤として良好なプロファイルを示すことが確認された。
これらのカプセルをヒトに投与した結果、同一薬物量の化合物(1a)を水に溶解させた製剤と比較し、Cmax低減及び24時間後の血中濃度の増大を示し、徐放製剤として良好なプロファイルを示すことが確認された。
本願発明は、化合物(1)などの薬理上活性な薬物を含有する、経口投与用徐放性マトリックス粒状製剤の製造に利用することができる。
Claims (17)
- (A)薬理上活性な薬物、
(B)ヒドロキシプロピルメチルセルロースアセテートサクシネート、
(C)可塑剤、及び、
(D)ポリエチレングリコール、
を混合した後、押し出し造粒して得られる徐放性製剤。 - 製剤中の(B)成分の含有率が40~90重量%である、請求項1に記載の製剤。
- 製剤中の(B)成分の含有率が50~80重量%である、請求項1に記載の製剤。
- (C)成分がクエン酸トリエチルである、請求項1~3いずれか1項に記載の製剤。
- 製剤中の(C)成分の含有率が5~30重量%である、請求項1~4のいずれか1項に記載の製剤。
- (D)成分が平均分子量4000~12000の範囲のポリエチレングリコールである、請求項1~5のいずれか1項に記載の製剤。
- (D)成分が平均分子量7000~10500の範囲のポリエチレングリコールである、請求項1~5のいずれか1項に記載の製剤。
- (D)成分がポリエチレングリコール6000である、請求項1~5いずれか1項に記載の製剤。
- 製剤中の(D)成分の含有率が1~10重量%である、請求項1~8のいずれか1項に記載の製剤。
- 製剤中の(D)成分の含有率が2~8重量%である、請求項1~8のいずれか1項に記載の製剤。
- さらに、結合剤及び/又は有機酸を含有する、請求項1~10のいずれか1項に記載の製剤。
- 結合剤がヒドロキシプロピルセルロースである、請求項11に記載の製剤。
- 有機酸が、フマル酸又はアルギン酸である、請求項11又は12に記載の製剤。
- 有機酸がフマル酸である、請求項11又は12に記載の製剤。
- (A)成分が塩基性薬物である、請求項1~14のいずれか1項に記載の製剤。
- (A)成分が、
(±)-1-(カルバゾール-4-イルオキシ)-3-[[2-(o-メトキシフェノキシ)エチル]アミノ]-2-プロパノール;
N1-(5-クロロピリジン-2-イル)-N2-((1S,2R,4S)-4-[(ジメチルアミノ)カルボニル]-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}シクロヘキシル)エタンジアミド、及び、
N1-(5-クロロピリジン-2-イル)-N2-[(1S,2R,4S)-2-{[(5-メチル-4,5,6,7-テトラヒドロチアゾロ[5,4-c]ピリジン-2-イル)カルボニル]アミノ}-4-([1,3,4]オキサジアゾール-2-イル)シクロヘキシル]エタンジアミド、
からなる群より選ばれる化合物、その薬理上許容される塩、又はそれらの水和物である、請求項1~14のいずれか1項に記載の製剤。 - 製剤中の(A)成分の含有率が0.1~50重量%である、請求項1~16のいずれか1項に記載の製剤。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012500676A JP5870023B2 (ja) | 2010-02-22 | 2011-02-21 | 経口用徐放性固形製剤 |
| EP20110744791 EP2540317A4 (en) | 2010-02-22 | 2011-02-21 | SOLID PREPARATION WITH DELAYED RELEASE FOR ORAL ADMINISTRATION |
| US13/591,949 US20130012535A1 (en) | 2010-02-22 | 2012-08-22 | Sustained-release solid preparation for oral use |
| US14/079,859 US20140070446A1 (en) | 2010-02-22 | 2013-11-14 | Sustained-release solid preparation for oral use |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-035883 | 2010-02-22 | ||
| JP2010035883 | 2010-02-22 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/591,949 Continuation US20130012535A1 (en) | 2010-02-22 | 2012-08-22 | Sustained-release solid preparation for oral use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011102505A1 true WO2011102505A1 (ja) | 2011-08-25 |
Family
ID=44483088
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/053643 WO2011102505A1 (ja) | 2010-02-22 | 2011-02-21 | 経口用徐放性固形製剤 |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US20130012535A1 (ja) |
| EP (1) | EP2540317A4 (ja) |
| JP (1) | JP5870023B2 (ja) |
| TW (1) | TW201132646A (ja) |
| WO (1) | WO2011102505A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013022059A1 (ja) | 2011-08-10 | 2013-02-14 | 第一三共株式会社 | ジアミン誘導体含有医薬組成物 |
| US9918975B2 (en) | 2010-03-19 | 2018-03-20 | Daiichi Sankyo Company, Limited | Method for improving dissolution of anticoagulant agent |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2540318B1 (en) | 2010-02-22 | 2018-10-24 | Daiichi Sankyo Company, Limited | Sustained-release solid preparation for oral use |
| ES2706994T3 (es) | 2012-09-03 | 2019-04-02 | Daiichi Sankyo Co Ltd | Composición farmacéutica de liberación prolongada de administración oral que contiene clorhidrato de hidromorfona |
| US10383944B2 (en) * | 2014-04-08 | 2019-08-20 | Dow Global Technologies Llc | Dispersion comprising an esterified cellulose ether |
| JP6426876B2 (ja) * | 2015-07-28 | 2018-11-21 | ダウ グローバル テクノロジーズ エルエルシー | エステル化セルロースエーテルを含む分散体 |
| TWI812602B (zh) * | 2016-12-01 | 2023-08-21 | 日商第一三共股份有限公司 | 含二胺衍生物之口腔崩散錠及其製造方法 |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63107933A (ja) * | 1986-04-30 | 1988-05-12 | Chugai Pharmaceut Co Ltd | シヨ糖硫酸エステルアルミニウム塩製剤の製造法 |
| JP2001513752A (ja) * | 1996-09-12 | 2001-09-04 | ロシュ ダイアグノスティクス ゲゼルシャフト ミット ベシュレンクテル ハフツング | 迅速崩壊ペレット |
| JP2001522794A (ja) * | 1997-11-12 | 2001-11-20 | ベーリンガー・マンハイム・ファーマシューティカルズ・コーポレイション−スミスクライン・ベックマン・コーポレイション・リミテッド・パートナーシップ・ナンバー1 | カルベジロールの新規経口剤形 |
| JP2001524131A (ja) * | 1997-05-09 | 2001-11-27 | セイジ、ファーマスーティカルズ、インク | 安定な経口医薬品剤形 |
| WO2003000680A1 (fr) | 2001-06-20 | 2003-01-03 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| WO2003000657A1 (fr) | 2001-06-20 | 2003-01-03 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| WO2003016302A1 (fr) | 2001-08-09 | 2003-02-27 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| JP2004026750A (ja) * | 2002-06-27 | 2004-01-29 | Nippon Shinyaku Co Ltd | 薬物の安定化法 |
| WO2004058715A1 (ja) | 2002-12-25 | 2004-07-15 | Daiichi Pharmaceutical Co., Ltd. | ジアミン誘導体 |
| WO2005000312A1 (ja) * | 2003-06-27 | 2005-01-06 | Otsuka Pharmaceutical Co., Ltd. | 薬物徐放性粒子及びその製法 |
| WO2006070781A1 (ja) * | 2004-12-27 | 2006-07-06 | Eisai R & D Management Co., Ltd. | 塩基性薬物又はその塩を含有するマトリックス型徐放性製剤およびその製造方法 |
| JP2007537175A (ja) * | 2004-05-11 | 2007-12-20 | バイエル・ヘルスケア・アクチェンゲゼルシャフト | バルデナフィルを含有する制御放出製剤 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0797987B1 (en) * | 1994-12-19 | 2003-10-15 | Daiichi Pharmaceutical Co., Ltd. | Sustained-release granular preparation and process for producing the same |
| EP0807433A4 (en) * | 1994-12-27 | 2005-12-28 | Akzo Nobel Nv | PREPARING WITH DELAYED RELEASE |
| DE69917618T2 (de) * | 1998-04-03 | 2005-06-23 | Egalet A/S | Zusammensetzung mit kontrollierter wirkstoff-freisetzung |
| EP1027885B1 (en) * | 1999-02-09 | 2008-07-09 | Pfizer Products Inc. | Basic drug compositions with enhanced bioavailability |
| JP4644397B2 (ja) * | 2001-09-05 | 2011-03-02 | 信越化学工業株式会社 | 難溶性薬物を含む医薬用固形製剤の製造方法 |
| US20040052844A1 (en) * | 2002-09-16 | 2004-03-18 | Fang-Hsiung Hsiao | Time-controlled, sustained release, pharmaceutical composition containing water-soluble resins |
| WO2004041252A1 (en) * | 2002-11-08 | 2004-05-21 | Egalet A/S | Controlled release carvedilol compositions |
| WO2005048979A2 (en) * | 2003-10-06 | 2005-06-02 | Torrent Pharmaceuticals Limited | Pharmaceutical composition having casing with multiple micro tablets |
| US8007826B2 (en) * | 2003-12-11 | 2011-08-30 | Acorda Therapeutics, Inc. | Sustained release aminopyridine composition |
| JP2007223903A (ja) * | 2004-07-09 | 2007-09-06 | Takeda Chem Ind Ltd | 新規な固体分散体およびその製造方法 |
| US20060008418A1 (en) * | 2004-07-12 | 2006-01-12 | Solidtech Animal Health, Inc. | Packaging and method for solid dose administration of an electronic identification chip and medicaments |
| GB2418854B (en) * | 2004-08-31 | 2009-12-23 | Euro Celtique Sa | Multiparticulates |
| US20060280789A1 (en) * | 2004-12-27 | 2006-12-14 | Eisai Research Institute | Sustained release formulations |
| US20080138404A1 (en) * | 2006-12-06 | 2008-06-12 | Biovail Laboratories International S.R.L. | Extended release formulations of carvedilol |
| BRPI0809205B8 (pt) * | 2007-03-29 | 2021-05-25 | Daiichi Sankyo Co Ltd | composição farmacêutica |
| SI22849A (sl) * | 2008-08-01 | 2010-02-26 | Krka, Tovarna Zdravil, D.D., Novo Mesto | Ropinirolni pripravek |
-
2011
- 2011-02-21 EP EP20110744791 patent/EP2540317A4/en not_active Withdrawn
- 2011-02-21 TW TW100105565A patent/TW201132646A/zh unknown
- 2011-02-21 JP JP2012500676A patent/JP5870023B2/ja active Active
- 2011-02-21 WO PCT/JP2011/053643 patent/WO2011102505A1/ja active Application Filing
-
2012
- 2012-08-22 US US13/591,949 patent/US20130012535A1/en not_active Abandoned
-
2013
- 2013-11-14 US US14/079,859 patent/US20140070446A1/en not_active Abandoned
Patent Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63107933A (ja) * | 1986-04-30 | 1988-05-12 | Chugai Pharmaceut Co Ltd | シヨ糖硫酸エステルアルミニウム塩製剤の製造法 |
| JP2001513752A (ja) * | 1996-09-12 | 2001-09-04 | ロシュ ダイアグノスティクス ゲゼルシャフト ミット ベシュレンクテル ハフツング | 迅速崩壊ペレット |
| JP2001524131A (ja) * | 1997-05-09 | 2001-11-27 | セイジ、ファーマスーティカルズ、インク | 安定な経口医薬品剤形 |
| JP2001522794A (ja) * | 1997-11-12 | 2001-11-20 | ベーリンガー・マンハイム・ファーマシューティカルズ・コーポレイション−スミスクライン・ベックマン・コーポレイション・リミテッド・パートナーシップ・ナンバー1 | カルベジロールの新規経口剤形 |
| WO2003000680A1 (fr) | 2001-06-20 | 2003-01-03 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| WO2003000657A1 (fr) | 2001-06-20 | 2003-01-03 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| WO2003016302A1 (fr) | 2001-08-09 | 2003-02-27 | Daiichi Pharmaceutical Co., Ltd. | Derives de diamine |
| JP2004026750A (ja) * | 2002-06-27 | 2004-01-29 | Nippon Shinyaku Co Ltd | 薬物の安定化法 |
| WO2004058715A1 (ja) | 2002-12-25 | 2004-07-15 | Daiichi Pharmaceutical Co., Ltd. | ジアミン誘導体 |
| WO2005000312A1 (ja) * | 2003-06-27 | 2005-01-06 | Otsuka Pharmaceutical Co., Ltd. | 薬物徐放性粒子及びその製法 |
| JP2007537175A (ja) * | 2004-05-11 | 2007-12-20 | バイエル・ヘルスケア・アクチェンゲゼルシャフト | バルデナフィルを含有する制御放出製剤 |
| JP2007537183A (ja) * | 2004-05-11 | 2007-12-20 | バイエル・ヘルスケア・アクチェンゲゼルシャフト | バルデナフィルを含有する制御放出製剤 |
| WO2006070781A1 (ja) * | 2004-12-27 | 2006-07-06 | Eisai R & D Management Co., Ltd. | 塩基性薬物又はその塩を含有するマトリックス型徐放性製剤およびその製造方法 |
Non-Patent Citations (16)
| Title |
|---|
| AMERICAN CHEMICAL SOCIETY-226TH NATIONAL MEETING, NEW YORK CITY, NY, USA, 2003 |
| D. KUBITZA ET AL.: "Multiple dose escalation study investigating the pharmacodynamics, safety, and pharmacokinetic of Bay59-7939, an oral, direct Factor Xa inhibitor, in healthy male subjects", BLOOD, 2003, pages 102 |
| GENERATING GREATER VALUE FROM OUR PRODUCTS AND PIPELINE. AVENTIS SA COMPANY PRESENTATION, 5 February 2004 (2004-02-05) |
| J. R. PRUITT ET AL., J. MED. CHEM., vol. 46, 2003, pages 5298 - 5313 |
| M. WILEY ET AL., 228TH ACS NATIONAL MEETING, PHILADELPHIA, 22 August 2004 (2004-08-22) |
| PINTO DJP; ORWAT MJ; LAM PYS ET AL.: "Discovery of 1-(4-Methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H-pyrazolo[3,4-c]pyridine-3-carboxamide (Apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa", J. MED. CHEM., vol. 50, no. 22, 2007, pages 5339 - 56, XP007915445, DOI: doi:10.1021/jm070245n |
| S. TAKEHANA ET AL., JAPANESE JOURNAL OF PHARMACOLOGY, vol. 82, no. 1, 2000, pages 213P |
| S. YOUNG, MEDICINAL CHEMISTRY-12TH RSC-SCI SYMPOSIUM, 7 September 2003 (2003-09-07) |
| SUSANNE R ET AL., J. MED. CHEM., vol. 48, 2005, pages 5900 - 5908 |
| T. KAYAHARA ET AL., JAPANESE JOURNAL OF PHARMACOLOGY, vol. 82, no. 1, 2000, pages 213P |
| WFO DRUG INFORMATION, vol. 16, no. 3, 2002, pages 257 |
| WFO DRUG INFORMATION, vol. 18, no. 3, 2004, pages 260 |
| WFO DRUG INFORMATION, vol. 20, no. 1, 2006, pages 38 |
| WFO DRUG INFORMATION, vol. 22, no. 3, 2008, pages 226 - 227 |
| XVIITH CONGRESS OF THE INTERNATIONAL SOCIETY FOR THROMBOSIS AND HAEMOSTASIS, WASHINGTON D. C., 14 August 1999 (1999-08-14) |
| ZHANG P; HUANG W; ZHU BY ET AL.: "Discovery of betrixaban (PRT054021), N-(5-chloropyridin-2-yl)-2-(4-(N,N-dimethylcarbamimidoyl)benzamido)-5-methoxybenzamide, a highly potent, selective, and orally efficacious factor Xa inhibitor", BIOORG MED. CHEM. LETT., vol. 19, no. 8, 2009, pages 2179 - 85, XP026079433, DOI: doi:10.1016/j.bmcl.2009.02.111 |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9918975B2 (en) | 2010-03-19 | 2018-03-20 | Daiichi Sankyo Company, Limited | Method for improving dissolution of anticoagulant agent |
| WO2013022059A1 (ja) | 2011-08-10 | 2013-02-14 | 第一三共株式会社 | ジアミン誘導体含有医薬組成物 |
| KR20140054029A (ko) | 2011-08-10 | 2014-05-08 | 다이이찌 산쿄 가부시키가이샤 | 디아민 유도체 함유 의약 조성물 |
| US9402907B2 (en) | 2011-08-10 | 2016-08-02 | Daiichi Sankyo Company, Limited | Pharmaceutical composition containing diamine derivative |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5870023B2 (ja) | 2016-02-24 |
| US20130012535A1 (en) | 2013-01-10 |
| TW201132646A (en) | 2011-10-01 |
| JPWO2011102505A1 (ja) | 2013-06-17 |
| EP2540317A1 (en) | 2013-01-02 |
| US20140070446A1 (en) | 2014-03-13 |
| EP2540317A4 (en) | 2014-04-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5870023B2 (ja) | 経口用徐放性固形製剤 | |
| TWI409064B (zh) | Pharmaceutical composition | |
| ES2605034T3 (es) | Composición farmacéutica con solubilidad mejorada | |
| JP5714562B2 (ja) | 経口用徐放性固形製剤 | |
| ES2601884T3 (es) | Procedimiento para mejorar la capacidad de disolución de un anticoagulante | |
| US9629808B2 (en) | Sustained-release solid preparation for oral use | |
| JP2018035171A (ja) | 医薬製剤 | |
| JPWO2013022059A1 (ja) | ジアミン誘導体含有医薬組成物 | |
| AU2016259762B2 (en) | Vortioxetine pyroglutamate | |
| AU2013365715B2 (en) | A pharmaceutical composition containing candesartan cilexetil and amlodipine | |
| JP6126780B2 (ja) | ロキソプロフェンナトリウム及びトラネキサム酸を含有する固形製剤 | |
| JP6129184B2 (ja) | ヒドロモルフォン塩酸塩含有の経口用徐放性医薬組成物 | |
| JP6181044B2 (ja) | カプセル剤 | |
| JP5534645B2 (ja) | 無包装状態において安定性に優れた塩酸サルポグレラート含有経口投与製剤 | |
| JPWO2003075919A1 (ja) | 塩酸ピルジカイニド含有錠剤(乾式) | |
| BR112015018952B1 (pt) | Composições farmacêuticas contendo dexcetoprofeno e tramadol, seu método de fabricação e seu uso | |
| JP2012140411A (ja) | ロキソプロフェンナトリウム及びdl−メチルエフェドリン塩酸塩を含有する固形製剤 | |
| JP2016204294A (ja) | セルトラリン及び/又はその薬学上許容しうる塩を含む製剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11744791 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012500676 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011744791 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |