WO2011012746A2 - Compuestos inhibidores de apaf-1 - Google Patents
Compuestos inhibidores de apaf-1 Download PDFInfo
- Publication number
- WO2011012746A2 WO2011012746A2 PCT/ES2010/000349 ES2010000349W WO2011012746A2 WO 2011012746 A2 WO2011012746 A2 WO 2011012746A2 ES 2010000349 W ES2010000349 W ES 2010000349W WO 2011012746 A2 WO2011012746 A2 WO 2011012746A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- aryl
- heteroaryl
- alkyl
- apoptosis
- ethyl
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims description 62
- 239000003112 inhibitor Substances 0.000 title description 9
- 230000006907 apoptotic process Effects 0.000 claims abstract description 28
- 238000011282 treatment Methods 0.000 claims abstract description 15
- 230000001575 pathological effect Effects 0.000 claims abstract description 13
- 230000004962 physiological condition Effects 0.000 claims abstract description 13
- 238000011321 prophylaxis Methods 0.000 claims abstract description 11
- -1 2- (4-fluorophenyl) ethyl Chemical group 0.000 claims description 38
- 238000000034 method Methods 0.000 claims description 32
- 125000000217 alkyl group Chemical group 0.000 claims description 28
- 125000003118 aryl group Chemical group 0.000 claims description 23
- 125000001072 heteroaryl group Chemical group 0.000 claims description 21
- 231100000135 cytotoxicity Toxicity 0.000 claims description 15
- 230000003013 cytotoxicity Effects 0.000 claims description 15
- 210000000056 organ Anatomy 0.000 claims description 13
- 150000003839 salts Chemical class 0.000 claims description 13
- 210000004027 cell Anatomy 0.000 claims description 12
- 230000007170 pathology Effects 0.000 claims description 11
- 206010012601 diabetes mellitus Diseases 0.000 claims description 10
- 238000004321 preservation Methods 0.000 claims description 10
- 125000001424 substituent group Chemical group 0.000 claims description 10
- 125000000623 heterocyclic group Chemical group 0.000 claims description 9
- 206010021143 Hypoxia Diseases 0.000 claims description 8
- 229910052736 halogen Inorganic materials 0.000 claims description 8
- 150000002367 halogens Chemical class 0.000 claims description 8
- 125000003342 alkenyl group Chemical group 0.000 claims description 7
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 7
- 230000007954 hypoxia Effects 0.000 claims description 7
- 239000003795 chemical substances by application Substances 0.000 claims description 6
- 208000010125 myocardial infarction Diseases 0.000 claims description 6
- 230000002265 prevention Effects 0.000 claims description 6
- 230000005855 radiation Effects 0.000 claims description 6
- 238000002054 transplantation Methods 0.000 claims description 6
- 208000030507 AIDS Diseases 0.000 claims description 5
- 206010011903 Deafness traumatic Diseases 0.000 claims description 5
- 206010012689 Diabetic retinopathy Diseases 0.000 claims description 5
- 208000010412 Glaucoma Diseases 0.000 claims description 5
- 206010061598 Immunodeficiency Diseases 0.000 claims description 5
- 208000029462 Immunodeficiency disease Diseases 0.000 claims description 5
- 206010061218 Inflammation Diseases 0.000 claims description 5
- 206010029155 Nephropathy toxic Diseases 0.000 claims description 5
- 208000002946 Noise-Induced Hearing Loss Diseases 0.000 claims description 5
- 208000007014 Retinitis pigmentosa Diseases 0.000 claims description 5
- 210000001744 T-lymphocyte Anatomy 0.000 claims description 5
- 206010064930 age-related macular degeneration Diseases 0.000 claims description 5
- 206010003246 arthritis Diseases 0.000 claims description 5
- 239000003124 biologic agent Substances 0.000 claims description 5
- 206010008118 cerebral infarction Diseases 0.000 claims description 5
- 208000026106 cerebrovascular disease Diseases 0.000 claims description 5
- 208000006454 hepatitis Diseases 0.000 claims description 5
- 231100000283 hepatitis Toxicity 0.000 claims description 5
- 230000007813 immunodeficiency Effects 0.000 claims description 5
- 208000015181 infectious disease Diseases 0.000 claims description 5
- 230000004054 inflammatory process Effects 0.000 claims description 5
- 230000003902 lesion Effects 0.000 claims description 5
- 208000002780 macular degeneration Diseases 0.000 claims description 5
- 230000001404 mediated effect Effects 0.000 claims description 5
- 201000006417 multiple sclerosis Diseases 0.000 claims description 5
- 230000007694 nephrotoxicity Effects 0.000 claims description 5
- 231100000417 nephrotoxicity Toxicity 0.000 claims description 5
- 230000004770 neurodegeneration Effects 0.000 claims description 5
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 5
- 201000008482 osteoarthritis Diseases 0.000 claims description 5
- 239000000126 substance Substances 0.000 claims description 5
- 238000001356 surgical procedure Methods 0.000 claims description 5
- 210000004153 islets of langerhan Anatomy 0.000 claims description 4
- 229910052757 nitrogen Inorganic materials 0.000 claims description 4
- 101100134922 Gallus gallus COR5 gene Proteins 0.000 claims description 3
- 241000700605 Viruses Species 0.000 claims description 3
- 239000004480 active ingredient Substances 0.000 claims description 2
- 239000003814 drug Substances 0.000 claims description 2
- 238000004519 manufacturing process Methods 0.000 claims description 2
- 125000000896 monocarboxylic acid group Chemical group 0.000 claims description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 230000001225 therapeutic effect Effects 0.000 claims description 2
- 108010062544 Apoptotic Protease-Activating Factor 1 Proteins 0.000 abstract description 4
- 102100034524 Apoptotic protease-activating factor 1 Human genes 0.000 abstract description 4
- BXRNXXXXHLBUKK-UHFFFAOYSA-N piperazine-2,5-dione Chemical class O=C1CNC(=O)CN1 BXRNXXXXHLBUKK-UHFFFAOYSA-N 0.000 abstract 1
- ZMXDDKWLCZADIW-UHFFFAOYSA-N dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 84
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 74
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 63
- 229920005989 resin Polymers 0.000 description 48
- 239000011347 resin Substances 0.000 description 48
- 239000000243 solution Substances 0.000 description 29
- 239000000543 intermediate Substances 0.000 description 26
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 21
- 239000000203 mixture Substances 0.000 description 20
- 238000006243 chemical reaction Methods 0.000 description 17
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 15
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- 239000002253 acid Substances 0.000 description 15
- 150000001412 amines Chemical class 0.000 description 14
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 230000002829 reductive effect Effects 0.000 description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 11
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 10
- 239000011541 reaction mixture Substances 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 9
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- 239000012071 phase Substances 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 239000007821 HATU Substances 0.000 description 6
- 230000030833 cell death Effects 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- 108010089941 Apoptosomes Proteins 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 150000001413 amino acids Chemical class 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- 239000000284 extract Substances 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- VHJKDOLGYMULOP-UHFFFAOYSA-N 2-(2,4-dichlorophenyl)ethanamine Chemical compound NCCC1=CC=C(Cl)C=C1Cl VHJKDOLGYMULOP-UHFFFAOYSA-N 0.000 description 4
- 101100439663 Arabidopsis thaliana CHR7 gene Proteins 0.000 description 4
- 102000011727 Caspases Human genes 0.000 description 4
- 108010076667 Caspases Proteins 0.000 description 4
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 4
- XXROGKLTLUQVRX-UHFFFAOYSA-N allyl alcohol Chemical compound OCC=C XXROGKLTLUQVRX-UHFFFAOYSA-N 0.000 description 4
- 230000001640 apoptogenic effect Effects 0.000 description 4
- 239000007822 coupling agent Substances 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 239000003643 water by type Substances 0.000 description 4
- 0 *N(CC(N(*)C1CC(O)=O)=O)C1=O Chemical compound *N(CC(N(*)C1CC(O)=O)=O)C1=O 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 102000004039 Caspase-9 Human genes 0.000 description 3
- 108090000566 Caspase-9 Proteins 0.000 description 3
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 3
- 238000003776 cleavage reaction Methods 0.000 description 3
- WBJINCZRORDGAQ-UHFFFAOYSA-N ethyl formate Chemical compound CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- 239000012074 organic phase Substances 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 230000007017 scission Effects 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 239000007790 solid phase Substances 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 238000000825 ultraviolet detection Methods 0.000 description 3
- UMRUUWFGLGNQLI-QFIPXVFZSA-N (2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-6-[(2-methylpropan-2-yl)oxycarbonylamino]hexanoic acid Chemical compound C1=CC=C2C(COC(=O)N[C@@H](CCCCNC(=O)OC(C)(C)C)C(O)=O)C3=CC=CC=C3C2=C1 UMRUUWFGLGNQLI-QFIPXVFZSA-N 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- JUIKUQOUMZUFQT-UHFFFAOYSA-N 2-bromoacetamide Chemical compound NC(=O)CBr JUIKUQOUMZUFQT-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 2
- ATHHXGZTWNVVOU-UHFFFAOYSA-N N-methylformamide Chemical compound CNC=O ATHHXGZTWNVVOU-UHFFFAOYSA-N 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 238000013375 chromatographic separation Methods 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 2
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 2
- 238000000132 electrospray ionisation Methods 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 230000032050 esterification Effects 0.000 description 2
- 238000005886 esterification reaction Methods 0.000 description 2
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 238000004896 high resolution mass spectrometry Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000037353 metabolic pathway Effects 0.000 description 2
- 230000006654 negative regulation of apoptotic process Effects 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- YBYRMVIVWMBXKQ-UHFFFAOYSA-N phenylmethanesulfonyl fluoride Chemical compound FS(=O)(=O)CC1=CC=CC=C1 YBYRMVIVWMBXKQ-UHFFFAOYSA-N 0.000 description 2
- 229920005990 polystyrene resin Polymers 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 238000006268 reductive amination reaction Methods 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 239000012047 saturated solution Substances 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 150000003512 tertiary amines Chemical class 0.000 description 2
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 239000003440 toxic substance Substances 0.000 description 2
- 230000026683 transduction Effects 0.000 description 2
- 238000010361 transduction Methods 0.000 description 2
- ZGYICYBLPGRURT-UHFFFAOYSA-N tri(propan-2-yl)silicon Chemical compound CC(C)[Si](C(C)C)C(C)C ZGYICYBLPGRURT-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 241000701161 unidentified adenovirus Species 0.000 description 2
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- QACMXJJLQXUOPQ-UHFFFAOYSA-N 1,2-dichloroethane;3-(ethyliminomethylideneamino)-n,n-dimethylpropan-1-amine Chemical compound ClCCCl.CCN=C=NCCCN(C)C QACMXJJLQXUOPQ-UHFFFAOYSA-N 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- JPRPJUMQRZTTED-UHFFFAOYSA-N 1,3-dioxolanyl Chemical group [CH]1OCCO1 JPRPJUMQRZTTED-UHFFFAOYSA-N 0.000 description 1
- XMOKZLVOWBOPCO-UHFFFAOYSA-N 1,4-diazepane-2,5-dione Chemical compound O=C1CCNC(=O)CN1 XMOKZLVOWBOPCO-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- JFLSOKIMYBSASW-UHFFFAOYSA-N 1-chloro-2-[chloro(diphenyl)methyl]benzene Chemical compound ClC1=CC=CC=C1C(Cl)(C=1C=CC=CC=1)C1=CC=CC=C1 JFLSOKIMYBSASW-UHFFFAOYSA-N 0.000 description 1
- DFPYXQYWILNVAU-UHFFFAOYSA-N 1-hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1.C1=CC=C2N(O)N=NC2=C1 DFPYXQYWILNVAU-UHFFFAOYSA-N 0.000 description 1
- MBVFGFFLHYMLNP-UHFFFAOYSA-N 2-[4-[2-(2,4-dichlorophenyl)ethyl]-1-(3,3-diphenylpropyl)-3,6-dioxopiperazin-2-yl]acetic acid Chemical compound O=C1CN(CCC=2C(=CC(Cl)=CC=2)Cl)C(=O)C(CC(=O)O)N1CCC(C=1C=CC=CC=1)C1=CC=CC=C1 MBVFGFFLHYMLNP-UHFFFAOYSA-N 0.000 description 1
- GCABUDPQUJGOPR-UHFFFAOYSA-N 2-[4-[2-(2,4-dichlorophenyl)ethyl]-3,6-dioxo-1-[[4-(trifluoromethyl)phenyl]methyl]piperazin-2-yl]acetic acid Chemical compound O=C1CN(CCC=2C(=CC(Cl)=CC=2)Cl)C(=O)C(CC(=O)O)N1CC1=CC=C(C(F)(F)F)C=C1 GCABUDPQUJGOPR-UHFFFAOYSA-N 0.000 description 1
- 125000000979 2-amino-2-oxoethyl group Chemical group [H]C([*])([H])C(=O)N([H])[H] 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- XPQIPUZPSLAZDV-UHFFFAOYSA-N 2-pyridylethylamine Chemical compound NCCC1=CC=CC=N1 XPQIPUZPSLAZDV-UHFFFAOYSA-N 0.000 description 1
- HVLUYXIJZLDNIS-UHFFFAOYSA-N 2-thiophen-2-ylethanamine Chemical compound NCCC1=CC=CS1 HVLUYXIJZLDNIS-UHFFFAOYSA-N 0.000 description 1
- KISZTEOELCMZPY-UHFFFAOYSA-N 3,3-diphenylpropylamine Chemical compound C=1C=CC=CC=1C(CCN)C1=CC=CC=C1 KISZTEOELCMZPY-UHFFFAOYSA-N 0.000 description 1
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 1
- CNPURSDMOWDNOQ-UHFFFAOYSA-N 4-methoxy-7h-pyrrolo[2,3-d]pyrimidin-2-amine Chemical compound COC1=NC(N)=NC2=C1C=CN2 CNPURSDMOWDNOQ-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010002660 Anoxia Diseases 0.000 description 1
- 241000976983 Anoxia Species 0.000 description 1
- 229940088872 Apoptosis inhibitor Drugs 0.000 description 1
- 108090000397 Caspase 3 Proteins 0.000 description 1
- 102100035904 Caspase-1 Human genes 0.000 description 1
- 108090000426 Caspase-1 Proteins 0.000 description 1
- 102100029855 Caspase-3 Human genes 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- VGCXGMAHQTYDJK-UHFFFAOYSA-N Chloroacetyl chloride Chemical compound ClCC(Cl)=O VGCXGMAHQTYDJK-UHFFFAOYSA-N 0.000 description 1
- 108050006400 Cyclin Proteins 0.000 description 1
- 102100030497 Cytochrome c Human genes 0.000 description 1
- 108010075031 Cytochromes c Proteins 0.000 description 1
- 230000005778 DNA damage Effects 0.000 description 1
- 231100000277 DNA damage Toxicity 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 101000726354 Equus caballus Cytochrome c Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- XLYMOEINVGRTEX-ARJAWSKDSA-N Ethyl hydrogen fumarate Chemical compound CCOC(=O)\C=C/C(O)=O XLYMOEINVGRTEX-ARJAWSKDSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 238000006957 Michael reaction Methods 0.000 description 1
- 108010057466 NF-kappa B Proteins 0.000 description 1
- 102000003945 NF-kappa B Human genes 0.000 description 1
- 208000008636 Neoplastic Processes Diseases 0.000 description 1
- 102000043276 Oncogene Human genes 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 102000009339 Proliferating Cell Nuclear Antigen Human genes 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 101100427180 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) RAD6 gene Proteins 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 101150016610 UBC2 gene Proteins 0.000 description 1
- 102100020695 Ubiquitin-conjugating enzyme E2 N Human genes 0.000 description 1
- 101710192923 Ubiquitin-conjugating enzyme E2 N Proteins 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- JXYRIQRQKAUQIY-UHFFFAOYSA-N acetic acid;oxolane Chemical compound CC(O)=O.C1CCOC1 JXYRIQRQKAUQIY-UHFFFAOYSA-N 0.000 description 1
- PBCJIPOGFJYBJE-UHFFFAOYSA-N acetonitrile;hydrate Chemical compound O.CC#N PBCJIPOGFJYBJE-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 230000007953 anoxia Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 238000003782 apoptosis assay Methods 0.000 description 1
- 239000000158 apoptosis inhibitor Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- HYTACLVSJIFYBY-UHFFFAOYSA-N azane;dichloromethane;methanol Chemical compound N.OC.ClCCl HYTACLVSJIFYBY-UHFFFAOYSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- KDPAWGWELVVRCH-UHFFFAOYSA-M bromoacetate Chemical compound [O-]C(=O)CBr KDPAWGWELVVRCH-UHFFFAOYSA-M 0.000 description 1
- 230000001964 calcium overload Effects 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 210000004413 cardiac myocyte Anatomy 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 230000010001 cellular homeostasis Effects 0.000 description 1
- 230000004637 cellular stress Effects 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 229940125890 compound Ia Drugs 0.000 description 1
- 210000004087 cornea Anatomy 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- SUYVUBYJARFZHO-RRKCRQDMSA-N dATP Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-RRKCRQDMSA-N 0.000 description 1
- SUYVUBYJARFZHO-UHFFFAOYSA-N dATP Natural products C1=NC=2C(N)=NC=NC=2N1C1CC(O)C(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-UHFFFAOYSA-N 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000003412 degenerative effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 230000001687 destabilization Effects 0.000 description 1
- 125000000723 dihydrobenzofuranyl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 description 1
- 125000004582 dihydrobenzothienyl group Chemical group S1C(CC2=C1C=CC=C2)* 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- LHWWETDBWVTKJO-UHFFFAOYSA-N et3n triethylamine Chemical compound CCN(CC)CC.CCN(CC)CC LHWWETDBWVTKJO-UHFFFAOYSA-N 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- 230000008622 extracellular signaling Effects 0.000 description 1
- 230000006624 extrinsic pathway Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- XLYMOEINVGRTEX-UHFFFAOYSA-N fumaric acid monoethyl ester Natural products CCOC(=O)C=CC(O)=O XLYMOEINVGRTEX-UHFFFAOYSA-N 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 150000002333 glycines Chemical class 0.000 description 1
- 125000003630 glycyl group Chemical group [H]N([H])C([H])([H])C(*)=O 0.000 description 1
- HHLFWLYXYJOTON-UHFFFAOYSA-N glyoxylic acid Chemical compound OC(=O)C=O HHLFWLYXYJOTON-UHFFFAOYSA-N 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- ALBYIUDWACNRRB-UHFFFAOYSA-N hexanamide Chemical compound CCCCCC(N)=O ALBYIUDWACNRRB-UHFFFAOYSA-N 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 230000006882 induction of apoptosis Effects 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000006623 intrinsic pathway Effects 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 102000006240 membrane receptors Human genes 0.000 description 1
- 108020004084 membrane receptors Proteins 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 210000001700 mitochondrial membrane Anatomy 0.000 description 1
- 230000009456 molecular mechanism Effects 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- GVOISEJVFFIGQE-YCZSINBZSA-N n-[(1r,2s,5r)-5-[methyl(propan-2-yl)amino]-2-[(3s)-2-oxo-3-[[6-(trifluoromethyl)quinazolin-4-yl]amino]pyrrolidin-1-yl]cyclohexyl]acetamide Chemical compound CC(=O)N[C@@H]1C[C@H](N(C)C(C)C)CC[C@@H]1N1C(=O)[C@@H](NC=2C3=CC(=CC=C3N=CN=2)C(F)(F)F)CC1 GVOISEJVFFIGQE-YCZSINBZSA-N 0.000 description 1
- UPSFMJHZUCSEHU-JYGUBCOQSA-N n-[(2s,3r,4r,5s,6r)-2-[(2r,3s,4r,5r,6s)-5-acetamido-4-hydroxy-2-(hydroxymethyl)-6-(4-methyl-2-oxochromen-7-yl)oxyoxan-3-yl]oxy-4,5-dihydroxy-6-(hydroxymethyl)oxan-3-yl]acetamide Chemical compound CC(=O)N[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1O[C@H]1[C@H](O)[C@@H](NC(C)=O)[C@H](OC=2C=C3OC(=O)C=C(C)C3=CC=2)O[C@@H]1CO UPSFMJHZUCSEHU-JYGUBCOQSA-N 0.000 description 1
- RIVIDPPYRINTTH-UHFFFAOYSA-N n-ethylpropan-2-amine Chemical compound CCNC(C)C RIVIDPPYRINTTH-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000004963 pathophysiological condition Effects 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 230000008823 permeabilization Effects 0.000 description 1
- 239000008194 pharmaceutical composition Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- FYRHIOVKTDQVFC-UHFFFAOYSA-M potassium phthalimide Chemical compound [K+].C1=CC=C2C(=O)[N-]C(=O)C2=C1 FYRHIOVKTDQVFC-UHFFFAOYSA-M 0.000 description 1
- 230000005522 programmed cell death Effects 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 235000019419 proteases Nutrition 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000007127 saponification reaction Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 229960005137 succinic acid Drugs 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 231100000167 toxic agent Toxicity 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/02—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings
- C07D241/06—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having one or two double bonds between ring members or between ring members and non-ring members
- C07D241/08—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings not condensed with other rings having one or two double bonds between ring members or between ring members and non-ring members with oxygen atoms directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention relates to compounds for the prophylaxis and / or treatment of disorders caused by cell death by apoptosis or for the prevention of degenerative processes caused by cell death by apoptosis.
- Apoptosis or programmed cell death, is a complex physiological phenomenon involved in the maintenance of cellular homeostasis. Apoptosis is regulated by multiple cellular control mechanisms because of its central role in maintaining health. Many pathologies are based on a dysfunction of apoptosis. Thus, an excess of cell death due to apoptosis can affect the functionality of the tissue (e.g. death of cardiomyocytes in cases of myocardial infarction), while an excessively inhibited apoptosis leads to cell survival.
- the cellular components that regulate apoptosis are in a constant dynamic equilibrium in a healthy cell.
- the extrinsic pathway is activated by extracellular signaling and requires the participation of specific membrane receptors.
- the intrinsic pathway responds to cellular stress, toxic agents, radiation, oxidizing agents, Ca 2+ overload, DNA damage; It is activated in response to oncogenes, and implies the destabilization of the mitochondria.
- pathophysiological conditions for example, anoxia in cells of organs that must be transplanted, treatment with toxic substances
- apoptosis is increased and the cells die excessively, making it impossible for the affected tissue to function and compromising survival in some cases.
- the molecular mechanisms of induction of apoptosis involve the activation of proteins with protease activity called caspases, also known as effectors of apoptosis. In order for them to be activated, the formation of a molecular complex called apoptosome is necessary.
- the apoptosome is formed by cytochrome c, procaspase-9 and the activating factor 1 of the apoptotic peptidase (Apaf-1, Apoptotic Peptidase Activating Factor 1). It has been shown that the inhibition of Apaf-1 inhibits Ia formation of the apoptosome complex and that this causes an inhibition of apoptosis (measured through the activation of caspase 3).
- inhibitors have been designed that act at different levels of the apoptotic cascade such as transcription factors, kinases, regulators of Ia
- Negative dominant of Apaf-1 by adenovirus showed to be more effective than the transduction by adenovirus of a negative dominant of Caspase-1.
- WO2007060524 describes the compounds derived from
- WO2008009758 describes the compounds of the attached formula, as inhibitors of UBC13-UEV interactions and that can be used in the preparation of pharmaceutical compositions directed to antitumor therapy or to the treatment and / or prophylaxis of diseases associated with metabolic pathways in which involves the enzyme UBC13, metabolic pathways in which the transcriptional factor NF-kB intervenes, or routes in which PCNA or RAD6 intervene. Although they can be considered structurally close to those of the present invention, they have a different use.
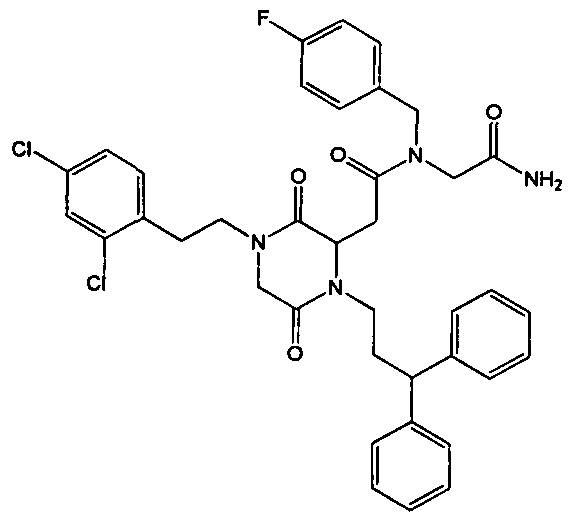
- the present invention provides new compounds derived from
- R1 and R2 are independently selected from -H, -Ci -5 alkyl, -C 2-5 alkenyl, - (CH 2 ) or -3-cycloalkyl, - (CH 2 ) i- 3- heterocycle, - (CH 2 ) o- 3 -aryl, - (CH 2 ) or- 3 -heteroaryl, - (CH 2 ) i -2 -CH (aryl) 2 , - (CH 2 ) and -2 -CH (aryl) (heteroaryl) and - (CH 2 ) i -2 - CH (heteroaryl) 2l
- R3 is selected from -H, -Ci -5 alkyl, -C 2-5 alkenyl, - (CH 2 ) 0-3 -cycloalkyl, - (CH 2 ) i -3- heterocycle, - (CH 2 ) i -3 -aryl, - (CH 2 ) 1-3 -heteroaryl, - (CH 2 ) i -3 -CONR5R6, - (CH 2 ) i -2 -CH (aryl) 2 , - (CH 2 ) i -2 -CH (aryl) (heteroaryl) and - (CH 2 ) 1-2 -CH (heteroaryl) 2 , R4 is selected from -H, -Ci -5 alkyl, - (CHR7) and -3 -CO-NR5R6, - (CHR7 ) i -3 -CO- OR5, - (CH 2 ) i -3 -NR5R6,
- R5 and R6 are independently selected from -H, -Ci -5 alkyl and - (CH 2 ) 0-3 -aryl,
- R7 is selected from -H, -Ci -5 alkyl, - (CH 2 ) i -3 -aryl and - (CH 2 ) i -3- heteroaryl, so that when m is greater than 1 the R7 substituents can be the same or different, where Ci -5 alkyl, C 2-5 alkenyl, cycloalkyl and heterocycle may be optionally substituted by one or more substituents independently selected from halogen, OR5, OCF 3, SH, SR5, NR5R6, NHCOR 5; COOH, COOR5, OCOR5, aryl and heteroaryl, where the aryl and heteroaryl groups may be optionally substituted by one or more substituents independently selected from halogen, CF3, OR5, OCF 3 , SH, SR5, NH 2 , NHCOR5; NO 2 , CN, COR5, COOR5,
- R1 is 2- (4-fluorophenyl) ethyl
- R3 is not 2- (4-methoxyphenyl) ethyl, 2- (2-pyridyl) ethyl or 2- (2,4-dichlorophenyl) ethyl
- R1 is 2- (2,4-dichlorophenyl) ethyl
- R3 is not 2- (4-methoxyphenyl) ethyl or 2- (2-pyridyl) ethyl.
- R1 is -Ci -5 alkyl or - (CH 2 ) 0-3 -aryl.
- R2 is -Ci -5 alkyl, - (CH 2 ) or -3- ahlo, - (CH 2 ) O-3 -heteroaryl or - (CH 2 ) and -2 -CH (aryl ) 2 .
- R3 is -H, -Ci -5 alkyl, - (CH 2 ) and -3 -heterocycle, - (CH 2 ) and -3 -aryl or - (CH 2 ) and -3 - heteroaryl.
- R4 is -H, - (CHR7) and -3 -CO-NR5R6, - (CHR7) and -3 -CO-OR5 or - (CH 2 ) and -3 -CO [NCHR7CO] m NH 2 .
- n 1
- n 1
- R5 is -H or -Ci -5 alkyl.
- R6 is -H.
- R7 is -H, -Ci -5 alkyl, - (CH 2 ) I -3 -aryl or - (CH 2 ) and -3- heteroaryl.
- a second aspect of the present invention relates to a compound of formula (I), or a pharmaceutically acceptable salt thereof for use as a pharmaceutical active ingredient, in particular for use in prophylaxis and / or treatment of a pathological and / or physiological condition associated with a
- cytotoxicity in particular cytotoxicity mediated by chemical substances, by physical agents such as radiation, acoustic trauma, burns, or by biological agents such as infection by the hepatitis virus
- pathologies due to hypoxia situations such as heart attack or cerebral infarction
- eye pathologies such as lesions caused by eye surgery, age-related macular degeneration, diabetic retinopathy, retinitis pigmentosa or glaucoma
- neurodegenerative diseases such as Alzheimer's, Huntington, Parkinson's or amyotrophic multiple sclerosis
- diabetes in particular preservation of islets of Langerhans or cytotoxicity associated with diabetes, such as nephrotoxicity; osteoarthritis; arthritis; inflammation or immunodeficiencies, such as depletion of CD4 + T lymphocytes associated with AIDS.
- Another aspect of the present invention relates to the use of a compound of formula (I) or a pharmaceutically acceptable salt thereof for the manufacture of a medicament intended for the prophylaxis and / or treatment of a pathological and / or physiological condition associated with an increase in apoptosis, in particular one of the conditions mentioned above.
- Another aspect of the present invention relates to a method of prophylaxis and / or treatment of an individual or organ that suffers or is susceptible to suffering from a pathological and / or physiological condition associated with an increase in apoptosis, in particular one of the conditions mentioned above, which comprises the administration to said individual or organ of a therapeutically effective amount of a compound of formula (I) or of a pharmaceutically acceptable salt thereof together with sufficient amounts of pharmaceutically acceptable excipients.
- cytotoxicity in particular cytotoxicity mediated by chemical substances, by physical agents such as radiation, acoustic trauma, burns, or by biological agents such as infection by the hepatitis virus
- pathologies due to hypoxia situations such as heart attack or cerebral infarction
- eye pathologies such as lesions caused by eye surgery, age-related macular degeneration, diabetic retinopathy, retinitis pigmentosa or glaucoma
- neurodegenerative diseases such as Alzheimer's, Huntington,
- Parkinson's or amyotrophic multiple sclerosis diabetes, in particular preservation of islets of Langerhans or cytotoxicity associated with diabetes, such as nephrotoxicity; osteoarthritis; arthritis; inflammation or immunodeficiencies, such as depletion of CD4 + T lymphocytes associated with AIDS.
- Ci -5 alkyl alone or in combination, means an alkyl group of straight or branched chain having 1 to 5 carbon atoms.
- C 2-5 alkenyl means a group having 2 to 5 carbon atoms, straight or branched chain and having one or more unsaturated bonds.
- cycloalkyl alone or in combination, refers to a stable 3- to 7-membered monocyclic radical, which is saturated or partially saturated, and which only consists of carbon and hydrogen atoms. Examples of cycloalkyl are the following: cyclopropyl, cyclopentyl, cyclohexyl, 1-cyclohexenyl, cycloheptyl.
- heterocycle alone or in combination, means a saturated or partially unsaturated heterocycle of 5 to 10 links, containing one or more heteroatoms chosen from nitrogen, oxygen and sulfur.
- the heterocycle may be a monocyclic or bicyclic ring system, which may include condensed ring systems.
- heterocycle groups are tetrahydrofuranyl (THF), dihydrofuranyl, dioxanyl, morphyl, piperazinyl, piperidinyl, 1,3-dioxolanyl, imidazolidinyl, midazolinyl, pyrrolidyl, pyrrolidinyl, tetrahydropyranyl, dihydropyranyl, and the like.
- aryl refers to a mono or polycyclic aromatic ring system containing carbon ring atoms. Preferred aryls are 5-10 member aromatic ring systems
- monocyclic or bicyclic such as phenyl or naphthyl which optionally carry one or more substituents, preferably one to three, independently selected from halogen, CF3, OH, OR5, OCF 3 , SH, SR5, NH 2 , NHCOR5; NO 2 , CN, COR5, COOR5, OCOR5, CONR5R6, - (CH 2 ) (W NR5R6, SO 2 NH 2 , NHSO 2 CH3, Ci -5 alkyl, aryl and heteroaryl.
- heteroaryl alone or in combination refers to an aromatic or partially aromatic heterocycle containing at least one ring heteroatom selected from O, S and N.
- the heteroaryls thus include heteroaryls fused to other kinds of rings, such as aryls , cycloalkyl and heterocycles that are not aromatic.
- heteroaryl groups include: pyrrolyl, isoxazolyl, isothiazolyl, pyrazolyl, pyridyl, oxazolyl, thiazolyl,
- substituents a group may be unsubstituted or substituted by one or more substituents, preferably by 1, 2, 3 or 4 substituents, provided that said group has 1, 2, 3 or 4 Possible positions to be substituted.
- pharmaceutically acceptable salts means those salts that retain the efficacy and biological properties of free bases or free acids and that are not bothersome in the biological sense or in any other.
- compositions are useful for the prophylaxis and / or treatment of a pathological and / or physiological condition associated with an increase in apoptosis through its activity as inhibitors of Apaf-1.
- all the technical and scientific terms used here have the same meaning as those commonly understood by a person skilled in the field of the invention. Methods and materials similar or equivalent to those described herein may be used in the practice of the present invention.
- the word "comprises” and its variants are not intended to exclude other technical characteristics, additives, components, steps or stereoisomers of the compounds involved. For those skilled in the art, other objects, advantages and characteristics of the invention will emerge partly from the description and partly from the practice of the invention.
- the compounds of formula (I) can be prepared following different methods known to any person skilled in the field of organic synthesis, in particular by the general procedures presented in the following schemes.
- the starting materials for the preparative methods are commercially available or can be prepared by methods of the literature.
- the groups R1, R2, R3, R4, R5, R6 and R7 have the meaning described in the general formula (I).
- the amine Il bound to the solid support is acylated with an acylating agent III, where X represents a leaving group, for example a halogen and Y represents OH or halogen.
- Y represents a halogen, for example chloroacetyl chloride
- the reaction can be carried out in the presence of a base such as triethylamine.
- Y represents -OH, for example bromoacetic acid
- the reaction can be carried out in the presence of a suitable coupling agent, for example ⁇ /, ⁇ / -diisopropylcarbodiimide.
- the reaction can be carried out in an inert solvent that is capable of swelling the resin, such as ⁇ /, ⁇ / -dimethylformamide or methylene chloride and at room temperature or under microwave irradiation, to minimize the reaction time.
- the IVa amine is coupled using a tertiary amine as the base. The reaction can be carried out at room temperature or by microwave irradiation.
- a carboxylic acid Vl, where GP represents a protective group, such as allyl, is reacted with the amine V to obtain the amide VII, using a coupling agent, such as for example the combination of N, N'-diisopropylcarbodiimide and 1-hydroxybenzotriazole.
- a coupling agent such as for example the combination of N, N'-diisopropylcarbodiimide and 1-hydroxybenzotriazole.
- an amine IVb is added, by reaction of Michael using a base and a solvent, such as ⁇ /, ⁇ / -dimethylformamide or dimethyl sulfoxide to obtain the HIV intermediate after the cleavage of the resin using a mixture of trifluoroacetic acid , dichloromethane and water.
- Intermediate VIII is cycled (intermediate IX) and deprotected in basic medium yielding acidic intermediate X.
- Intermediate X can be prepared alternatively to the solid phase according to scheme 2, where the amine V can be prepared from the amine IVa either by a reductive amination reaction with a glyoxylate in THF-AcOH using a reducing agent such as NaBH 3 CN, or alternatively by alkylation with a bromoacetate or a
- a compound of formula Ia can be obtained from intermediate X by coupling with an amine attached to a solid support Va or Va ', obtained according to the methodology indicated above, in the presence of a coupling agent such as, for example, the combination of HATU and HOBT.
- the compound of formula Ib is it can obtain analogously to the synthesis of compound Ia, except in the case of a solid phase in which the solid starting support (Mb) has a halogen group instead of an amino group, such as for example chlorotrityl resin, obtaining a acid after the cleavage of the resin.
- the ester Ic can be synthesized by esterification of the corresponding acid Ib by the usual esterification methods in organic synthesis, such as using methanol in an acidic medium such as sulfuric acid. In the case of Ib, it can be obtained by saponification of the ester Ic.
- the compounds of formula Id can be obtained by reacting intermediate X with a primary amine IVc.
- Peptide XIII and pseudopeptide XIV that will bind to acid X can be obtained by standard peptide synthesis reactions.
- the process can be repeated sequentially, after deprotection of the amine, to obtain peptide XIII.
- carboxylic acid X reacts with XIII to obtain compound Ie.
- An amine can be obtained by reaction of Mitsunobu starting from alcohol and potassium phthalimide in the presence of, for example, diethyl azodicarboxylate (DEAD) and triphenylphosphine in tetrahydrofuran as solvent and subsequent release with hydrazine hydrate.
- DEAD diethyl azodicarboxylate
- triphenylphosphine triphenylphosphine in tetrahydrofuran as solvent and subsequent release with hydrazine hydrate.
- the lic / -ubstituted glycines V and Xl can be synthesized by any of the methods shown below, such as, for example, reductive amination of the corresponding glycine with a suitable aldehyde (Scheme 5) using reducing agents such as NaBH 4 , NaBH 3 CN or NaBH (AcO) 3 or by nucleophilic substitution of an ester with an R-NH 2 amine (Scheme 6).
- the compounds were synthesized using an AM RAM polystyrene resin purchased from Rapp Polymere GmbH (Germany). Polystyrene syringes with a polyethylene disc were used in the reactions using an IKALabortechnik HS501 digital agitator. In the reactions carried out by microwaves, the CEM Discover model with 10 ml glass reactors was used. The products were analyzed by:
- Method B The products were analyzed using an Agilent 1100 HPLC device, equipped with a variable wavelength UV detector and a 1100 VL model mass spectrometer.
- the wavelength used for UV detection has been 210 nm, while the MS detector has operated in positive electrospray ionization mode and has performed a scan of m / z 100 to 1300.
- the column used it has been a Kromasil 100 C18 (4.0 x 40 mm, 3.5 ⁇ m) thermostated at 5O 0 C, and 5 ⁇ l have been injected.
- Solvent A consists of 0.2% formic acid in water, while B is 0.2% formic acid in acetonitrile.
- Method C using a Waters HPLC-UV-MS device, equipped with a serial diode detector and an EMD1000 model mass spectrometer. The wavelength used for UV detection has been 210 nm, while the MS detector has operated in positive electrospray ionization mode and has performed a scan of m / z 100 to 1000.
- the column used It has been a Kromasil C18 (2.1 x 50 mm, 3.5 ⁇ m) thermostated at 50 s C and 2 ⁇ l have been injected.
- the following gradient has been followed: 5 -100% B, 0-5 min; 100% B, 5-6.5 min; 5% B, 6.5-8 min.
- the flow rate of the mobile phase is 0.5 ml / min.
- High resolution mass spectrometry was carried out by UPLC-HRMS using a Waters Acquity UPLC device coupled to a spectrometer of time-of-flight masses with orthogonal acceleration model LCT Premier XE from Waters. Chromatographic analysis was performed using a Waters Acquity C18 column (10 x 2.1 mm, 1.7 ⁇ m).
- the reaction mixture was stirred for 2 min at 6O 0 C in a microwave reactor.
- the resin was filtered and washed with DMF (3 x 15 mL), isopropyl alcohol (3 x 15 mL) and DCM (3 x 15 mi).
- DMF 3 x 15 mL
- isopropyl alcohol 3 x 15 mL
- DCM 3 x 15 mi
- 12 ml of DMF was added to the resin and the suspension was stirred for 2 min. at 9O 0 C activated by microwave. The supernatant was removed and the reaction was repeated under the same conditions.
- the resin V obtained was filtered and washed with DMF (3 x 15 ml), isopropyl alcohol (3 x 15 ml) and DCM (3 x 15 ml). Then, the resin was treated with a solution of allyl ester of (Z) -2-butanedioic acid (Vl 1 957 mg, 5 eq.), HOBT (825 mg, 5 eq.) And DIC (770 ⁇ l_, 5 eq. ) in DCM: DMF (2: 1, 123 mi). The reaction mixture was stirred at room temperature for 30 min and filtered.
- the resin was dried and washed with DMF (3 x 15 mL), isopropyl alcohol (3 x 15 mL) and DCM (3 x 15 mL). Then, a solution of 3,3-diphenylpropylamine (IVb, 1, 29 g, 5 eq.) And triethylamine (0.85 ml, 5 eq.) In 12 ml of DMF was added to the resin and the suspension was stirred for 3 h at room temperature. The resin was filtered and the reaction was repeated for 16 h at the same temperature.
- the resin was dried and washed with DMF (3 x 3 mL), isopropyl alcohol (3 x 3 mL) and DCM (3 x 3 mL) and subsequently treated with a mixture of 60: 40: 2 TFA / DCM / water (5 mi) for 30 min at
- the Rink amide-Fmoc resin (II, 500 mg, 0.305 mmol) was deprotected with 5 ml of 20% piperidine in DMF by stirring in a microwave reactor for 2 min at 60 ° C. The resin was filtered and washed with DMF. (3 x 15 ml), isopropyl alcohol (3 x 15 ml) and DCM (3 x 15 ml). Then, the amino acid Fmoc-L-Lys (Boc) -OH (Xl, 286 mg, 2 eq.) was bound to the resin using HOBT (82 mg, 2 eq.) And DIC (96 ⁇ L, 2 eq.) in 5 ml of DMF.
- the mixture was stirred at room temperature for 1 h.
- the resin was filtered and washed with DMF (3 x 15 mL), isopropyl alcohol (3 x 15 mL) and DCM (3 x 15 mL). After removing the Fmoc group with 5 ml of 20% piperidine in DMF for 20 min, the resin was filtered and washed with DMF (3 x 15 ml), isopropyl alcohol (3 x 15 ml) and DCM (3 x 15 me).
- the resin was treated with a solution of acid X (181 mg, 1, 1 eq.), HATU (348 mg, 3 eq.), HOBT (123 mg, 3 eq.) And DIPEA (0.313 mi, 6 eq. ) in 5 ml of DMF.
- the reaction mixture was stirred at room temperature for 16 h.
- the resin was dried and washed with DMF (3 x 3 mL), isopropyl alcohol (3 x 3 mL) and DCM (3 x 3 mL) and subsequently treated with a mixture of 80: 20: 2.5: 2, 5 TFA / DCM / water / triisopropylsilane (5 ml) for 30 min at room temperature.
- the Rink amide-Fmoc resin (II, 800 mg, 0.42 mmol) was deprotected with 8 ml of 20% piperidine in DMF by stirring in a microwave reactor for 2 min at 60 ° C. The resin was filtered and washed. with DMF (3 x 15 ml), isopropyl alcohol (3 x 15 ml) and DCM (3 x 15 ml). Next, the amino acid Fmoc-L-Lys (Boc) -OH (XII, 497 mg, 2 eq.) was bound to the resin using HOBT (143 mg, 2 eq.) And DIC (165 ⁇ L, 2 eq.) in 8 ml of DMF.
- the mixture was stirred at room temperature for 1 h.
- the resin was filtered and washed with DMF (3 x 15 mL), isopropyl alcohol (3 x 15 mL) and DCM (3 x 15 mL). After removing the Fmoc group with 8 ml of 20% piperidine in DMF for 20 min, the resin was filtered and washed with DMF (3 x 15 ml), isopropyl alcohol (3 x 15 ml) and DCM (3 x 15 me).
- the resin was treated with a solution of bromoacetic acid (III, 295 mg, 4 eq.) And DIC (0.33 ml, 4 eq.) In DMF: DCM 1: 2 (8 mi) and the mixture was stirred for 20 min at room temperature.
- the resin was filtered and washed with DMF (3 x 15 mL), isopropyl alcohol (3 x 15 mL) and DCM (3 x 15 mL).
- the resin was dried and washed with DMF (3 x 3 mL), isopropyl alcohol (3 x 3 mL) and DCM (3 x 3 mL) and subsequently treated with a mixture of 80: 20: 2.5: 2, 5 TFA / DCM / water / triisopropylsilane (5 ml) for 30 min at room temperature.
- the resin was filtered and the filtrate was evaporated under reduced pressure.
- the obtained residue was purified by semi-preparative RP-HPLC using a gradient of a mixture of acetonitrile-water to obtain 128 mg of the desired compound (see 1.2, 34% yield, 99% purity).
- Recombinant Apaf-1 produced in insect cells was incubated in the presence (at a concentration of 10 ⁇ M) or absence (as a control) of the compounds to be evaluated in the assay buffer (20 mM Hepes-KOH pH 7.5, 10 mM KCI, 1.5 mM MgCI2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.1 mM PMSF) for 15 minutes at 30 0 C.
- the final concentration of rApaf-1 was 40 nM.
- dATP / Mg Sigma
- purified horse cytochrome c Sigma
- dATP / Mg Sigma
- purified horse cytochrome c Sigma
- the total assay volume was 200 ⁇ L.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Immunology (AREA)
- Diabetes (AREA)
- Ophthalmology & Optometry (AREA)
- Virology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Heart & Thoracic Surgery (AREA)
- Rheumatology (AREA)
- Cardiology (AREA)
- Psychology (AREA)
- AIDS & HIV (AREA)
- Molecular Biology (AREA)
- Urology & Nephrology (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Pain & Pain Management (AREA)
- Obesity (AREA)
- Tropical Medicine & Parasitology (AREA)
- Vascular Medicine (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
Abstract
Description














































































Claims

Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201080043254.1A CN102574818B (zh) | 2009-07-30 | 2010-07-29 | Apaf-1抑制剂化合物 |
| MX2012001339A MX2012001339A (es) | 2009-07-30 | 2010-07-29 | Compuestos inhibidores de apaf-1. |
| BR112012002134A BR112012002134B8 (pt) | 2009-07-30 | 2010-07-29 | compostos inibidores de apaf-1 |
| ES10803937T ES2727711T3 (es) | 2009-07-30 | 2010-07-29 | Compuestos inhibidores de Apaf-1 |
| KR1020127005131A KR101786761B1 (ko) | 2009-07-30 | 2010-07-29 | Apaf-1 억제제 화합물 |
| CA2769408A CA2769408C (en) | 2009-07-30 | 2010-07-29 | Apaf-1 inhibitor compounds |
| IN1378DEN2012 IN2012DN01378A (es) | 2009-07-30 | 2010-07-29 | |
| AU2010277505A AU2010277505B2 (en) | 2009-07-30 | 2010-07-29 | APAF-1 inhibitor compounds |
| JP2012522195A JP5694320B2 (ja) | 2009-07-30 | 2010-07-29 | Apaf−1阻害剤化合物 |
| US13/387,240 US9040701B2 (en) | 2009-07-30 | 2010-07-29 | Apaf-1 inhibitor compounds |
| NZ598125A NZ598125A (en) | 2009-07-30 | 2010-07-29 | Apaf-1 inhibitor compounds |
| EA201270215A EA021838B1 (ru) | 2009-07-30 | 2010-07-29 | Производные 2,5-пиперазиндиона в качестве ингибиторов apaf-1 |
| EP10803937.1A EP2460798B1 (en) | 2009-07-30 | 2010-07-29 | Apaf-1 inhibitor compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ESP200901757 | 2009-07-30 | ||
| ES200901757 | 2009-07-30 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2011012746A2 true WO2011012746A2 (es) | 2011-02-03 |
| WO2011012746A3 WO2011012746A3 (es) | 2011-07-14 |
Family
ID=43529761
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/ES2010/000349 WO2011012746A2 (es) | 2009-07-30 | 2010-07-29 | Compuestos inhibidores de apaf-1 |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US9040701B2 (es) |
| EP (1) | EP2460798B1 (es) |
| JP (1) | JP5694320B2 (es) |
| KR (1) | KR101786761B1 (es) |
| CN (1) | CN102574818B (es) |
| AU (1) | AU2010277505B2 (es) |
| BR (1) | BR112012002134B8 (es) |
| CA (1) | CA2769408C (es) |
| EA (1) | EA021838B1 (es) |
| ES (1) | ES2727711T3 (es) |
| IN (1) | IN2012DN01378A (es) |
| MX (1) | MX2012001339A (es) |
| NZ (1) | NZ598125A (es) |
| TR (1) | TR201907804T4 (es) |
| WO (1) | WO2011012746A2 (es) |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013063468A1 (en) * | 2011-10-27 | 2013-05-02 | Massachusetts Institute Of Technology | Amino acid derivates functionalized on the n- terminal capable of forming drug incapsulating microspheres |
| WO2013078413A1 (en) * | 2011-11-22 | 2013-05-30 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Modulators of lipid storage |
| WO2014013115A1 (es) * | 2012-07-19 | 2014-01-23 | Consejo Superior De Investigaciones Científicas (Csic) | Compuestos beta-lactámicos inhibidores de apaf1 |
| US8969353B2 (en) | 2008-11-07 | 2015-03-03 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US9006487B2 (en) | 2005-06-15 | 2015-04-14 | Massachusetts Institute Of Technology | Amine-containing lipids and uses thereof |
| US9181321B2 (en) | 2013-03-14 | 2015-11-10 | Shire Human Genetic Therapies, Inc. | CFTR mRNA compositions and related methods and uses |
| US9193827B2 (en) | 2010-08-26 | 2015-11-24 | Massachusetts Institute Of Technology | Poly(beta-amino alcohols), their preparation, and uses thereof |
| US9227917B2 (en) | 2012-08-13 | 2016-01-05 | Massachusetts Institute Of Technology | Amine-containing lipidoids and uses thereof |
| US9238716B2 (en) | 2011-03-28 | 2016-01-19 | Massachusetts Institute Of Technology | Conjugated lipomers and uses thereof |
| US9308281B2 (en) | 2011-06-08 | 2016-04-12 | Shire Human Genetic Therapies, Inc. | MRNA therapy for Fabry disease |
| US9315472B2 (en) | 2013-05-01 | 2016-04-19 | Massachusetts Institute Of Technology | 1,3,5-triazinane-2,4,6-trione derivatives and uses thereof |
| US9522176B2 (en) | 2013-10-22 | 2016-12-20 | Shire Human Genetic Therapies, Inc. | MRNA therapy for phenylketonuria |
| US9629804B2 (en) | 2013-10-22 | 2017-04-25 | Shire Human Genetic Therapies, Inc. | Lipid formulations for delivery of messenger RNA |
| US9840479B2 (en) | 2014-07-02 | 2017-12-12 | Massachusetts Institute Of Technology | Polyamine-fatty acid derived lipidoids and uses thereof |
| US9850269B2 (en) | 2014-04-25 | 2017-12-26 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US9957499B2 (en) | 2013-03-14 | 2018-05-01 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US10022455B2 (en) | 2014-05-30 | 2018-07-17 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10138213B2 (en) | 2014-06-24 | 2018-11-27 | Translate Bio, Inc. | Stereochemically enriched compositions for delivery of nucleic acids |
| US10201618B2 (en) | 2015-06-19 | 2019-02-12 | Massachusetts Institute Of Technology | Alkenyl substituted 2,5-piperazinediones, compositions, and uses thereof |
| US10561736B1 (en) | 2019-01-09 | 2020-02-18 | Spiral Therapeutics, Inc. | Apoptosis inhibitor formulations for prevention of hearing loss |
| US10576166B2 (en) | 2009-12-01 | 2020-03-03 | Translate Bio, Inc. | Liver specific delivery of messenger RNA |
| US11174500B2 (en) | 2018-08-24 | 2021-11-16 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US11173190B2 (en) | 2017-05-16 | 2021-11-16 | Translate Bio, Inc. | Treatment of cystic fibrosis by delivery of codon-optimized mRNA encoding CFTR |
| US11224642B2 (en) | 2013-10-22 | 2022-01-18 | Translate Bio, Inc. | MRNA therapy for argininosuccinate synthetase deficiency |
| US11253605B2 (en) | 2017-02-27 | 2022-02-22 | Translate Bio, Inc. | Codon-optimized CFTR MRNA |
| US11254936B2 (en) | 2012-06-08 | 2022-02-22 | Translate Bio, Inc. | Nuclease resistant polynucleotides and uses thereof |
| US12195505B2 (en) | 2018-11-21 | 2025-01-14 | Translate Bio, Inc. | Treatment of cystic fibrosis by delivery of nebulized mRNA encoding CFTR |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3781102A1 (en) | 2018-04-19 | 2021-02-24 | Spiral Therapeutics, Inc. | Inner ear drug delivery devices and methods of use |
| CN113766939B (zh) | 2019-02-25 | 2025-07-08 | 斯皮拉尔诊疗有限公司 | 用于输送药物的鼓室内注射器装置和针以及使用方法 |
| CA3165925A1 (en) | 2020-01-24 | 2021-07-29 | Eugene De Juan | Devices, systems, and methods for otology |
| EP4137131A1 (en) * | 2021-08-17 | 2023-02-22 | Centre National de la Recherche Scientifique (CNRS) | Novel serotonin analogues and their uses for treating iron-associated disorders |
| CN115124449B (zh) * | 2022-05-24 | 2024-02-02 | 上海英诺富成生物科技有限公司 | 一种吲哚化合物及制备方法与应用 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007060524A1 (en) | 2005-11-23 | 2007-05-31 | Laboratorios Salvat, S.A. | Compounds for the inhibition of apoptosis |
| WO2008009758A1 (es) | 2006-07-20 | 2008-01-24 | Consejo Superior De Investigaciones Científicas | Compuestos con actividad inhibidora de las interacciones ubc13-uev, composiciones farmacéuticas y aplicaciones terapéuticas |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3560503A (en) * | 1968-09-18 | 1971-02-02 | Council Scient Ind Res | Di-lower alkyl-substituted octahydropyrazinopyrimidinones |
| GB1284582A (en) | 1969-11-10 | 1972-08-09 | Council Scient Ind Res | Substituted piperazinopyrimidinones |
| JPH04234375A (ja) | 1990-12-27 | 1992-08-24 | Ajinomoto Co Inc | 新規2,5−ジオキソピペラジン化合物及びその製造法 |
| FI934894A7 (fi) * | 1991-05-07 | 1993-11-05 | Merck & Co Inc | Fibrinogeenireseptorin antagonisteja |
| JPH05117246A (ja) | 1991-10-23 | 1993-05-14 | Ajinomoto Co Inc | 新規2,5−ジオキソピペラジン化合物とその製造法及びα−L−アスパルチル−L−フエニルアラニンメチルエステル誘導体の製造法 |
| WO1996000391A1 (en) * | 1994-06-23 | 1996-01-04 | Affymax Technologies N.V. | Methods for the synthesis of diketopiperazines |
| AU2002248410B2 (en) * | 2001-01-19 | 2007-03-01 | Pharmacopeia, Inc. | Bisaryl derivatives having FSH receptor modulatory activity |
| NZ539735A (en) | 2002-10-02 | 2009-07-31 | Dmi Biosciences Inc | Diagnosis and monitoring of diseases using markers, such as diketopiperazines |
| JP4447557B2 (ja) | 2003-11-28 | 2010-04-07 | 協和発酵バイオ株式会社 | ジペプチドの製造法 |
-
2010
- 2010-07-29 EP EP10803937.1A patent/EP2460798B1/en active Active
- 2010-07-29 JP JP2012522195A patent/JP5694320B2/ja not_active Expired - Fee Related
- 2010-07-29 MX MX2012001339A patent/MX2012001339A/es active IP Right Grant
- 2010-07-29 ES ES10803937T patent/ES2727711T3/es active Active
- 2010-07-29 US US13/387,240 patent/US9040701B2/en active Active
- 2010-07-29 AU AU2010277505A patent/AU2010277505B2/en not_active Ceased
- 2010-07-29 NZ NZ598125A patent/NZ598125A/en not_active IP Right Cessation
- 2010-07-29 IN IN1378DEN2012 patent/IN2012DN01378A/en unknown
- 2010-07-29 WO PCT/ES2010/000349 patent/WO2011012746A2/es active Application Filing
- 2010-07-29 CN CN201080043254.1A patent/CN102574818B/zh not_active Expired - Fee Related
- 2010-07-29 CA CA2769408A patent/CA2769408C/en active Active
- 2010-07-29 BR BR112012002134A patent/BR112012002134B8/pt not_active IP Right Cessation
- 2010-07-29 KR KR1020127005131A patent/KR101786761B1/ko not_active Expired - Fee Related
- 2010-07-29 TR TR2019/07804T patent/TR201907804T4/tr unknown
- 2010-07-29 EA EA201270215A patent/EA021838B1/ru not_active IP Right Cessation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007060524A1 (en) | 2005-11-23 | 2007-05-31 | Laboratorios Salvat, S.A. | Compounds for the inhibition of apoptosis |
| WO2008009758A1 (es) | 2006-07-20 | 2008-01-24 | Consejo Superior De Investigaciones Científicas | Compuestos con actividad inhibidora de las interacciones ubc13-uev, composiciones farmacéuticas y aplicaciones terapéuticas |
Non-Patent Citations (3)
| Title |
|---|
| "March, Advanced Organic Chemistry", 1991, JOHN WILEY & SONS |
| MITSUNOBU, J. AM. CHEM. SOC., vol. 94, 1972, pages 679 - 680 |
| See also references of EP2460798A4 |
Cited By (94)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9006487B2 (en) | 2005-06-15 | 2015-04-14 | Massachusetts Institute Of Technology | Amine-containing lipids and uses thereof |
| US11414393B2 (en) | 2008-11-07 | 2022-08-16 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US10844028B2 (en) | 2008-11-07 | 2020-11-24 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US10189802B2 (en) | 2008-11-07 | 2019-01-29 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US8969353B2 (en) | 2008-11-07 | 2015-03-03 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US9556110B2 (en) | 2008-11-07 | 2017-01-31 | Massachusetts Institute Of Technology | Aminoalcohol lipidoids and uses thereof |
| US10576166B2 (en) | 2009-12-01 | 2020-03-03 | Translate Bio, Inc. | Liver specific delivery of messenger RNA |
| US9193827B2 (en) | 2010-08-26 | 2015-11-24 | Massachusetts Institute Of Technology | Poly(beta-amino alcohols), their preparation, and uses thereof |
| US9238716B2 (en) | 2011-03-28 | 2016-01-19 | Massachusetts Institute Of Technology | Conjugated lipomers and uses thereof |
| US10933139B2 (en) | 2011-03-28 | 2021-03-02 | Massachusetts Institute Of Technology | Conjugated lipomers and uses thereof |
| US10117934B2 (en) | 2011-03-28 | 2018-11-06 | Massachusetts Institute Of Technology | Conjugated lipomers and uses thereof |
| US11338044B2 (en) | 2011-06-08 | 2022-05-24 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11730825B2 (en) | 2011-06-08 | 2023-08-22 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11185595B2 (en) | 2011-06-08 | 2021-11-30 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11052159B2 (en) | 2011-06-08 | 2021-07-06 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11291734B2 (en) | 2011-06-08 | 2022-04-05 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US10888626B2 (en) | 2011-06-08 | 2021-01-12 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US9308281B2 (en) | 2011-06-08 | 2016-04-12 | Shire Human Genetic Therapies, Inc. | MRNA therapy for Fabry disease |
| US9597413B2 (en) | 2011-06-08 | 2017-03-21 | Shire Human Genetic Therapies, Inc. | Pulmonary delivery of mRNA |
| US12121592B2 (en) | 2011-06-08 | 2024-10-22 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US10507249B2 (en) | 2011-06-08 | 2019-12-17 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US10413618B2 (en) | 2011-06-08 | 2019-09-17 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US10350303B1 (en) | 2011-06-08 | 2019-07-16 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| US11951180B2 (en) | 2011-06-08 | 2024-04-09 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US11547764B2 (en) | 2011-06-08 | 2023-01-10 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US10238754B2 (en) | 2011-06-08 | 2019-03-26 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US11951179B2 (en) | 2011-06-08 | 2024-04-09 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for MRNA delivery |
| US11951181B2 (en) | 2011-06-08 | 2024-04-09 | Translate Bio, Inc. | Lipid nanoparticle compositions and methods for mRNA delivery |
| CN107522664A (zh) * | 2011-10-27 | 2017-12-29 | 麻省理工学院 | 能够形成药物包封微球的在n末端上官能化的氨基酸衍生物 |
| US11458158B2 (en) | 2011-10-27 | 2022-10-04 | Massachusetts Institute Of Technology | Amino acid-, peptide- and polypeptide-lipids, isomers, compositions, and uses thereof |
| US10682374B2 (en) | 2011-10-27 | 2020-06-16 | Massachusetts Intstitute Of Technology | Amino acid-, peptide- and polypeptide-lipids, isomers, compositions, and uses thereof |
| JP2023166457A (ja) * | 2011-10-27 | 2023-11-21 | マサチューセッツ インスティテュート オブ テクノロジー | 薬物内包微小球の形成が可能なn-末端で官能基化されたアミノ酸誘導体 |
| JP2014534220A (ja) * | 2011-10-27 | 2014-12-18 | マサチューセッツ インスティテュート オブ テクノロジー | 薬物内包微小球の形成が可能なn−末端で官能基化されたアミノ酸誘導体 |
| US10086013B2 (en) | 2011-10-27 | 2018-10-02 | Massachusetts Institute Of Technology | Amino acid-, peptide- and polypeptide-lipids, isomers, compositions, and uses thereof |
| KR20200037431A (ko) * | 2011-10-27 | 2020-04-08 | 메사추세츠 인스티튜트 오브 테크놀로지 | 약물 캡슐화 마이크로스피어를 형성할 수 있는, n-말단 상에 관능화된 아미노산 유도체 |
| KR20210082570A (ko) * | 2011-10-27 | 2021-07-05 | 메사추세츠 인스티튜트 오브 테크놀로지 | 약물 캡슐화 마이크로스피어를 형성할 수 있는, n-말단 상에 관능화된 아미노산 유도체 |
| EA032088B1 (ru) * | 2011-10-27 | 2019-04-30 | Массачусетс Инститьют Оф Текнолоджи | Аминокислотные производные, функционализованные на n-конце, способные образовывать микросферы, инкапсулирующие лекарственное средство |
| JP2021181463A (ja) * | 2011-10-27 | 2021-11-25 | マサチューセッツ インスティテュート オブ テクノロジー | 薬物内包微小球の形成が可能なn−末端で官能基化されたアミノ酸誘導体 |
| US9512073B2 (en) * | 2011-10-27 | 2016-12-06 | Massachusetts Institute Of Technology | Amino acid-, peptide-and polypeptide-lipids, isomers, compositions, and uses thereof |
| EP4074694A1 (en) * | 2011-10-27 | 2022-10-19 | Massachusetts Institute Of Technology | Amino acid-, peptide- an polypeptide-lipids, isomers, compositions, an uses thereof |
| JP7527650B2 (ja) | 2011-10-27 | 2024-08-05 | マサチューセッツ インスティテュート オブ テクノロジー | 薬物内包微小球の形成が可能なn-末端で官能基化されたアミノ酸誘導体 |
| AU2012328570B2 (en) * | 2011-10-27 | 2017-08-31 | Massachusetts Institute Of Technology | Amino acid derivatives functionalized on the n-terminus capable of forming drug encapsulating microspheres and uses thereof |
| CN107522664B (zh) * | 2011-10-27 | 2021-03-16 | 麻省理工学院 | 能够形成药物包封微球的在n末端上官能化的氨基酸衍生物 |
| KR20220139428A (ko) * | 2011-10-27 | 2022-10-14 | 메사추세츠 인스티튜트 오브 테크놀로지 | 약물 캡슐화 마이크로스피어를 형성할 수 있는, n-말단 상에 관능화된 아미노산 유도체 |
| WO2013063468A1 (en) * | 2011-10-27 | 2013-05-02 | Massachusetts Institute Of Technology | Amino acid derivates functionalized on the n- terminal capable of forming drug incapsulating microspheres |
| US20130158021A1 (en) * | 2011-10-27 | 2013-06-20 | Massachusetts Institute Of Technology | Amino acid-, peptide-and polypeptide-lipids, isomers, compositions, and uses thereof |
| WO2013078413A1 (en) * | 2011-11-22 | 2013-05-30 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Modulators of lipid storage |
| US11254936B2 (en) | 2012-06-08 | 2022-02-22 | Translate Bio, Inc. | Nuclease resistant polynucleotides and uses thereof |
| ES2443538A1 (es) * | 2012-07-19 | 2014-02-19 | Consejo Superior De Investigaciones Cientificas (Csic) | Compuestos beta-lactámicos inhibidores de APAF1 |
| WO2014013115A1 (es) * | 2012-07-19 | 2014-01-23 | Consejo Superior De Investigaciones Científicas (Csic) | Compuestos beta-lactámicos inhibidores de apaf1 |
| US9439968B2 (en) | 2012-08-13 | 2016-09-13 | Massachusetts Institute Of Technology | Amine-containing lipidoids and uses thereof |
| US9227917B2 (en) | 2012-08-13 | 2016-01-05 | Massachusetts Institute Of Technology | Amine-containing lipidoids and uses thereof |
| US9713626B2 (en) | 2013-03-14 | 2017-07-25 | Rana Therapeutics, Inc. | CFTR mRNA compositions and related methods and uses |
| US12234446B2 (en) | 2013-03-14 | 2025-02-25 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US10876104B2 (en) | 2013-03-14 | 2020-12-29 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US9181321B2 (en) | 2013-03-14 | 2015-11-10 | Shire Human Genetic Therapies, Inc. | CFTR mRNA compositions and related methods and uses |
| US10420791B2 (en) | 2013-03-14 | 2019-09-24 | Translate Bio, Inc. | CFTR MRNA compositions and related methods and uses |
| US11510937B2 (en) | 2013-03-14 | 2022-11-29 | Translate Bio, Inc. | CFTR MRNA compositions and related methods and uses |
| US9957499B2 (en) | 2013-03-14 | 2018-05-01 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US11692189B2 (en) | 2013-03-14 | 2023-07-04 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US11820977B2 (en) | 2013-03-14 | 2023-11-21 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US9315472B2 (en) | 2013-05-01 | 2016-04-19 | Massachusetts Institute Of Technology | 1,3,5-triazinane-2,4,6-trione derivatives and uses thereof |
| US11377642B2 (en) | 2013-10-22 | 2022-07-05 | Translate Bio, Inc. | mRNA therapy for phenylketonuria |
| US10959953B2 (en) | 2013-10-22 | 2021-03-30 | Translate Bio, Inc. | Lipid formulations for delivery of messenger RNA |
| US9522176B2 (en) | 2013-10-22 | 2016-12-20 | Shire Human Genetic Therapies, Inc. | MRNA therapy for phenylketonuria |
| US9629804B2 (en) | 2013-10-22 | 2017-04-25 | Shire Human Genetic Therapies, Inc. | Lipid formulations for delivery of messenger RNA |
| US11224642B2 (en) | 2013-10-22 | 2022-01-18 | Translate Bio, Inc. | MRNA therapy for argininosuccinate synthetase deficiency |
| US10493031B2 (en) | 2013-10-22 | 2019-12-03 | Translate Bio, Inc. | Lipid formulations for delivery of messenger RNA |
| US11890377B2 (en) | 2013-10-22 | 2024-02-06 | Translate Bio, Inc. | Lipid formulations for delivery of messenger RNA |
| US10208295B2 (en) | 2013-10-22 | 2019-02-19 | Translate Bio, Inc. | MRNA therapy for phenylketonuria |
| US10052284B2 (en) | 2013-10-22 | 2018-08-21 | Translate Bio, Inc. | Lipid formulations for delivery of messenger RNA |
| US9850269B2 (en) | 2014-04-25 | 2017-12-26 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US11059841B2 (en) | 2014-04-25 | 2021-07-13 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US11884692B2 (en) | 2014-04-25 | 2024-01-30 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US12060381B2 (en) | 2014-04-25 | 2024-08-13 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US10155785B2 (en) | 2014-04-25 | 2018-12-18 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US10912844B2 (en) | 2014-05-30 | 2021-02-09 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10022455B2 (en) | 2014-05-30 | 2018-07-17 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10286082B2 (en) | 2014-05-30 | 2019-05-14 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10293057B2 (en) | 2014-05-30 | 2019-05-21 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10493166B2 (en) | 2014-05-30 | 2019-12-03 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US11433144B2 (en) | 2014-05-30 | 2022-09-06 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US10286083B2 (en) | 2014-05-30 | 2019-05-14 | Translate Bio, Inc. | Biodegradable lipids for delivery of nucleic acids |
| US11104652B2 (en) | 2014-06-24 | 2021-08-31 | Translate Bio, Inc. | Stereochemically enriched compositions for delivery of nucleic acids |
| US10138213B2 (en) | 2014-06-24 | 2018-11-27 | Translate Bio, Inc. | Stereochemically enriched compositions for delivery of nucleic acids |
| US9840479B2 (en) | 2014-07-02 | 2017-12-12 | Massachusetts Institute Of Technology | Polyamine-fatty acid derived lipidoids and uses thereof |
| US10695444B2 (en) | 2015-06-19 | 2020-06-30 | Massachusetts Institute Of Technology | Alkenyl substituted 2,5-piperazinediones, compositions, and uses thereof |
| US10201618B2 (en) | 2015-06-19 | 2019-02-12 | Massachusetts Institute Of Technology | Alkenyl substituted 2,5-piperazinediones, compositions, and uses thereof |
| US11253605B2 (en) | 2017-02-27 | 2022-02-22 | Translate Bio, Inc. | Codon-optimized CFTR MRNA |
| US11173190B2 (en) | 2017-05-16 | 2021-11-16 | Translate Bio, Inc. | Treatment of cystic fibrosis by delivery of codon-optimized mRNA encoding CFTR |
| US12084702B2 (en) | 2018-08-24 | 2024-09-10 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US11174500B2 (en) | 2018-08-24 | 2021-11-16 | Translate Bio, Inc. | Methods for purification of messenger RNA |
| US12195505B2 (en) | 2018-11-21 | 2025-01-14 | Translate Bio, Inc. | Treatment of cystic fibrosis by delivery of nebulized mRNA encoding CFTR |
| US10561736B1 (en) | 2019-01-09 | 2020-02-18 | Spiral Therapeutics, Inc. | Apoptosis inhibitor formulations for prevention of hearing loss |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102574818A (zh) | 2012-07-11 |
| IN2012DN01378A (es) | 2015-06-05 |
| JP5694320B2 (ja) | 2015-04-01 |
| AU2010277505B2 (en) | 2016-01-14 |
| KR20120052354A (ko) | 2012-05-23 |
| US20120122868A1 (en) | 2012-05-17 |
| WO2011012746A3 (es) | 2011-07-14 |
| NZ598125A (en) | 2014-05-30 |
| AU2010277505A1 (en) | 2012-03-08 |
| KR101786761B1 (ko) | 2017-10-18 |
| TR201907804T4 (tr) | 2019-06-21 |
| JP2013500314A (ja) | 2013-01-07 |
| BR112012002134B1 (pt) | 2021-03-02 |
| EP2460798B1 (en) | 2019-02-27 |
| EP2460798A2 (en) | 2012-06-06 |
| BR112012002134B8 (pt) | 2021-05-25 |
| EA021838B1 (ru) | 2015-09-30 |
| MX2012001339A (es) | 2012-03-07 |
| CN102574818B (zh) | 2015-02-25 |
| BR112012002134A2 (pt) | 2016-05-31 |
| US9040701B2 (en) | 2015-05-26 |
| EP2460798A4 (en) | 2013-02-27 |
| CA2769408A1 (en) | 2011-02-03 |
| ES2727711T3 (es) | 2019-10-18 |
| CA2769408C (en) | 2017-09-05 |
| EA201270215A1 (ru) | 2012-11-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2727711T3 (es) | Compuestos inhibidores de Apaf-1 | |
| JP7659015B2 (ja) | 免疫調節剤としての1,2,4-オキサジアゾールおよびチアジアゾール化合物 | |
| TW201428002A (zh) | 用於抑制細胞凋亡抑制劑之巨環化合物 | |
| JP2002520335A (ja) | ドラスタチン15誘導体 | |
| WO1995003277A1 (fr) | Nouveau derive de pyrrolidine | |
| CZ300127B6 (cs) | Zpusob prípravy meziproduktu inhibujících proteázy retroviru | |
| CA2765150A1 (en) | 2, 3-dihydro-1h-indene compounds and their use to treat cancer | |
| US20020119962A1 (en) | Novel urea compounds, compositions and methods of use and preparation | |
| WO2015161745A1 (zh) | 鬼臼毒素衍生物、其制备方法、药物组合物及应用 | |
| US10287316B2 (en) | Process for preparation of nitrogen mustard derivatives | |
| CN102249987B (zh) | 一种考布他汀类化合物及其制备方法和用途 | |
| CN103387601A (zh) | 抗登革热病毒(denv)杂环肽类化合物及其制备方法和用途 | |
| NZ515026A (en) | Heterocyclic amidine derivatives useful as prodrugs of thrombin inhibitors | |
| CN113045550A (zh) | 一种三氮唑类衍生物及其制备方法和应用 | |
| WO2025082371A1 (zh) | 一种肝靶向化合物及其寡核苷酸缀合物、偶联方法与应用 | |
| FR2828884A1 (fr) | Hydrazinopeptoides et leurs utilisations dans le traitement des cancers | |
| CA2451768C (en) | Peptoid compounds | |
| CN114667136A (zh) | Trofinetide的组合物 | |
| WO2020022892A1 (en) | Tubulysin derivatives and methods for preparing the same | |
| CN106496132B (zh) | N-(4-取代苯基)-2-取代乙酰胺类化合物及其作为sirt2蛋白抑制剂的用途 | |
| CN101724016B (zh) | 一类肽化合物、其制备方法及用途 | |
| WO2016107541A1 (zh) | 吡咯酰胺类化合物及其制备方法与用途 | |
| CN106632405B (zh) | 一种鬼臼毒素与去甲斑蝥素的拼合物及其制备与应用 | |
| CN103626837A (zh) | 含生物可裂解二肽的糖原磷酸化酶抑制剂胆酸类衍生物、其制备方法及医药用途 | |
| CN109134295B (zh) | 蒽二酮衍生物及其制备方法和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080043254.1 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2769408 Country of ref document: CA Ref document number: 13387240 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012522195 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2012/001339 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010277505 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1378/DELNP/2012 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010803937 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20127005131 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201270215 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 2010277505 Country of ref document: AU Date of ref document: 20100729 Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112012002134 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112012002134 Country of ref document: BR Kind code of ref document: A2 Effective date: 20120130 |