WO2010107014A1 - 生分解性樹脂組成物の結晶化促進方法 - Google Patents
生分解性樹脂組成物の結晶化促進方法 Download PDFInfo
- Publication number
- WO2010107014A1 WO2010107014A1 PCT/JP2010/054402 JP2010054402W WO2010107014A1 WO 2010107014 A1 WO2010107014 A1 WO 2010107014A1 JP 2010054402 W JP2010054402 W JP 2010054402W WO 2010107014 A1 WO2010107014 A1 WO 2010107014A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- biodegradable resin
- resin composition
- crystallization
- acid
- acid amide
- Prior art date
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/0008—Organic ingredients according to more than one of the "one dot" groups of C08K5/01 - C08K5/59
- C08K5/0083—Nucleating agents promoting the crystallisation of the polymer matrix
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/20—Compounding polymers with additives, e.g. colouring
- C08J3/201—Pre-melted polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/20—Carboxylic acid amides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2300/00—Characterised by the use of unspecified polymers
- C08J2300/16—Biodegradable polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2367/00—Characterised by the use of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Derivatives of such polymers
- C08J2367/04—Polyesters derived from hydroxy carboxylic acids, e.g. lactones
Definitions
- the present invention relates to a method for promoting crystallization of a biodegradable resin composition. More specifically, a method for promoting crystallization of a biodegradable resin composition using a specific crystal nucleating agent, a biodegradable resin composition whose crystallization is promoted by the method, and molding of the biodegradable resin composition About the body.
- biodegradable resin When biodegradable resin is placed in soil, seawater, or the body of an animal, it begins to degrade in a few weeks due to the action of enzymes produced by microorganisms that inhabit the natural world. Disappears. Therefore, its use has attracted attention in recent years.
- Patent Document 1 as a method for obtaining a biodegradable resin composition having good crystallization speed and transparency, a group consisting of a biodegradable resin, a plasticizer, and an ester group, a hydroxyl group, and an amide group in the molecule.
- a production method is disclosed in which a crystal nucleating agent, which is an aliphatic compound having two or more selected at least one group, is melt-kneaded at a specific temperature and heat-treated.
- crystal nucleating agent examples include fatty acid esters such as aliphatic esters and hydroxy fatty acid esters; aliphatic amides such as hydroxy fatty acid monoamides, aliphatic bisamides, and hydroxy fatty acid bisamides; fatty acid metal salts such as hydroxy fatty acid metal salts. .
- the present invention [1] A method for promoting crystallization of a biodegradable resin composition, comprising a step of melt-kneading a raw material containing ethylenebis12-hydroxystearic acid amide having an amine value of 1.0 mgKOH / g or less and a biodegradable resin. , [2] A biodegradable resin composition in which crystallization is promoted by the crystallization promoting method described in [1], and [3] a biodegradable resin composition described in [2].
- the present invention relates to a biodegradable resin molded body.
- FIG. 1 is a view showing a molded body obtained using a vacuum / pressure forming machine.
- the present invention relates to a method for promoting crystallization of a biodegradable resin composition, a biodegradable resin composition whose crystallization is promoted by the method, and a molded article of the biodegradable resin composition.
- the biodegradable resin composition that has been crystallized by the method for promoting crystallization of the biodegradable resin composition of the present invention has a good crystallization rate, the time required for molding the composition is short. Thus, a molded body can be produced with high productivity.
- the method for promoting crystallization of a biodegradable resin composition of the present invention includes a step of melt-kneading a raw material containing a crystal nucleating agent and a biodegradable resin, and the crystal nucleating agent has a specific amine value.
- the ethylene bis 12-hydroxystearic acid amide has a great feature.
- Ethylene bis 12-hydroxystearic acid amide is used as a lubricant and an anti-blocking agent for thermoplastic resins. Further, it is a compound having two hydroxy groups contributing to dispersibility and two amide groups contributing to compatibility, and is also used as a crystal nucleating agent for polylactic acid resin and the like. This compound is obtained by subjecting 12-hydroxystearic acid and ethylenediamine to a dehydration condensation reaction. The resulting reaction product contains unreacted ethylenediamine, a reaction intermediate monoamide amine, a by-product amine such as imidazoline. It is.
- amines are not preferred because they cause coloring during storage and heating of ethylenebis12-hydroxystearic acid amide, and are also unfavorable for safety to the human body. It has been found that the biodegradable resin composition obtained using ethylene bis 12-hydroxystearic acid amide containing an amine causes a decrease in crystallization rate in addition to a deterioration in hue. Therefore, in the present invention, crystallization of the biodegradable resin composition can be promoted by using ethylenebis12-hydroxystearic acid amide having an amine value of 1.0 mgKOH / g or less as a crystal nucleating agent. .
- ethylene bis 12-hydroxystearic acid amide having an amine value of 1.0 mgKOH / g or less means a crystal nucleating agent comprising ethylene bis 12-hydroxystearic acid amide having an amine value of 1.0 mgKOH / g or less.
- the method for promoting crystallization of the biodegradable resin composition of the present invention comprises a raw material containing a crystal nucleating agent, ethylenebis 12-hydroxystearic acid amide having an amine value of 1.0 mgKOH / g or less, and a biodegradable resin. Including a step of melt kneading (hereinafter also referred to as a melt kneading step).
- Ethylene bis 12-hydroxystearic acid amide used in the present invention has an amine value of 1.0 mgKOH / g or less, preferably 0.5 mgKOH / g or less. Further, from the viewpoint of productivity, it is preferably 0.01 mgKOH / g or more. Therefore, the amine value of ethylenebis12-hydroxystearic acid amide used in the present invention is preferably 0.01 to 1.0 mgKOH / g, more preferably 0.01 to 0.5 mgKOH / g. In the present specification, the amine value means the total amount of amine and is also referred to as the total amine value. The amine value can be measured according to the method described in Examples described later.
- Ethylene bis 12-hydroxystearic acid amide may be a commercially available method as long as the amine value is 1.0 mg KOH / g or less (for example, the method described in JP-A-63-60956). ) May be synthesized.
- Ethylene bis 12-hydroxystearic acid amide can be obtained by using 12-hydroxystearic acid and ethylenediamine as raw materials and subjecting the raw materials to dehydration condensation reaction.
- the molar ratio of the raw materials used for the dehydration condensation reaction is adjusted, and the molar ratio of 12-hydroxystearic acid to ethylenediamine (12- (Hydroxystearic acid / ethylenediamine) is preferably 2.0 / 1 or more.
- the molar ratio is preferably 2.20 / 1 or less. 2.15 / 1 or less is more preferable. Accordingly, the molar ratio is preferably 2.0 / 1 to 2.20 / 1, and more preferably 2.0 / 1 to 2.15 / 1.
- the dehydration condensation reaction is performed under normal pressure (101.3 kPa) in an inert gas atmosphere such as nitrogen.
- the reaction temperature is preferably 180 to 230 ° C, more preferably 190 to 220 ° C. When the reaction temperature is 180 ° C. or higher, the reaction proceeds efficiently, and when it is 230 ° C. or lower, the hue of the obtained reaction product becomes good.
- the reaction time cannot be generally determined depending on the molar ratio of the raw materials and the reaction temperature, it is preferably a time during which 12-hydroxystearic acid and ethylenediamine can sufficiently react, and usually 3 to 7 hours are preferable.
- ethylene bis 12-hydroxystearic acid amide having an amine value of 1.0 mgKOH / g or less can be synthesized.
- ethylene bis 12-hydroxystearic acid amide having an amine value exceeding 1.0 mgKOH / g is purified according to a known method to reduce the amine value to 1.0 mgKOH / g or less. Also good. Furthermore, the ethylene bis 12-hydroxystearic acid amide obtained by the above synthesis method may be used by further purifying it to reduce the amine value.
- Ethylene bis 12-hydroxystearic acid amide is thermally washed and / or crystallized using at least one solvent selected from the group consisting of alcohol solvents, aromatic hydrocarbon solvents, ketone solvents, and ester solvents. Purify by analysis. In addition, when using 2 or more types of solvent, it can use as a mixed solvent.
- Alcohol solvents include methanol, ethanol, 1-propanol, 2-propanol, 1-butanol, 2-butanol, 2-methyl-1-propanol, 2-methyl-2-propanol, 1-pentanol, 2-pen Tanol, 3-pentanol, 2-methyl-1-butanol, 3-methyl-1-butanol, 2-methyl-2-butanol, 3-methyl-2-butanol, ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, etc. Is mentioned.
- methanol, ethanol, 1-propanol, 2-propanol, 1-butanol, 2-butanol, 2-methyl-1-propanol are used from the viewpoint of obtaining ethylenebis12-hydroxystearic acid amide having a low amine value.
- 2-methyl-2-propanol are preferred.
- 1-propanol, 2-propanol, 1-butanol, 2-butanol, 2-methyl-1-propanol, and 2-methyl-2-propanol is preferred.
- aromatic hydrocarbon solvent examples include toluene, o-xylene, m-xylene, p-xylene, ethylbenzene and the like, and among these, toluene is preferable.
- ketone solvent examples include acetone, methyl ethyl ketone, methyl isobutyl ketone, and the like, and methyl ethyl ketone and methyl isobutyl ketone are preferable.
- ester solvent examples include ethyl acetate, n-propyl acetate, n-butyl acetate, iso-butyl acetate, sec-butyl acetate and the like.
- ethylene bis 12-hydroxystearic acid amide (hereinafter also referred to as unpurified bis fatty acid amide) subjected to purification under normal pressure or pressure (0.1 to 10 MPa) It is suspended or dissolved by heating in at least one of the solvents. Thereafter, the suspension or solution is cooled and filtered under reduced pressure (0.001 to 0.09 MPa) or pressurized (0.11 to 0.5 MPa), and the filtrate is collected, washed and dried.
- the series of operations may be performed once, and only thermal cleaning may be performed multiple times, only crystallization may be performed multiple times, or a combination of thermal cleaning and crystallization may be performed, and the solvent used at that time is the same for each operation. But it can be different.
- the amount of the solvent used for suspending or dissolving the unpurified bisfatty acid amide is preferably 100 to 10,000 parts by weight, more preferably 200 to 3000 parts by weight, and more preferably 300 to 1500 parts by weight with respect to 100 parts by weight of the unpurified bisfatty acid amide. Part is more preferable. When it is 100 parts by weight or more, the viscosity of the suspension or solution does not become too high, and the workability of filtration is good. When the amount is 10,000 parts by weight or less, the loss of ethylenebis12-hydroxystearic acid amide obtained is small and economical.
- the heating performed during suspension or dissolution is preferably performed at a temperature of 50 to 180 ° C, more preferably 50 to 150 ° C, and even more preferably 80 to 130 ° C.
- the heating time is preferably 0.5 to 10 hours.
- the cooling after heating is preferably performed at a temperature of 5 to 40 ° C, more preferably 10 to 40 ° C, and even more preferably 15 to 40 ° C.
- the filtration method is not particularly limited and can be performed according to a known method.
- the filtrate contains unreacted raw materials (ethylenediamine, 12-hydroxystearic acid) and a reaction intermediate (monoamidoamine), and the amine content of the resulting residue is reduced.
- the filtration residue is washed because it contains the remaining filtrate.
- the washing method is not particularly limited, and the washing can be performed preferably at 10 to 80 ° C., more preferably at 10 to 60 ° C., using the same or different solvent as the solvent used for the suspension or dissolution.
- the number of washings is preferably 1 to 5 times, and more preferably 1 to 3 times.
- the amount of solvent used for one washing is preferably 10 to 2000 parts by weight, more preferably 100 to 1000 parts by weight, and even more preferably 100 to 500 parts by weight with respect to 100 parts by weight of the residue.
- Drying is desirably performed under normal pressure or preferably under reduced pressure of 15 kPa or less, more preferably 6.7 kPa or less.
- the drying temperature is preferably 10 to 180 ° C, more preferably 40 to 150 ° C, and further preferably 60 to 120 ° C.
- purified bis fatty acid amide ethylene bis 12-hydroxystearic acid amide having a reduced amine value
- unpurified bisfatty acid amide and purified bisfatty acid amide obtained by the above purification method are mixed by a known method, and the resulting mixture has an amine value of 1.0 mgKOH / g or less.
- the mixture may be used.
- the acid value of ethylenebis12-hydroxystearic acid amide having an amine value of 1.0 mgKOH / g or less is preferably 1.0 mgKOH / g or less, more preferably 0.6 mgKOH / g or less, and further preferably 0.4 mgKOH / g or less.
- ethylene bis 12-hydroxystearic acid amide having an acid value of 1.0 mgKOH / g or less means a crystal nucleating agent comprising ethylene bis 12-hydroxystearic acid amide having an acid value of 1.0 mgKOH / g or less.
- an acid value can be measured according to the method as described in the below-mentioned Example.
- ethylene bis 12-hydroxystearic acid amide obtained according to the method described in JP-A-63-609556 has a high purity with an amine value of 1.0 mgKOH / g or less.
- the method stops the reaction when the conversion rate of the raw material to bisamide reaches 85 to 98%, and then removes unreacted components including monoamidoamine by thin-film distillation, so that a high-purity fatty acid is obtained. Since this is a method for producing bisamide, the operation is complicated and the productivity is low.
- the hue of the resulting bisamide is not sufficient, in the present invention, even if ethylene bis 12-hydroxystearic acid amide having an amine value of 1.0 mg KOH / g or less is used, the ethylene bis purified by the above purification method is used. It is preferable to use 12-hydroxystearic amide.
- the hue of the purified bis-fatty acid amide is preferably 2 or less, more preferably 1 or less. In the present specification, the hue can be measured according to a method described in Examples described later.
- the freezing point of ethylenebis12-hydroxystearic acid amide used in the biodegradable resin composition is preferably higher.
- the freezing point of ethylenebis12-hydroxystearic acid amide having an amine value of 1.0 mgKOH / g or less is preferably 135 to 145 ° C, more preferably 136 to 144 ° C, and further preferably 138 to 143 ° C. 139 to 143 ° C is even more preferable.
- a crystallization method in which the freezing point of the purified product is increased is preferable.
- the freezing point is observed when the temperature is lowered to 25 ° C. at a rate of 10 ° C./min after melting at 200 ° C. for 2 minutes using a DSC apparatus (Perkin Elmer, Diamond DSC). It is obtained from the peak temperature of the crystallization exotherm.
- crystal nucleating agents other than the ethylenebis12-hydroxystearic acid amide may be contained within a range not impairing the effects of the present invention.
- the crystal nucleating agents described in JP-A-2008-174718 and JP-A-2008-115372 are preferable from the viewpoint of the bending strength and moldability of the biodegradable resin composition.
- crystal nucleating agent (2) a compound having a hydroxyl group and an amide group in the molecule, including ethylenebis12-hydroxystearic acid amide having an amine value of 1.0 mgKOH / g or less is used as the crystal nucleating agent ( 1)
- crystal nucleating agent (2) a compound having a hydroxyl group and an amide group in the molecule, including ethylenebis12-hydroxystearic acid amide having an amine value of 1.0 mgKOH / g or less.
- the crystal nucleating agent (1) other than ethylenebis12-hydroxystearic acid amide having an amine value of 1.0 mgKOH / g or less the moldability, heat resistance, impact resistance and crystal nucleating agent of the biodegradable resin composition
- hydroxy fatty acid bisamides such as hexamethylene bis 12-hydroxystearic acid amide, 12-hydroxystearic acid triglyceride are preferable, and ethylene bis 12-hydroxystearic acid amide is more preferable.
- the crystal nucleating agent (2) is preferably a phenylphosphonic acid metal salt from the viewpoint of crystallization speed.
- the phenylphosphonic acid metal salt is a metal salt of phenylphosphonic acid having a phenyl group which may have a substituent and a phosphonic group (—PO (OH) 2 ).
- phenylphosphonic acid examples include unsubstituted phenylphosphonic acid, methylphenylphosphonic acid, ethylphenylphosphonic acid, propylphenylphosphonic acid, butylphenylphosphonic acid, dimethoxycarbonylphenylphosphonic acid, and diethoxycarbonylphenylphosphonic acid. And unsubstituted phenylphosphonic acid is preferred.
- Examples of the metal of the metal salt of phenylphosphonic acid include lithium, sodium, magnesium, aluminum, potassium, calcium, barium, copper, zinc, iron, cobalt, nickel and the like, with zinc being preferred.
- crystal nucleating agents can be used alone or in combination of two or more.
- the weight ratio of the crystal nucleating agent (1) and the crystal nucleating agent (2) [crystal nucleating agent (1) / crystal nucleating agent (2)] 20/80 to 80/20 are preferred, 30/70 to 70/30 are more preferred, and 40/60 to 60/40 are even more preferred.
- the content of ethylenebis12-hydroxystearic acid amide having an amine value of 1.0 mgKOH / g or less in the crystal nucleating agent is preferably 80% by weight or more, more preferably 90% by weight or more, and substantially 100% by weight. More preferably it is.
- the content of the crystal nucleating agent is preferably 0.1 to 5 parts by weight, more preferably 0.5 to 3 parts by weight, and more preferably 0.5 to 3 parts by weight with respect to 100 parts by weight of the biodegradable resin from the viewpoint of moldability of the biodegradable resin composition. More preferred is ⁇ 2 parts by weight.
- “content” means “content or blending amount”.
- biodegradable resin Any biodegradable resin may be used as long as it has a biodegradability capable of being decomposed into low molecular weight compounds by being involved with microorganisms in the natural world.
- biodegradable resin polyhydroxybutyrate, polycaprolactone, polybutylene succinate, polybutylene succinate Nate / adipate, polyethylene succinate, polylactic acid resin, polymalic acid, polyglycolic acid, polydioxanone, poly (2-oxetanone) and other aliphatic polyesters; polybutylene succinate / terephthalate, polybutylene adipate / terephthalate, polytetramethylene adipate / Aliphatic aromatic copolyesters such as terephthalate; natural polymers such as starch, cellulose, chitin, chitosan, gluten, gelatin, zein, soy protein, collagen, keratin and the above aliphatic polyesters or Mixtures
- biodegradable means a property that can be decomposed into a low molecular weight compound by a microorganism in nature.
- JIS K6953 ISO 14855
- controlled aerobic composting conditions This means biodegradability based on the “Aerobic and Ultimate Biodegradation and Disintegration Test”.
- Polylactic acid resin is a polylactic acid obtained by polycondensing only a lactic acid component as a raw material monomer, and / or a lactic acid component and a hydroxycarboxylic acid component other than lactic acid as a raw material monomer (hereinafter also simply referred to as a hydroxycarboxylic acid component). And polylactic acid obtained by subjecting them to condensation polymerization.
- Lactic acid has optical isomers of L-lactic acid (L form) and D-lactic acid (D form).
- the lactic acid component either or both of the optical isomers may be contained, but from the viewpoint of moldability of the biodegradable resin composition, any of the optical isomers is a main component. It is preferable to use lactic acid having a high optical purity.
- the “main component” refers to a component whose content in the lactic acid component is 50 mol% or more.
- examples of the hydroxycarboxylic acid component include hydroxycarboxylic acid compounds such as glycolic acid, hydroxybutyric acid, hydroxyvaleric acid, hydroxypentanoic acid, and hydroxycaproic acid, which can be used alone or in combination of two or more. .
- glycolic acid and hydroxycaproic acid are preferable from the viewpoints of heat resistance and transparency of the biodegradable resin composition.
- the dimer of the lactic acid and the hydroxycarboxylic acid compound may be contained in each component, and as a preferred example, from the viewpoint of heat resistance and transparency of the biodegradable resin composition. From D-lactide and L-lactide.
- the dimer of lactic acid may be contained in the lactic acid component in any case where only the lactic acid component is subjected to polycondensation and in the case where the lactic acid component and the hydroxycarboxylic acid component are polycondensed.
- the content of the dimer of lactic acid is preferably 80 to 100 mol% and more preferably 90 to 100 mol% in the lactic acid component from the viewpoint of heat resistance of the biodegradable resin composition.
- the content of the dimer of the hydroxycarboxylic acid compound is preferably 80 to 100 mol%, more preferably 90 to 100 mol% in the hydroxycarboxylic acid component, from the viewpoint of heat resistance of the biodegradable resin composition.
- the polycondensation reaction of only the lactic acid component and the polycondensation reaction of the lactic acid component and the hydroxycarboxylic acid component are not particularly limited and can be performed using a known method.
- polylactic acid composed of 85 mol% or more and less than 100 mol% of either L-lactic acid or D-lactic acid and more than 0 mol% and 15 mol% or less of the hydroxycarboxylic acid component can be obtained.
- polylactic acid obtained by using lactide, which is a cyclic dimer of lactic acid, glycolide, which is a cyclic dimer of glycolic acid, and caprolactone, as raw material monomers is preferable.
- the optical purity of polylactic acid is preferably 95% or more, and more preferably 98% or more, from the viewpoints of heat resistance and transparency of the biodegradable resin composition.
- the optical purity of the polylactic acid resin is the D-form described in “Voluntary Standards for Food Containers and Packaging Made of Synthetic Resins such as Polyolefins Third Edition Revised Edition 6 June 2004 Supplement Part 3 Hygiene Test Law P12-13” It can obtain
- the polylactic acid is composed of two types of polylactic acid obtained using lactic acid components mainly composed of different isomers from the viewpoint of heat resistance and transparency of the biodegradable resin composition.
- Stereocomplex polylactic acid may be used.
- polylactic acid (A) contains 90 to 100 mol% of L isomer and 0 to 10 mol% of other components including D isomer.
- the other polylactic acid (hereinafter referred to as polylactic acid (B)) contains 90 to 100 mol% of D isomer and 0 to 10 mol% of other components including L isomer.
- dicarboxylic acid, polyhydric alcohol, hydroxycarboxylic acid, lactone, etc. having a functional group capable of forming two or more ester bonds are exemplified, and unreacted Polyester, polyether, polycarbonate or the like having two or more of the functional groups in the molecule may be used.
- the weight ratio of polylactic acid (A) to polylactic acid (B) [polylactic acid (A) / polylactic acid (B)] is preferably 10/90 to 90/10, and 20/80 to 80 / 20 is more preferable, and 40/60 to 60/40 is more preferable.
- the melting point (Tm) (° C.) of polylactic acid is preferably 140 to 250 ° C. from the viewpoint of dispersibility of plasticizers and crystal nucleating agents, and from the viewpoint of bending strength, deterioration and productivity of the biodegradable resin composition. More preferably, the temperature is 150 to 240 ° C, still more preferably 160 to 230 ° C.
- fusing point of resin is measured by the method as described in the below-mentioned Example.
- the content of polylactic acid in the polylactic acid resin is preferably 80% by weight or more, more preferably 90% by weight or more, and further preferably substantially 100% by weight.
- the content of the polylactic acid resin is not particularly limited, but is preferably 50% by weight or more, more preferably 60% by weight or more, and further preferably 70% by weight or more in the biodegradable resin composition.
- Polylactic acid can be synthesized by the above method, but commercially available products include, for example, “Lacia series” such as Lacia H-100, H-280, H-400, and H-440 (Mitsui Chemicals, Inc.). Manufactured), 3001D, 3051D, 4032D, 4042D, 6201D, 6251D, 7000D, 7032D, etc. “Nature Works” (manufactured by Nature Works), eco-plastic U'z S-09, S-12, S-17, etc. Eco plastic U'z series "(manufactured by Toyota Motor Corporation).
- Laissia H-100, H-280, H-400, H-440 manufactured by Mitsui Chemicals
- 3001D, 3051D, 4032D, 4042D, 6201D 6251D, 7000D, 7032D manufactured by Nature Works
- Ecoplastic U'z S-09, S-12, S-17 manufactured by Toyota Motor Corporation
- an ester compound having two or more ester groups in the molecule in addition to the ethylenebis12-hydroxystearic acid amide and the biodegradable resin, an ester compound having two or more ester groups in the molecule, it is preferable that at least one of the alcohol components constituting the ester compound contains a plasticizer which is an ester compound which is an alcohol obtained by adding an average of 0.5 to 5 moles of alkylene oxide having 2 to 3 carbon atoms per hydroxyl group. .
- the plasticizer in the present invention is an ester compound having two or more ester groups in the molecule, wherein at least one of the alcohol components constituting the ester compound is an alkylene oxide having 2 to 3 carbon atoms per hydroxyl group
- An ester compound which is an alcohol with an average of 0.5 to 5 moles added include plasticizers described in JP-A-2008-174718 and JP-A-2008-115372. Among them, an ester compound having two or more ester groups in the molecule, and an alcohol component constituting the ester compound is added with an average of 0.5 to 5 moles of alkylene oxide having 2 to 3 carbon atoms per hydroxyl group.
- a compound which is an alcohol is preferably a polyhydric alcohol ester having two or more ester groups in the molecule, a polycarboxylic acid ether ester, or a polyester of a dicarboxylic acid and a diol, and the alcohol component constituting the ester is a hydroxyl group
- a compound which is an alcohol obtained by adding an average of 0.5 to 5 moles of ethylene oxide per one is more preferable.
- the plasticizer having the structure is preferably a compound having two or more ester groups in the molecule and an average added mole number of ethylene oxide of 3 to 9 from the viewpoint of moldability, plasticity, and bleed resistance. More preferred is at least one selected from the group consisting of an ester of succinic acid or adipic acid and polyethylene glycol monomethyl ether, and an ester of acetic acid and glycerin or an ethylene oxide adduct of ethylene glycol. Succinic acid or adipic acid and polyethylene glycol are more preferred. More preferred are esters with monomethyl ether.
- an ester of acetic acid and an glycerin ethylene oxide average adduct of 3 to 9 mol, an average addition mol number of succinic acid and ethylene oxide Is an ester of polyethylene glycol monomethyl ether with a polyethylene glycol monomethyl ether of 2-4, an ester of polyvalent carboxylic acid such as an ester of polyethylene glycol monomethyl ether with an average addition mole number of adipic acid and ethylene oxide of 2-3, and polyethylene glycol monomethyl ether preferable.
- an ester of succinic acid and ethylene oxide with polyethylene glycol monomethyl ether having an average addition mole number of 2-3, adipic acid and diethylene glycol monomethyl More preferred are esters with ethers.
- an ester of succinic acid and triethylene glycol monomethyl ether is more preferable.
- the plasticizer used in the present invention contains an ester compound composed of an aromatic alcohol in which the average of more than 0 to 1.5 or less of the two or more ester groups.
- a diester of adipic acid and diethylene glycol monomethyl ether / benzyl alcohol 1/1 mixture is preferred.
- the average molecular weight of the plasticizer is preferably from 250 to 700, more preferably from 300 to 600, still more preferably from 350 to 550, and even more preferably from 400 to 500, from the viewpoint of bleed resistance and volatility resistance. is there.
- the ester is preferably a saturated ester that is all esterified from the viewpoint of sufficiently exerting the function as a plasticizer.
- the content of the plasticizer in the biodegradable resin composition in the present invention is preferably 5 to 30 parts by weight, preferably 7 to 30 parts by weight with respect to 100 parts by weight of the biodegradable resin, from the viewpoint of obtaining a sufficient crystallization rate. Part is more preferable, and 10 to 30 parts by weight is still more preferable.
- the raw material in the present invention may contain additives such as fillers, hydrolysis inhibitors and flame retardants.
- the filler is preferably blended from the viewpoint that a biodegradable resin composition excellent in mechanical properties, moldability, heat resistance, and the like can be obtained, and is usually a fibrous material used as a filler for a thermoplastic resin composition. Plates, granules, and powders can be used. Specifically, silicates such as talc, smectite, kaolin, mica, montmorillonite, inorganic compounds such as silica, magnesium oxide, titanium oxide, calcium carbonate, glass fiber, carbon fiber, graphite fiber, wollastonite, potassium titanate Examples thereof include fibrous inorganic fillers such as whiskers and silicon-based whiskers, and organic fillers such as nylon fibers and acrylic fibers.
- silicates such as talc, smectite, kaolin, mica, montmorillonite
- inorganic compounds such as silica, magnesium oxide, titanium oxide, calcium carbonate, glass fiber, carbon fiber, graphite fiber, wollastonite, potassium titanate
- inorganic filler fibers, plates, granules, and powders that are usually used for reinforcing thermoplastic resins can be used. Specifically, glass fiber, asbestos fiber, carbon fiber, graphite fiber, metal fiber, potassium titanate whisker, aluminum borate whisker, magnesium whisker, silicon whisker, wollastonite, sepiolite, asbestos, slag fiber, gypsum fiber , Silica fiber, silica / alumina fiber, zirconia fiber, boron nitride fiber, silicon nitride fiber and boron fiber and other inorganic fillers, glass flakes, graphite, metal foil, ceramic beads, talc, clay, mica, sericite, Zeolite, bentonite, dolomite, kaolin, finely divided silicic acid, feldspar powder, potassium titanate, calcium carbonate, magnesium carbonate, barium sulfate, calcium oxide, aluminum oxide, titanium oxide, aluminum silicate, silicon
- the aspect ratio of the fibrous filler is preferably 5 or more, more preferably 10 or more, and further preferably 20 or more.
- the inorganic filler may be coated or focused with a thermoplastic resin such as an ethylene / vinyl acetate copolymer, or a thermosetting resin such as an epoxy resin, and may be a coupling agent such as aminosilane or epoxysilane. It may be processed.
- a thermoplastic resin such as an ethylene / vinyl acetate copolymer, or a thermosetting resin such as an epoxy resin, and may be a coupling agent such as aminosilane or epoxysilane. It may be processed.
- Organic filler a chip-like, fiber-like, plate-like or powder-like one usually used for reinforcing thermoplastic resins can be used. Specific examples include rice husks, wood chips, okara, waste paper pulverized materials, chip-shaped materials such as clothing pulverized materials, cotton fibers, hemp fibers, bamboo fibers, wood fibers, kenaf fibers, jute fibers, banana fibers, coconut fibers.
- Plant fibers such as, or pulps and cellulose fibers processed from these plant fibers and fibrous materials such as animal fibers such as silk, wool, Angola, cashmere, camel, pulp powder, paper powder, wood powder, bamboo powder, Cellulose powder, rice husk powder, fruit husk powder, chitin powder, chitosan powder, protein, starch, etc. are mentioned, and from the viewpoint of moldability, paper powder, wood powder, bamboo powder, cellulose powder, kenaf powder, Powdered powder such as rice husk powder, fruit shell powder, chitin powder, chitosan powder, protein powder and starch is preferable, and paper powder, wood powder, bamboo powder, cellulose powder and kenaf powder are preferred. Ri preferred.
- an organic filler in the form of amorphous powdered cellulose using a vibrating rod mill, a bead mill or the like.
- the amorphous cellulose described in International Publication No. 2010/010961 pamphlet is used.
- the content of the filler is preferably 1 to 300 parts by weight and more preferably 5 to 150 parts by weight with respect to 100 parts by weight of the biodegradable resin from the viewpoint of obtaining sufficient heat resistance and impact resistance.
- hydrolysis inhibitor examples include carbodiimide compounds such as polycarbodiimide compounds and monocarbodiimide compounds, monocarbodiimide compounds are preferred from the viewpoint of moldability of the biodegradable resin composition, and the heat resistance of the biodegradable resin composition, From the viewpoint of impact resistance and bleed resistance of the crystal nucleating agent, a polycarbodiimide compound is preferred.
- polycarbodiimide compound examples include poly (4,4′-diphenylmethanecarbodiimide), poly (4,4′-dicyclohexylmethanecarbodiimide), poly (1,3,5-triisopropylbenzene) polycarbodiimide, poly (1,3,3). 5-triisopropylbenzene and 1,5-diisopropylbenzene) polycarbodiimide and the like, and examples of the monocarbodiimide compound include N, N′-di-2,6-diisopropylphenylcarbodiimide and the like.
- the carbodiimide compound may be used alone or in combination of two or more in order to satisfy the durability, impact resistance and moldability of the biodegradable resin composition.
- Poly (4,4′-dicyclohexylmethanecarbodiimide) is obtained by converting carbodilite LA-1 (manufactured by Nisshinbo Chemical Co., Ltd.), poly (1,3,5-triisopropylbenzene) polycarbodiimide and poly (1,3,5-trimethyl).
- the content of the hydrolysis inhibitor is preferably 0.05 to 15 parts by weight, more preferably 0.10 to 10 parts by weight, more preferably 0.20 with respect to 100 parts by weight of the biodegradable resin, from the viewpoint of inhibiting hydrolysis of the biodegradable resin. More preferred is 10 parts by weight.
- flame retardants include halogenated compounds containing bromine or chlorine such as tetrabromobisphenol-A-epoxy oligomer, tetrabromobisphenol-A-carbonate oligomer, brominated epoxy resin, antimony trioxide, zinc borate, etc.
- Inorganic flame retardants silicone flame retardants such as silicone resins and silicone oils, inorganic hydrates such as aluminum hydroxide and magnesium hydroxide (in terms of physical properties, surface treatment with silane coupling agent, especially isocyanate silane And phosphoric flame retardants such as triaryl isopropylates, condensed phosphates, melamine polyphosphates, piperazine polyphosphates and phosphazene compounds, and nitrogen-containing flame retardants such as melamine cyanurate. .
- the flame retardant is preferably a phosphorus-based flame retardant from the viewpoint of improving the flame retardancy of the biodegradable resin composition, and at least one selected from the group consisting of condensed phosphates, phosphates and condensed phosphates. Is preferred.
- an inorganic hydrate or a phosphorus flame retardant is preferable from the viewpoint of safety, and a combination of an inorganic hydrate and a phosphorus compound is preferable from the viewpoint of physical properties.
- the content of the flame retardant is preferably 10 to 60 parts by weight and more preferably 15 to 55 parts by weight with respect to 100 parts by weight of the biodegradable resin.
- the biodegradable resin composition of the present invention can contain high-strength organic synthetic fibers from the viewpoint of improving physical properties such as strength, heat resistance and impact resistance.
- the high-strength organic synthetic fibers include aramid fibers, polyarylate fibers, PBO fibers, and the like, and aramid fibers are preferable from the viewpoint of heat resistance.
- the content of the high-strength organic synthetic fiber is preferably 3 to 20 parts by weight and more preferably 5 to 10 parts by weight with respect to 100 parts by weight of the biodegradable resin.
- the biodegradable resin composition of the present invention may contain other resins from the viewpoint of improving physical properties such as rigidity, flexibility, heat resistance and durability.
- resins include polyethylene, polypropylene, polystyrene, ABS resin, AS resin, acrylic resin, polyamide, polyphenylene sulfide, polyether ether ketone, polyester, polyacetal, polysulfone, polyphenylene oxide, polyimide, polyetherimide,
- thermoplastic resins such as ethylene / glycidyl methacrylate copolymer, polyester elastomer, polyamide elastomer, soft thermoplastic resin such as ethylene / propylene terpolymer, ethylene / butene-1 copolymer, phenol resin, melamine resin, unsaturated Thermosetting resins such as polyester resins, silicone resins, and epoxy resins can be mentioned.
- amide bonds and S are preferred from the viewpoint of compatibility with biodegradable resins.
- the resin having a bond containing a carbonyl group of a carbonate bond or the like, structurally biodegradable resin is preferable because inter alia polylactic acid resin and affinity tends to be high.
- the biodegradable resin composition of the present invention may contain a core-shell type rubber from the viewpoint of improving physical properties such as impact resistance and toughness.
- a core-shell type rubber from the viewpoint of improving physical properties such as impact resistance and toughness.
- Specific examples include (core; silicone / acrylic polymer, shell; methyl methacrylate polymer), (core; silicone / acrylic polymer, shell; methyl methacrylate / glycidyl methacrylate polymer), (core; butanediene / Styrene polymer, shell; methyl methacrylate polymer), (core; acrylic polymer, shell; methyl methacrylate polymer) and the like.
- the content of the core-shell type rubber is preferably 2 to 30 parts by weight and more preferably 3 to 20 parts by weight with respect to 100 parts by weight of the biodegradable resin.
- the biodegradable resin composition of the present invention may be a hindered phenol or phosphite antioxidant, an aliphatic amide, a fatty acid metal salt, a hydrocarbon wax, or an anionic surfactant.
- Certain lubricants and the like can be contained.
- the content of each of the antioxidant and the lubricant is preferably 0.05 to 3 parts by weight and more preferably 0.10 to 2 parts by weight with respect to 100 parts by weight of the biodegradable resin.
- additives such as an antibacterial agent and a foaming agent, as a composition raw material in the range which does not prevent achievement of the objective of this invention.
- the melt kneading of the raw material is not particularly limited, and can be performed using a known kneader such as a closed kneader, a single or twin screw extruder, and an open roll kneader.
- the raw materials are preferably mixed in advance using a Henschel mixer, a super mixer, etc., and then subjected to melt kneading.
- the melt kneading temperature is not less than the melting point (Tm) of the biodegradable resin from the viewpoint of dispersibility of plasticizers, crystal nucleating agents, etc., preferably in the range of Tm to Tm + 100 ° C., more preferably Tm to Tm + 50 ° C. Range.
- the temperature is preferably 170 to 240 ° C, and more preferably 170 to 220 ° C.
- the melt-kneading time cannot be generally determined depending on the melt-kneading temperature and the type of the kneader, but is preferably 15 to 900 seconds.
- the present invention includes a step of cooling the melt-kneaded product after the melt-kneading step (hereinafter also referred to as a cooling step) from the viewpoint of further improving the crystallization speed of the melt-kneaded product obtained by the melt-kneading step.
- the cooling temperature is preferably 60 ° C. or more, more preferably 70 ° C. or more lower than the melt-kneading temperature, specifically 20 to 120 ° C., more preferably 20 to 100 ° C.
- the cooling time is preferably 2 to 90 seconds, more preferably 5 to 60 seconds.
- the melt-kneaded product may be cooled according to a known method.
- a step of holding at 50 to 120 ° C., more preferably 60 to 100 ° C., preferably 30 to 180 seconds, more preferably 30 to 120 seconds, further preferably 30 to 60 seconds (hereinafter referred to as “after cooling”). , Also referred to as a holding step).
- the temperature in the holding step may be the same as or different from the temperature in the cooling step.
- a biodegradable resin composition that is a raw material melt-kneaded material containing ethylene bis 12-hydroxystearic acid amide as a crystal nucleating agent and a biodegradable resin, with accelerated crystallization can be obtained. Therefore, the present invention provides a biodegradable resin composition whose crystallization is accelerated by the crystallization promoting method of the present invention.
- crystallization is promoted means a state where “crystallization speed is improved” and “crystallization is easy to proceed”.
- Specific crystallization methods include a melt crystallization method in which a biodegradable resin composition is crystallized by cooling from a molten state to a mold temperature as in injection molding and holding in the mold, and a glass transition.
- Examples thereof include a cold crystallization method in which a biodegradable resin composition in an amorphous state at a temperature or lower is crystallized by heating. Since the biodegradable resin composition of the present invention has an excellent crystallization rate, when the biodegradable resin composition contains a plasticizer, the half crystallization time at 100 ° C. in the melt crystallization method is 30 seconds. Or less, more preferably 27 seconds or less, and even more preferably 24 seconds or less.
- the half crystallization time (melt crystallization) at 100 ° C. can be determined by the method shown in the examples.
- the biodegradable resin composition of the present invention has an excellent crystallization rate, when the biodegradable resin composition contains a plasticizer, at 80 ° C. in the cold crystallization method (vacuum molding method).
- the half-crystallization time is preferably 24 seconds or less, more preferably 21 seconds or less, and even more preferably 18 seconds or less.
- the half crystallization time (cold crystallization) at 80 ° C. can be determined by the method shown in Examples.
- the biodegradable resin composition of the present invention has a good hue, a high crystallization rate, and excellent moldability, so that it can be processed at a low temperature of, for example, 200 ° C. or less, and can be molded into a film or sheet, Can be used for applications.
- the present invention also provides a method for producing the biodegradable resin composition of the present invention.
- the production method of the present invention is a production method including a step of melt-kneading a raw material containing ethylenebis12-hydroxystearic acid amide having an amine value of 1.0 mgKOH / g or less and a biodegradable resin, and the production conditions, etc. Is as described above.
- the present invention provides a biodegradable resin molded product obtained by molding the biodegradable resin composition of the present invention.
- the biodegradable resin molded article of the present invention may be any molded article of the biodegradable resin composition of the present invention, and specifically, obtained by the method for promoting crystallization of the biodegradable resin composition of the present invention.
- the resulting melt-kneaded product can be prepared by filling a mold using an injection molding machine or the like and molding it.
- the mold temperature is preferably 20 to 120 ° C., more preferably 20 to 100 ° C., and further preferably 20 to 80 ° C. from the viewpoint of improving the crystallization speed and improving workability.
- the holding time in the mold is preferably within 90 seconds, more preferably within 60 seconds, and further preferably within 30 seconds from the viewpoint of improving productivity.
- the biodegradable resin molded product of the present invention may be a product obtained by molding the biodegradable resin composition of the present invention into a sheet shape.
- a method for forming the sheet methods described in JP-A-2007-152760, JP-A-2007-130893, and JP-A-2007-130895 can be used.
- the biodegradable resin molded product of the present invention is prepared by molding the biodegradable resin composition of the present invention with accelerated crystallization, but uses a crystal nucleating agent with a reduced amine value.
- generation of the resin degradation material in is suppressed, and the mold release property from the metal mold
- the present invention will be specifically described with reference to examples, but the present invention is not limited to the following examples.
- the part in an example is a weight part.
- Weight average molecular weight of polylactic acid resin (Mw) The weight average molecular weight (Mw) is measured by GPC (gel permeation chromatography) under the following measurement conditions. ⁇ Measurement conditions> Column: GMHHR-H + GMHHR-H Column temperature: 40 ° C Detector: RI Eluent: Chloroform Flow rate: 1.0 mL / min Sample concentration: 1 mg / mL Injection volume: 0.1 mL Conversion standard: Polystyrene
- optical purity of polylactic acid The optical purity was determined according to the D-body content measurement method described in “Voluntary Standard for Food Containers and Packaging Made of Synthetic Resins such as Polyolefins, Third Edition, Revised June 2004, Part 3 Sanitation Test Method P12-13”. Measure under the measurement conditions. Specifically, sodium hydroxide / methanol is added to precisely weighed polylactic acid, set in a water bath shaker set at 65 ° C., and hydrolyzed until the resin content becomes a homogeneous solution.
- Dilute hydrochloric acid is added to the completed alkaline solution to neutralize it, and the decomposition solution is made up to volume with pure water, and then a fixed volume is separated into a volumetric flask and diluted with a high-performance liquid chromatography (HPLC) mobile phase solution to obtain a pH. Is adjusted to be in the range of 3-7, filtered to a volumetric flask, filtered through a membrane filter (0.45 ⁇ m), and the prepared solution is analyzed by quantifying D-lactic acid and L-lactic acid by HPLC. Obtain the optical purity of lactic acid.
- HPLC high-performance liquid chromatography
- the melting point of the polylactic acid resin is determined from the crystal melting endothermic peak temperature by the temperature rising method of differential scanning calorimetry (DSC, manufactured by Perkin Elmer, Diamond DSC) based on JIS-K7121. The melting point is measured by raising the temperature from 20 ° C. to 250 ° C. at a heating rate of 10 ° C./min.
- the obtained diester had an acid value of 0.2 (mgKOH / g), a saponification value of 276 (mgKOH / g), a hydroxyl value of 1 or less (mgKOH / g), and a hue of APHA200.
- ⁇ Plasticizer Production Example 2> (Triester compound with an ethylene oxide adduct obtained by adding 6 mol of ethylene oxide to acetic acid and glycerin) An autoclave is charged with a specified amount of 6 moles of ethylene oxide to 1 mole of concentrated glycerin for cosmetics manufactured by Kao Co., Ltd., and a constant pressure of 0.3 MPa is added using 1 mole% of KOH as a catalyst until the pressure becomes constant. After reacting at 0 ° C., the mixture was cooled to 80 ° C. to obtain a catalyst-unneutralized product.
- Kyoward 600S manufactured by Kyowa Chemical Industry Co., Ltd.
- an adsorption treatment was performed at 80 ° C. for 1 hour in the presence of nitrogen. Further, the liquid after the treatment is The adsorbent was filtered with a Nutse pre-coated with Radiolite # 900 on the filter paper No. 2 to obtain an adduct with 6 mol of glycerin ethylene oxide (hereinafter referred to as POE (6) glycerin). This was charged into a four-necked flask, heated to 105 ° C.
- POE (6) glycerin 6 mol of glycerin ethylene oxide
- the obtained slurry was used with a pressure filter equipped with a membrane filter (material: PTFE, manufactured by Advantech) with a pore size of 1 micron. Then, filtration was performed at a pressure of 0.2 MPa. The obtained residue was subjected to cake washing twice using 39 g of 1-butanol (25 ° C.) (100 parts by weight with respect to 100 parts by weight of residue) at a time, and then reduced in pressure (133 Pa) at 60 ° C. Drying was carried out for 12 hours to obtain purified ethylenebis12-hydroxystearic acid amide (refined product (1)). The quality of the unpurified product and the purified product (1) was evaluated according to the following method. The results are shown in Table 1.
- ⁇ Purification example 3 of crystal nucleating agent> Purification of ethylenebis12-hydroxystearic acid amide; combined use of crystallization method and thermal washing method
- a 1-liter cylindrical separable flask equipped with an anchor-type stirrer and thermometer was charged with 50 g of purified ethylenebis12-hydroxystearic acid amide (refined product (1)) obtained in Purification Example 1 and 500 g of 1-butanol. And stirred at 90 ° C. for 30 minutes. Thereafter, it was cooled to 25 ° C.
- the obtained cake was dried at 70 ° C. for 2 hours under reduced pressure (1 kPa) to obtain purified ethylenebis12-hydroxystearic acid amide (refined product (3)).
- the quality of the purified product (3) was evaluated according to the following method. The results are shown in Table 1.
- Crystal Nucleating Agents A to G Using purified products (1) to (3) of ethylenebis 12-hydroxystearic acid amide shown in Table 1 and unrefined products, or 12-hydroxystearic acid (produced by Ito Oil Co., Ltd., acid value 181 mgKOH / g), After blending at the ratio shown in Table 2, the mixture was mixed in a mortar for 10 minutes to obtain crystal nucleating agents A to G, respectively. Quality was evaluated in the same manner as the purified product (1). The results are shown in Table 2.
- Examples 1 to 9 and Comparative Examples 1 to 6 As the polylactic acid resin composition, the composition raw materials shown in Tables 3 and 4 were kneaded for 10 minutes with a 180 ° C kneader (Toyo Seiki Co., Ltd., Labo Plast Mill), and then thickened with a 190 ° C press molding machine. The sheet was molded into a sheet having a thickness of 0.3 mm, cooled to 25 ° C., held as it was for 60 seconds, and further held at 90 ° C. for 1 minute to obtain a sheet (molded body).
- a 180 ° C kneader Toyo Seiki Co., Ltd., Labo Plast Mill
- the yellowness (b value) of the sheet is measured using a color difference meter Spectro Color Meter SE 2000 manufactured by Nippon Denshoku Industries Co., Ltd. The larger the value is on the + side, the stronger the yellow color.
- ⁇ Semi-crystallization time> By cutting out the sheet, 7.5 mg of the test piece is precisely weighed, sealed in an aluminum pan, melted at 200 ° C. for 5 minutes using a DSC device (Perkin Elmer, Diamond DSC), and at a rate of ⁇ 500 ° C./min. The temperature is lowered to each holding temperature (90 ° C., 100 ° C., 110 ° C.) to obtain half time for crystal saturation (time for half crystallinity of saturated crystal; half crystallization time; t 1/2 ) . t 1/2 is calculated by setting the time when the sample temperature reaches the holding temperature as 0 minutes.
- the sheet of the example has lower transparency and yellowishness and a shorter half-crystallization time than the sheet of the comparative example.
- melt crystallization that is cooled from the high temperature side is used, but the molded body has a heat capacity, so even if there is a relatively small difference in half-crystallization time, the molding speed during actual molding It appears as a big difference. Therefore, the resin composition whose crystallization is promoted by the crystallization promoting method of the present invention is preferable in terms of productivity because the half crystallization time is short and the time required for molding is short.
- Examples 10 to 11 and Comparative Example 7 evaluation by vacuum forming
- the composition raw materials shown in Table 5 were kneaded for 10 minutes in a 180 ° C. kneader (Toyo Seiki Co., Ltd., Labo Plast Mill), The sheet was molded into a sheet of 150 mm length ⁇ 150 mm width ⁇ 0.4 mm thickness with a 190 ° C. press molding machine, cooled to 25 ° C. and held for 60 seconds to obtain a sheet (molded body).
- the obtained sheet was subjected to vacuum forming using a vacuum / pressure forming machine (FVS-500 type manufactured by Wakisaka Engineering Co., Ltd.) to obtain a formed body (see FIG. 1).
- Preheating was performed by holding the upper and lower heaters in a heater box set at 400 ° C. for 7 seconds, and instantaneously transferred to the molding zone to perform vacuum forming at a mold temperature of 90 ° C.
- the half crystallization time tcc 1/2 that is an index of the crystallization speed was measured by the following measurement method close to the crystallization method (cold crystallization) at the time of vacuum forming. Further, when molding the molded body, the molding time required for releasing the molded body was evaluated according to the following criteria. Furthermore, about the transparency of a molded object, the center flat part was cut out and it measured with the following method. These results are shown in Table 5.
- ⁇ Semi-crystallization time (cold crystallization)> By cutting out the sheet, 7.5 mg of the test piece is precisely weighed, sealed in an aluminum pan, melted at 200 ° C. for 5 minutes using a DSC device (Perkin Elmer, Diamond DSC), and at a rate of ⁇ 500 ° C./min. After cooling rapidly to 25 ° C and holding for 1 minute, the temperature is raised to each holding temperature (70 ° C, 80 ° C, 90 ° C) at a rate of 500 ° C / minute, and half the time for crystal saturation (half the saturation crystal) Time to reach crystallinity; semi-crystallization time (cold crystallization); tcc 1/2 ) was determined. tcc 1/2 was calculated by setting the time when the sample temperature reached the holding temperature as 0 minute.
- the mold holding time required for mold release is the shortest time during which the mold can be taken out without deformation.
- the mold holding time was measured by setting the time when the sheet obtained after press molding was attached to the mold as the starting point of the mold holding time, and the time for removing the mold from the sheet as the end point of the mold holding time. The shorter the mold holding time, the faster the cold crystallization rate of the resin composition and the better the moldability.
- the semi-crystallization time is shorter for the sheet of the example than for the sheet of the comparative example even in cold crystallization.
- cold crystallization is used in which heating is performed from the room temperature side.
- the resin composition whose crystallization has been promoted by the crystallization promotion method of the present invention is cooled.
- the half crystallization time is short and the time required for thermoforming is shortened, which is preferable in terms of productivity. Further, the transparency of the molded body can be improved.
- the biodegradable resin composition obtained by the crystallization promoting method of the present invention can be suitably used for various industrial uses such as daily goods, home appliance parts, automobile parts and the like.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Biological Depolymerization Polymers (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
Abstract
アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドと生分解性樹脂とを含有する原料を溶融混練する工程を含む、生分解性樹脂組成物の結晶化促進方法。本発明の結晶化促進方法により得られる生分解性樹脂組成物は、良好な結晶化速度を有するため、該組成物を成形する際に要する時間が短くなり、生産性良く成形体を製造することができ、例えば、日用雑貨品、家電部品、自動車部品等の様々な工業用途に好適に使用することができる。
Description
本発明は、生分解性樹脂組成物の結晶化促進方法に関する。さらに詳しくは、特定の結晶核剤を用いた生分解性樹脂組成物の結晶化促進方法、該方法により結晶化が促進された生分解性樹脂組成物、及び該生分解性樹脂組成物の成形体に関する。
生分解性樹脂は、土壌、海水中、あるいは動物の体内などに置かれた場合、自然界に生息する微生物の産出する酵素の働きによって、数週間で分解が始まり、約1年から数年の間に消滅する。従って、近年、その利用が注目されている。
例えば、特許文献1では、結晶化速度と透明性が良好な生分解性樹脂組成物を得る方法として、生分解性樹脂、可塑剤、及び、分子中にエステル基、水酸基及びアミド基からなる群より選ばれる少なくとも1種の基を2つ以上有する脂肪族化合物である結晶核剤を、特定の温度で溶融混練して熱処理を行う製造方法が開示されている。前記結晶核剤としては、脂肪族エステル、ヒドロキシ脂肪酸エステルなどの脂肪酸エステル;ヒドロキシ脂肪酸モノアミド、脂肪族ビスアミド、ヒドロキシ脂肪酸ビスアミドなどの脂肪族アミド;ヒドロキシ脂肪酸金属塩などの脂肪酸金属塩が例示されている。
本発明は、
〔1〕 アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドと生分解性樹脂とを含有する原料を溶融混練する工程を含む、生分解性樹脂組成物の結晶化促進方法、
〔2〕 前記〔1〕記載の結晶化促進方法により結晶化が促進されてなる、生分解性樹脂組成物、ならびに
〔3〕 前記〔2〕記載の生分解性樹脂組成物を成形してなる、生分解性樹脂成形体
に関する。
〔1〕 アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドと生分解性樹脂とを含有する原料を溶融混練する工程を含む、生分解性樹脂組成物の結晶化促進方法、
〔2〕 前記〔1〕記載の結晶化促進方法により結晶化が促進されてなる、生分解性樹脂組成物、ならびに
〔3〕 前記〔2〕記載の生分解性樹脂組成物を成形してなる、生分解性樹脂成形体
に関する。
1 透明性の評価に用いるサンプリング面
近年の市場のさらなる要求により、結晶化速度のさらなる高速化が求められており、従来の技術では十分ではないことが判明した。
本発明は、生分解性樹脂組成物の結晶化を促進させる方法、該方法により結晶化が促進された生分解性樹脂組成物、及び該生分解性樹脂組成物の成形体に関する。
本発明の生分解性樹脂組成物の結晶化促進方法により結晶化が促進された生分解性樹脂組成物は、良好な結晶化速度を有するため、該組成物を成形する際に要する時間が短くなり、生産性良く成形体を製造することができる。
本発明の生分解性樹脂組成物の結晶化促進方法は、結晶核剤と生分解性樹脂とを含有する原料を溶融混練する工程を含むものであって、結晶核剤が特定のアミン価を有するエチレンビス12-ヒドロキシステアリン酸アミドであることに大きな特徴を有する。
エチレンビス12-ヒドロキシステアリン酸アミドは、熱可塑性樹脂の滑剤、ブロッキング防止剤として用いられている。また、分散性に寄与するヒドロキシ基と、相溶性に寄与するアミド基をそれぞれ2個有する化合物であり、ポリ乳酸樹脂などの結晶核剤としても使用されている。この化合物は、12-ヒドロキシステアリン酸とエチレンジアミンを脱水縮合反応させることにより得られるが、得られる反応物には、未反応のエチレンジアミン、反応中間体のモノアミドアミン、副生物のイミダゾリンなどのアミンが含まれる。これらのアミンは、エチレンビス12-ヒドロキシステアリン酸アミドの保存時や加熱時の着色の原因となるほか、人体への安全性も懸念されるため好ましくないが、詳細な理由は不明なるも、該アミンを含有するエチレンビス12-ヒドロキシステアリン酸アミドを用いて得られる生分解性樹脂組成物は、色相の悪化に加えて、結晶化速度の低下も引き起こすことが判明した。そこで、本発明では、アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドを結晶核剤として用いることにより、生分解性樹脂組成物の結晶化を促進することが可能となる。なお、本発明において、アミン価が1.0mgKOH/g以下のエチレンビス12-ヒドロキシステアリン酸アミドとは、エチレンビス12-ヒドロキシステアリン酸アミドからなるアミン価が1.0mgKOH/g以下の結晶核剤を意味するものである。
本発明の生分解性樹脂組成物の結晶化促進方法は、結晶核剤である、アミン価が1.0mgKOH/g以下のエチレンビス12-ヒドロキシステアリン酸アミドと、生分解性樹脂とを含有する原料を溶融混練する工程(以降、溶融混練工程ともいう)を含む。
<結晶核剤>
本発明で用いるエチレンビス12-ヒドロキシステアリン酸アミドは、アミン価が1.0mgKOH/g以下、好ましくは0.5mgKOH/g以下である。また、生産性の観点から、好ましくは0.01mgKOH/g以上である。従って、本発明で用いるエチレンビス12-ヒドロキシステアリン酸アミドのアミン価は、0.01~1.0mgKOH/gが好ましく、0.01~0.5mgKOH/gがより好ましい。本明細書において、アミン価とはアミンの総量のことを意味し、全アミン価ともいう。アミン価は、後述の実施例に記載の方法に従って、測定することができる。
本発明で用いるエチレンビス12-ヒドロキシステアリン酸アミドは、アミン価が1.0mgKOH/g以下、好ましくは0.5mgKOH/g以下である。また、生産性の観点から、好ましくは0.01mgKOH/g以上である。従って、本発明で用いるエチレンビス12-ヒドロキシステアリン酸アミドのアミン価は、0.01~1.0mgKOH/gが好ましく、0.01~0.5mgKOH/gがより好ましい。本明細書において、アミン価とはアミンの総量のことを意味し、全アミン価ともいう。アミン価は、後述の実施例に記載の方法に従って、測定することができる。
エチレンビス12-ヒドロキシステアリン酸アミドは、アミン価が1.0mgKOH/g以下であれば、市販品であっても、当該分野で公知の方法(例えば、特開昭63-60956号公報に記載の方法)により合成されたものであってもよい。
以下に、アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドの合成方法について具体例を挙げる。
エチレンビス12-ヒドロキシステアリン酸アミドは、12-ヒドロキシステアリン酸とエチレンジアミンを原料として用い、該原料を脱水縮合反応させることにより得られるが、得られる反応物における未反応のエチレンジアミンや反応中間体のモノアミドアミン等のアミン含有量を低減してアミン価を1.0mgKOH/g以下とするために、前記脱水縮合反応に供する原料のモル比を調整し、12-ヒドロキシステアリン酸のエチレンジアミンに対するモル比(12-ヒドロキシステアリン酸/エチレンジアミン)は2.0/1以上が好ましい。また、得られる反応物における未反応の12-ヒドロキシステアリン酸含有量を低減して、12-ヒドロキシステアリン酸の縮合物の副生を抑制する観点から、前記モル比は、2.20/1以下が好ましく、2.15/1以下がより好ましい。従って、前記モル比は2.0/1~2.20/1が好ましく、2.0/1~2.15/1がより好ましい。
脱水縮合反応は、窒素などの不活性ガス雰囲気中、常圧下(101.3kPa)にて行われる。反応温度は、180~230℃が好ましく、190~220℃がより好ましい。反応温度が180℃以上であると反応が効率よく進み、230℃以下であると得られる反応物の色相が良好となる。反応時間は原料のモル比、反応温度によって一概には決定できないが、12-ヒドロキシステアリン酸とエチレンジアミンが十分に反応できる時間であることが好ましく、通常3~7時間が好ましい。
かくして、アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドを合成することができる。
また、本発明では、アミン価が1.0mgKOH/gを超えるエチレンビス12-ヒドロキシステアリン酸アミドを、公知の方法に従って精製することにより、アミン価を1.0mgKOH/g以下に低減させたものを用いてもよい。またさらに、前記合成方法により得られたエチレンビス12-ヒドロキシステアリン酸アミドを、さらに精製することにより、アミン価を低減させて用いてもよい。
以下に、エチレンビス12-ヒドロキシステアリン酸アミドの精製方法について具体例を挙げる。
エチレンビス12-ヒドロキシステアリン酸アミドは、アルコール系溶媒、芳香族炭化水素系溶媒、ケトン系溶媒、及びエステル系溶媒からなる群より選ばれる少なくとも1種の溶媒を用いて、熱洗浄及び/又は晶析することにより精製する。なお、2種以上の溶媒を用いる場合は、混合溶媒として用いることができる。
アルコール系溶媒としては、メタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール、2-ブタノール、2-メチル-1-プロパノール、2-メチル-2-プロパノール、1-ペンタノール、2-ペンタノール、3-ペンタノール、2-メチル-1-ブタノール、3-メチル-1-ブタノール、2-メチル-2-ブタノール、3-メチル-2-ブタノール、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル等が挙げられる。熱洗浄による精製では、アミン価の低いエチレンビス12-ヒドロキシステアリン酸アミドを得る観点から、メタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール、2-ブタノール、2-メチル-1-プロパノール、及び2-メチル-2-プロパノールが好ましい。また、晶析による精製では、アミン価の低いエチレンビス12-ヒドロキシステアリン酸アミドを得る観点から、1-プロパノール、2-プロパノール、1-ブタノール、2-ブタノール、2-メチル-1-プロパノール、及び2-メチル-2-プロパノールが好ましい。
芳香族炭化水素系溶媒としては、トルエン、o-キシレン、m-キシレン、p-キシレン、エチルベンゼン等が挙げられ、なかでも、トルエンが好ましい。
ケトン系溶媒としては、アセトン、メチルエチルケトン、メチルイソブチルケトン等が挙げられ、なかでも、メチルエチルケトン、及びメチルイソブチルケトンが好ましい。
エステル系溶媒としては酢酸エチル、酢酸n-プロピル、酢酸n-ブチル、酢酸iso-ブチル、酢酸sec-ブチル等を挙げることができる。
熱洗浄及び晶析はいずれも、常圧又は加圧(0.1~10MPa)下で、精製に供されるエチレンビス12-ヒドロキシステアリン酸アミド(以降、未精製ビス脂肪酸アミドともいう)を、上記の溶媒のうち少なくとも1種の溶媒中に、加熱して懸濁又は溶解させる。その後、該懸濁液又は溶解液を冷却し、減圧(0.001~0.09MPa)又は加圧(0.11~0.5MPa)下でろ過して、ろ過物を回収し、洗浄、乾燥して行う。なお、これら一連の操作を1回として、熱洗浄のみを複数回、晶析のみを複数回、又は熱洗浄と晶析を組み合わせて行ってもよく、その際に用いる溶媒は、操作毎に同一でも異なっていてもよい。
未精製ビス脂肪酸アミドの懸濁又は溶解に用いられる溶媒量は、未精製ビス脂肪酸アミド100重量部に対して、100~10000重量部が好ましく、200~3000重量部がより好ましく、300~1500重量部がさらに好ましい。100重量部以上であると、懸濁又は溶解液の粘度が高くなり過ぎず、ろ過の作業性がよい。10000重量部以下であると、得られるエチレンビス12-ヒドロキシステアリン酸アミドの損失が少なく、経済的である。
懸濁又は溶解に際して行う加熱は、好ましくは50~180℃、より好ましくは50~150℃、さらに好ましくは80~130℃の温度で行うことが望ましい。加熱時間は、0.5~10時間が好ましい。
加熱後の冷却は、好ましくは5~40℃、より好ましくは10~40℃、さらに好ましくは15~40℃の温度で行うことが望ましい。
ろ過方法としては、特に限定はなく、公知の方法に従って行うことができる。なお、ろ液には、未反応原料(エチレンジアミン、12-ヒドロキシステアリン酸)や、反応中間体(モノアミドアミン)が含まれており、得られる残渣のアミン含有量は低減されている。
ろ過残渣には、ろ液の残液が含まれるために洗浄を行う。洗浄方法としては、特に限定はなく、前記懸濁又は溶解に用いた溶媒と同一又は異なる溶媒を用いて、好ましくは10~80℃、より好ましくは10~60℃で行うことができる。洗浄回数は、1~5回が好ましく、1~3回がより好ましい。
1回の洗浄に用いる溶媒量は、残渣100重量部に対して、10~2000重量部が好ましく、100~1000重量部がより好ましく、100~500重量部がさらに好ましい。
乾燥は、常圧下又は、好ましくは15kPa以下、より好ましくは6.7kPa以下の減圧下で行われることが望ましい。乾燥温度は、10~180℃が好ましく、40~150℃がより好ましく、60~120℃がさらに好ましい。
かくして、アミン価が低減された、エチレンビス12-ヒドロキシステアリン酸アミドの精製物(以降、精製ビス脂肪酸アミドともいう)が得られる。なお、本発明においては、未精製ビス脂肪酸アミドと前記精製方法により得られた精製ビス脂肪酸アミドとを公知の方法により混合し、得られた混合物のアミン価が1.0mgKOH/g以下となるのであれば、該混合物を用いてもよい。
アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドの酸価は、1.0mgKOH/g以下が好ましく、0.6mgKOH/g以下がより好ましく、0.4mgKOH/g以下がさらに好ましい。なお、本発明において、酸価が1.0mgKOH/g以下のエチレンビス12-ヒドロキシステアリン酸アミドとは、エチレンビス12-ヒドロキシステアリン酸アミドからなる酸価が1.0mgKOH/g以下の結晶核剤を意味するものである。本明細書において、酸価は、後述の実施例に記載の方法に従って、測定することができる。
なお、特開昭63-60956号公報に記載の方法に従って得られるエチレンビス12-ヒドロキシステアリン酸アミドは、アミン価が1.0mgKOH/g以下であり、高純度なものである。しかし、該方法は、原料のビスアミドへの転化率が85~98%になった時点で反応を停止させて、次いで、薄膜蒸留によってモノアミドアミンを含む未反応成分を除くことによって、高純度な脂肪酸ビスアミドを製造する方法であるため、操作が煩雑であり生産性が低い。また、得られるビスアミドの色相が十分ではないために、本発明では、アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドであっても、前記精製方法によって精製されたエチレンビス12-ヒドロキシステアリン酸アミドを用いることが好ましい。精製ビス脂肪酸アミドの色相は、2ガードナー以下が好ましく、1ガードナー以下がより好ましい。本明細書において、色相は、後述の実施例に記載の方法に従って、測定することができる。
また、本発明においては、生分解性樹脂組成物を溶融結晶化、すなわち射出成形のように溶融状態から金型温度まで冷却し、さらに金型内で保持することで結晶化させる場合は、結晶化を促進させた生分解性樹脂組成物を得る観点から、生分解性樹脂組成物に用いるエチレンビス12-ヒドロキシステアリン酸アミドの凝固点は高い方が好ましい。このような観点から、アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドの凝固点は、135~145℃が好ましく、136~144℃がより好ましく、138~143℃がさらに好ましく、139~143℃がさらにより好ましい。また、前記精製方法によりエチレンビス12-ヒドロキシステアリン酸アミドを精製する場合には、結晶化を促進させた生分解性樹脂組成物を得る観点から、精製品の凝固点が高くなる晶析法が好ましい。なお、本明細書において、凝固点は、DSC装置(パーキンエルマー社製、ダイアモンドDSC)を用いて、200℃で2分間溶融した後、10℃/分の速度で25℃まで降温した際に観察される、結晶化の発熱のピーク温度より求められる。
本発明においては、本発明の効果を損なわない範囲で、前記エチレンビス12-ヒドロキシステアリン酸アミド以外のその他の結晶核剤を含有してもよい。
その他の結晶核剤としては、生分解性樹脂組成物の曲げ強度と成形性の観点から、特開2008-174718号公報及び特開2008-115372号公報に記載の結晶核剤が好ましく、具体的には、アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドを除く分子中に水酸基とアミド基を有する化合物、フェニルホスホン酸金属塩、フタロシアニン、リン酸エステルの金属塩、芳香族スルホン酸ジアルキルエステルの金属塩、ロジン酸類の金属塩、芳香族カルボン酸アミド、ロジン酸アミド、カルボヒドラジド類、N-置換尿素類、メラミン化合物の塩及びウラシル類からなる群より選ばれる少なくとも1種であることが好ましい。なお、本明細書において、結晶核剤の中で、アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドを含む、分子中に水酸基とアミド基を有する化合物を結晶核剤(1)、それ以外を結晶核剤(2)と記載することもある。
アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミド以外の結晶核剤(1)としては、生分解性樹脂組成物の成形性、耐熱性、耐衝撃性及び結晶核剤の耐ブルーム性の観点から、ヘキサメチレンビス12-ヒドロキシステアリン酸アミド等のヒドロキシ脂肪酸ビスアミド、12-ヒドロキシステアリン酸トリグリセライドが好ましく、エチレンビス12-ヒドロキシステアリン酸アミドがより好ましい。
結晶核剤(2)は、結晶化速度の観点から、フェニルホスホン酸金属塩が好ましい。フェニルホスホン酸金属塩は、置換基を有しても良いフェニル基とホスホン基(-PO(OH)2)を有するフェニルホスホン酸の金属塩であり、フェニル基の置換基としては、炭素数1~10のアルキル基、アルコキシ基の炭素数が1~10のアルコキシカルボニル基等が挙げられる。フェニルホスホン酸の具体例としては、無置換のフェニルホスホン酸、メチルフェニルホスホン酸、エチルフェニルホスホン酸、プロピルフェニルホスホン酸、ブチルフェニルホスホン酸、ジメトキシカルボニルフェニルホスホン酸、ジエトキシカルボニルフェニルホスホン酸等が挙げられ、無置換のフェニルホスホン酸が好ましい。
フェニルホスホン酸の金属塩の金属としては、リチウム、ナトリウム、マグネシウム、アルミニウム、カリウム、カルシウム、バリウム、銅、亜鉛、鉄、コバルト、ニッケル等が挙げられ、亜鉛が好ましい。
これらの結晶核剤は、単独で又は2種以上組み合わせて用いることができる。
結晶核剤(1)と結晶核剤(2)を併用する場合の結晶核剤(1)と結晶核剤(2)の重量比〔結晶核剤(1)/結晶核剤(2)〕は、20/80~80/20が好ましく、30/70~70/30がより好ましく、40/60~60/40がさらに好ましい。
結晶核剤における、アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドの含有量は、80重量%以上が好ましく、90重量%以上がより好ましく、実質的に100重量%であることがさらに好ましい。
結晶核剤の含有量は、生分解性樹脂組成物の成形性の観点から、生分解性樹脂100重量部に対して、0.1~5重量部が好ましく、0.5~3重量部がより好ましく、0.5~2重量部がさらに好ましい。なお、本明細書において「含有量」とは、「含有量もしくは配合量」のことを意味する。
<生分解性樹脂>
生分解性樹脂としては、自然界において微生物が関与して低分子化合物に分解される生分解性を有していればよく、例えば、ポリヒドロキシブチレート、ポリカプロラクトン、ポリブチレンサクシネート、ポリブチレンサクシネート/アジペート、ポリエチレンサクシネート、ポリ乳酸樹脂、ポリリンゴ酸、ポリグリコール酸、ポリジオキサノン、ポリ(2-オキセタノン)等の脂肪族ポリエステル;ポリブチレンサクシネート/テレフタレート、ポリブチレンアジペート/テレフタレート、ポリテトラメチレンアジペート/テレフタレート等の脂肪族芳香族コポリエステル;デンプン、セルロース、キチン、キトサン、グルテン、ゼラチン、ゼイン、大豆タンパク、コラーゲン、ケラチン等の天然高分子と上記の脂肪族ポリエステルあるいは脂肪族芳香族コポリエステルとの混合物等が挙げられる。これらのなかでも、加工性、経済性、入手性、及び物性の観点から、ポリ乳酸樹脂が好ましい。なお、本明細書において「生分解性」とは、自然界において微生物によって低分子化合物に分解され得る性質のことであり、具体的には、JIS K6953(ISO14855)「制御された好気的コンポスト条件の好気的かつ究極的な生分解度及び崩壊度試験」に基づいた生分解性のことを意味する。
生分解性樹脂としては、自然界において微生物が関与して低分子化合物に分解される生分解性を有していればよく、例えば、ポリヒドロキシブチレート、ポリカプロラクトン、ポリブチレンサクシネート、ポリブチレンサクシネート/アジペート、ポリエチレンサクシネート、ポリ乳酸樹脂、ポリリンゴ酸、ポリグリコール酸、ポリジオキサノン、ポリ(2-オキセタノン)等の脂肪族ポリエステル;ポリブチレンサクシネート/テレフタレート、ポリブチレンアジペート/テレフタレート、ポリテトラメチレンアジペート/テレフタレート等の脂肪族芳香族コポリエステル;デンプン、セルロース、キチン、キトサン、グルテン、ゼラチン、ゼイン、大豆タンパク、コラーゲン、ケラチン等の天然高分子と上記の脂肪族ポリエステルあるいは脂肪族芳香族コポリエステルとの混合物等が挙げられる。これらのなかでも、加工性、経済性、入手性、及び物性の観点から、ポリ乳酸樹脂が好ましい。なお、本明細書において「生分解性」とは、自然界において微生物によって低分子化合物に分解され得る性質のことであり、具体的には、JIS K6953(ISO14855)「制御された好気的コンポスト条件の好気的かつ究極的な生分解度及び崩壊度試験」に基づいた生分解性のことを意味する。
ポリ乳酸樹脂は、原料モノマーとして乳酸成分のみを縮重合させて得られるポリ乳酸、及び/又は、原料モノマーとして乳酸成分と乳酸以外のヒドロキシカルボン酸成分(以下、単に、ヒドロキシカルボン酸成分ともいう)とを用い、それらを縮重合させて得られるポリ乳酸を含有する。
乳酸には、L-乳酸(L体)、D-乳酸(D体)の光学異性体が存在する。本発明では、乳酸成分として、いずれかの光学異性体のみ、又は双方を含有してもよいが、生分解性樹脂組成物の成形性の観点から、いずれかの光学異性体を主成分とする光学純度が高い乳酸を用いることが好ましい。なお、本明細書において「主成分」とは、乳酸成分中の含有量が50モル%以上である成分のことをいう。
一方、ヒドロキシカルボン酸成分としては、グリコール酸、ヒドロキシ酪酸、ヒドロキシ吉草酸、ヒドロキシペンタン酸、ヒドロキシカプロン酸等のヒドロキシカルボン酸化合物が挙げられ、1種又は2種以上を組み合わせて利用することができる。これらのなかでも、生分解性樹脂組成物の耐熱性、及び透明性の観点から、グリコール酸、ヒドロキシカプロン酸が好ましい。
また、本発明においては、前記乳酸及びヒドロキシカルボン酸化合物の2量体が、それぞれの成分に含有されてもよく、好適例としては、生分解性樹脂組成物の耐熱性、及び透明性の観点から、D-ラクチド及びL-ラクチドが挙げられる。なお、乳酸の2量体は、乳酸成分のみを縮重合させる場合、及び乳酸成分とヒドロキシカルボン酸成分とを縮重合させる場合のいずれの場合の乳酸成分に含有されていてもよい。
乳酸の2量体の含有量は、生分解性樹脂組成物の耐熱性の観点から、乳酸成分中、80~100モル%が好ましく、90~100モル%がより好ましい。
ヒドロキシカルボン酸化合物の2量体の含有量は、生分解性樹脂組成物の耐熱性の観点から、ヒドロキシカルボン酸成分中、80~100モル%が好ましく、90~100モル%がより好ましい。
乳酸成分のみの縮重合反応、及び、乳酸成分とヒドロキシカルボン酸成分との縮重合反応は、特に限定はなく、公知の方法を用いて行うことができる。
かくして、原料モノマーを選択することにより、例えば、L-乳酸又はD-乳酸いずれかの成分85モル%以上100モル%未満とヒドロキシカルボン酸成分0モル%超15モル%以下からなるポリ乳酸が得られるが、なかでも、乳酸の環状二量体であるラクチド、グリコール酸の環状二量体であるグリコリド及びカプロラクトンを原料モノマーとして用いて得られるポリ乳酸が好ましい。なお、ポリ乳酸の光学純度は、生分解性樹脂組成物の耐熱性、及び透明性の観点から、95%以上が好ましく、98%以上がより好ましい。本明細書において、ポリ乳酸樹脂の光学純度は、「ポリオレフィン等合成樹脂製食品容器包装等に関する自主基準 第3版改訂版 2004年6月追補 第3部 衛生試験法 P12-13」記載のD体含有量の測定方法に従って求めることができる。具体的には、後述の実施例に記載の方法により測定される。
また、本発明において、ポリ乳酸として、生分解性樹脂組成物の耐熱性、及び透明性の観点から、異なる異性体を主成分とする乳酸成分を用いて得られた2種類のポリ乳酸からなるステレオコンプレックスポリ乳酸を用いてもよい。
ステレオコンプレックスポリ乳酸を構成する一方のポリ乳酸〔以降、ポリ乳酸(A)と記載する〕は、L体90~100モル%、D体を含むその他の成分0~10モル%を含有する。他方のポリ乳酸〔以降、ポリ乳酸(B)と記載する〕は、D体90~100モル%、L体を含むその他の成分0~10モル%を含有する。なお、L体及びD体以外のその他の成分としては、2個以上のエステル結合を形成可能な官能基を持つジカルボン酸、多価アルコール、ヒドロキシカルボン酸、ラクトン等が挙げられ、また、未反応の前記官能基を分子内に2つ以上有するポリエステル、ポリエーテル、ポリカーボネート等であってもよい。
ステレオコンプレックスポリ乳酸における、ポリ乳酸(A)とポリ乳酸(B)の重量比〔ポリ乳酸(A)/ポリ乳酸(B)〕は、10/90~90/10が好ましく、20/80~80/20がより好ましく、40/60~60/40がさらに好ましい。
ポリ乳酸の融点(Tm)(℃)は、可塑剤及び結晶核剤等の分散性の観点、ならびに生分解性樹脂組成物の曲げ強度、劣化、生産性の観点から、好ましくは140~250℃、より好ましくは150~240℃、さらに好ましくは160~230℃である。なお、本明細書において、樹脂の融点は、後述の実施例に記載の方法により測定される。
ポリ乳酸樹脂における、ポリ乳酸の含有量は、好ましくは80重量%以上、より好ましくは90重量%以上、さらに好ましくは実質的に100重量%である。
また、ポリ乳酸樹脂の含有量は、特に限定されないが、生分解性樹脂組成物中、50重量%以上が好ましく、60重量%以上がより好ましく、70重量%以上がさらに好ましい。
なお、ポリ乳酸は、前記方法により合成することができるが、市販の製品としては、例えば、レイシアH-100、H-280、H-400、H-440等の「レイシアシリーズ」(三井化学社製)、3001D、3051D、4032D、4042D、6201D、6251D、7000D、7032D等の「Nature Works」(ネイチャーワークス社製)、エコプラスチックU'z S-09、S-12、S-17等の「エコプラスチックU'zシリーズ」(トヨタ自動車社製)が挙げられる。これらのなかでも、生分解性樹脂組成物の耐熱性の観点から、レイシアH-100、H-280、H-400、H-440(三井化学社製)、3001D、3051D、4032D、4042D、6201D、6251D、7000D、7032D(ネイチャーワークス社製)、エコプラスチックU'z S-09、S-12、S-17(トヨタ自動車社製)が好ましい。
本発明においては、生分解性樹脂組成物の原料として、前記エチレンビス12-ヒドロキシステアリン酸アミド及び生分解性樹脂以外に、分子内に2個以上のエステル基を有するエステル化合物であって、該エステル化合物を構成するアルコール成分の少なくとも1種が水酸基1個当たり炭素数2~3のアルキレンオキサイドを平均0.5~5モル付加したアルコールであるエステル化合物、である可塑剤を含有することが好ましい。
<可塑剤>
本発明における可塑剤としては、分子内に2個以上のエステル基を有するエステル化合物であって、該エステル化合物を構成するアルコール成分の少なくとも1種が水酸基1個当たり炭素数2~3のアルキレンオキサイドを平均0.5~5モル付加したアルコールであるエステル化合物が挙げられ、具体的には、特開2008-174718号公報及び特開2008-115372号公報に記載の可塑剤が例示される。なかでも、分子内に2個以上のエステル基を有するエステル化合物であって、該エステル化合物を構成するアルコール成分が水酸基1個当たり炭素数2~3のアルキレンオキサイドを平均0.5~5モル付加したアルコールである化合物が好ましく、分子内に2個以上のエステル基を有する多価アルコールエステル、多価カルボン酸エーテルエステル、又はジカルボン酸とジオールとのポリエステルで、該エステルを構成するアルコール成分が水酸基1個当たりエチレンオキサイドを平均0.5~5モル付加したアルコールである化合物がより好ましい。
本発明における可塑剤としては、分子内に2個以上のエステル基を有するエステル化合物であって、該エステル化合物を構成するアルコール成分の少なくとも1種が水酸基1個当たり炭素数2~3のアルキレンオキサイドを平均0.5~5モル付加したアルコールであるエステル化合物が挙げられ、具体的には、特開2008-174718号公報及び特開2008-115372号公報に記載の可塑剤が例示される。なかでも、分子内に2個以上のエステル基を有するエステル化合物であって、該エステル化合物を構成するアルコール成分が水酸基1個当たり炭素数2~3のアルキレンオキサイドを平均0.5~5モル付加したアルコールである化合物が好ましく、分子内に2個以上のエステル基を有する多価アルコールエステル、多価カルボン酸エーテルエステル、又はジカルボン酸とジオールとのポリエステルで、該エステルを構成するアルコール成分が水酸基1個当たりエチレンオキサイドを平均0.5~5モル付加したアルコールである化合物がより好ましい。
前記構造を有する可塑剤としては、成形性、可塑性、耐ブリード性の観点から、分子内に2個以上のエステル基を有し、エチレンオキサイドの平均付加モル数が3~9の化合物が好ましく、コハク酸又はアジピン酸とポリエチレングリコールモノメチルエーテルとのエステル、及び酢酸とグリセリン又はエチレングリコールのエチレンオキサイド付加物とのエステルからなる群より選ばれる少なくとも1種がより好ましく、コハク酸又はアジピン酸とポリエチレングリコールモノメチルエーテルとのエステルがさらに好ましい。
前記構造を有する可塑剤としては、生分解性樹脂組成物の成形性に優れる観点から、酢酸とグリセリンのエチレンオキサイド平均3~9モル付加物とのエステル、コハク酸とエチレンオキサイドの平均付加モル数が2~4のポリエチレングリコールモノメチルエーテルとのエステル、アジピン酸とエチレンオキサイドの平均付加モル数が2~3のポリエチレングリコールモノメチルエーテルとのエステル等の多価カルボン酸とポリエチレングリコールモノメチルエーテルとのエステルが好ましい。生分解性樹脂組成物の成形性、及び可塑剤の耐ブリード性に優れる観点から、コハク酸とエチレンオキサイドの平均付加モル数が2~3のポリエチレングリコールモノメチルエーテルとのエステル、アジピン酸とジエチレングリコールモノメチルエーテルとのエステルがより好ましい。生分解性樹脂組成物の成形性、ならびに可塑剤の耐ブリード性、耐揮発性及び耐刺激臭の観点から、コハク酸とトリエチレングリコールモノメチルエーテルとのエステルがさらに好ましい。
また、耐揮発性の観点から、本発明に用いられる可塑剤は、2個以上のエステル基のうち平均0を超え~1.5以下のエステル基は芳香族アルコールから構成されるエステル化合物を含有してもよいが、アジピン酸とジエチレングリコールモノメチルエーテル/ベンジルアルコール=1/1混合物とのジエステルが好ましい。
可塑剤の平均分子量は耐ブリード性及び耐揮発性の観点から、好ましくは250~700であり、より好ましくは300~600であり、さらに好ましくは350~550であり、さらに好ましくは400~500である。尚、平均分子量は、JIS K0070に記載の方法で鹸化価を求め、次式より計算で求めることができる。
平均分子量=56108×(エステル基の数)/鹸化価
平均分子量=56108×(エステル基の数)/鹸化価
尚、前記エステルは、可塑剤としての機能を十分発揮させる観点から、全てエステル化された飽和エステルであることが好ましい。
本発明における生分解性樹脂組成物における可塑剤の含有量は、十分な結晶化速度を得る観点から、生分解性樹脂100重量部に対して、5~30重量部が好ましく、7~30重量部がより好ましく、10~30重量部がさらに好ましい。
本発明における原料は、前記以外に、充填剤、加水分解抑制剤、難燃剤等の添加剤を含有してもよい。
充填剤は、機械特性、成形性、及び耐熱性等に優れた生分解性樹脂組成物が得られるという観点から配合することが好ましく、通常、熱可塑性樹脂組成物の充填剤として用いられる繊維状、板状、粒状、粉末状のものを用いることができる。具体的には、タルク、スメクタイト、カオリン、マイカ、モンモリロナイト等のケイ酸塩、シリカ、酸化マグネシウム、酸化チタン、炭酸カルシウム等の無機化合物や、ガラス繊維、炭素繊維、グラファイト繊維、ワラスナイト、チタン酸カリウムウィスカー、珪素系ウィスカー等の繊維状無機充填剤、ナイロン繊維、アクリル繊維等の有機充填剤等が挙げられる。
[無機充填剤]
無機充填剤としては、通常熱可塑性樹脂の強化に用いられる繊維状、板状、粒状、粉末状のものを用いることができる。具体的には、ガラス繊維、アスベスト繊維、炭素繊維、グラファイト繊維、金属繊維、チタン酸カリウムウイスカー、ホウ酸アルミニウムウイスカー、マグネシウム系ウイスカー、珪素系ウイスカー、ワラステナイト、セピオライト、アスベスト、スラグ繊維、石膏繊維、シリカ繊維、シリカ・アルミナ繊維、ジルコニア繊維、窒化硼素繊維、窒化硅素繊維及び硼素繊維などの繊維状無機充填剤、ガラスフレーク、グラファイト、金属箔、セラミックビーズ、タルク、クレー、マイカ、セリサイト、ゼオライト、ベントナイト、ドロマイト、カオリン、微粉ケイ酸、長石粉、チタン酸カリウム、炭酸カルシウム、炭酸マグネシウム、硫酸バリウム、酸化カルシウム、酸化アルミニウム、酸化チタン、ケイ酸アルミニウム、酸化ケイ素、石膏及び白土などの板状や粒状の無機充填剤が挙げられる。これらの無機充填剤の中では、炭素繊維、ガラス繊維、ワラステナイト、マイカ、タルク及びカオリンが好ましい。また、繊維状充填剤のアスペクト比は5以上であることが好ましく、10以上であることがより好ましく、20以上であることがさらに好ましい。
無機充填剤としては、通常熱可塑性樹脂の強化に用いられる繊維状、板状、粒状、粉末状のものを用いることができる。具体的には、ガラス繊維、アスベスト繊維、炭素繊維、グラファイト繊維、金属繊維、チタン酸カリウムウイスカー、ホウ酸アルミニウムウイスカー、マグネシウム系ウイスカー、珪素系ウイスカー、ワラステナイト、セピオライト、アスベスト、スラグ繊維、石膏繊維、シリカ繊維、シリカ・アルミナ繊維、ジルコニア繊維、窒化硼素繊維、窒化硅素繊維及び硼素繊維などの繊維状無機充填剤、ガラスフレーク、グラファイト、金属箔、セラミックビーズ、タルク、クレー、マイカ、セリサイト、ゼオライト、ベントナイト、ドロマイト、カオリン、微粉ケイ酸、長石粉、チタン酸カリウム、炭酸カルシウム、炭酸マグネシウム、硫酸バリウム、酸化カルシウム、酸化アルミニウム、酸化チタン、ケイ酸アルミニウム、酸化ケイ素、石膏及び白土などの板状や粒状の無機充填剤が挙げられる。これらの無機充填剤の中では、炭素繊維、ガラス繊維、ワラステナイト、マイカ、タルク及びカオリンが好ましい。また、繊維状充填剤のアスペクト比は5以上であることが好ましく、10以上であることがより好ましく、20以上であることがさらに好ましい。
前記無機充填剤は、エチレン/酢酸ビニル共重合体などの熱可塑性樹脂や、エポキシ樹脂などの熱硬化性樹脂で被覆又は集束処理されていてもよく、アミノシランやエポキシシランなどのカップリング剤などで処理されていても良い。
[有機充填剤]
有機充填剤としては、通常熱可塑性樹脂の強化に用いられるチップ状、繊維状、板状、粉末状のものを用いることができる。具体例としては、籾殻、木材チップ、おから、古紙粉砕材、衣料粉砕材などのチップ状のもの、綿繊維、麻繊維、竹繊維、木材繊維、ケナフ繊維、ジュート繊維、バナナ繊維、ココナッツ繊維などの植物繊維もしくはこれらの植物繊維から加工されたパルプやセルロース繊維及び絹、羊毛、アンゴラ、カシミヤ、ラクダなどの動物繊維などの繊維状のもの、パルプ粉、紙粉、木粉、竹粉、セルロース粉末、籾殻粉末、果実殻粉末、キチン粉末、キトサン粉末、タンパク質、澱粉などの粉末状のものが挙げられ、成形性の観点から、紙粉、木粉、竹粉、セルロース粉末、ケナフ粉末、籾殻粉末、果実殻粉末、キチン粉末、キトサン粉末、タンパク質粉末、澱粉などの粉末状のものが好ましく、紙粉、木粉、竹粉、セルロース粉末、ケナフ粉末がより好ましい。また靱性向上の観点から、振動ロッドミル、ビーズミル等で、セルロースを非晶化した粉末の有機充填剤を用いることが好ましく、具体的には国際公開第2010/010961号パンフレット記載の非晶化セルロースであることが好ましい。
有機充填剤としては、通常熱可塑性樹脂の強化に用いられるチップ状、繊維状、板状、粉末状のものを用いることができる。具体例としては、籾殻、木材チップ、おから、古紙粉砕材、衣料粉砕材などのチップ状のもの、綿繊維、麻繊維、竹繊維、木材繊維、ケナフ繊維、ジュート繊維、バナナ繊維、ココナッツ繊維などの植物繊維もしくはこれらの植物繊維から加工されたパルプやセルロース繊維及び絹、羊毛、アンゴラ、カシミヤ、ラクダなどの動物繊維などの繊維状のもの、パルプ粉、紙粉、木粉、竹粉、セルロース粉末、籾殻粉末、果実殻粉末、キチン粉末、キトサン粉末、タンパク質、澱粉などの粉末状のものが挙げられ、成形性の観点から、紙粉、木粉、竹粉、セルロース粉末、ケナフ粉末、籾殻粉末、果実殻粉末、キチン粉末、キトサン粉末、タンパク質粉末、澱粉などの粉末状のものが好ましく、紙粉、木粉、竹粉、セルロース粉末、ケナフ粉末がより好ましい。また靱性向上の観点から、振動ロッドミル、ビーズミル等で、セルロースを非晶化した粉末の有機充填剤を用いることが好ましく、具体的には国際公開第2010/010961号パンフレット記載の非晶化セルロースであることが好ましい。
充填剤の含有量は、十分な耐熱性及び耐衝撃性を得る観点から、生分解性樹脂100重量部に対して、1~300重量部が好ましく、5~150重量部がより好ましい。
加水分解抑制剤としては、ポリカルボジイミド化合物やモノカルボジイミド化合物等のカルボジイミド化合物が挙げられ、生分解性樹脂組成物の成形性の観点からモノカルボジイミド化合物が好ましく、生分解性樹脂組成物の耐熱性、耐衝撃性及び結晶核剤の耐ブリード性の観点から、ポリカルボジイミド化合物が好ましい。
ポリカルボジイミド化合物としては、ポリ(4,4’-ジフェニルメタンカルボジイミド)、ポリ(4,4’-ジシクロヘキシルメタンカルボジイミド)、ポリ(1,3,5-トリイソプロピルベンゼン)ポリカルボジイミド、ポリ(1,3,5-トリイソプロピルベンゼン及び1,5-ジイソプロピルベンゼン)ポリカルボジイミド等が挙げられ、モノカルボジイミド化合物としては、N,N’-ジ-2,6-ジイソプロピルフェニルカルボジイミド等が挙げられる。
前記カルボジイミド化合物は、生分解性樹脂組成物の耐久性、耐衝撃性及び成形性を満たすために、単独で又は2種以上組み合わせて用いてもよい。また、ポリ(4,4’-ジシクロヘキシルメタンカルボジイミド)はカルボジライトLA-1(日清紡ケミカル社製)を、ポリ(1,3,5-トリイソプロピルベンゼン)ポリカルボジイミド及びポリ(1,3,5-トリイソプロピルベンゼン及び1,5-ジイソプロピルベンゼン)ポリカルボジイミドは、スタバクゾールP及びスタバクゾールP-100(Rhein Chemie社製)を、N,N’-ジ-2,6-ジイソプロピルフェニルカルボジイミドはスタバクゾールILF(Rhein Chemie社製)をそれぞれ購入して使用することができる。
加水分解抑制剤の含有量は、生分解性樹脂の加水分解抑制の観点から、生分解性樹脂100重量部に対して、0.05~15重量部が好ましく、0.10~10重量部がより好ましく、0.20~10重量部がさらに好ましい。
難燃剤の具体例としては、テトラブロムビスフェノール-A-エポキシオリゴマー、テトラブロムビスフェノール-A-カーボネートオリゴマー、ブロム化エポキシ樹脂等の臭素又は塩素を含有するハロゲン系化合物、三酸化アンチモン、ホウ酸亜鉛等の無機系難燃剤、シリコーン樹脂、シリコーンオイル等のシリコーン系難燃剤、水酸化アルミニウム、水酸化マグネシウム等の無機水和物(物性の観点からシランカップリング剤、なかでもイソシアネートシランで表面処理されていることが好ましい)、リン酸トリアリールイソプロピル化物、縮合リン酸エステル、ポリリン酸メラミン、ポリリン酸ピペラジン、ホスファーゼン化合物等のリン系難燃剤、及びメラミンシアヌレート等の含窒素系難燃剤などが挙げられる。
難燃剤としては、生分解性樹脂組成物の難燃性を向上させる観点から、リン系難燃剤が好ましく、縮合リン酸エステル、リン酸塩及び縮合リン酸塩からなる群より選ばれる少なくとも1種が好ましい。また、安全性の観点から、無機水和物又はリン系難燃剤が好ましく、物性の観点から無機水和物とリン化合物の併用が好ましい。難燃剤の含有量は、生分解性樹脂100重量部に対して、10~60重量部が好ましく、15~55重量部がより好ましい。
本発明の生分解性樹脂組成物は、強度、耐熱性、耐衝撃性等の物性向上の観点から、高強度有機合成繊維を含有することができる。高強度有機合成繊維の具体例としては、アラミド繊維、ポリアリレート繊維、PBO繊維等が挙げられ、耐熱性の観点からアラミド繊維が好ましい。高強度有機合成繊維の含有量は、生分解性樹脂100重量部に対して、3~20重量部が好ましく、5~10重量部がより好ましい。
本発明の生分解性樹脂組成物は、剛性、柔軟性、耐熱性、耐久性等の物性向上の観点から、その他の樹脂を含んでもよい。その他の樹脂の具体例としては、ポリエチレン、ポリプロピレン、ポリスチレン、ABS樹脂、AS樹脂、アクリル樹脂、ポリアミド、ポリフェニレンサルファイド、ポリエーテルエーテルケトン、ポリエステル、ポリアセタール、ポリスルホン、ポリフェニレンオキサイド、ポリイミド、ポリエーテルイミドなど、あるいはエチレン/グリシジルメタクリレート共重合体、ポリエステルエラストマー、ポリアミドエラストマー、エチレン/プロピレンターポリマー、エチレン/ブテン-1共重合体などの軟質熱可塑性樹脂などの熱可塑性樹脂や、フェノール樹脂、メラミン樹脂、不飽和ポリエステル樹脂、シリコーン樹脂、エポキシ樹脂などの熱硬化性樹脂などが挙げられるが、中でも生分解性樹脂との相溶性の観点からアミド結合、エステル結合、カーボネート結合等のカルボニル基を含む結合を有する樹脂が、構造的に生分解性樹脂、なかでもポリ乳酸樹脂と親和性が高い傾向があるため好ましい。
本発明の生分解性樹脂組成物は、耐衝撃性、靱性等の物性向上の観点から、コアシェル型ゴムを含有しても良い。具体例としては、(コア;シリコーン/アクリル重合体、シェル;メタクリル酸メチル重合体)、(コア;シリコーン/アクリル重合体、シェル;メタクリル酸メチル/メタクリル酸グリシジル重合体)、(コア;ブタンジエン/スチレン重合体、シェル;メタクリル酸メチル重合体)、(コア;アクリル重合体、シェル;メタクリル酸メチル重合体)等が挙げられる。透明性の観点から、市販品として、三菱レイヨン社製;メタブレンS-2006、S-2100、S-2200、ローム・アンド・ハース社製;パラロイドBPM-500が好ましい。コアシェル型ゴムの含有量は、生分解性樹脂100重量部に対して、2~30重量部が好ましく、3~20重量部がより好ましい。
本発明の生分解性樹脂組成物は、前記以外に、更にヒンダードフェノール又はホスファイト系の酸化防止剤、又は脂肪族アミド類、脂肪酸金属塩、炭化水素系ワックス類やアニオン型界面活性剤である滑剤等を含有することができる。酸化防止剤、滑剤のそれぞれの含有量は、生分解性樹脂100重量部に対し、0.05~3重量部が好ましく、0.10~2重量部がより好ましい。
また、本発明においては、前記添加剤以外に、安定剤(紫外線吸収剤、光安定剤等)、離形剤、染料及び顔料を含む着色剤、帯電防止剤、防曇剤、防カビ剤、抗菌剤、発泡剤等の添加剤を、本発明の目的達成を妨げない範囲で組成物原料として配合してもよい。
原料の溶融混練は、特に限定はなく、密閉式ニーダー、1軸もしくは2軸の押出機、オープンロール型混練機等の公知の混練機を用いて行うことができる。なお、原料は、予めヘンシェルミキサー、スーパーミキサー等を用いて均一に混合した後に、溶融混練に供することが好ましい。
溶融混練温度は、可塑剤、結晶核剤等の分散性の観点から、生分解性樹脂の融点(Tm)以上であり、好ましくはTm~Tm+100℃の範囲であり、より好ましくはTm~Tm+50℃の範囲である。例えば、好ましくは170~240℃であり、より好ましくは170~220℃である。溶融混練時間は、溶融混練温度、混練機の種類によって一概には決定できないが、15~900秒間が好ましい。
また、本発明は、前記溶融混練工程により得られた溶融混練物の結晶化速度をより向上させる観点から、溶融混練工程後に、溶融混練物を冷却する工程(以降、冷却工程ともいう)を含んでもよい。冷却温度は、溶融混練温度より、好ましくは60℃以上、より好ましくは70℃以上低い温度であり、具体的には、20~120℃が好ましく、20~100℃がより好ましい。冷却時間は、2~90秒間が好ましく、5~60秒間がより好ましい。なお、該冷却工程に際して、溶融混練物を公知の方法に従って成形してから冷却してもよい。
またさらに、冷却後には、好ましくは50~120℃、より好ましくは60~100℃で、好ましくは30~180秒、より好ましくは30~120秒、さらに好ましくは30~60秒保持する工程(以降、保持工程ともいう)を含んでもよい。なお、保持工程での温度は、冷却工程での温度と同一であっても異なっていてもよい。
かくして、結晶核剤であるエチレンビス12-ヒドロキシステアリン酸アミドと、生分解性樹脂とを含有する原料の溶融混練物であって、結晶化が促進された生分解性樹脂組成物が得られる。従って、本発明は、本発明の結晶化促進方法により結晶化が促進された生分解性樹脂組成物を提供する。なお、ここでいう「結晶化が促進された」とは、「結晶化速度が向上した」、「結晶化が進行し易い」状態のことを意味する。
具体的な結晶化方法としては、生分解性樹脂組成物を射出成形のように溶融状態から金型温度まで冷却し、さらに金型内で保持することで結晶させる溶融結晶化方法と、ガラス転移温度以下にある非晶状態にある生分解性樹脂組成物を加熱することで結晶させる冷結晶化方法が挙げられる。本発明の生分解性樹脂組成物は、優れた結晶化速度を有することから、生分解性樹脂組成物が可塑剤を含有する場合、溶融結晶化方法における100℃における半結晶化時間が30秒以下であることが好ましく、27秒以下であることがより好ましく、24秒以下であることがさらに好ましい。なお、100℃における半結晶化時間(溶融結晶化)は、実施例に示す方法により求めることができる。また、本発明の生分解性樹脂組成物は、優れた結晶化速度を有することから、生分解性樹脂組成物が可塑剤を含有する場合、冷結晶化方法(真空成形方法)における80℃における半結晶化時間が24秒以下であることが好ましく、21秒以下がより好ましく、18秒以下がさらに好ましい。なお、80℃における半結晶化時間(冷結晶化)は、実施例に示す方法により求めることができる。
本発明の生分解性樹脂組成物は、色相が良好で、結晶化速度が高く、成形性に優れるため、例えば200℃以下の低温で加工することができ、フィルムやシートに成形して、各種用途に用いることができる。
本発明はまた、本発明の生分解性樹脂組成物の製造方法を提供する。
本発明の製造方法は、アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドと生分解性樹脂とを含有する原料を溶融混練する工程を含む製造方法であり、製造条件等は前記記載の通りである。
またさらに、本発明は、本発明の生分解性樹脂組成物を成形した生分解性樹脂成形体を提供する。
本発明の生分解性樹脂成形体は、本発明の生分解性樹脂組成物を成形したものであればよく、具体的には、本発明の生分解性樹脂組成物の結晶化促進方法により得られる溶融混練物を、射出成形機等を用いて金型に充填し、成形して調製することができる。
金型温度は、結晶化速度向上及び作業性向上の観点から、好ましくは20~120℃であり、より好ましくは20~100℃であり、さらに好ましくは20~80℃である。
金型内での保持時間は、生産性向上の観点から、90秒以内が好ましく、60秒以内がより好ましく、30秒以内がさらに好ましい。
また、本発明の生分解性樹脂成形体は、本発明の生分解性樹脂組成物をシート状に成形したものであってもよい。シート状に成形する方法としては、特開2007-152760号公報、特開2007-130893号公報、特開2007-130895号公報に記載の方法を利用することができる。
本発明の生分解性樹脂成形体は、結晶化が促進された本発明の生分解性樹脂組成物を成形して調製するが、アミン価が低減された結晶核剤を用いているため成形体における樹脂劣化物の生成が抑制され、得られる成形体の金型からの離型性が優れるものとなる。
以下、実施例を示して本発明を具体的に説明するが、本発明は下記実施例に制限されるものではない。なお、例中の部は、特記しない限り重量部である。
〔ポリ乳酸樹脂の重量平均分子量(Mw)〕
重量平均分子量(Mw)は、GPC(ゲルパーミエーションクロマトグラフィー)により、下記の測定条件で測定する。
<測定条件>
カラム:GMHHR-H+GMHHR-H
カラム温度:40℃
検出器:RI
溶離液:クロロホルム
流速:1.0mL/min
サンプル濃度:1mg/mL
注入量:0.1mL
換算標準:ポリスチレン
重量平均分子量(Mw)は、GPC(ゲルパーミエーションクロマトグラフィー)により、下記の測定条件で測定する。
<測定条件>
カラム:GMHHR-H+GMHHR-H
カラム温度:40℃
検出器:RI
溶離液:クロロホルム
流速:1.0mL/min
サンプル濃度:1mg/mL
注入量:0.1mL
換算標準:ポリスチレン
〔ポリ乳酸の光学純度〕
光学純度は、「ポリオレフィン等合成樹脂製食品容器包装等に関する自主基準 第3版改訂版 2004年6月追補 第3部 衛生試験法 P12-13」記載のD体含有量の測定方法に従って、下記の測定条件で測定する。具体的には、精秤したポリ乳酸に水酸化ナトリウム/メタノールを加え、65℃に設定した水浴振とう器にセットして、樹脂分が均一溶液になるまで加水分解を行い、さらに加水分解が完了したアルカリ溶液に希塩酸を加え中和し、その分解溶液を純水にて定容した後、一定容量をメスフラスコに分液して高速液体クロマトグラフィー(HPLC)移動相溶液により希釈し、pHが3~7の範囲になるように調整してメスフラスコに定容、メンブランフィルター(0.45μm)によりろ過し、この調整溶液をHPLCにてD-乳酸、L-乳酸を定量することによってポリ乳酸の光学純度を求める。
<HPLC測定条件>
カラム :光学分割カラム
スミキラルOA6100(46mmφ×150mm、5μm)、住化分析センター社製
プレカラム:光学分割カラム
スミキラルQA6100(4mmφ×10mm、5μm)、住化分析センター社製
カラム温度:25℃
移動相 :2.5%メタノール含有1.5mM硫酸銅水溶液
移動相流量:1.0mL/分
検出器 :紫外線検出器(UV254nm)
注入量 :20μL
光学純度は、「ポリオレフィン等合成樹脂製食品容器包装等に関する自主基準 第3版改訂版 2004年6月追補 第3部 衛生試験法 P12-13」記載のD体含有量の測定方法に従って、下記の測定条件で測定する。具体的には、精秤したポリ乳酸に水酸化ナトリウム/メタノールを加え、65℃に設定した水浴振とう器にセットして、樹脂分が均一溶液になるまで加水分解を行い、さらに加水分解が完了したアルカリ溶液に希塩酸を加え中和し、その分解溶液を純水にて定容した後、一定容量をメスフラスコに分液して高速液体クロマトグラフィー(HPLC)移動相溶液により希釈し、pHが3~7の範囲になるように調整してメスフラスコに定容、メンブランフィルター(0.45μm)によりろ過し、この調整溶液をHPLCにてD-乳酸、L-乳酸を定量することによってポリ乳酸の光学純度を求める。
<HPLC測定条件>
カラム :光学分割カラム
スミキラルOA6100(46mmφ×150mm、5μm)、住化分析センター社製
プレカラム:光学分割カラム
スミキラルQA6100(4mmφ×10mm、5μm)、住化分析センター社製
カラム温度:25℃
移動相 :2.5%メタノール含有1.5mM硫酸銅水溶液
移動相流量:1.0mL/分
検出器 :紫外線検出器(UV254nm)
注入量 :20μL
〔ポリ乳酸樹脂の融点〕
ポリ乳酸樹脂の融点は、JIS-K7121に基づく示差走査熱量測定(DSC、パーキンエルマー社製、ダイアモンドDSC)の昇温法による結晶融解吸熱ピーク温度より求められる。融点の測定は、昇温速度10℃/分で20℃から250℃まで昇温して行う。
ポリ乳酸樹脂の融点は、JIS-K7121に基づく示差走査熱量測定(DSC、パーキンエルマー社製、ダイアモンドDSC)の昇温法による結晶融解吸熱ピーク温度より求められる。融点の測定は、昇温速度10℃/分で20℃から250℃まで昇温して行う。
<可塑剤の製造例1>(コハク酸とトリエチレングリコールモノメチルエーテルとのジエステル)
攪拌機、温度計、脱水管を備えた3Lフラスコに、無水コハク酸500g、トリエチレングリコールモノメチルエーテル2463g、パラトルエンスルホン酸一水和物9.5gを仕込み、空間部に窒素(500mL/分)を吹き込みながら、減圧下(4~10.7kPa)、110℃で15時間反応させた。反応液の酸価は1.6(mgKOH/g)であった。反応液に吸着剤キョーワード500SH(協和化学工業社製)27gを添加して80℃、2.7kPaで45分間攪拌してろ過した後、液温115~200℃、圧力0.03kPaでトリエチレングリコールモノメチルエーテルを留去し、80℃に冷却後、残液を減圧ろ過して、ろ液として、コハク酸とトリエチレングリコールモノメチルエーテルとのジエステルを得た。得られたジエステルは、酸価0.2(mgKOH/g)、鹸化価276(mgKOH/g)、水酸基価1以下(mgKOH/g)、色相APHA200であった。
攪拌機、温度計、脱水管を備えた3Lフラスコに、無水コハク酸500g、トリエチレングリコールモノメチルエーテル2463g、パラトルエンスルホン酸一水和物9.5gを仕込み、空間部に窒素(500mL/分)を吹き込みながら、減圧下(4~10.7kPa)、110℃で15時間反応させた。反応液の酸価は1.6(mgKOH/g)であった。反応液に吸着剤キョーワード500SH(協和化学工業社製)27gを添加して80℃、2.7kPaで45分間攪拌してろ過した後、液温115~200℃、圧力0.03kPaでトリエチレングリコールモノメチルエーテルを留去し、80℃に冷却後、残液を減圧ろ過して、ろ液として、コハク酸とトリエチレングリコールモノメチルエーテルとのジエステルを得た。得られたジエステルは、酸価0.2(mgKOH/g)、鹸化価276(mgKOH/g)、水酸基価1以下(mgKOH/g)、色相APHA200であった。
<可塑剤の製造例2>(酢酸とグリセリンにエチレンオキサイドを6モル付加させたエチレンオキサイド付加物とのトリエステル化合物)
オートクレーブに花王社製化粧品用濃グリセリン1モルに対しエチレンオキサイド6モルのモル比で規定量仕込み、1モル%のKOHを触媒として反応圧力0.3MPaの定圧付加し、圧力が一定になるまで150℃で反応した後、80℃まで冷却し、触媒未中和の生成物を得た。この生成物に触媒の吸着剤としてキョーワード600S(協和化学工業社製)を触媒重量の8倍添加し、窒素存在下で80℃、1時間吸着処理をおこなった。さらに処理後の液をNo.2のろ紙にラヂオライト#900をプレコートしたヌッツェで吸着剤を濾過し、グリセリンエチレンオキサイド6モル付加物(以下POE(6)グリセリンという)を得た。これを四つ口フラスコに仕込み、105℃に昇温して300r/minで攪拌し、無水酢酸をPOE(6)グリセリン1モルに対し7.2モルの比率で規定量を約1時間で滴下し反応させた。滴下後110℃で2時間熟成し、さらに120℃で1時間熟成した。熟成後、減圧下で未反応の無水酢酸及び副生の酢酸をトッピングし、さらにスチーミングして、POE(6)グリセリントリアセテートを得た。得られたPOE(6)グリセリントリアセテートは平均分子量490であった。
オートクレーブに花王社製化粧品用濃グリセリン1モルに対しエチレンオキサイド6モルのモル比で規定量仕込み、1モル%のKOHを触媒として反応圧力0.3MPaの定圧付加し、圧力が一定になるまで150℃で反応した後、80℃まで冷却し、触媒未中和の生成物を得た。この生成物に触媒の吸着剤としてキョーワード600S(協和化学工業社製)を触媒重量の8倍添加し、窒素存在下で80℃、1時間吸着処理をおこなった。さらに処理後の液をNo.2のろ紙にラヂオライト#900をプレコートしたヌッツェで吸着剤を濾過し、グリセリンエチレンオキサイド6モル付加物(以下POE(6)グリセリンという)を得た。これを四つ口フラスコに仕込み、105℃に昇温して300r/minで攪拌し、無水酢酸をPOE(6)グリセリン1モルに対し7.2モルの比率で規定量を約1時間で滴下し反応させた。滴下後110℃で2時間熟成し、さらに120℃で1時間熟成した。熟成後、減圧下で未反応の無水酢酸及び副生の酢酸をトッピングし、さらにスチーミングして、POE(6)グリセリントリアセテートを得た。得られたPOE(6)グリセリントリアセテートは平均分子量490であった。
<結晶核剤の精製例1>(エチレンビス12-ヒドロキシステアリン酸アミドの精製;晶析法)
アンカー型攪拌機、温度計付き1L容の筒型セパラブルフラスコに、未精製エチレンビス12-ヒドロキシステアリン酸アミド(日本化成社製、スリパックスH)(未精製品)13g、及び1-ブタノール130g(未精製エチレンビス12-ヒドロキシステアリン酸アミド100重量部に対して1000重量部)を加え、100r/minで、90℃で1時間加熱攪拌した。その後、3時間かけて25℃まで冷却し、25℃でさらに1時間攪拌後、得られたスラリーを孔径1ミクロンのメンブランフィルター(材質:PTFE、アドバンテック社製)を設置した加圧ろ過器を用いて、圧力0.2MPaでろ過を行った。得られた残渣は、1回あたり39gの1-ブタノール(25℃)(残渣100重量部に対して100重量部)を用いて、ケーク洗浄を2回行った後、60℃で減圧(133Pa)乾燥を12時間行って、精製エチレンビス12-ヒドロキシステアリン酸アミド(精製品(1))を得た。未精製品及び精製品(1)の品質を、下記の方法に従って評価した。結果を表1に示す。
アンカー型攪拌機、温度計付き1L容の筒型セパラブルフラスコに、未精製エチレンビス12-ヒドロキシステアリン酸アミド(日本化成社製、スリパックスH)(未精製品)13g、及び1-ブタノール130g(未精製エチレンビス12-ヒドロキシステアリン酸アミド100重量部に対して1000重量部)を加え、100r/minで、90℃で1時間加熱攪拌した。その後、3時間かけて25℃まで冷却し、25℃でさらに1時間攪拌後、得られたスラリーを孔径1ミクロンのメンブランフィルター(材質:PTFE、アドバンテック社製)を設置した加圧ろ過器を用いて、圧力0.2MPaでろ過を行った。得られた残渣は、1回あたり39gの1-ブタノール(25℃)(残渣100重量部に対して100重量部)を用いて、ケーク洗浄を2回行った後、60℃で減圧(133Pa)乾燥を12時間行って、精製エチレンビス12-ヒドロキシステアリン酸アミド(精製品(1))を得た。未精製品及び精製品(1)の品質を、下記の方法に従って評価した。結果を表1に示す。
<結晶核剤の精製例2>(エチレンビス12-ヒドロキシステアリン酸アミドの精製;熱洗浄法)
アンカー型攪拌機、温度計付き3Lの筒型セパラブルフラスコに精製例1で用いた未精製エチレンビス12-ヒドロキシステアリン酸アミド(日本化成社製、スリパックスH)400g、メタノール2000gを仕込み、100r/minで、60℃で1時間加熱攪拌した。内容物を0.5時間かけて25℃まで冷却した後、スラリーを孔径1ミクロンのメンブランフィルター(材質:PTFE、アドバンテック社製)を設置した加圧ろ過器に移し、圧力0.1MPaでろ過を行った。得られた残渣は1回あたり280gのメタノール(25℃)を用いて、ケーク洗浄を3回行った後、60℃で減圧(4kPa)乾燥を6h行って、精製エチレンビス12-ヒドロキシステアリン酸アミド(精製品(2))を得た。得られた精製品(2)の品質を下記の方法に従って評価した。結果を表1に示す。
アンカー型攪拌機、温度計付き3Lの筒型セパラブルフラスコに精製例1で用いた未精製エチレンビス12-ヒドロキシステアリン酸アミド(日本化成社製、スリパックスH)400g、メタノール2000gを仕込み、100r/minで、60℃で1時間加熱攪拌した。内容物を0.5時間かけて25℃まで冷却した後、スラリーを孔径1ミクロンのメンブランフィルター(材質:PTFE、アドバンテック社製)を設置した加圧ろ過器に移し、圧力0.1MPaでろ過を行った。得られた残渣は1回あたり280gのメタノール(25℃)を用いて、ケーク洗浄を3回行った後、60℃で減圧(4kPa)乾燥を6h行って、精製エチレンビス12-ヒドロキシステアリン酸アミド(精製品(2))を得た。得られた精製品(2)の品質を下記の方法に従って評価した。結果を表1に示す。
<結晶核剤の精製例3>(エチレンビス12-ヒドロキシステアリン酸アミドの精製;晶析法と熱洗浄法の併用)
アンカー型攪拌機、温度計付き1Lの筒型セパラブルフラスコに精製例1で得られた精製エチレンビス12-ヒドロキシステアリン酸アミド(精製品(1))50g、1-ブタノール500gを仕込み、100r/minで、90℃で30分間加熱攪拌した。その後、2時間かけて25℃まで冷却し、スラリーを孔径1ミクロンのメンブランフィルター(材質:PTFE、アドバンテック社製)を設置した加圧ろ過器に移し、圧力0.1MPaでろ過を行った。得られた残渣を用いて同様の操作を繰り返した。次に、この残渣をもう一度、筒型セパラブルフラスコに戻した後、メタノール300gを仕込み、100r/minで、60℃で30分間加熱攪拌した。内容物を0.5時間かけて25℃まで冷却した後、スラリーを同様の操作でろ過した。得られた残渣を再度筒型セパラブルフラスコに戻して、メタノール300gを仕込み同様の操作を繰り返してろ過ケークを得た。得られたケークを70℃で2h、減圧(1kPa)乾燥し、精製エチレンビス12-ヒドロキシステアリン酸アミド(精製品(3))を得た。精製品(3)の品質を下記の方法に従って評価した。結果を表1に示す。
アンカー型攪拌機、温度計付き1Lの筒型セパラブルフラスコに精製例1で得られた精製エチレンビス12-ヒドロキシステアリン酸アミド(精製品(1))50g、1-ブタノール500gを仕込み、100r/minで、90℃で30分間加熱攪拌した。その後、2時間かけて25℃まで冷却し、スラリーを孔径1ミクロンのメンブランフィルター(材質:PTFE、アドバンテック社製)を設置した加圧ろ過器に移し、圧力0.1MPaでろ過を行った。得られた残渣を用いて同様の操作を繰り返した。次に、この残渣をもう一度、筒型セパラブルフラスコに戻した後、メタノール300gを仕込み、100r/minで、60℃で30分間加熱攪拌した。内容物を0.5時間かけて25℃まで冷却した後、スラリーを同様の操作でろ過した。得られた残渣を再度筒型セパラブルフラスコに戻して、メタノール300gを仕込み同様の操作を繰り返してろ過ケークを得た。得られたケークを70℃で2h、減圧(1kPa)乾燥し、精製エチレンビス12-ヒドロキシステアリン酸アミド(精製品(3))を得た。精製品(3)の品質を下記の方法に従って評価した。結果を表1に示す。
<全アミン価>
ASTM D 2074に従って行う。
ASTM D 2074に従って行う。
<酸価>
JIS K 0070に従って行う。
JIS K 0070に従って行う。
<色相>
JIS K 0071-2に従って行う。
JIS K 0071-2に従って行う。
<凝固点>
サンプル7.5mgを精秤し、アルミパンに封入後、DSC装置(パーキンエルマー社製ダイアモンドDSC)を用い、200℃で2分間溶融した後、10℃/分の速度で25℃まで降温し、発熱のピーク温度を求めた。
サンプル7.5mgを精秤し、アルミパンに封入後、DSC装置(パーキンエルマー社製ダイアモンドDSC)を用い、200℃で2分間溶融した後、10℃/分の速度で25℃まで降温し、発熱のピーク温度を求めた。

<結晶核剤の製造例>(結晶核剤A~G)
表1に示すエチレンビス12-ヒドロキシステアリン酸アミドの精製品(1)~(3)、及び未精製品、あるいは12-ヒドロキシステアリン酸(伊藤製油社製、酸価181mgKOH/g)を使用し、表2に示す比率で配合した後、乳鉢で10分間混合し、それぞれ結晶核剤A~Gを得た。品質は精製品(1)と同様に評価した。結果を表2に示す。
表1に示すエチレンビス12-ヒドロキシステアリン酸アミドの精製品(1)~(3)、及び未精製品、あるいは12-ヒドロキシステアリン酸(伊藤製油社製、酸価181mgKOH/g)を使用し、表2に示す比率で配合した後、乳鉢で10分間混合し、それぞれ結晶核剤A~Gを得た。品質は精製品(1)と同様に評価した。結果を表2に示す。

実施例1~9及び比較例1~6
ポリ乳酸樹脂組成物として、表3、4に示す組成物原料を、180℃の混練機(東洋精機社製、ラボプラストミル)にて10分間混練した後、190℃のプレス成形機にて厚さ0.3mmのシート状に成形し、25℃に冷却してそのまま60秒間保持後、さらに90℃で1分間保持してシート(成形体)を得た。
ポリ乳酸樹脂組成物として、表3、4に示す組成物原料を、180℃の混練機(東洋精機社製、ラボプラストミル)にて10分間混練した後、190℃のプレス成形機にて厚さ0.3mmのシート状に成形し、25℃に冷却してそのまま60秒間保持後、さらに90℃で1分間保持してシート(成形体)を得た。
なお、表3、4における原料は以下の通りである。
<ポリ乳酸樹脂>
*1:ポリ乳酸樹脂(ネイチャーワークスLLC社製ポリ-L-乳酸、NatureWorks 4032D、光学純度98.5%、融点160℃、重量平均分子量180000)
<結晶核剤>
表2に記載
<可塑剤>
*2:(MeEO3)2SA、前記の製造例1で得られたコハク酸とトリエチレングリコールモノメチルエーテルとのジエステル化合物、平均分子量410
*3:DAIFATTY-101、アジピン酸と、ジエチレングリコールモノメチルエーテル/ベンジルアルコール=1/1混合物とのジエステル(大八化学工業社製)、平均分子量338
*4:(AcEO2)3Gly、前記の製造例2で得られた酢酸とグリセリンにエチレンオキサイドを6モル付加させたエチレンオキサイド付加物とのトリエステル化合物、平均分子量490
<加水分解抑制剤>
*5:カルボジライトLA-1(日清紡績社製)
<ポリ乳酸樹脂>
*1:ポリ乳酸樹脂(ネイチャーワークスLLC社製ポリ-L-乳酸、NatureWorks 4032D、光学純度98.5%、融点160℃、重量平均分子量180000)
<結晶核剤>
表2に記載
<可塑剤>
*2:(MeEO3)2SA、前記の製造例1で得られたコハク酸とトリエチレングリコールモノメチルエーテルとのジエステル化合物、平均分子量410
*3:DAIFATTY-101、アジピン酸と、ジエチレングリコールモノメチルエーテル/ベンジルアルコール=1/1混合物とのジエステル(大八化学工業社製)、平均分子量338
*4:(AcEO2)3Gly、前記の製造例2で得られた酢酸とグリセリンにエチレンオキサイドを6モル付加させたエチレンオキサイド付加物とのトリエステル化合物、平均分子量490
<加水分解抑制剤>
*5:カルボジライトLA-1(日清紡績社製)
得られたシートの特性を、下記の方法に従って評価した。結果を表3、4に示す。
<透明性>
JIS K 7105に従って、村上色彩技術研究所製、ヘイズメータHM-150を用いて、透明性(Haze%)を測定する。数値が小さい程、透明性が高いことを意味する。
JIS K 7105に従って、村上色彩技術研究所製、ヘイズメータHM-150を用いて、透明性(Haze%)を測定する。数値が小さい程、透明性が高いことを意味する。
<黄色味>
JIS Z 8722に従って、日本電色工業社製、色差計Spectro Color Meter SE 2000を用いてシートの黄色味(b値)を測定する。数値が+側に大きくなる程、黄色味が強いことを意味する。
JIS Z 8722に従って、日本電色工業社製、色差計Spectro Color Meter SE 2000を用いてシートの黄色味(b値)を測定する。数値が+側に大きくなる程、黄色味が強いことを意味する。
<半結晶化時間>
シートの切出しにより、試験片7.5mgを精秤し、アルミパンに封入後、DSC装置(パーキンエルマー社製、ダイアモンドDSC)を用いて、200℃で5分間溶融し、-500℃/分の速度で各保持温度(90℃、100℃、110℃)まで降温し、結晶飽和となる半分の時間(飽和結晶の半分の結晶化度になる時間;半結晶化時間;t1/2)を求める。t1/2は、サンプル温度が保持温度に達したときの時間を0分として算出する。
シートの切出しにより、試験片7.5mgを精秤し、アルミパンに封入後、DSC装置(パーキンエルマー社製、ダイアモンドDSC)を用いて、200℃で5分間溶融し、-500℃/分の速度で各保持温度(90℃、100℃、110℃)まで降温し、結晶飽和となる半分の時間(飽和結晶の半分の結晶化度になる時間;半結晶化時間;t1/2)を求める。t1/2は、サンプル温度が保持温度に達したときの時間を0分として算出する。

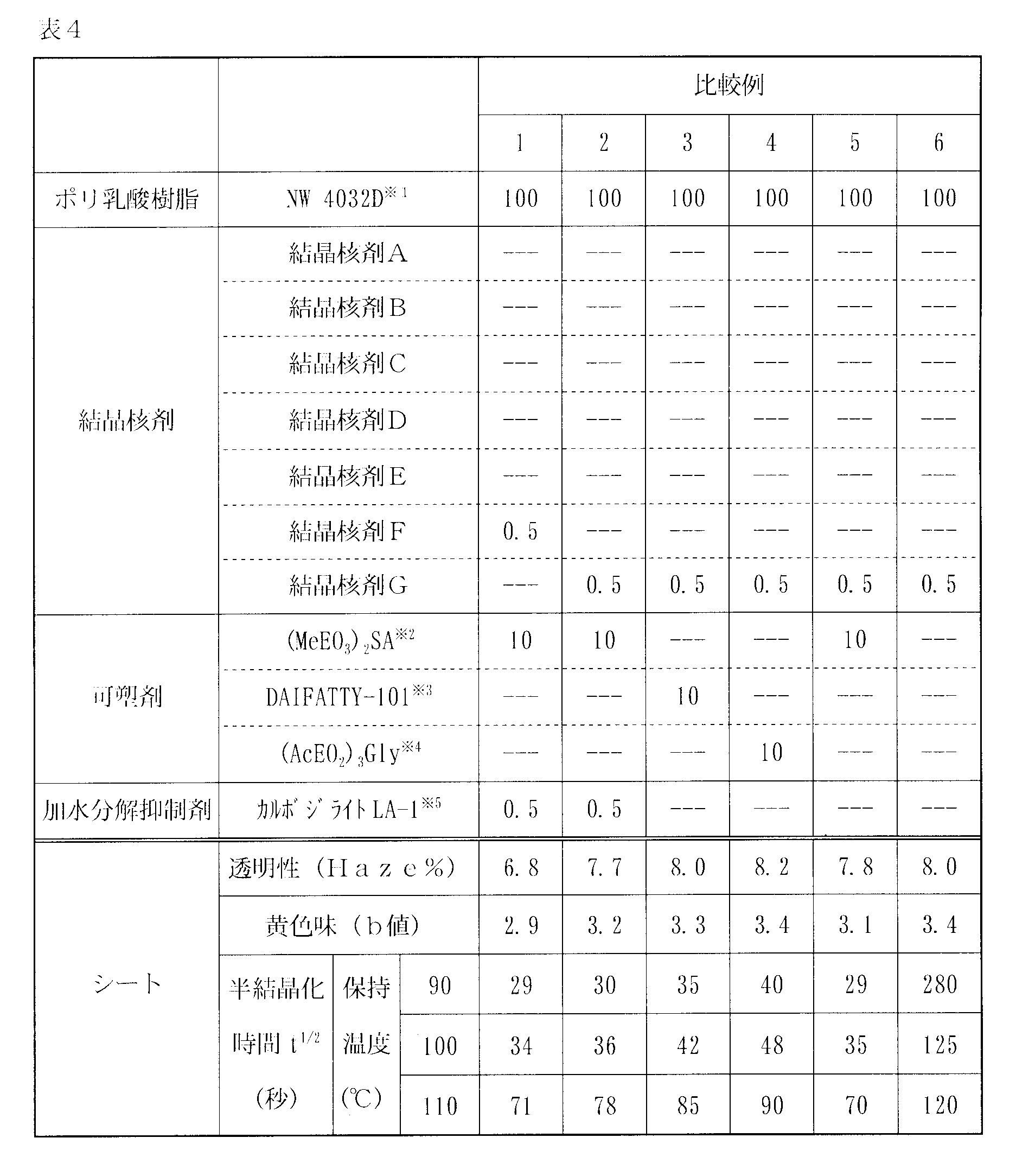
表3、4の結果から明らかなように、実施例のシートは比較例のシートに比べて、透明性及び黄色味が低く、半結晶化時間が短いことが分かる。射出成形やシート成形の多くの場合、高温側から冷却する溶融結晶化が用いられるが、成形体は熱容量があるため半結晶化時間の比較的小さな差であっても、実際の成形時には成形速度の大きな違いとなって表れる。従って、本発明の結晶化促進方法により結晶化が促進された樹脂組成物は、半結晶化時間が短く、成形に要する時間が短くなり、生産性上好ましい。また、溶融結晶化における半結晶化時間を短くするためには、アミン価を低減することによる効果が最も大きいことが示唆されるが、アミン価が1.0mgKOH/g以下の場合は、凝固点が高い方がより半結晶化時間が短くなり好ましいことが分かる。
実施例10~11及び比較例7(真空成形での評価)
ポリ乳酸樹脂組成物として、表5に示す組成物原料(各原料は表3、4と同じ)を、180℃の混練機(東洋精機社製、ラボプラストミル)にて10分間混練した後、190℃のプレス成形機にて縦150mm×横150mm×厚さ0.4mmのシート状に成形し、25℃に冷却してそのまま60秒間保持し、シート(成形体)を得た。得られたシートを、真空圧空成形機(脇坂エンジニアリング社製 FVS-500型)を用いて真空成形を行って、成形体を得た(図1参照)。予備加熱は上下ヒーターを400℃に設定したヒーターボックスに7秒間保持して行い、瞬時に成形ゾーンに移送して金型温度90℃で真空成形を行った。
ポリ乳酸樹脂組成物として、表5に示す組成物原料(各原料は表3、4と同じ)を、180℃の混練機(東洋精機社製、ラボプラストミル)にて10分間混練した後、190℃のプレス成形機にて縦150mm×横150mm×厚さ0.4mmのシート状に成形し、25℃に冷却してそのまま60秒間保持し、シート(成形体)を得た。得られたシートを、真空圧空成形機(脇坂エンジニアリング社製 FVS-500型)を用いて真空成形を行って、成形体を得た(図1参照)。予備加熱は上下ヒーターを400℃に設定したヒーターボックスに7秒間保持して行い、瞬時に成形ゾーンに移送して金型温度90℃で真空成形を行った。
真空成形時の結晶化方法(冷結晶化)に近い下記の測定方法で、結晶化速度の指標である半結晶化時間tcc1/2を測定した。また、前記成形体を成形する際に、前記成形体の離型に必要な成形時間を下記の基準で評価した。さらに、成形体の透明性について、中央の平坦部を切り出して下記の方法で測定した。これらの結果を表5に示す。
<半結晶化時間(冷結晶化)>
シートの切出しにより、試験片7.5mgを精秤し、アルミパンに封入後、DSC装置(パーキンエルマー社製、ダイアモンドDSC)を用いて、200℃で5分間溶融し、-500℃/分の速度で25℃まで急冷し1分間保持した後、500℃/分の速度で各保持温度(70℃、80℃、90℃)まで昇温し、結晶飽和となる半分の時間(飽和結晶の半分の結晶化度になる時間;半結晶化時間(冷結晶化);tcc1/2)を求めた。tcc1/2は、サンプル温度が保持温度に達したときの時間を0分として算出した。
シートの切出しにより、試験片7.5mgを精秤し、アルミパンに封入後、DSC装置(パーキンエルマー社製、ダイアモンドDSC)を用いて、200℃で5分間溶融し、-500℃/分の速度で25℃まで急冷し1分間保持した後、500℃/分の速度で各保持温度(70℃、80℃、90℃)まで昇温し、結晶飽和となる半分の時間(飽和結晶の半分の結晶化度になる時間;半結晶化時間(冷結晶化);tcc1/2)を求めた。tcc1/2は、サンプル温度が保持温度に達したときの時間を0分として算出した。
<離型に必要な金型保持時間の評価基準>
離型に必要な金型保持時間とは、脱型時に変形がなく取り出しが可能となる最短時間である。該金型保持時間は、プレス成型後に得られたシートを金型に張り付けた時点を金型保持時間の開始点とし、シートから金型を取り外す時間を金型保持時間の終点とし測定した。金型保持時間が短いほど、樹脂組成物の冷結晶化速度が速く、成形性に優れることを示す。
離型に必要な金型保持時間とは、脱型時に変形がなく取り出しが可能となる最短時間である。該金型保持時間は、プレス成型後に得られたシートを金型に張り付けた時点を金型保持時間の開始点とし、シートから金型を取り外す時間を金型保持時間の終点とし測定した。金型保持時間が短いほど、樹脂組成物の冷結晶化速度が速く、成形性に優れることを示す。
<透明性>
JIS K 7105に従って、村上色彩技術研究所製、ヘイズメータHM-150を用いて、透明性(Haze%)を測定する。数値が小さい程、透明性が高いことを意味する。
JIS K 7105に従って、村上色彩技術研究所製、ヘイズメータHM-150を用いて、透明性(Haze%)を測定する。数値が小さい程、透明性が高いことを意味する。

表5の結果から明らかなように、冷結晶化においても、実施例のシートは比較例のシートに比べて、半結晶化時間が短いことが分かる。熱成形の多くの場合、室温側から加熱する冷結晶化が用いられるが、表5の結果から明らかなように、本発明の結晶化促進方法により結晶化が促進された樹脂組成物は、冷結晶化においても半結晶化時間が短く、熱成形に要する時間が短くなり、生産性上好ましい。また、成形体の透明性も向上することができる。
本発明の結晶化促進方法により得られる生分解性樹脂組成物は、例えば、日用雑貨品、家電部品、自動車部品等の様々な工業用途に好適に使用することができる。
Claims (7)
- アミン価が1.0mgKOH/g以下であるエチレンビス12-ヒドロキシステアリン酸アミドと生分解性樹脂とを含有する原料を溶融混練する工程を含む、生分解性樹脂組成物の結晶化促進方法。
- エチレンビス12-ヒドロキシステアリン酸アミドの酸価が1.0mgKOH/g以下である、請求項1記載の結晶化促進方法。
- 原料が、さらに、分子内に2個以上のエステル基を有するエステル化合物であって、該エステル化合物を構成するアルコール成分の少なくとも1種が水酸基1個当たり炭素数2~3のアルキレンオキサイドを平均0.5~5モル付加したアルコールであるエステル化合物を含有してなる、請求項1又は2記載の結晶化促進方法。
- 請求項1~3いずれか記載の結晶化促進方法により結晶化が促進されてなる、生分解性樹脂組成物。
- 生分解性樹脂がポリ乳酸樹脂である、請求項4記載の生分解性樹脂組成物。
- 請求項4又は5記載の生分解性樹脂組成物を成形してなる、生分解性樹脂成形体。
- アミン価が0.01~1.0mgKOH/gであるエチレンビス12-ヒドロキシステアリン酸アミドと生分解性樹脂とを含有する原料を溶融混練する工程を含む、生分解性樹脂組成物の製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10753506.4A EP2410018B1 (en) | 2009-03-19 | 2010-03-16 | Method for promoting crystallization of biodegradable resin composition |
| US13/256,974 US8455579B2 (en) | 2009-03-19 | 2010-03-16 | Method for promoting crystallization of biodegradable resin composition |
| CN2010800196267A CN102414273B (zh) | 2009-03-19 | 2010-03-16 | 生物降解性树脂组合物的结晶化促进方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009-068523 | 2009-03-19 | ||
| JP2009068523 | 2009-03-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010107014A1 true WO2010107014A1 (ja) | 2010-09-23 |
Family
ID=42739674
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/054402 WO2010107014A1 (ja) | 2009-03-19 | 2010-03-16 | 生分解性樹脂組成物の結晶化促進方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8455579B2 (ja) |
| EP (1) | EP2410018B1 (ja) |
| JP (1) | JP5627255B2 (ja) |
| CN (1) | CN102414273B (ja) |
| WO (1) | WO2010107014A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012071481A (ja) * | 2010-09-28 | 2012-04-12 | Panasonic Corp | 成形品の製造方法、及び成形品 |
| US20140378595A1 (en) * | 2013-06-19 | 2014-12-25 | Lotte Chemical Corporation | Polylactic acid stereocomplex resin composition having improved crystallization rate and method for molding the same |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013040281A (ja) * | 2011-08-17 | 2013-02-28 | Dai Ichi Kogyo Seiyaku Co Ltd | ポリ乳酸樹脂組成物およびその成形体 |
| JP5882710B2 (ja) * | 2011-12-12 | 2016-03-09 | 第一工業製薬株式会社 | ポリ乳酸樹脂組成物およびその樹脂成形体 |
| JP5882709B2 (ja) * | 2011-12-12 | 2016-03-09 | 第一工業製薬株式会社 | ポリ乳酸樹脂組成物およびその樹脂成形体 |
| JP5936953B2 (ja) * | 2011-12-12 | 2016-06-22 | 第一工業製薬株式会社 | ポリ乳酸樹脂組成物およびその樹脂成形体 |
| ITMI20120250A1 (it) | 2012-02-20 | 2013-08-21 | Novamont Spa | Composizione polimerica biodegradabile per la realizzazione di articoli aventi elevata temperatura di inflessione sotto carico. |
| FR3012457B1 (fr) * | 2013-10-31 | 2016-01-01 | Arkema France | Additifs a base de diamides gras pour compositions reticulables sensibles aux nucleophiles |
| CA3118564A1 (en) * | 2018-11-06 | 2020-05-14 | Advanced Extrusion, Inc. | Compostable material for packaging |
| CN116751573B (zh) * | 2023-08-21 | 2023-11-14 | 大庆科讯油田技术服务有限公司 | 一种石油纳米解堵剂及其制备方法 |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6360956A (ja) | 1986-08-30 | 1988-03-17 | Nippon Oil & Fats Co Ltd | 脂肪酸ビスアミドの製造法 |
| JP2006176747A (ja) | 2004-05-11 | 2006-07-06 | Kao Corp | 生分解性樹脂組成物 |
| JP2007130895A (ja) | 2005-11-10 | 2007-05-31 | Kao Corp | 生分解性樹脂成形品の製造法。 |
| JP2007130893A (ja) | 2005-11-10 | 2007-05-31 | Kao Corp | 生分解性樹脂成形品の製造法。 |
| JP2007152760A (ja) | 2005-12-06 | 2007-06-21 | Kao Corp | ペレットの製造法 |
| JP2008031376A (ja) * | 2006-07-31 | 2008-02-14 | Mitsui Chemicals Inc | 乳酸系樹脂組成物、成形体、熱成形体および熱成形体の製造方法 |
| JP2008115372A (ja) | 2006-10-11 | 2008-05-22 | Kao Corp | 生分解性樹脂組成物 |
| JP2008174718A (ja) | 2006-12-19 | 2008-07-31 | Kao Corp | 生分解性樹脂組成物 |
| WO2009004769A1 (ja) * | 2007-06-29 | 2009-01-08 | Unitika Ltd. | 結晶性ポリ乳酸樹脂組成物およびそれからなる成形体 |
| JP2009024081A (ja) * | 2007-07-19 | 2009-02-05 | Unitika Ltd | 結晶性ポリ乳酸樹脂組成物およびそれからなる成形体 |
| JP2009185244A (ja) * | 2008-02-08 | 2009-08-20 | Unitika Ltd | 樹脂組成物およびそれを成形してなる成形体 |
| JP2010001338A (ja) * | 2008-06-19 | 2010-01-07 | Kao Corp | ポリ乳酸樹脂組成物 |
| WO2010010961A1 (en) | 2008-07-22 | 2010-01-28 | Kao Corporation | Biodegradable resin composition |
| JP2010031203A (ja) * | 2008-07-31 | 2010-02-12 | Kao Corp | ポリ乳酸樹脂組成物の製造方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1746136B1 (en) * | 2004-05-11 | 2015-04-22 | Kao Corporation | Biodegradable resin composition |
| JP5661997B2 (ja) * | 2006-02-14 | 2015-01-28 | 日本電気株式会社 | ポリ乳酸系樹脂組成物及び成形体 |
| KR101346751B1 (ko) | 2006-10-11 | 2013-12-31 | 가오 가부시키가이샤 | 생분해성 수지조성물 |
-
2010
- 2010-03-16 CN CN2010800196267A patent/CN102414273B/zh active Active
- 2010-03-16 US US13/256,974 patent/US8455579B2/en active Active
- 2010-03-16 JP JP2010058890A patent/JP5627255B2/ja active Active
- 2010-03-16 EP EP10753506.4A patent/EP2410018B1/en not_active Not-in-force
- 2010-03-16 WO PCT/JP2010/054402 patent/WO2010107014A1/ja active Application Filing
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6360956A (ja) | 1986-08-30 | 1988-03-17 | Nippon Oil & Fats Co Ltd | 脂肪酸ビスアミドの製造法 |
| JP2006176747A (ja) | 2004-05-11 | 2006-07-06 | Kao Corp | 生分解性樹脂組成物 |
| JP2007130895A (ja) | 2005-11-10 | 2007-05-31 | Kao Corp | 生分解性樹脂成形品の製造法。 |
| JP2007130893A (ja) | 2005-11-10 | 2007-05-31 | Kao Corp | 生分解性樹脂成形品の製造法。 |
| JP2007152760A (ja) | 2005-12-06 | 2007-06-21 | Kao Corp | ペレットの製造法 |
| JP2008031376A (ja) * | 2006-07-31 | 2008-02-14 | Mitsui Chemicals Inc | 乳酸系樹脂組成物、成形体、熱成形体および熱成形体の製造方法 |
| JP2008115372A (ja) | 2006-10-11 | 2008-05-22 | Kao Corp | 生分解性樹脂組成物 |
| JP2008174718A (ja) | 2006-12-19 | 2008-07-31 | Kao Corp | 生分解性樹脂組成物 |
| WO2009004769A1 (ja) * | 2007-06-29 | 2009-01-08 | Unitika Ltd. | 結晶性ポリ乳酸樹脂組成物およびそれからなる成形体 |
| JP2009024081A (ja) * | 2007-07-19 | 2009-02-05 | Unitika Ltd | 結晶性ポリ乳酸樹脂組成物およびそれからなる成形体 |
| JP2009185244A (ja) * | 2008-02-08 | 2009-08-20 | Unitika Ltd | 樹脂組成物およびそれを成形してなる成形体 |
| JP2010001338A (ja) * | 2008-06-19 | 2010-01-07 | Kao Corp | ポリ乳酸樹脂組成物 |
| WO2010010961A1 (en) | 2008-07-22 | 2010-01-28 | Kao Corporation | Biodegradable resin composition |
| JP2010031203A (ja) * | 2008-07-31 | 2010-02-12 | Kao Corp | ポリ乳酸樹脂組成物の製造方法 |
Non-Patent Citations (2)
| Title |
|---|
| "Poriorefin-toh Gosei-jushi-sei Shokuhin Youki Houso-toh ni Kansuru Jishukijun,Revised Third Edition", June 2004, article "Eisei Shikenho", pages: 12 - 13 |
| See also references of EP2410018A4 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012071481A (ja) * | 2010-09-28 | 2012-04-12 | Panasonic Corp | 成形品の製造方法、及び成形品 |
| US20140378595A1 (en) * | 2013-06-19 | 2014-12-25 | Lotte Chemical Corporation | Polylactic acid stereocomplex resin composition having improved crystallization rate and method for molding the same |
| US9085689B2 (en) * | 2013-06-19 | 2015-07-21 | Lotte Chemical Corporation | Polylactic acid stereocomplex resin composition having improved crystallization rate and method for molding the same |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102414273A (zh) | 2012-04-11 |
| EP2410018A4 (en) | 2015-03-11 |
| CN102414273B (zh) | 2013-08-07 |
| JP5627255B2 (ja) | 2014-11-19 |
| US20120016065A1 (en) | 2012-01-19 |
| EP2410018A1 (en) | 2012-01-25 |
| JP2010242068A (ja) | 2010-10-28 |
| EP2410018B1 (en) | 2015-11-11 |
| US8455579B2 (en) | 2013-06-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5627255B2 (ja) | 生分解性樹脂組成物の結晶化促進方法 | |
| JP5794731B2 (ja) | 樹脂組成物 | |
| JP4130695B2 (ja) | 生分解性樹脂組成物 | |
| JP4112568B2 (ja) | 生分解性樹脂組成物 | |
| JP5654501B2 (ja) | ポリ乳酸樹脂組成物の製造方法 | |
| JP5107873B2 (ja) | ポリ乳酸樹脂組成物 | |
| JP4130696B2 (ja) | 生分解性樹脂組成物 | |
| JP5264314B2 (ja) | ポリ乳酸樹脂組成物 | |
| JP4871891B2 (ja) | 生分解性樹脂組成物 | |
| JP4611214B2 (ja) | 生分解性樹脂組成物 | |
| JP2009270087A (ja) | ポリ乳酸樹脂組成物 | |
| JP5143374B2 (ja) | 生分解性樹脂組成物 | |
| EP2264103B1 (en) | Polylactic acid resin composite | |
| JP5107793B2 (ja) | ポリ乳酸樹脂射出成形体の製造方法 | |
| JP5107792B2 (ja) | ポリ乳酸樹脂射出成形体の製造方法 | |
| JP2012188657A (ja) | ポリ乳酸樹脂組成物 | |
| JP2010037438A (ja) | ポリ乳酸樹脂組成物 | |
| JP2007262422A (ja) | 生分解性樹脂組成物 | |
| JP5273847B2 (ja) | ポリ乳酸樹脂組成物 | |
| JP5225550B2 (ja) | ポリ乳酸樹脂組成物 | |
| JP2011116954A (ja) | ポリ乳酸系樹脂組成物および成形体 | |
| JP5525830B2 (ja) | ポリ乳酸樹脂組成物 | |
| JP2010037437A (ja) | ポリ乳酸樹脂組成物 | |
| JP5261006B2 (ja) | ポリ乳酸樹脂組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080019626.7 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10753506 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13256974 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010753506 Country of ref document: EP |