WO2010100936A1 - 吸水性樹脂の製造方法 - Google Patents
吸水性樹脂の製造方法 Download PDFInfo
- Publication number
- WO2010100936A1 WO2010100936A1 PCT/JP2010/001521 JP2010001521W WO2010100936A1 WO 2010100936 A1 WO2010100936 A1 WO 2010100936A1 JP 2010001521 W JP2010001521 W JP 2010001521W WO 2010100936 A1 WO2010100936 A1 WO 2010100936A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- water
- mixer
- absorbing resin
- absorbent resin
- particulate
- Prior art date
Links
- 239000011347 resin Substances 0.000 title claims abstract description 502
- 229920005989 resin Polymers 0.000 title claims abstract description 502
- 238000000034 method Methods 0.000 title claims abstract description 147
- 230000008569 process Effects 0.000 title abstract description 27
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 379
- 239000000203 mixture Substances 0.000 claims abstract description 125
- 239000003431 cross linking reagent Substances 0.000 claims abstract description 119
- 238000010438 heat treatment Methods 0.000 claims abstract description 42
- 239000000463 material Substances 0.000 claims abstract description 18
- 239000002250 absorbent Substances 0.000 claims description 253
- 239000002245 particle Substances 0.000 claims description 189
- 238000002156 mixing Methods 0.000 claims description 172
- 239000007788 liquid Substances 0.000 claims description 115
- 230000002745 absorbent Effects 0.000 claims description 99
- 238000004519 manufacturing process Methods 0.000 claims description 79
- 238000003756 stirring Methods 0.000 claims description 64
- 238000004132 cross linking Methods 0.000 claims description 63
- 238000010521 absorption reaction Methods 0.000 claims description 60
- 230000035699 permeability Effects 0.000 claims description 40
- 238000005192 partition Methods 0.000 claims description 37
- 150000003839 salts Chemical class 0.000 claims description 24
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 16
- 239000010419 fine particle Substances 0.000 claims description 11
- 229910052751 metal Inorganic materials 0.000 claims description 9
- 239000002184 metal Substances 0.000 claims description 9
- 238000010924 continuous production Methods 0.000 claims description 6
- 229920000768 polyamine Polymers 0.000 claims description 6
- 229920000058 polyacrylate Polymers 0.000 abstract 1
- 230000000704 physical effect Effects 0.000 description 77
- 239000000178 monomer Substances 0.000 description 58
- 238000006116 polymerization reaction Methods 0.000 description 55
- 239000000843 powder Substances 0.000 description 55
- 239000007864 aqueous solution Substances 0.000 description 49
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 48
- 239000000654 additive Substances 0.000 description 48
- 238000001035 drying Methods 0.000 description 45
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 42
- 230000000996 additive effect Effects 0.000 description 42
- 239000011230 binding agent Substances 0.000 description 39
- 229920000642 polymer Polymers 0.000 description 35
- DIZPMCHEQGEION-UHFFFAOYSA-H aluminium sulfate (anhydrous) Chemical compound [Al+3].[Al+3].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O DIZPMCHEQGEION-UHFFFAOYSA-H 0.000 description 30
- 239000011780 sodium chloride Substances 0.000 description 29
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 26
- 239000000017 hydrogel Substances 0.000 description 26
- 150000001875 compounds Chemical class 0.000 description 24
- 229910052782 aluminium Inorganic materials 0.000 description 23
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 23
- 239000000377 silicon dioxide Substances 0.000 description 23
- 239000000499 gel Substances 0.000 description 21
- -1 hydroxyacrylamide compound Chemical class 0.000 description 21
- 239000007787 solid Substances 0.000 description 20
- 230000000052 comparative effect Effects 0.000 description 19
- 230000000694 effects Effects 0.000 description 19
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 18
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 17
- WERYXYBDKMZEQL-UHFFFAOYSA-N butane-1,4-diol Chemical compound OCCCCO WERYXYBDKMZEQL-UHFFFAOYSA-N 0.000 description 16
- 239000006096 absorbing agent Substances 0.000 description 15
- 239000003505 polymerization initiator Substances 0.000 description 15
- 239000000243 solution Substances 0.000 description 15
- 239000006185 dispersion Substances 0.000 description 14
- 238000006297 dehydration reaction Methods 0.000 description 13
- 239000003795 chemical substances by application Substances 0.000 description 10
- 239000011521 glass Substances 0.000 description 10
- 229920001223 polyethylene glycol Polymers 0.000 description 10
- 238000010298 pulverizing process Methods 0.000 description 10
- 239000012756 surface treatment agent Substances 0.000 description 10
- 238000004381 surface treatment Methods 0.000 description 9
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 8
- 238000009826 distribution Methods 0.000 description 8
- 239000003960 organic solvent Substances 0.000 description 8
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 7
- 239000002202 Polyethylene glycol Substances 0.000 description 7
- 229940048053 acrylate Drugs 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- 238000005516 engineering process Methods 0.000 description 7
- 238000010528 free radical solution polymerization reaction Methods 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 150000005846 sugar alcohols Polymers 0.000 description 7
- 239000004094 surface-active agent Substances 0.000 description 7
- CYDQOEWLBCCFJZ-UHFFFAOYSA-N 4-(4-fluorophenyl)oxane-4-carboxylic acid Chemical compound C=1C=C(F)C=CC=1C1(C(=O)O)CCOCC1 CYDQOEWLBCCFJZ-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- 238000012545 processing Methods 0.000 description 6
- 239000001540 sodium lactate Substances 0.000 description 6
- 229940005581 sodium lactate Drugs 0.000 description 6
- 235000011088 sodium lactate Nutrition 0.000 description 6
- 238000005507 spraying Methods 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 5
- 239000011324 bead Substances 0.000 description 5
- 239000000835 fiber Substances 0.000 description 5
- 235000010746 mayonnaise Nutrition 0.000 description 5
- 239000008268 mayonnaise Substances 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- 239000003973 paint Substances 0.000 description 5
- 230000000379 polymerizing effect Effects 0.000 description 5
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 4
- IZXIZTKNFFYFOF-UHFFFAOYSA-N 2-Oxazolidone Chemical class O=C1NCCO1 IZXIZTKNFFYFOF-UHFFFAOYSA-N 0.000 description 4
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- 206010021639 Incontinence Diseases 0.000 description 4
- 229920002125 Sokalan® Polymers 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- 238000009835 boiling Methods 0.000 description 4
- 238000004040 coloring Methods 0.000 description 4
- 229920006037 cross link polymer Polymers 0.000 description 4
- XXMIOPMDWAUFGU-UHFFFAOYSA-N hexane-1,6-diol Chemical compound OCCCCCCO XXMIOPMDWAUFGU-UHFFFAOYSA-N 0.000 description 4
- 239000004570 mortar (masonry) Substances 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- 239000004584 polyacrylic acid Substances 0.000 description 4
- 230000002441 reversible effect Effects 0.000 description 4
- 239000007921 spray Substances 0.000 description 4
- 238000010557 suspension polymerization reaction Methods 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- LCPVQAHEFVXVKT-UHFFFAOYSA-N 2-(2,4-difluorophenoxy)pyridin-3-amine Chemical compound NC1=CC=CN=C1OC1=CC=C(F)C=C1F LCPVQAHEFVXVKT-UHFFFAOYSA-N 0.000 description 3
- 239000004593 Epoxy Substances 0.000 description 3
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 3
- 229910052783 alkali metal Inorganic materials 0.000 description 3
- 239000003242 anti bacterial agent Substances 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 239000002738 chelating agent Substances 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 230000018044 dehydration Effects 0.000 description 3
- 239000002781 deodorant agent Substances 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 238000007602 hot air drying Methods 0.000 description 3
- 230000002706 hydrostatic effect Effects 0.000 description 3
- 230000001771 impaired effect Effects 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 230000001678 irradiating effect Effects 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 238000000691 measurement method Methods 0.000 description 3
- 235000013372 meat Nutrition 0.000 description 3
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 3
- 239000002504 physiological saline solution Substances 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- CHQMHPLRPQMAMX-UHFFFAOYSA-L sodium persulfate Substances [Na+].[Na+].[O-]S(=O)(=O)OOS([O-])(=O)=O CHQMHPLRPQMAMX-UHFFFAOYSA-L 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 230000003068 static effect Effects 0.000 description 3
- 230000008961 swelling Effects 0.000 description 3
- 238000010998 test method Methods 0.000 description 3
- 210000002700 urine Anatomy 0.000 description 3
- DNIAPMSPPWPWGF-VKHMYHEASA-N (+)-propylene glycol Chemical compound C[C@H](O)CO DNIAPMSPPWPWGF-VKHMYHEASA-N 0.000 description 2
- ORTVZLZNOYNASJ-UPHRSURJSA-N (z)-but-2-ene-1,4-diol Chemical compound OC\C=C/CO ORTVZLZNOYNASJ-UPHRSURJSA-N 0.000 description 2
- YPFDHNVEDLHUCE-UHFFFAOYSA-N 1,3-propanediol Substances OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 description 2
- UWFRVQVNYNPBEF-UHFFFAOYSA-N 1-(2,4-dimethylphenyl)propan-1-one Chemical compound CCC(=O)C1=CC=C(C)C=C1C UWFRVQVNYNPBEF-UHFFFAOYSA-N 0.000 description 2
- 125000003504 2-oxazolinyl group Chemical class O1C(=NCC1)* 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 229920000742 Cotton Polymers 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical group C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 239000002211 L-ascorbic acid Substances 0.000 description 2
- 235000000069 L-ascorbic acid Nutrition 0.000 description 2
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 2
- ALQSHHUCVQOPAS-UHFFFAOYSA-N Pentane-1,5-diol Chemical compound OCCCCCO ALQSHHUCVQOPAS-UHFFFAOYSA-N 0.000 description 2
- 239000004743 Polypropylene Substances 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- 238000010793 Steam injection (oil industry) Methods 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 238000005273 aeration Methods 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 229960005070 ascorbic acid Drugs 0.000 description 2
- 239000012752 auxiliary agent Substances 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 239000002981 blocking agent Substances 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 230000006866 deterioration Effects 0.000 description 2
- 125000004386 diacrylate group Chemical group 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- SZXQTJUDPRGNJN-UHFFFAOYSA-N dipropylene glycol Chemical compound OCCCOCCCO SZXQTJUDPRGNJN-UHFFFAOYSA-N 0.000 description 2
- JBKVHLHDHHXQEQ-UHFFFAOYSA-N epsilon-caprolactam Chemical compound O=C1CCCCCN1 JBKVHLHDHHXQEQ-UHFFFAOYSA-N 0.000 description 2
- 235000019441 ethanol Nutrition 0.000 description 2
- 239000012632 extractable Substances 0.000 description 2
- 239000002657 fibrous material Substances 0.000 description 2
- 238000009775 high-speed stirring Methods 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 239000011261 inert gas Substances 0.000 description 2
- 238000007603 infrared drying Methods 0.000 description 2
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 238000006386 neutralization reaction Methods 0.000 description 2
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 description 2
- 150000002978 peroxides Chemical class 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- 229920001451 polypropylene glycol Polymers 0.000 description 2
- 229920000166 polytrimethylene carbonate Polymers 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 238000007717 redox polymerization reaction Methods 0.000 description 2
- 238000013341 scale-up Methods 0.000 description 2
- 239000002689 soil Substances 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 229910001220 stainless steel Inorganic materials 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000008400 supply water Substances 0.000 description 2
- 238000005979 thermal decomposition reaction Methods 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- PQUXFUBNSYCQAL-UHFFFAOYSA-N 1-(2,3-difluorophenyl)ethanone Chemical compound CC(=O)C1=CC=CC(F)=C1F PQUXFUBNSYCQAL-UHFFFAOYSA-N 0.000 description 1
- VILCJCGEZXAXTO-UHFFFAOYSA-N 2,2,2-tetramine Chemical compound NCCNCCNCCN VILCJCGEZXAXTO-UHFFFAOYSA-N 0.000 description 1
- JCTXKRPTIMZBJT-UHFFFAOYSA-N 2,2,4-trimethylpentane-1,3-diol Chemical compound CC(C)C(O)C(C)(C)CO JCTXKRPTIMZBJT-UHFFFAOYSA-N 0.000 description 1
- NOBUYVZUAMYLSQ-UHFFFAOYSA-N 2,3,4-trimethylpentane-1,3-diol Chemical compound CC(C)C(C)(O)C(C)CO NOBUYVZUAMYLSQ-UHFFFAOYSA-N 0.000 description 1
- JAHNSTQSQJOJLO-UHFFFAOYSA-N 2-(3-fluorophenyl)-1h-imidazole Chemical compound FC1=CC=CC(C=2NC=CN=2)=C1 JAHNSTQSQJOJLO-UHFFFAOYSA-N 0.000 description 1
- WFUGQJXVXHBTEM-UHFFFAOYSA-N 2-hydroperoxy-2-(2-hydroperoxybutan-2-ylperoxy)butane Chemical compound CCC(C)(OO)OOC(C)(CC)OO WFUGQJXVXHBTEM-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- QENRKQYUEGJNNZ-UHFFFAOYSA-N 2-methyl-1-(prop-2-enoylamino)propane-1-sulfonic acid Chemical compound CC(C)C(S(O)(=O)=O)NC(=O)C=C QENRKQYUEGJNNZ-UHFFFAOYSA-N 0.000 description 1
- CYUZOYPRAQASLN-UHFFFAOYSA-N 3-prop-2-enoyloxypropanoic acid Chemical compound OC(=O)CCOC(=O)C=C CYUZOYPRAQASLN-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 229910002012 Aerosil® Inorganic materials 0.000 description 1
- 229910002016 Aerosil® 200 Inorganic materials 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 240000007124 Brassica oleracea Species 0.000 description 1
- 235000003899 Brassica oleracea var acephala Nutrition 0.000 description 1
- 235000012905 Brassica oleracea var viridis Nutrition 0.000 description 1
- ASTTVEMLEHKCSP-UHFFFAOYSA-N CCC(CO)(CO)CO.OC(=O)CCOC(=O)C=C.OC(=O)CCOC(=O)C=C.OC(=O)CCOC(=O)C=C Chemical compound CCC(CO)(CO)CO.OC(=O)CCOC(=O)C=C.OC(=O)CCOC(=O)C=C.OC(=O)CCOC(=O)C=C ASTTVEMLEHKCSP-UHFFFAOYSA-N 0.000 description 1
- 229920003043 Cellulose fiber Polymers 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- RPNUMPOLZDHAAY-UHFFFAOYSA-N Diethylenetriamine Chemical compound NCCNCCN RPNUMPOLZDHAAY-UHFFFAOYSA-N 0.000 description 1
- BRLQWZUYTZBJKN-UHFFFAOYSA-N Epichlorohydrin Chemical compound ClCC1CO1 BRLQWZUYTZBJKN-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- CTKINSOISVBQLD-UHFFFAOYSA-N Glycidol Chemical compound OCC1CO1 CTKINSOISVBQLD-UHFFFAOYSA-N 0.000 description 1
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical class OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920002845 Poly(methacrylic acid) Polymers 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 229920002873 Polyethylenimine Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 229920001131 Pulp (paper) Polymers 0.000 description 1
- 229920000297 Rayon Polymers 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- OUUQCZGPVNCOIJ-UHFFFAOYSA-M Superoxide Chemical compound [O-][O] OUUQCZGPVNCOIJ-UHFFFAOYSA-M 0.000 description 1
- UWHCKJMYHZGTIT-UHFFFAOYSA-N Tetraethylene glycol, Natural products OCCOCCOCCOCCO UWHCKJMYHZGTIT-UHFFFAOYSA-N 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- QYKIQEUNHZKYBP-UHFFFAOYSA-N Vinyl ether Chemical class C=COC=C QYKIQEUNHZKYBP-UHFFFAOYSA-N 0.000 description 1
- 229920002978 Vinylon Polymers 0.000 description 1
- ORLQHILJRHBSAY-UHFFFAOYSA-N [1-(hydroxymethyl)cyclohexyl]methanol Chemical compound OCC1(CO)CCCCC1 ORLQHILJRHBSAY-UHFFFAOYSA-N 0.000 description 1
- XDODWINGEHBYRT-UHFFFAOYSA-N [2-(hydroxymethyl)cyclohexyl]methanol Chemical compound OCC1CCCCC1CO XDODWINGEHBYRT-UHFFFAOYSA-N 0.000 description 1
- LXEKPEMOWBOYRF-UHFFFAOYSA-N [2-[(1-azaniumyl-1-imino-2-methylpropan-2-yl)diazenyl]-2-methylpropanimidoyl]azanium;dichloride Chemical compound Cl.Cl.NC(=N)C(C)(C)N=NC(C)(C)C(N)=N LXEKPEMOWBOYRF-UHFFFAOYSA-N 0.000 description 1
- 150000008062 acetophenones Chemical class 0.000 description 1
- 239000002390 adhesive tape Substances 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- LFVGISIMTYGQHF-UHFFFAOYSA-N ammonium dihydrogen phosphate Chemical compound [NH4+].OP(O)([O-])=O LFVGISIMTYGQHF-UHFFFAOYSA-N 0.000 description 1
- 229910000387 ammonium dihydrogen phosphate Inorganic materials 0.000 description 1
- ROOXNKNUYICQNP-UHFFFAOYSA-N ammonium peroxydisulfate Substances [NH4+].[NH4+].[O-]S(=O)(=O)OOS([O-])(=O)=O ROOXNKNUYICQNP-UHFFFAOYSA-N 0.000 description 1
- VAZSKTXWXKYQJF-UHFFFAOYSA-N ammonium persulfate Chemical compound [NH4+].[NH4+].[O-]S(=O)OOS([O-])=O VAZSKTXWXKYQJF-UHFFFAOYSA-N 0.000 description 1
- 229910001870 ammonium persulfate Inorganic materials 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 125000000751 azo group Chemical group [*]N=N[*] 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- ISAOCJYIOMOJEB-UHFFFAOYSA-N benzoin Chemical class C=1C=CC=CC=1C(O)C(=O)C1=CC=CC=C1 ISAOCJYIOMOJEB-UHFFFAOYSA-N 0.000 description 1
- 150000008366 benzophenones Chemical class 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- LWMFAFLIWMPZSX-UHFFFAOYSA-N bis[2-(4,5-dihydro-1h-imidazol-2-yl)propan-2-yl]diazene Chemical compound N=1CCNC=1C(C)(C)N=NC(C)(C)C1=NCCN1 LWMFAFLIWMPZSX-UHFFFAOYSA-N 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- LLSDKQJKOVVTOJ-UHFFFAOYSA-L calcium chloride dihydrate Chemical compound O.O.[Cl-].[Cl-].[Ca+2] LLSDKQJKOVVTOJ-UHFFFAOYSA-L 0.000 description 1
- 229940052299 calcium chloride dihydrate Drugs 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 238000003889 chemical engineering Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000000701 coagulant Substances 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- LDHQCZJRKDOVOX-NSCUHMNNSA-N crotonic acid Chemical compound C\C=C\C(O)=O LDHQCZJRKDOVOX-NSCUHMNNSA-N 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 239000012024 dehydrating agents Substances 0.000 description 1
- LSXWFXONGKSEMY-UHFFFAOYSA-N di-tert-butyl peroxide Chemical compound CC(C)(C)OOC(C)(C)C LSXWFXONGKSEMY-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- 238000007599 discharging Methods 0.000 description 1
- GKIPXFAANLTWBM-UHFFFAOYSA-N epibromohydrin Chemical compound BrCC1CO1 GKIPXFAANLTWBM-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000007863 gel particle Substances 0.000 description 1
- 239000003349 gelling agent Substances 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 238000003898 horticulture Methods 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- YAMHXTCMCPHKLN-UHFFFAOYSA-N imidazolidin-2-one Chemical compound O=C1NCCN1 YAMHXTCMCPHKLN-UHFFFAOYSA-N 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 230000001788 irregular Effects 0.000 description 1
- 229940035429 isobutyl alcohol Drugs 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 229940050906 magnesium chloride hexahydrate Drugs 0.000 description 1
- DHRRIBDTHFBPNG-UHFFFAOYSA-L magnesium dichloride hexahydrate Chemical compound O.O.O.O.O.O.[Mg+2].[Cl-].[Cl-] DHRRIBDTHFBPNG-UHFFFAOYSA-L 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 125000005395 methacrylic acid group Chemical group 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- LVHBHZANLOWSRM-UHFFFAOYSA-N methylenebutanedioic acid Natural products OC(=O)CC(=C)C(O)=O LVHBHZANLOWSRM-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 235000019837 monoammonium phosphate Nutrition 0.000 description 1
- LSHROXHEILXKHM-UHFFFAOYSA-N n'-[2-[2-[2-(2-aminoethylamino)ethylamino]ethylamino]ethyl]ethane-1,2-diamine Chemical compound NCCNCCNCCNCCNCCN LSHROXHEILXKHM-UHFFFAOYSA-N 0.000 description 1
- RQAKESSLMFZVMC-UHFFFAOYSA-N n-ethenylacetamide Chemical compound CC(=O)NC=C RQAKESSLMFZVMC-UHFFFAOYSA-N 0.000 description 1
- 239000004745 nonwoven fabric Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000002921 oxetanes Chemical class 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- WXZMFSXDPGVJKK-UHFFFAOYSA-N pentaerythritol Chemical compound OCC(CO)(CO)CO WXZMFSXDPGVJKK-UHFFFAOYSA-N 0.000 description 1
- 229960003330 pentetic acid Drugs 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- USHAGKDGDHPEEY-UHFFFAOYSA-L potassium persulfate Chemical compound [K+].[K+].[O-]S(=O)(=O)OOS([O-])(=O)=O USHAGKDGDHPEEY-UHFFFAOYSA-L 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 239000002964 rayon Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000004064 recycling Methods 0.000 description 1
- 238000007670 refining Methods 0.000 description 1
- 238000002407 reforming Methods 0.000 description 1
- 239000005871 repellent Substances 0.000 description 1
- 239000011342 resin composition Substances 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 239000004576 sand Substances 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 238000010334 sieve classification Methods 0.000 description 1
- 238000004513 sizing Methods 0.000 description 1
- 229940047670 sodium acrylate Drugs 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000003516 soil conditioner Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- FAGUFWYHJQFNRV-UHFFFAOYSA-N tetraethylenepentamine Chemical compound NCCNCCNCCNCCN FAGUFWYHJQFNRV-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- LDHQCZJRKDOVOX-UHFFFAOYSA-N trans-crotonic acid Natural products CC=CC(O)=O LDHQCZJRKDOVOX-UHFFFAOYSA-N 0.000 description 1
- ZIBGPFATKBEMQZ-UHFFFAOYSA-N triethylene glycol Chemical compound OCCOCCOCCO ZIBGPFATKBEMQZ-UHFFFAOYSA-N 0.000 description 1
- SOBHUZYZLFQYFK-UHFFFAOYSA-K trisodium;hydroxy-[[phosphonatomethyl(phosphonomethyl)amino]methyl]phosphinate Chemical compound [Na+].[Na+].[Na+].OP(O)(=O)CN(CP(O)([O-])=O)CP([O-])([O-])=O SOBHUZYZLFQYFK-UHFFFAOYSA-K 0.000 description 1
- NLVXSWCKKBEXTG-UHFFFAOYSA-N vinylsulfonic acid Chemical compound OS(=O)(=O)C=C NLVXSWCKKBEXTG-UHFFFAOYSA-N 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 229920003176 water-insoluble polymer Polymers 0.000 description 1
- 229920003169 water-soluble polymer Polymers 0.000 description 1
- 210000002268 wool Anatomy 0.000 description 1
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
- C08J3/245—Differential crosslinking of one polymer with one crosslinking type, e.g. surface crosslinking
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01F—MIXING, e.g. DISSOLVING, EMULSIFYING OR DISPERSING
- B01F27/00—Mixers with rotary stirring devices in fixed receptacles; Kneaders
- B01F27/05—Stirrers
- B01F27/11—Stirrers characterised by the configuration of the stirrers
- B01F27/115—Stirrers characterised by the configuration of the stirrers comprising discs or disc-like elements essentially perpendicular to the stirrer shaft axis
- B01F27/1152—Stirrers characterised by the configuration of the stirrers comprising discs or disc-like elements essentially perpendicular to the stirrer shaft axis with separate elements other than discs fixed on the discs, e.g. vanes fixed on the discs
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01F—MIXING, e.g. DISSOLVING, EMULSIFYING OR DISPERSING
- B01F27/00—Mixers with rotary stirring devices in fixed receptacles; Kneaders
- B01F27/80—Mixers with rotary stirring devices in fixed receptacles; Kneaders with stirrers rotating about a substantially vertical axis
- B01F27/87—Mixers with rotary stirring devices in fixed receptacles; Kneaders with stirrers rotating about a substantially vertical axis the receptacle being divided into superimposed compartments
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F20/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride, ester, amide, imide or nitrile thereof
- C08F20/02—Monocarboxylic acids having less than ten carbon atoms, Derivatives thereof
- C08F20/04—Acids, Metal salts or ammonium salts thereof
- C08F20/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/04—Acids; Metal salts or ammonium salts thereof
- C08F220/06—Acrylic acid; Methacrylic acid; Metal salts or ammonium salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2333/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical, or of salts, anhydrides, esters, amides, imides, or nitriles thereof; Derivatives of such polymers
- C08J2333/02—Homopolymers or copolymers of acids; Metal or ammonium salts thereof
Definitions
- the present invention relates to a method for producing a water-absorbent resin that is excellent in physical properties and can be obtained efficiently while ensuring high productivity at a low cost. More specifically, the present invention relates to a method for reforming and producing a water absorbent resin in which a mixing device having a specific structure is used and / or mixed under specific conditions when mixing a particulate water absorbent resin and an additive.
- Water-absorbing resins are widely used in various applications such as paper diapers, sanitary napkins, sanitary materials such as adult incontinence products, and soil water retention agents because of their ability to absorb large amounts of aqueous solution, several times to several hundred times their own weight. Are produced and consumed in large quantities.
- a water-absorbing resin also referred to as a highly water-absorbing resin or a water-absorbing polymer
- JIS Japanese Industrial Standard
- the water-absorbing resin is produced by drying a water-containing gel-like polymer obtained by polymerizing an aqueous solution containing a hydrophilic monomer and a crosslinking agent, and performing a surface treatment.
- a hydrophilic monomer for example, poly (meth) acrylic acid (salt) is well known as the hydrophilic monomer.
- the hydrogel polymer obtained by polymerizing this monomer is obtained as a mass or an aggregate of hydrogel particles and is usually about 1 to 10 mm using a pulverizer such as a kneader or meat chopper. Coarsely pulverized (coarse crushing) to a particle size. The coarsely crushed (coarse crushed) hydrogel is dried to a solid content of about 95% by weight.
- pulverization is performed by a pulverizer so that the value of the weight average particle diameter is 150 ⁇ m or more and 850 ⁇ m or less, and a particulate water-absorbing resin is obtained. At this time, particles outside the target particle size (particle size) range are also included. Accordingly, the pulverized product after drying is sieved with a classifier to prepare a particulate water-absorbing resin having a size within the target particle size range.
- the particulate water-absorbing resin used for hygiene products those having a particle diameter in the range of 150 ⁇ m or more and less than 850 ⁇ m are preferably used.
- the particulate water-absorbing resin is subjected to a surface treatment process to obtain physical properties such as water absorption capacity and liquid permeability under pressure that are desirable for sanitary agents (sanitary products) and the like.
- the surface treatment step is usually a step of providing a highly crosslinked layer in the vicinity of the surface of the particulate water-absorbing resin by reacting the particulate water-absorbing resin with a surface crosslinking agent or a polymerizable monomer by light or heat (surface Cross-linking step) or additives (surface treatment agents) that impart functionality such as liquid permeability improvers, deodorants, anti-coloring agents, antibacterial agents, anti-blocking agents, etc. to the particulate water-absorbing resin
- the process (addition process) of providing an additive layer in the surface vicinity of particulate water-absorbing resin is said.
- the surface cross-linking agent contains a cross-linking agent having a functional group capable of reacting with a carboxyl group or a polymerizable monomer.
- the surface cross-linking techniques that have been studied so far include, for example, a technique regarding the combined use of a surface cross-linking agent (Patent Document 1), a technique regarding an apparatus for mixing a water-absorbing resin and a surface cross-linking agent (Patent Document 2), Technology for a heating device for reacting a resin and a surface cross-linking agent (Patent Document 3), Technology for temperature increase control of a heating temperature for reacting a water absorbent resin and a surface cross-linking agent (Patent Document 4), The technique (patent document 5) etc. about the surface cross-linking process of the water-absorbing resin having a high water content can be mentioned.
- a technique (patent documents 6 and 7) that modifies a water-absorbent resin by applying heat without using a surface cross-linking agent, unlike ordinary surface cross-linking is also known.
- oxazoline compounds Patent Document 8
- vinyl ether compounds Patent Document 9
- epoxy compounds Patent Document 10
- oxetane compounds Patent Document 11
- polyhydric alcohol compounds Patent.
- Document 12 polyamide polyamine-epihalo adduct
- Patent Document 15 hydroxyacrylamide compound
- Patent Document 16 oxazolidinone compound
- Patent Document 17 2-oxo Tetrahydro-1,3-oxazolidine compounds
- Patent Document 19 alkylene carbonate compounds
- Patent Documents 20 and 21 a technique for polymerizing monomers to crosslink the surface
- Patent Document 22 a technique for radical crosslinking with persulfate
- Patent Document 21 and 22 describe drying by heating in superheated steam atmosphere.
- Patent Documents 24 and 25 a water-soluble cation such as an aluminum salt
- Patent Document 26 an alkali
- Patent Document 27 a technique that uses a specific mixer as a mixer for the surface cross-linking agent is also known.
- Patent Document 29 a technique for performing surface crosslinking twice
- Patent Document 30 a technique for using a plurality of heat treatment apparatuses
- Patent Document 31 a technique for heating a water-absorbing resin before surface crosslinking in advance
- Patent Document 31 a technique for heating a water-absorbing resin before surface crosslinking in advance
- the speed at which the particulate water-absorbing resin moves toward the discharge port in the mixing tank can be changed depending on the direction of the stirring blades in the dispersion process and the mixing process of the particulate water-absorbing resin.
- Patent Document 35 the ratio of the particulate water-absorbing resin moving toward the discharge port and the particulate water-absorbing resin moving toward the inlet in the opposite direction is adjusted by a stirring blade in the mixer. Thus, it is described that the mixing property is improved. Furthermore, since the particulate water-absorbing resin to which the surface treatment agent is adhered aggregates and deteriorates the mixing property, the particulate water-absorbing resin has sufficient kinetic energy so that the particulate water-absorbing resin does not aggregate in the mixing process. This is described in Patent Document 35.
- Patent Document 35 as a mixer used for mixing a particulate water-absorbing resin and an additive, a Patterson-Kelly mixer, a DRAIS turbulent mixer, a Lodige mixer, a Luberg ( Ruberg) mixers, screw mixers, pan mixers, fluidized bed mixers, MTI mixers, Shugi mixers and the like are described.
- Patent Document 2 discloses a technique of using a water-repellent substrate on the inner wall of a mixer in order to prevent the particulate water-absorbing resin from adhering and to improve the mixing property.
- a mixer used for mixing the particulate water-absorbing resin and the surface cross-linking agent Henschel mixer (Mitsui Miike Machinery Co., Ltd.), New Speed Mixer (Okada Seiko Co., Ltd.), Heavy Duty Matrix (Nara) Machine Manufacturing Co., Ltd.), Turbulizer, Sand Turpo (both manufactured by Hosokawa Micron Corporation) and the like are described.
- a horizontal mixer that is, a mixer in which the rotating shaft of the stirring blade is in the horizontal direction, the height is 50% or less of the rotating circumferential diameter.
- Patent Document 36 describes that by installing a weir near the outlet or between stirring blades, the residence time in the mixing tank can be controlled, and a short pass can be prevented.
- particulate water-absorbing resin having a particle size smaller than the target particle size range generated in the production process of a series of water-absorbing resins is called fine powder.
- fine powder having a particle size smaller than 150 ⁇ m is used in absorbent articles such as diapers. Since clogging causes a decrease in liquid permeability, it is not suitable for use in hygiene agents (sanitary products).
- a binder such as an aqueous solution or a fine particle aqueous dispersion is preferable as the fine powder binder from the viewpoints of efficiency, safety, production cost, and the like.
- a binder such as an aqueous solution or a fine particle aqueous dispersion
- a high-speed stirring type mixer for example, a turbulizer (manufactured by Hosokawa Micron), a redige mixer (manufactured by Lodige), and a mortar mixer (manufactured by West Japan Testing Machine Co., Ltd.) is used for mixing fine powder and binder.
- the method which bind
- Patent Documents 8 to 19 many of the above-mentioned surface cross-linking agents (Patent Documents 8 to 19) and their combined use (Patent Document 1), their mixing devices (Patent Documents 2 and 28), surface cross-linking aids (Patent Documents 24 to 27), and their heating
- Patent Documents 3, 4, 29 to 34 many technologies such as treatment methods
- these surface crosslinking technologies alone provide the absorption capacity of the water-absorbent resin under pressure and the passing through of the user. It has been difficult to meet increasing demands for physical properties such as liquidity.
- the above method shows a certain effect in small-scale production at the laboratory level and batch-type (batch-type) production, but in the continuous production on the industrial scale (for example, 1 t or more per unit time), it is as small as the small scale. In some cases, the effect was not shown.
- the additive is a surface cross-linking agent
- the surface cross-linked layer will form only a part of the surface of the particulate water-absorbent resin.
- the physical properties such as properties are greatly impaired.
- the use amount of other surface treatment agents is increased in order to exert the effect, and physical properties are impaired.
- the fine powder has a large surface area relative to the weight, and is particularly difficult to mix uniformly.
- the binding force of the obtained binder is weak and easily collapses. Therefore, in order to spread the binder throughout the fine powder, the amount of water added as a binder is large in the conventional technology, the moisture content of the binder exceeds 50% by weight, and the energy required for drying is high. It was a problem to grow.
- the method of binding using only water vapor as in Patent Document 40 can reduce the moisture content of the binder, but in the disclosed method, the fine powder is sufficiently bound for a long time. They are mixed and difficult to operate continuously.
- the present invention has been made in view of the above-described conventional problems, and the object thereof is to efficiently obtain a surface-crosslinked water-absorbing resin having excellent physical properties while ensuring high productivity at low cost.
- Another object of the present invention is to provide a method for producing a water-absorbent resin, particularly a method for mixing a water-absorbent resin and an additive.
- a surface cross-linking agent and water are added to a particulate water absorbent resin in a mixer.
- a method for producing surface-crosslinked water-absorbent resin particles comprising sequentially a step and a step of reacting the water-absorbent resin mixture taken out of the mixer with a surface-crosslinking agent by heating or irradiation with active energy rays in a reactor.
- a part or all of the water added in the mixer is added with water vapor.
- the physical properties of the water-absorbent resin after surface cross-linking for example, water absorption capacity under pressure (AAP), without using a change of the surface cross-linking agent or a new auxiliary agent, Liquid permeability (SFC) and the like can be improved.
- AAP water absorption capacity under pressure
- SFC Liquid permeability
- the physical properties are reduced during the scale-up in the production process.
- the physical properties are hardly deteriorated even during the continuous production or the scale-up.
- the method for producing a water absorbent resin of the present invention efficiently obtains a particulate water absorbent resin having excellent physical properties by uniformly mixing the particulate water absorbent resin and an additive while ensuring high productivity. be able to.
- FIG. 3 is a cross-sectional view showing a schematic configuration of a mixer used in Examples 1 to 6, 8 to 11, and 14 to 37 and Comparative Example 3. It is sectional drawing which shows schematic structure of the mixer used in Example 12 and 13 and the comparative example 6. FIG. It is sectional drawing which shows schematic structure of the horizontal mixer used by the comparative examples 2, 5, and 7.
- Embodiments of the present invention will be described below.
- the definition of terms is [1], and a typical method for producing a water-absorbing resin is shown in [2] below, but the step of surface treatment, which is a characteristic part of the present invention, is shown in (2-6).
- the step of performing surface cross-linking is described in (2-6-1) below, and the step of adding an additive for imparting functionality such as a liquid permeability improver is performed in (2-6-2).
- the steps of binding are shown in (2-7) below.
- Each of these steps may be carried out batchwise or continuously, but industrially, it is preferable that the steps are connected and continuously produced as a whole. In particular, the mixing of the particulate water-absorbing resin and the additive is continuously performed.
- Water absorbent resin The “water-absorbing resin” in the present invention means a water-swellable water-insoluble polymer gelling agent.
- Water swellability means that the CRC (water absorption capacity under no pressure) specified in ERT441.2-02 is usually 5 [g / g] or more, and “water-insoluble” means Ext (water soluble content) specified by ERT470.2-02 is usually 0 to 50% by weight (particularly 20% by weight or less).
- the water-absorbent resin can be appropriately designed according to its use and is not particularly limited, but may be a hydrophilic cross-linked polymer obtained by cross-linking an unsaturated monomer having a carboxyl group. preferable. Further, the total amount (100% by weight) is not limited to the form of a polymer, and may contain additives and the like within a range that maintains the above performance.
- polyacrylic acid (salt) water-absorbing resin means a water-absorbing resin mainly composed of acrylic acid and / or a salt thereof (hereinafter referred to as acrylic acid (salt)) as a repeating unit. To do.
- the total monomer (excluding the crosslinking agent) used in the polymerization refers to a polymer usually containing 30 to 100 mol%, preferably 50 to 100 mol% of acrylic acid (salt), preferably Means a water-absorbing resin (water-swellable / water-insoluble crosslinking agent polymer) containing 70 to 100 mol%, more preferably 90 to 100 mol%, particularly preferably substantially 100 mol%.
- EDANA European Disposables and Nonwovens Associations
- ERT is an abbreviation for a method for measuring water-absorbent resin (EDANA Recommended Test Method), which is a European standard (almost world standard). is there.
- EDANA Recommended Test Method European Standard (almost world standard).
- CRC is an abbreviation for Centrifugation Retention Capacity (centrifuge retention capacity) and means water absorption capacity without pressure (hereinafter also referred to as “water absorption capacity”). Specifically, the water absorption capacity (unit: [g / g]) after 30 minutes of free swelling with respect to a 0.9 wt% sodium chloride aqueous solution and further drained with a centrifuge.
- AAP is an abbreviation for Absorption against Pressure, which means water absorption capacity under pressure. Specifically, the water absorption capacity (unit: [g / g]) after swelling under a load of 2.06 kPa for 1 hour with respect to a 0.9% by weight sodium chloride aqueous solution is 1 hour in the present invention. It was set as the water absorption capacity (unit: [g / g]) under a load of 4.83 kPa.
- Extractables is an abbreviation for Extractables and means a water-soluble component (water-soluble component amount). Specifically, it is a value (unit:% by weight) measured by pH titration after stirring 1 g of water absorbent resin for 16 hours with respect to 200 g of 0.9 wt% sodium chloride aqueous solution.
- liquid permeability The flow of the liquid flowing between the particles of the swollen gel under load or no load is called “liquid permeability”.
- Typical measurement methods for this “liquid permeability” include SFC (Saline Flow Conductivity) and GBP (Gel Bed Permeability).
- SFC Seline Flow Inducibility
- GBP refers to the permeability of 0.69 wt% physiological saline to the water-absorbent resin under load or free expansion. It is measured according to the GBP test method described in International Publication No. 2005/016393 pamphlet.
- X to Y indicating a range means “X or more and Y or less”, and “(meth) acryl” used for (meth) acrylic acid and the like. Means acrylic or methacrylic.
- t (ton) which is a unit of weight means “Metric ton”, and unless otherwise noted, “ppm” means “ppm by weight” or “ppm by mass”. "Means.
- acrylic acid alone, or a combination of acrylic acid and a monomer other than acrylic acid, or only from a monomer other than acrylic acid a water absorbent resin is appropriately used.
- a water absorbent resin is appropriately used.
- acrylic acid and / or a salt thereof is preferable.
- acrylic acid (salt) composed of 1 to 50 mol% of acrylic acid and 50 to 99 mol% of an alkali metal salt of acrylic acid is most preferably used. Is done.
- the acid group is a monovalent salt, preferably an alkali metal salt or an ammonium salt, more preferably an alkali metal salt, particularly preferably.
- Sodium salt is used.
- the acid group is neutralized before or after polymerization in the range of 0 to 100 mol%, preferably 20 to 100 mol%, more preferably 50 to 99 mol%, and still more preferably 60 to 90 mol%.
- crosslinking agent examples include N, N′-methylenebis (meth) acrylamide, (poly) ethylene glycol di (meth) acrylate, (poly) propylene glycol di (meth) acrylate, ( Polyoxyethylene) trimethylolpropane tri (meth) acrylate, trimethylolpropane di (meth) acrylate, polyethylene glycol di ( ⁇ -acryloyloxypropionate), trimethylolpropane tri ( ⁇ -acryloyloxypropionate), poly Compounds having at least two polymerizable double bonds in the molecule such as (meth) allyloxyalkane; polyglycidyl ether (ethylene glycol diglycidyl ether), polyol (ethylene glycol, polyethylene glycol, glycerin) One or more compounds capable of forming a covalent bond by reacting with the carboxyl groups of sorbitol) and the like.
- crosslinking agent it is preferable to essentially use a compound having at least two polymerizable double bonds in the molecule in consideration of the absorption characteristics of the resulting water-absorbent resin.
- the crosslinking agent is used in the range of 0.0001 to 5 mol%, preferably 0.005 to 2 mol%, based on the physical properties, with respect to the monomer.
- (C) Concentration These monomers are usually polymerized in an aqueous solution, and the monomer concentration is usually 10 to 90% by weight, preferably 20 to 80% by weight, more preferably 30 to 70% by weight, particularly The range is preferably 30 to 60% by weight.
- the aqueous solution contains a surfactant, polyacrylic acid (salt) or a crosslinked product thereof (water absorbent resin), a polymer compound such as starch, polyvinyl alcohol, various chelating agents, various additives, and the like. You may use together in 0 to 30weight% or less with respect to 0 weight%.
- the aqueous solution includes a dispersion exceeding the saturation concentration, but is preferably polymerized at a saturation concentration or less.
- the water-absorbent resin of the present invention is produced by crosslinking and polymerizing the unsaturated monomer to obtain a hydrogel polymer.
- Polymerization is usually spray polymerization, drop polymerization, aqueous solution polymerization or reverse phase suspension polymerization because of performance and ease of control of polymerization, and in particular, aqueous solution that has been difficult to control the particle size because of its irregularly shaped particles. Polymerization and further continuous aqueous polymerization are performed.
- Reverse phase suspension polymerization is a polymerization method in which an aqueous monomer solution is suspended in a hydrophobic organic solvent.
- aqueous solution polymerization is a method of polymerizing an aqueous monomer solution without using a dispersion solvent.
- aqueous solution polymerization or reverse phase suspension polymerization preferably aqueous solution polymerization, more preferably continuous aqueous solution polymerization, particularly preferably continuous belt polymerization or continuous kneader polymerization are applied.
- the solid content is 0.1% by weight or more, preferably 1 to 40% by weight before and after the polymerization. %, More preferably 2 to 30% by weight, particularly preferably 3 to 20% by weight.
- the rise in solid content is appropriately determined by the temperature at the time of polymerization (for example, polymerization at the boiling point), the air flow and the shape (particle size and sheet thickness of the polymer gel) and the like.
- polymerizations can be carried out in an air atmosphere, but are carried out in an inert gas atmosphere such as nitrogen or argon, for example, at an oxygen concentration of 1% by volume or less.
- the monomer component is preferably used for polymerization after the dissolved oxygen is sufficiently substituted with an inert gas and the oxygen concentration becomes less than 1 [mg / L] (ppm).
- the effect is exhibited by controlling the particle size in the production and pulverization of a real scale, especially a huge scale, rather than a laboratory scale. Therefore, in particular, the unsaturated monomer aqueous solution is polymerized in one line to make the water-absorbing resin 1 [t / hr] or more, further 2 [t / hr] or more, and further 5 [t / hr] or more.
- the present invention can be suitably employed in continuous polymerization and continuous pulverization produced or pulverized on a huge scale of 10 [t / hr] or more. The upper limit of production is appropriately determined, for example, 100 [t / hr].
- continuous kneader polymerization for example, US Pat. Nos. 6,987,151 and 6,710,141, US Patent Application Publication No. 2008/0080300
- continuous belt polymerization for example, US Pat. Nos. 4,893,999 and 6,241,928,.
- US Patent Application Publication No. 2005/215734 for example, US Pat. Nos. 6,987,151 and 6,710,141, US Patent Application Publication No. 2008/0080300
- Examples of the polymerization method for aqueous solution polymerization include a static polymerization method in which a monomer aqueous solution is polymerized in a static state, and a stirring polymerization method in which polymerization is performed in a stirring device.
- a static polymerization method a method using an endless belt is preferable.
- a stirring polymerization method a uniaxial stirrer can be used, but a multi-stirrer stirrer such as a kneader is preferably used.
- the polymerization method in the present invention there is a continuous polymerization method at a high monomer concentration using an endless belt as described in JP-A No. 2005-307195.
- Such continuous belt polymerization or continuous kneader polymerization is also suitably applied to the present invention.
- high temperature start for example, the temperature of the monomer is 30 ° C. or higher, more preferably 35 ° C. or higher, further preferably 40 ° C. or higher, particularly preferably 50 ° C. or higher, and the upper limit is the boiling point.
- High monomer concentration for example, 30% by weight or more, more preferably 35% by weight or more, further preferably 40% by weight or more, particularly preferably 45% by weight or more, and the upper limit is a saturation concentration). Is a preferred example.
- the polymerization initiator used by this invention is suitably selected by the form of superposition
- a polymerization initiator a water-soluble polymerization initiator, preferably a photodecomposition polymerization initiator, a thermal decomposition polymerization initiator, a redox polymerization initiator, and the like can be exemplified.
- photodegradable polymerization initiator examples include benzoin derivatives, benzyl derivatives, acetophenone derivatives, benzophenone derivatives, azo compounds, and the like.
- thermal decomposition type polymerization initiator examples include persulfates such as sodium persulfate, potassium persulfate and ammonium persulfate; peroxides such as hydrogen peroxide, t-butyl peroxide and methyl ethyl ketone peroxide; azonitrile compounds , Azoamidine compounds, cyclic azoamidine compounds, azoamide compounds, alkylazo compounds, 2,2′-azobis (2-amidinopropane) dihydrochloride, 2,2′-azobis [2- (2-imidazolin-2-yl) propane] dihydro And azo compounds such as chloride.
- persulfates such as sodium persulfate, potassium persulfate and ammonium persulfate
- peroxides such as hydrogen peroxide, t-butyl peroxide and methyl ethyl ketone peroxide
- azonitrile compounds Azoamidine compounds, cyclic azoamidine
- Examples of the redox polymerization initiator include a system in which a reducing compound such as L-ascorbic acid or sodium bisulfite is used in combination with the persulfate or peroxide, and the both are combined.
- the amount of the polymerization initiator is 0.0001 to 1 mol%, preferably 0.001 to 0.5 mol%, based on the monomer.
- the hydrogel polymer before drying is finely granulated during or after polymerization. Preferably it is.
- the hydrogel polymer (hydrous cross-linked polymer) obtained by aqueous solution polymerization (especially when continuous belt polymerization is used) in the present invention for example, in the form of a lump or sheet, is pulverized by a pulverizer to form a particulate hydrogel. And then dried.
- a particulate water-containing gel is obtained by polymerization, but the particulate water-containing gel after polymerization may be dried as it is, and if necessary, further pulverization or The particle size may be adjusted by binding.
- the weight average particle size (D50) determined by standard sieve classification is preferably in the range of 0.5 to 10 mm, and preferably in the range of 1 to 5 mm. Is more preferably 1 to 3 mm, particularly preferably 1 to 2 mm.
- the water-containing gel polymer preferably the particulate water-containing gel polymer, is dried until the solid content becomes pulverizable.
- the form of the hydrophilic cross-linked polymer (hydrogel polymer) used in the drying step is a coarsely crushed hydrogel, agglomerate thereof, and a sheet hydrogel by a kneader, meat chopper, cutter, or the like. You may put the crushing process and the crushing process of an aggregate suitably in this drying process. As such a technique, for example, US Pat. No. 6,187,902 is adopted.
- a drying method in the present invention various methods can be adopted so as to achieve a desired moisture content, and the drying with hot drying, hot air drying, reduced pressure drying, infrared drying, microwave drying, and hydrophobic organic solvent can be used. Examples include dehydration by boiling and high-humidity drying using high-temperature steam.
- a conduction heat transfer dryer eg, a radiation heat transfer dryer (eg, infrared drying), a hot air heat transfer dryer, a dielectric heating dryer (eg, microwave drying). ), And combinations thereof.
- a hot air heat transfer type dryer is preferably used from the viewpoint of drying efficiency.
- the hot air drying method there are a method of drying in a stationary state, a method of drying in a stirring state, a method of drying in a vibrating state, a method of drying in a fluidized state, a method of drying in an air current, and the like.
- hot air drying using fluidized bed drying or stationary drying (further aeration band drying) and continuous stationary drying (continuous aeration band drying) is used from the viewpoint of efficiency.
- the drying temperature is usually 60 to 250 ° C., preferably 100 to 250 ° C., more preferably 100 to 220 ° C., further preferably 120 to 200 ° C., particularly preferably 150 to 190 ° C. (especially hot air temperature). Is called.
- the drying time depends on the surface area of the polymer, the moisture content, the type of the dryer, and the air volume, and is selected so as to achieve the desired moisture content.
- the drying time may be appropriately selected within the range of 1 minute to 5 hours and 1 minute to 1 hour.
- the solid content of the hydrophilic crosslinked polymer is preferably increased to 70 to 95% by weight, more preferably 80 to 95% by weight, still more preferably 85 to 95% by weight, and particularly preferably 90 to 95% by weight. .
- the weight average particle diameter is preferably 100 to 1000 ⁇ m, more preferably 200 to 800 ⁇ m, and particularly preferably 300 to 600 ⁇ m.
- those having a particle size in the range of 150 ⁇ m or more and less than 850 ⁇ m are preferably 80% by weight or more, and more preferably 90% by weight or more.
- This particulate water-absorbing resin is sent to “(2-6) surface treatment step” described later.
- the fine powder having a particle size of 150 ⁇ m or less generated in this step deteriorates the physical properties of the water-absorbent resin, and is also a problem in safety and hygiene, so it is classified and removed.
- the step of classifying and removing fine powder may be performed during or after the heating and drying step, as will be described later.
- the fine powder is appropriately collected and subjected to a process such as being formed again into a granular shape or recovered in a monomer aqueous solution.
- the surface cross-linking step is a characteristic part of the present invention. That is, the method for producing a water absorbent resin of the present invention includes a step of adding a surface cross-linking agent and water to a particulate water absorbent resin in a mixer, and the water absorbent resin mixture taken out of the mixer is heated or activated in a reactor.
- a method for producing surface-crosslinked water-absorbing resin particles which sequentially includes a step of reacting with a surface-crosslinking agent by irradiation with energy rays, wherein a part or all of water addition in a mixer is added with water vapor.
- various organic crosslinking agents or inorganic crosslinking agents can be exemplified.
- known crosslinking agents exemplified in the above-mentioned documents 1-34.
- An agent can be used.
- the surface cross-linking agent can be used without particular limitation as long as it can cross-link the water-absorbent resin.
- a crosslinking technique Patent Document 22
- Patent Document 22 can also be used or included as a crosslinking agent.
- a carboxyl group of the water-absorbent resin particularly neutralized or unneutralized carboxyl group of polyacrylic acid
- a reactive surface cross-linking agent can be used.
- examples of the surface crosslinking agent include compounds having a hydroxyl group, an amino group, or a derivative thereof.
- polyhydric alcohol compounds, epoxy compounds, polyvalent amine compounds or their condensates with haloepoxy compounds, oxazoline compounds, mono-, di- or polyoxazolidinone compounds, polyvalent metal salts, alkylene carbonate compounds and the like can be mentioned.
- the cross-linking agent in which the surface cross-linking agent can dehydrate with carboxyl groups, especially A dehydration reactive crosslinking agent selected from polyhydric alcohol compounds (Patent Document 12), oxazolidinone compounds (Patent Documents 16 to 18), alkylene carbonate (Patent Document 19), and oxetane (Patent Document 11) can be preferably used.
- Such a dehydration-reactive crosslinking agent forms a covalent bond through a dehydration reaction with the carboxyl group of the water-absorbent resin, it does not substantially react when water vapor is added (water addition). That is, after that, the water-absorbing resin mixture taken out from the mixer is heated or irradiated with active energy in the reactor to perform a dehydration reaction, thereby providing an excellent water-absorbing resin.
- examples of the surface cross-linking agent capable of dehydrating with a carboxyl group include compounds exemplified in US Pat. Nos. 6,228,930, 6071976, 6254990, and the like.
- mono, di, tri, tetra or polyethylene glycol monopropylene glycol, 1,3-propanediol, dipropylene glycol, 2,3,4-trimethyl-1,3-pentanediol, polypropylene glycol, glycerin, polyglycerin , 2-butene-1,4-diol, 1,4-butanediol, 1,3-butanediol, 1,5-pentanediol, 1,6-hexanediol, 1,2-cyclohexanedimethanol, etc.
- Alcohol compounds Epoxy compounds such as ethylene glycol diglycidyl ether and glycidol; Multivalents such as ethylenediamine, diethylenetriamine, triethylenetetramine, tetraethylenepentamine, pentaethylenehexamine, polyethyleneimine, and polyamidepolyamine Haloepoxy compounds such as epichlorohydrin, epibromohydrin, ⁇ -methylepichlorohydrin; condensates of the above polyvalent amine compounds and the above haloepoxy compounds; oxazolidinone compounds such as 2-oxazolidinone; ethylene carbonate, etc. An alkylene carbonate compound; an oxetane compound; and a cyclic urea compound such as 2-imidazolidinone.
- Multivalents such as ethylenediamine, diethylenetriamine, triethylenetetramine, tetraethylenepentamine, pentaethylenehexamine, polyethyleneimine, and polyamidepolyamine
- polyamine and polyvalent metal salt can be used as a surface cross-linking agent capable of ionic cross-linking with the carboxyl group of the particulate water-absorbing resin, and can also be used as a liquid permeability improver described later.
- These surface cross-linkings may be performed once or may be performed a plurality of times on the particulate water-absorbing resin by using the same or different surface cross-linking agents.
- dehydration reactive cross-linking agents not disclosed in the above-mentioned Patent Document 33, in particular, polyhydric alcohols, (mono or polyvalent) oxazolidinones, (mono or polyvalent) alkylene carbonates, (mono or polyvalent) Oxetane is exemplified, and these dehydration-reactive cross-linking agents are AAP not disclosed in Patent Document 33 because the cross-linking agent or the ring-opened product of the hydroxyl group or amino group undergoes dehydration reaction and cross-linking with the carboxy group of the water absorbent resin. And SFC can be improved.
- these dehydration-reactive crosslinking agents are preferably added to the particulate water-absorbing resin as a solution, particularly an aqueous solution, thereby improving AAP and SFC not disclosed in Patent Document 33. be able to.
- a crosslinking agent particularly a dehydration-reactive crosslinking agent
- a dehydration reaction by heat treatment added to the water-absorbent resin as a solution, particularly as an aqueous solution, so that AAP or SFC can be obtained.
- the dehydration reaction hardly (or at all) proceeds in the presence of water, the progress of the dehydration reaction can be easily confirmed by a decrease in water content of the water-absorbent resin particles before and after surface cross-linking (synonyms; increase in solid content).
- the water content of the water-absorbing resin particles after the surface cross-linking is reduced, in particular 3% by weight or less, 2% by weight or less, 1% by weight or less, 0. This can be confirmed by a decrease to 5 wt% or less.
- the use of water vapor and the increase in the weight of the water-absorbent resin with water vapor are necessary for eliminating unevenness due to the use of an aqueous solution in surface crosslinking.
- the amount of the surface cross-linking agent used depends on the compounds used and combinations thereof, but is preferably in the range of 0.001 to 10 parts by weight, preferably 0.01 to 5 parts per 100 parts by weight of the particulate water-absorbing resin. More preferably within the range of parts by weight.
- water is used together with the surface cross-linking agent.
- the amount of water used is preferably in the range of 0.5 to 20 parts by weight, more preferably 0.5 to 10 parts by weight with respect to 100 parts by weight of the particulate water-absorbing resin.
- a hydrophilic organic solvent can be used in addition to water.
- the amount of the hydrophilic organic solvent used is in the range of more than 0 parts by weight and 10 parts by weight or less, preferably more than 0 parts by weight and 5 parts by weight or less with respect to 100 parts by weight of the particulate water-absorbing resin. It is a range.
- hydrophilic organic solvent for example, lower alcohols such as methyl alcohol, ethyl alcohol, n-propyl alcohol, isopropyl alcohol, n-butyl alcohol, isobutyl alcohol, and t-butyl alcohol; ketones such as acetone; Ethers such as dioxane, tetrahydrofuran, methoxy (poly) ethylene glycol; amides such as ⁇ -caprolactam and N, N-dimethylformamide; sulfoxides such as dimethyl sulfoxide; ethylene glycol, diethylene glycol, propylene glycol, triethylene glycol, tetra Ethylene glycol, polyethylene glycol, 1,3-propanediol, dipropylene glycol, 2,2,4-trimethyl-1,3-pentanediol, polypropylene Glycol, glycerin, polyglycerin, 2-butene-1,4-diol, 1,
- the polyhydric alcohols may be used as a crosslinking agent by appropriately selecting the temperature and time, or may be used as a solvent without reacting at all, or a plurality of polyhydric alcohols having these properties respectively. You may use together. These solutions including water are used at 0 to 100 ° C., preferably 5 to 50 ° C. in consideration of the mixing property, although depending on the freezing point and boiling point.
- the range does not hinder the effect of the present invention, for example, within the range of more than 0% by weight and not more than 10% by weight, preferably more than 0% by weight and more than 5% by weight.
- the water-insoluble fine particle powder and the surfactant may coexist in the following range, more preferably in the range of more than 0% by weight and 1% by weight or less.
- Preferred surfactants and methods for using them are exemplified in US Pat. No. 7,381,775, for example.
- the particulate water-absorbing resin supplied to the surface cross-linking step The particulate water-absorbing resin that has undergone the above “(2-5) step of controlling the particle size” is preferably temporarily stored in a heated or warmed storage facility. And is quantitatively supplied to the surface cross-linking step by a feeder.
- a preferred feeder is a circle feeder or a screw feeder.
- the temperature of the particulate water-absorbing resin supplied to the mixer is preferably lower than the temperature of water vapor, more preferably 10 to 100 ° C., further preferably 30 to 90 ° C., particularly 50 to 80 ° C. It is preferable.
- the temperature of the particulate water-absorbing resin supplied to the surface cross-linking step is preferably 30 to 150 ° C., more preferably 40 to 120 ° C., even more preferably when it is charged into the mixer. It is 30 to 90 ° C, particularly preferably 40 to 80 ° C, most preferably 50 to 70 ° C.
- the water-absorbent resin is likely to adhere and agglomerate, and if the temperature of the particulate water-absorbent resin to be added is lower than 30 ° C., the water-absorbent resin is likely to adhere and may be clogged due to the growth of the adhering matter during long time operation.
- the temperature is higher than 150 ° C., the particulate water-absorbing resin may be deteriorated, and the mixing property of the particulate water-absorbing resin may be deteriorated depending on the additive.
- the water is efficiently absorbed by the particulate water-absorbing resin by setting the temperature of the particulate water-absorbing resin lower than the water vapor.
- the temperature of the particulate water-absorbing resin is excessively lower than the temperature of the water vapor, for example, 110 ° C. or higher, and further 150 ° C. or higher, the aggregation of the particulate water-absorbing resin occurs, and the subsequent heat treatment, That is, the water-absorbent resin mixture taken out from the mixer may be disadvantageous for heating in the reactor to form a dehydration reaction.
- the temperature of the particulate water-absorbing resin can be measured by taking out the particulate water-absorbing resin put into the mixer and quickly bringing it into contact with a general contact thermometer.
- the first production method of the present invention is to supply water vapor into the mixer.
- saturated steam having a vapor pressure higher than 1 atm is supplied to the mixer through a steam line.
- a device for supplying a gas such as a blower becomes unnecessary, and water vapor can be efficiently supplied.
- the preferable vapor pressure (gauge pressure) of the supplied water vapor is 0.01 to 1 MPa, more preferably 0.05 to 0.9 MPa, and still more preferably 0.1 to 0.8 MPa. If it is less than 0.01 MPa, the mixing property is deteriorated. On the other hand, when the pressure is higher than 1 MPa, high-pressure steam is opened in the mixer, which is dangerous. Moreover, the temperature of water vapor
- the vapor pressure of saturated water vapor can be read as the temperature of saturated water vapor by using a table described in page 400 of Chemical Engineering Handbook 6th edition (issued by Maruzen).
- the preferred water vapor temperature is about 100-180 ° C.
- the dew point in the mixer is preferably 60 to 100 ° C, more preferably 70 to 100 ° C, and particularly preferably 80 to 100 ° C. When the dew point in the mixer is lower than 60 ° C., the effect of the present invention is reduced.
- (D) Mixer In the present invention, after adding a surface cross-linking agent to the particulate water-absorbing resin and mixing in the mixer, heating or active energy rays are used to react the surface cross-linking agent with the particulate water-absorbing resin. Irradiate.
- the reactor for reacting the surface cross-linking agent and the particulate water-absorbing resin may be of the same type as the apparatus for mixing the surface cross-linking agent and the particulate water-absorbing resin, or may be of another type. Good. However, since it is necessary for the mixer to quickly mix the particulate water-absorbing resin and the surface cross-linking agent, it is difficult to achieve a device structure that is heated or irradiated with active energy for a sufficient time to promote the cross-linking reaction. . Accordingly, the mixer and the reactor are preferably different types.
- the average residence time of the particulate water-absorbing resin in the mixer is preferably 1 second or more and less than 5 minutes, more preferably 1 second or more and less than 1 minute.
- the average residence time of the water-absorbing resin mixture in the connected reactor is appropriately determined depending on the reactivity of the cross-linking agent, and is, for example, 1 minute or longer, usually 6 minutes to 10 hours, further 10 minutes to 2 hours. is there.
- the mixing device used when mixing the particulate water-absorbing resin and the surface cross-linking agent has a large mixing force in order to mix these substances uniformly and reliably.
- Examples of the mixing apparatus include a cylindrical mixer, a double wall conical mixer, a high-speed stirring mixer, a V-shaped mixer, a ribbon mixer, a screw mixer, a double-arm kneader, and a pulverizing kneader.
- Rotating mixers, airflow mixers, turbulators, batch-type Redige mixers, continuous-type Redige mixers, and the like are suitable.
- a more preferred mixer is a vertical mixer having a cylindrical mixing tank and a paddle rotating about a central axis.
- the vertical type means that the rotation axis is in the vertical direction (vertical direction)
- the horizontal type means that the rotation axis is in the horizontal direction.
- the water-absorbing resin that is moistened easily accumulates in the lower part of the mixing tank, and there is a possibility that large water-absorbing resin agglomerates may be formed or the water-absorbing resin adheres to the paddles, resulting in poor mixing. .
- a vertical mixing apparatus that satisfies the following conditions (i) to (iii) for mixing the particulate water-absorbing resin and an additive such as a surface cross-linking agent. .
- the stirring blade rotates at 300 to 3000 rpm.
- At least one of the rotating shafts of the stirring blades is in the vertical direction (vertical mixer).
- the mixing tank is divided into two or more chambers at the top and bottom by a partition having an opening degree of 5 to 70%.
- the above vertical mixing apparatus has been studied by the present inventors.
- a conventional vertical mixer that is, a mixer whose rotating shaft is in the vertical direction, a stirring blade is used.
- a stirring blade is used.
- the temperature of the particulate water-absorbing resin is set to 30 to 150 ° C. in advance, and the following conditions (i) to (iii) ), (I)
- the stirring blade rotates at 300 to 3000 rpm.
- At least one rotating shaft of the stirring blade is in the vertical direction.
- the mixing tank is divided into two or more chambers at the top and bottom by a partition having an opening degree of 5 to 70%.
- uniform mixing can be performed in the mixing of the particulate water-absorbing resin and the additive, and the resulting mixture has preferable physical properties depending on the purpose of the additive. Furthermore, it is also suitable for the production of a water-absorbing resin with a scale exceeding 1 t / hr. That is, in the above method, the mixing of the particulate water-absorbing resin and the additive has high mixing properties, high processing power, and stable operability.
- the preferable shape of the mixing tank in the vertical mixer is a drum type, and has a rotating shaft of a stirring blade in the center. Such a barrel portion of the mixing tank may be swollen or constricted to the extent that the present invention is not hindered.
- the particulate water-absorbing resin charged into the vertical mixer is discharged through two processes: (I) dispersion and (II) stirring and mixing.
- the particulate water-absorbing resin obtains a centrifugal force and is dispersed toward the side wall by the stirring blade and the air flow generated by the rotation of the stirring blade.
- the rotational speed of the vertical mixer is 300 to 3000 rpm, preferably 500 to 3000 rpm.
- the maximum rotating diameter of the stirring blade is usually about 0.1 to 1 m, and the diameter of the mixing tank is usually about 0.15 to 1.2 m. If the rotational speed is slower than 300 rpm, an air flow sufficient for dispersion cannot be obtained. If the rotational speed is faster than 3000 rpm, the water-absorbent resin may be damaged by collision with the stirring blades, resulting in a decrease in physical properties and an increase in fine powder.
- At least one of the rotating shafts of the stirring blade is in the vertical direction.
- the vertical direction may not be strict, and may be inclined to the extent that does not impair the operation of the mixer.
- the rotating shaft of the stirring blade can be 1 to 5, and is usually one.
- the particulate water-absorbing resin is charged into the mixing tank from the charging port provided on the upper surface of the mixing tank.
- a partition is installed to prevent the particulate water-absorbing resin from falling toward the discharge port before being sufficiently dispersed.
- the partition is preferably used under the inlet.
- the partition of the continuous mixing apparatus has an opening degree of 5 to 70%, preferably 10 to 50%, particularly preferably 10 to 30%.
- the degree of opening refers to the area (S1) of the interior surrounded by the inner wall of the casing, the structure surrounded by the inner wall of the casing, and the particulate water absorption in a plane (horizontal plane in the present invention) perpendicular to the rotation axis. It is a value obtained from the following mathematical formula with respect to the area (S2) of the hollow portion inside the structure (for example, the hollow inside the rotating shaft) where the functional resin cannot enter.
- the shape and location of the opening are determined as appropriate, and the number of openings may be one or more.
- the outer periphery of the rotating shaft, particularly the inner wall is 1 to 5 locations, more preferably 1 to 3 locations, Installed at.
- a suitable opening has a partition structure as described below. Such a partition is preferably formed by a plate having an area of S2, particularly a disk, having an opening with an area (S1-S2), and upper and lower partitions.
- the size of one opening is larger than the water-absorbent resin particles, and the area is preferably at least twice, more preferably at least 10 times, at least 100 times the cross-sectional area of the weight average particle diameter (D50). And / or the cross-sectional area of one opening is 1 cm 2 or more, further 5 cm 2 or more.
- the upper limit of the aperture is properly determined by the processing amount 6000 cm 2 or less, usually about 2 1000 cm.
- partitions There are one or more partitions, and two or more partitions may be installed on the top and bottom, and the mixing tank is divided into two or more rooms on the top and bottom.
- the volume of any room is preferably 10% or more of the volume of the mixing tank, and the division method can be appropriately selected within this range.
- the partition may be a fixed type attached to the side wall of the mixing tank, but a partition that rotates about the rotation axis of the stirring blade can be used.
- a partition that rotates about the rotation axis of the stirring blade can be used.
- a plate-like structure may be attached to the rotating shaft, or a part of the rotating shaft may be thickened and used as the partition.
- such a partition may be integrated with the stirring blade. That is, instead of attaching the stirring blade to the shaft, the stirring blade can be attached to the partition.
- the additive to be mixed by the continuous mixing apparatus has properties of liquid, dispersion, or solid fine particles.
- the mixer used in the present invention is particularly effective in terms of preventing adhesion, etc., but even if the additive is a solid fine particle, good mixing properties are denied. It is not a thing, and since it mixes rapidly, the damage given to a particulate water-absorbing resin can be reduced.
- the amount of the additive depends on the purpose of use and properties, but is preferably 100 parts by weight or less, more preferably 70 parts by weight or less, and more preferably 0.001 parts by weight or more with respect to 100 parts by weight of the particulate water-absorbing resin. Preferably there is.
- the addition method of the additive is dripping or spraying, and preferably spraying in order to uniformly mix with the water absorbent resin.
- a preferable addition position of the additive is in the middle of the “(I) dispersion” process or after the completion of the “(I) dispersion” process.
- the particulate water-absorbing resin to which the above additives are attached enters the above-mentioned “(II) stirring and mixing” process.
- the area where “(I) dispersion” and “(II) stirring and mixing” are performed is divided by a large partition, thereby preventing a short pass.
- a plurality of the partitions may be installed.
- a partition is provided between the stirring blade and the other stirring blade.
- the particulate water-absorbing resin to which the additive has adhered is in a state of forming a staying layer while rotating along the side wall due to centrifugal force. As the stirring blade passes through the staying layer, the particulate water-absorbing resin and the additive are vigorously mixed, and uniform mixing is achieved. In this process, when the fine powder and the binder are mixed, the fine powders are further bound to each other to form a binder having a particle diameter of 150 ⁇ m or more.
- the particulate water-absorbing resin is retained on the side wall by centrifugal force, and in order to perform stirring, it is preferable to provide a discharge port on the rotating shaft side from the track drawn by the tip of at least one stirring blade. More preferably, in order to control the retention amount for the purpose of improving physical properties, a weir structure whose length and / or angle can be changed is provided at the discharge port.
- This weir structure has an inner wall of the casing in which the horizontal length ( ⁇ ) from the side wall (inner wall) to the rotation axis side is preferably the maximum radius of the mixing tank (the plane perpendicular to the rotation axis (horizontal plane in the present invention)).
- the maximum radius of the enclosed interior is 1 to 40%, and the angle ( ⁇ ) made with respect to the horizontal plane is preferably 10 to 80 °. If the weir angle exceeds 80 °, or the overhang length is less than 1% of the maximum radius of the mixing tank, the particulate water-absorbing resin may not form a staying layer and the mixing property may deteriorate. . On the other hand, if the weir angle is less than 10 °, or the overhang length exceeds 40% of the maximum radius of the mixing tank, the discharge performance may be deteriorated.
- the height of the mixing tank in the rotation axis direction may be low.
- the height of the mixing tank in the direction of the rotation axis is H, and the diameter of the maximum portion of the mixing tank (maximum diameter (maximum inside the plane surrounded by the inner wall of the casing in the plane perpendicular to the rotation axis (horizontal plane in the present invention)).
- the value of H / D is preferably 0.1 to 1.0, particularly preferably 0.1 to 0.5, where D is the diameter)).
- the maximum diameter of the mixing tank is preferably 0.15 to 1.2 m, and the height of the mixing tank is preferably 0.03 to 1 m. Therefore, although it is a mixer having a very compact shape, which has not been heretofore, the amount of the water-absorbing resin charged into the mixer is preferably 10 to 300 kg / hr, more preferably 10 to 300 kg / hr per mixing tank volume of the mixer. Is mixed with the particulate water-absorbing resin at a high processing force of 10 to 150 kg / hr. At this time, the processing power of one mixer is preferably 50 to 30000 kg / hr, although it depends on the size of the mixer.
- the inner wall is preferably heated or kept warm.
- the inner wall includes an inner surface of the casing, a shaft, a partition, and a stirring blade.
- steam or warm water inside a shaft or a partition is also preferable.
- the temperature of the inner wall is preferably 50 to 150 ° C. If it is lower than this range, it may adhere to the inner wall depending on the additive, and if it is higher than this range, the particulate water-absorbing resin may be deteriorated.
- the mixer according to the present invention is preferably coated with a material to which the inner wall is difficult to adhere.
- the inner wall can be coated using a material (base material) having a contact angle with water of 60 ° or more.
- a fluororesin can be used as such a material. Since the continuous mixing apparatus is compact, coating is easy. In particular, since the direction of the rotation axis can be shortened, it is advantageous that the area of the side wall that is heavily worn by friction with the particulate water-absorbent resin is small.
- water vapor may be introduced in mixing the particulate water-absorbing resin and the additive.
- water vapor By introducing water vapor, it may be possible to suppress adhesion of the particulate water-absorbing resin to the inner wall and control the permeability of the additive.
- the mixing of the fine powder and the binder is preferable because water vapor becomes water on the surface of the fine powder and has an effect of binding the fine powder.
- the water vapor supplied to the mixer is saturated water vapor and is preferably opened in the mixer.
- the gauge pressure of water vapor is 0.1 to 2.0 MPa, preferably 0.1 to 1.0 MPa, more preferably 0.1 to 0.5 MPa. If the gauge pressure is lower than 0.1 MPa, the effect of water vapor is not exhibited. If the gauge pressure is higher than 2.0 MPa, the particulate water-absorbing resin may be deteriorated.
- the supply amount of the water vapor is 1 to 100 kg / hr, preferably 1 to 50 kg / hr, more preferably 1 to 30 kg / hr with respect to 100 kg / hr of the particulate water absorbent resin. If the supply amount of the water vapor is less than 1 kg / hr with respect to the supply amount of the particulate water-absorbing resin 100 kg / hr, the effect of the water vapor is not exhibited, and if it is more than 100 kg / hr, the mixed state may be deteriorated.
- the water vapor absorbed by the particulate water-absorbent resin is only a part of the supplied water vapor.
- the mixer which is a feature of the present invention, is less suitable for adhesion when steam is introduced, and is therefore suitable for mixing with steam.
- the preferred rotation speed of the paddle in the mixer is 100 rpm or more and less than 5000 rpm, more preferably 300 rpm or more and less than 2000 rpm. If the rotational speed of the paddle is less than 100 rpm, large water-absorbent resin aggregates are likely to be formed in the mixing tank. Further, if the rotational speed of the paddle is 5000 rpm or more, the water absorbent resin may be crushed due to the collision between the paddle and the water absorbent resin.
- the residence time of the particulate water-absorbing resin in the mixer is preferably 1 second or more and less than 5 minutes. More preferably, it is 1 second or more and less than 1 minute. If the residence time is less than 1 second, sufficient mixing cannot be obtained, and physical properties after surface cross-linking, such as absorption capacity under pressure, may be deteriorated. On the other hand, if the residence time is 5 minutes or longer, the water-absorbent resin is damaged due to the collision between the paddle and the water-absorbent resin, and the physical properties after surface crosslinking may be deteriorated.
- an exhaust device is preferably provided from the mixer to the reactor inlet.
- the mixer preferably has an exhaust device. More preferably, an exhaust device is provided near the outlet of the water absorbent resin after mixing. This is to prevent excessive water vapor from staying. For this reason, it is preferable that the exhaust device is kept warm or heated.
- the pressure of the exhaust line is -0.01 to -1 kPa (gauge pressure), more preferably -0.05 to -0.5 kPa. If the exhaust pressure is less than -0.01 kPa, the water absorbent resin tends to form large agglomerates. If the exhaust pressure is higher than ⁇ 1 kPa, the water-absorbing resin may enter the exhaust line, which may cause a loss or reduce the capacity of the exhaust device.
- the temperature of the inner surface of the mixer (such as the inner wall and a stirring blade installed if necessary) is preferably lower than water vapor, more preferably 10 to 100 ° C., further preferably 30 to 90 ° C., particularly preferably 50 ⁇ 80 ° C lower.
- the temperature of the mixer is excessively lower than the temperature of the water vapor, for example, when the temperature is 110 ° C. or higher, and further 150 ° C. or higher, the water-absorbing resin particles are agglomerated, and the subsequent heat treatment, that is, mixing The water-absorbing resin mixture taken out from the machine is heated in the reactor, which is disadvantageous for forming a dehydration reaction.
- the temperature on the inner surface of the mixer is set low, water vapor is condensed on the inner surface of the mixer, and the particulate water-absorbing resin contacts the condensed inner surface, so that uniform water is supplied to the particulate water-absorbing resin. It is estimated that the addition is promoted. However, in the present embodiment, it does not matter whether water addition to the particulate water-absorbing resin is absorption of water vapor directly or absorption of condensed water of water vapor.
- FIG. 1 is a cross-sectional view illustrating a schematic configuration of an example of a vertical mixing apparatus according to the present embodiment
- FIG. 2 is a cross-sectional view illustrating a schematic configuration of another example of the vertical mixing apparatus according to the present embodiment
- FIG. 3 is a cross-sectional view showing a schematic configuration of still another example of the vertical mixing apparatus according to the present embodiment.
- the vertical mixing apparatus is provided in a mixing layer 10 with a rotating shaft 6 installed in the vertical direction for stirring, and the rotating shaft 6. And a plate-like partition 7 provided on the side wall 1 of the mixed layer 10 that divides the mixing tank 10 into two or more chambers.
- an inlet 2 for introducing water-absorbing resin particles and an additive inlet 4 for introducing a surface cross-linking agent are provided at the upper part of the mixed layer 10.
- a discharge port 3 for discharging the mixture of the particulate water-absorbing resin and the additive is provided at the lower part of the mixed layer 10, and the size of the discharge port 3 is changed by the weir 8. Can be made.
- the particulate water-absorbing resin supplied from the inlet 2 and the additive such as the surface cross-linking agent supplied from the additive inlet 4 are mixed in the mixed layer 10.
- the mixed layer 10 is divided into two chambers by the partition 7 in the vertical direction, the particulate water-absorbing resin is prevented from falling toward the discharge port before being sufficiently mixed. Then, a sufficiently mixed mixture of the particulate water-absorbing resin and the additive is discharged from the discharge port 3.
- the partition 7 is installed on the side wall 1, but as shown in FIGS. 2 and 3, the partition 7 is installed on the stirring shaft 6 and the rotating shaft of the stirring blade 5 is rotated.
- the structure which rotates to the center may be sufficient. 2 and 3, the stirring blade 5 is installed in the partition 7.
- the amount of water supplied to the mixer as water vapor and taken into the particulate water-absorbing resin is normally taken into 100% particulate water-absorbing resin. Good. That is, in the present invention, the amount of water supplied to the mixer as water vapor and taken into the particulate water-absorbent resin is the amount of water supplied to the mixer as liquid water from the increase in moisture in the particulate water-absorbent resin. It shall be subtracted.
- the amount of water taken into the particulate water absorbent resin as liquid water is preferably in the range of 0.5 to 20 parts by weight, more preferably 0.5 to 10 parts by weight with respect to 100 parts by weight of the particulate water absorbent resin. is there.
- the amount of water supplied as water vapor and taken into the particulate water-absorbing resin is preferably 0.1 to 10 parts by weight, more preferably 0.5 to 5 parts by weight with respect to 100 parts by weight of the particulate water-absorbing resin. Part range.
- the amount of water increased by incorporating water vapor is less than 0.1 parts by weight, the effect of the present invention is small.
- the amount exceeds 10 parts by weight there is a possibility that stable operation is difficult due to a large amount of aggregates of the particulate water-absorbing resin.
- the supplied water vapor is taken into the particulate water absorbent resin, it is preferably 1.1 to 5 times, more preferably 1.5 to 3 times the amount of water taken into the particulate water absorbent resin.
- water vapor must be supplied to the mixer. The amount of water vapor supplied to the mixer can be measured with a commercially available flow meter.
- the water added to the particulate water-absorbing resin is preferably a combination of liquid and water vapor.
- the liquid water may be water alone or may be added as a mixture of water and an organic solvent.
- the water added to the particulate water-absorbing resin is A crosslinker aqueous solution and water vapor are used in combination.
- 0.1 to 10 parts by weight of water is mixed with 100 parts by weight of the particulate water-absorbing resin, and the amount of water supplied as a liquid is 0 to 95% by weight of the total supply water, Is preferably 20 to 90% by weight, particularly 40 to 80% by weight.
- Patent Documents 20, 21, 33, and 34 a technique (Patent Documents 20, 21, 33, and 34) using water vapor for a heating reaction in the heat treatment of a water absorbent resin mixture after mixing a monomer and a surface cross-linking agent has also been proposed.
- a technique of using water vapor for granulating a water-absorbent resin Japanese Patent Laid-Open No. 2005-054151 has also been proposed.
- a water-absorbing resin having high physical properties is achieved by using water vapor, preferably water vapor and liquid water (particularly, an aqueous solution of the crosslinking agent) when mixing the surface crosslinking agent.
- (F) Step of reacting the surface cross-linking agent The water absorbent resin mixture after mixing the surface cross-linking agent is heated or irradiated with active energy in the reactor.
- the water-absorbent resin mixture is subjected to a heat treatment and, if necessary, a cooling treatment thereafter.
- the heating temperature is preferably in the range of 70 to 300 ° C, more preferably 120 to 250 ° C, still more preferably 150 to 250 ° C.
- the heating time is preferably 1 minute or longer, usually 6 minutes to 10 hours, more preferably 10 minutes to 2 hours.
- the heat treatment can be performed using a normal dryer or a heating furnace. Preferably, by using a dryer with a paddle, aggregate formation and heat unevenness can be prevented.
- a polymerizable or radical-reactive surface cross-linking agent such as in Patent Documents 20 to 22, heating or active energy ray irradiation may be performed in a reactor.
- the surface cross-linking method of the present invention may be less effective in small kales and batch reactions, and can be suitably used for continuous production on a continuous huge scale, and usually 0.1 [t / hr] or more, preferably Can be suitably used for continuous production of 1 [t / hr], further 2 to 100 [t / hr].
- the obtained water absorbent resin mixture (usually particles)
- the temperature of the surface water-absorbing resin mixed with 0.001 to 10 parts by weight of a surface cross-linking agent and 0.5 to 10 parts by weight of water) is increased by 2 ° C. or more, and further 3 to 60 It is preferable to raise the temperature by 4 ° C., raise the temperature by 4-50 ° C., raise the temperature by 5-40 ° C., and raise the temperature by 6-30 ° C.
- This temperature control is performed by controlling the amount of steam added, the residence time in the mixer, and the temperature of the inner wall of the mixer.
- steam is used as the first production method, and the inner wall of the mixer is heated to the above range.
- the temperature of the water-absorbing resin mixture taken out from the mixer is preferably 50 to 140 ° C., more preferably 60 to 110 ° C., and particularly preferably 70 to 95 ° C. It is.
- the present invention provides, as a second production method, a step of adding a surface cross-linking agent and water to a particulate water-absorbing resin in a mixer, and the water-absorbing resin mixture taken out from the mixer is heated or reacted in a reactor.
- a method of producing surface-crosslinked water-absorbent resin particles which sequentially comprises a step of reacting with a surface cross-linking agent by irradiation with active energy rays, wherein the surface cross-linking agent and water are added in a mixer in the form of particles
- a production method in which the temperature of the resulting water absorbent resin mixture is increased by 2 ° C. or more with respect to the temperature of the water absorbent resin.
- the preferred temperature rise is the use of water vapor, but in addition, “the temperature of the inner wall of the mixer and the residence time may be controlled.
- the surface cross-linking treatment in the present invention there is a method of performing surface cross-linking treatment by irradiating active energy after adding a treatment liquid containing a radical polymerizable compound to the particulate water-absorbing resin.
- Japanese Patent Application “Japanese Patent Application No. 2003-303306” US Pat. No. 7,201,941).
- a surface active agent can also be added to the said process liquid, and an active energy can be irradiated and surface crosslinking can also be performed.
- the water absorbent resin particles obtained by the method for producing a water absorbent resin of the present invention are further added with a liquid permeability improver at the same time as surface crosslinking or after surface crosslinking. It is preferable. By adding a liquid permeability improver, the difference from the prior art appears more remarkably, and the present invention is clarified. When the liquid permeability improving agent is added, the water absorbent resin particles have a liquid permeability improving agent layer. Thereby, the water absorbent resin particles are further excellent in liquid permeability.
- the particulate water-absorbing resin can be further added with other function-imparting agents such as deodorants, anti-coloring agents, antibacterial agents, anti-blocking agents, etc. at the same time or in separate steps.
- function-imparting agents such as deodorants, anti-coloring agents, antibacterial agents, anti-blocking agents, etc. at the same time or in separate steps.
- liquid permeability improver examples include polyamines, polyvalent metal salts, water-insoluble fine particles, and water-dispersed fine particles. Particularly preferred are polyvalent metal salts such as aluminum sulfate, particularly water-soluble polyvalent metal salts. Nos. 7179862, 7157141, 6831142, U.S. Patent Application Publication Nos. 2004/176557, 2006/204755, 2006/73969, 2007/1060113, and European Patent No. 1165631 The described techniques apply. Polyamines and water-insoluble fine particles are exemplified in International Publication Nos. 2006/082188, 2006/082189, and 2006/082197.
- Polyamines and polyvalent metal salts can also be used as ion-reactive surface cross-linking agents that can be ion-cross-linked with the carboxyl groups of the particulate water-absorbing resin.
- the surface cross-linking may be performed once, or may be performed a plurality of times by separately using an ion-reactive cross-linking agent after use or before use.
- the amount of the liquid permeability improver used is preferably in the range of 0.001 to 5 parts by weight, more preferably in the range of 0.01 to 1 part by weight, with respect to 100 parts by weight of the water-absorbent resin particles.
- the amount of the liquid permeability improver used is within the above range, the water absorption capacity under pressure (AAP) and physiological saline flow conductivity (SFC) of the surface-crosslinked water absorbent resin particles can be improved.
- the addition of the liquid permeability improver is preferably a method of mixing or dispersing in advance with water and / or a hydrophilic organic solvent, if necessary, and then spraying or dropping and mixing the water-absorbent resin particles, more preferably a spraying method.
- the addition of the liquid permeability improver is preferably performed in a cooling step in the fluidized bed of the water absorbent resin particles.
- the addition of the liquid permeation improver may be performed simultaneously with the addition of the surface cross-linking agent, or may be performed after the heat treatment or the cooling treatment in the surface cross-linking step, and the mixing method of the present invention is applicable in either case.
- Step of Binding Fine Powders In the step of binding fine powders, which is another embodiment of the present invention, fine powder is produced by the above-described vertical mixing apparatus satisfying (i) to (iii). And a binder are preferably mixed and discharged as a binder.
- fine powder is fine powder generated by pulverization in “(2-5) Step of controlling particle size” or process damage in “(2-6) Surface treatment step”.
- the fine powder used in the step of binding the fine powders contains 50 to 100% by weight, preferably 70 to 100% by weight, having a particle size of 150 ⁇ m or less.
- the binder preferably contains 90 to 100% by weight of water. Moreover, you may mix and use an inorganic metal salt, an inorganic fine particle, and an organic solvent for this liquid. Furthermore, it is preferable to put water vapor into the mixer.
- the amount of the binder added is preferably 100 parts by weight or less, more preferably 70 parts by weight or less, and preferably 10 parts by weight or more with respect to 100 parts by weight of the particulate water-absorbing resin.
- the fine powder is bound by the binder, and a binder having a particle size of 150 ⁇ m or more can be formed.
- This binder can be confirmed by the fact that a plurality of individual particles are gathered and aggregated while maintaining the shape by an optical microscope, and the fact that they are expanded as a plurality of discontinuous particles upon liquid absorption.
- This binder has a solid content of 50 to 90% by weight, preferably 60 to 90% by weight, particularly preferably 60 to 80% by weight.
- the vertical mixing device is a mixer with good mixing properties
- the amount of the binder may be smaller than that of the prior art and the solid content of the binder can be increased by using the vertical mixing device. For this reason, less energy is required to dry the binder.
- this mixer can have a high processing power of 10 to 300 kg / hr, more preferably 10 to 150 kg / hr per liter of mixing tank volume.
- the binder moves to roll on the side wall of the mixer and is therefore generally spherical.
- a feature of the vertical mixing apparatus is that the weight average particle diameter of the binder can be controlled in the range of 0.5 to 5 mm by the weir structure of the discharge port.
- the above-mentioned binders are “(2-2) polymerization step”, “(2-3) step of refining the hydrogel polymer”, “(2-4) drying step”, “(2-5) particle size” It is preferable that the process is returned to any one of the steps of “controlling” and “(2-6) surface treatment step” and reused.
- this binder is pulverized or classified so as to be dried and become a particulate water-absorbing resin having a weight average particle diameter of 300 ⁇ m or more and 600 ⁇ m or less. Since the binder dried in this step has a small ratio of returning to a fine powder again, it is possible to confirm the difficulty of collapsing the binder, which is an effect of the present invention.
- the surface-crosslinked water-absorbing resin particles are used during or after the polymerization, such as a lubricant, chelating agent, deodorant, antibacterial agent, water, Surfactants, water-insoluble fine particles, antioxidants, reducing agents, and the like can be added to and mixed with the water-absorbent resin particles at about 0 to 30% by weight, further about 0.01 to 10% by weight.
- Chelating agents that can be suitably used are exemplified in US Pat. No. 6,599,989 and International Publication No. 2008/090961, and surfactants and lubricants are exemplified in US Pat. Nos. 6,107,358 and 7,473,739.
- the addition and mixing can be performed before drying, after drying, before pulverization, or after pulverization.
- the water absorbent resin particles may be added with other substances as long as the properties of the water absorbent resin are not impaired.
- the method for adding other substances is not particularly limited. In the present invention, even when the water-absorbent resin contains a small amount of additives (for example, more than 0 and 30% by weight), that is, when it is a water-absorbent resin composition, it is generically called a water-absorbent resin.
- [3] Physical properties of the water-absorbent resin For the purpose of sanitary materials, particularly paper diapers, at least one of the preferred physical property ranges listed in the following (a) to (h), and further, AAP is added by the above polymerization and surface crosslinking. It is preferable to control so as to satisfy two or more, particularly three or more. When the following range is not satisfied, sufficient performance may not be exhibited with a high-concentration diaper described below.
- the water-absorbing resin is excellent in initial coloring.
- the L value Lightness
- the value is preferably 85 or more, more preferably 87 or more, still more preferably 89 or more
- b The value is -5 to 10, more preferably -5 to 5, further preferably -4 to 4, and the value a is -2 to 2, at least -1 to 1, preferably -0.5 to 1, Most preferably, it is 0-1.
- the YI value is 10 or less, further 8 or less, particularly 6 or less
- the WB value is 70 or more, further 75 or more, particularly 77 or more.
- such a water-absorbent resin is excellent in coloring over time, and exhibits sufficient whiteness even at high temperature and high humidity, which is an accelerated test (model) for long-term storage.
- AAP Absorption capacity under pressure
- AAP indicates the water absorption capacity of the water-absorbent resin under a load.
- the water absorption capacity (AAP) with respect to a 0.9 mass% sodium chloride aqueous solution under a pressure of 1.9 kPa and further under a pressure of 4.8 kPa is preferably 10 [g / g] or more, more preferably 15 [g / g] or more, further preferably 20 [g / g] or more, more preferably 22 [g / g] or more, and further preferably 24 [g / g] or more. g / g] or more.
- the upper limit is preferably 28 [g / g] or less, more preferably 27 [g / g] or less, and particularly preferably 26 [g / g] from the balance with other physical properties.
- SFC Saline flow conductivity
- the SFC which is a liquid flow characteristic under pressure, is 1 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ] or more. 10 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ] or more, preferably 20 [ ⁇ 10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ] or more, more preferably 50 [ ⁇ 10 ⁇ 7].
- the water absorption capacity (CRC) under no pressure is preferably 10 [g / g] or more, more preferably 20 [g / g] or more, still more preferably 25 [g / g] or more, particularly preferably 30 [g. / G] or more.
- the higher the CRC, the more preferable the upper limit value is not particularly limited, but from the balance with other physical properties, it is usually 100 [g / g] or less, preferably 50 [g / g] or less, more preferably 45 [g / g] or less. More preferably, it is 40 [g / g] or less.
- the water-soluble content is preferably 0 to 35% by mass or less, more preferably 25% by mass or less, further preferably 15% by mass or less, and particularly preferably 10% by mass or less.
- the amount of residual monomer is usually 500 ppm or less, preferably 0 to 400 ppm, more preferably 0 to 300 ppm, particularly preferably 0 to 200 ppm.
- the water absorption rate and impact resistance are preferably adjusted so that a predetermined amount of water remains (for example, a water content of 0.1 to 10% by weight, more preferably 1 to 8% by weight).
- solid content (% by weight) 100-water content (% by weight)
- the solid content is defined as 85 to 99.9% by weight, more preferably 90 to 99.9% by weight, and still more preferably 95 to 99.9% by weight. If the solid content is out of the above range, the physical properties may deteriorate.
- Weight average particle diameter (D50) The final water-absorbent resin that has undergone the above-described steps and the like has a weight average particle diameter (D50) of preferably 300 to 600 ⁇ m, more preferably 350 to 500 ⁇ m, from the viewpoint of physical properties.
- the ratio of 850 to 150 ⁇ m is preferably controlled to 90 to 100% by weight, more preferably 95 to 100% by weight, particularly 98 to 100% by weight.
- the water-absorbent resin according to the present invention is used for applications intended to absorb water and is widely used as an absorbent or absorbent article, but is particularly suitable as a sanitary material for absorbing bodily fluids such as urine and blood. Used. In particular, it is used for high-concentration diapers (a large amount of water-absorbent resin is used for one diaper), which has been problematic in the past due to odor, coloring, etc., especially in the absorbent upper layer in the absorbent article. When used in parts, particularly excellent performance is exhibited.
- the absorber is an absorbent molded with a particulate water absorbent (water absorbent resin) and hydrophilic fibers as main components.
- the absorbent body is manufactured by being molded into, for example, a film shape, a cylindrical shape, or a sheet shape using a particulate water-absorbing agent and hydrophilic fibers.
- the content (core concentration) of the particulate water-absorbing agent with respect to the total mass of the particulate water-absorbing agent and the hydrophilic fiber is 20 to 100% by weight, 30 to 100% by weight, 40 to 100% by weight, 50 -100% by weight, 60-100% by weight, and 70-100% by weight are preferable in this order, and 75-95% by weight is most preferable.
- the core concentration of the particulate water-absorbing agent is higher, the effect of reducing the absorption characteristics of the particulate water-absorbing agent at the time of production of the absorbent body or paper diaper becomes more prominent.
- the absorber is preferably thin with a thickness of 0.1 to 5 mm.
- the said absorbent article is an absorbent article provided with the said absorber, the surface sheet which has liquid permeability, and the back sheet
- the manufacturing method of the said absorbent article first produces an absorber (absorption core) by blending or sandwiching, for example, a fiber material and a particulate water-absorbing agent. Next, the absorbent body is sandwiched between a liquid-permeable top sheet and a liquid-impermeable back sheet, and if necessary, equipped with an elastic member, a diffusion layer, an adhesive tape, etc.
- Absorbent articles especially adult paper diapers and sanitary napkins.
- the absorbent body is used by being compression-molded in a range of density 0.06 to 0.50 g / cc and basis weight 0.01 to 0.20 g / cm 2 .
- the fiber material used include hydrophilic fibers such as pulverized wood pulp, cotton linters and crosslinked cellulose fibers, rayon, cotton, wool, acetate, and vinylon. Preferably, they are airlaid.
- the above water-absorbent article exhibits excellent absorption characteristics.
- Specific examples of such absorbent articles include diapers for children, sanitary napkins, sanitary materials such as so-called incontinence pads, as well as adult paper diapers that have been growing rapidly in recent years. However, it is not limited to them.
- the above-mentioned water-absorbent article has a small amount of return due to the excellent absorption characteristics of the particulate water-absorbing agent present in the absorbent article, has a remarkably dry feeling, and greatly reduces the burden on the wearer and the caregiver. be able to.
- weir structure of the mixer which is a feature of the present invention, the horizontal length from the side wall of the weir structure to the rotating shaft side is simply the weir length ( ⁇ ), and the angle formed with respect to the horizontal plane is simply the weir. Described as an angle ( ⁇ ).
- the particulate water-containing gel particle size before drying was in accordance with the method described in Japanese Patent Publication No. 3175790. That is, 25 g of the sampled hydrogel polymer (solid content ⁇ wt%) was put into 1200 g of a 20 wt% sodium chloride aqueous solution, and the stirrer chip was rotated at 300 rpm and stirred for 60 minutes.
- the dispersion is put into a sieve (aperture 9.5 mm, 8.0 mm, 4.0 mm, 2.0 mm, 0.85 mm, 0.60 mm, 0.30 mm, 0.075 mm), and from above 6000 g of 20% by weight aqueous sodium chloride solution was slowly poured to classify the particulate hydrogel polymer.
- the particulate hydrogel polymer on each classified sieve was thoroughly drained and weighed.
- the sieve opening was converted into a sieve opening R (100) corresponding to a solid content of 100% by weight of the hydrogel polymer according to the following formula.
- the weight average particle diameter (D50) is a particle diameter of a standard sieve corresponding to 50% by weight of the whole particle with a standard sieve having a constant opening, as described in US Pat. No. 5,051,259.
- the logarithmic standard deviation ( ⁇ ) of the particle size distribution is expressed by the following equation, and the smaller the value of ⁇ , the narrower the particle size distribution.
- the particulate water-absorbing resin in an amount corresponding to 0.2 g in solid content is used, and the solid content correction is performed at the time of CRC calculation. The above method was followed.
- AAP Absorption capacity under pressure
- a glass filter with a diameter of 90 mm (manufactured by Mutual Chemical Glass Co., Ltd., pore diameter: 100 to 120 ⁇ m) is placed inside a petri dish with a diameter of 150 mm, and a 0.90% by weight sodium chloride aqueous solution (20 to 25 ° C.). ) was added to the same level as the top surface of the glass filter.
- a sheet of 90 mm diameter filter paper (trade name: “JIS P 3801 No. 2”, manufactured by ADVANTEC Toyo Co., Ltd., thickness 0.26 mm, reserved particle size 5 ⁇ m) was placed so that the entire surface was wetted. Excess liquid was removed.
- the set of measuring devices was placed on the wet filter paper, and the liquid was absorbed under load. After 1 hour (60 minutes), the measuring device set was lifted and its mass W 3 (g) was measured. And from these masses W 2 and W 3 , the absorption capacity under pressure (AAP) (g / g) was calculated according to the formula (2).
- the absorption capacity under pressure (AAP) under the pressure (under load) of 4.83 kPa (0.7 Psi) was used when the baby was sleeping or sitting in the absorbent body or disposable diaper, etc. This assumes the situation where absorbent articles are used.
- a glass tube is inserted in the tank.
- the glass tube is arranged by adjusting the position of the lower end so that the level of the 0.69 wt% sodium chloride aqueous solution in the cell is maintained at a height of 5 cm above the bottom of the swollen gel.
- the 0.69 wt% sodium chloride aqueous solution in the tank is supplied to the cell through an L-shaped tube with a cock.
- a collection container for collecting the liquid that has passed is arranged under the cell, and the collection container is placed on an upper pan balance.
- the inner diameter of the cell is 6cm.
- a 400 stainless steel wire mesh (aperture 38 ⁇ m) is installed.
- the cell is placed on a table on which the cell is placed, and this table is installed on a stainless steel wire mesh so as not to prevent the liquid from passing therethrough.
- the artificial urine is composed of 0.25 g of calcium chloride dihydrate, 2.0 g of potassium chloride, 0.50 g of magnesium chloride hexahydrate, 2.0 g of sodium sulfate, 0.85 g of ammonium dihydrogen phosphate, 2 hydrogen phosphates. What added 0.15g of ammonium and 994.25g of pure waters is used.
- the SFC test was performed at room temperature (20 to 25 ° C.). Using a computer and a balance, the amount of liquid passing through the gel layer at 20 second intervals as a function of time was recorded for 10 minutes.
- the flow rate Fs (t) passing through the swollen gel (mainly between the particles) was determined in units of g / s by dividing the increased mass (g) by the increased time (s).
- the unit of the liquid passing rate under pressure is (10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ).
- Fs (t 0): flow rate expressed in g / s L 0 : height of gel layer expressed in cm ⁇ : density of NaCl solution (1.003 g / cm 3 ) A: Area above the gel layer in the cell (28.27 cm 2 ) ⁇ P: hydrostatic pressure applied to the gel layer (4920 dyne / cm 2 ) [Production Example 1] In a kneader equipped with two sigma blades, a monomer aqueous solution (monomer concentration: 39 wt%, neutralization rate: 75 mol%) consisting of an aqueous sodium acrylate solution and acrylic acid and water was prepared. Polyethylene glycol diacrylate (average number of ethylene oxide units: 9) was dissolved in the monomer aqueous solution so as to be 0.07 mol% (with respect to the monomer).
- Nitrogen gas was blown into the monomer aqueous solution to reduce dissolved oxygen in the monomer aqueous solution and to replace the entire inside of the kneader with nitrogen.
- cold water of 10 ° C. was circulated through the jacket, and the temperature of the monomer aqueous solution was adjusted to 20 ° C.
- the obtained hydrogel polymer was dried in a hot air drier at 170 ° C. for 60 minutes.
- the obtained dried product was roughly crushed and then sieved with a JIS standard sieve having an opening of 850 ⁇ m.
- the dried product remaining on the sieve was pulverized with a roll mill.
- the obtained pulverized product was classified using a sieve having openings of 850 ⁇ m and 180 ⁇ m.
- the unpassed product of the sieve having an opening of 850 ⁇ m was pulverized again with a roll mill and classified in the same manner as described above.
- the passing material (fine powder a) classified by a sieve having an opening of 180 ⁇ m was about 15 wt% of the entire dried product.
- the particulate water-absorbing resin (A-1) obtained by the above classification and having an opening between a sieve having an opening of 850 ⁇ m and a sieve having a diameter of 180 ⁇ m has a water content of 4.9% by weight, a water absorption capacity without pressure (CRC ) was 35 [g / g], and the weight average particle diameter (D50) was 420 ⁇ m.
- the fine powder (a) of the water-absorbent resin has a weight-absorbing capacity (CRC) of 34 [g / g] under no pressure, a weight average particle diameter (D50) of 88 ⁇ m, and a sieve passing through a sieve of 150 ⁇ m is about 80 wt%. Met.
- Example 1 Surface cross-linking with water vapor and vertical mixer of the present application
- three stirring blades 5 are provided above the partition, three below, and three on the side of the partition.
- Vertical rotating disk type mixer with an internal volume of 5 L (maximum diameter (D) of mixing tank 300 mm (maximum radius 150 mm), mixing tank height (H) 70 mm, opening degree 20%, weir length ( ⁇ ) 21 mm, Using a weir angle ( ⁇ ) of 45 ° and an inner wall of fluororesin coating, the stirring blades are rotated at 1000 rpm, and the above particulate water-absorbing resin (A- 1) was supplied to the mixer at 200 kg / hr.
- composition liquid having 1,4-butanediol / propylene glycol / water 0.4 parts by weight / 0.6 parts by weight / 3.0 parts by weight as an aqueous surface crosslinking agent solution with respect to 100 parts by weight of the particulate water-absorbing resin.
- B-1) 8 [kg / hr] and water vapor (gauge pressure 0.6 MPa, mixer internal release, 5 [kg / hr]) are continuously mixed while being injected into the mixer, and the water-absorbing resin A mixture (C-1) was obtained.
- the vertical mixer has three stirring blades above the rotating disk, three below, and three on the side of the disk.
- the mixing tank has a diameter (D) of 300 mm (radius 150 mm), mixing
- the height of the tank is (H) 70 mm.
- the vertical mixer has an exhaust facility above the discharge port.
- the particulate water-absorbing resin (A-1) was sampled at the outlet of the quantitative feeder, and a temperature measured by inserting a contact thermometer into this was 58 ° C.
- the temperature of the composition liquid (B-1) was 26 ° C.
- the temperature of the water absorbent resin mixture (C-1) obtained by the above mixing was 76 ° C.
- the water content was 9.4 wt%
- the flow rate was 212 [kg / hr]. Therefore, the water supplied by water vapor is 4 [kg / hr].
- the flow rate of the water-absorbent resin mixture was obtained by taking the mixture for 10 minutes in a bag and measuring the weight.
- the dew point near the outlet of the mixer was 100 ° C.
- mixing was stopped 30 minutes after the start of mixing and the inside of the mixer was inspected, there was no adhesion.
- the water-absorbent resin mixture (C-1) was heat-treated at 210 ° C. (oil bath temperature) for 40 minutes while stirring with a mortar mixer (manufactured by West Japan Testing Machine Co., Ltd.). Further, the particles were pulverized until passing through a sieve having an opening of 850 ⁇ m. Thus, surface-crosslinked water-absorbing resin particles (D-1) were obtained. Table 1 shows the physical properties of the surface-crosslinked water-absorbing resin particles (D-1). The water content of the surface-crosslinked water-absorbing resin particles (D-1) was 1%.
- Example 2 Surface cross-linking with water vapor and vertical mixer of the present application Particulate water absorption was performed in the same manner as in Example 1 except that the amount of water vapor injection was changed from 5 [kg / hr] to 15 [kg / hr].
- a mixture (C-2) of the water-soluble resin (A-1) and the composition liquid (B-1) and surface-crosslinked water-absorbing resin particles (D-2) were obtained.
- the temperature of the above mixture (C-2) was 79 ° C.
- the water content was 10.2 wt%
- the flow rate was 214 [kg / hr]. Therefore, the water supplied by water vapor is 6 [kg / hr].
- Table 1 shows the physical properties of the surface-crosslinked water-absorbent resin particles (D-2).
- Example 3 Surface cross-linking in the vertical mixer of the present application
- the particulate water-absorbing resin (A-1) and the composition liquid (B -1) mixture (E-1) and surface-crosslinked water-absorbing resin particles (F-1) were obtained.
- the temperature of the mixture (E-1) was 58 ° C.
- Table 1 shows the physical properties of the surface-crosslinked water-absorbent resin particles (F-1).
- Example 4 Surface cross-linking in the vertical mixer of the present application
- the composition liquid (B-2) in which the amount of water is increased is used instead of the composition liquid (B-1) without injecting water vapor into the mixer. Except for the above, in the same manner as in Example 1, the mixture (E-2) of the particulate water-absorbing resin (A-1) and the composition liquid (B-1), and the surface-crosslinked water-absorbing resin particles (F- 2) was obtained. The water content of the surface-crosslinked water-absorbing resin particles (F-2) was 1%.
- the temperature of the composition liquid (B-2) was 25 ° C.
- the temperature of the mixture (E-2) was 58 ° C., and many aggregates were observed.
- Table 1 shows the physical properties of the surface-crosslinked water-absorbent resin particles (F-2).
- Example 5 Surface cross-linking with water vapor and vertical mixer of the present application
- the mixture (E-1) of the particulate water-absorbing resin (A-1) and the composition liquid (B-1) of Example 3 The water-absorbing resin (A-1) and the composition liquid (B-1) were added again to the mixer, and steam was injected into the mixer at 5 [kg / hr] and heated with steam.
- the water content of the obtained mixture (C-3) was 9.0% by weight.
- the mixture (C-3) was heat-treated in the same manner as in Example 1 to obtain surface-crosslinked water-absorbing resin particles (D-3).
- Table 1 shows the physical properties of the surface-crosslinked water-absorbing resin particles (D-3).
- Example 6 Surface crosslinking with water vapor without using liquid water
- the composition liquid (B-3) in which the amount of water was 0 parts by weight was used. Except for the above, in the same manner as in Example 1, the mixture (C-4) of the particulate water-absorbing resin (A-1) and the composition liquid (B-3), and the surface-crosslinked water-absorbing resin particles (D- 4) was obtained.
- composition liquid (B-3) was 25 ° C.
- temperature of the mixture (C-4) was 57 ° C., and many aggregates were observed.
- Table 1 shows the physical properties of the surface-crosslinked water-absorbent resin particles (D-4).
- Example 7 Surface cross-linking with water vapor and horizontal type mixer 5.00 kg of particulate water-absorbing resin (A-1) adjusted to 60 ° C. obtained in Production Example 1 and composition liquid (B-1) 0.20 kg, horizontal type mixer (made by Ledge Mixer, Ledige) with a 20L internal volume, equipped with a vertical blade, crushing blade, spray nozzle (one-fluid spray nozzle, manufactured by Ikeuchi Co., No.
- the mixture (C-5) of the particulate water-absorbing resin (A-1) and the composition liquid (B-1) obtained by the above mixing has more aggregates than the mixture (C-1) of Example 1. It was.
- the temperature of the mixture (C-5) was 82 ° C., and the water content was 10.1% by weight. Note that 5.35 kg of the mixture (C-5) was obtained, and therefore, the water supplied by water vapor was 0.15 kg.
- the mixture (C-5) was heat-treated with the mortar mixer of Example 1, and the resulting particles were crushed until passing through a sieve having an opening of 850 ⁇ m, and surface-crosslinked water-absorbent resin particles (D-5) Got. Table 1 shows the physical properties of the surface-crosslinked water-absorbent resin particles (D-5).
- the water absorption obtained in Production Example 1 is obtained by rotating the stirring blade at 1300 rpm and supplying from the supply port (input port) 2 provided at the right end of the rotating shaft 6 in the casing.
- Resin particles (A-1) were supplied at a rate of 200 kg / hr using a quantitative feeder (manufactured by Accurate Inc.).
- air is supplied from one end of the horizontal continuous mixer, that is, a supply port (input port) 2 provided at the right end in FIG. 6, and the pressure in the casing in the horizontal continuous mixer is reduced.
- An additive supply port provided at a position 200 mm from the right end of the casing when the total length of the rotary shaft 6 existing in the casing of the horizontal continuous mixer is 490 mm while maintaining a reduced pressure of 5 mmH 2 O or less.
- the powder and the surface treatment agent were continuously mixed while spraying the surface treatment agent 8 kg / hr from the additive inlet 4).
- the particulate water-absorbing resin (A-1) was sampled at the outlet of the quantitative feeder, and when a temperature was measured by inserting a contact thermometer into this, it was 58 ° C.
- the obtained mixture (G-2) was heat-treated with a mortar under stirring at 200 ° C. for 40 minutes to obtain surface-crosslinked water-absorbent resin particles (H-2).
- Table 1 shows the physical properties of the surface-crosslinked water-absorbent resin particles (H-2).
- Example 3 Effect of powder temperature of water-absorbent resin in vertical mixer of the present application The same operation as in Example 3 was performed except that the operation of heating the particulate water-absorbent resin in an oven was not performed. At this time, when the particulate water-absorbing resin (A-1) was collected from the outlet of the quantitative feeder and the temperature was measured with a contact thermometer, it was 24 ° C. When mixing was stopped 30 minutes after the start of mixing and the inside was inspected, adhesion of water-absorbent resin was observed on the side wall of the mixer.
- Example 7 use of a horizontal mixer, steam injection (or increase the temperature of the resulting water absorbent resin mixture by 2 ° C. or more) )
- SFC saline flow conductivity
- Example 2 From the comparison between Example 1 and Example 4 in which the vertical mixer of the present application was used and the same 5 parts by weight as the amount of moisture uptake, the water absorption capacity under pressure (AAP) was 22 [g / g] (Comparative Example 2) to 24 [g / g] (Example 1), Saline flow conductivity (SFC) is 75 [10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ] (Comparative Example 2) Thus, it was confirmed that the density improved dramatically to 105 [10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ] (Example 1).
- AAP water absorption capacity under pressure
- SFC Saline flow conductivity
- Example 3 use of the vertical mixer of the present application
- Comparative Examples 1 and 2 use of the horizontal mixer
- SFC physiological saline flow conductivity Both
- Example 1 both the water absorption capacity under pressure (AAP) and the saline flow conductivity (SFC) were improved in Example 1. Therefore, it is preferable that the addition of water is performed using a combination of liquid water and water vapor rather than water vapor alone.
- AAP water absorption capacity under pressure
- SFC saline flow conductivity
- Example 1 improved both the absorption capacity under pressure (AAP) and the saline flow conductivity (SFC). Therefore, it is preferable that water is added as water vapor at the same time as the surface cross-linking agent is added.
- AAP absorption capacity under pressure
- SFC saline flow conductivity
- Example 8 Further use of aluminum sulfate In Example 1 in which water vapor and the vertical mixer of the present application were used for surface crosslinking, aluminum sulfate was further used. That is, 35 g of surface-crosslinked water-absorbent resin particles (D-1) and 10 g of glass beads obtained in Example 1 were placed in a 225 ml mayonnaise bottle, and a paint shaker (Toyo And shaken with Seiki Co., Ltd. for 30 minutes.
- Example 9 Further use of aluminum sulfate
- aluminum sulfate was further used. That is, the same procedure as in Example 8 was performed except that the surface-crosslinked water-absorbent resin particles (D-1) in Example 8 were changed to surface-crosslinked water-absorbent resin particles (F-1). A treated product (I-2) was obtained. Table 2 shows the physical properties of the aluminum surface-treated product (I-2).
- Example 10 Further use of aluminum sulfate In Example 4 in which the vertical mixer of the present application was used for surface crosslinking, aluminum sulfate was further used. That is, the same procedure as in Example 8 was conducted except that the surface-crosslinked water-absorbent resin particles (D-1) in Example 8 were changed to surface-crosslinked water-absorbent resin particles (F-2). A treated product (I-3) was obtained. Table 2 shows the physical properties of the aluminum surface-treated product (I-3).
- Example 11 Continuous mixing of aluminum sulfate in the vertical mixer of the present application
- the surface-crosslinked water-absorbing resin particles (F-1) obtained in Example 3 were put in an oven in a state of being packed in a polypropylene bag and 100 ° C. Heated.
- 500 kg / hr of surface crosslinked water-absorbing resin particles (F-1) surface treatment comprising aluminum sulfate 14-18 hydrate, sodium lactate and water.
- the mixture was continuously mixed while injecting 5 kg / hr of the agent.
- the particulate water-absorbing resin (F-1) was sampled at the outlet of the quantitative feeder, and the temperature was 94 ° C. when a contact-type thermometer was inserted therein to measure the temperature.
- the obtained mixture was heat-treated at 100 ° C. for 10 minutes with stirring to obtain water-absorbing resin particles (K-1).
- Table 2 shows the physical properties of the surface-crosslinked water-absorbent resin particles (K-1).
- the obtained mixture was heat-treated at 100 ° C. for 10 minutes with stirring to obtain water-absorbing resin particles (L-1).
- Table 2 shows the physical properties of the aluminum surface-treated water-absorbent resin particles (L-1).
- Table 2 shows the results of using a liquid permeability improver (aluminum sulfate) in the surface crosslinking of Table 1.
- a liquid permeability improver aluminum sulfate
- the SFC of Example 8 is 140 [10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ] (increase: 35 [10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ]).
- the SFC was 90 [10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ] (increase: 15 [10 ⁇ 7 ⁇ cm 3 ⁇ s ⁇ g ⁇ 1 ]).
- a method of adding part or all of the water added in the mixer with water vapor (or the temperature of the resulting water-absorbent resin mixture is 2 ° C. or less).
- the saline flow conductivity (SFC) is high, and a great effect of improving the saline flow conductivity (SFC) by the liquid permeability improver can be obtained. confirmed.
- Example 11 using the vertical mixer of this application has a large increase in SFC, as compared to Comparative Example 5 using a horizontal mixer as a mixing device of the surface-crosslinked water-absorbent resin particles and aluminum sulfate. It was. This indicates that the vertical mixer of the present application is excellent in mixing properties.
- Example 12 Binding of fine powder in vertical mixer of the present application
- the fine powder (a) of the water-absorbent resin obtained in Production Example 1 was put in an oven and heated to 60 ° C in a bag-packed state.
- FIG. 5 schematically shows a 5 liter vertical rotary disk mixer (maximum diameter of the mixing tank) having three stirring blades above the partition, three below the partition, and nine on the side of the partition.
- D 300 mm (maximum radius 150 mm), mixing tank height (H) 70 mm, opening degree 20%, weir length ( ⁇ ) 0.5 mm, weir angle ( ⁇ ), inner wall is coated with fluororesin)
- the stirring blade was rotated at 1100 rpm.
- Fine powder (a) was supplied at a rate of 500 kg / hr using a quantitative feeder (manufactured by Accurate Inc.) and continuously mixed while injecting 167 kg / hr of water.
- the binder was taken out from the mixer, dried in a hot air dryer at 170 ° C. for 60 minutes, and the obtained dried product was pulverized until it passed through a sieve having an opening of 850 ⁇ m.
- the water-absorbent resin particles (a1) obtained by the above pulverization have a non-pressurized absorption capacity (CRC) of 33 g / g, a weight average particle diameter (D50) of 370 ⁇ m, and the content of powder having a particle diameter of 150 ⁇ m or less is 18 wt%. Met.
- Example 13 Binding of fine powder in the vertical mixer of the present application Example 12 except that the conditions of the weir structure of the mixer were the weir length ( ⁇ ) 21 mm and the weir angle ( ⁇ ) 20 °.
- a binder and its pulverized product (a2) were obtained.
- the above-mentioned binder was spherical, and was quickly classified to measure the particle size distribution.
- the weight average particle size (D50) of the binder was 4.0 mm, and the logarithmic standard deviation ( ⁇ ) was 0.54.
- the pulverized product (a2) had an absorption capacity without load (CRC) of 34 g / g, a weight average particle diameter (D50) of 400 ⁇ m, and a content of a particulate water-absorbing resin having a particle diameter of 150 ⁇ m or less was 13 wt%. .
- air is supplied from one end of the horizontal continuous mixer, that is, the supply port provided at the right end in FIG. 6, and the pressure in the casing of the horizontal continuous mixer is 5 mmH 2 O or less.
- the degree of vacuum when the total length of the rotary shaft 6 existing in the casing of the horizontal continuous mixer is 490 mm, from the additive supply port 4 provided at a position 200 mm from the right end of the casing, 167 kg / water While spraying hr, the powder and the surface treatment agent were continuously mixed.
- the particulate water-absorbing resin (A-1) was sampled at the outlet of the quantitative feeder, and when a temperature was measured by inserting a contact thermometer into this, it was 58 ° C. Ten minutes after the start of mixing, the mixing was stopped because the current value of the mixer increased. When the inside of the mixer was inspected, severe adhesion and coarse aggregates were found inside.
- a 1.0% by weight sodium persulfate aqueous solution was further added to a concentration of 0.05 g / mol (with respect to the monomer), and then the temperature was maintained at about 100 ° C.
- a monomer aqueous solution was continuously supplied to an endless belt running at a speed of minutes. The monomer aqueous solution continuously supplied onto the belt started to polymerize rapidly, and a band-shaped hydrogel sheet (hydrogel polymer) was obtained.
- This hydrogel sheet is continuously finely granulated using a meat chopper having a screen with a diameter of 9.5 mm (manufactured by Hiraga Works), and a hydrogel gel having a weight average particle diameter (D50) of about 2.0 mm as a polymer. A polymer was obtained.
- the obtained hydrogel polymer was dried in a hot air drier at 170 ° C. for 60 minutes.
- the obtained dried product was roughly crushed and then sieved with a JIS standard sieve having an opening of 850 ⁇ m.
- the dried product remaining on the sieve was pulverized with a roll mill.
- the obtained pulverized product was classified using a sieve having openings of 850 ⁇ m and 180 ⁇ m.
- the unpassed product of the sieve having an opening of 850 ⁇ m was pulverized again with a roll mill and classified in the same manner as described above.
- the passing material classified by a sieve having an opening of 180 ⁇ m was about 15 wt% of the entire dried material.
- the particulate water-absorbing resin (A-2) obtained by the above classification and having a mesh size between a sieve having an opening of 850 ⁇ m and a sieve having a diameter of 180 ⁇ m has a water content of 5.1% by weight and a water absorption capacity without pressure (CRC).
- CRC water absorption capacity without pressure
- Example 14 Example 1 except that the particulate water-absorbing resin (A-1) was changed to the particulate water-absorbing resin (A-2) and the heat treatment conditions were 212 ° C. (oil bath temperature) and 35 minutes in Example 1.
- surface-crosslinked water-absorbing resin particles (M-1) were obtained.
- Table 3 shows the physical properties of the surface-crosslinked water-absorbent resin particles (M-1).
- the temperature increase of the obtained water absorbent resin mixture was 18 ° C. with respect to the temperature of the particulate water absorbent resin (A-2). .
- Table 3 shows the physical properties of the surface-crosslinked water-absorbent resin particles (D-5).
- the temperature increase of the obtained water absorbent resin mixture was 19 ° C. with respect to the temperature of the particulate water absorbent resin (A-2). .
- Table 3 shows the physical properties of the surface-crosslinked water-absorbent resin particles (D-6).
- the temperature increase of the obtained water absorbent resin mixture was 17 ° C. with respect to the temperature of the particulate water absorbent resin (A-2). .
- Surface-crosslinked water-absorbent resin particles (M-4) were obtained in the same manner as in Example 14 except that the heat treatment conditions were changed to 218 ° C. (oil bath temperature) and 20 minutes. Table 3 shows the physical properties of the surface-crosslinked water-absorbent resin particles (M-4).
- the temperature increase of the obtained water absorbent resin mixture was 18 ° C. with respect to the temperature of the particulate water absorbent resin (A-2). .
- Example 18 surface crosslinking was performed in the same manner as in Example 14 except that the composition liquid (B-2) was used instead of the composition liquid (B-1) without injecting water vapor into the mixer. Water-absorbing resin particles (N-1) were obtained. Table 3 shows the physical properties of the surface-crosslinked water-absorbent resin particles (N-1). In the step of adding the composition liquid (B-2) in the mixer, the temperature increase of the obtained water absorbent resin mixture was 0 ° C. with respect to the temperature of the particulate water absorbent resin (A-2). .
- Example 19 In Example 15, a composition liquid (B ⁇ ) in which the amount of water was increased by an amount corresponding to absorption from water vapor in Example 15 instead of the composition liquid (B-4) without injecting water vapor into the mixer.
- Surface-crosslinked water-absorbing resin particles (N-2) were obtained in the same manner as in Example 15 except that 7) was used.
- Table 3 shows the physical properties of the surface-crosslinked water-absorbent resin particles (N-2). In the step of adding the composition liquid (B-7) in the mixer, the temperature increase of the obtained water absorbent resin mixture was 0 ° C. with respect to the temperature of the particulate water absorbent resin (A-2). .
- Example 20 the composition liquid (B ⁇ ) was obtained by increasing the amount of water by an amount corresponding to absorption from water vapor in Example 11 instead of the composition liquid (B-5) without injecting water vapor into the mixer.
- Surface-crosslinked water-absorbent resin particles (N-3) were obtained in the same manner as in Example 16 except that 8) was used.
- Table 3 shows the physical properties of the surface-crosslinked water-absorbing resin particles (N-3).
- the temperature increase of the obtained water absorbent resin mixture was 0 ° C. with respect to the temperature of the particulate water absorbent resin (A-2). .
- Example 21 the composition liquid (B ⁇ ) was prepared by increasing the amount of water by an amount corresponding to absorption from water vapor in Example 17 instead of the composition liquid (B-6) without injecting water vapor into the mixer.
- Surface-crosslinked water-absorbent resin particles (N-4) were obtained in the same manner as in Example 17 except that 9) was used.
- Table 3 shows the physical properties of the surface-crosslinked water-absorbing resin particles (N-4).
- the temperature increase of the obtained water absorbent resin mixture was 0 ° C. with respect to the temperature of the particulate water absorbent resin (A-2). .
- Example 23 In Example 15, further aluminum sulfate was used. That is, in the same manner as in Example 22 except that the surface-crosslinked water-absorbing resin particles (M-1) in Example 22 were changed to surface-crosslinked water-absorbing resin particles (M-2), an aluminum surface-treated product was obtained. (O-2) was obtained. Table 4 shows the physical properties of the aluminum surface treated product (O-2).
- Example 24 aluminum sulfate was further used. That is, in Example 22, except that the surface-crosslinked water-absorbent resin particles (M-1) were changed to surface-crosslinked water-absorbent resin particles (M-3), an aluminum surface-treated product was obtained. (O-3) was obtained. Table 4 shows the physical properties of the aluminum surface-treated product (O-3).
- Example 25 aluminum sulfate was further used. That is, in Example 22, except that the surface-crosslinked water-absorbing resin particles (M-1) were changed to surface-crosslinked water-absorbing resin particles (M-4), an aluminum surface-treated product was obtained. (O-4) was obtained. Table 4 shows the physical properties of the aluminum surface treated product (O-4).
- Example 26 In Example 18, further aluminum sulfate was used. That is, in Example 22, except that the surface-crosslinked water-absorbent resin particles (M-1) were changed to surface-crosslinked water-absorbent resin particles (N-1), the aluminum surface-treated product was obtained. (P-1) was obtained. Table 4 shows the physical properties of the aluminum surface-treated product (P-1).
- Example 27 aluminum sulfate was further used. That is, in Example 22, except that the surface-crosslinked water-absorbent resin particles (M-1) were changed to surface-crosslinked water-absorbent resin particles (N-2), an aluminum surface-treated product was obtained. (P-2) was obtained. Table 4 shows the physical properties of the aluminum surface-treated product (P-2).
- Example 28 In Example 20, further aluminum sulfate was used. That is, in Example 22, except that the surface-crosslinked water-absorbing resin particles (M-1) were changed to surface-crosslinked water-absorbing resin particles (N-3), the aluminum surface-treated product was obtained. (P-3) was obtained. Table 4 shows the physical properties of the aluminum surface-treated product (P-3).
- Example 29 In Example 21, further aluminum sulfate was used. That is, in Example 22, except that the surface-crosslinked water-absorbent resin particles (M-1) were changed to surface-crosslinked water-absorbent resin particles (N-4), an aluminum surface-treated product was obtained. (P-4) was obtained. Table 4 shows the physical properties of the aluminum surface-treated product (P-4).
- Example 30 In Example 14, further silica was used. That is, 35 g of the surface-crosslinked water-absorbent resin particles (M-1) obtained in Example 14 and 10 g of glass beads were placed in a 225 ml mayonnaise bottle, and then for 30 minutes with a paint shaker (manufactured by Toyo Seiki Co., Ltd.). Shake. Next, silica (AEROSIL200 manufactured by Nippon Aerosil Co., Ltd.) as a liquid permeability improver is added to the water-absorbing resin particles after shaking, and 0.5 parts by weight is added to 100 parts by weight of the water-absorbing resin particles and mixed. A surface-treated product (Q-1) was obtained. Table 5 shows the physical properties of the silica surface-treated product (Q-1).
- AEROSIL200 manufactured by Nippon Aerosil Co., Ltd.
- Example 31 In Example 15, further silica was used. That is, in Example 30, except that the surface-crosslinked water-absorbent resin particles (M-1) were changed to surface-crosslinked water-absorbent resin particles (M-2), a silica surface-treated product was obtained. (Q-2) was obtained. Table 5 shows the physical properties of the silica surface-treated product (Q-2).
- Example 32 In Example 16, further silica was used. That is, in Example 30, except that the surface-crosslinked water-absorbent resin particles (M-1) were changed to surface-crosslinked water-absorbent resin particles (M-3), a silica surface-treated product was obtained. (Q-3) was obtained. Table 5 shows the physical properties of the silica surface-treated product (Q-3).
- Example 33 In Example 17, further silica was used. That is, in Example 30, except that the surface-crosslinked water-absorbent resin particles (M-1) were changed to surface-crosslinked water-absorbent resin particles (M-4), a silica surface-treated product was obtained. (Q-4) was obtained. Table 5 shows the physical properties of the silica surface-treated product (Q-4).
- Example 34 In Example 18, further silica was used.
- the silica surface-treated product was obtained in the same manner as in Example 30 except that the surface-crosslinked water-absorbent resin particles (M-1) were changed to surface-crosslinked water-absorbent resin particles (N-1) in Example 30. (R-1) was obtained.
- Table 5 shows the physical properties of the silica surface-treated product (R-1).
- Example 35 In Example 19, further silica was used. That is, in Example 30, the surface-treated water-absorbent resin particles (M-1) were changed to the surface-crosslinked water-absorbent resin particles (N-2) in the same manner as in Example 30, (R-2) was obtained. Table 5 shows the physical properties of the silica surface-treated product (R-2).
- Example 36 In Example 20, further silica was used. That is, in Example 30, the surface-treated water-absorbing resin particles (M-1) were replaced with the surface-crosslinked water-absorbing resin particles (N-3) in the same manner as in Example 30, (R-3) was obtained. Table 5 shows the physical properties of the silica surface-treated product (R-3).
- Example 37 In Example 21, further silica was used.
- the silica surface-treated product was obtained in the same manner as in Example 30 except that the surface-crosslinked water-absorbent resin particles (M-1) were changed to surface-crosslinked water-absorbent resin particles (N-4) in Example 30. (R-4) was obtained.
- Table 5 shows the physical properties of the silica surface-treated product (R-4).
- each of the vertical mixers of the present application was used, and each surface cross-linking agent was used in the same manner as when aluminum sulfate was added to the surface-treated silica. It was confirmed that the effect of improving the saline flow conductivity (SFC) is increased by using the water or raising the temperature of the mixture as compared with the case without water vapor or without raising the temperature of the mixture.
- SFC saline flow conductivity
- a water-absorbing resin comprising: a step of reacting water-absorbing resin particles having surface cross-linking, wherein part or all of the water added in the mixer is added with water vapor. Manufacturing method.
- 0.1 to 10 parts by weight of water is mixed with 100 parts by weight of the particulate water-absorbing resin, and the water supplied as a liquid is 0 to 95% by weight of the total amount of the supplied water.
- a continuous apparatus in which the mixer and the reactor are connected, the average residence time of the particulate water-absorbing resin in the mixer is 1 second to 5 minutes, and the water absorption in the reactor The production method according to any one of (1) to (6), wherein an average residence time of the resin mixture is 6 minutes to 10 hours.
- the temperature of the obtained water absorbent resin mixture is raised by 3 to 60 ° C. compared to the temperature of the particulate water absorbent resin by the step of adding the surface cross-linking agent and water in the mixer, (1) The manufacturing method according to any one of (15) to (15).
- a method for producing a water-absorbent resin wherein the temperature of the particulate water-absorbent resin is set to 30 to 150 ° C. in the mixing of the particulate water-absorbent resin and the additive, and the following conditions (a) to (C), (A) The stirring blade rotates at 300 to 3000 rpm. (B) At least one of the rotating shafts of the stirring blade is in the vertical direction. (C) The mixing tank is divided into two or more chambers at the top and bottom by a partition having an opening degree of 5 to 70%. Using a continuous mixing device that satisfies:
- the discharge port has a dam structure
- the dam structure has an angle with respect to the horizontal plane of 10 to 80 °
- the horizontal length from the side wall to the rotating shaft is 1 to the maximum radius of the mixing tank.
- a value (H / D) obtained by dividing the height (H) inside the mixing tank by the maximum diameter (D) of the mixing tank is 0.1 to 1, (1) to (10 ).
- the amount of the particulate water-absorbing resin charged into the mixer is 10 to 300 kg / hr per liter of mixing tank volume of the mixer, according to any one of (1) to (11). Method.
- the water absorbent resin obtained by the production method of the present invention exhibits excellent absorption characteristics and the like (water absorption magnification under pressure, liquid permeability, etc.).
- a particulate water-absorbing resin is, for example, a flocculant, a coagulant, a soil conditioner, as an absorbent for hygiene materials such as adult paper diapers, children's diapers, sanitary napkins, so-called incontinence pads, It can be widely used as a water-soluble polymer suitably used for soil stabilizers, thickeners, etc., or as a water retaining agent, dehydrating agent, etc. in the fields of agriculture and horticulture and civil engineering.
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
(1-1)「吸水性樹脂」
本発明における「吸水性樹脂」とは、水膨潤性水不溶性の高分子ゲル化剤を意味する。なお、「水膨潤性」とは、ERT441.2-02で規定するCRC(無加圧下吸水倍率)が通常5[g/g]以上であることをいい、また、「水不溶性」とは、ERT470.2-02で規定するExt(水可溶分)が通常0~50重量%(特には20重量%以下)であることをいう。
本発明における「ポリアクリル酸(塩)系吸水性樹脂」とは、繰り返し単位として、アクリル酸および/またはその塩(以下、アクリル酸(塩)と称する)を主成分とする吸水性樹脂を意味する。
「EDANA」は、欧州不織布工業会(European Disposables and Nonwovens Associations)の略称であり、「ERT」は、欧州標準(ほぼ世界標準)である吸水性樹脂の測定方法(EDANA Recomeded Test Method)の略称である。なお、本発明においては、特に断りのない限り、ERT原本(公知文献:2002年改定)に準拠して、吸水性樹脂等の物性を測定する。
「CRC」は、Centrifuge Retention Capacity(遠心分離機保持容量)の略称であり、無加圧下吸水倍率(以下、「吸水倍率」と称することもある)を意味する。具体的には、0.9重量%塩化ナトリウム水溶液に対する30分間の自由膨潤後さらに遠心分離機で水切りした後の吸水倍率(単位;[g/g])である。
「AAP」は、Absorption Against Pressureの略称であり、加圧下吸水倍率を意味する。具体的には、0.9重量%塩化ナトリウム水溶液に対する1時間、2.06kPaでの荷重下膨潤後の吸水倍率(単位;[g/g])であるが、本発明においては、1時間、4.83kPa荷重下での吸水倍率(単位;[g/g])とした。
「Ext」は、Extractablesの略称であり、水可溶分(水可溶成分量)を意味する。具体的には、0.9重量%塩化ナトリウム水溶液200gに対して、吸水性樹脂1gを16時間攪拌した後、溶解したポリマー量をpH滴定で測定した値(単位;重量%)である。
荷重下または無荷重下における膨潤ゲルの粒子間を流れる液の流れを「通液性」という。この「通液性」の代表的な測定方法として、SFC(Saline Flow Conductivity)や、GBP(Gel Bed Permeability)がある。
本明細書において、範囲を示す「X~Y」は、「X以上、Y以下」であることを意味し、(メタ)アクリル酸等に用いられる「(メタ)アクリル」はアクリルまたはメタクリルを意味する。また、重量の単位である「t(トン)」は、「Metric ton(メトリック トン)」であることを意味し、さらに、特に注釈のない限り、「ppm」は「重量ppm」または「質量ppm」を意味する。
(2-1)アクリル酸(塩)水溶液
(a)単量体
本発明で使用できる不飽和単量体として、アクリル酸、メタクリル酸、(無水)マレイン酸、フマル酸、クロトン酸、イタコン酸、ビニルスルホン酸、2-(メタ)アクリルアミド-2-メチルプロパンスルホン酸、(メタ)アクリロキシアルカンスルホン酸、N-ビニル-2-ピロリドン、N-ビニルアセトアミド、(メタ)アクリルアミド、N-イソプロピル(メタ)アクリルアミド、N,N-ジメチル(メタ)アクリルアミド、2-ヒドロキシエチル(メタ)アクリレート、メトキシポリエチレングリコール(メタ)アクリレート、ポリエチレングリコール(メタ)アクリレート等の親水性モノマー類、並びにそれらの塩が挙げられる。
任意に使用できる架橋剤としては、例えば、N,N’-メチレンビス(メタ)アクリルアミド、(ポリ)エチレングリコールジ(メタ)アクリレート、(ポリ)プロピレングリコールジ(メタ)アクリレート、(ポリオキシエチレン)トリメチロールプロパントリ(メタ)アクリレート、トリメチロールプロパンジ(メタ)アクリレート、ポリエチレングリコールジ(β-アクリロイルオキシプロピオネート)、トリメチロールプロパントリ(β-アクリロイルオキシプロピオネート)、ポリ(メタ)アリロキシアルカン等の分子内に重合性二重結合を少なくとも2個有する化合物;ポリグリシジルエーテル(エチレングリコールジグリシジルエーテル)、ポリオール(エチレングリコール、ポリエチレングリコール、グリセリン、ソルビトール)等のカルボキシル基と反応して共有結合を形成し得る化合物の1種または2種以上が挙げられる。
これらの単量体は、通常、水溶液で重合され、その単量体濃度は、通常10~90重量%、好ましくは20~80重量%、より好ましくは30~70重量%、特に好ましくは30~60重量%の範囲である。
(a)重合方法
本発明の吸水性樹脂は、前記不飽和単量体を架橋重合し、含水ゲル状重合体を得ることにより製造される。重合は、性能面や重合の制御の容易さから、通常、噴霧重合、滴下重合、水溶液重合または逆相懸濁重合、特に、従来、その不定形状粒子のために粒度制御が困難であった水溶液重合、さらには連続水溶液重合で行われる。
本発明で使用される重合開始剤は、重合の形態によって適宜選択される。このような重合開始剤としては、好ましくは水溶性重合開始剤、さらには、光分解型重合開始剤、熱分解型重合開始剤、レドックス系重合開始剤等を例示することができる。また、本発明においては、光分解型重合開始剤と熱分解型重合開始剤とを併用することも好ましい。
乾燥効率、乾燥後の粉砕効率並びに物性の面から、乾燥前の含水ゲル状重合体が、重合中ないし重合後に細粒化されていることが好ましい。
上記の含水ゲル重合体、好ましくは粒子状含水ゲル重合体は、粉砕が可能となる固形分量になるまで乾燥される。ここで、乾燥工程に供される親水性架橋重合体(含水ゲル状重合体)の形態は、ニーダー、ミートチョッパーおよびカッターなどによる粗砕含水ゲル並びにその凝集物、シート状含水ゲルである。この乾燥工程の中に適宜、凝集物の解砕工程や粗砕工程を入れてもよい。このような技術として、例えば、米国特許第6187902号が採用される。
乾燥により得られた乾燥物は、粒径制御のため、粉砕、および必要により分級される。これらの方法については、例えば、国際公開第2004/69915号(米国特許出願公開第2006/024755号)に記載されている。
(2-6-1)表面架橋工程
表面架橋工程は本発明の特徴的な部分である。すなわち、本発明の吸水性樹脂の製造方法は、粒子状吸水性樹脂に表面架橋剤および水を混合機中で添加する工程、混合機から取り出した吸水性樹脂混合物を反応機中で加熱または活性エネルギー線照射で表面架橋剤と反応させる工程を順次含む、表面架橋された吸水性樹脂粒子の製造方法であって、混合機中での水の添加の一部または全部を水蒸気で添加することを特徴とする。
本発明において表面架橋剤としては、種々の有機架橋剤または無機架橋剤を例示することができるが、例えば、上記文献1~34に例示の公知の架橋剤を用いることができる。本発明では、表面架橋剤は吸水性樹脂の架橋を行うものであれば特に制限なく使用でき、単量体を重合して表面架橋する技術(特許文献20、21)や過硫酸塩などでラジカル架橋する技術(特許文献22)も架橋剤として使用ないし包含できる。物性や取り扱い性の観点から、好ましくは、吸水性樹脂のカルボキシル基(特にポリアクリル酸の中和または未中和のカルボキシル基)と反応、特に共有結合またはイオン結合で反応、さらには共有結合で反応する表面架橋剤が使用できる。
上記「(2-5)粒度を制御する工程」を経た粒子状吸水性樹脂は、好ましくは、加熱もしくは保温された貯蔵設備に一時的に保存され、フィーダーによって定量的に表面架橋工程に供給される。好ましいフィーダーはサークルフィーダーまたはスクリューフィーダーである。
本発明の第一の製造方法は、混合機中に水蒸気を供給することである。好ましくは1気圧より高い蒸気圧の飽和水蒸気を蒸気ラインにより混合機に供給する。1気圧より高い蒸気圧とすることで、ブロワー等、気体を供給する装置が不要となり、効率よく水蒸気を供給することができる。
本発明において、混合機中で粒子状吸水性樹脂に表面架橋剤を添加し、混合した後、表面架橋剤と粒子状吸水性樹脂とを反応させるために加熱または活性エネルギー線の照射を行う。
(ii)撹拌羽根の回転軸は少なくとも1本は鉛直方向(縦型混合機)である、
(iii)開口度が5~70%の仕切りにより、混合槽が上下に2室以上に分けられている。
(i)300~3000rpmで撹拌羽根が回転する、
(ii)撹拌羽根の回転軸は少なくとも1本は鉛直方向である、
(iii)開口度が5~70%の仕切りにより、混合槽が上下に2室以上に分けられている、
を満たす連続混合装置を使用することを特徴とする方法を提供する。

粒子状吸水性樹脂中の水分の増加は、液体の水として混合機に供給されて粒子状吸水性樹脂に取り込まれる水分量と、水蒸気として混合機に供給されて粒子状吸水性樹脂に取り込まれる水分量との和である。なお、水蒸気として混合機に供給されて粒子状吸水性樹脂に取り込まれる水分には、直接的な水蒸気の吸収と、水蒸気の結露水の吸収とがあり得るが、本発明では特に問わない。
表面架橋剤を混合後の吸水性樹脂混合物は、反応機中で加熱または活性エネルギー照射が行われる。好ましくは、吸水性樹脂混合物は加熱処理され、必要によりその後に冷却処理される。加熱温度は、好ましくは70~300℃、より好ましくは120~250℃、さらに好ましくは150~250℃の範囲である。また、加熱時間は、好ましくは1分以上、通常6分~10時間、さらに好ましくは10分~2時間である。
本発明の表面架橋方法では小ケールやバッチ反応では効果が小さい場合もあり、連続巨大スケールの連続生産に好適に使用でき、通常、0.1[t/hr]以上、好ましくは、1[t/hr]、さらには2~100[t/hr]の連続生産に好適に使用できる。
本発明では上記水蒸気添加を達成手段の一例として、表面架橋剤および水を混合機中に添加する工程において、粒子状吸水性樹脂の温度に対して、得られた吸水性樹脂混合物(通常、粒子状吸水性樹脂100重量部に対して、表面架橋剤0.001~10重量部、水0.5~10重量部を混合したもの)の温度を、2℃以上昇温、さらには3~60℃昇温、4~50℃昇温、5~40℃昇温、6~30℃昇温させることが好ましい。
本発明の吸水性樹脂の製造方法により得られた吸水性樹脂粒子は、表面架橋と同時または表面架橋後に、さらに通液性向上剤が添加されることが好ましい。通液性向上剤を添加することにより、より従来技術との差が顕著に現れ、本発明が明確化される。通液性向上剤が添加されることによって、上記吸水性樹脂粒子は通液性向上剤層を有することになる。これにより、上記吸水性樹脂粒子は、さらに、通液性に優れることになる。
本発明のもう一つの実施形態である微粉同士を結着する工程では、上述した、(i)~(iii)を満たす縦型混合装置により、微粉と結着剤とが混合され、結着物となって排出されることが好ましい。
表面架橋された吸水性樹脂粒子は、重合中または重合後に、滑剤、キレート剤、消臭剤、抗菌剤、水、界面活性剤、水不溶性微粒子、酸化防止剤、還元剤等が吸水性樹脂粒子に0~30重量%、さらには0.01~10重量%程度で添加混合されうる。好適に使用できるキレート剤は、米国特許第6599989号、国際公開第2008/090961号等に、界面活性剤や滑剤は、米国特許第6107358号、同第7473739号等に例示されている。
上記の工程以外に、必要により、整粒工程、微粉除去工程、微粉リサイクル工程等を設けてもよい。例えば、米国特許第5264495号、米国特許第5369148号、米国特許第5478879号、米国特許第6228930号、米国公開公報第2006/247351号、国際公開公報第2006/101271号等に記載の工程が挙げられる。
衛生材料、特に紙オムツを目的とする場合、上記重合や表面架橋をもって、下記(a)~(h)に挙げる好ましい物性の範囲の少なくとも1つ、さらにはAAPを含め2つ以上、特に3つ以上を満たすように制御されることが好ましい。下記範囲を満たさない場合、後述の高濃度おむつでは十分な性能を発揮しないことがある。
かかる吸水性樹脂は初期着色に優れ、例えば、ハンターLab表面色系において、L値(Lightness)が好ましくは85以上、より好ましくは87以上、さらに好ましくは89以上であり、b値が-5~10、より好ましくは-5~5、さらに好ましくは-4~4であり、また、a値は-2~2、少なくとも-1~1、好ましくは-0.5~1、最も好ましくは0~1である。YI値は10以下、さらには8以下、特に6以下であり、WB値は70以上、さらには75以上、特に77以上である。さらに、かかる吸水性樹脂は経時着色にも優れ、長期保存の促進試験(モデル)である高温高湿でも十分な白色度を示す。
加圧下吸水倍率(AAP)は、荷重をかけた状態での、吸水性樹脂の吸水倍率を示している。
生理食塩水流れ誘導性(SFC)は、吸水性樹脂の膨潤時の通液性を示す値であり、その値が大きいほど高い通液性を有することを示している。
無加圧下吸水倍率(CRC)は、好ましくは10[g/g]以上であり、より好ましくは20[g/g]以上、さらに好ましくは25[g/g]以上、特に好ましくは30[g/g]以上に制御される。CRCは高いほど好ましく上限値は特に限定されないが、他の物性とのバランスから、通常100[g/g]以下、好ましくは50[g/g]以下、より好ましくは45[g/g]以下、さらに好ましくは40[g/g]以下である。
水可溶分は、好ましくは0~35質量%以下、より好ましくは25質量%以下であり、さらに好ましくは15質量%以下、特に好ましくは10質量%以下である。
上記重合を達成手段の一例として、残存モノマー(残存単量体)量は通常500ppm以下、好ましくは0~400ppm、より好ましくは0~300ppm、特に好ましくは0~200ppmを示す。
吸水速度や耐衝撃性からも好ましくは所定量の水が残存(例えば、含水率0.1~10重量%、さらに好ましくは1~8重量%)するように調整される。
固形分量(重量%)=100-含水率(重量%)
で定義される固形分量は、85~99.9重量%が好ましく、90~99.9重量%がより好ましく、95~99.9重量%がさらに好ましい。固形分量が上記範囲を外れると、物性が低下することがある。
上記工程等を経た、最終的な吸水性樹脂としては、物性面から重量平均粒子径(D50)が300~600μmであること好ましく、350~500μmであることがさらに好ましい。850~150μmの割合が90~100重量%、さらには95~100重量%特に98~100重量%に制御されることが好ましい。
本発明に係る吸水性樹脂は、吸水を目的とした用途に用いられ、吸収体や吸収性物品として広く使用されるが、特に、尿や血液等の体液を吸収するための衛生材料として好適に用いられる。特に、従来、原料由来の臭気、着色等が問題になっていた高濃度オムツ(1枚のオムツに多量の吸水性樹脂を使用したもの)に使用され、特に前記吸収性物品中の吸収体上層部に使用された場合に、特に優れた性能が発揮される。
以下に、製造例、実施例、比較例によって本発明をより具体的に説明するが、本発明はこれらに限定されるものではない。異なる実施例にそれぞれ開示された技術的手段を適宜組み合わせて得られる実施例についても、本発明の範囲に含まれる。
<粒径>
粒径の分布および重量平均粒子径(D50)は、以下で説明するように、試料を標準篩にかけることにより測定した。


<含水率>
含水ゲル状重合体ないし粒子状吸水性樹脂1gを6cmのアルミ皿に薄く広げて、180℃の無風オーブンで3時間乾燥することで、その乾燥前の重量と乾燥後の重量とを測定し、下記式に代入することにより含水率(重量%)を測定した。なお、固形分(重量%)は、(100-含水率)(重量%)で規定される。

粒子状吸水性樹脂0.2gを不織布製の袋(60mm×60mm、南国パルプ工業(製)ヒートロンペーパー GS-22)に均一に入れ、ヒートシール後、0.9重量%塩化ナトリウム水溶液(生理食塩水)中に浸漬した。30分後に袋を引き上げ、遠心分離器を用いて250×9.81m/s2(250G)で3分間水切りを行った後、袋の質量W1(g)を測定した。また、同様の操作を、粒子状吸水性樹脂を用いないで行い、そのときの質量W0(g)を測定した。そして、これら質量W1、W0から、式(1)に従って無加圧下吸収倍率(CRC)を算出した。

4.83kPa(0.7Psi)の圧力になるように調製した荷重を準備した。そして、底に400メッシュ(目開き38μm)の金網を貼着した直径60mmのプラスチック円筒の金網上に、粒子状吸水性樹脂0.90gを均一に散布した。その上に、上記荷重を載せて、この測定装置一式の質量W2(g)を測定した。

ERT470.2-02に従って測定した。
生理食塩水流れ誘導性(SFC)は、米国公開特許第2004-0106745号明細書、特表平09-509591号公報の塩水流れ誘導性(SFC)試験に準じて行った。

L0:cmで表したゲル層の高さ
ρ:NaCl溶液の密度(1.003g/cm3)
A:セル中のゲル層上側の面積(28.27cm2)
ΔP:ゲル層にかかる静水圧(4920dyne/cm2)
〔製造例1〕
2本のシグマ型ブレードを備えたニーダーに、アクリル酸ナトリウム水溶液、アクリル酸および水からなる単量体水溶液(単量体濃度:39wt%、中和率:75モル%)を調製し、さらにこの単量体水溶液に、ポリエチレングリコールジアクリレート(平均エチレンオキシドユニット数:9)を0.07モル%(対単量体)となるように溶解させた。
図4で模式的に示された、撹拌羽根5を仕切りの上方に3枚、下方に3枚、仕切りの側面に3枚備えた内容積5Lの縦型回転円盤型混合機(混合槽の最大直径(D)300mm(最大半径150mm)、混合槽の高さ(H)70mm、開口度20%、堰長さ(α)21mm、堰角度(β)45°、内壁をフッ素樹脂コート)を用いて、1000rpmで攪拌羽根を回転させ、定量供給機(アキュレートInc.製)を用いて、上記の粒子状吸水性樹脂(A-1)を200kg/hrで上記混合機に供給した。
水蒸気注入量を5[kg/hr]から15[kg/hr]に変更したこと以外は実施例1と同様にして、粒子状吸水性樹脂(A-1)と組成液(B-1)の混合物(C-2)、および表面架橋された吸水性樹脂粒子(D-2)を得た。混合開始の30分後に混合を止めて混合機の内部を点検したところ、付着はなかった。また、上記の混合物(C-2)の温度は79℃で、含水率は10.2重量%、流量は214[kg/hr]であった。従って、水蒸気により供給された水は6[kg/hr]となる。表面架橋された吸水性樹脂粒子(D-2)の物性を表1に示す。
混合機内部に水蒸気を注入しなかったこと以外は実施例1と同様にして、粒子状吸水性樹脂(A-1)と組成液(B-1)の混合物(E-1)、および表面架橋された吸水性樹脂粒子(F-1)を得た。混合物(E-1)の温度は58℃であった。表面架橋された吸水性樹脂粒子(F-1)の物性を表1に示す。
混合機内部に水蒸気を注入せずに、組成液(B-1)の代わりに水の量を増やした組成液(B-2)を使用したこと以外は実施例1と同様にして、粒子状吸水性樹脂(A-1)と組成液(B-1)の混合物(E-2)、および表面架橋された吸水性樹脂粒子(F-2)を得た。なお、表面架橋された吸水性樹脂粒子(F-2)の含水率は1%であった。
実施例3の、粒子状吸水性樹脂(A-1)と組成液(B-1)との混合物(E-1)を、粒子状吸水性樹脂(A-1)と組成液(B-1)との混合に用いた混合機に再度投入し、水蒸気を5[kg/hr]で上記混合機に注入して水蒸気加熱した。得られた混合物(C-3)の含水率は9.0重量%であった。この混合物(C-3)を実施例1と同様に加熱処理することにより表面架橋された吸水性樹脂粒子(D-3)を得た。表面架橋された吸水性樹脂粒子(D-3)の物性を表1に示す。
実施例1において、組成液(B-1)の代わりに水の量を0重量部とした組成液(B-3)を使用したこと以外は実施例1と同様にして、粒子状吸水性樹脂(A-1)と組成液(B-3)の混合物(C-4)、および表面架橋された吸水性樹脂粒子(D-4)を得た。なお、組成液(B-3)は粒子状吸水性樹脂100重量部に対し、1,4-ブタンジオール/プロピレングリコール/水=0.4重量部/0.6重量部/0重量部であった。また、組成液(B-3)の温度は25℃であった。混合物(C-4)の温度は57℃で、多数の凝集物が見られた。表面架橋された吸水性樹脂粒子(D-4)の物性を表1に示す。
製造例1で得られた、60℃に調温された粒子状吸水性樹脂(A-1)5.00kgと組成液(B-1)0.20kgとを、鋤型羽根、解砕羽根、噴霧ノズル(一流体噴霧ノズル、いけうち社製、No.6)およびジャケットを備えた内容積20Lの横型混合機(Ledige Mixer、Ledige社製)を用い、ノズル(内径3mmの直管)から0.6kPa(ゲージ圧)流量5[kg/hr]の飽和水蒸気(撹拌機内部開放)を供給しながら30秒間撹拌した。なお、混合機の回転数は200rpm、解砕羽根の回転速度は2000rpm、ジャケット温度は60℃であった。
水蒸気を供給しなかったこと以外は実施例7と同様にして、粒子状吸水性樹脂(A-1)と組成液(B-1)との混合物(G-1)、および表面架橋された吸水性樹脂粒子(H-1)を得た。混合物(G-1)の温度は61℃であった。表面架橋された吸水性樹脂粒子(H-1)の物性を表1に示す。
製造例1で得られた粒子状吸水性樹脂(A-1)を、袋詰めした状態でオーブンに入れて60℃に加熱した。
粒子状吸水性樹脂をオーブンで加熱する操作を行わなかった以外は、実施例3と同様の操作を行った。このとき定量供給機出口から粒子状吸水性樹脂(A-1)を採取し、接触型温度計で温度を測定すると24℃であった。混合を始めて30分後に混合を止めて内部点検したところ、混合機の側壁に吸水性樹脂の付着が見られた。

表1に示すように、比較例1、2(横型混合機の使用)に対し、実施例7(横型混合機の使用、水蒸気注入(または得られる吸水性樹脂混合物の温度を2℃以上昇温))では生理食塩水流れ誘導性(SFC)が向上することが確認された。なお、本願縦型混合機を使用し、水分取り込み量として同じ5重量部である実施例1と実施例4との比較から、水蒸気の使用によって、加圧下吸水倍率(AAP)が22[g/g](比較例2)から24[g/g](実施例1)、生理食塩水流れ誘導性(SFC)が75[10-7×cm3×s×g-1](比較例2)から105[10-7×cm3×s×g-1](実施例1)へと飛躍的に向上することが確認された。また、比較例1、2(横型混合機の使用)に対し、実施例3(本願縦型混合機の使用)では加圧下吸水倍率(AAP)が向上することが確認された。さらに実施例1および2(本願縦型混合機の使用、水蒸気注入(または得られる吸水性樹脂混合物の温度を2℃以上昇温))では加圧下吸水倍率(AAP)と生理食塩水流れ誘導性(SFC)がともに向上することが確認された。
水蒸気および本願縦型混合機を表面架橋に使用した実施例1において、さらに硫酸アルミニウムを使用した。すなわち、実施例1で得られた、表面架橋された吸水性樹脂粒子(D-1)35gとガラスビーズ10gとを、225mlのマヨネーズ瓶に入れ、プロセス上の耐衝撃モデルとして、ペイントシェイカー(東洋精機(株)製)で30分間振とうした。次いで、振とう後の吸水性樹脂粒子30gに、該吸水性樹脂粒子100重量部に対して、50%硫酸アルミニウム水溶液/プロピレングリコール/乳酸ナトリウム=1.0重量部/0.025重量部/0.3重量部からなる通液性向上剤0.3gを添加・混合後、60℃の乾燥機で30分間硬化を行った。硬化後、ガラスビーズ10gの入った225mlのマヨネーズ瓶に入れ、ペイントシェイカーで10分間振とうし、アルミ表面処理物(I-1)を得た。アルミ表面処理物(I-1)の物性を表2に示す。
本願縦型混合機を表面架橋に使用した実施例3において、さらに硫酸アルミニウムを使用した。すなわち、上記実施例8で表面架橋された吸水性樹脂粒子(D-1)を表面架橋された吸水性樹脂粒子(F-1)に変更したこと以外は実施例8と同様に行い、アルミ表面処理物(I-2)を得た。アルミ表面処理物(I-2)の物性を表2に示す。
本願縦型混合機を表面架橋に使用した実施例4において、さらに硫酸アルミニウムを使用した。すなわち、上記実施例8で表面架橋された吸水性樹脂粒子(D-1)を表面架橋された吸水性樹脂粒子(F-2)に変更したこと以外は実施例8と同様に行い、アルミ表面処理物(I-3)を得た。アルミ表面処理物(I-3)の物性を表2に示す。
水蒸気および縦型混合機を使用しない比較例1において、さらに硫酸アルミニウムを使用した。すなわち、表面架橋された吸水性樹脂粒子(D-1)を表面架橋された吸水性樹脂粒子(H-1)に変更したこと以外は実施例8と同様に行い、アルミ表面処理物(J-1)を得た。アルミ表面処理物(J-1)の物性を表2に示す。
実施例3で得た表面架橋吸水性樹脂粒子(F-1)を、ポリプロピレン袋に袋詰めした状態でオーブンに入れて100℃に加熱した。実施例1と同様の縦型回転円盤型混合機を用いて、表面架橋吸水性樹脂粒子(F-1)500kg/hrに、硫酸アルミニウム14-18水和物、乳酸ナトリウムおよび水からなる表面処理剤5kg/hrを注入しながら連続的に混合した。なお、定量供給機出口で粒子状吸水性樹脂(F-1)を採取し、これに接触型温度計を差し込んで温度を測定すると94℃であった。
実施例3で得た表面架橋吸水性樹脂粒子(F-1)を、ポリプロピレン袋に袋詰めした状態でオーブンに入れて100℃に加熱した。比較例2と同様の横型連続式混合機を用いて、表面架橋吸水性樹脂粒子(F-1)500kg/hrに、硫酸アルミニウム14-18水和物、乳酸ナトリウムおよび水からなる表面処理剤5kg/hrを注入しながら連続的に混合した。なお、定量供給機出口で粒子状吸水性樹脂(D-5)を採取し、これに接触型温度計を差し込んで温度を測定すると95℃であった。また、上記表面処理剤中の各成分の組成比(質量比)は、吸水性樹脂100質量部に対し、硫酸アルミウム14-18水和物/乳酸ナトリウム/水=0.4/0.1/0.5である。得られた混合物を、撹拌下、100℃で10分間加熱処理し、吸水性樹脂粒子(L-1)を得た。アルミ表面処理された吸水性樹脂粒子(L-1)の物性を表2に示す。

表2では、表1の表面架橋においてさらに通液性向上剤(硫酸アルミニム)を使用した結果を示す。表2に示すように、水分取り込み量として同じ5重量部である実施例8と実施例10との比較から、表面架橋後の通液性向上剤(硫酸アルミニウム)添加量が同じであっても、水蒸気使用の有無によって、生理食塩水流れ誘導性(SFC)およびその上昇度に影響を及ぼすことが分かる。すなわち、実施例8のSFCが140[10-7×cm3×s×g-1](上昇度;35[10-7×cm3×s×g-1])に対して、実施例10では、SFCが90[10-7×cm3×s×g-1](上昇度;15[10-7×cm3×s×g-1])であった。
製造例1で得られた吸水性樹脂の微粉(a)を、袋詰めした状態でオーブンに入れて60℃に加熱した。
混合機の堰構造の条件を堰長さ堰長さ(α)21mm、堰角度(β)20°としたこと以外は実施例12と同様にして、結着物およびその粉砕物(a2)を得た。上記結着物は球状で、速やかに分級して粒子径分布を測定したところ、結着物の重量平均粒子径(D50)は4.0mm、対数標準偏差(σζ)は0.54であった。上記粉砕物(a2)は、無加圧下吸収倍率(CRC)が34g/g、重量平均粒子径(D50)が400μm、粒子径150μm以下の粒子状吸水性樹脂の含有率が13wt%であった。
微粉をオーブンで加熱する操作を行わなかったこと以外は、実施例12と同様の操作を行った。このとき定量供給機出口から粒子状吸水性樹脂(a)を採取し、接触型温度計で温度を測定すると23℃であった。混合を始めて30分後に混合を止めて内部点検したところ、混合機の投入口と側壁に吸水性樹脂の付着が見られた。
製造例1で得られた吸水性樹脂の微粉(a)を、袋詰めした状態でオーブンに入れて60℃に加熱した。図6に示す横型連続式混合機において、1300rpmで攪拌翼を回転させ、上記ケーシング内における回転軸6の右端に設けられた供給口から、製造例1で得られた吸水性樹脂の微粉(a)を、定量供給機(アキュレートInc.製)を用いて、500kg/hrで供給した。
48.5重量%水酸化ナトリウム水溶液、アクリル酸、および水を単量体濃度45wt%、中和率:70モル%なるように供給し、さらに単量体水溶液に、ポリエチレングリコールジアクリレート(平均エチレンオキシドユニット数:9)を0.07モル%(対単量体)、1重量%ジエチレントリアミン5酢酸3ナトリウム水溶液を100ppm(対単量体)となるように上記単量体に加え、連続的に混合した。このとき、単量体水溶液の温度は95℃であった。
実施例1において粒子状吸水性樹脂(A-1)を粒子状吸水性樹脂(A-2)に、加熱処理の条件を212℃(オイルバス温度)、35分とした以外は、実施例1と同様にして、表面架橋された吸水性樹脂粒子(M-1)を得た。表面架橋された吸水性樹脂粒子(M-1)の物性を表3に示す。なお、組成液(B-1)を混合機中で添加する工程において、粒子状吸水性樹脂(A-2)の温度に対して、得られる吸水性樹脂混合物の温度上昇は18℃であった。
実施例14において、組成液(B-1)を粒子状吸水性樹脂100重量部に対し、1,4-ブタンジオール/水=0.9重量部/3.0重量部である組成液(B-4)に変更し、加熱処理の条件を218℃(オイルバス温度)、25分とした以外は、実施例14と同様にして、表面架橋された吸水性樹脂粒子(M-2)を得た。表面架橋された吸水性樹脂粒子(D-5)の物性を表3に示す。なお、組成液(B-4)を混合機中で添加する工程において、粒子状吸水性樹脂(A-2)の温度に対して、得られる吸水性樹脂混合物の温度上昇は19℃であった。
実施例14において、組成液(B-1)を粒子状吸水性樹脂100重量部に対し、1,6-ヘキサンジオール/水=0.9重量部/3.0重量部である組成液(B-5)に変更し、加熱処理の条件を218℃(オイルバス温度)、35分とした以外は、実施例14と同様にして、表面架橋された吸水性樹脂粒子(M-3)を得た。表面架橋された吸水性樹脂粒子(D-6)の物性を表3に示す。なお、組成液(B-5)を混合機中で添加する工程において、粒子状吸水性樹脂(A-2)の温度に対して、得られる吸水性樹脂混合物の温度上昇は17℃であった。
実施例14において、組成液(B-1)を粒子状吸水性樹脂100重量部に対し、炭酸エチレン/水=0.9重量部/3.0重量部である組成液(B-6)に変更し、加熱処理の条件を218℃(オイルバス温度)、20分とした以外は、実施例14と同様にして、表面架橋された吸水性樹脂粒子(M-4)を得た。表面架橋された吸水性樹脂粒子(M-4)の物性を表3に示す。なお、組成液(B-6)を混合機中で添加する工程において、粒子状吸水性樹脂(A-2)の温度に対して、得られる吸水性樹脂混合物の温度上昇は18℃であった。
実施例14において、混合機内部に水蒸気を注入せずに、組成液(B-1)の代わりに組成液(B-2)を使用した以外は実施例14と同様にして、表面架橋された吸水性樹脂粒子(N-1)を得た。表面架橋された吸水性樹脂粒子(N-1)の物性を表3に示す。なお、組成液(B-2)を混合機中で添加する工程において、粒子状吸水性樹脂(A-2)の温度に対して、得られる吸水性樹脂混合物の温度上昇は0℃であった。
実施例15において、混合機内部に水蒸気を注入せずに、組成液(B-4)の代わりに実施例15で水蒸気からの吸収に相当する分だけ水の量を増やした組成液(B-7)を使用した以外は実施例15と同様にして、表面架橋された吸水性樹脂粒子(N-2)を得た。なお、組成液(B-7)は粒子状吸水性樹脂100重量部に対し、1,4-ブタンジオール/水=0.9重量部/5.0重量部であった。表面架橋された吸水性樹脂粒子(N-2)の物性を表3に示す。なお、組成液(B-7)を混合機中で添加する工程において、粒子状吸水性樹脂(A-2)の温度に対して、得られる吸水性樹脂混合物の温度上昇は0℃であった。
実施例16において、混合機内部に水蒸気を注入せずに、組成液(B-5)の代わりに実施例11で水蒸気からの吸収に相当する分だけ水の量を増やした組成液(B-8)を使用した以外は実施例16と同様にして、表面架橋された吸水性樹脂粒子(N-3)を得た。なお、組成液(B-8)は粒子状吸水性樹脂100重量部に対し、1,6-ヘキサンジオール/水=0.9重量部/5.0重量部であった。表面架橋された吸水性樹脂粒子(N-3)の物性を表3に示す。なお、組成液(B-8)を混合機中で添加する工程において、粒子状吸水性樹脂(A-2)の温度に対して、得られる吸水性樹脂混合物の温度上昇は0℃であった。
実施例17において、混合機内部に水蒸気を注入せずに、組成液(B-6)の代わりに実施例17で水蒸気からの吸収に相当する分だけ水の量を増やした組成液(B-9)を使用した以外は実施例17と同様にして、表面架橋された吸水性樹脂粒子(N-4)を得た。なお、組成液(B-9)は粒子状吸水性樹脂100重量部に対し、炭酸エチレン/水=0.9重量部/5.0重量部であった。表面架橋された吸水性樹脂粒子(N-4)の物性を表3に示す。なお、組成液(B-9)を混合機中で添加する工程において、粒子状吸水性樹脂(A-2)の温度に対して、得られる吸水性樹脂混合物の温度上昇は0℃であった。
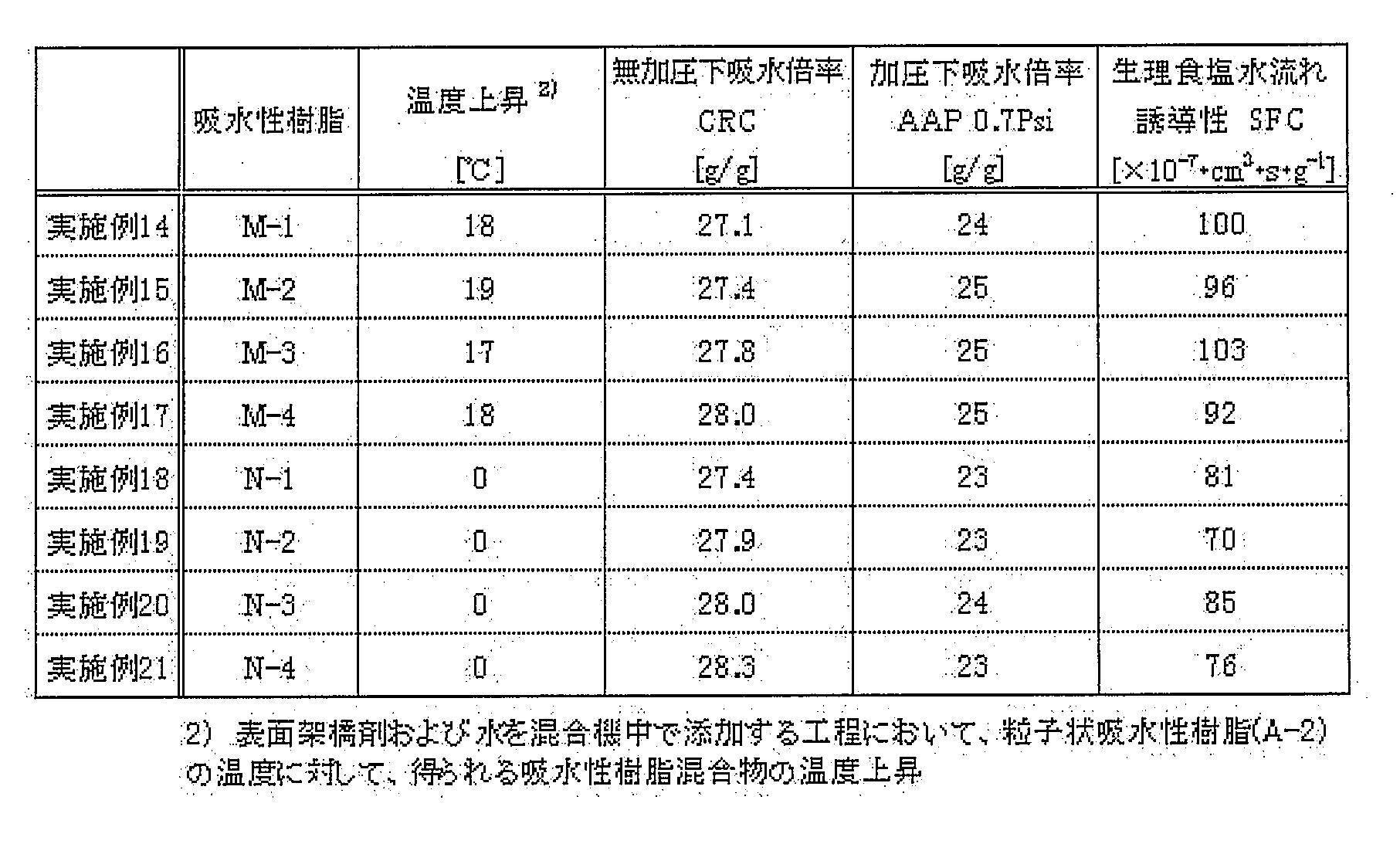
表3に示すように、いずれも本願の縦型混合機を用い、各表面架橋剤の使用に対し、表面架橋剤混合中の水蒸気の使用または混合物の昇温により、表面架橋された吸水性樹脂粒子の加圧下吸水倍率(AAP)または生理食塩水流れ誘導性(SFC)、特にSFCについて物性が向上することが確認された。
実施例14において、さらに硫酸アルミニウムを使用した。すなわち、実施例14で得られた、表面架橋された吸水性樹脂粒子(M-1)35gとガラスビーズ10gとを、225mlのマヨネーズ瓶に入れ、プロセス上の耐衝撃モデルとして、ペイントシェイカー(東洋精機(株)製)で30分間振とうした。次いで、振とう後の吸水性樹脂粒子30gに、該吸水性樹脂粒子100重量部に対して、50%硫酸アルミニウム水溶液/プロピレングリコール/乳酸ナトリウム=1.0重量部/0.025重量部/0.3重量部からなる通液性向上剤0.3gを添加・混合後、60℃の乾燥機で30分間硬化を行った。硬化後、ガラスビーズ10gの入った225mlのマヨネーズ瓶に入れ、ペイントシェイカーで10分間振とうし、アルミ表面処理物(O-1)を得た。アルミ表面処理物(O-1)の物性を表4に示す。
実施例15において、さらに硫酸アルミニウムを使用した。すなわち、実施例22において表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(M-2)に変更した以外は実施例22と同様に行い、アルミ表面処理物(O-2)を得た。アルミ表面処理物(O-2)の物性を表4に示す。
実施例16において、さらに硫酸アルミニウムを使用した。すなわち実施例22において、表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(M-3)に変更した以外は実施例22と同様に行い、アルミ表面処理物(O-3)を得た。アルミ表面処理物(O-3)の物性を表4に示す。
実施例17において、さらに硫酸アルミニウムを使用した。すなわち実施例22において、表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(M-4)に変更した以外は実施例22と同様に行い、アルミ表面処理物(O-4)を得た。アルミ表面処理物(O-4)の物性を表4に示す。
実施例18において、さらに硫酸アルミニウムを使用した。すなわち実施例22において、表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(N-1)に変更した以外は実施例22と同様に行い、アルミ表面処理物(P-1)を得た。アルミ表面処理物(P-1)の物性を表4に示す。
実施例19において、さらに硫酸アルミニウムを使用した。すなわち実施例22において、表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(N-2)に変更した以外は実施例22と同様に行い、アルミ表面処理物(P-2)を得た。アルミ表面処理物(P-2)の物性を表4に示す。
実施例20において、さらに硫酸アルミニウムを使用した。すなわち実施例22において、表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(N-3)に変更した以外は実施例22と同様に行い、アルミ表面処理物(P-3)を得た。アルミ表面処理物(P-3)の物性を表4に示す。
実施例21において、さらに硫酸アルミニウムを使用した。すなわち実施例22において、表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(N-4)に変更した以外は実施例22と同様に行い、アルミ表面処理物(P-4)を得た。アルミ表面処理物(P-4)の物性を表4に示す。

表4に示すように、いずれも本願の縦型混合機を用い、各表面架橋剤の使用に対し、表面架橋後の通液向上剤(硫酸アルミニウム)の添加量が同じであっても、表面架橋剤混合中の水蒸気の使用または混合物の昇温により、水蒸気なしまたは混合物の昇温なしの場合に比べ、生理食塩水流れ誘導性(SFC)の向上効果が大きくなることが確認された。
実施例14において、さらにシリカを使用した。すなわち、実施例14で得られた表面架橋された吸水性樹脂粒子(M-1)35gとガラスビーズ10gとを、225mlのマヨネーズ瓶に入れ、ペイントシェイカー(東洋精機(株)製)で30分間振とうした。次いで振とう後の吸水性樹脂粒子に通液性向上剤としてシリカ(AEROSIL200 日本アエロジル(株)社製)を、該吸水性樹脂粒子100重量部に対し0.5重量部加えて混合し、シリカ表面処理物(Q-1)を得た。シリカ表面処理物(Q-1)の物性を表5に示す。
実施例15において、さらにシリカを使用した。すなわち実施例30において、表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(M-2)に変更した以外は実施例30と同様に行い、シリカ表面処理物(Q-2)を得た。シリカ表面処理物(Q-2)の物性を表5に示す。
実施例16において、さらにシリカを使用した。すなわち実施例30において、表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(M-3)に変更した以外は実施例30と同様に行い、シリカ表面処理物(Q-3)を得た。シリカ表面処理物(Q-3)の物性を表5に示す。
実施例17において、さらにシリカを使用した。すなわち実施例30において、表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(M-4)に変更した以外は実施例30と同様に行い、シリカ表面処理物(Q-4)を得た。シリカ表面処理物(Q-4)の物性を表5に示す。
実施例18において、さらにシリカを使用した。すなわち実施例30において、表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(N-1)に変更した以外は実施例30と同様に行い、シリカ表面処理物(R-1)を得た。シリカ表面処理物(R-1)の物性を表5に示す。
実施例19において、さらにシリカを使用した。すなわち実施例30において、表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(N-2)に変更した以外は実施例30と同様に行い、シリカ表面処理物(R-2)を得た。シリカ表面処理物(R-2)の物性を表5に示す。
実施例20において、さらにシリカを使用した。すなわち実施例30において、表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(N-3)に変更した以外は実施例30と同様に行い、シリカ表面処理物(R-3)を得た。シリカ表面処理物(R-3)の物性を表5に示す。
実施例21において、さらにシリカを使用した。すなわち実施例30において、表面架橋された吸水性樹脂粒子(M-1)を表面架橋された吸水性樹脂粒子(N-4)に変更した以外は実施例30と同様に行い、シリカ表面処理物(R-4)を得た。シリカ表面処理物(R-4)の物性を表5に示す。

表5で示すように、いずれも本願の縦型混合機を用い、各表面架橋剤の使用に対し、シリカ表面処理物は、硫酸アルミニウムを添加した場合と同様に、表面架橋剤混合中の水蒸気の使用または混合物の昇温により、水蒸気なしまたは混合物の昇温なしの場合に比べ、生理食塩水流れ誘導性(SFC)の向上効果が大きくなることが確認された。
(a)300~3000rpmで撹拌羽根が回転する、
(b)撹拌羽根の回転軸は少なくとも1本は鉛直方向である、
(c)開口度が5~70%の仕切りにより、混合槽が上下に2室以上に分けられている、
を満たす連続混合装置を使用することを特徴とする方法。
2 投入口
3 排出口
4 添加剤投入口
5 撹拌羽根
6 回転軸
7 仕切り
8 堰(堰に連続して点線で囲まれた部分は堰の長さが可変であることを示す。)
9 水蒸気投入口
10 混合層(ケーシング)
Claims (31)
- 粒子状吸水性樹脂に、表面架橋剤および水を混合機中で添加する工程と、混合機から取り出した吸水性樹脂混合物を反応機中で加熱または活性エネルギー線照射で表面架橋剤と反応させる工程とを順次含む、表面架橋された吸水性樹脂粒子の製造方法であって、
上記混合機中で添加する水の一部または全部を水蒸気で添加することを特徴とする、吸水性樹脂の製造方法。 - 粒子状吸水性樹脂に、表面架橋剤および水を混合機中で添加する工程と、混合機から取り出した吸水性樹脂混合物を反応機中で加熱または活性エネルギー線照射で表面架橋剤と反応させる工程とを順次含む、表面架橋された吸水性樹脂粒子の製造方法であって、上記表面架橋剤および水を混合機中で添加する工程において、粒子状吸水性樹脂の温度に対して、得られる吸水性樹脂混合物の温度を2℃以上昇温させることを特徴とする、吸水性樹脂の製造方法。
- 上記粒子状吸水性樹脂に添加される水の一部または全部を水蒸気で添加する、請求項2に記載の製造方法。
- 上記粒子状吸水性樹脂に添加される水として、液体および水蒸気が併用される、請求項1~3のいずれか1項に記載の製造方法。
- 上記粒子状吸水性樹脂に添加される水として、表面架橋剤水溶液および水蒸気が併用される、請求項1~4のいずれか1項記載の製造方法。
- 上記粒子状吸水性樹脂100重量部に対して水0.1~10重量部が混合され、かつ、液体として供給された水が供給水全体の0~95重量%である、請求項1~3のいずれか1項に記載の製造方法。
- 上記混合機に供給される粒子状吸水性樹脂の温度が、水蒸気の温度より10~100℃低い、請求項1~6のいずれか1項に記載の製造方法。
- 上記混合機の内面の温度が、水蒸気の温度より10~100℃低い、請求項1~7のいずれか1項に記載の製造方法。
- 上記混合機と反応機とは、互いが連結された、上記各工程を連続的に行う装置であり、
混合機中の粒子状吸水性樹脂の平均滞留時間が1秒~5分であり、かつ、反応機中の吸水性樹脂混合物の平均滞留時間が6分~10時間である、請求項1~8のいずれか1項に記載の製造方法。 - 上記表面架橋剤が、カルボキシル基と脱水反応しうる架橋剤である、請求項1~9のいずれか1項に記載の製造方法。
- 上記水蒸気の供給により粒子状吸水性樹脂に取り込まれた水分量が、混合機へ供給される粒子状吸水性樹脂100重量部に対して、0.1~10重量部である、請求項1および3~10のいずれか1項に記載の製造方法。
- 上記供給される水蒸気の圧力が0.01~1MPa(ゲージ圧)である、請求項1および3~11のいずれか1項に記載の製造方法。
- 上記混合機内の露点が60~100℃である、請求項1~12のいずれか1項に記載の製造方法。
- 上記混合機に供給される粒子状吸水性樹脂の温度が30~90℃である、請求項1~13のいずれか1項に記載の製造方法。
- 上記混合機が縦型混合機である、請求項1~14のいずれか1項に記載の製造方法。
- 共有結合性表面架橋剤による表面架橋と同時または表面架橋後に、さらに、ポリアミン、多価金属塩、および水不溶性微粒子からなる群から選ばれる1種以上の通液性向上剤が添加される、請求項1~15のいずれか1項に記載の製造方法。
- 上記混合機へ投入する粒子状吸水性樹脂の温度に比べて、得られた吸水性樹脂混合物の温度を3~60℃昇温させる、請求項1~16のいずれか1項に記載の製造方法。
- 1時間あたりの処理量が1t以上の連続生産である、請求項1~17のいずれか1項に記載の製造方法。
- 上記混合機から取り出した吸水性樹脂混合物の温度が50~140℃である、請求項1~18のいずれか1項に記載の製造方法。
- 上記混合機から反応機の入口までに、保温または加温された排気装置が設けられている、請求項1~19のいずれか1項に記載の製造方法。
- 粒子状吸水性樹脂に、表面架橋剤および水を混合機中で添加する工程において、予め粒子状吸水性樹脂の温度を30~150℃とすること、および以下の条件(i)~(iii)、
(i)300~3000rpmで撹拌羽根が回転する、
(ii)撹拌羽根の回転軸は少なくとも1本は鉛直方向である、
(iii)開口度が5~70%の仕切りにより、混合槽が上下に2室以上に分けられている、を満たす連続混合装置を使用する、請求項1~13のいずれか1項に記載の製造方法。 - 粒子状吸水性樹脂に、表面架橋剤および水を混合機中で添加する工程において、予め粒子状吸水性樹脂の温度を30~150℃とすること、および以下の条件(i)~(iii)、
(i)300~3000rpmで撹拌羽根が回転する、
(ii)撹拌羽根の回転軸は少なくとも1本は鉛直方向である、
(iii)開口度が5~70%の仕切りにより、混合槽が上下に2室以上に分けられている、を満たす連続混合装置を使用する製造方法。 - 粒子状吸水性樹脂に、表面架橋剤および水を混合機中で添加する工程において、予め粒子状吸水性樹脂の温度を40~120℃とする、請求項21または22に記載の方法。
- 上記混合機は内壁が加熱または保温されている、請求項21~23のいずれか1項に記載の方法。
- 上記混合機は内壁が50~150℃に加熱されている、請求項21~24のいずれか1項に記載の方法。
- 上記混合機の内壁の一部または全面に、水に対する接触角が60°以上であり熱変形温度が70℃以上の材料を用いる、請求項21~25のいずれか1項に記載の方法。
- 上記混合機は、開口度が5~70%であり、撹拌羽根の回転軸を中心に回転する仕切りを備えている、請求項21~26のいずれか1項に記載の方法。
- 上記混合機において、少なくとも一つの撹拌羽根の先端が描く軌道より回転軸側に排出口がある、請求項21~27のいずれか1項に記載の方法。
- 上記混合機において、排出口に堰構造を備え、その堰構造は水平面に対する角度が10~80°、側壁から回転軸側への水平方向の長さが混合槽の最大半径の1~40%で、上記範囲内で角度および/または長さを変更できる、請求項21~28のいずれか1項に記載の方法。
- 上記混合機において、混合槽内部の高さ(H)を混合槽の最大直径(D)で除した値(H/D)が0.1~1である、請求項21~29のいずれか1項に記載の方法。
- 上記混合機に投入される粒子状吸水性樹脂の量が、混合機の混合槽容積1Lあたり10~300kg/hrである、請求項21~30のいずれか1項に記載の方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201080010035.3A CN102341435B (zh) | 2009-03-04 | 2010-03-04 | 吸水性树脂的制造方法 |
| US13/254,573 US8648150B2 (en) | 2009-03-04 | 2010-03-04 | Method for producing water absorbent resin |
| JP2011502661A JP5615801B2 (ja) | 2009-03-04 | 2010-03-04 | 吸水性樹脂の製造方法 |
| EP20100748535 EP2404954B1 (en) | 2009-03-04 | 2010-03-04 | Process for producing water-absorbing resin |
| US14/136,339 US9796820B2 (en) | 2009-03-04 | 2013-12-20 | Method for producing water absorbent resin |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009050381 | 2009-03-04 | ||
| JP2009-050381 | 2009-03-04 | ||
| JP2009197091 | 2009-08-27 | ||
| JP2009-197091 | 2009-08-27 | ||
| JP2010022690 | 2010-02-04 | ||
| JP2010-022690 | 2010-02-04 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/254,573 A-371-Of-International US8648150B2 (en) | 2009-03-04 | 2010-03-04 | Method for producing water absorbent resin |
| US14/136,339 Division US9796820B2 (en) | 2009-03-04 | 2013-12-20 | Method for producing water absorbent resin |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010100936A1 true WO2010100936A1 (ja) | 2010-09-10 |
Family
ID=42709502
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/001521 WO2010100936A1 (ja) | 2009-03-04 | 2010-03-04 | 吸水性樹脂の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US8648150B2 (ja) |
| EP (1) | EP2404954B1 (ja) |
| JP (2) | JP5615801B2 (ja) |
| CN (1) | CN102341435B (ja) |
| WO (1) | WO2010100936A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012102407A1 (ja) | 2011-01-28 | 2012-08-02 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法 |
| JP2015526572A (ja) * | 2012-08-29 | 2015-09-10 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | 吸水性ポリマー粒子の製造方法 |
| JP2016529368A (ja) * | 2013-08-27 | 2016-09-23 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| JP2020520390A (ja) * | 2018-04-03 | 2020-07-09 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| US10829630B2 (en) | 2016-03-11 | 2020-11-10 | Lg Chem, Ltd. | Super absorbent polymer |
| WO2021187323A1 (ja) * | 2020-03-18 | 2021-09-23 | 住友精化株式会社 | 吸水性樹脂粒子を製造する方法 |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ITCZ20080010A1 (it) * | 2008-10-30 | 2010-04-30 | Cit Di Tassone Giuseppe | Dispositivo per la miscelazione ed il confezionamento di materiali in polvere di qualsiasi granulometria |
| NZ593495A (en) * | 2011-06-16 | 2014-02-28 | David Kenneth Pinches | Disc for industrial plants |
| JP6002773B2 (ja) | 2012-09-11 | 2016-10-05 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水剤の製造方法及びその吸水剤 |
| CN104822740B (zh) | 2012-12-03 | 2020-08-11 | 株式会社日本触媒 | 聚丙烯酸(盐)系吸水性树脂及其制造方法 |
| US10662300B2 (en) * | 2013-05-10 | 2020-05-26 | Nippon Shokubai Co., Ltd. | Method for producing polyacrylic acid (salt)-based water-absorbent resin |
| US9868800B2 (en) | 2014-04-25 | 2018-01-16 | Nippon Shokubai Co., Ltd. | Method for producing polyacrylic acid (salt)-based water-absorbent resin |
| EP3009474B1 (de) | 2014-10-16 | 2017-09-13 | Evonik Degussa GmbH | Herstellverfahren für wasserlösliche Polymere |
| CN104356256B (zh) * | 2014-11-25 | 2016-06-08 | 中广核达胜加速器技术有限公司 | 一种电子束辐照喷淋法合成高性能吸水树脂的方法 |
| KR101919985B1 (ko) * | 2015-06-10 | 2018-11-19 | 주식회사 엘지화학 | 내파쇄성 고흡수성 수지 및 그 제조방법 |
| KR102429796B1 (ko) | 2016-03-31 | 2022-08-04 | 스미토모 세이카 가부시키가이샤 | 흡수성 수지 입자의 제조 장치 |
| AT519978B1 (de) * | 2017-12-19 | 2018-12-15 | Sonderhoff Eng Gmbh | Vorrichtung zur Herstellung von Kunststoffteilen |
| JP7257090B2 (ja) * | 2018-03-29 | 2023-04-13 | Sdpグローバル株式会社 | 吸水性樹脂粒子及びその製造方法 |
| KR102500281B1 (ko) | 2018-12-12 | 2023-02-15 | 주식회사 엘지화학 | 고흡수성 수지의 제조 방법 |
| CN111377654B (zh) * | 2018-12-29 | 2021-11-09 | 江苏苏博特新材料股份有限公司 | 一种杂化型无碱速凝剂及其制备方法 |
| CN110302712A (zh) * | 2019-07-18 | 2019-10-08 | 南京汇科高分子材料有限公司 | 一种玻璃包边材料生产用预处理釜 |
| JP7270828B2 (ja) * | 2020-02-14 | 2023-05-10 | 株式会社日本触媒 | 吸水性樹脂およびその製造方法 |
| CN111895272A (zh) * | 2020-07-24 | 2020-11-06 | 南通华林科纳半导体设备有限公司 | 抛光液dsp供应系统 |
Citations (104)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4093776A (en) | 1976-10-07 | 1978-06-06 | Kao Soap Co., Ltd. | Process for preparation of spontaneously-crosslinked alkali metal acrylate polymers |
| US4286082A (en) | 1979-04-06 | 1981-08-25 | Nippon Shokubai Kagaku Kogyo & Co., Ltd. | Absorbent resin composition and process for producing same |
| US4367323A (en) | 1980-12-03 | 1983-01-04 | Sumitomo Chemical Company, Limited | Production of hydrogels |
| US4446261A (en) | 1981-03-25 | 1984-05-01 | Kao Soap Co., Ltd. | Process for preparation of high water-absorbent polymer beads |
| US4625001A (en) | 1984-09-25 | 1986-11-25 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Method for continuous production of cross-linked polymer |
| US4683274A (en) | 1984-10-05 | 1987-07-28 | Seitetsu Kagaku Co., Ltd. | Process for producing a water-absorbent resin |
| US4734478A (en) | 1984-07-02 | 1988-03-29 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Water absorbing agent |
| JPS6399211A (ja) * | 1986-06-04 | 1988-04-30 | Hayashikane Zosen Kk | 改質吸水性樹脂の製造方法 |
| US4755562A (en) | 1986-06-10 | 1988-07-05 | American Colloid Company | Surface treated absorbent polymers |
| US4783510A (en) | 1986-06-04 | 1988-11-08 | Taiyo Fishery Co., Ltd. | Process for improving a water absorbent polyacrylic acid polymer and an improved polymer produced by said process |
| JPH01113406A (ja) | 1987-07-16 | 1989-05-02 | Lion Corp | 高吸水性ポリマーの製造方法 |
| US4873299A (en) | 1986-03-21 | 1989-10-10 | Basf Aktiengesellschaft | Batchwise preparation of crosslinked, finely divided polymers |
| JPH01297430A (ja) | 1988-05-24 | 1989-11-30 | Nippon Shokubai Kagaku Kogyo Co Ltd | 吸水性樹脂の表面処理方法 |
| US4893999A (en) | 1985-12-18 | 1990-01-16 | Chemische Fabrik Stockhausen Gmbh | Apparatus for the continuous production of polymers and copolymers of water-soluble monomers |
| JPH02160814A (ja) | 1988-12-14 | 1990-06-20 | Kazuo Saotome | 吸水性樹脂の改質方法 |
| US4973632A (en) | 1988-06-28 | 1990-11-27 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Production process for water-absorbent resin |
| US4985518A (en) | 1981-10-26 | 1991-01-15 | American Colloid Company | Process for preparing water-absorbing resins |
| JPH03152104A (ja) | 1989-09-15 | 1991-06-28 | Dow Chem Co:The | 水性流体吸収性微粒子の再循環方法および装置 |
| US5051259A (en) | 1987-12-15 | 1991-09-24 | Coloplast A/S | Skin barrier product with discontinuous adhesive layer |
| US5124416A (en) | 1988-05-23 | 1992-06-23 | Nippon Shokubai Kagaku Kogyo, Co., Ltd. | Method for production of absorbent polymer |
| JPH04214734A (ja) | 1990-04-02 | 1992-08-05 | Nippon Shokubai Co Ltd | 吸水性樹脂の表面処理方法 |
| JPH04227705A (ja) | 1990-04-27 | 1992-08-17 | Nippon Shokubai Co Ltd | 耐塩性吸水性樹脂の製造方法 |
| US5145906A (en) | 1989-09-28 | 1992-09-08 | Hoechst Celanese Corporation | Super-absorbent polymer having improved absorbency properties |
| US5206205A (en) | 1991-08-15 | 1993-04-27 | Kimberly-Clark Corporation | Thermal treatment of superabsorbents to enhance their rate of absorbency under load |
| US5244735A (en) | 1988-06-28 | 1993-09-14 | Nippon Shokubai Kagaku Kabushiki Kaisha | Water-absorbent resin and production process |
| US5250640A (en) | 1991-04-10 | 1993-10-05 | Nippon Shokubai Co., Ltd. | Method for production of particulate hydrogel polymer and absorbent resin |
| US5264495A (en) | 1990-04-27 | 1993-11-23 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Method for production of salt-resistant absorbent resin |
| US5275773A (en) | 1991-02-01 | 1994-01-04 | Nippon Shokubai Co., Ltd. | Method for production of particulate hydrated gel polymer and absorbent resin |
| EP0603292A1 (en) | 1991-09-09 | 1994-06-29 | The Dow Chemical Company | Superabsorbent polymers and process for producing |
| US5369148A (en) | 1990-04-27 | 1994-11-29 | Nippon Shokubai Co., Ltd. | Method for continuous agglomeration of an absorbent resin powder and apparatus therefor |
| US5380808A (en) | 1990-07-17 | 1995-01-10 | Sanyo Chemical Industries, Ltd. | Process for producing water-absorbing resins |
| US5422405A (en) | 1992-12-16 | 1995-06-06 | Nippon Shokubai Co., Ltd. | Method for production of absorbent resin |
| JPH07224204A (ja) | 1994-02-10 | 1995-08-22 | Toagosei Co Ltd | 吸水性樹脂の製造方法 |
| JPH07242709A (ja) | 1994-03-03 | 1995-09-19 | Toagosei Co Ltd | 吸水性樹脂の製造方法 |
| US5478879A (en) | 1991-01-22 | 1995-12-26 | Nippon Shokubai Co., Ltd. | Method for production of absorbent resin |
| US5610208A (en) | 1994-02-17 | 1997-03-11 | Nippon Shokubai Co., Ltd. | Water-absorbent agent, method for production thereof, and water-absorbent composition |
| US5669894A (en) | 1994-03-29 | 1997-09-23 | The Procter & Gamble Company | Absorbent members for body fluids having good wet integrity and relatively high concentrations of hydrogel-forming absorbent polymer |
| JPH09509591A (ja) | 1994-02-17 | 1997-09-30 | ザ、プロクター、エンド、ギャンブル、カンパニー | 改良された吸収特性を有する吸収性材料およびその製造方法 |
| US5672633A (en) | 1993-09-29 | 1997-09-30 | Chemische Fabrik Stockhausen Gmbh | Powdery polymers capable of absorbing aqueous liquids, a process for their production and their use as absorbents |
| EP0811636A1 (en) | 1996-06-05 | 1997-12-10 | Nippon Shokubai Co., Ltd. | Method for production of cross-linked polymer |
| EP0922717A1 (en) | 1997-12-10 | 1999-06-16 | Nippon Shokubai Co., Ltd. | Production process of water-absorbent resin |
| EP0955086A2 (en) | 1998-04-28 | 1999-11-10 | Nippon Shokubai Co., Ltd. | Method for production of shaped hydrogel of absorbent resin |
| JPH11349625A (ja) * | 1998-06-10 | 1999-12-21 | Sanyo Chem Ind Ltd | 吸水剤の製造法および吸水剤 |
| US6071976A (en) | 1995-12-27 | 2000-06-06 | Nippon Shokubai Co., Ltd. | Water absorbing agent, manufacturing method thereof, and manufacturing machine thereof |
| JP2000189794A (ja) | 1998-12-28 | 2000-07-11 | Nippon Shokubai Co Ltd | 吸水材の製造方法 |
| US6100305A (en) | 1996-10-24 | 2000-08-08 | Nippon Shokubai Co., Ltd. | Method of production of water-absorbing resin |
| US6107358A (en) | 1996-08-23 | 2000-08-22 | Nippon Shokubai Co., Ltd. | Water-absorbent resin and method for production thereof |
| US6133193A (en) | 1994-10-26 | 2000-10-17 | Nippon Shokubai Co., Ltd. | Absorbent resin composition and method for production thereof |
| US6140395A (en) | 1997-12-25 | 2000-10-31 | Nippon Shokubai Co., Ltd. | Method of producing hydrophilic resin |
| US6187902B1 (en) | 1997-12-25 | 2001-02-13 | Nippon Shokubai Co., Ltd. | Production process of hydrophilic crosslinked polymer |
| US6228930B1 (en) | 1997-06-18 | 2001-05-08 | Nippon Shokubai Co., Ltd. | Water-absorbent resin granule-containing composition and production process for water-absorbent resin granule |
| US6239230B1 (en) | 1999-09-07 | 2001-05-29 | Bask Aktiengesellschaft | Surface-treated superabsorbent polymer particles |
| US6241928B1 (en) | 1998-04-28 | 2001-06-05 | Nippon Shokubai Co., Ltd. | Method for production of shaped hydrogel of absorbent resin |
| JP3175790B2 (ja) | 1991-04-10 | 2001-06-11 | 株式会社日本触媒 | 粒子状含水ゲル状重合体および吸水性樹脂の製造方法 |
| US6254990B1 (en) | 1998-02-18 | 2001-07-03 | Nippon Shokubai Co., Ltd. | Surface-crosslinking process for water-absorbent resin |
| US6265488B1 (en) | 1998-02-24 | 2001-07-24 | Nippon Shokubai Co., Ltd. | Production process for water-absorbing agent |
| US6297319B1 (en) | 1998-11-05 | 2001-10-02 | Nippon Shokubai Co., Ltd. | Water-absorbing agent and production process therefor |
| EP1165631A1 (de) | 1999-03-05 | 2002-01-02 | STOCKHAUSEN GmbH & CO. KG | Pulverförmige, vernetzte, wässrige flüssigkeiten sowie blut absorbierende polymere, verfahren zu ihrer herstellung und ihre verwendung |
| EP1178059A2 (en) | 2000-08-03 | 2002-02-06 | Nippon Shokubai Co., Ltd. | Water-absorbent resin, hydropolymer, process for producing them, and uses of them |
| US6372852B2 (en) | 1998-03-31 | 2002-04-16 | Nippon Shokubai Co., Ltd | Water-absorbing composition and production process for water-absorbing agent |
| JP2002121291A (ja) * | 2000-02-29 | 2002-04-23 | Nippon Shokubai Co Ltd | 吸水性樹脂粉末およびその製造方法 |
| JP2002201290A (ja) * | 2000-09-20 | 2002-07-19 | Nippon Shokubai Co Ltd | 吸水性樹脂およびその製造方法 |
| US6472478B1 (en) | 1998-02-21 | 2002-10-29 | Basf Aktiengesellschaft | Process for crosslinking hydrogels with bis- and poly-2- oxazolidinones |
| US6514615B1 (en) | 1999-06-29 | 2003-02-04 | Stockhausen Gmbh & Co. Kg | Superabsorbent polymers having delayed water absorption characteristics |
| US6559239B1 (en) | 1998-11-26 | 2003-05-06 | Basf Aktiengesellschaft | Method for the secondary cross-linking of hydrogels with N-acyl-2-oxazolidinones |
| US6599989B2 (en) | 1998-03-03 | 2003-07-29 | Nippon Skokubai Co., Ltd. | Water-absorbent agents containing polycarboxylic amine chelating agents |
| US6620899B1 (en) | 1998-10-15 | 2003-09-16 | E. I. Du Pont De Nemours And Company | Polymers, containing a fluorocyclobutyl ring and their preparation |
| JP2003303306A (ja) | 2002-04-08 | 2003-10-24 | Nec Soft Ltd | プリペイド方式による商品代金支払システム |
| US6657015B1 (en) | 1998-11-26 | 2003-12-02 | Basf Aktiengesellschaft | Method for the secondary cross-linking of hydrogels with 2-oxotetrahydro-1,3-oxazines |
| JP2004037900A (ja) | 2002-07-04 | 2004-02-05 | Ricoh Co Ltd | 中間転写装置及び画像形成装置 |
| US6710141B1 (en) | 1999-11-20 | 2004-03-23 | Basf Aktiengesellschaft | Method for continuously producing cross-linked fine-particle geleous polymerizates |
| US20040069915A1 (en) | 2002-09-20 | 2004-04-15 | Alain Guennec | Satellite antenna holder |
| US20040106745A1 (en) | 2001-06-08 | 2004-06-03 | Yasuhisa Nakashima | Water-absorbing agent and production process therefor, and sanitary material |
| US20040176557A1 (en) | 2000-09-04 | 2004-09-09 | Richard Mertens | Pulverulent, crosslinked polymers which absorb aqueous liquids and blood |
| US6809158B2 (en) | 2000-10-20 | 2004-10-26 | Nippon Shokubai Co., Ltd. | Water-absorbing agent and process for producing the same |
| US20040234607A1 (en) | 2003-04-25 | 2004-11-25 | Nippon Shokubai Co.,Ltd. | Method for disintegrating hydrate polymer and method for production of water-absorbent resin |
| JP2004352941A (ja) | 2003-05-30 | 2004-12-16 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造法 |
| JP2004352940A (ja) | 2003-05-30 | 2004-12-16 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造方法および鋤型混合装置 |
| JP2005016393A (ja) | 2003-06-25 | 2005-01-20 | Toyota Motor Corp | 内燃機関の排気浄化システム |
| US20050048221A1 (en) | 2003-08-27 | 2005-03-03 | Yoshio Irie | Process for production of surface-treated particulate water-absorbent resin |
| JP2005054151A (ja) | 2003-08-07 | 2005-03-03 | Nippon Shokubai Co Ltd | 粒子状吸水性樹脂の製法 |
| US20050046069A1 (en) | 2003-09-01 | 2005-03-03 | Masazumi Sasabe | Process for production of hydrogel particles and process for cutting of high-concentration hydrogel sheet |
| US6875511B2 (en) | 2002-05-30 | 2005-04-05 | Nippon Shokubai Co., Ltd. | Production process for particulate water-absorbent resin |
| JP2005097585A (ja) * | 2003-08-27 | 2005-04-14 | Nippon Shokubai Co Ltd | 表面処理された粒子状吸水性樹脂の製造方法 |
| US20050215734A1 (en) | 2004-03-24 | 2005-09-29 | Yorimichi Dairoku | Method for continuous production of water-absorbent resin |
| JP2005307195A (ja) | 2004-03-24 | 2005-11-04 | Nippon Shokubai Co Ltd | 吸水性樹脂の連続製造方法 |
| US6987151B2 (en) | 2001-09-12 | 2006-01-17 | Dow Global Technologies Inc. | Continuous polymerization process for the manufacture of superabsorbent polymers |
| US20060024755A1 (en) | 2001-04-30 | 2006-02-02 | George Jackowski | Biopolymer marker indicative of disease state having a molecular weight of 1424 daltons |
| US20060073969A1 (en) | 2003-02-10 | 2006-04-06 | Kazushi Torii | Vater-absorbent resin composition and its production process |
| US20060101271A1 (en) | 2004-11-10 | 2006-05-11 | Michael Thomas | Method and system for conveying alternate acceptable canonicalizations of a digitally signed piece of electronic mail |
| WO2006082197A1 (en) | 2005-02-01 | 2006-08-10 | Basf Aktiengesellschaft | Polyamine-coated superabsorbent polymers |
| WO2006082188A1 (en) | 2005-02-01 | 2006-08-10 | Basf Aktiengesellschaft | Polyamine-coated superabsorbent polymers |
| WO2006082189A1 (en) | 2005-02-01 | 2006-08-10 | Basf Aktiengesellschaft | Polyamine-coated superabsorbent polymers |
| US20060204755A1 (en) | 2003-02-10 | 2006-09-14 | Kazushi Torii | Walter-absorbing agent |
| US20060247351A1 (en) | 2005-03-14 | 2006-11-02 | Kazushi Torii | Water-absorbing agent and its production process |
| US7157141B2 (en) | 2000-03-31 | 2007-01-02 | Stockhausen Gmbh | Pulverulent polymers crosslinked on the surface |
| US7179862B2 (en) | 1999-03-05 | 2007-02-20 | Stockhausen Gmbh | Powdery, cross-linked absorbent polymers method for the production thereof and their use |
| US20070106013A1 (en) | 2003-06-24 | 2007-05-10 | Yoshifumi Adachi | Water absorbent resin composition and production method thereof |
| US20070149760A1 (en) | 2005-12-22 | 2007-06-28 | Kenji Kadonaga | Method for surface crosslinking water-absorbing resin and method for manufacturing water-absorbing resin |
| EP1824910A2 (en) | 2004-12-10 | 2007-08-29 | Nippon Shokubai Co.,Ltd. | Method for production of modified water absorbent resin |
| JP2008038128A (ja) * | 2005-12-22 | 2008-02-21 | Nippon Shokubai Co Ltd | 吸水性樹脂の表面架橋方法および吸水性樹脂の製造方法 |
| US20080080300A1 (en) | 2004-09-28 | 2008-04-03 | Basf Aktiengesellschaft | Mixing Kneader and Process for Preparing Poly(Meth)Acrylates Using the Mixing Kneader |
| US20080090961A1 (en) | 2006-10-13 | 2008-04-17 | Bayer Materialscience Llc | Impact resistant, flame retardant thermoplastic molding composition |
| US7473739B2 (en) | 2004-02-05 | 2009-01-06 | Nippon Shokubai Co., Ltd. | Particulate water absorbent agent and production method thereof, and water absorbent article |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS522877A (en) | 1975-06-24 | 1977-01-10 | Noubi Kogyo Kk | Process and apparatus for granulating of high molecular coagulating ag ents |
| DE3308420A1 (de) | 1983-03-09 | 1984-09-13 | Wolff Walsrode Ag, 3030 Walsrode | Verfahren zur kontinuierlichen granulierung von carboxymethylcellulose |
| EP0386897B1 (en) | 1989-02-28 | 1995-05-03 | Nippon Shokubai Co., Ltd. | Process for producing quality-improved water-absorbent polymers and products |
| DE4131045C1 (ja) * | 1991-09-18 | 1992-11-19 | Cassella Ag, 6000 Frankfurt, De | |
| JP3742098B2 (ja) * | 1995-12-27 | 2006-02-01 | 株式会社日本触媒 | 吸水剤の製造方法並びに吸水性樹脂への水性液の混合方法 |
| DE60112630T3 (de) | 2000-02-29 | 2016-03-03 | Nippon Shokubai Co., Ltd. | Verfaren zur Herstellung eines wasserabsorbierenden Harzpulvers |
| US6720389B2 (en) * | 2000-09-20 | 2004-04-13 | Nippon Shokubai Co., Ltd. | Water-absorbent resin and production process therefor |
| AU2003296558A1 (en) | 2002-10-25 | 2004-05-13 | Stockhausen Gmbh | Absorbent polymer structure provided with an improved retention capacity and permeability |
| TWI302541B (en) | 2003-05-09 | 2008-11-01 | Nippon Catalytic Chem Ind | Water-absorbent resin and its production process |
| CN101242891B (zh) * | 2005-08-24 | 2011-05-11 | 巴斯夫欧洲公司 | 生产吸水性聚合物颗粒的方法 |
| DE102007024080A1 (de) * | 2007-05-22 | 2008-11-27 | Evonik Stockhausen Gmbh | Verfahren zum schonenden Mischen und Beschichten von Superabsorbern |
-
2010
- 2010-03-04 EP EP20100748535 patent/EP2404954B1/en active Active
- 2010-03-04 JP JP2011502661A patent/JP5615801B2/ja active Active
- 2010-03-04 WO PCT/JP2010/001521 patent/WO2010100936A1/ja active Application Filing
- 2010-03-04 US US13/254,573 patent/US8648150B2/en active Active
- 2010-03-04 CN CN201080010035.3A patent/CN102341435B/zh active Active
-
2013
- 2013-12-20 US US14/136,339 patent/US9796820B2/en active Active
-
2014
- 2014-09-10 JP JP2014184643A patent/JP5847260B2/ja active Active
Patent Citations (110)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4093776A (en) | 1976-10-07 | 1978-06-06 | Kao Soap Co., Ltd. | Process for preparation of spontaneously-crosslinked alkali metal acrylate polymers |
| US4286082A (en) | 1979-04-06 | 1981-08-25 | Nippon Shokubai Kagaku Kogyo & Co., Ltd. | Absorbent resin composition and process for producing same |
| US4367323A (en) | 1980-12-03 | 1983-01-04 | Sumitomo Chemical Company, Limited | Production of hydrogels |
| US4446261A (en) | 1981-03-25 | 1984-05-01 | Kao Soap Co., Ltd. | Process for preparation of high water-absorbent polymer beads |
| US4985518A (en) | 1981-10-26 | 1991-01-15 | American Colloid Company | Process for preparing water-absorbing resins |
| US4734478A (en) | 1984-07-02 | 1988-03-29 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Water absorbing agent |
| US4625001A (en) | 1984-09-25 | 1986-11-25 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Method for continuous production of cross-linked polymer |
| US4683274A (en) | 1984-10-05 | 1987-07-28 | Seitetsu Kagaku Co., Ltd. | Process for producing a water-absorbent resin |
| US4893999A (en) | 1985-12-18 | 1990-01-16 | Chemische Fabrik Stockhausen Gmbh | Apparatus for the continuous production of polymers and copolymers of water-soluble monomers |
| US4873299A (en) | 1986-03-21 | 1989-10-10 | Basf Aktiengesellschaft | Batchwise preparation of crosslinked, finely divided polymers |
| JPS6399211A (ja) * | 1986-06-04 | 1988-04-30 | Hayashikane Zosen Kk | 改質吸水性樹脂の製造方法 |
| US4783510A (en) | 1986-06-04 | 1988-11-08 | Taiyo Fishery Co., Ltd. | Process for improving a water absorbent polyacrylic acid polymer and an improved polymer produced by said process |
| US4824901A (en) | 1986-06-10 | 1989-04-25 | American Colloid Company | Surface treated absorbent polymers |
| US4755562A (en) | 1986-06-10 | 1988-07-05 | American Colloid Company | Surface treated absorbent polymers |
| JPH01113406A (ja) | 1987-07-16 | 1989-05-02 | Lion Corp | 高吸水性ポリマーの製造方法 |
| US5051259A (en) | 1987-12-15 | 1991-09-24 | Coloplast A/S | Skin barrier product with discontinuous adhesive layer |
| US5124416A (en) | 1988-05-23 | 1992-06-23 | Nippon Shokubai Kagaku Kogyo, Co., Ltd. | Method for production of absorbent polymer |
| JPH01297430A (ja) | 1988-05-24 | 1989-11-30 | Nippon Shokubai Kagaku Kogyo Co Ltd | 吸水性樹脂の表面処理方法 |
| US4973632A (en) | 1988-06-28 | 1990-11-27 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Production process for water-absorbent resin |
| US5244735A (en) | 1988-06-28 | 1993-09-14 | Nippon Shokubai Kagaku Kabushiki Kaisha | Water-absorbent resin and production process |
| JPH02160814A (ja) | 1988-12-14 | 1990-06-20 | Kazuo Saotome | 吸水性樹脂の改質方法 |
| JPH03152104A (ja) | 1989-09-15 | 1991-06-28 | Dow Chem Co:The | 水性流体吸収性微粒子の再循環方法および装置 |
| US5145906A (en) | 1989-09-28 | 1992-09-08 | Hoechst Celanese Corporation | Super-absorbent polymer having improved absorbency properties |
| JPH04214734A (ja) | 1990-04-02 | 1992-08-05 | Nippon Shokubai Co Ltd | 吸水性樹脂の表面処理方法 |
| US5264495A (en) | 1990-04-27 | 1993-11-23 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Method for production of salt-resistant absorbent resin |
| JPH04227705A (ja) | 1990-04-27 | 1992-08-17 | Nippon Shokubai Co Ltd | 耐塩性吸水性樹脂の製造方法 |
| US5369148A (en) | 1990-04-27 | 1994-11-29 | Nippon Shokubai Co., Ltd. | Method for continuous agglomeration of an absorbent resin powder and apparatus therefor |
| US5380808A (en) | 1990-07-17 | 1995-01-10 | Sanyo Chemical Industries, Ltd. | Process for producing water-absorbing resins |
| US5478879A (en) | 1991-01-22 | 1995-12-26 | Nippon Shokubai Co., Ltd. | Method for production of absorbent resin |
| US5275773A (en) | 1991-02-01 | 1994-01-04 | Nippon Shokubai Co., Ltd. | Method for production of particulate hydrated gel polymer and absorbent resin |
| US5250640A (en) | 1991-04-10 | 1993-10-05 | Nippon Shokubai Co., Ltd. | Method for production of particulate hydrogel polymer and absorbent resin |
| JP3175790B2 (ja) | 1991-04-10 | 2001-06-11 | 株式会社日本触媒 | 粒子状含水ゲル状重合体および吸水性樹脂の製造方法 |
| US5206205A (en) | 1991-08-15 | 1993-04-27 | Kimberly-Clark Corporation | Thermal treatment of superabsorbents to enhance their rate of absorbency under load |
| EP0603292A1 (en) | 1991-09-09 | 1994-06-29 | The Dow Chemical Company | Superabsorbent polymers and process for producing |
| US5422405A (en) | 1992-12-16 | 1995-06-06 | Nippon Shokubai Co., Ltd. | Method for production of absorbent resin |
| US5672633A (en) | 1993-09-29 | 1997-09-30 | Chemische Fabrik Stockhausen Gmbh | Powdery polymers capable of absorbing aqueous liquids, a process for their production and their use as absorbents |
| JPH07224204A (ja) | 1994-02-10 | 1995-08-22 | Toagosei Co Ltd | 吸水性樹脂の製造方法 |
| US5610208A (en) | 1994-02-17 | 1997-03-11 | Nippon Shokubai Co., Ltd. | Water-absorbent agent, method for production thereof, and water-absorbent composition |
| JPH09509591A (ja) | 1994-02-17 | 1997-09-30 | ザ、プロクター、エンド、ギャンブル、カンパニー | 改良された吸収特性を有する吸収性材料およびその製造方法 |
| JPH07242709A (ja) | 1994-03-03 | 1995-09-19 | Toagosei Co Ltd | 吸水性樹脂の製造方法 |
| US5669894A (en) | 1994-03-29 | 1997-09-23 | The Procter & Gamble Company | Absorbent members for body fluids having good wet integrity and relatively high concentrations of hydrogel-forming absorbent polymer |
| US6133193A (en) | 1994-10-26 | 2000-10-17 | Nippon Shokubai Co., Ltd. | Absorbent resin composition and method for production thereof |
| US6071976A (en) | 1995-12-27 | 2000-06-06 | Nippon Shokubai Co., Ltd. | Water absorbing agent, manufacturing method thereof, and manufacturing machine thereof |
| EP0811636A1 (en) | 1996-06-05 | 1997-12-10 | Nippon Shokubai Co., Ltd. | Method for production of cross-linked polymer |
| US6107358A (en) | 1996-08-23 | 2000-08-22 | Nippon Shokubai Co., Ltd. | Water-absorbent resin and method for production thereof |
| US6100305A (en) | 1996-10-24 | 2000-08-08 | Nippon Shokubai Co., Ltd. | Method of production of water-absorbing resin |
| US6458921B1 (en) | 1997-06-18 | 2002-10-01 | Nippon Shokubai Co., Ltd. | Water-absorbent resin granule-containing composition and production process for water-absorbent resin granule |
| US6228930B1 (en) | 1997-06-18 | 2001-05-08 | Nippon Shokubai Co., Ltd. | Water-absorbent resin granule-containing composition and production process for water-absorbent resin granule |
| EP0922717A1 (en) | 1997-12-10 | 1999-06-16 | Nippon Shokubai Co., Ltd. | Production process of water-absorbent resin |
| US6140395A (en) | 1997-12-25 | 2000-10-31 | Nippon Shokubai Co., Ltd. | Method of producing hydrophilic resin |
| US6187902B1 (en) | 1997-12-25 | 2001-02-13 | Nippon Shokubai Co., Ltd. | Production process of hydrophilic crosslinked polymer |
| US6254990B1 (en) | 1998-02-18 | 2001-07-03 | Nippon Shokubai Co., Ltd. | Surface-crosslinking process for water-absorbent resin |
| US6472478B1 (en) | 1998-02-21 | 2002-10-29 | Basf Aktiengesellschaft | Process for crosslinking hydrogels with bis- and poly-2- oxazolidinones |
| US6265488B1 (en) | 1998-02-24 | 2001-07-24 | Nippon Shokubai Co., Ltd. | Production process for water-absorbing agent |
| US6599989B2 (en) | 1998-03-03 | 2003-07-29 | Nippon Skokubai Co., Ltd. | Water-absorbent agents containing polycarboxylic amine chelating agents |
| US6372852B2 (en) | 1998-03-31 | 2002-04-16 | Nippon Shokubai Co., Ltd | Water-absorbing composition and production process for water-absorbing agent |
| US6241928B1 (en) | 1998-04-28 | 2001-06-05 | Nippon Shokubai Co., Ltd. | Method for production of shaped hydrogel of absorbent resin |
| EP0955086A2 (en) | 1998-04-28 | 1999-11-10 | Nippon Shokubai Co., Ltd. | Method for production of shaped hydrogel of absorbent resin |
| JPH11349625A (ja) * | 1998-06-10 | 1999-12-21 | Sanyo Chem Ind Ltd | 吸水剤の製造法および吸水剤 |
| US6620899B1 (en) | 1998-10-15 | 2003-09-16 | E. I. Du Pont De Nemours And Company | Polymers, containing a fluorocyclobutyl ring and their preparation |
| US6297319B1 (en) | 1998-11-05 | 2001-10-02 | Nippon Shokubai Co., Ltd. | Water-absorbing agent and production process therefor |
| US6559239B1 (en) | 1998-11-26 | 2003-05-06 | Basf Aktiengesellschaft | Method for the secondary cross-linking of hydrogels with N-acyl-2-oxazolidinones |
| US6657015B1 (en) | 1998-11-26 | 2003-12-02 | Basf Aktiengesellschaft | Method for the secondary cross-linking of hydrogels with 2-oxotetrahydro-1,3-oxazines |
| JP2000189794A (ja) | 1998-12-28 | 2000-07-11 | Nippon Shokubai Co Ltd | 吸水材の製造方法 |
| US7179862B2 (en) | 1999-03-05 | 2007-02-20 | Stockhausen Gmbh | Powdery, cross-linked absorbent polymers method for the production thereof and their use |
| EP1165631A1 (de) | 1999-03-05 | 2002-01-02 | STOCKHAUSEN GmbH & CO. KG | Pulverförmige, vernetzte, wässrige flüssigkeiten sowie blut absorbierende polymere, verfahren zu ihrer herstellung und ihre verwendung |
| US6605673B1 (en) | 1999-03-05 | 2003-08-12 | Stockhausen Gmbh & Co., Kg | Powdery, cross-linked polymers which absorb aqueous liquids and blood, method for the production thereof and their use |
| US6514615B1 (en) | 1999-06-29 | 2003-02-04 | Stockhausen Gmbh & Co. Kg | Superabsorbent polymers having delayed water absorption characteristics |
| US6239230B1 (en) | 1999-09-07 | 2001-05-29 | Bask Aktiengesellschaft | Surface-treated superabsorbent polymer particles |
| US6710141B1 (en) | 1999-11-20 | 2004-03-23 | Basf Aktiengesellschaft | Method for continuously producing cross-linked fine-particle geleous polymerizates |
| JP2002121291A (ja) * | 2000-02-29 | 2002-04-23 | Nippon Shokubai Co Ltd | 吸水性樹脂粉末およびその製造方法 |
| US7157141B2 (en) | 2000-03-31 | 2007-01-02 | Stockhausen Gmbh | Pulverulent polymers crosslinked on the surface |
| US6906159B2 (en) | 2000-08-03 | 2005-06-14 | Nippon Shokubai Co., Ltd. | Water-absorbent resin, hydropolymer, process for producing them, and uses of them |
| EP1178059A2 (en) | 2000-08-03 | 2002-02-06 | Nippon Shokubai Co., Ltd. | Water-absorbent resin, hydropolymer, process for producing them, and uses of them |
| US6831142B2 (en) | 2000-09-04 | 2004-12-14 | Stockhausen Gmbh & Co. Kg | Pulverulent, crosslinked polymers which absorb aqueous liquids and blood |
| US20040176557A1 (en) | 2000-09-04 | 2004-09-09 | Richard Mertens | Pulverulent, crosslinked polymers which absorb aqueous liquids and blood |
| JP2002201290A (ja) * | 2000-09-20 | 2002-07-19 | Nippon Shokubai Co Ltd | 吸水性樹脂およびその製造方法 |
| US6809158B2 (en) | 2000-10-20 | 2004-10-26 | Nippon Shokubai Co., Ltd. | Water-absorbing agent and process for producing the same |
| US20060024755A1 (en) | 2001-04-30 | 2006-02-02 | George Jackowski | Biopolymer marker indicative of disease state having a molecular weight of 1424 daltons |
| US20040106745A1 (en) | 2001-06-08 | 2004-06-03 | Yasuhisa Nakashima | Water-absorbing agent and production process therefor, and sanitary material |
| US6987151B2 (en) | 2001-09-12 | 2006-01-17 | Dow Global Technologies Inc. | Continuous polymerization process for the manufacture of superabsorbent polymers |
| JP2003303306A (ja) | 2002-04-08 | 2003-10-24 | Nec Soft Ltd | プリペイド方式による商品代金支払システム |
| US6875511B2 (en) | 2002-05-30 | 2005-04-05 | Nippon Shokubai Co., Ltd. | Production process for particulate water-absorbent resin |
| JP2004037900A (ja) | 2002-07-04 | 2004-02-05 | Ricoh Co Ltd | 中間転写装置及び画像形成装置 |
| US20040069915A1 (en) | 2002-09-20 | 2004-04-15 | Alain Guennec | Satellite antenna holder |
| US20060073969A1 (en) | 2003-02-10 | 2006-04-06 | Kazushi Torii | Vater-absorbent resin composition and its production process |
| US20060204755A1 (en) | 2003-02-10 | 2006-09-14 | Kazushi Torii | Walter-absorbing agent |
| US20040234607A1 (en) | 2003-04-25 | 2004-11-25 | Nippon Shokubai Co.,Ltd. | Method for disintegrating hydrate polymer and method for production of water-absorbent resin |
| JP2004352941A (ja) | 2003-05-30 | 2004-12-16 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造法 |
| JP2004352940A (ja) | 2003-05-30 | 2004-12-16 | Nippon Shokubai Co Ltd | 吸水性樹脂の製造方法および鋤型混合装置 |
| US20070106013A1 (en) | 2003-06-24 | 2007-05-10 | Yoshifumi Adachi | Water absorbent resin composition and production method thereof |
| JP2005016393A (ja) | 2003-06-25 | 2005-01-20 | Toyota Motor Corp | 内燃機関の排気浄化システム |
| JP2005054151A (ja) | 2003-08-07 | 2005-03-03 | Nippon Shokubai Co Ltd | 粒子状吸水性樹脂の製法 |
| US20050048221A1 (en) | 2003-08-27 | 2005-03-03 | Yoshio Irie | Process for production of surface-treated particulate water-absorbent resin |
| US7201941B2 (en) | 2003-08-27 | 2007-04-10 | Nippon Shokubai Co., Ltd. | Process for production of surface-treated particulate water-absorbent resin |
| JP2005097585A (ja) * | 2003-08-27 | 2005-04-14 | Nippon Shokubai Co Ltd | 表面処理された粒子状吸水性樹脂の製造方法 |
| US20050046069A1 (en) | 2003-09-01 | 2005-03-03 | Masazumi Sasabe | Process for production of hydrogel particles and process for cutting of high-concentration hydrogel sheet |
| US7473739B2 (en) | 2004-02-05 | 2009-01-06 | Nippon Shokubai Co., Ltd. | Particulate water absorbent agent and production method thereof, and water absorbent article |
| US20050215734A1 (en) | 2004-03-24 | 2005-09-29 | Yorimichi Dairoku | Method for continuous production of water-absorbent resin |
| JP2005307195A (ja) | 2004-03-24 | 2005-11-04 | Nippon Shokubai Co Ltd | 吸水性樹脂の連続製造方法 |
| US20080080300A1 (en) | 2004-09-28 | 2008-04-03 | Basf Aktiengesellschaft | Mixing Kneader and Process for Preparing Poly(Meth)Acrylates Using the Mixing Kneader |
| US20060101271A1 (en) | 2004-11-10 | 2006-05-11 | Michael Thomas | Method and system for conveying alternate acceptable canonicalizations of a digitally signed piece of electronic mail |
| EP1824910A2 (en) | 2004-12-10 | 2007-08-29 | Nippon Shokubai Co.,Ltd. | Method for production of modified water absorbent resin |
| WO2006082189A1 (en) | 2005-02-01 | 2006-08-10 | Basf Aktiengesellschaft | Polyamine-coated superabsorbent polymers |
| WO2006082188A1 (en) | 2005-02-01 | 2006-08-10 | Basf Aktiengesellschaft | Polyamine-coated superabsorbent polymers |
| WO2006082197A1 (en) | 2005-02-01 | 2006-08-10 | Basf Aktiengesellschaft | Polyamine-coated superabsorbent polymers |
| US20060247351A1 (en) | 2005-03-14 | 2006-11-02 | Kazushi Torii | Water-absorbing agent and its production process |
| JP2008038128A (ja) * | 2005-12-22 | 2008-02-21 | Nippon Shokubai Co Ltd | 吸水性樹脂の表面架橋方法および吸水性樹脂の製造方法 |
| US20070149760A1 (en) | 2005-12-22 | 2007-06-28 | Kenji Kadonaga | Method for surface crosslinking water-absorbing resin and method for manufacturing water-absorbing resin |
| US20080090961A1 (en) | 2006-10-13 | 2008-04-17 | Bayer Materialscience Llc | Impact resistant, flame retardant thermoplastic molding composition |
Non-Patent Citations (1)
| Title |
|---|
| "Chemical Engineering Handbook", MARUZEN COMPANY, LIMITED. |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012102407A1 (ja) | 2011-01-28 | 2012-08-02 | 株式会社日本触媒 | ポリアクリル酸(塩)系吸水性樹脂粉末の製造方法 |
| JP2015526572A (ja) * | 2012-08-29 | 2015-09-10 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | 吸水性ポリマー粒子の製造方法 |
| JP2016529368A (ja) * | 2013-08-27 | 2016-09-23 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| US10829630B2 (en) | 2016-03-11 | 2020-11-10 | Lg Chem, Ltd. | Super absorbent polymer |
| JP2020520390A (ja) * | 2018-04-03 | 2020-07-09 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| US11161942B2 (en) | 2018-04-03 | 2021-11-02 | Lg Chem, Ltd. | Method for preparing superabsorbent polymer |
| JP7008718B2 (ja) | 2018-04-03 | 2022-01-25 | エルジー・ケム・リミテッド | 高吸水性樹脂の製造方法 |
| WO2021187323A1 (ja) * | 2020-03-18 | 2021-09-23 | 住友精化株式会社 | 吸水性樹脂粒子を製造する方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US8648150B2 (en) | 2014-02-11 |
| US9796820B2 (en) | 2017-10-24 |
| US20140107293A1 (en) | 2014-04-17 |
| JP2015014002A (ja) | 2015-01-22 |
| JP5847260B2 (ja) | 2016-01-20 |
| CN102341435A (zh) | 2012-02-01 |
| JPWO2010100936A1 (ja) | 2012-09-06 |
| EP2404954A4 (en) | 2013-06-19 |
| CN102341435B (zh) | 2016-04-20 |
| JP5615801B2 (ja) | 2014-10-29 |
| US20110319518A1 (en) | 2011-12-29 |
| EP2404954A1 (en) | 2012-01-11 |
| EP2404954B1 (en) | 2015-04-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5847260B2 (ja) | 吸水性樹脂の製造方法 | |
| JP6385993B2 (ja) | ポリアクリル酸(塩)系吸水剤 | |
| JP5507254B2 (ja) | 吸水性樹脂の製造方法および吸水性樹脂並びにそれらの利用 | |
| EP3085439B1 (en) | Water absorbing agent based on polyacrylic acid and/or a salt thereof | |
| EP2896645B1 (en) | Method for manufacturing polyacrylic acid (polyacrylate)-based water-absorbent agent, and water-absorbent agent | |
| EP1677845B2 (en) | Water absorbent and producing method of same | |
| JP5064032B2 (ja) | 吸水性樹脂造粒物の製造方法および吸水性樹脂造粒物 | |
| WO2016204302A1 (ja) | ポリ(メタ)アクリル酸(塩)系粒子状吸水剤及び製造方法 | |
| JP5367364B2 (ja) | 吸水性樹脂を主成分として含む吸水剤およびその製造方法 | |
| WO2009125849A1 (ja) | 吸水性樹脂の表面処理方法および吸水性樹脂の製造方法 | |
| JP2005344103A (ja) | 吸水剤およびその製造方法 | |
| JP2010053296A (ja) | 吸水性樹脂の製造方法 | |
| JP4722545B2 (ja) | 吸水性樹脂組成物とその製造方法 | |
| JP4722546B2 (ja) | 吸水性樹脂組成物とその製造方法 | |
| JP2015016450A (ja) | 吸水剤及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080010035.3 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10748535 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011502661 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13254573 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010748535 Country of ref document: EP |