, , , - UND PYRAZOLOPYRIDIN DERIVATE ALS STIMULATOREN DER GUANYLATCYCLASE ZUR HERZ-KREISLAUFERKRANKUNGEN
Die vorliegende Anmeldung betrifft neue aza-bicyclische Verbindungen, Verfahren zu ihrer Herstellung, ihre Verwendung allein oder in Kombinationen zur Behandlung und/oder Prävention von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prävention von Krankheiten, insbesondere zur Behandlung und/oder Prävention von Herz-Kreislauf-Erkrankungen.
Eines der wichtigsten zellulären Übertragungssysteme in Säugerzellen ist das cyclische Guanosin- monophosphat (cGMP). Zusammen mit Stickstoffmonoxid (NO), das aus dem Endothel freigesetzt wird und hormonelle und mechanische Signale überträgt, bildet es das NO/cGMP-System. Die Guanylatcyclasen katalysieren die Biosynthese von cGMP aus Guanosintriphosphat (GTP). Die bisher bekannten Vertreter dieser Familie lassen sich sowohl nach strukturellen Merkmalen als auch nach der Art der Liganden in zwei Gruppen aufteilen: Die partikulären, durch natriuretische Peptide stimulierbaren Guanylatcyclasen und die löslichen, durch NO stimulierbaren Guanylatcyclasen. Die löslichen Guanylatcyclasen bestehen aus zwei Untereinheiten und enthalten höchst- wahrscheinlich ein Häm pro Heterodimer, das ein Teil des regulatorischen Zentrums ist. Dieses hat eine zentrale Bedeutung für den Aktivierungsmechanismus. NO kann an das Eisenatom des Häms binden und so die Aktivität des Enzyms deutlich erhöhen. Hämfreie Präparationen lassen sich hingegen nicht durch NO stimulieren. Auch Kohlenmonoxid (CO) ist in der Lage, an das Eisen- Zentralatom des Häms zu binden, wobei die Stimulierung durch CO deutlich geringer ist als die durch NO.
Durch die Bildung von cGMP und der daraus resultierenden Regulation von Phosphodiesterasen, Ionenkanälen und Proteinkinasen spielt die Guanylatcyclase eine entscheidende Rolle bei unterschiedlichen physiologischen Prozessen, insbesondere bei der Relaxation und Proliferation glatter Muskelzellen, der Plättchenaggregation und -adhäsion, der neuronalen Signalübertragung sowie bei Erkrankungen, welche auf einer Störung der vorstehend genannten Vorgänge beruhen. Unter pathophysiologischen Bedingungen kann das NO/cGMP-System supprimiert sein, was zum Beispiel zu Bluthochdruck, einer Plättchenaktivierung, einer vermehrten Zellproliferation, endothelialer Dysfunktion, Atherosklerose, Angina pectoris, Herzinsuffizienz, Myokardinfarkt, Thrombosen, Schlaganfall und sexueller Dysfunktion führen kann.
Eine auf die Beeinflussung des cGMP-Signalweges in Organismen abzielende NO-unabhängige Behandlungsmöglichkeit für derartige Erkrankungen ist aufgrund der zu erwartenden hohen Effizienz und geringen Nebenwirkungen ein vielversprechender Ansatz.
Zur therapeutischen Stimulation der löslichen Guanylatcyclase wurden bisher ausschließlich Verbindungen wie organische Nitrate verwendet, deren Wirkung auf NO beruht. Dieses wird durch Biokonversion gebildet und aktiviert die lösliche Guanylatcyclase durch Angriff am Eisen-Zentralatom des Häms. Neben den Nebenwirkungen gehört die Toleranzentwicklung zu den entscheiden- den Nachteilen dieser Behandlungsweise.
In den letzten Jahren wurden einige Substanzen beschrieben, die die lösliche Guanylatcyclase direkt, d.h. ohne vorherige Freisetzung von NO stimulieren, wie beispielsweise 3-(5'-Hydroxy- methyl-2'-furyl)-l-benzylindazol [YC-I; Wu et al., Blood 84 (1994), 4226; Mülsch et al., BHt. J. Pharmacol. 120 (1997), 681], Fettsäuren [Goldberg et al., J. Biol. Chem. 252 (1977), 1279], Diphenyliodonium-hexafluorophosphat [Pettibone et al., Eur. J. Pharmacol. 116 (1985), 307], Iso- liquiritigenin [Yu et al., BHt. J. Pharmacol. 114 (1995), 1587] sowie verschiedene substituierte Pyrazol-Derivate (WO 98/16223).
Anellierte Pyrazol-Derivate sind unter anderem in WO 98/16507, WO 98/23619, WO 00/06567, WO 00/06569, WO 02/42299, WO 02/42300, WO 02/42301, WO 02/42302, WO 02/092596, WO 03/004503 und WO 03/095451 als Stimulatoren der löslichen Guanylatcyclase beschrieben. Allerdings zeigte es sich, dass diese Verbindungen zum Teil bezüglich ihrer physikochemischen Eigenschaften, wie beispielsweise ihrer Löslichkeit, oder hinsichtlich ihrer in vj'vo-Eigenschaften, wie beispielsweise ihrem Verhalten in der Leber, ihrem pharmakokinetischen Verhalten, ihrer Dosis- Wirkungsbeziehung und/oder ihrem Metabolisierungsweg, Nachteile aufweisen.
Weiterhin werden in US 5 593 997, WO 01/57024, V/O 03/035005 sowie WO 2005/030121 verschiedene anellierte Pyrazol-Derivate zur Behandlung von Erkrankungen offenbart.
Aufgabe der vorliegenden Erfindung war die Bereitstellung neuer Substanzen, die als Stimulatoren der löslichen Guanylatcyclase wirken und ein verbessertes therapeutisches Profil gegenüber den aus dem Stand der Technik bekannten Verbindungen aufweisen.
Gegenstand der vorliegenden Erfindung sind Verbindungen der allgemeinen Formel (I)
L— CH- M— Q (I),
in welcher
L für Phenyl, Pyridyl, Furyl, Thienyl, Thiazolyl, Oxazolyl, Isothiazolyl oder Isoxazolyl, die jeweils bis zu zweifach, gleich oder verschieden, mit Halogen, Cyano, (Ci-C4)-Alkyl, Tri- fluormethyl und/oder (C2-C4)- Alkinyl substituiert sein können,
oder
für (C5-C7)-Cycloalkyl, das bis zu zweifach, gleich oder verschieden, mit Fluor und/oder (Ci-C4)-Alkyl substituiert sein kann,
steht,
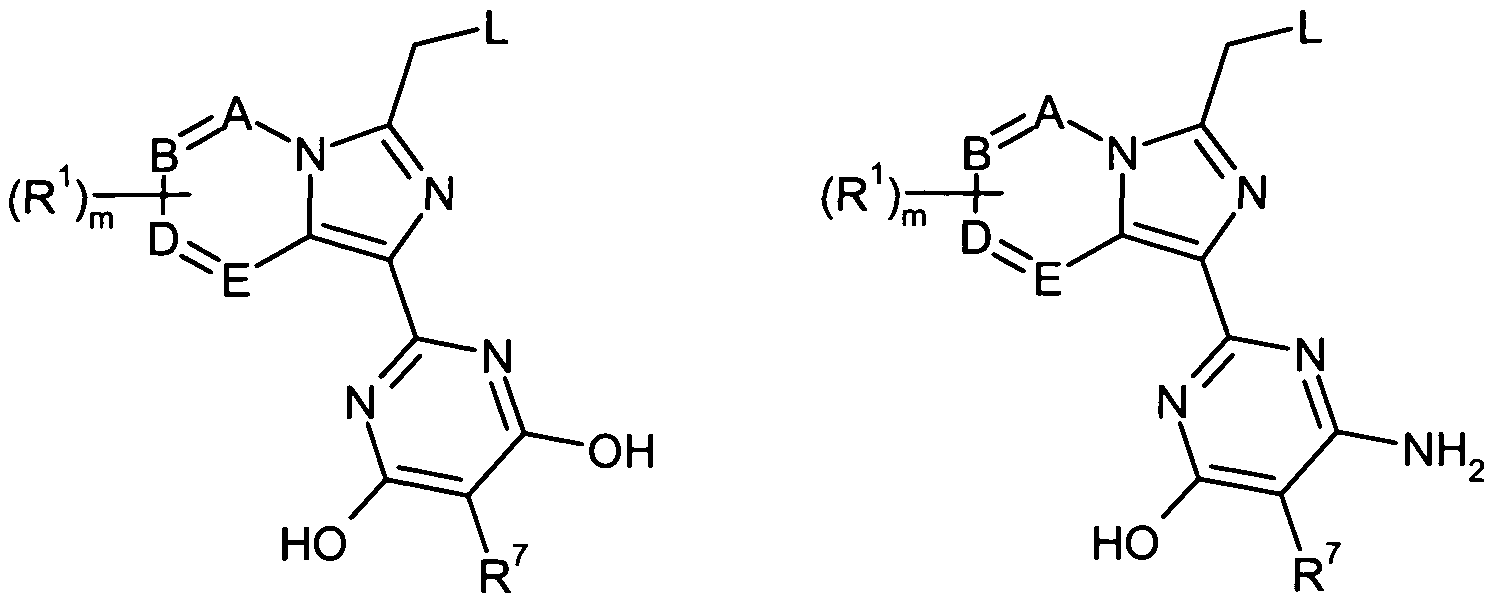
M für eine bicyclische Heteroaryl -Gruppe der Formel (a-1), (b-1) oder (c-1)
steht, worin
* die Verknüpfungsstelle mit der Gruppe -CH2-L bedeutet,
** die Verknüpfungsstelle mit der Gruppe Q bedeutet,
A, B, D und E jeweils CH, CR1 oder N bedeuten, wobei maximal zwei der Ringglieder A, B, D und E zugleich für N stehen,
R1 einen Substituenten ausgewählt aus der Reihe Halogen, Cyano, (CrC4)-Alkyl, Tri- fluormethyl, Amino, (Ci-C4)-Alkoxy und Trifluormethoxy bedeutet
und
m die Zahl 0, 1 oder 2 bedeutet,
wobei für den Fall, dass der Substituent R1 zweifach auftritt, seine Bedeutungen gleich oder verschieden sein können,
und
Q für einen ungesättigten oder aromatischen 5- oder 6-gliedrigen Heterocyclus mit bis zu vier Heteroatomen aus der Reihe N, O und/oder S steht, der bis zu vierfach, gleich oder ver- schieden, substituiert sein kann mit Resten ausgewählt aus der Gruppe bestehend aus
Halogen, Azido, Nitro, Cyano, Oxo, Thioxo, -R2, -C(=O)-R2, -C(=O)-OR2, -C(=O)-NR2R3, -0-(C=O)n-R2, -O-C(=O)-OR2, -O-C(=O)-NR2R3, -S(O)P-R2, -SO2-OR2, -SO2-NR2R3, -NR2-(C=O)n-R3, -NR2-SO2-R\ -NR2-C(=O)-OR3, -NR4-C(=O)-NR2R3 und -NR4-SO2- NR2R3, worin
n die Zahl 0 oder 1 bedeutet,
p die Zahl 0, 1 oder 2 bedeutet,
R2, R3 und R4 gleich oder verschieden sind und unabhängig voneinander Wasserstoff, (C1- C6)-Alkyl, (C2-C6)-Alkenyl, (C3-C8)-Cycloalkyl, (C3-C8)-Cycloalkenyl, (C6-C10)- Aryl, 4- bis 8-gliedriges Heterocyclyl oder 5- bis 10-gliedriges Heteroaryl bedeuten
und/oder
R2 und R3 oder R2 und R4 gemeinsam mit dem Rest, an den sie jeweils beide gebunden sind, einen 4- bis 8-gliedrigen Heterocyclus bilden können,
wobei R2, R3 und R4 ihrerseits gegebenenfalls bis zu fünffach, gleich oder verschieden, mit Resten ausgewählt aus der Gruppe bestehend aus Halogen, Azido, Nitro, Cyano, (C1-Co)-AIlCyI, Trifluormethyl, (C1-Co)-ACyI, Hydroxycarbonyl, (Q- C6)-Alkoxycarbonyl, Aminocarbonyl, Mono- und Di-(C1-C6)-alkylaminocarbonyl, Hydroxy, (C1-Ce)-AIkOXy, Trifluormethoxy, (CrC6)-Acyloxy, Oxo, Mercapto, (Cr C6)-Alkylthio, Amino, Mono- und Di-(Ci-C6)-alkylamino, (C1-C6)-Acylamino, (C1-
C6)-Alkoxycarbonylamino, (C3-C8)-Cycloalkyl, (C3-C8)-Cycloalkenyl sowie 4- bis 8-gliedriges Heterocyclyl substituiert sein können,
sowie ihre N-Oxide, Salze, Solvate, Salze der /V-Oxide und Solvate der N-Oxide und Salze.
Erfϊndungsgemäße Verbindungen sind die Verbindungen der Formel (I) und deren Salze, Solvate und Solvate der Salze, die von Formel (I) umfassten Verbindungen der nachfolgend genannten Formeln und deren Salze, Solvate und Solvate der Salze sowie die von Formel (I) umfassten, nachfolgend als Ausführungsbeispiele genannten Verbindungen und deren Salze, Solvate und Solvate der Salze, soweit es sich bei den von Formel (I) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt.
Erfindungsgemäße Verbindungen sind ebenso N-Oxide der Verbindungen der Formel (I) sowie deren Salze, Solvate und Solvate der Salze.
Die erfmdungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in stereoisomeren Formen (Enantiomere, Diastereomere) existieren. Die vorliegende Erfindung umfasst deshalb die Enantiomeren oder Diastereomeren und ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren.
Sofern die erfindungsgemäßen Verbindungen in tautomeren Formen vorkommen können, umfasst die vorliegende Erfindung sämtliche tautomere Formen.
Als Salze sind im Rahmen der vorliegenden Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt. Umfasst sind auch Salze, die für pharmazeutische Anwendungen selbst nicht geeignet sind, jedoch beispielsweise für die Isolierung oder Reinigung der erfindungsgemäßen Verbindungen verwendet werden können.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasserstoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure, Ethan- sulfonsäure, Toluolsulfonsäure, Benzolsulfonsäure, Naphthalindisulfonsäure, Essigsäure, Trifluor- essigsäure, Propionsäure, Milchsäure, Weinsäure, Äpfelsäure, Zitronensäure, Fumarsäure, Maleinsäure und Benzoesäure.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen auch Salze üblicher Basen, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kalium- salze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze) und Ammoniumsalze, abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C-Atomen, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, Ethyldiisopropylamin, Monoethanolamin, Diethanolamin, Trisethanolamin, Dicyclohexylamin, Dimethylaminoethanol, Prokain, Dibenzylamin, N-Methyl- morpholin, Arginin, Lysin, Ethylendiamin und N-Methylpiperidin.
Als Solvate werden im Rahmen der Erfindung solche Formen der erfindungsgemäßen Verbindungen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungsmittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt. Als Solvate sind im Rahmen der vorliegenden Erfindung Hydrate bevorzugt.
Außerdem umfasst die vorliegende Erfindung auch Prodrugs der erfindungsgemäßen Verbindungen. Der Begriff "Prodrugs" umfaßt Verbindungen, welche selbst biologisch aktiv oder inaktiv sein können, jedoch während ihrer Verweilzeit im Körper zu erfindungsgemäßen Verbindungen umgesetzt werden (beispielsweise metabolisch oder hydrolytisch).
Im Rahmen der vorliegenden Erfindung haben die Substituenten, soweit nicht anders spezifiziert, die folgende Bedeutung:
(Cj_-C(Q-Alkyl und (C1-Q)-AIkVl stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkylrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen. Bevorzugt ist ein geradkettiger
- o - oder verzweigter Alkylrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methyl, Ethyl, w-Propyl, Isopropyl, n-Butyl, iso-Buty\, sec.-Butyl, tert. -Butyl, 1-Ethyl- propyl, «-Pentyl und H-Hexyl.
(C2-Cfi)-Alkenyl und (Co-CaVAlkenyl stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkenylrest mit 2 bis 6 bzw. 2 bis 4 Kohlenstoffatomen und einer oder zwei Doppelbindungen. Bevorzugt ist ein geradkettiger oder verzweigter Alkenylrest mit 2 bis 4 Kohlenstoffatomen und einer Doppelbindung. Beispielhaft und vorzugsweise seien genannt: Vinyl, Allyl, Isopropenyl und «-But-2-en-l-yl.
(C2-CV)-AIkJnVl steht im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkinyl- rest mit 2 bis 4 Kohlenstoffatomen und einer Dreifachbindung. Bevorzugt ist ein geradkettiger Alkinylrest mit 2 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Ethinyl, n-Prop-1-in-l-yl, «-Prop-2-in-l-yl, n-But-1-in-l-yl, «-But-2-in-l-yl und «-But-3-in-l-yl.
(C1-CV)-AIkOXv und (C1-Cd)-AIkOXV stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkoxyrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen. Bevorzugt ist ein gerad- kettiger oder verzweigter Alkoxyrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methoxy, Ethoxy, «-Propoxy, Isopropoxy, «-Butoxy, tert.-Butoxy, «-Pentoxy und n-Hexoxy.
(C1-Cs)-AIkVUhJo und (G-QVAlkylthio stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkylthiorest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen. Bevorzugt ist ein geradkettiger oder verzweigter Alkylthiorest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methylthio, Ethylthio, «-Propylthio, Isopropylthio, «-Butylthio, tert.- Butylthio, n-Pentylthio und n-Hexylthio.
Mono-(C
1-Cfi)-alkylamino und Mono-fC-C^-alkylamino stehen im Rahmen der Erfindung für eine Amino-Gruppe mit einem geradkettigen oder verzweigten Alkylsubstituenten, der 1 bis 6 bzw. 1 bis 4 Kohlenstoffatome aufweist. Bevorzugt ist ein geradkettiger oder verzweigter Monoalkyl- amino-Rest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methyl- amino, Ethylamino, n-Propylamino, Isopropylamino, n-Butylamino, tert.-Butylamino, «-Pentyl- amino und n-Hexylamino.

stehen im Rahmen der Erfindung für eine Amino-Gruppe mit zwei gleichen oder verschiedenen geradkettigen oder verzweigten Alkylsubstituenten, die jeweils 1 bis 6 bzw. 1 bis 4 Kohlenstoffatome aufweisen. Bevorzugt sind gerad- kettige oder verzweigte Dialkylamino-Reste mit jeweils 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: NN-Dimethylamino, N,N-Diethylamino, N-Ethyl-N-methyl-
amino, N-Methyl-N-w-propylamino, N-Isopropyl-N-«-propylamino, NN-Diisopropylamino, N-M- Butyl-N-methylamino, N-terf.-Butyl-N-methylamino, N-Methyl-N-n-pentylamino und N-n-Hexyl- N-methylamino.
(C
rCg)-Acyl und (C
1-Ca)-AcVl [(C
rC
6)-Alkanoyl und (C,-C
4)-Alkanoyl] stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkylrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen, der in der 1 -Position ein doppelt gebundenes Sauerstoffatom trägt und über die 1 -Position verknüpft ist. Bevorzugt ist ein Acylrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Formyl, Acetyl, Propionyl, «-Butyryl, wo-Butyryl, n-Pentanoyl, Pivaloyl und «-Hexanoyl.
stehen im Rahmen der Erfindung für eine Amino- Gruppe mit einem geradkettigen oder verzweigten Acyl-Substituenten, der 1 bis 6 bzw. 1 bis 4 Kohlenstoffatome aufweist und über die Carbonylgruppe mit dem Ν-Atom verknüpft ist. Beispielhaft und vorzugsweise seien genannt: Formylamino, Acetylamino, Propionylamino, w-Butyryl- amino, zso-Butyrylamino und Pivaloylamino.
(C
1-Cg)-AcVlOXv und (C
1-Cd)-ACyIoXy stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkyl-Rest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen, der in der 1 -Position ein doppelt gebundenes Sauerstoffatom trägt und in der 1 -Position über ein weiteres Sauerstoffatom verknüpft ist. Beispielhaft und vorzugsweise seien genannt: Acetoxy, Propionoxy, rc-Butyroxy, iso- Butyroxy und Pivaloyloxy.
stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkoxyrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen, der über eine Carbonylgruppe verknüpft ist. Bevorzugt ist ein geradkettiger oder verzweigter Alkoxy- carbonyl-Rest mit 1 bis 4 Kohlenstoffatomen in der Alkoxygruppe. Beispielhaft und vorzugsweise seien genannt: Methoxycarbonyl, Ethoxycarbonyl, n-Propoxycarbonyl, Isopropoxycarbonyl, n- Butoxycarbonyl und terΛ-Butoxycarbonyl.
stehen im Rahmen der Erfindung für eine Amino-Gruppe mit einem geradkettigen oder verzweigten Alkoxycarbonyl-Substi- tuenten, der 1 bis 6 bzw. 1 bis 4 Kohlenstoffatome in der Alkoxygruppe aufweist und über die Carbonylgruppe mit dem Ν-Atom verknüpft ist. Beispielhaft und vorzugsweise seien genannt: Methoxycarbonylamino, Ethoxycarbonylamino, n-Propoxycarbonylamino, Isopropoxycarbonyl- amino, n-Butoxycarbonylamino und tert.-Butoxycarbonylamino.
Mono- bzw. Di-(C1-Cfi)-alkylaminocarbonyl und Mono- bzw. Di-(C1-C4)-alkylaminocarbonyl stehen im Rahmen der Erfindung für eine Amino-Gruppe, die über eine Carbonylgruppe verknüpft
- o - ist und die einen geradkettigen oder verzweigten bzw. zwei gleiche oder verschiedene geradkettige oder verzweigte Alkylsubstituenten mit jeweils 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen aufweist. Bevorzugt ist ein Mono- bzw. Dialkylaminocarbonyl-Rest mit 1 bis 4 Kohlenstoffatomen in der Alkylgruppe. Beispielhaft und vorzugsweise seien genannt: Methylaminocarbonyl, Ethylamino- carbonyl, «-Propylaminocarbonyl, Isopropylaminocarbonyl, n-Butylaminocarbonyl, tert.-Butyl- aminocarbonyl, N,N-Dimethylaminocarbonyl, NN-Diethylaminocarbonyl, N-Ethyl-N-methyl- aminocarbonyl, N-Methyl-N-«-propylaminocarbonyl, N-rc-Butyl-N-methylaminocarbonyl und N- ter/.-Butyl-N-methylaminocarbonyl.
(CrCgVCvcloalkyl. (C3-C7VCvClOaIkVl. (Q-CUVCvcloalkyl und (Cs-Cfi-Cvcloalkyl stehen im Rah- men der Erfindung für einen monocyclischen, gesättigten Carbocyclus mit 3 bis 8, 3 bis 7, 3 bis 6 bzw. 5 bis 7 Ring-Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl, Cycloheptyl und Cyclooctyl.
(C^-C^-Cycloalkenyl und (C2-C7)-Cycloalkenyl stehen im Rahmen der Erfindung für einen monocyclischen Carbocyclus mit 3 bis 8 bzw. 3 bis 7 Ring-Kohlenstoffatomen und einer Doppelbin- düng. Beispielhaft und vorzugsweise seien genannt: Cyclobutenyl, Cyclopentenyl, Cyclohexenyl, Cycloheptenyl und Cyclooctenyl.
(Cή-Cui)-Aryl steht im Rahmen der Erfindung für einen aromatischen Carbocyclus mit 6 oder 10 Ring-Kohlenstoffatomen. Bevorzugte Arylreste sind Phenyl und Νaphthyl.
5- bis 10-gliedriges Heteroaryl steht im Rahmen der Erfindung für einen mono- oder gegebenen- falls bicyclischen aromatischen Heterocyclus (Heteroaromaten) mit insgesamt 5 bis 10 Ringatomen, der bis zu drei gleiche oder verschiedene Ring-Heteroatome aus der Reihe Ν, O und/oder S enthält und über ein Ring-Kohlenstoffatom oder gegebenenfalls über ein Ring-Stickstoffatom verknüpft ist. Beispielhaft seien genannt: Furyl, Pyrrolyl, Thienyl, Pyrazolyl, Imidazolyl, Thia- zolyl, Oxazolyl, Isoxazolyl, Isothiazolyl, Triazolyl, Oxadiazolyl, Thiadiazolyl, Pyridyl, Pyrimi- dinyl, Pyridazinyl, Pyrazinyl, Triazinyl, Benzofuranyl, Benzothienyl, Benzimidazolyl, Benzoxa- zolyl, Benzothiazolyl, Benzotriazolyl, Indolyl, Indazolyl, Chinolinyl, Isochinolinyl, Νaphthyri- dinyl, Chinazolinyl, Chinoxalinyl, Phthalazinyl, Pyrazolo[3,4-b]pyridinyl. Bevorzugt sind mono- cyclische 5- oder 6-gliedrige Heteroaryl-Reste mit bis zu drei Ring-Heteroatomen aus der Reihe Ν, O und/oder S wie beispielsweise Furyl, Thienyl, Thiazolyl, Oxazolyl, Isothiazolyl, Isoxazolyl, Pyrazolyl, Imidazolyl, Triazolyl, Oxadiazolyl, Thiadiazolyl, Pyridyl, Pyrimidinyl, Pyridazinyl, Pyrazinyl, Triazinyl.
Ein 4- bis 8-gliedriger Heterocvclus steht im Rahmen der Erfindung für einen monocyclischen, gesättigten Heterocyclus mit insgesamt 4 bis 8 Ringatomen, der ein oder zwei Ring-Heteroatome
aus der Reihe N, O, S, SO und/oder SO2 enthält und über ein Ring-Kohlenstoffatom oder gegebenenfalls ein Ring-Stickstoffatom verknüpft ist. Bevorzugt ist ein 5- bis 7-gliedriger Heterocvclus mit ein oder zwei Ring-Heteroatomen aus der Reihe N, O und/oder S, besonders bevorzugt ein 5; oder 6-gliedriger Heterocvclus mit ein oder zwei Ring-Heteroatomen aus der Reihe N und/oder O. Beispielhaft seien genannt: Azetidinyl, Oxetanyl, Pyrrolidinyl, Pyrazolidinyl, Tetrahydrofuranyl, Thiolanyl, Piperidinyl, Piperazinyl, Tetrahydropyranyl, Tetrahydrothiopyranyl, Morpholinyl, Thio- morpholinyl, Hexahydroazepinyl und Hexahydro-l,4-diazepinyl. Bevorzugt sind Pyrrolidinyl, Tetrahydrofuranyl, Piperidinyl, Piperazinyl, Tetrahydropyranyl und Morpholinyl.
Ein ungesättigter oder aromatischer 5- oder 6-gliedriger Heterocvclus steht im Rahmen der Erfin- düng für einen monocyclischen Heterocyclus mit insgesamt 5 oder 6 Ringatomen, der bis zu vier Ring-Heteroatome aus der Reihe N, O und/oder S enthält, über ein Ring-Kohlenstoffatom oder gegebenenfalls ein Ring-Stickstoffatom verknüpft ist und im Falle des Fünfrings eine Doppelbindung enthält oder aromatisch ist und im Falle des Sechsrings eine oder zwei Doppelbindungen enthält oder aromatisch ist. Beispielhaft seien genannt: Pyrrolinyl, Dihydropyrazolyl, Imidazolinyl, Dihydrooxazolyl, Dihydroisoxazolyl, Dihydro-l,2,4-triazolyl, Dihydro-l,2,4-oxadiazolyl, Dihydro- 1,3,4-oxadiazolyl, Dihydro-l,2,4-thiadiazolyl, Dihydropyranyl, 1 ,4-Dihydropyridyl, Tetrahydro- pyrimidinyl, 1,3-Oxazinyl, Furyl, Pyrrolyl, Thienyl, Pyrazolyl, Imidazolyl, Thiazolyl, Oxazolyl, Isoxazolyl, Isothiazolyl, Triazolyl, Oxadiazolyl, Thiadiazolyl, Tetrazolyl, Pyridyl, Pyrimidinyl, Pyridazinyl, Pyrazinyl, Triazinyl.
Halogen schließt im Rahmen der Erfindung Fluor, Chlor, Brom und Iod ein. Bevorzugt sind Chlor oder Fluor.
Wenn Reste in den erfindungsgemäßen Verbindungen substituiert sind, können die Reste, soweit nicht anders spezifiziert, ein- oder mehrfach substituiert sein. Im Rahmen der vorliegenden Erfindung gilt, dass für alle Reste, die mehrfach auftreten, deren Bedeutung unabhängig voneinander ist. Eine Substitution mit ein, zwei oder drei gleichen oder verschiedenen Substituenten ist bevorzugt. Ganz besonders bevorzugt ist die Substitution mit einem Substituenten.
Bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
L für Phenyl oder Thienyl, die jeweils bis zu zweifach, gleich oder verschieden, mit Fluor,
Chlor, Cyano, Methyl und/oder Trifluormethyl substituiert sein können,
oder
für Cyclohexyl oder Cycloheptyl, die jeweils bis zu zweifach, gleich oder verschieden, mit Fluor und/oder Methyl substituiert sein können,
steht,
M für eine bicyclische Heteroaryl-Gruppe der Formel (a-2), (b-2) oder (c-2)
steht, worin
* die Verknüpfungsstelle mit der Gruppe -CH2-L bedeutet,
** die Verknüpfungsstelle mit der Gruppe Q bedeutet,
A und E unabhängig voneinander CH, CR1 oder N bedeuten,
R1 einen Substituenten ausgewählt aus der Reihe Fluor, Chlor, Brom, Cyano, (Ci-C4)- Alkyl, Trifluormethyl, Amino, (Ci-C4)-Alkoxy und Trifluormethoxy bedeutet
und
m die Zahl 0, 1 oder 2 bedeutet,
wobei für den Fall, dass der Substituent R1 zweifach auftritt, seine Bedeutungen gleich oder verschieden sein können,
und
Q für eine Gruppe der Formel
steht, worin
# die VerknüpfUngsstelle mit der Gruppe M bedeutet,
G CH oder N bedeutet,
J CR7 oder N bedeutet,
Z O oder S bedeutet,
R5, R6 und R7 gleich oder verschieden sind und unabhängig voneinander einen Rest ausgewählt aus der Gruppe bestehend aus Halogen, Nitro, Cyano, -R2, -C(=O)-R2, -C(=O)-OR2, -C(=O)-NR2R3, -0-(C=O)n-R2, -O-C(=O)-OR2, -O-C(=O)-NR2R3, -S(O)P-R2, -SO2-OR2, -SO2-NR2R3, -NR2-(C=O)n-R3, -NR2-SO2-R3, -NR2-C(=0)-
OR3, -NR4-C(=O)-NR2R3 und -NR4-SO2-NR2R3 bedeuten, worin
n die Zahl O oder 1 darstellt,
p die Zahl O oder 2 darstellt,
R2, R3 und R4 gleich oder verschieden sind und unabhängig voneinander Wasser- stoff, (C,-C6)-Alkyl, (C2-C6)-Alkenyl, (C3-C7)-Cycloalkyl, (C3-C7)-Cyclo- alkenyl, Phenyl, 5- bis 7-gliedriges Heterocyclyl oder 5- oder 6-gliedriges Heteroaryl darstellen
und/oder
R2 und R3 oder R2 und R4 gemeinsam mit dem Rest, an den sie jeweils beide gebunden sind, einen 5- bis 7-gliedrigen Heterocyclus bilden können,
wobei R2, R3 und R4 ihrerseits gegebenenfalls bis zu dreifach, gleich oder verschieden, mit Resten ausgewählt aus der Gruppe bestehend aus Fluor, Chlor, Cyano, (C,-C4)-Alkyl, Trifluormethyl, Hydroxy, (CrC4)-Alkoxy, Trifluormethoxy, Oxo, Amino, Mono-(Ci-C4)-alkylamino und Di-(Ci-C4)- alkylamino substituiert sein können,
R8 Wasserstoff, (Ci-C6)-Alkyl oder (C3-C7)-Cycloalkyl bedeutet,
wobei (C]-C6)-Alkyl bis zu fünffach mit Fluor und bis zu zweifach, gleich oder verschieden, mit (C3-C7)-Cycloalkyl, Hydroxy, (Ci-C4)-Alkoxy, Trifluormethoxy, (Ci-C4)-Acyloxy, Amino, Mono-(Ci-C4)-alkylamino, Di^Q-GO-alkylammo, (Ci- C4)-Acylamino, Hydroxycarbonyl, (CrC4)-Alkoxycarbonyl, Aminocarbonyl,
Mono-(Ci-C4)-aminocarbonyl, Di-(Ci -C4)-alkylaminocarbonyl und/oder einem 5- oder 6-gliedrigen Heterocyclus substituiert sein kann,
R9 (C,-C4)-Alkyl bedeutet, das mit Hydroxy, (CrC4)-Alkoxy, Amino, Mono-(CrC4)- alkylamino, Di-(C i-C4)-alkylamino oder bis zu dreifach mit Fluor substituiert sein kann,
R10 die oben angegebene Bedeutung von R8 hat
und
R1 ' Wasserstoff oder (C, -CVAlkyl bedeutet,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Besonders bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
L für Phenyl steht, das bis zu zweifach mit Fluor substituiert sein kann,
M für eine bicyclische Heteroaryl-Gruppe der Formel (a-3), (b-3) oder (c-3)
* die Verknüpfungsstelle mit der Gruppe -CH2-L bedeutet,
** die Verknüpfungsstelle mit der Gruppe Q bedeutet
und
A und E unabhängig voneinander CH oder N bedeuten,
und
Q für eine Gruppe der Formel
steht, worin
# die Verknüpfungsstelle mit der Gruppe M bedeutet,
J CR7 oder N bedeutet,
R5 und R6 unabhängig voneinander Wasserstoff oder Amino bedeuten,
R7 Wasserstoff, Fluor, Chlor, Brom, (C,-C4)-Alkyl, (C3-C6)-Cycloalkyl, Pyridyl oder -NR12R13 bedeutet, worin
R12 Wasserstoff oder (Ci-C4)-Alkyl, das mit Hydroxy, Methoxy oder bis zu dreifach mit Fluor substituiert sein kann, darstellt,
R13 Wasserstoff, (CrC4)-Alkyl, das mit Hydroxy, Methoxy oder bis zu dreifach mit Fluor substituiert sein kann, (d-C4)-Acyl, (CrC4)-Alkoxycarbo- nyl oder Mono- oder Di-(Ci-C4)-alkylaminocarbonyl darstellt
oder
R12 und R13 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 5- bis 7-ghedπgen Heterocyclus, der mit Oxo substituiert sein kann, bilden,
und
R8 Wasserstoff oder (Ci-C4)-Alkyl, das bis zu dreifach mit Fluor substituiert sein kann, bedeutet,
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Die in den jeweiligen Kombinationen bzw. bevorzugten Kombinationen von Resten im einzelnen angegebenen Reste-Definitionen werden unabhängig von den jeweiligen angegebenen Kombina- tionen der Reste beliebig auch durch Reste-Definitionen anderer Kombinationen ersetzt.
Besonders bevorzugt sind Kombinationen von zwei oder mehreren der oben genannten Vorzugsbereiche.
Ganz besonders bevorzugt im Rahmen der vorliegenden Erfindung sind die folgenden Verbindungen:
6-[3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-Z>]pyridin-l-yl]-l,3,5-triazin-2,4-diamin;
6-[3-(2-Fluorbenzyl)-lH-mdazol-l-yl]-l,3,5-tπazm-2,4-diamm:
6-[3-(2-Fluorbenzyl)imidazo[l,5-α]pyπdin-l-yl]-l,3,5-triazin-2,4-diarrun;
6-[8-(2-Fluorbenzyl)imidazo[l,5-α]pyπmidm-6-yl]-l,3,5-tπazin-2,4-diamin;
6-[3-(2-Fluorbenzyl)-lH-pvrazolo[3,4-6]pyπdm-l-yl]-l,3,5-tπazm-2,4-diamin;
6-[3-(2-Fluorbenzyl)-lH-pyrazolo[3,4-6]pyrazin-l-yl]-l,3,5-triazm-2,4-diamm;
2-[8-(2-Fluorbenzyl)imidazo[l,5-α]pyrimidin-6-yl]pyrimidin-4,6-diamin;
2-[3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-ό]pyridin-l-yl]pyrimidm-4,5,6-tπamin;
Methyl {4,6-diammo-2-[3-(2-fluorbenzyl)-lH-pyrazolo[4,3-ό]pyridin-l-yl]pyrimidin-5-yl}- carbamat;
5-[3-(2-Fluorbenzyl)imidazo[l,5-α]pyridm-l-yl]-l,3,4-oxadiazol-2(3H)-on;
und
3-(2-Fluorbenzyl)- 1 -(lH-tetrazol-5-yl)imidazo[ 1 ,5-α]pyridin
sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Die erfindungsgemäßen Verbindungen der Formel (I) können in Analogie zu in der Literatur be- schriebenen Methoden beispielsweise dadurch hergestellt werden, dass man
[A] eine Verbindung der Formel (II)
in welcher A, B, D, E, L, R1 und m jeweils die oben angegebenen Bedeutungen haben und
T für (C,-C4)-Alkyl steht,
mit einer Verbindung der Formel (HI)
in welcher J, R5 und R6 jeweils die oben angegebenen Bedeutungen haben,
zu einer Verbindung der Formel (I- A)
(i-A),
- - in welcher A, B, D, E, J, L, R
1, R
5, R
6 und m jeweils die oben angegebenen Bedeutungen haben,
kondensiert,
oder
[B] eine Verbindung der Formel (IV)
in welcher A, B, D, E, L, R1 und m jeweils die oben angegebenen Bedeutungen haben,
mit einer Verbindung der Formel (V)
in welcher J und R6 die oben angegebenen Bedeutungen haben,
zu einer Verbindung der Formel (I-B)
in welcher A, B, D, E, J, L, R1, R6 und m jeweils die oben angegebenen Bedeutungen haben,
umsetzt,
oder
[C] eine Verbindung der Formel (VI)
in welcher A, B, D, E, L, R1 und m jeweils die oben angegebenen Bedeutungen haben,
mit einer Verbindung der Formel (VIT)
in welcher G, J, R5 und R6 jeweils die oben angegebenen Bedeutungen haben und
X für eine geeignete Abgangsgruppe wie beispielsweise Halogen, Mesylat, Tosylat oder Triflat steht,
in eine Verbindung der Formel (I-C)
in welcher A, B, D, E, G, J, L, R1, R5, R6 und m jeweils die oben angegebenen Bedeutungen haben,
überfuhrt,
oder
[D] eine Verbindung der Formel (VHI)
- -
in welcher A, B, D, E, L, R1 und m jeweils die oben angegebenen Bedeutungen haben,
mit einer Verbindung der Formel (DCa), (Kb), (IXc) oder (DCd)
in welchen R7 und T die zuvor angegebenen Bedeutungen haben und
Y für Amino, Mono- oder Di-(C1-C4)-alkylamino, Piperidino, Morpholino, Hydroxy, (Q-GO-Alkoxy oder (CrC4)-Acyloxy steht,
zu einer Verbindung der Formel (I-D), (I-E), (I-F) bzw. (I-G)
(I-D) (I-E)
- -
(I-F) (I-G)
in welchen A, B, D, E, L, R , R und m jeweils die oben angegebenen Bedeutungen haben,
umsetzt,
oder
[E] eine Verbindung der Formel (IV)
in welcher A, B, D, E, L, R1 und m jeweils die oben angegebenen Bedeutungen haben,
mit einem Alkali-Azid in Gegenwart einer Säure oder mit Trimethylsilylazid in Gegenwart eines Katalysators wie Dibutylzinnoxid in eine Verbindung der Formel (I-H)
in welcher A, B, D, E, L, R1 und m jeweils die oben angegebenen Bedeutungen haben,
überfuhrt,
- - oder
[F] eine Verbindung der Formel (IV)
in welcher A, B, D, E, L, R1 und m jeweils die oben angegebenen Bedeutungen haben,
zunächst mit Hydroxylamin in eine Verbindung der Formel (X)
in welcher A, B, D, E, L, R1 und m jeweils die oben angegebenen Bedeutungen haben,
überfuhrt und diese dann mit Phosgen oder einem Phosgen-Äquivalent wie NN'-Carbonyl- diimidazol oder einem Chlorformiat zu einer Verbindung der Formel (I- J)
in welcher A, B, D, E, L, R1 und m jeweils die oben angegebenen Bedeutungen haben,
umsetzt,
oder
- - eine Verbindung der Formel (II)
in welcher A, B, D, E, L, T, R1 und m jeweils die oben angegebenen Bedeutungen haben,
zunächst mit Hydrazin in eine Verbindung der Formel (XI)
in welcher A, B, D, E, L, R1 und m jeweils die oben angegebenen Bedeutungen haben,
überfuhrt und diese dann mit Phosgen oder einem Phosgen-Äquivalent wie NN'-Carbonyl- diimidazol oder einem Chlorformiat zu einer Verbindung der Formel (I-K)
in welcher A, B, D, E, L, R1 und m jeweils die oben angegebenen Bedeutungen haben,
umsetzt,
- - gegebenenfalls die resultierenden Verbindungen der Formeln (I-A), (I-B), (I-C), (I-D), (I-E), (I-F), (I-G), (I-H), (I- J) bzw. (I-K) nach literaturüblichen Verfahren weiter im oben angegebenen Bedeutungsumfang der einzelnen Substituenten und Reste modifiziert
und die so erhaltenen erfindungsgemäßen Verbindungen gegebenenfalls mit den entsprechenden (i) Lösungsmitteln und/oder (n) Säuren oder Basen in ihre Solvate, Salze und/oder Solvate der Salze überführt.
Die Verbindungen der Formeln (II), (IV), (VI) und (VIQ) können in Analogie zu literaturbekannten Verfahren ausgehend von kommerziell erhältlichen oder in der Literatur beschriebenen Verbindungen hergestellt werden (vgl. nachfolgende Reaktionsschemata 1-7). Die Verbindungen der Formeln (m), (V), (VIT), (IXa), (DCb), (DCc) und (DCd) sind kommerziell erhältlich, literaturbekannt oder nach literaturübhchen Methoden herstellbar.
Die Herstellung der erfϊndungsgemäßen Verbindungen kann durch die folgenden Syntheseschemata beispielhaft veranschaulicht werden:
Schema 1
b)
[a): CuI, Pd(PPh3)2Cl2, NEt3; b): Hydrazinhydrat; c): Pd2dba3, XPHOS, Cs2CO3].
- -
Schema 2
[d): LiHMDS; e): NaCl, H2O, DMSO; f): Hydrazinhydrat, DMAP, Pyridin; g): H2, Pd/C; h): Pd2dba3, XPHOS, Cs2CO3].
- -
[i): Pd2dba3, XPHOS, Cs2CO3; j): H2, PdC; k): Chlorameisensäuremethylester].
- -
[I): LiHMDS; m): NaCl, H2O, DMSO; n): Hydrazinhydrat; o): Pd2dba3, XPHOS, Cs2CO3].
- -
P) q)
[p): KOH; q): MeI, K2CO3, Aceton; r): Dioxan; s): MeOH/EtOH; t): Biguanidin].
- -
Schema 6
[u): (2-Fluoφhenyl)-essigsäurechlorid; v): Phosphorylchlorid; w): Biguanidin].
Schema 7
[x): Pd(PPh3)2Cl2! CuI, NEt3; y): Pd2dba3, XPHOS, Cs2CO3].
Die erfindungsgemäßen Verbindungen besitzen wertvolle pharmakologische Eigenschaften und können zur Vorbeugung und Behandlung von Erkrankungen bei Menschen und Tieren verwendet werden. Die erfindungsgemäßen Verbindungen eröffnen eine weitere Behandlungsalternative und stellen somit eine Bereicherung der Pharmazie dar.
Die erfindungsgemäßen Verbindungen bewirken eine Gefäßrelaxation und eine Hemmung der Thrombozytenaggregation und fuhren zu einer Blutdrucksenkung sowie zu einer Steigerung des koronaren Blutflusses. Diese Wirkungen sind über eine direkte Stimulation der löslichen Guanylat- cyclase und einen intrazellulären cGMP-Anstieg vermittelt. Außerdem verstärken die erfindungs- gemäßen Verbindungen die Wirkung von Substanzen, die den cGMP-Spiegel steigern, wie beispielsweise EDRF (endothelium-derived relaxing factor), NO-Donatoren, Protoporphyrin DC, Arachidon- säure oder Phenylhydrazin-Derivate.
Die erfindungsgemäßen Verbindungen können daher in Arzneimitteln zur Behandlung von kardiovaskulären Erkrankungen wie beispielsweise zur Behandlung des Bluthochdrucks und der Herz- insuffizienz, stabiler und instabiler Angina pectoris, pulmonaler Hypertonie, peripheren und kardialen Gefäßerkrankungen, Arrhythmien, zur Behandlung von thromboembolischen Erkrankungen und Ischämien wie Myokardinfarkt, Hirnschlag, transistorischen und ischämischen Attacken, peripheren Durchblutungsstörungen, Reperfusionsschäden, zur Verhinderung von Restenosen wie nach Thrombolysetherapien, percutan-transluminalen Angioplastien (PTA), percutan-translumina- len Koronarangioplastien (PTCA) und Bypass, sowie zur Behandlung von Arteriosklerose, asthmatischen Erkrankungen, Krankheiten des Urogenitalsystems wie beispielsweise Prostatahypertrophie, erektile Dysfunktion, weibliche sexuelle Dysfunktion und Inkontinenz, von Osteoporose, Glaukom und Gastroparese eingesetzt werden.
Außerdem können die erfindungsgemäßen Verbindungen zur Behandlung von primärem und sekundärem Raynaud-Phänomen, von Mikrozirkulationsstörungen, Claudicatio, peripheren und autonomen Neuropathien, diabetischen Mikroangiopathien, diabetischer Retinopathie, diabetischen Geschwüren an den Extremitäten, Gangren, CREST-Syndrom, Erythematose, Onychomykose, rheumatischen Erkrankungen sowie zur Förderung der Wundheilung verwendet werden.
Ferner eignen sich die erfindungsgemäßen Verbindungen zur Behandlung von akuten und chronischen Lungenkrankheiten, wie den Respiratory Distress-Syndromen (ALI, ARDS) und chronisch-obstruktiven Atemwegserkrankungen (COPD), sowie zur Behandlung von akuter und chronischer Niereninsuffizienz.
Die in der vorliegenden Erfindung beschriebenen Verbindungen stellen auch Wirkstoffe zur Bekämpfung von Krankheiten im Zentralnervensystem dar, die durch Störungen des NO/cGMP- Systems gekennzeichnet sind. Insbesondere sind sie geeignet zur Verbesserung der Wahrnehmung, Konzentrationsleistung, Lernleistung oder Gedächtnisleistung nach kognitiven Störungen, wie sie insbesondere bei Situationen/Krankheiten/Syndromen auftreten wie "Mild cognitive impairment", altersassoziierten Lern- und Gedächtnisstörungen, altersassoziierten Gedächtnisverlusten, vaskulärer Demenz, Schädel-Hirn-Trauma, Schlaganfall, Demenz, die nach Schlaganfällen auftritt ("post stroke dementia"), post-traumatischem Schädel-Hirn-Trauma, allgemeinen Konzentrationsstörungen, Konzentrationsstörungen bei Kindern mit Lern- und Gedächtnisproblemen, Alzhei- mer'scher Krankheit, Demenz mit Lewy-Körperchen, Demenz mit Degeneration der Frontallappen einschliesslich des Pick's-Syndroms, Parkinson'scher Krankheit, progressiver nuclear palsy, Demenz mit corticobasaler Degeneration, Amyolateralsklerose (ALS), Huntington'scher Krankheit, Multipler Sklerose, Thalamischer Degeneration, Creutzfeld-Jacob-Demenz, HIV-Demenz, Schizophrenie mit Demenz oder Korsakoff-Psychose. Sie eignen sich auch zur Behandlung von Erkrankungen des Zentralnervensystems wie Angst-, Spannungs- und Depressionszuständen, zentral-nervös bedingten Sexualdysfunktionen und Schlafstörungen sowie zur Regulierung krankhafter Störungen der Nahrungs-, Genuss- und Suchtmittelaufhahme.
Weiterhin eignen sich die erfindungsgemäßen Verbindungen auch zur Regulation der cerebralen Durchblutung und stellen wirkungsvolle Mittel zur Bekämpfung von Migräne dar. Auch eignen sie sich zur Prophylaxe und Bekämpfung der Folgen cerebraler Infarktgeschehen (Apoplexia cerebri) wie Schlaganfall, cerebraler Ischämien und des Schädel-Hirn-Traumas. Ebenso können die erfindungsgemäßen Verbindungen zur Bekämpfung von Schmerzzuständen eingesetzt werden.
Zudem besitzen die erfindungsgemäßen Verbindungen antimflammatorische Wirkung und können daher als entzündungshemmende Mittel eingesetzt werden.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prävention von Erkran- kungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen, unter Verwendung einer wirksamen Menge von mindestens einer der erfindungsgemäßen Verbindungen.
Die erfindungsgemäßen Verbindungen können allein oder bei Bedarf in Kombination mit anderen Wirkstoffen eingesetzt werden. Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, enthaltend mindestens eine der erfindungsgemäßen Verbindungen und einen oder mehrere weitere Wirkstoffe, insbesondere zur Behandlung und/oder Prävention der zuvor genannten Er- krankungen. Als geeignete Kombinationswirkstoffe seien beispielhaft und vorzugsweise genannt:
• organische Nitrate und NO-Donatoren, wie beispielsweise Natriumnitroprussid, Nitroglycerin, Isosorbidmononitrat, Isosorbiddinitrat, Molsidomin oder SIN-I, sowie inhalatives NO;
• Verbindungen, die den Abbau von cyclischem Guanosinmonophosphat (cGMP) inhibieren, wie beispielsweise Inhibitoren der Phosphodiesterasen (PDE) 1, 2 und/oder 5, insbesondere
PDE 5 -Inhibitoren wie Sildenafil, Vardenafil und Tadalafil;
• antithrombotisch wirkende Mittel, beispielhaft und vorzugsweise aus der Gruppe der Thrombozytenaggregationshemmer, der Antikoagulantien oder der profibrinolytischen Substanzen;
• den Blutdruck senkende Wirkstoffe, beispielhaft und vorzugsweise aus der Gruppe der Calcium-Antagonisten, Angiotensin Aü-Antagonisten, ACE-Hemmer, Endothelin-Antagonisten, Renin-Inhibitoren, alpha-Rezeptoren-Blocker, beta-Rezeptoren-Blocker, Mineralocor- ticoid-Rezeptor-Antagonisten sowie der Diuretika; und/oder
• den Fettstoffwechsel verändernde Wirkstoffe, beispielhaft und vorzugsweise aus der Gruppe der Thyroidrezeptor-Agonisten, Cholesterinsynthese-Inhibitoren wie beispielhaft und vorzugsweise HMG-CoA-Reduktase- oder Squalensynthese-Inhibitoren, der ACAT-Inhibitoren, CETP-Inhibitoren, MTP-Inhibitoren, PPAR-alpha-, PPAR-gamma- und/oder PPAR-delta- Agonisten, Cholesterin-Absorptionshemmer, Lipase-Inhibitoren, polymeren Gallensäure- adsorber, Gallensäure-Reabsorptionshemmer und Lipoprotein(a)-Antagonisten.
Unter antithrombotisch wirkenden Mittel werden vorzugsweise Verbindungen aus der Gruppe der Thrombozytenaggregationshemmer, der Antikoagulantien oder der profibrinolytischen Substanzen verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thrombozytenaggregationshemmer, wie beispielhaft und vor- zugsweise Aspirin, Clopidogrel, Ticlopidin oder Dipyridamol, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfϊndungsgemäßen Verbindungen in Kombination mit einem Thrombin-Inhibitor, wie beispielhaft und vorzugsweise Ximela- gatran, Melagatran, Bivalirudin oder Clexane, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem GPIIb/πia-Antagonisten, wie beispielhaft und vorzugsweise Tirofiban oder Abciximab, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Faktor Xa-Inhibitor, wie beispielhaft und vorzugsweise Riva- roxaban (BAY 59-7939), DU- 176b, Apixaban, Otamixaban, Fidexaban, Razaxaban, Fondaparinux, Idraparinux, PMD-3112, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 oder SSR-128428, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit Heparin oder einem low molecular weight (LMW)-Heparin-Derivat verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Vitamin K-Antagonisten, wie beispielhaft und vorzugsweise Coumarin, verabreicht.
Unter den Blutdruck senkenden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der Calcium-Antagonisten, Angiotensin AH-Antagonisten, ACE-Hemmer, Endothelin-Antagonisten, Renin-Inhibitoren, alpha-Rezeptoren-Blocker, beta-Rezeptoren-Blocker, Mineralocorticoid-Rezep- tor-Antagonisten sowie der Diuretika verstanden.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Calcium-Antagonisten, wie beispielhaft und vorzugsweise Nifedipin, Amlodipin, Verapamil oder Diltiazem, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem alpha- 1 -Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Prazosin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem beta-Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Propranolol, Atenolol, Timolol, Pindolol, Alprenolol, Oxprenolol, Penbutolol, Bupranolol, Meti- pranolol, Nadolol, Mepindolol, Carazalol, Sotalol, Metoprolol, Betaxolol, Celiprolol, Bisoprolol,
Carteolol, Esmolol, Labetalol, Carvedilol, Adaprolol, Landiolol, Nebivolol, Epanolol oder Bucin- dolol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Angiotensin Aü-Antagonisten, wie beispielhaft und vorzugs- weise Losartan, Candesartan, Valsartan, Telmisartan oder Embursatan, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACE-Hemmer, wie beispielhaft und vorzugsweise Enalapril, Captopril, Lisinopril, Ramipril, Delapril, Fosinopril, Quinopril, Perindopril oder Trandopril, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Endothelin-Antagonisten, wie beispielhaft und vorzugsweise Bosentan, Darusentan, Ambrisentan oder Sitaxsentan, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Renin-Inhibitor, wie beispielhaft und vorzugsweise Aliskiren, SPP-600 oder SPP-800, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Mineralocorticoid-Rezeptor-Antagonisten, wie beispielhaft und vorzugsweise Spironolacton oder Eplerenon, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Diuretikum, wie beispielhaft und vorzugsweise Furosemid, verabreicht.
Unter den Fettstoffwechsel verändernden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der CETP-Inhibitoren, Thyroidrezeptor-Agonisten, Cholesterinsynthese-Inhibitoren wie HMG-CoA-Reduktase- oder Squalensynthese-Inhibitoren, der ACAT-Inhibitoren, MTP-Inhibi- toren, PPAR-alpha-, PPAR-gamma- und/oder PPAR-delta-Agonisten, Cholesterin-Absorptions- hemmer, polymeren Gallensäureadsorber, Gallensäure-Reabsorptionshemmer, Lipase-Inhibitoren sowie der Lipoprotein(a)-Antagonisten verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem CETP-Inhibitor, wie beispielhaft und vorzugsweise Torcetrapib (CP-529 414), JJT-705 oder CETP-vaccine (Avant), verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thyroidrezeptor-Agonisten, wie beispielhaft und vorzugsweise D-Thyroxin, 3,5,3'-Triiodothyronin (T3), CGS 23425 oder Axitirome (CGS 26214), verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem HMG-CoA-Reduktase-Inhibitor aus der Klasse der Statine, wie beispielhaft und vorzugsweise Lovastatin, Simvastatin, Pravastatin, Fluvastatin, Atorvastatin, Rosuvastatin, Cerivastatin oder Pitavastatin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Squalensynthese-Inhibitor, wie beispielhaft und vorzugsweise BMS-188494 oder TAK-475, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACAT-Inhibitor, wie beispielhaft und vorzugsweise Avasimibe, Melinamide, Pactimibe, Eflucimibe oder SMP-797, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem MTP-Inhibitor, wie beispielhaft und vorzugsweise Implitapide, BMS-201038, R-103757 oder JTT-130, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPAR-gamma-Agonisten, wie beispielhaft und vorzugsweise Piogiitazone oder Rosiglitazone, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPAR-delta-Agonisten, wie beispielhaft und vorzugsweise GW 501516 oder BAY 68-5042, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Cholesterin-Absorptionshemmer, wie beispielhaft und vorzugs- weise Ezetimibe, Tiqueside oder Pamaqueside, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Lipase-Inhibitor, wie beispielhaft und vorzugsweise Orlistat, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem polymeren Gallensäureadsorber, wie beispielhaft und vorzugsweise Cholestyramin, Colestipol, Colesolvam, CholestaGel oder Colestimid, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Gallensäure-Reabsorptionshemmer, wie beispielhaft und vorzugsweise ASBT (= IBAT)-Inhibitoren wie z.B. AZD-7806, S-8921, AK-105, BARI-1741, SC-435 oder SC-635, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Lipoprotein(a)-Antagonisten, wie beispielhaft und vorzugsweise Gemcabene calcium (CI-1027) oder Nicotinsäure, verabreicht.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nicht- toxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Die erfindungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctivae otisch oder als Implantat bzw. Stent.
Für diese Applikationswege können die erfindungsgemäßen Verbindungen in geeigneten Applikationsformen verabreicht werden.
Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende, die erfin- dυngsgemäßen Verbindungen schnell und/oder modifiziert abgebende Applikationsformen, die die erfindungsgemäßen Verbindungen in kristalliner und/oder amorphisierter und/oder gelöster Form enthalten, wie z.B. Tabletten (nicht-überzogene oder überzogene Tabletten, beispielsweise mit magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die Freisetzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfallende Tabletten oder Filme/Oblaten, Filme/Lyophylisate, Kapseln (beispielsweise Hart- oder Weich- gelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (z.B. intramuskulär, subcutan, intracutan, percutan oder intraperitoneal). Für die par- enterale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusionszubereitungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern.
Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulverinhalatoren, Nebulizer), Nasentropfen, -lösungen oder -sprays, lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapseln, Suppositorien, Ohren- oder Augen- präparationen, Vaginalkapseln, wäßrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspensionen, Salben, Cremes, transdermale therapeutische Systeme (z.B. Pflaster), Milch, Pasten, Schäume, Streupuder, Implantate oder Stents.
Bevorzugt sind die orale oder parenterale Applikation, insbesondere die orale Applikation.
Die erfmdungsgemäßen Verbindungen können in die angeführten Applikationsformen überführt werden. Dies kann in an sich bekannter Weise durch Mischen mit inerten, nichttoxischen, pharma- zeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a. Trägerstoffe (beispielsweise mikrokristalline Cellulose, Lactose, Mannitol), Lösungsmittel (z.B. flüssige PoIy- ethylenglycole), Emulgatoren und Dispergier- oder Netzmittel (beispielsweise Natriumdodecyl- sulfat, Polyoxysorbitanoleat), Bindemittel (beispielsweise Polyvinylpyrrolidon), synthetische und natürliche Polymere (beispielsweise Albumin), Stabilisatoren (z.B. Antioxidantien wie beispiels- weise Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisenoxide) und Geschmacks- und/oder Geruchskorrigentien.
Im Allgemeinen hat es sich als vorteilhaft erwiesen, bei parenteraler Applikation Mengen von etwa 0.001 bis 1 mg/kg, vorzugsweise etwa 0.01 bis 0.5 mg/kg Körpergewicht zur Erzielung wirksamer Ergebnisse zu verabreichen. Bei oraler Applikation beträgt die Dosierung etwa 0.01 bis 100 mg/kg, vorzugsweise etwa 0.01 bis 20 mg/kg und ganz besonders bevorzugt 0.1 bis 10 mg/kg Körpergewicht.
Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindestmenge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die nachfolgenden Ausführungsbeispiele erläutern die Erfindung. Die Erfindung ist nicht auf die Beispiele beschränkt.
Die Prozentangaben in den folgenden Tests und Beispielen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
- -
- -
A. Beispiele
Abkürzungen und Akronyme:
aq. wässrige Lösung ber. berechnet
DCI direkte chemische Ionisation (bei MS)
DMAP 4-NN-Dimethylaminopyridm
DMF Dimethylformamid
DMSO Dimethylsulfoxid d. Th. der Theorie (bei Ausbeute) eq. Äquivalent(e)
ESI Elektrospray-Ionisation (bei MS)
Et Ethyl gef. gefunden h Stunde(n)
HPLC Hochdruck-, Hochleistungsflüssigchromatographie
HRMS hochaufgelöste Massenspektrometrie konz. konzentriert
LC/MS Flüssigchromatographie-gekoppelte Massenspektrometrie
LiHMDS Lithiumhexamethyldisilazid
Me Methyl min Minute(n)
MS Massenspektrometrie
NMR Kernresonanzspektrometrie
Pd2dba3 Tris-(dibenzylidenaceton)-dipalladium
Ph Phenyl
RT Raumtemperatur
R. Retentionszeit (bei HPLC) THF Tetrahydrofuran
UV Ultraviolett-Spektrometrie v/v Volumen zu Volumen-Verhältnis (einer Lösung) XPHOS Dicyclohexyl-(2',4',6'-triisopropylbiphenyl-2-yl)-phosphin
LC/MS- und HPLC-Methoden:
Methode 1 (LC/MS):
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV DAD; Säule: Phenomenex Gemini 3μ 30 mm x 3.00 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2.5 min 30% A → 3.0 min 5% A -> 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 2 (LC/MS):
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Phenomenex Syn- ergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A — > 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 3 (LC/MS):
Instrument: Micromass Platform LCZ mit HPLC Agilent Serie 1100; Säule: Thermo Hypersil GOLD 3μ 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 100% A -> 0.2 min 100% A → 2.9 min 30% A → 3.1 min 10% A → 5.5 min 10% A; Ofen: 500C; Fluss: 0.8 ml/min; UV-Detektion: 210 nm.
Methode 4 (LC/MS):
Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Gemini 3μ 30 mm x 3.00 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 208-400 nm.
Methode 5 (LC/MS):
Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2.5 min
30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 208-400 nm.
Methode 6 (HPLO:
Instrument: HP 1100 mit DAD-Detektion; Säule: Kromasil 100 RP-18, 60 mm x 2.1 mm, 3.5 μm; Eluent A: 5 ml HClO4 (70%-ig) / Liter Wasser, Eluent B: Acetonitril; Gradient: 0 min 2% B → 0.5 min 2% B → 4.5 min 90% B → 6.5 min 90% B → 6.7 min 2% B → 7.5 min 2% B; Fluss: 0.75 ml/min; Säulentemperatur: 300C; UV-Detektion: 210 nm.
Methode 7 (LC/MS):
Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Onyx Monolithic C18, 100 mm x 3 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2 min 65% A → 4.5 min 5% A → 6 min 5% A; Fluss: 2 ml/min; Ofen: 400C; UV-Detektion: 208-400 nm.
Ausgangsverbindungen und Intermediate:
Beispiel IA
3-(3,5-Dichloφyridin-2-yl)-2-(2-fluoφhenyl)-3-oxopropansäuremethylester
Man legt 50 ml THF bei -78 0C vor und gibt 28.5 ml (28.5 mmol) einer 1 M Lösung von LiHMDS in Hexan hinzu. Dann wird eine Lösung von 4.00 g (23.8 mmol) (2-Fluorphenyl)-essigsäure- methylester in 10 ml THF zugetropft. Man läßt 1 h bei -78°C nachrühren und gibt dann portionsweise 6.00 g (28.5 mmol) 3,5-Dichlorpyridin-2-carbonsäurechlorid hinzu. Nach einer weiteren Stunde läßt man auf RT kommen und versetzt tropfenweise mit gesättigter Ammoniumchlorid- Lösung. Es wird mit Wasser verdünnt und mit Ethylacetat extrahiert. Die organische Phase wird mit gesättigter Natriumchlorid-Lösung gewaschen und über Natriumsulfat getrocknet. Man engt im Vakuum ein und reinigt den Rückstand durch Chromatographie an Kieselgel (Eluens: Dichlor-
methan/Methanol 50:1). Man erhält 4.46 g (46% d. Th.) der gewünschten Verbindung als gelbliches Öl.
LC/MS (Methode 5): Rt = 2.77, 2.82 min; MS (ESIpos): m/z = 340 (35Cl2), 342 (35Cl37Cl), 344 (37Cl2) [M+H]+.
Beispiel 2A
l-(3,5-Dichloφyridin-2-yl)-2-(2-fluorphenyl)ethanon
Man erhitzt eine Mischung aus 4.50 g (13.2 mmol) 3-(3,5-Dichlorpyridin-2-yl)-2-(2-fluorphenyl)- 3-oxopropansäuremethylester aus Beispiel IA, 845 mg (14.5 mmol) Natriumchlorid, 474 mg (26.3 mmol) Wasser und 13.5 ml DMSO in der Mikrowelle für 10 min auf 1500C, rührt dann in Wasser ein und extrahiert mit Ethylacetat. Die organische Phase wird über Natriumsulfat getrocknet und eingeengt. Den Rückstand reinigt man durch Chromatographie an Kieselgel (Eluens: Di- chlormethan) und erhält ein gelbes Öl, das allmählich durchkristallisiert und 3.18 g (85% d. Th.) der Titelverbindung ergibt.
1H-NMR (400 MHz, DMSO-d6): δ = 4.50 (s, 2H), 7.14-7.22 (m, 2H), 7.31-7.38 (m, 2H), 8.43 (d, J = 2.0 Hz, IH), 8.79 (d, J = 2.0 Hz, IH).
LC/MS (Methode 1): Rt = 2.76 min; MS (ESIpos): m/z = 283 (35Cl2), 285 (35Cl37Cl), 287 (37Cl2) [M+H]+.
Beispiel 3A
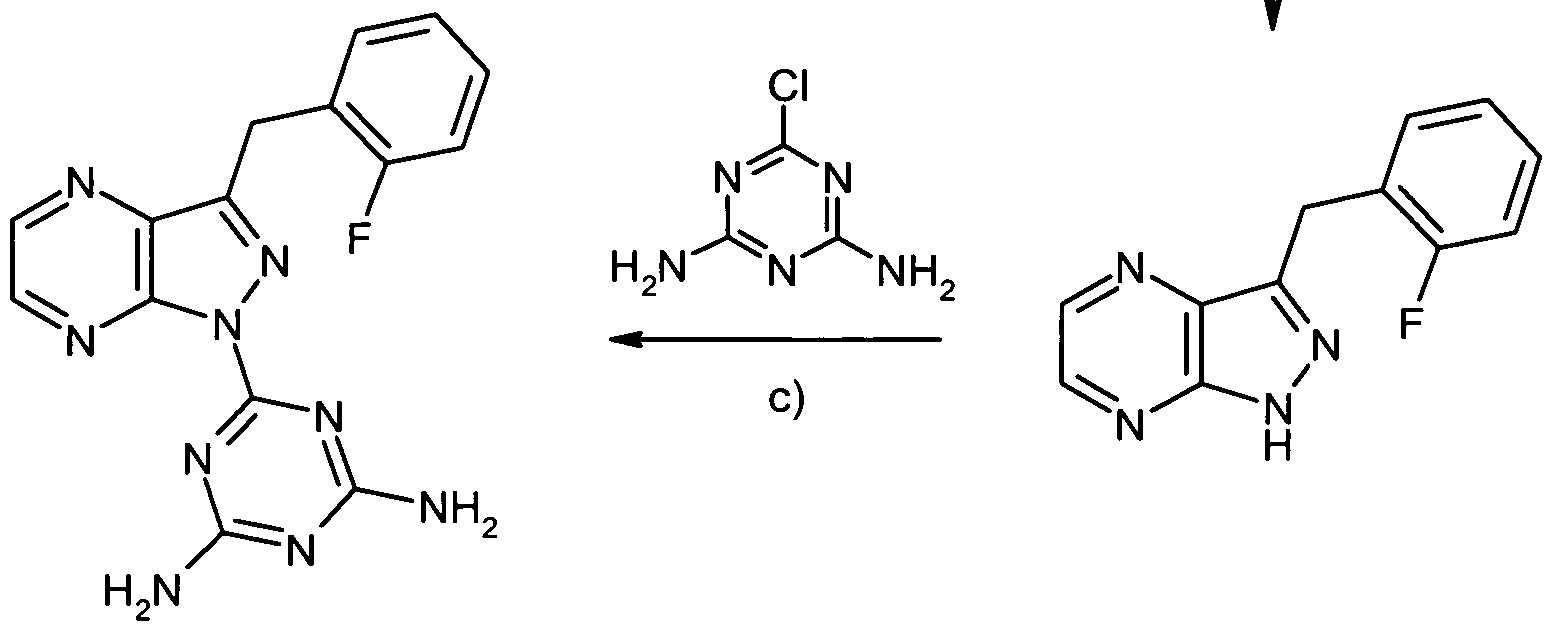
6-Chlor-3-(2-fluorbenzyl)-lH-pyrazolo[4,3-ό]pyridin
- -
Zu einer Lösung von 1.35 g (4.75 mmol) l-(3,5-Dichlθφyπdm-2-yl)-2-(2-fluorphenyl)ethanon aus Beispiel 2 A in 12 ml Pyridin werden etwas DMAP sowie 238 mg (4.75 mmol) Hydrazmhydrat gegeben. Man erhitzt in der Mikrowelle im geschlossenen Gefäß 20 min lang auf 1600C, engt dann im Vakuum ein und reinigt den Rückstand durch Chromatographie an Kieselgel (Eluens: Dichlor- methan/Methanol 100:3). Man erhält 507 mg (41% d. Th.) der Titelverbmdung und 388 mg des nicht cychsierten Hydrazons. Letzteres wird in 5 ml DMF erneut in der Mikrowelle 1.5 h lang auf 2000C erhitzt. Die Lösung wird eingeengt und der Rückstand mittels präparativer HPLC gereinigt. Man erhält so weitere 176 mg (12% d. Th.) der Titelverbmdung.
1H-NMR (400 MHz, DMSOd6): δ = 4.34 (s, 2H), 7.09 (dt, J= 7.3, 1.0 Hz, IH), 7.16 (ddd, J = 10.0, 8.3, 1.0 Hz, IH), 7.22-7.29 (m, IH), 7.31 (dt, J= 7.6, 1.5 Hz, IH), 8.15 (d, J= 2.0 Hz, IH), 8.48 (d, J= 2.0 Hz, IH), 13.21 (br. s, IH).
13C-NMR (125 MHz, DMSOd6): δ = 24.2 (d, 3JC,F = 3.5 Hz), 115.0 (d, 2JC,F = 21.7 Hz), 117.6, 124.2 (d, 4Jc,F = 3.5 Hz), 125.6 (d, 2JC,F = 15.6 Hz), 128.1, 128.2 (d, 3JC>F = 8.1 Hz), 131.1 (d, 3JC,F = 4.4 Hz), 133.1, 137.3, 143.3, 143.4, 160.1 (d, 1Jc1R = 244 Hz).
HRMS: ber. für C13H9ClFN3 261.0469; gef. 261.0466.
LC/MS (Methode 4): R, = 2.16 mm; MS (ESIpos): m/z = 263 (35Cl), 265 (37Cl) [M+H]+.
Beispiel 4A
3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-ö]pyπdin
Man löst 411 mg (1.57 mmol) 6-Chlor-3-(2-fluorbenzyl)-lH-pyrazolo[4,3-&]pyπdin aus Beispiel 3A in einer Mischung aus 4 ml Ethanol und 4 ml TΗF, gibt 159 mg (1.57 mmol) Tπethylamin und
- -
140 mg 10% Palladium auf Kohle hinzu und hydriert für 2 h in einer Wasserstoffatmosphäre bei normalem Druck. Dann wird vom Katalysator abfiltriert, im Vakuum eingeengt und in Wasser aufgenommen. Man extrahiert mit Dichlormethan und trocknet die organische Phase über Natriumsulfat. Nach dem Einengen erhält man 334 mg (94% d. Th.) der Titel Verbindung als weiße Kristalle.
1H-NMR (400 MHz, DMSOd6): δ = 4.35 (s, 2H), 7.08 (dt, J = 7.3, 1.2 Hz, IH), 7.15 (ddd, J = 10.3, 8.3, 1.2 Hz, IH), 7.22-7.28 (m, IH), 7.32 (dt, J= 7.6, 1.5 Hz, IH), 7.35 (dd, J= 8.6, 4.4 Hz, IH), 7.95 (dd, J = 8.6, 1.2 Hz, IH), 8.49 (dd, J= 4.4, 1.2 Hz, IH), 13.02 (br. s, IH).
13C-NMR (125 MHz, DMSOd6): δ = 24.2 (d, 3JC>F = 3.5 Hz), 115.0 (d, 2JC,F = 21.8 Hz), 118.2, 120.8, 124.1 (d, 4JC,F = 3.4 Hz), 126.0 (d, 2JC,F = 15.6 Hz), 128.1 (d, 3JC,F = 8.0 Hz), 131.2 (d, Vc1F = 4.4 Hz), 133.0, 138.8, 143.1, 144.5, 160.1 (d, 1Jc,,, = 244 Hz).
HRMS: ber. für C13H10FN3 227.0859; gef. 227.0856.
LC/MS (Methode 4): R, = 1.66 min; MS (ESIpos): m/z = 228 [M+H]+.
Beispiel 5A
3-(2-Fluorbenzyl)-lH-indazol
Unter Argon werden 200 mg (0.86 mmol) 2-Iodphenylhydrazin, 133 mg (1.11 mmol) 2-Fluor- phenylacetylen, 30 mg (0.04 mmol) Bis-(triphenylphosphin)-palladium(II)chlorid sowie 8 mg (0.04 mmol) Kupfer(I)iodid in einer Mischung aus 1.5 ml Triethylamin und 3.5 ml Benzol vorge- legt. Man erhitzt 4 h auf Rückfluss, verdünnt dann mit Ethylacetat und filtriert. Das Filtrat wird eingeengt und der Rückstand mittels präparativer ΗPLC aufgereinigt. Man erhält 36 mg (18% d. Th.) des gewünschten Produktes.
1H-NMR (500 MHz, DMSOd6): δ = 4.29 (s, 2H), 7.04 (dd, J= 8.1, 6.8 Hz, IH), 7.11 (ddd, J = 8.3, 6.8, 1.0 Hz, IH), 7.16 (ddd, J= 10.3, 8.3, 1.0 Hz, IH), 7.23-7.28 (m, IH), 7.31 (ddd, J= 8.3, 7.1, 1.0 Hz, 2H), 7.47 (d, J= 8.3 Hz, IH), 7.60 (d, J= 8.1 Hz, IH), 12.76 (br. s, IH).
- -
13C-NMR (125 MHz, DMSOd6): δ = 25.9 (d, 3JC,F = 3.3 Hz), 110.1, 115.2 (d, 2JC,F = 21.7 Hz), 119.68, 119.70, 121.3, 124.3 (d, 4JC,F = 3.4 Hz), 125.9, 126.3 (d, 2JC,F = 15.7 Hz), 128.4 (d, 3JC,F = 8.1 Hz), 131.2 (d, 3JC,F = 4.5 Hz), 140.9, 142.7, 160.3 (d, 'JC,F = 244 Hz).
HRMS: ber. für C14H11FN2 + [H+] 227.0980; gef. 227.0984.
LC/MS (Methode 2): Rt = 2.09 min; MS (ESIpos): m/z = 227 [M+H]+.
Beispiel 6A
Ethyl {[(2-fluorphenyl)acetyl]amino}(pyπdm-2-yl)acetat
Man legt 2.50 g (12.1 mmol) Ethyl amino(pyπdin-2-yl)acetat [G. van ZyI et al., J Org Chem 1961, 26, 3373] in 20 ml Dichlormethan vor, gibt 4.9 ml (60.3 mmol) Pyπdm hinzu und kühlt auf O0C. Dann gibt man langsam eine Lösung von 1.86 g (12 1 mmol) (2-Fluorphenyl)-εssigsäure- chloπd hinzu und rührt 30 mm bei 00C und dann 2 h bei RT nach. Man verdünnt mit Ethylacetat, wäscht mit Natπumhydrogencarbonat-Lösung und trocknet über Natriumsulfat. Das Rohprodukt wird durch Chromatographie an Kieselgel (Eluens: Cyclohexan/Ethylacetat 1 :1) gereinigt. Man er- hält 2.9 g (76% d. Th.) des gewünschten Produkts.
1H-NMR (400 MHz, DMSOd6): δ = 1.11 (t, J= 7.1 Hz, 3H), 3.60-3.69 (m, 2H), 4.04-4.16 (m, 2H), 5.60 (d, J= 7.6 Hz, IH), 7.11-7.17 (m, 2H), 7.25-7.32 (m, IH), 7.34 (dt, J= 7.7, 0.7 Hz, IH), 7.39 (ddd, J= 7.6, 4.9, 1.0 Hz, IH), 7.50 (d, J= 7.8 Hz, IH), 7.85 (dt, J= 7.8, 1.5 Hz, IH), 8.56 (d, J= 4.9 Hz, IH), 8.95 (d, J= 7.6 Hz, IH).
13C-NMR (125 MHz, DMSOd6): δ = 13.8, 34.5 (d, 3JC,F = 2.1 Hz), 57.9, 60.8, 114.8 (d, 2JC,F = 21.6 Hz), 122.9, 123.0 (d, 2JC,F = 16.0 Hz), 123.4, 124.0 (d, 4JC,F = 3.4 Hz), 128.5 (d, 3JC,F = 8.1 Hz), 131.6 (d, 3JC,F = 4.4 Hz), 137.3, 149.0, 155.3, 160.4 (d, 'JC,F = 244 Hz), 169.2, 169.5.
HRMS: ber. für C17H17FN2O3 + [H+] 317.1296; gef. 317.1286.
LC/MS (Methode 5): R, = 2.03 min; MS (ESIpos): m/z = 317 [M+H]+.
Beispiel 7A
Ethyl 3-(2-fluorbenzyl)imidazo[l,5-α]pyπdin-l-carboxylat
Man legt 2.72 g (8.60 mmol) Ethyl {[(2-fluorphenyl)acetyl]amino}(pyndm-2-yl)acetat aus Beispiel 6A in 30 ml 1 ,2-Dichlorethan vor und versetzt mit 4.81 ml (51.6 mmol) Phosphorylchloπd. Man erhitzt 9 h auf Rückfluss, engt dann im Vakuum ein und nimmt den Rückstand in Ethylacetat auf. Man wäscht mit gesättigter Natriumcarbonat-Lösung, trocknet über Natriumsulfat und reinigt durch Chromatographie an Kieselgel (Eluens: Cyclohexan/Ethylacetat 2: 1). Man erhält 2.16 g (84% d. Th.) der gewünschten Verbindung als dunkles Öl.
1H-NMR (400 MHz, DMSO-d6): δ = 1.32 (t, J= 7.1 Hz, 3H), 4.29 (q, J= 7.1 Hz, 2H), 4.49 (s, 2H), 6.98 (dt, J= 6.4, 1.0 Hz, IH), 7.12-7.35 (m, 5H), 8.04 (d, J= 9.2 Hz, IH), 8.42 (d, J= 7.1 Hz, IH).
13C-NMR (125 MHz, DMSOd6): δ = 14.4, 25.5 (d, 3JC,F = 3.2 Hz), 59.3, 113.9, 115.3 (d, 2JC,F = 21.4 Hz), 118.5, 119.1, 123.3, 123.4 (d, 2JC,F = 15.6 Hz), 124.5 (d, 4JC,F = 3.4 Hz), 124.8, 128.9 (d, 3JCF = 8.1 Hz), 130.9 (d, 3JC,F = 4.3 Hz), 134.2, 137.5, 160.4 (d, 1Jc1F = 245 Hz), 162.5.
HRMS: ber. für C17H15FN2O2 + [H+] 299.1191; gef. 299.1184.
LC/MS (Methode 2): R, = 2.04 min; MS (ESIpos): m/z = 299 [M+H]+.
Beispiel 8A
2-Amino-3-(2-fluorphenyl)propanmtπl
- -
Unter Argon wird bei 00C eine Lösung von 3.99 g (21.1 mmol) l-(Brornrnethyl)-2-fluorbenzol in 85 ml Dichlormethan zu einer Suspension von 5.00 g (22.7 mmol) N-(Diphenylmethylen)-ammo- acetonitril und 1.23 g (21.9 mmol) Kaliumhydroxid in 85 ml Dichlormethan getropft. Man läßt 20 min nachrühren, filtriert dann und engt im Vakuum ein. Der Rückstand wird mit 200 ml Di- ethylether und 200 ml 1 Ν Salzsäure versetzt und 10 h bei RT gerührt. Man trennt dann die wäss- rige Phase ab, stellt sie mit konz. Natronlauge alkalisch und nimmt das entstehende Öl in Dichlormethan auf. Die organische Phase wird über Natriumsulfat getrocknet und eingeengt. Man erhält 2.4 g (64% d. Th.) des gewünschten Produkts als gelbes Öl.
1H-NMR (500 MHz, DMSOd6): δ = 2.46 (d, J = 7.1 Hz, 2H), 2.97 (d, J = 7.6 Hz, 2H), 3.94 (tt, J = 7.6, 7.1 Hz, IH), 7.15-7.21 (m, 2H), 7.29-7.35 (m, IH), 7.39 (dt, J= 7.6, 1.5 Hz, IH).
13C-NMR (125 MHz, DMSO-d6): δ = 33.9 (d, 3JC,F = 1.6 Hz), 43.8 (d, 4JC,F = 1.2 Hz), 115.1 (d, 2Jc1F = 21.9 Hz), 122.3, 123.4 (d, 2JC,F = 15.5 Hz), 124.3 (d, 4JC,F = 3.4 Hz), 129.0 (d, 3JC,F = 8.2 Hz), 131.8 (d, 3JC,F = 4.4 Hz), 160.6 (d, 'JC,F = 244 Hz).
HRMS: ber. für C9H9FN2 164.0750; gef. 164.0749.
LC/MS (Methode 3): Rt = 1.89 min; MS (DCI): m/z = 182 [M+NH4]+.
Beispiel 9A
Ethyl 5-amino-4-(2-fluorbenzyl)-lH-imidazol-2-carboxylat
Man rührt eine Lösung aus 0.90 g (5.45 mmol) 2-Amino-3-(2-fluorphenyl)propannitril aus Beispiel 8A und 2.20 g (9.27 mmol) Ethyl-imino(methylthio)acetat [D. Catarzi et al., J. Med. Chem. 1995,
- -
38, 2196-2201] in 10 ml Dioxan zwei Tage lang bei RT. Danach wird die Lösung im Vakuum eingeengt und der Rückstand durch präparative HPLC aufgereinigt. Man erhält 770 mg (54% d. Th.) des gewünschten Produkts.
1H-NMR (400 MHz, DMSO-d6): δ = 1.24, 1.26 (t, J = 7.1 Hz, 3H), 3.77, 3.89 (s, 2H), 4.17, 4.21 (q, J= 7.1 Hz, 2H), 4.46, 5.00 (s, 2H), 7.02-7.27 (m, 4H), 12.05, 12.67 (br. s, IH) [das NMR zeigt zwei Signalsätze der beiden tautomeren Formen des Produkts].
13C-NMR (125 MHz, DMSOd6): δ = 14.3, 24.4 (d, 3JC,F = 3.2 Hz), 59.5, 120.7, 114.7 (d, 2JC,F = 21.7 Hz), 124.0 (d, 4JC,F= 3.3 Hz), 127.6 (d, 2JC,F = 15.8 Hz), 127.0, 127.5 (d, 3JC,F = 8.0 Hz), 130.6 (d, 3Jc,F = 4.8 Hz), 138.9, 157.9, 160.1 (d, 1Jc1F = 243 Hz); Minderkomponente: δ = 14.2, 21.9 (d, 3Jc1F = 3.5 Hz), 59.8, 111.6, 114.8 (d, 2JC,F = 21.5 Hz), 124.2 (d, 4JC,F = 3.3 Hz), 126.2 (d, 2JC,F = 15.7 Hz), 129.9, 128.0 (d, 3JC,F = 8.0 Hz), 130.0 (d, 3JC,F = 4.5 Hz), 146.2, 158.0, 160.0 (d, 1JcF = 244 Hz) [das NMR zeigt zwei Signalsätze der beiden tautomeren Formen des Produkts] .
HRMS: ber. TUr C13H14FN3O2 263.1070; gef. 263.1070.
LC/MS (Methode 1): R, = 1.37 min; MS (ESIpos): m/z = 264 [M+H]+.
Beispiel IQA
Ethyl 8-(2-fluorbenzyl)imidazo[l,5-α]pyrimidm-6-carboxylat
50 mg (0.19 mmol) Ethyl 5-amino-4-(2-fluorbenzyl)-lH-imidazol-2-carboxylat aus Beispiel 9A werden in 0.5 ml Ethanol auf Rückfluss erhitzt und tropfenweise mit einer Lösung von 34 mg (0.21 mmol) 1,1,3,3-Tetramethoxypropan in 1.0 ml Methanol versetzt. Im Anschluß läßt man noch 45 min bei Rückfluss rühren. Das Produkt wird direkt durch präparative ΗPLC aufgereinigt. Man erhält 37 mg (65% d. Th.) der gewünschten Verbindung.
1H-NMR (400 MHz, DMSO-d6): δ = 1.35 (t, J= 7.1 Hz, 3H), 4.32 (s, 2H), 4.38 (q, J= 7.1 Hz, 2H), 7.06-7.20 (m, 3H), 7.22-7.30 (m, 2H), 8.51 (dd, J= 3.9, 1.7 Hz, IH), 9.39 (dd, J= 7.3, 1.7 Hz, IH).
13C-NMR (125 MHz, DMSO-d6): δ = 14.1, 24.9 (d, Vc1F = 3.5 Hz), 60.7, 1 11.4, 115.0 (d, 2JC,F = 21.6 Hz), 122.8, 124.2 (d, VC,F= 3.5 Hz), 126.2 (d, VC>F= 15.5 Hz), 128.2 (d, VC,F= 8.1 Hz), 130.7, 131.1 (d, Vc1F= 4.4 Hz), 132.4, 137.6, 148.5, 158.5, 160.1 (d, 'JC,F= 234 Hz).
HRMS: ber. für C16H14FN3O2 299.1070; gef. 299.1067.
LC/MS (Methode 1): R, = 2.30 min; MS (ESIpos): m/z = 300 [M+H]+.
Beispiel IIA
l-(2-Chloφyridin-3-yl)-2-(2-fluorphenyl)ethanon
Eine Lösung von 5.00 g (29.7 mmol) (2-Fluorphenyl)-essigsäuremethylester in 80 ml THF wird in eine auf -78°C gekühlte 1 N Lösung von LiHMDS in Hexan (35.7 ml, 35.7 mmol) getropft. Man läßt 1 h bei -78°C nachrühren, gibt dann 6.28 g (35.7 mmol) 2-Chlornicotinsäurechlorid hinzu und rührt eine weitere Stunde nach. Es wird auf RT erwärmt und gesättigte Ammoniumchlorid-Lösung zugesetzt. Man verdünnt mit Wasser und extrahiert mit Diethylether. Die organische Phase wird über Natriumsulfat getrocknet und im Vakuum eingeengt. Der Rückstand wird in 24 ml DMSO gelöst und mit 0.95 g (52.7 mmol) Wasser sowie 1.70 g (29.0 mmol) Natriumchlorid versetzt. In 8 Portionen wird die Lösung jeweils 10 min lang auf 1500C erhitzt. Dann wird mit Wasser verdünnt und mit tert.-Butylmethylether extrahiert. Man trocknet die organische Phase über Natriumsulfat und engt im Vakuum ein. Der Rückstand wird durch Chromatographie an Kieselgel (Eluens: Cyclohexan/Ethylacetat 2: 1) gereinigt. Man erhält 3.00 g (46% d. Th.) der gewünschten Verbin- düng.
1H-NMR (400 MHz, DMSO-d6): δ = 4.42 (s, 2H), 7.16-7.22 (m, 2H), 7.32-7.39 (m, 2H), 7.59 (dd, J= 7.6, 4.7 Hz, IH), 8.26 (dd, J= 7.6, 2.0 Hz, IH), 8.55 (dd, J= 4.7, 2.2 Hz, IH).
13C-NMR (125 MHz, DMSO-d6): δ = 42.4 (d, VC,F = 1.2 Hz), 115.0 (d, 2JC,F = 21.4 Hz), 121.0 (d, 2JCF = 16.3 Hz), 123.1, 124.3 (d, 4JC,F = 3.4 Hz), 129.3 (d, VC,F = 8.1 Hz), 132.3 (d, VC,F = 4.5 Hz), 134.3, 138.3, 145.8, 151.4, 160.7 (d, VC,F= 245 Hz), 197.7.
HRMS: ber. für C13H9ClFNO + [H+] 250.0430; gef. 250.0427.
HPLC (Methode 6): R, = 4.24 min.
Beispiel 12A
3-(2-Fluorbenzyl)-lH-pyrazolo[3,4-6]pyridin
Man erhitzt eine Lösung von 2.00 g (8.01 mmol) l-(2-Chloφyridin-3-yl)-2-(2-fluorphenyl)ethanon aus Beispiel I IA und 560 mg (11.2 mmol) Ηydrazinhydrat in 6 ml 1-Butanol in der Mikrowelle für 10 min auf 2000C. Man verdünnt danach mit ter/.-Butylmethylether, wäscht mit gesättigter Natriumhydrogencarbonat-Lösung und trocknet über Natriumsulfat. Man engt die Lösung im Vakuum ein. Der kristalline Rückstand wird mit etwas tert.-Butylmethylether verrührt und abge- saugt. Die Mutterlauge wird eingeengt und durch Chromatographie an Kieselgel (Eluens: Cyclo- hexan/Ethylacetat 2: 1) gereinigt. Man erhält insgesamt 1.40 g (77% d. Th.) des gewünschten Produkts als gelbe Kristalle.
1H-NMR (400 MHz, DMSOd6): δ = 4.31 (s, 2H), 7.10-7.20 (m, 3H), 7.25-7.31 (m, IH), 7.35 (dt, 7= 7.8, 1.5 Hz, IH), 8.04 (d, J= 7.8 Hz, 1H); 8.48 (dd, 7= 4.6, 1.5 Hz, IH), 13.36 (s, IH).
13C-NMR (125 MHz, DMSO-d6): δ = 26.3 (d, 37C,F = 3.2 Hz), 113.1, 115.2 (d, 37C,F = 21.6 Hz), 116.1, 124.3 (d, 47C,F = 3.5 Hz), 125.7 (d, 27C,F= 15.6 Hz), 128.3 (d, 37C)F = 8.1 Hz), 129.1, 131.2 (d, Vc1F = 4.4 Hz), 142.5, 148.6, 152.3, 160.2 (d, '7C>F= 244 Hz).
HRMS: ber. für C13H10FN3 227.0859; gef. 227.0855.
HPLC (Methode 6): R, = 3.61 min.
Beispiel 13A
2-Chlor-3-[(2-fiuorphenyl)ethinyl]pyrazin
- -
Man fugt 49 mg (0.26 mmol) Kupfer(I)iodid und 179 mg (0.26 mmol) Bis-(tπphenylphosphin)- palladium(II)chloπd zu einer Lösung von 760 mg (5.10 mmol) 2,3-Dichlorpyrazin in 27 ml Tn- ethylamin und kühlt auf 00C. Dann tropft man 919 mg (7.65 mmol) 2-Fluorphenylacetylen hinzu und erhitzt 3 h auf 800C. Danach wird filtriert und eingeengt. Der Rückstand wird durch präpara- tive HPLC aufgereinigt. Man erhält 717 mg (60% d. Th.) der Titelverbindung als hellbeige Kristalle.
1H-NMR (500 MHz, DMSOd6): δ = 7.35 (t, J = 7.6 Hz, IH), 7.40-7.46 (m, IH), 7.59-7.65 (m, IH), 7.76 (dt, J = 7.6, 1.5 Hz, IH), 8.56 (d, J= 2.2 Hz, IH), 8.74 (d, J= 2.2 Hz, IH).
13 C/- -NMR (125 MHz, DMSO-d6): δ = 89.1 (d, VC,F = 3.2 Hz), 89.4, 108.7 (d, VClF= 15.3 Hz), 116.0 (d, 2Jc1F = 20.1 Hz), 125.1 (d, 4JC,F = 3.6 Hz), 132.9 (d, 3JC,F = 8.3 Hz), 133.8, 137.5, 2 x 143.5, 149.4, 162.2 (d, 1Jc1F= 252 Hz).
HRMS: ber. für C12H6ClFN2 + [H+] 233.0277; gef. 233.0288.
LC/MS (Methode 2): R, = 2 32 mm; MS (ESIpos): m/z = 233 [M+K]+.
Beispiel 14A
3-(2-Fluorbenzyl)-lH-pyrazolo[3,4-ό]pyrazin
Eine Lösung von 550 mg (2.36 mmol) 2-Chlor-3-[(2-fluorphenyl)ethinyl]pyrazin aus Beispiel 13A und 592 mg (11.8 mmol) Ηydrazinhydrat in 12 ml n-Butanol wird in der Mikrowelle 30 mm lang auf 1400C erhitzt. Danach wird die Lösung im Vakuum eingeengt und der Rückstand durch präpa- rative ΗPLC aufgereinigt. Man erhält 150 mg (26% d. Th.) der gewünschten Verbindung.
1H-NMR (500 MHz, DMSOd6): δ = 4.36 (s, 2H), 7.09 (t, J= 7.4 Hz, IH), 7.14-7.19 (m, IH), 7.22-7.28 (m, IH), 7.33 (t, J= 7.6 Hz, IH), 8.48 (s, 2H).
13C-NMR (125 MHz, DMSO-d6): δ = 25.0 (d, 3JC,F = 3.2 Hz), 115.1 (d, 2JC,F = 21.7 Hz), 124.3 (d, 4Jc1F = 3.5 Hz), 126.0 (d, 2JC,F = 15.5 Hz), 128.3 (d, 3JC,F = 7.9 Hz), 131.4 (d, 3Jc1F = 4.4 Hz), 131.6, 139.2, 142.1, 142.3, 146.3, 160.3 (d, 1Jc1F= 244 Hz).
HRMS: ber. für C12H9FN4 228.0811; gef. 228.0814.
LC/MS (Methode 1): R, = 1.87 min; MS (ESIpos): m/z = 229 [M+H]+.
Beispiel 15A
2-[3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-ό]pyridin-l-yl]-5-nitropyrimidm-4,6-diamin
Man erhitzt eine Lösung von 600 mg (2.64 mmol) 3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-ό]pyridin aus Beispiel 4A, 501 mg (2.64 mmol) 2-Chlor-5-nitropyrimidin-4,6-diamin [Bitterli et al., HeIv. Chim. Acta 1951, 34, 835], 48.4 mg (0.053 mmol) Tris-(dibenzylidenaceton)-dipalladium, 75.5 mg (0.158 mmol) Dicyclohexyl-(2',4',6'-triisopropylbiphenyl-2-yl)-phosphm (XPΗOS) sowie 1.20 g (3.70 mmol) Cäsiumcarbonat in einer entgasten Mischung aus 10 ml Toluol und 10 ml DMF 4 h lang auf 900C. Nach Abkühlen auf Raumtemperatur saugt man ab und wäscht mit TΗF nach. Der Feststoff wird mit 100 ml Wasser verrührt und erneut abgesaugt. Man erhält 572 mg (57% d. Th.) der gewünschten Verbindung.
1H-NMR (400 MHz, DMSO-d6): δ = 4.43 (s, 2H), 7.11 (ddd, J= 7.6, 7.6, 0.8 Hz, IH), 7.18 (ddd, J= 9.7, 8.5, 0.8 Hz, IH), 7.25-7.31 (m, IH), 7.36 (ddd, J= 7.7, 7.6, 1.3 Hz, IH), 7.56 (dd, J= 8.6, 4.4 Hz, IH), 8.66 (dd, J= 4.4, 1.2 Hz, IH), 8.76 (s, 2H), 8.94 (s, 2H), 9.32 (dd, J= 8.6, 1.2 Hz, IH).
13C-NMR (125 MHz, DMSO-d6): δ = 24.4 (d, 3JC>F = 3.7 Hz), 109.8, 115.3 (d, 2JC,F = 21.7 Hz), 122.8, 124.4 (d, VC,F = 3.5 Hz), 124.8 (d, 2JC,F= 15.5 Hz), 125.2, 128.6 (d, 3JC,F= 8.1 Hz), 131.2 (d, 3Jc,F= 4.2 Hz), 133.5, 142.0, 147.1, 148.7, 155.6, 160.0, 160.2 (d, 1Jc1P= 244 Hz).
HRMS: ber. für C17H13FN8O2 380.1146; gef. 380.1138.
LC/MS (Methode 1): Rt = 2.09 min; MS (ESIpos): m/z = 381 [M+H]+.
Beispiel 16A
3-(2-Fluorbenzyl)imidazo[l,5-α]pyridin-l-carbohydrazid
Man legt 150 mg (0.50 mmol) Ethyl 3-(2-fluorbenzyl)imidazo[l,5-α]pyridin-l-carboxylat aus Bei- spiel 7A in einer Mischung aus 1 ml Methanol und 0.5 ml THF vor und setzt 503 mg (10.1 mmol) Hydrazinhydrat hinzu. Man erhitzt zunächst 4 h auf 65°C, dann 10 h auf 900C. Danach wird zur Trockene eingeengt. Das erhaltene Rohprodukt (157 mg, quantitativ) wird ohne weitere Aufreinigung umgesetzt.
1H-NMR (400 MHz, DMSO-d6): δ = 4.33 (br. s, 2H), 4.48 (s, 2H), 6.83-6.88 (m, IH), 7.05-7.14 (m, 3H), 7.17-7.23 (m, IH), 7.27-7.34 (m, IH), 8.10 (d, J= 9.3 Hz, IH), 8.27 (d, J= 7.1 Hz, IH), 8.94 (br. s, IH).
LC/MS (Methode 2): R, = 1.46 min.; MS (ESIpos): m/z = 285 [M+H]+.
Beispiel 17A
3-(2-Fluorbenzyl)imidazo[l,5-α]pyridin-l-carboxamid
- -
560 mg (1.88 mmol) Ethyl 3-(2-fluorbenzyl)imidazo[l,5-α]pyridin-l-carboxylat aus Beispiel 7 A werden in 55 ml 33%-iger wässriger Ammoniak-Lösung in der Mikrowelle für 3 h auf 1300C erhitzt. Man verdünnt mit Wasser, setzt etwas Methanol hinzu und extrahiert mit Ethylacetat. Die organische Phase wird über Natriumsulfat getrocknet und eingeengt. Man erhält 229 mg (87.5% Reinheit, 40% d. Th.) der gewünschten Verbindung als grünliche Kristalle, die ohne weitere Aufreinigung umgesetzt werden.
1H-NMR (400 MHz, DMSO-d6): δ = 4.49 (s, 2H), 6.86 (ddd, J= 7.2, 6.1, 0.5 Hz, IH), 7.04-7.16 (m, 4H), 7.18-7.23 (m, IH), 7.25-7.34 (m, 2H), 8.12 (d, J= 9.2 Hz, IH), 8.29 (d, J = 7.2 Hz, IH).
LC/MS (Methode 7): R, = 2.28 min.; MS (ESIpos): m/z = 270 [M+H]+.
Beispiel 18A
3-(2-Fluorbenzyl)imidazo[l,5-α]pyridin-l-carbonitril
Man löst 269 mg (1.00 mmol) 3-(2-Fluorbenzyl)imidazo[l,5-α]pyridm-l-carboxamid aus Beispiel 17A in 2.5 ml THF und versetzt mit 200 mg (2.50 mmol) Pyridin sowie 525 mg (2.50 mmol) Tri- fluoressigsäureanhydrid. Man läßt 15 h bei RT rühren, gibt dann Wasser hinzu und extrahiert mit Ethylacetat. Die organische Phase wird mit gesättigter Natriumhydrogencarbonat-Lösung und 1 N Salzsäure gewaschen, über Natriumsulfat getrocknet und eingeengt. Das gewonnene Rohprodukt (188 mg, 61% Reinheit, 45% d. Th.) wird ohne weitere Aufreinigung umgesetzt.
LC/MS (Methode 7): R, = 2.93 min.; MS (ESIpos): m/z = 252 [M+H]+.
Ausführungsbeispiele:
Beispiel 1
6-[3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-ό]pyridin4-yl]-l,3,5-1riazin-2,4-diamin
Eine Lösung von 100 mg (0.44 mmol) 3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-ό]pyridin aus Beispiel 4A, 64 mg (0.44 mmol) 6-Chlor-l,3,5-triazin-2,4-diamin, 8.1 mg (0.009 mmol) Tris-(dibenzyliden- aceton)-dipalladium, 13 mg (0.026 mmol) Dicyclohexyl-(2',4',6'-triisopropylbiphenyl-2-yl)-phos- phin (XPΗOS) und 201 mg (0.62 mmol) Cäsiumcarbonat in 3 ml entgastem Toluol wird 20 h auf 900C erhitzt. Man verdünnt danach mit Ethylacetat und Methanol, filtriert und engt im Vakuum ein. Man kristallisiert den Rückstand aus Methanol und erhält 42 mg (28% d. Th.) der Titelverbindung als hellgelbe Kristalle.
1H-NMR (400 MHz, DMSO-d6): δ = 4.42 (s, 2H), 6.93 (br. s, 2H), 7.12 (dt, J = 7.3, 1.0 Hz, IH), 7.15-7.32 (m, 4H), 7.37 (dt, J= 7.8, 1.5 Hz, IH), 7.55 (dd, J= 8.6, 4.4 Hz, IH), 8.64 (dd, J = 4.4, 1.2 Hz, IH), 9.16 (dd, J= 8.6, 1.2 Hz, IH).
13C-NMR (125 MHz, DMSO-d6): δ = 24.4 (d, 3JC,F = 3.3 Hz), 115.2 (d, 2JC,F = 21.6 Hz), 122.4, 124.3 (d, 4Jc,F = 3.3 Hz), 124.5, 124.9 (d, 2JC,F= 15.5 Hz), 128.5 (d, 3JC,F = 8.0 Hz), 131.3 (d, 3JC,F = 4.2 Hz), 133.1, 141.6, 146.6, 147.7, 160.2 (d, 1JcF = 244 Hz), 163.1, 167.5.
HRMS: ber. für C16H13FN8 + [H+] 337.1320; gef. 337.1307.
LC/MS (Methode 3): Rt = 2.87 min; MS (ESIpos): m/z = 337 [M+H]+.
Beispiel 2
6- [3 -(2-Fluorbenzyl)- 1 H-indazol- 1 -yl] - 1 , 3 ,5 -triazin-2 ,4-diamin
Unter Argon werden 130 mg (0.58 mmol) 3-(2-Fluorbenzyl)-lH-indazol aus Beispiel 5A, 84 mg (0.58 mmol) 6-Chlor-l,3,5-triazin-2,4-diamin, 11 mg (0.011 mmol) Tris-(dibenzylidenaceton)-di- palladium, 16 mg (0.034 mmol) Dicyclohexyl-(2',4',6'-triisopropylbiphenyl-2-yl)-phosphin (XPΗOS) sowie 262 mg (0.80 mmol) Cäsiumcarbonat in 4 ml entgastem Toluol vorgelegt und 20 h auf 900C erhitzt. Man verdünnt danach mit Ethylacetat und Methanol, filtriert und engt im Vakuum ein. Man reinigt den Rückstand durch präparative ΗPLC und erhält 36 mg (19% d. Th.) der Titelverbindung. Daneben werden 65 mg der Ausgangsverbindung wiedergewonnen.
1H-NMR (500 MHz, DMSO-d6): δ = 4.37 (s, 2H), 6.86 (br. s, 2H), 7.12-7.22 (m, 4H), 7.25-7.32 (m, 2H), 7.36 (t, J = 7.7 Hz, IH), 7.50 (t, J = 1.1 Hz, IH), 7.68 (d, J= 8.0 Hz, IH), 8.86 (d, J = 8.5 Hz, IH).
13C-NMR (125 MHz, DMSOd6): δ = 25.9 (d, 3JC,F = 3.2 Hz), 115.3 (d, 2JC,F = 21.6 Hz), 116.6, 120.0, 122.6, 124.5 (d, 4JC,F = 3.4 Hz), 124.6, 125.0 (d, 2JC,F = 15.6 Hz), 127.7, 128.7 (d, 3JC,F = 8.1 Hz), 131.2 (d, 3JC,F= 4.3 Hz), 139.9, 147.2, 160.2 (d, 1JcP= 244 Hz), 163.4, 167.5.
HRMS: ber. für C17H14FN7 335.1295; gef. 335.1295.
LC/MS (Methode 2): R, = 1.84 min; MS (ESIpos): m/z = 336 [M+H]+.
Beispiel 3
6-[3-(2-Fluorbenzyl)imidazo[l,5-α]pyridin-l-yl]-l,3,5-triazin-2,4-diamin
- -
Man legt 437 mg (2.51 mmol) Biguanidin-Dihydrochlorid in 10 ml Methanol vor und gibt 1.0 ml (5.53 mmol) einer 30%-igen methanolischen Natriummethanolat-Lösung hinzu. Die Mischung wird für 30 min auf 500C erwärmt. Ausgefallenes Natriumchlorid wird abfϊltriert und mit 3 ml Methanol nachgewaschen. Zum Filtrat werden dann 500 mg (1.68 mmol) Ethyl 3-(2-fluorbenzyl)- imidazo[l,5-α]pyridin-l-carboxylat aus Beispiel 7A zugesetzt, und man erhitzt über Nacht auf Rückfluss. Zur Aufarbeitung verdünnt man mit Dichlormethan und wäscht mit Natriumcarbonat- Lösung, wobei sich ein Niederschlag bildet. Die organische Phase wird abgetrennt und mit dem Niederschlag vereinigt. Man engt im Vakuum ein und verrührt den Rückstand mit Methanol. Die so erhaltenen Kristalle werden aus DMF umkristallisiert, wodurch man 55 mg (10% d. Th.) des gewünschten Produktes erhält. Aus der Mutterlauge werden weitere 128 mg (23% d. Th.) an etwas verunreinigtem Material erhalten.
1H-NMR (400 MHz, DMSO-d6): δ = 4.46 (s, 2H), 6.56 (br. s, 4H), 6.86 (t, J = 6.5 Hz, IH), 7.05 (dd, J = 8.8, 6.6 Hz, IH), 7.12-7.25 (m, 3H), 7.28-7.37 (m, IH), 8.32 (d, J= 7.1 Hz, IH), 8.69 (d, J= 9.0 Hz, IH).
13C-NMR (125 MHz, DMSOd6): δ = 25.6 (d, 3JC,F = 3.1 Hz), 113.2, 115.3 (d, 2JC,F = 21.4 Hz), 121.1, 121.8, 122.5, 123.8 (d, 2JC,F = 15.7 Hz), 124.5 (d, 4JC,F = 3.4 Hz), 126.5, 128.7 (d, 3JC,F = 8.0 Hz), 131.0 (d, 3Jc,F= 4.3 Hz), 132.2, 136.5, 160.4 (d, 'JC,F= 244 Hz), 167.0, 167.9.
HRMS: ber. für C17H14FN7 + [H+] 336.1368; gef. 336.1363.
LC/MS (Methode 4): R, = 1.24 min; MS (ESIpos): m/z = 336 [M+H]+.
Beispiel 4
6-[8-(2-Fluorbenzyl)imidazo[l,5-α]pyrimidin-6-yl]-l,3,5-triazin-2,4-diamin
- -
Man legt 126 mg (0.70 mmol) Biguanidin-Dihydrochlorid in 3 ml Methanol vor und gibt 0.3 ml (1.66 mmol) einer 30%-igen methanolischen Natriummethanolat-Lösung hinzu. Die Mischung wird für 30 min auf 500C erwärmt. Man läßt danach auf RT abkühlen, setzt eine Lösung von 124 mg (0.41 mmol) Ethyl 8-(2-fluorbenzyl)imidazo[l,5-α]pyrimidin-6-carboxylat aus Beispiel 10A in 1.0 ml Methanol hinzu und erhitzt 3 h auf Rückfluss. Nach dem Abkühlen wird mit Wasser verrührt und abgesaugt. Man erhält 88 mg (63% d. Th.) des gewünschten Produkts als gelbe Kristalle.
1H-NMR (500 MHz, DMSOd6): δ = 4.31 (s, 2H), 6.78 (br. s, 2H), 6.99 (dd, J= 7.4, 3.8 Hz, IH), 7.05 (br. s, 2H), 7.10 (t, J= 7.4 Hz, IH), 7.15 (dd, J= 10.0, 8.4 Hz, IH), 7.22-7.28 (m, IH), 7.29 (t, J= 7.7 Hz, IH), 8.35-8.38 (m, IH), 10.21 (da, J= 7 A, 1.0 Hz, IH).
13C-NMR (125 MHz, DMSOd6): δ = 25.0 (d, 3JC,F = 3.4 Hz), 109.8, 115.1 (d, 2JC,F = 21.7 Hz), 124.3 (d, 4Jc1F= 3.4 Hz), 126.8 (d, 2JC,F= 15.5 Hz), 128.1 (d, 3JC,F= 8.0 Hz), 129.3, 130.1, 131.3 (d, 3JCF= 4.5 Hz), 134.2, 136.8, 146.8, 160.2 (d, 'JC,F= 244 Hz), 163.7, 166.8.
HRMS: ber. für C16H13FN8 336.1247; gef. 336.1236.
LC/MS (Methode 4): Rt = 1.51 min; MS (ESIpos): m/z = 337 [M+H]+.
Beispiel 5
6-[3-(2-Fluorbenzyl)-lH-pyrazolo[3,4-ό]pyridin-l-yl]-l,3,5-triazin-2,4-diamin
- -
Eine Lösung von 150 mg (0.66 mmol) 3-(2-Fluorbenzyl)-lH-pyrazolo[3,4-6]pyridin aus Beispiel 12A, 96 mg (0.66 mmol) 6-Chlor-l,3,5-triazin-2,4-diamin, 12 mg (0.013 mmol) Tris-(dibenzyli- denaceton)-dipalladium, 19 mg (0.040 mmol) Dicyclohexyl-(2',4',6'-triisopropylbiphenyl-2-yl)- phosphin (XPΗOS) sowie 300 mg (0.92 mmol) Cäsiumcarbonat in 4 ml entgastem Toluol wird 20 h lang auf 900C erhitzt. Danach verdünnt man mit Wasser und etwas Methanol und extrahiert mit Ethylacetat. Die organische Phase wird im Vakuum eingeengt und der Rückstand mit Methanol verrührt und abgesaugt. Man erhält so 62 mg (28% d. Th.) der gewünschten Verbindung als beigefarbene Kristalle.
1H-NMR (400 MHz, DMSOd6): δ = 4.38 (s, 2H), 6.91 (br. s, 2H), 7.11 (br. s, 2H), 7.14-7.22 (m, 2H), 7.28-7.34 (m, 2H), 7.39 (dt, J= 7.8, 1.5 Hz, IH), 8.13 (dd, J= 8.1, 1.5 Hz, IH), 8.62 (dd, J = 4.4, 1.5 Hz, IH).
13C-NMR (125 MHz, DMSO-d6): δ = 26.3 (d, 3JC,F = 3.1 Hz), 115.3 (d, 2JC,F = 21.5 Hz), 116.1, 118.2, 124.5 (d, 4JC,F = 3.4 Hz), 124.8 (d, 2JC,F = 15.6 Hz), 128.8 (d, 3JC,F= 8.5 Hz), 129.7, 131.3 (d, 3Jc1F= 4.3 Hz), 145.1, 149.4, 151.0, 160.3 (d, 'JC,F = 244 Hz), 163.0, 167.7.
HRMS: ber. für C16H13FN8 + [H+] 337.1320; gef. 337.1328.
LC/MS (Methode 2): R, = 1.45 min; MS (ESIpos): m/z = 337 [M+H]+.
Beispiel 6
6-[3-(2-Fluorbenzyl)-lH-pyrazolo[3,4-£]pyrazin-l-yl]-l,3,5-triazin-2,4-diamin
- o -
Eine Lösung von 144 mg (0.63 mmol) 3-(2-Fluorbenzyl)-lH-pyrazolo[3,4-6]pyrazin aus Beispiel 14A, 92 mg (0.63 mmol) 6-Chlor-l,3,5-triazm-2,4-diamin, 12 mg (0.013 mmol) Tris-(dibenzyli- denaceton)-dipalladium, 18 mg (0.038 mmol) Dicyclohexyl-(2',4l,6'-triisopropylbiphenyl-2-yl)- phosphin (XPΗOS) sowie 287 mg (0.88 mmol) Cäsiumcarbonat in 4 ml entgastem Toluol wird 20 h auf 900C erhitzt. Danach verdünnt man mit Ethylacetat und etwas Methanol und saugt ab. Der Feststoff wird erneut mit Methanol verrührt und abermals abgesaugt. Die Filtrate werden dann vereinigt und eingeengt und der Rückstand durch präparative ΗPLC aufgereinigt. Die gewonnene Produktfraktion wird eingeengt, nochmals mit Methanol verrührt und abgesaugt. Man erhält so 13 mg (6% d. Th.) des gewünschten Produkts.
1H-NMR (500 MHz, DMSO-d6): δ = 4.44 (s, 2H), 7.02 (br. s, 2H), 7.14 (t, J= 7.4 Hz, IH), 7.17- 7.22 (m, IH), 7.24 (br. s, 2H), 7.27-7.33 (m, IH), 7.39 (t, J= 7.5 Hz, IH), 8.74 (s, 2H).
13C-NMR (125 MHz, DMSO-d6): δ = 25.0 (d, 3JC,F = 3.4 Hz), 115.4 (d, 2JC,F = 21.4 Hz), 124.5 (d, 4JCF = 3.5 Hz), 124.6 (d, 2JC,F = 15.4 Hz), 128.9 (d, 3JC,F = 8.0 Hz), 131.5 (d, 3JC,F = 4.1 Hz), 133.8, 141.9, 144.0, 144.4, 145.8, 160.3 (d, 1Jc1P= 245 Hz), 162.8, 167.8.
HRMS: ber. für C15H12FN9 337.1200; gef. 337.1198.
LC/MS (Methode 1): R, = 1.66 min; MS (ESIpos): m/z = 338 [M+H]+.
Beispiel 7
2-[8-(2-Fluorbenzyl)imidazo[l,5-α]pyrimidin-6-yl]pyrimidin-4,6-diamin
- -
Man legt 147 mg (0.85 mmol) 1,3-Propandiimidamid-Dihydrochlorid in 2 ml Methanol vor und gibt 0.37 ml (2.00 mmol) einer 30%-igen methanolischen Natriummethanolat-Lösung hinzu. Die Mischung wird 30 min auf 500C erwärmt. Man läßt danach auf RT abkühlen, setzt eine Lösung von 150 mg (0.50 mmol) Ethyl 8-(2-fluorbenzyl)imidazo[l,5-α]pyrimidin-6-carboxylat aus Beispiel 10A in 2.0 ml Methanol hinzu und erhitzt 3 h auf Rückfluss. Nach dem Abkühlen wird mit Wasser verdünnt und mit Ethylacetat ausgeschüttelt. Man trocknet die organische Phase über Natriumsulfat und entfernt das Lösungsmittel im Vakuum. Man reinigt den Rückstand durch prä- parative HPLC und erhält 36 mg (21% d. Th.) des gewünschten Produkts als gelbe Kristalle.
1H-NMR (400 MHz, DMSOd6): δ = 4.30 (s, 2H), 5.37 (s, IH), 6.32 (br. s, 4H), 6.89 (dd, J= 7.5, 3.8 Hz, IH), 7.06-7.11 (m, IH), 7.12-7.17 (m, IH), 7.20-7.30 (m, 2H), 8.27 (dd, J= 3.8, 1.7 Hz, IH), 10.21 (dd, J= 7.5, 1.7 Hz, IH).
LC/MS (Methode 2): Rt = 1.38 min; MS (ESIpos): m/z = 336 [M+H]+.
Beispiel 8
2-[3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-ό]pyridin-l-yl]pyrimidm-4,5,6-triamin
690 mg (1.42 mmol) 2-[3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-ό]pyridin-l-yl]-5-nitropyrimidin-4,6- diamin aus Beispiel 15A werden in 120 ml Pyridin gelöst. Man setzt 270 mg 10%-iges Palladium auf Aktivkohle hinzu und hydriert 15 h lang bei 3.5 bar Wasserstoffdruck. Danach wird vom Katalysator abfiltriert und mit Ethanol nachgewaschen. Man engt bis zur Trockene ein, verrührt den Rückstand bei 500C mit Ethanol und saugt ab. Es werden 378 mg (76% d. Th.) des gewünschten Produkts erhalten. Weitere 92 mg (18% d. Th.) der Zielverbindung lassen sich aus dem Filtrat durch Reinigung mittels präparativer ΗPLC gewinnen.
1H-NMR (400 MHz, DMSO-d6): δ = 3.75 (br. s, 2H), 4.41 (s, 2H), 6.05 (s, 4H), 7.07-7.12 (m, IH), 7.14-7.20 (m, IH), 7.23-7.29 (m, IH), 7.34 (ddd, J = 7.7, 7.6, 1.2 Hz, IH), 7.46 (dd, J = 8.6, 4.4 Hz, IH), 8.57 (dd, J= 4.4, 1.2 Hz, IH), 9.06 (dd, J= 8.6, 1.2 Hz, IH).
13C-NMR (125 MHz, DMSO-d6): δ = 24.3 (d, 3JC,F = 3.7 Hz), 103.4, 115.2 (d, 2JCjF = 21.5 Hz), 121.7, 123.7, 124.3 (d, 4JC,F = 3.5 Hz), 125.6 (d, 2JC,F = 15.5 Hz), 128.4 (d, 3JC>F = 7.9 Hz), 131.2 (d, 3Jc,F= 4.4 Hz), 132.2, 140.8, 144.8, 145.7, 148.4, 152.7, 160.2 (d, 1JcF= 244 Hz).
HRMS: ber. für C17H15FN8 350.1404; gef. 350.1395.
LC/MS (Methode 1): Rt = 1.55 min; MS (ESIpos): m/z = 351 [M+H]+.
Beispiel 9
Methyl {4,6-diamino-2-[3-(2-fluorbenzyl)-lH-pyrazolo[4,3-&]pyridin-l-yl]pyrimidin-5-yl}- carbamat
200 mg (0.57 mmol) 2-[3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-ό]pyridm-l-yl]pyrimidin-4,5,6-triamin aus Beispiel 8 werden in 10 ml Pyridin gelöst. Man kühlt auf 00C und gibt 81 mg (0.86 mmol) Chlorameisensäuremethylester hinzu. Die Reaktionsmischung wird über Nacht bei RT gerührt,
dann im Vakuum eingeengt, mit Ethylacetat versetzt und zweimal mit Wasser und mit gesättigter Natriumchlorid-Lösung gewaschen. Die organische Phase wird über Natriumsulfat getrocknet und im Vakuum eingeengt. Man reinigt den Rückstand durch präparative HPLC und erhält 125 mg (54% d. Th.) der Titelverbindung als hellbeigen Feststoff.
1H-NMR (400 MHz, DMSOd6): δ = 3.54 und 3.61 (2 br. s, zus. 3H), 4.42 (s, 2H), 6.40 (br. s, 4H), 7.07-7.12 (m, IH), 7.15-7.21 (m, IH), 7.24-7.30 (m, IH), 7.32-7.37 (m, IH), 7.50 (dd, J = 8.6, 4.4 Hz, IH), 7.60 und 7.90 (2 br. s, zus. IH), 8.59 (dd, J = 4.4, 0.8 Hz, IH), 9.16 (dd, J= 8.6, 0.8 Hz, IH).
13C-NMR (125 MHz, DMSO-d6): δ = 24.3 (d, 3JC,F = 3.5 Hz), 51.7, 92.0, 115.2 (d, 2JC,F = 21.7 Hz), 122.1, 124.3, 124.4 (d, 4Jc1F = 3.7 Hz), 125.4 (d, 2JC,F = 15.5 Hz), 128.4 (d, 3Jc1F = 8.1 Hz), 131.2 (d, 3Jc1F = 4.2 Hz), 132.8, 141.2, 146.0, 146.1, 154.1, 155.3, 160.2 (d, 'Jc,F = 244 Hz), 161.0.
HRMS: ber. für C19H17FN8O2 408.1459; gef. 408.1459.
LC/MS (Methode 2): Rt = 1.52 min; MS (ESIpos): m/z = 409 [M+H]+.
Beispiel 10
5-[3-(2-Fluorbenzyl)imidazo[l,5-α]pyridin-l-yl]-l,3,4-oxadiazol-2(3H)-on
155 mg (0.55 mmol) 3-(2-Fluorbenzyl)imidazo[l,5-α]pyridin-l-carbohydrazid aus Beispiel 16A werden in 3 ml Methanol gelöst. Man setzt 106 mg (0.65 mmol) N,N'-Carbonyldiimidazol hinzu und erhitzt 2 h auf Rückfluss. Man reinigt danach direkt durch präparative ΗPLC und erhält 98 mg (58% d. Th.) der gewünschten Verbindung als hellbeigen Feststoff.
1H-NMR (400 MHz, DMSOd6): δ = 4.50 (s, 2H), 6.94 (t, J= 6.7 Hz, IH), 7.12-7.26 (m, 4H), 7.29-7.36 (m, IH), 7.86 (d, J= 9.1 Hz, IH), 8.41 (d, J= 7.2 Hz, IH), 12.30 (br. s, IH).
LC/MS (Methode 1): Rt = 1.92 min; MS (ESIpos): m/z = 311 [M+H]+.
- -
Beispiel 11
3-(2-Fluorbenzyl)-l-(lH-tetrazol-5-yl)imidazo[l,5-α]pyridin
Man erhitzt eine Lösung von 188 mg (0.75 mmol) 3-(2-Fluorbenzyl)imidazo[l,5-α]pyridin-l- carbonitril aus Beispiel 18A, 18.6 mg (0.075 mmol) Dibutylzinnoxid und 172 mg (1.50 mmol) Tri- methylsilylazid in 5 ml Toluol für 20 h unter Rückfluss. Nach dem Abkühlen setzt man 5 ml Ethanol hinzu und rührt 15 h bei RT nach. Man engt danach ein, versetzt mit Wasser und extrahiert mit Ethylacetat. Die organische Phase wird im Vakuum eingeengt und der Rückstand durch präparative ΗPLC gereinigt. Das Produkt wird in Ethylacetat aufgenommen und mit Aktivkohle geklärt. Der nach dem Einengen erhaltene Feststoff wird aus Dichlormethan kristallisiert. Man erhält 40 mg (18% d. Th.) der Titelverbindung als schwach-rötliche Kristalle.
1H-NMR (400 MHz, DMSO-d6): δ = 4.58 (s, 2H), 6.98 (ddd, J= 7.1, 6.4, 0.7 Hz, IH), 7.11-7.25 (m, 4H), 7.29-7.36 (m, IH), 8.20 (d, J= 9.2 Hz, IH), 8.40 (d, J= 7.1, IH), 16.6 (br. s, IH).
LC/MS (Methode 7): Rt = 2.88 min; MS (ESIpos): m/z = 295 [M+H]+.
- -
B. Bewertung der pharmakologischen Wirksamkeit
Die pharmakologische Wirkung der erfindungsgemäßen Verbindungen kann in folgenden Assays gezeigt werden:
B-I. Gefäßrelaxierende Wirkung in vitro
Kaninchen werden durch Nackenschlag betäubt und entblutet. Die Aorta wird entnommen, von anhaftendem Gewebe befreit, in 1.5 mm breite Ringe geteilt und einzeln unter einer Vorspannung in 5 ml-Organbäder mit 370C warmer, Carbogen-begaster Krebs-Henseleit-Lösung folgender Zusammensetzung gebracht (jeweils mM): NaCl: 119; KCl: 4.8; CaCl2 x 2 H2O: 1; MgSO4 x 7 H2O: 1.4; KH2PO4: 1.2; NaHCO3: 25; Glucose: 10. Die Kontraktionskraft wird mit Statham UC2-Zellen erfasst, verstärkt und über A/D-Wandler (DAS-1802 HC, Keithley Instruments München) digitalisiert sowie parallel auf Linienschreiber registriert. Zur Erzeugung einer Kontraktion wird Phenylephrin dem Bad kumulativ in ansteigender Konzentration zugesetzt. Nach mehreren Kontrollzyklen wird die zu untersuchende Substanz in jedem weiteren Durchgang in jeweils steigender Dosierung zugesetzt und die Höhe der Kontraktion mit der Höhe der im letzten Vordurchgang erreichten Kontraktion verglichen. Daraus wird die Konzentration errechnet, die erforderlich ist, um die Höhe des Kontrollwertes um 50% zu reduzieren (IC50- Wert). Das Standardapplikationsvolumen beträgt 5 μl, der DMSO-Anteil in der Badlösung entspricht 0.1%.
Repräsentative IC50- Werte für die erfindungsgemäßen Verbindungen sind in der nachstehenden Tabelle wiedergegeben:
B-2. Wirkung an rekombinanter Guanylatcvclase-Reporterzellhnie
Die zelluläre Wirkung der erfindungsgemäßen Verbindungen wird an einer rekombinanten Guanylat- cyclase-Reporterzellhnie, wie in F. Wunder et al., Anal. Bwchem 339, 104-112 (2005) beschrieben, bestimmt.
B-3. Bestimmung pharmakokinetischer Kenngrößen nach intravenöser und oraler Gabe
Die zu untersuchende Substanz wird Tieren (z.B. Maus, Ratte, Hund) intravenös als Lösung appliziert, die orale Applikation erfolgt als Lösung oder Suspension über eine Schlundsonde. Nach Substanzgabe wird den Tieren zu festgelegten Zeitpunkten Blut entnommen. Dieses wird hepaπni- siert, anschließend wird daraus durch Zentrifugation Plasma gewonnen. Die Substanz wird im Plasma über LC/MS-MS analytisch quantifiziert. Aus den so ermittelten Plasmakonzentration- Zeit- Verläufen werden die pharmakokinetischen Kenngrößen wie AUC, C013x, T1/2 (Halbwertszeit) und CL (Clearance) mittels eines vahdierten pharmakokinetischen Rechenprogramms berechnet.
B-4. Bestimmung der Löshchkeit
Benotigte Reagenzien:
• PBS-Puffer pH 7.4: 90.00 g NaCl p.a. (z.B. Fa. Merck, Art.-Nr. 1.06404.1000), 13.61 g KH2PO4 p.a. (z.B. Fa. Merck, Art.-Nr. 1.04873.1000) und 83.35 g 1 N NaOH (z.B. Fa. Bernd Kraft GmbH, Art.-Nr. 01030.4000) in einen 1 Liter-Messkolben einwiegen, mit Wasser auffüllen und ca. 1 Stunde rühren:
• Acetatpuffer pH 4 6: 5.4 g Natriumacetat x 3 H2O p.a. (z.B. Fa. Merck, Art.-Nr. 1.06267.0500) m einen 100 ml-Messkolben einwiegen, in 50 ml Wasser lösen, mit 2.4 g Eisessig versetzen, auf 100 ml mit Wasser auffüllen, pH- Wert überprüfen und falls notwendig auf pH 4.6 einstellen;
• Dimethylsulfoxid (z.B. Fa. Baker, Art -Nr. 7157.2500);
• destilliertes Wasser.
Herstellung der Kalibrierlösungen:
Herstellung der Ausgangslösung für Kahbrierlösungen (Stammlösung) • In ein 2 ml Eppendorf- Safe-Lock Tube (Fa. Eppendorf, Art.-Nr. 0030 120.094) werden ca. 0.5 mg der Testsubstanz genau eingewogen, zu einer Konzentration von 600 μg/ml mit DMSO versetzt (z.B. 0.5 mg Substanz + 833 μl DMSO) und bis zur vollständigen Lösung mittels eines Vortexers geschüttelt.
- -
Kalibrierlösung 1 (20 μg/ml): 34.4 μl der Stammlösung werden mit 1000 μl DMSO versetzt und homogenisiert.
Kalibrierlösung 2 (2.5 μg/ml): 100 μl der Kalibrierlösung 1 werden mit 700 μl DMSO versetzt und homogenisiert.
Herstellung der Probenlösungen:
Probenlösung för Löslichkeit bis 10 g/l in PBS-Puffer pH 7.4: In ein 2 ml Eppendorf-Safe-Lock Tube (Fa. Eppendorf, Art.-Nr. 0030 120.094) werden ca. 5 mg der Testsubstanz genau eingewogen und zu einer Konzentration von 5 g/l mit PBS-Puffer pH 7.4 versetzt (z.B. 5 mg Substanz + 500 μl PBS-Puffer pH 7.4).
Probenlösung för Löslichkeit bis 10 g/l in Acetatpuffer pH 4.6: In ein 2 ml Eppendorf-Safe-Lock Tube (Fa. Eppendorf, Art.-Nr. 0030 120.094) werden ca. 5 mg der Testsubstanz genau eingewogen und zu einer Konzentration von 5 g/l mit Acetatpuffer pH 4.6 versetzt (z.B. 5 mg Substanz + 500 μl Acetatpuffer pH 4.6).
Probenlösung för Löslichkeit bis 10 g/l in Wasser: In ein 2 ml Eppendorf-Safe-Lock Tube (Fa. Eppendorf, Art.-Nr. 0030 120.094) werden ca. 5 mg der Testsubstanz genau eingewogen und zu einer Konzentration von 5 g/l mit Wasser versetzt (z.B. 5 mg Substanz + 500 μl Wasser).
Durchführung:
Die so hergestellten Probenlösungen werden 24 Stunden bei 1400 rpm mittels eines temperierbaren Schüttlers (z.B. Fa. Eppendorf Thermomixer comfort Art.-Nr. 5355 000.011 mit Wechsel- block Art.-Nr. 5362.000.019) bei 200C geschüttelt. Von diesen Lösungen werden jeweils 180 μl abgenommen und in Beckman Polyallomer Centrifuge Tubes (Art.-Nr. 343621) überführt. Diese Lösungen werden 1 Stunde mit ca. 223.000 x g zentrifugiert (z.B. Fa. Beckman Optima L-90K Ultracentrifuge mit Type 42.2 Ti Rotor bei 42.000 rpm). Von jeder Probenlösung werden 100 μl des Überstandes abgenommen und 1 :5, 1 : 100 und 1 : 1000 mit dem jeweils verwendeten Lösungs- mittel (Wasser, PBS-Puffer 7.4 oder Acetatpuffer pH 4.6) verdünnt. Es wird von jeder Verdünnung eine Abfüllung in ein geeignetes Gefäß für die HPLC -Analytik vorgenommen.
Analytik:
Die Proben werden mittels RP-HPLC analysiert. Quantifiziert wird über eine Zwei-Punkt-Kalibra- tionskurve der Testverbindung in DMSO. Die Löslichkeit wird in mg/1 ausgedrückt. Analysen- sequenz: 1) Kalibrierlösung 2.5 mg/ml; 2) Kalibrierlösung 20 μg/ml; 3) Probenlösung 1 :5; 4) Probenlösung 1 :100; 5) Probenlösung 1: 1000.
- -
HPLC-Methode fiir Säuren:
Agilent 1100 mit DAD (Gl 315A), quat. Pumpe (G1311A), Autosampier CTC HTS PAL, Degaser (G1322A) und Säulenthermostat (G1316A); Säule: Phenomenex Gemini C18, 50 mm x 2 mm, 5 μ; Temperatur: 40°C; Eluent A: Wasser/Phosphorsäure pH 2; Eluent B: Acetonitril; Flussrate: 0.7 ml/min; Gradient: 0-0.5 min 85% A, 15% B; Rampe: 0.5-3 min 10% A, 90% B; 3-3.5 min 10% A, 90% B; Rampe: 3.5-4 min 85% A, 15% B; 4-5 min 85% A, 15% B.
HPLC-Methode für Basen:
Agilent 1100 mit DAD (G1315A), quat. Pumpe (G131 IA), Autosampier CTC HTS PAL, Degaser (G1322A) und Säulenthermostat (G1316A); Säule: VDSoptilab Kromasil 100 C18, 60 mm x 2.1 mm, 3.5 μ; Temperatur: 300C; Eluent A: Wasser + 5 ml Perchlorsäure/l; Eluent B: Acetonitril; Flussrate: 0.75 ml/min; Gradient: 0-0.5 min 98% A, 2% B; Rampe: 0.5-4.5 min 10% A, 90% B; 4.5-6 min 10% A, 90% B; Rampe: 6.5-6.7 min 98% A, 2% B; 6.7-7.5 min 98% A, 2% B.
C. Ausführungsbeispiele für pharmazeutische Zusammensetzungen
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überfuhrt werden:
Tablette:
Zusammensetzung :
100 mg der erfindungsgemäßen Verbindung, 50 mg Lactose (Monohydrat), 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrrolidon (PVP 25) (Fa. BASF, Ludwigshafen, Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg. Durchmesser 8 mm, Wölbungsradius 12 mm.
Herstellung:
Die Mischung aus erfindungsgemäßer Verbindung, Lactose und Stärke wird mit einer 5%-igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat 5 Minuten gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse verpresst (Format der Tablette siehe oben). Als Richtwert für die Verpressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzung:
1000 mg der erfindungsgemäßen Verbindung, 1000 mg Ethanol (96%), 400 mg Rhodigel® (Xanthan gum der Firma FMC, Pennsylvania, USA) und 99 g Wasser.
Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 10 ml orale Suspension.
Herstellung:
Das Rhodigel wird in Ethanol suspendiert, die erfindungsgemäße Verbindung wird der Suspension zugefügt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluß der Quellung des Rhodigels wird ca. 6 h gerührt.
- -
Oral applizierbare Lösung:
Zusammensetzung:
500 mg der erfindungsgemäßen Verbindung, 2.5 g Polysorbat und 97 g Polyethylenglycol 400. Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 20 g orale Lösung.
Herstellung:
Die erfindungsgemäße Verbindung wird in der Mischung aus Polyethylenglycol und Polysorbat unter Rühren suspendiert. Der Rührvorgang wird bis zur vollständigen Auflösung der erfindungsgemäßen Verbindung fortgesetzt.
i.v.-Lösung:
Die erfindungsgemäße Verbindung wird in einer Konzentration unterhalb der Sättigungslöslichkeit in einem physiologisch verträglichen Lösungsmittel (z.B. isotonische Kochsalzlösung, Glucose- lösung 5% und/oder PEG 400-Lösung 30%) gelöst. Die Lösung wird steril filtriert und in sterile und pyrogenfreie Injektionsbehältnisse abgefüllt.