WO2012059548A1 - Benzyl-substituierte carbamate und ihre verwendung - Google Patents
Benzyl-substituierte carbamate und ihre verwendung Download PDFInfo
- Publication number
- WO2012059548A1 WO2012059548A1 PCT/EP2011/069344 EP2011069344W WO2012059548A1 WO 2012059548 A1 WO2012059548 A1 WO 2012059548A1 EP 2011069344 W EP2011069344 W EP 2011069344W WO 2012059548 A1 WO2012059548 A1 WO 2012059548A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- salts
- solvates
- substituted
- Prior art date
Links
- 0 *c1n[n]c2cc(*)cnc12 Chemical compound *c1n[n]c2cc(*)cnc12 0.000 description 1
- IVRIRQXJSNCSPQ-UHFFFAOYSA-N CC(C)OC(Cl)=O Chemical compound CC(C)OC(Cl)=O IVRIRQXJSNCSPQ-UHFFFAOYSA-N 0.000 description 1
- VULKFUIMVAGNAI-UHFFFAOYSA-N CC(C)OC(N(Cc1ccccc1)c1c(N)nc(-[n](c2c3)nc(Cc(cccc4)c4F)c2ncc3F)nc1N)=O Chemical compound CC(C)OC(N(Cc1ccccc1)c1c(N)nc(-[n](c2c3)nc(Cc(cccc4)c4F)c2ncc3F)nc1N)=O VULKFUIMVAGNAI-UHFFFAOYSA-N 0.000 description 1
- GLSXNUREHMLIJC-UHFFFAOYSA-N CC(C)OC(Nc1c(N)nc(-[n](c2c3)nc(Cc(cccc4)c4F)c2ncc3F)nc1N)=O Chemical compound CC(C)OC(Nc1c(N)nc(-[n](c2c3)nc(Cc(cccc4)c4F)c2ncc3F)nc1N)=O GLSXNUREHMLIJC-UHFFFAOYSA-N 0.000 description 1
- RBVQKTIVBFULIG-UHFFFAOYSA-N Fc1c(Cc2n[nH]c3cccnc23)cccc1 Chemical compound Fc1c(Cc2n[nH]c3cccnc23)cccc1 RBVQKTIVBFULIG-UHFFFAOYSA-N 0.000 description 1
- QVNDRKGDPMVWFG-UHFFFAOYSA-N Nc(nc(nc1N)Cl)c1[N+]([O-])=O Chemical compound Nc(nc(nc1N)Cl)c1[N+]([O-])=O QVNDRKGDPMVWFG-UHFFFAOYSA-N 0.000 description 1
- YZXGPEFZAFYFSA-UHFFFAOYSA-N Nc1c(N)nc(-[n](c2c3)nc(Cc(cccc4)c4F)c2ncc3F)nc1N Chemical compound Nc1c(N)nc(-[n](c2c3)nc(Cc(cccc4)c4F)c2ncc3F)nc1N YZXGPEFZAFYFSA-UHFFFAOYSA-N 0.000 description 1
- DPXJHGQKMRVZLE-UHFFFAOYSA-N Nc1c(N)nc(-[n]2nc(Cc(cccc3)c3F)c3ncccc23)nc1N Chemical compound Nc1c(N)nc(-[n]2nc(Cc(cccc3)c3F)c3ncccc23)nc1N DPXJHGQKMRVZLE-UHFFFAOYSA-N 0.000 description 1
- XEPQBQQUQQRSJX-UHFFFAOYSA-N Nc1nc(-[n]2nc(Cc(cccc3)c3F)c3ncccc23)nc(N)c1[N+]([O-])=O Chemical compound Nc1nc(-[n]2nc(Cc(cccc3)c3F)c3ncccc23)nc(N)c1[N+]([O-])=O XEPQBQQUQQRSJX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/14—Vasoprotectives; Antihaemorrhoidals; Drugs for varicose therapy; Capillary stabilisers
Definitions
- the present application relates to novel benzyl-substituted carbamates, processes for their preparation, their use alone or in combinations for the treatment and / or prophylaxis of diseases and their use for the preparation of medicaments for the treatment and / or prophylaxis of diseases, in particular for treatment and / or prophylaxis of cardiovascular disease.
- cyclic guanosine monophosphate cGMP
- NO nitric oxide
- the guanylate cyclases catalyze the biosynthesis of cGMP from guanosine triphosphate (GTP).
- GTP guanosine triphosphate
- the previously known members of this family can be divided into two groups according to both structural features and the nature of the ligands: the particulate guanylate cyclases stimulable by natriuretic peptides and the soluble guanylate cyclases stimulable by NO.
- the soluble guanylate cyclases consist of two subunits and most likely contain one heme per heterodimer that is part of the regulatory center. This is central to the activation mechanism. NO can bind to the iron atom of the heme and thus significantly increase the activity of the enzyme. On the other hand, heme-free preparations can not be stimulated by NO. Also, carbon monoxide (CO) is able to bind to the central iron atom of the heme, with stimulation by CO being significantly less than by NO.
- CO carbon monoxide
- guanylate cyclase plays a crucial role in various physiological processes, in particular in the relaxation and proliferation of smooth muscle cells, platelet aggregation and adhesion, neuronal signaling and diseases based on a disturbance of the above operations.
- the NO / cGMP system may be suppressed, which may, for example, lead to hypertension, platelet activation, increased cell proliferation, endothelial dysfunction, arteriosclerosis, angina pectoris, heart failure, myocardial infarction, thrombosis, stroke and sexual dysfunction.
- a NO-independent treatment option for such diseases which is aimed at influencing the cGMP pathway in organisms, is a promising approach on account of the expected high efficiency and low side effects.
- WO 2008/03 15 13 discloses inter alia 1H-pyrazolo [4,3-b] pyridines as stimulators of soluble guanylate cyclase for the treatment of cardiovascular diseases.
- WO 2005/030121 describes fused pyrazoles for the treatment of cancers.
- WO 02/42300 describes pyrazolopyridines with carbamate substituents for the treatment of cardiovascular diseases.
- WO 2011/119518 and WO 2011/115804 disclose carbamate-substituted pyrimidines for the treatment of cardiovascular diseases.
- the object of the present invention was to provide new substances which act as potent stimulators of soluble guanylate cyclase, and are therefore suitable for the treatment and / or prophylaxis of cardiovascular diseases.
- the present invention relates to compounds of the general formula (I)
- R 1 is hydrogen or fluorine
- R 2 is hydrogen or amino
- R 3 is (C 1 -C 4 ) -alkyl
- R 4 is hydrogen, (C 1 -C 4 ) -alkyl or (C 1 -C 4 ) -alkoxycarbonyl
- R 5 is phenyl, tetrahydronaphthalenyl, naphthyl or 5- to 10-membered heteroaryl, wherein phenyl, tetrahydronaphthalenyl, naphthyl and 5- to 10-membered heteroaryl having 1 to 3 substituents independently of one another selected from the group halogen, nitro, cyano, difluoromethyl, trifluoromethyl, (C 1 -C 6 ) -alkyl, hydroxy, difluoromethoxy, trifluoromethoxy, (C 1 -C 4 ) - Alkoxy, amino, mono- (C 1 -C 4) -alkylamino, di- (C 1 -C 4) -alkylamino, hydroxycarbonyl, (C 1 -C 4) -alkoxycarbonyl
- R 6 is (C 1 -C 6 ) -alkyl or benzyl, where (C 1 -C 6 ) -alkyl is substituted by a substituent trifluoromethyl, where (C 1 -C 6 ) -alkyl may be substituted by 1 to 3 substituents of fluorine, and wherein benzyl is substituted by 1 to 3 substituents fluorine, and their N-oxides, salts, solvates, salts of N-oxides and solvates of N-oxides and salts.
- Compounds according to the invention are the compounds of the formula (I) and their salts, solvates and solvates of the salts comprising the compounds of the formulas below and their salts, solvates and solvates of the salts and of the formula (I) encompassed by formula (I), subsequently as Embodiments mentioned compounds and their salts, solvates and solvates of the salts, as far as the compounds of formula (I), mentioned below are not already salts, solvates and solvates of the salts.
- Salts which are preferred in the context of the present invention are physiologically acceptable salts of the compounds according to the invention. Also included are salts which are not suitable for pharmaceutical applications themselves, but can be used, for example, for the isolation or purification of the compounds according to the invention.
- Physiologically acceptable salts of the compounds of the invention include acid addition salts of mineral acids, carboxylic acids and sulfonic acids, e.g. Salts of hydrochloric, hydrobromic, sulfuric, phosphoric, methanesulfonic, ethanesulfonic, toluenesulfonic, benzenesulfonic, naphthalenedisulfonic, formic, acetic, trifluoroacetic, propionic, lactic, tartaric, malic, citric, fumaric, maleic and benzoic acids.
- Salts of hydrochloric, hydrobromic, sulfuric, phosphoric, methanesulfonic, ethanesulfonic, toluenesulfonic, benzenesulfonic, naphthalenedisulfonic formic, acetic, trifluoroacetic, propionic, lactic, tartaric, malic, citric, fumaric, maleic and benzoic
- Physiologically acceptable salts of the compounds according to the invention also include salts of customary bases, such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts) and ammonium salts derived from ammonia or organic amines having 1 to 16 carbon atoms, such as, by way of example and by way of illustration, ethylamine, diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, arginine, lysine, ethylenediamine and N-methylpiperidine.
- customary bases such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts
- Solvates in the context of the invention are those forms of the compounds according to the invention which form a complex in the solid or liquid state by coordination with solvent molecules. Hydrates are a special form of solvates that coordinate with water. As solvates, hydrates are preferred in the context of the present invention.
- the compounds according to the invention may exist in different stereoisomeric forms, ie in the form of configurational isomers or optionally also as conformational isomers (enantiomers and / or diastereomers, including those in the case of atropisomers).
- the present invention therefore encompasses the enantiomers and diastereomers and their respective mixtures. From such mixtures of enantiomers and / or diastereomers, the stereoisomerically uniform components can be isolated in a known manner; Preferably, chromatographic methods are used for this, in particular HPLC chromatography on achiral or chiral phase. If the compounds according to the invention can occur in tautomeric forms, the present invention encompasses all tautomeric forms.
- the present invention also includes all suitable isotopic variants of the compounds of the invention.
- An isotopic variant of a compound according to the invention is understood to mean a compound in which at least one atom within the compound according to the invention is exchanged for another atom of the same atomic number but with a different atomic mass than the atomic mass that usually or predominantly occurs in nature.
- isotopes that can be incorporated into a compound of the invention are those of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, chlorine, bromine and iodine, such as 2 H (deuterium), H (tritium), 1 C , 14 C, 15 N, 17 0, 18 0, 2 P, P, S, 4 S, 5 S, 6 S, 18 F, 6 Cl, 82 Br, 12 I, 124 I, 129 I and 1 1 I.
- isotopic variants of a compound of the invention such as those in which one or more radioactive isotopes are incorporated, may be useful, for example, for the study of the mechanism of action or distribution of drug in the body; because of the comparatively easy production and detectability, compounds labeled with H or 14 C isotopes are particularly suitable for this purpose.
- isotopes such as deuterium may result in certain therapeutic benefits as a result of greater metabolic stability of the compound, such as prolonging the body's half-life or reducing the required effective dose;
- modifications of the compounds according to the invention may therefore possibly also constitute a preferred embodiment of the present invention.
- Isotopic variants of the compounds according to the invention can be prepared by the processes known to the person skilled in the art, for example by the methods described below and the rules given in the exemplary embodiments, by using appropriate isotopic modifications of the respective reagents and / or starting compounds.
- the present invention also includes prodrugs of the compounds of the invention.
- prodrugs refers to compounds which themselves may be biologically active or inactive, but are converted during their residence time in the body to compounds of the invention (for example metabolically or hydrolytically). Unless otherwise specified, in the context of the present invention, the substituents have the following meaning:
- alkyl is a linear or branched alkyl radical having 1 to 6 or 1 to 4 carbon atoms.
- alkyl is a linear or branched alkyl radical having 1 to 6 or 1 to 4 carbon atoms.
- Alkoxy in the context of the invention is a linear or branched alkoxy radical having 1 to 4 carbon atoms. Examples which may be mentioned are: methoxy, ethoxy, n-propoxy, isopropoxy, 1-methylpropoxy, n-butoxy, isobutoxy and tert-butoxy.
- Alkoxycarbonyl in the context of the invention are a linear or branched alkoxy radical having 1 to 4 carbon atoms and an oxygen-bonded carbonyl group.
- alkoxycarbonyl in the context of the invention are a linear or branched alkoxy radical having 1 to 4 carbon atoms and an oxygen-bonded carbonyl group.
- Mono-alkylamino in the context of the invention represents an amino group having a linear or branched alkyl substituent which has 1 to 4 carbon atoms. Examples which may be mentioned are: methylamino, ethylamino, n-propylamino, isopropylamino and tert-butylamino.
- Di-alkylamino in the context of the invention represents an amino group having two identical or different linear or branched alkyl substituents, each having 1 to 4 carbon atoms.
- Mono-alkylaminocarbonyl in the context of the invention represents an amino group which is linked via a carbonyl group and which has a linear or branched alkyl substituent having 1 to 4 carbon atoms.
- Di-alkylaminocarbonyl is in the context of the invention an amino group which is linked via a carbonyl group and which has two identical or different linear or branched alkyl substituents each having 1 to 4 carbon atoms.
- N N-dimethylaminocarbonyl, N, N-diethylaminocarbonyl, N-ethyl-N-methylaminocarbonyl, N-methyl-N- ⁇ -propylaminocarbonyl, N- ⁇ -butyl-N-methylaminocarbonyl and N- tert-butyl-N-methylaminocarbonyl.
- Cycloalkylaminocarbonyl in the context of the invention is an amino group which is linked via a carbonyl group and which has a monocyclic, saturated carbocycle having 3 to 7 carbon atoms.
- cyclopropylaminocarbonyl cyclobutylaminocarbonyl, cyclopentylaminocarbonyl, cyclohexylaminocarbonyl, and cycloheptylaminocarbonyl.
- Heteroaryl is in the context of the invention for a mono- or optionally bicyclic aromatic heterocycle (heteroaromatic) having a total of 5 to 10 ring atoms containing up to three identical or different ring heteroatoms from the series N, O and / or S and via a ring -Coryl atom or optionally via a ring nitrogen atom is linked.
- heterocycle aromatic heterocycle
- Examples which may be mentioned are: furyl, pyrrolyl, thienyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isoxazolyl, isothiazolyl, triazolyl, oxadiazolyl, thiadiazolyl, pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, triazinyl, benzofuranyl, benzothienyl, benzimidazolyl, benzoxazolyl, benzothiazolyl, Benzotriazolyl, indolyl, indazolyl, quinolinyl, isoquinolinyl, naphthyridinyl, quinazolinyl, quinoxalinyl, phthalazinyl, pyrazolo [3,4-b] pyridinyl.
- Halogen is in the context of the invention for fluorine, chlorine, bromine and iodine.
- radicals are substituted in the compounds according to the invention, the radicals can, unless otherwise specified, be monosubstituted or polysubstituted. In the context of the present invention, the meaning is independent of each other for all radicals which occur repeatedly. Substitution with one, two or three identical or different substituents is preferred.
- R 1 is hydrogen or fluorine
- R 2 is hydrogen or amino
- R 3 is (C 1 -C 3 ) -alkyl
- R 4 is hydrogen or methyl
- R 5 is phenyl, thienyl, thiazolyl, pyridyl or quinolyl, where phenyl, thienyl, thiazolyl, pyridyl and quinolyl having 1 to 3 substituents independently of one another selected from the group fluorine, chlorine, nitro, cyano, Difluoromethyl, trifluoromethyl, (C 1 -C 6 ) -alkyl, hydroxy, difluoromethoxy, trifluoromethoxy, methoxy, ethoxy, amino, methylamino, ethylamino, dimethylamino, diethylamino, methoxycarbonyl, ethoxycarbonyl, aminocarbonyl, methylaminocarbonyl, ethylaminocarbonyl, dimethylaminocarbonyl, diethylaminocarbonyl, cyclopropylaminocarbonyl and cyclobutylaminocarbonyl in
- R 6 is 3,3,3-trifluoroprop-1-yl, 3,3,4,4,4-pentafluorobut-1-yl or benzyl, wherein benzyl is substituted by 1 or 2 substituents fluorine, and their salts, solvates and solvates of salts.
- R 1 is hydrogen or fluorine
- R 2 is hydrogen or amino
- R 3 is methyl, ethyl or iso-propyl
- R 4 is hydrogen
- R 5 is phenyl, where phenyl may be substituted by 1 substituent fluorine
- R 6 is 2-fluorobenzyl, and their salts, solvates and solvates of the salts.
- R 5 is phenyl, where phenyl may be substituted by 1 substituent fluorine, and also their salts, solvates and solvates of the salts.
- R 6 is 3,3,3-trifluoroprop-1-yl or 3,3,4,4,4-pentafluorobut-1-yl, and their salts, solvates and solvates of salts.
- the invention further provides a process for the preparation of the compounds of the formula (I) according to the invention which comprises reacting a compound of the formula (II)
- X 1 is a suitable leaving group, such as mesylate, tosylate or halogen, in particular bromine or iodine, is reacted, and optionally the resulting compounds of formula (I) optionally with the appropriate (i) solvents and / or (ii) acids or Bases are converted into their solvates, salts and / or solvates of the salts.
- the reaction (II) + (III) (IV) can be carried out in an inert solvent or without solvent.
- Inert solvents for process step (II) + (III) -> (IV) are, for example Alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, halogenated hydrocarbons such as dichloromethane, trichloromethane, carbon tetrachloride, trichlorethylene or chlorobenzene, hydrocarbons such as benzene, xylene , Toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), NN'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), acetonitrile or
- Suitable bases for process step (II) + (III) - »(IV) are alkali metal hydrides such as sodium hydride, alkali hydroxides such as lithium, sodium or potassium hydroxide, alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate, Alkahhydrogencarbonate such as sodium or potassium bicarbonate, alkali metal alcoholates such as sodium or potassium methoxide, sodium or potassium ethoxide or potassium tert-butoxide, amides such as sodium amide, lithium, sodium or potassium bis (trimethylsilyl) amide or lithium diisopropylamide, organometallic compounds such as butyllithium or phenyllithium, or organic amines such as triethylamine, diisopropylethylamine, pyridine, l, 8-diazabicyclo [5.4.0] undec-7-ene (DBU) or l, 5-diazabicyclo [4.3.0] non-5-ene (DBN).
- the reaction (II) + (III) -> (IV) is generally carried out in a temperature range of -10 ° C to + 30 ° C, preferably at 0 ° C to + 20 ° C.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., from 0.5 to 5 bar). Generally, one works at normal pressure.
- Inert solvents for process step (IV) + (V) - »(I) are, for example, alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or Diethylene glycol dimethyl ether, halogenated carbons such as dichloromethane, trichloromethane, carbon tetrachloride, trichlorethylene or chlorobenzene, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), NN'-dimethylpropyleneurea (DMPU ), N-methylpyrrolidone ( ⁇ ), acetonitrile or
- Suitable bases for process step (IV) + (V) - »(I) are alkali metal hydrides such as sodium hydride, alkali metal hydroxides such as lithium, sodium or potassium hydroxide, alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate, Alkahhydrogencarbonate such as sodium or potassium bicarbonate, alkali metal such as sodium or potassium methoxide, sodium or Potassium ethoxide or potassium tert-butoxide, amides such as sodium amide, lithium, sodium or potassium bis (trimethylsilyl) amide or lithium diisopropylamide, organometallic compounds such as butyl lithium or phenyllithium, or organic amines such as triethylamine, diisopropylethylamine, pyridine, l, 8 -Diazabicyclo [5.4.0] undec-7-ene (DBU) or l, 5-d
- the reaction (IV) + (V) -> (I) is generally carried out in a temperature range of -10 ° C to + 30 ° C, preferably at 0 ° C to + 20 ° C.
- the reaction may be carried out at normal, elevated or reduced pressure (e.g., from 0.5 to 5 bar). Generally, one works at normal pressure.
- normal pressure e.g., from 0.5 to 5 bar. Generally, one works at normal pressure.
- the compounds of the formula (III) and (V) are commercially available, known from the literature or can be prepared analogously to processes known in the literature.
- the compound of formula (II) can be prepared by reacting the compound of formula (VI)
- X 2 is a suitable leaving group such as, for example, halogen, mesylate, tosylate or triflate, to give a compound of the formula (VIII)
- Inert solvents for process step (VI) + (VII) -> (VIII) are, for example, alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers, such as diethyl ether, dioxane, tetrahydrofuran, Glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), NN'-dimethylpropyleneurea (DMPU), dimethylacetamide, N-methyl
- solvents for process step (VI) + (VII) -> (VIII) are, for example, alcohols, such as methanol, ethanol, n-propanol, isoprop
- Suitable bases for process step (VI) + (VII) - »(VIII) are alkali metal hydrides such as sodium hydride, alkali metal hydroxides such as lithium, sodium or potassium hydroxide, alkali metal carbonates such as lithium, sodium, potassium or cesium carbonate, alkali metal bicarbonates such as sodium or potassium bicarbonate, alkali metal alcoholates such as sodium or potassium methoxide, sodium or potassium ethoxide or potassium tert-butoxide, amides such as sodium amide, lithium, sodium or potassium bis (trimethylsilyl) amide or lithium diisopropylamide, organometallic compounds such as butyllithium or phenyllithium, or organic amines such as triethylamine, diisopropylethylamine, pyridine, 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU)
- the reaction (VI) + (VII) -> (VIII) is generally carried out in a temperature range from 0 ° C to + 120 ° C, preferably at + 20 ° C to + 80 ° C, optionally in a microwave.
- the reaction can be carried out at normal, elevated or at reduced pressure (for example from 0.5 to 5 bar). Generally, one works at normal pressure.
- the reduction (VIII) - »(II) is carried out in the presence of a suitable catalyst in an inert solvent, in a temperature range from + 20 ° C to + 40 ° C under normal hydrogen pressure.
- Inert solvents for the reduction (VIII) - »(II) are, for example, alcohols such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, or others Solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine, acetonitrile or even water. It is likewise possible to use mixtures of the solvents mentioned. Preferred are DMF and pyridine.
- Suitable catalysts for the reaction (VIII) - »(II) are, for example, palladium on activated carbon, platinum on carbon, palladium hydroxide or Raney nickel.
- the reduction (VIII) - »(II) may alternatively be treated with a metal or metal salt such as iron, zinc or stannous chloride in a suitable acid such as hydrochloric acid / hydrochloric acid, sulfuric acid, phosphoric acid or acetic acid in a temperature range of +20 ° C to + 140 ° C.
- a metal or metal salt such as iron, zinc or stannous chloride in a suitable acid such as hydrochloric acid / hydrochloric acid, sulfuric acid, phosphoric acid or acetic acid in a temperature range of +20 ° C to + 140 ° C.
- the compounds of the invention act as potent stimulators of soluble guanylate cyclase, have valuable pharmacological properties, and are therefore suitable for the treatment and / or prophylaxis of diseases in humans and animals.
- the compounds of the invention cause vasorelaxation and inhibition of platelet aggregation and lead to a reduction in blood pressure and to an increase in coronary blood flow. These effects are mediated via direct stimulation of soluble guanylate cyclase and intracellular cGMP increase.
- the compounds according to the invention enhance the action of substances which increase cGMP levels, such as, for example, EDRF (endothelium-derived relaxing factor), NO donors, protoporphyrin IX, arachidonic acid or phenylhydrazine derivatives.
- the compounds according to the invention can therefore be used in medicaments for the treatment and / or prophylaxis of cardiovascular diseases such as hypertension, acute and chronic heart failure, coronary heart disease, stable and unstable angina pectoris, peripheral and cardiac vascular diseases, arrhythmias, arrhythmia of the atria and the chambers and conduction disorders such for example atrio-ventricular blockades grade I-III (AB-B lock I-III), supraventricular tachyarrhythmia, atrial fibrillation, atrial flutter, ventricular fibrillation, ventricular flutter, ventricular tachyarrhythmia, torsades de pointes tachycardia, atrial and ventricular extrasystoles, AV-junctional extrasystoles , Sick sinus syndrome, syncope, AV node reentrant tachycardia, Wolff-Parkinson-White syndrome, acute coronary syndrome (ACS), autoimmune heart disease (pericarditis, endocarditis,
- cardiac insufficiency also encompasses more specific or related forms of disease such as acute decompensated heart failure, right heart failure, left heart failure, global insufficiency, ischemic cardiomyopathy, dilated cardiomyopathy, hypertrophic cardiomyopathy, idiopathic cardiomyopathy, congenital heart defects, valvular heart failure, cardiac valvulopathy, mitral valve stenosis, mitral valve insufficiency, Aortic valve stenosis, aortic valve insufficiency, tricuspid stenosis, tricuspid regurgitation, pulmonary valve stenosis, pulmonary valvular insufficiency, combined heart valve defects, myocarditis, chronic myocarditis, acute myocarditis, viral myocarditis, diabetic heart failure, alcoholic cardiomyopathy, cardiac storage disorders, diastolic heart failure and systolic heart failure.
- ischemic cardiomyopathy dilated cardiomyopathy
- the compounds according to the invention may also be used for the treatment and / or prophylaxis of arteriosclerosis, lipid metabolism disorders, hypolipoproteinemias, dyslipidemias, hypertriglyceridemias, hyperlipidemias, hypercholesterolemias, abetelipoproteinaemia, sitosterolemia, xanthomatosis, Tangier's disease, obesity (obesity) and combined hyperlipidemias and the metabolic syndrome.
- the compounds of the invention may be used for the treatment and / or prophylaxis of primary and secondary Raynaud's phenomenon, microcirculatory disorders, claudication, peripheral and autonomic neuropathies, diabetic microangiopathies, diabetic retinopathy, diabetic ulcers on the extremities, gangrenous, CREST syndrome, erythematosis, onychomycosis , rheumatic diseases and to promote wound healing.
- the compounds according to the invention are suitable for the treatment of urological diseases such as benign prostatic syndrome (BPS), benign prostatic hyperplasia (BPH), benign prostate enlargement (BPE), bladder emptying disorder (BOO), lower urinary tract syndromes (LUTS, including Feiine's urological syndrome ( FUS)), diseases of the urogenital system including neurogenic overactive bladder (OAB) and (IC), incontinence (UI) such as mixed, urge, stress, or overflow incontinence (MUI, UUI, SUI, OUI), Pelvic pain, benign and malignant diseases of the organs of the male and female urogenital system.
- BPS benign prostatic syndrome
- BPH benign prostatic hyperplasia
- BPE benign prostate enlargement
- BOO bladder emptying disorder
- LUTS lower urinary tract syndromes
- FUS lower urinary tract syndromes
- UI incontinence
- MUI mixed, urge, stress, or overflow incontinence
- UUI UUI
- SUI S
- kidney diseases in particular of acute and chronic renal insufficiency, as well as of acute and chronic renal failure.
- renal insufficiency includes both acute and chronic manifestations of renal insufficiency, as well as underlying or related renal diseases such as renal hypoperfusion, intradialytic hypotension, obstructive uropathy, glomerulopathies, glomerulonephritis, acute glomerulonephritis, glomerulosclerosis, tubulo-interstitial disorders, nephropathic disorders such as primary and congenital kidney disease, nephritis, immunological kidney diseases such as renal transplant rejection, immune complex-induced kidney disease, nephropathy induced by toxic substances, contrast agent-induced nephropathy, diabetic and non-diabetic nephropathy, pyelonephritis, renal cysts, nephrosclerosis, hyperten
- the present invention also encompasses the use of the compounds of the invention for the treatment and / or prophylaxis of sequelae of renal insufficiency, such as pulmonary edema, cardiac insufficiency, uremia, anemia, electrolyte disorders (e.g., hyperkalemia, hyponatremia), and disorders in bone and carbohydrate metabolism.
- sequelae of renal insufficiency such as pulmonary edema, cardiac insufficiency, uremia, anemia, electrolyte disorders (e.g., hyperkalemia, hyponatremia), and disorders in bone and carbohydrate metabolism.
- the compounds according to the invention are also suitable for the treatment and / or prophylaxis of asthmatic diseases, pulmonary arterial hypertension (PAH) and other forms of pulmonary hypertension (PH), chronic obstructive pulmonary disease (COPD), acute respiratory tract syndrome (ARDS) acute lung injury (ALI), the alpha-1-antitrypsin Deficiency (AATD), pulmonary fibrosis, pulmonary emphysema (eg, cigarette smoke-induced pulmonary emphysema) and cystic fibrosis (CF).
- PAH pulmonary arterial hypertension
- PH chronic obstructive pulmonary disease
- ARDS acute respiratory tract syndrome
- ALI acute lung injury
- AATD alpha-1-antitrypsin Deficiency
- pulmonary fibrosis pulmonary emphysema (eg, cigarette smoke-induced pulmonary emphysema) and cystic fibrosis (CF).
- the compounds described in the present invention are also agents for controlling diseases in the central nervous system, which are characterized by disorders of the NO / cGMP system.
- they are suitable for improving the perception, concentration performance, learning performance or memory performance after cognitive disorders such as occur in situations / diseases / syndromes such as mild cognitive impairment, age-associated learning and memory disorders, age-associated memory loss, vascular dementia, cranial brain -Trauma, stroke, post-stroke dementia, post-traumatic traumatic brain injury, generalized concentration disorder, difficulty concentrating in children with learning and memory problems, Alzheimer's disease, dementia with Lewy bodies , Dementia with degeneration of the frontal lobes including Pick's syndrome, Parkinson's disease, progressive nuclear palsy, dementia with corticobasal degeneration, amyolateral sclerosis (ALS), Huntington's disease, demyelinization, multiple sclerosis, thalamic degeneration, Creutzfeld-Jacob dementia, HIV dementia, schizophrenia with dementia or Korsakoff's psychosis.
- the compounds according to the invention are also suitable for the treatment and / or prophylaxis of diseases of the central nervous system such as states of anxiety, tension and depression, central nervous conditional sexual dysfunctions and sleep disorders as well as for the regulation of pathological disorders of food, consumption and addiction absorption.
- the compounds according to the invention are also suitable for regulating cerebral blood flow and are effective agents for combating migraine. They are also suitable for the prophylaxis and control of the consequences of cerebral infarct events (Apoplexia cerebri) such as stroke, cerebral ischaemias and craniocerebral trauma , Likewise, the compounds of the invention can be used to combat pain and tinnitus.
- the compounds of the invention have anti-inflammatory action and can therefore be used as anti-inflammatory agents for the treatment and / or prophylaxis of sepsis (SIRS), multiple organ failure (MODS, MOF), inflammatory diseases of the kidney, chronic enteritis (IBD, Crohn s Disease, UC), Pancreatitis, peritonitis, rheumatoid diseases, inflammatory skin diseases and inflammatory eye diseases.
- SIRS sepsis
- MODS multiple organ failure
- IBD chronic enteritis
- Crohn s Disease UC
- Pancreatitis peritonitis
- rheumatoid diseases inflammatory skin diseases and inflammatory eye diseases.
- the compounds of the invention can also be used for the treatment and / or prophylaxis of autoimmune diseases.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of fibrotic disorders of the internal organs such as, for example, the lung, the heart, the kidney, the bone marrow and in particular the liver, as well as dermatological fibroses and fibrotic disorders of the eye.
- fibrotic disorders includes in particular the following terms: liver fibrosis, cirrhosis, pulmonary fibrosis, endomyocardial fibrosis, nephropathy, glomerulonephritis, interstitial renal fibrosis, fibrotic damage as a result of diabetes, bone marrow fibrosis and similar fibrotic disorders, scleroderma, morphea, keloids, hypertrophic scarring (also after surgical procedures), nevi, diabetic retinopathy and proliferative vitroretinopathy.
- the compounds according to the invention are suitable for combating postoperative scar formation, for example as a consequence of glaucoma operations.
- the compounds according to the invention can likewise be used cosmetically for aging and keratinizing skin.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of hepatitis, neoplasm, osteoporosis, glaucoma and gastroparesis.
- Another object of the present invention is the use of the compounds of the invention for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases.
- the present invention furthermore relates to the compounds according to the invention for use in a method for the treatment and / or prophylaxis of cardiac insufficiency, angina pectoris, hypertension, pulmonary hypertension, ischaemias, vascular disorders, thromboembolic disorders and arteriosclerosis.
- Another object of the present invention is the use of the compounds of the invention for the manufacture of a medicament for the treatment and / or prophylaxis of Erkrankun- gene, in particular the aforementioned diseases.
- Another object of the present invention is a method for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases, using an effective amount of at least one of the compounds of the invention.
- the compounds of the invention may be used alone or as needed in combination with other agents.
- Another object of the present invention are pharmaceutical compositions containing at least one of the compounds of the invention and one or more further Active ingredients, in particular for the treatment and / or prophylaxis of the aforementioned diseases.
- suitable combination active ingredients may be mentioned by way of example and preferably:
- organic nitrates and NO donors such as sodium nitroprusside, nitroglycerin, isosorbide mononitrate, isosorbide dinitrate, molsidomine or SIN-1, and inhaled NO;
- Compounds which inhibit the degradation of cyclic guanosine monophosphate (cGMP) such as inhibitors of phosphodiesterases (PDE) 1, 2 and / or 5, in particular PDE 5 inhibitors such as sildenafil, vardenafil and tadalafil;
- Antithrombotic agents by way of example and preferably from the group of platelet aggregation inhibitors, anticoagulants or profibrinolytic substances;
- the blood pressure lowering agents exemplarily and preferably from the group of calcium antagonists, angiotensin AII antagonists, ACE inhibitors, endothelin antagonists, renin inhibitors, alpha-receptor blockers, beta-receptor blockers, mineralocorticoid receptor Antagonists and diuretics; and or
- Lipid metabolism-altering agents by way of example and preferably from the group of thyroid receptor agonists, cholesterol synthesis inhibitors as exemplified and preferably
- HMG-CoA reductase or squalene synthesis inhibitors include ACAT inhibitors, CETP inhibitors, MTP inhibitors, PPAR-alpha, PPAR-gamma and / or PPAR-delta agonists, cholesterol absorption inhibitors, lipase inhibitors , polymeric bile acid adsorbents, bile acid inhibitor inhibitors, and lipoprotein (a) antagonists.
- Antithrombotic agents are preferably understood as meaning compounds from the group of platelet aggregation inhibitors, anticoagulants or profibrinolytic substances.
- the compounds according to the invention are administered in combination with a platelet aggregation inhibitor, such as, by way of example and by way of preference, aspirin, clopidogrel, ticlopidine or dipyridamole.
- a platelet aggregation inhibitor such as, by way of example and by way of preference, aspirin, clopidogrel, ticlopidine or dipyridamole.
- the compounds according to the invention are administered in combination with a thrombin inhibitor, such as, by way of example and by way of preference, ximelagatran, dabigatran, melagatran, bivalirudin or Clexane.
- a thrombin inhibitor such as, by way of example and by way of preference, ximelagatran, dabigatran, melagatran, bivalirudin or Clexane.
- the compounds according to the invention are administered in combination with a GPIIb / IIIa antagonist, such as, by way of example and by way of preference, tirofiban or abciximab.
- the compounds according to the invention are used in combination with a factor Xa inhibitor, such as by way of example and preferably rivaroxaban (BAY 59-7939), DU-176b, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux, PMD 31 12, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 or SSR-128428.
- a factor Xa inhibitor such as by way of example and preferably rivaroxaban (BAY 59-7939), DU-176b, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux, PMD 31 12, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-10
- the compounds according to the invention are administered in combination with heparin or a low molecular weight (LMW) heparin derivative.
- LMW low molecular weight
- the compounds according to the invention are administered in combination with a vitamin K antagonist, such as by way of example and preferably coumarin.
- antihypertensive agents are preferably compounds from the group of calcium antagonists, angiotensin AII antagonists, ACE inhibitors, endothelin antagonists, renin inhibitors, alpha-receptor blocker, beta-receptor blocker, mineralocorticoid receptor Antagonists and diuretics understood.
- the compounds according to the invention are administered in combination with a calcium antagonist, such as, by way of example and by way of preference, nifedipine, amlodipine, verapamil or diltiazem.
- a calcium antagonist such as, by way of example and by way of preference, nifedipine, amlodipine, verapamil or diltiazem.
- the compounds according to the invention are administered in combination with an alpha-1-receptor blocker, such as by way of example and preferably prazosin.
- the compounds according to the invention are used in combination with a beta-receptor blocker, such as by way of example and preferably propranolol, atenolol, timolol, pindolol, alprenolol, oxprenolol, penbutolol, bupranolol, metipranolol, nadolol, mepindolol, carazalol, sotalol, Metoprolol, betaxolol, celiprolol, bisoprolol, carteolol, esmolol, labetalol, carvedilol, adaprolol, landiolol, nebivolol, epanolol or bucindolol.
- a beta-receptor blocker such as by way of example and preferably propranolol, atenolol, timolol
- the compounds according to the invention are administered in combination with an angiotensin AII antagonist, such as by way of example and preferably losartan, candesartan, valsartan, telmisartan or embursatan.
- an ACE inhibitor such as, by way of example and by way of preference, enalapril, captopril, lisinopril, ramipril, delapril, fosinopril, quinopril, perindopril or trandopril.
- the compounds according to the invention are administered in combination with an endothelin antagonist such as, by way of example and by way of preference, bosentan, darusentan, ambrisentan or sitaxsentan.
- an endothelin antagonist such as, by way of example and by way of preference, bosentan, darusentan, ambrisentan or sitaxsentan.
- the compounds of the invention are administered in combination with a renin inhibitor, such as by way of example and preferably aliskiren, SPP-600 or SPP-800.
- a renin inhibitor such as by way of example and preferably aliskiren, SPP-600 or SPP-800.
- the compounds according to the invention are administered in combination with a mineralocorticoid receptor antagonist, such as by way of example and preferably spironolactone or eplerenone.
- the compounds according to the invention are administered in combination with a diuretic, such as by way of example and preferably furosemide.
- a diuretic such as by way of example and preferably furosemide.
- the lipid metabolizing agents are preferably compounds from the group of CETP inhibitors, thyroid receptor agonists, cholesterol synthesis inhibitors such as HMG-CoA reductase or squalene synthesis inhibitors, ACAT inhibitors, MTP inhibitors, PPAR-alpha, PPAR gamma and / or PPAR delta agonists, cholesterol absorption inhibitors, polymeric bile acid adsorbers, bile acid inhibition inhibitors, lipase inhibitors, and the lipoprotein (a) antagonists.
- the compounds according to the invention are administered in combination with a CETP inhibitor, by way of example and with preference dalcetrapib, BAY 60-5521, anacetrapib or CETP vaccine (CETi-1).
- a CETP inhibitor by way of example and with preference dalcetrapib, BAY 60-5521, anacetrapib or CETP vaccine (CETi-1).
- the compounds of the invention are administered in combination with a thyroid receptor agonist such as, by way of example and by way of preference, D-thyroxine, 3,5,3'-triiodothyronine (T3), CGS 23425 or axitirome (CGS 26214).
- a thyroid receptor agonist such as, by way of example and by way of preference, D-thyroxine, 3,5,3'-triiodothyronine (T3), CGS 23425 or axitirome (CGS 26214).
- the compounds according to the invention are administered in combination with an HMG-CoA reductase inhibitor from the class of statins, such as by way of example and preferably lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastatin or pitavastatin.
- statins such as by way of example and preferably lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastatin or pitavastatin.
- the compounds according to the invention are administered in combination with a squalene synthesis inhibitor, such as by way of example and preferably BMS-188494 or TAK-475.
- the compounds according to the invention are administered in combination with an ACAT inhibitor, such as by way of example and preferably avasimibe, melinamide, pactimibe, eflucimibe or SMP-797.
- an ACAT inhibitor such as by way of example and preferably avasimibe, melinamide, pactimibe, eflucimibe or SMP-797.
- the compounds according to the invention are administered in combination with an MTP inhibitor such as, for example and preferably, implitapide, BMS-201038, R-103757 or JTT-130.
- an MTP inhibitor such as, for example and preferably, implitapide, BMS-201038, R-103757 or JTT-130.
- the compounds of the invention are administered in combination with a PPAR-gamma agonist such as, by way of example and by way of preference, pioglitazone or rosiglitazone.
- the compounds according to the invention are administered in combination with a PPAR delta agonist, such as by way of example and preferably GW 501516 or BAY 68-5042.
- the compounds according to the invention are administered in combination with a cholesterol absorption inhibitor, such as by way of example and preferably ezetimibe, tiqueside or pamaqueside.
- a cholesterol absorption inhibitor such as by way of example and preferably ezetimibe, tiqueside or pamaqueside.
- the compounds according to the invention are administered in combination with a lipase inhibitor, such as, for example and preferably, orlistat.
- a lipase inhibitor such as, for example and preferably, orlistat.
- the compounds of the invention are administered in combination with a polymeric bile acid adsorbent such as, by way of example and by way of preference, cholestyramine, colestipol, colesolvam, cholesta gel or colestimide.
- a polymeric bile acid adsorbent such as, by way of example and by way of preference, cholestyramine, colestipol, colesolvam, cholesta gel or colestimide.
- ASBT IBAT
- AZD-7806 S-8921
- AK-105 AK-105
- BARI-1741 AK-105
- SC-435 SC-635.
- the compounds of the invention are administered in combination with a lipoprotein (a) antagonist such as, by way of example and by way of preference, gemcabene calcium (CI-1027) or nicotinic acid.
- a lipoprotein (a) antagonist such as, by way of example and by way of preference, gemcabene calcium (CI-1027) or nicotinic acid.
- Another object of the present invention are pharmaceutical compositions containing at least one compound of the invention, usually together with one or more inert, non-toxic, pharmaceutically suitable excipients, and their use for the purposes mentioned above.
- the compounds according to the invention can act systemically and / or locally.
- the compounds according to the invention can be administered in suitable administration forms.
- the compounds of the invention rapidly and / or modified donating application forms containing the compounds of the invention in crystalline and / or amorphized and / or dissolved form, such.
- Tablets uncoated or coated tablets, for example with enteric or delayed-release or insoluble coatings which control the release of the compound of the invention
- tablets or films / wafers rapidly breaking down in the oral cavity, films / lyophilisates
- capsules e.g. Soft gelatin capsules
- dragees granules, pellets, powders, emulsions, suspensions, aerosols or solutions.
- Parenteral administration can be accomplished by bypassing a resorption step (e.g., intravenously, intraarterially, intracardially, intraspinal, or intralumbar) or by resorting to absorption (e.g., intramuscularly, subcutaneously, intracutaneously, percutaneously, or intraperitoneally).
- a resorption step e.g., intravenously, intraarterially, intracardially, intraspinal, or intralumbar
- absorption e.g., intramuscularly, subcutaneously, intracutaneously, percutaneously, or intraperitoneally.
- parenteral administration are suitable as application forms u.a. Injection and infusion preparations in the form of solutions, suspensions, emulsions, lyophilisates or sterile powders.
- Inhalation medicaments including powder inhalers, nebulizers
- nasal drops solutions or sprays
- lingual, sublingual or buccal tablets films / wafers or capsules
- suppositories ear or eye preparations
- vaginal capsules aqueous suspensions (lotions, shake mixtures)
- lipophilic suspensions ointments
- creams transdermal therapeutic systems (eg plasters)
- milk pastes, foams, powdered powders, implants or stents.
- compositions according to the invention can be converted into the stated administration forms. This can be done in a conventional manner by mixing with inert, non-toxic, pharmaceutically suitable excipients.
- excipients e.g., microcrystalline cellulose, lactose, mannitol
- solvents e.g, liquid polyethylene glycols
- emulsifiers and dispersing or wetting agents e.g., sodium dodecylsulfate, polyoxysorbitanoleate
- binders e.g., polyvinylpyrrolidone
- synthetic and natural polymers e.g.
- Albumin e.g antioxidants such as ascorbic acid
- dyes eg inorganic pigments such as iron oxides
- Instrument MS Waters
- instrument HPLC Waters (column Waters X-Bridge C18, 18 mm x 50 mm, 5 ⁇
- eluent A water + 0.05% triethylamine
- eluent B acetonitrile (ULC) + 0.05% triethylamine
- UV detection DAD, 210 - 400 nm).
- the compound was prepared analogously to Example 13A using ethyl chloroformate. 77 mg of the target compound were obtained (21% of theory).
- the compound was prepared analogously to Example 1 using benzyl bromide. 7 mg of the target compound were obtained (13% of theory).
- Example 2A The compound was prepared in analogy to Example 1 from 89 mg (0.22 mmol) of the compound in Example 2A.
- the purification was carried out by preparative RP-HPLC (acetonitrile: water (+0.1% formic acid) gradient). 80 mg of the target compound were obtained (67% of theory).
- the compound was prepared in analogy to Example 1 from 100 mg (0.245 mmol) of the compound in Example 2A and 31 (0.25 mmol) 4-fluorobenzyl bromide.
- the purification was carried out by preparative RP-HPLC (acetonitrile / water (+0.1% formic acid) gradient). 92 mg of the target compound were obtained (72% of theory).
- the compound was prepared in analogy to Example 1 from 100 mg (0.245 mmol) of the compound in Example 2A and 29 (0.25 mmol) of benzyl bromide.
- the purification was carried out by preparative RP-HPLC (acetonitrile / water (+0.1% formic acid) gradient). 79 mg of the target compound were obtained (63% of theory).
- the compound was prepared in analogy to Example 1 from 108 mg (0.238 mmol) of the compound in Example 13A and 29 (0.24 mmol) of benzyl bromide.
- the purification was carried out by preparative RP-HPLC (acetonitrile / water (+0.1% formic acid) gradient). 67 mg of the target compound were obtained (52% of theory).
- the compound was prepared in analogy to Example 1 from 61 mg (0.14 mmol) of the compound in Example 14A and 17 (0.14 mmol) of benzyl bromide.
- the purification was carried out by preparative RP-HPLC (acetonitrile / water (+0.1% formic acid) gradient). 17 mg of the target compound were obtained (22% of theory).
- the compound was prepared in analogy to Example 1 from 86 mg (0.13 mmol) of the compound in Example 9A and 16 (0.13 mmol) of benzyl bromide.
- the purification was carried out by preparative RP-HPLC (acetonitrile / water (+0.1% formic acid) gradient). 50 mg of the target compound were obtained (73% of theory).
- the force of contraction is detected with Statham UC2 cells, amplified and digitized via A / D converters (DAS-1802 HC, Keithley Instruments Munich) and registered in parallel on chart recorders.
- DAS-1802 HC A / D converters
- phenylephrine is added cumulatively to the bath in increasing concentration.
- the substance to be examined is added in each subsequent course in increasing dosages and the height of the contraction is compared with the height of the contraction achieved in the last predistortion. This is used to calculate the concentration required to reduce the level of the control value by 50% (IC 50 value).
- the standard application volume is 5 ⁇ , the DMSO content in the bath solution corresponds to 0.1%.
- the system consists of 3 main components:
- Implantable transmitters Physiotel® telemetry transmitters
- Receiver Physiotel® Receiver
- DSI Data Exchange Matrix DSI Data Exchange Matrix
- Data acquisition computer are connected.
- the telemetry system allows a continuous recording of blood pressure heart rate and body movement on awake animals in their habitual habitat.
- the experimental animals are kept individually in macroion cages type 3 after transmitter implantation. You have free access to standard food and water. The day - night rhythm in the experimental laboratory is changed by room lighting at 6:00 in the morning and at 19:00 in the evening.
- the TAI 1 PA - C40 telemetry transmitters are surgically implanted into the experimental animals under aseptic conditions at least 14 days before the first trial.
- the animals so instrumented are repeatedly used after healing of the wound and ingrowth of the implant.
- the fasting animals are anesthetized with pentobabital (Nembutal, Sanofi: 50 mg / kg i.p.) and shaved and disinfected on the ventral side.
- pentobabital Nembutal, Sanofi: 50 mg / kg i.p.
- tissue adhesive VetBonD TM, 3M.
- the transmitter housing is fixed intraperitoneally to the abdominal wall musculature and the wound is closed in layers.
- a solvent-treated group of animals is used as a control.
- the existing telemetry measuring device is configured for 24 animals. Each trial is registered under a trial number (VYear month day).
- the instrumented rats living in the plant each have their own receiving antenna (1010 receivers, DSI).
- the implanted transmitters can be activated externally via a built-in magnetic switch. They will be put on the air during the trial run.
- the emitted signals can be recorded online by a data acquisition system (Dataquest TM A.R.T. for Windows, DSI) and processed accordingly. The storage of the data takes place in each case in a folder opened for this purpose which carries the test number.
- DBP Diastolic blood pressure
- MAP Arterial mean pressure
- the measured value acquisition is repeated computer-controlled in 5-minute intervals.
- the absolute value of the source data is corrected in the diagram with the currently measured barometric pressure (Ambient Pressure Reference Monitor, APR-1) and stored in individual data. Further technical details can be found in the extensive documentation of the manufacturer (DSI). Unless otherwise stated, the administration of the test substances will take place at 9 o'clock on the day of the experiment. Following the application, the parameters described above are measured for 24 hours.
- the data is smoothed over a presettable time by averaging (15 minutes average) and transferred as a text file to a disk.
- the presorted and compressed measured values are transferred to Excel templates and displayed in tabular form.
- the filing of the collected data takes place per experiment day in a separate folder that bears the test number. Results and test reports are sorted in folders and sorted by paper. literature
- the compounds according to the invention can be converted into pharmaceutical preparations as follows:
- composition
- the mixture of compound of the invention, lactose and starch is granulated with a 5% solution (m / m) of the PVP in water.
- the granules are mixed after drying with the magnesium stearate for 5 minutes.
- This mixture is compressed with a conventional tablet press (for the tablet format see above).
- a pressing force of 15 kN is used as a guideline for the compression.
- the rhodigel is suspended in ethanol, the compound according to the invention is added to the suspension. While stirring, the addition of water. Until the completion of the swelling of Rhodigels is stirred for about 6 h.
- the compound of the invention is suspended in the mixture of polyethylene glycol and polysorbate with stirring. The stirring is continued until complete dissolution of the compound according to the invention.
- i.v. solution The compound of the present invention is dissolved in a concentration below the saturation solubility in a physiologically acceptable solvent (e.g., isotonic saline, glucose solution 5%, and / or PEG 400 solution 30%). The solution is sterile filtered and filled into sterile and pyrogen-free injection containers.
- a physiologically acceptable solvent e.g., isotonic saline, glucose solution 5%, and / or PEG 400 solution 30%.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Epidemiology (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Hospice & Palliative Care (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Die vorliegende Anmeldung betrifft neue benzyl-substituierte Carbamate, Verfahren zu ihrer Herstellung, ihre Verwendung allein oder in Kombinationen zur Behandlung und/oder Prophylaxe von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prophylaxe von Krankheiten, insbesondere zur Behandlung und/oder Prophylaxe von Herz-Kreislauf-Erkrankungen.
Description
Benzyl-substituierte Carbamate und ihre Verwendung
Die vorliegende Anmeldung betrifft neue benzyl-substituierte Carbamate, Verfahren zu ihrer Herstellung, ihre Verwendung allein oder in Kombinationen zur Behandlung und/oder Prophylaxe von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prophylaxe von Krankheiten, insbesondere zur Behandlung und/oder Prophylaxe von Herz-Kreislauf-Erkrankungen.
Eines der wichtigsten zellulären Übertragungssysteme in Säugerzellen ist das cyclische Guanosin- monophosphat (cGMP). Zusammen mit Stickstoffmonoxid (NO), das aus dem Endothel freigesetzt wird und hormonelle und mechanische Signale überträgt, bildet es das NO/cGMP-System. Die Guanylatcyclasen katalysieren die Biosynthese von cGMP aus Guanosintriphosphat (GTP). Die bisher bekannten Vertreter dieser Familie lassen sich sowohl nach strukturellen Merkmalen als auch nach der Art der Liganden in zwei Gruppen aufteilen: Die partikulären, durch natriuretische Peptide stimulierbaren Guanylatcyclasen und die löslichen, durch NO stimulierbaren Guanylatcyclasen. Die löslichen Guanylatcyclasen bestehen aus zwei Untereinheiten und enthalten höchstwahrscheinlich ein Häm pro Heterodimer, das ein Teil des regulatorischen Zentrums ist. Dieses hat eine zentrale Bedeutung für den Aktivierungsmechanismus. NO kann an das Eisenatom des Häms binden und so die Aktivität des Enzyms deutlich erhöhen. Hämfreie Präparationen lassen sich hingegen nicht durch NO stimulieren. Auch Kohlenmonoxid (CO) ist in der Lage, an das Eisen-Zentralatom des Häms zu binden, wobei die Stimulierung durch CO deutlich geringer ist als die durch NO. Durch die Bildung von cGMP und der daraus resultierenden Regulation von Phosphodiesterasen, Ionenkanälen und Proteinkinasen spielt die Guanylatcyclase eine entscheidende Rolle bei unterschiedlichen physiologischen Prozessen, insbesondere bei der Relaxation und Proliferation glatter Muskelzellen, der Plättchenaggregation und -adhäsion, der neuronalen Signalübertragung sowie bei Erkrankungen, welche auf einer Störung der vorstehend genannten Vorgänge beruhen. Unter patho- physiologischen Bedingungen kann das NO/cGMP-System supprimiert sein, was zum Beispiel zu Bluthochdruck, einer Plättchenaktivierung, einer vermehrten Zellproliferation, endothelialer Dysfunktion, Arteriosklerose, Angina pectoris, Herzinsuffizienz, Myokardinfarkt, Thrombosen, Schlaganfall und sexueller Dysfunktion führen kann.
Eine auf die Beeinflussung des cGMP-Signalweges in Organismen abzielende NO-unabhängige Behandlungsmöglichkeit für derartige Erkrankungen ist aufgrund der zu erwartenden hohen Effizienz und geringen Nebenwirkungen ein vielversprechender Ansatz.
Zur therapeutischen Stimulation der löslichen Guanylatcyclase wurden bisher ausschließlich Verbindungen wie organische Nitrate verwendet, deren Wirkung auf NO beruht. Dieses wird durch Bio-
konversion gebildet und aktiviert die lösliche Guanylatcyclase durch Angriff am Eisen-Zentralatom des Häms. Neben den Nebenwirkungen gehört die Toleranzentwicklung zu den entscheidenden Nachteilen dieser Behandlungsweise.
In den letzten Jahren wurden einige Substanzen beschrieben, die die lösliche Guanylatcyclase direkt, d.h. ohne vorherige Freisetzung von NO stimulieren, wie beispielsweise 3-(5'-Hydroxymethyl-2'- furyl)-l-benzylindazol [YC-1 ; Wu et al., Blood 84 (1994), 4226; Mülsch et al, Brit. J. Pharmacol. 120 (1997), 681], Fettsäuren [Goldberg et al, J. Biol. Chem. 252 (1977), 1279], Diphenyl- iodonium-hexafluorophosphat [Pettibone et al., Eur. J. Pharmacol. 116 (1985), 307], Iso- liquiritigenin [Yu et al., Brit. J. Pharmacol. 114 (1995), 1587] sowie verschiedene substituierte Pyrazol-Derivate (WO 98/16223).
WO 2008/03 15 13 offenbart unter anderem lH-Pyrazolo[4,3-b]pyridine als Stimulatoren der löslichen Guanylatcyclase zur Behandlung von Herzkreislauferkrankungen. In WO 2005/030121 werden annellierte Pyrazole zur Behandlung von Krebserkrankungen beschrieben. WO 02/42300 beschreibt Pyrazolopyridine mit Carbamat-Substituenten zur Behandlung kardiovaskulärer Erkrankungen. WO 2011/119518 und WO 2011/115804 offenbaren Carbamat-substituierte Pyrimidine zur Behandlung von Herzkreislauferkrankungen.
Aufgabe der vorliegenden Erfindung war die Bereitstellung neuer Substanzen, die als potente Stimulatoren der löslichen Guanylatcyclase wirken, und sich daher zur Behandlung und/oder Prophylaxe von kardiovaskulären Erkrankungen eignen.
Gegenstand der vorliegenden Erfindung sind Verbindungen der allgemeinen Formel (I)

R4 für Wasserstoff, (Ci-C4)-Alkyl oder (d-C4)-Alkoxycarbonyl steht, R5 für Phenyl, Tetrahydronaphthalenyl, Naphthyl oder 5- bis 10-gliedriges Heteroaryl steht, wobei Phenyl, Tetrahydronaphthalenyl, Naphthyl und 5- bis 10-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig von einander ausgewählt aus der Gruppe Halogen, Nitro, Cyano, Difluormethyl, Trifluormethyl, (Ci-C6)-Alkyl, Hydroxy, Difluormethoxy, Trifluor- methoxy, (Ci-C4)-Alkoxy, Amino, Mono-(Ci-C )-alkylamino, Di-(Ci-C )-alkylamino, Hydroxycarbonyl, (Ci-C )-Alkoxycarbonyl, Aminocarbonyl, Mono-(Ci-C )-alkylamino- carbonyl, Di-(Ci-C )-alkylaminocarbonyl, (C3-C7)-Cycloalkylaminocarbonyl, Phenyl- sulfonylmethyl, Phenyl und Phenoxy substituiert sein können, worin (Ci-C6)-Alkyl mit einem Substituenten unabhängig voneinander ausgewählt aus der Gruppe Hydroxycarbonyl und (Ci-C )-Alkoxycarbonyl substituiert sein kann, und worin Phenyl und Phenoxy mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen und Cyano substituiert sein können,
R6 für (Ci-C6)-Alkyl oder Benzyl steht, wobei (Ci-C6)-Alkyl mit einem Substituenten Trifluormethyl substituiert ist, wobei (Ci-C6)-Alkyl mit 1 bis 3 Substituenten Fluor substituiert sein kann, und wobei Benzyl mit 1 bis 3 Substituenten Fluor substituiert ist, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
Erfindungsgemäße Verbindungen sind die Verbindungen der Formel (I) und deren Salze, Solvate und Solvate der Salze, die von Formel (I) umfassten Verbindungen der nachfolgend genannten Formeln und deren Salze, Solvate und Solvate der Salze sowie die von Formel (I) umfassten, nachfolgend als
Ausführungsbeispiele genannten Verbindungen und deren Salze, Solvate und Solvate der Salze, soweit es sich bei den von Formel (I) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt.
Als Salze sind im Rahmen der vorliegenden Erfindung physiologisch unbedenkliche Salze der erfin- dungsgemäßen Verbindungen bevorzugt. Umfasst sind auch Salze, die für pharmazeutische Anwendungen selbst nicht geeignet sind, jedoch beispielsweise für die Isolierung oder Reinigung der erfmdungsgemäßen Verbindungen verwendet werden können.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasserstoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure, Ethansulfonsäure, Toluolsulfonsäure, Benzolsulfonsäure, Naphthalindisulfonsäure, Ameisensäure, Essigsäure, Trifluoressigsäure, Propionsäure, Milchsäure, Weinsäure, Äpfelsäure, Zitronensäure, Fumarsäure, Maleinsäure und Benzoesäure.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen auch Salze üblicher Basen, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kaliumsalze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze) und Ammoniumsalze, abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C-Atomen, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, Ethyldiisopropylamin, Monoethanolamin, Diethanolamin, Triethanolamin, Dicyclohexylamin, Dimethylaminoethanol, Prokain, Dibenzylamin, N-Methyl- morpholin, Arginin, Lysin, Ethylendiamin und N-Methylpiperidin.
Als Solvate werden im Rahmen der Erfindung solche Formen der erfindungsgemäßen Verbindungen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungsmittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt. Als Solvate sind im Rahmen der vorliegenden Erfindung Hydrate bevorzugt.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in unterschiedlichen stereoisomeren Formen existieren, d.h. in Gestalt von Konfigurationsisomeren oder gegebenenfalls auch als Konformationsisomere (Enantiomere und/oder Diastereomere, einschließlich solcher bei Atropisomeren). Die vorliegende Erfindung umfasst deshalb die Enantiomere und Diastereo- mere und ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/ oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren; vorzugsweise werden hierfür chromatographische Verfahren verwendet, insbesondere die HPLC- Chromatographie an achiraler bzw. chiraler Phase.
Sofern die erfindungsgemäßen Verbindungen in tautomeren Formen vorkommen können, umfasst die vorliegende Erfindung sämtliche tautomere Formen.
Die vorliegende Erfindung umfasst auch alle geeigneten isotopischen Varianten der erfindungsgemäßen Verbindungen. Unter einer isotopischen Variante einer erfindungsgemäßen Verbindung wird hierbei eine Verbindung verstanden, in welcher mindestens ein Atom innerhalb der erfindungsgemäßen Verbindung gegen ein anderes Atom der gleichen Ordnungszahl, jedoch mit einer anderen Atommasse als der gewöhnlich oder überwiegend in der Natur vorkommenden Atommasse ausgetauscht ist. Beispiele für Isotope, die in eine erfindungsgemäße Verbindung inkorporiert werden können, sind solche von Wasserstoff, Kohlenstoff, Stickstoff, Sauerstoff, Phosphor, Schwefel, Fluor, Chlor, Brom und Iod, wie 2H (Deuterium), H (Tritium), 1 C, 14C, 15N, 170, 180, 2P, P, S, 4S, 5S, 6S, 18F, 6C1, 82Br, 12 I, 124I, 129I und 1 1I. Bestimmte isotopische Varianten einer erfindungsgemäßen Verbindung, wie insbesondere solche, bei denen ein oder mehrere radioaktive Isotope inkorporiert sind, können von Nutzen sein beispielsweise für die Untersuchung des Wirkmechanismus oder der Wirkstoff- Verteilung im Körper; aufgrund der vergleichsweise leichten Herstell- und Detektierbarkeit sind hierfür insbesondere mit H- oder 14C-Isotopen markierte Verbindungen geeignet. Darüber hinaus kann der Einbau von Isotopen, wie beispielsweise von Deuterium, zu bestimmten therapeutischen Vorteilen als Folge einer größeren metabolischen Stabilität der Verbindung führen, wie beispielsweise eine Verlängerung der Halbwertszeit im Körper oder eine Reduktion der erforderlichen Wirkdosis; solche Modifikationen der erfindungsgemäßen Verbindun- gen können daher gegebenenfalls auch eine bevorzugte Ausführungsform der vorliegenden Erfindung darstellen. Isotopische Varianten der erfindungsgemäßen Verbindungen können nach den dem Fachmann bekannten Verfahren hergestellt werden, so beispielsweise nach den weiter unten beschriebenen Methoden und den bei den Ausführungsbeispielen wiedergegebenen Vorschriften, indem entsprechende isotopische Modifikationen der jeweiligen Reagentien und/oder Ausgangsverbindungen eingesetzt werden.
Außerdem umfasst die vorliegende Erfindung auch Prodrugs der erfindungsgemäßen Verbindungen. Der Begriff "Prodrugs" bezeichnet hierbei Verbindungen, welche selbst biologisch aktiv oder inaktiv sein können, jedoch während ihrer Verweilzeit im Körper zu erfindungsgemäßen Verbindungen umgesetzt werden (beispielsweise metabolisch oder hydrolytisch). Im Rahmen der vorliegenden Erfindung haben die Substituenten, soweit nicht anders spezifiziert, die folgende Bedeutung:
Alkyl steht im Rahmen der Erfindung für einen linearen oder verzweigten Alkylrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methyl, Ethyl, n-Propyl,
Isopropyl, n-Butyl, iso-Butyl, 1-Methylpropyl, tert.-Butyl, n-Pentyl, iso-Pentyl, 1-Ethylpropyl, 1- Methylbutyl, 2-Methylbutyl, 3-Methylbutyl, n-Hexyl, 1-Methylpentyl, 2-Methylpentyl, 3- Methylpentyl, 4-Methylpentyl, 3,3-Dimethylbutyl, 1-Ethylbutyl und 2-Ethylbutyl.
Alkoxy steht im Rahmen der Erfindung für einen linearen oder verzweigten Alkoxyrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methoxy, Ethoxy, n-Propoxy, Isopropoxy, 1-Methylpropoxy, n-Butoxy, iso-Butoxy und tert.-Butoxy.
Alkoxycarbonyl stehen im Rahmen der Erfindung für einen linearen oder verzweigten Alkoxyrest mit 1 bis 4 Kohlenstoffatomen und einer am Sauerstoff angebundenen Carbonylgruppe. Beispielhaft und vorzugsweise seien genannt: Methoxycarbonyl, Ethoxycarbonyl, n-Propoxycarbonyl, Isopropoxycarbonyl und tert.-Butoxycarbonyl.
Mono-alkylamino steht im Rahmen der Erfindung für eine Amino-Gruppe mit einem linearen oder verzweigten Alkylsubstituenten, der 1 bis 4 Kohlenstoffatome aufweist. Beispielhaft und vorzugsweise seien genannt: Methylamino, Ethylamino, n-Propylamino, Isopropylamino und tert.- Butylamino. Di-alkylamino steht im Rahmen der Erfindung für eine Amino-Gruppe mit zwei gleichen oder verschiedenen linearen oder verzweigten Alkylsubstituenten, die jeweils 1 bis 4 Kohlenstoffatome aufweisen. Beispielhaft und vorzugsweise seien genannt: N,N-Dimethylamino, N,N-Diethylamino, N- Ethyl-N-methylamino, N-Methyl-N-n-propylamino, N-Isopropyl-N-n-propylamino und N-tert.-Butyl- N-methy lamino . Mono-alkylaminocarbonyl steht im Rahmen der Erfindung für eine Amino-Gruppe, die über eine Carbonylgruppe verknüpft ist und die einen linearen oder verzweigten Alkylsubstituenten mit 1 bis 4 Kohlenstoffatomen aufweist. Beispielhaft und vorzugsweise seien genannt: Methylaminocarbonyl, Ethylaminocarbonyl, «-Propylaminocarbonyl, Isopropylaminocarbonyl, «-Butylaminocarbonyl und tert. -Butylaminocarbonyl . Di-alkylaminocarbonyl steht im Rahmen der Erfindung für eine Amino-Gruppe, die über eine Carbonylgruppe verknüpft ist und die zwei gleiche oder verschiedene lineare oder verzweigte Alkylsubstituenten mit jeweils 1 bis 4 Kohlenstoffatomen aufweist. Beispielhaft und vorzugsweise seien genannt: N,N-Dimethylaminocarbonyl, N,N-Diethylaminocarbonyl, N-Ethyl-N-methylamino- carbonyl, N-Methyl-N-«-propylaminocarbonyl, N-«-Butyl-N-methylaminocarbonyl und N-tert.- Butyl-N-methylaminocarbonyl.
Cycloalkylaminocarbonyl steht im Rahmen der Erfindung für eine Amino-Gruppe, die über eine Carbonylgruppe verknüpft ist und die einen monocyclischen, gesättigten Carbocyclus mit 3 bis 7 Kohlenstoffatomen aufw e i s t . B e i s p i e l h aft un d v o rzu g s w e i s e s e i e n g e n annt : Cyclopropylaminocarbonyl, Cyclobutylaminocarbonyl, Cyclopentylaminocarbonyl, Cyclohexyl- aminocarbonyl und Cycloheptylaminocarbonyl.
Heteroaryl steht im Rahmen der Erfindung für einen mono- oder gegebenenfalls bicyclischen aromatischen Heterocyclus (Heteroaromaten) mit insgesamt 5 bis 10 Ringatomen, der bis zu drei gleiche oder verschiedene Ring-Heteroatome aus der Reihe N, O und/oder S enthält und über ein Ring-Kohlenstoffatom oder gegebenenfalls über ein Ring-Stickstoffatom verknüpft ist. Beispielhaft seien genannt: Furyl, Pyrrolyl, Thienyl, Pyrazolyl, Imidazolyl, Thiazolyl, Oxazolyl, Isoxazolyl, Iso- thiazolyl, Triazolyl, Oxadiazolyl, Thiadiazolyl, Pyridyl, Pyrimidinyl, Pyridazinyl, Pyrazinyl, Triazinyl, Benzofuranyl, Benzothienyl, Benzimidazolyl, Benzoxazolyl, Benzothiazolyl, Benzotriazolyl, Indolyl, Indazolyl, Chinolinyl, Isochinolinyl, Naphthyridinyl, Chinazolinyl, Chinoxalinyl, Phthalazinyl, Pyrazolo[3,4-b]pyridinyl. Bevorzugt sind monocyclische 5- oder 6- gliedrige Heteroaryl-Reste mit bis zu drei Ring-Heteroatomen aus der Reihe N, O und/oder S wie beispielsweise Furyl, Thienyl, Thiazolyl, Oxazolyl, Isothiazolyl, Isoxazolyl, Pyrazolyl, Imidazolyl, Triazolyl, Oxadiazolyl, Thiadiazolyl, Pyridyl, Pyrimidinyl, Pyridazinyl, Pyrazinyl, Triazinyl.
Halogen steht im Rahmen der Erfindung für Fluor, Chlor, Brom und Iod.
Wenn Reste in den erfindungsgemäßen Verbindungen substituiert sind, können die Reste, soweit nicht anders spezifiziert, ein- oder mehrfach substituiert sein. Im Rahmen der vorliegenden Erfindung gilt, dass für alle Reste, die mehrfach auftreten, deren Bedeutung unabhängig voneinander ist. Eine Substitution mit ein, zwei oder drei gleichen oder verschiedenen Substituenten ist bevorzugt.
Bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
R1 für Wasserstoff oder Fluor steht, R2 für Wasserstoff oder Amino steht,
R3 für (d-C3)-Alkyl steht,
R4 für Wasserstoff oder Methyl steht,
R5 für Phenyl, Thienyl, Thiazolyl, Pyridyl oder Chinolyl steht, wobei Phenyl, Thienyl, Thiazolyl, Pyridyl und Chinolyl mit 1 bis 3 Substituenten unabhängig von einander ausgewählt aus der Gruppe Fluor, Chlor, Nitro, Cyano,
Difluormethyl, Trifluormethyl, (Ci-C6)-Alkyl, Hydroxy, Difluormethoxy, Trifluormethoxy, Methoxy, Ethoxy, Amino, Methylamino, Ethylamino, Dimethylamino, Diethylamino, Methoxycarbonyl, Ethoxycarbonyl, Aminocarbonyl, Methylaminocarbonyl, Ethylamino- carbonyl, Dimethylaminocarbonyl, Diethylaminocarbonyl, Cyclopropylaminocarbonyl und Cyclobutylaminocarbonyl substituiert sein können, worin (Ci-C6)-Alkyl mit einem Substituenten unabhängig voneinander ausgewählt aus der Gruppe Hydroxycarbonyl, Methoxycarbonyl und Ethoxycarbonyl substituiert sein kann,
R6 für 3,3,3-Trifluorprop-l-yl, 3,3,4,4,4-Pentafluorbut-l-yl oder Benzyl steht, wobei Benzyl mit 1 oder 2 Substituenten Fluor substituiert ist, sowie ihre Salze, Solvate und Solvate der Salze.
Besonders bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
R1 für Wasserstoff oder Fluor steht, R2 für Wasserstoff oder Amino steht, R3 für Methyl, Ethyl oder iso-Propyl steht, R4 für Wasserstoff steht, R5 für Phenyl steht, wobei Phenyl mit 1 Substituenten Fluor substituiert sein kann, R6 für 2-Fluorbenzyl steht, sowie ihre Salze, Solvate und Solvate der Salze.
Bevorzugt im Rahmen der vorliegenden Erfindung sind auch Verbindungen der Formel (I), in welcher R1 für Fluor steht, sowie ihre Salze, Solvate und Solvate der Salze.
Bevorzugt im Rahmen der vorliegenden Erfindung sind auch Verbindungen der Formel (I), in welcher R1 für Wasserstoff steht, sowie ihre Salze, Solvate und Solvate der Salze.
Bevorzugt im Rahmen der vorliegenden Erfindung sind auch Verbindungen der Formel (I), in welcher R6 für 2-Fluorbenzyl steht, sowie ihre Salze, Solvate und Solvate der Salze.
Bevorzugt im Rahmen der vorliegenden Erfindung sind auch Verbindungen der Formel (I), in welcher R5 für Phenyl steht, wobei Phenyl mit 1 Substituenten Fluor substituiert sein kann, sowie ihre Salze, Solvate und Solvate der Salze.
Bevorzugt im Rahmen der vorliegenden Erfindung sind auch Verbindungen der Formel (I), in welcher R6 für 3,3,3-Trifluorprop-l-yl oder 3,3,4,4,4-Pentafluorbut-l-yl steht, sowie ihre Salze, Solvate und Solvate der Salze.
Die in den jeweiligen Kombinationen bzw. bevorzugten Kombinationen von Resten im einzelnen angegebenen Reste-Definitionen werden unabhängig von den jeweiligen angegebenen Kombinationen der Reste beliebig auch durch Reste-Definitionen anderer Kombinationen ersetzt.
Besonders bevorzugt sind Kombinationen von zwei oder mehreren der oben genannten Vorzug bereiche.
Weiterer Gegenstand der Erfindung ist ein Verfahren zur Herstellung der erfindungsgemäßen Verbindungen der Formel (I) dadurch gekennzeichnet, dass man eine Verbindung der Formel (II)




X1 für eine geeignete Abgangsgruppe, wie beispielsweise Mesylat, Tosylat oder Halogen, insbesondere Brom oder Iod, steht, umsetzt, und gegebenenfalls die resultierenden Verbindungen der Formel (I) gegebenenfalls mit den entsprechenden (i) Lösungsmitteln und/oder (ii) Säuren oder Basen in ihre Solvate, Salze und/oder Solvate der Salze überführt. Die Umsetzung (II) + (III) (IV) kann in einem inerten Lösungsmittel oder ohne Lösungsmittel erfolgen. Inerte Lösungsmittel für den Verfahrensschritt (II) + (III) — » (IV) sind beispielsweise
Alkohole wie Methanol, Ethanol, n-Propanol, Isopropanol, n-Butanol oder tert.-Butanol, Ether wie Diethylether, Dioxan, Tetrahydrofuran, Glykoldimethylether oder Diethylenglykoldimethylether, Halogenkohlenwasserstoffe wie Dichlormethan, Trichlormethan, Tetrachlormethan, Trichlorethylen oder Chlorbenzol, Kohlenwasserstoffe wie Benzol, Xylol, Toluol, Hexan, Cyclohexan oder Erdöl- fraktionen, oder andere Lösungsmittel wie Dimethylformamid (DMF), Dimethylsulfoxid (DMSO), NN'-Dimethylpropylenharnstoff (DMPU), N-Methylpyrrolidon (ΝΜΡ), Acetonitril oder auch Wasser. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt sind Dimethylformamid und Toluol sowie ein Gemisch aus Dimethylformamid und Toluol.
Geeignete Basen für den Verfahrensschritt (II) + (III)— » (IV) sind Alkalihydride wie Natriumhydrid, Alkalihydroxide wie beispielsweise Lithium-, Natrium- oder Kaliumhydroxid, Alkalicarbonate wie Lithium-, Natrium-, Kalium- oder Cäsiumcarbonat, Alkahhydrogencarbonate wie Natrium- oder Kaliumhydrogencarbonat, Alkalialkoholate wie Natrium- oder Kaliummethanolat, Natrium- oder Kaliumethanolat oder Kalium-tert.-butylat, Amide wie Natriumamid, Lithium-, Natrium- oder Kalium-bis-(trimethylsilyl)amid oder Lithiumdiisopropylamid, metallorganische Verbindungen wie Butyllithium oder Phenyllithium, oder organische Amine wie Triethylamin, Diisopropylethylamin, Pyridin, l,8-Diazabicyclo[5.4.0]undec-7-en (DBU) oder l ,5-Diazabicyclo[4.3.0]non-5-en (DBN). Bevorzugt ist Pyridin.
Die Reaktion (II) + (III)— > (IV) wird im Allgemeinen in einem Temperaturbereich von -10°C bis +30°C, bevorzugt bei 0°C bis +20°C durchgeführt. Die Umsetzung kann bei normalem, erhöhtem oder bei erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Inerte Lösungsmittel für den Verfahrensschritt (IV) + (V)— » (I) sind beispielsweise Alkohole wie Methanol, Ethanol, n-Propanol, Isopropanol, n-Butanol oder tert.-Butanol, Ether wie Diethylether, Dioxan, Tetrahydrofuran, Glykoldimethylether oder Diethylenglykoldimethylether, Halogenkohlen- Wasserstoffe wie Dichlormethan, Trichlormethan, Tetrachlormethan, Trichlorethylen oder Chlorbenzol, Kohlenwasserstoffe wie Benzol, Xylol, Toluol, Hexan, Cyclohexan oder Erdölfraktionen, oder andere Lösungsmittel wie Dimethylformamid (DMF), Dimethylsulfoxid (DMSO), NN'- Dimethylpropylenharnstoff (DMPU), N-Methylpyrrolidon (ΝΜΡ), Acetonitril oder auch Wasser. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt ist THF. Geeignete Basen für den Verfahrensschritt (IV) + (V)— » (I) sind Alkalihydride wie Natriumhydrid, Alkalihydroxide wie beispielsweise Lithium-, Natrium- oder Kaliumhydroxid, Alkalicarbonate wie Lithium-, Natrium-, Kalium- oder Cäsiumcarbonat, Alkahhydrogencarbonate wie Natrium- oder Kaliumhydrogencarbonat, Alkalialkoholate wie Natrium- oder Kaliummethanolat, Natrium- oder
Kaliumethanolat oder Kalium-tert.-butylat, Amide wie Natriumamid, Lithium-, Natrium- oder Kalium-bis-(trimethylsilyl)amid oder Lithiumdiisopropylamid, metallorganische Verbindungen wie Butyllithium oder Phenyllithium, oder organische Amine wie Triethylamin, Diisopropylethylamin, Pyridin, l,8-Diazabicyclo[5.4.0]undec-7-en (DBU) oder l,5-Diazabicyclo[4.3.0]non-5-en (DBN). Bevorzugt ist Lithium-bis-(trimethylsilyl)amid oder Natriumhydrid.
Die Reaktion (IV) + (V)— > (I) wird im Allgemeinen in einem Temperaturbereich von -10°C bis +30°C, bevorzugt bei 0°C bis +20°C durchgeführt. Die Umsetzung kann bei normalem, erhöhtem oder bei erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck. Die Verbindungen der Formel (III) und (V) sind kommerziell erhältlich, literaturbekannt oder können in Analogie zu literturbekannten Verfahren hergestellt werden.
Das zuvor beschriebene Herstellverfahren wird durch das nachfolgende Syntheseschema (Schema 1) beispielhaft verdeutlicht:
Schema 1

[a): Pyridin; b): NaH, THF].
Die Verbindung der Formel (II) kann hergestellt werden, indem man die Verbindung der Formel (VI)


X2 für eine geeignete Abgangsgruppe wie beispielsweise Halogen, Mesylat, Tosylat oder Triflat steht, zu einer Verbindung der Formel (VIII)

Die Reduktion (VIII)— » (II) erfolgt in Gegenwart eines geeigneten Katalysators in einem inerten Lösungsmittel, in einem Temperaturbereich von +20°C bis +40°C unter Wasserstoff-Normaldruck.
Inerte Lösungsmittel für die Reduktion (VIII)— » (II) sind beispielsweise Alkohole wie Methanol, Ethanol, n-Propanol, Isopropanol, n-Butanol oder tert.-Butanol, Ether wie Diethylether, Dioxan, Tetrahydrofuran, Glykoldimethylether oder Diethylenglykoldimethylether, oder andere Lösungsmittel wie Dimethylformamid (DMF), Dimethylsulfoxid (DMSO), N,N'-Dimethylpropylenharnstoff (DMPU), N-Methylpyrrolidon (ΝΜΡ), Pyridin, Acetonitril oder auch Wasser. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt sind DMF und Pyridin.
Geeignete Katalysatoren für die Umsetzung (VIII) — » (II) sind beispielsweise Palladium auf Aktivkohle, Platin auf Kohle, Palladiumhydroxid oder Raney-Nickel.
Die Reduktion (VIII)— » (II) kann alternativ mit einem Metall bzw. Metallsalz wie beispielsweise Eisen, Zink oder Zinn(II)chlorid in einer geeigneten Säure wie beispielsweise Chlorwasserstoff/ Salzsäure, Schwefelsäure, Phosphorsäure oder Essigsäure in einem Temperaturbereich von +20°C bis +140°C erfolgen.
Die Verbindungen der Formel (VI) sind literaturbekannt (siehe z.B. WO 2008/031513 Beispiel 4A), können in Analogie zu literaturbekannten Verfahren oder wie im vorliegenden experimentellen Teil beschrieben (Beispiel 3A bis 6A) hergestellt werden.
Das zuvor beschriebene Verfahren wird durch das nachstehende Syntheseschema (Schema 2) beispielhaft verdeutlicht:
Schema 2:

Die erfindungsgemäßen Verbindungen wirken als potente Stimulatoren der löslichen Guanylatcyclase, besitzen wertvolle pharmakologische Eigenschaften, und eignen sich daher zur Behandlung und/ oder Prophylaxe von Erkrankungen bei Menschen und Tieren.
Die erfindungsgemäßen Verbindungen bewirken eine Gefäßrelaxation und eine Hemmung der Thrombozytenaggregation und fuhren zu einer Blutdrucksenkung sowie zu einer Steigerung des koronaren Blutflusses. Diese Wirkungen sind über eine direkte Stimulation der löslichen Guanylatcyclase und einen intrazellulären cGMP-Anstieg vermittelt. Außerdem verstärken die erfindungsgemäßen Verbindungen die Wirkung von Substanzen, die den cGMP-Spiegel steigern, wie beispielsweise EDRF (endothelium-derived relaxing factor), NO-Donatoren, Protoporphyrin IX, Arachidon- säure oder Phenylhydrazin-Derivate.
Die erfindungsgemäßen Verbindungen können daher in Arzneimitteln zur Behandlung und/oder Prophylaxe von kardiovaskulären Erkrankungen wie beispielsweise Bluthochdruck, akute und chronische Herzinsuffizienz, koronare Herzerkrankung, stabile und instabile Angina pectoris, periphere und kardiale Gefäßerkrankungen, Arrhythmien, Rhythmusstörungen der Vorhöfe und der Kammern sowie Überleitungsstörungen wie beispielsweise atrio-ventrikuläre Blockaden Grad I-III (AB-B l o c k I-III), supraventrikuläre Tachyarrhythmie, Vorhofflimmern, Vorhoffflattern, Kammerflimmern, Kammerflattern, ventrikuläre Tachyarrhytmie, Torsade de pointes-Tachykardie, Extrasystolen des Vorhoffs und des Ventrikels, AV-junktionale Extrasystolen, Sick-Sinus Syndrom, Synkopen, AV-Knoten-Reentrytachykardie, Wolff-Parkinson-White-Syndrom, von akutem Koronar- Syndrom (ACS), autoimmune Herzerkrankungen (Perikarditis, Endokarditis, Valvolitis, Aortitis, Kardiomyopathien), kardiogenem Schock, Aneurysmen, Boxerkardiomyopathie (premature
ventricular contraction (PVC)), zur Behandlung und/oder Prophylaxe von thromboembolischen Erkrankungen und Ischämien wie myokardiale Ischämie, Myokardinfarkt, Hirnschlag, Schock, Herzhypertrophie, transistorischen und ischämischen Attacken, Präeklampsie, entzündliche kardiovaskuläre Erkrankungen, Spasmen der Koronararterien und peripherer Arterien, Ödembildung wie beispielsweise pulmonales Ödem, Hirnödem, renales Ödem oder Herzinsuffizienz-bedingtes Ödem, peripheren Durchblutungsstörungen, Reperfusionsschäden, arterielle und venöse Thrombosen, Mikroalbuminurie, Herzmuskelschwäche, endotheliale Dysfunktion, zur Verhinderung von Restenosen wie nach Thrombolysetherapien, percutan-transluminalen Angioplastien (PTA), transluminalen Koronarangioplastien (PTCA), Herztransplantationen und Bypass-Operationen, sowie mikro- und makrovaskuläre Schädigungen (Vasculitis), erhöhte Spiegel von Fibrinogen und von LDL geringer Dichte sowie erhöhte Konzentrationen von Plasminogenaktivator-Inhibitor 1 (PAI-1), sowie zur Behandlung und/oder Prophylaxe von erektiler Dysfunktion und weiblicher sexueller Dysfunktion eingesetzt werden.
Im Sinne der vorliegenden Erfindung umfasst der Begriff Herzinsuffizienz auch spezifischere oder verwandte Krankheitsformen wie akut dekompensierte Herzinsuffizienz, Rechtsherzinsuffizienz, Linksherzinsuffizienz, Globalinsuffizienz, ischämische Kardiomyopathie , dilatative Kardiomyopathie, hypertrophe Kardiomyopathie, idiopathische Kardiomyopathie, angeborene Herzfehler, Herzklappenfehler, Herzinsuffizienz bei Herzklappenfehlern, Mitralklappenstenose, Mitralklappeninsuffizienz, Aortenklappenstenose, Aortenklappeninsuffizienz, Trikuspidalstenose, Trikuspidalinsuffizienz, Pulmonalklappenstenose, Pulmonalklappeninsuffizienz, kombinierte Herzklappenfehler, Herzmuskelentzündung (Myokarditis), chronische Myokarditis, akute Myokarditis, virale Myokarditis, diabetische Herzinsuffizienz, alkoholtoxische Kardiomyopathie, kardiale Speichererkrankungen, diastolische Herzinsuffizienz sowie systolische Herzinsuffizienz.
Darüber hinaus können die erfindungsgemäßen Verbindungen auch zur Behandlung und/oder Prophylaxe von Arteriosklerose, Lipidstoffwechselstörungen, Hypolipoproteinämien, Dyslipidämien, Hypertriglyceridämien, Hyperlipidämien, Hypercholesterolämien, Abetelipoproteinämie, Sitosterolämie, Xanthomatose, Tangier Krankheit, Fettsucht (Adipositas), Fettleibigkeit (Obesitas) und von kombinierten Hyperlipidämien sowie des Metabolischen Syndroms eingesetzt werden.
Außerdem können die erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von primärem und sekundärem Raynaud-Phänomen, von Mikrozirkulationsstörungen, Claudicatio, peripheren und autonomen Neuropathien, diabetischen Mikroangiopathien, diabetischer Retinopathie, diabetischen Geschwüren an den Extremitäten, Gangren, CREST-Syndrom, Erythematose, Onychomykose, rheumatischen Erkrankungen sowie zur Förderung der Wundheilung verwendet werden.
Weiterhin eignen sich die erfindungsgemäßen Verbindungen zur Behandlung urologischer Erkrankungen wie beispielsweise benignes Prostata-Syndrom (BPS), benigne Prostata-Hyperplasie (BPH), benigne Prostata Vergrösserung (BPE), Blasenentleerungsstörung (BOO), untere Harnwegssyndrome (LUTS, einschließlich Feiines Urologisches Syndrom (FUS)), Erkrankungen des Urogenital-Systems einschliesslich neurogene überaktive Blase (OAB) und (IC), Inkontinenz (UI) wie beispielsweise Misch-, Drang-, Stress-, oder Überlauf-Inkontinenz (MUI, UUI, SUI, OUI), Beckenschmerzen, benigne und maligne Erkrankungen der Organe des männlichen und weiblichen Urogenital-Systems .
Weiterhin eignen sich die erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von Nierenerkrankungen, insbesondere von aktuer und chronischer Niereninsuffizienz, sowie von akutem und chronischem Nierenversagen. Im Sinne der vorliegenden Erfindung umfasst der Begriff Niereninsuffizienz sowohl akute als auch chronische Erscheinungsformen der Niereninsuffizienz, wie auch zugrundeliegende oder verwandte Nierenerkrankungen wie renale Hypoperfusion, intradialytische Hypotonie, obstruktive Uropathie, Glomerulopathien, Glomerulonephritis, akute Glomerulonephritis, Glomerulosklerose, tubulointerstitielle Erkrankungen, nephropathische Erkrankungen wie primäre und angeborene Nierenerkrankung, Nierenentzündung, immunlogische Nierenerkrankungen wie Nierentransplantatabstoßung, Immunkomplex-induzierte Nierenerkrankungen, durch toxische Substanzen induzierte Nephropathie, Kontrastmittel-induzierte Nephropathie, diabetische und nicht-diabetische Nephropathie, Pyelonephritis, Nierenzysten, Nephrosklerose, hypertensive Nephrosklerose und nephrotisches Syndrom, welche diagnostisch beispielsweise durch abnorm verminderte Kreatinin- und/oder Wasser-Ausscheidung, abnorm erhöhte Blutkonzentrationen von Harnstoff, Stickstoff, Kalium und/oder Kreatinin, veränderte Aktivität von Nierenenzymen wie z.B. Glutamylsynthetase, veränderte Urinosmolarität oder Urinmenge, erhöhte Mikroalbuminurie, Makroalbuminurie, Läsionen an Glomerula und Arteriolen, tubuläre Dilatation, Hyperphosphatämie und/oder die Notwendigkeit zur Dialyse charakterisiert werden können. Die vorliegende Erfindung umfasst auch die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von Folgeerscheinungen einer Niereninsuffizienz, wie beispielsweise Lungenödem, Herzinsuffizienz, Urämie, Anämie, Elektrolytstörungen (z.B. Hyperkalämie, Hyponaträmie) und Störungen im Knochen- und Kohlen- hydrat-Metabolismus.
Weiterhin eignen sich die erfindungsgemäßen Verbindungen auch zur Behandlung und/oder Prophylaxe von asthmatischen Erkrankungen, pulmonaler arterieller Hypertonie (PAH) und anderen Formen der pulmonalen Hypertonie (PH), der chronisch-obstruktive Lungenerkrankung (COPD), des akuten Atemwegssyndrom (ARDS), der akuten Lungenschädigung (ALI), der alpha- 1 -Antitrypsin-
Defizienz (AATD), der Lungenfibrose, des Lungenemphysem (z.B. durch Zigarettenrauch induziertes Lungenemphysem) und der zystischen Fibrose (CF).
Die in der vorliegenden Erfindung beschriebenen Verbindungen stellen auch Wirkstoffe zur Bekämpfung von Krankheiten im Zentralnervensystem dar, die durch Störungen des NO/cGMP- Systems gekennzeichnet sind. Insbesondere sind sie geeignet zur Verbesserung der Wahrnehmung, Konzentrationsleistung, Lernleistung oder Gedächtnisleistung nach kognitiven Störungen, wie sie insbesondere bei Situationen/Krankheiten/Syndromen auftreten wie "Mild cognitive impairment", altersassoziierten Lern- und Gedächtnisstörungen, altersassoziierten Gedächtnisverlusten, vaskulärer Demenz, Schädel-Hirn-Trauma, Schlaganfall, Demenz, die nach Schlaganfällen auftritt ("post stroke dementia"), post-traumatischem Schädel-Hirn-Trauma, allgemeinen Konzentrationsstörungen, Konzentrationsstörungen bei Kindern mit Lern- und Gedächtnisproblemen, Alzheimer'scher Krankheit, Demenz mit Lewy-Körperchen, Demenz mit Degeneration der Frontallappen einschliesslich des Pick's-Syndroms, Parkinson'scher Krankheit, progressiver nuclear palsy, Demenz mit corticobasaler Degeneration, Amyolateralsklerose (ALS), Huntington'scher Krankheit, Demyelinisation, Multipler Sklerose, Thalamischer Degeneration, Creutzfeld- Jacob-Demenz, HIV- Demenz, Schizophrenie mit Demenz oder Korsakoff-Psychose. Sie eignen sich auch zur Behandlung und/oder Prophylaxe von Erkrankungen des Zentralnervensystems wie Angst-, Spannungs- und Depressionszuständen, zentral-nervös bedingten Sexualdysfunktionen und Schlafstörungen sowie zur Regulierung krankhafter Störungen der Nahrungs-, Genuss- und Suchtmittelaufhahme. Weiterhin eignen sich die erfindungsgemäßen Verbindungen auch zur Regulation der cerebralen Durchblutung und stellen wirkungsvolle Mittel zur Bekämpfung von Migräne dar. Auch eignen sie sich zur Prophylaxe und Bekämpfung der Folgen cerebraler Infarktgeschehen (Apoplexia cerebri) wie Schlaganfall, cerebraler Ischämien und des Schädel-Hirn-Traumas. Ebenso können die erfindungsgemäßen Verbindungen zur Bekämpfung von Schmerzzuständen und Tinnitus eingesetzt werden.
Zudem besitzen die erfindungsgemäßen Verbindungen antiinflammatorische Wirkung und können daher als entzündungshemmende Mittel zur Behandlung und/oder Prophylaxe von Sepsis (SIRS), multiplem Organversagen (MODS, MOF), entzündlichen Erkrankungen der Niere, chronischen Darmentzündungen (IBD, Crohn s Disease, UC), Pankreatitis, Peritonitis, rheumatoiden Erkrankun- gen, entzündlichen Hauterkrankungen sowie entzündlichen Augenerkrankungen eingesetzt werden.
Des weiteren können die erfindungsgemäßen Verbindungen ebenfalls zur Behandlung und/ oder Prophylaxe von Autoimmunerkrankungen eingesetzt werden.
Weiterhin sind die erfindungsgemäßen Verbindungen zur Behandlung und/ oder Prophylaxe fibrotischer Erkrankungen der inneren Organe, wie beispielsweise der Lunge, des Herzens, der Niere, des Knochenmarks und insbesondere der Leber, sowie dermatologischer Fibrosen und fibrotischer Erkrankungen des Auges, geeignet. Im Sinne der vorliegenden Erfindungen umfasst der Begriff fibrotischer Erkrankungen insbesondere die folgenden Begriffe Leberfibrose, Leberzirrhose, Lungenfibrose, Endomyocardfibrose, Nephropathie, Glomerulonephritis, interstitielle Nierenfibrose, fibrotische Schäden in Folge von Diabetes, Knochenmarksfibrose und ähnliche fibrotische Erkrankungen, Sklerodermie, Morphaea, Keloide, hypertrophe Narbenbildung (auch nach chirurgischen Eingriffen), Naevi, diabetische Retinopathie und proliferative Vitroretinopathie. Weiterhin eignen sich die erfindungsgemäßen Verbindungen zur Bekämpfung postoperativer Narbenbildung, z.B. in Folge von Glaukom-Operationen.
Die erfindungsgemäßen Verbindungen können ebenfalls kosmetisch bei alternder und verhornender Haut eingesetzt werden.
Außerdem sind die erfindungsgemäßen Verbindungen zur Behandlung und/ oder Prophylaxe von Hepatitis, Neoplasma, Osteoporose, Glaukom und Gastroparese geeignet.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung sind die erfindungsgemäßen Verbindungen zur Verwendung in einem Verfahren zur Bhandlung und/ oder Prophylaxe von Herzinsuffizienz, Angina pectoris, Hypertonie, pulmonaler Hypertonie, Ischämien, Gefäßerkrankungen, thromboembolischen Erkrankungen und Arteriosklerose.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von Erkrankun- gen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen, unter Verwendung einer wirksamen Menge von mindestens einer der erfindungsgemäßen Verbindungen.
Die erfindungsgemäßen Verbindungen können allein oder bei Bedarf in Kombination mit anderen Wirkstoffen eingesetzt werden. Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, enthaltend mindestens eine der erfindungsgemäßen Verbindungen und einen oder mehrere weitere
Wirkstoffe, insbesondere zur Behandlung und/oder Prophylaxe der zuvor genannten Erkrankungen. Als geeignete Kombinationswirkstoffe seien beispielhaft und vorzugsweise genannt:
• organische Nitrate und NO-Donatoren, wie beispielsweise Natriumnitroprussid, Nitroglycerin, Isosorbidmononitrat, Isosorbiddinitrat, Molsidomin oder SIN-1, sowie inhalatives NO; · Verbindungen, die den Abbau von cyclischem Guanosinmonophosphat (cGMP) inhibieren, wie beispielsweise Inhibitoren der Phosphodiesterasen (PDE) 1, 2 und/oder 5, insbesondere PDE 5- Inhibitoren wie Sildenafil, Vardenafil und Tadalafil;
• antithrombotisch wirkende Mittel, beispielhaft und vorzugsweise aus der Gruppe der Thrombozytenaggregationshemmer, der Antikoagulantien oder der profibrinolytischen Substanzen; · den Blutdruck senkende Wirkstoffe, beispielhaft und vorzugsweise aus der Gruppe der Calcium- Antagonisten, Angiotensin AII-Antagonisten, ACE-Hemmer, Endothelin-Antagonisten, Renin- Inhibitoren, alpha-Rezeptoren-Blocker, beta-Rezeptoren-Blocker, Mineralocorticoid-Rezeptor- Antagonisten sowie der Diuretika; und/oder
• den Fettstoffwechsel verändernde Wirkstoffe, beispielhaft und vorzugsweise aus der Gruppe der Thyroidrezeptor-Agonisten, Cholesterinsynthese-Inhibitoren wie beispielhaft und vorzugsweise
HMG-CoA-Reduktase- oder Squalensynthese-Inhibitoren, der ACAT-Inhibitoren, CETP- Inhibitoren, MTP-Inhibitoren, PPAR-alpha-, PPAR-gamma- und/oder PPAR-delta-Agonisten, Cholesterin-Abso tionshemmer, Lipase-Inhibitoren, polymeren Gallensäureadsorber, Gallensäure-Reabsoiptionshemmer und Lipoprotein(a)-Antagonisten. Unter antithrombotisch wirkenden Mittel werden vorzugsweise Verbindungen aus der Gruppe der Thrombozytenaggregationshemmer, der Antikoagulantien oder der profibrinolytischen Substanzen verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thrombozytenaggregationshemmer, wie beispielhaft und vorzugsweise Aspirin, Clopidogrel, Ticlopidin oder Dipyridamol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thrombin-Inhibitor, wie beispielhaft und vorzugsweise Ximelagatran, Dabigatran, Melagatran, Bivalirudin oder Clexane, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem GPIIb/IIIa-Antagonisten, wie beispielhaft und vorzugsweise Tirofiban oder Abciximab, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Faktor Xa-Inhibitor, wie beispielhaft und vorzugsweise Rivaroxaban (BAY 59-7939), DU-176b, Apixaban, Otamixaban, Fidexaban, Razaxaban, Fondaparinux, Idrapa- rinux, PMD-31 12, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 oder SSR-128428, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit Heparin oder einem low molecular weight (LMW)-Heparin-Derivat verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Vitamin K-Antagonisten, wie beispielhaft und vorzugsweise Coumarin, verabreicht.
Unter den Blutdruck senkenden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der Calcium- Antagonisten, Angiotensin AII-Antagonisten, ACE-Hemmer, Endothelin- Antagonisten, Renin-Inhibitoren, alpha-Rezeptoren-Blocker, beta-Rezeptoren-Blocker, Mineralocorticoid-Rezep- tor-Antagonisten sowie der Diuretika verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Calcium-Antagonisten, wie beispielhaft und vorzugsweise Nifedipin, Amlodipin, Verapamil oder Diltiazem, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem alpha- 1 -Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Prazosin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem beta-Rezeptoren-Blocker, wie beispielhaft und vorzugsweise Propranolol, Atenolol, Timolol, Pindolol, Alprenolol, Oxprenolol, Penbutolol, Bupranolol, Metipranolol, Nadolol, Mepindolol, Carazalol, Sotalol, Metoprolol, Betaxolol, Celiprolol, Bisoprolol, Carteolol, Esmolol, Labetalol, Carvedilol, Adaprolol, Landiolol, Nebivolol, Epanolol oder Bucindolol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Angiotensin AII-Antagonisten, wie beispielhaft und vorzugsweise Losartan, Candesartan, Valsartan, Telmisartan oder Embursatan, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACE-Hemmer, wie beispielhaft und vorzugsweise Enalapril, Captopril, Lisinopril, Ramipril, Delapril, Fosinopril, Quinopril, Perindopril oder Trandopril, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Endothelin-Antagonisten, wie beispielhaft und vorzugsweise Bosentan, Darusentan, Ambrisentan oder Sitaxsentan, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Renin-Inhibitor, wie beispielhaft und vorzugsweise Aliskiren, SPP-600 oder SPP-800, verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Mineralocorticoid-Rezeptor-Antagonisten, wie beispielhaft und vorzugsweise Spironolacton oder Eplerenon, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Diuretikum, wie beispielhaft und vorzugsweise Furosemid, verabreicht. Unter den Fettstoffwechsel verändernden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der CETP-Inhibitoren, Thyroidrezeptor-Agonisten, Cholesterinsynthese-Inhibitoren wie HMG-CoA-Reduktase- oder Squalensynthese-Inhibitoren, der ACAT-Inhibitoren, MTP -Inhibitoren, PPAR-alpha-, PPAR-gamma- und/oder PPAR-delta-Agonisten, Cholesterin-Absoiptionshemmer, polymeren Gallensäureadsorber, Gallensäure-Reabsoiptionshemmer, Lipase-Inhibitoren sowie der Lipoprotein(a)-Antagonisten verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem CETP -Inhibitor, wie beispielhaft und vorzugsweise Dalcetrapib, BAY 60- 5521, Anacetrapib oder CETP-vaccine (CETi-1), verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Thyroidrezeptor-Agonisten, wie beispielhaft und vorzugsweise D- Thyroxin, 3,5,3'-Triiodothyronin (T3), CGS 23425 oder Axitirome (CGS 26214), verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem HMG-CoA-Reduktase-Inhibitor aus der Klasse der Statine, wie beispielhaft und vorzugsweise Lovastatin, Simvastatin, Pravastatin, Fluvastatin, Atorvastatin, Rosuvastatin oder Pitavastatin, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Squalensynthese-Inhibitor, wie beispielhaft und vorzugsweise BMS- 188494 oder TAK-475, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACAT-Inhibitor, wie beispielhaft und vorzugsweise Avasimibe, Melinamide, Pactimibe, Eflucimibe oder SMP-797, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem MTP-Inhibitor, wie beispielhaft und vorzugsweise Implitapide, BMS- 201038, R-103757 oder JTT-130, verabreicht. Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPAR-gamma-Agonisten, wie beispielhaft und vorzugsweise Pioglitazone oder Rosiglitazone, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPAR-delta-Agonisten, wie beispielhaft und vorzugsweise GW 501516 oder BAY 68-5042, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Cholesterin-Absorptionshemmer, wie beispielhaft und vorzugsweise Ezetimibe, Tiqueside oder Pamaqueside, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Lipase-Inhibitor, wie beispielhaft und vorzugsweise Orlistat, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem polymeren Gallensäureadsorber, wie beispielhaft und vorzugsweise Cholestyramin, Colestipol, Colesolvam, CholestaGel oder Colestimid, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Gallensäure-Reabsorptionshemmer, wie beispielhaft und vorzugsweise ASBT (= IBAT)-Inhibitoren wie z.B. AZD-7806, S-8921, AK-105, BARI-1741, SC-435 oder SC- 635, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Lipoprotein(a)-Antagonisten, wie beispielhaft und vorzugsweise Gemcabene calcium (CI-1027) oder Nicotinsäure, verabreicht.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken. Die erfindungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctival, otisch oder als Implantat bzw. Stent.
Für diese Applikationswege können die erfindungsgemäßen Verbindungen in geeigneten Applika- tionsformen verabreicht werden.
Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende, die erfindungsgemäßen Verbindungen schnell und/oder modifiziert abgebende Applikationsformen, die die erfindungsgemäßen Verbindungen in kristalliner und/oder amorphisierter und/oder gelöster Form enthalten, wie z.B. Tabletten (nicht-überzogene oder überzogene Tabletten, beispielsweise mit magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die Freisetzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfallende Tabletten oder Filme/Oblaten, Filme/Lyophylisate, Kapseln (beispielsweise Hart- oder Weichgelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen. Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (z.B. intramuskulär, subcutan, intracutan, percutan oder intraperitoneal). Für die parenterale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusionszubereitungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern. Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulverinhalatoren, Nebulizer), Nasentropfen, -lösungen oder -sprays, lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapseln, Suppositorien, Ohren- oder Augen- präparationen, Vaginalkapseln, wäßrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspensionen, Salben, Cremes, transdermale therapeutische Systeme (z.B. Pflaster), Milch, Pasten, Schäume, Streupuder, Implantate oder Stents.
Bevorzugt sind die orale oder parenterale Applikation, insbesondere die orale Applikation.
Die erfindungsgemäßen Verbindungen können in die angeführten Applikationsformen überführt werden. Dies kann in an sich bekannter Weise durch Mischen mit inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a. Trägerstoffe (beispielsweise mikrokristalline Cellulose, Lactose, Mannitol), Lösungsmittel (z.B. flüssige Poly- ethylenglycole), Emulgatoren und Dispergier- oder Netzmittel (beispielsweise Natriumdodecylsulfat, Polyoxysorbitanoleat), Bindemittel (beispielsweise Polyvinylpyrrolidon), synthetische und natürliche Polymere (beispielsweise Albumin), Stabilisatoren (z.B. Antioxidantien wie beispielsweise Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisenoxide) und Geschmacks- und/oder Geruchskorrigentien. Im Allgemeinen hat es sich als vorteilhaft erwiesen, bei parenteraler Applikation Mengen von etwa 0.001 bis 1 mg/kg, vorzugsweise etwa 0.01 bis 0.5 mg/kg Körpergewicht zur Erzielung wirksamer Ergebnisse zu verabreichen. Bei oraler Applikation beträgt die Dosierung etwa 0.01 bis 100 mg/kg, vorzugsweise etwa 0.01 bis 20 mg/kg und ganz besonders bevorzugt 0.1 bis 10 mg/kg Körpergewicht. Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindestmenge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die nachfolgenden Ausführungsbeispiele erläutern die Erfindung. Die Erfindung ist nicht auf die Beispiele beschränkt.
Die Prozentangaben in den folgenden Tests und Beispielen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
A. Beispiele
Abkürzungen und Akronyme: aq. wässrige Lösung
ber. Berechnet
DC Dünnschichtchromatographie
DCI direkte chemische Ionisation (bei MS)
DMF Dimethylformamid
DMSO Dimethylsulfoxid
d. Th. der Theorie (bei Ausbeute)
eq. Äquivalent(e)
ESI Elektrospray-Ionisation (bei MS)
Et Ethyl
gef. Gefunden
h Stunde(n)
HPLC Hochdruck-, Hochleistungsflüssigchromatographie
HRMS hochaufgelöste Massenspektrometrie
konz. konzentriert
LC/MS Flüssigchromatographie-gekoppelte Massenspektrometrie
LiHMDS Lithiumhexamethyldisilazid
Me Methyl
min Minute(n)
MS Massenspektrometrie
MTBE Methyl-tert.-butylether
NMR Kernresonanzspektrometrie
Pd2dba3 Tris-(dibenzylidenaceton)-dipalladium
Ph Phenyl
RP Reversed phase
RT Raumtemperatur
Rt Retentionszeit (bei HPLC)
THF Tetrahydrofuran
UV Ultraviolett-Spektrometrie
v/v Volumen zu Volumen- Verhältnis (einer Lösung)
XPHOS Dicyclohexyl-(2',4',6'-triisopropylbiphenyl-2-yl)-phosphin
LC/MS-Methoden:
Methode 1 (LC-MS):
Instrument: Waters ACQUITY SQD UPLC System; Säule: Waters Acquity UPLC HSS T3 1,8μ 50 x 1mm; Eluent A: 1 1 Wasser + 0.25 ml 99%ige Ameisensäure , Eluent B: 1 1 Acetonitril + 0.25 ml 99%ige Ameisensäure; Gradient: 0.0 min 90% A -> 1.2 min 5% A -> 2.0 min 5% A; Ofen: 50°C; Fluss: 0.40 ml/min; UV-Detektion: 210 - 400 nm.
Methode 2 (LC-MS):
Instrument: Micromass Quattro Micro MS mit HPLC Agilent Serie 1100; Säule: Thermo Hypersil GOLD 3μ 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 100% A -> 3.0 min 10% A -> 4.0 min 10% A -> 4.01 min 100% A (Fluss 2.5 ml/min) -> 5.00 min 100% A; Ofen: 50°C; Fluss: 2 ml/min; UV-Detektion: 210 nm.
Methode 3 (LC-MS):
Instrument MS: Waters SQD; Instrument HPLC: Waters UPLC; Säule: Zorbax SB-Aq (Agilent), 50 mm x 2.1 mm, 1.8 μιη; Eluent A: Wasser + 0.025% Ameisensäure, Eluent B: Acetonitril (ULC) + 0.025% Ameisensäure; Gradient: 0.0 min 98%A - 0.9 min 25%A - 1.0 min 5%A - 1.4 min 5%A - 1.41 min 98%A - 1.5 min 98%A; Ofen: 40°C; Fluss: 0.600 ml/min; UV-Detektion: DAD; 210 nm.
Methode 4 (LC-MS):
Instrument MS: Waters, Instrument HPLC: Waters (Säule Waters X-Bridge C18, 18 mm x 50 mm, 5 μιη, Eluent A: Wasser + 0.05% Triethylamin, Eluent B: Acetonitril (ULC) + 0.05% Triethylamin, Gradient: 0.0 min 95 %A - 0.15 min 95 %A - 8.0 min 5%A - 9.0 min 5%A; Fluss: 40 ml/min; UV- Detektion: DAD; 210 - 400 nm).
Methode 5 (LC-MS):
Instrument MS: Waters, Instrument HPLC: Waters (Säule Phenomenex Luna 5μ C 18(2) 100A, AXIA Tech. 50 x 21.2 mm, Eluent A: Wasser + 0.05% Ameisensäure, Eluent B: Acetonitril (ULC) + 0.05% Ameisensäure, Gradient: 0.0 min 95%A - 0.15 min 95%A - 8.0 min 5%A - 9.0 min 5%A; Fluss: 40 ml/min; UV-Detektion: DAD; 210 - 400 nm).
Ausgangsverbindungen und Intermediate:
Beispiel 1A
2-[3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-4,5,6-triamin

Die Synthese dieser Verbindung ist beschrieben in WO 2008/031513, Beispiel 8.
Beispiel 2A
Methyl-{4,6-diamino-2-[3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-5-yl}carbamat

Die Synthese dieser Verbindung ist beschrieben in WO 2008/031513, Beispiel 9.
Beispiel 3A
3 , 5 -Difluorpyridin-2-carbonylchlorid

Eine Suspension von 5.00 g (31.4 mmol) 3, 5-Difluorpyridin-2 -carbonsäure in Thionylchlorid (21 mL) wurde 5 h zum Rückfluss erhitzt. Die Lösung wurde eingeengt, der Rückstand zweimal in wenig Toluol aufgenommen und wieder eingeengt. Man erhielt 3.80 g eines Feststoffs, der ohne weitere Reinigung direkt weiter umgesetzt wurde.
Beispiel 4A
Methyl-3 -(3 ,5 -difluo yridin-2-yl)-2-(2-fluorphenyl)-3 -oxopropanoat

In THF (30 mL) wurden unter Argon 21.4 mL (21.4 mmol) Lithiumhexamethyldisilazid (1.0 M in THF) vorgelegt und bei -78°C eine Lösung von 3.00 g (17.8 mmol) 2-Fluorphenylessigsäure- methylester in THF (15 mL) zugetropft. Die Reaktionsmischung wurde 1 h bei -78°C gerührt und anschließend eine Lösung von 3.80 g (21.4 mmol) der Verbindung aus Beispiel 3A in THF (15 mL) zugetropft. Die Lösung wurde lh bei -78°C gerührt, danach auf RT gebracht und portionsweise mit gesättigter wässriger Ammoniumchlorid-Lösung versetzt. Die Mischung wurde mit Wasser verdünnt und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mit MTBE verrührt, der Feststoff abfiltriert und das Filtrat eingeengt. Kieselgelchromatographie (Laufmittel: Cyclohexan- Ethylacetat 30: 1, 20: 1) des Rückstands lieferte 3.66 g (87%-ige Reinheit, 57 % d. Th.) der Titelverbindung. Das Rohprodukt wurde ohne weitere Reinigung umgesetzt.
LC-MS (Methode 1): R, = 1.05 min; MS (ESIpos): m/z = 310 (M+H)+
Ή-NMR (400 MHz, DMSO-d6): δ = 3.66 (s, 3H), 6.25 (s, 1H), 7.20 - 7.28 (m, 4H), 7.31 - 7.38 (m, 1H), 8.15 - 8.23 (m, 1H), 8.68 - 8.71 (m, 1H).
Beispiel 5A l-(3,5-Difluo yridin-2-yl)-2-(2-fluo henyl)ethanon

In DMSO (37 mL) wurden 1 1.65 g (37.67 mmol) der Verbindung aus Beispiel 4A vorgelegt. Anschließend wurden 2.42 g (41.44 mmol) Natriumchlorid sowie Wasser (7 mL) zugefügt und die Mischung 30 min bei 150°C in der Mikrowelle gerührt. Die Reaktionsmischung wurde mit Ethylacetat verdünnt, die organische Phase dreimal mit Wasser sowie einmal mit gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Man erhielt 9.07 g (89%ig, 85 % d. Th.) der gewünschten Verbindung als Feststoff, der ohne weitere Reinigung umgesetzt wurde.
LC-MS (Methode 1): R, = 1.05 min; MS (ESIpos): m/z = 252 (M+H)+
1H-NMR (400 MHz, DMSO-d6): δ = 4.53 (s, 2H), 7.15 - 7.22 (m, 2H), 7.30 - 7.37 (m, 2H), 8.1 1 - 8.18 (m, 1H), 8.70 - 8.72 (m, 1H).
Beispiel 6A
6-Fluor-3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin

LC-MS (Methode 2): R, = 1.87 min; MS (ESIpos): m/z = 246 (M+H)+
Ή-NMR (400 MHz, DMSO-d6): δ = 4.33 (s, 2H), 7.06 - 7.12 (m, 1H), 7.12 - 7.19 (m, 1H), 7.22 - 7.29 (m, 1H), 7.29 - 7.35 (m, 1H), 7.87 (dd, 1H), 7.84 - 7.89 (m, 1H), 8.48 - 8.51 (br. s, 1H).
Beispiel 7A 2-[6-Fluor-3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]-5-nitropyrimidin-4,6-diamin

In DMF (3.5 mL) wurden 156 mg (ca. 66% ige Reinheit, 0.636 mmol) der Verbindung aus Beispiel 6A gelöst, anschließend 28 mg (0.70 mmol) Natriumhydrid (60%ig in Mineralöl) zugefügt und die Mischung 2 h bei RT gerührt. Danach wurden 115 mg (0.604 mmol) 2-Chlor-5-nitropyrimidin-4,6- diamin (Synthese: Helvetica Chimica Acta (1951), 34, 835-40) zugefügt und die Reaktionsmischung für 1 h bei 80°C nachgerührt. Nach Abkühlen auf RT wurde die Mischung auf Wasser gegeben. Die so erhaltene Suspension wurde filtriert, der Feststoff mehrfach mit Wasser
gewaschen und anschließend im Hochvakuum getrocknet. Man erhielt 196 mg (77 % d. Th.) der Titelverbindung als Feststoff.
LC-MS (Methode 2): R, = 2.13 min; MS (ESIpos): m/z = 399 (M+H)+ Beispiel 8A 2-[6-Fluor-3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-4,5,6-triamin

In Pyridin (22 mL) wurden 196 mg (0.492 mmol) der Verbindung aus Beispiel 7A vorgelegt, anschließend 74 mg Palladium auf Kohle (10%ig w/w) zugefügt und die Mischung über Nacht bei Wasserstoff-Normaldruck hydriert. Anschließend wurde die Reaktionsmischung durch Kieselgur filtriert, mit Ethanol nachgewaschen und das Filtrat eingeengt. Der Rückstand wurde mit Ethanol bei 50°C verrührt, der Feststoff abfiltriert und im Hochvakuum getrocknet. Man erhielt 107 mg (58 % d. Th.) der Titelverbindung.
LC-MS (Methode 1): R, = 0.85 min; MS (ESIpos): m/z = 369 (M+H)+
Ή-NMR (400 MHz, DMSO-d6): δ = 3.76 (s, 2H), 4.39 (s, 2H), 6.12 (s, 4H), 7.07 - 7.14 (m, 1H), 7.14 - 7.21 (m, 1H), 7.23 - 7.31 (m, 1H), 7.31 - 7.37 (m, 1H), 8.58 - 8.62 (m, 1H), 8.94 (dd, 1H).
Beispiel 9A
Methyl-{4,6-diamino-2-[6-fluor-3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-5- yl}carbamat

In Pyridin (55 mL) wurden unter Argon 1.00 g (2.72 mmol) der Verbindung aus Beispiel 8A vorgelegt, die Mischung auf 0°C abgekühlt und anschließend 228 (2.72 mmol) Chlorameisensäuremethyle ster (Lösung in 10 mL Dichlormethan) zugetropft. Die Reaktionsmischung wurde über Nacht bei RT weiter gerührt und anschließend eingeengt. Der Rückstand wurde mit Ethanol ausgerührt, der Feststoff abfiltriert und bei 50°C im Hochvakuum getrocknet. Man erhielt 873 mg (75 % d. Th.) der Zielverbindung als Feststoff. Das Filtrat wurde erneut eingeengt und präparativer RP-HPLC (AcetonitrikWasser (+0.1% Ameisensäure) - Gradient) des Rückstands lieferte weitere 180 mg (16 % d. Th.) der Zielverbindung. LC-MS (Methode 1): R, = 0.88 min; MS (ESIpos): m/z = 427 (M+H)+
1H-NMR (400 MHz, DMSO-d6, Rotamerengemisch): δ [ppm]= 3.48 - 3.67 (m, 3H), 4.40 (s, 2H), 6.45 (br. s, 4H), 7.08 - 7.14 (m, 1H), 7.15 - 7.21 (m, 1H), 7.24 - 7.31 (m, 1H), 7.31 - 7.37 (m, 1H), 7.60 (br. s, 0.2H), 7.90 (br. s, 0.8H), 8.61 - 8.65 (m, 1H), 9.04 (dd, 1H).
Beispiel 10A 3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin

Beispiel IIA
2-[3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]-5-nitropyrimidin-4-amin

Beispiel 12A
2-[3-(2-Fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-4,5-diamin

In Pyridin (97 mL) wurden 793 mg (2.17 mmol) der Verbindung aus Beispiel I IA vorgelegt, anschließend 328 mg Palladium auf Kohle (10%ig w/w) zugefügt und die Mischung über Nacht bei Wasserstoff-Normaldruck hydriert. Anschließend wurde die Reaktionsmischung durch Kieselgur filtriert, mit Ethanol nachgewaschen und das Filtrat eingeengt. Der Rückstand wurde an Kieselgel (Dichlormethan: Methanol 30: 1 —► 20: 1) chromatographiert und die Produktfraktionen eingeengt. Man erhielt 330 mg (83%-ige Reinheit) der gewünschten Verbindung. Das Rohprodukt wurde ohne weitere Reinigung umgesetzt.
LC-MS (Methode 1): R, = 0.75 min; MS (EIpos): m/z = 336 [M+H]+. Beispiel 13A
Isopropyl-{4,6-diamino-2-[6-fluor-3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-5- yl}carbamat

LC-MS (Methode 1): R, = 0.97 min; MS (EIpos): m/z = 455 [M+H]+.
Ή-NMR (400 MHz, DMSO-d6, Rotamerengemisch): δ [ppm] = 1.06 - 1.31 (m, 6H), 4.40 (s, 2H), 4.77 - 4.85 (m, 1H), 6.39 (br. s, 4H), 7.08 - 7.14 (m, 1H), 7.15 - 7.21 (m, 1H), 7.24 - 7.31 (m, 1H), 7.31 - 7.38 (m, 1H), 7.43 - 7.52 (m, 0.25H), 7.78 - 7.86 (m, 0.75H), 8.61 - 8.65 (m, 1H), 9.01 - 9.09 (m, 1H).
Beispiel 14A
Ethyl-{4,6-diamino-2-[6-fluor-3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-5- yl}carbamat

Die Verbindung wurde analog zu Beispiel 13A unter Verwendung von Chlorameisensäureethylester hergestellt. Es wurden 77 mg der Zielverbindung erhalten (21 % d. Th.).
LC-MS (Methode 1): R, = 0.92 min; MS (EIpos): m/z = 441 [M+H]+.
Ή-NMR (400 MHz, DMSO-d6, Rotamerengemisch): δ [ppm] = 1.06 - 1.29 (m, 3H), 3.98 - 4.11 (m, 2H), 4.40 (s, 2H), 6.44 (br. s, 4H), 7.07 - 7.14 (m, 1H), 7.15 - 7.21 (m, 1H), 7.24 - 7.31 (m, 1H), 7.31 - 7.37 (m, 1H), 7.54 (br. s, 0.25H), 7.88 (br. s, 0.75H), 8.61 - 8.65 (m, 1H), 9.02 - 9.08 (m, 1H).
Beispiel 15A
Methyl-{4-amino-2-[3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-5-yl}carbamat

In Pyridin (12 mL) wurden unter Argon 330 mg (0.98 mmol) der Verbindung aus Beispiel 12A vorgelegt, die Mischung auf 0°C abgekühlt und anschließend 84 ( 1 .08 mmol) Chlorameisensäuremethylester zugetropft. Die Reaktionsmischung wurde über Nacht bei RT weiter gerührt und anschließend mit Ethylacetat versetzt. Die organische Phase wurde zweimal mit gesättigter wässriger Natriumhydrogencarbonatlösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer RP-HPLC (AcetonitrikWasser (+0.1% Ameisensäure) - Gradient) getrennt und lieferte 241 mg (62 % d. Th.) der Zielverbindung als farblosen Feststoff.
LC-MS (Methode 1): R, = 0.85 min; MS (EIpos): m/z = 394 [M+H]+.
Ή-NMR (400 MHz, DMSO-d6): δ [ppm] = 3.67 (s, 3H), 4.44 (s, 2H), 7.07 - 7.14 (m, 1H), 7.15 - 7.22 (m, 1H), 7.23 - 7.41 (m, 3H), 7.51 - 7.58 (m, 1H), 8.24 (br. s, 1H), 8.61 - 8.66 (m, 1H), 8.75 (br. s, 1H), 8.98 - 9.08 (m, 1H).
Ausführungsbeispiele:
Beispiel 1
Methyl-{4-amino-2-[3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-5-yl}(2- fluorbenzyl)carbamat

47 mg (0.12 mmol) der Verbindung aus Beispiel 15A wurden in 3 ml THF vorgelegt und auf 0°C abgekühlt. Danach wurden 5 mg (0.12 mmol) Natriumhydrid (60 %ig in Mineralöl) zugegeben und die Mischung weitere 30 min bei 0°C nachgerührt. Anschließend wurden 14 (0.12 mmol) 2- Fluorbenzylbromid zugetropft und die Mischung über Nacht bei RT gerührt. Die Reaktionsmischung wurde mit Ethylacetat verdünnt und die organische Phase zweimal mit gesättigter wässriger Natriumhydrogencarbonatlösung gewaschen. Die organische Phase wurde anschließend über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels präparativer RP- HPLC gereinigt (AcetonitrikWasser (+0.1 % Ameisensäure) - Gradient). Das so erhaltene Rohprodukt wurde mittels präparativer Dünnschichtchromatographie (Laufmittel: Dichlormethan- Methanol 20: 1) aufgereinigt. Es wurden 8 mg der Zielverbindung erhalten (13 % d. Th.).
LC-MS (Methode 1): R, = 1.10 min; MS (EIpos): m/z = 502 [M+H]+.
Ή-NMR (400 MHz, DMSO-d6, Rotamerengemisch): δ [ppm] = 3.56 - 3.79 (m, 3H), 4.31 - 4.39 (m, 1H), 4.42 (s, 2H), 5.06 - 5.16 (m, 1H), 7.07 - 7.21 (m, 4H), 7.23 - 7.39 (m, 3H), 7.43 - 7.50 (m, 1H), 7.53 (dd, 1H), 7.56 - 7.74 (m, 2H), 7.64 - 7.70 (m, 1H), 8.60 - 8.65 (m, 1H), 8.97 - 9.02 (m, 1H).
Beispiel 2
Methyl-{4-amino-2-[3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-5-yl}benzyl- carbamat

LC-MS (Methode 1): R, = 1.11 min; MS (EIpos): m/z = 484 [M+H]+.
Ή-NMR (400 MHz, DMSO-d6, Rotamerengemisch): δ [ppm] = 3.57 - 3.81 (m, 3H), 4.12 - 4.21 (m, 1H), 4.41 (s, 1H), 5.06 - 5.15 (m, 1H), 7.07 - 7.13 (m, 1H), 7.14 - 7.21 (m, 1H), 7.23 - 7.38 (m, 7H), 7.52 (dd, 1H), 7.55 - 7.66 (m, 2H), 7.57 - 7.63 (m, 1H), 8.60 - 8.64 (m, 1H), 8.96 - 9.00 (m, 1H).
Beispiel 3
Methyl-{4,6-diamino-2-[3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-5-yl}(2- fluorbenzyl)carbamat

Die Verbindung wurde in Analogie zu Beispiel 1 aus 89 mg (0.22 mmol) der Verbindung in Beispiel 2A hergestellt. Die Reinigung erfolgte mittels präparativer RP-HPLC (Acetonitril: Wasser (+0.1 % Ameisensäure) - Gradient). Es wurden 80 mg der Zielverbindung erhalten (67 % d. Th.). LC-MS (Methode 1): R, = 1.01 min; MS (EIpos): m/z = 517 [M+H]+.
Ή-NMR (400 MHz, DMSO-d6, Rotamerengemisch): δ [ppm] = 3.57 - 3.79 (m, 3H), 4.40 (s, 2H), 4.65 - 4.73 (m, 2H), 6.34 (br. s, 4H), 7.03 - 7.21 (m, 4H), 7.22 - 7.36 (m, 3H), 7.44 - 7.50 (m, 1H), 7.51 - 7.59 (m, 1H), 8.56 - 8.62 (m, 1H), 9.09 - 9.16 (m, 1H).
Beispiel 4 Methyl-{4,6-diamino-2-[3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-5-yl}(4- fluorbenzyl)carbamat

Die Verbindung wurde in Analogie zu Beispiel 1 aus 100 mg (0.245 mmol) der Verbindung in Beispiel 2A und 31 (0.25 mmol) 4-Fluorbenzylbromid hergestellt. Die Reinigung erfolgte mittels präparativer RP-HPLC (AcetonitriLWasser (+0.1 % Ameisensäure) - Gradient). Es wurden 92 mg der Zielverbindung erhalten (72 % d. Th.).
LC-MS (Methode 1): R, = 1.03 min; MS (EIpos): m/z = 517 [M+H]+.
Ή-NMR (400 MHz, DMSO-d6, Rotamerengemisch): δ [ppm] = 3.60 (s, 2H), 3.75 (s, 1H), 4.36 - 4.42 (m, 2H), 4.54 - 4.62 (m, 2H), 6.34 (br. s, 4H), 7.00 - 7.12 (m, 3H), 7.14 - 7.21 (m, 1H), 7.22 - 7.36 (m, 2H), 7.36 - 7.44 (m, 2H), 7.44 - 7.50 (m, 1H), 8.55 - 8.61 (m, 1H), 9.07 - 9.14 (m, 1H). Beispiel 5
Methyl-benzyl {4,6-diamino-2-[3 -(2-fluorbenzyl)- IH-pyrazolo [4,3 -b]pyridin- 1 -yl]pyrimidin-5 - yl}carbamat

Die Verbindung wurde in Analogie zu Beispiel 1 aus 100 mg (0.245 mmol) der Verbindung in Beispiel 2A und 29 (0.25 mmol) Benzylbromid hergestellt. Die Reinigung erfolgte mittels präparativer RP-HPLC (AcetonitriLWasser (+0.1 % Ameisensäure) - Gradient). Es wurden 79 mg der Zielverbindung erhalten (63 % d. Th.).
LC-MS (Methode 1): R, = 1.01 min; MS (EIpos): m/z = 499 [M+H]+.
Ή-NMR (400 MHz, DMSO-d6, Rotamerengemisch): δ [ppm] = 3.60 (s, 2H), 3.76 (s, 1H), 4.39 (s, 2H), 4.55 - 4.63 (m, 2H), 6.30 (br. s., 3H), 7.05 - 7.12 (m, 1H), 7.24 (br. s, 8H), 7.43 - 7.50 (m, 1H), 8.55 - 8.60 (m, 1H), 9.07 - 9.14 (m, 1H). Beispiel 6
Isopropyl-benzyl{4,6-diamino-2-[6-fluor-3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l- yl]pyrimidin-5 -yl } carbamat

Die Verbindung wurde in Analogie zu Beispiel 1 aus 108 mg (0.238 mmol) der Verbindung in Beispiel 13A und 29 (0.24 mmol) Benzylbromid hergestellt. Die Reinigung erfolgte mittels präparativer RP-HPLC (AcetonitriLWasser (+0.1 % Ameisensäure) - Gradient). Es wurden 67 mg der Zielverbindung erhalten (52 % d. Th.).
LC-MS (Methode 1): R, = 1.01 min; MS (EIpos): m/z = 545 [M+H]+.
1H-NMR (400 MHz, DMSO-d6, Rotamerengemisch): δ [ppm] = 1.07 - 1.18 (m, 4H), 1.27 - 1 (m, 2H), 4.38 (s, 2H), 4.51 - 4.62 (m, 2H), 4.83 - 4.92 (m, 1H), 6.29 (br. s, 4H), 7.06 - 7.13 1H), 7.14 - 7.21 (m, 1H), 7.21 - 7.40 (m, 7H), 8.60 - 8.64 (m, 1H), 8.98 - 9.04 (m, 1H). Beispiel 7
Ethyl-benzyl {4,6-diamino-2-[6-fluor-3 -(2-fluorbenzyl)- IH-pyrazolo [4,3 -b]pyridin- 1 -yl]pyrimidin-5 - yl}carbamat

Die Verbindung wurde in Analogie zu Beispiel 1 aus 61 mg (0.14 mmol) der Verbindung in Beispiel 14A und 17 (0.14 mmol) Benzylbromid hergestellt. Die Reinigung erfolgte mittels präparativer RP-HPLC (AcetonitriLWasser (+0.1 % Ameisensäure) - Gradient) . Es wurden 17 mg der Zielverbindung erhalten (22 % d. Th.).
LC-MS (Methode 1): R, = 1.13 min; MS (EIpos): m/z = 531 [M+H]+.
Ή-NMR (400 MHz, DMSO-d6, Rotamerengemisch): δ [ppm] = 1.11 (t, 2H), 1.28 - 1.37 (m, 1H), 4.08 (q, 1.5H), 4.13 - 4.23 (m, 0.5H), 4.38 (s, 2H), 4.54 - 4.62 (m, 2H), 6.35 (br. s, 4H), 7.07 - 7.12 (m, 1H), 7.14 - 7.20 (m, 1H), 7.21 - 7.40 (m, 7H), 8.60 - 8.63 (m, 1H), 8.97 - 9.02 (m, 1H). Beispiel 8
Memyl-benzyl{4,6-diamino-2-[6-fluor-3-(2-fluorte
5-yl}carbamat

Die Verbindung wurde in Analogie zu Beispiel 1 aus 86 mg (0.13 mmol) der Verbindung in Beispiel 9A und 16 (0.13 mmol) Benzylbromid hergestellt. Die Reinigung erfolgte mittels präparativer RP-HPLC (AcetonitriLWasser (+0.1 % Ameisensäure) - Gradient) . Es wurden 50 mg der Zielverbindung erhalten (73 % d. Th.).
LC-MS (Methode 1): R, = 1.09 min; MS (EIpos): m/z = 516 [M+H]+.
Ή-NMR (400 MHz, DMSO-d6, Rotamerengemisch): δ [ppm] = 3.60 (s, 2H), 3.76 (s, 1H), 4.37 (s, 2H), 4.55 - 4.63 (m, 2H), 6.39 (br. s, 4H), 7.06 - 7.13 (m, 1H), 7.14 - 7.20 (m, 1H), 7.21 - 7.40 (m, 7H), 8.59 - 8.64 (m, 1H), 8.95 - 9.02 (m, 1H). Beispiel 9
Memyl-{4,6-diamino-2-[6-fluor-3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-5- yl } { 2- [(phenylsulfonyl)methyl]benzyl } carbamat

39 mg (0.12 mmol) l-(Bromomethyl)-2-[(phenylsulfonyl)methyl]benzen wurden in einem well einer 96er deep well Multititerplatte vorgelegt und mit einer Lösung von 426 mg (0.1 mmol) Methyl-{4,6- diamino-2-[6-fluor-3-(2-fluorbenzyl)-lH-pyrazolo[4,3-b]pyridin-l-yl]pyrimidin-5-yl } carbamat in 0.6 ml DMF versetzt. Zu diesem Gemisch wurden 130 mg (0.4 mmol) Cäsiumcarbonat gegeben. Die Multititerplatte wurde abgedeckt und bei 60 °C über Nacht geschüttelt. Dann wurde abfiltriert und das Filtrat direkt via präparativer LC-MS (Methode 4 bzw. 5) gereinigt. Die produkthaltigen Fraktionen wurden mittels Zentrifugaltrockner im Vakuum eingeengt. Der Rückstand der einzelnen Fraktionen wurde in je 0.6 ml DMSO gelöst und vereint. Anschließend wurde im Zentrifügaltrockner das Lösemittel vollständig abgedampft. Es wurden 17.3 mg (26% d. Th.) Zielprodukt erhalten.
LC-MS (Methode 3): R, = 1.21 min; MS (ESIpos): m/z = 671 (M+H)+.
In Analogie zu Beispiel 9 wurden folgende Verbindungen in Tabelle 1 hergestellt.
Tabelle 1




















B. Bewertung der pharmakologischen Wirksamkeit
Die pharmakologische Wirkung der erfindungsgemäßen Verbindungen kann in folgenden Assays gezeigt werden:
B-l . Gefäßrelaxierende Wirkung in vitro Kaninchen werden durch Nackenschlag betäubt und entblutet. Die Aorta wird entnommen, von anhaftendem Gewebe befreit, in 1.5 mm breite Ringe geteilt und einzeln unter einer Vorspannung in 5 ml-Organbäder mit 37°C warmer, Carbogen-begaster Krebs-Henseleit-Lösung folgender Zusammensetzung gebracht (jeweils mM): Natriumchlorid: 119; Kaliumchlorid: 4.8; Calciumchlorid- Dihydrat: 1; Magnesiumsulfat-Heptahydrat: 1.4; Kaliumdihydrogenphosphat: 1 . 2 ; Natriumhydrogencarbonat: 25; Glucose: 10. Die Kontraktionskraft wird mit Statham UC2-Zellen erfasst, verstärkt und über A/D-Wandler (DAS-1802 HC, Keithley Instruments München) digitalisiert sowie parallel auf Linienschreiber registriert. Zur Erzeugung einer Kontraktion wird Phenylephrin dem Bad kumulativ in ansteigender Konzentration zugesetzt. Nach mehreren Kontrollzyklen wird die zu untersuchende Substanz in jedem weiteren Durchgang in jeweils steigender Dosierung zugesetzt und die Höhe der Kontraktion mit der Höhe der im letzten Vordurchgang erreichten Kontraktion verglichen. Daraus wird die Konzentration errechnet, die erforderlich ist, um die Höhe des Kontrollwertes um 50% zu reduzieren (IC50-Wert). Das Standardapplikationsvolumen beträgt 5 μΐ, der DMSO-Anteil in der Badlösung entspricht 0.1%.
Repräsentative IC50-Werte für die erfindungsgemäßen Verbindungen sind in der nachstehenden Tabelle (Tabelle 1) wiedergegeben:
Tabelle 1 :
Beispiel Nr. IC50 [nM]
2 0.705
3 0.399
4 0.359
5 0.185
6 0.745
Beispiel Nr. IC50 [nM]
8 0.154
B-2. Wirkung an rekombinanter Guanylatcyclase-Reporterzelllinie
Die zelluläre Wirkung der erfindungsgemäßen Verbindungen wird an einer rekombinanten Guanylat- cyclase-Reporterzelllinie, wie in F. Wunder et al., Anal. Biochem. 339. 104-112 (2005) beschrieben, bestimmt. Repräsentative Werte (MEC = minimal effektive Konzentration) für die erfindungsgemäßen Verbindungen sind in der nachstehenden Tabelle (Tabelle 2) wiedergegeben:
Tabelle 2:

B-3. Radiotelemetrische Blutdruckmessung an wachen, spontan hvpertensiven Ratten Für die im Folgenden beschriebene Blutdruckmessung an wachen Ratten wird ein im Handel erhältliches Telemetriesystem der Firma DATA SCIENCES INTERNATIONAL DSI, USA eingesetzt.
Das System besteht aus 3 Hauptkomponenten:
- Implantierbare Sender (Physiotel® Telemetrietransmitter)
- Empfanger (Physiotel® Receiver), die über einen Multiplexer (DSI Data Exchange Matrix ) mit einem
- Datenakquisitionscomputer verbunden sind. Die Telemetrieanlage ermöglicht eine kontinuierliche Erfassung von Blutdruck Herzfrequenz und Körperbewegung an wachen Tieren in ihrem gewohnten Lebensraum.
Tiermaterial
Die Untersuchungen werden an ausgewachsenen weiblichen spontan hypertensiven Ratten (SHR Okamoto) mit einem Körpergewicht von >200 g durchgeführt. SHR/NCrl von Okamoto Kyoto School of Medicine, 1963 wurden aus männlichen Wistar Kyoto Ratten mit stark erhöhtem Blutdruck und weiblichen mit leicht erhöhtem Blutdruck gekreuzt und in der F 13 an die U. S . National Institutes of Health abgegeben.
Die Versuchstiere werden nach Senderimplantation einzeln in Makroion - Käfigen Typ 3 gehalten. Sie haben freien Zugang zu Standardfutter und Wasser. Der Tag - Nacht - Rhythmus im Versuchslabor wird per Raumbeleuchtung um 6:00 Uhr morgens und um 19:00 Uhr abends gewechselt.
Senderimplantation
Die eingesetzten Telemetriesender TAI 1 PA - C40 werden den Versuchstieren mindestens 14 Tage vor dem ersten Versuchseinsatz unter aseptischen Bedingungen chirurgisch implantiert. Die so instrumentierten Tiere sind nach Abheilen der Wunde und Einwachsen des Implantats wiederholt einsetzbar.
Zur Implantation werden die nüchternen Tiere mit Pentobabital (Nembutal, Sanofi: 50mg/kg i.p. ) narkotisiert und an der Bauchseite weiträumig rasiert und desinfiziert. Nach Eröffnung des Bauchraumes entlang der Linea alba wird der flüssigkeitsgefüllte Meßkatheter des Systems oberhalb der Bifurcation nach cranial in die Aorta descendens eingesetzt und mit Gewebekleber (VetBonD TM, 3M) befestigt. Das Sendergehäuse wird intraperitoneal an der Bauchwandmuskulatur fixiert und die Wunde wird schichtweise verschlossen.
Postoperativ wird zur Infektionsprophylaxe ein Antibiotikum verabreicht (Tardomyocel COMP Bayer 1ml/kg s.c.)
Substanzen und Lösungen
Wenn nicht anders beschrieben werden die zu untersuchenden Substanzen jeweils einer Gruppe von Tieren (n = 6 ) per Schlundsonde oral verabreicht. Entsprechend einem Applikationsvolumen von 5 ml/kg Körpergewicht werden die Testsubstanzen in geeigneten Lösungsmittelgemischen gelöst oder in 0.5%-iger Tylose suspendiert.
Eine Lösungsmittel- behandelte Gruppe von Tieren wird als Kontrolle eingesetzt.
Versuchsablauf
Die vorhandene Telemetrie - Meßeinrichtung ist für 24 Tiere konfiguriert. Jeder Versuch wird unter einer Versuchsnummer registiert (VJahr Monat Tag). Den in der Anlage lebenden instrumentierten Ratten ist jeweils eine eigene Empfangsantenne zugeordnet (1010 Receiver, DSI ).
Die implantierten Sender sind über einen eingebauten Magnetschalter von außen aktivierbar. Sie werden bei Versuchsvorlauf auf Sendung geschaltet. Die ausgestrahlten Signale können durch ein Datenakquisitionssystem (Dataquest TM A.R.T. for WINDOWS, DSI ) online erfasst und entsprechend aufgearbeitet werden. Die Ablage der Daten erfolgt jeweils in einem hierfür eröffneten Ordner der die Versuchsnummer trägt.
Im Standardablauf werden über je 10 Sekunden Dauer gemessen:
- Systolischer Blutdruck (SBP)
- Diastolischer Blutdruck (DBP) - Arterieller Mitteldruck (MAP)
- Herzfrequenz (HR)
- Aktivität (ACT).
Die Messwerterfassung wird rechnergesteuert in 5 Minuten Abständen wiederholt. Die als Absolutwert erhobenen Quelldaten werden im Diagramm mit dem aktuell gemessenen Barometerdruck (Ambient Pressure Reference Monitor; APR-1) korrigiert und in Einzeldaten abgelegt. Weitere technische Details sind der umfangreichen Dokumentation der Herstellerfirma (DSI) zu entnehmen.
Wenn nicht anders beschrieben erfolgt die Verabreichung der Prüfsubstanzen am Versuchstag um 9.00 Uhr. Im Anschluss an die Applikation werden die oben beschriebenen Parameter 24 Stunden gemessen.
Auswertung Nach Versuchsende werden die erhobenen Einzeldaten mit der Analysis-Software (DATAQUEST TM A. R.T. TM ANALYSIS) sortiert. Als Leerwert werden hier 2 Stunden vor Applikation angenommen, so dass der selektierte Datensatz den Zeitraum von 7:00 Uhr am Versuchstag bis 9:00 Uhr am Folgetag umfasst.
Die Daten werden über eine voreinstellbare Zeit durch Mittelwertbestimmung geglättet (15 Minuten Average) und als Textdatei auf einen Datenträger übertragen. Die so vorsortierten und komprimierten Messwerte werden in Excel-Vorlagen übertragen und tabellarisch dargestellt. Die Ablage der erhobenen Daten erfolgt pro Versuchstag in einem eigenen Ordner, der die Versuchsnummer trägt. Ergebnisse und Versuchsprotokolle werden in Papierform nach Nummern sortiert in Ordnern abgelegt. Literatur
Klaus Witte, Kai Hu, Johanna Swiatek, Claudia Müssig, Georg Ertl and Bj örn Lemmer: Experimental heart failure in rats: effects on cardiovascular circadian rhythms and on myocardial ß- adrenergic signaling. Cardiovasc Res 47 (2): 203-405, 2000; Kozo Okamoto: Spontaneous hypertension in rats. Int Rev Exp Pathol 7: 227- 270, 1969; Maarten van den Buuse: Circadian Rhythms of Blood Pressure, Heart Rate, and Locomotor Activity in Spontaneously Hypertensive Rats as Measured With Radio-Telemetry. Physiology & Behavior 55(4): 783-787, 1994
C. Ausführungsbeispiele für pharmazeutische Zusammensetzungen
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überführt werden:
Tablette: Zusammensetzung :
100 mg der erfindungsgemäßen Verbindung, 50 mg Lactose (Monohydrat), 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrrolidon (PVP 25) (Fa. BASF, Ludwigshafen, Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg. Durchmesser 8 mm, Wölbungsradius 12 mm. Herstellung:
Die Mischung aus erfindungsgemäßer Verbindung, Lactose und Stärke wird mit einer 5%-igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat 5 Minuten gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse verpresst (Format der Tablette siehe oben). Als Richtwert für die Verpressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzung :
1000 mg der erfindungsgemäßen Verbindung, 1000 mg Ethanol (96%), 400 mg Rhodigel® (Xanthan gum der Firma FMC, Pennsylvania, USA) und 99 g Wasser. Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 10 ml orale Suspension.
Herstellung:
Das Rhodigel wird in Ethanol suspendiert, die erfindungsgemäße Verbindung wird der Suspension zugefügt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluß der Quellung des Rhodigels wird ca. 6 h gerührt.
Oral applizierbare Lösung:
Zusammensetzung :
500 mg der erfindungsgemäßen Verbindung, 2.5 g Polysorbat und 97 g Polyethylenglycol 400. Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 20 g orale Lösung. Herstellung:
Die erfindungsgemäße Verbindung wird in der Mischung aus Polyethylenglycol und Polysorbat unter Rühren suspendiert. Der Rührvorgang wird bis zur vollständigen Auflösung der erfindungsgemäßen Verbindung fortgesetzt. i.v.-Lösung: Die erfindungsgemäße Verbindung wird in einer Konzentration unterhalb der Sättigungslöslichkeit in einem physiologisch verträglichen Lösungsmittel (z.B. isotonische Kochsalzlösung, Glucoselösung 5% und/oder PEG 400-Lösung 30%) gelöst. Die Lösung wird steril filtriert und in sterile und pyrogenfreie Injektionsbehältnisse abgefüllt.
Claims
1. Verbindung der Formel (I)
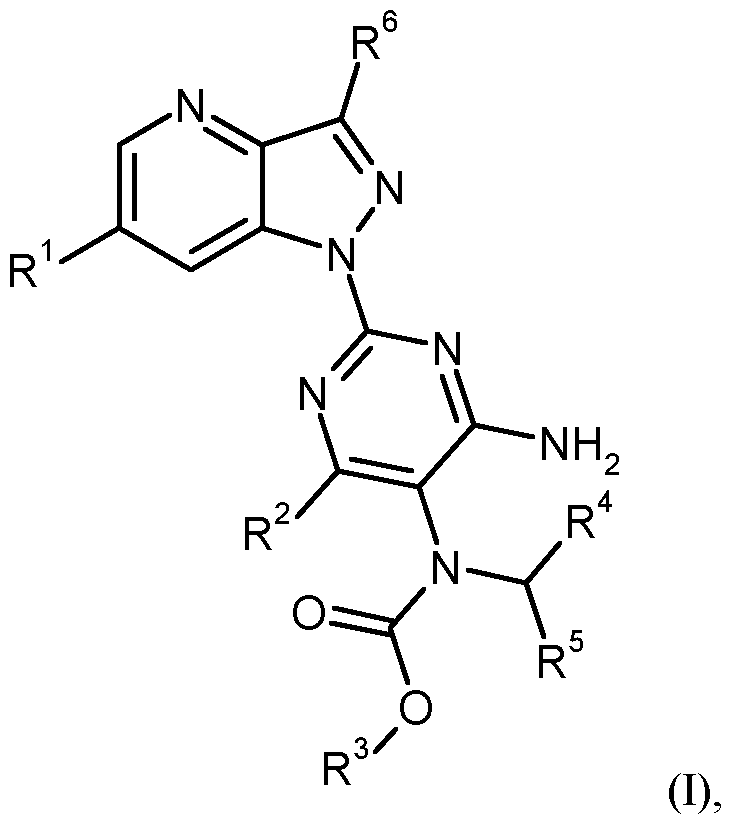
R1 für Wasserstoff oder Fluor steht, R2 für Wasserstoff oder Amino steht, R3 für (C1-C4)-Alkyl steht,
R4 für Wasserstoff, (Ci-C4)-Alkyl oder (d-C4)-Alkoxycarbonyl steht,
R5 für Phenyl, Tetrahydronaphthalenyl, Naphthyl oder 5- bis 10-gliedriges Heteroaryl steht, wobei Phenyl, Tetrahydronaphthalenyl, Naphthyl und 5- bis 10-gliedriges Heteroaryl mit 1 bis 3 Substituenten unabhängig von einander ausgewählt aus der Gruppe Halogen, Nitro, Cyano, Difluormethyl, Trifluormethyl, (Ci-C6)-Alkyl, Hydroxy, Difluormethoxy, Trifluormethoxy, (Ci-C4)-Alkoxy, Amino, Mono- (Ci-C )-alkylamino, Di-(Ci-C )-alkylamino, Hydroxycarbonyl, (Ci-C )-Alkoxy- carbonyl, Aminocarbonyl, Mono-(Ci-C )-alkylaminocarbonyl, Di-(Ci-C )-alkyl- aminocarbonyl, (C3-C7)-Cycloalkylaminocarbonyl, Phenylsulfonylmethyl, Phenyl und Phenoxy substituiert sein können, worin (Ci-C6)-Alkyl mit einem Substituenten unabhängig voneinander ausgewählt aus der Gruppe Hydroxycarbonyl und (Ci-C4)-Alkoxycarbonyl substituiert sein kann, und worin Phenyl und Phenoxy mit 1 bis 3 Substituenten unabhängig voneinander ausgewählt aus der Gruppe Halogen und Cyano substituiert sein können,
R6 für (Ci-C6)-Alkyl oder Benzyl steht, wobei (Ci-C6)-Alkyl mit einem Substituenten Trifluormethyl substituiert ist, wobei (Ci-C6)-Alkyl mit 1 bis 3 Substituenten Fluor substituiert sein kann, und wobei Benzyl mit 1 bis 3 Substituenten Fluor substituiert ist, sowie ihre N-Oxide, Salze, Solvate, Salze der N-Oxide und Solvate der N-Oxide und Salze.
2. Verbindung der Formel (I) nach Anspruch 1, in welcher R1 für Wasserstoff oder Fluor steht,
R2 für Wasserstoff oder Amino steht,
R3 für (d-C3)-Alkyl steht,
R4 für Wasserstoff oder Methyl steht,
R5 für Phenyl, Thienyl, Thiazolyl, Pyridyl oder Chinolyl steht, wobei Phenyl, Thienyl, Thiazolyl, Pyridyl und Chinolyl mit 1 bis 3 Substituenten unabhängig von einander ausgewählt aus der Gruppe Fluor, Chlor, Nitro, Cyano, Difluormethyl, Trifluormethyl, (Ci-C6)-Alkyl, Hydroxy, Difluormethoxy, Trifluor- methoxy, Methoxy, Ethoxy, Amino, Methylamino, Ethylamino, Dimethylamino, Diethylamino, Methoxycarbonyl, Ethoxycarbonyl, Aminocarbonyl, Methylamino- carbonyl, Ethylaminocarbonyl, Dimethylaminocarbonyl, Diethylaminocarbonyl,
Cyclopropylaminocarbonyl und Cyclobutylaminocarbonyl substituiert sein können, worin (Ci-C6)-Alkyl mit einem Substituenten unabhängig voneinander ausgewählt aus der Gruppe Hydroxycarbonyl, Methoxycarbonyl und Ethoxycarbonyl substituiert sein kann,
R6 für 3,3,3-Trifluorprop-l-yl, 3,3,4,4,4-Pentafluorbut-l-yl oder Benzyl steht, wobei Benzyl mit 1 oder 2 Substituenten Fluor substituiert ist, sowie ihre Salze, Solvate und Solvate der Salze.
3. Verbindung der Formel (I) nach Anspruch 1 oder 2, in welcher
R1 für Wasserstoff oder Fluor steht,
R2 für Wasserstoff oder Amino steht, R3 für Methyl, Ethyl oder iso-Propyl steht,
R4 für Wasserstoff steht,
R5 für Phenyl steht, wobei Phenyl mit 1 Substituenten Fluor substituiert sein kann,
R6 für 2-Fluorbenzyl steht, sowie ihre Salze, Solvate und Solvate der Salze.
4. Verfahren zur Herstellung von Verbindungen der Formel (I), wie in den Ansprüchen 1 bis 3 definiert, dadurch gekennzeichnet, dass man eine Verbindung der Formel (II)


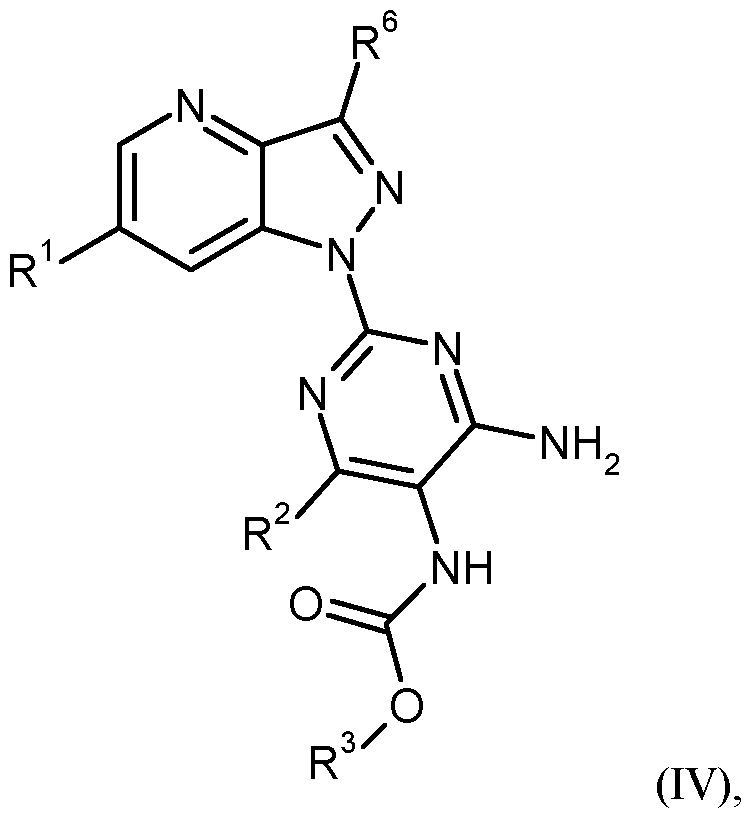

X1 für eine geeignete Abgangsgruppe, wie beispielsweise Mesylat, Tosylat oder Halogen, insbesondere Brom oder Iod, steht, umsetzt, und die resultierenden Verbindungen der Formel (I) gegebenenfalls mit den entsprechenden (i) Lösungsmitteln und/oder (ii) Säuren oder Basen in ihre Solvate, Salze und/oder Solvate der Salze überführt.
5. Verbindung der Formel (I), wie in einem der Ansprüche 1 bis 3 definiert, zur Behandlung und/oder Prophylaxe von Krankheiten.
6. Verbindung der Formel (I), wie in einem der Ansprüche 1 bis 3 definiert, zur Verwendung in einem Verfahren zur Behandlung und/ oder Prophylaxe von Herzinsuffizienz, Angina pectoris, Hypertonie, pulmonaler Hypertonie, Ischämien, Gefäßerkrankungen, Niereninsuffizienz, thromboembolischen Erkrankungen, fibrotischen Erkrankungen und Arterio- sklerose.
7. Verwendung einer Verbindung der Formel (I), wie in einem der Ansprüche 1 bis 3 definiert, zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von Herzinsuffizienz, Angina pectoris, Hypertonie, pulmonaler Hypertonie, Ischämien, Gefäßerkrankungen, Niereninsuffizienz, thromboembolischen Erkrankungen, fibrotischen Erkrankungen und Arteriosklerose.
8. Arzneimittel enthaltend eine Verbindung der Formel (I), wie in einem der Ansprüche 1 bis 3 definiert, in Kombination mit einem inerten, nicht-toxischen, pharmazeutisch geeigneten Hilfsstoff.
9. Arzneimittel enthaltend eine Verbindung der Formel (I), wie in einem der Ansprüche 1 bis 3 definiert, in Kombination mit einem weiteren Wirkstoff ausgewählt aus der Gruppe bestehend aus organischen Nitraten, NO-Donatoren, cGMP-PDE-Inhibitoren, antithrombotisch wirkenden Mitteln, den Blutdruck senkenden Mitteln sowie den Fettstoffwechsel verändernden Mitteln.
10. Arzneimittel nach Anspruch 8 oder 9 zur Behandlung und/oder Prophylaxe von Herz- Insuffizienz, Angina pectoris, Hypertonie, pulmonaler Hypertonie, Ischämien, Gefäßerkrankungen, Niereninsuffizienz, thromboembolischen Erkrankungen, fibrotischen Erkrankungen und Arteriosklerose.
11. Verfahren zur Behandlung und/oder Prophylaxe von Herzinsuffizienz, Angina pectoris, Hypertonie, pulmonaler Hypertonie, Ischämien, Gefäßerkrankungen, Niereninsuffizienz, thromboembolischen Erkrankungen, fibrotischen Erkrankungen und Arteriosklerose bei
Menschen und Tieren unter Verwendung einer wirksamen Menge mindestens einer Ver- bindung der Formel (I), wie in einem der Ansprüche 1 bis 3 definiert, oder eines Arzneimittels, wie in einem der Ansprüche 8 bis 10 definiert.
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/882,123 US9090609B2 (en) | 2010-11-04 | 2011-11-03 | Benzyl-substituted carbamates and use thereof |
| CA2816639A CA2816639A1 (en) | 2010-11-04 | 2011-11-03 | Benzyl-substituted carbamates and use thereof |
| ES11782103.3T ES2536894T3 (es) | 2010-11-04 | 2011-11-03 | Carbamatos sustituidos con bencilo y su uso |
| EP11782103.3A EP2635576B1 (de) | 2010-11-04 | 2011-11-03 | Benzyl-substituierte carbamate und ihre verwendung |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102010043380.2 | 2010-11-04 | ||
| DE102010043380A DE102010043380A1 (de) | 2010-11-04 | 2010-11-04 | Benzyl-substituierte Carbamate und ihre Verwendung |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012059548A1 true WO2012059548A1 (de) | 2012-05-10 |
Family
ID=44947075
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2011/069344 WO2012059548A1 (de) | 2010-11-04 | 2011-11-03 | Benzyl-substituierte carbamate und ihre verwendung |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9090609B2 (de) |
| EP (1) | EP2635576B1 (de) |
| CA (1) | CA2816639A1 (de) |
| DE (1) | DE102010043380A1 (de) |
| ES (1) | ES2536894T3 (de) |
| WO (1) | WO2012059548A1 (de) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014084312A1 (ja) | 2012-11-30 | 2014-06-05 | アステラス製薬株式会社 | イミダゾピリジン化合物 |
| WO2016177660A1 (en) | 2015-05-06 | 2016-11-10 | Bayer Pharma Aktiengesellschaft | The use of sgc stimulators, sgc activators, alone and combinations with pde5 inhibitors for the treatment of digital ulcers (du) concomitant to systemic sclerosis (ssc) |
| WO2017013010A1 (de) | 2015-07-23 | 2017-01-26 | Bayer Pharma Aktiengesellschaft | Stimulatoren und/oder aktivatoren der löslichen guanylatzyklase (sgc) in kombination mit einem inhibitor der neutralen endopeptidase (nep inhibitor) und/oder einem angiotensin aii-antagonisten und ihre verwendung |
| WO2018069126A1 (de) | 2016-10-11 | 2018-04-19 | Bayer Pharma Aktiengesellschaft | Kombination enthaltend sgc stimulatoren und mineralocorticoid-rezeptor-antagonisten |
| WO2019219672A1 (en) | 2018-05-15 | 2019-11-21 | Bayer Aktiengesellschaft | 1,3-thiazol-2-yl substituted benzamides for the treatment of diseases associated with nerve fiber sensitization |
| WO2020165010A1 (en) | 2019-02-13 | 2020-08-20 | Bayer Aktiengesellschaft | Process for the preparation of porous microparticles |
| US11331308B2 (en) | 2016-10-11 | 2022-05-17 | Bayer Pharma Aktiengesellschaft | Combination containing sGC activators and mineralocorticoid receptor antagonists |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2504334B1 (de) | 2009-11-27 | 2014-10-01 | Bayer Intellectual Property GmbH | Verfahren zur reinigung von methyl-{4,6-diamino-2-[1-(2-fluorobenzyl)-1h-pyrazolo[3,4-b]pyridin-3-yl]pyrimidin-5-yl}methylcarbamat |
| DE102010021637A1 (de) | 2010-05-26 | 2011-12-01 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 5-Fluor-1H-Pyrazolopyridine und ihre Verwendung |
| WO2012004258A1 (de) | 2010-07-09 | 2012-01-12 | Bayer Pharma Aktiengesellschaft | Annellierte pyrimidine und triazine und ihre verwendung zur behandlung bzw. prophylaxe von herz-kreislauf-erkrankungen |
| US9090610B2 (en) * | 2011-04-21 | 2015-07-28 | Bayer Intellectual Property Gmbh | Fluoroalkyl-substituted pyrazolopyridines and use thereof |
| BR122020001312B1 (pt) | 2011-11-25 | 2022-02-15 | Adverio Pharma Gmbh | Processo para preparação de 5-flúor-1h-pirazolopiridinas substituídas, e compostos intermediários |
| DE102012200349A1 (de) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituierte annellierte Pyrimidine und Triazine und ihre Verwendung |
| ES2644781T3 (es) | 2012-03-06 | 2017-11-30 | Bayer Intellectual Property Gmbh | Azabiciclos sustituidos y su uso |
| CA2901636A1 (en) | 2013-02-21 | 2014-08-28 | Adverio Pharma Gmbh | Forms of methyl {4,6-diamino-2-[1-(2-fluorobenzyl)-1h-pyrazolo[3,4-b]pyridino-3-yl]pyrimidino-5-yl}methyl carbamate |
| KR20150121007A (ko) | 2013-03-01 | 2015-10-28 | 바이엘 파마 악티엔게젤샤프트 | 트리플루오르메틸-치환된 고리-융합된 피리미딘 및 그의 용도 |
| JP2016523944A (ja) | 2013-07-10 | 2016-08-12 | バイエル・ファルマ・アクティエンゲゼルシャフト | ベンジル−1H−ピラゾロ[3,4−b]ピリジンおよびその使用 |
| ES2924359T3 (es) | 2017-04-11 | 2022-10-06 | Sunshine Lake Pharma Co Ltd | Compuestos de indazol sustituidos con flúor y usos de los mismos |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998016223A1 (de) | 1996-10-14 | 1998-04-23 | Bayer Aktiengesellschaft | Verwendung von 1-benzyl-3-(substituiertes-hetaryl)-kondensierten pyrazol-derivaten zur belandlung von speziellen erkrankungen des herz-kreislaufsystems und des zentralnervensystems |
| WO2002042300A1 (de) | 2000-11-22 | 2002-05-30 | Bayer Aktiengesellschaft | Neue carbamat-substituierte pyrazolopyridinderivate |
| WO2005030121A2 (en) | 2003-06-30 | 2005-04-07 | Hif Bio, Inc. | Compounds, compositions and methods |
| WO2008031513A1 (de) | 2006-09-15 | 2008-03-20 | Bayer Schering Pharma Aktiengesellschaft | Pyrazolopyridin, indazol, imidazopyridin, imidazopyrimidin, pyrazolopyrazin und pyrazolopyridin derivate als stimulatoren der guanylatcyclase zur herz-kreislauferkrankungen |
| WO2010079120A1 (de) * | 2009-01-09 | 2010-07-15 | Bayer Schering Pharma Aktiengesellschaft | Benzimidazol-und pyrazolopyridin-derivate zur behandlung und/oder prävention von herz-kreislauf-erkrankungen |
| WO2011115804A1 (en) | 2010-03-17 | 2011-09-22 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| WO2011119518A1 (en) | 2010-03-25 | 2011-09-29 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6451805B1 (en) | 1997-11-14 | 2002-09-17 | Bayer Aktiengesellschaft | Substituted pyrazole derivatives for the treatment of cardiocirculatory diseases |
| DE19834044A1 (de) | 1998-07-29 | 2000-02-03 | Bayer Ag | Neue substituierte Pyrazolderivate |
| DE19834047A1 (de) | 1998-07-29 | 2000-02-03 | Bayer Ag | Substituierte Pyrazolderivate |
| JP4295505B2 (ja) | 2000-11-22 | 2009-07-15 | バイエル アクチェンゲゼルシャフト | 新規なラクタム置換ピラゾロピリジン誘導体 |
| AR031176A1 (es) | 2000-11-22 | 2003-09-10 | Bayer Ag | Nuevos derivados de pirazolpiridina sustituidos con piridina |
| DE10122894A1 (de) | 2001-05-11 | 2002-11-14 | Bayer Ag | Neue Sulfonat-substituierte Pyrazolopyridinderivate |
| DE10132416A1 (de) | 2001-07-04 | 2003-01-16 | Bayer Ag | Neue Morpholin-überbrückte Pyrazolopyridinderivate |
| DE10220570A1 (de) | 2002-05-08 | 2003-11-20 | Bayer Ag | Carbamat-substituierte Pyrazolopyridine |
| DE10232571A1 (de) | 2002-07-18 | 2004-02-05 | Bayer Ag | 4-Aminosubstituierte Pyrimidinderivate |
| DE10232572A1 (de) | 2002-07-18 | 2004-02-05 | Bayer Ag | Neue 2,5-disubstituierte Pyrimidinderivate |
| DE10351903A1 (de) | 2003-11-06 | 2005-06-09 | Bayer Healthcare Ag | Neue Kombination |
| WO2009000832A2 (en) | 2007-06-25 | 2008-12-31 | Boehringer Ingelheim International Gmbh | Chemical compounds |
| MX2010001137A (es) | 2007-07-31 | 2010-03-31 | Vertex Pharma | Procesopara preparar 5-fluoro-1h-pirazol[3,4-b]piridin-3-amina y derivados del mismo. |
| WO2009145814A2 (en) | 2008-03-10 | 2009-12-03 | Vertex Pharmaceuticals Incorporated | Pyrimidines and pyridines useful as inhibitors of protein kinases |
| UY33040A (es) | 2009-11-27 | 2011-06-30 | Bayer Schering Pahrma Akitengesellschaft | Nuevas formas polimorfas de {4,6diamino-2-[1-(2-fluorobencil-1h-pirazol[3,4-b]piridin-3-il]pirimidin-5-il}carbamato de metilo |
| UY33041A (es) | 2009-11-27 | 2011-06-30 | Bayer Schering Pharma Aktienegesellschaft | Procedimiento para la preparaciòn de {4,6-diamino-2-[1-(2-fluorobencil)-1h-pirazolo[3,4-b]piridin-3-il]pirimidin-5-il}carbamato de metilo y su purificaciòn para el uso como principio activo farmacèutico |
| EP2504334B1 (de) | 2009-11-27 | 2014-10-01 | Bayer Intellectual Property GmbH | Verfahren zur reinigung von methyl-{4,6-diamino-2-[1-(2-fluorobenzyl)-1h-pyrazolo[3,4-b]pyridin-3-yl]pyrimidin-5-yl}methylcarbamat |
| DE102010021637A1 (de) | 2010-05-26 | 2011-12-01 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 5-Fluor-1H-Pyrazolopyridine und ihre Verwendung |
| DE102010040234A1 (de) | 2010-09-03 | 2012-03-08 | Bayer Schering Pharma Aktiengesellschaft | Verfahren zur Herstellung von 5-Flour-1H-pyrazolo[3,4-b]pyridin-3-carbonitril |
| DE102010040233A1 (de) | 2010-09-03 | 2012-03-08 | Bayer Schering Pharma Aktiengesellschaft | Bicyclische Aza-Heterocyclen und ihre Verwendung |
| DE102010043379A1 (de) | 2010-11-04 | 2012-05-10 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 6-Fluor-1H-Pyrazolo[4,3-b]pyridine und ihre Verwendung |
| BR122020001312B1 (pt) | 2011-11-25 | 2022-02-15 | Adverio Pharma Gmbh | Processo para preparação de 5-flúor-1h-pirazolopiridinas substituídas, e compostos intermediários |
-
2010
- 2010-11-04 DE DE102010043380A patent/DE102010043380A1/de not_active Withdrawn
-
2011
- 2011-11-03 US US13/882,123 patent/US9090609B2/en not_active Expired - Fee Related
- 2011-11-03 EP EP11782103.3A patent/EP2635576B1/de not_active Not-in-force
- 2011-11-03 ES ES11782103.3T patent/ES2536894T3/es active Active
- 2011-11-03 WO PCT/EP2011/069344 patent/WO2012059548A1/de active Application Filing
- 2011-11-03 CA CA2816639A patent/CA2816639A1/en not_active Abandoned
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998016223A1 (de) | 1996-10-14 | 1998-04-23 | Bayer Aktiengesellschaft | Verwendung von 1-benzyl-3-(substituiertes-hetaryl)-kondensierten pyrazol-derivaten zur belandlung von speziellen erkrankungen des herz-kreislaufsystems und des zentralnervensystems |
| WO2002042300A1 (de) | 2000-11-22 | 2002-05-30 | Bayer Aktiengesellschaft | Neue carbamat-substituierte pyrazolopyridinderivate |
| WO2005030121A2 (en) | 2003-06-30 | 2005-04-07 | Hif Bio, Inc. | Compounds, compositions and methods |
| WO2008031513A1 (de) | 2006-09-15 | 2008-03-20 | Bayer Schering Pharma Aktiengesellschaft | Pyrazolopyridin, indazol, imidazopyridin, imidazopyrimidin, pyrazolopyrazin und pyrazolopyridin derivate als stimulatoren der guanylatcyclase zur herz-kreislauferkrankungen |
| WO2010079120A1 (de) * | 2009-01-09 | 2010-07-15 | Bayer Schering Pharma Aktiengesellschaft | Benzimidazol-und pyrazolopyridin-derivate zur behandlung und/oder prävention von herz-kreislauf-erkrankungen |
| WO2011115804A1 (en) | 2010-03-17 | 2011-09-22 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| WO2011119518A1 (en) | 2010-03-25 | 2011-09-29 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
Non-Patent Citations (11)
| Title |
|---|
| F. WUNDER ET AL., ANAL. BIOCHEM., vol. 339, 2005, pages 104 - 112 |
| GOLDBERG ET AL., J BIOL. CHEM., vol. 252, 1977, pages 1279 |
| HELVETICA CHIMICA ACTA, vol. 34, 1951, pages 835 - 40 |
| J. MED. CHEM., vol. 35, 1992, pages 4455 - 4463 |
| KLAUS WITTE, KAI HU, JOHANNA SWIATEK, CLAUDIA MÜSSIG, GEORG ERTL, BJÖRN LEMMER: "Experimental heart failure in rats: effects on cardiovascular circadian rhythms and on myocardial ßadrenergic signaling", CARDIOVASC RES, vol. 47, no. 2, 2000, pages 203 - 405 |
| KOZO OKAMOTO: "Spontaneous hypertension in rats", INT REV EXP PATHOL, vol. 7, 1969, pages 227 - 270 |
| MAARTEN VAN DEN BUUSE: "Circadian Rhythms of Blood Pressure, Heart Rate, and Locomotor Activity in Spontaneously Hypertensive Rats as Measured With Radio-Telemetry", PHYSIOLOGY & BEHAVIOR, vol. 55, no. 4, 1994, pages 783 - 787 |
| MÜLSCH ET AL., BRIT. J PHARMACOL., vol. 120, 1997, pages 681 |
| PETTIBONE ET AL., EUR. J PHARMACOL., vol. 116, 1985, pages 307 |
| WU ET AL., BLOOD, vol. 84, 1994, pages 4226 |
| YU ET AL., BRIT. J PHARMACOL., vol. 114, 1995, pages 1587 |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014084312A1 (ja) | 2012-11-30 | 2014-06-05 | アステラス製薬株式会社 | イミダゾピリジン化合物 |
| WO2016177660A1 (en) | 2015-05-06 | 2016-11-10 | Bayer Pharma Aktiengesellschaft | The use of sgc stimulators, sgc activators, alone and combinations with pde5 inhibitors for the treatment of digital ulcers (du) concomitant to systemic sclerosis (ssc) |
| WO2017013010A1 (de) | 2015-07-23 | 2017-01-26 | Bayer Pharma Aktiengesellschaft | Stimulatoren und/oder aktivatoren der löslichen guanylatzyklase (sgc) in kombination mit einem inhibitor der neutralen endopeptidase (nep inhibitor) und/oder einem angiotensin aii-antagonisten und ihre verwendung |
| US11166932B2 (en) | 2015-07-23 | 2021-11-09 | Bayer Pharma Aktiengesellschaft | Stimulators and/or activators of soluble guanylate cyclase (sGC) in combination with an inhibitor of neutral endopeptidase (NEP inhibitor) and/or an angiotensin AII antagonist and the use thereof |
| WO2018069126A1 (de) | 2016-10-11 | 2018-04-19 | Bayer Pharma Aktiengesellschaft | Kombination enthaltend sgc stimulatoren und mineralocorticoid-rezeptor-antagonisten |
| US10918639B2 (en) | 2016-10-11 | 2021-02-16 | Bayer Pharma Aktiengesellschaft | Combination containing SGC stimulators and mineralocorticoid receptor antagonists |
| US11331308B2 (en) | 2016-10-11 | 2022-05-17 | Bayer Pharma Aktiengesellschaft | Combination containing sGC activators and mineralocorticoid receptor antagonists |
| US11684621B2 (en) | 2016-10-11 | 2023-06-27 | Bayer Pharma Aktiengesellschaft | Combination containing sGC stimulators and mineralocorticoid receptor antagonists |
| WO2019219672A1 (en) | 2018-05-15 | 2019-11-21 | Bayer Aktiengesellschaft | 1,3-thiazol-2-yl substituted benzamides for the treatment of diseases associated with nerve fiber sensitization |
| WO2020165010A1 (en) | 2019-02-13 | 2020-08-20 | Bayer Aktiengesellschaft | Process for the preparation of porous microparticles |
Also Published As
| Publication number | Publication date |
|---|---|
| DE102010043380A1 (de) | 2012-05-10 |
| US9090609B2 (en) | 2015-07-28 |
| EP2635576A1 (de) | 2013-09-11 |
| ES2536894T3 (es) | 2015-05-29 |
| EP2635576B1 (de) | 2015-02-25 |
| CA2816639A1 (en) | 2012-05-10 |
| US20150065533A1 (en) | 2015-03-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2635576B1 (de) | Benzyl-substituierte carbamate und ihre verwendung | |
| EP2635577B1 (de) | Substituierte 6-fluor-1h-pyrazolo[4,3-b]pyridine und ihre verwendung | |
| EP2590987B1 (de) | Annellierte 4-aminopyrimidine und ihre verwendung als stimulatoren der löslichen guanylatcyclase | |
| EP2576547B1 (de) | Substituierte 5-fluor-1h-pyrazolopyridine und ihre verwendung | |
| EP2822951B1 (de) | Substituierte azabicyclen und ihre verwendung | |
| EP2802580B1 (de) | Substituierte triazine derivate und ihre verwendung als stimulatoren der löslichen guanylatcyclase | |
| EP2379071B1 (de) | Neue aliphatisch substituierte pyrazolopyridine und ihre verwendung | |
| DE102012200352A1 (de) | Substituierte, annellierte Imidazole und Pyrazole und ihre Verwendung | |
| WO2012004258A9 (de) | Annellierte pyrimidine und triazine und ihre verwendung zur behandlung bzw. prophylaxe von herz-kreislauf-erkrankungen | |
| DE102012200349A1 (de) | Substituierte annellierte Pyrimidine und Triazine und ihre Verwendung | |
| EP2961755A1 (de) | Trifluormethyl-substituierte annellierte pyrimidine und ihre verwendung | |
| WO2012152630A1 (de) | Substituierte imidazopyridazine und ihre verwendung | |
| WO2012010578A1 (de) | Substituierte methyl-pyrimidin-5-ylcarbamate und ihre verwendung | |
| WO2012010577A1 (de) | Substituierte oxazolidinone und oxazinanone und ihre verwendung | |
| WO2012010576A1 (de) | Carbamat-substituierte diaminopyrimidine und ihre verwendung | |
| WO2017121692A1 (de) | Substituierte sulfamide und ihre verwendung | |
| DE102011075399A1 (de) | Substituierte Imidazopyridine und Imidazopyridazine und ihre Verwendung | |
| DE102011007891A1 (de) | Annellierte 4-Aminopyrimidine und ihre Verwendung | |
| DE102011007890A1 (de) | Fluoralkyl-substituierte Pyrazolopyridine und ihre Verwendung | |
| DE102010031148A1 (de) | Annellierte 4-Aminopyrimidine und ihre Verwendung | |
| DE102011078715A1 (de) | Heteroaryl-substituierte Pyrazolopyridine und ihre Verwendung | |
| DE102012200351A1 (de) | Substituierte annellierte Pyrimidine und ihre Verwendung | |
| DE102012200354A1 (de) | Heteroaryl-substituierte Pyrazolopyridine und ihre Verwendung | |
| DE102010031149A1 (de) | Annellierte Pyrimidine und Triazine und ihre Verwendung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11782103 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011782103 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2816639 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13882123 Country of ref document: US |