Substituierte Chromanol-Derivate und ihre Verwendung
Die vorliegende Anmeldung betrifft substituierte Chromanol-Derivate, Verfahren zu ihrer Herstellung, ihre Verwendung allein oder in Kombinationen zur Behandlung und/oder Prävention von Krankheiten sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prävention von Krankheiten, insbesondere zur Behandlung und/oder Prävention von kardiovaskulären Erkrankungen.
Durch Arteriosklerose bedingte koronare Herzerkrankungen gehören zu den Haupttodesursachen in der modernen Gesellschaft. In einer Vielzahl von Studien konnte gezeigt werden, dass niedrige Plasmaspiegel des HDL-Cholesterins einen wichtigen Risikofaktor für die Entwicklung von Arteriosklerose darstellen [Barter und Rye, Atherosclerosis 121, 1-12 (1996)]. HDL (high density lipoprotein) stellt neben LDL (low density lipoprotein) und VLDL (very low density lipoprotein) eine Klasse von Lipoproteinen dar, deren wichtigste Funktion der Transport von Lipiden, wie zum Beispiel Cholesterin, Cholesterinester, Triglyceriden, Fettsäuren oder Phospholipiden, im Blut ist. Hohe LDL-Cholesterinspiegel (>160 mg/dl) sowie niedrige HDL-Cholesterinspiegel (<40 mg/dl) tragen wesentlich zur Entwicklung von Arteriosklerose bei [ATP HI Guidelines, Report of the NCEP Expert Panel]. Neben koronaren Herzerkrankungen werden auch periphäre Gefäßerkrankungen sowie Schlaganfall durch ungünstige HDL/LDL-Verhältnisse in ihrer Entstehung gefördert. Neue Methoden zur Erhöhung von HDL-Cholesterin im Plasma stellen demzufolge eine therapeutisch sinnvolle Bereicherung bei der Vorbeugung und Behandlung von Arteriosklerose und der damit verbundenen Krankheiten dar.
Cholesterinester-Transfer-Protein (CETP) mediiert den Austausch von Cholesterinestern und Triglyceriden zwischen den verschiedenen Lipoproteinen im Blut [TaIl, J. Lipid Res. 34, 1255-74 (1993)]. Von besonderer Bedeutung ist dabei der Transfer von Cholesterinestern vom HDL auf das LDL, der zu einer Senkung der HDL-Cholesterin-Plasmaspiegel führt. Die Inhibition von CETP sollte demzufolge eine Erhöhung der Plasmaspiegel des HDL-Cholesterins und eine Absenkung der Plasmaspiegel des LDL-Cholesterins bewirken und damit zu einer therapeutisch nützlichen Beeinflussung des Lipidprofils im Plasma führen [McCarthy, Medicinal Res. Rev. 1_3, 139-59 (1993); Sitori, Pharmac. Ther. 67, 443-47 (1995); Swenson. J Biol. Chem. 264, 14318 (1989)].
Substituierte Tetrahydronaphthaline und Tetrahydrochinoline mit CETP-inhibitorischer Wirkung sind aus EP 0 818 448-A1, WO 99/14174, WO 99/14215, WO 99/15504 und WO 03/028727 bekannt. Heterocyclisch kondensierte Pyridine als CETP-Inhibitoren werden in EP 0 818 197-A1 offenbart. In WO 98/04528 werden substituierte Pyridin- und Phenyl-Derivate als Glucagon-Anta- gonisten zur Behandlung von Diabetes beschrieben.
Aufgabe der vorliegenden Erfindung war die Bereitstellung neuer Substanzen zur Bekämpfung von Erkrankungen, insbesondere von kardiovaskulären Erkrankungen, die ein verbessertes therapeutisches Profil aufweisen.
Gegenstand der vorliegenden Erfindung sind Verbindungen der allgemeinen Formel (I)
in welcher
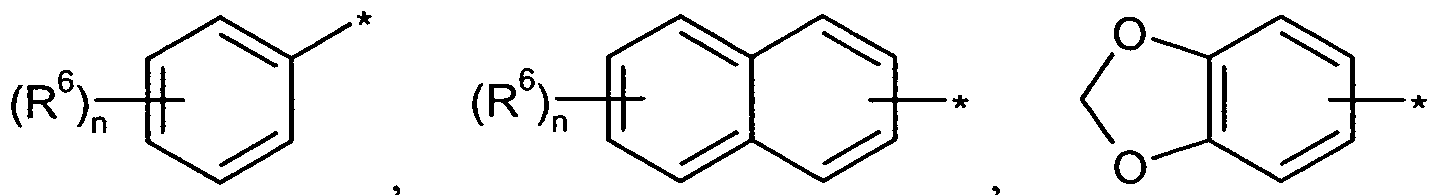
für eine Gruppe der Formel
steht, worin
die Verknüpfungsstelle mit der CR R -Gruppierung bedeutet,
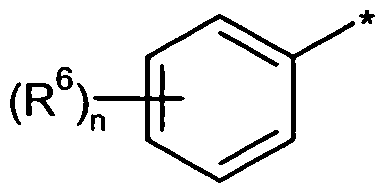
R6 für einen Substituenten ausgewählt aus der Reihe Halogen, Cyano, (CrC6)-Alkyl und (CrC6)-Alkoxy steht, wobei Alkyl und Alkoxy ihrerseits bis zu fünffach mit Fluor substituiert sein können,
und
die Zahl 0, 1, 2 oder 3 bedeutet,
wobei für den Fall, dass der Substituent R6 mehrfach auftritt, seine Bedeutungen gleich oder verschieden sein können,
D für (C3-Cg)-AIkVl, (C4-C8)-Cycloalkyl, (C4-C8)-Cycloalkenyl, (C6-C 10)-Aryl, 5- oder 6- gliedriges Heteroaryl, Tetrahydrofuranyl oder Tetrahydropyranyl steht, wobei
Aryl und Heteroaryl ihrerseits mit Halogen, Cyano, (CrC6)-Alkyl, (CrC6)-Alkoxy, Tri- fluormethyl oder Trifluormethoxy
und
Cycloalkyl und Cycloalkenyl ihrerseits mit Fluor oder (C1-Ce)-AIkVl substituiert sein können,
R1 für Wasserstoff, Fluor, Hydroxy, Methoxy, Mercapto oder Methyl steht,
R2 für Wasserstoff steht
oder
R1 und R2 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Carbonylgruppe bilden,
R3 für (C,-C6)-Alkyl oder (C3-C7)-Cycloalkyl steht
und
R4 und R5 unabhängig voneinander für Wasserstoff oder (Ci-C4)-Alkyl stehen oder gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, einen spiro-verknüpften 3- bis 5-gliedri- gen Cycloalkyl-Ring bilden,
sowie ihre Salze, Solvate und Solvate der Salze.
Erfindungsgemäße Verbindungen sind die Verbindungen der Formel (I) und deren Salze, Solvate und Solvate der Salze, die von Formel (I) umfassten Verbindungen der nachfolgend genannten Formeln und deren Salze, Solvate und Solvate der Salze sowie die von Formel (I) umfassten, nachfolgend als Ausführungsbeispiele genannten Verbindungen und deren Salze, Solvate und Solvate der Salze, soweit es sich bei den von Formel (I) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in stereoisomeren Formen (Enantiomere, Diastereomere) existieren. Die vorliegende Erfindung umfasst
deshalb die Enantiomeren oder Diastereomeren und ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren.
Sofern die erfindungsgemäßen Verbindungen in tautomeren Formen vorkommen können, umfasst die vorliegende Erfindung sämtliche tautomere Formen.
Als Salze sind im Rahmen der vorliegenden Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt. Umfasst sind auch Salze, die für pharmazeutische Anwendungen selbst nicht geeignet sind, jedoch beispielsweise für die Isolierung oder Reinigung der erfindungsgemäßen Verbindungen verwendet werden können.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasserstoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure, Ethan- sulfonsäure, Toluolsulfonsäure, Benzolsulfonsäure, Naphthalindisulfonsäure, Essigsäure, Trifluor- essigsäure, Propionsäure, Milchsäure, Weinsäure, Äpfelsäure, Zitronensäure, Fumarsäure, Malein- säure und Benzoesäure.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen auch Salze üblicher Basen, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kaliumsalze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze) und Ammoniumsalze, abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C- Atomen, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, Ethyldiisopropylamin, Monoethanolamin, Diethanolamin, Trisethanolamin, Dicyclohexylamin, Dimethylaminoethanol, Prokain, Dibenzylamin, N-Methyl- morpholin, Arginin, Lysin, Ethylendiamin und N-Methylpiperidin.
Als Solvate werden im Rahmen der Erfindung solche Formen der erfindungsgemäßen Verbindungen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungs- mittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt. Als Solvate sind im Rahmen der vorliegenden Erfindung Hydrate bevorzugt.
Außerdem umfasst die vorliegende Erfindung auch Prodrugs der erfindungsgemäßen Verbindungen. Der Begriff "Prodrugs" umfaßt Verbindungen, welche selbst biologisch aktiv oder inaktiv sein können, jedoch während ihrer Verweilzeit im Körper zu erfindungsgemäßen Verbindungen umgesetzt werden (beispielsweise metabolisch oder hydrolytisch).
Im Rahmen der vorliegenden Erfindung haben die Substituenten, soweit nicht anders spezifiziert, die folgende Bedeutung:
(C1-CgVAIkVl. (C2-Cg)-AIkVl. (CrCg)-Alkyl und (C-C^-Alkyl stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkylrest mit 1 bis 6, 3 bis 8, 3 bis 6 bzw. 1 bis 4 Kohlenstoffatomen. Im Falle der Gruppen D und R3 ist ein in 1 -Position verzweigter Alkylrest mit
3 bis 6 Kohlenstoffatomen bevorzugt. In den anderen Fällen ist ein geradkettiger oder verzweigter
Alkylrest mit 1 bis 4 Kohlenstoffatomen bevorzugt. Beispielhaft und vorzugsweise seien genannt:
Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, ώo-Butyl, sec-Butyl, tert.-Butyl, n-Pentyl, 1-Methyl- butyl, 1 -Ethylpropyl, 2,2-Dimethylpropyl, n-Hexyl, 1-Methylpentyl, 1 -Ethylbutyl, n-Heptyl, 1 -Ethylpentyl, 1 -Propylbutyl und n-Octyl .
(C1-Cs)-AIkOXy und (C1-Ca)-AIkOXv stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkoxyrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen. Bevorzugt ist ein geradkettiger oder verzweigter Alkoxyrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methoxy, Ethoxy, n-Propoxy, Isopropoxy, n-Butoxy, tert. -Butoxy, n-Pentoxy und n-Hexoxy.
(Q-CaVCycIoalkvK (Ci-CTVCycloalkyl und (C^-C^-Cycloalkyl stehen im Rahmen der Erfindung für eine monocyclische, gesättigte Cycloalkylgruppe mit 4 bis 8, 3 bis 7 bzw. 3 bis 6 Kohlenstoffatomen. Im Falle der Gruppe D ist ein Cycloalkylrest mit 5 oder 6 Kohlenstoffatomen und im Falle der Gruppe R3 ist ein Cycloalkylrest mit 4 bis 6 Kohlenstoffatomen bevorzugt. Beispielhaft und vorzugsweise seien genannt: Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl, Cycloheptyl und Cyclooctyl.
(C4-CR)-Cycloalkenyl steht im Rahmen der Erfindung für eine monocyclische Cycloalkylgruppe mit 4 bis 8 Kohlenstoffatomen und einer Doppelbindung. Bevorzugt ist ein Cycloalkenylrest mit 5 oder 6 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Cyclobutenyl, Cyclo- pentenyl, Cyclohexenyl, Cycloheptenyl und Cyclooctenyl.
(Cfi-Cjn)-Aryl steht im Rahmen der Erfindung für einen aromatischen Carbocyclus mit 6 oder 10 Ring-Kohlenstoffatomen. Bevorzugte Arylreste sind Phenyl und Naphthyl.
5- oder 6-gliedriges Heteroaryl steht im Rahmen der Erfindung für einen aromatischen Hetero- cyclus (Heteroaromaten) mit insgesamt 5 bzw. 6 Ringatomen, der ein oder zwei Ring-Heteroatome aus der Reihe N, O und/oder S enthält und über ein Ring-Kohlenstoffatom oder gegebenenfalls ein Ring-Stickstoffatom verknüpft ist. Beispielhaft seien genannt: Furyl, Pyrrolyl, Thienyl, Pyrazolyl, Imidazolyl, Thiazolyl, Oxazolyl, Isoxazolyl, Isothiazolyl, Pyridyl, Pyrimidinyl, Pyridazinyl und
Pyrazinyl. Bevorzugt sind 5-gliedrige Heteroaryl-Reste wie insbesondere Furyl, Thienyl, Thia- zolyl, Oxazolyl, Isoxazolyl und Isothiazolyl.
Halogen schließt im Rahmen der Erfindung Fluor, Chlor, Brom und Iod ein. Bevorzugt sind Chlor und Fluor.
Wenn Reste in den erfindungsgemäßen Verbindungen substituiert sind, können die Reste, soweit nicht anders spezifiziert, ein- oder mehrfach substituiert sein. Im Rahmen der vorliegenden Erfindung gilt, dass für alle Reste, die mehrfach auftreten, deren Bedeutung unabhängig voneinander ist. Eine Substitution mit ein, zwei oder drei gleichen oder verschiedenen Substituenten ist bevorzugt. Ganz besonders bevorzugt ist die Substitution mit einem Substituenten.
Bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
A für eine Gruppe der Formel
steht, worin
* die Verknüpfungsstelle mit der CR]R2-Gruppierung bedeutet,
R6 für einen Substituenten ausgewählt aus der Reihe Fluor, Chlor, Cyano, (Ci-C4)-
Alkyl und (CrC4)-Alkoxy steht, wobei Alkyl und Alkoxy ihrerseits bis zu fünffach mit Fluor substituiert sein können,
und
n die Zahl 0, 1, 2 oder 3 bedeutet,
wobei für den Fall, dass der Substiruent R6 mehrfach auftritt, seine Bedeutungen gleich oder verschieden sein können,
D für Phenyl, Thienyl, Furyl, Cyclopentyl, Cyclohexyl, Cyclopentenyl oder Cyclohexenyl steht, wobei
Phenyl, Thienyl und Furyl ihrerseits mit Fluor, Chlor, Cyano, (Ci-C4)-Alkyl, (C1-C4)- Alkoxy, Trifluormethyl oder Trifluormethoxy
- T - und
Cyclopentyl, Cyclohexyl, Cyclopentenyl und Cyclohexenyl ihrerseits mit Fluor oder (Ci - C4)-Alkyl substituiert sein können,
R1 für Wasserstoff, Fluor, Hydroxy oder Methyl steht,
R2 für Wasserstoff steht
oder
R1 und R2 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Carbonylgruppe bilden,
R3 für (C3-C6)-Alkyl oder (C3-C6)-Cycloalkyl steht
und
R4 und R5 unabhängig voneinander für Wasserstoff oder Methyl stehen oder gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, einen spiro-verknüpften 3- bis 5-gliedrigen Cycloalkyl-Ring bilden,
sowie ihre Salze, Solvate und Solvate der Salze.
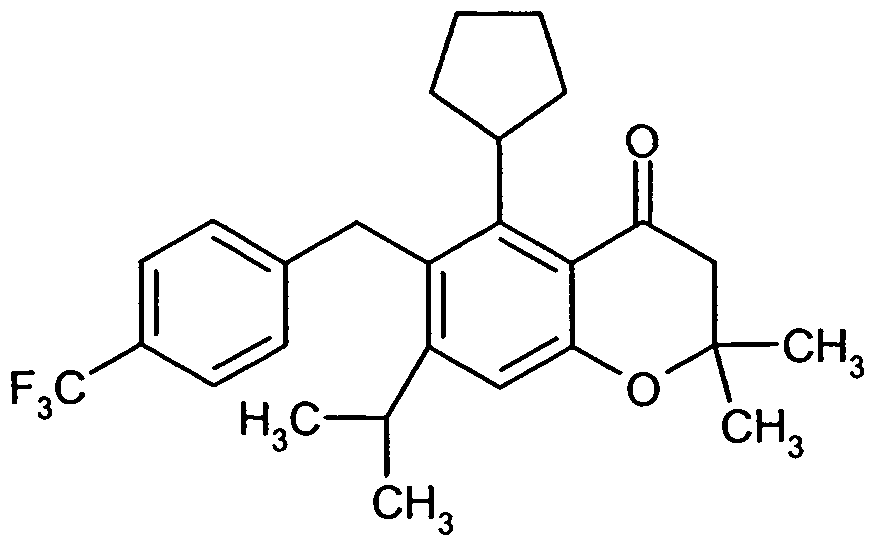
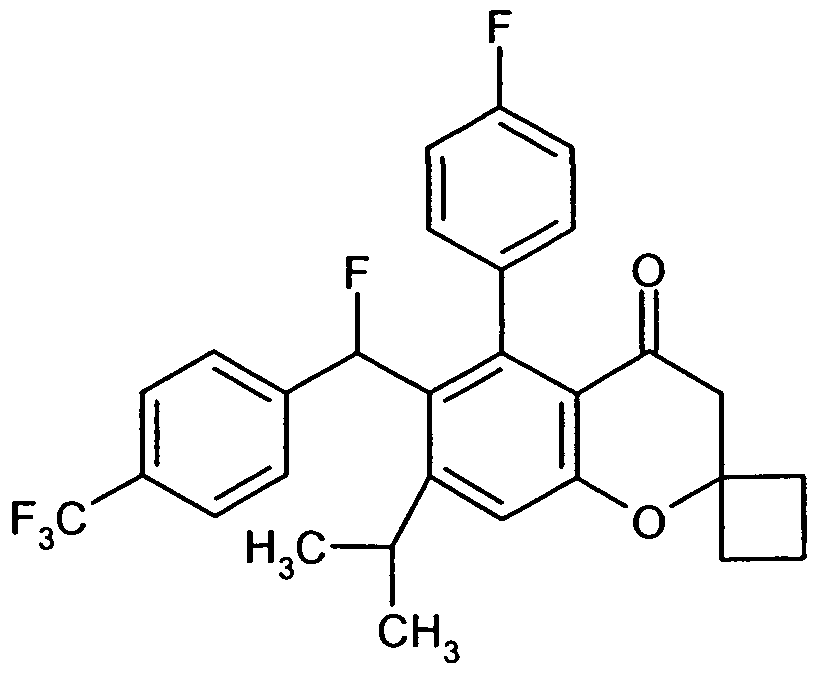
Besonders bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
A für eine Gruppe der Formel
steht, worin
* die Verknüpfungsstelle mit der CR1R2 -Gruppierung bedeutet
und
R6 Trifluormethyl, Trifluormethoxy oder tert.-Butyl bedeutet,
D für Phenyl, 4-Fluorphenyl, Cyclopentyl, Cyclohexyl, Cyclopent-1-en-l-yl oder Cyclohex- 1-en-l-yl steht,
R1 für Wasserstoff, Fluor oder Hydroxy steht,
R2 für Wasserstoff steht
oder
R1 und R2 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Carbonylgruppe bilden,
R3 für Isopropyl oder Cyclopentyl steht
und
R4 und R5 für Methyl stehen oder gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, einen spiro-verknüpften Cyclopropyl- oder Cyclobutyl-Ring bilden,
sowie ihre Salze, Solvate und Solvate der Salze.
Die in den jeweiligen Kombinationen bzw. bevorzugten Kombinationen von Resten im einzelnen angegebenen Reste-Definitionen werden unabhängig von den jeweiligen angegebenen Kombinationen der Reste beliebig auch durch Reste-Definitionen anderer Kombinationen ersetzt.
Ganz besonders bevorzugt sind Kombinationen von zwei oder mehreren der oben genannten Vor- zugsbereiche.
Weiterer Gegenstand der Erfindung ist ein Verfahren zur Herstellung der erfϊndungsgemäßen Verbindungen der Formel (I), dadurch gekennzeichnet, dass man entweder
[A] eine Verbindung der Formel (Ei)
in welcher R3, R4 und R5 jeweils die oben angegebenen Bedeutungen haben
und
R7 für Wasserstoff, Methyl oder eine übliche Hydroxy-Schutzgruppe wie beispielsweise AHyI, Benzyl, Tetrahydropyranyl oder Trialkylsilyl steht,
zunächst in einem inerten Lösungsmittel gegebenenfalls in Gegenwart eines Katalysators mit einer metallorganischen Verbindung der Formel (III)
A-Q (III),
in welcher A die oben angegebene Bedeutung hat
und
Q für Li, -MgBr, -ZnBr oder -B(OH)2 steht,
zu einer Verbindung der Formel (IV)
in welcher A, R3, R4, R5 und R7 jeweils die oben angegebenen Bedeutungen haben,
kuppelt, diese dann zu einer Verbindung der Formel (V)
in welcher A, R3, R4, R5 und R7 jeweils die oben angegebenen Bedeutungen haben,
oxidiert, anschließend im Falle, dass R
7 für Methyl oder eine Hydroxy-Schutzgruppe steht, diesen Rest nach üblichen Methoden abspaltet, die resultierende Verbindung der Formel (Va)
in welcher A, R , R und R jeweils die oben angegebenen Bedeutungen haben,
nach Standardmethoden in eine Verbindung der Formel (VI)
in welcher A, R3, R4 und R5 jeweils die oben angegebenen Bedeutungen haben
und
X für eine Abgangsgruppe wie beispielsweise Chlor, Brom, Iod, Tosylat, Mesylat oder Triflat steht,
überführt, danach in einem inerten Lösungsmittel in Gegenwart einer Base und eines geeigneten Palladium-Katalysators mit einem Boronsäure-Derivat der Formel (VE)
O-Ra
D-B (vπ),
O-Rö
in welcher D die oben angegebene Bedeutung hat
und
R8 für Wasserstoff oder (CrC4)-Alkyl steht oder beide Reste zusammen eine -C(CH3)2-C(CH3)2-Brücke bilden,
zu einer Verbindung der Formel (Vm)
in welcher A, D, R3, R4 und R5 jeweils die oben angegebenen Bedeutungen haben,
kuppelt und diese dann durch asymmetrische Reduktion zu einer Verbindung der Formel (I-A)
in welcher A, D, R , R und R jeweils die oben angegebenen Bedeutungen haben,
umsetzt
oder in veränderter Reihenfolge der Reaktionsschritte
[B] eine Verbindung der Formel (IIa)
in welcher R , R und R jeweils die oben angegebenen Bedeutungen haben,
zunächst nach Standardmethoden in eine Verbindung der Formel (EX)
in welcher X, R , R und R jeweils die oben angegebenen Bedeutungen haben,
überführt, dann in einem inerten Lösungsmittel in Gegenwart einer Base und eines geeigneten Palladium-Katalysators mit einem Boronsäure-Derivat der Formel (VII) zu einer Verbindung der Formel (X)
in welcher D, R >3 , r R>4 und R jeweils die oben angegebenen Bedeutungen haben,
kuppelt, anschließend in einem inerten Lösungsmittel gegebenenfalls in Gegenwart eines Katalysators mit einer metallorganischen Verbindung der Formel (JS) zu einer Verbindung der Formel (XI)
in welcher A, D, R i 3 , r R>4 und R jeweils die oben angegebenen Bedeutungen haben,
umsetzt und diese dann durch asymmetrische Reduktion in eine Verbindung der Formel (I-B)
in welcher A, D, R , R und R jeweils die oben angegebenen Bedeutungen haben,
überfuhrt
oder die Verbindung der Formel (XI) zunächst mit Hilfe eines Fluorierungsmittels zu einer Verbindung der Formel (XIT)
in welcher A, D, R , R und R jeweils die oben angegebenen Bedeutungen haben,
und nachfolgend durch asymmetrische Reduktion zu einer Verbindung der Formel (I-C)
in welcher A, D, R , R und R jeweils die oben angegebenen Bedeutungen haben,
umsetzt
und die so erhaltenen erfindungsgemäßen Verbindungen gegebenenfalls mit den entsprechenden (i) Lösungsmitteln und/oder (ii) Basen oder Säuren in ihre Solvate, Salze und/oder Solvate der Salze überführt.
Die aus den oben beschriebenen Verfahren resultierenden Verbindungen der Formeln (I- A), (I-B), (XI) und (XII) können gegebenenfalls nach üblichen Methoden durch Reduktion, Oxidation, Fluorierung und/oder Methylierung im Bedeutungsumfang von R1 und R2 modifiziert werden, wodurch weitere erfindungsgemäße Verbindungen der Formel (I) erhältlich sind (siehe auch nachfolgende Reaktionsschemata 1-8).
Die Chromanol-Hydroxygruppe kann bei diesen Transformationen, falls erforderlich oder zweckmäßig, temporär durch eine übliche Hydroxy-Schutzgruppe geschützt werden. Bevorzugt wird hierfür eine Trialkylsilyl-Gruppe verwendet; besonders bevorzugt ist tert.-Butyldimethylsilyl. Die Einführung und Entfernung solcher Schutzgruppen wird nach bekannten Methoden durchgeführt [siehe z.B. T.W. Greene und P.G.M. Wuts, Protective Groups in Organic Synthesis, Wiley, New York, 1999].
Zur Einführung der tert.-Butyldimethylsilyl-Gruppe wird bevorzugt tert.-Butyldimethylsilylchlorid oder ter/.-Butyldimethylsilyltrifluormethansulfonat in Verbindung mit Triethylamin, NN-Diiso- propylethylamin, Pyridin, 2,6-Lutidin oder 4-N,N-Dimethylaminopyridin als Base verwendet. Die
Abspaltung der ter/.-Butyldimethylsilyl-Gruppe erfolgt vorzugsweise mit Hilfe von Tetra-n-butyl- ammoniumfluorid (TBAP).
Geeignete Reduktionsmittel für die Reduktion von Ketonen zu sekundären Alkoholen sind beispielsweise komplexe Aluminium- oder Borhydride wie Lithium-, Natrium-, Kalium-, Zinkbor- hydrid, Lithiumaluminiumhydrid, Diisobutylaluminiumhydrid (DIBAL-H), Natrium-bis-(2-meth- oxyethoxy)-aluminiumdihydrid, Lithiumtrialkylborhydride oder Lithiumtrialkoxyaluminium- hydride, oder Boran-Komplexe wie Boran-Tetrahydrofuran-, Boran-Dimethylsulfid- oder Boran- NN-Diethylanilin-Komplex.
Inerte Lösungsmittel für die Verfahrensschritte (U) + (Iu) → (TV) und (X) + (UT) → (XI) sind bei- spielsweise Ether wie Diethylether, Diisopropylether, Dioxan, Tetrahydrofuran, Glykoldimethyl- ether oder Diethylenglykoldimethylether, oder Kohlenwasserstoffe wie Benzol, Xylol, Toluol, Pentan, Hexan, Cyclohexan oder Erdölfraktionen. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt ist Tetrahydrofuran.
Die Umsetzungen (U) + (UT) → (IV) und (X) + (UI) → (XI) können gegebenenfalls vorteilhaft unter Zusatz von Dialkylzink- Verbindungen oder Palladium- oder Rhodium-Phosphin-Komplexen als Katalysatoren durchgeführt werden [vgl. z.B. M. Ueda und Ν. Miyaura, J. Org. Chem. 65, 4450-4452 (2000) und dort zitierte Literatur].
Die Reaktionen erfolgen im Allgemeinen in einem Temperaturbereich von -800C bis +500C, bevorzugt bei -800C bis 00C.
Als Oxidationsmittel im Verfahrensschritt (IV) -> (V) sind beispielsweise Mangan(IV)-oxid, Pyri- diniumchlorochromat (PCC), N-Methylmorpholin-N-oxid, 2,2,6,6-Tetramethylpiperidin-l-yloxy- Radikal (TEMPO) oder Dess-Martin-Periodinan (l,l-Dihydro-l,l,l-triacetoxy-l,2-benziodoxol- 3(1H)-On) geeignet. Bevorzugt werden Mangan(IV)-oxid oder Dess-Martin-Periodinan verwendet.
Als Abgangsgruppe X wird vorzugsweise eine Trifiat-Gruppe (Trifluormethylsulfonat) eingesetzt. Zur Einführung dieser Gruppe in den Verfahrensschritten (Va) — > (VI) und (IIa) — > (IX) wird das Phenol-Derivat (Va) bzw. (Ha) in einem inerten Lösungsmittel wie Dichlormethan oder Dimethyl- formamid mit Trifluormethansulfonsäureanhydrid oder bevorzugt mit N,N-Bis(trifluormethansul- fonyl)anilin in Gegenwart einer Base wie Kaliumcarbonat, Pyridin, 2,6-Lutidin, 4-NN-Dimethyl- aminopyridin (DMAP), Triethylamin oder N,N-Diisopropylethylamin umgesetzt.
Inerte Lösungsmittel für die Verfahrensschritte (VI) + (VII) → (VTfl) und (DC) + (VU) → (X) sind beispielsweise Alkohole wie Methanol, Ethanol, n-Propanol, Isopropanol, n-Butanol oder tert.- Butanol, Ether wie Diethylether, Dioxan, Tetrahydrofuran, Glykoldimethylether oder Diethylen-
glykoldimethylether, Kohlenwasserstoffe wie Benzol, Xylol, Toluol, Hexan, Cyclohexan oder Erdölfraktionen, oder andere Lösungsmittel wie Dimethylformamid, Dimethylsulfoxid, NN- Dimethylpropylenharnstoff (DMPU), N-Methylpyrrolidon (ΝMP), Pyridin, Acetonitril oder auch Wasser. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt ist Dioxan.
Als Basen für die Verfahrensschritte (VI) + (VII) → (Vffl) und (DC) + (VIT) → (X) eignen sich übliche anorganische Basen. Hierzu gehören insbesondere Alkalihydroxide wie beispielsweise Lithium-, Natrium- oder Kaliumhydroxid, Alkalihydrogencarbonate wie Natrium- oder Kalium- hydrogencarbonat, Alkali- oder Erdalkalicarbonate wie Lithium-, Natrium-, Kalium-, Calcium- oder Cäsiumcarbonat, Alkalihydrogenphosphate wie Dinatrium- oder Dikaliumhydrogenphosphat, oder Alkaliphosphate wie Trinatrium- oder Trikaliumphosphat. Bevorzugt wird Trikaliumphosphat verwendet.
Als Palladium-Katalysator für die Verfahrensschritte (VI) + (VII) → (VTTT) und (DC) + (VIT) → (X) ["Suzuki-Kupplung"] sind beispielsweise Palladium(π)-acetat, Tetrakis-(triphenylphosphin)- palladium(O), Bis-(triphenylphosphm)-palladium(H)-chlorid, Bis-(acetomtril)-palladium(II)-chlorid oder [I,r-Bis(diphenylphosphino)ferrocen]dichloφalladium(π)-Dichlormethan-Komplex geeignet [vgl. z.B. J. Hassan et al., Chem. Rev. 102, 1359-1469 (2002)].
Die Reaktionen (VI) + (VTT) → (VTTT) und (DC) + (VIT) → (X) werden im Allgemeinen in einem Temperaturbereich von +200C bis +1500C, bevorzugt bei +600C bis +1200C durchgeführt.
Die asymmetrische Reduktion zum (S)-Chromanol in den Verfahrensschritten (VTTT) -> (I-A), (XI) — > (I-B) und (XTT) — > (I-C) sowie analogen Reaktionen erfolgt in Gegenwart katalytischer Mengen (0.01 bis 0.3 Mol-Äquivalente) von enantiomerenreinem (7R,25)-l-Aminoindan-2-ol als chiralem Induktor. Als Reduktionsmittel wird hierbei bevorzugt Boran-NN-Diethylanilin-Komplex eingesetzt. Die Reaktion wird im Allgemeinen in Toluol oder in einem Ether wie Diethylether, Diiso- propylether, Dioxan, Tetrahydrofuran, Glykoldimethylether oder Diethylenglykoldimethylether, vorzugsweise in Tetrahydrofuran, in einem Temperaturbereich von -800C bis +500C, bevorzugt bei 00C bis +300C, durchgeführt.
Die Fluorierung im Verfahrensschritt (XT) — > (XTT) und analogen Reaktionen wird im Allgemeinen in einem Kohlenwasserstoff wie Benzol, Toluol, Xylol, Pentan, Hexan oder Cyclohexan, oder in einem Halogenkohlenwasserstoff wie Dichlormethan, Trichlormethan, Tetrachlormethan, 1,2-Di- chlorethan, Trichlorethylen oder Chlorbenzol als Lösungsmittel durchgeführt. Bevorzugt ist Toluol oder Dichlormethan. Als Fluorierungsreagenz wird vorzugsweise Diethylaminoschwefeltrifluorid
(DAST) oder Morpholinoschwefeltrifluorid eingesetzt. Die Reaktion erfolgt im Allgemeinen in einem Temperaturbereich von -800C bis +400C, bevorzugt bei -600C bis +200C.
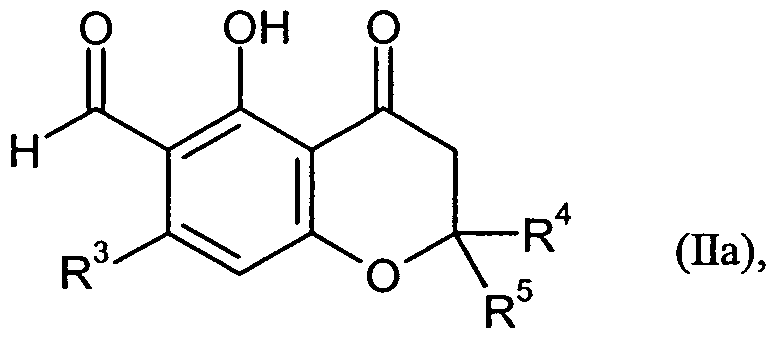
Die Verbindungen der Formel (Ha) können hergestellt werden, indem man eine Verbindung der Formel (XIII)
in welcher R4 und R5 die oben angegebenen Bedeutungen haben,
zunächst nach Standardmethoden in eine Verbindung der Formel (XIV)
in welcher R4 und R5 die oben angegebenen Bedeutungen haben
und
X1 für eine Abgangsgruppe wie Mesylat, Tosylat oder insbesondere Triflat steht,
überführt, anschließend in einem inerten Lösungsmittel in Gegenwart eines geeigneten Katalysators mit einer Zink-organischen Verbindung der Formel (XVa) oder (XVb)
(R3J2Zn R3ZnBr
(XVa) (XVb),
in welchen R3 die oben angegebene Bedeutung hat,
zu einer Verbindung der Formel (XVI)
in welcher R , R und R jeweils die oben angegebenen Bedeutungen haben,
umsetzt und diese dann mit Dichlormethyl-methylether in Gegenwart einer Lewis-Säure zur Verbindung der Formel (Da) formyliert (vgl. Reaktionsschema 4).
Verbindungen der Formel (XIII) sind durch Lewis-Säure-katalysierte Reaktion von Phloroglucinol (XVn) mit einem Acrylsäure-Derivat der Formel (XVIH)
(XVII) (xvπi),
in welcher R4 und R5 die oben angegebenen Bedeutungen haben,
erhältlich (vgl. Reaktionsschemata 2 und 4).
Verbindungen der Formel (II), in welcher R7 für Methyl oder eine Hydroxy-Schutzgruppe steht, können nach üblichen Verfahren aus den Verbindungen der Formel (Ha) erhalten werden.
Als Abgangsgruppe X1 wird vorzugsweise eine Triflat-Gruppe (Trifluormethylsulfonat) eingesetzt. Zur Einführung dieser Gruppe im Verfahrensschritt (XIH) — > (XIV) wird das Phenol-Derivat (XHI) in einem inerten Lösungsmittel wie Dichlormethan oder Dimethylformamid mit Trifluor- methansulfonsäureanhydrid oder bevorzugt mit NN-Bis(trifluormethansulfonyl)anilin in Gegenwart einer Base wie Kaliumcarbonat, Pyridin, 2,6-Lutidin, 4-N,N-Dimethylaminopyridin (DMAP), Triethylamin oder NN-Diisopropylethylamin umgesetzt.
Inerte Lösungsmittel für den Verfahrensschritt (XIV) + (XVa) bzw. (XVb) → (XVI) sind beispielsweise Ether wie Diethylether, Diisopropylether, Methyl-tert.-butylether, Dioxan, Tetrahydro- furan, Glykoldimethylether oder Diethylenglykoldimethylether, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan, Cyclohexan oder Erdölfraktionen, oder andere Lösungsmittel wie Dimethylformamid, Dimethylsulfoxid, NN'-Dimethylpropylenharnstoff (DMPU) oder N-Methylpyrrolidon
(NMP). Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt wird Dimethylformamid verwendet.
Als Katalysator für den Verfahrensschritt (XIV) + (XVa) bzw. (XVb) → (XVI) ["Negishi- Kumada-Kupplung"] sind beispielsweise Bis(diphenylphosphino)ferrocen-palladium(II)-chlorid oder Palladium(II)-acetat in Verbindung mit Triphenylphosphin, unter Zusatz von Ko-Katalysa- toren wie Kupfer(I)-iodid oder Lithiumchlorid, geeignet [vgl. z.B. A. Weichert et al., Synlett, Al?> (1996) und dort zitierte Literatur].
Die Reaktion wird im Allgemeinen in einem Temperaturbereich von -200C bis +1200C, bevorzugt bei 00C bis +600C durchgeführt.
Bei der Formylierung mit Dichlormethyl-methylether im Verfahrensschritt (XVI) →- (Ha) ist als Lewis-Säure beispielsweise Titan(IV)-chlorid, Titan(IV)-isopropylat, Zmn(II)-chlorid oder Magnesiumchlorid geeignet. Bevorzugt wird Titan(IV)-chlorid verwendet.
Im Verfahrensschritt (XVII) + (XVIII) — » (XHI) ist als Lewis-Säure insbesondere Bortrifluorid geeignet. Alternativ können auch Agentien wie Methansulfonsäure oder Phosphorpentoxid eingesetzt werden. Die Reaktion kann in einem Kohlenwasserstoff wie Benzol, Toluol, Xylol, Hexan oder Cyclohexan, oder in einem Halogenkohlenwasserstoff wie Dichlormethan, Trichlormethan, Tetrachlormethan, 1,2-Dichlorethan, Trichlorethylen oder Chlorbenzol als Lösungsmittel oder auch ohne Lösungsmittel durchgeführt werden.
Verbindungen der Formel (II), in welcher R4 und R7 jeweils für Methyl, R5 für Wasserstoff oder (CrC4)-Alkyl und R3 beispielsweise für Cyclopentyl stehen, können auch dadurch hergestellt werden, dass man eine Verbindung der Formel (XIX)
in welcher
R5A für Wasserstoff oder (C,-C4)-Alkyl steht,
zunächst durch Ozonolyse oder Dichromat-Oxidation in ein o-Hydroxybenzaldehyd-Derivat der Formel (XX)
in welcher R die oben angegebene Bedeutung hat,
überfuhrt, anschließend nach Standardmethoden zu einer Verbindung der Formel (XXI)
in welcher R5A die oben angegebene Bedeutung hat
und
X2 für eine Abgangsgruppe wie Tosylat, Mesylat oder insbesondere Triflat steht,
umsetzt, diese dann mit Cyclopenten in Gegenwart eines geeigneten Katalysators und einer Base zu einer Verbindung der Formel (XXII)
in welcher R5A die oben angegebene Bedeutung hat,
kuppelt und nachfolgend in Gegenwart eines geeigneten Katalysators zum Cyclopentan-Derivat
in welcher R5A die oben angegebene Bedeutung hat,
hydriert (vgl. Reaktionsschema 1).
Verbindungen der Formel (XDC) sind über eine 1,4-Addition von Trialkylaluminium-Verbindun- gen oder Alkylcupraten an Visnagin (5-Methoxy-2-methylfuranochromon) (XXTV)
bzw. über eine partielle Reduktion von (XXIV) zugänglich (vgl. Reaktionsschema 1).
Als Abgangsgruppe X2 wird vorzugsweise eine Triflat-Gruppe (Trifluormethylsulfonat) eingesetzt. Zur Einführung dieser Gruppe im Verfahrensschritt (XX) → (XXI) wird das Phenol-Derivat (XX) in einem inerten Lösungsmittel wie Dichlormethan oder Dimethylformamid mit Trifluormethan- sulfonsäureanhydrid oder bevorzugt mit NN-Bis(trifluormethansulfbnyl)anilin in Gegenwart einer Base wie Kaliumcarbonat, Pyridin, 2,6-Lutidin, 4-N,N-Dimethylaminopyridin (DMAP), Triethyl- amin oder N,N-Düsopropylethylamin umgesetzt.
Inerte Lösungsmittel für den Verfahrensschritt (XXI) — > (XXII) sind beispielsweise Ether wie Diethylether, Diisopropylether, Methyl-tert.-butylether, Dioxan, Tetrahydrofuran, Glykoldimethyl- ether oder Diethylenglykoldimethylether, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan,
Cyclohexan oder Erdölfraktionen, oder andere Lösungsmittel wie Dimethylformamid, Dimethyl- sulfoxid, NN'-Dimethylpropylenharnstoff (DMPU), N-Methylpyrrolidon (ΝMP) oder Acetonitril.
Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt wird Aceto- nitril verwendet.
Als Katalysator für den Verfahrensschritt (XXI) → (XXS) ["Heck-Kupplung"] sind beispielsweise Palladium(π)-acetat oder Palladium(π)-trifluoracetat, in Verbindung mit Triphenyl- oder Tritolyl-
phosphin, oder Bis-(dibenzylidenaceton)-palladium(0) geeignet. Die Umsetzung erfolgt unter Zusatz einer Base wie Kaliumcarbonat oder NN-Diisopropylethylamin.
Die Reaktion wird im Allgemeinen in einem Temperaturbereich von +2O0C bis +1200C, bevorzugt bei +400C bis +1000C durchgeführt.
Alle oben beschriebenen Umsetzungen können bei normalem, erhöhtem oder bei erniedrigtem Druck durchgeführt werden (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man jeweils bei Normaldruck.
Die Verbindungen der Formeln (JS), (VII), (XVa), (XVb), (XVII), (XVm) und (XXIV) sind kommerziell erhältlich, literaturbekannt oder können in Analogie zu literaurbekannten Verfahren her- gestellt werden.
Die Herstellung der erfindungsgemäßen Verbindungen kann durch die folgenden Syntheseschemata 1-8 veranschaulicht werden:
Schema 1
[a): AlMe3, Ni(acac)2, THF/Et2O, -200C → 00C; b): O3, CH2Cl2, -78°C; c): PhNTf2, Et3N, CH2Cl2, 00C → RT; d): Pd(OAc)2, P(o-Tol)3, /-Pr2EtN, CH3CN, 45°C; e): H2, 10% Pd/C, EtOAc, RT; f):
THF, -78°C → -200C; g): MnO2, CH2Cl2, RT; h): BBr3, CH2Cl2, -78°C; i): PhNTf2, K2CO3, DMF, -200C → RT; k): Pd(PPh3)4, K3PO4, Dioxan, 1000C; 1): (1Λ,2S)-Aminoindanol, Boran-N,N-diethyl- anilin-Komplex, THF, 00C → RT].
Schema 2
[a): LiOH, H2O, RT; b): PhCOOH, Toluol, 800C].
Schema 3
[a): TBSOTf, Lutidin, Toluol, -200C → 00C; b): DIBAL-H, Toluol, -78°C; c): DAST, CH2Cl2, RT; d): TBAF, THF, RT; e): DIBAL-H, Toluol, -780C].
Schema 4
-MgBr
[a): BF3 x Et2O, 700C; b): PhNTf2, K2CO3, DMF, -200C; c): (R3)2Zn oder R3ZnBr, PdCl2(dppf), LiCl, DMF, 00C; d): TiCl4, -700C → RT; e): PhNTf2, K2CO3, DMF, -200C → RT; f): Pd(PPh3)4, K3PO4, Dioxan, 1000C; g): THF, -78°C → RT].
Schema 5
a)
b)
[a): Dess-Martin-Periodinan, Pyridin, CH2Cl2, -300C → 00C; b): (1R,2S)-Aminoindanol, Boran- NN-diethylanilin-Komplex, THF, RT].
Schema 6
d)
[a): DAST, Toluol, -78°C → -600C; b): (1R,2S)-Aminoindanol, Boran-NN-diethylanilin-Komplex, THF, RT (bei R6 = OCF3 oder CMe3); c): 1. SOCl2, Et3N, THF, RT; 2. (1Λ,25)-Aminoindanol, Boran-NN-diethylanilin-Komplex, THF, RT; 3. LiAlH4, THF, RT; d): (1Λ,25)-Aminoindanol, Boran-NN-diethylanilin-Komplex, THF, RT (bei R6 = CF3)].
Schema 7
[a): (1R,2S)-Aminoindanol, Boran-N.N-diethylanilin-Komplex, THF, RT; b): H2, Pd/C, EtOH].
Schema 8
[a): H2, 5% Rh-Kat., EtOH, RT; b): DAST, CH2Cl2, -780C → -15°C; c): (1R,2S)-Aminoindanol, Boran-NN-diethylanilin-Komplex, THF, RT].
Die erfmdungsgemäßen Verbindungen besitzen wertvolle pharmakologische Eigenschaften und können zur Vorbeugung und Behandlung von Erkrankungen bei Menschen und Tieren verwendet werden.
Die erfindungsgemäßen Verbindungen eröffnen eine weitere Behandlungsalternative und stellen eine Bereicherung der Pharmazie dar. Im Vergleich zu bisher eingesetzten oder aus dem Stand der Technik bekannten Substanzen zeigen die erfindungsgemäßen Verbindungen ein verbessertes Wirkungsspektrum. Sie zeichnen sich durch große Spezifität und gute Verträglichkeit aus. Ein Vorteil der erfindungsgemäßen Verbindungen ist insbesondere ihre hohe Aktivität in humanem Plasma. Gleichzeitig weisen sie als weiteren Vorteil ein verringertes Ablagerungsverhalten im Fettgewebe auf.
Die erfindungsgemäßen Verbindungen sind hochwirksame Inhibitoren des Cholesterin-Ester- Transfer-Proteins (CETP) und stimulieren den reversen Cholesterin-Transport. Sie bewirken eine Erhöhung des HDL-Cholesterinspiegels im Blut. Die erfindungsgemäßen Verbindungen sind insbesondere zur Behandlung und zur primären oder sekundären Prävention koronarer Herzerkran- kungen, z.B. von Myokardinfarkt, Angina pectoris, Herzinsuffizienz, Herzmuskelschwäche, pulmonaler Hypertonie sowie Ischämie-bedingten Schädigungen des Herzens (akutes Koronarsyndrom) einsetzbar. Die erfindungsgemäßen Verbindungen können zudem zur Behandlung und Prävention von Arteriosklerose, periphären Gefäßerkrankungen, Restenose, Schlaganfallen sowie der Alzheimer'schen Krankheit verwendet werden. Darüber hinaus können die erfindungsgemäßen Verbindungen auch zur Behandlung und Prävention von Hypolipoproteinämien, Dyslipidämien, Hypertriglyceridämien, Hyperlipidämien, Hypercholesterolämien, Fettsucht (Adipositas), Fettleibigkeit (Obesitas), Pankreatitis, Insulin-abhängigem und nicht-Insulin-abhängigem Diabetes, diabetischen Spätfolgen, wie beispielsweise Retinopathie, Nephropathie und Neuropathie, von kombinierten Hyperlipidämien sowie des Metabolischen Syndroms eingesetzt werden.
Die pharmakologische Wirkung der erfindungsgemäßen Verbindungen kann mittels der im Folgenden aufgeführten CETP-Inhibitions-Tests ermittelt werden.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prävention von Erkrankungen, insbesondere der zuvor genannten Erkrankungen, unter Verwendung einer wirksamen Menge von mindestens einer der erfindungsgemäßen Verbindungen.
Die erfindungsgemäßen Verbindungen können allein oder bei Bedarf in Kombination mit anderen Wirkstoffen eingesetzt werden. Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine der erfindungsgemäßen Verbindungen sowie einen oder mehrere weitere Wirkstoffe enthalten, zur Behandlung und/oder Prävention der zuvor genannten Erkran- kungen. Als geeignete Kombinationswirkstoffe seien beispielhaft und vorzugsweise genannt:
• Antidiabetika,
• antithrombotisch wirkende Substanzen,
• blutdrucksenkende Substanzen,
• den Fettstoffwechsel verändernde Substanzen,
• antientzündlich wirkende Substanzen,
• den arteriosklerotischen Plaque stabilisierende Substanzen.
Die erfindungsgemäßen Verbindungen können vorzugsweise mit einem oder mehreren
• Wirkstoffen aus der Klasse der Antidiabetika, die in der Roten Liste 2002/π, Kapitel 12 genannt sind,
• antithrombotisch wirkenden Mitteln, beispielhaft und vorzugsweise aus der Gruppe der Thrombozytenaggregationshemmer oder der Antikoagulantien,
• den Blutdruck senkenden Wirkstoffen, beispielhaft und vorzugsweise aus der Gruppe der Calcium-Antagonisten, Angiotensin AH-Antagonisten, ACE-Hemmer, Renin-Inhibitoren, beta-Rezeptoren-Blocker, alpha-Rezeptoren-Blocker, Phosphodiesterase-Inhibitoren, Stimula- toren der löslichen Guanylatcyclase, Verstärker der cGMP-Spiegel, Adenosin-Rezeptor- Agonisten, Aldosteron-Antagonisten, Mineralocorticoid-Rezeptor-Antagonisten, Endothelin- Antagonisten, ECE-Inhibitoren, Vasopeptidase-Inhibitoren sowie der Diuretika, und/oder
• den Fettstoffwechsel verändernden Wirkstoffen, beispielhaft und vorzugsweise aus der Gruppe der Thyroidrezeptor-Agonisten, der Cholesterinsynthese-Inhibitoren wie HMG-CoA- Reduktase-Inhibitoren, Squalensynthase-Inhibitoren, Squalenepoxidase-Inhibitoren oder Oxi- dosqualencyclase-Inhibitoren, der ACAT-Inhibitoren, MTP-Inhibitoren, PPAR-Agonisten, Fibrate, Lipase-Inhibitoren, Cholesterinabsorptionshemmer, Gallensäure-Reabsorptionshem- mer, polymeren Gallensäureadsorber, Lipoprotein(a)-Antagonisten, RXR-Modulatoren, FXR- Modulatoren, LXR-Modulatoren, ATP-Citrat-Lyase-Inhibitoren, Cannabinoid-Rezeptor 1- Antagonisten, Leptin-Rezeptor-Agonisten, Bombesin-Rezeptor-Agonisten, Histamin-Rezep- tor-Agonisten und der Antioxidantien / Radikalfanger
kombiniert werden.
Unter Antidiabetika werden beispielhaft und vorzugsweise Insulin und Insulinderivate sowie oral wirksame hypoglykämische Wirkstoffe verstanden.
Insulin und Insulinderivate umfasst hierbei sowohl Insuline tierischen, menschlichen oder biotechnologischen Ursprungs als auch Gemische hieraus.
Die oral wirksamen hypoglykämischen Wirkstoffe umfassen beispielhaft und vorzugsweise SuI- phonylharnstoffe, Biguadine, Meglitinid-Derivate, Oxadiazolidinone, Thiazolidindione, Glukosi- dase-Inhibitoren, Glukagon-Antagonisten, GLP-I -Agonisten, DPPIV-Inhibitoren, Ghrelin-Rezep- tor- Antagonisten, CCK 1 -Rezeptor- Agonisten, Leptin-Rezeptor- Agonisten, Insulin-Sensitizer, In- hibitoren von Leberenzymen, die an der Stimulation der Glukoneogenese und/oder Glykogenolyse beteiligt sind, Modulatoren der Glukoseaufhahme sowie Kaliumkanalöffher, wie z.B. diejenigen, die in WO 97/26265 und WO 99/03861 offenbart sind.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfϊndungsgemäßen Verbindungen in Kombination mit Insulin verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Sulphonylharnstoff, wie beispielhaft und vorzugsweise Tolbutamid, Glibenclamid, Glimepirid, Glipizid oder Gliclazid, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Biguanid, wie beispielhaft und vorzugsweise Metformin, ver- abreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Meglitinid-Derivat, wie beispielhaft und vorzugsweise Repa- glinid oder Nateglinid, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfmdungsgemäßen Verbin- düngen in Kombination mit einem PPARgamma-Agonisten beispielsweise aus der Klasse der Thiazolidindione, wie beispielhaft und vorzugsweise Pioglitazon oder Rosiglitazon, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem gemischten PPARalpha/gamma-Agonisten, wie beispielhaft und vorzugsweise GI-262570 (Farglitazar), GW 2331, GW 409544, AVE 8042, AVE 8134, AVE 0847, MK-0767 (KRP-297) oder AZ-242, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Glukosidase-Lihibitor, wie beispielhaft und vorzugsweise Acar- bose, Adiposin, Voglibose oder Miglitol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem DPPIV-Inhibitor, wie beispielhaft und vorzugsweise Vilda- glipitin oder Sitaglipitin, verabreicht.
Unter antithrombotisch wirkenden Mitteln werden vorzugsweise Verbindungen aus der Gruppe der Thrombozytenaggregationshemmer, wie beispielhaft und vorzugsweise Aspirin, Clopidogrel, Ticlopidin oder Dipyridamol, oder der Antikoagulantien verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Thrombin-Inhibitor, wie beispielhaft und vorzugsweise Ximela- gatran, Melagatran, Dabigatran, Tanogitran, Bivalirudin oder Clexane, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfϊndungsgemäßen Verbindungen in Kombination mit einem GPÜb/IIIa-Antagomsten, wie beispielhaft und vorzugsweise Tirofiban oder Abciximab, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Faktor Xa-Inhibitor, wie beispielhaft und vorzugsweise Apixa- ban, Razaxaban, Otamixaban oder Rivaroxaban, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit Heparin oder einem low molecular weight (LMW)-Heparin-Derivat verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Vitamin K-Antagonisten, wie beispielhaft und vorzugsweise Coumarin, verabreicht.
Unter blutdrucksenkenden Mitteln werden beispielhaft und vorzugsweise Verbindungen aus der Gruppe der Calcium-Antagonisten, wie beispielhaft und vorzugsweise die Verbindungen Nifedipin, Amlodipin, Nitrendipin, Nisoldipin, Verapamil oder Diltiazem, der Angiotensin Aü-Antago- nisten, ACE-Hemmer, Renin-Inhibitoren, beta-Blocker, alpha-Blocker sowie der Diuretika verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit Reserpin, Minoxidil, Diazoxid, Dihydralazin, Hydralazin oder Stickoxid freisetzenden Stoffen, wie beispielhaft und vorzugsweise Glycerinnitrat oder Nitroprussid- natrium, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Angiotensin Aü-Antagonisten, wie beispielhaft und vorzugs- weise Losartan, Valsartan, Candesartan, Telmisartan, Embusartan, Irbesartan, Olmesartan, Taso- sartan oder Saprisartan, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACE-Hemmer, wie beispielhaft und vorzugsweise Enalapril, Captopril, Ramipril, Delapril, Fosinopril, Quinopril, Perindopril oder Trandolapril, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination "mit einem Renin-Inhibitor, wie beispielhaft und vorzugsweise Aliskiren, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem beta-Blocker, wie beispielhaft und vorzugsweise Propranolol oder Atenolol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Diuretikum, wie beispielhaft und vorzugsweise Furosemid, verabreicht.
Unter den Fettstoffwechsel verändernden Mitteln werden beispielhaft und vorzugsweise Verbindungen aus der Gruppe der Thyroidrezeptor-Agonisten, der Cholesterinsynthese-Inhibitoren wie HMG-CoA-Reduktase-Inhibitoren oder Squalensynthese-Inhibitoren, der ACAT-Inhibitoren, MTP-Inhibitoren, PPAR-Agonisten, Fibrate, Cholesterinabsorptionshemmer, Gallensäure-Reab- sorptionshemmer, Lipase-Inhibitoren, polymeren Gallensäureadsorber, Lipoproteine-Antagonisten sowie der Cannabinoid-Rezeptor 1 -Antagonisten verstanden.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Thyroidrezeptor-Agonisten, wie beispielhaft und vorzugsweise D-Thyroxin, 3,5,3'-Triiodothyronm (T3), CGS 23425 oder Axitirome (CGS 26214), verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Squalensynthese-Inhibitor, wie beispielhaft und vorzugsweise BMS-188494 oder TAK 475, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem ACAT-Inhibitor, wie beispielhaft und vorzugsweise Avasimibe, Eflucimibe oder CS-505, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Cholesterinabsorptionshemmer, wie beispielhaft und vorzugs- weise Ezetimibe, Tiqueside oder Pamaqueside, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Gallensäure-Reabsorptionshemmer, wie beispielhaft und vorzugsweise Barixibat, AZD 7508, SC 435, SC 635, S-8921, 264W94 oder HM 1453, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem MTP-Inhibitor, wie beispielhaft und vorzugsweise Implitapide, BMS-201038 oder R-103757, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPARalpha-Agonisten, wie z.B. die Fibrate Fenofibrat, Clofibrat, Bezafibrat, Ciprofibrat oder Gemfibrozil, oder wie beispielhaft und vorzugsweise GW 9578, GW 7647, LY-518674 oder NS-220, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem PPARdelta-Agonisten, wie beispielhaft und vorzugsweise GW 501516, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem gemischten PPARalpha/gamma-Agonisten, wie beispielhaft und vorzugsweise GI-262570 (Farglitazar), GW 2331, GW 409544, AVE 8042, AVE 8134, AVE 0847, MK-0767 (KRP-297) oder AZ-242, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem gemischten PPARalpha/gamma/delta-Agonisten, wie beispiel- haft und vorzugsweise MCC-555, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Lipase-Inhibitor aus der Gruppe der endothelialen Lipase- Lihibitoren, der pankreatischen Lipase-Inhibitoren, der gastrischen Lipase-Inhibitoren, der Hormon-sensitiven Lipase-Inhibitoren oder der hepatischen Lipase-Inhibitoren verabreicht.
Bei einer besonders bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Inhibitor der pankreatrischen Lipase, vorzugsweise aus der Klasse der Lipstatine wie beispielhaft Orlistat, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem polymeren Gallensäureadsorber, wie beispielhaft und vorzugs- weise Cholestyramin, Colestipol, Colesolvam, CholestaGel oder Colestimide, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Lipoprotein(a)-Antagonisten, wie beispielhaft und vorzugsweise Gemcabene calcium (CI- 1027) oder Nicotinsäure, verabreicht.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbin- düngen in Kombination mit einem Cannabinoid-Rezeptor 1 -Antagonisten, wie beispielhaft und vorzugsweise Rimonabant, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Antagonisten des Niacin-Rezeptors, wie beispielhaft und vorzugsweise Niaspan, Acipimox oder Niceritrol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Antioxidans, wie beispielhaft und vorzugsweise Probucol, AGI 1067 oder Bo 653, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem LDL-Rezeptor-Inducer, wie beispielhaft Lifibrol, verabreicht.
Bei einer bevorzugten Ausführungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem HMG-CoA-Reduktase-Inhibitor aus der Klasse der Statine, wie beispielhaft und vorzugsweise Lovastatin, Simvastatin, Pravastatin, Fluvastatin, Atorvastatin, Rosuvastatin, Cerivastatin oder Pitavastatin, verabreicht.
Ein weiterer Gegenstand der vorliegenden Erfindung sind Kombinationen der erfindungsgemäßen Verbindungen mit Substanzen, die eine Senkung der HMG-CoA-Reduktase-Genexpression bewirken. Derartige Substanzen können beispielsweise Inhibitoren der HMG-CoA-Reduktasetranskrip- tion oder der HMG-CoA-Reduktasetranslation sein. Eine Inhibition der HMG-CoA-Reduktase- Genexpression kann beispielsweise durch Inhibition der SlP (Site-l)-Protease oder durch Senkung der SREBP (sterol receptor binding protein)-Spiegel hervorgerufen werden.
Ein weiterer Gegenstand der vorliegenden Erfindung sind Kombinationen der erfindungsgemäßen Verbindungen mit Substanzen, die antientzündlich und/oder den arteriosklerotischen Plaque stabilisierend wirken. Derartige Substanzen können beispielsweise Wirkstoffe aus der Klasse der NSAIDs, der Lp-PLA2-Antagonisten oder der Chemokin-Rezeptor-Antagonisten, wie beispielhaft IL-8-Rezeptor- Antagonisten oder MCP-I -Antagonisten, darstellen.
Bei einer bevorzugten Ausfuhrungsform der Erfindung werden die erfindungsgemäßen Verbindungen in Kombination mit einem Lp-PLA2-Antagonisten, wie beispielhaft und vorzugsweise Darapladib oder Goxalapladib, verabreicht.
Die erfindungsgemäßen Wirkstoffkombinationen besitzen wertvolle pharmakologische Eigen- Schäften und können zur Vorbeugung und Behandlung von Erkrankungen verwendet werden.
Die erfindungsgemäßen Wirkstoffkombinationen sind insbesondere zur Behandlung und zur primären oder sekundären Prävention koronarer Herzerkrankungen, z.B. von Myokardinfarkt, Angina pectoris, Herzinsuffizienz, Herzmuskelschwäche, pulmonaler Hypertonie sowie Ischämie-bedingten Schädigungen des Herzens (akutes Koronarsyndrom) einsetzbar. Sie können zudem zur Be- handlung und Prävention von Arteriosklerose, periphären Gefäßerkrankungen, Restenose, Schlaganfällen sowie der Alzheimer'schen Krankheit verwendet werden. Darüber hinaus können die genannten Wirkstoffkombinationen auch zur Behandlung und Prävention von Hypolipoproteinämien, Dyslipidämien, Hypertriglyceridämien, Hyperlipidämien, Hypercholesterolämien, Fettsucht (Adi- positas), Fettleibigkeit (Obesitas), Pankreatitis, Insulin-abhängigem und nicht-Insulin-abhängigem Diabetes, diabetischen Spätfolgen, wie beispielsweise Retinopathie, Nephropathie und Neuropathie, von kombinierten Hyperlipidämien sowie des Metabolischen Syndroms eingesetzt werden. Ferner eignen sich die erfindungsgemäßen Wirkstoffkombinationen zur Behandlung von Bluthochdruck sowie von entzündlichen Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Die erfindungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctival, otisch oder als Implantat bzw. Stent.
Für diese Applikationswege können die erfindungsgemäßen Verbindungen in geeigneten Applikationsformen verabreicht werden.
Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende, die erfindungsgemäßen Verbindungen schnell und/oder modifiziert abgebende Applikationsformen, die die erfindungsgemäßen Verbindungen in kristalliner und/oder amorphisierter und/oder gelöster
Form enthalten, wie z.B. Tabletten (nicht-überzogene oder überzogene Tabletten, beispielsweise
mit magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die Freisetzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfallende Tabletten oder Filme/Oblaten, Filme/Lyophylisate, Kapseln (beispielsweise Hart- oder Weichgelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (z.B. intramuskulär, subcutan, intracutan, percutan oder intraperitoneal). Für die parenterale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusions- Zubereitungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern.
Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulverinhalatoren, Nebulizer), Nasentropfen, -lösungen oder -sprays, lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapseln, Suppositorien, Ohren- oder Augen- präparationen, Vaginalkapseln, wäßrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspensionen, Salben, Cremes, transdermale therapeutische Systeme (z.B. Pflaster), Milch, Pasten, Schäume, Streupuder, Implantate oder Stents.
Bevorzugt sind die orale oder parenterale Applikation, insbesondere die orale Applikation.
Die erfϊndungsgemäßen Verbindungen können in die angeführten Applikationsformen überführt werden. Dies kann in an sich bekannter Weise durch Mischen mit inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a. Trägerstoffe (beispielsweise mikrokristalline Cellulose, Lactose, Mannitol), Lösungsmittel (z.B. flüssige PoIy- ethylenglycole), Emulgatoren und Dispergier- oder Netzmittel (beispielsweise Natriumdodecyl- sulfat, Polyoxysorbitanoleat), Bindemittel (beispielsweise Polyvinylpyrrolidon), synthetische und natürliche Polymere (beispielsweise Albumin), Stabilisatoren (z.B. Antioxidantien wie beispielsweise Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisenoxide) und Geschmacks- und/oder Geruchskorrigentien.
Im Allgemeinen hat es sich als vorteilhaft erwiesen, bei parenteraler Applikation Mengen von etwa 0.001 bis 1 mg/kg, vorzugsweise etwa 0.01 bis 0.5 mg/kg Körpergewicht zur Erzielung wirksamer Ergebnisse zu verabreichen. Bei oraler Applikation beträgt die Dosierung etwa 0.01 bis 100 mg/kg, vorzugsweise etwa 0.01 bis 20 mg/kg und ganz besonders bevorzugt 0.1 bis 10 mg/kg Körpergewicht.
.
- 39 -
Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindest- menge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die nachfolgenden Ausführungsbeispiele erläutern die Erfindung. Die Erfindung ist nicht auf die Beispiele beschränkt.
Die Prozentangaben in den folgenden Tests und Beispielen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
A. Beispiele
Abkürzungen: abs. absolut
Ac Acetyl acac Acetylacetonat analyt. analytisch
BSA bovines Serumalbumin
CE Cholesterinester
CETP Cholesterinester-Transferprotein
DCI direkte chemische Ionisation (bei MS) d Tag(e)
DAST Diethylammoschwefeltrifluorid de Diastereomerenüberschuss
DIBAL-H Diisobutylaluminiumhydrid
DMAP 4-NN-Dirnethylaminopyridin
DMF N,N-Dimethylformamid
DMP Dess-Martin-Periodinan-Reagens
DMSO Dimethylsulfoxid d. Th. der Theorie (bei Ausbeute)
EDTA Ethylendiamin-NNN',N'-tetraessigsäure ee Enantiomerenüberschuss
EI Elektronenstoß-Ionisation (bei MS) eq. Äquivalent(e)
ESI Elektrospray-Ionisation (bei MS)
Et Ethyl
EtOAc Ethylacetat
GC/MS Gaschromatographie-gekoppelte Massenspektrometrie h Stunde(n)
HDL high density lipoprotein
HPLC Hochdruck-, Hochleistungsflüssigchromatographie
Kat. Katalysator
LC/MS Flüssigchromatographie-gekoppelte Massenspektrometrie
LDL low density lipoprotein
Me Methyl min Minute(n)
MS Massenspektrometrie
MTBE Methyl-ter/.-butylether
NMR Kernresonanzspektrometrie
PBS phosphate buffered saline
PdCl2(dppf) Bis(diphenylphosphino)ferrocen-palladium(II)chlorid
Ph Phenyl
Pr Propyl präp. präparativ rac racemisch
RT Raumtemperatur
Rt Retentionszeit (bei HPLC)
SPA scintülation proximity assay
TBAF Tetra-/i-butylammoniumfluorid
TBS tert.-Butyldimethylsilyl
Tf Triflat (Trifluormethylsulfonyl)
THF Tetrahydrofuran
ToI Tolyl
Tris Tris(hydroxymethyl)aminornethan
UV Ultraviolett-Spektrometrie v/v Volumen zu Volumen-Verhältnis (einer Lösung) w/v Gewicht zu Volumen-Verhältnis (einer Lösung)
LC/MS-, GC/MS- und HPLC-Methoden:
Methode 1 (präp. HPLC):
Gerätetyp: Abimed Gilson 305; Säule: YMC GEL ODS-AQS-5 / 15 μm, 250 mm x 30 mm; Lauf- mittel: Gradient Acetonitril/Wasser 50:50 → 80:20 (15 min) → 95:5 (27 min); Fluss: 40 ml/min; UV-Detektion: 210 nm.
Methode 2 (präp. HPLC, chiral):
Säule: Chiralpak AD-H, 250 mm x 20 mm; Laufmittel: Isohexan/Isopropanol 97:3 (20 min); Fluss: 15 ml/min; Temperatur: 24°C; UV-Detektion: 254 nm.
Methode 3 (präp. HPLC):
Säule: Kromasil 100 Cl 8 5 μm, 250 mm x 20 mm; Laufmittel: Acetonitril/Wasser 60:40 (9 min); Fluss: 25 ml/min; Temperatur: 400C; UV-Detektion: 280 nm.
Methode 4 (analyt. HPLC, chiral): Gerätetyp: HP 1100; Säule: Chiralpak IA, 250 mm x 4.6 mm; Laufmittel: Isopropanol/Isohexan 3:97; Fluss: 1.5 ml/min; Temperatur: 24°C; UV-Detektion: 254 nm.
Methode 5 (analyt. HPLC, chiral):
Gerätetyp: HP 1100; Säule: Chiralpak IA, 250 mm x 4.6 mm; Laufmittel: Isopropanol/Isohexan 3:97; Fluss: 1 ml/min; Temperatur: 24°C; UV-Detektion: 260 nm.
Methode 6 (analyt. HPLC, chiral):
Gerätetyp: HP 1100; Säule: Chiralpak IA, 250 mm x 4.6 mm, Laufmittel: Isopropanol/Isohexan 3:97; Fluss: 2 ml/min; Temperatur: 24°C; UV-Detektion: 254 nm.
Methode 7 (LOMS):
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A — > 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 8 (LC/MS): Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A -» 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 208-400 nm.
Methode 9 (LC/MS):
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV DAD; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisen-
säure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A → 2.5 min 30% A → 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 500C; UV-Detektion: 210 nm.
Methode 10 (GCVMS): Instrument: Micromass GCT, GC 6890; Säule: Restek RTX-35MS, 30 m x 250 μm x 0.25 μm; konstanter Fluss mit Helium: 0.88 ml/min; Ofen: 600C; Inlet: 25O0C; Gradient: 600C (0.30 min halten), 50°C/min → 1200C, 16°C/min → 2500C, 30°C/min → 3000C (1.7 min halten).
Methode 11 (analyt. HPLC):
Instrument: HP 1100 mit DAD-Detektion; Säule: Kromasil 100 RP-18, 60 mm x 2.1 mm, 3.5 μm; Eluent A: 5 ml HClO4 (70%-ig) / Liter Wasser, Eluent B: Acetonitril; Gradient: 0 min 2% B → 0.5 min 2% B → 4.5 min 90% B → 6.5 min 90% B → 6.7 min 2% B → 7.5 min 2% B; Fluss: 0.75 ml/min; Temperatur: 300C; UV-Detektion: 210 nm.
Methode 12 (präp. HPLC):
Säule: Kromasil Cl 8, 250 mm x 20, 25, 30 oder 40 mm; Eluent A: Wasser + 1% Ameisensäure, Eluent B: Acetonitril; Gradient: 90-95% A → 95% B; Fluss: 10-50 ml/min; Raumtemperatur; UV- Detektion: 210-254 nm.
Ausgangsverbindungen und Intermediate:
Beispie] IA
4-Methoxy-7,7-dimethyl-6,7-dihydro-5H-nαro[3,2-g]chromen-5-on
Unter Argon werden 25 g Visnagin (108.6 mmol) in 500 ml abs. Diethylether und 50 ml abs. Tetrahydrofuran suspendiert, mit 2.23 g (8.7 mmol) Nickel(II)acetylacetonat versetzt und auf -200C abgekühlt. Bei dieser Temperatur wird langsam mit 81.44 ml (162.9 mmol) Trimethylalumi- nium (2 M Lösung in Hexan) versetzt, dann langsam auf 00C erwärmt und weitergerührt. Nach einer Stunde wird der Ansatz mit 500 ml gesättigter Kalium/Natriumtartrat-Lösung versetzt (starke Gasentwicklung) und danach mit 500 ml Essigsäureethylester verdünnt. Die organische Phase wird abgetrennt und die wässrige Phase erneut mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden zweimal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und zur Trockene eingedampft. Der Rückstand wird über Kieselgel chro- matographiert (Laufmittel: Cyclohexan/Essigsäureethylester 5:1). Es werden 23.28 g (86% d. Th.) der Titelverbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ = 7.42 (d, IH), 6.91 (d, IH), 6.72 (s, IH), 4.15 (s, 3H), 2.71 (s, 2H), 1.44 (s, 6H).
MS (ESIpos): m/z = 247 (M+H)+, 269 (M+Na)+
HPLC (Methode 11): R1= 4.30 min.
Beispiel 2A
7-Hydroxy-5-methoxy-2,2-dimethyl-4-oxochroman-6-carbaldehyd
500 mg (2.03 mmol) 4-Methoxy-7,7-dimethyl-6,7-dihydro-5H-füro[3,2-g]chromen-5-on (Beispiel IA) und 666 mg (2.54 mmol) Triphenylphosphin werden vorgelegt und mit 50 ml Dichlormethan versetzt. Es wird auf -78°C abgekühlt und dann ca. 5 min lang Ozon eingeleitet. Nach Blaufärbung der Lösung wird überschüssiges Ozon mit Sauerstoff ausgetrieben. Es wird zwei Stunden weitergerührt, wobei die Lösung langsam auf Raumtemperatur erwärmt wird. Der Ansatz wird eingeengt und der verbleibende Rückstand über eine Kieselgelsäule gereinigt (Laufmittel: Cyclohexan/Essig- säureethylester 5:1). Es werden 370 mg (72% d. Th.) der Titelverbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ = 12.31 (s, IH), 10.15 (s, IH), 6.20 (s, IH), 4.00 (s, 3H), 2.69 (s, 2H), 1.46 (s, 6H).
MS (ESIpos): m/z = 251 (M+H)+.
Beispiel 3A
6-Formyl-5-methoxy-2,2-dimethyl-4-oxo-3,4-dihydro-2H-chromen-7-yl-trifluormethansulfonat
Unter Argon werden 9.1 g (36.36 mmol) 7-Hydroxy-5-methoxy-2,2-dimethyl-4-oxochroman-6- carbaldehyd (Beispiel 2A) und 10.14 ml (72.73 mmol) Triethylamin in 180 ml Dichlormethan gelöst. Es wird auf 00C abgekühlt, mit 15.59 g (43.64 mmol) N,N-Bis(trifluormethansulfonyl)- anilin und einer Spatelspitze DMAP versetzt, die Kühlung entfernt und der Ansatz bei Raumtemperatur 4 h lang weitergerührt. Danach wird mit Dichlormethan verdünnt und je zweimal mit Wasser und gesättigter Νatriumchlorid-Lösung gewaschen. Die organische Phase wird über Magnesiumsulfat getrocknet, filtriert und zur Trockene eingeengt. Der Rückstand wird über Kieselgel
chromatographiert (Laufmittel: Gradient Cyclohexan -> Cyclohexan/Essigsäureethylester 10:1 → 5:1 → 3:1). Es werden 13.65 g (98% d. Th.) der Titelverbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ = 10.27 (s, IH), 6.68 (s, IH), 4.01 (s, 3H), 2.77 (s, 2H), 1.51 (s, 6H).
MS (ESIpos): m/z = 383 (M+H)+.
Beispiel 4A und Beispiel 5A
7-Cyclopent-2-en-l -yl-5-methoxy-2,2-dimethyl-4-oxochroman-6-carbaldehyd (Beispiel 4 A) und
7-Cyclopent-3-en-l-yl-5-methoxy-2,2-dimethyl-4-oxochroman-6-carbaldehyd (Beispiel 5A)
Unter Argon werden 5 g (13.08 mmol) 6-Formyl-5-methoxy-2,2-dimethyl-4-oxo-3,4-dihydro-2H- chromen-7-yl-trifluormethansulfonat (Beispiel 3A), 46 ml (523.14 mmol) Cyclopenten und 2.73 ml (15.7 mmol) NN-Diisopropylethylamin in 125 ml Acetonitril gelöst. Es werden 29.4 mg (0.13 mmol) Palladium(II)acetat und 79.6 mg (0.26 mmol) Tri-o-tolylphosphin hinzugegeben, Argon- Gas durch die Lösung geleitet und anschließend der Ansatz unter Rückfluss (42-450C) erhitzt. Nach Rühren über Nacht werden nochmals die gleichen Mengen an Palladium(II)acetat und Tri-o- tolylphosphin zugesetzt und die Mischung weiter unter Rückfluss gerührt. Nach insgesamt 54 h wird der Ansatz abgekühlt, über eine Kieselgelschicht filtriert und gut mit Essigsäureethylester nachgewaschen. Die Lösung wird mit Essigsäureethylester verdünnt, anschließend zweimal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der erhaltene Rückstand wird über Kieselgel chromatographiert (Laufmittel: Cyclohexan/ Essigsäureethylester 10:1). Es werden 3.35 g (85% d. Th.) eines Gemischs der Produkte 7-Cyclo- pent-2-en-l -yl-5-methoxy-2,2-dimethyl-4-oxochroman-6-carbaldehyd und 7-Cyclopent-3-en-l-yl- 5-methoxy-2,2-dimethyl-4-oxochroman-6-carbaldehyd im Verhältnis 7:3 erhalten.
1H-NMR (300 MHz, CDCl3): δ - 10.46 (s, IH), 6.72/6.66 (s, IH), 6.05-5.98 (m, IH), 5.79-5.63 (m, IH), 4.88-4.78 und 4.57-4.47 (m, IH), 3.96/3.95 (s, 3H), 2.92-2.80 und 1.62-1.49 (m, IH), 2.71 (s, 2H), 2.66-2.53 (m, IH), 2.47-2.30 (m, 2H), 1.48 (s, 6H).
MS (DCI): m/z = 301 (M+H)+, 318 (M+NH4)+.
Beispiel 6A
7-Cyclopentyl-5-methoxy-2,2-dimethyl-4-oxochroman-6-carbaldehyd
Unter Argon werden 3.3 g (11 mmol) des Gemisches von 7-Cyclopent-2-en-l-yl-5-methoxy-2,2-di- methyl-4-oxochroman-6-carbaldehyd und 7-Cyclopent-3-en-l-yl-5-methoxy-2,2-dimethyl-4-oxo- chroman-6-carbaldehyd (Beispiel 4A/5A) in 200 ml Essigsäureethylester gelöst, mit 500 mg Palladium auf Kohle (10%) versetzt und bei Raumtemperatur unter Normaldruck mit Wasserstoff-Gas beschickt. Nach einer Stunde wird über eine Kieselgelschicht filtriert, mit Essigsäureethylester gut nachgewaschen und das Filtrat eingeengt. Es werden 3.3 g (99% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, CDCl3): 5 = 10.42 (s, IH), 6.78 (s, IH), 4.04 (heptett, IH), 3.92 (s, 3H), 2.71 (s, 2H), 2.12-2.03 (m, 2H), 1.83-1.68 (m, 4H), 1.57-1.43 (m, 8H).
MS (DCI): m/z = 303 (M+H)+, 320 (M+NH4)+.
Beispiel 7A
7-Cyclopentyl-6-{hydroxy[4-(trifluormethyl)phenyl]methyl}-5-methoxy-2,2-dimethyl-2,3-dihydro- 4H-chromen-4-on
Unter Argon werden 3.7 g (12.24 mmol) 7-Cyclopentyl-5-methoxy-2,2-dimethyl-4-oxochrornan-6- carbaldehyd (Beispiel 6A) in 150 ml Tetrahydrofuran suspendiert und auf -78°C abgekühlt. Es werden 29.4 ml (14.68 mmol) einer zuvor frisch hergestellten 0.5 M Lösung von Brom[4-(rrifluor- methyl)phenyl]magnesium in Tetrahydrofuran langsam hinzugefügt. Anschließend wird der An- satz auf -20
0C erwärmt und bei dieser Temperatur 30 min nachgerührt. Es werden bei -20
0C nochmals 12.3 ml (6.15 mmol) der obigen Grignard-Lösung zugesetzt und die Mischung weitere 45 min gerührt. Anschließend wird mit 5%-iger Natriumhydrogencarbonat-Lösung hydrolysiert und dann mehrfach mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden zweimal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und ein- geengt. Das Rohprodukt wird über Kieselgel chromatographiert (Laufmittel: Cyclohexan/Essig- säureethylester 20:1 -> 2:1). Es werden 2.35 g (43% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, CDCl3): δ = 7.59 (d, 2H), 7.44 (d, 2H), 6.74 (s, IH), 6.16 (d, IH), 3.72 (d, IH), 3.27-3.16 (m, 4H), 2.72-2.61 (m, 2H), 2.06-1.95 (m, IH), 1.91-1.55 (m, 7H), 1.48 (s, 3H), 1.47 (s, 3H).
MS (DCI): m/z = 449 (M+H)+, 466 (M+NH4)+.
Beispiel 8A
7-Cyclopentyl-5-methoxy-2,2-dimethyl-6-[4-(trifluormethyl)benzoyl]-2,3-dihydro-4H-chromen-4- on
100 mg (0.22 mmol) 7-Cyclopentyl-6-{hydroxy[4-(trifluormethyl)phenyl]methyl}-5-methoxy-2,2- dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 7A) werden in 5 ml Dichlormethan gelöst, mit 194 mg (2.23 mmol) Mangan(IV)oxid versetzt und die Mischung über Nacht bei Raumtemperatur gerührt. Es wird über eine Kieselgelschicht filtriert, gut mit Essigsäureethylester nachgewaschen und das Filtrat zur Trockene eingeengt. Das Rohprodukt wird über Kieselgel chromatographiert (Laufmittel: Cyclohexan/Essigsäureethylester 10:1). Es werden 82.5 mg (83% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, CDCl3): δ = 7.93 (d, 2H), 7.70 (d, 2H), 6.78 (s, IH), 3.64 (s, 3H), 2.78-2.68 (m, 3H), 1.92-1.81 (m, 2H), 1.79-1.70 (m, 2H), 1.62-1.50 (m, 4H), 1.49 (s, 6H).
MS (DCI): m/z = 447 (M+H)+.
Beispiel 9A 7-Cyclopentyl-5-hydroxy-2,2-dimethyl-6-[4-(trifluormethyl)benzoyl]-2,3-dihydro-4H-chromen-4- on
Unter Argon werden 3.10 g (6.94 mmol) 7-Cyclopentyl-5-methoxy-2,2-dimethyl-6-[4-(trifluor- methyl)benzoyl]-2,3-dihydro-4H-chromen-4-on (Beispiel 8A) in 30 ml abs. Dichlormethan gelöst. Es wird auf -78°C abgekühlt, mit 6.25 ml (6.25 mmol) Bortribromid (1 M in Dichlormethan) versetzt und die gelbliche Lösung bei -78°C gerührt. Nach 1.5 h werden nochmals 6.25 ml (6.25 mmol) Bortribromid (1 M in Dichlormethan) hinzugefügt und die Mischung weiter bei -78°C gerührt. Nach 30 min werden erneut 1.39 ml (1.39 mmol) Bortribromid (1 M in Dichlormethan) zugesetzt. Nach 30 min wird mit 100 ml Wasser versetzt und 30 min lang gerührt, wobei der Ansatz auf Raumtemperatur kommt. Danach wird mehrmals mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden einmal mit gesättigter Natriumhydrogencarbonat-Lösung und einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über eine Kiesel gelsäule gereinigt (Laufmittel: Cyclohexan/ Essigsäureethylester 25:1). Es werden 1.85 g (62% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, CDCl3): δ = 11.79 (s, IH), 7.99 (d, 2H), 7.72 (d, 2H), 6.49 (s, IH), 2.82 (heptett, IH), 2.76 (s, 2H), 1.94-1.82 (m, 2H), 1.81-1.70 (m, 2H), 1.62-1.47 (m, 10H).
MS (DCI): m/z = 433 (M+H)+, 450 (MfNH4)*.
Beispiel 10A
7-Cyclopentyl-2,2-dimethyl-4-oxo-6-[4-(trifluormethyl)benzoyl]-3,4-dihydro-2H-chromen-5-yl- trifluormethansulfonat
Unter Argon werden 390 mg (0.90 mmol) 7-Cyclopentyl-5-hydroxy-2,2-dimethyl-6-[4-(trifluor- methyl)benzoyl]-2,3-dihydro-4H-chromen-4-on (Beispiel 9A) in 3 ml abs. Dimethylformamid gelöst. Es wird auf 00C abgekühlt, mit 137 mg (0.99 mmol) Kaliumcarbonat versetzt, 15 min lang gerührt und anschließend auf -20°C abgekühlt. Dann tropft man eine Lösung von 338 mg (0.95 mmol) NN-Bis(trifluormethansulfonyl)anilm in 1.5 ml abs. Dimethylformamid langsam hinzu. Der Ansatz wird eine Stunde bei -200C gerührt, danach langsam auf Raumtemperatur erwärmt und weiter gerührt. Nach 3 h werden nochmals 20 mg (0.06 mmol) N,N-Bis(trifluormethansulfonyl)- anilin zugesetzt und die Mischung weiter bei Raumtemperatur gerührt. Nach 2 h wird der Ansatz mit Ammoniumchlorid-Lösung versetzt, mit Wasser und Essigsäureethylester verdünnt, die organische Phase abgetrennt und die wässrige Phase zweimal mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden zweimal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wird über Kieselgel chromatographiert (Laufmittel: Cyclohexan -> Cyclohexan/Essigsäureethylester 10:1). Es wer- den 482 mg (95% d. Th.) der Titelverbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ = 7.89 (d, 2H), 7.72 (d, 2H), 7.09 (s, IH), 2.89 (heptett, IH), 2.80 (s, 2H), 2.10-1.45 (m, 14H).
MS (ESIpos): m/z = 565 (M+H)+, 582 (M+NH4)+.
Beispiel IIA 7-Cyclopentyl-5-(4-fluorphenyl)-2,2-dimethyl-6-[4-(trifluormethyl)benzoyl]-2,3-dihydro-4H- chromen-4-on
In einem ausgeheizten Kolben werden 250 mg (0.44 mmol) 7-Cyclopentyl-2,2-dimethyl-4-oxo-6- [4-(trifluormethyl)benzoyl]-3,4-dihydro-2H-chromen-5-yl-trifluormethansulfonat (Beispiel 10A), 81 mg (0.58 mmol) 4-Fluorphenylboronsäure, 160 mg (0.75 mmol) Kaliumphosphat und 56 mg (0.05 mmol) Tetrakis(triphenylphosphin)palladium(0) vorgelegt und die Apparatur durch mehrmaliges Evakuieren und Belüften mit Argon gespült. Anschließend werden 4 ml Dioxan zugesetzt, die Apparatur verschlossen und das Reaktionsgemisch über Nacht unter Rückfluss erhitzt. Der Ansatz wird abgekühlt, über eine Kieselgelschicht filtriert, gut mit Essigsäureethylester nachgewaschen und das Filtrat eingeengt. Das Rohprodukt wird über Kiesel gel gereinigt (Laufmittel: Cyclohexan -> Cyclohexan/Essigsäureethylester 10:1 -> 5:1). Es werden 194 mg (86% d. Th.) der Titelverbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ = 7.60-7.49 (m, 4H), 7.15-6.45 (m, 5H), 2.81-2.60 (m, 3H), 2.18- 1.96 (br. s, IH), 1.84-1.45 (m, 13H).
MS (DCI): m/z = 511 (M+H)+, 528 (M+NH4)+.
Beispiel 12A
[(4S)-7-Cyclopentyl-5-(4-fluorphenyl)-4-hydroxy-2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl][4- (trifluormethyl)phenyl]methanon
Unter Argon werden 111 mg (0.71 mmol) (lR,2,S)-l-Aminoindan-2-ol in 100 ml abs. Tetrahydro- furan vorgelegt, unter Rühren mit 3.36 ml (18.89 mmol) Boran-N,N-diethylanilin-Komplex versetzt und die Mischung 30 min lang nachgerührt. Danach wird auf 00C abgekühlt und 2.41 g (4.72 mmol) 7-Cyclopentyl-5-(4-fluoφhenyl)-2,2-dimethyl-6-[4-(trifluormethyl)benzoyl]-2,3-dihy- dro-4H-chromen-4-on (Beispiel I IA), in 150 ml abs. Tetrahydrofuran gelöst, hinzugegeben. Der Ansatz wird bei auftauendem Eisbad langsam auf Raumtemperatur erwärmt. Nach Rühren über Nacht wird der Ansatz mit Methanol versetzt, zur Trockene eingeengt und der Rückstand in Essig- säureethylester aufgenommen. Es wird zweimal mit 1 N Salzsäure, zweimal mit gesättigter Natriumhydrogencarbonat-Lösung und zweimal mit gesättigter Natriumchlorid-Lösung gewaschen. Die organische Phase wird über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wird über Säulenchromatographie an Kieselgel gereinigt (Laufmittel: Cyclohexan/ Essig- säureethylester 10:1). Es werden 1.72 g (71% d. Th.) der Titelverbindung mit einem ee-Wert von 92% erhalten. Anschließende chromatographische Enantiomerentrennung an chiraler Phase [Säule: Chiralpak AD-Η, 250 x 20 mm; Eluent: iso-Propanol/iso-Ηexan 3:97; Fluss: 15 ml/min; 24 0C; Detektion: 254 nm] liefert 1.3 g der gewünschten, enantiomerenreinen Verbindung.
Rt = 14.62 min [Säule: Chiralpak ID, 250 x 4.6 mm; Eluent: iso-Propanol/iso-Ηexan 3:97; Fluss: 1.0 ml/min; Detektion: 254 nm].
1H-NMR (300 MHz, CDCl3): δ = 7.61 (d, 2H), 7.54 (d, 2H), 7.20-6.74 (m, 5H), 4.70 (br. s, IH), 2.72 (heptett, IH), 2.02 (d, 2H), 2.00-1.41 (m, 14H).
MS (ESIpos): m/z = 513 (M+H)+.
Beispiel 13A
[(4S)-4-{[tert.-Butyl(dimethyl)silyl]oxy}-7-cyclopentyl-5-(4-fluoφhenyl)-2,2-dimethyl-3,4-di- hydro-2H-chromen-6-yl][4-(trifluormethyl)phenyl]methanon
Unter Argon werden 200 mg (0.39 mmol) [(4S)-7-Cyclopentyl-5-(4-fluorphenyl)-4-hydroxy-2,2- dimethyl-3,4-dihydro-2H-chromen-6-yl][4-(trifluormethyl)phenyl]methanon (Beispiel 12A) und 180 μl (1.56 mmol) 2,6-Dimethylpyridin in 1.25 ml Toluol gelöst und auf -200C abgekühlt. Es wird eine Lösung von 0.18 ml (0.78 mmol) Trifluormethansulfonsäure-(tert.-butyldimethylsilyl- ester) in 1.25 ml Toluol zugetropft, 15 min bei -200C gerührt, anschließend auf 00C erwärmt und 1 h nachgerührt. Es werden nochmals 18 μl (0.078 mmol) Trifluormethansulfonsäure-(tert.-butyl- dimethylsilylester) zugesetzt und weitere 1.5 h gerührt. Der Ansatz wird mit 5 ml 0.1 N Salzsäure versetzt und mehrfach mit Essigsäureethylester extrahiert. Die organischen Phasen werden mit einer 1 : 1 -Mischung aus gesättigter Natriumhydrogencarbonat-Lösung und gesättigter Natriumchlorid-Lösung sowie mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wird über Säulenchromatographie an Kieselgel gereinigt (Laufmittel: Cyclohexan/Essigsäureethylester 15:1). Es werden 227 mg (93% d. Th.) der Titel Verbindung erhalten.
1H-NMR (400 MHz, CDCl3): δ = 7.75-7.30 (br. s, 4H), 7.20-6.47 (m, 5H), 4.36 (br. s, IH), 2.71 (br. s, IH), 2.09 (dd, IH), 2.00-1.90 (m, IH), 1.80-1.61 (m, 4H), 1.59-1.41 (m, 10H), 0.69 (s, 9H), -0.12 (s, 3H), -0.73 (s, 3H).
MS (DCI): m/z = 644 (M+NH4)+.
Beispiel 14A [(45)-4-{[tert.-Butyl(dimethyl)silyl]oxy}-7-cyclopentyl-5-(4-fluorphenyl)-2,2-dimethyl-3,4-di- hydro-2H-chromen-6-yl][4-(trifluormethyl)phenyl]methanol
Unter Argon werden 102 mg (0.16 mmol) [(4ιS)-4-{[ter/.-Butyl(dimethyl)silyl]oxy}-7-cyclopentyl- 5-(4-fluorphenyl)-2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl][4-(trifluormethyl)phenyl]methanon (Beispiel 13A) in 2 ml abs. Toluol vorgelegt und auf -78°C abgekühlt. Es werden langsam 250 μl (0.25 mmol) einer Diisobutylaluminiumhydrid-Lösung (1 M in Hexan) zugetropft und der Ansatz bei -78°C gerührt. Nach 30 min werden nochmals 80 μl (0.08 mmol) Diisobutylaluminiumhydrid- Lösung (1 M in Hexan) zugetropft und das Gemisch für weitere 30 min gerührt. Es wird mit 20%- iger Natrium/Kaliumtartrat-Lösung versetzt und mehrfach mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Das erhaltene Rohprodukt wird durch präparative Dickschichtchromatographie an Kieselgel gereinigt (Laufmittel: Cyclohexan/Essigsäureethylester 5:1). Es werden 47 mg (46% d. Th.) der Titelverbindung erhalten, welche ohne weitere Charakterisierung in der nächsten Stufe eingesetzt wird.
Beispiel 15A tert. -Butyl[((4S)-7-cyclopentyl-5-(4-fluoφhenyl)-6- {fluor[4-(trifluormethyl)phenyl]methyl} -2,2- dimethyl-3,4-dihydro-2H-chromen-4-yl)oxy]dimethylsilan
Unter Argon werden 47 mg (0.07 mmol) [(4S)-4-{[tert.-Butyl(dimethyl)silyl]oxy}-7-cyclopentyl- 5-(4-fluoφhenyl)-2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl][4-(trifluormethyl)phenyl]methanol (Beispiel 14A) in 500 μl Dichlormethan gelöst und bei Raumtemperatur langsam mit 16.3 μl (0.12 mmol) Diethylaminoschwefeltrifluorid versetzt. Der Ansatz wird 1 h bei Raumtemperatur gerührt, dann mit Wasser versetzt und mehrfach mit Dichlormethan extrahiert. Die vereinigten organischen Phasen werden einmal mit gesättigter Natriumhydrogencarbonat-Lösung und zweimal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird durch präparative Dickschichtchromatographie an Kieselgel gereinigt (Laufmittel: Cyclohexan/Essigsäureethylester 5:1). Es werden 21 mg (45% d. Th.) der Titelverbindung erhalten.
MS (ESIpos): m/z = 499 [M-OSi(CΗ3)2C(CΗ3)3]+.
Beispiel 16A
5-Hydroxy-2,2-dimethyl-4-oxo-3,4-dihydro-2H-chromen-7-yl-trifluormethansulfonat
Zu einer Lösung von 6.69 g (32.1 mmol) 5,7-Dihydroxy-2,2-dimethyl-2,3-dihydro-4H-chromen-4- on [hergestellt nach L. Xie, Y. Takeuchi, M. Cosentino, A.T. McPhail, K.-Η. Lee, J. Med. Chem. 44, 664-671 (2001)] in 80 ml Dimethylformamid werden bei 00C 4.88 g (35.3 mmol) Kalium- carbonat gegeben, und die Mischung wird für 15 min bei dieser Temperatur gerührt. Dann wird auf -200C abgekühlt und langsam eine Lösung von 11.48 g (31.1 mmol) N-Phenyl-bis(trifluormethan- sulfonimid) in 50 ml Dimethylformamid zugetropft. Nach Rühren bei dieser Temperatur für 3 h werden 10 ml gesättigte Natriumhydrogencarbonat-Lösung und 1000 ml Wasser zugesetzt, und es wird zweimal mit je 500 ml Essigsäureethylester extrahiert. Nach Waschen der vereinigten organischen Phasen mit 200 ml Wasser und 200 ml gesättigter Natriumhydrogencarbonat-Lösung sowie Trocknen über Natriumsulfat wird das Lösungsmittel im Vakuum entfernt. Reinigung des Rück- Stands mittels Säulenchromatographie an Kieselgel (Laufmittel: Gradient Cyclohexan — > Cyclo- hexan/Essigsäureethylester 20:1) liefert das Zielprodukt.
Ausbeute: 8.50 g (78% d. Th.)
LC/MS (Methode 9): R, = 2.94 min.
MS (ESIpos): m/z = 341 (M+H)+
1H-NMR (CDCl3, 300 MHz): δ = 1.49 (s, 6H), 2.78 (s, 2H), 6.34 (d, IH), 6.39 (d, IH).
Beispiel 17A
5-Hydroxy-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on
Zu einer Lösung von 4.25 g (12.5 mmol) 5-Hydroxy-2,2-dimethyl-4-oxo-3,4-dihydro-2H-chromen- 7-yl-trifluormethansulfonat (Beispiel 16A) und 1.59 g (37.5 mmol) Lithiumchlorid in 80 ml entgastem Dimethylformamid werden bei 00C 450 mg (550 μmol) Bis(diphenylphosphino)ferrocen- palladium(II)chlorid-Dichlormethan-Komplex gegeben. Danach werden langsam 24.98 ml (24.98 mmol) einer 1 M Lösung von Diisopropylzink in Toluol zugetropft. Nach Rühren für 10 min bei 00C wird die Mischung auf Raumtemperatur erwärmt und bei dieser Temperatur für weitere 5 h gerührt. Die Reaktionslösung wird vorsichtig mit Wasser hydrolysiert, mit 1 M Salzsäure sauer gestellt, mit 500 ml Wasser verdünnt und mit zweimal mit je 500 ml Essigsäureethylester extrahiert. Nach Waschen mit 100 ml Wasser und 50 ml gesättigter Natriumchlorid-Lösung werden die vereinigten organischen Phasen über Magnesiumsulfat getrocknet und im Vakuum eingeengt. Chromatographie des Rückstands an Kieselgel (Laufmittel: Cyclohexan/Essigsäureethylester 10:1) liefert das Zielprodukt im 2: 1-Gemisch mit dem Isomeren 5-Ηydroxy-2,2-dimethyl-7-propyl-2,3- dihydro-4H-chromen-4-on.
Ausbeute: 2.06 g (66% d. Th.)
LC/MS (Methode 7): Rt = 2.60 min (Ηauptisomer), R, = 2.65 min (Nebenisomer)
MS (ESIpos): Ηauptisomer: m/z = 235 (M+Η)+; Nebenisomer: m/z = 235 (M+H)+
1H-NMR (CDCl3, 400 MHz): Hauptisomer: δ = 1.21 (d, 6H), 1.46 (s, 6H), 2.71 (s, 2H), 2.73-2.85 (m, IH), 6.28 (d, IH), 6.37 (d, IH), 11.62 (s, IH); Nebenisomer: δ = 0.94 (t, 3H), 1.46 (s, 6H), 1.58-1.71 (m, 2H), 2.48 (t, 2H), 2.72 (s, 2H), 6.22 (d, IH), 6.30 (d, IH), 11.64 (s, IH).
Das Isomerengemisch wird mittels präparativer HPLC (Methode 3) aufgetrennt und man erhält 1.03 g isomerenreines 5-Hydroxy-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on:
R, = 3.35 min (HP 1100; Kromasil C18 5 μm, 250 mm x 4 mm; 400C; Fluss: 1 ml/min; Detektion: 280 nm; Laufmittel: Acetonitril/Wasser 70:30).
Beispiel 18A
5-Hydroxy-7-isopropyl-2,2-dimethyl-4-oxochroman-6-carbaldehyd
Zu einer Lösung von 1.03 g (4.40 mmol) 5-Hydroxy-7-isopropyl-2,2-dimethyl-2,3-dihydro^4H- chromen-4-on (Beispiel 17A) in 50 ml Dichlormethan werden bei -500C 13.19 ml (13.19 mmol) einer 1 M Lösung von Titan(IV)chlorid in Dichlormethan getropft, und die Mischung wird für 5 min bei dieser Temperatur gerührt. Anschließend werden langsam 437 μl (4.84 mmol) Dichlor- methylmethylether zugetropft. Man lässt die Mischung im Verlauf von 2.5 h auf -25°C auftauen. Dann wird mit Essigsäureethylester und 1 M Salzsäure verdünnt, die wässrige Phase mit Essig- säureethylester extrahiert und die vereinigten organischen Phasen über Natriumsulfat getrocknet. Das Lösungsmittel wird im Vakuum entfernt und der Rückstand wird mittels Säulenchromatographie an Kieselgel gereinigt (Laufmittel: Cyclohexan/Essigsäureethylester 16:1).
Ausbeute: 1.05 g (91% d. Th.)
LC/MS (Methode 9): R, = 2.72 min.
MS (ESIpos): m/z = 263 (M+Η)+
1H-NMR (CDCl3, 300 MHz): δ = 1.19 (d, 6H), 1.52 (s, 6H), 2.78 (s, 2H), 4.02-4.17 (m, IH), 6.46 (s, IH), 10.49 (s, IH), 12.70 (br. s, IH).
Beispiel 19A
6-Formyl-7-isopropyl-2,2-dimethyl-4-oxo-3,4-dihydro-2H-chromen-5-yl-trifluormethansulfonat
Zu einer Lösung von 2.33 g (8.88 mmol) 5-Hydroxy-7-isopropyl-2,2-dimethyl-4-oxochroman-6- carbaldehyd (Beispiel 18A) in 20 ml Dimethylformamid werden bei 00C 1.35 g (9.77 mmol) Kaliumcarbonat gegeben, und die Mischung wird für 15 min bei 00C gerührt. Dann wird bei -200C eine Lösung von 3.49 g (9.77 mmol) N-Phenyl-bis(trifluormethansulfonimid) in 15 ml Dimethylformamid zugetropft, und der Ansatz wird für 1 h bei dieser Temperatur gerührt. Danach läßt man im Verlauf von 3 h auf 00C auftauen und gibt anschließend gesättigte Ammoniumchlorid-Lösung sowie 200 ml Wasser hinzu. Nach Extraktion mit 2 x 150 ml Essigsäureethylester, Waschen der vereinigten organischen Phasen mit 150 ml gesättigter Natriumchlorid-Lösung und Trocknen über Magnesiumsulfat wird das Lösungsmittel im Vakuum entfernt. Kristallisation des Rückstands aus Cyclohexan liefert das Zielprodukt.
Ausbeute: 2.65 g (76% d. Th.)
LC/MS (Methode 7): R, = 2.76 min.
MS (ESIpos): m/z = 395 (M+H)+
1H-NMR (CDCl3, 300 MHz): δ = 1.24 (d, 6H), 1.52 (s, 6H), 2.79 (s, 2H), 3.88-4.03 (m, IH), 7.04 (s, IH), 10.38 (s, IH).
Beispiel 2OA
5-(4-Fluoφhenyl)-7-isopropyl-2,2-dimethyl-4-oxochroman-6-carbaldehyd
Eine Lösung von 800 mg (2.03 mmol) 6-Formyl-7-isopropyl-2,2-dimethyl-4-oxo-3,4-dihydro-2H- chromen-5-yl-trifluormethansulfonat (Beispiel 19A), 369 mg (2.64 mmol) 4-Fluorphenylboron- säure, 164 mg (142 μmol) Tetrakis(triphenylphosphin)palladium und 731 mg (3.45 mmol) Kalium- phosphat in 10 ml entgastem Dioxan wird über Nacht bei 1000C gerührt. Nach Abkühlen auf Raumtemperatur wird die Mischung mit Ammoniumchlorid-Lösung versetzt und zweimal mit Essigsäureethylester extrahiert. Nach Trocknen der vereinigten organischen Phasen über Natriumsulfat wird das Lösungsmittel im Vakuum entfernt und der Rückstand mittels Säulenchromato- graphie (Laufmittel: Gradient Cyclohexan — > Cyclohexan/Essigsäureethylester 20:1) gereinigt.
Ausbeute: 616 mg (87% d. Th.)
LC/MS (Methode 7): R, = 2.79 min.
MS (ESIpos): m/z = 341 (M+Η)+
1H-NMR (CDCl3, 400 MHz): δ = 1.28 (d, 6H), 1.49 (s, 6H), 2.64 (s, 2H), 3.87-4.02 (m, IH), 7.02- 7.24 (m, 2H), 9.60 (s, IH).
Beispiel 21 A rac-5-(4-Fluorphenyl)-6-{hydroxy[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3- dihydro-4H-chromen-4-on
Zu einer Lösung von 300 mg (881 μmol) 5-(4-Fluorphenyl)-7-isopropyl-2,2-dimethyl-4-oxo- chroman-6-carbaldehyd (Beispiel 20A) in 7 ml Tetrahydrofuran werden bei -78°C 2.29 ml (1.15 mmol) einer frisch hergestellten 0.5 M Lösung von Brom[4-(trifluormethyl)phenyl]magnesiurn in Tetrahydrofuran langsam zugetropft. Danach lässt man langsam auf -200C auftauen und rührt für 45 min bei dieser Temperatur. Anschließend wird 10%-ige Natriumhydrogencarbonat-Lösung zugegeben, dreimal mit Essigsäureethylester extrahiert und die vereinigten organischen Phasen mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phasen über Magnesiumsulfat wird das Lösungsmittel im Vakuum entfernt und der Rückstand mittels Säulenchro- matographie an Kieselgel (Laufmittel: Gradient Cyclohexan — > Cyclohexan/Essigsäureethylester 10:1) gereinigt.
Ausbeute: 400 mg (85% d. Th.)
LC/MS (Methode 7): Rt = 3.06 min
MS (ESIpos): m/z = 503 (M+H)+
1H-NMR (CDCl3, 400 MHz): δ = 0.66 (d, 3H), 1.18 (d, 3H), 1.47 (s, 3H), 1.49 (s, 3H), 2.12 (d, IH), 2.57 (d, IH), 2.68 (d, IH), 3.02-3.18 (m, IH), 5.68-5.77 (m, IH), 6.98 (s, IH), 6.99-7.20 (m, 4H), 7.32 (d, 2H), 7.53 (d, 2H).
Beispiel 22A rac-5-(4-Fluoφhenyl)-6-{fluor[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3- dihydro-4H-chromen-4-on
Zu einer Lösung von 170 mg (350 μmol) rac-5-(4-Fluoφhenyl)-6-{hydroxy[4-(trifluormethyl)- phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 21A) in 4.5 ml Dichlormethan wird bei -78°C langsam eine Lösung von 51 μl (380 μmol) Diethylaminoschwefel- trifluorid in 1 ml Dichlormethan getropft und die Mischung für 2.5 h bei dieser Temperatur gerührt. Dann lässt man langsam auf -200C auftauen. Anschließend wird Wasser zugegeben und zweimal mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden über Natriumsulfat getrocknet und im Vakuum eingeengt. Kristallisation des Rückstands aus Cyclo- hexan/Essigsäureethylester (10:1) liefert 77 mg des Zielprodukts. Durch Säulenchromatographie der eingeengten Mutterlauge an Kieselgel (Laufmittel: Cyclohexan/Essigsäureethylester 20:1) erhält man weitere 60 mg der Zielverbindung.
Ausbeute: 137 mg (78% d. Th.)
LC/MS (Methode 8): R, = 3.10 min
MS (ESIpos): m/z = 489 (M+Η)+
1H-NMR (CDCl3, 300 MHz): δ = 0.74 (d, 3H), 1.18 (d, 3H), 1.48 (s, 3H), 1.51 (s, 3H), 2.62 (d, IH), 2.68 (d, IH), 2.82-2.98 (m, IH), 6.33 (d, IH), 6.97 (d, 2H), 7.03 (s, IH), 7.04-7.22 (m, 4H), 7.57 (d, 2H).
Beispiel 23A
5-(4-Fluoφhenyl)-7-isopropyl-2,2-dimethyl-6-[4-(trifluormethyl)benzoyl]-2,3-dihydro-4H- chromen-4-on
Zu einer Lösung von 100 mg (207 μmol) rac-5-(4-Fluorphenyl)-6-{hydroxy[4-(trifluormethyl)- phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 21A) in 4.5 ml Dichlormethan werden bei 00C 174 mg (410 μmol) l,l-Dihydro-l,l,l-triacetoxy-l,2-benziodoxol- 3(lH)-on gegeben und die Mischung für 4 h bei dieser Temperatur gerührt. Dann wird mit Dichlormethan verdünnt und dreimal mit 1 M Natronlauge gewaschen. Nach Trocknen der organischen Phase über Magnesiumsulfat wird im Vakuum eingeengt und der Rückstand mittels Säulenchromatographie an Kieselgel (Laufmittel: Cyclohexan/Essigsäureethylester 20:1) gereinigt.
Ausbeute: 86 mg (86% d. Th.)
LC/MS (Methode 9): R, = 3.32 min
MS (ESIpos): m/z = 485 (M+Η)+
1H-NMR (CDCl3, 300 MHz): δ = 0.98-1.37 (m, 6H), 1.39-1.64 (m, 6H), 2.57-2.79 (m, 3H), 6.44- 7.19 (m, 4H), 7.03 (s, IH), 7.53 (d, 2H), 7.58 (d, 2H).
Beispiel 24A rαc-5-(4-Fluoφhenyl)-6-{hydroxy[4-(trifluormethoxy)phenyl]methyl}-7-isopropyl-2,2-dimethyl- 2, 3 -dihydro-4H-chromen-4-on
Zu einer Lösung von 338 mg (990 μmol) eines 2:1-Gemischs von rac-5-(4-Fluorphenyl)-7-iso- propyl-2,2-dimethyl-4-oxochroman-6-carbaldehyd (Beispiel 20A) und rac-5-(4-Fluorphenyl)-7-/j- propyl-2,2-dimethyl-4-oxochroman-6-carbaldehyd in 8 ml Tetrahydrofuran werden bei -78°C 2.35 ml (1.19 mmol) einer frisch hergestellten 0.5 M Lösung von Brom[4-(trifiuormethoxy)phenyl]- magnesium in Tetrahydrofuran langsam zugetropft. Danach lässt man langsam auf -200C auftauen und rührt für 45 min bei dieser Temperatur. Anschließend wird 10%-ige Natriumhydrogencarbo- nat-Lösung zugegeben, dreimal mit Essigsäureethylester extrahiert und die vereinigten organischen Phasen mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phasen über Magnesiumsulfat wird das Lösungsmittel im Vakuum entfernt und der Rückstand mittels prä- parativer HPLC (Methode 1) gereinigt.
Ausbeute: 293 mg (59% d. Th.)
LC/MS (Methode 7): R, = 3.06 min
MS (ESIpos): m/z = 503 (M+H)+
1H-NMR (CDCl3, 300 MHz): δ = 0.66 (d, 3H), 1.18 (d, 3H), 1.47 (s, 3H), 1.49 (s, 3H), 2.12 (d, IH), 2.57 (d, IH), 2.68 (d, IH), 3.02-3.18 (m, IH), 5.68-5.77 (m, IH), 6.98 (s, IH), 6.99-7.18 (m, 6H), 7.22 (d, 6H).
Beispiel 25A rαc-5-(4-Fluorphenyl)-6-{fluor[4-(trifluormethoxy)phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3- dihydro-4H-chromen-4-on
Zu einer Lösung von 157 mg (310 μmol) rαc-5-(4-Fluoφhenyl)-6-{hydroxy[4-(trifluormethoxy)- phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 24A) in 4 ml Dichlormethan wird bei -78°C langsam eine Lösung von 45 μl (340 μmol) Diethylaminoschwefel- trifluorid in 1 ml Dichlormethan getropft und die Mischung für 2.5 h bei dieser Temperatur gerührt. Dann lässt man langsam auf -15°C auftauen und rührt für weitere 1.5 h. Anschließend wird Wasser zugegeben und zweimal mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden über Natriumsulfat getrocknet und im Vakuum eingeengt. Säulenchromatographie des Rückstands an Kieselgel (Laufmittel: Gradient Cyclohexan → Cyclohexan/Essigsäureethyl- ester 10:1 → 5:1) liefert die Zielverbindung.
Ausbeute: 145 mg (92% d. Th.)
LC/MS (Methode 9): Rt = 3.42 min
MS (ESIpos): m/z = 505 (M+Η)+.
Beispiel 26A 5-(4-Fluoφhenyl)-7-isopropyl-2,2-dimethyl-6-[4-(trifluormethoxy)benzoyl]-2,3-dihydro-4H- chromen-4-on
Zu einer Lösung von 110 mg (220 μmol) 5-(4-Fluorphenyl)-6-{hydroxy[4-(trifluormethoxy)- phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 24A) in 4.5 ml Dichlormethan werden bei 00C 185 mg (439 μmol) l,l-Dihydro-l,l,l-triacetoxy-l,2-benziodoxol- 3(lH)-on gegeben und die Mischung für 4 h bei dieser Temperatur gerührt. Dann wird mit Dichlormethan verdünnt und dreimal mit 1 M Natronlauge gewaschen. Nach Trocknen der organischen Phase über Magnesiumsulfat wird im Vakuum eingeengt und der Rückstand mittels Säulen- chromatographie an Kieselgel (Laufmittel: Gradient Cyclohexan — > Cyclohexan/Essigsäureethyl- ester 10:1) gereinigt.
Ausbeute: 80 mg (68% d. Th.)
LC/MS (Methode 9): R, = 3.29 min
MS (ESIpos): m/z = 467 (M+Η)+
1H-NMR (CDCl3, 300 MHz): δ = 1.03-1.38 (m, 6H), 1.39-1.63 (m, 6H), 2.52-2.82 (m, 3H), 6.51- 7.18 (m, 7H), 7.54 (d, 2H).
Beispiel 27A rac-6-[(4-rert.-Butylphenyl)(hydroxy)methyl]-5-(4-fluorphenyl)-7-isopropyl-2,2-dimethyl-2,3-di- hydro-4H-chromen-4-on
Zu einer Lösung von 300 mg (880 μmol) 5-(4-Fluorphenyl)-7-isopropyl-2,2-dimethyl-4-oxo- chroman-6-carbaldehyd (Beispiel 20A) in 7 ml Tetrahydrofuran werden bei -78°C 2.12 ml (1.06 mmol) einer frisch hergestellten 0.5 M Lösung von Brom(4-tert.-butylphenyl)magnesium in Tetrahydrofuran langsam zugetropft. Danach lässt man langsam auf -200C auftauen und rührt für 45 min bei dieser Temperatur. Anschließend wird 10%-ige Natriumhydrogencarbonat-Lösung zugegeben, dreimal mit Essigsäureethylester extrahiert und die vereinigten organischen Phasen mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phasen über Magnesiumsulfat wird das Lösungsmittel im Vakuum entfernt und der Rückstand mittels Säulenchromatographie an Kieselgel (Laufmittel: Gradient Cyclohexan -> Cyclohexan/Essigsäureethylester 10:1) gereinigt.
Ausbeute: 325 mg (78% d. Th.)
LC/MS (Methode 7): Rt = 3.29 min
MS (ESIpos): m/z = 475 (M+H)+
1H-NMR (CDCl3, 300 MHz): δ = 0.62 (d, 3H), 1.18 (d, 3H), 1.29 (s, 9H), 1.46 (s, 3H), 1.49 (s, 3H), 2.03 (d, IH), 2.58 (d, IH), 2.66 (d, IH), 3.07-3.23 (m, IH), 5.72 (d, IH), 6.96 (s, IH), 6.97- 7.13 (m, 6H), 7.28-7.32 (m, 2H).
Beispiel 28A
/■αc-6-[(4-/er/.-Butylphenyl)(fluor)methyl]-5-(4-fluoφhenyl)-7-isopropyl-2,2-dimethyl-2,3-di- hydro-4H-chromen-4-on
Zu einer Lösung von 150 mg (320 μmol) rac-6-[(4-tert.-Butylphenyl)(hydroxy)methyl]-5-(4- fluorphenyl)-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 27A) in 4.5 ml Di- chlormethan wird bei -78°C langsam eine Lösung von 46 μl (350 μmol) Diethylaminoschwefel- trifluorid in 1 ml Dichlormethan getropft und die Mischung für 2.5 h bei dieser Temperatur gerührt. Dann lässt man langsam auf -15°C auftauen. Anschließend wird Wasser zugegeben und zweimal mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden über Natriumsulfat getrocknet und im Vakuum eingeengt. Säulenchromatographie des Rückstands an Kieselgel (Laufmittel: Gradient Cyclohexan → Cyclohexan/Essigsäureethylester 10: 1 → 5:1) liefert die Zielverbindung.
Ausbeute: 127 mg (84% d. Th.)
LC/MS (Methode 7): R, = 3.42 min
MS (ESIpos): m/z = 476 (M)+
1H-NMR (CDCl3, 300 MHz): δ = 0.73 (d, 3H), 1.16 (d, 3H), 1.29 (s, 9H), 1.48 (s, 3H), 1.50 (s, 3H), 2.59 (d, IH), 2.68 (d, IH), 2.95-3.11 (m, IH), 6.30 (d, IH), 6.89-7.33 (m, 9H).
Beispiel 29A
6-(4-/er/.-Butylbenzoyl)-5-(4-fluoφhenyl)-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on
Zu einer Lösung von 190 mg (400 μmol) eines 2:1 -Gemisches von rαc-6-[(4-tert.-Butylphenyl)- (hydroxy)methyl]-5-(4-fluorphenyl)-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 27A) und rαc-6-[(4-terΛ-Butylphenyl)(hydroxy)methyl]-5-(4-fluorphenyl)-7-τi-propyl-2,2-di- methyl-2,3-dihydro-4H-chromen-4-on in 7.5 ml Dichlormethan werden bei 00C 340 mg (800 μmol) l,l-Dihydro-l,l,l-triacetoxy-l,2-benziodoxol-3(lH)-on gegeben und die Mischung für 4 h bei dieser Temperatur gerührt. Dann wird mit Dichlormethan verdünnt und dreimal mit 1 M Natronlauge gewaschen. Nach Trocknen der organischen Phase über Magnesiumsulfat wird im Vakuum eingeengt und der Rückstand mittels präparativer ΗPLC (Methode 1) und nachfolgender Säulenchromatographie an Kieselgel (Laufmittel: Gradient Cyclohexan → Cyclohexan/Dichlor- methan/Essigsäureethylester 20:20:0.5) gereinigt.
Ausbeute: 54 mg (29% d. Th.)
LC/MS (Methode 8): Rt = 3.31 min
MS (ESIpos): m/z = 473 (M+Η)+
1H-NMR (CDCl3, 300 MHz): δ = 1.14 (d, 3H), 1.25 (d, 3H), 1.28 (s, 9H), 1.48 (s, 3H), 1.54 (s, 3H), 2.53-2.82 (m, 3H), 6.47-6.61 (m, IH), 6.62-6.73 (m, IH), 6.83-6.98 (m, IH), 7.03 (s, IH), 7.04-7.16 (m, IH), 7.28 (d, 2H), 7.43 (d, 2H).
Beispiel 3OA
7-Isopropyl-2,2-dimethyl-4-oxo-5-phenylchroman-6-carbaldehyd
Eine Lösung von 350 mg (890 μmol) eines Gemisches von 6-Formyl-7-isopropyl-2,2-dimethyl-4- oxo-3,4-dihydro-2H-chromen-5-yl-trifluormethansulfonat (Beispiel 19A) und 6-Formyl-7-n-pro- pyl-2,2-dimethyl-4-oxo-3,4-dihydro-2H-chromen-5-yl-trifluormethansulfonat, 162 mg (1.33 mmol) Phenylboronsäure, 51 mg (40 μmol) Tetrakis(triphenylphosphin)palladium und 320 mg (1.51 mmol) Kaliumphosphat in 6 ml entgastem Dioxan wird über Nacht bei 1000C gerührt. Nach Abkühlen auf Raumtemperatur wird die Mischung über Kieselgur filtriert, mit Essigsäureethylester nachgewaschen und dann das Filtrat im Vakuum eingeengt. Chromatographie des Rückstands an Kieselgel (Laufmittel: Cyclohexan/Essigsäureethylester 9:1) liefert die Zielverbindung als 2:1- Gemisch mit dem entsprechenden n-Propyl-Isomer.
Ausbeute: 263 mg (92% d. Th.)
LC/MS (Methode 8): Rt = 2.95 min (Ηauptisomer), R, = 2.99 min (Nebenisomer)
MS (ESIpos): Ηauptisomer: m/z = 323 (M+Η)+; Nebenisomer: m/z = 323 (M+H)+
1H-NMR (CDCl3, 400 MHz): Hauptisomer (Zielverbindung): δ = 1.26 (d, 6H), 1.48 (s, 6H), 2.63 (s, 2H), 3.81-4.04 (m, IH), 7.04 (s, IH), 7.13-7.21 (m, 2H), 7.37-7.46 (m, 3H), 9.58 (s, IH); Nebenisomer: δ = 1.02 (t, 3H), 1.53 (s, 6H), 2.72 (s, 2H), 1.57-1.72 (m, 2H), 2.91-3.01 (m, 2H), 6.88 (s, IH), 7.13-7.21 (m, 2H), 7.37-7.46 (m, 3H), 9.56 (s, IH).
Beispiel 31A rac-6-{Hydroxy[4-(trifluormethoxy)phenyl]methyl}-7-isopropyl-2,2-dimethyl-5-phenyl-2,3-di- hydro-4H-chromen-4-on
Zu einer Lösung von 138 mg (430 μmol) eines 2:1-Gemisches von 7-Isopropyl-2,2-dimethyl-4- oxo-5-phenylchroman-6-carbaldehyd (Beispiel 30A) und 7-n-Propyl-2,2-dimethyl-4-oxo-5-phenyl- chroman-6-carbaldehyd in 4 ml Tetrahydrofuran werden bei -78°C 1028 μl (510 μmol) einer frisch hergestellten 0.5 M Lösung von Brom[4-(trifluorrnethoxy)phenyl]magnesium in Tetrahydrofuran langsam zugetropft. Danach läßt man langsam auf -20°C auftauen und rührt für 45 min bei dieser Temperatur. Zur Vervollständigung der Reaktion werden weitere 342 μl (205 μmol) der obigen Grignard-Lösung zugetropft und es wird wiederum für 45 min nachgerührt. Danach wird 10%-ige Natriumhydrogencarbonat-Lösung zugegeben, dreimal mit Essigsäureethylester extrahiert und die vereinigten organischen Phasen mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phasen über Magnesiumsulfat wird das Lösungsmittel im Vakuum entfernt und der Rückstand mittels präparativer HPLC (Methode 1) gereinigt, wobei das n-Propyl -Isomer vom Zielprodukt abgetrennt wird.
Ausbeute: 107 mg (52% d. Th.)
LC/MS (Methode 9): Rt = 3.23 min
MS (ESIpos): m/z = 485 (M+H)+
1H-NMR (CDCl3, 300 MHz): δ = 0.63 (d, 3H), 1.18 (d, 3H), 1.48 (s, 3H), 1.49 (s, 3H), 2.16 (d, IH), 2.58 (d, IH), 2.67 (d, IH), 2.97-3.13 (m, IH), 5.70-5.80 (m, IH), 6.98 (s, IH), 7.07-7.25 (m, 5H), 7.28-7.44 (m, 4H).
Beispiel 32A
7-Isopropyl-2,2-dimethyl-5-phenyl-6-[4-(trifluormethoxy)benzoyl]-2,3-dihydro-4H-chromen-4-on
Zu einer Lösung von 95 mg (197 μmol) rαc-6-{Hydroxy[4-(trifluormethoxy)phenyl]methyl}-7-iso- propyl-2,2-dimethyl-5-phenyl-2,3-dihydro-4H-chromen-4-on (Beispiel 31A) in 3 ml Dichlor- methan werden bei 00C 166 mg (390 μmol) l,l-Dihydro-l,l,l-triacetoxy-l,2-benziodoxol-3(lH)- on gegeben und die Mischung für 4 h bei dieser Temperatur gerührt. Dann wird mit Dichlormethan verdünnt und dreimal mit 1 M Natronlauge gewaschen. Nach Trocknen der organischen Phase über Magnesiumsulfat wird im Vakuum eingeengt und der Rückstand mittels Säulenchromatographie an Kieselgel (Laufmittel: Gradient Cyclohexan — > Cyclohexan/Essigsäureethylester 10:1) gereinigt.
Ausbeute: 61 mg (64% d. Th.)
LC/MS (Methode 9): Rt = 3.23 min
MS (ESIpos): m/z = 483 (M+Η)+
1H-NMR (CDCl3, 300 MHz): δ = 1.07-1.38 (m, 6H), 1.39-1.63 (m, 6H), 2.53-2.82 (m, 3H), 6.51- 6.78 (m, IH), 6.81-6.97 (m, IH), 7.00-7.32 (m, 6H), 7.51 (d, 2H).
Beispiel 33 A
5-Cyclohex-l-en-l-yl-7-isopropyl-2,2-dimethyl-4-oxochroman-6-carbaldehyd
Eine Lösung von 365 mg (930 μmol) 6-Formyl-7-isopropyl-2,2-dimethyl-4-oxo-3,4-dihydro-2H- chromen-5-yl-trifluormethansulfonat (Beispiel 19A), 152 mg (1.20 mmol) Cyclohex-1-en-l-yl- boronsäure, 75 mg (60 μmol) Tetrakis(triphenylphosphin)palladium und 334 mg (1.57 mmol) Kaliumphosphat in 5.5 ml entgastem Dioxan wird über Nacht bei 100
0C gerührt. Nach Abkühlen auf Raumtemperatur wird die Mischung mit Ammoniumchlorid-Lösung versetzt und zweimal mit Essigsäureethylester extrahiert. Nach Trocknen der vereinigten organischen Phasen über Natriumsulfat wird das Lösungsmittel im Vakuum entfernt und der Rückstand mittels Säulenchromatographie an Kieselgel (Laufmittel: Cyclohexan/Essigsäureethylester 20:1) gereinigt.
Ausbeute: 203 mg (60% d. Th.)
LC/MS (Methode 8): Rt = 3.21 min
MS (ESIpos): m/z = 327 (M+Η)+
1H-NMR (CDCl3, 300 MHz): δ = 1.19 (d, 3H), 1.25 (d, 3H), 1.44 (s, 3H), 1.49 (s, 3H), 1.56-1.73 (m, IH), 1.73-2.07 (m, 4H), 2.12-2.27 (m, 2H), 2.42-2.48 (m, IH), 2.62 (d, IH), 2.73 (d, IH), 3.84- 4.00 (m, IH), 5.38-5.48 (m, IH), 6.91 (s, IH), 10.09 (s, IH).
Beispiel 34A
5 -Cyclopent- 1 -en- 1 -yl-7-isopropyl-2,2-dimethyl-4-oxochroman-6-carbaldehyd
Eine Lösung von 550 mg (1.39 mmol) 6-Formyl-7-isopropyl-2,2-dimethyl-4-oxo-3,4-dihydro-2H- chromen-5-yl-trifluormethansulfonat (Beispiel 19A), 202 mg (1.81 mmol) Cyclopent- 1-en-l-yl- boronsäure, 112 mg (100 μmol) Tetrakis(triphenylphosphin)palladium und 503 mg (2.37 mmol) Kaliumphosphat in 8 ml entgastem Dioxan wird über Nacht bei 1000C gerührt. Nach Abkühlen auf Raumtemperatur wird die Mischung mit Ammoniumchlorid-Lösung versetzt und zweimal mit Essigsäureethylester extrahiert. Nach Trocknen der vereinigten organischen Phasen über Natriumsulfat wird das Lösungsmittel im Vakuum entfernt und der Rückstand mittels Säulenchromato- graphie an Kieselgel (Laufmittel: Cyclohexan/Essigsäureethylester 20:1) gereinigt.
Ausbeute: 235 mg (43% d. Th.)
LCAlS (Methode 8): R, = 3.11 min
MS (ESIpos): m/z = 313 (M+H)+
1H-NMR (CDCl3, 300 MHz): δ = 1.22 (d, 6H), 1.47 (s, 6H), 2.06-2.23 (m, 2H), 2.49-2.62 (m, 4H), 2.68 (s, 2H), 3.83-4.00 (m, IH), 5.51-5.58 (m, IH), 6.92 (s, IH), 9.98 (s, IH).
Beispiel 35A
5-Cyclohex-l -en-1 -yl-6- {hydroxy[4-(trifluormethyl)phenyl]methyl} -7-isopropyl-2,2-dimethyl-2,3- dihydro-4H-chromen-4-on
Zu einer Lösung von 255 mg (780 μmol) 5-Cyclohex-l-en-l-yl-7-isopropyl-2,2-dimethyl-4-oxo- chroman-6-carbaldehyd (Beispiel 33A) in 6 ml Tetrahydrofuran werden bei -78°C 2.03 ml (1.02 mmol) einer frisch hergestellten 0.5 M Lösung von Brom[4-(trifluormethyl)phenyl]magnesium in Tetrahydrofuran langsam zugetropft. Danach lässt man langsam auf -200C auftauen und rührt für 45 min bei dieser Temperatur. Man gibt weitere 300 μl (193 μmol) der obigen Grignard-Lösung hinzu und rührt eine weitere Stunde, wobei man die Lösung langsam auf 00C auftaut. Danach wird 10%-ige Natriumhydrogencarbonat-Lösung zugegeben, dreimal mit Essigsäureethylester extrahiert und die vereinigten organischen Phasen mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phasen über Magnesiumsulfat wird das Lösungsmittel im Vakuum entfernt. Der Rückstand wird in einer Mischung aus Cyclohexan und Essigsäureethylester (10:1) gelöst, worauf das Zielprodukt auskristallisiert und abiϊltriert wird. Das Filtrat wird eingeengt und der Rückstand mittels Säulenchromatographie an Kieselgel gereinigt (Laufmittel: Gradient Cyclohexan → Cyclohexan/Essigsäureethylester 20:1 → 10:1). Das hierbei erhaltene Zielprodukt wird mit den Kristallen vereinigt. Die Verbindung liegt in Form von zwei Atropisomeren (1 :1) vor.
Ausbeute: 304 mg (82% d. Th.)
LC/MS (Methode 8): R, = 3.34 min (Isomer I), Rt = 3.37 min (Isomer II)
MS (ESIpos): Isomer I: m/z = 473 (M+H)+; Isomer II: m/z = 473 (M+H)+
1H-NMR (CDCl3, 300 MHz, Isomer I [Isomer II]): δ = 0.54 [0.57] (d, 3H), 1.12 [1.14] (d, 3H), 1.42 [1.46] (s, 3H), 1.48 [1.50] (s, 3H), 1.64-2.03 (m, 5H), 2.06-2.25 (m, 2H), 2.28-2.49 (m, IH), 2.58 [2.63] (d, IH), 2.72 [2.79] (d, IH), 2.89-3.09 (m, IH), 5.33-5.40 [5.40-5.47] (m, IH), 6.28- 6.37 (m, IH), 6.82 [6.83] (s, IH), 7.34-7.46 (m, 2H), 7.54 (d, 2H).
Beispiel 36A rαc-5-Cyclopent-l -en-1 -yl-6- {hydroxy[4-(rrifluormethyl)phenyl]methyl} -7-isopropyl-2,2-di- methyl-2,3-dihydro-4H-chromen-4-on
Zu einer Lösung von 94 mg (301 μmol) 5-Cyclopent-l-en-l-yl-7-isopropyl-2,2-dimethyl-4-oxo- chroman-6-carbaldehyd (Beispiel 34A) in 2.3 ml Tetrahydrofuran werden bei -78°C 780 μl (391 μmol) einer frisch hergestellten 0.5 M Lösung von Brom[4-(trifluormethyl)phenyl]magnesium in Tetrahydrofuran langsam zugetropft. Danach läßt man langsam auf -200C auftauen und rührt für 45 min bei dieser Temperatur. Dann wird 10%-ige Natriumhydrogencarbonat-Lösung zugegeben, dreimal mit Essigsäureethylester extrahiert und die vereinigten organischen Phasen mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phasen über Magnesiumsulfat wird das Lösungsmittel im Vakuum entfernt und der Rückstand mittels Säulenchromatographie an Kieselgel (Laufmittel: Gradient Cyclohexan — > Cyclohexan/Essigsäureethylester 20: 1 — » 10: 1) und anschließend mittels präparativer HPLC (Methode 1) gereinigt.
Ausbeute: 119 mg (79% d. Th.)
LC/MS (Methode 8): Rt = 3.30 min
MS (ESIpos): m/z = 459 (M+H)+
1H-NMR (CDCl3, 300 MHz): δ = 0.48-0.69 (m, 3H), 1.06-1.21 (m, 3H), 1.38-1.61 (m, 6H), 1.85- 2.08 (m, IH), 2.06-2.90 (m, 8H), 2.62-3.12 (m, IH), 5.38-5.52 (m, IH), 6.18 (s, IH), 6.83 (s, IH), 7.33-7.44 (m, 2H), 7.56 (d, 2H).
Beispiel 37A 5-Cyclohex-l-en-l-yl-7-isopropyl-2,2-dimethyl-6-[4-(trifluormethyl)benzoyl]-2,3-dihydro-4H- chromen-4-on
Zu einer Lösung von 85 mg (180 μmol) 5-Cyclohex-l-en-l-yl-6-{hydroxy[4-(trifluormethyl)- phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 35A) in 4 ml Dichlormethan werden bei 00C 153 mg (360 μmol) l,l-Dihydro-l,l,l-triacetoxy-l,2-benziodoxol- 3(lH)-on gegeben und die Mischung für 4 h bei dieser Temperatur gerührt. Dann wird mit Dichlormethan verdünnt und dreimal mit 1 M Natronlauge gewaschen. Nach Trocknen der organischen Phase über Magnesiumsulfat wird im Vakuum eingeengt und der Rückstand mittels Säulenchromatographie an Kieselgel (Laufmittel: Gradient Cyclohexan -» Cyclohexan/Dichlormethan/ Essigsäureethylester 20:20:0.5) gereinigt.
Ausbeute: 61 mg (76% d. Th.)
LC/MS (Methode 9): R, = 3.21 min
MS (ESIpos): m/z = 471 (M+Η)+
1H-NMR (CDCl3, 300 MHz): δ = 0.65-1.10 (m, 2H), 1.15 (d, 3H), 1.19-1.70 (m, 12H), 1.71-2.25 (m, 3H), 2.64-2.92 (m, 3H), 4.87 und 5.47 (m, IH), 6.94 (s, IH), 7.65-7.97 (m, 4H).
Beispiel 38A rac-5-Cyclopent- 1 -yl-6- {hydroxy[4-(trifluormethyl)phenyl]methyl } -7-isopropyl-2,2-dimethyl-2,3- dihydro-4H-chromen-4-on
Zu einer Lösung von 4.9 mg (33 μmol) (li?,2S)-l-Aminoindan-2-ol in 5.0 ml Tetrahydrofuran werden langsam 155 μl (872 μmol) Boran-N,N-diethylanilin-Komplex gegeben und die Mischung für 30 min gerührt. Anschließend wird sehr langsam eine Lösung von 100 mg (218 μmol) 5-Cyclo- pent-l-en-l-yl-6-{hydroxy[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3-dihy- dro-4H-chromen-4-on (Beispiel 36A) in 5.0 ml Tetrahydrofuran zugetropft und das Gemisch für 4 h gerührt. Dann wird Methanol zugegeben und im Vakuum eingeengt. Der Rückstand wird in Essigsäureethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Νatriumhydrogencarbonat-Lösung sowie einmal mit gesättigter Νatriumchlorid-Lösung gewa- sehen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Die Reinigung des Rohproduktes erfolgt mittels präparativer ΗPLC (Methode 1). Neben der Titelverbindung werden hierbei auch die als Ηerstellungsbeispiel 22 und 23 beschriebenen Verbindungen isoliert (Ausbeute und analytische Daten siehe dort).
Ausbeute: 35 mg (35% d. Th.)
LC/MS (Methode 7): R, = 3.31 min
MS (ESIpos): m/z = 461 (M+Η)+
1H-NMR (CDCl3, 400 MHz): δ = 0.66 (br. m, 3H), 1.13 (d, 3H), 1.46 (s, 6H), 1.50-1.93 (m, 8H), 2.07 (m, IH), 2.22 (d, IH), 2.76 (s, 2H), 3.00 (s, IH), 6.29 (m, IH), 6.78 (s, IH), 7.39 (d, 2H), 7.56 (d, 2H).
Beispiel 39A
5-Cyclopentyl-7-isopropyl-2,2-dimethyl-6-[4-(trifluormethyl)benzyl]-2,3-dihydro-4H-chromen-4- on
Eine Mischung von 60 mg (130 μmol) 5-Cyclopent-l-en-l-yl-6-{hydroxy[4-(trifluormethyl)- phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 36A) und 14 mg Palladium auf Aktivkohle (10%) in 10 ml Ethanol wird über Nacht unter einer Wasserstoff- atmosphäre bei Normaldruck gerührt. Die Suspension wird über Celite filtriert, mit Ethanol nachgewaschen und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird mittels Säulenchromatographie an Kieselgel (Laufmittel: Gradient Cyclohexan — » Cyclohexan/Essigsäureethylester 40:1) gereinigt.
Ausbeute: 23 mg (40% d. Th.)
LC/MS (Methode 7): R, = 3.42 min
MS (ESIpos): m/z = 444 (M+Η)+.
Beispiel 40A
Cyclobutylidenessigsäure
Es werden 313 g (2.48 mol) Methyl-cyclobutylidenacetat [hergestellt nach A. Goti et al., Tetrahedron 48 (25), 5283-5300 (1992)] vorgelegt, bei Raumtemperatur mit einer Lösung von 208 g (4.96 mol) Lithiumhydroxid-Monohydrat in 4.38 Liter Wasser versetzt und die Mischung 3.5 h lang bei Raumtemperatur gerührt. Danach wird auf 00C abgekühlt und mit konzentrierter Salzsäure auf pH 3.5 eingestellt. Anschließend wird abgesaugt, mit wenig kaltem Wasser gewa- sehen und im Vakuum getrocknet. Es werden 213 g (76% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, CDCl3): δ = 11.80 (br. s, IH), 5.61-5.58 (m, IH), 3.14 (t, 2H), 2.85 (t, 2H), 2.10 (quin, 2H).
MS (ESIpos): m/z = 113 (M+H)+.
Beispiel 41 A
Methyl-cyclopropylidenacetat
Eine Suspension von 100 g (574 mmol) [(1-Ethoxycyclopropyl)oxy](trimethyl)silan, 250 g (746 mmol) Methyl-(triphenylphosphoranyliden)acetat und 9.1 g (75 mmol) Benzoesäure in 1.64 Liter Toluol wird über Nacht bei ca. 800C gerührt. Anschließend wird die Mischung direkt über eine Kieselgelsäule chromatographiert (Laufmittel: 20 Liter Petrolether -> 20 Liter Dichlormethan). Die Produktfraktionen werden vereinigt und bei 400 mbar und 45°C eingeengt. Es werden 63 g (86% d. Th.) der Titelverbindung als farbloses Öl erhalten [siehe auch F. Seyed-Mahdavi et al., Tetrahedron Lett. 27 (51), 6185-6188 (1986)].
1H-NMR (300 MHz, CDCl3): δ = 6.21 (s, IH), 3.72 (s, 3H), 1.42 (t, 2H), 1.20 (t, 2H).
GC/MS (Methode 10): R, = 3.03 min
MS (EIpos): m/z = 112 (M)+.
Beispiel 42 A
5,7-Dihydroxyspiro[chromen-2,r-cyclobutan]-4(3H)-on
Es werden 8 g (49.34 mmol) 1,3,5-Trihydroxybenzol-Dihydrat und 6.64 g (59.21 mmol) Cyclo- butylidenessigsäure (Beispiel 40A) vorgelegt, mit 25 ml (197.4 mmol) Bortrifluorid-Diethylether- Komplex versetzt und dann auf 700C erhitzt. Nach drei Stunden wird der Ansatz abgekühlt, auf 600 ml Eiswasser gegossen, mit 6 N Salzsäure angesäuert und mehrmals mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden zweimal mit gesättigter Natriumchlorid- Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird in
Dichlormethan aufgenommen, verrührt und anschließend vom unlöslichen Feststoff abfiltriert. Das Filtrat wird mit Kieselgel versetzt, eingeengt und der Rückstand über Kieselgel chromatographisch gereinigt (Laufmittel: Cyclohexan — > Cyclohexan/Essigsäureethylester 5:1). Es werden 4.7 g (43% d. Th.) der Titelverbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ = 12.00 (s, IH), 6.25 (br. s, IH), 5.96 (d, IH), 5.93 (d, IH), 2.86 (s, 2H), 2.40-2.25 (m, 2H), 2.22-2.10 (m, 2H), 2.00-1.86 (m, IH), 1.80-1.62 (m, IH).
MS (DCI): m/z = 221 (M+H)+, 238 (M+NH4)+.
Beispiel 43A
5 ,7-Dihydroxyspiro [chromen-2 , 1 '-cyclopropan]-4(3H)-on
Unter Argon werden 112 g (690 mmol) 1,3,5-Trihydroxybenzol-Dihydrat in 230 ml abs. Dimethyl- formamid gelöst, mit 28.82 g (230 mmol) Methyl-cyclopropylidenacetat (Beispiel 41A) und 20 g 4Ä-Molekularsieb (als Pulver) versetzt und bei 1300C Badtemperatur über Nacht gerührt. Danach wird mit 1 Liter 1 N Salzsäure versetzt und mehrfach mit Essigsäureethylester extrahiert. Die ver- einigten organischen Phasen werden einmal mit Wasser und einmal mit gesättigter Natriumchlorid- Lösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Das erhaltene Produkt wird mit 230 ml (2.99 mol) Trifluoressigsäure versetzt, auf 75°C erwärmt und 8 Stunden gerührt. Danach wird abgekühlt, mit Wasser versetzt und mehrfach mit Essigsäureethylester extrahiert. Die vereinten organischen Phasen werden zweimal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wird über Kieselgel gereinigt (Laufmittel: Dichlormethan/Methanol 100:1 -> 100:3). Die Produktfraktionen werden vereinigt und eingeengt. Der erhaltene Rückstand wird mit Dichlormethan versetzt, kurz verrührt und der Niederschlag abgesaugt und im Hochvakuum getrocknet. Es werden so 2.25 g (5% d. Th.) der Zielverbindung erhalten. Die Mutterlauge wird anschließend nochmals über eine Kieselgelsäule gerei- nigt (Laufmittel: Dichlormethan/Methanol 100:1). Auf diese Weise werden weitere 3.41 g (7% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, CDCl3): δ = 12.08 (s, IH), 6.61 (br. s, IH), 5.99 (d, IH), 5.88 (d, IH), 2.77 (s, 2H), 1.08-1.04 (m, 2H), 0.71-0.66 (m, 2H).
MS (ESIpos): m/z = 207 (M+H)+.
Beispiel 44A
5-Hydroxy-4-oxo-3,4-dihydrospiro[chromen-2,r-cyclobutan]-7-yl-trifluormethansulfonat
Unter Argon werden 21.96 g (99.7 mmol) 5,7-Dihydroxyspiro[chromen-2,r-cyclobutan]-4(3H)-on (Beispiel 42A) in 600 ml abs. Dimethylformamid gelöst. Es wird auf 00C abgekühlt, mit 15.16 g (109.7 mmol) Kaliumcarbonat versetzt, 15 min nachgerührt und anschließend auf -200C abgekühlt. Dann tropft man eine Lösung von 37.41 g (104.7 mmol) N,N-Bis(trifluormethansulfonyl)anilin in 200 ml abs. Dimethylformamid langsam hinzu. Nach insgesamt 5 Stunden Rühren wird der Ansatz mit Ammoniumchlorid-Lösung versetzt. Es wird mit Wasser und Essigsäureethylester verdünnt, die organische Phase abgetrennt und die wässrige Phase noch zweimal mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden zweimal mit gesättigter Natriumchlorid- Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wird auf Kieselgel aufgezogen und über eine Kieselgelsäule gereinigt (Laufmittel: Cyclohexan/Essig- säureethylester 30: 1 ). Es werden 28 g (80% d. Th.) der Titelverbindung erhalten.
1H-NMR (500 MHz, CDCl3): δ = 11.89 (s, IH), 6.41 (s, IH), 6.40 (s, IH), 2.96 (s, 2H), 2.42-2.31 (m, 2H), 2.26-2.15 (m, 2H), 2.04-1.92 (m, IH), 1.80-1.68 (m, IH).
MS (ESIneg): m/z = 351 (M-H)".
Beispiel 45A 5-Hydroxy-4-oxo-3,4-dihydrospiro[chromen-2, 1 '-cyclopropan]-7-yl-trifluormethansulfonat
Die Darstellung der Titelverbindung erfolgt analog zu Beispiel 44A aus 5.5 g der Verbindung aus Beispiel 43 A. Es werden 5.4 g (60% d. Th.) des Zielprodukts erhalten.
1H-NMR (400 MHz, CDCl3): δ = 11.95 (s, IH), 6.48 (s, IH), 6.33 (s, IH), 2.88 (s, 2H), 1.18-1.09 (m, 2H), 0.80-0.72 (m, 2H).
Beispiel 46A
5-Hydroxy-7-isopropylspiro[chromen-2, 1 '-cyclobutan]-4(3H)-on
Unter Argon werden 16 g (45.42 mmol) 5-Ηydroxy-4-oxo-3,4-dihydrospiro[chromen-2,r-cyclo- butan]-7-yl-trifluormethansulfonat (Beispiel 44A), 3.71 g (4.54 mmol) [l,l'-Bis(diphenylphos- phmo)ferrocen]dichlorpalladium(II) und 5.78 g (136.25 mmol) Lithiumchlorid in 400 ml abs. Di- methylformamid suspendiert. Es wird auf 00C abgekühlt, mit 90.8 ml (90.8 mmol) Diisopropylzink (I M Lösung in Toluol) versetzt und 4 Stunden bei dieser Temperatur gerührt. Anschließend wird der Ansatz bei 00C mit gesättigter Ammoniumchlorid-Lösung versetzt. Es wird mit Wasser, 1 N Salzsäure und Essigsäureethylester verdünnt, die organische Phase abgetrennt und die wässrige Phase mehrfach mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden zweimal mit Wasser und einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natrium- sulfat getrocknet, filtriert und eingeengt. Der Rückstand wird auf Kieselgel aufgezogen und über eine Kieselgelsäule gereinigt (Laufmittel: Cyclohexan — > Cyclohexan/Essigsäureethylester 20:1). Es werden 9.6 g (86% d. Th.) der Titelverbindung erhalten.
1H-NMR (500 MHz, CDCl3): δ = 11.59 (s, IH), 6.38 (s, IH), 6.32 (s, IH), 2.88 (s, 2H), 2.80 (heptett, IH), 2.38-2.29 (m, 2H), 2.20-2.12 (m, 2H), 1.98-1.89 (m, IH), 1.78-1.68 (m, IH), 1.21 (d, 6H).
MS (ESIpos): m/z = 247 (M+H)+.
Beispiel 47A
5-Hydroxy-7-isopropylspiro[chromen-2, 1 '-cyclopropan]-4(3H)-on
Die Darstellung der Titelverbindung erfolgt analog zu Beispiel 46A aus 5.4 g 5-Hydroxy-4-oxo- S^-dihydrospirofchromen^l'-cyclopropan^-yl-irifluormethansulfonat (Beispiel 45A). Es werden 5.4 g (89% d. Th.) des Zielprodukts mit einer Reinheit von ca. 87% erhalten.
1H-NMR (400 MHz, CDCl3): δ = 11.67 (s, IH), 6.42 (s, IH), 6.27 (s, IH), 2.83-2.73 (m, 3H), 1.21 (d, 6H), 1.11-1.05 (m, 2H), 0.72-0.66 (m, 2H).
MS (DCI): m/z = 233 (M+H)+, 250 (M+NH4)+.
Als Hauptverunreinigung ist das «-Propyl-Isomer enthalten:
5-Hydroxy-7-propylspiro[chromen-2, 1 '-cyclopropan]-4(3H)-on
1H-NMR (400 MHz, CDCl3): δ = 11.68 (s, IH), 6.38 (s, IH), 6.22 (s, IH), 2.80 (s, 2H), 2.49 (t, 2H), 1.68-1.57 (m, 2H), 1.11-1.05 (m, 2H), 0.93 (t, 3H), 0.72-0.66 (m, 2H).
Beispiel 48A
7-Cyclopentyl-5-hydroxyspiro[chromen-2,r-cyclobutan]-4(3H)-on
Unter Argon werden 8.45 g (24.0 mmol) 5-Hydroxy-4-oxo-3,4-dihydrospiro[chromen-2,r-cyclo- butan]-7-yl-trifluormethansulfonat (Beispiel 44A), 3.92 g (4.8 mmol) [l,l'-Bis(diphenylphos-
phino)ferrocen]dichlorpalladium(II) und 6.10 g (144.0 mmol) Lithiumchlorid in 250 ml abs. Di- methylformamid suspendiert. Es wird auf 00C abgekühlt, mit 216 ml (108 mmol) (Cyclopentyl)- zinkbromid (0.5 M Lösung in Tetrahydrofuran) versetzt und über Nacht bei Raumtemperatur gerührt. Anschließend wird der Ansatz mit gesättigter Ammoniumchlorid-Lösung versetzt. Es wird mit Wasser, 1 N Salzsäure und Essigsäureethylester verdünnt, die organische Phase abgetrennt und die wässrige Phase mehrfach mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden zweimal mit Wasser und einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wird auf Kieselgel aufgezogen und über eine Kieselgelsäule gereinigt (Laufmittel: Cyclohexan — »■ Cyclohexan/Essigsäure- ethylester 10:1). Es werden 6.2 g (94% d. Th.) der Titelverbindung in einer Reinheit von 90% erhalten.
1H-NMR (300 MHz, CDCl3): δ = 11.60 (s, IH), 6.38 (s, IH), 6.32 (s, IH), 2.97-2.83 (m, 3H), 2.40- 2.27 (m, 2H), 2.22-2.11 (m, 2H), 2.10-1.88 (m, 3H), 1.86-1.50 (m, 7H).
MS (ESIpos): m/z = 273 (M+H)+.
Beispiel 49A
5-Hydroxy-7-isopropyl-4-oxo-3,4-dihydrospiro[chromen-2,l'-cyclobutan]-6-carbaldehyd
Unter Argon werden 9.60 g (39 mmol) 5-Hydroxy-7-isopropylspiro[chromen-2,l'-cyclobutan]- 4(3H)-on (Beispiel 46A) in 400 ml abs. Dichlormethan gelöst und auf -700C abgekühlt. Bei dieser Temperatur werden 97.44 ml (97.44 mmol) Titan(IV)chlorid (1 M Lösung in Dichlormethan) in dem Maße zugetropft, dass die Temperatur nicht über -65°C steigt. Es wird kurz bei -700C nachgerührt, dann 3.88 ml (42.87 mmol) Dichlormethylmethylether zugesetzt und der Ansatz anschließend auf 00C erwärmt. Nach 3 Stunden bei dieser Temperatur wird die Reaktionsmischung vorsichtig auf Eiswasser gegeben und viermal mit Dichlormethan extrahiert. Die vereinigten organischen Phasen werden dreimal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und zur Trockene eingeengt. Das erhaltene Rohprodukt wird auf Kieselgel aufgezogen und über eine Kieselgelsäule gereinigt (Laufmittel: Cyclohexan -» Cyclohexan/
Essigsäureethylester 30:1). Es werden 8.1 g (76% d. Th.) der Titelverbindung in einer Reinheit von 77% erhalten.
1H-NMR (400 MHz, CDCl3): δ = 12.80-12.55 (br. s, IH), 10.51 (s, IH), 6.50 (s, IH), 4.18-4.05 (m, IH), 2.96 (s, 2H), 2.48-2.32 (m, 2H), 2.28-2.17 (m, 2H), 2.06-1.92 (m, IH), 1.83-1.68 (m, IH), 1.21 (d, 6H).
MS (DCI): m/z = 275 (M+H)+, 292 (M+NH4)+.
Als Hauptverunreinigung ist das «-Propyl-Isomer enthalten:
5-Hydroxy-4-oxo-7-propyl-3,4-dihydrospiro[chromen-2,l'-cyclobutan]-6-carbaldehyd
1H-NMR (400 MHz, CDCl3): δ = 12.80-12.55 (br. s, IH), 10.49 (s, IH), 6.31 (s, IH), 2.98-2.92 (m, 4H), 2.48-2.32 (m, 2H), 2.28-2.17 (m, 2H), 2.06-1.92 (m, IH), 1.83-1.68 (m, IH), 1.65-1.58 (m, 2H), 1.00 (t, 3H).
Beispiel 5OA
6-Formyl-7-isopropyl-4-oxo-3,4-dihydrospiro[chromen-2,r-cyclobutan]-5-yl-trifluormethan- sulfonat
Unter Argon werden 2.16 g (7.87 mmol) 5-Hydroxy-7-isopropyl-4-oxo-3,4-dihydrospiro[chromen-
2,l'-cyclobutan]-6-carbaldehyd (Beispiel 49A) in 40 ml abs. Dimethylformamid gelöst. Es wird auf
00C abgekühlt, mit 1.2 g (8.66 mmol) Kaliumcarbonat versetzt, 15 min gerührt und anschließend bei -200C eine Lösung von 3.16 g (8.66 mmol) N,N-Bis(trifluormethansulfonyl)anilin in 38 ml abs.
Dimethylformamid langsam zugetropft. Der Ansatz wird kurz unter Kühlung nachgerührt und dann langsam auf Raumtemperatur erwärmt. Nach insgesamt 4 Stunden wird der Ansatz mit Ammoniumchlorid-Lösung versetzt, mit Wasser und Essigsäureethylester verdünnt und die organische Phase abgetrennt. Die wässrige Phase wird dreimal mit Essigsäureethylester extrahiert und die vereinigten organischen Phasen zweimal mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wird auf Kieselgel aufgezogen und über eine Kieselgelsäule gereinigt (Laufmittel: Cyclohexan/Essigsäureethylester 50:1 — > 20:1). Es werden 1.42 g (42% d. Th.) der Titelverbindung erhalten. Eine Mischfraktion wird nochmals über Kieselgel, wie oben beschrieben, aufgereinigt. Auf diese Weise werden weitere 1.94 g (39% d. Th.) der Zielverbindung mit einer Reinheit von 65% erhalten.
1H-NMR (300 MHz, CDCl3): δ = 10.36 (s, IH), 7.10 (s, IH), 4.02-3.87 (m, IH), 2.96 (s, 2H), 2.48- 2.33 (m, 2H), 2.29-2.19 (m, 2H), 2.06-1.90 (m, IH), 1.85-1.68 (m, IH), 1.24 (d, 6H).
MS (DCI): m/z = 424 (M+NIL,)+.
Beispiel 51A 5-(4-Fluorphenyl)-7-isopropyl-4-oxo-3,4-dihydrospiro[chromen-2,r-cyclobutan]-6-carbaldehyd
Unter Argon werden 4.00 g (9.84 mmol) 6-Formyl-7-isopropyl-4-oxo-3,4-dihydrospiro[chromen- 2,r-cyclobutan]-5-yl-trifluormethansulfonat (Beispiel 50A), 1.79 g (12.8 mmol) 4-Fluorphenyl- boronsäure, 3.55 g (16.73 mmol) Trikaliumphosphat und 1.25 g (1.08 mmol) Tetrakis(triphenyl- phosphin)palladium(O) vorgelegt und die Apparatur durch mehrmaliges Evakuieren und Belüften mit Argon gespült. Anschließend wird mit 100 ml abs. Dioxan versetzt, die Apparatur verschlossen und das Gemisch unter Rückfluss erhitzt. Nach Rühren über Nacht wird der Ansatz abgekühlt, über eine Kieselgelschicht filtriert, gut mit Essigsäureethylester nachgewaschen und das Filtrat eingeengt. Der Rückstand wird auf Kieselgel aufgezogen und über eine Kieselgelsäule chromato- graphiert (Laufmittel: Cyclohexan → Cyclohexan/Essigsäureethylester 25:1). Das erhaltene Roh-
produkt wird mit heissem Petrolether verrührt, langsam abgekühlt und der Niederschlag abgesaugt. Es werden 3.2 g der Zielverbindung in einer Reinheit von ca. 90% erhalten.
Als Hauptverunreinigung ist das «-Propyl-Isomer enthalten:
5 -(4-Fluorphenyl)-4-oxo-7-propyl-3 ,4-dihydrospiro [chromen-2, 1 '-cyclobutan] -6-carbaldehyd
Anschließende chromatographische Trennung an chiraler Phase [Säule: Daicel Chiralpak AD-H, 250 x 20 mm; Eluent: Ethanol/iso-Hexan 30:70; Fluss: 15 ml/min; 22°C; Detektion: 260 nm] liefert 2.78 g (80% d. Th.) der reinen Titelverbindung.
R, = 4.12 min [Säule: Chiralpak AD-H, 250 x 4.6 mm; Eluent: Ethanol/iso-Hexan 30:70; Fluss: 1 ml/min; Detektion: 260 nm].
1H-NMR (400 MHz, CDCl3): δ = 9.59 (s, IH), 7.16-7.07 (m, 5H), 3.98-3.88 (m, IH), 2.81 (s, 2H), 2.46-2.34 (m, 2H), 2.26-2.16 (m, 2H), 2.02-1.89 (m, IH), 1.78-1.60 (m, IH), 1.28 (d, 6H).
MS (DCI): m/z = 353 (M+H)+, 370 (M+NH4)+.
Beispiel 52A 5-(4-Fluorphenyl)-6- {hydroxy[4-(trifluormethyl)phenyl]methyl} -7-isopropylspiro[chromen-2, 1 '- cyclobutan]-4(3H)-on
Unter Argon werden 0.70 g (1.99 mmol) 5-(4-Fluorphenyl)-7-isopropyl-4-oxo-3,4-dihydrospiro- [chromen-2,r-cyclobutan]-6-carbaldehyd (Beispiel 51A) in 30 ml Tetrahydrofuran suspendiert und auf -78°C abgekühlt. Es werden 5.25 ml (2.62 mmol) einer frisch hergestellten 0.5 M Lösung von [4-(Trifluormethyl)phenyl]magnesiurnbromid in Tetrahydrofuran langsam hinzugefügt. Der Ansatz wird kurz bei -78°C nachgerührt und dann auf 00C erwärmt. Nach ca. 30 min wird mit Natrium- hydrogencarbonat-Lösung hydrolysiert, mit Wasser verdünnt und mehrfach mit Essigsäureethyl- ester extrahiert. Die vereinigten organischen Phasen werden zweimal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird portionsweise über präparative HPLC (Methode 12) gereinigt. Es werden 0.635 g (64% d. Th.) der Titelverbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ = 7.55 (d, 2H), 7.32 (d, 2H), 7.15-6.95 (m, 5H), 5.72 (s, IH), 3.12- 2.98 (m, IH), 2.79 (s, 2H), 2.49-2.30 (m, 2H), 2.29 (d, IH), 2.26-2.12 (m, 2H), 2.01-1.86 (m, IH), 1.78-1.60 (m, IH), 1.20 (d, 3H), 0.62 (d, 3H).
MS (DCI): m/z = 499 (M+H)+, 516 (M+NH4)+.
Beispiel 53A
5-(4-Fluorphenyl)-6- {fluor[4-(trifluormethyl)phenyl]methyl} -7-isopropylspiro[chromen-2, 1 '- cyclobutan] -4(3H)-on
Unter Argon werden 180 mg (0.32 mmol) 5-(4-Fluorphenyl)-6-{hydroxy[4-(trifluormethyl)- phenyl]methyl}-7-isopropylspiro[chromen-2,l'-cyclobutan]-4(3H)-on (Beispiel 52A) in 3.2 ml Toluol gelöst, bei -78°C langsam mit 52 μl (0.39 mmol) Diethylaminoschwefeltrifluorid versetzt und die Mischung 1 h bei -78°C gerührt. Anschließend wird die Temperatur langsam auf -600C erhöht. Nach 2 h wird der Ansatz mit Wasser versetzt und zweimal mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden einmal mit gesättigter Natriumhydrogencarbo- nat-Lösung und einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über eine Kieselgelsäule gereinigt (Lauf- mittel: Cyclohexan/Essigsäureethylester 12:1). Es werden 107 mg (66% d. Th.) der Titelverbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ = 7.59 (d, 2H), 7.26 (d, 2H), 7.22-7.04 (m, 3H), 6.99-6.95 (m, 2H), 6.32 (d, IH), 2.95-2.83 (m, IH), 2.80 (s, 2H), 2.49-2.30 (m, 2H), 2.25-2.14 (m, 2H), 2.02-1.86 (m, IH), 1.78-1.62 (m, IH), 1.19 (d, 3H), 0.73 (d, 3H).
MS (DCI): m/z = 501 (M+H)+, 518 (M+NH4)+.
Beispiel 54A 5-(4-Fluoφhenyl)-7-isopropyl-6-[4-(trifluormethyl)benzoyl]spiro[chromen-2,r-cyclobutan]-4(3H)- on
107 mg (0.25 mmol) l,l-Dihydro-l,l,l-triacetoxy-l,2-benziodoxol-3(lH)-on werden in 1 ml abs. Dichlormethan gelöst und auf -300C gekühlt. Es werden 14 μl (0.17 mmol) Pyridin hinzugefügt und anschließend mit 84 mg (0.17 mmol) 5-(4-Fluorphenyl)-6-{hydroxy[4-(trifluormethyl)phenyl]- methyl}-7-isopropylspiro[chrornen-2,r-cyclobutan]-4(3H)-on (Beispiel 52A), gelöst in 0.6 ml abs. Dichlormethan, tropfenweise versetzt. Der Ansatz wird langsam auf 00C erwärmt und bei dieser Temperatur 1.5 h lang gerührt. Es wird mit Essigsäureethylester verdünnt, mit 5 ml 1 N Natronlauge versetzt und anschließend zweimal mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden einmal mit 1 N Salzsäure und zweimal mit gesättigter Natriumchlorid- Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Das erhaltene Rohprodukt wird im Hochvakuum getrocknet und anschließend ohne weitere Reinigung umgesetzt. Es werden 89 mg (>100% d. Th.) der Titelverbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ = 7.58 (q, 4H), 7.12-6.48 (m, 5H), 2.82 (s, 2H), 2.78-2.68 (m, IH), 2.49-2.36 (m, 2H), 2.28-2.15 (m, 2H), 2.03-1.90 (m, IH), 1.78-1.68 (m, IH), 1.22 (br. s, 6H).
MS (DCI): m/z = 497 (M+H)+, 514 (M+NIL,)*.
Beispiel 55A
6-[(4-tert. -Butylphenyl)(hydroxy)methyl]-5-(4-fluoφhenyl)-7-isopropylspiro[chromen-2, 1 '-cyclo- butan]-4(3H)-on
Unter Argon werden 0.30 g (0.851 mmol) 5-(4-Fluorphenyl)-7-isopropyl-4-oxo-3,4-dihydrospiro- [chrornen-2,r-cyclobutan]-6-carbaldehyd (Beispiel 51A) in 6 ml Tetrahydrofuran suspendiert und auf -78°C abgekühlt. Es werden 2.54 ml (1.28 mmol) einer frisch hergestellten 0.5 M Lösung von [4-(ter/.-Butyl)phenyl]magnesiumbromid in Tetrahydrofuran langsam hinzugefügt. Der Ansatz wird kurz bei -78°C nachgerührt und dann auf 00C erwärmt. Nach ca. 1.5 h wird mit gesättigter Ammoniumchlorid-Lösung versetzt, mit wenig 1 N Salzsäure verdünnt und mehrfach mit Essig- säureethylester extrahiert. Die vereinigten organischen Phasen werden zweimal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird portionsweise über präparative HPLC (Methode 12) gereinigt. Es werden 0.239 g (58% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, CDCl3): δ = 7.29 (d, 2H), 7.13-6.95 (m, 7H), 5.72 (s, IH), 3.18 (heptett, IH), 2.79 (s, 2H), 2.46-2.30 (m, 2H), 2.23-2.12 (m, 2H), 2.09-1.88 (m, 2H), 1.76-1.62 (m, IH), 1.30 (s, 9H), 1.19 (d, 3H), 0.63 (d, 3H).
LC/MS (Methode 9): Rt = 3.44 min
MS (ESIpos): m/z = 487 (M+H)+.
Beispiel 56A
6-[(4-/ert.-Butylphenyl)(fluor)methyl]-5-(4-fluoφhenyl)-7-isopropylspiro[chromen-2,r-cyclo- butan]-4(3H)-on
Unter Argon werden 110 mg (0.23 mmol) 6-[(4-tert.-Butylphenyl)(hydroxy)methyl]-5-(4-fluor- phenyl)-7-isopropylspiro[chromen-2,r-cyclobutan]-4(3H)-on (Beispiel 55A) in 2.3 ml Toluol gelöst, bei -78°C langsam mit 36 μl (0.27 mmol) Diethylaminoschwefeltrifluorid versetzt und die Mischung 1 h bei -78°C gerührt. Anschließend wird die Temperatur langsam auf -6O0C erhöht. Nach 2 h wird der Ansatz mit Wasser versetzt und zweimal mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden einmal mit gesättigter Natriumhydrogencarbonat- Lösung und einmal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über eine Kieselgelsäule gereinigt (Laufmittel: Cyclohexan/Essigsäureethylester 12: 1). Es werden 92 mg (83% d. Th.) der Titelverbindung erhalten.
1H-NMR (300 MHz, CDCl3): δ = 7.30 (d, 2H), 7.22-7.14 (m, IH), 7.12-7.01 (m, 4H), 6.99-6.94 (m, 2H), 6.31 (d, IH), 3.10-2.98 (m, IH), 2.79 (s, 2H), 2.48-2.30 (m, 2H), 2.25-2.13 (m, 2H), 2.00-1.86 (m, IH), 1.76-1.63 (m, IH), 1.29 (s, 9H), 1.18 (d, 3H), 0.72 (d, 3H).
Beispiel 57A
6-(4-re/-/.-Butylbenzoyl)-5-(4-fluoφhenyl)-7-isopropylspiro[chromen-2,r-cyclobutan]-4(3H)-on
91 mg (0.21 mmol) l,l-Dihydro-l,l,l-triacetoxy-l,2-benziodoxol-3(lH)-on werden in 1.4 ml abs. Dichlormethan gelöst und auf -30
0C abgekühlt. Es werden 12 μl (0.14 mmol) Pyridin hinzugefügt und anschließend mit 68 mg (0.14 mmol) 6-[(4-/ert.-Butylphenyl)(hydroxy)methyl]-5-(4-fluor- phenyl)-7-isopropylspiro[chromen-2,r-cyclobutan]-4(3H)-on (Beispiel 55A), gelöst in 0.6 ml abs. Dichlormethan, tropfenweise versetzt. Der Ansatz wird langsam auf 0
0C erwärmt und bei dieser Temperatur 1.5 h gerührt. Es wird mit Essigsäureethylester verdünnt, mit 5 ml 1 N Natronlauge versetzt und anschließend zweimal mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden einmal mit 1 N Salzsäure und zweimal mit gesättigter Natriumchlorid- Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird im Hochvakuum getrocknet und anschließend ohne weitere Reinigung umgesetzt. Es werden 81 mg (>100% d. Th.) der Titelverbindung erhalten.
Beispiel 58A
5-(4-Fluorphenyl)-6- {hydroxy [4-(trifluormethoxy)phenyl]methyl } -7-isopropylspiro[chromen-2, 1 ' cyclobutan] -4(3H)-on
Unter Argon werden 0.60 g (1.70 mmol) 5-(4-Fluorphenyl)-7-isopropyl-4-oxo-3,4-dihydrospiro- [chromen-2,l'-cyclobutan]-6-carbaldehyd (Beispiel 51A) in 15 ml Tetrahydrofuran suspendiert und auf -700C abgekühlt. Es werden 4.3 ml (2.13 mmol) einer frisch hergestellten 0.5 M Lösung von [4-(Trifluormethoxy)phenyl]magnesiumbromid in Tetrahydrofuran langsam hinzugefügt. Der Ansatz wird kurz bei -78°C nachgerührt und dann auf 00C erwärmt. Nach 1 h wird mit Natrium- hydrogencarbonat-Lösung hydrolysiert, mit Wasser verdünnt und mehrfach mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden zweimal mit gesättigter Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über eine Kieselgelsäule gereinigt (Laufmittel: Cyclohexan/Essigsäureethylester 25:1 -> 10: 1). Es werden 710 mg (81% d. Th.) der Titelverbindung in einer Reinheit von 70% erhalten.
1H-NMR (400 MHz, CDCl3): δ = 7.22 (d, 2H), 7.15-6.97 (m, 7H), 5.71 (d, IH), 3.11 (heptett, IH), 2.79 (s, 2H), 2.47-2.30 (m, 2H), 2.23-2.13 (m, 2H), 2.11 (d, IH), 1.99-1.88 (m, IH), 1.76-1.63 (m, IH), 1.19 (d, 3H), 0.64 (d, 3H).
MS (DCI): m/z = 515 (M+H)+, 532 (M+NH4)+.
Beispiel 59A
5-(4-Fluoφhenyl)-6-{fluor[4-(trifluormethoxy)phenyl]methyl}-7-isopropylspiro[chromen-2,r- cyclobutan] -4(3H)-on
Unter Argon werden 159 mg (0.31 mmol) 5-(4-Fluorphenyl)-6-{hydroxy[4-(trifluormethoxy)- phenyl]methyl}-7-isopropylspiro[chromen-2,r-cyclobutan]-4(3H)-on (Beispiel 58A) in 3.1 ml Toluol gelöst, bei -78°C langsam mit 49 μl (0.37 mmol) Diethylaminoschwefeltrifluorid versetzt und die Mischung 2 h bei -78°C gerührt. Anschließend wird der Ansatz mit Wasser versetzt und zweimal mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden einmal mit gesättigter Natriumhydrogencarbonat-Lösung und einmal mit gesättigter Natriumchlorid- Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über eine Kieselgelsäule gereinigt (Laufmittel: Cyclohexan/Essigsäureethylester 15:1). Es werden 118 mg (74% d. Th.) der Titelverbindung erhalten.
1H-NMR (400 MHz, CDCl3): δ = 7.20-7.04 (m, 7H), 7.02-6.92 (m, 2H), 6.31 (d, IH), 2.94 (heptett, IH), 2.79 (s, 2H), 2.48-2.31 (m, 2H), 2.25-2.13 (m, 2H), 2.00-1.89 (m, IH), 1.78-1.62 (m, IH), 1.18 (d, 3H), 0.78 (d, 3H).
MS (DCI): m/z = 517 (M+H)+, 534 (M+NH4)+.
Beispiel 60A
5-(4-Fluoφhenyl)-7-isopropyl-6-[4-(trifluormethoxy)benzoyl]spiro[chromen-2,r-cyclobutan]- 4(3H)-on
68 mg (0.16 mmol) l,l-Dihydro-l,l,l-triacetoxy-l,2-benziodoxol-3(lH)-on werden in 0.6 ml abs. Dichlormethan gelöst und auf -300C abgekühlt. Es werden 9 μl (0.11 mmol) Pyridin hinzugefügt und anschließend mit 55 mg (0.11 mmol) 5-(4-Fluorphenyl)-6-{hydroxy[4-(trifluormethoxy)- phenyl]methyl}-7-isopropylspiro-[chromen-2,r-cyclobutan]-4(3H)-on (Beispiel 58A), gelöst in 0.4 ml abs. Dichlormethan, tropfenweise versetzt. Der Ansatz wird langsam auf 00C erwärmt und bei dieser Temperatur 6 h gerührt. Es wird mit Essigsäureethylester verdünnt, mit 5 ml 1 N Natronlauge versetzt und anschließend zweimal mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden einmal mit 1 N Salzsäure und zweimal mit gesättigter Natriumchlorid- Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird im Hochvakuum getrocknet und anschließend ohne weitere Reinigung umgesetzt. Es werden 54 mg (98% d. Th.) der Titelverbindung erhalten.
Beispiel 61A
5-Cyclopent-l-en-l-yl-7-isopropyl-4-oxo-3,4-dihydrospiro[chromen-2,r-cyclobutan]-6-carb- aldehyd
Die Titelverbindung wird analog zur Vorschrift in Beispiel 34A hergestellt.
LC/MS (Methode 8): R, = 3.20 min
MS (ESIpos): m/z = 325 (M+H)+
1H-NMR (CDCl3, 300 MHz): δ = 1.23 (d, 6H), 1.65-1.78 (m, IH), 1.85-2.00 (m, 2H), 2.05-2.73 (m, 9H), 2.86 (s, 2H), 3.86-3.97 (m, IH), 5.51-5.58 (m, IH), 6.97 (s, IH), 9.97 (s, IH).
Beispiel 62A rαc-5-Cyclopent-l-en-l-yl-6-{hydroxy[4-(trifluormethyl)phenyl]methyl}-7-isopropylspiro- [chromen-2, 1 '-cyclobutan]-4(3H)-on
Die Titelverbindung wird analog zur Vorschrift in Beispiel 36A hergestellt.
LC/MS (Methode 8): R, = 3.35 min
MS (ESIpos): m/z = 471 (M+H)+
MS (DCI): m/z = 471 (M+H)+, 488 (M+NH4)+
1H-NMR (CDCl3, 300 MHz): δ = 0.44-0.70 (m, 3H), 1.06-1.21 (m, 3H), 1.60-1.80 (m, IH), 1.83- 2.05 (m, 2H), 2.06-2.64 (m, 9H), 2.69-2.93 (m, 3H), 2.94-3.12 (m, IH), 5.38-5.51 (m, IH), 6.14- 6.24 (m, IH), 6.89 (s, IH), 7.31-7.47 (m, 2H), 7.56 (d, 2H).
Beispiel 63A rac-5-Cyclopentyl-6- {hydroxy[4-(trifluormethyl)phenyl]methyl} -7-isopropylspiro[chromen-2, 1 '- cyclobutan] -4(3H)-on
Eine Lösung von 289 mg (0.61 mmol) rαc-5-Cyclopent-l-en-l-yl-6-{hydroxy[4-(trifluormethyl)- phenyl]methyl}-7-isopropylspiro[chromen-2,r-cyclobutan]-4(3H)-on (Beispiel 62A) in 10 ml Ethanol wird mit 63 mg (0.031 mmol) Rhodium auf Aktivkohle (5%) versetzt und 18 h unter einem Wasserstoffdruck von 60 bar bei Raumtemperatur hydriert. Zur Aufarbeitung wird die Suspension über Kieselgur filtriert, mit Essigsäureethylester gewaschen und das Filtrat unter vermindertem Druck eingeengt. Der Rückstand wird in 10 ml Ethanol aufgenommen, erneut mit der gleichen Menge Rhodium auf Aktivkohle versetzt und weitere 18 h unter einem Wasserstoffdruck von 60 bar bei Raumtemperatur hydriert. Die Suspension wird wiederum über Kieselgur filtriert, mit Essigsäureethylester gewaschen und das Filtrat unter vermindertem Druck eingeengt. Das Rohprodukt wird durch Chromatographie an Kieselgel (Laufmittel: Cyclohexan/Essigsäureethylester 15:1) gereinigt.
Ausbeute: 116 mg (40% der Th.)
LC/MS (Methode 7): R, = 3.23 min
MS (ESIpos): m/z = 473 (M+Η)+
1H-NMR (CDCl3, 300 MHz): δ = 0.52-0.78 (m, 3H), 1.13 (d, 3H), 1.58-2.45 (m, 15H), 2.92 (s, 2H), 2.94-3.07 (m, IH), 4.29-4.67 (br. m, IH), 6.25-6.32 (m, IH), 6.82 (s, IH), 7.39 (d, 2H), 7.56 (d, 2H).
Beispiel 64A rac-S-Cyclopentyl-ό- {fluor[4-(trifluormethyl)phenyl]methyl} -7-isopropylspiro[chromen-2, 1 '- cyclobutan] -4(3H)-on
Die Titelverbindung wird analog zur Vorschrift in Beispiel 28A hergestellt.
Ausbeute: 192 mg (96% d. Th.)
LC/MS (Methode 7): R1 = 3.42 min
MS (ESIpos): m/z = 475 (M+H)+
1H-NMR (CDCl3, 400 MHz): δ = 0.72 (d, 3H), 1.11 (d, 3H), 1.58-2.45 (m, 14H), 2.79-2.99 (m, 3H), 4.38-4.53 (m, IH), 6.84 (s, IH), 6.96 (d, IH), 7.33 (d, 2H), 7.59 (d, 2H).
Ausführungsbeispiele:
Beispiel 1 und Beispiel 2
(45)-7-Cyclopentyl-5-(4-fluoφhenyl)-6-{(S)-hydroxy[4-(trifluoπnethyl)phenyl]methyl}-2,2- dimethylchroman-4-ol {Beispiel J)
und
(4S)-7-Cyclopentyl-5-(4-fluorphenyl)-6-{(R)-hydroxy[4-(trifluormethyl)phenyl]methyl}-2,2- dimethylchroman-4-ol (Beispiel 2)
Unter Argon werden 120 mg (0.23 mmol) [(4S)-7-Cyclopentyl-5-(4-fluorphenyl)-4-hydroxy-2,2- dimethyl-3,4-dihydro-2H-chromen-6-yl][4-(trifluormethyl)phenyl]methanon (Beispiel 12A) in 2 ml abs. Toluol vorgelegt und auf -78°C gekühlt. Bei dieser Temperatur werden 0.59 ml (0.59 mmol) Diisobutylaluminiumhydrid-Lösung (1 M in Toluol) langsam zugetropft und die Mischung anschließend 2 h bei -78°C weitergerührt. Danach wird noch soviel Diisobutylaluminiumhydrid zu- gesetzt, bis kein Edukt mehr nachweisbar ist. Der Ansatz wird dann mit gesättigter Kaliumnatrium- tartrat-Lösung versetzt, mit Wasser verdünnt und mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden einmal mit gesättigter Natriumchlorid-Lösung gewaschen. Die
orgamsche Phase wird über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wird über Dickschichtchromatographie (Laufrnittel: Cyclohexan/Essigsäureethylester 5:1) gereinigt, wobei die Diastereomere getrennt werden.
Beispiel 1: Ausbeute: 62 mg (52% d. Th.)
1H-NMR (400 MHz, CDCl3): δ = 7.51 (d, 2H), 7.35-7.27 (m, 3H), 7.18-7.02 (m, 3H), 6.89 (s, IH), 5.69 (br. s, IH), 4.57-4.52 (m, IH), 3.03 (heptett, IH), 2.09-1.95 (m, 4H), 1.78-1.53 (m, 5H), 1.52 (s, 3H), 1.42 (s, 3H), 1.36-1.24 (m, 2H).
MS (DCI): m/z = 532 (M+NH4)+.
Beispiel 2:
Ausbeute: 56 mg (46% d. Th.)
1H-NMR (400 MHz, CDCl3): δ = 7.50 (d, 2H), 7.38-7.30 (m, IH), 7.28 (d, 2H), 7.22-7.18 (m, IH), 7.17-7.08 (m, 2H), 6.89 (s, IH), 5.62 (br. s, IH), 4.57-4.52 (m, IH), 3.03 (heptett, IH), 2.11 (d, IH), 2.09-1.95 (m, 3H), 1.78-1.51 (m, 5H), 1.47 (s, 3H), 1.43 (s, 3H), 1.30-1.20 (m, 2H).
MS (DCI): m/z = 532 (M+NH4)+.
Beispiel 3
(4S)-7-Cyclopentyl-5-(4-fluoφhenyl)-6-{(S)-fluor[4-(triiluoπnethyl)phenyl]methyl}-2,2-dimethyl- chroman-4-ol
Unter Argon werden 21 mg (0.033 mmol) ter£-Butyl[((4S)-7-cyclopentyl-5-(4-fluorphenyl)-6- {fluor[4-(trifluormethyl)phenyl]methyl}-2,2-dimethyl-3,4-dihydro-2H-chromen-4-yl)oxy]dime-
thylsilan (Beispiel 15A) in 0.5 ml Tetrahydrofuran gelöst, mit 0.17 ml (0.17 mmol) TBAF-Lösung (1 M in Tetrahydrofuran) versetzt und bei Raumtemperatur gerührt. Nach 1 h wird der Ansatz mit 1.5 ml 0.2 N Salzsäure versetzt und mehrfach mit Essigsäureethylester extrahiert. Die vereinigten organischen Phasen werden zweimal mit gesättigter Natriumchlorid-Lösung gewaschen, über _ Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über präparative HPLC gereinigt. Es werden 11 mg (63% d. Th.) der Zielverbindung mit einem de-Wert von 88% erhalten. Anschließende chromatographische Isomerentrennung an chiraler Phase [Säule: Chiralpak AD-H, 250 x 8 mm; Eluent: iso-Propanol/iso-Hexan 10:90; Fluss: 3 ml/min; 24°C; Detektion: 254 nm] liefert 4 mg der diastereomerenreinen Verbindung.
Rt = 4.54 min [Chiralpak IA, 250 x 4.6 mm; Eluent: iso-Propanol/iso-Hexan 3:97; Fluss: 1.5 ml/min; Detektion: 254 nm].
1H-NMR (300 MHz, CDCl3): δ = 7.52 (d, 2H), 7.42-7.35 (m, IH), 7.20 (d, 2H), 7.18-7.06 (m, 3H), 6.92 (s, IH), 6.28 (d, IH), 4.57 (q, IH), 2.92 (heptett, IH), 2.13-1.98 (m, 3H), 1.80-1.42 (m, HH), 1.33-1.24 (m, 2H).
MS (ESIpos): m/z = 499 (M-H2O)+.
Beispiel 4
[(45)-5-(4-Fluorphenyl)-4-hydroxy-7-isopropyl-2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl][4- (trifluormethyl)phenyl]methanon
Zu einer Lösung von 3.79 mg (30 μmol) (li?,2,S)-l-Aminomdan-2-ol in 4 ml Tetrahydrofuran werden langsam 120 μl (680 μmol) Boran-N,N-diethylanilin-Komplex gegeben. Dann wird die Mischung auf 00C gekühlt und eine Lösung von 82 mg (170 μmol) 5-(4-Fluorphenyl)-7-isopropyl- 2,2-dimethyl-6-[4-(trifluormethyl)benzoyl]-2,3-dihydro-4H-chromen-4-on (Beispiel 23A) in 4 ml Tetrahydrofuran langsam zugetropft. Man lässt die Mischung langsam auftauen und rührt über
Nacht bei Raumtemperatur. Dann wird Methanol zugegeben und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird in Essigsäureethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Natriumhydrogencarbonat-Lösung sowie einmal mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands mittels Säulenchromatographie an Kieselgel (Laufmittel: Gradient Cyclohexan — > Cyclohexan/Essigsäureethylester 10:1) liefert die Zielverbindung.
Ausbeute: 65 mg (79% d. Th., 91% ee)
HPLC (Methode 4): R, = 3.83 min
LC/MS (Methode 8): R, = 2.81 min
MS (ESIpos): m/z = 487 (M+H)+
1H-NMR (CDCl3, 300 MHz): δ = 1.15 (d, 3H), 1.21 (d, 3H), 1.45 (s, 3H), 1.51 (s, 3H), 2.03 (d, 2H), 2.59-2.77 (m, IH), 4.61-4.78 (m, IH), 6.68-7.22 (m, 4H), 6.93 (s, IH), 7.53 (d, 2H), 7.63 (d, 2H).
Beispiel 5 und Beispiel 6 (4S)-5-(4-Fluoφhenyl)-6-{(6)-hydroxy[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-di- methylchroman-4-ol (Beispiel 5)
und
(4S)-5-(4-Fluorphenyl)-6-{(i?)-hydroxy[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-di- methylchroman-4-ol (Beispiel 6)
Zu einer Lösung von 10.58 mg (70 μmol) (lR,2ιS)-l-Aminoindan-2-ol in 11 ml Tetrahydrofuran werden langsam 340 μl (1.89 mmol) Boran-N,N-diethylanilin-Komplex gegeben und die Mischung noch 30 min nachgerührt. Dann wird eine Lösung von 230 mg (0.47 mmol) rac-5-(4-Fluorphenyl)- 6-{hydroxy[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen- 4-on (Beispiel 21A) in 11 ml Tetrahydrofuran sehr langsam zugetropft und das Gemisch über Nacht gerührt. Danach wird Methanol zugegeben und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird in Essigsäureethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Natriumhydrogencarbonat-Lösung sowie einmal mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands mittels Säulenchromatographie an Kieselgel (Laufmittel: Gradient Cyclohexan/Dichlormethan/Essigsäureethylester 40:40: 1 — > 2:2:1) liefert die diastereomerenreinen Zielverbindungen .
Beispiel 5: Ausbeute: 119 mg (52% d. Th., 91% ee)
ΗPLC (Methode 6): R, = 12.30 min
LC/MS (Methode 9): R, = 3.10 min
MS (DCI): m/z = 506 (M+NΗ4)+
1H-NMR (CDCl3, 300 MHz): δ = 0.72 (d, 3H), 1.18 (d, 3H), 1.42 (s, 3H), 1.49 (s, 3H), 1.99 (d, 2H), 2.03 (d, IH), 2.90-3.08 (m, IH), 4.54 (dd, IH), 5.61-5.69 (m, IH), 6.89 (s, IH), 7.01-7.22 (m, 3H), 7.28-7.41 (m, 3H), 7.52 (d, 2H).
Beispiel 6:
Ausbeute: 120 mg (52% d. Th., 91% ee)
HPLC (Methode 6): Rt = 14.45 min
LC/MS (Methode 9): R, = 3.16 min .
MS (DCI): m/z = 506 (M+NH4)+
1H-NMR (CDCl3, 300 MHz): δ = 0.62 (d, 3H), 1.18 (d, 3H), 1.44 (s, 3H), 1.47 (s, 3H), 1.95-2.18 (m, 3H), 2.92-3.08 (m, IH), 4.51-4.62 (m, IH), 5.58-5.68 (m, IH), 6.88 (s, IH), 7.03-7.42 (m, 6H), 7.50 (d, 2H).
Beispiel 7 und Beispiel 8 (4S)-5-(4-Fluoφhenyl)-6-{(S)-fluor[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-dimethyl- chroman-4-ol {Beispiel 7)
und
(4S)-5-(4-Fluoφhenyl)-6-{(R)-fluor[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-dimethyl- chroman-4-ol (Beispiel 8)
Zu einer Lösung von 3.5 mg (20 μmol) (li?,2S)-l-Aminoindan-2-ol in 4 ml Tetrahydrofuran werden langsam 110 μl (630 μmol) Boran-N^V-diethylanilin-Komplex gegeben und die Mischung fiir 30 min bei Raumtemperatur nachgerührt. Dann wird eine Lösung von 77 mg (160 μmol) 5-(4- Fluoφhenyl)-6-{(S)-fluor[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3-dihydro- 4H-chromen-4-on (Beispiel 22A) in 4 ml Tetrahydrofuran langsam zugetropft und das Gemisch für 6.5 h bei Raumtemperatur gerührt. Danach wird Methanol zugegeben und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird in Essigsäureethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Νatriumhydrogencarbonat-Lösung sowie einmal mit gesättigter Νatriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands mittels Säulenchromatographie an Kieselgel (Laufmittel: Cyclohexan/Essigsäureethylester 20:1) liefert die diastereomerenreinen Zielverbindungen.
Beispiel 7: Ausbeute: 11 mg (14% d. Th., 82% ee)
ΗPLC (Methode 4): Rt = 3.52 min
LC/MS (Methode 8): R, = 2.96 min
MS (DCI): m/z = 508 (M+NΗ4)+
1H-NMR (CDCl3, 300 MHz): δ = 0.82 (d, 3H), 1.14 (d, 3H), 1.41 (s, 3H), 1.49 (s, 3H), 1.98 (d, 2H), 2.78-2.93 (m, IH), 4.58 (dd, IH), 6.26 (d, IH), 6.91 (s, IH), 6.97-7.28 (m, 6H), 7.52 (d, 2H).
Beispiel 8:
Ausbeute: 10 mg (13% d. Th., 86% ee)
HPLC (Methode 4): R, = 3.63 min
LC/MS (Methode 8): R1 = 2.93 min
MS (DCI): m/z = 508 (M+NHL,)*
1H-NMR (CDCl3, 300 MHz): δ = 0.73 (d, 3H), 1.18 (d, 3H), 1.45 (s, 3H), 1.48 (s, 3H), 1.97-2.03 (m, 2H), 2.78-2.96 (m, IH), 4.55 (dd, IH), 6.28 (d, IH), 6.92 (s, IH), 7.02-7.44 (m, 6H), 7.52 (d, 2H).
Beispiel 9
(4S)-5-(4-Fluoφhenyl)-7-isopropyl-2,2-dimethyl-6-[4-(trifluormethyl)benzyl]chroman-4-ol
Zu einer Lösung von 2.8 mg (20 μmol) (lR,2S)-l-Aminoindan-2-ol in 3 ml Tetrahydrofuran werden langsam 87 μl (490 μmol) Boran-N,N-diethylamlin-Komplex gegeben und die Mischung für 30 min bei Raumtemperatur nachgerührt. Dann wird eine Lösung von 60 mg (120 μmol) 5-(4- Fluorphenyl)-6-{(5)-fluor[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3-dihydro- 4H-chromen-4-on (Beispiel 22A) in 3 ml Tetrahydrofuran langsam zugetropft und das Gemisch für 3 Tage bei Raumtemperatur gerührt. Danach wird Methanol zugegeben und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird in Essigsäureethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Νatriumhydrogencarbonat-Lösung sowie einmal mit gesättigter Νatriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands mittels Säulenchromatographie an Kiesel- gel (Laufmittel : Cyclohexan/Essigsäureethylester 20 : 1 ) liefert die Zielverbindung.
Ausbeute: 22 mg (36% d. Th.)
ΗPLC (Methode 4): Rt = 3.38 min
LC/MS (Methode 9): Rt = 3.42 min
MS (ESIpos): m/z = 473 (M+H)+
1H-NMR (CDCl3, 300 MHz): δ = 1.12 (d, 3H), 1.16 (d, 3H), 1.43 (s, 3H), 1.48 (s, 3H), 1.98 (d, 2H), 2/78-2.98 (m, IH), 3.71 (d, IH), 3.80 (d, IH), 4.56 (dd, IH), 6.97-7.28 (m, 6H), 7.52 (d, 2H).
Beispiel 10
[(4S)-5-(4-Fluoφhenyl)-4-hydroxy-7-isopropyl-2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl][4-(tri- fluormethoxy)phenyl]methanon
Zu einer Lösung von 3.04 mg (20 μmol) (lR,2S)-l-Arninoindan-2-ol in 2.5 ml Tetrahydrofuran werden langsam 97 μl (540 μmol) Boran-N^V-diethylanilin-Komplex gegeben. Dann wird die Mischung auf 00C gekühlt und eine Lösung von 68 mg (140 μmol) 5-(4-Fluorphenyl)-7-isopropyl- 2,2-dimethyl-6-[4-(trifluormethoxy)benzoyl]-2,3-dihydro-4H-chromen-4-on (Beispiel 26A) in 4 ml Tetrahydrofuran langsam zugetropft. Man lässt die Mischung langsam auftauen und rührt über Nacht bei Raumtemperatur. Zur Vervollständigung der Reaktion wird die Reaktionsmischung wie- der auf 00C gekühlt und weitere 97 μl (540 μmol) Boran-N^V-diethylanilin-Komplex zugegeben. Anschließend wird langsam auf Raumtemperatur aufgetaut und über Nacht bei dieser Temperatur gerührt. Danach wird Methanol zugegeben und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird in Essigsäureethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Natriumhydrogencarbonat-Lösung sowie einmal mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands durch präparative ΗPLC (Methode 1) und nachfolgende chirale ΗPLC (Methode 2) liefert die Zielverbindung.
Ausbeute: 30 mg (44% d. Th., 99.5% ee)
HPLC (Methode 4): R, = 5.20 min
LC/MS (Methode 8): R, = 3.28 min
MS (ESIpos): m/z = 503 (M+H)+
1H-NMR (CDCl3, 300 MHz): δ = 1.17 (d, 3H), 1.21 (d, 3H), 1.46 (s, 3H), 1.51 (s, 3H), 2.02 (d, 2H), 2.62-2.79 (m, IH), 4.71 (s, IH), 6.61-7.23 (m, 7H), 7.56 (d, 2H).
Daneben wird in einer zweiten Fraktion das Enantiomer des Zielprodukts, [(4i?)-5-(4-Fluor- phenyl)-4-hydroxy-7-isopropyl-2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl][4-(trifluormethoxy)- phenyljmethanon, erhalten:
Ausbeute: 9 mg (13% d. Th., 88% ee)
ΗPLC (Methode 4): R, = 8.06 min.
Beispiel 11 und Beispiel 12
(4S)-5-(4-Fluoφhenyl)-6-{(5)-hydroxy[4-(trifluormethoxy)phenyl]methyl}-7-isopropyl-2,2-di- methylchroman-4-ol {Beispiel 11)
und
(4S)-5-(4-Fluoφhenyl)-6-{(R)-hydroxy[4-(trifluormethoxy)phenyl]methyl}-7-isopropyl-2,2-di- methylchroman-4-ol {Beispiel 12)
Zu einer Lösung von 4.45 mg (30 μmol) (lΛ,25)-l-Aminoindan-2-ol in 5 ml Tetrahydrofuran werden langsam 140 μl (800 μmol) Boran-N^V-diethylanilin-Komplex gegeben. Dann wird die Mischung auf 0°C gekühlt und eine Lösung von 100 mg (200 μmol) rαc-5-(4-Fluorphenyl)-6- {hydroxy[4-(trifluormethoxy)phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen- 4-on (Beispiel 24A) in 5 ml Tetrahydrofuran langsam zugetropft. Man lässt die Mischung langsam auftauen und rührt über Nacht bei Raumtemperatur. Danach wird Methanol zugegeben und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird in Essigsäureethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Natriumhydrogencarbonat-Lösung sowie einmal mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands mittels präparativer ΗPLC (Methode 1) liefert die diastereomerenreinen Zielverbindungen.
Beispiel 11:
Ausbeute: 39 mg (37% d. Th., 86% ee)
ΗPLC (Methode 6): R, = 11.97 min
LC/MS (Methode 7): R, = 2.96 min
MS (ESIneg): m/z = 549 (M-Η+ΗCOOΗy
1H-NMR (CDCl3, 300 MHz): δ = 0.75 (d, 3H), 1.18 (d, 3H), 1.42 (s, 3H), 1.48 (s, 3H), 1.82-2.04 (m, 3H), 2.97-3.10 (m, IH), 4.53 (dd, IH), 5.62 (d, IH), 6.88 (s, IH), 7.02-7.18 (m, 5H), 7.19-7.33 (m, 3H).
Beispiel 12:
Ausbeute: 32 mg (32% d. Th., 84% ee)
HPLC (Methode 6): Rt = 14.05 min
LC/MS (Methode 7): Rt = 3.02 min
MS (ESIneg): m/z = 549 (M-H+HCOOH)-
1H-NMR (CDCl3, 300 MHz): δ = 0.65 (d, 3H), 1.18 (d, 3H), 1.44 (s, 3H), 1.48 (s, 3H), 1.99 (d, 2H), 2.07 (d, IH), 2.97-3.12 (m, IH), 4.55 (dd, IH), 5.60 (d, IH), 6.88 (s, IH), 7.03-7.38 (m, 8H).
Beispiel 13
(45)-5-(4-Fluorphenyl)-7-isopropyl-2,2-dimethyl-6-[4-(trifluormethoxy)benzyl]chroman-4-ol
Zu einer Lösung von 7.23 mg (50 μmol) (lR,2£)-l-Aminoindan-2-ol in 8 ml Tetrahydrofuran wer- den langsam 230 μl (1.29 mmol) Boran-N^N-diethylanilin-Komplex gegeben und die Mischung für 30 min nachgerührt. Dann wird eine Lösung von 163 mg (320 μmol) rac-5-(4-Fluorphenyl)-6- {fluor[4-(trifluormethoxy)phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 25A) in 8 ml Tetrahydrofuran langsam zugetropft. Nach Rühren über Nacht bei Raumtemperatur wird Methanol zugegeben und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird in Essigsäureethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Natriumhydrogencarbonat-Lösung sowie einmal mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands mittels präparativer ΗPLC (Methode 1) liefert das Zielprodukt.
Ausbeute: 35 mg (22% d. Th., 80% ee)
ΗPLC (Methode 4) : R, = 3.44 min
LC/MS (Methode 9): Rt = 3.42 min
MS (ESIpos): m/z = 471 (M+H-H2O)+
1H-NMR (CDCl3, 300 MHz): δ = 1.12 (d, 3H), 1.17 (d, 3H), 1.42 (s, 3H), 1.47 (s, 3H), 1.98 (d, 2H), 2.85-3.02 (m, IH), 3.67 (d, IH), 3.73 (d, IH), 4.54 (dd, IH), 6.80 (d, 2H), 6.88 (s, IH), 6.91- 7.17 (m, 6H).
Beispiel 14
(4-/ert.-Butylphenyl)[(4S)-5-(4-fluorphenyl)-4-hydroxy-7-isopropyl-2,2-dimethyl-3,4-dihydro-2H- chromen-6-yl]methanon
Zu einer Lösung von 2.46 mg (20 μmol) (lR,2S)-l-Aminoindan-2-ol in 2 ml Tetrahydrofuran werden langsam 78 μl (540 μmol) Boran-N,N-diethylanilin-Komplex gegeben. Dann wird die Mischung auf 00C gekühlt und eine Lösung von 52 mg (110 μmol) 6-(4-fert-Butylbenzoyl)-5-(4- fluorphenyl)-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 29A) in 3 ml Tetrahydrofuran langsam zugetropft. Man lässt die Mischung langsam auftauen und rührt über Nacht bei Raumtemperatur. Danach wird Methanol zugegeben und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird in Essigsäureethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Natriumhydrogencarbonat-Lösung sowie einmal mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands durch präparative ΗPLC (Methode 1) und nachfolgende chirale ΗPLC (Methode 2) liefert die Zielverbindung.
Ausbeute: 22 mg (42% d. Th., 99.5% ee)
ΗPLC (Methode 5): R, = 5.71 min
LC/MS (Methode 7): Rt = 3.22 min
MS (ESIpos): m/z = 473 (M+Η)+
1H-NMR (CDCl3, 300 MHz): δ = 1.03-1.38 (m, 6H), 1.29 (s, 9H), 1.46 (s, 3H), 1.49 (s, 3H), 2.02 (d, 2H), 2.62-2.83 (m, IH), 4.72 (br. s, IH), 6.53-7.24 (m, 7H), 7.37-7.59 (m, 2H).
Daneben wird in einer zweiten Fraktion das Enantiomer des Zielprodukts, (4-ter/.-Butylphenyl)- [(4R)-5-(4-fluoφhenyl)-4-hydroxy-7-isopropyl-2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl]metha- non, erhalten:
Ausbeute: 3 mg (5% d. Th., 65% ee)
ΗPLC (Methode 5): Rt = 8.26 min.
Beispiel 15 und Beispiel 16
(4S)-6-[(5)-(4-te^.-Butylphenyl)(hydroxy)methyl]-5-(4-fluorphenyl)-7-isopropyl-2,2-dimethyl- chroman-4-ol {Beispiel 15)
und
(4S)-6-[(R)-(4-/er/.-Butylphenyl)(hydroxy)methyl]-5-(4-fluoφhenyl)-7-isopropyl-2,2-dimethyl- chroman-4-ol (Beispiel 16)

Zu einer Lösung von 7.07 mg (50 μmol) (li?,2S)-l-Aminoindan-2-ol in 7 ml Tetrahydrofuran werden sehr langsam 220 μl (1.26 mmol) Boran-N,N-diethylanilin-Komplex gegeben und das Gemisch für 30 min bei Raumtemperatur nachgerührt. Dann wird eine Lösung von 150 mg (320 μmol) rαc- 6-[(4-/er/.-Butylphenyl)(hydroxy)methyl]-5-(4-fluoφhenyl)-7-isopropyl-2,2-dimethyl-2,3-dihydro- 4H-chromen-4-on (Beispiel 27A) in 7 ml Tetrahydrofuran sehr langsam innerhalb von 30 min zugetropft. Nach weiterem Rühren für 6 h wird Methanol zugegeben und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird in Essigsäureethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Natriumhydrogencarbonat-Lösung sowie einmal mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands mittels Säulenchromatographie an Kieselgel (Laufmittel: Gradient Cyclohexan -> Cyclohexan/Essigsäureethylester 10:1 -> 5:1) liefert die diastereomerenreinen Zielprodukte.
Beispiel 15:
Ausbeute: 45 mg (30% d. Th., 93% ee)
ΗPLC (Methode 6): Rt = 13.02 min
LC/MS (Methode 9): Rt = 3.29 min
MS (ESIneg): m/z = 521 (M-Η+ΗCOOΗ)"
1H-NMR (CDCl3, 300 MHz): δ = 0.72 (d, 3H), 1.17 (d, 3H), 1.29 (s, 9H), 1.42 (s, 3H), 1.49 (s, 3H), 1.91-2.02 (m, 3H), 3.02-3.18 (m, IH), 4.55 (dd, IH), 5.63 (d, IH), 6.98 (s, IH), 7.00-7.22 (m, 5H), 7.22-7.37 (m, 3H).
Beispiel 16:
Ausbeute: 49 mg (32% d. Th., 96% ee)
HPLC (Methode 6): R, = 9.87 min
LC/MS (Methode 9): Rt = 3.35 min
MS (ESIneg): m/z = 521 (M-H+HCOOH)-
1H-NMR (CDCl3, 300 MHz): δ = 0.63 (d, 3H), 1.18 (d, 3H), 1.27 (s, 9H), 1.43 (s, 3H), 1.47 (s, 3H), 1.93-2.08 (m, 3H), 3.02-3.18 (m, IH), 4.58 (dd, IH), 5.62 (d, IH), 6.88 (s, IH), 7.01-7.15 (m, 6H), 7.18-7.39 (m, 2H).
Beispiel 17 (4S)-6-(4-terΛ-Butylbenzyl)-5-(4-iluoφhenyl)-7-isopropyl-2,2-dimethylchroman-4-ol
Zu einer Lösung von 4.70 mg (30 μmol) (li?,25)-l-Aminoindan-2-ol in 5 ml Tetrahydrofuran werden langsam 150 μl (840 μmol) Boran-N,N-diethylanilin-Komplex gegeben und die Mischung für 30 min nachgerührt. Dann wird eine Lösung von 100 mg (210 μmol) rac-6-[(4-/ert.-Butylphenyl)- (fluor)methyl]-5-(4-fluoφhenyl)-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 28A) in 5 ml Tetrahydrofuran langsam zugetropft. Nach 3 h Rühren bei Raumtemperatur wird Methanol zugegeben und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird in Essig- säureethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Natrium- hydrogencarbonat-Lösung sowie einmal mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands mittels Säulenchromatographie an Kieselgel (Laufmittel: Gradient Cyclohexan → Cyclohexan/Essigsäureethylester 10:1 -» 5:1) liefert das Zielprodukt.
Ausbeute: 37 mg (38% d. Th., 90% ee)
HPLC (Methode 5): R, = 4.76 min
LC/MS (Methode 9): R, = 3.54 min
MS (ESIpos): m/z = 443 (M+H-H2O)+
MS (DCI): m/z = 478 (M+NH4)+
1H-NMR (CDCl3, 300 MHz): δ = 1.10 (d, 3H), 1.13 (d, 3H), 1.27 (s, 9H), 1.42 (s, 3H), 1.48 (s, 3H), 1.97 (d, 2H), 2.93-3.08 (m, IH), 3.61 (d, IH), 3.70 (d, IH), 4.57 (dd, IH), 6.72 (d, 2H), 6.88 (s, IH), 6.92-7.03 (m, 3H), 7.04-7.12 (m, IH), 7.16 (d, 2H).
Beispiel 18 [(4S)-4-Hydroxy-7-isopropyl-2,2-dimethyl-5-phenyl-3 ,4-dihydro-2H-chromen-6-yl] [4-(trifluor- methoxy)phenyl]methanon
Zu einer Lösung von 2.3 mg (20 μmol) (li?,2S)-l-Ammoindan-2-ol in 1.5 ml Tetrahydrofuran werden langsam 72 μl (410 μmol) Boran-NN-diethylanilin-Komplex gegeben und die Mischung für 30 min nachgerührt. Dann wird auf 00C gekühlt und eine Lösung von 49 mg (100 μmol) 7-Isopro- pyl-2,2-dimethyl-5-phenyl-6-[4-(trifluormethoxy)benzoyl]-2,3-dihydro-4H-chromen-4-on (Beispiel 32A) in 3.5 ml Tetrahydrofuran langsam zugetropft. Man lässt die Mischung langsam auftauen und rührt über Nacht bei Raumtemperatur. Danach wird Methanol zugegeben und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird in Essigsäureethylester aufgenommen und jeweils zwei- mal mit 1 M Salzsäure und gesättigter Natriumhydrogencarbonat-Lösung sowie einmal mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands mittels präparativer ΗPLC (Methode 1) liefert das Zielprodukt.
Ausbeute: 41 mg (83% d. Th., 88% ee)
HPLC (Methode 6): R, = 4.08 min
LC/MS (Methode 8): R, = 3.16 min
MS (ESIpos): m/z = 485 (M+H)+
1H-NMR (CDCl3, 400 MHz): δ = 1.15 (d, 3H), 1.23 (d, 3H), 1.45 (s, 3H), 1.49 (s, 3H), 1.94-2.09 (m, 2H), 2.64-2.81 (m, IH), 4.74 (s, IH), 6.92 (s, IH), 6.93-7.33 (m, 7H), 7.48-7.66 (m, 2H).
Beispiel 19
(5-Cyclohex-l-en-l-yl-4-hydroxy-7-isopropyl-2,2-dimethyl-3,4-dihydro-2H-chromen-6-yl)[4-(tri- fluormethyl)phenyl]methanon
Zu einer Lösung von 2.51 mg (17 μmol) (lR,2S)-l-Ammoindan-2-ol in 2.6 ml Tetrahydrofuran werden langsam 80 μl (450 μmol) Boran-N,N-diethylanilm-Komplex gegeben. Dann wird die Mischung auf 00C gekühlt und eine Lösung von 53 mg (112 μmol) 5-Cyclohex-l-en-l-yl-7-iso- propyl-2,2-dimethyl-6-[4-(trifluormethyl)benzoyl]-2,3-dihydro-4H-chromen-4-on (Beispiel 37A) in 2.6 ml Tetrahydrofuran langsam zugetropft. Man lässt die Mischung langsam auftauen und rührt über Nacht bei Raumtemperatur. Danach wird Methanol zugegeben und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird in Essigsäureethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Natriumhydrogencarbonat-Lösung sowie einmal mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands mittels Säulenchromatographie an Kieselgel (Laufmittel: Cyclohexan -> Cyclohexan/Essigsäureethylester 20:1) und nachfolgender chiraler ΗPLC (Methode 2) liefert die Zielverbindung.
Ausbeute: 15 mg (28% d. Th., >99% ee)
HPLC (Methode 2): R, = 3.62 min
LC/MS (Methode 7): R, = 3.15 min
MS (ESIpos): m/z = 473 (M+H)+.
Beispiel 20 und Beispiel 21 (45)-5-Cyclohex-l-en-l-yl-6-{(S)-hydroxy[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-di- methylchroman-4-ol (Beispiel 20)
und
(4S)-5-Cyclohex-l-en-l-yl-6-{(i?)-hydroxy[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-di- methylchroman-4-ol (Beispiel 21)
Zu einer Lösung von 7.10 mg (50 μmol) (li?,2iS)-l-Aminoindan-2-ol in 7.5 ml Tetrahydrofuran werden sehr langsam 230 μl (1.27 mmol) Boran-N,N-diethylanilin-Komplex gegeben und die Mischung für 30 min bei Raumtemperatur nachgerührt. Dann wird eine Lösung von 150 mg (320 μmol) 5-Cyclohex-l-en-l-yl-6-{hydroxy[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2- dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 35A) in 7.5 ml Tetrahydrofuran sehr langsam innerhalb von 30 min zugetropft. Nach Rühren über Nacht wird Methanol zugegeben und das
Lösungsmittel im Vakuum entfernt. Der Rückstand wird in Essigsäureethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Natriumhydrogencarbonat-Lösung sowie einmal mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands mittels Säulenchromato- graphie an Kieselgel (Laufmittel: Gradient Cyclohexan — > Cyclohexan/Essigsäureethylester 20:1 -> 10:1 -> 1 :1) und nachfolgender präparativer HPLC (Methode 1) liefert die diastereomeren- reinen Zielverbindungen, welche ihrerseits als Mischung zweier Rotamere vorliegen.
Beispiel 20:
Ausbeute: 53 mg (36% d. Th.)
HPLC (Methode 6): R, = 6.31 min
LC/MS (Methode 9): R1 = 3.24 min
MS (ESIneg): m/z = 519 (M-H+HCOOH)"
1H-NMR (CDCl3, 400 MHz): δ = 0.57 [0.62] (d, 3H), 1.08 [1.12] (d, 3H), 1.40 [1.42] (s, 3H), 1.50 [1.53] (s, 3H), 1.57-1.91 (m, 6H), 1.94-2.43 (m, 6H), 2.59 (s, IH), 2.77-2.88 [2.89-3.03] (m, IH), 4.68-4.77 [5.02-5.11] (m, IH), 5.63-5.72 [5.78-5.87] (m, IH), 6.03-6.11 [6.11-6.18] (m, IH), 6.74 (s, IH), 7.38-7.49 (m, 2H), 7.58 (d, 2H).
Beispiel 21:
Ausbeute: 47 mg (26% d. Th.)
HPLC (Methode 6): R. = 9.92 min
LC/MS (Methode 9): Rt = 3.28 min
MS (ESIneg): m/z = 519 (M-H+HCOOH)"
1H-NMR (CDCl3, 400 MHz): δ = 0.47 [0.59] (d, 3H), 1.12 [1.10] (d, 3H), 1.42 [1.45] (s, 3H), 1.47 [1.50] (s, 3H), 1.57-1.97 (m, 6H), 1.99-2.59 (m, 7H), 2.98 [2.86] (m, IH), 5.08 [4.71] (m, IH), 5.68 [5.88] (m, IH), 6.09 [6.12] (m, IH), 6.71 [6.76] (s, IH), 7.34-7.43 (m, 2H), 7.48-7.58 (d, 2H).
Beispiel 22 und Beispiel 23
(45)-5-Cyclopent-l-en-l-yl-6-{(5)-hydroxy[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-di- methylchroman-4-ol (Beispiel 22)
und
(4S)-5-Cyclopent-l -en-1 -yl-6- {(i?)-hydroxy[4-(trifluormethyl)phenyl]methyl} -7-isopropyl-2,2-di- methylchroman-4-ol {Beispiel 23)
Die Titelverbindungen werden als Nebenprodukte bei der Herstellung des Beispiels 38A erhalten.
Beispiel 22:
Ausbeute: 2.2 mg (2% d. Th.)
HPLC (Methode 6): R, = 10.67 min
LC/MS (Methode 7): R, = 2.97 min
MS (ESIneg): m/z = 505 (M-H+HCOOH)"
1H-NMR (CDCl3, 400 MHz): δ = 0.61 (d, 3H), 1.14 (d, 3H), 1.42 (s, 3H), 1.51 (s, 3H), 1.84-2.16 (m, 6H), 2.35-3.05 (m, 5H), 4.65-4.72 (m, IH), 5.82-5.90 (m, IH), 5.97-6.04 (m, IH), 6.76 (s, IH), 7.42 (d, 2H), 7.56 (d, 2H).
Beispiel 23:
Ausbeute: 7.3 mg (7% d. Th.)
HPLC (Methode 6): R, = 8.75 min
LC/MS (Methode 7): R, = 3.05 min
MS (ESIneg): m/z = 505 (M-H+HCOOH)-
1H-NMR (CDCl3, 400 MHz): δ = 0.59 (d, 3H), 1.12 (d, 3H), 1.45 (s, 3H), 1.49 (s, 3H), 1.97-2.25 (m, 5H), 2.34-3.06 (m, 6H), 4.64-4.72 (m, IH), 5.82-5.93 (m, IH), 5.99-6.07 (m, IH), 6.77 (s, IH), 7.39 (d, 2H), 7.54 (d, 2H).
Beispiel 24 und Beispiel 25
(4S)-5-Cyclopent-l-yl-6-{(5)-hydroxy[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-dimethyl- chroman-4-ol {Beispiel 24)
und
(4S)-5-Cyclopent-l-yl-6-{(i?)-hydroxy[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-dimethyl- chroman-4-ol {Beispiel 25)
Zu einer Lösung von 0.1 mg (6.7 μmol) (lR,2S)-l-Aminoindan-2-ol in 1.0 ml Tetrahydrofuran werden 23 μl (130 μmol) Boran-N,N-diethylanilin-Komplex gegeben und die Mischung für 30 min gerührt. Anschließend wird eine Lösung von 15 mg (33 μmol) 5-Cyclopent-l-yl-6-{hydroxy[4-
(trifluormethyl)phenyl]methyl}-7-isopropyl-2,2-dimethyl-2,3-dihydro-4H-chromen-4-on (Beispiel 38A) in 1.0 ml Tetrahydrofuran zugetropft und die Mischung für 2 h gerührt. Danach wird Methanol zugegeben und der Ansatz im Vakuum eingeengt. Der Rückstand wird in Essigsäure- ethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Natriumhydrogen- carbonat-Lösung sowie einmal mit gesättigter Natriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rohproduktes mittels präparativer ΗPLC (Methode 1) ergibt die diastereomerenreinen Zielverbindungen.
Beispiel 24:
Ausbeute: 5.4 mg (36% d. Th.)
ΗPLC (Methode 6): R, = 10.47 min
LC/MS (Methode 7): R, = 2.91 min
MS (ESIneg): m/z = 507 (M-Η+ΗCOOΗ)-
1H-NMR (CDCl3, 400 MHz): δ = 0.58 (br. d, 3H), 1.09 (d, 3H), 1.42 (s, 3H), 1.50 (s, 3H), 1.52- 1.96 (m, 8H), 2.02-2.21 (m, 4H), 2.90 (m, IH), 3.81 (m, IH), 5.04 (m, IH), 6.19 (m, IH), 6.72 (s, IH), 7.42 (d, 2H), 7.56 (d, 2H).
Beispiel 25:
Ausbeute: 5.9 mg (36% d. Th.)
HPLC (Methode 6): R, = 18.45 min
LC/MS (Methode 7): R, = 3.18 min
MS (ESIneg): m/z = 507 (M-H+HCOOH)"
1H-NMR (CDCl3, 400 MHz): δ = 0.59 (br. m, 3H), 1.09 (d, 3H), 1.44 (s, 3H), 1.49 (s, 3H), 1.52- 2.31 (m, 12H), 2.79-2.97 (m, IH), 3.72- 3.92 (m, IH), 4.97-5.06 (m, IH), 6.19 (m, IH), 6.73 (s, IH), 7.41 (d, 2H), 7.55 (d, 2H).
Beispiel 26 (4
ιS)-5-Cyclopentyl-7-isopropyl-2,2-dimethyl-6-[4-(trifluormethyl)benzyl]chroman-4-ol
Zu einer Lösung von 1.2 mg (10 μmol) (lR,2S)-l-Aminoindan-2-ol in 1.5 ml Tetrahydrofuran werden langsam 40 μl (210 μmol) Boran-NN-diethylanilin-Komplex gegeben und die Mischung für 30 min nachgerührt. Anschließend wird sehr langsam eine Lösung von 23 mg (50 μmol) 5-Cyclo- pentyl-7-isopropyl-2,2-dimethyl-6-[4-(trifluormethyl)benzyl]-2,3-dihydro-4H-chromen-4-on (Beispiel 39A) in 1.5 ml Tetrahydrofuran zugetropft und die Mischung für 2 h gerührt. Danach wird Methanol zugegeben und der Ansatz im Vakuum eingeengt. Der Rückstand wird in Essigsäure- ethylester aufgenommen und jeweils zweimal mit 1 M Salzsäure und gesättigter Νatriumhydrogen- carbonat-Lösung sowie einmal mit gesättigter Νatriumchlorid-Lösung gewaschen. Nach Trocknen der organischen Phase über Natriumsulfat wird im Vakuum eingeengt. Reinigung des Rückstands mittels präparativer ΗPLC (Methode 1) liefert das Zielprodukt.
Ausbeute: 17 mg (71% d. Th., 70% ee)
ΗPLC (Methode 5): R, = 6.87 min
LC/MS (Methode 8): R, = 3.39 min
MS (ESIpos): m/z = 429 (M+Η-Η2O)+
1H-NMR (CDCl3, 400 MHz): δ = 1.0 (d, 6H), 1.42 (s, 3H), 1.51 (s, 3H), 1.52-2.01 (m, 8H), 2.08 (dd, IH), 2.19 (dd, IH), 2.63-2.78 (m, IH), 3.61-3.78 (m, IH), 4.13 (s, 2H), 5.02 (s, IH), 4.98-5.08 (m, IH), 6.72 (s, IH), 7.09 (d, 2H), 7.48 (d, 2H).
Beispiel 27 [(45)-5-(4-Fluoφhenyl)-4-hydroxy-7-isopropyl-3,4-dihydrospiro[chromen-2, 1 '-cyclobutan]-6-yl]- [4-(trifluormethyl)phenyl]rnethanon
Unter Argon werden 2.7 mg (0.02 mmol) (li?,25)-l-Aminoindan-2-ol in 2 ml abs. Tetrahydrofuran gelöst, mit 130 μl (0.72 mmol) Boran-NN-diethylanilin-Komplex versetzt und 30 min bei Raumtemperatur gerührt. Dann werden 90 mg (0.18 mmol) 5-(4-Fluorphenyl)-7-isopropyl-6-[4-(trifluor- methyl)benzoyl]spiro[chromen-2,r-cyclobutan]^(3H)-on (Beispiel 54A), gelöst in 2.5 ml abs. Tetrahydrofuran, bei Raumtemperatur über 10 min hinzugefugt und anschließend der Ansatz für 5 h bei Raumtemperatur gerührt. Es wird mit 2 ml Methanol versetzt, 15 min gerührt und anschließend zur Trockene eingeengt. Der Rückstand wird in Essigsäureethylester aufgenommen, zweimal mit 1 Ν Salzsäure, zweimal mit gesättigter Νatriumhydrogencarbonat-Lösung und zweimal mit gesättigter Νatriumchlorid-Lösung gewaschen. Die organische Phase wird über Νatriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über präparative ΗPLC aufgereinigt. Es werden 49 mg (54% d. Th.) der Titelverbindung mit einem ee-Wert von 70% erhalten.
Rt = 5.54 min [Chiralpak IA, 250 x 4.6 mm; Eluent: iso-Propanol/iso-Ηexan 3:97; Fluss: 2 ml/min; Detektion: 260 nm].
1H-NMR (400 MHz, CDCl3): δ = 7.62 (d, 2H), 7.52 (d, 2H), 7.22-6.70 (m, 5H), 4.62 (br. s, IH), 2.75-2.59 (m, 3H), 2.48 (q, IH), 2.33-2.23 (m, 2H), 2.22-2.15 (m, IH), 2.07-1.92 (m, 2H), 1.81- 1.69 (m, IH), 1.19 (dd, 6H).
MS (DCI): m/z = 498 (M+H)+, 516 (M+NH4)+.
Beispiel 28 und Beispiel 29 (45)-5-(4-Fluorphenyl)-6-{(5)-hydroxy[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-3,4-dihydro- spiro[chromen-2,l'-cyclobutan]-4-ol {Beispiel 28)
und
(4S)-5-(4-Fluorphenyl)-6-{(i?)-hydroxy[4-(trifluoπnethyl)phenyl]methyl}-7-isopropyl-3,4-dihydro- spiro[chromen-2,l'-cyclobutan]-4-ol {Beispiel 29)
Unter Argon werden 1.6 mg (0.011 mmol) (lR,25)-l-Aminoindan-2-ol in 1 ml abs. Tetrahydro- furan gelöst, mit 77 μl (0.43 mmol) Boran-NN-diethylanilin-Komplex versetzt und 30 min bei Raumtemperatur gerührt. Nach dieser Zeit werden 60 mg (0.108 mmol) 5-(4-Fluorphenyl)-6- {hydroxy[4-(trifluormethyl)phenyl]methyl} -7-isopropylspiro[chromen-2, 1 '-cyclobutan]-4(3H)-on (Beispiel 52A), gelöst in 2 ml abs. Tetrahydrofuran, bei Raumtemperatur über 10 min hinzugefügt und anschließend der Ansatz 18 h bei Raumtemperatur gerührt. Es wird mit 2 ml Methanol versetzt, 15 min gerührt und anschließend zur Trockene eingeengt. Der Rückstand wird in Essigsäure- ethylester aufgenommen, zweimal mit 1 N Salzsäure, zweimal mit gesättigter Natriumhydrogen- carbonat-Lösung und zweimal mit gesättigter Natriumchlorid-Lösung gewaschen. Die organische Phase wird über Natriumsulfat getrocknet, filtriert und eingeengt. Das erhaltene Rohprodukt wird über präparative Dickschichtchromatographie (Laufmittel: Cyclohexan/Essigsäureethylester 4:1) gereinigt, wobei die Diastereomere getrennt werden.
.
- 124 -
Beispiel 28:
Ausbeute: 22 mg (41% d. Th.)
1H-NMR (300 MHz, CDCl3): δ = 7.51 (d, 2H), 7.38-7.27 (m, 3H), 7.19-7.02 (m, 3H), 6.91 (s, IH), 5.62 (br. s, IH), 4.54-4.47 (m, IH), 2.99 (heptett, IH), 2.72-2.60 (m, IH), 2.50-2.37 (m, IH), 2.35- 1.86 (m, 6H), 1.79-1.68 (m, IH), 1.48~(br. s, IH), 1.19 (d, 3H), 0.70 (d, 3H).
MS (DCI): m/z = 501 (M+H)+, 518 (M+NH4)+.
Beispiel 29:
Ausbeute: 22 mg (41% d. Th.)
1H-NMR (400 MHz, CDCl3): δ = 7.50 (d, 2H), 7.35-7.27 (m, 3H), 7.21-7.05 (m, 3H), 6.91 (s, IH), 5.62 (br. s, IH), 4.50 (br. s, IH), 2.99 (heptett, IH), 2.72-2.62 (m, IH), 2.49-2.38 (m, IH), 2.35- 1.88 (m, 6H), 1.80-1.68 (m, IH), 1.40 (d, IH), 1.21 (d, 3H), 0.66 (d, 3H).
MS (DCI): m/z = 501 (M+H)+, 518 (M+NK,)*.
Beispiel 30 und Beispiel 31
(4.S)-5-(4-Fluorphenyl)-6-{(5)-fluor[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-3,4-dihydro- Spiro [chromen-2, 1 '-cyclobutan]-4-ol {Beispiel 30)
und
(4S)-5 -(4-Fluorphenyl)-6- { (i?)-fluor [4-(trifluormethyl)phenyl]methyl } -7-isopropyl-3 ,4-dihydro- spiro[chromen-2,r-cyclobutan]-4-ol (Beispiel 31)
Unter Argon werden 3.1 mg (0.02 mmol) (lR,2S)-l-Aminoindan-2-ol in 3 ml abs. Tetrahydrofuran gelöst, mit 0.15 ml (0.84 mmol) Boran-N,N-diethylanilin-Komplex versetzt und 30 min bei Raumtemperatur gerührt. Nach dieser Zeit werden 105 mg (0.21 mmol) 5-(4-Fluorphenyl)-6-{fluor[4- (trifluormethyl)phenyl]rnethyl} -7-isopropylspiro[chromen-2, 1 '-cyclobutan]-4(3H)-on (Beispiel 53A), gelöst in 4 ml abs. Tetrahydrofuran, bei Raumtemperatur über 10 min hinzugefügt und anschließend der Ansatz 6 h bei Raumtemperatur gerührt. Es wird mit 3 ml Methanol versetzt, 15 min gerührt und anschließend zur Trockene eingeengt. Der Rückstand wird in Essigsäureethyl- ester aufgenommen, zweimal mit 1 N Salzsäure, zweimal mit gesättigter Natriumhydrogen- carbonat-Lösung und zweimal mit gesättigter Natriumchlorid-Lösung gewaschen. Die organische Phase wird über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über eine Kieselgelsäule (Laufmittel: Cyclohexan → Cyclohexan/Essigsäureethylester 10: 1) aufgereinigt. Es werden 110 mg eines Gemischs der beiden Zielverbindungen erhalten. Anschließende chromatographische Trennung an chiraler Phase [Säule: chiraler Kieselgel-Selektor basierend auf PoIy(N- methacryloyl-L-leucin-tert.-butylamid), 670 mm x 40 mm; Eluent: MTBE/iso-Ηexan 15:85; Fluss: 80 ml/min; 24°C; Detektion: 280 nm] liefert 36 mg (36% d. Th., 98% ee) des Beispiels 30 und 45 mg (43% d. Th., >99% ee) des Beispiels 31.
Beispiel 30:
Rt = 3.81 min [Säule: chiraler Kieselgel-Selektor basierend auf Poly(N-methacryloyl-L-leucm- tert.-butylamid), 250 mm x 4.6 mm; Eluent: MTBE/iso-Ηexan 15:85; Fluss: 2 ml/min; Detektion: 270 nm].
1H-NMR (400 MHz, CDCl3): δ = 7.54 (d, 2H), 7.27-7.14 (m, 4H), 7.12-6.99 (m, 2H), 6.96 (s, IH), 6.28 (d, IH), 4.57-4.50 (m, IH), 2.94-2.83 (m, IH), 2.71-2.62 (m, IH), 2.50-2.39 (m, IH), 2.35- 2.22 (m, 2H), 2.21-2.10 (m, IH), 2.00-1.89 (m, 2H), 1.80-1.68 (m, IH), 1.42 (d, IH), 1.18 (d, 3H), 0.81 (d, 3H).
MS (DCI): nVz = 502 (M)+, 520 (M+NH4)+.
Beispiel 31 :
Rt = 3.09 min [Säule: chiraler Kieselgel-Selektor basierend auf Poly(N-methacryloyl-L-leucin- /er/.-butylamid), 250 mm x 4.6 mm; Eluent: MTBE/iso-Hexan 15:85; Fluss: 2 ml/min; Detektion: 270 nra].
1H-NMR (400 MHz, CDCl3): δ = 7.53 (d, 2H), 7.38-7.32 (m, IH), 7.21 (d, 2H), 7.11-7.00 (m, 3H), 6.96 (s, IH), 6.28 (d, IH), 4.53-4.48 (m, IH), 2.88 (heptett, IH), 2.72-2.63 (m, IH), 2.50-2.39 (m, IH), 2.36-2.29 (m, IH), 2.28-2.11 (m, 2H), 2.01-1.89 (m, 2H), 1.80-1.69 (m, IH), 1.40 (d, IH), 1.19 (d, 3H), 0.76 (d, 3H).
MS (DCI): m/z = 502 (M)+, 520 (M+NH4)+.
Beispiel 32
(4S)-5-(4-Fluorphenyl)-7-isoproρyl-6-[4-(trifluormethyl)benzyl]-3,4-dihydrospiro[chromen-2,r- cyclobutan] -4-ol
Unter Argon werden 50 mg (0.10 mmol) 5-(4-Fluoφhenyl)-6-{hydroxy[4-(trifluormethyl)phenyl]- methyl}-7-isopropylspiro[chromen-2,l'-cyclobutan]-4(3H)-on (Beispiel 52A) in 1 ml Tetrahydro- furan gelöst und mit 22 μl (0.16 mmol) Triethylamin versetzt. Danach werden langsam 10 μl (0.13 mmol) Thionylchlorid zugetropft und der Ansatz 1.5 h lang gerührt. In einem separaten Kolben werden in der Zwischenzeit unter Argon 1.5 mg (0.01 mmol) (lR,25)-l-Aminoindan-2-ol in 2.7 ml abs. Tetrahydrofuran gelöst, mit 72 μl (0.40 mmol) Boran-NN-diethylanilin-Komplex versetzt und 30 min bei Raumtemperatur gerührt. Anschließend wird diese Lösung zum ersten Ansatz hinzugetropft und die Reaktionsmischung über Nacht bei Raumtemperatur gerührt. Danach werden in einem weiteren Kolben unter Argon abermals 1.5 mg (0.01 mmol) (li?,25)-l-Aminoindan-2-ol in
1 ml abs. Tetrahydrofuran gelöst, mit 54 μl (0.30 mmol) Boran-N,N-diethylanilin-Komplex versetzt, 30 min bei Raumtemperatur nachgerührt und dann wiederum zur Reaktionslösung hinzugetropft. Die Reaktionsmischung wird anschließend nochmals über Nacht bei Raumtemperatur gerührt.
Nach dieser Zeit werden 0.5 ml (0.5 mmol) Lithiumaluminiumhydrid-Lösung (1 M in Tetrahydrofuran) bei Raumtemperatur zum Reaktionsgemisch hinzugetropft. Nach 2 und 4 h werden nochmals jeweils die gleichen Mengen Lithiumaluminiumhydrid-Lösung hinzugefügt. Nach einer weiteren Stunde wird der Ansatz mit Tetrahydrofuran verdünnt, auf Eiswasser gegeben und mit 5 ml 6 N Salzsäure versetzt. Die wässrige Phase wird zweimal mit Essigsäureethylester extrahiert. Die organischen Phasen werden einmal mit 1 N Salzsäure, zweimal mit gesättigter Natriumhydrogen- carbonat-Lösung und zweimal mit gesättigter Natriumchlorid-Lösung gewaschen. Die organische Phase wird über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird durch prä- parative Dickschichtchromatographie (Laufmittel: Cyclohexan/Essigsäureethylester 5:1) aufgereinigt. Anschließende chromatographische Enantiomerentrennung an chiraler Phase [Säule: Chiral- pak AD-H, 250 x 20 mm; Eluent: iso-Propanol/iso-Hexan 3:97; Fluss: 1.5 ml/min; 24°C; Detek- tion: 260 nm] liefert 8 mg (16% d. Th.) der gewünschten, enantiomerenreinen Verbindung.
Rt = 4.34 min [Säule: Chiralpak IA, 250 x 4.6 mm; Eluent: iso-Propanol/iso-Hexan 3:97; Fluss: 1.5 ml/min; Detektion: 260 nm].
1H-NMR (400 MHz, CDCl3): δ = 7.40 (d, 2H), 7.11-7.06 (m, IH), 7.02-6.88 (m, 6H), 4.54-4.48 (m, IH), 3.81-3.69 (m, 2H), 2.89 (heptett, IH), 2.70-2.61 (m, IH), 2.49-2.38 (m, IH), 2.32-2.10 (m, 3H), 2.00-1.89 (m, 2H), 1.80-1.66 (m, IH), 1.41 (d, IH), 1.17 (d, 3H), 1.12 (d, 3H).
MS (DCI): m/z = 484 (M)+, 502 (M+NH4)+.
Beispiel 33
(4-tert. -Butylphenyl) [(4S)-5 -(4-fluorphenyl)-4-hydroxy-7-isopropyl-3 ,4-dihydrospiro [chromen- 2,1 '-cyclobutan]-6-yl]methanon
Unter Argon werden 2.1 mg (0.014 mmol) (li?,2S)-l-Aminoindan-2-ol in 1.5 ml abs. Tetrahydro- furan gelöst, mit 99 μl (0.56 mmol) Boran-NN-diethylanilin-Komplex versetzt und 30 min bei Raumtemperatur gerührt. Nach dieser Zeit werden 81.5 mg (0.14 mmol) 6-(4-tert.-Butylbenzoyl)- 5-(4-fluorphenyl)-7-isopropylspiro[chromen-2,r-cyclobutan]-4(3H)-on (Beispiel 57A), gelöst in 2 ml abs. Tetrahydrofuran, bei Raumtemperatur über 10 min hinzugegeben und der Ansatz anschließend 4 h bei Raumtemperatur gerührt. Es wird mit 2 ml Methanol versetzt, 15 min nachgerührt und anschließend zur Trockene eingeengt. Der Rückstand wird in Essigsäureethylester aufgenommen und zweimal mit 1 N Salzsäure, zweimal mit gesättigter Natriumhydrogencarbonat- Lösung und zweimal mit gesättigter Natriumchlorid-Lösung gewaschen. Die organische Phase wird über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über präparative ΗPLC aufgereinigt. Es werden 41 mg (60% d. Th.) der Titelverbindung mit einem ee-Wert von 87% erhalten.
R, = 8.46 min [Säule: Chiralpak IA, 250 x 4.6 mm; Eluent: iso-Propanol/iso-Ηexan 3:97; Fluss: 1.5 ml/min; Detektion: 230 nm].
1H-NMR (400 MHz, CDCl3): δ = 7.48 (br. s, 2H), 7.28 (s, 2H), 7.24-6.55 (m, 5H), 4.65 (br. s, IH), 2.80-2.56 (m, 2H), 2.46 (q, IH)3 2.33-2.12 (m, 3H), 2.07-1.90 (m, 2H), 1.80-1.69 (m, IH), 1.30 (s, 9H), 1.25-1.09 (m, 6H).
MS (DCI): m/z = 487 (M+H)+, 504 (M+NH4)+.
Beispiel 34 und Beispiel 35
(4S)-6-[(S)-(4-/e^.-Butylphenyl)(hydroxy)methyl]-5-(4-fluoφhenyl)-7-isopropyl-3,4-dihydrospiro- [chromen-2,l'-cyclobutan]-4-ol {Beispiel 34)
und
(4S)-6-[(R)-(4-/er/.-Butylphenyl)(hydroxy)methyl]-5-(4-fluorphenyl)-7-isopropyl-3,4-dihydrospiro- [chromen-2,l'-cyclobutan]-4-ol {Beispiel 35)
Unter Argon werden 2.1 mg (0.014 mmol) (li?,2.S)-l-Aminoindan-2-ol in 3 ml abs. Tetrahydro- furan gelöst, mit 102 μl (0.575 mmol) Boran-NN-diethylanilin-Komplex versetzt und 30 min bei Raumtemperatur gerührt. Nach dieser Zeit werden 70 mg (0.144 mmol) 6-[(4-tert.-Butylphenyl)- (hydroxy)methyl]-5-(4-fluoφhenyl)-7-isopropylspiro[chromen-2,r-cyclobutan]-4(3H)-on (Beispiel 55A), gelöst in 3.25 ml abs. Tetrahydrofuran, bei Raumtemperatur über 10 min hinzugegeben und der Ansatz anschließend 18 h bei Raumtemperatur gerührt. Es wird mit 2 ml Methanol versetzt, 15 min nachgerührt und anschließend zur Trockene eingeengt. Der Rückstand wird in Essigsäure- ethylester aufgenommen und zweimal mit 1 N Salzsäure, zweimal mit gesättigter Natriumhydro- gencarbonat-Lösung und zweimal mit gesättigter Natriumchlorid-Lösung gewaschen. Die organi- sehe Phase wird über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über präparative Dickschichtchromatographie (Laufmittel: Cyclohexan/Essigsäureethylester 5:1) aufgereinigt. Es werden 26 mg (37% d. Th., 86% ee) des Beispiels 34 und 29 mg (42% d. Th., 89% ee) des Beispiels 35 erhalten. Anschließende chromatographische Trennung von 15 mg des Beispiels
34 an chiraler Phase [Säule: Chiralpak AD-H, 250 x 20 mm; Eluent: iso-Propanol/iso-Hexän 5:95; Fluss: 15 ml/min; 24°C; Detektion: 260 nm] liefert 10.6 mg des Beispiels 34 mit einem ee-Wert von >99%.
Beispiel 34: Rt = 9.34 min [Säule: Chiralpak IA, 250 x 4.6 mm; Eluent: iso-Propanol/iso-Hexan 5:95; Fluss: 2 ml/min; Detektion: 260 nm].
1H-NMR (300 MHz, CDCl3): δ = 7.36-7.27 (m, 3H), 7.20-7.00 (m, 5H), 6.91 (s, IH), 5.62 (d, IH), 4.53-4.47 (m, IH), 3.11 (heptett, IH), 2.73-2.60 (m, IH), 2.49-2.09 (m, 4H), 2.02-1.87 (m, 3H), 1.82-1.68 (m, IH), 1.45 (d, IH), 1.30 (s, 9H), 1.18 (d, 3H), 0.71 (d, 3H).
MS (DCI): m/z = 506 (M+NH4)+.
Beispiel 35:
Rt = 10.98 min [Säule: Chiralpak IA, 250 x 4.6 mm; Eluent: iso-Propanol/iso-Hexan 5:95; Fluss: 2 ml/min; Detektion: 260 nm].
1H-NMR (300 MHz, CDCl3): δ = 7.33-7.18 (m, 4H), 7.12-7.03 (m, 4H), 6.91 (s, IH), 5.62 (d, IH), 4.54-4.48 (m, IH), 3.11 (heptett, IH), 2.73-2.61 (m, IH), 2.50-2.38 (m, IH), 2.35-2.09 (m, 3H), 2.03-1.88 (m, 3H), 1.81-1.67 (m, IH), 1.39 (d, IH), 1.27 (s, 9H), 1.20 (d, 3H), 0.65 (d, 3H).
MS (DCI): m/z = 506 (M+NH4)+.
Beispiel 36
(4S)-6-(4-teri.-Butylbenzyl)-5-(4-fluorphenyl)-7-isopropyl-3,4-dihydrospiro[chromen-2,r-cyclo- butan]-4-ol

Unter Argon werden 1.4 mg (0.009 mmol) (li?,2S)-l-Aminoindan-2-ol in 2 ml abs. Tetrahydro- furan gelöst, mit 66 μl (0.37 mmol) Boran-N,N-diethylanilin-Komplex versetzt und 30 min bei Raumtemperatur gerührt. Nach dieser Zeit werden 45 mg (0.092 mmol) 6-[(4-ter/.-Butylphenyl)- (fluor)methyl]-5-(4-fluoφhenyl)-7-isopropylspiro[chromen-2,r-cyclobutan]-4(3H)-on (Beispiel 55A), gelöst in 2 ml abs. Tetrahydrofuran, bei Raumtemperatur über 10 min hinzugegeben und der Ansatz anschließend 15 h bei Raumtemperatur gerührt. Es wird mit 2 ml Methanol versetzt, 15 min gerührt und anschließend zur Trockene eingeengt. Der Rückstand wird in Essigsäureethylester aufgenommen und zweimal mit 1 N Salzsäure, zweimal mit gesättigter Natriumhydrogencarbonat- Lösung und zweimal mit gesättigter Natriumchlorid-Lösung gewaschen. Die organische Phase wird über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über Dickschichtchromatographie (Laufmittel: Cyclohexan/Essigsäureethylester 10:1) aufgereinigt. Es werden 33.5 mg (77% d. Th., 80% ee) der Titelverbindung erhalten. Chromatographische Trennung von 60 mg des Beispiels 36 an chiraler Phase [Säule: Chiralpak AD-Η, 250 x 20 mm; Eluent: iso- Propanol/iso-Ηexan 3:97; Fluss: 15 ml/min; 24°C; Detektion: 260 nm] liefert 45 mg der Titelver- bindung mit einem ee-Wert von >99%.
Rt = 5.51 min [Säule: Chiralpak IA, 250 x 4.6 mm; Eluent: iso-Propanol/iso-Ηexan 3:97; Fluss: 2 ml/min; Detektion: 230 nm].
1H-NMR (300 MHz, CDCl3): δ = 7.17 (d, 2H), 7.11-7.03 (m, IH), 7.00-6.89 (m, 4H), 6.71 (d, 2H), 4.53-4.48 (m, IH), 3.68 (dd, 2H), 3.02 (heptett, IH), 2.72-2.59 (m, IH), 2.50-2.36 (m, IH), 2.32- 2.08 (m, 3H), 2.00-1.86 (m, 2H), 1.80-1.66 (m, IH), 1.42 (d, IH), 1.17 (d, 3H), 1.11 (d, 3H).
MS (ESIpos): m/z = 455 (M+H-H2O)+.
Beispiel 37
[(4S)-5-(4-Fluoφhenyl)-4-hydroxy-7-isopropyl-3,4-dihydrospiro[chromen-2,r-cyclobutan]-6-yl]- [4-(trifluormethoxy)phenyl]methanon

Unter Argon werden 1.6 mg (0.01 mmol) (lΛ,2iS)-l-Aminoindan-2-ol in 1.5 ml abs. Tetrahydro- furan gelöst, mit 76 μl (0.43 mmol) Boran-NN-diethylanilin-Komplex versetzt und 30 min bei Raumtemperatur gerührt. Nach dieser Zeit werden 54.5 mg (0.11 mmol) 5-(4-Fluorphenyl)-7-iso- propyl-6-[4-(trifluormethoxy)benzoyl]spiro[chromen-2, 1 '-cyclobutan]-4(3H)-on (Beispiel 60A), gelöst in 2.5 ml abs. Tetrahydrofuran, bei Raumtemperatur über 10 min hinzugegeben und der Ansatz anschließend 5 h bei Raumtemperatur gerührt. Es wird mit 2 ml Methanol versetzt, 15 min gerührt und anschließend zur Trockene eingeengt. Der Rückstand wird in Essigsäureethylester aufgenommen und zweimal mit 1 N Salzsäure, zweimal mit gesättigter Natriumhydrogencarbonat- Lösung und zweimal mit gesättigter Natriumchlorid-Lösung gewaschen. Die organische Phase wird über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über präparative ΗPLC aufgereinigt. Es werden 40 mg (73% d. Th.) der Titelverbindung mit einem ee-Wert von 91% erhalten.
R, = 6.02 min [Säule: Chiralpak IA, 250 x 4.6 mm; Eluent: iso-Propanol/iso-Ηexan 3:97; Fluss: 2 ml/min; Detektion: 260 nm].
1H-NMR (400 MHz, CDCl3): δ = 7.58 (d, 2H), 7.22-6.60 (m, 7H), 4.62 (br. s, IH), 2.78-2.61 (m, 2H), 2.47 (q, IH), 2.33-2.12 (m, 3H), 2.08-1.91 (m, 2H), 1.81-1.69 (m, IH), 1.28-1.09 (m, 6H).
MS (DCI): m/z = 515 (M+H)+, 532 (M+NH4)+.
Beispiel 38 und Beispiel 39
(4S)-5-(4-Fluorphenyl)-6-{(.S)-hydroxy[4-(trifluormethoxy)phenyl]methyl}-7-isopropyl-3,4-di- hydrospiro[chromen-2,l'-cyclobutan]-4-ol {Beispiel 38)
und
(4S)-5-(4-Fluoφhenyl)-6-{(R)-hydroxy[4-(trifluormethoxy)phenyl]methyl}-7-isopropyl-3,4-di- hydrospiro[chromen-2,r-cyclobutan]-4-ol (Beispiel 39)
Unter Argon werden 1.5 mg (0.01 mmol) (IR^1S)-I -Aminoindan-2-ol in 1.5 ml abs. Tetrahydro- furan gelöst, mit 69 μl (0.39 mmol) Boran-NN-diethylanilin-Komplex versetzt und 30 min bei Raumtemperatur gerührt. Nach dieser Zeit werden 50 mg (0.10 mmol) 5-(4-Fluorphenyl)-6- {hydroxy[4-(trifluormethoxy)phenyl]methyl} -7-isopropylspiro[chromen-2, 1 '-cyclobutan]-4(3H)-on (Beispiel 58A), gelöst in 2 ml abs. Tetrahydrofuran, bei Raumtemperatur über 10 min hinzugegeben und der Ansatz anschließend über Nacht bei Raumtemperatur gerührt. Es wird mit 2 ml Methanol versetzt, 15 min gerührt und anschließend zur Trockene eingeengt. Der Rückstand wird in Essigsäureethylester aufgenommen und zweimal mit 1 N Salzsäure, zweimal mit gesättigter Natriumhydrogencarbonat-Lösung und zweimal mit gesättigter Natriumchlorid-Lösung gewaschen. Die organische Phase wird über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über präparative Dickschichtchromatographie (Laufmittel: Cyclohexan/Essigsäure- ethylester 5:1) gereinigt. Es werden 13.5 mg (27% d. Th., 78% ee) des Beispiels 38 und 16.5 mg (33% d. Th.) des Beispiels 39 erhalten.
Beispiel 38:
R, = 7.70 min [Säule: Chiralpak IA, 250 x 4.6 mm; Eluent: iso-Propanol/iso-Ηexan 3:97; Fluss: 2 ml/min; Detektion: 254 nm].
1H-NMR (300 MHz, CDCl3): δ = 7.33-7.30 (m, 3H), 7.18-7.03 (m, 5H), 6.91 (s, IH), 5.62 (d, IH), 4.53-4.47 (m, IH), 3.05 (heptett, IH), 2.73-2.60 (m, IH), 2.49-2.37 (m, IH), 2.33-2.09 (m, 3H), 2.00-1.88 (m, 2H), 1.81-1.68 (m, IH), 1.44 (d, IH), 1.18 (d, 3H), 0.71 (d, 3H).
LC/MS (Methode 7): Rt = 3.06 min
MS (ESIpos): m/z = 499 (M+H-H2O)+.
Beispiel 39:
1H-NMR (300 MHz, CDCl3): δ = 7.32-7.25 (m, IH), 7.21-7.04 (m, 7H), 6.91 (s, IH), 5.62 (d, IH), 4.53-4.48 (m, IH), 3.04 (quin, IH), 2.72-2.60 (m, IH), 2.50-2.38 (m, IH), 2.36-2.09 (m, 3H), 2.03- 1.88 (m, 2H), 1.81-1.68 (m, IH), 1.40 (d, IH), 1.20 (d, 3H), 0.68 (d, 3H).
LC/MS (Methode 7): R, = 3.13 min
MS (ESIpos): m/z = 499 (M+H-H2O)+.
Beispiel 40
(4S)-5-(4-Fluorphenyl)-6-{fluor[4-(trifluormethoxy)phenyl]methyl}-7-isopropyl-3,4-dihydrospiro- [chromen-2, 1 '-cyclobutan]-4-ol
Unter Argon werden 0.6 mg (0.004 mmol) (lR,2S)-l-Aminoindan-2-ol in 0.5 ml abs. Tetrahydro- furan gelöst, mit 27.5 μl (0.15 mmol) Boran-N,N-diethylanilin-Komplex versetzt und 30 min bei Raumtemperatur gerührt. Nach dieser Zeit werden 20 mg (0.04 mmol) 5-(4-Fluorphenyl)-6-{fluor- [4-(trifluormethoxy)phenyl]methyl} -7-isopropylspiro[chromen-2, 1 '-cyclobutan]-4(3H)-on (Beispiel 59A), gelöst in 1 ml abs. Tetrahydrofuran, bei Raumtemperatur über 10 min hinzugegeben und der Ansatz anschließend über Nacht bei Raumtemperatur gerührt. Es wird mit 3 ml Methanol versetzt, 15 min gerührt und anschließend zur Trockene eingeengt. Der Rückstand wird in Essig- säureethylester aufgenommen und zweimal mit 1 N Salzsäure, zweimal mit gesättigter Natrium- hydrogencarbonat-Lösung und zweimal mit gesättigter Natriumchlorid-Lösung gewaschen. Die organische Phase wird über Natriumsulfat getrocknet, filtriert und eingeengt. Das Rohprodukt wird über eine Dickschichtchromatographie (Laufmittel: Cyclohexan → Cyclohexan/Essigsäureethyl- ester 5:1) aufgereinigt. Anschließend erfolgt eine weitere Reinigung mittels präparativer ΗPLC. Es werden 5.3 mg (26% d. Th.) der Titelverbindung als Diastereomerengemisch erhalten.
1H-NMR (300 MHz, CDCl3): δ = 7.38-6.90 (m, 9H), 6.23 (d, IH), 4.57-4.47 (m, IH), 2.98-2.87 (m, IH), 2.73-2.60 (m, IH), 2.51-2.10 (m, 4H), 2.03-1.87 (m, 2H), 1.80-1.68 (m, IH), 1.45-1.39 (m, IH), 1.20-1.12 (m, 3H), 0.88-0.73 (m, 3H).
MS (ESIpos): m/z = 501 (M+H-H2O)+.
Beispiel 41
(4S)-5-(4-Fluorphenyl)-7-isopropyl-6-[4-(trifluormethoxy)benzyl]-3,4-dihydrospiro[chromen-2,r- cyclobutan]-4-ol
Die Titelverbindung wird als Nebenprodukt bei der Darstellung von Beispiel 40 erhalten.
Ausbeute: 5.5 mg (28% d. Th.)
1H-NMR (300 MHz, CDCl3): δ = 7.13-7.03 (m, IH), 7.02-6.90 (m, 6H), 6.80 (d, 2H), 4.53-4.47 (m, IH), 3.70 (dd, 2H), 2.93 (heptett, IH), 2.70-2.60 (m, IH), 2.49-2.38 (m, IH), 2.32-2.10 (m, 3H), 1.99-1.89 (m, 2H), 1.78-1.68 (m, IH), 1.41 (d, IH), 1.17 (d, 3H), 1.12 (d, 3H).
MS (DCI): m/z = 501 (M+H)+, 518 (M+NH4)+.
Beispiel 42 und Beispiel 43
(4S)-5-Cyclopentyl-6-{(S)-fluor[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-3,4-dihydrospiro- [chroman-2, l'-cyclobutan]-4-ol (Beispiel 42)
und
(4S)-5-Cyclopentyl-6-{(R)-fluor[4-(trifluormethyl)phenyl]methyl}-7-isopropyl-3,4-dihydrospiro- [chroman-2,r-cyclobutan]-4-ol (Beispiel 43)
Die Titelverbindungen werden analog zur Vorschrift von Beispiel 7 und Beispiel 8 hergestellt.
Beispiel 42:
Ausbeute: 45 mg (26% d. Th.)
LC/MS (Methode 7): Rt = 3.29 min
HPLC (Methode 4): R, = 5.18 min
MS (ESIpos): m/z = 459 (M+H-H20)+
1H-NMR (CDCl3, 300 MHz): δ = 0.68 (d, 3H), 1.06 (d, 3H), 1.59-2.32 (m, 12H), 2.36-2.53 (m, 2H), 2.60-2.82 (m, 2H), 3.78 (quin, IH), 5.01-5.10 (m, IH), 6.78 (s, IH), 6.94 (d, IH), 7.37 (d, 2H), 7.59 (d, 2H).
Beispiel 43:
Ausbeute: 26 mg (15% d. Th.)
LC/MS (Methode 7): R, = 3.32 min
HPLC (Methode 4): Rt = 5.84 min
MS (ESIpos): m/z = 459 (M+H-H2O)+
1H-NMR (CDCl3, 300 MHz): δ = 0.67 (d, 3H), 1.08 (d, 3H), 1.58-2.28 (m, 12H), 2.38-2.59 (m, 2H), 2.61-2.82 (m, 2H), 3.79 (quin, IH), 4.98-5.05 (m, IH), 6.80 (s, IH), 6.89 (d, IH), 7.33 (d, 2H), 7.58 (d, 2H).
Die in der folgenden Tabelle 1 aufgeführten Beispiele werden in Analogie zu den oben beschriebenen Verfahren aus Beispiel 48A hergestellt:
Tabelle 1
Die in der folgenden Tabelle 2 aufgeführten Beispiele werden in Analogie zu den oben beschriebenen Verfahren aus Beispiel 47A hergestellt:
Tabelle 2
Zur chromatographischen Trennung der beiden Diastereomere Beispiel 57 und Beispiel 58 (227 mg) wird folgende chirale Phase verwendet: Kromasil TBB, 250 mm x 20 mm; Eluent: MTBE/iso-Hexan 10:90; Fluss: 25 ml/min; 24°C; Detektion: 250 nm. Es werden 56 mg (23% d. Th., 98% ee) des Diastereomers Beispiel 57 und 90 mg (37% d. Th., 97% ee) des Diastereomers Beispiel 58 erhalten.
B. Bewertung der pharmakologischen Wirksamkeit
Die pharmakologische Wirkung der erfindungsgemäßen Verbindungen kann in folgenden Assays gezeigt werden:
B-I. CETP-Inhibitions-Testung
B-Ll. Gewinnung von CETP
CETP wird aus humanem Plasma durch Differential-Zentrifugation und Säulenchromatographie in partiell gereinigter Form gewonnen und zum Test verwendet. Dazu wird humanes Plasma mit NaBr auf eine Dichte von 1.21 g pro ml eingestellt und 18 h bei 50.000 Upm und 4°C zentrifugiert. Die Bodenfraktion (d >1.21 g/ml) wird auf eine Phenyl-Sepharose 26/10 HP fast flow-Säule (Fa. Pharma- cia) aufgetragen, mit PBS-Puffer gewaschen und anschließend mit destilliertem Wasser eluiert. Das Eluat wird mit 10 Teilen PBS-Puffer und 1% (w/v) BSA versetzt. Die CETP-aktiven Fraktionen werden gepoolt.
B-I.2. CETP-Fluoreszenztest
Messung der CETP-katalysierten Übertragung eines fluoreszierenden Cholesterinesters zwischen Liposomen [modifiziert nach der Vorschrift von Bisgaier et al., J. LipidRes. 34, 1625 (1993)]:
Zur Herstellung der Donorliposomen wird 1 mg Cholesteryl-4,4-difluor-5,7-dimethyl-4-bora-3a,4a- diaza-s-indacen-3-dodecanoat (Cholesteryl BODIPY® FL Ci2, Fa. Molecular Probes) mit 5.35 mg Triolein und 6.67 mg Phosphatidylcholin in 2 ml Chloroform gelöst. Das Lösungsmittel wird bei mittlerer Temperatur in einer SpeedVac abgezogen und der Rückstand 1 h im Hochvakuum getrocknet. Der Rückstand wird anschließend am Ultraschallbad unter leichtem Erwärmen in 600 μl Dioxan gelöst und diese Lösung sehr langsam unter Ultrabeschallung zu 63 ml 50 mM TrisHCl / 150 mM NaCl / 2 mM EDTA-Puffer pH 7.3 bei Raumtemperatur gegeben. Die Suspension wird anschließend unter N2-Atmosphäre 30 min im Branson-Ultraschallbad bei ca. 50 Watt beschallt, wobei die Temperatur auf ca. 200C gehalten wird.
Die Akzeptorliposomen werden analog aus 86 mg Cholesteryloleat, 20 mg Triolein und 100 mg Phosphatidylcholin, in 1.2 ml Dioxan und 114 ml des obigen Puffers gelöst, durch 30-minütige Ultrabeschallung bei 50 Watt (200C) gewonnen.
B-L2.1.CETP-FIuoreszenztest mit angereichertem CETP
Zur Testung wird ein Testmix bestehend aus 1 Teil obigen Puffers, 1 Teil Donorliposomen und 2 Teilen Akzeptorliposomen verwendet.
50 μl Testmix werden mit 48 μl angereicherter CETP-Fraktion (1-3 μg), gewonnen über hydrophobe Chromatographie aus Humanplasma, sowie 2 μl einer Lösung der zu untersuchenden Substanz in DMSO versetzt und 4 h bei 37°C inkubiert.
Die Veränderung der Fluoreszenz bei 485/535 nm ist ein Maß für den CE-Transfer; die Hemmung des Transfers im Vergleich zum Kontrollansatz ohne Substanz wird ermittelt. Repräsentative Wirkdaten (IC50- Werte) zu den erfindungsgemäßen Verbindungen sind in Tabelle 3 aufgeführt:
Tabelle 3
B-I.2.2.CETP-FIuoreszenztest mit humanem Plasma
42 μl (86% v/v) humanes Plasma (Sigma P9523) werden mit 6 μl (12% v/v) Donorliposomen sowie 1 μl (2% v/v) einer Lösung der zu untersuchenden Substanz in DMSO versetzt und 24 h bei 37°C inkubiert.
Die Veränderung der Fluoreszenz bei 510/520 nm (Spaltbreite 2.5 nm) ist ein Maß für den CE- Transfer; die Hemmung des Transfers im Vergleich zum Kontrollansatz ohne Substanz wird ermittelt.
B-I.2.3.£r y/vo-CETP-Fluoreszenztest 80 μl Testmix werden mit 10 μl Puffer sowie 2 μl Serum versetzt und 4 h bei 370C inkubiert.
Die Veränderung der Fluoreszenz bei 485/535 nm ist ein Maß für den CE-Transfer; die Hemmung des Transfers im Vergleich zum Kontrollansatz ohne Substanz wird ermittelt.
B-I.3. CETP-SPA-Test
Zur Testung der CETP-Aktivität wird die Übertragung von 3H-Cholesterolester von humanen HD- Lipoproteinen auf biotinylierte LD-Lipoproteine gemessen. Die Reaktion wird durch Zugabe von Streptavidin-SPA®-beads (Fa. Amersham) beendet und die übertragene Radioaktivität direkt im Liquid Scintillation Counter bestimmt.
Im Testansatz werden 10 μl HDL-3H-Cholesterolester (ca. 50.000 cpm) mit 10 μl Biotin-LDL (Fa.
Amersham) in 50 mM Hepes / 0.15 M NaCl / 0.1% Rinderserumalbumin (RSA) / 0.05% NaN3 pH 7.4 mit 10 μl CETP (1 mg/ml) und 3 μl einer Lösung der zu prüfenden Substanz in 10% DMSO /
1% RSA für 18 h bei 37°C inkubiert. Anschließend werden 200 μl der SPA-Streptavidin-bead- Lösung (TRKQ 7005) zugesetzt, 1 h unter Schütteln weiter inkubiert und dann im Scintillations- zähler gemessen. Als Kontrollen dienen entsprechende Inkubationen mit 10 μl Puffer, 10 μl CETP bei 4°C sowie 10 μl CETP bei 37°C.
Die in den Kontrollansätzen mit CETP bei 37°C übertragene Aktivität wird als 100% Übertragung gewertet. Die Substanzkonzentration, bei der diese Übertragung auf die Hälfte reduziert ist, wird als IC50- Wert angegeben. Repräsentative Wirkdaten zu den erfindungsgemäßen Verbindungen sind in Tabelle 4 aufgeführt:
Tabelle 4
B-II. Bestimmung der ex vivo- und in vivo- Wirkung
B-II.1. Messung der ex v/vo-Aktivität an transgenen hCETP-Mäusen
Zur Prüfung auf CETP-inhibitorische Aktivität werden die Substanzen transgenen hCETP-Mäusen aus eigener Zucht [Dinchuk et al., BBA, 1295-1301 (1995)] oral mit der Schlundsonde verabreicht.
Dazu werden männliche Tiere einen Tag vor Versuchsbeginn randomisiert Gruppen mit gleicher
Tierzahl, in der Regel n = 4, zugeordnet. Vor der Substanzapplikation wird jeder Maus zur Bestimmung ihrer basalen CETP-Aktivität im Serum Blut durch Punktion des retroorbitalen Venen- plexus entnommen (Zeitpunkt Tl). Anschließend wird den Tieren die Testsubstanz mit der Schlundsonde peroral verabreicht. Hierfür werden die Substanzen in 10% Solutol HS 15 / 10% Ethanol / 80% einer 0.9%-igen Natriumchlorid-Lösung gelöst; das Applikationsvolumen beträgt in der Regel 10 ml pro kg Körpergewicht. Zu bestimmten Zeiten nach Applikation der Testsubstanz wird den Tieren ein zweites Mal Blut durch Punktion entnommen (Zeitpunkt T2), in der Regel 16 oder 24 h nach Substanzapplikation; gegebenenfalls kann dies aber auch zu einem anderen Zeitpunkt erfolgen.
Um die Hemmaktivität einer Substanz bewerten zu können, wird für jeden Zeitpunkt, also 16 bzw. 24 Stunden, eine entsprechende Kontrollgruppe eingesetzt, deren Tiere nur das Formulierungsmittel ohne Substanz erhalten. Bei den Kontrolltieren erfolgen die zwei Blutentnahmen pro Tier wie bei den substanzbehandelten Tieren, um die Veränderung der CETP-Aktivität ohne Inhibitor über den entsprechenden Versuchszeitraum (16 bzw. 24 h) bestimmen zu können.
Die Blutproben werden nach Abschluß der Gerinnung zentrifugiert und das Serum wird abpipettiert. Zur Bestimmung der CETP-Aktivität wird der Cholesterylester-Transport über 4 h bestimmt. Dazu werden im Testansatz in der Regel 2 μl Serum eingesetzt; der Test wird wie unter B-I.2.3. beschrieben durchgeführt.
Die Differenzen im Cholesterylester-Transport [pM CE/h (T2) - pM CE/h (Tl)] werden für jedes Tier berechnet und in den Gruppen gemittelt. Eine Substanz, die zu einem der Zeitpunkte den Cholesterylester-Transport um >20% herabsetzt, wird als wirksam angesehen.
B-II.2. Messung der in vivo-Wirksamkeit an Syrischen Goldhamstern
Zur Bestimmung der oralen Wirkung von CETP-Inhibitoren auf Serum-Lipoproteine und -Triglyceride werden 150-200 g schwere weibliche syrische Goldhamster aus eigener Zucht (Stamm BAYrDSN) verwendet. Die Tiere werden pro Käfig zu sechst gruppiert und zwei Wochen bei Futter und Wasser ad libitum akklimatisiert.
Unmittelbar vor Versuchsbeginn und nach Abschluss der Substanzapplikation wird mittels retro- orbitaler Punktion des Venenplexus Blut entnommen, woraus nach 30 min Inkubation bei Raumtemperatur und 20 min Zentrifugation bei 30.000 g Serum gewonnen wird. Die Substanzen werden in 20% Solutol / 80% Wasser gelöst und mit Hilfe einer Schlundsonde peroral verabreicht. Die Kontrolltiere erhalten identische Volumina Lösungsmittel ohne Testsubstanz.
Die Bestimmung von Triglyceriden, Gesamt-Cholesterin, HDL-Cholesterin und LDL-Cholesterin erfolgt mit Hilfe des Analysegeräts COBAS INTEGRA 400 plus (Fa. Roche Diagnostics) nach Angaben des Herstellers. Aus den Messwerten wird für jeden Parameter die durch Substanzbehandlung hervorgerufene prozentuale Veränderung für jedes Tier berechnet und als Mittelwert mit Standardabweichung pro Gruppe (n = 6 oder n = 12) angegeben. Sind die Substanzeffekte im Vergleich zur Lösungsmittel-behandelten Gruppe signifikant, wird der mittels t-Test ermittelte p-Wert hinzugefügt (* p <0.05; ** p <0.01; *** p <0.005).
B-II.3. Messung der in vi'vo-Wirksamkeit an transgenen hCETP-Mäusen
Zur Bestimmung der oralen Wirkung auf Lipoproteine und Triglyceride wird transgenen Mäusen [Dinchuk et al., BBA, 1295-1301 (1995)] Testsubstanz an 3 Tagen einmal täglich mit der Schlundsonde peroral verabreicht. Die Substanzen werden dazu in 10% Solutol HS 15 / 10% Ethanol / 80% einer 0.9%-igen Natriumchlorid-Lösung gelöst; das Applikationsvolumen beträgt in der Regel 10 ml pro kg Körpergewicht. Vor Versuchsbeginn wird den Mäusen retroorbital Blut entnommen, um Cholesterin und Triglyceride im Serum zu bestimmen. Das Serum wird, wie oben für Hamster beschrieben, durch Inkubation bei Raumtemperatur für 30 min und anschließende Zentrifugation bei 6.000 g gewonnen. Am Tag nach der letzten Substanzapplikation wird den Mäusen wieder Blut entnommen, um Lipoproteine und Triglyceride erneut zu bestimmen. Die Veränderungen der gemessenen Parameter werden als prozentuale Veränderung gegenüber dem Ausgangswert ausgedrückt.
Repräsentative Wirkdaten zu den erfindungsgemäßen Verbindungen sind in Tabelle 5 aufgeführt:
Tabelle 5
C. Ausführungsbeispiele für pharmazeutische Zusammensetzungen
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überfuhrt werden:
Tablette:
Zusammensetzung:
100 mg der erfϊndungsgemäßen Verbindung, 50 mg Lactose (Monohydrat), 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrrolidon (PVP 25) (Fa. BASF, Ludwigshafen, Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg. Durchmesser 8 mm, Wölbungsradius 12 mm.
Herstellung:
Die Mischung aus erfϊndungsgemäßer Verbindung, Lactose und Stärke wird mit einer 5%-igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat 5 Minuten gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse verpresst (Format der Tablette siehe oben). Als Richtwert für die Verpressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzung:
1000 mg der erfmdungsgemäßen Verbindung, 1000 mg Ethanol (96%), 400 mg Rhodigel® (Xanthan gum der Firma FMC, Pennsylvania, USA) und 99 g Wasser.
Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 10 ml orale Suspension.
Herstellung:
Das Rhodigel wird in Ethanol suspendiert, die erfϊndungsgemäße Verbindung wird der Suspension zugefügt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluß der Quellung des Rhodigels wird ca. 6 h gerührt.
Oral applizierbare Lösung:
Zusammensetzung:
500 mg der erfindungsgemäßen Verbindung, 2.5 g Polysorbat und 97 g Polyethylenglycol 400. Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 20 g orale Lösung.
Herstellung:
Die erfmdungsgemäße Verbindung wird in der Mischung aus Polyethylenglycol und Polysorbat unter Rühren suspendiert. Der Rührvorgang wird bis zur vollständigen Auflösung der erfindungsgemäßen Verbindung fortgesetzt.
i.v.-Lösung:
Die erfindungsgemäße Verbindung wird in einer Konzentration unterhalb der Sättigungslöslichkeit in einem physiologisch verträglichen Lösungsmittel (z.B. isotonische Kochsalzlösung, Glucose- lösung 5% und/oder PEG 400-Lösung 30%) gelöst. Die Lösung wird steril filtriert und in sterile und pyrogenfreie Injektionsbehältnisse abgefüllt.