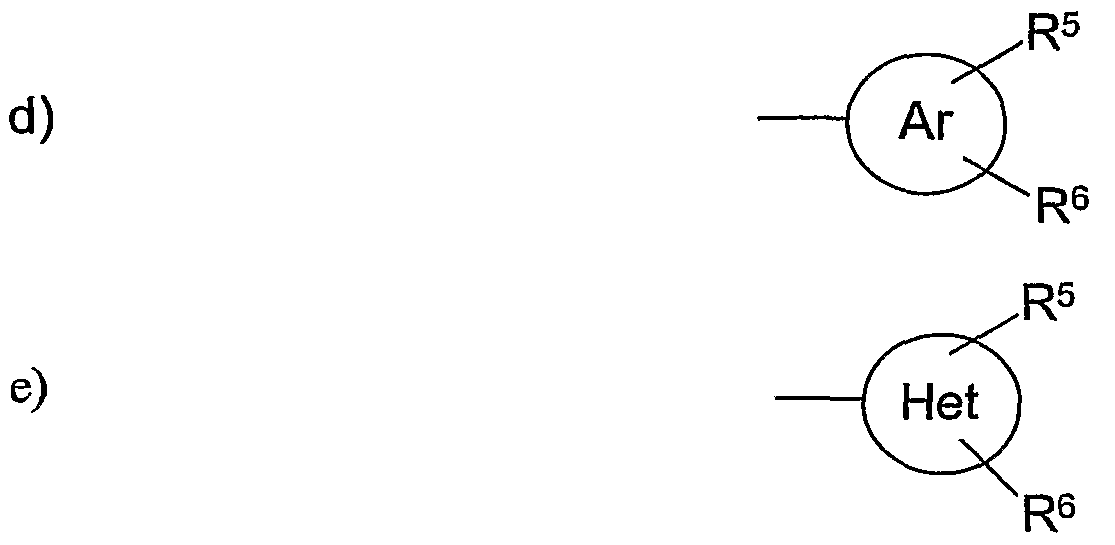2-Thio-substituierte Imidazolderivate und ihre Verwendung in der Pharmazie
Die vorliegende Erfindung betrifft 2-Thio-substituierte Imidazolderivate mit immun- modulierender und die Cytokinfreisetzung hemmender Wirkung, pharmazeutische Mittel, welche diese Verbindungen enthalten, und ihre Verwendung in der Pharmazie.
Pharmakologisch wirksame Imidazolverbindungen mit antiinflammatorischer Aktivität sind bereits bekannt. So wurden unter anderem Verbindungen mit 4,5-Di(hetero)aryl- imidazol-Elementen näher untersucht und verschiedene pharmazeutische Wirkungen davon beschrieben. Es sind auch Verbindungen bekannt, die an der 2-Position eine Substitution aufweisen. Das US-Patent 4,585,771 offenbart 4,5-Diphenylimidazolderi- vate, die an der 2-Position mit einem Pyrrolyl-, Indolyl-, Imidazolyl- oder Thiazolylrest substituiert sind und eine antiinflammatorische und antiallergische Aktivität besitzen. Die US-Patente 4,528,298 und 4,402,960 beschreiben 4,5-Di(hetero)arylimidazol- derivate, die an der 2-Position über eine Thio-, Sulfinyl- oder Sulfonylgruppe mit. einem Phenyl-, Pyridyl-, N-Oxypyridyl-, Pyrimidyl-, Thiazolyl- oder Thienylrest substituiert sind, und eine antiinflammatorische und antiallergische Aktivität besitzen. Die US-Patente 4,461 ,770, 4,528,298 und 4,584,310 (EP 004 648 A) beschreiben 4-(5- Aryl)-5-(4-heteroaryl)-imidazolderivate, die an der 2-Position über eine Thio-, Sulfinyl- oder Sulfonylgruppe mit einem substituierten oder nicht-substituierten aliphatischen Kohlenwasserstoff substituiert sind, und unter anderem eine antiinflammatorische Wirkung haben. Imidazolverbindungen mit immunmodulierender und die Cytokinfrei- setzung hemmender Wirkung sind beschrieben in WO 02/066458, WO 02/076951 und DE 102 22 103. Weitere Imidazolverbindungen mit antiinflammatorischer Wirkung sind in der WO 96/03387, EP 005 545 (US 4,440,776; 4,355,039; 4,269,847), EP 236 628 (US 4,686,231), DE 35 04 678, US 4,190,666, US 4,402,960 und US 4,585,771 beschrieben. Die EP 372 445 (US 5,318,984; US 5,166,214) und US 5,364,875 beschreiben Imidazolverbindungen mit antihypercholesterolämischer Aktivität.
Die WO 00/17192 (DE 198 42 833) betrifft 4-Heteroaryl-5-phenylimidazolderivate, die an der 2-Position mit einer Phenylalkylthiogruppe substituiert sind. Diese Verbindun-
gen wirken als Entzündungshemmstoffe und Hemmstoffe der Cytokinfreisetzung. Die WO 99/03837 und WO 93/14081 beschreiben 2-substituierte Imidazole, die die Synthese einer Anzahl inflammatorischer Cytokine hemmen. Die in der WO 93/14081 beschriebenen Verbindungen weisen in der 2-Position einen über ein Schwefelatom gebundenen, phosphorhaltigen Substituenten oder einen Aryl- oder Heteroarylsubsti- tuenten auf. Die WO 91/10662 und WO 91/13876 beschreiben Imidazolderivate, welche die Acyl-Coenzyme A:Cholesterol-0-acyltransferase und die Bindung von Thromboxan TxA2 inhibieren. Die WO 95/00501 beschreibt Imidazolderivate, die als Cyclooxygenaseinhibitoren brauchbar sind.
J. Med. Chem. 1996, 39, 3927-37 beschreibt Verbindungen mit 5-Lipoxygenase- und Cyclooxygenase-inhibierender Wirkung, wobei 2-(4-Methylsulfinylphenyl)-4-(4~ fluorphenyl-5-(pyrid-4-yl)-imidazol auch eine Cytokin-inhibierende Wirkung besitzt.
Weitere 2-Thio-substituierte Imidazolderivate sind beschrieben in JP 01-040 467, SU 1 415 725, Acta Chim. 1969, 61 , 69 - 77, J. prakt. Chem. 1972, 314, 785 - 792 und DE 101 14 775, Indian J. Chem., Sect. B, 1983, 22B(3), 268-269, Bioorganic & Me- dicinal Chem. Lett., Vol 5, No. 2, 177-180, 1995, Phosphorus Sulfur 1988, 35(1-2), 83-88, Arch. Biochem. Biophys. Vol. 297, 258-264, 1992, J. Med. Chem. 1995, 38, 1067-1083, Helv. Chim. Acta 82, 1999, 290-296, Helv. Chim. Acta 81 , 1998, 1585- 1595.
Trotz der zahlreichen bekannten Verbindungen besteht daher weiterhin ein Bedarf nach Verbindungen mit antiinflammatorischer Wirkung, die die Cytokinfreisetzung hemmen.
Die Aufgabe der Erfindung ist die Bereitstellung solcher Verbindungen.
Überraschenderweise wurde nun gefunden, dass bestimmte 2-substituierte Imida- zolderivate eine hohe immunmodulierende und/oder die Cytokinfreisetzung hemmende Wirksamkeit aufweisen.
Gegenstand der vorliegenden Erfindung sind daher die 2-Thio-substituierten Imidazolderivate der Formel I
worin
R1 für CrC6-Alkyl, C3-C7-Cycloalkyl oder Aryl steht, das gegebenenfalls durch ein Halogenatom, CrCe-Alkyl oder Halogen-Cι-C6-Alkyl substituiert ist;
R2 ausgewählt ist unter
a) Aryl-Cι-C4-alkyl, wobei der Arylrest ein, zwei oder drei Substituenten aufweisen kann, die unabhängig voneinander ausgewählt sind unter CrC6-Alkyl, Cι-C6-Alkoxy, Halogen, CrC6- Alkylsulfanyl, CrC6-Alkylsulfinyl, C-ι-C6-Alkylsulfonyl und Hydro- xy, und
b) CrC6-Alkyl, das gegebenenfalls durch CN oder Halogen substituiert ist;
R° ausgewählt ist unter
a) NR4R10;
b) NR7COR10;
c) NR7COOR10;
d) NR7CONR7R10;
e) NR7CONR7COR10;
f) OR10;
g) S(0)mR10;
h) Halogen;
i) OH;
j) N3
k) NH2
I) SH;
wobei R3 nicht für OH, Halogen, CrC6-Alkylthio oder Cι-C6-Alkoxy steht, wenn R2 für Phenyl-C C4-alkyl steht und der Phenylrest einen CrC6-Alkylsulfanyl-, Ci-Cβ-Alkylsulfinyl- oder CrC6-Alkylsulfonyl-Substituenten aufweist;
R4 für H oder eine physiologisch abspaltbare Gruppe steht,
R5 und R6, die gleich oder verschieden sein können, für H, Halogen, OH, d- C6-Alkoxy, CrCe-Alkyl, Halogen-C C6-Alkyl, Cι-C6-Alkylsulfanyl, NH2, d-C6- Alkylamino oder Di-CrC6-alkylamino stehen;
R7 für R4, CrCe-Alkyl oder Benzyl steht;
R10 eine der folgenden Bedeutungen besitzt:
a) A — B
f) d-Cβ-Alkyl, das mit 2 oder 3 Phenylgruppen substituiert ist;
g) Trifluormethyl (insbesondere, wenn R3 für die Reste b) bis f) steht)
A für geradkettiges oder verzweigtes d-C6-Alkylen, C2-C6-Alkenylen oder C3-
Alkinylen steht;
B ausgewählt ist unter
a) H
f) Od-Ce-Alkyl; g) NR
11R
12; h) OH; i) Halogen; j) d-Ce-Alkylsulfanyl
R11 und R12, die gleich oder verschieden sein können, für H, CrCe-Alkyl oder Phenyl stehen;
Hy für einen 3- bis 10-gliedrigen nicht-aromatischen mono-, bi- oder tricycli- schen Carbocyclus steht, der gegebenenfalls mit einem Benzolring anelliert sein kann;
Ar für einen 5- oder 6-gliedrigen aromatischen Heterocyclus steht, der 1, 2 oder 3 Heteroatome aufweist, die unabhängig voneinander ausgewählt sind unter 0, S und N und der gegebenenfalls mit einem Benzolring anelliert sein kann;
Het für einen 5- oder 6-gliedrigen nicht-aromatischen Heterocyclus steht, der 1 , 2 oder 3 Heteroatome aufweist, die unabhängig voneinander ausgewählt sind unter O, S und N, der gegebenenfalls mit einem Benzolring anelliert sein kann oder der gegebenenfalls auch bicyclisch oder tricyclisch überbrückt sein kann;
m für 0,1 oder 2 steht; n für 1 , 2, 3, 4 oder 5 steht;
und die Tautomere, optischen Isomere und physiologisch verträglichen Salze davon.
Soweit die erfindungsgemäßen Verbindungen Asymmetriezentren aufweisen, sind Racemate sowie optische Isomere (Enantiomere, Diastereomere) umfasst. Bei den erfindungsgemäßen Verbindungen kann folgendes tautomeres Gleichgewicht vorliegen:
Die Erfindung umfasst beide tautomeren Formen.
Die Erfindung umfasst auch die physiologisch verträglichen Salze der Verbindungen der Formel I. Dabei handelt es sich im vorliegenden Fall insbesondere um Säureadditionssalze. Für Säureadditionssalze werden anorganische Säuren wie Salzsäure, Schwefelsäure oder Phosphorsäure oder organische Säuren wie Weinsäure, Citro- nensäure, Maleinsäure, Fumarsäure, Äpfelsäure, Mandelsäure, Ascorbinsäure, Glu- consäure und dergleichen, eingesetzt.
Der Ausdruck „Alkyl" (auch in Verbindung mit anderen Gruppen, wie Phenylalkyl, Al- kylsulfonyl, Alkoxy etc.) umfasst geradkettige und verzweigte Alkylgruppen mit 1 bis 6 bzw. 1 bis 4 C-Atomen, wie Methyl, Ethyl, n- und i-Propyl, n-, i- und t-Butyl, sec- Butyl, n-Pentyl, Isoamyi, Neopentyl und n-Hexyl. Entsprechendes gilt für "CrC6- Alkylen".
Der Ausdruck „Carbocyclus" umfasst gesättigte oder ungesättigte, nicht-aromatische, monocyclische, bicyclische und tricyclische Kohlenwasserstoffe. Die Kohlenwasserstoffe können mit einem oder zwei Benzolringen anelliert sein. Monocyclische Kohlenwasserstoffe sind C3-C6-Cycloalkyl, wie Cyclopropyl, Cyclopentyl, Cyclohexyl. Beispiele für bi- und tricyclische Kohlenwasserstoffe sowie benzoanellierte Carbocyc- len sind Indanyl, Decalinyl, Tetralinyl, Fluorenyl, Dihydroanthracenyl, Dibenzosube-
renyl, Norbomyl oder Adamantyl. Beispiele für substituierte Carbocyclen sind Methyl- cyclopropyl oder Methylcyclohexyl. Bevorzugt sind unsubstituierte Reste.
Der Ausdruck „Aryl" umfasst aromatische Ringsysteme wie Phenyl oder Naphthyl.
Der Ausdruck „Halogen" steht für ein Fluor-, Chlor-, Brom- oder lodatom, insbesondere ein Fluor - oder Chloratom.
Der Ausdruck "Halogen-CrC6-alkyl" umfasst geradkettige und verzweigte Alkylgrup- pen mit 1 bis 6 und insbesondere 1 bis 4 Kohlenstoff atomen, die mono- oder polyha- logeniert sind. Vorzugsweise sind 1 , 2, 3, 4 oder 5 Halogenatome vorhandeln. Bevorzugte Halogenatome sind F und Cl. Beispiele für Halogen-Cι-C6-alkyl sind -CH2CI, -CH2CH2CI, -CH2CCI3, -CF3, -CHF2, -CH2F, -CH2CF3und -CF2CF3. CF3 ist bevorzugt.
Eine physiologisch abspaltbare Gruppe ist eine Gruppe, die sich unter physiologischen Bedingungen vom Rest des Moleküls enzymatisch oder chemisch abspalten lässt. Beispiele sind -COR14, -C02R14, -CONH2, -CONHR14, -CHR16-OR14, -CHR16-0- COR14, -COC(R16)2-OH, -COR15, SO2R15 und -S02R14, worin R14 für d-C6-Alkyl oder CF3, R15 für Phenyl oder Tolyl (insbesondere p-Tolyl) und R16 für H oder d-Ce-Alkyl steht.
Bei dem aromatischen, 5- oder 6-gliedrigen Heterocyclus handelt es sich insbesondere um unsubstituiertes (R5, R6 = H) oder substituiertes 2-Pyridyl, 3-Pyridyl, 4- Pyridyl, 2- oder 3-Thienyl, 2- oder 3-Furyl, Thiazolyl, Imidazolyl, Oxazolyl, Isothiazo- lyl, Pyrazolyl, Isoxazolyl, Triazolyl oder Pyrimidyl. Bevorzugte Substituenten sind eine oder zwei Gruppen, die unabhängig voneinander ausgewählt sind unter Halogen, insbesondere Cl oder C C6-Alkyl. Der oder die Substituenten sind dabei an ein C- Atom oder N-Atom des aromatischen Restes gebunden. Bevorzugt sind unsubstitu- ierte Reste. Beispiele für substituierte Reste sind Chlorthienyl, insbesondere 5-
Chlorthien-2-yl, Chlorfuryl, insbesondere 5-Chlor-fur-2-yl, Beispiele für anellierte Reste sind Benzofuranyl, Benzothiazolyl und Benzothiophen.
Der nicht-aromatische 5- oder 6-gliedrige Heterocyclus kann gesättigt oder ungesättigt sein. Vorzugsweise handelt es sich um unsubstituiertes oder substituiertes Tetra- hydrofuranyl, Tetrahydropyranyl, Pyrrolidinyl, N-Methylpyrrolidinyl, N-Ethylpyrrolidinyl, Piperazinyl oder Morpholinyl, wobei der Heterocyclus über ein N-Heteroatom oder Ring-C-Atom gebunden oder substituiert sein kann. Bevorzugte Substituenten sind ein oder zwei Reste, die unabhängig voneinander ausgewählt sind unter Halogen, insbesondere Cl, oder d-Cβ-Alkyl. Bevorzugt sind unsubstituierte Reste.
steht für substituiertes oder unsubstituiertes Cyclopropyl, Cyclobutyl, Cycloheptyl
un insbesondere Cyclopentyl und Cyc- lohexyl. R
5 und R
6 stehen vorzugsweise und unabhängig voneinander für H, Halogen oder CrCe-Alkyl.
Beispiele für substituierte Cycloalkylgruppen sind Methylcyclopropyl oder Methylcyc- lohexyl. Bevorzugt sind unsubstituierte Reste.
Phenyl-d-d-alkyl bedeutet insbesondere Benzyl, 1 -Phenylethyl oder 2-Phenylethyl.
R1 steht vorzugsweise für einen Phenylrest und insbesondere für einen Halogen-, CF3- oder CrCβ-Alkyl-substituierten Phenylrest, wobei ein Fluor-substituierter Phenylrest besonders bevorzugt ist. Der Substituent steht vorzugsweise in 3- und insbe- sondere in 4-Position. Beispiele für substituierte Phenylreste sind 4-Fluorphenyl, 2,4- Difluorphenyl, 3-Trifluormethyl, 3-Tolyl oder 3-Chlorphenyl.
R2 steht vorzugsweise für einen Benzyl-, C3-C6-Cycloalkyl, C4-C7-Methyl-cycloalkyl oder d-Cβ-Alkylrest, wobei die Phenylgruppe des Benzylrests wie oben angegebe- nen substituiert sein kann. Bevorzugte Substituenten der Phenylgruppe des Benzyl- restes sind CrC6-Alkylsulfanyl, d-Cβ-Alkylsulfinyl und d-C6-Alkylsulfonyl. Beispiele für R2 sind CH3) CH3CH2, (CH3)2CH, CH2CN, CH2CF3, CF3 und Cyclopropyl.
R3 steht vorzugsweise für den Rest der Formel
worin R
4, R
5 und R
6 sowie A die oben angegebenen Bedeutungen besitzen. R
5 und R
6 stehen vorzugsweise für H, Methyl, Methoxy oder Chlor. Wenn der Phenylring dieser Gruppe substituiert ist, stehen die Reste R
5 und R
6 vorzugsweise in 3- und/oder 4-Position.
Weiterhin bevorzugt steht R3 für
a) NR4R10, wobei R10 für Cyclopropyl, Cyclopropylmethyl, Cyclopentyl, Cy- clohexyl oder Cycloheptyl steht;
b) NR4R10, wobei R10 für d-Ce-Alkyl, insbesondere Methyl, Ethyl oder I- sopropyl, oder für 3,3-Diphenylpropyl oder 1 ,3-Diphenylprop-2-yl steht;
c) NR4R10, wobei R10 für A — B und B für OH, C C6-Alkyoxy, NR11R12 oder Phenyl stehen;
d) NR7COR10- wobei R10 für A — B und B für Phenyl stehen;
e) NR7COOR10, wobei R10 für d-C6-Alkyl steht.
A steht vorzugsweise für d-C2-Alkylen und insbesondere für Ethyliden.
m steht vorzugsweise für 0.
Eine besonders bevorzugte Ausführungsform sind die Verbindungen der Formel I, worin R1 für 4-Fluorphenyl steht, R2 für CrC6-Alkyl oder Benzyl steht, wobei die Phenylgruppe des Benzylrests wie oben angegeben substituiert sein kann; R3 für den Rest der Formel
steht, wobei R
4, R
5, R
6 und A die oben angegebenen Bedeutungen besitzen und m für 0 steht.
Eine weitere bevorzugte Ausführungsform sind Verbindungen der Formel I, worin R2 für CrCe-Alkyl, insbesondere Methyl, steht und R1 für Halogenphenyl oder Halogen- CrC6-Alkylphenyl, insbesondere 4-Fluorphenyl, 2,4-Difluorphenyl, 4- Trifluormethylphenyl oder 3-Trifluormethylphenyl steht. R3 besitzt dann vorzugsweise folgende Bedeutungen:
a) Halogen, insbesondere F oder Cl;
b) OH oder O Cι-C6-Alkyl, insbesondere Methoxy und Isopropoxy,
c) Phenylamino;
d) Phenylgruppe durch Phenyl- oder Naphthyl-d-d-alkylamino 1 oder 2 Halo gen, insbesondere F oder Cl, CrC6-Alkoxy oder CrC6-Alkyl substituiert sein kann. Die Aminogruppe kann zusätzlich durch CrC6-Alkyl substituiert sein. Beispiele für derartige Reste sind Benzylamino, 4-Methoxybenzylamino, 4- Methylbenzylamino, 4-Chlorbenzylamino, 3,4-Dichlorbenzylamino, 2-
Phenylethylamino, 1-Phenylethylamino, 1-Naphth-1-yl-amino 1-Naphth-2- ylamino; 1-Phenylprop-3-yl-amino, 3-Phenylpropylamino, 1-(4- lsobutylphenyl)ethylamino;
worin A für CrC2-Alkylen, R5 und R6 für H und
für Thienyl, Furyl, 2-, 3- oder 4-Pyridyl, Thiazolyl, Oxazolyl, Benzothiophenyl oder Benzofuranyl stehen, wobei die heterozyklischen Reste durch Halogen, insbesondere F oder Cl, oder d-C
6-Alkyl substituiert sein können.
f)
worin A, R5, R6 und
die unter e) genannten Bedeutungen besitzen.
g) NH-d-Ce-Alkyl, das durch 2 oder 3 Phenylgruppen substituiert ist, z. B. 3,3- Diphenylpropylamino, 1 ,3-Diphenylprop-2-ylamino;
h)
worin A für Cι-C2-Alkylen, R 5ü und i R D6 für H und
für Tetrahydrofuranyl, Tetrahydropyranyl, Pyrrolidin, N-Methyl- oder N- Ethylpyrrolidin stehen;
i)
worin R5, Rδ und
die unter h) genannten Bedeutungen besitzen;
j) -NH-COOR10, worin R10 für C3-C6-Cycloalkyl steht;
k) -NH-CO-NHR1ß, worin R10 für C3-C6-Cycloalkyl steht;
I) -NH-COR10, worin R10 für C3-C6-Cycloalkyl-CrC4-alkyl steht, z. B. Cyclopen- tylmethyl, Cyclohexylmethyl;
m) NH-CONH CO Phenyl;
n) -A-C3-C6-Cycloalkyl, worin A für d-C2-Alkylen steht, z. B. Cyclopentylmethyl- amino, Cyclohexylmethylamino.
Die Herstellung der erfindungsgemäßen Verbindungen kann in entsprechender Weise nach den im eingangs erwähnten Stand der Technik, insbesondere der WO 00/17192 beschriebenen Verfahren erfolgen. Als besonders zweckmäßig hat sich die Herstellung gemäß folgendem zweistufigen Verfahren erwiesen. Im ersten Schritt wird zunächst ein substituiertes lmidazol-2-thion der allgemeinen Formel II hergestellt. Dieses wird dann im zweiten Schritt so umgesetzt, dass unter Einführung des gewünschten Substituenten R
2 die 2-thio-substituierten Imidazolderivate der Formel I erhalten werden.
1) Herstellung des lmidazol-2-thions 5
Zur Herstellung von imidazol-2-thionen mit R3 = H, Halogen (Br, Cl, F), O-Alkyl oder S-Alkyl wird nach Verfahren A oder B vorgegangen. Das Verfahren A wird beispielhaft an Verbindungen erläutert, worin R1 für 4-Fluorphenyl- und R3für H, Verfahren B an Verbindungen, worin R1 für 4-Fluorphenyl- und R3 für Cl, (25a), F (25b) oder O-
L0 Alkyl (25c, 25d) stehen (die Zahlen in Klammern beziehen sich auf die Nummern der Beispiele). 2-Thio-substituierte-lmidazolderivate mit R3 = NR4R10 werden nicht aus entsprechenden lmidazol-2-thionen mit R3 = NR4R10 sondern nach dem Verfahren C in anderer Weise hergestellt. 2-Thio-substituierte-lmidazolderivate mit R3 = O-Alkyl oder S-Alkyl können sowohl nach Verfahren C oder als auch nach Verfahren B her-
15 gestellt werden.
Verfahren A
0 Die Synthese der substituierten lmidazol-2-thione mit R3 = H erfolgt ausgehend von Isonicotinsäureethylester und 4-Fluorphenylacetonitril nach dem Reaktionsablauf in Schema 1.
Die Ausgangsstoffe werden in einer Kondensationsreaktion mit Hilfe von metalli- 5 sehen Natrium in einem Alkohol, z. B. Ethanol, zu 2-Cyano-2-(4-fluorphenyl)-1-(4- pyridyl)-ethanon (lila) umgesetzt. Anschließend wird die Cyanogruppe durch Hydrolyse, z. B. mit Bromwasserstoffsäure, und Decarboxylierung entfernt, so dass 2-(4- Fluorphenyl)-1-(4-pyridyl)-ethanon (IVa) entsteht. Im nächsten Schritt wird die IVa durch Behandlung mit Ammoniumchlorid/Natriumacetat in einem alkoholischen Lö- 30 sungsmittel, wie Methanol, in das Oxim (Va) überführen. Dieses wird durch Umsetzung mit p-Toluolsulfochlorid in Pyridin in das Tosylat (Via) überführt. Die Thion-
Verbindung (lla) erhält man aus dem Tosylat durch Behandlung mit Natriumethylat und Umsetzung des gebildeten Aziren-Intermediats mit Kaliumthiocyanat.
Schema 1:
Svntheseweq der erfindunαsqemäßen Thione nach Verfahren A
Verfahren B:
Die Herstellung derjenigen erfindungsgemäßen Verbindungen, bei denen der Pyri- dinrest einen Halogen-, O-Alkyl-, oder S-Alkylsubstituenten aufweist, erfolgt gemäß Schema 2 über entsprechende 2-Halogenpyridyl-substituierte Imidazolthione (Verfahren B). Die Herstellung dieser Imidazolthione wird am Beispiel der 2-Fluor- substituierten Pyridinverbindungen (R3 = 2-F) mit R1 = p-Fluorphenyl erläutert. Imida- zolthiole, die in Position 4 Alkyl und Cycloalkyl-Reste tragen (R1 = C C4-Alkyl, C3-C7- Cycloalkyl), werden ausgehend von den entsprechend substituierten 2-Fluor-γ- Picolin-Ketonen in analoger Weise gewonnen.
Schema 2:
(Vlllb)
γ-Picolin (R3 = H), sowie die mit Halogen- (R3 = F, Cl, Br, I), Methoxy- (R3 = OCH3), und Methylthio-(24, R3 = SCH3) substituierten γ-Picoline werden unter Ausschluss von Feuchtigkeit in dafür geeigneten Lösemitteln, wie Kohlenwasserstoffen, Ethern und deren Gemische (z. B. Hexan, Tetrahydrofuran, Ethylenglykoldimethylether), mit Lithium-diisopropylamid (LDA) in der γ-Methylgruppe lithiiert und anschließend mit geeigneten Carbonsäurederivaten (R1-COOR, R1-CONR2, R1-CN) kondensiert. Als besonders geeignet zeigen sich hierbei die Amide des N,0-Dimethyl-hydroxylamins (R1-CONCH3(OCH3), 20). Die gebildeten γ-Picolylketone (IVb) werden mit Salpetrigsäureestern und Basen, z. B. Amylnitrit/Natriummethylat oder mit Alkalinitrit und Säure in der γ-Picolylposition nitrosiert. Als besonders vorteilhaft erweist sich die Umset- zung des in Eisessig gelösten γ-Picolylketons mit wässriger Natriumnitrit-Lösung. Die Nitrosoketone wandeln sich dabei vollständig in die tautomeren Oximketone (VI Ib) um.
Die Oximketone werden in alkoholischer Lösung in Gegenwart von Wasserstoff und mineralischen Säuren, z. B. HCI, durch Palladium auf Aktivkohle in die Ammoniumsalze der Aminketone (Vlllb) reduziert (23b).
Alternativ können andere Oximketone in alkoholischer Lösung in Gegenwart von mi- neralischen Säuren, z. B. H2Sθ4, mit Zinkstaub zu den entsprechenden Ammonium- ketonen reduziert werden (23f).
Aus diesen Ammoniumketon-Verbindungen erhält man durch Einwirkung von Alka- lithiocyanaten, z. B. Kaliumrhodanid in trockenem Dimethylformamid (DMF) beim Erhitzen unter Rückfluss die Imidazolthione der allgemeinen Formel Mb, mit R3 = F , Cl, Br, O-Alkyl, oder S-Alkyl als gelbgefärbte Feststoffe (24b).
Die Herstellung derjenigen erfindungsgemäßen Verbindungen, bei denen der Pyri- dinrest einen Ether- (R3 = OR 0), Thioether- (R3 = SR10) oder Aminosubstituent (R3 = NR4R10) aufweist, erfolgt gemäß Schema 4 bzw. Schema 5 über entsprechende 2- Halogenpyridyl-substituierte Imidazolthione (Verfahren C s.u.).
2) Herstellung der 2-Thio-imidazolverbindung
Die gemäß Verfahren A oder B erhaltenen Imidazolthion-Verbindungen der allgemei- nen Formel II werden durch Substitution des Schwefelatoms in der 2-Position zu den erfindungsgemäßen Verbindungen der Formel I umgesetzt. Die Substitutionen, wie beispielhaft für einige Verbindungen in Schema 3 gezeigt, erfolgen in bekannter Weise durch eine nukleophile Substitutionsreaktion. Die Verbindung lla bzw. Ilb wird dabei mit R2-X in einem inerten polaren Lösungsmittel, wie einem Alkohol, umgesetzt. X steht für eine leicht austauschbare Gruppe, wie Hai, insbesondere Cl, Br, I, Methyl- sulfonyl, Tosyl etc. Geeignete Verfahren sind dem Fachmann bekannt und beispielsweise beschrieben in WO 00/17192, EP 0 372 445 und US 4,440,776. Die Verbindungen R2-X sind bekannt oder können nach bekannten Verfahren, wie sie z. B. in der WO 00/17192 beschrieben sind, hergestellt werden.
Schema 3:
3. Substitution des Schwefels durch Alkyl- und Arylalkylhalogenide oder Alkohol- sulfonate.
3.1.
3.2.
Verfahren C:
Die erfindungsgemäßen Verbindungen, bei denen in R3 für einen Amino- Substituenten steht (R3 = NR4R10), werden ausgehend von den 2-Thioimidazolen mit 4(5)-(2-Halogenpyridin-4-yl)-Substitution hergestellt. Das Verfahren (Verfahren C) ist am Beispiel der 2-Benzylamino- (R3 = NH-CH2Ph) mit R1 = p-Fluorphenyl in Schema 4 erläutert (25f).
Die Ausgangsverbindungen (lb) können nach den oben beschriebenen Verfahren hergestellt werden.
Schema 4:
4.1. 4-(2-Amino-pyridin- yl)-substituierte 2-Thioimidazole
4.2. 4-(2-Aryloxy-pyridin-4yl)-substituierte 2-Thioimidazole
4.3. 4-(2-Arylthio-pyridin-4yl)-substituierte 2-Thioimidazole
4.4. 4-(2-Arylsulfonyl-pyridin^yl)-substituierte 2-Thioimidazole
Die Umsetzung erfolgt zweckmäßigerweise in dem jeweiligen Amin das man vorzugsweise in einer Menge von 5 bis 20 Moläquivalenten pro Moläquivalent der Verbindung (lb) verwendet. Die Reaktionstemperatur liegt im Allgemeinen im Bereich von 100 bis 200 °C. Gewünschtenfalls kann auch ein inertes Lösungsmittel, wie Dio- xan, Dimethylformamid, Diethylacetamid, Tetraethylharnstoff, Methylpyrrolidon, etc. und entsprechende Zusätze wie Alkalicarbonate oder einwertige Kupferhalogenide, zur Neutralisation freiwerdender Säureäquivalente oder zur Katalyse des Halogenaustrittes, verwendet werden.
Die erfindungsgemäßen Verbindungen, bei denen R3 für einen Alkoxy-Substituenten oder Alkylthio-Substituenten steht (R3 = 0-d-C6-Alkyl, S-d-Ce-Alkyl), können nicht nur nach Verfahren B (ausgehend von entsprechend substituierten Picolinen), sondern auch nach Verfahren C ausgehend von den 4(5)-(2-Halogenpyridin-4-yl)- substitutierten 2-Thioimidazolen hergestellt werden.
Verfahren D:
Die erfindungsgemäßen Verbindungen, bei denen R3 für einen Alkoxy-Substituenten steht (R3 = 0-CrC6-Alkyl), können nicht nur nach Verfahren B oder C, sondern auch nach Verfahren D ausgehend von den 4(5)-(2-Halogenpyridin-4-yl)-substitutierten 2-
Thioimidazolen hergestellt werden. Das Verfahren ist am Beispiel der 2-
Isopropyloxy-Pyridinverbindungen (R3 = OCH(CH3)2) mit R1 = p-Fluorphenyl in Schema 5 erläutert.
Die Ausgangsverbindungen (lb) können nach den oben beschriebenen Verfahren hergestellt werden.
Schema 5:
4-(2-Alkoxy-pyridin-4yl)-substituierte 2-Thioimidazole
Die Umsetzung erfolgt zweckmäßigerweise in dem Alkohol, den man vorzugsweise in einer Menge von 5 bis 20 Moläquivalenten pro Moläquivalent der Verbindung (lb) verwendet, bei niederen Alkoholen auch bis zum Hundertfachen, in Anwesenheit ei- ner starken Säure wie HCI oder Trifluoressigsäure, Methansulfonsäure etc.. Die Reaktionstemperatur liegt im Allgemeinen im Siedebereich der niederen Alkohole, bei höheren Alkoholen im Bereich von 100 bis 200 °C. Es hat sich als günstig erwiesen den Alkohol, z. B. mit gasförmigem HCI zu sättigen oder während der Umsetzung nachzusättigen.
Der Austausch von Fluor gegen Alkoxy in Position 2 des Pyridylsubstituenten kann alternativ auf einer früheren Synthesezwischenstufe ausgeführt werden zB auf der Stufe der Oximketone oder der Aminketone. Die Umsetzungen laufen in diesen Fällen unter Bedingungen vergleichbar zu denen der eben beschriebenen der Zwi- schenstufe lb (22c).
Verfahren E, F und G:
Die erfindungsgemäßen Verbindungen, bei denen R3 für einen Amido-Substituenten steht (R3 = NR7COR10), werden zum einen ausgehend von den 2-Thioimidazolen mit 4(5)-(2-Halogenpyridin-4-yl)-Substitution hergestellt. Das Verfahren (Verfahren E) ist am Beispiel der 2-Benzoylamido- (R3 = NH-COPh) mit R1 = p-Fluorphenyl in Schema 6.1 erläutert. Zum zweiten können weitere Amido-substituenten nach Hydrolyse der Amide zu den Amin-substituierten (R3 = NR7H, NHR10) 2-Thioimidazolen und deren erneute Acylierung oder Derivatisierung zu Amiden, Harnstoffen und Urethanen erhalten werden (Verfahren F). Dies ist in Schema 6.2 erläutert. Zum dritten können 2- Amino-pyridin-Vorläufer-Verbindungen aus 4(5)-(2-Halogenpyridin-4-yl)-Verbindun- gen über den Umweg der 4(5)-(2-Azidopyridin-4-yl)-Verbindungen erhalten werden (Verfahren G). Bei dieser Variante wird das Halogen nukleophil durch Alkali-Azid substituiert und anschließend die Azidgruppe durch Reduktionsmethoden zur Ami- nogruppe umgewandelt, siehe Schema 6.3.
Verfahren H:
Diese interesssante Variante eröffnet auch den Zugang zu alkylierten Aminen aus Aldehyd- und Keton-vorläufern. Wird die Umwandlung der Azid-Gruppe in die Amino- Gruppe unter Hydrierungsbedingungen mit einem Hydrierkatalysators in Gegenwart dieser Aldehyde und Ketone vorgenommen, werden alkylierte Amine mit R3 = NHCH2-B bzw. NHCH(AIkyl)-B erhalten (Verfahren H, Schema 6.4). Zum gleichen Ergebnis gelangt man, wenn das Azid mit einem Phosphin zum Phosphimid gespalten und diese nach einer Aza-Wittig-Reaktion mit einem Aldehyd (oder Keton) erhaltenen Imide mit komplexen Hydriden zu den Aminen reduziert werden (Verfahren H, Schema 6.5).
Die Ausgangsverbindungen (lb) können nach den oben beschriebenen Verfahren hergestellt werden.
Schema 6:
6.1: 4-(2-Amido-pyridin-4yl)-substituierte 2-Thioimidazole
Die Umsetzung erfolgt zweckmäßigerweise in dem jeweiligen Amid das man vorzugsweise in einer Menge von 5 bis 20 Mόläquivalenten pro Moläquivalent der Ver- bindung (lb) verwendet. Die Reaktionstemperatur liegt im Allgemeinen im Bereich von 100 bis 200 °C. Gewünschtenfalls kann auch ein inertes Lösungsmittel, wie Dio- xan, Dimethylformamid, Diethylacetamid, Tetraethylharnstoff, Methylpyrrolidon, etc. und entsprechende Zusätze wie Alkalicarbonate oder einwertige Kupferhalogenide, zur Neutralisation freiwerdender Säureäquivalente oder zur Katalyse des Halogen- austrittes, verwendet werden.
Die 2-Aminopyridin-Verbindungen können aus 2-Amidoacyl-Pyridinen durch Hydrolyse erhalten werden (6.2) oder aber durch Azid-Substitution der 2-Fluorverbindungen und nachfolgende Reduktion der 2-Azido-Pyridine (6.3) z. B. mittels Hydrierung auf Palladium-Aktivkohle in alkoholischen Lösemitteln.
6.2
(lb) (Id) (le)
6.3
(lb) (li) (Ik)
6.4
(Id) (Id)
6.5
Weitere Umwandlungen der erhaltenen Amine (Ik, Id) durch Derivatisierung sind möglich (Verfahren F). Umsetzungen der Amine Id und Ik sowohl mit Säureanhydriden und Säurechloriden zu weiteren Amiden, als auch Umsetzungen mit Chloamei- sensäureestern zu Urethanen, mit Isocyanaten zu Harnstoffe und mit Acyl- isocyanaten zu Acylharnstoffen kommen zur Anwendung. Diese Derivatbildungen sind die in Schema 7 erläutert.
Schema 7
Umwandlungen der Amine (Ik, Id) zu Amiden, Urethanen, und Harnstoffen
Die erfindungsgemäßen Verbindungen zeigen in vitro und in vivo immunmodulierende und die Cytokinfreisetzung hemmende Wirkung. Cytokine sind Proteine wie TNF-α und IL-ß, die eine wichtige Rolle bei zahlreichen inflammatorischen Erkrankungen spielen. Die erfindungsgemäßen Verbindungen eignen sich durch ihre die Cytokinfreisetzung hemmende Wirkung zur Behandlung von Erkrankungen, die im Zusammenhang mit einer Störung des Immunsystems stehen. Sie eignen sich beispielsweise zur Behandlung von Autoimmunerkrankungen, Krebs, rheumatoider Ar- thritis, Gicht, septischem Schock, Osteoporosis, neuropathischem Schmerz, HIV- Ausbreitung, HIV-Demenz, viraler Myokarditis, insulinabhängiger Diabetis, Periodon- talerkrankungen, Restenosis, Alopezie, T-Zell-Depletion bei HIV-Infektionen oder AIDS, Psoriasis, akuter Pankreatitis, Abstoßungsreaktionen bei allogenen Transplantaten, allergisch bedingter Lungenentzündung, Artheriosklerose, Multipler Skle- rose, Kachexie, Alzheimer Erkrankung, Schlaganfall, Iktus, Colitis uicerosa, Morbus Crohn, Inflammatory-Bowel-Disease (IBD), Ischämie, kongestive Herzinsuffizienz,
Lungen-Fibrose, Hepatitis, Glioblastom, Guillain-Barre-Syndrom, systematischer Lupus erythematodes, Adult-respiratory-distress-Syndrom (ARDS) und Atemnot- syndrom.
Die erfindungsgemäßen Verbindungen können entweder als einzelne therapeutische Wirkstoffe oder als Mischungen mit anderen therapeutischen Wirkstoffen verabreicht werden. Die Verbindungen können alleine verabreicht werden, im Allgemeinen werden sie jedoch in Form pharmazeutischer Mittel dosiert und verabreicht, d. h. als Mischungen der Wirkstoffe mit geeigneten pharmazeutischen Trägern oder Verdün- nungsmittel. Die Verbindungen oder Mittel können oral oder parenteral verabreicht werden, vorzugsweise werden sie in oralen Dosierungsformen gegeben.
Die Art des pharmazeutischen Mittels oder Trägers bzw. des Verdünnungsmittels hängt von der gewünschten Verabreichungsform ab. Orale Mittel können beispiels- weise als Tabletten oder Kapseln vorliegen und können übliche Exzipienzien enthalten, wie Bindemittel (z. B. Sirup, Akazia, Gelatine, Sorbit, Tragant oder Polyvinylpyr- rolidon), Füllstoffe (z. B. Lactose, Zucker, Maisstärke, Calciumphosphat, Sorbit oder Glycin), Gleitmittel (z. B. Magnesiumstearat, Talcum, Polyethylenglycol oder Silici- umdioxid), desintegrierende Mittel (z. B. Stärke) oder Netzmittel (z. B. Natriumlauryl- sulfat). Flüssige Oralpräparate können in Form wässriger oder öliger Suspensionen, Lösungen, Emulsionen, Sirupe, Elixiere oder Sprays und dergleichen sein. Sie können auch als Trockenpulver vorliegen, das zur Rekonstitution mit Wasser oder einem anderen geeigneten Träger aufbereitet wird. Derartige flüssige Präparate können übliche Additive, beispielsweise Suspendiermittel, Geschmacksstoffe, Verdünnungsmit- tel oder Emulgatoren enthalten. Für die parenterale Verabreichung kann man Lösungen oder Suspensionen mit üblichen pharmazeutischen Trägern einsetzen.
Die erfindungsgemäßen Verbindungen oder Mittel können an Säuger (Mensch oder Tier) in einer Dosis von etwa 0,5 mg bis 100 mg pro kg Körpergewicht pro Tag verab- reicht werden. Sie können in einer Einzeldosis oder in mehreren Dosen gegeben werden. Das Wirkungsspektrum der Verbindungen als Inhibitoren der Cytokinfreisetzung wurde anhand nachstehender Testsysteme untersucht (C. Donat und S. Laufer in Arch. Pharm. Pharm. Med. Chem. 333, Suppl. 1 , 1 - 40, 2000).
//7-wϊ o-Testverfahren mit humanem Vollblut
Proben aus humanem Kalium-EDTA-Vollblut (ä 400 μl) werden mit der Testsubstanz versetzt und 15 min bei 37 °C in einem C02-lnkubator (5 % C02; 95 % feuchtigkeits- gesättigte Luft) vorinkubiert. Danach werden die Proben 4 Stunden mit 1 μg/ml LPS (E.coli 026:B6) bei 37 °C in einem C02-lnkubator (5 % C02; 95 % feuchtigkeitsgesättigte Luft) stimuliert. Die Reaktion wird gestoppt, indem die Proben auf Eis gestellt, mit DPBS-Puffer versetzt und anschließend 15 min bei 1000*g zentrifugiert werden. Anschließend wird die Menge IL-1ß und TNFα im Plasmaüberstand mittels ELISA ermittelt.
/n- Yro-Testverfahren mit PBMCs
1 ) Aus 1 :3 verdünntem humanen Kalium-EDTA-Vollblut werden mittels Dichte- gradientenzentrifugation (Histopaque®-1,077) die mononukleären Zellen
(PBMCs) isoliert. Diese werden 2-mal mit DPBS-Puffer gewaschen, in Makrophagen-SFM-Medium resuspendiert und auf eine Zellzahl von 1*106 Zellen/ml eingestellt.
Die erhaltene PBMCs-Suspension (ä 390 μl Proben) wird mit der Testsubstanz 15 min bei 37 °C in einem C02-lnkubator (5 % C02; 95 % feuchtigkeitsgesättigte Luft) vorinkubiert. Danach werden die Proben 4 Stunden mit jeweils 1 μl/ml LPS (E.coli 026:B6) bei 37 °C in einem C02-lnkubator (5 % C02: 95 % feuchtigkeitsgesättigte Luft) stimuliert. Die Reaktion wird gestoppt, indem die Proben auf Eis gestellt, mit DPBS-Puffer versetzt und anschließend 12 min bei 15880*g zentrifugiert werden. Anschließend wird die Menge IL-1 ß und TNFα im Plasmaüberstand mittels ELISA ermittelt.
2) Kinase-Assay
Mikro-Titerplatten wurden mit 50 μl ATF2-Lösung (20 μg/ml) eine Stunde bei 37 °C beschichtet. Nach dreimaligem Waschen mit Wasser wurden 50 μl Ki- nase-Mischung (50 mM tris-HCI 10mM MgCfe, 10 mM ß-Glyzerolphosphat, 10 μg/ml BSA, 1 mM DTT, 100 μM ATP, 100 μM Na3V04, 10 ng aktiviertes
p38α) mit oder ohne Inhibitor in die Vertiefungen gegeben und 1 Stunde bei 37 °C inkubiert. Nach dreimaligem Waschen wurden die Platten mit Phosphor- ATF-2-Antikörper eine Stunde bei 37 °C inkubiert. Nach erneutem dreimaligem Waschen wurde mit alkalischer Phosphatase markiertes Ziege-anti- Kaninchen-IgG eine Stunde bei 37 °C zugegeben (um den Antikörper phos- phoryliertes Protein-Substrat-Komplex festzuhalten). Nach dreimaligem Waschen wurde die alkalische Phosphatase-Substratlösung (3mM 4-NPP, 50 mM NaHC03, 50 mM MgCI2> 100 μl/Vertiefung) 1,5 Stunden bei 37 °C zugegeben. Die Bildung von 4-Nitrophenolat wurde bei 405 nm unter Verwendung eines Mikrotiterplatten-Lesers gemessen. Die ICso-Werte wurden berechnet.
ebnisse der in-vitro-Tests sind in der nachstehenden Tabelle 1 gezeigt.
Tabelle 1 : Testergebnisse
Die nachfolgenden Beispiele erläutern die Erfindung, ohne sie zu begrenzen.
Beispiel 1
a) 4-(4-Fluorphenyl)-5-pyridin-4-yl-1,3-dihydro-imidazol-2-thion
2-(4-Fluorphenyl)-3-hydroxy-3-pyridin-4-yl-acrylonitril (a1)
Zu einer Lösung von metallischem Natrium (17,3 g; 0,7 mol) in absolutem Ethanol (250 mL) wurde ein Gemisch von Isonicotinsäureethylester (75,8 g; 0,5 mol) und 4- Fluorphenylacetonitril (67,6 g; 0,5 mol) zugetropft. Der Reaktionsansatz wurde 15 min bei 100 °C gerührt. Anschließend wurde im Eisbad abgekühlt und mit 600 mL destilliertem H20 versetzt. Beim Ansäuern mit konzentriertem HCI (90 mL) fiel das Hydrochlorid von a1 bei pH 1 als gelber Niederschlag aus. Der Niederschlag wurde abfiltriert, mit H20 gewaschen und im Vakuum über P205 getrocknet. Fp. 226 °C
2-(4-Fluorphenyl)-1-pyridin-4-yl-ethanon (a2)
Eine Lösung von a1 (40,6 g; 0,15 mol) in 48%iger Bromwasserstoffsäure (130 mL) wurde 19 h unter Rückfluss gerührt. Der Ansatz wurde im Eisbad abgekühlt, der sich abscheidende Niederschlag (4-Fluorphenylessigsäure) abfiltriert und mit H20 gewa- sehen. Beim Neutralisieren des Filtrats mit Ammoniakwasser (80 mL) fiel a2 als dunkelgrüner Niederschlag aus, welcher abfiltriert, mit H20 gewaschen und im Vakuum über P2O5 getrocknet wurde: hellgrau-beiges Pulver. Fp. 215 °C
2-(4-Fluorphenyl)-1-pyridin-4-yl-ethanon-oxim (a3)
In eine Suspension von a2 (21 ,5 g; 0,1 mol) in 50%igem Methanol (350 mL) wurden Natriumacetat (36,1 g; 0,44 mol) und Hydroxylaminhydrochlorid (22,0 g; 0,32 mol) eingetragen. Das Reaktionsgemisch wurde 1 h unter Rückfluss gerührt. Beim Abküh- len der klaren Lösung im Eisbad fiel a3 als beigefarbener Niederschlag aus, welcher abfiltriert, mit H20 gewaschen und im Vakuum über P2O5 getrocknet wurde. Fp. 155 °C
2-(4-Fluorphenyl)-1 -pyridin-4-yl-ethanon, 0-[(4-Methylphenyl)su(fonyl]oxim (a4)
Unter Argonatmosphäre wurde a3 (10,1 g; 0,04 mol) in absolutem Pyridin (50 mL) gelöst. Die Lösung wurde auf 6 °C abgekühlt und portionsweise mit Toluolsulfonsäu- rechlorid (10,1 g; 0,05 mol) versetzt. Nach beendeter Zugabe wurde das Reaktionsgemisch 20 h bei Raumtemperatur gerührt. Anschließend wurde der Ansatz auf 500 mL Eiswasser gegossen. Der Niederschlag (a4) wurde abfiltriert, mit H20 kalt gewaschen und im Trockenschrank bei 50 °C getrocknet. Fp. 201 °C
4-(4-Fluorphenyl)-5-pyridin-4-yl-1 ,3-dihydro-imidazol-2-thion (1 a).
Unter Argonatmosphäre wurde eine Lösung von a4 (10,0 g; 0,03 mol) in absolutem Ethanol (56 mL) auf 5 °C abgekühlt und tropfenweise mit einer frisch hergestellten Lösung von metallischem Natrium (0,75 g; 0,03 mol) in absolutem Ethanol (30 mL) versetzt. Das Reaktionsgemisch wurde 5 h bei 5 °C gerührt. Nach Zugabe von Diethylether (500 mL) wurde weitere 30 min. gerührt. Der Niederschlag (TosOH) wurde abfiltriert und mit Diethylether (4 * 50 mL) gewaschen. Die vereinigte etheri- sche Phase wurde mit 10%iger Salzsäure (3 * 90 mL) extrahiert. Der wässrige Extrakt wurde auf ein Volumen von ca. 40 mL eingeengt und mit Kaliumthiocyanat (5,0 g; 0,05 mol) versetzt. Das Reaktionsgemisch wurde 1 h unter Rückfluss gerührt. Beim Neutralisieren mit 5%iger Natriumhydrogencarbonat-Lösung (270 mL) fiel a5 als beiger Niederschlag aus, welcher abfiltriert, mit H20 gewaschen und im Trockenschrank bei 60 °C getrocknet wurde. Ausbeute 5,6 g (79 %); Fp. 382 °C
1H-NMR (DMSO-of6): δ (ppm) 7,1 (m, 2H, 4-F-Ph), 7,3 (m, 2H, 4-Pyr), 7,5 (m, 2H, 4-F-Ph), 8,5 (m, 2H, 4-Pyr), 12,7 (d, 2H, austauschbar, NH)
Auf entsprechende Weise wurden erhalten:
1b 3-(4-Fluorphenyl)-5-pyridin-4-yl-1 ,3-dihydroimidazol-2-thion 1c 4-(4-Chlorphenyl)-5-pyridin-4-yl-1 ,3-dihydroimidazol-2-thion 1d 4-(4-Bromphenyl)-5-pyridin-4-yl-1 ,3-dihydroimidazol-2-thion
1e 4-Phenyl-5-pyridin-4-yl-1 ,3-dihydroimidazol-2-thion
Beispiel 2
1 -Chlormethyl-4-methylsulfanyl-benzol (2)
4-Methylsulfanylbenzylalkohol (30,5 g; 0,2 mol) wurde in Dichlormethan (180 mL) gelöst. Der unter Rückfluss gehaltenen Vorlage wurde eine Lösung von Thionylchlorid (23,8 g; 0,2 mol) in Dichlormethan (120 mL) zugetropft. Das Reaktionsgemisch wurde weitere 2 h unter Rückfluss gerührt. Die Lösung wurde auf Raumtemperatur abgekühlt, mit H20 (2 x 250 mL) gewaschen, über Na2S04 getrocknet und eingeengt. Der ölige Rückstand (6) wurde säulenchromatographisch gereinigt (Al203, CH2CI2).
1H-NMR (CDCI3): δ (ppm) 2,46 (s, 3H, CH3), 4,5 (s, 2H, CH2), 7,2-7,3 (q, 4H, 4-MeS- Ph)
Beispiel 3
1 -Chlormethyl-4-methansulfinyl-benzol (3)
Eine Lösung von 2 (17,3 g; 0,1 mol) in Eisessig (150 mL) wurde auf 10 °C abgekühlt. Der Vorlage wurde eine Lösung von H202 (35%ige Lösung; 13,1 g; 0,13 mol) in Eisessig (50 mL) zugetropft. Das Reaktionsgemisch wurde 2 h bei Raumtemperatur gerührt. Der Ansatz wurde im Eisbad abgekühlt, mit Eis (200 g) versetzt und mit Ammoniakwasser (290 mL) neutralisiert. Die wässrige Phase wurde mit Ethylacetat (2 * 300 mL) extrahiert. Die organische Phase wurde mit H20 (2 * 300 mL) gewaschen, über Na2S0 getrocknet und eingeengt. Aus dem öligen Rückstand wurde 3 durch Anreiben und Abkühlen in kristalliner Form gewonnen.
1H-NMR (CDCI3): δ (ppm) 2,73 (s, 3H, CH3), 4,6 (s, 2H, CH2), 7,5 (d, 2H, 4-MeS(0)- Ph), 7,6 (d, 2H, 4-MeS(0)-Ph)
Beispiel 4
1 -Chlormethyl-4-methansulfonyl-benzol (4)
In eine Lösung von 3 (3,0 g; 0,02 mol) in Chloroform (50 mL) wurde m-Chlorper- benzoesäure (70 %; 8,6 g; 0,04 mol) eingetragen. Das Reaktionsgemisch wurde 4 h unter Rückfluss gerührt. Der Ansatz wurde auf Raumtemperatur abgekühlt und filtriert. Das Filtrat wurde mit gesättigter NaHC03-Lösung gewaschen (2 *) und über Na2S04 getrocknet. Nach dem Einengen der organischen Phase blieb 8 als kristalli- ner, weißer Feststoff zurück. Fp. 102 °C
1H-NMR (CDCl3): d (ppm) 3,07 (s, 3H, CH3), 4,6 (s, 2H, CH2), 7,6 (d, 2H, 4-MeS02- Ph), 7,9 (d, 2H, 4-MeS02-Ph)
Beispiel 5
5-Chlorsulfonyl-2-hydroxy-benzoesäuremethylester (5a)
5a wurde ausgehend von Salicylsäuremethylester (10,0 g; 65,7 mmol) nach der in der Synthese von 5c beschriebenen Methode dargestellt.
1H-NMR (CDCI3): δ (ppm) 4,05 (s, 3H, CH3), 7,18 (d, 1H, 8,9 Hz, C3-H), 8,09 (dd, 1H, 2,5/9,0 Hz, C4-H), 8,57 (d, 1H, 2,5 Hz, C6-H), 11,55 (s, 1H, austauschbar, Phenol- OH)
5-Chlor-3-chlorsulfonyl-2-hydroxy-benzoesäuremethylester (5b)
5b wurde ausgehend von 5-Chlorsalicylsäuremethylester (16,0 g; 85,7 mmol) nach der in der Synthese von 5c beschriebenen Methode dargestellt.
1H-NMR (CDCI3): δ(ppm) 4,06 (s, 3H, CH3), 8,11 (d, 1 H, 2,7 Hz, C6-H), 8,19 (d,1 H, 2,7 Hz, C4-H), 12,09 (s, 1H, austauschbar, Phenol-OH)
3-Chlorsulfonyl-4-methoxy-benzoesäureethylester (5c)
Eine Lösung von 4-Methoxy-benzoesäureethylester (15,7 g; 87,2 mmol) in CCI4 (60 mL) wurde auf -15 °C abgekühlt und innerhalb von 15 min tropfenweise mit Chlorsulfonsäure (17,5 mL; 263 mmol) versetzt, wobei die Temperatur auf -10 °C anstieg. Nach beendeter Zugabe wurde das Reaktionsgemisch 2 h bei Raumtemperatur gerührt und anschließend auf 50 °C erwärmt, bis das Edukt dünnschichtchro- matographisch nicht mehr nachweisbar war. Das Reaktionsgemisch wurde unter Eiskühlung und kräftigem Rühren auf eine Suspension von Eis (50 g) in CCU (100 mL) gegeben. Es wurde 3 min kräftig gerührt. Die organische Phase wurde abgetrennt und die wässrige Phase mit CH2CI2 (3 * 100 mL) extrahiert. Die vereinigten organischen Extrakte wurden mit gesättigter NaCI-Lösung gewaschen (3 x), über Na2S04 getrocknet und eingeengt. Beim Verreiben des öligen, braunen Rückstands mit Diethylether fiel 5c als kristalliner, weißer Feststoff an.
1H-NMR (CDCI3): δ (ppm) 1,41 (t, 3H, 7,1 Hz, CH3), 4,14 (s, 3H, CH3), 4,42 (q, 2H, 7,1 Hz, CH2), 7,18 (d, 1 H, 8,8 Hz, C5-H), 8,37 (dd, 1 H,2,1/8,8 Hz, C6-H), 8,63 (d, 1 H, 2,1 Hz, C2-H)
Beispiel e
2-Hydroxy-5-mercapto-benzoesäure (6a)
6a wurde ausgehend von 6a (0,50 g; 2,0 mmol) nach der in der Synthese von 7c be- schriebenen Methode unter Verzicht auf die Alkylierung mit Dimethylsulfat erhalten.
1H-NMR (DMSO-dβ): δ (ppm) 5,39 (bs, 1H, austauschbar, Carboxyl-OH), 6,90 (d, 1H, 8,7 Hz, C3-H), 7,45 (dd, 1H, 2,5/8,6 Hz, C -H), 7,75 (d,1H, 2,5 Hz, C6-H), Phenol-OH nicht sichtbar
Beispiel 7
2-Hydroxy-5-methylsulfanyl-benzoesäure (7a)
7a wurde ausgehend von 5a (10,0 g; 40,0 mmol) nach der in der Synthese von 7c beschriebenen Methode dargestellt.
1H-NMR (CDCI3): δ (ppm) 2,48 (s, 3H, CH3), 6,97 (d, 1H, 8,7 Hz, C3-H), 7,51 (dd, 1H, 2,5/8,7 Hz, C4-H), 6,97 (d, 1H, 8,7 Hz, C3-H), 7,87(d, 1H, 2,4 Hz, C6-H), 10,26 (bs, 1 H, Phenol-OH), COzH nicht sichtbar
5-Chloro-2-hydroxy-3-methylsulfanyl-benzoesäure (7b)
7b wurde ausgehend von 5b (13,0 g; 45,6 mmol) nach der in der Synthese von 7c beschriebenen Methode dargestellt.
1H-NMR (DMSO-ds): δ (ppm) 2,47 (s, 3H, CH3), 7,33 (d, 1H, 2,4 Hz, C6-H), 7,52 (d, 1H, 2,4 Hz, C4-H), Phenol-OH und C02H nicht sichtbar
4-Methoxy-3-methylsulfanyl-benzoesäure (7c)
In eine Lösung von 5c (5,1 g; 18,3 mmol) in Toluol (50 mL) wurde portionsweise Tri- phenylphosphan (20,5 g; 78,2 mmol) eingetragen. Das Reaktionsgemisch wurde 4,5 h bei Raumtemperatur gerührt. Vom Niederschlag (Triphenylphosphinoxid) wurde abfiltriert und das gelbe Filtrat mit 10%iger Natronlauge extrahiert (4 x). Zum vereinigten wässrigen Extrakt wurde Dimethylsulfat (2 mL) zugegeben und das Reaktionsgemisch 2 h bei Raumtemperatur gerührt. Der sich abscheidende Niederschlag wurde durch Erwärmen auf Rückflusstemperatur aufgelöst. Die klare Lösung wurde abgekühlt und mit 20%iger Salzsäure auf pH 1 eingestellt. Der Niederschlag (7c) wurde abfiltriert, mit H2O gewaschen und im Vakuum über CaCI2 getrocknet.
1H-NMR (CD3OD): δ (ppm) 2,43 (s, 3H, S-CH3), 3,93 (s, 3H, O-CH3), 6,98 (d, 1 H, 8,4 Hz, C5-H), 7,79-7,86 (m, 2H, C2-/C6-H)
4-Hydroxy-3-methylsulfanyl-benzoesäure (7d)
Eine Suspension von 7c (0,5 g; 2,5 mmol) in Eisessig/Bromwasserstoffsäure 48 % (1+1, 7 mL) wurde 6 h unter Rückfluss gerührt. Das Reaktionsgemisch wurde abge- kühlt, auf H20 (20 mL) gegeben und mit 10%iger Na2C03-Lösung auf pH 2 eingestellt. Die wässrige Lösung wurde mit Diethylether extrahiert (4 * 20 mL). Der vereinigte organische Extrakt wurde mit gesättigter NaCI-Lösung gewaschen (2 x), über Na2S04 getrocknet und eingeengt. Beim Stehenlassen bei Raumtemperatur kristallisierte der schmutzig-braune, ölige Rückstand (7d) aus. Das Kristallisat wurde mit H2O ausgerührt, abfiltriert und getrocknet.
1H-NMR (CDCI3): δ (ppm) 2,38 (s, 3H, CH3), 7,05 (d, 1 H, 8,5 Hz, C5-H), 8,02 (dd, 1 H, 2,2/8,5 Hz, C6-H), 8,29 (d, 1H, 2,2 Hz, C2-H), Phenol-OH und C02H nicht sichtbar
Beispiel 8
2-Hydroxymethyl-4-methylsulfanyl-phenol (8a)
8a wurde ausgehend von 7a (1 ,5 g; 8,1 mmol) nach der in der Synthese von 8c be- schriebenen Methode dargestellt.
1H-NMR (CDCI3): δ ppm) 2,42 (s, 3H, CH3), 4,79 (s, 2H, CH2), 6,81 (d, 1 H, 8,4 Hz, C6-H), 7,01 (d, 1 H, 2,1 Hz, C3-H), 7,17 (dd, 1 H, 2,3/8,4 Hz, C3-H), OH nicht sichtbar
4-Chloro-2-hydroxymethyl-6-methylsulfanyl-phenol (8b)
8b wurde ausgehend von 7b (2,2 g; 10,1 mmol) nach der in der Synthese von 8c beschriebenen Methode dargestellt.
1H-NMR (DMSO-cfe): δ (ppm) 2,38 (s, 3H, CH3), 4,52 (s, 2H, CH2), 5,3-5,5 (bs, 1H, austauschbar, Hydroxyl-OH), 7,03 (d, 1H, 2,6 Hz, C5-H), 7,11 (d, 2,4 Hz, C3-H), 9,02 (bs, 1H, austauschbar, Phenol-OH)
4-Hydroxymethyl-2-methylsulfanyl-phenol (8c)
Unter Eiskühlung wurde eine Lösung von 7d (1 ,37 g; 7,4 mmol) in abs. Tetrahydrofu- ran (THF; 15 mL) zu einer Suspension von 95%igem LiAIH (0,55 g; 14 mmol) in ab- solutem THF (10 mL) in einem ausgeheizten und mit Argon gespülten Dreihalskolben (DHK) so zugetropft, dass nur mäßige Gasentwicklung erfolgte. Nach beendeter Zugabe wurde die Kühlung entfernt und das Reaktionsgemisch 30 min bei Raumtemperatur und weitere 21 h bei 55 - 65 °C gerührt. Das Reaktionsgemisch wurde unter Eiskühlung mit Eiswasser versetzt. Der Niederschlag von AI(OH)3 wurde durch Zu- satz von 10%iger Schwefelsäure aufgelöst und die wässrig-saure Lösung (pH 1) mit Diethylether (3 50 mL) extrahiert. Der vereinigte etherische Extrakt wurde mit 10%iger Natronlauge extrahiert (2 25 mL). Die vereinigte natronalkalische Lösung wurde mit 20%iger Salzsäure neutralisiert. Der Niederschlag (8c) wurde abfiltriert, mit H2O gewaschen und getrocknet. Eine weitere Menge an 8c wurde durch Extrak- tion der neutralen, wässrigen Lösung mit Diethylether gewonnen. Der etherische Extrakt wurde mit gesättigter NaCI-Lösung gewaschen, über Na2S04 getrocknet und eingeengt: kristalliner, weißer Rückstand.
1H-NMR (CDCI3): δ (ppm) 2,34 (s, 3H, CH3), 4,60 (s, 2H, CH2), 6,97 (d, 1 H, 8,3 Hz, C6-H), 7,24 (dd, 1 H, 2,0/8,4 Hz, C5-H), 7,50 (d, 1 H, 2,0 Hz, C3-H), OH nicht sichtbar
Beispiel 9
2-Hydroxy-5-methylsulfanyl-benzaldehyd (9a)
Die Titelverbindung fiel als Nebenprodukt in der Synthese von 8a an.
1H-NMR (CDCI3): δ (ppm) 2,48 (s, 3H, CH3), 6,96 (d, 1H, 9,8 Hz, C3-H), 7,48-7,54 (m, 2H, C4-/C6-H), 9,87 (s, 1H, austauschbar, OH), 10,91 (s, 1H, Aldehyd-H)
Beispiel 10
4-(3-Chlorethyl)-benzolsulfonylchlorid (10a)
Unter Eiskühlung wurde innerhalb von 40 min (2-Chlorethyl)benzol (14,0 g; 0,1 mol) zu Chlorsulfonsäure (72 g) zugetropft. Die braune Lösung wurde 24 h bei Raumtemperatur gerührt, im Eisbad abgekühlt und portionsweise auf Eis gegeben, wobei sich eine zähe, nicht filtrierbare Masse abschied. Die wässrige Lösung wurde mit Ethyl- acetat extrahiert (3 x). Der vereinigte organische Extrakt wurde mit 10%iger NaH- Cθ3-Lösung gewaschen, über Na2Sθ4 getrocknet und eingeengt Der ölige Rückstand wurde in tert-Butylmethylether/Petrolether aufgenommen. Die Lösung wurde mit einem Glasstab angerieben und abgekühlt. Das weiße Kristallisat wurde abfiltriert und getrocknet. Weiteres Reaktionsprodukt wurde aus der Mutterlauge gewonnen. Das Rohprodukt wurde ohne weitere Reinigung in der Synthese von 11a eingesetzt.
1H-NMR (CDCI3): δ (ppm) 3,20 (t, 2H, 6,8 Hz, CH2), 3,79 (t, 2H, 6,8 Hz, CH2), 7,46- 7,53 (m, 2H, Phenyl), 7,97-8,04 (m, 2H, Phenyl)
4-(3-Chlorpropyl)-benzolsulfonylchlorid (1 Ob)
10b wurde ausgehend von (3-Chlorpropyl)benzol (15,5 g; 0,1 mol) nach der in der Synthese von 10a beschriebenen Methode dargestellt. Das Rohprodukt wurde ohne weitere Reinigung in der Synthese von 11b eingesetzt.
MS: m/z (%) 253 (90, M+), 217 (100, M+-Cl), 189 (35), 153 (97, M+-S02CI), 125 (94), 119 (65, Phenylpropylcarbenium+), 91(90), 77 (29, Phenyl+)
Beispiel 11
1 -(3-Chlorethyl)-4-methylsulfanyl-benzol (11 a)
11a wurde ausgehend von 10a (12,0 g; 0,05 mol) nach der in der Synthese von 11b beschriebenen Methode dargestellt.
1H-NMR (CDCI3): δ (ppm) 2,47 (s, 3H, CH3), 3,02 (t, 2H, 7,4 Hz, CH2), 3,68 (t, 2H, 7,5 Hz, CH2), 7,11-7,25 (m, 4H, Phenyl)
1 -(3-Chlorpropyl)-4-methylsulfanyl-benzol (11 b)
Bei Raumtemperatur wurde zu einer Suspension von LiAIH4 (2,9 g; 7,6 mmol) in Diethylether (50 mL) eine Lösung von 10b (12,7 g; 5,0 mmol) in Diethylether (75 mL) innerhalb von 2,5 h zugetropft. Nach beendeter Zugabe wurde das Reaktionsgemisch bei Raumtemperatur und gelegentlicher Zugabe von LiAIH4 gerührt, bis das Edukt dünnschichtchromatographisch nicht mehr nachweisbar war (2,5 h). In den Reaktionsansatz wurde unter Eiskühlung Eis eingetragen und die wässrige Phase mit 10%iger Salzsäure angesäuert (pH 1). Die organische Phase wurde abgetrennt und die wässrige Phase mit Diethylether extrahiert (3 x). Der vereinigte organische Extrakt wurde mit 10%iger Natronlauge (4 x 50 mL) bis zur weitgehenden Farblosig- keit gewaschen. Der vereinigte natronalkalische Auszug wurde mit Dimethylsulfat (9,0 g; 7,0 mmol) versetzt und 16,5 h bei Raumtemperatur gerührt. Die ölige Abscheidung wurde in Diethylether aufgenommen. Die organische Phase wurde abgetrennt und die wässrige Phase erneut mit Diethylether extrahiert (2 x). Der vereinigte organische Extrakt wurde über Na2S04 getrocknet und eingeengt. Der braune, ölige Rückstand wurde am Kugelrohr destilliert (0,2 mbar, 250 °C).
1H-NMR (CDCI3): δ (ppm) 2,01-2,11 (m, 2H, CH2), 2,46 (s, 3H, CH3), 2,73 (t, 2H, 7,1 Hz, CH2), 3,51 (t, 2H, 6,5 Hz, CH2), 7,09-7,25 (m, 4H, Phenyl)
Beispiel 12
1 -(2-Chlorethyl)-4-methansulfinyl-benzol (12a)
Zu einer Lösung von 11a (1 ,5 g; 8,0 mmol) in Eisessig (20 mL) wurde unter Kühlung eine 35%ige Lösung von H2O2 (0,9 g; 9,3 mmol) zugegeben. Das Reaktionsgemisch wurde nach beendeter Zugabe 2,5 h bei Raumtemperatur gerührt, unter Kühlung mit Eiswasser verdünnt und mit 25%igem Ammoniakwasser auf pH 8 eingestellt. Die ölige, weiße Abscheidung wurde mit Diethylether aufgenommen und die wässrige Pha-
se mit Diethylether extrahiert (3 x). Der vereinigte organische Extrakt wurde über Na2Sθ getrocknet und eingeengt.
1H-NMR (CDCI3): δ (ppm) 2,73 (s, 3H, CH3), 3,14 (t, 2H, 7,1 Hz, CH2), 3,76 (t, 2H, 7,1 Hz, CH2), 7,38-7,42 (m, 2H, Phenyl), 7,60-7,64 (m, 2H, Phenyl)
1 -(3-Chlorpropyl)-4-methansulfinyl-benzol (12b)
12b wurde ausgehend von 11b (2,0 g; 10,0 mmol) nach der in der Synthese von 12a beschriebenen Methode dargestellt. Fp: 46 °C
Allgemeine Methoden zur Herstellung der Verbindungen der Formel I:
Herstellung der 2-Arylalkyl- oder Alkylsulfanylimidazole (Allgemeine Methode A)
Eine Suspension des jeweiligen lmidazol-2-thions (1 Äquivalent), der jeweiligen Base (1 ,2 Äquivalente) und des jeweiligen Arylalkyl- oder Alkylhalogenids (1 Äquivalent) in Ethanol/THF (8+2) wurde solange unter Rückfluss gerührt, bis das lmidazol-2-thion dünnschichtchromatographisch nicht mehr nachweisbar war. Das Reaktionsgemisch wurde auf Raumtemperatur abgekühlt und filtriert. Das meist rot-orange gefärbte Filtrat wurde eingeengt und der Rückstand durch Säulenchromatographie, Umkristallisa- tion oder Ausrühren gereinigt. Auf diese Weise wurden die Verbindungen 13a - c, 14a - c sowie 17a - m hergestellt.
Herstellung der 2-Benzylsulfanylimidazole mit phenolischer Funktionalität im Rest R2 (Allgemeine Methode B)
Durch Zusatz von 10%iger Salzsäure (10 - 15 Tropfen) wurde lmidazol-2-thion 1a (1 Äquivalent) in Eisessig (5 mL) aufgelöst. Der jeweilige Benzylalkohol (1 Äquivalent) wurde zur hellgelb gefärbten Vorlage zugegeben und das Reaktionsgemisch bei geeigneter Temperatur solange gerührt (Temperatur/Zeit), bis 1a dünnschichtchromatographisch nicht mehr nachweisbar war. Im Falle der Sulfoxide 18g-i wurde eine 35%ige H202-Lösung zugegeben und das Reaktionsgemisch weitere 4 h bei Raum-
temperatur gerührt. Das Reaktionsgemisch wurde mit H20 (5 mL) verdünnt und mit 25%igem Ammoniakwasser auf pH 8 eingestellt. Der Niederschlag wurde abfiltriert und mit Wasser gewaschen. Das Rohprodukt wurde durch Säulenchromatographie, Umkristallisation oder Ausrühren gereinigt. Auf diese Weise wurden die lmidazol-2- ylsulfanylmethyl-phenole 18a-i hergestellt.
Herstellung N-substituierter 2-Aminopyridine (Allgemeine Methode C)
Unter Argon wurde das jeweilige 5-(2-Halogenopyridin-4-yl)-imidazol (1 Äquivalent) in dem jeweiligen Amin (ca. 10 Äquivalente) suspendiert. Das Reaktionsgemisch wurde bei der jeweiligen Temperatur solange gerührt, bis das Edukt dünnschichtchromatographisch nicht mehr nachweisbar war. Das Reaktionsgemisch wurde auf Raumtemperatur abgekühlt und in 10 % Zitronensäure aufgenommen, die zuvor mit 20 % NaOH auf pH 5 eingestellt worden war. Die wässrige Emulsion wurde mit Ethylacetat extrahiert (3 x). Der vereinigte organische Extrakt wurde mit 10 % Zitronensäure/pH 5 (1 x), 10 % Na2C03-Lösung (2 x) und gesättigter NaCI-Lösung (1 x) gewaschen, über Na2S04 getrocknet und eingeengt. Der ölige Rückstand wurde säu- lenchromatographisch getrennt. Auf diese Weise wurden die Aminopyridine 25f-p, 26c-e und 27c-d hergestellt.
Beispiel 13
3-[5-(4-Fluorphenyl)-2-(4-methylsulfanyl-benzylsulfanyl)-3H-imidazol-4-yl]- pyridin (13a)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1b (0,42 g; 1 ,5 mmol) und 2 (0,25 g; 1 ,4 mmol) nach 4,5-stündiger Reaktionszeit und säulenchromatographischer Trennung (Si02 60, CH2CI2/Et0H 9+1) erhalten. Fp. 163 °C
|R (ATR) (attenuated total reflection) 1506, 1493, 1222 (C-F), 837, 806 cm -1
1H-NMR (DMSO-d6 ): δ (ppm) 2,45 (s, 3H, CH3), 4,38 (s, 2H, CH2), 7,19-7,49 (m, 10H, 3-Pyr, 4-F-Ph und 4-MeS-Ph), 7,78-7,82 (m, 1H, 3-Pyr), 8,45-8,47 (m, 1H, 3- Pyr), 8,61 (s, 1H, 3-Pyr), 12,71 (bs, 1H, austauschbar, NH)
3-[5-(4-Fluorphenyl)-2-(4-methansulfinyl-benzylsulfanyl)-3H-imidazol-4-yl]- pyridin (13b).
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1b (0,42 g; 1 ,5 mmol) und 3 (0,27 g; 1 ,5 mmol) nach 8-stündiger Reaktionszeit und säu- lenchromatographischer Trennung (Si02 60, CH2CI2/EtOH 9+1) erhalten. Fp. 127 °C
IR (ATR): 1506, 1222 (C-F), 1027 (S=0), 1013, 838, 811 cm"1
1H-NMR (DMSO-c β ): δ (ppm) 3,19 (s, 3H, CH3), 4,46 (s, 2H, CH2), 7,16-7,46 (m, 5H, 3-Pyr und 4-F-Ph), 7,56-7,66 (m, 4H, 4-MeS(0)-Ph), 7,72-7,81 (m, 1 H, 3-Pyr), 8,41- 8.62 (m, 2H, 3-Pyr), 12,77 (bs, 1H, austauschbar, NH)
3-[5-(4-Fluorphenyl)-2-(4-methansulfonyl-benzylsulfanyl)-3H-imidazol-4-yl]- pyridin (13c).
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1b (0,42 g; 1 ,5 mmol) und 4 (0,29 g; 1,43 mmol) und unter Zusatz von Na2C03 (0,43 g; 4,1 mmol) nach 6,5-stündiger Reaktionszeit und Ausrühren mit heißem Ethylacetat erhalten. Fp. 129 °C
IR (ATR): 1506, 1296 (S02), 1222 (C-F), 1145 (S02), 1089, 839, 812 cm-1
1H-NMR (DMSO-de): δ (ppm) 3,19 (s, 3H, CH3), 4,50 (s, 2H, CH2), 7,17-7.45 (m, 5H, 3-Pyr und 4-F-Ph), 7,64-7,90 (m, 5H, 3-Pyr und 4-MeSO2-Ph), 8,43-8,61 (m, 2H, 3- Pyr), 12,78 (bs, 1 H, austauschbar, NH)
Beispiel 14
4-[5-(4-Chlorphenyl)-2-(4-methylsulfanyl-benzylsulfanyl)-3H-imidazol-4-yl]- pyridin (14a)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1c (0,26 g; 0,9 mmol) und 6 (0,15 g; 0,87 mmol) und unter Zusatz von Na2C03 (zwei Spatelspitzen) nach 6,5-stündiger Reaktionszeit und säulenchromatographischer Trennung (SiQ2 60, CH2CI2/EtOH 9+1) erhalten. Fp. 236 °C
IR (ATR): 1600, 1492, 1094, 1005, 968, 829, 684 (C-Cl), 561 cm'1
1H-NMR (DMSO-dβ): δ (ppm) 2,44 (s, 3H, CH3), 4,38 (s, 2H, CH2), 7,18-7,56 (m, 10H, 4-Pyr, 4-CI-Ph und 4-MeS-Ph), 8,45-8,55 (m, 2H, 4-Pyr), 12,86 (bs, 1H, austausch- bar, NH)
4-[5-(4-Chlorphenyl)-2-(4-methansulfinyl-benzylsulfanyl)-3H-imidazol-4-yl]- pyridin (14b)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1c (0,26 g; 0,9 mmol) und 3 (0,16 g; 0,85 mmol) und unter Zusatz von 6,5-stündiger Reaktionszeit und säulenchromatographischer Trennung (Si02 60, CH2CI2/EtOH 9+1 ) erhalten. Fp. 224 °C
IR (ATR): 1600, 1510, 1490, 1033 (S=0), 1001 , 967, 829, 677 cm"1 (C-Cl)
1H-NMR (DMSO-d6): δ (ppm) 2,70 (s, 3H, CH3), 4,47 (s, 2H, CH2), 7,31-7,65 (m, 10H, 4-Pyr, 4-CI-Ph und 4-MeS(0)-Ph), 8,44-8,54 (m, 2H, 4-Pyr), 12,87 (bs, 1H, austauschbar, NH)
4-[5-(4-Chlorphenyl)-2-(4-methansulfonyl-benzylsulfanyl)-3H-imidazol-4-yl]- pyridin (14c)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1c (0,26 g; 0,9 mmol) und 4 (0,18 g; 0,9 mmol) und unter Zusatz von Na2C03 (zwei Spatelspitzen) nach 6,5-stündiger Reaktionszeit und säulenchromatographischer Trennung (Si02 60, CH2CI2/EtOH 9+1 ) erhalten. Fp. 232 °C
IR (ATR): 1603, 1490, 1300 (S02), 1141 (S02), 1086, 1002, 952, 829, 681 (C-Cl), 550 cm"1
1H-NMR (DMSO-d6): δ (ppm) 3,19 (s, 3H, CH3), 4,52 (s, 2H, CH2), 7,32-7,58 (m, 6H, 4-Pyr und 4-CI-Ph), 7,67 (d, 2H, 8,2 Hz, 4-MeS02-Ph), 7,88 (d, 2H, 8,3 Hz, 4-MeS02- Ph), 8,45-8,55 (m, 2H, 4-Pyr), 12,89 (bs, 1H, austauschbar, NH)
Beispiel 15
4-[5-(4-Bromphenyl)-2-(4-methylsulfanyl-benzylsulfanyl)-3H-imidazol-4-yl]- pyridin (15a)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1d (0,25 g; 0,75 mmol) und 2 (0,13 g; 0,72 mmol) nach 5-stündiger Reaktionszeit und säulenchromatographischer Trennung (Si02 60, CH2CI2/EtOH 9+1) erhalten.
IR (ATR): 1600, 1517, 1490, 1089, 1069, 1003, 968, 826 cm"1
1H-NMR (DMSO-d6): δ (ppm) 2,43 (s, 3H, CH3), 4,36 (s, 2H, CH2), 7,16-7,87 (m, 10H, 4-Pyr, 4-Br-Ph und 4-MeS-Ph), 8,45-8,55 (m, 2H, 4-Pyr), 12,90 (bs, 1H, austauschbar, NH)
4-[5-(4-Bromphenyl)-2-(4-methansuIfinyl-benzylsulfanyl)-3H-imidazol-4-yl]- pyridin (15b)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1d (0,25 g; 0,75 mmol) und 3 (0,14 g; 0,72 mmol) nach 10-stündiger Reaktionszeit und säulenchromatographischer Trennung (Si02 60, CH2CI2/EtOH 9+1) erhalten. Fp. 222 °C
IR (ATR): 1604, 1487, 1035 (S=0), 1010, 1000, 966, 822 cm"1
1H-NMR (DMSO-d6): δ (ppm) 2,71 (s, 3H, CH3), 4,48 (s, 2H, CH2), 7,40-7,62 (m, 20H, 4-Pyr, 4-Br-Ph und 4-MeS(0)-Ph), 8,49-8,57 (m, 2H, 4-Pyr), 12,90 (bs, 1H, austauschbar, NH)
4-[5-(4-Bromphenyl)-2-(4-methansulfonyl-benzylsulfanyl)-3H-imidazol-4-yl]- pyridin (15c)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1d (0,25 g; 0,75 mmol) und 4 (0,15 g; 0,72 mmol) nach 5-stündiger Reaktionszeit und säulenchromatographischer Trennung (Si02 60, CH2CI2/EtOH 9+1 ) erhalten. Fp. 226 °C
IR (ATR): 1605, 1318, 1303 (S02), 1145 (S02), 1003, 967, 957, 827, 822 cm"1
1H-NMR (DMSO-d6): δ (ppm) 3,18 (s, 3H, CH3), 4,50 (s, 2H, CH2), 7,33-7,89 (m, 10H, 4-Pyr, 4-Br-Ph und 4-MeS02-Ph), 8,45-8,54 (m, 2H, 4-Pyr), 12,91 (bs, 1 H, austauschbar, NH)
Beispiel 16
4-[2-(4-Methylsulfanyl-benzylsulfanyl)-5-phenyl-3H-imidazol-4-yl]-pyridin (16a)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1e (0,38 g; 1 ,5 mmol) und 2 (0,25 g; 1 ,4 mmol) nach 5,75-stündiger Reaktionszeit und säulenchromatographischer Trennung (Si02 60, CH2CI2/Et0H 9+1) erhalten. Fp. 213 °C
IR (ATR): 1601, 1491, 1417, 1094, 1004, 967, 828, 771, 700 cm"1
1H-NMR (DMSO-d6): δ (ppm) 2,44 (s, 3H, CH3), 4,38 (s, 2H, CH2), 7,18-7,58 (m, 11 H, 4-Pyr, Ph und 4-MeS-Ph), 8,44-8,47 (m, 2H, 4-Pyr), 12,82 (bs, 1 H, austauschbar, NH)
4-[2-(4-Methansulfinyl-benzylsulfanyl)-5-phenyl-3H-imidazol-4-yl]-pyridin (16b)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1e (0,38 g; 1 ,5 mmol) und 3 (0,27 g; 1 ,43 mmol) nach 5,5-stündiger Reaktionszeit und säulenchromatographischer Trennung (Si0260, CH2CI2/EtOH 9+1 ) erhalten. Fp. 189 °C
IR (ATR): 1603, 1494, 1051 (S=0), 1003, 833, 701 cm"1
1H-NMR (DMSO-de): δ (ppm) 2,71 (s, 3H, CH3), 4,48 (s, 2H, CH2), 7,32-7,52 (m, 7H, 4-Pyr und Ph), 7,57-7,67 (m, 4H, 4-MeS(0)-Ph), 8.45-8,54 (m, 2H, 4-Pyr), 12,84 (bs, 1 H, austauschbar, NH)
4-[2-(4-Methansulfonyl-benzylsulfanyl)-5-phenyl-3H-imidazol-4-yl]-pyridin (16c)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1e (0,38 g; 1 ,5 mmol) und 4 (0,29 g; 1 ,43 mmol) nach 4,25-stündiger Reaktionszeit und säulenchromatographischer Trennung (Si02 60, CH2CI2/EtOH 9+1 ) erhalten. Fp. 247 °C
IR (ATR): 1602, 1298 (S02), 1145 (S02), 1006, 953, 827, 775, 701 cm"1
1H-NMR (DMSO-d6): δ (ppm) 3,21 (s, 3H, CH ), 4,54 (s, 2H, CH2), 7,31-7,58 (m, 7H, 4-Pyr und Ph), 7,70 (d, 2H, 8,3 Hz, 4-MeS02-Ph), 7,91 (d, 2H, 8,3 Hz, 4-MeS02-Ph), 8,45-8,59 (m, 2H, 4-Pyr), 12,87 (bs, 1 H, austauschbar, NH)
Beispiel 17
4-{5-(4-Fluorphenyl)-2-[2-(4-methansulfinyl-phenyl)-ethylsulfanyl]-1H-imidazol- 4-yl}-pyridin (17a)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1a (0,25 g; 0,9 mmol) und 12a (0,22 g; 1,1 mmol) und unter Zusatz von Na2C03 (1 Spatelspitze) sowie einer katalytischen Menge Nal nach 50-stündiger Reaktionszeit und säulenchromatographischer Reinigung (Si02 60, CH2CI2/EtOH 9+1 ) erhalten. Fp. 177 °C
IR (ATR): 1221 (C-F), 1032 cm"1 (S=0)
1H-NMR (DMSO-de): δ (ppm) 2,71 (s, 3H, CH3), 3,06-3,13 (m, 2H, CH2), 3,42-3,49 (m, 2H, CH2), 7,25-7,65 (m, 10H, 4-Pyr, 4-F-Ph und 4-MeS(0)-Ph), 8,40-8,58 (m, 2H, 4-Pyr), 12,80 (bs, 1 H, austauschbar, NH)
4-{5-(4-FIuorphenyl)-2-[2-(4-methansulfinyl-phenyl)-propylsulfanyl]-1H- imidazol-4-yl}-pyridin (17b)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1a (0,25 g; 0,9 mmol) und 12b (0,22 g; 1,0 mmol) und unter Zusatz von Na2C03 (1 Spatelspitze) sowie einer katalytischen Menge Nal nach 40-stündiger Reaktionszeit und säulenchromatographischer Reinigung (Si02 60, CH2CI2/EtOH 9+1 ) erhalten. Fp. 142 °C
IR (ATR): 1222 (C-F), 1043 cm"1 (S=0)
1H-NMR (DMSO-d6): δ (ppm) 1,95-2,09 (m, 2H, CH2), 2,71 (s, 3H, CH3), 2,82 (t, 2H, 7,4 Hz, CH2), 3,15 (t, 2H, 7,0 Hz, CH2), 7,25-7,62 (m, 10H, 4-Pyr, 4-F-Ph und 4- MeS(O)-Ph), 8,46-8,49 (m, 2H, 4-Pyr), 12,86 (bs, 1H, austauschbar, NH)
4-[2-Benzylsulfanyl-5-(4-f luorphenyl)-1 H-imidazol-4-yl]-pyridin (17c)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1a (0,28 g; 1 ,0 mmol) und 1 -Chlormethylbenzol (0,13 g; 1 ,0 mmol) nach 6-stündiger Reaktionszeit und Ausrühren mit MeOH erhalten. Fp. 223 °C
IR (ATR): 1233 cm -"11 (C-F)
1H-NMR (DMSO-d6): δ (ppm) 4,41 (s, 2H, CH2), 7,23-7,51 (m, 11H, 4-Pyr, 4-F-Ph und Bz), 8,44-8,47 (m, 2H, 4-Pyr), 12,82 (bs, 1H, austauschbar, NH)
4-[5-(4-Fluorphenyl)-2-phenethylsulfanyl-1 H-imidazol-4-yl3«pyridm (17d)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1a (0,5 g; 1 ,9 mmol) und 2-Chlorethylbenzol (0,28 g; 2,0 mmol) und unter Zusatz von Na2C03 (1 Spatelspitze) sowie einer katalytischen Menge Nal nach 70-stündiger Reaktionszeit und Ausrühren mit EtOH erhalten. Fp. 257 °C
IR (ATR): 1223 cm"1 (C-F)
1H-NMR (DMSO-de): δ (ppm) 2,99 (t, 2H, 7,4 Hz, CH2), 3,40 (t, 2H, 7,5 Hz, CH2), 7,17-7,53 (m, 11H, 4-Pyr, 4-F-Ph und Bz), 8.44-8,46 (m, 2H, 4-Pyr), NH nicht sichtbar
4-[5-(4-Fluorphenyl)-2-(3-phenyl-propylsulfanyl)-1 H-imidazol-4-yl]-pyridin (17e)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1a (0,5 g; 1 ,9 mmol) und 3-Chlorpropylbenzol (0,31 g; 2,0 mmol) und unter Zusatz von Na2C03 (1 Spatelspitze) sowie einer katalytischen Menge Nal nach 70-stündiger Reaktionszeit und Ausrühren mit EtOH erhalten. Fp. 183 °C
IR (ATR): 1226 cm"1 (C-F)
1H-NMR (DMSO-d6): δ (ppm) 1,90-2,04 (m, 2H, CH2), 2,72 (t, 2H, 7,4 Hz, CH2). 3,12 (t, 2H, 7,0 Hz, CH2), 7,18-7,51 (m, 11 H, 4-Pyr, 4-F-Ph und Bz), 8,37-8,44 (m, 2H, 4- Pyr), 12,82 (bs, 1H, austauschbar, NH)
[5-(4-Fluorphenyl)-4-pyridin-4-yl-1 H-imidazol-2-ylsulfanyl]-acetonitril (17f)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1a (1,1 g; 4,0 mmol) und Chloracetonitril (0,30; 4,0 mmol) nach 18-stündiger Reaktionszeit und säulenchromatographischer Reinigung (Si02 60, Ethylacetat) erhalten. Fp. 219 °C
IR (ATR): 2243 (CN), 1226 cm"1 (C-F)
1H-NMR (DMSO-d6): δ (ppm) 4,32 (s, 2H, CH2), 7,34-7,57 (m, 6H, 4-Pyr und 4-F-Ph), 8,50-8,52 (m, 2H, 4-Pyr), 13,20 (bs, 1H, austauschbar, NH)
4-[5-(4-Fluorphenyl)-2-(naphthalen-1-ylmethylsulfanyl)-1H-imidazol-4-yl]-pyridin (17g)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1a (0,28 g; 1 ,0 mmol) und 1-Chlormethylnaphthol (0,18 g;, 1 ,0 mmol) nach 6,5-stündiger Reaktionszeit und säulenchromatographischer Reinigung (Si02 60, Ethylacetat) er- halten. Fp. 364 °C
IR (ATR): 1225 cm"1 (C-F)
1H-NMR (DMSO-d6): δ (ppm) 4,90 (s, 2H, CH2), 7,25-7,62 (m, 10H, 4-Pyr, 4-F-Ph und Naphthyl), 7,80-7,98 (m, 2H, Naphthyl), 8,20-8,23 (m, 1 H, Naphthyl), 8,48-8,52 (m, 2H, 4-Pyr), 12, (bs, 1 H, austauschbar, NH)
4-[2-Cyclohexylmethylsulfanyl-5-(4-fluorphenyl)-1 H-imidazol-4-yl]-pyπ'din (17h)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1a (0,25 g; 0,9 mmol) und 1 -Chlormethylcyclohexan (0,18 g; 1,0 mmol) und unter Zusatz von Na2C03 (1 Spatelspitze) sowie einer katalytischen Menge Nal nach 47-stündiger Reaktionszeit und Ausrühren mit EtOH erhalten. Fp. 235 °C
IR (ATR): 2922, 2852 (c-Hex), 1222 cm"1 (C-F)
1H-NMR (DMSO-de): δ (ppm) 0,95-1,23 (m, 5H, cyc/o-Hex), 1,51-1,85 (m, 6H, cyc/o- Hex), 3,06 (d, 2H, 6,7 Hz, CH2), 7,22-7,51 (m, 6H, 4-Pyr und 4-F-Ph), 8,43-8,45 (m, 2H, 4-Pyr), 12,76 (bs, 1H, austauschbar, NH)
4-[5-(4-Fluorphenyl)-2-methylsulfanyl-1 H-imidazol-4-yl]-pyridin (17i)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1a (0,41 g; 1 ,5 mmol) und Methyliodid (0,27 g; 1 ,9 mmol) nach 8-stündiger Reaktionszeit und Ausrühren mit EtOH erhalten. Fp. 263 °C
IR (ATR): 1226 cm"1 (C-F)
H-NMR (DMSO-d6): δ (ppm) 2,61 (s, 3H, CH3), 7,22-7,51 (m, 6H, 4-Pyr und 4-F-Ph), 8,42-8,45 (m, 2H, 4-Pyr), NH nicht sichtbar
4-[5-(4-Fluorphenyl)-2-(2-methylsulfanyl-benzylsulfanyl)-1 H-imidazol-4-yl]- pyridin (17j)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1a (0,28 g; 1,0 mmol) und 1-Chlormethy!-2-methylsu!fanylbenzol (0,17 g; 1 ,0 mmol) nach 5,5-stündiger Reaktionszeit und säulenchromatographischer Reinigung (Si02 60, Ethylacetat) erhalten. Fp. 223 °C
IR (ATR): 1228 cm"1 (C-F)
1H-NMR (CD3OD): δ (ppm) 2,51 (s, 3H, CH3), 4,44 (s, 2H, CH2), 7,13-7,48 (m, 10H, 4-Pyr, 4-F-Ph und 2-MeS-Ph), 8,43-8,46 (m, 2H, 4-Pyr)
4-[5-(4-Fluoφhenyl)-2-(2-methansulfinyl-benzylsulfanyl)-1H-imidazol-4-yl]- pyridin (17k)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1a (0,28 g; 1 ,0 mmol) und 1-Chlormethyl-2-methansulfinylbenzol (0,18 g; 1 ,0 mmol) nach 4-stündiger Reaktionszeit und Umkristallisation aus Methanol/Ethylacetat (1+1) erhalten. Fp. 205 °C
IR (KBr): 1213 (C-F), 1033 cm"1 (S=0)
1H-NMR (CD3OD): δ (ppm) 2,87 (s, 3H, CH3), 4,50 (d, 1H, 13,6 Hz, CH2), 4,62 (d, 1H, 13,6 Hz, CH2), 7,24-7,33 (m, 2H, 4-F-Ph), 7,47-7,62 (m, 5H, 4-F-Ph, C4-/C5-/C6-H 2- MeS(O)-Ph), 7,95 (d, 1 H, 7,2 Hz, C3-H 2-MeS(0)-Ph), 7,99-8,03 (m, 2H, 4-Pyr), 8,55- 8,58 (m, 2H, 4-Pyr)
4-[5-(4-Fluorphenyl)-2-(3-methylsulfanyl-benzylsulfanyl)-1H-imidazol-4-yl]- pyridin (171)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 1a (1 ,1 g; 4,1 mmol) und 1-Chlormethyl-3-methylsulfanylbenzol (0,7 g; 4,1 mmol) nach 11 -stündiger Reaktionszeit und Umkristallisation aus EtOH erhalten. Fp. 218 °C
IR (KBr): 1225 cm"1 (C-F)
1H-NMR (DMSO-d6): δ (ppm) 2,40 (s, 3H, CH3), 4,46 (s, 2H, CH2), 7,16-7,43 (m, 6H, 4-F-Ph und 3-MeS-Ph), 7,56-7,63 (m, 2H, 4-F-Ph), 7,90-7,93 (m, 2H, 4-Pyr), 8,66- 8,69 (m, 2H, 4-Pyr), NH nicht sichtbar
4-[5-(4-Fluoιphenyl)-2-(3-methansulfinyl-benzylsulfanyl)-1H-imidazol-4-yl]- pyridin (17m)
Zu einer Suspension von 171 (0,50 g; 1 ,2 mmol) in Eisessig (7 mL) wurde eine 35%ige Lösung von H O2 (0,13 mL; 1 ,3 mmol) zugetropft. Das Reaktionsgemisch wurde 20,5 h bei Raumtemperatur gerührt, mit H20 (5 mL) verdünnt, mit 25%igem Ammoniakwasser auf pH 9 eingestellt und mit Ethylacetat extrahiert (3 x). Der vereinigte organische Extrakt wurde mit gesättigter NaCI-Lösung gewaschen (3 x) und über Na2S04 getrocknet. Das nach Entfernen des Lösemittels erhaltene ölige Roh- produkt wurde mit Diethylether/Ethylacetat (1+1) verrieben und der halbfeste Rückstand säulenchromatographisch gereinigt (RP-18, MeOH). Fp. 171 °C
IR (KBr): 1228 (C-F), 1019 cm"1 (S=0)
1H-NMR (CD3OD): δ (ppm) 2,67 (s, 3H, CH3), 4,37 (s, 2H, CH2), 7,13-7,21 (m, 2H, 4- F-Ph), 7,37-7,58 (m, 8H, 4-Pyr, 4-F-Ph und 3-MeS(0)-Ph), 8,40-8,43 (m, 2H, 4-Pyr)
Beispiel 18
2-[5-(4-Fluorphenyl)-4-pyridin-4-yl-1 H-imidazol-2-ylsulfanylmethyl]-phenol (18a)
Nach der Allgemeinen Methode B (23 h, Raumtemperatur) wurde die Titelverbindung ausgehend von 1a (0,20 g; 0,7 mmol) und 2-Hydroxymethylphenol (0,10 g; 0,8 mmol) nach Ausrühren mit EtOH erhalten. Fp. 200 °C (Zersetzung)
IR (ATR): 1266 (OH bending), 1222 (C-F), 1005 (C-O)
1H-NMR (DMSO-d6): δ (ppm) 4,37 (s, 2H, CH2), 6,70-6,85 (m, 2H, 2-HO-Ph), 7,05- 7,14 (m, 1 H, 2-HO-Ph), 7,23-7,53 (m, 7H, 4-Pyr, 4-F-Ph und 2-HO-Ph), 8,46-8,49 (m, 2H, 4-Pyr), 9,95 (bs, 1H, austauschbar, OH), 12.81 (bs, 1H, austauschbar, NH)
3-[5-(4-Fluorphenyl)-4-pyridin-4-yl-1 H-imidazol-2-ylsulfanylmethyl]-phenol (18b)
Nach der Allgemeinen Methode B (9 h, Rückfluss) wurde die Titelverbindung ausgehend von 1a (0,20 g; 0,7 mmol) und 3-Hydroxymethylphenol (0,10 g; 0,8 mmol) nach säulenchromatographischer Reinigung (Si02 60, CH2CI /EtOH 9+1) erhalten. Fp. 230 °C
IR (ATR): 1287 (OH bending), 1241 (C-F), 1007 cm"1 (C-O)
1H-NMR (DMSO-de): δ (ppm) 4,34 (s, 2H, CH2), 6,65 (dd, 1H, 1 ,4/8,0 Hz, 3-HO-Ph C4-H), 6,79-6,82 (m, 2H, 3-HO-Ph C2-/C6-H), 7,07-7,15 (m, 1 H, 3-HO-Ph C5-H), 7,27- 7,53 (m, 6H, 4-Pyr und 4-F-Ph), 9,45 (s, 1H, austauschbar, OH), 12,83 (bs, 1H, austauschbar, NH)
4-[5-(4-Fluorphenyl)-4-pyridin-4-yl-1 H-imidazol-2-ylsulfanylmethyl]-phenol (18c)
Nach der Allgemeinen Methode B (14 h, Raumtemperatur) wurde die Titelverbindung ausgehend von 1a (0,20 g; 0,7 mmol) und 4-Hydroxymethylphenol (0,10 g; 0,8 mmol) nach säulenchromatographischer Reinigung (Si02 60, CH2CI2/EtOH 9+1) erhalten. Fp. 250 °C (Zersetzung)
IR (ATR): 1271 (OH bending), 1232 (C-F), 1004 cm"1 (C-O)
1H-NMR (DMSO-de): δ (ppm) 4,32 (s, 2H, CH2), 6,69 (d, 2H, 7,5 Hz, 4-HO-Ph), 7,19 (d, 2H, 7,9 Hz, 4-HO-Ph), 7,27-7,51 (m, 6H, 4-Pyr und 4-F-Ph), 8,43-8,53 (m, 2H, 4- Pyr), 9,41 (s, 1H, austauschbar, OH), 12,79 (bs, 1H, austauschbar, NH)
2-[5-(4-Fluorphenyl)-4-pyridin-4-yl-1H-imidazol-2-ylsulfanylmethyl]-4-methyl- sulfanyl-phenol (18d)
Nach der Allgemeinen Methode B (1 h, Raumtemperatur) wurde die Titelverbindung ausgehend von 1a (0,50 g; 2,9 mmol) und 8a (0,50 g; 2.9 mmol) nach Ausrühren mit MeOH erhalten. Fp. 243°C
IR (KBr): 1275 (OH bending), 1230 (C-F), 1005 cm"1 (C-O)
1H-NMR (DMF-d7): δ (ppm) 2,36 (s, 3H, CH3), 4,46 (s, 2H, CH2), 6,90 (d, 1H, 8,4 Hz, 2-HO-Ph C3-H), 7,13 (dd, 1H, 2,3/8.3 Hz, 2-HO-Ph C4-H),7,27-7,35 (m, 3H, 4-F-Ph und 2-HO-Ph C6-H), 7,51-7,53 (m, 2H, 4-Pyr), 7,58-7,65 (m, 2H, 4-F-Ph), 8,52-8,55 (m, 2H, 4-Pyr), 10,30-10,70 (bs, 1H, austauschbar, NH), OH nicht sichtbar
4-Chlor-2-[5-(4-fluQrphenyl)-4-pyridin-4-yl-1H-imidazol-2-ylsulfanylmethyl]-6- methylsulfanylphenol (18e)
Nach der Allgemeinen Methode B (1.5 h, 75 °C) wurde die Titelverbindung ausge- hend von 1a (0,80 g; 3,0 mmol) und 8b (0,60 g; 3,0 mmol) nach Ausrühren mit Me- OH erhalten. Fp. 220 °C (Zersetzung)
IR (KBr): 1259 (OH bending), 1225 (C-F), 1007 cm"1 (C-O)
1H-NMR (DMSO-de): δ (ppm) 2,34 (s, 3H, CH3), 4,38 (s, 2H, CH2), 6,97 (d, 1H, 2,3 Hz, 3-CI-Ph C2-H), 7,17 (d, 1 H, 2,3 Hz, 3-CI-Ph C4-H), 7,23-7,51 (m, 6H, 4-Pyr und 4- F-Ph), 8,48-8,50 (m, 2H, 4-Pyr), 12,74 (bs, 1H, austauschbar, NH), OH nicht sichtbar
4-[5-(4-Fluorphenyl)-4-pyridin-4-yl-1H-imidazol-2-ylsulfanylmethyl]-2-methyl- sulfanyl-phenol (18f)
Nach der Allgemeinen Methode B (2 h, Raumtemperatur) wurde die Titelverbindung ausgehend von 1a (0,20 g; 0,7 mmol) und 8c (0.14 g, 0.8 mmol) nach Ausrühren mit MeOH erhalten. Fp. 230 °C (Zersetzung)
IR (KBr): 1227 (C-F), 1019 cm"1 (C-O)
1H-NMR (CD3OD): δ (ppm) 2,21 (s, 3H, CH3), 4,17 (s, 2H, CH2), 6,69 (d, 1H, 8,0 Hz, 4-HO-Ph C3-H), 6,90-7,01 (m, 2H, 4-HO-Ph C2-/C6-H), 7,12-7,21 (m, 2H, 4-F-Ph), 7,32-7,53 (m, 4H, 4-Pyr und 4-F-Ph), 8,39-8,43 (m, 2H, 4-Pyr)
2-[5-(4-Fluorphenyl)-4-pyridin-4-yl-1H-imidazol-2-ylsulfanylmethyl]-4-methan- sulfinyiphenol (18g)
Nach der Allgemeinen Methode B (1 h, Raumtemperatur) wurde die Titelverbindung ausgehend von 1a (0,27 g; 1,0 mmol) und 8a (0,17 g; 1 ,0 mmol) unter Zusatz von 35%iger H202-Lösung nach Umkristallisation aus Toluol/THF (1+1) erhalten. Fp. 216 °C
IR (KBr): 1278 (OH bending), 1232 (C-F), 1031 (S=0), 1003 cm"1 (C-O)
1H-NMR (CD3OD): δ (ppm) 2,60 (s, 3H, CH3), 4,33 (s, 2H, CH2), 6,96 (d, 1H, 8,2 Hz, 2-HO-Ph C3-H), 7,11-7,21 (m, 2H, 4-F-Ph), 7,41-7,47 (m, 6H, 4-Pyr, 4-F-Ph und 2- HO-Ph C4-/C6-H), 8,39-8,42 (m, 2H, 4-Pyr)
4-Chlor-2-[5-(4-fluorphenyl)-4-pyridin-4-yl-1H-imidazol-2-ylsulfanylmethyl]-6- methansulfinyl-phenol (18h)
Nach der Allgemeinen Methode B (1 ,5 h, 75 °C) wurde die Titelverbindung ausgehend von 1a (0,27 g; 1 ,0 mmol) und 8b (0,21 g, 1 ,0 mmol) unter Zusatz von 35%iger H2θ2-Lösung nach säulenchromatographischer Reinigung (Si02 60, Aceton) erhalten. Fp. 175 °C (Zersetzung)
IR (KBr): 1265 (OH bending), 1236 (C-F), 1051 (S=0), 1005 cm"1 (C-O)
1H-NMR (CD3OD): δ (ppm) 2,72 (s, 3H, CH3), 4,39 (s, 2H, CH2), 7,14-7,23 (m,
2H, 4-F-Ph), 7,39 (d, 1H, 2,6 Hz, 3-CI-Ph C2-H), 7,42-7,49 (m, 6H, 4-Pyr, 4-F-Ph und 3-CI-Ph C4-H), 8,43-8,46 (m, 2H, 4-Pyr)
Beispiel 19
4-[5-(4-Fluorphenyl)-4-pyridin-4-yl-1H-imidazol-2-ylsulfanylmethyl]-2-methan- sulfinyl-phenol (19)
Nach der Allgemeinen Methode B (2,5 h, Raumtemperatur) wurde die Titelverbindung ausgehend von 1a (0.20 g; 0,7 mmol) und 8c (0,14 g; 0,8 mmol) unter Zusatz von 35%iger H202-Lösung nach Ausrühren mit Aceton erhalten. Fp. 185 °C (Zersetzung)
IR (KBr): 1296 (OH bending), 1230 (C-F), 1062 (S=0), 1013 cm"1 (C-O)
1H-NMR (CD3OD): δ (ppm) 2,70 (s, 3H, CH3), 4,28 (s, 2H, CH2), 6,78 (d, 1 H, 8,3 Hz, 4-HO-Ph C3-H), 7,12-7,21 (m, 2H, 4-F-Ph), 7,28 (dd, 1H, 2,2/8,3 Hz, 4-HO-Ph C2-H), 7,39-7,46 (m, 5H, 4-Pyr, 4-F-Ph und 4-HO-Ph C6-H), 8,40 (m, 2H, 4-Pyr)
Beispiel 20
4-Fluor-N-methoxy-N-methyl-benzamid (20)
Eine Suspension von 4-Fluorbenzoesäure (20 g, 143 mmol) in Thionylchlorid (130 g; 1,1 mol) wurde 6 h unter Rückfluss gerührt: Heftige Gasentwicklung, klare Lösung nach ca. 10 min, Farbvertiefung von Gelb nach Orange. Das überschüssige Thio- nylchlorid wurde destillativ entfernt (zunächst Normaldruck/40 °C, dann Membran- pumpenvakuum/40 °C). Aus dem Destillationsrückstand wurde 4-Fluorbenzoe- säurechlorid im Membranpumpenvakuum bei 90 °C über eine kurze Kolonne abdes- tilliert. Das Reaktionsprodukt kristallisierte bei Aufbewahrung im Kühlschrank aus (Π20D 1,5315; F 9 °C, Ausbeute 20 g/89 %). Zu einer Suspension von N,0-Dimethyl- hydroxylaminhydrochlorid (9,0 g; 92 mmol) in CH2CI2 (75 mL) wurde frisch destilliertes Triethylamin (29 mL) zugegeben. Das Reaktionsgemisch wurde 2 h bei Raumtemperatur gerührt und anschließend auf -10 °C abgekühlt. Innerhalb von 6 min wurde 4-Fluorbenzoesäurechlorid (13,5 g; 85 mmol) unter Kühlung zur Vorlage zugetropft. Nach beendeter Zugabe wurde die Kühlung entfernt und das Reaktionsge- misch 1 ,5 h bei Raumtemperatur nachgerührt. Die hellbraune Suspension wurde auf H2O (100 mL) gegeben. Die organische Phase wurde abgetrennt und die wässrige Phase mit Diethylether extrahiert (2 x). Der vereinigte organische Extrakt wurde mit gesättigter NaCI-Lösung gewaschen, über NaS04 getrocknet und eingeengt. Der ölige, braune Rückstand kristallisierte beim Abkühlen und Anreiben. Das Rohprodukt
wurde an der Ölpumpe getrocknet (Triethylamin-Reste!) und ohne weitere Reinigung umgesetzt.
1H-NMR (CDCI3): δ (ppm) 3,37 (s, 3H, NCH3), 3,54 (s, 3H, OCH3), 7,04-7,13 (m, 2H, 4-F-Ph), 7,71-7,78 (m, 2H, 4-F-Ph)
Beispiel 21
2-(2-Chlorpyridin-4-yl)-1 -(4-fluorphenyl)-ethanon (21a)
Zu einer auf -85 °C abgekühlten Lösung von Diisopropylamin (15 mL, 106 mmol) in THF abs. (150 mL) wurde in einem ausgeheizten und mit Argon gespülten DHK n- BuLi (15%ige Lösung in n-Hexan, 45 mL, 104 mmol) zugetropft: Temperaturanstieg auf -50 °C. Nach beendeter Zugabe wurde die hellgelbe Lösung 55 min bei -85 °C gerührt. Zu dieser Vorlage wurde bei -85 °C eine Lösung von 2-Chlor-4-methyl- pyridin (2-Chlor-γ-picolin, 8,6 g; 68 mmol) in THF abs. (75 mL) zugetropft: Temperaturanstieg auf -50 °C, sofortige Purpurfärbung. Nach beendeter Zugabe wurde das Reaktionsgemisch 1 h bei -85 °C gerührt und bei dieser Temperatur innerhalb von 3 min mit einer Lösung von 20 (12,4 g; 68 mmol) in THF abs. (75 mL) versetzt: Tempe- raturanstieg auf -60 °C. Das purpurfarbene, breiige Reaktionsgemisch wurde 1 h bei -85 °C gerührt und anschließend innerhalb von 1 h auf 0 °C erwärmt. Der Ansatz wurde auf gesättigte NaCI-Lösung (300 mL) gegeben, die mit Ethylacetat (300 mL) überschichtet war. Die organische Phase wurde abgetrennt und die wässrige Phase mit Ethylacetat extrahiert (2 * 250 mL), wobei sich an der Phasengrenzfläche nur wenig brauner, schaumiger Niederschlag von 1 ,3-Bis-(2-chlorpyridin-4-yl)-2-(4- fluorphenyl)-propan-2-ol abschied. Der vereinigte organische Extrakt wurde mit gesättigter NaCI-Lösung gewaschen, über NaS04 getrocknet und eingeengt. Der ölige Rückstand wurde in etwas tert-Butylmethylether aufgenommen und über Nacht bei 4 °C gelagert. Das Kristallisat wurde abfiltriert und getrocknet.
1H-NMR (CDCI3): δ (ppm) 4,26 (s, 2H, CH2), 7,11-7,26 (m, 4H, C -/C5-H 2-CI-Pyr und 4-F-Ph), 7,99-8,06 (m, 2H, 4-F-Ph), 8,35 (dd, 1 H, 0,6/5,1 Hz, C6-H 2-CI-Pyr)
1 -(4-Fluorphenyl)-2-(2-f luorpyι idin-4-yl)-ethanon (21 b)
21b wurde ausgehend von 2-Fluor-4-methyl-pyridin (13,9 g; 125 mmol) nach der in der Synthese von 21a beschriebenen Methode dargestellt.
1H-NMR (CDCI3): δ (ppm) 4,32 (s, 2H, CH2), 6,85-6,86 (m, 1H, C3-H 2-F-Pyr), 7,08- 7,19 (m, 3H, C5-H 2-F-Pyr und 4-F-Ph), 8,00-8,07 (m, 2H, 4-F-Ph), 8,18 (d, 1H, 5,1 Hz, C6-H 2-F-Pyr)
2-(2-Brompyridin-4-yl)-1-(4-fluorphenyl)-ethanon (21c)
21c wurde ausgehend von 2-Brom-4-methyl-pyridin (9,6 g; 56 mmol) nach der in der Synthese von 21a beschriebenen Methode dargestellt.
1H-NMR (CDCI3): δ (ppm) 4,35 (s, 2H, CH2), 7,17-7,37 (m, 3H, 2-Br-Pyr und 4-F-Ph), 7,50 (s, 1H, C3-H 2-Br-Pyr), 8,07-8,15 (m, 2H, 4-F-Ph), 8,42 (d, 1H, 5,1 Hz, C6-H 2- Br-Pyr)
Beispiel 22
1 -(2-Chlorpyridin-4-yl)-2-(4-fluorphenyl)-ethan-1 ,2-dion-1 -oxim (22a)
Eine Lösung von 21a (3,0 g; 12 mmol) in Eisessig (30 mL) wurde unter Rühren und Kühlung im Wasserbad (ca. 10 °C) innerhalb von 2,5 min tropfenweise mit einer Lö- sung von NaN02 (0,85 g; 12,3 mmol) in H20 (10 mL) versetzt. Nach beendeter Zugabe wurde das Reaktionsgemisch 0,5 h bei Raumtemperatur gerührt, mit H20 (60 mL) ergänzt und weitere 3 h bei Raumtemperatur gerührt. Der hellbeige Niederschlag wurde abfiltriert, mit Wasser gewaschen und im Vakuum über CaCI2 getrocknet.
1H-NMR (DMSO-de): δ (ppm) 7,34-7,52 (m, 4H, C3-/C5-H 2-CI-Pyr und 4-F-Ph), 7,93- 8,00 (m, 2H, 4-F-Ph), 8,47 (d, 1 H, 5,2 Hz, C6-H 2-CI-Pyr), 12,71 (bs, 1H, austauschbar, OH)
1 -(2-Fluorpyridin-4-yl)-2-(4-fluorphenyl)-ethan-1 ,2-dion-1 -oxim (22b)
22b wurde ausgehend von 21b (10,0 g; 43 mmol) nach der in der Synthese von 22a beschriebenen Methode dargestellt.
1H-NMR (DMSO-de): δ (ppm) 7,19-7,20 (m, 1H, C3-H 2-F-Pyr), 7,35-7,47 (m, 3H, C5- H 2-F-Pyr und 4-F-Ph), 7,91-7,98 (m, 2H, 4-F-Ph), 8,29 (d, 1H, 5,3 Hz, C6-H 2-F- Pyr), 12,69 (s, 1H, austauschbar, OH)
1 -(4-Fluorphenyl)-2-(2-isopropoxypyridin-4-yl)-ethan-1 ,2-dion-2-oxim (22c)
Eine Lösung von 22b (200 mg; 0,76 mmol) in HCI-gesättigtem Isopropanol (15 mL) wurde 2,5 h unter Rückfluss gerührt. Die Lösung wurde eingeengt und der gelblichweiße Rückstand mit wenig Ethanol ausgerührt, abfiltriert und getrocknet.
1H-NMR (DMSO-de): δ (ppm) 1,24 (d, 6H, 6,2 Hz, 2 x CH3), 5,15-5,27 (m, 1H, Me- thin-H), 6,54 (s, 1H, C3-H 2-lso-O-Pyr), 7,08 (dd, 1H, 1 ,2/5,3 Hz, C5-H 2-lso-O-Pyr), 7,36-7,49 (m, 2H, 4-F-Ph), 7,88-7,97 (m, 2H, 4-F-Ph), 8,19 (d, 1 H, 5,4 Hz, C6-H 2- Iso-O-Pyr), 12,44 (bs, 1H, austauschbar, OH)
1 -(2-Brompyridin-4-yl)-2-(4-f luorphenyl)-ethan-1 ,2-dion-1 -oxim (22d)
22d wurde ausgehend von 21c (5,0 g; 17 mmol) nach der in der Synthese von 22a beschriebenen Methode dargestellt.
1H-NMR (DMSO-d6): δ (ppm) 7,40-7,48 (m, 3H, C3-H 2-Br-Pyr und 4-F-Ph), 7,65 (d, 1H, 0,8 Hz, C5-H 2-Br-Pyr), 7,93-8,01 (m, 2H, 4-F-Ph), 8,45 (d, 1H, 5,2 Hz, C6-H 2- Br-Pyr), 12,72 (bs, 1H, austauschbar, OH)
Beispiel 23
2-Amino-2-(2-chlorpyridin-4-yl)-1-(4-fluorphenyl)-ethanon-hydrochlorid (23a)
22a (1 ,5 g; 5,4 mmol) wurde unter leichtem Erwärmen in Methanol (15 mL) gelöst. Die Lösung wurde auf Raumtemperatur abgekühlt, mit salzsaurem Methanol (20 mL) ergänzt und in einen DHK überführt. In die Vorlage wurde Pd-C 10 % (150 mg) eingetragen. Das Reaktionsgefäß wurde an der Ölpumpe evakuiert und anschließend über eine Gaseinleitungskapillare mit H beschickt (4 x). Die Suspension wurde bei Raumtemperatur im abgeschlossenen Dreihalskolben in H2-Atmosphäre geschüttelt (240 Hub/min), bis das Edukt dünn-schichtchromatographisch nicht mehr nachweisbar war (6 h). Die Suspension wurde filtriert und der Katalysator mit reichlich Methanol gewaschen. Das vereinigte Filtrat wurde eingeengt und der senffarbene, festamorphe Rückstand an der Ölpumpe getrocknet. Das Rohprodukt wurde ohne weite- re Aufreinigung im nächsten Reaktionsschritt eingesetzt.
1H-NMR (DMSO-d6): δ (ppm) 6,53 (bs, 1H, Methin-H), 7,35-7,45 (m, 2H, 4-F-Ph), 7,59 (dd, 1 H, 1 ,5/5,2 Hz, C5-H 2-CI-Pyr), 7,85 (d, 1H, 0,9 Hz,C3-H 2-CI-Pyr), 8,17- 8,25 (m, 2H, 4-F-Ph), 8,49 (d, 1H, 4,9 Hz, C6-H 2-CI-Pyr), 9,33 (bs, 3H, austausch- bar, NH3 +)
2-Amino-2-(2-fluorpyridin-4-yl)-1-(4-fluorphenyl)-ethanon-hydrochlorid (23b)
22b (5,0 g; 19 mmol) wurde unter leichtem Erwärmen in salzsaurem Isopropanol (IsOH/HCI-gesättigter IsOH 1 +1 , 60 mL) gelöst. Die gelbliche Lösung wurde auf Raumtemperatur abgekühlt und in einen DHK (100 mL) überführt. In die Vorlage wurde Pd-C 10 % (1,5 g) eingetragen. Das Reaktionsgefäß wurde an der Ölpumpe evakuiert und anschließend über eine Gaseinleitungskapillare mit H2 beschickt (4 x). Bei Raumtemperatur wurde die Suspension im abgeschlossenen Dreihalskolben in H2-Atmosphäre geschüttelt (240 Hub/min), bis das Edukt dünnschichtchromatographisch nicht mehr nachweisbar war (6,5 h). Es wurde vom Katalysator abfiltriert. Der Filtrationsrückstand wurde mit reichlich Methanol gewaschen (ca. 800 mL). Die vereinigten Filtrate wurden eingeengt und der fest-amorphe Rückstand an der Öl-
pumpe getrocknet. Das Rohprodukt wurde ohne weitere Aufreinigung im nächsten Reaktionsschritt eingesetzt.
1H-NMR (DMSO-de): δ (ppm) 6,58 (bs, 1H, Methin-H), 7,33-7,41 (m, 2H, 4-F-Ph), 7,54 (m, 2H, C3-/C5-H 2-F-Pyr), 8,14-8,25 (m, 2H, 4-F-Ph), 8,30 (d, 1 H, 5,5 Hz, C6-H 2-F-Pyr), 9,40 (bs, 3H, austauschbar, NH3 +)
2-Amino-1-(4-fluorphenyl)-2-(2-isopropoxypyridin-4-yl)-ethanon-hydrochlorid (23c)
23c wurde ausgehend von 22c (2,0 g; 7,6 mmol) nach der in der Synthese von 23a beschriebenen Methode dargestellt.
1H-NMR (DMSO-de): δ (ppm) 1 ,23 (d, 6H, 5,6 Hz, 2 x CH3), 5,09-5,22 (m, 1 H, Me- thin-H CH(CH3)2), 6,38-6,41 (bs, 1H, Methin-H CH-NH3 +), 7,00-7,08 (m, 2H, 2-lso-O- Pyr), 7,33-7,46 (m, 2H, 4-F-Ph), 8,14-8,23 (m, 3H, 2-lso-O-Pyr und 4-F-Ph), 9,21 (bs, 3H, austauschbar, NH3 +)
2-Amino-1-(4-fluorphenyl)-2-(2-methoxypyridin-4-yl)-ethanon-hydrochlorid (23d)
23d entstand durch Behandlung von 22b (7,5 g; 29 mmol) unter den in der Synthese von 23a beschriebenen Bedingungen.
1H-NMR (DMSO-de): δ (ppm) 3,83 (s, 3H, CH3), 6,44 (bs, 1H, Methin-H), 7,13-7,16 (m, 2H, C3-/C5-H 2-MeO-Pyr), 7,34-7,46 (m, 2H, 4-F-Ph), 8,16-8,25 (m, 3H, C6-H 2- MeO-Pyr und 4-F-Ph), 9,29 (bs, 3H, austauschbar, NH3 +)
2-Amino-1-(4-fluorphenyl)-2-pyridin-4-yl-ethanon-hydrochlorid (23e)
23e entstand durch Behandlung von 22c (4,0 g; 12,4 mmol) unter den in der Synthese von 23b beschriebenen Bedingungen.
1H-NMR (DMSO-de): δ (ppm) 6,78 (bs, 1 H, Methin-H), 7,32-7,38 (m, 2H, 4-F-Ph), 8,07-8,13 (m, 2H, 4-Pyr), 8,17-8,27 (m, 2H, 4-F-Ph), 8,92-8,95 (m, 2H, 4-Pyr), 9,43 (bs, 3H, austauschbar, NH3 +)
2-Amino-2-(2-brompyridin-4-yl)-1 -(4-fluorphenyl)-ethanon-hydrochlorid (23f)
Eine Lösung von 22d (1 ,8 g; 5,6 mmol) in Ethanol absolut (30 mL) wurde auf -10 °C abgekühlt und mit konzentrierter Schwefelsäure (1 ,3 mL) versetzt. Unter Kühlung wurde Zink-Staub (1 ,1 g) portionsweise in die Vorlage eingetragen. Das Reaktions- gemisch wurde 30 min bei -10 °C gerührt und anschließend auf Raumtemperatur erwärmt. Die grau-grüne Suspension wurde filtriert und der weiße Rückstand (ZnS04) mit reichlich Ethanol gewaschen. Das vereinigte, gelbgefärbte Filtrat wurde eingeengt und der feste, gelbliche Rückstand an der Ölpumpe getrocknet.
1H-NMR (DMSO-de): δ (ppm) 6,39 (bs, 1H, Methin-H), 7,35-7,44 (m, 2H, 4-F-Ph), 7,56 (dd, 1 H, 1 ,4/5,1 Hz, C5-H 2-Br-Pyr), 7,91 (s, 1H, C3-H2-Br-Pyr), 8,12-8,19 (m, 2H, 4-F-Ph), 8,46 (d, 1H, 5,1 Hz, C6-H2-Br-Pyr), 8,94 (bs, 3H, austauschbar, NH3 +)
Beispiel 24
4-(2-Chlorpyridin-4-yl)-5-(4-fluorphenyl)-1,3-dihydro-imidazol-2-thion (24a)
23a (2,9 g; ca. 9,6 mmol) wurde unter leichtem Erwärmen in DMF absolut (75 mL) gelöst. In die klare, orange-rote Lösung wurde Kaliumrhodanid (1 ,9 g; 19,6 mmol) eingetragen: Sofortige Trübung und Farbaufhellung. Das Reaktionsgemisch wurde 1 ,5 h unter Rückfluss gerührt. Die Suspension wurde auf Raumtemperatur abgekühlt und unter H20-Kühlung tropfenweise mit H20 (ca. 140 mL) verdünnt. Der gelbe Niederschlag wurde abfiltriert, mit H20 gewaschen und im Vakuum über CaCI2 getrocknet.
1H-NMR (DMSO-de): δ (ppm) 7,12-7,52 (m, 6H, C3-/C5-H 2-CI-Pyr und 4-F-Ph), 8,27 (d, 1H, 5,2 Hz, C6-H 2-CI-Pyr), 12,82 (bs, 2H, austauschbar, 2 NH)
4-(4-Fluorphenyl)-5-(2-fluorpyridin-4-yl)-1,3-dihydro-imidazol-2-thion (24b)
24b wurde ausgehend von 23b (6,1 g; 20 mmol) nach der in der Synthese von, 24a beschriebenen Methode dargestellt.
1H-NMR (DMSO-d6): δ (ppm) 7,12-7,16 (m, 2H, C3-/C5-H 2-F-Pyr), 7,28-7,27 (m, 2H, 4-F-Ph), 7,46-7,55 (m, 2H, 4-F-Ph), 8,13 (d, 1 H, 5,1 Hz, C6-H 2-F-Pyr), 12,85 (bs, 2H, austauschbar, 2 x NH)
4-(4-Fluorphenyl)-5-(2-isopropoxypyridin-4-yl)-1,3-dihydro-imidazol-2-thion (24c)
24c wurde ausgehend von 23c (2,5 g; 7,6 mmol) nach der in der Synthese von 24a beschriebenen Methode dargestellt.
H-NMR (DMSO-de): δ (ppm) 1,24 (d, 6H, 6,2 Hz, 2 CH3), 5,10-5,19 (m, 1H, Methin-H), 6,69-6,76 (m, 2H, 2-lso-O-Pyr), 7,24-7,32 (m, 2H, 4-F-Ph), 7,42-7,49 (m ,2H, 4-F-Ph), 8,02 (d, 1H, 5,5 Hz, C6-H 2-lso-O-Pyr), 12,68 (bs, 2H, austauschbar, 2 NH)
4-(4-Fluorphenyl)-5-(2-methoxypyridin-4-yl)-1 ,3~dihydro-imidazol-2-thion (24d)
In eine Lösung von 23d (3,2 g; 10,8 mmol) in 10%iger Salzsäure (50 mL) wurde Ka- liumrhodanid (2 g; 20,6 mmol) eingetragen. Das Reaktionsgemisch wurde 30 min unter Rückfluss gerührt. Die orangefarbene Lösung wurde abgekühlt und mit 10%iger NaHC03-Lösung neutralisiert. Der Niederschlag wurde abfiltriert, mit H20 gewaschen und im Vakuum über CaCl2 getrocknet. Das Rohprodukt wurde mit Ethanol ausgerührt und vom unlöslichen Anteil abfiltriert. Aus dem ethanolischen Filtrat fiel 24d beim Stehenlassen aus.
1H-NMR (DMSO-de): δ (ppm) 3,81 (s, 3H, OCH3), 6,79-6,82 (m, 2H, C3-/C5-H2-MeO- Pyr), 7,26-7,50 (m, 4H, 4-F-Ph), 8.06 (d, 1H, 5,3 Hz, C6-H 2-MeO-Pyr), 12,65 (bs, 2H, austauschbar, 2 NH)
Beispiel 25
2-Chlor-4-[5-(4-fluorphenyl)-2-methylsulfanyl-3H-imidazoI-4-yl]-pyridin (25a)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 24a (0,5 g; 1 ,6 mmol) und Methyliodid (0,35 g; 2,5 mmol) nach 12-stündiger Reaktionszeit und säulenchromatographischer Reinigung (Al203, CH2CI2/Ethylacetat 1+1) erhalten. Fp. 236 °C
IR (ATR): 3126, 3057, 2929, 1591, 1529, 1499, 1389, 1231 (C-F), 1159, 996, 976, 844, 780 cm"1
H-NMR (DMSO-de): δ (ppm) 2,62 (s, 1H, CH3), 7,27-7,36 (m, 3H, 2-CI-Pyr und 4-F- Ph), 7,45-7,55 (m, 3H, 2-CI-Pyr und 4-F-Ph), 8,24 (d, 1H, 5,1 Hz, C6-H 2-CI-Pyr), 12,85 (bs, 1H, austauschbar, NH)
2-Fluor-4-[5-(4-fluorphenyl)-2-methylsulfanyl-3H-imidazol-4-yl]-pyridin (25b)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 24b (0,95 g; 3,3 mmol) und Methyliodid (1 ,4 g; 9,9 mmol) nach 40-stündiger Reaktionszeit erhalten. Das Rohprodukt wurde mit CH2CI2/Ethylacetat (1+1) ausgekocht. Der vereinigte organische Extrakt wurde mit AI2O3 entfärbt und der nach Einengen des Filtrates erhaltene Rückstand mit wenig EtOH ausgerührt. Fp. 224 °C
IR (ATR): 3073, 1609, 1497, 1421 , 1234, 1219 (C-F), 1159, 1002, 883, 851 , 833, 815 cm"1
1H-NMR (DMSO-de): δ (ppm) 2,62 (s, 3H, CH3), 7,08 (s, 1H, C3-H 2-F-Pyr), 7,26-7,35 (m, 3H, C5-H 2-F-Pyr und 4-F-Ph), 7,46-7,54 (m, 2H, 4-F-Ph), 8,08 (d, 1H, 5,3 Hz, C6-H 2-F-Pyr), 12,85 (bs, 1 H, austauschbar, NH)
4-[5-(4-Fluorphenyl)-2-methylsulfanyl-3H-imidazol-4-yl]-2-isopropoxypyridin (25c)
In eine Lösung von 24c (4,0 g; 13,8 mmol) in THF absolut (60 mL) wurde NaH (55 - 65 %; 1 ,0 g; ca. 23 mmol) eingetragen. Diese Vorlage wurde 5 min bei Raumtemperatur gerührt und tropfenweise und unter H20-Kühlung mit einer Lösung von Methyliodid (2,2 g; 17,3 mmol) in THF absolut (5 mL) versetzt. Das Reaktionsgemisch wurde 1 h bei Raumtemperatur gerührt. Die klare, braune Lösung wurde eingeengt und der Rückstand in H2O aufgenommen. Die wässrige Lösung wurde mit 10%iger Salz- säure neutralisiert und mit Ethylacetat extrahiert (2 x). Der vereinigte organische Extrakt wurde mit gesättigter NaCI-Lösung gewaschen, über Na2S04 getrocknet und eingeengt. Der halbfeste Rückstand wurde mit fert-Butylmethylether ausgekocht (2 x) und filtriert. Das klare, etherische Filtrat wurde eingeengt und der feste Rückstand mit wenig fert-Butylmethylether ausgerührt, abfiltriert und getrocknet. Weiteres Reak- tionsprodukt ließ sich durch säulenchromatographische Trennung der Mutterlauge gewinnen (Si0260, CH2CI2/Ethylacetat 1+1). Fp. 141 °C
IR (ATR): 2928, 1610, 1544, 1509, 1412, 1314, 1222 (C-F), 1104, 1005, 954, 865, 843, 816 cm"1
1H-NMR (CD3OD): δ (ppm) 1 ,28 (d, 6H, 6,1 Hz, 2 CH3), 2,63 (s, 3H, SCH3), 5,08- 5,14 (m, 1H, Methin-H), 6,76 (s, 1H, C3-H 2-lso-O-Pyr), 6,88 (dd, 1H, 1 ,4/5,4 Hz, C5- H 2-lso-O-Pyr), 7,10-7,19 (m, 2H, 4-F-Ph), 7,40-7,47 (m, 2H, 4-F-Ph), 7,95 (dd, 1H, 0,7/5,4 Hz, C6-H 2-lso-O-Pyr)
4-[5-(4-Fluorphenyl)-2-methylsulfanyl-3H-imidazoI-4-yl]-2-methoxypyridin (25d)
Eine Lösung von 24d (1 ,0 g; 3,3 mmol) und Methyliodid (5,6 g; 39 mmol) in Methanol (50 mL) wurde 3 h unter Rückfluss gerührt. Das Reaktionsgemisch wurde abgekühlt und filtriert. Das Filtrat wurde eingeengt und der Rückstand in Ethanol aufgenommen. Vom unlöslichen Anteil wurde abfiltriert und das Filtrat eingeengt. Der Rückstand wurde in CH2CI2/EtOH (9+1) aufgenommen. Vom unlöslichen Anteil wurde abfiltriert und das Filtrat säulenchromatographisch getrennt (Si02 60, CH2CI2/EtOH 9+1 ). Fp. 158 °C
IR (ATR): 1618, 1608, 1497, 1391, 1222 (C-F), 1212, 1036, 835, 825 cm"1
1H-NMR (CD3OD): δ (ppm) 2,67 (s, 3H, SCH3), 3,90 (s, 3H, OCH3), 6,87-6,89 (m, 1H, C3-H 2-MeO-Pyr), 6,98 (dd, 1H, 1,5/5,5 Hz, C5-H2-MeO-Pyr), 7,16-7,24 (m, 2H, 4-F- Ph), 7,46-7,53 (m, 2H, 4-F-Ph), 8,03 (dd, 1H, 0,7/5,5 Hz, C6-H 2-MeO-Pyr)
4-[5-(4-Fluorphenyl)-2-methylsulfanyl-3H-imidazol-4-yl]-1H-pyridin-2-on(25e)
25e fiel als einziges Reaktionsprodukt bei der Behandlung von 23d (8,8 g; 31 mmol) mit Kaliumrhodanid in DMF in der Siedehitze analog der für 24a beschriebenen Methode an. Fp. 314 °C (Zersetzung). Nach Zyklisierung zum 1 ,3-dihydro-imidazolthion erfolgt Übertragung der Methylgruppe aus dem Methoxysubstituenten auf das nukle- ophile Schwefelatom des Thions unter Bildung des 2-methylsulfanyl-3H-imidazols ei- nerseits und des 2-Hydroxypyridin/1H-pyridin-2-ons andererseits.
IR (ATR): 1634 (Pyridon I), 1610, 1557 (Pyridon II), 1493, 1220 (C-F), 968, 837, 800 cm"1
1H-NMR (DMSO-de): δ (ppm) 2,61 (s, 3H, SCH3), 6,16 (bs, 1 H, C3-H Pyridon), 6,34 (s, 1 H, C5-H Pyridon), 7,25-7,33 (m, 3H, C6-H Pyridon und 4-F-Ph), 7,46-7,53 (m, 2H, 4-F-Ph), 11 ,38 (bs, 1H, austauschbar, Pyridon-NH), 12,71 (bs, 1 H, austauschbar, Imidazol-NH)
Benzyl-{4-[5-(4-fluorphenyl)-2-methylsulfanyl-3H-imidazol-4-yl]-pyridin-2-yl}- amin (25f)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 25b (0,2 g; 0,7 mmol) und Benzylamin (0,8 g; 7,5 mmol) nach 5-stündiger Umsetzung bei 160 °C und säulenchromatographischer Trennung (Al2θ3, CH2CI2/Ethylacetat 1+1) erhalten. Fp. 152 °C (Zersetzung)
IR (ATR): 3234 (NH), 3006, 2916, 1601 , 1583, 1501, 1451 , 1432, 1353, 1225 (C-F), -1074, 844, 813, 729, 695 cm"1
1H-NMR (CD3OD): δ (ppm) 2,59 (s, 3H, CH3), 4,37 (s, 2H, CH2), 6,56-6,59 (m, 2H, C3-/C5-H 2-Amino-Pyr), 7,04-7,44 (m, 9H, Ph und 4-F-Ph), 7,83 (d, 1H, 5,6 Hz, C6-H 2-Amino-Pyr)
{4-[5-(4-Fluorphenyl)-2-methylsulfanyl-3H-imidazol-4-yl]-pyridin-2-yl>-(4- methoxybenzyl)-amin (25g)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 25b (0,44 g; 1 ,5 mmol) und 4-Methoxybenzylamin (2,0 g; 14,6 mmol) nach 7-stündiger Umsetzung bei 160 °C und säulenchromatographischer Trennung (Si02, CH2CI2/EtOH 9+1) erhalten. Fp. 207 °C
IR (ATR): 1598, 1558, 1510, 1244, 1217 (C-F), 846, 812 cm"1
1H-NMR (CD3OD): δ (ppm) 2,61 (s, 3H, SCH3), 3,75 (s, 3H, OCH3), 4,30 (s, 2H, CH2), 6,56-6,59 (m, 2H, C3-/C5-H 2-Amino-Pyr), 6,81-7,30 (m, 6H, 4-MeO-Ph und 4- F-Ph), 7,39-7,46 (m, 2H, 4-F-Ph), 7,84 (d, 1H, 6,0 Hz, C6-H 2-Amino-Pyr)
{4-[5-(4-Fluorphenyl)-2-methylsulfanyl-3H-imidazol-4-yl]-pyridin-2-yl}-(4-methyl- benzyl)-amin (25h)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 25b (0,2 g; 0,7 mmol) und 4-Methylbenzylamin (0,85 g; 7,0 mmol) nach 6-stündiger Um- setzung bei 160 °C und säulenchromatographischer Trennung (Si02 60, CH2CI2/EtOH 9+1) erhalten. Fp.185 °C
IR (ATR): 1600, 1559, 1502, 1427, 1218 (C-F), 844, 809 cm"1
1H-NMR (CD3OD): δ (ppm) 2.29 (s, 3H, CH3), 2.60 (s, 3H, SCH3), 4.32 (s, 2H, CH2), 6.57-6.60 (m, 2H, C3-/C5-H 2-Amino-Pyr), 7.05-7.50 (m, 8H, 4-Me-Ph und 4-F-Ph), 7.83 (d, 1 H, 5.3 Hz, C6-H2-Amino-Pyr)
(4-Chlorbenzyl)-{4-[5-(4-fluorphenyl)-2-methylsulfanyl-3H-imidazol-4-yl]-pyridin -2-yl}-amin (25i)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 25b (0,2 g; 0,7 mmol) und 4-Chlorbenzylamin (1 ,0 g; 7,0 mmol) nach 5,5-stündiger Umsetzung unter Rückfluss und säulenchromatographischer Trennung (Si02 60, CH2CI2/EtOH 9+1) erhalten. Fp. 195 °C
IR (ATR): 3409, 1597, 1549, 1502, 1489, 1422, 1218 (C-F), 843, 814, 793 cm"1
1H-NMR (CD3OD): δ (ppm) 2,60 (s, 3H, SCH3), 4,38 (s, 2H, CH2), 6,57-6,60 (m, 2H, C3-/C5-H 2-Amino-Pyr), 7,05-7,14 (m, 2H, 4-F-Ph), 7,22-7,30 (m, 4H, 4-CI-Ph), 7,38- 7,45 (m, 2H, 4-F-Ph), 7,83 (d, 1H, 5,7 Hz, C6-H 2-Amino-Pyr)
(3,4-Dichlorbenzyl)-{4-[5-(4-fluorphenyl)-2-methylsulfanyl-3H-imidazol-4-yl]- pyridin-2-yl}-amin (25j)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 25b (0,2 g; 0,7 mmol) und 3,4-Dichlorbenzylamin (1 ,2 g; 6,8 mmol) nach 7,5-stündiger Umsetzung bei 160 °C und säulenchromatographischer Trennung (Si02 60, CH2CI2/EtOH 9+1) erhalten. Fp. 212 °C
IR (ATR): 3409, 1600, 1552, 1509, 1490, 1424, 1225 (C-F), 842, 827, 813 cm"1
1H-NMR (CD3OD): δ (ppm) 2,60 (s, 3H, SCH3), 4,39 (s, 2H, CH2), 6,56-6,62 (m, 2H, C3-/C5-H 2-Amino-Pyr), 7,06-7,50 (m, 7H, 3,4-Di-CI-Ph und 4-F-Ph), 7,84 (d, 1H, 5,5 Hz, C6-H 2-Amino-Pyr)
{4-[5-(4-Fluorphenyl)-2-methylsulfanyl-3H-imidazol-4-yl]-pyridin-2-yl}-phenyl- amin (25k)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 25b (0,2 g; 0,7 mmol) und Anilin (0,65 g; 7,0 mmol) nach 6-stündiger Umsetzung unter
Rückfluss und säulenchromatographischer Trennung (Si02 60, CH2CI2/EtOH 9+1) erhalten. Fp. 228 °C
IR (ATR): 3031 , 1610, 1590, 1561, 1504, 1433, 1265, 1225 (C-F), 839, 827, 749, 695 cm 1
1H-NMR (DMSO-de): δ (ppm) 2,62 (s, 3H, CH3), 5,95-6,13 (m, 2H, C3-/C5-H2-Amino- Pyr), 6,68-7,60 (m, 9H, Ph und 4-F-Ph), 7,97-8,01 (m, 1H, C6-H 2-Amino-Pyr), 8,99 (bs, 1 H, austauschbar, Anilino-NH), 12,68 (bs, 1H, austauschbar, Imidazol-NH)
{4-[5-(4-Fluorphenyl)-2-methylsulfanyl-3H-imidazol-4-yl]-pyridin-2-yl}-phen- ethylamin (251)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 25b (0,2 g; 0,7 mmol) und 2-Phenylethylamin (0,85 g; 7,0 mmol) nach 5,5-stündiger Umsetzung bei 160 °C und säulenchromatographischer Trennung (Si02 60, CH2CI2/EtOH 9+1 ) erhalten. Fp. 99 °C
IR (ATR): 3409, 1604, 1546, 1504, 1220 (C-F), 838, 813, 698 cm -1
1H-NMR (CD3OD): δ (ppm) 2,61 (s, 3H, SCH3), 2,81 (t, 2H, 7,7 Hz, NCH2), 3,41 (t, 2H, 7,7 Hz, CH2Ph), 6,55-6,57 (m, 2H, C3-/C5-H 2-Amino-Pyr), 7,08-7,26 (m, 7H, Ph und 4-F-Ph), 7,42-7,49 (m, 2H, 4-F-Ph), 7,82 (d, 1H, 6,1 Hz, C6-H 2-Amino-Pyr)
(RS)-{4-[5-(4-Fluorphenyl)-2-methylsulfanyl-3H-imidazoI-4-yl]-pyridin-2-yl}-(1 - phenylethyl)-amin (25m).
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 25b (0,2 g; 0,7 mmol) und (RS)-1 -Phenylethylamin (0,80 g; 6,6 mmol) nach 7-stündiger Umsetzung bei 160 °C und säulenchromatographischer Trennung (Si02 60, CH2CI2/EtOH 9+1) erhalten. Fp. 117 - 119 °C
|R (ATR): 2926, 1607, 1547, 1502, 1434, 1221 (C-F), 1157, 838, 814, 699 cm"1
1H-NMR (DMSO-de): δ (ppm) 1 ,37 (d, 3H, 5,5 Hz, CH3), 2,58 (s, 3H, SCH3), 4,82- 5,03 (m, 1H, Methin-H), 6,39-7,74 (m, 12H, Ph, 2-Amino-Pyr und 4-F-Ph), 12,57 (bs, 1H, austauschbar, NH)
(R)-{4-[5-(4-Fluorphenyl)-2-methylsulfanyl-3H-imidazol-4-yl]-pyridin-2-yl}-(1- phenylethyl)-amin (25n)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 25b (0,2 g; 0,7 mmol) und R)-I-Phenylethylamin (0,80 g; 6,6 mmol) nach 7-stündiger Umsetzung bei 170 °C und säulenchromatographischer Trennung (Si02 60, CH2CI2/Ethylacetat 1+1) erhalten. Fp. 117 - 119 °C
IR (ATR): 2926, 1607, 1547, 1502, 1434, 1221 (C-F), 1157, 838, 814, 699 cm"1
1H-NMR (CD3OD): δ (ppm) 1 ,44 (d, 3H, 6,9 Hz, CH3), 2,59 (s, 3H, SCH3), 4,62-4,69 (m, 1 H, Methin-H), 6,47-6,57 (m, 2H, C3-/C5-H2-Amino-Pyr), 7,05-7,42 (m, 9H, Ph und 4-F:Ph), 7,80 (d, 1H, 5,5 Hz, C6-H 2-Amino-Pyr)
(S)-{4-[5-(4-Fluorphenyl)-2-methylsulfanyl-3H-imidazol-4-yl]-pyridin-2-yl}-(1- phenylethyl)-amin (25o)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 25b (0,2 g; 0,7 mmol) und (S)-1 -Phenylethylamin (0,80 g; 6,6 mmol) nach 13-stündiger Umsetzung bei 170 °C und säulenchromatographischer Trennung (Si02 60, CH2CI2/Ethylacetat 1+1) erhalten. Fp. 117 - 119 °C
IR (ATR): 2926, 1607, 1547, 1502, 1434, 1221 (C-F), 1157, 838, 814, 699 cm"1
1H-NMR (CD3OD): δ (ppm) 1,44 (d, 3H, 6,9 Hz, CH3), 2,59 (s, 3H, SCH3), 4,62-4,69 (m, 1H, Methin-H), 6,47-6,57 (m, 2H, C3-/C5-H2-Amino-Pyr), 7,05-7,42 (m, 9H, Ph und 4-F-Ph), 7,80 (dd, 1 H, 0,5/5,5 Hz, C6-H 2-Amino-Pyr)
Benzyl-{4-[5-(4-fluorphenyl)-2-methyIsulfanyl-3H-imidazol-4-yl]-pyridin-2-yl}- methylamin (25p)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 25b (0,2 g; 0,7 mmol) und N-Methylbenzylamin (0,85 g; 7,0 mmol) nach 7-stündiger Umsetzung bei 180 °C und zweimaliger säulenchromatographischer Trennung (SiO2 60, CH2CI2/Ethylacetat 1+1) erhalten. Fp. 79 °C
IR (ATR): 2924, 1601 , 1494, 1407, 1219 (C-F), 837, 810, 730, 696 cm"1
1H-NMR (CD3OD): δ (ppm) 2,60 (s, 3H, SCH3), 2,97 (s, 3H, NCH3), 4,64 (s, 2H, CH2), 6,64-6,66 (m, 2H, C3-/C5-H 2-Amino-Pyr), 7,02-7,45 (m, 9H, Ph und 4-F-Ph), 7,96 (d, 1H, 5,0 Hz, C6-H 2-Amino-Pyr)
Nach der obigen Methode wurden die in der nachfolgenden Tabelle 2 zusammengestellten Verbindungen erhalten:
Tabelle 2:
Bsp. VerName Struktur fahren
25q (4-Fluoro-phenyl)-{4-[5-(4-fluoro- phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridin-2-yl}-amine
25r (4-Chloro-phenyl)-{4-[5-(4-fluoro- phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridin-2-yl}-amine
25s {4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]- pyridin-2-yl}-m-tolyl-amine
4-[2-Benzylsulfanyl-5-(4-fluorphenyl)-3H-imidazol-4-yl]-2-chlorpyridin (26a)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 24a (0,3 g; 1 ,0 mmol) und Benzylchlorid (0,12 g; 1 ,0 mmol) nach 6-stündiger Reaktionszeit und säulenchromatographischer Reinigung (Si02 60, CH2CI2/EtOH 9+1 ) erhalten. Fp. 223 °C
IR (ATR): 2939, 1591, 1530, 1505, 1233 (C-F), 997, 838, 782, 700 cm"1
1H-NMR (DMSO-de): δ (ppm) 4,43 (s, 2H, CH2), 7,27-7,47 (m, 11 H, 2-CI-Pyr, Ph und 4-F-Ph), 8,26 (d, 1H, 5,2 Hz, C6-H 2-CI-Pyr), 12,94 (bs, 1H, austauschbar, NH)
4-[2-Benzylsulfanyl-5-(4-f luorphenyl)-3H-imidazol-4-yl]-2-fluorpyridin (26b)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 24b (5,1 g; 17,6 mmol) und Benzylbromid (9,2 g; 54 mmol) nach 1 ,5-stündiger Reaktionszeit und säulenchromatographischer Trennung (Al203, CH2CI2/Ethylacetat 1+1 ) erhal- ten. Fp. 174 °C
IR (ATR): 3028, 2948, 1611 , 1496, 1413, 1228 (C-F), 1203, 1003, 879, 838, 698 cm"1
1H-NMR (DMSO-de): δ (ppm) 4,43 (s, 2H, CH2), 7,11 (s, 1H, C3-H 2-F-Pyr), 7,25-7,51 (m, 10H, C5-H 2-F-Pyr, Ph und 4-F-Ph), 8,10 (d, 1 H, 5,3 Hz, C6-H 2-F-Pyr), 12,93 (bs, 1H, austauschbar, NH)
Benzyl-{4-[2-benzylsulfanyl-5-(4-fluorphenyl)-3H-imidazol-4-yl]-pyridin-2-yI}- amin (26c)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 26b (0,2 g; 0,53 mmol) und Benzylamin (0,60 g; 5,6 mmol) nach 6-stündiger Umsetzung bei 180 °C und säulenchromatographischer Trennung (Si02 60, CH2CI2/Ethylacetat 1 +1) erhalten. Fp. 185 °C
IR (ATR): 3407 (NH), 3025, 2855, 2713, 1599, 1550, 1489, 1356, 1220 (C-F), 1155, 840, 814, 693 cm"1
1
H-NMR (CD3OD): δ (ppm) 4,21 (s, 2H, NCH2), 4,38 (s, 2H, SCH2), 6,52-6,55 (m, 2H, C3-/C5-H 2-Amino-Pyr), 7,03-7,38 (m, 9H, Ph und 4-F-Ph), 7,83 (d, 1H, 5,7 Hz, C6-H 2-Amino-Pyr)
(RS)-{4-[2-Benzylsulfanyi-5-(4-fluorphenyl)-3H-imidazol-4-yl]-pyridin-2-yl}-(1- phenylethyl)-amin (26d)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 26b (0,2 g; 0,53 mmol) und (RS)-l-Phenylethylamin (0,65 g; 5,4 mmol) nach 15-stündiger Umsetzung bei 150 °C und säulenchromatographischer Trennung (Si02 60, CH2CI2/Ethylacetat 1+1) erhalten. Fp. 145 °C
IR (ATR): 3028, 1606, 1546, 1494, 1450, 1221 (C-F), 1157, 837, 813, 697 cm -1
1H-NMR (CD3OD): δ (ppm) 1,44 (d, 3H, 6,8 Hz, CH3), 4,22 (s, 2H, CH2), 6,44-6,54 (m, 2H, C3-/C5-H 2-Amino-Pyr), 7,04-7,35 (m, 9H, Ph und 4-F-Ph), 7,80 (d, 1H, 5,4 Hz, C6-H 2-Amino-Pyr)
{4-[2-Benzylsulfanyl-5-(4-fluorphenyl)-3H-imidazol-4-yl]-pyridin-2-yl}-(4- methoxybenzyl)-amin (26e)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 26a (0.2 g; 0,5 mmol) und 4-Methoxybenzylamin (2,0 g; 14,6 mmol) nach 22-stündiger Umsetzung unter Rückfluss und säulenchromatographischer Trennung (Al203, CH2CI2/Ethylacetat 1+1) erhalten. Fp. 196 - 200 °C
IR (ATR): 1605, 1574, 1507, 1245, 1225 (C-F), 843, 814, 698
1H-NMR (DMSO-d6): δ (ppm) 4,29 (s, 2H Isomere „A" + „B", NCH2), 4,35 (s, 2H „A" + „B", SCH2), 6,43-6,47 (m, 1 H „A" + 2H „B", C5-H "A" und C3-/C5-H "B" 2-Amino-Pyr),
6,65 (s, 1H "A", C3-H2-Amino-Pyr), 6,80-6,84 (m, 2H „A" + „B", 4-MeO-Ph), 7,14-7,51 (m, 11 H „A" + „B", 4-MeO-Ph, Ph und 4-F-Ph), 7,79 (d, 1H "B", 5,4 Hz, C6-H 2- Amino-Pyr), 7,91 (d, 1H "A", 5,4 Hz, C6-H 2-Amino-Pyr), 12,67 (bs, 1 H, austauschbar, Imidazol-NH), Amino-NH nicht sichtbar
4-[2-Benzylsulfanyl-5-(4-fluorphenyl)-3H-imidazol-4yl]-2-methoxy-pyridin (26f)
Eine Suspension von 25a (0,1 g; 0,25 mmol) in methanolischer NaOCH3-Lösung (30 %, 2 mL) wurde mit Methanol (5 mL) verdünnt und 13 h unter Rückfluss gerührt. Das Reaktionsgemisch wurde mit H20 verdünnt und die wässrige Lösung mit CH2CI2 extrahiert (3χ). Der vereinigte organische Extrakt wurde mit gesättigter NaCI-Lösung gewaschen, über Na2S04 getrocknet und eingeengt. Der ölige Rückstand wurde säu- lenchromatographisch gereinigt (Si02 60, CH2CI2/Ethylacetat 1+1).
1H-NMR (CDCI3): δ (ppm) 3,91 (s, 3H, OCH3), 4,29 (s, 2H, CH2), 6,91-6,95 (m, 1 H, 2- MeO-Pyr), 7,02-7,11 (m, 2H, 4-F-Ph), 7,27-7,38 (m, 7H, Ph und 4-F-Ph), 8,05 (d, 1H, 5,4 Hz, C6-H 2-MeO-Pyr), NH nicht sichtbar
Beispiel 27
2-Chlor-4-[5-(4-fluorphenyl)-2-(4-methansulfinyl-benzylsulfanyl)-3H-imidazol-4- yl]-pyridin (27a)
In eine Lösung von 24a (0,31 g; 1,0 mmol) in THF absolut (15 mL) wurde NaH (55 - 65 %; 0,1 g; ca. 2 mmol) eingetragen. Die Vorlage wurde 5 min bei Raumtemperatur gerührt und mit 4-Methylsulfinyl-benzylchlorid (3, 0,19 g; 1 ,0 mmol) versetzt. Das Reaktionsgemisch wurde 2 h bei Raumtemperatur gerührt. Die gelb-braune Lösung wurde mit H20 verdünnt und mit 10%iger Zitronensäure neutralisiert. Das THF wurde entfernt und die wässrige Lösung mit Ethylacetat (2 x) extrahiert. Der vereinigte or- ganische Extrakt wurde mit gesättigter NaCI-Lösung gewaschen (2 x), über Na2S04 getrocknet und eingeengt. Der feste Rückstand wurde säulenchromatographisch gereinigt (SiQ2 60, CH2CI2/EtOH 9,5+0,5). Fp. 179 °C
IR (ATR): 3049, 1592, 1505, 1374, 1224 (C-F), 1086, 1030 (S=0), 1014, 989, 839, 816, 781 cm"1 (C-Cl)
1H-NMR (DMSO-d6): δ (ppm) 2,71 (s, 3H, CH3), 4,48 (s, 2H, CH2), 7,25-8,24 (m, 10H, 2-CI-Pyr, 4-MeS(0)-Ph und 4-F-Ph), 8,26 (d, 1H, 5,3 Hz, C6-H 2-CI-Pyr), 12,94 (bs, 1H, austauschbar, NH)
2-Fluor-4-[5-(4-fluorphenyl)-2-(4-methansulfinyl-benzylsulfanyl)-3H-imidazol-4- yl]-pyridin (27b)
Nach der Allgemeinen Methode A wurde die Titelverbindung ausgehend von 24b (4,2 g; 14,5 mmol) und 3 (4,1 g; 22 mmol) nach 2-stündiger Reaktionszeit und säulenchromatographischer Trennung (1. Al203, CH2CI2/Ethylacetat 1+1 , 2. Si02 60, CH2CI2/Et0H 9+1 ) erhalten. Fp. 150 °C
IR (ATR): 3061 , 1610, 1506, 1408, 1227 (C-F), 1030 (S=0), 1016, 995, 978, 882, 839, 815 cm"1
1H-NMR (DMSO-de): δ (ppm) 2,71 (s, 3H, CH3), 4,49 (s, 2H, CH2), 7,10 (s, 1 H, C3- H2-F-Pyr), 7,30-7,37 (m, 3H, C5-H 2-F-Pyr und 4-F-Ph), 7,47-7,67 (m, 6H, 4-F-Ph und 4-MeS(0)-Ph), 8,11 (d, 1H, 4,8 Hz, C6-H 2-F-Pyr), 12,95 (bs, 1H, austauschbar, NH)
Benzyl-{4-[5-(4-fluorphenyl)-2-(4-methansulfinyl-benzylsulfanyl)-3H-imidazol-4- yl]-pyridin-2-yl}-amin (27c)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 27b (0,3 g; 0,68 mmol) und Benzylamin (0,75 g; 7,0 mmol) nach 7-stündiger Umsetzung bei 170 °C und säulenchromatographischer Trennung (Si02 60, CH2CI2/Ethanol 19+1) erhalten. Fp. 149 °C
IR (ATR): 3238, 3064, 1600, 1558, 1514, 1495, 1227 (C-F), 1034 (S=0), 1006, 982, 839, 814 cm"1
1H-NMR (CD3OD): δ (ppm) 2,70 (s, 3H, CH3), 4,21 (s, 2H, NCH2), 4,32 (s, 2H, SCH2), 6,51-6,55 (m, 2H, C3-/C5-H 2-Amino-Pyr), 7,03-7,42 (m, 13H. Ph, 4-MeS(0)-Ph und 4-F-Ph), 7,82 (d, 1H, 5,5 Hz, C6-H 2-Amino-Pyr)
(RS)-{4-[5-(4-Fluorphenyl)-2-(4-methansulfinyl-benzylsulfanyl)-3H-imidazol-4- yl]-pyridin-2-yl}-(1 -phenylethyl)-amin (27d)
Nach der Allgemeinen Methode C wurde die Titelverbindung ausgehend von 27b (0,3 g; 0,68 mmol) und (ΗSJ-l-Phenylethylamin (0,85 g; 7,0 mmol) nach 10-stündiger Umsetzung bei 170 °C und säulenchromatographischer Trennung (Si02 60, CH2CI2/Ethanol 9+1) erhalten. Fp. 193 °C
IR (ATR): 2967, 1606, 1547, 1502, 1221 (C-F), 1085, 1031 (S=0), 1014, 838, 814, 670 cm"1
1H-NMR (CD3OD): δ (ppm) 1,45 (d, 3H, 6,8 Hz, CH3), 2,67 (s, 3H, S(O)CH3), 4,28 (s, 2H, CH2), 4,62-4,73 (m, 1 H, Methin-H), 6,42-6,53 (m, 2H, C3-/C5-H 2-Amino-Pyr), 7,09-7,44 (m, 9H, Ph und 4-F-Ph), ), 8,21 (d, 1 H, 5,0 Hz, C6-H 2-F-Pyr).
In der nachfolgenden Tabelle 3 sind Verbindungen zusammengestellt, die nach Verfahren C erhalten werden (Verbindung Nr. 31 jedoch nach Verfahren D):
Tabelle 3: 1H-NMR
M/43166-PCT
M/43166-PCT
Beispiel 37a
Cyclohexyl-{4-[5-(4-fluoro-phenyl)-2-methylsulfanyl-1H-imidazol-4-yl]-pyridin-2- yl}-amine (37a)
Ausgehend von 2-Fluoro-4-[5-(4-fluoro-phenyl)-2-methylsulfanyl-1 H-imidazol-4-yl]- pyridine (1 ,2 g, 4 mMol ), zuvor in einen mit Argon begasten 25 ml Einhalsrundkolben eingewogen, wurde in Cyclohexylamin (3,97g, 0,04 Mol) suspendiert mit Argon über- schichten, und anschliessend 48h in einem auf 160 °C temperierten Ölbad unter Rückfluss des Amins erhitzt. Die braune Suspension lässt man auf RT abkühlen. Dann werden 30 ml Na-Citrat-Lösung (10%ige Citronensäurelösung pH 5 mit konz. NaOH) zugegeben, 10 min. gerührt, mit zweimal 30 ml Ethylacetat extrahiert Vereinigte organische Phasen werden noch 2 mal mit je 30 ml Na-Citrat-Lösung (10%ige Citronensäurelösung pH 5 mit konz. NaOH) und anschliessend mit 30 ml NaHC03-Lösung extrahieren und 1Mal mit gesättigter NaCI -Lösung extrahiert. Die organische Phase wird über Na2S04 getrocknet am Rotationsverdampfer eingeengt und der Rückstand aus 10 ml kristallisiert.
Die Umkristallisation erfolgt aus Isopropanol / Wasser (ca. 5 ml ISOH erwärmen, abkühlen lassen langsam ca. 5 ml dest. Wasser zugeben). Ausbeute: 0,76g (49,7%) mit Reinheit (HPLC) 96,6%.
Nach dieser Methode wurden die in der folgenden Tabelle 4 zusammengestellten Verbindungen der Formel I erhalten:
Tabelle 4:
Nach den obigen Verfahren wurden die in der nachfolgenden Tabelle 5 zusammengestellten Verbindungen erhalten:
Tabelle 5:
Bsp. VerName Struktur fahren
38 4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl-amine
39 Methyl-{4-[5-(4-fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-amine
40 Ethyl-{4-[5-(4-fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]- pyridin-2-yl}-amine
41 lsopropyl-{4-[5-(4-fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-amine
Methoxy-ethyl-{4-[5-(4-fluoro- phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridin-2-yl}-amine
N,N-Dimethylamino-ethyl-{4-[5-(4- fluoro-phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridin-2-yl}-amine
Hydroxypropyl-{4-[5-(4-fluoro- phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridin-2-yl}-amine
N'-{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]- pyridin-2-yl}-N,N-diphenyl-ethane-
1 ,2-diamine -
Cinnamyl-{4-[5-(4-fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyhdin-2-yl}-amine -
Cyclopropyl-{4-[5-(4-fluoro-phenyl)-
2-methylsulfanyl-1H-imidazol-4-yl]- pyridin-2-yl}-amine
Cyclopropylmethyl-{4-[5-(4-fluoro- phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridin-2-yl}-amine
Cycloheptyl-{4-[5-(4-fluoro-phenyl)-
2-methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-amine
Bicyclo[2.2.1]hept-2-yl-{4-[5-(4- fluoro-phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridin-2-yl}-amine
Adamantan-1 -yl-{4-[5-(4-fluoro- phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridin-2-yl}-amine
Adamantan-2-yl-{4-[5-(4-fluoro- phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridin-2-yl}-amine
{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]- pyridin-2-yl}-(tetrahydro-furan-3-yl)- amine-
{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]- pyridin-2-yl}-(tetrahydro-pyran-4-yl)- amine
(1-Ethyl-pyrrolidin-2-ylmethyl)-{4-[5-
(4-fluoro-phenyl)-2-methylsulfanyl-
1 H-imidazol-4-yl]-pyridin-2-yl}- amine
H [1-(5-Chloro-thiophen-2-yl)-ethyl]-
{4-[5-(4-fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-amine
{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]- pyridin-2-yl}-thiophen-3-ylmethyl- amine
Benzo[b]thiophen-2-ylmethyl-{4-[5-
(4-fluoro-phenyl)-2-methylsulfanyl-
1 H-imidazol-4-yl]-pyridin-2-yl}- amine
Benzofuran-2-ylmethyl-{4-[5-(4- fluoro-phenyl)-2-methylsulfanyl-1H- imidazol-4-yl]-pyridin-2-yl}-amine
H (1-Benzofuran-2-yl-ethyl)-{4-[5-(4- fluoro-phenyl)-2-methylsulfanyl-1H- imidazol-4-yl]-pyridin-2-yl}-amine
(2,3-Dihydro-benzofuran-2- ylmethyl)-{4-[5-(4-fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-amine
{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-oxazol-2-ylmethyl- amine
77 Benzooxazol-2-ylmethyl-{4-[5-(4- fluoro-phenyl)-2-methylsulfanyl-1H- imidazol-4-yl]-pyridin-2-yl}-amine
78 2-({4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-ylamino}-methyl)-4- isopropyl-oxazol-5-ol
79 N-{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-N'-(2-methylsulfanyl- vinyl)-formamidine
80 N-{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-N'-(1 -methyl-2- methylsulfanyl-propenyl)- formamidine
81 N-{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-N'-(2-methylsulfanyl- phenyl)-formamidine
82 {4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-(2-methyl-thiazol-5- ylmethyl)-amine
83 {4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]- pyridin-2-yl}-(2-methyl-thiazol-4- ylmethyl)-amine
84 {4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-[2-(2-methyleneamino- phenylsulfanyl)-ethyl]-amine
85 B 2-Bromo-4-[5-(4-fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridine
86 2-Azido-4-[5-(4-fluoro-phenyl)-2- methylsuIfanyl-1 H-imidazol-4-yl]- pyridine
87 D 2-Ethoxy-4-[5-(4-fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridine
88 4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]-2- p-tolyloxy-pyridine
89 2-(2,6-Dichlόro-phenoxy)-4-[5-(4- fluoro-phenyl)-2-methylsulfanyl-1H- imidazol-4-yl]-pyridine
90 D 2-Benzyloxy-4-[5-(4-fluoro-phenyl)-
2-methylsulfanyl-1 H-imidazol-4-ylj- pyridine
91 D 4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]-2- phenethyloxy-pyridine
92 D 2-Cyclohexyloxy-4-[5-(4-fluoro- phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridine
93 4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]-2- (tetrahydro-furan-3-yloxy)-pyridine
94 D 6-{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]- pyridin-2-yloxy}-hexahydro-furo[3,2- bjfuran-3-ol
95 4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]-2-
(tetrahyd ro-f u ran-2-yl methoxy)- pyridine
96 4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]-2-
(tetrahydro-pyran-2-ylmethoxy)- pyridine
97 2-(Benzofuran-2-ylmethoxy)-4-[5-
(4-fluoro-phenyl)-2-methylsu!fanyl-
1 H-imidazol-4-yl]-pyridine-
98 4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]-2-
(furan-2-ylmethoxy)-pyridine
99 4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]-2-
(thiophen-2-ylmethoxy)-pyridine
100 4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]-2-
(5-chloro-thiophen-2-ylmethoxy)- pyridine
101 4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]-2-
(thiophen-3-ylmethoxy)-pyridine -
102 4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]-2-
(thiazol-2-ylmethoxy)-pyridine
103 Bicyclo[2.2.1]hept-2-yl-{4-[5-(4- fluoro-phenyl)-2-methylsulfanyl-1H- imidazol-4-yl]-pyridin-2-yl}-amine
104 3-{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]- pyridin-2-yloxy}-1 -aza- bicyclo[2.2.2]octane
4-[5-(4-Fluoro-phenyl)-2-
105 methylsulfanyl-1H-imidazol-4-yl]-2- methylsulfanyl-pyridine
106 2-Benzenesulfonyl-4-[5-(4-fluoro- phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridine
107 4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]-2- phenylsulfanyl-pyridine
108 2-Ethylsulfanyl-4-[5-(4-fluoro- phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridine
109 2-{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-ylsulfanyl}-ethanol
110 3-{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1H-imidazol-4-yl]- pyridin-2-ylsulfanyl}-propan-1-ol
110a N-{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-formamide
111 N-{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-benzamide
112 4-Chloro-N-{4-[5-(4-fluoro-phenyl)-
2-methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-benzamide
120 2-(4-Fluoro-phenyl)-N-{4-[5-(4- fluoro-phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridin-2-yl}- acetamide
121 2-(2-Fluoro-phenyl)-N-{4-[5-(4- fluoro-phenyl)-2-methylsulfanyl-1 H- imidazol-4-yl]-pyridin-2-yl}- acetamide
122 N-{4-[5-(4-Fluoro-phenyl)-2- methylsuϊfanyl-1H-imidazol-4-yl]- pyridin-2-yl}-3-phenyl-propionamide
123 N-{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-3-phenyl-acrylamide
124 {4_[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-carbamic acid benzyl ester
125 1-Benzoyl-3-{4-[5-(4-fluoro-phenyl)
-2-methylsulfanyl-1 H-imidazol-4-yl]
-pyridin-2-yl}-urea
126 N-{4-[5-(4-Fluoro-phenyl)-2- methylsulfanyl-1 H-imidazol-4-yl]- pyridin-2-yl}-acetamide