WO2001059143A1 - Verfahren zur herstellung von monoglycosidierten flavonoiden - Google Patents
Verfahren zur herstellung von monoglycosidierten flavonoiden Download PDFInfo
- Publication number
- WO2001059143A1 WO2001059143A1 PCT/EP2001/001447 EP0101447W WO0159143A1 WO 2001059143 A1 WO2001059143 A1 WO 2001059143A1 EP 0101447 W EP0101447 W EP 0101447W WO 0159143 A1 WO0159143 A1 WO 0159143A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- enzyme
- solution
- reaction
- particles
- rutin
- Prior art date
Links
- ODYJCLZVHOXGIZ-UHFFFAOYSA-N CC(C(C(C1O)O)O)OC1OCC(C(C(C1O)O)O)OC1OC Chemical compound CC(C(C(C1O)O)O)OC1OCC(C(C(C1O)O)O)OC1OC ODYJCLZVHOXGIZ-UHFFFAOYSA-N 0.000 description 1
- 0 C[C@@]([C@@]([C@@]([C@@]1O)O)O)O[C@]1OC[C@]([C@]([C@@]([C@]1O)O)O)O[C@]1OCC1=C(c(cc2)cc(*)c2O)Oc2cc(O)cc(O)c2*1=O Chemical compound C[C@@]([C@@]([C@@]([C@@]1O)O)O)O[C@]1OC[C@]([C@]([C@@]([C@]1O)O)O)O[C@]1OCC1=C(c(cc2)cc(*)c2O)Oc2cc(O)cc(O)c2*1=O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/14—Preparation of compounds containing saccharide radicals produced by the action of a carbohydrase (EC 3.2.x), e.g. by alpha-amylase, e.g. by cellulase, hemicellulase
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/44—Preparation of O-glycosides, e.g. glucosides
- C12P19/60—Preparation of O-glycosides, e.g. glucosides having an oxygen of the saccharide radical directly bound to a non-saccharide heterocyclic ring or a condensed ring system containing a non-saccharide heterocyclic ring, e.g. coumermycin, novobiocin
Definitions
- the present invention relates to a process for the preparation of monoglycosidated flavonoids by enzymatic hydrolysis of rutinosides.
- the rhamno residue of the rutinosides is cleaved off enzymatically.
- rutinosides are compounds which contain a sugar-free component in which a radical of the formula (I)
- the rutinosides are flavonoids with the bisgylcosidic unit shown in formula (I).
- Rhamnose and / or the corresponding glucopyranosides can be obtained from the rutinosides.
- the glucopyranosides are derived from the rutinosides in that instead of the residue of the formula (I) they contain a residue of the formula (I *)
- both rhamnose and isoquercetin can be obtained from rutin.
- Rhamnose is a monosaccharide, which is widespread in nature, but mostly occurs only in small amounts.
- An important source for rhamnose are, for example, the glycosidic residues of natural flavonoids, such as rutin, from which the rhamnose can be obtained by glycoside cleavage.
- Rhamnose for example, plays an important role as a raw material for the representation of artificial flavors, such as furaneol.
- Isoquercetin is a monoglycosidated flavonoid of the following structural formula (II)
- the sugar-free component of the flavonoids is the so-called agiykon.
- isoquercetin is a glycoside of the aglycon quercetin (2- (3,4-dihydrophenyl) -3,5,7-trihydroxy-4H-1-benzopyran-4-one), which differs from flavone in that it contains five hydroxyl groups.
- isoquercetin the carbohydrate residue glucose is bound to the hydroxyl group in position 3 of quercetin.
- isoquercetin is used as quercetin-3-O-ß-D-glucopyranoside or 2- (3,4-dihydroxyphenyl) -3- (ß-D-glucopyranosyloxy) -5,7-dihydroxy-4H-1-benzopyran-4- called on.
- flavonoids or flavonoid mixtures are used, for example, in the food and cosmetics industry and are becoming increasingly important there.
- monoglycosidated flavonoids, such as isoquercetin are characterized by their good absorption capacity in the human body.
- rutin which has the following structural formula (III):
- rutin is also a glycoside of the aglycone quercetin, the carbohydrate residue rutinose being bound to the hydroxyl group in position 3 of the quercetin.
- the carbohydrate residue in rutin consists of a glucose unit linked in the 1- and 6-position and a terminally bound rhamnose or 6-deoxymannose unit.
- Rutin is used, for example, as quercetin-3-O-ß-D-rutinoside or 2- (3,4-dihydroxyphenyl) -3- ⁇ [6-O- (6-deoxy- ⁇ -mannopyranosol) -ß-D-glucopyranosyl] oxy!
- Rutin With three molecules of water of crystallization, rutin forms pale yellow to greenish needles.
- Anhydrous rutin has the property of a weak acid, turns brown at 125 ° C and decomposes at 214-215 ° C.
- Rutin which is found in many plant species - often as a companion of vitamin C - occurs, for example, in citrus, in yellow pansies, forsythia, acacia, various Solanum and Nicotiana species, capers, linden flowers, St. John's wort, tea, etc., was isolated in 1842 from the garden rhombus (Ruta graveolens). Rutin can also be obtained from the leaves of buckwheat and the East Asian coloring drug Wei-Fa (Sophora japonica, Fabaceae), which contains 13-27% rutin.
- EP-A-0317033 describes a process for the preparation of L-rhamnose, the rhamnosidic binding of glycosides which keep rhamnose bound in the terminal position is achieved by enzymatic hydrolysis.
- the substrate is usually carried out as a suspension in an aqueous medium.
- these reactions are mostly not very selective.
- due to the bisglycosidic structure of the carbohydrate residue in rutin a mixture of the two monosaccharides glucose and rhamnose is often formed.
- Agiykon quercetin there is usually a high proportion of Agiykon quercetin and other undesirable by-products.
- immobilization is, however, not suitable for all enzyme processes and has so far only been used to a limited extent.
- immobilized enzymes are used on an industrial scale: glucose isomerization with immobilized glucose isomerase and peniciliin G cleavage with immobilized penicillin amidase.
- immobilized enzyme processes cannot compete with free enzymes or chemical processes.
- the enzymes or the reaction conditions are often also unsuitable for immobilization. So there is no universal method for immobilization, and each enzyme must be viewed individually.
- This object is achieved by a process for the production of monoglycosidated flavonoids by enzymatic hydrolysis of rutionosides, in which an enzyme which is immobilized on a support is used for the enzymatic hydrolysis.
- the process can be carried out continuously or discontinuously and with a high effectiveness in comparison with the reaction with the native enzyme.
- the process according to the invention is characterized in particular by the fact that high automation of the entire process, including the recycling of the solvent and the monitoring of the enzyme activity, is possible.
- Figure 1 shows the continuous production of isoquercetin from rutin as an example of the process according to the invention.
- Suitable rutinosides for the process according to the invention are those which contain a 2-phenyl-4H-1-benzopyran-4-one base body as a sugar-free constituent or agiycon, which bears a radical of the formula (I) in position 3 and whose phenyl group pen, apart from position 3, can also be substituted one or more times by -OH or -0 (CH 2 ) n -H, where n is 1 to 8, n is preferably 1.
- substitution of the 2-phenyl-4H-1-benzopyran-one base body by -OH and / or -0 (CH 2 ) n -H preferably occurs in positions 5, 7, 3 'and / or 4'.
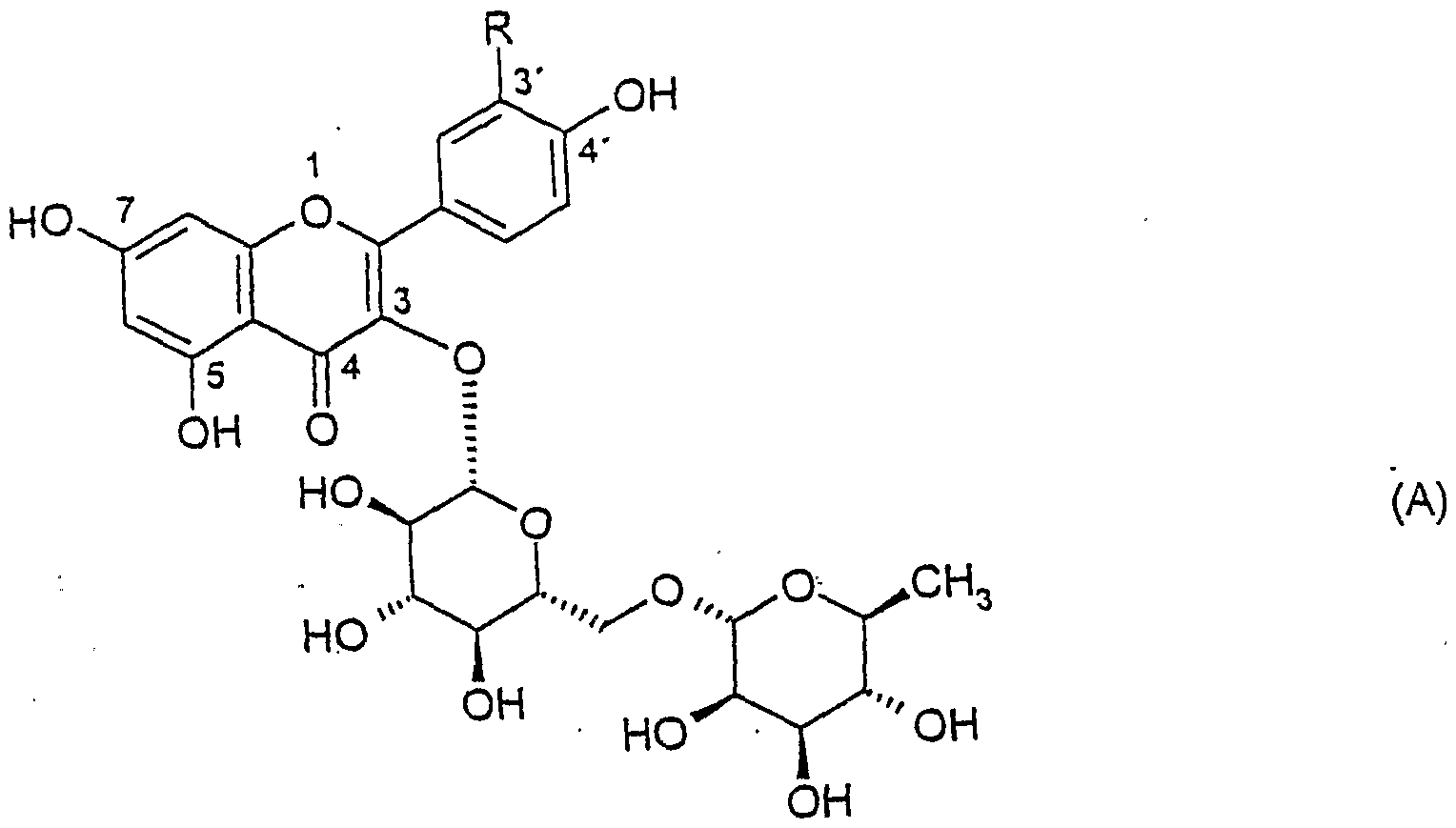
- Rutinosides which are represented by the formula (A), are particularly preferably used:
- RH, OH or OCH represents 3 .
- the compound in which RH represents is referred to as fighter olrutinoside; the rutinoside, in which R represents OCH 3 , is known as isorhamnetin rutinoside.
- the compound in which R is OH is called rutin. Accordingly, rhamnose and fighter olglucoside can be obtained from fighter olrutinoside, rhamnose and isoquercetin from rutin rutinoside and rhamnose and isorhamnetin glycoside from isorhamnetin rutinoside.
- the rutinoside rutin is particularly preferably used.
- the rutinosides can be used in pure form or as mixtures of rutinosides as starting material for the process according to the invention.
- the rutinosides can also be contaminated with other flavonoids or with residues from rutinoside production without the reaction being adversely affected.
- Usual hydrolases which are able to split off the rhamnose residue of the rutinosides can be used as enzymes for the enzymatic hydrolysis of the rutinosides. Hydrolases originating from the strain Penicillium decumbens are preferred won, used.
- ⁇ -L-Rhamnosidases are particularly preferably used as enzymes, since they have a high selectivity for the hydrolysis of the rhamnose residue. Suitable ⁇ -L-rhamnosidases are, for example, hesperidinase, naringinase, and those described in Kurosawa et al. (1973), J. Biochem., Vol. 73: 31-37. It is particularly preferred to use the enzyme hesperidinase.
- Both the rutinosides and the enzymes for the process according to the invention can be purchased as commercial products. It is also possible to obtain or produce the starting materials and enzymes by generally known methods.
- the enzyme is immobilized on a suitable carrier.
- suitable carriers such as silica gel, for example commercially available spherical or commercially available broken silica gels, for example Lichrosorb ® , Lichroprep ® , Lichrospher ® and Trisoperl ® , and commercially available polymeric carriers, for example Eupergit ® , Fractogel ® , in particular Fractogel epoxy ® , and Fractoprep ® , are used.
- Silica gel is the preferred carrier material.
- magnetic particles can also be used as carriers. These are preferably carrier materials with a magnetic core. This core is usually coated with an inorganic oxide.
- the inorganic oxide is preferably silica gel. Examples of such magnetic carriers include MagneSil TM (Promega Corp., Madison, Wisconsin, US), MagPrep TM (Merck) and AGOWAmag TM (AGOWA GmbH, Berlin, DE).
- Magnetic glass particles e.g. MPG (CPG Inc., Lincoln Park, New Jersey, US)
- magnetite-containing pigments e.g. Microna Matte, Mica Black, Colorona Blackstar (all Merck)
- Non-porous magnetic particles are particularly well suited because they cannot block pores, which would drastically impair the enzyme activity.
- the enzyme carrier usually has the following properties:
- the particle size of the carrier is preferably 0.005 to 1 mm, more preferably 0.01 to 0.5 mm.
- the pore diameter is usually in the range from 10 to 4000 nm, a pore diameter from 30 to 100 nm being particularly preferred.
- a sufficiently large pore size can ensure that the enzyme fits on the support without loss of activity.
- the particle surface is advantageously 40 to 100 m 2 / g, and the pore volume is preferably selected from a range of 0.5 to 3 ml / g. In some cases, a very large pore diameter of 2 to 20 ⁇ m can also be suitable.
- the enzyme can be coupled covalently or adsorptively.
- a covalent coupling is preferable.
- Examples of a covalent coupling include epoxidation, a carbodiimide method, silanization, a cyanogen bromide method, a glutaraldehyde crosslinking or a dicresyl chloride method (see Biotransformations and Enzyme Reactions, Bommarius, AS, Biotechnology (2nd ed.), Vol. 3, p. 427-465, compiled by Stephanopoulos, G., VCH Weinheim, Germany 1993, Walt, DR et al., Trends in analytical chemistry, Vol. 13, No. 10, 1994, NH Park, HN Chang; J. Ferment.
- the enzymatic hydrolysis takes place in a suitable reactor.
- a commercially available column is particularly suitable for a continuous implementation of the method according to the invention.
- a column such as that used for preparative HPLC can be used.
- the reactor, especially the column should have a high hydraulic efficiency. This can be quantified by the number of theoretical floors. It is therefore advantageous that the raw material solution comes into intensive contact with the surface of the property in order to ensure effective use of the enzyme and to achieve high productivity.
- the preparative HPLC column mentioned fulfills these requirements. and is also equipped with the appropriate technology with peripherals (pumps, valves, control). It is also advantageous that detection techniques such as UV or RI detection techniques have already been developed for this, so that, if desired, the measurement and regulation of the degree of conversion of the reaction can be carried out automatically.
- tubular reactors are usually used with a device which keeps the magnetic particles in suspension, e.g. electromagnetic coils that generate a largely homogeneous magnetic field in the flow tube, the field lines of which run parallel to the direction of flow (Helmholtz magnetic field).
- a device which keeps the magnetic particles in suspension e.g. electromagnetic coils that generate a largely homogeneous magnetic field in the flow tube, the field lines of which run parallel to the direction of flow (Helmholtz magnetic field).
- MSFB Magnetically Stabilized Fluidized Bed
- This technology can also be used advantageously for catalytic reactions in viscous reaction media.
- a conventional container which is preferably equipped with a stirring device, is suitable for carrying out the process discontinuously.
- a round bottom flask with a stirrer can be used on a small scale and a stirred kettle on a large scale.
- the immobilizate is packed into the reactor in the usual way before the reaction.
- the rutinoside to be reacted is usually fed into the reactor, for example a column, such as a fixed bed column, in the form of a solution or suspension. If the reactor is a fixed bed reactor, the rutinoside solution should be completely free of solids. It is advantageous to pre-dissolve the rutinoside with the solvent in a tank, preferably with stirring and / or heating, in order to achieve optimal solubility. If necessary, pre-filtration of the solution can also be performed to remove any solids.
- the solvent is preferably an aqueous system to ensure enzyme activity and to prevent possible denaturation.
- NEN additional solvents are added.
- the process according to the invention is preferably carried out in the presence of a solvent mixture of water and at least one organic solvent.
- the organic solvent or solvents additionally present include both organic solvents which are miscible with water and organic solvents which are not miscible with water.
- Suitable solvents for the process according to the invention are nitriles, such as acetonitrile, amides, such as dimethylformamide, esters, such as acetic acid esters, in particular methyl acetate or ethyl acetate, alcohols, such as methanol or ethanol, ethers, such as tetrahydrofuran or methyl tert-butyl ether and hydrocarbons, such as Toluene.
- the process according to the invention is preferably carried out in the presence of one or more of the organic solvents acetic acid ester, methanol, ethanol, methyl tert-butyl ether or toluene.
- the process according to the invention is particularly preferably carried out in the presence of one or more acetic acid esters, in particular in the presence of methyl acetate, in addition to water.
- Suitable volume ratios of water to organic solvent for the process according to the invention are ratios from 1:99 to 99: 1.
- the process according to the invention is preferably carried out with volume ratios of water to organic solvent from 20:80 to 80:20, in particular with volume ratios from 50:50 to 70:30.
- the amount of rutinoside in the solvent or solvent mixture for the process according to the invention depends on the solubility of the rutinoside in the solvent or solvent mixture. In order to carry out the process according to the invention optimally, the rutinoside should be readily soluble. It is therefore preferable to work with an undersaturated solution.
- the amount of rutinoside in the solvent or solvent mixture is usually 0.001 to 5 g / l, preferably 0.05 to 2 g / l, more preferably 0.1 to 1.5 g / l.
- the ratio of rutinoside to immobilizate or enzyme depends on the life of the enzyme in the column and its activity in immobilized form.
- the reaction is usually carried out at a temperature of 15 to 80 ° C.
- a temperature of 30 to 60 ° C. is preferred, in particular a temperature of 40 to 50 ° C. is advantageous in order to counteract possible destruction of the enzyme and at the same time to ensure a high solubility of the rutinoside
- reaction temperature If the reaction temperature is too low, the reaction proceeds at an inappropriately slow reaction rate due to decreasing enzyme activity. In addition, the solubility of the rutinoside is reduced in such a way that unnecessarily high amounts of solvent are necessary. On the other hand, if the reaction temperature is too high, the enzyme, which is a protein, is denatured and thus deactivated.
- the reactor can be provided with a temperature control device.
- Common temperature control devices include a heating coil system or double jacket. It is furthermore advantageous if the rutinoside to be reacted and in particular the rutinoside solution is tempered before entering the reactor.
- the rutinoside solution it is common for the rutinoside solution to be fed from a temperature-controlled tank which is set to the temperature desired for the reaction. Alternatively, the solution to be fed can be passed through a heatable hose in order to set the desired temperature before entering the reactor. Possible tempering out of the rutinoside can also be avoided by the temperature control.
- Suitable pH values for the method according to the invention are pH values between 3 and 8.
- the method according to the invention is preferably carried out at pH values from 3 to 7, in particular at pH values from 3 to 6.
- further preferred pH values can be used vary within the given limits depending on the enzyme used.
- pH values from 3.8 to 4.3 are extremely preferred when using the enzyme hesperidinase.
- the method is preferably carried out in such a way that the pH is adjusted with the aid of a buffer system. In principle, all common buffer systems that are suitable for setting the above-mentioned pH values can be used. However, aqueous citrate buffer is preferably used.
- the rutinoside mixture which may be in the form of a solution or a suspension, is added to the reactor containing the immobilizate to carry out the enzymatic hydrolysis.
- This reaction can be batch, i.e. be carried out batchwise or continuously.
- a rutinoside suspension is usually added to the reactor.
- the degree of conversion is determined by the amount of rutinoside and immobilizate.
- the ratio of rutinoside to immobilizate is usually 100: 1 to 1: 1000, preferably 10: 1 to 1: 100, more preferably 1: 1 to 1:20.
- the ratio of the immobilizate to the total volume of the suspension is usually 1: 1000 to 1: 1, preferably 1: 100 to 1: 2, more preferably 1:50 to 1: 5.
- the residence time in the reactor is normally 1 hour to 10 days, preferably 8 hours to 4 days, more preferably 1 to 2 days.
- a rutinoside solution is usually continuously and continuously conveyed through the reactor, preferably a column or MSFB reactor, using a suitable pump.
- the flow rate By setting the flow rate accordingly, any desired degree of conversion can be achieved. Normally, a flow rate of 0.001 to 1 mm / s, based on the empty pipe cross-section of the column, is used.
- the activity of the enzyme in the system decreases over time. It is therefore necessary for the immobilisate to be replaced in whole or in part at regular intervals.
- the reaction solution After the reaction solution has emerged, the product obtained can be separated.
- the reaction mixture consists mainly of solvent, unreacted rutinoside, rhamnose, the desired monoglycosidated flavonoid and possibly other additives, such as buffers.
- the monoglycosidated flavonoid usually precipitates as soon as the solubility limit is reached and gradually accumulates as a solid.
- the immobilized enzyme can be separated from the product suspension in a simple manner with the aid of a magnetic separation device after the reaction has ended.
- a strong permanent magnet in plate form can be used on a laboratory scale.
- Such a system can e.g. consist of a vertical flow tube, which contains a package of fine stainless steel wires.
- electromagnetic coils By means of suitably arranged electromagnetic coils, high magnetic field gradients are generated on the wires and a very effective separation of even the smallest particles in the nanometer range is achieved. If the magnetic particles are superparamagnetic, i.e. In the absence of an external magnetic field, there is no remanence magnetization, after switching off the magnetic field, it can be completely removed again from the separator by repeated rinsing with water.
- the desired reaction product is isolated by customary methods with customary workup.
- the product is preferably precipitated when concentrated. If the solvent contains a mixed solvent containing at least one organic solvent, it is preferred that the organic solvent be distilled off under reduced pressure.
- the crystallizing monoglycosidated flavonoid is usually separated from the remaining reaction mixture by solid / liquid separation, such as suction or filtration under reduced pressure or by centrifuging off the precipitated Crystals, separated. The solid is then washed, preferably with water and then dried.
- the entire reactor contents can first be filtered off.
- the filter cake containing the product is then treated with a solvent or a buffer-solvent mixture in which the product is soluble.
- the reaction product is extracted from the filter cake.
- the catalyst, the immobilisate, which is insoluble in this mixture remains.
- the solvent or the buffer-solvent mixture do not have an adverse effect on the enzyme.
- the immobilized enzyme e.g. Naringinase or hesperidinase, in certain solvent-buffer mixtures or under moderately alkaline conditions, has no or only a fraction of the original activity, but the activity can be practically completely restored if the enzyme is subsequently carefully mixed with a buffer in the pH range 4 -6 is rinsed; it is therefore a temporary loss of activity, not an irreversible denaturation of the enzyme.
- Extraction agents which are very suitable for this procedure are tetrahydrofuran buffer mixtures - preferably with a tetrahydrofuran content of 10-25%, in particular also at a slightly elevated temperature.
- Other suitable extractant components are e.g. 1-propanol, 2-propanol, 1, 4-dioxane and methyl acetate.
- the product can be very easily recovered from the extract by distilling off the solvent under reduced pressure and then cooling the aqueous product solution to 0 to 10 ° C. The reaction product crystallizes out of the mother liquor in very high purity.
- a dilute ammonia or soda solution can also be used as the extraction agent, since the reaction product has phenolic OH groups which are already deprotonated in a weakly basic medium; the anion of the reaction product is comparatively readily soluble, but is also very sensitive to oxidation, which is the result of a gradual discoloration of the extract from yellow to brown. Therefore, this variant should be operated quickly, ie the extraction should be completed within 10 minutes to 6 hours, preferably 20 minutes to 2 hours. It is preferable to work in a protective gas atmosphere.
- the enzyme activity is not lost even by treatment with weakly basic extraction agents, such as aqueous solutions of alkali or ammonium salts of acetic acid, oxalic acid, citric acid, phosphoric acid, boric acid or carbonic acid, or aqueous solutions of alkylamines, piperidine or pyridine.
- weakly basic extraction agents such as aqueous solutions of alkali or ammonium salts of acetic acid, oxalic acid, citric acid, phosphoric acid, boric acid or carbonic acid, or aqueous solutions of alkylamines, piperidine or pyridine.
- the purity of the monoglycosidated flavonoid obtained using pure rutinoside is normally greater than 94%.
- the end product can, for example, be recrystallized from suitable solvents, e.g. from water or from solvent mixtures consisting of toluene and methanol or water and methyl acetate.
- the amount of solvent obtained after the reaction is preferably recovered in order to ensure the economy of the process according to the invention.
- This recirculation is usually continuous and automated. Commercial evaporator systems with appropriate controls are available for this.
- the solvent to be used is a solvent mixture of water and at least one organic solvent, it is generally not possible to immediately use the distillate again for the process, since the solvent ratios have changed as a result of the organic solvent being distilled off. The desired solvent ratio can be restored by automatic quality measurement and correction and the solvent can thus be returned.
- silica gel 250 g are mixed with sufficient HCI (7%) in a 1 L bottle and left overnight to moisten the silica gel.
- the silica gel suspension is then washed chloride-free with demineralized water. To do this, you have to test the supernatant with nitric acid and silver nitrate after each wash. Because of the properties of the silica gel particles, the washing is carried out on an approximately 24 cm diameter ceramic funnel.
- the acid-treated silica gel is mixed with enough water in a 2 L three-necked flask equipped with a reflux condenser and a dropping funnel, so that it becomes stirrable.
- 1 mmol / g carrier of the 3-aminopropyltrimethoxysilane (for 250 g of silica gel, 135 ml of solution is required) is added dropwise to the silica gel suspension at a rate of about 5 drops / s.
- the suspension is then stirred at 90 ° C. for 2 hours.
- the suspension is then cooled with ice.
- the supernatant must be checked for possible residues of 3-aminopropyltrimethoxysilane by means of pH measurement.
- the bead suspension is washed with demineralized water until the pH remains constant.
- Glutardialdehyde is added to the silica gel suspension obtained in a concentration of 1 mmol / g of carrier (13 ml of 50% GDA solution are required for 250 g of carrier).
- the suspension (plus some water) is rolled in a 1 L bottle at room temperature for 2 hours. At the beginning the suspension is colored yellow and at the end it turns dark red.
- the enzyme solution is poured into the 1 L bottle mentioned above, and the enzyme solution is rolled at room temperature.
- Biomex BSA bovine serum albumin powder
- Approx. 20 g of the silica gel suspension obtained under 1.4 are mixed with the protein solution in a 0.5 L bottle, and 60 ⁇ l ProClin300 are added.
- the protein content of a solution is determined using the Bradford test.
- the standard assay is performed. 20 ⁇ l sample are mixed in 1 ml Bradford dye reagent (diluted 1: 5) and measured after 15 min in a photometer at 595 nm.
- the microassay must be carried out at very low protein concentrations. 0.8 ml of sample are mixed in 0.2 ml of Bradford dye reagent (concentrated) and measured after 15 min in a photometer at 595 nm. 3.2) Activity
- the activity of a solution is measured by the reaction with a replacement substrate. For each sample one takes:
- This 1 ml solution is mixed in an Eppendorf reaction vessel. After an incubation time of 2 and 5 min at 40 ° C in a shaker, 100 ⁇ l of the reaction mixture are mixed with 1 ml of 1 M soda solution. Then the concentration of p-nitrophenol is measured in the photometer at 400 nm. The activity is calculated by changing the concentration of p-nitrophenol per time.
- PROTEIN CONTENT and ACTIVITY VALUES (FREE HESPERIDINASE)
- PROTEIN CONTENT and ACTIVITY VALUES IMOBILIZED HESPERIDINASE
- the solution is fed to the piston metering pump (7) via a bag filter (5) and a candle filter (6).
- the bag filter has the task of stopping the larger amount of undissolved components, while the candle filter cleans the solution with a fineness of 0.2 ⁇ m.
- the piston metering pump (7) conveys the solution through a heatable hose, which adjusts the solution to a temperature of 40 ° C at the entrance of the column using a thermometer, with a flow of 1 l / min into the column (9) (100x400 mmj.
- the column contains 1.5 kg of immobilized material, since the electrically heated hose cannot cool the solution, the temperature in the stirred tank (1) must be selected so that cooling on the way to the pump leads to a maximum pump flow of up to 40 ° C.
- a sample can be taken from the solution after passing through the column via a manual valve (10), with which the temperature and the degree of conversion of the reaction can be measured off-line again. If the measured degree of conversion is lower than required, the flow rate of the pump is reduced accordingly.
- the reaction After passing through the column, the reaction is complete, so that the solution can be fed into the collecting vessel (11). There the solution is about 10-20% of the volume is removed from the capacitor (12). This significantly reduces the propanol content, which greatly reduces the solubility of the isoquercetin. Subsequent cooling further reduces the solubility, so that the product precipitates and can be separated in the prey filter (13). From there it is transferred to a drying cabinet (14) for further drying. The mother liquor and the distilled-off condensate are returned together in the stirred tank (1) for fresh preparation.
- silica gel eg LiChrospher Sl 300, Merck, Darmstadt
- 250 g of silica gel were poured with 400 ml of 10% HCl in a closable vessel, degassed with ultrasound for 10 minutes and left to stand at RT for 24 hours.
- the silica gel was then filtered off and demineralized with several liters. Washed water until the pH was> 4.5 and chloride ions were no longer detectable in the filtrate (spot reaction with acetic acid AgN0 3 solution).
- the acid-treated moist silica gel was then transferred to a 4 1 three-necked flask equipped with KPG stirrer, reflux condenser and 100 ml dropping funnel and slurried with 3 l of deionized water. With stirring, 100 ml of aminopropyltrimethoxysilane (ABCR, Düsseldorf) were added via the dropping funnel over the course of 15 minutes. The suspension was then heated and stirred at 90 ° C. for 90 minutes. The cooled suspension was filtered and eight times with 1 l of demin. Washed water.
- the amino-activated silica gel was suspended in 3 I using ultrasonically degassed water in a 4 I three-necked flask equipped with KPG stirrer and 100 ml dropping funnel; the pH was lowered to 8.0 with a few drops of 2 M acetic acid. Then 100 ml of 50% glutardialdehyde solution (Merck, Darmstadt) were added dropwise within 1 h and the suspension was stirred for a further 2.5 h at RT. The activated silica gel is filtered again and so long with ice-cold demin. Washed water until Wash water no more glutaraldehyde could be detected (spot reaction with sulfuric acid 2,4-dinitrophenylhydrazine solution).
- the silica gel modified with aldehyde groups was deminized in 500 ml. Water is suspended in a 4 liter flask by stirring with a KPG stirrer. 13 g of naringinase (Sigma, Deisenhofen) were dissolved in 2.5 1 0.25 M phosphate buffer, pH 8.0. The enzyme solution was added to the silica gel suspension and stirred at RT for 96 h. The immobilizate was then filtered off and washed several times, first with 0.2 M sodium chloride solution and then with 50 ml of citrate buffer, pH 4.0.
- the rhamnosidase activity of the immobilizate was determined using p-nitrophenyl-L- ⁇ -rhamnopyranoside (Sigma, Deisenhofen) as substrate by the Kurosawa method; it was 120 U / g.
- Eupergit 50 g of Eupergit (Röhm, Rothstadt) were mixed in a screw-on 500 ml glass bottle with 300 ml of 0.8 M potassium phosphate buffer, pH 8.5, and left to stand for 30 minutes. Then 5.0 g of hesperidinase (Amano) were added and the mixture was stirred for 120 hours at RT on a roller mixer. The Eupergit was filtered off by means of a glass frit and washed several times first with 0.2 M sodium chloride solution, then twice with 1 I 0.1 M citrate buffer, pH 4.0.
- the rhamnosidase activity of the immobilizate was determined using p-nitrophenyl-L- rhamnopyranoside (Sigma, Deisenhofen) as substrate by the Kurosawa method; it was 15 U / g, based on dry immobilizate, or 4.2 U / g, based on moist immobilisate.
- the filter cake was returned to the round bottom flask and stirred for 30 minutes in a mixture of 400 ml of 50 mM citrate buffer, pH 4.0 and 100 ml of tetrahydrofuran at 40 ° C., with most of the isoquercetin dissolving.
- the mixture was filtered warm and the filter cake extracted again with 500 ml of buffer tetrahydrofuran mixture for 30 minutes.
- the two isoquercetin extracts were combined with the first filtrate and the tetrahydrofuran was removed using a rotary evaporator.
- the aqueous isoquercetin solution was cooled to 4 ° C. After filtration and drying in a desiccator, a yield of 5.8 g of product was obtained, which was composed of 98% isoquercetin and 2% rutin.
- the moist eupergite was washed once with cold tetrahydrofuran buffer mixture, then repeatedly with 50 mM citrate buffer, pH 4.0, until the smell of tetrahydrofuran was barely perceptible.
- the activity of the enzyme was still 3.6 U / g, which corresponds to an activity loss of 14%.
- the moist filter cake was put back into the round bottom flask and stirred at RT in 300 ml of 50 mM sodium carbonate buffer, pH 10.0 for 5 min, during which part of the isoquercetin dissolved with an intensely yellow color.
- the suspension was filtered and the filter cake was immediately extracted again with carbonate buffer. After a total of 7 extraction cycles, the eupergite was largely colorless and the isoquercetin was almost completely dissolved.
- the extracts were combined, carefully acidified with dilute hydrochloric acid until the pH was approximately 3 and then cooled to 4 ° C. After filtration and drying in a desiccant A yield of 4.9 g of product was obtained, which was composed of 98% isoquercetin and 2% rutin.
- the moist eupergite was washed twice with 50 mM citrate buffer and was then ready for further reactions.
- the activity of the enzyme was still 3.9 U / g, which corresponds to a loss of activity of 7%.
- a suspension of 30 g magnetic silica particles (MagPrep, Merck, Darmstadt) in 600 ml water was placed in a 1 liter three-necked flask equipped with a KPG stirrer, dropping funnel and reflux condenser.
- a mixture of 20 ml of aminopropyltriethoxysilane (ABCR, Düsseldorf) and 20 ml of isopropanol was added dropwise with stirring over the course of 30 minutes. The mixture was then heated to 85 ° C. and stirred at this temperature for 1 hour. After cooling, the suspension was transferred to a beaker, the particles were collected at the bottom of the vessel using a strong permanent magnet, and the supernatant was decanted. The particles were repeated with demin.
- the particles were then resuspended in 600 ml of water and the pH was adjusted to about 8 with a few drops of acetic acid; after the addition of 24 ml of 50% glutardialdehyde solution, the suspension was stirred at RT for 4 h and the particles thereafter with demin. Washed water until no more glutaraldehyde could be detected in the wash water (spot reaction with sulfuric acid 2,4-dinitrophenylhydrazine solution).
- the aldehyde-derivatized particles were resuspended in a 1 liter round-bottom flask in 600 ml of 0.2 M potassium phosphate buffer, pH 9. After adding a solution of 1 g of naringinase (Sigma, Deisenhofen) in 100 ml of 50 mM sodium chloride solution, the mixture was stirred at RT for 2 days using a KPG stirrer. The particles were then separated using a permanent magnet and repeated first with 0.2 M sodium hydroxide. rium chloride solution, then washed with 50 mM citrate buffer, pH 4.0.
- the rhamnosidase activity of the immobilizate was determined using p-nitrophenyl-L- rhamnopyranoside (Sigma, Deisenhofen) as a substrate using the Kurosawa method (Kurosawa, Ikeda, Egami, J. Biochem. 73, 31-37 (1973) : ⁇ -L-Rhamnosidases of the liver of Turbo cornutus and Aspergillus niger); it was 162 U / g.
- a suspension of 30 g of magnetic silica particles (MagPrep, Merck, Darmstadt) in 600 ml of 50 mM sodium acetate solution was placed in a 1 liter three-necked flask equipped with a KPG stirrer, dropping funnel and reflux condenser.
- the rhamnosidase activity of the immobilizate was determined using p-nitrophenyl-L- ⁇ -rhamnopyranoside (Sigma, Deisenhofen) determined as substrate by the Kurosawa method; it was 102 U / g.
- a suspension of 30 g magnetic silica particles (MagPrep, Merck, Darmstadt) in 600 ml water was placed in a 1 liter three-necked flask equipped with a KPG stirrer, dropping funnel and reflux condenser.
- the suspension was transferred to a beaker, the particles were collected at the bottom of the vessel using a strong permanent magnet, and the supernatant was decanted.
- the particles were demineralized three times. Water, once with a 2 M acetic acid solution and then repeatedly with demin. Washed water until the pH of the wash water remained constant.
- the particles were then separated using a permanent magnet and repeatedly washed first with 0.2 M sodium chloride solution, then with 50 mM citrate buffer, pH 4.0.
- the rhamnosidase activity of the immobilizate was determined using p-nitrophenyl-L- ⁇ -rhamnopyranoside (Sigma, Deisenhofen) as substrate by the Kurosawa method; it was 71 U / g. 4. Modification of magnetic mica pigments with aldehyde groups and immobilization of hesperidinase on these particles
- a suspension of 30 g of magnetic mica pigments ("Colorado Blackstar Green", Merck, Darmstadt) in 300 ml of water was placed in a 1 liter three-necked flask equipped with a KPG stirrer, dropping funnel and reflux condenser.
- a mixture of 20 ml of aminopropyltriethoxysilane (ABCR, Düsseldorf) and 20 ml of isopropanol was added dropwise with stirring over the course of 30 minutes. The mixture was then heated to 85 ° C. and stirred at this temperature for 1 hour. After cooling, the suspension was transferred to a beaker, the particles were collected at the bottom of the vessel using a permanent magnet, and the supernatant was decanted. The particles repeated with demin.
- the particles were then resuspended in 300 ml of water and the pH was adjusted to about 8 with a few drops of acetic acid; after addition of 25 ml of 50% glutardialdehyde solution, the suspension was stirred for 4 hours at RT and the particles thereafter with demin. Washed water until no more glutaraldehyde could be detected in the wash water (spot reaction with sulfuric acid 2,4-dinitrophenylhydrazine solution).
- the rhamnosidase activity of the immobilizate was determined using p-nitrophenyl-L- ⁇ -rhamnopyranoside (Sigma, Deisenhofen) as substrate by the Kurosawa method; it was 10 U / g. 5. Conversion of rutin to isoguercetin with immobilized naringinase in the stirred tank reactor and separation of the magnetic biocatalyst with a permanent magnet
- the reactor contents were passed through an electromagnetic HGMS system using a peristaltic pump with a flow rate of 25 ml / min, whereby the magnetic particles were completely separated on the wire matrix (technical data of the separation system: glass tube with an inner diameter of 20 mm and 200 mm Length, empty volume 65 ml, weight of the wire packing made of SS alloy 15 g, 4 coils connected in series, current 6 A, magnet. Field strength of the Helmholtz field 25 mT). Subsequently, 100 ml of citrate buffer were pumped through the column with the magnetic field switched on in order to rinse the magnetic particles.
- the methyl acetate was first removed from the product mixture on a rotary evaporator; then the product was filtered off, washed several times with ice water and dried in a desiccator.
- the product yield was 2.9 g; the product consisted of 86% isoquercetin and 14% rutin.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Microbiology (AREA)
- General Chemical & Material Sciences (AREA)
- Biotechnology (AREA)
- Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Saccharide Compounds (AREA)
Abstract
Description





Claims

Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001558479A JP2003522532A (ja) | 2000-02-11 | 2001-02-09 | モノグリコシド化フラボノイドの製造方法 |
| EP01903733A EP1259632A1 (de) | 2000-02-11 | 2001-02-09 | Verfahren zur herstellung von monoglycosidierten flavonoiden |
| AU2001231726A AU2001231726A1 (en) | 2000-02-11 | 2001-02-09 | Method for producing monoglycosidated flavonoids |
| CA002400014A CA2400014A1 (en) | 2000-02-11 | 2001-02-09 | Preparation of monoglycosidated flavonoids |
| BR0108273-6A BR0108273A (pt) | 2000-02-11 | 2001-02-09 | Preparação de flavonóides monoglicosidados |
| US10/203,537 US20030157653A1 (en) | 2000-02-11 | 2001-09-02 | Method for producing monoglycosidated flavonoids |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10006147A DE10006147A1 (de) | 2000-02-11 | 2000-02-11 | Verfahren zur Herstellung von monoglycosidierten Flavonoiden |
| DE10006147.8 | 2000-02-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2001059143A1 true WO2001059143A1 (de) | 2001-08-16 |
Family
ID=7630616
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2001/001447 WO2001059143A1 (de) | 2000-02-11 | 2001-02-09 | Verfahren zur herstellung von monoglycosidierten flavonoiden |
Country Status (9)
| Country | Link |
|---|---|
| EP (1) | EP1259632A1 (de) |
| JP (1) | JP2003522532A (de) |
| KR (1) | KR20030013371A (de) |
| CN (1) | CN1416470A (de) |
| AU (1) | AU2001231726A1 (de) |
| BR (1) | BR0108273A (de) |
| CA (1) | CA2400014A1 (de) |
| DE (1) | DE10006147A1 (de) |
| WO (1) | WO2001059143A1 (de) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004027074A2 (en) * | 2002-09-23 | 2004-04-01 | Her Majesty The Queen In Right Of Canada, As Represented By The Minister Of Agriculture | Extraction, purification and conversion of flavonoids from plant biomass |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101100683B (zh) * | 2006-09-08 | 2010-05-12 | 颜廷和 | 一种甙元型黄酮生物转化及纯化的方法 |
| KR20080033705A (ko) * | 2006-10-13 | 2008-04-17 | (주)아모레퍼시픽 | 캄페롤-3-o-루티노사이드의 제조방법 |
| JP5184786B2 (ja) * | 2007-01-19 | 2013-04-17 | サントリーホールディングス株式会社 | フラボノイド類の配糖化方法 |
| KR101105846B1 (ko) * | 2009-08-17 | 2012-01-13 | 주식회사 젬백스앤카엘 | 흡착제 교체가 용이한 필터 여재 |
| CN101670010B (zh) * | 2009-10-10 | 2012-06-20 | 杭州富春食品添加剂有限公司 | 一种黄酮苷类物质的提取方法 |
| CN102286576B (zh) * | 2011-09-14 | 2013-11-06 | 江苏科技大学 | 强化酶法合成异槲皮苷的介质工程方法 |
| CN105779473A (zh) * | 2014-12-19 | 2016-07-20 | 上海交通大学 | 鼠李糖异黄酮的合成基因簇及其应用 |
| KR102528136B1 (ko) * | 2018-01-02 | 2023-05-02 | 최병국 | 이소케르세틴 및 알파-글리코실이소케르세틴의 제조방법 |
| KR102615753B1 (ko) * | 2022-11-30 | 2023-12-19 | 주식회사 다인소재 | 항균성 및 항산화 활성이 우수한 이소케르세틴의 제조방법 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0317033A1 (de) * | 1987-11-20 | 1989-05-24 | Unilever N.V. | Verfahren zur Herstellung von L-Rhamnose |
| US4971812A (en) * | 1989-08-31 | 1990-11-20 | National Science Council | Immobilized penicillium sp. naringnase and its use in removing naringin and limonin from fruit juice |
| US5641659A (en) * | 1992-11-27 | 1997-06-24 | Hoechst Aktiengesellschaft | α-l-rhamnosidase for obtaining rhamnose, a process for its preparation and its use |
-
2000
- 2000-02-11 DE DE10006147A patent/DE10006147A1/de not_active Withdrawn
-
2001
- 2001-02-09 WO PCT/EP2001/001447 patent/WO2001059143A1/de not_active Application Discontinuation
- 2001-02-09 EP EP01903733A patent/EP1259632A1/de not_active Withdrawn
- 2001-02-09 AU AU2001231726A patent/AU2001231726A1/en not_active Abandoned
- 2001-02-09 BR BR0108273-6A patent/BR0108273A/pt not_active IP Right Cessation
- 2001-02-09 JP JP2001558479A patent/JP2003522532A/ja active Pending
- 2001-02-09 CN CN01806409A patent/CN1416470A/zh active Pending
- 2001-02-09 KR KR1020027010449A patent/KR20030013371A/ko active IP Right Grant
- 2001-02-09 CA CA002400014A patent/CA2400014A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0317033A1 (de) * | 1987-11-20 | 1989-05-24 | Unilever N.V. | Verfahren zur Herstellung von L-Rhamnose |
| US4971812A (en) * | 1989-08-31 | 1990-11-20 | National Science Council | Immobilized penicillium sp. naringnase and its use in removing naringin and limonin from fruit juice |
| US5641659A (en) * | 1992-11-27 | 1997-06-24 | Hoechst Aktiengesellschaft | α-l-rhamnosidase for obtaining rhamnose, a process for its preparation and its use |
Non-Patent Citations (8)
| Title |
|---|
| APPL. BIOCHEM. BIOTECHNOL. (1986), 13(1), 1-13 * |
| BIOTECHNOLOGY TECHNIQUES, vol. 12, no. 1, January 1998 (1998-01-01), pages 63 - 65, ISSN: 0951-208X * |
| DATABASE BIOSIS [online] BIOSCIENCES INFORMATION SERVICE, PHILADELPHIA, PA, US; 1989, TSEN H-Y ET AL: "FIBER ENTRAPMENT OF NARINGINASE FROM PENICILLIUM-SP AND APPLICATION TO FRUIT JUICE DEBITTERING", XP002168066, Database accession no. PREV198987114493 * |
| DATABASE BIOSIS [online] BIOSCIENCES INFORMATION SERVICE, PHILADELPHIA, PA, US; January 1998 (1998-01-01), ELLENRIEDER G ET AL: "Hydrolysis of supersaturated naringin solutions by free and immobilized naringinase.", XP002168067, Database accession no. PREV199800134334 * |
| DATABASE CHEMABS [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; TURECEK, P. ET AL: "Simple enzyme reactors suitable for the byproduct-free preparation of the aglycones of naturally occurring glycosides under mild conditions", XP002168068, retrieved from STN Database accession no. 105:95983 * |
| DATABASE CHEMABS [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; TURECEK, P. L. ET AL: "Applications of enzyme immobilization in the analysis of naturally occurring compounds, for example,.alpha.-L-rhamnosides", XP002168065, retrieved from STN Database accession no. 109:34782 * |
| JOURNAL OF FERMENTATION AND BIOENGINEERING, vol. 67, no. 3, 1989, pages 186 - 189, ISSN: 0922-338X * |
| SCI. PHARM. (1987), 55(4), 275-83 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004027074A2 (en) * | 2002-09-23 | 2004-04-01 | Her Majesty The Queen In Right Of Canada, As Represented By The Minister Of Agriculture | Extraction, purification and conversion of flavonoids from plant biomass |
| WO2004027074A3 (en) * | 2002-09-23 | 2004-09-23 | Ca Minister Agriculture & Food | Extraction, purification and conversion of flavonoids from plant biomass |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2001231726A1 (en) | 2001-08-20 |
| KR20030013371A (ko) | 2003-02-14 |
| JP2003522532A (ja) | 2003-07-29 |
| CN1416470A (zh) | 2003-05-07 |
| EP1259632A1 (de) | 2002-11-27 |
| CA2400014A1 (en) | 2001-08-16 |
| BR0108273A (pt) | 2003-03-05 |
| DE10006147A1 (de) | 2001-08-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE2413220C3 (de) | Selektiv adsorbierende Partikel und deren Verwendung zum Trennen von organischen Verbindungen | |
| DE69420390T2 (de) | Hydrolyse von Kaffee mit immobilisierter Beta-Mannanase | |
| EP1259632A1 (de) | Verfahren zur herstellung von monoglycosidierten flavonoiden | |
| DE2717965A1 (de) | Poroese zelluloseperlen | |
| DE3017861C2 (de) | Verfahren zur enzymatischen Herstellung von L-Tryptophan | |
| DE69311134T2 (de) | Bioreaktor mit immobilisierten milchsäurebakterien und seine verwendung | |
| DE2519382A1 (de) | Verfahren zur gewinnung einer gereinigten alpha-galactosidase aus einer alpha-galactosidase enthaltenden fluessigkeit | |
| EP1124981B1 (de) | Verfahren zur enzymatischen spaltung von rutinosiden | |
| DE19912799B4 (de) | Superparamagnetisches Adsorptionsmaterial und seine Verwendung | |
| DE19823332C2 (de) | Verfahren zur enzymatischen Herstellung von Betalactam-Antibiotika | |
| DE1966427C3 (de) | Verfahren zur Herstellung von 6-AminopenicUlansäure | |
| US20030157653A1 (en) | Method for producing monoglycosidated flavonoids | |
| DE3922278C2 (de) | Verfahren zur herstellung von freiem (epsilon)-polylysin | |
| CN110791537A (zh) | 一种制备异烟酸的方法 | |
| JP2717227B2 (ja) | 固定化ウレアーゼとそれを用いた酒類の製造方法 | |
| CN115490738B (zh) | 一种槐糖脂的脱色方法 | |
| EP0259751A2 (de) | Annomycin, ein Antibiotikum; mikrobiologisches Verfahren zu dessen Herstellung und seine Verwendung | |
| CN105384678A (zh) | 一种发酵液提取l-色氨酸的方法 | |
| RU2114173C1 (ru) | Способ получения кристаллического тилозина | |
| DE102005015447A1 (de) | Verfahren zur Spaltung von Rutinosiden | |
| DE19821038A1 (de) | Neue Glykoside des Acarviosins, Synthese eines neuen Saccharase-Inhibitors durch Biotransformation der Acarbose | |
| JP2690779B2 (ja) | L―アスコルビン酸誘導体及びその製造法 | |
| SU1255642A1 (ru) | Способ получени раствора полисахарида из бактериальной суспензии | |
| EP2021456A2 (de) | Verfahren zur submersen kultivierung von filamentös wachsenden organismen | |
| EP2762561A1 (de) | Verfahren zur Herstellung von Nanobiokatalysatoren |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2400014 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2001 558479 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020027010449 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001903733 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 018064094 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001903733 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10203537 Country of ref document: US |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020027010449 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020027010449 Country of ref document: KR |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2001903733 Country of ref document: EP |