TW202100534A - 矽烷化合物之製造方法 - Google Patents
矽烷化合物之製造方法 Download PDFInfo
- Publication number
- TW202100534A TW202100534A TW109109215A TW109109215A TW202100534A TW 202100534 A TW202100534 A TW 202100534A TW 109109215 A TW109109215 A TW 109109215A TW 109109215 A TW109109215 A TW 109109215A TW 202100534 A TW202100534 A TW 202100534A
- Authority
- TW
- Taiwan
- Prior art keywords
- compound
- siloxane
- carbonate
- aforementioned
- producing
- Prior art date
Links
- -1 silane compound Chemical class 0.000 title claims abstract description 183
- 229910000077 silane Inorganic materials 0.000 title claims abstract description 64
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 62
- 150000001875 compounds Chemical class 0.000 claims abstract description 97
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 claims abstract description 56
- 238000000354 decomposition reaction Methods 0.000 claims abstract description 48
- 239000003054 catalyst Substances 0.000 claims abstract description 41
- 239000000203 mixture Substances 0.000 claims abstract description 28
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims abstract description 22
- 150000007514 bases Chemical class 0.000 claims abstract description 21
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 46
- 125000001424 substituent group Chemical group 0.000 claims description 38
- ROORDVPLFPIABK-UHFFFAOYSA-N diphenyl carbonate Chemical compound C=1C=CC=CC=1OC(=O)OC1=CC=CC=C1 ROORDVPLFPIABK-UHFFFAOYSA-N 0.000 claims description 36
- 229920001296 polysiloxane Polymers 0.000 claims description 34
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 27
- 229910052799 carbon Inorganic materials 0.000 claims description 27
- 125000003118 aryl group Chemical group 0.000 claims description 23
- 125000000217 alkyl group Chemical group 0.000 claims description 19
- 125000003342 alkenyl group Chemical group 0.000 claims description 18
- 238000004821 distillation Methods 0.000 claims description 16
- 229910052757 nitrogen Inorganic materials 0.000 claims description 14
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 13
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 13
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 13
- 238000001953 recrystallisation Methods 0.000 claims description 13
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 9
- 125000003277 amino group Chemical group 0.000 claims description 8
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 claims description 8
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 7
- 125000003545 alkoxy group Chemical group 0.000 claims description 6
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 6
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 6
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 6
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 5
- 229920002554 vinyl polymer Polymers 0.000 claims description 5
- 150000008044 alkali metal hydroxides Chemical class 0.000 claims description 4
- 229910000288 alkali metal carbonate Inorganic materials 0.000 claims description 3
- 150000008041 alkali metal carbonates Chemical class 0.000 claims description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 3
- 239000003513 alkali Substances 0.000 claims description 2
- 125000001183 hydrocarbyl group Chemical group 0.000 claims 2
- 230000001568 sexual effect Effects 0.000 claims 1
- 230000007613 environmental effect Effects 0.000 abstract description 5
- 238000010438 heat treatment Methods 0.000 abstract description 4
- 239000003518 caustics Substances 0.000 abstract description 3
- 239000003960 organic solvent Substances 0.000 abstract description 3
- 239000002253 acid Substances 0.000 abstract description 2
- 150000007513 acids Chemical class 0.000 abstract 1
- XMSXQFUHVRWGNA-UHFFFAOYSA-N Decamethylcyclopentasiloxane Chemical compound C[Si]1(C)O[Si](C)(C)O[Si](C)(C)O[Si](C)(C)O[Si](C)(C)O1 XMSXQFUHVRWGNA-UHFFFAOYSA-N 0.000 description 61
- SWLVAJXQIOKFSJ-UHFFFAOYSA-N dimethyl(diphenoxy)silane Chemical compound C=1C=CC=CC=1O[Si](C)(C)OC1=CC=CC=C1 SWLVAJXQIOKFSJ-UHFFFAOYSA-N 0.000 description 58
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 50
- 229910052710 silicon Inorganic materials 0.000 description 43
- 239000010703 silicon Substances 0.000 description 43
- 238000006243 chemical reaction Methods 0.000 description 38
- 238000005160 1H NMR spectroscopy Methods 0.000 description 36
- 239000011541 reaction mixture Substances 0.000 description 32
- 238000004458 analytical method Methods 0.000 description 29
- RJPCFFMEJWSVLK-UHFFFAOYSA-N [dimethyl(phenoxy)silyl]oxy-dimethyl-phenoxysilane Chemical compound C=1C=CC=CC=1O[Si](C)(C)O[Si](C)(C)OC1=CC=CC=C1 RJPCFFMEJWSVLK-UHFFFAOYSA-N 0.000 description 20
- 239000012299 nitrogen atmosphere Substances 0.000 description 19
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 18
- VSIKJPJINIDELZ-UHFFFAOYSA-N 2,2,4,4,6,6,8,8-octakis-phenyl-1,3,5,7,2,4,6,8-tetraoxatetrasilocane Chemical compound O1[Si](C=2C=CC=CC=2)(C=2C=CC=CC=2)O[Si](C=2C=CC=CC=2)(C=2C=CC=CC=2)O[Si](C=2C=CC=CC=2)(C=2C=CC=CC=2)O[Si]1(C=1C=CC=CC=1)C1=CC=CC=C1 VSIKJPJINIDELZ-UHFFFAOYSA-N 0.000 description 13
- 239000000843 powder Substances 0.000 description 13
- 238000000034 method Methods 0.000 description 10
- 150000004756 silanes Chemical class 0.000 description 10
- 239000000047 product Substances 0.000 description 9
- 239000013078 crystal Substances 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 8
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- 239000004205 dimethyl polysiloxane Substances 0.000 description 7
- 235000013870 dimethyl polysiloxane Nutrition 0.000 description 7
- YLDKQPMORVEBRU-UHFFFAOYSA-N diphenoxy(diphenyl)silane Chemical compound C=1C=CC=CC=1O[Si](C=1C=CC=CC=1)(C=1C=CC=CC=1)OC1=CC=CC=C1 YLDKQPMORVEBRU-UHFFFAOYSA-N 0.000 description 7
- JULIZFWJALMDGQ-UHFFFAOYSA-N phenoxy-[phenoxy(diphenyl)silyl]oxy-diphenylsilane Chemical compound C=1C=CC=CC=1O[Si](C=1C=CC=CC=1)(C=1C=CC=CC=1)O[Si](C=1C=CC=CC=1)(C=1C=CC=CC=1)OC1=CC=CC=C1 JULIZFWJALMDGQ-UHFFFAOYSA-N 0.000 description 7
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 7
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 125000004122 cyclic group Chemical group 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- 239000002994 raw material Substances 0.000 description 5
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 4
- 230000000052 comparative effect Effects 0.000 description 4
- 125000004093 cyano group Chemical group *C#N 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 150000002430 hydrocarbons Chemical group 0.000 description 4
- DRXHEPWCWBIQFJ-UHFFFAOYSA-N methyl(triphenoxy)silane Chemical compound C=1C=CC=CC=1O[Si](OC=1C=CC=CC=1)(C)OC1=CC=CC=C1 DRXHEPWCWBIQFJ-UHFFFAOYSA-N 0.000 description 4
- 125000003396 thiol group Chemical group [H]S* 0.000 description 4
- IXJNGXCZSCHDFE-UHFFFAOYSA-N triphenoxy(phenyl)silane Chemical compound C=1C=CC=CC=1O[Si](C=1C=CC=CC=1)(OC=1C=CC=CC=1)OC1=CC=CC=C1 IXJNGXCZSCHDFE-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 125000005843 halogen group Chemical group 0.000 description 3
- HTDJPCNNEPUOOQ-UHFFFAOYSA-N hexamethylcyclotrisiloxane Chemical compound C[Si]1(C)O[Si](C)(C)O[Si](C)(C)O1 HTDJPCNNEPUOOQ-UHFFFAOYSA-N 0.000 description 3
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 3
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- HMMGMWAXVFQUOA-UHFFFAOYSA-N octamethylcyclotetrasiloxane Chemical compound C[Si]1(C)O[Si](C)(C)O[Si](C)(C)O[Si](C)(C)O1 HMMGMWAXVFQUOA-UHFFFAOYSA-N 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 238000005292 vacuum distillation Methods 0.000 description 3
- MFGOFGRYDNHJTA-UHFFFAOYSA-N 2-amino-1-(2-fluorophenyl)ethanol Chemical compound NCC(O)C1=CC=CC=C1F MFGOFGRYDNHJTA-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- JPMDJMXICBMIIB-UHFFFAOYSA-N [diphenoxy(phenyl)silyl]oxy-diphenoxy-phenylsilane Chemical compound C=1C=CC=CC=1O[Si](C=1C=CC=CC=1)(O[Si](OC=1C=CC=CC=1)(OC=1C=CC=CC=1)C=1C=CC=CC=1)OC1=CC=CC=C1 JPMDJMXICBMIIB-UHFFFAOYSA-N 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 150000001341 alkaline earth metal compounds Chemical class 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- HUCVOHYBFXVBRW-UHFFFAOYSA-M caesium hydroxide Inorganic materials [OH-].[Cs+] HUCVOHYBFXVBRW-UHFFFAOYSA-M 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- GGTRGAJZTXLYAO-UHFFFAOYSA-N methyl-[methyl(diphenoxy)silyl]oxy-diphenoxysilane Chemical compound C=1C=CC=CC=1O[Si](O[Si](C)(OC=1C=CC=CC=1)OC=1C=CC=CC=1)(C)OC1=CC=CC=C1 GGTRGAJZTXLYAO-UHFFFAOYSA-N 0.000 description 2
- 150000002989 phenols Chemical class 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- 125000005017 substituted alkenyl group Chemical group 0.000 description 2
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 1
- 229910002808 Si–O–Si Inorganic materials 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001339 alkali metal compounds Chemical class 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000004703 alkoxides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- 125000005587 carbonate group Chemical group 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 230000006837 decompression Effects 0.000 description 1
- GQNWJCQWBFHQAO-UHFFFAOYSA-N dibutoxy(dimethyl)silane Chemical compound CCCCO[Si](C)(C)OCCCC GQNWJCQWBFHQAO-UHFFFAOYSA-N 0.000 description 1
- QLVWOKQMDLQXNN-UHFFFAOYSA-N dibutyl carbonate Chemical compound CCCCOC(=O)OCCCC QLVWOKQMDLQXNN-UHFFFAOYSA-N 0.000 description 1
- MROCJMGDEKINLD-UHFFFAOYSA-N dichlorosilane Chemical class Cl[SiH2]Cl MROCJMGDEKINLD-UHFFFAOYSA-N 0.000 description 1
- ZXPDYFSTVHQQOI-UHFFFAOYSA-N diethoxysilane Chemical class CCO[SiH2]OCC ZXPDYFSTVHQQOI-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- LIKFHECYJZWXFJ-UHFFFAOYSA-N dimethyldichlorosilane Chemical compound C[Si](C)(Cl)Cl LIKFHECYJZWXFJ-UHFFFAOYSA-N 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000005055 methyl trichlorosilane Substances 0.000 description 1
- JLUFWMXJHAVVNN-UHFFFAOYSA-N methyltrichlorosilane Chemical compound C[Si](Cl)(Cl)Cl JLUFWMXJHAVVNN-UHFFFAOYSA-N 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 238000009790 rate-determining step (RDS) Methods 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- ZDHXKXAHOVTTAH-UHFFFAOYSA-N trichlorosilane Chemical class Cl[SiH](Cl)Cl ZDHXKXAHOVTTAH-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
- C07F7/1872—Preparation; Treatments not provided for in C07F7/20
- C07F7/188—Preparation; Treatments not provided for in C07F7/20 by reactions involving the formation of Si-O linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0834—Compounds having one or more O-Si linkage
- C07F7/0838—Compounds with one or more Si-O-Si sequences
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Silicon Polymers (AREA)
Abstract
本發明提供一種不伴隨酸等腐蝕性物質的產生,且不使用有機溶劑之與減低環境負荷相關連之矽烷化合物的以下之製造方法。
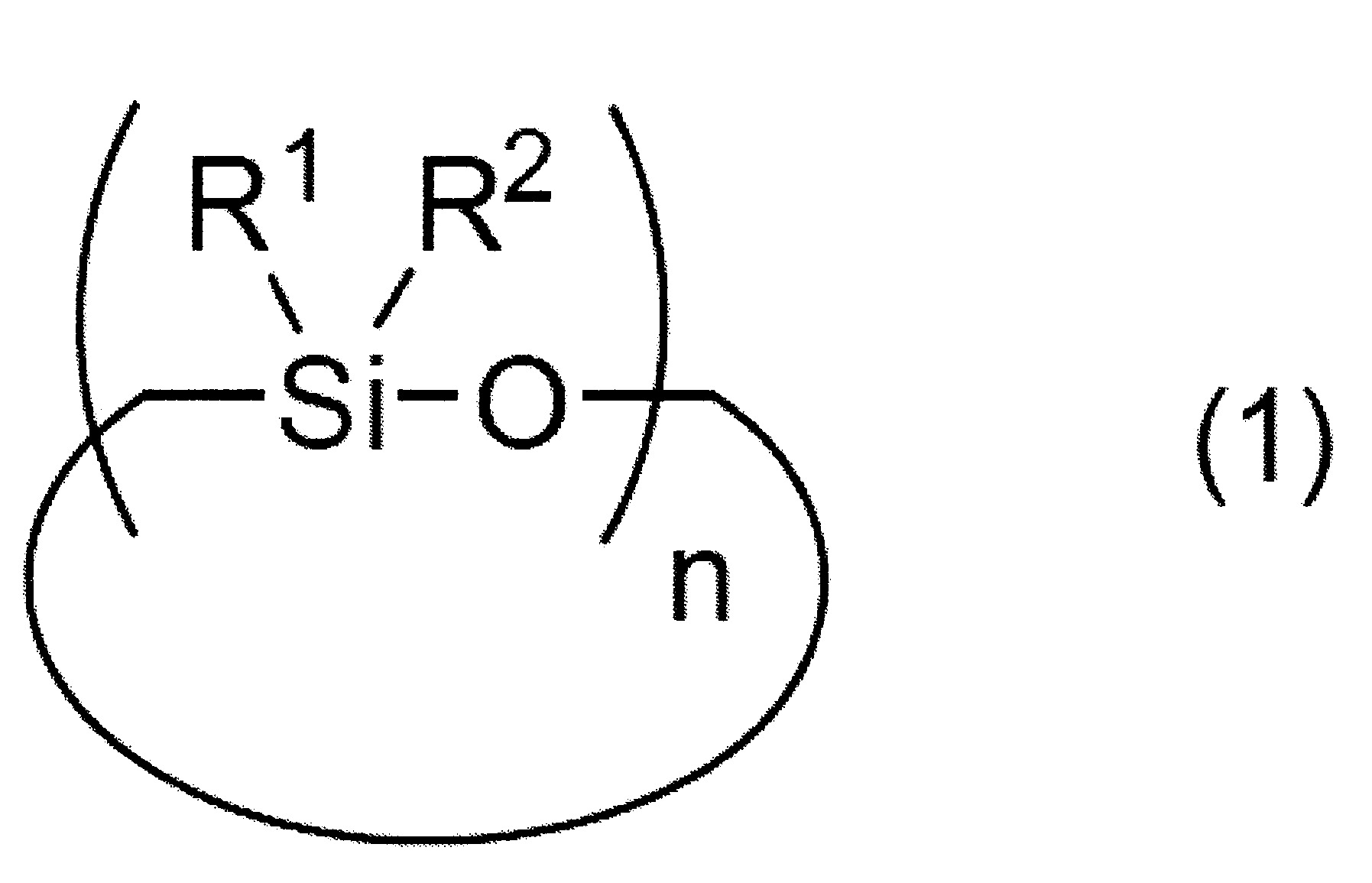
該矽烷化合物之製造方法係具有下述D)矽氧烷分解步驟:該D)矽氧烷分解步驟係將含有A)矽氧烷化合物,其係下述式(1)表示之A-1)環狀矽氧烷化合物、下述式(2)表示之A-2)直鏈狀矽氧烷化合物及/或將矽氧烷鍵作為主鏈骨架之下述式(3)表示之倍半矽氧烷化合物;B)碳酸酯化合物,其包含二芳基碳酸酯、二烷基碳酸酯及單烷基單芳基碳酸酯之至少任一者;以及C)鹼性化合物觸媒之混合物進行加熱,而使A)之矽氧烷化合物烷氧基化及/或芳氧基化,
 (式中,R1
~R5
、X、n、m及p係如同本案說明書所記載)。
(式中,R1
~R5
、X、n、m及p係如同本案說明書所記載)。
Description
本發明係關於一種矽烷化合物之製造方法,尤其是芳氧基矽烷化合物、烷氧基矽烷化合物等的矽烷化合物之製造方法。
作為二芳氧基矽烷化合物、二烷氧基矽烷化合物等的矽烷化合物之製造方法,一般已知有對於以二氯矽烷化合物為代表之二鹵化矽烷化合物及以二乙醯氧基矽烷化合物為代表之二醯氧基矽烷化合物,使苯酚衍生物或各種醇化合物進行反應的手法。
例如,專利文獻1中揭示了來自二甲基二氯矽烷與苯酚的二甲基二苯氧基矽烷之製造方法。本手法中,需要將與產生二甲基二苯氧基矽烷的同時副生成之氯化氫氣體進行處理之步驟、對進行蒸餾純化前之產物使用鹼進行洗淨之洗淨步驟、使用有機溶劑之萃取步驟等之各種的步驟。進而,廢液處理步驟亦為必要之專利文獻1所記載之上述製程,環境負荷大。
又,專利文獻2中雖揭示了由聚二甲基矽氧烷與苯酚獲得二甲基二苯氧基矽烷之製造方法,但本反應中亦使用溶劑,並且不能得到充分的產率。
作為三芳氧基矽烷化合物、三烷氧基矽烷化合物等的矽烷化合物之製造方法,一般已知有對於以三氯矽烷化合物為代表之三鹵化矽烷化合物,使苯酚衍生物或各種醇化合物進行反應的手法。
例如,專利文獻3中揭示了由甲基三氯矽烷與苯酚獲得甲基三苯氧基矽烷之製造方法。由於此反應為平衡反應,因此如何將副生成之氯化氫去除至反應系外成為反應的速率決定步驟。作為目標物之三芳氧基矽烷化合物亦與上述二芳氧基矽烷化合物同樣地,在酸性水溶液中不安定的情況多,為了抑制水解反應或縮合反應,去除氯化氫之步驟是必要的。
如上所述,在過往之製法中,並不能說安全且有效率地製造矽烷化合物一定是容易的。
[先前技術文獻]
[專利文獻]
[專利文獻1] US2012/184702號公報
[專利文獻2] 日本特開2008-297227號公報
[專利文獻3] 日本特開平10-218885號公報
[發明欲解決之課題]
提供一種不伴隨鹽酸或醋酸等腐蝕性物質的產生,且不使用有機溶劑之與減低環境負荷相關連之二芳氧基矽烷化合物、二烷氧基矽烷化合物、三芳氧基矽烷化合物、三烷氧基矽烷化合物等的矽烷化合物之製造方法。
[用以解決課題之手段]
本發明係使環境負荷減低並且可有效率地生成目標化合物之以下所記載的矽烷化合物之製造方法。
[1] 一種矽烷化合物之製造方法,其係包含二芳氧基矽烷化合物、二烷氧基矽烷化合物、單芳氧基單烷氧基矽烷化合物、三芳氧基矽烷化合物及三烷氧基矽烷化合物之至少任一者的矽烷化合物之製造方法,其具有下述D)矽氧烷分解步驟:
該D)矽氧烷分解步驟係將含有
A)矽氧烷化合物,其係下述式(1)表示之A-1)環狀矽氧烷化合物、下述式(2)表示之A-2)直鏈狀矽氧烷化合物及/或將矽氧烷鍵作為主鏈骨架之下述式(3)表示之倍半矽氧烷化合物;
B)碳酸酯化合物,其包含二芳基碳酸酯、二烷基碳酸酯及單烷基單芳基碳酸酯之至少任一者;以及
C)鹼性化合物觸媒
之混合物進行加熱,而使前述A)之矽氧烷化合物烷氧基化及/或芳氧基化,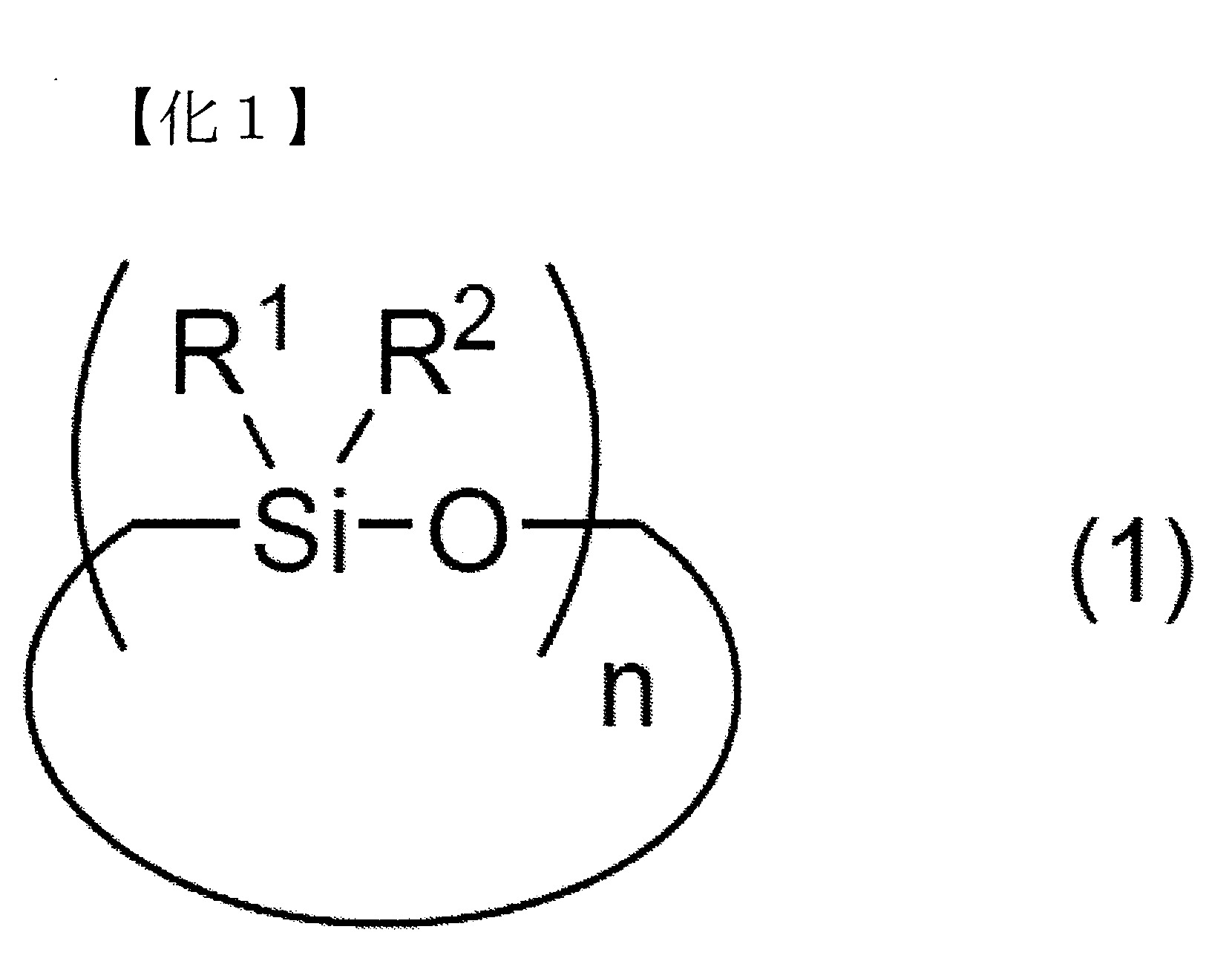 (式中,R1
及R2
各自獨立地表示可具有取代基之烷基、烯基或芳基,
n表示3以上30以下之整數)
(式中,R1
及R2
各自獨立地表示可具有取代基之烷基、烯基或芳基,
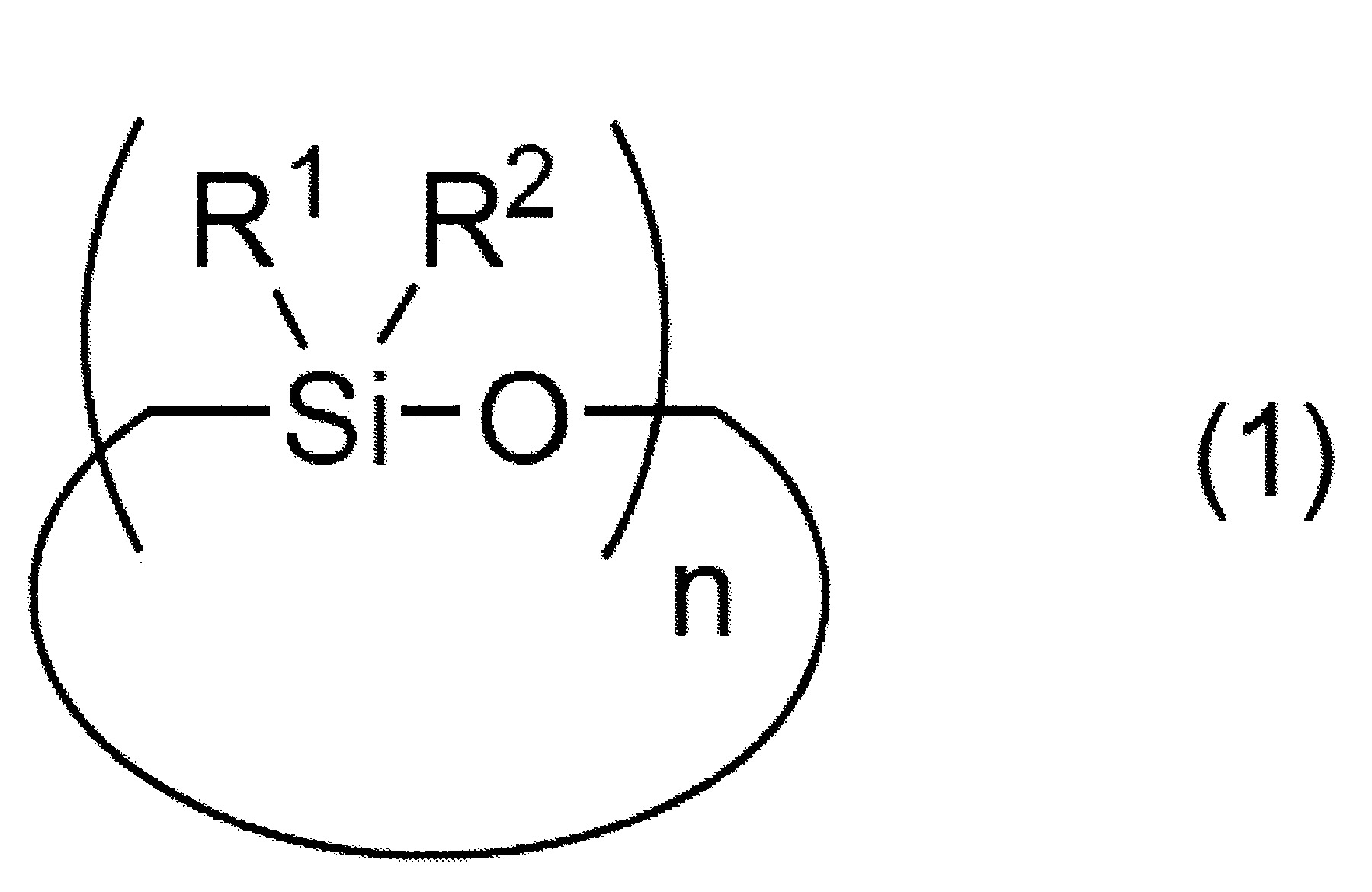
n表示3以上30以下之整數) (式中,R3
及R4
各自獨立地表示可具有取代基之烷基、烯基或芳基,
m表示2以上10000以下之整數,
X各自獨立地表示氫原子、羥基、可具有取代基之合計碳數1~10之烷氧基、可具有取代基且可具有氧原子或氮原子之合計碳數1~10之烴基、或可具有取代基之胺基)
(式中,R3
及R4
各自獨立地表示可具有取代基之烷基、烯基或芳基,
m表示2以上10000以下之整數,
X各自獨立地表示氫原子、羥基、可具有取代基之合計碳數1~10之烷氧基、可具有取代基且可具有氧原子或氮原子之合計碳數1~10之烴基、或可具有取代基之胺基)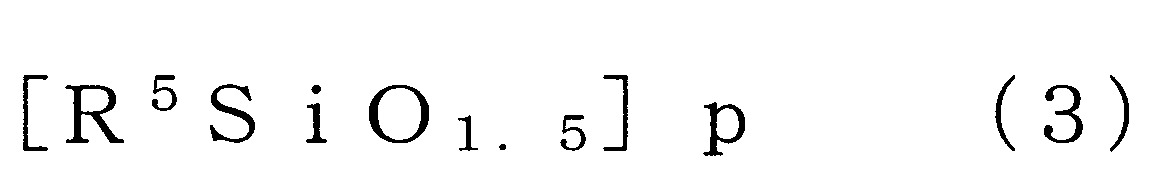 (式中,R5
表示可具有取代基之碳數1~4之烷基、碳數2~4之烯基或碳數6~12之芳基,
p為4以上24以下之數)。
[2] 一種矽烷化合物之製造方法,其係包含二芳氧基矽烷化合物、二烷氧基矽烷化合物及單芳氧基單烷氧基矽烷化合物之至少任一者的矽烷化合物之製造方法,其具有下述D)矽氧烷分解步驟:
該D)矽氧烷分解步驟係將含有
A)下述式(1)表示之A-1)環狀矽氧烷化合物及/或下述式(2)表示之A-2)直鏈狀矽氧烷化合物;
B)碳酸酯化合物,其包含二芳基碳酸酯、二烷基碳酸酯及單烷基單芳基碳酸酯之至少任一者;以及
C)鹼性化合物觸媒
之混合物進行加熱,而使前述A)之矽氧烷化合物烷氧基化及/或芳氧基化,
(式中,R5
表示可具有取代基之碳數1~4之烷基、碳數2~4之烯基或碳數6~12之芳基,
p為4以上24以下之數)。
[2] 一種矽烷化合物之製造方法,其係包含二芳氧基矽烷化合物、二烷氧基矽烷化合物及單芳氧基單烷氧基矽烷化合物之至少任一者的矽烷化合物之製造方法,其具有下述D)矽氧烷分解步驟:
該D)矽氧烷分解步驟係將含有
A)下述式(1)表示之A-1)環狀矽氧烷化合物及/或下述式(2)表示之A-2)直鏈狀矽氧烷化合物;
B)碳酸酯化合物,其包含二芳基碳酸酯、二烷基碳酸酯及單烷基單芳基碳酸酯之至少任一者;以及
C)鹼性化合物觸媒
之混合物進行加熱,而使前述A)之矽氧烷化合物烷氧基化及/或芳氧基化, (式中,R1
及R2
各自獨立地表示可具有取代基之烷基、烯基或芳基,
n表示3以上30以下之整數)
(式中,R1
及R2
各自獨立地表示可具有取代基之烷基、烯基或芳基,
n表示3以上30以下之整數)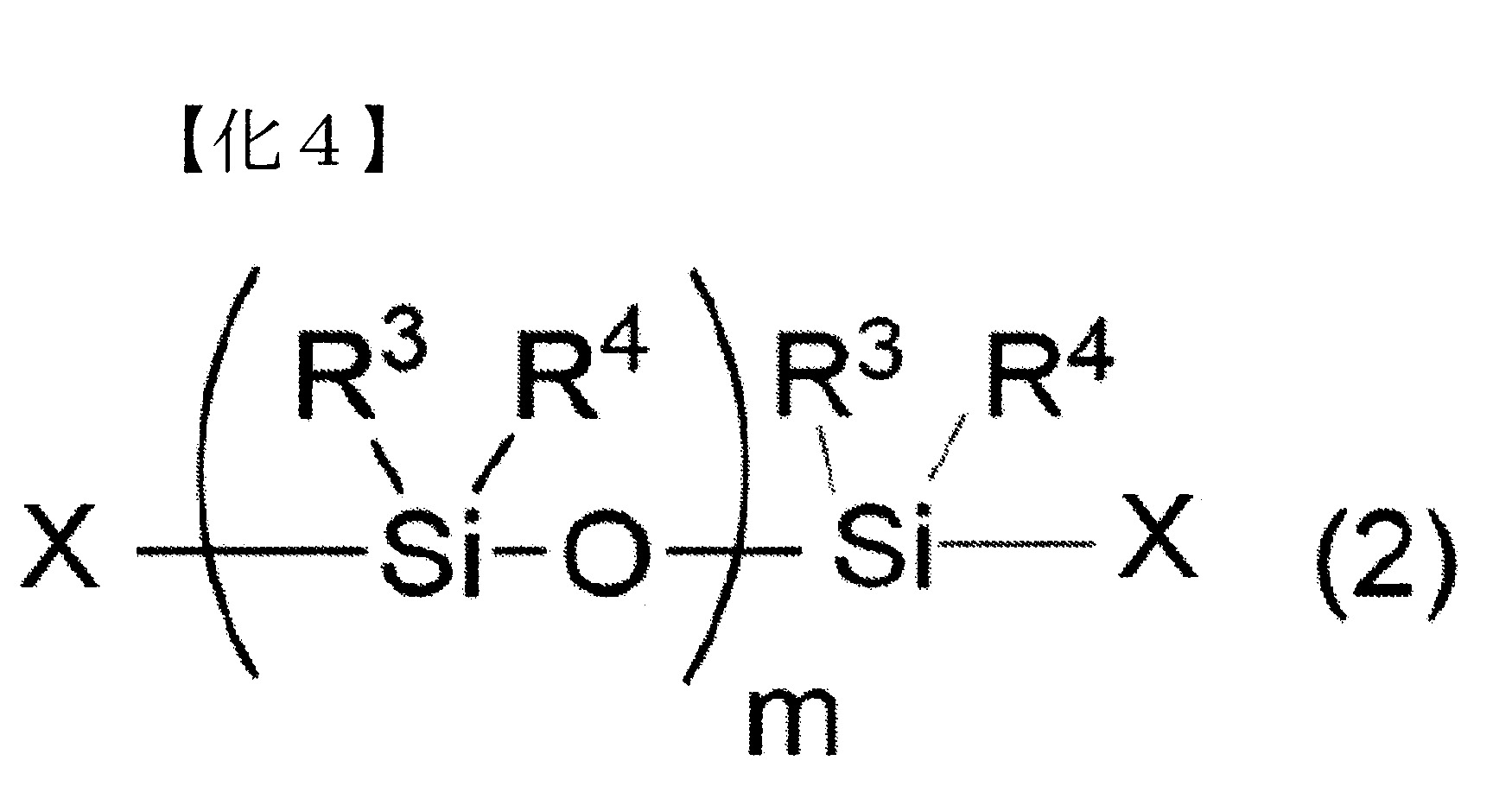 (式中,R3
及R4
各自獨立地表示可具有取代基之烷基、烯基或芳基,
m表示2以上10000以下之整數,
X各自獨立地表示氫原子、羥基、可具有取代基之合計碳數1~10之烷氧基、可具有取代基且可具有氧原子或氮原子之合計碳數1~10之烴基、或可具有取代基之胺基)。
[3] 如上述[1]或[2]之矽烷化合物之製造方法,其中,前述R1
~R4
各自獨立地表示可具有取代基之合計碳數1~8之烷基、烯基或合計碳數6~30之芳基。
[4] 如上述[3]之矽烷化合物之製造方法,其中,前述R1
~R4
各自獨立地為選自由甲基、苯基、乙烯基及丙基所成群中之任一種。
[5] 如上述[1]~[4]中任一項之矽烷化合物之製造方法,其中,前述B)二芳基碳酸酯包含二苯基碳酸酯。
[6] 如上述[1]~[4]中任一項之矽烷化合物之製造方法,其中,前述B)二烷基碳酸酯中之烷基的碳數為4以下。
[7] 如上述[1]~[6]中任一項之矽烷化合物之製造方法,其中,前述C)鹼性化合物觸媒包含鹼金屬碳酸鹽或鹼金屬氫氧化物。
[8] 如上述[7]之矽烷化合物之製造方法,其中,前述C)鹼性化合物觸媒包含碳酸銫及碳酸鉀之至少任一者。
[9] 如上述[1]~[8]中任一項之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,前述B)碳酸酯化合物的莫耳量相對於前述A)矽氧烷化合物的Si莫耳量之比x為x≧1。
[10] 如上述[1]~[8]中任一項之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,前述B)碳酸酯化合物的莫耳量相對於前述A)矽氧烷化合物的前述Si莫耳量之比x為0.8<x<1.6。
[11] 如上述[1]~[10]中任一項之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,前述C)鹼性化合物觸媒的莫耳量相對於前述A)矽氧烷化合物的前述Si莫耳量之比y為0.0001 mmol/mol≦y≦20 mmol/mol。
[12] 如上述[1]~[11]中任一項之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,使前述矽氧烷化合物分解之溫度為50℃以上300℃以下。
[13] 如上述[12]之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,前述溫度為50℃以上150℃以下。
[14] 如上述[1]之矽烷化合物之製造方法,其中,前述式(1)表示之前述A-1)環狀矽氧烷化合物之分子量為2,000以下,前述式(2)表示之前述A-2)直鏈狀矽氧烷化合物之分子量為60,000以下,前述式(3)表示之前述A-3)倍半矽氧烷化合物之分子量為3,500以下。
[15] 如上述[1]~[14]中任一項之矽烷化合物之製造方法,其進而具有E)蒸餾步驟/再結晶步驟,
該E)蒸餾步驟/再結晶步驟係,蒸餾由前述D)矽氧烷分解步驟所生成之前述矽烷化合物之蒸餾步驟,與再結晶由前述D)矽氧烷分解步驟所生成之前述矽烷化合物之再結晶步驟的至少任一者。
[16] 如上述[15]之矽烷化合物之製造方法,其中,前述E)之前述蒸餾步驟中之壓力為1hPa以上20hPa以下。
[17] 如上述[1]~[16]中任一項之矽烷化合物之製造方法,其中,進而具有對含有前述B)碳酸酯化合物及前述C)鹼性化合物觸媒之混合物,滴下前述A)矽氧烷化合物之F)滴下步驟。
[發明的效果]
(式中,R3
及R4
各自獨立地表示可具有取代基之烷基、烯基或芳基,
m表示2以上10000以下之整數,
X各自獨立地表示氫原子、羥基、可具有取代基之合計碳數1~10之烷氧基、可具有取代基且可具有氧原子或氮原子之合計碳數1~10之烴基、或可具有取代基之胺基)。
[3] 如上述[1]或[2]之矽烷化合物之製造方法,其中,前述R1
~R4
各自獨立地表示可具有取代基之合計碳數1~8之烷基、烯基或合計碳數6~30之芳基。
[4] 如上述[3]之矽烷化合物之製造方法,其中,前述R1
~R4
各自獨立地為選自由甲基、苯基、乙烯基及丙基所成群中之任一種。
[5] 如上述[1]~[4]中任一項之矽烷化合物之製造方法,其中,前述B)二芳基碳酸酯包含二苯基碳酸酯。
[6] 如上述[1]~[4]中任一項之矽烷化合物之製造方法,其中,前述B)二烷基碳酸酯中之烷基的碳數為4以下。
[7] 如上述[1]~[6]中任一項之矽烷化合物之製造方法,其中,前述C)鹼性化合物觸媒包含鹼金屬碳酸鹽或鹼金屬氫氧化物。
[8] 如上述[7]之矽烷化合物之製造方法,其中,前述C)鹼性化合物觸媒包含碳酸銫及碳酸鉀之至少任一者。
[9] 如上述[1]~[8]中任一項之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,前述B)碳酸酯化合物的莫耳量相對於前述A)矽氧烷化合物的Si莫耳量之比x為x≧1。
[10] 如上述[1]~[8]中任一項之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,前述B)碳酸酯化合物的莫耳量相對於前述A)矽氧烷化合物的前述Si莫耳量之比x為0.8<x<1.6。
[11] 如上述[1]~[10]中任一項之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,前述C)鹼性化合物觸媒的莫耳量相對於前述A)矽氧烷化合物的前述Si莫耳量之比y為0.0001 mmol/mol≦y≦20 mmol/mol。
[12] 如上述[1]~[11]中任一項之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,使前述矽氧烷化合物分解之溫度為50℃以上300℃以下。
[13] 如上述[12]之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,前述溫度為50℃以上150℃以下。
[14] 如上述[1]之矽烷化合物之製造方法,其中,前述式(1)表示之前述A-1)環狀矽氧烷化合物之分子量為2,000以下,前述式(2)表示之前述A-2)直鏈狀矽氧烷化合物之分子量為60,000以下,前述式(3)表示之前述A-3)倍半矽氧烷化合物之分子量為3,500以下。
[15] 如上述[1]~[14]中任一項之矽烷化合物之製造方法,其進而具有E)蒸餾步驟/再結晶步驟,
該E)蒸餾步驟/再結晶步驟係,蒸餾由前述D)矽氧烷分解步驟所生成之前述矽烷化合物之蒸餾步驟,與再結晶由前述D)矽氧烷分解步驟所生成之前述矽烷化合物之再結晶步驟的至少任一者。
[16] 如上述[15]之矽烷化合物之製造方法,其中,前述E)之前述蒸餾步驟中之壓力為1hPa以上20hPa以下。
[17] 如上述[1]~[16]中任一項之矽烷化合物之製造方法,其中,進而具有對含有前述B)碳酸酯化合物及前述C)鹼性化合物觸媒之混合物,滴下前述A)矽氧烷化合物之F)滴下步驟。
[發明的效果]
依據本發明之矽烷化合物之製造方法,不會副生成酸等之腐蝕性物質,且可抑制必要之步驟數而有效率地生成矽烷化合物。因此,依據本發明,可提供一種減低環境負荷,並且有效率地製造目標之矽烷化合物的方法。
本發明之矽烷化合物之製造方法具有下述矽氧烷分解步驟:
該矽氧烷分解步驟係將含有
A)詳如後述之環狀矽氧烷化合物、直鏈狀矽氧烷化合物及/或倍半矽氧烷化合物;
B)碳酸酯化合物,其包含二芳基碳酸酯、二烷基碳酸酯及單烷基單芳基碳酸酯之至少任一者;
C)鹼性化合物觸媒
之混合物進行加熱,而使前述A)之矽氧烷化合物烷氧基化及/或芳氧基化。
依據本發明,可製造二芳氧基矽烷化合物、二烷氧基矽烷化合物、單芳氧基單烷氧基矽烷化合物、及三芳氧基矽烷化合物、三烷氧基矽烷化合物之至少任一者的矽烷化合物。
以下,詳細地說明關於本發明之矽烷化合物之製造方法。
[A)矽氧烷化合物]
A-1)環狀矽氧烷化合物
於矽氧烷分解步驟所使用的矽氧烷化合物之中,環狀矽氧烷化合物係以下述式(1)表示。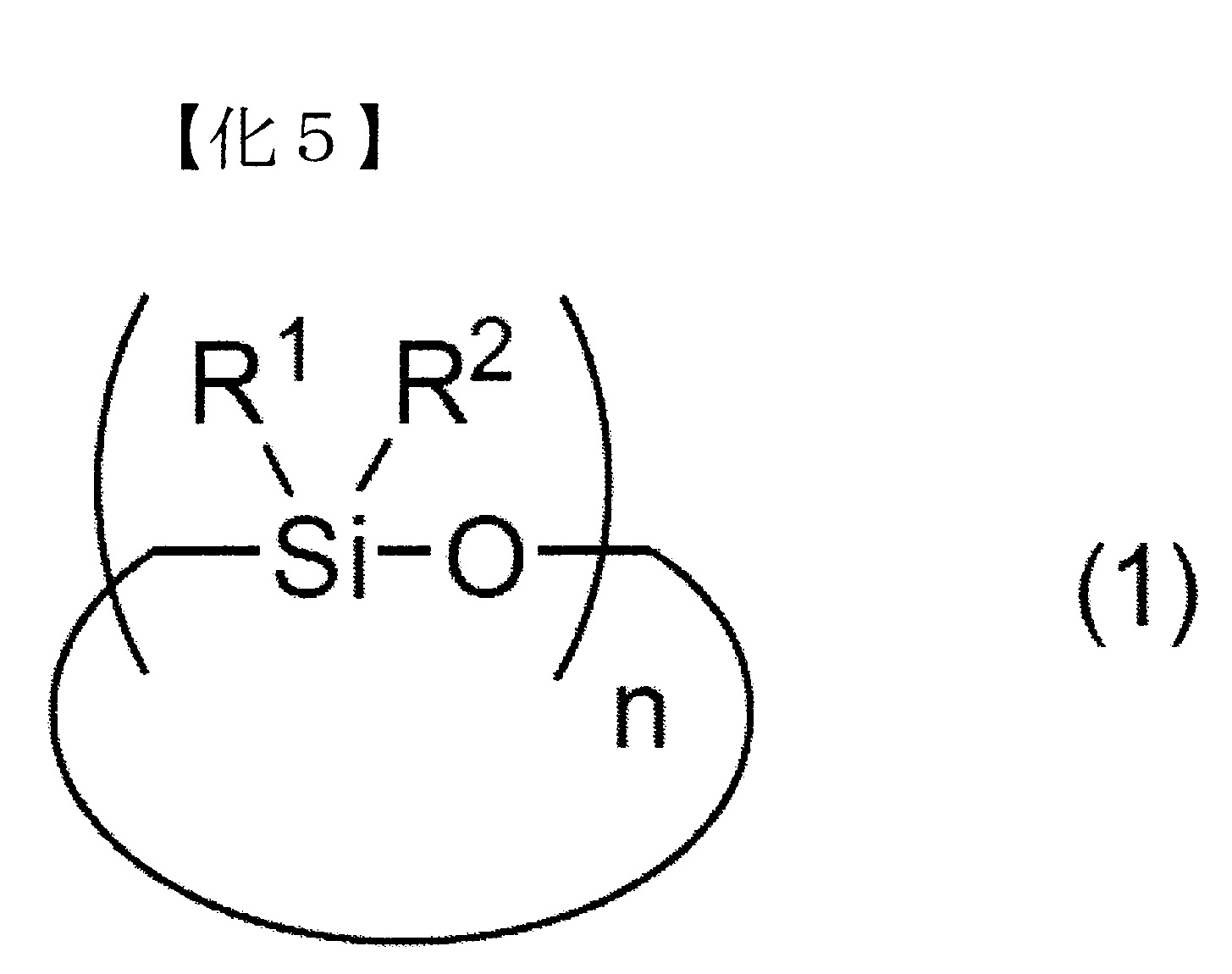 式(1)中,R1
及R2
各自獨立地表示可具有取代基之烷基、烯基或芳基。式(1)中之R1
及R2
各自為可具有取代基之合計碳數1~8之烷基、烯基或合計碳數6~30之芳基較佳。
當R1
及R2
為可具有取代基之烷基或可具有取代基之烯基時,較佳合計碳數為1~6,更佳合計碳數為1~4,特佳合計碳數為1或2。
又,當R1
及R2
為可具有取代基之芳基時,較佳合計碳數為6~20,更佳合計碳數為6~12,特佳合計碳數為6~8。
作為上述之取代基,可列舉羥基、鹵素、胺基、乙烯基、羧基、氰基、(甲基)丙烯醯氧基、縮水甘油基氧基(glycidyloxy)、巰基等。
作為式(1)中之R1
及R2
之較佳具體例,可列舉甲基、苯基、乙烯基及丙基。
式(1)中,R1
及R2
各自獨立地表示可具有取代基之烷基、烯基或芳基。式(1)中之R1
及R2
各自為可具有取代基之合計碳數1~8之烷基、烯基或合計碳數6~30之芳基較佳。
當R1
及R2
為可具有取代基之烷基或可具有取代基之烯基時,較佳合計碳數為1~6,更佳合計碳數為1~4,特佳合計碳數為1或2。
又,當R1
及R2
為可具有取代基之芳基時,較佳合計碳數為6~20,更佳合計碳數為6~12,特佳合計碳數為6~8。
作為上述之取代基,可列舉羥基、鹵素、胺基、乙烯基、羧基、氰基、(甲基)丙烯醯氧基、縮水甘油基氧基(glycidyloxy)、巰基等。
作為式(1)中之R1
及R2
之較佳具體例,可列舉甲基、苯基、乙烯基及丙基。
式(1)中,n表示3以上30以下之整數。式(1)中之n值較佳為3以上15以下,更佳為3以上10以下,再更佳為3以上8以下,特佳為3以上5以下。
又,式(1)表示之環狀矽氧烷化合物亦可為n值不同者的混合物、分子構造不同者的混合物、n值與分子構造皆不同者的混合物。
式(1)表示之環狀矽氧烷化合物之分子量較佳為2,000以下,更佳為1,600以下,再更佳為1,200以下,特佳為1,000以下。又,式(1)表示之環狀矽氧烷化合物之分子量,例如為100以上,較佳為150以上,更佳為200以上。
A-2)直鏈狀矽氧烷化合物
於矽氧烷分解步驟所使用的矽氧烷化合物之中,直鏈狀矽氧烷化合物係以下述式(2)表示。 式(2)中,R3
及R4
各自獨立地表示可具有取代基之烷基、烯基或芳基。式(2)中之R3
及R4
各自為可具有取代基之合計碳數1~8之烷基、烯基或合計碳數6~30之芳基較佳。
當R3
及R4
為可具有取代基之烷基或可具有取代基之烯基時,較佳合計碳數為1~6,更佳合計碳數為1~4,特佳合計碳數為1或2。
又,當R3
及R4
為可具有取代基之芳基時,較佳合計碳數為6~20,更佳合計碳數為6~12,特佳合計碳數為6~8。
作為上述之取代基,可列舉羥基、鹵素、胺基、乙烯基、羧基、氰基、(甲基)丙烯醯氧基、縮水甘油基氧基、巰基等。
作為式(2)中之R3
及R4
之較佳具體例,可列舉甲基、苯基、乙烯基及丙基。
式(2)中,R3
及R4
各自獨立地表示可具有取代基之烷基、烯基或芳基。式(2)中之R3
及R4
各自為可具有取代基之合計碳數1~8之烷基、烯基或合計碳數6~30之芳基較佳。
當R3
及R4
為可具有取代基之烷基或可具有取代基之烯基時,較佳合計碳數為1~6,更佳合計碳數為1~4,特佳合計碳數為1或2。
又,當R3
及R4
為可具有取代基之芳基時,較佳合計碳數為6~20,更佳合計碳數為6~12,特佳合計碳數為6~8。
作為上述之取代基,可列舉羥基、鹵素、胺基、乙烯基、羧基、氰基、(甲基)丙烯醯氧基、縮水甘油基氧基、巰基等。
作為式(2)中之R3
及R4
之較佳具體例,可列舉甲基、苯基、乙烯基及丙基。
式(2)中,m表示2以上10,000以下之整數。式(2)中之m值較佳為10以上7,000以下,更佳為20以上2,000以下,再更佳為30以上1,000以下,特佳為40以上800以下。
又,式(2)表示之直鏈狀矽氧烷化合物亦可為m值不同者的混合物、分子構造不同者的混合物、m值與分子構造皆不同者的混合物。
式(2)中,X各自獨立地表示氫原子、羥基、可具有取代基之合計碳數1~10之烷氧基、可具有取代基且可具有氧原子或氮原子之合計碳數1~10之烴基、或可具有取代基之胺基。X較佳各自獨立地為氫原子、羥基、可具有取代基之合計碳數1~10之烷氧基、可具有取代基且可具有氧原子或氮原子之合計碳數1~10之烴基的任一者,更佳為羥基或可具有取代基之合計碳數1~10之烷基,進而更佳為羥基或合計碳數1~5之烷基。
作為上述X之取代基,可列舉羥基、鹵素、胺基、乙烯基、羧基、氰基、(甲基)丙烯醯氧基、縮水甘油基氧基、巰基等。
式(2)表示之直鏈狀矽氧烷化合物之分子量較佳為60,000以下,更佳為56,000以下,再更佳為50,000以下,特佳為45,000以下。又,式(2)表示之直鏈狀矽氧烷化合物之分子量,例如為1,000以上,較佳為2,000以上,更佳為3,000以上。
A-3)倍半矽氧烷化合物
於矽氧烷分解步驟所使用的矽氧烷化合物之中,倍半矽氧烷化合物為將矽氧烷鍵(Si-O-Si鍵)作為主鏈骨架之矽氧烷系的化合物,且係以下述式(3)表示。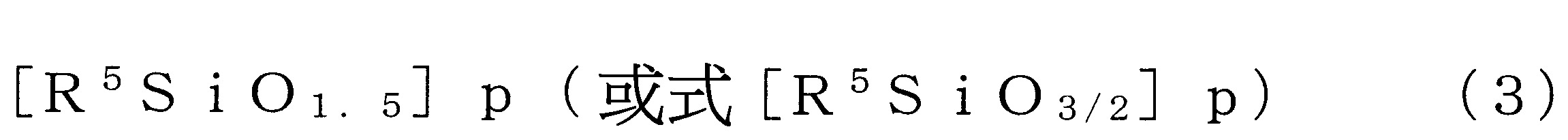
作為式(3)之倍半矽氧烷,例如可使用階梯型、網型、籠型、無規型等。就以該等中對於碳酸酯化合物的溶解性的觀點而言,籠型之倍半矽氧烷較佳。例如,作為籠型之倍半矽氧烷可列舉以下之式(3’)表示者(式(3’)中之R為與下述R5
相同之取代基)。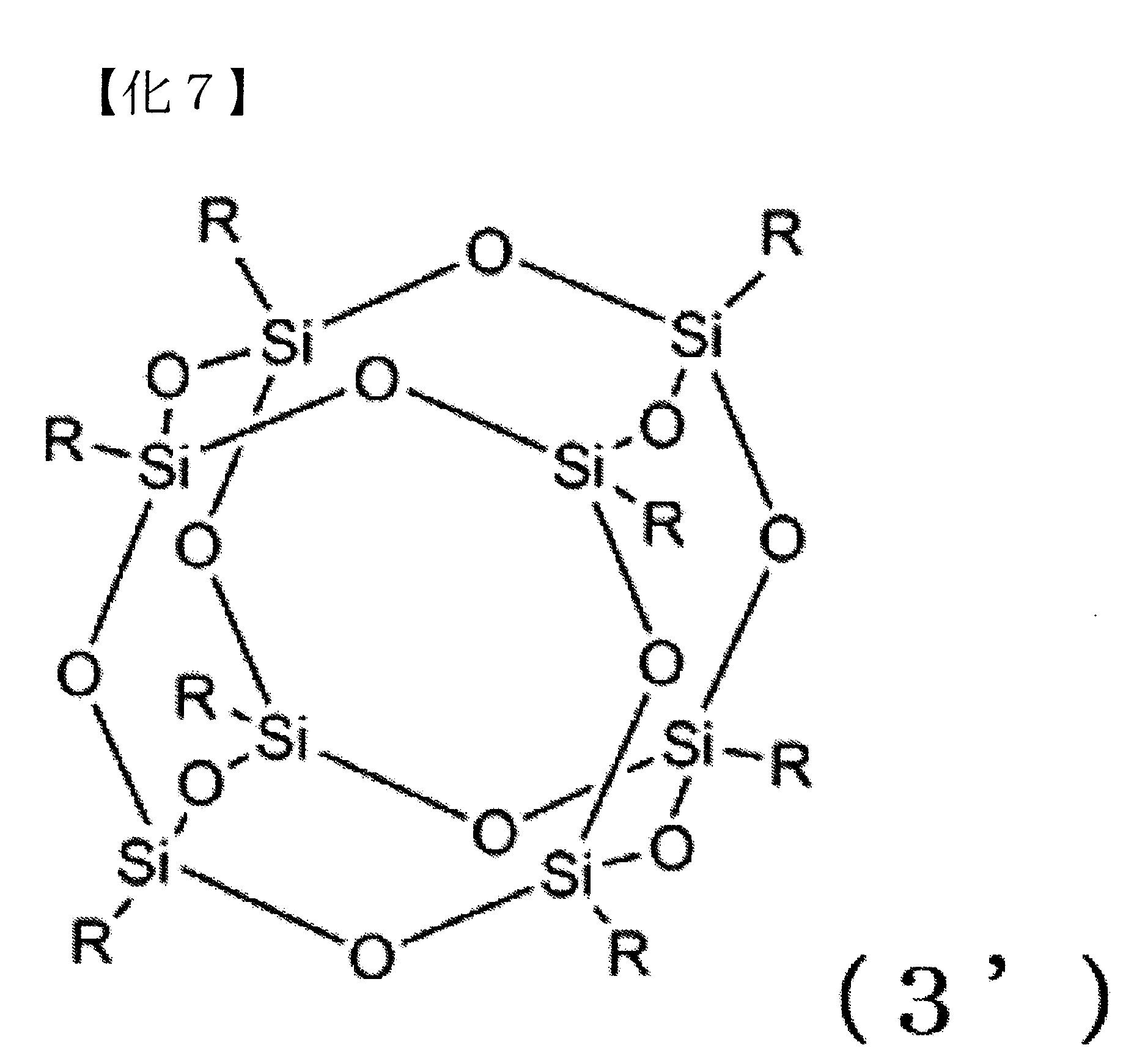 式(3)中,R5
為可具有取代基之碳數1~4之烷基、碳數2~4之烯基或碳數6~12之芳基。
式(3)中之R5
各自為可具有取代基之合計碳數1或2之烷基、可具有取代基之合計碳數2或3之烯基或合計碳數6~10之芳基較佳。又,當R5
為可具有取代基之芳基時,較佳合計碳數為6~8。
作為上述之取代基,可列舉羥基、鹵素、胺基、乙烯基、羧基、氰基、(甲基)丙烯醯氧基、縮水甘油基氧基、巰基等。
作為式(3)中之R5
之較佳具體例,可列舉甲基、苯基、乙烯基及丙基。
式(3)中,R5
為可具有取代基之碳數1~4之烷基、碳數2~4之烯基或碳數6~12之芳基。
式(3)中之R5
各自為可具有取代基之合計碳數1或2之烷基、可具有取代基之合計碳數2或3之烯基或合計碳數6~10之芳基較佳。又,當R5
為可具有取代基之芳基時,較佳合計碳數為6~8。
作為上述之取代基,可列舉羥基、鹵素、胺基、乙烯基、羧基、氰基、(甲基)丙烯醯氧基、縮水甘油基氧基、巰基等。
作為式(3)中之R5
之較佳具體例,可列舉甲基、苯基、乙烯基及丙基。
式(3)中,p表示4以上24以下之整數。式(3)中之p值較佳為5以上20以下,更佳為6以上18以下。p值為8~14更佳,尤其是為8、10、12或14。式(3)之倍半矽氧烷具有該等上述之p值且為籠型較佳。
又,式(3)表示之倍半矽氧烷化合物亦可為p值不同者的混合物、分子構造不同者的混合物、p值與分子構造皆不同者的混合物。
式(3)之倍半矽氧烷化合物中,作為取代基之R5
的種類與倍半矽氧烷化合物之分子量具有以下之表所示之關係。並且如同上述,作為式(3)之倍半矽氧烷化合物較佳為p值係8~14者,因此定為具有代表之取代基之式(3)之倍半矽氧烷化合物之分子量的較佳範圍。
亦即,式(3)表示之倍半矽氧烷化合物之分子量較佳為3,500以下,更佳為3,100以下,再更佳為1,900以下,再更佳為1,600以下,特佳為1,300以下。又,式(3)表示之式(3)之倍半矽氧烷化合物之分子量,例如為100以上,較佳為150以上,更佳為200以上,再更佳為250以上。
可僅使用上述式(1)之環狀矽氧烷化合物、式(2)表示之直鏈狀矽氧烷化合物及、式(3)之倍半矽氧烷化合物之中任一者的矽氧烷化合物,亦可作為混合物而使用環狀矽氧烷化合物、直鏈狀矽氧烷化合物及倍半矽氧烷化合物的任一者。該等矽氧烷化合物可藉由公知的手法而合成,且亦可使用市售者。
尚,式(1)之環狀矽氧烷化合物及式(2)表示之直鏈狀矽氧烷化合物是適用於二芳氧基矽烷化合物、二烷氧基矽烷化合物、單芳氧基單烷氧基矽烷化合物之任一的矽烷化合物的製造。又,式(3)之倍半矽氧烷化合物是適用於三芳氧基矽烷化合物及三烷氧基矽烷化合物之至少任一的矽烷化合物的製造。
[B)碳酸酯化合物]
作為矽氧烷分解步驟中所使用的碳酸酯化合物,可使用二芳基碳酸酯、二烷基碳酸酯及單烷基單芳基碳酸酯之至少任一者。
二芳基碳酸酯及單烷基單芳基碳酸酯較佳具有合計碳數6~30之芳基,且芳基之合計碳數較佳為6~20,更佳合計碳數為6~12,特佳合計碳數為6~8。
又,作為上述芳基之較佳具體例,可列舉苯基,並且當使用二芳基碳酸酯作為碳酸酯化合物時,較佳至少含有二苯基碳酸酯。
又,作為碳酸酯化合物之二烷基碳酸酯及單烷基單芳基碳酸酯,較佳具有合計碳數1~8之烷基,更佳烷基之合計碳數為1~6,再更佳合計碳數為1~4,亦即4以下之烷基。
尚,上述碳酸酯化合物可藉由公知的手法而合成,且亦可使用市售者。
[C)鹼性化合物觸媒]
作為於矽氧烷分解步驟中所使用的鹼性化合物觸媒,可列舉鹼金屬化合物、鹼土類金屬化合物等,作為此般之化合物可使用鹼金屬及鹼土類金屬化合物等之有機酸鹽、無機鹽、氧化物、氫氧化物、氫化物或烷氧化物、4級氫氧化銨及該等之鹽、胺類等,該等化合物可單獨或組合複數種類而使用。為了矽氧烷分解步驟之反應的效率化,鹼性化合物觸媒較佳包含鹼金屬碳酸鹽或鹼金屬氫氧化物。
作為鹼性化合物觸媒之較佳具體例,可列舉碳酸銫、碳酸鉀、碳酸鈉、碳酸氫鈉、氫氧化銫、氫氧化鉀、氫氧化鈉等。
尚,上述鹼性化合物觸媒可藉由公知的手法而調製,且亦可使用市售者。又,可將鹼性化合物觸媒作為水溶液添加至反應系,亦可作為粉體添加至反應系。
[D)矽氧烷分解步驟]
矽氧烷分解步驟中,係在上述C)之鹼性化合物觸媒的存在下,將含有上述A)之環狀矽氧烷化合物及/或直鏈狀矽氧烷化合物與上述B)之碳酸酯化合物之混合物進行加熱,而使上述A)之矽氧烷化合物烷氧基化及/或芳氧基化,以進行矽氧烷分解。如此般,藉由上述A)之矽氧烷化合物的矽氧烷分解反應,可生成導入有上述B)之碳酸酯化合物的芳基及/或烷基之矽烷化合物。
矽氧烷分解步驟中,使用二芳基碳酸酯作為上述B)之碳酸酯化合物的情況下,所生成之矽烷化合物為二芳氧基矽烷化合物;使用二烷基碳酸酯作為上述B)之碳酸酯化合物的情況下,所生成之矽烷化合物為二烷氧基矽烷化合物;使用單烷基單芳基碳酸酯作為上述B)之碳酸酯化合物的情況下,所生成之矽烷化合物為單芳氧基單烷氧基矽烷化合物。
例如,使用二苯基碳酸酯作為上述B)之碳酸酯化合物,且使用十甲基環五矽氧烷作為上述A)之環狀矽氧烷化合物時之矽氧烷分解反應,係以下式(4)表示,可得到二甲基二苯氧基矽烷(以下,亦稱為DMDPS)作為主要產物。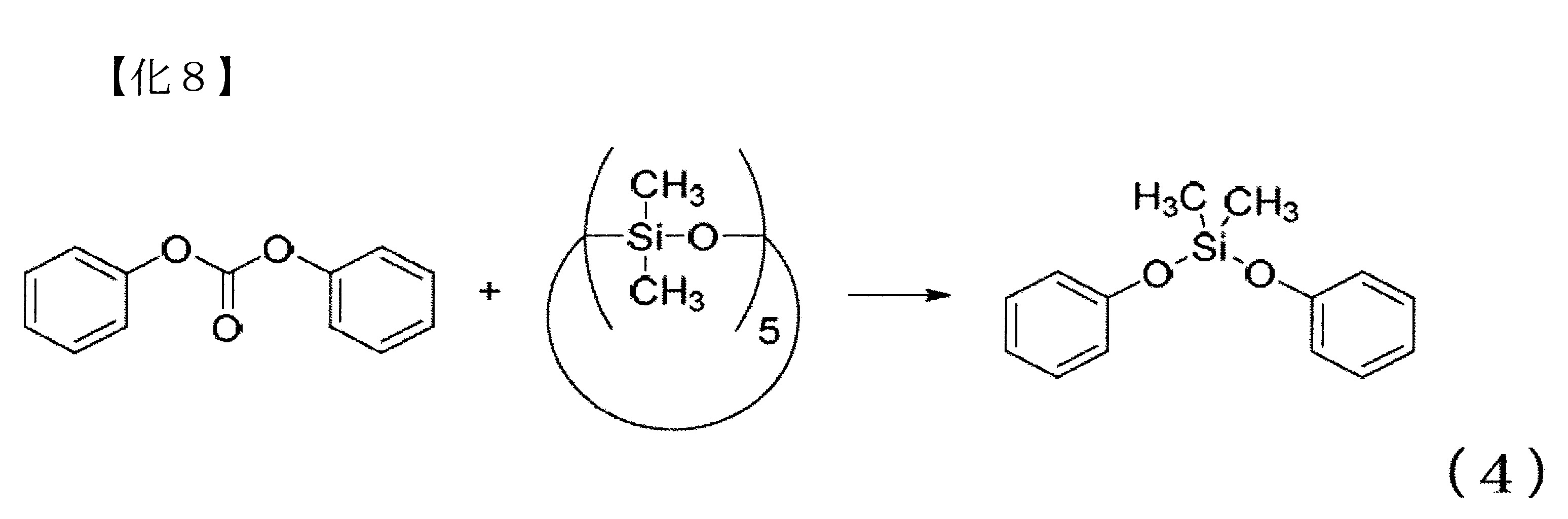
式(4)是概略地表示矽氧烷分解反應,其省略了會生成之一些的副產物,且並未調整二苯基碳酸酯、十甲基環五矽氧烷及二甲基二苯氧基矽烷之各化合物間的莫耳比。
矽氧烷分解步驟中,為了促進矽氧烷化合物之分解反應,所使用之上述B)之碳酸酯化合物的莫耳量較佳為上述A)之矽氧烷化合物的Si莫耳量,即矽氧烷化合物中之矽原子的莫耳量以上。亦即,前述B)碳酸酯化合物的莫耳量相對於A)矽氧烷化合物的Si莫耳量之比x(即,A)矽氧烷化合物的Si莫耳量/B)碳酸酯化合物的莫耳量之值)滿足x≧1的關係較佳。又,碳酸酯化合物的莫耳量可為矽氧烷化合物的Si莫耳量之1.1倍以上(x≧1.1),例如1.5倍左右。
又,為了矽氧烷化合物之分解反應的效率化,上述比x較佳為0.8<x<2.0,更佳為0.8<x<1.8,再更佳為0.8<x< 1.6,特佳為0.9<x<1.5。此情況下,對於原料之矽氧烷化合物,可抑制未反應而殘餘之殘量。
尚,可依據原料之矽氧烷化合物的種類來調整上述比x之值。
例如,作為原料,使用上述A-1)環狀矽氧烷化合物或A-2)直鏈狀矽氧烷化合物時,碳酸酯化合物的莫耳量與矽氧烷化合物的Si莫耳量之理想的比率為1:1,因此,上述比x之值可為0.8<x<1.2左右,較佳為0.9<x<1.1左右。又,以上述A-3)倍半矽氧烷化合物作為原料時,碳酸酯化合物的莫耳量與矽氧烷化合物的Si莫耳量之理想的比率為1:1.5,因此,上述比x之值可為1.0<x<1.8左右,較佳為1.3<x<1.7左右。
矽氧烷分解步驟中,就分解反應之效率化與抑制過剩之觸媒使用的觀點而言,上述C)之鹼性化合物觸媒相對於上述A)之矽氧烷化合物的Si莫耳量之比y(即,C)之鹼性化合物觸媒的莫耳量/A)矽氧烷化合物的Si莫耳量之值)較佳為0.0001 mmol/mol≦y≦20 mmol/mol(y之值為0.0001 mmol/mol以上20 mmol/mol以下),更佳為0.001 mmol/mol≦y≦15 mmol/mol(y之值為0.001 mmol/mol以上15 mmol/mol以下),再更佳為0.1 mmol/mol≦y≦8 mmol/mol(y之值為0.1 mmol/mol以上8 mmol/mol以下),特佳為1 mmol/mol≦y≦5 mmol/mol(y之值為1 mmol/mol以上5 mmol/mol以下)。
矽氧烷分解步驟中之矽氧烷分解反應的溫度,即分解A)矽氧烷化合物之溫度,就分解反應之效率化與產率的觀點而言,較佳為50℃以上300℃以下,亦可為50℃以上150℃以下。矽氧烷分解步驟中之溫度,更佳為70℃以上250℃以下,再更佳為80℃以上240℃以下,特佳為90℃以上220℃以下。
矽氧烷分解步驟中之矽氧烷分解反應可於常壓下進行,且不需要加壓或減壓之壓力的調整。
[E)蒸餾步驟/再結晶步驟]
於本發明之矽烷化合物之製造方法中,為了提升矽烷化合物之純度,較佳進而具有蒸餾由矽氧烷分解步驟所生成之矽烷化合物之蒸餾步驟,與再結晶由矽氧烷分解步驟所生成之矽烷化合物之再結晶步驟的至少任一者。
為了有效率的蒸餾,蒸餾步驟中之壓力較佳為1hPa以上20hPa以下,更佳為2hPa以上10hPa以下,再更佳為3hPa以上5hPa以下。
又,為了有效率的蒸餾,蒸餾步驟中之溫度較佳為50℃以上300℃以下,更佳為70℃以上250℃以下,再更佳為100℃以上200℃以下,特佳為120℃以上180℃以下。
再結晶步驟中,使用因應生成之矽烷化合物的性質,特別是溶解度等而選擇之再結晶溶劑,惟作為再結晶溶劑,就與產物之溶解性的觀點而言,較佳為庚烷、辛烷、甲苯、苯、環己烷、己烷等。又,較佳為在50℃以上120℃以下之溫度下使矽烷化合物溶解於溶劑,且將溶液以0.5℃/min以上1℃/min以下進行冷卻。
[F)滴下步驟]
矽烷化合物之製造方法中,較佳為預先混合A)矽氧烷化合物以外之原料與觸媒,並對於所得到之混合物,主要是混合溶液,滴下A)矽氧烷化合物。如此般,藉由進而設置對至少含有B)碳酸酯化合物及C)鹼性化合物觸媒之混合物,逐漸地滴下A)矽氧烷化合物之F)滴下步驟,可更提高矽烷化合物之製造方法的安全性。
F)滴下步驟中之A)矽氧烷化合物的滴下速度,雖可依據反應規模(scale)而調整,但通常為0.1ml/分~10 l(升)/分,較佳為1.0ml/分~7.0 l/分,更佳為10ml/分~5.0 l/分,特佳為100ml/分~1.0 l/分。又,A)矽氧烷化合物的滴下速度,例如可為1~100ml/分左右。
例如,在使用約250g之二苯基碳酸酯的情況下,較佳以滴下速度1ml/分左右而添加0.23mol左右的矽氧烷化合物,滴下時間的合計為90分左右。F)滴下步驟中之最佳滴下速度雖因應反應規模而不同,但滴下所花費之較佳合計時間變化不大,通常較佳為30分~3小時左右,更佳為1小時~2小時左右,例如為90分左右。
又,可使A)矽氧烷化合物溶解於適宜的溶劑中,而容易調整滴下速度。
如此般,藉由對含有B)碳酸酯化合物與C)鹼性化合物觸媒之混合物,通常為進而包含溶劑之溶液,逐漸地添加A)矽氧烷化合物,可抑制急劇之CO2
產生,提升安全性。又,在工業規模之矽烷化合物之製造步驟中,亦藉由採用滴下步驟而可防止副產物之CO2
所致之反應釜內之急劇的加壓,安全性提高。
[實施例]
<評估項目>
NMR:以下1
H-NMR分析中之化學位移是以CDCl3
的波峰之7.24ppm作為基準。
(實施例1)
將十甲基環五矽氧烷7.5g(20.2 mmol,Si莫耳量:101.0 mmol)、二苯基碳酸酯21.6g(101.0 mmol)及作為觸媒之碳酸銫33mg(0.1 mmol),置換為氮氣環境下,並以200℃攪拌60分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於十甲基環五矽氧烷之矽上的甲基之質子的波峰,確認十甲基環五矽氧烷的轉化率為100%。
接著,將反應混合物冷卻至40℃後,藉由在減壓度4hPa、150℃下進行減壓蒸餾,得到23.7g之無色油狀成分。
以1
H-NMR分析得到之油狀成分的結果,作為矽上的甲基之質子的波峰,僅觀測到基於二甲基二苯氧基矽烷(DMDPS)之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為98.6:1.4。
由得到之油狀成分的重量與上述莫耳比率所求出之二甲基二苯氧基矽烷之莫耳量的產率,相對於十甲基環五矽氧烷所含之矽原子的莫耳量而言,為96.0%。
二甲基二苯氧基矽烷(1
H-NMR(CDCl3
,500MHz,δ;ppm)=0.378(s;6H)、6.942、6.944(d;4H)、6.959、6.961、6.995(t;2H)、7.230、7.245、7.257(t;4H))。
尚,本實施例之矽氧烷分解反應係以下式(4-1)表示,除了主產物之DMDPS以外,可得到作為副產物之1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷。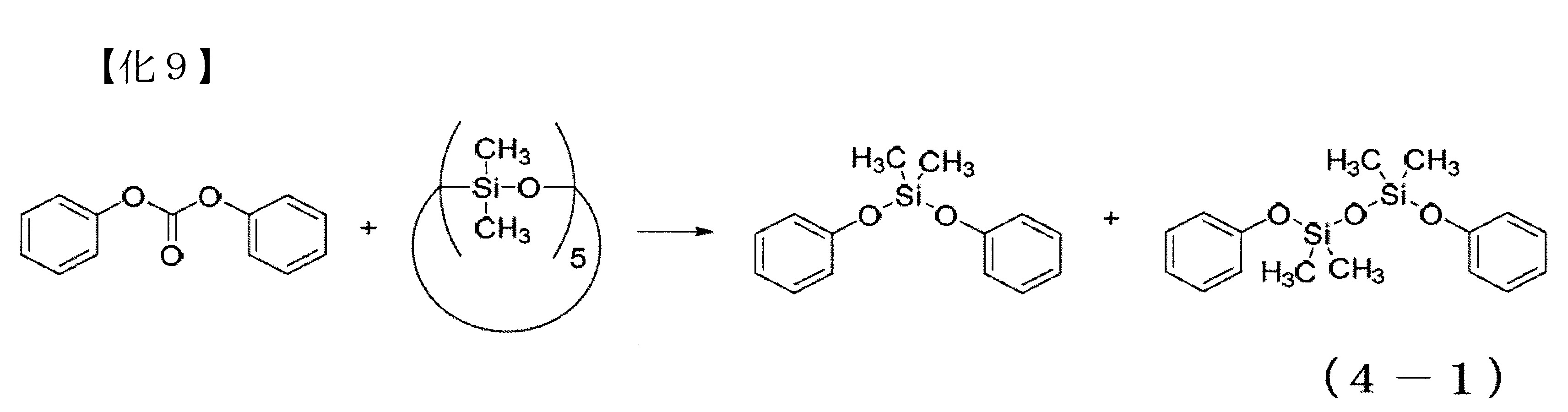
(實施例2)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯23.6g(110.0 mmol)及作為觸媒之碳酸銫33mg(0.1 mmol),置換為氮氣環境下,並以200℃攪拌60分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於十甲基環五矽氧烷之矽上的甲基之質子的波峰,確認十甲基環五矽氧烷的轉化率為100%。
接著,將反應混合物冷卻至40℃後,藉由在減壓度4hPa、150℃下進行減壓蒸餾,得到25.1g之無色油狀成分。
以1
H-NMR分析得到之油狀成分的結果,作為矽上的甲基之質子的波峰,僅觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為99.8:0.2。又,亦觀測到基於二苯基碳酸酯之與碳酸酯基位於間位之質子的波峰7.406ppm。
從得到之油狀成分的重量減去由源自混入之二苯基碳酸酯的波峰面積所算出之二苯基碳酸酯的重量,由上述莫耳比率求出之二甲基二苯氧基矽烷之莫耳量的產率,相對於十甲基環五矽氧烷所含之矽原子的莫耳量而言,為97.7%。
(實施例3)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯32.1g(150.0 mmol)及作為觸媒之碳酸銫33mg(0.1 mmol),置換為氮氣環境下,並以200℃攪拌60分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於十甲基環五矽氧烷之矽上的甲基之質子的波峰,確認十甲基環五矽氧烷的轉化率為100%。
作為矽上的甲基之質子的波峰,僅觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為98.8:1.2。
(實施例4)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯19.3g(90.0 mmol)及作為觸媒之碳酸銫33mg(0.1 mmol),置換為氮氣環境下,並以200℃攪拌60分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於十甲基環五矽氧烷之矽上的甲基之質子的波峰,十甲基環五矽氧烷的轉化率為100%。
作為矽上的甲基之質子的波峰,僅觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為93.2:6.8。
(實施例5)
將下述式(5)表示之八苯基環四矽氧烷(熔點:197℃,分子量:793.17)14.9g(18.8 mmol,Si莫耳量:75.0 mmol)、二苯基碳酸酯16.1g(75.0 mmol)及作為觸媒之碳酸銫33mg(0.1 mmol),置換為氮氣環境下,並以200℃攪拌10分鐘。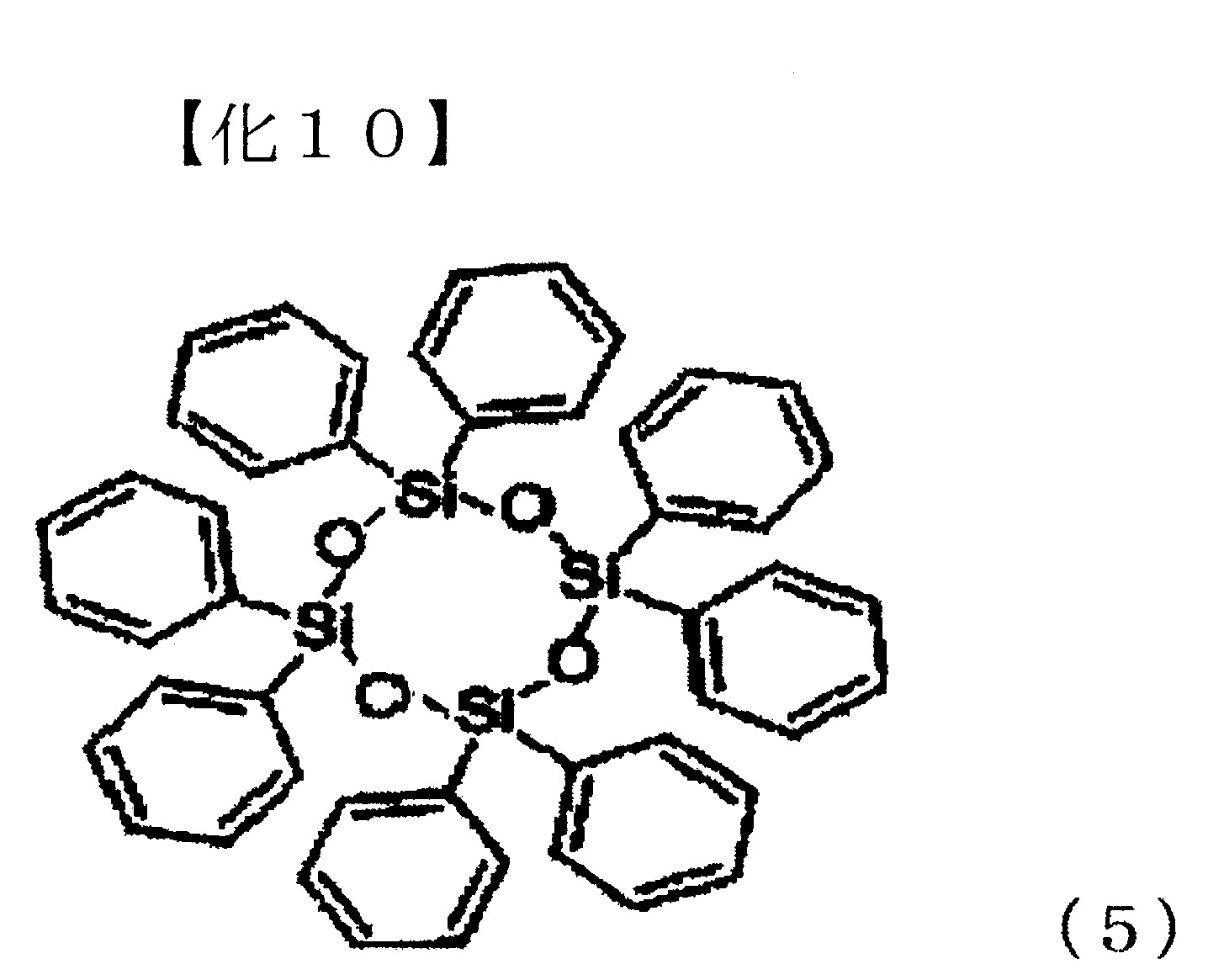
以1
H-NMR分析反應混合物的結果,未觀測到基於八苯基環四矽氧烷之對位之質子的波峰,確認八苯基環四矽氧烷的轉化率為100%。
室溫冷卻後,於經固化之反應混合物中加入20g之庚烷,至90℃後進行熱過濾。藉由將得到之濾液於室溫下放置3天,而使白色結晶析出。過濾進而加入冷卻至5℃的庚烷10g之混合物,取出藉此而得到之濾紙上的結晶,在40℃、減壓度1hPa下乾燥45小時,結果得到24.1g之白色粉末。以1
H-NMR分析得到之粉末的結果,觀測到源自二苯基二苯氧基矽烷與1,3-二苯氧基-1,1,3,3-四苯基二矽氧烷的波峰。由波峰面積比率求出二甲基二苯氧基矽烷與1,3-二苯氧基-1,1,3,3-四苯基二矽氧烷之莫耳比率,為95.4:4.6。
由得到之白色粉末重量與上述莫耳比率所求出之二苯基二苯氧基矽烷之莫耳量的產率,相對於八苯基環四矽氧烷所含之矽原子的莫耳量而言,為81.1%。
二苯基二苯氧基矽烷(1
H-NMR(CDCl3,500MHz,δ;ppm)=6.915、6.927、6.939、6.952、6.965(p;6H)、7.142、7.155、7.169(t;4H)、7.354、7.366、7.379(t;4H)、7.425、7.437、7.449(t;2H))、7.750、7.762(d;4H)。
(實施例6)
將八苯基環四矽氧烷14.9g(18.8 mmol,Si莫耳量:75.0 mmol)、二苯基碳酸酯16.9g(78.8 mmol)及作為觸媒之碳酸銫33mg(0.1 mmol) ,置換為氮氣環境下,並以200℃攪拌10分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於八苯基環四矽氧烷之對位之質子的波峰,確認八苯基環四矽氧烷的轉化率為100%。
室溫冷卻後,於經固化之反應混合物中加入20g之庚烷,至90℃後進行熱過濾。藉由將得到之濾液於室溫下放置3天,而使白色結晶析出。過濾進而加入冷卻至5℃的庚烷10g之混合物,取出藉此而得到之濾紙上的結晶,在40℃、減壓度1hPa下乾燥45小時,結果得到24.4g之白色粉末。
以1
H-NMR分析得到之粉末的結果,觀測到源自二苯基二苯氧基矽烷與1,3-二苯氧基-1,1,3,3-四苯基二矽氧烷的波峰。由波峰面積比率求出二甲基二苯氧基矽烷與1,3-二苯氧基-1,1,3,3-四苯基二矽氧烷之莫耳比率,為99.5:0.5。
由得到之白色粉末重量與上述莫耳比率所求出之二苯基二苯氧基矽烷之莫耳量的產率,相對於八苯基環四矽氧烷所含之矽原子的莫耳量而言,為87.6%。
(實施例7)
將八苯基環四矽氧烷14.9g(18.8 mmol,Si莫耳量:75.0 mmol)、二苯基碳酸酯17.7g(82.6 mmol)及作為觸媒之碳酸銫33mg(0.1 mmol),置換為氮氣環境下,並以200℃攪拌60分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於八苯基環四矽氧烷之對位之質子的波峰,確認八苯基環四矽氧烷的轉化率為100%。
室溫冷卻後,於經固化之反應混合物中加入20g之庚烷,至90℃後進行熱過濾。藉由將得到之濾液於室溫下放置3天,而使白色結晶析出。過濾進而加入冷卻至5℃的庚烷10g之混合物,取出藉此而得到之濾紙上的結晶,在40℃、減壓度1hPa下乾燥45小時,結果得到23.1g之白色粉末。
以1
H-NMR分析得到之粉末的結果,觀測到源自二苯基二苯氧基矽烷與1,3-二苯氧基-1,1,3,3-四苯基二矽氧烷的波峰。由波峰面積比率求出二甲基二苯氧基矽烷與1,3-二苯氧基-1,1,3,3-四苯基二矽氧烷之莫耳比率,為99.6:0.4。
由得到之白色粉末重量與上述莫耳比率所求出之二苯基二苯氧基矽烷之莫耳量的產率,相對於八苯基環四矽氧烷所含之矽原子的莫耳量而言,為83.1%。
(實施例8)
將下述式(6)表示之二甲基聚矽氧烷(完成羥基終端處理,分子量:4,200)7.4g(1.8 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g(100.0 mmol)及作為觸媒之碳酸銫65mg(0.2 mmol),置換為氮氣環境下,並以220℃攪拌30分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於二甲基聚矽氧烷之質子的波峰,確認二甲基聚矽氧烷的轉化率為100%。
作為矽上的甲基之質子的波峰,僅觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為95.0:5.0。
(實施例9)
將下述式(7)表示之二甲基聚矽氧烷(信越化學工業製,KF-96-3000cs,三甲基矽烷氧基末端,分子量:40,000)7.4g(0.19 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g(100.0 mmol)及作為觸媒之碳酸銫33mg(0.1 mmol),置換為氮氣環境下,並以200℃攪拌60分鐘。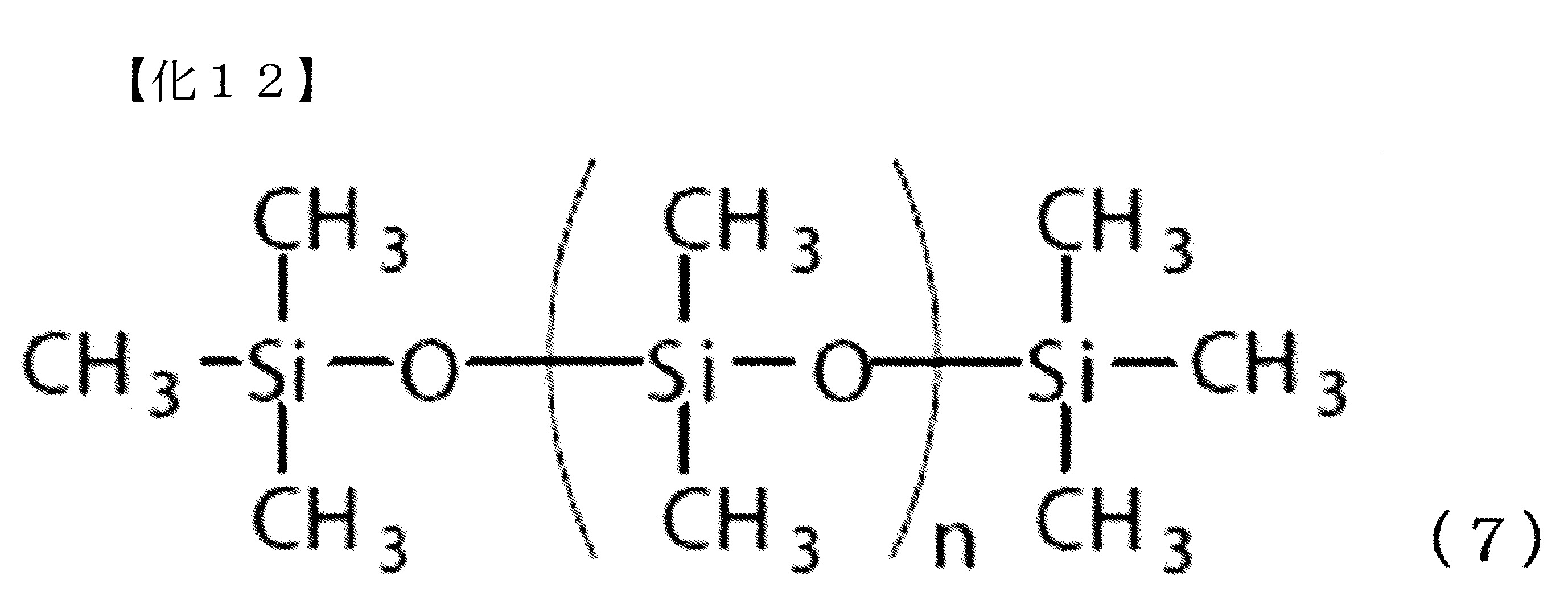 以1
H-NMR分析反應混合物的結果,未觀測到基於二甲基聚矽氧烷之質子的波峰,確認二甲基聚矽氧烷的轉化率為100%。
作為矽上的甲基之質子的波峰,僅觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為98.1:1.9。
以1
H-NMR分析反應混合物的結果,未觀測到基於二甲基聚矽氧烷之質子的波峰,確認二甲基聚矽氧烷的轉化率為100%。
作為矽上的甲基之質子的波峰,僅觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為98.1:1.9。
(實施例10)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二丁基碳酸酯17.4g(100.0 mmol)及作為觸媒之碳酸銫33mg(0.1 mmol),置換為氮氣環境下,並以200℃攪拌60分鐘。
以1
H-NMR分析反應混合物的結果,作為矽上的甲基之質子的波峰,觀測到基於十甲基環五矽氧烷之質子的波峰0.055ppm及基於二甲基二丁氧基矽烷之波峰0.070ppm。由兩波峰面積之比率算出之十甲基環五矽氧烷的轉化率為69%。
(實施例11)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g(100.0 mmol)及作為觸媒之碳酸銫33mg(0.1 mmol),置換為氮氣環境下,並以150℃攪拌60分鐘。
以1
H-NMR分析反應混合物的結果,作為矽上的甲基之質子的波峰,觀測到基於十甲基環五矽氧烷之質子的波峰0.055ppm及基於二甲基二苯氧基矽烷之質子的波峰0.378ppm。由兩波峰面積之比率算出之十甲基環五矽氧烷的轉化率為71%。
作為矽上的甲基之質子的波峰,觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由兩波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為78.7:21.3。
(實施例12)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g(100.0 mmol)及作為觸媒之碳酸銫65mg(0.2 mmol),置換為氮氣環境下,並以220℃攪拌30分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於十甲基環五矽氧烷之矽上的甲基之質子的波峰,確認十甲基環五矽氧烷的轉化率為100%。
作為矽上的甲基之質子的波峰,僅觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為99.6:0.4。
(實施例13)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g(100.0 mmol)及作為觸媒之碳酸鉀28mg(0.2 mmol),置換為氮氣環境下,並以220℃攪拌30分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於十甲基環五矽氧烷之矽上的甲基之質子的波峰,十甲基環五矽氧烷的轉化率為100%。
作為矽上的甲基之質子的波峰,僅觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為99.7:0.3。
(實施例14)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g(100.0 mmol)及作為觸媒之碳酸鈉21mg(0.2 mmol),置換為氮氣環境下,並以220℃攪拌30分鐘。
以1
H-NMR分析反應混合物的結果,作為矽上的甲基之質子的波峰,觀測到基於十甲基環五矽氧烷之質子的波峰0.055ppm及基於二甲基二苯氧基矽烷之質子的波峰0.378ppm。由兩波峰面積之比率算出之十甲基環五矽氧烷的轉化率為89%。
作為矽上的甲基之質子的波峰,觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由兩波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為94.3:5.7。
(實施例15)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g(100.0 mmol)及作為觸媒之碳酸氫鈉17mg(0.2 mmol),置換為氮氣環境下,並以220℃攪拌30分鐘。
以1
H-NMR分析反應混合物的結果,作為矽上的甲基之質子的波峰,觀測到基於十甲基環五矽氧烷之質子的波峰0.055ppm及基於二甲基二苯氧基矽烷之質子的波峰0.378ppm。由兩波峰面積之比率算出之十甲基環五矽氧烷的轉化率為87%。
作為矽上的甲基之質子的波峰,觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由兩波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為97.4:2.6。
(實施例16)
將下述式(8)表示之六甲基環三矽氧烷7.4g(33.3 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g (100.0 mmol)及作為觸媒之碳酸銫65mg(0.2 mmol),置換為氮氣環境下,並以220℃攪拌30分鐘。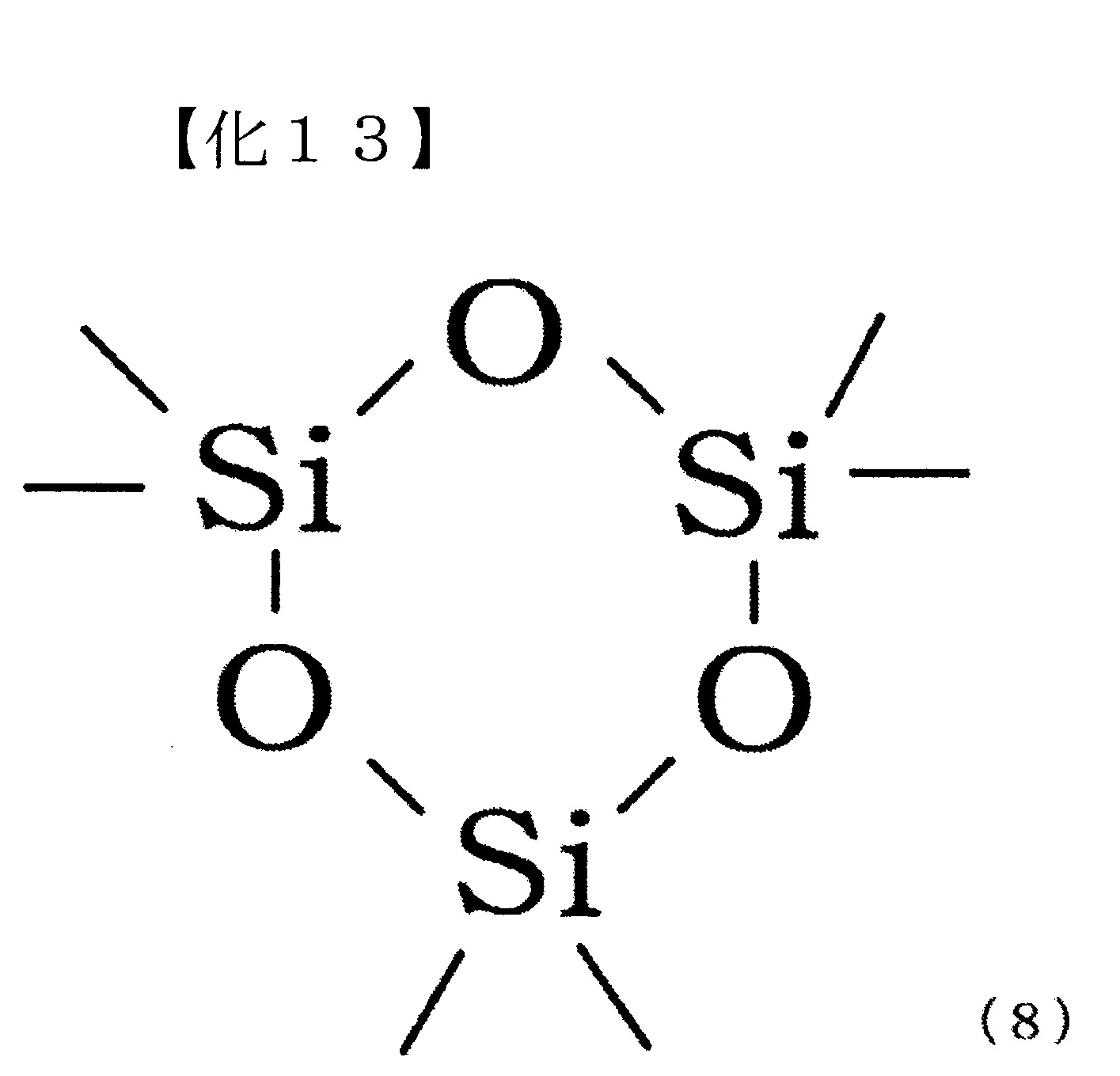
以1
H-NMR分析反應混合物的結果,未觀測到基於六甲基環三矽氧烷之矽上的甲基之質子的波峰,六甲基環三矽氧烷的轉化率為100%。
作為矽上的甲基之質子的波峰,僅觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由兩波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為99.3:0.7。
(實施例17)
將下述式(9)表示之八甲基環四矽氧烷7.4g(25.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g (100.0 mmol)及作為觸媒之碳酸銫65mg(0.2 mmol),置換為氮氣環境下,並以220℃攪拌30分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於八甲基環四矽氧烷之矽上的甲基之質子的波峰,八甲基環四矽氧烷的轉化率為100%。
作為矽上的甲基之質子的波峰,僅觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由兩波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為99.4:0.6。
(實施例18)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g(100.0 mmol)及作為觸媒之氫氧化銫75mg(0.5 mmol),置換為氮氣環境下,並以200℃攪拌60分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於十甲基環五矽氧烷之矽上的甲基之質子的波峰,十甲基環五矽氧烷的轉化率為100%。
作為矽上的甲基之質子的波峰,僅觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由兩波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為98.2:1.8。
(實施例19)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g(100.0 mmol)及作為觸媒之氫氧化鉀11mg(0.2 mmol),置換為氮氣環境下,並以200℃攪拌60分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於十甲基環五矽氧烷之矽上的甲基之質子的波峰,十甲基環五矽氧烷的轉化率為100%。
作為矽上的甲基之質子的波峰,僅觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由兩波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為97.6:2.4。
(實施例20)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g(100.0 mmol)及作為觸媒之氫氧化鈉12mg(0.3 mmol),置換為氮氣環境下,並以200℃攪拌60分鐘。
以1
H-NMR分析反應混合物的結果,作為矽上的甲基之質子的波峰,觀測到基於十甲基環五矽氧烷之質子的波峰0.055ppm及基於二甲基二苯氧基矽烷之質子的波峰0.378ppm。由兩波峰面積之比率算出之十甲基環五矽氧烷的轉化率為66%。
作為矽上的甲基之質子的波峰,觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由兩波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為94.4:5.6。
(實施例21)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g(100.0 mmol)及作為觸媒之碳酸銫33mg(0.1 mmol),置換為氮氣環境下,並以120℃攪拌60分鐘。
以1
H-NMR分析反應混合物的結果,作為矽上的甲基之質子的波峰,觀測到基於十甲基環五矽氧烷之質子的波峰0.055ppm及基於二甲基二苯氧基矽烷之質子的波峰0.378ppm。由兩波峰面積之比率算出之十甲基環五矽氧烷的轉化率為67%。
作為矽上的甲基之質子的波峰,觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由兩波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為82.0:18.0。
(實施例22)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g(100.0 mmol)及作為觸媒之碳酸銫33mg(0.1 mmol),置換為氮氣環境下,並以90℃攪拌60分鐘。
以1
H-NMR分析反應混合物的結果,作為矽上的甲基之質子的波峰,觀測到基於十甲基環五矽氧烷之質子的波峰0.055ppm及基於二甲基二苯氧基矽烷之質子的波峰0.378ppm。由兩波峰面積之比率算出之十甲基環五矽氧烷的轉化率為2%。
作為矽上的甲基之質子的波峰,觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由兩波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為33.3:66.7。
(實施例23)
將1,3,5,7,9,11,13,15-八甲基五環[9.5.1.13,9
.15,15
.17,13
]八矽氧烷2.06g(3.83 mmol,Si莫耳量:30.7 mmol)、二苯基碳酸酯9.85g(46.0 mmol)及作為觸媒之碳酸銫15mg(0.05 mmol),置換為氮氣環境下,並以180℃攪拌150分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於1,3,5,7,9,11,13,15-八甲基五環[9.5.1.13,9
.15,15
.17,13
]八矽氧烷之矽上的甲基之質子的波峰,1,3,5,7,9,11,13,15-八甲基五環[9.5.1.13,9
.15,15
.17,13
]八矽氧烷的轉化率為100%。
作為矽上的甲基之質子的波峰,僅觀測到基於甲基三苯氧基矽烷之波峰0.483ppm、及基於1,3-二甲基-1,1,3,3-四苯氧基二矽氧烷之波峰0.289ppm。由兩波峰面積比率求出甲基三苯氧基矽烷與1,3-二甲基-1,1,3,3-四苯氧基二矽氧烷之莫耳比率,為93.7:6.3。
將作為產物之三芳氧基矽烷化合物等的化學式表示如以下。下述式(10)表示甲基三苯氧基矽烷,下述式(11)表示1,3-二甲基-1,1,3,3-四苯氧基二矽氧烷。
(實施例24)
將1,3,5,7,9,11,13,15-八苯基五環[9.5.1.13,9
.15,15
.17,13
]八矽氧烷3.96g(3.83 mmol,Si莫耳量:30.6 mmol)、二苯基碳酸酯9.85g(46.0 mmol)及作為觸媒之碳酸銫15mg(0.05 mmol),置換為氮氣環境下,並以180℃攪拌150分鐘。
以1
H-NMR分析反應混合物的結果,未觀測到基於1,3,5,7,9,11,13,15-八苯基五環[9.5.1.13,9
.15,15
.17,13
]八矽氧烷之對位之質子的波峰,確認1,3,5,7,9,11,13,15-八苯基五環[9.5.1.13,9
.15,15
.17,13
]八矽氧烷的轉化率為100%。
室溫冷卻後,於經固化之反應混合物中加入10g之庚烷,至90℃後進行熱過濾。藉由將得到之濾液於室溫下放置3天,而使白色結晶析出。過濾進而加入冷卻至5℃的庚烷5g之混合物,取出藉此而得到之濾紙上的結晶,在40℃、減壓度1hPa下乾燥45小時,結果得到9.42g之白色粉末。
以1
H-NMR分析得到之粉末的結果,觀測到源自苯基三苯氧基矽烷與1,3-二苯基-1,1,3,3-四苯氧基二矽氧烷的波峰。由波峰面積比率求出苯基三苯氧基矽烷與1,3-二苯氧基-1,1,3,3-四苯基二矽氧烷之莫耳比率,為96.0:4.0。
由得到之白色粉末重量與上述莫耳比率所求出之苯基三苯氧基矽烷之莫耳量的產率,相對於八苯基環四矽氧烷所含之矽原子的莫耳量而言,為76.8%。
將作為產物之三芳氧基矽烷化合物等的化學式表示如以下。下述式(12)表示苯基三苯氧基矽烷,下述式(13)表示1,3-二苯基-1,1,3,3-四苯氧基二矽氧烷。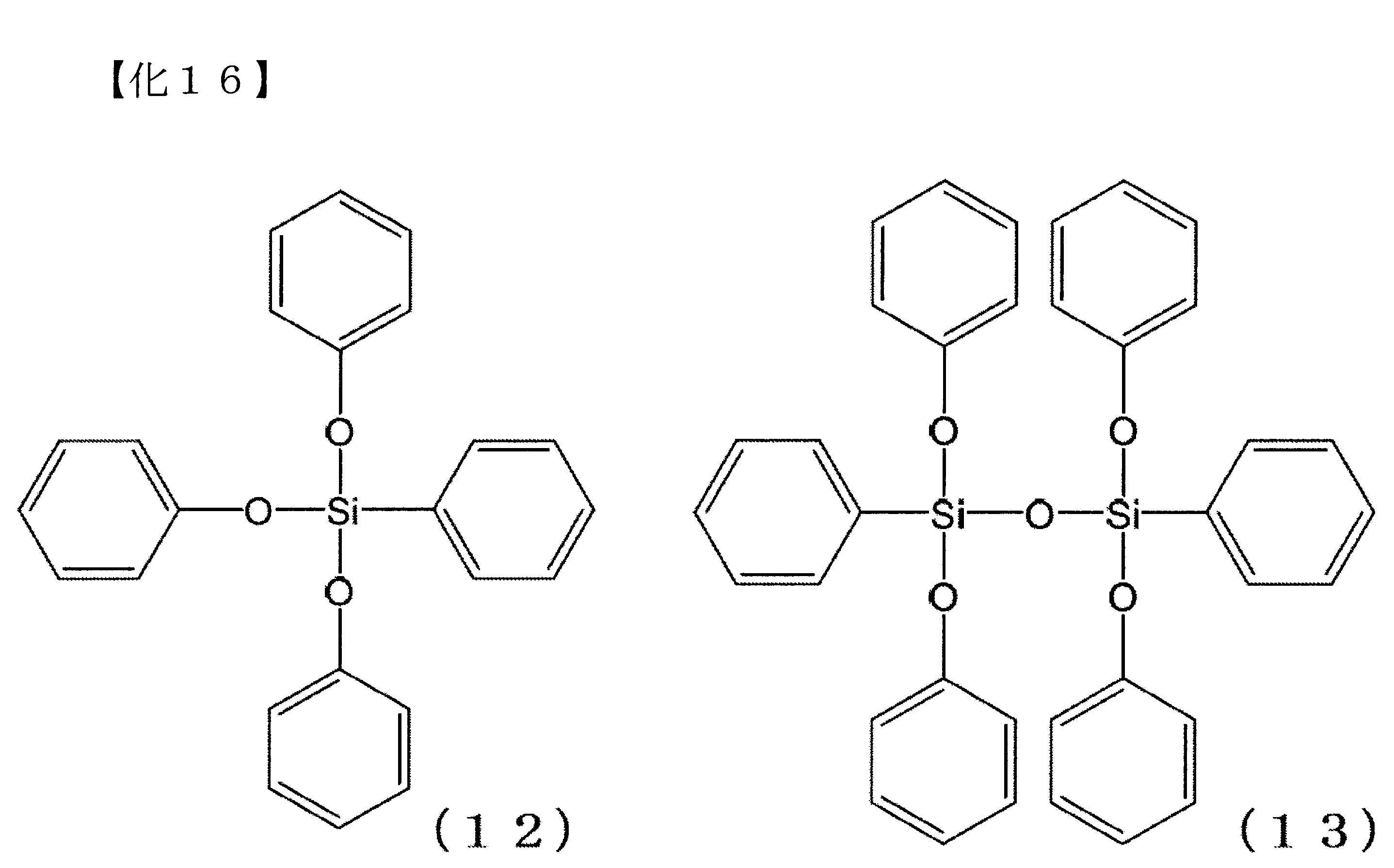
(實施例25)
將滴液漏斗、溫度計、冷卻管安裝至500ml四口燒瓶,並饋入二苯基碳酸酯246.35g,1.15mol)及作為觸媒之碳酸銫0.3748g(1.15 mmol),不攪拌下加熱至180℃。於100℃附近二苯基碳酸酯溶解。到達180℃後,花費90分鐘將十甲基環五矽氧烷85.30g(0.23mol)以滴下速度1ml/分而滴下。滴下十甲基環五矽氧烷後即產生CO2
氣體。在十甲基環五矽氧烷的滴下結束後升溫至200℃,以200℃加熱90分後停止加熱,結束反應。
接著,將反應混合物冷卻至40℃後,藉由在減壓度4hPa、150℃下進行減壓蒸餾,得到270g與實施例1同樣之無色油狀成分。
(比較例1)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)、二苯基碳酸酯21.4g(100.0 mmol)及作為觸媒之碳酸銫33mg(0.1 mmol),置換為氮氣環境下,並在室溫攪拌60分鐘。
以1
H-NMR分析混合物的結果,作為矽上的甲基之質子的波峰,僅觀測到基於十甲基環五矽氧烷之質子的波峰,十甲基環五矽氧烷的轉化率為0%。
(比較例2)
將十甲基環五矽氧烷7.4g(20.0 mmol,Si莫耳量:100.0 mmol)及二苯基碳酸酯21.4g(100.0 mmol),置換為氮氣環境下,並以200℃攪拌60分鐘。
以1
H-NMR分析混合物的結果,作為矽上的甲基之質子的波峰,僅觀測到基於十甲基環五矽氧烷之質子的波峰,十甲基環五矽氧烷的轉化率為0%。
(比較例3)
將十甲基環五矽氧烷0.37g(1.0 mmol,Si莫耳量:5.0 mmol)、苯酚0.94g(10.0 mmol)及作為觸媒之碳酸銫3.3mg (0.01 mmol),置換為氮氣環境下,並以175℃攪拌90分鐘。
以1
H-NMR分析反應混合物的結果,作為矽上的甲基之質子的波峰,觀測到基於十甲基環五矽氧烷之質子的波峰0.055ppm及基於二甲基二苯氧基矽烷之質子的波峰0.378ppm。由兩波峰面積之比率算出之十甲基環五矽氧烷的轉化率為4%。
作為矽上的甲基之質子的波峰,觀測到基於二甲基二苯氧基矽烷之波峰0.378ppm、及基於1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之波峰0.222ppm。由兩波峰面積比率求出二甲基二苯氧基矽烷與1,1,3,3-四甲基-1,3-二苯氧基二矽氧烷之莫耳比率,為59.6:40.4。
將各實施例及比較例之結果示於以下之表2、表3。

Claims (17)
- 一種矽烷化合物之製造方法,其係包含二芳氧基矽烷化合物、二烷氧基矽烷化合物、單芳氧基單烷氧基矽烷化合物、三芳氧基矽烷化合物及三烷氧基矽烷化合物之至少任一者的矽烷化合物之製造方法,其具有下述D)矽氧烷分解步驟: 該D)矽氧烷分解步驟係將含有 A)矽氧烷化合物,其係下述式(1)表示之A-1)環狀矽氧烷化合物、下述式(2)表示之A-2)直鏈狀矽氧烷化合物及/或將矽氧烷鍵作為主鏈骨架之下述式(3)表示之倍半矽氧烷化合物; B)碳酸酯化合物,其包含二芳基碳酸酯、二烷基碳酸酯及單烷基單芳基碳酸酯之至少任一者;以及 C)鹼性化合物觸媒 之混合物進行加熱,而使A)之矽氧烷化合物烷氧基化及/或芳氧基化,(式中,R1 及R2 各自獨立地表示可具有取代基之烷基、烯基或芳基, n表示3以上30以下之整數)
 (式中,R3 及R4 各自獨立地表示可具有取代基之烷基、烯基或芳基, m表示2以上10000以下之整數, X各自獨立地表示氫原子、羥基、可具有取代基之合計碳數1~10之烷氧基、可具有取代基且可具有氧原子或氮原子之合計碳數1~10之烴基、或可具有取代基之胺基)
(式中,R3 及R4 各自獨立地表示可具有取代基之烷基、烯基或芳基, m表示2以上10000以下之整數, X各自獨立地表示氫原子、羥基、可具有取代基之合計碳數1~10之烷氧基、可具有取代基且可具有氧原子或氮原子之合計碳數1~10之烴基、或可具有取代基之胺基)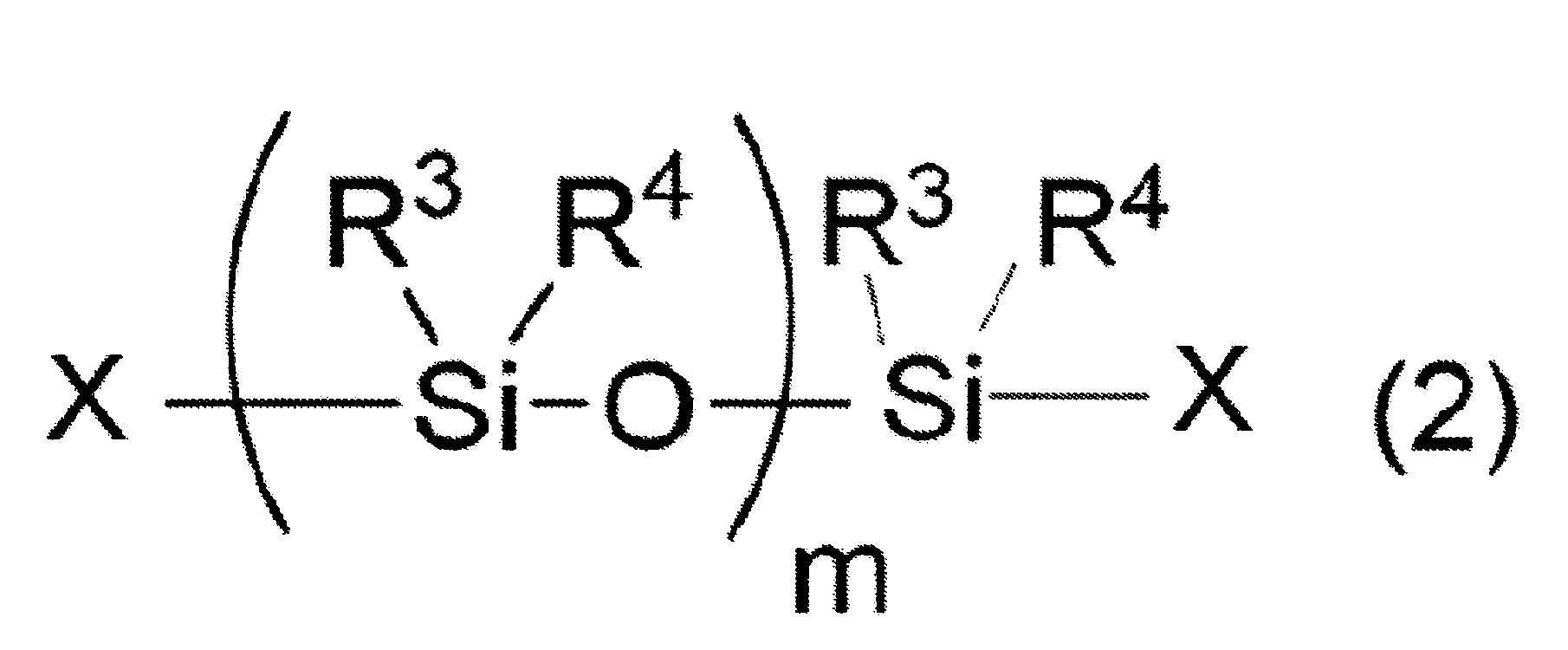 (式中,R5 表示可具有取代基之碳數1~4之烷基、碳數2~4之烯基或碳數6~12之芳基, p為4以上24以下之數)。
(式中,R5 表示可具有取代基之碳數1~4之烷基、碳數2~4之烯基或碳數6~12之芳基, p為4以上24以下之數)。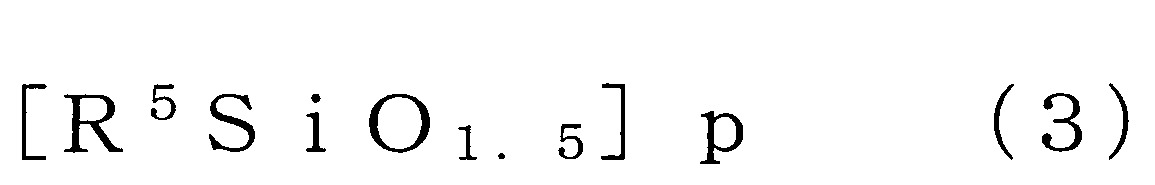
- 一種矽烷化合物之製造方法,其係包含二芳氧基矽烷化合物、二烷氧基矽烷化合物及單芳氧基單烷氧基矽烷化合物之至少任一者的矽烷化合物之製造方法,其具有下述D)矽氧烷分解步驟: 該D)矽氧烷分解步驟係將含有 A)下述式(1)表示之A-1)環狀矽氧烷化合物及/或下述式(2)表示之A-2)直鏈狀矽氧烷化合物; B)碳酸酯化合物,其包含二芳基碳酸酯、二烷基碳酸酯及單烷基單芳基碳酸酯之至少任一者;以及 C)鹼性化合物觸媒 之混合物進行加熱,而使A)之矽氧烷化合物烷氧基化及/或芳氧基化,(式中,R1 及R2 各自獨立地表示可具有取代基之烷基、烯基或芳基, n表示3以上30以下之整數)
 (式中,R3 及R4 各自獨立地表示可具有取代基之烷基、烯基或芳基, m表示2以上10000以下之整數, X各自獨立地表示氫原子、羥基、可具有取代基之合計碳數1~10之烷氧基、可具有取代基且可具有氧原子或氮原子之合計碳數1~10之烴基、或可具有取代基之胺基)。
(式中,R3 及R4 各自獨立地表示可具有取代基之烷基、烯基或芳基, m表示2以上10000以下之整數, X各自獨立地表示氫原子、羥基、可具有取代基之合計碳數1~10之烷氧基、可具有取代基且可具有氧原子或氮原子之合計碳數1~10之烴基、或可具有取代基之胺基)。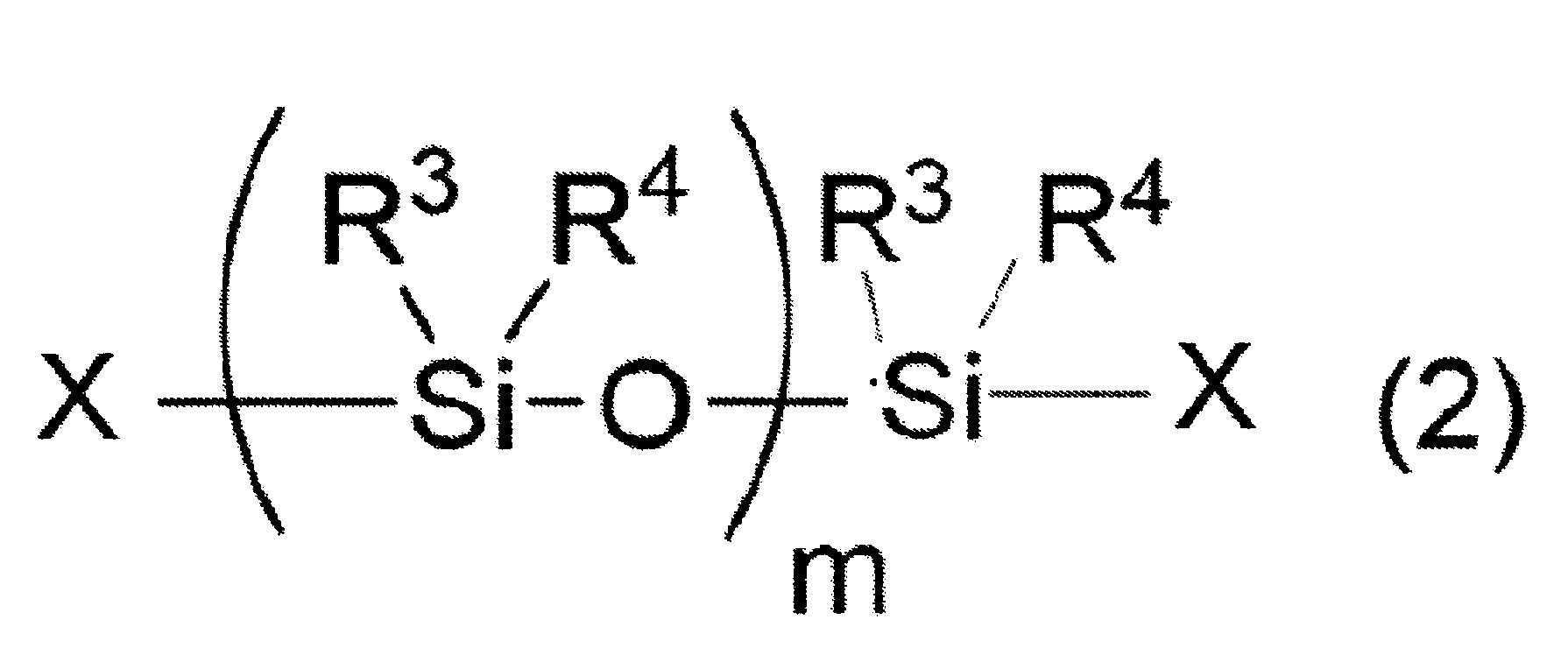
- 如請求項1或2之矽烷化合物之製造方法,其中,前述R1 ~R4 各自獨立地表示可具有取代基之合計碳數1~8之烷基、烯基或合計碳數6~30之芳基。
- 如請求項3之矽烷化合物之製造方法,其中,前述R1 ~R4 各自獨立地為選自由甲基、苯基、乙烯基及丙基所成群中之任一種。
- 如請求項1~4中任一項之矽烷化合物之製造方法,其中,前述B)二芳基碳酸酯包含二苯基碳酸酯。
- 如請求項1~4中任一項之矽烷化合物之製造方法,其中,前述B)二烷基碳酸酯中之烷基的碳數為4以下。
- 如請求項1~6中任一項之矽烷化合物之製造方法,其中,前述C)鹼性化合物觸媒包含鹼金屬碳酸鹽或鹼金屬氫氧化物。
- 如請求項7之矽烷化合物之製造方法,其中,前述C)鹼性化合物觸媒包含碳酸銫及碳酸鉀之至少任一者。
- 如請求項1~8中任一項之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,前述B)碳酸酯化合物的莫耳量相對於前述A)矽氧烷化合物的Si莫耳量之比x為x≧1。
- 如請求項1~8中任一項之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,前述B)碳酸酯化合物的莫耳量相對於前述A)矽氧烷化合物的前述Si莫耳量之比x為0.8<x<2.0。
- 如請求項1~10中任一項之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,前述C)鹼性化合物觸媒的莫耳量相對於前述A)矽氧烷化合物的前述Si莫耳量之比y為0.0001 mmol/mol≦y≦20 mmol/mol。
- 如請求項1~11中任一項之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,使前述A)矽氧烷化合物分解之溫度為50℃以上300℃以下。
- 如請求項12之矽烷化合物之製造方法,其中,前述D)矽氧烷分解步驟中,前述溫度為50℃以上150℃以下。
- 如請求項1之矽烷化合物之製造方法,其中,式(1)表示之前述A-1)環狀矽氧烷化合物之分子量為2,000以下,式(2)表示之前述A-2)直鏈狀矽氧烷化合物之分子量為60,000以下,式(3)表示之前述A-3)倍半矽氧烷化合物之分子量為3,500以下。
- 如請求項1~14中任一項之矽烷化合物之製造方法,其進而具有E)蒸餾步驟/再結晶步驟, 該E)蒸餾步驟/再結晶步驟係,蒸餾由前述D)矽氧烷分解步驟所生成之前述矽烷化合物之蒸餾步驟,與再結晶由前述D)矽氧烷分解步驟所生成之前述矽烷化合物之再結晶步驟的至少任一者。
- 如請求項14之矽烷化合物之製造方法,其中,前述E)之前述蒸餾步驟中之壓力為1hPa以上20hPa以下。
- 如請求項1~16中任一項之矽烷化合物之製造方法,其中,進而具有對含有前述B)碳酸酯化合物及前述C)鹼性化合物觸媒之混合物,滴下前述A)矽氧烷化合物之F)滴下步驟。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019055617 | 2019-03-22 | ||
| JP2019-055617 | 2019-03-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| TW202100534A true TW202100534A (zh) | 2021-01-01 |
| TWI850354B TWI850354B (zh) | 2024-08-01 |
Family
ID=
Also Published As
| Publication number | Publication date |
|---|---|
| EP3943471A1 (en) | 2022-01-26 |
| US20220185830A1 (en) | 2022-06-16 |
| CN113574034B (zh) | 2024-03-08 |
| WO2020196344A1 (ja) | 2020-10-01 |
| CN113574034A (zh) | 2021-10-29 |
| KR20210143714A (ko) | 2021-11-29 |
| EP3943471A4 (en) | 2022-05-04 |
| JPWO2020196344A1 (zh) | 2020-10-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3616757B2 (ja) | 環状シラザン、その製法およびビスアミノアルキル−末端ジシロキサンの製法 | |
| JP2002128896A (ja) | アミノ官能性オルガノシロキサンの製法 | |
| CN107250219A (zh) | 高纯度的氨基硅氧烷 | |
| JP2695751B2 (ja) | 有機珪素基を有する酸素含有クロルホスファゼン、その製法及び有機珪素化合物の縮合法及び平衡化法 | |
| JP2007106894A (ja) | ポリシランの製造方法 | |
| JP2009263316A (ja) | 不完全縮合オリゴシルセスキオキサンの製造方法 | |
| TW202100534A (zh) | 矽烷化合物之製造方法 | |
| TWI850354B (zh) | 矽烷化合物之製造方法 | |
| JP2000103859A (ja) | エポキシ基含有オルガノポリシロキサン又はエポキシ基含有オルガノシランの製造方法 | |
| JP2005517749A (ja) | アミノメチレン官能性シロキサン | |
| JP5420751B2 (ja) | 1,3−ビス(アミノアルキル)ジシロキサンの合成方法 | |
| WO2007007597A1 (ja) | 新規な有機ケイ素化合物及びその製造方法 | |
| EP0423686A2 (en) | Silacyclobutanes and process for preparation | |
| JPH0493326A (ja) | アルコキシ官能性オルガノポリシロキサンの製造方法 | |
| JP3236409B2 (ja) | オルガノシロキシ置換ポリシラン及びその製造方法 | |
| JP2012107011A (ja) | アルコキシ置換された1,2−ビス−シリル−エタンの製造法 | |
| JPH07196805A (ja) | フェノール基を有するシロキサン化合物 | |
| US7847116B2 (en) | Method of manufacturing an aminoaryl-containing organosilicon compound and method of manufacturing an intermediate product of the aforementioned compound | |
| JP2000072781A (ja) | アリールシリルエーテル化合物の脱シリル化方法およびフェノール基含有ケイ素化合物の製造方法 | |
| JPH1121289A (ja) | 3−アミノプロピルシロキサンの製造方法 | |
| JP2619086B2 (ja) | アミノシラン化合物の製造方法 | |
| JP3486203B2 (ja) | アルコキシシランの製造方法 | |
| JP2652888B2 (ja) | α―クロロ―ω―ハイドロジェンオルガノポリシロキサンの製造法 | |
| JP4172342B2 (ja) | 環状有機ケイ素化合物及びその製造方法 | |
| JPH11315082A (ja) | 嵩高い炭化水素基を有するジオルガノジアルコキシシランの製造方法 |