TW202010771A - 球狀聚甲基倍半矽氧烷粒子 - Google Patents
球狀聚甲基倍半矽氧烷粒子 Download PDFInfo
- Publication number
- TW202010771A TW202010771A TW108123231A TW108123231A TW202010771A TW 202010771 A TW202010771 A TW 202010771A TW 108123231 A TW108123231 A TW 108123231A TW 108123231 A TW108123231 A TW 108123231A TW 202010771 A TW202010771 A TW 202010771A
- Authority
- TW
- Taiwan
- Prior art keywords
- particles
- particle
- spherical polymethylsilsesquioxane
- spherical
- mass
- Prior art date
Links
- 239000002245 particle Substances 0.000 title claims abstract description 520
- 229920003217 poly(methylsilsesquioxane) Polymers 0.000 title claims abstract description 175
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims abstract description 70
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 51
- 229910052710 silicon Inorganic materials 0.000 claims abstract description 49
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 41
- 238000000034 method Methods 0.000 claims abstract description 36
- 230000001186 cumulative effect Effects 0.000 claims abstract description 30
- 125000004430 oxygen atom Chemical group O* 0.000 claims abstract description 29
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims abstract description 24
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 13
- 238000001179 sorption measurement Methods 0.000 claims abstract description 8
- 238000000790 scattering method Methods 0.000 claims abstract description 7
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 114
- 229910052799 carbon Inorganic materials 0.000 claims description 41
- 239000003795 chemical substances by application Substances 0.000 claims description 32
- 230000002209 hydrophobic effect Effects 0.000 claims description 23
- 238000004381 surface treatment Methods 0.000 claims description 15
- -1 trimethylsiloxy group Chemical group 0.000 claims description 15
- 125000005389 trialkylsiloxy group Chemical group 0.000 claims description 12
- 125000004432 carbon atom Chemical group C* 0.000 claims description 11
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 9
- 238000004448 titration Methods 0.000 claims description 7
- 239000007788 liquid Substances 0.000 description 113
- 239000006185 dispersion Substances 0.000 description 112
- 239000007864 aqueous solution Substances 0.000 description 101
- 239000003960 organic solvent Substances 0.000 description 98
- 239000000843 powder Substances 0.000 description 76
- 238000011282 treatment Methods 0.000 description 73
- 238000006068 polycondensation reaction Methods 0.000 description 54
- 239000012295 chemical reaction liquid Substances 0.000 description 43
- 238000005481 NMR spectroscopy Methods 0.000 description 41
- 238000005259 measurement Methods 0.000 description 41
- 239000007787 solid Substances 0.000 description 37
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 36
- 230000032683 aging Effects 0.000 description 34
- 239000011259 mixed solution Substances 0.000 description 32
- 239000000243 solution Substances 0.000 description 32
- 230000000052 comparative effect Effects 0.000 description 31
- IUVCFHHAEHNCFT-INIZCTEOSA-N 2-[(1s)-1-[4-amino-3-(3-fluoro-4-propan-2-yloxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]ethyl]-6-fluoro-3-(3-fluorophenyl)chromen-4-one Chemical compound C1=C(F)C(OC(C)C)=CC=C1C(C1=C(N)N=CN=C11)=NN1[C@@H](C)C1=C(C=2C=C(F)C=CC=2)C(=O)C2=CC(F)=CC=C2O1 IUVCFHHAEHNCFT-INIZCTEOSA-N 0.000 description 30
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 30
- 150000001721 carbon Chemical group 0.000 description 30
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 28
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 28
- 239000002994 raw material Substances 0.000 description 25
- 238000006243 chemical reaction Methods 0.000 description 24
- 238000001035 drying Methods 0.000 description 24
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 24
- 238000002156 mixing Methods 0.000 description 24
- 238000000967 suction filtration Methods 0.000 description 24
- 230000000694 effects Effects 0.000 description 22
- 238000004519 manufacturing process Methods 0.000 description 21
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 18
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 18
- 230000015572 biosynthetic process Effects 0.000 description 17
- 238000004364 calculation method Methods 0.000 description 17
- 239000012736 aqueous medium Substances 0.000 description 14
- 239000002243 precursor Substances 0.000 description 13
- 239000003054 catalyst Substances 0.000 description 11
- 239000011521 glass Substances 0.000 description 11
- 239000000203 mixture Substances 0.000 description 11
- 239000000523 sample Substances 0.000 description 11
- 238000006460 hydrolysis reaction Methods 0.000 description 10
- BFXIKLCIZHOAAZ-UHFFFAOYSA-N methyltrimethoxysilane Chemical compound CO[Si](C)(OC)OC BFXIKLCIZHOAAZ-UHFFFAOYSA-N 0.000 description 10
- 238000009826 distribution Methods 0.000 description 9
- 230000002431 foraging effect Effects 0.000 description 9
- 238000001819 mass spectrum Methods 0.000 description 9
- 239000000463 material Substances 0.000 description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 7
- 235000011114 ammonium hydroxide Nutrition 0.000 description 7
- 239000011248 coating agent Substances 0.000 description 7
- 239000000413 hydrolysate Substances 0.000 description 7
- 239000002244 precipitate Substances 0.000 description 7
- 238000011084 recovery Methods 0.000 description 7
- 239000012756 surface treatment agent Substances 0.000 description 7
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 6
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 6
- 235000011054 acetic acid Nutrition 0.000 description 6
- 125000000217 alkyl group Chemical group 0.000 description 6
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 5
- 239000000654 additive Substances 0.000 description 5
- 150000001875 compounds Chemical class 0.000 description 5
- 239000011347 resin Substances 0.000 description 5
- 229920005989 resin Polymers 0.000 description 5
- 125000006850 spacer group Chemical group 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 150000001298 alcohols Chemical class 0.000 description 4
- 238000000576 coating method Methods 0.000 description 4
- SWXVUIWOUIDPGS-UHFFFAOYSA-N diacetone alcohol Chemical compound CC(=O)CC(C)(C)O SWXVUIWOUIDPGS-UHFFFAOYSA-N 0.000 description 4
- 229920001971 elastomer Polymers 0.000 description 4
- 239000000945 filler Substances 0.000 description 4
- 239000010419 fine particle Substances 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 239000012535 impurity Substances 0.000 description 4
- 239000004973 liquid crystal related substance Substances 0.000 description 4
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 4
- 239000005060 rubber Substances 0.000 description 4
- 238000010079 rubber tapping Methods 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- 125000004469 siloxy group Chemical group [SiH3]O* 0.000 description 4
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000011247 coating layer Substances 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 239000011344 liquid material Substances 0.000 description 3
- 239000003973 paint Substances 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 230000009257 reactivity Effects 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- CPUDPFPXCZDNGI-UHFFFAOYSA-N triethoxy(methyl)silane Chemical compound CCO[Si](C)(OCC)OCC CPUDPFPXCZDNGI-UHFFFAOYSA-N 0.000 description 3
- BGJSXRVXTHVRSN-UHFFFAOYSA-N 1,3,5-trioxane Chemical compound C1OCOCO1 BGJSXRVXTHVRSN-UHFFFAOYSA-N 0.000 description 2
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 description 2
- 230000001133 acceleration Effects 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 150000001335 aliphatic alkanes Chemical group 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- DCFKHNIGBAHNSS-UHFFFAOYSA-N chloro(triethyl)silane Chemical compound CC[Si](Cl)(CC)CC DCFKHNIGBAHNSS-UHFFFAOYSA-N 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 2
- 229960001231 choline Drugs 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- 239000002537 cosmetic Substances 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 229940113088 dimethylacetamide Drugs 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 230000007613 environmental effect Effects 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 238000000445 field-emission scanning electron microscopy Methods 0.000 description 2
- 239000010408 film Substances 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- TZMQHOJDDMFGQX-UHFFFAOYSA-N hexane-1,1,1-triol Chemical compound CCCCCC(O)(O)O TZMQHOJDDMFGQX-UHFFFAOYSA-N 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 238000007561 laser diffraction method Methods 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 238000000691 measurement method Methods 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 239000000123 paper Substances 0.000 description 2
- 230000000704 physical effect Effects 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- 229920001296 polysiloxane Polymers 0.000 description 2
- 238000011085 pressure filtration Methods 0.000 description 2
- 230000035484 reaction time Effects 0.000 description 2
- 238000001878 scanning electron micrograph Methods 0.000 description 2
- 239000003566 sealing material Substances 0.000 description 2
- 229910000077 silane Inorganic materials 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 229920002545 silicone oil Polymers 0.000 description 2
- 239000012798 spherical particle Substances 0.000 description 2
- 230000003068 static effect Effects 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- WGTYBPLFGIVFAS-UHFFFAOYSA-M tetramethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)C WGTYBPLFGIVFAS-UHFFFAOYSA-M 0.000 description 2
- 125000004665 trialkylsilyl group Chemical group 0.000 description 2
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 238000001132 ultrasonic dispersion Methods 0.000 description 2
- 238000005303 weighing Methods 0.000 description 2
- YVHUUEPYEDOELM-UHFFFAOYSA-N 2-ethylpropanedioic acid;piperidin-1-id-2-ylmethylazanide;platinum(2+) Chemical compound [Pt+2].[NH-]CC1CCCC[N-]1.CCC(C(O)=O)C(O)=O YVHUUEPYEDOELM-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 235000002597 Solanum melongena Nutrition 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 239000003377 acid catalyst Substances 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000007605 air drying Methods 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 238000000149 argon plasma sintering Methods 0.000 description 1
- 230000001174 ascending effect Effects 0.000 description 1
- 229910000062 azane Inorganic materials 0.000 description 1
- 230000008033 biological extinction Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- SXPLZNMUBFBFIA-UHFFFAOYSA-N butyl(trimethoxy)silane Chemical compound CCCC[Si](OC)(OC)OC SXPLZNMUBFBFIA-UHFFFAOYSA-N 0.000 description 1
- KOPOQZFJUQMUML-UHFFFAOYSA-N chlorosilane Chemical class Cl[SiH3] KOPOQZFJUQMUML-UHFFFAOYSA-N 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 239000000805 composite resin Substances 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 125000006165 cyclic alkyl group Chemical group 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- JJQZDUKDJDQPMQ-UHFFFAOYSA-N dimethoxy(dimethyl)silane Chemical compound CO[Si](C)(C)OC JJQZDUKDJDQPMQ-UHFFFAOYSA-N 0.000 description 1
- LIKFHECYJZWXFJ-UHFFFAOYSA-N dimethyldichlorosilane Chemical compound C[Si](C)(Cl)Cl LIKFHECYJZWXFJ-UHFFFAOYSA-N 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical class [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 239000004744 fabric Substances 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- POPACFLNWGUDSR-UHFFFAOYSA-N methoxy(trimethyl)silane Chemical compound CO[Si](C)(C)C POPACFLNWGUDSR-UHFFFAOYSA-N 0.000 description 1
- HLXDKGBELJJMHR-UHFFFAOYSA-N methyl-tri(propan-2-yloxy)silane Chemical compound CC(C)O[Si](C)(OC(C)C)OC(C)C HLXDKGBELJJMHR-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 238000006011 modification reaction Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000003961 organosilicon compounds Chemical class 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 238000005498 polishing Methods 0.000 description 1
- 229920000555 poly(dimethylsilanediyl) polymer Polymers 0.000 description 1
- 239000002861 polymer material Substances 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 230000010349 pulsation Effects 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 238000004064 recycling Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 239000004065 semiconductor Substances 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 238000004904 shortening Methods 0.000 description 1
- 150000003377 silicon compounds Chemical class 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229940073455 tetraethylammonium hydroxide Drugs 0.000 description 1
- LRGJRHZIDJQFCL-UHFFFAOYSA-M tetraethylazanium;hydroxide Chemical compound [OH-].CC[N+](CC)(CC)CC LRGJRHZIDJQFCL-UHFFFAOYSA-M 0.000 description 1
- HUZZQXYTKNNCOU-UHFFFAOYSA-N triethyl(methoxy)silane Chemical compound CC[Si](CC)(CC)OC HUZZQXYTKNNCOU-UHFFFAOYSA-N 0.000 description 1
- 239000005051 trimethylchlorosilane Substances 0.000 description 1
- OLTVTFUBQOLTND-UHFFFAOYSA-N tris(2-methoxyethoxy)-methylsilane Chemical compound COCCO[Si](C)(OCCOC)OCCOC OLTVTFUBQOLTND-UHFFFAOYSA-N 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 239000002966 varnish Substances 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/045—Polysiloxanes containing less than 25 silicon atoms
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/14—Polysiloxanes containing silicon bound to oxygen-containing groups
- C08G77/18—Polysiloxanes containing silicon bound to oxygen-containing groups to alkoxy or aryloxy groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/06—Preparatory processes
- C08G77/08—Preparatory processes characterised by the catalysts used
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/12—Powdering or granulating
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/12—Powdering or granulating
- C08J3/16—Powdering or granulating by coagulating dispersions
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Dispersion Chemistry (AREA)
- Silicon Polymers (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
本發明提供具有以前所沒有的較大的比表面積比率之新穎的球狀聚甲基倍半矽氧烷粒子;該球狀聚甲基倍半矽氧烷粒子具有如下特徵:具有至少含有由矽原子(n)和與矽原子(n)結合的氧原子(n)形成之交聯網狀結構、與矽原子(n)結合之甲基、與矽原子(n)結合之烷氧基的粒子主體,並且,滿足以下的算式(1);
算式(1) S1
/S2
≥8.0
在算式(1)中,S1
係透過氮吸附BET單點法測量之比表面積(m2
/g),S2
係6/(ρ×D50)且其單位為(m2
/g),ρ係粒子密度(g/m3
),D50係透過雷射繞射散射法測量之體積基準的累積50%粒徑(m)。
Description
本發明係有關於球狀聚甲基倍半矽氧烷粒子(spherical polymethylsilses quioxane particle)。
粒子表面和內部具有疏水性的烷基之聚甲基倍半矽氧烷粉末,一般其環境穩定性、分散性、耐熱性、耐溶劑性、憎水性等出色。因此,聚甲基倍半矽氧烷粉末使用於高分子材料用改性劑或塗覆材料、調色劑用外添劑、液晶用密封材料之間隔物(spacer)、塗料或化妝品的添加劑等的各種用途中。
關於聚甲基倍半矽氧烷粉末及其製造方法,已提案有各種。例如,在專利文獻1中,作為塗料的添加劑,報告有如下內容,即:使甲基三烷氧基矽烷水解或縮合,將其生成物水洗後乾燥,從而得到聚甲基倍半矽氧烷粉末。另外,在專利文獻2中,報告有如下內容,即:使有機三烷氧基矽烷之水解或部分縮合物在鹼性水溶液中進行縮聚反應,從而得到聚有機倍半矽氧烷微粒。
在專利文獻3中,作為液晶顯示器之防反射薄膜的用途,報告有如下內容,即:使有機三烷氧基矽烷水解或部分縮合,將透過靜置分層的下層之部分縮合物的醇混合物與鹼性水溶液混合,從而得到球狀聚有機倍半矽氧烷微粒。另外,在專利文獻4中,揭示有:作為調色劑用外添劑等有用的中值粒徑在0.05μm~0.3μm範圍內之疏水化球狀聚有機倍半矽氧烷微粒。
[先前技術文獻]
[專利文獻]
專利文獻1:日本專利特開昭60-13813號公報
專利文獻2:日本專利特開平1-217039號公報
專利文獻3:日本專利特開2008-208158號公報
專利文獻4:第2015/107961號國際公開 小冊子
在上述聚甲基倍半矽氧烷粉末等的各種二氧化矽類粉末的市場中,存在有如下(ⅰ)和(ⅱ)的市場,其中,(ⅰ)的市場是指:根據使用用途設計粉末的各種特性之需求對應型的市場,(ⅱ)的市場是指:在開發出具有以前所沒有的新穎特性之粉末的基礎上,著眼於該粉末的新穎特性,開拓新用途,或者,代替在規定用途中已經使用之現有產品(市場上出售的粉末)等的需求開拓型的市場。而且,在二氧化矽類粉末中,實際情況是,在大多數情況下,透過需求開拓型的市場,創造或者獲得一個以上的新市場。因此,在不被既存之需求束縛的情況下,開發出具有以前所沒有的新穎特性之聚甲基倍半矽氧烷粉末,是極其重要的。
另一方面,本發明人們對專利文獻1~4等中所例示之現有的聚甲基倍半矽氧烷粉末進行了研究後,掌握了如下情況,即:關於實際比表面積相對於根據粒徑等計算出的比表面積(理論比表面積)之比率(以下,有時稱為「比表面積比率」),在專利文獻4所揭示之粉末中具有較大的值,但並不存在進一步超過該值的粉末。
本發明是鑒於上述情況而完成的,其課題在於提供一種具有以前所沒有的較大的比表面積比率之新穎的球狀聚甲基倍半矽氧烷粒子。
上述課題透過以下的本發明實現。即,
本發明之球狀聚甲基倍半矽氧烷粒子,具有如下特徵。
具有至少含有由矽原子(n)和與矽原子(n)結合的氧原子(n)形成之交聯網狀結構、與矽原子(n)結合之甲基、與矽原子(n)結合之烷氧基的粒子主體,並且,滿足以下的算式(1);
算式(1) S1
/S2
≥8.0
(在算式(1)中,S1
係透過氮吸附BET單點法測量之比表面積(m2
/g),S2
係6/(ρ×D50)且其單位為(m2
/g),ρ係粒子密度(g/m3
),D50係透過雷射繞射散射法測量之體積基準的累積50%粒徑(m)。)
在本發明之球狀聚甲基倍半矽氧烷粒子的一實施方式中,較佳係:算式(1)中所示之比率S1
/S2
為10.0以上。
在本發明之球狀聚甲基倍半矽氧烷粒子的另一實施方式中,較佳係:粒子主體之表面未被實施表面處理。
在本發明之球狀聚甲基倍半矽氧烷粒子的另一實施方式中,較佳係:透過甲醇滴定法測量之疏水度為25 vol%~45% vol%。
在本發明之球狀聚甲基倍半矽氧烷粒子的另一實施方式中,較佳係:粒子主體之表面利用疏水劑進行表面處理。
在本發明之球狀聚甲基倍半矽氧烷粒子的另一實施方式中,較佳係:粒子主體之至少表面附近含有與矽原子(n)結合之三烷基矽氧基。
在本發明之球狀聚甲基倍半矽氧烷粒子的另一實施方式中,較佳係:三烷基矽氧基是三甲基矽氧基。
在本發明之球狀聚甲基倍半矽氧烷粒子的另一實施方式中,較佳係:透過甲醇滴定法測量之疏水度超過45 vol%且70 vol%以下。
在本發明之球狀聚甲基倍半矽氧烷粒子的另一實施方式中,較佳係:滿足以下的算式(2);
算式(2) 0.016≤A/B≤0.030
(在算式(2)中,A和B係透過13
C DDMAS NMR測量之峰的面積,A係源於烷氧基中含有的碳原子(該碳原子是與烷氧基中所含有之氧原子(tg)結合的碳原子)之峰的面積,B係源於與矽原子(n)結合之甲基中所含有碳原子之峰的面積。)
在本發明之球狀聚甲基倍半矽氧烷粒子的另一實施方式中,較佳係:算式(2)中所示之比率A/B為0.020~0.030。
在本發明之球狀聚甲基倍半矽氧烷粒子的另一實施方式中,較佳係:累積50%粒徑是0.14μm~2.0μm。
在本發明之球狀聚甲基倍半矽氧烷粒子的另一實施方式中,較佳係:累積50%粒徑超過0.30μm且2.0μm以下。
在本發明之球狀聚甲基倍半矽氧烷粒子的另一實施方式中,較佳係:累積50%粒徑超過0.30μm。
在本發明之球狀聚甲基倍半矽氧烷粒子的另一實施方式中,較佳係:烷氧基是甲氧基。
(發明功效)
根據本發明,能夠提供一種具有以前所沒有的較大的比表面積比率之新穎的球狀聚甲基倍半矽氧烷粒子。
本實施方式之球狀聚甲基倍半矽氧烷粒子(spherical polymethyl silsesquioxane particle)(以下,有時僅簡稱為「粒子」)具有如下特徵,即:具有至少含有由矽原子(n)和與矽原子(n)結合的氧原子(n)形成之交聯網狀結構、與矽原子(n)結合之甲基以及與矽原子(n)結合之烷氧基的粒子主體,並且,滿足以下的算式(1)。
算式(1) S1
/S2
≥8.0
(在算式(1)中,S1
係透過氮吸附BET單點法(nitrogen adsorption BET single point method)測量之比表面積(m2
/g),S2
係6/(ρ×D50)且其單位為(m2
/g),ρ係粒子密度(g/m3
),D50係透過雷射繞射散射法(Laser diffraction scattering method)測量之體積基準的累積50%粒徑(m)。)
在此,在算式(1)中,S1
係本實施方式之粒子的實際比表面積(BET比表面積),S2
係本實施方式之粒子的根據粒徑等計算出的比表面積(理論比表面積),因此,S1
/S2
係該二個比表面積之比率。因此,比表面積比率S1
/S2
的值越接近1,則表示粒子的表面更加平滑,比表面積比率S1
/S2
的值越大,則表示粒子的表面變得不平滑,換而言之,表示粒子的表面變得更加粗糙(例如,粒子的表面上形成有很多微細的凹凸等),以及/或者,在粒子之至少表面附近存在有很多可氮滲透之微細孔。而且,在本實施方式之球狀聚甲基倍半矽氧烷粒子中,比表面積比率S1
/S2
為8.0以上,這樣的比表面積比率S1
/S2
的值,是現有的球狀聚甲基倍半矽氧烷粒子中所無法實現的值。
另外,關於比表面積比率S1
/S2
,可以在8.0以上的範圍內,根據本實施方式之粒子的用途等適當地進行選擇,但一般而言,較佳係10.0以上,更較佳係12.0以上,進一步較佳係15.0以上。另外,比表面積比率S1
/S2
之上限值並沒有特別限定,但一般而言,較佳係100以下,更較佳係50以下。另外,比表面積S1
並沒有特別限定,但較佳係30m2
/g~500m2
/g,更較佳係50m2
/g~350m2
/g。
另外,在具有8.0以上的比表面積比率S1
/S2
之本實施方式之粒子中,例如,可以期待發揮以下所列舉之各種作用功效。
(a)與現有的粒子相比,在本實施方式之粒子中,當使本實施方式之粒子附著於其他固體部件表面上時,能夠使接觸面積變大。因此,能夠使本實施方式之粒子相對於其他固體部件表面的密合性變得更大。
(b)當使本實施方式之粒子分散於其他硬化性液態材料中之後,使硬化性液態材料硬化時,本實施方式之粒子固定在硬化性液態材料硬化而成的矩陣(matrix)中。另一方面,與現有的粒子相比,本實施方式之粒子的表面更加粗糙。因此,本實施方式之粒子,利用更大的錨固效應(anchor effect)牢固地固定在矩陣中。
(c)當假設現有粒子的粒徑和本實施方式之粒子的粒徑相同時,與現有的粒子相比,本實施方式之粒子的實際表面積更大。因此,在本實施方式之粒子中,能夠更多地吸附或者保持與粒子表面的親和性高的物質。
(d)與現有粒子相比,本實施方式之粒子的表面更加粗糙。因此,本實施方式之粒子能夠更加有效地散射從外部入射的光。
因此,透過至少著眼於上述(a)~(d)中所例示之主要因為較大的比表面積比率S1
/S2
而產生之各種作用功效、或者主要因為只是比表面積比率S1
/S2
較大而產生之各種作用功效,可以期待將本實施方式之粒子利用於以下所例示之各種產業上的用途中。此外,當為現有粒子已經被利用之用途時,可以期待在該用途中將本實施方式之粒子用作現有粒子之代替品。
(A)作為至少著眼於上述(a)中所示作用功效之產業上的利用例,可以舉出以將二個固體部件之間的間隔保持為大致固定間隔等的目的而配置於二個固體部件之間之間隔物(spacer)。當將本實施方式之粒子用作間隔物時,可以期待容易抑制本實施方式之粒子相對於與固體部件表面平行之方向發生位置偏移。作為這樣的間隔物用途之代表性的具體例,可以舉出用於將形成於二個基板間的液晶層之厚度保持為大致固定厚度之液晶用間隔物。
(B)作為至少著眼於上述(b)中所示作用功效之產業上的利用例,可以舉出:在分散含有粒子之固體部件的使用中,不希望粒子從固體部件主體剝離脫落的部件。作為這種用途的具體例,可以舉出作為研磨劑分散含有本實施方式之粒子的研磨墊、或者作為填料分散含有本實施方式之粒子的齒科用複合樹脂等。
(C)作為至少著眼於上述(c)中所示作用功效之產業上的利用例,可以舉出將本實施方式之粒子用作各種吸附劑或者載體劑。
(D)作為至少著眼於上述(d)中所示作用功效之產業上的利用例,可以舉出對使用本實施方式之粒子的部件賦予因為較大的光散射性而生成之光學特性(例如,消光、不透明性等)的情況。作為這種用途的具體例,可以舉出作為填料或光學特性調整材料含有本實施方式之粒子的橡膠部件、樹脂部件或塗料、或者使用本實施方式之粒子的化妝品等。
(E)作為至少著眼於只是比表面積比率S1
/S2
較大的情況之產業上的利用例,例如,可以舉出將本實施方式之粒子用作研磨劑。本實施方式之球狀聚甲基倍半矽氧烷粒子與現有的球狀聚甲基倍半矽氧烷粒子相比,比表面積比率S1
/S2
較大地不同。這表示:在進行研磨時,粒子和固體表面之間的接觸滑動形態較大地不同。因此,與粒徑和材質實質上相同之現有的球狀聚甲基倍半矽氧烷粒子相比,根據本實施方式之粒子,可以期待獲得與現有粒子不同的新穎的研磨特性。
需要說明的是,上述(a)~(d)中所示之作用功效僅僅是其一例,因為比表面積比率S1
/S2
在8.0以上而產生之作用功效,並不限制存在有上述(a)~(d)中所例示以外之其他未知的作用功效。另外,上述(A)~(E)中所示之本實施方式之粒子的產業上的利用例,僅僅是舉例說明,其並不是用來限制本實施方式之粒子的產業上的其他可利用性。作為本實施方式之粒子的利用例或用途,也可以是上述(A)~(E)中所例示以外的其他利用例或用途,例如,還可以舉出半導體封固材料或薄膜等的填料、塑料、橡膠、紙等的改性用添加劑等。
另外,在將本實施方式之粒子利用於產業上的各種用途中的情況下,(ⅰ)在某一個用途中,可以主要著眼於較大的比表面積比率S1
/S2
或因此而產生之作用功效而利用本實施方式之粒子,(ⅱ)在某一個用途中,可以著眼於較大的比表面積比率S1
/S2
或因此而產生之作用功效、和其他特性或者其作用功效之複合性組合而利用本實施方式之粒子,(ⅲ)在某一個用途中,主要著眼於其他特性或者其作用功效而利用本實施方式之粒子。需要說明的是,在上述(ⅲ)中所示之情況下,(ⅲa)可以與本實施方式之粒子的利用者的意圖無關地,只是在其結果上,較大的比表面積比率S1
/S2
或因此而產生之作用功效,貢獻於某一個用途中的某種特性的改善,或者,(ⅲb)可以與本實施方式之粒子的利用者的意圖無關地,只是在其結果上,較大的比表面積比率S1
/S2
或因此而產生之作用功效、和其他特性或者其作用功效之複合性組合,貢獻於某一個用途中的某種特性的改善。
另外,算式(1)中所示之BET比表面積S1
係透過氮吸附BET單點法測量。該測量是按照以下步驟實施。首先,在稱重後的測量容器內,稱量放入0.12g左右的球狀聚甲基倍半矽氧烷粉末。接著,在將填充有球狀聚甲基倍半矽氧烷粉末之測量容器設置在覆套式電阻加熱器(mantle heater)內之後,實施在對覆套式電阻加熱器內部進行氮置換的同時以200℃加熱80分鐘的預處理。之後,將冷卻至室溫之測量容器設置在BET比表面積測量裝置(日本柴田化學株式會社製的SA-1000)內,使用液態氮,使氮氣吸附在球狀聚甲基倍半矽氧烷粉末之表面上。然後,根據該吸附量並透過BET單點法得到BET表面積Sx。測出填充有測量後之球狀聚甲基倍半矽氧烷粉末之測量容器的質量,從該值減去之前預先測量之測量容器自身之質量,從而計算出去掉透過預處理脫離之水分等的質量部分之後的球狀聚甲基倍半矽氧烷粉末之質量m。將上述BET表面積Sx除以該質量m而求出比表面積S1
。在進行該計算時,對於BET表面積Sx,是在將單位標注為m2
時的數值之小數點後第1位四捨五入之後使用於計算中,關於質量m,是在將單位標注為g時的數值之小數點後第3位四捨五入之後使用於計算中。而且,將在該計算中得到的數值之小數點後第2位四捨五入後得到的值,作為BET比表面積S1
。
另外,算式(1)中所示之比表面積S2
的計算中使用之ρ和D50,分別按照如下方式進行了測量。在此,粒子密度ρ是使用乾式自動密度計進行了測量。該測量是按照以下步驟實施。
首先,將在120℃下減壓乾燥處理24小時後之球狀聚甲基倍半矽氧烷粉末,在10ml試料容器中稱重至0.0001g的單位。接著,在將試料容器設置在乾式自動密度計(日本株式會社島津製作所製的AccuPly 1330)之測量室內之後,在使He氣體在測量室內流動的同時,以測量溫度25℃測量了粒子密度。在上述乾式自動密度計中,當輸入提供給測量之粉末質量時,粒子密度是以g/cm3
為單位顯示至小數點後5位。因此,作為比表面積S2
的計算中使用之粒子密度ρ,使用的是如下數值,即:將標注為g/cm3
之粒子密度的小數點後第3位四捨五入後得到的數值,進一步換算為g/m3
單位的數值。
D50是透過雷射繞射散射法測量。該測量是按照以下步驟實施。首先,在將乾燥的0.1g球狀聚甲基倍半矽氧烷粉末放入到玻璃製容器(內徑4㎝、高度11㎝)內之後,進一步添加50g的2-丙醇,從而得到了混合液。接著,在將超音波分散機之探針(probe)(前端的內徑7mm)中之、從其前端起4.5cm為止的部分浸入混合液中之狀態下,以20W的輸出功率進行15分鐘的超音波分散處理,從而得到了分散液。接著,使用該分散液,並使用利用了雷射繞射散射法之粒度分佈測量裝置(美國貝克曼庫爾特有限公司(Beckman Coulter, Inc.)製的LS13 320)測量了體積基準的累積50%粒徑(D50)。另外,在本案說明書中,透過與D50相同的測量方法,還測量出體積基準的累積10%粒徑(D10)以及體積基準的累積90%粒徑(D90)。另外,在上述美國貝克曼庫爾特有限公司製造的LS13 320中,粒徑是以μm為單位標記至小數點後6位。但是,在本案說明書中,是將標記至小數點後6位之粒徑的小數點後第3位四捨五入後得到的數值,採用為粒徑的值。因此,比表面積S2
的計算中使用之粒徑(D50)的值,是將上述四捨五入後之μm單位的數值換算為m單位後使用。
另外,本實施方式之粒子的形狀為球狀。在此,在本案說明書中,「粒子為球狀」是指:在利用電子顯微鏡觀察粒子時,能夠辨別為粒子的形狀實質上呈真球狀之程度,具體而言,圓度較佳係0.80以上,更較佳係0.90以上,進一步較佳係0.92以上。而且,「粒子為球狀」的定義中使用之「圓度」是指累積50%圓度。在此,累積50%圓度是指:在對100個粒子分別測量各個粒子的圓度之後,將100個粒子的圓度按照昇冪排列時,從具有最小值之第一個圓度數起第50個圓度。另外,累積50%圓度的值越接近1,則表示粒子的形狀越接近於真球狀。當累積50%圓度小於0.80時,有時會導致各個粒子彼此的附著性變高,從而各個粒子凝集而形成之凝集塊的破碎性變差。
累積50%圓度的確定所需之各個粒子的圓度,是根據以下算式(2)進行計算。另外,各個粒子的圓度的計算所需之粒子的周長I以及投影面積S3
,是透過利用圖像分析軟體對透過FE-SEM得到之圖像資料進行分析而測量。
算式(2) 圓度=4π×(S3
/I2
)
(在算式(2)中,I係利用場發射掃描電子顯微鏡(Field Emission Scanning Electron Microscope)對粒子進行拍攝得到之SEM圖像上的粒子的周長(nm),S3
係SEM圖像上之粒子的投影面積(nm2
)。)
本實施方式之球狀聚甲基倍半矽氧烷粒子具有至少包括以下(a)~(c)中所示之結構以及基的粒子主體。
(a)由矽原子(n)和與矽原子(n)結合的氧原子(n)形成之交聯網狀結構。
(b)與矽原子(n)結合之甲基(以下,有時僅稱為「(b)甲基」)。
(c)與矽原子(n)結合之烷氧基(以下,有時僅稱為「(c)烷氧基」)。
另外,在本案說明書中,將構成交聯網狀結構之矽原子記載為「矽原子(n)」,將構成交聯網狀結構之氧原子記載為「氧原子(n)」。另外,關於與構成交聯網狀結構之矽原子(n)結合之基(末端基)中含有之矽原子和氧原子,為了與矽原子(n)和氧原子(n)進行區別,分別記載為矽原子(tg)和氧原子(tg)。例如,可以將(c)烷氧基中含有之氧原子記載為「氧原子(tg)」。在此,矽原子(n)與三個氧原子結合,氧原子(n)在交聯網狀結構中與二個矽原子(n)結合。而且,透過這些矽原子(n)和氧原子(n)以三維交聯之方式結合,形成交聯網狀結構。另外,關於與上述矽原子(n)結合之氧原子,並不限於一定是氧原子(n),有時還可以是構成上述(c)烷氧基或者後述之(d)羥基或(e)其他基之氧原子。
另外,關於(c)與矽原子(n)結合之烷氧基,並沒有特別限定,但較佳係碳數1~5之烷氧基,更較佳係碳數1~3之烷氧基,進一步較佳係甲氧基或乙氧基,最較佳係甲氧基。
另外,作為參考說明的話,現有的一般的球狀聚甲基倍半矽氧烷粒子,具有以下通式(1)中所示之基本結構。即,與現有的一般的球狀聚甲基倍半矽氧烷粒子相比,本實施方式之粒子的結構上的主要特徵在於,存在有(c)與矽原子(n)結合之烷氧基。
CH3
SiO3/2
通式(1)
另外,在粒子主體中,除了(a)~(c)所示之結構以及基以外,還可以進一步含有:(d)與矽原子(n)結合之羥基(以下,有時僅稱為「(d)羥基」),以及/或者,(e)與矽原子(n)結合之其他基(以下,有時僅稱為「(e)其他基」)。在此,(e)其他基是指:(b)甲基、(c)烷氧基以及(d)羥基以外的基。另外,關於(d)羥基以及(e)其他基,可以透過適當地選擇粒子主體之粒子形成步驟或者粒子形成步驟完成後之表面處理步驟,將其導入粒子主體中。例如,當採用後述之粒子形成步驟(第一步驟~第三步驟)製造本實施方式之粒子時,在完成第三步驟的時點上,粒子主體中含有(a)~(d)所示之結構以及基。另外,當對包括(a)~(d)所示之結構以及基的粒子主體進行使用了表面處理劑之表面處理(例如,使用了疏水劑的疏水化處理)時,粒子主體之表面附近進一步含有(e)其他基(源於表面處理劑/疏水劑的基)。
在本實施方式之粒子中,粒子主體只要至少包括(a)~(c)所示之結構以及基,便沒有特別限制,作為粒子主體中含有之結構以及基的較佳組合,可以舉出以下所例示之三個形態。
<第一形態>
粒子主體實質上僅含有(a)交聯網狀結構、(b)甲基以及(c)烷氧基之第一形態。
<第二形態>
粒子主體實質上僅含有(a)交聯網狀結構、(b)甲基、(c)烷氧基以及(d)羥基之第二形態。
<第三形態>
粒子主體至少含有(a)交聯網狀結構、(b)甲基、(c)烷氧基以及(e)其他基之第三形態。
在第一形態中,並不排除粒子主體中不可避免地含有微量的(d)羥基以及/或者(e)其他基的情況,另外,在第二形態中,並不排除粒子主體中不可避免地含有微量的(e)其他基的情況。另外,作為粒子主體中不可避免地含有微量的(d)羥基或者(e)其他基的情況的一例,可以舉出粒子主體之粒子形成過程中的不純物的存在、或者與存在於粒子主體之保管環境中的污染成分等的反應等。另外,第一形態和第二形態的粒子主體,通常是透過直接使用經過粒子形成步驟得到之粒子主體,換而言之,是透過對粒子形成步驟結束後之粒子主體的表面不實施任何表面處理而得到。
另外,在本案說明書中,「表面處理」是指:透過使經過粒子形成步驟得到之粒子主體的表面和表面處理劑進行反應,利用表面處理劑對粒子主體之表面進行化學修飾。另外,在進行表面處理時,透過適當地選擇表面處理條件(表面處理劑之種類或濃度、反應溫度或時間等),可以實現所希望的表面修飾反應。作為代表性的表面處理劑,可以舉出疏水劑。
在第三形態中,粒子主體中還可以進一步含有(d)羥基。另外,(e)其他基也可以是存在於粒子主體之至少表面附近的基。該情況下,作為(e)其他基的具體例,可以舉出透過表面處理形成的基(例如,三烷基矽氧基(trialkylsilyloxy group)等的透過疏水化處理形成的基等)。另外,作為第三形態之較佳變化,可以舉出在粒子主體之至少表面附近,作為(e)其他基含有三烷基矽氧基的情況。在此,構成三烷基矽氧基之烷基的碳數較佳係1~3,更較佳係1(換而言之,其他基為三甲基矽氧基)。
另外,在以下的說明中,有時將對粒子主體未實施任何表面處理的本實施方式之粒子稱為「非表面處理粒子」,將利用疏水化處理劑對粒子主體實施表面處理之粒子稱為「疏水化處理粒子」。
在本實施方式之粒子中,粒子主體至少含有(b)甲基以及(c)烷氧基。而且,b)甲基以及(c)烷氧基之烷基部分示出疏水性,因此,非表面處理粒子具有示出疏水性的傾向。另外,相對於通式(1)所示之現有的一般的球狀聚甲基倍半矽氧烷粒子(未實施任何表面處理之粒子),非表面處理粒子還含有(c)烷氧基。因此,與現有的一般的球狀聚甲基倍半矽氧烷粒子(未實施任何表面處理之粒子)相比,非表面處理粒子具有更加不易受到濕度影響,從而在耐氣候性或環境穩定性方面有利的傾向。
另外,關於本實施方式之粒子,根據其需要,可以利用矽油等的包覆劑對粒子主體之表面實施包覆處理,但也可以不實施任何包覆處理。即,關於本實施方式之粒子,(ⅰ)可以由粒子主體和包覆粒子主體表面之包覆層構成,其中,該粒子主體由非表面處理粒子或疏水化處理粒子構成,(ⅱ)也可以僅由粒子主體構成(換而言之,不具有包覆層),其中,該粒子主體由非表面處理粒子或疏水化處理粒子構成。另外,在本案說明書中,「包覆處理」是指利用由包覆劑構成的包覆層包覆粒子主體之表面。在此,包覆劑與表面處理劑不同,是指不與粒子主體之表面進行任何化學反應之材料。
進而,在本實施方式之粒子中,如上所述,作為與形成(a)交聯網狀結構之矽原子(n)結合的基,粒子主體中至少必須含有(b)甲基以及(c)烷氧基,進而,可以含有(d)羥基或(e)其他基。在此,在本實施方式之粒子中,通常,(b)甲基和(c)烷氧基較佳係:還存在於粒子主體之徑向上的任意位置處。與此同樣地,當本實施方式之粒子中進一步含有(d)羥基時,(d)羥基也較佳係:還存在於粒子主體之徑向上的任意位置處。這樣的結構,能夠透過後述之第一或第二製造方法容易地實現。
另一方面,本實施方式之粒子的透過甲醇滴定法測量之疏水度(M值),可以根據粒子的使用用途適當地進行選擇。關於粒子的疏水度,透過適當地選擇粒子形成條件、疏水化處理之有無、實施疏水化處理時的疏水化處理條件,能夠將其在20vol%~70vol%左右的範圍內進行控制。例如,非表面處理粒子具有在上述範圍內示出相對低的疏水度(例如,25vol%~45vol%的範圍)的傾向,而疏水化處理粒子具有在上述範圍內示出相對高的疏水度(例如,超過45vol%且70vol%以下的範圍)的傾向。另外,關於疏水化處理粒子的疏水度,其下限值更較佳在50vol%以上,進一步較佳在55vol%以上,其上限值較佳在65vol%以下。
另外,在含有(d)羥基的粒子中,存在著容易生成透過氫鍵粒子彼此牢固地結合而成的凝集體之傾向。但是,在疏水化處理粒子中,能夠抑制這樣的凝集體的生成。因此,疏水化處理粒子的流動性出色,並且,當使疏水化處理粒子分散含有在各種材料(尤其是樹脂材料)中時,其分散性出色。另外,為了更佳有效地表達這種效果,其有效的方法是:使疏水度變得更大,或者,使粒子主體的表面附近所含有之透過疏水化處理形成的基(例如,三烷基矽氧基等)的量變得更多。
疏水度(M值)是透過甲醇滴定法測量。該測量是按照以下步驟進行。首先,在容積200ml的容器(燒杯)中放入50ml純水和0.2g球狀聚甲基倍半矽氧烷粉末。接著,直到成為球狀聚甲基倍半矽氧烷粉末全部被浸濕且分散於液體中的狀態為止,對容器內之容納物進行攪拌,與此同時從滴定管(burette)向容器內滴加甲醇。將所滴加甲醇量相對於滴加結束時的純水(50ml)和所滴加甲醇的總量之體積百分率的值設為疏水度(M值)。疏水度(M值)值越高,則表示疏水性越高,其值越低,則表示親水性越高。
關於(a)交聯網狀結構、(b)甲基、(c)烷氧基、(d)羥基以及(d)其他基、或者構成這些結構和基的原子,能夠透過NMR測量或FT-IR測量等的公知分析手段進行確定。以下,對利用NMR的測量之詳細情況進行說明。
<13
C DDMAS NMR>
透過利用13
C DDMAS NMR測量,能夠得到有關包含粒子主體中所含有之碳的基((b)甲基、(c)烷氧基、(e)其他基(含有三烷基矽氧基等的碳原子之基的情況))的資訊。關於測量條件的詳細情況之後進行說明,當將外部標準作為甘氨酸的羰基的峰(peak)(176.03ppm)進行測量時,能夠得到以下所例示之各種資訊。
<ⅰ> 在-4ppm附近檢測出(b)甲基的碳原子的峰。
<ⅱ> 當(c)烷氧基為甲氧基時,在-50ppm附近檢測出源於與(c)烷氧基的氧原子(tg)結合之碳原子的峰。
<ⅲ> 當(e)其他基為三甲基矽氧基時,在-1ppm附近檢測出源於與三甲基矽氧基的矽原子(tg)結合之碳原子的峰。
即使是相同的碳原子,其檢測峰也不同,是因為:受到了與該碳原子結合之原子的種類、數量、結合狀態等不同的影響。例如,<ⅰ>(b)甲基的碳原子與形成交聯網狀結構之一個矽原子(n)、以及甲基中所含有之三個氫原子結合。另外,<ⅱ>作為(e)其他基的三甲基矽氧基的碳原子,與三甲基矽氧基中所含有之一個矽原子(tg)、以及三甲基矽氧基的甲基部分中所含有之三個氫原子結合。<ⅰ>和<ⅱ>所示之碳原子,在與一個矽原子和三個氫原子結合這一點上是相同的。但是,與<ⅰ>所示之碳原子結合的矽原子(n),與二個或三個氧原子(n)結合,與<ⅱ>所示之碳原子結合的矽原子(tg)與三個碳原子和一個氧原子(tg)結合。
另外,作為參考,圖1和圖2中示出本實施方式之粒子的透過13
C DDMAS NMR測量得到之NMR質譜(spectre)之一例。另外,圖1的NMR測量中使用之粒子是後述之實施例A2的粒子(非表面處理粒子),圖2的NMR測量中使用之粒子是後述之實施例B1的粒子(疏水化處理粒子)。在圖1所示之NMR質譜中,能夠在-4ppm附近確認源於(b)甲基之碳原子的峰,能夠在-50ppm附近確認源於(c)甲氧基之碳原子的峰。另外,在圖2所示之NMR質譜中,能夠在-4ppm附近確認源於(b)甲基之碳原子的峰,能夠在-50ppm附近確認源於(c)甲氧基之碳原子的峰,能夠在-1ppm附近確認源於(e)三甲基矽氧基之碳原子的峰。
如以上所說明,透過利用13
C DDMAS NMR測量,能夠辨別(b)甲基、(c)烷氧基、含有碳原子之(e)其他基(例如,三烷基矽氧基等)。另外,在以下的說明中,在實施13
C DDMAS NMR測量時,將源於(c)烷氧基中含有的碳原子(該碳原子是與(c)烷氧基中所含有之氧原子(tg)結合的碳原子)之峰a的面積設為A,將源於(b)甲基中所含有的碳原子之峰b的面積設為B,並且,當(e)其他基為三烷基矽氧基時,將源於與三烷基矽氧基的矽原子(tg)結合的碳原子之峰c的面積設為C。
該情況下,峰面積(peak area)之比率(A/B)係顯示將(b)甲基的總量作為基準值時的(c)烷氧基之總量的相對比例的參數。在本實施方式之粒子中,關於比率A/B,只要超過0便沒有特別限制,但較佳係在0.016~0.030的範圍,更較佳係在0.020~0.030的範圍。另外,在疏水化處理粒子中,當將峰面積之比率(C/(A+B+C))標記為%時,較佳係在2.0%以上,更較佳係在4.0%以上,進一步較佳係在6.0%以上。
<29
Si DDMAS NMR>
透過利用29
Si DDMAS NMR測量,能夠得到有關包含粒子主體中所含有之矽的交聯網狀結構和基((a)交聯網狀結構、(e)其他基(包含三烷基矽氧基等的矽原子(tg)的基的情況))的資訊。關於測量條件的詳細情況之後進行說明,當將外部標準作為聚二甲基矽烷的峰(34ppm)進行測量時,能夠得到以下所例示之各種資訊。
<ⅰ> 形成滿足以下所示條件之(a)交聯網狀結構的矽原子(n)的峰,是在-56ppm附近檢測出。
即,在形成(a)交聯網狀結構之矽原子(n)的四個結合鍵(atomic bonding)中,(1)二個結合鍵與形成(a)交聯網狀結構之氧原子(n)結合;(2)一個結合鍵與(b)甲基的碳原子結合;(3)一個結合鍵與作為(c)烷氧基的甲氧基的氧原子(tg),或者,(d)羥基的氧原子(tg)結合。
<ⅱ> 形成滿足以下所示條件之(a)交聯網狀結構的矽原子(n)的峰,是在-65ppm附近檢測出。
即,在形成(a)交聯網狀結構之矽原子(n)的四個結合鍵中,(1)三個結合鍵與形成(a)交聯網狀結構之氧原子(n)結合;(2)一個結合鍵與(b)甲基的碳原子結合。
<ⅲ> 當(e)其他基為三甲基矽氧基時,在-8ppm附近檢測出源於三甲基矽氧基的矽原子(tg)的峰。
另外,上述<ⅰ>中所示之矽原子(n)對應於以下結構式(1)中以符號D所示之矽原子,上述<ⅱ>中所示之矽原子(n)對應於以下結構式(2)中以符號E所示之矽原子。在此,在以下結構式(1)中,R1
係(c)烷氧基或者(d)羥基。
[化學式1]
[化學式2]
另外,作為參考,圖3和圖4中示出本實施方式之粒子的透過29
Si DDMAS NMR測量得到之NMR質譜之一例。另外,圖3的NMR測量中所使用之粒子是後述之實施例A2的粒子(非表面處理粒子),圖4的NMR測量中所使用之粒子是後述之實施例B1的粒子(疏水化處理粒子)。在圖3所示之NMR質譜中,能夠在-56ppm附近確認出上述<ⅰ>中所示之形成(a)交聯網狀結構之矽原子(n)的峰,能夠在-65ppm附近確認出上述<ⅱ>中所示之形成(a)交聯網狀結構之矽原子(n)的峰。另外,在圖4所示之NMR質譜中,能夠在-56ppm附近確認出上述<ⅰ>中所示之形成(a)交聯網狀結構之矽原子(n)的峰,能夠在-65ppm附近確認出上述<ⅱ>中所示之形成(a)交聯網狀結構之矽原子(n)的峰,能夠在-8ppm附近確認出(e)三甲基矽氧基的矽原子(tg)的峰。
如以上所說明,透過利用29
Si DDMAS NMR測量,能夠在(a)交聯網狀結構中,辨別結合狀態互相不同之矽原子(n),或者,能夠辨別含有矽原子之(e)其他基(例如,三烷基矽氧基等)。另外,在以下的說明中,在實施29
Si DDMAS NMR測量時,將源於上述<ⅰ>所示之矽原子(n)的峰d的面積設為D,將源於上述<ⅱ>所示之矽原子(n)的峰e的面積設為E。
另外,透過適當地組合峰a~e的面積A~E,能夠得到具有以下所例示之各種技術意義的參數(parameter)。
(1)D+E
<ⅰ>和<ⅱ>所示之矽原子(n)一定與一個(b)甲基結合。因此,二個峰面積之和(D+E)還是與(b)甲基的總量對應的參數。
(2)F=D/(D+E)
<ⅰ>所示之矽原子(n)一定與(c)烷氧基或(d)羥基結合。因此,面積D還是與(c)烷氧基和(d)羥基的總量對應的參數。而且,根據上述(1),F=D/(D+E)係顯示將(b)甲基的總量作為基準值時的(c)烷氧基和(d)羥基之總量的相對比例的參數(官能團比率)。
(3)G=(A/B)/F
A/B係顯示將(b)甲基的總量作為基準值時的(c)烷氧基之總量的相對比例的參數,F係顯示將(b)甲基的總量作為基準值時的(c)烷氧基和(d)羥基之總量的相對比例的參數(官能團比率)。因此,A/B除以F得到的值G係顯示將(c)烷氧基和(d)羥基之總量作為基準值時的(c)烷氧基之總量的相對比例的參數。因此,表示:G的值越大,則(c)烷氧基的相對比例越大,G的值越小,則(d)羥基的相對比例越大。另外,A/B和F,分別係根據透過不同的測量方法得到之峰面積獲得的參數。因此,在計算G時,需要預先計算A/B的值和F的值。
在本實施方式之粒子中,G可以在超過0且1以下的範圍內取任意的值,但較佳係0.05~0.15的範圍,更較佳係0.06~0.15的範圍,進一步較佳係0.09~0.13的範圍。
本實施方式之粒子的粒徑(累積50%粒徑(D50))並沒有特別限定,可以根據粒子的使用用途適當地進行選擇。但是,根據粒子的可製造性、處理性、可設想的各種用途,粒徑可以在0.1μm~10μm左右的範圍內適當地進行選擇。另外,一般而言,粒徑的下限值較佳係0.14μm以上,更較佳係0.16μm以上、0.18μm以上或者0.25μm以上,進一步較佳係超過0.3μm,尤其較佳係0.35μm以上。另外,一般而言,粒徑的上限值較佳係2.0μm以下,更較佳係1.5μm以下,進一步較佳係1.1μm以下,尤其較佳係1.0μm以下。另外,作為粒徑的上限值和下限值之較佳組合範圍,只要在適當地組合上述所列舉之上限值和下限值的範圍內即可,例如,可以例示出0.14μm~2.0μm的範圍、或者超過0.30μm且2.0μm以下的範圍、0.35μm~1.1μm的範圍等。
另外,與本實施方式之粒子的使用用途或目的無關地,粒徑(D50)和S1
/S2
,一般而言,較佳係滿足以下算式(3)中所示之關係,更較佳係滿足以下算式(4)。
算式(3) S1
/S2
≥44.5×D50+2.07
算式(4) S1
/S2
≥57.5×D50+1.47
在此,在算式(3)和算式(4)中,D50的單位為μm,S1
/S2
的單位為無量綱(dimensionless)。另外,在算式(3)中,D50的下限值較佳係0.14μm以上,更較佳係0.2μm以上,尤其較佳係超過0.3μm。另外,在算式(4)中,D50的下限值較佳係0.12μm以上,更較佳係0.2μm以上,尤其較佳係超過0.3μm。
另外,關於粒徑,還可以著眼於本實施方式之粒子的作用功效、或者本實施方式之粒子的用途,適當地進行選擇。例如,當將本實施方式之粒子用作各種橡膠或樹脂的填料時,從確保將本實施方式之粒子添加到熔融狀態之樹脂等中時的黏度或流動性的觀點出發,粒徑較佳係0.14μm以上。另外,在製造分散含有本實施方式之粒子的橡膠材料或樹脂材料時,從確保這些部件的透明性的觀點出發,粒徑較佳係2.0μm以下。
另外,從提高粒子相對於固體表面的密合性之觀點出發,較佳係滿足上述算式(3),更較佳係滿足算式(4)。該情況下,在算式(3)中,D50的下限值較佳係0.14μm以上,更較佳係0.2μm以上,尤其較佳係超過0.3μm。另外,在算式(4)中,D50的下限值較佳係0.12μm以上,更較佳係0.2μm以上,尤其較佳係超過0.3μm。
另外,本實施方式之粒子的粒度分佈之寬度,能夠利用累積90%粒徑(D90)相對於累積10%粒徑(D10)之比率(D90/D10)進行表達。D90/D10的值越小,則表示粒度分佈的寬度窄,單分散性(monodispersity)高。在本實施方式之粒子中,D90/D10的值並沒有特別限制,可以根據用途適當地進行選擇,但一般而言,較佳係其值較小,具體而言,較佳係1.5~7.0,更較佳係5.5以下。
接下來,對本實施方式之粒子的製造方法進行說明。本實施方式之粒子的製造方法並沒有特別限制,當本實施方式之粒子為非表面處理粒子時,較佳係以下所示之第一製造方法,當本實施方式之粒子為疏水化粒子時,較佳係以下所示之第二製造方法。
在此,在第一製造方法中,透過從實施第一步驟~第三步驟(粒子形成步驟)後得到之球狀聚甲基倍半矽氧烷粒子分散液中回收固形物,可以得到非表面處理粒子。另外,在第二製造方法中,透過從實施第一步驟~第三步驟(粒子形成步驟),之後進一步實施第四步驟(疏水化處理)後得到之疏水化處理後之球狀聚甲基倍半矽氧烷粒子分散液中回收固形物,可以得到疏水化處理粒子。另外,第一步驟~第四步驟的概要如下。
(第一步驟)
得到含有粒子前驅體(precursor)和有機溶劑(第一有機溶劑)之原料溶液,其中,該粒子前驅體選自由(ⅰ)甲基三烷氧基矽烷的水解物、(ⅱ)上述水解物的部分縮合物以及(ⅲ)上述水解物和上述部分縮合物的混合物構成之群中。
(第二步驟)
透過將上述原料溶液和含有有機溶劑(第二有機溶劑)之鹼性水系介質進行混合,並使上述粒子前驅體發生縮聚反應,從而得到縮聚反應液。
(第三步驟)
接著,透過將上述縮聚反應液和含有有機溶劑(第三有機溶劑)之水性溶液混合,從而得到使球狀聚甲基倍半矽氧烷粒子分散之球狀聚甲基倍半矽氧烷粒子分散液,其中,該有機溶劑(第三有機溶劑)的有機溶劑濃度為5質量%以上且48質量%以下。在此,縮聚反應液和水性溶液的混合被實施為:將縮聚反應液和水性溶液混合得到的混合溶液中所含有之有機溶劑(第一~第三溶劑的總計)的比例成為超過35質量%且60質量%以下之比例。
(第四步驟)
在球狀聚甲基倍半矽氧烷粒子分散液中混合疏水劑,對球狀聚甲基倍半矽氧烷粒子之表面進行疏水化處理。
關於第一製造方法,除了第三步驟的實施條件和有沒有實施第四步驟這一點上不同之外,與專利文獻4中所揭示之製造方法實質上相同,關於第二製造方法,除了第三步驟之實施條件不同之外,與專利文獻4中所揭示之製造方法實質上相同。但是,關於第三步驟,在第一及第二製造方法和專利文獻4所揭示之製造方法中,以下(ⅰ)、(ⅱ)及其組合明顯不同。以下,對各步驟的詳細情況進行說明。
(ⅰ)使用含有有機溶劑濃度為5質量%以上且48質量%以下之有機溶劑的水性溶液。
(ⅱ)將縮聚反應液和水性溶液混合得到的混合溶液中之有機溶劑的比例超過35質量%且60質量%以下。
在第一步驟中,將作為原料之甲基三烷氧基矽烷在酸催化劑的存在下進行水解,得到含有粒子前驅體和有機溶劑之原料溶液,其中,上述粒子前驅體選自由(ⅰ)甲基三烷氧基矽烷的水解物、(ⅱ)水解物的部分縮合物以及(ⅲ)水解物和部分縮合物的混合物構成之群中。
在此,甲基三烷氧基矽烷係由以下通式(2)所表達之化合物。
CH3
Si(OR2
)3
通式(2)
(在通式(2)中,R2
係烷基或環狀烷基中之任意一者。)
作為上述甲基三烷氧基矽烷,可以例示出:甲基三甲氧基矽烷、甲基三乙氧基矽烷、甲基三異丙氧基矽烷、甲基三(甲氧乙氧基)矽烷等。這些物質可以使用一種,也可以同時使用二種或二種以上。
作為催化劑,可以適當地使用公知的催化劑。具體而言,可以舉出:鹽酸、硝酸、硫酸等的無機酸;甲酸、乙酸、乙二酸、檸檬酸、丙酸等的有機酸。
關於催化劑的使用量,只要根據甲基三烷氧基矽烷和酸的種類適當地進行調整即可,可以在相對於將甲基三烷氧基矽烷水解時所使用之水量100質量份而在1×10-3
質量份~1質量份的範圍內進行選擇。當催化劑之使用量少於1×10-3
質量份時,無法充分地進行反應,而當超過1質量份時,不僅作為不純物而殘留於粒子中的濃度變高,而且所生成之水解物容易縮合。關於水的使用量,較佳係:相對於1摩爾之甲基三烷氧基矽烷為2摩爾~15摩爾。當水量不足2摩爾時,無法充分地進行水解反應,而當水量超過15摩爾時,存在生產率變差的情況。
反應溫度並沒有特別限制,可以在常溫或加熱狀態下進行,但是,從可以在短時間內得到水解物,且能夠抑制所生成之水解物發生部分縮合反應的觀點出發,較佳係在保持為10℃~60℃的狀態下進行反應。反應時間並沒有特別限制,只要在考慮到所使用之甲基三烷氧基矽烷的反應性、或者由甲基三烷氧基矽烷、酸以及水調製成的反應液之組成、生產率後適當地進行選擇即可,但一般為10分鐘~10小時左右。
透過進行這樣的操作,甲基三烷氧基矽烷的至少一部分之烷氧基(alkoxy)進行水解而生成乙醇。因此,所得到之原料溶液中,除了透過甲基三烷氧基矽烷的水解和縮合生成之粒子前驅體之外,還含有該乙醇(第一有機溶劑)。另外,甲基三烷氧基矽烷的水解,也可以在另行添加的有機溶劑的存在下進行。該情況下,另行添加的有機溶劑也包含在第一有機溶劑中。
在第二步驟中,將上述第一步驟中得到之原料溶液和含有有機溶劑(第二有機溶劑)之鹼性水系介質進行混合,使粒子前驅體進行縮聚反應,由此得到縮聚反應液。在此,鹼性水系介質是將鹼性成分、水、有機溶劑混合得到之液體。
鹼性水系介質中所使用之鹼性成分,其水溶液示出鹼性,其可以作為第一步驟中使用之酸的中和劑、或者作為第二步驟之縮聚反應的催化劑而發揮作用。作為該鹼性成分,可以例示出:氫氧化鋰、氫氧化鈉、氫氧化鉀等的鹼金屬氫氧化物;氨;以及甲胺、二甲胺等的有機胺類;四甲基氫氧化銨、四乙基氫氧化銨、膽鹼(choline)等的氫氧化季銨等。
在該第二步驟中,為了調製出鹼性水系介質,除了鹼性成分和水之外,還使用有機溶劑。關於該有機溶劑,只要是相對於水具有相溶性便沒有特別限制,可以使用在常溫常壓下每100g中溶解10g以上水的有機溶劑。作為上述溶劑,可以例示出:甲醇(methanol)、乙醇(ethanol)、正丙醇、2-丙醇、丁醇等的醇;乙二醇、二甘醇、丙二醇、甘油、三羥甲基丙烷、己三醇等的多元醇;丙酮、甲乙酮、二丙酮醇等的酮;乙二醇單乙醚、二乙醚、四氫呋喃等的醚;二甲基甲醯胺、二甲基乙醯胺、N-甲基吡咯烷酮等的醯胺化合物等。關於有機溶劑的含有比例,較佳係50質量%以上,且較佳係90質量%以下。
鹼性成分的使用量是將酸進行中和且作為縮聚反應的催化劑有效發揮作用的量,例如,當作為鹼性成分使用氨時,通常,相對於水和有機溶劑之混合物100質量份,在0.01質量%以上且12.5質量%以下的範圍內進行選擇。當鹼性成分的使用量少於0.01質量%時,在接下來的第三步驟中不易獲得球狀聚甲基倍半矽氧烷粒子,產量容易降低。另外,當鹼性成分的使用量超過12.5質量%時,容易生成析出物,因而不易得到均勻的反應液,從而在接下來的第三步驟中,存在無法穩定地生成球狀聚甲基倍半矽氧烷粒子的情況。另外,廢液的處理也容易變得複雜。
該鹼性水系介質的混合量,較佳係在粒子前驅體的濃度成為1質量%~20質量%程度的範圍。另外,該粒子前驅體的濃度,是假設作為原料之甲基三烷氧基矽烷全部完全反應而計算。
縮聚反應液的反應時間,是在考慮到反應溫度、原料溶液的組成、鹼性水系介質的組成等後適當地確定,具體為混合之後不久的透明的反應液中產生渾濁的時間至反應液中開始產生析出物的時間為止的範圍內。當混合時間過短或過長時,容易發生如下的不良情況,即:在接下來的第三步驟中得到之球狀聚甲基倍半矽氧烷粒子的球形度降低、或者生成凝集物。
在第三步驟中,透過將上述第二步驟中得到之縮聚反應液和含有有機溶劑(第三有機溶劑)之水性溶液混合,從而得到分散有球狀聚甲基倍半矽氧烷粒子之球狀聚烷基倍半矽氧烷分散液。
關於與縮聚反應液混合之含有有機溶劑之水性溶液中使用的有機溶劑,只要是相對於水具有相溶性,便沒有特別限制,可以使用常溫常壓下每100g中溶解10g以上水的有機溶劑。作為上述有機溶劑,可以例示出:甲醇、乙醇、正丙醇、2-丙醇、丁醇等的醇;乙二醇、二甘醇、丙二醇、甘油、三羥甲基丙烷、己三醇等的多元醇;丙酮、甲乙酮、二丙酮醇等的酮;乙二醇單乙醚、丙酮、二乙醚、四氫呋喃等的醚;二甲基甲醯胺、二甲基乙醯胺、N-甲基吡咯烷酮等的醯胺化合物等。
在此,為了得到本實施方式之粒子,需要同時滿足以下(ⅰ)和(ⅱ)中所示之條件。由此,容易得到S1
/S2
為8.0以上的粒子。
(ⅰ)使用含有有機溶劑濃度為5質量%以上且48 質量%以下之有機溶劑的水性溶液。
(ⅱ)將縮聚反應液和水性溶液混合得到的混合溶液中之有機溶劑的比例超過35 質量%且60 質量%以下。
在此,混合溶液中之有機溶劑的含有量,對應於第一步驟中調製的原料溶液中所含有之有機溶劑(第一有機溶劑)、第二步驟中使用的鹼性水系介質中所含有之有機溶劑(第二有機溶劑)、以及第三步驟中使用的水性溶液中所含有之有機溶劑(第三有機溶劑)的總計含有量。另外,在混合溶液中,通常,除了液體成分之外,還含有隨著時間的經過而生成之粒子(固體成分)。另外,混合溶液的總量(100質量%)對應於第一步驟至第三步驟之間所使用之所有原料的總計量。
另外,水性溶液的有機溶劑濃度較佳係10 質量%~48 質量%,更較佳係18質量%~48 質量%,進一步較佳係25 質量%~45 質量%。另外,縮聚反應液和水性溶液的總量混合結束後的混合溶液中之有機溶劑的比例,較佳係38 質量%以上,更較佳係45 質量%以上。另外,在滿足上述條件(ⅱ)的範圍內,含有有機溶劑之水性溶液的混合量,較佳係在相對於縮聚反應液為0.25質量倍~10質量倍的範圍內。
另外,在第三步驟中,具有如下傾向,即:水性溶液的有機溶劑濃度以及/或者混合溶液中之有機溶劑的比例越多,則後述之熟化處理後得到的粒子的粒徑(D50)更大。
將縮聚反應液和含有有機溶劑之水性溶液混合的方法,並沒有特別限制,可以採用公知的方法。但是,混合較佳係按照以下所示之形態實施。
即,按照將縮聚反應液與含有有機溶劑的水性溶液混合後之混合溶液的組成隨著時間的經過而始終保持固定不變之方式,將縮聚反應液與含有有機溶劑之水性溶液混合。由此,能夠容易使所得到之球狀聚甲基倍半矽氧烷粒子分散液中的球狀聚甲基倍半矽氧烷粉末之粒度分佈變得足夠窄。
另外,當縮聚反應液和含有有機溶劑之水性溶液的混合不充分時,所生成之球狀聚甲基倍半矽氧烷粒子發生凝集,由此,容易使S1
/S2
小於8.0。
作為混合方法的具體例,例如,可以舉出:ⅰ)向空容器內,從縮聚反應液供給管供給縮聚反應液,與此同時,從水性溶液供給管供給水性溶液,並在容器內混合縮聚反應液和水性溶液的方法,或者,ⅱ)使用三叉管混合縮聚反應液和水性溶液的方法等。
其中,較佳係使用由Y字管、T字管等的三叉管構成之管式反應器的混合方法。該情況下,從流量的穩定性等觀點出發,較佳係使用如日本專利特開2003-221222號公報或者日本專利特許第6116711號公報所揭示那樣的、在朝向連接部供給液體的流道(第一流道和第二流道)中設有縮徑部的Y字狀的管式反應器。
圖5係顯示本實施方式之球狀聚烷基倍半矽氧烷粒子的製造方法中所使用之反應裝置的一例之概略模式圖,圖6係顯示圖5中所示管式反應器之剖面結構的一例之放大剖視圖。圖5中所示之反應裝置10具備:Y字狀的三叉管式反應器20,其具有第一流道110、第二流道120、第三流道130、以及將這三條流道110、120、130各自的一端相互連接的連接部140;第一泵30,其與第一流道110的入口側(與連接部140呈相反側的端側)相連;第二泵32,其與第二流道120的入口側(與連接部140呈相反側的端側)相連;第一原料箱40,其與第一泵30相連;第二原料箱42,其與第二泵相連;以及回收箱50,其與第三流道130的出口側(與連接部140呈相反側的端側)相連。
在使用圖5中所示之反應裝置10實施第三步驟時,例如,將儲藏在第一原料箱40中之縮聚反應液經由第一泵30以固定的流量連續地向第一流道110供給,並且,將儲藏在第二原料箱42中之水性溶液經由第二泵32以固定的流量連續地向第二流道120供給。由此,在連接部140中,縮聚反應液與水性溶液在相互碰撞的同時混合。然後,縮聚反應液與水性溶液的混合液,作為球狀聚烷基倍半矽氧烷粒子分散液,從連接部140側經由第三流道130被回收至回收箱50內。另外,也可以使從第三流道130排出的混合液流入靜止型混合器等中,進一步進行攪拌混合。該情況下,能夠將管式反應器中之縮聚反應液和水性溶液的流速設定為更高的速度,因而能夠獲得高生產率。
另外,關於將縮聚反應液和水性溶液朝向三叉管式反應器20輸送的方法,除了泵30、32以外,也可以無限制地採用加壓輸送等公知的液體輸送方法。其中,較佳係:能夠連續且均勻地使縮聚反應液與水性溶液碰撞混合的加壓輸送。另外,在使用泵30、32時,較佳係:使用不會產生脈動之多聯式往復泵、或者設有儲能器(accumulator)等的緩衝裝置的泵。
另外,從容易得到具有所希望之粒徑以及粒度分佈之球狀聚烷基倍半矽氧烷微粒的觀點出發,較佳係如圖6中所示那樣,在第一流道110內和第二流道120內分別設有能夠調整流速之縮徑部112、114。該情況下,從縮徑部112、114的流出口側(連接部140側)至連接部140的中心點C為止的距離R,較佳係:縮徑部112的縮徑部直徑d1、縮徑部114的縮徑部直徑d2:1倍~25倍,更較佳係1倍~9倍。另外,較佳係:從縮徑部112的流出口側(連接部140側)至中心點C為止的距離R與從縮徑部114的流出口側(連接部140側)至中心點C為止的距離R相等。
使縮聚反應液和含有有機溶劑之水性溶液混合得到之混合溶液,需要暫時靜置(熟化處理)。透過熟化處理,混合溶液中生成之球狀聚甲基倍半矽氧烷粒子變得穩定化,並且,能夠得到S1
/S2
更大的粒子。
關於熟化溫度或熟化時間,只要能夠得到S1
/S2
為8.0以上的粒子,便沒有特別限定,只要根據本實施方式之粒子的用途調整為具有理想的物性即可。但是,關於熟化溫度,從必須是混合溶液之凍結溫度以上的溫度且不需要強制冷卻的觀點出發,較佳係10℃以上,更較佳係15℃以上,尤其較佳係18℃以上。另外,從預防混合溶液中所含有之成分揮發掉的觀點出發,熟化溫度較佳係60℃以下,更較佳係50℃以下,進而,從不需要加熱的觀點出發,尤其較佳係40℃以下。另外,例如,關於在20℃的溫度下進行熟化時的熟化時間,從能夠更加可靠地得到S1
/S2
為8.0以上之粒子的觀點出發,尤其較佳係至少在6小時以上。另外,關於在20℃的溫度下進行熟化時的熟化時間,更較佳係15小時以上,進一步較佳係20小時以上,尤其較佳係24小時以上,更進一步較佳係160小時以上。熟化處理結束後,能夠得到使球狀聚甲基倍半矽氧烷粒子分散後之球狀聚甲基倍半矽氧烷粒子分散液(熟化處理結束後之混合溶液)。
在得到非表面處理粒子時,在實施第一步驟~第三步驟(粒子形成步驟)之後,無需實施第四步驟(疏水化處理)等的任何表面處理,對第三步驟中得到之球狀聚甲基倍半矽氧烷粒子分散液進行固液分離處理。透過固液分離處理回收到的固形物(非表面處理粒子)可以直接利用於各種用途中。但是,為了得到不純物更少的非表面處理粒子,較佳係:對固形物進行乾燥處理。關於固形物之乾燥方法,並沒有特別限制,可以從鼓風乾燥或者減壓乾燥等的公知的方法中進行選擇。其中,尤其較佳係減壓乾燥,根據該減壓乾燥,能夠得到易於破碎的乾燥粉末。關於乾燥溫度,只要是非表面處理粒子中所含有之烷基等的官能團不會分解的溫度,便沒有特別限制,可以在65℃~350℃範圍、尤其是80℃~250℃的範圍內適當地設定合適的溫度。另外,乾燥時間並沒有特別限制,但是,透過將乾燥時間設定為2小時~48小時,能夠得到充分乾燥的非表面處理粒子。
另一方面,在得到疏水化處理粒子時,是在實施第一步驟~第三步驟(粒子形成步驟)之後,進一步實施第四步驟(疏水化處理)。
在第四步驟中,在球狀聚甲基倍半矽氧烷粒子分散液中進一步混合疏水劑,從而對球狀聚甲基倍半矽氧烷粒子(非表面處理粒子)之表面進行疏水化處理。由此,與非表面處理粒子相比,能夠使所得到之疏水化處理粒子之疏水度(M值)更大。
另外,作為疏水化處理方法,例如,可以舉出:將從第三步驟中得到之球狀聚甲基倍半矽氧烷粒子分散液中回收的球狀聚甲基倍半矽氧烷粒子(非表面處理粒子)提供給疏水化處理的方法。但是,在該方法中,在使非表面處理粒子固液分離的步驟或者進一步乾燥的步驟中,粒子彼此凝集而容易生成凝集塊。因此,疏水化處理後得到之疏水化處理粒子,具有:粒度分佈的寬度寬,且破碎性容易降低之傾向。
相對於此,當如上所述那樣直接將疏水劑混合在球狀聚甲基倍半矽氧烷粒子分散液中進行疏水化處理時,即使所得到之疏水化處理粒子形成凝集塊,其破碎性也是出色的。
上述疏水化處理中所使用之疏水劑,通常,可以使用有機矽化合物。作為該有機矽化合物,並沒有特別限制,舉例說明的話,可以舉出:六甲基二矽氮烷等的烷基矽氮烷類化合物;二甲基二甲氧基矽烷、二乙基二乙氧基矽烷、三甲基甲氧基矽烷、三乙基甲氧基矽烷、甲基三甲氧基矽烷、丁基三甲氧基矽烷等的烷基烷氧基矽烷類化合物;二甲基二氯矽烷、三甲基氯矽烷、三乙基氯矽烷等的氯矽烷類化合物;或者,矽油、矽酮漆(silicone varnish)等。這些疏水劑,可以單獨使用一種,也可以混合二種或二種以上使用,還可以利用有機溶劑等稀釋後使用。
在上述疏水劑中,較佳係:能夠將三烷基矽基(trialkylsilyl group)導入球狀聚甲基倍半矽氧烷粒子之疏水劑,更較佳係:能夠將各個烷基的碳數為1至3的三烷基矽基導入球狀聚甲基倍半矽氧烷粒子之疏水劑。另外,從反應性良好且易於處理等方面出發,更較佳係:使用選自由烷基矽氮烷類化合物或(三烷基)烷氧基矽烷類化合物構成之群中的至少一種,從所得到之疏水化處理粒子的流動性更高的觀點出發,最較佳係:使用六甲基二矽氮烷。
疏水劑的使用量並沒有特別限制,但是,當疏水劑的使用量過少時,疏水化處理可能不充分,而當疏水劑的使用量過多時,後續處理變複雜。因此,疏水劑的混合量,較佳係:相對於成為疏水化處理對象之非表面處理粒子的(乾燥重量)100質量份為0.01質量份~300質量份,更較佳係1質量份~200質量份。
向球狀聚甲基倍半矽氧烷粒子分散液中混合疏水劑的混合方法,並沒有特別限制。例如,當在常溫、常壓下使用液態疏水劑時,可以將疏水劑滴加至球狀聚甲基倍半矽氧烷粒子分散液中,也可以向球狀聚甲基倍半矽氧烷粒子分散液之液面噴射疏水劑。但從操作簡便方面出發,較佳係滴加方式。
處理溫度並沒有特別限制,只要在考慮到所使用疏水劑之反應性後確定即可,例如,可以設定為0度~100度。另外,處理時間例如可以設定為0.1小時~72小時。但是,從縮短處理時間的觀點出發,處理溫度越高越好,具體而言,較佳係:設定為第二步驟中使用之有機溶劑的沸點前後的溫度。
透過利用疏水劑對非表面處理粒子之表面進行疏水化處理,所得到的疏水化處理粒子,通常懸浮在添加疏水劑後的球狀聚甲基倍半矽氧烷粒子分散液之上層部分(以下,將該液體稱為「粉體懸浮液」)。從粉體懸浮液中回收疏水化處理粒子的方法,能夠無特別限制地使用公知的方法。例如,可以採用撈取懸浮粉體的方法,也可以採用過濾法,但是,從操作簡便方面出發,較佳係採用過濾法。過濾的方法並沒有特別限制,可以選擇減壓過濾或離心過濾、加壓過濾等公知的方法。過濾中所使用之濾紙或過濾器(filter)、濾布等,只要是能夠從工業渠道獲得便沒有特別限制,可以根據所使用的裝置適當地進行選擇。在進行過濾時,也可以根據需要,利用純水或甲醇等對固形物進行清洗。
回收後之固形物(疏水化處理粒子)可以直接利用於各種用途中。但是,為了得到不純物更少的疏水化處理粒子,較佳係:對固形物進行乾燥處理。乾燥處理的方法和具體條件並沒有特別限制,可以選擇與對第三步驟結束後回收到之固形物(非表面處理粒子)的乾燥處理相同的方法和條件。
[實施例]
以下,透過實施例和比較例進一步詳細地說明本發明,但本發明並不限於此。另外,關於所製造的試料之諸物性,是根據以下的方法進行了評價。另外,實施例、比較例中的粒子的製造,均在室溫20℃下管理的室內進行。
1.粒子的各種評價方法
(1)累積10%粒徑(D10)、累積50%粒徑(D50)、累積90%粒徑(D90)的測量以及D90/D10的計算
體積基準的累積10%粒徑(D10)、累積50%粒徑(D50)以及累積90%粒徑(D90),是透過基於雷射繞射散射法之測量求出。該測量是按照以下步驟實施。首先,在將乾燥的0.1g的球狀聚甲基倍半矽氧烷粉末放入到玻璃製容器(內徑4㎝、高度11㎝)內之後,進一步添加50g的2-丙醇,從而得到了混合液。接著,在將超音波分散機之探針(probe)(前端的內徑7mm)中之、從其前端起4.5cm為止的部分浸入混合液中的狀態下,以20W的輸出功率進行15分鐘的超音波分散處理,從而得到了分散液。接著,使用該分散液,並使用利用了雷射繞射散射法之粒度分佈測量裝置(美國貝克曼庫爾特有限公司(Beckman Coulter, Inc.)製造的LS13 320)測量並算出了體積基準的累積10%粒徑(D10)、累積50%粒徑(D50)、累積90%粒徑(D90)以及D90/D10。D90/D10係顯示粒度分佈寬度之寬度,其值越小,則表示粒度分佈的寬度窄。
(2)13
C DDMAS NMR測量
透過13
C DDMAS NMR測量了以下所示之三種峰及其面積。測量裝置使用的是瑞士布魯克拜厄斯賓有限公司(Bruker Biospin)製的AVANCE Ⅱ。
<ⅰ> 源於(c)烷氧基中含有的碳原子(該碳原子是與(c)烷氧基中含有之氧原子(tg)結合的碳原子)之峰a及其面積A。
<ⅱ> 源於甲基中所含有的碳原子之峰b及其面積B。
<ⅲ> 源於與作為(e)其他基的三烷基矽氧基的矽原子(tg)結合的碳原子之峰c及其面積C。
測量條件如下,即:使用固體測量用探針(直徑4㎜),測量原子核素(nuclide)13C、MAS轉速7kHz、脈衝程式(pulse program)hpdec、重複時間10sec、積算次數6000次以上、外部標準為甘氨酸的羰基的峰(176.03ppm)。另外,各峰的面積,使用峰波形分離程式計算。另外,在後述之表示NMR測量結果的表中,將峰a的面積A、峰b的面積B以及峰c的面積C的總計記載為100%。
另外,在以上述測量條件進行NMR測量的情況下,當(c)烷氧基為甲氧基時,在-50ppm附近觀察到峰a,當為(b)甲基時,在-4ppm附近觀察到峰b,當(e)其他基為三甲基矽基(trimethylsilyl group)時,在-1ppm附近觀察到峰c。因此,在後述的NMR測量結果中,峰a的存在表示被測量粒子含有烷氧基,峰b的面積小於100%,表示存在著源於聚甲基倍半矽氧烷的甲基以外的碳,峰c的面積C表示源於因為疏水化處理而粒子表面附近所含有的三烷基矽氧基之碳的存在比例。
(3)BET比表面積(S1
)的測量
BET比表面積(S1
)透過氮吸附BET單點法測量。該測量是按照以下步驟實施。首先,在稱重後的測量容器內,稱量放入0.12g左右的球狀聚甲基倍半矽氧烷粉末。接著,在將填充有球狀聚甲基倍半矽氧烷粉末之測量容器設置在覆套式電阻加熱器(mantle heater)內之後,實施在對覆套式電阻加熱器內部進行氮置換的同時以200℃加熱80分鐘的預處理。之後,將冷卻至室溫的測量容器設置於BET比表面積測量裝置(日本柴田化學株式會社製的SA-1000)內,使用液態氮,使氮氣吸附在球狀聚甲基倍半矽氧烷粉末之表面上。然後,根據該吸附量並透過BET單點法得出BET表面積Sx。測出填充有測量後的球狀聚甲基倍半矽氧烷粉末之測量容器的質量,從該值減去之前預先測量之測量容器自身的質量,從而計算出除去透過預處理脫離之水分等的質量部分之後的球狀聚甲基倍半矽氧烷粉末的質量m。將上述BET表面積Sx除以該質量m而求出比表面積S1
。在進行該計算時,對於BET表面積Sx,是在將單位標注為m2
時的數值之小數點後第1位四捨五入之後使用於計算中,關於質量m,是在將單位標注為g時的數值之小數點後第3位四捨五入之後使用於計算中。而且,將在該計算中得到的數值之小數點後第2位四捨五入後得到的值,作為BET比表面積S1
。
(4)粒子密度(ρ)的測量、以及理論比表面積(S2
)的計算
粒子密度(ρ)是利用日本株式會社島津製作所製的乾式自動密度計AccuPyc 1330進行了測量。該測量是按照以下步驟實施。首先,將在120℃下減壓乾燥處理24小時後之球狀聚甲基倍半矽氧烷粉末,在10ml試料容器中稱量至0.0001g的單位。接著,在將試料容器設置在乾式自動密度計之測量室內之後,在使He氣體在測量室內流動的同時以測量溫度25℃測量了粒子密度。在上述乾式自動密度計中,當輸入提供給測量中之粉末質量時,粒子密度是以g/cm3
為單位顯示至小數點後5位。因此,作為比表面積S2
的計算中使用之粒子密度ρ,使用的是如下數值,即:將標注為g/cm3
之粒子密度的小數點後第3位四捨五入後得到的數值,進一步換算為g/m3
單位的數值。
使用D50以及ρ的測量值並根據以下算式(5),計算出理論比表面積S2
(m2
/g)。此時,將基於以下算式(5)之右邊計算出的數值之小數點後第2位四捨五入後的值,作為理論比表面積S2
。
算式(5) S2
=6/ρd
(在算式(5)中,d為D50(m)的值,ρ為粒子密度(g/m3
)。另外,在本案說明書中,通常,D50的值標注為μm,ρ的值標注為g/cm3
。但是,在計算理論比表面積S2
時,D50以及ρ的值,分別換算為標注成m和g/m3
的值。)
(5)疏水度(M值)的測量
疏水度(M值)是透過甲醇滴定法測量。該測量是按照以下步驟實施。首先,在容積200ml的容器(燒杯)中放入50ml純水和0.2g球狀聚甲基倍半矽氧烷粉末。接著,直到成為球狀聚甲基倍半矽氧烷粉末全部被浸濕且分散於液體中的狀態為止,對容器內之容納物進行攪拌,與此同時,從滴定管向容器內滴加甲醇。將所滴加甲醇量相對於滴加結束時的純水(50ml)和所滴加甲醇的總量之體積百分率的值設為疏水度(M值)。疏水度(M值)值越高,則表示疏水性越高,其值越低,則表示親水性越高。
(7)29
Si DDMAS NMR測量
透過29
Si DDMAS NMR測量了源於結構式(1)中以符號D所示的矽原子(n)之峰d的面積D、以及源於結構式(2)中以符號E所示的矽原子(n)之峰e的面積E。測量裝置使用的是瑞士布魯克拜厄斯賓有限公司(Bruker Biospin)製的AVANCE Ⅱ。
測量條件如下,即:使用固體測量用探針(直徑4㎜),測量原子核素(nuclide)29Si、MAS轉速8kHz、脈衝程式hpdec、重複時間20sec、積算次數4000次以上、外部標準為聚二甲基矽烷的峰(34ppm),由此進行了測量。另外,各峰的面積是利用峰波形分離程式進行了計算。另外,在後述之表示NMR測量結果的表中,示出將峰面積E作為100%(基準值)時的峰面積D的相對比例(%)。在-56ppm附近觀察到的峰為峰d,在-65ppm附近觀察到的峰為峰e。
(7)碳量的測量
球狀聚甲基倍半矽氧烷中所含有之碳原子的總量(碳量),是利用日本株式會社住化分析中心製的SUMIGRAPH NC-22F測量。具體而言,稱重30mg的球狀聚甲基倍半矽氧烷粉末,在反應爐溫度900℃下進行燃燒,在還原爐溫度600℃下測量了碳量。
(8)粒子相對於固體表面的附著量(黏著量)的測量
粒子相對於固體表面的附著量(黏著量)的測量是按照以下的步驟實施。首先,在將乾燥的1g的球狀聚甲基倍半矽氧烷粉末放入到玻璃製容器(內徑4㎝、高度11㎝)內之後,進一步添加50g的2-丙醇,從而得到了混合液。接著,在將超音波分散機之探針(前端的內徑為7mm)中之、從其前端起4.5cm為止的部分浸入混合液中的狀態下,以20W的輸出功率進行15分鐘的超音波分散處理,從而得到了分散液。將該分散液4cc均勻地滴加到質量W1(g)的玻璃載片(slide glass)(日本松波硝子工業株式會社製,品號:S1111、品名:白緣磨No.1、寬度26㎜、長度76㎜)的一面上之後,將玻璃載片在120℃下真空乾燥了3小時。使用日本細川密克朗株式會社(Hosokawa Micron Corporation)製造的Powder Tester PT-X,將真空乾燥處理後之玻璃載片,以行程長度18㎜、輕敲速度(tapping speed)60次/min進行了10次輕敲。然後,測量了輕敲後之玻璃載片的重量W2(g)。在此,對於玻璃的黏著量,根據以下算式(6)計算。
算式(6) 對於玻璃的黏著量(g/m2
)=(W2-W1)/0.001976
2.粒子的製造
(實施例A1)
<第一步驟>
在1000ml茄形燒瓶(eggplant flask)中裝入56g水和0.01g作為催化劑的乙酸,並在30℃下進行攪拌。在其中加入70g甲基三甲氧基矽烷並攪拌1小時,從而得到了126g原料溶液。此時,透過甲基三甲氧基矽烷的水解反應而生成之甲醇量為49.5g。另外,該醇量為100%水解時的理論計算值,以下的各實施例和比較例中也與此相同。
<第二步驟>
在1000ml茄形燒瓶中裝入2.9g的25%氨水、91.2g水、313.8g甲醇,並在30℃下進行攪拌,從而調製出鹼性水系介質。將在第一步驟中得到的126.0g原料溶液在1分鐘內滴加至該鹼性水系介質中。將該原料溶液滴加後之混合液攪拌25分鐘,使粒子前驅體進行縮聚反應,從而得到了533.9g(液溫30℃)縮聚反應液。
<第三步驟>
第三步驟是使用圖5中所示之反應裝置10實施。另外,在所使用之Y字狀三叉管式反應器20中,第一流道110的中心軸與第二流道120的中心軸所形成之角度(以下,有時稱為「分叉角度」)為90度,從縮徑部112(和縮徑部114)的流出口側至中心點C為止的距離R與縮徑部直徑d1(和d2)之比R/d1(和R/d2)為12.5。
在此,在以連接部140附近處的流速為1.9m/s之方式,從第一流道110的入口側供給533.9g縮聚反應液的同時,以連接部140附近處的流速為1.9m/s(水性溶液的流速/縮聚反應液的流速=1,以下稱為「流速比」)之方式,從第二流道120的入口側供給作為水性溶液的10質量%甲醇水溶液533.9g(液溫25℃),使縮聚反應液與水性溶液在連接部140處碰撞混合。然後,得到1067.8g從第三流道130排出的混合溶液、即含有球狀聚甲基倍半矽氧烷粒子之分散液。該分散液(混合溶液)中所含有之球狀聚甲基倍半矽氧烷粒子為3.2質量%,另外,分散液(混合溶液)中所含有之有機溶劑的含有量為39 質量%。
在將該分散液在20℃下靜置(熟化處理)24小時之後,透過吸濾(suction filtration)回收球狀聚甲基倍半矽氧烷粒子的粉末,在120℃下減壓乾燥24小時,從而得到34g白色的球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例A2)
除了將第三步驟中使用之水性溶液變更為20 質量%甲醇水溶液以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為44質量%。
(實施例A3)
除了將第三步驟中使用之水性溶液變更為25質量%甲醇水溶液以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為47質量%。
(實施例A4)
除了將第三步驟中使用之水性溶液變更為30質量%甲醇水溶液以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為49質量%。
(實施例A5)
除了將第三步驟中使用之水性溶液變更為35質量%甲醇水溶液以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為52質量%。
(實施例A6)
除了將第三步驟中使用之水性溶液變更為40質量%甲醇水溶液以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為54質量%。
(實施例A7)
除了將第三步驟中使用之水性溶液變更為45質量%甲醇水溶液以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為57質量%。
(實施例A8)
除了將第三步驟中使用之水性溶液變更為20質量%乙醇(ethanol)水溶液以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為44質量%。
(實施例A9)
除了將第三步驟中使用之水性溶液變更為30質量%乙醇水溶液,且將第三步驟中得到之分散液的靜置時間(熟化時間)變更為144小時以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為49質量%。
(實施例A10)
除了將第三步驟中使用之水性溶液變更為20質量%異丙醇(isopropyl alcohol)水溶液以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為44質量%。
(實施例A11)
除了將第三步驟中使用之水性溶液變更為25質量%異丙醇水溶液,且將第三步驟中得到之分散液的靜置時間(熟化時間)變更為144小時以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為47質量%。
(實施例A12)
除了將第三步驟中使用之水性溶液變更為15質量%丙酮水溶液以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為42質量%。
(實施例A13)
除了將第三步驟中使用之水性溶液變更為25質量%丙酮水溶液,且將第三步驟中得到之分散液的靜置時間(熟化時間)變更為144小時以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為47質量%。
(實施例A14)
除了將第三步驟中使用之水性溶液變更為15質量%四氫呋喃水溶液以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為42質量%。
(實施例A15)
<第一步驟>
在1000ml茄形燒瓶中裝入62g水和0.55g作為催化劑的乙酸,並在40℃下進行攪拌。在其中加入98.3g甲基三乙氧基矽烷並攪拌90分鐘,從而得到160.9g原料溶液。此時,透過甲基三乙氧基矽烷的水解反應而生成之乙醇量為76g。
<第二步驟>
在1000ml茄形燒瓶中裝入4.3g的25%氨水、91.2g水、313.8g乙醇,並在30℃下進行攪拌,從而調製出鹼性水系介質。將在第一步驟中得到的160.9g原料溶液在1分鐘內滴加至該鹼性水系介質中。將該原料溶液滴加後之混合液攪拌25分鐘,使粒子前驅體進行縮聚反應,從而得到570.2g(液溫30℃)的縮聚反應液。
<第三步驟>
第三步驟是使用圖5中所示之反應裝置10實施。另外,在所使用之Y字狀三叉管式反應器20中,第一流道110的中心軸與第二流道120的中心軸所形成之角度(以下,有時稱為「分叉角度」)為90度,從縮徑部112(和縮徑部114)的流出口側至中心點C為止的距離R與縮徑部直徑d1(和d2)之比R/d1(和R/d2)為12.5。
在此,在以連接部140附近處的流速為1.9m/s之方式,從第一流道110的入口側供給570.2g縮聚反應液的同時,以連接部140附近處的流速為1.9m/s(水性溶液的流速/縮聚反應液的流速=1,以下稱為「流速比」)之方式,從第二流道120的入口側供給作為水性溶液的20質量%乙醇水溶液570.2g(液溫25℃),使縮聚反應液與水性溶液在連接部140處碰撞混合。然後,得到1140.4g從第三流道130排出的混合溶液、即含有球狀聚甲基倍半矽氧烷粒子之分散液。該分散液(混合溶液)中所含有之球狀聚甲基倍半矽氧烷粒子為3.3質量%,另外,分散液(混合溶液)中所含有之有機溶劑的含有量為44 質量%。
在將該分散液在20℃下靜置(熟化處理)72小時之後,與實施例A1同樣地,透過吸濾回收球狀聚甲基倍半矽氧烷粒子的粉末,在120℃下減壓乾燥24小時,從而得到白色的球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(比較例A1)
除了將第三步驟中使用之水性溶液變更為50質量%甲醇水溶液以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為59質量%。在將該分散液在20℃下靜置24小時而進行熟化後,嘗試透過吸濾回收球狀聚甲基倍半矽氧烷粒子的粉末。但是,所回收之固形物為凝膠狀(gel-like)的塊體。另外,關於該凝膠狀的塊體,即使透過FE-SEM進行確認,也沒有發現粒子成分。
(比較例A2)
與專利文獻4的實施例2-7同樣地,依次實施了第一步驟、第二步驟以及第三步驟。在該粒子形成步驟中,縮聚反應液的液溫為30℃,水性溶液的液溫為25℃。另外,第三步驟中所使用之水性溶液僅由水構成(有機溶劑濃度0質量%),第三步驟中得到之分散液中所含有之有機溶劑的含有量為49 質量%。
對於所得到的分散液,與實施例A1同樣地實施熟化處理、基於吸濾的回收以及減壓乾燥,從而得到白色的球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
另外,作為參考,專利文獻4的實施例2-7和比較例A2之製造步驟上的實質上的不同點在於,有沒有實施第四步驟(疏水化處理)以及有沒有在20℃下實施熟化處理24小時。另外,專利文獻4的實施例2-7,是在專利文獻4中所揭示之所有實施例中,透過第三步驟得到之分散液中所含有的有機溶劑之含有量最大的實施例。
(比較例A3)
與專利文獻4的實施例2-25同樣地,依次實施了第一步驟、第二步驟以及第三步驟。在該粒子形成步驟中,縮聚反應液的液溫為30℃,水性溶液的液溫為25℃。另外,第三步驟中所使用之水性溶液僅由水構成(有機溶劑濃度0質量%),第三步驟中得到之分散液中所含有的有機溶劑之含有量為48 質量%。
對於所得到的分散液,與實施例A1同樣地實施熟化處理、基於吸濾的回收以及減壓乾燥,從而得到白色的球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
另外,作為參考,專利文獻4的實施例2-7和比較例A3之製造步驟上的實質上的不同點在於,有沒有實施第四步驟(疏水化處理)以及有沒有在20℃下實施熟化處理24小時。另外,專利文獻4的實施例2-25,是在專利文獻4中所揭示之所有實施例中,透過第三步驟得到之分散液中所含有的有機溶劑之含有量第二大的實施例。
(比較例A4)
除了將第三步驟中使用之水性溶液變更為不含有有機溶劑的水以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。第三步驟中得到之分散液的有機溶劑之含有量為34質量%。
(比較例A5)
除了在第三步驟中,將分叉管反應器20的連接部140處之碰撞混合條件變更為以下所示之條件以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。
<碰撞混合條件>
(1)從第一流道100的入口側供給之縮聚反應液的供給量及其流速:
設置為與實施例A1相同。
(2)從第二流道120的入口側供給之10質量%甲醇水溶液的供給量及其流速:
將供給量變更為1016g(液溫25℃),將流速變更為3.6m/s。
(3)流速比
將流速比變更為1.9。
另外,透過實施第三步驟,得到含有球狀聚甲基倍半矽氧烷粒子之分散液1550g。另外,該分散液(混合溶液)中所含有之球狀聚甲基倍半矽氧烷粒子為2.2質量%,另外,分散液(混合溶液)中所含有之有機溶劑的含有量為30 質量%。
(比較例A6)
除了將第三步驟中得到之分散液(混合溶液)的熟化時間變更為0.5小時以外,與實施例A1同樣地製造了球狀聚甲基倍半矽氧烷粒子。
(比較例A7)
除了將第三步驟中得到之分散液(混合溶液)的熟化時間變更為0.5小時以外,與實施例A2同樣地製造了球狀聚甲基倍半矽氧烷粒子。但是,即使吸濾熟化處理後之分散液,實際上也無法回收固形物,由此知道了未充分形成粒子。
(比較例A8)
向200ml茄形燒瓶中裝入54.0g水和0.01g作為催化劑的乙酸,並在30℃下進行攪拌。向其中加入68.0g甲基三甲氧基矽烷並攪拌4小時,從而得到122.0g反應溶液。此時,透過甲基三甲氧基矽烷的水解反應而生成之甲醇量為48.1g。
當將該122.0g反應溶液添加到由14g的25%氨水和498g的水構成之25℃的混合液中時,溶液立即乳化。該混合液中之有機溶劑的含有量為7.6質量%。當將乳化的混合液在30℃下攪拌16小時之後靜置5分鐘時,在燒瓶的底部生成了白色的沉澱物。透過吸濾回收該沉澱物,將所得到之濾餅(cake)在120℃下減壓乾燥24小時,從而得到球狀聚甲基倍半矽氧烷粒子的粉末。
(比較例A9)
除了將比較例A8中之25%氨水的量變更為2.8g,將水的量變更為511g以外,進行與比較例A8相同的操作,從而得到球狀聚甲基倍半矽氧烷粒子的粉末。該混合液中之有機溶劑的含有量為7.6 質量%。
(實施例B1)
<第一步驟>
在1000ml茄形燒瓶中裝入56g水和0.01g作為催化劑的乙酸,並在30℃下進行攪拌。在其中加入70g甲基三甲氧基矽烷並攪拌1小時,從而得到126g原料溶液。此時,透過甲基三甲氧基矽烷的水解反應而生成之甲醇量為49.5g。另外,該醇量為100%水解時的理論計算值,以下的各實施例和比較例中也與此相同。
<第二步驟>
在1000ml茄形燒瓶中裝入2.9g的25%氨水、91.2g水、313.8g甲醇,並在30℃下進行攪拌,從而調製出鹼性水系介質。將在第一步驟中得到之126.0g原料溶液在1分鐘內滴加至該鹼性水系介質中。將該原料溶液滴加後之混合液攪拌25分鐘,使粒子前驅體進行縮聚反應,從而得到533.9g(液溫30℃)的縮聚反應液。
<第三步驟>
第三步驟是使用圖5中所示之反應裝置10實施。另外,在所使用之Y字狀三叉管式反應器20中,第一流道110的中心軸與第二流道120的中心軸所形成之角度(以下,有時稱為「分叉角度」)為90度,從縮徑部112(和縮徑部114)的流出口側至中心點C為止的距離R與縮徑部直徑d1(和d2)之比R/d1(和R/d2)為12.5。
在此,在以連接部140附近處的流速為1.9m/s之方式,從第一流道110的入口側供給533.9g縮聚反應液的同時,以連接部140附近處的流速為1.9m/s(水性溶液的流速/縮聚反應液的流速=1,以下稱為「流速比」)之方式,從第二流道120的入口側供給作為水性溶液的10質量%甲醇水溶液533.9g(液溫25℃),使縮聚反應液與水性溶液在連接部140處碰撞混合。然後,得到1067.8g從第三流道130排出之混合溶液、即含有球狀聚甲基倍半矽氧烷粒子之分散液。該分散液(混合溶液)中所含有之微粒為3.2質量%,另外,有機溶劑的含有量為39 質量%。
將該分散液在20℃下靜置(熟化處理)24小時。
<第四步驟>
將熟化處理後之分散液升溫至65℃後,作為疏水劑添加17.2g的六甲基二矽氮烷並攪拌3小時。透過吸濾回收浮出至液體上層部分之疏水化球狀聚甲基倍半矽氧烷粒子的粉體,並在120℃下減壓乾燥24小時,從而得到34g白色的疏水化球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B2)
除了將第三步驟中使用之水性溶液變更為20質量%甲醇水溶液,且將第三步驟中得到之分散液的靜置時間(熟化時間)變更為15小時以外,與實施例B1同樣地得到了球狀聚甲基倍半矽氧烷粒子分散液。第三步驟中得到之分散液的有機溶劑之含有量為44質量%。接著,將熟化處理後之分散液升溫至65℃後,作為疏水劑添加8.6g的六甲基二矽氮烷並攪拌3小時。之後,與實施例B1同樣地進行吸濾以及減壓乾燥,從而得到球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B3)
除了將第三步驟中使用之水性溶液變更為25質量%甲醇水溶液以外,與實施例B1同樣地得到了球狀聚甲基倍半矽氧烷粒子分散液。第三步驟中得到之分散液的有機溶劑之含有量為47質量%。接著,在將該分散液與實施例B1同樣地進行熟化處理,並進一步升溫至65℃之後,作為疏水劑添加17.2g的六甲基二矽氮烷並攪拌3小時。之後,與實施例B1同樣地進行吸濾和減壓乾燥,從而得到球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B4)
除了將第三步驟中使用之水性溶液變更為30質量%甲醇水溶液以外,與實施例B1同樣地得到了球狀聚甲基倍半矽氧烷粒子分散液。第三步驟中得到之分散液的有機溶劑之含有量為49質量%。在將該分散液在20℃下靜置24小時而進行熟化後,與實施例B2同樣地實施疏水化處理、吸濾以及減壓乾燥,從而得到球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B5)
除了將疏水化處理時的六甲基二矽氮烷之添加量變更為3.4g以外,與實施例B4同樣地得到了球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B6)
將與實施例B4同樣地製造之球狀聚甲基倍半矽氧烷粒子分散液,在20℃下靜置168小時而進行熟化處理。接著,使用熟化處理後之分散液,與實施例B2同樣地實施疏水化處理,從而得到球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B7)
除了將疏水化處理時的六甲基二矽氮烷之添加量變更為3.4g以外,與實施例B6同樣地得到了球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B8)
除了將第三步驟中使用之水性溶液變更為35質量%甲醇水溶液以外,與實施例B1同樣地得到了球狀聚甲基倍半矽氧烷粒子分散液。第三步驟中得到之分散液的有機溶劑之含有量為52質量%。在將該分散液在20℃下靜置24小時而進行熟化後,與實施例B1同樣地實施疏水化處理、吸濾以及減壓乾燥,從而得到球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B9)
除了將第三步驟中使用之水性溶液變更為40質量%甲醇水溶液以外,與實施例B1同樣地得到了球狀聚甲基倍半矽氧烷粒子分散液。第三步驟中得到之分散液的有機溶劑之含有量為54質量%。在將該分散液在20℃下靜置24小時而進行熟化處理後,與實施例B2同樣地實施疏水化處理、吸濾以及減壓乾燥,從而得到球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B10)
將與實施例B9同樣地製造之球狀聚甲基倍半矽氧烷粒子分散液,在20℃下靜置168小時而進行了熟化處理。接著,使用熟化處理後之分散液,與實施例B2同樣地實施疏水化處理、吸濾以及減壓乾燥,從而得到球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B11)
除了將第三步驟中使用之水性溶液變更為45質量%甲醇水溶液以外,與實施例B1同樣地得到了球狀聚甲基倍半矽氧烷粒子分散液。第三步驟中得到之分散液的有機溶劑之含有量為57質量%。在將該分散液在20℃下靜置24小時而進行熟化處理後,與實施例B1同樣地實施疏水化處理、吸濾以及減壓乾燥,從而得到球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B12)
除了將第三步驟中使用之水性溶液變更為30質量%乙醇水溶液以外,與實施例B1同樣地得到了球狀聚甲基倍半矽氧烷粒子分散液。第三步驟中得到之分散液的有機溶劑之含有量為49質量%。在將該分散液在20℃下靜置144小時而進行熟化處理後,與實施例B1同樣地實施疏水化處理、吸濾以及減壓乾燥,從而得到球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B13)
除了將第三步驟中使用之水性溶液變更為20質量%異丙醇水溶液以外,與實施例B1同樣地得到了球狀聚甲基倍半矽氧烷粒子分散液。第三步驟中得到之分散液的有機溶劑之含有量為44質量%。在將該分散液在20℃下靜置24小時而進行熟化處理後,與實施例B1同樣地實施疏水化處理、吸濾以及減壓乾燥,從而得到球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B14)
除了將第三步驟中使用之水性溶液變更為25質量%異丙醇水溶液以外,與實施例B1同樣地得到了球狀聚甲基倍半矽氧烷粒子分散液。第三步驟中得到之分散液的有機溶劑之含有量為47質量%。在將該分散液在20℃下靜置144小時而進行熟化處理後,與實施例B1同樣地實施疏水化處理、吸濾以及減壓乾燥,從而得到球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B15)
除了將第三步驟中使用之水性溶液變更為25質量%丙酮水溶液以外,與實施例B1同樣地得到了球狀聚甲基倍半矽氧烷粒子分散液。第三步驟中得到之分散液的有機溶劑之含有量為47質量%。在將該分散液在20℃下靜置144小時而進行熟化處理後,與實施例B1同樣地實施疏水化處理、吸濾以及減壓乾燥,從而得到球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(實施例B16)
直至第三步驟為止進行與實施例A15相同的操作,得到含有球狀聚甲基倍半矽氧烷粒子之分散液1140.4g。另外,第三步驟中得到之分散液的有機溶劑之含有量為與實施例A15相同的44質量%。在將該分散液在20℃下靜置(熟化處理)72小時之後,將其升溫至65℃,作為疏水劑添加17.2g的六甲基二矽氮烷並攪拌3小時。透過吸濾回收浮出至液體上層部分中之疏水化球狀聚甲基倍半矽氧烷粒子的粉體,並在120℃下減壓乾燥24小時,從而得到38g白色的疏水化球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(比較例B1)
作為專利文獻4的實施例2-7的再現實驗,依次實施了第一步驟、第二步驟、第三步驟以及第四步驟。在該粒子形成步驟中,縮聚反應液的液溫為30℃,水性溶液的液溫為25℃。另外,第三步驟中使用之水性溶液僅由水構成(有機溶劑濃度0質量%),第三步驟中得到之分散液中所含有的有機溶劑之含有量為49 質量%。
對於透過第四步驟所得到的分散液,與實施例A1同樣地實施基於吸濾的回收以及減壓乾燥,從而得到白色的球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
另外,未實施積極的熟化處理,但是,由於在第三步驟結束後且第四步驟實施(六甲基二矽氮烷的添加)為止需要約30分鐘的作業時間,因此,熟化時間為0.5小時。
(比較例B2)
除了在第三步驟結束後且第四步驟實施前,在20℃下進行12小時的熟化處理以外,進行與比較例B1相同的操作,從而得到白色的球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(比較例B3)
作為專利文獻4的實施例2-25的再現實驗,依次實施了第一步驟、第二步驟、第三步驟以及第四步驟。在該粒子形成步驟中,縮聚反應液的液溫為30℃,水性溶液的液溫為25℃。另外,第三步驟中使用之水性溶液僅由水構成(有機溶劑濃度0質量%),第三步驟中得到之分散液中所含有的有機溶劑之含有量為48 質量%。
對於透過第四步驟所得到的分散液,與實施例A1同樣地實施基於吸濾的回收以及減壓乾燥,從而得到白色的球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
另外,未實施積極的熟化處理,但是,由於在第三步驟結束後且第四步驟實施(六甲基二矽氮烷的添加)為止需要約30分鐘的作業時間,因此,熟化時間為0.5小時。
(比較例B4)
除了在第三步驟結束後且第四步驟實施前,在20℃下進行12小時的熟化以外,進行與比較例B3相同的操作,從而得到白色的球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(比較例B5)
直至第三步驟結束為止進行與比較例B4相同的操作,從而得到含有球狀聚甲基倍半矽氧烷粒子之分散液(混合溶液)。
對於所得到的分散液,與實施例B1同樣地,在20℃下靜置(熟化處理)24小時。然後,將熟化處理後之分散液升溫至65℃後,作為疏水劑添加8.6g的六甲基二矽氮烷並攪拌3小時,由此實施了第四步驟(疏水化處理)。在疏水化處理之後,與實施例B1同樣地實施基於吸濾的回收以及減壓乾燥,從而得到白色的球狀聚甲基倍半矽氧烷粒子的乾燥粉末。
(比較例B6)
向200ml茄形燒瓶中裝入54.0g水和0.01g作為催化劑的乙酸,並在30℃下進行攪拌。向其中加入68.0g甲基三甲氧基矽烷並攪拌4小時,從而得到122.0g反應溶液。此時,透過甲基三甲氧基矽烷的水解反應而生成之甲醇量為48.1g。
當將該122.0g反應溶液添加到由14g的25%氨水和498g的水構成之25℃的混合液中時,溶液立即乳化。該混合液中之有機溶劑的含有量為7.6質量%。將乳化的混合液在30℃下攪拌16小時之後添加15.2g的六甲基二矽氮烷並攪拌了48小時。將透過吸濾回收沉澱物而得到的濾餅在120℃下減壓乾燥24小時,從而得到球狀聚甲基倍半矽氧烷粒子的粉末,其中,該沉澱物是添加六甲基二矽氮烷並攪拌後混合液中生成之沉澱物。
3.實驗結果
表1~表9中示出各實施例和比較例中之粒子的主要製造條件以及各種評價結果。另外,在表中,標注*1~*9的語句之含義如下。
*1:在第三步驟中,與縮聚反應液混合的水性溶液。
*2:在第三步驟中,將縮聚反應液和水性溶液混合後之混合溶液(分散液)。
*3:利用六甲基二矽氮烷進行的表面處理(疏水化處理)的有無。
*4:透過13
C DDMAS NMR測量得到之峰面積的相對比例(將面積A、B、C之總計值作為100%(基準值)時的值)。
*5:透過29
Si DDMAS NMR測量得到之峰面積的相對比例(將面積D的值作為100%(基準值)時的值)。
*6:表示[{(c)烷氧基+(d)羥基}/(b)甲基]的比率。
*7:表示[(c)烷氧基/{(c)烷氧基+(d)羥基)}]的比率。
另外,圖7係顯示各實施例和比較例之粒子相對於固體表面的附著量和粒徑(D50)之三次方的1000倍值之間關係的圖表。在此,關於粒徑的三次方,可以認為是與粒子欲從固體表面脫離之力(脫離力)實質上呈比例的參數(關於其理由,若需要,請參照後述之備考欄)。因此,當假設粒子相對於固體表面的密合力為固定不變的力時,隨著圖7中之橫軸的值(1000×D503
)變大,粒子對於固體表面的附著量必然變小。另外,在1000×D503
的值相同的粒子A和粒子B中,當粒子對於固體表面的附著量存在顯著差異時,可以認為該附著量之差表示的是粒子A和粒子B相對於固體表面的密合力之差。
另一方面,參照圖7可知,S1
/S2
為8.0以上的實施例的粒子,與S1
/S2
小於8.0的比較例的粒子相比,粒子相對於固體表面的附著量普遍變大。另外,從實施例A系列(series)和實施例B系列(series)的比較中可知,即使是S1
/S2
為8.0以上的粒子,實施表面處理(疏水化處理)的粒子中,粒子相對於固體表面的附著量普遍變大。
<備考>
從表2、5、8明確可知,可以視為各實施例和比較例之粒子的密度是大致固定的。另外,實質上呈球狀之粒子的體積是與粒徑的三次方呈比例的值,粒子的質量m是粒子的體積和密度之乘積。因此,作為圖7中的橫軸所示參數之粒徑的三次方,是與粒子的質量m呈比例的值。另一方面,作用於粒子的力f,是以粒子的質量m和加速度a之乘積來表達。而且,在測量粒子相對於固體表面的附著量時,是透過將玻璃載片進行輕敲(tapping)而實施。因此,可以認為:在進行輕敲時,力f是與粒子質量m的大小無關地,對任意的粒子均以實質相同的加速度a發揮作用。因此,粒子的質量m(粒徑的三次方的值)越大,則輕敲時作用於粒子的力f(粒子欲從固體表面脫離之力)越大。
[表1]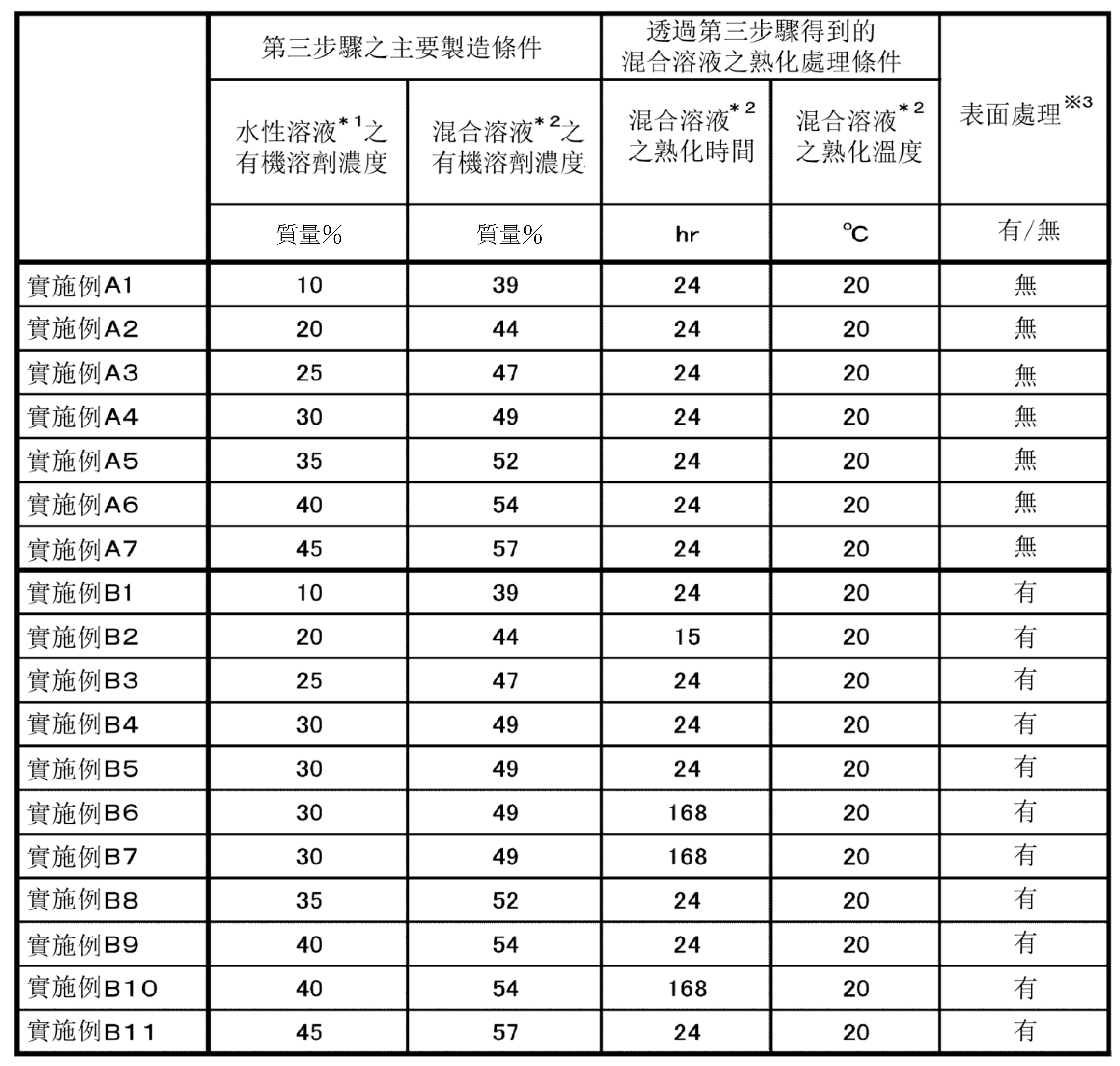
[表2]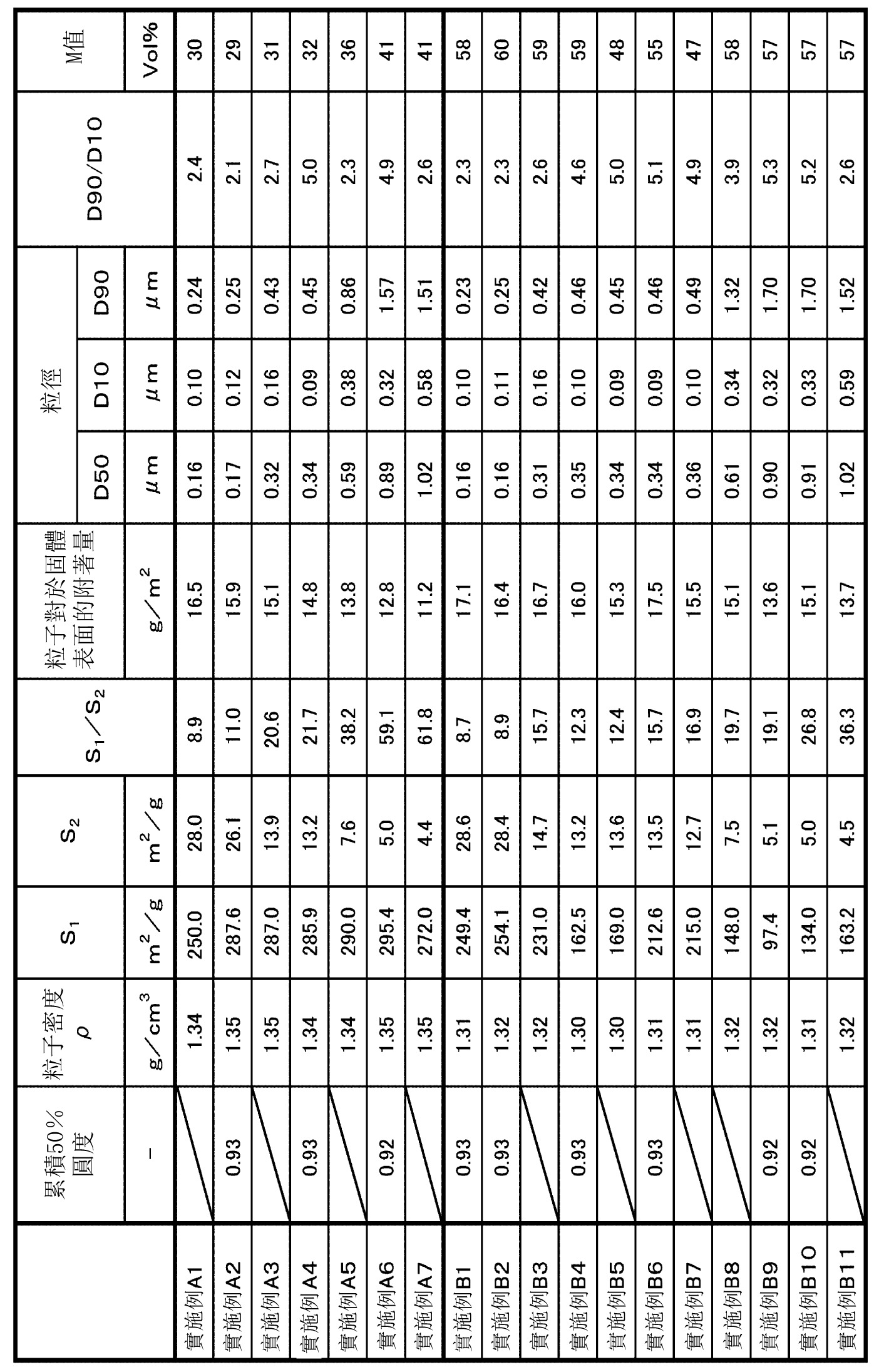
[表3]
[表4]
[表5]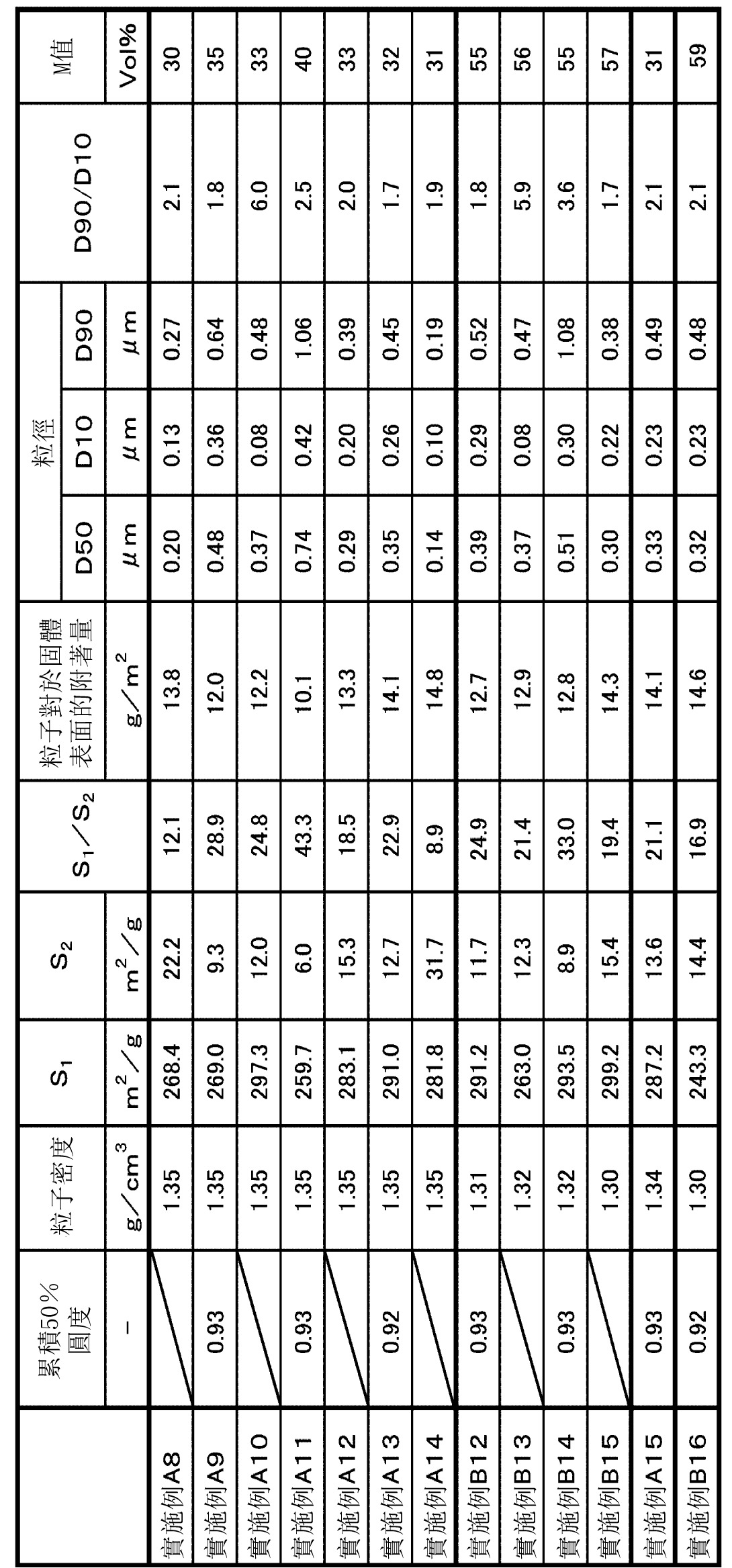
[表6]
[表7]
[表8]
[表9]
10:反應裝置
20:三叉管式反應器
30:第一泵
32:第二泵
40:第一原料箱
42:第二原料箱
50:回收箱
110:第一流道
112、114:縮徑部
120:第二流道
130:第三流道
140:連接部
圖1係顯示實施例A2的球狀聚甲基倍半矽氧烷粒子(非表面處理粒子)之透過13
C DDMAS NMR測量得到之NMR質譜。
圖2係顯示實施例B1的球狀聚甲基倍半矽氧烷粒子(疏水化處理粒子)之透過13
C DDMAS NMR測量得到之NMR質譜。
圖3係顯示實施例A2的球狀聚甲基倍半矽氧烷粒子(非表面處理粒子)之透過29
Si DDMAS NMR測量得到之NMR質譜。
圖4係顯示實施例B1的球狀聚甲基倍半矽氧烷粒子(疏水化處理粒子)之透過29
Si DDMAS NMR測量得到之NMR質譜。
圖5係顯示本實施方式之球狀聚甲基倍半矽氧烷粒子的製造中所使用之反應裝置的一例之概略模式圖。
圖6係顯示圖5中所示之管式反應管的剖面結構的一例之放大剖視圖。
圖7係顯示各實施例和比較例之粒子相對於固體表面的附著量和粒徑(D50)的三次方的1000倍值之間關係之圖表。
Claims (14)
- 一種球狀聚甲基倍半矽氧烷粒子,其特徵在於, 具有至少含有由矽原子(n)和與所述矽原子(n)結合的氧原子(n)形成之交聯網狀結構、與所述矽原子(n)結合之甲基、與所述矽原子(n)結合之烷氧基的粒子主體,並且,滿足以下的算式(1); 算式(1) S1 /S2 ≥8.0 在所述算式(1)中,S1 係透過氮吸附BET單點法測量之比表面積(m2 /g),S2 係6/(ρ×D50)且其單位為(m2 /g),ρ係粒子密度(g/m3 ),D50係透過雷射繞射散射法測量之體積基準的累積50%粒徑(m)。
- 如申請專利範圍第1項所述之球狀聚甲基倍半矽氧烷粒子,其中,所述算式(1)中所示之比率S1 /S2 為10.0以上。
- 如申請專利範圍第1或2項所述之球狀聚甲基倍半矽氧烷粒子,其中,所述粒子主體之表面未被實施表面處理。
- 如申請專利範圍第1~3項中任一項所述之球狀聚甲基倍半矽氧烷粒子,其中,透過甲醇滴定法測量之疏水度為25 vol%~45 vol%。
- 如申請專利範圍第1或2項所述之球狀聚甲基倍半矽氧烷粒子,其中,所述粒子主體之表面利用疏水劑實施表面處理。
- 如申請專利範圍第1、2或5項中任一項所述之球狀聚甲基倍半矽氧烷粒子,其中,所述粒子主體之至少表面附近含有與所述矽原子(n)結合之三烷基矽氧基。
- 如申請專利範圍第6項所述之球狀聚甲基倍半矽氧烷粒子,其中,所述三烷基矽氧基是三甲基矽氧基。
- 如申請專利範圍第1、2、5~7項中任一項所述之球狀聚甲基倍半矽氧烷粒子,其中,透過甲醇滴定法測量之疏水度超過45vol%且70 vol%以下。
- 如申請專利範圍第1~8項中任一項所述之球狀聚甲基倍半矽氧烷粒子,其中,滿足以下的算式(2); 算式(2) 0.016≤A/B≤0.030 在所述算式(2)中,A和B係透過13 C DDMAS NMR測量之峰的面積,A係源於所述烷氧基中含有的碳原子之峰的面積,其中,該碳原子是與所述烷氧基中所含有之氧原子(tg)結合的碳原子,B係源於與所述矽原子(n)結合的甲基中所含有的碳原子之峰的面積。
- 如申請專利範圍第9項所述之球狀聚甲基倍半矽氧烷粒子,其中,所述算式(2)中所示之比率A/B為0.020~0.030。
- 如申請專利範圍第1~10項中任一項所述之球狀聚甲基倍半矽氧烷粒子,其中,所述累積50%粒徑是0.14μm~2.0μm。
- 如申請專利範圍第11項所述之球狀聚甲基倍半矽氧烷粒子,其中,所述累積50%粒徑超過0.30μm且2.0μm以下。
- 如申請專利範圍第1~11項中任一項所述之球狀聚甲基倍半矽氧烷粒子,其中,所述累積50%粒徑超過0.30μm。
- 如申請專利範圍第1~13項中任一項所述之球狀聚甲基倍半矽氧烷粒子,其中,所述烷氧基是甲氧基。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018126534 | 2018-07-03 | ||
| JP2018-126534 | 2018-07-03 | ||
| JP2018128713 | 2018-07-06 | ||
| JP2018-128713 | 2018-07-06 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| TW202010771A true TW202010771A (zh) | 2020-03-16 |
| TWI793343B TWI793343B (zh) | 2023-02-21 |
Family
ID=69060362
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW108123231A TWI793343B (zh) | 2018-07-03 | 2019-07-02 | 球狀聚甲基倍半矽氧烷粒子 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US12054587B2 (zh) |
| EP (1) | EP3819327B1 (zh) |
| JP (1) | JP7284167B2 (zh) |
| KR (1) | KR102746472B1 (zh) |
| CN (1) | CN112004860B (zh) |
| TW (1) | TWI793343B (zh) |
| WO (1) | WO2020009068A1 (zh) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7209609B2 (ja) * | 2018-10-23 | 2023-01-20 | 株式会社トクヤマ | 球状ポリメチルシルセスオキサンからなる液晶用スペーサー |
| CN111302436A (zh) * | 2020-03-02 | 2020-06-19 | 东南大学 | 一种用于清洗废液油水分离的吸附型破乳剂的制备方法 |
Family Cites Families (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5874472A (ja) | 1981-10-29 | 1983-05-04 | Murata Mach Ltd | スパン糸の糸継ぎ装置 |
| JPS6013813A (ja) | 1983-07-05 | 1985-01-24 | Toshiba Silicone Co Ltd | ポリメチルシルセスキオキサンの製造方法 |
| JPH0618879B2 (ja) * | 1988-02-26 | 1994-03-16 | 東芝シリコーン株式会社 | ポリオルガノシルセスキオキサン微粒子 |
| JP2502664B2 (ja) * | 1988-03-25 | 1996-05-29 | 東芝シリコーン株式会社 | クラスタ―状ポリメチルシルセスキオキサン微粉末およびその製造方法 |
| JPH0657777B2 (ja) * | 1988-03-31 | 1994-08-03 | 信越化学工業株式会社 | ゴム組成物 |
| CA2004297A1 (en) * | 1988-12-02 | 1990-06-02 | Hiroshi Kimura | Polyorganosiloxane fine particles |
| JPH0655828B2 (ja) * | 1988-12-15 | 1994-07-27 | 信越化学工業株式会社 | 表面変性ポリメチルシルセスキオキサン球状微粒子及びその製造方法 |
| FR2656317B1 (fr) * | 1989-12-27 | 1994-02-04 | Rhone Poulenc Chimie | Microspheres magnetisables a base de polysilsesquioxane, leur procede de preparation et leur application en biologie. |
| JP3262570B2 (ja) * | 1991-09-06 | 2002-03-04 | ジーイー東芝シリコーン株式会社 | シリコーン系感圧接着剤組成物 |
| JPH06179751A (ja) * | 1992-12-14 | 1994-06-28 | Tokuyama Soda Co Ltd | ポリオルガノシルセスキオキサンの製法 |
| JPH07103238B2 (ja) * | 1993-09-27 | 1995-11-08 | 東芝シリコーン株式会社 | ポリオルガノシルセスキオキサン微粒子の製造方法 |
| JP3970449B2 (ja) * | 1998-12-21 | 2007-09-05 | モメンティブ・パフォーマンス・マテリアルズ・ジャパン合同会社 | 球状ポリメチルシルセスキオキサン微粒子の製造方法 |
| TR200101846T2 (tr) * | 1998-12-22 | 2001-12-21 | Firmenich Sa | Yüzerme özellikleri olan gözenekli polimetilsilseskioksan |
| WO2001012731A1 (en) | 1999-08-19 | 2001-02-22 | Ppg Industries Ohio, Inc. | Hydrophobic particulate inorganic oxides and polymeric compositions containing same |
| JP4014882B2 (ja) | 2002-01-30 | 2007-11-28 | 株式会社トクヤマ | シリカゾルの製造方法 |
| US7125937B2 (en) * | 2004-01-27 | 2006-10-24 | Fina Technology, Inc. | Polyorganosilsesquioxane supported metallocene catalysts |
| JP5153163B2 (ja) | 2007-02-23 | 2013-02-27 | 日興リカ株式会社 | 球状ポリオルガノシルセスキオキサン微粒子の製造方法 |
| KR101077274B1 (ko) * | 2007-05-28 | 2011-10-27 | 코오롱인더스트리 주식회사 | 폴리알킬실세스퀴옥산 미립자 및 그 제조방법 |
| JP5607001B2 (ja) * | 2011-08-11 | 2014-10-15 | 信越化学工業株式会社 | シリコーン微粒子及びその製造方法 |
| JP2013092748A (ja) | 2011-10-26 | 2013-05-16 | Cabot Corp | 複合体粒子を含むトナー添加剤 |
| JP5844889B2 (ja) * | 2012-04-27 | 2016-01-20 | 積水化成品工業株式会社 | 重合体粒子、その製造方法、及び、その用途 |
| JP2015232054A (ja) * | 2012-10-02 | 2015-12-24 | 株式会社カネカ | 活性エネルギー線硬化性コーティング用樹脂組成物 |
| JP5859478B2 (ja) | 2013-04-26 | 2016-02-10 | 信越化学工業株式会社 | シリコーン複合粒子及びその製造方法 |
| EP3095805B1 (en) * | 2014-01-14 | 2018-10-24 | Tokuyama Corporation | Hydrophobized spherical poly (alkyl silsesquioxane) microparticles, external additive for toner, dry electrophotography toner, and method for manufacturing hydrophobized spherical poly (alkyl silsesquioxane) microparticles |
-
2019
- 2019-07-01 KR KR1020207033363A patent/KR102746472B1/ko active Active
- 2019-07-01 US US17/051,696 patent/US12054587B2/en active Active
- 2019-07-01 EP EP19830015.4A patent/EP3819327B1/en active Active
- 2019-07-01 CN CN201980027309.0A patent/CN112004860B/zh active Active
- 2019-07-01 WO PCT/JP2019/026135 patent/WO2020009068A1/ja not_active Ceased
- 2019-07-01 JP JP2020528855A patent/JP7284167B2/ja active Active
- 2019-07-02 TW TW108123231A patent/TWI793343B/zh active
Also Published As
| Publication number | Publication date |
|---|---|
| CN112004860A (zh) | 2020-11-27 |
| JP7284167B2 (ja) | 2023-05-30 |
| US20210238359A1 (en) | 2021-08-05 |
| KR102746472B1 (ko) | 2024-12-23 |
| KR20210025518A (ko) | 2021-03-09 |
| US12054587B2 (en) | 2024-08-06 |
| EP3819327A4 (en) | 2022-03-30 |
| EP3819327A1 (en) | 2021-05-12 |
| TWI793343B (zh) | 2023-02-21 |
| CN112004860B (zh) | 2022-09-06 |
| WO2020009068A1 (ja) | 2020-01-09 |
| JPWO2020009068A1 (ja) | 2021-09-02 |
| EP3819327B1 (en) | 2024-10-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6116711B2 (ja) | 疎水化球状ポリアルキルシルセスキオキサン微粒子、トナー用外添剤、電子写真用乾式トナー、および、疎水化球状ポリアルキルシルセスキオキサン微粒子の製造方法 | |
| CN107075289B (zh) | 表面改性金属氧化物粒子分散液、硅酮树脂复合组合物及复合物、光学构件及发光装置 | |
| JP5795840B2 (ja) | シリカ粒子材料、シリカ粒子材料含有組成物、およびシリカ粒子の表面処理方法 | |
| US20080268362A1 (en) | Hydrophobic spherical silica microparticles having a high degree of flowability, method of producing same, electrostatic image developing toner external additive using same, and organic resin composition containing same | |
| JP3988936B2 (ja) | シラン表面処理球状シリカチタニア系微粒子、その製造方法、および、それを用いた静電荷像現像用トナー外添剤 | |
| CN1246222C (zh) | 具有甲硅烷基化剂均匀层的硅石 | |
| JP2010143806A (ja) | 表面処理シリカ系粒子及びその製造方法 | |
| JP2009513741A (ja) | 高い充填剤含量を有するシラン製剤 | |
| JP2013075822A (ja) | 表面処理シリカ系粒子の製造方法 | |
| JP2012214554A (ja) | 熱可塑性樹脂組成物 | |
| TWI793343B (zh) | 球狀聚甲基倍半矽氧烷粒子 | |
| JP4060241B2 (ja) | 疎水性球状シリカ系微粒子、その製造方法、および、それを用いた静電荷像現像用トナー外添剤 | |
| JP2013204029A (ja) | 光学用樹脂組成物 | |
| JP6053444B2 (ja) | 金属酸化物ナノ粒子分散液の製造方法 | |
| JP5687785B2 (ja) | シリカ粒子の表面処理方法 | |
| JP2018016502A (ja) | シリカ粒子、シリカ粒子含有組成物、オルガノゾル及びシリカ粒子の製造方法 | |
| JP7209609B2 (ja) | 球状ポリメチルシルセスオキサンからなる液晶用スペーサー | |
| US20220185739A1 (en) | Silica-titania composite oxide powder |