KR20130108997A - 치환된 헤테로시클릴 벤질 피라졸 및 그의 용도 - Google Patents
치환된 헤테로시클릴 벤질 피라졸 및 그의 용도 Download PDFInfo
- Publication number
- KR20130108997A KR20130108997A KR1020127032069A KR20127032069A KR20130108997A KR 20130108997 A KR20130108997 A KR 20130108997A KR 1020127032069 A KR1020127032069 A KR 1020127032069A KR 20127032069 A KR20127032069 A KR 20127032069A KR 20130108997 A KR20130108997 A KR 20130108997A
- Authority
- KR
- South Korea
- Prior art keywords
- formula
- compound
- salts
- mmol
- solvates
- Prior art date
Links
- LQKJPBCCNFJUCX-UHFFFAOYSA-N CC(C)C(C(C)=O)N Chemical compound CC(C)C(C(C)=O)N LQKJPBCCNFJUCX-UHFFFAOYSA-N 0.000 description 1
- 0 CN(C)CC(*)=O Chemical compound CN(C)CC(*)=O 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4245—Oxadiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Epidemiology (AREA)
- Diabetes (AREA)
- Rheumatology (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Dermatology (AREA)
- Ophthalmology & Optometry (AREA)
- Hospice & Palliative Care (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pain & Pain Management (AREA)
- Pulmonology (AREA)
- Vascular Medicine (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Obesity (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
본원은 신규한 치환된 1-[3-(헤테로시클릴)벤질]-1H-피라졸 유도체, 그의 제조 방법, 질환의 치료 및/또는 예방을 위한 그의 용도, 및 질환, 특히 과다증식성 및 혈관신생 질환뿐만 아니라 및 저산소 상태에 대한 대사적 적응에 의해 유발되는 질환의 치료 및/또는 예방에 사용되는 의약의 제조를 위한 그의 용도에 관한 것이다. 그러한 치료는 단일요법으로서 또는 다른 의약 또는 다른 치료적 처치와 조합하여 시행될 수 있다.
Description
본원은 신규한 치환된 1-[3-(헤테로시클릴)벤질]-1H-피라졸 유도체, 그의 제조 방법, 질환의 치료 및/또는 예방을 위한 그의 용도 및 질환의 치료 및/또는 예방용, 더 특히 과다증식성 및 혈관신생 질환 및 저산소 상태에 대한 대사적 적응에 기인하는 그러한 질환의 치료 및/또는 예방용 의약의 제조를 위한 그의 용도에 관한 것이다. 그러한 치료는 단일요법의 형태로 또는 아니면 다른 의약 또는 추가의 치료적 처치와 조합하여 수행될 수 있다.
암은 매우 다양한 상이한 조직의 제어되지 않은 세포 성장의 결과이다. 다수의 경우에 새로운 세포는 기존 조직 내로 침투하거나 (침습성 성장), 이들은 먼 기관으로 전이한다. 암은 매우 다양한 상이한 기관에서 발생하고 흔히 조직에 특이적인 방식으로 진행된다. 따라서 용어 "암"은 총칭으로서 상이한 기관, 조직 및 세포 유형의 정의된 질환의 대형 군을 기술한다.
2002년에, 전 세계 440만명의 사람들이 유방, 장, 난소, 폐 또는 전립선 종양으로 진단받았다. 동년도에, 대략 250만명이 이들 질환의 결과로 사망한 것으로 추정되었다 (문헌 [Globocan 2002 Report)]. 미국에서만, 2005년에, 125만 초과의 신규 암 발생 및 암으로부터 500,000건 초과의 사망이 예측되었다. 이들 신규한 암 발생의 대다수는 장암 (~ 100,000), 폐암 (~ 170,000), 유방암 (~ 210,000) 및 전립선암 (~ 230,000)과 관련된다. 다음 10년에 걸쳐 대략 15%의 암의 추가의 증가가 예상된다 (문헌 [American Cancer Society, Cancer Facts and Figures 2005]).
초기 단계에서 일부 암은 수술 및 방사선 처치에 의해 제거될 수 있다. 전이된 종양은 일반적으로 화학요법제에 의해 임시방편으로만 치료될 수 있다. 여기서의 목적은 삶의 질의 개선 및 생명의 연장의 최적 조합을 달성하는 것이다.
화학요법은 흔히 세포독성 의약의 조합으로 이루어진다. 이들 물질의 대다수는 그의 작용 메카니즘으로서 튜불린에 대한 결합을 갖거나, 이들은 핵산의 형성 및 처리와 상호작용하는 화합물이다. 최근의 것으로서, 이들은 또한 후성적 DNA 변형 또는 세포 주기 진행을 간섭하는 효소 억제제 (예를 들어 히스톤 데아세틸라제 억제제, 오로라 키나제 억제제)를 포함한다. 이러한 요법은 독성이기 때문에, 최근 세포내에서의 특이 과정이 높은 수준의 독성 스트레스 없이 차단되는 표적 요법에 대한 집중이 증가되고 있다. 이들은 특히 수용체의 인산화 및 신호 전달 분자를 억제하는 키나제 억제제를 포함한다. 이의 한 예는 이마티닙이고, 이는 만성 골수성 백혈병 (CML) 및 위장관 간질 종양 (GIST)의 치료에 매우 성공적으로 사용된다. 추가의 예는 EGFR 키나제 및 HER2를 차단하는 물질, 예컨대 에를로티닙, 및 VEGFR 키나제 억제제, 예컨대 소라페닙 및 수니티닙이고, 이들은 신장 세포 암종, 간 암종 및 진행된 단계의 GIST에 대해 사용된다.
VEGF에 대한 항체를 사용하여, 결장직장 암종 환자의 수명을 연장하는 것이 가능해왔다. 베바시주맙은 혈관의 성장을 억제하는데, 이는 종양의 급속 팽창에 대한 장해물이며, 그 이유는 종양이 공급 및 처분 기능을 지속적으로 하기 위해 혈관계로의 접속을 필요로 하기 때문이다.
혈관신생에 대한 한 자극은 저산소증이고, 이는 고형 종양으로 되풀이되어 일어나는데, 그 이유는 혈액 공급이 조절되지 않은 성장 때문에 불충분하기 때문이다. 산소의 결핍이 있는 경우, 세포는 그의 대사 작용을 산화적 인산화에서 해당작용으로 전환시켜, 세포 중 ATP 수준이 안정화되도록 한다. 당해 과정은 세포 중 산소 함량에 의존하여 상향조절되는 전사 인자에 의해 제어된다. "저산소증-유도성 인자(HIF)"로 칭해지는 이러한 전사 인자는 통상적으로 급속 분해에 의해 번역후 제거되고 세포 핵으로 이송되지 못하도록 한다. 이는 산소-분해성 도메인(ODD) 중 2개의 프롤린 단위 및 효소 프롤릴 데히드로게나제 및 FIH("HIF 억제 인자")에 의한 C 말단 근처의 1개의 아스파라긴 단위의 히드록실화에 의해 완수된다. 프롤린 단위의 변형 후, HIF는 프로테아솜 장치를 통해 히펠-린다우(Hippel-Lindau) 단백질 (유비퀴틴-E3-리가제 복합체의 부분)에 의한 매개로 분해될 수 있다 (문헌 [Maxwell, Wiesener et al ., 1999]). 산소 결핍의 경우, 분해는 일어나지 않고 단백질은 무조절되어 다수의 (100개 초과) 다른 단백질의 전사가 야기되거나 당해 전사의 차단이 야기된다 (문헌 [Semenza and Wang, 1992]; [Wang and Semenza, 1995]).
전사 인자 HIF는 조절된 α-서브유닛 및 구성적으로 존재하는 β-서브유닛에 의해 형성된다 (ARNT, 아릴 탄화수소 수용체 핵 전이체). 3종의 상이한 종의 α-서브유닛인 1α, 2α 및 3α가 있고, 후자는 오히려 억제인자인 것으로 추정된다 (문헌 [Makino, Cao et al ., 2001]). HIF 서브유닛은 그의 HLH 및 PAS (Per-Arnt-Sim) 도메인을 통해 이량체화하는 bHLH (염기성 헬릭스 루프 헬릭스 (basic helix loop helix)) 단백질이고, 이로써 그의 전사활성화 활성이 개시된다 (문헌 [Jiang, Rue et al., 1996]).
가장 중요한 종양 실체에서, HIF1α 단백질의 과다발현은 증가되는 혈관 밀도 및 증진된 VEGF 발현과 상관관계가 있다. (문헌 [Hirota and Semenza, 2006]). 동시에, 글루코스 대사는 해당작용으로 이동되고, 크렙스 회로는 세포 단위의 생성을 위하여 감소된다. 이는 또한 지질 대사의 변화를 의미한다. 이러한 변화는 종양의 생존을 보장하는 것으로 보인다. 한편, HIF의 활성이 이제 억제될 경우, 종양의 발전은 결과적으로 억제될 수 있을 것이다. 이는 이미 다양한 실험 모델에서 관찰되었다 (문헌 [Chen, Zhao et al ., 2003]; [Stoeltzing, McCarty et al ., 2004]; [Li, Lin et al ., 2005]; [Mizukami, Jo et al ., 2005]; [Li, Shi et al ., 2006]). 따라서 HIF-제어된 대사의 특이적 억제제는 종양 치료제로서 적합할 수 있다.
WO 2004/089303-A2에는 디아릴-치환된 피라졸이 정신 장애의 치료에 대한 mGluR5 조절제로서 기재되어 있다. WO 2010/072352-A1 및 WO 2010/085584-A1에는 3-페닐-5-(1H-피라졸-4-일)-1,2,4-옥사디아졸 유도체가 자가면역 및 혈관 장애의 치료용 스핑고신-1-포스페이트 효능제로서 개시되어 있다.
WO 2005/030121-A2 및 WO 2007/065010-A2에는 종양 세포에서 HIF 및 HIF-조절 유전자의 발현 억제를 위한 특정 피라졸 유도체의 유용성이 기재되어 있다. WO 2008/141731-A2에는 헤테로아릴-치환 N-벤질피라졸이 암 치료용 HIF 조절 경로의 억제제로서 개시되어 있다. 그러나, 다수의 이들 화합물은, 동물 모델에서 그의 약동학적 특성을 기반으로, 충분한 억제 활성 또는 기타의 것을 갖지 않고, 인간 신체에서 긴 반감기 (>48 h)를 갖는 것으로 기대되므로 1일 1회 반복 투여 후 상당한 물질 축적의 개연성이 있는 것으로 밝혀졌다.
따라서 본 발명의 목적은 첫째로 전사 인자 HIF의 전사활성화 작용의 강력한 억제제로서 작용하고 둘째로 임상적으로 관련되는 축적의 동시 발생 없이 1일 1회 반복 투여를 가능하게 하는 약동학적 프로파일을 갖는 신규 화합물을 발견하고 제공하는 것이다. 이러한 특성은 또한 이들 HIF 억제제의 임상 이용가능성을 전반적으로 확대하고 더 특히는 다른 활성 성분, 예를 들어 통상적인 종양 화학요법제와의 조합성을 촉진할 수 있을 것이다.
이러한 목적은 이하에 기재된 본 발명의 화합물에 의해 달성된다. 구조적 측면에서, 이러한 신규한 군의 N-벤질피라졸 유도체는 벤질 헤드 기(head group)의 3 위치에 히드록실- 또는 시아노-치환된 헤테로시클릴 라디칼을 특징으로 하고, 이는 놀랍게도 화합물의 특성의 개선된 프로파일을 초래한다.
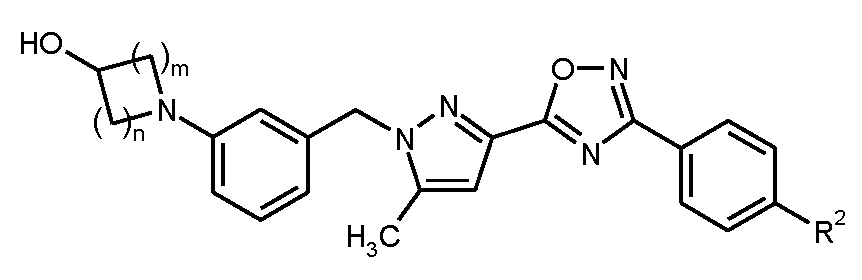
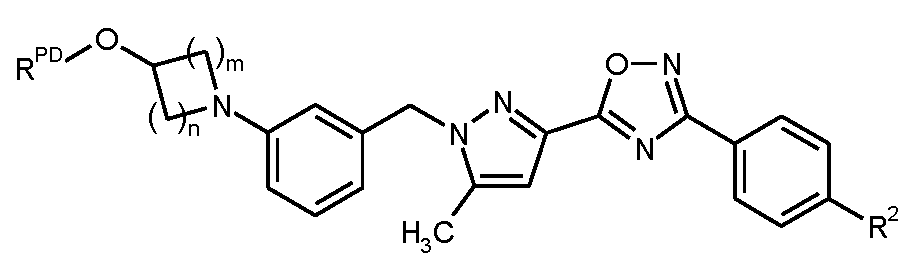
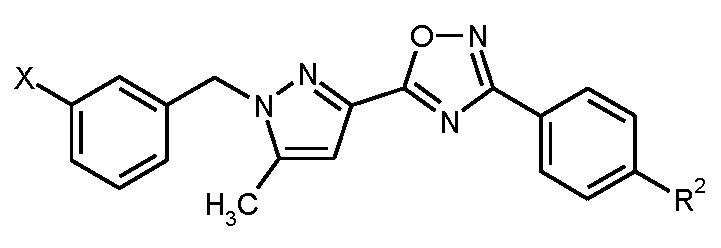
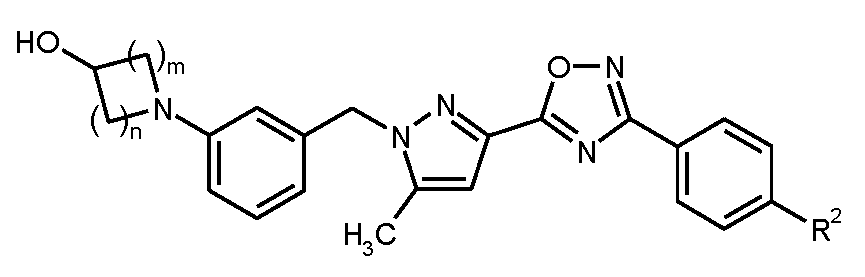
본 발명은 구체적으로 화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물에 관한 것이다.
<화학식 I>

상기 식에서,
m은 1 또는 2이고,
n은 1, 2 또는 3이고,
R1은 히드록실 또는 시아노이고,
R2는 트리플루오로메톡시, 트리플루오로메틸술파닐, 트리플루오로메틸술포닐, 펜타플루오로술파닐 또는 화학식  의 기이고, 여기서
의 기이고, 여기서
*는 페닐 고리에 대한 결합 부위를 나타내고,
R3A 및 R3B는 각각 독립적으로 플루오린 또는 메틸이거나, 또는
서로 결합하여, 이들이 결합된 탄소 원자와 함께, 시클로프로판-1,1-디일, 시클로부탄-1,1-디일, 시클로펜탄-1,1-디일, 시클로헥산-1,1-디일, 옥세탄-3,3-디일 또는 테트라히드로-2H-피란-4,4-디일 고리를 형성하고,
R4는 수소, 플루오린, 메틸, 트리플루오로메틸 또는 메톡시이다.
본 발명의 맥락에서 바람직한 것은
m 및 n이 각각 독립적으로 1 또는 2이고,
R1이 히드록실 또는 시아노이고,
R2가 트리플루오로메틸, 트리플루오로메톡시, 트리플루오로메틸술파닐 또는 화학식  의 기이고, 여기서
의 기이고, 여기서
*가 페닐 고리에 대한 결합 부위를 나타내고,
R3A 및 R3B가 둘 다 메틸이거나, 또는 서로 결합하여, 이들이 결합된 탄소 원자와 함께, 시클로프로판-1,1-디일, 시클로부탄-1,1-디일, 옥세탄-3,3-디일 또는 테트라히드로-2H-피란-4,4-디일 고리를 형성하고,
R4가 수소, 플루오린, 메틸 또는 트리플루오로메틸인 화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물이다.
본 발명의 맥락에서 특히 바람직한 것은
m 및 n이 둘 다 1 또는 2이고,
R1이 히드록실 또는 시아노이고,
R2가 트리플루오로메톡시 또는 화학식 의 기이고, 여기서 *가 페닐 고리에 대한 결합 부위를 나타내는 것인 화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물이다.
본 발명의 특정 실시양태는
R1이 히드록실이고,
m, n 및 R2 각각이 상기 정의된 바와 같은 것인 화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물이다.
이 후자의 실시양태는 화학식 I-A의 화합물, 및 그의 염, 용매화물 및 염의 용매화물에 상응한다.
<화학식 I-A>

상기 식에서, m, n 및 R2 각각은 상기 정의된 바와 같다.
추가 측면에서, 본 발명은 또한 화학식 I-A의 화합물의 특정 전구약물에 관한 것이다. 일반적으로, 용어 "전구약물"는 본원에서 화학식 I-A의 화합물의 공유 유도체를 지칭하며, 이는 그 자체로 생물학적으로 활성 또는 비활성일 수 있지만, 체내에 존재하는 동안, 예를 들어 대사 또는 가수분해 경로에 의해, 화학식 I-A의 화합물로 전환된다.
따라서, 본 발명은 추가로 화학식 I-PD의 화합물, 및 그의 염, 용매화물 및 염의 용매화물을 제공한다.
<화학식 I-PD>

상기 식에서,
m, n 및 R2 각각은 상기 정의된 바와 같고,
RPD는 화학식  의 전구약물 기이고, 여기서
의 전구약물 기이고, 여기서
#는 산소 원자에 대한 결합 부위를 나타내고,
R5는 수소 또는 (C1-C4)-알킬이고,
R6A 및 R6B는 각각 독립적으로 수소 또는 메틸이다.
본 발명의 맥락에서 바람직한 것은
RPD가 화학식  의 전구약물 기이고, 여기서 #가 산소 원자에 대한 결합 부위를 나타내는 것인 화학식 I-PD의 화합물, 및 그의 염, 용매화물 및 염의 용매화물이다.
의 전구약물 기이고, 여기서 #가 산소 원자에 대한 결합 부위를 나타내는 것인 화학식 I-PD의 화합물, 및 그의 염, 용매화물 및 염의 용매화물이다.
화학식 I-PD의 화합물은 수성 또는 다른 생리학상 상용성 매질에 우수한 용해도를 갖는 화학식 I-A의 화합물의 전구약물이고; 게다가 이들은 적절한 산과의 염 형성의 가능성을 제공하며, 이는 용해도의 추가의 증가를 초래할 수 있다. 따라서, 화학식 I-PD의 화합물 및 그의 염은 특히 정맥내 투여 형태 또는 아니면 변형된 방출 특성을 갖는 고체 제제에 특히 적합하다. 또한 이는 이들 화합물을 사용하는 추가의 치료 분야를 일으킬 수 있을 것이다.
따라서 본 발명의 화합물은, 화학식 I, I-A 및 I-PD에 포함되고 이하에 특정된 화합물이 아직 염, 용매화물 및 염의 용매화물이 아닌 정도까지, 화학식 I, I-A 및 I-PD의 화합물 및 그의 염, 용매화물 및 염의 용매화물, 화학식 I, I-A 및 I-PD에 포함되고 이하에 특정된 화학식의 화합물 및 그의 염, 용매화물 및 염의 용매화물, 및 화학식 I, I-A 및 I-PD에 포함되고 실시예로서 이하에 특정된 화합물 및 그의 염, 용매화물 및 염의 용매화물이다.
그의 구조에 의존하여, 본 발명의 화합물은 상이한 입체이성질체 형태로, 즉 배위 이성질체의 형태로 또는 적절한 경우 또한 형태 이성질체 (거울상이성질체 및/또는 부분입체이성질체 (회전장애이성질체의 경우의 것을 포함함))로서 존재할 수 있다. 따라서 본 발명은 거울상이성질체 또는 부분입체이성질체 및 그의 각각의 혼합물을 포함한다. 입체 이성질체적으로 균질한 구성요소는 공지된 방식으로 거울상이성질체 및/또는 부분입체이성질체의 그러한 혼합물로부터 단리될 수 있고; 이러한 목적으로 바람직하게는 크로마토그래피 과정, 특히 비키랄(achiral) 또는 키랄 상에 대해 HPLC 크로마토그래피가 사용된다.
본 발명의 화합물이 호변이성질체 형태로 존재할 수 있는 경우, 본 발명은 모두 호변이성질체 형태를 포함한다.
본 발명은 또한 본 발명의 화합물의 모든 적합한 동위원소 변이체를 포함한다. 본 발명의 화합물의 동위원소 변이체는 본원에서 본 발명의 화합물 내에서 1종 이상의 원자가 동일 원자 번호를 가지나 자연에서 통상 또는 주로 존재하는 원자 질량과는 상이한 원자 질량을 갖는 또 다른 원자로 교환된 화합물을 의미하는 것으로 이해된다. 본 발명의 화합물로 혼입될 수 있는 동위원소의 예는 수소, 탄소, 질소, 산소, 인, 황, 플루오린, 염소, 브로민 및 아이오딘의 동위원소, 예컨대 2H (중수소), 3H (삼중수소), 13C, 14C, 15N, 17O, 18O, 32P, 33P, 33S, 34S, 35S, 36S, 18F, 36Cl, 82Br, 123I, 124I, 129I 및 131I이다. 본 발명의 화합물의 특정 동위원소 변이체, 특히 1종 이상의 방사성 동위원소가 혼입된 것은, 예를 들어 작용 메카니즘, 또는 체내 활성 성분 분포의 검사에 유익할 수 있고; 비교적 용이한 제조가능성 및 검출가능성으로 인해, 특히 3H 또는 14C 동위원소로 표지된 화합물은 이러한 목적에 적합하다. 게다가, 동위원소, 예를 들어 중수소의 혼입은 화합물의 더 큰 대사적 안정성의 결과로서 특별한 치료 이익, 예를 들어 체내 반감기의 연장 또는 필요한 활성 용량의 감소를 초래할 수 있고; 따라서 본 발명의 화합물의 이러한 변형은 일부 경우에 또한 본 발명의 바람직한 실시양태를 구성할 수 있다. 본 발명의 화합물의 동위원소 변이체는 당업자에게 공지된 일반적으로 통상적인 공정에 의해, 예를 들어 하기 기재된 방법 및 실시예에 기재된 방법에 의해, 거기서 특정 시약 및/또는 출발 화합물의 상응하는 동위원소 변형을 사용함으로써 제조할 수 있다.
본 발명의 맥락에서, 바람직한 염은 본 발명의 화합물의 생리학상 허용되는 염이다. 또한, 그 자체는 제약 적용에 적합하지 않지만, 예를 들어 본 발명의 화합물의 단리, 정제 또는 저장용으로 사용될 수 있는 염도 포함된다.
본 발명의 화합물의 생리학상 허용되는 염에는 특히 무기 산, 카르복실산 및 술폰산의 산 부가염, 예를 들어 염산, 브로민화수소산, 황산, 인산, 메탄술폰산, 에탄술폰산, 톨루엔술폰산, 벤젠술폰산, 나프탈렌술폰산, 아세트산, 트리플루오로아세트산, 프로피온산, 락트산, 타르타르산, 말산, 시트르산, 푸마르산, 말레산 및 벤조산의 염이 포함된다.
본 발명의 맥락에서, 용매화물은 고체 또는 액체 상태로 용매 분자와의 배위에 의한 착체를 형성하는 본 발명의 화합물의 그러한 형태를 지칭한다. 수화물은 배위가 물과 함께인 용매화물의 구체적 형태이다. 본 발명의 맥락에서 바람직한 용매화물은 수화물이다.
본 발명의 맥락에서, 달리 명시되는 않는 한, 치환기는 다음과 같이 정의된다:
본 발명의 맥락에서 ( C 1 - C 4 )- 알킬은 1 내지 4개의 탄소 원자를 갖는 직쇄 또는 분지형 알킬 라디칼이다. 바람직한 예에는 메틸, 에틸, n-프로필, 이소프로필, n-부틸, 이소부틸, sec-부틸, tert-부틸이 포함된다.
본 발명의 맥락에서, 1회 초과로 존재하는 모든 라디칼은 서로 독립적으로 정의된다. 본 발명의 화합물 중 라디칼이 치환될 경우, 달리 명시되지 않는 한, 라디칼은 일- 또는 다중치환될 수 있다. 1개 또는 2개의 동일하거나 상이한 치환기에 의한 치환이 바람직하다. 특히 바람직한 것은 1개의 치환기에 의한 치환이다.
라디칼의 특정 조합 또는 바람직한 조합에서 특정된 개별적인 라디칼 정의는 특정된 라디칼의 특정 조합과 독립적으로, 또한 원하는 바에 따라 다른 조합의 라디칼 정의에 의해 대체된다.
매우 특히 바람직한 것은 상기 언급된 바람직한 범위 중 2개 이상의 조합이다.
본 발명은 추가로, 하기 화학식 II의 화합물을 적합한 팔라듐 촉매 및 염기의 존재하에 하기 화학식 III의 화합물과 커플링시키고, 생성된 화학식 I의 화합물을 임의로 그의 거울상이성질체 및/또는 부분입체이성질체로 분리시키고/시키거나 적절한 (i) 용매 및/또는 (ii) 산을 사용하여 그의 용매화물, 염 및/또는 염의 용매화물로 전환시키는 것을 특징으로 하는, 화학식 I의 화합물의 제조 방법을 제공한다.
<화학식 II>

상기 식에서,
R2는 상기 정의된 바와 같고
X는 브로민 또는 아이오딘이다.
<화학식 III>

상기 식에서,
m, n 및 R1 각각은 상기 정의된 바와 같다.
반응 (II) + (III) -> (I)에 대한 적합한 불활성 용매는, 예를 들어, 방향족 탄화수소, 예컨대 벤젠, 톨루엔 또는 크실렌, 에테르, 예컨대 디에틸 에테르, 디이소프로필 에테르, 메틸 tert-부틸 에테르, 1,2-디메톡시에탄, 비스(2-메톡시에틸) 에테르, 테트라히드로푸란 또는 1,4-디옥산, 또는 이중극성 비양성자성 용매, 예컨대 아세토니트릴, N,N-디메틸포름아미드 (DMF), N,N-디메틸아세트아미드 (DMA), 디메틸 술폭시드 (DMSO), N, N'-디메틸프로필렌우레아 (DMPU), N-메틸피롤리논 (NMP) 또는 피리딘이다. 마찬가지로 이들 용매의 혼합물을 사용하는 것이 가능하다. 1,2-디메톡시에탄, 테트라히드로푸란, N,N-디메틸포름아미드 또는 톨루엔을 사용하는 것이 바람직하다.
커플링 반응 (II) + (III) -> (I)은 전이 금속 촉매를 활용하여 수행한다. 이러한 목적으로 특히 적합한 것은, 임의로 추가의 포스핀 리간드, 예컨대 2-디시클로헥실포스피노-2'-(N,N-디메틸아미노)비페닐, 2-디시클로헥실포스피노-2',4',6'-트리이소프로필비페닐 (X-Phos) 또는 4,5-비스(디페닐포스피노)-9,9-디메틸크산텐 (Xantphos)과 조합된, 팔라듐(0) 촉매, 예를 들어 비스(디벤질리덴아세톤)팔라듐(0), 트리스(디벤질리덴아세톤)디팔라듐(0) 또는 테트라키스(트리페닐포스피노)팔라듐(0)이다 [예를 들어 문헌 [J. Hassan et al., Chem . Rev . 102, 1359-1469 (2002)] 참조]. 트리스(디벤질리덴아세톤)디팔라듐(0)을 2-디시클로헥실포스피노-2',4',6'-트리이소프로필비페닐 (X-Phos)과 함께 사용하는 것이 바람직하다.
커플링은 일반적으로 염기를 가하여 수행한다. 이러한 목적으로 특히 적합한 것은 알칼리 금속 탄산염, 탄산수소염, 인산염, 인산수소염 또는 tert-부톡시드, 예컨대 탄산나트륨, 탄산칼륨, 탄산세슘, 탄산수소나트륨, 탄산수소칼륨, 인산삼칼륨, 인산수소이나트륨, 인산수소이칼륨, 나트륨 tert-부톡시드 또는 칼륨 tert-부톡시드이다. 탄산세슘 또는 나트륨 tert-부톡시드를 사용하는 것이 바람직하다.
반응 (II) + (III) -> (I)은 일반적으로 +50℃ 내지 +200℃, 바람직하게는 +100℃ 내지 +150℃의 온도 범위 내에서 표준압에서 수행한다. 그러나, 감압 또는 승압 (예를 들어 0.5 내지 5 bar)에서의 수행도 가능하다. 동시 마이크로파 조사를 사용하여 전환을 수행하는 것이 유용할 수 있다.
R1 라디칼이 히드록실인 경우, 상기 기재된 커플링 반응 (II) + (III) -> (I)에서 보호된 상기 히드록실 기를 일시적으로 유지하는 것이 유리할 수 있다. 이러한 목적으로, 화학식 III의 실릴 에테르 유도체, 예를 들어, 상응하는 트리메틸실릴, 트리이소프로필실릴, tert-부틸디메틸실릴 또는 tert-부틸(디페닐)실릴 에테르를 사용하는 것이 바람직하고, 이들은 공지된 방법으로 화학식 III의 화합물로부터 용이하게 수득될 수 있다. 커플링의 완료시, 그 다음 이들 보호기를 통상적인 방법에 의해, 예를 들어 테트라-n-부틸암모늄 플루오라이드로 처리하여 다시 탈리시킬 수 있다.
화학식 II의 화합물 그 자체는 먼저 하기 화학식 IV의 N'-히드록시아미딘을 하기 화학식 V의 피라졸카르복실산과 축합시켜 하기 화학식 VI의 1,2,4-옥사디아졸 유도체를 수득한 다음 화학식 VI의 화합물을 염기의 존재하에 하기 화학식 VII의 화합물과 반응시킴으로써 제조할 수 있다.
<화학식 IV>

상기 식에서,
R2는 상기 정의된 바와 같다.
<화학식 V>

<화학식 VI>

상기 식에서,
R2는 상기 정의된 바와 같다.
<화학식 VII>
상기 식에서,
X는 상기 정의된 바와 같고,
Y는 이탈기, 예를 들어 염소, 브로민, 아이오딘, 메실레이트, 트리플레이트 또는 토실레이트이다.
축합 반응 (IV) + (V) -> (VI)은 바람직하게는 카르보디이미드, 예컨대 N'-(3-디메틸아미노프로필)-N-에틸카르보디이미드 (EDC)를 활성 에스테르 성분으로서의 1-히드록시-1H-벤조트리아졸 (HOBt)과 함께 활용하거나, 고비점 이중극성 비양성자성 용매, 예를 들어 N,N-디메틸포름아미드 또는 디메틸 술폭시드 중 포스겐 유도체, 예컨대 1,1'-카르보닐디이미다졸 (CDI)을 활용하여 수행한다.
이 반응의 초기 커플링 단계는 일반적으로 0℃ 내지 +50℃의 온도 범위 내에서 수행한 다음; 1,2,4-옥사디아졸을 수득하기 위한 고리화는 반응 혼합물을 +100℃ 내지 +150℃의 온도로 후속적으로 가열함으로써 완수한다. 반응은 표준압, 승압 또는 감압 (예를 들어 0.5 내지 5 bar)에서 수행할 수 있고; 일반적으로, 표준압을 사용한다.
공정 단계 (VI) + (VII) -> (II)에 대한 불활성 용매는, 예를 들어, 할로탄화수소, 예컨대 디클로로메탄, 트리클로로메탄, 테트라클로로메탄, 트리클로로에틸렌 또는 클로로벤젠, 에테르, 예컨대 디에틸 에테르, 디이소프로필 에테르, 메틸 tert-부틸 에테르, 테트라히드로푸란 또는 1,4-디옥산, 1,2-디메톡시에탄 또는 비스(2-메톡시에틸) 에테르, 탄화수소, 예컨대 벤젠, 톨루엔, 크실렌, 펜탄, 시클로헥산 또는 미네랄 오일 분획, 또는 이중극성 비양성자성 용매, 예컨대 아세톤, 메틸 에틸 케톤, 에틸 아세테이트, 아세토니트릴, N,N-디메틸포름아미드 (DMF), N,N-디메틸아세트아미드 (DMA), 디메틸 술폭시드 (DMSO), N, N'-디메틸프로필렌우레아 (DMPU), N-메틸피롤리논 (NMP) 또는 피리딘이다. 마찬가지로 언급된 용매의 혼합물을 사용하는 것이 가능하다. 테트라히드로푸란 또는 1,4-디옥산을 사용하는 것이 바람직하다.
반응 (VI) + (VII) -> (II)에 적합한 염기는 통상적인 무기 또는 유기 염기이다. 이들에는 바람직하게는 알칼리 금속 수산화물, 예를 들어 수산화리튬, 수산화나트륨 또는 수산화칼륨, 알칼리 금속 알콕시드, 예컨대 나트륨 또는 칼륨 메톡시드, 나트륨 또는 칼륨 에톡시드 또는 나트륨 또는 칼륨 tert-부톡시드, 알칼리 금속 수소화물, 예컨대 수소화나트륨 또는 수소화칼륨, 또는 아미드, 예컨대 나트륨 아미드, 리튬 또는 칼륨 비스(트리메틸실릴)아미드 또는 리튬 디이소프로필아미드가 포함된다. 칼륨 tert-부톡시드를 사용하는 것이 바람직하다. 알킬화 촉매, 예를 들어 브로민화리튬, 아이오딘화나트륨, 테트라-n-부틸암모늄 브로마이드 또는 벤질트리에틸암모늄 클로라이드의 첨가가 유리할 수 있다.
반응은 일반적으로 -20℃ 내지 +100℃, 바람직하게는 0℃ 내지 +60℃의 온도 범위 내에서 수행한다. 반응은 표준압, 승압 또는 감압 (예를 들어 0.5 내지 5 bar 범위)에서 수행할 수 있고; 일반적으로, 표준압을 사용한다.
본 발명은 추가로, 하기 화학식 I-A의 화합물을 하기 화학식 VIII의 화합물 또는 상기 화합물의 활성화 형태와 함께 통상적인 방법에 의해 에스테르화하고, 생성된 화학식 I-PD의 화합물을 임의로 그의 거울상이성질체 및/또는 부분입체이성질체로 분리시키고/시키거나 적절한 (i) 용매 및/또는 (ii) 산을 사용하여 그의 용매화물, 염 및/또는 염의 용매화물로 전환시키는 것을 특징으로 하는, 화학식 I-PD의 화합물의 제조 방법을 제공한다.
<화학식 I-A>
상기 식에서,
m, n 및 R2 각각은 상기 정의된 바와 같다.
<화학식 VIII>
상기 식에서,
RPD는 상기 정의된 바와 같다.
전구약물 기 RPD의 도입에 적합한 화학식 VIII의 화합물의 활성화 형태는, 예를 들어, 상응하는 클로라이드 또는 무수물 (혼합 무수물을 포함함), 또는 아니면 특정 에스테르 또는 아미드 유도체이다. RPD 라디칼에 존재하는 유리 아미노 기는 적절하게는 여기서 일시적으로 보호된 형태로 존재한 다음, 에스테르화 반응 말미에 친숙한 방법에 의해 다시 방출된다. 사용되는 이러한 보호는 바람직하게는 tert-부톡시카르보닐 기의 형태이고, 이는 강산, 예컨대 염화수소 또는 트리플루오로아세트산을 사용한 처리에 의해 탈리될 수 있다 [예를 들어, 문헌 [M. Bodanszky and A. Bodanszky, The Practice of Peptide Synthesis, Springer-Verlag, Berlin, 1984]; [M. Bodanszky, Principles of Peptide Synthesis, Springer-Verlag, Berlin, 1993]; [T.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, Wiley, New York, 1999] 참조].
화학식 I-A의 화합물 그 자체는 상기 기재된 커플링 반응 (II) + (III) -> (I)을 통해 수득될 수 있다.
화학식 III, IV, V, VII 및 VIII의 화합물은 상업적으로 입수가능하거나 문헌에서의 것과 같이 기재되어 있거나, 이들은 당업자에게 자명한 방법으로 문헌에 공개된 방법과 유사하게 제조할 수 있다. 출발 물질의 제조에 대한 다수의 상세한 방법 및 문헌 정보는 또한 출발 화합물 및 중간체의 제조에 대한 섹션의 실험 부분에서 찾을 수 있다.
본 발명의 화합물의 제조는 하기 반응식에 의해 설명될 수 있다.
<반응식 1>

<반응식 2>

<반응식 3>
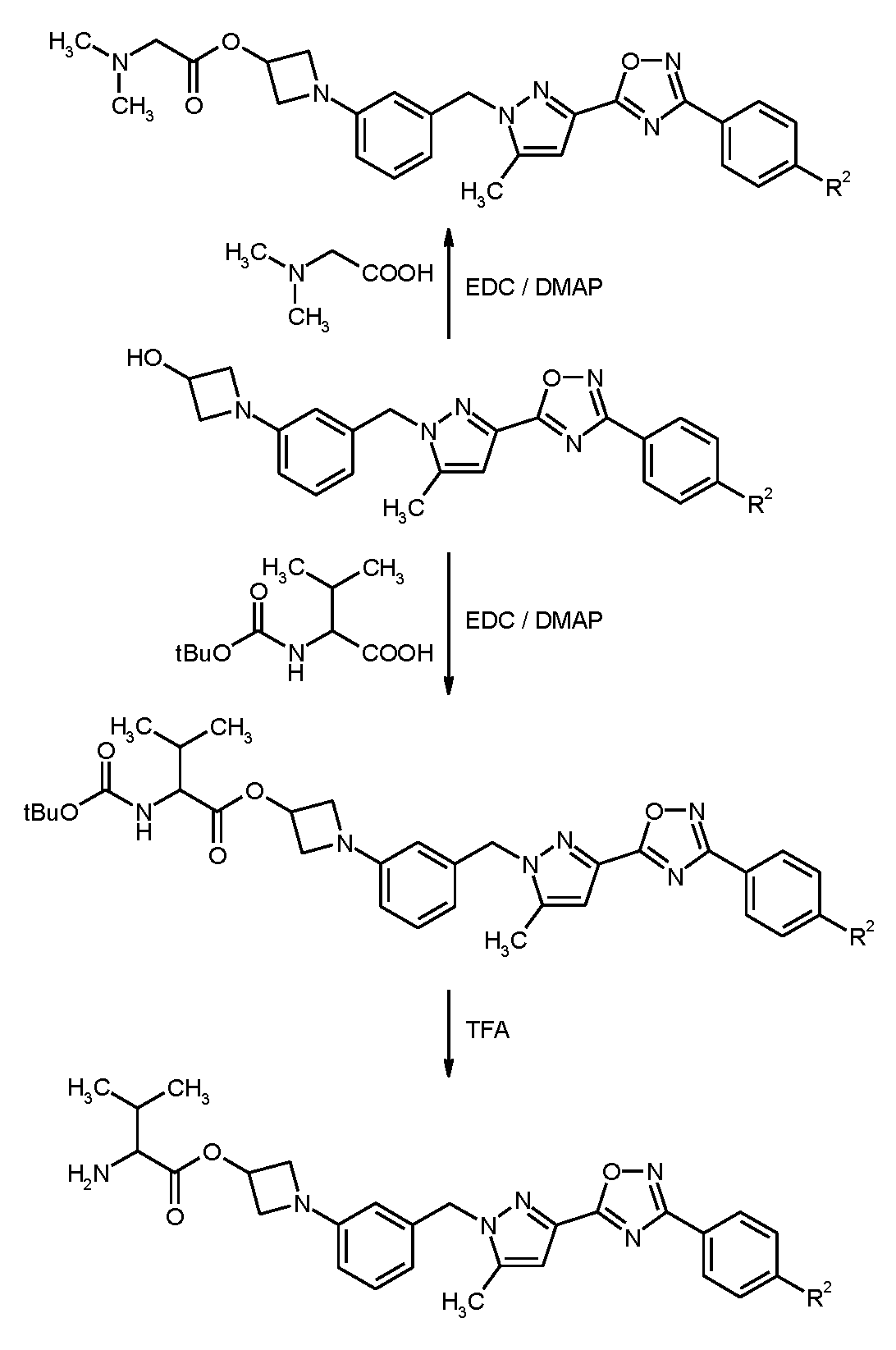
본 발명의 화합물은 유익한 약리 특성을 갖고 있어 인간 및 동물의 질환의 예방 및 치료에 사용될 수 있다.
본 발명의 화합물은 HIF 조절 경로의 매우 강력한 억제제이다. 게다가, 본 발명의 화합물은 그의 분포 용적 및/또는 그의 클리어런스(clearance), 및 그로부터 유래된 반감기에 관해 유리한 약동학적 특성을 가지므로, 이를 1일 1회 반복 투여에 적합하게 된다.
그의 작용 프로파일을 기반으로, 본 발명의 화합물은 일반적으로 인간 및 포유 동물의 과다증식성 질환의 치료에 특히 적합하다. 화합물은 세포 증식 및 세포 분열을 억제, 차단, 감소 또는 저하시킬 수 있고, 다음으로 아폽토시스를 증가시킨다.
본 발명의 화합물을 사용하여 치료될 수 있는 과다증식성 질환에는, 건선, 켈로이드, 반흔의 형성 및 피부의 다른 증식성 질환, 양성 질환, 예컨대 양성 전립선 비대증 (BPH), 특히 종양 질환 군이 포함된다. 본 발명의 맥락에서, 이들은 특히 하기 질환을 의미하지만 이에 제한되지 않는 것으로 이해된다: 유방 암종 및 유방 종양 (유관 및 소엽 형태(또한 상피내)), 기도의 종양 (소세포성(parvicellular) 및 비-소세포성 암종, 기관지 암종), 뇌 종양 (예를 들어 뇌간 및 시상하부의 종양, 성상세포종, 수모세포종, 뇌실막세포종 및 신경-외배엽 및 송과체 종양), 소화 기관 (식도, 위, 담낭, 소장, 대장, 직장)의 종양, 간 종양 (간세포 암종, 담관세포 암종 및 혼합 간세포 및 담관세포 암종 포함), 두경부 영역 (후두, 하인두, 비인두, 중인두, 구순 및 구강)의 종양, 피부 종양 (편평 상피 암종, 카포시(Kaposi) 육종, 악성 흑색종, 메르켈(Merkel) 세포 피부암 및 비흑색종 피부암), 연조직의 종양 (연조직 육종, 골육종, 악성 섬유성 조직구종, 림프육종 및 횡문근육종 포함), 눈의 종양 (안내 흑색종 및 망막모세포종 포함), 내분비선 및 외분비선 (예를 들어 갑상선 및 부갑상선, 췌장 및 타액선)의 종양, 요로의 종양 (방광, 음경, 신장, 신우 및 요관의 종양) 및 생식 기관의 종양 (여성의 자궁내막, 자궁경부, 난소, 질, 음문 및 자궁의 암종 및 남성의 전립선 및 고환의 암종). 이들에는 또한 고형 형태로 및 순환 혈액 세포로서 증식성 혈액 질환, 예컨대 림프종, 백혈병 및 골수증식성 질환, 예를 들어 급성 골수성, 급성 림프모구성, 만성 림프구성, 만성 골수성 및 유모세포 백혈병, 및 AIDS-관련 림프종, 호지킨(Hodgkin) 림프종, 비-호지킨 림프종, 피부 T 세포 림프종, 버킷(Burkitt) 림프종 및 중추신경계의 림프종이 포함된다.
이러한 인간에서 익히 기재된 질환은 또한 다른 포유동물에서 비교할 만한 병인학으로 존재할 수 있고 거기서 본 발명의 화합물로 치료될 수 있다.
본 발명의 맥락에서, 용어 "치료" 또는 "치료하다"는 통상적인 의미로 사용되고 질환 또는 건강 이상을 퇴치, 감소, 경감 또는 완화시키는 것을 목적으로 환자를 시중들고, 돌보고 간호하는 것, 및 예를 들어 암의 사례에서와 같이 상기 질환에 의해 손상된 생활 여건을 개선시키는 것을 의미한다.
본 발명의 화합물은 HIF 조절 경로의 조절제로서 작용하며 따라서 또한 HIF 전사 인자의 유해한 발현과 관련된 질환의 치료에 적합하다. 이는 특히 전사 인자 HIF-1α 및 HIF-2α에 적용된다. 용어 "HIF의 유해한 발현"은 여기서 HIF 단백질의 비정상적 생리학상 존재를 의미한다. 이는 단백질의 과도한 합성 (mRNA- 또는 번역-관렴됨)에, 감소된 분해에 또는 전사 인자의 기능화에서 불충분한 반대-조절(counter-regulation)에 기인할 수 있다.
HIF-1α 및 HIF-2α는 100개 초과의 유전자를 조절한다. 이는 혈관신생에서 역할을 하며 따라서 종양, 및 또한 글루코스, 아미노산 및 지질 대사, 및 세포 이동, 전이 및 DNA 복구에 영향을 주거나, 아폽토시스를 억제하여 종양 세포의 종양을 개선시키는 것에 직접 관련되는 단백질에 적용된다. 기타는 면역 반응의 억제 및 염증 세포 중 혈관신생 인자의 상향 조절을 통해 더 간접 작용한다. HIF는 또한, 상승된 HIF 수준을 갖는 것으로 보고된, 줄기 세포, 및 여기서 특히 종양 줄기 세포에서 중요한 역할을 한다. 따라서 본 발명의 화합물에 의한 HIF 조절 경로의 억제는 또한, 높은 증식 속도를 갖지 않고 따라서 세포독성 물질에 의해 단지 불충분하게 영향을 받는 종양 줄기 세포에 대해 치료 효과를 나타낸다 (문헌 [ Semenza, 2007]; [Weidemann and Johnson, 2008] 참조).
HIF에 의한 세포 대사의 변화는 종양에만 국한된 것이 아니고, 만성 또는 일과성이든 다른 저산소증 병리생리학적 과정에서도 일어난다. HIF 억제제 -예컨대 본 발명의 화합물 - 는 예를 들어, 손상된 세포는 의도된 바와 같이 기능하지 않을 경우 추가의 손상을 유발할 수 있기 때문에, 추가적 손상이 세포의 저산소 상황으로의 적응에 기인하는 그러한 맥락에서 치료상 유용하다. 이의 한 예는 졸중에 이은 부분적으로 파괴된 조직에서의 간질 초점의 형성이다. 혈전색전증 사례, 염증, 상처, 중독 또는 다른 원인의 결과로서 허혈 과정이 심장 또는 뇌에서 일어날 경우 유사한 상황이 심혈관 질환의 경우에서 발견된다. 이들은 손상, 예컨대 국소 지연된 활동 전위를 초래할 수 있고, 이는 결국 부정맥 또는 만성 심부전을 야기할 수 있다. 일과성 형태에서, 예를 들어 무호흡의 결과로서, 특정 상황하에 본태성 고혈압일 수 있고, 이는 공지된 후유증, 예를 들어 졸중 및 심근경색을 야기할 수 있다.
따라서 본 발명의 화합물에 의해 달성되는 바와 같은 HIF 조절 경로의 억제는 또한 심부전, 부정맥, 심근경색, 무호흡-유도 고혈압, 폐고혈압, 이식 허혈, 재관류 손상, 졸중 및 황반 변성과 같은 질환의 사례, 및 또한 외상성 손상 또는 절단 후 신경 기능의 회복에 유용할 수 있다.
HIF는 상피 세포에서 중간엽 세포 유형으로의 전이를 제어하는 인자 중 하나이고, 이는 폐 및 신장에 특히 중요하기 때문에, 본 발명의 화합물은 또한 HIF와 관련된 폐 및 신장의 섬유증을 예방하거나 제어하는데 사용할 수 있다.
본 발명의 화합물을 사용하여 치료될 수 있는 추가의 질환은, 염증성 관절 질환, 예컨대 다양한 형태의 관절염, 및 염증성 장 질환, 예를 들어, 크론(Crohn) 병이다.
추바시(Chuvash) 다혈구증은 다른 기관 중에서 비장에서 적혈구 생성 동안 HIF-2α 활성에 의해 매개된다. 따라서, HIF 조절 경로의 억제제로서, 본 발명의 화합물은 또한 여기서 과도한 적혈구 형성을 억제하고 따라서 상기 질환의 영향을 완화하는데 적합하다.
본 발명의 화합물은 또한 과도한 또는 비정상적 혈관신생과 관련된 질환 질환의 치료에 사용될 수 있다. 이들에는 당뇨병성 망막병증, 허혈성 망막 정맥 폐쇄 및 미숙아에서의 망막병증 (문헌 [Aiello et al ., 1994]; [Peer et al ., 1995)] 참조), 노인성 황반 변성 (AMD; 문헌 [Lopez et al ., 1996] 참조), 신생혈관 녹내장, 건선, 후수정체 섬유증식증, 혈관섬유종, 염증, 류마티스 관절염 (RA), 재협착, 스텐트 내 재협착, 및 혈관 이식에 따른 재협착이 포함된다.
증가된 혈액 공급은 추가로 암성의 신생 조직과 관련되고 여기서 종양 성장을 가속화시킨다. 더욱이, 새로운 혈관 및 림프관의 성장은 전이의 형성 및 결과적으로 종양의 확산을 촉진시킨다. 새로운 림프관 및 혈관은 또한 면역 특권 조직, 예컨대 눈에서 동종 이식에 유해하며, 이는 예를 들어 거부 반응에 대한 취약성을 증가시킨다. 따라서 본 발명의 화합물은 또한 예를 들어 혈관의 성장의 억제 또는 혈관수의 감소에 의해 상기 언급된 질환 중 하나의 치료에 사용될 수 있다. 이는 내피 세포 증식의 억제 또는 혈관의 형성을 예방 또는 경감시키는 다른 메카니즘을 통해 및 아폽토시스에 의한 신생물 세포의 감소를 통해 달성될 수 있다.
비만의 경우, HIF-1α는 지방 조직에 풍부하게 되고, 결과적으로 해당작용의 방향으로 이화작용에서 HIF-매개 이동을 야기하며, 따라서 에너지 운반체로서 증가된 양의 글루코스가 소비된다. 이는 동시에 지질 대사를 감소시키고 따라서 조직 내 지질이 저장된다. 따라서 본 발명의 물질은 또한, 특히 비만의 경우 조직 내 HIF-1α-매개 지질 풍부화의 치료에 적합하다.
본 발명은 추가로 장애, 특히 상기 언급된 장애의 치료 및/또는 예방을 위한 본 발명의 화합물의 용도를 제공한다.
본 발명은 추가로 장애, 특히 상기 언급된 장애의 치료 및/또는 예방용 의약의 제조를 위한 본 발명의 화합물의 용도를 제공한다.
본 발명은 더욱이 장애, 특히 상기 언급된 장애의 치료 및/또는 예방 방법에서 본 발명의 화합물의 용도를 제공한다.
본 발명은 추가로 유효량의 하나 이상의 본 발명의 화합물을 사용하는, 장애, 특히 상기 언급된 장애의 치료 및/또는 예방 방법을 제공한다.
본 발명의 화합물은 단독으로 또는, 필요한 경우, 1종 이상의 다른 약리학상 활성 물질과 조합하여 사용될 수 있되, 단, 이러한 조합은 바람직하지 못하고 허용되지 않는 부작용을 초래하지 않아야 한다. 따라서 본 발명은 추가로, 특히 상기 언급된 장애의 치료 및/또는 예방을 위한 하나 이상의 본 발명의 화합물 및 1종 이상의 추가의 활성 성분을 포함하는 의약을 제공한다.
예를 들어, 본 발명의 화합물을 암 치료를 위한 공지된 항과다증식성, 세포증식억제 또는 세포독성 물질과 조합할 수 있다. 따라서 본 발명에 따른 화합물과 암 요법에 통상적으로 사용되는 다른 물질과의 또는 방사선요법과의 조합이 특히 적절하며, 그 이유는 종양의 저산소 영역은 언급된 통상적인 요법에 단지 약하게 반응하며, 한편 본 발명의 화합물은 특히 거기서 그의 활성을 나타내기 때문이다.
조합에 적합한 활성 성분의 예에는 다음이 포함된다:
알데스류킨, 알렌드로산, 알파페론, 알리트레티노인, 알로퓨리놀, 알로프림, 알록시, 알트레타민, 아미노글루테티미드, 아미포스틴, 암루비신, 암사크린, 아나스트로졸, 안즈메트, 아라네스프, 아르글라빈, 삼산화비소, 아로마신, 5-아자시티딘, 아자티오프린, BCG 또는 티스-BCG, 베스타틴, 베타메타손 아세테이트, 베타메타손 나트륨 포스페이트, 벡사로텐, 블레오마이신 술페이트, 브록수리딘, 보르테조밉, 부술판, 칼시토닌, 캄파트, 카페시타빈, 카르보플라틴, 카소텍스, 세페손, 셀모류킨, 세루비딘, 클로람부실, 시스플라틴, 클라드리빈, 클로드론산, 시클로포스파미드, 시타라빈, 다카르바진, 닥티노마이신, 다우녹솜, 데카드론, 데카드론 포스페이트, 델레스트로겐, 데니류킨 디프티톡스, 데포메드롤, 데슬로렐린, 덱스라족산, 디에틸스틸베스트롤, 디플루칸, 도세탁셀, 독시플루리딘, 독소루비신, 드로나비놀, DW-166HC, 엘리가르드, 엘리텍, 엘렌스, 에멘드, 에피루비신, 에포에틴-알파, 에포겐, 에프타플라틴, 에르가미솔, 에스트라세, 에스트라디올, 에스트라무스틴 나트륨 포스페이트, 에티닐에스트라디올, 에티올, 에티드론산, 에토포포스, 에토포시드, 파드로졸, 파르스톤, 필그라스팀, 피나스테리드, 플리그라스팀, 플록수리딘, 플루코나졸, 플루다라빈, 5-플루오로데옥시우리딘 모노포스테이트, 5-플루오르우라실(5-FU), 플루옥시메스테론, 플루타미드, 포르메스탄, 포스테아빈, 포테무스틴, 풀베스트란트, 감마가르드, 겜시타빈, 겜투주맙, 글리벡, 글리아델, 고세렐린, 그라니세트론 히드로클로라이드, 히스트렐린, 히캄틴, 히드로코르톤, 에리트로-히드록시노닐아데닌, 히드록시우레아, 이브리투모마브 티욱세탄, 이다루비신, 이포스파미드, 인터페론-알파, 인터페론-알파-2, 인터페론-알파-2α, 인터페론-알파-2β, 인터페론-알파-n1, 인터페론-알파-n3, 인터페론-베타, 인터페론-감마-1α, 인터류킨-2, 인트론 A, 이레사, 이리노테칸, 키트릴, 렌티난 술페이트, 레트로졸, 류코보린, 류프롤리드, 류프롤리드 아세테이트, 레바미솔, 레보폴산 칼슘 염, 레보트로이드, 레복실, 로무스틴, 로니다민, 마리놀, 메클로레타민, 메코발라민, 메드록시프로게스테론 아세테이트, 메게스트롤 아세테이트, 멜팔란, 메네스트, 6-메르캅토퓨린, 메스나, 메토트렉세이트, 메트빅스, 밀테포신, 미노시클린, 미토마이신 C, 미토탄, 미톡산트론, 모드레날, 미오세트, 네다플라틴, 뉴라스타, 뉴메가, 뉴포겐, 닐루타미드, 놀바덱스, NSC-631570, OCT-43, 옥트레오티드, 온단세트론 히드로클로라이드, 오라프레드, 옥살리플라틴, 파클리탁셀, 페디아프레드, 페가스파르가제, 페가시스, 펜토스타틴, 피시바닐, 필로카르핀 히드로클로라이드, 피라루비신, 플리카마이신, 포르피머 나트륨, 프레드니무수틴, 프로드니솔론, 프레드시손, 프레마린, 프로카르바진, 프로크리트, 랄티트렉세드, 레비프, 레늄-186 에티드로네이트, 리툭시마브, 로페론-A, 로무르티드, 살라겐, 산도스타틴, 사르그라모스팀, 세무스틴, 시조피란, 소부족산, 솔루-메드롤, 스트렙토조신, 스트론튬-89 클로라이드, 신트로이드, 타목시펜, 탐술로신, 타소네르민, 타스토락톤, 탁소테르, 테세류킨, 테모졸로미드, 테니포시드, 데스토스테론 프로피오네이트, 테스트레드, 티오구아닌, 티오테파, 티로트로핀, 틸루드론산, 토포테칸, 토레미펜, 토시투모마브, 타스투주맙, 테오술판, 트레티노인, 트렉살, 트리메틸멜라민, 트리메트렉세이트, 트립토렐린 아세테이트, 트립토렐린 파모에이트, UFT, 우리딘, 발루비신, 베스나리논, 빈블라스틴, 빈크리스틴, 빈데신, 비노렐빈, 비룰리진, 지네카르드, 지노스타틴-스티말라머, 조프란; ABI-007, 아콜비펜, 악티문, 아피니탁, 아미노프테린, 아르족시펜, 아소프리스닐, 아타메스탄, 아트라센탄, 아바스틴, CCI-779, CDC-501, 셀레브렉스, 세툭시마브, 크리스나톨, 시프로테론 아세테이트, 데시타빈, DN-101, 독소루비신-MTC, dSLIM, 두타스테리드, 에도테카린, 에플로르니틴, 엑사테칸, 펜레티니드, 히스타민 디히드로클로라이드, 히스트렐린 히드로겔 임플란트, 홀뮴-166 DOTMP, 이반드론산, 인터페론-감마, 인트론-PEG, 익사베필론, 키홀 림페트 헤모시아닌, L-651582, 란레오티드, 라소폭시펜, 리브라, 노나파르닙, 미프록시펜, 미노드로네이트, MS-209, 리포소말 MTP-PE, MX-6, 나파렐린, 네모루비신, 네오바스타트, 놀라트렉세드, 오블리메르센, 온코-TCS, 오시뎀, 파클리탁셀 폴리글루타메이트, 파미드로네이트 이나트륨, PN-401, QS-21, 쿠아제팜, R-1549, 랄록시펜, 란피르나세, 레고라페닙, 13-시스-레티노산, 사트라플라틴, 세오칼시톨, 소라페닙, T-138067, 타르세바, 탁소프렉신, 티모신-알파-1, 티아조퓨린, 티피파르닙, 티라파자민, TLK-286, 토레미펜, 트랜스MID-107R, 발스포다르, 바프레오티드, 바탈라닙, 베르테포르핀, 빈플루닌, Z-100, 졸레드론산 및 그의 조합.
바람직한 실시양태에서, 본 발명의 화합물은 항과다증식제와 조합될 수 있고, 이는 예로서 다음과 같을 수 있으나 이러한 기재로 국한되는 것은 아니다:
아미노글루테티미드, L-아스파라기나제, 아자티오프린, 5-아자시티딘, 블레오마이신, 부술판, 캄포테신, 카르보플라틴, 카르무수틴, 클로람부실, 시스플라틴, 콜라스파제, 시클로포스파미드, 시타라빈, 다카르바진, 닥티노마이신, 다우노루비신, 디에틸스틸베스트롤, 2',2'-디플루오로데옥시시티딘, 도세탁셀, 독소루비신 (아드리아마이신), 에피루비신, 에포틸론 및 그의 유도체, 에리트로히드록시노닐 아데닌, 에티닐에스트라디올, 에토포시드, 플루다라빈 포스페이트, 5-플루오로데옥시우리딘, 5-플루오로데옥시우리딘 모노포스페이트, 5-플루오로우라실, 플루옥시메스테론, 플루타미드, 헥사메틸멜라민, 히드록시우레아, 히드록시프로게스테론 카프로에이트, 이다루비신, 이포스파미드, 인터페론, 이리노데칸, 루코보린, 로무스틴, 메클로르에타민, 메드록시프로게스테론 아세테이트, 메게스트롤 아세테이트, 멜팔란, 6-메르캅토퓨란, 메스나, 메토트렉세이트, 미토마이신 C, 미토탄, 미토잔트론, 파클리탁셀, 펜토스타틴, N-포스포노아세틸-L-아스파르테이트 (PALA), 플리카마이신, 프레드니솔론, 프리드니손, 프로카르바진, 랄록시펜, 세무스틴, 스트렙토조신, 타목시펜, 테니포시드, 테스토스테론 프로피오네이트, 티오구아닌, 티오테파, 토포테칸, 트리메틸멜라민, 우리딘, 빈블라스틴, 빈크리스틴, 빈데신 및 비노렐빈.
본 발명의 화합물은 또한 매우 유망한 방식으로 생물학적 치료제, 예컨대 항체 (예를 들어 아바스틴, 리툭산, 에르비툭스, 헤르셉틴) 및 재조합 단백질과 조합될 수 있고, 이는 HIF 신호 경로 전달의 억제 효과를 상가적으로 또는 상승적으로 향상시킨다.
HIF 조절 경로의 억제제, 예컨대 본 발명의 화합물은 또한 혈관신생에 대한 다른 치료제, 예를 들어 아바스틴, 악시니팁, 레센틴, 레고라페닙, 소라페닙 또는 수니티닙과 조합하여 긍정적인 효과를 달성할 수 있다. 프로테아솜 및 mTOR의 억제제 및 항호르몬 및 스테로이드 대사 효소 억제제와의 조합은 그의 부작용의 유익한 프로파일 때문에 특히 적합하다.
일반적으로, 본 발명의 화합물과 다른 세포증식억제 또는 세포독성 활성제와의 조합을 사용하여 다음 목적을 추구할 수 있다:
· 개별 활성 성분으로의 치료와 비교하여, 종양의 성장을 지연시키거나, 그의 크기를 감소시키거나 심지어 그의 완전한 제거에서 향상된 효능;
· 단독요법의 경우보다 더 낮은 투여량으로 사용된 화학요법제의 사용 가능성:
· 개별 투여와 비교하여 더 적은 부작용으로 더 용인되는 요법의 가능성;
· 보다 넓은 범위의 종양의 치료 가능성;
· 요법에 대한 더 높은 반응률의 달성;
· 현대 표준 요법과 비교하여 환자의 더 긴 생존 시간.
게다가, 본 발명의 화합물은 또한 방사선요법 및/또는 외과적 처치와 함께 사용할 수 있다.
본 발명은 추가로 하나 이상의 본 발명의 화합물을, 전형적으로 1종 이상의 불활성, 비독성의 제약상 적합한 부형제와 함께 포함하는 의약, 및 상기 언급된 목적을 위한 그의 용도를 제공한다.
본 발명의 화합물은 전신 및/또는 국소 작용할 수 있다. 이러한 목적으로, 이는 적합한 방식으로, 예를 들어 경구, 비경구, 폐, 비강, 설하, 설측, 협측, 직장, 피부, 경피, 결막, 귀(otic) 경로에 의해, 또는 임플란트 또는 스텐트로서 투여될 수 있다.
본 발명의 화합물은 이들 투여 경로에 적합한 투여 형태로 투여될 수 있다.
경구 투여에 적합한 투여 형태는, 선행 기술에 따라 작용하고, 본 발명의 화합물을 급속히 및/또는 변형된 방식으로 방출하고, 본 발명에 따른 화합물을 결정질 및/또는 무정화 및/또는 용해된 형태, 예를 들어 정제 (비코팅된 또는 코팅된 정제, 예를 들어 본 발명의 화합물의 방출을 제어하는 위액-내성 또는 지연-용해 또는 불용성 코팅이 사용됨), 구강 내에서 급속히 붕해하는 정제 또는 필름/편구(oblate), 필름/동결건조물 또는 캡슐 (예를 들어 경질 또는 연질 젤라틴 캡슐), 당의정, 과립, 환제, 분말, 에멀젼, 현탁액, 에어로졸 또는 용액으로 함유하는 것이다.
비경구 투여는 흡수 단계를 우회하거나 (예를 들어 정맥내, 동맥내, 심장내, 척수강내 또는 요추내) 흡수를 포함한다 (예를 들어 근육내, 피하, 피내, 경피 또는 복강내). 비경구 투여에 적합한 투여 형태에는 용액, 현탁액, 에멀젼, 동결건조물 또는 멸균 분말 형태의 주사 및 주입용 제제가 포함된다.
다른 투여 경로에 대해, 적합한 예는 흡입가능 의약 형태 (분말 흡입기, 분무기 포함), 비강 점적제, 용액 또는 스프레이, 설측, 설하 또는 협측 투여용 정제, 필름/편구 또는 캡슐, 좌제, 귀 또는 눈용 제제, 질내 캡슐, 수성 현탁액 (로션, 쉐이킹(shaking) 혼합물), 친유성 현탁액, 연고, 크림, 경피 치료 시스템 (예를 들어 패치), 밀크, 페이스트, 포옴, 스프링클링(sprinkling) 분말, 임플란트 또는 스텐트이다.
경구 및 비경구 투여, 특히 경구 및 정맥내 투여가 바람직하다.
본 발명의 화합물은 언급된 투여 형태로 전환시킬 수 있다. 이는 그 자체가 공지된 방식으로 불활성, 비독성의 제약상 적합한 부형제와 혼합함으로써 행할 수 있다. 이들 부형제에는 담체 (예를 들어 미세결정질 셀룰로스, 락토스, 만니톨), 용매 (예를 들어 액체 폴리에틸렌 글리콜), 유화제 및 분산 또는 습윤제 (예를 들어 나트륨 도데실술페이트, 폴리옥시소르비탄 올레에이트), 결합제 (예를 들어 폴리비닐피롤리돈), 합성 및 천연 중합체 (예를 들어 알부민), 안정화제 (예를 들어 항산화제, 예를 들어 아스코르브산), 염료 (예를 들어 무기 안료, 예를 들어 산화철) 및 향미제 및/또는 악취 중화제(odour correctant)가 포함된다.
일반적으로, 비경구 투여의 경우 유효한 결과를 달성하기 위하여 약 0.001 내지 1 mg/kg, 바람직하게는 약 0.01 내지 0.5 mg/kg(체중)의 양을 투여하는 것이 유리한 것으로 밝혀졌다. 경구 투여의 경우, 투여량은 약 0.01 내지 100 mg/kg, 바람직하게는 약 0.01 내지 20 mg/kg이고 가장 바람직하게는 0.1 내지 10 mg/kg(체중)이다.
그럼에도 불구하고 적절한 경우, 구체적으로 체중의 함수로서 명시된 양, 투여 경로, 활성 성분에 대한 개별 반응, 제제의 특성 및 투여가 실시되는 시간 또는 간격에서 벗어나는 것이 필요할 수 있다. 예를 들어, 일부 경우에, 상기 언급된 최소량 미만이 충분할 수 있으며, 한편 다른 경우, 언급된 상한을 초과하여야만 한다. 비교적 많은 양의 투여의 경우, 이를 하루에 걸쳐 수 개의 개별 용량으로 분할하는 것이 바람직할 수 있다.
하기 실시예는 본 발명을 설명하는 것이다. 본 발명은 실시예로 제한되지 않는다.
하기 시험 및 실시예에서의 백분율은, 달리 지시되지 않는 한, 중량 백분율이고; 부는 중량부이다. 액체/액체 용액에 대한 용매 비, 희석률 및 농도 데이터는 각 경우 부피를 기준으로 한다.
A.
실시예
약어 및
두문자어
:
abs. 무수
Ac 아세틸
aq. 수성
Ex. 실시예
Bu 부틸
approx. 약, 대략
CDI 1,1'-카르보닐디이미다졸
CI 화학 이온화 (MS에서)
d 이중선 (NMR에서)
d 일(들)
DAST 디에틸아미노황 트리플루오라이드
TLC 박층 크로마토그래피
DCI 직접 화학 이온화 (MS에서)
dd 이중선의 이중선 (NMR에서)
DMAP 4-N,N-디메틸아미노피리딘
DME 1,2-디메톡시에탄
DMF N,N-디메틸포름아미드
DMSO 디메틸 술폭시드
dt 삼중선의 이중선 (NMR에서)
EDC N'-(3-디메틸아미노프로필)-N-에틸카르보디이미드 히드로클로라이드
ee 거울상이성질체 과잉률
EI 전자 충격 이온화 (MS에서)
eq. 당량(들)
ESI 전기분무 이온화 (MS에서)
Et 에틸
GC 기체 크로마토그래피
h 시간(들)
HOBt 1-히드록시-1H-벤조트리아졸 수화물
HPLC 고압 고성능 액체 크로마토그래피
iPr 이소프로필
LC-MS 액체 크로마토그래피-결합 질량 분광측정법
lit. 문헌 (참조)
m 다중선 (NMR에서)
Me 메틸
min 분(들)
MPLC 중압 액체 크로마토그래피 (실리카 겔 상에서; "섬광 크로마토그래피"
로도 칭해짐)
Ms 메탄술포닐 (메실)
MS 질량 분광측정법
NMP N-메틸-2-피롤리디논
NMR 핵 자기 공명 분광측정법
Pd/C 활성 탄소상 팔라듐
PEG 폴리에틸렌 글리콜
Pr 프로필
quart 사중선 (NMR에서)
quint 오중선 (NMR에서)
Rf 체류 지수 (TLC에서)
RT 실온
Rt 체류 시간 (HPLC에서)
s 단일선 (NMR에서)
sept 칠중선 (NMR에서)
t 삼중선 (NMR에서)
TBAF 테트라-n-부틸암모늄 플루오라이드
tBu tert-부틸
Tf 트리플루오로메틸술포닐 (트리플릴)
TFA 트리플루오로아세트산
THF 테트라히드로푸란
UV 자외선 분광측정법
v/v 부피 대 부피 비 (용액의)
HPLC
,
LC
/
MS
및
GC
/
MS
방법:
방법 1 (
LC
/
MS
):
MS 기기 유형: 마이크로매스(Micromass) ZQ; HPLC 기기 유형: HP 1100 시리즈; UV DAD; 칼럼: 페노메넥스 게미니(Phenomenex Gemini) 3 μ, 30 mm x 3.00 mm; 용리액 A: 1 l의 물 + 0.5 ml의 50% 포름산, 용리액 B: 1 l의 아세토니트릴 + 0.5 ml의 50% 포름산; 구배: 0.0분 90% A -> 2.5분 30% A -> 3.0분 5% A -> 4.5분 5% A; 유량: 0.0분 1 ml/분 -> 2.5분/3.0분/4.5분 2 ml/분; 오븐: 50℃; UV 검출: 210 nm.
방법 2 (
LC
/
MS
):
기기: HPLC 아질런트(Agilent) 시리즈 1100을 구비한 마이크로매스 콰트로 마이크로(Quattro Micro) MS; 칼럼: 써모 하이퍼실 골드(Thermo Hypersil GOLD) 3 μ, 20 mm x 4 mm; 용리액 A: 1 l의 물 + 0.5 ml의 50% 포름산, 용리액 B: 1 l의 아세토니트릴 + 0.5 ml의 50% 포름산; 구배: 0.0분 100% A -> 3.0분 10% A -> 4.0분 10% A -> 4.01분 100% A (유량: 2.5 ml/분) -> 5.00 분 100% A; 오븐: 50℃; 유량: 2 ml/분; UV 검출: 210 nm.
방법 3 (
LC
/
MS
):
MS 기기 유형: 마이크로매스 ZQ; HPLC 기기 유형: 워터스 얼라이언스(Waters Alliance) 2795; 칼럼: 페노메넥스 시네르기(Phenomenex Synergi) 2.5 μ MAX-RP 100A 머큐리(Mercury) 20 mm x 4 mm; 용리액 A: 1 l의 물 + 0.5 ml의 50% 포름산, 용리액 B: 1 l의 아세토니트릴 + 0.5 ml의 50% 포름산; 구배: 0.0분 90% A -> 0.1분 90% A -> 3.0분 5% A -> 4.0분 5% A -> 4.01분 90% A; 유량: 2 ml/분; 오븐: 50℃; UV 검출: 210 nm.
방법 4 (
LC
/
MS
):
기기: 워터스 UPLC 액퀴티(Acquity)를 구비한 마이크로매스 콰트로 프리미어(Premier); 칼럼; 써모 하이퍼실 골드 1.9 μ, 50 mm x 1 mm; 용리액 A: 1 l의 물 + 0.5 ml의 50% 포름산, 용리액 B: 1 l의 아세토니트릴 + 0.5 ml의 50% 포름산; 구배: 0.0분 90% A -> 0.1분 90% A -> 1.5분 10% A -> 2.2분 10% A (유량: 0.33 ml/분) 오븐: 50℃; UV 검출: 210 nm.
방법 5 (
LC
/
MS
):
기기: HPLC 아질런트 시리즈 1100을 구비한 마이크로매스 콰트로 LCZ; 칼럼: 페노메넥스 시네르기 2.5 μ MAX-RP 100A 머큐리 20 mm x 4 mm; 용리액 A: 1 l의 물 + 0.5 ml의 50% 포름산, 용리액 B: 1 l의 아세토니트릴 + 0.5 ml의 50% 포름산; 구배: 0.0분 90% A -> 0.1분 90% A -> 3.0분 5% A -> 4.0분 5% A -> 4.1분 90% A; 유량: 2 ml/분; 오븐: 50℃; UV 검출: 208-400 nm.
방법 6 (
LC
/
MS
):
기기: 워터스 액퀴티 SQD UPLC 시스템; 칼럼; 워터스 액퀴티 UPLC HSS T3 1.8 μ, 50 mm x 1 mm; 용리액 A: 1 l의 물 + 0.25 ml의 99% 포름산, 용리액 B: 1 l의 아세토니트릴 + 0.25 ml의 99% 포름산; 구배: 0.0분 90% A -> 1.2분 5% A -> 2.0분 5% A; 유량: 0.40 ml/분; 오븐: 50℃; UV 검출: 210-400 nm.
방법 7 (
LC
/
MS
):
MS 기기 유형: 워터스 ZQ; HPLC 기기 유형: 아질런트 1100 시리즈; UV DAD; 칼럼; 써모 하이퍼실 골드 3 μ, 20 mm x 4 mm; 용리액 A: 1 l의 물 + 0.5 ml의 50% 포름산, 용리액 B: 1 l의 아세토니트릴 + 0.5 ml의 50% 포름산; 구배: 0.0분 100% A -> 3.0분 10% A -> 4.0분 10% A -> 4.1분 100% A (유량: 2.5 ml/분); 오븐: 55℃; 유량: 2 ml/분; UV 검출: 210 nm.
방법 8 (
LC
/
MS
):
MS 기기 유형: 마이크로매스 ZQ; HPLC 기기 유형: HP 1100 시리즈; UV DAD, 칼럼: 페노메넥스 게미니 3 μ, 30 mm x 3.00 mm; 용리액 A: 1 l의 물 + 0.5 ml의 50% 포름산, 용리액 B: 1 l의 아세토니트릴 + 0.5 ml의 50% 포름산; 구배: 0.0분 90% A -> 2.5분 30% A -> 3.0분 5% A -> 4.5분 5% A; 유량: 0.0분 1 ml/분 -> 2.5분/3.0분/4.5분 2 ml/분; 오븐: 50℃; UV 검출: 210 nm.
방법 9 (
GC
/
MS
):
기기: 마이크로매스 GCT, GC 6890; 칼럼: 레스텍(Restek) RTX-35, 15 m x 200 μm x 0.33 μm; 헬륨의 일정 유량: 0.88 ml/분; 오븐: 70℃; 입구: 250℃; 구배: 70℃, 30℃/분 -> 310℃ (3분 동안 유지).
방법 10 (분석용
HPLC
):
기기: DAD 검출을 구비한 HP 1100: 칼럼: 크로마실(Kromasil) 100 RP-18, 60 mm x 2.1 mm, 3.5 μm; 용리액 A: 5 ml의 과염소산 (70%) / 1 l의 물, 용리액 B: 아세토니트릴; 구배: 0분 2% B -> 0.5분 2% B -> 4.5분 90% B -> 6.5분 90% B -> 6.7분 2% B -> 7.5분 2% B; 유량: 0.75 ml/분; 칼럼 온도: 30℃; UV 검출: 210 nm.
방법 11 (정제용
HPLC
):
칼럼: 그롬-실(Grom-Sil) 120 ODS-4 HE, 10 μm, 250 mm x 30 mm; 용리액 및 구배 프로그램: 아세토니트릴/0.1% 수성 포름산 10:90 (0-3분), 아세토니트릴/0.1% 수성 포름산 10:90 -> 95:5 (3-27분), 아세토니트릴/0.1% 수성 포름산 95:5 (27-34분), 아세토니트릴/0.1% 수성 포름산 10:90 (34-38분); 유량: 50 ml/분; 온도: 22℃; UV 검출: 254 nm.
방법 12 (정제용
HPLC
):
칼럼: 다이소(Daiso) C18 바이오 스프링(Bio Spring) 칼럼, 10 μm, 300 mm x 100 mm; 용리액 및 구배 프로그램: 물/메탄올 80:20 (0-6분), 물/메탄올 80:20 -> 20:80 (6-60분), 물/메탄올 20:80 (60-95분), 물/메탄올 10:90 (95-105분), 물/메탄올 80:20 (105-113분); 유량: 250 ml/분; 온도: 25℃; UV 검출: 240 nm.
방법 13 (정제용
HPLC
):
칼럼: 레프로실-푸르(Reprosil-Pur) C18, 10 μm, 250 mm x 30 mm; 용리액 및 구배 프로그램: 아세토니트릴/0.1% 수성 포름산 10:90 (0-3분), 아세토니트릴/0.1% 수성 포름산 10:90 -> 95:5 (3-27분), 아세토니트릴/0.1% 수성 포름산 95:5 (27-34분), 아세토니트릴/0.1% 수성 포름산 10:90 (34-38분); 유량: 50 ml/분; 온도: 22℃; UV 검출: 254 nm.
방법 14 (
LC
/
MS
):
기기: 워터스 액퀴티 SQD UPLC 시스템; 칼럼; 워터스 액퀴티 UPLC HSS T3 1.8 μ, 30 mm x 2 mm; 용리액 A: 1 l의 물 + 0.25 ml의 99% 포름산, 용리액 B: 1 l의 아세토니트릴 + 0.25 ml의 99% 포름산; 구배: 0.0분 90% A -> 1.2분 5% A -> 2.0분 5% A; 유량: 0.60 ml/분; 오븐: 50℃; UV 검출: 208-400 nm.
제조가 이하에 명시적으로 기재되지 않은 모든 반응물 또는 시약은 일반적으로 입수용이한 공급원으로부터 상업적으로 구매하였다. 마찬가지로 제조가 이하에 기재되지 않고 상업적으로 입수가능하지 않았거나 일반적으로 입수용이하지 않은 공급원으로부터 구입된 모든 다른 반응물 또는 시약에 대해, 그의 제조가 기재된 공개된 문헌을 참조한다.
출발 화합물 및 중간체:
실시예
1A
5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-[4-(트리플루오로메톡시)페닐]-1,2,4-옥사디아졸
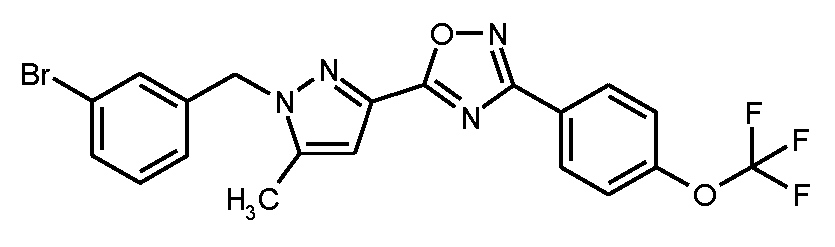
단계 1: 5-(5-메틸-1H-피라졸-3-일)-3-[4-(트리플루오로메톡시)페닐]-1,2,4-옥사디아졸
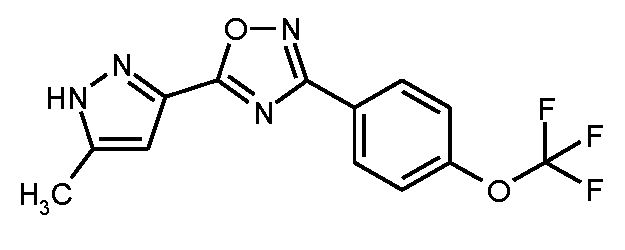
75 ml의 무수 DMF 중 20.25 g (125 mmol)의 1,1'-카르보닐디이미다졸의 현탁액에, 실온에서 15분 이내로, 75 ml의 무수 DMF 중 15.0 g (119 mmol)의 5-메틸-1H-피라졸-3-카르복실산의 용액을 적가하였다. 혼합물을 실온에서 1시간 45분 동안 교반한 후, 26.19 g (119 mmol)의 N'-히드록시-4-(트리플루오로메톡시)벤젠카르복스이미드 아미드를 가하였다. 후속적으로, 반응 혼합물을 4 h 동안 110℃로 가열하였다. 냉각 후, 대부분의 용매를 회전 증발기 상에서 제거하였다. 800 ml의 물을 잔류물에 가하고, 이를 수 분 동안 교반하였다. 미용해 생성물을 흡인으로 여과해 내고 디에틸 에테르로 세척하였다. 여과물로부터 에테르 상을 제거하고 수성 상을 디에틸 에테르로 1회 더 추출하였다. 25 ml의 메탄올을 합한 에테르 추출물에 가하고, 사전에 흡인으로 여과해 낸 생성물을 거기서 현탁화하였다. 수 분 동안 교반 후, 혼합물을 흡인으로 다시 여과해 냈다. 잔류물을 고진공 하에 건조시켜 제1 분획의 표제 화합물 (8.79 g)을 수득하였다. 동일한 양의 물을 여과물에 가하고, 이를 진탕에 의해 추출하였다. 상 분리 후, 유기 상을 포화 염화나트륨 용액으로 세척하고, 무수 황산마그네슘 상에서 건조시키고 최종적으로 회전 증발기 상에서 용매를 제거하였다. 잔류물을 고진공 하에 건조시킨 후, 제2 분획의 표제 화합물을 이러한 방법으로 수득하였다 (24.43 g). 이렇게 하여 총 33.32 g (이론치의 90%)의 표제 화합물을 수득하였다.

단계 2: 5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-[4-(트리플루오로메톡시)페닐]-1,2,4-옥사디아졸

6.58 g (58.7 mmol)의 고체 칼륨 tert-부톡시드를 실온에서 450 ml의 무수 디옥산 중 14.0 g (45.1 mmol)의 실시예 1A / 단계 1로부터의 화합물 및 13.54 g (54.2 mmol)의 3-브로모벤질 브로마이드의 용액에 가하였다. 반응 혼합물을 먼저 실온에서 16 h 동안 및 그 다음 45℃에서 추가의 2 h 동안 교반하여, 전환을 완료하였다. 후속적으로, 500 ml의 물을 가하고 혼합물을 매회 대략 300 ml의 에틸 아세테이트로 추출하였다. 합한 유기 추출물을 물 및 포화 염화나트륨 용액으로 연속적으로 세척하고, 무수 황산마그네슘 상에서 건조시키고 최종적으로 용매를 회전 증발기 상에서 제거하였다. 생성된 조 생성물을 525 ml의 펜탄과 35 ml의 디이소프로필 에테르의 혼합물과 함께 교반하면서 정제하였다. 고체를 흡인으로 여과해 내고 건조시킨 후, 19.08 g (이론치의 84%, 95% 순도)의 표제 화합물을 수득하였다.
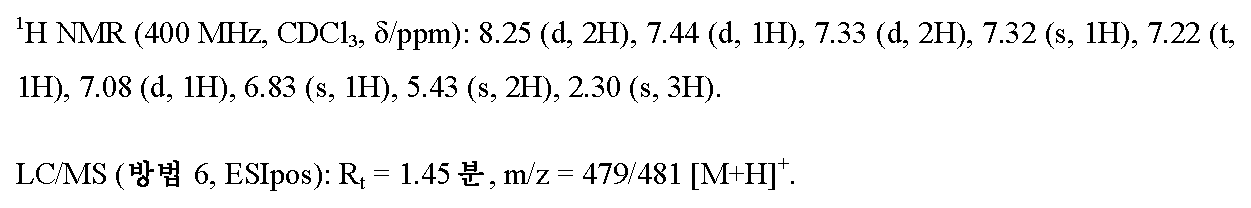
실시예
2A
5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-{4-[1-(트리플루오로메틸)시클로프로필]페닐}-1,2,4-옥사디아졸

단계 1: 1-브로모-4-[1-(트리플루오로메틸)시클로프로필]벤젠
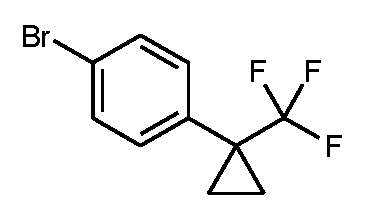
먼저, 몬모릴로나이트 상의 활성화 브로민화아연을 다음과 같이 제조하였다: 7.0 g (31.1 mmol)의 브로민화아연을 처음에 1 리터 플라스크에서 225 ml의 메탄올 중에 채우고, 28.2 g의 K10 몬모릴로나이트를 가하였다. 후속적으로, 현탁액을 실온에서 1 h 동안 교반하였다. 그 다음 혼합물을 회전 증발기 상에서 농축 건조시켰다. 잔존 미분을 약한 진공하에 (대략 500 mbar) 모래조에서 조 온도 200℃로 1 h 동안 가열한 다음 아르곤 하에 냉각하였다.
그 다음 표제 화합물을 다음과 같이 제조하였다: 49.63 g (267 mmol)의 1-페닐-1-(트리플루오로메틸)시클로프로판을 처음에 1.25 리터의 펜탄에 채우고, 상기 수득된 몬모릴로나이트 상의 활성화 브로민화아연을 가하였다. 그 다음 반응 용기 외부를 알루미늄 호일로 감싸서 광의 입사를 감소시켰다. 137 ml (2.67 mol)의 브로민을 교반하면서 서서히 적가하였다. 후속적으로, 반응 혼합물을 실온에서 16 h 동안 어둠 속에서 교반하였다. 그 다음, 얼음으로 냉각하면서, 1 리터의 포화 아황산나트륨 수용액을 적가하였다. 고체를 흡인으로 여과해 내고 펜탄으로 2회 세척하였다. 상 분리 후, 여과물을 매회 1 리터의 펜탄으로 2회 더 추출하였다. 합한 유기 추출물을 무수 황산나트륨 상에서 건조시키고, 여과하고 단지 약한 진공 하에 회전 증발기 상에서 용매를 제거하였다. 77.18 g (92% 순도, 이론치의 100%)의 표제 화합물을 수득하였다.

단계 2: 4-[1-(트리플루오로메틸)시클로프로필]벤조니트릴
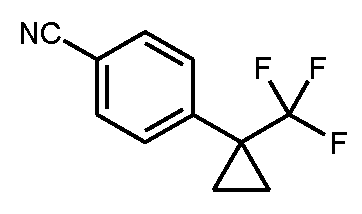
990 ml의 DMF와 10 ml의 물의 혼합물 중 75.0 g (283 mmol)의 실시예 2A / 단계 1로부터의 화합물의 용액에 약한 진공을 반복 적용하고 아르곤을 유입(admission)하면서 산소를 제거하였다. 그 다음 37.87 g (322 mmol)의 시안화아연 및 32.69 g (28.3 mmol)의 테트라키스(트리페닐포스핀)팔라듐(0)을 가하였다. 반응 혼합물을 후속적으로 120℃에서 5 h 동안 가열하였다. 실온으로 냉각 후, 불용성 물질을 여과해 내고 잔류물을 약간의 DMF로 세척하였다. 후속적으로 회전 증발기 상에서 여과물로부터 용매를 제거하였다. 생성된 조 생성물을 1.5 리터의 에틸 아세테이트에 용해시키고 혼합물을 매회 500 ml 포화 염화암모늄 용액으로 2회 및 500 ml의 포화 염화나트륨 용액으로 1회 세척하였다. 유기 상을 무수 황산마그네슘 상에서 건조시킨 후, 혼합물을 여과하고 여과물을 회전 증발기 상에서 농축하였다. 수득된 오일을 용리액으로서 40:1 시클로헥산/에틸 아세테이트를 사용하여 175 g의 실리카 겔을 통해 흡인 여과에 의해 정제하였다. 생성물 분획의 농축 및 고진공 하에 건조 후, 49.7 g (이론치의 83%)의 표제 화합물을 수득하였다.

단계 3: N'-히드록시-4-[1-(트리플루오로메틸)시클로프로필]벤젠카르복스이미드 아미드

14.48 g (208 mmol)의 히드록실암모늄 클로라이드 및 29 ml (208 mmol)의 트리에틸아민을 500 ml의 에탄올 중 20.0 g (94.7 mmol)의 실시예 2A / 단계 2로부터의 화합물의 용액에 가하였다. 반응 혼합물을 2 h 동안 환류 하에 가열하였다. 후속적으로, 용매의 약 절반을 회전 증발기 상에서 제거하였다. 1.5 리터의 물을 잔류물에 가하고, 생성된 현탁액을 실온에서 20분 동안 교반하였다. 그 다음 고체를 흡인으로 여과해 내고, 약간의 냉수로 세척하고 고진공 하에 건조시켰다. 추가의 정제용으로, 이를 120 ml의 펜탄과 30 ml의 디클로로메탄의 혼합물과 함께 교반하였다. 고체를 흡인으로 다시 여과해 내고 건조시킨 후, 15.79 g (이론치의 68%)의 표제 화합물을 수득하였다.

단계 4 : 5-(5-메틸-1H-피라졸-3-일)-3-{4-[1-(트리플루오로메틸)시클로프로필]페닐}-1,2,4-옥사디아졸

25 ml의 무수 DMF 중 6.75 g (41.6 mmol)의 1,1'-카르보닐디이미다졸의 현탁액에, 실온에서 15분 이내로, 25 ml의 무수 DMF 중 5.0 g (36.6 mmol)의 5-메틸-1H-피라졸-3-카르복실산의 용액을 적가하였다. 혼합물을 실온에서 1시간 45분 동안 교반한 후, 9.68 g (39.6 mmol)의 실시예 2A / 단계 3으로부터의 화합물을 가하였다. 후속적으로, 반응 혼합물을 2.5 h 동안 110℃로 가열하였다. 실온으로 냉각 후, 격렬하게 교반하면서 800 ml의 물을 서서히 첨가하고, 그 결과 생성물이 침전되었다. 고체를 흡인으로 여과해 내고 약간의 냉수로 세척하였다. 여전히 습기있는 조 생성물을 300 ml의 에탄올과 350 ml의 물의 비등 혼합물로부터 재결정화하였다. 11.03 g (이론치의 83%)의 표제 화합물을 수득하였다.

단계 5: 5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-{4-[1-(트리플루오로메틸)시클로프로필]페닐}-1,2,4-옥사디아졸

436 mg (3.89 mmol)의 고체 칼륨 tert-부톡시드를 0℃에서 30 ml의 무수 디옥산 중 1.0 g (2.99 mmol)의 실시예 2A / 단계 4로부터의 화합물 및 897 mg (3.59 mmol)의 3-브로모벤질 브로마이드의 용액에 가하였다. 그 다음 반응 혼합물을 실온에서 16 h 동안 교반하였다. 그 다음 대략 200 ml의 물을 가하고 혼합물을 매회 대략 100 ml의 에틸 아세테이트로 추출하였다. 합한 유기 추출물을 물 및 포화 염화나트륨 용액으로 연속적으로 세척하고, 무수 황산마그네슘 상에서 건조시키고 최종적으로 용매를 회전 증발기 상에서 제거하였다. 수득된 조 생성물을 먼저 용리액으로서 10:1 시클로헥산/에틸 아세테이트를 사용하여 실리카 겔을 통해 흡인 여과에 의해 정제하였다. 생성물을 추가로 50 ml의 펜탄과 2 ml의 디에틸 에테르의 혼합물과 함께 교반하면서 정제하였다. 고체를 흡인으로 여과해 내고 건조시킨 후, 1.34 g (이론치의 88%)의 표제 화합물을 수득하였다.
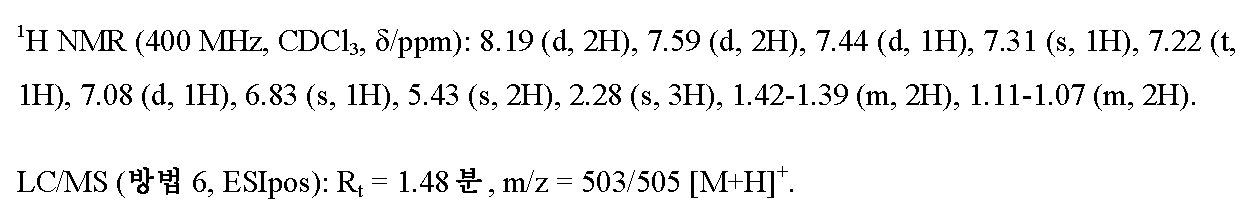
실시예
3A
5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-[4-(1,1,1-트리플루오로-2-메틸프로판-2-일)페닐]-1,2,4-옥사디아졸

단계 1: 2-(4-브로모페닐)-1,1,1-트리플루오로프로판-2-올

헵탄/디클로로메탄 혼합물 중 디클로로(디메틸)티타늄의 현탁액을 먼저 다음과 같이 제조하였다: 디클로로메탄 중 사염화티타늄의 1 M 용액 100 ml (100 mmol)를 -30℃로 냉각하고, 헵탄 중 디메틸아연의 1 M 용액 100 ml (100 mmol)를 적가하고 혼합물을 -30℃에서 추가의 30분 동안 교반하였다. 후속적으로, 당해 현탁액을 -40℃로 냉각하고 50 ml의 디클로로메탄 중 10 g (39.5 mmol)의 1-(4-브로모페닐)-2,2,2-트리플루오로에탄온의 용액을 가하였다. 혼합물을 -40℃에서 추가의 5분 동안 교반한 다음, 온도를 실온에 이르게 하고 혼합물을 추가의 2 h 동안 실온에서 교반하였다. 얼음으로 냉각하면서, 50 ml의 물을 서서히 적가한 다음, 혼합물을 추가의 300 ml의 물로 희석하였다. 혼합물을 디클로로메탄으로 2회 추출하고, 합한 디클로로메탄 상을 물로 1회 세척하고, 무수 황산마그네슘 상에서 건조시키고 여과하고, 용매를 회전 증발기 상에서 제거하였다. 잔류물을 실리카 겔 상에서 칼럼 크로마토그래피 (용리액: 85:15 시클로헥산/에틸 아세테이트)에 의해 정제하였다. 10.5 g (이론치의 100%)의 표제 화합물을 수득하였고, 이는 1H NMR에 따르면, 여전히 용매 잔류물을 함유하였다.

단계 2: 2-(4-브로모페닐)-1,1,1-트리플루오로프로판-2-일 메탄술포네이트

3.12 g (78.05 mmol, 미네랄 오일 중 60%)의 수소화나트륨을 처음에 아르곤 하에 45 ml의 THF에 채우고, 20 ml의 THF 중 10.5 g (39.03 mmol)의 실시예 3A /단계 1에서 수득된 화합물의 용액을 실온에서 적가하였다. 혼합물을 실온에서 1 h 동안 및 40℃에서 30분 동안 교반한 후, 45 ml의 THF 중 8.94 g (78.05 mmol)의 메탄술포닐 클로라이드의 용액을 적가하고 반응 혼합물을 40℃에서 추가의 60분 동안 교반하였다. 후속적으로, 50 ml의 물을 혼합물에 서서히 적가하고, 이를 포화 탄산수소나트륨 수용액으로 희석하고 에틸 아세테이트로 2회 추출하였다. 합한 에틸 아세테이트 상을 무수 황산마그네슘 상에서 건조시키고 여과하고, 용매를 회전 증발기 상에서 제거하였다. 잔류물을 헥산 중에서 교반하고 수득된 고체를 여과해 내고 감압 하에 건조시켰다. 12.4 g (이론치의 92%)의 표제 화합물을 수득하였다.
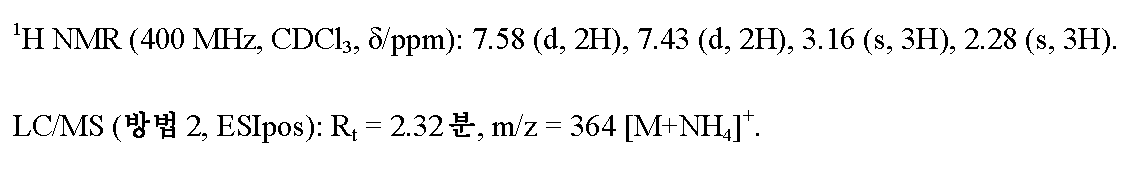
단계 3: 1-브로모-4-(1,1,1-트리플루오로-2-메틸프로판-2-일)벤젠

12.4 g (35.72 mmol)의 실시예 3A /단계 2에서 수득된 화합물을 처음에 250 ml의 디클로로메탄에 채우고 혼합물을 0℃로 냉각하였다. 그 다음 교반하면서 0℃에서 35.7 ml (71.44 mmol)의 트리메틸알루미늄의 2 M 용액을 서서히 적가한 다음, 혼합물을 실온에 이르게 하고 실온에서 추가의 1.5 h 동안 교반하였다. 120 ml 포화 탄산수소나트륨 수용액을 혼합물에 서서히 적가하고, 이어서 40 ml의 포화 염화나트륨 수용액을 적가하였다. 혼합물을 규조토를 통해 여과하고 규조토를 다시 디클르로메탄으로 2회 세척하였다. 합한 디클로로메탄 상을 포화 염화나트륨 수용액으로 1회 세척하고 무수 황산마그네슘 상에서 건조시키고, 용매를 회전 증발기 상에서 제거하였다. 8.69 g (이론치의 87%)의 표제 화합물을 순도 95%로 수득하였다.

단계 4: 4-(1,1,1-트리플루오로-2-메틸프로판-2-일)벤젠카르보니트릴

3.34 g (12.50 mmol)의 실시예 3A /단계 3에서 수득된 화합물을 처음에 아르곤 하에 2.5 ml의 탈기 DMF에 채우고, 881 mg (7.50 mmol)의 시안화아연 및 867 mg (0.75 mmol)의 테트라키스(트리페닐포스핀)팔라듐(0)을 가하고 혼합물을 80℃에서 밤새 교반하였다. 실온으로 냉각 후, 반응 혼합물을 에틸 아세테이트로 희석하고 고체 성분을 여과해 냈다. 여과물을 2 N 암모니아 수용액으로 2회 및 포화 염화나트륨 수용액으로 1회 세척하고, 무수 황산마그네슘 상에서 건조시키고 회전 증발기 상에서 용매를 제거하였다. 잔류물을 실리카 겔 상에서 칼럼 크로마토그래피 (용리액: 85:15 시클로헥산/에틸 아세테이트)에 의해 정제하였다. 2.08 g (이론치의 78%)의 표제 화합물을 수득하였다.
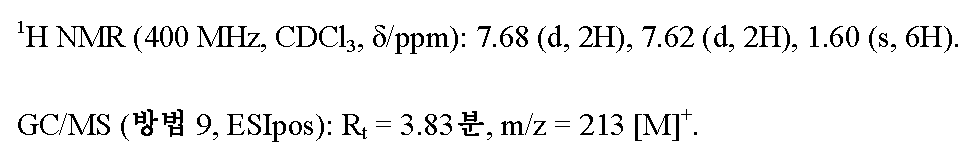
단계 5: N'-히드록시-4-(1,1,1-트리플루오로-2-메틸프로판-2-일)벤젠카르복스이미드 아미드

60 ml의 에탄올 중 2.40 g (11.26 mmol)의 실시예 3A / 단계 4로부터의 화합물, 1.72 g (24.77 mmol)의 히드록실아민 히드로클로라이드 및 3.45 ml (24.77 mmol)의 트리에틸아민의 혼합물을 환류 하에 1 h 동안 교반하였다. 실온으로 냉각 후, 용매를 회전 증발기 상에서 제거하였다. 에틸 아세테이트를 잔류물에 가하고 존재하는 고체를 여과해 냈다. 에틸 아세테이트 용액을 물 및 포화 염화나트륨 수용액으로 연속적으로 세척하고, 무수 황산마그네슘 상에서 건조시키고 여과하였다. 용매 제거 후, 수득된 오일을 석유 에테르로 연화처리하였다. 생성된 고체를 흡인으로 여과해 내고 고진공 하에 건조시켜, 2.65 g (이론치의 96%)의 표제 화합물을 수득하였다.

단계 6: 5-(5-메틸-1H-피라졸-3-일)-3-[4-(1,1,1-트리플루오로-2-메틸프로판-2-일)페닐]-1,2,4-옥사디아졸
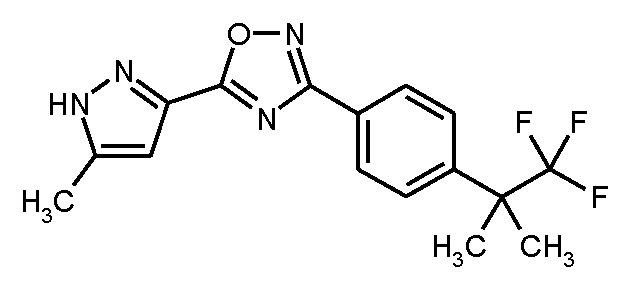
11.6 g (60.0 mmol)의 EDC, 8.13 g (60.0 mmol)의 HOBt 및 14.8 g (60.0 mmol)의 실시예 3A /단계 5로부터의 화합물을 실온에서 300 ml의 무수 DMF 중 7.57 g (60.0 mmol)의 5-메틸-1H-피라졸-3-카르복실산의 용액에 연속적으로 가하였다. 혼합물을 먼저 실온에서 2 h 동안 및 그 다음 140℃에서 5 h 동안 교반하였다. 냉각 후, 반응 혼합물을 900 ml의 얼음-물 내로 교반하였다. 침전된 생성물을 흡인으로 여과하고, 냉수로 세척한 다음 고진공 하에 건조시켰다. 14.7 g (이론치의 73%)의 표제 화합물을 수득하였다.

단계 7 : 5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-[4-(1,1,1-트리플루오로-2-메틸프로판-2-일)페닐]-1,2,4-옥사디아졸
실시예 2A / 단계 5에 기재된 공정과 유사하게, 750 mg (2.23 mmol)의 실시예 3A / 단계 6으로부터의 화합물 및 669 mg (2.68 mmol)의 3-브로모벤질 브로마이드를 사용하여 1.02 g (이론치의 84%, 94% 순도)의 표제 화합물을 수득하였다.
실시예
4A
5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-[4-(테트라히드로-2H-피란-4-일)페닐]-1,2,4-옥사디아졸

단계 1: 4-(테트라히드로-2H-피란-4-일)벤조니트릴
186 mg (0.594 mmol)의 아이오딘화니켈(II), 90 mg (0.594 mmol)의 트랜스-2-아미노시클로헥산올 히드로클로라이드 및 3.63 g (19.8 mmol)의 나트륨 헥사메틸디실라지드를 20 ml의 이소프로판올 중 2.91 g (19.8 mmol)의 4-시아노페닐보론산의 용액 [문헌 [M. Nishimura et al ., Tetrahedron 2002, 58 (29), 5779-5788]]에 가하였다. 이렇게 수득된 현탁액을 아르곤 분위기 하에 실온에서 5분 동안 교반하였다. 그 다음 2.1 g (9.90 mmol)의 4-아이오도테트라히드로피란 [문헌 [Heuberger et al ., J. Chem . Soc . 1952, 910]]을 가하였다. 반응 혼합물을 75℃ 온도에서 15 h 동안 교반 후, 이를 실온으로 냉각하고 대략 50 g의 실리카 겔을 통한 여과에 의해 디클로로메탄과의 무기 염을 실질적으로 제거하였다. 조 생성물을 MPLC (실리카 겔, 용리액: 디클로로메탄)에 의해 정제하였다. 986 mg (이론치의 53%)의 표제 화합물을 수득하였다.

단계 2: N'-히드록시-4-(테트라히드로-2H-피란-4-일)벤젠카르복스이미드 아미드

480 mg (2.56 mmol)의 실시예 4A / 단계 1로부터의 화합물, 392 mg (5.64 mmol)의 히드록실아민 히드로클로라이드 및 786 μl (5.64 mmol)의 트리에틸아민을 18 ml의 에탄올 중 16 h 동안 환류 하에 가열하였다. 후속적으로, 대부분의 휘발성 성분을 회전 증발기 상에서 제거하였다. 50 ml의 물을 잔존 잔류물에 가하고, 이를 실온에서 수 분 동안 교반하였다. 그 다음 고체를 흡인으로 여과해 내고, 약간의 냉수로 세척하고 최종적으로 고진공 하에 건조시켰다. 525 mg (이론치의 93%)의 표제 화합물을 수득하였다.

단계 3 : 5-(5-메틸-1H-피라졸-3-일)-3-[4-(테트라히드로-2H-피란-4-일)페닐]-1,2,4-옥사디아졸

785 mg (4.10 mmol)의 EDC, 627 mg (4.10 mmol)의 HOBt 및 820 mg (3.72 mmol)의 실시예 4A / 단계 2로부터의 화합물을 실온에서 15 ml의 무수 DMF 중 469 mg (3.72 mmol)의 5-메틸-1H-피라졸-3-카르복실산의 용액에 연속적으로 가하였다. 혼합물을 먼저 실온에서 16 h 동안 및 그 다음 140℃에서 20분 동안 교반하였다. 냉각 후, 100 ml의 물을 반응 혼합물에 가하고, 이를 매회 대략 100 ml의 에틸 아세테이트로 3회 추출하였다. 합한 유기 추출물을 포화 염화나트륨 용액으로 세척하고, 무수 황산마그네슘 상에서 건조시키고 최종적으로 회전 증발기 상에서 농축 건조시켰다. 잔존 잔류물을 사용하여 450 mg의 표제 화합물을 아세토니트릴로부터 추출 교반함으로써 수득하고, 모액을 정제용 HPLC (방법 13)에 의해 정제 후 추가의 97 mg의 표제 화합물을 수득하였다 (총 수율: 이론치의 47%).

단계 4: 5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-[4-(테트라히드로-2H-피란-4-일)페닐)-1,2,4-옥사디아졸

실시예 1A / 단계 2에 기재된 공정과 유사하게, 250 mg (0.806 mmol)의 실시예 4A / 단계 3으로부터의 화합물 및 242 mg (0.967 mmol)의 3-브로모벤질 브로마이드를 사용하여 338 mg (이론치의 87%)의 표제 화합물을 수득하였다. 여기서 생성물의 최종 정제를 10 ml의 펜탄/디이소프로필 에테르 (5:1)로부터 추출 교반함으로써 수행하고, 여기에 수 방울의 디클로로메탄을 가하였다.
실시예
5A
5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-[4-(4-플루오로테트라히드로-2H-피란-4-일)페닐]-1,2,4-옥사디아졸

단계 1: 4-4-(히드록시테트라히드로-2H-피란-4-일)벤젠카르보니트릴

불활성 조건 하에, 디에틸 에테르 중 이소프로필마그네슘 클로라이드의 2 M 용액 109 ml (218 mmol)를 -40℃에서 1000 ml의 무수 THF 중 50.0 g (218 mmol)의 4-아이오도벤조니트릴의 용액에 적가하였다. 혼합물을 동일 온도에서 1.5 h 동안 교반한 후, 250 ml의 무수 THF 중 32.8 g (327 mmol)의 테트라히드로-4H-피란-4-온의 용액을 가하였다. 첨가 종료 후, 반응 혼합물을 먼저 -40℃에서 10분 동안, 그 다음 0℃에서 30분 동안 및 최종적으로 실온에서 60분 동안 교반하였다. 그 다음 대략 20 ml의 포화 염화암모늄 수용액을 -20℃에서 가하였다. 후속적으로, 용매를 회전 증발기 상에서 실질적으로 제거하였다. 1000 ml의 물을 잔존 잔류물에 가하고, 이를 매회 대략 500 ml의 디클로로메탄으로 3회 추출하였다. 합한 유기 추출물을 물 및 포화 염화나트륨 용액으로 연속적으로 세척하였다. 무수 황산마그네슘 상에서 건조시킨 후, 혼합물을 여과하고 용매를 회전 증발기 상에서 제거하였다. 생성된 조 생성물을 10:1 시클로헥산/에틸 아세테이트와 함께 교반함으로써 정제하였다. 19.3 g (이론치의 43%)의 표제 화합물을 수득하였다.

단계 2: 4-(4-(플루오로테트라히드로-2H-피란-4-일)벤젠카르보니트릴
불활성 조건 하에, 58 ml의 디클로로메탄 중 6.19 g (38.4 mmol)의 디에틸아미노황 트리플루오라이드 (DAST)의 용액을 -78℃에서 800 ml의 디클로로메탄 중 6.5 g (31.98 mmol)의 실시예 5A / 단계 1로부터의 화합물의 현탁액에 적가하였다. -78℃에서 30분 후, 반응 혼합물을 얼음/물 조를 활용하여 매우 급속히 -20℃로 가온하였다. 대략 30초 후, 200 ml의 1 M 수산화나트륨 용액을 가하고 혼합물을 실온으로 가온하였다. 500 ml의 물로 희석 후, 혼합물을 매회 대략 200 ml의 디에틸에테르로 3회 추출하였다. 합한 유기 추출물을 무수 황산마그네슘 상에서 건조시키고, 여과 후, 용매를 회전 증발기 상에서 제거하였다. 조 생성물을 MPLC (실리카 겔, 용리액: 10:1 -> 5:1 -> 2:1 -> 1:1 시클로헥산/에틸 아세테이트)에 의해 정제하였다. 3.73 g (이론치의 57%)의 표제 화합물을 수득하였다.
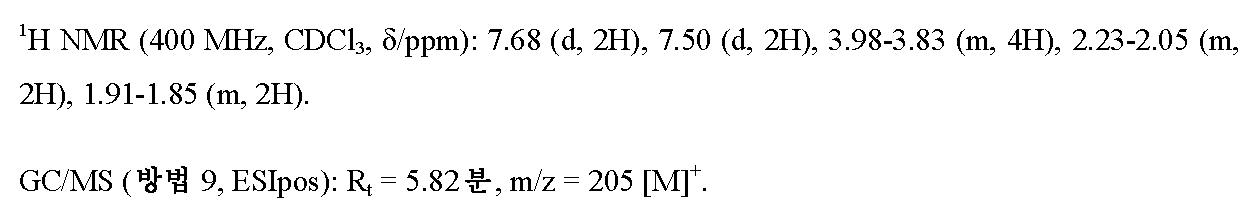
단계 3: 4-(4-(플루오로테트라히드로-2H-피란-4-일)-N'-히드록시벤젠카르복스이미드 아미드

실시예 4A / 단계 2에 기재된 공정에 의해, 3.5 g (17.05 mmol)의 실시예 5A / 단계 2로부터의 화합물을 사용하여 3.57 g (이론치의 88%)의 표제 화합물을 수득하였다. 이 경우 반응 시간은 2 h이었다

단계 4: 3-[4-(4-(플루오로테트라히드로-2H-피란-4-일)페닐]-5-(5-메틸-1H-피라졸-3-일)-1,2,4-옥사디아졸
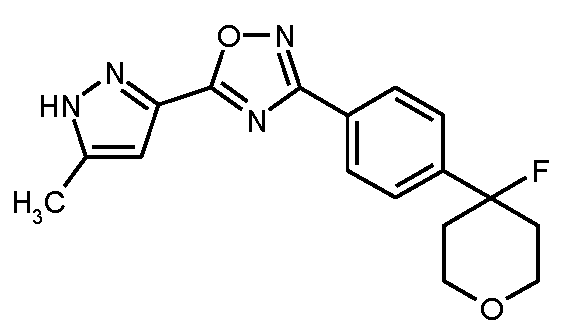
20 ml의 무수 DMF 중 4.30 g (34.1 mmol)의 5-메틸-1H-피라졸-3-카르복실산의 용액을 실온에서 대략 15분 이내에 25 ml의 무수 DMF 중 5.81 g (35.8 mmol)의 1,1'-카르보닐디이미다졸 (CDI)의 현탁액에 적가하였다. 혼합물을 실온에서 105분 동안 교반한 후, 8.12 g (34.1 mmol)의 실시예 5A / 단계 3으로부터의 화합물을 가하였다. 후속적으로, 반응 혼합물을 5 h 동안 110℃로 가열하였다. 실온으로 냉각 후, 혼합물을 800 ml의 물 내로 서서히 교반하였다. 이 과정에서, 생성물이 침전되었다. 이를 흡인으로 여과해 내고 물로 세척하였다. 후속적으로, 습기있는 조 생성물을 430 ml의 에탄올을 재결정화하였다. 여과하고 고체를 건조시킨 후, 8.31 g (이론치의 74%)의 표제 화합물을 수득하였다. 모액을 농축함으로써 추가의 분획을 수득하였다 (1.69 g, 85% 순도, 이론치의 13%).

단계 5: 5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-[4-(4-플루오로테트라히드로-2H-피란-4-일)페닐]-1,2,4-옥사디아졸

실시예 1A / 단계 2에 기재된 공정과 유사하게, 250 mg (0.761 mmol)의 실시예 5A / 단계 4로부터의 화합물 및 228 mg (0.914 mmol)의 3-브로모벤질 브로마이드를 사용하여 355 mg (이론치의 94%)의 표제 화합물을 수득하였다. 여기서 반응 혼합물을 45℃로 가열하는 것을 생략하는 것이 가능하였다. 생성물의 최종 정제를 10 ml의 펜탄/디이소프로필 에테르 (5:1)로부터 추출 교반함으로써 수행하고, 여기에 수 방울의 디클로로메탄을 가하였다.

실시예
6A
5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-[4-(3-플루오로옥세탄-3-일)페닐]-1,2,4-옥사디아졸

단계 1: 4-(3-히드록시옥세탄-3-일)벤젠카르보니트릴

불활성 조건 하에, 디에틸 에테르 중 이소프로필마그네슘 클로라이드의 2 M 용액 11 ml (21.8 mmol)를 -40℃에서 100 ml의 무수 THF 중 5.0 g (21.8 mmol)의 4-아이오도벤조니트릴의 용액에 적가하였다. 혼합물을 동일 온도에서 1.5 h 동안 교반한 후, 이를 -78℃로 냉각하고, 마찬가지로 -78℃로 냉각된, 100 ml의 무수 THF 중 2.95 g (32.7 mmol, 디클로로메탄 중 80%)의 3-옥소옥세탄의 용액 (문헌 [G. Wuitschik et al ., Angew . Chem . Int . Ed . Engl . 2006, 45 (46), 7736-7739])에 캐뉼라를 활용하여 서서히 가하였다. 첨가 종료 후, 반응 혼합물을 먼저 -78℃에서 10분 동안, 그 다음 0℃에서 2 h 동안 및 최종적으로 실온에서 30분 동안 교반하였다. 그 다음 수 ml의 포화 염화암모늄 수용액을 가하였다. 후속적으로, 용매를 회전 증발기 상에서 실질적으로 제거하였다. 수득된 잔류물을 200 ml의 물로 희석하고 매회 대략 200 ml의 에틸 아세테이트로 3회 추출하였다. 합한 유기 추출물을 물 및 포화 염화나트륨 용액으로 연속적으로 세척하였다. 무수 황산마그네슘 상에서 건조시킨 후, 혼합물을 여과하고 용매를 회전 증발기 상에서 제거하였다. 생성된 조 생성물을 10:1 시클로헥산/에틸 아세테이트로부터 재결정화에 의해 정제하였다. 2.42 g (이론치의 63%)의 표제 화합물을 수득하였다.
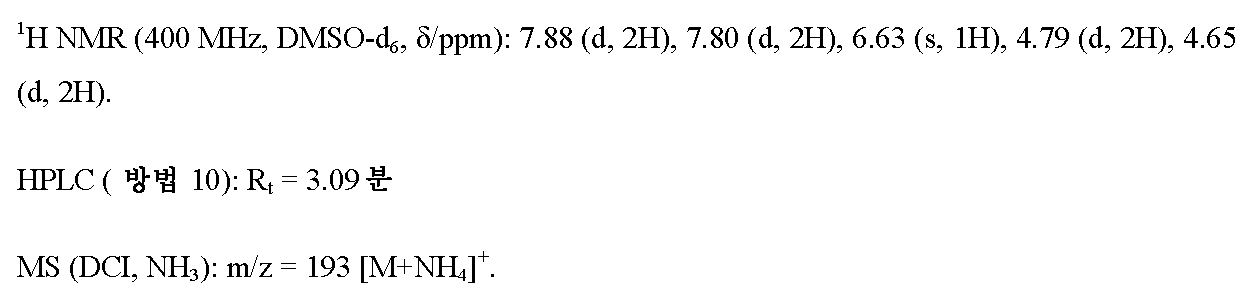
단계 2: 4-(3-(플루오로옥세탄-3-일)벤젠카르보니트릴

실시예 5A / 단계 2에 기재된 공정과 유사하게, 600 mg (3.43 mmol)의 실시예 6A / 단계 1로부터의 화합물 및 662 mg (4.11 mmol)의 디에틸아미노황 트리플루오라이드 (DAST)를 사용하여 495 mg (이론치의 82%)의 표제 화합물을 수득하였다. MPLC 정제용으로, 여기서 8:1 시클로헥산/에틸 아세테이트를 용리액으로서 사용하였다.
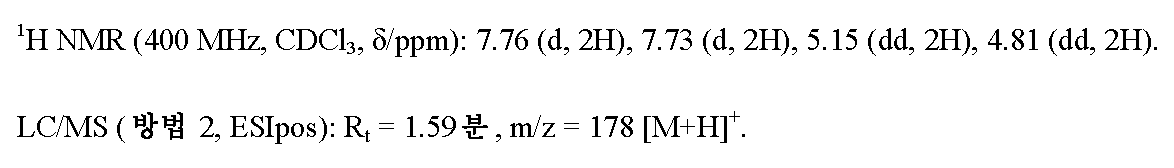
단계 3: 4-(3-(플루오로옥세탄-3-일)-N'-히드록시벤젠카르복스이미드 아미드

실시예 4A / 단계 2에 기재된 공정에 의해, 450 mg (2.54 mmol)의 실시예 6A / 단계 2로부터의 화합물을 사용하여 470 mg (이론치의 86%)의 표제 화합물을 수득하였다.

단계 4: 3-[4-(3-(플루오로옥세탄-3-일)페닐]-5-(5-메틸-1H-피라졸-3-일)-1,2,4-옥사디아졸

502 mg (2.62 mmol)의 EDC, 401 mg (2.62 mmol)의 HOBt 및 500 mg (2.38 mmol)의 실시예 6A / 단계 3으로부터의 화합물을 실온에서 10 ml의 무수 DMF 중 300 mg (2.38 mmol)의 5-메틸-1H-피라졸-3-카르복실산의 용액에 연속적으로 가하였다. 혼합물을 먼저 실온에서 16 h 동안 및 그 다음 140℃에서 45분 동안 교반하였다. 냉각 후, 용매를 회전 증발기 상에서 제거하였다. 120 ml의 물을 잔존 잔류물에 가하고, 이를 매회 대략 100 ml의 디에틸 에테르로 3회 추출하였다. 합한 유기 추출물을 포화 염화나트륨 용액으로 세척하고, 무수 황산마그네슘 상에서 건조시키고, 여과하고 최종적으로 회전 증발기 상에서 농축하였다. 조 생성물을 5 ml의 에탄올과 함께 실온에서 1 h 동안 교반하였다. 여과하고 미용해 고체를 고진공 하에 건조시킨 후, 제1 분획의 204 mg의 표제 화합물을 수득하였다. 모액을 농축 건조시켰다. 후속적으로, 정제용 HPLC (방법 13)에 의해 잔류물로부터 추가의 분획의 29 mg의 표제 화합물을 단리하였다. 이렇게 하여 총 233 mg (이론치의 33%)의 표제 화합물을 수득하였다.
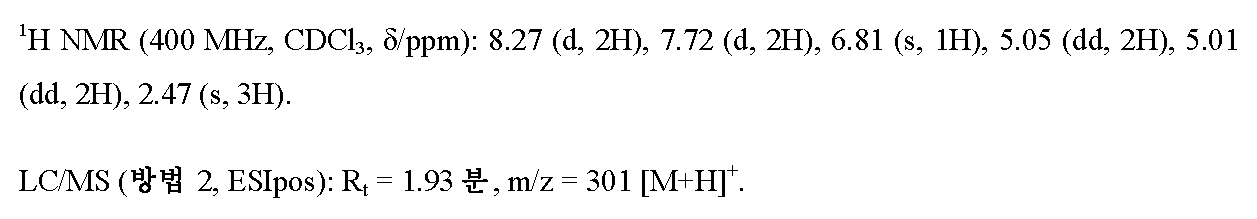
단계 5: 5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-[4-(3-(플루오로옥세탄-3-일)페닐]-1,2,4-옥사디아졸

실시예 1A / 단계 2에 기재된 공정과 유사하게, 250 mg (0.833 mmol)의 실시예 6A / 단계 4로부터의 화합물 및 250 mg (0.999 mmol)의 3-브로모벤질 브로마이드를 사용하여 347 mg (이론치의 89%)의 표제 화합물을 수득하였다. 여기서 반응 혼합물을 45℃로 가열하는 것을 생략하는 것이 가능하였다. 생성물의 최종 정제를 10 ml의 펜탄/디이소프로필 에테르 (5:1)로부터 추출 교반함으로써 수행하고, 여기에 수 방울의 디클로로메탄을 가하였다.

실시예
7A
5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-[4-(1-메톡시시클로부틸)페닐]-1,2,4-옥사디아졸

단계 1: 4-(1-히드록시시클로부틸)벤조니트릴

실시예 5A / 단계 1에 기재된 공정과 유사하게, 32.67 g (143 mmol)의 4-아이오도벤조니트릴, 디에틸 에테르 중 2 M 이소프로필마그네슘 클로라이드 용액 75 ml (150 mmol) 및 15.0 g (214 mmol)의 시클로부타논을 사용하여 13.31 g (이론치의 78%)의 표제 화합물을 수득하였다. 이 경우 실온에서 반응 시간은 16 h이었고, 수성 후처리 후, 조 생성물을 용리액으로서 10:1 -> 4:1 시클로헥산/에틸 아세테이트를 사용하여 150 g의 실리카 겔을 통해 흡인 여과에 의해 정제하였다.

단계 2: 4-(1-메톡시시클로부틸)벤조니트릴

508 mg (12.7 mmol)의 수소화나트륨 (미네랄 오일 중 60% 현탁액)을 0℃에서 40 ml의 무수 DMF 중 2.0 g (11.5 mmol)의 실시예 7A / 단계 1로부터의 화합물의 용액에 가하였다. 1 h 후, 863 μl (13.9 mmol)의 아이오도메탄을 가하고, 얼음-물 조를 제거하였다. 실온에서 16 h 동안 교반 후, 반응 혼합물을 120 ml의 물에 붓고 매회 대략 100 ml의 디에틸 에테르로 3회 추출하였다. 합한 유기 추출물을 물 및 포화 염화나트륨 용액으로 연속적으로 세척하고, 무수 황산마그네슘 상에서 건조시키고, 여과하고 최종적으로 회전 증발기 상에서 용매를 제거하였다. 잔존 잔류물을 용리액으로서 20:1 -> 4:1 시클로헥산/에틸 아세테이트를 사용하여 대략 100 g의 실리카 겔을 통해 흡인 여과에 의해 정제하였다. 1.27 g (이론치의 59%)의 표제 화합물을 수득하였다.

단계 3: N'-히드록시-4-(1-메톡시시클로부틸)벤젠카르복스이미드 아미드
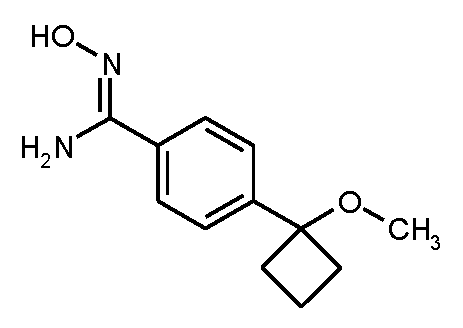
실시예 3A / 단계 5에 기재된 공정과 유사하게, 1.10 g (5.88 mmol)의 실시예 7A / 단계 2로부터의 화합물 및 612 mg (8.81 mmol)의 히드록실암모늄 클로라이드를 사용하여 1.28 g (이론치의 93%, 95% 순도)의 표제 화합물을 수득하였다. 이 경우 반응 시간은 16 h이었다.

단계 4 : 3-[4-(1-메톡시시클로부틸)페닐]-5-(5-메틸-1H-피라졸-3-일]-1,2,4-옥사디아졸
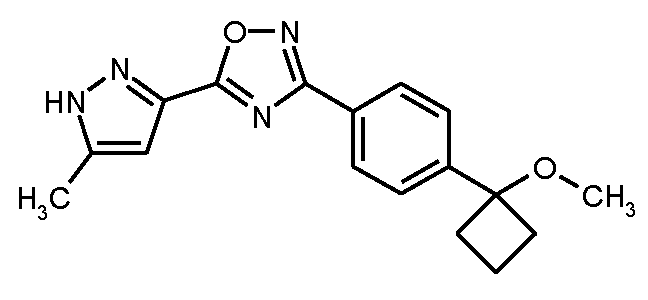
2 ml의 무수 DMF 중 491 mg (3.03 mmol)의 1,1'-카르보닐디이미다졸(CDI)의 현탁액에, 실온에서 15분 이내로, 2 ml의 무수 DMF 중 364 mg (2.88 mmol)의 5-메틸-1H-피라졸-3-카르복실산의 용액을 적가하였다. 혼합물을 실온에서 2 h 동안 교반한 후, 635 mg (2.88 mmol)의 실시예 7A / 단계 3으로부터의 화합물을 가하였다. 후속적으로, 반응 혼합물을 4.5 h 동안 110℃로 가열하였다. 실온으로 냉각 후, 반응 혼합물을 격렬하게 교반하면서 60 ml의 물 내로 서서히 교반하고, 이 과정에서 생성물이 침전되었다. 고체를 흡인으로 여과해 내고 약간의 냉수로 세척하였다. 고 진공 하에 건조 후, 640 mg (이론치의 68%, 95% 순도)의 표제 화합물을 수득하였다.

단계 5: 5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-[4-(1-메톡시시클로부틸)페닐]-1,2,4-옥사디아졸
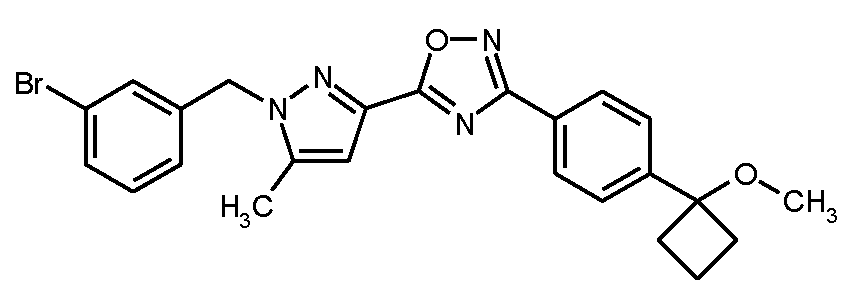
실시예 1A / 단계 2에 기재된 공정과 유사하게, 250 mg (0.806 mmol)의 실시예 7A / 단계 4로부터의 화합물 및 242 mg (0.967 mmol)의 3-브로모벤질 브로마이드를 사용하여 326 mg (이론치의 84%)의 표제 화합물을 수득하였다. 여기서 반응 혼합물을 45℃로 가열하는 것을 생략하는 것이 가능하였다.
실시예
8A
3-{[tert-부틸(디페닐)실릴]옥시}아제티딘

단계 1: tert-부틸 3-{[tert-부틸(디페닐)실릴]옥시}아제티딘-1-카르복실레이트
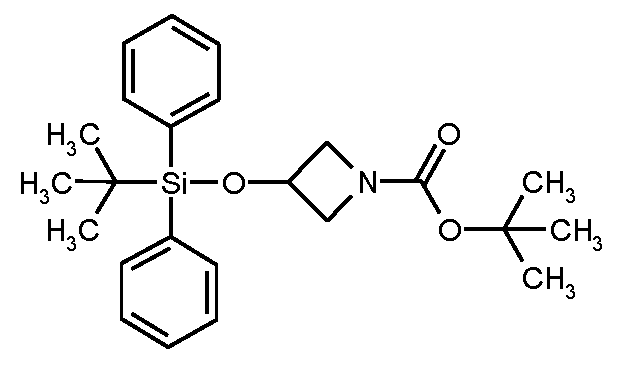
20.0 g (115 mmol)의 tert-부틸 3-히드록시아제티딘-1-카르복실레이트 및 9.43 g (139 mmol)의 이미다졸을 처음에 200 ml의 무수 DMF에 채우고, 34.91 g (127 mmol)의 tert-부틸(디페닐)실릴 클로라이드를 실온에서 가하였다. 반응 혼합물을 실온에서 18 h 동안 교반한 후, 이를 3.2 리터의 물에 부은 다음 매회 대략 1 리터의 디에틸 에테르로 3회 추출하였다. 합한 유기 추출물을 포화 탄산수소나트륨 용액, 물 및 포화 염화나트륨 용액으로 연속적으로 세척하였다. 무수 황산마그네슘 상에서 건조 후, 혼합물을 여과하고 용매를 회전 증발기 상에서 제거하였다. 잔존 잔류물을 100 ml의 펜탄과 함께 수 분 동안 교반하였다. 후속적으로, 고체를 흡인으로 여과해 내고 고진공 하에 건조시켰다. 29.18 g (이론치의 61%)의 표제 화합물을 수득하였다.
단계 2: 3-{[tert-부틸(디페닐)실릴]옥시}아제티딘

70 ml의 트리플루오로아세트산 (TFA)을 실온에서 70 ml의 디클로로메탄 중 20.0 g (48.6 mmol)의 실시예 8A / 단계 1로부터의 화합물의 용액에 적가하였다. 반응 혼합물을 실온에서 30분 동안 교반한 후, 모든 휘발성 성분을 회전 증발기 상에서 제거하였다. 1 리터의 1 M 수산화나트륨 용액을 잔존 잔류물에 가하고, 이를 매회 대략 200 ml의 디클로로메탄으로 3회 추출하였다. 합한 유기 추출물을 무수 황산마그네슘 상에서 건조시키고, 여과하고 최종적으로 회전 증발기 상에서 농축 건조시켰다. 잔류물을 고진공 하에 건조시킨 후, 14.85 g (이론치의 98%)의 표제 화합물을 수득하였다.
실시예
9A
4-{[tert-부틸(디페닐)실릴]옥시}피페리딘
단계 1: tert-부틸 4-{[tert-부틸(디페닐)실릴]옥시}피페리딘-1-카르복실레이트
10.0 g (49.7 mmol)의 tert-부틸 4-히드록시아제티딘-1-카르복실레이트 및 4.06 g (59.7 mmol)의 이미다졸을 처음에 100 ml의 무수 DMF에 채우고, 15.02 g (54.7 mmol)의 tert-부틸(디페닐)실릴 클로라이드를 0℃에서 가하였다. 반응 혼합물을 실온에서 48 h 동안 교반한 후, 이를 1.6 리터의 물에 부은 다음 매회 대략 500 ml의 디에틸 에테르로 3회 추출하였다. 합한 유기 추출물을 포화 탄산수소나트륨 용액, 물 및 포화 염화나트륨 용액으로 연속적으로 세척하였다. 무수 황산마그네슘 상에서 건조 후, 혼합물을 여과하고 용매를 회전 증발기 상에서 제거하였다. 잔존 잔류물을 흡인 여과에 의해 조악하게 정제하였다 (대략 300 g의 실리카 겔, 용리액: 시클로헥산 -> 2:1 시클로헥산/에틸 아세테이트). 22.21 g (이론치의 91%, 대략 90% 순도)의 표제 화합물을 수득하였다.

단계 2: 4-{[tert-부틸(디페닐)실릴]옥시}피페리딘

실시예 8A / 단계 2에 기재된 공정과 유사하게, 2.5 g (5.12 mmol, 90% 순도)의 실시예 9A / 단계 1로부터의 화합물을 사용하여 1.45 g (이론치의 83%)의 표제 화합물을 수득하였다. 이 경우, 생성물을 MPLC (대략 50 g의 실리카 겔, 용리액: 에틸 아세테이트 -> 9:1 에틸 아세테이트/트리에틸아민)에 의해 정제하였다.
실시예
10A
5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-{4-[(트리플루오로메틸)술파닐]페닐}-1,2,4-옥사디아졸

단계 1: N'-히드록시-4-[(트리플루오로메틸)술파닐]벤젠카르복스이미드 아미드

73 g (1.05 mol)의 히드록실암모늄 클로라이드를 1.4 리터의 에탄올 중 113 g (500 mmol)의 4-[(트리플루오로메틸)술파닐]벤젠카르보니트릴 및 147 ml (1.05 mol)의 트리에틸아민의 용액에 가한 다음, 혼합물을 30분 동안 환류 하에 가열하였다. 실온으로 냉각 후, 1 리터의 물을 가하고 혼합물을 총 1.4 리터의 에틸 아세테이트로 3회 추출하였다. 합한 유기 추출물을 물 및 포화 염화나트륨 용액으로 연속적으로 세척하였다. 무수 황산마그네슘 상에서 건조시킨 후, 혼합물을 여과하고 용매를 회전 증발기 상에서 제거하였다. 생성된 잔류물을 1 리터의 시클로헥산에 용해시키고, 40 ml의 디이소프로필 에테르를 가하고 혼합물을 실온에서 20분 동안 교반하였다. 생성된 고체를 흡인으로 여과해 내고, 고진공 하에 건조 후, 제1 분량의 78.6 g의 표제 화합물을 수득하였다. 여과물을 농축하고 잔류물을 후속적으로 100 ml의 시클로헥산 및 5 ml의 에틸 아세테이트의 혼합물 중에서 30분 동안 비등 교반하였다. 냉각 후, 고체를 흡인으로 여과해 내고 고진공 하에 건조시켰다. 이로써 제2 분량의 4.2 g의 표제 화합물을 수득하였다. 이렇게 하여 총 82.8 g (이론치의 70%)의 표제 화합물을 수득하였다.

단계 2 : 5-(5-메틸-1H-피라졸-3-일]-3-{4-[(트리플루오로메틸)술파닐]페닐}-1,2,4-옥사디아졸

235 ml의 무수 DMF 중 63.45 g (391 mmol)의 1,1'-카르보닐디이미다졸의 현탁액에, 실온에서 15분 이내로, 235 ml의 무수 DMF 중 47 g (373 mmol)의 5-메틸-1H-피라졸-3-카르복실산의 용액을 적가하였다. 혼합물을 실온에서 1시간 45분 동안 교반한 후, 88 g (373 mmol)의 실시예 10A / 단계 1로부터의 화합물을 가하였다. 후속적으로, 반응 혼합물을 4 h 동안 110℃로 가열하였다. 실온으로 냉각 후, 3.5 리터의 물을 격렬하게 교반하면서 서서히 가하고, 그 결과 생성물이 침전되었다. 고체를 흡인으로 여과해 내고 대략 1 리터의 냉수로 세척하였다. 조 생성물을 각각 500 ml의 아세토니트릴과 에탄올로 이루어진 용매 혼합물로부터 재결정화함으로써 정제하였다. 냉각, 여과 및 고진공 하에 건조 후, 제1 분량의 94.2 g의 표제 화합물을 수득하였다. 농축 및 150 ml의 에탄올로부터의 또 다른 결정화 후, 추가의 6.2 g의 표제 화합물을 수득하였다. 이렇게 하여 총 100.4 g (이론치의 83%)의 표제 화합물을 수득하였다.

단계 3: 5-[1-(3-브로모벤질)-5-메틸-1H-피라졸-3-일]-3-{4-[(트리플루오로메틸)술파닐]페닐}-1,2,4-옥사디아졸

2.24 g (19.9 mmol)의 고체 칼륨 tert-부톡시드를 실온에서 150 ml의 무수 디옥산 중 5.0 g (15.3 mmol)의 실시예 10A / 단계 2로부터의 화합물 및 4.60 g (18.4 mmol)의 3-브로모벤질 브로마이드의 용액에 가하였다. 반응 혼합물을 실온에서 16 h 동안 교반하였다. 후속적으로, 대략 50-100 ml의 용매를 회전 증발기 상에서 제거하였다. 잔존 잔류물을 900 ml의 에틸 아세테이트로 희석하고 매회 200 ml의 물로 3회 및 최종적으로 200 ml의 포화 염화나트륨 용액으로 1회 세척하였다. 무수 황산마그네슘 상에서 건조 및 여과 후, 용매를 회전 증발기 상에서 제거하였다. 생성된 조 생성물을 시클로헥산으로부터 재결정화하였다. 고체를 흡인으로 여과해 내고 건조시킨 후, 5.03 g (이론치의 66%)의 표제 화합물을 수득하였다.

실시예
11A
(S)-1-{3-(5-메틸-3-{3-[4-(트리플루오로메톡시)페닐]-1,2,4-옥사디아졸-5-일}-1H-피라졸-1-일)메틸]페닐}아제티딘-3-일 N-(tert-부톡시카르보닐)-L-발리네이트

117 mg (0.955 mmol)의 4-N,N-디메틸아미노피리딘 및 915 mg (4.77 mmol)의 EDC를 30 ml의 디클로로메탄 중 750 mg (1.59 mmol)의 실시예 1로부터의 화합물 (실시예에 대한 섹션 참조) 및 691 mg (3.18 mmol)의 (S)-N-(tert-부톡시카르보닐)-L-발린의 용액에 가하였다. 반응 혼합물을 실온에서 2 h 동안 교반한 후, 이를 100 ml의 디클로로메탄으로 희석하고 매회 100 ml의 물로 2회 및 100 ml의 포화 염화나트륨 용액으로 1회 세척하였다. 무수 황산마그네슘 상에서 건조, 여과 및 농축 후, 생성물을 MPLC (대략 100 g의 실리카 겔, 용리액: 4:1 -> 3:1 시클로헥산/에틸 아세테이트)에 의해 단리하였다. 671 mg (이론치의 90%)의 표제 화합물을 수득하였다.

실시예
:
실시예
1
1-{3-[(5-메틸-3-{3-[4-(트리플루오로메톡시)페닐]-1,2,4-옥사디아졸-5-일}-1H-피라졸-1-일)메틸]페닐}아제티딘-3-올
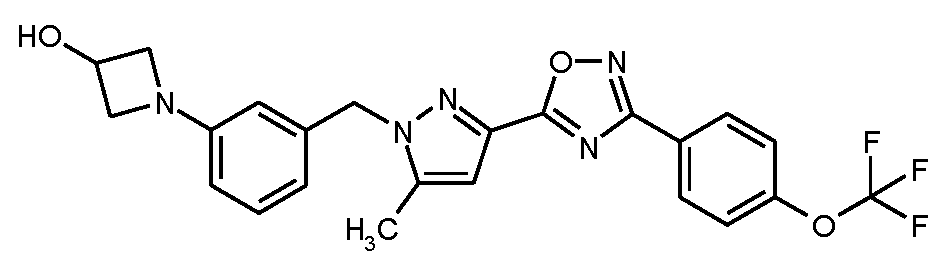
20 ml의 무수 DMF 중 2.0 g (4.17 mmol)의 실시예 1A로부터의 화합물, 1.95 g (6.26 mmol)의 실시예 8A로부터의 화합물, 256 mg (0.280 mmol)의 트리스(디벤질리덴아세톤)디팔라듐(0), 398 mg (0.835 mmol)의 2-디시클로헥실포스피노-2',4',6'-트리이소프로필비페닐 (X-Phos) 및 2.72 g (8.35 mmol)의 탄산세슘의 혼합물을 아르곤 하에 마이크로파 오븐 (비오티지 이니시에이터(Biotage Initiator), 입사 전력의 동적 제어)에서 120℃로 2.5 h 동안 가열하였다. 실온으로 냉각 후, 100 ml의 물을 반응 혼합물에 가하고, 이를 매회 대략 100 ml의 에틸 아세테이트로 3회 추출하였다. 합한 유기 추출물을 물 및 포화 염화나트륨 용액으로 연속적으로 세척하고, 무수 황산마그네슘 상에서 건조시키고, 여과하고 최종적으로 회전 증발기 상에서 용매를 제거하였다. 잔존 잔류물을 40 ml의 THF에 용해시키고, THF 중 테트라-n-부틸암모늄 플루오라이드 (TBAF)의 1 M 용액 6.3 ml (6.26 mmol)를 가하였다. 이 혼합물을 실온에서 1 h 동안 교반한 후, 이를 상기 기재된 바와 동일한 방법으로 수성 후처리에 적용하였다. 잔존 잔류물을 용리액으로서 2:1 (3 리터) -> 1:1 (3 리터) 시클로헥산/에틸 아세테이트를 사용하여 200 g의 실리카 겔을 통해 흡인 여과에 의해 정제하였다. 농축 및 고진공 하에 건조 후, 1.56 g (이론치의 79%)의 표제 화합물을 수득하였다.
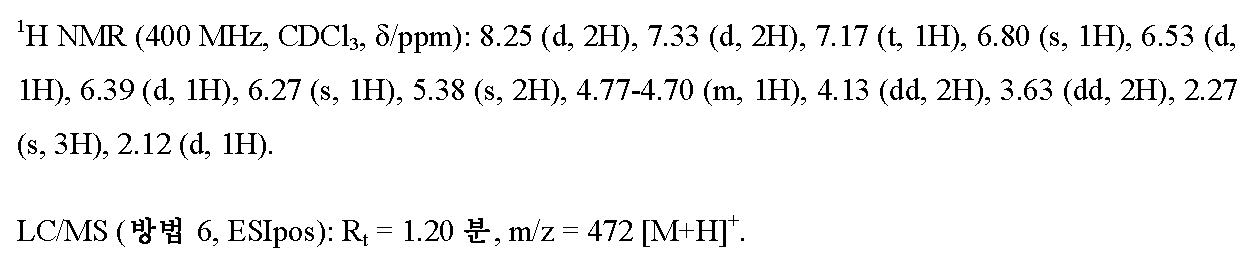
실시예
2
1-{3-{[5-메틸-3-(3-{4-(1-(트리플루오로메틸)시클로프로필]페닐}-1,2,4-옥사디아졸-5-일)-1H-피라졸-1-일]메틸}페닐)아제티딘-3-올

실시예 1에 기재된 공정과 유사하게, 500 mg (0.993 mmol)의 실시예 2A로부터의 화합물 및 464 mg (1.49 mmol)의 실시예 8A로부터의 화합물을 사용하여 289 mg (이론치의 58%)의 표제 화합물을 수득하였다. 여기서 공정의 제2 성분 단계 (TBAF를 사용한 실릴 에테르 절단)를 생략하는 것이 가능하였고, 그 이유는 보호기가 제1 성분 단계의 과정에서 이미 탈리되었기 때문이다. 조 생성물을 MPLC (실리카 겔, 용리액: 1:1 시클로헥산/에틸 아세테이트) 및 25 ml의 펜탄, 1 ml의 디이소프로필 에테르 및 125 μl의 디클로로메탄의 혼합물 중 후속 추출 교반에 의해 정제하였다.

실시예
3
1-{3-[(5-메틸-3-{3-[4-(1,1,1-트리플루오로-2-메틸프로판-2-일)페닐]-1,2,4-옥사디아졸-5-일}-1H-피라졸-1-일)메틸]페닐}아제티딘-3-올

실시예 1에 기재된 공정과 유사하게, 150 mg (0.297 mmol)의 실시예 3A로부터의 화합물 및 139 mg (0.445 mmol)의 실시예 8A로부터의 화합물을 사용하여 68 mg (이론치의 46%)의 표제 화합물을 수득하였다. 공정의 제1 성분 단계에서의 반응 시간은 1 h이었다. 조 생성물을 정제용 HPLC (방법 13)에 의해 정제하였다. 탄산수소염 카트리지 (폴리머랩스(Polymerlabs)로부터, 스트라토스피어스(Stratospheres) SPE, PL-HCO3 MP SPE, 용량 0.9 mmol)를 통해 염의 메탄올성 용액의 퍼콜레이션(percolation)에 의해 생성된 표제 화합물의 포르메이트 염으로부터 유리 염기를 수득하였다. 3 ml의 펜탄과 수 방울의 디이소프로필 에테르의 혼합물 중에서 추출 교반에 의해 최종 정제를 수행하였다.
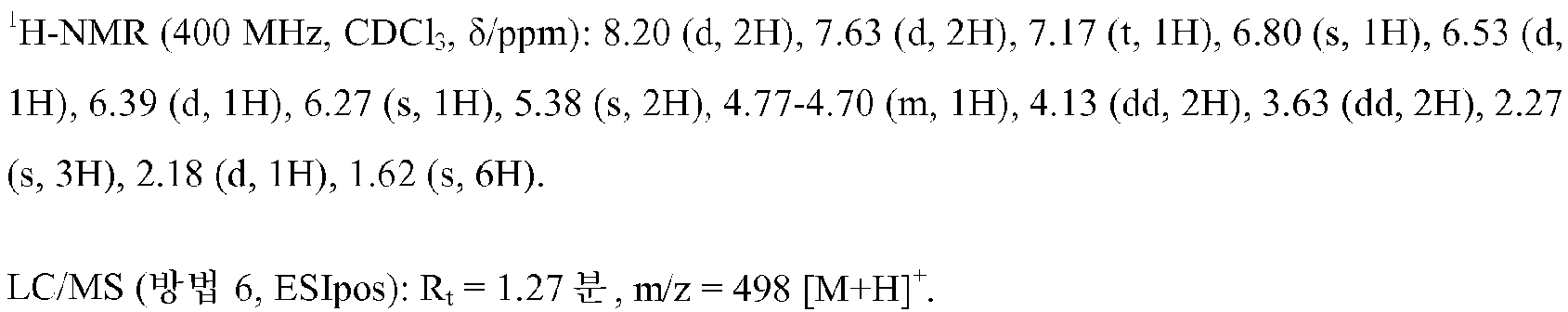
실시예
4
1-{3-[(5-메틸-3-{3-[4-(테트라히드로-2H-피란-4-일)페닐]-1,2,4-옥사디아졸-5-일}-1H-피라졸-1-일)메틸]페닐}아제티딘-3-올

실시예 3에 기재된 공정과 유사하게, 150 mg (0.313 mmol)의 실시예 4A로부터의 화합물 및 146 mg (0.469 mmol)의 실시예 8A로부터의 화합물을 사용하여 77 mg (이론치의 52%)의 표제 화합물을 수득하였다.

실시예
5
1-{3-[(3-{3-[4-(4-플루오로테트라히드로-2H-피란-4-일)페닐]-1,2,4-옥사디아졸-5-일}-5-메틸-1H-피라졸-1-일)메틸]페닐}아제티딘-3-올
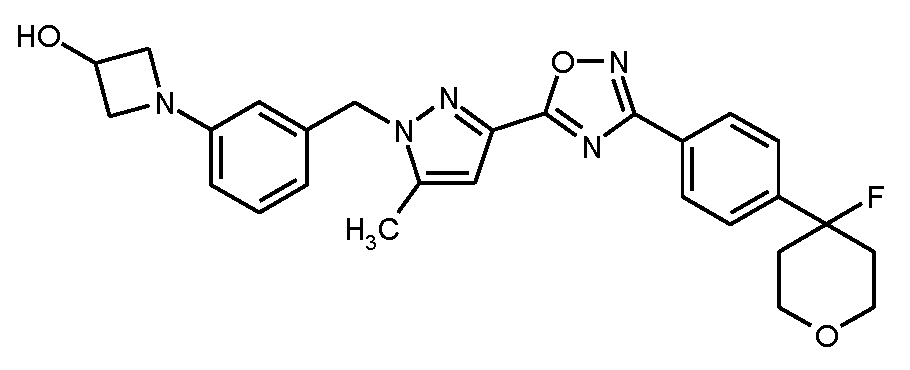
실시예 3에 기재된 공정과 유사하게, 150 mg (0.302 mmol)의 실시예 5A로부터의 화합물 및 141 mg (0.452 mmol)의 실시예 8A로부터의 화합물을 사용하여 62 mg (이론치의 42%)의 표제 화합물을 수득하였다.

실시예
6
1-{3-[(3-{3-[4-(3-플루오로옥세탄-3-일)페닐]-1,2,4-옥사디아졸-5-일}-5-메틸-1H-피라졸-1-일)메틸]페닐}아제티딘-3-올

실시예 3에 기재된 공정과 유사하게, 150 mg (0.320 mmol)의 실시예 6A로부터의 화합물 및 149 mg (0.479 mmol)의 실시예 8A로부터의 화합물을 사용하여 60 mg (이론치의 39%)의 표제 화합물을 수득하였다.

실시예
7
1-{3-[(3-{3-[4-(1-메톡시시클로부틸)페닐]-1,2,4-옥사디아졸-5-일}-5-메틸-1H-피라졸-1-일)메틸]페닐}아제티딘-3-올
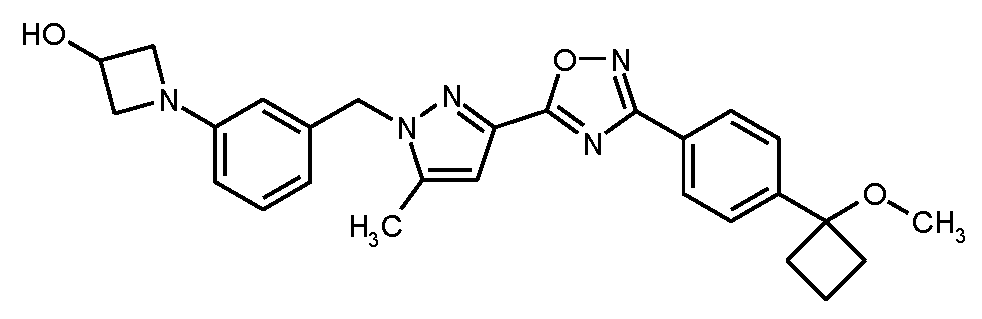
실시예 3에 기재된 공정과 유사하게, 150 mg (0.313 mmol)의 실시예 7A로부터의 화합물 및 146 mg (0.469 mmol)의 실시예 8A로부터의 화합물을 사용하여 53 mg (이론치의 36%)의 표제 화합물을 수득하였다.
실시예
8
1-{3-[(5-메틸-3-{3-[4-(트리플루오로메톡시)페닐]-1,2,4-옥사디아졸-5-일}-1H-피라졸-1-일)메틸]페닐}피페리딘-4-올

3 ml의 무수 DMF 중 150 mg (0.313 mmol)의 실시예 1A로부터의 화합물, 63 mg (0.626 mmol)의 4-히드록시피페리딘, 19 mg (0.021 mmol)의 트리스(디벤질리덴아세톤)디팔라듐(0), 30 mg (0.063 mmol)의 2-디시클로헥실포스피노-2',4',6'-트리이소프로필비페닐 (X-Phos) 및 254 mg (0.782 mmol)의 탄산세슘의 혼합물을 아르곤 하에 80℃에서 12 h 동안 교반하였다. 실온으로 냉각 후, 반응 혼합물을 약간의 셀라이트를 통해 여과한 다음 여과물을 정제용 HPLC (방법 11)에 의해 그의 성분으로 분리하였다. 생성물 분획의 농축 및 고진공 하에 건조 후, 36 mg (이론치의 23%)의 표제 화합물을 수득하였다.

실시예
9
1-{3-{[5-메틸-3-(3-{4-[1-(트리플루오로메틸)시클로프로필]페닐}-1,2,4-옥사디아졸-5-일)-1H-피라졸-1-일]메틸}페닐)피페리딘-4-올

실시예 3에 기재된 공정과 유사하게, 125 mg (0.248 mmol)의 실시예 2A로부터의 화합물 및 50 mg (0.497 mmol)의 4-히드록시피페리딘을 사용하여 20 mg (이론치의 16%)의 표제 화합물을 수득하였다. 공정의 제1 성분 단계를 여기서 130℃의 반응 온도에서 수행하였다. 정제용 HPLC 정제를 방법 11에 의해 수행하였다.

실시예
10
1-{3-[(5-메틸-3-{3-{4-(1,1,1-트리플루오로-2-메틸프로판-2-일)페닐]-1,2,4-옥사디아졸-5-일}-1H-피라졸-1-일)메틸]페닐}피페리딘-4-올
실시예 9에 기재된 공정과 유사하게, 125 mg (0.247 mmol)의 실시예 3A로부터의 화합물 및 50 mg (0.495 mmol)의 4-히드록시피페리딘을 사용하여 16 mg (이론치의 12%)의 표제 화합물을 수득하였다.

실시예
11
1-{3-[(5-메틸-3-{3-[4-(트리플루오로메톡시)페닐]-1,2,4-옥사디아졸-5-일}-1H-피라졸-1-일)메틸]페닐}피페리딘-4-카르보니트릴

실시예 8에 기재된 공정과 유사하게, 150 mg (0.313 mmol)의 실시예 1A로부터의 화합물 및 69 mg (0.626 mmol)의 4-시아노피페리딘을 사용하여 68 mg (이론치의 43%)의 표제 화합물을 수득하였다.

실시예
12
1-{3-{[5-메틸-3-(3-{4-[1-(트리플루오로메틸)시클로프로필]페닐}-1,2,4-옥사디아졸-5-일)-1H-피라졸-1-일)메틸}페닐)피페리딘-4-카르보니트릴

실시예 9에 기재된 공정과 유사하게, 50 mg (0.099 mmol)의 실시예 2A로부터의 화합물 및 22 mg (0.199 mmol)의 4-시아노피페리딘을 사용하여 23 mg (이론치의 43%)의 표제 화합물을 수득하였다.

실시예
13
1-{3-[(5-메틸-3-{3-[4-(1,1,1-트리플루오로-2-메틸프로판-2-일)페닐]-1,2,4-옥사디아졸-5-일}-1H-피라졸-1-일)메틸]페닐}피페리딘-4-카르보니트릴

실시예 9에 기재된 공정과 유사하게, 125 mg (0.247 mmol)의 실시예 3A로부터의 화합물 및 55 mg (0.495 mmol)의 4-시아노피페리딘을 사용하여 66 mg (이론치의 50%)의 표제 화합물을 수득하였다.

실시예
14
1-(3-{[5-메틸-3-(3-{4-[(트리플루오로메틸)술파닐]페닐]-1,2,4-옥사디아졸-5-일}-1H-피라졸-1-일)메틸}페닐)피페리딘-4-올

10 ml의 톨루엔 중 500 mg (1.01 mmol)의 실시예 10A로부터의 화합물, 514 mg (1.51 mmol)의 실시예 9A로부터의 화합물, 92 mg (0.101 mmol)의 트리스(디벤질리덴아세톤)디팔라듐(0), 96 mg (0.202 mmol)의 2-디시클로헥실포스피노-2',4',6'-트리이소프로필비페닐 (X-Phos) 및 194 mg (2.02 mmol)의 나트륨 tert-부톡시드의 혼합물을 아르곤 하에 마이크로파 오븐 (비오티지 이니시에이터, 입사 전력의 동적 제어)에서 3 h 동안 80℃로 가열하였다. 실온으로 냉각 후, 100 ml의 물을 반응 혼합물에 가하고, 이를 매회 대략 100 ml의 에틸 아세테이트로 3회 추출하였다. 합한 유기 추출물을 물 및 포화 염화나트륨 용액으로 연속적으로 세척하고, 무수 황산마그네슘 상에서 건조시키고, 여과하고 최종적으로 회전 증발기 상에서 용매를 제거하였다. 상응하는 실릴-보호된 중간체인, 생성된 조 생성물을 MPLC (대략 70 g의 실리카 겔, 용리액: 시클로헥산 -> 5:1 시클로헥산/에틸 아세테이트)에 의해 정제하였다. 후속적으로, 이 생성물을 3.5 ml의 THF에 용해시키고, THF 중 테트라-n-부틸암모늄 플루오라이드 (TBAF)의 1 M 용액 3.5 ml (3.5 mmol)를 가하였다. 이 혼합물을 실온에서 20분 동안 교반한 후, 이를 회전 증발기 상에서 농축시켰다. 표제 화합물을 MPLC (대략 20 g의 실리카 겔, 용리액: 2:1 -> 1:3 시클로헥산/에틸 아세테이트)에 의해 단리하였다. 농축 및 건조 후, 생성물을 펜탄/디에틸 에테르와 함께 1회 더 교반하였다. 65 mg (이론치의 12%)의 표제 화합물을 수득하였다.

실시예
15
1-{3-[(5-메틸-3-{3-[4-(트리플루오로메톡시)페닐]-1,2,4-옥사디아졸-5-일}-1H-피라졸-1-일)메틸]페닐}아제티딘-3-일 N,N-디메틸글리시네이트

86 mg (0.70 mmol)의 4-N,N-디메틸아미노피리딘 및 671 mg (3.50 mmol)의 EDC를 11 ml의 디클로로메탄 중 550 mg (1.17 mmol)의 실시예 1로부터의 화합물 및 361 mg (3.50 mmol)의 N,N-디메틸글리신의 용액에 가하였다. 반응 혼합물을 실온에서 8 h 동안 교반한 후, 모든 휘발성 성분을 회전 증발기 상에서 제거하였다. 수득된 잔류물을 200 ml의 에틸 아세테이트에 용해시키고 대략 100 ml의 물로 1회 세척하였다. 무수 황산마그네슘 상에서 건조, 여과 및 농축 후, 생성물을 MPLC (대략 30 g의 실리카 겔, 용리액: 25:25:1 시클로헥산/에틸 아세테이트/트리에틸아민)에 의해 단리하였다. 제1 분획의 328 mg의 표제 화합물을 수득하였다. 시클로펜틸 메틸 에테르로부터 재결정화에 의해 제2의 여전히-오염된 분획으로부터 추가의 115 mg의 표제 화합물을 수득하였다. 이렇게 하여 총 443 mg (이론치의 68%)의 표제 화합물을 수득하였다. 디이소프로필 에테르/시클로펜틸 메틸 에테르 (2:1, 100 mg 당 1.5 ml)로부터의 또 다른 결정화로부터 117℃의 융점을 갖는 표제 화합물을 수득하였다.

실시예
16
(S)-1-{3-[(5-메틸-3-{3-[4-(트리플루오로메톡시)페닐]-1,2,4-옥사디아졸-5-일}-1H-피라졸-1-일)메틸]페닐}아제티딘-3-일 L-발리네이트

25 ml의 트리플루오로아세트산 (TFA)을 0℃에서 45 ml의 디클로로메탄 중 965 mg (1.44 mmol)의 실시예 11A로부터의 화합물의 용액에 가하였다. 반응 혼합물을 실온에서 1 h 동안 교반하였다. 그 후, 모든 휘발성 성분을 회전 증발기 상에서 제거하고 수득된 잔류물을 500 ml의 디클로로메탄에 다시 용해시켰다. 생성물을 매회 대략 100 ml의 포화 탄산수소나트륨 용액으로 2회, 그 다음 100 ml의 물로 1회 및 최종적으로 100 ml의 포화 염화나트륨 용액으로 1회 세척하였다. 무수 황산마그네슘 상에서 건조시킨 후, 혼합물을 여과하고 용매를 회전 증발기 상에서 제거하였다. 생성된 조 생성물 (780 mg)을 40 ml의 펜탄, 2 ml의 디이소프로필 에테르 및 수 방울의 디클로로메탄의 혼합물 중에서 교반하였다. 고체를 흡인으로 여과해 내고 고진공 하에 건조시킨 후, 이렇게 하여 659 mg (이론치의 80%)의 표제 화합물을 수득하였다.

B.
약리 활성의 평가
본 발명의 화합물의 약리 활성은 당업자에게 공지된 바와 같이 시험관내 및 생체내 연구에 의해 입증될 수 있다. 본 발명의 물질의 유용성은 하기 기재된 바와 같이 시험관내 (종양) 세포 실험 및 생체내 종양 모델에 의해 예로서 설명될 수 있다. HIF 전사 활성의 억제와 종양 성장의 억제와의 관련성은 문헌에 기재된 다수의 연구에 의해 입증되어 있다 (예를 들어 문헌 [Warburg, 1956]; [Semenza, 2007] 참조).
B-1. HIF 루시퍼라제 검정
HCT 116 세포를 HIF-반응성 서열의 제어하에 루시퍼라제 리포터를 함유하는 플라스미드를 사용하여 안정적으로 형질감염시켰다. 이들 세포를 마이크로타이터 플레이트에 시딩하였다 [10% 소 태아 혈청 (FCS) 및 100 ㎍/ml의 히그로마이신을 함유하는 RPMI 1640 배지 중 20,000개 세포/캐비티]. 이들을 표준 조건 (5% CO2, 21% O2, 37℃, 습윤화됨)하에 밤새 인큐베이션하였다. 다음날 아침, 세포를 저산소 챔버 (1% O2)에서 상이한 농도의 시험 물질 (0 내지 10 μmol/l)과 함께 인큐베이션하였다. 24 h 후, 브라이트 글로(Bright Glo) 시약 (프로메가(Promega), 미국 위스콘신주)을 제조업체의 설명서에 따라 가하고, 5분 후에 발광을 측정하였다. 산소 정상 상태하에 인큐베이션된 세포는 배경 대조군의 역할을 하였다.
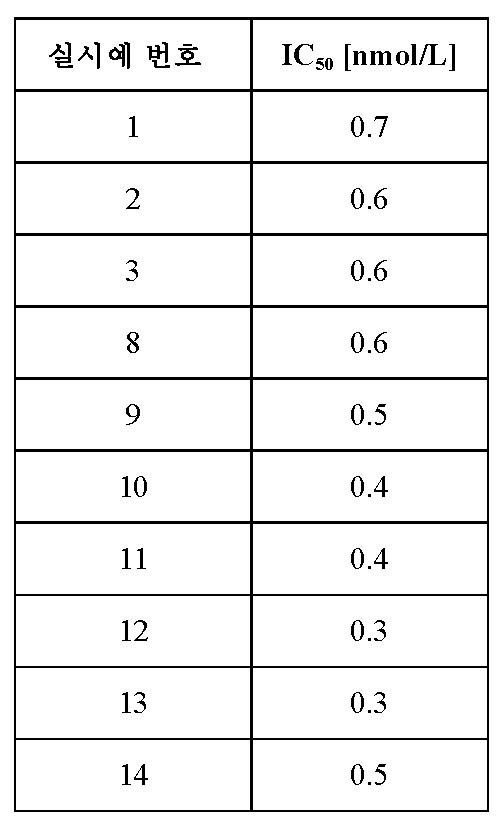
대표적인 실시예에 대한 상기 검정으로부터의 IC50 값을 하기 표에 기재하였다:
WO 2008/141731-A2의 실시예 1로서 기재되어 있고, 본 발명의 화합물과 달리, 벤질 헤드 기 상에 헤테로시클릴 치환기를 갖지 않는 화합물 5-[5-메틸-1-(4-메틸벤질)-1H-피라졸-3-일]-3-[4-(트리플루오로메톡시)페닐]-1,2,4-옥사디아졸은 당해 검정에서 30 nmol/l의 IC50 값을 나타내었다.
B-2. HIF 표적 유전자의 시험관내 억제:
인간 기관지 암종 세포 (H460 및 A549)를 가변적 농도의 시험 물질 (1 nM 내지 10 μM)과 함께 16 h 동안 산소 정상 조건하에 및 산소 분압 1% 하에 인큐베이션하였다 (HIF 루시퍼라제 검정 참조). 총 RNA를 세포로부터 단리하고 cDNA로 전사시키고 HIF 표적 유전자의 mRNA 발현을 실시간 PCR로 분석하였다. 활성 시험 물질은 산소 정상 조건하에, 그러나 특히 저산소 조건하에 비처리 세포와 비교하여 HIF 표적 유전자의 mRNA 발현을 이미 낮추었다.
B-3. 인간 이종이식 종양 모델:
면역결핍 마우스에서의 인간 종양 이종이식 모델을 사용하여 물질을 평가하였다. 이러한 목적으로, 종양 세포를 시험관내 배양하고 피하로 이식하거나, 종양 이종이식 단편을 추가로 피하 이식하였다. 종양이 확립된 후에 동물을 경구, 피하 또는 복강내 요법으로 처리하였다. 시험 물질의 활성을 단일요법 및 다른 약리학상 활성 물질과의 조합 요법으로 분석하였다. 또한, 진행된 크기 (대략 100 ㎟)의 종양에 대한 시험 물질의 종양 억제 효능을 특성화하였다. 동물의 건강 상태를 매일 확인하고, 동물 보호 규정에 따라 처리를 수행하였다. 종양 영역을 슬라이드 게이지 (길이 L, 폭 B = 더 짧은 치수)로 측정하였다. 종양 용적을 식 (L x B2)/2로부터 계산하였다. 연구의 말미에 종양 성장의 억제를 종양 영역 또는 종양 중량의 T/C 비율로서 및 TGI 값으로서 측정하였다 (종양 성장 억제, 식 [1-(T/C)] x 100으로부터 계산) (T = 처리군에서의 종양 크기; C = 비처리 대조군에서의 종양 크기).
처리 및 비처리 종양-보유 마우스에 대해 컴퓨터 미세단층촬영 및 초음파 극소부분연구(microstudy)를 활용하여, 종양 혈관 구조 및 종양 내 혈류에 대한 시험 물질의 영향을 확인하였다.
C. 약동학적 파라미터의 결정
정맥내 또는 경구 투여 후 본 발명의 화합물의 약동학적 파라미터를 다음과 같이 결정할 수 있었다:
조사할 물질을 동물 (예를 들어, 마우스 또는 래트)에게 용액 (예를 들어, 적은 첨가량의 DMSO를 함유하는 상응하는 혈장 중, 또는 PEG/에탄올/물 혼합물 중)으로서 정맥내로 투여하였고, 경구 투여는 용액 (예를 들어, 솔루톨/에탄올/물 또는 PEG/에탄올/물 혼합물 중)으로서 또는 현탁액 (예를 들어, 틸로스 중)으로서, 각 경우 위관 영양법을 통해 수행하였다. 물질의 투여 후, 혈액을 지정된 시점에 동물로부터 채취하였다. 혈액을 헤파린처리한 다음, 원심분리에 의해 그로부터 혈장을 수득하였다. LC-MS/MS를 통해 혈장에서의 물질을 분석적으로 정량화하였다. 이러한 방식으로 결정된 혈장 농도/시간 플롯으로부터, 약동학적 파라미터, 예컨대 AUC (농도/시간 곡선 이하의 면적), Cmax (최대 혈장 농도), t1 /2 (반감기), VSS (분포 용적) 및 CL (클리어런스), 및 절대적 및 상대적 생체이용률 F 및 Frel (정맥내/경구 비교, 또는 경구 투여 후 현탁액 대 용액 비교)을 내부 표준물을 사용하고 인증된 컴퓨터 프로그램을 활용하여 계산하였다.
전구약물 화합물로부터의 활성 성분 방출을 결정하기 위하여, 전구약물을 상기 기재된 바와 같이 정맥내 또는 경구 투여하고, 전구약물 및 방출된 활성 성분 둘 다의 농도를 처리된 혈장 중에서 정량화하였다.
C-1. 래트에서 정맥내 투여후 약동학적 파라미터:
조사할 물질을 래트에게 각 경우 2% 이하의 DMSO를 함유하는 혈장 중 용액으로서 0.3 내지 1.0 mg/kg의 양으로 정맥내 투여하였다. 실시예 1 및 14에 대해 결정된 동역학적 파라미터를 본 발명의 화합물에 대해 예로서 하기에 나타내었다 [CL혈장 = 혈장 클리어런스]:

WO 2008/141731-A2의 실시예 1로서 기재되어 있고, 본 발명의 화합물과 달리, 벤질 헤드 기 상에 헤테로시클릴 치환기를 갖지 않는 화합물 5-[5-메틸-1-(4-메틸벤질)-1H-피라졸-3-일]-3-[4-(트리플루오로메톡시)페닐]-1,2,4-옥사디아졸은 래트에서 정맥내 투여 후 상기 파라미터에 대해 하기 데이터를 나타내었다: t1 /2 = 30 h, CL혈장 = 0.4 l/h/kg, VSS = 6.9 l/kg.
C-2. 래트에서 경구 투여후 약동학적 파라미터:
조사할 물질을 래트에게 각 경우 솔루톨/에탄올/물 (40:10:50 또는 40:20:40)의 용액으로서 1 내지 3 mg/kg의 양으로 경구 투여하였다. 실시예 1에 대해 결정된 동역학적 파라미터를 본 발명의 화합물에 대해 예로서 하기에 나타내었다 [AUC정상 = 용량-표준화 노출부 (농도/시간 곡선 이하의 면적)]:

상기 표에서 제2열은 실시예 15로부터의 전구약물 화합물 (물 중 틸로스 현탁액으로서 1 mg/kg의 결정질 물질)의 경구 투여 후 수득된 바와 같은, 실시예 1에 대한 약동학적 파라미터를 제시한다. 전구약물 화합물 자체는 래트의 혈장에서 조사되는 시점 어느 때에서도 검출되지 않았다.
WO 2008/141731-A2의 실시예 1로서 기재되어 있고, 본 발명의 화합물과 달리, 벤질 헤드 기 상에 헤테로시클릴 치환기를 갖지 않는 화합물 5-[5-메틸-1-(4-메틸벤질)-1H-피라졸-3-일]-3-[4-(트리플루오로메톡시)페닐]-1,2,4-옥사디아졸은 래트에서 경구 투여 후 상기 파라미터에 대해 하기 데이터를 나타내었다: t1 /2 = 29 h, AUC정상 = 1.9 kg·h/l, F = 74%.
D.
안정성 특성의 결정
비특이적 가수분해에 대한 및 혈장 내에서의 본 발명의 전구약물 화합물의 안정성을 하기 기재된 시험으로 결정하였다:
D-1. pH -의존성 가수분해 안정성의 결정:
0.3 mg의 시험 물질 (전구약물)을 2 ml HPLC 바이알 내로 칭량하여 넣고, 0.6 ml의 아세토니트릴 또는 아세토니트릴/DMSO 혼합물 (20 부피% 이하의 DMSO 함유)을 가하였다. 물질을 용해시키기 위하여, 샘플 혈관을 초음파 조에 대략 10초 동안 위치시켰다. 후속적으로, 1.0 ml의 특정 수성 완충 용액을 가하고 (pH 2, 4, 6.5, 8 및 10의 상업적으로 입수가능한 완충 용액) 샘플을 초음파 조에서 다시 처리하였다. 37℃에서 24시간의 기간에 걸쳐, 5 μl의 샘플 용액을 가수분해에 의해 그로부터 방출된 미변화 전구약물 또는 활성 성분의 함량에 대해 HPLC에 의해 매시간 분석하였다. 상응하는 HPLC 피크의 면적 백분율을 통해 정량화를 수행하였다.
실시예 15에 대해 pH 6.5에서의 안정성 값을 하기에 기재하였다:

pH의 함수로서, 실시예 15에 대해 12 h 후 하기 값을 수득하였다:

D-2.
시험관내
혈장 안정성의 결정:
1 mg의 시험 물질 (전구약물)을 2 ml HPLC 바이알 내로 칭량하여 넣고, 1.5 ml의 DMSO 및 1 ml의 물을 가하였다. 물질을 용해시키기 위하여, 샘플 혈관을 초음파 조에 대략 10초 동안 위치시켰다. 37℃에서 0.5 ml의 래트 혈장 또는 인간 혈장을 0.5 ml의 상기 용액에 가하였다. 샘플을 교반하고 대략 10 μl를 제1 분석 (시간 t0) 용으로 취하였다. 인큐베이션 개시 후 2시간 이하의 기간 이내에, 4 내지 6개의 추가의 분취물을 정량화용으로 취하였다. 샘플을 37℃에서 시험 기간에 걸쳐 유지시켰다. 특성화 및 정량화를 HPLC에 의해 수행하였다.
실시예 15에 대한 래트 혈장에서의 안정화 값을 하기에 기재하였다:

인간 혈장에서, 실시예 15에 대해 하기 값을 수득하였다:

E.
제약 조성물에 대한
실시예
본 발명의 화합물은 다음과 같이 제약 제제로 전환시킬 수 있다:
정제:
조성:
100 mg의 본 발명의 화합물, 50 mg의 락토스 (일수화물), 50 mg의 옥수수 전분 (천연), 10 mg의 폴리비닐피롤리돈 (PVP 25) (바스프(BASF), 독일 루드비스하펜) 및 2 mg의 스테아르산마그네슘.
정제 중량 212 mg, 직경 8 mm, 곡률 반경 12 mm.
제조:
본 발명의 화합물, 락토스 및 전분의 혼합물을 물 중 5% (w/w) PVP 용액과 함께 과립화하였다. 건조 후, 과립을 스테아르산마그네슘과 5분 동안 혼합하였다. 이 혼합물을 통상적인 정제화 프레스를 사용하여 압착시켰다 (정제의 포맷에 대해서는 상기를 참조). 15 kN의 압착력이 압착에 대한 지침 값으로 사용되었다.
경구 투여용 현탁액:
조성:
1,000 mg의 본 발명의 화합물, 1,000 mg의 에탄올 (96%), 400 mg의 로디겔 (Rhodigel)® (FMC로부터의 크산탄 검, 미국 펜실베이니아주) 및 99 g의 물.
100 mg의 본 발명의 화합물의 단일 용량은 10 ml의 경구용 현탁액에 상응한다.
제조:
로디겔을 에탄올에 현탁시키고 본 발명의 화합물을 현탁액에 가하였다. 교반하면서 물을 가하였다. 로디겔의 팽윤이 종료될 때까지 혼합물을 대략 6 h 동안 교반하였다.
경구 투여용 용액:
조성:
500 mg의 본 발명의 화합물, 2.5 g의 폴리소르베이트 및 97 g의 폴리에틸렌 글리콜 400. 100 mg의 본 발명의 화합물의 단일 용량은 20 g의 경구용 용액에 상응한다.
제조:
본 발명의 화합물을 폴리에틸렌 글리콜과 폴리소르베이트의 혼합물에 교반하면서 현탁화하였다. 본 발명의 화합물의 용해가 완료될 때까지 교반 조작을 계속하였다.
정맥내
용액:
본 발명의 화합물을 생리학상 허용되는 용매 (예를 들어, 등장성 염수, 글루코스 용액 5% 및/또는 PEG 400 용액 30%) 중 포화 용해도 미만의 농도로 용해시켰다. 용액을 멸균 여과하고 멸균 및 발열원-비함유 주사 용기 내로 나누어 주입하였다.
F.
문헌



Claims (19)
- 화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물.
<화학식 I>

상기 식에서,
m은 1 또는 2이고,
n은 1, 2 또는 3이고,
R1은 히드록실 또는 시아노이고,
R2는 트리플루오로메톡시, 트리플루오로메틸술파닐, 트리플루오로메틸술포닐, 펜타플루오로술파닐 또는 화학식의 기이고, 여기서
*는 페닐 고리에 대한 결합 부위를 나타내고,
R3A 및 R3B는 각각 독립적으로 플루오린 또는 메틸이거나, 또는
서로 결합하여, 이들이 결합된 탄소 원자와 함께, 시클로프로판-1,1-디일, 시클로부탄-1,1-디일, 시클로펜탄-1,1-디일, 시클로헥산-1,1-디일, 옥세탄-3,3-디일 또는 테트라히드로-2H-피란-4,4-디일 고리를 형성하고,
R4는 수소, 플루오린, 메틸, 트리플루오로메틸 또는 메톡시이다. - 제1항에 있어서,
m 및 n이 각각 독립적으로 1 또는 2이고,
R1이 히드록실 또는 시아노이고,
R2가 트리플루오로메틸, 트리플루오로메톡시, 트리플루오로메틸술파닐 또는 화학식의 기이고, 여기서
*가 페닐 고리에 대한 결합 부위를 나타내고,
R3A 및 R3B가 둘 다 메틸이거나, 또는 서로 결합하여, 이들이 결합된 탄소 원자와 함께, 시클로프로판-1,1-디일, 시클로부탄-1,1-디일, 옥세탄-3,3-디일 또는 테트라히드로-2H-피란-4,4-디일 고리를 형성하고,
R4가 수소, 플루오린, 메틸 또는 트리플루오로메틸인
화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물. - 제1항 또는 제2항에 있어서,
m 및 n이 둘 다 1 또는 2이고,
R1이 히드록실 또는 시아노이고,
R2가 트리플루오로메틸 또는 화학식의 기이고, 여기서 *가 페닐 고리에 대한 결합 부위를 나타내는 것인
화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물. - 제1항 내지 제3항 중 어느 한 항에 있어서,
R1이 히드록실이고,
m, n 및 R2 각각이 제1항 내지 제3항 중 어느 한 항에 정의된 바와 같은 것인
화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물. - 화학식 I-PD의 화합물, 및 그의 염, 용매화물 및 염의 용매화물.
<화학식 I-PD>
상기 식에서,
m, n 및 R2 각각은 제1항 내지 제4항 중 어느 한 항에 정의된 바와 같고,
RPD는 화학식의 전구약물 기이고, 여기서
#는 산소 원자에 대한 결합 부위를 나타내고,
R5는 수소 또는 (C1-C4)-알킬이고,
R6A 및 R6B는 각각 독립적으로 수소 또는 메틸이다. - 제5항에 있어서,
RPD가 화학식의 전구약물 기이고, 여기서 #가 산소 원자에 대한 결합 부위를 나타내는 것인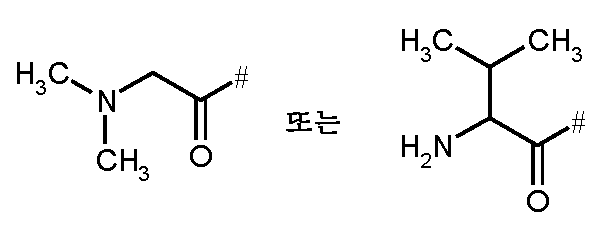
화학식 I-PD의 화합물, 및 그의 염, 용매화물 및 염의 용매화물. - 하기 화학식 II의 화합물을 적합한 팔라듐 촉매 및 염기의 존재하에 하기 화학식 III의 화합물과 커플링시키고, 생성된 화학식 I의 화합물을 임의로 그의 거울상이성질체 및/또는 부분입체이성질체로 분리시키고/시키거나 적절한 (i) 용매 및/또는 (ii) 산을 사용하여 그의 용매화물, 염 및/또는 염의 용매화물로 전환시키는 것을 특징으로 하는, 제1항 내지 제4항 중 어느 한 항에 따른 화학식 I의 화합물의 제조 방법.
<화학식 II>
상기 식에서,
R2는 제1항 내지 제4항 중 어느 한 항에 정의된 바와 같고
X는 브로민 또는 아이오딘이다.
<화학식 III>

상기 식에서,
m, n 및 R1 각각은 제1항 내지 제4항 중 어느 한 항에 정의된 바와 같다. - 하기 화학식 I-A의 화합물을 하기 화학식 VIII의 화합물 또는 상기 화합물의 활성화 형태와 함께 통상적인 방법에 의해 에스테르화하고, 생성된 화학식 I-PD의 화합물을 임의로 그의 거울상이성질체 및/또는 부분입체이성질체로 분리시키고/시키거나 적절한 (i) 용매 및/또는 (ii) 산을 사용하여 그의 용매화물, 염 및/또는 염의 용매화물로 전환시키는 것을 특징으로 하는, 제5항 또는 제6항에 따른 화학식 I-PD의 화합물의 제조 방법.
<화학식 I-A>
상기 식에서,
m, n 및 R2 각각은 제1항 내지 제4항 중 어느 한 항에 정의된 바와 같다.
<화학식 VIII>

상기 식에서,
RPD는 제5항 또는 제6항에 정의된 바와 같다. - 제1항 내지 제6항 중 어느 한 항에 있어서, 질환의 치료 및/또는 예방을 위한 화합물.
- 제1항 내지 제6항 중 어느 한 항에 있어서, 암 또는 종양의 치료 및/또는 예방 방법에 사용하기 위한 화합물.
- 제1항 내지 제6항 중 어느 한 항에 있어서, 허혈성 심혈관 질환, 심부전, 심근경색, 부정맥, 졸중, 폐고혈압, 신장 및 폐의 섬유성 질환, 건선, 당뇨병성 망막병증, 황반 변성, 류마티스 관절염 및 추바시(Chuvash) 다혈구증의 치료 및/또는 예방 방법에 사용하기 위한 화합물.
- 암 또는 종양의 치료 및/또는 예방용 의약의 제조를 위한 제1항 내지 제6항 중 어느 한 항에 따른 화합물의 용도.
- 허혈성 심혈관 질환, 심부전, 심근경색, 부정맥, 졸중, 폐고혈압, 신장 및 폐의 섬유성 질환, 건선, 당뇨병성 망막병증, 황반 변성, 류마티스 관절염 및 추바시 다혈구증의 치료 및/또는 예방용 의약의 제조를 위한 제1항 내지 제6항 중 어느 한 항에 따른 화합물의 용도.
- 제1항 내지 제6항 중 어느 한 항에 따른 화합물을 1종 이상의 불활성, 비독성의 제약상 적합한 부형제와 조합하여 포함하는 의약.
- 제1항 내지 제6항 중 어느 한 항에 따른 화합물을 1종 이상의 추가의 활성 성분과 조합하여 포함하는 의약.
- 제14항 또는 제15항에 있어서, 암 또는 종양의 치료 및/또는 예방을 위한 의약.
- 제14항 또는 제15항에 있어서, 허혈성 심혈관 질환, 심부전, 심근경색, 부정맥, 졸중, 폐고혈압, 신장 및 폐의 섬유성 질환, 건선, 당뇨병성 망막병증, 황반 변성, 류마티스 관절염 및 추바시 다혈구증의 치료 및/또는 예방을 위한 의약.
- 유효량의 하나 이상의 제1항 내지 제6항 중 어느 한 항에 따른 화합물, 또는 제14항 내지 제16항 중 어느 한 항에 따른 의약을 사용하는, 인간 및 동물의 암 또는 종양의 치료 및/또는 예방 방법.
- 활성량의 하나 이상의 제1항 내지 제6항 중 어느 한 항에 따른 화합물, 또는 제14항, 제15항 및 제17항 중 어느 한 항에 따른 의약을 사용하는, 인간 및 동물의 허혈성 심혈관 질환, 심부전, 심근경색, 부정맥, 졸중, 폐고혈압, 신장 및 폐의 섬유성 질환, 건선, 당뇨병성 망막병증, 황반 변성, 류마티스 관절염 및 추바시 다혈구증의 치료 및/또는 예방 방법.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10004855 | 2010-05-08 | ||
| EP10004855.2 | 2010-05-08 | ||
| PCT/EP2011/057021 WO2011141326A1 (de) | 2010-05-08 | 2011-05-03 | Substituierte heterocyclylbenzyl-pyrazole und ihre verwendung |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20130108997A true KR20130108997A (ko) | 2013-10-07 |
Family
ID=44063276
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020127032069A KR20130108997A (ko) | 2010-05-08 | 2011-05-03 | 치환된 헤테로시클릴 벤질 피라졸 및 그의 용도 |
Country Status (25)
| Country | Link |
|---|---|
| US (2) | US20130172311A1 (ko) |
| EP (1) | EP2569312B1 (ko) |
| JP (1) | JP5727002B2 (ko) |
| KR (1) | KR20130108997A (ko) |
| CN (1) | CN103003269B (ko) |
| AR (1) | AR081368A1 (ko) |
| AU (1) | AU2011252223A1 (ko) |
| BR (1) | BR112012028651A2 (ko) |
| CA (1) | CA2798375A1 (ko) |
| CO (1) | CO6640218A2 (ko) |
| CR (1) | CR20120570A (ko) |
| CU (1) | CU20120157A7 (ko) |
| DO (1) | DOP2012000284A (ko) |
| EA (1) | EA201291167A1 (ko) |
| EC (1) | ECSP12012288A (ko) |
| ES (1) | ES2462522T3 (ko) |
| IL (1) | IL222851A0 (ko) |
| MA (1) | MA34939B1 (ko) |
| MX (1) | MX2012012960A (ko) |
| PE (1) | PE20130228A1 (ko) |
| SG (1) | SG185459A1 (ko) |
| TN (1) | TN2012000531A1 (ko) |
| TW (1) | TW201200510A (ko) |
| UY (1) | UY33369A (ko) |
| WO (1) | WO2011141326A1 (ko) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5603232B2 (ja) * | 2007-05-01 | 2014-10-08 | ヒルズ・ペット・ニュートリシャン・インコーポレーテッド | ネコ類において骨関節炎を診断するための方法および組成物 |
| US8796253B2 (en) * | 2007-05-18 | 2014-08-05 | Bayer Intellectual Property Gmbh | Heteroaryl substituted pyrazole derivatives useful for treating hyper-proliferative disorders and diseases associated with angiogenesis |
| JP5727002B2 (ja) * | 2010-05-08 | 2015-06-03 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツングBayer Intellectual Property GmbH | 置換ヘテロサイクリルベンジルピラゾール類およびその使用 |
| US9040711B2 (en) | 2012-07-02 | 2015-05-26 | Monsanto Technology Llc | Processes for the preparation of 3,5-disubstituted-1,2,4-oxadiazoles |
| JP6453218B2 (ja) | 2012-08-24 | 2019-01-16 | ボード・オブ・リージエンツ,ザ・ユニバーシテイ・オブ・テキサス・システム | 疾患を治療するためのhif活性の複素環式修飾物質 |
| US9115120B2 (en) | 2012-08-24 | 2015-08-25 | Board Of Regents, The University Of Texas Systems | Heterocyclic modulators of HIF activity for treatment of disease |
| WO2014031928A2 (en) * | 2012-08-24 | 2014-02-27 | Philip Jones | Heterocyclic modulators of hif activity for treatment of disease |
| US9663504B2 (en) | 2014-02-25 | 2017-05-30 | Board Of Regents, The University Of Texas System | Salts of heterocyclic modulators of HIF activity for treatment of disease |
| US10191597B2 (en) | 2015-06-30 | 2019-01-29 | Synaptics Incorporated | Modulating a reference voltage to preform capacitive sensing |
| CN110167916B (zh) * | 2017-01-13 | 2022-11-11 | 日产化学株式会社 | 芳香族二胺化合物前体的制造方法 |
| CN115944626B (zh) * | 2022-12-08 | 2024-04-19 | 桂林医学院附属医院 | 小分子化合物在制备抗缺氧或缺血再灌注损伤药物中的应用 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6610726B2 (en) * | 2001-06-29 | 2003-08-26 | North Shore-Long Island Jewish Research Institute | Compositions and agents for modulating cellular proliferation and angiogenesis |
| CN100387594C (zh) | 2003-04-03 | 2008-05-14 | 麦克公司 | 二芳基取代的吡唑类代谢型谷氨酸受体-5调节剂 |
| JP4499721B2 (ja) | 2003-06-30 | 2010-07-07 | ヒフ バイオ,インク. | 化合物、組成物および方法 |
| CN1842332A (zh) * | 2003-06-30 | 2006-10-04 | Hif生物公司 | 化合物、组合物及方法 |
| GB0508472D0 (en) * | 2005-04-26 | 2005-06-01 | Glaxo Group Ltd | Compounds |
| WO2007065010A2 (en) | 2005-12-02 | 2007-06-07 | Hif Bio, Inc. | Anti-angiogenesis compounds |
| US8796253B2 (en) * | 2007-05-18 | 2014-08-05 | Bayer Intellectual Property Gmbh | Heteroaryl substituted pyrazole derivatives useful for treating hyper-proliferative disorders and diseases associated with angiogenesis |
| BRPI0921257A2 (pt) * | 2008-11-14 | 2016-02-23 | Bayer Schering Pharma Ag | composto de arila heterociclicamente subsittuidos como inibidores hif |
| DE102008057344A1 (de) * | 2008-11-14 | 2010-05-20 | Bayer Schering Pharma Aktiengesellschaft | Aminoalkyl-substituierte Aryl-Verbindungen und ihre Verwendung |
| WO2010054764A1 (de) * | 2008-11-14 | 2010-05-20 | Bayer Schering Pharma Aktiengesellschaft | Heteroaromatische verbindungen zur verwendung als hif-inhibitoren |
| EP2202232A1 (en) | 2008-12-26 | 2010-06-30 | Laboratorios Almirall, S.A. | 1,2,4-oxadiazole derivatives and their therapeutic use |
| ES2405054T3 (es) | 2009-01-23 | 2013-05-30 | Bristol-Myers Squibb Company | Derivados de pirazol-1,2,4-oxadiazol como agonistas de esfingosina-1-fosfato |
| JP5727002B2 (ja) * | 2010-05-08 | 2015-06-03 | バイエル・インテレクチュアル・プロパティ・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツングBayer Intellectual Property GmbH | 置換ヘテロサイクリルベンジルピラゾール類およびその使用 |
-
2011
- 2011-05-03 JP JP2013509505A patent/JP5727002B2/ja not_active Expired - Fee Related
- 2011-05-03 MA MA35350A patent/MA34939B1/fr unknown
- 2011-05-03 ES ES11716595.1T patent/ES2462522T3/es active Active
- 2011-05-03 AU AU2011252223A patent/AU2011252223A1/en not_active Abandoned
- 2011-05-03 US US13/696,878 patent/US20130172311A1/en not_active Abandoned
- 2011-05-03 BR BR112012028651A patent/BR112012028651A2/pt not_active IP Right Cessation
- 2011-05-03 KR KR1020127032069A patent/KR20130108997A/ko not_active Application Discontinuation
- 2011-05-03 PE PE2012002121A patent/PE20130228A1/es not_active Application Discontinuation
- 2011-05-03 EA EA201291167A patent/EA201291167A1/ru unknown
- 2011-05-03 SG SG2012082145A patent/SG185459A1/en unknown
- 2011-05-03 MX MX2012012960A patent/MX2012012960A/es active IP Right Grant
- 2011-05-03 WO PCT/EP2011/057021 patent/WO2011141326A1/de active Application Filing
- 2011-05-03 CN CN201180033637.5A patent/CN103003269B/zh not_active Expired - Fee Related
- 2011-05-03 CA CA2798375A patent/CA2798375A1/en not_active Abandoned
- 2011-05-03 EP EP11716595.1A patent/EP2569312B1/de active Active
- 2011-05-04 AR ARP110101533A patent/AR081368A1/es not_active Application Discontinuation
- 2011-05-04 UY UY0001033369A patent/UY33369A/es not_active Application Discontinuation
- 2011-05-06 TW TW100115869A patent/TW201200510A/zh unknown
- 2011-05-09 US US13/103,305 patent/US8470811B2/en not_active Expired - Fee Related
-
2012
- 2012-11-05 IL IL222851A patent/IL222851A0/en unknown
- 2012-11-07 DO DO2012000284A patent/DOP2012000284A/es unknown
- 2012-11-07 TN TNP2012000531A patent/TN2012000531A1/en unknown
- 2012-11-07 CU CU2012000157A patent/CU20120157A7/es unknown
- 2012-11-08 CO CO12201711A patent/CO6640218A2/es not_active Application Discontinuation
- 2012-11-08 EC ECSP12012288 patent/ECSP12012288A/es unknown
- 2012-11-08 CR CR20120570A patent/CR20120570A/es unknown
Also Published As
| Publication number | Publication date |
|---|---|
| UY33369A (es) | 2011-12-01 |
| CU20120157A7 (es) | 2013-03-27 |
| PE20130228A1 (es) | 2013-03-15 |
| CR20120570A (es) | 2013-04-22 |
| AR081368A1 (es) | 2012-08-29 |
| US20120028950A1 (en) | 2012-02-02 |
| MX2012012960A (es) | 2012-12-17 |
| MA34939B1 (fr) | 2014-03-01 |
| CN103003269A (zh) | 2013-03-27 |
| JP5727002B2 (ja) | 2015-06-03 |
| US8470811B2 (en) | 2013-06-25 |
| CA2798375A1 (en) | 2011-11-17 |
| EP2569312A1 (de) | 2013-03-20 |
| DOP2012000284A (es) | 2013-03-31 |
| ECSP12012288A (es) | 2012-12-28 |
| TN2012000531A1 (en) | 2014-04-01 |
| TW201200510A (en) | 2012-01-01 |
| BR112012028651A2 (pt) | 2016-08-09 |
| EP2569312B1 (de) | 2014-03-26 |
| ES2462522T3 (es) | 2014-05-23 |
| IL222851A0 (en) | 2012-12-31 |
| EA201291167A1 (ru) | 2013-05-30 |
| AU2011252223A1 (en) | 2012-12-06 |
| US20130172311A1 (en) | 2013-07-04 |
| CN103003269B (zh) | 2015-11-25 |
| CO6640218A2 (es) | 2013-03-22 |
| JP2013526497A (ja) | 2013-06-24 |
| SG185459A1 (en) | 2012-12-28 |
| WO2011141326A1 (de) | 2011-11-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR20130108997A (ko) | 치환된 헤테로시클릴 벤질 피라졸 및 그의 용도 | |
| JP5702138B2 (ja) | 過剰増殖障害および血管新生に関連する疾患の処置に有用なヘテロアリール置換ピラゾール誘導体 | |
| JP5829915B2 (ja) | 複素環式置換基を有するアリール化合物およびそれらの使用 | |
| US20140329797A1 (en) | Substituted oxadiazolyl pyridinones and oxadiazolyl pyridazinones as hif inhibitors | |
| US20130150325A1 (en) | 3-(Fluorovinyl)pyrazoles and their use | |
| KR20110082570A (ko) | Hif 억제제로서 사용하기 위한 헤테로방향족 화합물 | |
| AU2010294588A1 (en) | Substituted (heteroarylmethyl) thiohydantoins as anticancer drugs | |
| JP2012508702A (ja) | アミノアルキル置換基を有するアリール化合物およびそれらの使用 | |
| KR20130108998A (ko) | 히드록시알킬 벤질 피라졸, 및 과다증식성 및 혈관신생 질환의 치료를 위한 그의 용도 | |
| DE102008057364A1 (de) | Substituierte Aryl-Verbindungen und ihre Verwendung | |
| TW201311646A (zh) | 3-(氟乙烯基)吡唑類及其用途 | |
| DE102009041241A1 (de) | Substituierte Aryl-Verbindungen und ihre Verwendung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WITN | Application deemed withdrawn, e.g. because no request for examination was filed or no examination fee was paid |