JP7317841B2 - アルギナーゼインヒビタ及びその使用方法 - Google Patents
アルギナーゼインヒビタ及びその使用方法 Download PDFInfo
- Publication number
- JP7317841B2 JP7317841B2 JP2020543307A JP2020543307A JP7317841B2 JP 7317841 B2 JP7317841 B2 JP 7317841B2 JP 2020543307 A JP2020543307 A JP 2020543307A JP 2020543307 A JP2020543307 A JP 2020543307A JP 7317841 B2 JP7317841 B2 JP 7317841B2
- Authority
- JP
- Japan
- Prior art keywords
- tert
- butyl
- pyrrolidine
- mmol
- pharmaceutically acceptable
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
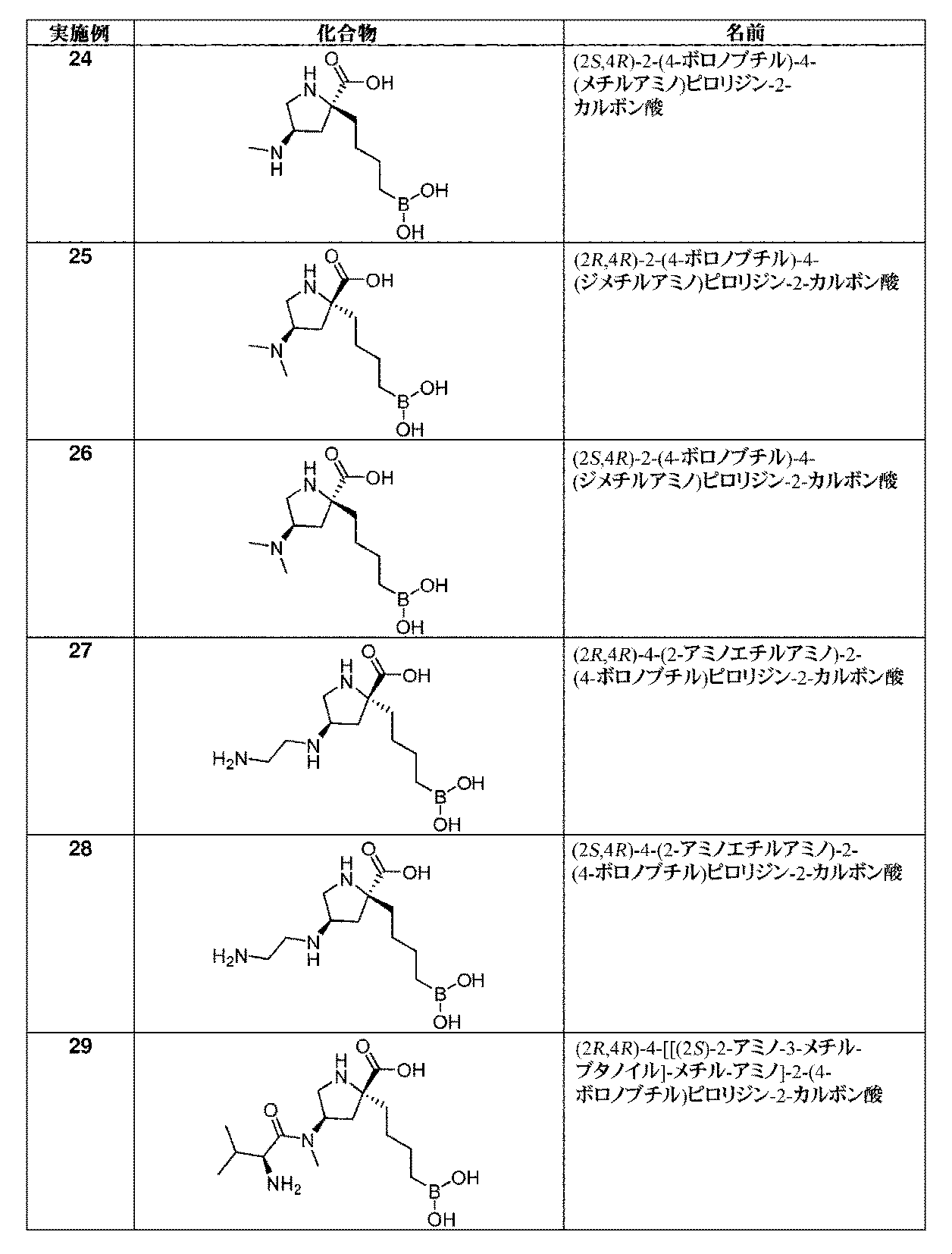
- C07F5/025—Boronic and borinic acid compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
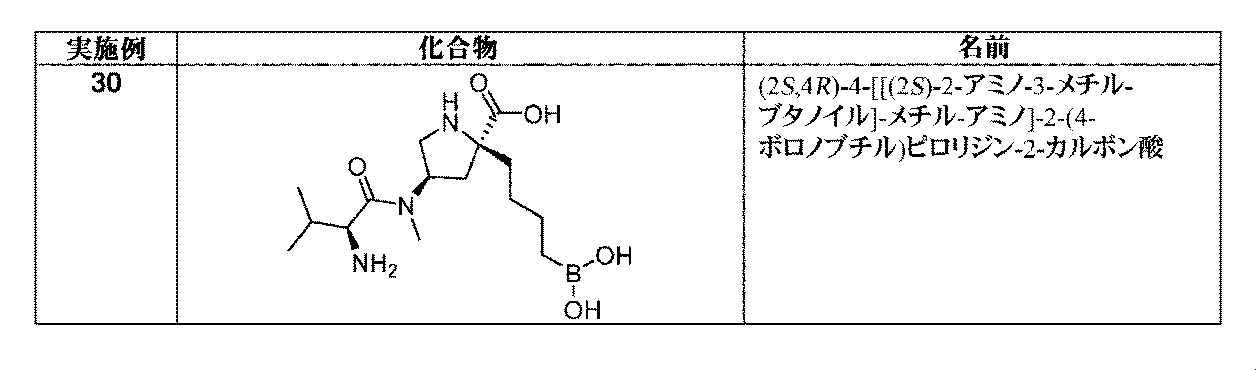
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/69—Boron compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pulmonology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
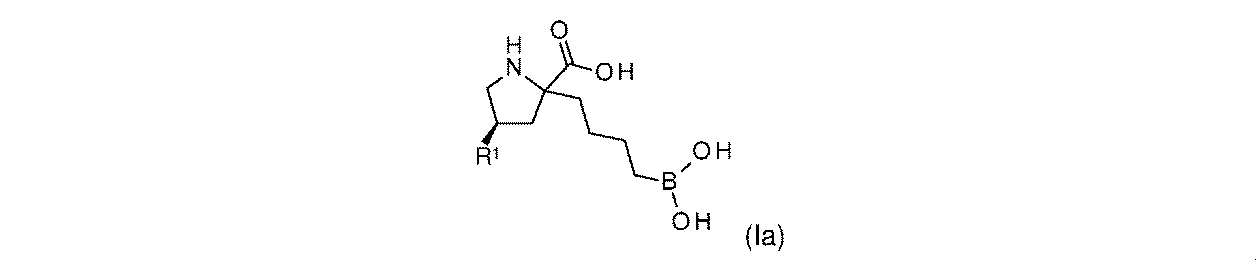
R1は-NHR1aであり;
R1aは、-H又は-C(O)CH(R1b)NH2であり;そして
R1bは、-CH3又は-CH(CH3)2である。

R1は-NHR1aであり;
R1aは、-H又は-C(O)CH(R1b)NHR1cであり;そして
R1bは、-H、-(C1~C4)アルキル、及びCH2OR1dから選択され、そしてR1cは-Hであり;又は
R1b及びR1cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R1dは、H又は-CH3である。

R1は-NHR1aであり;
R1aは、-H又は-C(O)CH(R1b)NHR1cであり;そして
R1bは、-H、-(C1~C4)アルキル、及びCH2OR1dから選択され、そしてR1cは-Hであり;又は
R1b及びR1cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R1dは、H又は-CH3である。

R2は、-OH又は-NHR2aであり;
R2aは、-H又は-C(O)CH(R2b)NH2であり;そして
R2bは、-CH3又は-CH(CH3)2である。

R2は、-OH又は-NHR2aであり;
R2aは、-H又は-C(O)CH(R2b)NHR2cであり;
R2bは、-H、-(C1~C4)アルキル、及びCH2OR2dから選択され、そしてR2cは-Hであり;又は
R2b及びR2cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R2dは、-H又は-CH3である。

R2は、-OH又は-NHR2aであり;
R2aは、-H又は-C(O)CH(R2b)NHR2cであり;
R2bは、-H、-(C1~C4)アルキル、及びCH2OR2dから選択され、そしてR2cは-Hであり;又は
R2b及びR2cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R2dは、-H又は-CH3である。


R3は、-H、-(C1~C4)アルキル、及びCH2OR3aから選択され;そして
R3aは、-H又は-CH3である。

R3は、-H、-(C1~C4)アルキル、及び-CH2OR3aから選択され;そして
R3aは、-H又は-CH3である。

R6aは、-H又は-CH3であり;
R6bは、-C(O)C(R6cR6d)NH2であるか;又は0若しくは1アミノ、若しくは-OR6eで置換されている-(C1~C3)アルキルであり;そして
R6cは、0若しくは1アミノ、又は-OR6fで置換されている-(C1~C3)アルキルであり;
R6dは、H又は-CH3であり;そして
R6e及びR6fは独立して、-H又は-CH3である。
R6aは、-H又は-CH3であり;
R6bは、-C(O)C(R6cR6d)NH2であるか;又は0若しくは1アミノ、若しくは-OR6eで置換されている-(C1~C3)アルキルであり;そして
R6cは、0若しくは1アミノ、又は-OR6fで置換されている-(C1~C3)アルキルであり;
R6dは、H又は-CH3であり;そして
R6e及びR6fは独立して、-H又は-CH3である。
一実施形態において、開示されるのは、式(I)の化合物、又はその薬学的に許容される塩であり:
R1は-NHR1aであり;
R1aは、-H又は-C(O)CH(R1b)NH2であり;そして
R1bは、-CH3又は-CH(CH3)2である。
R1は-NHR1aであり;
R1aは、-H又は-C(O)CH(R1b)NHR1cであり;そして
R1bは、-H、-(C1~C4)アルキル、及びCH2OR1dから選択され;そしてR1cは-Hであり;又は
R1b及びR1cは、これらが結合する原子と合わせて、5員環を形成し;そして
R1dは、H又は-CH3である。
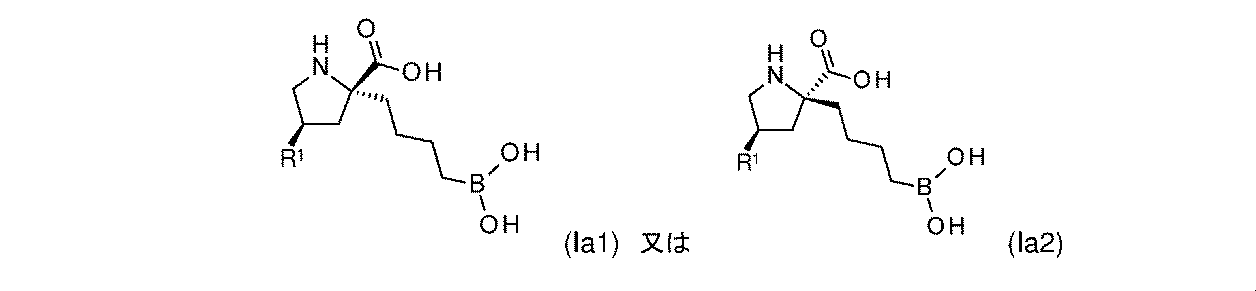
R1は-NHR1aであり;
R1aは、-H又は-C(O)CH(R1b)NHR1cであり;そして
R1bは、-H、-(C1~C4)アルキル、及びCH2OR1dから選択され;そしてR1cは-Hであり;又は
R1b及びR1cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R1dは、H又は-CH3である。

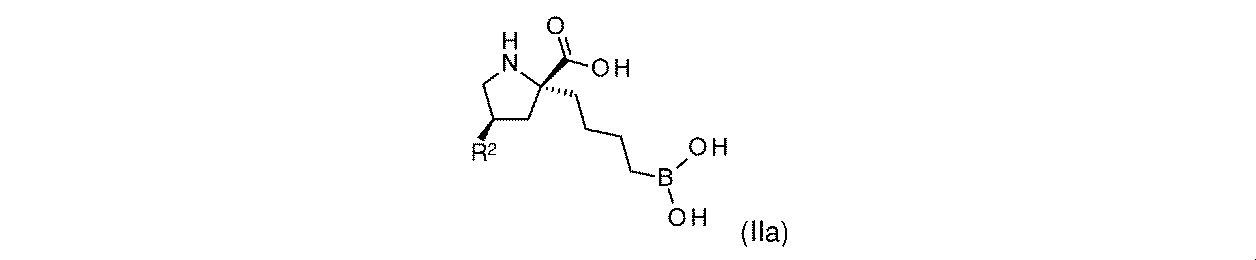
R2は、-OH又は-NHR2aであり;
R2aは、-H又は-C(O)CH(R2b)NH2であり;そして
R2bは、-CH3又は-CH(CH3)2である。
R2は、-OH又は-NHR2aであり;
R2aは、-H又は-C(O)CH(R2b)NHR2cであり;
R2bは、-H、-(C1~C4)アルキル、及びCH2OR2dから選択され;そしてR2cは-Hであり;又は
R2b及びR2cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R2dは、H又は-CH3である。
R2は、-OH又は-NHR2aであり;
R2aは、-H又は-C(O)CH(R2b)NHR2cであり;
R2bは、-H、-(C1~C4)アルキル、及びCH2OR2dから選択され;そしてR2cは-Hであり;又は
R2b及びR2cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R2dは、-H又は-CH3である。
R3は、-H、-(C1~C4)アルキル、及びCH2OR3aから選択され;そして
R3aは、-H又は-CH3である。
R3は、-H、-(C1~C4)アルキル、及び-CH2OR3aから選択され;そして
R3aは、-H又は-CH3である。
R6aは、-H又は-CH3であり;
R6bは、-C(O)C(R6cR6d)NH2であるか;又は0若しくは1アミノ、若しくは-OR6eで置換されている-(C1~C3)アルキルであり;そして
R6cは、0若しくは1アミノ、又は-OR6fで置換されている-(C1~C3)アルキルであり;
R6dは、H又は-CH3であり;そして
R6e及びR6fは独立して、-H又は-CH3である。
R6aは、-H又は-CH3であり;
R6bは、-C(O)C(R6cR6d)NH2であるか;又は0若しくは1アミノ、若しくは-OR6eで置換されている-(C1~C3)アルキルであり;そして
R6cは、0若しくは1アミノ、又は-OR6fで置換されている-(C1~C3)アルキルであり;
R6dは、H又は-CH3であり;そして
R6e及びR6fは独立して、-H又は-CH3である。
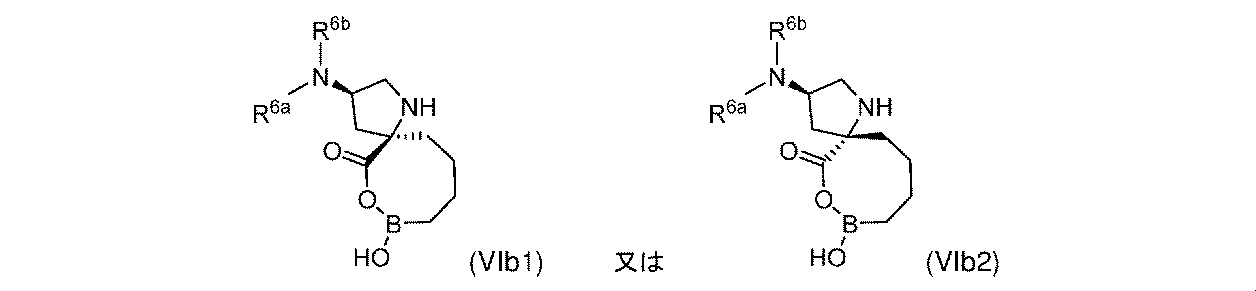
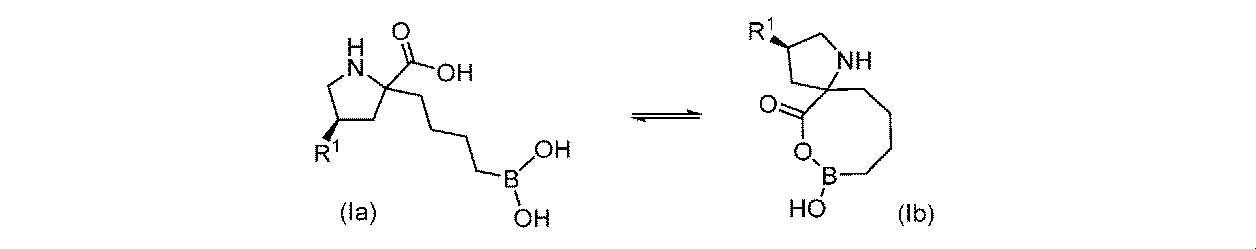

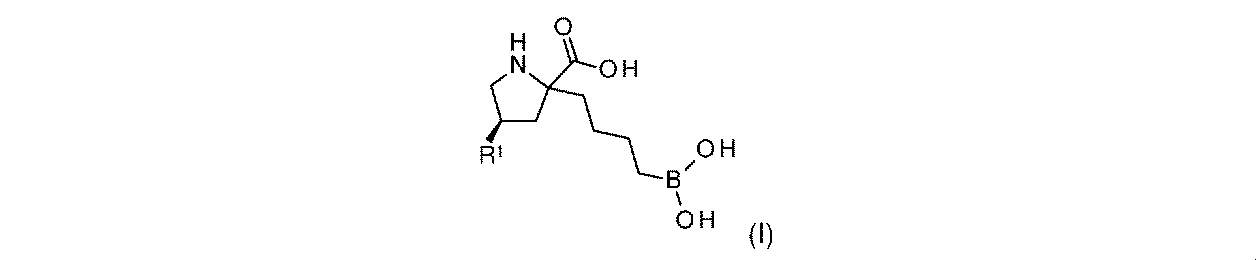
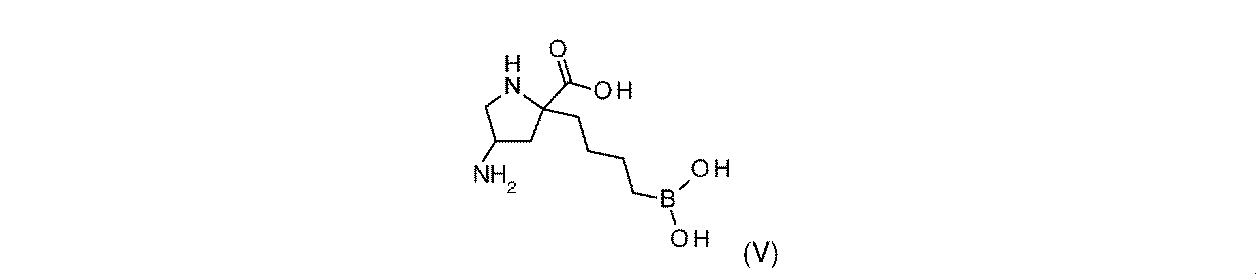
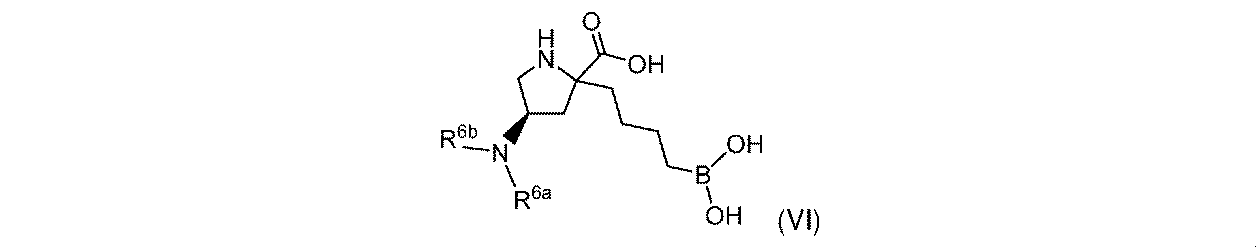
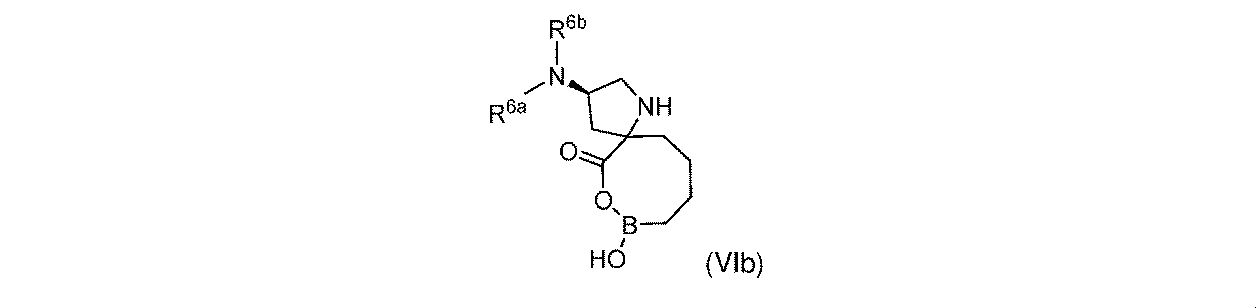
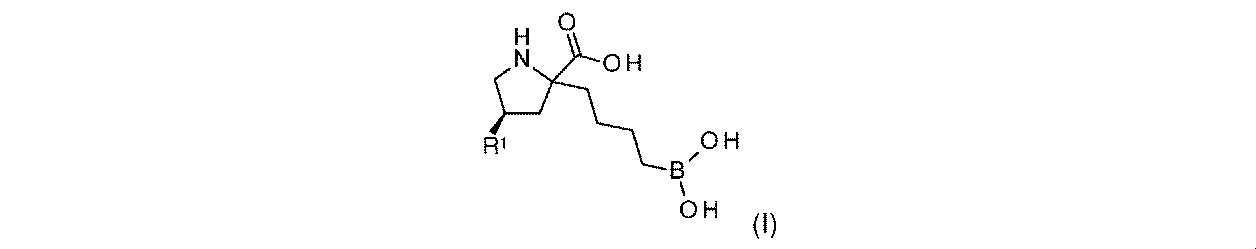
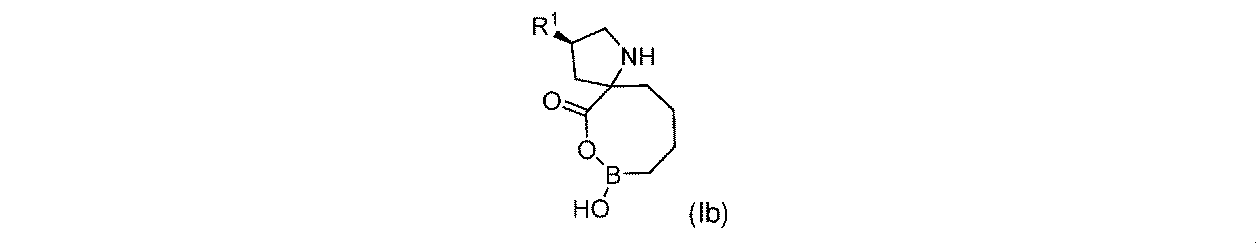
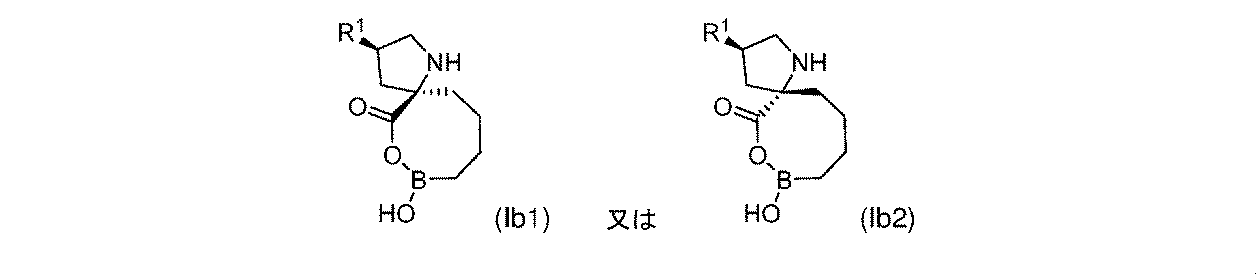
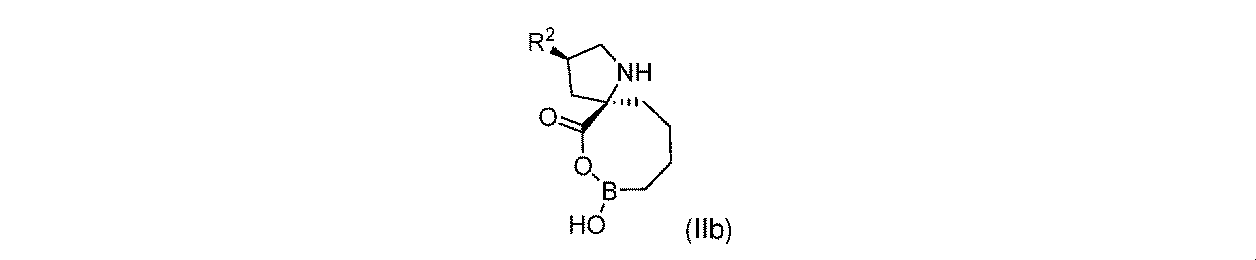
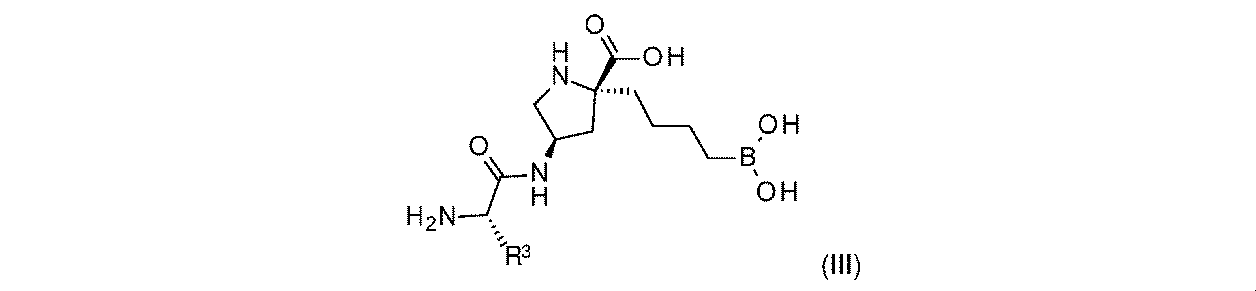
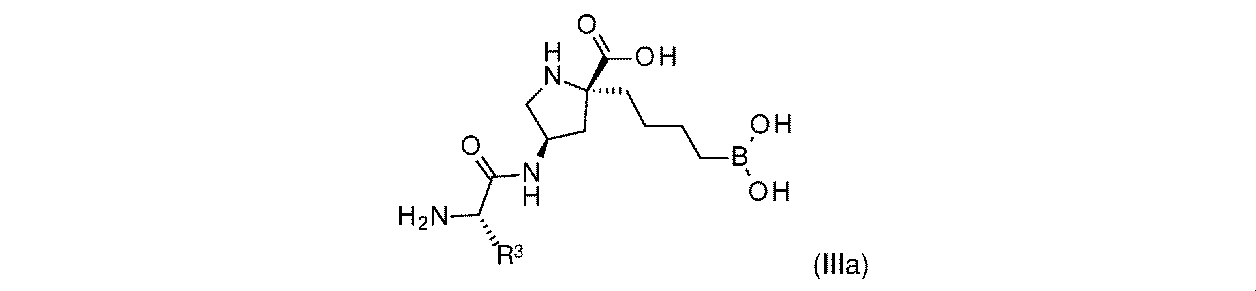
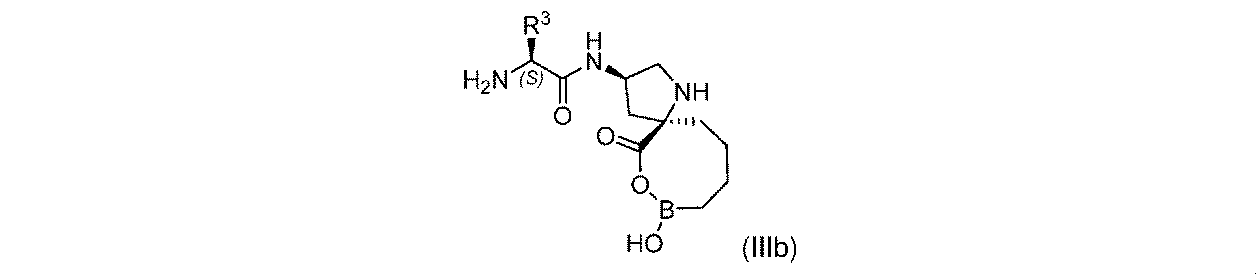
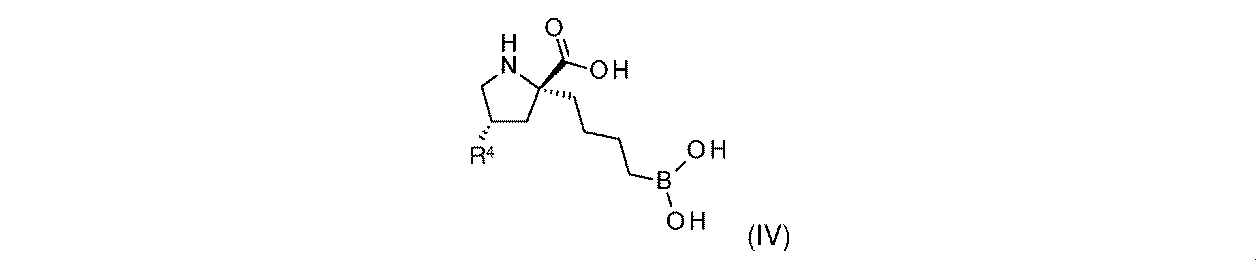
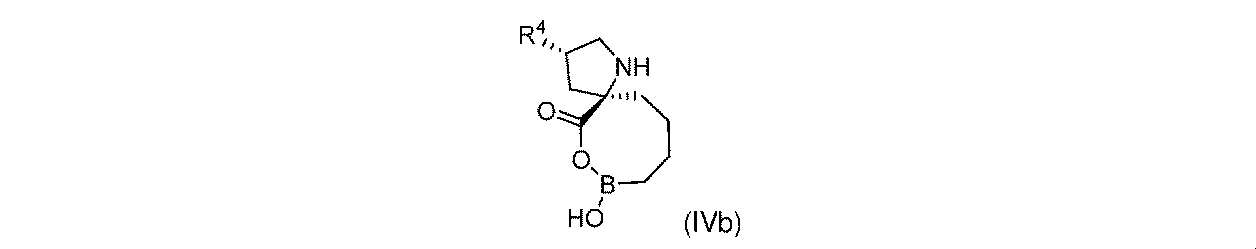
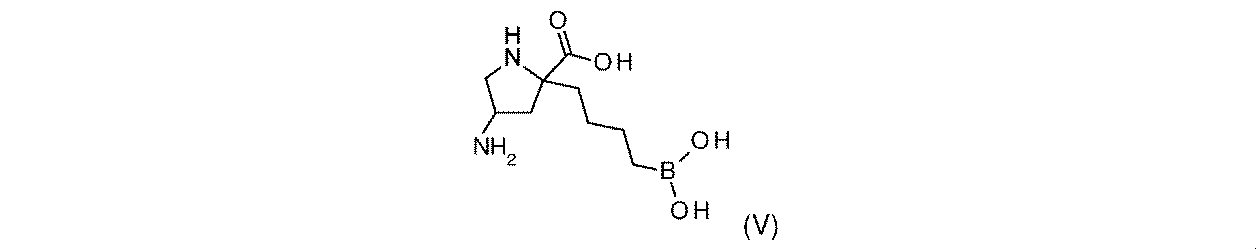
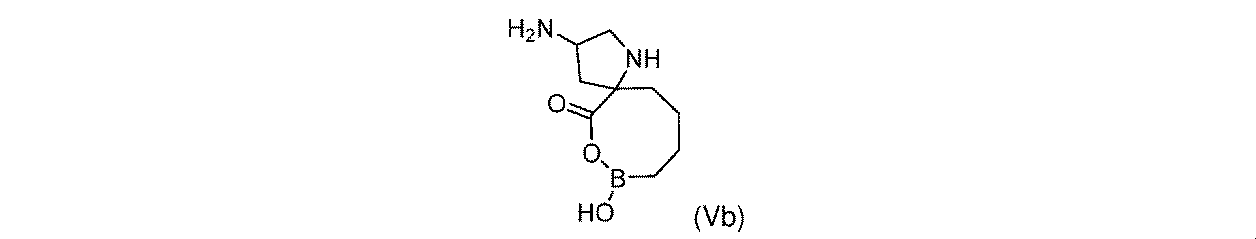
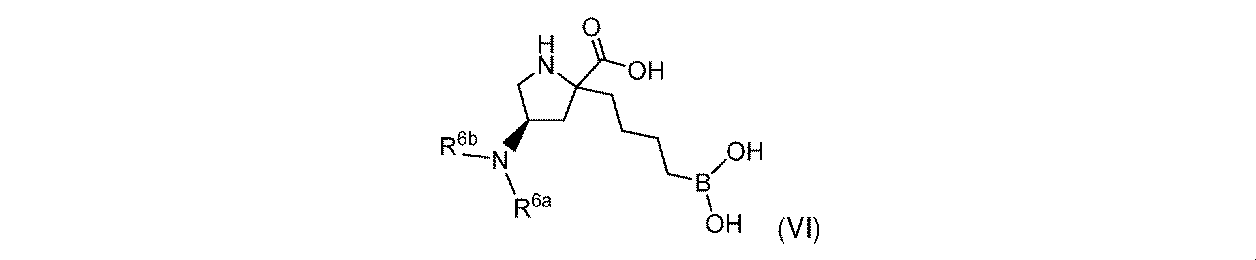
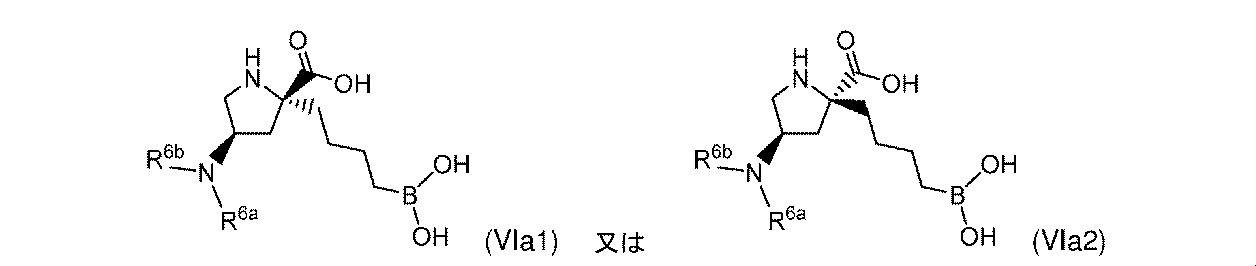
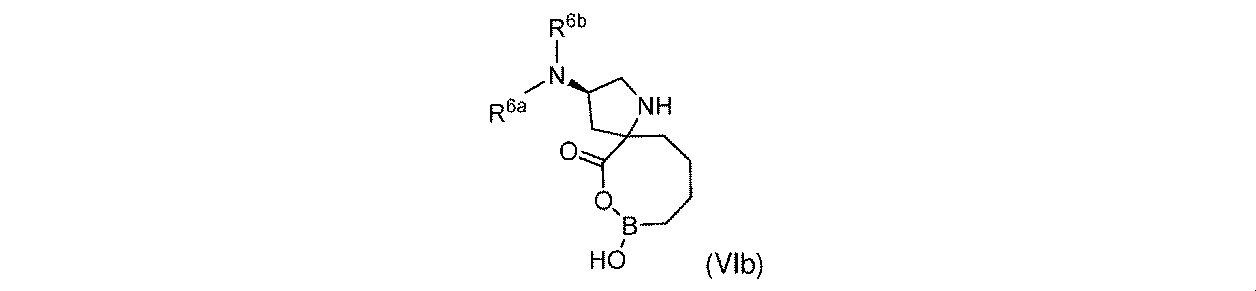
一部の実施形態において、開示されるのは、式(I)、(Ia)、(Ib)、(II)、(IIa)、(IIb)、(III)、(IIIa)、(IIIb)、(IV)、(IVb)、(V)、(Vb)、(VI)、(VIb)(それらのあらゆる亜属又は種を含む)、又は表1の化合物、及び薬学的に許容されるキャリアを含む医薬組成物である。
本化合物は、アルギナーゼインヒビタとして治療に有用である。
(i)全ての合成を、特に明記しない限り、周囲温度にて、すなわち17~25℃の範囲内で、そして不活性ガス、例えば窒素の雰囲気下で実行した;
(ii)蒸発を、ロータリエバポレーションによって、又はGenevac機器若しくはBiotage v10真空エバポレータを利用して実行し、そして仕上げ手順を、濾過による残留固体の除去後に実行した;
(iii)フラッシュクロマトグラフィ精製を、パック済みのRediSep Rf Gold(商標)Silica Columns(20~40μm、球形粒子)、GraceResolv(商標)Cartridges(Davisil(登録商標)シリカ)、又はSilicycleカートリッジ(40~63μm)を用いた自動化Teledyne Isco CombiFlash(登録商標)Rf又はTeledyne Isco CombiFlash(登録商標)Companion(登録商標)で実行した;
(iv)分取クロマトグラフィを、UVコレクションによるGilson prep HPLC機器で実行した;代わりに、分取クロマトグラフィを、MS-及びUV-トリガコレクションによるWaters AutoPurification HPLC-MS機器で実行した;
(v)キラル分取クロマトグラフィを、UVコレクション(233個のインジェクタ/画分コレクタ、333及び334個のポンプ、155個のUV検出器)によるGilson機器、又はGilson305インジェクションでランするVarian Prep Star機器(2×SD1ポンプ、325個のUV検出器、701個の画分コレクタ)ポンプで実行した;代わりに、キラル分取クロマトグラフィを、MS-及びUV-トリガコレクションによるWaters Prep 100 SFC-MS機器、又はUVコレクションによるThar MultiGram III SFC機器で実行した;
(vi)存在する場合、収量は、必ずしも、達成できる最大量であるわけではない;
(vii)一般に、式Iの最終生成物の構造を、核磁気共鳴(NMR)分光法によって確認した;NMR化学シフト値を、デルタスケールで測定した[プロトン磁気共鳴スペクトルを、Bruker Avance III 600(600MHz)、Bruker Avance 400(400MHz)、Bruker Avance 300(300MHz)、又はBruker DRX 500(500MHz)機器を用いて求めた];測定を、特に明記しない限り、周囲温度にて行った;以下の略語を用いた:s、シングレット;d、ダブレット;t、トリプレット;q、カルテット;m、マルチプレット;dd、ダブレットのダブレット;ddd、ダブレットのダブレットのダブレット;dt、トリプレットのダブレット;bs、広いシグナル。
(viii)また、一般に、式Iの最終生成物を、液体クロマトグラフィ(LCMS又はUPLC)後の質量分光法によって特徴付けた;Waters SQ質量分析計を取り付けたWaters UPLCを用いて(カラム温度40℃、UV=220~300nm又は190~400nm、Mass Spec=正/負スイッチングによるESI)、97%A+3%B~3%A+97%Bの溶媒系を用いて、1mL/分の流量にて、1.50分にわたって(平衡が出発条件その他に戻る総ランタイム、1.70分)、UPLCを実行した。ここで、A=水中0.1%ギ酸若しくは0.05%トリフルオロ酢酸(酸性研究について)、又は水中0.1%水酸化アンモニウム(塩基性研究について)、及びB=アセトニトリルである。酸性分析に用いたカラムは、Waters Acquity HSS T3(1.8μm、2.1×50mm)であり、塩基性分析に用いたカラムは、Waters Acquity BEH C18(1.7μm、2.1×50mm)であった。これ以外にも、Waters SQ質量分析計を取り付けたWaters UPLCを用いて(カラム温度30℃、UV=210~400nm、Mass Spec=正/負スイッチングによるESI)、2~98%Bの溶媒勾配を用いて1mL/分の流量にて、1.5分にわたって(平衡が出発条件に戻る総ランタイム、2分)、UPLCを実行した。ここで、A=水中0.1%ギ酸、及びB=アセトニトリル中0.1%ギ酸(酸性研究について)、又はA=水中0.1%水酸化アンモニウム及びB=アセトニトリル(塩基性研究について)である。酸性分析に用いたカラムは、Waters Acquity HSS T3(1.8μm、2.1×30mm)であり、塩基性分析に用いたカラムは、Waters Acquity BEH C18(1.7μm、2.1×30mm)であった;Waters ZQ ESCi質量分析計を取り付けたWaters Alliance(2795)及びPhenomenex Gemini-NX C18(5μm、110A、2.1×50mmカラム)を用いて、4分にわたる、0.5分の保持の、1.1mL/分の95%A~95%Bの流量にて、LCMSを実行した。ここで、A=アセトニトリル中0.1%ギ酸及びB=0.1%ギ酸(酸性研究について)、又はA=水中0.1%水酸化アンモニウム及びB=アセトニトリル(塩基性研究について)である。加えて、Shimadzu LCMS-2020質量分析計を取り付けたShimadzu UFLC、及びWaters HSS C18(1.8μm、2.1×50mm)、Shim-pack XR-ODS(2.2μm、3.0×50mm)、又はPhenomenex Gemini-NX C18(3μm、3.0×50mm)カラムを用いて、0.7mL/分(Waters HSS C18カラムについて)、1.0mL/分(Shim-pack XR-ODSカラムについて)、又は1.2mL/分(Phenomenex Gemini-NX C18について)の流量にて、2.2分にわたる、0.6分の保持の、95%A~95%Bにて、LCMSを実行した。ここで、A=水中0.1%ギ酸若しくは0.05%トリフルオロ酢酸(酸性研究について)、又は水中0.1%水酸化アンモニウム若しくは6.5mM炭酸アンモニウム(塩基性研究について)、及びB=アセトニトリルである。報告した分子イオンは、特に明記しない限り、[M+H]+に相当する;複数の同位体パターンがある分子(Br、Clその他)について、報告した値は、特に明記しない限り、最も低い同位体質量について得られたものである。
(ix)イオン交換精製を、通常、SCX-2(Biotage)カートリッジを用いて実行した。
(x)中間体の純度を、薄層クロマトグラフィ、質量分光法、LCMS、UPLC/MS、HPLC(高速液体クロマトグラフィ)、及び/又はNMR分析によって評価した;
(xi)以下の略語を用いた:
EtOH:エタノール
EtOAc:酢酸エチル
LDA:リチウムジイソプロピルアミド
MeOH:メタノール
TFA:トリフルオロ酢酸
MeCN:アセトニトリル
LCMS:液体クロマトグラフィ-質量分析
rt又はRT:室温
aq:水性
THF:テトラヒドロフラン
KHMDS:カリウムビス(トリメチルシリル)アミド
DCM:ジクロロメタン
DMF:ジメチルホルムアミド
HATU:(1-[ビス(ジメチルアミノ)メチレン]-1H-1,2,3-トリアゾロ[4,5-b]ピリジニウム3-オキシドヘキサフルオロホスファート)
BOC:tert-ブトキシカルボニル
DTNB:5,5’-ジチオビス(2-ニトロ安息香酸)
TNB:2-ニトロ-5-チオ安息香酸
HEPES:(4-(2-ヒドロキシエチル)-1-ピペラジンエタンスルホン酸)
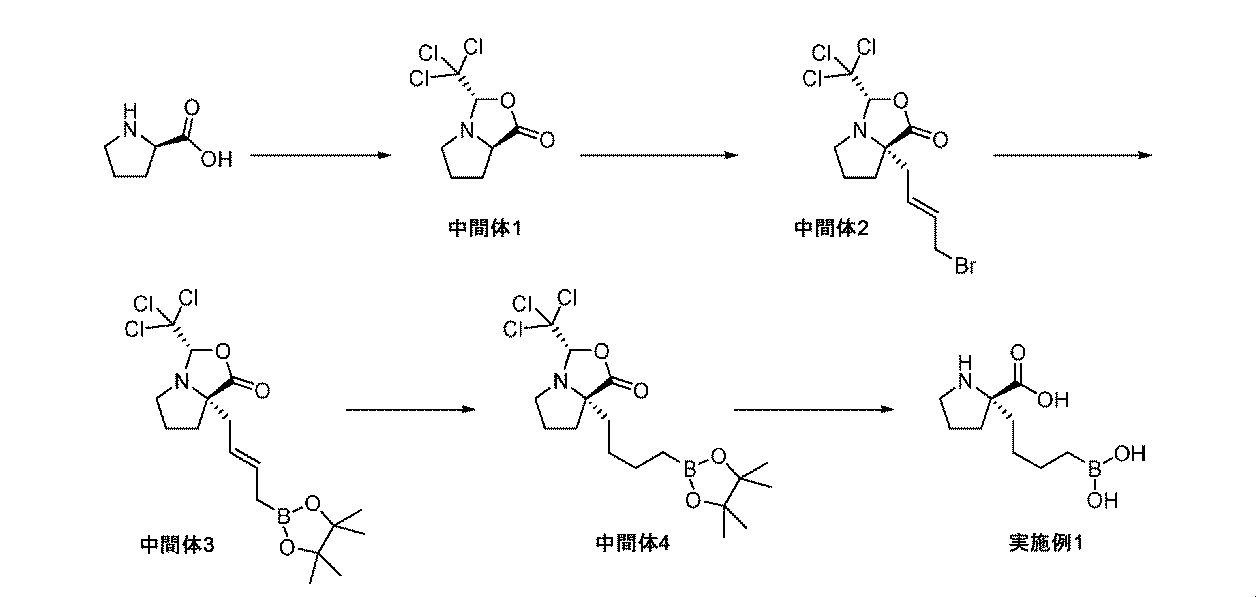
2,2,2-トリクロロエタン-1,1-ジオール(2.155g、13.03mmol)を、D-プロリン(1.00g、8.69mmol)のCHCl3溶液(100mL)に、窒素下で加えた。反応フラスコは、逆ディーンスタークトラップを備えた。反応混合液を加熱して、48時間、撹拌により還流させた。反応混合液を、室温に冷却して、DCM(100mL)で希釈して、水(2×200mL)及び飽和ブライン(2×200mL)で順次洗浄した。有機層を、Na2SO4上で乾燥させて、濾過して、蒸発させて、粗生成物を得た。粗物質を、EtOHから結晶化によって精製して、生成物(中間体1、1.13g、53.2%の収率)を白色の固体として得た。1H NMR(300MHz,CDCl3)δ 1.66-2.41(4H,m),3.05-3.20(1H,m),3.40-3.50(1H,m),4.10-4.20(1H,m),5.18(1H,s).
LDAの溶液(THF/ヘプタン/エチルベンゼン中2.0M、2.05mL、4.09mmol)を、(3S,7aR)-3-(トリクロロメチル)テトラヒドロピロロ[1,2-c]オキサゾール-1(3H)-オン(中間体1、1.00g、4.09mmol)のTHF溶液(500mL)に、-78℃にて、窒素雰囲気下で滴加した。生じた溶液を、-78℃にて20分間撹拌した。(E)-1,4-ジブロモブタ-2-エン(875mg、4.09mmol)を、THF溶液(2mL)として、反応混合液に滴加した。反応混合液を-78℃にて30分間撹拌してから、さらに2時間撹拌しながら室温に温めた。反応混合液を蒸発させて乾燥させて、生じた残留物を、EtOAc(20mL)中に希釈して、水(2×20mL)及び飽和ブライン(2×20mL)で順次洗浄した。有機層を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、生成物(中間体2、760mg、49%の収率)を得た。1H NMR(300MHz,CDCl3)δ 1.55-1.75(1H,m),1.85-2.25(3H,m),2.52-2.73(2H,m),3.14-3.32(2H,m),3.89-4.10(2H,m),5.01(1H,s),5.79-5.99(2H,m);m/z(ES+)[M+H]+=378.
Pd2(dba)3(85.0mg、0.0928mmol)を、(3S,7aS)-7a-((E)-4-ブロモブタ-2-エニル)-3-(トリクロロメチル)テトラヒドロピロロ[1,2-c]オキサゾール-1(3H)-オン(中間体2、700mg、1.85mmol)及びビス(ピナコラート)ジボロン(942mg、3.71mmol)のTHF溶液(30mL)に、窒素雰囲気下で加えた。生じた混合液を、60℃に加熱して、5時間撹拌した。反応混合液を室温に冷却して、濃縮して乾燥させた。生じた残留物を、EtOAc(50mL)で希釈して、水及び飽和ブラインで順次洗浄した。有機層を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。生じた粗物質を、シリカゲルクロマトグラフィによって精製して、生成物(中間体3、510mg、65%の収率)を得た。1H NMR(300MHz,CDCl3)δ 1.28(12H,s),1.58-1.80(2H,m),1.83-2.12(3H,m)2.42-2.65(1H,m),3.20(1H,dd),3.47(1H,q),3.71(1H,t),3.90(1H,t),4.98(1H,s),5.38-5.53(1H,m),5.64-5.83(1H,m);m/z(ES+)[M+H]+=424.
Pd/C(10wt%、125mg、0.12mmol)を、(3S,7aS)-7a-((E)-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブタ-2-エニル)-3-(トリクロロメチル)テトラヒドロピロロ[1,2-c]オキサゾール-1(3H)-オン(中間体3、500mg、1.18mmol)のMeOH溶液(5mL)に加えた。反応フラスコは、H2のバルーンを備えた。懸濁液を、室温にて30分間撹拌した。反応混合液を、珪藻土で濾過して、MeOHで洗浄した。濾液を濃縮して乾燥させて、粗生成物(中間体4、390mg、78%の収率)を得た。これを、更なる精製なしに用いた。m/z(ES+)[M+H]+=426.
水性濃HCl(1.00mL、12.0mmol)を、(3S,7aR)-7a-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)-3-(トリクロロメチル)テトラヒドロピロロ[1,2-c]オキサゾール-1(3H)-オン(中間体4、300mg、0.703mmol)及びフェニルボロン酸(172mg、1.41mmol)の1,4-ジオキサン溶液(20mL)に加えた。生じた溶液を、80℃に15時間加熱した。反応混合液を室温に冷却して、濃縮して乾燥させた。粗物質を、分取LCMS(XBridge Prep C18 OBDカラム、5μシリカ、19×150mm、H2O(w/0.05%TFA/MeCN)によって精製した。純粋な画分を収集して、濃縮して乾燥させて、(R)-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例1、85mg、37%の収率)を白色の固体として得た。1H NMR(400MHz,D2O)δ 0.63-0.74(2H,m),1.09-1.27(2H,m),1.27-1.37(2H,m),1.65-1.75(1H,m),1.77-2.08(4H,m),2.25-2.37(1H,m),3.21-3.37(2H,m);m/z(ES+)[M+H]+=216.
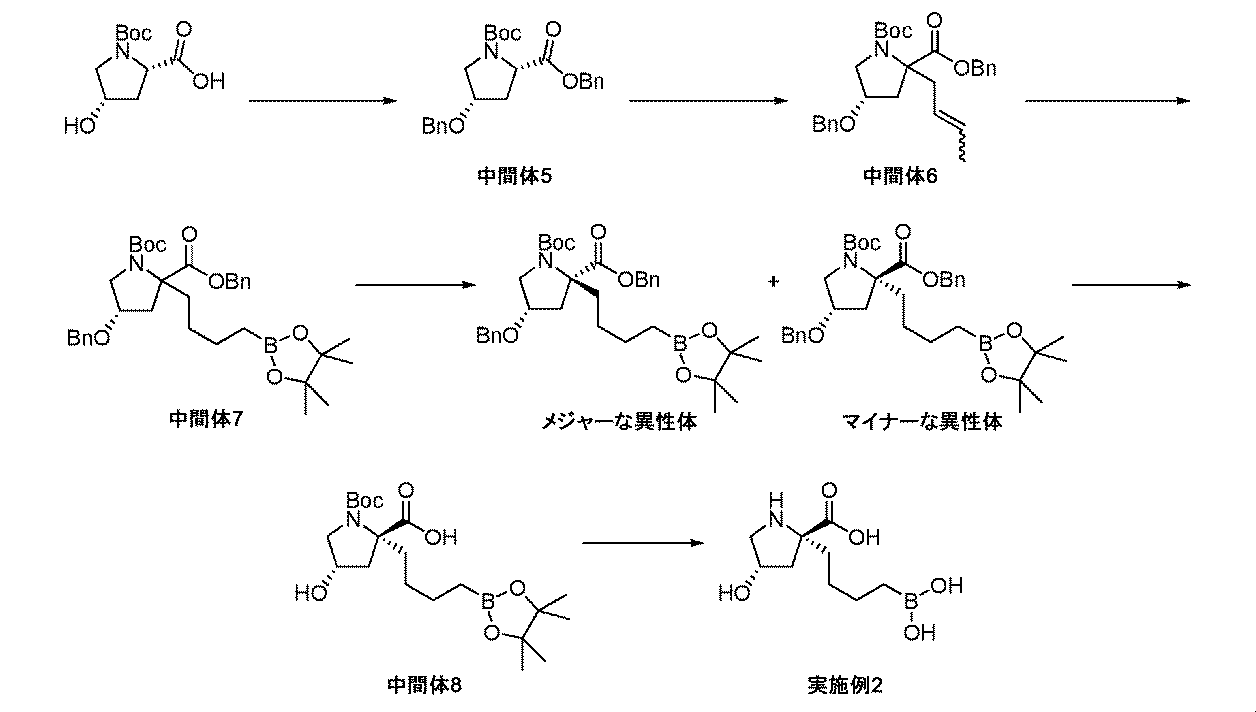
(2S,4S)-1-(tert-ブトキシカルボニル)-4-ヒドロキシピロリジン-2-カルボン酸(5.00g、21.6mmol)をDMF(73mL)中に溶解して、溶液を0℃に冷却した。水素化ナトリウム(鉱油中60%分散系)(1.81g、45.3mmol)を少量ずつ加えて、懸濁液を0℃にて1時間撹拌した。臭化ベンジル(12.9mL、108mmol)を加えて、反応混合液を一晩、RTにゆっくり温めながら撹拌した。粗反応混合液を、酢酸エチル(250mL)で希釈して、クエン酸(10%aq)及び水で順次洗浄した。有機質をNa2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗生成物を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、生成物(中間体5、5.5g、62%の収率)を回転異性体の混合物として得た。1H NMR(300MHz,DMSO-d6)δ 1.28-1.40(9H,s x2)回転異性体,2.20(1H,dd),2.36-2.45(1H,m),3.37(1H,dd),3.51-3.58(1H,m),4.14(1H,br s),4.33-4.50(3H,m),4.94-5.17(2H,m),7.25-7.32(10H,m);m/z(ES+)[M+H]+=412.
(2S,4S)-2-ベンジル1-tert-ブチル4-(ベンジルオキシ)ピロリジン-1,2-ジカルボキシラート(中間体5、2.75g、6.68mmol)及び臭化クロチル(1.03mL、10.0mmol)を、THF(45mL)中に溶解して、溶液を、N2雰囲気下で-78℃に冷却した。溶液を、KHMDSの溶液(トルエン中0.5M、20.1mL、10.0mmol)の滴加により処理した。反応混合液を、室温にゆっくり温めて、3時間撹拌した。粗反応混合液を水でクエンチして、揮発性物質を真空内で除去した。粗混合液を、DCM中に希釈して、層を分離させた。有機層を水で洗浄して、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、生成物(中間体6、2.54g、82%の収率)を、回転異性体及びE/Zオレフィンの混合物として得た。1H NMR(300MHz,DMSO-d6)δ 1.20-1.41(9H,s x2)回転異性体,1.54-1.62(3H,m),2.10-2.59(3H,m),2.67-2.97(1H,m),3.10-3.43(1H,m),3.50-3.78(1H,m),3.98-4.15(1H,m),4.34-4.49(2H,m),4.94-5.13(2H,m),5.18-5.30(1H,m),5.38-5.63(1H,m),7.25-7.36(10H,m);m/z(ES+)[M+H]+=466.
ビス(1,5-シクロオクタジエン)ジイリジウム(I)ジクロリド(0.366g、0.550mmol)及びビス(ジフェニルホスフィノ)メタン(0.419g、1.09mmol)を、オーブン乾燥した丸底フラスコに加えた。フラスコをシールして、N2でパージした。固体を、DCM(31mL)中に溶解して、4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(1.74mL、12.0mmol)を溶液にゆっくり加えた。反応液を、室温にて10分間撹拌した。(4S)-2-ベンジル1-tert-ブチル4-(ベンジルオキシ)-2-(ブタ-2-エニル)ピロリジン-1,2-ジカルボキシラート(中間体6、2.54g、5.46mmol)を、DCM溶液(21mL)としての反応液に加えて、反応混合液を一晩撹拌した。反応混合液をDCMで希釈して、水でクエンチした。層を分離させて、水性層をDCMで抽出した。組み合わせた有機質を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、(4S)-2-ベンジル1-tert-ブチル4-(ベンジルオキシ)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体7、2.0g、61%の収率)を回転異性体の混合物として得た。精製物質を、キラルSFC[(S,S)Whelk-O1カラム、21.2×250mm、5μm、温度=23℃、移動相=0~15%MeOH:CO2、UV検出@220nm、ローディング=33mg/inj、conc=MeOH中220ng/mL、流量=75mL/分、出口圧力=100bar]にかけて、2つの偏左右異性体を得た。立体化学は、メジャーな異性体について、アンチ付加生成物として割り当て、そしてマイナーな異性体について、シン付加生成物として割り当てた。マイナーな異性体(368mg、0.620mmol)を、酢酸エチル(6.2mL)中に溶解して、Pd/C(10wt%、132mg、0.124mmol)により処理した。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液を、珪藻土で濾過して、メタノールでリンスした。濾液を、減圧下で濃縮して、(2R,4S)-1-(tert-ブトキシカルボニル)-4-ヒドロキシ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体8、228mg、98%の収率)を回転異性体の混合物として得た。これを、更なる精製なしに用いた。1H NMR(300MHz,DMSO-d6)δ 0.57-0.74(2H,m),1.17(12H,s),1.24-1.47(13H,m),1.59-1.78(1H,m),1.78-1.96(1H,m),2.01-2.20(2H,m),2.84-3.09(1H,m),3.58-3.73(1H,m),4.14-4.31(1H,m),4.98-5.09(1H,m),12.20-12.60(1H,m);m/z(ES+)[M+H]+=414.
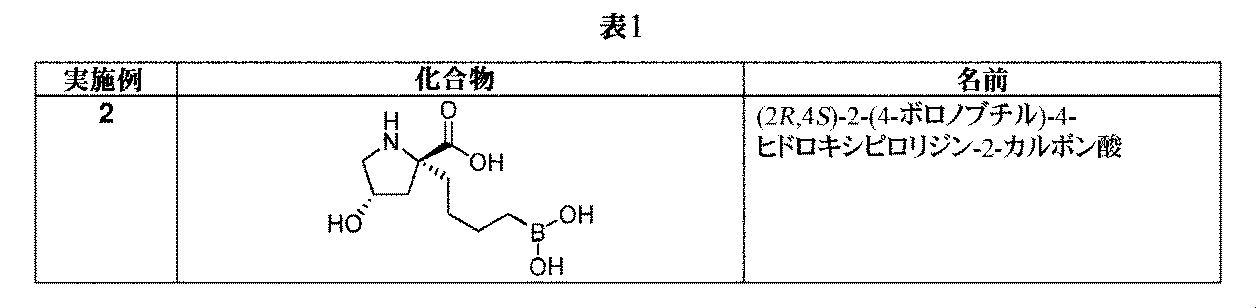
トリフルオロ酢酸(0.65mL、8.4mmol)を、(2R,4S)-1-(tert-ブトキシカルボニル)-4-ヒドロキシ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体8、175mg、0.423mmol)のDCM溶液(4mL)に加えた。生じた溶液を、室温にて1時間撹拌してから、真空下で濃縮した。粗アミノ酸を、Et2O(3mL)及び1M aq HCl(3mL)中に溶解した。フェニルボロン酸(103mg、0.847mmol)を加えて、澄明な二相性溶液を、室温にて1時間撹拌した。反応混合液を水で希釈して、Et2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 60ccカラム)によって精製した。所望の生成物を、カラムから、2Mアンモニア/メタノールを用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18Aq、水中0~10%~100%アセトニトリル)によって精製して、(2R,4S)-2-(4-ボロノブチル)-4-ヒドロキシピロリジン-2-カルボン酸(実施例2、33mg、33%の収率)を白色の固体として得た。1H NMR(400MHz,D2O)δ 0.67-0.78(2H,m),1.08-1.41(4H,m),1.81-2.12(3H,m),2.51(1H,dd),3.22-3.37(2H,m),4.46-4.56(1H,m);m/z(ES+)[M+H]+=232.
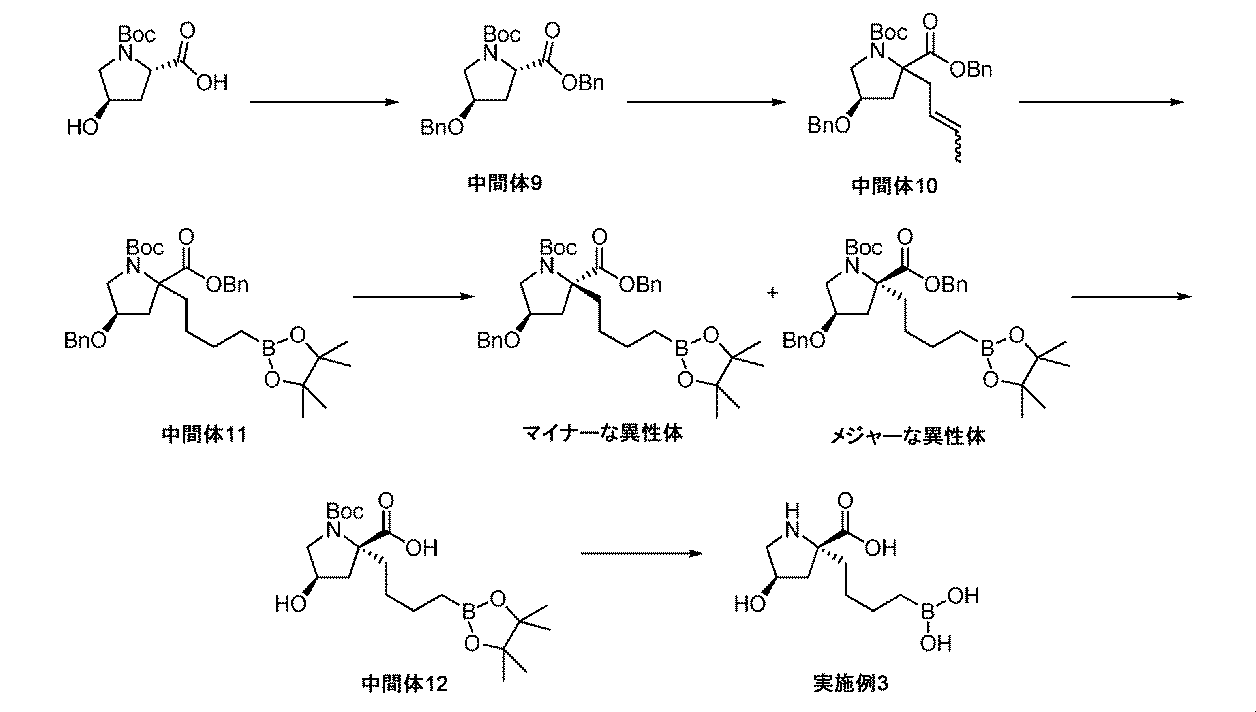
(2S,4R)-1-(tert-ブトキシカルボニル)-4-ヒドロキシピロリジン-2-カルボン酸(5.00g、21.6mmol)を、DMF(73mL)中に溶解して、溶液を0℃に冷却した。水素化ナトリウム(鉱油中60wt%、1.81g、45.4mmol)を少量ずつ加えて、懸濁液を0℃にて1時間撹拌した。臭化ベンジル(12.86mL、108.1mmol)を加えて、反応混合液を、RTにゆっくり温めながら一晩撹拌した。粗反応混合液を酢酸エチル(250mL)で希釈して、クエン酸(10%aq)及び水で順次洗浄した。有機質をNa2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、生成物(中間体9、5.9g、66%の収率)を回転異性体の混合物として得た。1H NMR(300MHz,DMSO-d6)δ 1.27-1.39(9H,s x2)回転異性体,1.95-2.08(1H,m),2.34-2.47(1H,m),3.41-3.53(2H,m),4.17(1H,br s),4.28(1H,q),4.43-4.55(2H,m),5.06-5.22(2H,m),7.25-7.41(10H,m);m/z(ES+)[M+H]+=412.
(2S,4R)-2-ベンジル1-tert-ブチル4-(ベンジルオキシ)ピロリジン-1,2-ジカルボキシラート(中間体9、2.75g、6.68mmol)及び臭化クロチル(1.03mL、10.0mmol)を、THF(45mL)中に溶解して、溶液を、N2雰囲気下で-78℃に冷却した。溶液を、KHMDSの溶液(トルエン中0.5M、20.1mL、10.0mmol)の滴加により処理した。反応混合液を、室温にゆっくり温めて、3時間撹拌した。粗反応混合液を水でクエンチして、揮発性物質を真空内で除去した。粗混合液をDCM中に希釈して、層を分離させた。有機層を水で洗浄して、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、生成物(中間体10、1.23g、40%の収率)を、回転異性体及びE/Zオレフィンの混合物として得た。1H NMR(300MHz,DMSO-d6)δ 1.25-1.34(9H,s x2)回転異性体,1.45-1.63(3H,m),2.12-2.64(2H,m),2.64-3.04(1H,m),3.06-3.19(1H,m),3.31-3.45(1H,m),3.46-3.81(1H,m),4.03-4.21(1H,m),4.30-4.55(2H,m),4.90-5.16(2H,m),5.16-5.34(1H,m),5.38-5.68(1H,m),7.25-7.41(10H,m).m/z(ES+)[M+H]+=466.
ビス(1,5-シクロオクタジエン)ジイリジウム(I)ジクロリド(177mg、0.264mmol)及びビス(ジフェニルホスフィノ)メタン(203mg、0.527mmol)を、オーブン乾燥した丸底フラスコに加えた。フラスコをシールして、N2でパージした。固体をDCM(15mL)中に溶解して、4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(0.84mL、5.8mmol)を溶液にゆっくり加えた。反応液を室温にて10分間撹拌した。(4R)-2-ベンジル1-tert-ブチル4-(ベンジルオキシ)-2-(ブタ-2-エニル)ピロリジン-1,2-ジカルボキシラート(中間体10、1.23g、2.64mmol)を、DCM溶液(10mL)としての反応液に加えて、反応混合液を一晩撹拌した。反応混合液をDCMで希釈して、水でクエンチした。層を分離させて、水性層をDCMで抽出した。組み合わせた有機質を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、(4R)-2-ベンジル1-tert-ブチル4-(ベンジルオキシ)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体11、950mg、60%の収率)を得た。精製した物質を、キラルSFC[(S,S)Whelk-O1カラム、21.2×250mm、5μm、温度=23℃、移動相=0~15%MeOH:CO2、UV検出@220nm、ローディング=33mg/inj、conc=MeOH中220ng/mL、流量=75mL/分、出口圧力=100bar]にかけて、2つの偏左右異性体を得た。立体化学は、メジャーな異性体について、アンチ付加生成物として割り当て、そしてマイナーな異性体について、シン付加生成物として割り当てた。メジャーな異性体(385mg、0.649mmol)を、酢酸エチル(6.4mL)中に溶解して、Pd/C(10wt%、138mg、0.130mmol)により処理した。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液を、珪藻土で濾過して、メタノールでリンスした。濾液を、減圧下で濃縮して、生成物(中間体12、249mg、93%の収率)を回転異性体の混合物として得た。1H NMR(300MHz,DMSO-d6)δ 0.61-0.73(2H,m),0.97-1.11(1H,m),1.12-1.23(12H,m),1.25-1.44(12H,m),1.51-1.71(1H,m),1.84-2.04(2H,m),2.05-2.19(2H,m),3.12-3.29(1H,m),3.37-3.59(1H,m),4.09-4.23(1H,m);m/z(ES+)[M+H]+=414.
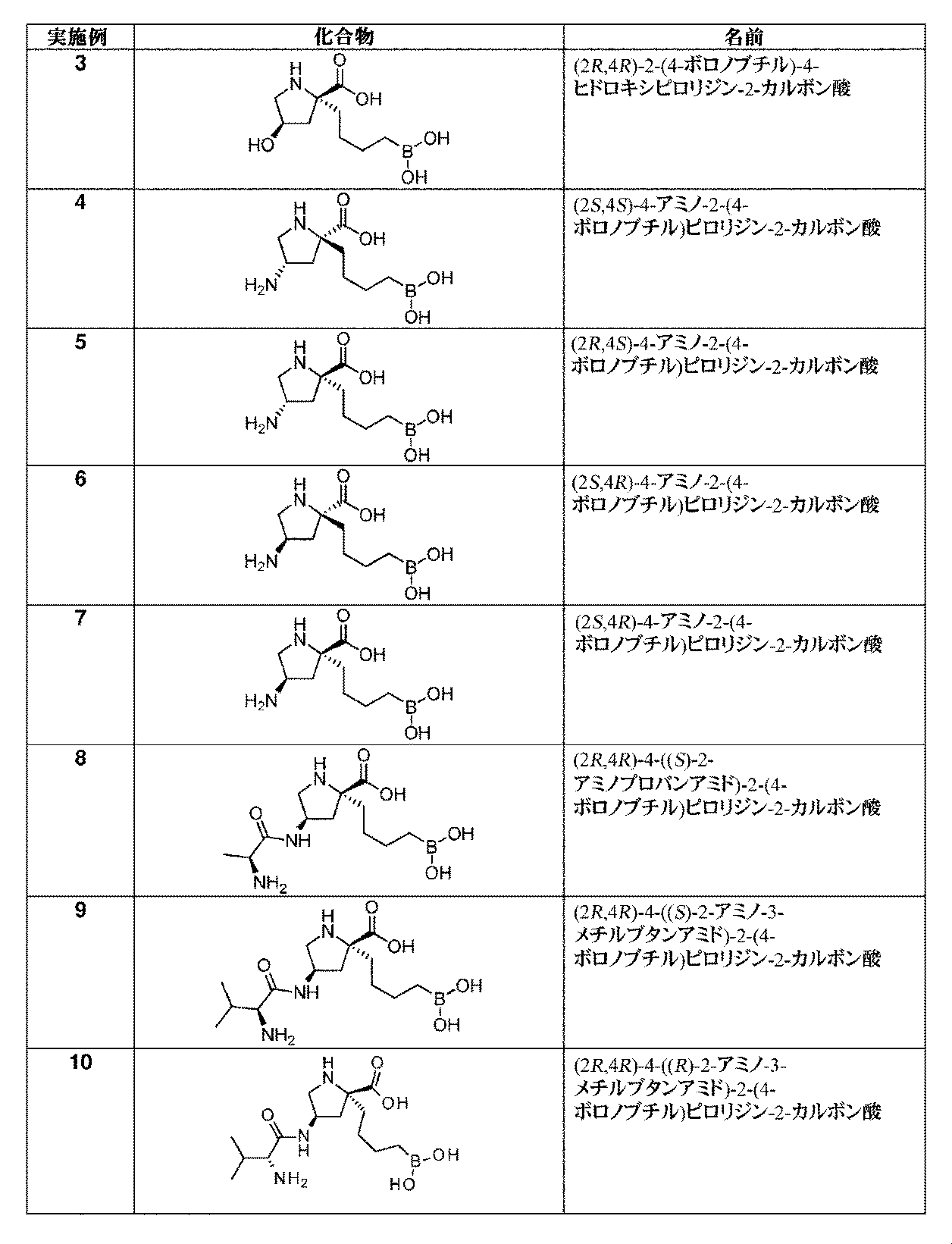
トリフルオロ酢酸(0.65mL、8.5mmol)を、(2R,4R)-1-(tert-ブトキシカルボニル)-4-ヒドロキシ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体12、197mg、0.179mmol)のDCM溶液(3mL)に加えた。生じた溶液を、室温にて1時間撹拌してから、真空下で濃縮した。粗アミノ酸を、Et2O(3mL)及び1M aq HCl(3mL)中に溶解した。フェニルボロン酸(102mg、0.837mmol)を加えて、澄明な二相性溶液を室温にて1時間撹拌した。反応混合液を水で希釈して、Et2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 60ccカラム)によって精製した。所望の生成物をカラムから、2Mアンモニア/メタノールを用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18Aq、水中0~10%アセトニトリル)によって精製して、(2R,4R)-2-(4-ボロノブチル)-4-ヒドロキシピロリジン-2-カルボン酸(実施例3、25mg、25%の収率)を白色の固体として得た。1H NMR(300MHz,D2O)δ 0.68-0.78(2H,m),1.13-1.43(4H,m),1.64-1.79(1H,m),1.94-2.14(2H,m),2.47(1H,d),3.39(2H,m),4.46-4.53(1H,m).m/z(ES+)[M+H]+=232.
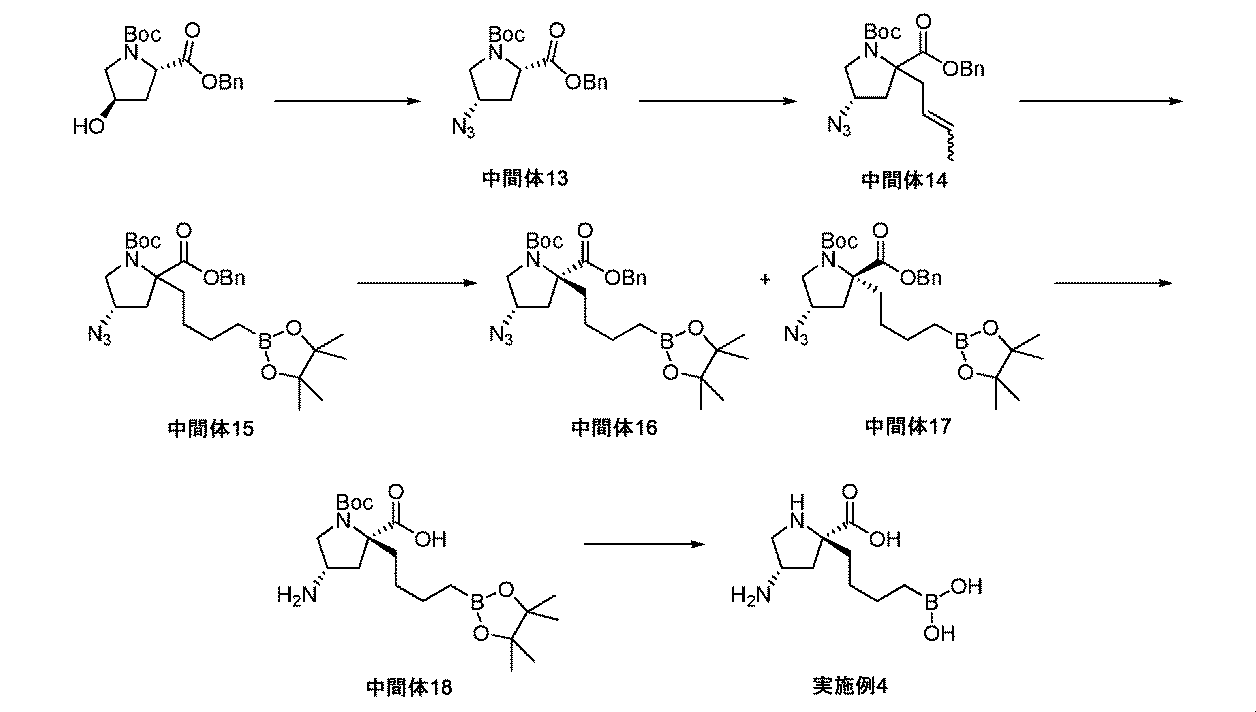
メタンスルホニルクロリド(0.71mL、9.2mmol)を、2-ベンジル1-(tert-ブチル)(2S,4R)-4-ヒドロキシピロリジン-1,2-ジカルボキシラート(2.45g、7.26mmol)及びトリエチルアミン(1.27mL、9.15mmol)のDCM溶液(9.6mL)に0℃にて滴加した。反応混合液を0℃にて1時間撹拌してから、さらに1時間撹拌しながら室温に温めた。反応混合液をジクロロメタンで希釈して、水で洗浄した。有機層を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させて、2-ベンジル1-(tert-ブチル)(2S,4R)-4-((メチルスルホニル)オキシ)ピロリジン-1,2-ジカルボキシラート(2.9g、95%の収率)を得た。これを、更なる精製なしに用いた。m/z(ES+)[M+H]+=400.アジ化ナトリウム(1.65g、25.4mmol)を、2-ベンジル1-(tert-ブチル)(2S,4R)-4-((メチルスルホニル)オキシ)ピロリジン-1,2-ジカルボキシラート(2.90g、7.26mmol)のDMF溶液(7.2mL)に加えた。反応混合液を50℃に加熱して、一晩撹拌した。反応混合液を室温に冷却して、濃縮した。生じた残留物を、EtOAcで希釈して、水で洗浄した。有機層を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、生成物(中間体13、2.00g、80%の収率)を回転異性体の混合物として得た。1H NMR(300MHz,DMSO-d6)δ 1.27及び1.40(9H,s x2)回転異性体,1.96-2.02(1H,m),2.53-2.63(1H,m),3.24-3.29(1H,m),3.58-3.66(1H,m),4.32-4.41(2H,m),5.06-5.22(2H,m),7.33-7.39(5H,m);m/z(ES+)[M+H]+=347.
(2S,4S)-2-ベンジル1-tert-ブチル4-アジドピロリジン-1,2-ジカルボキシラート(中間体13、1.00g、2.89mmol)及び臭化クロチル(0.44mL、4.3mmol)を、THF(20mL)中に溶解して、溶液を、N2雰囲気下で-78℃に冷却した。溶液を、KHMDSの溶液(トルエン中0.5M、8.66mL、4.33mmol)の滴加により処理した。反応混合液を、室温にゆっくり温めて、3時間撹拌した。粗反応混合液を水でクエンチして、揮発性物質を真空内で除去した。粗混合物をDCM中に希釈して、層を分離させた。有機層を水で洗浄して、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、生成物(中間体14、750mg、65%の収率)を、回転異性体及びE/Zオレフィンの混合物として得た。1H NMR(300MHz,DMSO-d6)δ 1.25-1.34(9H,s x2)rotomers,1.55-1.64(3H,m),1.99-2.15(1H,m),2.33-2.62(2H,m),2.73-3.10(1H,m),3.26-3.39(1H,m),3.52-3.84(1H,m),4.24-4.33(1H,m),5.03-5.21(2H,m),5.28-5.35(1H,m),5.49-5.65(1H,m),7.31-7.36(5H,m);m/z(ES+)[M+H]+=401.
ビス(1,5-シクロオクタジエン)ジイリジウム(I)ジクロリド(126mg、0.188mmol)及びビス(ジフェニルホスフィノ)メタン(144mg、0.375mmol)を、オーブン乾燥した丸底フラスコに加えた。フラスコをシールして、N2でパージした。固体を、DCM(10mL)中に溶解して、4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(0.60mL、4.1mmol)を溶液にゆっくり加えた。反応液を、室温にて10分間撹拌した。(4S)-2-ベンジル1-tert-ブチル4-アジド-2-(ブタ-2-エニル)ピロリジン-1,2-ジカルボキシラート(中間体14、750mg、1.87mmol)を、DCM溶液(8mL)としての反応液に加えて、反応混合液を室温にて一晩撹拌した。反応混合液をDCMで希釈して、水でクエンチした。層を分離させて、水性層をDCMで抽出した。組み合わせた有機質を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、(4S)-2-ベンジル1-tert-ブチル4-アジド-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体15、678mg、68%の収率)を得た。精製物質を、キラルSFC[(S,S)Whelk-O1カラム、21.2×250mm、5μm、温度=23℃、移動相=0~15%MeOH:CO2、UV検出@220nm、ローディング=33mg/inj、conc=MeOH中220ng/mL、流量=75mL/分、出口圧力=100bar]にかけて、2つの偏左右異性体を得た。立体化学は、メジャーな異性体について、アンチ付加生成物中間体16として割り当て、そしてマイナーな異性体について、シン付加生成物中間体17であった。
(2S,4S)-2-ベンジル1-tert-ブチル4-アジド-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体16、255mg、0.483mmol)を、酢酸エチル(5mL)及びメタノール(5mL)中に溶解して、Pd/C(10wt%、128mg、0.120mmol)により処理した。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液を珪藻土で濾過して、メタノールでリンスした。濾液を、減圧下で濃縮して、生成物(中間体18、190mg、95%の収率)を、回転異性体の混合物として得た。これを、更なる精製なしに用いた。1H NMR(300MHz,DMSO-d6)δ 0.66(2H,t),0.88-1.03(1H,m),1.16(12H,s),1.24-1.38(13H,m),1.40-1.56(1H,m),1.80-1.91(1H,m),2.00-2.15(2H,m),3.17-3.28(1H,m),3.58-3.61(1H,m),3.80(1H,dd),9.01(2H,br s);m/z(ES+)[M+H]+=413.
トリフルオロ酢酸(0.71mL、9.2mmol)を、(2S,4S)-4-アミノ-1-(tert-ブトキシカルボニル)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体18、190mg、0.461mmol)のDCM溶液(4mL)に加えた。生じた溶液を、室温にて1時間撹拌してから、真空下で濃縮した。粗アミノ酸を、Et2O(3mL)及び1M aq HCl(3mL)中に溶解した。フェニルボロン酸(112mg、0.919mmol)を加えて、澄明な二相性溶液を、室温にて1時間撹拌した。反応混合液を水で希釈して、Et2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 60ccカラム)によって精製した。所望の生成物をカラムから、2Mアンモニア/メタノールを用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18Aq、水中0~10%アセトニトリル)によって精製して、(2S,4S)-4-アミノ-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例4、40mg、37%の収率)を白色の固体として得た。1H NMR(300MHz,D2O)δ 0.73(2H,t),1.10-1.42(4H,m),1.69(1H,ddd),1.86-1.99(1H,m),2.10-2.30(2H,m),3.05(1H,dd),3.44(1H,dd),3.69(1H,quin);m/z(ES+)[M+H]+=231.
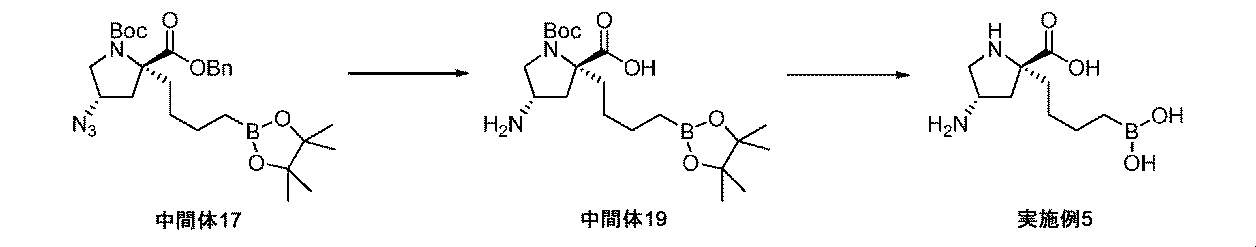
(2R,4S)-2-ベンジル1-tert-ブチル4-アジド-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体17、220mg、0.416mmol)を、酢酸エチル(5mL)及びメタノール(5mL)中に溶解して、Pd/C(10wt%、111mg、0.104mmol)により処理した。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液を、珪藻土で濾過して、メタノールでリンスした。濾液を減圧下で濃縮して、生成物(中間体19、150mg、87%の収率)を回転異性体の混合物として得た。これを、更なる精製なしに用いた。1H NMR(300MHz,DMSO-d6)δ 0.64-0.71(2H,m),1.17(12H,s),1.31-1.40(15H,m),1.49-1.93(3H,m),2.02-2.26(3H,m),3.38-3.47(1H,m),3.72-3.81(1H,m);m/z(ES+)[M+H]+=413.
トリフルオロ酢酸(0.56mL、7.3mmol)を、(2R,4S)-4-アミノ-1-(tert-ブトキシカルボニル)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体19、150mg、0.364mmol)のDCM溶液(3mL)に加えた。生じた溶液を、室温にて1時間撹拌してから、真空下で濃縮した。粗アミノ酸を、Et2O(2mL)及び1M aq HCl(2mL)中に溶解した。フェニルボロン酸(99mg、0.81mmol)を加えて、澄明な二相性溶液を室温にて1時間撹拌した。反応混合液を水で希釈して、Et2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 60ccカラム)によって精製した。所望の生成物をカラムから、2Mアンモニア/メタノールを用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18Aq、水中0~10%、0~100%アセトニトリル)によって精製して、(2R,4S)-4-アミノ-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例5、33mg、39%の収率)を白色の固体として得た。1H NMR(400MHz,D2O)δ 0.72(1H,m),1.11-1.39(3H,m),1.46-1.55(1H,m),1.63-1.79(2H,m),1.95-2.05(1H,m),2.58-2.65(1H,m),2.87-2.95(1H,m),3.48-3.58(3H,m);m/z(ES+)[M+H]+=231.

メタンスルホニルクロリド(2.86mL、36.7mmol)を、(2S,4S)-1-tert-ブチル2-メチル4-ヒドロキシピロリジン-1,2-ジカルボキシラート(7.50g、30.6mmol)及びトリエチルアミン(5.11mL、36.7mmol)のDCM溶液(38mL)に0℃にて滴加した。反応混合液を0℃にて1時間撹拌してから、さらに1時間撹拌しながら室温に温めた。反応混合液をジクロロメタンで希釈して、水で洗浄した。有機層を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させて、1-(tert-ブチル)2-メチル(2S,4S)-4-((メチルスルホニル)オキシ)ピロリジン-1,2-ジカルボキシラート(9.9g、100%の収率)を得た。これを、更なる精製なしに用いた。m/z(ES+)[M+NH4]+=341.
水酸化ナトリウム(5.28g、132mmol)の水溶液(22mL)を、(2S,4R)-1-tert-ブチル2-メチル4-アジドピロリジン-1,2-ジカルボキシラート(中間体20、5.95g、22.0mmol)のTHF(44mL)及びMeOH(22mL)溶液に0℃にて滴加した。反応混合液を、室温にゆっくり温めながら、一晩撹拌した。揮発性物質を真空内で除去して、水性層を5M HClで約3のpHに酸性化して、DCMで抽出した。組み合わせた有機質を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させて、(2S,4R)-4-アジド-1-(tert-ブトキシカルボニル)ピロリジン-2-カルボン酸(5.64g、100%の収率)を回転異性体の混合物として得た。これを、更なる精製なしに用いた。1H NMR(300MHz,DMSO-d6)δ 1.35及び1.40(9H,s x2)回転異性体,2.07-2.18(1H,m),2.26-2.38(1H,m),3.34-3.44(1H,m),3.48-3.63(1H,m),4.09-4.17(1H,m),4.30-4.37(1H,m);m/z(ES-)[M+HCOO]-=301.
(2S,4R)-2-ベンジル1-tert-ブチル4-アジドピロリジン-1,2-ジカルボキシラート(中間体21、5.09g、14.7mmol)及び臭化クロチル(2.27mL、22.0mmol)を、THF(100mL)中に溶解して、溶液を、N2雰囲気下で-78℃に冷却した。溶液を、KHMDSの溶液(トルエン中0.5M、44.1mL、22.0mmol)の滴加により処理した。反応混合液を室温にゆっくり温めて、3時間撹拌した。粗反応混合液を水でクエンチして、揮発性物質を真空内で除去した。粗混合液をDCMで希釈して、層を分離させた。有機層を水で洗浄して、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、生成物(中間体22、4.6g、78%の収率)を、回転異性体及びE/Zオレフィンの混合物として得た。1H NMR(300MHz,DMSO-d6)δ 1.26-1.43(9H,m),1.59-1.66(3H,m),2.07-2.17(1H,m),2.32-2.48(2H,m),2.57-3.12(2H,m),3.35-3.82(1H,m),4.20-4.38(1H,m),5.02-5.22(2H,m),5.24-5.41(1H,m),5.46-5.68(1H,m),7.28-7.42(5H,m);m/z(ES+)[M+H]+=401.
ビス(1,5-シクロオクタジエン)ジイリジウム(I)ジクロリド(772mg、1.15mmol)及びビス(ジフェニルホスフィノ)メタン(883mg、2.30mmol)を、オーブン乾燥した丸底フラスコに加えた。フラスコをシールして、N2でパージした。固体をDCM(66mL)中に溶解して、4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(3.67mL、25.3mmol)を溶液にゆっくり加えた。反応液を室温にて10分間撹拌した。(4R)-2-ベンジル1-tert-ブチル4-アジド-2-(ブタ-2-エニル)ピロリジン-1,2-ジカルボキシラート(中間体22、4.60g、11.5mmol)を、DCM溶液(44mL)としての反応液に加えて、反応混合液を一晩撹拌した。反応混合液をDCMで希釈して、水でクエンチした。層を分離させて、水性層をDCMで抽出した。組み合わせた有機質を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、(4R)-2-ベンジル1-tert-ブチル4-アジド-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体23、2.7g、44%の収率)を得た。精製物質を、キラルSFC(Chiralpak IGカラム、21.2×250mm、5μm、温度=23℃、移動相=0~7%MeOH(w/0.2%NH4OH):CO2、UV検出@220nm、ローディング=16.8mg/inj、conc=MeOH中112.5ng/mL、流量=70mL/分、出口圧力=100bar]にかけて、2つの偏左右異性体を得た。立体化学は、メジャーな偏左右異性体中間体25について、アンチ付加生成物として割り当て、そしてマイナーな偏左右異性体中間体24について、シン付加生成物であった。
(2S,4R)-2-ベンジル1-tert-ブチル4-アジド-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体24、236mg、0.447mmol)を、酢酸エチル(4.5mL)中に溶解して、Pd/C(10wt%、119mg、0.112mmol)により処理した。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液を、珪藻土で濾過して、メタノールでリンスした。濾液を減圧下で濃縮して、生成物(中間体26、275mg、100%の収率)を得た。これを、更なる精製なしに用いた。1H NMR(300MHz,DMSO-d6)δ 0.64-0.71(2H,M),1.17(12H,s),1.27-1.40(15H,m),1.57-1.82(4H,m),1.98-2.08(3H,m),3.70-3.78(1H,m);m/z(ES+)[M+H]+=413.
トリフルオロ酢酸(0.69mL、8.9mmol)を、(2S,4R)-4-アミノ-1-(tert-ブトキシカルボニル)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体26、184mg、0.446mmol)のDCM溶液(4mL)に加えた。生じた溶液を、室温にて1時間撹拌してから、真空下で濃縮した。粗アミノ酸を、Et2O(2mL)及び1M aq HCl(2mL)中に溶解した。フェニルボロン酸(109mg、0.894mmol)を加えて、澄明な二相性溶液を室温にて1時間撹拌した。反応混合液を水で希釈して、Et2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 60ccカラム)によって精製した。所望の生成物をカラムから、2Mアンモニア/メタノールを用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18Aq、水中0~100%アセトニトリル)によって精製して、(2S,4R)-4-アミノ-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例6、38mg、37%の収率)を白色の固体として得た。1H NMR(400MHz,D2O)δ 0.72(2H,td),1.09-1.19(1H,m),1.22-1.39(3H,m),1.65-1.76(2H,m),1.95-2.04(1H,m),2.58-2.64(1H,m),2.87-2.94(1H,m),3.48-3.57(2H,m);m/z(ES+)[M+H]+=231.
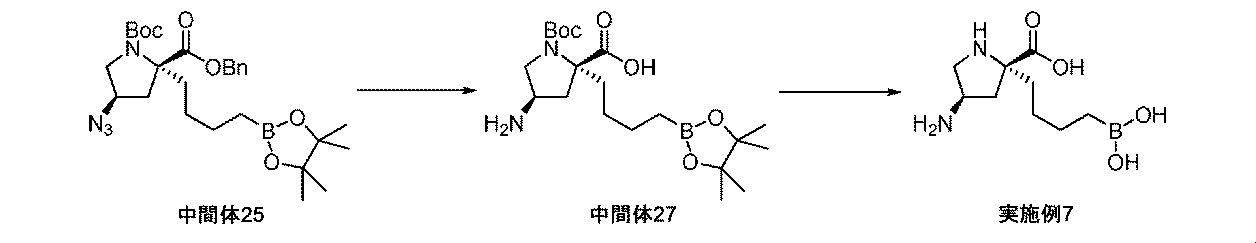
(2R,4R)-2-ベンジル1-tert-ブチル4-アジド-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体25、688mg、1.30mmol)を、酢酸エチル(13mL)及びメタノール(4mL)中に溶解して、Pd/C(10%wt、346mg、0.325mmol)により処理した。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液を、珪藻土で濾過して、メタノールでリンスした。濾液を減圧下で濃縮して、生成物(中間体27、500mg、93%の収率)を得た。これを、更なる精製なしに用いた。1H NMR(300MHz,DMSO-d6)δ 0.67(2H,t),0.94-1.00(1H,m),1.17(12H,s),1.22-1.38(11H,m),1.43-1.53(1H,m),1.85(1H,d),2.00-2.15(2H,m),3.23(2H,dd),3.58-3.61(1H,m),3.80-3.88(1H,m),8.96(2H,m);m/z(ES+)[M+H]+=413.
トリフルオロ酢酸(1.02mL、13.3mmol)を、(2R,4R)-4-アミノ-1-(tert-ブトキシカルボニル)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体27、275mg、0.667mmol)のDCM溶液(4mL)に加えた。生じた溶液を、室温にて1時間撹拌してから、真空下で濃縮した。粗アミノ酸を、Et2O(2mL)及び1M aq HCl(2mL)中に溶解した。フェニルボロン酸(163mg、1.34mmol)を加えて、澄明な二相性溶液を室温にて1時間撹拌した。反応混合液を水で希釈して、Et2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 60ccカラム)によって精製した。所望の生成物をカラムから、2Mアンモニア/メタノールを用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18Aq、水中0~10%~100%アセトニトリル)によって精製して、(2R,4R)-4-アミノ-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例7、53mg、34%の収率)を白色の固体として得た。1H NMR(400MHz,D2O)δ 0.76(2H,dt),1.10-1.46(4H,m),1.62-1.71(1H,m),1.84-1.96(1H,m),2.10-2.21(1H,m),2.22-2.32(1H,m),3.07(1H,dd),3.46(1H,dd),3.71(1H,quin);m/z(ES+)[M+H]+=231.
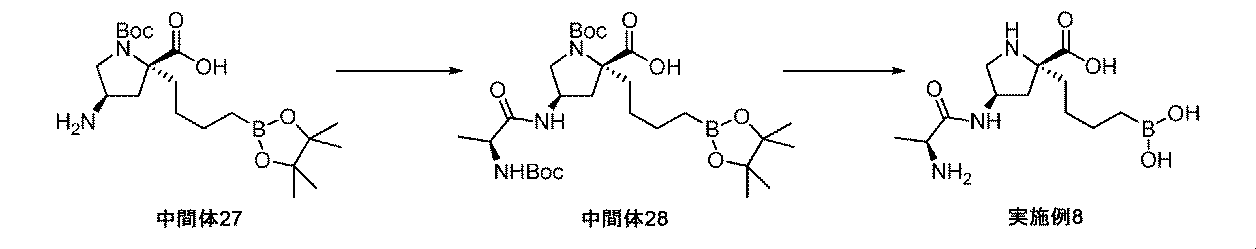
トリエチルアミン(0.18mL、1.3mmol)及びHATU(213mg、0.560mmol)を順次、Boc-Ala-OH(106mg、0.560mmol)のDMF溶液(2.4mL)に加えて、反応液を室温にて30分間撹拌した。(2R,4R)-4-アミノ-1-(tert-ブトキシカルボニル)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体27、210mg、0.509mmol)を、DMF溶液(2.4mL)としての反応混合液に加えた。反応液を、室温にて一晩撹拌した。粗反応混合液を濃縮して、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって直接精製して、生成物(中間体28、236mg、79%の収率)を回転異性体の混合物として得た。1H NMR(300MHz,DMSO-d6)δ 0.65-0.72(2H,m),1.11-1.18(18H,m),1.26-1.37(20H,m),1.63-1.73(1H,m),2.02-2.25(2H,m),3.09-3.20(1H,m),3.59-3.72(1H,m),3.83-3.94(1H,m),4.18-4.29(1H,m),6.80(1H,br s),7.96(1H,s),13.78(1H,br s);m/z(ES+)[M+H]+=584.
トリフルオロ酢酸(0.62mL、8.1mmol)を、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-2-(tert-ブトキシカルボニルアミノ)プロパンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体28、236mg、0.404mmol)のDCM溶液(4mL)に加えた。生じた溶液を、室温にて1時間撹拌してから、真空下で濃縮した。粗アミノ酸を、Et2O(2mL)及び1M aq HCl(2mL)中に溶解した。フェニルボロン酸(99mg、0.81mmol)を加えて、澄明な二相性溶液を室温にて1時間撹拌した。反応混合液を水で希釈して、Et2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 60ccカラム)によって精製した。所望の生成物をカラムから、2Mアンモニア/メタノールを用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18Aq、水中0~10%アセトニトリル)によって精製して、(2R,4R)-4((S)-2-アミノプロパンアミド)-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例8、18mg、15%の収率)を、白色の固体、及び回転異性体の混合物として得た。1H NMR(500MHz,D2O)δ 0.69(2H,dt),1.05-1.14(1H,m),1.21(3H,d),1.23-1.35(3H,m),1.65(1H,dt),1.91-1.96(1H,m),2.17(1H,dd),2.35(1H,dd),3.26(1H,dd),3.46-3.57(2H,m),4.29-4.34(1H,m);m/z(ES+)[M+H]+=302.
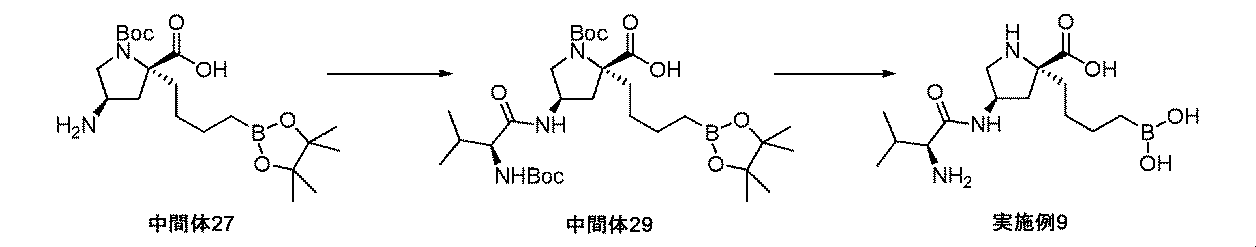
トリエチルアミン(0.21mL、1.5mmol)及びHATU(254mg、0.668mmol)を順次、Boc-Val-OH(145mg、0.668mmol)のDMF溶液(2.9mL)に加えて、反応液を室温にて30分間撹拌した。(2R,4R)-4-アミノ-1-(tert-ブトキシカルボニル)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体27、250mg、0.606mmol)を、DMF溶液(2.9mL)としての反応混合液に加えた。反応液を、室温にて一晩撹拌した。粗反応混合液を濃縮して、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって直接精製して、生成物(中間体29、250mg、67%の収率)を回転異性体の混合物として得た。1H NMR(300MHz,DMSO-d6)δ 0.64-0.73(2H,m),0.73-0.85(6H,m),1.13-1.14(1H,m),1.17(12H,s),1.22-1.42(22H,m),1.56-1.75(1H,m),1.79-1.97(1H,m),2.00-2.26(2H,m),3.08-3.24(1H,m),3.54-3.77(2H,m),4.12-4.36(1H,m),6.58(1H,t),7.96-8.03(2H,m);m/z(ES+)[M+H]+=584.
トリフルオロ酢酸(0.63mL、8.2mmol)を、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-2-(tert-ブトキシカルボニルアミノ)-3-メチルブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体29、250mg、0.409mmol)のDCM溶液(4mL)に加えた。生じた溶液を、室温にて1時間撹拌してから、真空下で濃縮した。粗アミノ酸を、Et2O(2mL)及び1M aq HCl(2mL)中に溶解した。フェニルボロン酸(99mg、0.81mmol)を加えて、澄明な二相性溶液を室温にて1時間撹拌した。反応混合液を水で希釈して、Et2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 60ccカラム)によって精製した。所望の生成物をカラムから、2Mアンモニア/メタノールを用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18Aq、水中0~10%アセトニトリル)によって精製して、(2R,4R)-4((S)-2-アミノ-3-メチルブタンアミド)-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例9、28mg、20%の収率)を、白色の固体、及び回転異性体の混合物として得た。1H NMR(300MHz,D2O)δ 0.66-0.76(2H,m),0.85(6H,dd),1.07-1.43(4H,m),1.55-1.68(1H,m),1.77-1.97(2H,m),2.13-2.33(2H,m),3.07(1H,d),3.08-3.16(1H,m),3.37-3.48(1H,m),4.27-4.40(1H,m);m/z(ES+)[M+H]+=330.
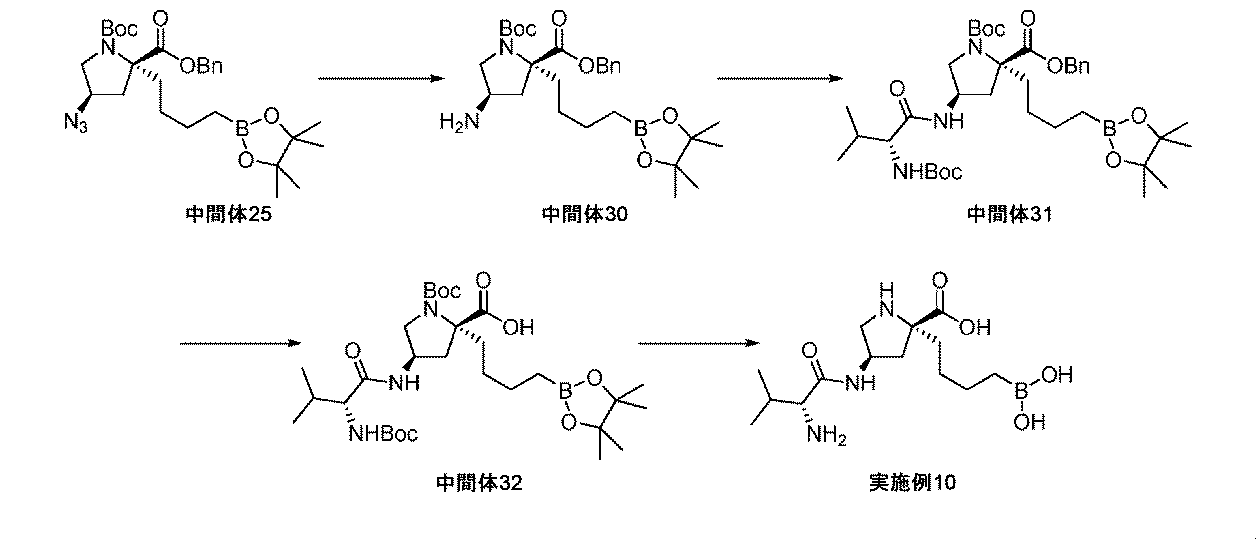
リンドラー触媒(5wt%、0.275g、2.58mmol)を、(2R,4R)-2-ベンジル1-tert-ブチル4-アジド-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体25、1.56g、2.95mmol)のTHF溶液(25mL)に加えた。懸濁液を、水素雰囲気(バルーン、フラスコを排気して、水素で充填し直し×3)下で、室温にて8.5時間撹拌した。反応混合液をMeOHで希釈して、珪藻土で濾過して、濾液を濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(DCM中1~15%MeOH)によって精製して、(2R,4R)-2-ベンジル1-tert-ブチル4-アミノ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体30、1.01g、68%の収率)をガムとして、そして回転異性体の混合物として得た。1H NMR(500MHz,CD2Cl2)δ 0.74(2H,q),1.20-1.22(14H,m),1.23-1.29(2H,m),1.32(6H,s),1.37-1.42(5H,m),1.74-1.83(1H,m),1.83-1.93(1H,m),2.11-2.19(0.6H,m),2.21-2.32(1.4H,m),3.19(0.4H,dd),3.28(0.6H,dd),3.44-3.51(1H,m),3.63(1H,dd),5.07-5.20(2H,m),7.28-7.34(1H,m),7.34-7.41(4H,m);m/z(ES+)[M+H]+=503.
N,N-ジイソプロピルエチルアミン(0.235mL、1.34mmol)を、HATU(245mg、0.64mmol)及びBoc-D-Val-OH(117mg、0.54mmol)の撹拌DMF溶液(2mL)に、室温にてゆっくり加えた。溶液を20分間撹拌してから、(2R,4R)-2-ベンジル1-tert-ブチル4-アミノ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体30、270mg、0.54mmol)のDMF溶液(2mL)を加えた。反応液を2.5時間撹拌して、DCM(30mL)で希釈して、水(3×25mL)及び飽和水性塩化ナトリウム(30mL)で順次洗浄した。有機層をMgSO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン中5~65%EtOAc)によって精製して、(2R,4R)-2-ベンジル1-tert-ブチル4-((R)-2-(tert-ブトキシカルボニルアミノ)-3-メチルブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体31、239mg、63%の収率)を無色の泡として、そして回転異性体の混合物として得た。1H NMR(500MHz,CD2Cl2)δ 0.74-0.81(2H,m),0.84(3H,d),0.87-0.94(3H,m),1.24(12H,s),1.26-1.34(2H,m),1.37(5H,s),1.40-1.43(2H,m),1.45(4H,s),1.46(9H,s),1.78-1.89(1H,m),1.95-2.07(2H,m),2.21-2.29(0.6H,m),2.31-2.46(1.4H,m),3.51-3.60(1.5H,m),3.65(0.5H,br d),3.72(1H,br dd),4.49-4.58(1H,m),5.01(1H,br d),5.19-5.29(2H,m),6.93-7.09(1H,m),7.36-7.40(1H,m),7.43(4H,app d);m/z(ES+)[M+H]+=702.
Pd/C(10wt%、25mg、0.23mmol)を、(2R,4R)-2-ベンジル1-tert-ブチル4-((R)-2-(tert-ブトキシカルボニルアミノ)-3-メチルブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体31、239mg、0.34mmol)のEtOAc溶液(4mL)に加えた。懸濁液を、水素雰囲気(バルーン、フラスコを排気して、水素で充填し直し×3)下で、室温にて2時間撹拌した。反応混合液をMeOHで希釈して、珪藻土で濾過して、濾液を濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(DCM中2~15%MeOH)によって精製して、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((R)-2-(tert-ブトキシカルボニルアミノ)-3-メチルブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体32、196mg、94%の収率)を白色の固体として、そして回転異性体の混合物として得た。1H NMR(500MHz,DMSO-d6)δ 0.63-0.71(2H,m),0.75-0.82(6H,m),1.15(12H,s),1.21-1.30(2H,m),1.32(6H,s),1.36(13H,br s),1.61-1.72(1H,m),1.81-1.90(1H,m),1.92-2.05(2H,m),2.05-2.13(0.6H,m),2.13-2.28(1.4H,m),3.03-3.14(1H,m),3.62(0.6H,t),3.66(1.4H,t),4.18-4.29(1H,m),6.59(1H,d),7.99(1H,br s),12.48(0.4H,br s),12.65(0.6H,br s);m/z(ES+)[M+H]+=612.
トリフルオロ酢酸(0.37mL、4.8mmol)を、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((R)-2-(tert-ブトキシカルボニルアミノ)-3-メチルブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体32、195mg、0.32mmol)の撹拌DCM溶液(2mL)に滴加した。反応溶液を室温にて22時間撹拌してから、減圧下で濃縮した。粗アミノ酸を、1M HCl aq(2mL)及びEt2O(2mL)中に溶解した。フェニルボロン酸(117mg、0.96mmol)を加えて、澄明な二相性溶液を室温にて5時間撹拌した。混合液をEt2O(20mL)及び水(5mL)で希釈して、層を分離させた。水性層をEt2Oで洗浄して、水性層を凍結乾燥した。生じた固体をMeOH(3mL)中に溶解して、イオン交換クロマトグラフィ(PoraPak Rxn CX 20ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア溶液(20mL)を用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18、水中0~80%アセトニトリル)によって精製して、(2R,4R)-4-((R)-2-アミノ-3-メチルブタンアミド)-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例10、46mg、44%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.73-0.80(2H,m),0.90(6H,app t),1.13-1.25(1H,m),1.26-1.35(1H,m),1.40(2H,quin),1.68-1.80(1H,m),1.85-1.96(1H,m),2.00(1H,td),2.29(1H,dd),2.37-2.45(1H,m),3.18(1H,d),3.28(1H,dd),3.59(1H,dd),4.36-4.49(1H,m);m/z(ES+)[M+H]+=330.
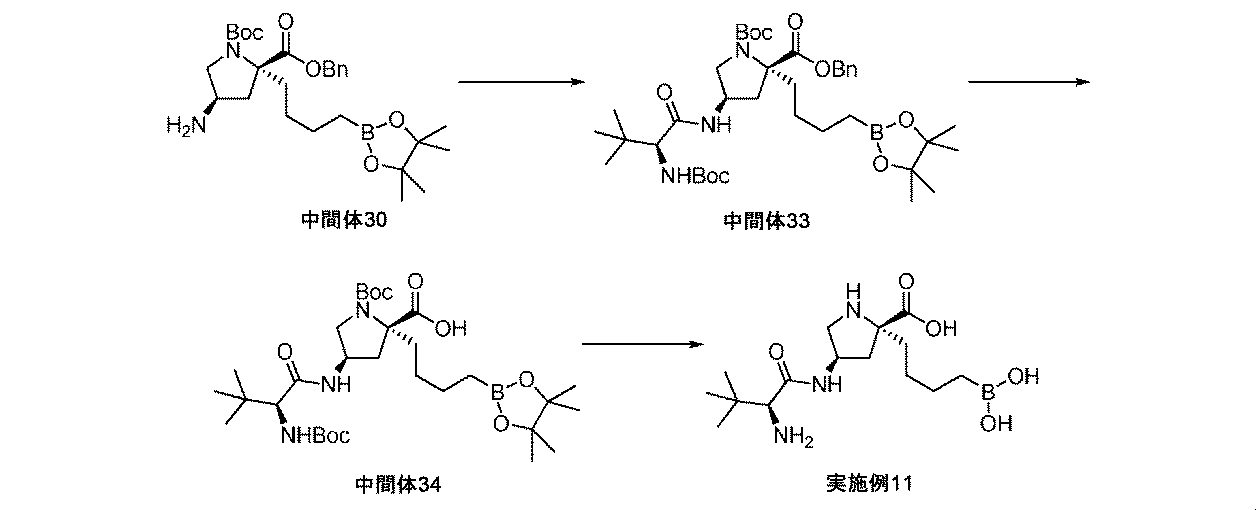
N,N-ジイソプロピルエチルアミン(0.165mL、0.95mmol)を、HATU(158mg、0.42mmol)及びBoc-Tle-OH(92mg、0.40mmol)の撹拌DMF溶液(1.5mL)に、室温にてゆっくり加えた。溶液を15分間撹拌してから、(2R,4R)-2-ベンジル1-tert-ブチル4-アミノ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体30、190mg、0.38mmol)のDMF溶液(1.5mL)を加えた。反応液を3時間撹拌して、EtOAc(30mL)で希釈して、水(3×25mL)、飽和水性NaHCO3(30mL)、及び飽和水性塩化ナトリウム(30mL)で順次洗浄した。有機層をMgSO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン中5~65%EtOAc)によって精製して、(2R,4R)-2-ベンジル1-tert-ブチル4-((S)-2-(tert-ブトキシカルボニルアミノ)-3,3-ジメチルブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体33、261mg、96%の収率)を白色の固体として、そして回転異性体の混合物として得た。1H NMR(500MHz,DMSO-d6)δ 0.69(2H,q),0.87(9H,s),1.17(12H,s),1.26(5H,s),1.29-1.32(1H,m),1.34(5H,s),1.38(9H,s),1.72-1.85(1H,m),1.90-2.08(2H,m),2.06-2.18(1H,m),2.22-2.35(2H,m),3.11-3.22(1H,m),3.68-3.81(2H,m),4.23-4.37(1H,m),5.06-5.19(2H,m),6.40(1H,t),7.31-7.40(5H,m),8.11(1H,d);m/z(ES+)[M+Na]+=738.
Pd/C(10wt%、25mg、0.23mmol)を、(2R,4R)-2-ベンジル1-tert-ブチル4-((S)-2-(tert-ブトキシカルボニルアミノ)-3,3-ジメチルブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体33、260mg、0.36mmol)のEtOAc溶液(4mL)に加えた。懸濁液を、水素雰囲気(バルーン、フラスコを排気して、水素で充填し直し×3)下で、室温にて15時間撹拌した。反応混合液をMeOHで希釈して、珪藻土で濾過して、濾液を濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(DCM中2~10%MeOH)によって精製して、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-2-(tert-ブトキシカルボニルアミノ)-3,3-ジメチルブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体34、207mg、91%の収率)を白色の固体として、そして回転異性体の混合物として得た。1H NMR(500MHz,DMSO-d6)δ 0.63-0.72(2H,m),0.86(9H,s),1.09-1.20(14H,m),1.21-1.30(2H,m),1.33(5H,s),1.35-1.38(13H,m),1.61-1.73(1H,m),1.89-2.11(2H,m),2.14-2.27(1H,m),3.06-3.14(1H,m),3.59-3.72(1H,m),3.72-3.80(1H,m),4.20-4.30(1H,m),6.35(1H,d),8.08(1H,br s),12.47(0.4H,br s),12.63(0.6H,br s);m/z(ES+)[M+H]+=626.
トリフルオロ酢酸(0.38mL、4.9mmol)を、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-2-(tert-ブトキシカルボニルアミノ)-3,3-ジメチルブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体34、206mg、0.33mmol)の撹拌DCM溶液(2mL)に滴加した。反応液を室温にて15時間撹拌してから、減圧下で濃縮した。粗アミノ酸を、1M HCl aq(4mL)及びEt2O(4mL)中に溶解した。フェニルボロン酸(120mg、0.99mmol)を加えて、澄明な二相性溶液を室温にて3時間撹拌した。混合液をEt2O(20mL)及び水(5mL)で希釈して、層を分離させた。水性層をEt2Oで洗浄して、水性層を凍結乾燥した。生じた固体をMeOH(3mL)中に溶解して、イオン交換クロマトグラフィ(PoraPak Rxn CX 20ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア溶液(20mL)を用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18、水中2~50%アセトニトリル)によって精製して、(2R,4R)-4-((S)-2-アミノ-3,3-ジメチルブタンアミド)-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例11、40mg、35%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.72(2H,td),0.89(9H,s),1.10-1.21(1H,m),1.22-1.30(1H,m),1.35(2H,quin),1.64-1.75(1H,m),1.90-2.02(1H,m),2.22-2.34(2H,m),3.04(1H,s),3.22(1H,dd),3.56(1H,dd),4.41(1H,quin);m/z(ES+)[M+H]+=344.
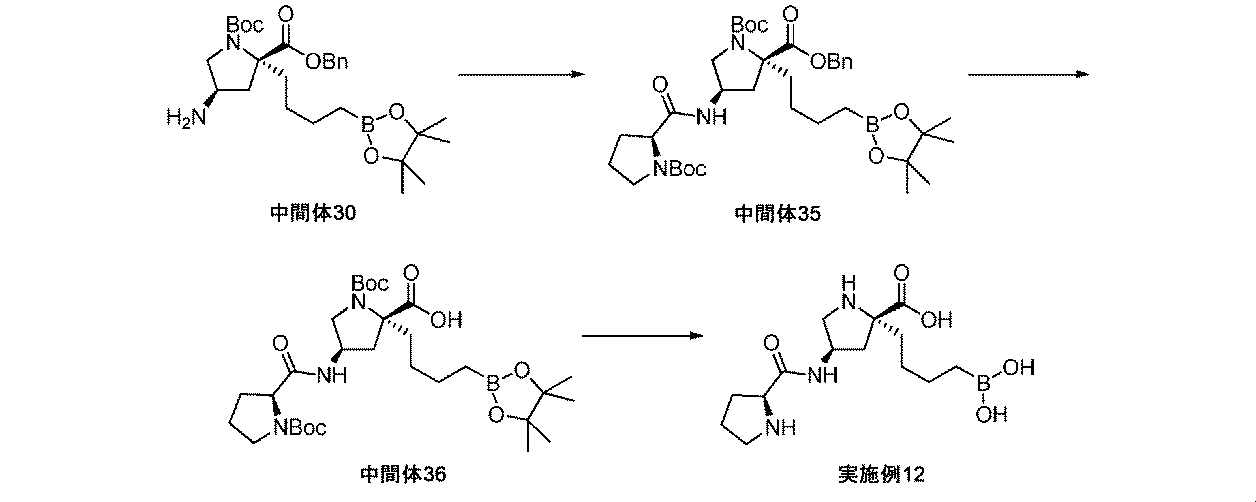
N,N-ジイソプロピルエチルアミン(0.182mL、1.04mmol)を、HATU(175mg、0.46mmol)及びBoc-Pro-OH(94mg、0.44mmol)の撹拌DMF溶液(1.5mL)に室温にてゆっくり加えた。溶液を20分間撹拌してから、(2R,4R)-2-ベンジル1-tert-ブチル4-アミノ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体30、210mg、0.42mmol)のDMF溶液(1.5mL)を加えた。反応液を2時間撹拌して、EtOAc(30mL)で希釈して、水(3×25mL)、飽和水性NaHCO3、及び飽和水性塩化ナトリウム(30mL)で順次洗浄した。有機層をMgSO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン中5~100%EtOAc)によって精製して、(2R,4R)-2-ベンジル1-tert-ブチル4-((S)-1-(tert-ブトキシカルボニル)ピロリジン-2-カルボキサミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体35、249mg、85%の収率)を無色の膜として、そして回転異性体の混合物として得た。1H NMR(500MHz,CD2Cl2)δ 0.68-0.79(2H,m),1.20(12H,s),1.31(5H,s),1.36-1.48(16H,m),1.74-1.87(3H,m),1.89-2.10(3H,m),2.13-2.46(2H,m),3.27-3.40(1H,m),3.44(2H,br s),3.50-3.64(2H,m),3.78-4.05(1H,m),4.49(1H,br s),5.10-5.27(2H,m),7.10(1H,br s),7.30-7.42(5H,m);m/z(ES+)[M+H]+=700.
Pd/C(10wt%、25mg、0.23mmol)を、(2R,4R)-2-ベンジル1-tert-ブチル4-((S)-1-(tert-ブトキシカルボニル)ピロリジン-2-カルボキサミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体35、249mg、0.36mmol)のEtOAc溶液(4mL)に加えた。懸濁液を、水素雰囲気(バルーン、フラスコを排気して、水素で充填し直し×3)下で、室温にて5時間撹拌した。反応混合液をMeOHで希釈して、珪藻土で濾過して、濾液を減圧下で濃縮して、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-1-(tert-ブトキシカルボニル)ピロリジン-2-カルボキサミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体36、207mg、87%の収率)を無色の膜として、そして回転異性体の混合物として得た。これを、更なる精製なしに用いた。1H NMR(500MHz,CD2Cl2)δ 0.66-0.83(2H,m),1.21(12H,s),1.34-1.51(21H,m),1.63-1.97(4H,m),2.06(1H,m),2.11-2.29(2H,m),2.33-2.67(1H,m),3.24-3.52(3H,m),3.53-3.67(1H,m),4.15-4.34(1H,m),4.47-4.74(1H,m),6.76-7.23(1H,m),7.17-7.69(1H,m),9.74(1H,br s);m/z(ES+)[M+H]+=610.
トリフルオロ酢酸(0.518mL、6.73mmol)を、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-1-(tert-ブトキシカルボニル)ピロリジン-2-カルボキサミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体36、205mg、0.34mmol)の撹拌DCM溶液(2mL)に滴加した。反応液を室温にて2時間撹拌してから、減圧下で濃縮した。粗アミノ酸を、1M HCl aq(4mL)及びEt2O(4mL)中に溶解した。フェニルボロン酸(123mg、1.01mmol)を加えて、澄明な二相性溶液を室温にて2時間撹拌した。混合液をEt2O(20mL)及び水(5mL)で希釈して、層を分離させた。水性層をEt2Oで洗浄して、水性層を凍結乾燥した。生じた固体をMeOH(3mL)中に溶解して、イオン交換クロマトグラフィ(PoraPak Rxn CX 20ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア溶液(20mL)を用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18、水中0~50%アセトニトリル)によって精製して、(2R,4R)-2-(4-ボロノブチル)-4-((S)-ピロリジン-2-カルボキサミド)ピロリジン-2-カルボン酸(実施例12、89mg、81%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.70-0.79(2H,m),1.11-1.23(1H,m),1.24-1.33(1H,m),1.34-1.42(2H,m),1.60-1.71(1H,m),1.81-1.91(3H,m),1.91-1.99(1H,m),2.18(1H,dd),2.22-2.29(1H,m),2.40(1H,dd),3.08-3.16(1H,m),3.16-3.22(1H,m),3.25(1H,dd),3.48(1H,dd),3.99(1H,dd),4.29-4.38(1H,m);m/z(ES+)[M+H]+=328.
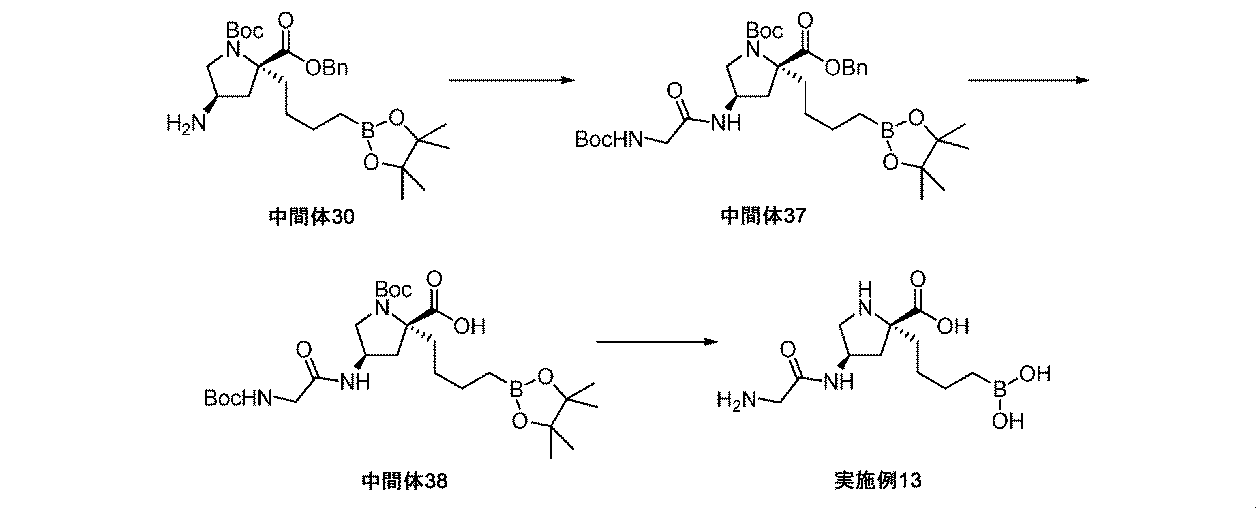
N,N-ジイソプロピルエチルアミン(0.182mL、1.04mmol)を、HATU(175mg、0.46mmol)及びBoc-Gly-OH(77mg、0.44mmol)の撹拌DMF溶液(1.5mL)に室温にてゆっくり加えた。溶液を20分間撹拌してから、(2R,4R)-2-ベンジル1-tert-ブチル4-アミノ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体30、210mg、0.42mmol)のDMF溶液(1.5mL)を加えた。反応溶液を2時間撹拌して、EtOAc(30mL)で希釈して、水(3×25mL)、飽和水性NaHCO3、及び飽和水性塩化ナトリウム(30mL)で順次洗浄した。有機層をMgSO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン中10~100%EtOAc)によって精製して、(2R,4R)-2-ベンジル1-tert-ブチル4-(2-(tert-ブトキシカルボニルアミノ)アセトアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体37、235mg、85%の収率)を無色の膜として、そして回転異性体の混合物として得た。1H NMR(500MHz,CD2Cl2)δ 0.70-0.78(2H,m),1.12-1.18(1H,m),1.20(12H,s),1.25-1.30(1H,m),1.33(5H,s),1.43(15H,s),1.74-1.86(1H,m),1.95(0.5H,br d),2.03(0.5H,br d),2.15-2.26(1H,m),2.26-2.37(1H,m),2.40(1H,dd),3.43-3.57(3H,m),3.57-3.64(1H,m),4.50(1H,br s),5.03(0.5H,br s),5.10(0.5H,br s),5.13-5.25(1H,m),7.01(1H,dd),7.32-7.37(1H,m),7.36-7.41(4H,m);m/z(ES+)[M+H]+=660.
Pd/C(10wt%、25mg、0.23mmol)を、(2R,4R)-2-ベンジル1-tert-ブチル4-(2-(tert-ブトキシカルボニルアミノ)アセトアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体37、233mg、0.35mmol)のEtOAc溶液(4mL)に加えた。懸濁液を、水素雰囲気(バルーン、フラスコを排気して、水素で充填し直し×3)下で、室温にて6時間撹拌した。反応混合液をMeOHで希釈して、珪藻土で濾過して、濾液を濃縮して乾燥させて、(2R,4R)-1-(tert-ブトキシカルボニル)-4-(2-(tert-ブトキシカルボニルアミノ)アセトアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体38、176mg、87%の収率)を無色の膜として、そして回転異性体の混合物として得た。これを、更なる精製なしに用いた。1H NMR(500MHz,CD2Cl2)δ 0.65-0.80(2H,m),1.11-1.18(1H,m),1.18-1.23(12H,m),1.25-1.32(1H,m),1.36-1.45(13H,m),1.47(7H,s),1.70-1.83(0.4H,m),1.84-1.95(0.6H,m),2.06-2.27(2H,m),2.33-2.47(0.4H,m),2.63(0.6H,br d),3.44-3.62(2H,m),3.63-3.82(2H,m),4.28(0.6H,br s),4.36-4.60(0.4H,m),5.26(0.6H,br s),5.58-5.90(0.3H,m),6.83(0.6H,br s),6.97-7.44(0.4H,m);m/z(ES+)[M+H]+=570.
トリフルオロ酢酸(0.476mL、6.18mmol)を、(2R,4R)-1-(tert-ブトキシカルボニル)-4-(2-(tert-ブトキシカルボニルアミノ)アセトアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体38、176mg、0.31mmol)の撹拌DCM溶液(2mL)に滴加した。反応溶液を室温にて2時間撹拌してから、減圧下で濃縮した。粗アミノ酸を、1M HCl aq(4mL)及びEt2O(4mL)中に溶解した。フェニルボロン酸(113mg、0.93mmol)を加えて、澄明な二相性溶液を室温にて2時間撹拌した。混合液をEt2O(20mL)及び水(5mL)で希釈して、層を分離させた。水性層をEt2Oで洗浄して、水性層を凍結乾燥した。生じた固体をMeOH(3mL)中に溶解して、イオン交換クロマトグラフィ(PoraPak Rxn CX 20ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア溶液(20mL)を用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18、水中0~40%アセトニトリル)によって精製して、(2R,4R)-4-(2-アミノアセトアミド)-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例13、62mg、70%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.76(2H,td),1.15-1.25(1H,m),1.26-1.34(1H,m),1.36-1.46(2H,m),1.69-1.79(1H,m),2.00(1H,ddd),2.27(1H,dd),2.44(1H,dd),3.33(1H,dd),3.42(2H,s),3.59(1H,dd),4.36-4.45(1H,m);m/z(ES+)[M+H]+=288.
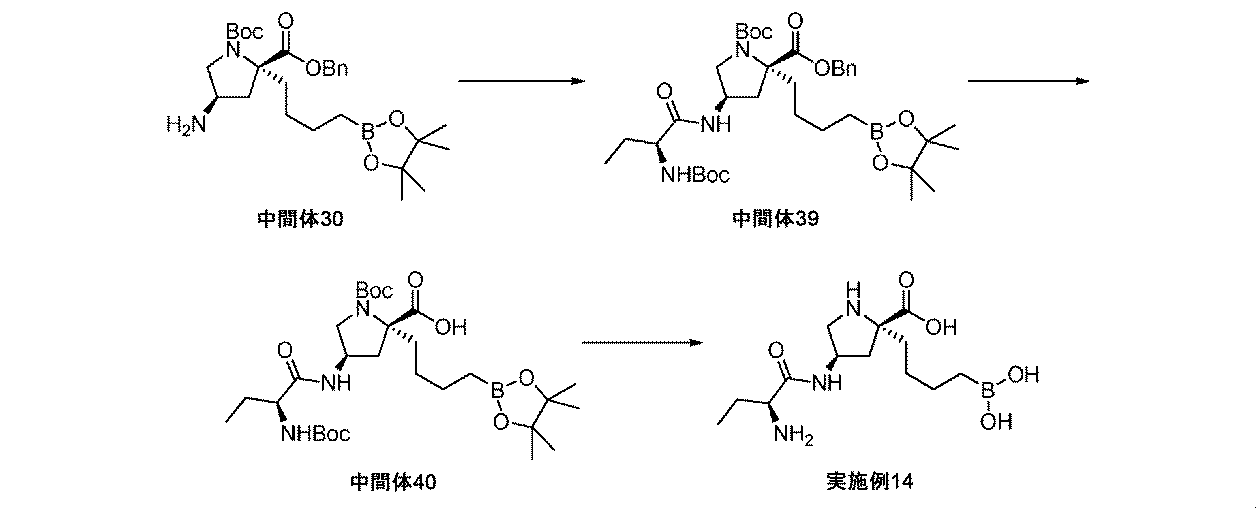
HATU(804mg、2.11mmol)を、Boc-Abu-OH(430mg、2.11mmol)のDMF溶液(4mL)に加えて、反応液を、室温にて10分間撹拌した。(2R,4R)-2-ベンジル1-tert-ブチル4-アミノ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体30、885mg、1.76mmol)を、DMF溶液(3mL)としての反応混合液に加えた。N,N-ジイソプロピルエチルアミン(0.75mL、4.3mmol)を加えて、反応液を室温にて一晩撹拌した。次に、反応液を、水(15mL)及びEt2O(10mL)で希釈した。層を分離させて、水性層をEt2Oで抽出した(2×10mL)。組み合わせた有機質を、5%水性塩化リチウム(10mL)で洗浄して、MgSO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、(2R,4R)-2-ベンジル1-tert-ブチル4-((S)-2-(tert-ブトキシカルボニルアミノ)ブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体39、766mg、63%の収率)を白色の固体として得た。1H NMR(500MHz,CDCl3)δ 0.70-0.84(5H,m),1.09-1.19(1H,m),1.21(12H,s),1.36-1.46(18H,m),1.46-1.97(7H,m),2.12-2.45(2H,m),3.45-3.60(1H,m),3.68(1H,br d),3.78-3.94(1H,m),4.42-4.64(1H,m),4.73-5.03(1H,m),5.07-5.33(2H,m),7.09(1H,br d),7.28-7.40(5H,m).m/z(ES+)[M+H]+=688.
(2R,4R)-2-ベンジル1-tert-ブチル4-((S)-2-(tert-ブトキシカルボニルアミノ)ブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体39、766mg、1.11mmol)を、EtOAc(11mL)中に溶解して、Pd/C(10wt%、119mg、0.11mmol)により処理した。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液を、珪藻土で濾過して、EtOAc及びメタノールでリンスした。濾液を、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-2-(tert-ブトキシカルボニルアミノ)ブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体40、470mg、70%の収率)を白色の泡として得た。1H NMR(500MHz,CDCl3)δ 0.67-0.82(2H,m),0.89(3H,br t),1.11-1.28(14H,m),1.37-1.51(20H,m),1.53-1.65(1H,m),1.65-1.94(2H,m),2.02-2.12(1H,m),2.13-2.31(1H,m),2.70(1H,br d),3.40-3.62(2H,m),3.88-4.04(1H,m),4.26(1H,br s),5.01(1H,br s),6.73(1H,br d);m/z(ES+)[M+H]+=598.
フェニルボロン酸(192mg、1.57mmol)を、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-2-(tert-ブトキシカルボニルアミノ)ブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体40、470mg、0.79mmol)の2M HCl aq溶液(5mL)に加えて、反応液を室温にて16時間撹拌した。反応液を、水(10mL)及びEt2O(10mL)で希釈して、層を分離させた。水性層を、Et2O(3×5mL)で洗浄してから凍結乾燥すると、泡になった。有機層を、真空下で濃縮した。生じた残留物を、ジオキサン中4M HCl(4mL、16mmol)中で希釈して、生じた溶液を、室温にて20時間撹拌した。反応液を、水(10mL)及びEt2O(10mL)で希釈して、層を分離させた。水性層をEt2O(3×5mL)で洗浄してから凍結乾燥すると、泡になった。これらの2つの操作由来の泡を組み合わせて、生じた粗アミノ酸を、イオン交換クロマトグラフィ(Silicycle SiliaSep SPE-R51230B-20X 5gカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア溶液を用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18Aq、水中0~25%アセトニトリル)によって精製して、(2R,4R)-4-((S)-2-アミノブタンアミド)-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例14、96mg、39%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.55-0.82(2H,m),0.88(3H,t),1.12-1.48(4H,m),1.56-1.81(3H,m),1.86-2.10(1H,m),2.12-2.53(2H,m),3.15-3.37(1H,m),3.41-3.53(1H,m),3.62(1H,dd),4.35-4.52(1H,m);m/z(ES+)[M-H2O+H]+=298.
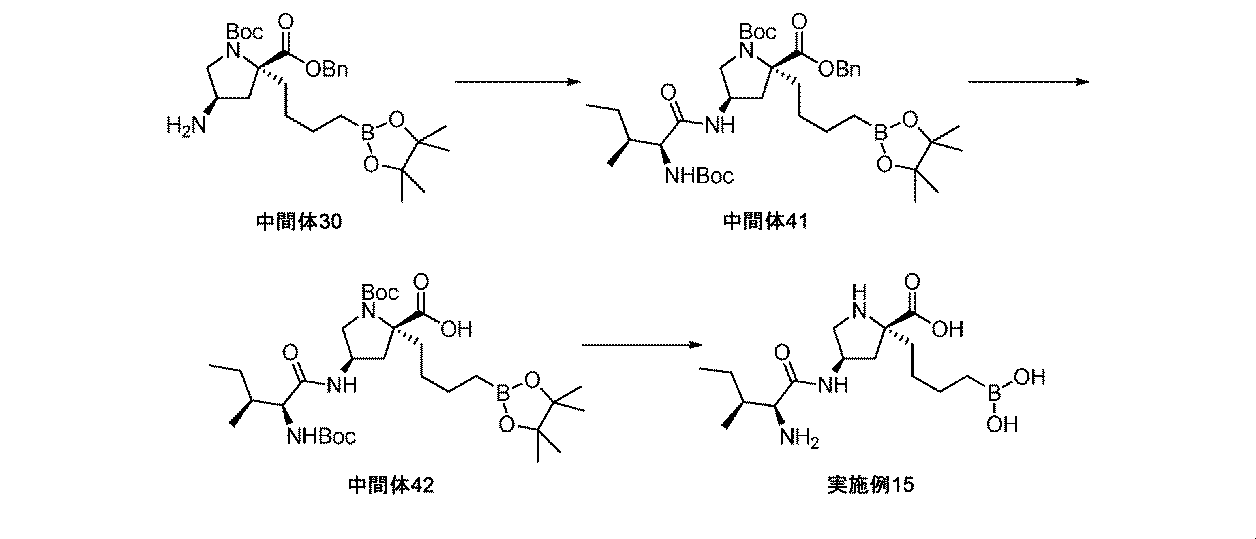
HATU(804mg、2.11mmol)を、Boc-Ile-OH(489mg、2.11mmol)のDMF溶液(4mL)に加えて、反応液を、室温にて10分間撹拌した。(2R,4R)-2-ベンジル1-tert-ブチル4-アミノ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体30、885mg、1.76mmol)を、DMF溶液(3mL)としての反応混合液に加えた。N,N-ジイソプロピルエチルアミン(0.75mL、4.3mmol)を加えて、反応液を室温にて一晩撹拌した。次に、反応液を、水(15mL)及びEt2O(10mL)で希釈した。層を分離させて、水性層をEt2Oで抽出した(2×10mL)。組み合わせた有機層を、5%水性塩化リチウム(10mL)で洗浄して、MgSO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、(2R,4R)-2-ベンジル1-tert-ブチル4-((2S,3S)-2-(tert-ブトキシカルボニルアミノ)-3-メチルペンタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体41、707mg、56%の収率)を白色の固体として得た。1H NMR(500MHz,CDCl3)δ 0.79(2H,br t),0.83-1.05(8H,m),1.12-1.21(1H,m),1.24(12H,s),1.32-1.61(20H,m),1.62-2.02(4H,m),2.18-2.55(2H,m),3.49-3.65(1H,m),3.65-3.78(1H,m),3.90(1H,br s),4.53-4.72(1H,m),4.95(1H,br s),5.07-5.43(2H,m),7.16(1H,br d),7.30-7.44(5H,m).m/z(ES+)[M+H]+=716.
(2R,4R)-2-ベンジル1-tert-ブチル4-((2S,3S)-2-(tert-ブトキシカルボニルアミノ)-3-メチルペンタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体41、707mg、0.99mmol)を、EtOAc(10mL)中に溶解して、Pd/C(10wt%、105mg、0.10mmol)により処理した。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液を、珪藻土で濾過して、EtOAc及びメタノールでリンスした。濾液を濃縮して乾燥させて、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((2S,3S)-2-(tert-ブトキシカルボニルアミノ)-3-メチルペンタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体42、603mg、98%の収率)を得た。これを、更なる精製なしに用いた。1H NMR(500MHz,CDCl3)δ 0.68-0.80(2H,m),0.83-0.93(6H,m),1.05-1.14(1H,m),1.21(12H,s),1.28-1.36(1H,m),1.37-1.55(22H,m),1.71-1.96(2H,m),2.18-2.31(1H,m),2.72(1H,br d),3.42-3.51(2H,m),3.52-3.63(1H,m),3.86-4.04(1H,m),4.16-4.34(1H,m),4.98(1H,br d),6.69(1H,br s);m/z(ES+)[M+H]+=626.
トリフルオロ酢酸(1.10mL、14.3mmol)を、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((2S,3S)-2-(tert-ブトキシカルボニルアミノ)-3-メチルペンタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体42、603mg、0.96mmol)のDCM溶液(6mL)に加えた。生じた溶液を、室温にて16時間撹拌してから、真空下で濃縮した。粗アミノ酸を、Et2O(10mL)中に溶解して、真空下で再濃縮した。この再溶解再濃縮プロセスを、さらに2回繰り返した。次に、粗アミノ酸を、Et2O(6mL)及び1M HCl aq(6mL)中に溶解した。フェニルボロン酸(235mg、1.93mmol)を加えて、澄明な二相性溶液を室温にて3時間撹拌した。反応混合液を水で希釈して、Et2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(Silicycle SiliaSep SPE-R51230B-20X 5gカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア溶液を用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18Aq、水中0~25%アセトニトリル)によって精製して、(2R,4R)-4-((2S,3S)-2-アミノ-3-メチルペンタンアミド)-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例15、136mg、41%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.55-0.81(2H,m),0.89(6H,dd),1.07-1.52(6H,m),1.63-1.83(2H,m),1.86-2.09(1H,m),2.13-2.51(2H,m),3.11-3.40(2H,m),3.45-3.67(1H,m),4.39-4.54(1H,m);m/z(ES+)[M+H]+=344.
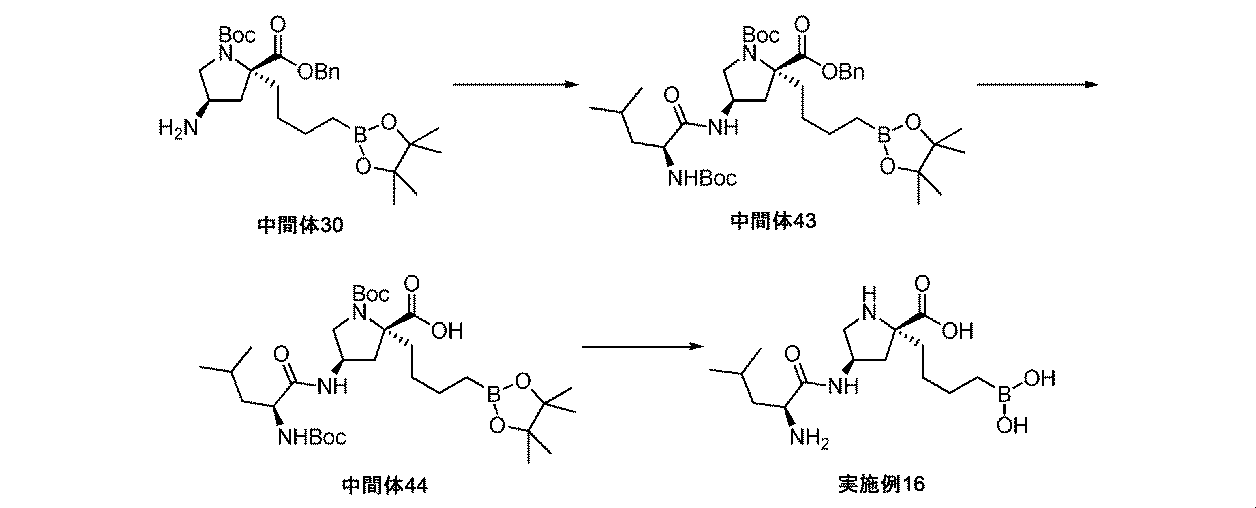
HATU(247mg、0.65mmol)を、Boc-Leu-OH(125mg、0.54mmol)のDCM溶液(2mL)に加えて、反応液を、室温にて10分間撹拌した。(2R,4R)-2-ベンジル1-tert-ブチル4-アミノ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体30、272mg、0.54mmol)を、DCM溶液(2mL)としての反応混合液に加えた。N,N-ジイソプロピルエチルアミン(0.19mL、1.1mmol)を加えて、反応液を室温にて1時間撹拌した。次に、反応液を、DCM(20mL)で希釈して、水(25mL)及び飽和水性塩化ナトリウム(30mL)で順次洗浄した。有機層を、MgSO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、(2R,4R)-2-ベンジル1-tert-ブチル4-((S)-2-(tert-ブトキシカルボニルアミノ)-4-メチルペンタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体43、210mg、54%の収率)を無色の泡として、そして回転異性体の混合物として得た。1H NMR(500MHz,CDCl3)δ 0.77(2H,t),0.91(6H,d),1.06-1.19(1H,m),1.19-1.24(12H,m),1.31-1.51(20H,m),1.50-1.63(2H,m),1.73-2.02(2H,m),2.17-2.55(2H,m),3.35-3.75(2H,m),3.84-4.06(1H,m),4.35-4.75(2H,m),5.00-5.46(2H,m),7.08-7.22(1H,m),7.28-7.42(5H,m);m/z(ES+)[M+H]+=716.
(2R,4R)-2-ベンジル1-tert-ブチル4-((S)-2-(tert-ブトキシカルボニルアミノ)-4-メチルペンタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体43、201mg、0.28mmol)を、EtOAc(4mL)中に溶解して、Pd/C(10wt%、100mg、0.094mmol)により処理した。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて2時間撹拌した。反応混合液を、珪藻土で濾過して、EtOAc及びメタノールでリンスした。濾液を、濃縮して乾燥させて、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-2-(tert-ブトキシカルボニルアミノ)-4-メチルペンタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体44、170mg、97%の収率)を白色の固体として、そして回転異性体の混合物として得た。1H NMR(500MHz,CDCl3)δ 0.78(2H,t),0.93(6H,d),1.15-1.25(12H,m),1.25-1.31(2H,m),1.39-1.51(19H,m),1.55-1.72(2H,m),1.73-1.89(1H,m),2.01-2.11(1H,m),2.18-2.36(1H,m),2.47-2.83(1H,m),3.37-3.74(2H,m),3.96-4.10(1H,m),4.17-4.32(1H,m),4.82-5.31(1H,m),6.62-7.12(1H,m);m/z(ES+)[M+H]+=626.
トリフルオロ酢酸(1.00mL、13.0mmol)を、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-2-(tert-ブトキシカルボニルアミノ)-4-メチルペンタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体44、170mg、0.27mmol)のDCM溶液(2mL)に加えた。生じた溶液を、室温にて2時間撹拌してから、真空下で濃縮した。次に、粗アミノ酸を、Et2O(5mL)及び水(4mL)中に溶解した。フェニルボロン酸(66mg、0.54mmol)を加えて、澄明な二相性溶液を室温にて2時間撹拌した。反応混合液を水(5mL)及びEt2O(20mL)で希釈して、層を分離させた。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 2gカラム)によって精製して、(2R,4R)-4-((S)-2-アミノ-4-メチルペンタンアミド)-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例16、88mg、94%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.46-0.68(2H,m),0.73-0.82(6H,m),1.03-1.13(1H,m),1.13-1.23(1H,m),1.23-1.42(4H,m),1.42-1.52(1H,m),1.54-1.66(1H,m),1.76-1.93(1H,m),2.07-2.19(1H,m),2.29(1H,dd),3.13(1H,q),3.34(1H,t),3.48(1H,q),4.23-4.40(1H,m);m/z(ES+)[M+H]+=344.
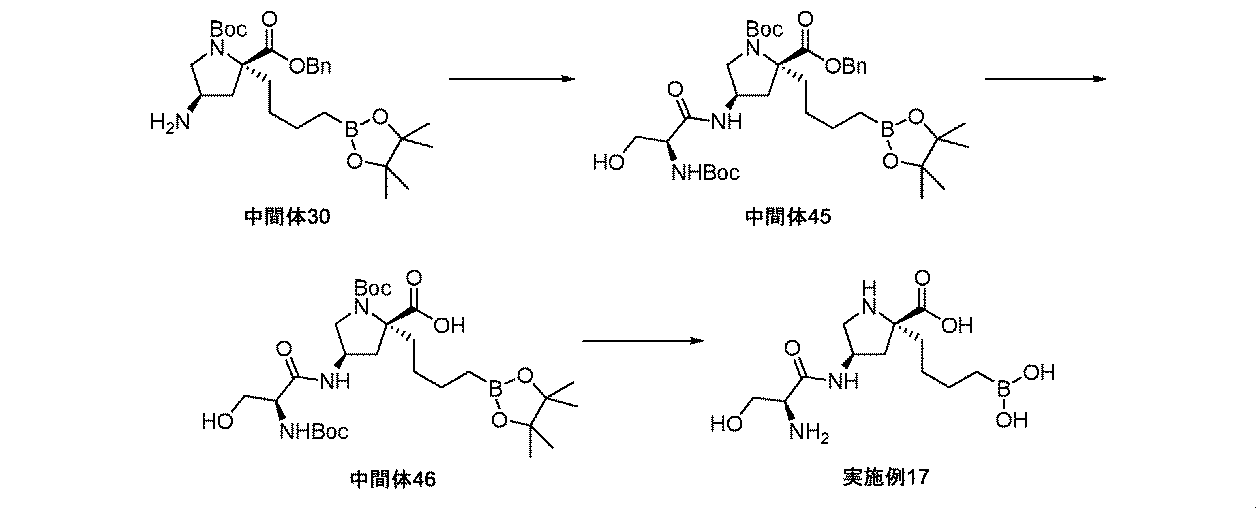
N,N-ジイソプロピルエチルアミン(0.108mL、0.62mmol)を、COMU(292mg、0.68mmol)、(2R,4R)-2-ベンジル1-tert-ブチル4-アミノ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体30、311mg、0.62mmol)、及びBoc-Ser-OH(133mg、0.65mmol)の撹拌DMF溶液(5mL)に室温にて加えた。反応液を3時間撹拌して、水(80mL)及びEtOAc(15mL)で希釈した。相を分離させて、水性相をさらに飽和水性NaHCO3で希釈してから、EtOAcで抽出した(2×20mL)。組み合わせた有機質を、飽和水性NaCl(2×10mL)で洗浄して、MgSO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、(2R,4R)-2-ベンジル1-tert-ブチル4-((S)-2-(tert-ブトキシカルボニルアミノ)-3-ヒドロキシプロパンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体45、358mg、84%の収率)を無色の乾いた膜として、そして回転異性体の混合物として得た。1H NMR(500MHz,CDCl3)δ 0.76(2H,t),1.16(1H,m),1.21(12H,s),1.31(6H,s),1.35-1.42(5H,m),1.43(11H,s),1.73-1.87(1H,m),1.87-2.02(2H,m),2.14-2.24(1H,m),2.29-2.41(1H,m),3.43-3.52(0.4H,m),3.52-3.61(2H,m),3.66(0.6H,d),3.79-3.92(1H,m),3.92-4.04(1H,m),4.51(1H,br s),5.06-5.26(2H,m),5.36(1H,br s),7.30-7.40(5H,m);m/z(ES+)[M+H]+=690.
(2R,4R)-2-ベンジル1-tert-ブチル4-((S)-2-(tert-ブトキシカルボニルアミノ)-3-ヒドロキシプロパンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体45、358mg、0.52mmol)を、EtOAc(4mL)中に溶解して、Pd/C(10wt%、50mg、0.047mmol)により処理した。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて3.5時間撹拌した。反応混合液を、MeOHで希釈して、珪藻土で濾過して、濃縮して乾燥させて、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-2-(tert-ブトキシカルボニルアミノ)-3-ヒドロキシプロパンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体46、303mg、97%の収率)を白色の固体として、そして回転異性体の混合物として得た。これを、更なる精製なしに用いた。1H NMR(500MHz,CD2Cl2)δ 0.67-0.91(2H,m),1.25(14H,s),1.41-1.47(3H,m),1.48(9H,s),1.53(10H,s),1.82-1.90(2H,m),2.06-2.14(1H,m),2.24-2.33(1H,m),2.76-2.91(1H,m),3.48-3.54(1H,m),3.59(1H,dd),3.70(1H,dd),3.93(1H,d),4.03-4.17(1H,m),4.29(1H,d),6.79-6.98(1H,m);m/z(ES+)[M+H]+=600.
トリフルオロ酢酸(0.771mL、10.01mmol)を、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-2-(tert-ブトキシカルボニルアミノ)-3-ヒドロキシプロパンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体46、300mg、0.50mmol)の撹拌DCM溶液(4mL)に室温にて滴加した。1.5時間後、溶液を減圧下で濃縮して、生じた残留物を、1M HCl aq(4mL、4.00mmol)及びEt2O(4mL)中に溶解した。フェニルボロン酸(183mg、1.50mmol)を加えて、澄明な二相性溶液を、室温にて3時間撹拌した。混合液をEt2O(20mL)及び水(5mL)で希釈して、層を分離させた。水性層をEt2Oで洗浄して、層を分離させて、水性層を凍結乾燥した。生じた固体をMeOH(3mL)中に溶解して、イオン交換クロマトグラフィ(PoraPak Rxn CX 20ccカラム)にかけた。所望の生成物をカラムから、MeOH中5%アンモニア溶液(20mL)を用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18、水中0~30%アセトニトリル)によって精製して、(2R,4R)-4-((S)-2-アミノ-3-ヒドロキシプロパンアミド)-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例17、94mg、59%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.77(2H,td),1.16-1.26(1H,m),1.26-1.35(1H,m),1.35-1.45(2H,m),1.76(1H,ddd),2.02(1H,ddd),2.29(1H,dd),2.46(1H,dd),3.35(1H,dd),3.48(1H,t),3.62(1H,dd),3.66-3.77(2H,m),4.40-4.50(1H,m);m/z(ES+)[M+H]+=318.
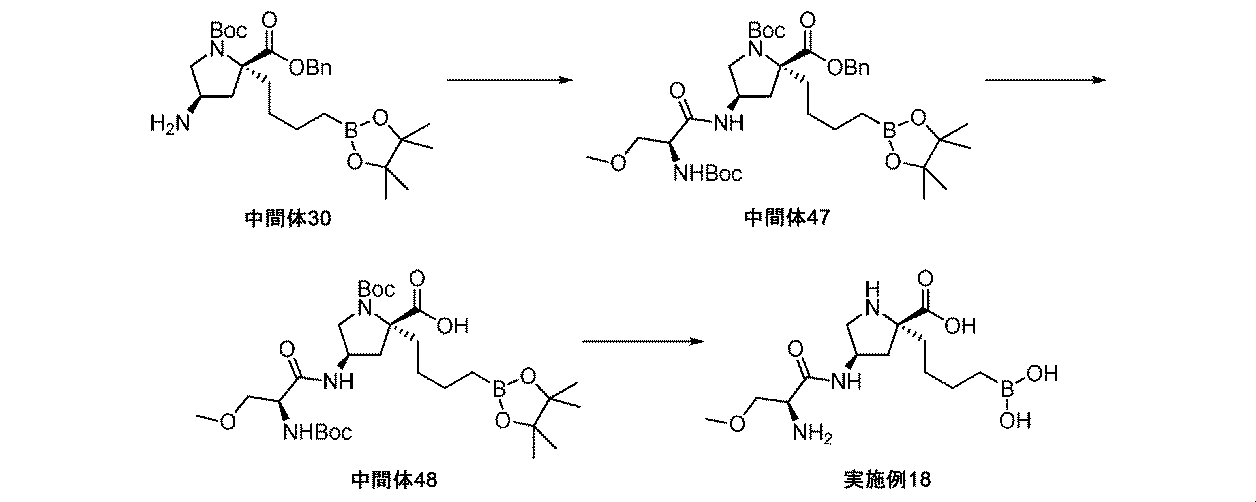
N,N-ジイソプロピルエチルアミン(0.082mL、0.47mmol)を、COMU(220mg、0.51mmol)、(2R,4R)-2-ベンジル1-tert-ブチル4-アミノ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体30、235mg、0.47mmol)、及びN-Boc-O-メチル-L-セリン(108mg、0.49mmol)の撹拌DMF溶液(3mL)に室温にて加えた。反応液を2時間撹拌してから、水(60mL)及びDCM(15mL)で希釈した。相を分離させて、水性相をDCM(2×20mL)で抽出した。組み合わせた有機質を、飽和水性NaHCO3(30mL)、飽和水性NaCl(2×10mL)で洗浄して、MgSO4上で乾燥させて、濾過して、減圧下で濃縮した。生じた残留物を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、(2R,4R)-2-ベンジル1-tert-ブチル4-((S)-2-(tert-ブトキシカルボニルアミノ)-3-メトキシプロパンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体47、145mg、44%の収率)を無色の乾いた膜として、そして回転異性体の混合物として得た。1H NMR(500MHz,CDCl3)δ 0.78(2H,t),1.18(1H,br dd),1.22-1.26(13H,m),1.33(6H,s),1.44(5H,br s),1.46(9H,s),1.57-1.74(1H,m),1.74-1.85(1H,m),1.94(0.4H,d),2.01(0.6H,d),2.18-2.27(0.6H,m),2.32-2.47(1.4H,m),3.32(3H,s),3.39-3.46(1H,m),3.49-3.55(0.4H,m),3.60(1H,dd),3.65-3.76(1.6H,m),4.15(1H,br d),4.51-4.64(1H,m),5.06-5.22(2H,m),5.23-5.34(1H,m),7.32-7.39(5H,m);m/z(ES+)[M+H]+=704.
(2R,4R)-2-ベンジル1-tert-ブチル4-((S)-2-(tert-ブトキシカルボニルアミノ)-3-メトキシプロパンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体47、145mg、0.21mmol)を、EtOAc(2mL)中に溶解して、Pd/C(10wt%、22mg、0.021mmol)により処理した。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて4時間撹拌した。反応混合液を、MeOHで希釈して、珪藻土で濾過して、濃縮して乾燥させて、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-2-(tert-ブトキシカルボニルアミノ)-3-メトキシプロパンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体48、126mg、100%の収率)を白色の固体として、そして回転異性体の混合物として得た。これを、更なる精製なしに用いた。1H NMR(500MHz,CD2Cl2)δ 0.65-0.80(2H,m),1.14-1.29(14H,m),1.35-1.43(5H,m),1.44(7H,s),1.46-1.60(8H,m),1.77-1.95(1H,m),2.03-2.14(1H,m),2.14-2.26(1H,m),2.68(1H,br d),3.33(3H,s),3.38-3.47(1H,m),3.47-3.60(2H,m),3.65-3.76(1H,m),4.02-4.15(1H,m),4.24(1H,br s),5.38(1H,br s),7.06(1H,br s);m/z(ES+)[M+H]+=614.
トリフルオロ酢酸(0.25mL、3.26mmol)を、(2R,4R)-1-(tert-ブトキシカルボニル)-4-((S)-2-(tert-ブトキシカルボニルアミノ)-3-メトキシプロパンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-2-カルボン酸(中間体48、100mg、0.16mmol)の撹拌DCM溶液(2mL)に周囲温度にて滴加した。1時間後、溶液を減圧下で濃縮して、生じた残留物を、1M HCl aq(2mL、2.00mmol)及びEt2O(2mL)中に溶解した。フェニルボロン酸(60mg、0.49mmol)を加えて、澄明な二相性溶液を、室温にて3時間撹拌した。混合液をEt2O(20mL)及び水(5mL)で希釈して、層を分離させた。水性層をEt2Oで洗浄して、層を分離させて、水性層を凍結乾燥した。生じた固体をMeOH(3mL)中に溶解して、イオン交換クロマトグラフィ(PoraPak Rxn CX 20ccカラム)にかけた。所望の生成物をカラムから、MeOH中5%アンモニア溶液(20mL)を用いて溶出した。得られた物質をさらに、逆相クロマトグラフィ(RediSep Rf Gold(登録商標)C18、水中0~20%アセトニトリル)によって精製した。生成物の画分を凍結乾燥して、生じた物質を再度、逆相フラッシュクロマトグラフィ(RediSep Rf Gold(登録商標)C18、水中0~2%アセトニトリル)にかけた。生成物の画分を凍結乾燥して、生じた物質を再度、逆相フラッシュクロマトグラフィ(RediSep Rf Gold(登録商標)C18、水中0~5%アセトニトリル)によって精製して、(2R,4R)-4-((S)-2-アミノ-3-メトキシプロパンアミド)-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例18、18mg、35%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.77(2H,td),1.14-1.27(1H,m),1.27-1.35(1H,m),1.35-1.45(2H,m),1.72-1.81(1H,m),2.02(1H,ddd),2.30(1H,dd),2.41(1H,dd),3.31-3.38(4H,m),3.55-3.60(3H,m),3.63(1H,dd),4.42-4.51(1H,m);m/z(ES+)[M+H]+=332.
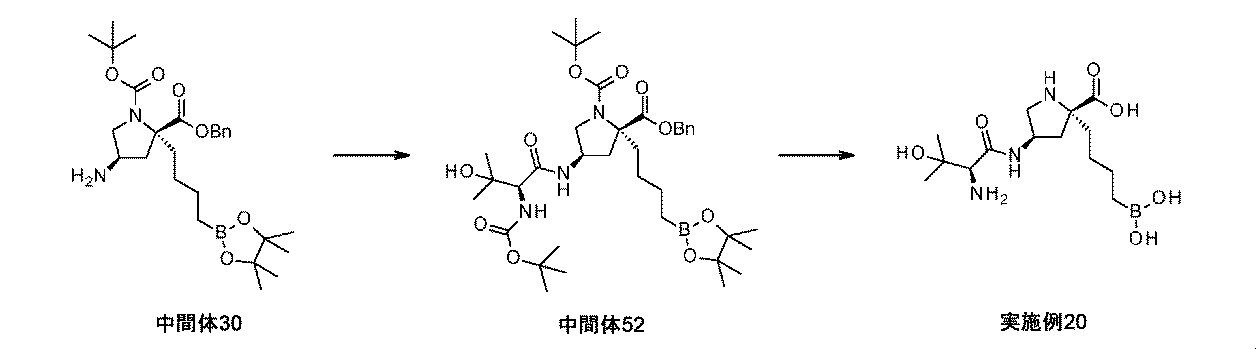
N,N-ジイソプロピルエチルアミン(0.13mL、0.76mmol)を、(S)-N-アルファ-t-ブチルオキシカルボニル-3,3-ジメチル-セリン(106mg、0.454mmol)及びHATU(0.173g、0.454mmol)のDMF溶液(2.6mL)に0℃にて加えて、反応液を15分間撹拌した。(2R,4R)-2-ベンジル1-tert-ブチル4-アミノ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体30、190mg、0.38mmol)のDMF溶液(1mL)を加えて、反応液を、室温にゆっくり温めながら、2時間撹拌した。反応混合液を、EtOAc(40mL)で希釈して、飽和水性NH4Cl(2×20mL)、飽和水性NaHCO3(2×20mL)、及びブライン(20mL)で洗浄した。有機層を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン中0~50%EtOAc)によって精製して、2-ベンジル1-(tert-ブチル)(2R,4R)-4-((S)-2-((tert-ブトキシカルボニル)アミノ)-3-ヒドロキシ-3-メチルブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体52、223mg、82%の収率)を白色の泡として、そして回転異性体の混合物として得た。1H NMR(500MHz,MeOH-d4)δ 0.70-0.83(m,2H)1.14-1.27(m,19H)1.31(s,6H)1.36-1.42(m,5H)1.44(s,10H)1.81-1.93(m,1H)2.02-2.14(m,1H)2.14-2.30(m,1H)2.36-2.49(m,1H)3.39-3.48(m,1H)3.73-3.83(m,1H)3.87-3.97(m,1H)4.43-4.53(m,1H)5.09-5.25(m,2H)7.28-7.45(m,5H);m/z(ES+)[M+H]+=718.
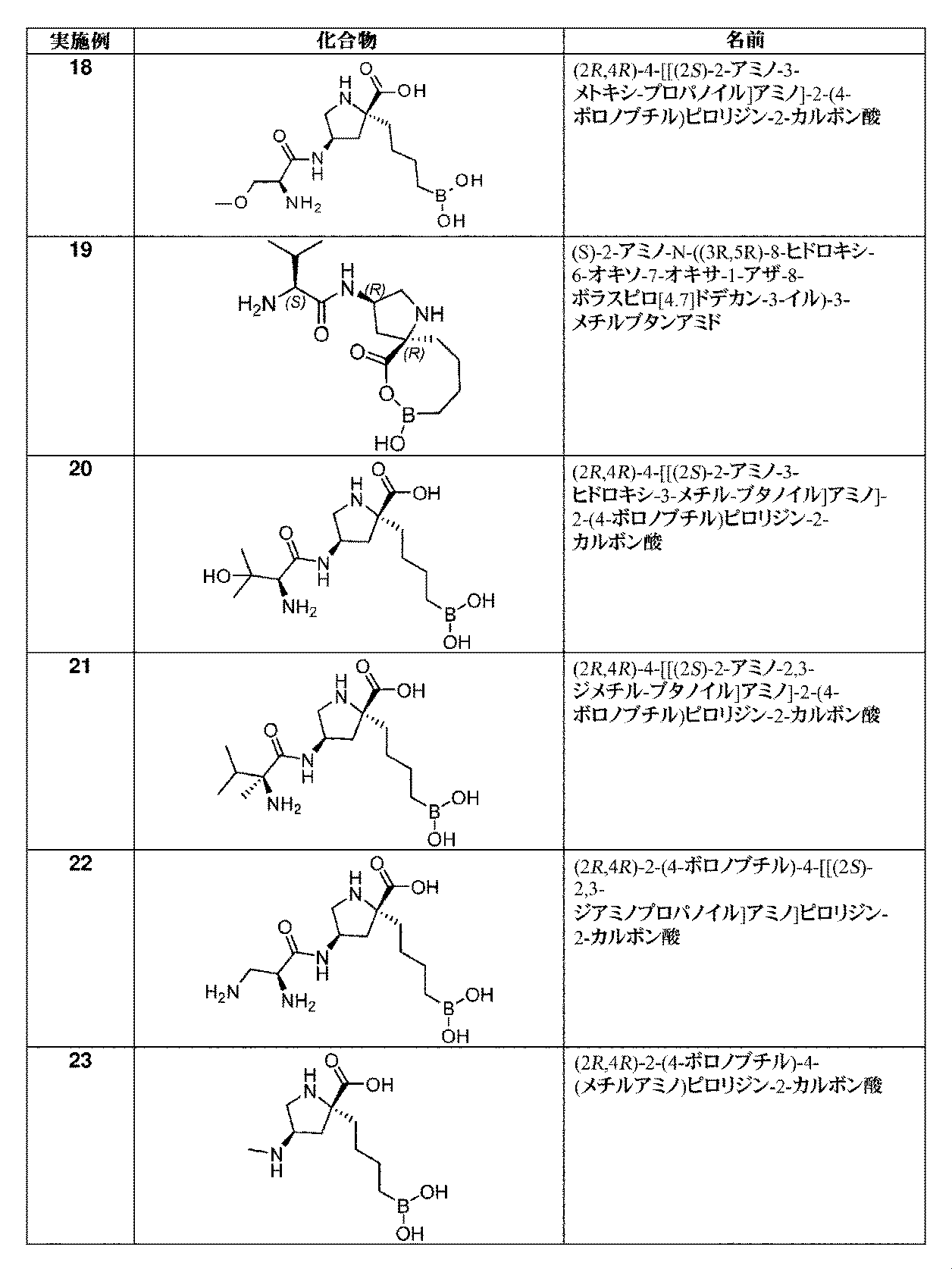
Pd/C(10wt%、100mg、0.09mmol)を、2-ベンジル1-(tert-ブチル)(2R,4R)-4-((S)-2-((tert-ブトキシカルボニル)アミノ)-3-ヒドロキシ-3-メチルブタンアミド)-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体52、220mg、0.31mmol)のEtOAc溶液(3mL)に加えた。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて3時間撹拌した。反応混合液を、MeOHで希釈して、珪藻土で濾過して、濃縮して乾燥させた。白色の固体を、DCM(1mL)及びトリフルオロ酢酸(0.50mL、6.5mmol)中に溶解して、反応液を、室温にて2時間撹拌した。溶液を濃縮して、生じた残留物を、Et2O(2mL)及び1M HCl aq(2mL)中に溶解した。フェニルボロン酸(100mg、0.82mmol)を加えて、澄明な二相性溶液を、室温にて2時間撹拌した。反応混合液を水で希釈して、Et2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 20ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア(20mL)を用いて溶出して、(2R,4R)-4-[[(2S)-2-アミノ-3-ヒドロキシ-3-メチル-ブタノイル]アミノ]-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例20、92mg、87%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.70(2H,t),1.20(4H,s),1.28(4H,s),1.31-1.40(2H,m),1.77-1.88(1H,m),2.04-2.14(1H,m),2.40-2.47(1H,m),2.48-2.54(1H,m),3.40(1H,dd),3.74(1H,s),3.75-3.80(1H,m),4.47-4.55(1H,m);m/z:(ES+)[M+H]+=346.
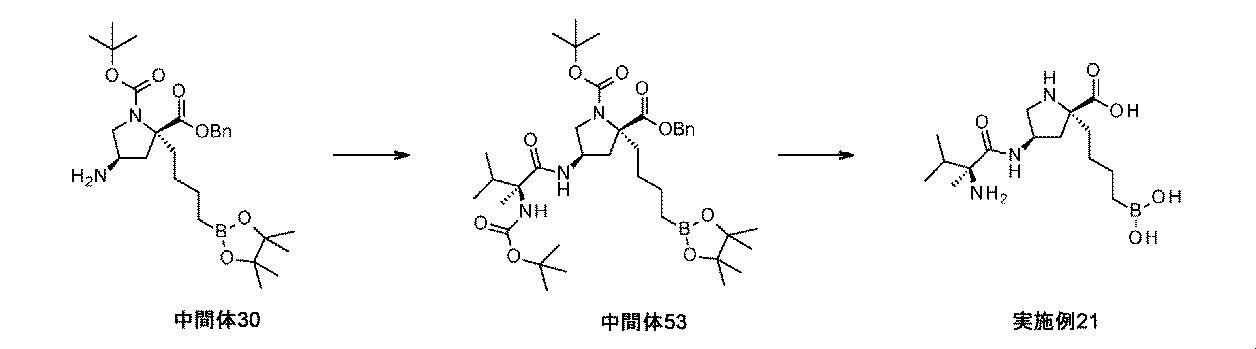
N,N-ジイソプロピルエチルアミン(0.14mL、0.82mmol)を、(S)-2-((tert-ブトキシカルボニル)アミノ)-2,3-ジメチルブタン酸(0.113g、0.489mmol)及びHATU(0.186g、0.489mmol)のDMF溶液(3mL)に0℃にて加えて、反応液を15分間撹拌した。(2R,4R)-2-ベンジル1-tert-ブチル4-アミノ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体30、205mg、0.408mmol)のDMF溶液(1mL)を加えて、反応液を、室温にゆっくり温めながら、3時間撹拌した。反応混合液を、EtOAc(50mL)で希釈して、飽和水性NH4Cl(2×25mL)、飽和水性NaHCO3(2×25mL)、及びブライン(20mL)で洗浄した。有機層を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン中0~50%EtOAc)によって精製して、2-ベンジル1-tert-ブチル(2R,4R)-4-[[(2S)-2-(tert-ブトキシカルボニルアミノ)-2,3-ジメチル-ブタノイル]アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体53、211mg、72%の収率)を白色の泡として、そして回転異性体の混合物として得た。1H NMR(500MHz,MeOH-d4)δ 0.72-0.79(2H,m),0.80-0.93(6H,m),1.17-1.25(13H,m),1.31(9H,d),1.36-1.47(16H,m),1.82-1.92(2H,m),1.97-2.09(1H,m),2.15-2.30(1H,m),2.34-2.51(1H,m),3.61-3.75(1H,m),4.42-4.56(1H,m),5.08-5.31(2H,m),7.29-7.46(5H,m);m/z:(ES+)[M+H]+=716.
Pd/C(10wt%、90mg、0.08mmol)を、2-ベンジル1-tert-ブチル(2R,4R)-4-[[(2S)-2-(tert-ブトキシカルボニルアミノ)-2,3-ジメチル-ブタノイル]アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体53、210mg、0.29mmol)のEtOAc溶液(4mL)に加えた。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて3時間撹拌した。反応混合液を、MeOHで希釈して、珪藻土で濾過して、濃縮して乾燥させた。白色の固体を、DCM(1mL)及びトリフルオロ酢酸(0.50mL、6.5mmol)中に溶解して、反応液を、室温にて2時間撹拌した。溶液を濃縮して、生じた残留物を、Et2O(2mL)及び1M HCl aq(2mL)中に溶解した。フェニルボロン酸(100mg、0.82mmol)を加えて、澄明な二相性溶液を、室温にて2時間撹拌した。反応混合液を水で希釈して、Et2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 20ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア(20mL)を用いて溶出して、(2R,4R)-4-[[(2S)-2-アミノ-2,3-ジメチル-ブタノイル]アミノ]-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例21、90mg、89%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.74-0.78(2H,m),0.79(3H,d),0.89(3H,d),1.17-1.27(4H,m),1.28-1.35(1H,m),1.36-1.47(2H,m),1.70-1.81(1H,m),1.93-2.07(2H,m),2.27(1H,dd),2.43(1H,dd),3.27-3.39(1H,m),3.60(1H,dd),4.39-4.48(1H,m);m/z:(ES+)[M+H]+=344.
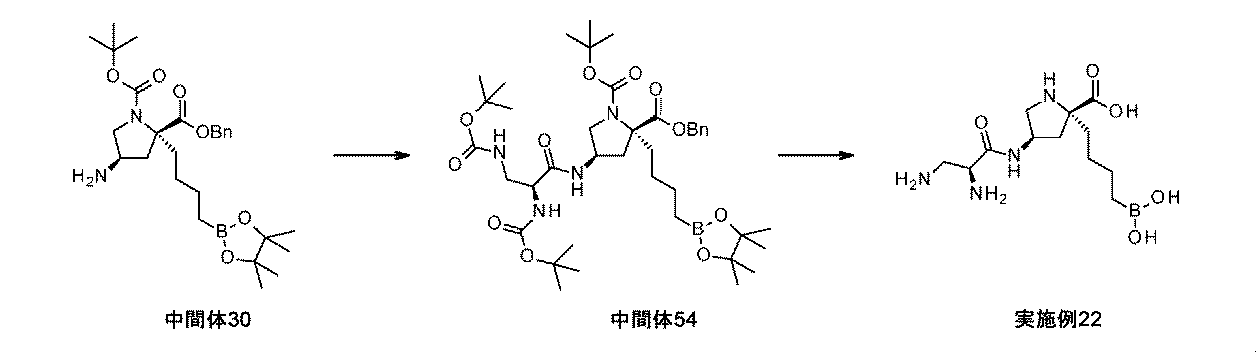
N,N-ジイソプロピルエチルアミン(0.14mL、0.82mmol)を、Boc-Dap(Boc)-OH・DCHA(0.238g、0.489mmol)及びHATU(0.186g、0.489mmol)のDMF溶液(3mL)に0℃にて加えて、反応液を15分間撹拌した。(2R,4R)-2-ベンジル1-tert-ブチル4-アミノ-2-(4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル)ピロリジン-1,2-ジカルボキシラート(中間体30、205mg、0.408mmol)のDMF溶液(1mL)を加えて、反応液を、室温にゆっくり温めながら、16時間撹拌した。反応混合液を、EtOAc(50mL)で希釈して、飽和水性NH4Cl(2×25mL)、飽和水性NaHCO3(2×25mL)、及びブライン(20mL)で洗浄した。有機層を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン中0~50%EtOAc)によって精製して、2-ベンジル1-tert-ブチル(2R,4R)-4-[[(2S)-2,3-ビス(tert-ブトキシカルボニルアミノ)プロパノイル]アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体54、214mg、66%の収率)を白色の泡として、そして回転異性体の混合物として得た。1H NMR(500MHz,MeOH-d4)δ 0.76(2H,q),1.19-1.26(14H,m),1.27-1.34(6H,m),1.37-1.52(24H,m),1.76-1.93(1H,m),2.06-2.31(2H,m),2.50(1H,s),3.19-3.27(2H,m),3.66-3.84(1H,m),4.00-4.13(1H,m),4.35-4.50(1H,m),5.04-5.25(2H,m),7.26-7.50(5H,m);m/z:(ES+)[M+H]+=789.
Pd/C(10wt%、57mg、0.053mmol)を、2-ベンジル1-tert-ブチル(2R,4R)-4-[[(2S)-2,3-ビス(tert-ブトキシカルボニルアミノ)プロパノイル]アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体54、210mg、0.27mmol)のEtOAc溶液(4mL)に加えた。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて3時間撹拌した。反応混合液を、MeOHで希釈して、珪藻土で濾過して、濃縮して乾燥させた。白色の固体を、DCM(1mL)及びトリフルオロ酢酸(0.50mL、6.5mmol)中に溶解して、反応液を、室温にて2時間撹拌した。溶液を濃縮して、生じた残留物を、Et2O(2mL)及び1M HCl aq(2mL)中に溶解した。フェニルボロン酸(100mg、0.82mmol)を加えて、澄明な二相性溶液を、室温にて3時間撹拌した。反応混合液を水で希釈して、Et2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 20ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア(20mL)を用いて溶出して、(2R,4R)-2-(4-ボロノブチル)-4-[[(2S)-2,3-ジアミノプロパノイル]アミノ]ピロリジン-2-カルボン酸(実施例22、74mg、88%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.71(2H,t),1.11-1.24(1H,m),1.26-1.41(3H,m),1.75-1.89(1H,m),2.00-2.13(1H,m),2.31-2.48(1H,m),2.51-2.67(1H,m),3.39-3.53(3H,m),3.68-3.80(1H,m),4.27(1H,t),4.41-4.52(1H,m);m/z:(ES+)[M+H]+=317.
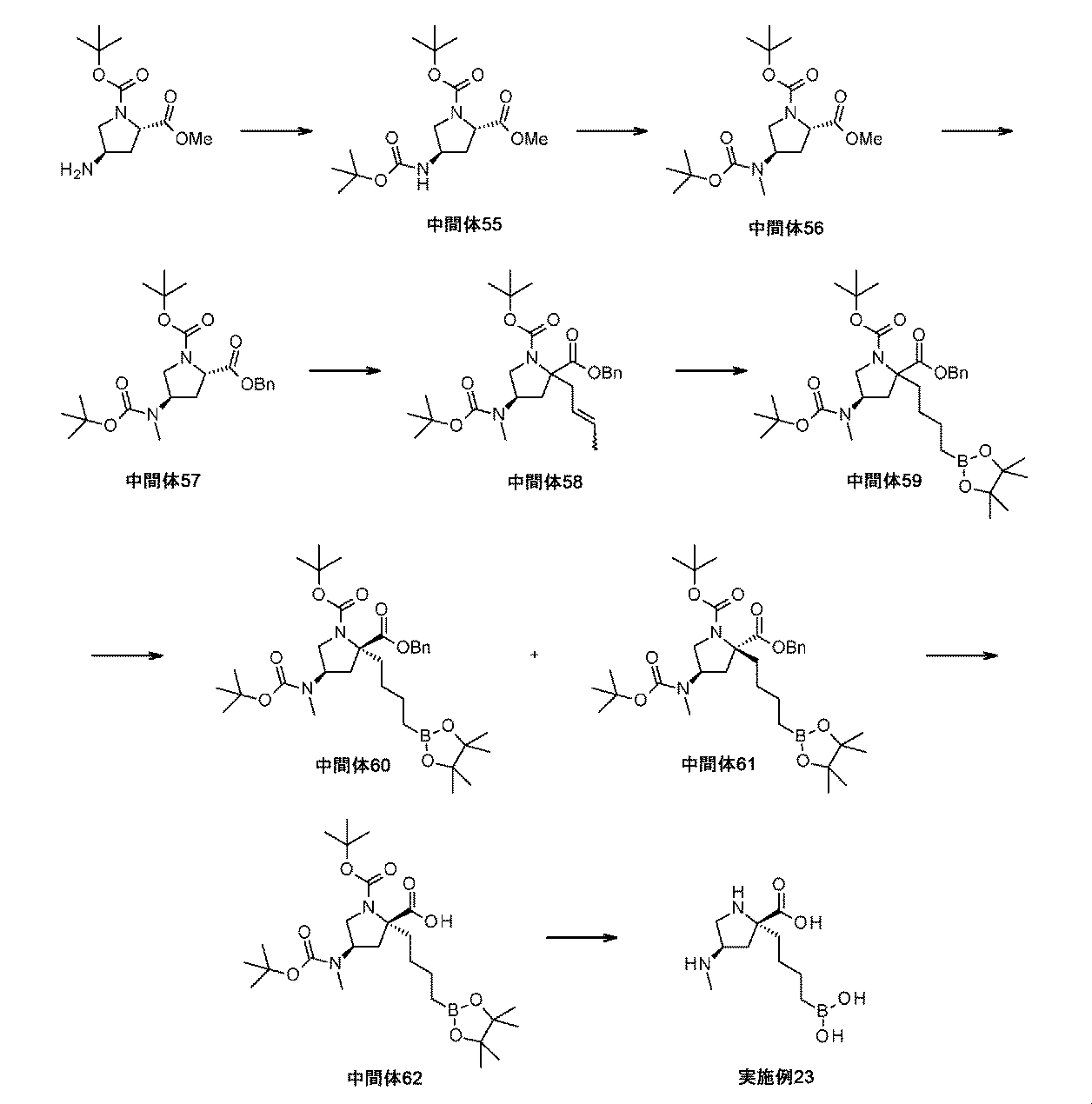
ジ-tert-ブチルジカルボナート(4.41g、20.2mmol)を、1-(tert-ブチル)2-メチル(2S,4R)-4-アミノピロリジン-1,2-ジカルボキシラートシュウ酸塩(4.50g、13.5mmol)及びトリエチルアミン(5.63mL、40.4mmol)のDCM溶液(57mL)に加えて、反応液を、N2雰囲気下で室温にて一晩撹拌した。粗反応混合液をDCM(200mL)で希釈して、0.5M HCl(aq)、飽和重炭酸ナトリウム、及びブラインで順次洗浄した。有機層を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(DCM/MeOH)によって精製して、1-tert-ブチル2-メチル(2S,4R)-4-(tert-ブトキシカルボニルアミノ)ピロリジン-1,2-ジカルボキシラート(中間体55、3.66g、79%の収率)を白色の固体として得た。1H NMR(500MHz,DMSO-d6)δ 1.29-1.42(18H,m),1.96-2.17(2H,m),3.12-3.17(1H,m),3.45-3.56(1H,m),3.62-3.69(3H,m),3.96-4.06(1H,m),4.24-4.34(1H,m),7.17-7.26(1H,m);m/z:(ES+)[M+H]+=345.
水素化ナトリウム(鉱油中60%分散系)(0.491g、12.3mmol)を、1-tert-ブチル2-メチル(2S,4R)-4-(tert-ブトキシカルボニルアミノ)ピロリジン-1,2-ジカルボキシラート(中間体55、3.66g、10.7mmol)のDMF溶液(35mL)に少量ずつ加えた。添加後、反応液を10分間撹拌してから、ヨウ化メチル(0.715ml、11.4mmol)を加えて、反応液をさらに3時間撹拌した。反応混合液を0℃に冷却して、水でクエンチした。混合液をEtOAc(200mL)で希釈して、層を分離させた。有機層を水及びブラインで順次洗浄して、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、1-tert-ブチル2-メチル(2S,4R)-4-[tert-ブトキシカルボニル(メチル)アミノ]ピロリジン-1,2-ジカルボキシラート(中間体56、3.13g、82%の収率)を無色の油として得た。1H NMR(500MHz,DMSO-d6)δ 1.28-1.41(18H,m),1.87-2.02(1H,m),2.29-2.44(1H,m),2.69(3H,s),3.13-3.25(1H,m),3.44-3.57(1H,m),3.60-3.68(3H,m),4.23-4.32(1H,m),4.62(1H,br s);m/z:(ES+)[M+H]+=359.
水酸化ナトリウム(2.10g、52.4mmol)の水溶液(11mL)を、1-tert-ブチル2-メチル(2S,4R)-4-[tert-ブトキシカルボニル(メチル)アミノ]ピロリジン-1,2-ジカルボキシラート(中間体56、3.13g、8.73mmol)のTHF(22mL)及びMeOH(11mL)溶液に0℃にて加えた。反応混合液を、室温にゆっくり温めながら、3時間撹拌した。揮発性物質を減圧下で除去して、水性層を5M HCl(aq)で約3のpHに酸性化して、DCMで抽出した。組み合わせた有機質を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させて、粗カルボン酸を白色の固体として得た。これを、更なる精製なしに用いた。
2-ベンジル1-tert-ブチル(2S,4R)-4-[tert-ブトキシカルボニル(メチル)アミノ]ピロリジン-1,2-ジカルボキシラート(中間体57、3.05g、7.02mmol)及び臭化クロチル(1.08mL、10.5mmol)を、THF(25mL)中に溶解して、溶液を、N2雰囲気下で-78℃に冷却した。KHMDSの溶液(トルエン中0.5M、21.0mL、10.5mmol)を反応混合液に滴加して、反応液を、室温にゆっくり温めながら、17時間撹拌した。粗反応混合液を水でクエンチして、揮発性物質を減圧下で除去した。粗混合液をDCM中に希釈して、層を分離させた。有機層を、水で洗浄して、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲル精製(ヘキサン/EtOAc)によって精製して、2-ベンジル1-tert-ブチル(4R)-2-(ブタ-2-エニル)-4-[tert-ブトキシカルボニル(メチル)アミノ]ピロリジン-1,2-ジカルボキシラート(中間体58、1.26g、37%の収率)を黄色の油として、そして偏左右異性体、E/Zオレフィン異性体、及び回転異性体の混合物として得た。1H NMR(500MHz,DMSO-d6)δ 1.23-1.44(18H,m),1.51-1.70(3H,m),2.00-2.26(2H,m),2.38-2.47(1H,m),2.57-2.68(3H,m),2.71-3.00(1H,m),3.00-3.25(1H,m),3.40-3.74(1H,m),4.54-4.80(1H,m),5.01-5.27(2H,m),5.28-5.47(1H,m),5.49-5.72(1H,m),7.25-7.42(5H,m);m/z:(ES+)[M+H]+=489.
ビス(1,5-シクロオクタジエン)ジイリジウム(I)ジクロリド(269mg、0.400mmol)及びビス(ジフェニルホスフィノ)メタン(308mg、0.801mmol)を、オーブン乾燥した丸底フラスコに加えた。フラスコをシールして、N2でパージした。固体を、DCM(11mL)中に溶解して、4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(1.28mL、8.82mmol)を溶液にゆっくり加えた。反応液を、室温にて10分間撹拌した。2-ベンジル1-tert-ブチル(4R)-2-(ブタ-2-エニル)-4-[tert-ブトキシカルボニル(メチル)アミノ]ピロリジン-1,2-ジカルボキシラート(中間体58、1.96g、4.01mmol)を、DCM溶液(7.5mL)としての反応液に加えて、反応混合液を室温にて16時間撹拌した。反応混合液を0℃に冷却して、MeOH及び水で注意深くクエンチした。層を分離させて、水性層をDCMで抽出した。組み合わせた有機質を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。生じた残留物を、フラッシュシリカクロマトグラフィ(ヘキサン/EtOAc)によって精製して、2-ベンジル1-tert-ブチル(4R)-4-[tert-ブトキシカルボニル(メチル)アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体59、2.5g、100%の収率)を黄色の油として、そして偏左右異性体及び回転異性体の混合物として得た。精製物質を、キラルSFC[(S,S)Whelk-O1カラム、30mm×250mm、5μm、温度=20℃、移動相=0~30%MeOH:CO2、UV検出@220nm、ローディング=31mg/inj、conc=MeOH中125mg/mL、流量=75mL/分、出口圧力=100bar]にかけて、2つの偏左右異性体を得た。各偏左右異性体の立体化学を、他の例示する化合物と一致するように、実施例23及び実施例24の酵素効力に基づいて遡及的に割り当てた。
Pd/C(10wt%、165mg、0.155mmol)を、2-ベンジル1-tert-ブチル(2R,4R)-4-[tert-ブトキシカルボニル(メチル)アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体60、637mg、1.03mmol)のEtOAc溶液(7mL)に加えた。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液を、MeOHで希釈して、珪藻土で濾過した。濾液を濃縮して乾燥させて、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、(2R,4R)-1-tert-ブトキシカルボニル-4-[tert-ブトキシカルボニル(メチル)アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体62、350mg、64%の収率)を白色の固体として得た。1H NMR(500MHz,DMSO-d6)δ 0.63-0.73(2H,m),1.14-1.19(12H,m),1.22-1.42(21H,m),1.63-1.77(1H,m),1.99-2.18(3H,m),2.63-2.67(3H,m),3.21-3.27(2H,m),3.41-3.55(1H,m),4.57-4.80(1H,m),12.32-12.75(1H,m);m/z:(ES+)[M+H]+=527.
トリフルオロ酢酸(0.51mL、6.7mmol)を、(2R,4R)-1-tert-ブトキシカルボニル-4-[tert-ブトキシカルボニル(メチル)アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体62、350mg、0.66mmol)の撹拌DCM溶液(4mL)に室温にて滴加した。1時間後、溶液を減圧下で濃縮して、生じた残留物を、1M HCl aq(5mL)及びEt2O(5mL)中に溶解した。フェニルボロン酸(162mg、1.33mmol)を加えて、澄明な二相性溶液を、室温にて1時間撹拌した。混合液をEt2O及び水で希釈して、層を分離させた。水性層をEt2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 60ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア(60mL)を用いて溶出して、(2R,4R)-2-(4-ボロノブチル)-4-(メチルアミノ)ピロリジン-2-カルボン酸(実施例23、140mg、86%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.72-0.82(2H,m),1.09-1.43(4H,m),1.62-1.77(1H,m),1.83-1.95(1H,m),2.23(2H,d),2.46(3H,s),3.02-3.11(1H,m),3.37-3.49(1H,m),3.49-3.61(1H,m);m/z:(ES+)[M+H]+=245.
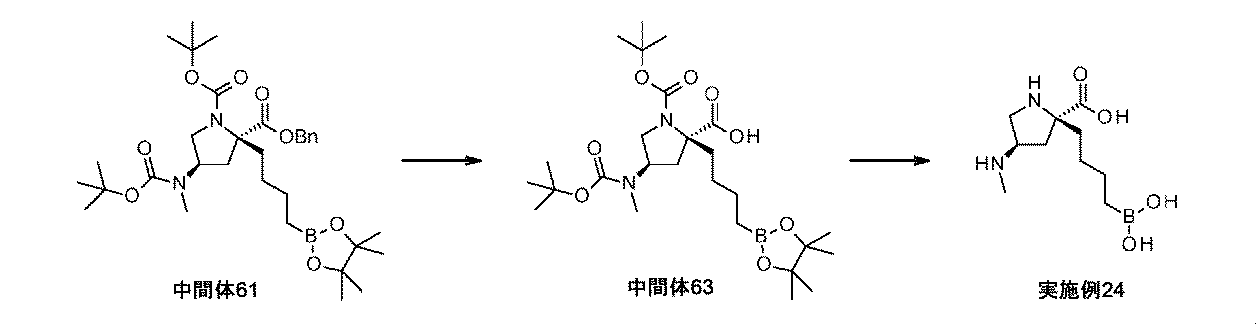
Pd/C(10wt%、223mg、0.209mmol)を、2-ベンジル1-tert-ブチル(2S,4R)-4-[tert-ブトキシカルボニル(メチル)アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体61、860mg、1.39mmol)のEtOAc溶液(9.3mL)に加えた。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液を、MeOHで希釈して、珪藻土で濾過した。濾液を濃縮して乾燥させて、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、(2S,4R)-1-tert-ブトキシカルボニル-4-[tert-ブトキシカルボニル(メチル)アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体63、520mg、71%の収率)を白色の固体として得た。1H NMR(500MHz,DMSO-d6)δ 0.60-0.75(2H,m),1.16(12H,s),1.28-1.45(22H,m),1.56-1.74(1H,m),1.84-1.95(1H,m),2.01-2.22(2H,m),2.69(3H,s),3.01-3.17(1H,m),3.56-3.70(1H,m),4.52-4.73(1H,m),12.35-12.77(1H,m);m/z:(ES+)[M+H]+=527.
トリフルオロ酢酸(1.23mL、16.0mmol)を、(2S,4R)-1-tert-ブトキシカルボニル-4-[tert-ブトキシカルボニル(メチル)アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体63、520mg、0.80mmol)の撹拌DCM溶液(8.5mL)に室温にて滴加した。1時間後、溶液を減圧下で濃縮して、生じた残留物を、1M HCl aq(5mL)及びEt2O(5mL)中に溶解した。フェニルボロン酸(196mg、1.60mmol)を加えて、澄明な二相性溶液を、室温にて1時間撹拌した。混合液をEt2O及び水で希釈して、層を分離させた。水性層をEt2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 60ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア(60mL)を用いて溶出して、(2S,4R)-2-(4-ボロノブチル)-4-(メチルアミノ)ピロリジン-2-カルボン酸(実施例24、163mg、83%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.70-0.90(2H,m),1.10-1.46(4H,m),1.65-1.77(2H,m),1.96-2.07(1H,m),2.43(3H,s),2.68-2.77(1H,m),2.96-3.10(1H,m),3.37-3.50(1H,m),3.50-3.60(1H,m);m/z:(ES+)[M+H]+=245.
トリフェニルホスフィン(7.87g、30.0mmol)及び水(0.54mL、30.0mmol)を、2-ベンジル1-(tert-ブチル)(2R,4R)-4-アジドピロリジン-1,2-ジカルボキシラート(5.20g、15.0mmol)のTHF溶液(68mL)に室温にて加えた。反応液を60℃に加熱して、6時間撹拌した。反応混合液を室温に冷却して、EtOAcで希釈して、水(2×100mL)及び飽和水性塩化ナトリウム(100mL)で順次洗浄した。有機層を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(DCM/MeOH)によって精製して、2-ベンジル1-tert-ブチル(2R,4R)-4-アミノピロリジン-1,2-ジカルボキシラート(中間体64、3.2g、67%の収率)を無色の油として得た。1H NMR(500MHz,DMSO-d6)δ 1.20-1.49(9H,m),1.53-1.64(1H,m),1.65-1.79(2H,m),2.26-2.40(1H,m),2.85-3.01(1H,m),3.31-3.42(1H,m),3.45-3.56(1H,m),4.13-4.24(1H,m),4.99-5.25(2H,m),7.36(5H,s);m/z:(ES+)[M+H]+=321.
ナトリウムトリアセトキシボロヒドリド(6.35g、29.9mmol)を、2-ベンジル1-tert-ブチル(2R,4R)-4-アミノピロリジン-1,2-ジカルボキシラート(中間体64、3.20g、9.99mmol)及びホルムアルデヒド(H2O中37wt%、4.46mL、59.9mmol)のMeOH溶液(79mL)に少量ずつ加えた。添加後、反応液を室温にて17時間撹拌した。揮発性物質を減圧下で除去して、生じた残留物をDCMで希釈した。固体を濾過によって除去して、濾液を濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc(NH4OH入り))によって精製して、2-ベンジル1-tert-ブチル(2R,4R)-4-(ジメチルアミノ)ピロリジン-1,2-ジカルボキシラート(中間体65、3.00g、86%の収率)を得た。1H NMR(500MHz,DMSO-d6)δ 1.24(9H,s),1.53-1.67(1H,m),2.10(6H,s),2.40-2.48(1H,m),2.53-2.73(1H,m),2.83-3.06(1H,m),3.59-3.69(1H,m),4.15-4.29(1H,m),5.02-5.21(2H,m),7.26-7.42(5H,m);m/z:(ES+)[M+H]+=350.
2-ベンジル1-tert-ブチル(2R,4R)-4-(ジメチルアミノ)ピロリジン-1,2-ジカルボキシラート(中間体65、3.00g、8.61mmol)及び臭化クロチル(1.33mL、12.9mmol)を、THF(18mL)中に溶解して、溶液を、N2雰囲気下で-78℃に冷却した。KHMDSの溶液(トルエン中0.5M、25.8mL、12.9mmol)を、反応混合液に滴加して、反応液を、室温にゆっくり温めながら、17時間撹拌した。粗反応混合液を水でクエンチして、揮発性物質を減圧下で除去した。粗混合液をDCM中で希釈して、層を分離させた。有機層を水で洗浄して、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲル精製(ヘキサン/EtOAc)によって精製して、2-ベンジル1-tert-ブチル(4R)-2-(ブタ-2-エニル)-4-(ジメチルアミノ)ピロリジン-1,2-ジカルボキシラート(中間体66、1.55g、45%の収率)を黄色の油として、そして偏左右異性体、E/Zオレフィン異性体、及び回転異性体の混合物として得た。1H NMR(500MHz,DMSO-d6)δ 1.21-1.44(9H,m),1.51-1.70(3H,m),1.79-1.96(1H,m),2.05(6H,s),2.08-2.24(1H,m),2.31-2.43(1H,m),2.54-2.74(1H,m),2.75-3.08(2H,m),3.56-3.86(1H,m),5.00-5.23(2H,m),5.23-5.42(1H,m),5.43-5.73(1H,m),7.23-7.41(5H,m);m/z:(ES+)[M+H]+=403.
ビス(1,5-シクロオクタジエン)ジイリジウム(I)ジクロリド(240mg、0.36mmol)及びビス(ジフェニルホスフィノ)メタン(275mg、0.715mmol)を、オーブン乾燥した丸底フラスコに加えた。フラスコをシールして、N2でパージした。固体を、DCM(10mL)中に溶解して、4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(1.14mL、7.87mmol)を溶液にゆっくり加えた。反応液を、室温にて10分間撹拌した。2-ベンジル1-tert-ブチル(4R)-2-(ブタ-2-エニル)-4-(ジメチルアミノ)ピロリジン-1,2-ジカルボキシラート(中間体66、1.44g、3.58mmol)を、DCM溶液(6.7mL)としての反応液に加えて、反応混合液を室温にて16時間撹拌した。反応混合液を0℃に冷却して、MeOH及び水で注意深くクエンチした。層を分離させて、水性層をDCMで抽出した。組み合わせた有機質を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。生じた残留物を、フラッシュシリカクロマトグラフィ(ヘキサン/EtOAc)によって精製して、2-ベンジル1-tert-ブチル(4R)-4-(ジメチルアミノ)-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体67、1.5g、79%の収率)を黄色の油として、そして偏左右異性体及び回転異性体の混合物として得た。精製物質を、キラルSFC[(S,S)Whelk-O1カラム、30mm×250mm、5μm、温度=20℃、移動相=0~15%MeOH:CO2、UV検出@220nm、ローディング=32mg/inj、conc=MeOH中80mg/mL、流量=120mL/分、出口圧力=100bar]にかけて、2つの偏左右異性体を得た。各偏左右異性体の立体化学を、他の例示する化合物と一致するように、実施例25及び実施例26の酵素効力に基づいて遡及的に割り当てた。
Pd/C(10wt%、57mg、0.054mmol)を、2-ベンジル1-tert-ブチル(2R,4R)-4-(ジメチルアミノ)-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体68、189mg、0.356mmol)のEtOAc溶液(2.4mL)に加えた。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液を、MeOHで希釈して、珪藻土で濾過した。濾液を濃縮して乾燥させて、(2R,4R)-1-tert-ブトキシカルボニル-4-(ジメチルアミノ)-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体70、150mg、96%の収率)を白色の固体として得た。これを、更なる精製なしに用いた。1H NMR(500MHz,DMSO-d6)δ 0.53-0.70(2H,m),1.11-1.20(12H,m),1.21-1.41(14H,m),1.56-1.71(1H,m),1.92-2.06(2H,m),2.20(7H,s),2.69-2.78(1H,m),3.37-3.51(1H,m),3.67-4.21(1H,m);m/z:(ES+)[M+H]+=441.
トリフルオロ酢酸(0.26mL、3.4mmol)を、(2R,4R)-1-tert-ブトキシカルボニル-4-(ジメチルアミノ)-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体70、150mg、0.34mmol)の撹拌DCM溶液(2mL)に室温にて滴加した。1時間後、溶液を減圧下で濃縮して、生じた残留物を、1M HCl aq(5mL)及びEt2O(5mL)中に溶解した。フェニルボロン酸(83mg、0.68mmol)を加えて、澄明な二相性溶液を、室温にて1時間撹拌した。混合液をEt2O及び水で希釈して、層を分離させた。水性層をEt2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 20ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア(20mL)を用いて溶出して、(2R,4R)-2-(4-ボロノブチル)-4-(ジメチルアミノ)ピロリジン-2-カルボン酸(実施例25、73mg、83%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.62-0.73(2H,m),1.14-1.36(4H,m),1.79-1.90(1H,m),1.99-2.10(1H,m),2.57-2.73(2H,m),2.82-2.90(6H,m),3.48-3.56(1H,m),3.89-3.98(1H,m),4.12-4.20(1H,m);m/z:(ES+)[M+H]+=259.
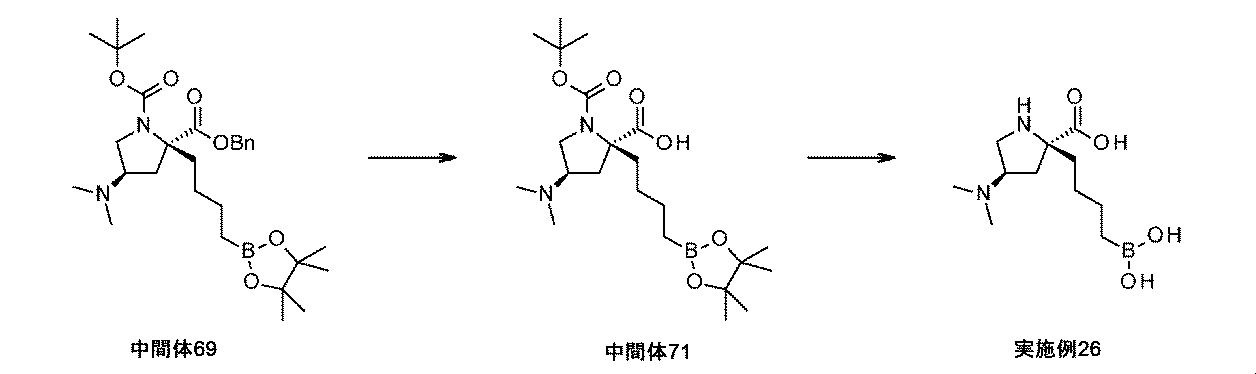
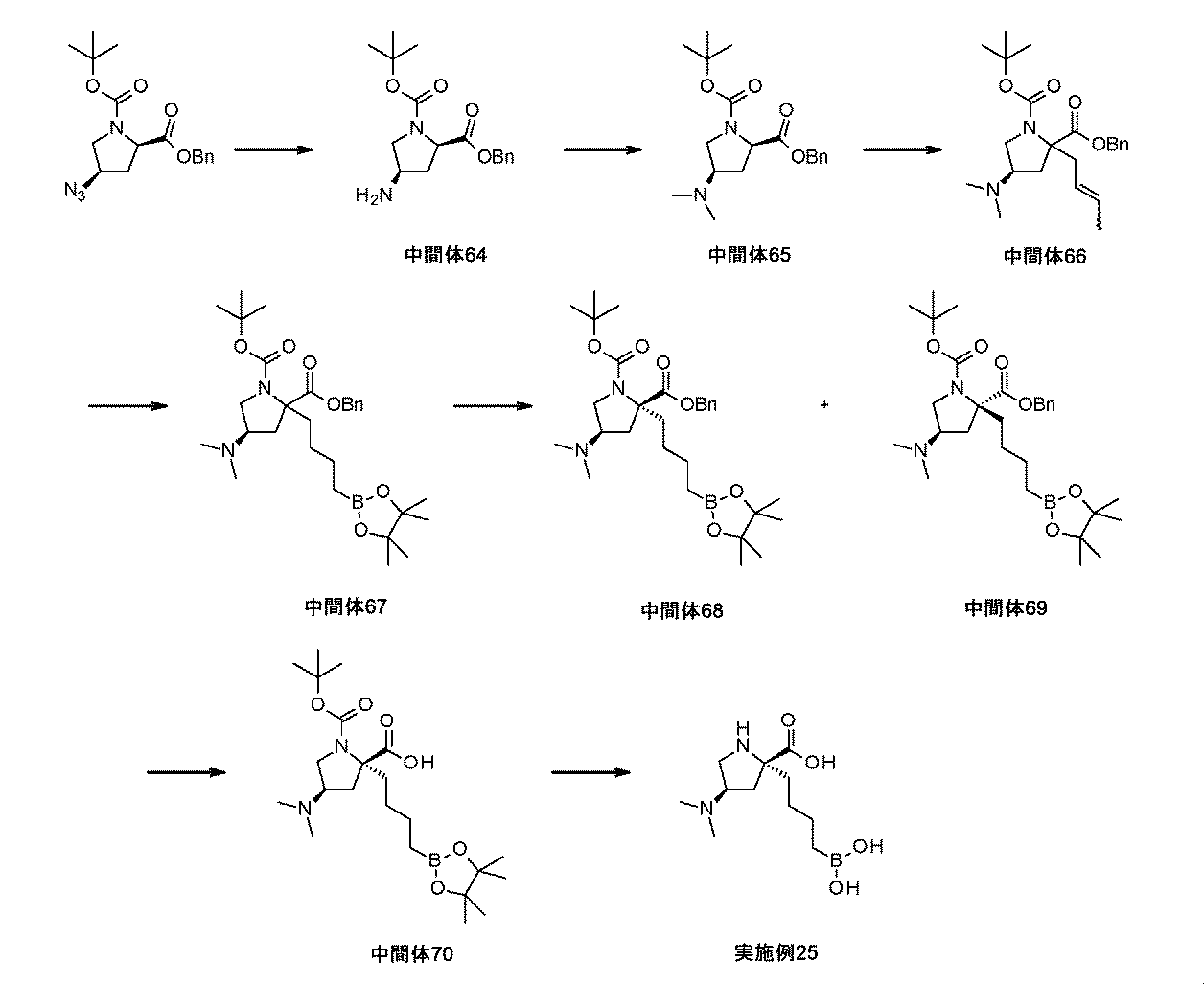
Pd/C(10wt%、148mg、0.139mmol)を、2-ベンジル1-tert-ブチル(2S,4R)-4-(ジメチルアミノ)-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体69、490mg、0.93mmol)のEtOAc溶液(6.7mL)に加えた。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液をMeOHで希釈して、珪藻土で濾過した。濾液を濃縮して乾燥させて、(2S,4R)-1-tert-ブトキシカルボニル-4-(ジメチルアミノ)-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体71、405mg、99%の収率)を白色の固体として得た。これを、更なる精製なしに用いた。1H NMR(500MHz,DMSO-d6)δ 0.52-0.71(2H,m),0.83-1.06(1H,m),1.16(12H,s),1.32(12H,s),1.53-1.67(1H,m),1.75-1.88(1H,m),2.01-2.13(7H,m),2.54-2.69(1H,m),2.79-2.95(1H,m),3.66-3.83(1H,m),7.24-7.46(1H,m),11.86-12.88(1H,m);m/z:(ES+)[M+H]+=441.
トリフルオロ酢酸(1.0mL、13mmol)を、(2S,4R)-1-tert-ブトキシカルボニル-4-(ジメチルアミノ)-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体71、405mg、0.919mmol)の撹拌DCM溶液(5.1mL)に室温にて滴加した。1時間後、溶液を減圧下で濃縮して、生じた残留物を、1M HCl aq(5mL)及びEt2O(5mL)中に溶解した。フェニルボロン酸(224mg、1.84mmol)を加えて、澄明な二相性溶液を、室温にて1時間撹拌した。混合液をEt2O及び水で希釈して、層を分離させた。水性層をEt2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 60ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア(60mL)を用いて溶出して、(2S,4R)-2-(4-ボロノブチル)-4-(ジメチルアミノ)ピロリジン-2-カルボン酸(実施例26、206mg、87%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.71-0.89(2H,m),1.12-1.44(4H,m),1.70-1.82(2H,m),1.97-2.11(1H,m),2.29(6H,s),2.62-2.71(1H,m),2.95-3.10(2H,m),3.61-3.68(1H,m);m/z:(ES+)[M+H]+=259.
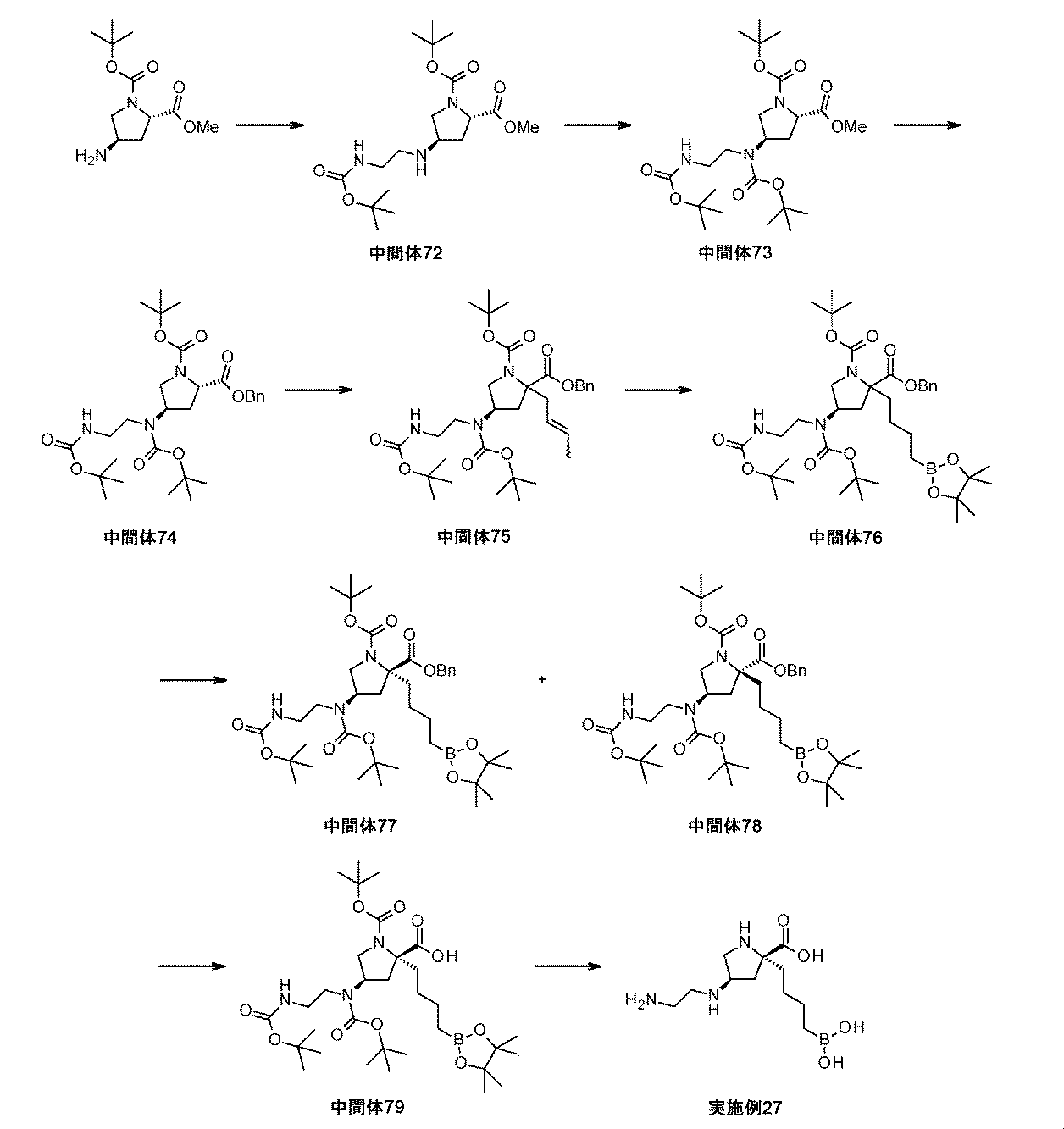
酢酸(42μL、0.73mmol)を、1-(tert-ブチル)2-メチル(2S,4R)-4-アミノピロリジン-1,2-ジカルボキシラート(7.48g、30.6mmol)及びtert-ブチル(2-オキソエチル)カルバメート(4.64g、29.2mmol)のMeOH溶液(194mL)に加えて、反応液を室温にて3時間撹拌した。溶液を0℃に冷却して、ナトリウムトリアセトキシボロヒドリド(9.27g、43.7mmol)を少量ずつ加えた。反応液を、室温にゆっくり温めながら、一晩撹拌した。反応混合液を減圧下で濃縮して、生じた残留物をDCMで希釈して、飽和重炭酸ナトリウム、水、及び飽和塩化ナトリウムで順次洗浄した。有機層をNa2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(DCM/EtOAc/MeOH)によって精製して、1-tert-ブチル2-メチル(2S,4R)-4-[2-(tert-ブトキシカルボニルアミノ)エチルアミノ]ピロリジン-1,2-ジカルボキシラート(中間体72、4.77g、42%の収率)を無色の油として得た。1H NMR(500MHz,DMSO-d6)δ 1.25-1.48(20H,m),1.84-2.07(3H,m),2.88-2.98(2H,m),3.02-3.17(1H,m),3.17-3.27(1H,m),3.40-3.51(1H,m),3.64(3H,s),4.14-4.24(1H,m),6.63-6.74(1H,m);m/z:(ES+)[M+H]+=388.
ジ-tert-ブチルジカルボナート(4.29mL、18.5mmol)を、1-tert-ブチル2-メチル(2S,4R)-4-[2-(tert-ブトキシカルボニルアミノ)エチルアミノ]ピロリジン-1,2-ジカルボキシラート(中間体72、4.77g、12.3mmol)及びN,N-ジイソプロピルエチルアミン(4.30mL、24.6mmol)のDCM溶液(53mL)に室温にて加えて、反応液を17時間撹拌した。反応混合液をDCMで希釈して、水及び飽和水性塩化ナトリウムで順次洗浄した。有機層をNa2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、1-tert-ブチル2-メチル(2S,4R)-4-[tert-ブトキシカルボニル-[2-(tert-ブトキシカルボニルアミノ)エチル]アミノ]ピロリジン-1,2-ジカルボキシラート(中間体73、4.25mg、71%の収率)を得た。1H NMR(500MHz,DMSO-d6)δ 1.21-1.27(1H,m),1.27-1.42(27H,m),1.99-2.07(1H,m),2.33-2.46(1H,m),2.93-3.14(3H,m),3.17-3.24(1H,m),3.48-3.61(1H,m),3.61-3.69(3H,m),4.20-4.44(2H,m),6.79-6.94(1H,m);m/z:(ES+)[M+H]+=488.
水酸化ナトリウム(2.09g、52.3mmol)の水溶液(11mL)を、1-tert-ブチル2-メチル(2S,4R)-4-[tert-ブトキシカルボニル-[2-(tert-ブトキシカルボニルアミノ)エチル]アミノ]ピロリジン-1,2-ジカルボキシラート(中間体73、4.25g、8.72mmol)のTHF(22mL)及びMeOH(11mL)溶液に0℃にて加えた。反応混合液を、室温にゆっくり温めながら、6時間撹拌した。揮発性物質を減圧下で除去して、水性層を5M HCl(aq)で約3のpHに酸性化して、DCMで抽出した。組み合わせた有機質を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させて、粗カルボン酸を白色の固体として得た。これを、更なる精製なしに用いた。
2-ベンジル1-tert-ブチル(2S,4R)-4-[tert-ブトキシカルボニル-[2-(tert-ブトキシカルボニルアミノ)エチル]アミノ]ピロリジン-1,2-ジカルボキシラート(中間体74、3.91g、6.94mmol)及び臭化クロチル(0.93mL、9.0mmol)を、THF(10.6mL)中に溶解して、溶液を、N2雰囲気下で-78℃に冷却した。KHMDSの溶液(トルエン中0.5M、34.7mL、17.3mmol)を反応混合液に滴加して、反応液を、室温にゆっくり温めながら、17時間撹拌した。粗反応混合液を水でクエンチして、揮発性物質を減圧下で除去した。粗混合液をDCM中に希釈して、層を分離させた。有機層を、水で洗浄して、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲル精製(ヘキサン/EtOAc)によって精製して、2-ベンジル1-tert-ブチル(4R)-2-(ブタ-2-エニル)-4-[tert-ブトキシカルボニル-[2-(tert-ブトキシカルボニルアミノ)エチル]アミノ]ピロリジン-1,2-ジカルボキシラート(中間体75、1.34g、31%の収率)を黄色の油として、そして偏左右異性体、E/Zオレフィン異性体、及び回転異性体の混合物として得た。1H NMR(500MHz,DMSO-d6)δ 1.21-1.45(27H,m),1.53-1.71(3H,m),1.91-2.32(2H,m),2.37-2.47(1H,m),2.69-2.86(1H,m),2.86-3.11(4H,m),3.46-3.58(1H,m),3.58-3.77(1H,m),4.29-4.57(1H,m),5.00-5.20(2H,m),5.28-5.46(1H,m),5.46-5.71(1H,m),6.83-7.00(1H,m),7.19-7.43(5H,m);m/z:(ES+)[M+H]+=618.
ビス(1,5-シクロオクタジエン)ジイリジウム(I)ジクロリド(222mg、0.331mmol)及びビス(ジフェニルホスフィノ)メタン(254mg、0.661mmol)を、オーブン乾燥した丸底フラスコに加えた。フラスコをシールして、N2でパージした。固体を、DCM(9mL)中に溶解して、4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(1.05mL、7.26mmol)を溶液にゆっくり加えた。反応液を、室温にて10分間撹拌した。2-ベンジル1-tert-ブチル(4R)-2-(ブタ-2-エニル)-4-[tert-ブトキシカルボニル-[2-(tert-ブトキシカルボニルアミノ)エチル]アミノ]ピロリジン-1,2-ジカルボキシラート(中間体75、2.04g、3.30mmol)を、DCM溶液(6.1mL)としての反応液に加えて、反応混合液を室温にて16時間撹拌した。反応混合液を0℃に冷却して、MeOH及び水で注意深くクエンチした。層を分離させて、水性層をDCMで抽出した。組み合わせた有機質を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。生じた残留物を、フラッシュシリカクロマトグラフィ(ヘキサン/EtOAc)によって精製して、2-ベンジル1-tert-ブチル(4R)-4-[tert-ブトキシカルボニル-[2-(tert-ブトキシカルボニルアミノ)エチル]アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体76、1.14g、46%の収率)を黄色の油として、そして偏左右異性体及び回転異性体の混合物として得た。精製物質を、キラルSFC[(S,S)Whelk-O1カラム、30mm×250mm、5μm、温度=20℃、移動相=0~20%IPA(0.2%NH4OH入り):CO2、UV検出@220nm、ローディング=18mg/inj、conc=MeOH中46mg/mL、流量=120mL/分、出口圧力=100bar]にかけて、2つの偏左右異性体を得た。各偏左右異性体の立体化学を、他の例示する化合物と一致するように、実施例27及び実施例28の酵素効力に基づいて遡及的に割り当てた。
Pd/C(10wt%、99mg、0.093mmol)を、2-ベンジル1-tert-ブチル(2R,4R)-4-[tert-ブトキシカルボニル-[2-(tert-ブトキシカルボニルアミノ)エチル]アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体77、347mg、0.465mmol)のEtOAc溶液(2.3mL)に加えた。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液をMeOHで希釈して、珪藻土で濾過した。濾液を濃縮して乾燥させて、(2R,4R)-1-tert-ブトキシカルボニル-4-[tert-ブトキシカルボニル-[2-(tert-ブトキシカルボニルアミノ)エチル]アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体79、287mg、94%の収率)を白色の固体として得た。これを、更なる精製なしに用いた。1H NMR(500MHz,DMSO-d6)δ 0.65-0.74(2H,m),1.14-1.21(12H,m),1.31-1.43(31H,m),1.66-1.76(1H,m),2.00-2.27(3H,m),2.96-3.12(5H,m),3.37-3.42(1H,m),3.49-3.61(1H,m),4.34-4.49(1H,m),6.84-6.93(1H,m);m/z:(ES+)[M+H]+=656.
トリフルオロ酢酸(0.67mL、8.8mmol)を、(2R,4R)-1-tert-ブトキシカルボニル-4-[tert-ブトキシカルボニル-[2-(tert-ブトキシカルボニルアミノ)エチル]アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体79、287mg、0.438mmol)の撹拌DCM溶液(3.7mL)に室温にて滴加した。1時間後、溶液を減圧下で濃縮して、生じた残留物を、1M HCl aq(5mL)及びEt2O(5mL)中に溶解した。フェニルボロン酸(107mg、0.878mmol)を加えて、澄明な二相性溶液を、室温にて1時間撹拌した。混合液をEt2O及び水で希釈して、層を分離させた。水性層をEt2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 20ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア(20mL)を用いて溶出して、(2R,4R)-4-(2-アミノエチルアミノ)-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例27、106mg、89%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.63-0.77(2H,m),1.13-1.23(1H,m),1.37(3H,br d),1.63-1.76(1H,m),1.92-2.02(1H,m),2.06-2.14(1H,m),2.24-2.33(1H,m),2.78(2H,s),2.96(2H,s),3.08-3.19(1H,m),3.28-3.43(1H,m),3.43-3.51(1H,m);m/z:(ES+)[M+H]+=274.
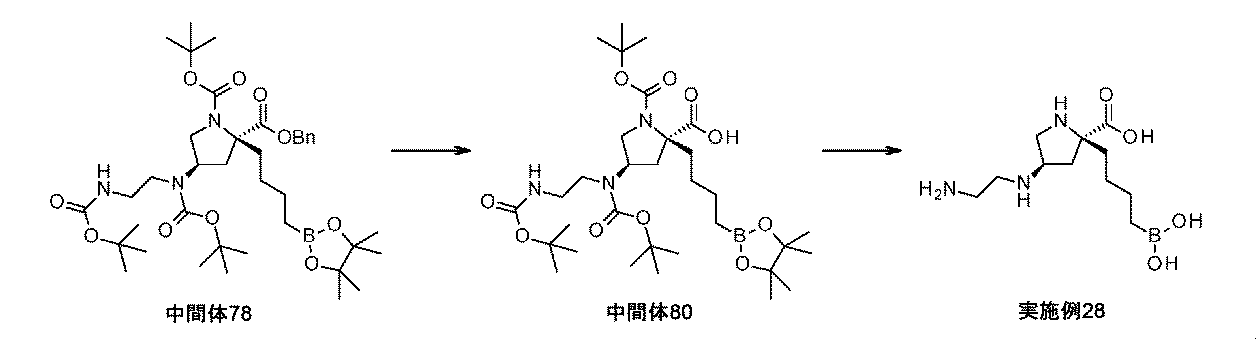
Pd/C(10wt%、137mg、0.129mmol)を、2-ベンジル1-tert-ブチル(2S,4R)-4-[tert-ブトキシカルボニル-[2-(tert-ブトキシカルボニルアミノ)エチル]アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体78、480mg、0.64mmol)のEtOAc溶液(7.2mL)に加えた。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液をMeOHで希釈して、珪藻土で濾過した。濾液を濃縮して乾燥させて、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって精製して、(2S,4R)-1-tert-ブトキシカルボニル-4-[tert-ブトキシカルボニル-[2-(tert-ブトキシカルボニルアミノ)エチル]アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体80、170mg、40%の収率)を白色の固体として得た。1H NMR(500MHz,DMSO-d6)δ 0.58-0.73(2H,m),1.15(12H,s),1.37(31H,br d),1.58-1.74(2H,m),2.18-2.33(2H,m),2.92-3.16(6H,m),3.59-3.73(1H,m),4.31-4.50(1H,m),6.84-6.95(1H,m);m/z:(ES+)[M+H]+=656.
トリフルオロ酢酸(1.00mL、13.0mmol)を、(2S,4R)-1-tert-ブトキシカルボニル-4-[tert-ブトキシカルボニル-[2-(tert-ブトキシカルボニルアミノ)エチル]アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体80、170mg、0.26mmol)の撹拌DCM溶液(5mL)に室温にて滴加した。1時間後、溶液を減圧下で濃縮して、生じた残留物を、1M HCl aq(5mL)及びEt2O(5mL)中に溶解した。フェニルボロン酸(63mg、0.52mmol)を加えて、澄明な二相性溶液を、室温にて1時間撹拌した。混合液をEt2O及び水で希釈して、層を分離させた。水性層をEt2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 20ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア(20mL)を用いて溶出して、(2S,4R)-4-(2-アミノエチルアミノ)-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例28、67mg、95%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.65-0.77(2H,m),1.11-1.24(1H,m),1.25-1.34(1H,m),1.34-1.42(2H,m),1.62-1.79(2H,m),1.97-2.06(1H,m),2.61-2.70(1H,m),2.72-2.84(2H,m),2.93(3H,s),3.30-3.41(1H,m),3.47-3.55(1H,m);m/z:(ES+)[M+H]+=274.
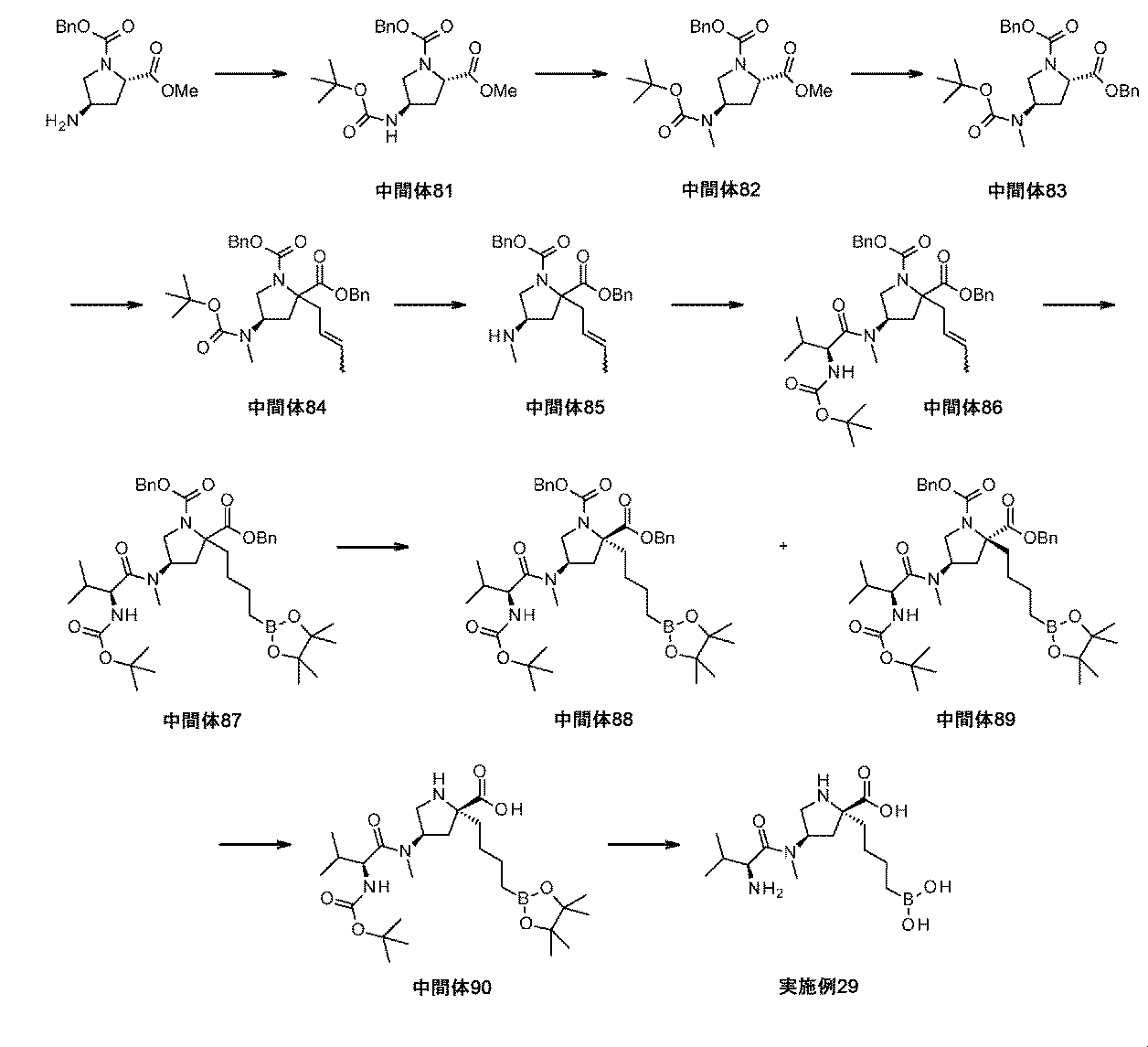
ジ-tert-ブチルジカルボナート(6.27g、28.8mmol)を、1-ベンジル2-メチル(2S,4R)-4-アミノピロリジン-1,2-ジカルボキシラート(5.00g、18.0mmol)及びトリエチルアミン(5.00mL、35.9mmol)のDCM溶液(78ml)に加えて、反応液を室温にて16時間撹拌した。反応混合液をDCMで希釈して、0.5MHCl(aq)、飽和重炭酸ナトリウム、及び飽和塩化ナトリウムで順次洗浄した。有機層をNa2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲルクロマトグラフィ(DCM/MeOH)によって精製して、1-ベンジル2-メチル(2S,4R)-4-(tert-ブトキシカルボニルアミノ)ピロリジン-1,2-ジカルボキシラート(中間体81、5.4g、79%の収率)を白色の固体として得た。1H NMR(500MHz,DMSO-d6)δ 1.31-1.42(9H,m),1.99-2.23(2H,m),3.22-3.28(1H,m),3.52-3.65(4H,m),3.96-4.10(1H,m),4.30-4.48(1H,m),4.92-5.13(2H,m),7.18-7.41(5H,m);m/z:(ES+)[M+H]+=379.
水酸化ナトリウム(鉱油中60%分散系)(0.423g、12.3mmol)を、1-ベンジル2-メチル(2S,4R)-4-(tert-ブトキシカルボニルアミノ)ピロリジン-1,2-ジカルボキシラート(中間体81、3.20g、8.46mmol)のDMF溶液(33mL)に0℃にて少量ずつ加えた。反応混合液を、0℃にて1時間撹拌してから、さらに1時間撹拌しながら室温に温めた。ヨウ化メチル(0.63ml、10.2mmol)を加えて、反応液を室温にて16時間撹拌した。反応混合液を0℃に冷却して、水でクエンチした。揮発性物質を減圧下で除去して、懸濁液をDCM(200mL)で希釈した。層を分離させて、有機層を水(4×50mL)及び飽和水性塩化ナトリウム(2×50mL)で順次洗浄した。有機層を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させて、1-ベンジル2-メチル(2S,4R)-4-[tert-ブトキシカルボニル(メチル)アミノ]ピロリジン-1,2-ジカルボキシラート(中間体82、2.33g、70%の収率)を無色の油として得た。これを、更なる精製なしに用いた。1H NMR(500MHz,DMSO-d6)δ 1.25-1.47(9H,m),1.89-2.08(1H,m),2.31-2.46(1H,m),2.69(3H,d),3.22-3.39(2H,m),3.65(4H,s),4.22-4.49(1H,m),4.87-5.17(2H,m),7.18-7.46(5H,m);m/z:(ES+)[M+H]+=393.
水酸化ナトリウム(1.425g、35.62mmol)の水溶液(7.5mL)を、1-ベンジル2-メチル(2S,4R)-4-[tert-ブトキシカルボニル(メチル)アミノ]ピロリジン-1,2-ジカルボキシラート(中間体82、2.33g、5.94mmol)のTHF(15mL)及びMeOH(7.5mL)溶液に0℃にて加えた。反応混合液を、室温にゆっくり温めながら、6時間撹拌した。揮発性物質を減圧下で除去して、水性層を5M HCl(aq)で約3のpHに酸性化して、DCMで抽出した(4×50mL)。組み合わせた有機質を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させて、粗カルボン酸を白色の固体として得た。これを、更なる精製なしに用いた。
ジベンジル(2S,4R)-4-[tert-ブトキシカルボニル(メチル)アミノ]ピロリジン-1,2-ジカルボキシラート(中間体83、2.45g、5.23mmol)及び臭化クロチル(0.81mL、7.8mmol)を、THF(18mL)中に溶解して、溶液を、N2雰囲気下で-78℃に冷却した。KHMDSの溶液(トルエン中0.5M、15.7mL、7.85mmol)を反応混合液に滴加して、反応液を、室温にゆっくり温めながら、17時間撹拌した。粗反応混合液を水でクエンチして、揮発性物質を減圧下で除去した。粗混合液をDCM中に希釈して、層を分離させた。有機層を、水で洗浄して、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。粗物質を、シリカゲル精製(ヘキサン/EtOAc)によって精製して、ジベンジル(4R)-2-(ブタ-2-エニル)-4-[tert-ブトキシカルボニル(メチル)アミノ]ピロリジン-1,2-ジカルボキシラート(中間体84、2.10g、86%の収率)を黄色の油として、そして偏左右異性体、E/Zオレフィン異性体、及び回転異性体の混合物として得た。1H NMR(500MHz,DMSO-d6)δ 1.31-1.40(9H,m),1.46-1.68(3H,m),1.99-2.31(2H,m),2.40-2.47(1H,m),2.55-2.68(3H,m),2.69-3.00(1H,m),3.30(1H,s),3.51-3.77(1H,m),4.55-4.74(1H,m),4.79-5.21(4H,m),5.26-5.41(1H,m),5.42-5.71(1H,m),7.16-7.42(10H,m);m/z:(ES+)[M+H]+=523.
トリフルオロ酢酸(3.84mL、50.2mmol)を、ジベンジル(4R)-2-(ブタ-2-エニル)-4-[tert-ブトキシカルボニル(メチル)アミノ]ピロリジン-1,2-ジカルボキシラート(中間体84、2.10g、4.02mmol)のDCM溶液(32mL)に滴加して、反応液を、室温にて2時間撹拌した。反応混合液を、濃縮して乾燥させて、ジベンジル(4R)-2-(ブタ-2-エニル)-4-(メチルアミノ)ピロリジン-1,2-ジカルボキシラートのTFA塩(中間体85、2.55g、100%の収率)を無色の油として得た。これを、精製なしに用いた。1H NMR(500MHz,DMSO-d6)δ 1.45-1.74(3H,m),2.14-2.33(1H,m),2.34-2.46(1H,m),2.57-2.65(3H,m),2.66-3.07(1H,m),3.15-3.56(1H,m),3.66-3.77(1H,m),3.81-4.13(1H,m),4.79-5.22(4H,m),5.26-5.73(2H,m),7.20-7.41(10H,m),8.62-8.87(2H,m);m/z:(ES+)[M+H]+=423.
N,N-ジイソプロピルエチルアミン(0.70mL、4.0mmol)を、HATU(1.52、4.02mmol)及びBoc-Val-OH(873mg、4.02mmol)のDMF溶液(15mL)に加えて、反応液を室温にて30分間撹拌した。ジベンジル(4R)-2-(ブタ-2-エニル)-4-(メチルアミノ)ピロリジン-1,2-ジカルボキシラートTFA塩(中間体85、2.16g、4.02mmol)のDMF(15mL)及びN,N-ジイソプロピルエチルアミン(0.70mL、4.0mmol)溶液を加えて、反応液を室温にてさらに17時間撹拌した。反応混合液を濃縮して、シリカゲルクロマトグラフィ(ヘキサン/EtOAc)によって直接精製して、ジベンジル(4R)-2-(ブタ-2-エニル)-4-[[(2S)-2-(tert-ブトキシカルボニルアミノ)-3-メチル-ブタノイル]-メチル-アミノ]ピロリジン-1,2-ジカルボキシラート(中間体86、1.97g、79%の収率)を回転異性体の混合物として得た。1H NMR(500MHz,DMSO-d6)δ 1H NMR(500MHz,DMSO-d6)0.61-0.92(6H,m),1.35(9H,br s),1.62(3H,br d),1.79-1.92(1H,m),2.03-2.33(2H,m),2.57-2.76(2H,m),2.76-3.00(3H,m),3.30(1H,s),3.49-3.77(1H,m),4.04-4.16(1H,m),4.78-5.20(5H,m),5.26-5.72(2H,m),6.72-7.03(1H,m),7.17-7.38(10H,m);m/z:(ES+)[M+H]+=622.
ビス(1,5-シクロオクタジエン)ジイリジウム(I)ジクロリド(213mg、0.317mmol)及びビス(ジフェニルホスフィノ)メタン(244mg、0.635mmol)を、オーブン乾燥した丸底フラスコに加えた。フラスコをシールして、N2でパージした。固体を、DCM(8.9mL)中に溶解して、4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン(1.01mL、6.97mmol)を溶液にゆっくり加えた。反応液を、室温にて10分間撹拌した。ジベンジル(4R)-2-(ブタ-2-エニル)-4-[[(2S)-2-(tert-ブトキシカルボニルアミノ)-3-メチル-ブタノイル]-メチル-アミノ]ピロリジン-1,2-ジカルボキシラート(中間体86、1.97g、3.17mmol)を、DCM溶液(5.9mL)としての反応液に加えて、反応混合液を室温にて16時間撹拌した。反応混合液を0℃に冷却して、MeOH及び水で注意深くクエンチした。層を分離させて、水性層をDCMで抽出した。組み合わせた有機質を、Na2SO4上で乾燥させて、濾過して、濃縮して乾燥させた。生じた残留物を、フラッシュシリカクロマトグラフィ(ヘキサン/EtOAc)によって精製して、ジベンジル(4R)-4-[[(2S)-2-(tert-ブトキシカルボニルアミノ)-3-メチル-ブタノイル]-メチル-アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体87、1.5g、63%の収率)を黄色の油として、そして偏左右異性体及び回転異性体の混合物として得た。精製物質を、キラルSFC[(S,S)Whelk-O1カラム、30mm×250mm、5μm、温度=20℃、移動相=0~30%MeOH:CO2、UV検出@220nm、ローディング=31mg/inj、conc=MeOH中125mg/mL、流量=75mL/分、出口圧力=100bar]にかけて、2つの偏左右異性体を得た。各偏左右異性体の立体化学を、他の例示する化合物と一致するように、実施例29及び実施例30の酵素効力に基づいて遡及的に割り当てた。
Pd/C(10wt%、99mg、0.093mmol)を、ジベンジル(2R,4R)-4-[[(2S)-2-(tert-ブトキシカルボニルアミノ)-3-メチル-ブタノイル]-メチル-アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体88、280mg、0.37mmol)のEtOAc溶液(1.8mL)に加えた。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて4時間撹拌した。反応混合液をMeOHで希釈して、珪藻土で濾過した。濾液を濃縮して乾燥させて、(2R,4R)-4-[[(2S)-2-(tert-ブトキシカルボニルアミノ)-3-メチル-ブタノイル]-メチル-アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体90、180mg、92%の収率)を白色の固体として得た。これを、更なる精製なしに用いた。1H NMR(500MHz,DMSO-d6)δ 0.59-0.69(2H,m),0.74-0.87(6H,m),1.16(12H,s),1.22-1.30(3H,m),1.35(9H,s),1.49-1.65(1H,m),1.65-1.82(1H,m),1.82-1.95(2H,m),2.15-2.26(1H,m),2.63-2.91(3H,m),2.92-3.21(3H,m),4.05-4.22(1H,m),4.80-5.16(1H,m),6.76(1H,s),7.56-8.05(1H,m);m/z:(ES+)[M+H]+=526.
トリフルオロ酢酸(0.53mL、6.9mmol)を、(2R,4R)-4-[[(2S)-2-(tert-ブトキシカルボニルアミノ)-3-メチル-ブタノイル]-メチル-アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体90、180mg、0.34mmol)の撹拌DCM溶液(10mL)に室温にて滴加した。1時間後、溶液を減圧下で濃縮して、生じた残留物を、1M HCl aq(5mL)及びEt2O(5mL)中に溶解した。フェニルボロン酸(84mg、0.69mmol)を加えて、澄明な二相性溶液を、室温にて1時間撹拌した。混合液をEt2O及び水で希釈して、層を分離させた。水性層をEt2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 60ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア(60mL)を用いて溶出して、(2R,4R)-4-[[(2S)-2-アミノ-3-メチル-ブタノイル]-メチル-アミノ]-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例29、98mg、86%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.72-0.83(2H,m),0.85-1.01(6H,m),1.10-1.45(4H,m),1.61-1.84(1H,m),1.85-2.03(2H,m),2.19-2.34(1H,m),2.34-2.49(1H,m),3.00(3H,s),3.16-3.51(2H,m),3.76-3.96(1H,m),4.95-5.10(1H,m);m/z:(ES+)[M+H]+=344.
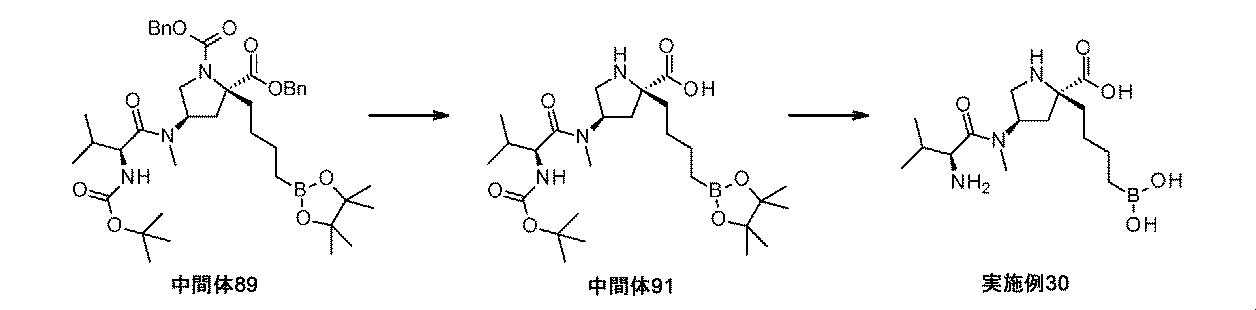
Pd/C(10wt%、209mg、0.196mmol)を、ジベンジル(2S,4R)-4-[[(2S)-2-(tert-ブトキシカルボニルアミノ)-3-メチル-ブタノイル]-メチル-アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-1,2-ジカルボキシラート(中間体89、590mg、0.79mmol)のEtOAc溶液(4mL)に加えた。フラスコは、H2のバルーンを備えた。懸濁液を、室温にて一晩撹拌した。反応混合液をMeOHで希釈して、珪藻土で濾過した。濾液を濃縮して乾燥させて、(2S,4R)-4-[[(2S)-2-(tert-ブトキシカルボニルアミノ)-3-メチル-ブタノイル]-メチル-アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体91、397mg、96%の収率)を白色の固体として得た。これを、更なる精製なしに用いた。1H NMR(500MHz,DMSO-d6)δ 0.58-0.67(2H,m),0.73-0.87(6H,m),1.16(12H,s),1.22-1.31(3H,m),1.35(9H,s),1.51-1.67(1H,m),1.68-1.95(3H,m),2.28-2.37(2H,m),2.95(4H,s),4.05-4.20(1H,m),4.66-4.79(1H,m),6.65-6.90(1H,m),7.85-8.15(1H,m);m/z:(ES+)[M+H]+=526.
トリフルオロ酢酸(1.17mL、15.2mmol)を、(2S,4R)-4-[[(2S)-2-(tert-ブトキシカルボニルアミノ)-3-メチル-ブタノイル]-メチル-アミノ]-2-[4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)ブチル]ピロリジン-2-カルボン酸(中間体91、397mg、0.755mmol)の撹拌DCM溶液(10mL)に室温にて滴加した。1時間後、溶液を減圧下で濃縮して、生じた残留物を、1M HCl aq(5mL)及びEt2O(5mL)中に溶解した。フェニルボロン酸(186mg、1.52mmol)を加えて、澄明な二相性溶液を、室温にて1時間撹拌した。混合液をEt2O及び水で希釈して、層を分離させた。水性層をEt2Oで洗浄した。水性層を凍結乾燥して、イオン交換クロマトグラフィ(PoraPak Rxn CX 60ccカラム)によって精製した。所望の生成物をカラムから、MeOH中5%アンモニア(60mL)を用いて溶出して、(2S,4R)-4-[[(2S)-2-アミノ-3-メチル-ブタノイル]-メチル-アミノ]-2-(4-ボロノブチル)ピロリジン-2-カルボン酸(実施例30、163mg、92%の収率)を白色の固体として得た。1H NMR(500MHz,D2O)δ 0.68-0.78(2H,m),0.83-0.94(6H,m),1.10-1.41(4H,m),1.66-2.13(4H,m),2.49-2.68(1H,m),3.02(3H,s),3.22-3.30(1H,m),3.43-3.60(1H,m),3.70-3.88(1H,m),4.53-4.66(1H,m);m/z:(ES+)[M+H]+=344.
大腸菌(E.coli)から生成した組換えアルギナーゼ1又はアルギナーゼ2を用いて、チオアルギニン(thioarginine)からのチオール基の形成を測定することによって、ヒトアルギナーゼ1の活性及びアルギナーゼ2活性に及ぼす実施例1~実施例30の阻害効果を定量化した。チオール基を、エルマン試薬、5,5’-ジチオビス(2-ニトロ安息香酸)(DTNB)で検出した。DTNBは、チオールと反応して混合ジスルフィド及び2-ニトロ-5-チオ安息香酸(TNB)を与え、これをアニオン(TNB2-)の412nmでの吸光度によって定量化する。
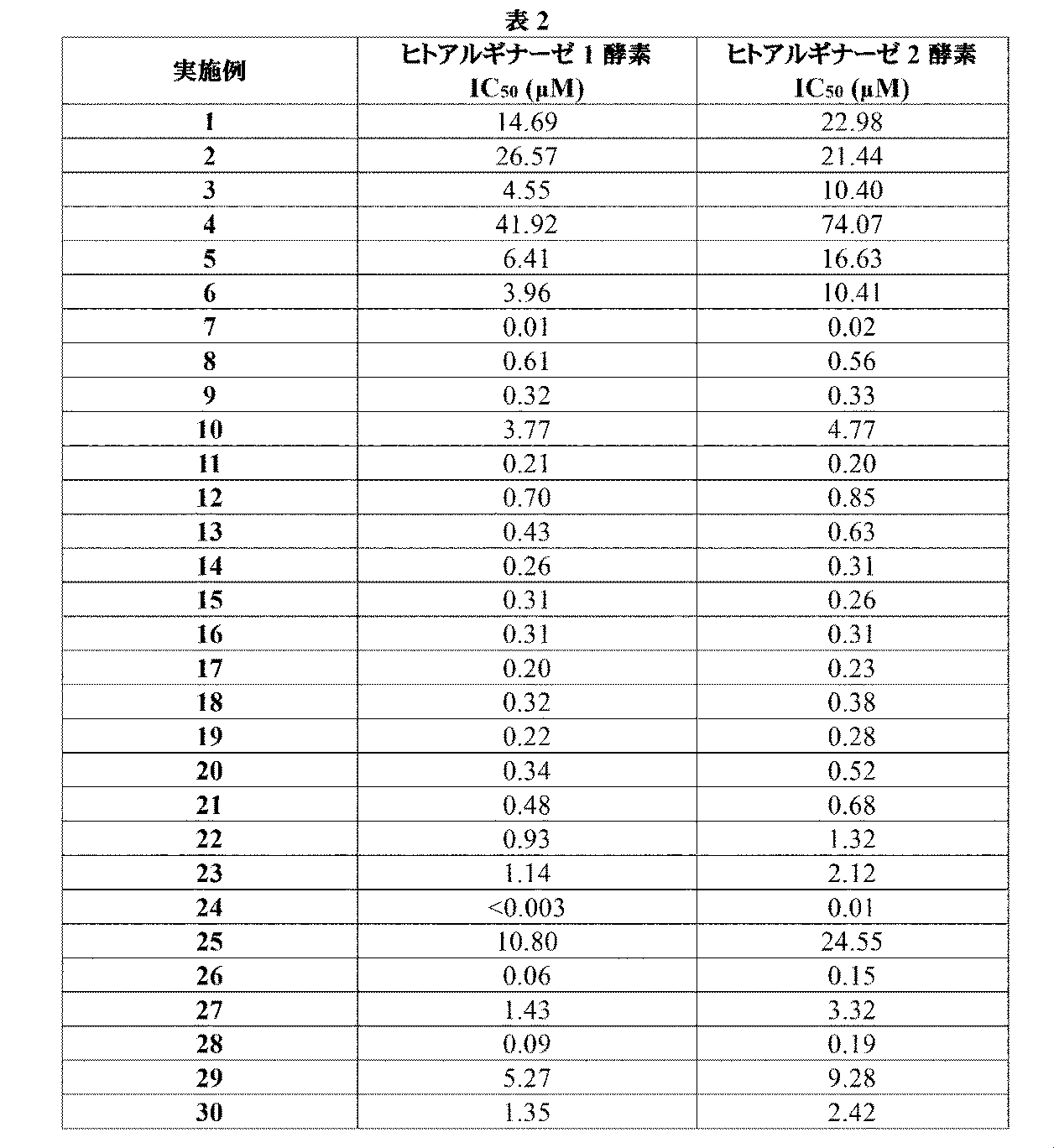
実施例8、9、及び13~20は、実施例7のプロドラッグ形態である。以下の薬物動態研究は、実施例8から実施例7の生体利用性を実証するために実行した。実施例8は、IV投薬用に、0.9w/v%生理食塩水pH4(1M HClで調整)中で製剤化した。製剤を、2頭の雄ラットへの大腿骨カテーテルによって2mg/kgにてそれぞれ投薬した(170~250g)。頸静脈カテーテル連続血液サンプルを、投薬の0.033、0.083、0.167、0.5、1、2、4、8、及び24時間後に採った。PO投薬用に、実施例8を、脱イオン水pH4(1M HClで調整)中で製剤化して、2頭の雄ラットへの経口強制飼養によって5mg/kgにてそれぞれ投薬した(170~250g)。連続血液サンプルを、投薬の0.25、0.5、1、1.5、2、3、4、8、及び24時間後に、頸静脈カテーテルによって採った。血漿サンプルを血液から、低速遠心分離を用いて生じさせた。実施例7及び実施例8を含有する較正標準の単一のセットを、ブランクの血漿をスパイクすることによって調製した。サンプル及び標準を、2容量のアセトニトリルによる析出に続く遠心分離によって抽出した。得られた結果を用いて、実施例7及び実施例8の双方について、Cl(mL/min/kg)、Vdss(L/kg)、Cmax(μM)、AUC(μMh)、tmax(h)、及び%Fを求めた。実施例8として投薬した場合の実施例7のPO用量標準化AUCを、実施例7として投薬した場合の実施例7の用量標準化IV AUCに対して比較することによって、絶対的な生体利用性を求めた。必要に応じて、公称でない測定された用量を用いて、生体利用性を算出した。類似して、同じ手順を、実施例9及び実施例13~実施例20について繰り返した。結果を、表3~表12に示す。これらの結果は、特定のアミノ酸部分をプロドラッグとして組み込むことによって生体利用性が増大し得ることを示している。

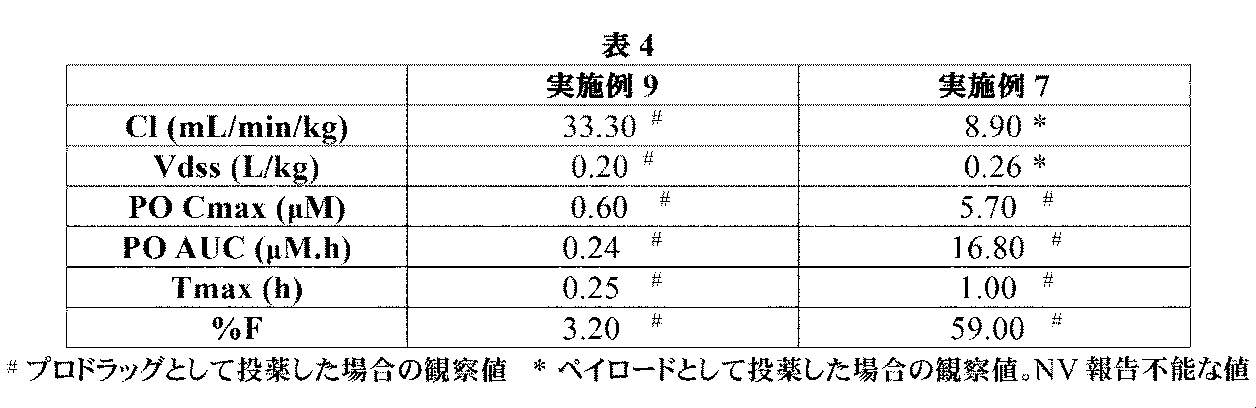
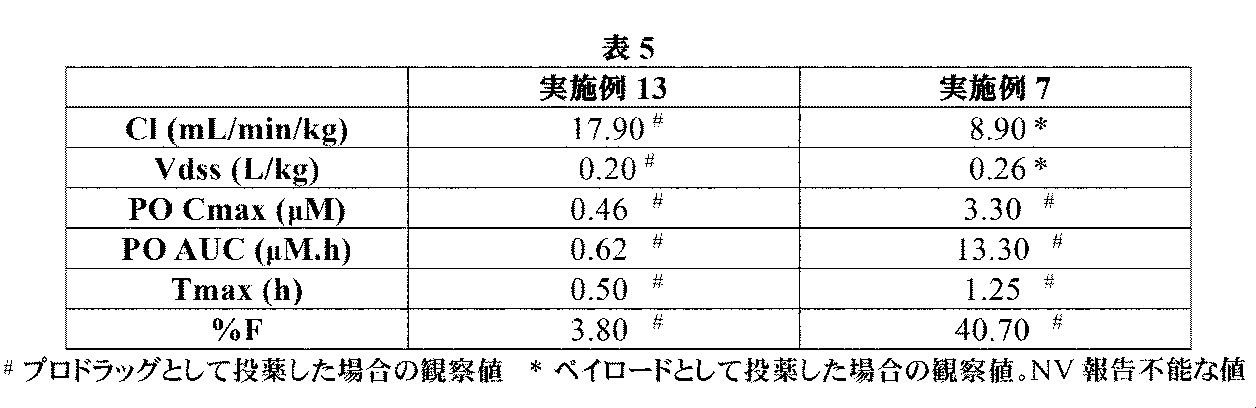



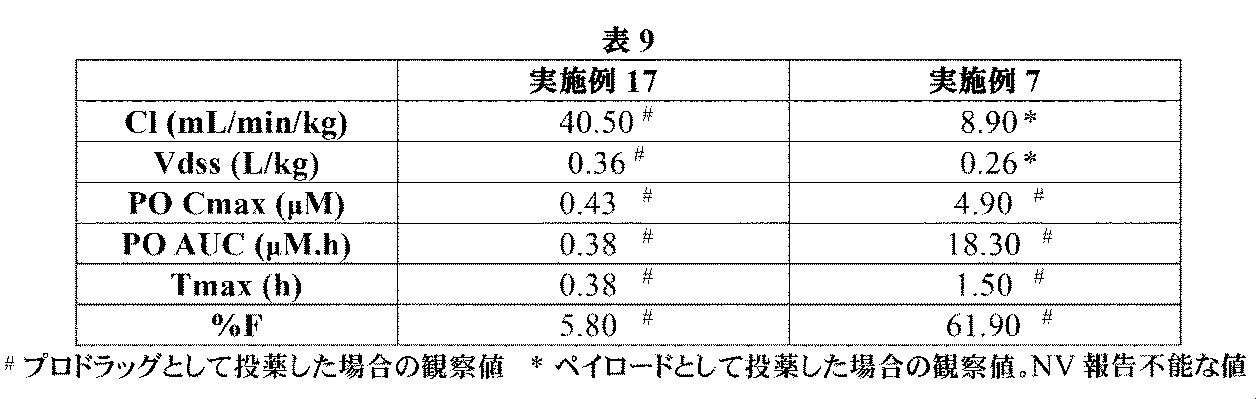

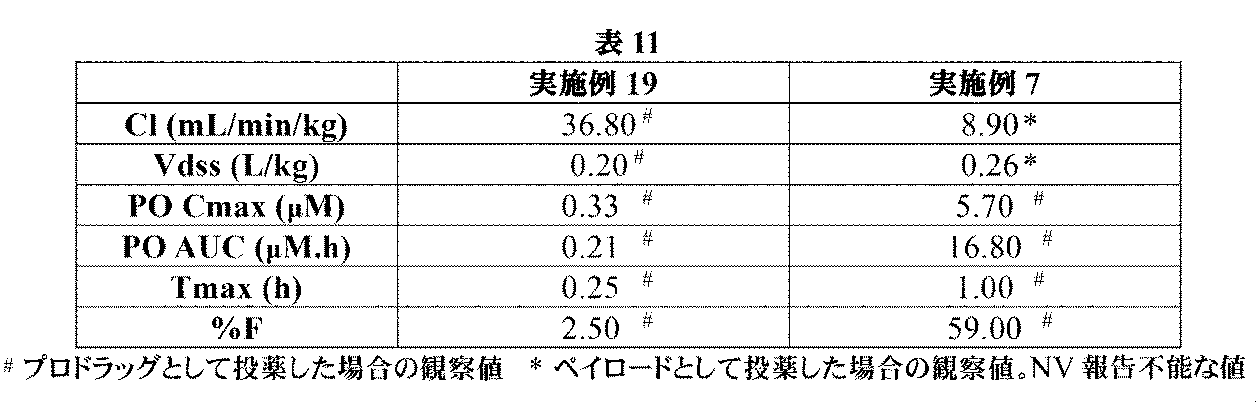
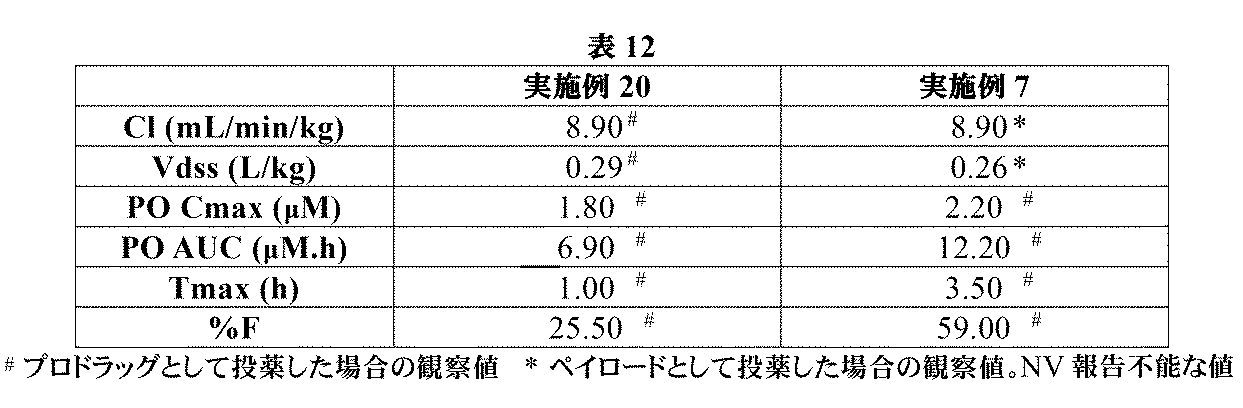
[項1]
式(I)の化合物、又はその薬学的に許容される塩であって:
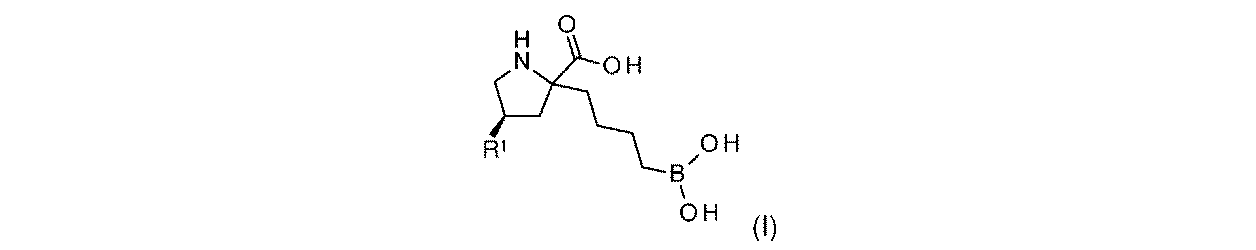
R1は-NHR1aであり;
R1aは、-H又は-C(O)CH(R1b)NHR1cであり;そして
R1bは、-H、-(C1~C4)アルキル、及びCH2OR1dから選択され、そしてR1cは-Hであり;又は
R1b及びR1cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R1dは、H又は-CH3である、式(I)の化合物、又はその薬学的に許容される塩。
[項2]
式(II)の化合物、又はその薬学的に許容される塩であって:
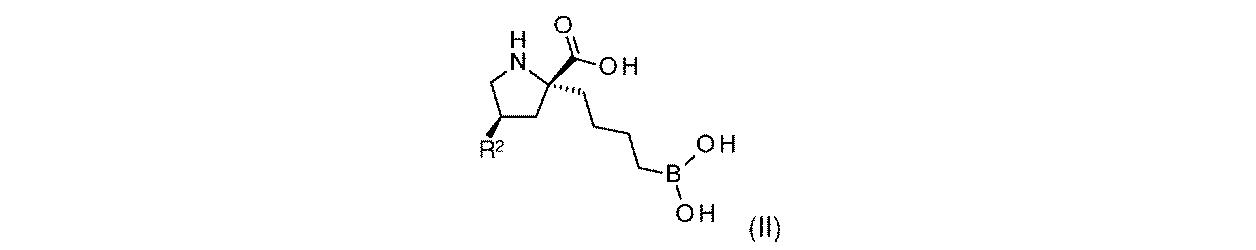
R2は、-OH又は-NHR2aであり;
R2aは、-H又は-C(O)CH(R2b)NHR2cであり;
R2bは、-H、-(C1~C4)アルキル、及びCH2OR2dから選択され、そしてR2cは-Hであり;又は
R2b及びR2cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R2dは、-H又は-CH3である、式(II)の化合物、又はその薬学的に許容される塩。
[項3]
式(III)の化合物、又はその薬学的に許容される塩であって:
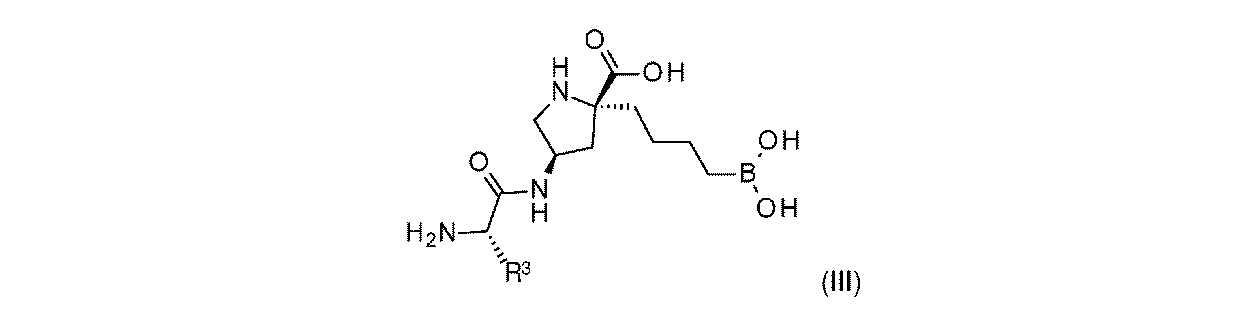
R3は、-H、-(C1~C4)アルキル、及び-CH2OR3aから選択され;そして
R3aは、-H又は-CH3である、式(III)の化合物、又はその薬学的に許容される塩。
[項4]
式(IV)の化合物、又はその薬学的に許容される塩であって:
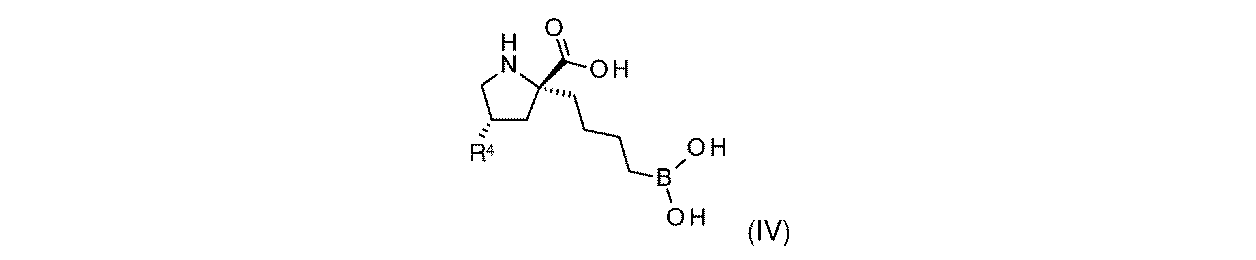
[項5]
式(V)の化合物、又はその薬学的に許容される塩:
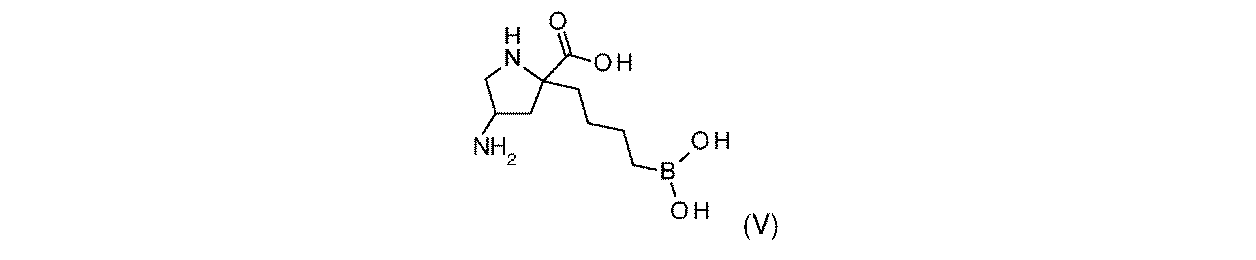
[項6]
式(VI)の化合物、又はその薬学的に許容される塩であって:
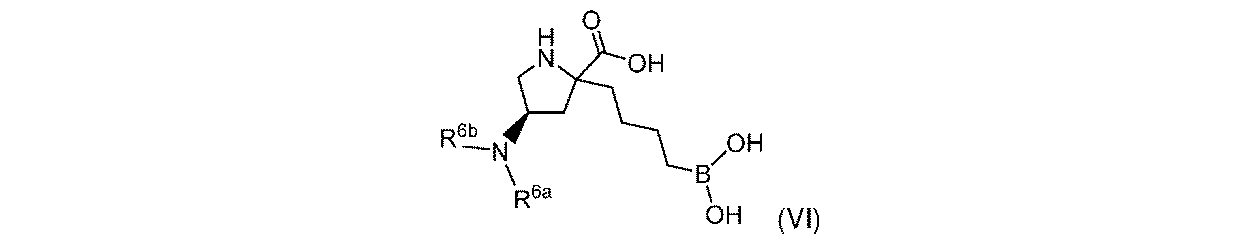
R6aは、-H又は-CH3であり;
R6bは、-C(O)C(R6cR6d)NH2であるか;又は0若しくは1アミノ、若しくは-OR6eで置換されている-(C1~C3)アルキルであり;そして
R6cは、0若しくは1アミノ、又は-OR6fで置換されている-(C1~C3)アルキルであり;
R6dは、H又は-CH3であり;そして
R6e及びR6fは独立して、-H又は-CH3である、式(VI)の化合物、又はその薬学的に許容される塩。
[項7]
式(VI)の化合物、又はその薬学的に許容される塩であって:
R6aは、-H又は-CH3であり;そしてR6bは、0又は1アミノで置換されている-(C1~C3)アルキルであり;又は代わりに、
R6aは、-H又は-CH3であり;R6bは、-C(O)C(R6cR6d)NH2であり;R6cは、0若しくは1アミノ、又は-OHで置換されている-(C1~C3)アルキルであり;そしてR6dは、H又は-CH3である、式(VI)の化合物、又はその薬学的に許容される塩。
[項8]
式(Ib)の化合物、又はその薬学的に許容される塩であって:
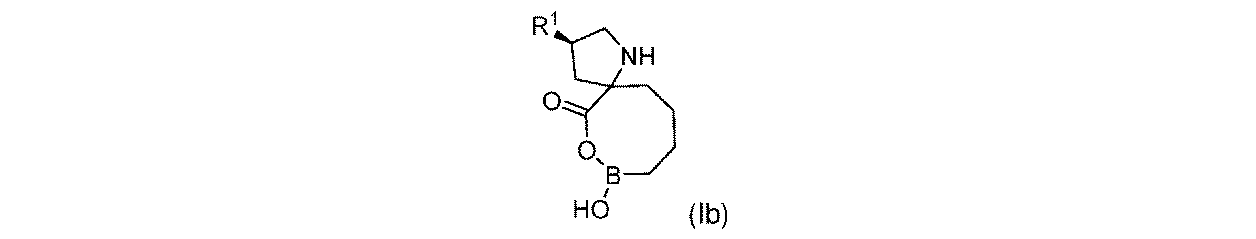
R1は-NHR1aであり;
R1aは、-H又は-C(O)CH(R1b)NHR1cであり;そして
R1bは、-H、-(C1~C4)アルキル、及びCH2OR1dから選択され、そしてR1cは-Hであり;又は
R1b及びR1cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R1dは、H又は-CH3である、式(Ib)の化合物、又はその薬学的に許容される塩。
[項9]
式(IIb)の化合物、又はその薬学的に許容される塩であって:
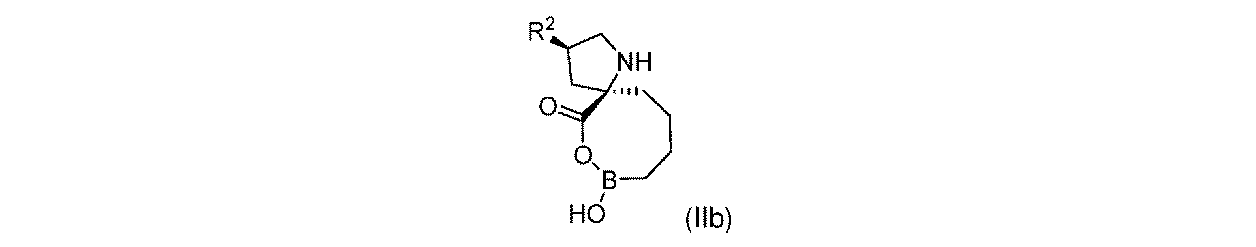
R2は、-OH又は-NHR2aであり;
R2aは、-H又は-C(O)CH(R2b)NHR2cであり;
R2bは、-H、-(C1~C4)アルキル、及びCH2OR2dから選択され、そしてR2cは-Hであり;又は
R2b及びR2cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R2dは、-H又は-CH3である、式(IIb)の化合物、又はその薬学的に許容される塩。
[項10]
式(IIIb)の化合物、又はその薬学的に許容される塩であって:

R3は、-H、-(C1~C4)アルキル、及び-CH2OR3aから選択され;そして
R3aは、-H又は-CH3である、式(IIIb)の化合物、又はその薬学的に許容される塩。
[項11]
式(IVb)の化合物、又はその薬学的に許容される塩であって:
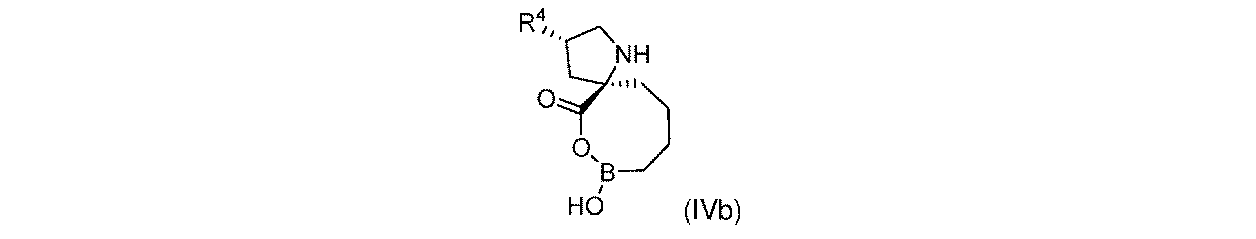
[項12]
式(Vb)の化合物、又はその薬学的に許容される塩:
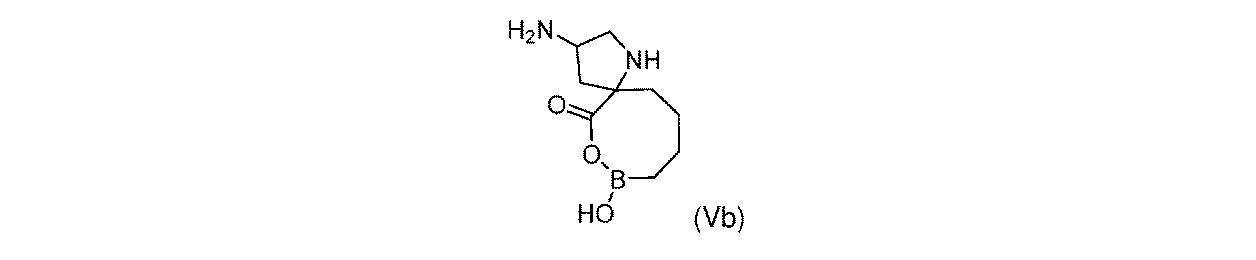
[項13]
式(VIb)の化合物、又はその薬学的に許容される塩であって:
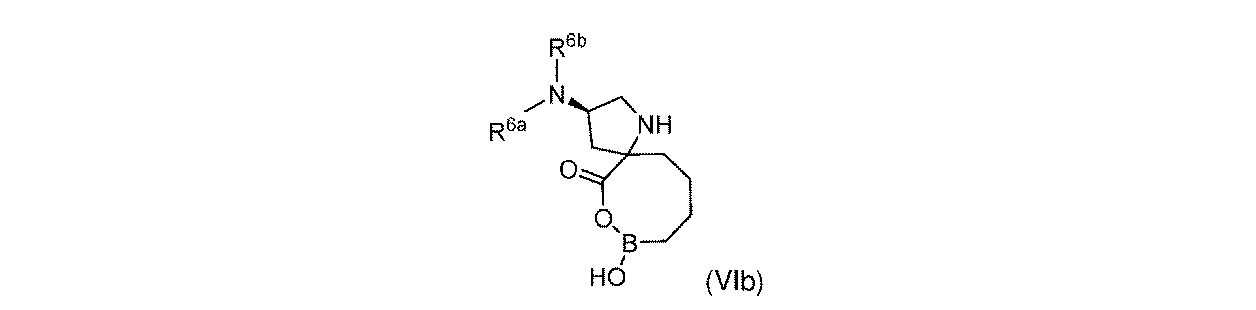
R6aは、-H又は-CH3であり;
R6bは、-C(O)C(R6cR6d)NH2であるか;又は0若しくは1アミノ、若しくは-OR6eで置換されている-(C1~C3)アルキルであり;そして
R6cは、0若しくは1アミノ、又は-OR6fで置換されている-(C1~C3)アルキルであり;
R6dは、H又は-CH3であり;そして
R6e及びR6fは独立して、-H又は-CH3である、式(VIb)の化合物、又はその薬学的に許容される塩。
[項14]
式(VIb)の化合物、又はその薬学的に許容される塩であって:
R6aは、-H又は-CH3であり;そしてR6bは、0又は1アミノで置換されている-(C1~C3)アルキルであり;又は代わりに、
R6aは、-H又は-CH3であり;R6bは、-C(O)C(R6cR6d)NH2であり;R6cは、0若しくは1アミノ、又は-OHで置換されている-(C1~C3)アルキルであり;そしてR6dは、H又は-CH3である、式(VIb)の化合物、又はその薬学的に許容される塩。
[項15]
表1の化合物、又はその医薬的に許容される塩。
[項16]
項1~15のいずれかに記載の化合物、又はその薬学的に許容される塩、及び薬学的に許容されるキャリアを含む医薬組成物。
[項17]
癌を処置する方法であって、項1~15のいずれかに記載の化合物、又はその薬学的に許容される塩を含む方法。
[項18]
癌を処置するための、項1~15のいずれかに記載の化合物、又はその薬学的に許容される塩。
[項19]
癌を処置するのに用いられる医薬の製造における、項1~15のいずれかに記載の化合物、又はその薬学的に許容される塩。
[項20]
癌を処置するのに用いられる、項1~15のいずれかに記載の化合物、又はその薬学的に許容される塩を含む医薬組成物。
[項21]
呼吸器炎症性疾患を処置する方法であって、項1~15のいずれかに記載の化合物、又はその薬学的に許容される塩を含む方法。
[項22]
呼吸器炎症性疾患を処置するための、項1~15のいずれかに記載の化合物、又はその薬学的に許容される塩。
[項23]
呼吸器炎症性疾患を処置するのに用いられる医薬の製造における、項1~15のいずれかに記載の化合物、又はその薬学的に許容される塩。
[項24]
呼吸器炎症性疾患を処置するのに用いられる、項1~15のいずれかに記載の化合物、又はその薬学的に許容される塩を含む医薬組成物。
Claims (27)
- 式(I)の化合物、又はその薬学的に許容される塩であって:
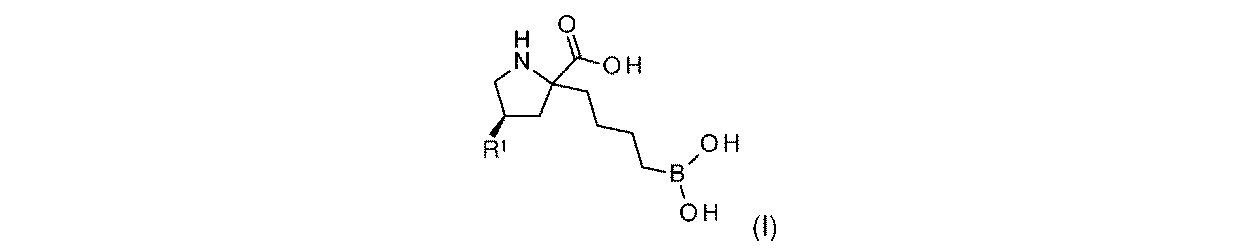
R1は-NHR1aであり;
R1aは、-H又は-C(O)CH(R1b)NHR1cであり;そして
R1bは、-H、-(C1~C4)アルキル、及びCH2OR1dから選択され、そしてR1cは-Hであり;又は
R1b及びR1cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R1dは、H又は-CH3である、式(I)の化合物、又はその薬学的に許容される塩。 - R 1 が-NHR 1a であり;
R 1a が-H又は-C(O)CH(R 1b )NH 2 であり;そして
R 1b が-CH 3 又は-CH(CH 3 ) 2 である、
請求項1に記載の化合物、またはその薬学的に許容される塩。 - 式(II)の化合物、又はその薬学的に許容される塩であって:
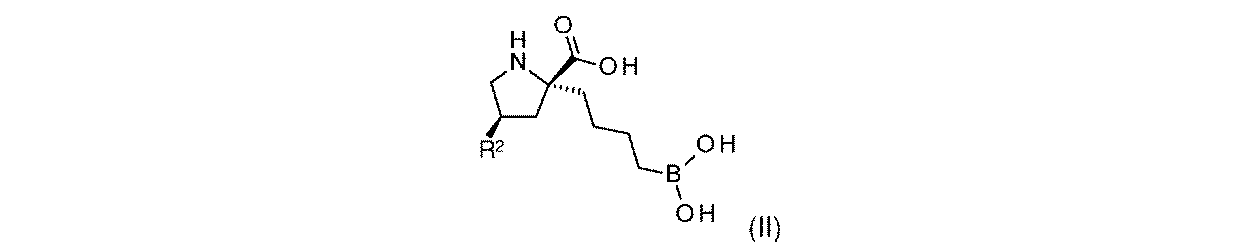
R2は、-OH又は-NHR2aであり;
R2aは、-H又は-C(O)CH(R2b)NHR2cであり;
R2bは、-H、-(C1~C4)アルキル、及びCH2OR2dから選択され、そしてR2cは-Hであり;又は
R2b及びR2cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R2dは、-H又は-CH3である、式(II)の化合物、又はその薬学的に許容される塩。 - R 2 が-OH又は-NHR 2a であり;
R 2a が-H又は-C(O)CH(R 2b )NH 2 であり;そして
R 2b が-CH 3 又は-CH(CH 3 ) 2 である、
請求項3に記載の化合物、またはその薬学的に許容される塩。 - 式(III)の化合物、又はその薬学的に許容される塩であって:
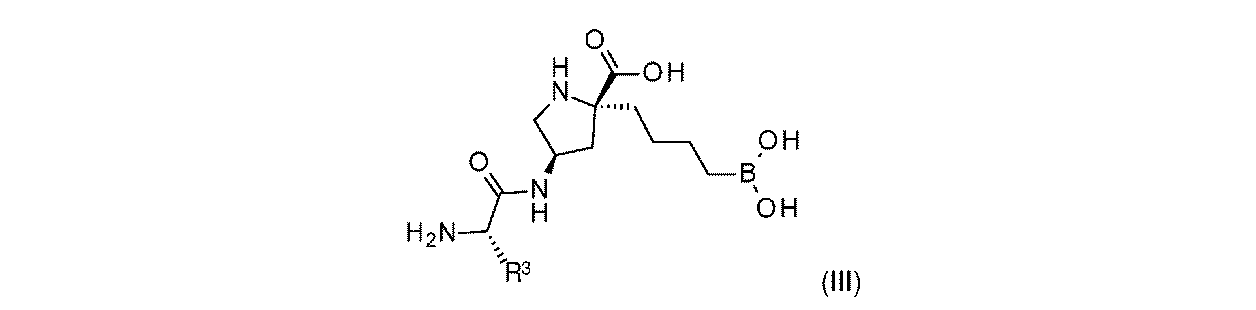
R3は、-H、-(C1~C4)アルキル、及び-CH2OR3aから選択され;そして
R3aは、-H又は-CH3である、式(III)の化合物、又はその薬学的に許容される塩。 - R 3 が-CH 3 又は-CH(CH 3 ) 2 である、請求項5に記載の化合物、またはその薬学的に許容される塩。
- 式(IV)の化合物、又はその薬学的に許容される塩であって:
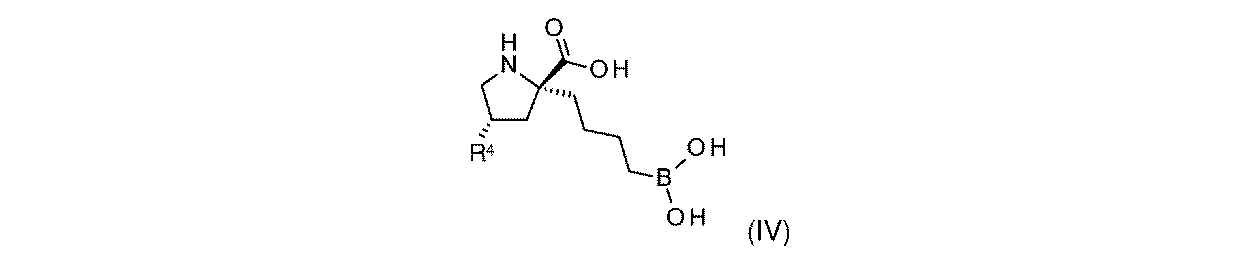
- 式(V)の化合物、又はその薬学的に許容される塩:
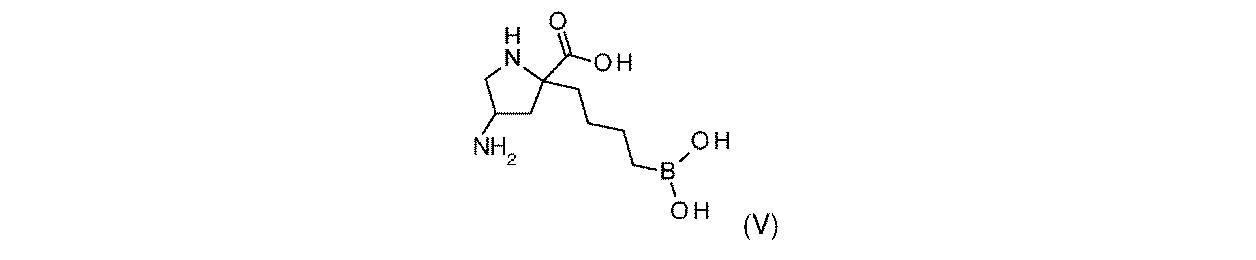
- 式(VI)の化合物、又はその薬学的に許容される塩であって:

R6aは、-H又は-CH3であり;
R6bは、-C(O)C(R6cR6d)NH2であるか;又は0若しくは1アミノ、若しくは-OR6eで置換されている-(C1~C3)アルキルであり;そして
R6cは、0若しくは1アミノ、又は-OR6fで置換されている-(C1~C3)アルキルであり;
R6dは、H又は-CH3であり;そして
R6e及びR6fは独立して、-H又は-CH3である、式(VI)の化合物、又はその薬学的に許容される塩。 - R 6a が、-H又は-CH3であり;そしてR6b が、0又は1アミノで置換されている-(C1~C3)アルキルであり;又は代わりに、
R6a が、-H又は-CH3であり;R6b が、-C(O)C(R6cR6d)NH2であり;R6c が、0若しくは1アミノ、又は-OHで置換されている-(C1~C3)アルキルであり;そしてR6d が、H又は-CH3である、
請求項9に記載の化合物、又はその薬学的に許容される塩。 - 式(Ib)の化合物、又はその薬学的に許容される塩であって:

R1は-NHR1aであり;
R1aは、-H又は-C(O)CH(R1b)NHR1cであり;そして
R1bは、-H、-(C1~C4)アルキル、及びCH2OR1dから選択され、そしてR1cは-Hであり;又は
R1b及びR1cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R1dは、H又は-CH3である、式(Ib)の化合物、又はその薬学的に許容される塩。 - 式(IIb)の化合物、又はその薬学的に許容される塩であって:

R2は、-OH又は-NHR2aであり;
R2aは、-H又は-C(O)CH(R2b)NHR2cであり;
R2bは、-H、-(C1~C4)アルキル、及びCH2OR2dから選択され、そしてR2cは-Hであり;又は
R2b及びR2cは、これらが結合する原子と合わせて、5員複素環を形成し;そして
R2dは、-H又は-CH3である、式(IIb)の化合物、又はその薬学的に許容される塩。 - 式(IIIb)の化合物、又はその薬学的に許容される塩であって:
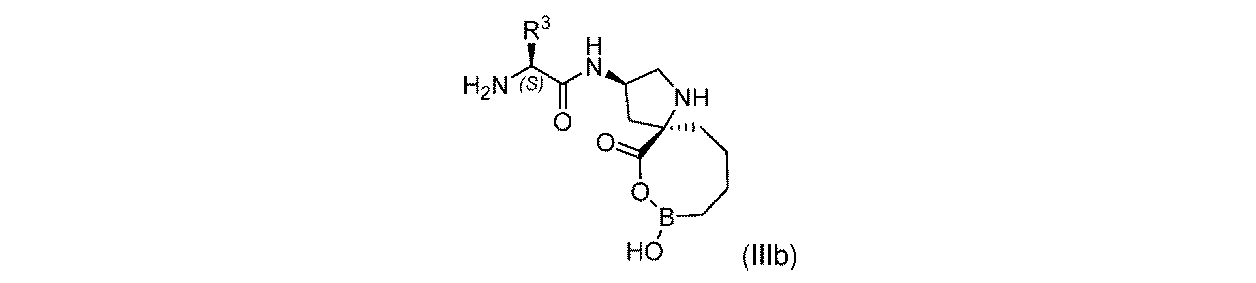
R3は、-H、-(C1~C4)アルキル、及び-CH2OR3aから選択され;そして
R3aは、-H又は-CH3である、式(IIIb)の化合物、又はその薬学的に許容される塩。 - 式(IVb)の化合物、又はその薬学的に許容される塩であって:

- 式(Vb)の化合物、又はその薬学的に許容される塩:

- 式(VIb)の化合物、又はその薬学的に許容される塩であって:
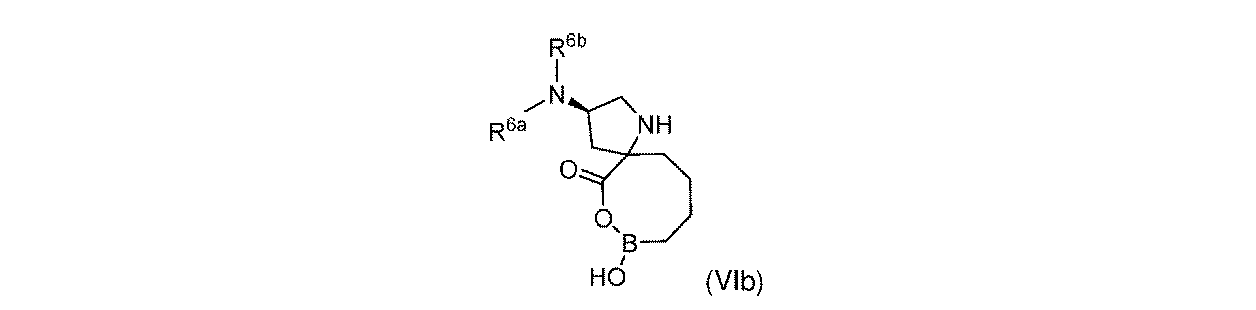
R6aは、-H又は-CH3であり;
R6bは、-C(O)C(R6cR6d)NH2であるか;又は0若しくは1アミノ、若しくは-OR6eで置換されている-(C1~C3)アルキルであり;そして
R6cは、0若しくは1アミノ、又は-OR6fで置換されている-(C1~C3)アルキルであり;
R6dは、H又は-CH3であり;そして
R6e及びR6fは独立して、-H又は-CH3である、式(VIb)の化合物、又はその薬学的に許容される塩。 - R 6a が、-H又は-CH3であり;そしてR6b が、0又は1アミノで置換されている-(C1~C3)アルキルであり;又は代わりに、
R6a が、-H又は-CH3であり;R6b が、-C(O)C(R6cR6d)NH2であり;R6c が、0若しくは1アミノ、又は-OHで置換されている-(C1~C3)アルキルであり;そしてR6d が、H又は-CH3である、
請求項16に記載の化合物、又はその薬学的に許容される塩。 - 下記:





- 化合物が、下記:

- 化合物が、下記:

- 請求項1~20のいずれか一項に記載の化合物、又はその薬学的に許容される塩、及び薬学的に許容されるキャリアを含む医薬組成物。
- 癌を処置するための、請求項1~20のいずれか一項に記載の化合物、又はその薬学的に許容される塩。
- 癌を処置するのに用いられる医薬の製造における、請求項1~20のいずれか一項に記載の化合物、又はその薬学的に許容される塩。
- 癌を処置するのに用いられる、請求項1~20のいずれか一項に記載の化合物、又はその薬学的に許容される塩を含む医薬組成物。
- 呼吸器炎症性疾患を処置するための、請求項1~20のいずれか一項に記載の化合物、又はその薬学的に許容される塩。
- 呼吸器炎症性疾患を処置するのに用いられる医薬の製造における、請求項1~20のいずれか一項に記載の化合物、又はその薬学的に許容される塩。
- 呼吸器炎症性疾患を処置するのに用いられる、請求項1~20のいずれか一項に記載の化合物、又はその薬学的に許容される塩を含む医薬組成物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023117406A JP7699171B2 (ja) | 2018-02-17 | 2023-07-19 | アルギナーゼインヒビタ及びその使用方法 |
Applications Claiming Priority (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862631659P | 2018-02-17 | 2018-02-17 | |
| US62/631,659 | 2018-02-17 | ||
| US201862671576P | 2018-05-15 | 2018-05-15 | |
| US62/671,576 | 2018-05-15 | ||
| US201862721113P | 2018-08-22 | 2018-08-22 | |
| US62/721,113 | 2018-08-22 | ||
| US201862778002P | 2018-12-11 | 2018-12-11 | |
| US62/778,002 | 2018-12-11 | ||
| PCT/IB2019/051236 WO2019159120A1 (en) | 2018-02-17 | 2019-02-15 | Arginase inhibitors and methods of use thereof |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023117406A Division JP7699171B2 (ja) | 2018-02-17 | 2023-07-19 | アルギナーゼインヒビタ及びその使用方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2021513964A JP2021513964A (ja) | 2021-06-03 |
| JP7317841B2 true JP7317841B2 (ja) | 2023-07-31 |
Family
ID=65763693
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020543307A Active JP7317841B2 (ja) | 2018-02-17 | 2019-02-15 | アルギナーゼインヒビタ及びその使用方法 |
| JP2023117406A Active JP7699171B2 (ja) | 2018-02-17 | 2023-07-19 | アルギナーゼインヒビタ及びその使用方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023117406A Active JP7699171B2 (ja) | 2018-02-17 | 2023-07-19 | アルギナーゼインヒビタ及びその使用方法 |
Country Status (34)
| Country | Link |
|---|---|
| US (4) | US11420984B2 (ja) |
| EP (2) | EP4678238A2 (ja) |
| JP (2) | JP7317841B2 (ja) |
| KR (1) | KR102657919B1 (ja) |
| CN (3) | CN117186134A (ja) |
| AU (2) | AU2019221896B2 (ja) |
| BR (1) | BR112020015973A2 (ja) |
| CA (1) | CA3091365A1 (ja) |
| CL (1) | CL2020002096A1 (ja) |
| CR (1) | CR20200418A (ja) |
| DK (1) | DK3752251T3 (ja) |
| DO (1) | DOP2020000145A (ja) |
| EC (1) | ECSP20058183A (ja) |
| ES (1) | ES3058584T3 (ja) |
| FI (1) | FI3752251T3 (ja) |
| HR (1) | HRP20251592T1 (ja) |
| IL (1) | IL276711B2 (ja) |
| JO (1) | JOP20200197A1 (ja) |
| LT (1) | LT3752251T (ja) |
| MA (1) | MA51837B1 (ja) |
| MX (1) | MX2020008570A (ja) |
| NI (1) | NI202000055A (ja) |
| PE (1) | PE20210176A1 (ja) |
| PL (1) | PL3752251T3 (ja) |
| PT (1) | PT3752251T (ja) |
| RS (1) | RS67559B1 (ja) |
| SG (1) | SG11202007739XA (ja) |
| SI (1) | SI3752251T1 (ja) |
| SM (1) | SMT202500499T1 (ja) |
| TW (1) | TWI803574B (ja) |
| UA (1) | UA129083C2 (ja) |
| UY (1) | UY38096A (ja) |
| WO (1) | WO2019159120A1 (ja) |
| ZA (2) | ZA202305542B (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2021535111A (ja) * | 2018-08-22 | 2021-12-16 | アストラゼネカ・アクチエボラーグAstrazeneca Aktiebolag | アルギナーゼインヒビタ及びその使用方法 |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117186134A (zh) * | 2018-02-17 | 2023-12-08 | 阿斯利康(瑞典)有限公司 | 精氨酸酶抑制剂及其使用方法 |
| TWI877770B (zh) | 2018-02-27 | 2025-03-21 | 美商英塞特公司 | 作為a2a / a2b抑制劑之咪唑并嘧啶及三唑并嘧啶 |
| WO2019186497A1 (en) | 2018-03-29 | 2019-10-03 | Oncoarendi Therapeutics S.A. | Dipeptide piperidine derivatives |
| EP3810610A1 (en) | 2018-05-18 | 2021-04-28 | Incyte Corporation | Fused pyrimidine derivatives as a2a / a2b inhibitors |
| WO2020010197A1 (en) | 2018-07-05 | 2020-01-09 | Incyte Corporation | Fused pyrazine derivatives as a2a / a2b inhibitors |
| TWI829857B (zh) | 2019-01-29 | 2024-01-21 | 美商英塞特公司 | 作為a2a / a2b抑制劑之吡唑并吡啶及三唑并吡啶 |
| PH12021551902A1 (en) * | 2019-02-08 | 2022-08-01 | Astrazeneca Ab | Arginase inhibitors and methods of use thereof |
| CN114599372A (zh) * | 2019-11-04 | 2022-06-07 | 阿斯利康(瑞典)有限公司 | 用于治疗癌症的组合疗法 |
| TW202228720A (zh) | 2020-12-22 | 2022-08-01 | 波蘭商昂科艾倫迪治療法股份公司 | 精胺酸酶抑制劑及其使用方法 |
| JP2025523364A (ja) * | 2022-06-08 | 2025-07-23 | デウン ファーマシューティカル カンパニー リミテッド | アルギナーゼ阻害剤およびこれを含む薬学的組成物 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013525364A (ja) | 2010-04-22 | 2013-06-20 | マーズ インコーポレイテッド | アルギナーゼ阻害剤およびそれらの治療用途 |
| JP2014506253A (ja) | 2010-12-31 | 2014-03-13 | コリドー ファーマシューティカルズ インコーポレイテッド | アルギナーゼ阻害剤およびその使用法 |
| JP2015516397A (ja) | 2012-04-18 | 2015-06-11 | マーズ インコーポレイテッド | アルギナーゼ阻害剤としての環拘束性類似体 |
| WO2016210106A1 (en) | 2015-06-23 | 2016-12-29 | Calithera Biosciences, Inc. | Compositions and methods for inhibiting arginase activity |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HRP20160305T1 (hr) | 2010-10-26 | 2016-07-01 | Mars, Incorporated | Borati kao inhibitori arginaze |
| PL410665A1 (pl) | 2014-12-29 | 2016-07-04 | Oncoarendi Therapeutics Spółka Z Ograniczoną Odpowiedzialnością | Inhibitory arginazy oraz ich zastosowania terapeutyczne |
| SG11201802961PA (en) | 2015-10-30 | 2018-05-30 | Calithera Biosciences Inc | Compositions and methods for inhibiting arginase activity |
| PL417066A1 (pl) | 2016-05-04 | 2017-11-06 | Oncoarendi Therapeutics Spółka Z Ograniczoną Odpowiedzialnością | Inhibitory arginazy oraz ich zastosowania terapeutyczne |
| JP2020502259A (ja) * | 2016-11-08 | 2020-01-23 | カリセラ バイオサイエンシズ,インコーポレイテッド | アルギナーゼ阻害剤併用療法 |
| CN114989205A (zh) | 2016-12-22 | 2022-09-02 | 卡里塞拉生物科学股份公司 | 用于抑制精氨酸酶活性的组合物和方法 |
| CN111491937A (zh) | 2017-12-22 | 2020-08-04 | 广东新契生物医药科技有限公司 | 作为精氨酸酶抑制剂的杂环化合物 |
| CN117186134A (zh) * | 2018-02-17 | 2023-12-08 | 阿斯利康(瑞典)有限公司 | 精氨酸酶抑制剂及其使用方法 |
| EP3765006A4 (en) | 2018-03-13 | 2022-02-23 | Merck Sharp & Dohme Corp. | ARGINASE INHIBITORS AND METHODS OF USE |
| CN111770756B (zh) | 2018-04-27 | 2023-03-31 | 四川科伦博泰生物医药股份有限公司 | 非天然氨基酸类衍生物、包含其的药物组合物及其制备方法和用途 |
| WO2019218904A1 (zh) | 2018-05-18 | 2019-11-21 | 四川科伦博泰生物医药股份有限公司 | 非天然氨基酸类衍生物、其制备方法及用途 |
| WO2019245890A1 (en) | 2018-06-20 | 2019-12-26 | Merck Sharp & Dohme Corp. | Arginase inhibitors and methods of use |
| PH12021551902A1 (en) * | 2019-02-08 | 2022-08-01 | Astrazeneca Ab | Arginase inhibitors and methods of use thereof |
-
2019
- 2019-02-15 CN CN202311155200.XA patent/CN117186134A/zh active Pending
- 2019-02-15 ES ES19710796T patent/ES3058584T3/es active Active
- 2019-02-15 SI SI201931003T patent/SI3752251T1/sl unknown
- 2019-02-15 BR BR112020015973-5A patent/BR112020015973A2/pt unknown
- 2019-02-15 UA UAA202005697A patent/UA129083C2/uk unknown
- 2019-02-15 AU AU2019221896A patent/AU2019221896B2/en active Active
- 2019-02-15 MX MX2020008570A patent/MX2020008570A/es unknown
- 2019-02-15 LT LTEPPCT/IB2019/051236T patent/LT3752251T/lt unknown
- 2019-02-15 CR CR20200418A patent/CR20200418A/es unknown
- 2019-02-15 SG SG11202007739XA patent/SG11202007739XA/en unknown
- 2019-02-15 EP EP25194463.3A patent/EP4678238A2/en active Pending
- 2019-02-15 UY UY0001038096A patent/UY38096A/es active IP Right Grant
- 2019-02-15 US US16/969,739 patent/US11420984B2/en active Active
- 2019-02-15 KR KR1020207026580A patent/KR102657919B1/ko active Active
- 2019-02-15 JP JP2020543307A patent/JP7317841B2/ja active Active
- 2019-02-15 HR HRP20251592TT patent/HRP20251592T1/hr unknown
- 2019-02-15 PT PT197107964T patent/PT3752251T/pt unknown
- 2019-02-15 JO JOP/2020/0197A patent/JOP20200197A1/ar unknown
- 2019-02-15 MA MA51837A patent/MA51837B1/fr unknown
- 2019-02-15 WO PCT/IB2019/051236 patent/WO2019159120A1/en not_active Ceased
- 2019-02-15 CN CN201980013045.3A patent/CN111712302B/zh active Active
- 2019-02-15 EP EP19710796.4A patent/EP3752251B1/en active Active
- 2019-02-15 DK DK19710796.4T patent/DK3752251T3/da active
- 2019-02-15 TW TW108105043A patent/TWI803574B/zh active
- 2019-02-15 RS RS20251277A patent/RS67559B1/sr unknown
- 2019-02-15 PE PE2020001241A patent/PE20210176A1/es unknown
- 2019-02-15 CN CN202311157009.9A patent/CN117180290A/zh active Pending
- 2019-02-15 FI FIEP19710796.4T patent/FI3752251T3/fi active
- 2019-02-15 PL PL19710796.4T patent/PL3752251T3/pl unknown
- 2019-02-15 CA CA3091365A patent/CA3091365A1/en active Pending
- 2019-02-15 SM SM20250499T patent/SMT202500499T1/it unknown
- 2019-02-15 IL IL276711A patent/IL276711B2/en unknown
-
2020
- 2020-07-22 DO DO2020000145A patent/DOP2020000145A/es unknown
- 2020-08-13 CL CL2020002096A patent/CL2020002096A1/es unknown
- 2020-08-14 NI NI202000055A patent/NI202000055A/es unknown
- 2020-09-16 EC ECSENADI202058183A patent/ECSP20058183A/es unknown
-
2022
- 2022-06-07 AU AU2022203938A patent/AU2022203938B2/en active Active
- 2022-07-15 US US17/812,736 patent/US12195485B2/en active Active
-
2023
- 2023-05-23 ZA ZA2023/05542A patent/ZA202305542B/en unknown
- 2023-07-10 US US18/349,336 patent/US11912727B2/en active Active
- 2023-07-19 JP JP2023117406A patent/JP7699171B2/ja active Active
-
2024
- 2024-10-21 ZA ZA2024/07941A patent/ZA202407941B/en unknown
- 2024-12-13 US US18/979,749 patent/US20250313578A1/en active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013525364A (ja) | 2010-04-22 | 2013-06-20 | マーズ インコーポレイテッド | アルギナーゼ阻害剤およびそれらの治療用途 |
| JP2014506253A (ja) | 2010-12-31 | 2014-03-13 | コリドー ファーマシューティカルズ インコーポレイテッド | アルギナーゼ阻害剤およびその使用法 |
| JP2015516397A (ja) | 2012-04-18 | 2015-06-11 | マーズ インコーポレイテッド | アルギナーゼ阻害剤としての環拘束性類似体 |
| WO2016210106A1 (en) | 2015-06-23 | 2016-12-29 | Calithera Biosciences, Inc. | Compositions and methods for inhibiting arginase activity |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2021535111A (ja) * | 2018-08-22 | 2021-12-16 | アストラゼネカ・アクチエボラーグAstrazeneca Aktiebolag | アルギナーゼインヒビタ及びその使用方法 |
| JP7441210B2 (ja) | 2018-08-22 | 2024-02-29 | アストラゼネカ・アクチエボラーグ | アルギナーゼインヒビタ及びその使用方法 |
| US11952392B2 (en) | 2018-08-22 | 2024-04-09 | Astrazeneca Ab | Arginase inhibitors and methods of use thereof |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7699171B2 (ja) | アルギナーゼインヒビタ及びその使用方法 | |
| AU2020218651B2 (en) | Arginase inhibitors and methods of use thereof | |
| AU2020319132B2 (en) | Arginase inhibitors and methods of use thereof | |
| HK40037636A (en) | Arginase inhibitors and methods of use thereof | |
| EA042598B1 (ru) | Ингибиторы аргиназы и способы их применения | |
| HK40037636B (zh) | 精氨酸酶抑制剂及其使用方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20220204 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20230119 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20230207 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20230427 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20230620 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20230719 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7317841 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |