JP5184806B2 - 透明熱可塑性フィルムの製造方法および透明熱可塑性フィルム - Google Patents
透明熱可塑性フィルムの製造方法および透明熱可塑性フィルム Download PDFInfo
- Publication number
- JP5184806B2 JP5184806B2 JP2007088851A JP2007088851A JP5184806B2 JP 5184806 B2 JP5184806 B2 JP 5184806B2 JP 2007088851 A JP2007088851 A JP 2007088851A JP 2007088851 A JP2007088851 A JP 2007088851A JP 5184806 B2 JP5184806 B2 JP 5184806B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- film
- stretching
- preferable
- transparent thermoplastic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B23/00—Layered products comprising a layer of cellulosic plastic substances, i.e. substances obtained by chemical modification of cellulose, e.g. cellulose ethers, cellulose esters, viscose
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/18—Manufacture of films or sheets
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2301/00—Characterised by the use of cellulose, modified cellulose or cellulose derivatives
- C08J2301/08—Cellulose derivatives
- C08J2301/10—Esters of organic acids
-
- G—PHYSICS
- G02—OPTICS
- G02B—OPTICAL ELEMENTS, SYSTEMS OR APPARATUS
- G02B5/00—Optical elements other than lenses
- G02B5/30—Polarising elements
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Shaping By String And By Release Of Stress In Plastics And The Like (AREA)
- Polarising Elements (AREA)
- Manufacture Of Macromolecular Shaped Articles (AREA)
Description
(1)横延伸ゾーン長(L2)と予熱ゾーン長(L1)の比(L2/L1)が0.5〜30の範囲で、1%〜200%横延伸することを含む、透明熱可塑性フィルムの製造方法。
(2)横延伸ゾーン内の拡幅角を5度〜45度とすることを含む、(1)に記載の透明熱可塑性フィルムの製造方法。
(3)横延伸温度をT1としたとき、横延伸後にT1−50℃〜T1−2℃で熱処理することを含む、(1)または(2)に記載の透明熱可塑性フィルムの製造方法。
(4)横延伸温度をT1としたとき、横延伸前にT1+2℃〜T1+50℃で熱処理することを含む、(1)〜(3)のいずれか1項に記載の透明熱可塑性フィルムの製造方法。
(5)縦/横比(L/W)が0.01を越え0.3未満の範囲、又は2を超え50以下で、1%〜100%に縦延伸することを含む、(1)〜(4)のいずれか1項に記載の透明熱可塑性フィルムの製造方法。
(6)縦延伸後および横延伸後の少なくとも一方において、前記透明熱可塑性フィルムのガラス転移温度をTgとしたとき、Tg−30℃〜Tg+30℃の温度範囲において、縦方向および横方向の緩和量の合計が1%〜20%となるように緩和することを含む、(1)〜(5)のいずれか1項に記載の透明熱可塑性フィルムの製造方法。
(7)タッチロールを用いて溶融製膜することを含む、(1)〜(6)のいずれか1項に記載の透明熱可塑性フィルムの製造方法。
(8)透明熱可塑性フィルムであって、該フィルムの一辺30cmの正方形の内部の熱収縮分布および弾性率分布がいずれも10%以下である透明熱可塑性フィルム。
(9)横延伸ゾーン長(L2)と予熱ゾーン長(L1)の比(L2/L1)が0.5〜30の範囲で、1%〜200%横延伸してなる、(8)に記載の透明可塑性フィルム。
(10)(8)または(9)に記載の透明熱可塑性フィルムであって、該フィルムの幅全域に亘り配向角が0°±5°以内、または90°±5°以内である、透明熱可塑性フィルム。
(11)面内のレターデーション(Re)が20nm〜400nm、厚み方向のレターデーション(Rth)が50nm〜400nmである、(8)〜(10)のいずれか1項に記載の透明熱可塑性フィルム。
(12)下記式を満足するセルロースアシレートを含む、(8)〜(11)のいずれか1項に記載の透明熱可塑性フィルム。
2.0≦A+B<3.0
0.1≦B<3
A:アセテート基の置換度、
B:プロピオネート基、ブチレート基、ペンタノイル基の置換度の総和
(13)下記式(T−1)および(T−2)を満たす組成を有するセルロースアシレートからなることを特徴とする(8)〜(12)のいずれか1項に記載の熱可塑性フィルム。
式(T−1):2.5≦A+C<3.0
式(T−2):0.1≦C<2
(式中、Aは、アセテート基の置換度を示し、Cは置換もしくは無置換の芳香族アシル基を示す。)
(14)飽和ノルボルネン樹脂を含む、(8)〜(13)のいずれか1項に記載の透明熱可塑性フィルム。
(15)残留溶剤が0.01質量%以下である、(8)〜(14)のいずれか1項に記載の透明熱可塑性フィルム。
このようなRe、Rth等の光学特性に影響を与えやすいのが、熱収縮率、弾性率であり、これらはそれぞれ、一辺30cmの正方形内の分布が10%以下であるのが好ましく、5%以下であるのがより好ましく、3%以下であるのがさらに好ましい。特に、熱収縮率および弾性率の両方について、該要件を満たすことが好ましい。
次に影響を与えやすいのがフィルム面内の配向角である。配向角の方向あるいはその直交方向に分子が並んでいるため、配向角に沿って、あるいは直交方向に最大弾性率が発現する。このため配向角が斜めの場合、熱収縮応力が斜めに発生し、これが面内の光学特性の不均一性を発現する。配向角は製膜幅全域に亘り0°±5°以内、または90°±5°以内であることが好ましく、より好ましくは0°±4°以内、または90°±4°以内であり、さらに好ましくは0°±3°以内、または90°±3°以内である。
さらに本発明では、Reが20nm〜400nmであることが好ましく、30nm〜250nmであることがより好ましく、40nm〜150nmであることがさらに好ましい。一方、Rthは、50nm〜400nmであることが好ましく、70nm〜350nmであることがより好ましく、100nm〜300nmであることがさらに好ましい。特に、本発明では、Reが20nm〜400nm、かつ、Rthが50nm〜400nmであることが好ましく、より好ましくはReが30nm〜250nm、かつ、Rthが70nm〜350nmであり、さらに好ましくはReが40nm〜150nm、かつ、Rthが100nm〜300nmである。
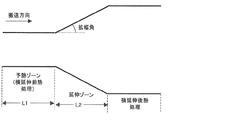
横延伸ゾーン長(L2)と予熱ゾーン長(L1)の比(L2/L1)が0.5〜30であることが好ましく、0.7〜20であることがより好ましく、1〜10であることがさらに好ましい(図1)。通常L2/L1は50以上で使用されることが多いが、本発明ではL2/L1を小さくしていることを特徴としている。即ちL1を長くし予熱時間を十分取ることでフィルム全面の温度むらを無くし均一な延伸を達成したものである。
横延伸ゾーンでの延伸倍率は1%〜200%が好ましく、10%〜150%がより好ましく、40%〜120%がさらに好ましい。
ここで、図1は、横延伸の予熱ゾーン長、延伸ゾーン長、拡幅角を示す図である。 また、拡幅角は図1に示すように拡幅(延伸)が開始する部分の角度を示す。さらに、後述するとおり、横延伸後に横延伸後熱処理ゾーンを設け熱処理することで、ボーイングを抑制することができる。
本発明における延伸倍率は以下式(1)で定義される。
式(1)
延伸倍率(%)=100×(延伸後の長さ−延伸前の長さ)/延伸前の長さ
このような延伸は、透明熱可塑性フィルムのガラス転移温度Tgとすると、Tg−10℃〜Tg+50℃で実施するのが好ましく、より好ましくはTg℃〜Tg+40℃であり、さらに好ましくはTg+5℃〜Tg+30℃である。
通常の延伸では拡幅角は60度以上の急激な角度で延伸するが、本発明では上記のような緩やかな角度で延伸することを特徴としている。これにより延伸むらが発生せず、均一な延伸を達成したことを特徴としている。
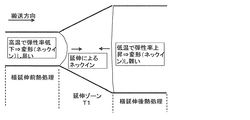
このように延伸前の温度を高くすることで、フィルムの弾性率を低下させ、延伸前(テンター入口側)に発生するネックインを発生し易くしている。これにより、延伸後にネックインを発生し難くさせる上記横延伸処理後の対策とバランスさせることでより配向角を小さくする効果を出すことができる。
ここで、図2は、横延伸前熱処理、横延伸後熱処理により配向角を低減する機構を示す図である。具体的には、延伸ゾーンでは幅方向に延伸されるため、直交方向(搬送方向)にネックインが起こり細くなろうとする。このため延伸ゾーンの両側に搬送方向に弓状に変形しようとする力が働く。入口側(横延伸前熱処理温度)を高温にし、出口側(横延伸後熱処理)温度を低くすると、出口側より入口側が柔らかくなり変形し易くなる。この結果、入口側のみ弓状に変形し出口側の変形は抑制され、横延伸後の弓状の変形(ボーイング)を小さくでき、端部の配向角の変化の小さな(より均一な)フィルムを得ることができる。
ここで、短スパン延伸法(斜め延伸法)はニップロール間を斜めにフィルムパスさせることにより、Lを短くでき縦横比の小さい縦延伸を実現し易くできる方法である。さらに縦延伸に引き続き縦緩和を行うことが好ましい。例えば、縦緩和は出口側ニップロールの搬送速度より、ニップロール後の搬送ロールの搬送速度を遅くすることで達成できる。
一方、通常の延伸法では、ニップロール間の隙間以上にLを小さくできないため、縦/横比を小さくし難い。
このような縦延伸の倍率は1%〜100%とすることが好ましく、より好ましくは2%〜50%、さらに好ましくは3%〜30%である。ここで言う延伸倍率は上述の式(1)で定義されるものである。
縦延伸温度はTg−20℃〜Tg+50℃で実施するのが好ましく、より好ましくはTg〜Tg+40℃、さらに好ましくはTg+5℃〜Tg+30℃である。
図3は、長スパン延伸を行う場合の、熱可塑性フィルムを溶融製膜で製造する場合のフィルム製造装置10の構成概略図である。
フィルム製造装置10は、液晶表示装置等に使用できる熱可塑性フィルムFを製造する装置である。熱可塑性フィルムFの原材料であるペレット状のセルロースアシレート樹脂またはシクロオレフィン樹脂を乾燥機12に導入して乾燥させた後、このペレットを押出機14によって押し出し、ギアポンプ16によりフィルタ18に供給する。次いで、フィルタ18により異物が濾過され、ダイ20から押し出される。その後、キャストドラム28とタッチロール24で挟まれ、キャストドラム28とロール26の間を通過して固化し、所定の表面粗さの未延伸フィルムFaが形成される。そして、この未延伸フィルムFaが長スパン延伸を行う縦延伸部30に供給される。
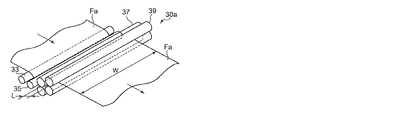
縦延伸部30では、未延伸フィルムFaが入口側ニップロール32及び出口側ニップローラ34間で搬送方向に延伸され、縦延伸フィルムFbとされる。なお、図4は、縦延伸部30の斜視説明図であり、縦延伸の縦/横比(L/W)は、入口側ニップロール32及び出口側ニップローラ34間の距離Lと、入口側ニップロール32及び出口側ニップローラ34の長さ方向の幅Wとによって規定される。次いで、縦延伸フィルムFbは、予熱部36を通過することで所定の予熱温度に調整された後、横延伸部42に供給される。
横延伸部42では、縦延伸フィルムFbが搬送方向と直交する幅方向に延伸され、横延伸フィルムFcとされる。そして、横延伸フィルムFcは、熱固定部44に供給され、巻取部46によって巻き取られることで、配向角、レターデーションが調整された最終製品である熱可塑性フィルムFが製造される。なお、横延伸フィルムFcには熱固定部44を通過した後、さらに熱緩和処理を施してもよい。
このフィルム製造装置10aでは、未延伸フィルムFaが予熱ロール33、35によって所定の温度まで予熱された後、二組のニップロール37、39間に供給されて縦延伸が行われる。この場合、ニップロール37、39は、未延伸フィルムFaの搬送方向に近接して配置されるとともに、上下方向に所定距離だけ高さが異なるように配置されている。ニップロール37、39をこのように配置することにより、縦延伸部30aにおける未延伸フィルムFaの搬送距離を確保できるとともに、縦延伸部30aの前後に配置される機構間の距離を短縮して、フィルム製造装置10aの小型化を図ることができる。
なお、図6は、縦延伸部30aの斜視説明図であり、縦延伸の縦/横比(L/W)は、ニップロール37、39によってニップされる未延伸フィルムFaの搬送方向の距離Lと、ニップロール37、39の長さ方向の幅Wとによって規定される。
延伸に伴いフィルムは伸張されるが、この時フィルムは体積変化を小さくしようと厚み、および幅を減少させる。このときニップロールとフィルム間の摩擦により幅方向の収縮が制限される。このためニップロール間隔を大きくすると幅方向に収縮しやすくなり厚み減少を抑制できる。厚み減少が大きいとフィルムが厚み方向に圧縮されたことと同じ効果があり、フィルム面内に分子配向が進みRthが大きくなり易い。縦横比が大きく厚み減少が少ないと、逆にRthは発現し難く、低いRthを実現できる。
さらに縦横比が長いと幅方向の均一性を向上することができる。これは以下の理由による。
・縦延伸に伴いフィルムは幅方向に収縮しようとする。幅方向中央部では、その両側も幅方向に収縮しようとするため、綱引き状態となり自由に収縮できない。
・フィルム幅方向の端部は片側としか綱引き状態とならず、比較的自由に収縮できる。
・この両端と中央部の延伸に伴う収縮挙動の差が幅方向の延伸ムラとなる。
このような両端と中央部の不均一性により、幅方向のレターデーションむら、軸ズレ(遅相軸の配向角分布)が発生する。これに対し、長スパン延伸は長い2本のニップロール間でゆっくり延伸されるため、延伸中にこれらの不均一性の均一化(分子配向が均一になる)が進行する。これに対し、通常の縦延伸(縦横比=0.7〜2)では、このような均一化は発生しない。
このような縦横比が2を越え50以下の延伸は、所定の距離だけ離れて設けられた2対のニップロールの間でフィルムを加熱して延伸すればよく、加熱方法はヒーター加熱法(赤外線ヒーター、ハロゲンヒーター、パネルヒーター等をフィルム上や下に設置し輻射熱で加熱)でもよく、ゾーン加熱法(熱風等を吹き込み所定の温度に調温したゾーン内で加熱)でもよい。本発明では延伸温度の均一性の観点からゾーン加熱法が好ましい。この時、ニップロールは延伸ゾーン内に設置しても良く、ゾーンの外に出しても良いが、フィルムとニップロールの粘着を防止するためにはゾーンの外に出すのが好ましい。このような延伸の前にフィルムを予熱することも好ましく、予熱温度は(Tg−80)℃〜(Tg+100)℃である。
縦方向の緩和(縦緩和)は、例えば、低張力(好ましくは20Kg/m以下、より好ましくは10kg/m以下)で搬送しながら上記温度で熱処理することで達成させることができる。緩和量は、フィルムの長手方向に一定間隔でつけた印が緩和後にどれだけ縮んだかで計測できる。縦緩和は縦延伸と横延伸の間、縦延伸および横延伸終了後等いずれの段階で実施しても良い。
横方向の緩和(横緩和)は延伸後にテンター幅を1%〜20%、より好ましくは2%〜15%狭めることで達成される。横緩和の温度は上記の範囲が好ましく、このような横緩和は、延伸後熱処理と併せて実施しても良く、延伸後熱処理の前で実施しても良く、延伸後熱処理の後で実施しても良い。
これらの縦方向の緩和(縦緩和)、横方向の緩和(横緩和)はどちらか一方だけでも効果があり、両方実施することも好ましい。
また、本発明で用いる熱可塑性フィルムの数平均重合度は15〜3000であり、より好ましくは30〜600である。熱可塑性樹脂がセルロースアシレート樹脂の場合の数平均重合度は、110〜270がより好ましく、より好ましくは120〜260であり、さらに好ましくは140〜250である。この範囲にすることで、延伸むらを抑制でき、熱収縮分布、弾性率分布を小さくすることができる。
本発明で用いる透明熱可塑性フィルムとしては、セルロースアシレート樹脂、ポリカーボネート樹脂、および、飽和ノルボルネン樹脂のいずれかを含む透明熱可塑性フィルムが好ましい。中でも好ましいのがセルロースアシレート樹脂および飽和ノルボルネン樹脂のいずれかを含む透明熱可塑性フィルムである。
本発明で用いるセルロースアシレートは以下の特徴を有するものが好ましい。(Aはアセテート基の置換度、Bはプロピオネート基、ブチレート基、ペンタノイル基の置換度の総和を示す)。
2.0≦A+B<3.0
0.1≦B<3
より好ましくは、Bの1/2以上がプロピオネート基の場合
2.6≦A+B≦2.95
2.0≦B≦2.95
Bの1/2未満がプロピオネート基の場合
2.6≦A+B≦2.95
1.3≦B≦2.5
さらに好ましくは、Bの1/2以上がプロピオネート基の場合
2.7≦A+B≦2.95
2.4≦B≦2.9
Bの1/2未満がプロピオネート基の場合
2.7≦A+B≦2.95
1.3≦B≦2.0
本発明では、アシル基の中に占めるアセテート基の置換度を少なくし、プロピオネート基、ブチレート基、ペンタノイル基の置換度の総和を多くしていることが特徴である。これにより、より延伸しやすくし、延伸に伴う不均一性(むら)をより軽減できる。これはアセテート基より長いこれらの基を多くすることでフィルムの柔軟性を向上させ延伸性を高めることができるためである。しかし、アシル基を上記のものより長くすると、ガラス転移温度や弾性率を低下させすぎる傾向にある。このためアセチル基より大きなプロピオネート基、ブチレート基、ペンタノイル基が好ましく、より好ましくはプロピオネート基、ブチレート基であり、さらに好ましくはプロピオネート基である。
また、本発明で用いるセルロースアシレートのTgとしては、100〜180℃が好ましく、120〜160℃がより好ましい。
セルロース原料としては、広葉樹パルプ、針葉樹パルプ、綿花リンター由来のものが好ましく用いられる。セルロース原料としては、α−セルロース含量が92質量%〜99.9質量%の高純度のものを用いることが好ましい。
セルロース原料がフィルム状や塊状である場合は、あらかじめ解砕しておくことが好ましく、セルロースの形態はフラッフ状になるまで解砕が進行していることが好ましい。
セルロース原料はアシル化に先立って、活性化剤と接触させる処理(活性化)を行うことが好ましい。活性化剤としては、カルボン酸または水を用いることができるが、水を用いた場合には、活性化の後に酸無水物を過剰に添加して脱水を行ったり、水を置換するためにカルボン酸で洗浄したり、アシル化の条件を調節したりするといった工程を含むことが好ましい。活性化剤はいかなる温度に調節して添加してもよく、添加方法としては噴霧、滴下、浸漬などの方法から選択することができる。
本発明におけるセルロースアシレートを製造する方法においては、セルロースにカルボン酸の酸無水物を加え、ブレンステッド酸またはルイス酸を触媒として反応させることで、セルロースの水酸基をアシル化することが好ましい。
セルロース混合アシレートを得る方法としては、アシル化剤として2種のカルボン酸無水物を混合または逐次添加により反応させる方法、2種のカルボン酸の混合酸無水物(例えば、酢酸・プロピオン酸混合酸無水物)を用いる方法、カルボン酸と別のカルボン酸の酸無水物(例えば、酢酸・プロピオン酸無水物)を原料として反応系内で混合酸無水物(例えば、酢酸・プロピオン酸混合酸無水物)を合成してセルロースと反応させる方法、置換度が3に満たないセルロースアシレートを一旦合成し、酸無水物や酸ハライドを用いて、残存する水酸基をさらにアシル化する方法などを用いることができる。
カルボン酸の酸無水物として、好ましくはカルボン酸としての炭素数が2〜7であり、例えば、無水酢酸、プロピオン酸無水物、酪酸無水物、2−メチルプロピオン酸無水物、吉草酸無水物、3−メチル酪酸無水物、2−メチル酪酸無水物、2,2−ジメチルプロピオン酸無水物(ピバル酸無水物)、ヘキサン酸無水物、2−メチル吉草酸無水物、3−メチル吉草酸無水物、4−メチル吉草酸無水物、2,2−ジメチル酪酸無水物、2,3−ジメチル酪酸無水物、3,3−ジメチル酪酸無水物、シクロペンタンカルボン酸無水物、ヘプタン酸無水物、シクロヘキサンカルボン酸無水物、安息香酸無水物などを挙げることができる。
より好ましくは、無水酢酸、プロピオン酸無水物、酪酸無水物、吉草酸無水物、ヘキサン酸無水物、ヘプタン酸無水物などの無水物であり、より好ましくは、無水酢酸、プロピオン酸無水物、酪酸無水物である。
混合エステルを調製する目的で、これらの酸無水物を併用して使用することが好ましく行われる。その混合比は目的とする混合エステルの置換比に応じて決定することが好ましい。酸無水物は、セルロースに対して、通常は過剰当量添加する。すなわち、セルロースの水酸基に対して1.2〜50当量添加することが好ましく、1.5〜30当量添加することがより好ましく、2〜10当量添加することが特に好ましい。
本発明におけるセルロースアシレートの製造に用いるアシル化の触媒には、ブレンステッド酸またはルイス酸を使用することが好ましい。ブレンステッド酸およびルイス酸の定義については、例えば、「理化学辞典」第五版(2000年)に記載されている。好ましいブレンステッド酸の例としては、硫酸、過塩素酸、リン酸、メタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸などを挙げることができる。好ましいルイス酸の例としては、塩化亜鉛、塩化スズ、塩化アンチモン、塩化マグネシウムなどを挙げることができる。
触媒としては、硫酸または過塩素酸がより好ましく、硫酸が特に好ましい。触媒の好ましい添加量は、セルロースに対して0.1〜30質量%であり、より好ましくは1〜15質量%であり、特に好ましくは3〜12質量%である。
アシル化を行う際には、粘度、反応速度、攪拌性、アシル置換比などを調整する目的で、溶媒を添加してもよい。このような溶媒としては、ジクロロメタン、クロロホルム、カルボン酸、アセトン、エチルメチルケトン、トルエン、ジメチルスルホキシド、スルホランなどを用いることもできるが、好ましくはカルボン酸であり、例えば、炭素数2〜7のカルボン酸(例えば、酢酸、プロピオン酸、酪酸、2−メチルプロピオン酸、吉草酸、3−メチル酪酸、2−メチル酪酸、2,2−ジメチルプロピオン酸(ピバル酸)、ヘキサン酸、2−メチル吉草酸、3−メチル吉草酸、4−メチル吉草酸、2,2−ジメチル酪酸、2,3−ジメチル酪酸、3,3−ジメチル酪酸、シクロペンタンカルボン酸)などを挙げることができる。さらに好ましくは、酢酸、プロピオン酸、酪酸などを挙げることができる。これらの溶媒は混合して用いてもよい。
アシル化を行う際には、酸無水物と触媒、さらに、必要に応じて溶媒を混合してからセルロースと混合してもよく、またこれらを別々に逐次セルロースと混合してもよいが、通常は、酸無水物と触媒との混合物、または、酸無水物と触媒と溶媒との混合物をアシル化剤として調整してからセルロースと反応させることが好ましい。アシル化の際の反応熱による反応容器内の温度上昇を抑制するために、アシル化剤は予め冷却しておくことが好ましい。冷却温度としては、−50℃〜20℃が好ましく、−35℃〜10℃がより好ましく、−25℃〜5℃がさらに好ましい。アシル化剤は液状で添加しても、凍結させて結晶、フレーク、またはブロック状の固体として添加してもよい。
アシル化剤はさらに、セルロースに対して一度に添加しても、分割して添加してもよい。また、アシル化剤に対してセルロースを一度に添加しても、分割して添加してもよい。アシル化剤を分割して添加する場合は、同一組成のアシル化剤を用いても、複数の組成の異なるアシル化剤を用いても良い。好ましい例として、1)酸無水物と溶媒の混合物をまず添加し、次いで、触媒を添加する、2)酸無水物、溶媒と触媒の一部の混合物をまず添加し、次いで、触媒の残りと溶媒の混合物を添加する、3)酸無水物と溶媒の混合物をまず添加し、次いで、触媒と溶媒の混合物を添加する、4)溶媒をまず添加し、酸無水物と触媒との混合物あるいは酸無水物と触媒と溶媒との混合物を添加する、などを挙げることができる。
反応の最低温度は−50℃以上が好ましく、−30℃以上がより好ましく、−20℃以上がさらに好ましい。好ましいアシル化時間は0.5時間〜24時間であり、1時間〜12時間がより好ましく、1.5時間〜6時間が特に好ましい。0.5時間以下では通常の反応条件では反応が十分に進行せず、24時間を越えると、工業的な製造のために好ましくない。
本発明に用いられるセルロースアシレートを製造する方法においては、アシル化反応の後に、反応停止剤を加えることが好ましい。
反応停止剤としては、酸無水物を分解するものであればいかなるものでもよく、好ましい例として、水、アルコール(例えば、エタノール、メタノール、プロパノール、イソプロピルアルコールなど)またはこれらを含有する組成物などを挙げることができる。また、反応停止剤には、後述の中和剤を含んでいても良い。反応停止剤の添加に際しては、反応装置の冷却能力を超える大きな発熱が生じて、セルロースアシレートの重合度を低下させる原因となったり、セルロースアシレートが望まない形態で沈殿したりする場合があるなどの不都合を避けるため、水やアルコールを直接添加するよりも、酢酸、プロピオン酸、酪酸等のカルボン酸と水との混合物を添加することが好ましく、カルボン酸としては酢酸が特に好ましい。カルボン酸と水の組成比は任意の割合で用いることができるが、水の含有量が5質量%〜80質量%、さらには10質量%〜60質量%、特には15質量%〜50質量%の範囲であることが好ましい。
反応停止剤は、アシル化の反応容器に添加しても、反応停止剤の容器に反応物を添加してもよい。反応停止剤は3分〜3時間かけて添加することが好ましい。反応停止剤の添加時間が3分以上であれば、発熱が大きくなりすぎて重合度低下の原因となったり、酸無水物の加水分解が不十分になったり、セルロースアシレートの安定性を低下させたりするなどの不都合が生じないので好ましい。また反応停止剤の添加時間が3時間以下であれば、工業的な生産性の低下などの問題も生じないので好ましい。反応停止剤の添加時間として、好ましくは4分〜2時間であり、より好ましくは5分〜1時間であり、特に好ましくは10分〜45分である。反応停止剤を添加する際には反応容器を冷却しても冷却しなくてもよいが、解重合を抑制する目的から、反応容器を冷却して温度上昇を抑制することが好ましい。また、反応停止剤を冷却しておくことも好ましい。
アシル化の反応停止工程あるいはアシル化の反応停止工程後に、系内に残存している過剰の無水カルボン酸の加水分解、カルボン酸およびエステル化触媒の一部または全部の中和のために、中和剤(例えば、カルシウム、マグネシウム、鉄、アルミニウムまたは亜鉛の炭酸塩、酢酸塩、水酸化物または酸化物)またはその溶液を添加してもよい。中和剤の溶媒としては、水、アルコール(例えば、エタノール、メタノール、プロパノール、イソプロピルアルコールなど)、カルボン酸(例えば、酢酸、プロピオン酸、酪酸など)、ケトン(例えば、アセトン、エチルメチルケトンなど)、ジメチルスルホキシドなどの極性溶媒、およびこれらの混合溶媒を好ましい例として挙げることができる。
このようにして得られたセルロースアシレートは、全置換度がほぼ3に近いものであるが、所望の置換度のものを得る目的で、少量の触媒(一般には、残存する硫酸などのアシル化触媒)と水との存在下で、20〜90℃に数分〜数日間保つことによりエステル結合を部分的に加水分解し、セルロースアシレートのアシル置換度を所望の程度まで減少させること(いわゆる熟成)が一般的に行われる。部分加水分解の過程でセルロースの硫酸エステルも加水分解されることから、加水分解の条件を調節することにより、セルロースに結合した硫酸エステルの量を削減することができる。
所望のセルロースアシレートが得られた時点で、系内に残存している触媒を、前記のような中和剤またはその溶液を用いて完全に中和し、部分加水分解を停止させることが好ましい。反応溶液に対して溶解性が低い塩を生成する中和剤(例えば、炭酸マグネシウム、酢酸マグネシウムなど)を添加することにより、溶液中あるいはセルロースに結合した触媒(例えば、硫酸エステル)を効果的に除去することも好ましい。
セルロースアシレート中の未反応物、難溶解性塩、その他の異物などを除去または削減する目的として、反応混合物(ドープ)のろ過を行うことが好ましい。ろ過は、アシル化の完了から再沈殿までの間のいかなる工程において行ってもよい。ろ過圧や取り扱い性の制御の目的から、ろ過に先立って適切な溶媒で希釈することも好ましい。
このようにして得られたセルロースアシレート溶液を、水もしくはカルボン酸(例えば、酢酸、プロピオン酸など)水溶液のような貧溶媒中に混合するか、セルロースアシレート溶液中に、貧溶媒を混合することにより、セルロースアシレートを再沈殿させ、洗浄および安定化処理により目的のセルロースアシレートを得ることができる。再沈殿は連続的に行っても、一定量ずつバッチ式で行ってもよい。セルロースアシレート溶液の濃度および貧溶媒の組成をセルロースアシレートの置換様式あるいは重合度により調整することで、再沈殿したセルロースアシレートの形態や分子量分布を制御することも好ましい。
生成したセルロースアシレートは洗浄処理することが好ましい。洗浄溶媒はセルロースアシレートの溶解性が低く、かつ、不純物を除去することができるものであればいかなるものでも良いが、通常は水または温水が用いられる。洗浄水の温度は、好ましくは25℃〜100℃であり、さらに好ましくは30℃〜90℃であり、特に好ましくは40℃〜80℃である。洗浄処理はろ過と洗浄液の交換を繰り返すいわゆるバッチ式で行っても、連続洗浄装置を用いて行ってもよい。再沈殿および洗浄の工程で発生した廃液を再沈殿工程の貧溶媒として再利用したり、蒸留などの手段によりカルボン酸などの溶媒を回収して再利用することも好ましい。
洗浄の進行はいかなる手段で追跡を行ってよいが、水素イオン濃度、イオンクロマトグラフィー、電気伝導度、ICP、元素分析、原子吸光スペクトルなどの方法を好ましい例として挙げることができる。
このような処理により、セルロースアシレート中の触媒(硫酸、過塩素酸、トリフルオロ酢酸、p−トルエンスルホン酸、メタンスルホン酸、塩化亜鉛など)、中和剤(例えば、カルシウム、マグネシウム、鉄、アルミニウムまたは亜鉛の炭酸塩、酢酸塩、水酸化物または酸化物など)、中和剤と触媒との反応物、カルボン酸(酢酸、プロピオン酸、酪酸など)、中和剤とカルボン酸との反応物などを除去することができ、このことはセルロースアシレートの安定性を高めるために有効である。
温水処理による洗浄後のセルロースアシレートは、安定性をさらに向上させたり、カルボン酸臭を低下させるために、弱アルカリ(例えば、ナトリウム、カリウム、カルシウム、マグネシウム、アルミニウムなどの炭酸塩、炭酸水素塩、水酸化物、酸化物など)の水溶液などで処理することも好ましい。
残存不純物の量は、洗浄液の量、洗浄の温度、時間、攪拌方法、洗浄容器の形態、安定化剤の組成や濃度により制御できる。本発明においては、残留硫酸根量(硫黄原子の含有量として)が0〜500ppmになるようにアシル化、部分加水分解および洗浄の条件を設定する。
本発明においてセルロースアシレートの含水率を好ましい量に調整するためには、セルロースアシレートを乾燥することが好ましい。乾燥の方法については、目的とする含水率が得られるのであれば特に限定されないが、加熱、送風、減圧、攪拌などの手段を単独または組み合わせで用いることで効率的に行うことが好ましい。乾燥温度として好ましくは0〜200℃であり、さらに好ましくは40〜180℃であり、特に好ましくは50〜160℃である。本発明のセルロースアシレートは、その含水率が2質量%以下であることが好ましく、1質量%以下であることがさらに好ましく、0.7質量%以下であることが特に好ましい。
本発明のセルロースアシレートは粒子状、粉末状、繊維状、塊状など種々の形状を取ることができるが、フィルム製造の原料としては粒子状または粉末状であることが好ましいことから、乾燥後のセルロースアシレートは、粒子サイズの均一化や取り扱い性の改善のために、粉砕や篩がけを行っても良い。セルロースアシレートが粒子状であるとき、使用する粒子の90質量%以上は、0.5〜5mmの粒子サイズを有することが好ましい。また、使用する粒子の50質量%以上が1〜4mmの粒子サイズを有することが好ましい。セルロースアシレート粒子は、なるべく球形に近い形状を有することが好ましい。また、本発明のセルロースアシレート粒子は、見かけ密度が好ましくは0.5〜1.3、さらに好ましくは0.7〜1.2、特に好ましくは0.8〜1.15である。見かけ密度の測定法に関しては、JIS K−7365に規定されている。
本発明のセルロースアシレート粒子は安息角が10〜70度であることが好ましく、15〜60度であることがさらに好ましく、20〜50度であることが特に好ましい。
本発明で好ましく用いられるセルロースアシレートの数平均重合度は110〜270であり、好ましくは120〜260であり、さらに好ましくは140〜250である。数平均重合度は、本発明では後述のゲル浸透クロマトグラフィー (GPC)を用いた方法で測定される。
本発明においては、セルロースアシレートのGPCによる重量平均重合度/数平均重合度が1.6〜3.6であることが好ましく、1.7〜3.3であることがさらに好ましく、1.8〜3.2であることがさらに好ましい。
これらのセルロースアシレートは1種類のみを用いてもよく、2種以上混合しても良い。また、セルロースアシレート以外の高分子成分を適宜混合したものでもよい。混合される高分子成分はセルロースエステルと相溶性に優れるものが好ましく、フィルムにしたときの透過率が80%以上、さらに好ましくは90%以上、さらに好ましくは92%以上である。
本発明では、下記式(T−1)および(T−2)を満たす組成を有する芳香族アシル化セルロースアシレートを用いることも好ましい。
式(T−1):2.5≦A+C<3.0
式(T−2):0.1≦C<2
より好ましくは、
式(T−3):2.6≦A+C<2.95
式(T−4):0.1≦C<1.5
さらに好ましくは、
式(T−3):2.7≦A+C<2.95
式(T−4):0.1≦C<1.0
である。
尚、式中Aは、アセチル基の置換度を示し、Cは置換もしくは無置換の芳香族アシル基を示す。 ここで置換もしくは無置換の芳香族アシル基としては下記一般式(I)で表される基があげられる。

上記アルキル基は、環状構造または分岐構造を有していてもよい。アルキル基の炭素原子数は、1〜20であることが好ましく、1〜12であることがより好ましく、1〜6であることがさらに好ましく、1〜4であることが最も好ましい。アルキル基が置換基を有する場合は、該置換基の炭素原子数も含めた数が、前記炭素原子数であることが好ましい(以下、他の基についても同じ)。アルキル基の例には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert−ブチル基、ヘキシル基、シクロヘキシル基、オクチル基および2−エチルヘキシル基が含まれる。
上記アルコキシ基は、環状構造または分岐を有していてもよい。アルコキシ基の炭素原子数は、1〜20であることが好ましく、1〜12であることがより好ましく、1〜6であることがさらに好ましく、1〜4であることが最も好ましい。アルコキシ基は、さらに別のアルコキシ基で置換されていてもよい。アルコキシ基の例には、メトキシ基、エトキシ基、2−メトキシエトキシ基、2−メトキシ−2−エトキシエトキシ基、ブチルオキシ基、ヘキシルオキシ基およびオクチルオキシ基が含まれる。
上記アリールオキシ基の例には、フェノキシ基およびナフトキシ基が含まれる。上記アシル基の炭素原子数は、1〜20であることが好ましく、1〜12であることがさらに好ましい。
上記アシル基の例には、ホルミル基、アセチル基およびベンゾイル基が含まれる。
上記カルボンアミド基の炭素原子数は、1〜20であることが好ましく、1〜12であることがより好ましい。カルボンアミド基の例には、アセトアミド基およびベンズアミド基が含まれる。上記スルホンアミド基の炭素原子数は、1〜20であることが好ましく、1〜12であることがさらに好ましい。
上記スルホンアミド基の例には、メタンスルホンアミド基、ベンゼンスルホンアミド基およびp−トルエンスルホンアミド基が含まれる。
上記ウレイド基の炭素原子数は、1〜20であることが好ましく、1〜12であることがさらに好ましい。ウレイド基の例には、(無置換)ウレイド基が含まれる。
上記アルコキシカルボニル基の例には、メトキシカルボニル基が含まれる。上記アリールオキシカルボニル基の炭素原子数は、7〜20であることが好ましく、7〜12であることがより好ましい。アリールオキシカルボニル基の例には、フェノキシカルボニル基が含まれる。上記アラルキルオキシカルボニル基の炭素原子数は、8〜20であることが好ましく、8〜12であることがより好ましい。アラルキルオキシカルボニル基の例には、ベンジルオキシカルボニル基が含まれる。上記カルバモイル基の炭素原子数は、1〜20であることが好ましく、1〜12であることがより好ましい。カルバモイル基の例には、(無置換)カルバモイル基およびN−メチルカルバモイル基が含まれる。上記スルファモイル基の炭素原子数は、20以下であることが好ましく、12以下であることがより好ましい。スルファモイル基の例には、(無置換)スルファモイル基およびN−メチルスルファモイル基が含まれる。
上記アシルオキシ基の炭素原子数は、1〜20であることが好ましく、2〜12であることがさらに好ましい。アシルオキシ基の例には、アセトキシ基およびベンゾイルオキシ基が含まれる。
上記アルキニル基の例には、チエニル基が含まれる。上記アルキルスルホニル基の炭素原子数は、1〜20であることが好ましく、1〜12であることがさらに好ましい。上記アリールスルホニル基の炭素原子数は、6〜20であることが好ましく、6〜12であることがさらに好ましい。
上記アルキルオキシスルホニル基の炭素原子数は、1〜20であることが好ましく、1〜12であることがより好ましい。
上記アリールオキシスルホニル基の炭素原子数は、6〜20であることが好ましく、6〜12であることがより好ましい。
上記アルキルスルホニルオキシ基の炭素原子数は、1〜20であることが好ましく、1〜12であることがより好ましい。上記アリールオキシスルホニル基の炭素原子数は、6〜20であることが好ましく、6〜12であることがより好ましい。
(可塑剤)
さらに本発明ではセルロースアシレートに可塑剤を添加することで、延伸歪を軽減しやすく好ましい。可塑剤としては、例えば、アルキルフタリルアルキルグリコレート類、リン酸エステルやカルボン酸エステル等が挙げられる。
アルキルフタリルアルキルグリコレート類として例えばメチルフタリルメチルグリコレート、エチルフタリルエチルグリコレート、プロピルフタリルプロピルグリコレート、ブチルフタリルブチルグリコレート、オクチルフタリルオクチルグリコレート、メチルフタリルエチルグリコレート、エチルフタリルメチルグリコレート、エチルフタリルプロピルグリコレート、メチルフタリルブチルグリコレート、エチルフタリルブチルグリコレート、ブチルフタリルメチルグリコレート、ブチルフタリルエチルグリコレート、プロピルフタリルブチルグリコレート、ブチルフタリルプロピルグリコレート、メチルフタリルオクチルグリコレート、エチルフタリルオクチルグリコレート、オクチルフタリルメチルグリコレート、オクチルフタリルエチルグリコレート等が挙げられる。
リン酸エステルとしては、例えばトリフェニルホスフェート、トリクレジルホスフェート、フェニルジフェニルホスフェート等を挙げることができる。さらに特表平6−501040号公報の請求項3〜7に記載のリン酸エステル系可塑剤を用いることが好ましい。
カルボン酸エステルとしては、例えばジメチルフタレート、ジエチルフタレート、ジブチルフタレート、ジオクチルフタレートおよびジエチルヘキシルフタレート等のフタル酸エステル類、およびクエン酸アセチルトリメチル、クエン酸アセチルトリエチル、クエン酸アセチルトリブチル等のクエン酸エステル類、ジメチルアジペート、ジブチルアジペート、ジイソブチルアジペート、ビス(2−エチルヘキシル)アジペート、ジイソデシルアジペート、ビス(ブチルジグリコールアジペート)等のアジピン酸エステルを挙げることができる。またその他、オレイン酸ブチル、リシノール酸メチルアセチル、セバシン酸ジブチル、トリアセチン等を単独あるいは併用するのが好ましい。
これらの可塑剤はセルロースアシレートフィルムに対し0質量%〜20質量%が好ましく、より好ましくは1質量%〜20質量%、さらに好ましくは2質量%〜15質量%である。これらの可塑剤は必要に応じて、2種類以上を併用して用いてもよい。
具体的なグリセリンエステルとして、グリセリンジアセテートステアレート、グリセリンジアセテートパルミテート、グリセリンジアセテートミスチレート、グリセリンジアセテートラウレート、グリセリンジアセテートカプレート、グリセリンジアセテートノナネート、グリセリンジアセテートオクタノエート、グリセリンジアセテートヘプタノエート、グリセリンジアセテートヘキサノエート、グリセリンジアセテートペンタノエート、グリセリンジアセテートオレート、グリセリンアセテートジカプレート、グリセリンアセテートジノナネート、グリセリンアセテートジオクタノエート、グリセリンアセテートジヘプタノエート、グリセリンアセテートジカプロエート、グリセリンアセテートジバレレート、グリセリンアセテートジブチレート、グリセリンジプロピオネートカプレート、グリセリンジプロピオネートラウレート、グリセリンジプロピオネートミスチレート、グリセリンジプロピオネートパルミテート、グリセリンジプロピオネートステアレート、グリセリンジプロピオネートオレート、グリセリントリブチレート、グリセリントリペンタノエート、グリセリンモノパルミテート、グリセリンモノステアレート、グリセリンジステアレート、グリセリンプロピオネートラウレート、グリセリンオレートプロピオネートなどが挙げられるがこれに限定されず、これらを単独もしくは併用して使用することができる。
この中でも、グリセリンジアセテートカプリレート、グリセリンジアセテートペラルゴネート、グリセリンジアセテートカプレート、グリセリンジアセテートラウレート、グリセリンジアセテートミリステート、グリセリンジアセテートパルミテート、グリセリンジアセテートステアレート、グリセリンジアセテートオレートが好ましい。
この中でも、ジグリセリンテトラアセテート、ジグリセリンテトラプロピオネート、ジグリセリンテトラブチレート、ジグリセリンテトラカプリレート、ジグリセリンテトララウレートが好ましい。
ポリアルキレングリコールの水酸基にアシル基が結合した化合物の具体的な例として、ポリオキシエチレンアセテート、ポリオキシエチレンプロピオネート、ポリオキシエチレンブチレート、ポリオキシエチレンバリレート、ポリオキシエチレンカプロエート、ポリオキシエチレンヘプタノエート、ポリオキシエチレンオクタノエート、ポリオキシエチレンノナネート、ポリオキシエチレンカプレート、ポリオキシエチレンラウレート、ポリオキシエチレンミリスチレート、ポリオキシエチレンパルミテート、ポリオキシエチレンステアレート、ポリオキシエチレンオレート、ポリオキシエチレンリノレート、ポリオキシプロピレンアセテート、ポリオキシプロピレンプロピオネート、ポリオキシプロピレンブチレート、ポリオキシプロピレンバリレート、ポリオキシプロピレンカプロエート、ポリオキシプロピレンヘプタノエート、ポリオキシプロピレンオクタノエート、ポリオキシプロピレンノナネート、ポリオキシプロピレンカプレート、ポリオキシプロピレンラウレート、ポリオキシプロピレンミリスチレート、ポリオキシプロピレンパルミテート、ポリオキシプロピレンステアレート、ポリオキシプロピレンオレート、ポリオキシプロピレンリノレートなどが挙げられるがこられに限定されず、これらを単独もしくは併用して使用することができる。
本発明では、安定剤としてホスファイト系化合物、亜リン酸エステル系化合物のいずれか、もしくは両方を用いることが好ましい。これらの安定剤の配合量は、0.005〜0.5重量%であるのが好ましく、より好ましくは0.01〜0.4重量%であり、さらに好ましくは0.02〜0.3重量%である。
具体的なホスファイト系安定剤は、特に限定されないが、下記式(2)〜式(4)で示されるホスファイト系安定剤が好ましい。
亜リン酸エステル系安定剤は、例えば、サイクリックネオペンタンテトライルビス(オクタデシル)ホスファイト、サイクリックネオペンタンテトライルビス(2,4−ジ−tert−ブチルフェニル)ホスファイト、サイクリックネオペンタンテトライルビス(2,6−ジ−tert−ブチル−4−メチルフェニル)ホスファイト、2,2−メチレンビス(4,6−ジ−tert−ブチルフェニル)オクチルホスファイト、トリス(2,4−ジ−tert−ブチルフェニル)ホスファイト等が挙げられる。
弱有機酸、チオエーテル系化合物、エポキシ化合物等を安定剤として配合しても良い。
弱有機酸とは、pKaが1以上のものであり、本発明の作用を妨害せず、着色防止性、物性劣化防止性を有するものであれば特に限定されない。例えば、酒石酸、クエン酸、リンゴ酸、フマル酸、シュウ酸、コハク酸、マレイン酸などが挙げられる。これらは単独で用いても良いし、2種以上を併用して用いても良い。
チオエーテル系化合物としては、例えば、ジラウリルチオジプロピオネート、ジトリデシルチオジプロピオネート、ジミリスチルチオジプロピオネート、ジステアリルチオジプロピオネート、パルミチルステアリルチオジプロピオネートが挙げられ、これらは単独で用いても良いし、2種以上を併用して用いても良い。
エポキシ化合物としては、例えばエピクロルヒドリンとビスフェノールAより誘導されるものが挙げられ、エピクロルヒドリンとグリセリンからの誘導体やビニルシクロヘキセンジオキサイドや3,4−エポキシ−6−メチルシクロヘキシルメチル−3,4−エポキシ−6−メチルシクロヘキサンカルボキシレートの如き環状のものも用いることができる。また、エポキシ化大豆油、エポキシ化ヒマシ油や長鎖−α−オレフィンオキサイド類なども用いることができる。これらは単独で用いても良いし、2種以上を併用して用いても良い。
マット剤として微粒子を加えることが好ましい。本発明に使用される微粒子としては、二酸化珪素、二酸化チタン、酸化アルミニウム、酸化ジルコニウム、炭酸カルシウム、タルク、クレイ、焼成カオリン、焼成珪酸カルシウム、水和ケイ酸カルシウム、ケイ酸アルミニウム、ケイ酸マグネシウムおよびリン酸カルシウムを挙げることができる。
これらの微粒子は、通常平均粒子サイズが0.1〜3.0μmの2次粒子を形成し、これらの微粒子はフィルム中では、1次粒子の凝集体として存在し、フィルム表面に0.1〜3.0μmの凹凸を形成させる。2次平均粒子サイズは0.2μm〜1.5μmが好ましく、0.4μm〜1.2μmがさらに好ましく、0.6μm〜1.1μmが最も好ましい。1次、2次粒子サイズはフィルム中の粒子を走査型電子顕微鏡で観察し、粒子に外接する円の直径をもって粒子サイズとした。また、場所を変えて粒子200個を観察し、その平均値をもって平均粒子サイズとした。
好ましい微粒子の量はセルロースアシレートに対し重量比で1ppm〜5000ppmが好ましく、より好ましくは5ppm〜1000ppm、さらに好ましくは10ppm〜500ppmである。
微粒子はケイ素を含むものが濁度を低くでき好ましく、特に二酸化珪素が好ましい。二酸化珪素の微粒子は、1次平均粒子サイズが20nm以下であり、かつ見かけ比重が70g/リットル以上であるものが好ましい。1次粒子の平均径が5〜16nmと小さいものがフィルムのヘイズを下げることができより好ましい。見かけ比重は90〜200g/リットル以上が好ましく、100〜200g/リットル以上がさらに好ましい。見かけ比重が大きい程、高濃度の分散液を作ることが可能になり、ヘイズ、凝集物が良化するため好ましい。
二酸化珪素の微粒子は、例えば、アエロジルR972、R972V、R974、R812、200、200V、300、R202、OX50、TT600(以上日本アエロジル(株)製)などの市販品を使用することができる。酸化ジルコニウムの微粒子は、例えば、アエロジルR976およびR811(以上日本アエロジル(株)製)の商品名で市販されており、使用することができる。
これらの中でアエロジル200V、アエロジルR972Vが1次平均粒子サイズが20nm以下であり、かつ見かけ比重が70g/リットル以上である二酸化珪素の微粒子であり、光学フィルムの濁度を低く保ちながら、摩擦係数をさげる効果が大きいため特に好ましい。
光学調整剤、界面活性剤および臭気トラップ剤(アミン等)など)を加えることができる。これらの詳細は、発明協会公開技法公技番号2001−1745号(2001年3月15日発行、発明協会)、p.17〜22に詳細に記載されている素材が好ましく用いられる。
赤外吸収染料としては例えば特開2001−194522号公報のものが使用でき、紫外線吸収剤としては、例えば、特開2001−151901号公報に記載のものが使用でき、それぞれセルロースアシレートに対して0.001〜5質量%含有させることが好ましい。
光学調整剤としてはレターデーション調整剤を挙げることができ、例えば、特開2001−166144号、特開2003−344655号、特開2003−248117号、特開2003−66230号各公報記載のものを使用することができ、これにより面内のレターデーション(Re)、厚み方向のレターデーション(Rth)を制御できる。好ましい添加量は0〜10質量%であり、より好ましくは0〜8質量%、さらに好ましくは0〜6質量%である。
本発明は、飽和ノルボルネン樹脂を用いることも好ましい。飽和ノルボルネン樹脂として、下記飽和ノルボルネン樹脂−A、飽和ノルボルネン樹脂−Bいずれも好ましく用いることができる。
本発明で使用する飽和ノルボルネン系樹脂としては、例えば、(1)ノルボルネン系モノマーの開環(共)重合体を、必要に応じてマレイン酸付加、シクロペンタジエン付加のごときポリマー変性を行なった後に、水素添加した樹脂、(2)ノルボルネン系モノマーを付加型重合させた樹脂、(3)ノルボルネン系モノマーとエチレンやα−オレフィンなどのオレフィン系モノマーと付加型共重合させた樹脂などが挙げることができる。重合方法および水素添加方法は、常法により行なうことができる。
ノルボルネン系モノマーとしては、例えば、ノルボルネン、およびそのアルキルおよび/またはアルキリデン置換体、例えば、5−メチル−2−ノルボルネン、5−ジメチル−2−ノルボルネン、5−エチル−2−ノルボルネン、5−ブチル−2−ノルボルネン、5−エチリデン−2−ノルボルネン等、これらのハロゲン等の極性基置換体;ジシクロペンタジエン、2,3−ジヒドロジシクロペンタジエン等;ジメタノオクタヒドロナフタレン、そのアルキルおよび/またはアルキリデン置換体、およびハロゲン等の極性基置換体、例えば、6−メチル−1,4:5,8−ジメタノ−1,4,4a,5,6,7,8,8a−オクタヒドロナフタレン、6−エチル−1,4:5,8−ジメタノ−1,4,4a,5,6,7,8,8a−オクタヒドロナフタレン、6−エチリデン−1,4:5,8−ジメタノ−1,4,4a,5,6,7,8,8a−オクタヒドロナフタレン、6−クロロ−1,4:5,8−ジメタノ−1,4,4a,5,6,7,8,8a−オクタヒドロナフタレン、6−シアノ−1,4:5,8−ジメタノ−1,4,4a,5,6,7,8,8a−オクタヒドロナフタレン、6−ピリジル−1,4:5,8−ジメタノ−1,4,4a,5,6,7,8,8a−オクタヒドロナフタレン、6−メトキシカルボニル−1,4:5,8−ジメタノ−1,4,4a,5,6,7,8,8a−オクタヒドロナフタレン等;シクロペンタジエンとテトラヒドロインデン等との付加物;シクロペンタジエンの3〜4量体、例えば、4,9:5,8−ジメタノ−3a,4,4a,5,8,8a,9,9a−オクタヒドロ−1H−ベンゾインデン、4,11:5,10:6,9−トリメタノ−3a,4,4a,5,5a,6,9,9a,10,10a,11,11a−ドデカヒドロ−1H−シクロペンタアントラセン;等が挙げられる。
また、飽和ノルボルネン樹脂として、下記式(12)〜(15)で表わされるものを挙げることができ、これらのうち、下記式(12)で表されるものが特に好ましい。
これらの飽和ノルボルネン樹脂の重量平均分子量としては、5,000〜1,000,000が好ましく、より好ましくは8,000〜200,000である。また、数平均分子量としては、2000〜500000が好ましく、より好ましくは4000〜100000である。
本発明の飽和ノルボルネン系樹脂としては、例えば、特開昭60−168708号公報、特開昭62−252406号公報、特開昭62−252407号公報、特開平2−133413号公報、特開昭63−145324号公報、特開昭63−264626号公報、特開平1−240517号公報、特公昭57−8815号公報などに記載されている樹脂などを挙げることができる。
これらの樹脂の中でも、ノルボルネン系モノマーの開環重合体を水素添加して得られる水添重合体が特に好ましい。
これらの飽和ノルボルネン樹脂のガラス転移温度(Tg)は120℃以上であることが好ましく、さらに好ましくは140℃以上である。また飽和ノルボルネン樹脂の飽和吸水率は1重量%以下であることが好ましく、さらに好ましくは0.8重量%以下である。上記式(12)〜(15)で表わされる飽和ノルボルネン樹脂のガラス転移温度(Tg)および飽和吸水率は、置換基A、B、C、Dの種類を選択することにより制御することができる。
これらのノルボルネン系樹脂は、クロロホルム中、30℃で測定される固有粘度(ηinh)が、0.1〜1.5dl/gであることが好ましく、さらに好ましくは0.4〜1.2dl/gである。また、水添重合体の水素添加率としては、60MHz、1H−NMRで測定した値が50%以上とされ、好ましくは90%以上、さらに好ましくは98%以上である。水素添加率が高いほど、得られる飽和ノルボルネンフィルムは、熱や光に対する安定性が優れたものとなる。該水添重合体中に含まれるゲル含有量が5重量%以下であることが好ましく、さらに好ましくは1重量%以下である。
[A−1]:炭素数が2〜20のα-オレフィンと下記式(II)で表される環状オレフィンとのランダム共重合体の水素添加物、
[A−2]:下記式(II)で表される環状オレフィンの開環重合体または共重合体の水素添加物などを挙げることができる。
上記式(II)中、nは0または1であり、mは0または1以上の整数であり、qは0または1である。なお、qが1の場合には、R a およびR b は、それぞれ独立に、下記に示す原子または炭化水素基であり、qが0の場合には、それぞれの結合手が結合して5員環を形成する。
また、これらの飽和ノルボルネン樹脂の合成法については、特開2001−114836号公報の段落番号0039〜0068を参考に実施することができる。
このようなシクロオレフィン(共)重合体は、特開平10−168201号公報の段落番号0019〜0020に従い合成することができる。
これらの酸化防止剤の添加量は、飽和ノルボルネン系樹脂100重量部に対して、例えば、0.1〜3重量部であり、好ましくは0.2〜2重量部である。
さらに飽和ノルボルネン系樹脂には、所望により、フェノール系やリン系などの老化防止剤、耐電防止剤、紫外線吸収剤、上述の易滑剤などの各種添加剤を添加してもよい。特に、液晶は、通常、紫外線により劣化するので、ほかに紫外線防護フィルターを積層するなどの防護手段を取らない場合は、紫外線吸収剤を添加することが好ましい。紫外線吸収剤としては、ベンゾフェノン系紫外線吸収剤、ベンゾトリアゾル系紫外線吸収剤、アクリルニトリル系紫外線吸収剤などを用いることができ、それらの中でもベンゾフェノン系紫外線吸収剤が好ましく、添加量は、例えば、10〜100,000ppm、好ましくは100〜10,000ppmである。また、溶液流延法によりシートを作製する場合は、表面粗さを小さくするため、レベリング剤の添加が好ましい。レベリング剤としては、例えば、フッ素系ノニオン界面活性剤、特殊アクリル樹脂系レベリング剤、シリコーン系レベリング剤など塗料用レベリング剤を用いることができ、それらの中でも溶媒との相溶性の良いものが好ましく、添加量は、例えば、5〜50,000ppm、好ましくは10〜20,000ppmである。
本発明の製膜工程において、残留溶剤が好ましくは0.5質量%以下、より好ましくは0.01質量%以下、さらに好ましくは0質量%である。残留溶剤は局所的に残留し易く、フィルム面内の不均一性が発生し易い。このような残留溶剤は溶液製膜で発生し易いため、本発明では溶融製膜のほうが好ましい。
(1)ペレット化
上記透明熱可塑性樹脂と添加物は溶融製膜に先立ち混合しペレット化するのが好ましい。
ペレット化を行うにあたり透明熱可塑性樹脂および添加物は事前に乾燥を行うことが好ましいが、ベント式押出機を用いることで、これを代用することもできる。乾燥を行う場合は、乾燥方法として、加熱炉内にて90℃で8時間以上加熱する方法等を用いることができるが、この限りではない。ペレット化は上記透明熱可塑性樹脂と添加物を2軸混練押出機を用い150℃〜250℃で溶融後、ヌードル状に押出したものを水中で固化し裁断することで作製することができる。また、押出機による溶融後水中に口金より直接押出ながらカットする、アンダーウオーターカット法等によりペレット化を行ってもよい。
押出機は十分な、溶融混練が得られる限り、任意の公知の単軸スクリュー押出機、非かみ合い型異方向回転二軸スクリュー押出機、かみ合い型異方向回転二軸スクリュー押出機、かみ合い型同方向回転二軸スクリュー押出機などを用いることができる。
好ましいペレットの大きさは断面積が1mm2〜300mm2、長さが1mm〜30mmがこのましく、より好ましくは断面積が2mm2〜100mm2、長さが1.5mm〜10mmである。
またペレット化を行う時に、上記添加物は押出機の途中にある原料投入口やベント口から投入する異もできる。
押出機の回転数は10rpm〜1000rpmが好ましく、より好ましくは、20rpm〜700rpm、さらにより好ましくは30rpm〜500rpmである。これより、回転速度が遅くなると滞留時間が長くなり、熱劣化により分子量が低下したり、黄色味が悪化しやすくなる為、好ましくない。また回転速度が速すぎると剪断により分子の切断がおきやすくなり、分子量低下を招いたり、架橋ゲルの発生は増加するなどの問題が生じやすくなる。
ペレット化における押出滞留時間は、好ましくは10秒〜30分、より好ましくは15秒〜10分、さらに好ましくは30秒〜3分である。十分に溶融ができれば、滞留時間は短い方が、樹脂劣化、黄色み発生を抑えることができる点で好ましい。
溶融製膜に先立ちペレット中の水分を減少させることが好ましい。乾燥の方法については、除湿風乾燥機を用いて乾燥する事が多いが、目的とする含水率が得られるのであれば特に限定されない(加熱、送風、減圧、攪拌などの手段を単独または組み合わせて効率的に行うことが好ましい、さらに好ましくは、乾燥ホッパーを断熱構造にすることが好ましい)。乾燥温度として好ましくは0〜200℃であり、さらに好ましくは40〜180℃であり、特に好ましくは60〜150℃である。乾燥温度をこのような範囲にすることにより、含有水分率を好ましい範囲に保ちつつ、樹脂が粘着してブロッキングするのを効果的に抑止でき好ましい。乾燥風量として好ましくは20〜400m3/時間であり、さらに好ましくは50〜300m3/時間であり、特に好ましくは100〜250m3/時間である。乾燥風量を20m3/時間以上とすることにより、乾燥効率がより好ましい傾向にある。一方、風量を400m3/時間以下とすることにより、十分な乾燥効果を保ちつつ、経済的にも好ましくなる。エアーの露点として、好ましくは0〜−60℃であり、さらに好ましくは−10〜−50℃であり、特に好ましくは−20〜−40℃である。乾燥時間は、通常15分以上必要であり、好ましくは1時間以上であり、特に好ましくは2時間以上である。一方、乾燥時間を50時間以下とすることにより、十分な水分率の低減効果が得られつつ、樹脂の熱劣化を抑止することができるため好ましい。本発明の透明熱可塑性樹脂は、その含水率が1.0質量%以下であることが好ましく、0.1質量%以下であることがさらに好ましく、0.01質量%以下であることが特に好ましい。
上述した透明熱可塑性樹脂は押出機の供給口を介してシリンダー内に供給される。シリンダー内は供給口側から順に、供給口から供給した透明熱可塑性樹脂を定量輸送する供給部(領域A)と透明熱可塑性樹脂を溶融混練・圧縮する圧縮部(領域B)と溶融混練・圧縮された透明熱可塑性樹脂を計量する計量部(領域C)とで構成される。樹脂は上述の方法により水分量を低減させるために、乾燥することが好ましいが、残存する酸素による溶融樹脂の酸化を防止するために、押出機内を不活性(窒素等)気流中、あるいはベント付き押出し機を用い真空排気しながら実施するのがより好ましい。押出機のスクリュー圧縮比は2.5〜4.5に設定され、L/Dは20〜70に設定されている。ここでスクリュー圧縮比とは供給部Aと計量部Cとの容積比、即ち供給部Aの単位長さあたりの容積÷計量部Cの単位長さあたりの容積で表され、供給部Aのスクリュー軸の外径d1、計量部Cのスクリュー軸の外径d2、供給部Aの溝部径a1、および計量部Cの溝部径a2とを使用して算出される。また、L/Dとはシリンダー内径に対するシリンダー長さの比である。また、押出温度は190〜240℃に設定される。押出機内での温度が230℃を超える場合には、押出機とダイとの間に冷却機を設ける様にするとよい。
また、押出温度は上述の温度範囲にすることが好ましい。このようにして得た透明熱可塑性フィルムは、例えば、ヘイズが2.0%以下、イエローインデックス(YI値)が10以下である特性値を有している。
ここで、ヘイズは押出温度が低過ぎないかの指標、換言すると製造後の透明熱可塑性フィルムに残存する未溶解異物の多少を知る指標になり、ヘイズが2.0%を超えると、製造後の透明熱可塑性フィルムの強度低下と延伸時の破断が発生しやすい傾向にある。また、イエローインデックス(YI値)は押出温度が高過ぎないかを知る指標となり、イエローインデックス(YI値)が10以下であれば、黄色味が殆ど発生しない傾向にある。
なお、好ましいスクリューの直径は目標とする単位時間あたりの押出量によって異なるが、例えば、10mm〜300mm、より好ましくは20mm〜250mm、さらに好ましくは30mm〜150mmである。
樹脂中の異物濾過のためや異物によるギアポンプ損傷を避けるため押し出し機出口にフィルター濾材を設けるいわゆるブレーカープレート式の濾過を行うことが好ましい。またさらに精度高く異物濾過をするために、ギアポンプ通過後にいわゆるリーフ型ディスクフィルターを組み込んだ濾過装置を設けることが好ましい。濾過は、濾過部を1カ所設けて行うことができ、また複数カ所設けて行う多段濾過でも良い。フィルター濾材の濾過精度は高い方が好ましいが、濾材の耐圧や濾材の目詰まりによる濾圧上昇から、濾過精度は15μmm〜3μmmが好ましく、10μmm〜3μmmがより好ましい。特に最終的に異物濾過を行うリーフ型ディスクフィルター装置を使用する場合では品質の上で濾過精度の高い濾材を使用することが好ましく、耐圧、フィルターライフの適性を確保するために装填枚数にて調整することが可能である。濾材の種類は、高温高圧下で使用される点から鉄鋼材料を用いることが好ましく、鉄鋼材料の中でも特にステンレス鋼、スチールなどを用いることが好ましく、腐食の点から特にステンレス鋼を用いることが望ましい。濾材の構成としては、線材を編んだものの他に、例えば金属長繊維あるいは金属粉末を焼結し形成する焼結濾材が使用でき、濾過精度、フィルターライフの点から焼結濾材が好ましい。
厚み精度を向上させるためには、吐出量の変動を減少させることが重要であり、押出機出機とダイスの間にギアポンプを設けて、ギアポンプから一定量のセルロースアシレート樹脂を供給することは効果がある。ギアポンプとは、ドライブギアとドリブンギアとからなる一対のギアが互いに噛み合った状態で収容され、ドライブギアを駆動して両ギアを噛み合い回転させることにより、ハウジングに形成された吸引口から溶融状態の樹脂をキャビティ内に吸引し、同じくハウジングに形成された吐出口からその樹脂を一定量吐出するものである。押出機先端部分の樹脂圧力が若干の変動があっても、ギアポンプを用いることにより変動を吸収し、製膜装置下流の樹脂圧力の変動は非常に小さなものとなり、厚み変動が改善される。ギアポンプを用いることにより、ダイ部分の樹脂圧力の変動巾を±1%以内にすることが可能である。
ギアポンプによる定量供給性能を向上させるために、スクリューの回転数を変化させて、ギアポンプ前の圧力を一定に制御する方法も用いることができる。また、ギアポンプのギアの変動を解消した3枚以上のギアを用いた高精度ギアポンプも有効である。
ギアポンプを用いるその他のメリットとしては、スクリュー先端部の圧力を下げて製膜できることから、エネルギー消費の軽減・樹脂温上昇の防止・輸送効率の向上・押出機内での滞留時間の短縮・押出機のL/Dを短縮が期待できる。また、異物除去のために、フィルターを用いる場合には、ギアポンプが無いと、ろ圧の上昇と共に、スクリューから供給される樹脂量が変動したりすることがあるが、ギアポンプを組み合わせて用いることにより解消が可能である。一方、ギアポンプのデメリットとしては、設備の選定方法によっては、設備の長さが長くなり、樹脂の滞留時間が長くなることと、ギアポンプ部のせん断応力によって分子鎖の切断を引き起こすことがあり、注意が必要である。
樹脂が供給口から押出機に入ってからダイスから出るまでの樹脂の好ましい滞留時間は2分〜60分であり、より好ましくは3分〜40分であり、さらに好ましくは4分〜30分である。
ギアポンプの軸受循環用ポリマーの流れが悪くなることにより、駆動部と軸受部におけるポリマーによるシールが悪くなり、計量および送液押し出し圧力の変動が大きくなったりする問題が発生するため、透明熱可塑性樹脂の溶融粘度に合わせたギアポンプの設計(特にクリアランス)が必要である。また、場合によっては、ギアポンプの滞留部分が透明熱可塑性樹脂の劣化の原因となるため、滞留のできるだけ少ない構造が好ましい。押出機とギアポンプあるいはギアポンプとダイ等をつなぐポリマー管やアダプタについても、できるだけ滞留の少ない設計が必要であり、且つ溶融粘度の温度依存性の高い透明熱可塑性樹脂の押出圧力安定化のためには、温度の変動をできるだけ小さくすることが好ましい。一般的には、ポリマー管の加熱には設備コストの安価なバンドヒーターが用いられることが多いが、温度変動のより少ないアルミ鋳込みヒーターを用いることがより好ましい。さらに上述のように押出し機内で、押出し機のバレルを3〜20に分割したヒーターで加熱し溶融することが好ましい。
上記の如く構成された押出機によって透明熱可塑性樹脂が溶融され、必要に応じ濾過機、ギアポンプを経由して溶融樹脂がダイに連続的に送られる。ダイはダイス内の溶融樹脂の滞留が少ない設計であれば、一般的に用いられるTダイ、フィッシュテールダイ、ハンガーコートダイの何れのタイプでも構わない。また、Tダイの直前に樹脂温度の均一性アップのためのスタティックミキサーを入れることも問題ない。Tダイ出口部分のクリアランスは、通常、フィルム厚みの1.0〜5.0倍が良く、好ましくは1.2〜3倍であり、さらに好ましくは1.3〜2倍である。リップクリアランスを、フィルム厚みの1.0倍以上とすることにより、より面状の良好なシートを得ることができる。また、リップクリアランスを、フィルム厚みの5.0倍以下とすることにより、シートの厚み精度が向上する傾向にあり好ましい。ダイはフィルムの厚み精度を決定する非常に重要な設備であり、厚み調整がシビアにコントロールできるものが好ましい。通常厚み調整は40〜50mm間隔で調整可能であるが、好ましくは35mm間隔以下、さらに好ましくは25mm間隔以下でフィルム厚み調整が可能なタイプが好ましい。また、製膜フィルムの均一性を向上するために、ダイの温度むらや巾方向の流速むらのできるだけ少ない設計が重要である。また、下流のフィルム厚みを計測して、厚み偏差を計算し、その結果をダイの厚み調整にフィードバックさせる自動厚み調整ダイも長期連続生産の厚み変動の低減に有効である。
フィルムの製造は設備コストの安い単層製膜装置が一般的に用いられるが、場合によっては機能層を外層に設けために多層製膜装置を用いて2種以上の構造を有するフィルムの製造も可能である。一般的には機能層を表層に薄く積層することが好ましいが、特に層比を限定するものではない。
上記方法にて、ダイよりシート上に押し出された溶融樹脂をキャスティングドラム上で冷却固化し、フィルムを得る。この時、静電印加法、エアナイフ法、エアーチャンバー法、バキュームノズル法、タッチロール法等の方法を用い、キャスティングドラムと溶融押出ししたシートの密着を上げることが好ましい。このような密着向上法は、溶融押出しシートの全面に実施してもよく、一部に実施しても良い。
タッチロールは通常剛直な素材を用いるが、剛直すぎるとダイから出たメルトをロール間で挟む時に残留歪が発生し易く、一層部分的な歪を助長する。このためタッチロールの材質は、弾性を有するものが好ましい。これにより過剰な面圧はタッチロールが変形することで吸収し歪を抑制する。ロールに弾性を付与するためには、ロールの外筒厚みを通常のロールよりも薄くすることが必要であり、外筒の肉厚Zは、0.05mm〜7.0mmが好ましく、より好ましくは0.2mm〜5.0mmである。さらに好ましくは0.3mm〜3.5mmである。タッチロールは金属シャフトの上に設置し、その間に熱媒(流体)を通しても良く、外筒と金属シャフトの上に間に弾性体層を設け、外筒の間に熱媒(流体)を満たしたものがあげられる。
このようにタッチロールは低弾性であるため、キャスティングロールとに接触させるとその押圧で凹状に弾性変形する。従って、タッチロールとキャスティングロールは冷却ロールと面接触するため押圧が分散され、低い面圧を達成できる。このためこの間に挟まれたフィルムに残留歪を残すことなく、均一な冷却を達成できる。好ましいタッチロールの線圧は、1kg/cm 〜100kg/cm、より好ましくは2kg/cm 〜80kg/cm、さらに好ましくは3kg/cm〜60kg/cmである。ここでいう線圧とはタッチロールに加える力をダイの吐出口の幅で割った値である。線圧は1kg/cm以上とすることにより、タッチロールの押し付けが弱くなって面内が不均一性になるのを効果的に是正でき、一方、100kg/cm以下とすることにより、全幅に亘り均一な線圧を加えることができ、すなわち、ロールがたわみ両端or中央に線圧が集中するのをより効果的に抑止でき、面内が不均一性になるのをより効果的に是正できる。
タッチロールは、好ましくは60℃〜160℃、より好ましくは70℃〜150℃、さらに好ましくは80℃〜140℃に設定するのが好ましい。このような温度制御はこれらのロール内部に温調した液体、気体を通すことで達成できる。
タッチロール、キャスティングロールは、表面が鏡面であることが好ましく、算術平均高さRaが好ましくは100nm以下、より好ましくは50nm以下、さらに好ましくは25nm以下である。具体的には例えば特開平11−314263号、特開2002−36332号、特開平11−235747号、特開2004−216717号、特開2003−145609号各公報、国際公開WO97/28950号パンフレット等に記載のものを利用できる。
この後、キャスティングドラムから剥ぎ取り、ニップロールを経た後巻き取る。巻き取り速度は10m/分〜100m/分が好ましく、より好ましくは15m/分〜80m/分、さらに好ましくは20m/分〜70m/分である。
製膜幅は、好ましくは0.7m〜5m、より好ましくは1m〜4m、さらに好ましくは1.3m〜3mが好ましい。このようにして得られた未延伸フィルムの厚みは30μm〜400μmが好ましく、より好ましくは40μm〜300μm、さらに好ましくは50μm〜200μmである。
また巻取り前に片端あるいは両端に厚みだし加工(ナーリング処理)を行うことも好ましい。厚みだし加工による凹凸の高さは1μm〜200μmが好ましく、より好ましくは10μm〜150μm、さらに好ましくは20μm〜100μmである。厚みだし加工は両面に凸になるようにしても、片面に凸になるようにしても構わない。厚みだし加工の幅は1mm〜50mmが好ましく、より好ましくは3mm〜30mm、さらに好ましくは5mm〜20mmである。押出し加工は、通常、室温(例えば、18℃)〜300℃で実施できる。
好ましい巻き取り張力は1kg/m幅〜50kg/幅であり、より好ましくは2kg/m幅〜40kg/幅、さらに好ましくは3kg/m幅〜20kg/幅である。巻き取り張力を1kg/m幅以上とすることにより、フィルムを均一に巻き取りやすい傾向にある。逆に、巻き取り張力を50kg/幅以下とすることにより、フィルムが堅巻きになったり、巻き外観が悪化するのを抑止できる傾向にあり、また、フィルムのコブの部分がクリープ現象により延びてフィルムの波うちの原因になったり、フィルムの伸びによる残留複屈折が生じたりするのを抑止できる傾向にある。巻き取り張力は、ラインの途中のテンションコントロールにより検知し、一定の巻き取り張力になるようにコントロールされながら巻き取ることが好ましい。製膜ラインの場所により、フィルム温度に差がある場合には熱膨張により、フィルムの長さが僅かに異なる場合があるため、ニップロール間のドロー比率を調整し、ライン途中でフィルムに規定以上の張力がかからない様にすることが必要である。
巻き取り張力はテンションコントロールの制御により、一定張力で巻き取ることもできるが、巻き取った直径に応じてテーパーをつけ、適正な巻取り張力にすることがより好ましい。一般的には巻き径が大きくなるにつれて張力を少しずつ小さくするが、場合によっては、巻き径が大きくなるにしたがって張力を大きくする方が好ましい場合もある。
上記において、溶融製膜の方法を説明したが、本発明においては、セルロースアシレート樹脂やポリカーボネートなどを溶液製膜によって行うこともできる。この場合の溶剤としては、(a)塩素系溶剤および(b)非塩素系溶剤のいずれも用いることができる。
塩素系有機溶媒としては、ジクロロメタンおよびクロロホルムが好ましく、ジクロロメタンがより好ましい。また、塩素系有機溶媒以外の有機溶媒を混合してもよい。この場合は、ジクロロメタンが50質量%以上であることが好ましい。
本発明の併用される非塩素系有機溶媒について以下に記す。すなわち、好ましい非塩素系有機溶媒としては、炭素数が3〜12のエステル、ケトン、エーテルおよびアルコール、炭化水素から選ばれる溶媒が好ましい。エステル、ケトン、エーテルおよびアルコールは、環状構造を有していてもよい。エステル、ケトンおよびエーテルの官能基(すなわち、−O−、−CO−および−COO−)のいずれかを2つ以上有する化合物も溶媒として用いることができ、例えば、アルコール性水酸基のような他の官能基を同時に有していてもよい。2種類以上の官能基を有する溶媒の場合、その炭素数はいずれかの官能基を有する化合物の規定範囲内であればよい。炭素数が3〜12のエステル類の例には、エチルホルメート、プロピルホルメート、ペンチルホルメート、メチルアセテート、エチルアセテートおよびペンチルアセテートが挙げられる。炭素数が3〜12のケトン類の例には、アセトン、メチルエチルケトン、ジエチルケトン、ジイソブチルケトン、シクロペンタノン、シクロヘキサノンおよびメチルシクロヘキサノンが挙げられる。炭素数が3〜12のエーテル類の例には、ジイソプロピルエーテル、ジメトキシメタン、ジメトキシエタン、1,4−ジオキサン、1,3−ジオキソラン、テトラヒドロフラン、アニソールおよびフェネトールが挙げられる。2種類以上の官能基を有する有機溶媒の例には、2−エトキシエチルアセテート、2−メトキシエタノールおよび2−ブトキシエタノールが挙げられる。
塩素系有機溶媒と併用される非塩素系有機溶媒については、特に限定されないが、酢酸メチル、酢酸エチル、蟻酸メチル、蟻酸エチル、アセトン、ジオキソラン、ジオキサン、炭素数が4〜7のケトン類またはアセト酢酸エステル、炭素数が1〜10のアルコールまたは炭化水素から選ばれる。なお好ましい併用される非塩素系有機溶媒は、酢酸メチル、アセトン、蟻酸メチル、蟻酸エチル、メチルエチルケトン、シクロペンタノン、シクロヘキサノン、アセチル酢酸メチル、メタノール、エタノール、1−プロパノール、2−プロパノール、1−ブタノール、2−ブタノール、およびシクロヘキサノール、シクロヘキサン、ヘキサンを挙げることができる。
・ジクロロメタン/メタノール/エタノール/ブタノール(80/10/5/5)
・ジクロロメタン/アセトン/メタノール/プロパノール(80/10/5/5)
・ジクロロメタン/メタノール/ブタノール/シクロヘキサン(80/10/5/5)
・ジクロロメタン/メチルエチルケトン/メタノール/ブタノール(80/10/5/5)
・ジクロロメタン/アセトン/メチルエチルケトン/エタノール/イソプロパノール(72/9/9/4/6)
・ジクロロメタン/シクロペンタノン/メタノール/イソプロパノール(80/10/5/5)
・ジクロロメタン/酢酸メチル/ブタノール(80/10/10)
・ジクロロメタン/シクロヘキサノン/メタノール/ヘキサン(70/20/5/5)
・ジクロロメタン/メチルエチルケトン/アセトン/メタノール/エタノール(50/20/20/5/5)
・ジクロロメタン/1,3ジオキソラン/メタノール/エタノール(70/20/5/5)
・ジクロロメタン/ジオキサン/アセトン/メタノール/エタノール (60/20/10/5/5)
・ジクロロメタン/アセトン/シクロペンタノン/エタノール/イソブタノール/シクロヘキサン(65/10/10/5/5/5)
・ジクロロメタン/メチルエチルケトン/アセトン/メタノール/エタノール (70/10/10/5/5)
・ジクロロメタン/アセトン/酢酸エチル/エタノール/ブタノール/ヘキサン (65/10/10/5/5/5)
・ジクロロメタン/アセト酢酸メチル/メタノール/エタノール(65/20/10/5)
・ジクロロメタン/シクロペンタノン/エタノール/ブタノール(65/20/10/5)
好ましい非塩素系有機溶媒は、炭素数が3〜12のエステル、ケトンおよびエーテルから選ばれる溶媒が好ましい。エステル、ケトンおよびエーテルは、環状構造を有していてもよい。エステル、ケトンおよびエーテルの官能基(すなわち、−O−、−CO−および−COO−)のいずれかを2つ以上有する化合物も、主溶媒として用いることができ、例えば、アルコール性水酸基のような他の官能基を有していてもよい。2種類以上の官能基を有する主溶媒の場合、その炭素数はいずれかの官能基を有する化合物の規定範囲内であればよい。炭素数が3〜12のエステル類の例には、エチルホルメート、プロピルホルメート、ペンチルホルメート、メチルアセテート、エチルアセテートおよびペンチルアセテートが挙げられる。炭素数が3〜12のケトン類の例には、アセトン、メチルエチルケトン、ジエチルケトン、ジイソブチルケトン、シクロペンタノン、シクロヘキサノンおよびメチルシクロヘキサノンが挙げられる。炭素数が3〜12のエーテル類の例には、ジイソプロピルエーテル、ジメトキシメタン、ジメトキシエタン、1,4−ジオキサン、1,3−ジオキソラン、テトラヒドロフラン、アニソールおよびフェネトールが挙げられる。2種類以上の官能基を有する有機溶媒の例には、2−エトキシエチルアセテート、2−メトキシエタノールおよび2−ブトキシエタノールが挙げられる。
第1の溶媒は、さらに好ましくは酢酸メチル、アセトン、蟻酸メチル、蟻酸エチルあるいはこれらの混合物である。第2の溶媒は、メチルエチルケトン、シクロペンタノン、シクロヘキサノン、アセチル酢酸メチルが好ましく、これらの混合液であってもよい。 第3の溶媒であるアルコールは、直鎖、分枝若しくは環状のいずれでもよく、その中でも飽和脂肪族炭化水素であることが好ましい。アルコールの水酸基は、第一級〜第三級のいずれであってもよい。アルコールの例には、メタノール、エタノール、1−プロパノール、2−プロパノール、1−ブタノール、2−ブタノール、tert−ブタノール、1−ペンタノール、2−メチル−2−ブタノールおよびシクロヘキサノールが含まれる。尚、アルコールとしては、フッ素系アルコールも用いられる。例えば、2−フルオロエタノール、2,2,2−トリフルオロエタノール、2,2,3,3−テトラフルオロ−1−プロパノールなども挙げられる。さらに炭化水素は、直鎖、分岐、環状のいずれであってもよい。また、芳香族炭化水素と脂肪族炭化水素のいずれも用いることができる。脂肪族炭化水素は、飽和であっても不飽和であってもよい。炭化水素の例には、シクロヘキサン、ヘキサン、ベンゼン、トルエンおよびキシレンが含まれる。
これら第3の溶媒であるアルコールおよび炭化水素は単独でもよいし2種類以上の混合物でもよく特に限定されない。第3の溶媒としては、好ましい具体的化合物は、アルコールとしてはメタノール、エタノール、1−プロパノール、2−プロパノール、1−ブタノール、2−ブタノール、シクロヘキサノール、シクロヘキサン、ヘキサンを挙げることができ、特にはメタノール、エタノール、1−プロパノール、2−プロパノール、1−ブタノールである。
・酢酸メチル/アセトン/メタノール/エタノール/ブタノール(75/10/5/5/5)
・酢酸メチル/アセトン/メタノール/エタノール/プロパノール(75/10/5/5/5)
・酢酸メチル/アセトン/メタノール/ブタノール/シクロヘキサン(75/10/5/5/5)
・酢酸メチル/アセトン/エタノール/ブタノール(81/8/7/4)
・酢酸メチル/アセトン/エタノール/ブタノール(82/10/4/4)
・酢酸メチル/アセトン/エタノール/ブタノール(80/10/4/6)
・酢酸メチル/メチルエチルケトン/メタノール/ブタノール(80/10/5/5)
・酢酸メチル/アセトン/メチルエチルケトン/エタノール/イソプロパノール(75/8/8/4/5)
・酢酸メチル/シクロペンタノン/メタノール/イソプロパノール(80/10/5/5)
・酢酸メチル/アセトン/ブタノール(85/10/5)
・酢酸メチル/シクロペンタノン/アセトン/メタノール/ブタノール(60/15/15/5/5)
・酢酸メチル/シクロヘキサノン/メタノール/ヘキサン(70/20/5/5)
・酢酸メチル/メチルエチルケトン/アセトン/メタノール/エタノール(50/20/20/5/5)
・酢酸メチル/1、3ジオキソラン/メタノール/エタノール(70/20/5/5)
・酢酸メチル/ジオキサン/アセトン/メタノール/エタノール(60/20/10/5/5)
・酢酸メチル/アセトン/シクロペンタノン/エタノール/イソブタノール/シクロヘキサン(65/10/10/5/5/5)
・ギ酸メチル/メチルエチルケトン/アセトン/メタノール/エタノール(50/20/20/5/5)
・ギ酸メチル/アセトン/酢酸エチル/エタノール/ブタノール/ヘキサン(65/10/10/5/5/5)
・アセトン/アセト酢酸メチル/メタノール/エタノール(65/20/10/5)
・アセトン/シクロペンタノン/エタノール/ブタノール(65/20/10/5)
・アセトン/1,3ジオキソラン/エタノール/ブタノール(65/20/10/5)
・1、3ジオキソラン/シクロヘキサノン/メチルエチルケトン/メタノール/ブタノール(55/20/10/5/5/5)
・酢酸メチル/アセトン/エタノール/ブタノール(81/8/7/4)でセルロースアシレート溶液を作製し、ろ過・濃縮後に2質量部のブタノールを追加添加
・酢酸メチル/アセトン/エタノール/ブタノール(81/10/4/2)でセルロースアシレート溶液を作製し、ろ過・濃縮後に4質量部のブタノールを追加添加
・酢酸メチル/アセトン/エタノール(84/10/6)でセルロースアシレート溶液を作製し、ろ過・濃縮後に5質量部のブタノールを追加添加
本発明では、塩素系、非塩素系溶剤いずれの場合でも、溶媒にセルロースアシレートを10〜40質量%溶解していることが好ましく、より好ましくは13〜35質量%であり、特には15〜30質量%である。溶解に先立ち、0℃〜50℃、0.1時間〜100時間膨潤させることが好ましい。なお、種々の添加剤は膨潤工程の前に添加しても良く、膨潤工程中あるいは後でもよく、さらには、この後冷却溶解中あるいは後でも構わない。
さらに本発明のドープは、濃縮およびろ過を実施することが好ましく、これらは発明協会公開技報(公技番号 2001−1745、2001年3月15日発行、発明協会)にて25頁に詳細に記載されているものを使用できる。
ドープを貯蔵釜から、例えば、回転数によって高精度に定量送液できる加圧型定量ギヤポンプを通して加圧型ダイに送り、ドープを加圧型ダイの口金(スリット)からエンドレスに走行している流延部の金属支持体(バンド、ドラム)の上に均一に流延され、金属支持体がほぼ一周した剥離点で、生乾きのドープ膜(ウェブとも呼ぶ)を金属支持体から剥離する。剥離したウェブの両端をクリップで挟み、幅保持しながらテンターで搬送して乾燥し、続いて乾燥装置のロール群で搬送し乾燥した後、トリミングした後ナーリング(型押し)加工を行った後巻き取り機で所定の長さに巻き取る。巻取り長は1000m〜5000mが好ましく、より好ましくは1500m〜4000mである。巻取り幅は0.5m〜4mが好ましく、より好ましくは1m〜3mである。厚みは20μm〜300μmが好ましく、より好ましくは30μm〜200μmである。なお、乾燥温度は100℃〜200℃が好ましく、より好ましくは120℃〜180℃である。好ましい乾燥時間は10分〜200分であり、より好ましく15分〜80分である。
また、流延工程では1種類のセルロースアシレート溶液を単層流延してもよいし、2種類以上のセルロースアシレート溶液を同時およびまたは逐次共流延してもよい。
このような溶液製膜は公開技法(公技番号 2001−1745、2001年3月15日発行、発明協会)に記載の方法も用いることができる。
本発明では、上述のように製膜した透明熱可塑性フィルムを、上述の方法で縦延伸/横延伸、緩和することが好ましい。縦延伸、横延伸はいずれか一方でも良く、両方実施しても良い。また縦延伸、横延伸は各々1回で行っても良く、複数回に亘って実施しても良く、同時に縦、横に延伸しても良い。縦延伸、横延伸の順序は特に限定されないが、縦延伸後に横延伸を行うのが好ましい。これらの縦延伸、横延伸は製膜と切り離して実施しても良く(製膜後、一端巻き取ったものを再度送り出して延伸)、連続して行っても良い(製膜後、そのまま連続して延伸する)。
緩和は縦延伸、横延伸の少なくとも一方の後に実施するのが好ましく、より好ましくは(1)縦延伸後に縦緩和を行ない、横延伸後に横緩和を行う、(2)縦延伸/横延伸後に絶て緩和、横緩和を実施する。
(1)縦延伸
上記の条件(縦横比、温度、倍率)で実施することが好ましい。
(2)横延伸
上記の条件(横延伸ゾーン長/予熱ゾーン長、延伸前熱処理温度)で予熱、延伸前熱処理を行うことが好ましい。この後、上記条件(拡幅角、温度、倍率)で横延伸する。この後、上記条件で延伸後熱処理を行う。
(3)緩和
縦延伸、横延伸の一方または両方の後に上述の方法で縦緩和、横緩和を実施することが好ましい。
このようにして得られた延伸フィルムの厚みは30μm〜200μmが好ましく、より好ましくは40μm〜150μm、さらに好ましくは50μm〜120μmである
このようにして得た透明熱可塑性フィルム単独で使用してもよく、これらと偏光板と組み合わせて使用してもよく、これらの上に液晶層や屈折率を制御した層(低反射層)やハードコート層を設けて使用しても良い。これらは以下の工程により達成できる。
1.表面処理
(1)セルロースアシレートフィルム
表面処理を行うことによって、各機能層(例えば、下塗層およびバック層)との接着の向上させることができる。例えばグロー放電処理、紫外線照射処理、コロナ処理、火炎処理、酸またはアルカリ処理を行うことができる。ここでいうグロー放電処理とは、10-3〜20Torrの低圧ガス下でおこる低温プラズマでもよく、さらにまた大気圧下でのプラズマ処理も好ましい。プラズマ励起性気体とは上記のような条件においてプラズマ励起される気体をいい、アルゴン、ヘリウム、ネオン、クリプトン、キセノン、窒素、二酸化炭素、テトラフルオロメタンの様なフロン類およびそれらの混合物などが挙げられる。これらについては、詳細が発明協会公開技報(公技番号 2001−1745、2001年3月15日発行、発明協会)の30頁〜32頁に詳細に記載されている。なお、近年注目されている大気圧でのプラズマ処理は、例えば、10〜1000Kev下で20〜500Kgyの照射エネルギーが用いられ、好ましくは30〜500Kev下で20〜300Kgyの照射エネルギーが用いられる。
これらの中でも特に好ましくは、アルカリ鹸化処理である。
塗布方法の場合、ディップコーティング法、カーテンコーティング法、エクストルージョンコーティング法、バーコーティング法およびE型塗布法を用いることができる。アルカリ鹸化処理塗布液の溶媒は、鹸化液の透明支持体に対して塗布するために濡れ性が良く、また鹸化液溶媒によって透明支持体表面に凹凸を形成させずに、面状を良好なまま保つ溶媒を選択することが好ましい。具体的には、アルコール系溶媒が好ましく、イソプロピルアルコールが特に好ましい。また、界面活性剤の水溶液を溶媒として使用することもできる。アルカリ鹸化塗布液のアルカリは、上記溶媒に溶解するアルカリが好ましく、KOH、NaOHがさらに好ましい。鹸化塗布液のpHは10以上が好ましく、12以上がさらに好ましい。アルカリ鹸化時の反応条件は、室温で1秒〜5分が好ましく、5秒〜5分がさらに好ましく、20秒〜3分が特に好ましい。アルカリ鹸化反応後、鹸化液塗布面を水洗あるいは酸で洗浄したあと水洗することが好ましい。また、塗布式鹸化処理と後述の配向膜解塗設を、連続して行うことができ、工程数を減少できる。これらの鹸化方法は、具体的には、例えば、特開2002−82226号公報、国際公開WO02/46809号パンフレットに内容の記載が挙げられる。
機能層との接着のため下塗り層を設けることも好ましい。この層は上記表面処理をした後、塗設しても良く、表面処理なしで塗設しても良い。下塗層についての詳細は、発明協会公開技報(公技番号 2001−1745、2001年3月15日発行、発明協会)の32頁に記載されている。
これらの表面処理、下塗り工程は、製膜工程の最後に組み込むこともでき、単独で実施することもでき、後述の機能層付与工程の中で実施することもできる。
飽和ノルボルネンフィルム等の透明熱可塑性フィルムは、グロー放電処理、紫外線照射処理、コロナ処理、火炎処理、酸またはアルカリ処理を用いることができる。ここでいうグロー放電処理とは、10-3〜20Torrの低圧ガス下でおこる低温プラズマでもよく、さらにまた大気圧下でのプラズマ処理も好ましい。プラズマ励起性気体とは上記のような条件においてプラズマ励起される気体をいい、アルゴン、ヘリウム、ネオン、クリプトン、キセノン、窒素、二酸化炭素、テトラフルオロメタンの様なフロン類およびそれらの混合物などがあげられる。これらについては、詳細が発明協会公開技報(公技番号 2001−1745、2001年3月15日発行、発明協会)の30頁〜32頁に詳細に記載されている。なお、近年注目されている大気圧でのプラズマ処理は、例えば10〜1000Kev下で20〜500Kgyの照射エネルギーが用いられ、好ましくは30〜500Kev下で20〜300Kgyの照射エネルギーが用いられる。
これらの中でも特に好ましくは、グロー放電処理、コロナ処理、火炎処理である。
機能層との接着のため下塗り層を設けることも好ましい。この層は上記表面処理をした後、塗設しても良く、表面処理なしで塗設しても良い。下塗層についての詳細は、発明協会公開技報(公技番号 2001−1745、2001年3月15日発行、発明協会)の32頁に記載されている。
これらの表面処理、下塗り工程は、製膜工程の最後に組み込むこともでき、単独で実施することもでき、後述の機能層付与工程の中で実施することもできる。
本発明の透明熱可塑性フィルムに、発明協会公開技報(公技番号 2001−1745、2001年3月15日発行、発明協会)の32頁〜45頁に詳細に記載されている機能性層を組み合わせることが好ましい。中でも好ましいのが、偏光層の付与(偏光板)、光学補償層の付与(光学補償シート)、反射防止層の付与(反射防止フィルム)である。
(1)偏光層の付与(偏光板の作製)
(1−1)使用素材
現在、市販の偏光層は、延伸したポリマーを、浴槽中のヨウ素もしくは二色性色素の溶液に浸漬し、バインダー中にヨウ素、もしくは二色性色素を浸透させることで作製されるのが一般的である。偏光子は、Optiva Inc.に代表される塗布型偏光子も利用できる。偏光子におけるヨウ素および二色性色素は、バインダー中で配向することで偏向性能を発現する。二色性色素としては、アゾ系色素、スチルベン系色素、ピラゾロン系色素、トリフェニルメタン系色素、キノリン系色素、オキサジン系色素、チアジン系色素あるいはアントラキノン系色素が用いられる。二色性色素は、水溶性であることが好ましい。二色性色素は、親水性置換基(例えば、スルホ基、アミノ基、ヒドロキシル基)を有することが好ましい。例えば、発明協会公開技法、公技番号2001−1745号、58頁(発行日2001年3月15日)に記載の化合物が挙げられる。
バインダー厚みの下限は、10μmであることが好ましい。厚みの上限は、液晶表示装置の光漏れの観点からは、薄ければ薄い程よい。現在市販の偏光板(約30μm)以下であることが好ましく、25μm以下が好ましく、20μm以下がさらに好ましい。
偏光子のバインダーは架橋していてもよい。架橋性の官能基を有するポリマー、モノマーをバインダー中に混合しても良く、バインダーポリマー自身に架橋性官能基を付与しても良い。架橋は、光、熱あるいはpH変化により行うことができ、架橋構造をもったバインダーを形成することができる。架橋剤については、米国再発行特許23297号明細書に記載がある。また、ホウ素化合物(例えば、ホウ酸、硼砂)も、架橋剤として用いることができる。バインダーの架橋剤の添加量は、バインダーに対して、0.1〜20質量%が好ましい。偏光素子の配向性、偏光子の耐湿熱性が良好となる。
架橋反応が終了後でも、未反応の架橋剤は1.0質量%以下であることが好ましく、0.5質量%以下であることがさらに好ましい。このようにすることで、耐候性が向上する。
偏光子は、偏光子を延伸するか(延伸法)、もしくはラビングした(ラビング法)後に、ヨウ素、二色性染料で染色することが好ましい。
延伸法の場合、延伸倍率は2.5〜30.0倍が好ましく、3.0〜10.0倍がさらに好ましい。延伸は、空気中でのドライ延伸で実施できる。また、水に浸漬した状態でのウェット延伸を実施してもよい。ドライ延伸の延伸倍率は、2.5〜5.0倍が好ましく、ウェット延伸の延伸倍率は、3.0〜10.0倍が好ましい。延伸はMD方向に平行に行っても良く(平行延伸)、斜め方向におこなっても良い(斜め延伸)。これらの延伸は、1回で行っても、数回に分けて行ってもよい。数回に分けることによって、高倍率延伸でもより均一に延伸することができる。
a)平行延伸法
延伸に先立ち、PVAフィルムを膨潤させる。膨潤度は1.2〜2.0倍(膨潤前と膨潤後の重量比)である。この後、ガイドロール等を介して連続搬送しつつ、水系媒体浴内や二色性物質溶解の染色浴内で、15〜50℃、就中17〜40℃の浴温で延伸する。延伸は2対のニップロールで把持し、後段のニップロールの搬送速度を前段のそれより大きくすることで達成できる。延伸倍率は、延伸後/初期状態の長さ比(以下同じ)に基づくが前記作用効果の点より好ましい延伸倍率は1.2〜3.5倍、就中1.5〜3.0倍である。この後、50℃から90℃において乾燥させて偏光子を得る。
これには特開2002−86554号公報に記載の斜め方向に傾斜め方向に張り出したテンターを用い延伸する方法を用いることができる。この延伸は空気中で延伸するため、事前に含水させて延伸しやすくすることが必用である。好ましい含水率は5%〜100%、より好ましくは10%〜100%である。
延伸時の温度は40℃〜90℃が好ましく、より好ましくは50℃〜80℃である。湿度は50%相対湿度(RH)〜100%RH以下が好ましく、より好ましくは70%RH〜100%RH、さらに好ましくは80%RH〜100%RH以下である。長手方向の進行速度は、1m/分以上が好ましく、より好ましくは3m/分以上である。
延伸の終了後、好ましくは50℃〜100℃、より好ましくは60℃〜90℃で、0.5分〜10分乾燥する。より好ましくは1分〜5分である。
このようにして得られた偏光子の吸収軸は10度〜80度が好ましく、より好ましくは30度〜60度であり、さらに好ましくは実質的に45度(例えば、40度〜50度)である。
上記表面処理後の透明熱可塑性フィルムと、延伸して調製した偏光層を貼り合わせ偏光板を調製する。張り合わせる方向は、透明熱可塑性フィルムの流延軸方向と偏光板の延伸軸方向が45度になるように行うのが好ましい。
貼り合わせの接着剤は特に限定されないが、ポリビニルアルコール(PVA)系樹脂(アセトアセチル基、スルホン酸基、カルボキシル基、オキシアルキレン基等の変性PVAを含む)やホウ素化合物水溶液等が挙げられ、中でもPVA系樹脂が好ましい。接着剤層厚みは乾燥後に0.01〜10μmが好ましく、0.05〜5μmが特に好ましい。
このようにして得た偏光板の光線透過率は高い方が好ましく、偏光度も高い方が好ましい。偏光板の透過率は、波長550nmの光において、30〜50%の範囲にあることが好ましく、35〜50%の範囲にあることがより好ましく、40〜50%の範囲にあることがさらに好ましい。偏光度は、波長550nmの光において、90〜100%の範囲にあることが好ましく、95〜100%の範囲にあることがより好ましく、99〜100%の範囲にあることがさらに好ましい。
さらに、このようにして得た偏光板はλ/4板と積層し、円偏光を作製することができる。この場合λ/4の遅相軸と偏光板の吸収軸を45度になるように積層する。この時、λ/4は特に限定されないが、より好ましくは低波長ほどレターデーションが小さくなるような波長依存性を有するものがより好ましい。さらには長手方向に対し20度〜70度傾いた吸収軸を有する偏光子、および液晶性化合物からなる光学異方性層から成るλ/4板を用いることが好ましい。
光学異方性層は、液晶表示装置の黒表示における液晶セル中の液晶化合物を補償するためのものであり、透明熱可塑性フィルムの上に配向膜を形成し、さらに光学異方性層を付与することで形成される。
(2−1)配向膜
上記表面処理した透明熱可塑性フィルム上に配向膜を設ける。この膜は、液晶性分子の配向方向を規定する機能を有する。しかし、液晶性化合物を配向後にその配向状態を固定してしまえば、配向膜はその役割を果たしているために、本発明の構成要素としては必ずしも必須のものではない。即ち、配向状態が固定された配向膜上の光学異方性層のみを偏光子上に転写して本発明の偏光板を作製することも可能である。
配向膜は、有機化合物(好ましくはポリマー)のラビング処理、無機化合物の斜方蒸着、マイクログルーブを有する層の形成、あるいはラングミュア・ブロジェット法(LB膜)による有機化合物(例えば、ω−トリコサン酸、ジオクタデシルメチルアンモニウムクロライド、ステアリル酸メチル)の累積のような手段で設けることができる。さらに、電場の付与、磁場の付与あるいは光照射により、配向機能が生じる配向膜も知られている。
配向膜は、ポリマーのラビング処理により形成することが好ましい。配向膜に使用するポリマーは、原則として、液晶性分子を配向させる機能のある分子構造を有する。
本発明では、液晶性分子を配向させる機能に加えて、架橋性官能基(例えば、二重結合)を有する側鎖を主鎖に結合させるか、あるいは、液晶性分子を配向させる機能を有する架橋性官能基を側鎖に導入することが好ましい。
液晶性分子を配向させる機能を有する側鎖は、一般に疎水性基を官能基として有する。具体的な官能基の種類は、液晶性分子の種類および必要とする配向状態に応じて決定する。例えば、変性ポリビニルアルコールの変性基としては、共重合変性、連鎖移動変性またはブロック重合変性により導入できる。変性基の例には、親水性基(カルボン酸基、スルホン酸基、ホスホン酸基、アミノ基、アンモニウム基、アミド基、チオール基等)、炭素数10〜100個の炭化水素基、フッ素原子置換の炭化水素基、チオエーテル基、重合性基(不飽和重合性基、エポキシ基、アジリニジル基等)、アルコキシシリル基(トリアルコキシ基、ジアルコキシ基、モノアルコキシ基)等が挙げられる。これらの変性ポリビニルアルコール化合物の具体例として、例えば特開2000−155216号公報の[0022]〜[0145]、同2002−62426号公報の[0018]〜[0022]に記載のもの等が挙げられる。
架橋性官能基を有する側鎖を配向膜ポリマーの主鎖に結合させるか、あるいは、液晶性分子を配向させる機能を有する側鎖に架橋性官能基を導入すると、配向膜のポリマーと光学異方性層に含まれる多官能モノマーとを共重合させることができる。その結果、多官能モノマーと多官能モノマーとの間だけではなく、配向膜ポリマーと配向膜ポリマーとの間、そして多官能モノマーと配向膜ポリマーとの間も共有結合で強固に結合される。従って、架橋性官能基を配向膜ポリマーに導入することで、光学補償シートの強度を著しく改善することができる。
架橋剤としては、アルデヒド、N−メチロール化合物、ジオキサン誘導体、カルボキシル基を活性化することにより作用する化合物、活性ビニル化合物、活性ハロゲン化合物、イソオキサゾールおよびジアルデヒド澱粉が含まれる。二種類以上の架橋剤を併用してもよい。具体的には、例えば特開2002−62426号公報明細書中の段落番号[0023]〜[0024]記載の化合物等が挙げられる。反応活性の高いアルデヒド、特にグルタルアルデヒドが好ましい。
架橋剤の添加量は、ポリマーに対して0.1〜20質量%が好ましく、0.5〜15質量%がさらに好ましい。配向膜に残存する未反応の架橋剤の量は、1.0質量%以下であることが好ましく、0.5質量%以下であることがさらに好ましい。このように調節することで、配向膜を液晶表示装置に長期使用、或は高温高湿の雰囲気下に長期間放置しても、レチキュレーション発生のない充分な耐久性が得られる。
配向膜は、基本的に、配向膜形成材料である上記ポリマー、架橋剤を含む透明支持体上に塗布した後、加熱乾燥(架橋させ)し、ラビング処理することにより形成することができる。架橋反応は、前記のように、透明支持体上に塗布した後、任意の時期に行って良い。ポリビニルアルコールのような水溶性ポリマーを配向膜形成材料として用いる場合には、塗布液は消泡作用のある有機溶媒(例えば、メタノール)と水の混合溶媒とすることが好ましい。その比率は質量比で水:メタノールが0:100〜99:1が好ましく、0:100〜91:9であることがさらに好ましい。これにより、泡の発生が抑えられ、配向膜、さらには光学異方層の層表面の欠陥が著しく減少する。
配向膜は、透明支持体上または上記下塗層上に設けられる。配向膜は、上記のようにポリマー層を架橋したのち、表面をラビング処理することにより得ることができる。
前記ラビング処理は、LCDの液晶配向処理工程として広く採用されている処理方法を適用することができる。即ち、配向膜の表面を、紙やガーゼ、フェルト、ゴムあるいはナイロン、ポリエステル繊維などを用いて一定方向に擦ることにより、配向を得る方法を用いることができる。一般的には、長さおよび太さが均一な繊維を平均的に植毛した布などを用いて数回程度ラビングを行うことにより実施される。
工業的に実施する場合、搬送している偏光層のついたフィルムに対し、回転するラビングロールを接触させることで達成するが、ラビングロールの真円度、円筒度、振れ(偏芯)はいずれも30μm以下であることが好ましい。ラビングロールへのフィルムのラップ角度は、0.1〜90°が好ましい。ただし、特開平8−160430号公報に記載されているように、360°以上巻き付けることで、安定なラビング処理を得ることもできる。フィルムの搬送速度は1〜100m/minが好ましい。ラビング角は0〜60°の範囲で適切なラビング角度を選択することが好ましい。液晶表示装置に使用する場合は、40〜50°が好ましい。45°が特に好ましい。
このようにして得た配向膜の膜厚は、0.1〜10μmの範囲にあることが好ましい。
光学異方性層に用いる液晶性分子には、棒状液晶性分子および円盤状液晶性分子が含まれる。棒状液晶性分子および円盤状液晶性分子は、高分子液晶でも低分子液晶でもよく、さらに、低分子液晶が架橋され液晶性を示さなくなったものも含まれる。
棒状液晶性分子としては、アゾメチン類、アゾキシ類、シアノビフェニル類、シアノフェニルエステル類、安息香酸エステル類、シクロヘキサンカルボン酸フェニルエステル類、シアノフェニルシクロヘキサン類、シアノ置換フェニルピリミジン類、アルコキシ置換フェニルピリミジン類、フェニルジオキサン類、トラン類およびアルケニルシクロヘキシルベンゾニトリル類が好ましく用いられる。
なお、棒状液晶性分子には、金属錯体も含まれる。また、棒状液晶性分子を繰り返し単位中に含む液晶ポリマーも、棒状液晶性分子として用いることができる。言い換えると、棒状液晶性分子は、(液晶)ポリマーと結合していてもよい。
棒状液晶性分子については、季刊化学総説第22巻液晶の化学(1994)日本化学会編の第4章、第7章および第11章、および液晶デバイスハンドブック日本学術振興会第142委員会編の第3章に記載がある。
棒状液晶性分子の複屈折率は、0.001〜0.7の範囲にあることが好ましい。
棒状液晶性分子は、その配向状態を固定するために、重合性基を有することが好ましい。重合性基は、ラジカル重合性不飽基或はカチオン重合性基が好ましく、具体的には、例えば特開2002−62427号公報の[0064]〜[0086]記載の重合性基、重合性液晶化合物が挙げられる。
円盤状(ディスコティック)液晶性分子には、C.Destradeらの研究報告、Mol.Cryst.71巻、111頁(1981年)に記載されているベンゼン誘導体、C.Destradeらの研究報告、Mol.Cryst.122巻、141頁(1985年)、Physics lett,A,78巻、82頁(1990)に記載されているトルキセン誘導体、B.Kohneらの研究報告、Angew.Chem.96巻、70頁(1984年)に記載されたシクロヘキサン誘導体およびJ.M.Lehnらの研究報告、J.Chem.Commun.,1794頁(1985年)、J.Zhangらの研究報告、J.Am.Chem.Soc.116巻、2655頁(1994年)に記載されているアザクラウン系やフェニルアセチレン系マクロサイクルが含まれる。
円盤状液晶性分子としては、分子中心の母核に対して、直鎖のアルキル基、アルコキシ基、置換ベンゾイルオキシ基が母核の側鎖として放射線状に置換した構造である液晶性を示す化合物も含まれる。分子または分子の集合体が、回転対称性を有し、一定の配向を付与できる化合物であることが好ましい。円盤状液晶性分子から形成する光学異方性層は、最終的に光学異方性層に含まれる化合物が円盤状液晶性分子である必要はなく、例えば、低分子の円盤状液晶性分子が熱や光で反応する基を有しており、結果的に熱、光で反応により重合または架橋し、高分子量化し液晶性を失った化合物も含まれる。円盤状液晶性分子の好ましい例は、特開平8−50206号公報に記載されている。また、円盤状液晶性分子の重合については、特開平8−27284公報に記載がある。
円盤状液晶性分子を重合により固定するためには、円盤状液晶性分子の円盤状コアに、置換基として重合性基を結合させる必要がある。円盤状コアと重合性基は、連結基を介して結合する化合物が好ましく、これにより重合反応においても配向状態を保つことができる。例えば、特開2000−155216号公報の[0151]〜[0168]に記載の化合物等が挙げられる。
偏光子側の円盤状液晶性分子の長軸の平均方向は、一般に円盤状液晶性分子あるいは配向膜の材料を選択することにより、またはラビング処理方法の選択することにより、調整することができる。また、表面側(空気側)の円盤状液晶性分子の長軸(円盤面)方向は、一般に円盤状液晶性分子あるいは円盤状液晶性分子と共に使用する添加剤の種類を選択することにより調整することができる。円盤状液晶性分子と共に使用する添加剤の例としては、可塑剤、界面活性剤、重合性モノマーおよびポリマーなどを挙げることができる。長軸の配向方向の変化の程度も、上記と同様に、液晶性分子と添加剤との選択により調整できる。
上記の液晶性分子と共に、可塑剤、界面活性剤、重合性モノマー等を併用して、塗工膜の均一性、膜の強度、液晶分子の配向性等を向上することができる。液晶性分子と相溶性を有し、液晶性分子の傾斜角の変化を与えられるか、あるいは配向を阻害しないことが好ましい。
重合性モノマーとしては、ラジカル重合性若しくはカチオン重合性の化合物が挙げられる。好ましくは、多官能性ラジカル重合性モノマーであり、上記の重合性基含有の液晶化合物と共重合性のものが好ましい。例えば、特開2002−296423号公報の[0018]〜[0020]に記載のものが挙げられる。上記化合物の添加量は、円盤状液晶性分子に対して一般に1〜50質量%の範囲にあり、5〜30質量%の範囲にあることが好ましい。
界面活性剤としては、従来公知の化合物が挙げられるが、特にフッ素系化合物が好ましい。具体的には、例えば特開2001−330725号公報の[0028]〜[0056]に記載の化合物が挙げられる。
円盤状液晶性分子とともに使用するポリマーは、円盤状液晶性分子に傾斜角の変化を与えられることが好ましい。
ポリマーの例としては、セルロースエステルを挙げることができる。セルロースエステルの好ましい例としては、特開2000−155216号公報の[0178]に記載のものが挙げられる。液晶性分子の配向を阻害しないように、上記ポリマーの添加量は、液晶性分子に対して0.1〜10質量%の範囲にあることが好ましく、0.1〜8質量%の範囲にあることがより好ましい。
円盤状液晶性分子のディスコティックネマティック液晶相−固相転移温度は、70〜300℃が好ましく、70〜170℃がさらに好ましい。
光学異方性層は、液晶性分子および必要に応じて後述の重合性開始剤や任意の成分を含む塗布液を、配向膜の上に塗布することで形成できる。
塗布液の調製に使用する溶媒としては、有機溶媒が好ましく用いられる。有機溶媒の例には、アミド(例えば、N,N−ジメチルホルムアミド)、スルホキシド(例えば、ジメチルスルホキシド)、ヘテロ環化合物(例えば、ピリジン)、炭化水素(例えば、ベンゼン、ヘキサン)、アルキルハライド(例えば、クロロホルム、ジクロロメタン、テトラクロロエタン)、エステル(例えば、酢酸メチル、酢酸ブチル)、ケトン(例えば、アセトン、メチルエチルケトン)、エーテル(例えば、テトラヒドロフラン、1,2−ジメトキシエタン)が含まれる。アルキルハライドおよびケトンが好ましい。二種類以上の有機溶媒を併用してもよい。
塗布液の塗布は、公知の方法(例えば、ワイヤーバーコーティング法、押し出しコーティング法、ダイレクトグラビアコーティング法、リバースグラビアコーティング法、ダイコーティング法)により実施できる。
光学異方性層の厚さは、0.1〜20μmであることが好ましく、0.5〜15μmであることがさらに好ましく、1〜10μmであることが最も好ましい。
配向させた液晶性分子を、配向状態を維持して固定することができる。固定化は、重合反応により実施することが好ましい。重合反応には、熱重合開始剤を用いる熱重合反応と光重合開始剤を用いる光重合反応とが含まれる。光重合反応が好ましい。
光重合開始剤の例には、α−カルボニル化合物(米国特許2367661号、同2367670号の各明細書記載)、アシロインエーテル(米国特許2448828号明細書記載)、α−炭化水素置換芳香族アシロイン化合物(米国特許2722512号明細書記載)、多核キノン化合物(米国特許3046127号、同2951758号の各明細書記載)、トリアリールイミダゾールダイマーとp−アミノフェニルケトンとの組み合わせ(米国特許3549367号明細書記載)、アクリジンおよびフェナジン化合物(特開昭60−105667号公報、米国特許4239850号明細書記載)およびオキサジアゾール化合物(米国特許4212970号明細書記載)が含まれる。
光重合開始剤の使用量は、塗布液の固形分の0.01〜20質量%の範囲にあることが好ましく、0.5〜5質量%の範囲にあることがさらに好ましい。
液晶性分子の重合のための光照射は、紫外線を用いることが好ましい。
照射エネルギーは、20mJ/cm2〜50J/cm2 の範囲にあることが好ましく、20〜5000mJ/cm2 の範囲にあることがより好ましく、100〜800mJ/cm2 の範囲にあることがさらに好ましい。また、光重合反応を促進するため、加熱条件下で光照射を実施してもよい。
保護層を、光学異方性層の上に設けてもよい。
偏光層と光学補償層の傾斜角度は、LCDを構成する液晶セルの両側に貼り合わされる2枚の偏光板の透過軸と液晶セルの縦または横方向のなす角度にあわせるように延伸することが好ましい。通常の傾斜角度は45°である。しかし、最近は、透過型、反射型および半透過型LCDにおいて必ずしも45°でない装置が開発されており、延伸方向はLCDの設計にあわせて任意に調整できることが好ましい。
このような光学補償フィルムが用いられる各液晶モードについて説明する。
(TNモード液晶表示装置)
カラーTFT液晶表示装置として最も多く利用されており、多数の文献に記載がある。TNモードの黒表示における液晶セル中の配向状態は、セル中央部で棒状液晶性分子が立ち上がり、セルの基板近傍では棒状液晶性分子が寝た配向状態にある。
(OCBモード液晶表示装置)
棒状液晶性分子を液晶セルの上部と下部とで実質的に逆の方向に(対称的に)配向させるベンド配向モードの液晶セルである。ベンド配向モードの液晶セルを用いた液晶表示装置は、米国特許4583825号、同5410422号の各明細書に開示されている。棒状液晶性分子が液晶セルの上部と下部とで対称的に配向しているため、ベンド配向モードの液晶セルは、自己光学補償機能を有する。そのため、この液晶モードは、OCB(Optically Compensatory Bend) 液晶モードとも呼ばれる。
OCBモードの液晶セルもTNモード同様、黒表示においては、液晶セル中の配向状態は、セル中央部で棒状液晶性分子が立ち上がり、セルの基板近傍では棒状液晶性分子が寝た配向状態にある。
(VAモード液晶表示装置)
電圧無印加時に棒状液晶性分子が実質的に垂直に配向しているのが特徴であり、VAモードの液晶セルには、(1)棒状液晶性分子を電圧無印加時に実質的に垂直に配向させ、電圧印加時に実質的に水平に配向させる狭義のVAモードの液晶セル(特開平2−176625号公報記載)に加えて、(2)視野角拡大のため、VAモードをマルチドメイン化した(MVAモードの)液晶セル(SID97、Digest of tech. Papers(予稿集)28(1997)845記載)、(3)棒状液晶性分子を電圧無印加時に実質的に垂直配向させ、電圧印加時にねじれマルチドメイン配向させるモード(n−ASMモード)の液晶セル(日本液晶討論会の予稿集58〜59(1998)記載)および(4)SURVAIVALモードの液晶セル(LCDインターナショナル98で発表)が含まれる。
(IPSモード液晶表示装置)
電圧無印加時に棒状液晶性分子が実質的に面内に水平に配向しているのが特徴であり、これが電圧印加の有無で液晶の配向方向を変えることでスイッチングするのが特徴である。具体的には特開2004−365941号、特開2004−12731号、特開2004−215620号、特開2002−221726号、特開2002−55341号、特開2003−195333号各公報に記載のものなどを使用できる。
(その他液晶表示装置)
ECBモードおよびSTNモードに対しても、上記と同様の考え方で光学的に補償することができる。
反射防止膜は、一般に、防汚性層でもある低屈折率層、および低屈折率層より高い屈折率を有する少なくとも一層の層(即ち、高屈折率層、中屈折率層)とを透明基体上に設けて成る。
屈折率の異なる無機化合物(金属酸化物等)の透明薄膜を積層させた多層膜として、化学蒸着(CVD)法や物理蒸着(PVD)法、金属アルコキシド等の金属化合物のゾルゲル方法でコロイド状金属酸化物粒子皮膜を形成後に後処理(紫外線照射:特開平9−157855号公報、プラズマ処理:特開2002−327310号公報)して薄膜を形成する方法が挙げられる。
一方、生産性が高い反射防止膜として、無機粒子をマトリックスに分散されてなる薄膜を積層塗布してなる反射防止膜が各種提案されている。
上述したような塗布による反射防止フィルムに最上層表面が微細な凹凸の形状を有する防眩性を付与した反射防止層から成る反射防止フィルムも挙げられる。
本発明の透明熱可塑性フィルムは上記いずれの方式にも適用できるが、特に好ましいのが塗布による方式(塗布型)である。
基体上に少なくとも中屈折率層、高屈折率層、低屈折率層(最外層)の順序の層構成から成る反射防止膜は、以下の関係を満足する屈折率を有する様に設計される。
高屈折率層の屈折率>中屈折率層の屈折率>透明支持体の屈折率>低屈折率層の屈折率
また、透明支持体と中屈折率層の間に、ハードコート層を設けてもよい。さらには、中屈折率ハードコート層、高屈折率層および低屈折率層からなってもよい。
例えば、特開平8−122504号公報、同8−110401号公報、同10−300902号公報、特開2002−243906号公報、特開2000−111706号公報等が挙げられる。 また、各層に他の機能を付与させてもよく、例えば、防汚性の低屈折率層、帯電防止性の高屈折率層としたもの(例えば、特開平10−206603号公報、特開2002−243906号公報等)等が挙げられる。
反射防止膜のヘイズは、5%以下あることが好ましく、3%以下がさらに好ましい。又膜の強度は、JIS K5400に従う鉛筆硬度試験でH以上であることが好ましく、2H以上であることがさらに好ましく、3H以上であることが最も好ましい。
反射防止膜の高い屈折率を有する層は、平均粒子サイズ100nm以下の高屈折率の無機化合物超微粒子およびマトリックスバインダーを少なくとも含有する硬化性膜から成る。
高屈折率の無機化合物微粒子としては、屈折率1.65以上の無機化合物が挙げられ、好ましくは屈折率1.9以上のものが挙げられる。例えば、Ti、Zn、Sb、Sn、Zr、Ce、Ta、La、In等の酸化物、これらの金属原子を含む複合酸化物等が挙げられる。
このような超微粒子とするには、粒子表面が表面処理剤で処理されること(例えば、シランカップリング剤等:特開平11−295503号公報、同11−153703号公報、特開2000−9908、アニオン性化合物或は有機金属カップリング剤:特開2001−310432号公報等)、高屈折率粒子をコアとしたコアシェル構造とすること:特開2001−166104等)、特定の分散剤併用(例えば、特開平11−153703号公報、特許番号US6210858B1、特開2002−2776069号公報等)等が挙げられる。
マトリックスを形成する材料としては、従来公知の熱可塑性樹脂、硬化性樹脂皮膜等が挙げられる。
さらに、ラジカル重合性および/またはカチオン重合性の重合性基を少なくとも2個以上含有の多官能性化合物含有組成物、加水分解性基を含有の有機金属化合物およびその部分縮合体組成物から選ばれる少なくとも1種の組成物が好ましい。例えば、特開2000−47004号公報、同2001−315242号公報、同2001−31871号公報、同2001−296401号公報等に記載の化合物が挙げられる。
また、金属アルコキドの加水分解縮合物から得られるコロイド状金属酸化物と金属アルコキシド組成物から得られる硬化性膜も好ましい。例えば、特開2001−293818号公報等に記載されている。
高屈折率層の屈折率は、−般に1.70〜2.20である。高屈折率層の厚さは、5nm〜10μmであることが好ましく、10nm〜1μmであることがさらに好ましい。
中屈折率層の屈折率は、低屈折率層の屈折率と高屈折率層の屈折率との間の値となるように調整する。中屈折率層の屈折率は、1.50〜1.70であることが好ましい。
低屈折率層は、高屈折率層の上に順次積層して成る。低屈折率層の屈折率は1.20〜1.55である。好ましくは1.30〜1.50である。
耐擦傷性、防汚性を有する最外層として構築することが好ましい。耐擦傷性を大きく向上させる手段として表面への滑り性付与が有効で、従来公知のシリコーンの導入、フッ素の導入等から成る薄膜層の手段を適用できる。
含フッ素化合物の屈折率は1.35〜1.50であることが好ましい。より好ましくは1.36〜1.47である。また、含フッ素化合物はフッ素原子を35〜80質量%の範囲で含む架橋性若しくは重合性の官能基を含む化合物が好ましい。
例えば、特開平9−222503号公報の[0018]〜[0026]、同11−38202号公報の[0019]〜[0030]、特開2001−40284号公報の[0027]〜[0028]、特開2000−284102号公報等に記載の化合物が挙げられる。
シリコーン化合物としてはポリシロキサン構造を有する化合物であり、高分子鎖中に硬化性官能基あるいは重合性官能基を含有して、膜中で橋かけ構造を有するものが好ましい。例えば、反応性シリコーン(例えば、サイラプレーン(チッソ(株)製等)、両末端にシラノール基含有のポリシロキサン(特開平11−258403号公報等)等が挙げられる。
架橋または重合性基を有する含フッ素および/またはシロキサンのポリマーの架橋または重合反応は、重合開始剤、増感剤等を含有する最外層を形成するための塗布組成物を塗布と同時または塗布後に光照射や加熱することにより実施することが好ましい。
また、シランカップリング剤等の有機金属化合物と特定のフッ素含有炭化水素基含有のシランカップリング剤とを触媒共存下に縮合反応で硬化するゾルゲル硬化膜も好ましい。
例えば、ポリフルオロアルキル基含有シラン化合物またはその部分加水分解縮合物(特開昭58−142958号公報、同58−147483号公報、同58−147484号公報、特開平9−157582号公報、同11−106704号公報記載等記載の化合物)、フッ素含有長鎖基であるポリ「パーフルオロアルキルエーテル」基を含有するシリル化合物(特開2000−117902号公報、同2001−48590号公報、同2002−53804号公報記載の化合物等)等が挙げられる。
低屈折率層は、上記以外の添加剤として充填剤(例えば、二酸化珪素(シリカ)、含フッ素粒子(フッ化マグネシウム、フッ化カルシウム、フッ化バリウム)等の一次粒子平均径が1〜150nmの低屈折率無機化合物、特開平11−3820公報の[0020]〜[0038]に記載の有機微粒子等)、シランカップリング剤、滑り剤、界面活性剤等を含有することができる。
低屈折率層が最外層の下層に位置する場合、低屈折率層は気相法(真空蒸着法、スパッタリング法、イオンプレーティング法、プラズマCVD法等)により形成されても良い。安価に製造できる点で、塗布法が好ましい。
低屈折率層の膜厚は、30〜200nmであることが好ましく、50〜150nmであることがより好ましく、60〜120nmであることがさらに好ましい。
ハードコート層は、反射防止フィルムに物理強度を付与するために、透明支持体の表面に設ける。特に、透明支持体と前記高屈折率層の間に設けることが好ましい。
ハードコート層は、光および/または熱の硬化性化合物の架橋反応、または、重合反応により形成されることが好ましい。 硬化性官能基としては、光重合性官能基が好ましく、又加水分解性官能基含有の有機金属化合物は有機アルコキシシリル化合物が好ましい。
これらの化合物の具体例としては、高屈折率層で例示したと同様のものが挙げられる。
ハードコート層の具体的な構成組成物としては、例えば、特開2002−144913号公報、同2000−9908号公報、国際公開WO0/46617号パンフレット等記載のものが挙げられる。
高屈折率層はハードコート層を兼ねることができる。このような場合、高屈折率層で記載した手法を用いて微粒子を微細に分散してハードコート層に含有させて形成することが好ましい。
ハードコート層は、平均粒子サイズ0.2〜10μmの粒子を含有させて防眩機能(アンチグレア機能)を付与した防眩層(後述)を兼ねることもできる。
ハードコート層の膜厚は用途により適切に設計することができる。ハードコート層の膜厚は、0.2〜10μmであることが好ましく、より好ましくは0.5〜7μmである。
ハードコート層の強度は、JIS K5400に従う鉛筆硬度試験で、H以上であることが好ましく、2H以上であることがより好ましく、3H以上であることがさらに好ましい。また、JIS K5400に従うテーバー試験で、試験前後の試験片の摩耗量が少ないほど好ましい。
前方散乱層は、液晶表示装置に適用した場合の、上下左右方向に視角を傾斜させたときの視野角改良効果を付与するために設ける。上記ハードコート層中に屈折率の異なる微粒子を分散することで、ハードコート機能と兼ねることもできる。
例えば、前方散乱係数を特定化した特開11−38208号公報、透明樹脂と微粒子の相対屈折率を特定範囲とした特開2000−199809号公報、ヘイズ値を40%以上と規定した特開2002−107512号公報等が挙げられる。
(3−6)その他の層
上記の層以外に、プライマー層、帯電防止層、下塗り層や保護層等を設けてもよい。
(3−7)塗布方法
反射防止フィルムの各層は、ディップコート法、エアーナイフコート法、カーテンコート法、ローラーコート法、ワイヤーバーコート法、グラビアコート、マイクログラビア法やエクストルージョンコート法(米国特許2681294号明細書)により、塗布により形成することができる。
(3−8)アンチグレア機能
反射防止膜は、外光を散乱させるアンチグレア機能を有していてもよい。アンチグレア機能は、反射防止膜の表面に凹凸を形成することにより得られる。反射防止膜がアンチグレア機能を有する場合、反射防止膜のヘイズは、3〜30%であることが好ましく、5〜20%であることがさらに好ましく、7〜20%であることが最も好ましい。
反射防止膜表面に凹凸を形成する方法は、これらの表面形状を充分に保持できる方法であればいずれの方法でも適用できる。例えば、低屈折率層中に微粒子を使用して膜表面に凹凸を形成する方法(例えば、特開2000−271878号公報等)、低屈折率層の下層(高屈折率層、中屈折率層またはハードコート層)に比較的大きな粒子(例えば、粒子サイズ0.05〜2μm)を少量(例えば、0.1〜50質量%)添加して表面凹凸膜を形成し、その上にこれらの形状を維持して低屈折率層を設ける方法(例えば、特開2000−281410号公報、同2000−95893号公報、同2001−100004号公報、同2001−281407号公報等)、最上層(防汚性層)を、塗設後の表面に物理的に凹凸形状を転写する方法(例えば、エンボス加工方法として、特開昭63−278839号公報、特開平11−183710号公報、特開2000−275401号公報等記載)等が挙げられる。
実開平3−110418号公報、特開平5−119216号公報、特開平5−162261号公報、特開平5−182518号公報、特開平5−19115号公報、特開平5−196819号公報、特開平5−264811号公報、特開平5−281411号公報、特開平5−281417号公報、特開平5−281537号公報、特開平5−288921号公報、特開平5−288923号公報、特開平5−311119号公報、特開平5−339395号公報、特開平5−40204号公報、特開平5−45512号公報、特開平6−109922号公報、特開平6−123805号公報、特開平6−160626号公報、特開平6−214107号公報、特開平6−214108号公報、特開平6−214109号公報、特開平6−222209号公報、特開平6−222353号公報、特開平6−234175号公報、特開平6−235810号公報、特開平6−258520号公報、特開平6−264030号公報、特開平6−305270号公報、特開平6−331826号公報、特開平6−347641号公報、特開平6−75110号公報、特開平6−75111号公報、特開平6−82779号公報、特開平6−93133号公報、特開平7−104126号公報、特開平7−134212号公報、特開平7−181322号公報、特開平7−188383号公報、特開平7−230086号公報、特開平7−290652号公報、特開平7−294903号公報、特開平7−294904号公報、特開平7−294905号公報、特開平7−325219号公報、特開平7−56014号公報、特開平7−56017号公報、特開平7−92321号公報、特開平8−122525号公報、特開平8−146220号公報、特開平8−171016号公報、特開平8−188661号公報、特開平8−21999号公報、特開平8−240712号公報、特開平8−25575号公報、特開平8−286179号公報、特開平8−292322号公報、特開平8−297211号公報、特開平8−304624号公報、特開平8−313881号公報、特開平8−43812号公報、特開平8−62419号公報、特開平8−62422号公報、特開平8−76112号公報、特開平8−94834号公報、特開平9−137143号公報、特開平9−197127号公報、特開平9−251110号公報、特開平9−258023号公報、特開平9−269413号公報、特開平9−269414号公報、特開平9−281483号公報、特開平9−288212号公報、特開平9−288213号公報、特開平9−292525号公報、特開平9−292526号公報、特開平9−294959号公報、特開平9−318817号公報、特開平9−80233号公報、特開平10−10320号公報、特開平10−104428号公報、特開平10−111403号公報、特開平10−111507号公報、特開平10−123302号公報、特開平10−123322号公報、特開平10−123323号公報、特開平10−176118号公報、特開平10−186133号公報、特開平10−264322号公報、特開平10−268133号公報、特開平10−268134号公報、特開平10−319408号公報、特開平10−332933号公報、特開平10−39137号公報、特開平10−39140号公報、特開平10−68821号公報、特開平10−68824号公報、特開平10−90517号公報、特開平11−116903号公報、特開平11−181131号公報、特開平11−211901号公報、特開平11−211914号公報、特開平11−242119号公報、特開平11−246693号公報、特開平11−246694号公報、特開平11−256117号公報、特開平11−258425号公報、特開平11−263861号公報、特開平11−287902号公報、特開平11−295525号公報、特開平11−295527号公報、特開平11−302423号公報、特開平11−309830号公報、特開平11−323552号公報、特開平11−335641号公報、特開平11−344700号公報、特開平11−349947号公報、特開平11−95011号公報、特開平11−95030号公報、特開平11−95208号公報、特開2000−109780号公報、特開2000−110070号公報、特開2000−119657号公報、特開2000−141556号公報、特開2000−147208号公報、特開2000−17099号公報、特開2000−171603号公報、特開2000−171618号公報、特開2000−180615号公報、特開2000−187102号公報、特開2000−187106号公報、特開2000−191819号公報、特開2000−191821号公報、特開2000−193804号公報、特開2000−204189号公報、特開2000−206306号公報、特開2000−214323号公報、特開2000−214329号公報、特開2000−230159号公報、特開2000−235107号公報、特開2000−241626号公報、特開2000−250038号公報、特開2000−267095号公報、特開2000−284122号公報、特開2000−304927号公報、特開2000−304928号公報、特開2000−304929号公報、特開2000−309195号公報、特開2000−309196号公報、特開2000−309198号公報、特開2000−309642号公報、特開2000−310704号公報、特開2000−310708号公報、特開2000−310709号公報、特開2000−310710号公報、特開2000−310711号公報、特開2000−310712号公報、特開2000−310713号公報、特開2000−310714号公報、特開2000−310715号公報、特開2000−310716号公報、特開2000−310717号公報、特開2000−321560号公報、特開2000−321567号公報、特開2000−338309号公報、特開2000−338329号公報、特開2000−344905号公報、特開2000−347016号公報、特開2000−347017号公報、特開2000−347026号公報、特開2000−347027号公報、特開2000−347029号公報、特開2000−347030号公報、特開2000−347031号公報、特開2000−347032号公報、特開2000−347033号公報、特開2000−347034号公報、特開2000−347035号公報、特開2000−347037号公報、特開2000−347038号公報、特開2000−86989号公報、特開2000−98392号公報、特開2001−100012号公報、特開2001−108805号公報、特開2001−108806号公報、特開2001−133627号公報、特開2001−133628号公報、特開2001−142062号公報、特開2001−142072号公報、特開2001−174630号公報、特開2001−174634号公報、特開2001−174637号公報、特開2001−179902号公報、特開2001−183526号公報、特開2001−188103号公報、特開2001−188124号公報、特開2001−188125号公報、特開2001−188225号公報、特開2001−188231号公報、特開2001−194505号公報、特開2001−228311号公報、特開2001−228333号公報、特開2001−242461号公報、特開2001−242546号公報、特開2001−247834号公報、特開2001−26061号公報、特開2001−264517号公報、特開2001−272535号公報、特開2001−278924号公報、特開2001−2797号公報、特開2001−287308号公報、特開2001−305345号公報、特開2001−311827号公報、特開2001−350005号公報、特開2001−356207号公報、特開2001−356213号公報、特開2001−42122号公報、特開2001−42323号公報、特開2001−42325号公報、特開2001−4819号公報、特開2001−4829号公報、特開2001−4830号公報、特開2001−4831号公報、特開2001−4832号公報、特開2001−4834号公報、特開2001−4835号公報、特開2001−4836号公報、特開2001−4838号公報、特開2001−4839号公報、特開2001−51118号公報、特開2001−51119号公報、特開2001−51120号公報、特開2001−51273号公報、特開2001−51274号公報、特開2001−55573号公報、特開2001−66431号公報、特開2001−66597号公報、特開2001−74920号公報、特開2001−81469号公報、特開2001−83329号公報、特開2001−83515号公報、特開2002−162628号公報、特開2002−169024号公報、特開2002−189421号公報、特開2002−201367号公報、特開2002−20410号公報、特開2002−258046号公報、特開2002−275391号公報、特開2002−294174号公報、特開2002−311214号公報、特開2002−311246号公報、特開2002−328233号公報、特開2002−338703号公報、特開2002−363266号公報、特開2002−365164号公報、特開2002−370303号公報、特開2002−40209号公報、特開2002−48917号公報、特開2002−6109号公報、特開2002−71950号公報、特開2003−105540号公報、特開2003−114331号公報、特開2003−131036号公報、特開2003−139952号公報、特開2003−172819号公報、特開2003−35819号公報、特開2003−43252号公報、特開2003−50318号公報、特開2003−96066号公報、特開2006−45501号公報、特開2006−45502号公報、特開2006−45499号公報、特開2006−45500号公報、特開2006−182008号公報、特開2006−241433号公報、特開2006−348123号公報、特開2005−325258、特開2006−2026、特開2006−2025、特開2006−183005号公報、特開2006−183004号公報、特開2006−143873号公報、特開2006−257204号公報、特開2006−205472号公報、特開2006−241428号公報、特開2006−251746号公報、特開2007−1198号公報、特開2007−1238号公報、国際公開WO2005/103122号公報、特開2006−176736号公報、特開2006−243688号公報、特開2006−327105号公報、特開2006−124642号公報、特開2006−205708号公報、特開2006−341443号公報、特開2006−199913号公報、特開2006−335050号公報、特開2007−8154号公報、特開2006−334840号公報、特開2006−341450号公報、特開2006−327162号公報、特開2006−341510号公報、特開2006−327161号公報、特開2006−327107号公報、特開2006−327160号公報、特開2006−328316号公報、特開2006−334839号公報、特開2007−8151号公報、特開2007−1286号公報、特開2006−327106号公報、特開2006−334841号公報、特開2006−334842号公報、特開2005−330411号公報、特開2006−116945号公報、特開2005−301225号公報、特開2007−1287号公報、特開2006−348268号公報、国際公開WO2006/132367号パンフレット、国際公開WO20
06/132367号パンフレット、特開2005−178194号公報、特開2006−336004号公報、特開2006−249418号公報、特開2007−2216号公報、特開2006−28345号公報、特開2006−215535号公報、特開2006−28387号公報、特開2007−2215号公報、特開2006−343479号公報、特開2006−263992号公報、特開2000−352620号公報、特開2005−088578号公報、特開2005−300978号公報、特開2005−342929号公報、特開2006−021459号公報、特開2006−030425号公報、特開2006−036840号公報、特開2006−045306号公報、特開2006−045307号公報、特開2006−058825号公報、特開2006−063169号公報、特開2006−77067号公報、特開2006−77113号公報、特開2006−82261号公報、特開2006−91035号公報、特開2006−91078号公報、特開2006−104374号公報、特開2006−106247号公報、特開2006−111796号公報、特開2006−111797号公報、特開2006−113175号公報、特開2006−113551号公報、特開2006−113567号公報、特開2006−116904号公報、特開2006−117714号公報、特開2006−119182号公報、特開2006−119183号公報、特開2006−123513号公報、特開2006−123177号公報、特開2006−124629号公報、特開2006−137821号公報、特開2006−142800号公報、特開2006−163033号公報、特開2006−163034号公報、特開2006−171404号公報、特開2006−178020号公報、特開2006−182020号公報、特開2006−182865号公報、特開2006−188663号公報、特開2006−195407号公報、特開2006−208934号公報、特開2006−219615号公報、特開2006−220814号公報、特開2006−224589号公報、特開2006−249221号公報、特開2006−256082号公報、特開2006−272616号公報、特開2006−290929号公報、特開2006−293201号公報、特開2006−301500号公報、特開2006−301592号公報。
一辺30cmの正方形内の熱収縮分布
本発明では、「一辺30cmの正方形内部」として、一辺30cmの正方形の中から5cm×15cmサンプルを、下記のように切り出したものを採用した。
30cmの一辺(A辺)に平行に15cm長をとったサンプルを6枚
A辺と直交するもう一辺(B辺)に平行に15cm長をとったサンプルを6枚
これらのサンプルを25℃60%RHで5時間以上調湿後、10cm基長のピンゲージを用い測長した(L1)。これを80℃の恒温槽にて無張力で500時間放置(サーモ処理)した。恒温槽から取り出した後、25℃60%RHで5時間以上調湿後、10cm基長のピンゲージを用い測長した(L2)。下記式で各サンプルの熱収縮率を測定する。
熱収縮率(%)=100×(L1−L2)/L1
12点に亘って測定した熱収縮率のうち最大値(M)と最小値(S)と12点の平均値(A)から下記式で熱収縮分布を求める。
熱収縮分布(%)=100×(M−S)/A
一辺30cmの正方形の中から1cm×15cmサンプルを、下記のように切り出した。
30cmの一辺(A辺)に平行に15cm長をとったサンプルを30枚
A辺と直交するもう一辺(B辺)に平行に15cm長をとったサンプルを30枚
これらを25℃60%RHで5時間以上調湿後、チャック間10cmで10mm/分の引張り速度で引張り試験を行ない弾性率を測定した。
60点に亘って測定した弾性率のうち最大値(M)と最小値(S)と60点の平均値(A)から下記式で熱収縮分布を求める。
弾性率分布(%)=100×(M−S)/A
延伸後の透明熱可塑性フィルムを全幅にわたって等間隔で50点、製膜方向に平行にサンプルを切り出し、製膜方向と直交する辺がサンプルホルダーと平行になるようにセットした。これらのサンプルの自動複屈折計(KOBRA−21ADH、王子計測機器(株))を用い25℃60%RHで遅相軸を測定し、配向角とした。
(4)Re、Rth
サンプルフィルムを25℃60%RHに5時間以上調湿後、自動複屈折計(KOBRA−21ADH:王子計測器(株)製)を用いて、25℃60%RHにおいて、サンプルフィルム表面に対し垂直方向および、フィルム面法線から±40°傾斜させて方向から波長550nmにおけるレターデーション値を測定する。垂直方向から面内のレターデーション(Re)、垂直方向、±40°方向の測定値から厚み方向のレターデーション(Rth)を算出する。これらをRe、Rthとする。
(5)セルロースアシレートの置換度
セルロースアシレートのアシル置換度は、Carbohydr.Res.273(1995)83−91(手塚他)に記載の方法で13C−NMRにより求めた。
(6)残留溶剤
サンプルフィルム300mgを酢酸メチル30mlに溶解したもの(サンプルA)、およびジクロロメタン30mlに溶解したもの(サンプルB)を作製する。
次いで、これらをガスクロマトグラフィー(GC)を用い、下記条件で測定する。
カラム:DB−WAX(0.25mmφ×30m、膜厚0.25μm)
カラム温度:50℃
キャリアーガス:窒素
分析時間:15分間
サンプル注入量:1μml
下記方法で溶剤量を求めることができる。
サンプルAで溶剤(酢酸メチル)以外の各ピークについて検量線を用い含率を求め、その総和をSaとする。サンプルBで、サンプルAにおいて溶剤ピークで隠れていた領域の各ピークについて検量線を用い含率を求め、その総和をSbとし、SaとSbの和を残留溶剤量とする。
1.セルロースアシレート樹脂
(1)セルロースアセテートプロピオネート(CAP)の合成
セルロース(広葉樹パルプ)150重量部、酢酸75重量部を、還流装置を付けた反応容器に取り、60℃に加熱しながら2時間激しく攪拌した。このような前処理を行ったセルロースは膨潤、解砕されてフラッフ状を呈した。反応容器を2℃の氷水浴に30分間置き冷却した。
別途、アシル化剤としてプロピオン酸無水物1410重量部、硫酸10.5重量部の混合物を作製し、−30℃に冷却した後に、上記の前処理を行ったセルロースを収容する反応容器に一度に加えた。30分経過後、外設温度を徐々に上昇させ、アシル化剤の添加から2時間経過後に内温が25℃になるように調節した。反応容器を5℃の氷水浴にて冷却し、アシル化剤の添加から0.5時間後に内温が10℃、2時間後に内温が23℃になるように調節し、内温を23℃に保ってさらに3時間攪拌した。反応容器を5℃の氷水浴にて冷却し、5℃に冷却した25質量%含水酢酸120重量部を1時間かけて添加した。内温を40℃に上昇させ、2時間攪拌した(熟成)。次いで反応容器に、50質量%含水酢酸に酢酸マグネシウム4水和物を硫酸の2倍モル溶解した溶液を添加し、30分間攪拌した。25質量%含水酢酸1000重量部、33質量%含水酢酸500重量部、50質量%含水酢酸1000重量部、水1000重量部をこの順に加え、セルロースアセテートプロピオネートを沈殿させた。得られたセルロースアセテートプロピオネートの沈殿は温水にて洗浄を行った。洗浄後、20℃の0.005質量%水酸化カルシウム水溶液中で0.5時間攪拌し、洗浄液のpHが7になるまで、さらに水で洗浄を行った後、70℃で真空乾燥させた。
NMRおよび、GPC測定によれば、得られたセルロースアセテートプロピオネートは、アセチル(Ac)化度0.45、プロピオニル(Pr)化度2.45、重合度160であった。なお組成は上記のNMR法で測定し、重合度はGPCを用いて下記のように測定した。
THFを溶離液として、単分散ポリスチレンを標準分子量として、数平均分子量(Mn)を求める。NMRで求めた組成から1セグメントあたりの分子量を分子量(m)を求める。Mnをmで割り、重合度(数平均重合度:DPn)を求める。
セルロース(綿花リンター)100重量部、酢酸135重量部を還流装置を付けた反応容器に取り、60℃に加熱しながら、1時間放置した。その後、60℃で加熱しながら1時間激しく攪拌した。このような前処理を行ったセルロースは膨潤、解砕されてフラッフ状を呈した。反応容器を5℃の氷水浴に1時間置き、セルロースを十分に冷却した。
別途、アシル化剤として酪酸無水物1080重量部、硫酸10.0重量部の混合物を作製し、−20℃に冷却した後に、前処理を行ったセルロースを収容する反応容器に一度に加えた。30分経過後、外設温度を20℃まで上昇させ、5時間反応させた。反応容器を5℃の氷水浴にて冷却し、約5℃に冷却した12.5質量%含水酢酸2400重量部を1時間かけて添加した。内温を30℃に上昇させ1.5時間攪拌した(熟成)。次いで反応容器に、酢酸マグネシウム4水和物の50質量%水溶液を100重量部添加し、30分間攪拌した。酢酸1000重量部、50質量%含水酢酸2500重量部を徐々に加え、セルロースアセテートブチレートを沈殿させた。得られたセルロースアセテートブチレートの沈殿は温水にて洗浄を行った。洗浄後、0.005質量%水酸化カルシウム水溶液中で0.5時間攪拌し、さらに、洗浄液のpHが7になるまで水で洗浄を行った後、70℃で乾燥させた。得られたセルロースアセテートブチレートはアセチル(Ac)化度0.84、ブチリル(Bu)化度2.12、重合度180であった。
アシル化剤の種類、量を変えることで置換度を変え、熟成時間を変えることで重合度を変え、表1記載のCAP、CAB以外のセルロースアシレートを合成した。
また、置換もしくは無置換の芳香族アシル基を結合したセルロースアシレートとして、特開2002−32201号公報の実施例1に準じて安息香酸と酢酸でエステル化したセルロースアシレートを合成した。但し、原料のセルロースアシレートを2.0置換、2.45置換のセルロースアセテートを用いた。この結果、酢酸置換度が2.0、安息香酸置換度が1.0、および酢酸置換度が2.45、安息香酸置換度が0.55の芳香族アシル基置換セルロースアシレートを得た。
これ以外に特開2002−32201号公報の実施例2〜7に記載の芳香族アシル基を結合したセルロースアシレートを合成、製膜、延伸したが、いずれも上記安息香酸置換したセルロースアシレートと同様に良好な結果を得た。
(1)セルロースアシレートのペレット化
上記セルロースアシレート100重量部、安定剤(住友化学(株)製スミライザーGP)0.3重量部、二酸化珪素部粒子(アエロジルR972V)0.05重量部、紫外線吸収剤(2−(2'−ヒドロキシー3',5−ジ−tert−ブチルフェニル)−ベンゾトリアゾール0.05重量部、2,4−ヒドロキシ−4−メトキシ−ベンゾフェノン0.1重量部)を混合した。
これらを100℃で3時間乾燥し含水率を0.1質量%以下にした後、2軸混練機を用い180℃で溶融した後、60℃の温水中に押し出しストランドとした後裁断し、直径3mm長さ5mmの円柱状のペレットに成形した。
上記方法で調製したセルロースアシレートペレットを、露点温度−40℃の脱湿風を用いて100℃で5時間乾燥し含水率を0.01質量%以下にした。これを80℃のホッパーに投入し、180℃(入口温度)から230℃(出口温度)に調整した溶融押出し機で溶融した。なお、これに用いたスクリューの直径は60mm、L/D=50、圧縮比4であった。溶融押出機から押出された樹脂はギアポンプで一定量計量され送り出されるが、この時ギアポンプ前の樹脂圧力が10MPaの一定圧力で制御できる様に、押出機の回転数を変更させた。ギアポンプから送り出されたメルト樹脂は濾過精度5μmmのリーフディスクフィルターにて濾過し、スタティックミキサーを経由してスリット間隔0.8mm、230℃のハンガーコートダイから、Tg−5℃、Tg℃、Tg−10℃の設定した3連のキャストロール上に押し出し、最上流側のキャストロールに表1記載の条件でタッチロールを接触させ、未延伸フィルムを製膜した。なお、タッチロールは特開平11−235747の実施例1に記載のもの(二重抑えロールと記載のあるもの)を用い、Tg−5℃に調温した(但し薄肉金属外筒厚みは3mmとした)。
固化したメルトをキャスティングドラムから剥ぎ取り、巻き取り直前に両端(全幅の各5%)をトリミングした後、両端に幅10mm、高さ50μmの厚みだし加工(ナーリング)をつけた後、30m/分で幅1.5m、長さ3000mの未延伸フィルムを得た。このTgはDSCを用いて下記方法で測定した。
(Tg測定)DSCの測定パンにサンプルを20mg入れる。これを窒素気流中で、10℃/分で30℃から250℃まで昇温した後(1st−run)、30℃まで−10℃/分で冷却する。この後、再度30℃〜250℃まで昇温する(2nd−run)。2nd−runでベースラインが低温側から偏奇し始める温度をガラス転移温度(Tg)とした。
表1記載の全水準のセルロースアシレート樹脂に可塑剤(トリフェニルフォスファイト/ビフェニル・ジフェニル・フォスフェート=重量比3/1)を12質量%(対樹脂)混合したものを、ジクロロメタン90質量%、メタノール5質量%、エタノール5質量%の混合溶剤に溶解し、バンド状に流延、剥ぎ取り後に乾燥を行ない、残留溶剤を0.1〜0.3質量%、厚み80μmおよび40μmのセルロースアシレートフィルムを調製した。これらについても表1記載の溶融製膜フィルムと同じ条件で延伸を行ったところ、本発明を実施したものは同様に良好な結果が得られた。
(1)縦(MD)延伸
2対のニップロールを用い、表1に記載の縦/横比(L/W)で、Tg+15℃で表1記載の倍率に縦延伸した。縦延伸後に縦緩和を行った。縦延伸後の縦緩和は、縦延伸のニップロール直後に配置した搬送ロールの速度を遅くすることで実施した。
(2)横(TD)延伸
縦延伸、縦緩和後に予熱ゾーンで横延伸前加熱を表1記載の温度で実施した。この時の予熱ゾーン長(L1)と横延伸ゾーン長(L2)の比(L2/L1)を表1に記載した。
予熱後、テンターを用いてTg+10℃で表1に記載の倍率、拡幅率で横方向に延伸した。この後、横延伸後熱処理を表1記載の温度で実施した。さらに、横延伸後熱処理の後、テンター幅を縮めることで横緩和した。さらにテンターから出た後低張力(3kg/m)で搬送しながら縦緩和を行った。これらの緩和はいずれも表1記載の温度で行ない、緩和率の総和を表1に記載した。
このようにして延伸したものの残留溶剤を上記の方法で測定したが、いずれも0%であった。このようにして得た延伸フィルの、30cmの正方形の熱収縮分布、弾性率分布、配向角、Re、Rth、残留溶剤を上記の方法で測定し、表1に記載した。
表面処理
延伸後のセルロースアシレートフィルムを下記の浸漬法で鹸化を行った。なお下記塗布鹸化も実施したが浸漬鹸化と同様の結果を得た。
(1)浸漬鹸化
NaOHの1.5規定水溶液を鹸化液として用いた。これを60℃に調温し、セルロースアシレートフィルムを2分間浸漬した。この後、0.1Nの硫酸水溶液に30秒浸漬した後、水洗浴を通した。
(2)塗布鹸化
イソプロパノール80重量部に水20重量部を加え、これにKOHを1.5規定となるように溶解し、これを60℃に調温したものを鹸化液として用いた。これを60℃のセルロースアシレートフィルム上に10g/m2塗布し、1分間鹸化した。この後、50℃の温水をスプレーを用い、10l/m2・分で1分間吹きかけ洗浄した。
特開2001−141926号公報の実施例1に従い、2対のニップロール間に周速差を与え、長手方向に延伸したで厚み20μmの偏光層を調製した。
このようにして得た偏光層を、上記方法で製膜、延伸、鹸化処理したセルロースアシレートフィルムを用い、下記構成となるようにPVA((株)クラレ製PVA−117H)3%水溶液を接着剤とし貼り合せ偏光板を作製した。なお、下記に記載したフジタック(富士写真フィルム製TD80)も上記の方法で鹸化処理を行った。
偏光板A:延伸セルロースアシレート/偏光層/フジタック
偏光板B:延伸セルロースアシレート/偏光層/未延伸セルロースアシレート
(偏光板Bでは延伸、未延伸セルロースアシレートは同じ種類のセルロースアシレートを用いた)
このようにして得た偏光板を、延伸セルロースアシレートを液晶側になるようにして、特開2000−154261号公報の図4〜9に記載の20インチVA型液晶表示装置液晶表示装置に取り付けた。これを全面黒表示とした状態で30℃の環境内に2時間設置した後、瞬時に10℃の環境内に移し3時間放置した後、目視で黒表示中のむら(光漏れ)の領域で計測し、表示部全面積に対する割合を表1に示した。本発明を実施したものは良好な性能が得られた。
一方、本発明の範囲外のものは、光学特性が低下した。特に、特開2005−300978号公報の実施例1に準じたもの(表1の比較例3)に比べ本発明は液晶表示むらが大幅に改善された。一方これに近い条件で本発明を実施したもの(表1の実施例40)は良好な性能を得た。
特開平11−316378号公報の実施例1の液晶層を塗布したセルロースアセテートフィルムの代わりに、本発明の延伸セルロースアシレートフィルムを使用した。これを上記と同様の方法で30℃から10℃に移した時のむら(光漏れ)を計測した(全体に占めるむらの発生領域を%で示した)。本発明を実施したものは良好な性能が得られた。
特開平7−333433号公報の実施例1の液晶層を塗布したセルロースアセテートフィルムに代わって、本発明の延伸セルロースアシレートフィルムに変更し光学補償フィルターフィルムを作製したものでも同様に良好な光学補償フィルムを作製できた。
本発明の延伸セルロースアシレートフィルムを発明協会公開技報(公技番号2001−1745)の実施例47に従い本発明の延伸セルロースアシレートフィルムを用いて低反射フィルムを作製したところ、良好な光学性能が得られた。
上記本発明の偏光板を、特開平10−48420号公報の実施例1に記載の液晶表示装置、特開平9−26572号公報の実施例1に記載のディスコティック液晶分子を含む光学的異方性層、ポリビニルアルコールを塗布した配向膜、特開2000−154261号公報の図4〜9に記載の20インチVA型液晶表示装置、特開2000−154261号公報の図30〜15に記載の20インチOCB型液晶表示装置、特開2004−12731号公報の図31に記載のIPS型液晶表示装置に用いた。さらに、本発明の低反射フィルムをこれらの液晶表示装置の最表層に貼り評価を行ったところ、良好な液晶表示素子を得た。
1.飽和ノルボルネン樹脂
(1)飽和ノルボルネン樹脂−A
6−メチル−1,4,5,8−ジメタノ−1,4,4a,5,6,7,8,8a−オクタヒドロナフタレンに、重合触媒としてトリエチルアルミニウムの15%シクロヘキサン溶液10部、トリエチルアミン5部、および四塩化チタンの20%シクロヘキサン溶液10部を添加して、シクロヘキサン中で開環重合し、得られた開環重合体をニッケル触媒で水素添加してポリマー溶液を得た。このポリマー溶液をイソプロピルアルコール中で凝固させ、乾燥し、粉末状の樹脂を得た。この樹脂の数平均分子量(Mn)は40,000、重量平均分子量(Mw)は、80000、水素添加率は99.8%以上、Tgは139℃であった。
8−メチル−8−メトキシカルボニルテトラシクロ[4.4.0.12.5,17.10]−3−ドデセン(特定単量体B)100部と、5−(4−ビフェニルカルボニルオキシ)ビシクロ[2.2.1]ヘプト−2−エン(特定単量体A)150部と、1−ヘキセン(分子量調節剤)18部と、トルエン750部とを窒素置換した反応容器に仕込み、この溶液を60℃に加熱した。次いで、反応容器内の溶液に、重合触媒としてトリエチルアルミニウム(1.5モル/l)のトルエン溶液0.62部と、t−ブタノールおよびメタノールで変性した六塩化タングステン(t−ブタノール:メタノール:タングステン=0.35モル:0.3モル:1モル)のトルエン溶液(濃度0.05モル/l)3.7部とを添加し、この系を80℃で3時間加熱攪拌することにより開環重合反応させて開環重合体溶液を得た。この重合反応における重合転化率は97%であり、得られた開環重合体について、30℃のクロロホルム中で測定した固有粘度(ηinh)は0.65dl/gであった。
このようにして得られた開環重合体溶液4,000部をオートクレーブに仕込み、この開環重合体溶液に、RuHCl(CO)[P(C6 H5 )3 ]3 0.48部を添加し、水素ガス圧100kg/cm2、反応温度165℃の条件下で、3時間加熱攪拌して水素添加反応を行った。得られた反応溶液(水素添加重合体溶液)を冷却した後、水素ガスを放圧した。この反応溶液を大量のメタノール中に注いで凝固物を分離回収し、これを乾燥して、水素添加重合体(特定の環状ポリオレフィン系樹脂)を得た。このようにして得られた水素添加重合体について400MHz、1H−NMRを用いてオレフィン性不飽和結合の水素添加率を測定したところ99.9%であった。GPC法(溶媒:テトラヒドロフラン)によりポリスチレン換算の数平均分子量(Mn)および重量平均分子量(Mw)を測定したところ、数平均分子量(Mn)は39,000、重量平均分子量(Mw)は126,000、分子量分布(Mw/Mn)は3.23であった。
特開2005−330465号公報の実施例2に記載の飽和ノルボルネン化合物(Tg:127℃)を採用した。該樹脂の数平均分子量は23000、重量平均分子量は48000である。
(4)飽和ノルボルネン樹脂−D
特表平8−507800号公報の実施例1に記載の飽和ノルボルネン化合物(Tg:181℃)を採用した。該樹脂の数平均分子量は28000、重量平均分子量は60000である。
(5)飽和ノルボルネン樹脂−E
三井化学(株)製APL6015T(Tg:145℃)を採用した。該樹脂の数平均分子量は25000、重量平均分子量は53000である。
(6)飽和ノルボルネン樹脂−F
ポリプラスチックス(株)製TOPAS6013(Tg:130℃)を採用した。該樹脂の数平均分子量は20000、重量平均分子量は41000である。
(7)飽和ノルボルネン樹脂−G
特許第3693803号公報の実施例1に記載の飽和ノルボルネン化合物(Tg:140℃)を採用した。該樹脂の数平均分子量は21500、重量平均分子量は45000である。
上記飽和ノルボルネン樹脂−A〜Gを直径3mm長さ5mmの円柱状のペレットに成形した。これを110℃の真空乾燥機で乾燥し、含水率を0.1%以下とした後、Tg−10℃になるように調整したホッパーに投入した。
溶融粘度が1000Pa・sとなるように溶融温度を調整し、この温度で5分間かけて1軸混練機を用い溶融した後、溶融温度より10℃高く設定したT−ダイからTg−5℃に設定したキャスティングドラム上に流延し固化しフィルムとした。この時、表2記載の条件でタッチロール製膜を実施した。固化したメルトを剥ぎ取り、巻き取った。なお、巻き取り直前に両端(全幅の各3%)をトリミングした後、両端に幅10mm、高さ50μmの厚みだし加工(ナーリング)をつけた。各水準とも、幅は1.5mで30m/分で3000m巻き取った。
上記溶融製膜で得た飽和ノルボルネンフィルムを実施例1と同様にして表2に記載の条件で縦延伸、横延伸、緩和処理を行なった。
このようにして製膜、延伸したものの残留溶剤は、いずれも0%であった。さらに上記の方法で熱収縮分布、弾性率分布、Re、Rthを測定し表1に記載した。
表面処理
いずれの水準も、表面の水との接触角が60度になるように、フィルム表面にコロナ処理を行った。
特開2001−141926号公報の実施例1に従い、2対のニップロール間に周速差を与え、長手方向に延伸したで厚み20μmの偏光層を調製した。
実施例−Aと同様にして下記偏光板を作製した。
偏光板A:延伸飽和ノルボルネン/偏光層/フジタック
このようにして得た偏光板を、実施例−Aと同様にしてVA型液晶表示装置液晶表示装置に取り付け30℃から10℃の環境内に移した際の黒表示中のむら(光漏れ)を計測し表2に示した。本発明を実施したものは良好な性能が得られた。
実施例−Aと同様にして光学補償フィルムを作製した。本発明を実施したものは良好な性能が得られた。
実施例−Aと同様にして低反射フィルムを作製したところ、本発明を実施したものは良好な光学性能が得られた。
実施例−Aと同様にして液晶表示素子を作製した。本発明を実施したものは、良好な液晶表示素子を得た。
Claims (13)
- 予熱ゾーン及び横延伸ゾーンが少なくともこの順で配置され、予熱ゾーンで熱処理を行い、前記熱処理後、横延伸ゾーンで横延伸を行うことを含む透明熱可塑性フィルムの製造方法であって、
横延伸温度をT1としたとき、前記熱処理は、T1+6℃〜T1+40℃であり、
横延伸ゾーン長(L2)と予熱ゾーン長(L1)の比(L2/L1)が0.5〜30の範囲で、1%〜200%横延伸することを含む、透明熱可塑性フィルムの製造方法。 - 横延伸ゾーン内の拡幅角を5度〜45度とすることを含む、請求項1に記載の透明熱可塑性フィルムの製造方法。
- 横延伸温度をT1としたとき、横延伸後にT1−50℃〜T1−2℃で熱処理することを含む、請求項1または2に記載の透明熱可塑性フィルムの製造方法。
- 縦/横比(L/W)が0.01を越え0.3未満の範囲、又は2を超え50以下で、1%〜100%に縦延伸することを含む、請求項1〜3のいずれか1項に記載の透明熱可塑性フィルムの製造方法:ただし、Lはニップロール間でフィルムがニップロールに接触していない部分の長さであり、Wは、延伸前のフィルムの幅である。
- 縦延伸後および横延伸後の少なくとも一方において、前記透明熱可塑性フィルムのガラス転移温度をTgとしたとき、Tg−30℃〜Tg+30℃の温度範囲において、縦方向および横方向の緩和量の合計が1%〜20%となるように緩和することを含む、請求項1〜4のいずれか1項に記載の透明熱可塑性フィルムの製造方法。
- タッチロールを用いて溶融製膜することを含む、請求項1〜5のいずれか1項に記載の透明熱可塑性フィルムの製造方法。
- 予熱ゾーン及び横延伸ゾーンが少なくともこの順で配置され、予熱ゾーンで熱処理を行い、前記熱処理後、横延伸ゾーンで横延伸を行うことで得られる透明熱可塑性フィルムであって、横延伸温度をT1としたとき、前記熱処理は、T1+6℃〜T1+40℃であり、横延伸ゾーン長(L2)と予熱ゾーン長(L1)の比(L2/L1)が0.5〜30の範囲で、1%〜200%横延伸してなる透明熱可塑性フィルム。
- 請求項7に記載の透明熱可塑性フィルムであって、該フィルムの幅全域に亘り配向角が0°±5°以内、または90°±5°以内である、透明熱可塑性フィルム。
- 面内のレターデーション(Re)が20nm〜400nm、厚み方向のレターデーション(Rth)が50nm〜400nmである、請求項7又は8に記載の透明熱可塑性フィルム。
- 下記式を満足するセルロースアシレートを含む、請求項7〜9のいずれか1項に記載の透明熱可塑性フィルム。
2.0≦A+B<3.0
0.1≦B<3
A:アセテート基の置換度、
B:プロピオネート基、ブチレート基、ペンタノイル基の置換度の総和 - 下記式(T−1)および(T−2)を満たす組成を有するセルロースアシレートからなることを特徴とする請求項7〜10のいずれか1項に記載の熱可塑性フィルム。
式(T−1):2.5≦A+C<3.0
式(T−2):0.1≦C<2
(式中、Aは、アセテート基の置換度を示し、Cは置換もしくは無置換の芳香族アシル基を示す。) - 飽和ノルボルネン樹脂を含む、請求項7〜11のいずれか1項に記載の透明熱可塑性フィルム。
- 残留溶剤が0.01質量%以下である、請求項7〜12のいずれか1項に記載の透明熱可塑性フィルム。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007088851A JP5184806B2 (ja) | 2006-04-11 | 2007-03-29 | 透明熱可塑性フィルムの製造方法および透明熱可塑性フィルム |
| US11/783,501 US20070286998A1 (en) | 2006-04-11 | 2007-04-10 | Method of producing transparent thermoplastic film and transparent thermoplastic film |
| US12/638,548 US20100090364A1 (en) | 2006-04-11 | 2009-12-15 | Method of producing transparent thermoplastic film and transparent thermoplastic film |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006108931 | 2006-04-11 | ||
| JP2006108931 | 2006-04-11 | ||
| JP2007088851A JP5184806B2 (ja) | 2006-04-11 | 2007-03-29 | 透明熱可塑性フィルムの製造方法および透明熱可塑性フィルム |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2007301978A JP2007301978A (ja) | 2007-11-22 |
| JP5184806B2 true JP5184806B2 (ja) | 2013-04-17 |
Family
ID=38822343
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007088851A Active JP5184806B2 (ja) | 2006-04-11 | 2007-03-29 | 透明熱可塑性フィルムの製造方法および透明熱可塑性フィルム |
Country Status (2)
| Country | Link |
|---|---|
| US (2) | US20070286998A1 (ja) |
| JP (1) | JP5184806B2 (ja) |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT1793187E (pt) * | 2004-09-21 | 2012-03-05 | Joan Vives Iglesias | Processo e máquina destinados à sinterização e/ou secagem de materiais em pó utilizando uma radiação infravermelha |
| JP5015705B2 (ja) * | 2007-09-18 | 2012-08-29 | ルネサスエレクトロニクス株式会社 | 層間絶縁膜形成方法、層間絶縁膜、半導体デバイス、および半導体製造装置 |
| JP4964794B2 (ja) * | 2008-01-22 | 2012-07-04 | 富士フイルム株式会社 | 光学フィルムおよびその製造方法 |
| JPWO2009119328A1 (ja) * | 2008-03-27 | 2011-07-21 | コニカミノルタオプト株式会社 | 光学フィルムの製造方法及び光学フィルム |
| JP5366426B2 (ja) * | 2008-04-04 | 2013-12-11 | 東芝機械株式会社 | 多孔性フィルムの製膜方法及び多孔性フィルム製膜用の逐次二軸延伸装置 |
| JP5906879B2 (ja) * | 2012-03-27 | 2016-04-20 | 日本ゼオン株式会社 | 位相差板の製造方法 |
| KR101555782B1 (ko) * | 2013-06-18 | 2015-09-24 | 주식회사 엘지화학 | 박형 편광판 및 그의 제조 방법 |
| CN104395791B (zh) * | 2013-06-18 | 2018-03-13 | Lg化学株式会社 | 薄偏光板及其制造方法 |
| JP6527505B2 (ja) * | 2014-03-31 | 2019-06-05 | 株式会社クラレ | 二軸延伸フィルムの製造方法 |
| US10611106B2 (en) * | 2015-02-15 | 2020-04-07 | Roger Wen Yi Hsu | Methods and systems for making an optical functional film |
| US10675598B2 (en) * | 2015-03-24 | 2020-06-09 | South Dakota Board Of Regents | High shear thin film machine for dispersion and simultaneous orientation-distribution of nanoparticles within polymer matrix |
| KR102043492B1 (ko) * | 2016-05-02 | 2019-11-11 | 주식회사 엘지화학 | 광학 필름의 제조 방법, 이를 이용하여 제조되는 광학 부재 및 광학 필름, 이를 포함하는 편광판 및 액정표시장치 |
| CN111670105B (zh) * | 2018-02-02 | 2022-06-07 | 三菱化学株式会社 | 三维造型用材料、三维造型用细丝、该细丝的卷绕体和三维打印机用盒 |
| WO2020105810A1 (ko) * | 2018-11-19 | 2020-05-28 | 효성화학 주식회사 | 셀룰로오스 에스테르 위상차 필름 |
| DE102018131830A1 (de) * | 2018-12-11 | 2020-06-18 | Reifenhäuser GmbH & Co. KG Maschinenfabrik | Folienanlage und verfahren zum herstellen einer folienbahn sowie verwendung einer folienmaterialschmelze verarbeitenden walzeneinrichtung |
| CN111605173A (zh) * | 2020-04-24 | 2020-09-01 | 张家刘 | 一种耐高温塑料袋及其制备方法 |
| CN114654709B (zh) * | 2022-03-23 | 2024-02-06 | 四川绵阳四0四医院 | 一种用于固定头颈肩部放疗的热塑膜预拉伸辅助装置 |
| CN114714608B (zh) * | 2022-03-23 | 2024-02-09 | 四川绵阳四0四医院 | 一种用于对头部放疗的热塑膜自动预成型装置 |
| CN117021527B (zh) * | 2023-07-31 | 2025-10-03 | 四川东方绝缘材料股份有限公司 | 一种厚型双向拉伸液晶聚合物薄膜的制备方法 |
Family Cites Families (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3794547A (en) * | 1965-01-28 | 1974-02-26 | Unitika Ltd | Polyamide films |
| GB1572463A (en) * | 1976-04-15 | 1980-07-30 | Agfa Gevaert | Flatness control of thermoplastic film |
| TW384327B (en) * | 1996-08-26 | 2000-03-11 | Nippon Petrochemicals Co Ltd | Web expansion apparatus |
| JPH10249933A (ja) * | 1997-03-10 | 1998-09-22 | Fuji Photo Film Co Ltd | 横延伸熱可塑性ポリマーフィルムの製造方法 |
| US5885501A (en) * | 1997-06-24 | 1999-03-23 | E. I. Du Pont De Nemours And Company | Process for preparing dimensionally stabilized biaxially stretched thermoplastic film |
| US6045894A (en) * | 1998-01-13 | 2000-04-04 | 3M Innovative Properties Company | Clear to colored security film |
| US6179948B1 (en) * | 1998-01-13 | 2001-01-30 | 3M Innovative Properties Company | Optical film and process for manufacture thereof |
| JP2002036266A (ja) * | 2000-07-21 | 2002-02-05 | Konica Corp | フィルム製造方法、フィルムおよび偏光板用フィルム |
| TWI243264B (en) * | 2000-12-04 | 2005-11-11 | Fuji Photo Film Co Ltd | Optical compensating sheet and process for producing it, polarizing plate and liquid crystal display device |
| US6657690B2 (en) * | 2000-12-25 | 2003-12-02 | Fuji Photo Film Co., Ltd. | Optical compensatory sheet comprising optically uniaxial or biaxial transparent stretched film |
| JP4818531B2 (ja) * | 2001-05-07 | 2011-11-16 | 日東電工株式会社 | 配向フィルムの製造方法、偏光フィルム、偏光板および液晶表示装置 |
| JP4178810B2 (ja) * | 2001-12-25 | 2008-11-12 | Jsr株式会社 | 熱可塑性ノルボルネン系樹脂系光学用フィルム |
| JP4218305B2 (ja) * | 2002-10-21 | 2009-02-04 | コニカミノルタホールディングス株式会社 | ポリマーフィルムの製造方法及び位相差フィルム |
| US6936209B2 (en) * | 2002-11-27 | 2005-08-30 | 3M Innovative Properties Company | Methods and devices for processing polymer films |
| JP2005004096A (ja) * | 2003-06-13 | 2005-01-06 | Sekisui Chem Co Ltd | 位相差補償フィルム、複合偏光板、偏光板、液晶表示装置及び位相差補償フィルムの製造方法 |
| JP2005254812A (ja) * | 2004-02-12 | 2005-09-22 | Nippon Zeon Co Ltd | 熱可塑性ノルボルネン系樹脂からなる延伸フィルムの製造方法及び位相差フィルム |
| US7820301B2 (en) * | 2004-03-19 | 2010-10-26 | Fujifilm Corporation | Cellulose acylate film and method for producing the same |
| JP4577033B2 (ja) * | 2004-04-19 | 2010-11-10 | コニカミノルタオプト株式会社 | 位相差フィルムの製造方法 |
| JP4721655B2 (ja) * | 2004-05-21 | 2011-07-13 | 富士フイルム株式会社 | 飽和ノルボルネンフィルムおよびその製造方法 |
| JP4807939B2 (ja) * | 2004-05-21 | 2011-11-02 | 富士フイルム株式会社 | セルロースアシレートフィルムおよびその製造方法 |
| JP2006027263A (ja) * | 2004-06-16 | 2006-02-02 | Fuji Photo Film Co Ltd | ポリマーフィルム及び溶液製膜方法 |
| JP4626757B2 (ja) * | 2004-07-14 | 2011-02-09 | 富士フイルム株式会社 | 熱可塑性フィルム及びその製造方法 |
| JP4731143B2 (ja) * | 2004-09-17 | 2011-07-20 | 富士フイルム株式会社 | セルロースアシレートフイルム、光学補償フイルム、偏光板および液晶表示装置 |
| KR101260718B1 (ko) * | 2005-06-09 | 2013-05-06 | 도레이 카부시키가이샤 | 2축 연신 폴리에스테르 필름의 제조방법 |
| WO2006132367A1 (ja) * | 2005-06-10 | 2006-12-14 | Fujifilm Corporation | セルロースアシレートフィルムおよびその製造方法、偏光板、位相差フィルム、光学補償フィルム、反射防止フィルム、並びに液晶表示装置 |
| JP4779646B2 (ja) * | 2005-12-27 | 2011-09-28 | 日本ゼオン株式会社 | フィルム延伸装置およびフィルム延伸方法 |
-
2007
- 2007-03-29 JP JP2007088851A patent/JP5184806B2/ja active Active
- 2007-04-10 US US11/783,501 patent/US20070286998A1/en not_active Abandoned
-
2009
- 2009-12-15 US US12/638,548 patent/US20100090364A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| JP2007301978A (ja) | 2007-11-22 |
| US20100090364A1 (en) | 2010-04-15 |
| US20070286998A1 (en) | 2007-12-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5184806B2 (ja) | 透明熱可塑性フィルムの製造方法および透明熱可塑性フィルム | |
| JP5230221B2 (ja) | 熱可塑性フィルムおよびその製造方法 | |
| JP4975504B2 (ja) | 透明熱可塑性フィルムおよびその製造方法 | |
| US8043714B2 (en) | Transparent thermoplastic film and a method of producing the same | |
| US8529241B2 (en) | Method for manufacturing thermoplastic resin film, and optical compensation film and polarization plate for liquid crystal display panel | |
| JP5192753B2 (ja) | 熱可塑性樹脂フィルムの製造方法及び液晶表示板用光学補償フィルム及び偏光板 | |
| KR20070116732A (ko) | 열가소성 수지 필름과 그 제조 방법, 편광판, 광학 보상필름, 반사 방지 필름 및 액정 표시 장치 | |
| JP2007062334A (ja) | セルロースアシレート樹脂フィルム及びその製造方法 | |
| CN101472725A (zh) | 纤维素类树脂膜及其制造方法 | |
| JP2008055890A (ja) | 熱可塑性樹脂フィルムおよびその製造方法、並びに、偏光板、光学補償フィルム、反射防止フィルムおよび液晶表示装置 | |
| JP5189785B2 (ja) | 熱可塑性フィルムおよびその製造方法、並びに、偏光板、光学補償フィルム、反射防止フィルム、および、液晶表示装置 | |
| CN101489758B (zh) | 酰化纤维素薄膜及饱和降冰片烯系树脂薄膜以及它们的制造方法 | |
| JP2008023986A (ja) | 熱可塑性フィルムおよびその製造方法、偏光板、反射防止フィルム、並びに、液晶表示装置 | |
| JP5483804B2 (ja) | シクロオレフィン樹脂フィルム、およびこれらを用いた偏光板、光学補償フィルム、反射防止フィルム、液晶表示装置、ならびに、シクロオレフィン樹脂フィルムの製造方法 | |
| JP5186122B2 (ja) | 熱可塑性フイルムの製造方法 | |
| JP5184810B2 (ja) | 熱可塑性樹脂組成物、熱可塑性樹脂フィルムおよびその製造方法、偏光板、光学補償フィルム、反射防止フィルム、並びに液晶表示装置 | |
| JP2006327107A (ja) | 熱可塑性フィルムの製造方法 | |
| JP2006327161A (ja) | 熱可塑性フィルムの製造方法 | |
| JP2008265167A (ja) | 熱可塑性フイルム及びその製造方法、並びに、偏光板、液晶表示板用光学補償フイルム、反射防止フイルム及び液晶表示装置 | |
| JP2010120304A (ja) | ノルボルネン系樹脂フィルム、ノルボルネン系樹脂フィルムの製造方法、偏光版、液晶表示板用光学補償フィルム及び反射防止フィルム | |
| JP2008273059A (ja) | シクロオレフィン樹脂フィルムの製造方法、シクロオレフィン樹脂フィルム、該フィルムを用いた偏光板、光学補償フィルム、反射防止フィルム、液晶表示装置 | |
| JP2008272983A (ja) | シクロオレフィン樹脂フィルムの製造方法およびシクロオレフィン樹脂フィルム、ならびに、偏光板、光学補償フィルム、反射防止フィルムおよび液晶表示装置 | |
| JP2006249418A (ja) | セルロースアシレートフィルム、ならびにこれを用いた偏光板、位相差フィルム、光学補償フィルム、反射防止フィルムおよび画像表示装置 | |
| JP2008273057A (ja) | シクロオレフィン樹脂フィルム、およびこれらを用いた偏光板、光学補償フィルム、反射防止フィルム、液晶表示装置 | |
| JP2006327160A (ja) | 熱可塑性フィルムの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090911 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20111012 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20111018 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20111212 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120117 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120313 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20121225 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20130117 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 Ref document number: 5184806 Country of ref document: JP |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20160125 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |