JP5158366B2 - 含水素フルオロオレフィン化合物の製造方法 - Google Patents
含水素フルオロオレフィン化合物の製造方法 Download PDFInfo
- Publication number
- JP5158366B2 JP5158366B2 JP2008299953A JP2008299953A JP5158366B2 JP 5158366 B2 JP5158366 B2 JP 5158366B2 JP 2008299953 A JP2008299953 A JP 2008299953A JP 2008299953 A JP2008299953 A JP 2008299953A JP 5158366 B2 JP5158366 B2 JP 5158366B2
- Authority
- JP
- Japan
- Prior art keywords
- reaction
- trialkylphosphine
- solvent
- perfluorocycloolefin
- ether
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
特許文献1では、オクタフルオロシクロペンテンを貴金属触媒存在下に水素化してオクタフルオロシクロペンタンを得、続いて、アルカリ処理することによりヘプタフルオロシクロペンテンを得ている。
特許文献2では、1−ハロゲノ−または1,2−ジハロゲノ−ヘキサフルオロシクロペンテンと水素ガスを気相において、銅、鉄、クロムまたはニッケルを含む触媒存在下で還元して1−ハロゲノ−ヘキサフルオロシクロペンテンまたは3,3,4,4,5,5−ヘキサフルオロシクロペンテンを製造する方法が開示されている。具体的には、1位や2位に結合するハロゲンとして塩素原子である化合物を還元し、1位に塩素原子を有する化合物又は3,3,4,4,5,5−ヘキサフルオロシクロペンテンを得ているだけで、炭素原子とフッ素原子と水素原子とのみからなる1H−ペンタフルオロシクロペンテンを得てはいない。この方法で炭素原子とフッ素原子と水素原子とのみからなる1H−ペンタフルオロシクロペンテンを得るには反応温度を高温にする必要があるが、触媒の耐久性が乏しくなり、連続的に反応を継続することが困難である。
非特許文献1においては、ヘキサフルオロシクロブテンを金属水素化物で処理することにより、ペンタフルオロシクロブテンが得られている。また、非特許文献2においてはジクロロヘキサフルオロシクロブタンを水素化リチウムアルミニウムヒドリドにより還元させて得られるヘキサフルオロシクロブタンをアルカリ処理することによりペンタフルオロシクロブテンを得ている。
また、非特許文献3においては、ヘキサフルオロシクロブテンをエーテル中で、トリフェニルホスフィンと反応させると、ヘキサフルオロシクロブテン−ホスホニウムイリドが生成する旨の記載がなされている。非特許文献4においては、同様にヘキサフルオロシクロブテン−ホスホニウムイリドを生成し、ハロゲン(塩素、臭素、ヨウ素)を反応させると、1,1−ジハロヘキサフルオロシクロブタンが生成するとの記載がなされている。
しかしながら、非特許文献1においては火災等の危険性を伴う金属水素化物を使用しており、工業的製造法とは言い難く、非特許文献2においてもヘキサフルオロシクロブタンをアルカリ処理を行うだけで操作は簡便であるが、原料と目的物であるペンタフルオロシクロブテンとの沸点差がほとんど無いがために精製が極めて困難と言わざるを得ない。
また、非特許文献3、および4においては、ヘキサフルオロシクロブテン−ホスホニウムイリドが生成する旨の記載がなされているが、還元体である水素化物が生成するとの記載はなされていない。
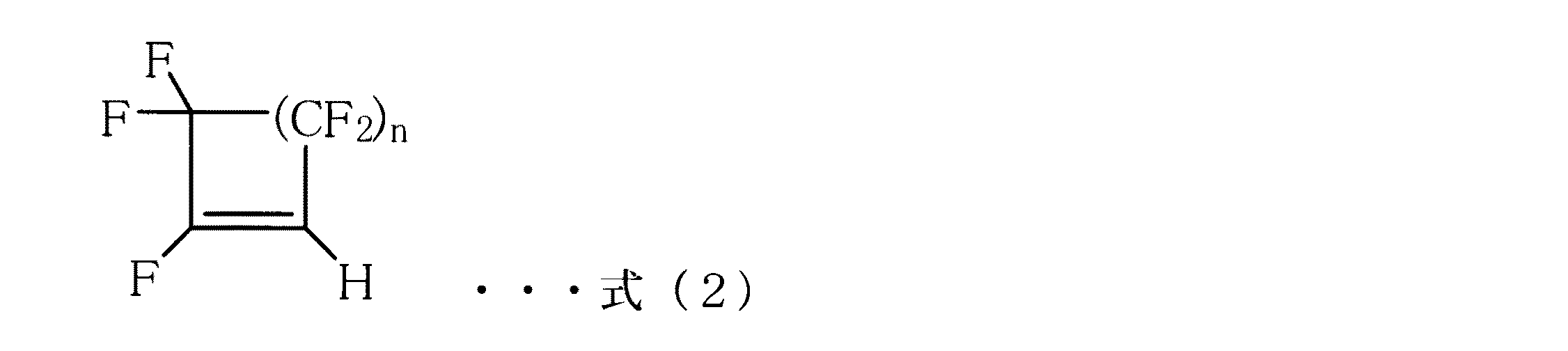
本発明において、前記トリアルキルホスフィンがトリ−n−ブチルホスフィンであることが好ましい。
また本発明において、前記式(2)で表される化合物が1,3,3,4,4−ペンタフルオロシクロブテン又は1,3,3,4,4,5,5−ヘプタフルオロシクロペンテンであることが好ましい。
これらトリアルキルホスフィンの添加量は原料に用いるパーフルオロシクロアルケン対して1〜5モル当量、1.2〜2モル当量がより好ましい。
反応開始時から溶媒中に水分を含有させる場合、パーフルオロシクロオレフィンのトリアルキルホスフィン付加体の加水分解を完全に行うために、パーフルオロシクロオレフィンの消費がほぼ停止した状態で、反応系に水を更に添加しても構わない。この操作を行うことで、目的とする1H−パーフルオロシクロオレフィンの収率向上に寄与する。
(1)ガスクロマトグラフィー分析(GC分析)
装置:G−5000(日立製作所社製)
カラム:TC−1(60m×I.D0.25μm、1.0μmdf) (GLサイエンス社製)
昇温プログラム:(1)50℃で10分保持し、次いで(2)20℃/分で昇温した後、(3)250℃で10分保持する。
インジェクション温度:200℃
キャリヤーガス:窒素ガス
検出器:FID
装置:アジレント社製ガスクロマトグラフ質量分析計「HP6890」
カラム:アジレントHP−1(長さ60m、内径250μm、膜厚1μm)
昇温プログラム:(1)40℃で10分保持し、次いで(2)20℃/分で昇温した後、(3)240℃で10分保持する。
インジェクション温度:150℃
ディテクター温度:150℃
キャリヤーガス:ヘリウムガス(282mL/分)
スプリット比:170/1
MS部分:アジレント5973ネットワーク(アジレント社製)
検出器:EI型(加速電圧:70eV)
装置:日本電子社製核磁気共鳴装置「JNM−ECA400型」
攪拌機、滴下ロートを付したガラス製反応器にオクタフルオロシクロペンテン 42部、約5重量%の水分を含むエチレングリコールジメチルエーテル87部を仕込み、反応器をドライアイス−エタノール浴に浸して−10℃に冷却した。滴下ロートより、トリ−n−ブチルホスフィン45部を10分間かけて滴下し、−10℃で30分間攪拌を継続した後、ドライアイス−エタノール浴を取り去り、室温で8時間攪拌した。反応液をガスクロマトグラフィーにて分析した結果、原料であるオクタフルオロシクロペンテンの消失を確認した。反応器にドライアイス−エタノール浴に浸したガラス製トラップをつなぎ、真空ポンプにて系内を減圧した。約30分後、系内を常圧に戻し、トラップ内に捕集された液体を分析したところ、目的物である、1,3,3,4,4,5,5−ヘプタフルオロシクロペンテン28部(収率72%)が得られた。
1,3,3,4,4,5,5−ヘプタフルオロシクロペンテンのスペクトルデータ
19F−NMR(CFCl3,CDCl3):δ−107.8(s,2F),−120.4(s,2F),−125.0(m,2F),−130.5(s,1F),1H−NMR(TMS,CDCl3):δ5.95(m、1H)
GC−MS(EI−MS):m/z 194,173,144
実施例1において、約5重量%の水分を含むエチレングリコールジメチルエーテル87部を約6重量%の水分を含むテトラヒドロフラン89部に変更したこと以外は実施例1と同様に反応を行った。その結果、1,3,3,4,4,5,5−ヘプタフルオロシクロペンテンが26部(収率69%)得られた。
実施例1において、トリ−n−ブチルホスフィン45部をトリ−n−オクチルホスフィン78部に変更したこと以外は実施例1と同様に反応を行った。その結果、1,3,3,4,4,5,5−ヘプタフルオロシクロペンテンが27部(収率70%)得られた。
攪拌機、ガス導入管を付したステンレス製オートクレーブに約5重量%の水分を含むエチレングリコールジメチルエーテル87部を仕込み、オートクレーブをドライアイス−エタノール浴に浸して−50℃に冷却した。あらかじめステンレス製シリンダーに秤量したヘキサフルオロシクロブテン32部をガス導入管からオートクレーブ内に吹き込み、ドライアイス−エタノール浴を−10℃まで昇温させた。トリ−n−ブチルホスフィン45部を10分間かけて注入し、−10℃で30分間攪拌を継続した後、ドライアイス−エタノール浴を取り去って氷水浴に変更し、約20℃で10時間攪拌した。オートクレーブにドライアイス−エタノール浴に浸したガラス製トラップをつなぎ、真空ポンプにて系内を減圧した。約30分後、系内を常圧に戻し、トラップ内に捕集された液体を分析したところ、目的物である、1,3,3,4,4−ペンタフルオロシクロブテン13部(収率45%)が得られた。
1,3,3,4,4−ペンタフルオロシクロブテンのスペクトルデータ
19F−NMR(CFCl3,CDCl3):δ−103.87(s、C=CF),−113.45(s、2F)、−118.88(s、2F)
1H−NMR(TMS,CDCl3):δ5.94(m、1H)
GC−MS(EI−MS):m/z 144,125,75
攪拌機、滴下ロートを付したガラス製反応器にデカフルオロシクロヘキセン52部、約5重量%の水分を含むエチレングリコールジメチルエーテル100部を仕込み、反応器をドライアイス−エタノール浴に浸して−10℃に冷却した。滴下ロートより、トリ−n−ブチルホスフィン41部を10分間かけて滴下し、−10℃で30分間攪拌を継続した後、ドライアイス−エタノール浴を取り去り、室温で6時間攪拌した。反応液をガスクロマトグラフィーにて分析した結果、原料であるデカフルオロシクロヘキセンの消失を確認した。反応器にドライアイス−エタノール浴に浸したガラス製トラップをつなぎ、真空ポンプにて系内を減圧した。約30分後、系内を常圧に戻し、トラップ内に捕集された液体を分析したところ、目的物である、1,3,3,4,4,5,5,6,6−ノナフルオロシクロヘキセン30部(収率62%)を得た。
1,3,3,4,4,5,5,6、6−ノナフルオロシクロヘキセンのスペクトルデータ
19F−NMR(CFCl3,CDCl3):δ〜121.7(s、C=CF),−121.7(s、2F)、−135.4(m、4F),−107.1(s、2F)、1H−NMR(TMS,CDCl3):δ3.90(m、1H)
GC−MS(EI−MS):m/z 244、225、175、144、113、75
実施例1において、反応温度を−20℃、反応時間6時間に変更したこと以外は実施例1と同様に反応を行った。ガスクロマトグラフィー分析を行った結果、原料のオクタフルオロシクロペンテンが95%残存しており、目的物である1,3,3,4,4,5,5−ヘプタフルオロシクロペンテンは2%しか生成していなかった。
実施例1において、トリ−n−ブチルホスフィン41部をトリフェニルホスフィン53部に変更したこと以外は、実施例1と同様に反応を行った。ガスクロマトグラフィー分析を行った結果、原料のオクタフルオロシクロペンテンはほぼ消失していたが、目的物である1,3,3,4,4,5,5−ヘプタフルオロシクロペンテンの生成は認められず、オクタフルオロシクロペンテンのトリフェニルホスフィン付加体の段階で、反応が停止していることが示唆された。
Claims (3)
- 式(1)で示されるパーフルオロシクロオレフィンと、トリアルキルホスフィンを含水エーテル溶媒存在下に、−10℃〜30℃の温度範囲で接触させて、式(2)で示される含水素フルオロシクロオレフィンを得ることを特徴とする製造方法。
- トリアルキルホスフィンがトリ−n−ブチルホスフィンであることを特徴とする請求項1に記載の製造方法。
- 式(2)で表される化合物が1,3,3,4,4−ペンタフルオロシクロブテン又は1,3,3,4,4,5,5−ヘプタフルオロシクロペンテンであることを特徴とする請求項1又は2に記載の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008299953A JP5158366B2 (ja) | 2008-11-25 | 2008-11-25 | 含水素フルオロオレフィン化合物の製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008299953A JP5158366B2 (ja) | 2008-11-25 | 2008-11-25 | 含水素フルオロオレフィン化合物の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010126452A JP2010126452A (ja) | 2010-06-10 |
| JP5158366B2 true JP5158366B2 (ja) | 2013-03-06 |
Family
ID=42327074
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008299953A Expired - Fee Related JP5158366B2 (ja) | 2008-11-25 | 2008-11-25 | 含水素フルオロオレフィン化合物の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5158366B2 (ja) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2014129488A1 (ja) | 2013-02-21 | 2017-02-02 | 日本ゼオン株式会社 | 高純度1h−ヘプタフルオロシクロペンテン |
| CN107365244B (zh) * | 2017-08-03 | 2020-05-15 | 北京宇极科技发展有限公司 | 有机溶剂提供氢源发生氢-卤交换反应制备1h-全卤环烯烃的方法 |
| JP7166911B2 (ja) * | 2018-12-25 | 2022-11-08 | ダイキン工業株式会社 | シクロブテンの製造方法 |
| JP6835060B2 (ja) * | 2018-12-27 | 2021-02-24 | ダイキン工業株式会社 | シクロブテンの製造方法 |
| CN115417745A (zh) * | 2022-11-04 | 2022-12-02 | 北京宇极科技发展有限公司 | 一种合成氢氟烯烃的方法 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4120043B2 (ja) * | 1998-04-01 | 2008-07-16 | 日本ゼオン株式会社 | フッ素化不飽和炭化水素の製造方法 |
| JP5477290B2 (ja) * | 2008-07-18 | 2014-04-23 | 日本ゼオン株式会社 | 含水素フルオロオレフィン化合物の製造方法 |
-
2008
- 2008-11-25 JP JP2008299953A patent/JP5158366B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2010126452A (ja) | 2010-06-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JPWO2007125972A1 (ja) | ヘキサフルオロ−1,3−ブタジエンの製造方法 | |
| JP5158366B2 (ja) | 含水素フルオロオレフィン化合物の製造方法 | |
| JP5267561B2 (ja) | 含フッ素エーテルの製造方法 | |
| JP2021119113A (ja) | 四フッ化硫黄の製造方法 | |
| KR102695286B1 (ko) | 시클로부텐의 제조 방법 | |
| JP4207001B2 (ja) | ポリフルオロアルキルエチルアイオダイドの製造方法 | |
| JP5311009B2 (ja) | 含水素フルオロオレフィン化合物の製造方法 | |
| KR20160122126A (ko) | 불소화 탄화수소의 제조 방법 | |
| EP2451798B1 (en) | Process for producing perfluorinated organic compounds | |
| US10737997B2 (en) | Method for the manufacture of fluorinated compounds | |
| Petrov et al. | New partially fluorinated epoxides by oxidation of olefins with sodium hypohalites under phase transfer catalysis | |
| US10975053B2 (en) | Production methods for 1,3-dioxolane compound and perfluoro(2,2-dimethyl-1,3-dioxole) | |
| JP2012131731A (ja) | フッ素化アルケンの製造法 | |
| US6596906B2 (en) | Process for preparing fluorine-containing benzaldehydes | |
| EP2643289B1 (en) | Process for producing perfluorinated organic compounds | |
| JP6003709B2 (ja) | 1,2−ビス(パーフルオロアルキル)−パーフルオロシクロアルケンの製造方法 | |
| JP5194500B2 (ja) | 高純度含フッ素アルキルエーテルの製造方法 | |
| JP5266648B2 (ja) | パーフルオロ(エキソメチレンシクロアルケン)化合物の製造方法 | |
| JP5126936B2 (ja) | フルオロ(アルキルビニルエーテル)およびその誘導体の製造方法 | |
| US20030125583A1 (en) | Process for preparing fluorine-containing benzaldehydes | |
| WO2018037999A1 (ja) | ブテン類の変換方法及びモノフルオロブタンの精製方法 | |
| CN121866274A (zh) | 卤代全氟脂肪族化合物及其制备和使用方法 | |
| TW202419431A (zh) | 氯氟丁烷之製造方法 | |
| JP4364545B2 (ja) | 新規なジフルオロテトラヒドロチオフェン1,1−ジオキシド及びその製造方法 | |
| JP2003146917A (ja) | パーフルオロアルキン化合物の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110912 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20120906 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120912 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120914 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20121114 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121127 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5158366 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151221 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |