JP5134183B2 - 予防または治療ワクチン接種および/或いはその他疾患の治療を目的とする、抗原提示細胞を標的とすることおよび/または活性化すること、並びに/またはカーゴ分子を運搬することへの生物学的活性hiv−1tat、その断片または誘導体の使用 - Google Patents
予防または治療ワクチン接種および/或いはその他疾患の治療を目的とする、抗原提示細胞を標的とすることおよび/または活性化すること、並びに/またはカーゴ分子を運搬することへの生物学的活性hiv−1tat、その断片または誘導体の使用 Download PDFInfo
- Publication number
- JP5134183B2 JP5134183B2 JP2003515259A JP2003515259A JP5134183B2 JP 5134183 B2 JP5134183 B2 JP 5134183B2 JP 2003515259 A JP2003515259 A JP 2003515259A JP 2003515259 A JP2003515259 A JP 2003515259A JP 5134183 B2 JP5134183 B2 JP 5134183B2
- Authority
- JP
- Japan
- Prior art keywords
- tat
- infection
- pharmaceutical composition
- cells
- composition according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 210000000612 antigen-presenting cell Anatomy 0.000 title claims description 23
- 238000011282 treatment Methods 0.000 title claims description 20
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 title claims description 18
- 230000001225 therapeutic effect Effects 0.000 title claims description 12
- 239000012634 fragment Substances 0.000 title description 18
- 201000010099 disease Diseases 0.000 title description 16
- 238000002255 vaccination Methods 0.000 title description 15
- 230000008685 targeting Effects 0.000 title description 8
- 230000000069 prophylactic effect Effects 0.000 title description 4
- 230000003213 activating effect Effects 0.000 title description 3
- 210000004027 cell Anatomy 0.000 claims description 156
- 239000000427 antigen Substances 0.000 claims description 88
- 102000036639 antigens Human genes 0.000 claims description 88
- 108091007433 antigens Proteins 0.000 claims description 88
- 210000002889 endothelial cell Anatomy 0.000 claims description 60
- 241000713772 Human immunodeficiency virus 1 Species 0.000 claims description 43
- 239000008194 pharmaceutical composition Substances 0.000 claims description 42
- 208000015181 infectious disease Diseases 0.000 claims description 38
- 108010044426 integrins Proteins 0.000 claims description 35
- 102000006495 integrins Human genes 0.000 claims description 35
- 230000000694 effects Effects 0.000 claims description 34
- 210000004443 dendritic cell Anatomy 0.000 claims description 32
- 206010028980 Neoplasm Diseases 0.000 claims description 31
- 239000002671 adjuvant Substances 0.000 claims description 26
- 210000002540 macrophage Anatomy 0.000 claims description 26
- 241000725303 Human immunodeficiency virus Species 0.000 claims description 25
- 244000052769 pathogen Species 0.000 claims description 23
- 230000035800 maturation Effects 0.000 claims description 21
- 238000004519 manufacturing process Methods 0.000 claims description 18
- 239000000178 monomer Substances 0.000 claims description 18
- 230000001717 pathogenic effect Effects 0.000 claims description 18
- 241000282414 Homo sapiens Species 0.000 claims description 15
- 241000700605 Viruses Species 0.000 claims description 14
- 210000004698 lymphocyte Anatomy 0.000 claims description 14
- 229960005486 vaccine Drugs 0.000 claims description 14
- 230000002265 prevention Effects 0.000 claims description 10
- 241000607142 Salmonella Species 0.000 claims description 7
- 210000004369 blood Anatomy 0.000 claims description 7
- 239000008280 blood Substances 0.000 claims description 7
- 241000186781 Listeria Species 0.000 claims description 6
- 230000001472 cytotoxic effect Effects 0.000 claims description 6
- 239000003814 drug Substances 0.000 claims description 6
- 208000006454 hepatitis Diseases 0.000 claims description 6
- 231100000283 hepatitis Toxicity 0.000 claims description 6
- 201000008827 tuberculosis Diseases 0.000 claims description 6
- 241001529453 unidentified herpesvirus Species 0.000 claims description 6
- 201000006082 Chickenpox Diseases 0.000 claims description 5
- 241001631646 Papillomaviridae Species 0.000 claims description 5
- 206010043376 Tetanus Diseases 0.000 claims description 5
- 206010046980 Varicella Diseases 0.000 claims description 5
- 210000004072 lung Anatomy 0.000 claims description 5
- 210000001519 tissue Anatomy 0.000 claims description 5
- 210000004881 tumor cell Anatomy 0.000 claims description 5
- 241000589989 Helicobacter Species 0.000 claims description 4
- 230000001154 acute effect Effects 0.000 claims description 4
- 210000000988 bone and bone Anatomy 0.000 claims description 4
- 210000000481 breast Anatomy 0.000 claims description 4
- 230000002124 endocrine Effects 0.000 claims description 4
- 210000004185 liver Anatomy 0.000 claims description 4
- 201000004792 malaria Diseases 0.000 claims description 4
- 210000001672 ovary Anatomy 0.000 claims description 4
- 210000000496 pancreas Anatomy 0.000 claims description 4
- 238000002360 preparation method Methods 0.000 claims description 4
- 210000004872 soft tissue Anatomy 0.000 claims description 4
- 210000004291 uterus Anatomy 0.000 claims description 4
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 claims description 3
- 206010000830 Acute leukaemia Diseases 0.000 claims description 3
- 241000222122 Candida albicans Species 0.000 claims description 3
- 206010007134 Candida infections Diseases 0.000 claims description 3
- 241000701022 Cytomegalovirus Species 0.000 claims description 3
- 206010019375 Helicobacter infections Diseases 0.000 claims description 3
- 208000001688 Herpes Genitalis Diseases 0.000 claims description 3
- 208000004898 Herpes Labialis Diseases 0.000 claims description 3
- 206010019973 Herpes virus infection Diseases 0.000 claims description 3
- 241000701044 Human gammaherpesvirus 4 Species 0.000 claims description 3
- 208000034762 Meningococcal Infections Diseases 0.000 claims description 3
- 206010067152 Oral herpes Diseases 0.000 claims description 3
- 206010031252 Osteomyelitis Diseases 0.000 claims description 3
- 208000035109 Pneumococcal Infections Diseases 0.000 claims description 3
- 206010039491 Sarcoma Diseases 0.000 claims description 3
- 208000019802 Sexually transmitted disease Diseases 0.000 claims description 3
- 206010041925 Staphylococcal infections Diseases 0.000 claims description 3
- 206010061372 Streptococcal infection Diseases 0.000 claims description 3
- 230000001772 anti-angiogenic effect Effects 0.000 claims description 3
- 239000002260 anti-inflammatory agent Substances 0.000 claims description 3
- 201000003984 candidiasis Diseases 0.000 claims description 3
- 208000024207 chronic leukemia Diseases 0.000 claims description 3
- 231100000433 cytotoxic Toxicity 0.000 claims description 3
- 206010014665 endocarditis Diseases 0.000 claims description 3
- 201000004946 genital herpes Diseases 0.000 claims description 3
- 206010022000 influenza Diseases 0.000 claims description 3
- 206010040872 skin infection Diseases 0.000 claims description 3
- 239000000126 substance Substances 0.000 claims description 3
- 208000006379 syphilis Diseases 0.000 claims description 3
- 241000712461 unidentified influenza virus Species 0.000 claims description 3
- 208000019206 urinary tract infection Diseases 0.000 claims description 3
- 230000009385 viral infection Effects 0.000 claims description 3
- 241000295644 Staphylococcaceae Species 0.000 claims description 2
- 201000006747 infectious mononucleosis Diseases 0.000 claims description 2
- 241000607626 Vibrio cholerae Species 0.000 claims 3
- 210000001072 colon Anatomy 0.000 claims 3
- 210000002307 prostate Anatomy 0.000 claims 3
- 210000003491 skin Anatomy 0.000 claims 3
- 229940118696 vibrio cholerae Drugs 0.000 claims 3
- 206010008631 Cholera Diseases 0.000 claims 2
- 241000193449 Clostridium tetani Species 0.000 claims 2
- 206010011831 Cytomegalovirus infection Diseases 0.000 claims 2
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 claims 2
- 206010024641 Listeriosis Diseases 0.000 claims 2
- 241000187479 Mycobacterium tuberculosis Species 0.000 claims 2
- 241000588650 Neisseria meningitidis Species 0.000 claims 2
- 241000191967 Staphylococcus aureus Species 0.000 claims 2
- 241000194017 Streptococcus Species 0.000 claims 2
- 241000193998 Streptococcus pneumoniae Species 0.000 claims 2
- 125000003275 alpha amino acid group Chemical group 0.000 claims 2
- 229940121363 anti-inflammatory agent Drugs 0.000 claims 2
- 239000002246 antineoplastic agent Substances 0.000 claims 2
- 230000003211 malignant effect Effects 0.000 claims 2
- 229940031000 streptococcus pneumoniae Drugs 0.000 claims 2
- 101710149951 Protein Tat Proteins 0.000 description 518
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 52
- 108090000623 proteins and genes Proteins 0.000 description 49
- 102000004127 Cytokines Human genes 0.000 description 45
- 108090000695 Cytokines Proteins 0.000 description 45
- 238000011534 incubation Methods 0.000 description 45
- 235000018102 proteins Nutrition 0.000 description 45
- 102000004169 proteins and genes Human genes 0.000 description 45
- 108090000765 processed proteins & peptides Proteins 0.000 description 42
- 230000000975 bioactive effect Effects 0.000 description 40
- 208000031886 HIV Infections Diseases 0.000 description 36
- 230000028993 immune response Effects 0.000 description 30
- 238000011533 pre-incubation Methods 0.000 description 27
- 239000002609 medium Substances 0.000 description 26
- 210000005087 mononuclear cell Anatomy 0.000 description 26
- 230000037361 pathway Effects 0.000 description 26
- 230000006870 function Effects 0.000 description 25
- 102000004196 processed proteins & peptides Human genes 0.000 description 24
- 150000001413 amino acids Chemical group 0.000 description 22
- 230000027455 binding Effects 0.000 description 21
- 239000000872 buffer Substances 0.000 description 21
- 102000005962 receptors Human genes 0.000 description 20
- 108020003175 receptors Proteins 0.000 description 20
- 210000001744 T-lymphocyte Anatomy 0.000 description 18
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 18
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 18
- 230000002757 inflammatory effect Effects 0.000 description 18
- 239000013642 negative control Substances 0.000 description 18
- 230000014509 gene expression Effects 0.000 description 17
- 230000001965 increasing effect Effects 0.000 description 17
- 239000012139 lysis buffer Substances 0.000 description 16
- 108010031318 Vitronectin Proteins 0.000 description 15
- 102100035140 Vitronectin Human genes 0.000 description 15
- 230000005764 inhibitory process Effects 0.000 description 15
- 230000004044 response Effects 0.000 description 15
- 238000000034 method Methods 0.000 description 14
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 13
- 208000030507 AIDS Diseases 0.000 description 13
- 102100037362 Fibronectin Human genes 0.000 description 13
- 108010067306 Fibronectins Proteins 0.000 description 13
- 108010065805 Interleukin-12 Proteins 0.000 description 13
- 102000013462 Interleukin-12 Human genes 0.000 description 13
- 208000007766 Kaposi sarcoma Diseases 0.000 description 13
- 241000699670 Mus sp. Species 0.000 description 13
- 230000000875 corresponding effect Effects 0.000 description 13
- 238000002474 experimental method Methods 0.000 description 13
- 230000036962 time dependent Effects 0.000 description 13
- 230000004071 biological effect Effects 0.000 description 12
- 230000004913 activation Effects 0.000 description 11
- 231100000673 dose–response relationship Toxicity 0.000 description 11
- 230000035755 proliferation Effects 0.000 description 11
- 238000010186 staining Methods 0.000 description 11
- 102000001327 Chemokine CCL5 Human genes 0.000 description 9
- 108010055166 Chemokine CCL5 Proteins 0.000 description 9
- 102000006354 HLA-DR Antigens Human genes 0.000 description 9
- 108010058597 HLA-DR Antigens Proteins 0.000 description 9
- 108010055044 Tetanus Toxin Proteins 0.000 description 9
- 235000018417 cysteine Nutrition 0.000 description 9
- 230000003053 immunization Effects 0.000 description 9
- 230000002779 inactivation Effects 0.000 description 9
- 230000003993 interaction Effects 0.000 description 9
- 230000003834 intracellular effect Effects 0.000 description 9
- 229940118376 tetanus toxin Drugs 0.000 description 9
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 8
- 108700012434 CCL3 Proteins 0.000 description 8
- 102000000013 Chemokine CCL3 Human genes 0.000 description 8
- 238000004458 analytical method Methods 0.000 description 8
- 230000002491 angiogenic effect Effects 0.000 description 8
- 238000002649 immunization Methods 0.000 description 8
- 210000000265 leukocyte Anatomy 0.000 description 8
- 230000003647 oxidation Effects 0.000 description 8
- 238000007254 oxidation reaction Methods 0.000 description 8
- 230000000242 pagocytic effect Effects 0.000 description 8
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 239000012679 serum free medium Substances 0.000 description 8
- 102000001902 CC Chemokines Human genes 0.000 description 7
- 108010040471 CC Chemokines Proteins 0.000 description 7
- 101150013553 CD40 gene Proteins 0.000 description 7
- 102100035793 CD83 antigen Human genes 0.000 description 7
- 102000001326 Chemokine CCL4 Human genes 0.000 description 7
- 108010055165 Chemokine CCL4 Proteins 0.000 description 7
- 208000035473 Communicable disease Diseases 0.000 description 7
- 108020004414 DNA Proteins 0.000 description 7
- 102000008055 Heparan Sulfate Proteoglycans Human genes 0.000 description 7
- 101000946856 Homo sapiens CD83 antigen Proteins 0.000 description 7
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 7
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 7
- 241000713311 Simian immunodeficiency virus Species 0.000 description 7
- 206010042602 Supraventricular extrasystoles Diseases 0.000 description 7
- 108090000054 Syndecan-2 Proteins 0.000 description 7
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 7
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 7
- 230000000735 allogeneic effect Effects 0.000 description 7
- 230000000139 costimulatory effect Effects 0.000 description 7
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 7
- 238000000684 flow cytometry Methods 0.000 description 7
- 230000006698 induction Effects 0.000 description 7
- 239000003446 ligand Substances 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 239000013641 positive control Substances 0.000 description 7
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 7
- 241000282693 Cercopithecidae Species 0.000 description 6
- 108010047852 Integrin alphaVbeta3 Proteins 0.000 description 6
- 230000030741 antigen processing and presentation Effects 0.000 description 6
- 230000011748 cell maturation Effects 0.000 description 6
- 230000002950 deficient Effects 0.000 description 6
- 230000004069 differentiation Effects 0.000 description 6
- 239000012091 fetal bovine serum Substances 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 230000001939 inductive effect Effects 0.000 description 6
- 208000027866 inflammatory disease Diseases 0.000 description 6
- 238000010212 intracellular staining Methods 0.000 description 6
- 230000007246 mechanism Effects 0.000 description 6
- 230000002829 reductive effect Effects 0.000 description 6
- 230000010076 replication Effects 0.000 description 6
- 101710177291 Gag polyprotein Proteins 0.000 description 5
- 241000713340 Human immunodeficiency virus 2 Species 0.000 description 5
- 206010061218 Inflammation Diseases 0.000 description 5
- 101710125418 Major capsid protein Proteins 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 241000283973 Oryctolagus cuniculus Species 0.000 description 5
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 5
- 230000035622 drinking Effects 0.000 description 5
- 230000004054 inflammatory process Effects 0.000 description 5
- 238000011081 inoculation Methods 0.000 description 5
- 230000009545 invasion Effects 0.000 description 5
- 239000002773 nucleotide Substances 0.000 description 5
- 125000003729 nucleotide group Chemical group 0.000 description 5
- IXQPRUQVJIJUEB-UHFFFAOYSA-N 7-(diethylamino)-n-[2-(2,5-dioxopyrrol-1-yl)ethyl]-2-oxochromene-3-carboxamide Chemical compound O=C1OC2=CC(N(CC)CC)=CC=C2C=C1C(=O)NCCN1C(=O)C=CC1=O IXQPRUQVJIJUEB-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- 241000282560 Macaca mulatta Species 0.000 description 4
- 101100256737 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) hlm-1 gene Proteins 0.000 description 4
- 108091028043 Nucleic acid sequence Proteins 0.000 description 4
- 108010047620 Phytohemagglutinins Proteins 0.000 description 4
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 4
- 230000005867 T cell response Effects 0.000 description 4
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 4
- 239000011651 chromium Substances 0.000 description 4
- 150000001875 compounds Chemical class 0.000 description 4
- 230000002519 immonomodulatory effect Effects 0.000 description 4
- 108010051618 macrophage stimulatory lipopeptide 2 Proteins 0.000 description 4
- 230000005012 migration Effects 0.000 description 4
- 238000013508 migration Methods 0.000 description 4
- 239000000203 mixture Substances 0.000 description 4
- 239000002953 phosphate buffered saline Substances 0.000 description 4
- 230000001885 phytohemagglutinin Effects 0.000 description 4
- 230000003449 preventive effect Effects 0.000 description 4
- 230000037452 priming Effects 0.000 description 4
- 230000006318 protein oxidation Effects 0.000 description 4
- 229940021747 therapeutic vaccine Drugs 0.000 description 4
- 241001430294 unidentified retrovirus Species 0.000 description 4
- 230000002792 vascular Effects 0.000 description 4
- 230000029812 viral genome replication Effects 0.000 description 4
- 230000003612 virological effect Effects 0.000 description 4
- 102100035875 C-C chemokine receptor type 5 Human genes 0.000 description 3
- 101710149870 C-C chemokine receptor type 5 Proteins 0.000 description 3
- 241000588724 Escherichia coli Species 0.000 description 3
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 3
- 102000014150 Interferons Human genes 0.000 description 3
- 108010050904 Interferons Proteins 0.000 description 3
- 102000015696 Interleukins Human genes 0.000 description 3
- 108010063738 Interleukins Proteins 0.000 description 3
- 230000029662 T-helper 1 type immune response Effects 0.000 description 3
- 230000000961 alloantigen Effects 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 239000003636 conditioned culture medium Substances 0.000 description 3
- 210000000805 cytoplasm Anatomy 0.000 description 3
- 230000007423 decrease Effects 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 238000009826 distribution Methods 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000012636 effector Substances 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 3
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 3
- 229960002897 heparin Drugs 0.000 description 3
- 229920000669 heparin Polymers 0.000 description 3
- 210000002865 immune cell Anatomy 0.000 description 3
- 238000010348 incorporation Methods 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 229940079322 interferon Drugs 0.000 description 3
- NIQQIJXGUZVEBB-UHFFFAOYSA-N methanol;propan-2-one Chemical compound OC.CC(C)=O NIQQIJXGUZVEBB-UHFFFAOYSA-N 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- JGVWCANSWKRBCS-UHFFFAOYSA-N tetramethylrhodamine thiocyanate Chemical compound [Cl-].C=12C=CC(N(C)C)=CC2=[O+]C2=CC(N(C)C)=CC=C2C=1C1=CC=C(SC#N)C=C1C(O)=O JGVWCANSWKRBCS-UHFFFAOYSA-N 0.000 description 3
- 230000032258 transport Effects 0.000 description 3
- DMWMUMWKGKGSNW-OPMCLZTFSA-N (2S)-6-amino-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S,3S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-4-amino-2-[[2-[[(2R)-2-amino-3-[(2R)-2,3-di(hexadecanoyloxy)propyl]sulfanylpropanoyl]amino]acetyl]amino]-4-oxobutanoyl]amino]-4-oxobutanoyl]amino]-3-carboxypropanoyl]amino]-4-carboxybutanoyl]amino]-3-hydroxypropanoyl]amino]-4-oxobutanoyl]amino]-3-methylpentanoyl]amino]-3-hydroxypropanoyl]amino]-3-phenylpropanoyl]amino]hexanoyl]amino]-4-carboxybutanoyl]amino]hexanoic acid Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](CSC[C@H](N)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(O)=O)OC(=O)CCCCCCCCCCCCCCC DMWMUMWKGKGSNW-OPMCLZTFSA-N 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 2
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 2
- 102000019034 Chemokines Human genes 0.000 description 2
- 108010012236 Chemokines Proteins 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101001027128 Homo sapiens Fibronectin Proteins 0.000 description 2
- 108010070875 Human Immunodeficiency Virus tat Gene Products Proteins 0.000 description 2
- 108010002352 Interleukin-1 Proteins 0.000 description 2
- 102000000589 Interleukin-1 Human genes 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- 108090000978 Interleukin-4 Proteins 0.000 description 2
- 241000282553 Macaca Species 0.000 description 2
- 102000009571 Macrophage Inflammatory Proteins Human genes 0.000 description 2
- 108010009474 Macrophage Inflammatory Proteins Proteins 0.000 description 2
- 101710151805 Mitochondrial intermediate peptidase 1 Proteins 0.000 description 2
- 102000016611 Proteoglycans Human genes 0.000 description 2
- 108010067787 Proteoglycans Proteins 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- 239000012980 RPMI-1640 medium Substances 0.000 description 2
- 230000024932 T cell mediated immunity Effects 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- 102000004142 Trypsin Human genes 0.000 description 2
- 108090000631 Trypsin Proteins 0.000 description 2
- 206010047115 Vasculitis Diseases 0.000 description 2
- 241000607598 Vibrio Species 0.000 description 2
- 208000026935 allergic disease Diseases 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 239000002870 angiogenesis inducing agent Substances 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 230000005875 antibody response Effects 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 230000020411 cell activation Effects 0.000 description 2
- 238000001516 cell proliferation assay Methods 0.000 description 2
- 230000002596 correlated effect Effects 0.000 description 2
- 150000001945 cysteines Chemical class 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 239000013024 dilution buffer Substances 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- -1 epicipients Substances 0.000 description 2
- 230000028996 humoral immune response Effects 0.000 description 2
- 230000036039 immunity Effects 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 230000019734 interleukin-12 production Effects 0.000 description 2
- 230000021633 leukocyte mediated immunity Effects 0.000 description 2
- 238000011068 loading method Methods 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 210000004897 n-terminal region Anatomy 0.000 description 2
- 210000004940 nucleus Anatomy 0.000 description 2
- 239000013612 plasmid Substances 0.000 description 2
- 230000023603 positive regulation of transcription initiation, DNA-dependent Effects 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 229940023867 prime-boost vaccine Drugs 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- 230000009696 proliferative response Effects 0.000 description 2
- NQLVQOSNDJXLKG-UHFFFAOYSA-N prosulfocarb Chemical compound CCCN(CCC)C(=O)SCC1=CC=CC=C1 NQLVQOSNDJXLKG-UHFFFAOYSA-N 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 238000013207 serial dilution Methods 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- 210000004989 spleen cell Anatomy 0.000 description 2
- 230000002269 spontaneous effect Effects 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 125000000341 threoninyl group Chemical group [H]OC([H])(C([H])([H])[H])C([H])(N([H])[H])C(*)=O 0.000 description 2
- 229940104230 thymidine Drugs 0.000 description 2
- 239000012588 trypsin Substances 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 230000003827 upregulation Effects 0.000 description 2
- 239000012646 vaccine adjuvant Substances 0.000 description 2
- 229940124931 vaccine adjuvant Drugs 0.000 description 2
- 208000019553 vascular disease Diseases 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- OBYNJKLOYWCXEP-UHFFFAOYSA-N 2-[3-(dimethylamino)-6-dimethylazaniumylidenexanthen-9-yl]-4-isothiocyanatobenzoate Chemical compound C=12C=CC(=[N+](C)C)C=C2OC2=CC(N(C)C)=CC=C2C=1C1=CC(N=C=S)=CC=C1C([O-])=O OBYNJKLOYWCXEP-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- 208000012124 AIDS-related disease Diseases 0.000 description 1
- 101710197241 Accessory gland-specific peptide 70A Proteins 0.000 description 1
- 208000028185 Angioedema Diseases 0.000 description 1
- 201000003076 Angiosarcoma Diseases 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 102100031151 C-C chemokine receptor type 2 Human genes 0.000 description 1
- 101710149815 C-C chemokine receptor type 2 Proteins 0.000 description 1
- 102100024167 C-C chemokine receptor type 3 Human genes 0.000 description 1
- 101710149862 C-C chemokine receptor type 3 Proteins 0.000 description 1
- 102100037853 C-C chemokine receptor type 4 Human genes 0.000 description 1
- 101710149863 C-C chemokine receptor type 4 Proteins 0.000 description 1
- 102100036301 C-C chemokine receptor type 7 Human genes 0.000 description 1
- 102100031650 C-X-C chemokine receptor type 4 Human genes 0.000 description 1
- 101100381481 Caenorhabditis elegans baz-2 gene Proteins 0.000 description 1
- 101100152433 Caenorhabditis elegans tat-1 gene Proteins 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 102000009410 Chemokine receptor Human genes 0.000 description 1
- 108050000299 Chemokine receptor Proteins 0.000 description 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 102000052510 DNA-Binding Proteins Human genes 0.000 description 1
- 108700020911 DNA-Binding Proteins Proteins 0.000 description 1
- 206010012689 Diabetic retinopathy Diseases 0.000 description 1
- 102100025012 Dipeptidyl peptidase 4 Human genes 0.000 description 1
- 208000035859 Drug effect increased Diseases 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 238000008157 ELISA kit Methods 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 108010054218 Factor VIII Proteins 0.000 description 1
- 102000001690 Factor VIII Human genes 0.000 description 1
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 208000010412 Glaucoma Diseases 0.000 description 1
- 208000024869 Goodpasture syndrome Diseases 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 208000001258 Hemangiosarcoma Diseases 0.000 description 1
- 102000008949 Histocompatibility Antigens Class I Human genes 0.000 description 1
- 108010088652 Histocompatibility Antigens Class I Proteins 0.000 description 1
- 101000716065 Homo sapiens C-C chemokine receptor type 7 Proteins 0.000 description 1
- 101000922348 Homo sapiens C-X-C chemokine receptor type 4 Proteins 0.000 description 1
- 101000908391 Homo sapiens Dipeptidyl peptidase 4 Proteins 0.000 description 1
- 241000598436 Human T-cell lymphotropic virus Species 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 208000004575 Infectious Arthritis Diseases 0.000 description 1
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 description 1
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 102000043129 MHC class I family Human genes 0.000 description 1
- 108091054437 MHC class I family Proteins 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 241001602730 Monza Species 0.000 description 1
- 241000581002 Murex Species 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 241000186359 Mycobacterium Species 0.000 description 1
- 206010029113 Neovascularisation Diseases 0.000 description 1
- 208000030852 Parasitic disease Diseases 0.000 description 1
- 208000034038 Pathologic Neovascularization Diseases 0.000 description 1
- 208000006311 Pyoderma Diseases 0.000 description 1
- 239000012979 RPMI medium Substances 0.000 description 1
- 101100372762 Rattus norvegicus Flt1 gene Proteins 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 208000021386 Sjogren Syndrome Diseases 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 201000009594 Systemic Scleroderma Diseases 0.000 description 1
- 206010042953 Systemic sclerosis Diseases 0.000 description 1
- 230000006044 T cell activation Effects 0.000 description 1
- 230000033540 T cell apoptotic process Effects 0.000 description 1
- 208000002474 Tinea Diseases 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 241000893966 Trichophyton verrucosum Species 0.000 description 1
- 101710162629 Trypsin inhibitor Proteins 0.000 description 1
- 229940122618 Trypsin inhibitor Drugs 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 1
- 108700005077 Viral Genes Proteins 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 125000000637 arginyl group Chemical group N[C@@H](CCCNC(N)=N)C(=O)* 0.000 description 1
- MXWJVTOOROXGIU-UHFFFAOYSA-N atrazine Chemical compound CCNC1=NC(Cl)=NC(NC(C)C)=N1 MXWJVTOOROXGIU-UHFFFAOYSA-N 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 244000052616 bacterial pathogen Species 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 230000001364 causal effect Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 239000002771 cell marker Substances 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 239000005482 chemotactic factor Substances 0.000 description 1
- OJYGBLRPYBAHRT-IPQSZEQASA-N chloralose Chemical compound O1[C@H](C(Cl)(Cl)Cl)O[C@@H]2[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]21 OJYGBLRPYBAHRT-IPQSZEQASA-N 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 238000003501 co-culture Methods 0.000 description 1
- 230000000112 colonic effect Effects 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 239000012228 culture supernatant Substances 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 229940127071 cytotoxic antineoplastic agent Drugs 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- BFMYDTVEBKDAKJ-UHFFFAOYSA-L disodium;(2',7'-dibromo-3',6'-dioxido-3-oxospiro[2-benzofuran-1,9'-xanthene]-4'-yl)mercury;hydrate Chemical compound O.[Na+].[Na+].O1C(=O)C2=CC=CC=C2C21C1=CC(Br)=C([O-])C([Hg])=C1OC1=C2C=C(Br)C([O-])=C1 BFMYDTVEBKDAKJ-UHFFFAOYSA-L 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 230000012202 endocytosis Effects 0.000 description 1
- 230000010595 endothelial cell migration Effects 0.000 description 1
- 230000003511 endothelial effect Effects 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 239000013604 expression vector Substances 0.000 description 1
- 230000010222 extracellular calcium influx Effects 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 229960000301 factor viii Drugs 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 239000012997 ficoll-paque Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 108010038082 heparin proteoglycan Proteins 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 229920001519 homopolymer Polymers 0.000 description 1
- 229960000900 human factor viii Drugs 0.000 description 1
- 230000005965 immune activity Effects 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 238000010166 immunofluorescence Methods 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 230000016784 immunoglobulin production Effects 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 230000002584 immunomodulator Effects 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 201000001371 inclusion conjunctivitis Diseases 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 230000002132 lysosomal effect Effects 0.000 description 1
- 241001515942 marmosets Species 0.000 description 1
- 201000006512 mast cell neoplasm Diseases 0.000 description 1
- 208000006971 mastocytoma Diseases 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 102000006240 membrane receptors Human genes 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 230000002297 mitogenic effect Effects 0.000 description 1
- 108010032806 molgramostim Proteins 0.000 description 1
- 230000000877 morphologic effect Effects 0.000 description 1
- 210000004877 mucosa Anatomy 0.000 description 1
- 230000016379 mucosal immune response Effects 0.000 description 1
- 210000005088 multinucleated cell Anatomy 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 210000000633 nuclear envelope Anatomy 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 230000030648 nucleus localization Effects 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 201000008482 osteoarthritis Diseases 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 230000008823 permeabilization Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 108010089520 pol Gene Products Proteins 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 230000000861 pro-apoptotic effect Effects 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 230000010837 receptor-mediated endocytosis Effects 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 229910052594 sapphire Inorganic materials 0.000 description 1
- 239000010980 sapphire Substances 0.000 description 1
- 201000000306 sarcoidosis Diseases 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 201000001223 septic arthritis Diseases 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- 206010043778 thyroiditis Diseases 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 206010044325 trachoma Diseases 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 239000002753 trypsin inhibitor Substances 0.000 description 1
- 231100000397 ulcer Toxicity 0.000 description 1
- 210000003606 umbilical vein Anatomy 0.000 description 1
- 239000013598 vector Substances 0.000 description 1
- 239000013603 viral vector Substances 0.000 description 1
- 108010047303 von Willebrand Factor Proteins 0.000 description 1
- 102100036537 von Willebrand factor Human genes 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
Images




























Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/39—Medicinal preparations containing antigens or antibodies characterised by the immunostimulating additives, e.g. chemical adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/515—Animal cells
- A61K2039/5154—Antigen presenting cells [APCs], e.g. dendritic cells or macrophages
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/53—DNA (RNA) vaccination
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55516—Proteins; Peptides
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16311—Human Immunodeficiency Virus, HIV concerning HIV regulatory proteins
- C12N2740/16322—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Virology (AREA)
- Immunology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Molecular Biology (AREA)
- Diabetes (AREA)
- Pulmonology (AREA)
- Endocrinology (AREA)
- Ophthalmology & Optometry (AREA)
- Epidemiology (AREA)
- Gastroenterology & Hepatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Rheumatology (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Tropical Medicine & Parasitology (AREA)
- Urology & Nephrology (AREA)
- Mycology (AREA)
- Microbiology (AREA)
- Physical Education & Sports Medicine (AREA)
- Dermatology (AREA)
- Neurology (AREA)
Description
前記HIV Tat、断片又は誘導体は:
(i)HIV−1 TatのRGDドメインを含み;
(ii)アジュバント活性を有し;且つ
(iii)下記の:
(a)腫瘍細胞の抗原;及び
(b)HIVではない病原体の抗原であって、前記病原体は、ウィルス、結核菌、リステリア、連鎖球菌、ブドウ球菌、肺炎双球菌、破傷風、マイコバクテリウム、髄膜炎菌、ヘリコバクター、サルモネラ、ビブリオ、カンジダ又は変形体である、抗原;
からなる群から選択された抗原分子と混合されていることを特徴とする医薬品組成物。
単離された未変性の実質モノマーである生物活性HIV Tat、若しくはその単離された断片又はこれらの単離された誘導体を含んでなり、
前記HIV Tat、断片又は誘導体は:
(i)HIV−1 TatのRGDドメインを含み;
(ii)アジュバント活性を有し;且つ
(iii)病原体の抗原からなる抗原分子と混合されていることを特徴とする医薬品組成物。
前記HIV Tat、断片又は誘導体は:
(i)HIV−1 TatのRGDドメインを含み;且つ
(ii)治療分子を含む支持体パーティクルと関連づけられることによって該治療分子と組み合わされ、又は治療分子に結合されており、
該治療分子は、
(a)抗炎症薬;
(b)抗血管新生分子;及び
(c)細胞傷害性抗腫瘍薬;
からなる群から選択されることを特徴とする医薬品組成物。
・予防および治療ワクチン接種または感染症、炎症性および血管新生疾患並びに腫瘍の治療を目的とする免疫反応を誘導するために、in vitroおよびin vivoにて抗原または活性化合物を、DC、内皮細胞およびマクロファージを含むα5β1およびαvβ3インテグリンを発現している抗原提示細胞に選択的に運搬すること。
・in vitroおよびin vivoに於いてDC、内皮細胞およびマクロファージを含むα5β1およびαvβ3インテグリンを発現している細胞の抗原提示機能を活性化または高め、HIV/AIDS、その他感染症および腫瘍に対するTh−1型免疫反応を誘導するアジュバントとして。
NH2−MEPVDPRLEPWKHPGSQPKTACTNCYCKKCCFHCQVCFITKALGISYGRKKRRQRRRPPQGSQTHQVSLSKQPTSQSRGDPTGPKE−COOH
そして生物活性型のHIV−2 Tatタンパク質は以下のアミノ酸配列(配列番号100)を持つもので、配列中、この位置に限定されるものではないが、例えば元のアミノ酸配列のアミノ酸92と93の間の位置にRGD配列が挿入されているもの:
NH2−METPLKAPESSLKSCNEPFSRTSEQDVATQELARQGEEILSQLYRPLETCNNSCYCKRCCYHCQMCFLNKGLGICYERKGRRRRTPKKTKTHRGDPSPTPDKSISTRTGDSQPTKKQKKTVEATVETDTGPGR−COOH
または位置105のスレオニンが欠失しているものであり(配列番号102):
NH2−METPLKAPESSLKSCNEPFSRTSEQDVATQELARQGEEILSQLYRPLETCNNSCYCKRCCYHCQMCFLNKGLGICYERKGRRRRTPKKTKTHPSPTPDKSISTRGDSQPTKKQKKTVEATVETDTGPGR−COOH
そして配列番号2、100または102に対し50%より高い;好ましくは60%より高い、さらに好ましくは70%より高い、より好ましくは80%より高い、更に好ましくは90%より高い配列相同性を有する実質モノマー型であるHIV−1およびHIV−2のその他Tat変異体は、ピコモルからナノモル濃度に於いて以下の判定基準の少なくとも1つを満たすことができる:
(i)ピコモル〜ナノモル濃度(0.01から1000ng/ml)、細胞への曝露5〜10分以内でDC、内皮細胞およびマクロファージを含む抗原提示細胞により選択的且つ効率的に取り込まれる。
(ii)ピコモル〜ナノモル濃度に於いてサイトカイン−活性化内皮細胞の移動(集合)、浸潤および増殖を促進する。
(iii)ナノモル濃度に於いてHIV遺伝子をトランス活性化するか、またはTat欠失HIVプロウイルスをレスキューする。
(iv)DCの成熟および抗原提示機能を活性化する。
(v)抗原提示細胞付加後の、それ自身または異種抗原に対するT−ヘルパーおよび/または細胞傷害活性を含むT細胞媒介免疫反応を高める。
5’ATGGAGCCAGTAGATCCTAGACTAGAGCCCTGGAAGCATCCAGGAAGTCAGCCTAAAACTGCTTGTACCAATTGCTATTGTAAAAAGTGTTGCTTTCATTGCCAAGTTTGTTTCATAACAAAAGCCTTAGGCATCTCCTATGGCAGGAAGAAGCGGAGACAGCGACGAAGACCTCCTCAAGGCAGTCAGACTCATCAAGTTTCTCTATCAAAGCAGCCCACCTCCCAATCCCGAGGGGACCCGACAGGCCCGAAGGAATGA3’
そしてHIV−2 tat DNAは以下に限定されるものではないが、例えば元の配列のヌクレオチド276と277の間にRGDをコードする配列が挿入されている以下のヌクレオチド配列で表される(配列番号99):
5’ATGGAGACACCCTTGAAGGCGCCAGAGAGCTCATTAAAGTCCTGCAACGAGCCCTTTTCACGCACTTCAGAGCAGGATGTGGCCACTCAAGAATTGGCCAGACAAGGGGAAATCCTCTCTCAGCTATACCGACCCCTAGAAACATGCAATAACTCATGCTATTGTAAGCGATGCTGCTACCATTGTCAGATGTGTTTTCTAAACAAGGGGCTCGGGATATGTTATGAACGAAAGGGCAGACGAAGAAGGACTCCAAAGAAAACTAAGACTCATCGAGGGGACCCGTCTCCTACACCAGACAAATCCATATCCAACAAGGACCGGGGACAGCCAGCCAACGAAGAAACAGAAGAAGACGGTGGAAGCAACGGTGGAGACAGATACTGGCCCTGGCCGATAG3’
または位置322〜324のスレオニンをコードする配列ACCが欠失している配列(配列番号101):
5’ATGGAGACACCCTTGAAGGCGCCAGAGAGCTCATTAAAGTCCTGCAACGAGCCCTTTTCACGCACTTCAGAGCAGGATGTGGCCACTCAAGAATTGGCCAGACAAGGGGAAATCCTCTCTCAGCTATACCGACCCCTAGAAACATGCAATAACTCATGCTATTGTAAGCGATGCTGCTACCATTGTCAGATGTGTTTTCTAAACAAGGGGCTCGGGATATGTTATGAACGAAAGGGCAGACGAAGAAGGACTCCAAAGAAAACTAAGACTCATCCGTCTCCTACACCAGACAAATCCATATCCAACAAGGACCGGGGACAGCCAGCCAACGAAGAAACAGAAGAAGACGGTGGAAGCAACGGTGGAGACAGATACTGGCCCTGGCCGATAG3
および配列番号1、99または101に対し40%より高い、好ましくは50%より高い、好ましくは60%より高い、より好ましくは70%より高い、更に好ましくは80%より高い、より好ましくは90%より高い配列相同性を持ついずれかのHIVタイプおよびサブタイプの他変異として表される。
配列番号3
5’CCCACCTCCCAATCCCGAGGGGACCCGACAGGCCCGAAGGAA3’
配列番号4
PTSQSRGDPTGPKE
配列番号5
5’ACCTCCCAATCCCGAGGGAGGGGACCCGACAGGCCCG3’
配列番号6
TSQSRGDPTGP
配列番号7
5’TCCCAATCCCGAGGGGACCCGACAGGC3’
配列番号8
SQSRGDPTG
配列番号9
5’CAATCCCGAGGGGACCCGACA3’
配列番号10
QSRGDPT
配列番号11
5’TCCCGAGGGGACCCG3’
配列番号12
SRGDP
配列番号13
5’TCCCGAGGGGACCCGACA3’
配列番号14
SRGDPT
配列番号15
5’TCCCGAGGGGACCCGACAGGC3’
配列番号16
SRGDPTG
配列番号17
5’CAATCCCGAGGGGACCCGACAGGC3’
配列番号18
QSRGDPTG
配列番号19
5’TGTACCAATTGCTATTGTAAAAAGTGTTGCTTTCATTGCCAAGTTTGT3’
配列番号20
CTNCYCKKCCFHCQVC
配列番号21
5’GGCAGGAAGAAGCGGAGACAGCGACGAAGACCTCCTCAAGGC3’
配列番号22
GRKKRRQRRRPPQG
配列番号23
5’TTCATAACAAAAGCCTTAGGCATCTCCTAT3’
配列番号24
FITKALGISY
配列番号25
5’ATGGAGCCAGTAGATCCTAGACTAGAGCCCTGGA亜GCATCCAGGAAGTCAGCCTAAAACT3’
配列番号26
MEPVDPRLEPWKHPGSQPKT
エピトープ1(aa1〜20):
5’ATGGAGCCAGTAGATCCTAGACTAGAGCCCTGGAAGCATCCAGGAAGTCAGCCTAAAACT3’(配列番号27)、
MEPVDPRLEPWKHPGSQPKE(配列番号28)、
エピトープ2(aa11〜24):
5’TGGAAGCATCCAGGAAGTCAGCCTAAAACTGCTTGTACCAAT3’(配列番号29)、
WKHPGSQPKTACTN(配列番号30)
エピトープ3(aa21〜40):
5’GCTTGTACCAATTGCTATTGTAAAAAGTGTTGCTTTCATTGCCAAGTTTGTTTCATAACA3’(配列番号31)、
ACTNCYCKKCCFHCQVCFIT(配列番号32)、
エピトープ4(aa36〜50):
5’GTTTGTTTCATAACAAAAGCCTTAGGCATCTCCTATGGCAGGAAG3’(配列番号33)、
VCFITKALGISYGRK(配列番号34)、
エピトープ5(aa83〜102):
5’GGCCCGAAGGAACAGAAGAAGAAGGTGGAGAGAGAGACAGAGACAGATCCGGTCCATCAG3’(配列番号35)、
5GPKEQKKKVERETETDPVHQ(配列番号36)、
エピトープ6(aa1〜15):
5’ATGGAGCCAGTAGATCCTAGACTAGAGCCCTGGAAGCATCCAGGA3’(配列番号37)
MEPVDPRLEPWKHPG(配列番号38)、
エピトープ7(aa6〜20):
5’CCTAGACTAGAGCCCTGGAAGCATCCAGGAAGTCAGCCTAAAACT3’(配列番号39)、
TCAGCCTAAAACT3’(配列番号40)、
エピトープ8(aa11〜25):
5’TGGAAGCATCCAGGAAGTCAGCCTAAAACTGCTTGTACCAATTGC3’(配列番号41)、
WKHPGSQPKTACTNC(配列番号42)、
エピトープ9(aa16〜30):
5’AGTCAGCCTAAAACTGCTTGTACCAATTGCTATTGTAAAAAGTGT3’(配列番号43)、
SQPKTACTNCYCKKC(配列番号44)、
エピトープ10(aa21〜35):
5’GCTTGTACCAATTGCTATTGTAAAAAGTGTTGCTTTCATTGCCAA3’(配列番号45)、
ACTNCYCKKCCFHCQ(配列番号46)、
エピトープ11(aa26〜40):
5’TATTGTAAAAAGTGTTGCTTTCATTGCCAAGTTTGTTTCATAACA3’(配列番号47)、
YCKKCCFHCQVCFIT(配列番号48)
エピトープ12(aa31〜45):
5’TGCTTTCATTGCCAAGTTTGTTTCATAACAAAAGCCTTAGGCATC3’(配列番号49)、
CFHCQVCFITKALGI(配列番号50)、
エピトープ13(aa36〜50):
5’TGCTTTCATTGCCAAGTTTGTTTCATAACAAAAGCCTTAGGCATC3’(配列番号51)、
VCFITKALGISYGRK(配列番号52)、
エピトープ14(aa41〜55):
5’AAAGCCTTAGGCATCTCCTATGGCAGGAAGAAGCGGAGACAGCGA3’(配列番号53)、
KALGISYGRKKRRQR(配列番号54)、
エピトープ15(aa46〜60):
5’TCCTATGGCAGGAAGAAGCGGAGACAGCGACGAAGACCTCCTCAA3’(配列番号55)、
SYGRKKRRQRRRPPQ(配列番号56)、
エピトープ16(aa51〜65):
5’AAGCGAGACAGCGACGAAGACCTCCTCAAGGCAGTCAGACTCAT3’(配列番号57)、
KRRQRRRPPQGSQTH(配列番号58)、
エピトープ17(aa56〜70):
5’CGAAGACCTCCTCAAGGCAGTCAGACTCATCAAGTTTCTCTATCA3’(配列番号59)、
RRPPQGSQTHQVSLS(配列番号60)、
エピトープ18(aa61〜75):
5’GGCAGTCAGACTCATCAAGTTTCTCTATCAAAGCAGCCCACCTCC3’(配列番号61)、
GSQTHQVSLSKQPTS(配列番号62)、
エピトープ19(aa66〜80):
5’CAAGTTTCTCTATCAAAGCAGCCCACCTCCCAATCCCGAGGGGAC3’(配列番号63)、
QVBSLSKQPTSQSRGD(配列番号64)、
エピトープ20(aa71〜85):
5’AAGCAGCCCACCTCCCAATCCCGAGGGGACCCGACAGGCCCGAAG3’(配列番号65)、
KQPTSQSRGDPTGPK(配列番号66)、
エピトープ21(aa76〜90):
5’CAGTCCCGAGGGGACCCGACAGGCCCGAAGGAACAGAAGAAGAAG3’(配列番号67)、
QSRGDPTGPKEQKKK(配列番号168)
エピトープ22(aa21〜29):
5’GCTTGTACCAATTGCTATTGTAAAAAG3’(配列番号69)、
ACTNCYCKK(配列番号70)、
エピトープ23(aa26〜34):
5’TATTGTAAAAAGTGTTGCTTTCATTGC3’(配列番号71)、
YCKKCCFHC(配列番号72)、
エピトープ24(aa31〜39):
5’TGCTTTCATTGCCAAGTTTGTTTCATA3’(配列番号73)、
CFHCQVCFI(配列番号74)、
エピトープ25(aa36〜44):
5’GTTTGTTTCATAACAAAAGCCTTAGGC3’(配列番号75)、
VCFITKALG(配列番号76)、
エピトープ26(aa41〜49):
5’AAAGCCTTAGGCATCTCCTATGGCAGG3’(配列番号77)
KALGISYGR(配列番号78)、
エピトープ27(aa46〜54):
5’TCCTATGGCAGGAAGAAGCGGAGACAG3’(配列番号79)、
SYGRKKRRQ(配列番号80)、
エピトープ28(aa51〜59):
5’AAGCGGAGACAGCGACGAAGACCTCCT3’(配列番号81)、
KRRQRRRPP(配列番号82)、
エピトープ29(aa56〜64):
5’CGAAGACCTCCTCAAGGCAGTCAGACT3’(配列番号83)、
RRPPQGSQT(配列番号84)
エピトープ30(aa61〜69):
5’GGCAGTCAGACTCATCAAGTTTCTCTA3’(配列番号85)、
GSQTHQVSL(配列番号86)、
エピトープ31(aa66〜74):
5’CAAGTTTCTCTATCAAAGCAGCCCACC3’(配列番号87)、
QVSLSKQPT(配列番号88)、
エピトープ32(aa71〜79):
5’AAGCAGCCCACCTCCCAATCCCGAGGG3’(配列番号89)、
KQPTSQSRG(配列番号90)、
エピトープ33(aa76〜84):
QSRGDPTGP(配列番号91)
5’CAATCCCGAGGGGACCCGACAGGCCCG3’(配列番号92)、
エピトープ34(aa81〜89):
5’CCGACAGGCCCGAAGGAACAGAAGAAG3’(配列番号93)。
PTGPKEQKK(配列番号94)。
cyc22突然変異体のヌクレオチド配列(配列番号95):
5’ATGGAGCCAGTAGATCCTAGACTAGAGCCCTGGAAGCATCCAGGAAGTCAGCCTAAAACTGCTGGTACCAATTGCTATTGTAAAAAGTGTTGCTTTCATTGCCAAGTTTGTTTCATAACAAAAGCCTTAGGCATCTCCTATGGCAGGAAGAAGCGGAGACAGCGACGAAGACCTCCTCAAGGCAGTCAGACTCATCAAGTTTCTCTATCAAAGCAGCCCACCTCCCAATCCCGAGGGGACCCGACAGGCCCGAAGGAATGA3’
cys22突然変異体のアミノ酸配列(配列番号96):
NH2−MEPVDPRLEPWKHPGSQPKTAGTNCYCKKCCFHCQVCFITKALGISYGRKKRRQRRRPPQGSQTHQVSLSKQPTSQSRGDPTGPKE−COOH
lys41のヌクレオチド配列(配列番号97):
5’ATGGAGCCAGTAGATCCTAGACTAGAGCCCTGGAAGCATCCAGGAAGTCAGCCTAAAACTGCTTGTACCAATTGCTATTGTAAAAAGTGTTGCTTTCATTGCCAAGTTTGTTTCATAACAAACGCCTTAGGCATCTCCTATGGCAGGAAGAAGCGGAGACAGCGACGAAGACCTCCTCAAGGCAGTCAGACTCATCAAGTTTCTCTATCAAAGCAGCCCACCTCCCAATCCCGAGGGGACCCGACAGGCCCGAAGGAATGA3’。
lys41のアミノ酸配列:
NH2−MEPVDPRLEPWKHPGSQPKTACTNCYCKKCCFHCQVCFITTALGISYGRKKRRQRRRPPQGSQTHQVSLSKQPTSQSRGDPTGPKE−COOH
−上記DNA配列と少なくとも60%の相同性を、好ましくは少なくとも70%の相同性を、より好ましくは少なくとも80%の、更に好ましくは少なくとも90%の相同性を有するDNA配列。
−上記アミノ酸配列と少なくとも40%の相同性を、好ましくは少なくとも50%の相同性を、好ましくは少なくとも60%の相同性を、好ましくは少なくとも70%の相同性を、より好ましくは少なくとも80%の、更に好ましくは少なくとも90%の相同性を有するアミノ酸配列。
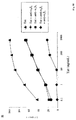
図1.生物活性Tatは用量−および時間−依存的な様式でMDDCに効率的且つ選択的に取り込まれるが、BLCLまたはTCBには取り込まれない。A、MDDCは14名の健康人ドナーの末梢血単核細胞から、確立された方法(Fanales Belasio 1997年)を用い得た。簡単に述べると、末梢血単核細胞(PBMC)は密度勾配分離法(Ficoll−Paque特級、Pharmacia Biotech、Uppsala、Sweden)を用い単離した。単核細胞は、抗CD14コーティングマイクロビーズ(Miltenyi Biotec、Bergisch Gladbach、Germany)と共にインキュベーションし、続いて磁石装置(MiniMacs Separation Unit、Miltenyi)を用いメーカー指示書に従いソーティングして更に精製した。単核細胞の純度は、フローサイトメトリー(FACScan、Becton Dickinson、S.Jose、CA)で評価した場合、常に>95%であった。GM−CSF(200ng/ml)(Leucomax、Novartis、Origgio、Italy)およびIL−4(100ng/ml)(Peprotech、London、UK)存在下に完全培地中で6日間培養し、単核細胞をDC(MDDC)に分化誘導した。DCへの分化は形態観察およびフローサイトメトリーによる特異的表面マーカー(HLA−DR、CD86、CD83、CD40、CD80)の検出によって評価した。次にMDDCを段階濃度(0.1から10,000ng/ml)の活性HIV−1 Tatまたは溶解バッファー若しくは培地のみ(陰性コントロール)存在下に、完全培地中2×105/mlの密度で5、10、30または60分間、37度、暗所で培養した。Tatタンパク質は既報の如くにE.coliから発現、精製し、使用前に既報の如くに細胞増殖アッセイとHIV−LTRトランス活性化を用い試験した。Tatタンパク質は0.1%ウシ血清アルブミンを含む脱気したリン酸緩衝生理食塩水(PBS−BSA)に再懸濁した。他所記載の様にTatが酸化され活性を失わないようにないように注意した。次に細胞を冷培地で洗浄し、10分間37度に於いてトリプシン−EDTA(Life−Technologies、Paisley、UK)で処理し、外部に結合したタンパク質を除いた。固定および透過処理後、MDDCをアフィニティー精製したウサギポリクローナル抗Tat IgG抗体(Ensoli 1993年;Chang 1997年;Ensoli 1994年)またはウサギIgGコントロール抗体(ICN Biomedicals、Opera、Italy)、続いてFITC−標識抗ウサギIg(Pierce、Rockford)で染色した。蛍光をフローサイトメトリーにより分析し、結果はイソタイプ染色サンプルと比較した場合の陽性細胞のパーセンテージとして表した。このタンパク質が細胞質内に特異局在していることを示すために、抗Tat抗体による染色については非透過処理MDDCについても必ず行った。陽性細胞のパーセンテージ(イソタイプ染色サンプルとの比較)をボックス内に示す。示したデータは試験した14名中の1ドナー名のものであるが、この名のTat取込レベルは、10分(0.1、1、10、100、1000および10、000ng/mlでは49%、52%、49%、70%、94%、98%容易性細胞)および30分(0.1、1、10、100、1000および10、000ng/mlでは49%、45%、54%、65%、95%、98%陽性細胞)いずれについても、試験した全ドナーに観察された値の中央値に近かった。B、MDDCを上記に従い活性Tatタンパク質(10〜1000ng/ml)と10または30分間インキュベーションして透過処理細胞または非透過処理細胞の両方についてFACSを用い分析し、Tatの特異的取込を立証した。C、BLCL(マル)は、2名の健康ドナーより得たヒトPBMCを、エプスタインバーウイルス産生株であるB95−8マーモセット細胞株より得た上清存在下に2時間培養し、更に既報の如くに少なくとも4週間更に拡大して作製した。TCB(三角)は4名の健康人ドナーから得たヒトPBMCを既報の如く1μg/mlのフィトヘマグルチニン(PHA)(Murex Diagnostics、Chatillon、France)で3日間刺激し、更に10 IU/mlのrIL−2(Becton Dickinson Labware、Bedford、MA)を加えた完全培地で2週間拡大して得た。BLCLおよびTCBを上記に従い5×105/mlで、100から10、000ng/mlの濃度の活性Tatを含む完全培地、溶解緩衝液、または培地にて30または60分間、37℃、暗所で培養してから染色し細胞内Tatを検出した。データを同一時間および同一用量のタンパク質を用い培養したMDDSのデータ(正方形)と比較し、このドナーのTat取込みレベルが試験した11名のドナーの中央値に非常に近かったため代表とした(10、100および1、000ng/mlのTat、30分培養後の陽性細胞のそれぞれ54%、範囲17〜91%、65%、範囲27〜91%、および95%、範囲83〜99%)。
パネルA、緩衝液と前インキュベーションした後、BSAとインキュベーションした。
パネルB、緩衝液と前インキュベーションした後、Tatとインキュベーションした。
パネルC、1μg/mlの未標識Tatと前インキュベーションした後、Tatとインキュベーションした。
パネルA、緩衝液と前インキュベーションした後、BSAとインキュベーションした。
パネルB、緩衝液と前インキュベーションした後、Tatとインキュベーションした。
パネルC、抗α5および抗β1モノクローナル抗体と前インキュベーションした後、Tatとインキュベーションした。
パネルD、抗αvおよび抗β3モノクローナル抗体と前インキュベーションした後、Tatとインキュベーションした。
パネルE、抗FVIII抗体(コントロール抗体)と前インキュベーションした後、Tatとインキュベーションした。
パネルA、緩衝液と前インキュベーションした後、BSAとインキュベーションした。
パネルB、緩衝液と前インキュベーションした後、Tatとインキュベーション。
パネルC、α5β1インテグリンのα鎖およびβ鎖に対するモノクローナル抗体と前インキュベーションした後Tatとインキュベーションした。
パネルD、αvβ3インテグリンのα鎖およびβ鎖に対するモノクローナル抗体と前インキュベーションした後、Tatとインキュベーションした。
パネルE、抗ヒト第VIII因子抗体(コントロール抗体)と前インキュベーションした後、Tatとインキュベーションした。
パネルA、緩衝液と前インキュベーションした後、BSAとインキュベーションした。
パネルB、緩衝液と前インキュベーションした後、Tatとインキュベーション。
パネルC、抗α5および抗β1モノクローナル抗体と前インキュベーションした後、Tatとインキュベーションした。
パネルD、抗αvおよび抗β3モノクローナル抗体と前インキュベーションした後、Tatとインキュベーションした。
パネルA、緩衝液と前インキュベーションした後、BSAとインキュベーションした。
パネルB、抗α5および抗β1モノクローナル抗体と前インキュベーションした後、Tatとインキュベーションした。
パネルC、抗αvおよび抗β3モノクローナル抗体と前インキュベーションした後、Tatとインキュベーションした。
活性TatはMDDCにより効率的に取込まれるが、TCBおよびBLCLには取り込まれない。
MDDCによる活性Tatの取込は細胞の成熟に伴い増加し、タンパク質の酸化および不活性化により消失する
未成熟なMDDCは抗原を貪食反応および飲反応によって取込む。成熟したDCはこれら活性を失うが、その一方で強い抗原提示能を獲得する。細胞の成熟が未変性Tatの取込みに影響するか実証するために、LPSを用いてMDDCを成熟させた。未成熟MDDCと比較し、成熟MDDCは高レベルのHLA−DR、CD83およびCD86表面マーカーを発現した。次に未成熟または成熟細胞を使用して取込み実験を行った。試験した全てのタンパク質濃度に於いて、MDDCの成熟によりTatの取込みは大きく増加した(図2A)。実際、成熟MDDCと低濃度のTatとのインキュベーションでは、最高濃度のTatと未成熟細胞とのインキュベーションに観察されたレベルの細胞内染色が認められた(図1Aおよび2A)。

抗α5β1および抗αvβ3モノクローナル抗体またはその天然リガンドであるFN若しくはVNによるMDDCの活性Tat取込み阻止
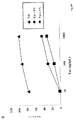
MDDCによる未変性Tatの取込みが特異的受容体介在エンドサイトーシス経路を辿るか検証するために、インテグリン受容体α5β1およびαvβ3に対する特異的モノクローナル抗体または競合リガンドを用いて実験を行った。これら受容体は各種タイプの細胞にある複数のTat結合受容体から選ばれたものであるが、それはこれら受容体が活性化された内皮細胞やKS細胞で強く発現しており、そしてこれら細胞がTat存在下に増殖、移動および接着し、それがα5β1およびαvβ3インテグリンへのTatのRGD領域の結合を介し行われているからである。図3A、Bに示すように、MDDCによるTatの取込みは、抗α5β1および抗αvβ3モノクローナル抗体により阻害され、この阻害はこれら抗体が両方存在するとき完全である(図3A、B)。これら受容体の天然リガンド、即ちFNおよびVNは同様の様式でTat取込みを阻止する(図4A、B)。即ち、α5β1およびαvβ1インテグリンはMDDCに於けるTatの侵入を介在する。この取込み経路はピコモル〜ナノモルのタンパク質濃度の時に主流であるが、これより高いTat濃度ではまだこのTat侵入阻止は認められるものの完全ではない(図3AおよびB)。このことはMDDCがTat取込み関して少なくとも2つの経路を利用しており、低Tat濃度(ピコモル〜ナノモル)で機能する第一経路はインテグリン介在エンドサイトーシスであり、一方より高いTat濃度では低親和性の細胞表面相互作用も共存する。
活性TatはMDMおよびMDDCに用量−および時間−依存的様式で効率的に取り込まれるが、単核細胞には取り込まれない。
MDDCは単核細胞由来であること、そして単核細胞およびMDMはリンパ細胞に対し抗原を効率的に提示することが知られていることから、これら細胞からの活性Tatの取込みを分析し、同一ドナーより得たMDDCの取込みと比較した。単核細胞を末梢血から精製し、MDDCまたはMDMへ分化誘導した(図1および図5の記載の通りにして)。図5に示すように、単核細胞は活性Tatを最高用量時のみ取り込んだ(1、000および10、000ng/mlでそれぞれ45%および67%)が、同時に細胞表面への結合も認められた(非透過処理細胞でそれぞれ19%および33%)。MDMはTatを単核細胞に比べより効率的に取込み、比較的低容量(10ng/mlおよび100ng/mlでそれぞれ32%および43%)でも、そして表面へのタンパク質結合が観察されるものの(非透過処理細胞で30%)試験した最高Tat用量まで(10、000ng/mlで72%)染色が認められた。同一ドナーのMDDCは10分および30分の両時点において、試験した最低用量(0.1ng/ml)でも単核細胞およびMDMより効率的にTatを取込んだ(それぞれ22%および13%)。最高用量では(1000ng/ml)Tatへ曝露した10分および30分後に、MDDCの80%および73%が陽性であった。全てのタンパク質用量於いて、MDDCおよびMDMは単核細胞に比べより効率的にTatを顕著に取込んだ。MDDCおよびMDMは共に単核細胞から分化することから、これらデータは細胞分化が特異的にTatタンパク質を標的とし取込むメカニズムを持った細胞をもたらすことを示している。
HIV−1 Tatタンパク質の塩基性領域およびRGDドメインを包含するペプチドによる活性Tat取込み阻害。
Tatのどのドメインがこの生物活性タンパク質の取込みに関係しているか検証するために、MDDCをN末端領域N−末端(1〜15+6〜20)、富システイン領域(21〜35+26〜40)、塩基性領域(46〜60+51〜65)、RGD領域(66〜80+71〜85)(図6、パネルAおよびB)をカバーする各種Tatペプチド(15mer)と前インキュベーションして阻止実験を行った。N末端領域と富システイン領域をカバーするペプチドは試験したいずれの用量(0.1〜1、000ng/ml)のTat取込みにも影響しなかった(図7、パネルA)。塩基性領域をカバーするペプチドは高用量(100および1000ng/ml)のTatの取込みを大きく低下したが、低濃度(0.1〜10ng/ml)の生物活性Tatタンパク質の取込みには影響しなかった。これとは逆に、RGD領域をカバーするペプチドは10ng/mlまでの用量のTatの取込を無効にし、そして100と1000ng/mlの取込を顕著に低下させた。Tatペプチド46〜60と66〜80の組合せが10および100ng/ml用量の生物活性Tatの取込を無効にしたことに注意(図7、パネルBおよびC)。同一実験条件下に於いてN末端とシステイン領域の両方をカバーする長いTatペプチド(1〜38)(図6、パネルC)は生物活性Tatの取込みに影響しなかったが、システインと塩基性領域の両方をカバーするTatペプチド21〜58は、100および1000ng/mlのTat取込みを顕著に低下させ、そしてRGD領域もカバーするTatペプチド57〜102は100ng/ml以下の用量のTat取込みを無効にした(図7、パネルD)。従って、RGDモチーフを包含するペプチドによるα5β1およびαvβ3インテグリンの遮断は、ピコモル〜ナノモル濃度の生物活性Tatの取込みを無効にし、一方より高濃度(ナノからマイクロモル、10〜1、000ng/ml)濃度のTat取込みはヘパリン結合プロテオグリカンとの相互作用を介してTat塩基性領域により仲介されている。これと関連し、塩基性およびRGD領域を共に包含するペプチドの組合せは、生物活性Tatタンパク質の結合経路と取込み経路の両方を妨害することで、試験した用量全てに於いてTat取込みを完全に無効にする。従って、これらデータは最適なTat取込みには両方のドメインが必要であることを示すが、同時に効率的(即ち低濃度での)および選択的な{(即ちMDDCおよび活性化EC(以下参照)の様なα5β1およびαvβ3インテグリン発現型細胞}Tat取込みにとってはRGD領域が重要であり、塩基性領域は高濃度Tatの取込みのみに関係していること、そしてこれはあらゆるタイプの細胞に見られることから標的特異性を欠くものであることも示している。
炎症性サイトカインによる細胞活性化前後での初代内皮細胞による活性Tatの取込み
内皮細胞による活性Tatの取込みを分析するために、HUVECを炎症性サイトカインで活性化し、または活性化しないままローダミン標識Tatによる取込み試験に用いた。ローダミン標識Tatが未標識Tatと同一濃度域に於いてKS細胞の増殖をまだ促進できることから、ローダミン標識後もTatは活性であった。このことは蛍光ラベルの結合がその生物機能を害さないことを示している。細胞を広に濃度範囲のTatに曝露した。更に、取込み阻害実験(以下参照)に合わせるために、細胞をウシ胎児血清を含まない培地と4℃、2時間前インキュベーションした。この前インキュベーションはその後のローダミン標識Tatの取込みに影響しない。ローダミン標識Tatを用いた場合、活性化HUVEC(IC−HUVEC)の核または仁へのタンパク質の取込みおよび移動は10ng/mlローダミン標識Tatとのインキュベーション15分以内に明瞭に検出できるようになった(図8)。細胞内Tat活性は用量−依存的(図8)および時間−依存的様式(図9および10)で増加した。ローダミン標識BSAまたは緩衝液(陰性コントロール)は殆ど、または全く取込を示さなかった(図8、9、10)。
未標識タンパク質による活性Tatの取込み阻害
ローダミン標識Tatタンパク質のIC−HUVECによる取込が固有な飽和経路を介したものであるか検証するために、10ng/mlから1μg/mlの濃度域のローダミンTatとインキュベーションする前に、IC−HUVECを1μg/mlの未標識Tatとインキュベーションした。この操作によりローダミン標識Tatの取込みはほぼ完全に阻害され(図13および表II)、その蛍光レベルは陰性コントロール(BSA)のレベルと同等であった。このことは、IC−HUVECによるTatタンパク質の取込は特異的且つ飽和型であることを示しており、このプロセスに受容体が関係していることを示唆している。
α5β1およびαvβ3インテグリンに対するモノクローナル抗体、またはそれらのリガンドであるFNおよびVNによるピコモル濃度(10から100ng/ml)の活性Tatの取込み阻害。
1μg/mlの活性Tat取込へのインテグリン受容体の介在は部分的である
より高濃度(ナノモルからマイクロモル)の活性Tatの取込みへのインテグリンの関与を決定するために、IC−HUVECを1μg/mlのローダミン標識Tatとインキュベーションした。15分以内に細胞内に非常に強い蛍光シグナルが観察された(図8パネルD、図10パネルAおよび図16パネルB)。インキュベーションのこの時点で、細胞をα5β1(図16パネルC)またはαvβ3(図16パネルD)に対するモノクローナル抗体で前処理すると、若干のTat取込み阻害が見られたが、それは10または100ng/mlのTatに観察されたものより低かった。IC−HUVECを1μg/mlのローダミン標識Tatとより長時間インキュベーションすると(60分)、抗α5β1または抗αvβ3モノクローナル抗体と前インキュベーションしてもTat取込みは阻害されなかった(図17)。これらの結果は、この濃度とインキュベーション時間ではインテグリン依存型の取込は飽和しており、別のTat取込経路が主流であることを示している。
活性Tatの取込を介在するTatドメイン
どのTatドメインがピコモル〜ナノモルおよびマイクロモル濃度のタンパク質取込みを担っているか検証するために、IC−HUVECをRGD領域(Tat65〜80)および塩基性領域(Tat46〜60)をカバーするTatペプチドと前インキュベーションして阻害実験を行った(表IV)。Tatペプチド11〜24を陰性コントロールに使用した。ローダミン標識Tatを細胞に加えた場合、ピコモル濃度のTatの取込はTatペプチド65〜80によって阻害されたが、Tat46〜60または11〜24では阻害されなかった(表IV)。これに対し細胞外Tatが高濃度(1μg/ml)の場合、取込みはTat65〜80では阻害されなかったが、Tat46〜60に若干の阻害作用が認められた。このことから、ピコモル〜ナノモル濃度のTatはTat RGD領域とα5β1およびαvβ3インテグリンとの相互作用を介していることが確認される。これに対し高濃度(ナノからマイクロモル)のTatの取込は、ヘパリン結合プロテオグリカンとの相互作用を介してTat塩基性領域が媒介する。即ち、RGD領域は効率的なTat取込みとって重要であるのに対し、塩基性領域は高濃度のTat取込みに関係しているが、kの取込はいずれのタイプの細胞にも認められることから標的特異性を欠いたものである。
活性TatはMDDCの活性化および成熟化を誘導するが、酸化・不活性化Tatは誘導しない。
MDDCの活性化と成熟化への活性Tatタンパク質の作用を評価するために、MHC、HLA−ABCおよびHLA−DRの表面発現、並びに補助刺激分子CD40、CD80、CD86およびCD83の表面発現を、タンパク質、完全培地、溶解用緩衝液またはLPS(陽性コントロール)存在下に18時間培養した細胞をサイトフローメトリーで分析した。10名のドナーについて得られた結果からは、Tatが細胞毒性なしにMHCおよび補助刺激分子の発現増加を用量−依存的に誘導することが示された(表V、パネルA)。平均蛍光強度(MFI)の顕著な増加がHLA−ABC(3/6ドナー、平均37%)、HLA−DR(10/10、平均49%)、CD40(6/10、平均35%)、CD80(8/8、平均50%)、CD83(9/10、平均164%)およびCD86(10/10、平均140%)について観察された。溶解用緩衝液または培地単独では、分析した分子の発現レベルに変化は生じなかった。注目すべきことに、Tatの酸化および不活性化(表I)によって、このタンパク質が持つMDCCに対するMHCおよび補助刺激分子のアップレギュレーション能力が顕著に低下した(表V、パネルB)。即ち、活性TatのみがMDDCの活性化と成熟化を促進する。
細胞を未変性または酸化し不活性化したTat、溶解用緩衝液、完全培地、またはLPS(10μg/ml)に18時間曝露し、次の蛍光色素標識モノクローナル抗体:FITC−またはPE標識IgGイソタイプ、FITC標識抗CD14およびHLA−DR(Becton−Dickinson)、FITC標識抗−CD40、−CD80、−CD83抗体、PE標識抗CD86抗体(Pharmingen、San Diego、CA)で染色した後フローサイトメトリーを用い解析した。パネルAには10名のドナーより得たMDDCでの表面分子の発現がTat緩衝液(Tatについて)または培地(LPSについて)とインキュベーションした細胞の平均蛍光強度(MFI)と比較した場合のMFIの%増加として報告されている。1例の代表ドナーの酸化型と未変性Tat(20μg/ml)比較データをパネルBに示す。HLAおよび補助刺激分子の誘導に関しては、LPSと共に培養したMDDCを陽性コントロールとして使用した。
未変性または酸化Tat存在下に培養したMDDCは常に生存可能であり、培地または溶解用緩衝液で処理したMDDCと異なる点はなかった(データ未提示)。
未変性TatはMDDCを活性化し、MDDCによるTh−1型サイトカインおよびβ−ケモカインの産生を高めるが、酸化され不活性化されたTatはしない。
DC活性化に及ぼす活性Tatの影響を評価するために、免疫細胞を活性化しTh−1型反応を誘導することが知られているサイトカインIL−12およびTNF−αの産生、並びに免疫反応の既知メディエイターであるβ−ケモカインであるRANTES、MIP−1αおよびMIP−1βの産生を、タンパク質、溶解用緩衝液(陰性コントロール)またはLPS(陽性コントロール)と共に18時間培養した細胞の上清をELISAにかけ調べた。(図18)。活性TatとのインキュベーションはIL−12およびTNFαのレベルの用量依存的増加を誘導し、最高用量のTatでは緩衝液のみで処理した細胞に比べIL−12については23倍(p<0.02)、TNF−αについては20倍(p<0.03)増加した。同様に、Tatは用量依存的様式でPANTES(10倍、p<0.02)、MIP−1α(97倍、p<0.005)0.005)およびMIP−1β(15倍、p<0.01)の産生を顕著に高めた。溶解用緩衝液は影響を及ぼさなかったが、LPS(陽性コントロール)はサイトカインとβ−ケモカインの両方の産生を高めた。
活性TatはMDCCによる同種提示を増加する
MDDCの抗原提示能力に及ぼすTatの作用を評価するために、細胞をTatタンパク質、溶解用緩衝液、完全培地またはLPS(陽性コントロール)に曝露し、連続する細胞対細胞比で同種PBLと共培養した(図19)。このアッセイを選んだ理由は、特定抗原に特異的ではないが、MDDCの抗原提示機能全体について適切な情報を提供するかである。未処理のMDDCは同種リンパ細胞の増殖を、使用したAPC数に応じて若干増加した。しかし同種PBLの増殖反応はTatでパルス処理したMDDCにより顕著に増加し(3.3倍、p<0.01、最高DC/PBL比時)、LPSによる誘導(同一細胞対細胞比で3.8倍、p<0.005)と同レベルに達した。これに対しMDDCを溶解用緩衝液で処理した場合には同種リンパ細胞の増殖促進は観察されなかった(図19)。即ち、未変性TatはDCの提示機能を高める。
活性TatはMDDCの異種抗原提示および特異的T細胞反応を高める
MDDCの抗原特異的提示能力に及ぼす活性Tatの作用を、細胞をTatタンパク質、溶解用緩衝液または完全培地で一過性に処理し、追想抗原TT存在下に自己リンパ細胞と一緒に培養し評価した。図20に示すように、未処理MDDCは解析した3名中2名のドナーで自己リンパ細胞のTT特異的増殖を誘導し(それぞれS.I.15.4および7.3)、そしてこの作用が活性Tat処理により高まる(それぞれS.I. 27.9および12.5)が、溶解用緩衝液による処理では高まらなかった(それぞれS.I.13および5.9)。Tat処理MDDCはTTに反応しなかった被験者ではTTに対するリンパ細胞増殖を誘導しなかった。即ち、Tatは他の抗原に対する特異的T細胞反応を高めることができる。
SIV Gag+MALP−2粘膜アジュバントと組合わせて生物活性Tatでマウスを免疫すると、Gag+MALP−2によるワクチン接種に比べより強力な液性および細胞性免疫反応を誘導する。
MDDC活性に対する作用から明らかなように生物活性Tatには強力なアジュバント活性があることから、名目抗原と共に粘膜投与した場合に粘膜粘膜および全身免疫反応の両方を高めることが期待される(図21の2群)。この目的のために、Balb/cマウス(図21の2群)に生物活性Tat(10μg)と混合した10μgのSIV Gagタンパク質を用いて2回皮内プライミングを行った後、Tat+Gag+Malp−2で鼻内ブーストを2回行った。Elisaにより決定されたこれら動物に誘導された抗Gag抗体のレベルは、Gag単独で免疫された動物(図21および22の1群)に見られたレベルに比べ高かった(図22の2群)。より重要なことは、古典的な5時間の51クロム放出アッセイを用い、エフェクター細胞(外来性IL−12存在下にGagペプチドのプールと共に6日間培養した脾臓細胞)を加える前に3〜4時間Gagペプチド(標的)のプールでパルス処理されたp815マウス肥満細胞腫細胞の殺滅度を測定したところ、Th−1型T細胞免疫反応が誘導されたと判定されたことである。液性および細胞性免疫反応は共にTatなしに上記免疫を行ったコントロールグループに誘導されたものより強力であった(図23)。即ち、生物活性Tatを組合わせた名目抗原による免疫は、名目抗原のみで免疫したマウスに比べより優れた免疫反応を生じた。具体的には、実質的に高い名目抗原に対する血清IgGの力価およびCTL反応が認められた。同様に生物活性Tatによる粘膜免疫反応(即ち肺や膣洗浄液からの分泌性IgA)も増加することが予想される。
ベクターDNA単独または他のHIV抗原を発現するDNA結合タンパク質と組合せ発現させた生物活性Tatでサルを全身免疫すると、他抗原に対するTh−1反応を増し、ウイルス攻撃に対しより効果的な感染制御ができる。
Tat−DNA発現ベクター単独または他抗原発現DNAとの組合せによるサルの全身免疫は、免疫抗原に対するT細胞免疫反応およびIFNγElispot(Th−1型)を誘導し、そしてTat無しで他の抗原によりワクチン接種を行った場合に比べ他抗原に対するTh−1反応を増加することが期待される。更に、Env、GagおよびPolの様な他のHIV抗原とTatとの組合わせは、病原性SHIV89.6Pによる攻撃に対しTat単独でのワクチン接種に比べより優れた予防を提供することが期待される。
2.アルビーニ、エイら、Proc.Natl.Acad.Sci.USA 92:4838、1995年。
3.アルビーニ、エイら、Nat.Med.2:1371、1996年。
4.アルビーニ、エイら、Proc.Natl.Acad.Sci.USA 95:13153、1998年a。
5.アルビーニ、エイら、J.Biol.Chem.273:15895、1998年b。
7.アリャ、エス、ケイら、Science 229:69、1985年。
8.バドウ、エイら、J.Virol.74:10551、2000年
9.バンチェリュー、ジェイとアール、エム、シュタインマン.Nature 392:245、1998年。
10.バリラーリ、ジーら、J.Immunol.149:3727、1992年。
12.バリラーリ、ジーら、J.Immunol.163:1929、1999年a。
13.バリラーリ、ジーら、Blood 94:663、1999年b。
14.ベル、ディーら、Adv.Immunol.72:255、1999年。
15.ベネリー、アールら、AIDS 12:261、1998年。
17.ボイキンズ、アール、エイら、J.Immunol.163:15、1999年。
18.カファロ、エイら、Nat.Med.5:643、1999年。
19.カファロ、エイら、J.Med.Primatol.29:193、2000年。
20.カファロ、エイら、Vaccine 19:2862、2001年。
22.チャン、エイチ−ケイら、J.Biomed.Sci.2:189、1995年。
23.キルムール、エヌら、J.Virol.69:492、1995年。
24.コーエン、エス、エスら、Proc.Natl.Acad.Sci.USA 96:10842、1999年。
25.デロッシ、ディーら、Trends Cell.Biol.8:84、1998年。
27.エンゾーリ、ビーら、J.Virol.67:277、1993年。
28.エンゾーリ、ビーら、Nature 371:674、1994年。
29.ファナーレス−ベラッシオ、イーら、J.Immunol.159:2203、1997年。
30.ファーウェル、エスら、Proc.Natl.Acad.Sci.USA 91:664、1994年。
32.フィレーリ、ブイら、J.Immunol.162:1165、1999年。
33.フィシャー、エイ、ジーら、Nature 320:367、1986年。
34.フランケル、エイ、ディーとシー、オー、パボ、Cell 55:1189、1988年。
35.フランケル、エイ、ディーら、Proc.Natl.Acad.Sci.USA 86:7397、1989年。
37.フローベル、ケイ、エスら、AIDS Res.Hum.Retroviruses 10suppl。2:S83、1994年。
38.ギャロ、R.C.Proc.Natl.Acad.Sci.USA 96:8324、1999年。
39.ガンジュ、アール、ケイら、J.Virol.72:6131、1998年。
40.ゴールドシュタイン、ジーら、Vaccine 18:2789、2000年。
42.ホワン、エルら、J.Virol.72:8952、1998年
43.イトー、エムら、AIDS Res.Hum.Retroviruses 14:845、1998年。
44.キム、ディー、ティーら、J.Immunol.159:1166、1997年。
45.コルソン、ディー、エルら、AIDS Res.Hum.Retroviruses 9:677、1993年。
47.リー、シー、ジェイら、Science 268:429、1995年。
48.リー、シー、ジェイら、Proc.Natl.Acad.Sci.USA 94:8116,1997年。
49.マン、ディー、エイとエイ、ディー、フランケル、EMBO J.10:1733、1991年。
50.マソード、アールら、Biochem.Biophys.Res.Commun.202:374、1994年
52.ミシュレッティ、エフら、Eur.J.Immunol.29:2579,1999年。
53.ミラーニ、ディーら、J.Biol.Chem.271:22961,1999年。
54.ミトーラ、エスら、J.Virol.74:344、2000年。
55.ミトーラ、エスら、Blood 90:1365、1997年。
57.モーゼル、ビーとピー、レスチャー、Nat.Immunol.2:123、2001年。
58.モイ、ピーら、Mol.Biotechnol.6:105、1996年。
59.オスターハウス、エイ、ディーら、Vaccine 17:2713、1999年。
60.オッツ、エムら、Science 275:1481、1997年。
62.ピティス、エム、ジーら、Viral.Immunol。9:169、1996年。
63.ポーバー、ジェイ、エス、「内皮細胞」、ウナ、エス、リャン(編集) C.R.C.Press Inc.,Boca Raton、Florida、1998年。
64.ポギー、エイら、J.Biol.Chem.273:7025、1998年。
65.パルビス、エス、エフら、AIDS Res.Hum.Retroviruses 11:443、1995年。
67.リー、エム、シーら、J.Acquir, Immune Defic.Syndr.Hum.Retrovirol.10:408、1995年。
68.レイズ、ピーら、J.Med.Virol.30:163、1990年。
69.ロッドマン、ティー、シーら、Proc.Natl.Acad.Sci.USA 90:7719、1993年。
70.ロマーニャイ、エス、Immunol.Today 18:263、1997年。
72.ラスナッチ、エムら、J.Biol.Chem.273:16027、1998年。
73.サバティエ、ジェイ、エムら、J.Virol.65:961、1991年。
74.サファイ、ビーら、Ann.Intern.Med.103:744、1985年。
75サマニエゴ、エフら、Am.J.Pathol.152:433、1998年。
77.シュパイサー、ピー、ピーら、Methods Find Exp.Clin.Pharmacol.13:337、1991年。
78.サブラマンヤム、エムら、J.Immunol.150:2544、1993年。
79.タケウリ、エイチら、Adv.Drug Deliv.Rev.47:39、2001年。
80.チャギ、エムら、J.Biol.Chem.276:3254、2001年。
82.ベネット、エイら、J.Immunol.148:2889、1992年。
83.ビシディ、アール、ピーら、Science 246:1606、1989年。
84.フォーゲル、ビー、イーら、J.Cell.Biol.121:461、1993年。
85.ウイークス、ビー、エスら、J.Biol.Chem.268:5279、1993年。
87.ウエステンドロップ、エム、オーら、Nature 375:497、1995年。
88.レンジャー、エスら、FEBS Lett.383:145、1996年。
89.レンジャー、エスら、J.Biol.Chem.272:30283、1997年。
90.ウー、エム、エックスとエス、エフ、シュロスマン。Proc.Natl.Acad.Sci.USA 94:13832、1997年。
92.ザグリー、ジェイ、エフら、J.Hum.Virol.1:282、1998年。
93.ザウリ、ジーら、Cancer Res.53:4481、1993年。
94.ザウリ、ジーら、J.Acquir.Immune Defic.Syndr.Hum.Retrovirol.10:306、1995年a。
95.ザウリ、ジーら、Blood 86:3823、1995年b。
Claims (34)
- 単離された未変性の実質モノマーである生物活性HIV−1 Tatであって、配列番号2に記載のアミノ酸配列を有するHIV−1 Tatを含んでなる医薬品組成物であって、
前記HIV−1 Tatは:
(i)アジュバント活性を有し;且つ
(ii)下記の:
(a)腫瘍細胞の抗原;及び
(b)HIVではない病原体の抗原であって、前記病原体は、ウィルス、結核菌、リステリア、連鎖球菌、ブドウ球菌、肺炎双球菌、破傷風、髄膜炎菌、ヘリコバクター、サルモネラ、コレラ菌又はカンジダである、抗原;
からなる群から選択された抗原分子と混合されていることを特徴とする医薬品組成物。 - 前記HIV−1 Tatは、選択的に、α5β1および/またはαvβ3インテグリン発現抗原提示細胞を標的とし、該細胞に結合し、又は該細胞に侵入する、請求項1に記載の医薬品組成物。
- 前記抗原提示細胞は、樹状細胞、内皮細胞又はマクロファージである、請求項2に記載の医薬品組成物。
- 前記HIV−1 Tatは、抗原提示細胞の成熟又は抗原提示機能をさらに誘導する、請求項1〜3のいずれか一項に記載の医薬品組成物。
- 抗炎症薬、抗血管新生分子、及び細胞傷害性抗腫瘍薬からなる群から選択される治療分子をさらに含んで成る、請求項1〜4のいずれか一項に記載の医薬品組成物。
- 前記抗原分子は、腫瘍細胞の抗原である、請求項1〜5のいずれか一項に記載の医薬品組成物。
- 前記腫瘍細胞は、軟組織、骨、血液、肺、結腸、乳房、前立腺、皮膚、肝臓、膵臓、内分泌組織、子宮、卵巣又はリンパ細胞である、請求項6に記載の医薬品組成物。
- 医薬として使用するための、請求項1〜7のいずれか一項に記載の医薬品組成物。
- 腫瘍の処置又は予防に使用するための、請求項6又は7に記載の医薬品組成物。
- 前記腫瘍は、軟組織、骨又は血液の良性又は悪性の腫瘍である、請求項9に記載の医薬品組成物。
- 前記腫瘍は、肉腫である、請求項9に記載の医薬品組成物。
- 前記腫瘍は、急性若しくは慢性の白血病、又は肺、結腸、乳房、前立腺、皮膚、肝臓、膵臓、内分泌組織、子宮、卵巣、若しくはリンパ球の新生物である、請求項9に記載の医薬品組成物。
- 腫瘍の処置又は予防のためのワクチンの製造への、請求項6又は7に記載の医薬品組成物の使用。
- 前記腫瘍は、軟組織、骨又は血液の良性又は悪性の腫瘍である、請求項13に記載の使用。
- 前記腫瘍は、肉腫である、請求項13に記載の使用。
- 前記腫瘍は、急性若しくは慢性の白血病、又は肺、結腸、乳房、前立腺、皮膚、肝臓、膵臓、内分泌組織、子宮、卵巣、若しくはリンパ球の新生物である、請求項13に記載の使用。
- 前記抗原分子は、HIVではない病原体の抗原である、請求項1〜5のいずれか一項に記載の医薬品組成物。
- 前記病原体は、ウィルスである、請求項17に記載の医薬品組成物。
- 前記ウィルスは、ヘルペスウイルス、肝炎ウイルス、インフルエンザウイルス、水痘、エプスタインバーウィルス、ヒトヘルペス8型ウイルス、サイトメガロウイルス、又はパピローマウイルスである、請求項18に記載の医薬品組成物。
- 前記病原体は、結核菌、リステリア、連鎖球菌、ブドウ球菌、肺炎球菌、破傷風菌、髄膜炎菌、ヘリコバクター、サルモネラ菌、コレラ菌又はカンジダである、請求項17に記載の医薬品組成物。
- 医薬として使用するための、請求項17〜20のいずれか一項に記載の医薬品組成物。
- 病原体を原因とする感染症の処置又は予防に使用するための、請求項17〜20のいずれか一項に記載の医薬品組成物。
- 病原体を原因とする感染症の処置又は予防に使用するための、医薬品組成物であって、感染症が、ヘルペス感染、肝炎ウイルス感染、結核、マラリア、カンジダ症、リステリア感染、インフルエンザ感染、性感染症、心内膜炎、尿路感染、骨髄炎、皮膚感染、連鎖球菌感染、ブドウ球菌感染、肺炎球菌感染、破傷風、髄膜炎菌感染症、ヘリコバクター感染、サルモネラ、梅毒、水痘、伝染性単核球症、ヒトヘルペスウイルス8型感染、サイトメガロウイルス感染、口唇ヘルペス、陰部ヘルペス、パピローマウイルス原因感染、又はコレラ菌感染である、請求項17に記載の医薬品組成物。
- 病原体を原因とする感染症の処置又は予防のためのワクチンの調製への、請求項17〜20のいずれか一項に記載の医薬品組成物の使用。
- 病原体を原因とする感染症の処置又は予防に使用するためのワクチンの調製への、医薬品組成物の使用であって、感染症が、ヘルペス感染、肝炎ウイルス感染、結核、マラリア、カンジダ症、リステリア感染、インフルエンザ感染、性感染症、心内膜炎、尿路感染、骨髄炎、皮膚感染、連鎖球菌感染、ブドウ球菌感染、肺炎球菌感染、破傷風、髄膜炎菌感染症、ヘリコバクター感染、サルモネラ、梅毒、水痘、伝染性単核球症、ヒトヘルペスウイルス8型感染、サイトメガロウイルス感染、口唇ヘルペス、陰部ヘルペス、パピローマウイルス原因感染、又はコレラ菌感染である、請求項17に記載の医薬品組成物の使用。
- HIVでない病原体を原因とする感染症の処置又は予防に使用する医薬品組成物であって、単離された未変性の実質モノマーである生物活性HIV−1 Tatであって、配列番号2に記載のアミノ酸配列を有するHIV−1 Tatを含んでなり、
前記HIV−1 Tatは:
(i)アジュバント活性を有し;且つ
(ii)病原体の抗原からなる抗原分子と混合されていることを特徴とする医薬品組成物。 - 前記HIV−1 Tatは、選択的に、α5β1および/またはαvβ3インテグリン発現抗原提示細胞を標的とし、該細胞に結合し、又は該細胞に侵入する、請求項26に記載の医薬品組成物。
- 前記抗原提示細胞は、樹状細胞、内皮細胞又はマクロファージである、請求項27に記載の医薬品組成物。
- 前記HIV−1 Tatは、抗原提示細胞の成熟又は抗原提示機能をさらに誘導する、請求項26〜28のいずれか一項に記載の医薬品組成物。
- 抗炎症薬、抗血管新生分子、及び細胞傷害性抗腫瘍薬からなる群から選択される治療分子をさらに含んで成る、請求項26〜29のいずれか一項に記載の医薬品組成物。
- 前記病原体は、ウィルスである、請求項26〜30のいずれか一項に記載の医薬品組成物。
- 前記ウィルスは、ヘルペスウイルス、肝炎ウイルス、インフルエンザウイルス、水痘、エプスタインバーウィルス、ヒトヘルペス8型ウイルス、サイトメガロウイルス、又はパピローマウイルスである、請求項31に記載の医薬品組成物。
- 前記病原体は、結核菌、リステリア、連鎖球菌、ブドウ球菌、肺炎球菌、破傷風菌、髄膜炎菌、ヘリコバクター、サルモネラ菌、コレラ菌又はカンジダである、請求項26〜30のいずれか一項に記載の医薬品組成物。
- 病原体を原因とする感染症の処置又は予防のためのワクチンの調製への、請求項26〜33のいずれか一項に記載の医薬品組成物の使用。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP01118114A EP1279404A1 (en) | 2001-07-26 | 2001-07-26 | Use of HIV-1 tat, fragments or derivatives thereof, to target or to activate antigen-presenting cells, to deliver cargo molecules for vaccination or to treat other diseases |
| EP01118114.6 | 2001-07-26 | ||
| PCT/EP2002/008377 WO2003009867A1 (en) | 2001-07-26 | 2002-07-26 | Use of biologically active hiv-1 tat, fragments or derivates thereof, to target and/or to activate antigen-presenting cells, and/or to deliver cargo molecules for preventive or therapeutic vaccination and/or to treat other diseases |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2005505520A JP2005505520A (ja) | 2005-02-24 |
| JP5134183B2 true JP5134183B2 (ja) | 2013-01-30 |
Family
ID=8178141
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003515259A Expired - Fee Related JP5134183B2 (ja) | 2001-07-26 | 2002-07-26 | 予防または治療ワクチン接種および/或いはその他疾患の治療を目的とする、抗原提示細胞を標的とすることおよび/または活性化すること、並びに/またはカーゴ分子を運搬することへの生物学的活性hiv−1tat、その断片または誘導体の使用 |
Country Status (19)
| Country | Link |
|---|---|
| US (1) | US7811573B2 (ja) |
| EP (2) | EP1279404A1 (ja) |
| JP (1) | JP5134183B2 (ja) |
| CN (1) | CN1318091C (ja) |
| AP (1) | AP2004002986A0 (ja) |
| AT (1) | ATE510558T1 (ja) |
| AU (1) | AU2002331350B2 (ja) |
| BR (1) | BR0211474A (ja) |
| CA (1) | CA2455227C (ja) |
| DK (1) | DK1425035T3 (ja) |
| EA (1) | EA006850B1 (ja) |
| ES (1) | ES2367255T3 (ja) |
| HU (1) | HUP0400660A2 (ja) |
| IL (1) | IL160015A0 (ja) |
| MX (1) | MXPA04000287A (ja) |
| NZ (1) | NZ531364A (ja) |
| OA (1) | OA12701A (ja) |
| PL (1) | PL368243A1 (ja) |
| WO (1) | WO2003009867A1 (ja) |
Families Citing this family (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0323840D0 (en) * | 2003-10-10 | 2003-11-12 | Ist Superiore Sanita | Vaccines |
| GB0405480D0 (en) | 2004-03-11 | 2004-04-21 | Istituto Superiore Di Sanito | Novel tat complexes,and vaccines comprising them |
| WO2005090968A1 (en) | 2004-03-16 | 2005-09-29 | Inist Inc. | Tat-based immunomodulatory compositions and methods of their discovery and use |
| US9492400B2 (en) | 2004-11-04 | 2016-11-15 | Massachusetts Institute Of Technology | Coated controlled release polymer particles as efficient oral delivery vehicles for biopharmaceuticals |
| CN100429228C (zh) * | 2005-06-17 | 2008-10-29 | 中国人民解放军第三军医大学第三附属医院 | 重组人a20蛋白及其用途 |
| WO2007070682A2 (en) | 2005-12-15 | 2007-06-21 | Massachusetts Institute Of Technology | System for screening particles |
| JP2009534309A (ja) | 2006-03-31 | 2009-09-24 | マサチューセッツ インスティテュート オブ テクノロジー | 治療剤の標的化送達のためのシステム |
| JP5630998B2 (ja) | 2006-05-15 | 2014-11-26 | マサチューセッツ インスティテュート オブ テクノロジー | 機能的粒子のためのポリマー |
| WO2007150030A2 (en) | 2006-06-23 | 2007-12-27 | Massachusetts Institute Of Technology | Microfluidic synthesis of organic nanoparticles |
| JP2010500407A (ja) * | 2006-08-14 | 2010-01-07 | サイモン・エル・エル・シー | Hiv−1の複数の株及びサブタイプを治療及び予防するための組成物及び方法 |
| WO2008098165A2 (en) | 2007-02-09 | 2008-08-14 | Massachusetts Institute Of Technology | Oscillating cell culture bioreactor |
| WO2008124632A1 (en) | 2007-04-04 | 2008-10-16 | Massachusetts Institute Of Technology | Amphiphilic compound assisted nanoparticles for targeted delivery |
| AU2008314647B2 (en) | 2007-10-12 | 2013-03-21 | Massachusetts Institute Of Technology | Vaccine nanotechnology |
| GB0802224D0 (en) | 2008-02-06 | 2008-03-12 | Inst Superiore Di Sanito | Process for the production of vaccine components |
| CN101980738A (zh) | 2008-02-07 | 2011-02-23 | 华盛顿大学 | 圆周气雾设备 |
| EP2106803A1 (en) * | 2008-04-04 | 2009-10-07 | Fondazione Centro San Raffaele del Monte Tabor | Method to design and uses of overlapping peptides for monitoring T-cell responses in HIV patients |
| JP5576859B2 (ja) | 2008-05-14 | 2014-08-20 | ジャワハーラル ネール センター フォー アドヴァンスド サイエンティフィック リサーチ | Tatのdna配列、遺伝子コンストラクト、ワクチンおよびそれらの方法 |
| US8343497B2 (en) | 2008-10-12 | 2013-01-01 | The Brigham And Women's Hospital, Inc. | Targeting of antigen presenting cells with immunonanotherapeutics |
| US8591905B2 (en) | 2008-10-12 | 2013-11-26 | The Brigham And Women's Hospital, Inc. | Nicotine immunonanotherapeutics |
| US8343498B2 (en) | 2008-10-12 | 2013-01-01 | Massachusetts Institute Of Technology | Adjuvant incorporation in immunonanotherapeutics |
| US8277812B2 (en) | 2008-10-12 | 2012-10-02 | Massachusetts Institute Of Technology | Immunonanotherapeutics that provide IgG humoral response without T-cell antigen |
| CN102405057B (zh) | 2009-03-23 | 2016-05-25 | 那尼尔科斯治疗公司 | 用免疫刺激性Hiv Tat衍生物多肽治疗癌症 |
| EP2536838A4 (en) * | 2010-02-18 | 2013-08-21 | Univ Emory | VECTOR FOR THE EXPRESSION OF HIV ANTIGENES AND GM-CSF AND CORRESPONDING METHODS FOR GENERATING IMMUNE RESPONSE |
| US20110287045A1 (en) * | 2010-05-20 | 2011-11-24 | Zongdong Li | Enhanced immune response against infections |
| BR122021002471B8 (pt) | 2011-03-03 | 2022-10-25 | Impel Neuropharma Inc | Dispositivo de distribuição de droga nasal |
| CN103619485B (zh) | 2011-05-09 | 2017-08-08 | 英倍尔药业股份有限公司 | 用于鼻腔药物递送的喷嘴 |
| RU2670488C2 (ru) * | 2012-09-13 | 2018-10-23 | Юниверсите Де Женев | Новые проникающие в клетку пептиды |
| CN103880961A (zh) * | 2012-12-19 | 2014-06-25 | 邱书奇 | 一种诱导免疫耐受融合肽构建 |
| WO2014179228A1 (en) | 2013-04-28 | 2014-11-06 | Impel Neuropharma Inc. | Medical unit dose container |
| RU2695653C2 (ru) | 2013-10-04 | 2019-07-25 | Пин Фарма, Инк. | Иммуностимулирующие полипептидные производные тат вич для применения в лечении рака |
| EP3134107B1 (en) * | 2014-04-24 | 2020-05-27 | Janssen Sciences Ireland Unlimited Company | Use of a hiv derived accessory protein for the reactivation of latent hiv |
| NZ741171A (en) | 2015-09-10 | 2022-01-28 | Impel Neuropharma Inc | In-line nasal delivery device |
| JP7150611B2 (ja) | 2016-01-08 | 2022-10-11 | ジオバックス・インコーポレイテッド | 腫瘍関連抗原に対する免疫応答を誘起するための組成物及び方法 |
| CN105535937B (zh) * | 2016-03-11 | 2018-03-13 | 刘红 | 一种用于治疗类风湿性关节炎的皮下注射用原位凝胶 |
| WO2018050225A1 (en) * | 2016-09-15 | 2018-03-22 | Yu Di | T-cell immunotherapy |
| US11311612B2 (en) | 2017-09-19 | 2022-04-26 | Geovax, Inc. | Compositions and methods for generating an immune response to treat or prevent malaria |
| KR102131898B1 (ko) * | 2017-10-31 | 2020-07-08 | 주식회사 노벨티노빌리티 | Scf 및 갈렉틴-1을 표적으로 하는 이중표적항체 및 그 용도 |
| US11571532B2 (en) | 2017-11-21 | 2023-02-07 | Impel Pharmaceuticals Inc. | Intranasal device with dip tube |
| WO2019104192A1 (en) | 2017-11-21 | 2019-05-31 | Impel Neuropharma, Inc. | Intranasal device with inlet interface |
| CN111936140A (zh) | 2018-01-05 | 2020-11-13 | 英倍尔药业股份有限公司 | 通过精密鼻装置的双氢麦角胺的鼻内递送 |
| CA3087698A1 (en) | 2018-01-05 | 2019-07-11 | Impel Neuropharma, Inc. | Intranasal delivery of olanzapine by precision olfactory device |
| EP3823607A4 (en) | 2018-07-19 | 2022-04-06 | Impel NeuroPharma Inc. | ADMINISTRATION OF LEVODOPA AND DOPA DECARBOXYLASE INHIBITOR INTO THE RESPIRATORY TRACT FOR THE TREATMENT OF PARKINSON'S DISEASE |
| EA202191856A1 (ru) | 2019-01-03 | 2021-09-02 | Импел Ньюрофарма, Инк. | Устройство для назальной доставки лекарственных средств |
| JP7317379B2 (ja) * | 2019-01-04 | 2023-07-31 | 国立大学法人京都大学 | 潰瘍性大腸炎及び原発性硬化性胆管炎の検査方法 |
| CN114025816A (zh) | 2019-05-17 | 2022-02-08 | 英倍尔药业股份有限公司 | 一次性鼻腔给送装置 |
| TW202112394A (zh) * | 2019-06-06 | 2021-04-01 | 日商電化股份有限公司 | 以肽核酸為基礎之佐劑 |
| CN111450082A (zh) * | 2020-04-26 | 2020-07-28 | 天津大学 | 一种系统性红斑狼疮治疗纳米颗粒的合成方法 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9720585D0 (en) * | 1997-09-26 | 1997-11-26 | Smithkline Beecham Biolog | Vaccine |
| IT1297090B1 (it) * | 1997-12-01 | 1999-08-03 | Barbara Ensoli | Tat di hiv-1 o suoi derivati, da soli od in combinazione, a scopo vaccinale, profilattico e terapeutico, contro l'aids i tumori e le |
| FI105105B (fi) * | 1998-02-27 | 2000-06-15 | Finnish Immunotechnology Ltd O | Itsereplikoiva DNA-vektori immunisoimiseksi HIV:ta vastaan |
| AU4082300A (en) * | 1999-06-21 | 2001-01-09 | Government Of The United States Of America, As Represented By The Secretary Of The Department Of Health And Human Services, The | Hiv tat peptides and multiple peptide conjugate system |
| CA2376992A1 (en) | 1999-06-29 | 2001-01-04 | Smithkline Beecham Biologicals S.A. | Use of cpg as an adjuvant for hiv vaccine |
| EP1246647B1 (en) | 1999-08-12 | 2010-04-28 | InIst, Inc. | Non-immunosuppressant hiv tat |
| WO2001019393A1 (en) * | 1999-09-13 | 2001-03-22 | Cornell Research Foundation, Inc. | Delivering to eucaryotic cells bacterial proteins that are secreted via type iii secretion systems |
| KR20070073987A (ko) | 2000-01-31 | 2007-07-10 | 글락소스미스클라인 바이오로지칼즈 에스.에이. | Hiv에 대한 예방 또는 치료용 면역화를 위한 백신 |
| AU2001234616A1 (en) | 2000-01-31 | 2001-08-07 | Vic Jira | Vaccine composition, process and methods |
| US6544780B1 (en) | 2000-06-02 | 2003-04-08 | Genphar, Inc. | Adenovirus vector with multiple expression cassettes |
| AT410635B (de) | 2000-10-18 | 2003-06-25 | Cistem Biotechnologies Gmbh | Vakzin-zusammensetzung |
-
2001
- 2001-07-26 EP EP01118114A patent/EP1279404A1/en not_active Withdrawn
-
2002
- 2002-07-26 EP EP02767265A patent/EP1425035B1/en not_active Expired - Lifetime
- 2002-07-26 ES ES02767265T patent/ES2367255T3/es not_active Expired - Lifetime
- 2002-07-26 EA EA200400227A patent/EA006850B1/ru not_active IP Right Cessation
- 2002-07-26 CA CA2455227A patent/CA2455227C/en not_active Expired - Fee Related
- 2002-07-26 US US10/485,180 patent/US7811573B2/en not_active Expired - Fee Related
- 2002-07-26 AT AT02767265T patent/ATE510558T1/de not_active IP Right Cessation
- 2002-07-26 DK DK02767265.8T patent/DK1425035T3/da active
- 2002-07-26 MX MXPA04000287A patent/MXPA04000287A/es not_active Application Discontinuation
- 2002-07-26 WO PCT/EP2002/008377 patent/WO2003009867A1/en active IP Right Grant
- 2002-07-26 HU HU0400660A patent/HUP0400660A2/hu unknown
- 2002-07-26 NZ NZ531364A patent/NZ531364A/en not_active IP Right Cessation
- 2002-07-26 IL IL16001502A patent/IL160015A0/xx not_active IP Right Cessation
- 2002-07-26 JP JP2003515259A patent/JP5134183B2/ja not_active Expired - Fee Related
- 2002-07-26 PL PL02368243A patent/PL368243A1/xx unknown
- 2002-07-26 AP APAP/P/2004/002986A patent/AP2004002986A0/en unknown
- 2002-07-26 BR BR0211474-7A patent/BR0211474A/pt not_active IP Right Cessation
- 2002-07-26 AU AU2002331350A patent/AU2002331350B2/en not_active Ceased
- 2002-07-26 CN CNB028146581A patent/CN1318091C/zh not_active Expired - Fee Related
- 2002-07-26 OA OA1200400015A patent/OA12701A/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CN1318091C (zh) | 2007-05-30 |
| US20050036985A1 (en) | 2005-02-17 |
| AP2004002986A0 (en) | 2004-03-31 |
| EP1425035A1 (en) | 2004-06-09 |
| HUP0400660A2 (hu) | 2005-01-28 |
| EP1279404A1 (en) | 2003-01-29 |
| ATE510558T1 (de) | 2011-06-15 |
| CA2455227A1 (en) | 2003-02-06 |
| OA12701A (en) | 2006-06-23 |
| BR0211474A (pt) | 2004-07-13 |
| CA2455227C (en) | 2012-01-24 |
| PL368243A1 (en) | 2005-03-21 |
| US7811573B2 (en) | 2010-10-12 |
| IL160015A0 (en) | 2004-06-20 |
| CN1533284A (zh) | 2004-09-29 |
| NZ531364A (en) | 2005-12-23 |
| AU2002331350B2 (en) | 2008-06-12 |
| EA006850B1 (ru) | 2006-04-28 |
| MXPA04000287A (es) | 2005-03-07 |
| WO2003009867A1 (en) | 2003-02-06 |
| DK1425035T3 (da) | 2011-08-29 |
| ES2367255T3 (es) | 2011-10-31 |
| EA200400227A1 (ru) | 2004-06-24 |
| EP1425035B1 (en) | 2011-05-25 |
| JP2005505520A (ja) | 2005-02-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5134183B2 (ja) | 予防または治療ワクチン接種および/或いはその他疾患の治療を目的とする、抗原提示細胞を標的とすることおよび/または活性化すること、並びに/またはカーゴ分子を運搬することへの生物学的活性hiv−1tat、その断片または誘導体の使用 | |
| AU2002331350A1 (en) | Use of biologically active HIV-1 Tat, fragments or derivatives thereof, to target and/or to activate antigen-presenting cells, and/or to deliver cargo molecules for preventive or therapeutic vaccination and/or to treat other diseases | |
| JP5270500B2 (ja) | 外因性タンパク質をサイトゾルに運搬する方法およびその使用 | |
| US7744896B1 (en) | HIV-1 Tat compositions | |
| Fanales-Belasio et al. | Native HIV-1 Tat protein targets monocyte-derived dendritic cells and enhances their maturation, function, and antigen-specific T cell responses | |
| JP2008539751A (ja) | 新規組成物およびその使用 | |
| KR20190079628A (ko) | 백신으로 사용하기 위한 엑소좀-앵커링 단백질을 발현하는 뉴클레오티드 서열 | |
| Nardelli et al. | Cellular immune responses induced by in vivo priming with a lipid-conjugated multimeric antigen peptide. | |
| JP2008531530A (ja) | Hivエピトープ及び同じものを含む製薬組成物 | |
| HU210605A9 (en) | Immunostimulation | |
| CN117659140A (zh) | 新冠病毒hla-a2限制性表位肽及应用 | |
| AU2008203414B2 (en) | Methods of delivery of exogenous proteins to the cytosol and uses thereof | |
| CN117903264A (zh) | 新冠病毒SARS-CoV-2 HLA-A2限制性表位肽及应用 | |
| MXPA00005325A (en) | Hiv-1 tat, or derivatives thereof for prophylactic and therapeutic vaccination |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050427 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080813 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20081107 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20081114 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20081215 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20090128 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20090501 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090528 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20090713 |
|
| A912 | Re-examination (zenchi) completed and case transferred to appeal board |
Free format text: JAPANESE INTERMEDIATE CODE: A912 Effective date: 20090731 |
|
| RD01 | Notification of change of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7421 Effective date: 20110725 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20110725 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20110825 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20110831 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20110929 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20111004 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120920 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121109 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151116 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |