JP5129133B2 - 安定なnad/nadh誘導体 - Google Patents
安定なnad/nadh誘導体 Download PDFInfo
- Publication number
- JP5129133B2 JP5129133B2 JP2008523257A JP2008523257A JP5129133B2 JP 5129133 B2 JP5129133 B2 JP 5129133B2 JP 2008523257 A JP2008523257 A JP 2008523257A JP 2008523257 A JP2008523257 A JP 2008523257A JP 5129133 B2 JP5129133 B2 JP 5129133B2
- Authority
- JP
- Japan
- Prior art keywords
- dehydrogenase
- enzyme
- independently
- compound
- coenzyme
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- BOPGDPNILDQYTO-NNYOXOHSSA-N nicotinamide-adenine dinucleotide Chemical class C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 BOPGDPNILDQYTO-NNYOXOHSSA-N 0.000 title description 25
- 102000004190 Enzymes Human genes 0.000 claims description 57
- 108090000790 Enzymes Proteins 0.000 claims description 57
- 239000005515 coenzyme Substances 0.000 claims description 40
- 150000001875 compounds Chemical class 0.000 claims description 34
- 108010050375 Glucose 1-Dehydrogenase Proteins 0.000 claims description 30
- 238000012360 testing method Methods 0.000 claims description 30
- 239000012491 analyte Substances 0.000 claims description 29
- 238000001514 detection method Methods 0.000 claims description 28
- 238000005259 measurement Methods 0.000 claims description 27
- 239000003153 chemical reaction reagent Substances 0.000 claims description 17
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 16
- -1 alkyl compound Chemical class 0.000 claims description 16
- 229910052760 oxygen Inorganic materials 0.000 claims description 16
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 15
- 238000000034 method Methods 0.000 claims description 15
- 125000000217 alkyl group Chemical group 0.000 claims description 14
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 claims description 13
- 239000008103 glucose Substances 0.000 claims description 13
- 150000003839 salts Chemical class 0.000 claims description 13
- 229910052801 chlorine Inorganic materials 0.000 claims description 11
- 239000000758 substrate Substances 0.000 claims description 11
- 229910052717 sulfur Inorganic materials 0.000 claims description 11
- 229910052739 hydrogen Inorganic materials 0.000 claims description 10
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 9
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 claims description 9
- 230000008569 process Effects 0.000 claims description 9
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 claims description 7
- 229910052799 carbon Inorganic materials 0.000 claims description 7
- 238000006911 enzymatic reaction Methods 0.000 claims description 7
- 229930024421 Adenine Natural products 0.000 claims description 6
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 claims description 6
- 108010021809 Alcohol dehydrogenase Proteins 0.000 claims description 6
- 102000007698 Alcohol dehydrogenase Human genes 0.000 claims description 6
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 claims description 6
- 102000004420 Creatine Kinase Human genes 0.000 claims description 6
- 108010042126 Creatine kinase Proteins 0.000 claims description 6
- 102000003855 L-lactate dehydrogenase Human genes 0.000 claims description 6
- 108700023483 L-lactate dehydrogenases Proteins 0.000 claims description 6
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 claims description 6
- 229960000643 adenine Drugs 0.000 claims description 6
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 claims description 6
- 229910052731 fluorine Inorganic materials 0.000 claims description 6
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 claims description 6
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 claims description 6
- 108030000198 L-amino-acid dehydrogenases Proteins 0.000 claims description 5
- 125000002837 carbocyclic group Chemical group 0.000 claims description 5
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 5
- 230000001419 dependent effect Effects 0.000 claims description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 5
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 claims description 4
- 102000004008 5'-Nucleotidase Human genes 0.000 claims description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 claims description 4
- 101710088194 Dehydrogenase Proteins 0.000 claims description 4
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 claims description 4
- 235000001014 amino acid Nutrition 0.000 claims description 4
- 125000000623 heterocyclic group Chemical group 0.000 claims description 4
- 239000001630 malic acid Substances 0.000 claims description 4
- 235000011090 malic acid Nutrition 0.000 claims description 4
- 108010043671 prostatic acid phosphatase Proteins 0.000 claims description 4
- 125000006728 (C1-C6) alkynyl group Chemical group 0.000 claims description 3
- 108010050201 2-hydroxybutyrate dehydrogenase Proteins 0.000 claims description 3
- 101000892220 Geobacillus thermodenitrificans (strain NG80-2) Long-chain-alcohol dehydrogenase 1 Proteins 0.000 claims description 3
- 108010024636 Glutathione Proteins 0.000 claims description 3
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 claims description 3
- 108010009384 L-Iditol 2-Dehydrogenase Proteins 0.000 claims description 3
- 102100026974 Sorbitol dehydrogenase Human genes 0.000 claims description 3
- 235000010323 ascorbic acid Nutrition 0.000 claims description 3
- 229960005070 ascorbic acid Drugs 0.000 claims description 3
- 239000011668 ascorbic acid Substances 0.000 claims description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 claims description 3
- 239000004202 carbamide Substances 0.000 claims description 3
- 239000001569 carbon dioxide Substances 0.000 claims description 3
- 229910002092 carbon dioxide Inorganic materials 0.000 claims description 3
- 235000012000 cholesterol Nutrition 0.000 claims description 3
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 claims description 3
- 235000018417 cysteine Nutrition 0.000 claims description 3
- 229960002433 cysteine Drugs 0.000 claims description 3
- 235000019441 ethanol Nutrition 0.000 claims description 3
- 235000001727 glucose Nutrition 0.000 claims description 3
- 229960003180 glutathione Drugs 0.000 claims description 3
- 235000003969 glutathione Nutrition 0.000 claims description 3
- 235000011187 glycerol Nutrition 0.000 claims description 3
- 229910052736 halogen Inorganic materials 0.000 claims description 3
- 150000002367 halogens Chemical class 0.000 claims description 3
- 239000004310 lactic acid Substances 0.000 claims description 3
- 235000014655 lactic acid Nutrition 0.000 claims description 3
- 229910052757 nitrogen Inorganic materials 0.000 claims description 3
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 3
- 229960001860 salicylate Drugs 0.000 claims description 3
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 claims description 3
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 claims description 3
- 108700040097 Glycerol dehydrogenases Proteins 0.000 claims description 2
- 108010026217 Malate Dehydrogenase Proteins 0.000 claims description 2
- 102000013460 Malate Dehydrogenase Human genes 0.000 claims description 2
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 claims description 2
- 150000001721 carbon Chemical group 0.000 claims description 2
- 239000011535 reaction buffer Substances 0.000 claims description 2
- PEHVGBZKEYRQSX-UHFFFAOYSA-N 7-deaza-adenine Chemical compound NC1=NC=NC2=C1C=CN2 PEHVGBZKEYRQSX-UHFFFAOYSA-N 0.000 claims 8
- LHCPRYRLDOSKHK-UHFFFAOYSA-N 7-deaza-8-aza-adenine Chemical compound NC1=NC=NC2=C1C=NN2 LHCPRYRLDOSKHK-UHFFFAOYSA-N 0.000 claims 4
- 108020005199 Dehydrogenases Proteins 0.000 claims 3
- 125000006727 (C1-C6) alkenyl group Chemical group 0.000 claims 2
- HRYKDUPGBWLLHO-UHFFFAOYSA-N 8-azaadenine Chemical compound NC1=NC=NC2=NNN=C12 HRYKDUPGBWLLHO-UHFFFAOYSA-N 0.000 claims 2
- 238000006356 dehydrogenation reaction Methods 0.000 claims 2
- 125000004433 nitrogen atom Chemical group N* 0.000 claims 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 claims 1
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 74
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 54
- 229950006238 nadide Drugs 0.000 description 52
- BAWFJGJZGIEFAR-NNYOXOHSSA-N NAD zwitterion Chemical compound NC(=O)C1=CC=C[N+]([C@H]2[C@@H]([C@H](O)[C@@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 BAWFJGJZGIEFAR-NNYOXOHSSA-N 0.000 description 47
- 229940088598 enzyme Drugs 0.000 description 46
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 45
- 239000000243 solution Substances 0.000 description 43
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 39
- 239000000203 mixture Substances 0.000 description 29
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 20
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- 239000000047 product Substances 0.000 description 18
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 17
- 230000000694 effects Effects 0.000 description 16
- 239000008367 deionised water Substances 0.000 description 14
- 229910021641 deionized water Inorganic materials 0.000 description 14
- 239000002904 solvent Substances 0.000 description 13
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 12
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 12
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 12
- 238000000862 absorption spectrum Methods 0.000 description 12
- 239000000872 buffer Substances 0.000 description 12
- 238000004458 analytical method Methods 0.000 description 11
- 230000015572 biosynthetic process Effects 0.000 description 11
- 239000012043 crude product Substances 0.000 description 11
- 238000003756 stirring Methods 0.000 description 11
- 238000003786 synthesis reaction Methods 0.000 description 11
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 10
- 239000008346 aqueous phase Substances 0.000 description 10
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 239000000460 chlorine Substances 0.000 description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- AFABGHUZZDYHJO-UHFFFAOYSA-N 2-Methylpentane Chemical compound CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 8
- 229960000583 acetic acid Drugs 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical group NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 7
- 230000008859 change Effects 0.000 description 7
- 238000010828 elution Methods 0.000 description 7
- 239000000706 filtrate Substances 0.000 description 7
- 239000012362 glacial acetic acid Substances 0.000 description 7
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 6
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 6
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- 239000003480 eluent Substances 0.000 description 6
- 239000000463 material Substances 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- VKIGAWAEXPTIOL-UHFFFAOYSA-N 2-hydroxyhexanenitrile Chemical compound CCCCC(O)C#N VKIGAWAEXPTIOL-UHFFFAOYSA-N 0.000 description 5
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 5
- 238000010521 absorption reaction Methods 0.000 description 5
- 238000004040 coloring Methods 0.000 description 5
- 238000001816 cooling Methods 0.000 description 5
- 238000011161 development Methods 0.000 description 5
- 238000002189 fluorescence spectrum Methods 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- WVLBCYQITXONBZ-UHFFFAOYSA-N trimethyl phosphate Chemical compound COP(=O)(OC)OC WVLBCYQITXONBZ-UHFFFAOYSA-N 0.000 description 5
- GVNVAWHJIKLAGL-UHFFFAOYSA-N 2-(cyclohexen-1-yl)cyclohexan-1-one Chemical compound O=C1CCCCC1C1=CCCCC1 GVNVAWHJIKLAGL-UHFFFAOYSA-N 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- 101150065749 Churc1 gene Proteins 0.000 description 4
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- 102100038239 Protein Churchill Human genes 0.000 description 4
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- UDMBCSSLTHHNCD-KQYNXXCUSA-N adenosine 5'-monophosphate Chemical class C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O UDMBCSSLTHHNCD-KQYNXXCUSA-N 0.000 description 4
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 4
- 239000012300 argon atmosphere Substances 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 230000003287 optical effect Effects 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- 238000001228 spectrum Methods 0.000 description 4
- UYHMQTNGMUDVIY-UHFFFAOYSA-M 1-(2,4-dinitrophenyl)pyridin-1-ium;chloride Chemical compound [Cl-].[O-][N+](=O)C1=CC([N+](=O)[O-])=CC=C1[N+]1=CC=CC=C1 UYHMQTNGMUDVIY-UHFFFAOYSA-M 0.000 description 3
- 0 CC(C=*)=CC=CN**P(*P(C)(OCC1C(*)C(*)C(*)*1)=*)([U])=* Chemical compound CC(C=*)=CC=CN**P(*P(C)(OCC1C(*)C(*)C(*)*1)=*)([U])=* 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- 101000610640 Homo sapiens U4/U6 small nuclear ribonucleoprotein Prp3 Proteins 0.000 description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 3
- 101001110823 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L6-A Proteins 0.000 description 3
- 101000712176 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L6-B Proteins 0.000 description 3
- 229920005654 Sephadex Polymers 0.000 description 3
- 239000012507 Sephadex™ Substances 0.000 description 3
- 102100040374 U4/U6 small nuclear ribonucleoprotein Prp3 Human genes 0.000 description 3
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 238000011325 biochemical measurement Methods 0.000 description 3
- 238000006555 catalytic reaction Methods 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 239000007857 degradation product Substances 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 239000002274 desiccant Substances 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- 238000000295 emission spectrum Methods 0.000 description 3
- 230000005284 excitation Effects 0.000 description 3
- 238000000695 excitation spectrum Methods 0.000 description 3
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 3
- 235000019341 magnesium sulphate Nutrition 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- FEMOMIGRRWSMCU-UHFFFAOYSA-N ninhydrin Chemical compound C1=CC=C2C(=O)C(O)(O)C(=O)C2=C1 FEMOMIGRRWSMCU-UHFFFAOYSA-N 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 238000001308 synthesis method Methods 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 2
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 2
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- PWJFNRJRHXWEPT-UHFFFAOYSA-N ADP ribose Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OCC(O)C(O)C(O)C=O)C(O)C1O PWJFNRJRHXWEPT-UHFFFAOYSA-N 0.000 description 2
- SRNWOUGRCWSEMX-TYASJMOZSA-N ADP-D-ribose Chemical compound C([C@H]1O[C@H]([C@@H]([C@@H]1O)O)N1C=2N=CN=C(C=2N=C1)N)OP(O)(=O)OP(O)(=O)OC[C@H]1OC(O)[C@H](O)[C@@H]1O SRNWOUGRCWSEMX-TYASJMOZSA-N 0.000 description 2
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- 108010089254 Cholesterol oxidase Proteins 0.000 description 2
- 229940126657 Compound 17 Drugs 0.000 description 2
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 2
- 108010015776 Glucose oxidase Proteins 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- XXDDQPLHHUHJOU-IDTAVKCVSA-N N-(5'-adenylyl)morpholine Chemical compound C([C@H]1O[C@H]([C@@H]([C@@H]1O)O)N1C=2N=CN=C(C=2N=C1)N)OP(O)(=O)N1CCOCC1 XXDDQPLHHUHJOU-IDTAVKCVSA-N 0.000 description 2
- KUFMXPZUZBPFCT-UHFFFAOYSA-N P(O)(OCOP(O)=O)=O Chemical compound P(O)(OCOP(O)=O)=O KUFMXPZUZBPFCT-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- 102000003929 Transaminases Human genes 0.000 description 2
- 108090000340 Transaminases Proteins 0.000 description 2
- 239000002250 absorbent Substances 0.000 description 2
- 230000002745 absorbent Effects 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- 238000002983 circular dichroism Methods 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940125758 compound 15 Drugs 0.000 description 2
- 229940126142 compound 16 Drugs 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- SHFJWMWCIHQNCP-UHFFFAOYSA-M hydron;tetrabutylazanium;sulfate Chemical compound OS([O-])(=O)=O.CCCC[N+](CCCC)(CCCC)CCCC SHFJWMWCIHQNCP-UHFFFAOYSA-M 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 229910003002 lithium salt Inorganic materials 0.000 description 2
- 159000000002 lithium salts Chemical class 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 229960003966 nicotinamide Drugs 0.000 description 2
- 235000005152 nicotinamide Nutrition 0.000 description 2
- 239000011570 nicotinamide Substances 0.000 description 2
- 238000000853 optical rotatory dispersion Methods 0.000 description 2
- 238000005191 phase separation Methods 0.000 description 2
- RLOWWWKZYUNIDI-UHFFFAOYSA-N phosphinic chloride Chemical compound ClP=O RLOWWWKZYUNIDI-UHFFFAOYSA-N 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 229920001467 poly(styrenesulfonates) Polymers 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- AOJFQRQNPXYVLM-UHFFFAOYSA-N pyridin-1-ium;chloride Chemical compound [Cl-].C1=CC=[NH+]C=C1 AOJFQRQNPXYVLM-UHFFFAOYSA-N 0.000 description 2
- 150000003222 pyridines Chemical class 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- QEVHRUUCFGRFIF-MDEJGZGSSA-N reserpine Chemical compound O([C@H]1[C@@H]([C@H]([C@H]2C[C@@H]3C4=C(C5=CC=C(OC)C=C5N4)CCN3C[C@H]2C1)C(=O)OC)OC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 QEVHRUUCFGRFIF-MDEJGZGSSA-N 0.000 description 2
- 125000000548 ribosyl group Chemical group C1([C@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 230000006641 stabilisation Effects 0.000 description 2
- 238000013112 stability test Methods 0.000 description 2
- 238000011105 stabilization Methods 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- FHNKBDPGQXLKRW-KAZBKCHUSA-N (1r,2s,3r,5r)-3-amino-5-(hydroxymethyl)cyclopentane-1,2-diol Chemical compound N[C@@H]1C[C@H](CO)[C@@H](O)[C@H]1O FHNKBDPGQXLKRW-KAZBKCHUSA-N 0.000 description 1
- ALNDFFUAQIVVPG-NGJCXOISSA-N (2r,3r,4r)-3,4,5-trihydroxy-2-methoxypentanal Chemical compound CO[C@@H](C=O)[C@H](O)[C@H](O)CO ALNDFFUAQIVVPG-NGJCXOISSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- HEWZVZIVELJPQZ-UHFFFAOYSA-N 2,2-dimethoxypropane Chemical compound COC(C)(C)OC HEWZVZIVELJPQZ-UHFFFAOYSA-N 0.000 description 1
- DDUFYKNOXPZZIW-UHFFFAOYSA-N 3-azabicyclo[2.2.1]hept-5-en-2-one Chemical compound C1C2C(=O)NC1C=C2 DDUFYKNOXPZZIW-UHFFFAOYSA-N 0.000 description 1
- JNRLEMMIVRBKJE-UHFFFAOYSA-N 4,4'-Methylenebis(N,N-dimethylaniline) Chemical compound C1=CC(N(C)C)=CC=C1CC1=CC=C(N(C)C)C=C1 JNRLEMMIVRBKJE-UHFFFAOYSA-N 0.000 description 1
- XTWYTFMLZFPYCI-KQYNXXCUSA-N 5'-adenylphosphoric acid Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XTWYTFMLZFPYCI-KQYNXXCUSA-N 0.000 description 1
- JPFNWUUUXFFWOQ-UHFFFAOYSA-N 7h-purin-6-amine;1-pyridin-3-ylethanone Chemical compound CC(=O)C1=CC=CN=C1.NC1=NC=NC2=C1NC=N2 JPFNWUUUXFFWOQ-UHFFFAOYSA-N 0.000 description 1
- 102000000074 ADP-ribosyl Cyclase Human genes 0.000 description 1
- 108010080394 ADP-ribosyl Cyclase Proteins 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- 101100313763 Arabidopsis thaliana TIM22-2 gene Proteins 0.000 description 1
- 239000007989 BIS-Tris Propane buffer Substances 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- UDMBCSSLTHHNCD-UHFFFAOYSA-N Coenzym Q(11) Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(O)=O)C(O)C1O UDMBCSSLTHHNCD-UHFFFAOYSA-N 0.000 description 1
- FBPFZTCFMRRESA-KAZBKCHUSA-N D-altritol Chemical compound OC[C@@H](O)[C@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KAZBKCHUSA-N 0.000 description 1
- VYZAHLCBVHPDDF-UHFFFAOYSA-N Dinitrochlorobenzene Chemical compound [O-][N+](=O)C1=CC=C(Cl)C([N+]([O-])=O)=C1 VYZAHLCBVHPDDF-UHFFFAOYSA-N 0.000 description 1
- 239000004366 Glucose oxidase Substances 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- 229910021380 Manganese Chloride Inorganic materials 0.000 description 1
- GLFNIEUTAYBVOC-UHFFFAOYSA-L Manganese chloride Chemical compound Cl[Mn]Cl GLFNIEUTAYBVOC-UHFFFAOYSA-L 0.000 description 1
- LFTLOKWAGJYHHR-UHFFFAOYSA-N N-methylmorpholine N-oxide Chemical compound CN1(=O)CCOCC1 LFTLOKWAGJYHHR-UHFFFAOYSA-N 0.000 description 1
- JLEBZPBDRKPWTD-TURQNECASA-O N-ribosylnicotinamide Chemical group NC(=O)C1=CC=C[N+]([C@H]2[C@@H]([C@H](O)[C@@H](CO)O2)O)=C1 JLEBZPBDRKPWTD-TURQNECASA-O 0.000 description 1
- XJLXINKUBYWONI-NNYOXOHSSA-O NADP(+) Chemical compound NC(=O)C1=CC=C[N+]([C@H]2[C@@H]([C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-NNYOXOHSSA-O 0.000 description 1
- BHVJSLYJWDSJMS-UHFFFAOYSA-N O.O.NC1CCCCC1 Chemical compound O.O.NC1CCCCC1 BHVJSLYJWDSJMS-UHFFFAOYSA-N 0.000 description 1
- WSDRAZIPGVLSNP-UHFFFAOYSA-N O.P(=O)(O)(O)O.O.O.P(=O)(O)(O)O Chemical compound O.P(=O)(O)(O)O.O.O.P(=O)(O)(O)O WSDRAZIPGVLSNP-UHFFFAOYSA-N 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- 102100034233 Protein N-terminal asparagine amidohydrolase Human genes 0.000 description 1
- 101710097214 Protein N-terminal asparagine amidohydrolase Proteins 0.000 description 1
- 102100029683 Ribonuclease T2 Human genes 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- DFPAKSUCGFBDDF-ZQBYOMGUSA-N [14c]-nicotinamide Chemical compound N[14C](=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-ZQBYOMGUSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- LNQVTSROQXJCDD-UHFFFAOYSA-N adenosine monophosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(CO)C(OP(O)(O)=O)C1O LNQVTSROQXJCDD-UHFFFAOYSA-N 0.000 description 1
- 150000003838 adenosines Chemical class 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 125000005741 alkyl alkenyl group Chemical group 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 150000001414 amino alcohols Chemical class 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 238000012742 biochemical analysis Methods 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000010549 co-Evaporation Methods 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- NISGSNTVMOOSJQ-UHFFFAOYSA-N cyclopentanamine Chemical compound NC1CCCC1 NISGSNTVMOOSJQ-UHFFFAOYSA-N 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000006345 epimerization reaction Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- IRXSLJNXXZKURP-UHFFFAOYSA-N fluorenylmethyloxycarbonyl chloride Chemical compound C1=CC=C2C(COC(=O)Cl)C3=CC=CC=C3C2=C1 IRXSLJNXXZKURP-UHFFFAOYSA-N 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 238000005194 fractionation Methods 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 235000019420 glucose oxidase Nutrition 0.000 description 1
- 229940116332 glucose oxidase Drugs 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- FBPFZTCFMRRESA-UHFFFAOYSA-N hexane-1,2,3,4,5,6-hexol Chemical compound OCC(O)C(O)C(O)C(O)CO FBPFZTCFMRRESA-UHFFFAOYSA-N 0.000 description 1
- AFQIYTIJXGTIEY-UHFFFAOYSA-N hydrogen carbonate;triethylazanium Chemical compound OC(O)=O.CCN(CC)CC AFQIYTIJXGTIEY-UHFFFAOYSA-N 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 235000002867 manganese chloride Nutrition 0.000 description 1
- 239000011565 manganese chloride Substances 0.000 description 1
- 229940099607 manganese chloride Drugs 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 238000000816 matrix-assisted laser desorption--ionisation Methods 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- LZGUHMNOBNWABZ-UHFFFAOYSA-N n-nitro-n-phenylnitramide Chemical compound [O-][N+](=O)N([N+]([O-])=O)C1=CC=CC=C1 LZGUHMNOBNWABZ-UHFFFAOYSA-N 0.000 description 1
- 229940101270 nicotinamide adenine dinucleotide (nad) Drugs 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 229910000489 osmium tetroxide Inorganic materials 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 210000002381 plasma Anatomy 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- 239000008057 potassium phosphate buffer Substances 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 150000003138 primary alcohols Chemical class 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 229940076788 pyruvate Drugs 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000006479 redox reaction Methods 0.000 description 1
- 108090000446 ribonuclease T(2) Proteins 0.000 description 1
- 239000012488 sample solution Substances 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 238000009958 sewing Methods 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910001495 sodium tetrafluoroborate Inorganic materials 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000000967 suction filtration Methods 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 150000003536 tetrazoles Chemical class 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 239000002351 wastewater Substances 0.000 description 1
Images













Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/26—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving oxidoreductase
- C12Q1/32—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving oxidoreductase involving dehydrogenase
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/531—Production of immunochemical test materials
- G01N33/532—Production of labelled immunochemicals
- G01N33/533—Production of labelled immunochemicals with fluorescent label
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Biomedical Technology (AREA)
- Food Science & Technology (AREA)
- Medicinal Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Cell Biology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Saccharide Compounds (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Investigating Or Analysing Materials By The Use Of Chemical Reactions (AREA)
Description
(i)補酵素依存性酵素またはその種の酵素の基質および
(ii)補酵素として下記一般式(I)で表される化合物であって、

Tはそれぞれ独立してO、S、
Uはそれぞれ独立してOH、SH、BH3 -、BCNH2 -、
Vはそれぞれ独立してOHまたはフォスフェート基、
WはCOOR、CON(R)2、COR、CSN(R)2であって、Rはそれぞれ独立してHまたはC1〜C2アルキル、
X1、X2はそれぞれ独立してO、CH2、CHCH3、C(CH3)2、NH、NCH3、
YはNH、S、O、CH2、
Zおよびピリジン残基がグリコシド結合によらずに結合しているという条件で、
Zは必要に応じてO、SおよびNから選択されたヘテロ原子と必要に応じて1個以上の置換基を含む5C原子を有する環状基を含む残基、および、R4はそれぞれ独立してH、F、Cl、CH3であり、CR42が前記環状基およびX2に結合しているCR42残基
である化合物もしくはその塩または必要な場合にはその還元型
を含む試験エレメントによって達成される。
下記一般式(I’)で表される化合物であって、
Tはそれぞれ独立してO、S、
Uはそれぞれ独立してOH、SH、BH3 -、BCNH2 -、
Vはそれぞれ独立してOHまたはフォスフェート基、
WはCOOR、CON(R)2、COR、CSN(R)2であって、Rはそれぞれ独立してHまたはC1〜C2アルキル、
X1、X2はそれぞれ独立してO、CH2、CHCH3、C(CH3)2、NH、NCH3、
YはNH、S、O、CH2、
Zは飽和もしくは不飽和の炭素環またはヘテロ環の五員環、特に、一般式(II)の化合物であって、
R4はそれぞれ独立してH、F、Cl、CH3、
R5はCR42、
R5’とR5”の間に一重結合が存在する場合には、
R5’はO、S、NH、NC1〜C2アルキル、CR42、CHOH、CHOCH3、
R5”はCR42、CHOH、CHOCH3、
R5’とR5”の間に二重結合が存在する場合には、
R5’およびR5”はCR4、
R6、R6’はそれぞれ独立してCH、CCH3
である化合物もしくはその塩または必要な場合にはその還元型
を含む。
Aはアデニンまたはその類似体、
Tはそれぞれ独立してO、S、
Uはそれぞれ独立してOH、SH、BH3 -、BCNH2 -、
Vはそれぞれ独立してOHまたはフォスフェート基、
WはCOOR、CON(R)2、COR、CSN(R)2であって、Rはそれぞれ独立してHまたはC1〜C2アルキル、
X1、X2はそれぞれ独立してO、CH2、CHCH3、C(CH3)2、NH、NCH3、
YはNH、S、O、CH2、
Zは飽和もしくは不飽和の炭素環またはヘテロ環の五員環、特に、一般式(II)の化合物であって、
R4はそれぞれ独立してH、F、Cl、CH3、
R5はCR42、
R5’とR5”の間に一重結合が存在する場合には、
R5’はO、S、NH、NC1〜C2アルキル、CR42、CHOH、CHOCH3、
R5”はCR42、CHOH、CHOCH3、
R5’とR5”の間に二重結合が存在する場合には、
R5’およびR5”はCR4、
R6、R6’はそれぞれ独立してCH、CCH3、
ただし、R5がCH2、TがO、UがそれぞれOH、VがOH、WがCONH2、XがOおよびYがOのとき、R5’とR5”は同時にCHOHではないことを条件
とする化合物もしくはその塩または必要な場合にはその還元型。
(a)補酵素を含む本発明の試験エレメントまたは試薬キットと試料の接触
(b)例えば補酵素の変化に基づく被分析物の検出
の過程を含む、被分析物検出のための方法も本発明の対象である。
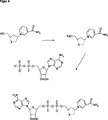
安定なNAD/NADH誘導体の製造について、例としてカルバNAD(化合物9、図1)とピロリドンNAD(化合物18、図5)に基づいて説明する。その他の誘導体はしかるべき合成方法によって製造することができる。出発試薬として使用される、しかるべきアミノアルコールは、以下のとおり文献から公知である。
Huryn, Donna M. ; Sluboski, Barbara C. ; Tam, Steve Y. ; Todaro, Louis J. ; Weigele, Manfred.、Tetrahedron Letters(1989年)、第30巻(第46号)、6259〜62頁。
Beres, J. ; Sagi, G. ; Tomoskozi, I. ; Gruber, L. ; Gulacsi, E. ; Otvos, L.、Tetrahedron Letters(1988年)、第29巻(第22号)、2681〜4頁。
I.1R−(−)−エキソ−シス−5,6−ジヒドロキシ−2−アザビシクロ[2.2.1]へプタン−3−オン(1)
80mlの脱イオン水に22.5g(167mmol)のN−メチルモルフォリン−N−オキシドを溶かした溶液を1l入り丸底フラスコに入れ、そこへ400mlのアセトンに16.4g(147mmol)の1R−(−)−2−アザビシクロ[2.2.1]ヘプト−5−エン−3−オンを溶かした溶液を加える。さらに、tert−ブタノールに溶かした2.5%オスミウムテトラオキシド溶液の15ml(1.2mmol)を、氷冷下15分以内で添加する。ついでこの混合物を室温で一晩撹拌する。
酢酸エチル/メタノール/氷酢酸 7:2:1、 Rf0.75(出発物質)、0.53(1)、TDMによる着色/塩素チェンバでの顕色
溶液1:40mlの氷酢酸と200mlの脱イオン水の混合液に10gのN,N,N’,N’−テトラメチル−4,4’−ジアミノジフェニルメタンを溶かした溶液。
溶液2:400mlの脱イオン水に20gの塩化カリウムを溶かした溶液。
溶液3:10mlの氷酢酸に0.3gのニンヒドリンを溶かし、90mlの脱イオン水を添加した溶液。
調製試薬:溶液1と2の混合液に6mlの溶液3を加えた溶液。
粗生成物1を200mlの無水エタノール中で環流下1時間沸騰させる。400ml(3.26mol)ジメトキシプロパンおよび250mg(2.2mmol)ピリジン塩酸塩の添加後、混合物を環流下でさらに15分間沸騰させる。10mlの炭酸水素ナトリウム飽和溶液の添加後、当該溶液をロータリーエバボレーターにおいて減圧濃縮乾固する。残渣に500mlのクロロフォルム、150mlの食塩飽和溶液および75mlの炭酸水素ナトリウム飽和溶液を加え、分液ロートに移し入れる。振盪抽出後一晩放置しておけば、相分離が起きる。
酢酸エチル/メタノール/氷酢酸 7:2:1
Rf0.84、TDMによる着色/塩素チェンバでの顕色
450mlの無水クロロフォルムに24,9g(135.7mmol)の粗生成物2を溶かした溶液に、アルゴン雰囲気下で41.5g(190mmol)のジ−tert−ブチルジカーボネートと0.83g(6.8mmol)の4−ジメチルアミノピリジンを添加する。この混合物を、気体の発生が停止するまで、環流撹拌下で沸騰させる。この混合物を、40gのSilica Gel 60が充填され、クロロフォルムで平衡化されたカラムを通して濾過する。つぎに、100mlのクロロフォルムで洗浄する。ロータリーエバボレーターにより、濾液から溶媒を減圧下で蒸留分離する。粗生成物を10mbar、40℃の条件で60分間乾燥させる。粗生成物はさらに精製することなく使用される。
酢酸エチル/ヘキサン3:2、 Rf=0.85、TDMによる着色/塩素チェンバでの顕色
粗生成物3を400mlのテトラヒドロフランに室温で撹拌しながら溶解し、80mlの脱イオン水を添加する。4℃に冷却後、5.3gの水素化ホウ素ナトリウムを一度に添加し、一晩撹拌する。その間、その混合物を室温までゆっくりと加温する。100mlのエタノールを加え、室温で6時間撹拌する。ロータリーエバボレーターで溶媒を減圧下蒸留分離する。300mlの塩化ナトリウム飽和溶液と650mlの酢酸エチルを添加し、分液ロートに移し入れる。有機相を分離し、水相を再び350mlの酢酸エチルで洗浄する。合わせた有機相は硫酸マグネシウム上で乾燥させる。乾燥剤の濾別後、ロータリーエバボレーターで溶媒を減圧下蒸留分離する。粗生成物(42.2g)をSilica Gel 60(カラムの高さ93cm、直径10cm)を充填したカラムクロマトグラフィーにより精製する(溶離剤THF/ヘキサン 当初1:3、ついで2:3、流速3l/時間)。40mlの画分を収集する。該画分はDC(Merck Silica Gel 60 F−254:酢酸エチル/ヘキサン 3:2、Rf=0.45、TDMによる着色/塩素チェンバでの顕色)により測定する。ロータリーエバボレーターにより、合わせた生成物画分から溶媒を真空下で蒸留分離させる。収量:24.9g。
11.09g(38.6mmol)の生成物4に、8mlの脱イオン水、ついで80mlのトリフルオロ酢酸を添加する。室温で6時間激しく撹拌すると、薄黄色の透明溶液が生成する。200mlの脱イオン水を追加し、ロータリーエバボレーターで真空下蒸発させる。さらに200mlの脱イオン水を加えて再度ロータリーエバボレーターで真空下蒸発させる。粗生成物を超音波槽で100mlの脱イオン水に溶かし、濾過する。濾液をDowex 1X8(100〜200メッシュ、OH型)イオン交換カラム(15×4.9cm)に添加し、水で溶出する。生成物の溶出は、容量約150mlを越えた時点から容量300mlまで行われる(pH10.4)。画分をDC(Merck Silica Gel 60 F−254:ブタノール/氷酢酸/水 5:2:3、Rf0.42、TDMによる着色/塩素チェンバでの顕色)により測定する。ロータリーエバボレーターにより、合わせた生成物画分から溶媒を真空下で蒸留分離させる。収量:5.2g、無色オイル。
窒素雰囲気下で58.6gのジニトロクロロベンゼンを融解させ、ついでこの融解物に29.32gのニコチンアミドを添加する。これを110℃で2.5時間加熱する。3:2(v/v)のエタノール/水混合物500mlを、環流冷却器を通して添加し、溶液となるまで環流下で沸騰させる。室温で一晩撹拌した後、150mlの50%エタノール/水および100mlの水を添加し、分液ロートに移し入れて、それぞれ500mlのクロロフォルムで3回洗浄する。分離した水相に300mlおよび50gの活性炭を加え、室温で1時間撹拌した後、Seitz Tiefenfilter K 700深層フィルターに通して濾過する。濾液をロータリーエバボレーターにより真空下で約100mlに濃縮するが、その間浴温は20℃を越えてはならない。つぎに、300mlの水で希釈して、撹拌下室温で70gのテトラフルオルホウ酸ナトリウムを添加する。沈殿物をメタノール/水で再結晶化させる。この結晶体を濾別し、少量のアセトンついでジエチルエーテルで洗浄して高真空下40℃で24時間乾燥させる(収量21.1g、23%)。画分をDC(Merck Silica Gel 60 F−254:ブタノール/氷酢酸/水 5:2:3、Rf=0.56)により測定する。
110mlの無水メタノールに4.5g(31mmol)のシクロペンチルアミン5を溶かした溶液を、110mlの無水メタノールに15.3g(40.7mmol)のZincke塩6を溶かした溶液に、撹拌下室温で90分以内に滴下する。1mlのジイソプロピルエチルアミンを添加し、その後室温で2日間撹拌する。500mlの水を添加し、分液ロートに移し入れて、それぞれ200mlの塩化メチレンで2回洗浄する。ロータリーエバボレーターにより、分離した水相から水を真空下で蒸留分離させる。残渣は100mlの水に入れて、Sephadex C25(Na+型)によるカラムクロマトグラフィーで精製する:カラム70×7.5cm、緩衝液A(脱イオン水)から緩衝液B(0.35M NaCl水溶液)への変更下での溶出、流速200ml/時間。15mlの画分を収集し、DC(Merck Silica gel 60 F−254:ブタノール/氷酢酸/水 5:2:3、Rf0.22)により測定する。
80mlの無水トリメチルリン酸エステルへ7g(27.7mmol)のカルバNMNを加えた懸濁液に、20mlのオキシ塩化リンと50mlのトリメチルリン酸エステルの混合物を0℃で添加する。これを0℃で2時間、ついで室温で2時間撹拌する。氷冷下で300mlの水を添加した後、この混合物をロータリーエバボレーターにより真空下で10mlに濃縮する。これを100mlの水に入れ、濾過後Sephadex C25(NEt3H+型)によるカラムクロマトグラフィーで精製する:カラム66×9cm、緩衝液A(脱イオン水)から緩衝液B(0.60M酢酸アンモニウム)への変更下での溶離、流速200ml/時間。15mlの画分を収集し、DC(Merck Silica gel 60 F−254 plates:イソ酪酸/アンモニア/水 66:1:33、Rf0.25)により測定する。ロータリーエバボレーターにより、合わせた生成物画分から溶媒を真空下で蒸留分離する。残渣を100mlの水に溶かし、凍結乾燥する。この操作を3回繰り返す。収量:4.0g。
40mlの無水DMFに3.31g(10mmol)のカルバNMNモノフォスフェートを溶かした溶液と無水アセトニトリルに78ml(39mmol)の3.5%テトラゾールを溶かした溶液の混合液に、40mlの無水DMFに1.25g(30mmol)のAMPモルフォリデートを溶かした溶液を室温にて1時間以内に滴下する。この混合物を室温で2日間撹拌する。
カルバNADまたはNADの10mM溶液を、pH8の0.1Mリン酸カリウム緩衝液に加えて負荷をかける。0.25、75および175時間後にHPLCクロマトグラフィーにより含有量を測定する。
緩衝液B:緩衝液A+アセトニトリル 1:1
流速 1.0ml/分、検出:254nm
RP18カラム:長さ125mm、直径4.6mm
グラジエント:40分で35%緩衝液Bに調整、2分間維持、ついで3分内で0%緩衝液Aに設定。
I.pNAD合成の第1過程(化合物10)
生成物Rf:0.78
MS ESI ES+242
*トランス−N−t−BOC−O−メシル−4−ヒドロキシ−L−プロリノールは、Sanochemia Pharmazeutika(株)からカタログ番号P−719として販売されている。
DC(KG60 F254 移動相 イソへキサン/酢酸エステル 2:1):RF0.13
MS ESI ES +439/+339
*収率は第1過程の溶離物を基準とする。
残渣=2.6g=31.3%
DC RP8 F254/MeOH/水 9/1
MS ESI ES−517.13
残渣=2.56g=100%
MS ESI ES−295
NH3カチオンを除去するため、残渣を2回ヒューニッヒ塩基中で溶解させ、各回それぞれ高真空下で再度蒸発させた。
残渣=1.60g=47.9%
DC RP8 254 MeOH/水 9/1
MS ES−400.1/ES+402.0 2倍の質量を示す場合もある。
さらに、生成物の同一性をNMR測定により確認した。
残渣=5.3g(理論値2.64g)**
MS、ESI ES−729.3は生成物、ES−415はAMPモルフォリデートのカチオン、ES−400.2/ES+402.1は化合物15(第6過程)の残基
DC RP8 F254 Rf0.085
*この溶液は、2516mgの無水MnCl2を100mlの99.99%フォルムアミド中で撹拌下溶解させて調製し、ついで4Aモレキュラーシーブを添加した。
**残渣は粗生成物としてさらに処理した。
MS ESI ES−729.24(NH3の添加が必要)
精製は、200mgと300mgに2分割の上、それぞれ下記の2分離過程を通して行った。
フラクトゲルEMD SO3−sカラム:直径(内径)14mm、長さ(外装)85mm
I.条件
(流速5ml/分) a) 100ml H2O
b) 200ml 0.25M H2SO4
c) 100ml H2O
d) 200ml 1Mアンモニア溶液
e) 100ml H2O
b)H2O→0.2M NH4HCO3溶液のグラジエントにより溶出させる。(移動溶媒Aは、250mlのH2Oをエルレンマイヤーフラスコに用意し、マグネチックスターラーで撹拌し、5ml/分の速度でカラムにポンピング供与する。移動溶媒Bは0.2M NH4HCO3溶液で、2.5ml/分の速度でAに対してポンピング供与する)。
b)不純物の第1ピーク。
c)約70mlの予備溶出後の第2ピークは物質。
b)100mlのH2O
Diaion HP20、カラム直径(内径)30mm、長さ(外装)130mm、100mlの水および100mlの水/5%イソプロパノールにより溶離させる。
分析HPLCにより3画分が得られた:F1=13.5mg
F2=5.5mg
F3=11.5mg
総計=30.5mg=12.2%
ピロリジニルNAD(化合物18)の同一性はNMR測定によって確認された。
グルコース脱水素酵素(GlucDH)のコファクターとしてのpNADの役割を調べるため、0.1Mトリス/0.2M NaCl(pH8.5)緩衝液中でGlucDH活性の分析を行った。グルコースの濃度は80mMであった。pNADおよびNAD濃度は0.05〜0.5mMを使用した。これに対し、10mg/ml(pNAD)または0.002mg/ml(NAD)[それぞれ83または0.017μM]のGlucDHを添加した。分析は室温で行い、酵素反応は規則的な時間間隔で吸収スペクトルを記録することにより測定した。表1に示した値は、4分後の吸収測定を対象としたものである。
図6Aおよび6BにはNAD、pNADおよびNADH、pNADHの吸収スペクトルが示されている。NADおよびpNADは260nmで吸収極大を示す。PNADH、すなわちGlucDH活性分析後のpNADは、NADHに比べて吸収極大が約10nm赤方偏移している(図6B)。
NADとの比較でpNADの安定性を調べるため、同量のNADとpNADをそれぞれ、0.15M KPO4、1M NaCl緩衝液(pH7.0)中に取り、50℃でインキュベートした。NADおよびpNADの分解はHPLCにより追跡した。図9には、ゼロ時間における(p)NAD量を基準として比較した(p)NAD量の面積率が%表示されている。図から明らかなように、pNADはNADに比較して非常に安定である。
項目B)でpNADについて記載したとおり、cNADに対するグルコース脱水素酵素活性分析をNADとの比較で行った。このために、グルコース脱水素酵素の使用濃度は0.1mg/ml(cNADの場合)または0.002mg/ml(NADの場合)[それぞれ0.83または0.017μM]とした。使用量および結果は表2に示した。
図10A、10Bおよび10Cは、NADとcNADの吸収スペクトルを示している。NADもcNADも260nmに吸収極大を有している。図10BはNADHおよびcNADHの吸収スペクトルを示している。そのスペクトルは、それぞれグルコース脱水素酵素活性分析後に記録したものである。cNADHの吸収極大は20nmの赤方偏移を示している。図10CにはNADHおよびcNADHの吸収スペクトルがさらに示されているが、注釈部分に記載されているように、それぞれのグルコース脱水素酵素活性分析では異なった条件が選択されている。
Claims (15)
- 被分析物測定のための試験用デバイスであって、
(i)補酵素依存性酵素またはその種の酵素の基質および
(ii)補酵素として下記一般式(I)で表される化合物であって、
Tはそれぞれ独立してO、S、
Uはそれぞれ独立してOH、SH、
Vはそれぞれ独立してOHまたはフォスフェート基、
WはCOOR、CON(R)2、COR、CSN(R)2であって、Rはそれぞれ独立してHまたはC1〜C2アルキル、
X1はO、
X2はO、
YはO、
Zは下記一般式(II)で表される飽和の炭素環またはヘテロ環の五員環であって、
上記式(II)のR6’が上記式(I)のピリジン環の窒素原子に結合し、
R5’とR5”との間が一重結合であり、
R4はそれぞれ独立してH、F、Cl、CH3、
R5はC(R4) 2、
R5’はNH、NC1〜C2アルキル、C(R4) 2、CHOH、CHOCH3、
R5”はC(R4) 2、CHOH、CHOCH3、
R6、R6’はそれぞれ独立してCH、CCH3
である化合物もしくはその塩または必要な場合にはその還元型化合物
を含み、前記補酵素依存性酵素がグルコース脱水素酵素であるグルコース測定のための試験用デバイス。 - 請求項1記載の試験用デバイスであって、WはCONH2またはCOCH3である試験用デバイス。
- 試験ストリップの形態である請求項1または2記載の試験用デバイス。
- 一般式(I”)で表わされる化合物であって、
Tはそれぞれ独立してO、S、
Uはそれぞれ独立してOH、SH、
Vはそれぞれ独立してOHまたはフォスフェート基、
WはCOOR、CON(R)2、COR、CSN(R)2であって、Rはそれぞれ独立してHまたはC1〜C2アルキル、
X1はO、
X2はO、
YはO、
Zは一般式(II)で表される飽和の炭素環またはヘテロ環の五員環であって、
上記式(II)のR6’が上記式(I”)のピリジン環の窒素原子に結合し、
R5’とR5”との間が一重結合であり、
R4はそれぞれ独立してH、F、Cl、CH3、
R5はC(R4) 2、
R5’はNH、NC1〜C2アルキル、C(R4) 2、CHOH、CHOCH3、
R5”はC(R4) 2、CHOH、CHOCH3、
R6、R6’はそれぞれ独立してCH、CCH3、
ただし、R5がCH2、TがO、UがそれぞれOH、VがOH、WがCONH2、X1、X2がOおよびYがOのとき、R5’とR5”は同時にCHOHではないことを条件
とする化合物もしくはその塩または必要な場合にはその還元型化合物。 - 請求項4記載の化合物であって、WがCONH2またはCOCH3である化合物。
- 請求項4または5記載の化合物であって、第1残基のVはOH基であり、第2残基のVはフォスフェート基であって、必要な場合には前記OH基およびフォスフェート基はこれらが結合する炭素原子と環を形成する化合物。
- 請求項4〜6のいずれか1項に記載の化合物であって、R5’がC(R4) 2もしくはCHOHまたはR5’がNHもしくはNC1〜C2アルキルである化合物。
- 然るべき酵素と請求項4〜7のいずれか1項に記載の化合物との組み合わせからなる酵素/補酵素複合体であって、前記酵素が、グルコース脱水素酵素(E.C.1.1.1.47)、乳酸脱水素酵素(E.C.1.1.1.27、 1.1.1.28)、りんご酸脱水素酵素(E.C.1.1.1.37)、グリセロール脱水素酵素(E.C.1.1.1.6)、アルコール脱水素酵素(E.C.1.1.1.1)、アルファ−ヒドロキシブチレート脱水素酵素、ソルビトール脱水素酵素またはL−アミノ酸脱水素酵素(E.C.1.4.1.5)から選択されたアミノ酸脱水素酵素から選択された脱水素酵素であることを特徴とする複合体。
- 酵素反応による試料中の被分析物の検出のための請求項4〜7のいずれか1項に記載の化合物または請求項8記載の複合体の使用。
- 請求項4〜7のいずれか1項に記載の化合物および必要な場合には、然るべき酵素または請求項8記載の酵素/補酵素複合体、さらには然るべき反応緩衝液を含む試薬キットであって、前記酵素が、グルコース脱水素酵素(E.C.1.1.1.47)、乳酸脱水素酵素(E.C.1.1.1.27、 1.1.1.28)、りんご酸脱水素酵素(E.C.1.1.1.37)、グリセロール脱水素酵素(E.C.1.1.1.6)、アルコール脱水素酵素(E.C.1.1.1.1)、アルファ−ヒドロキシブチレート脱水素酵素、ソルビトール脱水素酵素またはL−アミノ酸脱水素酵素(E.C.1.4.1.5)から選択されたアミノ酸脱水素酵素から選択された脱水素酵素であることを特徴とする試薬キット。
- グルコース測定のための、請求項1〜3のいずれか1項に記載の試験用デバイスの使用。
- グルコース、乳酸、りんご酸、グリセロール、アルコール、コレステロール、トリグリセリド、アスコルビン酸、システイン、グルタチオン、ペプチド、尿素、アンモニウム、サリチル酸塩、ピルビン酸塩、5’−ヌクレオチダーゼ、クレアチンキナーゼ(CK)、乳酸脱水素酵素(LDH)および二酸化炭素から選択された被分析物の測定のための、請求項10記載の試薬キットの使用。
- (a)試料と請求項1〜3のいずれか1項に記載の補酵素含有試験用デバイスとの接触、
(b)グルコースの検出
の過程を含む、グルコース検出のための方法。 - (a)試料と請求項10記載の補酵素含有試薬キットとの接触、
(b)被分析物の検出
の過程を含む、被分析物検出のための方法。 - 前記被分析物の検出が分光測定または蛍光測定により行われることを特徴とする請求項113または14記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102005035461.0 | 2005-07-28 | ||
| DE102005035461A DE102005035461A1 (de) | 2005-07-28 | 2005-07-28 | Stabile NAD/NADH-Derivate |
| PCT/EP2006/007493 WO2007012494A1 (de) | 2005-07-28 | 2006-07-28 | Stabile nad/nadh-derivate |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012172878A Division JP5650695B2 (ja) | 2005-07-28 | 2012-08-03 | 安定なnad/nadh誘導体 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2009502844A JP2009502844A (ja) | 2009-01-29 |
| JP2009502844A5 JP2009502844A5 (ja) | 2012-04-19 |
| JP5129133B2 true JP5129133B2 (ja) | 2013-01-23 |
Family
ID=37137552
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008523257A Active JP5129133B2 (ja) | 2005-07-28 | 2006-07-28 | 安定なnad/nadh誘導体 |
| JP2012172878A Active JP5650695B2 (ja) | 2005-07-28 | 2012-08-03 | 安定なnad/nadh誘導体 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012172878A Active JP5650695B2 (ja) | 2005-07-28 | 2012-08-03 | 安定なnad/nadh誘導体 |
Country Status (17)
| Country | Link |
|---|---|
| US (3) | US8809013B2 (ja) |
| EP (3) | EP1907408B1 (ja) |
| JP (2) | JP5129133B2 (ja) |
| KR (4) | KR101483899B1 (ja) |
| CN (2) | CN103122371B (ja) |
| AU (2) | AU2006274140B8 (ja) |
| BR (1) | BRPI0614217A2 (ja) |
| CA (2) | CA2737123A1 (ja) |
| DE (1) | DE102005035461A1 (ja) |
| DK (3) | DK1907408T3 (ja) |
| ES (3) | ES2453150T3 (ja) |
| HK (1) | HK1123308A1 (ja) |
| MX (1) | MX2008000946A (ja) |
| PL (3) | PL2364988T3 (ja) |
| PT (3) | PT2364989E (ja) |
| SG (1) | SG187511A1 (ja) |
| WO (1) | WO2007012494A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012224638A (ja) * | 2005-07-28 | 2012-11-15 | F Hoffmann-La Roche Ag | 安定なnad/nadh誘導体 |
Families Citing this family (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4786451B2 (ja) * | 2005-07-28 | 2011-10-05 | エフ ホフマン−ラ ロッシュ アクチェン ゲゼルシャフト | Nad/nadhの安定化 |
| EP2022859A1 (de) * | 2007-08-01 | 2009-02-11 | Roche Diagnostics GmbH | Verfahren und Vorrichtung zur Bestimmung der Konzentration eines Analyten mittels Fluoreszenzmessung |
| CN101497638B (zh) * | 2008-01-30 | 2012-09-26 | 中国科学院大连化学物理研究所 | 一种nad+类似物及其合成和应用 |
| EP2093284A1 (de) * | 2008-02-19 | 2009-08-26 | F.Hoffmann-La Roche Ag | Stabilisierung von Dehydrogenasen mit stabilen Coenzymen |
| WO2010094427A2 (de) | 2009-02-19 | 2010-08-26 | Roche Diagnostics Gmbh | Platzsparende magazinierung analytischer hilfsmittel |
| EP2398909B1 (de) | 2009-02-19 | 2015-07-22 | F. Hoffmann-La Roche AG | Schnelle reaktionskinetik von enzymen mit niedriger aktivität in trockenen chemieschichten |
| EP2226007A1 (de) | 2009-02-19 | 2010-09-08 | Roche Diagnostics GmbH | Testelementmagazin mit abgedeckten Testfeldern |
| EP2226008A1 (de) | 2009-02-19 | 2010-09-08 | Roche Diagnostics GmbH | Verfahren zur Herstellung eines analytischen Magazins |
| CA2750474C (en) * | 2009-02-19 | 2017-07-25 | F. Hoffmann-La Roche Ag | Fast reaction kinetics of enzymes having low activity in dry chemistry layers |
| JP5544423B2 (ja) | 2009-07-27 | 2014-07-09 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | N置換ピリジニウム化合物の調製のための方法及び物質 |
| CN102471274B (zh) * | 2009-07-27 | 2016-08-03 | 霍夫曼-拉罗奇有限公司 | 用于制备n-取代的吡啶鎓化合物的方法和物质 |
| WO2011012270A1 (en) * | 2009-07-27 | 2011-02-03 | Roche Diagnostics Gmbh | Enzymatic synthesis of carba-nad |
| EP2292751A1 (de) | 2009-08-20 | 2011-03-09 | Roche Diagnostics GmbH | Stabilisierung von Enzymen mit stabilen Coenzymen |
| EP2287605A1 (de) | 2009-08-20 | 2011-02-23 | Roche Diagnostics GmbH | Vereinfachte Magazinierung integrierter Systeme |
| EP2333544A1 (de) | 2009-12-11 | 2011-06-15 | F. Hoffmann-La Roche AG | Sterilisierbare Chemie für Testelemente |
| KR101506928B1 (ko) | 2009-12-16 | 2015-03-30 | 에프. 호프만-라 로슈 아게 | 보호된 분석대상물의 제어 방출에 의한 시험 요소에서 효소의 분해 검출 |
| KR101252351B1 (ko) * | 2010-05-24 | 2013-04-08 | 동국대학교 산학협력단 | 전기효소적 합성 반응을 위한 nad(p) 유도체의 전기화학적 환원 방법 |
| CN102453067B (zh) * | 2010-10-29 | 2015-08-05 | 中国科学院大连化学物理研究所 | 一种nad+类似物的制备方法及其应用 |
| ES2529023T3 (es) | 2011-01-26 | 2015-02-16 | F. Hoffmann-La Roche Ag | Método y sustancias para la preparación de compuestos de piridinio N-sustituidos |
| WO2012140027A1 (de) | 2011-04-12 | 2012-10-18 | Roche Diagnostics Gmbh | Analytisches hilfsmittel |
| US9752990B2 (en) | 2013-09-30 | 2017-09-05 | Honeywell International Inc. | Low-powered system for driving a fuel control mechanism |
| CN102863495A (zh) * | 2011-07-06 | 2013-01-09 | 上海执诚生物科技股份有限公司 | 一种含有nad+或nadh的稳定的组合物 |
| WO2013045487A1 (de) | 2011-09-28 | 2013-04-04 | F. Hoffmann-La Roche Ag | Azomediatoren |
| JP6128808B2 (ja) * | 2011-11-15 | 2017-05-17 | 栄研化学株式会社 | 酵素活性の検出、測定方法およびこれを利用したキット |
| EP2620507A1 (en) | 2012-01-25 | 2013-07-31 | Roche Diagnostics GmbH | Method for the evaluation of the quality of a test element |
| EP2636750A1 (en) | 2012-03-06 | 2013-09-11 | Roche Diagniostics GmbH | Compatible solute ectoine as well as derivatives thereof for enzyme stabilization |
| KR101728597B1 (ko) | 2012-04-19 | 2017-04-19 | 에프. 호프만-라 로슈 아게 | 혈액 내 분석물 농도를 결정하는 방법 및 디바이스 |
| US8920628B2 (en) | 2012-11-02 | 2014-12-30 | Roche Diagnostics Operations, Inc. | Systems and methods for multiple analyte analysis |
| US8921061B2 (en) * | 2012-11-02 | 2014-12-30 | Roche Diagnostics Operations, Inc. | Reagent materials and associated test elements |
| EP2936124B1 (en) | 2012-12-20 | 2017-03-15 | Roche Diabetes Care GmbH | Methods for evaluating medical measurement curves |
| KR101750638B1 (ko) | 2012-12-20 | 2017-06-23 | 에프. 호프만-라 로슈 아게 | 체액의 샘플을 분석하는 방법 |
| EP2770064A1 (de) | 2013-02-22 | 2014-08-27 | F. Hoffmann-La Roche AG | Hocheffiziente Herstellung von Blutglucose-Teststreifen |
| EP2781919A1 (en) | 2013-03-19 | 2014-09-24 | Roche Diagniostics GmbH | Method / device for generating a corrected value of an analyte concentration in a sample of a body fluid |
| WO2014180939A1 (en) | 2013-05-08 | 2014-11-13 | Roche Diagnostics Gmbh | Stabilization of enzymes by nicotinic acid |
| EP3004372A1 (en) * | 2013-06-04 | 2016-04-13 | Roche Diagnostics GmbH | Novel compounds useful for fret and methods related thereto |
| EP2811299A1 (en) | 2013-06-07 | 2014-12-10 | Roche Diagniostics GmbH | Test element for detecting at least one analyte in a body fluid |
| WO2015063025A1 (en) | 2013-10-29 | 2015-05-07 | F. Hoffmann-La Roche Ag | Nano-enzyme containers for test elements |
| EP3074524B1 (en) | 2013-11-27 | 2019-11-06 | Roche Diabetes Care GmbH | Composition comprising up-converting phosphors for detecting an analyte |
| EP2927319A1 (en) | 2014-03-31 | 2015-10-07 | Roche Diagnostics GmbH | High load enzyme immobilization by crosslinking |
| CA2945612C (en) | 2014-04-14 | 2019-09-10 | F. Hoffmann-La Roche Ag | Phenazinium mediators |
| WO2016022604A2 (en) * | 2014-08-05 | 2016-02-11 | Becton Dickinson And Company | Methods and compositions for analyzing glucose-6-phosphate dehydrogenase activity in blood samples |
| CN106573910B (zh) | 2014-08-22 | 2020-08-28 | 豪夫迈·罗氏有限公司 | 氧化还原指示剂 |
| ES2756714T3 (es) | 2014-08-25 | 2020-04-27 | Hoffmann La Roche | Tira reactiva de dos electrodos que compensan la interferencia |
| KR20180087329A (ko) | 2015-12-21 | 2018-08-01 | 에프. 호프만-라 로슈 아게 | 알칼리게네스 파에칼리스로부터의 돌연변이체 3-히드록시부티레이트 탈수소효소 뿐만 아니라 이의 관련 방법 및 용도 |
| JP6989528B2 (ja) | 2016-02-09 | 2022-01-05 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | ロドバクター・スフェロイデス(Rhodobacter sphaeroides)由来の変異型3−ヒドロキシブチレート脱水素酵素及びそれを含む方法 |
| CA3035874A1 (en) | 2016-10-05 | 2018-04-12 | F. Hoffmann-La Roche Ag | Detection reagents and electrode arrangements for multi-analyte diagnostic test elements, as well as methods of using the same |
| EP3339431A1 (en) | 2016-12-22 | 2018-06-27 | Roche Diabetes Care GmbH | Glucose dehydrogenase variants with improved properties |
| CN107966555A (zh) * | 2017-11-23 | 2018-04-27 | 中山市创艺生化工程有限公司 | 一种用于稳定保存nad或其衍生物的存储液 |
| WO2019166394A1 (en) | 2018-02-28 | 2019-09-06 | F. Hoffmann-La Roche Ag | Biocompatibility coating for continuous analyte measurement |
| CN111217744A (zh) * | 2018-11-26 | 2020-06-02 | 中国科学院大连化学物理研究所 | 一种d-氨基酸基nad+类似物及其合成和应用 |
| WO2022132963A1 (en) | 2020-12-15 | 2022-06-23 | Abbott Diabetes Care Inc. | Nad(p) depot for nad(p)-dependent enzyme-based sensors |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS437333Y1 (ja) | 1965-07-08 | 1968-04-01 | ||
| JPS53107486A (en) * | 1977-02-28 | 1978-09-19 | Wako Pure Chem Ind Ltd | Stabilized nadh aqueous solution |
| JPS61274636A (ja) | 1985-05-30 | 1986-12-04 | 西南自動車工業株式会社 | 活魚水槽 |
| US5801006A (en) | 1997-02-04 | 1998-09-01 | Specialty Assays, Inc. | Use of NADPH and NADH analogs in the measurement of enzyme activities and metabolites |
| JP4260245B2 (ja) * | 1998-06-25 | 2009-04-30 | 株式会社三菱化学ヤトロン | 還元型ニコチンアミドアデニンジヌクレオチド類の分解を防止する方法及び分解防止用試薬 |
| US6380380B1 (en) | 1999-01-04 | 2002-04-30 | Specialty Assays, Inc. | Use of nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucliotide phosphate (NADP) analogs to measure enzyme activities metabolites and substrates |
| WO2001094370A1 (fr) | 2000-06-07 | 2001-12-13 | Wako Pure Chemical Industries, Ltd. | Derives de coenzymes et enzymes appropries |
| DE10221845A1 (de) | 2002-05-16 | 2003-11-27 | Roche Diagnostics Gmbh | Verfahren und Reagenzsystem mit nicht-regenerierbarem Enzym-Coenzym-Komplex |
| DE50305687D1 (de) | 2002-05-16 | 2006-12-28 | Roche Diagnostics Gmbh | Verfahren zur herstellung von polymerschichten |
| DE102005035461A1 (de) * | 2005-07-28 | 2007-02-15 | Roche Diagnostics Gmbh | Stabile NAD/NADH-Derivate |
-
2005
- 2005-07-28 DE DE102005035461A patent/DE102005035461A1/de not_active Withdrawn
-
2006
- 2006-07-28 CN CN201210559220.9A patent/CN103122371B/zh active Active
- 2006-07-28 PT PT111674768T patent/PT2364989E/pt unknown
- 2006-07-28 PL PL11167471T patent/PL2364988T3/pl unknown
- 2006-07-28 KR KR20137030825A patent/KR101483899B1/ko not_active IP Right Cessation
- 2006-07-28 ES ES11167471.9T patent/ES2453150T3/es active Active
- 2006-07-28 BR BRPI0614217-6A patent/BRPI0614217A2/pt not_active IP Right Cessation
- 2006-07-28 CA CA2737123A patent/CA2737123A1/en active Pending
- 2006-07-28 KR KR1020117002057A patent/KR20110028522A/ko not_active Application Discontinuation
- 2006-07-28 CN CN2006800273591A patent/CN101233144B/zh active Active
- 2006-07-28 PL PL06776486T patent/PL1907408T3/pl unknown
- 2006-07-28 ES ES11167476.8T patent/ES2461624T3/es active Active
- 2006-07-28 DK DK06776486.0T patent/DK1907408T3/da active
- 2006-07-28 KR KR1020087004422A patent/KR101035524B1/ko not_active IP Right Cessation
- 2006-07-28 AU AU2006274140A patent/AU2006274140B8/en not_active Ceased
- 2006-07-28 PT PT67764860T patent/PT1907408E/pt unknown
- 2006-07-28 EP EP06776486.0A patent/EP1907408B1/de active Active
- 2006-07-28 PT PT111674719T patent/PT2364988E/pt unknown
- 2006-07-28 CA CA2614792A patent/CA2614792C/en active Active
- 2006-07-28 DK DK11167471.9T patent/DK2364988T3/da active
- 2006-07-28 SG SG2013005301A patent/SG187511A1/en unknown
- 2006-07-28 MX MX2008000946A patent/MX2008000946A/es active IP Right Grant
- 2006-07-28 KR KR20127005354A patent/KR101483829B1/ko not_active IP Right Cessation
- 2006-07-28 ES ES06776486.0T patent/ES2461266T3/es active Active
- 2006-07-28 DK DK11167476.8T patent/DK2364989T3/da active
- 2006-07-28 PL PL11167476T patent/PL2364989T3/pl unknown
- 2006-07-28 EP EP11167476.8A patent/EP2364989B1/de active Active
- 2006-07-28 EP EP11167471.9A patent/EP2364988B1/de active Active
- 2006-07-28 WO PCT/EP2006/007493 patent/WO2007012494A1/de active Application Filing
- 2006-07-28 JP JP2008523257A patent/JP5129133B2/ja active Active
-
2008
- 2008-01-25 US US12/020,244 patent/US8809013B2/en active Active
-
2009
- 2009-01-21 HK HK09100651.1A patent/HK1123308A1/xx not_active IP Right Cessation
-
2011
- 2011-02-04 AU AU2011200465A patent/AU2011200465B2/en not_active Revoked
-
2012
- 2012-08-03 JP JP2012172878A patent/JP5650695B2/ja active Active
-
2014
- 2014-03-23 US US14/222,645 patent/US9399790B2/en active Active
-
2016
- 2016-07-20 US US15/214,494 patent/US10167497B2/en active Active
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012224638A (ja) * | 2005-07-28 | 2012-11-15 | F Hoffmann-La Roche Ag | 安定なnad/nadh誘導体 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5129133B2 (ja) | 安定なnad/nadh誘導体 | |
| JP4786451B2 (ja) | Nad/nadhの安定化 | |
| AU2012201023B2 (en) | Stable NAD/NADH derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20100525 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20111101 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120201 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120208 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20120229 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20120403 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120803 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20120815 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120925 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120927 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20121023 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121101 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 Ref document number: 5129133 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151109 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |