JP5127043B2 - ホスファチジルセリンに結合するfc融合構築物およびそれらの治療用途 - Google Patents
ホスファチジルセリンに結合するfc融合構築物およびそれらの治療用途 Download PDFInfo
- Publication number
- JP5127043B2 JP5127043B2 JP2007552418A JP2007552418A JP5127043B2 JP 5127043 B2 JP5127043 B2 JP 5127043B2 JP 2007552418 A JP2007552418 A JP 2007552418A JP 2007552418 A JP2007552418 A JP 2007552418A JP 5127043 B2 JP5127043 B2 JP 5127043B2
- Authority
- JP
- Japan
- Prior art keywords
- tumor
- β2gpi
- construct
- binding
- phosphatidylserine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- ZWZWYGMENQVNFU-UHFFFAOYSA-N Glycerophosphorylserin Natural products OC(=O)C(N)COP(O)(=O)OCC(O)CO ZWZWYGMENQVNFU-UHFFFAOYSA-N 0.000 title claims description 222
- TZCPCKNHXULUIY-RGULYWFUSA-N 1,2-distearoyl-sn-glycero-3-phosphoserine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCCCC TZCPCKNHXULUIY-RGULYWFUSA-N 0.000 title claims description 219
- 230000001225 therapeutic effect Effects 0.000 title claims description 31
- 230000004927 fusion Effects 0.000 title description 2
- 206010028980 Neoplasm Diseases 0.000 claims description 318
- 239000003814 drug Substances 0.000 claims description 149
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 141
- 238000009739 binding Methods 0.000 claims description 137
- 230000027455 binding Effects 0.000 claims description 136
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 105
- 239000000203 mixture Substances 0.000 claims description 103
- 229920001184 polypeptide Polymers 0.000 claims description 94
- 229940124597 therapeutic agent Drugs 0.000 claims description 65
- 241000282414 Homo sapiens Species 0.000 claims description 52
- 239000002246 antineoplastic agent Substances 0.000 claims description 50
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 47
- 239000003443 antiviral agent Substances 0.000 claims description 44
- 201000010099 disease Diseases 0.000 claims description 44
- 201000011510 cancer Diseases 0.000 claims description 40
- 230000009385 viral infection Effects 0.000 claims description 39
- 208000036142 Viral infection Diseases 0.000 claims description 38
- 238000002560 therapeutic procedure Methods 0.000 claims description 33
- 239000000427 antigen Substances 0.000 claims description 25
- 108091007433 antigens Proteins 0.000 claims description 25
- 102000036639 antigens Human genes 0.000 claims description 25
- 239000012634 fragment Substances 0.000 claims description 24
- 239000004037 angiogenesis inhibitor Substances 0.000 claims description 23
- 239000000032 diagnostic agent Substances 0.000 claims description 20
- 102100030802 Beta-2-glycoprotein 1 Human genes 0.000 claims description 16
- 108010023562 beta 2-Glycoprotein I Proteins 0.000 claims description 16
- 238000004519 manufacturing process Methods 0.000 claims description 15
- 102000037865 fusion proteins Human genes 0.000 claims description 14
- 108020001507 fusion proteins Proteins 0.000 claims description 14
- 239000000126 substance Chemical group 0.000 claims description 11
- 238000003259 recombinant expression Methods 0.000 claims description 10
- 238000003384 imaging method Methods 0.000 claims description 8
- 230000003612 virological effect Effects 0.000 claims description 8
- 238000001959 radiotherapy Methods 0.000 claims description 4
- 239000012216 imaging agent Substances 0.000 claims 1
- 102000005962 receptors Human genes 0.000 description 143
- 108020003175 receptors Proteins 0.000 description 143
- 210000004027 cell Anatomy 0.000 description 122
- 238000000034 method Methods 0.000 description 91
- 238000011282 treatment Methods 0.000 description 91
- -1 anionic phospholipids Chemical class 0.000 description 86
- 239000000562 conjugate Substances 0.000 description 79
- 108090000623 proteins and genes Proteins 0.000 description 68
- 229940079593 drug Drugs 0.000 description 65
- 239000003795 chemical substances by application Substances 0.000 description 64
- 210000004204 blood vessel Anatomy 0.000 description 62
- 102000004169 proteins and genes Human genes 0.000 description 57
- 235000018102 proteins Nutrition 0.000 description 54
- 108010000499 Thromboplastin Proteins 0.000 description 53
- 102000002262 Thromboplastin Human genes 0.000 description 53
- 210000002889 endothelial cell Anatomy 0.000 description 53
- 241001465754 Metazoa Species 0.000 description 49
- 239000003446 ligand Substances 0.000 description 48
- 210000004881 tumor cell Anatomy 0.000 description 41
- 230000006870 function Effects 0.000 description 40
- 239000002502 liposome Substances 0.000 description 40
- 238000001727 in vivo Methods 0.000 description 36
- 102100032231 Caveolae-associated protein 2 Human genes 0.000 description 35
- 108050005259 Caveolae-associated protein 2 Proteins 0.000 description 35
- 230000014509 gene expression Effects 0.000 description 35
- 210000001519 tissue Anatomy 0.000 description 35
- 125000005647 linker group Chemical group 0.000 description 34
- 235000001014 amino acid Nutrition 0.000 description 33
- 150000001413 amino acids Chemical class 0.000 description 32
- 102000000412 Annexin Human genes 0.000 description 31
- 108050008874 Annexin Proteins 0.000 description 31
- 229940024606 amino acid Drugs 0.000 description 31
- 230000006907 apoptotic process Effects 0.000 description 31
- 239000000306 component Substances 0.000 description 30
- 239000003053 toxin Substances 0.000 description 29
- 231100000765 toxin Toxicity 0.000 description 29
- 108700012359 toxins Proteins 0.000 description 29
- 241000699670 Mus sp. Species 0.000 description 28
- 230000000694 effects Effects 0.000 description 28
- 210000004379 membrane Anatomy 0.000 description 27
- 238000000338 in vitro Methods 0.000 description 26
- 210000002540 macrophage Anatomy 0.000 description 26
- 239000012528 membrane Substances 0.000 description 26
- 230000002792 vascular Effects 0.000 description 26
- 102000004121 Annexin A5 Human genes 0.000 description 25
- 108090000672 Annexin A5 Proteins 0.000 description 25
- 102000004127 Cytokines Human genes 0.000 description 25
- 108090000695 Cytokines Proteins 0.000 description 25
- 229940127089 cytotoxic agent Drugs 0.000 description 25
- 230000006378 damage Effects 0.000 description 25
- 238000009472 formulation Methods 0.000 description 25
- 230000000840 anti-viral effect Effects 0.000 description 24
- 108090000790 Enzymes Proteins 0.000 description 23
- 102000004190 Enzymes Human genes 0.000 description 23
- 125000003275 alpha amino acid group Chemical group 0.000 description 23
- 239000000539 dimer Substances 0.000 description 23
- 229940088598 enzyme Drugs 0.000 description 23
- 241000699666 Mus <mouse, genus> Species 0.000 description 22
- 239000012636 effector Substances 0.000 description 22
- 239000002243 precursor Substances 0.000 description 22
- 230000033115 angiogenesis Effects 0.000 description 21
- 230000007246 mechanism Effects 0.000 description 21
- 239000008194 pharmaceutical composition Substances 0.000 description 21
- 230000008685 targeting Effects 0.000 description 21
- 241000700605 Viruses Species 0.000 description 20
- 230000000259 anti-tumor effect Effects 0.000 description 20
- 150000003904 phospholipids Chemical class 0.000 description 20
- 238000002360 preparation method Methods 0.000 description 20
- 210000003556 vascular endothelial cell Anatomy 0.000 description 20
- 229940039227 diagnostic agent Drugs 0.000 description 19
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 18
- 230000001640 apoptogenic effect Effects 0.000 description 18
- 210000003038 endothelium Anatomy 0.000 description 18
- 230000032258 transport Effects 0.000 description 18
- 230000015271 coagulation Effects 0.000 description 17
- 238000005345 coagulation Methods 0.000 description 17
- 238000002474 experimental method Methods 0.000 description 17
- 102000023884 phosphatidylserine binding proteins Human genes 0.000 description 17
- 108091008423 phosphatidylserine binding proteins Proteins 0.000 description 17
- 210000005166 vasculature Anatomy 0.000 description 17
- JZNWSCPGTDBMEW-UHFFFAOYSA-N Glycerophosphorylethanolamin Natural products NCCOP(O)(=O)OCC(O)CO JZNWSCPGTDBMEW-UHFFFAOYSA-N 0.000 description 16
- 239000003124 biologic agent Substances 0.000 description 16
- 150000008104 phosphatidylethanolamines Chemical class 0.000 description 16
- 230000008901 benefit Effects 0.000 description 15
- 102400000068 Angiostatin Human genes 0.000 description 14
- 108010079709 Angiostatins Proteins 0.000 description 14
- 102000015081 Blood Coagulation Factors Human genes 0.000 description 14
- 108010039209 Blood Coagulation Factors Proteins 0.000 description 14
- 108010094028 Prothrombin Proteins 0.000 description 14
- FZCSTZYAHCUGEM-UHFFFAOYSA-N aspergillomarasmine B Natural products OC(=O)CNC(C(O)=O)CNC(C(O)=O)CC(O)=O FZCSTZYAHCUGEM-UHFFFAOYSA-N 0.000 description 14
- 239000003114 blood coagulation factor Substances 0.000 description 14
- 229940127121 immunoconjugate Drugs 0.000 description 14
- 210000002966 serum Anatomy 0.000 description 14
- 239000004066 vascular targeting agent Substances 0.000 description 14
- 102000014914 Carrier Proteins Human genes 0.000 description 13
- 206010053567 Coagulopathies Diseases 0.000 description 13
- 239000004971 Cross linker Substances 0.000 description 13
- 230000035602 clotting Effects 0.000 description 13
- 238000002648 combination therapy Methods 0.000 description 13
- 230000001419 dependent effect Effects 0.000 description 13
- 230000003993 interaction Effects 0.000 description 13
- 102100023804 Coagulation factor VII Human genes 0.000 description 12
- 108010023321 Factor VII Proteins 0.000 description 12
- 108090000190 Thrombin Proteins 0.000 description 12
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 12
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 12
- 238000001994 activation Methods 0.000 description 12
- 230000004913 activation Effects 0.000 description 12
- 108091008324 binding proteins Proteins 0.000 description 12
- 210000004369 blood Anatomy 0.000 description 12
- 239000008280 blood Substances 0.000 description 12
- 239000000701 coagulant Substances 0.000 description 12
- 229940012413 factor vii Drugs 0.000 description 12
- 230000002209 hydrophobic effect Effects 0.000 description 12
- 150000002632 lipids Chemical class 0.000 description 12
- 230000001404 mediated effect Effects 0.000 description 12
- 238000006467 substitution reaction Methods 0.000 description 12
- 229960004072 thrombin Drugs 0.000 description 12
- 239000013598 vector Substances 0.000 description 12
- 241000283690 Bos taurus Species 0.000 description 11
- 102100031162 Collagen alpha-1(XVIII) chain Human genes 0.000 description 11
- 108010079505 Endostatins Proteins 0.000 description 11
- 230000009471 action Effects 0.000 description 11
- 150000001875 compounds Chemical class 0.000 description 11
- 230000021615 conjugation Effects 0.000 description 11
- 230000001939 inductive effect Effects 0.000 description 11
- 239000003550 marker Substances 0.000 description 11
- 230000001717 pathogenic effect Effects 0.000 description 11
- 102000005912 ran GTP Binding Protein Human genes 0.000 description 11
- 125000006850 spacer group Chemical group 0.000 description 11
- 108010054265 Factor VIIa Proteins 0.000 description 10
- 102100027378 Prothrombin Human genes 0.000 description 10
- 108010039491 Ricin Proteins 0.000 description 10
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 10
- 230000001772 anti-angiogenic effect Effects 0.000 description 10
- 230000001093 anti-cancer Effects 0.000 description 10
- 210000000170 cell membrane Anatomy 0.000 description 10
- 210000003989 endothelium vascular Anatomy 0.000 description 10
- 229940012414 factor viia Drugs 0.000 description 10
- 229940039716 prothrombin Drugs 0.000 description 10
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 9
- 241000282412 Homo Species 0.000 description 9
- 102000018071 Immunoglobulin Fc Fragments Human genes 0.000 description 9
- 108010091135 Immunoglobulin Fc Fragments Proteins 0.000 description 9
- 241000700159 Rattus Species 0.000 description 9
- 230000015572 biosynthetic process Effects 0.000 description 9
- 230000000295 complement effect Effects 0.000 description 9
- 210000004072 lung Anatomy 0.000 description 9
- 230000004614 tumor growth Effects 0.000 description 9
- 102100023700 C-C motif chemokine 16 Human genes 0.000 description 8
- 108010035532 Collagen Proteins 0.000 description 8
- 102000008186 Collagen Human genes 0.000 description 8
- 101000978375 Homo sapiens C-C motif chemokine 16 Proteins 0.000 description 8
- 239000004365 Protease Substances 0.000 description 8
- 241000700584 Simplexvirus Species 0.000 description 8
- 108060008245 Thrombospondin Proteins 0.000 description 8
- 102000002938 Thrombospondin Human genes 0.000 description 8
- 239000002253 acid Substances 0.000 description 8
- 230000004071 biological effect Effects 0.000 description 8
- 238000003776 cleavage reaction Methods 0.000 description 8
- 229920001436 collagen Polymers 0.000 description 8
- 231100000599 cytotoxic agent Toxicity 0.000 description 8
- 238000011161 development Methods 0.000 description 8
- 230000018109 developmental process Effects 0.000 description 8
- 239000003112 inhibitor Substances 0.000 description 8
- 230000004807 localization Effects 0.000 description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 8
- 230000007017 scission Effects 0.000 description 8
- 239000000243 solution Substances 0.000 description 8
- 108020004414 DNA Proteins 0.000 description 7
- 108010074860 Factor Xa Proteins 0.000 description 7
- 108060003951 Immunoglobulin Proteins 0.000 description 7
- 102000005741 Metalloproteases Human genes 0.000 description 7
- 108010006035 Metalloproteases Proteins 0.000 description 7
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 7
- 102000035195 Peptidases Human genes 0.000 description 7
- 108091005804 Peptidases Proteins 0.000 description 7
- 239000002202 Polyethylene glycol Substances 0.000 description 7
- 208000007536 Thrombosis Diseases 0.000 description 7
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 7
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 7
- 108090000183 alpha-2-Antiplasmin Proteins 0.000 description 7
- 102000003801 alpha-2-Antiplasmin Human genes 0.000 description 7
- 230000002491 angiogenic effect Effects 0.000 description 7
- 210000001772 blood platelet Anatomy 0.000 description 7
- 239000011575 calcium Substances 0.000 description 7
- 230000030833 cell death Effects 0.000 description 7
- 238000002512 chemotherapy Methods 0.000 description 7
- 239000003431 cross linking reagent Substances 0.000 description 7
- 231100000433 cytotoxic Toxicity 0.000 description 7
- 230000001472 cytotoxic effect Effects 0.000 description 7
- 229960003668 docetaxel Drugs 0.000 description 7
- 229960004679 doxorubicin Drugs 0.000 description 7
- 229940012426 factor x Drugs 0.000 description 7
- 102000018358 immunoglobulin Human genes 0.000 description 7
- 230000001965 increasing effect Effects 0.000 description 7
- 230000002757 inflammatory effect Effects 0.000 description 7
- 230000007935 neutral effect Effects 0.000 description 7
- 238000011275 oncology therapy Methods 0.000 description 7
- 229920001223 polyethylene glycol Polymers 0.000 description 7
- 230000003389 potentiating effect Effects 0.000 description 7
- LGZKGOGODCLQHG-CYBMUJFWSA-N 5-[(2r)-2-hydroxy-2-(3,4,5-trimethoxyphenyl)ethyl]-2-methoxyphenol Chemical compound C1=C(O)C(OC)=CC=C1C[C@@H](O)C1=CC(OC)=C(OC)C(OC)=C1 LGZKGOGODCLQHG-CYBMUJFWSA-N 0.000 description 6
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 6
- 108010014172 Factor V Proteins 0.000 description 6
- 108010014173 Factor X Proteins 0.000 description 6
- 108010088842 Fibrinolysin Proteins 0.000 description 6
- 108700004714 Gelonium multiflorum GEL Proteins 0.000 description 6
- 102000003886 Glycoproteins Human genes 0.000 description 6
- 108090000288 Glycoproteins Proteins 0.000 description 6
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 6
- 206010021143 Hypoxia Diseases 0.000 description 6
- 108010002350 Interleukin-2 Proteins 0.000 description 6
- 102000000588 Interleukin-2 Human genes 0.000 description 6
- 102100027998 Macrophage metalloelastase Human genes 0.000 description 6
- 108010076501 Matrix Metalloproteinase 12 Proteins 0.000 description 6
- 229930012538 Paclitaxel Natural products 0.000 description 6
- 102000013566 Plasminogen Human genes 0.000 description 6
- 108010051456 Plasminogen Proteins 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 208000027418 Wounds and injury Diseases 0.000 description 6
- 230000002022 anti-cellular effect Effects 0.000 description 6
- 230000001580 bacterial effect Effects 0.000 description 6
- 230000022534 cell killing Effects 0.000 description 6
- 230000004663 cell proliferation Effects 0.000 description 6
- LGZKGOGODCLQHG-UHFFFAOYSA-N combretastatin Natural products C1=C(O)C(OC)=CC=C1CC(O)C1=CC(OC)=C(OC)C(OC)=C1 LGZKGOGODCLQHG-UHFFFAOYSA-N 0.000 description 6
- 150000004814 combretastatins Chemical class 0.000 description 6
- 238000004132 cross linking Methods 0.000 description 6
- 239000002254 cytotoxic agent Substances 0.000 description 6
- 239000013604 expression vector Substances 0.000 description 6
- 230000002637 immunotoxin Effects 0.000 description 6
- 239000002596 immunotoxin Substances 0.000 description 6
- 229940051026 immunotoxin Drugs 0.000 description 6
- 231100000608 immunotoxin Toxicity 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 208000014674 injury Diseases 0.000 description 6
- 102000006495 integrins Human genes 0.000 description 6
- 108010044426 integrins Proteins 0.000 description 6
- 210000000265 leukocyte Anatomy 0.000 description 6
- 230000004576 lipid-binding Effects 0.000 description 6
- 229960001592 paclitaxel Drugs 0.000 description 6
- 239000002245 particle Substances 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 150000003839 salts Chemical class 0.000 description 6
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 6
- 230000005945 translocation Effects 0.000 description 6
- 230000029812 viral genome replication Effects 0.000 description 6
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 5
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 5
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 5
- 108010009906 Angiopoietins Proteins 0.000 description 5
- 102000009840 Angiopoietins Human genes 0.000 description 5
- 208000003343 Antiphospholipid Syndrome Diseases 0.000 description 5
- 102000019034 Chemokines Human genes 0.000 description 5
- 108010012236 Chemokines Proteins 0.000 description 5
- 241000701022 Cytomegalovirus Species 0.000 description 5
- 108010048049 Factor IXa Proteins 0.000 description 5
- 102100037362 Fibronectin Human genes 0.000 description 5
- 108010067306 Fibronectins Proteins 0.000 description 5
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 5
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 5
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 5
- 239000004472 Lysine Substances 0.000 description 5
- 206010028851 Necrosis Diseases 0.000 description 5
- 102000004211 Platelet factor 4 Human genes 0.000 description 5
- 108090000778 Platelet factor 4 Proteins 0.000 description 5
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 5
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- 108010069801 angiopoietin 4 Proteins 0.000 description 5
- 230000003429 anti-cardiolipin effect Effects 0.000 description 5
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 5
- 238000010913 antigen-directed enzyme pro-drug therapy Methods 0.000 description 5
- 230000023555 blood coagulation Effects 0.000 description 5
- 239000000969 carrier Substances 0.000 description 5
- 230000008859 change Effects 0.000 description 5
- 239000002299 complementary DNA Substances 0.000 description 5
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 5
- 235000018417 cysteine Nutrition 0.000 description 5
- 238000005516 engineering process Methods 0.000 description 5
- 238000001415 gene therapy Methods 0.000 description 5
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 5
- 230000007954 hypoxia Effects 0.000 description 5
- 229940072221 immunoglobulins Drugs 0.000 description 5
- 239000000411 inducer Substances 0.000 description 5
- 230000006698 induction Effects 0.000 description 5
- 208000015181 infectious disease Diseases 0.000 description 5
- 239000004615 ingredient Substances 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- 230000002601 intratumoral effect Effects 0.000 description 5
- 230000003211 malignant effect Effects 0.000 description 5
- 230000035772 mutation Effects 0.000 description 5
- 230000017074 necrotic cell death Effects 0.000 description 5
- 230000001338 necrotic effect Effects 0.000 description 5
- 230000001575 pathological effect Effects 0.000 description 5
- 125000001095 phosphatidyl group Chemical group 0.000 description 5
- 230000004962 physiological condition Effects 0.000 description 5
- 229940012957 plasmin Drugs 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 230000001737 promoting effect Effects 0.000 description 5
- 230000004044 response Effects 0.000 description 5
- 230000000284 resting effect Effects 0.000 description 5
- 241000894007 species Species 0.000 description 5
- 150000003431 steroids Chemical class 0.000 description 5
- 229960003433 thalidomide Drugs 0.000 description 5
- 125000003396 thiol group Chemical group [H]S* 0.000 description 5
- GKSPIZSKQWTXQG-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-[1-(pyridin-2-yldisulfanyl)ethyl]benzoate Chemical compound C=1C=C(C(=O)ON2C(CCC2=O)=O)C=CC=1C(C)SSC1=CC=CC=N1 GKSPIZSKQWTXQG-UHFFFAOYSA-N 0.000 description 4
- VEEGZPWAAPPXRB-BJMVGYQFSA-N (3e)-3-(1h-imidazol-5-ylmethylidene)-1h-indol-2-one Chemical compound O=C1NC2=CC=CC=C2\C1=C/C1=CN=CN1 VEEGZPWAAPPXRB-BJMVGYQFSA-N 0.000 description 4
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 4
- PORPENFLTBBHSG-MGBGTMOVSA-N 1,2-dihexadecanoyl-sn-glycerol-3-phosphate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(O)=O)OC(=O)CCCCCCCCCCCCCCC PORPENFLTBBHSG-MGBGTMOVSA-N 0.000 description 4
- 108010048154 Angiopoietin-1 Proteins 0.000 description 4
- 102000009088 Angiopoietin-1 Human genes 0.000 description 4
- 102000004145 Annexin A1 Human genes 0.000 description 4
- 108090000663 Annexin A1 Proteins 0.000 description 4
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 4
- 239000004475 Arginine Substances 0.000 description 4
- 102000004506 Blood Proteins Human genes 0.000 description 4
- 108010017384 Blood Proteins Proteins 0.000 description 4
- 206010006187 Breast cancer Diseases 0.000 description 4
- 208000026310 Breast neoplasm Diseases 0.000 description 4
- 201000009030 Carcinoma Diseases 0.000 description 4
- VWFCHDSQECPREK-LURJTMIESA-N Cidofovir Chemical compound NC=1C=CN(C[C@@H](CO)OCP(O)(O)=O)C(=O)N=1 VWFCHDSQECPREK-LURJTMIESA-N 0.000 description 4
- 108091026890 Coding region Proteins 0.000 description 4
- 102000029816 Collagenase Human genes 0.000 description 4
- 108060005980 Collagenase Proteins 0.000 description 4
- 108010073385 Fibrin Proteins 0.000 description 4
- 102000009123 Fibrin Human genes 0.000 description 4
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 4
- 102000013382 Gelatinases Human genes 0.000 description 4
- 108010026132 Gelatinases Proteins 0.000 description 4
- 239000004471 Glycine Substances 0.000 description 4
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 4
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 4
- 108010047761 Interferon-alpha Proteins 0.000 description 4
- 102000006992 Interferon-alpha Human genes 0.000 description 4
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 4
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 4
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 4
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 4
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 4
- 102000000422 Matrix Metalloproteinase 3 Human genes 0.000 description 4
- 108091028043 Nucleic acid sequence Proteins 0.000 description 4
- 108700020796 Oncogene Proteins 0.000 description 4
- 206010057249 Phagocytosis Diseases 0.000 description 4
- 108010029485 Protein Isoforms Proteins 0.000 description 4
- 102000001708 Protein Isoforms Human genes 0.000 description 4
- 108020004511 Recombinant DNA Proteins 0.000 description 4
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 4
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 4
- ATBOMIWRCZXYSZ-XZBBILGWSA-N [1-[2,3-dihydroxypropoxy(hydroxy)phosphoryl]oxy-3-hexadecanoyloxypropan-2-yl] (9e,12e)-octadeca-9,12-dienoate Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(COP(O)(=O)OCC(O)CO)OC(=O)CCCCCCC\C=C\C\C=C\CCCCC ATBOMIWRCZXYSZ-XZBBILGWSA-N 0.000 description 4
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 4
- 230000003213 activating effect Effects 0.000 description 4
- 239000012190 activator Substances 0.000 description 4
- 239000013543 active substance Substances 0.000 description 4
- 239000000443 aerosol Substances 0.000 description 4
- 239000000556 agonist Substances 0.000 description 4
- 235000004279 alanine Nutrition 0.000 description 4
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 4
- 125000000539 amino acid group Chemical group 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 238000010171 animal model Methods 0.000 description 4
- 230000002137 anti-vascular effect Effects 0.000 description 4
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 4
- WZPBZJONDBGPKJ-VEHQQRBSSA-N aztreonam Chemical compound O=C1N(S([O-])(=O)=O)[C@@H](C)[C@@H]1NC(=O)C(=N/OC(C)(C)C(O)=O)\C1=CSC([NH3+])=N1 WZPBZJONDBGPKJ-VEHQQRBSSA-N 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 229960000724 cidofovir Drugs 0.000 description 4
- 238000000576 coating method Methods 0.000 description 4
- AGVAZMGAQJOSFJ-WZHZPDAFSA-M cobalt(2+);[(2r,3s,4r,5s)-5-(5,6-dimethylbenzimidazol-1-yl)-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl] [(2r)-1-[3-[(1r,2r,3r,4z,7s,9z,12s,13s,14z,17s,18s,19r)-2,13,18-tris(2-amino-2-oxoethyl)-7,12,17-tris(3-amino-3-oxopropyl)-3,5,8,8,13,15,18,19-octamethyl-2 Chemical compound [Co+2].N#[C-].[N-]([C@@H]1[C@H](CC(N)=O)[C@@]2(C)CCC(=O)NC[C@@H](C)OP(O)(=O)O[C@H]3[C@H]([C@H](O[C@@H]3CO)N3C4=CC(C)=C(C)C=C4N=C3)O)\C2=C(C)/C([C@H](C\2(C)C)CCC(N)=O)=N/C/2=C\C([C@H]([C@@]/2(CC(N)=O)C)CCC(N)=O)=N\C\2=C(C)/C2=N[C@]1(C)[C@@](C)(CC(N)=O)[C@@H]2CCC(N)=O AGVAZMGAQJOSFJ-WZHZPDAFSA-M 0.000 description 4
- 229960002424 collagenase Drugs 0.000 description 4
- 230000009089 cytolysis Effects 0.000 description 4
- 239000000824 cytostatic agent Substances 0.000 description 4
- 230000007423 decrease Effects 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 229950003499 fibrin Drugs 0.000 description 4
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 229920000669 heparin Polymers 0.000 description 4
- 229960002897 heparin Drugs 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 206010025135 lupus erythematosus Diseases 0.000 description 4
- 230000010534 mechanism of action Effects 0.000 description 4
- 230000011278 mitosis Effects 0.000 description 4
- 230000037361 pathway Effects 0.000 description 4
- 210000001539 phagocyte Anatomy 0.000 description 4
- 230000008782 phagocytosis Effects 0.000 description 4
- 239000002831 pharmacologic agent Substances 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 4
- 150000003905 phosphatidylinositols Chemical class 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 230000002062 proliferating effect Effects 0.000 description 4
- 235000019419 proteases Nutrition 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 238000010186 staining Methods 0.000 description 4
- 108091007196 stromelysin Proteins 0.000 description 4
- 238000013268 sustained release Methods 0.000 description 4
- 239000012730 sustained-release form Substances 0.000 description 4
- 238000011287 therapeutic dose Methods 0.000 description 4
- 230000001732 thrombotic effect Effects 0.000 description 4
- RZWIIPASKMUIAC-VQTJNVASSA-N thromboxane Chemical compound CCCCCCCC[C@H]1OCCC[C@@H]1CCCCCCC RZWIIPASKMUIAC-VQTJNVASSA-N 0.000 description 4
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 4
- 230000008728 vascular permeability Effects 0.000 description 4
- 229960002555 zidovudine Drugs 0.000 description 4
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 3
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 3
- 102100026802 72 kDa type IV collagenase Human genes 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 102100025665 Angiopoietin-related protein 1 Human genes 0.000 description 3
- 108090001008 Avidin Proteins 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical group C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 108010078791 Carrier Proteins Proteins 0.000 description 3
- 108010067225 Cell Adhesion Molecules Proteins 0.000 description 3
- 102000016289 Cell Adhesion Molecules Human genes 0.000 description 3
- 102100022641 Coagulation factor IX Human genes 0.000 description 3
- 108010001463 Collagen Type XVIII Proteins 0.000 description 3
- 102000047200 Collagen Type XVIII Human genes 0.000 description 3
- 241000196324 Embryophyta Species 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 108010076282 Factor IX Proteins 0.000 description 3
- 108010074105 Factor Va Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 3
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 3
- 102100026720 Interferon beta Human genes 0.000 description 3
- 108090000467 Interferon-beta Proteins 0.000 description 3
- 102000014150 Interferons Human genes 0.000 description 3
- 108010050904 Interferons Proteins 0.000 description 3
- 102000000589 Interleukin-1 Human genes 0.000 description 3
- 108010002352 Interleukin-1 Proteins 0.000 description 3
- 102000013462 Interleukin-12 Human genes 0.000 description 3
- 108010065805 Interleukin-12 Proteins 0.000 description 3
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 3
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 3
- 206010025323 Lymphomas Diseases 0.000 description 3
- 206010027476 Metastases Diseases 0.000 description 3
- 101000605401 Mus musculus Plasminogen Proteins 0.000 description 3
- 101100481410 Mus musculus Tek gene Proteins 0.000 description 3
- MSHZHSPISPJWHW-UHFFFAOYSA-N O-(chloroacetylcarbamoyl)fumagillol Chemical compound O1C(CC=C(C)C)C1(C)C1C(OC)C(OC(=O)NC(=O)CCl)CCC21CO2 MSHZHSPISPJWHW-UHFFFAOYSA-N 0.000 description 3
- MSHZHSPISPJWHW-PVDLLORBSA-N O-(chloroacetylcarbamoyl)fumagillol Chemical compound C([C@H]([C@H]([C@@H]1[C@]2(C)[C@H](O2)CC=C(C)C)OC)OC(=O)NC(=O)CCl)C[C@@]21CO2 MSHZHSPISPJWHW-PVDLLORBSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 108090000526 Papain Proteins 0.000 description 3
- 229940123573 Protein synthesis inhibitor Drugs 0.000 description 3
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 3
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 3
- 206010068771 Soft tissue neoplasm Diseases 0.000 description 3
- 102100030416 Stromelysin-1 Human genes 0.000 description 3
- 210000001744 T-lymphocyte Anatomy 0.000 description 3
- 102000012753 TIE-2 Receptor Human genes 0.000 description 3
- 108010090091 TIE-2 Receptor Proteins 0.000 description 3
- 108700012411 TNFSF10 Proteins 0.000 description 3
- 102000004243 Tubulin Human genes 0.000 description 3
- 108090000704 Tubulin Proteins 0.000 description 3
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 3
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 3
- 206010054094 Tumour necrosis Diseases 0.000 description 3
- 108090000435 Urokinase-type plasminogen activator Proteins 0.000 description 3
- 102000003990 Urokinase-type plasminogen activator Human genes 0.000 description 3
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 3
- 125000000129 anionic group Chemical group 0.000 description 3
- 239000005557 antagonist Substances 0.000 description 3
- 239000003146 anticoagulant agent Substances 0.000 description 3
- 229940127219 anticoagulant drug Drugs 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 230000003143 atherosclerotic effect Effects 0.000 description 3
- XDHNQDDQEHDUTM-UHFFFAOYSA-N bafliomycin A1 Natural products COC1C=CC=C(C)CC(C)C(O)C(C)C=C(C)C=C(OC)C(=O)OC1C(C)C(O)C(C)C1(O)OC(C(C)C)C(C)C(O)C1 XDHNQDDQEHDUTM-UHFFFAOYSA-N 0.000 description 3
- 108700000707 bcl-2-Associated X Proteins 0.000 description 3
- 102000055102 bcl-2-Associated X Human genes 0.000 description 3
- 229960002685 biotin Drugs 0.000 description 3
- 235000020958 biotin Nutrition 0.000 description 3
- 239000011616 biotin Substances 0.000 description 3
- 230000017531 blood circulation Effects 0.000 description 3
- 210000004899 c-terminal region Anatomy 0.000 description 3
- 150000001720 carbohydrates Chemical group 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 238000010367 cloning Methods 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 108010047295 complement receptors Proteins 0.000 description 3
- 102000006834 complement receptors Human genes 0.000 description 3
- 229920001577 copolymer Polymers 0.000 description 3
- 230000003013 cytotoxicity Effects 0.000 description 3
- 231100000135 cytotoxicity Toxicity 0.000 description 3
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 3
- 230000002950 deficient Effects 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 238000003745 diagnosis Methods 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 239000002612 dispersion medium Substances 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 238000012377 drug delivery Methods 0.000 description 3
- 230000010595 endothelial cell migration Effects 0.000 description 3
- 239000002158 endotoxin Substances 0.000 description 3
- 230000002708 enhancing effect Effects 0.000 description 3
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 3
- 229960005420 etoposide Drugs 0.000 description 3
- 229960004222 factor ix Drugs 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 229960002989 glutamic acid Drugs 0.000 description 3
- 150000004676 glycans Chemical class 0.000 description 3
- 201000011066 hemangioma Diseases 0.000 description 3
- 230000001976 improved effect Effects 0.000 description 3
- 230000002779 inactivation Effects 0.000 description 3
- 230000008595 infiltration Effects 0.000 description 3
- 238000001764 infiltration Methods 0.000 description 3
- 230000028709 inflammatory response Effects 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 230000002452 interceptive effect Effects 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 230000003902 lesion Effects 0.000 description 3
- 208000032839 leukemia Diseases 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 238000012423 maintenance Methods 0.000 description 3
- 210000004962 mammalian cell Anatomy 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 102000006240 membrane receptors Human genes 0.000 description 3
- 206010061289 metastatic neoplasm Diseases 0.000 description 3
- 229960000485 methotrexate Drugs 0.000 description 3
- 244000005700 microbiome Species 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 238000012544 monitoring process Methods 0.000 description 3
- OHDXDNUPVVYWOV-UHFFFAOYSA-N n-methyl-1-(2-naphthalen-1-ylsulfanylphenyl)methanamine Chemical compound CNCC1=CC=CC=C1SC1=CC=CC2=CC=CC=C12 OHDXDNUPVVYWOV-UHFFFAOYSA-N 0.000 description 3
- 239000002105 nanoparticle Substances 0.000 description 3
- 150000007523 nucleic acids Chemical class 0.000 description 3
- 235000019834 papain Nutrition 0.000 description 3
- 229940055729 papain Drugs 0.000 description 3
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 229920001282 polysaccharide Polymers 0.000 description 3
- 239000005017 polysaccharide Substances 0.000 description 3
- 229920002451 polyvinyl alcohol Polymers 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 230000000861 pro-apoptotic effect Effects 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 230000000750 progressive effect Effects 0.000 description 3
- 230000035755 proliferation Effects 0.000 description 3
- 239000000007 protein synthesis inhibitor Substances 0.000 description 3
- 239000002510 pyrogen Substances 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 230000006641 stabilisation Effects 0.000 description 3
- 238000011105 stabilization Methods 0.000 description 3
- 239000013589 supplement Substances 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 229940126585 therapeutic drug Drugs 0.000 description 3
- DSNBHJFQCNUKMA-SCKDECHMSA-N thromboxane A2 Chemical compound OC(=O)CCC\C=C/C[C@@H]1[C@@H](/C=C/[C@@H](O)CCCCC)O[C@@H]2O[C@H]1C2 DSNBHJFQCNUKMA-SCKDECHMSA-N 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 241000701161 unidentified adenovirus Species 0.000 description 3
- 229960005356 urokinase Drugs 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- 229960003048 vinblastine Drugs 0.000 description 3
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 3
- 230000003442 weekly effect Effects 0.000 description 3
- NGGMYCMLYOUNGM-UHFFFAOYSA-N (-)-fumagillin Natural products O1C(CC=C(C)C)C1(C)C1C(OC)C(OC(=O)C=CC=CC=CC=CC(O)=O)CCC21CO2 NGGMYCMLYOUNGM-UHFFFAOYSA-N 0.000 description 2
- JWDFQMWEFLOOED-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-(pyridin-2-yldisulfanyl)propanoate Chemical compound O=C1CCC(=O)N1OC(=O)CCSSC1=CC=CC=N1 JWDFQMWEFLOOED-UHFFFAOYSA-N 0.000 description 2
- VKUYLANQOAKALN-UHFFFAOYSA-N 2-[benzyl-(4-methoxyphenyl)sulfonylamino]-n-hydroxy-4-methylpentanamide Chemical compound C1=CC(OC)=CC=C1S(=O)(=O)N(C(CC(C)C)C(=O)NO)CC1=CC=CC=C1 VKUYLANQOAKALN-UHFFFAOYSA-N 0.000 description 2
- 101710151806 72 kDa type IV collagenase Proteins 0.000 description 2
- 108091006112 ATPases Proteins 0.000 description 2
- 108010066676 Abrin Proteins 0.000 description 2
- 102000057290 Adenosine Triphosphatases Human genes 0.000 description 2
- 206010067484 Adverse reaction Diseases 0.000 description 2
- 101800002638 Alpha-amanitin Proteins 0.000 description 2
- 102100033402 Angiopoietin-4 Human genes 0.000 description 2
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 2
- 108700020463 BRCA1 Proteins 0.000 description 2
- 102000036365 BRCA1 Human genes 0.000 description 2
- 101150072950 BRCA1 gene Proteins 0.000 description 2
- 108010081589 Becaplermin Proteins 0.000 description 2
- 102100026189 Beta-galactosidase Human genes 0.000 description 2
- 102100021943 C-C motif chemokine 2 Human genes 0.000 description 2
- 101710155857 C-C motif chemokine 2 Proteins 0.000 description 2
- 108010074051 C-Reactive Protein Proteins 0.000 description 2
- 102100032752 C-reactive protein Human genes 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 108010006303 Carboxypeptidases Proteins 0.000 description 2
- 102000005367 Carboxypeptidases Human genes 0.000 description 2
- 102000003952 Caspase 3 Human genes 0.000 description 2
- 108090000397 Caspase 3 Proteins 0.000 description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 description 2
- 206010057248 Cell death Diseases 0.000 description 2
- 231100000023 Cell-mediated cytotoxicity Toxicity 0.000 description 2
- 206010057250 Cell-mediated cytotoxicity Diseases 0.000 description 2
- 241000282693 Cercopithecidae Species 0.000 description 2
- HVXBOLULGPECHP-WAYWQWQTSA-N Combretastatin A4 Chemical compound C1=C(O)C(OC)=CC=C1\C=C/C1=CC(OC)=C(OC)C(OC)=C1 HVXBOLULGPECHP-WAYWQWQTSA-N 0.000 description 2
- 102000010831 Cytoskeletal Proteins Human genes 0.000 description 2
- 108010037414 Cytoskeletal Proteins Proteins 0.000 description 2
- 241000271032 Daboia russelii Species 0.000 description 2
- 108010092160 Dactinomycin Proteins 0.000 description 2
- 101100481408 Danio rerio tie2 gene Proteins 0.000 description 2
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 2
- 241000702421 Dependoparvovirus Species 0.000 description 2
- 238000009007 Diagnostic Kit Methods 0.000 description 2
- 102100023471 E-selectin Human genes 0.000 description 2
- 241000588724 Escherichia coli Species 0.000 description 2
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 2
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 2
- 108010074864 Factor XI Proteins 0.000 description 2
- 201000006107 Familial adenomatous polyposis Diseases 0.000 description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- 108010000487 High-Molecular-Weight Kininogen Proteins 0.000 description 2
- 208000017604 Hodgkin disease Diseases 0.000 description 2
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 2
- 101500025915 Homo sapiens Angiostatin Proteins 0.000 description 2
- 101000935040 Homo sapiens Integrin beta-2 Proteins 0.000 description 2
- 101000605403 Homo sapiens Plasminogen Proteins 0.000 description 2
- 101000753253 Homo sapiens Tyrosine-protein kinase receptor Tie-1 Proteins 0.000 description 2
- 108090000144 Human Proteins Proteins 0.000 description 2
- 102000003839 Human Proteins Human genes 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 2
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 102100025390 Integrin beta-2 Human genes 0.000 description 2
- 102100037850 Interferon gamma Human genes 0.000 description 2
- 108010074328 Interferon-gamma Proteins 0.000 description 2
- 108090000978 Interleukin-4 Proteins 0.000 description 2
- 108010002616 Interleukin-5 Proteins 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- 102100035792 Kininogen-1 Human genes 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 2
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 2
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 2
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 2
- 102100025136 Macrosialin Human genes 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 102100030417 Matrilysin Human genes 0.000 description 2
- 108090000855 Matrilysin Proteins 0.000 description 2
- 102000000380 Matrix Metalloproteinase 1 Human genes 0.000 description 2
- 108010016113 Matrix Metalloproteinase 1 Proteins 0.000 description 2
- 108010015302 Matrix metalloproteinase-9 Proteins 0.000 description 2
- 206010073150 Multiple endocrine neoplasia Type 1 Diseases 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- 206010029260 Neuroblastoma Diseases 0.000 description 2
- 102000043276 Oncogene Human genes 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 2
- MHRARRIFWSILEQ-UHFFFAOYSA-N Pederin Natural products COCC(CC1OC2C(NC(=O)C(O)C3(CC(=O)C(C)C(C)O3)OC)OCOC2C(OC)C1(C)C)OC MHRARRIFWSILEQ-UHFFFAOYSA-N 0.000 description 2
- 108010081690 Pertussis Toxin Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 108090000216 Phospholipid Transfer Proteins Proteins 0.000 description 2
- 102000003867 Phospholipid Transfer Proteins Human genes 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 229920000954 Polyglycolide Polymers 0.000 description 2
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 2
- 102100021923 Prolow-density lipoprotein receptor-related protein 1 Human genes 0.000 description 2
- 101800004937 Protein C Proteins 0.000 description 2
- 102000017975 Protein C Human genes 0.000 description 2
- 229940096437 Protein S Drugs 0.000 description 2
- 108010066124 Protein S Proteins 0.000 description 2
- 102000029301 Protein S Human genes 0.000 description 2
- 241000589516 Pseudomonas Species 0.000 description 2
- 108010083644 Ribonucleases Proteins 0.000 description 2
- 102000006382 Ribonucleases Human genes 0.000 description 2
- RXGJTYFDKOHJHK-UHFFFAOYSA-N S-deoxo-amaninamide Natural products CCC(C)C1NC(=O)CNC(=O)C2Cc3c(SCC(NC(=O)CNC1=O)C(=O)NC(CC(=O)N)C(=O)N4CC(O)CC4C(=O)NC(C(C)C(O)CO)C(=O)N2)[nH]c5ccccc35 RXGJTYFDKOHJHK-UHFFFAOYSA-N 0.000 description 2
- 101800001700 Saposin-D Proteins 0.000 description 2
- 229920002684 Sepharose Polymers 0.000 description 2
- 101710084578 Short neurotoxin 1 Proteins 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 101710145796 Staphylokinase Proteins 0.000 description 2
- 101710108790 Stromelysin-1 Proteins 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- 101710182532 Toxin a Proteins 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 102100022007 Tyrosine-protein kinase receptor Tie-1 Human genes 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 2
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 2
- 229940122803 Vinca alkaloid Drugs 0.000 description 2
- 208000008383 Wilms tumor Diseases 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 230000009102 absorption Effects 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 2
- 239000000362 adenosine triphosphatase inhibitor Substances 0.000 description 2
- 229940009456 adriamycin Drugs 0.000 description 2
- 230000006838 adverse reaction Effects 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 230000002152 alkylating effect Effects 0.000 description 2
- 239000004007 alpha amanitin Substances 0.000 description 2
- CIORWBWIBBPXCG-SXZCQOKQSA-N alpha-amanitin Chemical compound O=C1N[C@@H](CC(N)=O)C(=O)N2C[C@H](O)C[C@H]2C(=O)N[C@@H]([C@@H](C)[C@@H](O)CO)C(=O)N[C@@H](C2)C(=O)NCC(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@H]1C[S@@](=O)C1=C2C2=CC=C(O)C=C2N1 CIORWBWIBBPXCG-SXZCQOKQSA-N 0.000 description 2
- CIORWBWIBBPXCG-UHFFFAOYSA-N alpha-amanitin Natural products O=C1NC(CC(N)=O)C(=O)N2CC(O)CC2C(=O)NC(C(C)C(O)CO)C(=O)NC(C2)C(=O)NCC(=O)NC(C(C)CC)C(=O)NCC(=O)NC1CS(=O)C1=C2C2=CC=C(O)C=C2N1 CIORWBWIBBPXCG-UHFFFAOYSA-N 0.000 description 2
- 230000000964 angiostatic effect Effects 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000000844 anti-bacterial effect Effects 0.000 description 2
- 230000001740 anti-invasion Effects 0.000 description 2
- 230000000340 anti-metabolite Effects 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 229940121375 antifungal agent Drugs 0.000 description 2
- 239000003429 antifungal agent Substances 0.000 description 2
- 229940100197 antimetabolite Drugs 0.000 description 2
- 239000002256 antimetabolite Substances 0.000 description 2
- 238000003782 apoptosis assay Methods 0.000 description 2
- 230000005775 apoptotic pathway Effects 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- 235000009582 asparagine Nutrition 0.000 description 2
- 229960001230 asparagine Drugs 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- 229930192649 bafilomycin Natural products 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 108010005774 beta-Galactosidase Proteins 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 239000003181 biological factor Substances 0.000 description 2
- 230000008827 biological function Effects 0.000 description 2
- 239000012472 biological sample Substances 0.000 description 2
- 230000000903 blocking effect Effects 0.000 description 2
- 210000002798 bone marrow cell Anatomy 0.000 description 2
- DQXBYHZEEUGOBF-UHFFFAOYSA-N but-3-enoic acid;ethene Chemical compound C=C.OC(=O)CC=C DQXBYHZEEUGOBF-UHFFFAOYSA-N 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- 230000003185 calcium uptake Effects 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 235000014633 carbohydrates Nutrition 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 230000022131 cell cycle Effects 0.000 description 2
- 230000005890 cell-mediated cytotoxicity Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 208000029664 classic familial adenomatous polyposis Diseases 0.000 description 2
- 229960001338 colchicine Drugs 0.000 description 2
- 238000011284 combination treatment Methods 0.000 description 2
- 229960005537 combretastatin A-4 Drugs 0.000 description 2
- 229930192183 combretastatin A1 Natural products 0.000 description 2
- YUSYSJSHVJULID-UHFFFAOYSA-N combretastatin A1 Z Natural products OC1=C(O)C(OC)=CC=C1C=CC1=CC(OC)=C(OC)C(OC)=C1 YUSYSJSHVJULID-UHFFFAOYSA-N 0.000 description 2
- HVXBOLULGPECHP-UHFFFAOYSA-N combretastatin A4 Natural products C1=C(O)C(OC)=CC=C1C=CC1=CC(OC)=C(OC)C(OC)=C1 HVXBOLULGPECHP-UHFFFAOYSA-N 0.000 description 2
- YUSYSJSHVJULID-WAYWQWQTSA-N combretastatin a-1 Chemical compound OC1=C(O)C(OC)=CC=C1\C=C/C1=CC(OC)=C(OC)C(OC)=C1 YUSYSJSHVJULID-WAYWQWQTSA-N 0.000 description 2
- 230000024203 complement activation Effects 0.000 description 2
- 230000004154 complement system Effects 0.000 description 2
- 239000012141 concentrate Substances 0.000 description 2
- 238000010276 construction Methods 0.000 description 2
- 239000000356 contaminant Substances 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 230000003436 cytoskeletal effect Effects 0.000 description 2
- 210000004292 cytoskeleton Anatomy 0.000 description 2
- 230000001086 cytosolic effect Effects 0.000 description 2
- 230000001085 cytostatic effect Effects 0.000 description 2
- 239000002619 cytotoxin Substances 0.000 description 2
- 229960000640 dactinomycin Drugs 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- 230000008021 deposition Effects 0.000 description 2
- 230000001066 destructive effect Effects 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- 238000006471 dimerization reaction Methods 0.000 description 2
- 206010013023 diphtheria Diseases 0.000 description 2
- ZGSPNIOCEDOHGS-UHFFFAOYSA-L disodium [3-[2,3-di(octadeca-9,12-dienoyloxy)propoxy-oxidophosphoryl]oxy-2-hydroxypropyl] 2,3-di(octadeca-9,12-dienoyloxy)propyl phosphate Chemical compound [Na+].[Na+].CCCCCC=CCC=CCCCCCCCC(=O)OCC(OC(=O)CCCCCCCC=CCC=CCCCCC)COP([O-])(=O)OCC(O)COP([O-])(=O)OCC(OC(=O)CCCCCCCC=CCC=CCCCCC)COC(=O)CCCCCCCC=CCC=CCCCCC ZGSPNIOCEDOHGS-UHFFFAOYSA-L 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 125000002228 disulfide group Chemical group 0.000 description 2
- 150000002019 disulfides Chemical class 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 230000007613 environmental effect Effects 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 150000002159 estradiols Chemical class 0.000 description 2
- 210000002744 extracellular matrix Anatomy 0.000 description 2
- 239000003527 fibrinolytic agent Substances 0.000 description 2
- 210000002950 fibroblast Anatomy 0.000 description 2
- 230000004907 flux Effects 0.000 description 2
- 229960000936 fumagillin Drugs 0.000 description 2
- NGGMYCMLYOUNGM-CSDLUJIJSA-N fumagillin Chemical compound C([C@H]([C@H]([C@@H]1[C@]2(C)[C@H](O2)CC=C(C)C)OC)OC(=O)\C=C\C=C\C=C\C=C\C(O)=O)C[C@@]21CO2 NGGMYCMLYOUNGM-CSDLUJIJSA-N 0.000 description 2
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 2
- 229960005277 gemcitabine Drugs 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 239000004220 glutamic acid Substances 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- 229960003180 glutathione Drugs 0.000 description 2
- 230000013595 glycosylation Effects 0.000 description 2
- 238000006206 glycosylation reaction Methods 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N hydrazine group Chemical group NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 229920001600 hydrophobic polymer Polymers 0.000 description 2
- 239000012642 immune effector Substances 0.000 description 2
- 230000036737 immune function Effects 0.000 description 2
- 230000005847 immunogenicity Effects 0.000 description 2
- 229940121354 immunomodulator Drugs 0.000 description 2
- 208000000509 infertility Diseases 0.000 description 2
- 230000036512 infertility Effects 0.000 description 2
- 208000021267 infertility disease Diseases 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 239000007972 injectable composition Substances 0.000 description 2
- 229940102223 injectable solution Drugs 0.000 description 2
- 229940047124 interferons Drugs 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 229960000310 isoleucine Drugs 0.000 description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 2
- 238000005304 joining Methods 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- RGLRXNKKBLIBQS-XNHQSDQCSA-N leuprolide acetate Chemical compound CC(O)=O.CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 RGLRXNKKBLIBQS-XNHQSDQCSA-N 0.000 description 2
- 230000003859 lipid peroxidation Effects 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 208000002780 macular degeneration Diseases 0.000 description 2
- 230000005291 magnetic effect Effects 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- 230000009401 metastasis Effects 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 239000000693 micelle Substances 0.000 description 2
- 238000001823 molecular biology technique Methods 0.000 description 2
- 210000001616 monocyte Anatomy 0.000 description 2
- 230000014508 negative regulation of coagulation Effects 0.000 description 2
- 201000008026 nephroblastoma Diseases 0.000 description 2
- 210000000440 neutrophil Anatomy 0.000 description 2
- 231100000956 nontoxicity Toxicity 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 230000008520 organization Effects 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 201000002528 pancreatic cancer Diseases 0.000 description 2
- 208000008443 pancreatic carcinoma Diseases 0.000 description 2
- 230000007170 pathology Effects 0.000 description 2
- ZNEZZONMADKYTB-VRCUBXEUSA-N pederin Chemical compound C1[C@@H](O)C(C)(C)[C@@H](C[C@@H](COC)OC)O[C@@H]1[C@H](OC)NC(=O)[C@@H](O)[C@]1(OC)O[C@H](C)[C@H](C)C(=C)C1 ZNEZZONMADKYTB-VRCUBXEUSA-N 0.000 description 2
- 239000000863 peptide conjugate Substances 0.000 description 2
- CTRLRINCMYICJO-UHFFFAOYSA-N phenyl azide Chemical compound [N-]=[N+]=NC1=CC=CC=C1 CTRLRINCMYICJO-UHFFFAOYSA-N 0.000 description 2
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 230000010118 platelet activation Effects 0.000 description 2
- 239000004633 polyglycolic acid Substances 0.000 description 2
- 230000009290 primary effect Effects 0.000 description 2
- 239000003805 procoagulant Substances 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 230000005522 programmed cell death Effects 0.000 description 2
- 235000019833 protease Nutrition 0.000 description 2
- 238000000159 protein binding assay Methods 0.000 description 2
- 229960000856 protein c Drugs 0.000 description 2
- 230000006337 proteolytic cleavage Effects 0.000 description 2
- 230000002685 pulmonary effect Effects 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 230000006798 recombination Effects 0.000 description 2
- 230000000717 retained effect Effects 0.000 description 2
- IGLNJRXAVVLDKE-UHFFFAOYSA-N rubidium atom Chemical compound [Rb] IGLNJRXAVVLDKE-UHFFFAOYSA-N 0.000 description 2
- 108010073863 saruplase Proteins 0.000 description 2
- 108010078070 scavenger receptors Proteins 0.000 description 2
- 102000014452 scavenger receptors Human genes 0.000 description 2
- 150000003335 secondary amines Chemical group 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 230000001568 sexual effect Effects 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 238000002741 site-directed mutagenesis Methods 0.000 description 2
- 239000003998 snake venom Substances 0.000 description 2
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 2
- 230000004936 stimulating effect Effects 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 230000035882 stress Effects 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 230000001502 supplementing effect Effects 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- 230000002885 thrombogenetic effect Effects 0.000 description 2
- 230000007888 toxin activity Effects 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 230000014599 transmission of virus Effects 0.000 description 2
- 238000011269 treatment regimen Methods 0.000 description 2
- PXSOHRWMIRDKMP-UHFFFAOYSA-N triaziquone Chemical compound O=C1C(N2CC2)=C(N2CC2)C(=O)C=C1N1CC1 PXSOHRWMIRDKMP-UHFFFAOYSA-N 0.000 description 2
- 230000001960 triggered effect Effects 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 230000005740 tumor formation Effects 0.000 description 2
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 2
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 2
- 241001430294 unidentified retrovirus Species 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- 239000004474 valine Substances 0.000 description 2
- 229910052720 vanadium Inorganic materials 0.000 description 2
- 201000011531 vascular cancer Diseases 0.000 description 2
- 230000004218 vascular function Effects 0.000 description 2
- 206010055031 vascular neoplasm Diseases 0.000 description 2
- 239000005526 vasoconstrictor agent Substances 0.000 description 2
- 230000007502 viral entry Effects 0.000 description 2
- 239000013603 viral vector Substances 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 229960005502 α-amanitin Drugs 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- NNJPGOLRFBJNIW-HNNXBMFYSA-N (-)-demecolcine Chemical compound C1=C(OC)C(=O)C=C2[C@@H](NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-HNNXBMFYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- XMQUEQJCYRFIQS-YFKPBYRVSA-N (2s)-2-amino-5-ethoxy-5-oxopentanoic acid Chemical compound CCOC(=O)CC[C@H](N)C(O)=O XMQUEQJCYRFIQS-YFKPBYRVSA-N 0.000 description 1
- YNZXLMPHTZVKJN-VBKCWIKWSA-N (3z,5e,7r,8r,9s,10s,11r,13e,15e,17s,18r)-18-[(2s,3r,4s)-4-[(2r,4r,5s,6r)-2,4-dihydroxy-5-methyl-6-[(e)-prop-1-enyl]oxan-2-yl]-3-hydroxypentan-2-yl]-9-ethyl-8,10-dihydroxy-3,17-dimethoxy-5,7,11,13-tetramethyl-1-oxacyclooctadeca-3,5,13,15-tetraen-2-one Chemical compound O1C(=O)\C(OC)=C\C(\C)=C\[C@@H](C)[C@@H](O)[C@@H](CC)[C@@H](O)[C@H](C)C\C(C)=C\C=C\[C@H](OC)[C@H]1[C@@H](C)[C@@H](O)[C@H](C)[C@]1(O)O[C@H](\C=C\C)[C@@H](C)[C@H](O)C1 YNZXLMPHTZVKJN-VBKCWIKWSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- MQLACMBJVPINKE-UHFFFAOYSA-N 10-[(3-hydroxy-4-methoxyphenyl)methylidene]anthracen-9-one Chemical compound C1=C(O)C(OC)=CC=C1C=C1C2=CC=CC=C2C(=O)C2=CC=CC=C21 MQLACMBJVPINKE-UHFFFAOYSA-N 0.000 description 1
- PNDPGZBMCMUPRI-HVTJNCQCSA-N 10043-66-0 Chemical compound [131I][131I] PNDPGZBMCMUPRI-HVTJNCQCSA-N 0.000 description 1
- FUFLCEKSBBHCMO-UHFFFAOYSA-N 11-dehydrocorticosterone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 FUFLCEKSBBHCMO-UHFFFAOYSA-N 0.000 description 1
- WUAPFZMCVAUBPE-NJFSPNSNSA-N 188Re Chemical compound [188Re] WUAPFZMCVAUBPE-NJFSPNSNSA-N 0.000 description 1
- WAUGGYPDCQZJKK-UHFFFAOYSA-N 1h-pyrrol-3-amine Chemical compound NC=1C=CNC=1 WAUGGYPDCQZJKK-UHFFFAOYSA-N 0.000 description 1
- 150000003923 2,5-pyrrolediones Chemical class 0.000 description 1
- FBUTXZSKZCQABC-UHFFFAOYSA-N 2-amino-1-methyl-7h-purine-6-thione Chemical compound S=C1N(C)C(N)=NC2=C1NC=N2 FBUTXZSKZCQABC-UHFFFAOYSA-N 0.000 description 1
- IZMMAJINAROYPF-UHFFFAOYSA-N 2H-thiophen-5-imine Chemical class N=C1SCC=C1 IZMMAJINAROYPF-UHFFFAOYSA-N 0.000 description 1
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 1
- KINMYBBFQRSVLL-UHFFFAOYSA-N 4-(4-phenoxybutoxy)furo[3,2-g]chromen-7-one Chemical compound C1=2C=COC=2C=C2OC(=O)C=CC2=C1OCCCCOC1=CC=CC=C1 KINMYBBFQRSVLL-UHFFFAOYSA-N 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- HRUYBRGMRSNLNW-UHFFFAOYSA-N 6-methoxy-2-[(4-methylphenyl)methylsulfanyl]-1h-benzimidazole Chemical compound N1C2=CC(OC)=CC=C2N=C1SCC1=CC=C(C)C=C1 HRUYBRGMRSNLNW-UHFFFAOYSA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 108010024878 Adenovirus E1A Proteins Proteins 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 208000005989 Arenaviridae Infections Diseases 0.000 description 1
- 241000712891 Arenavirus Species 0.000 description 1
- 102000009133 Arylsulfatases Human genes 0.000 description 1
- 101000669426 Aspergillus restrictus Ribonuclease mitogillin Proteins 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 208000037260 Atherosclerotic Plaque Diseases 0.000 description 1
- 206010003694 Atrophy Diseases 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 102000052609 BRCA2 Human genes 0.000 description 1
- 108700020462 BRCA2 Proteins 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 102100036597 Basement membrane-specific heparan sulfate proteoglycan core protein Human genes 0.000 description 1
- 102100023962 Bifunctional arginine demethylase and lysyl-hydroxylase JMJD6 Human genes 0.000 description 1
- 101800000285 Big gastrin Proteins 0.000 description 1
- 241000588832 Bordetella pertussis Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 101150008921 Brca2 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 229940127291 Calcium channel antagonist Drugs 0.000 description 1
- 102100029968 Calreticulin Human genes 0.000 description 1
- 108090000549 Calreticulin Proteins 0.000 description 1
- 101800000626 Canstatin Proteins 0.000 description 1
- 102400000730 Canstatin Human genes 0.000 description 1
- 102000053642 Catalytic RNA Human genes 0.000 description 1
- 108090000994 Catalytic RNA Proteins 0.000 description 1
- 108010084457 Cathepsins Proteins 0.000 description 1
- 102000005600 Cathepsins Human genes 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 102100024330 Collectin-12 Human genes 0.000 description 1
- ZSNYQENLWQYSRK-UHFFFAOYSA-N Combretastatin B1 Natural products OC1=C(O)C(OC)=CC=C1CCC1=CC(OC)=C(OC)C(OC)=C1 ZSNYQENLWQYSRK-UHFFFAOYSA-N 0.000 description 1
- SFWXNQDSPCKFAW-UHFFFAOYSA-N Combretastatin B3 Natural products COC1=C(OC)C(OC)=CC(CCC=2C=C(O)C(O)=CC=2)=C1 SFWXNQDSPCKFAW-UHFFFAOYSA-N 0.000 description 1
- PBRZRNRAYCSTKB-UHFFFAOYSA-N Combretastatin B4 Natural products COC1=CC(OC)=CC(CCC=2C=C(O)C(O)=CC=2)=C1 PBRZRNRAYCSTKB-UHFFFAOYSA-N 0.000 description 1
- UNKWNXAIWJJVFF-UHFFFAOYSA-N Combretastatin D-1 Natural products Oc1ccc2CCC(=O)OCC3OC3c4cccc(Oc1c2)c4 UNKWNXAIWJJVFF-UHFFFAOYSA-N 0.000 description 1
- QXDPFEIJLOEFJN-UHFFFAOYSA-N Combretastatin D2 Natural products Oc1ccc2CCC(=O)OCC=C/c3cccc(Oc1c2)c3 QXDPFEIJLOEFJN-UHFFFAOYSA-N 0.000 description 1
- 102100025877 Complement component C1q receptor Human genes 0.000 description 1
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 1
- MFYSYFVPBJMHGN-ZPOLXVRWSA-N Cortisone Chemical compound O=C1CC[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 MFYSYFVPBJMHGN-ZPOLXVRWSA-N 0.000 description 1
- MFYSYFVPBJMHGN-UHFFFAOYSA-N Cortisone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)(O)C(=O)CO)C4C3CCC2=C1 MFYSYFVPBJMHGN-UHFFFAOYSA-N 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 240000005109 Cryptomeria japonica Species 0.000 description 1
- JPVYNHNXODAKFH-UHFFFAOYSA-N Cu2+ Chemical compound [Cu+2] JPVYNHNXODAKFH-UHFFFAOYSA-N 0.000 description 1
- 108010058546 Cyclin D1 Proteins 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- 108010080611 Cytosine Deaminase Proteins 0.000 description 1
- 102000000311 Cytosine Deaminase Human genes 0.000 description 1
- 101710112752 Cytotoxin Proteins 0.000 description 1
- 125000000030 D-alanine group Chemical group [H]N([H])[C@](C([H])([H])[H])(C(=O)[*])[H] 0.000 description 1
- LEVWYRKDKASIDU-QWWZWVQMSA-N D-cystine Chemical compound OC(=O)[C@H](N)CSSC[C@@H](N)C(O)=O LEVWYRKDKASIDU-QWWZWVQMSA-N 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 101100126652 Danio rerio jmjd6 gene Proteins 0.000 description 1
- NNJPGOLRFBJNIW-UHFFFAOYSA-N Demecolcine Natural products C1=C(OC)C(=O)C=C2C(NC)CCC3=CC(OC)=C(OC)C(OC)=C3C2=C1 NNJPGOLRFBJNIW-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010012689 Diabetic retinopathy Diseases 0.000 description 1
- 108090000204 Dipeptidase 1 Proteins 0.000 description 1
- 108010053187 Diphtheria Toxin Proteins 0.000 description 1
- 102000016607 Diphtheria Toxin Human genes 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 229910052692 Dysprosium Inorganic materials 0.000 description 1
- 108010024212 E-Selectin Proteins 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 206010014513 Embolism arterial Diseases 0.000 description 1
- 206010014522 Embolism venous Diseases 0.000 description 1
- 102100037241 Endoglin Human genes 0.000 description 1
- 108010036395 Endoglin Proteins 0.000 description 1
- 102100038083 Endosialin Human genes 0.000 description 1
- 101710144543 Endosialin Proteins 0.000 description 1
- 229910052691 Erbium Inorganic materials 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- 102100027279 FAS-associated factor 1 Human genes 0.000 description 1
- 101150021185 FGF gene Proteins 0.000 description 1
- 108091008794 FGF receptors Proteins 0.000 description 1
- 108010029144 Factor IIa Proteins 0.000 description 1
- 108010054218 Factor VIII Proteins 0.000 description 1
- 102000001690 Factor VIII Human genes 0.000 description 1
- 108010061932 Factor VIIIa Proteins 0.000 description 1
- 108010080865 Factor XII Proteins 0.000 description 1
- 102000000429 Factor XII Human genes 0.000 description 1
- 108010071289 Factor XIII Proteins 0.000 description 1
- 108010000196 Factor XIIIa Proteins 0.000 description 1
- 108010071241 Factor XIIa Proteins 0.000 description 1
- 108010080805 Factor XIa Proteins 0.000 description 1
- 102000009109 Fc receptors Human genes 0.000 description 1
- 108010087819 Fc receptors Proteins 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- 102000044168 Fibroblast Growth Factor Receptor Human genes 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 102100024165 G1/S-specific cyclin-D1 Human genes 0.000 description 1
- 229910052688 Gadolinium Inorganic materials 0.000 description 1
- GYHNNYVSQQEPJS-OIOBTWANSA-N Gallium-67 Chemical compound [67Ga] GYHNNYVSQQEPJS-OIOBTWANSA-N 0.000 description 1
- GYHNNYVSQQEPJS-YPZZEJLDSA-N Gallium-68 Chemical compound [68Ga] GYHNNYVSQQEPJS-YPZZEJLDSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 208000010412 Glaucoma Diseases 0.000 description 1
- 102000006395 Globulins Human genes 0.000 description 1
- 108010044091 Globulins Proteins 0.000 description 1
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 1
- 102000005720 Glutathione transferase Human genes 0.000 description 1
- 108010070675 Glutathione transferase Proteins 0.000 description 1
- 206010018498 Goitre Diseases 0.000 description 1
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 1
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 1
- 208000012766 Growth delay Diseases 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 208000037357 HIV infectious disease Diseases 0.000 description 1
- 208000008899 Habitual abortion Diseases 0.000 description 1
- 102100021866 Hepatocyte growth factor Human genes 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 229910052689 Holmium Inorganic materials 0.000 description 1
- 101000793425 Homo sapiens Beta-2-glycoprotein 1 Proteins 0.000 description 1
- 101000869050 Homo sapiens Caveolae-associated protein 2 Proteins 0.000 description 1
- 101000909528 Homo sapiens Collectin-12 Proteins 0.000 description 1
- 101000933665 Homo sapiens Complement component C1q receptor Proteins 0.000 description 1
- 101000622123 Homo sapiens E-selectin Proteins 0.000 description 1
- 101000914654 Homo sapiens FAS-associated factor 1 Proteins 0.000 description 1
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 1
- 101000898034 Homo sapiens Hepatocyte growth factor Proteins 0.000 description 1
- 101001056180 Homo sapiens Induced myeloid leukemia cell differentiation protein Mcl-1 Proteins 0.000 description 1
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 description 1
- 101001046677 Homo sapiens Integrin alpha-V Proteins 0.000 description 1
- 101001015004 Homo sapiens Integrin beta-3 Proteins 0.000 description 1
- 101001076408 Homo sapiens Interleukin-6 Proteins 0.000 description 1
- 101001013150 Homo sapiens Interstitial collagenase Proteins 0.000 description 1
- 101000934372 Homo sapiens Macrosialin Proteins 0.000 description 1
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 1
- 101000596892 Homo sapiens Neurotrimin Proteins 0.000 description 1
- 101001131840 Homo sapiens Pregnancy zone protein Proteins 0.000 description 1
- 101001043564 Homo sapiens Prolow-density lipoprotein receptor-related protein 1 Proteins 0.000 description 1
- 101000668165 Homo sapiens RNA-binding motif, single-stranded-interacting protein 1 Proteins 0.000 description 1
- 101001104199 Homo sapiens Retinitis pigmentosa 9 protein Proteins 0.000 description 1
- 101000868152 Homo sapiens Son of sevenless homolog 1 Proteins 0.000 description 1
- 101000622304 Homo sapiens Vascular cell adhesion protein 1 Proteins 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 206010020608 Hypercoagulation Diseases 0.000 description 1
- 208000031226 Hyperlipidaemia Diseases 0.000 description 1
- 102000009490 IgG Receptors Human genes 0.000 description 1
- 108010073807 IgG Receptors Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 description 1
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 description 1
- 238000012404 In vitro experiment Methods 0.000 description 1
- 102100026539 Induced myeloid leukemia cell differentiation protein Mcl-1 Human genes 0.000 description 1
- 206010061216 Infarction Diseases 0.000 description 1
- 102100022338 Integrin alpha-M Human genes 0.000 description 1
- 102100022337 Integrin alpha-V Human genes 0.000 description 1
- 102100022297 Integrin alpha-X Human genes 0.000 description 1
- 108010047852 Integrin alphaVbeta3 Proteins 0.000 description 1
- 102100032999 Integrin beta-3 Human genes 0.000 description 1
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 description 1
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 1
- 108090000174 Interleukin-10 Proteins 0.000 description 1
- 102000003814 Interleukin-10 Human genes 0.000 description 1
- 108090000177 Interleukin-11 Proteins 0.000 description 1
- 108090000176 Interleukin-13 Proteins 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- 102000004889 Interleukin-6 Human genes 0.000 description 1
- 108010002586 Interleukin-7 Proteins 0.000 description 1
- 102000004890 Interleukin-8 Human genes 0.000 description 1
- 108090001007 Interleukin-8 Proteins 0.000 description 1
- 108010002335 Interleukin-9 Proteins 0.000 description 1
- ZCYVEMRRCGMTRW-AHCXROLUSA-N Iodine-123 Chemical compound [123I] ZCYVEMRRCGMTRW-AHCXROLUSA-N 0.000 description 1
- 108010092694 L-Selectin Proteins 0.000 description 1
- 102000016551 L-selectin Human genes 0.000 description 1
- 102000007547 Laminin Human genes 0.000 description 1
- 108010085895 Laminin Proteins 0.000 description 1
- 206010023927 Lassa fever Diseases 0.000 description 1
- 102100032352 Leukemia inhibitory factor Human genes 0.000 description 1
- 108090000581 Leukemia inhibitory factor Proteins 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- 108010058188 Low-Molecular-Weight Kininogen Proteins 0.000 description 1
- 102100040705 Low-density lipoprotein receptor-related protein 8 Human genes 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- 101710156482 Macrosialin Proteins 0.000 description 1
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 1
- WAEMQWOKJMHJLA-UHFFFAOYSA-N Manganese(2+) Chemical compound [Mn+2] WAEMQWOKJMHJLA-UHFFFAOYSA-N 0.000 description 1
- 108010016165 Matrix Metalloproteinase 2 Proteins 0.000 description 1
- 108010016160 Matrix Metalloproteinase 3 Proteins 0.000 description 1
- 102000002274 Matrix Metalloproteinases Human genes 0.000 description 1
- 108010000684 Matrix Metalloproteinases Proteins 0.000 description 1
- 102000001776 Matrix metalloproteinase-9 Human genes 0.000 description 1
- 102100030412 Matrix metalloproteinase-9 Human genes 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 1
- 101000793426 Mus musculus Beta-2-glycoprotein 1 Proteins 0.000 description 1
- 101001104198 Mus musculus Retinitis pigmentosa 9 protein homolog Proteins 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- 125000001429 N-terminal alpha-amino-acid group Chemical group 0.000 description 1
- 108091061960 Naked DNA Proteins 0.000 description 1
- 206010029113 Neovascularisation Diseases 0.000 description 1
- 102000005348 Neuraminidase Human genes 0.000 description 1
- 108010006232 Neuraminidase Proteins 0.000 description 1
- 102100035107 Neurotrimin Human genes 0.000 description 1
- VEQPNABPJHWNSG-UHFFFAOYSA-N Nickel(2+) Chemical compound [Ni+2] VEQPNABPJHWNSG-UHFFFAOYSA-N 0.000 description 1
- KYRVNWMVYQXFEU-UHFFFAOYSA-N Nocodazole Chemical compound C1=C2NC(NC(=O)OC)=NC2=CC=C1C(=O)C1=CC=CS1 KYRVNWMVYQXFEU-UHFFFAOYSA-N 0.000 description 1
- 229940122313 Nucleoside reverse transcriptase inhibitor Drugs 0.000 description 1
- COGWIGJGNQCZPK-UHFFFAOYSA-N O1C(=C2)C(O)=CC=C2CCC(=O)OCC2OC2C2=CC=C1C=C2 Chemical compound O1C(=C2)C(O)=CC=C2CCC(=O)OCC2OC2C2=CC=C1C=C2 COGWIGJGNQCZPK-UHFFFAOYSA-N 0.000 description 1
- 108090000630 Oncostatin M Proteins 0.000 description 1
- 102100031942 Oncostatin-M Human genes 0.000 description 1
- 108700026244 Open Reading Frames Proteins 0.000 description 1
- 102100025386 Oxidized low-density lipoprotein receptor 1 Human genes 0.000 description 1
- 101710199789 Oxidized low-density lipoprotein receptor 1 Proteins 0.000 description 1
- 108010035766 P-Selectin Proteins 0.000 description 1
- 102100023472 P-selectin Human genes 0.000 description 1
- 102000016387 Pancreatic elastase Human genes 0.000 description 1
- 108010067372 Pancreatic elastase Proteins 0.000 description 1
- 208000034038 Pathologic Neovascularization Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 108010073038 Penicillin Amidase Proteins 0.000 description 1
- 241001479489 Peponocephala electra Species 0.000 description 1
- 102100033616 Phospholipid-transporting ATPase ABCA1 Human genes 0.000 description 1
- 101710205202 Phospholipid-transporting ATPase ABCA1 Proteins 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 102000001938 Plasminogen Activators Human genes 0.000 description 1
- 108010001014 Plasminogen Activators Proteins 0.000 description 1
- 206010035664 Pneumonia Diseases 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 102000004005 Prostaglandin-endoperoxide synthases Human genes 0.000 description 1
- 108090000459 Prostaglandin-endoperoxide synthases Proteins 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 108010067787 Proteoglycans Proteins 0.000 description 1
- 102000016611 Proteoglycans Human genes 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 230000006819 RNA synthesis Effects 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 241000725643 Respiratory syncytial virus Species 0.000 description 1
- 206010062106 Respiratory tract infection viral Diseases 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 102100040073 Retinitis pigmentosa 9 protein Human genes 0.000 description 1
- 201000000582 Retinoblastoma Diseases 0.000 description 1
- 108700025701 Retinoblastoma Genes Proteins 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 1
- 238000011579 SCID mouse model Methods 0.000 description 1
- 108010005173 SERPIN-B5 Proteins 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 108010084592 Saporins Proteins 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 239000002262 Schiff base Substances 0.000 description 1
- 150000004753 Schiff bases Chemical class 0.000 description 1
- 241000242677 Schistosoma japonicum Species 0.000 description 1
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 1
- 102100030333 Serpin B5 Human genes 0.000 description 1
- 241000607720 Serratia Species 0.000 description 1
- 101001010097 Shigella phage SfV Bactoprenol-linked glucose translocase Proteins 0.000 description 1
- 108010061312 Sphingomyelin Phosphodiesterase Proteins 0.000 description 1
- 102000011971 Sphingomyelin Phosphodiesterase Human genes 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- 108090000787 Subtilisin Proteins 0.000 description 1
- 108090000054 Syndecan-2 Proteins 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 102100028866 TATA element modulatory factor Human genes 0.000 description 1
- 101710136628 TATA element modulatory factor Proteins 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- GKLVYJBZJHMRIY-OUBTZVSYSA-N Technetium-99 Chemical compound [99Tc] GKLVYJBZJHMRIY-OUBTZVSYSA-N 0.000 description 1
- 229910052771 Terbium Inorganic materials 0.000 description 1
- 108090001109 Thermolysin Proteins 0.000 description 1
- 101710097834 Thiol protease Proteins 0.000 description 1
- 108010069102 Thromboxane-A synthase Proteins 0.000 description 1
- 108090000848 Ubiquitin Proteins 0.000 description 1
- 102000044159 Ubiquitin Human genes 0.000 description 1
- GDXLZSYACWZHOC-ZNRYNLAGSA-N Ustiloxin D Chemical compound O[C@H]1[C@H](NC)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(=O)NCC(O)=O)[C@](CC)(C)OC2=CC1=CC=C2O GDXLZSYACWZHOC-ZNRYNLAGSA-N 0.000 description 1
- GDXLZSYACWZHOC-UHFFFAOYSA-N Ustiloxin D Natural products OC1C(NC)C(=O)NC(C(C)C)C(=O)NC(C(=O)NCC(O)=O)C(CC)(C)OC2=CC1=CC=C2O GDXLZSYACWZHOC-UHFFFAOYSA-N 0.000 description 1
- 102000011731 Vacuolar Proton-Translocating ATPases Human genes 0.000 description 1
- 108010037026 Vacuolar Proton-Translocating ATPases Proteins 0.000 description 1
- 108010034265 Vascular Endothelial Growth Factor Receptors Proteins 0.000 description 1
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 1
- 208000024248 Vascular System injury Diseases 0.000 description 1
- 208000012339 Vascular injury Diseases 0.000 description 1
- 206010053648 Vascular occlusion Diseases 0.000 description 1
- 101710181904 Venom factor Proteins 0.000 description 1
- 108700005077 Viral Genes Proteins 0.000 description 1
- 108010031318 Vitronectin Proteins 0.000 description 1
- 102100035140 Vitronectin Human genes 0.000 description 1
- 229910052769 Ytterbium Inorganic materials 0.000 description 1
- VWQVUPCCIRVNHF-OUBTZVSYSA-N Yttrium-90 Chemical compound [90Y] VWQVUPCCIRVNHF-OUBTZVSYSA-N 0.000 description 1
- JCABVIFDXFFRMT-DIPNUNPCSA-N [(2r)-1-[ethoxy(hydroxy)phosphoryl]oxy-3-hexadecanoyloxypropan-2-yl] octadec-9-enoate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OCC)OC(=O)CCCCCCCC=CCCCCCCCC JCABVIFDXFFRMT-DIPNUNPCSA-N 0.000 description 1
- PNDPGZBMCMUPRI-XXSWNUTMSA-N [125I][125I] Chemical compound [125I][125I] PNDPGZBMCMUPRI-XXSWNUTMSA-N 0.000 description 1
- 239000006096 absorbing agent Substances 0.000 description 1
- 230000035508 accumulation Effects 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 208000038016 acute inflammation Diseases 0.000 description 1
- 230000006022 acute inflammation Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 230000009218 additive inhibitory effect Effects 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 206010064930 age-related macular degeneration Diseases 0.000 description 1
- HXHWSAZORRCQMX-UHFFFAOYSA-N albendazole Chemical compound CCCSC1=CC=C2NC(NC(=O)OC)=NC2=C1 HXHWSAZORRCQMX-UHFFFAOYSA-N 0.000 description 1
- 229960002669 albendazole Drugs 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- HAXFWIACAGNFHA-UHFFFAOYSA-N aldrithiol Chemical compound C=1C=CC=NC=1SSC1=CC=CC=N1 HAXFWIACAGNFHA-UHFFFAOYSA-N 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 108010001818 alpha-sarcin Proteins 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229960003896 aminopterin Drugs 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 230000005911 anti-cytotoxic effect Effects 0.000 description 1
- 230000002482 anti-endothelial effect Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 230000001147 anti-toxic effect Effects 0.000 description 1
- 230000006023 anti-tumor response Effects 0.000 description 1
- 230000002155 anti-virotic effect Effects 0.000 description 1
- 238000009175 antibody therapy Methods 0.000 description 1
- 239000002842 anticancer formulation Substances 0.000 description 1
- 229940124572 antihypotensive agent Drugs 0.000 description 1
- 229940034982 antineoplastic agent Drugs 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 229940124522 antiretrovirals Drugs 0.000 description 1
- 239000003903 antiretrovirus agent Substances 0.000 description 1
- 229940121357 antivirals Drugs 0.000 description 1
- 210000000709 aorta Anatomy 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 229910052785 arsenic Inorganic materials 0.000 description 1
- RQNWIZPPADIBDY-UHFFFAOYSA-N arsenic atom Chemical compound [As] RQNWIZPPADIBDY-UHFFFAOYSA-N 0.000 description 1
- FIVPIPIDMRVLAY-UHFFFAOYSA-N aspergillin Natural products C1C2=CC=CC(O)C2N2C1(SS1)C(=O)N(C)C1(CO)C2=O FIVPIPIDMRVLAY-UHFFFAOYSA-N 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 230000037444 atrophy Effects 0.000 description 1
- 230000002358 autolytic effect Effects 0.000 description 1
- MIXLRUYCYZPSOQ-HXPMCKFVSA-N azatoxin Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=C(C4=CC=CC=C4N3)C[C@@H]3N2C(OC3)=O)=C1 MIXLRUYCYZPSOQ-HXPMCKFVSA-N 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 210000002469 basement membrane Anatomy 0.000 description 1
- 244000285940 beete Species 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 102000006635 beta-lactamase Human genes 0.000 description 1
- 230000001588 bifunctional effect Effects 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- 229910052797 bismuth Inorganic materials 0.000 description 1
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 description 1
- 208000002352 blister Diseases 0.000 description 1
- 239000012503 blood component Substances 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 239000012888 bovine serum Substances 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 210000004900 c-terminal fragment Anatomy 0.000 description 1
- AIYUHDOJVYHVIT-UHFFFAOYSA-M caesium chloride Chemical compound [Cl-].[Cs+] AIYUHDOJVYHVIT-UHFFFAOYSA-M 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- 239000003710 calcium ionophore Substances 0.000 description 1
- 108090001015 cancer procoagulant Proteins 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 210000000845 cartilage Anatomy 0.000 description 1
- 238000010523 cascade reaction Methods 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 230000032677 cell aging Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000005779 cell damage Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000007910 cell fusion Effects 0.000 description 1
- 208000037887 cell injury Diseases 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 230000005859 cell recognition Effects 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 230000007969 cellular immunity Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 229940106189 ceramide Drugs 0.000 description 1
- 150000001783 ceramides Chemical class 0.000 description 1
- 235000011222 chang cao shi Nutrition 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 238000012412 chemical coupling Methods 0.000 description 1
- 238000010382 chemical cross-linking Methods 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 230000003399 chemotactic effect Effects 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- BFGKITSFLPAWGI-UHFFFAOYSA-N chromium(3+) Chemical compound [Cr+3] BFGKITSFLPAWGI-UHFFFAOYSA-N 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 230000004087 circulation Effects 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 230000004186 co-expression Effects 0.000 description 1
- XLJKHNWPARRRJB-UHFFFAOYSA-N cobalt(2+) Chemical compound [Co+2] XLJKHNWPARRRJB-UHFFFAOYSA-N 0.000 description 1
- 229940000425 combination drug Drugs 0.000 description 1
- KTDUNELJBIXZMQ-UHFFFAOYSA-N combretastatin A-3 Natural products COc1ccc(C=C/c2cc(O)c(OC)c(O)c2)cc1OC KTDUNELJBIXZMQ-UHFFFAOYSA-N 0.000 description 1
- HRRAOGKGGZFKSW-UHFFFAOYSA-N combretastatin A2 Natural products COc1ccc(C=C/c2cc(O)c3OCOc3c2)cc1OC HRRAOGKGGZFKSW-UHFFFAOYSA-N 0.000 description 1
- VHHWNDDKJSLBGT-UHFFFAOYSA-N combretastatin B-2 Natural products COc1ccc(CCc2cc(O)c(OC)c(O)c2)cc1OC VHHWNDDKJSLBGT-UHFFFAOYSA-N 0.000 description 1
- DTZHRIFDDQELBE-UPHRSURJSA-N combretastatin d-2 Chemical compound O1C(=C2)C(O)=CC=C2CCC(=O)OC\C=C/C2=CC=C1C=C2 DTZHRIFDDQELBE-UPHRSURJSA-N 0.000 description 1
- 230000004540 complement-dependent cytotoxicity Effects 0.000 description 1
- 229930184793 concanamycin Natural products 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- RYGMFSIKBFXOCR-AKLPVKDBSA-N copper-67 Chemical compound [67Cu] RYGMFSIKBFXOCR-AKLPVKDBSA-N 0.000 description 1
- 229960004544 cortisone Drugs 0.000 description 1
- 244000038559 crop plants Species 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 239000002875 cyclin dependent kinase inhibitor Substances 0.000 description 1
- 229940043378 cyclin-dependent kinase inhibitor Drugs 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 229960003067 cystine Drugs 0.000 description 1
- JVHIPYJQMFNCEK-UHFFFAOYSA-N cytochalasin Natural products N1C(=O)C2(C(C=CC(C)CC(C)CC=C3)OC(C)=O)C3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 JVHIPYJQMFNCEK-UHFFFAOYSA-N 0.000 description 1
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 1
- 230000007402 cytotoxic response Effects 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 210000004443 dendritic cell Anatomy 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- UGMCXQCYOVCMTB-UHFFFAOYSA-K dihydroxy(stearato)aluminium Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[Al](O)O UGMCXQCYOVCMTB-UHFFFAOYSA-K 0.000 description 1
- 210000001840 diploid cell Anatomy 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 1
- AMRJKAQTDDKMCE-UHFFFAOYSA-N dolastatin Chemical compound CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)C)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 AMRJKAQTDDKMCE-UHFFFAOYSA-N 0.000 description 1
- 229930188854 dolastatin Natural products 0.000 description 1
- 230000005059 dormancy Effects 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- KBQHZAAAGSGFKK-UHFFFAOYSA-N dysprosium atom Chemical compound [Dy] KBQHZAAAGSGFKK-UHFFFAOYSA-N 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000009762 endothelial cell differentiation Effects 0.000 description 1
- 230000007515 enzymatic degradation Effects 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- UYAHIZSMUZPPFV-UHFFFAOYSA-N erbium Chemical compound [Er] UYAHIZSMUZPPFV-UHFFFAOYSA-N 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 239000005038 ethylene vinyl acetate Substances 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 231100000776 exotoxin Toxicity 0.000 description 1
- 239000002095 exotoxin Substances 0.000 description 1
- 239000013613 expression plasmid Substances 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 208000030533 eye disease Diseases 0.000 description 1
- 229960000301 factor viii Drugs 0.000 description 1
- 229940012444 factor xiii Drugs 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- MSNWSDPPULHLDL-UHFFFAOYSA-K ferric hydroxide Chemical compound [OH-].[OH-].[OH-].[Fe+3] MSNWSDPPULHLDL-UHFFFAOYSA-K 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 230000020764 fibrinolysis Effects 0.000 description 1
- 230000003480 fibrinolytic effect Effects 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- UIWYJDYFSGRHKR-UHFFFAOYSA-N gadolinium atom Chemical compound [Gd] UIWYJDYFSGRHKR-UHFFFAOYSA-N 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- 238000001476 gene delivery Methods 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 229940045109 genistein Drugs 0.000 description 1
- 235000006539 genistein Nutrition 0.000 description 1
- TZBJGXHYKVUXJN-UHFFFAOYSA-N genistein Natural products C1=CC(O)=CC=C1C1=COC2=CC(O)=CC(O)=C2C1=O TZBJGXHYKVUXJN-UHFFFAOYSA-N 0.000 description 1
- ZCOLJUOHXJRHDI-CMWLGVBASA-N genistein 7-O-beta-D-glucoside Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=C2C(=O)C(C=3C=CC(O)=CC=3)=COC2=C1 ZCOLJUOHXJRHDI-CMWLGVBASA-N 0.000 description 1
- FIVPIPIDMRVLAY-RBJBARPLSA-N gliotoxin Chemical compound C1C2=CC=C[C@H](O)[C@H]2N2[C@]1(SS1)C(=O)N(C)[C@@]1(CO)C2=O FIVPIPIDMRVLAY-RBJBARPLSA-N 0.000 description 1
- 125000000404 glutamine group Chemical group N[C@@H](CCC(N)=O)C(=O)* 0.000 description 1
- 230000036252 glycation Effects 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 229930182470 glycoside Natural products 0.000 description 1
- 150000002338 glycosides Chemical class 0.000 description 1
- CBMIPXHVOVTTTL-UHFFFAOYSA-N gold(3+) Chemical compound [Au+3] CBMIPXHVOVTTTL-UHFFFAOYSA-N 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 230000009931 harmful effect Effects 0.000 description 1
- 229910001385 heavy metal Inorganic materials 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 239000002634 heparin fragment Substances 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 238000010562 histological examination Methods 0.000 description 1
- KJZYNXUDTRRSPN-UHFFFAOYSA-N holmium atom Chemical compound [Ho] KJZYNXUDTRRSPN-UHFFFAOYSA-N 0.000 description 1
- 230000003284 homeostatic effect Effects 0.000 description 1
- 229920001519 homopolymer Polymers 0.000 description 1
- 102000045682 human PZP Human genes 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 1
- 150000007857 hydrazones Chemical class 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 230000003027 hypercoagulation Effects 0.000 description 1
- 230000001146 hypoxic effect Effects 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 229940124452 immunizing agent Drugs 0.000 description 1
- 238000003119 immunoblot Methods 0.000 description 1
- 238000010166 immunofluorescence Methods 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 238000011532 immunohistochemical staining Methods 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- APFVFJFRJDLVQX-AHCXROLUSA-N indium-111 Chemical compound [111In] APFVFJFRJDLVQX-AHCXROLUSA-N 0.000 description 1
- APFVFJFRJDLVQX-YPZZEJLDSA-N indium-113 Chemical compound [113In] APFVFJFRJDLVQX-YPZZEJLDSA-N 0.000 description 1
- 230000007574 infarction Effects 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 206010022000 influenza Diseases 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 229940076144 interleukin-10 Drugs 0.000 description 1
- 229940100601 interleukin-6 Drugs 0.000 description 1
- 229940096397 interleukin-8 Drugs 0.000 description 1
- XKTZWUACRZHVAN-VADRZIEHSA-N interleukin-8 Chemical compound C([C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@@H](NC(C)=O)CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CCSC)C(=O)N1[C@H](CCC1)C(=O)N1[C@H](CCC1)C(=O)N[C@@H](C)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC(O)=CC=1)C(=O)N[C@H](CO)C(=O)N1[C@H](CCC1)C(N)=O)C1=CC=CC=C1 XKTZWUACRZHVAN-VADRZIEHSA-N 0.000 description 1
- 238000007919 intrasynovial administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- PGLTVOMIXTUURA-UHFFFAOYSA-N iodoacetamide Chemical compound NC(=O)CI PGLTVOMIXTUURA-UHFFFAOYSA-N 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- 210000004153 islets of langerhan Anatomy 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- CZMAIROVPAYCMU-UHFFFAOYSA-N lanthanum(3+) Chemical compound [La+3] CZMAIROVPAYCMU-UHFFFAOYSA-N 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- 238000004246 ligand exchange chromatography Methods 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 108010031117 low density lipoprotein receptor-related protein 8 Proteins 0.000 description 1
- 101150084157 lrp-1 gene Proteins 0.000 description 1
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 108020004084 membrane receptors Proteins 0.000 description 1
- QSHDDOUJBYECFT-AHCXROLUSA-N mercury-197 Chemical compound [197Hg] QSHDDOUJBYECFT-AHCXROLUSA-N 0.000 description 1
- QSHDDOUJBYECFT-NJFSPNSNSA-N mercury-203 Chemical compound [203Hg] QSHDDOUJBYECFT-NJFSPNSNSA-N 0.000 description 1
- 239000003475 metalloproteinase inhibitor Substances 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000004530 micro-emulsion Substances 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 210000001589 microsome Anatomy 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 210000001700 mitochondrial membrane Anatomy 0.000 description 1
- 230000002297 mitogenic effect Effects 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 230000009456 molecular mechanism Effects 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 210000003098 myoblast Anatomy 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- 229920005615 natural polymer Polymers 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 230000027498 negative regulation of mitosis Effects 0.000 description 1
- QEFYFXOXNSNQGX-UHFFFAOYSA-N neodymium atom Chemical compound [Nd] QEFYFXOXNSNQGX-UHFFFAOYSA-N 0.000 description 1
- 201000003142 neovascular glaucoma Diseases 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 108010087904 neutravidin Proteins 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 229950006344 nocodazole Drugs 0.000 description 1
- 229940042402 non-nucleoside reverse transcriptase inhibitor Drugs 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 239000002726 nonnucleoside reverse transcriptase inhibitor Substances 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 239000003865 nucleic acid synthesis inhibitor Substances 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 230000014207 opsonization Effects 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 150000005623 oxindoles Chemical class 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 238000006213 oxygenation reaction Methods 0.000 description 1
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 231100000255 pathogenic effect Toxicity 0.000 description 1
- 230000007918 pathogenicity Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000002572 peristaltic effect Effects 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- 239000008024 pharmaceutical diluent Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 150000004633 phorbol derivatives Chemical class 0.000 description 1
- 239000002644 phorbol ester Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 108010077613 phosphatidylserine receptor Proteins 0.000 description 1
- 150000004713 phosphodiesters Chemical class 0.000 description 1
- DCWXELXMIBXGTH-QMMMGPOBSA-N phosphonotyrosine Chemical class OC(=O)[C@@H](N)CC1=CC=C(OP(O)(O)=O)C=C1 DCWXELXMIBXGTH-QMMMGPOBSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- DCWXELXMIBXGTH-UHFFFAOYSA-N phosphotyrosine Chemical compound OC(=O)C(N)CC1=CC=C(OP(O)(O)=O)C=C1 DCWXELXMIBXGTH-UHFFFAOYSA-N 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 230000015843 photosynthesis, light reaction Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 230000003169 placental effect Effects 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229940127126 plasminogen activator Drugs 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 231100000572 poisoning Toxicity 0.000 description 1
- 230000000607 poisoning effect Effects 0.000 description 1
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 description 1
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920002338 polyhydroxyethylmethacrylate Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 230000031915 positive regulation of coagulation Effects 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000001023 pro-angiogenic effect Effects 0.000 description 1
- RZIMNEGTIDYAGZ-HNSJZBNRSA-N pro-gastrin Chemical compound N([C@@H](CC(C)C)C(=O)NCC(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N1CCC[C@H]1C(=O)NCC(=O)NCC(=O)NCC(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CC(O)=O)C(=O)NCC(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N1CCC[C@H]1C(=O)NCC(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)NCC(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)C(=O)[C@@H]1CCC(=O)N1 RZIMNEGTIDYAGZ-HNSJZBNRSA-N 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 208000037821 progressive disease Diseases 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 150000003180 prostaglandins Chemical class 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 108010014806 prothrombinase complex Proteins 0.000 description 1
- 125000002294 quinazolinyl group Chemical class N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical class C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 238000000163 radioactive labelling Methods 0.000 description 1
- 230000009103 reabsorption Effects 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 108091006084 receptor activators Proteins 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 230000010837 receptor-mediated endocytosis Effects 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 208000034213 recurrent susceptibility to 1 pregnancy loss Diseases 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 238000007634 remodeling Methods 0.000 description 1
- 201000010174 renal carcinoma Diseases 0.000 description 1
- 230000008521 reorganization Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000020874 response to hypoxia Effects 0.000 description 1
- 229930002330 retinoic acid Natural products 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- WUAPFZMCVAUBPE-IGMARMGPSA-N rhenium-186 Chemical compound [186Re] WUAPFZMCVAUBPE-IGMARMGPSA-N 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 1
- 229960000329 ribavirin Drugs 0.000 description 1
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 1
- 108091092562 ribozyme Proteins 0.000 description 1
- 239000003419 rna directed dna polymerase inhibitor Substances 0.000 description 1
- 229910052701 rubidium Inorganic materials 0.000 description 1
- DOSGOCSVHPUUIA-UHFFFAOYSA-N samarium(3+) Chemical compound [Sm+3] DOSGOCSVHPUUIA-UHFFFAOYSA-N 0.000 description 1
- 150000004671 saturated fatty acids Chemical class 0.000 description 1
- 235000003441 saturated fatty acids Nutrition 0.000 description 1
- 238000007423 screening assay Methods 0.000 description 1
- 239000000932 sedative agent Substances 0.000 description 1
- 230000001624 sedative effect Effects 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 239000011669 selenium Substances 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 102000026456 serine binding proteins Human genes 0.000 description 1
- 108091014772 serine binding proteins Proteins 0.000 description 1
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 1
- 239000004017 serum-free culture medium Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 229940007115 shark cartilage extract Drugs 0.000 description 1
- 108010047846 soblidotin Proteins 0.000 description 1
- DZMVCVHATYROOS-ZBFGKEHZSA-N soblidotin Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)NCCC1=CC=CC=C1 DZMVCVHATYROOS-ZBFGKEHZSA-N 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 210000004872 soft tissue Anatomy 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 230000007480 spreading Effects 0.000 description 1
- 238000003892 spreading Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- PJANXHGTPQOBST-UHFFFAOYSA-N stilbene Chemical class C=1C=CC=CC=1C=CC1=CC=CC=C1 PJANXHGTPQOBST-UHFFFAOYSA-N 0.000 description 1
- 239000004575 stone Substances 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical class O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 108060007951 sulfatase Proteins 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 238000006557 surface reaction Methods 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000012622 synthetic inhibitor Substances 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- 231100000462 teratogen Toxicity 0.000 description 1
- 239000003439 teratogenic agent Substances 0.000 description 1
- GZCRRIHWUXGPOV-UHFFFAOYSA-N terbium atom Chemical compound [Tb] GZCRRIHWUXGPOV-UHFFFAOYSA-N 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 150000007979 thiazole derivatives Chemical class 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229940033663 thimerosal Drugs 0.000 description 1
- 206010043554 thrombocytopenia Diseases 0.000 description 1
- 229960000103 thrombolytic agent Drugs 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000167 toxic agent Toxicity 0.000 description 1
- 230000002110 toxicologic effect Effects 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000014616 translation Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 210000002993 trophoblast Anatomy 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 210000004981 tumor-associated macrophage Anatomy 0.000 description 1
- 230000007306 turnover Effects 0.000 description 1
- 150000004917 tyrosine kinase inhibitor derivatives Chemical class 0.000 description 1
- 108010033615 ustiloxin D Proteins 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- LEONUFNNVUYDNQ-UHFFFAOYSA-N vanadium atom Chemical compound [V] LEONUFNNVUYDNQ-UHFFFAOYSA-N 0.000 description 1
- 210000005167 vascular cell Anatomy 0.000 description 1
- 230000006459 vascular development Effects 0.000 description 1
- 208000019553 vascular disease Diseases 0.000 description 1
- 208000021331 vascular occlusion disease Diseases 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 208000004043 venous thromboembolism Diseases 0.000 description 1
- 230000007998 vessel formation Effects 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 230000006490 viral transcription Effects 0.000 description 1
- 210000002845 virion Anatomy 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- NAWDYIZEMPQZHO-UHFFFAOYSA-N ytterbium Chemical compound [Yb] NAWDYIZEMPQZHO-UHFFFAOYSA-N 0.000 description 1
Images















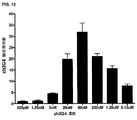













Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/775—Apolipopeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/1703—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- A61K38/1709—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6811—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a protein or peptide, e.g. transferrin or bleomycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6811—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a protein or peptide, e.g. transferrin or bleomycin
- A61K47/6817—Toxins
- A61K47/6831—Fungal toxins, e.g. alpha sarcine, mitogillin, zinniol or restrictocin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6891—Pre-targeting systems involving an antibody for targeting specific cells
- A61K47/6897—Pre-targeting systems with two or three steps using antibody conjugates; Ligand-antiligand therapies
- A61K47/6898—Pre-targeting systems with two or three steps using antibody conjugates; Ligand-antiligand therapies using avidin- or biotin-conjugated antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/0002—General or multifunctional contrast agents, e.g. chelated agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y5/00—Nanobiotechnology or nanomedicine, e.g. protein engineering or drug delivery
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3076—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells against structure-related tumour-associated moieties
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/44—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material not provided for elsewhere, e.g. haptens, metals, DNA, RNA, amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/62—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising only variable region components
- C07K2317/622—Single chain antibody (scFv)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/77—Internalization into the cell
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/30—Non-immunoglobulin-derived peptide or protein having an immunoglobulin constant or Fc region, or a fragment thereof, attached thereto
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Virology (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Zoology (AREA)
- Gastroenterology & Hepatology (AREA)
- Genetics & Genomics (AREA)
- Toxicology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Nanotechnology (AREA)
- Cell Biology (AREA)
- Biotechnology (AREA)
- Ophthalmology & Optometry (AREA)
- General Engineering & Computer Science (AREA)
- Medical Informatics (AREA)
- Crystallography & Structural Chemistry (AREA)
- Marine Sciences & Fisheries (AREA)
- Rheumatology (AREA)
- Microbiology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
Description
本発明は、ホスファチジルセリン生物学の分野、および腫瘍およびウイルス感染の治療に関する。本発明は、腫瘍血管標的輸送および癌治療、および、ウイルス感染およびその他の疾患治療用の、驚くべき新規構築物、組成物、方法および組み合わせを提供する。本発明は特に、複数の性質の驚くべき組み合わせを有する、新規ホスファチジルセリン結合構築物、およびその診断性および治療性接合体を提供する。この新規構築物は、ホスファチジルセリン疾患標的に効果的に結合し、その破壊を促進し、さらに、治療剤を特定部位に送達することが可能なので、癌、ウイルス感染、およびその他の疾患治療のための、ある範囲の方法を提供する。
化学療法剤に対する腫瘍細胞の耐性は、臨床腫瘍学において重要な問題となっている。腫瘍治療において対処しなければならないもう一つの大問題は、「細胞殲滅」、すなわち、無統制増殖能力を持つ、所謂「クローン性」悪性細胞の全てを殺し、治療によって除去することが可能な全ての腫瘍塊を置換したいという希求である。この分野では若干の進歩が見られるけれども、ヒト癌の主要形態の多くが、有効な化学療法介入に依然として抵抗するのには大きく二つの理由がある。
本発明は、腫瘍血管に標的輸送し、癌治療するため、かつ、ウイルス感染およびその他の疾患を治療するための、新規構築物、組成物、方法および組み合わせを提供することによって、従来技術における前述およびその他の要求に応える。本発明は、特に、疾患においてホスファチジルセリン標的に効果的に結合し、それら標的の破壊を、例えば、宿主エフェクター機能を増進することによって促進する、驚くべき特性の組み合わせを持つ、新規ホスファチジルセリン結合構築物を提供する。新規構築物が、さらに生物学的因子、診断剤、および治療剤に結合され、それらの薬剤の疾患部位に対する特異的送達が可能とされる、ある範囲の接合体組成物も提供される。本発明はさらに、腫瘍血管標的輸送、癌治療、および、ウイルス感染およびその他の疾患の治療における、新規構築物および接合体、およびそれらの組み合わせの効果的な使用法も提供する。
(a)検出可能標識または診断剤に対して作動可能に結合される、診断有効量の、本発明の構築物、レセプター本体、またはベータ体を含む第1の薬学的組成物;および、
(b)治療有効量の、本発明の構築物、レセプター本体、またはベータ体、好ましくは、治療有効量の、第1の薬学的組成物において使用されたものと同じ構築物、レセプター本体、またはベータ体を含む第2の薬学的組成物。
(a)生物学的有効量の、本発明の少なくとも第1の構築物、レセプター本体、またはベータ体、その際、該構築物、レセプター本体、またはベータ体は、少なくとも第1の酵素に、作動可能に連結、または共有的に結合または接合する;および、
(b)生物学的有効量の、少なくとも第1の、実質的に不活性な薬剤前駆体、該薬剤前駆体は、前記構築物、レセプター本体、またはベータ体に連結、結合、または接合される酵素によって、実質的に活性な薬剤に転換される、
を含むキットが提供される。
(a)腫瘍を抱える動物または患者に対し、抗腫瘍作用を及ぼすように設計された少なくとも第1の治療を施すこと;
(b)次いで、同じ動物または患者に対し、検出可能な標識または診断剤に作動可能に結合される、本発明の少なくとも第1の構築物、レセプター本体、またはベータ体の診断的有効量を投与し、腫瘍の検出可能な画像、好ましくは、腫瘍内の、前アポトーシスまたはアポトーシス腫瘍細胞または腫瘍血管内皮細胞の画像を形成すること;
(c)腫瘍の検出可能な画像、好ましくは、腫瘍内の、前アポトーシスまたはアポトーシス腫瘍細胞または腫瘍血管内皮細胞の画像を分析し、抗腫瘍作用を及ぼすように設計された少なくとも第1の治療の進行または効果を評価すること、
を含む。
(a)腫瘍を持つ動物または患者に対し、検出可能な標識または診断剤に作動可能に結合される、本発明の少なくとも第1の構築物、レセプター本体、またはベータ体の診断的最小量または有効量を投与することによって腫瘍の画像を形成し、該腫瘍の検出可能画像を形成すること;および、
(b)次いで、同じ動物または患者に対し、本発明の少なくとも第1の構築物、レセプター本体、またはベータ体、またはその接合体の治療的最適量または有効量を投与することによって抗腫瘍作用をもたらすこと、
を含む。
(a)腫瘍を抱える動物または患者に対し、少なくとも第1の酵素に対し作動可能に連結、共有的に結合または接合される、本発明の第1の構築物、レセプター本体、またはベータ体を含む、第1の薬学的組成物を投与すること;
(b)次いで、該動物または患者に対し、効果的な時間間隔の後、生物学的有効量の、少なくとも一つの実質的に不活性な薬剤前駆体を投与することを含み、その際、薬剤前駆体は、腫瘍内に局在化される、本発明の構築物、レセプター本体、またはベータ体に連結、結合、または接合される酵素によって実質的に活性な薬剤に転換される、
方法を提供する。
固形腫瘍およびガン腫は、ヒトにおける全てのガンの90%より多くに相当する。モノクローナル抗体および免疫毒素の使用は、リンパ腫および白血病の治療において検討されている(Vitettaら,1991)が、これらの因子は、ガン腫および他の固形腫瘍に対する臨床試験においては失望すべきことに有効ではなかった(AbramsおよびOldham,1985)。抗体に基づく処置が有効でないことについての主な理由は、高分子は固形腫瘍に容易に輸送されないということである。一旦、腫瘍塊内になっても、これらの分子は、腫瘍細胞、線維性間質、間質性圧力勾配および結合部位障壁の間の緊密な結合の存在に一様に起因して分布することができない(Denekamp,1990;Dvorakら,1991)。
本発明者らは、抗VCAM−1コアグリガンドが、心臓および肺における血管上で構成的に発現されるVCAM−1に対して結合するが、その血管において血栓は生じない能力の背景の機構を理解しようと努めた。心臓および肺における腫瘍環境のプロトロンビン性質および任意の線維素溶解性素因と一般的に結び付いた、この実験的な観察については多くの科学的な可能性がある。
ホスファチジルセリンは、通常、様々な細胞の原形質膜二重層の内面に分離されるが(Gaffet et al.,1995;Julien et al.,1995)、この脂質分離によって二重層間非対称が形成される(Williamson and Schlegel,1994)。本発明人達は以前に、PSは、腫瘍血管内皮細胞の表面に転位すること、かつ、この転位の少なくとも相当部分は、アポトーシスまたは他の細胞死機構とは無関係に起こることを明らかにした(米国特許第6,406,693号)。従って、腫瘍環境におけるPSの表面発現は、細胞死の結果ではなく、また、それが、直後に細胞破壊を誘発することでもない。各種硬組織腫瘍の未処置の血管内皮細胞にPS露出が一貫して検出されるけれども、腫瘍血管内皮は全くアポトーシスを呈することはなく、形態的には健全で(ただし、正常組織の形態とは異なるが)、代謝的には活発である。このことは、腫瘍血管内皮細胞におけるPSの外部膜への転位は十分に安定で、従って、PSは、治療(裸の抗体または治療接合体を用いる)の成功に必要な、標的輸送可能な実体となり得ることを意味するのであるから、PS標的輸送に基づく治療法にとって重要である。
本明細書で示すように、3G4は、有効で、よく受忍される抗腫瘍剤であり、最終的に腫瘍血管上のホスファチジルセリンに結合することによって作用し、宿主細胞介在性抗腫瘍作用をもたらす。PSは、ヒトおよびマウスにおいて同じ分子であり、同じ細胞分布を持ち、両動物種においてROSによって調整および誘発される(Balasubramanian and Schroit,2003;Whitworth et al.,1990)のであるから、上記実験はさらに、ヒトの癌治療にもホスファチジルセリンに結合する抗体および他の構築物を使用することを支持する。実際、3G4の、キメラ版およびヒト化版が、すでにこの目的のため、および他の目的のために調製されている。
WO2004/006847に記載される内容と好対照をなす、3G4抗体、特に、3G4とβ2GPIとの相互作用に関する前述の詳細な分析もまた、本発明人達が行った、本発明のレセプター本体の開発に貢献した。本発明はまた、重要局面として、ホスファチジルセリンに結合し、宿主のエフェクター機能を惹起する「レセプター本体」と呼ばれる、ある範囲の構築物を提供する。レセプター本体の開発局面を後述する。
第2のに、3G4のような抗体は、PS発現腫瘍内皮細胞からの、PS−仲介「安静」シグナル、通常は、腫瘍血管および腫瘍細胞に結合するマクロファージによる炎症反応を抑える筈であるシグナルをブロックする可能性がある(図7)。同様の機構が、アポトーシス細胞に対する、マクロファージ系統細胞の炎症反応の欠如を説明するものと考えられる(Henson et al.,2001;Matzinger,1998;Gallucci and Matzinger,2001;Fadok et al.,2001b)。従って、3G4のような抗体は、マクロファージを刺激してTNF−α、IL−1、および、腫瘍上皮を直接傷害する他の炎症性サイトカインを分泌させることによって腫瘍血管傷害を誘発し、さらに宿主細胞を腫瘍の内部に招集する(Fadok et al.,1998)。
レセプター本体における結合性タンパク質またはポリペプチド、レセプター、リガンド、またはペプチドは、いくつかのホスファチジルセリン結合性タンパク質、例えば、細胞由来レセプター、因子V、プロトロンビン(因子II)等を含む結合性タンパク質の内の任意の一つであってもよい。β2−糖タンパク質Iの使用は非常に好ましい。
Fc領域は、ホスファチジルセリン結合タンパク質またはポリペプチド、レセプター、リガンド、またはペプチドに結合、結合、または接合されるが、結合、結合、または接合は、Fc領域を結合することによって、得られるレセプター本体の所望の活性は実質的に破壊されるように行われる。従来技術で既知の接合またはリンカー技術の任意のもの、例えば、本明細書の関連セクションに記載されるものの採用が可能である。要すれば、当業者にとっては今や標準実技となっている、例えば、これも本明細書に例示される分子生物学技術を用いて融合タンパク質を創成することも可能である。従って、レセプター本体は、化学的接合体または融合タンパク質として作製することが可能である。
Fc領域をホスファチジルセリン結合タンパク質またはポリペプチドに結合することは、本発明のレセプター本体の好ましい実施態様ではあるが、ホスファチジルセリン結合タンパク質はまた、生物学的に活性な分子に長い寿命を与えるために不活性な担体に結合されてもよい。従って、本発明はまた、少なくとも第1の不活性担体に作動可能に結合した、β2GPIを含む少なくとも第1のアミノリン脂質結合タンパク質を含む構築物を提供する。
本発明の構築物およびレセプター本体はまた、結合した治療剤を、特異的標的、例えば、腫瘍および腫瘍内血管、腫瘍細胞、ウイルス感染細胞、およびウイルス粒子に送達するために使用されてもよい。Fc領域を持つ構築物に関するものではないが、米国特許第6,312,694、6,783,760、および6,818,213号も、治療剤に結合するアミノリン脂質結合タンパク質に関する一般的教示に関する参照により本明細書に特異的に含められる。本発明の構築物およびレセプター本体はまた、安全で効果的な抗ウイルス治療に使用されるので、これらの構築物は、ある範囲の既知の抗ウイルス剤に結合されると好都合である。
特定の適用のために、治療剤は、細胞(特に、腫瘍内皮細胞、腫瘍細胞またはウイルスに感染した細胞)の増殖または細胞分裂を消失または抑制する能力を有する、細胞傷害性薬剤または薬理学的薬剤、特に細胞傷害性薬剤、細胞分裂抑制剤か、そうでなければ抗細胞性薬剤である。
サイトカインおよびケモカインは、本発明の構築物、レセプターボディ(receptorbody)またはベータボディ(betabody)に連結するための因子の特定の例である。ある範囲のサイトカインが使用され得、これには、IL−3、IL−4、IL−5、IL−7、IL−8、IL−9、IL−11、IL−13、TGF−β、M−CSF、G−CSF、TNFβ、LAF、TCGF、BCGF、TRF、BAF、BDG、MP、LIF、OSM、TMF、IFN−α、IFN−βが挙げられる。より好ましいサイトカインとしては、IL−1α、IL−1β、IL−2、IL−6、IL−10、GM−CSF、IFNγ、単球化学誘引物質タンパク質−1(MCP−1)、血小板由来増殖因子−BB(PDGF−BB)、およびC−反応性タンパク質(CRP)などが挙げられる。特に好ましい例は、TNFα、TNFα誘導因子IL−2、IL−12、IFN−α、IFN−β、IFN−γおよびLECである。
本発明の構築物、レセプターボディまたはベータボディは、コアグリガンドを形成するために、凝固を直接的または間接的に刺激し得る成分に連結され得る。米国特許第6,093,399号、同第6,004,555号、同第5,877,289号、および同第6,036,955号が、コアグリガンドを形成するために、抗体と凝固剤の作動可能な会合をさらに記載する目的で、本明細書中において具体的に援用される。
一連の薬物は、チューブリン活性との干渉を介してその効果を行使する。チューブリンの機能は、有糸分裂および細胞の生存能力に不可欠であるので、特定の「抗チューブリン薬」は、強力な化学療法剤である。「抗チューブリン薬」は、本明細書で使用される場合、好ましくは、細胞有糸分裂に必要なチューブリン活性(好ましくは、チューブリン重合もしくは脱重合)を直接的もしくは間接的に阻害することによって細胞有糸分裂を阻害する任意の因子、薬物、プロドラッグまたはそれらの組み合わせを意味する。
抗脈管形成剤は、本発明の構築物、レセプターボディまたはベータボディへの結合に有用である。多くの抗癌剤が、それらの作用の機構の一部として、抗脈管形成効果を有する。併用療法で使用するために記載されるこのような任意の1つ以上の薬剤(表Eに含まれる)がまた、本明細書中に記載されるように、本発明の構築物、レセプターボディまたはベータボディに結合体化され得る。特定の他の薬剤が、作用の一次的な機構として、抗脈管形成効果を有することが発見されるか、設計されるか、または選択された。このような薬剤の例は、以下に記載され、これらのうちの任意がまた、免疫結合体を調製するために使用され得るか、または本発明の併用療法において別々に使用され得る。

本発明はまた、任意の細胞(腫瘍細胞および腫瘍脈管内皮細胞およびウイルスに感染した細胞を含む)においてアポトーシスを誘発する因子を送達するために使用され得る。多くの抗癌剤は、その作用機構の一部として、アポトーシス誘発効果を有する。併用療法において使用するために記載された任意の1つ以上のこのような薬剤(表Fに含まれる)はまた、本明細書中に記載されるように、本発明の抗体に結合体化され得る。特定の他の薬剤が、一次的な機構として、アポトーシス効果を有することが発見されるか、設計されるか、または選択された。このような因子の例は、以下に記載され、これらのうちの任意がまた、免疫結合体を調製するために使用され得るか、または本発明の併用療法において別々に使用され得る。
PSおよび他のアニオン性リン脂質がウイスルに感染した細胞に曝露される場合、本発明の構築物、レセプターボディまたはベータボディもまた、任意の1以上の抗ウイルス薬剤に連結され得る。構築物、レセプターボディまたはベータボディに連結するための例示的な抗ウイルス薬剤としては、表Gのものが挙げられる。そのような抗ウイルス薬剤もまた、本発明の抗ウイルス併用療法において別々に使用され得る。
抗体およびエフェクターの等価物またはさらなる改良物が、今や、一般には上記に提供される物質を出発点として使用して作製され得る。改変および変化が、このような抗体の構造中になされ得、そしてさらに類似または他の様式の所望の特徴を有する分子を入手し得る。例えば、特定のアミノ酸が、相互作用性結合能(例えば、アミノリン脂質、PSおよびPEへの結合)のかなり大きな損失なしに、タンパク質構造において他のアミノ酸の代わりに置換され得る。これらの考察もまた、毒素、抗脈管形成剤、アポトーシス誘発剤および凝血薬などにあてはまる。
抗体Fc領域は、本発明の構築物、レセプターボディまたはベータボディを提供するために少なくとも第1のホルファチジルセリン結合タンパク質に作動可能に結合されるか、そのタンパク質と関連するか、または結合体化される。そのような構築物、レセプターボディまたはベータボディは、例えば、抗細胞性薬剤および細胞傷害性薬剤、凝血薬ならびに抗ウイルス薬剤にさらに結合体化または結合され得る。
上記に提供される一般の情報に加えて、特定の好ましい生化学架橋剤が使用され得る。架橋試薬は、2つの異なる分子の官能基を共に結ぶ分子架橋を形成するために使用される。段階的な様式にて2つの異なるタンパク質を連結するために、所望されないホモポリマー形成を排除するヘテロ二官能性架橋剤が、使用され得る。例示的なヘテロ二官能性架橋剤は、表Cにおいて参照される。
任意の結合部分は、血液において合理的な安定性を有し、疾患、例えば、腫瘍部位に標的化する前に、結合した薬剤の実質的な放出を予防することが、好ましいけれども、特定の局面において、生物学的に放出可能な結合および/または選択的に切断可能なスペーサーまたはリンカーの使用が、意図される。「生物学的に放出可能な結合」および「選択的に切断可能なスペーサーまたはリンカー」は、循環において合理的な安定性をなお有する。
構築物、レセプターボディまたはベータボディは、分子生物学技術を使用して融合タンパク質として調製され得る。任意の融合タンパク質が、任意の構築物、レセプターボディまたはベータボディならびに第二の薬剤(本明細書中に開示されるものおよび当該分野で公知のもの)を使用して設計および作製され得る。融合タンパク質技術は、他の改変(例えば、CDR配列の最適化、選択的に切断可能なペプチド配列を介する連結など)を有する融合タンパク質を調製するために容易に適合可能である。
本発明の治療剤は、一般に、薬学的組成物として処方される。薬学的組成物は、少なくとも本発明の第1の治療剤(薬学的に受容可能なキャリアまたは水性媒体中に、溶解または分散した)の生物学的または治療的有効量を含む。組み合わせ治療もまた意図され、そして同じ型の基礎をなす薬学的組成物を、単一医薬および組み合わせ医薬の両方について使用し得る。
本発明の治療剤は、しばしば、非経口投与(例えば、静脈内、筋肉内、皮下、経皮を介する注射のための処方、またはぜん動投与および腫瘍もしくは疾患部位(腔内投与)中への直接滴下を含む他の経路のための処方)のために処方される。抗体、免疫結合体またはペプチド結合体を活性成分として含有する水性組成物の調製は、本明細書の開示を参照すれば、当業者に公知である。代表的には、そのような組成物は、水溶液または懸濁液のいずれかにおいて、注射可能な薬剤として調製され得る;注射前の液体への添加において、溶液または懸濁液を調製するための使用に適切な固体形態もまた調製され得る;そしてその調製物はまた、乳化され得る。
処方物は、種々の投薬形態(例えば、上記の注射可能な溶液の型)において容易に投与されるが、他の薬学的に受容可能な形態(例えば、錠剤、ピル、カプセルまたは経口投与のための他の固体、座剤、腟坐薬、経鼻溶液またはスプレー、エアロゾル、吸入剤、リポソーム形態など)もまた意図される。投与形態の型は、処置される疾患または障害に適合される。
特定の実施形態において、リポソームおよび/またはナノカプセルもまた、治療剤とともに使用され得る。リポソームの処方および使用は、以下に要約されるように、当業者に、一般に公知である。本発明は、抗体、リポソームおよび化学療法剤の特定の組合せ(以下に記載される)を提供する。さらに、リポソーム処方物は、本発明の治療剤のいずれかの慣用的な成分として使用され得る。
眼の多くの疾患(特に、脈管形成成分を有するもの)は、本発明によって処置され得る。例えば、眼の脈管新生疾患、年齢関連黄斑変性症、糖尿病性網膜症、未熟成の網膜症、角膜移植拒絶、脈管新生緑内障、水晶体後線維増殖症、ならびに角膜脈管新生または網膜/脈絡膜脈管新生と関連する他の疾患(本明細書中で以下に記載される)。
広範な意味で、局所投与のための処方物は、口(頬側)を介してかつ皮膚を通して送達するための処方物を含む。「局所送達システム」はまた、投与される成分を含む経皮的パッチを含む。皮膚を通す送達は、所望の場合、イオン注入または電気的運搬によってさらに達成され得る。
鼻経路および呼吸器経路を介する局所送達は、種々の状態を処置するため(特に、本発明の抗ウイルス処置方法における使用のため)に意図される。これらの送達経路はまた、薬剤を全身性循環に送達するために適切である。従って、鼻性投与のための適切なキャリアにおける活性成分の処方物は、本発明内に含まれ、例えば、鼻性溶液、スプレー、エアロゾルおよび吸入抗原が挙げられる。キャリアが個体である場合、処方物は、例えば、20〜500ミクロンの範囲の粒子サイズを有する粗散剤が挙げられ、例えば、鼻に近接して維持される散剤のコンテナから鼻経路を通る急速な吸入によって投与される。
本発明が動物およびヒトの処置レジメンにおいて有意な有用性を有するものの、多くの他の特異的および信頼できる使用(多くのインビトロ実施形態における特定の使用を含む)も有する。これらの使用の特定のものは、抗体、ペプチドおよび免疫結合体の特異的結合特性に関連する。本発明の構築物の各々が、アミノリン脂質および/または陰イオン性リン脂質に結合する少なくとも1つの抗体またはペプチド成分を含む点で、これらは、種々の結合実施形態(有用な結合アッセイを含む)において使用され得る。
の結合特性および機能的特性が本明細書中に開示されるように特に特異的であるので、このような「コントロール」の使用は、実際に非常に価値がある。本発明のこのような実際的な適用からの利点があるアッセイとしては、例えば、細胞表面でのPSまたは陰イオン性リン脂質の検出に関するアッセイが挙げられる。
本発明はまた、処置方法、組合せ処置方法ならびに/または画像化および処置実施形態において使用するための、本発明の少なくとも1つの第1の構築物、レセプターボディまたはベータボディを含む診断キットおよび治療キットを提供する。このようなキットは、一般的に、少なくとも第1の適切な容器(または容器手段)、少なくとも1つの治療剤、アミノリン脂質または陰イオン性リン脂質に結合する抗体、免疫結合体またはペプチド結合体の薬学的に受容可能な処方物を含む。キットは、例えば、前臨床的、臨床的および/または獣医学的実施形態での使用のための記載された指示書または電子指示書を備え得る。
本発明はさらに、インビトロおよびインビボでの診断法および画像化法を提供する。このような方法は、診断情報、予後情報および/または画像情報(例えば、脈管形成疾患およびウイルス感染、好ましくは、腫瘍処置および画像化方法に関連する)を生成する際の使用に適用可能である。本発明の方法は、インビトロでの診断試験(例えば、サンプルが非侵襲的に得られ、好ましくは、高スループットアッセイにて試験され得るか、そして/または曖昧および確認における臨床診断が望まれる)を含む。インビボ診断および画像化の分野において、本発明の構築物、レセプターボディまたはベータボディは、1つ以上の検出可能な薬剤に連結され、そして必要に応じて、処置の前に第1の工程として、脈管形成部位または腫瘍の画像を形成するために使用される。
従って、本発明は、アミノリン脂質および陰イオン性リン脂質を結合、精製、除去、定量またはさもなければ一般には検出するため(例えば、活性化細胞およびアポトーシス細胞および関連する疾患の診断において使用するため)の、免疫検出法に関する。本発明の構築物、レセプターボディまたはベータボディが、インビボ(下記)にて、単離された組織サンプル、生検またはスワブにおいて、そして/またはホモジナイズされた組織サンプルにおいて、PSおよび陰イオン性リン脂質を検出するために使用され得る。このような免疫検出法は、明らかな診断的有用性を有するが、また、非臨床サンプルに(例えば、抗原サンプルの力価測定などにおいて)も適用を有する。
本発明は、種々のインビボ診断および画像化の実施形態を提供する。本発明の特定の局面は、インビボ診断および画像化について、新規な驚くべき有効な組成物に関する。例えば、本発明の構築物、レセプターボディまたはベータボディが、インビトロで検出可能な薬剤に連結されて、本発明の免疫診断結合体を形成する。抗体がこの分野において重要な発展を示しているものの、得られる免疫診断は、現在、アミノリン脂質および/または陰イオン性リン脂質の検出と連結されて、任意の以前に記載された診断実施形態または画像化実施形態において使用され得る。
インビボ診断および画像化に関して、本発明は、さらに、癌治療のための代理マーカーとして使用するための組成物および方法を提供する。このような実施形態は、インビボ検出可能な薬剤に連結された、本発明の構築物、レセプターボディまたはベータボディの使用に関する。
本発明の重要な局面は、悪性疾患、腫瘍および脈管新生化腫瘍の処置に関する。これは、脈管新生が多かれ少なかれ重要である腫瘍、およびプロトロンビン血管を有する腫瘍を含む。良性腫瘍の処置が本発明に含まれ、例えば、聴覚神経腫、神経線維腫、トラコーマ、化膿性肉芽腫およびBPHが挙げられる。血液の腫瘍(例えば、白血病)および骨髄の種々の急性または慢性の新生物形成疾患の処置もまた包含される。
本発明の処置方法は、患者が示す特定の腫瘍、疾患または障害の処置において一般に使用される任意の他の方法と組み合わされてもよい。特定の治療アプローチがそれ自体で患者の状態に対して有害であることが知られておらず、そして本発明の処置に有意に反作用しない限り、本発明とのその組み合わせが考えられる。
本明細書において用いる場合、本発明の「一次治療因子(primary therapeutic agents)」は、構築物、レセプターボディまたはベータボディである。本明細書において用いる場合、「二次治療因子(secondary therapeutic agents)」は、第二の、別個の治療因子または抗ガン剤、すなわち、一次治療因子「以外の(other than)」治療剤または抗ガン剤である。任意の二次治療因子は、本発明の併用療法において用いられ得る。また、二次治療因子または「二次抗ガン剤」は、以下の手引きに従って、相加作用、相加作用より大きい作用および潜在的に相乗的な作用を達成するという観点で選択され得る。
内毒素および解毒された内毒素誘導体は、併用処置において、好ましくは低用量で用いられ得る(本明細書において参考として詳細に援用される、PCT公開第WO03/028840号)。動物での使用のために、詳細にはヒトにおける使用のために好ましい、種々の解毒された内毒素が利用可能である。解毒および精練された内毒素、ならびにそれらの組み合わせは、各々が参考として本明細書において詳細に援用される、米国特許第4,866,034号;同第4,435,386号;同第4,505,899号;同第4,436,727号;同第4,436,728号;同第4,505,900号に記載される。
サイトカイン療法は、組み合わせの治療レジメンのための有効なパートナーであることが証明されている。本発明の組み合わせアプローチにおいて、種々のサイトカインが使用され得る。サイトカインの例としては、IL−1α、IL−1β、IL−2、IL−3、IL−4、IL−5、IL−6、IL−7、IL−8、IL−9、IL−10、IL−11、IL−12、IL−13、TGF−β、GM−CSF、M−CSF、G−CSF、TNFα、TNFβ、LAF、TCGF、BCGF、TRF、BAF、BDG、MP、LIF、OSM、TMF、PDGF、IFN−α、IFN−β、IFN−γが挙げられる。サイトカインは、患者の状態およびサイトカインの相対的な毒性のような、臨床的指標と整合する標準的レジメンに従って投与される。ウテログロビンはまた、転移を防止または阻害するために用いられ得る(米国特許第5,696,092号;参考として本明細書に援用される)。
TNFαおよびTNFαの誘導因子はまた、本発明と組み合わせて用いられ得る。TNFαは、脈管透過性を増大し、従って、腫瘍への抗ガン剤の浸透を促進することにおいて有用である。抗体局在化は、PSおよび陰イオン性リン脂質を標的化する場合には決して問題ではない。なぜなら本発明においては、TNFαの併用使用は、腫瘍への他の化学療法剤および免疫複合体の接近を容易にし得、そして遠くの腫瘍細胞への本発明の抗体の結合を増大さえするからである。
背景にある機構(単数または複数)にかかわらず、種々の化学療法剤が、本明細書に開示される併用処置方法において用いられ得る。併用療法について考えられる化学療法剤としては、例えば、タモキシフェン、タキソール、ビンブラスチン、エトポシド(VP−16)、アドリアマイシン、5−フルオロウラシル(5FU)、カンプトテシン、アクチノマイシン−D、マイトマイシンC、コンブレタスタチン(類)、さらに詳細には、ドセタキセル(タキソテール)、シスプラチン(CDDP)、シクロホスファミド、ドキソルビシン、メトトレキセート、パクリタキセル、およびビンクリスチン、ならびにその誘導体およびプロドラッグが挙げられる。
用語「脈管形成」とは、一般的に組織または器官への新規な脈管の生成をいう。通常の生理学的条件下で、ヒトまたは動物は、特定の制限された状況においてのみ脈管形成を受ける。例えば、脈管形成は、通常、組織治癒、胎児および胚の発生、および黄体、子宮内膜および胎盤の形成において観察される。しかし、新たな証拠として、脈管形成が、例えば、副腎組織、前立腺および卵巣における特定の正常な状態において重要であることが示される。本発明の治療剤(抗脈管形成が、作用の様式だけでない)は、従って、所望のまたは「生理学的」脈管形が、本発明を使用する場合、阻害されない点で、顕著な抗脈管形成治療に対する利点を、有する(例えば、抗体A4.6.1(Brem、1998;Bacaら、1997;Prestaら、1997)。
VEGFは、低酸素および発ガン性変異によって誘導される多機能性サイトカインである。VEGFは、胚形成における脈管ネットワークの発生および維持の最初の刺激物である。これは、強力な浸透性誘導剤、内皮細胞化学走化性剤、内皮生存因子、および内皮細胞増殖因子として機能する。その活性は、正常な胚発生に必要とされる。なぜなら、VEGFの一方または両方の対立遺伝子の標的化された破壊が、胚の死を生じるからである。
本発明の治療剤はまた、好ましくは、腫瘍(腫瘍細胞および腫瘍脈管内皮細胞を含む)内で任意の細胞のアポトーシスを誘導する処置方法と組み合わされる。アポトーシスを誘導する例示的な薬剤は、(免疫結合体とともに)上に列挙される。任意の1つ以上のこのようなアポトーシス誘導剤が、本発明の抗体に連結されること無しに、本発明の併用療法において使用され得る。
本発明はまた、標的タンパク質が腫瘍細胞のマーカー、腫瘍脈管構造または腫瘍支質に向けられる、他の免疫毒素またはコアグリガンドとともに使用され得る。PE結合ペプチドを標的化する際に使用するための本明細書中に記載される標的化剤のいずれかが、これらの実施形態において使用され得る。免疫毒素において、結合される薬剤は、抗細胞剤または細胞傷害剤、サイトカイン、放射線治療剤、抗脈管形成剤、アポトーシス誘導剤および抗チューブリン薬物を含む。コアグリガンドにおいて、結合される薬剤は、コアグリガンドである。米国特許第5,855,866号、同第5,965,132号、同第6,261,535号、同第6,051,230号、同第6,451,312号(免疫毒素)、同第6,093,399号、同第6,004,555号、同第5,877,289号、および同第6,036,955号(コアグリガンド)が、このような構築物を例示するために、本明細書中で具体的に参考として援用される。
本発明の構築物、レセプターボディまたはベータボディは、プロドラッグと組み合わせて使用され得、ここでこの抗体は、プロドラッグ活性化成分(例えば、抗体に接触した際にのみ、プロドラッグをより活性な形態へ変換するプロドラッグ活性化酵素)と作動可能に付随される。この技術は、一般に「ADEPT」と称され、そして例えば、WO95/13095;WO97/26918、WO97/24143および米国特許第4,975,278号および同第5,568,568号(これらは、各々具体的に本明細書中に参考として援用される)において記載される。
リポソーム処方物は、しばしば、治療剤および薬剤において使用される。しかし、最初の研究においてリポソームの生体分布は、このような処方物が、ヒトにおいて幅広く適用可能でないことを意味する。リポソームは、細網内皮系(RES)の食細胞(循環する単核食細胞ならびに肝臓および脾臓に位置する細胞を含む)によって迅速に取り込まれる。したがって、血液循環半減期は、数分と短くあり得る。
本発明はまた、異常な脈管形成が関与する他の疾患(プロトロンビン血管を有する疾患および障害を含む)の処置において使用され得る。治療機構のみではないが、本発明の抗体、免疫結合体およびペプチドベース治療剤はまた、異常な脈管形成(例えば、種々の疾患および障害に寄与する)を有する動物および患者を処置するために使用され得る。
本発明はさらに、ウイルス感染を処置するのにおける使用のために、必要に応じて抗ウイルス剤に結合体化された、ある範囲の構築物を提供する。この処置レジメン、および詳細には用量は一般に、本発明のガン処置局面について上記されたとおりであって、この適応性は本発明全体の利点である。作用の特定の機構(単数または複数)の理解は、本発明の抗ウイルス処置を行なうのに必要ではないが、本明細書の実施例によって支持されるとおり、ウイルス処置の基礎にある特定の理由は以下のとおりである。
脊椎動物のウイルス
けるウイルスの任意の1つ以上が本発明を用いて阻害され得、これによってその得られた感染および疾患が処置され得る。
ヒトにおけるウイルス疾患
(抗VCAM−1−tTFコアグリガンドを用いた腫瘍処置)
本実施例は、腫瘍保有動物に対する腫瘍脈管標的化凝固因子(「コアグリガンド(coaguligand)」)の投与後に生じるインビボにおける腫瘍脈管構造の特異的な凝固、および得られた抗腫瘍効果を示す。このコアグリガンドでは、VCAM−1(血管内皮接着分子−1、VCAM−1)に対する抗体を、腫瘍脈管構造に対して改変型のヒト凝固因子である短縮型組織因子(truncated Tissue Factor)(tTF)を送達するための標的因子として用いる。
腫瘍血管上のホスファチジルコリン発現
心臓および肺におけるVCAM−1陽性脈管構造上の抗VCAM−1・tTFの血栓性効果の欠失を説明するために、正常な血管と腫瘍血管との間の異なるアミノリン脂質および陰イオン性リン脂質、例えば、PSおよびPEの局在化という本発明者らの発展させた概念を確認した。詳細には、彼らは、正常な組織における内皮細胞が、アミノリン脂質および陰イオン性リン脂質、例えばPSおよびPEを、原形質膜のリン脂質二重相の内面に分離して、ここでPSは血栓性反応に関与することができないが;一方、腫瘍の内皮細胞は、原形質膜の外面に対してアミノリン脂質および陰イオン性リン脂質を転位させ、ここでPSはコアグリガンドの凝固作用を支持し得るという仮説をたてた。細胞表面でのPS発現によって、凝固が可能になる。なぜなら、PS発現によって、膜に対する凝固因子の結合が可能になり、凝固開始複合体のアセンブリが協調されるからである。
両方ともマウスのモノクローナルIgM抗体である、抗PS抗体および抗カルジオリピン抗体を、実施例IVに記載のとおり、Roteら(1993、本明細書において参考として援用される)によって生成して、特徴付けした。3SBの主な反応性はPSとの反応であるが、3SBはまた、正常な細胞における内部小葉部分に緊密にやはり分離される原形質膜の比較的少ない成分である、陰イオン性リン脂質、ホスファチジン酸との反応性も有する。
この免疫組織学研究によって、抗PS抗体は、VCAM−1を欠き得る腫瘍の中央領域の血管を含むほとんどの腫瘍血管に10分以内で局在したことが示された。VCAM−1が陽性であった血管はまたPSについても陽性であった。従って、腫瘍におけるVCAM−1発現血管上ではPSの同時発現が存在する。
アネキシンVはコアグリガンド活性をブロックする。
アネキシンVがコアグリガンドによって誘導される第Xa因子形成に影響を与える能力を、色素生産性アッセイによって決定した。IL−1α刺激したbEnd.3細胞を、抗VCAM−・tTFとともにインキュベートし、そしてサポニンで透過化処理した。アネキシンVを、0.1〜10μg/mlの範囲の濃度で添加し、そして細胞を、希釈したProlexTの添加前に30分間インキュベートした。アネキシンVの存在下または非存在下で生成した第Xa因子の量を、決定した。各処置を2連で行い、そして少なくとも2回繰り返した。
アネキシンVのインビボでコアグリガンド誘導性血栓症を阻害する能力を、L540ホジキン腫保有SCIDマウスにおいて試験した。腫瘍を、上記の実施例IIに記載のようにマウスにおいて増殖させた。1群あたり2匹のマウス(直径0.5cmの腫瘍サイズ)を、以下の試薬のいずれか1つで尾静脈を介して静脈内注射した:a)生理食塩水;b)100gのアネキシンV;c)40μgの抗VCAM−1・tTF;d)100μgのアネキシンV、続いて2時間後に40μgの抗VCAM−1・tTF。
アミノリン脂質、陰イオン性リン脂質および複合体に対する抗体の生成
本実施例は、腫瘍血管内皮細胞におけるアミノリン脂質および陰イオン性リン脂質の転位に対するその観察に照らして、本発明者らによって設計された、そしてアミノリン脂質および陰イオン性リン脂質の転位に対する抗体の生成において十分に機能するように開発された、免疫プロトコールを記載する。アミノリン脂質および陰イオン性リン脂質、例えばPSおよびPEと反応性の多数の抗体が得られた。本実施例および以下の実施例において、簡便性のために、PSと反応性の抗体を「抗PS抗体(anti−PS antibody)」と命名してもよいが、特定のこれらの抗体の結合はPSには限定されず、本明細書に示されるような特定の他のアミノリン脂質および陰イオン性リン脂質に拡大される。
さらに強力な免疫原として免疫系にアミノリン脂質および陰イオン性リン脂質を提示するために、アミノリン脂質および陰イオン性リン脂質を、アミノリン脂質陽性細胞および陰イオン性リン脂質陽性細胞として処方した。他の膜成分によって囲まれる、膜に挿入されたアミノリン脂質および陰イオン性リン脂質は、抗体を惹起するためのさらに良好な構成およびクリアランス速度を有する。
陰イオン性リン脂質、例えばPSと反応性の抗体の極度に高い力価を有するマウスを得た(表1)。マウスは、なんら毒性の兆候を示さなかった。この免疫プロトコールは、ラット全体よりもマウスで有効であったが、ラットの免疫は、有効であって、9D2抗体が産生された(以下を参照のこと)。
抗PS IgG抗体生成
免疫した動物由来の脾細胞とミエローマパートナーP3X63AG8.653細胞(ATCC,Rockville,MD)とを融合することによってハイブリドーマを得た。
抗体をELISAによってさらに研究して、3SBおよびD11と比較した。本研究で用いた抗PS ELISAは以下のとおり行なう。特別な相違が特定されない限り、これが本出願の研究全体を通じて用いたELISAの形式である。
抗PS抗体の相対的な親和性
2変換のために用いたMW:IgM−900kDa、IgG−150kDa、アネキシンV−36kDa
3PSに対するアネキシンVの親和性は、0.1nM〜1nMの範囲である。この表における値は、抗PS抗体についてと同じELISA条件を用いてストレプトアビジン−HRPによって検出された市販のビオチン化アネキシンVの結合に相当する。
2>>という記号は、同一の抗体濃度で試験した種々のリン脂質に対する結合における少なくとも10倍の相違を示す。
3G4抗体とプラスチック固定リン脂質との反応性をさらに試験した。リン脂質を、n−ヘキサン中に50μg/mlの濃度まで溶解させた。この溶液の100μlを96ウェルマイクロタイタープレートに添加した。空気中で溶媒をエバポレートしたのち、このプレートを、2mMのCa2+を含むDPBS中に溶解した10% FBS(結合緩衝液)で2時間ブロックした。3G4抗体を33nMの初期濃度で10%のウシ血清の存在下で結合緩衝液に希釈した。連続2倍希釈物をプレート中に調製した(1ウェルあたり100μl)。次いで、このプレートを2時間室温でインキュベートした。洗浄の後、HRPヤギ抗マウスIgG(1:1000希釈)を用いて、3G4を検出した。二次試薬を
発色基質OPDを用い、その後マイクロプレートリーダー(Molecular Devices,Palo Alto,CA)を用いて490nmでプレートを読み取ることによって検出した。
外面化したホスファチジルセリンは腫瘍血管の全体的なマーカーである。
これらの研究で用いた抗PS抗体は、3SBと命名されたマウスモノクローナルIgM抗体だった(実施例IV、Roteら、1993)。3SBは、主にPSに結合するがまた、PSのような分布を有する比較的わずかな陰イオン性リン脂質であるPAとも反応する。用いた抗CL抗体は、D11と命名されたマウスモノクローナルIgM抗体であった(実施例IV、Roteら、1993)。
膜の内面に対してPSを隔離する能力を腫瘍脈管構造が失う時点を推定するため、140〜1600mm3の容積におよぶL540腫瘍において、抗PS局在化を検査した。
中間および大きいサイズの腫瘍において検出されたPS外面化
†汎内皮Ab Meca 32を用いて血管の総数を決定した。PS陽性およびMeca陽性の血管を腫瘍の1断面あたり4領域でカウントした。同じ群内のPS陽性血管の範囲(%)を示す。
同じ抗PS(3SB)抗体および抗CL(D11)抗体を用いて、腫瘍および正常血管内皮上のPS露出を、さらなる3つの動物腫瘍モデル(全部で6つ)を用いるさらなる研究において試験した:L540ヒトホジキンリンパ腫、NCI H358ヒト非小細胞肺ガン(NSCLC)、HT29ヒト結腸直腸ガン、Colo26マウス結腸ガン、B16マウス黒色腫、および3LLマウス肺腫瘍。
腫瘍脈管に対する抗PS抗体の特異的局在化
†非抗原特異的尿細管染色は、抗PSおよび抗CLレシピエントの両方で可視であった。
腫瘍におけるアポトーシスマーカーの発現
†3LL腫瘍の壊死領域に時折存在する血管(100を超える内の1)は、アポトーシスの両方のマーカーを示した。
腫瘍血管内皮細胞上でのPS露出をまた、マウスで増殖しているMDA−MB−231ヒト乳房腫瘍において、および皮下で増殖しているマウスMeth A線維肉腫において検査した。これらの研究において用いた抗体は、実施例IVに記載のように生成された、9D2抗体であって、これは陰イオン性リン脂質と反応性である。
さらなる乳ガンモデルにおいては、腫瘍血管内皮上のPS露出を、マウスで増殖しているMDA−MB−435ヒト乳ガン細胞中で検査した。これらの研究で用いた抗体は、3G4抗体のキメラバージョン(ch3G4)である。3G4抗体生成は実施例IVに記載され、そしてキメラ3G4抗体の生成は、実施例XIXに詳細に記載される。MDA−MB−435モデルにおける腫瘍血管内皮に対するch3G4の局在化は、実施例XIXにさらに詳細に記載されており、図22に示される。
10番目のモデルについて、腫瘍血管内皮に対するPSの露出を、多段階の発現の「RIP−Tag」トランスジェニックマウスモデル(RIP1−Tag 2)において検査した。このトランスジェニックマウスモデルでは、あらゆるマウスが、インスリン産生β細胞におけるSV40 T抗原(Tag)ガン遺伝子の発現の結果として12〜14週齢までに膵臓の島腫瘍を発症する。腫瘍は、過剰増殖性島から多段階で発症して、悪性に進行するには血管形成性の切り替えを要する。マトリックスメタロプロテイナーゼ9が血管形成切り替えを制御する(REF)。
本発明者らは、多くの様々な腫瘍を有するマウスの腫瘍血管への、種々の抗PS抗体の局在化を研究した。そのような研究から合わせた結果が、以下の表6Bに要約される。抗PS抗体は、すべての腫瘍の腫瘍血管に局在化するが、脈管局在化は正常な器官では観察されない(表6B)。
陰イオン性リン脂質を腫瘍血管の表面上に露出させる。
1.材料
Na125Iは、Amersham(Arlington Heights,IL)から入手した。ダルベッコ改変イーグル組織培養培地、およびCa2+およびMg2+を含有するダルベッコPBSをGibcoから入手した(Grand Island,NY)。ウシ胎仔血清をHyclone(Logan,Utah)から入手した。L−α−ホスファチジルセリン、L−α−ホスファチジルコリン、カルジオリピン、L−α−ホスファチジルエタノールアミン、L−α−ホスファチジルイノシトール、スフィンゴミエリン、ホスファチジン酸、ホスファチジルグリセロール、O−フェニレンジアミン、過酸化水素およびトロンビンをSigma(St.Louis,MO)から入手した。24ウェルの平底プレートをFalcon(Becton DickinsonおよびCo.,Lincoln Park,NJ)から入手した。
l Type I)のインターフェロン(全てのタイプのインターフェロンの代わりになるハイブリッドタンパク質)をPBL Biomedical Laboratories(New Brunswick,NJ)から購入した。組み換えのヒト血管内皮増殖因子121(VEGF)、ヒト血小板由来増殖因子−BB、インターロイキン−6(IL−6)、インターロイキン−8(IL−8)、インターロイキン−10(IL−10)およびヒト線維芽細胞増殖因子−2(FGF−2)をPeproTech(Rocky Hill,NJ)から購入した。
MECA32、汎マウス内皮細胞抗体は、Dr.E.Butcher(Stanford University,CA)から入手して、免疫組織学的研究のための陽性コントロールとして用いた。この抗体の詳細は公表されている(Leppinkら、1989)。西洋ワサビペルオキシダーゼ(HRP)に結合体化したウサギ抗ラット免疫グロブリン二次抗体、ラット抗マウス免疫グロブリン二次抗体、ならびにヤギ抗マウス二次抗体および抗ラット二次抗体は、Daco(Carpinteria,CA)またはJackson Immunoreserarch Labs(West Grove,PA)のいずれかから購入した。
末期の疾患を有する患者由来のL540Cyホジキンリンパ腫細胞は、V.Diehl教授(Koeln,Germany)から提供された。NCI−H358ヒト非小細胞肺ガンは、Adi Gazdar博士(Southwestern Medical Center,Dallas,TX)から提供された。Meth Aマウス線維肉腫およびMDA−MB−231ヒト乳ガンは、American Type Cell Collection(Rockville,MD)から入手した。マウス脳内皮腫株bEnd.3は、Werner Risau教授(Max Plank Institution,Munich,Germany)から提供されて、10%FBSを有するDMEM中で維持した。成体ウシ大動脈内皮(ABAE)細胞は、Clonetics(San Diego,CA;Walkerville,MD)から購入した。ABAE細胞は、10%血清および2ng/mlのbFGFを含有するDMEM中で維持した。
bEnd.3,ABAE細胞およびL540Cyリンパ腫以外の全ての腫瘍細胞を、10%ウシ胎仔血清、2mM L−グルタミン、2単位/mlペニシリンGおよび2μg/mlストレプトマイシンを補充したDMEM中で維持した。L540Cy細胞は、同じ添加物を含むRPMI 1640中で維持した。細胞を週に1回継代培養した。0.2% EDTAを含有するPBSにおいて0.125%トリプシンを用いてbENd.3細胞のトリプシン処理を行なった。インビトロ研究については、24ウェルプレートにおいて1mlの培養培地に10×103細胞/mlの密度で内皮細胞を播種して、アッセイでの使用の前に48〜96時間インキュベートした。各々の研究の24時間前に培地を再生した。
リン脂質をn−ヘキサンに50μg/mlの濃度まで溶解した。この溶液の100μlを96ウェルマイクロタイタープレートのウェルに加えた。空気中の溶媒のエバポレーション後に、このプレートを、2mM Ca2+を含有するDPBSに溶解した10%ウシ胎仔血清(結合緩衝液)を用いて2時間ブロックした。
内皮細胞が約70%コンフルエンスに達するまで内皮細胞を増殖させた。PS露出を誘導するために、細胞をH2O2(200μM)を用いて37℃で1時間処置した。コントロールのスライドおよび処置したスライドをCa2+およびMg2+含有DPBSを用いて洗浄して、同じ緩衝液に希釈した0.25%グルタルアルデヒドで固定した。50mMのNH4Clとの5分間のインキュベーションによって過剰なアルデヒド基をクエンチした。リン脂質の検出に対する界面活性剤および有機溶媒の効果を試験するために、いくつかのスライドをアセトンを用いるか(5分)、または1%(v/v)TritonTMX−100を含有するPBSを用いてプレインキュベートした。
リン脂質認識の特異性をさらに、種々のリポソームを用いた競合アッセイによって確認した。リポソームを、5mgの単一のリン脂質を含有するクロロホルムの溶液から調製した。この溶液を窒素下で乾燥して、丸底ガラスフラスコ中に薄層を形成させた。次いで、10mlのTris緩衝液(0.1M、pH7.4)を添加して、フラスコを2分間に5回超音波処理した。9D2またはアネキシンV(6.66nM)を200μg/mlのリポソーム溶液とともに室温で1時間プレインキュベートした。この混合物をリン脂質コーティングしたプレートまたは内皮細胞単層に加えた。異なるリポソームの有無において固定されたリン脂質または細胞表面に対して9D2が結合する能力を上記のように決定した。
10倍モル過剰のN−ヒドロキシスクシンイミドビオチン(Sigma,MO)を用いて精製されたタンパク質を室温で1時間インキュベートすることによって、ビオチン化9D2抗体およびアネキシンVを調製した。遊離のビオチンをPBSに対する透析によって除去した。ビオチン化手順は、いずれのタンパク質のPS結合能力も損なうことはなかった。競合研究について、未改変のタンパク質およびビオチン化タンパク質を10倍モル過剰の未改変のタンパク質と事前混合した。次いで、この混合物をPSコーティングプレートに加えた。結合した試薬を、1:1000に希釈されたストレプトアビジン−HRP結合体によって検出した。競合物の非存在下での各々の試薬のPSへの結合を100%値として採用した。
局在化研究のために、2×107個のL540ヒトホジキンリンパ腫細胞または他の腫瘍タイプの1×107個の細胞をSCIDマウス(Charles River,Wilmington,MA)の右脇腹に皮下注射した。腫瘍を0.4〜0.7cm3の容積まで到達させた。1群あたり最小3匹の動物のを用いた。研究を少なくとも3回繰り返した。
雌性のnu/nuマウスまたはSCIDマウスをCharles Riverから購入した。MDA−MB−231ヒト乳腺ガン細胞を、公表されたプロトコール(Price,1996)に従って乳腺脂肪パッドに移植した。要するに、マウスを麻酔して外胸部にわたって皮膚に5mmの切開を作製した。乳腺パッドを露出させて、0.1mlの生理食塩水に再懸濁した1×107個のMDA−MB−231細胞の注射のための正確な部位を確認した。
9D2またはアネキシンVが凍結組織の切片に直接付与される免疫組織化学技術では、原形質膜の内部小葉構造と外部小葉構造との上の陰イオン性リン脂質の間が識別されない。外部に位置するリン脂質を検出するために、本質的に以前に記載されたような方法を行なった(実施例V;Ranら、1998)。腫瘍保有SCIDマウスに、50μgの9D2もしくはビオチン化9D2抗体、または100μgのビオチン化アネキシンVのいずれかを静脈内注射した。60分後にマウスを屠殺して、その血液循環を放血して、以前に記載されたようにヘパリン処理生理食塩水を用いて灌流させた(Burrowsら、1992)。全ての主な器官および腫瘍を回収して、凍結切片の調製のために急速凍結させた。
局在化した9D2抗体またはアネキシンVを有する構造を、DAB染色した切片上の形態学的外観によって、および凍結組織の連続切片上の汎内皮細胞マーカーMECA32を用いた同時染色によって、血管として同定した。腫瘍の連続切片においてMECA 32、9D2またはアネキシンVによって染色した血管をカウントすることによって、DAB染色した切片上の定量を行なった。9D2抗体、コントロールラットIgMまたはアネキシンVを注射した6匹のマウス由来の各々の腫瘍タイプの6つのスライドを試験した。1切片あたり少なくとも10個の無作為の視野(1視野あたり0.317mm2)を、2人の独立した観察者によって、盲検的にスコア付けした。9D2、アネキシンVまたはMECA32によって染色された血管の平均数および標準誤差を算出した。各々の腫瘍タイプ群において決定した9D2またはアネキシンVの陽性血管の平均数を、同じ腫瘍群におけるMECA32陽性血管の平均数と比較した。9D2またはアネキシンV陽性血管の割合を算出した。
1.9D2抗体およびアネキシンVのリン脂質特異性
9D2抗体は、ELISAにおいて、特異的に陰イオン性リン脂質(PS、PA、CL、PI、PG)を認識して、中性のリン脂質(PE、PCおよびSM)とは有意な反応性を有さなかった(図2A;表8)。ELISAにおけるリン脂質に対する9D2の結合の強度の順序は、PA>PS=CL>PG=PIであった。結合は抗原特異的であった。なぜなら無関係の特異性であるいくつかのコントロールラットIgMでは結合は観察されなかったからである。ELISAプレートに吸着された任意の陰イオン性リン脂質に対する9D2の結合は、任意の陰イオン性リン脂質から調製されたリポソームによってブロックされたが、任意の中性のリン脂質から調製されたリポソームによってはブロックされなかった。
9D2抗体およびアネキシンVがPSに対する結合について競合するか否かを試験するために、PSコーティングしたプレート上でビオチン化タンパク質を用いて、交差ブロック研究を行なった。ビオチン化9D2抗体およびアネキシンVの結合を、それぞれ10倍モル過剰の未改変9D2およびアネキシンVによってブロックした(表9)。しかし、未改変のアネキシンVは、ビオチン化9D2がPSプレートに対して結合する能力に影響しなかった。同様に、未改変の9D2抗体の添加は、ビオチン化アネキシンVがPSプレートに対して結合する能力に影響しなかった(表9)。
b競合物の非存在下でのビオチン化試薬の反応性を100%とした。三連の決定の平均値を示す。SDは、平均値の10%未満であった。
3.細胞表面上に外面化された陰イオン性リン脂質に対する結合
細胞表面に対する9D2抗体およびアネキシンVの結合を、マウスbEnd.3内皮細胞またはウシABAE細胞を用いて試験した。9D2もアネキシンVも、静止条件下でいずれの細胞タイプの非透過性単層にも結合しなかった。このことは、原形質膜の陰イオン性リン脂質のほとんどが細胞質ドメインに対して正常に隔離されることを示す。対照的に、内皮細胞の90〜100%においてアポトーシスを生じる条件下で細胞をTNFαおよびアクチノマイシンDとともにプレインキュベートした場合、強力な染色が観察された。
9D2またはアネキシンVが凍結組織の切片に直接加えられる、直接免疫組織化学技術では、原形質膜の内部小葉構造および外部小葉構造の上の陰イオン性リン脂質の間は識別されない。外側に位置するリン脂質を検出するために、9D2およびアネキシンVを腫瘍保有マウスに静脈内注射して、腫瘍血管に対する局在化を直接免疫組織化学によって決定した。
9D2抗体およびアネキシンVは、この研究に含まれた5つ全ての腫瘍において腫瘍血管に局在した(図4;表10)。腫瘍は以下であった:SCIDマウスの乳腺脂肪パッドにおいて正所性に増殖しているヒトMDA−MB−231乳ガン;皮下で増殖しているヒトL540ホジキン腫瘍;皮下で増殖しているヒトNCI−H358 NSCLC;皮下で増殖しているマウスB16黒色腫、および皮下で増殖しているマウスMeth A線維肉腫。
bアネキシンVの局在化は、ビオチン化アネキシンV注射と、それに続くストレプトアビジン−ペルオキシダーゼ結合体を用いる凍結切片上での検出によって決定された。
c非抗原特異的尿細管染色は、9D2およびコントロール抗体レシピエントの両方で可視であった。
正所性MDA−MB−231乳房腫瘍を保有するマウスにビオチン化9D2抗体、ビオチン化コントロールIgMまたはビオチン化アネキシンVを静脈内注射した二重染色研究を行なった。1時間後、マウスを屠殺して、その腫瘍を取り出して、凍結切片を切断した。次いで腫瘍切片をCy3結合体化ストレプトアビジンを用いて染色して、ビオチン化タンパク質を検出し、FITC結合体化MECA32を用いて染色して血管内皮細胞を検出した。この検出方法では赤および緑によってビオチン化タンパク質および血管内皮細胞を標識した。ビオチン化タンパク質が内皮に結合した場合、集束型画像(converged image)は黄色になる。
腫瘍環境における陰イオン性リン脂質膜転位
腫瘍血管内皮細胞に固有のインビボ表面マーカーとしてのアミノリン脂質および陰イオン性リン脂質の発見によって、このような分子の転位および外膜発現に対する腫瘍の微小環境の効果をさらに検討するように本発明者らは促された。本実施例は、腫瘍内の条件を模倣する特定の条件に対して細胞内皮をインビトロで暴露することによって、インタクトな生存可能な細胞において、初期に観察されたアミノリン脂質および陰イオン性リン脂質表面発現が再現されることを示す。
1.アネキシンVのヨウ素化
組み換えヒトアネキシンVは、ET12a−Panionic リン脂質1プラスミド(University of Washington,SeattleのJ.Tait博士から入手)で形質転換したE.coliから精製した。タンパク質の純度およびPSに対する結合を、それぞれSDS−PAGEおよびPSコーティングされたプラスチックで確認した。ウサギポリクローナル、アフィニティー精製抗アネキシンV抗体を用いてPSに結合したアネキシンVを検出した。アネキシンVは、Bocci(1964)によって記載されたとおりクロラミンTを用いて125Iで放射性標識した。比活性は、Brandfordアッセイ(1976)で測定した場合、タンパク質1μgあたり約1×106cpmであった。
表11に挙げた濃度のサイトカインまたは増殖因子を用いて内皮細胞を処置した。全ての試薬を10%の血清を含む培地中で希釈して、細胞とともに37℃で24時間インキュベートした。
上記の試薬を用いた処置後に、処置した細胞およびコントロール細胞を、7.1ピコモルの125I標識アネキシンV(200μl/ウェル)を含有する結合緩衝液とともにインキュベートした。室温での2時間のインキュベーション後、細胞を徹底的に洗浄して、0.5MのNaOHに溶解した。0.5mlの全体容積をプラスチックチューブに移して、ガンマカウンターでカウントした。5mM EDTAの存在下で非特異的結合を決定して、実験値から差し引いた。その結果を、1×106個の細胞について正規化した、細胞結合したアネキシンVの正味のピコモルとして表現した。
HUVEC細胞および腫瘍細胞を8ウェルチャンバースライド上で約70%コンフルエンスまで増殖させた。PS曝露を誘導するために、細胞を無血清培地中でH2O2(200μM)で37℃にて1時間処理した。細胞をDPBSで洗浄し、無血清培地中に希釈した2μg/mlの3G4抗体とともに室温で1時間インキュベートした。DPBSで穏やかに洗浄したのち、細胞をPBS中の4%(v/v)パラホルムアルデヒドで15分間固定した。
1.H2O2による誘導
マウスbEnd.3内皮細胞を、50,000細胞/ウェルの初期密度で播種した。24時間後に細胞を、漸増濃度(10μM〜500μM)のH2O2とともに37℃で1時間インキュベートするか、または未処置のままにした。インキュベーションの終わりに細胞を0.2%ゼラチン含有PBSで3回洗浄して、0.25%グルタルアルデヒドで固定した。同一のウェルを抗PS IgMで染色するか、またはトリプシン処理してトリパンブルー排除試験によって生存度について評価した。抗PS染色のために、2%ゼラチンを用いた10分間のブロッキング後に、細胞を2μg/mlの抗PS抗体とともにインキュベートし、続いて抗マウスIgM−HRP結合体を用いて検出した。
トロンビンはまた、PS発現を増大するが、H2O2と同じ程度までではないことが観察された。このデータはまた、本発明者らによって開発されたPS発現の腫瘍誘導モデルの欠くことのできない部分である:正常組織におけるトロンビン誘導性PS表面発現はまた、さらなる凝固物である。なぜなら、PS発現は凝固開始複合体のアセンブリを協調させるからである。
インビトロにおけるマウスbEnd.3またはウシABAE細胞を、多くの腫瘍の微小環境に存在する(Lichtenbeldら、1996;Harrisら、1996)種々の濃度の因子および条件、例えば、低酸素/再酸素化、トロンビン、酸性度、炎症性サイトカインおよび過酸化水素を用いて24時間処置した(表11)。
ABAEおよびbEnd.3細胞をIL−1αまたはTNFαの存在下で低酸素/再酸素化に供した場合、強化されたPSおよび陰イオン性リン脂質露出が観察された。サイトカインの非存在下で、低酸素/再酸素化は、ABAE細胞によるPS露出を、アポトーシス濃度のアクチノマイシンDおよびTNFαで処置した細胞について最大レベルの15%〜17.5%まで増大した。毒性未満の濃度のIL−1αまたはTNFαの存在下で、低酸素/再酸素化は、陰イオン性リン脂質露出をそれぞれ最大の26%および33%まで増大した(図5;表11)。低酸素/再酸素化の非存在下でのサイトカインの効果との比較によって、サイトカインおよび低酸素/再酸素化の組み合わせがPS露出に対して相加作用よりも大きい作用であったことが示される。同様の効果がbEnd.3細胞上で観察された。
H2O2処理した、および未処理のHUVECおよびMDA−MB−435細胞に対する3G4抗体の結合を、フローサイトメトリーによって分析した(図5A)。H2O2処理条件は、上記のように確立し、原形質膜の外面における陰イオン性リン脂質の曝露を誘導した。
アミノホスホリピドが腫瘍脈管構造の安定なマーカーであるという驚くべき知見はまた、抗体−治療剤構築物がガン治療に使用され得ることを意味する。標的剤としての抗体の使用に加えて、本発明者らは、アネキシン、および他のアミノホスホリピド結合タンパク質もまた、治療剤を腫瘍脈管構造に特異的に送達するのに使用され得ると結論付けた。以下のデータは、アネキシン−TF構築物のインビボ投与から生じる抗腫瘍効果を示す。
アネキシンV−tTF結合体を調製し、そして固形腫瘍を有するnu/nuマウスに投与した。この腫瘍は、少なくとも約1.2cm3の腫瘍を形成したヒトHT29結腸直腸腺ガン細胞から形成された。アネキシンV−tTFコアグリガンド(10μg)を静脈内投与し、そして24時間循環させた。生理食塩水で処置したマウスを、コントロールマウスとして別々に維持した。1日の処置期間の後、このマウスを屠殺し、そして放血させ、そしてこの腫瘍および主な器官を分析のために採取した。
アネキシンV−tTF結合体が、HT29腫瘍保有マウスにおいて特定の腫瘍血管凝固を誘導することを見出した。アネキシンv−tTF結合体処置した動物における腫瘍血管の約55%が、1回の注射後に血栓形成した。対照的に、コントロール動物の腫瘍脈管構造において血栓、最小限に現れた。
3SB抗PS抗体の抗腫瘍効果
本実施例は、同系および異種の腫瘍モデルを用いる抗PS抗体の抗腫瘍効果を示す。本研究で用いる3SB抗体は、PS(およびPA)に結合するが、本質的にPEとの反応性は欠く。この抗PS抗体は、血栓および腫瘍壊死を伴った腫瘍血管傷害を生じた。
陰イオン性リン脂質に対する抗体(9D2)の抗腫瘍効果
本実施例は、インビボにおける抗腫瘍研究において、PSおよび他の陰イオン性リン脂質に結合する、9D2抗体の効果を実証する。
2リンパ節における転移
さらなる研究では、9D2抗体を、L540腫瘍を有するマウスに1週あたり3回100μgの用量で腹腔内注射した。週に2回ノギスで腫瘍サイズを測定した。コントロール群に比較した抗腫瘍効果を図7に示す。カッコの数は、1群あたりのマウスの総数あたりの退行した腫瘍を有するマウスの数を示す。
抗PS抗体3G4の抗腫瘍効果
本実施例は、同系および異種の腫瘍モデルにおいて抗PS抗体3G4を用いて、さらなる抗腫瘍効果を実証する。本研究で用いる3G4抗体は、PSおよび他の陰イオン性リン脂質に結合するIgG抗体である(実施例IV)。
同系および異種の腫瘍モデルにおいて3G4の効果を検討した。動物腫瘍処置研究のための一般的プロトコールは以下のとおり行なう。特に相違を示さない限り、これは本出願の研究全体にわたって用いたプロトコールである。
キシラジン(20mg/ml);1ml アセプロマジン(10mg/ml);11ml滅菌水である。投与量は、30分間にわたりIP経路を介して体重20〜30グラムあたり0.1mlである。
同系モデルについては、Meth Aマウス線維肉腫腫瘍細胞を用いた。異種モデルの1つにおいて、ヒトMDA−MB−231乳ガン細胞を、乳腺脂肪パッドに播種した。別の異種モデルでは、細胞を注射することおよび腫瘍を処置の前に500mm3を超えるサイズまで増殖させることによって、大きいヒトホジキンリンパ腫L540異種移植片を樹立した。腫瘍保有マウス(1群あたり10匹)に、コントロールに対して、100μgの3G4抗PS抗体(IgG)を腹腔内注射した。処置は週に3回繰り返した。動物を腫瘍測定のために週に2回モニターした。
CMVに対する抗PS抗体の抗ウイルス効果
驚くべきことに、腫瘍脈管構造からウイルス感染へ分野を切り替えて、本発明者らは次に、アミノリン脂質および陰イオン性リン脂質に対する抗体がまた抗ウイルス効果を発揮する可能性が高いと判断した。本実施例は実際に、サイトメガロ(CMV)感染の処置において3G4抗体を最初に用いて、これが真実であることを示す。
1.インビトロにおけるCMV感染細胞の処置
6ウェルプレート中のヒト二倍体包皮線維芽細胞(HHF−R2)のコンフルエントな単層を、前に記載されたように、緑色蛍光タンパク質(GFP)を発現するヒトCMV AD169を用いて、MOI=0.01で感染させた(Bresnahanら、1996)。要するに、この細胞を、1ウェルあたり1mlの総容積でウイルスとともに、37℃で90分間インキュベートした。感染の間、プレートを30分ごとに穏やかにロッキングさせた。感染後、細胞上清を取り出してDMEM/10% FBS/pen−strep(1ウェルあたり2ml)を各々のウェルに添加した。
組み換えCMVは、SV40プロモーターの制御下でGFPを発現する。従って、感染した細胞は、蛍光顕微鏡下で緑になる。これらの研究では、抗体で処置したCMV感染細胞は、2日目、3日目および9日目に蛍光顕微鏡下で観察された。
標準的プロトコールと用いてプラークアッセイを行なった。要するに、凍結細胞細胞懸濁液を37℃で急速に融解して、1000rpmで1分間、遠心分離によって砕片を除去した。細胞上清の種々の希釈物を、6ウェルプレート中のHHF−R2細胞のサブコンルフエントな単層に加えて、その細胞を37℃で90分間インキュベートさせた(このプレートを30分ごとに穏やかにロッキングさせた)。感染後に、細胞上清を取り出して、2mlのDMEM/10% FBSと交換した。4日目に、各々のウェルの上清を取り出して、細胞に0.01%の低融点のアガロース/DMEM/10% FBSを重層した。このプレートを感染後、37℃で、全部で14日間インキュベートさせた。14日目に、感染した単層を10%緩衝化ホルマリンで固定して、メチレンブルーで染色して、プラークを可視化した。
1.3G4がCMVのウイルス伝播を阻害する
3G4がCMV感染および複製に対して阻害性効果を有するか否かを検討するために、コンフルエントなヒト線維芽細胞を、低いm.o.iでCMVを添加する前に、3G4で前処置した。これらの研究において用いられるCMVは、緑色蛍光タンパク質(GFP)を発現する。従って、感染された細胞は、蛍光顕微鏡下で観察された場合に緑になる。
低m.o.iでの抗ウイルス効果に必要な3G4の濃度を決定するために、感染した細胞を、異なる濃度の3G4およびコントロール抗体GV39Gで処置した。図10に示されるように、3G4を100μg/mlおよび50μg/mlで用いて細胞間の伝播の完全な阻害が観察される。細胞を25、12.5および6.25μg/mlの3G4で処置した場合、GFP陽性CMV感染細胞の数が増大される。3G4は、これらのさらに低濃度の一次感染細胞からのウイルス伝播を完全には妨げないが、依然として意味のある抗ウイルス効果を有する。なぜなら、GV39G処置したコントロールウェルに比較して3G4処置したウェルでは、みられるGFP陽性CMV感染細胞はわずかであるからである(図10)。
抗体処置後のウイルスロードを決定するプラークアッセイを行なうことによって3G4の抗ウイルス効果を定量した。このコントロールは、未処置の細胞、GV39G抗体、およびC44抗体を用いるさらなる抗体コントロール、コルヒチンに対するマウスIgG2aアイソタイプ抗体を含んだ。
3という高m.o.i.で感染された線維芽細胞の3G4処置によってまた、ウイルス力価の劇的な低下が生じる。100μg/mlでは、3G4での処置は、コントロールのGV39G処置細胞に比較してウイルス力価の5log10の低下が生じた(図11B)。50μg/mlでは、3G4が、GV39Gに比較して3logまでウイルス複製を阻害した(図11B)。
CMV複製サイクルのどの段階が3G4によってブロックされるかを決定するために、タイミング的な添加研究を行なった。このために、感染後種々の時点で高m.o.i.で感染した線維芽細胞に3G4を加えた。ウイルスロード(細胞および上清の両方において)を標準的なプラークアッセイを用いて定量した。
RSVに対する抗PS抗体の抗ウイルス効果
実施例XIIに示されるCMVに対する劇的な抗ウイルス効果に加えて、本実施例は、呼吸器合胞体ウイルス(RSV)複製の阻害における3つの異なる抗PS抗体の使用を実証する。
1.インビトロにおけるRSV感染細胞の処置
A549細胞を、3つのCoaster 12ウェル組織培養プレートにおいて100%コンフルエンスまで増殖させた。200μLの最小基本イーグル培地を全てのウェルに添加した。抗リン脂質抗体(Ab)を各々のプレートの9つのウェルに添加(100μLに100μg)して、30分後に最初の9ウェルのうちの6つにおける細胞をRSV長型株を含む100μLの容積を1のMOIで用いて感染させた。3つの残りのウェルを感染されていない、抗体処置ウェルのまま残した。抗体を有さない3つの他のウェルを、RSVを上記と同じMOIで用いて感染させた。
プラークアッセイを前に記載の通り行なった(Kischら、1963;Grahamら、1988)。要するに、凍結細胞の細胞上清を37℃で急速融解させた。未希釈の細胞上清から3段の10倍希釈を行なった:10−1、10−2および10−3。各々の希釈の100μLに加えて未希釈のサンプルを、全て3連の80%コンフルエントHep−2細胞株プレートに接種した。プレートを5%CO2,40℃インキュベーターに5日間入れた。5日目に、このプレートを発色させて、ヘマトキシリンおよびエオシンで染色して、各々のウェルにおけるプラークを明らかにした。このプラークを、解剖顕微鏡を用いてカウントして、pfu(プラーク形成単位)/mLにおけるRSVウイルスロードを算出した。
図12に示されるとおり、3SBまたは1B9のいずれかを用いるRSV感染細胞の処置によって、ウイルス複製の対数低下が生じた。感染した細胞を3G4で処置した場合、抗ウイルス効果が、さらに証明された。3G4を用いた処置によって、ウイルス力価において2log10の低下が得られた(図12)。阻害は、CMVで見られるよりも低く、これは3G4の濃度が低かった(25〜50μg/ml)ためである可能性が最も高い。
短鎖抗PS抗体
抗腫瘍因子単独として、腫瘍に対して結合した治療因子を送達するための標的化因子として、および抗ウイルス剤としての使用を含む、本明細書に記載される抗PS抗体の多くの使用を考慮すれば、本実施例は、短鎖(scFv)抗PS抗体を生成するのに適切な技術を記載しており、すなわち、ここではVHドメインおよびVLドメインがペプチドリンカーによって一般に結合される短鎖ポリペプチド鎖に存在する。
細菌ライブラリーの二次ストック(約1×1010クローン)を100μg/mlアンピシリンおよび1%グルコースを含む100ml 2×TYに接種した。これをODが600nmで0.5になるまで、37℃で振盪しながら増殖させた。
20μmolのホスファチジルイノシトールおよび20μmolのビオチン化ホスファチジルセリンを10mlのヘキサンに溶解した。この溶液を、回転エバポレーターを用いてフラスコの表面上に薄層に乾燥させた。2ml PBSを添加して、槽を4℃で30分間超音波処理した。
プレートからの個々のHB2151コロニー(4回の選択後)を、96ウェルプレート中に100μg/mlのアンピシリンおよび1%グルコースを含有する500μl 2×TY中に接種して、37℃で一晩振盪しながら(300rm.)増殖させた。このプレートからの5μlを、1ウェルあたり100μg/mlのアンピシリンを含有する500μl 2×TYを含有する第二の96ウェルプレートに移して、振盪しながら37℃で3時間増殖させた(OD600=0.9)。
4回のパンニング後、以下のクローンはPSプレート上の見込みのあるELISAシグナルを生じて、正確なサイズの挿入物を有する:3E5、3A2、G5、C8、E4および4D5。これらをサブクローニングさせたが、ここではE4は、5つの陽性のサブクローンを有して、4D5は、5つの陽性のサブクローンを有した(表14)。
PSプレートでのELISA
PE結合ペプチド誘導体の合成
本実施例は、腫瘍およびウイルス疾患を処置するのにおける使用のための例示的なPE結合ペプチド誘導体および結合体の設計および合成に関する。例示的なデュラマイシン誘導体の構造は、図13A〜図13Oのパネルに示されており、これは以下の説明に合致する。
0.387mlの0.1M NaHCO3を含有する水に溶解された0.5mg(0.25μmol)のデュラマイシンを0.113mg(0.25μmol)のNHS−LC−ビオチン(Sigma)に添加した。この反応混合物を室温で1時間、次いで4℃で一晩インキュベートさせた。サンプルをシリカカラムにロードして、0.1%トリフルオロ酢酸(TFA)を用いて洗浄して、0.1%TFAおよび70%CH3CNを用いて溶出させた。この溶出物を収集して、遠心分離によって濃縮した。総収量は0.5mgであった(図13A)。
0.286mlの0.1M NaHCO3を含有する水に溶解された0.5mg(0.25μmol)のデュラマイシンを0.034mg(0.25μmol)の2−イミノチオラン塩酸塩(2−IT)に添加した。この混合物を室温で1時間インキュベートさせた。0.13mg(0.26μmol)のヨードアセチル−LC−ビオチン(Pierce)を添加して、その反応物を室温で1時間、そして4℃で一晩インキュベートさせた。サンプルをシリカカラムにロードして、0.1% TFAを用いて洗浄して、0.1%TFAおよび70%CH3CNを用いて溶出させた。この溶出物を収集して、遠心分離によって濃縮した。総収量は0.5mgであった(図13B)。
0.5mlの0.1M NaHCO3を含有する水に1.9mg(0.94μmol)のデュラマイシンを溶解した。ここに、0.4mg(0.88μmol)のNHS−LC−ビオチン(Sigma)を含有する200μlジメチルホルムアミド(DMF)を添加した。この混合物を室温で4時間インキュベートさせた。10mg(0.17μmol)ニュートラビジン(NA)を含有する1mlをこの反応混合物に添加して、これを室温で2時間、次いで4℃で一晩インキュベートさせた。次いでこの反応混合物を、PBS緩衝液を含むG−25カラム(容積50ml)上にロードさせた。この画分を収集して、SDS PAGE(phastゲル)によって解析した。タンパク質含有画分(7〜16)を一緒にプールして、0.22μmフィルターを通した濾過によって滅菌して、280nmでの吸光度を測定することによって濃度を決定した。総収量は5.1mgであった。
0.61mgの(DLB)4NAを含有するPBS緩衝液を、0.005mgN−ヒドロキシスクシンイミジルフルオレセイン(NHS−フルオレセイン)(Sigma)を含有するDMFに添加した。この混合物を室温で1時間インキュベートした。次いで、この反応物をPD10カラム(10ml)に分画した。(DLB)4NA−Fを、タンパク質含有画分(3および4)に溶出させて、これを一緒にプールして、0.22μmフィルターを通した濾過によって滅菌した。総収量は0.5mgであった(図13D)。
ヒトIgG(HIgG)を最初に以下のとおり精製した:1.3ml HIgG(これは、1mM EDTA、pH9を有するホウ酸緩衝液に100mg/ml HIgG、22.5mg/mlグリシンおよび3mg/mlアルブミンを含んだ)を、FPLC(S200、250ml)カラムに加えた。この画分を収集して、phastゲル上でSDS PAGEによって解析した。単量体のIgGを含む画分(21〜32)を一緒にプールして、0.22μmフィルターを通した濾過によって滅菌した。280nmでの吸光度によって決定される総収量は111mgであった。
1mg(0.7ml)の(DIM)nHIgGを、5μlの、NHS−フルオレセインを含有するDMFに添加した。反応混合物を室温で1時間インキュベートして、PD−10カラムで脱塩した。タンパク質含有画分(2〜3)をプールして、0.22μmフィルターを通した濾過によって滅菌した。総収量は0.9mgであった(図13F)。
[(DIM)nHIgG]2のビオチン化誘導体を合成するために、0.66mg(1ml)の[(DIM)nHIgG]2を8μlの、1mg/mlのNHS−LC−ビオチン(Pierce)を含有するDMFに添加した。この混合物を室温で1時間インキュベートした。次いで、この反応混合物をPD−10カラムで脱塩した。タンパク質含有画分(3および4)をプールして、0.22μmフィルターを通した濾過によって滅菌した。最終の収量は0.46mgであった。
2mg(0.99μモル)のデュラマイシンを、0.5ml 0.1M NaHCO3に溶解して、0.136mg(0.99μmol)の2−ITに添加した。この反応混合物を室温で1時間インキュベートした。その後に、0.483mg(0.95μmol)のヨードアセチル−LC−ビオチン(Pierce)を添加してその反応混合物を室温で1時間インキュベートした。10mg(0.17μmol)のニュートラビジンを含む1mlのH2Oを添加して、4℃で一晩インキュベートした。反応混合物をFPLCによって分画した。3つの異なるピークを収集してプールした:[(DIB)4NA]3(画分17〜23);[(DIB)4NA]2(画分24〜33)および(DIB)4NA(画分35〜48)。全てのサンプルを0.22μmのフィルターを通した濾過によって滅菌した。得られた最終収量は以下であった:0.87mgの[(DIB)4NA]3;1.25mgの[(DIB)4NA]2;および1.83mgの(DIB)4NA(図13H)。
J.023mg(0.3μmol)の(DIB)4NAを、0.9μgのNHS−LC−ビオチン(Pierce)に添加した。この反応物を室温で1時間インキュベートして、次いでPD10カラムで脱塩した。総収量は0.04mgであった(図13I)。
0.5ml 0.1M NaHCO3を含有する水に溶解した5mg(2.5μmol)のデュラマイシンを、0.319mg(2.6μmol)の1,3プロパンスルトンに添加した。この混合物を4℃で一晩インキュベートした。このサンプルをシリカカラムにロードして、0.1%TFAで洗浄して、0.1% TFAおよび70% CH3CNを用いて溶出させた。この溶出物を収集して、減圧下での遠心分離によって濃縮した。総収量は5mgであった(図13J)。
0.3ml 0.1M NaHCO3を含有する水に溶解した1mg(0.497μmol)のデュラマイシンを、0.072mg(0.523μmol)の2−ITに添加した。この反応混合物を室温で1時間インキュベートした。0.125mg(0.49μmol)のSBF−塩化物(Pierce)を添加した。この反応混合物を室温で1時間および4℃で一晩インキュベートした。このペプチドをシリカカラムで精製した。この溶出物を収集して、減圧下での遠心分離によって濃縮した。総収量は1mgであった(図13K)。
0.4ml 0.1M NaHCO3を含有する水に溶解した1mg(0.497μmol)のデュラマイシンを、0.109mg(0.592μmol)の2−スルホ安息香酸環状無水物に添加した。この反応物を室温で1時間、4℃で一晩インキュベートした。このペプチドをシリカカラム上で精製した。この溶出物を収集して、減圧下での遠心分離によって濃縮した。総収量は1mgであった(図13L)。
0.5mlの、0.1M NaHCO3を含有する水に溶解した0.25mg(0.124μmol)のデュラマイシンを、0.017mg(0.124μmol)の2−ITに添加した。この反応混合物を室温で1時間インキュベートした。次いで、この混合物を0.049mg(0.124μmol)のEllman試薬に添加した。この混合物を室温で2時間および4℃で一晩インキュベートした。250μlの1mg/mlの4−アミノ−5−ヒドロキシ−2,7−ナフタレンジスルホン酸一ナトリウム塩水和物を100μlの1mg/ml 2−ITに添加した。この反応物を室温で1時間インキュベートした。50μlの反応混合物を前の反応物に添加して、室温で1時間インキュベートした。このペプチドをシリカカラムで精製した。溶出物を収集して、減圧下での遠心分離によって濃縮した(図13M)。
0.5ml 0.1M NaHCO3を含有する水に溶解した5mg(2.5μmol)のデュラマイシンを、0.356mg(2.6μmol)の1,3−ブタンスルトンに添加した。この混合物を4℃で一晩インキュベートした。このサンプルをシリカカラムにロードして、0.1%TFAで洗浄して、0.1% TFAおよび70% CH3CNで溶出した。この溶出物を収集して、減圧下での遠心分離によって濃縮した。総収量は5mgであった(図13N)。
0.5ml 0.1M NaHCO3を含有する水に溶解した0.25mg(0.124μmol)のデュラマイシンを、0.017mg(0.124μmol)の2−ITに添加した。この反応混合物を室温で1時間インキュベートした。次いで、この混合物を0.049mg(0.124μmol)のEllman試薬に添加した。この混合物を室温で2時間および4℃で一晩インキュベートした。このペプチドをシリカカラムで精製した。この溶出物を収集して、減圧下での遠心分離によって濃縮した(図13O)。
デュラマイシン誘導体はPEに特異的に結合する
本実施例は、実施例XVにおいて合成されたデュラマイシン誘導体がPSに特異的であり、従って細胞不浸透性の標的化または抗ウイルス剤に対して連結すること、ならびに腫瘍およびウイルス疾患の処置における使用によって設計されたとおり用いることができることが示される。
PE結合ペプチド誘導体の抗ウイルス効果
実施例XIIおよび実施例XIIIに示されるとおり、抗PS抗体によって媒介される抗ウイルス効果に加えて、本実施例は、他の共通のアミノリン脂質PEに特異的に結合するペプチド誘導体の抗ウイルス効果を実証する。
1.インビトロにおけるCMV感染細胞の処置
6ウェルプレートにおけるヒト二倍体包皮線維芽細胞(HHF−R2)のコンフルエント単層を、実施例XIIに記載されるように、MOI=0.01で、緑色蛍光タンパク質(GFP)を発現するヒトCMV AD169を用いて感染させた(Bresnahanら、1996)。この細胞を1ウェルあたりの1.5mlの総容積でウイルスとともに37℃で90分間インキュベートした。感染の間、30分ごとにこのプレートを穏やかにロッキングさせた。感染の後に、細胞上清取り出して、DMEM/10% FBS/pen−strep(1ウェルあたり2ml)を各々のウェルに添加した。
実施例XIIに記載のとおり、組み換えCMVは、SV40プロモーターの制御下でGFPを発現する。従って、感染した細胞は、蛍光顕微鏡下で緑にみえる。これらの研究では、デュラマイシン誘導体で処置したCMV感染した細胞は、4日および6日に蛍光顕微鏡下で観察された。
4日目に、未処理のウェルならびに(DLB)4NAおよび(DIM)nHIgGで処理したウェルに単一の感染したGFP陽性緑色細胞がある(図15、左パネル)。従って、これらのデュラマイシン誘導体でのHHF−R2細胞の処置は、細胞へのウイルスの侵入を阻害しないようである。デュラマイシン誘導体DS−1、DS−2およびDS−3が細胞へのウイルス進入を阻害するといういくつかの前段階的な証拠がある。
3G4抗体の利点
実施例IVに記載のように、本発明者らの固有のプロトコールによって開発された3G4抗体は、文献の抗PS抗体を上回る多くの利点を有するが、これには卓越した抗PS抗体である3SBが挙げられる(Roteら、(1993)。本実施例は、これらの特有の利点を記載する。
3G4はIgG抗体であり、一方3SBはIgMである。IgGクラスの抗体は、高い親和性、インビボにおける低いクリアランス速度、ならびに精製、改変および取り扱いの容易さを含む、IgMを超える多くの利点を有する。IgM抗体である3SBのPS結合の、3G4および別のIgG抗体との比較を図19Aおよび図19Bに示す。
3G4は、PSおよび他の陰イオン性リン脂質を外面化する、活性化された、分裂中の、傷害されたアポトーシスおよび悪性の細胞に結合する。3G4抗体は、インビトロにおいて内皮細胞の増殖を阻害して、静止期細胞と対照的に、分裂している内皮細胞の顕著な選択性阻害を示す。
3G4は、インビボにおいて腫瘍血管内皮細胞の表面に結合する。種々の腫瘍を保有するマウスに静脈内注射した場合、3G4は腫瘍に特異的かつ一貫して局在するが、正常な器官には局在しない。染色は腫瘍血管内皮(図22)、壊死領域および個々の悪性細胞で観察された。腫瘍には、腫瘍内皮および腫瘍細胞の両方の同時の標的を可能にする、3G4についてのマルチ結合部位が存在する。
3G4抗体は、文献で記載される抗リン脂質抗体とは異なる。代表的には、抗リン脂質抗体は、凝固カスケードを妨害する病原性抗体とみなされる。それらは、インビトロにおいて凝固反応を阻害してインビボで血栓を生じる。対照的に3G4、9D2および同様の抗体は、病原性効果のない治療抗体である。
血清の非存在下と同じく血清の存在下で強力にPSコーティングプレートに結合する抗体を同定するためのスクリーニングを用いて、好ましくは所望の抗体が選択されるべきであるという本発明者らの認識から、3G4、9D2および同様の抗体の基部の重要な局面が得られる。この新規な展開によってPSタンパク質および血清タンパク質の複合体を認識する抗体を同定して排除する能力が得られる。なぜならこのような複合体は、抗リン脂質症候群および関連の病態の原因であるかまたはその重要な要因であると考えられるからである。
3G4はマウスにおいて毒性を有さないか毒性が低いという最初の証拠は、3G4が毒性の証拠なしにマウスにおいてハイブリドーマとして増殖するという知見によってもたらされる。また、1mgの精製された3G4が腹腔内に注射された場合、毒性は観察されなかった。
ヒト化3G4抗体(以下の実施例XIXを参照のこと)もまた、安全性研究においてサルに投与され、有意な副作用は観察されていない。ヒト化3G4抗体を、100mg/kgまで単一のボーラスでカニクイザルにIV投与した。これは計算される治療用量(1mg/kg)の100倍である。
3G4抗体はまた、有意な抗ウイルス効果を発揮する。実施例XIIIに示されるとおり、3G4を用いたRSV感染細胞の処置は、3SBを用いて観察される効果よりも優れていた。従って、これらの結果によって、文献における目立った抗PS抗体である3SBを上回る3G4抗体の別の利点が強調される(Roteら(1993))。
3G4抗体、CDR配列およびキメラ
従って3G4抗体は、血管新生阻害、抗腫瘍血管、および抗ウイルス剤の合わせた特性を保有する。細胞分裂、血管新生、腫瘍増殖およびウイルス性感染に対する3G4の阻害活性は、明白な毒性の欠失とともに考慮すれば、血管新生障害、ガン、糖尿病およびウイルス性感染の処置を含む、この抗体についての広範な治療指標を示す。
抗体の可変領域のもとの配列を、3G4抗体を生成するハイブリドーマからRACEによって得て、そしてその配列を確証した。3G4抗体CDR1−3の重鎖(Vh)の可変領域の核酸およびアミノ酸の配列は、それぞれ配列番号1および配列番号2によって提示される。
マウス相補性決定領域およびヒト定常領域を含むキメラ構築物(ch3G4)を生成して、もとのマウス抗体と本質的に同じように挙動することが示された。
インビボでは、ch3G4は、腫瘍血管内皮細胞に局在して、抗腫瘍効果を発揮する。マウスにおいて増殖しているMDA−MB−435ヒト乳ガン細胞におけるch3G4の抗腫瘍効果は、実施例XIに記載されて、図8Gに示される。キメラ抗体を用いるMDA−MB−435腫瘍を有するマウスの処置は、コントロールとは対照的に腫瘍増殖を効率的に遅延させた。
マウス3G4抗体のヒトキメラ(ch3G4)は、ヒトIgG1アイソタイプ(hIgG1)である。ch3G4マウスIgGホモログは、マウスIgG2aアイソタイプ(mIgG2a)を必要とする。この構築物を作製し、試験し、親抗体と本質的に同じようにふるまうことを示した。
ドセタキセルとの併用療法における3G4抗体
本実施例は、3G4抗体および化学療薬ドセタキセルを用いる腫瘍処置のための併用療法に関する。これらの薬剤は、腫瘍脈管構造内皮細胞および腫瘍細胞区画を攻撃するように設計されて、毒性の低い相乗的な処置をもたらす。この結果によって、この併用療法は、処置効率を実際に有意に増強したことが示された。
インビトロでの腫瘍細胞に対する阻害効果について3G4抗体を試験した。腫瘍細胞に対する直接の阻害効果は観察されなかった。従って、3G4抗体の抗腫瘍効果は、Fcドメイン媒介性の免疫エフェクター機能の増強、例えば、抗体媒介性食作用、ADCC、CDCおよびサイトカイン産生の刺激、またはこれらの機構の組み合わせの増強を含む可能性が高い。
コントロールされた研究からの結果は、ドセタキセルが腫瘍脈管におけるPS曝露を増加させることを示す。そのような研究において、マウスのドセタキセル処置は、陰イオン性リン脂質を曝露する腫瘍脈管の割合を35%〜60%増大させた。正常組織の脈管では、ドセタキセルでの全身処置後でさえ、PSの誘導は観察されなかった。
従って、本発明者らは、臨床濃度未満のドセタキセルを用いた内皮細胞および腫瘍細胞の処置が3G4結合を有意に増大することを示した。それらによってまた、3G4抗体が、表面上にPSが露出されている腫瘍細胞のマクロファージ媒介性食作用を促進することが示された。従って、ドセタキセルによって媒介される3G4結合の増大は腫瘍細胞の食作用および3G4抗体のFcドメインによって媒介される他の抗腫瘍効果、例えば、NK細胞の溶解活性を増強して、さらに有効なADCCをもたらす。他の研究によってまた、ドセタキセルを用いた乳ガン患者の処置は血清のIFN−γ、IL−2、IL−6およびGM−CSFサイトカインのレベルの増大、ならびにNKおよびLAK細胞の活性の増強をもたらすことが示されている(Tsavarisら、2002)。
3G4抗体はまた、動物において薬物耐性の乳房腫瘍に対するシスプラチンの活性を増強する。マウスに薬物耐性の乳房腫瘍を接種し、20日目の後に処置した。処置群は、シスプラチン単独、3G4抗体単独、アイソタイプがマッチするコントロール抗体(BBG3)または、3G4抗体と併用したシスプラチンであった。
膵臓癌に対して改善された処置の緊急の必要性が存在する。ヒトにおいて、膵臓癌を有する患者についての全体的な長期生存率は、4%未満である。膵臓癌患者のうちの約70〜80%が、肝臓への転移に起因して治療に失敗し、現在、転移性疾患に対する有効な治療は存在しない。
肺癌を有するマウスの研究において、10Gyの焦点照射は、二重標識研究で示されるように、腫瘍血管内皮におけるPSの曝露を誘導した。処置段階において、照射および3G4抗体単独は、抗腫瘍効果を示した。この場合も、処置モダリティ(すなわち、照射および3G4抗体)の組み合わせは、最も高い抗腫瘍効果を示した。
3G4に加えてドセタキセルで処置したマウス由来の腫瘍はまた、コントロールの腫瘍に比較して異常な量のリンパ球を含んだ。この現象は腫瘍細胞を崩壊させることによる免疫細胞の代表的な化学誘引に相当しているが、これはまた、免疫エフェクター細胞上のFcγRに対するFc結合を通じて媒介された3G4による免疫系の活性化を反映し得る。
(抗PS抗体は、インビボにおいてCMV感染を処置する)
実施例XIIに示されるインビトロのCMVに対する抗ウイルス効果に従い、本実施例は、CMVウイルスのマウスバージョン、mCMVを感染させたマウスの生存の増大が実証される。
(インビボにおいてPE結合ペプチド誘導体はCMV感染を処置する)
実施例XVIIに示されるCMVに対するインビトロの抗ウイルス効果に加えて、本実施例によって、デュラマイシンビオチン誘導体であるDLBがmCMVで感染されたマウスの生存を向上させたことが実証される。
(抗PS抗体は、ウイルス感染した細胞に結合する)
本実施例によって、ウイルス感染が細胞表面でPS露出を誘導することおよび抗PS抗体がウイルス感染した細胞に結合することが示される。FACS解析において実証される細胞表面に対するキメラ3G4抗体の結合の増大によって示されるように、ワクシニアウイルスに感染した細胞はPS陽性になる。
(Pichindeウイルスに対する抗PS抗体の抗ウイルス効果)
CMVおよびRSVに対する抗ウイルス効果に加えて、本実施例によってさらに、抗PS抗体がインビトロでPichindeウイルス感染を阻害することが示される。Pichindeウイルスは新世界(New World)アレナウイルスであって、ヒトでは非病原性であり、ラッサ熱の動物モデルにおいて用いられる。
(PE結合ペプチド誘導体を用いる腫瘍処置)
デュラマイシン誘導体の抗ウイルス効果に加えて、インビトロおよびインビボの両方で、本実施例は、腫瘍脈管構造に対するデュラマイシン誘導体の局在化および関連の抗腫瘍効果を実証する。
ヒトIgG(HIgG)を実施例XVに記載されるように最初に精製した。精製したHIgGを、SIABリンカーを用いてデュラマイシンに連結して、得られた(D−SIAB)nHIgG結合体を精製した。
腫瘍サイズが500mm3に達したとき、上記と同じMethAマウス腫瘍モデルを用いて、100μg(D−SIAB)nHIgGを含有する100μlPBSを、尾静脈を介して注射した。同じ量のヒトIgGをコントロールとして注射した。4時間後、マウスを屠殺して正常な生理食塩水を用いて5分間、1%パラホルムアルデヒドを用いて10分間灌流させた。腫瘍および他の主な器官を切り取って液体窒素中で凍結させた。OCTに埋め込んだ後、組織を10μmの切片に凍結切片化してシラン処理したスライドにおいた。冷アセトン中での10分間の固定後に、ペルオキシダーゼ標識ヤギ抗ヒトIgGを用いてスライドを染色して、デュラマイシンHuIgGの体内分布を検出した。Meca32およびペルオキシダーゼ標識したヤギ抗ラットIgGを用いて組織の血管構造を検出した。
(デュラマイシン結合体の生体分布および特性)
本実施例は、インビトロにおける細胞不浸透性デュラマイシン誘導体の毒性の欠失、インビボで投与されたデュラマイシン誘導体の体内分布、およびデュラマイシン−抗体結合体がマクロファージによるアポトーシス細胞の食作用を増大する能力を実証する。
本発明のデュラマイシン誘導体および結合体は、親のデュラマイシン分子の非特異的な毒性効果を最小にするように設計する。多くの実施例では、これは細胞不浸透性基に対するデュラマイシンの連結によって達成される(実施例XV)。
ヒト乳ガン細胞株MDA−MB−435を増殖させて、対数期に回収して、DPBSに再懸濁した。約107個の細胞を、6〜8週齢の雌性無胸腺ヌードマウスの乳房の脂肪パッドに注射した。100μgデュラマイシン−ビオチンを含む100μl PBSを尾静脈を介して注射した。4時間後、マウスを屠殺して正常な生理食塩水を用いて5分間、1%パラホルムアルデヒドを用いて10分間灌流させた。心臓、肺、肝臓、腎臓、脳、小腸、胸腺および脾臓を含む主な器官を切り取って液体窒素中で凍結させた。OCTに埋め込んだ後、組織を10μmの切片に凍結切片化してシラン処理したスライドにおいた。冷アセトン中での10分間の固定後に、Cy3標識したストレプトアビジンを用いてスライドを染色してデュラマイシン−ビオチン構築物の体内分布を検出した。Meca32およびFITC標識したヤギ抗ラットIgGを用いて組織の血管構造を検出した。
デュラマイシン−抗体結合体(デュラマイシン−C44、DuC44)がアポトーシス細胞の食作用を増大する能力を次に検討した。
腫瘍治療時におけるマクロファージの浸潤
前述の実験に示すように、アミノリン脂質および陰イオン性リン脂質に対する抗体は癌治療に有効である。例えば、3G4抗体は、腫瘍血管に特異的に局在し、腫瘍血管の破壊をもたらし、かつ、複数のマウスモデルにおいて毒性をもたらすことなく腫瘍増殖を遅らせる(実施例XI;実施例XVIII)。本実施例は、腫瘍を抱える動物に対する3G4治療もまた、単球白血球の腫瘍血管内皮に対する結合をもたらし、マクロファージの腫瘍への浸潤を誘発することを示す。
8−10匹の雌性SCIDマウスから成るグループに、2×107個のL540細胞、または同所的に1×107個のMDA−MB−435またはMDA−MB−231細胞を皮下に注入した。BALB/Cマウスに1×106個のMeth A細胞を皮下に注入した。腫瘍を、平均直径0.8−1cm(L540)、0.6−0.7cm(MDA−MB−435)、0.5−0.7cm(MDA−MB−231)、または<0.1cm(Meth A)となるまで増殖させた。次に、マウスを、100μgの3G4抗体、またはコントロール抗体(BBG3)を週に3回2−3週腹腔に投与して処置した。週に3回、腫瘍サイズおよび体重について動物を監視した。コントロールマウスの腫瘍が直径1.5−2cmに達した時動物を屠殺した。
同所性MDA−MB−435およびMDA−MB−231腫瘍を抱えるマウスの3G4による治療は、単球白血球の、腫瘍血管内皮に対する結合、および腫瘍間隙組織への浸潤をもたらした(図6A、図6B、図6C、および6D)。この図において、赤色染色は内皮を示し、緑色染色はマクロファージを示し、青色染色は核を示す。
F(ab’)2断片は腫瘍治療に有効である
アミノリン脂質および陰イオン性リン脂質に対する抗体、例えば、3G4は、腫瘍治療に有効である(実施例XI;実施例XVIII)。本実施例は、3G4抗体のF(ab’)2断片も、元の3G4抗体と同程度に腫瘍治療に有効であることを示す。
アネキシンV二量体は、モノマーよりも優れて腫瘍血管に局在する
実施例XXVIIおよび実施例XXVIIIは、アミノリン脂質および陰イオン性リン脂質に対する抗体による腫瘍治療におけるマクロファージの役割に関する。これらのデータおよびその他の情報から、本発明人達は、全体発明の内のレセプター本体局面を発展させた。この実施態様を支えるものとして、アネキシンVのホモの二量体を作製したところ、対応するアネキシンVモノマーよりも優れて腫瘍血管に局在することが判明した。
3G4およびβ2GPIのPSに対する協調結合
本実施例は、3G4抗体とPSとの相互作用は、血漿タンパク質、β2−糖タンパク質I(β2GPI)に依存することを明らかにする。3G4はβ2GPIに対しドメインIIにおいて結合するが、このドメインIIは、APS患者から単離された病原性抗体(一般に、β2GPIドメインIを認識する)とは結合しない。データから、PS陽性細胞に対する結合を含むPS結合の強化には、3G4/β2GPIの2価の複合体が必要であることが示された。なぜなら、3G4 Fab’断片はこの活性を持たないからである。PS陽性細胞に対する、3G4/β2GPIの2価複合体の結合は、本発明のFc−β2GPI構築物の使用、すなわち同様に、二量体である該構築物の、PS陽性細胞を標的とし、従って癌およびウイルス感染のような疾患を治療するための使用を支持する。
1.材料
ダルベッコの改変イーグル培養液(DMEM)およびトリプシン/EDTAは、Mediatech,Inc.(Herndon、バージニア州)から入手した。ウシ胎児血清(FBS)、ヒトの正常血清、ラットの正常血清、およびマウスの正常血清は、Biomedia(Foster City、カリフォルニア州)から入手した。ヒトの新鮮血漿は、Carter Blood Care(ダラス、テキサス州)から入手した。血清無添加ハイブリドーマ培養液、Synthechol NS0添加剤、L−α−ホスファチジルセリン(PS)、ウシ血清アルブミン(BSA)、および、ニワトリ卵白由来のオボアルブミン(OVA)は、Sigma Chemical Co.,(セントルイス、ミズーリ州)から入手した。DEAEセルロース、ヘパリン−セファローズ、およびHybond−P膜は、Amersham Biosciences(Buckinghamshire,UK)から入手した。1−パルミトイル−2−ヒドロキシ−sn−グリセロ−3−フォスフォコリン[リゾフォスファチジルコリン(LPC)]は、Avanti Polar Lipids(Alabaster,アラバマ州)から入手した。96ウェルImmulon−1Bおよび−2HBマイクロタイタープレートは、Thermo Lab Systems(Franklin、マサチューセッツ州)から入手した。Tris−HCl勾配SDS−PAGEゲルおよびOpti−4CN基質キットは、Biorad(Hercules,カリフォルニア州)から入手した。8ウェル・ガラスチェンバーは、BD Biosciences(ベッドフォード、マサチューセッツ州)から入手した。
陰イオン性リン脂質PSに結合するよう免疫化された、3G4マウスモノクロナール抗体は、実施例IV、および本明細書の他の実施例に記載される抗体である(また、Ran et al.,2005も参照されたい)。3G4は、もともと、ハイブリドーマ上清において生産されたものであるが、その後、マウスIgG2a異性体に転換され(実施例XIX、D)、現在では、NS0マウス骨髄腫細胞系統において生産されている。
3G4 F(ab’)2は、プロテーアゼ、ペプシンとインキュベーションすることによって作製した。3G4 Fab、およびコントロールFab 7H11(抗アデノウイルス)は、プロテアーゼ、パパインとインキュベーションすることによって作製した。抗体切断産物は全てFPLCにて精製し、SDS−PAGEにて確認した。
ヒトβ2GPI(hβ2GP1)は、事実上前述の通りにヒトの血漿から精製した(Polz et al.,1980;Wurm et al.,1984、なお、引用によりそれぞれを本明細書に特異的に含める)。簡単に言うと、プールされた血漿に過塩素酸(70%)を、最終濃度が1.57%(v/v)となるように加えた。沈殿を捨て、上清を、飽和Na2CO3にてpH7.5に調整し、次いで、50mM Tris,pH8.0に対して十分な透析を行った。この物質を、50mM Tris,pH8.0で平衡させたDEAEセルロースカラムに対して与え汚染物質を除いた。次に、DEAEカラム流出物を、50mM Tris,pH8.0で平衡させたヘパリンセファロース・アフィニティーカラムに与え、結合タンパク質を、1.0M NaClを用いて溶出した。最後に、このβ2GPI調製品をPBSに対して透析し、タンパク質A/Gでさらに精製し、汚染性のIgGを除去した。最終調製品は、非還元性SDS/PAGEおよびクーマシー染色で示されるように、50kDaの所に均一なバンドを含んでいた。
β2GPIの純粋な組み換え全長形態、および欠失形態を作製するために、酵母のシャトル発現ベクターpPIC6αA(Invitrogen)および宿主株Mut+X−33(Invitrogen)を用いた。この発現ベクターは、アルコール(メタノール)オキシダーゼ遺伝子(AOX1)の5’プロモーターおよび3’転写終止配列を含む。ベクターはまた、組み換え異種タンパク質が培養液へ分泌されるようにするために、AOX1プロモーターの下流に、外来cDNAが融合可能な酵母α接合因子シグナル配列を持つ。P.pastorisにおける発現は、哺乳類細胞におけるものと同様の、グリコシル化およびジスルフィド結合形成を実現する。
5’プライマー;5’−GGAATTCGGACGGACCTGTCCCAAGC−3’(配列番号12)
(2)ドメイン1欠失(ドメイン2−5)
5’プライマー;5’−GGAATTCGTATGTCCTTTTGC−3’(配列番号13)
(3)ドメイン1および2欠失(ドメイン3−5)
5’プライマー;5’−GGAATTCGCTCCCATCATCTGC−3’(配列番号14)
(4)ドメイン1−3欠失(ドメイン4−5)
5’プライマー;5’−GGAATTCGTAAAATGCCCATTC−3’(配列番号15)、および、
(5)ドメイン5のみ
5’プライマー;5’−GGAATTCGCATCTTGTAAAGTAC−3’(配列番号16)
全ての断片のPCRにおいて共通の3’プライマー、5’−TTCTAGATTAGCATGGCTTTAC−3’(配列番号17)が用いられた。PCR増幅断片は、α接合因子シグナル配列の直接下流側の、pPICαAのEcoRIおよびXbaI制限部位の間にインフレームに挿入された。C−末端において組み換えタンパク質がc−mycエピトープまたはHisタグに融合することを防止するために、各断片の末端に終止コドンを導入した。プラスミド構築物を、100μg/mlのブラスチシディンの存在下に大腸菌に移し、制限分析とヌクレオチド配列によって確認した。構築物(1)、(2)、(3)、(4)、および(5)によって発現される組み換えタンパク質は、グリコシル化前、それぞれ、約36、29、24、16、および、9kDaのタンパク質をコードした。
ニック入りhβ2GPIは、ヒトの血漿から精製された元のhβ2GPIから前述のようにして調製した。hβ2GPIを、プラスミン被覆ビーズと共に37℃で17時間インキュベートした。ビーズを遠心で取り除き、切断されたタンパク質を含む上清を回収した。精製産物のウェスタンブロット分析により、このニック入りβ2GPI調製品は、プラスミン非含有であり、プラスミンの自己分解産物も含まないことが示された(抗プラスミンまたは抗アンギオスタチン抗体に対して無反応)。N−末端配列分析から、β2GPIのN−末端に対応するもの、および、Lys317/Thr318切断部位に生成された新規配列から成る2個のN−末端が明らかにされた。
アッセイは下記のようにして実行した(さらにRan et al.,2005を参照;なお、この文献を引用により本明細書に特異的に含める)。PS−塗布Immunlon 1Bマイクロタイタープレートを1%OVA(w/v)において一晩ブロックした。翌日、血清含有または血清非含有上清から精製した3G4について、初期濃度13.33nMから2倍ずつの連続希釈液を調製した。希釈は、1%OVA、または10%の、ウシ、ヒト、ラット、または、マウスの非加熱不活性化血清を用いて行った。プレートを37℃で1時間インキュベートし、3G4の結合を検出した。ELISA実験は全て少なくとも3回実行した。
アッセイは、前述の通りに、ただし下記の改変を加えて行った。hβ2GPI、ニック入りhβ2GPI、または組み換えhβ2GPIペプチドを、96ウェルImmunlon 2HBマイクロタイタープレート上に10μg/mlの濃度で塗布し一晩置いた。次に、プレートを、1%OVAに室温で1時間浸してブロックした。3G4、ch3G4、または抗β2GPIを、1%OVAにて13.33nMの初期濃度に希釈し、2倍ずつの連続希釈を調製した。プレートを37℃で1時間インキュベートし、抗体結合を検出した。ELISA実験は全て少なくとも3回実行した。
タンパク質サンプルは、非還元性SDSサンプルバッファーにおいて95℃で5分間加熱した。次にサンプルを、Tris−HCl 4−15%勾配のSDS−PAGEゲルに負荷し、Mini Protean II装置(Biorad)を用いて分離した。分離されたタンパク質をPVDF膜に移し、3%BSA(w/v)で一晩ブロックした。膜を、3%BSAにおいて1μg/mlに希釈した、抗β2GPI、3G4、またはコントロールマウスIgGによって探査し、十分に洗浄し、ペルオキシダーゼ標識ヤギ抗マウスIgGとインキュベートした。膜を、Opti−4CN基質キットを用いて現像した。
成熟ウシ大動脈内皮(ABAE)細胞を、10%FBSおよび2mM L−グルタミンを添加したDMEMにおいて維持した。ABAE細胞を、0.25%トリプシン/0.02%EDTAに短時間暴露することによって、下集密状態の培養体から取り出し、8−ウェル・チェンバースライドに、2×104細胞数/ウェルとなるように撒いた。一晩培養後、細胞をPBSで穏やかに洗浄し、200μMリゾフォスファチジルコリン(LPC)で処理してPS露出を誘発した。LPC−処理は、10%FBS、または10%正常マウス血清(MS)のいずれかにおいて、3G4、ch3G4、またはコントロールIgGの存在下に37℃で30分実行した。LPC−処理を10%MSで実行する場合、hβ2GPIを、補因子として加えた。これは、そうしないと、3G4/ch3G4が、MS中のPSに結合できないからである。
抗体結合面積は、画像中の輝いているピクセルの数を定量することが可能な、MetaVue画像分析ソフトウェアを用いて求めた。FITC蛍光画像を用いて抗体結合を定量した。DAPI蛍光の対応画像を用いて、視野に存在する細胞の数についてFITC画像を正規化した。小さいFITC/DAPI比は、小さい抗体結合面積を示し、一方、大きいFITC/DAPI比は、大きい結合面積を示す。FITC/DAPI比は、選択された条件下における抗体結合の基礎量に対する、抗体結合面積の増減を定めるのに用いられた。各分析には200×倍率の、5枚の画像を用いた。データは、標準偏差を表す誤差バー付きの、FITC/DAPI平均相対比として表した。
1.3G4は、PS−塗布マイクロタイタープレートに結合するのに血清因子を必要とする
血清含有培養液(SCM)、または血清非含有培養液(SFM)から精製した3G4抗体は、連続希釈を10%FBSで行った場合、PS−塗布マイクロタイタープレートに結合する(図9A、実線)。一方、連続希釈を1%OVA(ウシ血清タンパク質を欠如する)で行った場合、SFMから精製した3G4はPSに結合しない(図9A、点線、黒四角)。この所見は、ウシ血清に存在するある因子が、3G4とPSの間の相互作用を仲介することを示す。
他の哺乳動物種由来の血清が、3G4とPSの間の相互作用を仲介可能かどうかを決めるために、3G4の連続希釈を、10%マウス、ラット、ヒト、またはその他の血清で行った。3G4は、ウシ血清の存在下と全く同様に、ラットおよびヒト血清の存在下にPSに結合した(図9B)。しかしながら、3G4は、マウス血清の存在下ではPSに結合しなかった(図9B)。別の実験で、3G4は、ハムスター、フェレット、モルモット、ウサギ、およびサルの血清の存在下ではPSに結合した。従って、3G4によって認識される血清タンパク質のエピトープは、マウスを除き、試験した全ての哺乳動物種において保存されている。
β2GPIは、五つのドメインを持つが、その内の第5ドメインが、陰イオン性リン脂質に対する結合に与る。β2GPIのどのドメインが3G4抗体によって認識されるかを決めるために、種々のドメイン構造を持つ組み換えヒトβ2GPI構築物を作製し、全長のhβ2GPI組み換え体と共に調べた。これらのドメイン構築物は、N−末端からの連続短縮によって作製したので、各N−末端ドメインが下記のように順に欠如する。すなわち、全長hβ2GPI組み換え体は、ドメインI−Vを含む;ドメインIを欠失させたhβ2GPIはドメインII−Vを含む;ドメインIおよびIIを欠失させたhβ2GPIはドメインIII−Vを含む;ドメインI、II,およびIIIを欠失させたhβ2GPIはドメインIV−Vを含む;および、ドメインI、II、III、およびIVを欠失させたhβ2GPIはドメインVのみを含む。
β2GPI塗布マイクロタイタープレート使用による抗−β2GPI抗体結合の特徴解明は、タイプや、場合によってはマイクロタイタープレートのロットにまで依存して観察される不一致のために問題が多かった(de Laat et al.,2004a)。さらに生理的な条件下で上記所見を確かめるために、生細胞結合アッセイを開発した。このアッセイは、膜破壊剤であるリゾフォスファチジルコリン(LPC)による処理によってPS露出を誘発した後、内皮細胞膜表面に対する抗体の結合を検出し、測定する。
PS露出内皮細胞、および他の陰イオン性リン脂質に対するβ2GPIと3G4(およびch3G4)の協調結合にはβ2GPIの脂質結合領域が必要であることを確かめるために、「ニック入り」hβ2GPIによる生細胞結合アッセイを行った。ニック入りhβ2GPIは、ドメインVの脂質結合領域内におけるプラスミン介在性切断のために、PSおよび他の陰イオン性リン脂質に結合することができない(Hunt et al., 1993; Hunt and Krilis, 1994)。
Fc−β2GPIの調製と特徴解明
本実施例は、Fc−β2GPI構築物の調製および特徴解明に関する。この構築物では、マウスIgG2aのFc領域が、完全長のマウスのβ2GPI(ドメインI−V)に結合されて、マウスFc−β2GPI、すなわちFc−mβ2GPIを生成する。このマウスIgG2a Fcは、二量体形成ドメイン、およびエフェクター機能付与ドメインとして用いられる。得られたFc−β2GPIの二量体構築物は、いずれもPSを露出させる、腫瘍血管およびウイルス感染細胞に対する標的輸送を含む、様々な用途を持つ。Fc領域を用いる利点は、Fc−β2GPI構築物は、宿主の免疫システムによって攻撃の対象とされる標的細胞をマークすることができることである。
各種Fc−β2GPI構築物が設計されてきたけれども、本実施例の、例示のFc−β2GPI(Fc−mβ2GPI)構築物は、N−末端としてマウスのFc領域を用い、C−末端としてマウスのβ2GPIのドメインI−Vを用いる。このようにして、Fc−β2GPIは、2個の、全長マウスβ2GPIタンパク質のN−末端に結合した、マウスIgG2a由来の、ジスルフィド結合ヒンジ領域、および、Fc領域の2個のCH2およびCH3ドメインを含む(図16)。
Fc−mβ2GPIは、PS塗布プレートの上でPSに結合が可能であった。PSをコートさせたマイクロタイタープレートを用い、Fc−mβ2GPI細胞培養上清(#8および#18)が、濃度依存的にPSに結合することが確かめられた(図20)。
β2GPI二量体は、PS露出内皮細胞に結合する
実施例XXXに記載される結果に従い、さらに、実施例XXXIに記載される情報の正当性を跡づけるために、本実施例は、β2GPIの二量体は、3G4抗体の不在下でもPS露出内皮細胞に結合することを示すデータを提供する。
Claims (17)
- 2個のβ2−糖タンパク質I(β2GPI)ポリペプチドに、共有結合を介して、組換え発現により融合タンパク質として、または、ペプチドリンカーもしくは化学的リンカーを介して作動可能に結合した抗体Fc領域を含む構築物であって、
該β2GPIポリペプチドの各々は、β2GPIのインタクトなドメインVを少なくとも含み、該β2GPIポリペプチドは、該Fc領域に結合された場合に、ホスファチジルセリンに結合し、そして、該Fc領域は、抗原結合領域、抗原結合ドメインまたは抗原結合断片を含まない、構築物。 - 前記β2GPIポリペプチドが、それぞれ、少なくとも、β2GPIのインタクトなドメインVと、β2GPIの他の4つのドメインのうちの1つ以上を含む、請求項1に記載の構築物。
- 前記β2GPIポリペプチドが、それぞれ、少なくとも、β2GPIのインタクトなドメインVと、β2GPIのドメインIを含む、請求項2に記載の構築物。
- 前記β2GPIポリペプチドが、それぞれ、β2GPIのドメインI、ドメインII、ドメインIII、ドメインIV、およびインタクトなドメインVを含む、請求項2に記載の構築物。
- 前記β2GPIポリペプチドが、それぞれ、ヒトのβ2GPIポリペプチドである、請求項1〜4のいずれか1項に記載の構築物。
- 前記抗体Fc領域が、ヒトのIgG1(γ1)抗体由来のFc領域またはヒトのIgG3(γ3)抗体由来のFc領域である、請求項1〜5のいずれかに記載の構築物。
- 前記構築物が、少なくとも1つの診断剤、画像化剤または治療剤に作動可能にさらに結合されている、請求項1〜6のいずれかに記載の構築物。
- 前記構築物が、少なくとも1つの抗脈管形成剤、抗癌剤、または抗ウイルス剤に作動可能にさらに結合されている、請求項7に記載の構築物。
- 治療に用いるための、請求項1〜8のいずれかに記載の構築物を含む、組成物。
- 癌を治療するための、請求項1〜8のいずれかに記載の構築物を含む、組成物。
- 前記組成物が、第2の治療剤または抗癌剤と組み合わせて、あるいは、放射線療法と組み合わせて使用するためのものである、請求項10に記載の組成物。
- ウイルス感染またはウイルス疾患を治療するための、請求項1〜8のいずれかに記載の構築物を含む、組成物。
- 前記組成物が、第2の治療剤または抗ウイルス剤と組み合わせて使用するためのものである、請求項12に記載の組成物。
- 癌を治療するための医薬の製造における、請求項1から8のいずれか1項に記載の構築物の使用。
- 前記医薬が、第2の治療剤または抗癌剤と組み合わせて、あるいは、放射線療法と組み合わせて使用するためのものである、請求項14に記載の使用。
- ウイルス感染またはウイルス疾患を治療するための医薬の製造における、請求項1から8のいずれか1項に記載の構築物の使用。
- 前記医薬が、第2の治療剤または抗ウイルス剤と組み合わせて使用するためのものである、請求項16に記載の使用。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US64633305P | 2005-01-24 | 2005-01-24 | |
| US60/646,333 | 2005-01-24 | ||
| PCT/US2006/002964 WO2006079120A2 (en) | 2005-01-24 | 2006-01-24 | Fc-fusion constructs binding to phosphatidylserine and their therapeutic use |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012057880A Division JP2012111783A (ja) | 2005-01-24 | 2012-03-14 | ホスファチジルセリンに結合するfc融合構築物およびそれらの治療用途 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2008528521A JP2008528521A (ja) | 2008-07-31 |
| JP2008528521A5 JP2008528521A5 (ja) | 2009-03-12 |
| JP5127043B2 true JP5127043B2 (ja) | 2013-01-23 |
Family
ID=36693034
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007552418A Expired - Fee Related JP5127043B2 (ja) | 2005-01-24 | 2006-01-24 | ホスファチジルセリンに結合するfc融合構築物およびそれらの治療用途 |
| JP2012057880A Withdrawn JP2012111783A (ja) | 2005-01-24 | 2012-03-14 | ホスファチジルセリンに結合するfc融合構築物およびそれらの治療用途 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012057880A Withdrawn JP2012111783A (ja) | 2005-01-24 | 2012-03-14 | ホスファチジルセリンに結合するfc融合構築物およびそれらの治療用途 |
Country Status (14)
| Country | Link |
|---|---|
| US (3) | US8956616B2 (ja) |
| EP (1) | EP1853631B1 (ja) |
| JP (2) | JP5127043B2 (ja) |
| CN (3) | CN104277117A (ja) |
| AU (1) | AU2006206187B2 (ja) |
| CA (1) | CA2591914C (ja) |
| DK (1) | DK1853631T3 (ja) |
| ES (1) | ES2565543T3 (ja) |
| HK (2) | HK1198255A1 (ja) |
| IL (1) | IL184406A (ja) |
| NZ (1) | NZ556065A (ja) |
| SG (1) | SG158919A1 (ja) |
| SI (1) | SI1853631T1 (ja) |
| WO (1) | WO2006079120A2 (ja) |
Families Citing this family (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090291086A1 (en) * | 2001-02-21 | 2009-11-26 | Alavita Pharmaceuticals, Inc. | Compositions and Methods for Treating Cerebral Thrombosis and Global Cerebral Ischemia |
| US20050222030A1 (en) * | 2001-02-21 | 2005-10-06 | Anthony Allison | Modified annexin proteins and methods for preventing thrombosis |
| GB0707034D0 (en) | 2007-04-12 | 2007-05-23 | St Andrews The | Compounds |
| US9163091B2 (en) * | 2007-05-30 | 2015-10-20 | Lpath, Inc. | Compositions and methods for binding lysophosphatidic acid |
| US20110064744A1 (en) * | 2007-05-30 | 2011-03-17 | Sabbadini Roger A | Prevention and treatment of pain using antibodies to lysophosphatidic acid |
| EP2164992B1 (en) * | 2007-05-30 | 2016-05-04 | Lpath, Inc. | Compositions and methods for binding lysophosphatidic acid |
| US9446150B2 (en) | 2007-10-09 | 2016-09-20 | Washington University | Particles for imaging |
| US9468607B2 (en) | 2007-10-09 | 2016-10-18 | Washington University | Ligand directed toroidal nanoparticles for therapy and diagnostic imaging |
| WO2009065065A1 (en) * | 2007-11-15 | 2009-05-22 | Ceramoptec Industries, Inc. | Pegylated liposomal formulations for photodynamic treatment of inflammatory diseases |
| GB0723797D0 (en) * | 2007-12-05 | 2008-01-16 | Immunosolv Ltd | Method |
| WO2009089098A1 (en) * | 2008-01-03 | 2009-07-16 | Musc Foundation For Research Development | Methods for the treatment of cancers |
| KR20100127842A (ko) * | 2008-03-26 | 2010-12-06 | 유니버시티 오브 옥스퍼드 | 소포체 표적화 리포좀 |
| GB0819287D0 (en) | 2008-10-22 | 2008-11-26 | Univ Bradford | Compounds |
| JP5580329B2 (ja) | 2008-12-05 | 2014-08-27 | アブラクシス バイオサイエンス リミテッド ライアビリティー カンパニー | Sparc結合ペプチド及びその使用 |
| WO2011036467A1 (en) * | 2009-09-24 | 2011-03-31 | Russell David Keenan | Products useful in the treatment of haemophilia |
| WO2011069090A1 (en) * | 2009-12-04 | 2011-06-09 | Alavita Pharmaceuticals, Inc. | Inhibition of the activation of coagulation factor xii by ligands of phosphatidylserine |
| US9808500B2 (en) | 2009-12-17 | 2017-11-07 | Washington University | Antithrombotic nanoparticle |
| US9764043B2 (en) | 2009-12-17 | 2017-09-19 | Washington University | Antithrombotic nanoparticle |
| EP2369551B1 (en) * | 2010-03-25 | 2019-10-30 | Emory University | Imaging system and method |
| CA2796435C (en) | 2010-04-15 | 2019-05-07 | The Washington University | Prodrug compositions, prodrug nanoparticles, and methods of use thereof |
| US8691771B2 (en) * | 2010-05-21 | 2014-04-08 | Merrimack Pharmaceuticals, Inc. | Bi-specific fusion proteins for tissue repair |
| CN105524176B (zh) * | 2010-05-21 | 2021-03-19 | 银溪制药股份有限公司 | 双特异性融合蛋白 |
| WO2012030949A2 (en) * | 2010-09-01 | 2012-03-08 | Kansas State University Research Foundation | B2-glycoprotein i peptide inhibitors |
| EP2750662A4 (en) | 2011-08-31 | 2015-06-24 | Univ Georgia | NANOPARTICLES TARGETING APOPTOSIS |
| US9463217B1 (en) * | 2012-02-10 | 2016-10-11 | Institut National De La Santé Et De La Recherche Médicale (Inserm) | Methods and pharmaceutical compositions for treatment of retinal occlusion |
| JP6356614B2 (ja) | 2012-02-17 | 2018-07-11 | ユニバーシティ・オブ・ジョージア・リサーチ・ファウンデイション・インコーポレイテッド | 薬剤のミトコンドリア輸送のためのナノ粒子 |
| WO2014018535A1 (en) * | 2012-07-25 | 2014-01-30 | Salk Institute For Biological Studies | Regulating the interaction between tam ligands and lipid membranes with exposed phosphatidylserine |
| US20140287024A1 (en) * | 2013-03-08 | 2014-09-25 | Shu Wang | Nanoparticle-based delivery system with oxidized phospholipids as targeting ligands for the prevention, diagnosis and treatment of atherosclerosis |
| ES2756532T3 (es) * | 2013-03-15 | 2020-04-27 | Gladiator Biosciences Inc | Dominios de Gla como agentes de direccionamiento |
| GB2516882A (en) | 2013-08-02 | 2015-02-11 | Univ Bradford | Tumour-targeted theranostic |
| JP6685225B2 (ja) * | 2013-12-16 | 2020-04-22 | ザ・ユニヴァーシティ・オヴ・ノース・キャロライナ・アト・チャペル・ヒル | 形質細胞様樹状細胞の枯渇 |
| WO2015138992A1 (en) | 2014-03-14 | 2015-09-17 | University Of Georgia Research Foundation, Inc. | Mitochondrial delivery of 3-bromopyruvate |
| WO2015192020A1 (en) | 2014-06-13 | 2015-12-17 | Children's Medical Center Corporation | Products and methods to isolate mitochondria |
| US11000603B2 (en) | 2015-04-14 | 2021-05-11 | Benhealth Biopharmaceutic (Shenzhen) Co., Ltd. | Multi-specific binding conjugate, related pharmaceutical compositions and use |
| US20180154012A1 (en) * | 2015-05-05 | 2018-06-07 | Rubicon Biotechnology Llc | Cancer immunotherapeutic |
| WO2016201064A1 (en) * | 2015-06-09 | 2016-12-15 | The Board Of Regents Of The University Of Texas System | Diagnostic test for early stage cancer |
| EP3865147A1 (en) * | 2015-10-02 | 2021-08-18 | Silver Creek Pharmaceuticals, Inc. | Bi-specific therapeutic proteins for tissue repair |
| AU2016370648B2 (en) | 2015-12-15 | 2023-03-09 | OncoC4, Inc. | Chimeric and humanized anti-human CTLA4 monoclonal antibodies and uses thereof |
| EP3402490B1 (en) * | 2016-01-15 | 2022-06-01 | The Children's Medical Center Corporation | Therapeutic use of mitochondria and combined mitochondrial agents |
| WO2017133018A1 (zh) * | 2016-02-06 | 2017-08-10 | 法玛科技顾问股份有限公司 | 重组β2-糖蛋白胜肽及其于抗肿瘤的应用 |
| CN109195992A (zh) * | 2016-04-29 | 2019-01-11 | 本康生物制药(深圳)有限公司 | 多特异性结合偶联物、相关的药物组合物及应用 |
| EP3502142B1 (en) | 2016-06-22 | 2021-10-27 | Benhealth Biopharmaceutic (Shenzhen) Co., Ltd. | Bispecific antibody and antibody conjugate for tumour therapy and use thereof |
| EA038617B1 (ru) * | 2016-07-06 | 2021-09-23 | Онкоиммьюн, Инк. | Химерные и гуманизированные античеловеческие ctla-4 моноклональные антитела и их использование |
| KR20190057303A (ko) * | 2016-09-27 | 2019-05-28 | 온콜로지, 인크. | β2-당단백질 1의 수준에 기초한 바비툭시마브에 의한 암 치료 방법, 및 이를 위한 에세이 |
| MX2019003489A (es) | 2016-09-27 | 2020-01-23 | Cero Therapeutics Inc | Moleculas del receptor de envolvimiento quimerico. |
| EP3606960A1 (en) * | 2017-04-03 | 2020-02-12 | Oncologie, Inc. | Methods for treating cancer using ps-targeting antibodies with immuno-oncology agents |
| CN108727489B (zh) * | 2017-04-19 | 2021-02-09 | 广州市第八人民医院 | 一种单克隆抗体zk2c2及应用 |
| US11352404B2 (en) * | 2017-07-24 | 2022-06-07 | Rutgers, The State University Of New Jersey | Phosphatidylserine targeting fusion molecules and methods for their use |
| US11661447B2 (en) | 2017-08-03 | 2023-05-30 | The Cleveland Clinic Foundation | Human β2-glycoprotein I expression |
| CN111051500A (zh) * | 2017-09-05 | 2020-04-21 | 角斗士生物科学公司 | 有效载荷递送至干细胞 |
| JP7286658B2 (ja) | 2017-09-26 | 2023-06-05 | セロ・セラピューティクス・インコーポレイテッド | キメラエンガルフメント受容体分子および使用方法 |
| CN108021784B (zh) * | 2017-12-01 | 2021-05-11 | 大连理工大学 | 纳米金属氧化物光致产生活性氧物种的预测方法 |
| ES2727261B2 (es) * | 2018-04-12 | 2020-05-06 | Fundacion Para La Investig Biomedica Del Hospital 12 De Octubre | INMUNOCOMPLEJOS ARTIFICIALES Y SU USO COMO CALIBRES EN SISTEMAS DE DETECCION DE INMUNOCOMPLEJOS CIRCULANTES B2GP1-aB2GP1 |
| EP3876909A4 (en) * | 2018-11-08 | 2022-09-28 | Nasr, Mahmoud | NANODISCS FOR THE PREVENTION AND TREATMENT OF PATHOGENIC INFECTIONS AND METHODS OF USE THEREOF |
| TW202039583A (zh) | 2018-12-07 | 2020-11-01 | 瑞士商巴克斯歐塔有限公司 | 結合因子IXa及因子X的蛋白分子 |
| WO2020114614A1 (en) | 2018-12-07 | 2020-06-11 | Baxalta GmbH | Proteinaceous molecules binding factor ixa and factor x |
| EP3711772A1 (en) * | 2019-03-20 | 2020-09-23 | Oslo Universitetssykehus HF | Recombinant proteins and fusion proteins |
| US20220395578A1 (en) * | 2019-06-28 | 2022-12-15 | The Board Of Regents Of The University Of Oklahoma | Therapeutic annexin-drug conjugates and methods of use |
| US20220033426A1 (en) * | 2020-07-28 | 2022-02-03 | Yale University | Transition Metal Macrocyclics as MRI Contrast Agents for Molecular Imaging |
| CN116249525A (zh) * | 2020-08-21 | 2023-06-09 | 芝加哥大学 | 用于癌症疗法的含有多个可裂解前体药物的纳米颗粒 |
| JP2024512207A (ja) * | 2021-02-05 | 2024-03-19 | ヴィクィ インク. | 細胞の形態学的変化の早期検出のための機械学習 |
| CN114002133B (zh) * | 2021-10-25 | 2023-01-24 | 南京大学 | 血清中ps阳性外泌体作为乳腺癌化疗敏感性标志物的应用 |
| WO2023141524A2 (en) * | 2022-01-19 | 2023-07-27 | Lyndra Therapeutics, Inc. | Dosage forms for gastric retention |
| CN114288399B (zh) * | 2022-01-20 | 2023-10-27 | 武汉科技大学 | 一种嵌合抗原受体联合抗肿瘤药物组合物及其应用 |
| US20230324382A1 (en) * | 2022-02-01 | 2023-10-12 | Trustees Of Tufts College | Antiphospholipid antibodies for the diagnosis of lyme disease |
| WO2023150672A1 (en) * | 2022-02-04 | 2023-08-10 | Duke University | Compositions for and methods of treating hematological cancers |
Family Cites Families (53)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5245015A (en) * | 1991-04-26 | 1993-09-14 | Tanox Biosystems, Inc. | Monoclonal antibodies which neutralize HIV-1 through reaction with a conformational epitope in vitro |
| CA2058769C (en) | 1990-04-06 | 1999-03-02 | Steven Anthony Krilis | Methods for determining phospholipids and antibodies thereof |
| US5472883A (en) | 1992-02-05 | 1995-12-05 | Yamasa Corporation | Solid phase reagent and assay method for measuring antibodies specific to antiphospholipid syndrome |
| WO1994013323A1 (en) | 1992-12-04 | 1994-06-23 | The Liposome Company, Inc. | Method of inhibiting the clearance of liposomes from the circulation |
| FR2701263B1 (fr) | 1993-02-09 | 1995-04-21 | Elie Stefas | Procédé d'obtention d'une composition aqueuse protéinique, composition correspondante, glycoprotéine contenue et son utilisation pour la stabilisation de l'albumine et la détection ou le dosage d'anticorps. |
| FR2701319B1 (fr) | 1993-02-09 | 1995-04-21 | Elie Stefas | Procédé de détection et/ou de dosage de composés viraux et support portant une glycoprotéine. |
| CA2172247A1 (en) | 1993-09-29 | 1995-04-06 | Eiji Matsuura | Method of assaying oxidized lipoprotein and application thereof |
| WO1995014231A1 (fr) | 1993-11-16 | 1995-05-26 | Yamasa Corporation | Procede pour le dosage d'anticorps antiphospholipides et kit associe |
| FR2722991B1 (fr) | 1994-08-01 | 1996-10-11 | Orstom | Utilisation de la beta2-glycoproteine i sous au moins une de ses formes comme agent anti-infectueux et composition pharmaceutique corrrespondante |
| ES2179879T3 (es) | 1994-08-01 | 2003-02-01 | Inst Rech Developpement Ird | Procedimiento de separacion y/o de deteccion y/o de cuantificacion de compuesto(s) infeccioso(s) y soporte para la realizacion de dicho procedimiento. |
| US5817789A (en) | 1995-06-06 | 1998-10-06 | Transkaryotic Therapies, Inc. | Chimeric proteins for use in transport of a selected substance into cells |
| US5874409A (en) | 1995-06-07 | 1999-02-23 | La Jolla Pharmaceutical Company | APL immunoreactive peptides, conjugates thereof and methods of treatment for APL antibody-mediated pathologies |
| US6203980B1 (en) | 1998-02-02 | 2001-03-20 | University Of Pittsburgh | Identification of apolipoprotein H mutations and their diagnostic uses |
| US6858210B1 (en) | 1998-06-09 | 2005-02-22 | La Jolla Pharmaceutical Co. | Therapeutic and diagnostic domain 1 β2GPI polypeptides and methods of using same |
| IL125262A0 (en) | 1998-07-07 | 1999-03-12 | Yeda Res & Dev | Synthetic peptides and pharmaceutical compositions comprising them |
| JP2000028607A (ja) | 1998-07-10 | 2000-01-28 | Iatron Lab Inc | 新規なモノクローナル抗体及びニックβ2グリコプロテインIの免疫学的分析方法 |
| CA2331789C (en) | 1998-07-13 | 2013-09-10 | Board Of Regents, The University Of Texas System | Cancer treatment methods using therapeutic conjugates that bind to aminophospholipids |
| US6406693B1 (en) | 1998-07-13 | 2002-06-18 | Board Of Regents, The University Of Texas System | Cancer treatment methods using antibodies to aminophospholipids |
| US6818213B1 (en) | 1998-07-13 | 2004-11-16 | Board Of Regents, The University Of Texas System | Cancer treatment compositions comprising therapeutic conjugates that bind to aminophospholipids |
| WO2000002584A2 (en) | 1998-07-13 | 2000-01-20 | Board Of Regents, The University Of Texas System | Cancer treatment methods using antibodies to aminophospholipids |
| US20050148499A1 (en) | 1998-10-04 | 2005-07-07 | Dror Harats | Compositions containing beta 2-glycoprotein I for the prevention and/or treatment of atherosclerosis |
| US20050197283A1 (en) | 1998-10-04 | 2005-09-08 | Vascular Biogenics Ltd. | Compositions containing beta 2-glycoprotein I for the prevention and/or treatment of vascular disease |
| IL126447A (en) | 1998-10-04 | 2004-09-27 | Vascular Biogenics Ltd | An immune preparation that confers tolerance in oral administration and its use in the prevention and / or treatment of atherosclerosis |
| AUPQ272699A0 (en) | 1999-09-09 | 1999-09-30 | Unisearch Limited | Use of beta2GPI in diagnostic tests for autoimmune diseases |
| US7600039B2 (en) * | 2000-02-16 | 2009-10-06 | Motorola, Inc. | Label-based multiplexing |
| IL151655A0 (en) | 2000-03-08 | 2003-04-10 | Nat Jewish Med & Res Center | Phosphatidyl serine receptors and uses thereof |
| EP1292337A2 (en) | 2000-06-08 | 2003-03-19 | La Jolla Pharmaceutical | Multivalent platform molecules comprising high molecular weight polyethylene oxide |
| EP2357187A1 (en) * | 2000-12-12 | 2011-08-17 | MedImmune, LLC | Molecules with extended half-lives, compositions and uses thereof |
| EP1362127B1 (en) * | 2001-01-26 | 2007-11-07 | The General Hospital Corporation | Serpin drugs for treatment of hiv infection and method of use thereof |
| JP4351041B2 (ja) | 2001-06-13 | 2009-10-28 | ザ・ユニバーシティ・オブ・シドニー | 傷治癒のための処置および組成物 |
| FR2827047B1 (fr) | 2001-07-03 | 2003-09-26 | Apoh Technollgies Sa | Procede de separation et/ou detection et/ou identification et/ou quantification de proteines prions |
| JP4681175B2 (ja) | 2001-09-28 | 2011-05-11 | 三菱化学メディエンス株式会社 | 播種性血管内凝固症候群及びその発症前段階を検出する方法 |
| JP4070443B2 (ja) | 2001-10-11 | 2008-04-02 | 株式会社三菱化学ヤトロン | 脳梗塞を検出する方法 |
| US20030100036A1 (en) | 2001-11-08 | 2003-05-29 | Aristo Vojdani | Saliva immunoassay for detection of antibodies for cardiovascular disease |
| US20030138419A1 (en) | 2001-11-16 | 2003-07-24 | The University Of Tennessee Research Corporation | Recombinant antibody fusion proteins and methods for detection of apoptotic cells |
| US7329642B2 (en) * | 2002-05-17 | 2008-02-12 | Board Of Regents, University Of Texas System | β-2-glycoprotein is an inhibitor of angiogenesis |
| BR0312692A (pt) | 2002-07-15 | 2007-06-26 | Univ Texas | anticorpos selecionados e peptìdeos de duramicina que se ligam a fosfolipìdios aniÈnicos e aminofosfolipìdios e seus usos no tratamento de infecções virais e cáncer |
| US7611704B2 (en) | 2002-07-15 | 2009-11-03 | Board Of Regents, The University Of Texas System | Compositions and methods for treating viral infections using antibodies and immunoconjugates to aminophospholipids |
| US7790159B2 (en) | 2002-07-15 | 2010-09-07 | Board Of Regents, The University Of Texas System | Methods, combinations and kits for treating viral infections using immunoconjugates and antibodies to aminophospholipids |
| US7625563B2 (en) | 2002-07-15 | 2009-12-01 | Board Of Regents, The University Of Texas System | Cancer treatment methods using selected immunoconjugates for binding to aminophospholipids |
| US7247303B2 (en) | 2002-07-15 | 2007-07-24 | Board Of Regents, The University Of Texas System | Selected antibody CDRs for binding to aminophospholipids |
| US7622118B2 (en) | 2002-07-15 | 2009-11-24 | Board Of Regents, The University Of Texas System | Cancer treatment methods using selected antibodies to aminophospholipids |
| US7906115B2 (en) | 2002-07-15 | 2011-03-15 | Thorpe Philip E | Combinations kits and methods for treating viral infections using antibodies and immunoconjugates to aminophospholipids |
| US7455833B2 (en) | 2002-07-15 | 2008-11-25 | Board Of Regents, The University Of Texas System | Methods and compositions for treating viral infections using antibodies and immunoconjugates to aminophospholipids |
| US7572448B2 (en) | 2002-07-15 | 2009-08-11 | Board Of Regents, The University Of Texas System | Combined cancer treatment methods using selected antibodies to aminophospholipids |
| US7615223B2 (en) | 2002-07-15 | 2009-11-10 | Board Of Regents, The University Of Texas System | Selected immunoconjugates for binding to aminophospholipids |
| US7714109B2 (en) | 2002-07-15 | 2010-05-11 | Board Of Regents, The University Of Texas System | Combinations and kits for cancer treatment using selected antibodies to aminophospholipids |
| US7678386B2 (en) | 2002-07-15 | 2010-03-16 | Board Of Regents The University Of Texas | Liposomes coated with selected antibodies that bind to aminophospholipids |
| ATE512364T1 (de) | 2002-08-07 | 2011-06-15 | Ablynx Nv | Modulation der blutplättchen-adhäsion basierend auf dem oberflächen-exponierten beta-switch loop des blutplättchen-glycoproteins ib-alpha |
| EP2298347B1 (en) * | 2003-05-06 | 2015-09-30 | Biogen Hemophilia Inc. | Clotting factor chimeric proteins for treatment of a hemostatic disorder |
| WO2005019429A2 (en) | 2003-08-22 | 2005-03-03 | Potentia Pharmaceuticals, Inc. | Compositions and methods for enhancing phagocytosis or phagocyte activity |
| EP1874334A4 (en) | 2005-04-15 | 2011-03-30 | Vascular Biogenics Ltd | COMPOSITIONS WITH BETA 2-GLYCOPROTEIN I-PEPTIDES FOR THE PREVENTION AND / OR TREATMENT OF VASCULAR DISEASES |
| WO2006113720A2 (en) | 2005-04-18 | 2006-10-26 | Bio-Rad Laboratories, Inc. | Solid phase immobilization of phospholipids and cofactor proteins via covalent attachment |
-
2006
- 2006-01-24 CN CN201410333397.6A patent/CN104277117A/zh not_active Withdrawn
- 2006-01-24 SG SG201000541-1A patent/SG158919A1/en unknown
- 2006-01-24 EP EP06719706.1A patent/EP1853631B1/en not_active Not-in-force
- 2006-01-24 WO PCT/US2006/002964 patent/WO2006079120A2/en active Application Filing
- 2006-01-24 AU AU2006206187A patent/AU2006206187B2/en not_active Ceased
- 2006-01-24 US US11/339,392 patent/US8956616B2/en active Active
- 2006-01-24 DK DK06719706.1T patent/DK1853631T3/en active
- 2006-01-24 SI SI200632035T patent/SI1853631T1/sl unknown
- 2006-01-24 CN CNA2006800070648A patent/CN101146826A/zh active Pending
- 2006-01-24 NZ NZ556065A patent/NZ556065A/en unknown
- 2006-01-24 CA CA2591914A patent/CA2591914C/en not_active Expired - Fee Related
- 2006-01-24 CN CN201410301233.5A patent/CN104109209B/zh not_active Expired - Fee Related
- 2006-01-24 ES ES06719706.1T patent/ES2565543T3/es active Active
- 2006-01-24 JP JP2007552418A patent/JP5127043B2/ja not_active Expired - Fee Related
-
2007
- 2007-07-04 IL IL184406A patent/IL184406A/en not_active IP Right Cessation
-
2012
- 2012-03-14 JP JP2012057880A patent/JP2012111783A/ja not_active Withdrawn
-
2014
- 2014-11-21 HK HK14111797.6A patent/HK1198255A1/zh not_active IP Right Cessation
-
2015
- 2015-02-02 US US14/611,634 patent/US20160015826A1/en not_active Abandoned
- 2015-06-05 HK HK15105389.1A patent/HK1205133A1/xx unknown
-
2016
- 2016-07-12 US US15/207,955 patent/US20160311886A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| EP1853631B1 (en) | 2016-03-09 |
| HK1198255A1 (zh) | 2015-03-20 |
| US20060228299A1 (en) | 2006-10-12 |
| SI1853631T1 (sl) | 2016-04-29 |
| EP1853631A2 (en) | 2007-11-14 |
| CA2591914C (en) | 2017-04-25 |
| IL184406A0 (en) | 2008-12-29 |
| AU2006206187B2 (en) | 2011-03-10 |
| WO2006079120A2 (en) | 2006-07-27 |
| CN104109209B (zh) | 2016-06-08 |
| AU2006206187A1 (en) | 2006-07-27 |
| US20160311886A1 (en) | 2016-10-27 |
| JP2008528521A (ja) | 2008-07-31 |
| IL184406A (en) | 2014-11-30 |
| DK1853631T3 (en) | 2016-03-29 |
| CN104109209A (zh) | 2014-10-22 |
| US8956616B2 (en) | 2015-02-17 |
| HK1205133A1 (en) | 2015-12-11 |
| SG158919A1 (en) | 2010-02-26 |
| ES2565543T3 (es) | 2016-04-05 |
| WO2006079120A3 (en) | 2007-07-19 |
| CN101146826A (zh) | 2008-03-19 |
| US20160015826A1 (en) | 2016-01-21 |
| CA2591914A1 (en) | 2006-07-27 |
| JP2012111783A (ja) | 2012-06-14 |
| CN104277117A (zh) | 2015-01-14 |
| NZ556065A (en) | 2009-04-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5127043B2 (ja) | ホスファチジルセリンに結合するfc融合構築物およびそれらの治療用途 | |
| JP6188258B2 (ja) | 選択された抗体および処置におけるその使用 | |
| JP5721693B2 (ja) | アミノリン脂質に結合する治療結合体を用いる癌処置方法 | |
| JP4926320B2 (ja) | Vegfの選択的阻害による癌処置のための組成物および方法 | |
| JP2002543093A5 (ja) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090126 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090126 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110914 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20111116 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20111124 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120110 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120117 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120208 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120215 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120314 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20120314 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20121026 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121029 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 Ref document number: 5127043 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151109 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |