JP5115675B2 - プリプレグ、および繊維強化複合材料 - Google Patents
プリプレグ、および繊維強化複合材料 Download PDFInfo
- Publication number
- JP5115675B2 JP5115675B2 JP2012515833A JP2012515833A JP5115675B2 JP 5115675 B2 JP5115675 B2 JP 5115675B2 JP 2012515833 A JP2012515833 A JP 2012515833A JP 2012515833 A JP2012515833 A JP 2012515833A JP 5115675 B2 JP5115675 B2 JP 5115675B2
- Authority
- JP
- Japan
- Prior art keywords
- prepreg
- component
- constituent element
- epoxy resin
- fiber
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/14—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by a layer differing constitutionally or physically in different parts, e.g. denser near its faces
- B32B5/145—Variation across the thickness of the layer
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/06—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material
- B32B27/08—Layered products comprising a layer of synthetic resin as the main or only constituent of a layer, which is next to another layer of the same or of a different material of synthetic resin
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B38/00—Ancillary operations in connection with laminating processes
- B32B38/08—Impregnating
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B5/00—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts
- B32B5/16—Layered products characterised by the non- homogeneity or physical structure, i.e. comprising a fibrous, filamentary, particulate or foam layer; Layered products characterised by having a layer differing constitutionally or physically in different parts characterised by features of a layer formed of particles, e.g. chips, powder or granules
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B7/00—Layered products characterised by the relation between layers; Layered products characterised by the relative orientation of features between layers, or by the relative values of a measurable parameter between layers, i.e. products comprising layers having different physical, chemical or physicochemical properties; Layered products characterised by the interconnection of layers
- B32B7/02—Physical, chemical or physicochemical properties
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/4007—Curing agents not provided for by the groups C08G59/42 - C08G59/66
- C08G59/4014—Nitrogen containing compounds
- C08G59/4021—Ureas; Thioureas; Guanidines; Dicyandiamides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/42—Polycarboxylic acids; Anhydrides, halides or low molecular weight esters thereof
- C08G59/4215—Polycarboxylic acids; Anhydrides, halides or low molecular weight esters thereof cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/50—Amines
- C08G59/5033—Amines aromatic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/04—Reinforcing macromolecular compounds with loose or coherent fibrous material
- C08J5/0405—Reinforcing macromolecular compounds with loose or coherent fibrous material with inorganic fibres
- C08J5/042—Reinforcing macromolecular compounds with loose or coherent fibrous material with inorganic fibres with carbon fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/241—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres
- C08J5/243—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs using inorganic fibres using carbon fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/24—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs
- C08J5/249—Impregnating materials with prepolymers which can be polymerised in situ, e.g. manufacture of prepregs characterised by the additives used in the prepolymer mixture
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K7/00—Use of ingredients characterised by shape
- C08K7/02—Fibres or whiskers
- C08K7/04—Fibres or whiskers inorganic
- C08K7/06—Elements
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L25/00—Compositions of, homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring; Compositions of derivatives of such polymers
- C08L25/02—Homopolymers or copolymers of hydrocarbons
- C08L25/04—Homopolymers or copolymers of styrene
- C08L25/06—Polystyrene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L75/00—Compositions of polyureas or polyurethanes; Compositions of derivatives of such polymers
- C08L75/04—Polyurethanes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2363/00—Characterised by the use of epoxy resins; Derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2425/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring; Derivatives of such polymers
- C08J2425/02—Homopolymers or copolymers of hydrocarbons
- C08J2425/04—Homopolymers or copolymers of styrene
- C08J2425/06—Polystyrene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2475/00—Characterised by the use of polyureas or polyurethanes; Derivatives of such polymers
- C08J2475/04—Polyurethanes
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/249921—Web or sheet containing structurally defined element or component
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/26—Web or sheet containing structurally defined element or component, the element or component having a specified physical dimension
- Y10T428/269—Web or sheet containing structurally defined element or component, the element or component having a specified physical dimension including synthetic resin or polymer layer or component
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Inorganic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Reinforced Plastic Materials (AREA)
Description
[B]:1分子内に2個以上のエポキシ基を有するエポキシ樹脂
[C]:構成要素[B]を硬化しうる化合物
[D]:屈折率が1.49〜1.61の範囲にある架橋樹脂粒子
[E]:カーボンブラック
条件(I):構成要素[B]および構成要素[C]からなる樹脂組成物を硬化して得られるエポキシ樹脂硬化物の屈折率n0と、構成要素[D]の屈折率n1との関係が、0.92≦n1/n0≦1.05である。
条件(i):構成要素[B]中に構成要素[F]を配合し160℃の温度で30分加熱して得られる樹脂組成物中に、0.1μm以上の構成要素[F]の固形物が存在しない。
条件(II):構成要素[B]、[C]および[F]からなる樹脂組成物を硬化して得られるエポキシ樹脂硬化物の屈折率n0’と、構成要素[D]の屈折率n1との関係が、0.92≦n1/n0’≦1.05である。
本発明のプリプレグは、少なくとも次の構成要素[A]〜[E]から構成される。
[B]:1分子内に2個以上のエポキシ基を有するエポキシ樹脂
[C]:構成要素[B]を硬化しうる化合物
[D]:屈折率が1.49〜1.61の範囲にある架橋樹脂粒子
[E]:カーボンブラック。
例えば、ジシアンジアミドに、尿素誘導体やイミダゾール類を組み合わせて好適に用いることができる。ジシアンジアミド単独では硬化に170〜180℃程度の温度が必要であるのに対し、かかる組み合わせを用いた繊維強化複合材料用エポキシ樹脂組成物は80〜150℃の温度で硬化可能になる。
<強化繊維(構成要素[A])>
・炭素繊維(“トレカ(登録商標)”T700SC−12K、引張強度4.9GPa、引張弾性率230GPa、東レ(株)製)。
・液状ビスフェノールA型エポキシ樹脂(“エポトート(登録商標)”YD128、新日鐵化学(株)製)
・固体ビスフェノールA型エポキシ樹脂(“jER(登録商標)”1001、三菱化学(株)製)
・フェノールノボラック型エポキシ樹脂(“jER(登録商標)”154、三菱化学(株)製)。
・ジシアンジアミド(Dicy7、三菱化学(株)製)
・ウレア化合物(“オミキュア(登録商標)”24、CVC Specialty Chemicals,Inc.製)
・3,3’−ジアミノジフェニルスルホン(3,3’−DAS、小西化学工業(株)製)
・メチルヘキサヒドロ無水フタル酸/ヘキサヒドロ無水フタル酸=70/30(質量比)の混合物(“リカシッド(登録商標)”MH−700、新日本理化(株))
・トリフェニルホスフィン(TPP、北興化学工業(株)製)。
・架橋ポリウレタン(“ダイミックビーズ(登録商標)”UCN−5051D、平均粒子径15μm、屈折率1.49、大日精化工業(株)製)
・スチレン・ジビニルベンゼン共重合体(“テクポリマー(登録商標)”SBX−12、平均粒子径12μm、屈折率1.59、積水化成品工業(株)製)
・スチレン・ジビニルベンゼン共重合体(“ケミスノー(登録商標)”SGP−150C、平均粒子径55μm、屈折率1.59、綜研化学(株)製)
・中空ガラス粒子(“Sphericel(登録商標)”110P8、平均粒子径12μm、屈折率1.51、ポッターズ・バロティーニ(株)製)
・シリカ粒子(“アドマファイン(登録商標)”SO−C6、平均粒子径2.2μm、屈折率1.46、アドマテックス(株)製)
・アルミナ粒子(“アドマファイン(登録商標)”AO−509、平均粒子径10μm、屈折率1.76、アドマテックス(株)製)。
・“三菱(登録商標)”カーボンブラックRCF#10(平均粒子径75nm、三菱化学(株)製)
・“三菱(登録商標)”カーボンブラックRCF#40(平均粒子径24nm、三菱化学(株)製)
・“三菱(登録商標)”カーボンブラックMCF#990(平均粒子径16nm、三菱化学(株)製)。
・“ビニレック(登録商標)”K(ポリビニルホルマール、チッソ(株)製)。
混練装置中に、構成要素[B]のエポキシ樹脂および組成に応じて構成要素[F]の熱可塑性樹脂を投入し、160℃の温度まで昇温させ、160℃の温度で30分間加熱混練を行った。ここで、構成要素[F]の熱可塑性樹脂を配合した場合は、後述する方法(8)によって、得られた樹脂組成物中に熱可塑性樹脂の固形物が存在しないことを確認した。その後、混錬を続けたまま60〜80℃の温度まで降温させ、組成に応じて構成要素[D]の架橋樹脂粒子および構成要素[E]のカーボンブラックを加えて30分間撹拌した。混錬を続けたまま55〜65℃の温度まで降温させ、構成要素[C]の硬化剤を加えて30分間撹拌し、エポキシ樹脂組成物を得た。
〈プリプレグの作製方法1〉
構成要素[B]、構成要素[C]、構成要素[D]、構成要素[E]および組成に応じて構成要素[F]を上記方法にて混練し、得られたエポキシ樹脂組成物を、ナイフコーターを用いて離型紙上に塗布し、樹脂目付けが37g/m2の樹脂フィルムを2枚作製した。次に、単位面積当たりの繊維が149g/m2になるようにシート状に一方向に配列させた構成要素[A]である炭素繊維に、得られた樹脂フィルム2枚を炭素繊維の両面から重ね、温度90℃、圧力2MPaの条件で加熱加圧してエポキシ樹脂組成物を含浸させ、Wfが67質量%の一方向プリプレグを得た。
構成要素[B]、構成要素[C]、構成要素[E]および組成に応じて構成要素[F]を上記方法にて混練し、得られたエポキシ樹脂組成物1を、ナイフコーターを用いて離型紙上に塗布し、樹脂目付けが17g/m2の樹脂フィルム1を2枚作製した。次に、単位面積当たりの繊維が149g/m2になるようにシート状に一方向に配列させた構成要素[A]である炭素繊維に、得られた樹脂フィルム1を2枚、炭素繊維の両面から重ね、温度90℃、圧力2MPaの条件で加熱加圧してエポキシ樹脂組成物を含浸させ、Wfが79質量%のプリプレグ前駆体を得た。
上記方法(3)で得られた一方向プリプレグ6枚を、炭素繊維の配列方向として、[0°/90°/0°/0°/90°/0°]構成で積層し、予備積層体とした。得られた予備積層体をオートクレーブにセットして0.6MPaの圧力で、室温から120℃の温度まで1分間に1.7℃ずつ昇温し、120℃の温度下で1時間かけて硬化して繊維強化複合材料を得た。
構成要素[D]の架橋樹脂粒子の屈折率n1を、JIS K 7142(1996)に記載のあるベッケ線法に従って測定した。
また、構成要素[D]および構成要素[E]を含まない以外は、上記方法(2)と同様にして得られたエポキシ樹脂組成物を、1mm厚の空隙を持った所定の型枠内に注入し、熱風オーブン中で室温から120℃の温度まで1分間に1.5℃ずつ昇温した後、120℃の温度下で1時間かけて硬化した。得られたエポキシ樹脂組成物の硬化物を縦20mm、横8mmに切り出し、JIS K 7142(1996)に記載のあるアッベ屈折計を用いて、屈折率n0(構成要素[F]を配合しない樹脂組成)またはn0’(構成要素[F]を配合した樹脂組成)を測定した。
各測定結果よりn1/n0またはn1/n0’を算出した。
上記方法(3)で得られたプリプレグを、2枚の表面の平滑なポリ四フッ化エチレン樹脂板間に挟持して密着させ、7日間かけて徐々に150℃迄温度を上昇させてゲル化および硬化させて板状の樹脂硬化物を作製した。硬化後、密着面と垂直な方向から切断し、その断面を研磨後、光学顕微鏡で200倍以上に拡大し、プリプレグの上下面が視野内に納まるようにして写真撮影した。断面写真の横方向の5ヵ所でポリ四フッ化エチレン樹脂板間の間隔を測定し、その平均値をプリプレグの平均厚みとした。
上記方法(3)で得られたプリプレグを、2枚の表面の平滑なポリ四フッ化エチレン樹脂板間に挟持して密着させ、7日間かけて徐々に150℃迄温度を上昇させてゲル化および硬化させて板状の樹脂硬化物を作製した。硬化後、密着面と垂直な方向から切断し、その断面を研磨後、光学顕微鏡で200倍以上に拡大し、プリプレグの上下面が視野内に納まるようにして写真撮影した。断面写真の横方向の5ヵ所でポリ四フッ化エチレン樹脂板間の間隔を測定し、その平均値をプリプレグの平均厚みとした。
混練装置中で構成要素[B]のエポキシ樹脂に、構成要素[F]の熱可塑性樹脂を配合し、160℃の温度まで昇温させ、160℃の温度で30分間加熱混練を行い、樹脂組成物を得た。得られた樹脂組成物の適量をスライドガラスに採り、カバーガラスで覆った後、透過観察型の光学顕微鏡を使用し、倍率5倍以上に拡大して、構成要素[F]の0.1μm以上の固形物の有無を確認した。
上記方法(2)で得られたエポキシ樹脂組成物の粘度は、動的粘弾性装置ARES−2KFRTN1−FCO−STD(ティー・エイ・インスツルメント社製)を用い、上下部測定冶具に直径40mmの平板のパラレルプレートを用い、上部と下部の冶具間距離が1mmとなるように該エポキシ樹脂組成物をセット後、ねじりモード(測定周波数:0.5Hz)で、測定温度範囲40〜140℃を昇温速度1.5℃/分で測定した。
上記方法(3)で得られたプリプレグのタック値を、以下のように測定した。18×18mmのカバーガラスを3.9Nの力で5秒間プリプレグに圧着し、タックテスタ(PICMAタックテスタII、東洋精機(株)製)を用いて、30mm/分の速度にて引っ張り、剥がれる際の抵抗力をタック値として測定した。測定数はn=7とし、最大および最小の2点を外した5点の平均値でタック値を評価した。
上記方法(4)で得られた繊維強化複合材料の外観品位を目視にて“ユラギ”や“イラツキ”などの外観斑の有無を評価した。
次の組成にて、上記方法(2)にてエポキシ樹脂組成物を調合した;
構成要素[B]として、“エポトート(登録商標)”YD128を20質量部、“jER(登録商標)”1001を50質量部、および“jER(登録商標)”154を30質量部、
構成要素[C]としてDICY7を3.5質量部および“オミキュア(登録商標)”24を2質量部、
構成要素[D]として、“ダイミックビーズ(登録商標)”UCN−5051Dを10質量部、
構成要素[E]として、“三菱(登録商標)”カーボンブラックRCF#40を1質量部。
表1および表2に示すように組成を変更した以外は実施例1と同様に、エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料を得た。結果を表1および表2に示す。
次の組成にて、上記方法(2)にてエポキシ樹脂組成物1を調合した;
構成要素[B]として、“エポトート(登録商標)”YD128を20質量部、“jER(登録商標)”1001を50質量部、および“jER(登録商標)”154を30質量部、
構成要素[C]としてDICY7を3.5質量部および“オミキュア(登録商標)”24を2質量部、
構成要素[E]として、“三菱(登録商標)”カーボンブラックRCF#40を1質量部。
構成要素[C]としてDICY7を3.5質量部および“オミキュア(登録商標)”24を2質量部、
構成要素[D]として、“ダイミックビーズ(登録商標)”UCN−5051Dを20質量部、
構成要素[E]として、“三菱(登録商標)”カーボンブラックRCF#40を1質量部。
表3および表4に示すように組成を変更した以外は実施例13と同様に、エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料を得た。結果を表5に示す。
(比較例1〜6)
表6に示すように組成を変更した以外は実施例1と同様に、エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料を得た。
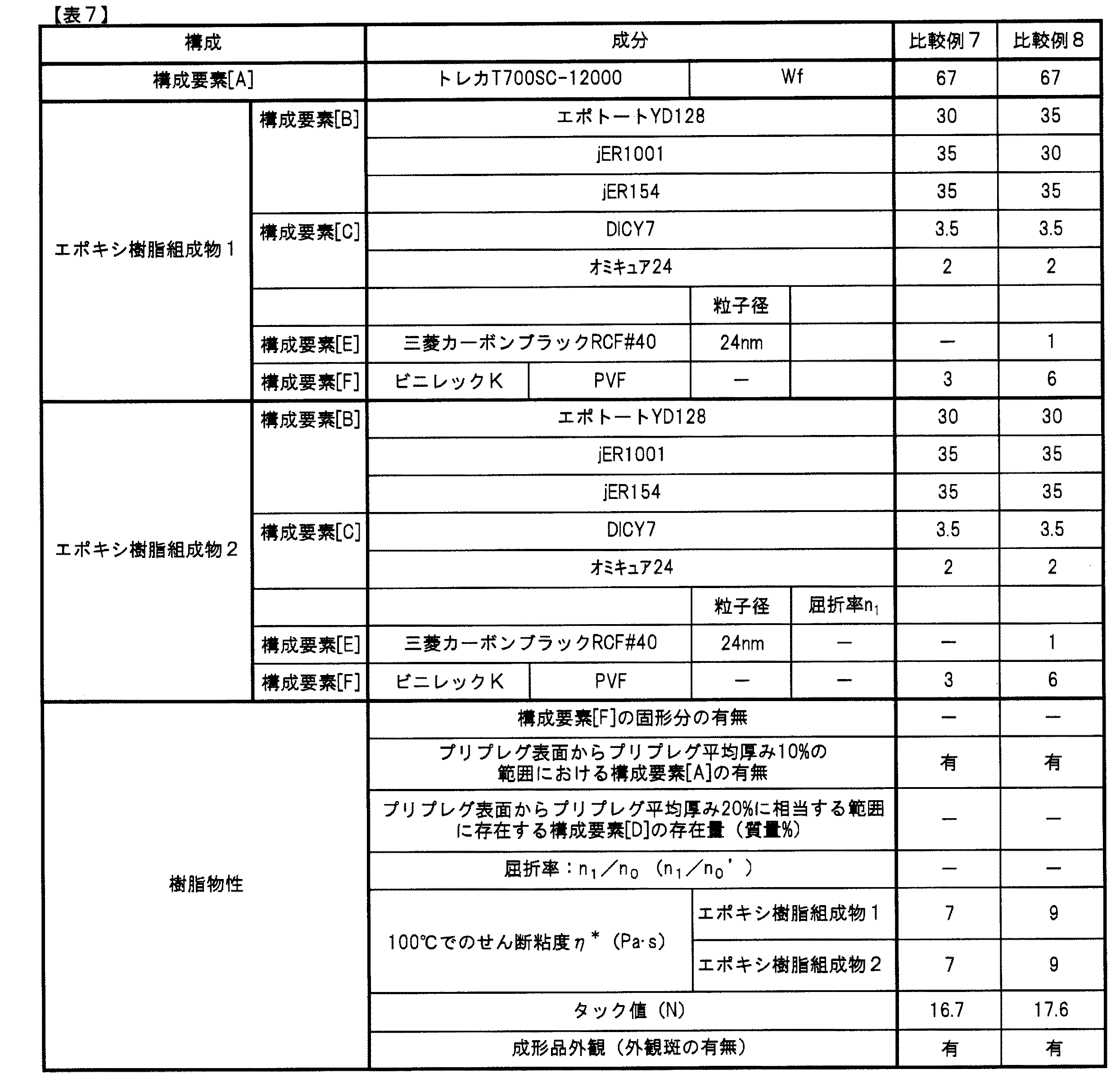
(比較例7、8)
表7に示すように組成を変更した以外は実施例13と同様に、エポキシ樹脂組成物、プリプレグおよび繊維強化複合材料を得た。
Claims (13)
- 下記構成要素[A]〜[E]を含むプリプレグであって、該プリプレグの表面から該プリプレグの平均厚み10%に相当する範囲は、構成要素[B]〜[E]で構成されており、かつ、下記条件(I)を満たしているプリプレグ;
[A]:強化繊維
[B]:1分子内に2個以上のエポキシ基を有するエポキシ樹脂
[C]:構成要素[B]を硬化しうる化合物
[D]:屈折率が1.49〜1.61の範囲にある架橋樹脂粒子
[E]:カーボンブラック
条件(I):構成要素[B]および構成要素[C]からなる樹脂組成物を硬化して得られるエポキシ樹脂硬化物の屈折率n0と、構成要素[D]の屈折率n1との関係が、0.92≦n1/n0≦1.05である。 - 構成要素[D]の85質量%以上が、前記プリプレグの表面から該プリプレグの平均厚み20%に相当する範囲に存在する、請求項1に記載のプリプレグ。
- さらに構成要素[F]を含むプリプレグであって、該プリプレグの表面から該プリプレグの平均厚み10%に相当する範囲は、構成要素[B]〜[F]で構成されており、かつ、下記条件(II)を満たしている、請求項1または2に記載のプリプレグ;
[F]:下記条件(i)を満たす熱可塑性樹脂
条件(i):構成要素[B]中に構成要素[F]を配合し160℃の温度で30分加熱して得られる樹脂組成物中に、0.1μm以上の構成要素[F]の固形物が存在しない;
条件(II):構成要素[B]、構成要素[C]および構成要素[F]からなる樹脂組成物を硬化して得られるエポキシ樹脂硬化物の屈折率n0’と、構成要素[D]の屈折率n1との関係が、0.92≦n1/n0’≦1.05である。 - 構成要素[B]〜[E]、および必要に応じて構成要素[F]からなるエポキシ樹脂組成物の100℃でのせん断粘度η*が2〜20Pa・sの範囲にある請求項1〜3のいずれかに記載のプリプレグ。
- 構成要素[D]の体積平均粒子径が10〜60μmの範囲にある、請求項1〜4のいずれかに記載のプリプレグ。
- 構成要素[E]の体積平均粒子径が1〜80nmの範囲にある、請求項1〜5のいずれかに記載のプリプレグ。
- 構成要素[D]が、エポキシ樹脂、ポリスチレンおよびポリウレタンからなる群から選ばれた少なくとも一種である、請求項1〜6のいずれかに記載のプリプレグ。
- 構成要素[D]を、構成要素[B]100質量部に対し、10〜70質量部含む、請求項1〜7のいずれかに記載のプリプレグ。
- 構成要素[E]を、構成要素[B]100質量部に対し、1〜5質量部含む、請求項1〜8のいずれかに記載プリプレグ。
- 構成要素[F]を、構成要素[B]100質量部に対し、1〜10質量部含む、請求項3〜9のいずれかに記載プリプレグ。
- 構成要素[A]が炭素繊維である、請求項1〜10のいずれかに記載のプリプレグ。
- まず構成要素[B]、構成要素[C]、構成要素[E]および必要に応じて構成要素[F]からなり、構成要素[D]を含まない樹脂フィルム1を、構成要素[A]の両側あるいは片側から含浸させプリプレグ前駆体を得た後、構成要素[B]〜[E]および必要に応じて構成要素[F]からなる樹脂フィルム2を該プリプレグ前駆体の両側あるいは片側に貼付することでプリプレグを得る、請求項1〜11のいずれかに記載のプリプレグの製造方法。
- 請求項1〜11のいずれかに記載のプリプレグを硬化させて得られる繊維強化複合材料。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012515833A JP5115675B2 (ja) | 2011-03-25 | 2012-03-22 | プリプレグ、および繊維強化複合材料 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011067357 | 2011-03-25 | ||
| JP2011067357 | 2011-03-25 | ||
| JP2012515833A JP5115675B2 (ja) | 2011-03-25 | 2012-03-22 | プリプレグ、および繊維強化複合材料 |
| PCT/JP2012/057310 WO2012133096A1 (ja) | 2011-03-25 | 2012-03-22 | プリプレグ、および繊維強化複合材料 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP5115675B2 true JP5115675B2 (ja) | 2013-01-09 |
| JPWO2012133096A1 JPWO2012133096A1 (ja) | 2014-07-28 |
Family
ID=46930824
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012515833A Expired - Fee Related JP5115675B2 (ja) | 2011-03-25 | 2012-03-22 | プリプレグ、および繊維強化複合材料 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20130316169A1 (ja) |
| EP (1) | EP2690128B1 (ja) |
| JP (1) | JP5115675B2 (ja) |
| KR (1) | KR101425334B1 (ja) |
| CN (1) | CN103443173B (ja) |
| TW (1) | TWI455972B (ja) |
| WO (1) | WO2012133096A1 (ja) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI602671B (zh) * | 2013-01-28 | 2017-10-21 | 東邦特耐克絲歐洲股份有限公司 | 浸漬強化纖維紗及其於製造複合材料之用途 |
| FR3001458B1 (fr) * | 2013-01-31 | 2015-04-10 | Centre Nat Rech Scient | Materiau composite conducteur et methode de fabrication dudit materiau composite conducteur |
| JP5966969B2 (ja) * | 2013-02-26 | 2016-08-10 | 東レ株式会社 | プリプレグの製造方法 |
| DE102013214801A1 (de) * | 2013-07-29 | 2015-01-29 | Bayerische Motoren Werke Aktiengesellschaft | Verfahren zum Herstellen einer Faserverbundanordnung mit einem Faserverbundbauteil und einem verbundenen Stützelement |
| CN105431475B (zh) * | 2013-08-01 | 2018-05-08 | 帝人株式会社 | 纤维增强复合材料和其制造方法 |
| ITMI20131648A1 (it) * | 2013-10-04 | 2015-04-05 | Saati Spa | Metodo per realizzare parti in composito avanzato, ad elevata finitura superficiale, e prodotto in composito avanzato, realizzato con tale metodo. |
| DE102014207785A1 (de) * | 2014-04-25 | 2015-10-29 | Evonik Degussa Gmbh | Verfahren zur Herstellung von lagerstabilen Epoxy-Prepregs und daraus hergestellte Composites auf Basis von radikalisch polymerisierbaren Säuren und Epoxiden |
| US9963588B2 (en) * | 2014-05-12 | 2018-05-08 | Diversified Chemical Technologies, Inc. | Sprayable, carbon fiber-epoxy material and process |
| EP3208078B1 (en) | 2014-10-17 | 2024-12-04 | Toray Industries, Inc. | Method for producing fiber-reinforced composite material, resin base and preform |
| JP6642423B2 (ja) * | 2015-02-27 | 2020-02-05 | 東レ株式会社 | 樹脂供給材料、強化繊維の使用方法、プリフォーム、および繊維強化樹脂の製造方法 |
| KR101786189B1 (ko) | 2015-08-07 | 2017-11-15 | 현대자동차주식회사 | 경량화 투명 복합재료 조성물, 상기 복합재료의 제조방법 및 그에 따른 복합재료 |
| CN109952182A (zh) * | 2016-10-26 | 2019-06-28 | 东丽株式会社 | 预浸料坯层叠体、纤维增强复合材料及纤维增强复合材料的制造方法 |
| JP7124687B2 (ja) * | 2018-12-20 | 2022-08-24 | トヨタ自動車株式会社 | 樹脂成形品 |
| CN115315471B (zh) * | 2020-04-01 | 2024-11-01 | 三菱瓦斯化学株式会社 | 预浸料、纤维增强复合材料、高压气体贮藏罐、预浸料的制造方法和高压气体贮藏罐的制造方法 |
| JPWO2022113976A1 (ja) * | 2020-11-27 | 2022-06-02 | ||
| US12528275B2 (en) | 2021-01-15 | 2026-01-20 | Toray Industries, Inc. | Laminate, method for producing same and prepreg |
| CN117319534A (zh) * | 2023-09-07 | 2023-12-29 | 深圳市零壹创新科技有限公司 | 一种电子设备保护壳 |
| WO2025254178A1 (ja) * | 2024-06-05 | 2025-12-11 | クラレノリタケデンタル株式会社 | 歯科用樹脂組成物 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001049013A (ja) * | 1999-05-31 | 2001-02-20 | Toray Ind Inc | プリプレグ及び炭素繊維強化複合材料 |
| JP2001114915A (ja) * | 1999-10-13 | 2001-04-24 | Toray Ind Inc | プリプレグ及び繊維強化複合材料 |
| JP2008214547A (ja) * | 2007-03-06 | 2008-09-18 | Toray Ind Inc | 繊維強化複合材料用プリプレグおよび繊維強化複合材料 |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4957801A (en) * | 1989-05-17 | 1990-09-18 | American Cyanamid Company | Advance composites with thermoplastic particles at the interface between layers |
| JPH03119038A (ja) * | 1989-09-29 | 1991-05-21 | Sumitomo Chem Co Ltd | 着色プリプレグ及び着色繊維強化樹脂成形体 |
| US5413847A (en) * | 1992-03-30 | 1995-05-09 | Toray Industries, Inc. | Prepreg and composite |
| ES2290051T3 (es) * | 1999-10-13 | 2008-02-16 | Toray Industries, Inc. | Preimpregnado y material compuesto reforzado con fibras. |
| JP2003048263A (ja) | 2001-08-07 | 2003-02-18 | Toray Ind Inc | 繊維強化複合材料およびその製造方法 |
| US20040034142A1 (en) * | 2002-05-13 | 2004-02-19 | Techno Polymer Co., Ltd., | Laser-marking thermoplastic resin composition |
| JP2004099814A (ja) | 2002-09-12 | 2004-04-02 | Toray Ind Inc | プリプレグおよび繊維強化複合材料 |
| US8021752B2 (en) * | 2004-02-27 | 2011-09-20 | Toray Industries, Inc. | Epoxy resin composition for carbon-fiber-reinforced composite material, prepreg, integrated molding, fiber-reinforced composite sheet, and casing for electrical/electronic equipment |
| WO2005083002A1 (ja) * | 2004-03-02 | 2005-09-09 | Toray Industries, Inc. | 繊維強化複合材料用エポキシ樹脂組成物、プリプレグ、および繊維強化複合材料 |
| JP2006328183A (ja) * | 2005-05-25 | 2006-12-07 | Toray Ind Inc | プリプレグおよび繊維強化複合材料 |
| JP4141487B2 (ja) * | 2006-04-25 | 2008-08-27 | 横浜ゴム株式会社 | 繊維強化複合材料用エポキシ樹脂組成物 |
| KR101426755B1 (ko) * | 2006-10-11 | 2014-08-06 | 스미토모 베이클라이트 가부시키가이샤 | 투명 복합 시트 |
| JP4829766B2 (ja) * | 2006-12-13 | 2011-12-07 | 横浜ゴム株式会社 | 繊維強化複合材料用エポキシ樹脂組成物 |
| US7790637B2 (en) | 2007-10-31 | 2010-09-07 | Apple Inc. | Composite laminate having an improved cosmetic surface and method of making same |
| EP2275489B1 (en) * | 2008-01-28 | 2019-11-13 | Kaneka Corporation | Rubbery polymer-containing resin composition, cured product thereof, and production method thereof |
| JP5478266B2 (ja) * | 2008-02-15 | 2014-04-23 | 株式会社クラレ | 硬化性樹脂組成物および樹脂硬化物 |
| US8658736B2 (en) * | 2008-09-29 | 2014-02-25 | Toray Industries, Inc. | Epoxy resin composition, prepreg and fiber-reinforced composite material |
| ES2531305T3 (es) * | 2009-03-24 | 2015-03-12 | Toray Industries | Composición de resina epoxi para material compuesto reforzado con fibra, impregnado previamente, y material compuesto reforzado con fibra |
| JP2013019796A (ja) * | 2011-07-12 | 2013-01-31 | Canon Inc | 放射線検出器 |
-
2012
- 2012-03-22 CN CN201280014282.XA patent/CN103443173B/zh not_active Expired - Fee Related
- 2012-03-22 KR KR1020137026878A patent/KR101425334B1/ko not_active Expired - Fee Related
- 2012-03-22 EP EP12763086.1A patent/EP2690128B1/en not_active Not-in-force
- 2012-03-22 JP JP2012515833A patent/JP5115675B2/ja not_active Expired - Fee Related
- 2012-03-22 WO PCT/JP2012/057310 patent/WO2012133096A1/ja not_active Ceased
- 2012-03-22 US US13/983,397 patent/US20130316169A1/en not_active Abandoned
- 2012-03-23 TW TW101110096A patent/TWI455972B/zh not_active IP Right Cessation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001049013A (ja) * | 1999-05-31 | 2001-02-20 | Toray Ind Inc | プリプレグ及び炭素繊維強化複合材料 |
| JP2001114915A (ja) * | 1999-10-13 | 2001-04-24 | Toray Ind Inc | プリプレグ及び繊維強化複合材料 |
| JP2008214547A (ja) * | 2007-03-06 | 2008-09-18 | Toray Ind Inc | 繊維強化複合材料用プリプレグおよび繊維強化複合材料 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20140016328A (ko) | 2014-02-07 |
| EP2690128A1 (en) | 2014-01-29 |
| TW201247752A (en) | 2012-12-01 |
| CN103443173A (zh) | 2013-12-11 |
| CN103443173B (zh) | 2014-08-27 |
| US20130316169A1 (en) | 2013-11-28 |
| WO2012133096A1 (ja) | 2012-10-04 |
| EP2690128A4 (en) | 2014-09-24 |
| JPWO2012133096A1 (ja) | 2014-07-28 |
| KR101425334B1 (ko) | 2014-08-01 |
| EP2690128B1 (en) | 2015-10-28 |
| TWI455972B (zh) | 2014-10-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5115675B2 (ja) | プリプレグ、および繊維強化複合材料 | |
| JP5159990B2 (ja) | プリプレグ及びその製造方法 | |
| JP5454138B2 (ja) | エポキシ樹脂組成物、繊維強化複合材料、およびその製造方法 | |
| CN104245804B (zh) | 纤维强化复合材料 | |
| JP5532549B2 (ja) | プリプレグおよび繊維強化複合材料の成形方法 | |
| JP5739361B2 (ja) | 繊維強化複合材料 | |
| TWI723188B (zh) | 預浸漬物及其製造方法 | |
| CN104220497B (zh) | 纤维强化复合材料 | |
| JP5723505B2 (ja) | 樹脂組成物、硬化物、プリプレグ、および繊維強化複合材料 | |
| WO2014030638A1 (ja) | エポキシ樹脂組成物、及びこれを用いたフィルム、プリプレグ、繊維強化プラスチック | |
| CN103857731A (zh) | 纤维强化树脂复合材料及其制造方法 | |
| WO2013046434A1 (ja) | ベンゾオキサジン樹脂組成物及び繊維強化複合材料 | |
| TWI793219B (zh) | 碳纖維之製造方法 | |
| CN104583284B (zh) | 纤维增强复合材料 | |
| JP7143473B2 (ja) | プリプレグ及びその製造方法、並びに繊維強化複合材料 | |
| JP2021513578A (ja) | プリプレグシート、及び低ボイド含有量繊維強化複合材料の製造に有用であるプリプレグスタック | |
| JP2021516705A (ja) | プリプレグ、積層体、繊維強化複合材料、及び繊維強化複合材料の製造方法 | |
| JP2015108078A (ja) | エポキシ樹脂組成物、プリプレグ及び繊維強化複合材料 | |
| JP2006219513A (ja) | エポキシ樹脂組成物・プリプレグ・繊維強化複合材料 | |
| JP2014111772A (ja) | 繊維強化複合材料の成形方法 | |
| JP4480854B2 (ja) | プリプレグ及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120918 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121001 |
|
| R151 | Written notification of patent or utility model registration |
Ref document number: 5115675 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R151 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151026 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |