JP4635356B2 - カルボシラン及びポリカルボシラン - Google Patents
カルボシラン及びポリカルボシラン Download PDFInfo
- Publication number
- JP4635356B2 JP4635356B2 JP2001071336A JP2001071336A JP4635356B2 JP 4635356 B2 JP4635356 B2 JP 4635356B2 JP 2001071336 A JP2001071336 A JP 2001071336A JP 2001071336 A JP2001071336 A JP 2001071336A JP 4635356 B2 JP4635356 B2 JP 4635356B2
- Authority
- JP
- Japan
- Prior art keywords
- carbon atoms
- group
- polycarbosilane
- carbosilane
- independently
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images












Landscapes
- Silicon Polymers (AREA)
- Materials For Photolithography (AREA)
Description
【発明の属する技術分野】
本発明は、新規なカルボシラン、それを用いて合成した新規なポリカルボシラン及びその製造方法に関する。
【0002】
【従来の技術】
オリゴカルボシランは従来Grinard法若しくはWurtz法によって合成されている。例えば、下記に示す反応により合成することが可能であると報告されている(K.Nate, M. Ishikawa, H.Ni, H.Watanabe, Y.Saheki, Organometallics, 6, 1673 (1987))。
【化4】
【発明が解決しようとする課題】
しかし、上述した方法では重合度のコントロールが困難であるため所望の分子量の化合物がなかなか得られないという欠点があった。本発明者はこの様な従来技術の欠点を改良することを目的に研究を進めた結果、特定の有機金属基を末端に有するカルボシランを原料として用いることにより従来技術の欠点を克服することが可能であることを知り本発明に到達した。また、本発明の有機金属基を末端に有する本発明のカルボシラン若しくはポリカルボシランはそれ自体有用な有機材料であると共に、マクロモノマーとしてこれを原料とした新規な材料への展開が期待されるものである。
【0004】
本発明の方法が重合度のコントロールされたポリカルボシランの合成を可能にしたのは以下の理由による。
(1)原料として分子の末端にスズ等の特定の有機金属基を有する本発明のカルボシランを用いたこと。
(2)開始剤としてブチルリチウム等の有機典型金属化合物を用いたこと。
この両化合物を組合せて用いることによって、重合反応を行った後でも分子末端の有機金属基は安定に保存されている。従って、更に、開始剤である有機典型金属化合物を添加すると再び反応が始まりポリカルボシランの重合度を増加させることができる。
あるいは、反応時間を変化させることによりポリカルボシランの重合度は変えられる。これらの方法により重合度をコントロールすることができる。
【0005】
【課題を解決するための手段】
本発明は以下の構成よりなる。
[1]下記一般式(1)で表わされるカルボシラン。
【化5】
【0006】
[2]前記一般式(1)において、Aが1,4−フェニレン、2,5−ジメチル−1,4−フェニレン、1,4−キシリレン、2,5−チオフェン若しくは4,4′−ビフェニレンである[1]項に記載のカルボシラン。
[3]前記一般式(1)において、R1、R2、R3、R4、R5、R6、R7、R8、R9及びR10が何れもメチル基である[1]または[2]項に記載のカルボシラン。
[4]前記一般式(2)で示される、ポリスチレン換算重量平均分子量が800〜100,000であるポリカルボシラン。
【0007】
【化6】
【0008】
[5]下記一般式(3)で示される、ポリスチレン換算重量平均分子量が800〜100,000であるポリカルボシラン。
【化7】
【0009】
[6]前記一般式(2)または(3)においてAが1,4−フェニレン、2,5−ジメチル−1,4−フェニレン、1,4−キシリレン、2,5−チオフェン若しくは4,4′−ビフェニレンである[4]または[5]項に記載のポリカルボシラン。
[7]前記[1]項に記載の同種若しくは異種のカルボシランを有機典型金属化合物を開始剤として反応させることを特徴とする[4]項若しくは[5]項に記載のポリカルボシランの製造方法。
【0010】
【発明の実施の形態】
本発明の一般式(1)で示されるカルボシランは例えば以下の式に従って合成することができる。更にそれを有機典型金属化合物を開始剤として反応させることにより本発明のポリカルボシランを得ることができる。尚、本発明で言うポリカルボシランはいわゆるオリゴマーからポリマーまで含み、GPCにより測定したポリスチレン換算重量平均分子量が800〜100,000までのものを言う。
【0011】
【化8】
【0012】
尚、上式では式を単純化するためカルボシラン及びポリカルボシランの化学式が一般式(1)及び(2)と完全には同一でないが、反応させる塩化シラン及び有機典型金属化合物の種類を2種類とすることにより一般式(1)及び(2)のR1〜R10が互いに異なる基からなる化合物も得ることはできる。
【0013】
一般式(1)、(2)若しくは(3)で示される化合物の珪素若しくは4価の金属の置換基であるR1、R2、R3、R4、R5、R6、R7、R8、R9、R10、R41、R51、R91及びR101については、既に述べたが、それらのうちでもアルキル基は炭素数1〜6のものが好ましく、アルケニル基若しくはアルキニル基は炭素数2〜6のものが好ましく、アリール基は炭素数6〜12のものが好ましい。4価の金属の置換基であるR1、R2、R3、R6、R7及びR8の場合はこれらのうちの少なくとも1個がメチル基若しくはエチル基であることが好ましく、すべてがメチル基若しくはエチル基であることがより好ましい。
【0014】
一般式(1)、(2)若しくは(3)においてMはSn、Ge若しくはPb原子を表わすが、Snが最も好ましい。
更に2価の基であるA及びA1は既に述べたが、ここで、炭素環式基とは脂環式基若しくは芳香族炭化水素基等の単環式炭化水素基、縮合多環式炭化水素基及び橋かけ環式炭化水素基の内の少なくとも1種を必須成分とし、必要によりこれらに任意の置換基若しくは橋かけ基、例えば、炭化水素基、または珪素、酸素、硫黄及び窒素原子の1種以上を含有する炭化水素基あるいはスルホン基等のようにこれらの原子のみから構成される基により置換されあるいは橋かけされている基をいう。
【0015】
具体的には1,4−フェニレン、2,5−ジメチル−1,4−フェニレン、1,4−キシリレン、2,5−チオフェン若しくは4,4′−ビフェニレンが好ましく、これらのうちでは2,5−チオフェンが溶媒に対する溶解性に優れている。
【0016】
一般式(1)で示されるカルボシランの具体例として以下の式で表わされる化合物を挙げることができる。
【化9】
上述したような方法で合成した末端に有機金属基を有するカルボシランを有機典型金属化合物を開始剤として重合反応させることにより本発明のポリカルボシランを得ることができる。既に述べたように本発明のポリカルボシランは反応終了後もその末端の有機金属基は安定に存在しているために、開始剤を添加すると更に反応は進行する。あるいは反応時間を変化させることによっても生成するポリカルボシランの重合度をコントロールすることができる。
【0018】
重合体は単一の原料から合成することも可能であるが、2種類以上のカルボシランを最初から混合することによるランダムポリマーの合成を行うことができる。この様な方法により溶媒に対する溶解性に優れたポリマーを合成することができる。あるいは2種類のポリマーが別々に保有する異なる物性上の特徴を併せ持つポリマーを得るためには、予め両ポリマーを別々に合成しておき、それらを混合し、開始剤の存在下反応を行うことによりブロックポリマーを合成することもできる。
【0019】
重合反応用の開始剤として用いられる有機典型金属化合物の有機基は炭素数1〜20のアルキル基または炭素数2〜20のアルケニル基若しくはアルキニル基であり、好ましくは炭素数1〜6のアルキル基、炭素数2〜6のアルケニル基若しくはアルキニル基または炭素数6〜12のアリール基である。その典型金属はリチウム、ナトリウム、マグネシウム、カルシウム若しくは亜鉛であり、好ましくはリチウムである。リチウム化合物の中でもブチルリチウム、メチルリチウムが好ましい。多価金属の場合、置換基の少なくとも1個が上述した有機基であればよい。
【0020】
本発明の製造方法において、反応は溶媒中で行うことができる。原料化合物と反応せず、且つ原料化合物を溶解することができるものなら特に限定されないが、例えば、汎用溶媒であるテトラヒドロフラン、ジオキサン、エチレングリコールジエチルエーテル、ジエチレングリコールジメチルエーテル等のエーテル類、ヘキサン、ヘプタン、シクロヘキサン等の脂肪族、若しくは脂環式炭化水素、トルエン、キシレン等の芳香族炭化水素等を単一で若しくは混合して使用することができる。反応温度は−40〜30℃、好ましくは−20〜10℃である。
【0021】
このようにして合成された本発明のカルボシランあるいはポリカルボシランの用途としては液晶化合物、導電性材料、光スイッチ素子、メモリー素子のようなセンサー材料、有機感光材料、光メモリー材料等の機能性材料あるいはセラミック前駆体等が考えられる。
以下、本発明を実施例により具体的に開示するが、本発明は、これらの具体例に限定されるものではない。
【0022】
【実施例】
実施例1
【化10】
(1) 1,4−ビス(ジメチルヒドロシリル)ベンゼンの調製
滴下ロート2本と還流冷却器付き500ml三口フラスコに削状Mg(5.35g、220mmol)を入れ窒素置換した。窒素雰囲気下で無水THF(150ml)を入れ、一方の滴下ロートに1,4−ジブロモベンゼン(23.6g、100mmol)と無水THF(25.0ml)、もう一方の滴下ロートにジメチルクロロシラン(25.5ml、230mmol)を入れ均一溶液にし、激しく撹拌した。約3.0mlを滴下し、ドライヤーで加熱した。約10分後、還流が始まり、保温しながら残りを滴下した。滴下終了後、ホッチングマグネチックスターラーを用いて約30分間加熱還流を行った。氷浴を用いてフラスコを冷却し、エーテル(60.0ml)を加えて、飽和NH4Cl水溶液(100ml)で中和、加水分解を行った。その後、セライト−エーテルを用いた濾過でマグネシウム塩を除去し、エーテル(100ml)で分液抽出し、無水MgSO4で乾燥した。乾燥後、乾燥剤を濾別し、常圧蒸留及び減圧濃縮で溶媒を除去後、精留ビーズを詰めた蒸留塔を用いて減圧蒸留により目的物である1,4−ビス(ジメチルヒドロシリル)ベンゼンを得た。IR測定により2100cm-1にSi−Hの伸縮振動が確認された。蒸留条件は102〜104℃/16mmHg、収量は11.3g、収率は41%であった。
【0024】
(2) ヒドロシリル基の塩素化反応
窒素置換した還流冷却器付き100ml丸底フラスコに1,4−ビス(ジメチルヒドロシリル)ベンゼンを入れ、窒素雰囲気下で脱水CCl4(45.0ml)を加え均一溶液とした。PdCl2(0.021g、0.12mmol)を加え、オイルバスを用いて加熱還流した。約3時間加熱還流後、室温に冷却し、反応溶液の一部をマイクロシリンジで抜き取りGC分析で反応終了を確認した。減圧で溶媒を除去後、クーゲルロール蒸留装置を用いて塩素化物の単離を行った。外気にできる限り触れないよう窒素置換した受器を用いて蒸留することにより目的物である1,4−ビス(ジメチルクロロシリル)ベンゼンを得た。蒸留終了後、窒素バルーンをつけて次の反応まで保存した。蒸留条件は80〜110℃/0.9mmHg、収率は90%であった。なお、次の反応の反応器への移動については受器を反応器に連結して無水THFで流し込んだ。
【0025】
(3) トリメチルスタニルリチウムの調製
100ml滴下ロートと温度計を付けた200ml三つ口フラスコを窒素置換して、Arガス雰囲気下で無水THF(80ml)を入れ、塊状金属Li(400mmol)を細かく切り入れた。滴下ロートにMe3SnCl(1mol/LのTHF溶液100ml)を入れ、氷塩浴を用いて−3〜0℃の範囲にフラスコを冷却した。約5.0ml滴下して20〜30分後、0〜1℃まで温度上昇して溶液が黄緑色へ変化した。その後、約1.5時間−3〜0℃に温度を保ち、残りを滴下した。
【0026】
(4) ビス(スタニルシリル)化合物の調製
(a) 1,4−ビス(ジメチルトリメチルスタニルシリル)ベンゼンの調製
窒素置換した100ml滴下ロート付き300ml三つ口フラスコにArガス雰囲気下でフラスコへ、無水THF(60ml)と1,4−ビス(ジメチルクロロシリル)ベンゼン(50mmol)を入れ、均一溶液にした。滴下ロートに注射器でスタニルリチウムのTHF溶液を移し、氷塩液を用いて約0℃にフラスコを冷却し、0℃を越えないように滴下した。滴下終了後、20〜30分間撹拌放置し、反応溶液の一部をマイクロシリンジで抜き取りGC分析で反応終了を確認した。減圧蒸留で溶媒を除去後、セライト−ヘキサン(100ml)を用いた濾過で塩化リチウムを除去し、溶液に無水MgSO4を入れ乾燥した。乾燥後、減圧濃縮で溶媒を除去し、精留ビーズを詰めた蒸留塔で減圧蒸留して、沸点98〜130℃/1.3mmHgの白色固体である1,4−ビス(ジメチルトリメチルスタニルシリル)ベンゼン15.6g(収率60%)を得た。また、得られた生成物の1H−NMR分析、13C−NMR分析、IR分析及びGC分析から構造を決定した。ヘキサンを用いて再結晶化して、白色結晶化物を得た。融点は97〜98℃であった。
【0027】
実施例2〜4
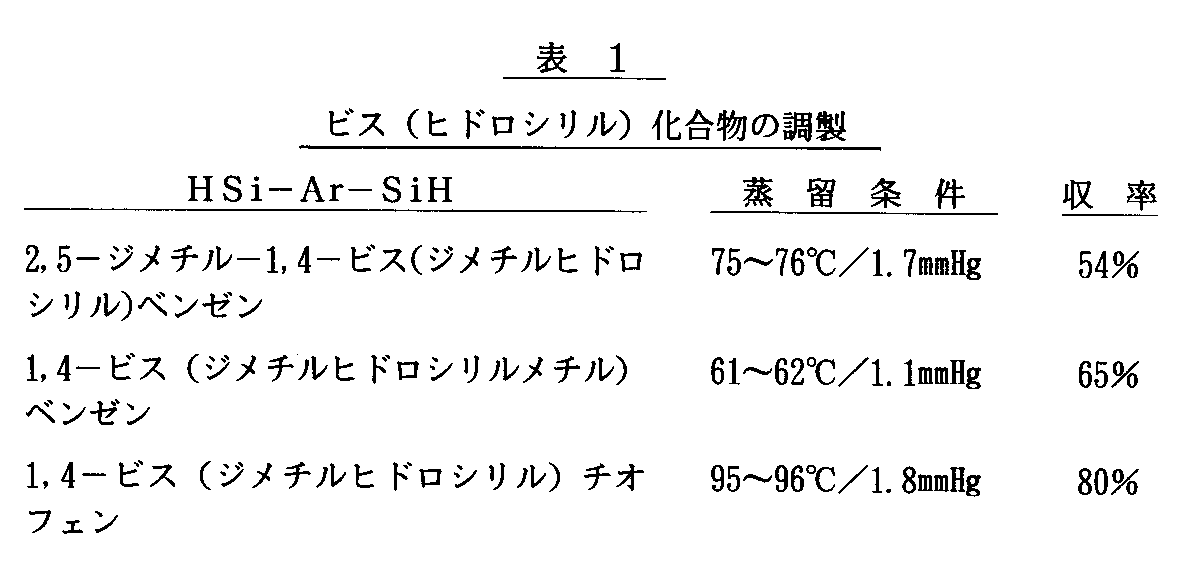
実施例1(1)の1,4−ジブロモベンゼンに代えて2,5−ジメチル−1,4−ジブロモベンゼン、1,4−ビス(ブロモメチル)ベンゼン、及び1,4−ジブロモチオフェンをそれぞれ用いて同様に反応を行うことにより、2,5−ジメチル−1,4−ビス(ジメチルヒドロシリル)ベンゼン、1,4−ビス(ジメチルヒドロシリルメチル)ベンゼン及び1,4−ビス(ジメチルヒドロシリル)チオフェンをそれぞれ表1に示す収率で得た。
【0028】
【表1】
更に、実施例1(2)の1,4−ビス(ジメチルヒドロシリル)ベンゼンに代えて2,5−ジメチル−1,4−ビス(ジメチルヒドロシリル)ベンゼン、1,4−ビス(ジメチルヒドロシリルメチル)ベンゼン、及び1,4−ビス(ジメチルヒドロシリル)チオフェンをそれぞれ用いて同様に反応を行うことにより、2,5−ジメチル−1,4−ビス(ジメチルクロロシリル)ベンゼン、1,4−ビス(ジメチルクロロシリルメチル)ベンゼン及び1,4−ビス(ジメチルクロロシリル)チオフェンをそれぞれ表2に示す収率で得た。
【0030】
【表2】
更に、実施例1(4)(a)における1,4−ビス(ジメチルクロロシリル)ベンゼンに代えて2,5−ジメチル−1,4−ビス(ジメチルクロロシリル)ベンゼン、1,4−ビス(ジメチルクロロシリルメチル)ベンゼン、及び1,4−ビス(ジメチルクロロシリル)チオフェンをそれぞれ用いて同様に反応を行うことにより、2,5−ジメチル−1,4−ビス(ジメチルトリメチルスタニルシリル)ベンゼン、1,4−ビス(ジメチルトリメチルスタニルシリルメチル)ベンゼン及び1,4−ビス(ジメチルトリメチルスタニルシリル)チオフェンをそれぞれ表3に示す収率で得た。
【0032】
【表3】
実施例5 ビス(スタニルシリル)化合物の重合反応
【化11】
【0034】
実施例6〜9
実施例5の1,4−ビス(ジメチルトリメチルスタニルシリル)ベンゼンに代えて、それぞれ2,5−ジメチル−1,4−ビス(ジメチルトリメチルスタニルシリル)ベンゼン(実施例6)、1,4−ビス(ジメチルトリメチルスタニルシリルメチル)ベンゼン(実施例7)、1,4−ビス(ジメチルトリメチルスタニルシリル)チオフェン(実施例8)または4,4′−ビス(ジメチルトリメチルスタニルシリル)ビフェニル(実施例9)を用いて実施例5と同様に反応を行い各ポリマーを得た。結果は表4に示す。
【0035】
尚、実施例8では表4に示す所定の反応を行った後、更に開始剤を追加して重合を行った結果得られたポリマーの分子量が増加していた。GPCにより、測定したポリスチレン換算分子量の測定結果は以下の通りである(溶媒THF)。
【表4】
尚、実施例7で得られたポリマーの1H−NMR分析チャートを図3、13C−NMR分析チャートを図4に示す。更に、実施例8で1段目の反応で得られたポリマーの1H−NMR分析チャートを図5に、2段目の反応で得られたポリマーの1H−NMR分析チャートを図6に、13C−NMR分析チャートを図7に、実施例9で得られたポリマーの1H−NMR分析チャートを図8に、13C−NMR分析チャートを図9に、UV分析チャートを図10に、それぞれ示す。
【0038】
実施例10
【化12】
【0039】
【発明の効果】
特定の有機金属基末端を有する本発明の新規なカルボシランを原料として有機典型金属化合物を開始剤として合成することにより、重合度の制御されたポリカルボシランを合成することが可能になり、また、本発明の新規なカルボシラン及びポリカルボシランはそれ自体有用な工業材料であると共に、マクロモノマーとして有用な高分子材料の原料となる可能性を有している化合物である。
【図面の簡単な説明】
【図1】実施例5で得られた本発明のポリカルボシランの1H−NMR分析チャートである。
【図2】実施例5で得られた本発明のポリカルボシランの13C−NMR分析チャートである。
【図3】実施例7で得られた本発明のポリカルボシランの1H−NMR分析チャートである。
【図4】実施例8で得られた本発明のポリカルボシランの13C−NMR分析チャートである。
【図5】実施例8の最初の重合で得られた本発明のポリカルボシランの1H−NMR分析チャートである。
【図6】実施例8の追加の重合で得られた本発明のポリカルボシランの1H−NMR分析チャートである。
【図7】実施例8の追加の重合で得られた本発明のポリカルボシランの13C−NMR分析チャートである。
【図8】実施例9の重合で得られた本発明のポリカルボシランの1H−NMR分析チャートである。
【図9】実施例9の重合で得られた本発明のポリカルボシランの13C−NMR分析チャートである。
【図10】実施例9の重合で得られた本発明のポリカルボシランのUV分析チャートである。
【図11】実施例10の共重合で得られた本発明のポリカルボシラン共重合体の1H−NMR分析チャートである。
【図12】実施例10の共重合で得られた本発明のポリカルボシランの13C−NMR分析チャートである。
Claims (7)
- 下記一般式(1)で表わされるカルボシラン。
- 前記一般式(1)において、Aが1,4−フェニレン、2,5−ジメチル−1,4−フェニレン、1,4−キシリレン、2,5−チオフェン若しくは4,4′−ビフェニレンである請求項1に記載のカルボシラン。
- 前記一般式(1)において、R1、R2、R3、R4、R5、R6、R7、R8、R9及びR10が何れもメチル基である請求項1または2に記載のカルボシラン。
- 下記一般式(2)で示される、ポリスチレン換算重量平均分子量が800〜100,000であるポリカルボシラン。
- 下記一般式(3)で示される、ポリスチレン換算重量平均分子量が800〜100,000であるポリカルボシラン。
- 前記一般式(2)または(3)においてAが1,4−フェニレン、2,5−ジメチル−1,4−フェニレン、1,4−キシリレン、2,5−チオフェン若しくは4,4′−ビフェニレンである請求項4または5に記載のポリカルボシラン。
- 請求項1に記載の同種若しくは異種のカルボシランを有機典型金属化合物を開始剤として反応させることを特徴とする請求項4若しくは5に記載のポリカルボシランの製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001071336A JP4635356B2 (ja) | 2000-03-14 | 2001-03-14 | カルボシラン及びポリカルボシラン |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000070561 | 2000-03-14 | ||
| JP2000-70561 | 2000-03-14 | ||
| JP2001071336A JP4635356B2 (ja) | 2000-03-14 | 2001-03-14 | カルボシラン及びポリカルボシラン |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2001328991A JP2001328991A (ja) | 2001-11-27 |
| JP4635356B2 true JP4635356B2 (ja) | 2011-02-23 |
Family
ID=26587452
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2001071336A Expired - Fee Related JP4635356B2 (ja) | 2000-03-14 | 2001-03-14 | カルボシラン及びポリカルボシラン |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4635356B2 (ja) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100521809B1 (ko) * | 2002-10-30 | 2005-10-14 | 삼성전자주식회사 | 포토레지스트용 폴리카르보메틸실란 유도체 및 이를포함한 포토레지스트 조성물 |
| JP5614933B2 (ja) * | 2006-02-16 | 2014-10-29 | ユニバーシティ オブ コネチカット | 前駆体ポリマからの導電性ポリマ、その作製方法およびその使用 |
| JP5152783B2 (ja) * | 2007-10-31 | 2013-02-27 | 公立大学法人大阪市立大学 | Si−Si結合を有する高分子化合物の製造方法 |
| US20120108761A1 (en) | 2009-05-22 | 2012-05-03 | Lintec Corporation | Formed article, method of producing same, electronic device member, and electronic device |
| TWI552883B (zh) | 2011-07-25 | 2016-10-11 | Lintec Corp | Gas barrier film laminates and electronic components |
| KR102064143B1 (ko) | 2011-11-04 | 2020-01-08 | 린텍 가부시키가이샤 | 가스 배리어 필름 및 그 제조 방법, 가스 배리어 필름 적층체, 전자 디바이스용 부재, 그리고 전자 디바이스 |
| EP2830068B1 (en) | 2012-03-22 | 2018-05-23 | LINTEC Corporation | Transparent conductive laminate and electronic device or module |
| CN113226746B (zh) | 2018-12-27 | 2023-08-11 | 琳得科株式会社 | 阻气性层叠体 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000053770A (ja) * | 1998-08-05 | 2000-02-22 | Chisso Corp | ケイ素−スズ結合を有する化合物を用いたケイ素−ケイ素結合を有する化合物の製造方法及び新規なケイ素−スズ結合を有する化合物 |
-
2001
- 2001-03-14 JP JP2001071336A patent/JP4635356B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2001328991A (ja) | 2001-11-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JPH023417B2 (ja) | ||
| JP4635356B2 (ja) | カルボシラン及びポリカルボシラン | |
| JP6246225B2 (ja) | 液状スズ(ii)アルコキシドの製造方法 | |
| JPH0583568B2 (ja) | ||
| US5633312A (en) | Process for preparing polyorganosilane | |
| US5874514A (en) | Siloxane unsaturated hydrocarbon based polymers | |
| US6596833B2 (en) | Carbosilane and polycarbosilane | |
| JP2721108B2 (ja) | 片末端ビニル重合性シリコーンの製造方法 | |
| Xu et al. | Synthesis and characterization of oligomeric silsesquioxane with pendent carboxylic acid groups | |
| US5489662A (en) | Process for the preparation of organosilicon polymer | |
| CN109320443B (zh) | 化合物的制备方法及包含其的聚合物的制备方法 | |
| JP4797440B2 (ja) | 芳香族化合物の製造方法 | |
| US5288832A (en) | Poly(silethynylenedisiloxane) and method for the manufacture thereof | |
| RU2466156C2 (ru) | Дендронизованные полиарилсиланы и способ их получения | |
| Malanga et al. | Head‐to‐head polymers. XXIV. Synthesis of head‐to‐head polyisobutylene by Grignard coupling reaction | |
| Lozano et al. | Aromatic polyamides and polyimides derived from 3, 3′‐diaminobiphenyl: Synthesis, characterization, and molecular simulation study | |
| JP3083614B2 (ja) | 有機ポリゲルマンの製法 | |
| JP3673856B2 (ja) | ビナフチルシロキサン系ポリマーおよびその製造方法 | |
| JP3053736B2 (ja) | ケイ素含有重合体の製造方法 | |
| Koopmann et al. | Synthesis and properties of poly (dimethyldiphenylsilylenemethylene) | |
| Li et al. | SYNTHESIS AND CHARACTERIZATION OF A NOVEL REACTIVE LADDER-LIKE POLYSILSESQUIOXANE WITH LATERAL BROMOPHENYL GROUPS. | |
| JP3017323B2 (ja) | 珪素を有する新規化合物及びその製造方法 | |
| JP3636904B2 (ja) | カルボラン含有ケイ素系重合体及びその製造方法 | |
| JPS63162687A (ja) | チオフエン系化合物及びその製造方法 | |
| JP2003252998A (ja) | カルボシランボラジン系共重合ポリマー及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20070926 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100810 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20100811 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101004 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20101026 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20101108 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131203 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4635356 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| R371 | Transfer withdrawn |
Free format text: JAPANESE INTERMEDIATE CODE: R371 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131203 Year of fee payment: 3 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131203 Year of fee payment: 3 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |