JP4635162B2 - 芳香族ジアミンの製造方法及び芳香族ジアミン化合物 - Google Patents
芳香族ジアミンの製造方法及び芳香族ジアミン化合物 Download PDFInfo
- Publication number
- JP4635162B2 JP4635162B2 JP2000128431A JP2000128431A JP4635162B2 JP 4635162 B2 JP4635162 B2 JP 4635162B2 JP 2000128431 A JP2000128431 A JP 2000128431A JP 2000128431 A JP2000128431 A JP 2000128431A JP 4635162 B2 JP4635162 B2 JP 4635162B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- aromatic
- aromatic diamine
- general formula
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- NLTCSYNRJNXHJM-UHFFFAOYSA-N CC(C)[N](C)(C)C Chemical compound CC(C)[N](C)(C)C NLTCSYNRJNXHJM-UHFFFAOYSA-N 0.000 description 1
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Compounds Containing Sulfur Atoms (AREA)
- Furan Compounds (AREA)
- Pyrrole Compounds (AREA)
Description
【発明の属する技術分野】
本発明は、芳香族ポリアミンの製造方法に関する。さらに詳しくは、医薬品や感光性ポリマー合成などの中間体として重要な少なくとも1つのシスー共役ジエニル基を有する芳香族ポリアミンの製造方法に関する。さらに本発明は、医薬品や感光性ポリマー合成などの中間体として芳香族ジアミン化合物に関する。
【0002】
【従来の技術】
芳香族アミノ化合物の製造法としては白金、ニッケル、パラジウムなどの貴金属触媒を用いた接触還元、亜鉛、鉄、スズ、塩化スズなどによる還元、硫化ナトリウム、硫化アンモニウムなど硫化物による還元などがよく用いられている。しかし、これらの製造方法は多くの欠点や問題点があり適用範囲が極めて狭い。すなわち、欠点とは、
1)アゾベンゼン誘導体など副生成物ができ易く低収率である。
2)還元反応後に生成してくる硫黄などの分離が困難で後処理が煩雑。
3)ハロゲン原子、カルボニル基、ニトリル基などの官能基が共存すると該官能基が反応したり脱離したりしてニトロ基を選択的に還元することが困難。
【0003】
従って、少なくとも2つ以上のニトロ基を有する芳香族ポリニトロ化合物にこれらの還元法を適用して、少なくとも2つ以上のアミノ基を有する芳香族ポリアミノ化合物が製造できるのは極く単純な芳香族ポリニトロ化合物に限定され、エステル基、アルケニル基、ジエニル基などの官能基が共存する芳香族ポリニトロ化合物への適用はほとんど不可能に近い状況であった。
【0004】
最近、ハロゲン原子、カルボニル基、ニトリル基などの官能基が共存する芳香族モノニトロ化合物を温和な条件でニトロ基のみを選択的に還元する方法が開発されている。例えば、
1)塩化第二鉄触媒共存下、N,N−ジメチルヒドラジンによる4−ニトロ安息香酸メチルなどの4−アミノ安息香酸メチルへの還元。
S. R. Boothroyd and M. A. Kerr, Tetrahedron Lett., 36, 2411(1995).
2)酸化鉄−酸化マグネシウム触媒共存下、ヒドラジンによる4−クロルニトロベンゼンなどの4−クロルアニリンへの還元。
P. S. Kumbhar, J. Sanchez-Valente, and F. Figueras, Tetrahedron Lett., 39, 2573-2574(1998).
3)ジエチルクロロホスファイト/3級アミンによる4−ニトロベンズアルデヒドなどの4−アミノベンズアルデヒドへの還元。
B. Fischer and L. Sheihet, J. Org. Chem., 63, 393-395(1998).
さらには、
4)塩化アンモニウム飽和水溶液を含むエチルアルコール中、インジウム金属粉末による、ハロゲン原子、カルボキシル基、カルボン酸エステル、ニトリル基などが共存するニトロベンゼン誘導体のアニリン誘導体への還元。
C. D. Moody and M. R. Pitts, Synlett., 1028(1998).
【0005】
しかし、これらの還元法の芳香族ジニトロ化合物への適用は報告されておらず、エステル基、アルケニル基、ジエニル基などの官能基が共存する芳香族ポリニトロ化合物へ適用できる還元法の出現が強く求められている。
【0006】
【発明が解決しようとする課題】
本発明は、医薬品や感光性ポリマー合成などの中間体である芳香族ポリアミン、特に、少なくとも1つのシスー共役ジエニル基またはフェニル基を有する芳香族ジアミンを、相当する芳香族ポリニトロ化合物、特に芳香族ジニトロ化合物から、温和な条件の還元反応により高収率で製造する方法を提供することを目的とする。
さらに本発明の目的は、医薬品や感光性ポリマー合成などの中間体である新規芳香族ジアミン化合物を提供することにある。
【0007】
【課題を解決するための手段】
本発明者らは、上記の課題を解決すべく鋭意研究を重ねた結果、芳香族ジニトロ化合物を非プロトン性極性溶媒中、4級アンモニウム塩水溶液共存下、インジウムメタルにより還元することにより、温和な条件下で、従来の合成法と比べて、極めて高収率で芳香族ジアミン等の芳香族ポリアミンが得られることを見出し、この知見に基づいて本発明を完成するに至った。
【0008】
すなわち、本発明は、非プロトン性極性溶媒中、4級アンモニウム塩水溶液共存下インジウムメタルにより、少なくとも2つのニトロ基を有する芳香族ポリニトロ化合物を還元して少なくとも2つのアミノ基を有する芳香族ポリアミンを製造することを特徴とする芳香族ポリアミンの製造方法に関する。
【0009】
上記製造方法において、芳香族ポリアミンは、少なくとも1つのシスー共役ジエニル基を有するものであることができる。
【0010】
さらに、上記製造方法において、芳香族ポリアミンは一般式[1]又は一般式[2]で表される芳香族ジアミンであることができる。
【0011】
【化4】
【0012】
【化5】
【0013】
上記製造方法においてシスー共役ジエニル基は、フリル基、チエニル基又はピロリル基であることができる。
【0014】
また、上記製造方法は、還元反応を55℃以下で行い、芳香族ポリニトロ化合物の消失を確認した後、60℃以上で還元反応を完結させることができる。
【0015】
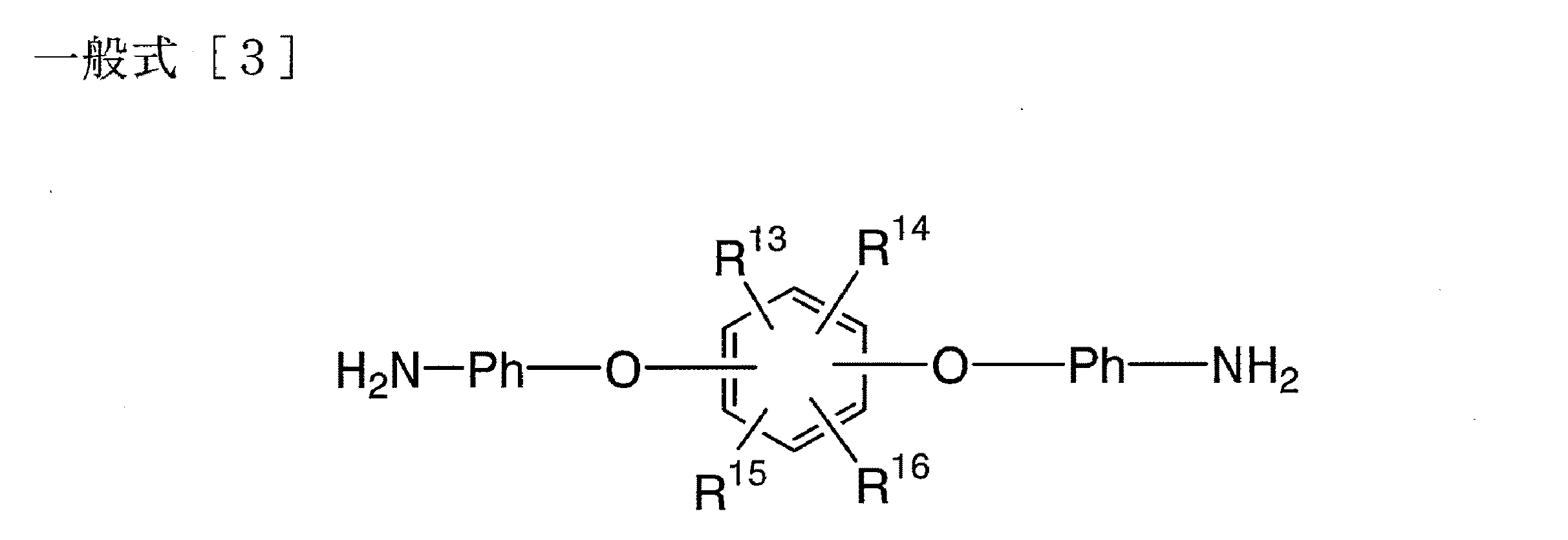
さらに本発明は下記一般式[3]で表される芳香族ジアミン化合物に関する。
【化6】
【0016】
【発明の実施の形態】
本発明の製造方法は、原料として少なくとも2つのニトロ基を有する芳香族ポリニトロ化合物を用いる。少なくとも2つのニトロ基を有する芳香族ポリニトロ化合物としては、例えば、一般式[4]又は一般式[5]で表される芳香族ポリニトロ化合物を挙げることができる。
【化7】
【0017】
上記芳香族ポリニトロ化合物は、例えば、以下の方法により合成することができる。
(1)市販の3,5−ジニトロベンゾイルクロリドなどのポリニトロ置換ベンゾイルクロリドとフルフリルアルコールなどのシス−共役ジエニル基を含むアルコールとのエステル化反応。
(2)市販の3,5−ジニトロベンジルアルコールなどのポリニトロアレーン構造を有するアルコールと2-フロイルクロリドなどのシス−共役ジエニル基を含む酸クロリドとのエステル化反応。
(3)市販の3,5−ジヒドロキシベンジルアルコールなどのポリヒドロキシベンジルアルコール誘導体と4-フルオロニトロベンゼンなどのハロゲン置換ニトロベンゼンとのエーテル化反応の後、残存するアルコールの上記(2)のエステル化反応による共役ジエニル基の導入。
【0018】
本発明の製造方法においては、芳香族ポリアミンは、少なくとも1つのシスー共役ジエニル基を有するものであることができる。シスー共役ジエニル基を有する芳香族ポリニトロ化合物は、一般に、シスー共役ジエニル基を還元することなしにニトロ基のみを選択的に還元することが難しい。しかるに、本発明の製造方法であれば、シスー共役ジエニル基を有する芳香族ポリニトロ化合物であっても、シスー共役ジエニル基を還元することなしにニトロ基のみを選択的に還元することができる。
【0019】
シスー共役ジエニル基としては、フリル基、チエニル基、ピロリル基、ピラニル基、イソベンゾフラニル基、インドリジニル基、キノリジニル基等を挙げることができ、中でも、フリル基、チエニル基またはピロリル基であることが好ましい。
【0020】
さらに、本発明の製造方法において、芳香族ポリアミンは上記一般式[1]又は一般式[2]で表される芳香族ジアミンであることができる。
一般式[1]中、R1、R2、R3及びR4の少なくとも1つはシスー共役ジエニル基を含む1価の有機基であり、残余は、それぞれ独立に水素、炭素数1〜20のアルキル基又は炭素数1〜20のアルコキシル基である。また、一般式[2]中、R5、R6、R7、R8、R9、R10、R11及びR12の少なくとも1つはシスー共役ジエニル基を含む1価の有機基であり、残余は、それぞれ独立に水素、炭素数1〜20のアルキル基又は炭素数1〜20のアルコキシル基である。
一般式[1]及び一般式[2]において、シスー共役ジエニル基を含む1価の有機基としては、例えば、−CH2O−CO−D、−O−CO−D、−CO−O−CH2−D、−CH2O−CH2−D、−O−CH2−D、−NH−CO−D、−CO−NH−CH2−D等を挙げることができる。但し、Dはシスー共役ジエニル基を表す。
炭素数1〜20のアルキル基としては、例えば、メチル基、エチル基、イソプロピル基、ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、デシル基、ラウリル基、等を挙げることができる。これらの中でも、メチル基、エチル基、イソプロピル基、ブチル基、ペンチル基等であることが好ましい。
炭素数1〜20のアルコキシル基としては、例えば、メトキシ基、エトキシ基、イソプロピルオキシ基、ブトキシ基、ペンチルオキシ基、ヘキシルオキシ基、ラウリルオキシ基等を挙げることができる。これらの中でも、メトキシ基、エトキシ基、イソプロピルオキシ基、ブトキシ基、ペンチルオキシ基等であることが好ましい。
【0021】
一般式[1]中、X1及びX2はそれぞれ独立に酸素若しくは硫黄、又は炭素数1〜4の置換基を有していてもよいアルキレン基、アルキリデン基若しくはアルキレンオキシ基である。一般式[2]中、Yは酸素又は硫黄であるか、炭素数1〜4の置換基を有していてもよいアルキレン基、アルキリデン基又はアルキレンオキシ基である。炭素数1〜4の置換基を有していてもよいアルキレン基、アルキリデン基又はアルキレンオキシ基としては、例えば、メチレン基、エチレン基、プロピレン基、イソプロピリデン基、ブチレン基、1,1,1,3,3,3-ヘキサフルオロイソプロピリデン基、メチレンオキシ基、エチレンオキシ基、プロピレンオキシ基、ブチレンオキシ基等を挙げることができる。
【0022】
一般式[1]中、Ar1及びAr2はそれぞれ独立に2価の芳香族基であり、2価の芳香族基としては、例えば、フェニレン基(1,4-又は1,3-)、ナフチレン基(1,4-)等を挙げることができる。
一般式[1]中、l1、l2、m1及びm2はそれぞれ独立に0又は1である。ただし、l1及び/又はl2が1のとき,m1及び/又はm2も1である。l1、l2、m1及びm2は、好ましくはl1=l2=m1=m2=1またはl1=l2=m1=m2=0である。
一般式[2]中、n1は0又は1である。
【0023】
一般式[1]又は一般式[2]で表される芳香族ジアミンの具体例を以下に示す。
【化8】
本発明に用いられる非プロトン性極性溶媒としては、例えば、N,N−ジメチルホルムアミド(DMF)、N,N−ジメチルアセトアミド、N−メチル−2−ピロリドン、γ−ブチロラクトン、ジメチルスルホキシドなどを挙げることができ、これらを単独又は混合して用いることができる。非プロトン性極性溶媒としては、DMFが好ましい。
本発明に用いられる4級アンモニウム塩としては、塩化アンモニウム、臭化アンモニウム、よう化アンモニウム、硫酸アンモニウム、硝酸アンモニウム、テトラメチルアンモニウムクロリド、テトラメチルアンモニウムブロミド、テトラエチルアンモニウムクロリド、テトラエチルアンモニウムブロミドなどを挙げることができ、これらを単独又は混合して用いることができる。4級アンモニウム塩としては、塩化アンモニウムが好ましい。
4級アンモニウム塩水溶液の量は、インジウムメタル1g当り、4級アンモニウム塩飽和水溶液として1〜10ml、より好ましくは、2〜5mlとすることが適当である。
【0025】
本発明の方法では、インジウムメタルにより芳香族ポリニトロ化合物を還元する。インジウムメタル量の量は、芳香族ポリニトロ化合物1g当り、0.6〜20g、好ましくは1.5〜10g、より好ましくは3〜6gとすることが適当である。
還元温度は、室温〜85℃、好ましくは40〜75℃、より好ましくは45〜55℃とすることが適当である。40℃以上とすることで還元反応の速度が速くなり比較的短時間に反応を終了させることができ、75℃以下であれば副反応も起こりにくく、高い収量で芳香族ポリアミン化合物を得ることができる。還元反応は、芳香族ポリニトロ化合物が反応して消失したことを確認後、60〜70℃にて反応を完結させるのが良い。芳香族ポリニトロ化合物の消失は、例えば、薄層クロマトグラフィー(TLC)により確認することができる。なお、生成した芳香族ポリアミン化合物の酸化や着色を防ぐため、上記還元反応は、不活性ガス、例えば、アルゴンガスあるいは窒素ガス、雰囲気下で行うことが望ましい。
【0026】
本発明は、下記一般式[3]で表される新規芳香族ジアミン化合物を包含する。
【化11】
本発明の新規芳香族ジアミン化合物の具体例は、前記1-(9)〜1-(16)で表される化合物である。特に、実施例3、4及び6で合成された化合物が好ましい。
【0027】
本発明の新規芳香族ジアミン化合物は、下記一般式[6]に示す芳香族ジニトロ化合物を原料として前記本発明の芳香族ポリアミンの製造方法を用いて製造することができる。
【0028】
【化12】
一般式[6]で表されるジニトロ化合物は、前記一般式[4]で示される芳香族ポリニトロ化合物と同様の方法で、市販または公知の化合物を原料として合成することができる。
【0029】
【実施例】
以下、実施例により本発明を具体的に説明するが、本発明はこれら実施例のみに限定されるものではない。
下記の表に実施例で得られた芳香族ポリアミンの構造式と収率を示す。
【0030】
【表1】
実施例1:3,5−ジアミノベンジル−2−フロエートの製造
3,5−ジニトロベンジル−2−フロエートの製造:攪拌機、水冷式還流冷却管、温度計及び滴下ロートを備えた500mlセパラブルフラスコ中で、3,5−ジニトロベンジルアルコール37.0gをピリジン100mlに溶解した。0〜5℃に冷却しながら、2−フロイルクロリド53.1gを滴下した。滴下終了後、反応液を室温に戻し、2時間攪拌した。400mlの水を加え、析出した固体をろ過した。得られた個体をエチルアルコール/水(80/20、容積比)から再結晶し、3,5−ジニトロベンジル−2−フロエート47gを得た。収率86%。融点116.0〜117.7℃。
【0032】
3,5−ジアミノベンジル−2−フロエートの製造:攪拌機、水冷式還流冷却管、温度計及び窒素ガス導入管を備えた1リットルのセパラブルフラスコ中で、3,5−ジニトロベンジル−2−フロエート23.6gを300mlのN,N−ジメチルホルムアミド(DMF)に溶解した。この溶液にインジウム金属粉末130gを分散し、窒素気流下、195mlの塩化アンモニウム飽和水溶液を加え52℃で4時間還元反応を行った。反応は薄層クロマトグラフィー(TLC)で追跡した。反応終了後、反応液に酢酸エチル800ml及び水400mlを加え、固体をろ別した。ろ液を分液し、酢酸エチル層を200mlの水及び150mlの飽和食塩水で洗浄した後、硫酸マグネシウムで乾燥した。減圧下、酢酸エチルを留去し、3,5−ジアミノベンジル−2−フロエートの粗結晶15.03gを得た。収率80%。イソプロピルアルコールから再結晶し、融点119.0〜121.1℃の淡黄色プリズム状結晶を得た。
【0033】
IR(KBr disc): 3419, 3349, 3218,3142,1707, 1600, 1476, 1353, 1209, 1174, 1122, 1077, 947, 771 and 754 cm-1
1HNMR(500MHz, CDCl3): δ 3.61(bs, NH2), 5,16(s, 2H), 6.00(t, J=1.66Hz, 1H), 6.18(d, J=1.66Hz, 2H), 6.50(dd, J=3.32, 1.66Hz, 1H), 7.21(dd J= 3.32, 0.66Hz, 1H), 7.58(dd, J=1.66, 0.66Hz, 1H)
13CNMR(500MHz, CDCl3): δ 66.6, 101.6, 105.7, 111.8, 118.1, 137.9, 144.7, 146.3, 147.7, 158.6
MS: 232(M+)
実施例2:3,5−ジアミノ安息香酸フルフリルエステルの製造
3,5−ジニトロ安息香酸フルフリルエステルの製造:攪拌機、水冷式還流冷却管、温度計及び滴下ロートを備えた1リットルのセパラブルフラスコ中で、フルフリルアルコール29.4gをDMF200ml及びピリジン80mlに溶解した。15〜17℃に冷却しながら、250mlのDMFに3,5−ジニトロベンゾイルクロリド69.15gを溶解した溶液を滴下した。滴下終了後、反応液を室温に戻し、3時間半攪拌した。1.5規定塩酸800mlを加え析出した固体をろ過した。得られた個体を少量の冷メタノールで洗浄後、減圧乾燥して82.02gの3,5−ジニトロ安息香酸フルフリルエステルを得た。収率94%。
【0035】
3,5−ジアミノ安息香酸フルフリルエステルの製造:攪拌機、水冷式還流冷却管、温度計及び窒素ガス導入管を備えた2リットルのセパラブルフラスコ中で、3,5−ジニトロ安息香酸フルフリルエステル43.8gを500mlのDMFに溶解した。この溶液にインジウム金属粉末200gを分散し、窒素気流下、300mlの塩化アンモニウム飽和水溶液を加え52〜53℃で2時間還元反応を行ない、TLCで3,5−ジニトロ安息香酸フルフリルエステルが完全に反応したことを確認した後、さらに、62〜63℃で1時間反応を続けた。反応終了後、実施例1と同様の後処理を行い、3,5−ジアミノ安息香酸フルフリルエステルの粗結晶31.4gを得た。収率90%。イソプロピルアルコールから再結晶し、融点132.6〜134.3℃の淡黄色針状結晶を得た。
【0036】
IR(KBr disc): 3431, 3332, 3212, 3117(w), 3061(w), 2955(w), 1713(s), 1628, 1501, 1465, 1379, 1351, 1293, 1228(s), 1198, 1152, 1097, 1015, 981, 918, 861, 822(w), 769 cm-1
1HNMR(500MHz, CDCl3): δ 3.66(s, NH2), 5,25(s, 2H), 6.17(t, J=1.98Hz, 1H), 6.37(dd, J=3.30, 1.98Hz, 1H), 6.46(d, J=3.30Hz, 1H), 6.78(d, J= 1.98Hz, 2H), 7.44(dd, J=1.98, 0.66Hz, 1H)
13CNMR(500MHz, CDCl3): δ 58.4, 105.7, 107.0, 110.5, 110.6, 131.7, 143.2, 147.4, 149.6, 166.4(-CO-O)
実施例3:3,5−ビス(4−アミノフェノキシ)ベンジル−2−フロエートの製造
3,5−ビス(4−ニトロフェノキシ)ベンジルアルコールの製造:攪拌機、水冷式還流冷却管、温度計及び滴下ロートを備えた500mlのセパラブルフラスコに、3,5−ジヒロキシベンジルアルコール28g,炭酸カリウム60.72g及びN,N−ジメチルアセタミド300mlを加え、83〜84℃に加熱し、4−フルオロニトロベンゼン56.4gを5時間かけて滴下した。滴下終了後、さらに4時間半85℃で攪拌し反応を完結させた。反応終了後、析出した固体をろ別し、酢酸エチルでよく洗浄した。ろ液と酢酸エチル洗浄液とを合わせ、減圧蒸留により酢酸エチル及び大部分のN,N−ジメチルアセタミドを留去して粘調な油状液体を得た。油状液体に酢酸エチル500mlを加え飽和食塩水100mlで3回洗浄した後、硫酸ナトリウムで乾燥した。減圧下、酢酸エチルを留去して粗生成物77.45gの固体を得た。固体にメタノール750mlを加え5時間還流した後、メタノールに不溶な固体をろ別した。ろ液からメタノールを減圧下に留去して3,5−ビス(4−ニトロフェノキシ)ベンジルアルコール54.54gを得た。収率71%。融点137〜140℃。
3,5−ビス(4−ニトロフェノキシ)ベンジル−2−フロエートの製造:攪拌機、水冷式還流冷却管、温度計及び滴下ロートを備えた1リットルのセパラブルフラスコ中で、3,5−ビス(4−ニトロフェノキシ)ベンジルアルコール38.2gをDMF330ml及びピリジン100mlに溶解した。11〜12℃に冷却しながら、2−フロイルクロリド15.66gを滴下した。滴下終了後、反応液を室温に戻し、4時間攪拌した。実施例2の3,5−ジニトロ安息香酸フルフリルエステルの製造の時と同様の後処理を行い、3,5−ビス(4−ニトロフェノキシ)ベンジル−2−フロエート46.85gを得た。収率98%。融点121〜124℃
【0038】
3,5−ビス(4−アミノフェノキシ)ベンジル−2−フロエートの製造:攪拌機、水冷式還流冷却管、温度計及び窒素ガス導入管を備えた1リットルのセパラブルフラスコ中で、3,5−ビス(4−ニトロフェノキシ)ベンジル−2−フロエート46.85gを300mlのDMFに溶解した。この溶液にインジウム金属粉末136gを分散し、窒素気流下、204mlの塩化アンモニウム飽和水溶液を加え50〜51℃で12時間還元反応を行ない、TLCで3,5−ビス(4−ニトロフェノキシ)ベンジル−2−フロエートが完全に反応したことを確認した後、さらに、68〜70℃で1時間半反応を続けた。反応終了後、実施例1と同様の後処理を行い、41.0gの油状粗生成物を得た。油状粗生成物をカラムクロマトグラフィーにより精製し、3,5−ビス(4−アミノフェノキシ)ベンジル−2−フロエート31.95gを得た。収率80%。イソプロピルアルコールから再結晶し、融点117〜121℃の淡黄色プリズム状結晶を得た。
【0039】
IR(KBr disc): 3417, 3332, 3231, 1719, 1613, 1592, 1507, 1475, 1453, 1400, 1305, 1213, 1122, 1008, 965, 838 and 763 cm-1
1HNMR(500MHz, CDCl3): δ 3.59(bs, -NH2), 5.18(s, 2H), 6.49(t, J=2.37Hz, 1H), 6.50(dd, J=3.72, 1.69Hz, 1H), 6.61(d, J=2.37Hz, 2H), 6.66 & 6.86(ABq, J=8.79Hz, 4H,4H), 7.16(dd, J=3.72, 1.01Hz, 1H), 7.57(dd, J=1.69, 1.01Hz, 1H)
13CNMR(500MHz, CDCl3): δ 65.9, 105.9, 111.8, 116.2, 118.2, 121.5, 138.1, 143.0, 144.4, 146.5, 147.9, 158.3, 160.3
MS: 416(M+)
実施例4:3,5−ビス(4−アミノフェノキシ)ベンジルフルフリルエーテルの製造
3,5−ビス(4−ニトロフェノキシ)ベンジルヨージドの製造:攪拌機、水冷式還流冷却管及び滴下ロートを備えた1リットルのセパラブルフラスコ中に、実施例3で製造した3,5−ビス(4−ニトロフェノキシ)ベンジルアルコール51.34g、テトラエチルアンモニウムヨージド68.85g及びクロロホルム400mlを加えた。この混合液を攪拌しながら室温下、三弗化ほう素ジエチルエーテル錯体28.52gを滴下した。滴下後、6時間半還流した。反応液を室温に冷却し、炭酸水素ナトリウムの飽和水溶液180mlを加え、クロロホルム層を分液した。クロロホルム層を10%チオ硫酸ナトリウム水溶液200mlで2回、飽和食塩水200mlで2回洗浄した後、硫酸ナトリウムで乾燥した。クロロホルムを減圧下で留去し、粗生成物59.68gの固体を得た。粗生成物をシリカゲルカラムクロマトグラフィーにより精製し、3,5−ビス(4−ニトロフェノキシ)ベンジルヨージド43.33gの薄黄色結晶を得た。収率66%。融点159.5〜162.5℃
【0041】
3,5−ビス(4−ニトロフェノキシ)ベンジルフルフリルエーテルの製造:攪拌機、水冷式還流冷却管及び温度計を備えた1リットルのセパラブルフラスコ中に、3,5−ビス(4−ニトロフェノキシ)ベンジルヨージド43.33g、フルフリルアルコール8.62g及び使用直前に蒸留したテトラヒドロフラン400mlを加えた。この混合液に水素化ナトリウム2.33gの粉末を20℃に保ちながら加えていった。反応液を室温に戻し1時間半攪拌した。反応液からテトラヒドロフランを減圧下留去し、残さに酢酸エチル300mlを加え、10%チオ硫酸ナトリウム水溶液120mlで1回、飽和食塩水120mlで2回洗浄した後、硫酸マグネシウムで乾燥した。減圧下酢酸エチルを留去して3,5−ビス(4−ニトロフェノキシ)ベンジルフルフリルエーテル39.28gの粘調液体を得た。収率97%。
【0042】
3,5−ビス(4−アミノフェノキシ)ベンジルフルフリルエーテルの製造:攪拌機、水冷式還流冷却管、温度計及び窒素ガス導入管を備えた1リットルのセパラブルフラスコ中で、3,5−ビス(4−ニトロフェノキシ)ベンジルフルリルエーテル39.20gを300mlのDMFに溶解した。この溶液にインジウム金属粉末136gを分散し、窒素気流下、204mlの塩化アンモニウム飽和水溶液を加え51〜53℃で11時間還元反応を行ない、TLCで3,5−ビス(4−ニトロフェノキシ)ベンジルフルフリルエーテルが完全に反応したことを確認した後、さらに、62〜64℃で1時間反応を続けた。反応終了後、実施例1と同様の後処理を行い、33.28gの油状粗生成物を得た。油状粗生成物をカラムクロマトグラフィーにより精製し、3,5−ビス(4−アミノフェノキシ)ベンジルフルフリルエーテル24.53gを得た。収率72%。イソプロピルアルコールから再結晶し、融点82〜84℃の淡黄色プリズム状結晶を得た。
【0043】
IR(KBr disc): 3429, 3327, 3220, 3116, 2921(vw), 2860(vw), 1635, 1595, 1507(vs), 1458, 1355, 1323, 1301, 1275, 1213, 1150, 1122, 1070, 994, 912, 838 and 743 cm-1
1HNMR(500MHz, CDCl3): δ3.54(bs, 4H), 4.39(s, 2H), 4.43(s, 2H), 6.27(d, J=3.1Hz, 1H), 6.32(dd, J=3.1, 1.7Hz, 1H), 6.47(t, J=2.1Hz, 1H), 6.55(d,J=2.1Hz, 2H), 6.65&6.85(Abq, J=8.6Hz, 4H,4H), 7.39(dd, J=1.7, 0.7Hz, 1H)
13CNMR(500MHz, CDCl3): δ 63.8, 71.4, 105.7, 109.4, 109.9, 110.2, 116.2, 121.2, 140.6, 142.7, 142.8, 148.1, 151.5, 160.2
実施例5:3,3′−ジアミノ−4,4′−ジ−2−フロイルアミノビフェニルの製造
3,3′−ジニトロ−4,4′−ジ−2−フロイルアミノビフェニルの製造:攪拌機、水冷式還流冷却管、温度計及び滴下ロートを備えた500mlのセパラブルフラスコ中で、3,3′−ジニトロ−4,4′−ジアミノビフェニル10.96gをDMF200mlに溶解したのち、ピリジン20mlを加えた。13〜14℃に冷却しながら、2−フロイルクロリド12.53gを滴下した。滴下終了後、反応溶液を室温に戻し、5時間攪拌を続けた。2規定塩酸80ml、次いで水100mlを加え、析出した固体をろ過した・固体をよく水洗した後、減圧乾燥して3,3′−ジニトロ−4,4′−ジ−2−フロイルアミノビフェニル15.0gを得た。収率81%
【0045】
3,3′−ジアミノ−4,4′−ジ−2−フロイルアミノビフェニルの製造:攪拌機、水冷式還流冷却管、温度計及び窒素ガス導入管を備えた1リットルのセパラブルフラスコ中で、3,3′−ジニトロ−4,4′−ジ−2−フロイルアミノビフェニル14.8g、インジウム金属粉末52g及び400mlのDMFを仕込んだ。この分散溶液に、窒素気流下、78mlの塩化アンモニウム飽和水溶液を加え52〜53℃で20時間還元反応を行ない、TLCで3,3′−ジニトロ−4,4′−ジ−2−フロイルアミノビフェニルが完全に反応したことを確認した後、さらに、64〜65℃で2時間反応を続けた。反応終了後、実施例1と同様の後処理を行い、12.0gの粉末状粗生成物を得た。粗生成物をカラムクロマトグラフィーにより精製し、3,3′−ジアミノ−4,4′−ジ−2−フロイルアミノビフェニル9.02gを得た。収率70%。N−メチル−2−ピロリドン/酢酸エチル(25/75、容積比)から再結晶し、分解点256〜259℃の黄色針状結晶を得た。
【0046】
IR(KBr disc): 3439, 3340, 3198, 3129, 1649, 1587, 1465, 1409, 1310, 1223, 1167, 1013, 883, 860, 797, 759 cm-1
1HNMR(400MHz, d6-DMSO): δ 5.01(s, -NH2), 6.68(dd, J=3.42, 1.95 Hz, 2H), 6.80(m,2H), 7.00(d, J=1.95 Hz, 2H), 7.22 (d, J=8.3 Hz, 2H), 7.29(d, J=3.41 Hz, 2H), 7.91(d, J=1.95, 2H), 9.58(s, -CONH-)
13CNMR(400MHz, d6-DMSO): δ 112.0, 114.0, 114.4, 114.7, 121.9, 126.8, 138.8, 143.1, 145.4, 147.7, 156.4
MS; 402(M+)
実施例6:3,5−ビス(4−アミノフェノキシ)ベンジルベンゾエートの製造
3,5−ビス(4−ニトロフェノキシ)ベンジルベンゾエートの製造:攪拌機、水冷式還流冷却管、温度計及び滴下ロートを備えた200mlの四つ口フラスコ中で、実施例3で製造した3,5−ビス(4−ニトロフェノキシ)ベンジルアルコール5.43gをDMF40mlに溶解したのち、ピリジン10mlを加えた。混合液を18℃に冷却しながら、ベンゾイルクロリド2.4gを滴下した。滴下終了後、反応溶液を室温に戻し、2時間攪拌を続けた。2規定塩酸50ml、次いで水100mlを加え、析出した固体をろ過した・固体をよく水洗した後、減圧乾燥して3,5−ビス(4−ニトロフェノキシ)ベンジルベンゾエート6.35gを得た。収率93%
【0048】
3,5−ビス(4−アミノフェノキシ)ベンジルベンゾエートの製造:攪拌機、水冷式還流冷却管、温度計及び窒素ガス導入管を備えた200mlの四つ口フラスコ中で、3,5−ビス(4−ニトロフェノキシ)ベンジルベンゾエート5.13g、インジウム金属粉末16.9g及び45mlのDMFを仕込んだ。この分散溶液に、窒素気流下、25.4mlの塩化アンモニウム飽和水溶液を加え50〜51℃で6時間還元反応を行ない、TLCで3,5−ビス(4−ニトロフェノキシ)ベンジルベンゾエートが完全に反応したことを確認した後、さらに、61〜62℃で1時間半反応を続けた。反応終了後、実施例1と同様の後処理を行い、4.38gの油状粗生成物を得た。粗生成物をカラムクロマトグラフィーにより精製し、ガラス状の3,5−ビス(4−アミノフェノキシ)ベンジルベンゾエート3.16gを得た。収率70%。
【0049】
IR(KBr disc): 3445, 3364, 3219, 3041, 1715, 1616, 1594, 1506, 1450, 1373, 1274, 1211, 1122, 1070, 1025, 966, 832 and 713 cm-1
1HNMR(500MHz, CDCl3): δ 3.60(bs, -NH2), 5,21(s, 2H), 6.49(bs, 1H), 6.63(d, J=1.66Hz, 2H), 6.66 & 6.87(ABq, J=8.64Hz, 4H, 4H), 7.44(t J=7.64Hz, 2H), 7.56(t, J=7.64Hz, 1H), 8.02(d, J=7.64Hz, 2H)
13CNMR(500MHz, CDCl3): δ 66.0, 105.7, 109.6, 116.2, 121.3, 128.3, 129.7, 129.9, 133.0, 138.5, 143.0, 147.9, 160.3, 166.2
実施例7:3,5−ビス(4−アミノフェノキシ)ベンジルアルコールの製造
攪拌機、水冷式還流冷却管、温度計及び窒素ガス導入管を備えた200mlの四つ口フラスコ中で、実施例3で製造した3,5−ビス(4−ニトロフェノキシ)ベンジルアルコール5.73g、インジウム金属粉末20g及び60mlのDMFを仕込んだ。この分散溶液に、窒素気流下、30mlの塩化アンモニウム飽和水溶液を加え52〜53℃で7時間還元反応を行ない、TLCで3,5−ビス(4−ニトロフェノキシ)ベンジルアルコールが完全に反応したことを確認した後、さらに、62〜63℃で2時間反応を続けた。反応終了後、実施例1と同様の後処理を行い、4.92gの油状粗生成物を得た。粗生成物をカラムクロマトグラフィーにより精製し、ガラス状の3,5−ビス(4−アミノフェノキシ)ベンジルアルコール3.89gを得た。収率81%。室温で放置しておくと結晶化した。
イソプロピルアルコールより再結晶し、融点100.2〜101.9℃の淡黄色プリズム状結晶を得た。
【0051】
IR(KBr disc): 3411, 3334, 3200, 1612, 1599, 1504, 1451, 1316, 1211, 1117, 1017, 970 and 841 cm-1
1HNMR(500MHz, CDCl3): δ 2.17(s, -OH), 3.55(bs, -NH2), 4.52(s, 2H), 6.48 (t, J=1.83 Hz, 1H), 6.53(d, J=1,83 Hz, 2H), 6.65 & 6.85 (ABq, J=8.71 Hz, 4H, 4H)
13CNMR(500MHz, CDCl3): δ 64.9, 105.5, 108.6, 116.2, 121.3, 142.9, 143.6, 148.0, 160.3
比較例1
実施例3の3,5−ビス(4−ニトロフェノキシ)ベンジル−2−フロエートの還元において3,5−ビス(4−ニトロフェノキシ)ベンジル−2−フロエート11.9gをDMFに代えてエチルアルコール130mlと酢酸エチル100mlに溶解し、インジウム金属粉末40gと塩化アンモニウム飽和水溶液60mlを加え還流下7時間還元反応を行った。実施例3と同様の後処理を行い黒色タール状粗生成物10gを得た。TLCから多数の生成物の混合物であることが判明した。粗生成物のシリカゲルカラムクロマトグラフィー精製を2回繰り返し、2.6gの3,5−ビス(4−アミノフェノキシ)ベンジル−2−フロエートを得た。
収率25%。
【0053】
比較例2
実施例5の3,3′−ジニトロ−4,4′−ジ−2−フロイルアミノビフェニルの還元においてDMFに代えてエチルアルコール150mlとN−メチル−2−ピロリドン200mlに溶解し80℃にて1時間攪拌し還元反応を行った。TLCで3,3′−ジニトロ−4,4′−ジ−2−フロイルアミノビフェニルが完全に反応したことを確認した。反応溶液を室温まで冷却した後、固形物をろ別した。ろ液を減圧下約1/2に濃縮した後、水600mlを加え、析出した固体をろ過して粗生成物6.0gを得た。粗生成物をシリカゲルカラムクロマトグラフィーにより精製し、3,3′−ジアミノ−4,4′−ジ−2−フロイルアミノビフェニル4.0gを得た。収率23%。
【0054】
【発明の効果】
本発明の製造方法によれば、医薬品や感光性ポリマー合成などの中間体である芳香族ポリアミン、特に、少なくとも1つのシスー共役ジエニル基を含有する芳香族ジアミンを、相当する芳香族ポリニトロ化合物、特に芳香族ジニトロ化合物から、温和な条件の還元反応により高収率で製造することができる。
さらに、本発明によれば、医薬品や感光性ポリマー合成などの中間体として有用な新規芳香族ジアミンを提供することができる。
Claims (5)
- 非プロトン性極性溶媒中、4級アンモニウム塩水溶液共存下、インジウムメタルにより、芳香族ジニトロ化合物を還元して下記一般式[7]、下記一般式[8]又は下記一般式[9]で表される芳香族ジアミンを製造することを特徴とする芳香族ジアミンの製造方法。
- 非プロトン性極性溶媒中、4級アンモニウム塩水溶液共存下、インジウムメタルにより、少なくとも2つのニトロ基を有する芳香族ジニトロ化合物を還元して2つのアミノ基を有する下記式で表される芳香族ジアミンを製造することを特徴とする芳香族ジアミンの製造方法。
- 還元反応を55℃以下で行い、芳香族ジニトロ化合物の消失を確認した後、60℃以上で還元反応を完結させることを特徴とする請求項1または2に記載の製造方法。
- 下記一般式[7]で表される芳香族ジアミン化合物。
- 下記式で表される芳香族ジアミン化合物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000128431A JP4635162B2 (ja) | 2000-04-27 | 2000-04-27 | 芳香族ジアミンの製造方法及び芳香族ジアミン化合物 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000128431A JP4635162B2 (ja) | 2000-04-27 | 2000-04-27 | 芳香族ジアミンの製造方法及び芳香族ジアミン化合物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2001302598A JP2001302598A (ja) | 2001-10-31 |
| JP4635162B2 true JP4635162B2 (ja) | 2011-02-16 |
Family
ID=18637880
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2000128431A Expired - Fee Related JP4635162B2 (ja) | 2000-04-27 | 2000-04-27 | 芳香族ジアミンの製造方法及び芳香族ジアミン化合物 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4635162B2 (ja) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2436676A1 (en) * | 2002-06-12 | 2012-04-04 | Symphony Evolution, Inc. | Human adam-10 inhibitors |
| KR100530345B1 (ko) * | 2002-10-30 | 2005-11-22 | 한국과학기술연구원 | 인듐 금속 와이어를 사용하여 니트로 화합물로부터 아민 화합물을 제조하는 방법 |
| US8063238B2 (en) | 2006-09-01 | 2011-11-22 | Tokai University Educational System | Diamine compound having phosphorylcholine group, polymer thereof, and process for producing the polymer |
| KR101742838B1 (ko) * | 2009-12-14 | 2017-06-01 | 닛산 가가쿠 고교 가부시키 가이샤 | 액정 배향 처리제 및 그것을 사용한 액정 표시 소자 |
| WO2014204686A1 (en) * | 2013-06-19 | 2014-12-24 | Dow Global Technologies Llc | Method for making anion exchange and chelant resins including aliphatic amino functional groups |
| CN108191674A (zh) * | 2018-02-11 | 2018-06-22 | 邵玉田 | 一种联苯胺化合物的合成方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2417763A1 (de) * | 1974-04-11 | 1975-10-30 | Bayer Ag | Carbonsaeureamide, verfahren zu ihrer herstellung sowie ihre verwendung als arzneimittel |
| JPH0390052A (ja) * | 1989-09-01 | 1991-04-16 | Wakayama Seika Kogyo Kk | 新規なエーテルジアミンおよびその製造方法 |
| JPH06157428A (ja) * | 1992-11-27 | 1994-06-03 | Mitsui Toatsu Chem Inc | 芳香族ジニトロ化合物、芳香族ジアミノ化合物及びそれらの製造方法 |
| JP3899556B2 (ja) * | 1996-08-07 | 2007-03-28 | 日産化学工業株式会社 | ビス置換フェノキシフェニレンジアミン誘導体及びその製造法 |
| JP4016301B2 (ja) * | 1997-09-24 | 2007-12-05 | 日立化成工業株式会社 | 新規なアゾ基含有芳香族化合物及びその製造法 |
| JP2000080272A (ja) * | 1998-09-03 | 2000-03-21 | Toray Ind Inc | 低誘電率重合体組成物 |
| JP4258690B2 (ja) * | 1999-05-26 | 2009-04-30 | 独立行政法人理化学研究所 | 感光性樹脂組成物 |
-
2000
- 2000-04-27 JP JP2000128431A patent/JP4635162B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2001302598A (ja) | 2001-10-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN112552285A (zh) | 4-(2,2,2-三氯乙基)-β-内酰胺衍生物的合成方法 | |
| JPH0262854A (ja) | 置換フェノキシアセトアルデヒドオキシム類の製造方法 | |
| JP4635162B2 (ja) | 芳香族ジアミンの製造方法及び芳香族ジアミン化合物 | |
| CN110437129A (zh) | 一种合成3-醚基异吲哚啉酮类化合物的简单方法 | |
| CN112194548B (zh) | α-氨基-γ-丁内酯类化合物及其制备方法 | |
| CN104774183B (zh) | 一种甲酰基瑞舒伐汀钙中间体的制备方法 | |
| NO853953L (no) | R¯ntgenkontrastmidler. | |
| CN116023359A (zh) | 一种氨基噻吩类化合物的合成方法及氨基噻吩类化合物 | |
| JP2002030050A (ja) | アミン化合物、中間体、製造法および光学分割剤 | |
| JPH02138243A (ja) | トラニラスト中間生成物及びこのものの製造方法 | |
| JPS62198678A (ja) | フルオル化された↓0−ジアミノベンゾ−1,4−ジオキセン類の製造法 | |
| JPS6317850A (ja) | 3−フエノキシカテコ−ル類の製造方法 | |
| CN102531954A (zh) | 一种芳香偶氮化合物的合成方法 | |
| CN119264028B (zh) | 一种1-三氟甲硫基吲哚类化合物的合成方法 | |
| KR100567449B1 (ko) | 아이오디사놀 유도체의 제조방법 | |
| JP2706554B2 (ja) | 4―トリフルオロメチルアニリン誘導体及びその製造法 | |
| JPH02255673A (ja) | 4―アリールオキシー1,3―ベンゾジオキソール類およびその製造方法 | |
| CN1332936C (zh) | 一种酰基芳胺的合成方法 | |
| JP2003041236A (ja) | 有機ゲル化剤 | |
| JPH02212490A (ja) | 20,21―セコリゾキシン誘導体 | |
| JP4016301B2 (ja) | 新規なアゾ基含有芳香族化合物及びその製造法 | |
| JP2893906B2 (ja) | 不飽和ケトン化合物の製造方法 | |
| CN120329310A (zh) | 一类苯并呋喃氮杂环化合物及其制备方法 | |
| CN117304052A (zh) | 一种3-氨基联苯类化合物的合成方法 | |
| CN107406385A (zh) | 二胺化合物及其中间体的制造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20031201 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20040415 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20070410 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20090602 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20090605 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20100315 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100406 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100604 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100629 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100809 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100928 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20101025 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131203 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |