JP4632016B2 - 非水電解質電池 - Google Patents
非水電解質電池 Download PDFInfo
- Publication number
- JP4632016B2 JP4632016B2 JP2003143261A JP2003143261A JP4632016B2 JP 4632016 B2 JP4632016 B2 JP 4632016B2 JP 2003143261 A JP2003143261 A JP 2003143261A JP 2003143261 A JP2003143261 A JP 2003143261A JP 4632016 B2 JP4632016 B2 JP 4632016B2
- Authority
- JP
- Japan
- Prior art keywords
- negative electrode
- battery
- active material
- electrode active
- same manner
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Landscapes
- Silicon Compounds (AREA)
- Secondary Cells (AREA)
- Battery Electrode And Active Subsutance (AREA)
Description
【発明の属する技術分野】
本発明は非水電解質電池に関する。
【0002】
【従来の技術】
近年、携帯用電話、PDA、デジタルカメラ等の電源として、高エネルギー密度を有する非水電解質電池が広く用いられている。電子機器のコードレス化が進む中で、非水電解質電池が担う役割は今後ますます大きくなることが予想される。
【0003】
現在、非水電解質電池の負極活物質には黒鉛が、正極活物質にはリチウム遷移金属酸化物が主に用いられている。しかし、そのエネルギー密度は次世代電子機器用電源としては不十分である。このため近年、活物質単位重量当たりの放電容量を大きくする研究が盛んにおこなわれている。負極活物質に関してはリチウムと合金化しうる材料である、ケイ素、スズ、アルミニウム等の金属またはこれらの酸化物がとくに注目されている。中でもケイ素酸化物は大きな放電容量を示すため、次世代リチウム二次電池用負極活物質としてとくに注目されている(例えば、特許文献1または非特許文献1参照)。
【0004】
また、このケイ素酸化物中に種々の金属を含有させることによって、それを負極活物質として備えた電池の特性がさらに向上することが報告されている(例えば、特許文献2参照)。あるいはまた、ケイ素の酸化物と共に、鉄、ニッケルなどの金属を共存させることが可能であることが報告されている(例えば、特許文献3参照)。
【0005】
【特許文献1】
特許第2997741号公報
【特許文献2】
特許第3010226号公報
【特許文献3】
特開平10−270088号公報
【非特許文献1】
第38回電池討論会講演要旨集、1997年、p179
【発明が解決しようとする課題】
しかしながら、ケイ素酸化物を負極活物質として備えた電池について本発明者が鋭意努力して調べた結果、それを充電状態およびその状態でさらに60℃以上の高温で保存すると、電池の膨れが大きいことがわかった。
すなわち、ケイ素酸化物を用いた電池には、それを充電状態およびその状態でさらに高温で保存した場合に、電池の膨れを抑制するという課題があった。本発明はこの課題を解決しようとするものである。
【0006】
【課題を解決するための手段】
上記課題である電池の膨れ抑制について検討したところ、膨れの程度がケイ素酸化物中の鉄(Fe)、クロム(Cr)およびニッケル(Ni)のそれぞれの含有率の総和によること、さらにその含有率がある一定の値を超えると、膨れが著しく増大することがわかった。すわなち、例えばFe、Niなどは従来ケイ素の酸化物と共に共存させることが可能であるとされていた(特許文献3参照)のではあるが、充電状態およびその状態でさらに高温で保存した場合に電池の膨れを抑制するためには、ケイ素酸化物中における前記遷移金属の含有率の総和を制御することが非常に重要であることがわかったのである。
【0007】
すなわち上記課題を解決する本願請求項1の発明は、SiとOとを含み、Siに対するOの原子比xが0<x<2で表され、Fe、CrおよびNiを含み、Niの含有率が45〜200ppmで、Fe、CrおよびNiの含有率の総和が250ppm以下である物質(A)を負極活物質として備えた非水電解質電池である。請求項1の発明によれば、この負極活物質を備えた電池では、充電状態でさらに高温で保存した場合における電池の膨れが抑制される。
【0008】
好ましくは、前記負極活物質を備えた負極合剤層の多孔度を20〜50%とした前記非水電解質電池である。これにより、充電状態の電池の膨れ、および充電状態でさらに60℃以上の高温で保存した電池の膨れをさらに抑制することができる。
【0009】
【発明の実施の形態】
Siに対するOの原子比をxとするとき、SiとOとを含む本発明の物質(A)の組成式はSiOxとして表される。xの値を、固体NMR、元素分析、エネルギー分散型エックス線検出器(FESEM/EDS)等から計算することができる。
【0010】
本発明者は、物質(A)を負極活物質として備えた電池を充電状態にし、さらに60℃以上の高温で保存した場合の電池の厚みと、前記物質(A)に含まれるFe、CrあるいはNiの含有率との関係を調べた。その結果、これら遷移金属の含有率の総和が250ppmを越えない場合に電池の膨れが著しく小さくなることがわかった。そして、含有量が少ないほど、膨れが小さくなることが分かった。電池が膨れる主な原因は電池内部におけるガス発生であることから、この結果は上記遷移金属の含有率の総和を250ppm以下まで低くすることによって、負極上でのガス発生量が低減されたからと考えられる。
【0011】
上記金属元素(Fe、CrあるいはNi)の含有率を高周波誘導結合プラズマ(ICP)発光分光分析によって求めることができる。また、その含有率はSi、Oおよびこれら金属元素の総質量を基に計算される。
【0012】
さて、非水電解質電池を充電すると負極活物質表面上に電解液の分解生成物が形成され、これが不活性被膜となって以後電解液の分解が抑制されることが知られている。したがって、上記遷移金属表面上の不活性被膜は、SiOx表面上のそれとくらべて、電解液の分解を抑制する能力が低いことが推察される。また、上記金属元素の含有率の総和が100ppm以下の場合、電池の膨れがさらに抑制されるので、とくに好ましい。
【0013】
また、非水電解質電池で用いられる負極は負極合剤層および負極集電体からなる。ここで、負極合剤層は負極活物質を含んでおり、結着剤や導電剤等を含むことが好ましい。本発明者は、負極合剤層の多孔度と、充電状態の電池の膨れおよび充電状態でさらに60℃以上の高温で保存した電池の膨れとの関係をそれぞれ調べた。その結果、多孔度が50%を超えると、充電状態でさらに60℃以上の高温で保存した電池の膨れが著しく大きくなることがわかった。この理由は明確ではないが、多孔度が50%を超えると、負極合剤層中の電解液と負極活物質との接触面積が増大することによって、活物質表面における電解液の分解とそれにともなうガス発生が促進され、その結果電池が大きく膨れるからと考えられる。また、負極合剤層の多孔度が20%を下回ると、2CmA以上の高い電流値における放電性能が著しく低下することがわかった。これは、負極合剤層内に保持する電解液量が減少するため、液抵抗が大きくなるからと考えられる。したがって、負極合剤層の多孔度は、高率放電性能を考慮して20%以上であることが好ましい。また、負極合剤層のさらに好適な多孔度は25〜35%であり、この場合電池の膨れが小さく、さらに高率放電性能も良好である。
【0014】
特開平7−312219では、非水電解質電池の多孔度を10〜60%とすることによって、電池の放電電位が高くなり、そして放電容量が大きくなり、さらにサイクル性能が向上することが報告されている。しかし、本発明負極活物質を用いた電池で、その電池の膨れが抑制されて、さらに高率放電性能が良好となる多孔度の値についてはこれまで明らかではなかった。そこで、本発明者が鋭意努力して研究した結果、その好適な値の範囲は上述したように20〜50%であって、これは従来の公知例から予想できないものである。
【0015】
負極合剤層の多孔度の計算方法としては、活物質、導電剤、結着剤の密度および質量、および負極層の厚みから計算する方法が例示される。
【0016】
本発明の物質(A)は、SiO1.5(Si2O3)、SiO1.33(Si3O4)、SiOなどの化学量論組成の化合物、およびxが0より大きく2未満である任意の組成の化合物が例示される。また、この組成で表されるならばSiとSiOx(0<x≦2)とを任意の割合で含む物質でもよい。xの範囲が0<x≦0.5の場合、物質(A)中のSi含有量が多くなり、その結果充電時における活物質の体積膨張が著しくなり、電池の膨れが大きくなる。したがって、本発明負極活物質(A)のさらに好ましい組成は、SiOx(0.5<x<2)である。
【0017】
本発明電池に用いる非水電解質としてフッ素含有塩を用いた場合、本発明負極活物質(A)は、好ましくはSiとSiOx(1<x≦2)との両相を含む物質であって、さらにその物質(A)のCuKα線を用いたX線回折測定において、Si(111)面およびSi(220)面の各回折ピークの半値幅のうち、すくなくとも一方が3°未満であることが好ましい。その理由は、両回折ピークの半値幅が3°以上である物質(以後、非晶質物質(X)と呼ぶ)を備えた電池、およびそのいずれか一方の半値幅が3°未満である物質(以後、結晶質物質(Y)と呼ぶ)を備えた電池とをそれぞれ充電状態にして、さらに60℃以上の高温で保存すると、後者の電池の膨れが著しく抑制されるからである。ただし、両電池にはフッ素含有塩を非水電解質として用いた。
【0018】
本発明者は、この原因はフッ素含有塩を含む非水電解質中に存在するフッ化水素酸(HF)と負極活物質との反応性が、負極活物質の種類によって異なることにあると推定し、非晶質物質(X)と結晶質物質(Y)との間で60℃以上の温度に保ったフッ化水素酸への溶解性を調べた。その結果、後者の物質の方がフッ化水素酸への溶解性が著しく低く、また溶解とともに発生するガスの量がはるかに少ないことがわかった。また、上記回折ピークの半値幅が小さくなりすぎると、ケイ素とその酸化物とに相分離した物質の電子伝導性が著しく低下するために、その結果電池の放電容量が小さくなる。したがって、電池の膨れと放電容量との観点から、Si(111)面およびSi(220)面の各回折ピークの少なくとも一方の半値幅B(2θ)がとくに0.6°<B<2.0°であることが好ましい。
【0019】
ケイ素とその酸化物とに相分離した物質は、少なくとも電池組み込み前に、Si(111)面およびSi(220)面回折ピークを示す。しかし、充放電後の活物質に関してはその限りではない。すなわち、充放電後の電池を解体した後、負極活物質を取り出して、そのX線回折パターンをした場合、上記回折ピークが観察されなくても、または異なる角度にピークが現れてもよい。
【0020】
本発明負極活物質(A)は、その表面の一部または全面に炭素材料(B)を備えることが好ましい。その理由は、炭素材料を備えることによって負極活物質(A)の電子伝導性が著しく高くなり、その結果活物質(A)の利用率が向上し、電池の放電容量が大きくなるからである。炭素材料(B)としては黒鉛および低結晶性炭素が好ましい。また、炭素の形状は薄膜または粒子のいずれでもよい。
【0021】
本発明負極活物質(A)の表面に炭素材料(B)を備えて得られる物質を負極活物質(C)とする。負極活物質(C)に関してX線光電子分光法で表面観察した場合、90〜110eVの範囲でSi−O−Siに由来するSi2pスペクトルが観察されないことが好ましい。また、この場合においても負極活物質(C)を例えばアルゴンイオンで、炭素のC1sピーク強度がエッチング前におけるそのピーク強度と比較して半分以下となるまで十分エッチングし、その表面をX線光電子分光法で観察すると上記Si2pスペクトルが明瞭に観察される。この強度をIX1とし、エッチングする前の表面におけるスペクトルの強度をIX2とすると、IX2/IX1<0.1であることが好ましい。なぜなら、これらいずれの場合も、炭素材料(B)は負極活物質(A)のほぼ全面を被覆しているからである。
【0022】
炭素材料の結晶性をラマン分光法で特徴づけることができる。アルゴンイオンレーザーを用いたラマン分光分析の結果、1575〜1620cm−1および1340〜1370cm−1の範囲に現れるピークの強度をそれぞれIAおよびIBとすると、炭素材料(B)としては、IB/IA<1.5を満たす炭素材料を用いることが好ましい。より好ましくはIB/IA<0.5であり、この場合炭素の結晶性が高いため、負極活物質(A)の電子伝導性が著しく向上する。
【0023】
負極活物質(A)の電子伝導性および利用率は、炭素材料(B)の含有量が負極活物質(C)の全質量に対して3質量%以上である場合に著しく向上し、その結果電池の放電容量が大きくなる。また、活物質(A)の利用率は、炭素含有量を15〜30質量%とすることにより著しく向上し、その結果電池の放電容量がとくに大きくなる。一方、炭素含有量が50質量%をこえる場合、負極活物質(A)の放電容量とくらべて炭素材料(B)のそれが小さいことに起因して、電池の放電容量が小さくなる。したがって、電池の放電容量の観点から、炭素材料(B)の好適な含有量は、負極活物質(C)の全質量に対して3〜50%であり、さらに好ましくは15〜30%である。
【0024】
本発明非水電解質電池が備える負極は、上記本発明負極活物質と炭素材料(D)とを混合したものを用いることが好ましい。この混合物を用いることによって、活物質間の接触導電性が向上し、その結果電池の放電容量が大きくなるからである。
【0025】
本発明負極活物質と炭素材料(D)との合計質量に対して炭素材料(D)の混合量が5%以上の場合、活物質間の接触導電性が向上することによって負極放電容量が大きくなり、その結果電池の放電容量が大きくなる。そして、その混合割合が20%以上のとき、電池の放電容量がさらに大きくなる。
【0026】
また、その混合割合が50質量%以上の場合、0℃以下の低温においてサイクル試験を繰り返した場合に電池の膨れが著しく抑制される。この原因は、活物質間の接触導電性が向上することによって負極中の電流分布がより均一化され、充電時におけるリチウムの電析が大幅に抑制されるからである。
【0027】
さらに、その混合割合が95質量%を超えると、炭素材料(D)の質量当たりの放電容量が本発明負極活物質のそれとくらべて小さいために、電池の放電容量が小さくなる。したがって、電池の放電容量および低温サイクル時における電池の膨れの観点から、炭素材料(D)の混合量が、本発明負極活物質と炭素材料(D)との合計質量に対して5〜95%、さらには20〜95%であることが好ましく、より好適な範囲は50〜95%である。この場合、本発明負極活物質がその表面に上述した炭素材料(B)を備えていても、備えていなくてもどちらでもよい。炭素材料(B)を備えている場合、本発明負極活物質と炭素材料(D)との合計質量に、この炭素材料(B)の質量も含まれる。
【0028】
炭素材料(D)は、天然黒鉛、人造黒鉛、メソカーボンマイクロビーズ(MCMB)、アセチレンブラック、気相成長炭素繊維(VGCF)、低結晶性炭素および難黒鉛化性炭素からなる群から選ばれた少なくとも1種類の炭素材料を用いることができる。好ましくは、天然黒鉛、人造黒鉛、メソカーボンマイクロビーズ(MCMB)、アセチレンブラック、気相成長炭素繊維(VGCF)である。その理由は、これらの炭素材料を用いた方が、低結晶性炭素および難黒鉛化性炭素を用いた場合よりも負極の不可逆容量が小さくなり、その結果電池のエネルギー密度が高くなるからである。
【0029】
天然黒鉛、人造黒鉛の数平均粒径r(μm)およびBET比表面積S(m2/g)に関しては、0.5<r<50、0.05<S<30の範囲が好ましい。なお、粒子の数平均粒径は、それを15秒間超音波分散した後、レーザー法によって求められる値である。とくに好適な数平均粒径および比表面積は1<r<30および0.1<S<10である。数平均粒径および比表面積を上記範囲とすることによって、黒鉛表面上における電解液の分解を抑制し、不可逆容量を少なくし、さらに電池のエネルギー密度を高くすることができる。
【0030】
人造黒鉛としては、コークス等の易黒鉛化性炭素を焼成することによって得られるものや、黒鉛を硫酸溶液と処理した後、熱処理することによって得られる膨張化黒鉛が例示される。
【0031】
VGCFの長軸径が長い場合、セパレーターを貫通することによって正極活物質と短絡する恐れがある。したがって、その長軸径はセパレーターの厚さ以下であることが好ましい。電池に用いられているセパレーターの厚さが約20μmの場合には、VGCFの好適な長軸径は20μm以下が好ましい。
【0032】
本発明負極活物質(A)および(B)の比表面積S(m2/g)は、好ましくは1<S<30、さらに好ましくは1<S<10である。S<30の場合、高温保存時における活物質表面上での電解液の分解を小さくすることができ、その結果電池の膨れが抑制される。また、この効果はS<10の場合とくに顕著である。一方、S≦1の場合、2CmA以上の高い電流値において電池の放電容量が著しく低下する。したがって、1<Sが好ましい。
【0033】
本発明負極活物質(A)および(B)の形態としては、板、薄膜、粒子および繊維が例示される。この活物質を粒子として用いる場合、その数平均粒径r(μm)がr<15であることが好ましい。その理由は、15≦rの場合、活物質粒子中のリチウム拡散に起因する抵抗が大きくなるため、0℃以下の低温においてサイクル試験を繰り返した場合に負極上に多くのリチウムが析出する。リチウムが析出すると電池の膨れが大きくなる。したがって、r<15を満たす負極活物質を用いることにより、低温サイクル時における電池の膨れを抑制することができる。
【0034】
さらにrが5よりも小さい場合、電池の膨れがさらに抑制されるので好ましい。したがって、本発明負極活物質の数平均粒径r(μm)は好ましくはr<15であり、さらに好ましくはr<5である。
【0035】
炭素を備えた本発明負極活物質の合成方法としては、メタン、エタン、エチレン、アセチレン、ブタン、ベンゼン、トルエン、キシレンのような有機化合物を気相中分解し、その分解性生物をSiOx(0<x<2)の表面に付着させる方法(CVD法)や、ピッチ、タールまたはフルフリルアルコールなどの熱可塑性樹脂をSiOx(0<x<2)表面に塗布した後にそれらを焼成する方法、SiOx(0<x<2)粒子と黒鉛粒子とを造粒し、この造粒体表面上にCVDで炭素を付着させる方法、および機械的方法によってSiOx(0<x<2)と炭素材料とを付着させる方法が例示される。
【0036】
機械的方法には、メカニカルミリング法、メカノフュージョン法、およびハイブリダイゼーション法が例示される。これら種々の合成方法のなかでも、SiOx(0<x<2)の表面上に炭素材料を均一に被覆することができるCVD法がとくに好ましい。
【0037】
SiOx(0<x<2)の合成方法としては下に示す方法が例示される。(1)SiO2とSiとを混合し、非酸化性雰囲気中または真空中で加熱することによりガスを発生させ、つぎに冷却する方法、(2)SiO2を水素等の還元性ガス中または炭素と混合して、つぎに加熱して還元する方法、(3)Siを酸素ガス中または酸化物と混合して、つぎに加熱して酸化する方法。
【0038】
本発明負極活物質(A)は上記方法の中で、例えば、原料として用いるSiO2およびSiに含まれるFe、CrおよびNiの含有率の総和を250ppm以下とすることにより合成することができる。また、これら金属元素は、上記方法で得られた生成物を採取する際にその生成物中に取り込まれるおそれがあるため、上記含有率を100ppm以下とすることが好ましい。
【0039】
前述したように、本発明物質(A)は、好ましくはSiとSiOx(1<x≦2)との両相を含む物質である。この物質の製造方法を例示すると、SiOx(0<x<2)を非酸化性雰囲気中または真空中、温度T(800<T(℃))で熱処理する工程を経て作製される。熱処理時間としては30分以上、好ましくは1時間以上である。また、前記工程で得られた化合物を、さらにフッ素含有化合物またはアルカリ水溶液と反応させることがより好ましい。その理由は、SiOx(0<x<2)粒子表面上に生成した絶縁性のSiO2の量を低減することが可能であるからである。
【0040】
非酸化性雰囲気に用いるガスとしては、窒素、アルゴン、水素およびこれらの混合ガスが例示される。フッ素含有化合物には、フッ化水素、フッ化水素アンモニウム等、SiO2を溶解しうるいかなる化合物も用いることができる。また、これらフッ素含有化合物を単体もしくは水溶液として用いてもよい。さらに、アルカリ水溶液としては、アルカリ金属またはアルカリ土類金属を含む水酸化物を用いることができる。この水酸化物としては、水酸化リチウム、水酸化ナトリウム、水酸化カリウムが例示される。SiO2の溶解を促進するために、アルカリ水溶液の温度が40℃以上であることが好ましい。
【0041】
フッ素含有化合物またはアルカリ水溶液の濃度が高すぎないことが好ましい。また、前記化合物または溶液による反応時間が長すぎないことが好ましい。その理由は、それらの濃度が高すぎる、または反応時間が長すぎる場合、SiO2の溶解以外にSiの溶解も促進されるため、活物質中のSi含有率が大きく減少するからである。Si含有率が減少すると、それを用いた負極の放電容量が低下する。好適な濃度および反応時間はそれぞれ1gのSiOx(0<x<2)当たり5mol以下、24h以下であり、とくに好ましくは0.5mol以下、6h以下である。
【0042】
また、上記で述べたように、本発明負極活物質の製造方法においては、SiOx(0<x<2)の熱処理は非酸化性雰囲気中または減圧下でおこなわれるが、ここにおける減圧下についてさらに好適な条件を記述すると、より好ましくは30Torr以下であり、さらに好ましくは3Torr以下であり、さらに好ましくは0.3Torr以下である。ただし、言うまでもなく、30Torrよりも高い圧力下であっても、減圧下であれば本発明の効果は得られる。
【0043】
本発明負極活物質中に、B、C、N、P、F、Cl、Br、I等の典型非金属元素、Li、Na、Mg、Al、K、Ca、Zn、Ga、Ge等の典型金属元素、Sc、Ti、V、Mn、Co、Cu等の遷移金属元素を含んでいてもよい。Fe、CrおよびNiの含有率の総和が250ppm以下であるならば、その他の金属元素の含有率はそれ以上でもよい。
【0044】
本発明非水電解質電池の正極活物質としては、二酸化マンガン、五酸化バナジウムのような遷移金属化合物や、硫化鉄、硫化チタンのような遷移金属カルコゲン化合物、リチウム含有オリビン形化合物、およびリチウム遷移金属酸化物を用いることができる。リチウム遷移金属酸化物としては、LixM1yM2zO2(M1、M2は、Ti、V、Cr、Mn、Fe、Co、Ni、Cuを表し、0.5≦x≦1、y+z=1)、LixM3yMn2−yO4(M3は、Ti、V、Cr、Fe、Co、Ni、Cuを表し、0.9≦x≦1.1、0.4≦y≦0.6)が例示される。さらに、これらの化合物や酸化物にAl、P、B、またはそれ以外の典型非金属元素、典型金属元素を含有した物質を使用することができる。これら正極活物質のなかでも、リチウムとコバルトとの複合酸化物や、リチウム、コバルトおよびニッケルを含む複合酸化物が好ましい。その理由は、これらの正極活物質を用いることにより、高電圧、高エネルギー密度および良好なサイクル性能をもつ電池が得られるからである。
【0045】
上述したように、本発明の非水電解質電池で用いられる負極は、負極活物質を含む負極合剤層および負極集電体からなる。負極合剤層は、負極活物質および結着剤を溶媒中混合し、得られたスラリーを負極集電体に塗布し、さらに乾燥することにより製造することができる。また、負極合剤層中に、負極活物質とは別に導電剤が含まれていることが好ましい。
【0046】
負極活物質としては、本発明活物質を単独で用いてもよいし、リチウムイオンを吸蔵・放出することが可能な物質および金属リチウムの中ですくなくとも一種と本発明活物質との混合物を用いてもよい。リチウムイオンを吸蔵・放出することが可能な物質には、炭素材料、酸化物、Li3−PMPN(ただし、Mは遷移金属、0≦P≦0.8)などの窒化物およびリチウム合金が例示される。炭素材料としては、コークス、メソカーボンマイクロビーズ(MCMB)、メソフェーズピッチ系炭素繊維、熱分解気相成長炭素繊維等の易黒鉛化性炭素、フェノール樹脂焼成体、ポリアクリロニトリル系炭素繊維、擬等方性炭素、フルフリルアルコール樹脂焼成体等の難黒鉛化性炭素、天然黒鉛、人造黒鉛、黒鉛化MCMB、黒鉛化メソフェーズピッチ系炭素繊維、黒鉛ウイスカー等の黒鉛質材料、さらに、これらの混合物を用いることができる。リチウム合金としては、リチウムとアルミニウム、亜鉛、ビスマス、カドミウム、アンチモン、シリコン、鉛、錫、ガリウム、インジウムとの合金を用いることができる。酸化物としては、前記リチウム合金の酸化物を用いることができる。
【0047】
本発明の非水電解質電池で用いられる正極は、正極活物質を含む正極合剤層および正極集電体からなる。正極合剤層は、正極活物質および結着剤を溶媒中混合し、得られたスラリーを正極集電体に塗布し、さらに乾燥することにより製造することができる。また、正極合剤層中に、正極活物質とは別に導電剤が含まれていることが好ましい。
【0048】
正極または負極に用いられる導電剤としては、上記炭素材料(D)の他、種々の炭素材料を用いることができ、天然黒鉛、人造黒鉛等の黒鉛や、アセチレンブラック等のカーボンブラック、ニードルコークス等の無定形炭素が例示される。
【0049】
正極または負極に用いられる結着剤としては、例えば、PVdF(ポリフッ化ビニリデン)、P(VdF/HFP)(ポリフッ化ビニリデン−ヘキサフルオロプロピレン共重合体)、PTFE(ポリテトラフルオロエチレン)、フッ素化ポリフッ化ビニリデン、EPDM(エチレン−プロピレン−ジエン三元共重合体)、SBR(スチレン−ブタジエンゴム)、NBR(アクリロニトリル−ブタジエンゴム)、フッ素ゴム、ポリ酢酸ビニル、ポリメチルメタクリレート、ポリエチレン、ニトロセルロース、またはこれらの誘導体を、単独でまたは混合して用いることができる。
【0050】
正極活物質または負極活物質と結着剤とを混合する際に用いる溶媒としては、結着剤を溶解または分散する溶媒を用いることができる。その溶媒としては、非水溶媒または水溶液を用いることができる。非水溶媒には、N―メチル−2−ピロリドン、ジメチルホルムアミド、ジメチルアセトアミド、メチルエチルケトン、シクロヘキサノン、酢酸メチル、アクリル酸メチル、ジエチルトリアミン、N−N−ジメチルアミノプロピルアミン、エチレンオキシド、テトラヒドロフラン等をあげることができる。一方、水溶液には、水、または分散剤、増粘剤等を加えた水溶液を用いることができる。後者の水溶液中で、SBR等のラテックスと活物質とを混合し、それらをスラリー化することができる。
【0051】
正極または負極の集電体としては、鉄、銅、アルミニウム、ステンレス、ニッケルを用いることができる。また、その形状としては、シート、発泡体、焼結多孔体、エキスパンド格子が例示される。さらに、集電体として、前記集電体に任意の形状で穴を開けたものを用いてもよい。
【0052】
本発明の非水電解質電池用セパレーターには、微多孔性高分子膜を用いることができ、その材質としては、ナイロン、セルロースアセテート、ニトロセルロース、ポリスルホン、ポリアクリロニトリル、ポリフッ化ビニリデン、およびポリプロピレン、ポリエチレン、ポリブテン等のポリオレフィンが例示される。これらの中では、ポリオレフィンの微多孔性膜がとくに好ましい。または、ポリエチレンとポリプロピレンとを積層した微多孔製膜を用いてもよい。
【0053】
本発明の非水電解質電池で用いられる非水電解質としては、非水電解液、高分子固体電解質、ゲル状電解質、無機固体電解質を用いることができる。電解質には孔があってもよい。非水電解液は、非水溶媒および溶質から構成される。
非水電解液に用いられる溶媒としては、エチレンカーボネート、プロピレンカーボネート、ジメチルカーボネート、ジエチルカーボネート、メチルエチルカーボネート、γ−ブチロラクトン、スルホラン、ジメチルスルホキシド、アセトニトリル、ジメチルホルムアミド、ジメチルアセトアミド、1、2−ジメトキシエタン、1、2−ジエトキシエタン、テトラヒドロフラン、2−メチルテトラヒドロフラン、ジオキソラン、メチルアセテート、酢酸メチル等の溶媒、およびこれらの混合溶媒が例示される。
【0054】
非水電解液に用いられる溶質としては、LiPF6、LiBF4、LiAsF6、LiClO4、LiSCN、LiCF3CO2、LiCF3SO3、LiN(SO2CF3)2、LiN(SO2CF2CF3)2、LiN(COCF3)2およびLiN(COCF2CF3)2等の塩、およびこれらの混合物が例示される。
【0055】
高分子固体電解質としては、ポリエチレンオキサイド、ポリプロビレンオキサイド、ポリエチレンイミド等の高分子、およびこれらの混合物を用いることができる。また、ゲル状電解質としては、上記高分子に、上記の溶媒および溶質を加えて得られる物質を用いることができる。
【0056】
無機固体電解質としては、結晶質または非晶質の固体電解質を用いることができる。前者には、LiI、Li3N、Li1+xMxTi2−x(PO4)3(M=Al、Sc、Y、La)、Li0.5−3xR0.5+xTiO3(R=La、Pr、Nd、Sm)、およびLi4−xGe1−xPxS4に代表されるチオLISICONを用いることができ、後者にはLiI−Li2O−B2O5系、Li2O−SiO2系等の酸化物ガラス、およびLiI−Li2S−B2S3系、LiI−Li2S−SiS2系、Li2S−SiS2−Li3PO4系等の硫化物ガラスを用いることができる。また、これらの混合物を用いることができる。
【0057】
また、負極の利用率向上を目的として、上記溶媒中に、エチレンサルファイド(ES)、フッ化水素(HF)、トリアゾール系環状化合物、フッ素含有エステル系溶媒、テトラエチルアンモニウムフルオライドのフッ化水素錯体(TEAFHF)、またはこれらの誘導体、または、CO2、NO2、CO、SO2等のガスを、添加剤として加えてもよい。
【0058】
以下に、本発明非水電解質電池を実施例に基づいて、さらに詳細に説明する。しかしながら、本発明は、以下の実施例によって限定されるものではない。
【0059】
[実施例1]
SiO2粉末とSi粉末とを1:1のモル比で混合した。SiO2粉末中にはFe、Cr、Niがそれぞれ50ppm含まれていた。この混合物をセラミック製の反応器に入れ、さらに内部の圧力および温度をそれぞれ0.05Torrおよび1400℃に保持することによってSiOガスを発生させた。つぎに、このガスを、水冷した上記反応器の壁面上で冷却することにより、物質を析出させた。最後に、この析出物をタングステンカーバイド(WC)製のボールで粉砕することにより、粒子状生成物を得た。このようにして得られた粒子の組成はFESEM/EDSで求めた結果SiO1.1であった。この粒子を以後SiO粒子と呼ぶ。また、粒度分析装置(島津製作所(株)製SALD2000J)およびBET比表面積測定装置(島津製作所(株)製ジェミニ2375)を用いて測定した結果、その数平均粒径は12μmBET比表面積は5m2/gであった。
【0060】
このSiOに関してX線回折測定をおこなうと、ブロードな回折パターンが得られ、その結晶構造が無定形であることがわかった。この無定形のSiO粒子を物質(X)とする。この物質を負極活物質(e1)に用いて、非水電解質電池を製作した。まず、負極活物質(e1)70質量%と、アセチレンブラック10質量%と、ポリビニリデンフルオライド(PVdF)20質量%とを、N−メチル−2−ピロリドン(NMP)中で分散させることによりペーストを作製した。このペーストを厚さ15μmの銅箔上に塗布し、つぎに、150℃で乾燥することにより、NMPを蒸発させた。この作業を銅箔の両面に対しておこない、さらに、両面をロールプレスで圧縮成型した。このようにして、両面に負極層を備えた負極を製作した。負極合剤層の多孔度は35%であった。
【0061】
つぎに、コバルト酸リチウム90質量%と、アセチレンブラック5質量%と、PVdF5質量%とをNMP中で分散させることによりペーストを作製した。このペーストを厚さ20μmのアルミニウム箔上に塗布し、つぎに150℃で乾燥することにより、NMPを蒸発させた。以上の操作をアルミニウム箔の両面におこない、さらに両面をロールプレスで圧縮成型した。このようにして、両面に正極合剤層を備えた正極を製作した。
【0062】
このようにして準備した正極および負極を、厚さ20μm、多孔度40%の連通多孔体であるポリエチレンセパレータを間に挟んで重ねて巻き、高さ48mm、幅30mm、厚さ4.2mmの容器中に挿入して、角形電池を組み立てた。最後に、この電池の内部に非水電解液を注入することによって、実施例電池(E1)を得た。この非水電解液として、エチレンカーボネート(EC)とジエチルカーボネート(DEC)との体積比1:1の混合溶媒に1mol/lのLiPF6を溶解したものを用いた。
【0063】
[実施例2]
原料として用いるSiO2粉末中にFe、Cr、Niがそれぞれ50ppm、50ppm、60ppm含まれていたこと以外は実施例1と同様にしてSiO粒子を得た。つぎに、この物質を負極活物質(e2)に用いたこと以外は実施例1と同様にして実施例電池(E2)を得た。
【0064】
[実施例3]
原料として用いるSiO2粉末中にFe、Cr、Niがそれぞれ50ppm、50ppm、150ppm含まれていたこと以外は実施例1と同様にしてSiO粒子を得た。つぎに、この物質を負極活物質(e3)に用いたこと以外は実施例1と同様にして実施例電池(E3)を得た。
【0065】
[実施例4]
原料として用いるSiO2粉末中にFe、Cr、Niがそれぞれ50ppm、50ppm、200ppm含まれていたこと以外は実施例1と同様にしてSiO粒子を得た。つぎに、この物質を負極活物質(e4)に用いたこと以外は実施例1と同様にして実施例電池(E4)を得た。
【0066】
[実施例5]
原料として用いるSiO2粉末中にFe、Cr、Niがそれぞれ50ppm、50ppm、250ppm含まれていたこと以外は実施例1と同様にしてSiO粒子を得た。つぎに、この物質を負極活物質(e5)に用いたこと以外は実施例1と同様にして実施例電池(E5)を得た。
【0067】
[実施例6]
負極合剤層の多孔度が50%であったこと以外は実施例1と同様にして実施例電池(E6)を得た。
【0068】
[実施例7]
負極合剤層の多孔度が40%であったこと以外は実施例1と同様にして実施例電池(E7)を得た。
【0069】
[実施例8]
負極合剤層の多孔度が30%であったこと以外は実施例1と同様にして実施例電池(E8)を得た。
【0070】
[実施例9]
負極合剤層の多孔度が25%であったこと以外は実施例1と同様にして実施例電池(E9)を得た。
【0071】
[実施例10]
負極合剤層の多孔度が20%であったこと以外は実施例1と同様にして実施例電池(E10)を得た。
【0072】
[実施例11]
物質(X)をアルゴン雰囲気中830℃で熱処理したこと以外は実施例1と同様にして、負極活物質(e11)および実施例電池(E11)を得た。
【0073】
[実施例12]
物質(X)をアルゴン雰囲気中900℃で熱処理したこと以外は実施例1と同様にして、負極活物質(e12)および実施例電池(E12)を得た。
【0074】
[実施例13]
物質(X)をアルゴン雰囲気中1000℃で熱処理したこと以外は実施例1と同様にして、負極活物質(e13)および実施例電池(E13)を得た。
【0075】
[実施例14]
物質(X)をアルゴン雰囲気中1050℃で熱処理したこと以外は実施例1と同様にして、負極活物質(e14)および実施例電池(E14)を得た。
【0076】
[実施例15]
物質(X)をアルゴン雰囲気中1100℃で熱処理したこと以外は実施例1と同様にして、負極活物質(e15)および実施例電池(E15)を得た。
【0077】
[実施例16]
物質(X)をアルゴン雰囲気中1200℃で熱処理したこと以外は実施例1と同様にして、負極活物質(e16)および実施例電池(E16)を得た。
【0078】
[実施例17]
SiO粒子の数平均粒径が15μmであったこと以外は実施例1と同様にして、負極活物質(e17)および実施例電池(E17)を得た。
【0079】
[実施例18]
SiO粒子の数平均粒径が5μmであったこと以外は実施例1と同様にして、負極活物質(e18)および実施例電池(E18)を得た。
【0080】
[実施例19]
SiO粒子の数平均粒径が4μmであったこと以外は実施例1と同様にして、負極活物質(e19)および実施例電池(E19)を得た。
【0081】
[実施例20]
SiO粒子の数平均粒径が2μmであったこと以外は実施例1と同様にして、負極活物質(e20)および実施例電池(E20)を得た。
【0082】
[実施例21]
ハイブリダイゼーション法によって負極活物質(e1)と数平均粒径5μmの人造黒鉛とを混合し、前者の表面に黒鉛を担持させた。このようにして得られた物質(z1)に含まれる負極活物質(e1)と黒鉛との質量比率は99.5:0.5であった。つぎに、アルゴン雰囲気中、この物質(z1)表面上でトルエンガスを1000℃で熱分解することによって、負極活物質(e21)を作製した。負極活物質(e1)に担持した炭素の量は、負極活物質(e21)の全質量に対して1%であった。つぎに、負極活物質(e21)を用いたこと以外は実施例1と同様にして実施例電池(E21)を得た。
【0083】
[実施例22]
ハイブリダイゼーション法によって負極活物質(e1)と数平均粒径5μmの人造黒鉛とを混合し、前者の表面に黒鉛を担持させた。このようにして得られた物質(z2)に含まれる負極活物質(e1)と黒鉛との質量比率は99:1であった。つぎに、アルゴン雰囲気中、この物質(z2)表面上でトルエンガスを1000℃で熱分解することによって、負極活物質(e22)を作製した。負極活物質(e1)に担持した炭素の量は、負極活物質(e22)の全質量に対して3%であった。つぎに、負極活物質(e22)を用いたこと以外は実施例1と同様にして実施例電池(E22)を得た。
【0084】
[実施例23]
ハイブリダイゼーション法によって負極活物質(e1)と数平均粒径5μmの人造黒鉛とを混合し、前者の表面に黒鉛を担持させた。このようにして得られた物質(z3)に含まれる負極活物質(e1)と黒鉛との質量比率は92:8であった。つぎに、アルゴン雰囲気中、この物質(z3)表面上でトルエンガスを1000℃で熱分解することによって、負極活物質(e23)を作製した。負極活物質(e1)に担持した炭素の量は、負極活物質(e23)の全質量に対して10%であった。つぎに、負極活物質(e23)を用いたこと以外は実施例1と同様にして実施例電池(E23)を得た。
【0085】
[実施例24]
ハイブリダイゼーション法によって負極活物質(e1)と数平均粒径5μmの人造黒鉛とを混合し、前者の表面に黒鉛を担持させた。このようにして得られた物質(z4)に含まれる負極活物質(e1)と黒鉛との質量比率は87:3であった。つぎに、アルゴン雰囲気中、この物質(z1)表面上でトルエンガスを1000℃で熱分解することによって、負極活物質(e24)を作製した。負極活物質(e1)に担持した炭素の量は、負極活物質(e24)の全質量に対して15%であった。つぎに、負極活物質(e24)を用いたこと以外は実施例1と同様にして実施例電池(E24)を得た。
【0086】
[実施例25]
ハイブリダイゼーション法によって負極活物質(e1)と数平均粒径5μmの人造黒鉛とを混合し、前者の表面に黒鉛を担持させた。このようにして得られた物質(z5)に含まれる負極活物質(e1)と黒鉛との質量比率は82:18であった。つぎに、アルゴン雰囲気中、この物質(z1)表面上でトルエンガスを1000℃で熱分解することによって、負極活物質(e25)を作製した。負極活物質(e1)に担持した炭素の量は、負極活物質(e25)の全質量に対して20%であった。つぎに、負極活物質(e25)を用いたこと以外は実施例1と同様にして実施例電池(E25)を得た。
【0087】
[実施例26]
ハイブリダイゼーション法によって負極活物質(e1)と数平均粒径5μmの人造黒鉛とを混合し、前者の表面に黒鉛を担持させた。このようにして得られた物質(z6)に含まれる負極活物質(e1)と黒鉛との質量比率は77:23であった。つぎに、アルゴン雰囲気中、この物質(z1)表面上でトルエンガスを1000℃で熱分解することによって、負極活物質(e26)を作製した。負極活物質(e1)に担持した炭素の量は、負極活物質(e26)の全質量に対して25%であった。つぎに、負極活物質(e26)を用いたこと以外は実施例1と同様にして実施例電池(E26)を得た。
【0088】
[実施例27]
ハイブリダイゼーション法によって負極活物質(e1)と数平均粒径5μmの人造黒鉛とを混合し、前者の表面に黒鉛を担持させた。このようにして得られた物質(z7)に含まれる負極活物質(e1)と黒鉛との質量比率は72:28であった。つぎに、アルゴン雰囲気中、この物質(z1)表面上でトルエンガスを1000℃で熱分解することによって、負極活物質(e27)を作製した。負極活物質(e1)に担持した炭素の量は、負極活物質(e27)の全質量に対して30%であった。つぎに、負極活物質(e27)を用いたこと以外は実施例1と同様にして実施例電池(E27)を得た。
【0089】
[実施例28]
ハイブリダイゼーション法によって負極活物質(e1)と数平均粒径5μmの人造黒鉛とを混合し、前者の表面に黒鉛を担持させた。このようにして得られた物質(z8)に含まれる負極活物質(e1)と黒鉛との質量比率は52:48であった。つぎに、アルゴン雰囲気中、この物質(z1)表面上でトルエンガスを1000℃で熱分解することによって、負極活物質(e28)を作製した。負極活物質(e1)に担持した炭素の量は、負極活物質(e28)の全質量に対して50%であった。つぎに、負極活物質(e28)を用いたこと以外は実施例1と同様にして実施例電池(E28)を得た。
【0090】
[実施例29]
ハイブリダイゼーション法によって負極活物質(e1)と数平均粒径5μmの人造黒鉛とを混合し、前者の表面に黒鉛を担持させた。このようにして得られた物質(z9)に含まれる負極活物質(e1)と黒鉛との質量比率は42:58であった。つぎに、アルゴン雰囲気中、この物質(z1)表面上でトルエンガスを1000℃で熱分解することによって、負極活物質(e29)を作製した。負極活物質(e1)に担持した炭素の量は、負極活物質(e29)の全質量に対して60%であった。つぎに、負極活物質(e29)を用いたこと以外は実施例1と同様にして実施例電池(E29)を得た。
【0091】
[実施例30]
数平均粒径20μmの天然黒鉛粉末と負極活物質(e13)とを質量比で3:97であるように混合した。この混合物90質量%と、PVdF10質量%とをNMP中で分散させることによってペーストを作製した。このペーストを厚さ15μmの銅箔上に塗布し、つぎに、150℃で乾燥することにより、NMPを蒸発させた。この作業を銅箔の両面に対しておこない、さらに、両面をロールプレスで圧縮成型した。このようにして、両面に負極層を備えた負極を製作した。つぎに、この負極を用いたこと以外は実施例1と同様にして実施例電池(E30)を得た。
【0092】
[実施例31]
天然黒鉛粉末と負極活物質(e13)との混合質量比を5:95としたこと以外は実施例30と同様にして実施例電池(E31)を得た。
【0093】
[実施例32]
天然黒鉛粉末と負極活物質(e13)との混合質量比を10:90としたこと以外は実施例30と同様にして実施例電池(E32)を得た。
【0094】
[実施例33]
天然黒鉛粉末と負極活物質(e13)との混合質量比を30:70としたこと以外は実施例30と同様にして実施例電池(E33)を得た。
【0095】
[実施例34]
天然黒鉛粉末と負極活物質(e13)との混合質量比を40:60としたこと以外は実施例30と同様にして実施例電池(E34)を得た。
【0096】
[実施例35]
天然黒鉛粉末と負極活物質(e13)との混合質量比を50:50としたこと以外は実施例30と同様にして実施例電池(E35)を得た。
【0097】
[実施例36]
天然黒鉛粉末と負極活物質(e13)との混合質量比を60:40としたこと以外は実施例30と同様にして実施例電池(E36)を得た。
【0098】
[実施例37]
天然黒鉛粉末と負極活物質(e13)との混合質量比を70:30としたこと以外は実施例30と同様にして実施例電池(E37)を得た。
【0099】
[実施例38]
天然黒鉛粉末と負極活物質(e13)との混合質量比を80:20としたこと以外は実施例30と同様にして実施例電池(E38)を得た。
【0100】
[実施例39]
天然黒鉛粉末と負極活物質(e13)との混合質量比を90:10としたこと以外は実施例30と同様にして実施例電池(E39)を得た。
【0101】
[実施例40]
天然黒鉛粉末と負極活物質(e13)との混合質量比を95:5としたこと以外は実施例30と同様にして実施例電池(E40)を得た。
【0102】
[実施例41]
天然黒鉛粉末と負極活物質(e13)との混合質量比を99:1としたこと以外は実施例30と同様にして実施例電池(E41)を得た。
【0103】
[実施例42]
実施例13の負極製作工程でアセチレンブラックを用いなかったこと以外は実施例13と同様にして、実施例電池(E42)を得た。
【0104】
[実施例43]
実施例13の負極製作工程でアセチレンブラックの代わりに数平均粒径20μm、比表面積が5m2/gのメソカーボンマイクロビーズ(MCMB)を用いたこと以外は実施例13と同様にして、実施例電池(E43)を得た。
【0105】
[実施例44]
実施例13の負極製作工程でアセチレンブラックの代わりに数平均粒径20μm、比表面積が5m2/gの難黒鉛化性炭素粉末を用いたこと以外は実施例13と同様にして、実施例電池(E44)を得た。
【0106】
[実施例45]
負極合剤層の多孔度が55%であったこと以外は実施例1と同様にして実施例電池(E45)を得た。
【0107】
[実施例46]
負極合剤層の多孔度が18%であったこと以外は実施例1と同様にして実施例電池(E46)を得た。
【0108】
[比較例1]
原料として用いるSiO2粉末中にFe、Cr、Niがそれぞれ50ppm、50ppm、270ppm含まれていたこと以外は実施例1と同様にしてSiO粒子を得た。つぎに、この物質を負極活物質(r1)に用いたこと以外は実施例1と同様にして比較例電池(R1)を得た。
【0109】
高率放電性能を調べるために、上記実施例1、6、7、8、9、10、45および46において、同様の方法で電池を製作した。電池記号は、それぞれE1(HR)、E6(HR)、E7(HR)、E8(HR)、E9(HR)、E10(HR)、E45(HR)、E46(HR)で表される。
【0110】
低温サイクル試験を実施するために、上記実施例1、13、17、18、19、20、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45および46において、同様の方法で電池を製作した。電池記号は、それぞれE1(LT)、E13(LT)、E17(LT)、E18(LT)、E19(LT)、E20(LT)、E30(LT)、E31(LT)、E32(LT)、E33(LT)、E34(LT)、E35(LT)、E36(LT)、E37(LT)、E38(LT)、E39(LT)、E40(LT)、E41(LT)、E42(LT)、E43(LT)、E44(LT)、E45(LT)およびE46(LT)で表される。
【0111】
[X線回折測定]
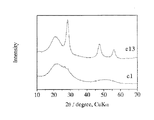
図1に、負極活物質(e1)および(e13)のX線回折パターンを示す。前者はブロードな回折パターンを示したのに対して、後者は約22°、28°、47°に明瞭な回折ピークを示した。約22°のピークはケイ素酸化物に、約28°、47°のピークはそれぞれSi(111)面ピークおよびSi(220)面ピークに由来する。したがって、負極活物質(e13)はケイ素およびその酸化物の両相を備えていることがわかった。また、負極活物質(e11)、(e12)、(e14)〜(e16)、(e21)〜(e29)も同様にして、Si(111)面ピークおよびSi(220)面ピークを示した。(e11)を除く上記負極活物質に関しては、Siに由来するこれら回折ピークの半値幅のいずれか一方は3°未満であった。なお、X線回折測定装置として理学電機(株)製RINT2400を用いた。また、発散スリット幅を1.0°、散乱スリット幅を1.0°、受光スリット幅を0.15mmとし、スキャンスピードを1°/minとした。
【0112】
[ICP発光分光分析]
実施例電池および比較例電池に用いたSiOに関してICP発光分光分析をおこなった結果、Fe、Crの含有率は、いずれのSiOにおいてもそれぞれ35ppm、15ppmであった。また、Niの含有率は、負極活物質(e1)、(e2)、(e3)、(e4)、(e5)、(r1)において、それぞれ45ppm、50ppm、130ppm、170ppm、200ppm、207ppmであった。上記負極活物質以外のSiOにおいては、Niの含有率はいずれも45ppmであった。なお、分析装置としては、日本ジャーレル・アッシュ(株)製IRIS/APを使用した。
【0113】
[充放電測定]
実施例電池E1〜E5および比較例電池R1を、25℃、1CmAの電流値で4.2Vまで充電し、続いて電圧を4.2Vで3時間一定とした。引き続いて、1CmAの電流値で2.5Vまで放電した。これを1サイクルとする。その結果、1サイクル目で得られた放電容量は全て400mAhであった。ここで1CmAは400mAに相当する。つぎに、25℃で1CmAの電流で4.2Vまで充電し、続いて電圧を4.2Vで3時間一定とした。その後、電池を80℃の恒温槽中で50時間保存した。保存後に電池の厚みを測定した。表1に、負極活物質に用いたSiO中のFe、CrおよびNiの含有率の総和(T)および80℃で保存後の電池厚みを示す。なお、電池厚みを、80℃で保存前に対する保存後の厚みを相対的に表す(百分率表示)。電池厚みが大きいほど、電池の膨れが大きいと判断できる。
【0114】
【表1】

表1から明らかなように、充電状態でさらに高温で保存した電池の膨れは、Tが250ppm以下の場合に著しく抑制された。また、Tが100ppm以下の場合、電池の膨れがさらに抑制された。したがって、電池の膨れの観点から、SiOx(0<x<2)中のFe、CrおよびNiの含有率の総和が250ppm以下であることが好ましく、とくにその含有率が100ppm以下であることが好ましい。なお、上記実施例では、Fe、Crの含有率が同じものを用いたが、これらの値をも変えて確認したところ、上記同様、これら3元素の含有率の総和が250ppmを越えない場合に、電池厚みの増加を抑制できた。
【0116】
実施例電池E6〜E10、E45、E46においてもE1と同様にして、上記の充放電試験をおこなった後、その電池を充電状態にして、さらに80℃の恒温槽中で50時間保存した。保存後に電池の厚みを測定した。その結果、1サイクル目で得られた放電容量は全て400mAhであった。表2.1に、負極合剤層の多孔度、80℃で保存後の電池厚みを示す。また、実施例電池E1(HR)、E6(HR)、E7(HR)、E8(HR)、E9(HR)、E10(HR)、E45(HR)、E46(HR)、比較例電池R1(HR)においてはE1と同様にして、上記の充放電試験をおこなった後、その電池を充電状態にして、つぎに0.2CmA(80mA)で放電した。その後、同様の方法で4.2Vで充電した。最後に、2CmA(800mA)で放電した。表2.2に、各電池の0.2CmAで得られた放電容量に対する2CmAで得られた容量の比(百分率表示)を示す。
【0117】
【表2】
表2.1
表2.2
表2.1から明らかなように、充電状態でさらに高温で保存した電池の膨れは、負極合剤層の多孔度を50%以下とすることにより著しく抑制された。また、表2.2から明らかなように、電池の高率放電性能は、負極合剤層の多孔度を20%以上とすることにより良好に保たれた。したがって、電池の膨れおよび高率放電性能の観点から、負極合剤層の多孔度が20〜50%であることが好ましい。
【0120】
表2.1および2.2から、負極合剤層の多孔度が25〜35%のとき、電池の膨れが小さく、さらに高率放電性能が良好であるため、とくに好ましいことがわかった。
【0121】
実施例電池E11〜E16および比較例電池R4においてもE1と同様にして、上記の充放電試験をおこなった後、その電池を充電状態にして、さらに80℃の恒温槽中で50時間保存した。表3に、負極活物質のX線回折パターンから得られたSi(111)面およびSi(220)面回折ピークの半値幅、80℃で保存後の電池厚みおよび1サイクル目で得られた放電容量を示す。
【0122】
【表3】
表3から明らかなように、充電状態でさらに高温で保存した電池の膨れは、Si(111)面およびSi(220)面の各回折ピークの少なくとも一方の半値幅B(2θ)が3°未満である場合、電池の膨れが著しく小さくなった。さらにこの効果は、半値幅が2°未満のときにとくに顕著であった。また、半値幅が0.6°以下の場合、放電容量が大きく低下した。したがって、電池の膨れおよび放電容量の観点から、Si(111)面およびSi(220)面の各回折ピークの少なくとも一方の半値幅B(2θ)が0.6°<B<2.0°であることが好ましい。
【0124】
実施例電池E17〜E20においてもE1と同様にして、上記の充放電試験をおこなった後、その電池を充電状態にして、さらに80℃の恒温槽中で50時間保存した。その結果、80℃で保存後の電池厚みは、保存前とくらべて全て110%であった。また、1サイクル目で得られた放電容量は全て400mAhであった。つぎに、実施例電池E1(LT)、E17(LT)、E18(LT)、E19(LT)、E20(LT)においてはE1と同様にして、上記の充放電試験をおこなった。充放電後の電池厚みをT(1)とする。つぎに、これらの電池を0℃に保存し、その温度で1CmAの電流値で充放電サイクル試験を20回繰り返した。サイクル試験の条件は1サイクル目と同様である。その後、電池を25℃に保存して、電池厚みを測定した。この厚みをT(2)とする。表4に、各電池の、低温サイクル試験前後における電池厚み比(T(2)/T(1))(百分率表示)を示す。
【0125】
【表4】
表4から明らかなように、SiOの数平均粒径を15μm未満とすることにより、低温サイクル試験後の電池の膨れが著しく抑制された。また、粒子径を5μm未満とすることにより、電池の膨れがさらに抑制された。したがって、低温サイクル試験後の電池の膨れを考慮して、SiOx(0<x<2)の数平均粒径が15μm未満であることが好ましく、とくに5μm未満であることが好ましい。
【0127】
実施例電池E21〜E29においてもE1と同様にして、上記の充放電試験をおこなった後、その電池を充電状態にして、さらに80℃の恒温槽中で50時間保存した。表5に、SiOに備えた炭素材料(B)の担持量および1サイクル目で得られた放電容量を示す。なお、80℃で保存後の電池厚みは、保存前とくらべて全て110%であった。
【0128】
【表5】
表5から明らかなように、SiOに備えた炭素材料(B)の担持量が両者を合わせた全質量に対して3質量%以上である場合に、電池の放電容量が著しく大きくなった。また、その担持量が15〜30質量%の場合、電池の放電容量がとくに大きくなった。しかし、その担持量が50質量%をこえると、電池の放電容量が低下した。したがって、電池の放電容量の観点から、SiOx(0<x<2)が、その表面に備える炭素材料(B)の担持量は、3〜50質量%であることが好ましく、とくに15〜30質量%であることが好ましい。
【0130】
実施例電池E21〜E29に用いた負極活物質に関して、波長が514.5nmのアルゴンイオンレーザーを用いたラマン分光分析をおこなった。その測定においては、出力を50mW、積算時間を300sとした。その結果、いずれの活物質についても、1575〜1620cm−1および1340〜1370cm−1の範囲に現れるピークの強度をそれぞれIAおよびIBとすると、IB/IA=0.8であった。
【0131】
実施例電池E30〜E44においてもE1と同様にして、上記の充放電試験をおこなった後、その電池を充電状態にして、さらに80℃の恒温槽中で50時間保存した。表6.1に、負極に添加した炭素材料材料(D)の混合割合および1サイクル目で得られた放電容量を示す。なお、80℃で保存後の電池厚みは、保存前とくらべて全て110%であった。また、実施例電池E13(LT)、30(LT)〜E44(LT)においてはE1(LT)と同様にして、0℃で低温サイクル試験を20回繰り返した。表6.2に、負極に添加した炭素材料(D)の混合割合、および各電池の低温サイクル試験前後における電池厚み比(T(2)/T(1))(百分率表示)を示す。
【0132】
【表6】
表6.1
表6.2
表6.1から明らかなように、SiOと混合した炭素材料(D)の混合割合が、両者を合わせた全質量に対して5質量%以上である場合に、電池の放電容量が著しく大きくなった。また、その混合量が20質量%以上の場合、電池の放電容量がとくに大きくなった。しかし、その混合量が95質量%をこえると、電池の放電容量が低下した。また、表6.2から明らかなように、低温サイクル時における電池の膨れは、炭素材料(D)の混合割合が50質量%以上の場合、著しく抑制された。したがって、電池の放電容量および低温サイクル時の電池膨れの観点から、SiOx(0<x<2)と混合した炭素材料(D)の混合割合が、両者を合わせた全質量に対して好ましくは50〜95%である。
【0135】
実施例電池E13、E42〜E44とを比較すると、SiOx(0<x<2)と炭素材料(D)とを混合したものを負極に用いることによって電池の放電容量が著しく大きくなることがわかる。
【0136】
充放電後の実施例電池E12〜E16、E21〜E44を解体し、負極活物質を取り出して、そのX線回折測定をおこなったところ、電池組み込み前に現れた約28°、47°におけるSi(111)面、Si(220)面の各回折ピークの強度が著しく低下することがわかった。両ピークの半値幅はともに3°(2θ)以上であった。したがって、本発明負極活物質にリチウムを挿入・脱離させると、ケイ素の結晶性が著しく低下することがわかった。
【0137】
【発明の効果】
以上述べたように、SiとOとを含み、Siに対するOの原子比xが0<x<2で表され、Fe、CrおよびNiを含み、Niの含有率が45〜200ppmで、さらにFe、CrおよびNiの含有率の総和が250ppm以下である物質(A)負極活物質として備えた非水電解質電池では、充電状態でさらに高温で保存した場合における電池の膨れが抑制される。
【図面の簡単な説明】
【図1】回折角(2θ)が10°〜70°の範囲における負極活物質(e1)および(e13)のX線回折パターンを示す図。
Claims (1)
- SiとOとを含み、Siに対するOの原子比xが0<x<2で表され、Fe、CrおよびNiを含み、Niの含有率が45〜200ppmで、Fe、CrおよびNiの含有率の総和が250ppm以下である物質(A)を負極活物質として備えた非水電解質電池。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003143261A JP4632016B2 (ja) | 2003-05-21 | 2003-05-21 | 非水電解質電池 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003143261A JP4632016B2 (ja) | 2003-05-21 | 2003-05-21 | 非水電解質電池 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004349057A JP2004349057A (ja) | 2004-12-09 |
| JP4632016B2 true JP4632016B2 (ja) | 2011-02-16 |
Family
ID=33531098
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003143261A Expired - Fee Related JP4632016B2 (ja) | 2003-05-21 | 2003-05-21 | 非水電解質電池 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4632016B2 (ja) |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4519592B2 (ja) * | 2004-09-24 | 2010-08-04 | 株式会社東芝 | 非水電解質二次電池用負極活物質及び非水電解質二次電池 |
| JP5230904B2 (ja) * | 2005-06-17 | 2013-07-10 | パナソニック株式会社 | 非水電解液二次電池 |
| JP2009076372A (ja) * | 2007-09-21 | 2009-04-09 | Shin Etsu Chem Co Ltd | 非水系二次電池 |
| WO2009054149A1 (ja) * | 2007-10-24 | 2009-04-30 | Panasonic Corporation | 電気化学素子用電極およびそれを用いた電気化学素子 |
| JP5143700B2 (ja) * | 2007-10-24 | 2013-02-13 | パナソニック株式会社 | 電気化学素子用電極およびそれを用いた電気化学素子 |
| JP5315665B2 (ja) | 2007-10-31 | 2013-10-16 | ソニー株式会社 | リチウムイオン二次電池用負極およびリチウムイオン二次電池 |
| JP5196149B2 (ja) * | 2008-02-07 | 2013-05-15 | 信越化学工業株式会社 | 非水電解質二次電池用負極材及びその製造方法並びにリチウムイオン二次電池及び電気化学キャパシタ |
| WO2009119093A1 (ja) * | 2008-03-26 | 2009-10-01 | パナソニック株式会社 | リチウム二次電池用電極およびその製造方法 |
| JP5184567B2 (ja) * | 2010-03-12 | 2013-04-17 | 信越化学工業株式会社 | 非水電解質二次電池用負極材並びにリチウムイオン二次電池及び電気化学キャパシタ |
| JP6020331B2 (ja) * | 2012-05-16 | 2016-11-02 | 信越化学工業株式会社 | 珪素酸化物粒子及びその製造方法、ならびにリチウムイオン二次電池及び電気化学キャパシタ |
| JP2013008696A (ja) * | 2012-09-18 | 2013-01-10 | Shin Etsu Chem Co Ltd | 非水電解質二次電池用負極材の製造方法 |
| JP5904071B2 (ja) * | 2012-09-18 | 2016-04-13 | 株式会社豊田自動織機 | 蓄電装置、及び電極の製造方法 |
| JP5737265B2 (ja) * | 2012-10-23 | 2015-06-17 | 信越化学工業株式会社 | 珪素酸化物及びその製造方法、負極、ならびにリチウムイオン二次電池及び電気化学キャパシタ |
| JP2014089855A (ja) * | 2012-10-30 | 2014-05-15 | Hitachi Maxell Ltd | 非水二次電池用負極活物質および非水二次電池 |
| JP6173969B2 (ja) * | 2014-05-16 | 2017-08-02 | 信越化学工業株式会社 | 粒状珪素酸化物及びその製造方法 |
| JP6445956B2 (ja) * | 2015-11-17 | 2018-12-26 | 信越化学工業株式会社 | 負極活物質、混合負極活物質材料、非水電解質二次電池用負極、リチウムイオン二次電池 |
| JP7251915B2 (ja) * | 2017-10-02 | 2023-04-04 | 株式会社大阪チタニウムテクノロジーズ | 負極材用粉末及び負極材料製造方法 |
| US10879735B2 (en) * | 2018-03-12 | 2020-12-29 | Omega Energy Systems, Llc | Solid-state energy harvester of transition metal suboxides |
| CN112310384B (zh) * | 2019-07-29 | 2022-03-11 | 宁德时代新能源科技股份有限公司 | 硅氧化合物及二次电池 |
| KR20240153758A (ko) * | 2023-04-17 | 2024-10-24 | 에스케이온 주식회사 | 리튬 이차 전지용 음극 및 이를 포함하는 리튬 이차 전지 |
| CN120413606A (zh) * | 2024-01-31 | 2025-08-01 | 珠海冠宇电池股份有限公司 | 一种负极片及锂离子电池 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6168313A (ja) * | 1984-09-07 | 1986-04-08 | Osaka Titanium Seizo Kk | シリコン切粉から高純度けい素の回収方法 |
| JPH07312219A (ja) * | 1994-03-24 | 1995-11-28 | Fuji Photo Film Co Ltd | 非水二次電池と充電法 |
| JP3809662B2 (ja) * | 1996-01-30 | 2006-08-16 | 宇部興産株式会社 | 非水二次電池 |
| JP3865033B2 (ja) * | 2000-02-04 | 2007-01-10 | 信越化学工業株式会社 | 酸化珪素粉末の連続製造方法及び連続製造装置 |
| JP2001226112A (ja) * | 2000-02-15 | 2001-08-21 | Shin Etsu Chem Co Ltd | 高活性な酸化珪素粉末及び製造方法 |
| JP2002260669A (ja) * | 2001-02-28 | 2002-09-13 | Shin Etsu Chem Co Ltd | 非水電解質二次電池 |
-
2003
- 2003-05-21 JP JP2003143261A patent/JP4632016B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2004349057A (ja) | 2004-12-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101038637B1 (ko) | 음극 활성 물질, 그것을 이용한 음극, 그것을 이용한비수성 전해질 전지, 및 음극 활성 물질의 제조 방법 | |
| JP4632016B2 (ja) | 非水電解質電池 | |
| KR102316341B1 (ko) | 실리콘계 복합체, 이를 포함하는 음극, 및 리튬 이차전지 | |
| EP2874211B1 (en) | Transition metal-pyrophosphate anode active material, manufacturing method therefor, and lithium secondary battery or hybrid capacitor comprising same | |
| KR101241810B1 (ko) | 리튬실리케이트계 화합물의 제조 방법, 이 제조 방법에 의해 얻어진 리튬실리케이트계 화합물로 이루어진 리튬 이온 이차 전지용 정극 활물질, 상기 화합물을 포함하는 리튬 이차 전지용 정극 및 리튬 이차 전지 | |
| JP4965790B2 (ja) | 非水電解質二次電池 | |
| KR101666878B1 (ko) | 리튬 이차 전지용 음극 활물질, 그 제조 방법, 및 이를 포함하는 리튬 이차 전지 | |
| EP3386015A1 (en) | Cathode active material for secondary battery, and secondary battery comprising same | |
| JP5614307B2 (ja) | 非水電解質二次電池 | |
| JP4332845B2 (ja) | 非水電解質電池 | |
| KR20190044444A (ko) | 리튬 이차전지용 양극 활물질, 이의 제조방법, 이를 포함하는 리튬 이차전지용 양극 및 리튬 이차전지 | |
| US9997781B2 (en) | Transition metal-metaphosphate anode active material, method of preparing the same, and lithium secondary battery or hybrid capacitor including the anode active material | |
| JP6176510B2 (ja) | シリコン材料及び二次電池の負極 | |
| JP5660403B2 (ja) | 負極活物質とその製造方法及び蓄電装置 | |
| CN111201200A (zh) | 制备二次电池用正极活性材料的方法和使用其的二次电池 | |
| KR101530963B1 (ko) | 리튬 이차 전지용 음극 활물질, 이의 제조 방법 및 리튬 이차 전지 | |
| KR101632358B1 (ko) | ZnO-MnO-C 복합체, 산화아연 및 산화망간을 포함하는 복합체의 제조방법 및 이를 포함하는 리튬 이차 전지용 음극 활물질 | |
| EP4329013B1 (en) | Method for preparing cathode additive for lithium secondary battery | |
| CN115119528B (zh) | 锂过渡金属氧化物、锂二次电池用正极添加剂以及包含其的锂二次电池 | |
| KR20150089389A (ko) | 양극 활물질, 이를 채용한 리튬전지 및 양극 활물질의 제조방법 | |
| JP4635409B2 (ja) | 非水電解質電池 | |
| JP6065678B2 (ja) | 負極活物質とその製造方法及び蓄電装置 | |
| KR20250031467A (ko) | 음극 활물질 및 이를 포함하는 리튬 이차 전지 | |
| WO2022045128A1 (ja) | 二次電池用負極活物質および二次電池 | |
| WO2026024357A2 (en) | Electroactive materials having hard carbon-silicon dioxide nanocomposites from bioderived byproducts for electrochemical cells, methods for making and use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20051213 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20060519 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20090805 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090818 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090924 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20100507 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20101020 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20101102 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131126 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |