JP4239343B2 - 酸化物の結晶成長方法、電界効果トランジスタの製造方法および強誘電体不揮発性メモリの製造方法 - Google Patents
酸化物の結晶成長方法、電界効果トランジスタの製造方法および強誘電体不揮発性メモリの製造方法 Download PDFInfo
- Publication number
- JP4239343B2 JP4239343B2 JP2000027362A JP2000027362A JP4239343B2 JP 4239343 B2 JP4239343 B2 JP 4239343B2 JP 2000027362 A JP2000027362 A JP 2000027362A JP 2000027362 A JP2000027362 A JP 2000027362A JP 4239343 B2 JP4239343 B2 JP 4239343B2
- Authority
- JP
- Japan
- Prior art keywords
- cerium oxide
- silicon substrate
- oxide
- cerium
- film
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images
























Description
【発明の属する技術分野】
この発明は、酸化物の結晶成長方法、セリウム酸化物、プロメチウム酸化物、酸化物積層構造、電界効果トランジスタの製造方法、電界効果トランジスタ、強誘電体不揮発性メモリの製造方法および強誘電体不揮発性メモリに関し、特に、シリコン基板上に展開される酸化物エレクトロニクスに適用して好適なものである。
【0002】
【従来の技術】
シリコン(Si)の熱酸化により形成した酸化シリコン(SiO2 )膜は、電気的絶縁性の高さ、界面準位密度の低さ、プロセス的容易さ、熱的安定性等により、これまで、MOS(Metal-Oxide-Semiconductor)−FETのゲート絶縁膜として専ら用いられてきた。ゲート絶縁膜として用いられるこの熱酸化によるSiO2 膜は、比誘電率が低い(εr 〜3.8)ため、Si基板上に極めて薄く形成する必要がある。しかしながら、集積化の要請から、ゲート絶縁膜の薄膜化、短チャネル化等が進むにつれ、ゲート絶縁膜の絶縁破壊、ソース・ドレイン電圧の影響によるチャネルのピンチオフ(短チャネル効果)等の問題が顕著となり、ゲート絶縁膜の材料的な限界が近づいている。このような理由により、サブ0.1ミクロン世代のMOS−FETの技術的課題として、リソグラフィー技術の進展は勿論のこと、高い誘電率を有する新たなゲート絶縁膜の必要性が叫ばれている(例えば、(1)MTL VLSI Seminar(Massachusetts Institute of Technology))。高い誘電率を有する材料を用いてゲート絶縁膜を形成することができれば、あまり薄膜化しないで済むため、ゲートリークの抑制が可能となり、また短チャネル効果も抑制することができると考えられる。
【0003】
一方、近年、強誘電体不揮発性メモリ(FeRAM)(例えば、(2)Appl.Phys.Lett.,48(1986)1439 、(3)IEDM Tech.Dig.,(1987)850 、(4)IEEE J.Solid State Circuits,23(1988)1171 、(5)1988 IEEE Int.Solid-State Circuits Conf.(ISSCC88)、(6)Digest of Technical Papers,THAM 10.6(1988)130、(7)Oyo Buturi,62(1993)1212)の研究が活発化している。この強誘電体不揮発性メモリで現在最も実用に近いと考えられているものは、DRAM類似構造の強誘電体不揮発性メモリ(2トランジスタ−2キャパシタ型メモリセルまたは1トランジスタ−1キャパシタ型メモリセルを用いたもの)である。この構造は、CMOSプロセスと強誘電体キャパシタプロセスとを層間絶縁膜の使用により分離することが可能であるため、Siプロセスとの干渉を比較的抑制し易い、という利点がある。しかしながら、この強誘電体不揮発性メモリでは、構造的にSiデバイスのスケーリング則を適用することができないことから、微細化が進むと、キャパシタの蓄積電荷量確保のため、構造を複雑にするか、さらに残留分極値の大きな材料を探索する必要がある。他方、この強誘電体不揮発性メモリと双璧をなす、MFS(Metal-Ferroelectrics-Semiconductor)−FET型メモリセル(MFMIS(Metal-Ferroelectrics-Metal-Insulator-Semiconductor)−FET型メモリセル、FCG(Ferroelectric Capacitor Gate)型メモリセル等も含む)を用いた強誘電体不揮発性メモリの研究に取り組んでいる研究機関も少なくない。この強誘電体不揮発性メモリは、スケーリング則にのる上、必要な残留分極が非常に小さく(〜0.1μC/cm2 程度)、また、1トランジスタのみで記憶することができるため、セルサイズも小さく、高集積化に有利である。さらに、非破壊読み出しであるため、破壊読み出しの2トランジスタ−2キャパシタ型メモリセルまたは1トランジスタ−1キャパシタ型メモリセルに比べて、強誘電体の本質的問題とも考えられる疲労に対しても有利であり、高速動作も可能である。このように優れた特性を期待することができるため、MFS−FET型強誘電体不揮発性メモリは究極のメモリと考えられている((8)Appl.Surf.Sci.113/114(1997)656)。
【0004】
このMFS−FET型強誘電体不揮発性メモリの実用化を阻害しているのは、プロセス的問題である。Si基板上に直接強誘電体を成長させるのは極めて困難であり、そのため、Si基板上への絶縁体からなるバッファ層の成長は、極めて重要な技術と位置付けられている。MFS−FET型強誘電体不揮発性メモリの一種であるMFIS(Metal-Ferroelectrics-Insulator-Semiconductor)−FET型強誘電体不揮発性メモリの場合には、絶縁層にもゲート電圧が分配されるため、書き込み電圧が高くなる欠点が生ずる。これを抑制するためには、高誘電率の絶縁層が必要になる。一方、強誘電体に要求される材料特性としては、低誘電率、適切な残留分極値(デバイス設計に依存するが、典型的には〜0.1μC/cm2 程度)、そして何よりも良好な角型比が挙げられる。また、より良好な界面を実現するためには、これらの材料を低温で成長させることができることが重要な条件である。このように、1トランジスタ−1キャパシタ方式とはまた異なった観点から、材料を選択、開発することが求められる。MFIS−FET型強誘電体不揮発性メモリの研究報告は多々あるが、界面特性が十分ではないため、リテンション(電荷保持特性)まで含めて実用化可能なものの報告例はほとんど皆無というのが現状である。一方で、ゲート絶縁膜として既存の熱酸化によるSiO2 膜を採用することができるMFMIS構造も検討されており((9)Jpn.J.Appl.Phys.,33(1994)5207) 、これは比較的実用段階に近いと考えられる。これをさらに進めて、強誘電体キャパシタを別に形成し、これと多結晶Siゲートとを配線で接続する方式も提案されている((10)特開平8−250608号公報、(11)特開平9−205181号公報)。この方法によれば、強誘電体とSiトランジスタとの素子分離が容易なだけでなく、キャパシタとゲートとの面積比の設計自由度が上がるため、キャパシタの相対面積を小さく設計することにより、低い書き込み電圧で十分な分極を得ることが可能になる。しかしながら、多結晶の強誘電体では必要な角型比を安定的に得るのは困難であり、また、微細化が進めば上述の熱酸化によるSiO2 膜の材料的限界に直面することに変わりはない。結局、MFS−FET型強誘電体不揮発性メモリ実現のキーテクノロジーも、Si基板上への高誘電率絶縁膜の成長に帰着する。それも、急峻な界面と熱酸化によるSiO2 膜並の低い界面準位密度とを実現するためには、格子整合性の良い材料をエピタキシャルに成長させることが有利であり、基板としてはSiの最大の易動度を有するSi〈110〉方向にチャネルを形成することができ、MOS−FET用基板として専ら用いられているSi(001)基板上である必要がある、という極めて高い技術的ハードルがある。
【0005】
一方、半導体産業にSiO2 以外の酸化物材料を導入することは、意義深いことである。1986年の高温超伝導材料の発見((12)Z.Phys.B.,64,189-193(1986)) は言うまでもなく、取り分け、ペロブスカイトまたはそれに関連した構造をとる酸化物材料は、強誘電性、高誘電率、超伝導性、超巨大磁気抵抗等、半導体デバイスにとって非常に重要な物性を有する((13)Mater.Sci.Eng.,B41(1996)166 、(14)J.Ceram.Soc.Japan,Int.Ed.,103(1995)1088)。例えば、上述の強誘電体不揮発性メモリのキャパシタの強誘電体材料としては、自発分極値が大きく、プロセス温度の低いジルコンチタン酸鉛(PZT)(例えば、(15)J.Appl.Phys.,70,382-388(1991))、駆動電圧が低く、分極反転に伴う自発分極値の劣化の少ないビスマスストロンチウムタンタレート(Bi2 SrTa2 O9 ((16)Nature,374(1995)627 、(17)Appl.Phys.Lett.,66(1995)221 、(18)Mater.Sci.Eng.,B32(1995)75、(19)Mater.Sci.Eng.,B32(1995)83、(20)Appl.Phys.Lett.,67(1995)572 、(21)J.Appl.Phys.,78(1995)5073 、(22)Appl.Phys.Lett.,68(1996)566 、(23)Appl.Phys.Lett.,68(1996)690 、(24)国際公開公報WO93/12542号)が双璧をなしている。さらに、磁場印加により抵抗率変化が何桁にもおよぶ超巨大磁気抵抗材料(CMR(Colossal Magnetoresistance)材料)がMn酸化物系で発見される((25)Phys.Rev.Lett.74(1995)5108)など、酸化物材料が如何に高い潜在力を有するかが注目され始めており((26)Mater.Sci.Eng.,B41(1996)166 、(27)J.Ceram.Soc.Japan,Int.Ed.,103(1995)1088)、酸化物を薄膜化する技術はここ10年ほどで驚異的に進展しつつある。
【0006】
このような極めて高機能な物性を有する酸化物材料を半導体産業の基盤であるSi上で展開することができれば、材料に高い市場性を付与することが可能になる。しかしながら、これらの機能性酸化物材料とSiとの間には、相互熱拡散、熱膨張率の違い等の問題があるため、直接これらの機能性酸化物材料をSi上に成長させるのは一般には容易でない。
【0007】
上述したように、これらの機能性酸化物材料群のほとんどは、ペロブスカイト構造を母体とする構造をとっている。中でも、液体窒素温度を超える臨界温度を有するイットリウム系超伝導材料、上述したBi2 SrTa2 O9 等、異方性の極めて大きな層状ペロブスカイトと称される構造をとるものが少なくない。これらの層状ペロブスカイト構造酸化物における超伝導電流経路、分極軸等は、特定の方向に限定されており、また、単純ペロブスカイト構造酸化物の場合も、上述のPZTのように分極軸が特定の方向に限定されているものも多い。そのため、デバイス化した場合、最大の特性を引き出すためには、酸化物を配向させるか、より好ましくは下地に対してエピタキシャル成長させることが重要である。
【0008】
さて、蛍石(フルオライト)構造を有するセリア(酸化セリウム:CeO2 )は、熱的安定性、高い比誘電率(εr 〜26)、Si基板との極めて良好な格子整合性(ミスフィット:約0.35%)から、熱酸化によるSiO2 膜に代わるサブ0.1ミクロン世代の高誘電率のゲート絶縁膜の材料の候補の一つであり、また、Si基板上でペロブスカイト関連酸化物をエピタキシャル成長させるための最も理想的なバッファ層材料の一つであると考えられている。実際、Si基板上にセリアをエピタキシャル成長させる研究が行われているが、その多くが、最原子稠密で成長しやすいCeO2 (111)/Si(111)構造のものである(例えば、(28)Jpn.J.Appl.Phys.34(1995),L688 、(29)特開平7−25698号公報)。しかしながら、応用上最も重要なSi(001)基板に関しては、エピタキシャルCeO2 (001)/Si(001)構造こそが理想であると認識され、積層構造、デバイス等も種々考案されていながら((30)特開平2−267104号公報、(31)特開平6−97452号公報、(32)特開平10−182292号公報等)、実現されなかった経緯がある。これまでは、Si(001)−2×1、1×2の表面再構成によるダイマー(dimer)構造を反映し、アンチフェーズドメイン(antiphase domain) を有するCeO2 (110)がエピタキシャル成長すると考えられてきた。また、成長温度に関しては、高真空中でSiO2 膜がSi表面に形成されない800℃程度((33)J.Vac.Sci.Technol.A13(1995)772) がエピタキシャル温度の下限であると考えられている((34)Jpn.J.Appl.Phys.33(1994),5219 、(35)Appl.Phys.Lett.56(1990),1332、(36)Appl.Phys.Lett.59(1991),3604、(37)Physica C 192(1992)154、(38)Jpn.J.Appl.Phys.36(1997),5253 、(39)特開平9−64206号公報)。また、急峻な界面を得るには成長温度を低温化することが必要と考えられ、このために電子線アシストエピタキシャル成長等が試みられているが、それでも報告されているエピタキシャル成長温度は710℃が下限である((40)1998年春季応用物理学会学術講演会講演予稿集28p−PA−1)。CeO2 (001)/Si(001)構造の報告例も皆無ではないが、それを示す実験データがない((41)特開平2−267104号公報)、基板回折との分離について全く議論されていない((42)Solid State Comm.108(1998)225)等、エピタキシャル成長が証明されたものはこれまで存在しない。Si(001)基板上で、エピタキシャルでCeO2 (001)を実現するため、ジルコニア(酸化ジルコニウム:ZrO2 )とセリアとの固溶体(Ce,Zr)O2 ((43)Jpn.J.Appl.Phys.35(1996),5150)、これをバッファ層としたCeO2 /(Ce,Zr)O2 /Si積層構造((44)Jpn.J.Appl.Phys.36(1997),5253,(45)1998年春季応用物理学会学術講演会講演予稿集29p−ZF−4)やCeO2 /SrTiO3 /Si構造((46)Jpn.J.Appl.Phys.30(1991)L1136)も提案されているが、Si(001)基板上に直接CeO2 (001)がエピタキシャル成長したという報告はこれまで皆無であった。
【0009】
C−希土構造(ビクスバイト(bixbyite) )をとるイットリア(酸化イットリウム:Y2 O3 )に関しても、セリアに準ずる材料特性を有するため、鋭意検討が進められているが、セリアと同様な困難があり、Si(001)基板上には、Y2 O3 (110)がエピタキシャル成長するのが通例である((47)Appl.Phys.Lett.71(1997),903)。
【0010】
一方、セリア(またはイットリア)上にはペロブスカイト型酸化物がエピタキシャル成長する例があり(例えば、(48)Appl.Phys.Lett.68(1996)553) 、したがってセリアの配向性を制御することができれば、機能性酸化物の特性を生かしたデバイスの実現が可能になる。
【0011】
【発明が解決しようとする課題】
このような状況下、応用上最も重要なSi(001)基板上にCeO2 (001)をエピタキシャル成長させる技術は、極めて重要となる。
【0012】
したがって、この発明の目的は、酸化セリウムや酸化イットリウム、さらにはこれらと同様な結晶構造を有する希土類酸化物を(001)面方位のシリコン基板上に(001)面方位でエピタキシャル成長させ、エピタキシャルな希土類酸化物(001)/シリコン(001)構造を実現することができる酸化物の結晶成長方法、セリウム酸化物、プロメチウム酸化物、酸化物積層構造、この酸化物の結晶成長方法をゲート絶縁膜の形成に適用した電界効果トランジスタの製造方法、電界効果トランジスタ、この酸化物の結晶成長方法を強誘電体キャパシタの形成に適用した強誘電体不揮発性メモリの製造方法および強誘電体不揮発性メモリを提供することにある。
【0013】
【課題を解決するための手段】
本発明者は、従来技術が有する上述の課題を解決するために、鋭意検討を行った。以下にその概要について説明する。ここでは、代表例として、酸化セリウムの成長について説明する。
【0014】
すなわち、上記の目的を達成するためには、少なくとも一旦はシリコン基板表面をステップ、テラスで構成された表面とすることが重要になる。このような表面は、例えば、石坂、白木等によって提案された手法((49)J.Electrochem.Soc.,133(1986)666)またはこれに準ずる手法により実現可能である。成長装置は、界面制御性に優れ、超高真空中で清浄な表面が保ちやすく、また、反射高速電子線回折(RHEED)等により表面観察可能な分子線エピタキシー(MBE)装置、レーザアブレーション装置、反応性蒸着装置等が望ましいが、所定の圧力、温度等を制御可能なものであれば基本的にはどのようなものでもよい。酸化セリウム(CeO2 )の成長の場合、セリウム原料としては、一般的には酸化セリウムそのものが用いられることが多いが、蒸気圧の高い酸素が選択的に揮発するため、低い酸素分圧を制御するためにはセリウム金属を用いることが望ましい。ただし、例えば成長室の真空排気にゲッタポンプ等を併用して高真空を保つ工夫をすれば、酸化物原料を用いてもよいと考えられる。セリウム金属は、融点が高く、蒸気圧が低いため、高温型クヌーセンセル、電子線蒸着、エキシマレーザ等により気化させることが望ましい。また、これらの気体または金属元素等と活性度の高いシリコン基板の表面との反応を抑制するため、低温でのプロセスが重要となる。これまでになされた報告の中には、表面処理に関しては特に記述はなく、セリウム金属を用い、450〜600℃の基板温度、(4〜6)×10-4Torrの酸素分圧(基板付近では10-2Torr台と推定)にて、RFプラズマにより活性化させて、エピタキシャルCeO2 をシリコン基板上に形成することができたとするものがある((50)特開平2−267104号公報)。しかしながら、エピタキシャル成長方位に関しては記述がなく、このような基板温度、酸素分圧では、シリコン基板表面にSiO2 膜が生成してしまい、エピタキシャルCeO2 (001)の成長には至らない。RFプラズマにより活性化させれば、このような反応がさらに促進されてしまうと考えられる。
【0015】
本発明者は、成長温度の低温化により、セリウムシリサイド生成、酸化シリコン生成等の速度を抑制し、さらにその他の観点から、シリコン(001)基板上で、CeO2 (001)をエピタキシャル成長させる条件の検討を行った。その結果、まず、シリコン(001)基板の表面を成長前に2×1、1×2表面再構成によるダイマー構造としておくことが基本的に重要であり、さらに、成長時の原料の供給の方法も重要であるという結論に至った。後者の原料の供給方法については、具体的には、シリコン(001)基板表面への酸素などの酸化性ガスの供給をまず開始し、その後にCe原料の供給を開始することが重要である。その理由は現時点では完全に解明されていないが、Ce原料の供給が開始される時点でシリコン(001)基板の表面が酸素などの酸化性ガスの分子あるいは原子で覆われていることがCeO2 の(001)エピタキシャル成長を促進するものと推測される。
【0016】
上記のことに加えて、CeO2 (001)をエピタキシャル成長させるためには、成長温度に対し、Ce原料の供給量に対する酸化性ガスの供給量の比、あるいは、Ce原料の供給量を一定とすれば酸化性ガスの分圧あるいは供給量を適切に選ぶことが重要である。図1に、成長温度Tと、Ce原料の供給量を一定としたときのCe原料の供給量に対する酸化性ガスの供給量の比O/Ceとの関係を示す。ただし、便宜上、図1の縦軸には、O/Ceの代わりにO2 流量[sccm]の値を示してある。図1より、CeO2 (001)をエピタキシャル成長させることができる領域は限られていることがわかる。また、成長温度の上限は約300℃付近にあることがわかる。さらに、成長温度をより低温にするにしたがってO/CeあるいはO2 分圧の幅が広がり、100℃より低くすることにより相当な幅が得られ、実用上好ましい。
【0017】
本発明者は、以上の検討および知見に加えて、様々な観点から検討を行い、この発明を案出するに至ったものである。
【0018】
すなわち、上記課題を解決するために、この発明の第1の発明は、
(001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
上記シリコン基板上に立方晶系または正方晶系の希土類酸化物を(001)面方位にエピタキシャル成長させる工程と
を有することを特徴とする酸化物の結晶成長方法である。
【0019】
この発明の第2の発明は、
(001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
酸化性ガスを含む雰囲気中で少なくとも一種以上の希土類元素を含む原料を用いて上記シリコン基板上に立方晶系または正方晶系の希土類酸化物を(001)面方位にエピタキシャル成長させる工程と
を有することを特徴とする酸化物の結晶成長方法である。
【0020】
この発明の第3の発明においては、典型的には、300℃未満の成長温度、好適には100℃以下の成長温度で希土類酸化物をエピタキシャル成長させる。
【0021】
この発明の第3の発明は、
(001)面方位のシリコン基板を1×10-6Torr以下の圧力の真空中で加熱することによりその表面の酸化シリコン膜を揮発させる工程と、
上記酸化シリコン膜を揮発させた上記シリコン基板上に立方晶系または正方晶系の希土類酸化物を(001)面方位にエピタキシャル成長させる工程と
を有することを特徴とする酸化物の結晶成長方法である。
【0022】
この発明の第4の発明は、ビクスバイト構造を有することを特徴とするセリウム酸化物である。
【0023】
この発明の第5の発明は、ビクスバイト構造を有することを特徴とするプロメチウム酸化物である。
【0024】
この発明の第6の発明は、
(001)面方位のシリコン基板と、
上記シリコン基板上に第1の成長温度で成長された第1のCeO2 膜と、
上記第1のCeO2 膜上に第1の成長温度より高い第2の成長温度でエピタキシャル成長された第2のCeO2 膜と
を有することを特徴とする酸化物積層構造である。
【0025】
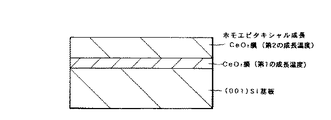
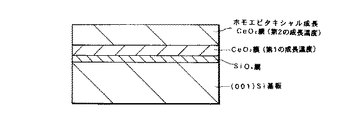
この発明の第6の発明において、典型的には、第2のCeO2 膜は(001)面方位である。また、第1の成長温度は例えば室温から300℃程度である。また、シリコン基板と第1のCeO2 膜との界面にはSiOx 膜が存在する場合もある。
【0026】
この発明の第7の発明は、
(001)面方位のシリコン基板と、
上記シリコン基板上のSiOx 膜と、
上記SiOx 膜上のアモルファスCeOy 膜と、
上記アモルファスCeOy 膜上の上記シリコン基板に対してエピタキシャルに配置した(001)面方位のCeO2 膜と
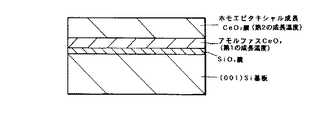
を有することを特徴とする酸化物積層構造である。
【0027】
この発明の第6および第7の発明において、SiOx 膜のxは通常は1≦x≦2であり、アモルファスCeOy 膜のyは通常は1.5≦x≦2である。
【0028】
この発明の第6の発明(シリコン基板と第1のCeO2 膜との界面にSiOx 膜が存在しない場合と存在する場合)および第7の発明による酸化物積層構造を図示すると、それぞれ図1、図2および図3のようになる。
【0029】
この発明の第8の発明は、
(001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
上記シリコン基板上に立方晶系または正方晶系の希土類酸化物を(001)面方位にエピタキシャル成長させることによりゲート絶縁膜を形成する工程と
を有することを特徴とする電界効果トランジスタの製造方法である。
【0030】
この発明の第8の発明においては、典型的には、300℃未満の成長温度、好適には100℃以下の成長温度で希土類酸化物をエピタキシャル成長させることによりゲート絶縁膜を形成する。また、シリコン基板を1×10-6Torr以下の圧力の真空中で加熱することによりその表面の酸化シリコン膜を揮発させてその表面をダイマー構造とし、このシリコン基板上に希土類酸化物をエピタキシャル成長させることによりゲート絶縁膜を形成する場合もある。
【0031】
この発明の第9の発明は、
(001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
酸化性ガスを含む雰囲気中で少なくとも一種以上の希土類元素を含む原料を用いて300℃未満の成長温度で上記シリコン基板上に立方晶系または正方晶系の希土類酸化物を(001)面方位にエピタキシャル成長させることによりゲート絶縁膜を形成する工程と
を有することを特徴とする電界効果トランジスタの製造方法である。
【0032】
この発明の第10の発明は、
(001)面方位のシリコン基板を1×10-6Torr以下の圧力の真空中で加熱することによりその表面の酸化シリコン膜を揮発させる工程と、
上記酸化シリコン層を揮発させた上記シリコン基板上に立方晶系または正方晶系の希土類酸化物を(001)面方位にエピタキシャル成長させることによりゲート絶縁膜を形成する工程と
を有することを特徴とする電界効果トランジスタの製造方法である。
【0033】
この発明の第11の発明は、
(001)面方位のシリコン基板と、
上記シリコン基板上にエピタキシャル成長された立方晶系または正方晶系の(001)面方位の希土類酸化物からなるゲート絶縁膜と、
上記ゲート絶縁膜上にエピタキシャル成長された強誘電体膜と
を有することを特徴とする電界効果トランジスタである。
【0034】
この発明の第12の発明は、
(001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
上記シリコン基板上に立方晶系または正方晶系の希土類酸化物を(001)面方位にエピタキシャル成長させる工程と、
上記希土類酸化物上に強誘電体膜をエピタキシャル成長させる工程と
を有することを特徴とする強誘電体不揮発性メモリの製造方法である。
【0035】
この発明の第13の発明は、
(001)面方位のシリコン基板と、
上記シリコン基板上にエピタキシャル成長された立方晶系または正方晶系の(001)面方位の希土類酸化物からなるゲート絶縁膜と、
上記ゲート絶縁膜上にエピタキシャル成長された強誘電体膜とを有する電界効果トランジスタを用いたことを特徴とする強誘電体不揮発性メモリである。
【0036】
この発明の第14の発明は、
(001)面方位のシリコン基板と、
上記シリコン基板の第1の領域の表面にエピタキシャル成長された立方晶系または正方晶系の(001)面方位の希土類酸化物と、
上記希土類酸化物上にエピタキシャル成長された強誘電体膜を用いたキャパシタと、
上記シリコン基板の第2の領域に形成されたMIS−FETとを有し、
上記キャパシタと上記MIS−FETのゲート電極とが配線により互いに接続されている
ことを特徴とする強誘電体不揮発性メモリである。
【0037】
この発明の第15の発明は、
単結晶絶縁体基板と、
上記単結晶絶縁体基板の第1の領域の表面にエピタキシャル成長された立方晶系または正方晶系の(001)面方位の希土類酸化物と、
上記希土類酸化物上にエピタキシャル成長された強誘電体膜を用いたキャパシタと、
上記単結晶絶縁体基板の第2の領域の表面にエピタキシャル成長されたシリコン膜に形成されたMIS−FETとを有し、
上記キャパシタと上記MIS−FETのゲート電極とが配線により互いに接続されている
ことを特徴とする強誘電体不揮発性メモリである。
【0038】
この発明において、(001)面方位のシリコン基板には、実質的に(001)面方位と同等と認められる範囲内で(001)面からオフしたシリコン基板も含まれる。
【0039】
この発明において、希土類酸化物は、具体的にはセリウム(Ce)、イットリウム(Y)等の希土類元素の酸化物であり、これには1種の希土類元素の酸化物のほかに、2種以上の希土類元素の酸化物も含まれる。この希土類酸化物をReOz (ただし、Reは希土類元素)と表すと、通常、0<z≦3である。また、この希土類酸化物は立方晶系または正方晶系であるが、立方晶系または正方晶系と認められる範囲内で多少歪んだものも含まれるものとする。この希土類酸化物は、典型的には、蛍石構造(CeO2 構造;フルオライト構造)またはC−希土構造(Y2 O3 構造;ビクスバイト(bixbyite)構造)をとる。
【0040】
この発明においては、希土類酸化物をより確実に(001)面方位にエピタキシャル成長させる観点から、好適には、希土類酸化物をエピタキシャル成長させる際に、シリコン基板の表面に酸化性ガスの供給を開始してから少なくとも一種以上の希土類元素を含む原料の供給を行う。この少なくとも一種以上の希土類元素を含む原料は少なくとも一種以上の希土類元素からなるものであっても、例えば希土類酸化物からなるものであってもよい。ここで、希土類元素は金属元素である。
【0041】
この発明においては、希土類酸化物をエピタキシャル成長させた後、1×10-6Torr以下の圧力の真空中において希土類酸化物の成長温度以上の温度で熱処理を行う工程をさらに有する場合もある。この真空中熱処理により、希土類酸化物から一部の酸素が引き抜かれる。ここで、特に、希土類酸化物がCeOz である場合、このCeOz をエピタキシャル成長させた後、1×10-6Torr以下の圧力の真空中においてCeOz の成長温度以上の温度で熱処理を行い、CeOz から一部のOを引き抜くことにより、酸素欠陥型蛍石構造またはC−希土構造(ビクスバイト構造)をとるCeOz-d を生成することができる。ここで、CeOz-d におけるz−dは通常、1.5≦z−d<2であり、典型的には例えば1.5≦z−d≦1.8である。希土類酸化物がPmOz である場合も同様にしてビクスバイト構造をとるCeOz-d を生成することができる。また、希土類酸化物をエピタキシャル成長させた後、上記希土類酸化物の成長温度より高い成長温度で上記希土類酸化物上に希土類酸化物をホモエピタキシャル成長させる工程をさらに有する場合もある。さらに、希土類酸化物をエピタキシャル成長させた後、希土類酸化物上に機能性酸化物をエピタキシャル成長させる工程をさらに有する場合もある。
【0042】
この発明においては、成長条件等によっては、希土類酸化物の成長後に、シリコン基板と希土類酸化物との界面に厚さ5nm以下の酸化シリコン膜または欠陥層が形成されることもある。
【0043】
この発明において、機能性酸化物は、典型的には、ペロブスカイト構造または層状ペロブスカイト構造である。このような機能性酸化物は、基本的にはどのようなものであってもよいが、具体的には、強誘電体、超伝導体、焦電体、圧電体などである。
【0044】
上述のように構成されたこの発明によれば、(001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とし、さらに、好適には、希土類酸化物をエピタキシャル成長させる際に、シリコン基板の表面に酸化性ガスの供給を開始してから少なくとも一種以上の希土類元素を含む原料の供給を行うことにより、(001)面方位のシリコン基板上に希土類酸化物を(001)面方位に良好にエピタキシャル成長させることができる。
【0045】
【発明の実施の形態】
以下、この発明の実施形態について図面を参照しながら説明する。なお、実施形態の全図において、同一または対応する部分には同一の符号を付す。
【0046】
まず、以下の実施形態においてCeO2 の成長に用いるMBE装置について説明する。このMBE装置は、通常の半導体成長用に設計されたMBE装置でも不可能ではないが、酸化性ガス、好ましくは酸素ガスを成長室に導入することができるようにノズル、配管等が設置されており、また、耐酸化性の部材で成長室、ヒータ、セル等が構成されている必要がある。また、基板表面の状態をモニタリングすることができるよう、基板表面に対して数度以内の角度で高速電子線を入射させることができ、また、その電子線回折像を観測することができるよう、蛍光体を塗布したスクリーンが配備されている(RHEEDシステム)。基板のサセプターはシリコンカーバイド(SiC)からなり、裏面よりSiCヒータで加熱されるようになっている。ソース原料の供給のため、分子線発生用の3基のクヌーセンセル、電子ビーム蒸着用の2基の電子ビーム(EB)ガンが設置されている。酸化性ガスは、マスフローコントローラにて流量制御された後、基板付近までノズルで供給され、基板に吹き付けられるようになっている。
【0047】
まず、この発明の第1の実施形態による酸化物の結晶成長方法について説明する。
【0048】
この第1の実施形態においては、まず、図5Aに示すように、(100)面方位のSi基板1を用意する。
【0049】
次に、この(100)Si基板1の表面処理を行う。この表面処理は、石坂、白木等によって提案された先に述べた手法またはこれに準ずる手法により、表面に揮発性の高いSiO2 膜を形成し、熱処理により表面を出すことにより行う。具体的には、Si基板1をRCA洗浄し、これに対し、120〜130℃に煮沸した濃硝酸中での10分間の処理(基板表面のエッチングとSiO2 膜の形成)と2.5%希フッ酸による10〜15秒程度の処理(SiO2 膜の除去)とを3〜4回繰り返し行うことにより平坦面を形成する。次に、それを90℃に加熱したアンモニア過水(NH4 OH:H2 O2 :H2 O=1:1:3)中に入れてSi基板1の表面に薄いSiO2 膜を形成し(10分間)、2.5%希フッ酸で10〜15秒程度処理(SiO2 膜の除去)する。それを、再び90℃に加熱したアンモニア過水(NH4 OH:H2 O2 :H2 O=1:1.25:3)中に入れて薄いSiO2 膜を形成し(10分間)、さらに抵抗率18MΩcm以上の超純水にてリンス後、窒素ガスで基板表面の水滴を除去してMBE装置の成長室に投入する。そして、この成長室において、真空中、600℃で1時間脱ガスした後、1000℃まで昇温して数秒保持し、Si基板1の表面からSiOを揮発させる(Si+SiO2 →2SiO↑)。この過程で、850℃前後まで昇温した時点で2×1、1×2再構成表面が得られる(図5B)。このようにして2×1、1×2表面再構成によるダイマー構造が形成されたSi基板1を成長温度まで冷却してCeO2 成長用基板とする。なお、本発明者の実験では、ヒータおよびサセプターにSiCを用いていることもあり、通常コンタミネーションであるとされるSiCと思われる島状の構造を完全に除去するには至らなかった。
【0050】
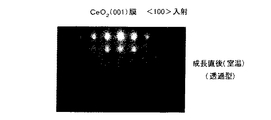
次に、図5Cに示すように、2×1、1×2の表面再構成によるダイマー構造が表面に形成されたSi基板1上に、100℃程度以下の低温の基板温度において、セリウム金属および酸素を原料に用い、それらを適切なレート(図1参照)で供給して、(001)面方位のCeO2 膜2をエピタキシャル成長させる。具体的には、例えば、基板温度を27℃とし、EBガン電流150mA、酸素流量0.05sccmにて、セリウム金属および酸素を成長室内に供給する。このとき重要なことは、まずSi基板1の表面への酸素の供給を開始してから、セリウム金属の供給を行うことである。このようにして成長を行うと、CeO2 (001)が、エピタキシャルに成長していることがRHEEDスクリーン上で確認される。図6および図7に、(001)CeO2 膜の〈100〉入射による電子線回折位置のシミュレーションパターンおよび実際に得られたRHEEDパターンをそれぞれ示す。また、図8および図9に、(001)CeO2 膜の〈110〉入射による電子線回折位置のシミュレーションパターンおよび実際に得られたRHEEDパターンを示す。上記の条件でCeO2 膜2を5nm程度以上の膜厚に厚く成長させると、膜厚の増加に連れて配向性が悪化する傾向を示した。このようにして得られるCeO2 (001)/Si(001)構造の透過型電子顕微鏡による断面格子像(〈110〉入射)の一例を図10に示す。図10より、Si基板1とCeO2 膜2との界面に2nm程度の膜厚の欠陥層(SiOx 膜と考えられる)が存在しているものの、CeO2 膜2の格子縞がSi基板1のそれに揃っていることから、CeO2 膜2はエピタキシャルに成長していることがわかる。
【0051】
以上のように、この第1の実施形態によれば、(001)Si基板1の表面を2×1、1×2表面再構成によるダイマー構造とした後、成長室にセリウム金属および酸素を適切なレートで供給し、この際酸素の供給を先行させ、100℃程度以下の低温の基板温度で成長を行っていることにより、CeO2 膜2を(001)面方位に良好にエピタキシャル成長させることができる。
【0052】
次に、この発明の第2の実施形態による酸化物の結晶成長方法について説明する。
【0053】
この第2の実施形態においては、まず、図11Aに示すように、第1の実施形態と同様にして、2×1、1×2の表面再構成によるダイマー構造が表面に形成された(001)Si基板1上に(001)CeO2 膜2をエピタキシャル成長させる。CeO2 膜2の膜厚は例えば5nmとする。
【0054】
次に、このCeO2 膜2をMBE装置の成長室において、真空中(例えば、10-9Torr台)で熱処理する。この真空中熱処理によるRHEEDパターンの変化を図12〜図17に示す。ここで、図12〜図14は〈100〉入射の場合で、図12は成長直後の室温におけるRHEEDパターン、図13は熱処理温度が300℃のときのRHEEDパターン、図14は熱処理温度が600℃のときのRHEEDパターンである。また、図15〜図17は〈110〉入射の場合で、図15は成長直後の室温におけるRHEEDパターン、図16は熱処理温度が300℃のときのRHEEDパターン、図17は熱処理温度が600℃のときのRHEEDパターンである。図12〜図17より、300℃程度の真空中熱処理により一旦平坦化し、500℃程度以上の温度の真空中熱処理により超格子スポットが現れているのがわかる。900℃まで昇温した後に室温まで冷却することにより、図11Bに示すように、ビクスバイト相の(001)CeO2-x 膜3が得られる。この試料の〈100〉入射によるRHEEDパターンを図19に、ビクスバイト相のCeO2-x (001)膜の〈100〉入射による電子線回折位置のシミュレーションパターンを図18に示す。また、同じ試料の〈110〉入射によるRHEEDパターンを図21に、ビクスバイト相のCeO2-x (001)膜の〈110〉入射による電子線回折位置のシミュレーションパターンを図20に示す。消滅則に対する考察から、空間群はIa3(206)と決定される(図18および図20)。これはY2 O3 と同形の酸素欠陥相CeO2-X であると考えられる((51)JCPDS:23−1048,44−1086)。このビクスバイト相は、酸素量に関しては不定比性があると考えられ、高真空中でのみ安定な新規物質であり、10-7Torr以上の酸素圧下では超格子パターンが消失する傾向を示した。
【0055】
以上のように、この第2の実施形態によれば、第1の実施形態と同様にして、2×1、1×2の表面再構成によるダイマー構造が表面に形成された(001)Si基板1上に(001)CeO2 膜2をエピタキシャル成長させた後、真空中熱処理を行うことにより、ビクスバイト相の(001)CeO2-x 膜3を得ることができる。
【0056】
次に、この発明の第3の実施形態による酸化物の結晶成長方法について説明する。
【0057】
この第3の実施形態においては、まず、図22Aに示すように、第2の実施形態と同様にして、(001)Si基板1上にビクスバイト相の(001)CeO2-x 膜3を形成する。ここで、このCeO2-x 膜3はCe2 O3 という定比でなくてもよい。
【0058】
次に、図22Bに示すように、CeO2-x 膜3上にCeO2 膜4を(001)面方位にホモエピタキシャル成長させる。このCeO2 膜4の成長は、具体的には、例えば、基板温度700℃にて、EBガン電流150mA、酸素流量0.25sccmの条件で行う。また、このCeO2 膜4の膜厚は例えば45nmとする。このようにしてホモエピタキシャル成長したCeO2 膜4のRHEEDパターンを図23および図24に示す。ここで、図23は〈100〉入射の場合、図24は〈110〉入射の場合である。第1の実施形態における条件ではCeO2 膜を5nm程度以上の膜厚に厚く成長させることは難しかったが、この第3の実施形態のようにホモエピタキシャル成長させることにより、700℃という高い基板温度でCeO2 膜4を厚く成長させることができる。このとき、ホモエピタキシャル成長の下地層は、10-7Torr以上の酸素圧下で超格子パターンの認められない相でもよいが、表面を極力平坦化し、CeO2 /Si界面への酸素拡散を緩和するためには、第2の実施形態において説明したようなビクスバイト相のCeO2-x 膜3を生成する工程を経るのが好ましいと考えられる。
【0059】
第1の実施形態によるエピタキシャルCeO2 膜2は、膜厚が薄いため、X線による解析が困難であったが、このホモエピタキシャルCeO2 膜4では、膜厚が十分に厚いため、X線による解析が可能になった。図25に、室温で成長されたCeO2-x 膜上に基板温度700℃で(001)CeO2 膜をホモエピタキシャル成長させたCeO2 (001)/Si(001)のθ/2θスキャンによるX線回折図形の測定結果を示す。ただし、この測定に使用されたディフラクトメータは、θ/2θ以外に、あおりαおよび面内回転βの自由度を有しており、軸立て、φスキャン等の測定が可能である。また、X線源はCuKα線である。
【0060】
図25に示すX線回折図形においては、良好な格子整合性を反映して、CeO2 (002)に極めて近接するSi基板1からの回折ピークが重畳する。ここでは、この回折ピークを便宜上Si(002)と表記する。これはダイヤモンド構造をとるSi結晶(空間群:Fd3m)では禁制反射である。この回折ピークの解釈については諸説あるが、特異な面内回転角依存性から、少なくとも主成分は例えば(111)、(−1−11)等の組み合わせによる二重回折であると考えられている((52)まてりあ第37巻第5号、p.421(1998))。このため、通常行われるように、Si(004)ピークで軸立てを行っても、Si(002)の強度には再現性がないのが普通である。一方、面内回転角依存性で、バックグラウンド成分も認められることから、結晶不完全性や、結晶として不完全である表面、界面等に起因して対称性が低下し、回折ピークとして観測される成分も存在している可能性がある。ここでは、このSi(002)ピークが出現する面内回転角に試料をセットした後、Si(004)の対称反射が最大強度をとるように軸立てを行い、結晶評価を行った。
【0061】
図25に示すX線回折図形のデータは、酸化性ガス導入ノズルが基板直近にあることから、CeO2 膜の成長時の酸素供給濃度に若干の分布があるため、試料のノズル先端付近の10mm角の領域を評価したものである。この領域では、CeO2 (111)、(220)等の回折はほとんど認められず、単相のCeO2 (001)が形成されているのがわかる。図25のSi(002)、CeO2 (002)ピーク付近を拡大したのが図26である。図26においては、ピークの重畳は避け難いが、CeO2 (002)ピークが明瞭に認められる。格子定数はバルク値(〜5.411Å)よりもやや小さく、〜5.392Å程度である。
【0062】
図25および図26の試料のCeO2 {204}および基板Si{202}ピークのφスキャンデータを図27に示す。図27より、CeO2 {204}、基板Si{202}の面内回転位相が揃っており、マクロな領域でCeO2 およびSiの結晶軸がキューブオンキューブ(cube on cube) に重なっていることを確認することができ、この結果からも、CeO2 膜4のエピタキシャル成長が確認される。
【0063】
図28に、(001)Si基板1上に室温でCeO2-x 膜3を成長させた後、このCeO2-x 膜3上にCeO2 膜4を(001)面方位に700℃で厚くホモエピタキシャル成長させた試料の透過型電子顕微鏡による断面格子像の一例を示す。ただし、図28中ではCeO2-x 膜3をCeOx と表記している。また、このCeOx はアモルファスであるので、a−CeOx と表記している。図28より、Si基板1とCeO2 膜4との界面に厚さ2.5nmのa−SiOx が存在しているものの、CeO2 膜4の格子縞がSi基板1のそれに揃っていることから、CeO2 膜4はエピタキシャルに成長していることがわかる。
【0064】
以上のように、この第3の実施形態によれば、第2の実施形態と同様にして、(001)Si基板1上にビクスバイト相の(001)CeO2-x 膜3を形成した後、このCeO2-x 膜3上にCeO2 膜4を(001)面方位にホモエピタキシャル成長させていることにより、十分な厚さのエピタキシャル(001)CeO2 膜4を得ることができる。
【0065】
次に、この発明の第4の実施形態による強誘電体不揮発性メモリの製造方法について説明する。この強誘電体不揮発性メモリは、CeO2 (001)/Si(001)エピタキシャル構造を用いた、MIS−FETおよび強誘電体キャパシタゲート(FCG)による強誘電体不揮発性メモリである。この強誘電体不揮発性メモリの完成状態を図29に示す。
【0066】
この第4の実施形態においては、まず、第1の実施形態と同様にして、(001)Si基板11のキャパシタ形成領域にエピタキシャル(001)CeO2 膜12を選択的に形成する。このCeO2 膜12を形成するには、Si基板11の表面を成長マスク(図示せず)で選択的に覆ってCeO2 膜12を選択的にエピタキシャル成長させてもよいし、Si基板11の全面にCeO2 膜12をエピタキシャル成長させた後、このCeO2 膜12をイオンミリング等でパターニングしてもよい。
【0067】
(001)CeO2 /(001)Si構造の形成以外のプロセスとしては、基本的に従来の半導体プロセスを用いることができる。すなわち、例えば、まず、LOCOS(Local Oxidation of Silicon) 法によりSi基板11の表面を選択的に熱酸化してSiO2 膜からなるフィールド絶縁膜13を形成する。次に、このフィールド絶縁膜13に囲まれた活性領域の表面に熱酸化法によりSiO2 膜からなるゲート絶縁膜14を形成する。次に、CVD(Chemical Vapor Deposition)法などにより全面に多結晶Si膜を形成し、この多結晶Si膜に不純物をドープして低抵抗化した後、この多結晶Si膜をエッチングしてパターニングすることによりゲート電極15を形成する。次に、このゲート電極15をマスクとしてSi基板1中に不純物をイオン注入し、必要に応じてさらに不純物の電気的活性化のための熱処理を行うことにより、ソース領域16およびドレイン領域17をゲート電極15に対して自己整合的に形成する。この不純物としては、nチャネルMIS−FETを形成する場合にはn型不純物、例えばヒ素(As)を用い、pチャネルMIS−FETを形成する場合にはp型不純物、例えばホウ素(B)を用いる。
【0068】
次に、(001)CeO2 膜12上に、例えば(001)SrRuO3 のような導電性酸化物からなる下部電極18、例えば(001)PZTからなる強誘電体膜19および例えば(001)SrRuO3 のような導電性酸化物からなる上部電極20を順次形成し、強誘電体キャパシタを形成する。ここで、下部電極18は、後に配線を接続することができるように、強誘電体膜19および上部電極20よりも大きめに形成する。これらの導電性酸化物や強誘電体膜は、スパッタリング法、CVD法等によりエピタキシャル成長させることができる。この際には、成長マスクを用いて選択的に成長を行ってもよいし、全面に成長を行った後にSiO2 膜などからなるマスクを用いてイオンミリング等で加工するようにしてもよい。
【0069】
次に、例えばCVD法により全面にSiO2 膜のような層間絶縁膜21を成膜した後、この層間絶縁膜21のうちゲート電極15上の所定部分、上部電極20上の所定部分および下部電極18上の所定部分をエッチング除去することによりコンタクトホール22、23、24を形成する。次に、例えば真空蒸着法、スパッタリング法等により全面に例えばアルミニウム(Al)膜を形成した後、このAl膜をエッチングによりパターニングして、ゲート電極15と上部電極20とを接続する局所配線25および下部電極18に接続された局所配線26を形成する。
【0070】
以上の工程により、図29に示すように、MIS−FETのゲート電極15に強誘電体キャパシタが接続されたMFMIS構造の強誘電体不揮発性メモリが製造される。
【0071】
この第4の実施形態による強誘電体不揮発性メモリによれば、上述したMFS(MFMIS)構造の強誘電体不揮発性メモリとして多くの利点を有しているのみならず、信頼性の高いSiO2 膜をゲート絶縁膜として用いることが可能であること、強誘電体キャパシタをMIS−FETと分離して形成しているため、ゲート/キャパシタ面積比の設計自由度が大きく、強誘電体キャパシタを相対的に小さく構成することで、低電圧でも十分な強誘電体の分極が得られること、そして何よりも強誘電体膜19をエピタキシャル単結晶薄膜として形成することができるため、良好な角型性が期待され、MFS構造の強誘電体不揮発性メモリの最大の課題であるリテンション、ディスターブ等に対して極めて有利であることが最大の特徴である。また、CeO2 (001)/Si(001)界面は、熱酸化プロセス等でSiOx 膜が形成される等、劣化の可能性があるが、本構造では、(001)CeO2 上に例えばSrRuO3 等の導電性酸化物がエピタキシャル成長しさえすれば、界面劣化が起きてもデバイス動作上問題はなく、プロセス的にも従来のMFIS構造の強誘電体不揮発性メモリに比べて有利である。
【0076】
以上、この発明の実施形態について具体的に説明したが、この発明は、上述の実施形態に限定されるものではなく、この発明の技術的思想に基づく各種の変形が可能である。
【0077】
例えば、上述の第1の実施形態においては、(001)CeO2 膜2の成長時には、石坂、白木等により提案された手法でSi基板1の表面を処理しているが、必ずしもそのようにする必要はなく、他の方法で処理してもよい。
【0078】
【発明の効果】
以上説明したように、この発明によれば、酸化セリウムなどの希土類酸化物を(001)面方位のシリコン基板上に(001)面方位でエピタキシャル成長させ、エピタキシャルな希土類酸化物(001)/シリコン(001)構造を実現することができる。そして、このエピタキシャル(001)希土類酸化物を用いて電界効果トランジスタのゲート絶縁膜や、強誘電体不揮発性メモリにおける強誘電体膜の下地層を形成することにより、良好な特性の電界効果トランジスタや強誘電体不揮発性メモリを得ることができる。
【図面の簡単な説明】
【図1】(001)Si基板上にCeO2 を成長させる場合における得られる面方位と成長条件との関係を説明するための略線図である。
【図2】(001)Si基板に形成されたエピタキシャル構造の一例を示す略線図である。
【図3】(001)Si基板に形成されたエピタキシャル構造の一例を示す略線図である。
【図4】(001)Si基板に形成されたエピタキシャル構造の一例を示す略線図である。
【図5】この発明の第1の実施形態による酸化物の結晶成長方法を説明するための断面図である。
【図6】この発明の第1の実施形態において成長された(001)CeO2 膜の〈100〉入射による電子線回折位置のシミュレーションパターンを示す略線図である。
【図7】この発明の第1の実施形態において成長された(001)CeO2 膜の〈100〉入射により実際に得られたRHEEDパターンを示す図である。
【図8】この発明の第1の実施形態において成長された(001)CeO2 膜の〈110〉入射による電子線回折位置のシミュレーションパターンを示す略線図である。
【図9】この発明の第1の実施形態において成長された(001)CeO2 膜の〈110〉入射により得られたRHEEDパターンを示す図である。
【図10】この発明の第1の実施形態により得られた試料の透過型電子顕微鏡による断面格子像を示す。
【図11】この発明の第2の実施形態による酸化物の結晶成長方法を説明するための断面図である。
【図12】この発明の第2の実施形態における(001)CeO2 膜の成長直後のRHEEDパターンを示す略線図である。
【図13】この発明の第2の実施形態において(001)CeO2 膜の成長後に300℃で真空中熱処理を行った後の〈100〉入射により得られたRHEEDパターンを示す略線図である。
【図14】この発明の第2の実施形態において(001)CeO2 膜の成長後に600℃で真空中熱処理を行った後の〈100〉入射により得られたRHEEDパターンを示す略線図である。
【図15】この発明の第2の実施形態における(001)CeO2 膜の成長直後の〈110〉入射により得られたRHEEDパターンを示す略線図である。
【図16】この発明の第2の実施形態において(001)CeO2 膜の成長後に300℃で真空中熱処理を行った後の〈110〉入射により得られたRHEEDパターンを示す略線図である。
【図17】この発明の第2の実施形態において(001)CeO2 膜の成長後に600℃で真空中熱処理を行った後の〈110〉入射により得られたRHEEDパターンを示す略線図である。
【図18】この発明の第2の実施形態において真空中熱処理を行った後に得られる(001)CeO2-x 膜の〈100〉入射による電子線回折位置のシミュレーションパターンを示す略線図である。
【図19】この発明の第2の実施形態において真空中熱処理を行った後に得られる(001)CeO2-x 膜の〈100〉入射により得られたRHEEDパターンを示す図である。
【図20】この発明の第2の実施形態において真空中熱処理を行った後に得られる(001)CeO2-x 膜の〈110〉入射による電子線回折位置のシミュレーションパターンを示す略線図である。
【図21】この発明の第2の実施形態において真空中熱処理を行った後に得られる(001)CeO2-x 膜の〈110〉入射により得られたRHEEDパターンを示す図である。
【図22】この発明の第3の実施形態による酸化物の結晶成長方法を説明するための断面図である。
【図23】この発明の第3の実施形態においてホモエピタキシャル成長された(001)CeO2 膜の〈100〉入射により得られたRHEEDパターンを示す図である。
【図24】この発明の第3の実施形態においてホモエピタキシャル成長された(001)CeO2 膜の〈110〉入射により得られたRHEEDパターンを示す図である。
【図25】この発明の第3の実施形態においてホモエピタキシャル成長された(001)CeO2 膜のX線回折図形を示す略線図である。
【図26】図25に示すX線回折図形の一部を拡大して示す略線図である。
【図27】この発明の第3の実施形態において(001)CeO2 膜がホモエピタキシャル成長された試料のCeO2 {204}および基板Si{202}ピークのφスキャンデータを示す略線図である。
【図28】この発明の第3の実施形態により得られた試料の透過型電子顕微鏡による断面格子像を示す。
【図29】この発明の第4の実施形態による強誘電体不揮発性メモリの製造方法を説明するための断面図である。
【符号の説明】
1、11・・・Si基板、2、4、12・・・CeO2 膜、3・・・CeO2-x 膜、14、54・・・ゲート絶縁膜、15、55・・・ゲート電極、18、58・・・下部電極、19、59・・・強誘電体膜、20、60・・・上部電極
Claims (14)
- (001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
上記シリコン基板上に酸化セリウムを(001)面方位にエピタキシャル成長させる工程とを有し、
上記酸化セリウムをエピタキシャル成長させる際に、上記シリコン基板の表面に酸化性ガスの供給を開始してからセリウムを含む原料の供給を行うようにした酸化物の結晶成長方法。 - 上記セリウムを含む原料はセリウムからなる請求項1記載の酸化物の結晶成長方法。
- 上記セリウムを含む原料は酸化セリウムからなる請求項1記載の酸化物の結晶成長方法。
- (001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
上記シリコン基板上に酸化セリウムを(001)面方位にエピタキシャル成長させる工程と、
上記酸化セリウムをエピタキシャル成長させた後、1×10-6Torr以下の圧力の真空中において上記酸化セリウムの成長温度以上の温度で熱処理を行う工程とを有し、
上記酸化セリウムをエピタキシャル成長させる際に、上記シリコン基板の表面に酸化性ガスの供給を開始してからセリウムを含む原料の供給を行うようにした酸化物の結晶成長方法。 - (001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
上記シリコン基板上に酸化セリウムを(001)面方位にエピタキシャル成長させる工程と、
上記酸化セリウムをエピタキシャル成長させた後、上記酸化セリウムの成長温度より高い成長温度で上記酸化セリウム上に酸化セリウムをホモエピタキシャル成長させる工程とを有し、
上記酸化セリウムをエピタキシャル成長させる際に、上記シリコン基板の表面に酸化性ガスの供給を開始してからセリウムを含む原料の供給を行うようにした酸化物の結晶成長方法。 - 上記酸化セリウムをエピタキシャル成長させた後、上記酸化セリウム上に酸化物からなる強誘電体、超伝導体、焦電体または圧電体をエピタキシャル成長させる工程をさらに有する請求項1〜4のいずれか一項記載の酸化物の結晶成長方法。
- 上記シリコン基板と上記酸化セリウムとの界面に厚さ5nm以下の酸化シリコン膜または欠陥層が形成された請求項1〜6のいずれか一項記載の酸化物の結晶成長方法。
- 上記酸化物からなる強誘電体、超伝導体、焦電体または圧電体はペロブスカイト構造または層状ペロブスカイト構造である請求項6記載の酸化物の結晶成長方法。
- (001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
上記シリコン基板上に酸化セリウムを(001)面方位にエピタキシャル成長させることによりゲート絶縁膜を形成する工程とを有し、
上記酸化セリウムをエピタキシャル成長させる際に、上記シリコン基板の表面に酸化性ガスの供給を開始してからセリウムを含む原料の供給を行うようにした電界効果トランジスタの製造方法。 - (001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
上記シリコン基板上に酸化セリウムを(001)面方位にエピタキシャル成長させることによりゲート絶縁膜を形成する工程と、
上記酸化セリウムをエピタキシャル成長させた後、1×10 -6 Torr以下の圧力の真空中において上記酸化セリウムの成長温度以上の温度で熱処理を行う工程とを有し、
上記酸化セリウムをエピタキシャル成長させる際に、上記シリコン基板の表面に酸化性ガスの供給を開始してからセリウムを含む原料の供給を行うようにした電界効果トランジスタの製造方法。 - (001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
上記シリコン基板上に酸化セリウムを(001)面方位にエピタキシャル成長させることによりゲート絶縁膜を形成する工程と、
上記酸化セリウムをエピタキシャル成長させた後、上記酸化セリウムの成長温度より高い成長温度で上記酸化セリウム上に酸化セリウムをホモエピタキシャル成長させる工程とを有し、
上記酸化セリウムをエピタキシャル成長させる際に、上記シリコン基板の表面に酸化性ガスの供給を開始してからセリウムを含む原料の供給を行うようにした電界効果トランジスタの製造方法。 - (001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
上記シリコン基板上に酸化セリウムを(001)面方位にエピタキシャル成長させる工程と、
上記酸化セリウム上に強誘電体膜をエピタキシャル成長させる工程とを有し、
上記酸化セリウムをエピタキシャル成長させる際に、上記シリコン基板の表面に酸化性ガスの供給を開始してからセリウムを含む原料の供給を行うようにした強誘電体不揮発性メモリの製造方法。 - (001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
上記シリコン基板上に酸化セリウムを(001)面方位にエピタキシャル成長させる工程と、
上記酸化セリウムをエピタキシャル成長させた後、1×10 -6 Torr以下の圧力の真空中において上記酸化セリウムの成長温度以上の温度で熱処理を行う工程と、
上記酸化セリウム上に強誘電体膜をエピタキシャル成長させる工程とを有し、
上記酸化セリウムをエピタキシャル成長させる際に、上記シリコン基板の表面に酸化性ガスの供給を開始してからセリウムを含む原料の供給を行うようにした強誘電体不揮発性メモリの製造方法。 - (001)面方位のシリコン基板の表面を2×1、1×2の表面再構成によるダイマー構造とする工程と、
上記シリコン基板上に酸化セリウムを(001)面方位にエピタキシャル成長させる工程と、
上記酸化セリウムをエピタキシャル成長させた後、上記酸化セリウムの成長温度より高い成長温度で上記酸化セリウム上に酸化セリウムをホモエピタキシャル成長させる工程と、
上記酸化セリウム上に強誘電体膜をエピタキシャル成長させる工程とを有し、
上記酸化セリウムをエピタキシャル成長させる際に、上記シリコン基板の表面に酸化性ガスの供給を開始してからセリウムを含む原料の供給を行うようにした強誘電体不揮発性メモリの製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000027362A JP4239343B2 (ja) | 1999-03-26 | 2000-01-31 | 酸化物の結晶成長方法、電界効果トランジスタの製造方法および強誘電体不揮発性メモリの製造方法 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP11-84919 | 1999-03-26 | ||
| JP8491999 | 1999-03-26 | ||
| JP2000027362A JP4239343B2 (ja) | 1999-03-26 | 2000-01-31 | 酸化物の結晶成長方法、電界効果トランジスタの製造方法および強誘電体不揮発性メモリの製造方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2000344599A JP2000344599A (ja) | 2000-12-12 |
| JP2000344599A5 JP2000344599A5 (ja) | 2006-02-16 |
| JP4239343B2 true JP4239343B2 (ja) | 2009-03-18 |
Family
ID=26425882
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2000027362A Expired - Fee Related JP4239343B2 (ja) | 1999-03-26 | 2000-01-31 | 酸化物の結晶成長方法、電界効果トランジスタの製造方法および強誘電体不揮発性メモリの製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4239343B2 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8099635B2 (en) | 2007-03-21 | 2012-01-17 | Arm Limited | Techniques for generating a trace stream for a data processing apparatus |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6569240B1 (en) | 1999-03-17 | 2003-05-27 | Matsushita Electric Industrial Co., Ltd. | Dielectric film and method for forming the same |
| JP5029539B2 (ja) * | 2007-09-04 | 2012-09-19 | 三菱マテリアル株式会社 | 多結晶シリコンの洗浄方法及び多結晶シリコンの製造方法 |
| JP5262068B2 (ja) * | 2007-11-01 | 2013-08-14 | 富士ゼロックス株式会社 | 画像形成装置 |
| JP2009224403A (ja) * | 2008-03-13 | 2009-10-01 | Toshiba Corp | 情報記録素子及びそれを備えた情報記録再生装置 |
| JP2010212391A (ja) * | 2009-03-10 | 2010-09-24 | Hitachi Kokusai Electric Inc | 半導体装置の製造方法及び基板処理装置 |
-
2000
- 2000-01-31 JP JP2000027362A patent/JP4239343B2/ja not_active Expired - Fee Related
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8099635B2 (en) | 2007-03-21 | 2012-01-17 | Arm Limited | Techniques for generating a trace stream for a data processing apparatus |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2000344599A (ja) | 2000-12-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6610548B1 (en) | Crystal growth method of oxide, cerium oxide, promethium oxide, multi-layered structure of oxides, manufacturing method of field effect transistor, manufacturing method of ferroelectric non-volatile memory and ferroelectric non-volatile memory | |
| US5753934A (en) | Multilayer thin film, substrate for electronic device, electronic device, and preparation of multilayer oxide thin film | |
| US5828080A (en) | Oxide thin film, electronic device substrate and electronic device | |
| US6387712B1 (en) | Process for preparing ferroelectric thin films | |
| JP3472087B2 (ja) | 膜構造体、電子デバイス、記録媒体および酸化物導電性薄膜の製造方法 | |
| JP3310881B2 (ja) | 積層薄膜、電子デバイス用基板、電子デバイスおよび積層薄膜の製造方法 | |
| US7368172B2 (en) | Membrane multi-layer structure, and actuator element, capacitive element and filter element using the same | |
| JP3363301B2 (ja) | 強誘電体薄膜被覆基板及びその製造方法及び強誘電体薄膜被覆基板によって構成された不揮発性メモリ | |
| US6307225B1 (en) | Insulating material, substrate covered with an insulating film, method of producing the same, and thin-film device | |
| JP4401300B2 (ja) | (001)配向したペロブスカイト膜の形成方法、およびかかるペロブスカイト膜を有する装置 | |
| JP4239343B2 (ja) | 酸化物の結晶成長方法、電界効果トランジスタの製造方法および強誘電体不揮発性メモリの製造方法 | |
| JP3095944B2 (ja) | 酸化物結晶薄膜の製造方法及び薄膜素子 | |
| US6749686B2 (en) | Crystal growth method of an oxide and multi-layered structure of oxides | |
| JP3994468B2 (ja) | 酸化物積層構造およびその製造方法ならびに強誘電体不揮発性メモリ | |
| JP4142128B2 (ja) | 積層薄膜およびその製造方法 | |
| JPH08109099A (ja) | 酸化物薄膜、電子デバイス用基板および酸化物薄膜の形成方法 | |
| JP3470068B2 (ja) | 誘電体膜の形成方法 | |
| JPH10182292A (ja) | 酸化物積層構造およびその製造方法 | |
| JPH11103024A (ja) | 強誘電体素子および半導体装置 | |
| JP3229911B2 (ja) | 強誘電体記憶素子 | |
| JP2004006960A (ja) | 誘電体膜の形成方法 | |
| JPH08181289A (ja) | 強誘電体薄膜と基体との複合構造体およびその製造方法 | |
| JP3251462B2 (ja) | Mis半導体デバイスおよびその製造方法 | |
| KR20040079884A (ko) | 갈륨나이트라이드를 기판으로한 페로브스카이트 구조의강유전체 박막트랜지스터 및 그 제조방법 | |
| JPH1197630A (ja) | 強誘電体薄膜の製造方法及び強誘電体メモリ素子 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20041222 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20050111 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20051227 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20051227 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20080516 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080527 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080630 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080902 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20081015 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20081202 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20081215 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120109 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120109 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |