JP3628708B2 - ポリエチレンナフタレートの製造方法 - Google Patents
ポリエチレンナフタレートの製造方法 Download PDFInfo
- Publication number
- JP3628708B2 JP3628708B2 JP51804897A JP51804897A JP3628708B2 JP 3628708 B2 JP3628708 B2 JP 3628708B2 JP 51804897 A JP51804897 A JP 51804897A JP 51804897 A JP51804897 A JP 51804897A JP 3628708 B2 JP3628708 B2 JP 3628708B2
- Authority
- JP
- Japan
- Prior art keywords
- esterification
- water
- nda
- reaction
- esterification reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/88—Post-polymerisation treatment
- C08G63/89—Recovery of the polymer
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/12—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from polycarboxylic acids and polyhydroxy compounds
- C08G63/16—Dicarboxylic acids and dihydroxy compounds
- C08G63/18—Dicarboxylic acids and dihydroxy compounds the acids or hydroxy compounds containing carbocyclic rings
- C08G63/181—Acids containing aromatic rings
- C08G63/185—Acids containing aromatic rings containing two or more aromatic rings
- C08G63/187—Acids containing aromatic rings containing two or more aromatic rings containing condensed aromatic rings
- C08G63/189—Acids containing aromatic rings containing two or more aromatic rings containing condensed aromatic rings containing a naphthalene ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/78—Preparation processes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/78—Preparation processes
- C08G63/82—Preparation processes characterised by the catalyst used
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polyesters Or Polycarbonates (AREA)
Description
本発明は、従来よりも短時間でポリエチレンナフタレートを製造することができるようなポリエチレンナフタレートの製造方法に関するものである。
背景技術
ポリエチレンナフタレートは、一般にナフタレンジカルボン酸またはそのエステル誘導体(たとえば低級アルキルエステル、フェニルエステルなど)と、エチレングリコールまたはそのエステル誘導体(たとえばモノカルボン酸エステルエチレンオキサイドなど)とをエステル化反応させ、ナフタレンジカルボン酸とエチレングリコールとのエステル化物を調製し、次いで重縮合触媒の存在下にエステル化物を重縮合させることにより製造されている。そして前記エステル化反応は、エチレングリコールが環流する条件下で、反応によって生成した水あるいはアルコールを系外に除去しながら実施される。
しかしながら、上記エステル化反応は完結するのに長時間を要するためポリエチレンナフタレートの製造に時間がかかり、また長時間の反応中にポリエチレンナフタレート成形体の外観等を低下させるような反応生成物が生成することがある。このためエステル化反応を従来より短時間で行うことができれば、ポリエチレンナフタレートの製造時間を短縮することができ、しかも製品の品質低下を招くような反応生成物の生成量を低下させることができるためその工業的価値は大きい。
また、ナフタレンジカルボン酸とエチレングリコールとをエステル化反応させると、カルボキシル−ヒドロキシエトキシカルボニルナフタレンおよびビス(ヒドロキシエトキシカルボニル)ナフタレンを含むエステル化反応生成物が得られるが、この反応生成物には、前記成分以外にジエチレングリコール骨格を有するナフタレンジカルボン酸のエステル(以下「NDA−DEG」ということがある。)が含まれている。
しかしながら、NDA−DEGは、得られるポリエチレンナフタレートの外観等の品質を低下させることがある。このためNDA−DEGの含有割合の少ないナフタレンジカルボン酸のエステル化反応生成物を得ることができれば、ポリエチレンナフタレートの品質を向上させることができるためその工業的価値は大きい。
本発明は、上記のような従来技術に鑑みてなされたものであって、従来より短時間でポリエチレンナフタレートを製造することができ、かつポリエチレンナフタレート成形体の外観等を低下させるような反応生成物の生成が少ないポリエチレンナフタレートの製造方法を提供することを目的としている。
発明の開示
本発明に係る第1のポリエチレンナフタレートの製造方法では、水の存在下でナフタレンジカルボン酸とエチレングリコールとをエステル化反応させた後、水を除去しながらさらにエステル化反応を行いナフタレンジカルボン酸とエチレングリコールとのエステル化物を調製し、次いでこのエステル化物の重縮合反応を行っている。
本発明では前記水の存在下で行うエステル化反応を、硝酸塩、カルボン酸塩、リン酸塩、リン水素酸塩およびアミンから選ばれる少なくとも1種の触媒の共存下で行うことができる。前記触媒としては、硝酸コバルト、酢酸マンガン、リン酸二水素ナトリウムおよびトリエチルアミンから選ばれる少なくとも1種の触媒が挙げられる。
前記水の存在下で行うエステル化反応は、エチレングリコールに対して0.03〜1.5倍重量、好ましくは0.1〜0.7倍重量の水の存在下で行うことが望ましい。
また、水の存在下でナフタレンジカルボン酸とエチレングリコールとをエステル化反応させた後のエステル化率が40〜95%であることが望ましい。
本発明によると従来より短時間でナフタレンジカルボン酸のエステル化反応を終了させることができる。このためポリエチレンナフタレートを従来より短時間で製造することができる。また本発明によるとNDA−DEGの生成を抑制できる。
【図面の簡単な説明】
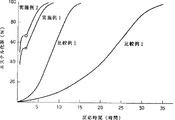
図1は、実施例1、2および比較例1、2における、反応時間とエステル化率との関係を示す図である。
図2は、実施例3、4および比較例3における、エステル化率とNDA−DEG含量の関係を示す図である。
発明を実施するための最良の形態
本発明に係る第1のポリエチレンナフタレートの製造方法は、水の存在下でナフタレンジカルボン酸とエチレングリコールとをエステル化反応させた後、水を除去しながらさらにエステル化反応を行いナフタレンジカルボン酸とエチレングリコールとのエステル化合物を調製し、次いでこのエステル化物の重縮合反応を行っている。
本発明では、まず水の存在下でナフタレンジカルボン酸とエチレングリコールとをエステル化反応〔エステル化反応(1)〕させる。このナフタレンジカルボン酸とエチレングリコールとのエステル化反応は、エチレングリコールに対して通常0.03〜1.5倍重量、好ましくは0.1〜0.7倍重量の水の存在下で行われる。そして、エチレングリコールはナフタレンジカルボン酸1モルに対して通常0.6〜30モル、好ましくは1.2〜15モルの量で用いられる。
ここで前記水としては、反応開始時から反応系に存在する水、およびナフタレンジカルボン酸とエチレングリコールとのエステル化反応により生成する水が含まれる。エステル化反応(1)では、エステル化反応の進行にともなって系内の水の量が増加するが、水を一部留去して系内の水の量を特定の範囲内に調整しながらエステル化反応を行うこともできる。なお、反応開始時には反応系に水を存在させず、エステル化反応により生成した水を系外に除去せずに、エステル化反応により生成した水の存在下に反応を行う態様もエステル化反応(1)に含まれる。
エステル化反応(1)は、反応温度が通常180〜280℃、好ましくは200〜260℃、圧力が通常0〜30kg/cm2、好ましくは0.5〜20kg/cm2の条件下で行われる。また反応時間は、反応条件にもよるが通常0.2〜6時間、好ましくは0.5〜3時間である。
このエステル化反応(1)終了時におけるエステル化率は、反応系に存在する水の割合にもよるが通常40〜95%、好ましくは45〜80%、特に好ましくは60〜80%である。
なお本明細書において、エステル化率(%)とは、下記式によって算出される値である。

上記のような触媒は、ナフタレンジカルボン酸とエチレングリコールとのエステル化反応を促進するとともに、NDA−DEGの生成を抑制する。
このようにエステル化反応を特定の触媒と、特定割合の水の存在下に行うと、生成した水を除去しながらエステル化する従来の方法と比較して、同一のエステル化率になるまでの時間を大幅に短縮することができ、しかも、NDA−DEGの生成量を少なくすることができる。
次に、前記水およびエステル化によって生成した水を系外に除去しながらさらにエステル化反応〔エステル化反応(2)〕を行う。このエステル化反応(2)は、エステル化によって生成した水を系外に除去しながら行われるめ、系内の水の量は、エチレングリコールに対して通常0.1重量%以下である。
エステル化反応(2)は、反応温度が通常180〜280℃、好ましくは220〜240℃、圧力(ゲージ圧)が通常0〜30kg/cm2、好ましくは1〜15kg/cm2の条件下で行われる。このエステル化反応(2)終了時におけるエステル化率は、通常93%以上、好ましくは97%以上である。
これらのエステル化反応(1)および(2)によりナフタレンジカルボン酸とエチレングリコールとのエステル化物(低次縮合物)が得られ、この低次縮合物の数平均分子量は、通常、500〜2000である。
このようなエステル化反応は、ナフタレンジカルボン酸およびエチレングリコール以外の添加物を添加せずに実施することも可能であり、また後述する重縮合触媒の共存下に実施することも可能であるが、さらに塩基性化合物を少量添加して実施することもできる。
本発明では、特定のエステル化率になるまで特定割合の水の存在下にエステル化反応〔エステル化反応(1)〕を行っているので、生成した水を除去しながらエステル化する従来の方法と比較して、同一のエステル化率になるまでの時間を大幅に短縮することができる。したがって、この後従来の方法と同様に、生成した水を除去しながらエステル化反応〔エステル化反応(2)〕を行ってもエステル化に要する時間を従来より短縮することができる。
また本発明の水の存在下にエステル化反応(1)を行うときのさらなる効果として、エステル化反応の際に副生して、後の重縮合反応によって得られるポリエチレンナフタレートの品質を損なう、下記に示すようなジエチレングリコール骨格を有するナフタレンジカルボン酸のエステル(NDA−DEG)の生成を抑制できる利点が挙げられる。なお、反応開始時から系内に水を存在させるとNDA−DEGの生成を抑制する効果が大きい。
NDA−DEGの色
このようにして得られたエステル化物は、次に重縮合反応が行われる。
重縮合反応は、従来公知の方法で行われ、重縮合触媒の存在下に減圧下で、得られるポリエチレンナフタレートの融点以上の温度に加熱し、この際生成するグリコールを系外に留去させながら重縮合させる。重縮合反応は、反応温度が通常、250〜290℃、好ましくは260〜280℃、圧力が通常、500Torr以下、好ましくは200Torr以下の条件下で行われる。
上記のような液相重縮合反応は、重縮合触媒の存在下、必要に応じて安定剤の共存下に行われる。
重縮合触媒としては、二酸化ゲルマニウム、ゲルマニウムテトラエトキシド、ゲルマニウムテトラn−ブトキシドなどのゲルマニウム化合物、三酸化アンチモンなどのアンチモン触媒またはチタニウムテトラブトキシドなどのチタン触媒を用いることができる。重縮合触媒は、ナフタレンジカルボン酸とエチレングリコールとの合計重量に対して、重縮合触媒中の金属重量換算で、0.0005〜0.2重量%、好ましくは0.001〜0.05重量%の割合で用いられることが望ましい。
次に、本発明に係るナフタレンジカルボン酸のエステル化反応生成物混合液およびその製造方法、ナフタレンジカルボン酸のエステル化反応生成混合物およびその製造方法、ならびに該混合物を用いるポリエチレンナフタレートの製造方法について説明する。
本発明に係るナフタレンジカルボン酸のエステル化反応生成物混合液は、反応開始時から系内に水を存在させながら、ナフタレンジカルボン酸とエチレングリコールとをエステル化反応させることにより得られる。
このナフタレンジカルボン酸とエチレングリコールとのエステル化反応は、エチレングリコールに対して通常0.03〜1.5倍重量、好ましくは0.1〜0.7倍重量の水の存在下で行われる。そして、エチレングリコールはナフタレンジカルボン酸1モルに対して通常0.6〜30モル、好ましくは1.2〜15モルの量で用いられる。本発明では、通常ナフタレンジカルボン酸、エチレングリコールおよび水を、上記の比率になるように混合しエステル化反応させる。
ナフタレンジカルボン酸とエチレングリコールとのエステル化反応により水が生成し、エステル化反応の進行にともなって系内の水の量が増加するが、水を一部留去して系内水の量を特定の範囲内に調整しながらエステル化反応を行うこともできる。
エステル化反応は、反応温度180〜280℃、好ましくは200〜260℃、圧力0〜30kg/cm2、好ましくは0.5〜20kg/cm2の条件下で行われる。また反応時間は、反応条件にもよるが通常0.2〜6時間、好ましくは0.5〜3時間である。
このエステル化反応終了時におけるエステル化率は、反応系に存在する水の割合にもよるが通常40〜95%、好ましくは45〜80%の範囲にある。エステル化率が80%を超えると、前記のようなジエチレングリコール骨格を有するナフタレンジカルボン酸のエステル(NDA−DEG)の含有割合が増加するため、ポリエチレンナフタレートを製造したときに得られるポリエチレンナフタレートの外観が低下することがある。
このようにして得られたナフタレンジカルボン酸のエステル化反応生成物混合液は、ナフタレンジカルボン酸、カルボキシル−ヒドロキシエトキシカルボニルナフタレンおよびビス(ヒドロキシエトキシカルボニル)ナフタレンを含有している。そしてこのナフタレンジカルボン酸の反応生成物混合液は、NDA−DEGの含有割合が低く、混合液中の全ナフタレンジカルボン酸成分に対して、通常3モル%以下、好ましくは1モル%以下、特に好ましくは0.5モル%以下である。なお、全ナフタレンジカルボン酸成分とは、エステル化していないナフタレンジカルボン酸とエステル化したナフタレンジカルボン酸との合計量を意味する。
このようにエステル化反応を、反応開始時から特定割合の水の存在下に行うと、生成した水を除去しながらエステル化する方法と比較して、NDA−DEGの副生を抑えることができる。
本発明では、前記エステル化反応を特定の触媒の共存下に行うことができ、このような触媒としては、硝酸塩、カルボン酸塩、リン酸塩、リン水素酸塩およびアミンから選ばれる少なくとも1種の触媒が挙げられ、より具体的には、硝酸コバルト、酢酸マンガン、リン酸二水素ナトリウムおよびトリエチルアミンから選ばれる少なくとも1種の触媒が挙げられる。前記触媒はナフタレンジカルボン酸とエチレングリコールとの合計重量に対して、10〜0.001重量%、好ましくは0.1〜0.01重量%の割合で用いられる。上記のような触媒は、ナフタレンジカルボン酸とエチレングリコールとのエステル化反応を促進するとともに、NDA−DEGの生成を抑制する。前記のような触媒の共存下に行うエステル化反応におけるナフタレンジカルボン酸、エチレングリコールおよび水の量、反応温度、反応時間、エステル化率等の反応条件は、前記と同様である。
このようにエステル化反応を、反応開始時から特定割合の水の存在下、特定の触媒の共存下に行うと、NDA−DEGの副生をさらに抑えることができる。
本発明に係るナフタレンジカルボン酸のエステル化反応生成混合物は、前記ナフタレンジカルボン酸のエステル化反応生成物混合液から結晶を分離することにより得られる。ナフタレンジカルボン酸のエステル化反応生成混合物を前記反応温度より冷却することにより、ナフタレンジカルボン酸、ナフタレンジカルボン酸エステルを含む混合物の結晶が析出する。
このようにして得られたナフタレンジカルボン酸のエステル化反応生成混合物は、ナフタレンジカルボン酸、カルボキシル−ヒドロキシエトキシカルボニルナフタレンおよびビス(ヒドロキシエトキシカルボニル)ナフタレンを含有している。
このナフタレンジカルボン酸のエステル化反応生成混合物は、前記のようなジエチレングリコール骨格を有するナフタレンジカルボン酸のエステル(NDA−DEG)の含有割合が低く、混合物中の全ナフタレンジカルボン酸成分に対して、通常3モル%以下、好ましくは1モル%以下、特に好ましくは0.5モル%以下である。
このナフタレンジカルボン酸のエステル化反応生成混合物は、NDA−DEGなどのポリエチレンナフタレートの品質を損なう成分の含有割合が少ない。このためこのようなナフタレンジカルボン酸のエステル化物混合物を用いてポリエチレンナフタレートを製造すると品質に優れたポリエチレンナフタレートを製造することができる。
本発明に係る第2のポリエチレンナフタレートの製造方法は、前記ナフタレンジカルボン酸のエステル化反応生成混合物に、必要に応じてエチレングリコールを加えて重縮合反応を行いポリエチレンナフタレートを製造する。
重縮合反応は、従来公知の方法で行われ、重縮合反応の条件は、前記の第1のポリエチレンナフタレートの製造方法と同様である。
発明の効果
本発明に係る第1のポリエチレンナフタレートの製造方法は、ナフタレンジカルボン酸とエチレングリコールとのエステル化の一部を特定量の水の存在下、必要に応じて特定の触媒の共存下で行っているので、エステル化反応が完結する時間が従来より短い。したがって従来より短時間でポリエチレンナフタレートを製造することができる。また、この方法によれば、NDA−DEG等の不純物の生成が従来より少ない。
本発明に係るナフタレンジカルボン酸のエステル化反応生成物混合液の製造方法は、反応開始時から系内に水を存在させながら、必要に応じて特定の触媒の共存下に、ナフタレンジカルボン酸とエチレングリコールとをエステル化しているので、ジエチレングリコール骨格を有するナフタレンジカルボン酸のエステル(NDA−DEG)などのポリエチレンナフタレートの品質を損なう成分の含有割合が少ないナフタレンジカルボン酸のエステル化反応生成物混合液が得られる。このようなナフタレンジカルボン酸のエステル化反応生成物混合液から品質に優れたポリエチレンナフタレートを製造することができる。
本発明に係るナフタレンジカルボン酸のエステル化反応生成混合物の製造方法は、前記ナフタレンジカルボン酸のエステル化反応生成物混合液から、結晶を分離しているので、NDA−DEGなどのポリエチレンナフタレートの品質を損なう成分の含有割合が少ないナフタレンジカルボン酸のエステル化反応生成混合物が得られる。このようなナフタレンジカルボン酸のエステル化反応生成混合物から品質に優れたポリエチレンナフタレートを製造することができる。
本発明に係る第2のポリエチレンナフタレートの製造方法は、前記のようなナフタレンジカルボン酸のエステル化物混合物を用いてポリエチレンナフタレートを製造するので、品質に優れたポリエチレンナフタレートを製造することができる。
実施例
以下、実施例に基づいて本発明をさらに具体的に説明するが、本発明はこれら実施例に限定されるものではない。
参考例1
エステル化反応(1)
2,6−ナフタレンジカルボン酸(以下「NDA」と記載する)を40g、エチレングリコール(以下「EG」と記載する)を120g、水を80g(EGに対して0.67倍重量)を500mlのオートクレーブに仕込み、窒素置換(10kg/cm2加圧)し、250℃で2時間反応させ、NDAのエステル化反応生成物混合液を得た。
その後、オートクレーブを冷却し、析出した結晶を液体とを分離し、44gの結晶(NDAのエステル化反応生成混合物)を回収した。この回収した結晶の組成を下記に示す。
NDA:23重量%
NDAモノエステル:46重量%
(2−カルボキシル−6−ヒドロキシエトキシカルボニルナフタレン)
NDAジエステル:25重量%
(2,6−ビス(ヒドロキシエトキシカルボニル)ナフタレン)
NDAジエステルのオリゴマー:6重量%
エステル化率:54%
エステル化反応(2)
次に上記の回収した結晶の全量(44g)と150gのEGを簡単な蒸留装置を備えたガラスフラスコに仕込み、80℃のオイルバスに浸し、30分間かけて225℃まで昇温した。この時、蒸留塔上部より留出する水を回収し、さらにエステル化の進行により生成する水が留出しなくなるまで加熱を続けたところ、80℃から加熱し始めてから6時間で水が留出しなくなった。この水が留出しなくなった時をエステル化反応の終点とした。
エステル化(エステル化工程(1)に要した時間+エステル化工程(2)に要した時間)に要した時間は8時間であった。
参考例2
エステル化反応(1)
NDAを40g、EGを160g、水を40g(EGに対して0.25倍重量)を500mlのオートクレーブに仕込み、窒素置換(10kg/cm2加圧)し、250℃で2時間反応させ、NDAのエステル化反応生成物混合液を得た。その後、オートクレーブを冷却し、析出した結晶と液体とを分離し、50gの結晶(NDAのエステル化反応生成混合物)を回収した。この回収した結晶の組成を下記に示す。
NDA:14重量%
NDAモノエステル:39重量%
NDAジエステル:28重量%
NDAジエステルのオリゴマー:19重量%
エステル化率:67%
エステル化反応(2)
次に上記の回収した結晶の全量(50g)と150gのEGを簡単な蒸留装置を備えたガラスフラスコに仕込み、80℃のオイルバスに浸し、30分間かけて225℃まで昇温した。この時、蒸留塔上部より留出する水を回収し、さらにエステル化の進行により生成する水が留出しなくなるまで加熱を続けたところ、80℃から加熱し始めてから4.5時間で水が留出しなくなった。この水が留出しなくなった時をエステル化反応の終点とした。エステル化に要した時間は6.5時間であった。
参考比較例1
NDAを40g、EGを160gを簡単な蒸留装置を備えたガラスフラスコに仕込み、80℃のオイルバスに浸し、30分間かけて225℃まで昇温した。この時、蒸留塔上部より留出する水を回収し、さらにエステル化の進行により生成する水が留出しなくなるまで加熱を続けたところ、80℃から加熱し始めてから35時間で水が留出しなくなった。この水が留出しなくなった時をエステル化反応の終点とした。エステル化に要した時間は35時間であった。
参考比較例2
NDAを40g、EGを160gを簡単な蒸留装置を備えたオートクレーブに仕込み、250℃まで昇温し、圧力を1.7kg/cm2に保ち、生成する水を蒸留塔より留出させながらエステル化反応を行った。加熱し始めてから15時間で水が留出しなくなった。この水が留出しなくなった時をエステル化反応の終点とした。エステル化に要した時間は15時間であった。
前記参考例1、2および参考比較例1、2における、反応時間とエステル化率との関係を図1に示す。図中の参考例1、2において前半部分はエステル化反応(1)、後半部分はエステル化反応(2)に対応している。
図1に示すように、本発明の方法である参考例1、2では、エステル化反応(2)の反応速度は参考比較例2とほとんど同じであるが、エステル化反応(1)の反応速度が速いため、全体としてエステル化速度が速い。
参考例3
NDA 40g、EG 120g、水 80g(EGに対する水の量は0.67倍重量)を500mlオートクレーブに仕込み、窒素置換(10kg/cm2加圧)し、加熱したオイルバスに浸し250℃に昇温し、250℃に到達した時から1,2,3,4時間後に反応溶液を取り出した。反応液を冷却後、反応液中に含まれるNDA−DEGを液クロマトグラフで定量した。それぞれの時間におけるNDA−DEG含量およびエステル化率を調べた結果を表1に示す。
反応終了後、オートクレーブを25℃まで冷却し、析出した結晶と液体とを分離して結晶を回収した。
参考例3において、EGを160g、水を40g(EGに対する水の量は0.25倍重量)とした以外は、参考例3と同様にして反応を行い、それぞれの時間におけるNDA−DEG含量およびエステル化率を測定し表2に示した。
反応終了後、オートクレーブを25℃まで冷却し、析出した結晶と液体とを分離して結晶を回収した。
NDA 100g、EG 320g、水 180g(EGに対して0.56倍重量)を1000mlのオートクレーブに仕込み、窒素置換(10kg/cm2加圧)し、240℃で3時間反応させ、NDAのエステル化反応生成物混合液を得た。
その後、反応溶液を取り出し1000mlの蒸留水を加え結晶を濾過回収し、乾燥して、NDAのエステル化反応生成混合物を129g回収した。この回収した結晶(NDAのエステル化反応生成混合物)の組成を下記に示す。
NDA:21.2モル%
NDAモノエステル:48.2モル%
NDAジエステル:28.9モル%
NDA−DEG:0.3モル%
NDAジエステルのオリゴマー:1.4モル%
エステル化率:54.7%
なお濾液中にNDA−DEGを0.7モル%検出した。
上記の操作を2回繰り返し、NDAのエステル化反応生成混合物を240g調製した。
このNDAのエステル化反応生成混合物を簡単な蒸留装置を備えたガラスフラスコに仕込み、80℃のオイルバスに浸し、30分間かけて225℃まで昇温し、その温度で加熱を続けた。この時、蒸留塔上部より留出する水を回収した。エステル化の進行により生成する水が留出しなくなった時をエステル化の終了とした。
前記エステル化反応により得られたNDAのエステル化物重合触媒として二酸化ゲルマニウムを21mg、安定剤としてテトラエチルアンモニウムヒドロキシド15mg、リン酸39mgを5gのEGに溶解させ添加した。この重合触媒と安定剤とを添加したNDAのエステル化物を260℃に昇温し、留出するEGを回収しながら1時間攪拌した。次に、1時間かけて280℃に昇温しながら反応系を1Torr以下にし、EGを留出させた。さらに280℃、1Torr以下の減圧下でEGを留出させながら1.5時間反応を続けた。その後、反応を停止し、生成したポリエチレンナフタレートを回収した。
回収したポリエチレンナフタレートの固有粘度は0.55d1/g(1:1のo−クロロフェノール/フェノール溶液に溶かし25℃で測定した)であり、示差走査型熱量計で測定したガラス転移温度(Tg)は118℃、溶融温度(Tm)は268℃であり、ポリエチレンナフタレート中のNDA−DEG含量は0.94重量%であった。
参考比較例3
水を仕込まないで、NDA 40g、EG 200gを500mlオートクレーブに仕込み、窒素置換(10kg/cm2加圧)し、加熱したオイルバスに浸し250℃に昇温し、250℃に到達した時から20分、40分および1,2時間後に反応溶液を取り出した。なお、該エステル化反応では、反応中生成した水を系外に除去しないで反応を行った。反応液を冷却後、反応液中に含まれる副生成物であるNDA−DEGを液クロマトグラフで定量した。それぞれの反応時間におけるNDA−DEG含量およびエステル化率を表3に示した。
反応終了後、オートクレーブを25℃まで冷却し、析出した結晶と液体とを分離して結晶を回収した。
図2に示すように、エステル化反応の初期から水を存在させてエステル化を行う本発明の方法では、エステル化率が増加しても、NDA−DEG含量の増加は少ない。これに対して、参考比較例3のように水を仕込まないでエステル化反応を行うと、エステル化反応中に生成する水を系外に除去しないで反応を行っても系内の生成水による水の量が少ないのでNDA−DEGの副生量が多くなる。
実施例2
NDA 100g、EG 220g、水 40g(EGに対して0.18倍重量)を1000mlのオートクレーブに仕込み、窒素置換(10kg/cm2加圧)し、250℃で3時間反応させた。得られた反応溶液を冷却後、オートクレーブから取り出し、遠心分離器を用い固液分離して、エチレングリコール水溶液45重量%とNDAのエステル化反応生成混合物55重量%とからなるエステル化反応生成混合物がエチレングリコール水溶液で湿っている反応生成物混合体を212g回収した。
この反応生成物混合体を液体クロマトグラフィーで分析したところ、エステル化率は55.6%であった。また、この反応生成物混合体をガスクロマトグラフィーで分析したところ、NDA−DEG中のジエチレングリコール単位をジエチレングリコールに換算したものと、ジエチレングリコールとの合計含量は0.46重量%であった。
上記の操作を2回繰り返し、反応生成物混合体を420g調製した。
この反応生成物混合体を簡単な蒸留装置を備えたガラスフラスコに仕込み、80℃のオイルバスに浸し、30分間かけて225℃まで昇温し、その温度で加熱を続けた。この時、蒸留塔上部より留出する水を回収した。エステル化の進行により生成する水が留出しなくなった時をエステル化の終了とした。
前記エステル化反応により得られたNDAのエステル化物に、重合触媒として二酸化ゲルマニウムを21mg、安定剤としてテトラエチルアンモニウムヒドロキシド15mg、リン酸39mgを5gのEGに溶解させ添加した。この重合触媒と安定剤とを添加したNDAのエステル化物を260℃に昇温し、留出するEGを回収しながら1時間攪拌した。次に、1時間かけて280℃に昇温しながら反応系を1Torr以下にし、EGを留出させた。さらに280℃、1Torr以下の減圧下でEGを留出させながら1.5時間反応を続けた。その後、反応を停止し、生成したポリエチレンナフタレートを回収した。
回収したポリエチレンナフタレートの固有粘度は0.55d1/g(1:1のo−クロロフェノール/フェノール溶液に溶かし25℃で測定した)であり、示差走査型熱量計で測定したガラス転移温度(Tg)は118℃、溶融温度(Tm)は267℃であり、ポリエチレンナフタレート中のジエチレングリコール単位含量は1.6重量%であった。
参考例5
NDA 2g、EG 6.5g、水 3.5g(EGに対して0.54倍重量)、リン酸二水素ナトリウム 0.5gを50mlのオートクレーブに仕込み、窒素置換(10kg/cm2加圧)し、250℃で3時間反応させ、NDAのエステル化反応生成物混合液を得た。
その後、前記混合液を取り出し100mlの蒸留水を加え結晶を濾過回収し、乾燥して、NDAのエステル化反応生成混合物を2.2g得た。このNDAのエステル化反応生成混合物の組成およびNDA−DEGを液体クロマトグラフィーで分析した結果を表4に示す。
参考例6
参考例5において、リン酸二水素ナトリウムを硝酸コバルトに代えた以外は、参考例5と同様にしてNDAのエステル化反応生成物混合液およびNDAのエステル化反応生成混合物を得た。結果を表4に示す。
参考例7
参考例5において、リン酸二水素ナトリウムを酢酸マンガンに代えた以外は、参考例5と同様にしてNDAのエステル化反応生成物混合液およびNDAのエステル化反応生成混合物を得た。結果を表4に示す。
参考例8
参考例5において、リン酸二水素ナトリウムをトリエチルアミンに代えた以外は、参考例5と同様にしてNDAのエステル化反応生成物混合液およびNDAのエステル化反応生成混合物を得た。結果を表4に示す。
参考例9
参考例5において、リン酸二水素ナトリウムを用いなかった以外は、実施例7と同様にしてNDAのエステル化反応生成物混合液およびNDAのエステル化反応生成混合物を得た。結果を表4に示す。
参考例5において、リン酸二水素ナトリウムの添加量を0.25gとした以外は、参考例5と同様にしてNDAのエステル化反応生成物混合液およびNDAのエステル化反応生成混合物を得た。結果を表5に示す。
参考例11
参考例5において、リン酸二水素ナトリウムの添加量を0.05gとした以外は、参考例5と同様にしてNDAのエステル化反応生成物混合液およびNDAのエステル化反応生成混合物を得た。結果を表5に示す。
NDA 100g、EG 320g、水 180g(EGに対して0.56倍重量)、リン酸二水素ナトリウム、1.0gを1000mlのオートクレーブに仕込み、窒素置換(10kg/cm2加圧)し、250℃で3時間反応させた。その後、反応溶液を取り出し1000mlの蒸留水を加え結晶を濾過回収し、乾燥してNDAのエステル化反応生成混合物を130g回収した。この回収した結晶(NDAのエステル化反応生成混合物)の組成およびNDA−DEGを液体クロマトグラフィーで分析した結果、エステル化率は55.2%であり、NDA−DEGは0.3モル%であった。
上記の操作を2回繰り返し、NDAのエステル化反応生成混合物を240g調製した。
このNDAのエステル化反応生成混合物を簡単な蒸留装置を備えたガラスフラスコに仕込み、80℃のオイルバスに浸し、30分間かけて225℃まで昇温し、その温度で加熱を続けた。この時、蒸留塔上部より留出する水を回収した。エステル化の進行により生成する水が留出しなくなった時をエステル化の終了とした。
前記エステル化反応により得られたNDAのエステル化物に重合触媒として二酸化ゲルマニウムを21mg、安定剤としてテトラエチルアンモニウムヒドロキシド 15mg、リン酸 39mgを5gのEGに溶解させ添加した。この重合触媒と安定剤とを添加したNDAのエステル化物を260℃に昇温し、留出するEGを回収しながら1時間攪拌した。次に、1時間かけて280℃に昇温しながら反応系を1Torr以下にし、EGを留出させた。さらに280℃、1Torr以下の減圧下でEGを留出させながら1.5時間反応を続けた。その後、反応を停止し、生成したポリエチレンナフタレートを回収した。
回収したポリエチレンナフタレートの固有粘度は0.55d1/g(1:1のo−クロロフェノール/フェノール溶液に溶かし25℃で測定した)であり、示差走査型熱量計で測定したガラス転移温度(Tg)は118℃、溶融温度(Tm)は268℃であり、ポリエチレンナフタレート中のDEG含量は0.94重量%であった。
Claims (6)
- 水の存在下でナフタレンジカルボン酸とエチレングリコールとをエステル化反応させた後、水を除去しながらさらにエステル化反応を行いナフタレンジカルボン酸とエチレングリコールとのエステル化物を調製し、次いでこのエステル化物の重縮合反応を行うことを特徴とするポリエチレンナフタレートの製造方法。
- 前記水の存在下で行うエステル化反応を、硝酸塩、カルボン酸塩、リン酸塩、リン水素酸塩およびアミンから選ばれる少なくとも1種の触媒の共存下で行う請求の範囲第1項に記載のポリエチレンナフタレートの製造方法。
- 前記触媒は、硝酸コバルト、酢酸マンガン、リン酸二水素ナトリウムおよびトリエチルアミンから選ばれる少なくとも1種の触媒である請求の範囲第2項に記載のポリエチレンナフタレートの製造方法。
- 前記水の存在下で行うエステル化反応を、エチレングリコールに対して0.03〜1.5倍重量の水の存在下で行う請求の範囲第1〜3項のいずれかに記載のポリエチレンナフタレートの製造方法。
- 前記水の存在下で行うエステル化反応を、エチレングリコールに対して0.1〜0.7倍重量の水の存在下で行う請求の範囲第1〜3項のいずれかに記載のポリエチレンナフタレートの製造方法。
- 水の存在下でナフタレンジカルボン酸とエチレングリコールとをエステル化反応させた後のエステル化率が40〜95%である請求の範囲第1〜5項のいずれかに記載のポリエチレンナフタレートの製造方法。
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP29310295 | 1995-11-10 | ||
| JP7/293102 | 1995-11-10 | ||
| JP23662596 | 1996-09-06 | ||
| JP8/236626 | 1996-09-06 | ||
| JP8/236625 | 1996-09-06 | ||
| JP23662696 | 1996-09-06 | ||
| PCT/JP1996/003116 WO1997017391A1 (en) | 1995-11-10 | 1996-10-25 | Process for producing polyethylene naphthalate |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004298965A Division JP3720040B2 (ja) | 1995-11-10 | 2004-10-13 | ナフタレンジカルボン酸のエステル化反応生成物混合液または混合物およびこれらの製造方法、ならびにポリエチレンナフタレートの製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH10511138A JPH10511138A (ja) | 1998-10-27 |
| JP3628708B2 true JP3628708B2 (ja) | 2005-03-16 |
Family
ID=27332386
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP51804897A Expired - Lifetime JP3628708B2 (ja) | 1995-11-10 | 1996-10-25 | ポリエチレンナフタレートの製造方法 |
| JP2004298965A Expired - Fee Related JP3720040B2 (ja) | 1995-11-10 | 2004-10-13 | ナフタレンジカルボン酸のエステル化反応生成物混合液または混合物およびこれらの製造方法、ならびにポリエチレンナフタレートの製造方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004298965A Expired - Fee Related JP3720040B2 (ja) | 1995-11-10 | 2004-10-13 | ナフタレンジカルボン酸のエステル化反応生成物混合液または混合物およびこれらの製造方法、ならびにポリエチレンナフタレートの製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US5811513A (ja) |
| EP (1) | EP0802938B1 (ja) |
| JP (2) | JP3628708B2 (ja) |
| KR (1) | KR100231406B1 (ja) |
| CA (1) | CA2209994A1 (ja) |
| DE (1) | DE69625466T2 (ja) |
| WO (1) | WO1997017391A1 (ja) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR100286251B1 (ko) * | 1999-02-24 | 2001-03-15 | 박호군 | 폴리에틸렌 나프탈레이트계 중합체의 제조 방법 |
| US6300462B1 (en) | 2000-02-18 | 2001-10-09 | Eastman Chemical Company | Process for preparing poly(ethylene-2,6-naphthalene dicarboxylate |
| KR100326660B1 (ko) * | 2000-02-26 | 2002-03-02 | 박호군 | 폴리에틸렌 나프탈레이트계 중합체의 제조 방법 |
| US6551675B2 (en) | 2001-05-14 | 2003-04-22 | Nan Ya Plastics Corporation | Manufacturing method of a copolyester containing ethylene naphthalate unit (EN) and its application |
| US7276625B2 (en) | 2002-10-15 | 2007-10-02 | Eastman Chemical Company | Process for production of a carboxylic acid/diol mixture suitable for use in polyester production |
| US7193109B2 (en) | 2003-03-06 | 2007-03-20 | Eastman Chemical Company | Process for production of a carboxylic acid/diol mixture suitable for use in polyester production |
| KR100663255B1 (ko) * | 2003-08-22 | 2007-01-02 | 주식회사 효성 | 에틸렌글리콜을 사용한 폴리에틸렌나프탈레이트의 고상중합 |
| US7214760B2 (en) * | 2004-01-15 | 2007-05-08 | Eastman Chemical Company | Process for production of a carboxylic acid/diol mixture suitable for use in polyester production |
| JP4890958B2 (ja) * | 2006-06-16 | 2012-03-07 | 帝人デュポンフィルム株式会社 | 二軸配向ポリエステルフィルム |
| KR100939024B1 (ko) * | 2007-11-30 | 2010-01-27 | 주식회사 효성 | 2,6-나프탈렌디카르복실산을 이용한폴리에틸렌나프탈레이트의 제조 방법 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1472777A (en) * | 1974-07-08 | 1977-05-04 | Teijin Ltd | Process for preparing polyethylene-2,6-naphthalate |
| JP2824716B2 (ja) * | 1992-07-22 | 1998-11-18 | 富士写真フイルム株式会社 | ポリエチレンナフタレートの製造方法 |
| US5294695A (en) * | 1993-03-15 | 1994-03-15 | Skc Limited | Process for preparing polyethylene naphthalate |
| US5331082A (en) * | 1993-06-16 | 1994-07-19 | Amoco Corporation | Process for manufacture of high molecular weight polyester resins from 2,6-naphthalene dicarboxylic acid |
-
1996
- 1996-10-25 KR KR1019970704673A patent/KR100231406B1/ko not_active IP Right Cessation
- 1996-10-25 DE DE69625466T patent/DE69625466T2/de not_active Expired - Fee Related
- 1996-10-25 WO PCT/JP1996/003116 patent/WO1997017391A1/en active IP Right Grant
- 1996-10-25 US US08/860,701 patent/US5811513A/en not_active Expired - Fee Related
- 1996-10-25 EP EP96935441A patent/EP0802938B1/en not_active Expired - Lifetime
- 1996-10-25 CA CA002209994A patent/CA2209994A1/en not_active Abandoned
- 1996-10-25 JP JP51804897A patent/JP3628708B2/ja not_active Expired - Lifetime
-
2004
- 2004-10-13 JP JP2004298965A patent/JP3720040B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| US5811513A (en) | 1998-09-22 |
| EP0802938A1 (en) | 1997-10-29 |
| EP0802938B1 (en) | 2002-12-18 |
| DE69625466D1 (de) | 2003-01-30 |
| JP3720040B2 (ja) | 2005-11-24 |
| DE69625466T2 (de) | 2003-10-02 |
| KR100231406B1 (ko) | 1999-11-15 |
| WO1997017391A1 (en) | 1997-05-15 |
| KR19980701284A (ko) | 1998-05-15 |
| JPH10511138A (ja) | 1998-10-27 |
| JP2005133093A (ja) | 2005-05-26 |
| CA2209994A1 (en) | 1997-05-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100517853B1 (ko) | 고분자량 폴리에스테르의 개선된 제조 방법 | |
| JP3628708B2 (ja) | ポリエチレンナフタレートの製造方法 | |
| EP3350246B1 (en) | Process for the preparation of a polyester | |
| US20120123147A1 (en) | Ketocarboxylic acids, methods of manufacture and uses thereof | |
| JP2005133093A6 (ja) | ナフタレンジカルボン酸のエステル化反応生成物混合液または混合物およびこれらの製造方法、ならびにポリエチレンナフタレートの製造方法 | |
| JP2002535461A (ja) | ポリエステルの調製中におけるリサイクルされる1,3−プロパンジオールの精製 | |
| US20060041158A1 (en) | Method for the production of an ester | |
| WO1998011047A1 (fr) | Procede de purification d'acide naphtalenedicarboxylique brut, et procede de preparation de polyethylene naphtalate | |
| TW201245208A (en) | Method for preparing poly(1,4:3,6-dianhydrohexitol esters) | |
| JP4306038B2 (ja) | ポリブチレンテレフタレートの製造法 | |
| JPH01110650A (ja) | 精製された2,6−ナフタレジカルボン酸ビス(2−ヒドロキシエチル)エステルの製造方法 | |
| JP2001288180A (ja) | 精製β−ヒドロキシエトキシ酢酸塩類化合物及びその製造方法、並びに、精製2−p−ジオキサノン及びその製造方法 | |
| JPH10310557A (ja) | ナフタレンジカルボン酸のエチレングリコールエステル混合物の製造方法およびポリエチレンナフタレートの製造方法 | |
| JP3578433B2 (ja) | ポリエチレンナフタレートの製造方法 | |
| JPS63107947A (ja) | 複合エステルの製造方法 | |
| JP2020528467A (ja) | 添加剤を用いてポリエステルを調製するための方法 | |
| KR102576090B1 (ko) | 비스-하이드록시알킬렌 디카르복실레이트를 생산하기 위한 공정 | |
| JP3553404B2 (ja) | 大環状エステルまたはラクトンの製造方法 | |
| SU899582A1 (ru) | Способ получени полиэфиров | |
| CN118234714A (zh) | 呋喃二羧酸类化合物、呋喃二羧酸类化合物的制备方法、聚酯以及聚酯的制备方法 | |
| JP3775953B2 (ja) | 芳香族ジカルボン酸ジアリールエステルの製造方法 | |
| JPH0621114B2 (ja) | フタル酸ジフェニルの精製方法 | |
| JPH0710981A (ja) | ポリブチレンテレフタレートの製造方法 | |
| JP3725974B2 (ja) | ポリエステル製造用触媒溶液の製造方法 | |
| JPH0372653B2 (ja) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20040817 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20041013 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20041130 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20041209 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20081217 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20091217 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20101217 Year of fee payment: 6 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20111217 Year of fee payment: 7 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20111217 Year of fee payment: 7 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20121217 Year of fee payment: 8 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20121217 Year of fee payment: 8 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131217 Year of fee payment: 9 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |